
Cáncer Infantilcampus.usal.es/~ogyp/Clases teoricas 2012 2013...Las leucemias agudas son...
80
Cáncer Infantil Dra. M. Muriel
Transcript of Cáncer Infantilcampus.usal.es/~ogyp/Clases teoricas 2012 2013...Las leucemias agudas son...

Cáncer Infantil
Dra. M. Muriel

Cáncer infantil
El cáncer infantil es la 2ª causa de muerte, tras los accidentes, en los niños mayores de 1 año y la tercera en los menores de 1 año tras las malformaciones congénitas y los accidentes.

Incidencia de Cáncer en los niños
145 casos /millón de niños /año Se diagnostican 1.350 casos nuevos cada
año en España en niños y adolescentes 57% de los afectados son varones y 43%
mujeres Tendencia a aumentar: 1% anual Supervivencia global 75% frente al 60% que
ocurre en los adultos Uno de cada 1.000 jóvenes que alcance 20
años será superviviente de cáncer infantil
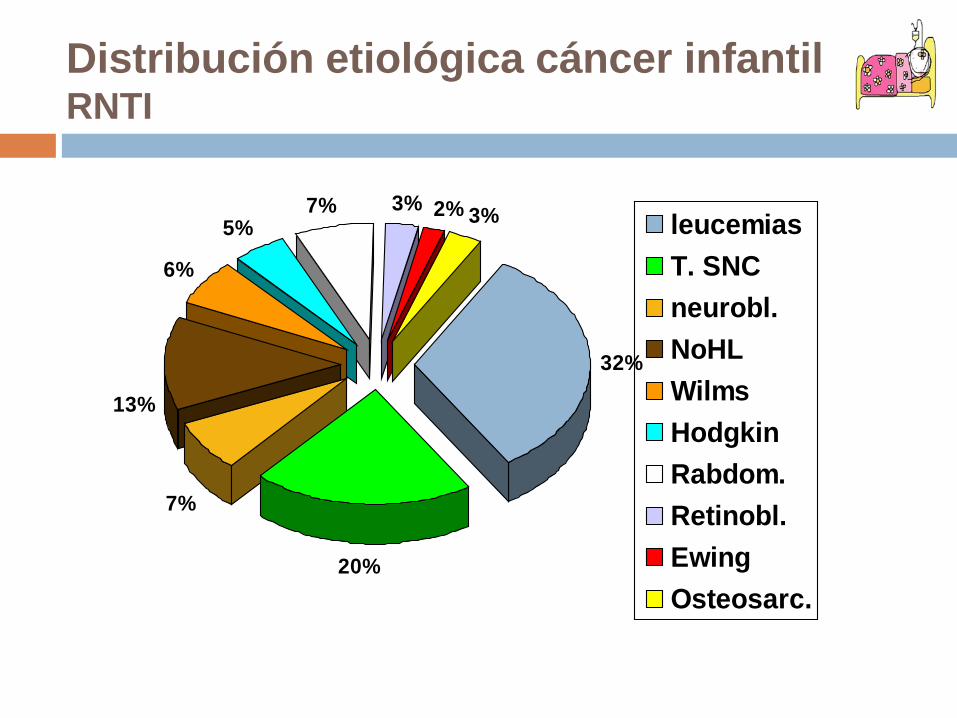
Distribución etiológica cáncer infantilRNTI
32%
20%
7%
13%
6%
5%7% 3% 2% 3% leucemias
T. SNCneurobl.NoHLWilmsHodgkinRabdom.Retinobl.EwingOsteosarc.

LEUCEMIAS EN LA INFANCIA

Leucemias Agudas en la InfanciaConcepto
Las leucemias agudas son proliferaciones clonalesmalignas (blastos) de las células hematopoyéticas en distintos grados de diferenciación.
Las células leucémicas infiltran medula ósea con desplazamiento de células normales → síntomas
(anemia, trombo y neutropenia)
Infiltración secundaria de órganos → síntomasextramedulares

Leucemias Agudas en la InfanciaClasificación
LEUCEMIAAGUDA (LA)
32% T.infantiles
LINFOIDE (LLA)80%
MIELOIDE(LMA)20%

Leucemia Linfoblástica AgudaConcepto y epidemiologia
EPIDEMIOLOGIA La leucemia más frecuente en la infancia
(80% de todas las leucemias agudas) Pico de incidencia entre los 3 - 5 años Mas frecuente raza blanca Incidencia parece ir en aumento Supervivencia global 75%
Proliferación clonal de célula linfoide inmadura

Leucemia Linfoblastica AgudaClasificación
MORFOLOGICA (FAB)Aspectos de los blastos al M. opticoINMUNOLOGICAInmunofenotipo de blastos por citometria de
flujoCITOGENETICAAnomalias cromosómicas y moleculares de
los blastos

Leucemia Linfoblastica AgudaClasificación Morfológica (FAB)
Linfoblástos pleomórficos 13%
Linfoblástos pequeños poco dismórficos. 85%
Linfoblástos grandes, nucleos prominente. Tipo Burkitt. 2%

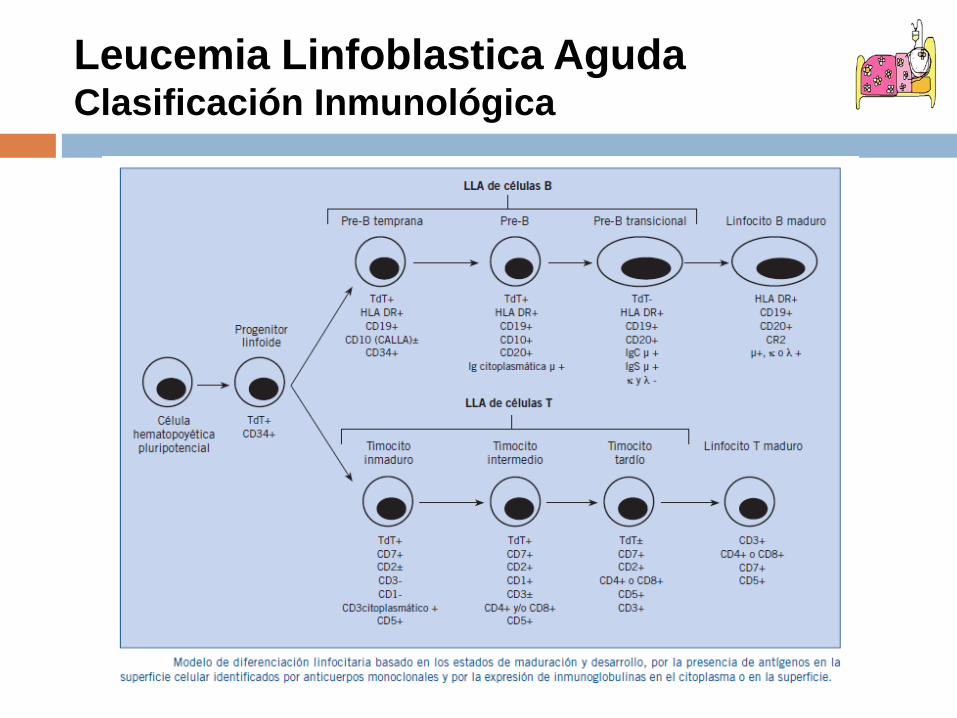
Leucemia Linfoblastica AgudaClasificación Inmunológica
•Es la clasificación mas utilizada•Implicaciones pronósticas y terapéuticas•Identifica antígenos en membrana o citoplasma celular mediante Ac Monoclonales•Define estirpes celulares•Establece subgrupos •Define poblaciones
Leucemia Linfoblastica AgudaClasificación Inmunológica

Leucemia Linfoblastica AgudaClasificación Inmunológica (EGIL)
LLA células B: CD19+/CD22+ProB: CD10-Común: CD10+PreB: Cadenas µ citoplasmáticas B madura: Inmunoglobulinas de
superficieLLA células T: CD3 citoplasma+ProT: CD7+, CD2-, CD5-, CD8-, CD1a-Pre T: CD7+ y/o CD5+ y/o CD8+,
CD1a-T cortical: CD1a+, CD3 membrana+/-T madura: CD3 superficie +, CD1a-

Leucemia Linfoblastica AgudaClasificación Citogenética
Traslocaciones mas frecuentes en LLA
Traslocacion Genes afectados
t(1:19)t(9:22)t(4:11)t(12:21)
E2A-PBX1BCR-ABLMLL-AF4TEL-AML
Basada en alteraciones cromosómicas de los linfoblástos.
Alteraciones numéricas:Hiperdiploidias > 50 cromosomas (buen pronóstico)Hipodiploidias < 50 cromosomas (prnóstico desfavorable)
Alteraciones estructurales:

LLAPro B
LLA Bcomún
LLA
Pre-B
LLA
B
LLA
T
Tdt + + + - +
Ia + + + + -
CD19 + + + + --
CD 10 - + + +/- -
cIg - - + +/- -
Alt. cromosomica
t(1,19) t(8,14)t(2,28)
t(8,22)
t(11,14)
Frecuencia 5% Niño (60%)
15% 3% (15%)
Leucemia Linfoblastica AgudaClasificación

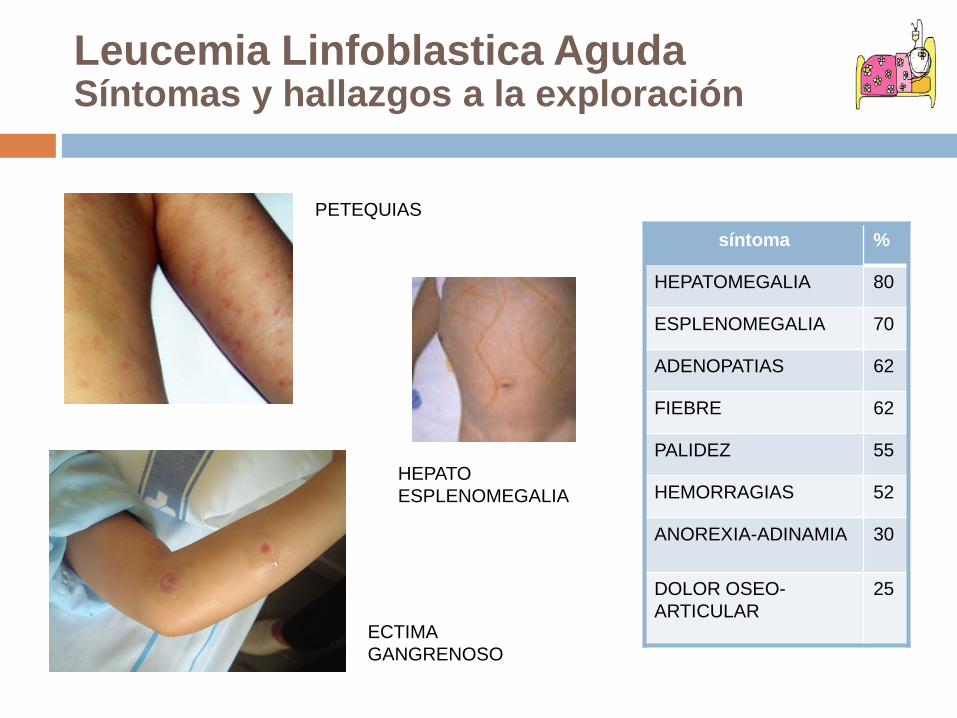
Leucemia Linfoblastica AgudaSíntomas y hallazgos a la exploración
S. anémicoPalidez, cansancio, anorexia S. InfecciosoFiebre, infección tórpidaS hemorrágicoPetequias y hemorragiasInfiltración tumoralHepatomegalia, esplenomegalia, adenopatías, masa mediastínica, afectación testicular,
afectación meníngeaDolores articulares
Leucemia Linfoblastica AgudaSíntomas y hallazgos a la exploración
HEPATOESPLENOMEGALIA
PETEQUIAS
ECTIMA GANGRENOSO
síntoma %
HEPATOMEGALIA 80
ESPLENOMEGALIA 70
ADENOPATIAS 62
FIEBRE 62
PALIDEZ 55
HEMORRAGIAS 52
ANOREXIA-ADINAMIA 30
DOLOR OSEO-ARTICULAR
25

Leucemia Linfoblastica AgudaCélulas T
CARACTERISTICAS:•Linfoadenopatias•Esplenomegalia masiva•Masa mediastÍnica anterior•Afectación SNC•Hiperleucocitosis > 100.000•Riesgo lisis tumoral (>LDH,K,P y ac.úrico) Radiografía tórax:
Niño afecto LLA células T

Traslocación Genes afectados
Caracteristicas
t(1:19)
t(9:22)
t(4:11)
t(12:21)
E2A-PBX1
BCR-ABL
MLL-AF4
TEL-AML
Fenotipo pre-B hiperleucocitosis. Necesario tratamiento intensivoCromosoma Philadelfia.Tratamiento imatinib. HiperleucocitosisEstirpe B. Asociada a LLA lactante.Hiperleucocitosis. Pronóstico pobre Fenotipo B. Buen pronóstico.Marcada sensibilidad a la asparraginasa
Leucemia Linfoblastica AgudaCorrelación clínico-inmuno-citogenética

Leucemia Linfoblastica AgudaDiagnóstico
Analítica básica Hemograma (anemia, leucocitosis/leucopenia, trombopenia Examen morfológico celular (frotis /blastos sangre periférica) Bioquímica básica (↑LDH, ↑uratos, fosforo, ↑calcio) Función hepática Estudio coagulación Estudio morfológico /citométrico LCRAspirado médula ósea (infiltración blástica > 20 %) Citomorfologia (FAB) Citometria de flujo (fenotipo inmunológico) Citogenética- Biología MolecularEstudios de imagen Radiografía tórax: Ecografía abdominal; Ecografía testicular Ecocardiografía

Leucemia Linfoblastica AgudaGrupos de Riesgo
Riesgoestandar
Edad: entre 1-9 añosInmunofenotipo Común: CD19+, CD10+Leucocitos<20x10⁹/LAusencia a.citogenética desfavorablePresencia <5% blastos M.O/dia+14 ttoEMR<0.1% fin tto inducción
Alto riesgo Edad: > 10 añosInmunofenotipo cualquiera que no sea el comúnLeucocitos 20-200 x 10⁹/LCitogenética desfavorableAfectación extramedular (SNC o testes)Riesgo estándar >5% blastos M.O/dia+14 ttoRiesgo estándar EMR >0.1% fin tto inducción
Muy altoriesgo
Leucocitos > 200 x 10⁹/LPresencia de t(9,22) o BCR/ABLPresencia de t(4,11) o MLLAploidia (24/29 cromosomas)Alto riesgo con > 5% blastos en médula ósea /dia+14/+21Alto riesgo con EMR > 0.1% fin tto. consolidación
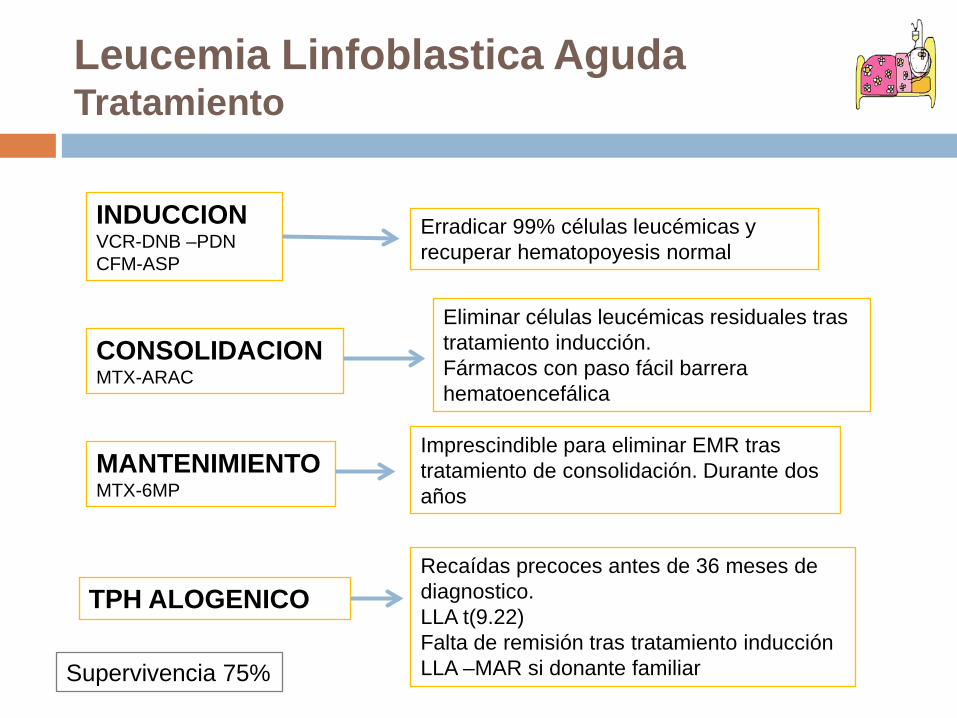
Leucemia Linfoblastica AgudaTratamiento
INDUCCIONVCR-DNB –PDNCFM-ASP
CONSOLIDACIONMTX-ARAC
MANTENIMIENTOMTX-6MP
TPH ALOGENICO
Erradicar 99% células leucémicas y recuperar hematopoyesis normal
Eliminar células leucémicas residuales tras tratamiento inducción.Fármacos con paso fácil barrera hematoencefálica
Imprescindible para eliminar EMR tras tratamiento de consolidación. Durante dos años
Recaídas precoces antes de 36 meses de diagnostico. LLA t(9.22)Falta de remisión tras tratamiento inducciónLLA –MAR si donante familiarSupervivencia 75%

CASO CLÍNICO
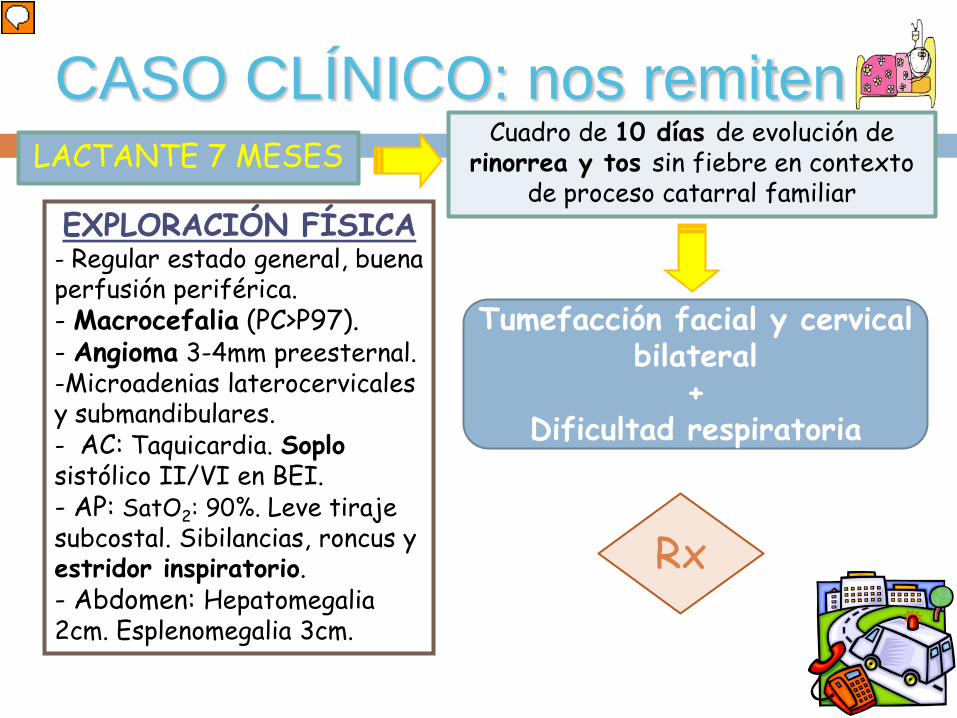
CASO CLÍNICO: nos remitenLACTANTE 7 MESES
Cuadro de 10 días de evolución de rinorrea y tos sin fiebre en contexto
de proceso catarral familiar
Tumefacción facial y cervical bilateral
+Dificultad respiratoria
EXPLORACIÓN FÍSICA- Regular estado general, buena perfusión periférica.- Macrocefalia (PC>P97).- Angioma 3-4mm preesternal.-Microadenias laterocervicales y submandibulares.- AC: Taquicardia. Soplosistólico II/VI en BEI.- AP: SatO2: 90%. Leve tiraje subcostal. Sibilancias, roncus y estridor inspiratorio. - Abdomen: Hepatomegalia 2cm. Esplenomegalia 3cm.
Rx
Moderador
Notas de la presentación
Lactante de 7 meses trasladada a nuestro hospital porque tras cuadro de diez días de evolución de tos y rinorrea sin fiebre en el contexto de proceso catarral familiar ha comenzado con tumefacción facial y cervical bilateral además de dificultad respiratoria. En la exploración física se aprecia regular estado general, buena perfusión periférica, macrocefalia (Perímetro craneal>P97). Angioma de 3-4mm en región preesternal. Adenopatías laterocervicales bilaterales y subangulomandobibulares. Taquicardia con soplo sistólico II/VI. Sibiliancias aisladas y roncus diseminados con estridor inspiratorio. Leve tiraje. Saturación de Oxígeno del 90%. Hepatomegalia 2cm y esplenomegalia 3cm.
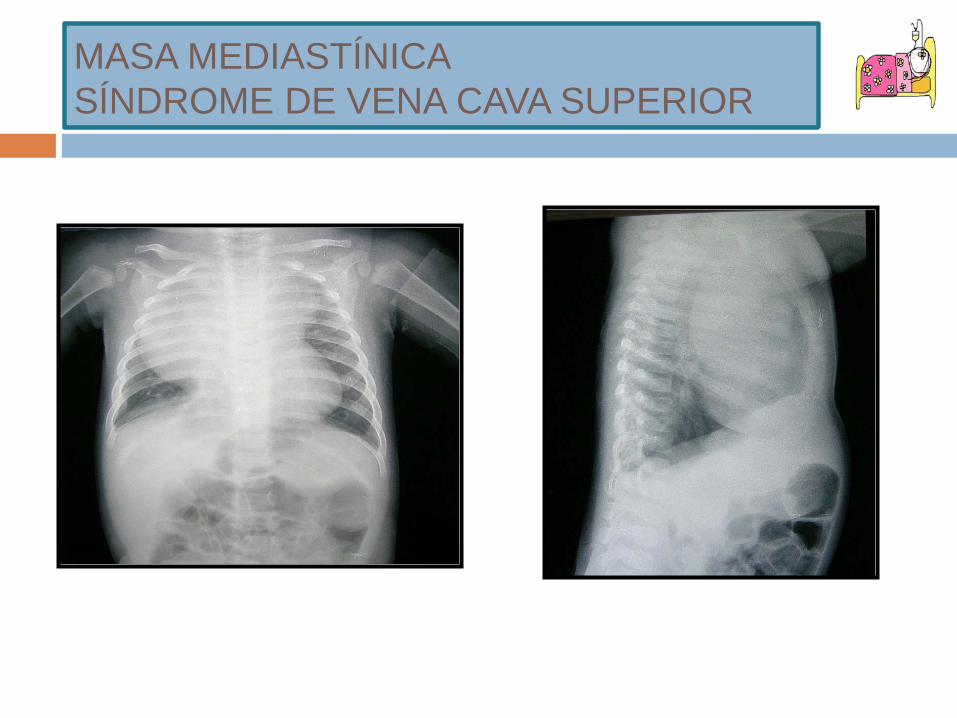
MASA MEDIASTÍNICASÍNDROME DE VENA CAVA SUPERIOR

SÍNDROME DE VENA CAVA SUPERIOR
CONCEPTO: Conjunto de síntomas causados por el deterioro del flujo sanguíneo a través de la vena cava superior a la aurícula derecha.
SOSPECHA CLÍNICA: SÍNTOMAS: disnea, tos. SIGNOS: distensión venas cuello y tórax, edema de cara o EESS, plétora y taquipnea. Más
raros: cianosis, s. Horner. URGENCIA ONCOLÓGICA.
DIAGNÓSTICO: Anamnesis + Exploración física + Rx de tórax. ETIOLOGÍA EN NIÑOS:
Poco común (12% de pacientes con tumores malignos mediastínicos). Causa maligna + frec: LNH. Causa no maligna: Trombosis.
TRATAMIENTO: Corticoides Radioterapia Quimioterapia
Moderador
Notas de la presentación
El SVCS es una urgencia oncológica por compromiso de la vía aérea. Se presenta en el 12% de los pacientes pediátricos con tumores malignos mediastínicos (linfoma no Hodgkin el más frecuente).. Una vez que se reconoce el SVCS es importante la pronta atención clínica. Se deberá establecer un diagnóstico antes de iniciar la terapia debido a las siguientes razones:[3] 75% de los pacientes tienen síntomas y signos por más de 1 semana antes de que busquen la atención médica. Los pacientes con cáncer a quienes se diagnostica el SVCS no mueren del síndrome mismo sino del grado de su enfermedad subyacente. 3% a 5% de los pacientes en quienes se diagnostica el SVCS no tienen cáncer. En la ausencia de obstrucción traqueal, no es probable que el SVCS sea una emergencia oncológica que ponga la vida en peligro y no se justifica el tratamiento antes de un diagnóstico definitivo. Como se describe en otras secciones de este documento, el SVCS se refiere a los signos y síntomas asociados con la compresión u obstrucción de la VCS; la compresión de la traquea se dice síndrome mediastínico superior (SMS). Debido a que el SMS, y el compromiso respiratorio que resulta, ocurre frecuentemente en niños con SVCS, los dos síndromes han llegado a ser casi sinónimos en la práctica pediátrica.[26,27] En adultos, la traquea y el bronquio derecho principal son estructuras relativamente rígidas comparadas con la vena cava, pero en los niños estas estructuras son más susceptibles de compresión. Además, los diámetros intraluminales relativamente más pequeños de la traquea y bronquio de un niño pueden tolerar poco edema antes de que ocurran síntomas respiratorios. Debido a este componente respiratorio que le acompaña, el SVCS en niños difiere del síndrome en adultos y constituye una urgencia médica seria. El SVCS es poco común en niños y aparece al presentarse en 12% de los pacientes pediátricos con tumores malignos mediastínicos.[28,29] La etiología, diagnóstico y tratamiento del SVCS en niños difieren de los de adultos. Mientras que la causa más frecuente de SVCS en adultos es el carcinoma broncogénico,[3] en niños la causa maligna más frecuente del síndrome es el linfoma no Hodgkin. Como en los adultos, una causa frecuente no maligna es la trombosis por cateterización para acceso venoso. Si se sospecha de linfoma o de otra enfermedad maligna, es deseable obtener una muestra de tejido para diagnóstico. Sin embargo, el procedimiento para obtener la muestra podría implicar un riesgo significativo y podría no ser factible. Los niños con SVCS tienen una tolerancia precaria para la anestesia general necesaria, porque los cambios cardiovasculares y pulmonares que le acompañan agravan el SVCS, haciendo frecuentemente imposible la intubación. También la extubación puede ser difícil o imposible, requiriendo así una provisión prolongada de vía aérea (intubación). Una exploración de tomografía computarizada del tórax para determinar el tamaño traqueal, ecocardiografía vertical y supina, y un circuito de volumen de flujo pueden ayudar a evaluar el riesgo anestésico. Porque el uso de anestesia es un riesgo grave, el diagnóstico se deberá hacer con los medios menos invasivos posibles
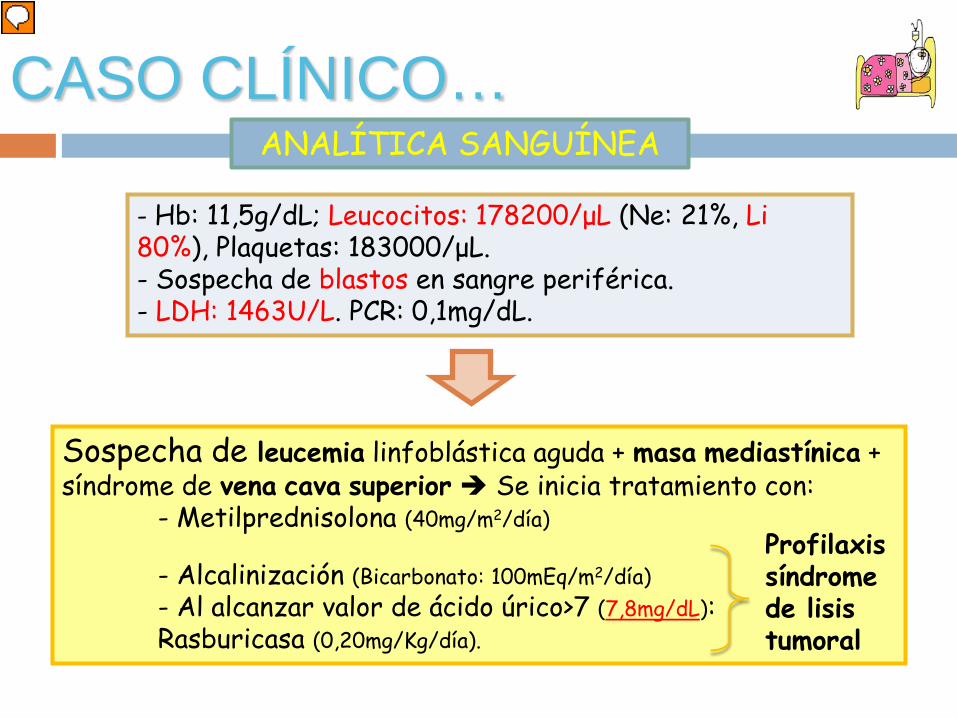
ANALÍTICA SANGUÍNEACASO CLÍNICO…
- Hb: 11,5g/dL; Leucocitos: 178200/μL (Ne: 21%, Li 80%), Plaquetas: 183000/μL.- Sospecha de blastos en sangre periférica. - LDH: 1463U/L. PCR: 0,1mg/dL.
Sospecha de leucemia linfoblástica aguda + masa mediastínica + síndrome de vena cava superior Se inicia tratamiento con:
- Metilprednisolona (40mg/m2/día)
- Alcalinización (Bicarbonato: 100mEq/m2/día)- Al alcanzar valor de ácido úrico>7 (7,8mg/dL):Rasburicasa (0,20mg/Kg/día).
Profilaxissíndrome de lisis tumoral
Moderador
Notas de la presentación
Al iniciar tto con dexametasona presentó TA límites que luego se normalizaron. SÍNDROME DE LISIS TUMORAL El síndrome de lisis tumoral es el término aplicado a un grupo de complicaciones metabólicas que puede ocurrir después del tratamiento de trastornos neoplásicos. Los hallazgos que pueden encontrarse son: hiperfosfatemia, hipocalcemia (causada por la precipitación de fosfato cálcico), hiperuricemia, hipercaliemia y fallo renal agudo. La rápida destrucción de las células leucémicas después de la quimioterapia puede causar sobreproducción y sobreexcreción de ácido úrico. La precipitación de ácido úrico en los túbulos puede llevar a fallo renal oligúrico o anúrico conocido como nefropatía por ácido úrico. En un estudio de 328 niños con LLA, los siguientes 4 factores se identificaron como predictores independientes de síndrome de lisis tumoral en un análisis multivariante: Edad >10años Esplenomegalia Masa mediastínica Recuento de leucocitos inicial superior a 20.000/mcL La ausencia de estos 4 factores de riesgo indica un riesgo bajo de desarrollar síndrome de lisis tumoral con un VPN del 98% y una sensibilidad del 96%. La profilaxis incluye la administración de medicamentos para reducir la producción de ácido úrico (alopurinol, uricasa) o aumentar su excreción (sueroterapia iv, diuréticos). La hemodiálisis puede ser necesaria para eliminar el exceso circulante de ácido úrico y fosfato en pacientes que desarrollan fallo renal y en los que no se logra una diuresis adecuada. La necesidad de hemodiálisis en niños con nefropatía por ácido úrico inducida por el tumor ha descendido con el uso de urato-oxidasa recombinante (uricasa),la enzima que cataliza la oxidación de ácido úrico a un componente mucho más soluble: alantoína. La uricasa (elitek, rasburicasa) fue aprobada por la FDA para el “manejo inicial de los niveles plasmáticos de ácido úrico en los pacientes pediátricos con leucemia, linfoma y tumores sólidos malignos que están recibiendo terapia anticancerosa que pueda dar lugar a un síndrome de lisis tumoral y subsecuente elevación plasmática del ácido úrico”. Los efectos adversos de la uricasa incluyen: reacciones alérgicas (incluyendo anafilaxia), hemólisis, hemoglobinuria, metahemoglobinemia e interferencia con las mediciones de ácido úrico. Este agente está contraindicado en pacientes con deficiencia de glucosa 6fosfato deshidrogenasa porque puede dar lugar a hemólisis severa.

CASO CLÍNICO…
EVALUACIÓN DIAGNÓSTICA: CMF SP: 41% células blásticas: Ag precursores: TdT, CD34, HLA-DR: Negativos. Ag B: CD19, CD10, CD20: Negativos. Ag T: CD2, CD3, CD3c, CD4, CD5, CD7, CD8 Y RCT alfa/beta: Positivos.
RCT gamma/delta: Negativo. Ag mieloides: CD45: Positivo. CD13, CD33, CD56, CD66, CD117: Negativos.
ESTUDIO M.O: CITOGENÉTICA: Pérdidas en el 13q. HIS: Sonda RB1 en 200 células: Positivo en 60% clonal (confirma pérdidas
del 13q). Reordenamiento MLL, BCR/ABL: negativos.
LCR: Sin blastos
LEUCEMIA LINFOBLÁSTICA AGUDA T
Moderador
Notas de la presentación
El estudio con sondas específicas de hibridación in situ para pérdidas del 13q confirma los hallazgos citogenéticos.

CASO CLÍNICO…
OTRAS PRUEBAS COMPLEMENTARIAS: ECOGRAFÍA ABDOMINAL: Poliesplenia. Hígado y
riñones normales. RMN CRANEAL (3m): Adelgazamiento del cuerpo
calloso. ECOGRAFÍA CEREBRAL (7m): Dilatación
biventricular principalmente derecha que sugiere atrofia cerebral de predominio frontal.
OFTALMOLOGÍA: Microftalmia, globo ocular con alteraciones en polo anterior. AV compatible con ceguera. Leucocoria bilateral (catarata congénita).
GAMMAGRAFÍA ÓSEA: Sin alteraciones.

FENOTIPO ESPECIAL…
CASO CLÍNICO…
- Macrocefalia (P>97)- Frente ancha- Aplanamiento parietal- Microftalmia bilateral- Ceguera congénita- Hipertelorismo- Raíz nasal hundida- Angioma preesternal (3-4mm)- Estenosis pulmonar (gradiente máximo: 17mmHg)-Poliesplenia

SÍNDROME CARDIO-FACIO-CUTÁNEO
Esporádico, prevalencia desconocida.
1986: estatura baja, defectos cardíacos congénitos, retraso mental, anormalidades ectodérmicas y apariencia facial característica.
DIAGNÓSTICO:Clínico.Genético: Mutaciones heterocigotas de
RAS, RAF y MEK (44%).
ASOCIACIÓN CON TUMORES: Se ha descrito asociación con leucemias agudas (mutación gen BRAF)
Moderador
Notas de la presentación
- El Síndrome cardio-facio-cutáneo fue descrito en 1986 como un síndrome que incluía: estatura baja, defectos cardíacos congénitos, retraso mental, anormalidades ectodérmicas y apariencia facial característica. Ninguna de las características del síndrome documentadas hasta la fecha es patognomónica ni obligatoria. El diagnóstico es clínico y se debe establecer diagnóstico diferencial con el síndrome de Noonan y el síndrome de Costello. - Esporádico. - Prevalencia desconocida. - CLÍNICA: alteraciones en la alimentación, hipotonía, dismorfia facial característica (frente alta y estrecha, hipertelorismo, raíz nasal hundida y narinas antevertidas) y anomalías de la piel (ictiocitosis, hiperqueratosis o placas de hiperpigmentación) y de faneras (cabellos ralos y rizados, ausencia de cejas). Se han descrito malformaciones cardíacas (estenosis pulmonar, comunicación interauricular o miocardiopatía hipertrófica). También se han descrito retraso del desarrollo y retardo estatural. SIGNOS Y SÍNTOMAS: Estatura baja Cuello corto Macrocefalia relativa Hipotonía Retraso del desarrollo (más comúnmente motor y del lenguaje) Defecto cardíaco congénito (con más frecuencia estenosis valvular pulmonar y defectos del septo auricular) Cara típica: frente ancha, constricción bitemporal, hipoplasia supraorbitaria, nariz pequeña con depresión del puente nasal y carinas antevertidas, orejas de implantación baja y anguladas posteriormente, hipertelorismo, ptosis, epicanto, hendidura palpebral inclinada hacia abajo. Alteraciones ectodérmicas: pelo ralo, rizado y de crecimiento lento, pestañas y cejas ralas o ausentes, queratosis folicular, ictiosis, hiperqueratosis, hiperpigmentación generalizada, piel hiperelástica y crecimiento lento de las uñas. - Mutaciones heterocigotas de RAS, RAF y MEK en sujetos que presentan un síndrome CFC. Se ha evidenciado anomalía molecular en 44-81% de los pacientes que se han estudiado. Estas mutaciones no se han encontrado en los padres de estos pacientes ni en sujetos control estudiados hablando del carácter esporádico de esta afección. Se han identificado mutaciones en KRAS y BRAF en pacientes con síndrome CFC, lo que sugiere que la desregulación de la vía RAS/RAF/MEK/ERK es la base molecular de este síndrome. DIAGNÓSTICO GENÉTICO: Para los pacientes en los que el diagnóstico clínico es incierto, se puede comenzar el estudio con la secuencia de HRAS porque es el de menos tamaño. Si ´ste análisis es negativo, analizar la secuencia de BRAF y MEK1, seguida del análisis de MEK2. Estudios adicionales pueden incluir la secuenciación de KRAS. Por último, se puede estudiar PTPN11 Y SOS1 ya que las mutaciones en estos genes se han relacionado con el síndrome de Noonan. - Relación con TUMORES: No se han descrito casos de tumores sólidos malignos asociados con el síndrome cardio-facio-cutáneo, pero si se ha visto una leucemia linfoblástica aguda en un paciente con una mutación en el gen BRAF. Caso asociado a Leucemia: 9años, mutación en BRAF E501G, LLA (1año y 9 meses)

• SE INICIA TRATAMIENTO CON PROTOCOLO PARA LLA DEL LACTANTE SHOP-02:– Tratamiento de inducción:
• Día +7: 22% células blásticas en SP.• Día +14: 12% células blásticas en SP.• Día +32 (final inducción): 2,6% de células blásticas en M.O por CMF
REMISIÓN COMPLETA– Tratamiento de consolidación.– Tramiento de intensificación (Bloques A, B y C)
– Profilaxis SNC.
CASO CLÍNICO…

CASO CLÍNICO…
INCIDENCIAS DURANTE EL TRATAMIENTO
COMPLICACIONES INFECCIOSAS
1. ITU por E.coli.2. Bronquiolitis VRS (+).3. GEA por Rotavirus.4. GEA por C.difficile.
COMPLICACIONES SECUNDARIAS AL TRATAMIENTO
1. Aplasia severa.2. Hepatotoxicidad grado 4.3. Dermatitis tóxica por
metrotexate.4. Reacción alérgica a L-Asparraginasa
con urticaria y dificultad respiratoria.
Moderador
Notas de la presentación
MTX: A las 72h de su administración comienza con rash cutáneo maculo-papuloso pruriginoso en EEII y área del pañal.
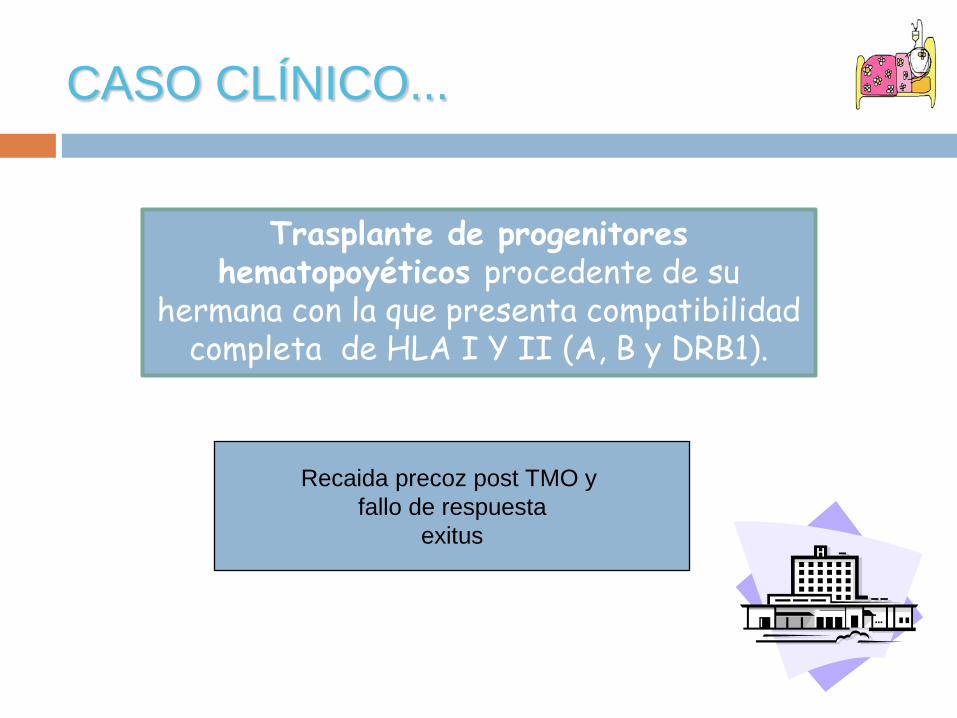
CASO CLÍNICO...
Trasplante de progenitores hematopoyéticos procedente de su
hermana con la que presenta compatibilidad completa de HLA I Y II (A, B y DRB1).
Recaida precoz post TMO y fallo de respuesta
exitus

Leucemia Mieloblástica Aguda Infantil
Grupo heterogéneo de leucemias que proceden de proliferación clonal de precursores mieloides, eritroides, megacariocitos y monocitos
EPIDEMIOLOGIA
•Frecuencia 15-20 % de todas las leucemias•Mayor incidencia < 1 año de edad y > diez años•Asociada a alteraciones genéticas (Sd Dowm, Sd Bloom, A.Blackfan-Diamon, Neurofibromatosis tipo I, Sd Kostman)•Segundo tumor si tratamiento con alquilantes, tenepósido, inhibidores topoisomerasa

Leucemia Mieloblástica Aguda InfantilClasificación morfológica (FAB)
La mayoría se desarrolla a partir de una célula progenitora ya diferenciada en una línea celular.
TIPOS:M0: indiferenciada (3%)M1: sin maduración (15-20%)M2: con maduración (25-30%),t(8,21),AML-1 (B)M3: promielocitica* (45%),t(15,17), RAR-alfa (B)M4: mielomonocitica (25%)11q23 M. Inversión C.16 (Eo)M5: monoblastica * (10%) 11q23M6: eritroleucemia (3%)M7: megacarioblástica (3%)

Leucemia Mieloblástica Aguda InfantilClasificación Inmunológica
InmunofenotipoCD13-CD33-CD117: Comunes a todos los tiposMarcador MPO : Común (excepto M0, M6, M7)CD14-CD11b (M4-M5)Glicoforina (M6) eritroleucemiaCD41-CD42-CD61 (M7) LA megacariocítica

Leucemia Mieloblástica Aguda InfantilClasificación Citogenética
CariotipoMonosomia del 5 y del 7 mal pronosticoReordenamientosAML1-ETO: t(8:21) M2CBFβ-MYH11: Inversión 16 M4MLL-DPT (alteración 11q23) MLL-AF9: t(9:11) M5PML-RAR: t(15:17) M3FLT3-ITD AR

Leucemia Mieloblástica Aguda InfantilClasificación OMS
LMA con alteraciones citogenéticasLMA con displasia multineal
Con Sd mielodisplásico previoSin Sd mielodisplásico previo
LMA secundaria a tratamientos Alquilantes / IrradiaciónInhibidores topoisomerasa II
LMA sin otras caracteristicasM0-M7 clasificación FABSarcoma granulocítico
Moderador
Notas de la presentación
clasificac

Leucemia Mieloblástica AgudaClínica I
Síntomas de insuficiencia medular Síndrome Hemorrágico: Trombocitopenia y/o
coagulopatia de consumo Síndrome anémico: Anemia, palidez, astenia Síndrome febril: Neutropenia/Procesos
infecciosos Dolor óseo: Expansión cavidad medular por
proliferación células malignas

Leucemia Mieloblástica AgudaClínica II
Por infiltracion extramedular: SNC: meningismo, cefalea, confusión, coma Menos frecuente hepato-esplenomegalia Infiltrados cutáneos, hipertrofia gingival
Leucostasis si leucocitos >100.000
Pulmones: infiltrados intersticiales. IRA SNC: cefalea, Ictus, coma
Liberación de sustancias: Tromboplásticas: CID Lisozima: IRA Sind. Lisis tumoral (↑ac. úrico, P, K) IRA
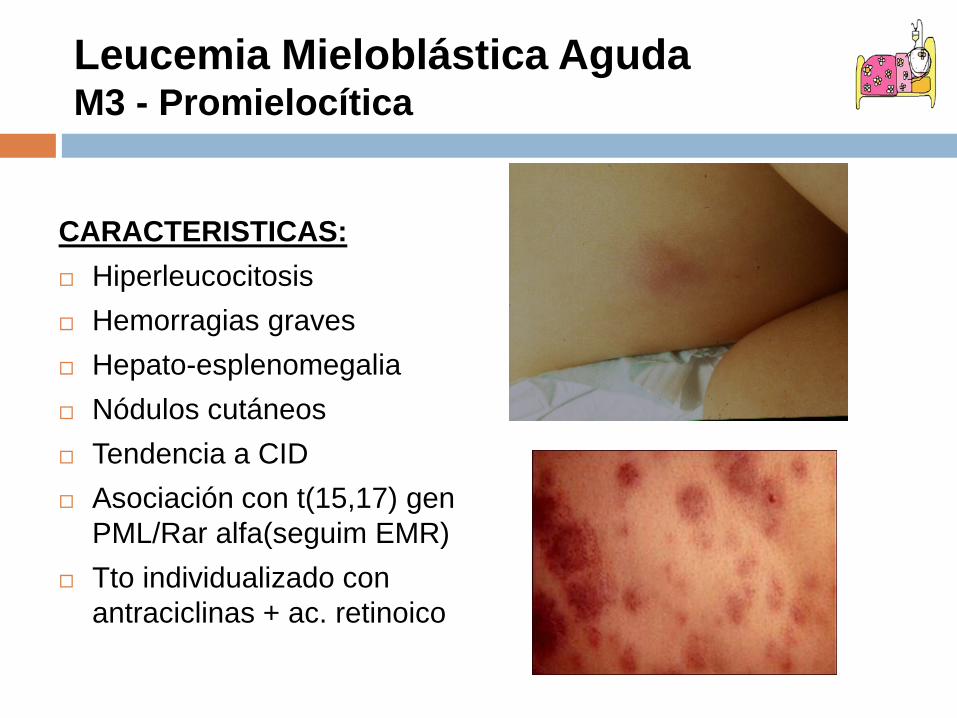
CARACTERISTICAS: Hiperleucocitosis Hemorragias graves Hepato-esplenomegalia Nódulos cutáneos Tendencia a CID Asociación con t(15,17) gen
PML/Rar alfa(seguim EMR) Tto individualizado con
antraciclinas + ac. retinoico
Leucemia Mieloblástica AgudaM3 - Promielocítica

Frecuente en <2 años Masas extramedulares por infiltración muy
frecuente (Piel, encías, meninges, N óptico, pulmones y hueso)
Asociación 11q23
Leucemia Mieloblástica AgudaM5 - Monoblástica

Leucemia Mieloblástica AgudaClínica
Cloroma orbitario Sarcoma granulocíticoHemorragia mucosas
Hipertrofia gingival Hemorragia conjuntival

Leucemia Mieloblástica AgudaDiagnóstico
Morfología: Blastos: sangre periférica y en MO (>20%):
Tinción con peroxidasas y alfa-naftilacetatoesterasa
Inmunofenotipo: Marcadores específicos de serie mieloide (CD13/CD33)
Citogenética: Alt cromosómicas en relación con el subtipo: M2: t(8,21) 10% M3: t(15,17)8% M4eos: inv16, t(16,16) (12%) M5a: 11q23 (20%) Monosom 7* Monosomia 5* Mutac FLT3* Anomalias múltiples >3* Nucleofosmina (NPM1)*
* desfavorable

Desfavorables Favorables
Cariotipo desfavorableLeucocitos >100.000Tardanza en la remision*M0.M5,M6, M7Fenotipo CD14+/DR-LAM secundaria
*EMR >1% tras 1º ciclo inducción SV 33%
Edad < 2 añosLeucocitos < 100.000M4-M5t(8:21), inv. 16, t(15:17)
Leucemia Mieloblástica AgudaFactores Pronósticos

Leucemia Mieloblástica AgudaTratamiento
Blastos +No blastos
INDUCCIONMO-dia+21 tto
CONSOLIDACION
Supervivencia 60%
ARAC+DN+etoposido
ARAC-C > dosis+mitoxantrone
AR y DonanteF.I.:Alo-T
No donante o BR
(>5%,>1% CMF)
2º MO en recuperación
MAR: Alo-T
2º INDUCCION
2ª consolidacion Auto-T

Leucemias AgudasTratamiento de soporte
Catéter central y sus cuidados Derivados hemáticos Factores de crecimiento celular Profilaxis de infecciones Apoyo nutricional Apoyo psicológico y escolar Diagnóstico de complicaciones vitales (SVCS, Lisis
tumoral, Coagulopatias, sepsis)

LINFOMAS INFANTILES

Linfomas Infantiles
Tumores malignos del sistema linfoide
Tercero en frecuencia despues de las leucemia y tumores del SNC. 14% de todos los tumores infantiles

Linfomas en la infanciaClasificación OMS
Linfoma Hodgkin
Linfoma no Hodgkin:L. Burkitt (L. de cls pequeñas): LBL.B difuso células grandesL. Primario Mediastínico cls. B grandesL. linfoblástico : Pre B – Pre TL. anaplásico de cls. grandes

Linfoma Hodgkin en niños
1832 Descripción anatómica detallada de la enfermedad
STERBERG 1898 Y REED 1902 DESCUBRIERON LA CELULA CARACTERISTICA MULTINUCLEADA

Linfoma Hodgkin en niños

Linfoma Hodgkin en niños
SLP de origen B (99%de los casos), se origina en los centros germinales linfoides y secretan
potentes citoquinas responsables de los síntomas B
. Incidencia: 10 casos / millón de niños/añoEdad: Neoplasia mas frecuente entre 15 – 19 años
En menores de 10 años: 4%Sexo: Mas frecuente en varones. Proporción 3/1
EPIDEMIOLOGIA

Linfoma HodgkinManifestaciones clínicas
Adenopatías en grupos o solitarias indoloras elásticas de crecimiento lento. Laterocervicales bajas, supraclaviculares
Sintomatología compresiva: Dolor retroesternal, tos, disnea → intratoracico Dolor lumbar en supino → intraabdominal
Sintomatología B: fiebre >38º,sudoración nocturna, pérdida de peso
>10% en los 6m últimos Prurito: al diagnóstico o a lo largo de la enfermedad
hasta 80%

Linfoma HodgkinManifestaciones clínicas
Adenopatías: elásticas, no dolorosas, mayores de 1 cm, o en paquetes, crecimiento lento Cervicales y supraclavicular 60-80% Axilares 10% Inguinales 6%
Masa mediastinica 75% de los casos al diagnóstico
Hepatomegalia rara al diagnóstico < 5% Esplenomegalia 20%
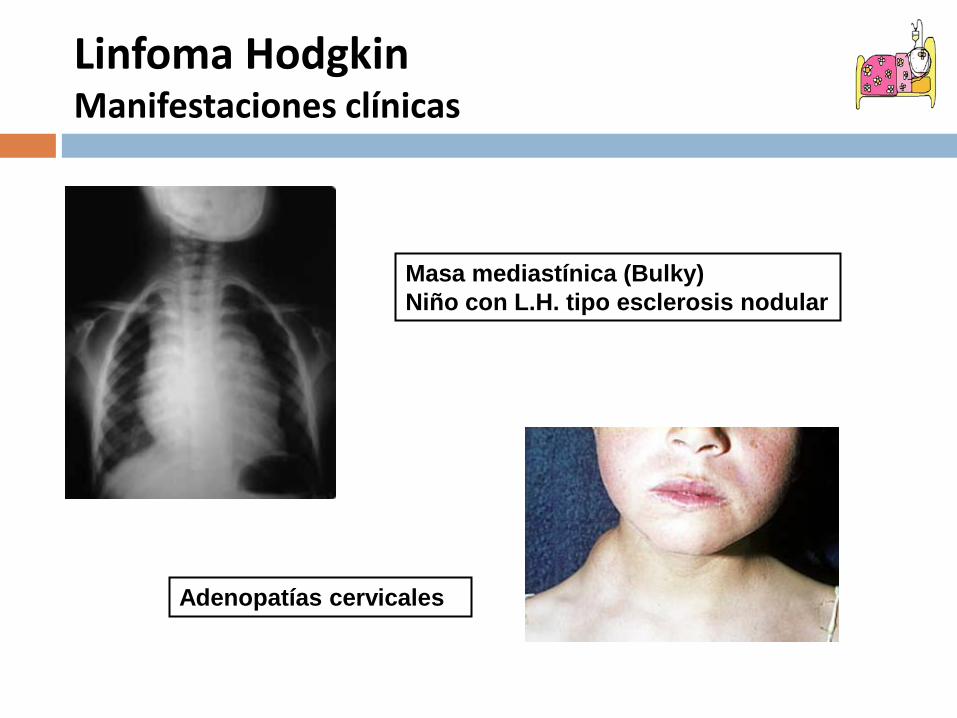
Linfoma HodgkinManifestaciones clínicas
Masa mediastínica (Bulky)Niño con L.H. tipo esclerosis nodular
Adenopatías cervicales
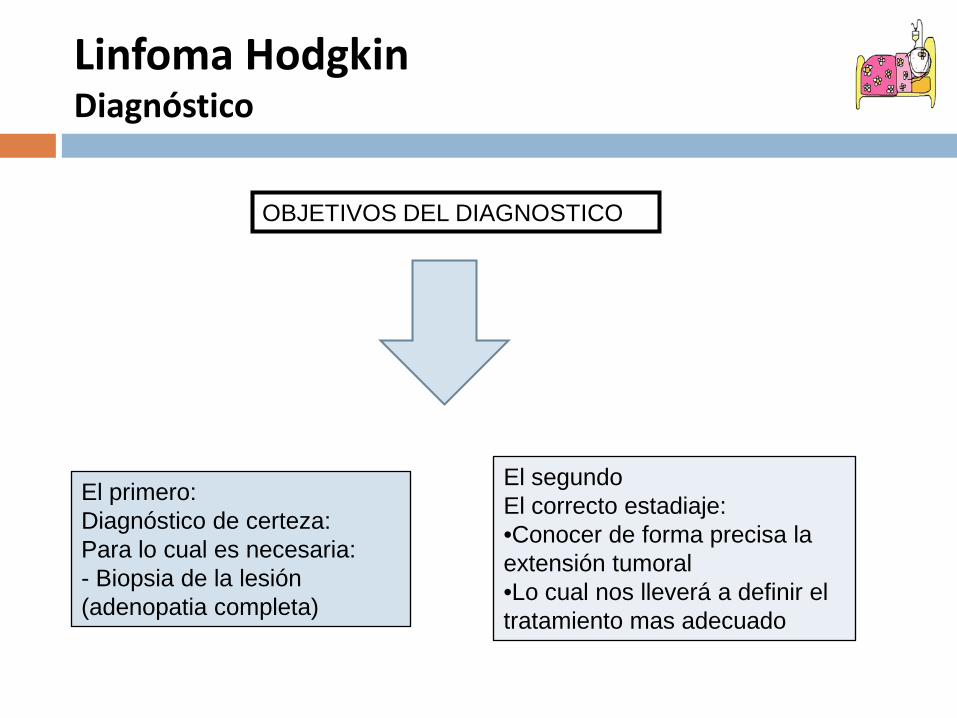
Linfoma HodgkinDiagnóstico
El primero:Diagnóstico de certeza:Para lo cual es necesaria:- Biopsia de la lesión (adenopatia completa)
El segundoEl correcto estadiaje:•Conocer de forma precisa la extensión tumoral•Lo cual nos lleverá a definir el tratamiento mas adecuado
OBJETIVOS DEL DIAGNOSTICO

Célula de Reed Stemberg: Grande, abundante citoplasma, núcleo lobulado o varios núcleos, membrana nuclear intensamente teñida. Célula clonal de origen B
Linfoma HodgkinDiagnóstico Histológico
Infiltrado pleomórfico con células de Reed Stemberg

Linfoma HodgkinDiagnóstico
Tipos anatomo-patológicos: LH Clásico (90-95%)
Predominio linfocítico*. 19%, buen pronóstico Celularidad mixta: mas frecuente en adultos. Cl. de
Stemberg típica CD Ia+ Deplección linfocítica: asoc. VEB, estadios avanzados.
Mal pronóstico Esclerosis nodular*: afección mediastínica, pronóstico
intermedio. CD30+, CD15+ (75%) LH Nodular de predominio linfocítico (5-10%)
Curso indolente. Enfermedad localizada sin afectacion mediastínica. CD15-, CD30-

Linfoma HodgkinInmunofenotipo
CLASICO NODULAR PRED.LINFOCITARIO
CD45-CD15+CD30+P53+
CD45+ (raro)CD20+, CD15-CD3+CD30-

Linfoma HodgkinDiagnóstico Extensión
Hemograma: anemia, eosinofilia, >VSG >LDH, f. hepática y renal, >PCR, F.alcalinas >Cobre, lisozyma, B2 microglobulina, interleukinas Medula ósea (estadios III, IV y/o síntomas B
PRUEBAS IMAGEN Rx. Tórax PA y lateral: Adenopatías mediastinicas Gammagrafía con galio: Captación en regiones afectas.
Falsos negativos PET/TAC en todos al diagnostico y seguimiento. Actividad
metabólica de región afectaSensible y útil para valorar respuesta
Gammagrafía con Tec. Si sospecha de afectación ósea RMN para el abdomen

Linfoma HodgkinDiagnóstico Extensión
Linfoma Hodgkin, estadio II. Afectacion supraclavicular bilateral, infraclavicular izquierda y mediastinica. PET-CT antes y despues de dos ciclos de QT. Persiste actividad paratraqueal y supraclavicular derecha

Linfoma Hodgkin Clasificación Ann Arbor
Estadio I: Unica región ganglionar o de un organo linfoide
Estadio II: Dos regiones ganglionares o estructuras linfoides en el mismo lado del diafragma
Estadio III: Regiones linfoides o estructuras a ambos lados del diafragma
Estadio IV: Localizaciones extranodales con o sin afectación ganglionar asociada (M.O, higado hueso, pleura)
Se añade letra A o B en función de la ausencia o presencia de síntomas B

Linfoma Hodgkin Tratamiento
QT + RT según extensión enfermedad (Protocolo Euronet)
Quimioterapia:2OEPA+ 2-4 COPP+/-RT
PET-TAC tras dos ciclos QT. Si positivo RT sobre campos ganglionares
inicialmente afectos
Supervivencia global 90 %

Linfoma HodgkinComplicaciones del Tratamiento
•Hipotiroidismo y alteración inmune de larga duración•Problemas psicosociales•Disfunción cardiopulmonar•Esterilidad•Anomalías en crecimiento óseo y tejidos blandos•Alteraciones tróficas secundarias a radioterapia•Segundas neoplásias

Linfomas no HodgkinClasificación
•L. Burkitt (L. de cls pequeñas): LB•L.B difuso células grandes•L. Primario Mediastínico cls. B grandes•L. linfoblástico : Pre B – Pre T•L. anaplásico de cls. grandes

Linfomas no Hodgkin
Proliferación clonal de precursores linfoides inmaduros. Engloba todos los linfomas malignos no clasificados, como Linfoma de Hodgkin
EPIDEMIOLOGIAFrecuencia: 7.5 % de los tumores infantilesIncidencia: 7.5 por millón de niños /año.Pico de incidencia: 7 a 10 añosSexo: Predominio varón 3/1Población de riesgo:
•Inmunodeficiencias congénitas (Ataxia telangiectasia, S. Wiskott Aldrich, SCIDS, XLP)•Inmunodeficiencias adquiridas (SIDA, tto inmunosupresor post-Trasplante)

Linfomas no HodgkinClinica
Los Linfomas no Hodgkin tienen un crecimiento rápido, con una progresión a diseminación rápida, pueden debutar como una emergencia médica
Afectación extra nodal afectando abdomen, mediastino o zona de cabeza y cuello
•Dolor abdominal•Crisis suboclusivas•Masa mediastinica•Derrame pleural •Insuficiencia respiratoria•Síndrome vena cava superior•Lesiones cutáneas•Afectación pares craneales
El comienzo de los sintomas puede ser explosivo

Linfoma no Hodgkin Clinica II

Linfoma no Hodgkin Linfoma de Burkitt
Célula B madura: CD10,CD19,CD20,CD22T(8:14)-C-MyC
•Masa abdominal (80 % casos). Fosa iliaca derecha•Invaginación intestinal•Derrame pleural•Ascitis•Medula ósea (afectación aislada blastos > 20%) = Leucemia cls Burkitt (L3)
Células monomórficas, de mediano tamaño, con citoplasma basofílico amplio, núcleo redondo, nucléolos múltiples y con altísimo índice de mitosis y cariorexis y apariencia de cielo estrellado. EBV principalmente en los casos africanos (L.B. endémico) y los asociados a inmunodeficiencias
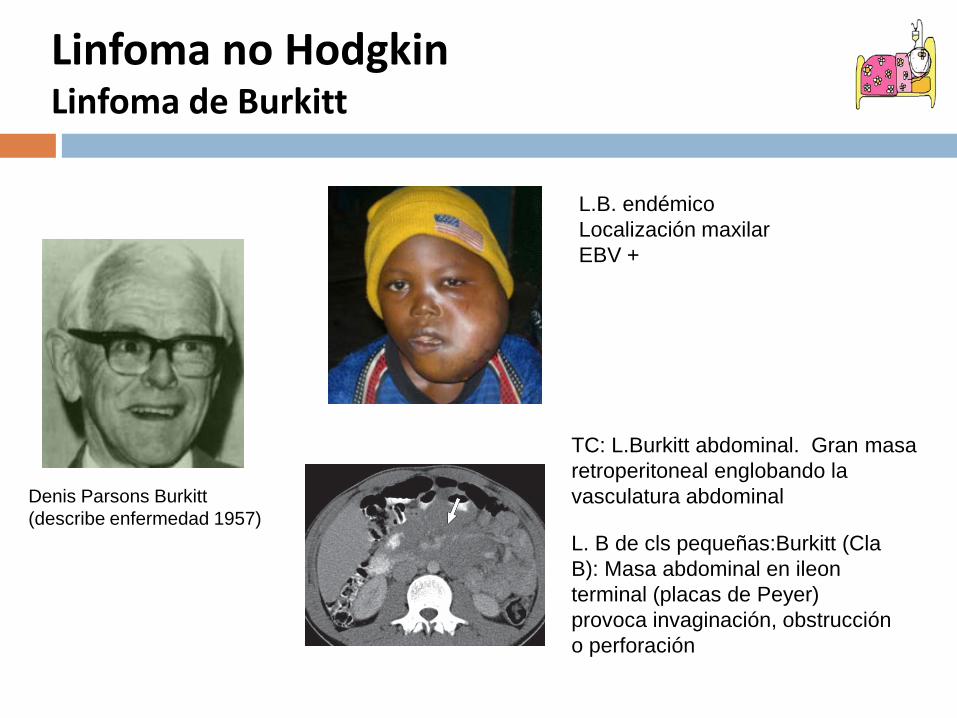
Linfoma no Hodgkin Linfoma de Burkitt
Denis Parsons Burkitt(describe enfermedad 1957)
TC: L.Burkitt abdominal. Gran masa retroperitoneal englobando la vasculatura abdominal
L. B de cls pequeñas:Burkitt (Cla B): Masa abdominal en ileon terminal (placas de Peyer) provoca invaginación, obstrucción o perforación
L.B. endémicoLocalización maxilar EBV +

Estudio de Biopsia de tejidos o líquidos (Pleural o ascítico). Histologia, con cariotipo y HIS.
Hemograma Función hepática / renal (LDH, ac.urico) Estudio de biopsia bicrestal de MO Estudio de LCR Estudio de imagen
Gammagrafías con Galio/ Tecnecio RM o TAC PET/TAC
Linfoma no HodgkinDiagnóstico

L. Burkitt:TC. engrosamiento concéntrico de la pared de las asas intestinales con dilataciones de varios segmentos intestinales sin obstrucción
Linfoma no HodgkinDiagnóstico Imagen

L. Burkitt: Múltiples lesiones hipodensas en riñones y una única lesión hipodensa en el hígadoL. Burkitt:
Masa en el cuadrante superior derecho con necrosis central.
Linfoma no HodgkinDiagnóstico Imagen
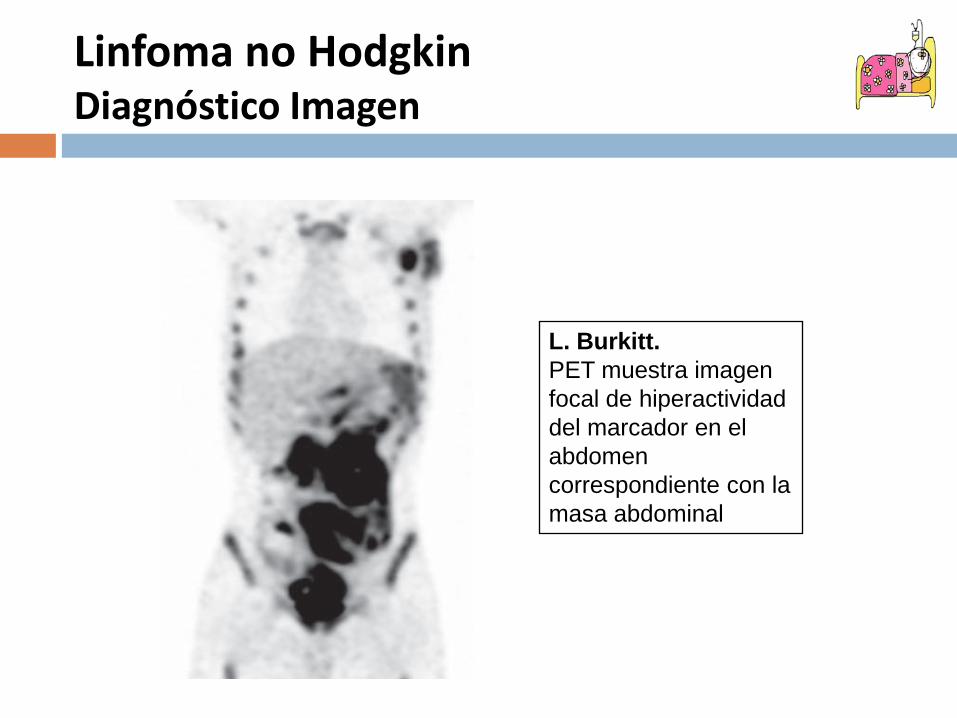
L. Burkitt.PET muestra imagen focal de hiperactividad del marcador en el abdomen correspondiente con la masa abdominal
Linfoma no HodgkinDiagnóstico Imagen
Linfoma no HodgkinDiagnóstico Imagen
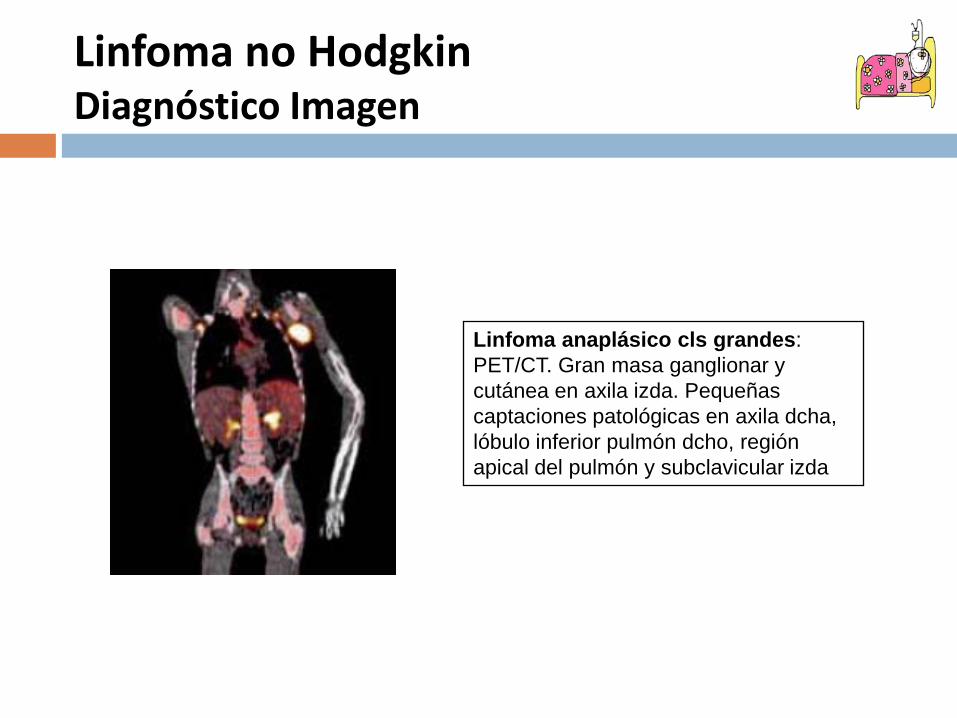
Linfoma anaplásico cls grandes:PET/CT. Gran masa ganglionar y cutánea en axila izda. Pequeñas captaciones patológicas en axila dcha, lóbulo inferior pulmón dcho, región apical del pulmón y subclavicular izda

Linfoma no HodgkinClasificación Murphy

Linfoma no HodgkinTratamiento
TRATAMIENTO TUMORALQuimioterapia intensiva MTX, ARAC, ADSupervivencia 70-90% depende tipo histológico y extensión
URGENCIAS ONCOLOGICASSíndrome “Vena cava superior” (QT)Síndrome “Lisis tumoral” (hiperhidratacion-rasburicasa)Síndrome “Compresión medular” (RT)

SOSPECHA MALIGNIDAD / ADENOPATIA
•Diámetro superior 2.5 cm•No respuesta tratamiento antiinflamatorio/antibiotico > 2 semanas•Localización supraclavicular, retroauricular o epitroclear•Asociación a síndrome constitucional y/o perdida peso >10%•Radiografía tórax patológica con ensanchamiento mediastínico


















