
Citometría de flujo en el diagnóstico y clasificación de...
35
Revista de Heematología Vol. 7, No.. 2, 2006 53 Solicitud de reimpresos: Q.F.B. Josefa Piedras-Ross. Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición SZ. Vasco de Quirooga 15, Colonnia Sección XVI, Delegación Tlalpan 14000, México, D.F., México. Tel.: 54 870 900 Ext. 2704 Citometría dee flujo en el diagnóstico y clasificación de padecimientos hematológicos: leucemias agudas, síndromes linfoproliferativos crónicos y glicoproteínas plaquetarias. Q.F.B. Josefa Piedras Departamento de Hematología y Oncologia. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México, D.F., México. los que usualmente incluyen mediciones de tamaño y granularidad junto con 4 a 8 colores de fluorescencia; cuando miden 8 parámetros es posible resolver hasta 64 poblaciones distintas de células con valores positivos o negativos para cada parámetro (2). Algunos citómetros de flujo pueden separar físicamente las células basado en diferencias de cualquier parámetro medible, y los instrumentos del futuro también permitirán medir microbios y organelos celulares. Una de las áreas más beneficiadas en CF con el análisis multiparamétrico de las células es precisamente la hemato-oncología médica. INMUNOFENOTIPO. En mayo del 2005 se llevó a cabo en la Ciudad de Querétaro, México, la segunda Conferencia de Consenso Latinoamericano para la inmunofenotipificación de padecimientos hematológicos malignos (3), cuyo objetivo fue el de actualizar las recomendaciones emanadas de la Primera Conferencia de Consenso (4). Se estableció la utilidad clínica del inmunofenotipo para la clasificación, pronóstico y seguimiento INTRODUCCIÓN. La citometría de flujo (CF) es la medición de las propiedades de las células que se encuentran suspendidas en un fluido y que interrumpen un haz de luz láser. El método permite el análisis cualitativo y cuantitativo de diferentes propiedades como tamaño, estructura y contenido (análisis multiparámetro), de poblaciones celulares en líquidos corporales, así como de cualquier partícula tan pequeña como 0.1 µm. La dispersión de la luz fue usada como un indicador de la presencia de una célula, y el reporte Coons y Kaplan de la conjugación de la fluoresceina a los anticuerpos constituyó un descubrimiento importante que permitió la identificación de antígenos tisulares por anticuerpos específicos usando fluorescencia (1). Desde la primera generación de los citómetros de flujo en 1980 ha habido grandes avances, y los citómetros actuales combinan una mezcla de tecnologías modernas tales como: mecánica de fluidos, rayos láser, óptica, electrónica análoga y digital y computer software. Hoy en día con los citómetros de flujo se pueden evaluar de 4 a 8 parámetros,
Transcript of Citometría de flujo en el diagnóstico y clasificación de...

Revista de Hematología Vol. 7, No. 2, 2006
53
Solicitud de reimpresos: Q.F.B. Josefa Piedras-Ross. Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición SZ. Vasco de Quiroga 15, Colonia Sección XVI, Delegación Tlalpan 14000, México, D.F., México. Tel.: 54 870 900 Ext. 2704
Citometría de flujo en el diagnóstico y clasificaciónde padecimientos hematológicos: leucemias agudas, síndromes linfoproliferativos crónicos y glicoproteínas
plaquetarias.
Q.F.B. Josefa Piedras
Departamento de Hematología y Oncologia. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México, D.F., México.
los que usualmente incluyen mediciones de tamaño y granularidad junto con 4 a 8 colores de fluorescencia; cuando miden 8 parámetros es posible resolver hasta 64 poblaciones distintas de células con valores positivos o negativos para cada parámetro (2). Algunos citómetros de flujo pueden separar físicamente las células basado en diferencias de cualquier parámetro medible, y los instrumentos del futuro también permitirán medir microbios y organelos celulares. Una de las áreas más beneficiadas en CF con el análisis multiparamétrico de las células es precisamente la hemato-oncología médica.
INMUNOFENOTIPO.En mayo del 2005 se llevó a cabo en
la Ciudad de Querétaro, México, la segunda Conferencia de Consenso Latinoamericano para la inmunofenotipificación de padecimientos hematológicos malignos (3), cuyo objetivo fue el de actualizar las recomendaciones emanadas de la Primera Conferencia de Consenso (4). Se estableció la utilidad clínica del inmunofenotipo para la clasificación, pronóstico y seguimiento
INTRODUCCIÓN.La citometría de flujo (CF) es la
medición de las propiedades de las células que se encuentran suspendidas en un fluido y que interrumpen un haz de luz láser. El método permite el análisis cualitativo y cuantitativo de diferentes propiedades como tamaño, estructura y contenido (análisis multiparámetro), de poblaciones celulares en líquidos corporales, así como de cualquier partícula tan pequeña como 0.1 µm. La dispersión de la luz fue usada como un indicador de la presencia de una célula, y el reporte Coons y Kaplan de la conjugación de la fluoresceina a los anticuerpos constituyó un descubrimiento importante que permitió la identificación de antígenos tisulares por anticuerpos específicos usando fluorescencia (1). Desde la primera generación de los citómetros de flujo en 1980 ha habido grandes avances, y los citómetros actuales combinan una mezcla de tecnologías modernas tales como: mecánica de fluidos, rayos láser, óptica, electrónica análoga y digital y computer software. Hoy en día con los citómetros de flujo se pueden evaluar de 4 a 8 parámetros,

54
Revista de Hematología Vol. 7, No. 2, 2006
J Piedras
de los pacientes con leucemia aguda (LA), y para el diagnóstico, clasificación, pronóstico y seguimiento de los pacientes con padecimientos linfoproliferativos crónicos (PLPC).
Preparación de la muestra. Se recomendó, para la mayoría de los casos, el empleo de muestra total de sangre o de médula ósea sometida a lisis de eritrocitos y fijación, y la separación celular por gradiente de densidad sólo en aquellas muestras contaminadas con células necróticas estromales o grasas. También se recomendó reducir el número de células teñidas con el objeto de disminuir el volumen de anticuerpo monoclonal utilizado, siempre y cuando la reducción sea validada experimentalmente.
Paneles de anticuerpos monoclona-les. Mediante el inmunofenotipo se pueden clasificar cuatro tipos de leucemias: a) leuce-mia aguda linfoblástica de estirpe T (LAL-T), b) leucemia aguda linfoblástica de precurso-res de células B (LAL-B), c) leucemia aguda mieloblástica (LAM) y d) leucemia aguda bifenotípica (LAB).
La combinación de los marcadores CD2, CD7 y CD3 citoplásmico se consideró la más apropiada para definir a la LAL-T, requiriendo la co-expresión de CD3 citoplásmico con cuando menos uno de los otros dos antígenos. Para establecer el grado de madurez se recomendó investigar la expresión de CD34, TdT y la intensidad de expresión del CD45. No se consideró de relevancia clínica una sub-clasificación de la LAL-T.
Para la clasificación de la LAL-B, además de la combinación de anticuerpos anotada en el cuadro 1 se decidió, por utilidad terapéutica y clínica, adoptar la sub-clasificación del EGIL (European Group of Immunophenotyping of Leukemias) en: Pro-B (B-I), Común (B-II), Pre-B (B-III) y LAL de células B maduras (B-IV) empleando los anticuerpos anotados en el cuadro 1.
Al igual que para la LAL-B, para la identificación de la LAM se consideró el uso de tres diferentes categorías de anticuerpos esenciales, junto con un cuarto grupo opcional (cuadro 2) (figura 1).
Cuadro 1Panel de anticuerpos para la inmunofenotipificación de la leucemia aguda
linfoblástica de precursor de células B.Linaje* Madurez Sub-clasificación Opcional**CD19CD79a (citoplásmico)
HLA-DRTdTCD34CD45
CD10Ig superficieCadenas µ citoplásmicas
CD20CD38
*Ambos marcadores CD19 y CD79a deben estar presentes.** La inclusión de CD20 y CD38 se consideró como informativa para la búsqueda de anormalidades genéticas comunes en la LAL-B.
Cuadro 2Panel de anticuerpos para la inmunofenotipificación de la leucemia aguda mieloide.
Linaje* Madurez Sub-clasificación OpcionalMPO (citoplásmica)CD13CD33CD117
HLA-DRCD34CD45
CD15 CD36CD64
* Los blastos deben expresar dos (si MPO+) o más (si MPO-) marcadores mieloides.

Revista de Hematología Vol. 7, No. 2, 2006
55Citometría de flujo en padecimientos hematológicos.
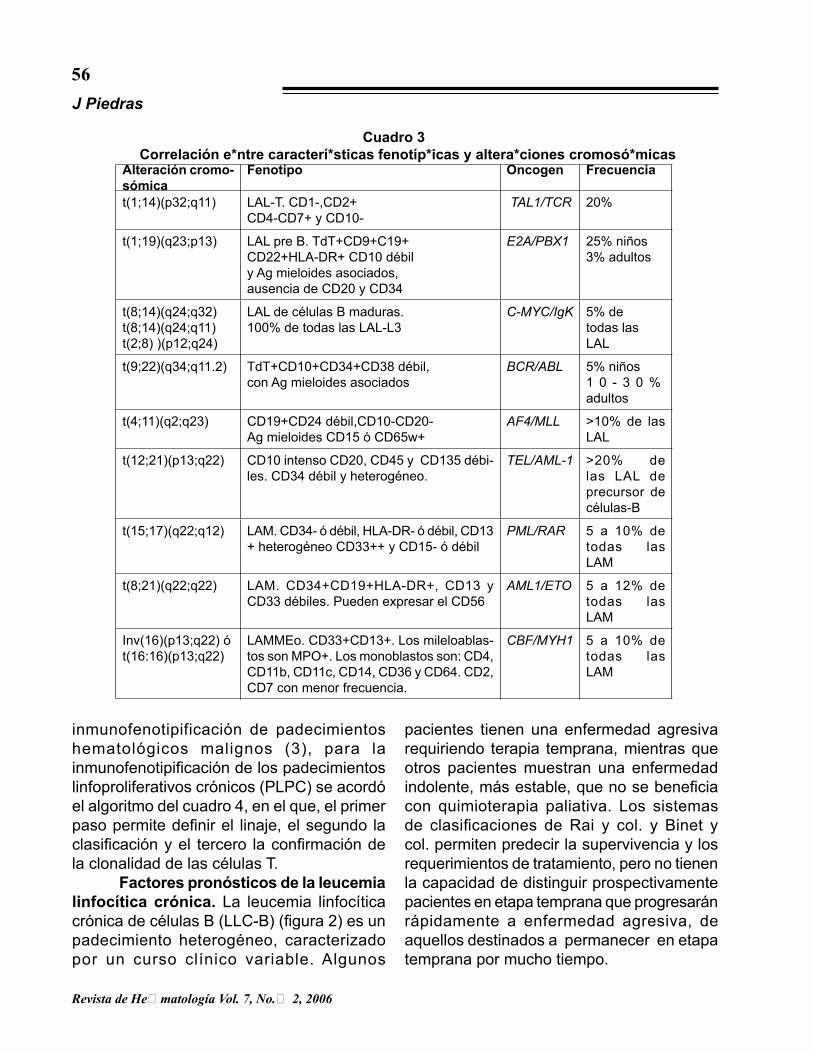
6)(cuadro 3). Estas observaciones, junto con la producción de nuevos anticuerpos que reconocen proteínas de fusión, o proteínas expresadas en células portadoras de una traslocación específica, le han dado mayor valor al inmunofenotipo en la clasificación y pronóstico de las leucemias.
PADECIMIENTOS LINFOPROLIFERATIVOS CRÓNICOS.
En la segunda Conferencia de Consenso Latinoamericano para la
Figura 1.- Paciente con leucemia mielomonocítica. Por expresión de CD45 se identifican dos poblaciones de blastos: la verde (mieloide) con expresión intensa de CD117, CD34, CD13, CD33 y expresión débil de CD15; la azul (monocítica) con expresión intensa de CD64/CD14, CD11b, CD13 y expresión débil de CD34.
La leucemia bifenotípica debe expresar antígenos de dos o más de los linajes mencionados. Los marcadores más específicos para cada estirpe son: CD3 para células T, CD19 y CD79b para células B y mieloperoxidasa para células mieloides.
Inmunofenotipo y alteraciones cromosómicas. En años recientes se ha demostrado la presencia, en las leucemias agudas, de correlación entre algunos de los patrones de expresión de antígenos asociados a la leucemia y traslocaciones genéticas (5,
56
Revista de Hematología Vol. 7, No. 2, 2006
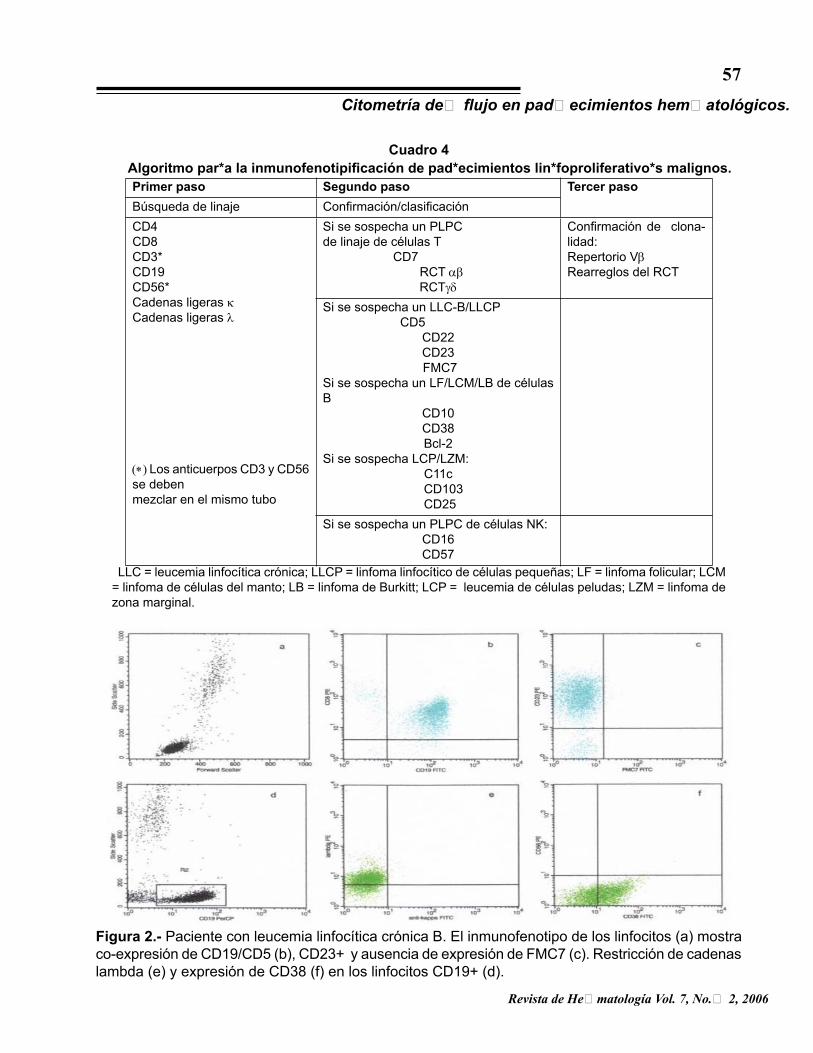
inmunofenotipificación de padecimientos hematológicos malignos (3), para la inmunofenotipificación de los padecimientos linfoproliferativos crónicos (PLPC) se acordó el algoritmo del cuadro 4, en el que, el primer paso permite definir el linaje, el segundo la clasificación y el tercero la confirmación de la clonalidad de las células T.
Factores pronósticos de la leucemia linfocítica crónica. La leucemia linfocítica crónica de células B (LLC-B) (figura 2) es un padecimiento heterogéneo, caracterizado por un curso clínico variable. Algunos
pacientes tienen una enfermedad agresiva requiriendo terapia temprana, mientras que otros pacientes muestran una enfermedad indolente, más estable, que no se beneficia con quimioterapia paliativa. Los sistemas de clasificaciones de Rai y col. y Binet y col. permiten predecir la supervivencia y los requerimientos de tratamiento, pero no tienen la capacidad de distinguir prospectivamente pacientes en etapa temprana que progresarán rápidamente a enfermedad agresiva, de aquellos destinados a permanecer en etapa temprana por mucho tiempo.
Cuadro 3Correlación entre características fenotípicas y alteraciones cromosómicas
Alteración cromo-sómica
Fenotipo Oncogen Frecuencia
t(1;14)(p32;q11) LAL-T. CD1-,CD2+CD4-CD7+ y CD10-
TAL1/TCR 20%
t(1;19)(q23;p13) LAL pre B. TdT+CD9+C19+CD22+HLA-DR+ CD10 débily Ag mieloides asociados,ausencia de CD20 y CD34
E2A/PBX1 25% niños3% adultos
t(8;14)(q24;q32)t(8;14)(q24;q11)t(2;8) )(p12;q24)
LAL de células B maduras.100% de todas las LAL-L3
C-MYC/IgK 5% detodas lasLAL
t(9;22)(q34;q11.2) TdT+CD10+CD34+CD38 débil, con Ag mieloides asociados
BCR/ABL 5% niños1 0 - 3 0 % adultos
t(4;11)(q2;q23) CD19+CD24 débil,CD10-CD20-Ag mieloides CD15 ó CD65w+
AF4/MLL >10% de las LAL
t(12;21)(p13;q22) CD10 intenso CD20, CD45 y CD135 débi-les. CD34 débil y heterogéneo.
TEL/AML-1 >20% de las LAL de precursor de células-B
t(15;17)(q22;q12) LAM. CD34- ó débil, HLA-DR- ó débil, CD13 + heterogéneo CD33++ y CD15- ó débil
PML/RAR 5 a 10% de todas las LAM
t(8;21)(q22;q22) LAM. CD34+CD19+HLA-DR+, CD13 y CD33 débiles. Pueden expresar el CD56
AML1/ETO 5 a 12% de todas las LAM
Inv(16)(p13;q22) ót(16:16)(p13;q22)
LAMMEo. CD33+CD13+. Los mileloablas-tos son MPO+. Los monoblastos son: CD4, CD11b, CD11c, CD14, CD36 y CD64. CD2, CD7 con menor frecuencia.
CBF/MYH1 5 a 10% de todas las LAM
J Piedras
Revista de Hematología Vol. 7, No. 2, 2006
57
Cuadro 4Algoritmo para la inmunofenotipificación de padecimientos linfoproliferativos malignos.Primer paso Segundo paso Tercer pasoBúsqueda de linaje Confirmación/clasificaciónCD4CD8CD3*CD19CD56*Cadenas ligeras κCadenas ligeras λ
(∗) Los anticuerpos CD3 y CD56 se debenmezclar en el mismo tubo
Si se sospecha un PLPC de linaje de células T CD7
RCT αβRCTγδ
Confirmación de clona-lidad:Repertorio VβRearreglos del RCT
Si se sospecha un LLC-B/LLCP CD5
CD22CD23 FMC7
Si se sospecha un LF/LCM/LB de células B
CD10CD38Bcl-2
Si se sospecha LCP/LZM:C11c
CD103 CD25
Si se sospecha un PLPC de células NK:CD16CD57
LLC = leucemia linfocítica crónica; LLCP = linfoma linfocítico de células pequeñas; LF = linfoma folicular; LCM = linfoma de células del manto; LB = linfoma de Burkitt; LCP = leucemia de células peludas; LZM = linfoma de zona marginal.
Figura 2.- Paciente con leucemia linfocítica crónica B. El inmunofenotipo de los linfocitos (a) mostra co-expresión de CD19/CD5 (b), CD23+ y ausencia de expresión de FMC7 (c). Restricción de cadenas lambda (e) y expresión de CD38 (f) en los linfocitos CD19+ (d).
Citometría de flujo en padecimientos hematológicos.
En 1999 dos grupos de investigadores (7, 8) demostraron que los pacientes con un inmunofenotipo de células de memoria con genes de inmunoglobulinas (IgVH) mutados, tenían un desenlace mucho más favorable y una baja probabilidad de desarrollar enfermedad progresiva, mientras que aquellos con genes IgVH no mutados, tenían
mayor probabilidad de desarrollar enfermedad progresiva y de tener una supervivencia más corta. Sin embargo, el análisis de la mutación de gen IgVH se basa en la secuenciación de ADN, técnicamente difícil y no disponible para uso clínico rutinario, por lo que, la identificación de marcadores apropiados subrogados para el estado mutacional ha
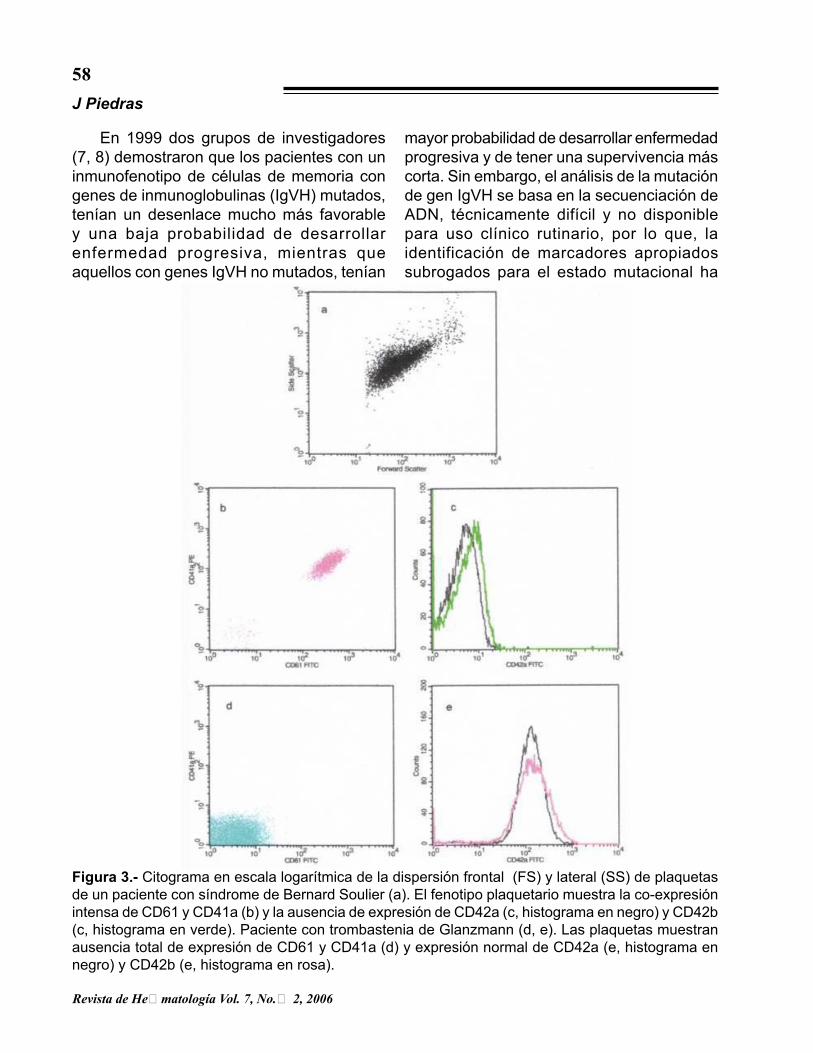
Figura 3.- Citograma en escala logarítmica de la dispersión frontal (FS) y lateral (SS) de plaquetas de un paciente con síndrome de Bernard Soulier (a). El fenotipo plaquetario muestra la co-expresión intensa de CD61 y CD41a (b) y la ausencia de expresión de CD42a (c, histograma en negro) y CD42b (c, histograma en verde). Paciente con trombastenia de Glanzmann (d, e). Las plaquetas muestran ausencia total de expresión de CD61 y CD41a (d) y expresión normal de CD42a (e, histograma en negro) y CD42b (e, histograma en rosa).
58
Revista de Hematología Vol. 7, No. 2, 2006
J Piedras

Revista de Hematología Vol. 7, No. 2, 2006
59
atraído la atención de muchos investigadores (9). Aún cuando hay una tendencia para que las anormalidades cariotípicas adversas se presenten principalmente en los pacientes con IgVH no mutado, la asociación no es absoluta y el estado mutacional del gen de la inmunoglobulina y el cariotipo son factores pronósticos independientes (10). En estudios con microarreglos de DNA (11) se demostró que las células con el gen IgVH no mutado se podían distinguir de aquellas con el gen mutado por la expresión diferencial de un pequeño número de genes, uno de los cuales codifica para la proteína zeta-asociada de 70 kDa (ZAP-70). La ZAP-70 es una molécula de señalización clave para linfocitos T y NK y aún cuando no se expresa en linfocitos B normales se asocia con incremento en la señalización intracelular del receptor de las Igs en las células de LLC-B. Tomando en cuenta las publicaciones recientes, el ZAP-70 es el marcador subrogado más promisorio para el estado de mutación del IgVH (12). El CD38 es una proteína membranal que marca la activación y maduración celular y tiene actividad de señalización. La expresión de CD38 en células de LLC-B se asocia con un pronóstico global de la enfermedad menos favorable y en los estudios iniciales se consideró como un marcador subrogado para los dos subgrupos importantes de la LLC-B : IgVH mutado y no mutado (8). Sin embargo, después de la publicación de un buen número de trabajos, se ha podido demostrar claramente que aún cuando la expresión de CD38 tiene cierto grado de correlación con el estado de mutación del IgVH, ambos parámetros se consideran variables pronósticas independientes en LLC-B (13, 14). La información pronóstica dada por la expresión de ZAP-70 y CD38 es complementaria. Bakke y col. (15) y Gibbs G y col. (16) demostraron que el método
citofluorográfico para identificar células ZAP-70 en LLC no está completamente estandarizado entre los diferentes laboratorios. En el trabajo de Bakke y col. (15) se analizó la expresión de ZAP-70 en células de LLC empleando 4 diferentes anticuerpos comerciales; con uno de ellos los porcentajes de expresión de ZAP-70 fueron consistentemente elevados, mientras que con los otros tres anticuerpos se obtuvo una amplia oscilación de porcentajes de expresión de ZAP-70 en las células tumorales, mostrando además una pobre correlación entre todos. En este estudio se concluyó que las estrategias analíticas previas no eran reproducibles, y se propone una relación métrica, con la cual se obtuvieron mejores correlaciones. Gibbs y col. (16) compararon la medición de la expresión de ZAP-70 en LLC empleando dos anticuerpos diferentes y dos métodos de tinción. Con el anticuerpo de Caltag y el reactivo de permeabilización de Fix and Perm se obtuvo 91% de concordancia entre la expresión de ZAP-70 y el estado de mutación del gen IgVH. Al igual que otros autores no hubo correlación entre la expresión de CD38 ni con ZAP-70 ni con el estado de IGVH. Los autores consideran que la expresión de ZAP-70 empleando la combinación anticuerpo Caltag/Fix and Perm podría ser introducida en el panel de inmunonofenotipificación de LLC-B.
IDENTIFICACIÓN DE GLICOPROTEÍNAS PLAQUETARIAS.
El estudio de pacientes con alteraciones genéticas de la función plaquetaria ha hecho posible entender la relación crítica estructura-función que determina la función normal de la plaqueta (17). La CF, empleando la dispersión de la luz y los marcadores inmunológicos, permite la identificación inequívoca de las plaquetas en sangre o en mezclas
Citometría de flujo en padecimientos hematológicos.

60
Revista de Hematología Vol. 7, No. 2, 2006
complejas. Mediante la CF se puede aislar electrónicamente a las plaquetas de la sangre total o del plasma rico en plaquetas, evitando la manipulación y en consecuencia la activación de las mismas. Esta metodología ha permitido identificar partículas en la sangre que son fácilmente confundidas con plaquetas, como fragmentos de eritrocitos o de leucocitos, complejos inmunes y microorganismos. La heterogeneidad en el tamaño de las plaquetas (de 1 µm a 4 ó 5 µm) excede al de cualquier otra célula hematológica, similar en tamaño a una bacteria y más grande que los complejos inmunes (18). En la preparación de plaquetas para análisis citofluorométrico es necesario adicionar EDTA al diluyente para evitar agregación de las plaquetas. Una aplicación directa de la CF en la evaluación clínica de las plaquetas es la cuantificación de las principales glicoproteínas de superficie de las mismas. El fenotipo plaquetario se ha usado para definir varios síndromes genéticos, resultantes de la expresión anormal de receptores de superficie, entre otras la trombastenia de Glanzmann y el síndrome de Bernard Soulier. La trombastenia de Glanzmann es una enfermedad autosómica recesiva, caracterizada por alteración en la agregación plaquetaria con ausencia de respuesta a los agonistas necesarios para que el fibrinógeno se una y forma microagregados. Se caracteriza por una anormalidad cualitativa y cuantitativa del complejo glicoproteíco IIb/IIIa en la membrana plaquetaria (19). Existen en el mercado diferentes anticuerpos monoclonales que se pueden usar con CF para demostrar el grado de deficiencia del complejo GP IIb/IIIa (CD41a y CD61, respectivamente) en la superficie de la plaqueta. La principal ventaja de usar la CF como prueba confirmatoria del diagnóstico es que los estudios a los miembros de la familia permiten establecer la severidad del defecto en el caso índice y
el grado de herocigocidad en los familiares. Además con la CF se puede determinar en los heterocigotos si la expresión de GPIIb/IIIa se encuentra uniformemente distribuida en todas las plaquetas, o si tienen una subpoblación de plaquetas con ausencia completa de la glicoproteína. En el síndrome de Bernard Soulier la anormalidad morfológica más consistente es la presencia de plaquetas gigantes (>10 um), aparentemente como resultado de activación in vitro de las mismas. En agregometría el único hallazgo anormal es la respuesta de las plaquetas Bernard-Soulier a trombina o a ristocetina más factor WV. En este síndrome la CF en sangre total permite el análisis de plaquetas sin necesidad de realizar el procedimiento técnicamente difícil de separar físicamente las plaquetas gigantes, de eritrocitos y de leucocitos de tamaño similar. La anormalidad bioquímica de las plaquetas en esta trombastenia, es la ausencia del complejo glicoproteico1b/IX/V en la membrana de la plaqueta y del megacariocito (20). La GPIX (CD42a) y la GP1bα,β (CD42b) son miembros de la familia de glicoproteínas ricas en leucina necesaria para la interacción del factor de Von Willebrand y la membrana plaquetaria esencial para una adhesión normal.
Detección de inmunoglobulinas asociadas a plaquetas. Varios investigadores empleando la CF han encontrado que las plaquetas de la mayoría de los pacientes con púrpura trombocitopénica inmunológica (PTI) tienen cantidades elevadas de inmunoglobulinas en la superficie de las plaquetas (21, 22). Sin embargo, en otros estudios empleando la misma metodología, se ha demostrado que otras formas de trombocitopenia también pueden tener concentraciones elevados de inmunoglobulinas en las plaquetas (23). En un estudio que incluyó, además de controles
J Piedras

Revista de Hematología Vol. 7, No. 2, 2006
61
normales pacientes con trombocitopenias no inmunológicas encontramos una sensibilidad de 90.3% con especificidad de 39.3%, en el diagnóstico de PTI, concluyéndose que la detección, por CF, de inmunoglobulinas asociadas a plaquetas, constituye un ensayo sensible pero no específico, haciéndolo innecesario e inapropiado para establecer el diagnóstico de PTI (24).
REFERENCIAS.1.- Coons AH, Kaplan MH. Localization of antigen in tissue cells. II. Improvements in a method for the detection of antigen by means of fluorescent antibody. J Exp Med 1950;91:1-13.
2.- Stewart CC, Goolsby Ch, Shackney SE. Emerging technology and future development in flow cytometry. Hematol Oncol Clin North Am 2002;16:477-95.
3.- Ruiz-Argüelles A, Duque RE, Orfao A. Report on the first Latin American consensus conference for flor cytometric immnophenotyping of leukemia. Cytometry 1998;343:39-42.
4.- Ruiz-Argüelles A, Rivadeneyra-Espinoza L, Duque RE, Orfao A. Report on the Second Latin American Consensus Conference for flow cytometric immunophenotyping of hematological malignancies. Cytometry B Clin Cytom 2006;70:39-44.
5.- Riley RS, Massey D, Jackson-Cook C, Idowu M, Romagnoli G. Immunophenotypic analysis of acute lymphocytic leukemia. Hematol Oncol Clin North Am 2002;16:245-99.
6.- Todd WM. Acute myeloid leukemia and related conditions. Hematol Oncol Clin North Am 2002;16:301-319.
7.- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848-54.
8.- Damle RN, Wasil T, Fais F, Ghiotto F, Jaletto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999;94:1840-7.
Citometría de flujo en padecimientos hematológicos.
9.- Montillo M, Hamblin T, Hallek M, Montserrat E, Morra E. Chronic lymphocytic leukemia: novel prognostic factors and their relevance for risk-adopted therapeutic strategies. Haematologica 2005;90:391-9.
10.- Krober A, Seller T, Benner A, Bullinger L, Bruckle E, Lichter P, et al. VH mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 2002;100:1410-6
11.- Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia . J Exp Med 2001;194:1639-47.
12.- Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003; 101:4944-51.
13.- Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003; 348:1764-75.
14.- Deaglio S, Capobianco A, Bergui L, Düring J, Morabito F, Dührsen U, et al. CD38 is a signaling molecule in B cell chronic lymphocytic leukemia cells. Blood 2003; 102:2146-55.
15.- Bakke AC, Purtzer Z, Leis J, Huang J. A robust ratio metric mehod for analysis of Zap-70 expression in chronic lymphocytic leukemia (CLL) Cytometry B Clin Cytom 2005; en prensa (diciembre 2005).
16.- Gibbs G, Bromidge T, Howe D, Hopkins J, Johnson S. Comparison of flow cytometric methods for he measurement of ZAP-70 expression in a routine diagnostic laboratory. Clin Lab Haematol 2005; 27:258-66.
17.- Parker S. Qualitative disorders of platelet function. En: Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F Glader B (eds.) Wintrobe’s Clinical Hematology 11th edition. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 1603-18.

18.- Hickerson DHM, Bode AP. Flow cytometry of platelets for clinical analysis. Hematol Oncol Clin N Am 2002;16:421-54.
19.- George JN, Caen JP, Nurden AT. Glansmann’s trombastenia: the spectrum of clinical disease. Blood 1990;75:1383-95.
20.- López JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndome. Blood 1998;91:4397-418.
21.- Ault KA,Flow cytometric measurement of platelet-associated immunoglobulin. Pathol Immunophatol Res 1988; 7:395-408.
22.- Rosenfeld CS, Nichols G, Bodensteiner DC. Flow cytometric measurement of anti-platelet antibodies. Am J Clin Pathol 1987;87:518-22.
23.- Helm M, Peterson B. Detection of platelet-associated immunoglobulin in immune thrombocytopenia by flow cytometry. Diagn Clin Immunol 1988; 5:309-13.
24.- Romero-Guzman LT, López-Karpovitch X, Paredes R, Barrales-Benitez O, Piedras J. Detection of platelet-associated immunoglobulin by flow cytometry for the diagnosis of immune thrombocytopenia: a prospective study and critical review Haematologica 2000; 85:627-31.
62
Revista de Hematología Vol. 7, No. 2, 2006
J Piedras

Revista de Hematología Vol. 7, No. 2, 2006
63
Citometría de flujo en el diagnóstico y clasificación de padecimientos hematológicos: hemoglobinuria paroxística
nocturna.
Dr. Xavier López-Karpovitch.
Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México, D.F., México.
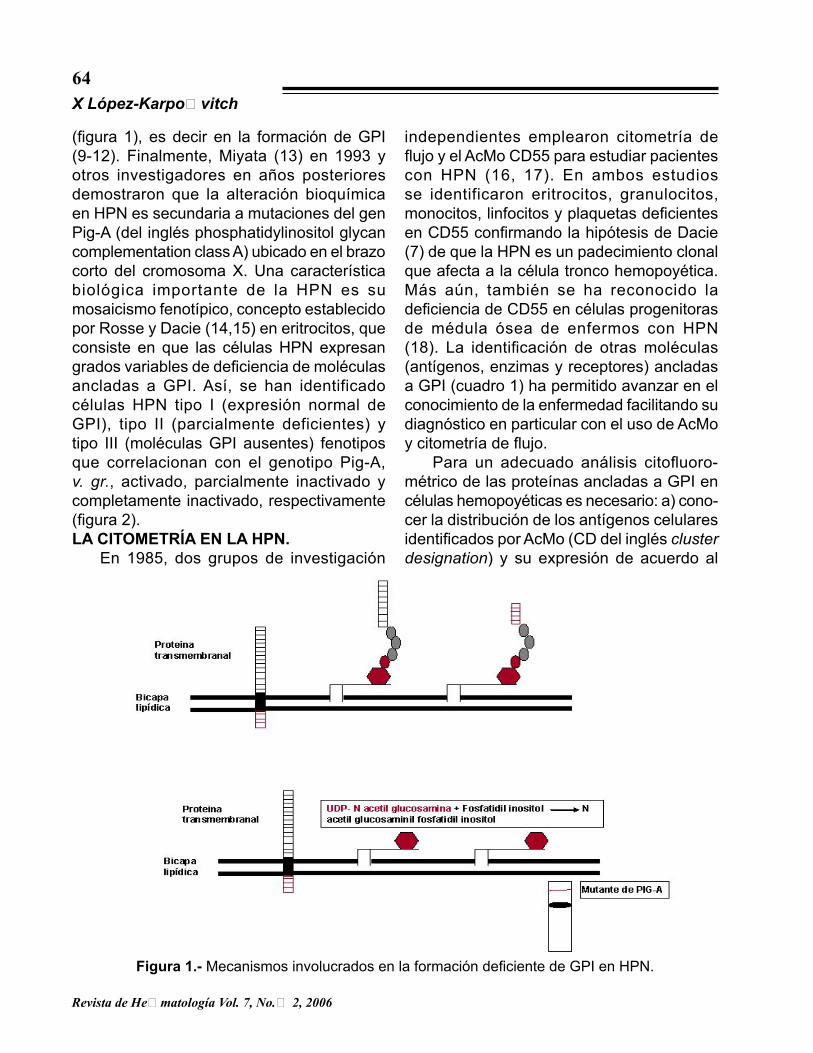
con síndrome mielodisplásico. Un estudio realizado en México en 164 enfermos con HPN mostró que 44 % de ellos presentaron aplasia, 29 % hemolisis, 25 % mielodisplasia y 2 % trombosis (3). Cincuenta años después de la primera descripción de la enfermedad, Ham y Dingle (4) demostraron que los eritrocitos HPN se destruían por presentar mayor sensibilidad al complemento al acidificar el suero y posteriormente otros autores encontraron que también los granulocitos y las plaquetas mostraban el mismo fenómeno (5-7). En 1983 se demostró la deficiencia del factor acelerador del decaimiento (del inglés decay acceleration factor [DAF] o CD55), proteína anclada a la superficie celular mediante GPI que inhibe la formación de convertasas de C3, en la membrana de los eritrocitos de pacientes con HPN (8). Este hallazgo culminó en la demostración de que todas las proteínas ancladas a GPI se encuentran disminuidas o ausentes en HPN y que el anclaje anómalo de las mismas se debe a una alteración bioquímica en la transferencia de N-acetilglucosamina a fosfatidilinositol
Solicitud de reimpresos: Dr. Xavier López-Karpovitch. Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición SZ. Vasco de Quiroga 15, Colonia Sección XVI, Delegación Tlalpan 14000, México, D.F., México. Tel.: 54 870 900 Ext. 2704
INTRODUCCIÓN. Dentro de la gran variedad de métodos de citometría de flujo empleados en la clínica existen pocos capaces de identificar enfermedades específicas. La detección de moléculas ancladas a glucofosfatidilinositol (GPI) en la superficie celular mediante anticuerpos monoclonales (AcMo) y citometría de flujo son la base de una prueba específica para el diagnostico de hemoglobinuria paroxística nocturna (HPN) (1).CARACTERÍSTICAS DE LA HPN. La HPN es un padecimiento clonal del tejido hemopoyético, adquirido y de etiología desconocida. El amplio espectro clínico y curso variable de la enfermedad son un reto para el clínico en términos de diagnóstico y manejo. La presentación clínica clásica del padecimiento es la anemia hemolítica, intravascular, lo que llevó a Strübing (2) a denominarlo como HPN por acompañarse de hemoglobinuria durante la noche. Además de la hemolisis, al diagnóstico esta entidad patológica puede presentarse como aplasia medular, trombosis e inclusive confundirse
64
Revista de Hematología Vol. 7, No. 2, 2006
(figura 1), es decir en la formación de GPI (9-12). Finalmente, Miyata (13) en 1993 y otros investigadores en años posteriores demostraron que la alteración bioquímica en HPN es secundaria a mutaciones del gen Pig-A (del inglés phosphatidylinositol glycan complementation class A) ubicado en el brazo corto del cromosoma X. Una característica biológica importante de la HPN es su mosaicismo fenotípico, concepto establecido por Rosse y Dacie (14,15) en eritrocitos, que consiste en que las células HPN expresan grados variables de deficiencia de moléculas ancladas a GPI. Así, se han identificado células HPN tipo I (expresión normal de GPI), tipo II (parcialmente deficientes) y tipo III (moléculas GPI ausentes) fenotipos que correlacionan con el genotipo Pig-A, v. gr., activado, parcialmente inactivado y completamente inactivado, respectivamente (figura 2).LA CITOMETRÍA EN LA HPN. En 1985, dos grupos de investigación
independientes emplearon citometría de flujo y el AcMo CD55 para estudiar pacientes con HPN (16, 17). En ambos estudios se identificaron eritrocitos, granulocitos, monocitos, linfocitos y plaquetas deficientes en CD55 confirmando la hipótesis de Dacie (7) de que la HPN es un padecimiento clonal que afecta a la célula tronco hemopoyética. Más aún, también se ha reconocido la deficiencia de CD55 en células progenitoras de médula ósea de enfermos con HPN (18). La identificación de otras moléculas (antígenos, enzimas y receptores) ancladas a GPI (cuadro 1) ha permitido avanzar en el conocimiento de la enfermedad facilitando su diagnóstico en particular con el uso de AcMo y citometría de flujo. Para un adecuado análisis citofluoro-métrico de las proteínas ancladas a GPI en células hemopoyéticas es necesario: a) cono-cer la distribución de los antígenos celulares identificados por AcMo (CD del inglés cluster designation) y su expresión de acuerdo al
X López-Karpovitch
Figura 1.- Mecanismos involucrados en la formación deficiente de GPI en HPN.

Revista de Hematología Vol. 7, No. 2, 2006
65
Cuadro 1Expresión de moléculas ancladas a GPI en
células hemopoyéticas.Antígeno Distribución celularCD14 Monocitos y granulocitosCD16 Neutrófilos, células NK y subpoblación de linfocitos TCD24 Células B y granulocitosCD48 Linfocitos y monocitosCD52 (Campath) Linfocitos y monolitosCD55 (DAF) Todas las células hemopoyéticasCD58 (LFA-3) Todas las células hemopoyéticasCD59 (MIRL) Todas las células hemopoyéticasCD66b GranulocitosCD66c GranulocitosCD66e GranulocitosCD73 Subpoblaciones de células T y BCD87 Células T, NK, neutrófilos y monolitosCD90 Subpoblaciones células tronco hemopoyéticas y TCD108 Eritrocitos y linfocitosCD109 Células T activadas, plaquetas, megacariocitos, subpoblaciones de células tronco hemopoyéticasCD157 Células estromales de médula ósea, monocitos y granulocitos
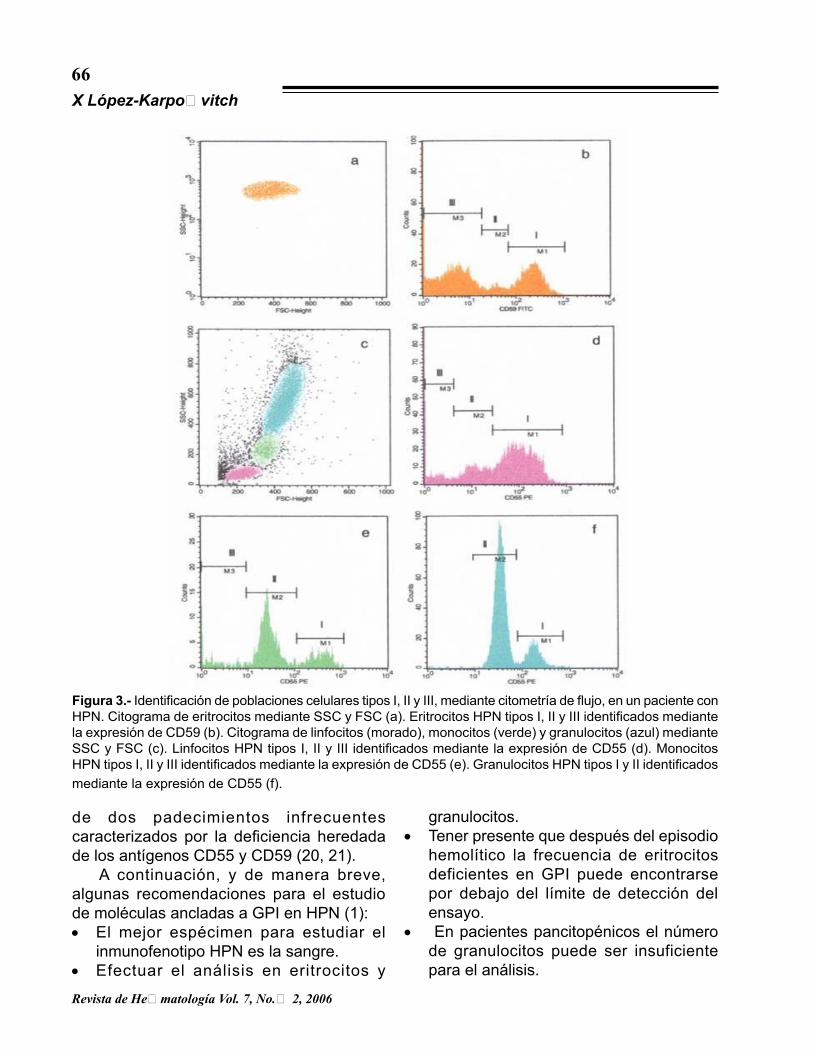
estadio de diferenciación de las células hemo-poyéticas, b) emplear AcMo conjugados con fluorocromos (fluorescencia directa) y c) estar familiarizado con las estrategias electrónicas de “ventaneo” (del inglés gatting), así como con los términos de dispersión lumínica frontal (FSC del inglés forward scatter que discrimina tamaño celular) y lateral (SSC del inglés side scatter que discrimina características intrace-lulares). La figura 3 ilustra un caso de HPN en el que se analizó la expresión del antígeno CD55 en linfocitos, monocitos y granulocitos, así como la expresión de CD59 en eritrocitos. Tanto en eritrocitos como en linfocitos y monocitos se identificaron poblaciones tipos I, II y III, mientras que en granulocitos se identificaron poblaciones tipos I y II. Aún cuando en la actualidad no se cuenta con lineamientos consensuados para establecer el diagnóstico definitivo de HPN mediante citometría de flujo, por ello, Hall y Rosse (19) recomiendan identificar la deficiencia de dos moléculas ancladas a GPI en por lo menos dos poblaciones celulares. Esta recomendación se da debido a la existencia
Citometría de flujo en el diagnóstico de la HPN.
Figura 2.- Representación esquemática del mosaicismo en HPN.
66
Revista de Hematología Vol. 7, No. 2, 2006
de dos padecimientos infrecuentes caracterizados por la deficiencia heredada de los antígenos CD55 y CD59 (20, 21). A continuación, y de manera breve, algunas recomendaciones para el estudio de moléculas ancladas a GPI en HPN (1):• El mejor espécimen para estudiar el
inmunofenotipo HPN es la sangre.• Efectuar el análisis en eritrocitos y
granulocitos.• Tener presente que después del episodio
hemolítico la frecuencia de eritrocitos deficientes en GPI puede encontrarse por debajo del límite de detección del ensayo.
• En pacientes pancitopénicos el número de granulocitos puede ser insuficiente para el análisis.
Figura 3.- Identificación de poblaciones celulares tipos I, II y III, mediante citometría de flujo, en un paciente con HPN. Citograma de eritrocitos mediante SSC y FSC (a). Eritrocitos HPN tipos I, II y III identificados mediante la expresión de CD59 (b). Citograma de linfocitos (morado), monocitos (verde) y granulocitos (azul) mediante SSC y FSC (c). Linfocitos HPN tipos I, II y III identificados mediante la expresión de CD55 (d). Monocitos HPN tipos I, II y III identificados mediante la expresión de CD55 (e). Granulocitos HPN tipos I y II identificados mediante la expresión de CD55 (f).
X López-Karpovitch

Revista de Hematología Vol. 7, No. 2, 2006
67
En resumen, el análisis de proteínas ancladas a GPI mediante citometría de flujo es una técnica específica y sensible para el diagnóstico de HPN. Además, con esta metodología es posible estimar el tamaño de la clona HPN (22).
REFERENCIAS.1.- Richards SJ, Rawstron AC, Hillmen P. Application of flow cytometry to the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry 2000;42:223-33.
2.- Strübing P. Paroxysmal hemoglobinuria. Dtsch Med Wochenschr 1882;8:1-3.
3.- Góngora-Biachi RA. Hemoglobinuria paroxística nocturna: Nuevos conceptos de una vieja enfermedad. En Góngora-Biachi RA, editor. Hematología: Actualización 2005. Mérida: Agrupación Mexicana para el Estudio de la Hematología, A.C.; 2005. p. 31-9.
4.- Ham TH, Dingle JH. Studies on the destruction of red blood cells . II. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria. Certain immunological aspects of the hemolytic mechanism with special reference to serum complement. J Clin Invest 1939;18:657-72.
5.- Beck WS, Valentine WN. Biochemical studies on leucocytes. II. Phosphate activity in chronic lymphatic leukemia acute and miscellaneous hematologic conditions. J Lab Clin Med 1951;38:245-53.
6.- Lewis SM, Dacie JV. Neutrophil (leucocyte) alkaline phosphatase in paroxysmal nocturnal haemoglobinuria. Br J Haematol 1965;11:549-56.
7.- Dacie JV. Paroxysmal nocturnal haemoglobinuria. Proc R Soc Med 1963;56:587-96.
8.- Nicholson-Weller A, March JP, Rosenfeld JP, Austen KF. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein decay acceleration factor. Proc Natl Acad Sci USA 1983;80:5066-70.
9.- Holguin MH, Frederick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of
paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989;84:7-17.
10.- Armstrong C, Schubert J, Ueda E, Knez JJ, Gelperin D, Hirose S, et al. Affected paroxysmal nocturnal hemoglobinuria T lymphocytes harbour a common defect in assembly of N-acetyl-D-glucosamine inositol phospholipids corresponding to that in class A Thy-1-murine lymphoma mutants. J Biol Chem 1992;267:25347-51.
11.- Hillmen P, Bessler M, Mason PJ, Watkins WM, Luzzatto I. Specific defect in N-acetylglucosamine incorporation in the GPI-anchor synthetic pathway in cloned cell lines from patients with paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA 1993;90:5272-6.
12.- Takahashi M, Takeda J, Hirose S, Hyman R, Inoue N, Miyata T, et al. Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate od glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. J Exp Med 1993;177:517-21.
13.- Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosíntesis. Science 1993;259:1318-20.
14.- Rosse WF. Variations in the red cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol 1973;24:327-42.
15.- Rosse WF, Dacie JV. The role of complement in the sensitivity of the paroxysmal nocturnal hemoglobinuria red cell to immune lysis. Bibl Haematol 1965;23:11-8.
16.- Kinoshita T, Medof ME, Silber R, Nussenzweig V. Distribution of decay-accelerating factor in the peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J Exp Med 1985;162:75-92.
17.- Nicholson-Weller A, Spicer DB, Austen KF. Deficiency of the complement regulatory protein “decay-accelerating factor” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. N Engl J Med 1985;312:1091-7.
18.- Kanamaru A, Okuda K, Ueda E, Kitani T, Kinoshita T, Nagai K. Different distribution of decay accelerating
Citometría de flujo en el diagnóstico de la HPN.

factor on hemopoietic progenitors from normal individuals and patients with paroxysmal nocturnal hemoglobinuria. Blood 1988;72:507-11.
19.- Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood 1996;87:5332-40.
20.- Reid ME, Mallinson G, Sim RB, Poole J, Pausch V, Merry AH, et al. Biochemical studies on red cells from a patient with Inab phenotype (decay-accelerating factor deficiency). Blood 1991;78:3291-7.
21.- Yamashina M, Ueda E, Kinoshita T, Takami T, Ojima A, Ono H, et al. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N. Engl J Med 1990;323:1184-9.
22.- Piedras J, López-Karpovitch X. Flow cytometry analysis of glycosylphosphatidyl-inositol-anchored proteins to assess paroxysmal nocturnal hemoglobinuria clone size. Cytometry 2000;42:234-8.
68
Revista de Hematología Vol. 7, No. 2, 2006
X López-Karpovitch

Revista de Hematología Vol. 7, No. 2, 2006
69
INTRODUCCIÓN En medicina el término quimera se emplea para designar a aquel individuo cuyo cuerpo contiene poblaciones celulares que provienen de diferentes individuos de la misma o de diferente especie, ya sea espontánea o artificialmente (1). La coexistencia de células de dos diferentes organismos en el cuerpo es llamado quimerismo. El trasplante de médula ósea (TMO) es una modalidad de tratamiento cuyo objetivo es proporcionar las condiciones para la reconstitución de la médula ósea dañada. En el TMO alogénico el donante pertenece a la misma especie que el receptor, presenta la mayor compatibilidad y generalmente es un familiar de primer grado (2-6). Se pueden presentar diferentes grados de quimerismo en el TMO. Cuando el paciente no presenta ninguna evidencia de células del receptor después de un tiempo del trasplante se considera quimerismo completo; mientras que si el paciente presenta evidencia de células tanto del receptor como células del donador se considera como quimerismo mixto (cuadro 1)(8). Con el surgimiento de procedimientos de trasplante cada vez más avanzados se
Quimerismo por microsatélites.
Olga Verónica Barrales-Benitez, Samuel Canizales-Quinteros.
Departamento de Hematología y Oncología. Departamento de Medicina Genómica y Biología Molecular, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán,
México, D.F., México.
Solicitud de reimpresos: Olga V. Barrales-Benitez. Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición SZ. Vasco de Quiroga 15, Colonia Sección XVI, Delegación Tlalpan, C.P. 14000, México, D.F., México. Tel.: 54 870 900 Ext. 2704
ha hecho necesario establecer el grado de quimerismo en los pacientes después del trasplante con el objeto de: a) verificar el éxito del trasplante, b) prevenir recaída de la enfermedad, c) iniciar inmunoterapia (linfocitos del donador) para evitar rechazo, d) observar el origen de las recaídas debidas y e) intentar nuevos esquemas de acondicionamiento (2, 5-7, 9, 10).
METODOLOGÍA. El fundamento en el que se basa la detección del grado de quimerismo es el empleo de diferentes marcadores genéticos polimórficos entre donador y receptor. La elección del marcador y de la técnica a emplear depende de la sensibilidad y especificidad, de si hay diferencia de género entre donador y receptor, y del tipo de enfermedad del paciente pre-trasplante. Las diferentes técnicas de rutina empleadas para detectar grado de quimerismo se muestran en el cuadro 2. A pesar de las diferencias en los protocolos de estas metodologías, todas se basan en que el receptor y el donador son estudiados antes del trasplante en la búsqueda de marcadores genéticos diferentes, los cuales se consideran marcadores “informativos” (figura1). Un
70
Revista de Hematología Vol. 7, No. 2, 2006
OV Barrales-Benitez, S Canizales-Quinteros
marcador genético se considera “informativo” cuando: a) se encuentra ampliamente distribuido en diferentes cromosomas; b) presenta gran variabilidad o polimorfismo, lo que permite demostrar múltiples alelos y por lo tanto heterocigocidad. A continuación estos marcadores son analizados en el paciente post TMO alogénico para medir y cuantificar
la cantidad de células del receptor y del donador, y establecer el grado de quimerismo (8). Uno de los métodos más empleado por diversos investigadores se basa en la amplificación por Reacción en Cadena de la Polimerasa (PCR) de un sistema STR/VNTR altamente polimórfico que se considera muy
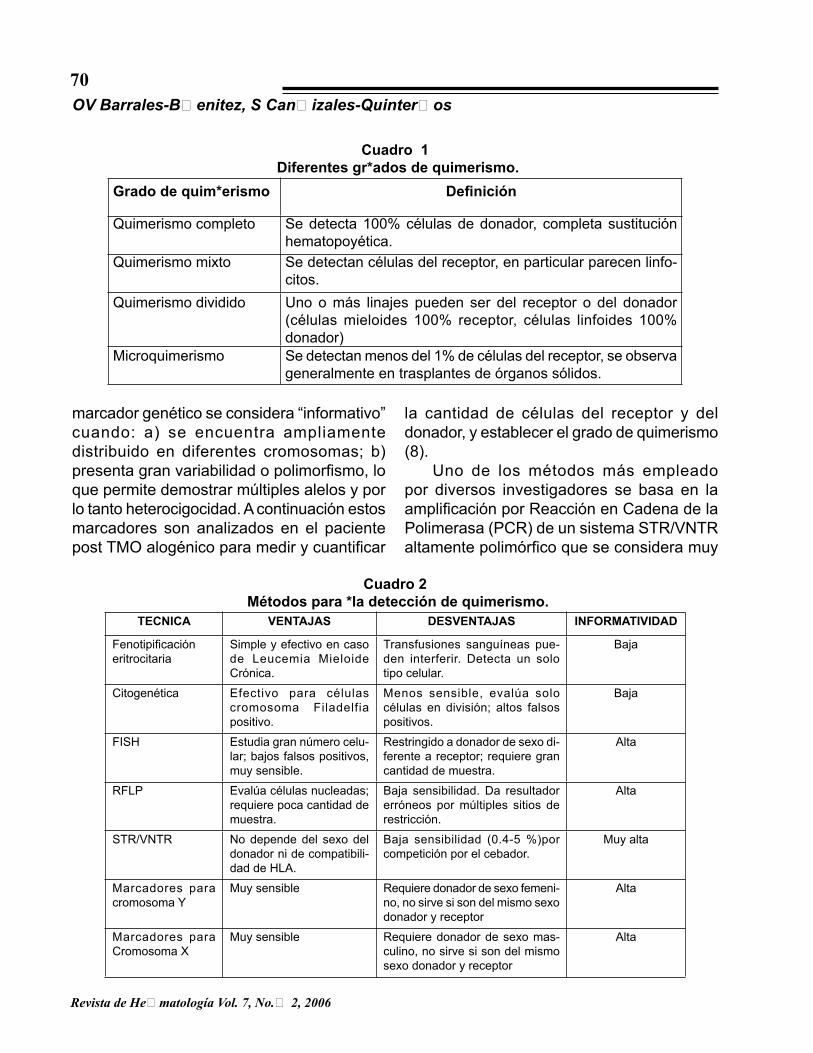
Cuadro 1Diferentes grados de quimerismo.
Grado de quimerismo Definición
Quimerismo completo Se detecta 100% células de donador, completa sustitución hematopoyética.
Quimerismo mixto Se detectan células del receptor, en particular parecen linfo-citos.
Quimerismo dividido Uno o más linajes pueden ser del receptor o del donador (células mieloides 100% receptor, células linfoides 100% donador)
Microquimerismo Se detectan menos del 1% de células del receptor, se observa generalmente en trasplantes de órganos sólidos.
Cuadro 2Métodos para la detección de quimerismo.
TECNICA VENTAJAS DESVENTAJAS INFORMATIVIDAD
Fenotipificacióneritrocitaria
Simple y efectivo en caso de Leucemia Mieloide Crónica.
Transfusiones sanguíneas pue-den interferir. Detecta un solo tipo celular.
Baja
Citogenética Efectivo para células cromosoma Filadelfia positivo.
Menos sensible, evalúa solo células en división; altos falsos positivos.
Baja
FISH Estudia gran número celu-lar; bajos falsos positivos, muy sensible.
Restringido a donador de sexo di-ferente a receptor; requiere gran cantidad de muestra.
Alta
RFLP Evalúa células nucleadas; requiere poca cantidad de muestra.
Baja sensibilidad. Da resultador erróneos por múltiples sitios de restricción.
Alta
STR/VNTR No depende del sexo del donador ni de compatibili-dad de HLA.
Baja sensibilidad (0.4-5 %)por competición por el cebador.
Muy alta
Marcadores para cromosoma Y
Muy sensible Requiere donador de sexo femeni-no, no sirve si son del mismo sexo donador y receptor
Alta
Marcadores para Cromosoma X
Muy sensible Requiere donador de sexo mas-culino, no sirve si son del mismo sexo donador y receptor
Alta

Revista de Hematología Vol. 7, No. 2, 2006
71
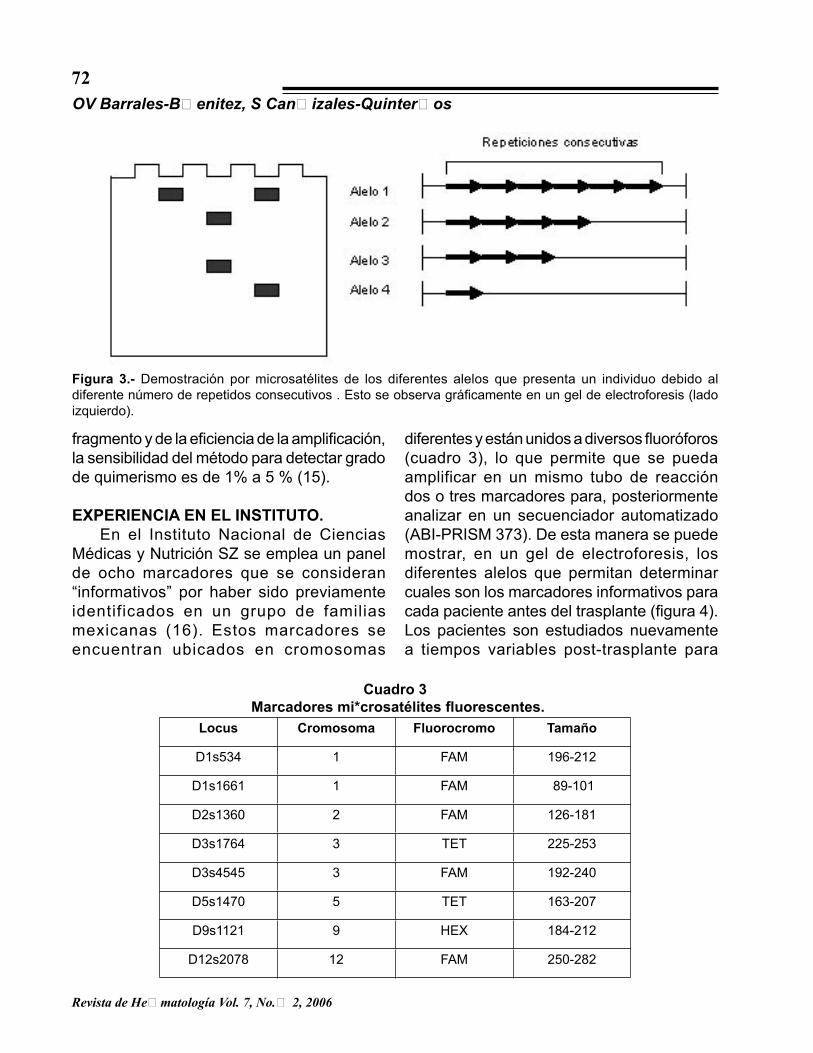
informativo y sensible (11). Los STR (del inglés short tandem repetition, repetidos cortos consecutivos) y los VNTR (del inglés variable number tandem repetition, número variable de repetidos consecutivos) son secuencias repetidas agrupadas de DNA que constituyen aproximadamente de un 10 a un 15% del genoma y consisten en formaciones de varias repeticiones cortas que se organizan consecutivamente (en tandem). Las familias de DNA satélite varían de acuerdo con su localización en el genoma, la extensión total de la formación consecutiva y la longitud de las unidades repetidas que constituyen la formación (12). Cuando la secuencia de repeticiones consecutivas tiene una longitud de 2 a 8 nucleótidos se denomina STR o microsatélite (figura 2); y si tiene de 15 a 50 nucleótidos se llama VNTR o minisatélite (13,14). Estas secuencias polimórficas son heredadas de manera mendeliana codominante. Existen tres características importantes de estas secuencias STR o VNTR que las hacen muy útiles para los estudios de ligamiento genético. Primero, un microsatélite con
frecuencia tiene varios alelos (extensiones de nucleótidos repetidas) presentes en la población, haciendo que la probabilidad de que un individuo sea heterocigoto sea mayor del 70% (figura 3). Los marcadores más informativos tienen hasta varias docenas de alelos o más y, por lo tanto, resulta poco probable que dos individuos no emparentados compartan los mismos alelos Segundo, a diferencia del análisis con RFLP (del ingles restriction fragment lenght polimorfism polimorfismo en la longitud del fragmento de restricción) o con VNTR, la genotipificación con microsatélites no requiere el uso de tediosas técnicas como Southern blot. Con la técnica de PCR se usan cebadores complementarios a secuencias específicas de DNA que flanquean los microsatélites y generan productos de amplificación que difieren en longitud, dependiendo de cuantos repetidos estén presentes (figura 3). Tercero, miles de microsatélites han sido totalmente identificados en el genoma humano, de manera que muy pocas regiones del genoma no pueden ser mapeadas usando estos marcadores (12) Por otra parte, el uso de amplificación del DNA permite determinar el grado de quimerismo con muy poca cantidad de muestra, una gran ventaja cuando el paciente esta rechazando el injerto y presenta severa leucopenia. Dependiendo de la longitud del
Figura 1.- Análisis de quimerismo para marcadores genéticos. A y C son marcadores informativos R = receptor; D = donador.
Figura 2.- Análisis de quimerismo por marcadores microsatélites. Repetidos consecutivos STR que muestran la diferencia en número de repetidos en dos cromosomas. F = cebador delantero, R = cebador inverso.
Quimerismo por microsatélites.
72
Revista de Hematología Vol. 7, No. 2, 2006
fragmento y de la eficiencia de la amplificación, la sensibilidad del método para detectar grado de quimerismo es de 1% a 5 % (15).
EXPERIENCIA EN EL INSTITUTO. En el Instituto Nacional de Ciencias Médicas y Nutrición SZ se emplea un panel de ocho marcadores que se consideran “informativos” por haber sido previamente identificados en un grupo de familias mexicanas (16). Estos marcadores se encuentran ubicados en cromosomas
Cuadro 3Marcadores microsatélites fluorescentes.
Locus Cromosoma Fluorocromo Tamaño
D1s534 1 FAM 196-212
D1s1661 1 FAM 89-101
D2s1360 2 FAM 126-181
D3s1764 3 TET 225-253
D3s4545 3 FAM 192-240
D5s1470 5 TET 163-207
D9s1121 9 HEX 184-212
D12s2078 12 FAM 250-282
Figura 3.- Demostración por microsatélites de los diferentes alelos que presenta un individuo debido al diferente número de repetidos consecutivos . Esto se observa gráficamente en un gel de electroforesis (lado izquierdo).
diferentes y están unidos a diversos fluoróforos (cuadro 3), lo que permite que se pueda amplificar en un mismo tubo de reacción dos o tres marcadores para, posteriormente analizar en un secuenciador automatizado (ABI-PRISM 373). De esta manera se puede mostrar, en un gel de electroforesis, los diferentes alelos que permitan determinar cuales son los marcadores informativos para cada paciente antes del trasplante (figura 4). Los pacientes son estudiados nuevamente a tiempos variables post-trasplante para
OV Barrales-Benitez, S Canizales-Quinteros
Revista de Hematología Vol. 7, No. 2, 2006
73
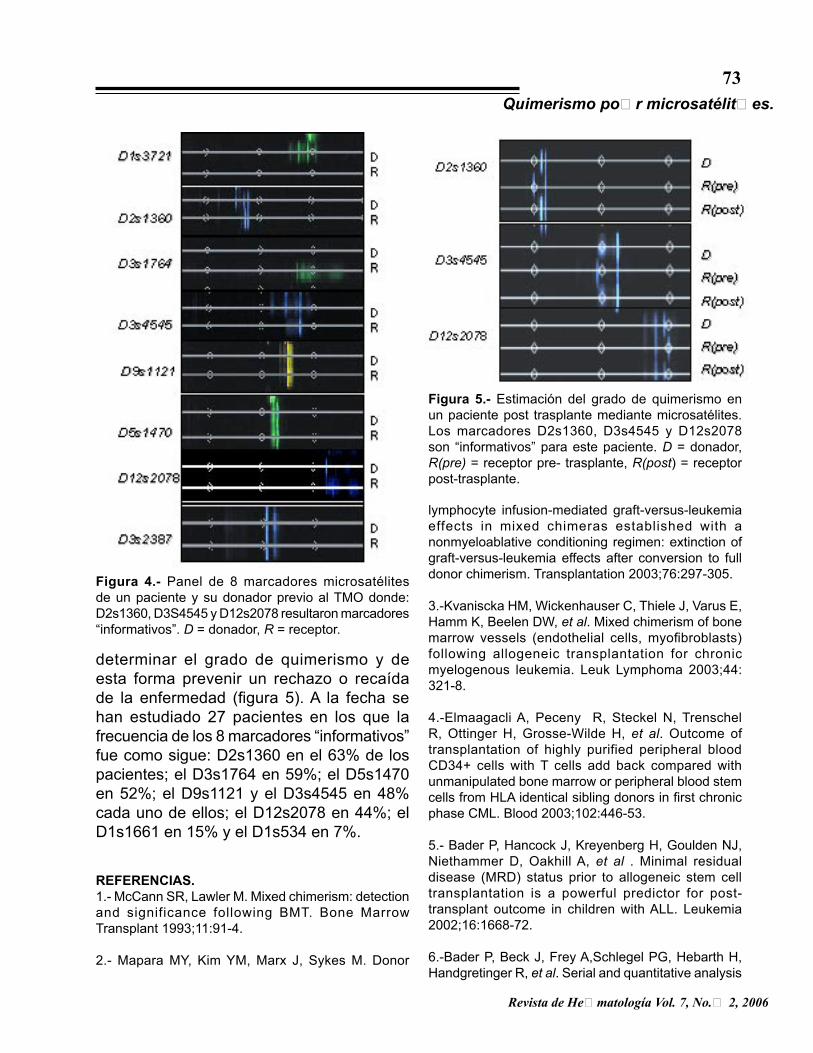
determinar el grado de quimerismo y de esta forma prevenir un rechazo o recaída de la enfermedad (figura 5). A la fecha se han estudiado 27 pacientes en los que la frecuencia de los 8 marcadores “informativos” fue como sigue: D2s1360 en el 63% de los pacientes; el D3s1764 en 59%; el D5s1470 en 52%; el D9s1121 y el D3s4545 en 48% cada uno de ellos; el D12s2078 en 44%; el D1s1661 en 15% y el D1s534 en 7%.
REFERENCIAS.1.- McCann SR, Lawler M. Mixed chimerism: detection and significance following BMT. Bone Marrow Transplant 1993;11:91-4.
2.- Mapara MY, Kim YM, Marx J, Sykes M. Donor
lymphocyte infusion-mediated graft-versus-leukemia effects in mixed chimeras established with a nonmyeloablative conditioning regimen: extinction of graft-versus-leukemia effects after conversion to full donor chimerism. Transplantation 2003;76:297-305.
3.-Kvaniscka HM, Wickenhauser C, Thiele J, Varus E, Hamm K, Beelen DW, et al. Mixed chimerism of bone marrow vessels (endothelial cells, myofibroblasts) following allogeneic transplantation for chronic myelogenous leukemia. Leuk Lymphoma 2003;44: 321-8.
4.-Elmaagacli A, Peceny R, Steckel N, Trenschel R, Ottinger H, Grosse-Wilde H, et al. Outcome of transplantation of highly purified peripheral blood CD34+ cells with T cells add back compared with unmanipulated bone marrow or peripheral blood stem cells from HLA identical sibling donors in first chronic phase CML. Blood 2003;102:446-53.
5.- Bader P, Hancock J, Kreyenberg H, Goulden NJ, Niethammer D, Oakhill A, et al . Minimal residual disease (MRD) status prior to allogeneic stem cell transplantation is a powerful predictor for post-transplant outcome in children with ALL. Leukemia 2002;16:1668-72.
6.-Bader P, Beck J, Frey A,Schlegel PG, Hebarth H, Handgretinger R, et al. Serial and quantitative analysis
Figura 4.- Panel de 8 marcadores microsatélites de un paciente y su donador previo al TMO donde: D2s1360, D3S4545 y D12s2078 resultaron marcadores “informativos”. D = donador, R = receptor.
Figura 5.- Estimación del grado de quimerismo en un paciente post trasplante mediante microsatélites. Los marcadores D2s1360, D3s4545 y D12s2078 son “informativos” para este paciente. D = donador, R(pre) = receptor pre- trasplante, R(post) = receptor post-trasplante.
Quimerismo por microsatélites.

74
Revista de Hematología Vol. 7, No. 2, 2006
of mixed hematopoietic chimerism by PCR in patients with acute leukemias allows the prediction of relapse after allogeneic BMT. Bone Marrow Transplant 1998; 2:487-95.
7.-Bader P, Holle W, Klingebiel T, Handgretinger R, Brenda N, Schlegel PG, et al. Mixed hematopoietic chimerism after allogeneic bone marrow transplantation: the impact of quantitative PCR analysis for prediction of relapses and graft rejection in children. Bone Marrow Transplant 1997; 19:697-702.
8.-Khan F, Agarwal A, Agrawal S. Significance of chimerism in hematopoietic stem cell transplantation: new variations on an old theme. Bone Marrow Transplant 2004; 34:1-12.
9.- Blazar BR, Lees CJ, Martin PJ, Noeller RJ, Kwon B, Murphy W, et al. Host T cells resist graft-versus-host disease mediated by donor leukocyte infusions. J Immunol 2000; 165:4901-9.
10.- Mackinnon S, Barnett L, Heller G, O’Reilly RJ. Minimal residual disease is more common in patients who have mixed T-cell chimerism after bone marrow transplantation for chronic myelogenous leukemia. Blood 1994; 83:3409-16.
11.- Choi SJ, Lee KH, Lee JH, Kim S, Chyng HJ, Lee JS, et al. Prognostic value of hematopoietic chimerism in patients with acute leukemia after allogeneic bone marrow transplantation: a prospective study. Bone Marrow Transplant 2000; 26:327-32.
12.- Nussbaum RL, McInnes RR, Willard HF. Thompson & Thompson. Genetics in medicine . 6ª. ed. Philadelphia: WB Saunders Company; 2001. p.31.
13.- Weber JL, May PE. Abundant class of DNA polymorphism which may be typed using the polymerase chain reaction. Am J Hum Genet 1989; 44:388-96.
14.- Jeffreys AJ, Wilson V, Neuman R, Keyte J. Amplification of human minisatellites by the polymerase chain reaction: Towards DNA fingerprinting of single cells. Nucleic Acids Res 1988; 16:10953-71.
15.- Thomas ED, Blume KG, Forman SJ. Hematopoietic cell transplantation. 2ª. ed. Massachussets: Blackwell Science; 1999. p. 201.
16.- Canizales-Quinteros S, Aguilar-Salinas CA, Reyes-Rodriguez E, Riba L, Rodríguez-Torres M, Ramirez-Jimenez S, et al. Locus on chromosome 6p linked to elevated HDL cholesterol serum levels and to protection against premature atherosclerosis in a kindred with familial hypercholesterolemia. Circ Res 2003; 92:569-76.
OV Barrales-Benitez, S Canizales-Quinteros

Revista de Hematología Vol. 7, No. 2, 2006
75
Talasemias: aspectos bioquímicos,moleculares y diagnósticos.
Beatriz Ibarra, Francisco José Perea.
División de Genética, Centro de Investigación Biomédica de Occidente, CMNO, Instituto Mexicano del Seguro Social, Guadalajara, Jalisco, México.
INTRODUCCIÓN. La Organización Mundial de la Salud (OMS) ha estimado que cerca del 7% de la población mundial son portadores de alguna hemoglobinopatía, y que al año nacen aproximadamente 370,000 niños homocigotos heterocigotos compuestos para anemia drepanocítica o para algún tipo de talasemia severa (1). Las talasemias son un grupo heterogéneo de anemias hemolíticas hereditarias que se caracterizan por la ausencia o disminución en la síntesis de una o más cadenas globínicas, que conducen a un desequilibrio en la relación de cadenas α/no-α. Las más comunes y clínicamente importantes son las talasemias α y β.
TALASEMIA ALFA. El término talasemia α (Tal-α) define los desórdenes hereditarios que afectan la síntesis de cadenas α globina. Debido a que las cadenas α están presentes tanto en hemoglobinas fetales y adultas, la deficiencia de su producción afectará la síntesis de la mayoría de las hemoglobinas. En la vida fetal la ausencia o reducción en la síntesis de
cadenas α resulta en un exceso de cadenas γ, la cual forma tetrámeros γ4 o hemoglobina Bart. En la vida adulta, el exceso de cadenas β, forma tetrámeros β4 o hemoglobina H (2). La familia de genes globínicos α se localiza en una extensión aproximada de 35 kilobases (Kb), región en la que se encuentran además aproximadamente 50 genes (factores de transcripción, dedos de zinc, enzimas, proteínas estructurales, etc.) y la conforman 4 genes, 3 de ellos funcionales: ζ que se expresa sólo en el período embrionario, los α2 y α1, que se sintetizan desde la etapa fetal. El gen θ se localiza en el extremo 3’ del gen α1, su expresión a nivel de ARNm se ha demostrado en saco vitelino, hígado fetal y células eritroides adultas, pero no hay evidencias de la formación de una proteína estable. En este bloque se encuentran también cuatro pseudogenes (ψζ, ψα2, ψα1 y ρ), 5 regiones hipervariables (secuencias de ADN altamente repetitivas) y 12 regiones Alu (secuencias medianamente repetitivas) (figura 1).
TALASEMIA BETA. La Talasemia β (Tal-β) muestra una
Solicitud de reimpresos: Beatriz Ibarra. División de Genética, Centro de Investigación Biomédica de Occidente, CMNO, Instituto Mexicano del Seguro Social. Sierra Mojada No. 800, Col. Independencia, C.P. 44340, Guadalajara, Jalisco, México. Tel.: 01 33 36 18 94 10

76
Revista de Hematología Vol. 7, No. 2, 2006
B Ibarra, FJ Perea
considerable heterogeneidad clínica y fenotípica revelada por los parámetros hematológicos y bioquímicos. Sí hay ausencia total de globina-β se llaman alelos β° y si sólo hay reducción de globina-β se denominan alelos β+. Con base en el estado clínico de los pacientes se clasifican en: a) Tal-β mayor o anemia de Cooley, b) Tal-β intermedia, y c) Tal-β menor (2-5). BASES MOLECULARES DE TALASEMIA β. Los genes globínicos no α o semejantes a β, se localizan en 11p15.5 tienen una extensión aproximada de 65 kilobases (Kb) y consta de 5 genes funcionales: ε, γG, γA, δ y β y de un pseudogen llamado ψβ (3). En todo el bloque se encuentran dos secuencias medianamente repetidas de la familia Kpn y ocho de la familia Alu (2, 5) (figura 2). Éstos genes globínicos tienen una estructura interna similar con una longitud
aproximada de 1.6 Kb (figura 2) y están constituidos del extremo 5’ a 3’ como se muestra en el esquema de la Figura 1 (3, 6). Hasta Agosto de 2005, se han descrito 238 alelos que causan Tal-β, la mayoría de ellos tienen debidos a una mutación puntual, como cambios de una base por otra, deleción o inserción de una o varias bases. Una lista completa de las mutaciones que originan talasemia beta puede ser consultada en las páginas de acceso libre en Internet como Globin Gene Server (http://globin.cse.psu.edu), en la Base de Datos inglesa Human Gene Mutation Database (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html), y en la pagina del Online Mendelian Inheritance in the Man (http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=141900), que regularmente las están actualizando. Los estudios poblacionales han mostrado que aproximadamente 25 mutaciones representan la mayor parte de los alelos
Figura 1.- Esquema de la familia de genes globínicos α.
Figura 2.- Esquema de la familia de genes globínicos β y organización interna del gen.

Revista de Hematología Vol. 7, No. 2, 2006
77
implicados en todas la poblaciones con alta frecuencia de Tal-β (7, 8), de manera que con base en su frecuencia se pueden agrupar en: a) alelos frecuentes o comunes, si se encuentran en frecuencias mayores a 10.0%; b) alelos poco frecuentes, con frecuencias entre 5.0% a 10.0% y c) alelos raros con frecuencia menor a 5.0% (cuadro 1).
TALASEMIA β EN MÉXICO. La Tal-β es la hemoglobinopatía más común en nuestro país, ya que se ha encontrado en cerca del 70 % de los pacientes con desórdenes de la hemoglobina estudiados en el Centro de Investigación Biomédica de Occidente y en los Laboratorios Clínicos de Puebla, respectivamente, tanto en estado homocigoto (βTal/βTal) como heterocigoto simple (βTal/β) o compuesto (βTal/βS, βTal/βD) (9, 10) . En nuestro Centro de Investigación hemos analizado la patología molecular de 80 cromosomas, entre los cuales hemos identificado 17 alelos distintos (β0 y β+), incluyendo una nueva mutación, deleción
Cuadro 1Ejemplos de Alelos observados en diferentes regiones geográficas del mundo (7).Frecuencia Mutaciones País o Región
Frecuentes
Sin sentido codón 39 C→TIVS1:1 G→ AIVS1: 6 T→ C
IVS1:110 G→A
África del Norte, Europa Occidental, Balca-nes, Mediterráneo, y Medio Oriente
IVS2:745 C→GIVS 2: 1 G→ A
Italia, Grecia, Turquía, Francia, España, Macedonia, Bulgaria
IVS1: 1 G→ TIVS1:5 G→C
DML codón 41/42 –CTTTDeleción de 619 pb
Oriente Medio, Asia y Sureste Asiático, Malasia
Poco frecuen-tes
-28 A→C Angola, Turquía, Kurdistan, México -87 C→G Italia, Grecia, Turquía, Francia, Yugoslavia
BulgariaSin sentido codón 17 A→T China y Tailandia
IVS1:5 G→A Italia, Turquía, Grecia, España
Raros(δβ)) - Talasemia tipo español
DML codón 11 –TEspaña, México
(δβ)) - Talasemia tipo sicilia Islas de Sicilia y Cerdeña
de una citosina en el codón 77 ó 78. Seis mutaciones constituyen el 78.4 % de los alelos observados. La mayoría de ellos son considerados de origen Mediterráneo (10 alelos), 3 de origen Asiático, 3 alelos privados, uno de ellos particularmente de origen Curdo, y el alelo restante, a la fecha, sólo se ha encontrado en nuestra población (11, 12) (cuadro 2).
El espectro de mutaciones observado en nuestra población, es característico de las poblaciones con baja frecuencia de talasemias. Sin embargo, hace patente la necesidad de considerar la presencia de talasemia al realizar el diagnóstico diferencial de anemia microcítica hipocrómica.
DIAGNÓSTICO DE TALASEMIA EN EL LABORATORIO. El diagnóstico de la talasemia se fundamenta en tres evaluaciones: hematológica, bioquímica y molecular. a) Hematológica. Citometría hemática. El análisis de
Talasemias.

78
Revista de Hematología Vol. 7, No. 2, 2006
los índices eritrocitarios obtenidos en equipo automatizado es fundamental, ya que permite sospechar la presencia de talasemia, en particular de Tal-β. Los valores para considerar el rasgo de Tal-β menores Volumen Corpuscular Medio (VCM) <78 fL y Hemoglobina Corpuscular Media (HCM) <27 pg (13, 14). Frotis sanguíneo. La elaboración de un frotis sanguíneo teñido con Wright-Giemsa ayuda a establecer la presencia de normoblastos, dianocitos, drepanocitos, o punteado basófilo. Para descartar la presencia de un componente hemolítico se recomienda realizar la cuenta de reticulocitos (13, 14).
Cuadro 2Distribución alélica observada en pacientes mexicanos con talasemia beta.
Total % Tipo de alelo1. Alelos Transcripcionales
-87 C→T 2 2.4 β+
-28 A→C 5 6.0 β+
2. Procesamiento ARN IVS1:1 G→A 12 14.5 β°IVS1:5 G→A 5 6.0 β+
IVS1:5 G→C 2 2.4 β+
IVS1:110 G→A 12 14.5 β+
IVS2:1 G→A 1 1.2 β°IVS2:745 C→G 1 1.2 β+
3. Alelos en la Traducción 3.1 Inicio codón
ATG→GTG (MET→VAL) 3 3.6 β°3.2 Sin sentidoCodon 39 C→T 26 31.4 β°Codon 17 A→T 1 1.2 β°
3.3 Desviación Marco de LecturaCodon 6 –A 2 2.4 β°Codon 11 –T 3 3.6 β°
Codon 77/78 –C 1 1.2 β°Codones 41/42 -TTCT 1 1.2 β°4. Alelos por Deleción
(δβ)0-talasemia tipo Español 5 6.0 β°5. Alelos por Fusión
Hb Lepore Washington Boston 1 1.2 β+
Total de genes analizados 83 100.0
Cuando se sospecha de talasemia alfa se puede realizar una incubación de 2 horas a 37 °C, con colorante para reticulocitos una alícuota enriquecida de eritrocitos jóvenes, para observar los cuerpos de inclusión generados por la precipitación de la HbH (15).b) Evaluación Bioquímica. Electroforesis de Hemoglobina. Por muchos años ha sido considerada la prueba ha realizar para la detección e identificación de variantes estructurales y de Tal-β. Este ensayo está basado en las propiedades electroforéticas y cromatográficas de las variantes en estructura. Convencionalmente la electroforesis se
B Ibarra, FJ Perea

Revista de Hematología Vol. 7, No. 2, 2006
79
realiza en acetato de celulosa a pH alcalino (8.6), si se observa algún cambio en la movilidad de las Hbs se realizan estudios confirmatorios a pH ácido (6.2). Con este esquema de trabajo es posible identificar las variantes estructurales más comunes como por ejemplo, HbS, HbC, Hb E, Hb Bart y HbH entre otras. En ocasiones es necesario realizar otras técnicas electroforéticas para identificar y caracterizar variantes estructurales. La alternativa más utilizada es la electroforesis de globinas en condiciones desnaturalizantes, para establecer cual es la globina variante y su comportamiento a pH alcalino y ácido. El isoelectroenfoque es una alternativa reciente con un gran poder resolutivo, pero por su alto costo no se implementa en laboratorios con pocos recursos. Cuantificación de las Hbs F y A2. La HbF se evalúa por métodos que se consideran de alta resistencia a desnaturalización alcalina. Esta Hb se encuentra elevada en diferentes formas de persistencia hereditaria de HbF, así como de Tal-β mayor, Tal-β intermedia y Tal-δβ. La evaluación de HbA2 se realiza por microcromatografía de intercambio aniónico y es fundamental para establecer el diagnóstico de Tal-β. Los métodos automatizados basados en cromatografía líquida de alta resolución permiten cuantificar los diferentes componentes de Hbs con el consiguiente ahorro de tiempo y esfuerzo, sin embargo, resultan demasiado onerosos para utilizarlos de forma convencional para el diagnóstico de hemoglobinopatía. Otros estudios. Para confirmar la presencia de una Hb inestable se realizan pruebas de estabilidad a calor, isopropanol y butanol. Como existen algunas variantes que co-migran con la HbS, además de inducción de cuerpos de Heinz es importante realizar
las pruebas de inducción a drepanocitos con metabisulfito de sodio y de solubilidad, que son positivas en presencia de HbS.c) Evaluación Molecular. La identificación de alelos frecuentes y raros en una población es esencial para la implementación de las estrategias metodológicas apropiadas para realizar la identificación de las mutaciones de manera oportuna. La técnica de Southern blot, que fue utilizada para la identificación de HbS y de algunos alelos de Tal-β, fue entrando en desuso con el advenimiento de la técnica de Reacción en Cadena de la Polimerasa (PCR). Sin embargo todavía en la actualidad se utiliza para detectar los alelos que conducen a Tal-α-1, debidos a deleción de grandes segmentos de ADN (13, 14). Amplificación de los genes globínicos por PCR. La PCR es fundamental para realizar la detección de mutaciones puntuales en los genes globínicos. El conocimiento de las mutaciones presentes en la población permite establecer una estrategia para el diagnóstico temprano de las hemoglobinopatías. Se emplean una gran variedad de técnicas, basadas en la amplificación con PCR, para la identificación de mutaciones, entre las que se incluyen análisis por hibridación en manchas o dot-blot, análisis reverso de hibridación por mancha (dt-blot reverso), amplificación con iniciadores alelo específico, análisis con enzimas de restricción y Gap-PCR. Cada método tiene sus ventajas y desventajas, y cada laboratorio elige los procedimientos que más convengan a sus capacidades técnicas. Búsqueda de mutaciones comunes en México. La técnica de amplificación con iniciadores alelo específico es ampliamente empleada para identificar mutaciones conocidas de Tal-β y Tal-α. Con esta técnica es posible buscar hasta 3 alelos diferentes en
Talasemias.

80
Revista de Hematología Vol. 7, No. 2, 2006
un tubo de reacción. La técnica se fundamenta en la utilización de un iniciador común y un iniciador que presenta la mutación buscada en el extremo 3’, de tal forma que si la mutación está presente es reconocida y se logra un fragmento amplificado. Pero si no está presente la mutación buscada, no amplifica. Esta técnica permite un procedimiento rápido, sencillo y económico para la identificación de mutaciones. El análisis de restricción es un procedimiento que se emplea cada vez menos en nuestro laboratorio para la búsqueda de mutaciones en el gen globínico beta, ya que siete de las mutaciones descritas son identificadas por enzimas de restricción. Estos dos procedimientos nos permiten identificar cerca del 80% de las mutaciones que conducen a talasemia beta en los pacientes mexicanos. El 20% restante entra a la búsqueda de mutaciones desconocidas. Búsqueda de deleciones conocidas. Actualmente se emplea la variación denominada Gap-PCR que consiste en utilizar iniciadores que hibridan en las zonas cercanas a los sitios de ruptura y unión de las deleciones. Con esta técnica se detectan cerca de 8 deleciones comunes de talasemia alfa, aproximadamente 5 deleciones de talasemia beta, 4 deleciones de tal-δβ y 5 deleciones de Persistencia Hereditaria de Hemoglobina fetal (13, 14). Búsqueda de mutaciones desconocidas. Para la identificación y caracterización de mutaciones desconocidas en los genes globínicos se han descrito varios procedimientos entre los que se encuentra polimorfismo conformacional de cadena sencilla, análisis de Heteroduplex, electroforesis en gel con gradiente desnaturalizante. Los fragmentos que muestran alguna anomalía en las técnicas antes mencionadas son después dirigidas a secuenciación de nucleótidos. En nuestro
laboratorio empleamos directamente la secuenciación de nucleótidos para detección y caracterización de mutaciones puntuales desconocidas. Finalmente, el estudio de las talasemias en México de una forma integral (hematológica, clínica, y molecular), permitirá definir con mayor certeza la evolución clínica del paciente, así como las bases moleculares de la fisiopatología. Estamos convencidos de que esta enfermedad hereditaria en nuestro país es mucho más compleja de lo que parece, por lo que se requiere realizar más investigación en este campo.
REFERENCIAS.1.- Watherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 2001;79:704-12.
2.- Weatherall DJ, Clegg JB, Higgs DR, Wood WG. The Hemoglobinopathies. In: The Metabolic & Molecular Bases of Inherited Disease. Scriver Ch R, Beaudet AR, Sly WS, Valle D, (Eds). McGraw Hill, New York, Vol III, pages 4571-4636, 2001a.
3.- Thein S. β- thalassaemia. En: Sickle cell disease and thalassaemia, G.P. Rodgers, (Ed) 11(1):91-126, 1998.
4.- Ibarra B, Perea FJ, Jaloma AR, Romero F, González-García JR. Genética y Hematología. En: Guízar-Vázquez J. (Editor). Genética Clínica. Diagnóstico y Manejo de las enfermedades hereditarias. México: Editorial El Manual Moderno; 2001. p. 512-7.
5.- Weatherall DJ: The Thalassaemias. In: William’s Hematology. Beutler E, Lichtman MA, Coller BS, Kipps TJ, Seligsohn U. (Eds) 6th edición. McGraw Hill, 547-580, 2001b.
6.- Bunn H, Forget B. Hemoglobin: Molecular Genetic and Clinical Aspect. Dyson J (ed). Philadelphia: W B Saunders Company; 1986.
7.- Huisman THJ, Carver MFM, Baysal E. A Syllabus of Thalassemia Mutations. Sickle Cell Anemia Foundation, GA, USA, 1997.
B Ibarra, FJ Perea

Revista de Hematología Vol. 7, No. 2, 2006
81
8.- Flint J, Harding E, Boyce A, Clegg JB. The population genetics of the haemoglobinopathies. Baillière´s Clinical Haematology 1998; 11:1-52.
9.- Ruiz-Reyes G, Ibarra-Cortés B, González-Martínez P, Lisker R. Anemias hereditarias en México. En Góngora-Biachi RA, editor. Hematología. Actualización 2004. Mérida: Agrupación Mexicana para el Estudio de la Hematología, A.C.; 2004. p. 15-25.
10.- Ruiz-Arguelles GJ, López Martínez B, Ruiz-Reyes G. Heterozygous beta-thalassemia: not infrequent in Mexico. Arch Med Res 2001; 32:293-5.
11.- Villalobos-Arámbula AR, Bustos R, Casa-Castañeda M, Gutiérrez E, Perea FJ, Thein LS, Ibarra B. β-Thalassemia and βA globin gene haplotypes in Mexican mestizos. Hum Genet 1997; 99:498-500.
12.- Perea FJ, Magaña MT, Cobian JG, Sanchez-Lopez JY, Chavez ML, Zamudio G, et al. Molecular spectrum of beta-thalassemia in the Mexican population. Blood Cells Mol Dis 2004; 33:150-2.
13.- Cao A. Carrier screening and genetic counselling in beta-thalassemia. Int J Hematol 2002; 76 (Suppl 2): 105-13.
14.- Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev 2003; 17:43-53.
15.- Pan LL, Eng HL, Kuo CY, Chen WJ, Huang HY. Usefulness of brilliant cresyl blue staining as an auxiliary method of screening for alpha-thalassemia. J Lab Clin Med 2005; 145:94-7.
Talasemias.

Revista de Hematología Vol. 7, No. 2, 2006
83
Diagnóstico en el laboratorio delanticoagulante lúpico.
Virginia Domínguez-Martínez, Rosario Villa-Márquez, Darinel Hernández-Hernández.
Laboratorio de Coagulación. Departamento de Hematología-Oncología. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México, D.F., México.
se emplean fosfolípidos con una conformación específica (v. gr.: fosfolípidos en fase hexagonal). Ambos procedimientos acortan el tiempo de coagulación en los plasmas que contienen AL (1). De acuerdo a este principio, el diagnóstico de laboratorio del AL debe seguir 4 pasos: 1) Presencia de los tiempos de coagulación dependiente de fosfolípidos alargados (escrutinio). 2) Presencia de un inhibidor demostrado con el procedimiento de mezcla de plasma problema con plasma normal sin corrección del tiempo de coagulación. 3) Confirmación de la naturaleza antifosfolípidica del inhibidor por corrección del defecto de la coagulación, mediante el uso de un procedimiento de neutralización del inhibidor (incremento de la cantidad de fosfolípidos). 4) Ausencia de inhibición específica sobre un factor de la coagulación. Los primeros 2 pasos son necesarios para probar la presencia de un inhibidor y los otros
INTRODUCCIÓN. El anticoagulante lúpico (AL) está compuesto de anticuerpos heterogéneos dirigidos en contra de los fosfolípidos con carga negativa. Esta reacción es dependiente de la presencia de ciertas proteínas plasmáticas como la β2-glicoproteína 1 (β2-GP1), protrombina, proteína S y anexina V. Desde los años 60 existe evidencia que indica que la presencia de AL se asocia con incremento en el riesgo de tromboembolismo arterial/venoso, en pacientes con o sin enfermedades autoinmunes. Además, se ha asociado a perdida fetal en mujeres “sanas”. Los procedimientos para detectar el AL se basan en la prolongación de los tiempos de coagulación de las pruebas dependientes de fosfolípidos, y se sospecha la presencia de éste cuando al agregar una cantidad igual de plasma normal al plasma problema, esta prolongación no revierte. Para la confirmación de la presencia del AL se realizan pruebas en las cuales se incrementa la concentración de fosfolípidos, o
Solicitud de reimpresos: Virginia Domínguez-Martínez. Departamento de Hematología y Oncología. Instituto Nacional de Ciencias Médicas y Nutrición SZ. Vasco de Quiroga 15, Colonia Sección XVI, Delegación Tlalpan, C.P. 14000, México, D.F., México. Tel.: 54 870 900 Ext. 2704

84
Revista de Hematología Vol. 7, No. 2, 2006
para diferenciar un AL de otro inhibidor de la coagulación (figura 1)(2, 3). A pesar de estos criterios, en pacientes con alteraciones diferentes a la presencia de AL, la interpretación de los resultados en las pruebas de detección de AL resulta
problemática (cuadro 1). Un ejemplo específico lo constituyen los pacientes con anticoagulación oral (ACO), por la dificultad en la interpretación de los resultados en las pruebas de coagulación dependientes de fosfolípidos. La interpretación de la
V Domínguez-Martínez, R Villa-Márquez, D Hernández-Hernández
Cuadro 1Diagnósticos diferenciales de inhibidores de la coagulación.
1. Inhibidores asociados con sangradoa) anti-factor VIIIb) anti-factor II, VII, IX, X, XIc) anti-fibrinógeno/fibrinad) anticoagulantes similares a la heparina
2. Inhibidores con o sin sangradoa) anti factor V
3. Inhibidores usualmente no asociados con sangradoa) anticoagulante lúdicob) antifactor XII
Figura 1.- Abordaje diagnóstico en el laboratorio para la detección de anticoagulante lúpico. ACO = anticoagulación oral. LA1 y LA2 = fosfolípidos a diferentes concentraciones.

Revista de Hematología Vol. 7, No. 2, 2006
85
magnitud de la corrección o acortamiento del tiempo de coagulación, que precisamente constituye la base de muchos procedimientos confirmatorios diseñados para detectar AL, se hace muy difícil, ya que la prolongación en el tiempo de coagulación causada por la ACO se traslapa con la inducida por el AL. Se han descrito procedimientos confirmatorios que aparentemente no son modificados por el ACO, sin embargo, su eficacia no está plenamente evaluada ni comprobada. En nuestro laboratorio, la causa principal de prolongación de los tiempos de coagulación esta dada por los pacientes con ACO. Muchos de estos pacientes se encuentran en estudio de una probable trombofilia, y entre estos estudios se incluye la búsqueda de AL. En esta situación, sería necesario esperar al término del periodo de anticoagulación para realizar la determinación, o suspenderla transitoriamente. Ambas alternativas pueden ser deletéreas para el paciente. Sin embargo, el conocer la condición respecto al AL ayudaría a la decisión de la duración e intensidad de la ACO, para prevenir recurrencia de la trombosis. Por las razones mencionadas, en los últimos 20 años se han desarrollado diversos tipos de pruebas de escrutinio y confirmación para la identificación de AL, no obstante hasta la fecha no hay una única prueba para detectar AL que sea 100% específica. Estas pruebas incluyen varios procedimientos o modificaciones de los mismos, como el tiempo de tromboplastina parcial activada (TTPa), tiempo de coagulación con caolín, tiempo de coagulación con sílice (SCT), tiempo de protrombina diluido o inhibición de la tromboplastina tisular (TPd) y tiempo de veneno de víbora de Russell diluido (TVVRd). Desafortunadamente, hay variaciones en la sensibilidad y especificidad de estas pruebas para la detección del AL o inhibidores similares, ya que la composición de fosfolípidos es un
determinante crítico para la sensibilidad de una prueba determinada; y como se ha mencionado previamente, esta variación se incrementa en los pacientes con ACO (3, 4). La mezcla de plasma del paciente con plasma normal es una forma de compensar la prolongación en el tiempo de coagulación en este tipo de muestras, sin embargo, este procedimiento consume tiempo y reactivo, por lo que resulta una solución cara (1, 5)
(figura1). Debido a que muchos de los pacientes a quienes se les solicita AL se encuentran anticoagulados, en estudios previos se ha reportado que ciertas pruebas como el SCT, realizado con concentraciones bajas y altas de fosfolípidos sin adición de plasma normal y el TVVRd, procesado con fosfolípidos y sin adición de plasma normal no son alteradas con la ACO y se sugiere que pueden ser usadas para la detección de AL en este tipo de pacientes (6). En el Instituto se realizó un estudio para establecer la utilidad diagnóstica del TPd (con una tromboplastina tisular recombinante), y del TVVRd (sin la adición de plasma normal) en la detección de AL en pacientes anticoagulados y no anticoagulados, empleando como “estándar de oro” la prueba de fosfolípidos hexagonales (FH) (1, 3). Se estudiaron 40 muestras de pacientes de la Clínica de Anticoagulantes del Instituto con ACO y cuadro(s) de trombosis previa(s). De estas muestras, 12 pertenecían a pacientes con diagnóstico de síndrome antifosfolípido (SAF) primario o secundario, 9 eran de pacientes con enfermedad inmunológica (lupus eritematoso generalizado (LEG), artritis reumatoide (AR), hepatitis autoinmune, enfermedad de Graves) y sospecha de SAF; 17 muestras eran de pacientes anticoagulados por otras razones (fibrilación auricular, prótesis valvulares, cáncer, etc.) y 2 de pacientes en estudios de trombofilia: 1 con antecedente
Aticoagulante lúpico.

de trombosis recurrente y otra con abortos recurrentes. Además se estudiaron 27 muestras de pacientes no anticoagulados, 20 de ellas para detección de AL, y el resto con prolongación del TTPa por otras razones (cirrosis hepática, metástasis hepáticas, cáncer hepático). Para la obtención de cifras de referencia se estudiaron 26 muestras de individuos clínicamente sanos (cuadro 2). Métodos. Las muestras de plasma citratado se almacenaron a –70°C hasta la realización de las pruebas. El TPd se realizó en un instrumento automatizado (BCS®, Dade Behring) en el modo de TTPa, usando tromboplastina tisular recombinante (Innovin®, Dade Behring), diluida 1:100 y 1:200 en buffer veronal. Los cocientes del TPd se calcularon dividiendo el tiempo obtenido en cada muestra problema entre la media del valor obtenido en los controles: 34.1 para la dilución 1:100 y 46.1 para la dilución 1:200. El TVVRd se realizó en el mismo equipo automatizado, empleando muestras de plasma pareadas, a las que se les agregó como activador el veneno de víbora (Dade Behring) y 2 concentraciones diferentes de fosfolípidos (LA1 y LA2). Los resultados se expresaron como porcentajes de corrección, de acuerdo a la siguiente formula:% corrección = [(VVRd1p /VVRd1n) - (VVRd2p /VVRd2n)] / ((VVRd1p /VVRd1n) x 100 Para la prueba de FH se empleo el reactivo Staclot® LA (Stago) procesándose como prueba pareada de TTPa sin (tiempo de coagulación 1) y con (tiempo de coagulación 2) fosfolípidos hexagonales (FH), agregados a
una mezcla de plasma problema con plasma normal, y de acuerdo a las instrucciones del fabricante. Los resultados se expresaron como la diferencia en segundos entre el tiempo de coagulación 1 y 2. Se consideró un plasma positivo para AL cuando la diferencia entre el tiempo de coagulación 1 y el tiempo de coagulación 2 estuvo por arriba del valor de corte de la población control. De los 67 plasmas analizados, (59.7%) trece correspondieron a pacientes anticoagulados. (32.5%) de éstos con diagnóstico de SAF primario o secundario. Los otros 27 plasmas eran de pacientes no anticoagulados: 20 fueron enviados para la detección de AL, la mayoría de ellos de pacientes con enfermedades inmunológicas y con TTPa prolongado que no corrigieron al mezclar con plasma normal, 8 plasmas con TTPa prolongado debido a falla hepática. La prueba de los FH fue positiva en 33 de los 67 plasmas analizados y de éstos 19 correspondieron a pacientes anticoagulados. La sensibilidad en la identificación de AL del TPd 1:100, 1:200 y el TVVRd en los pacientes con ACO fue muy similar a la encontrada en los pacientes sin ACO (cuadro 3), sin embargo, la especificidad de las tres pruebas fue menor en los pacientes con ACO. Con la prueba del TVVRd se obtuvieron las cifras más altos de sensibilidad (>90%) y especificidad (de 86% a 92%), mientras que con el empleo del TPd 1:100 y 1:200, en los pacientes con ACO la especificidad disminuyó a 33% y 57%, respectivamente. En forma similar de las 3 pruebas de laboratorio estudiadas, con el
Cuadro 2Valores de la percentila 95 de un grupo de 26 sujetos sanos para las pruebas
fosfolípidos hexagonales (FH), tiempo de protrombina diluido (TPd) y tiempo deveneno de Rusell diluido (TVVRd).
FH TPd !:100 TPd 1:200 TVVRd
Punto de corte 6.0 s 1.66 2.0 21.7%
86
Revista de Hematología Vol. 7, No. 2, 2006
V Domínguez-Martínez, R Villa-Márquez, D Hernández-Hernández

Revista de Hematología Vol. 7, No. 2, 2006
87
TVVRd se obtuvieron las cifras más altas de valor predictivo positivo y negativo en los 2 grupos de pacientes (cuadro 4). El AL se identificó en 78.5% de pacientes con SAF (11/14 pacientes con diagnóstico conocido de SAF; 2 de ellos no anticoagulados), en 69% de pacientes con enfermedad inmunológica y/o sospecha de SAF (20/29 muestras con solicitud de AL; 11 de ellos anticoagulados) y en 20.8% de pacientes con otras enfermedades (2 con cáncer, 2 con hipertensión arterial pulmonar e insuficiencia cardiaca y 1 con DM 2). Aunque el porcentaje de detección de AL en pacientes con diagnóstico conocido SAF es aceptable, existe la posibilidad que sean positivos para anticuerpos antifosfolípidos en fase sólida,
Cuadro 3Sensibilidad (sens.) y especificidad (esp.) del tiempo de protrombina diluido (TPd) y del tiempo de veneno de Rusell diluido (TVVRd) en la detección de anticoagulante lúpico en pacientes con y sin anticoagulación oral (ACO) y empleando como prueba diagnóstica los fosfolípidos
hexagonales.
PACIENTES TPd 1:!00 TPd 1:200 TVVRd
sens. % esp. % sens. % esp. % sens. % esp. %
Todos n=67 85 50 85 65 94 88
Con ACO n=40 84 33 84 57 95 86
Sin ACO n=27 86 77 86 77 93 92
Cuadro 4Valor predictivo positivo (vpp) y valor predictivo negativo (vpn) del tiempo de protrombina diluído (TPd) y del tiempo de veneno de Rusell diluído (TVVRd) en la detección de anticoagulante lúpico en pacientes con y sin anticoagulación oral (ACO) y empleando como prueba diagnóstica
los fosfolípidos hexagonales.
PACIENTES TPd 1:!00 TPd 1:200 TVVRd
vpp % vpn % vpp % vpn % vpp % vpn %
Todos n=67 62 77 70 81 89 94
Con ACO n=40 53 70 64 80 86 95
Sin ACO n=27 80 83 80 83 93 92
pero no para AL. Los resultados del estudio indican que el mejor balance de sensibilidad especificidad se observó en los pacientes no anticoagulados. Considerando ambos grupos de pacientes el TPd 1:100 y 1:200 mostró sensibilidad adecuada, pero pobre especificidad para la búsqueda de AL, y aunque en estudios previos se ha reportado que la dilución 1:200 es la que tiene mayor sensibilidad, en nuestra experiencia ésta fue muy similar a la obtenida con el TPd 1:100. En conclusión, de las tres pruebas analizadas, el TVVRd es una prueba útil para detectar AL en pacientes con o sin ACO. La ventaja de esta prueba, comparativamente con la de FH, se basa principalmente en que puede
Aticoagulante lúpico.

88
Revista de Hematología Vol. 7, No. 2, 2006
automatizarse, lo cual reduce el tiempo de realización, economiza reactivos, minimiza las variables pre-analíticas que pueden alterar los resultados y por lo tanto reduce los costos. La prueba de FH (Staclot® LA) es difícil de realizar en sistemas automatizados, ya que la cantidad de reactivos usados y los tiempos de incubación necesarios exceden la capacidad de la mayoría de los instrumentos automatizados. Además, el costo institucional por paciente con cada prueba de Staclot® LA es de aproximadamente $300 pesos, mientras que el de TVVR es de $100 pesos. Estos hallazgos han hecho que en el Instituto se emplee el TVVRd, sin añadir plasma normal, como primera elección para la búsqueda de AL en pacientes con ACO. Sin embargo, el diagnóstico de AL sigue basándose en la realización de varias pruebas de escrutinio y de confirmación que implican ambas vías de la coagulación. Aunque hay lineamientos internacionales para tratar de estandarizarlas, la principal limitante es la ausencia de controles internacionalmente aceptados, lo que obliga a llevar el control de calidad interno en las muestras positivas de pacientes, lo cual dificulta la estandarización.
REFERENCIAS.1.- 1.Tripodi A, Chantarangkul V, Clerici M. Laboratory diagnosis of lupus anticoagulants for patients on oral anticoagulant treatment. Thromb Haemost 2002:8;583-6.
2.- Arnout J, Vanrusselt M, Huybrechts E. Optimization of the dilute prothrombin time for the detection of the lupus anticoagulant by use of a recombinant tissue thromboplastin. Br J Haematol 1994;87:94-9.
3.- Wong RC, Adelstein S, Gillis D, Favaloro EJ. Development of consensus guidelines for anticardiolipin and lupus anticoagulant testing. Semin Thromb Hemost 2005; 31:39-48.
4.- Arnout J. Antiphospholipid syndrome: Diagnostic aspects of lupus anticoagulants. Thromb Haemost 2001; 86:83-91.
5.- Chantarangkul V, Tripodi A, Arbini A. Silica clotting time (SCT) as a screening and confirmatory test for detection of the lupus anticoagulants. Thromb Res 1992; 67:355-65.
6.- Lawrie AS, Purdy G, Mackie IJ. Monitoring of oral anticoagulant therapy in lupus anticoagulant positive patients with the antiphospholipid syndrome. Br J Haematol 1997; 98:887-92.
V Domínguez-Martínez, R Villa-Márquez, D Hernández-Hernández


















