
Casos Clínicos Interdisciplinarioscdb.hospitalclinic.org/media/upload/pdf//interdisc...CASO...
21
Coordinación Dra. Irene Madrigal Bajo Edición Esther Fernández Galán Casos Clínicos Interdisciplinarios Servicio de Bioquímica y Genética Molecular
Transcript of Casos Clínicos Interdisciplinarioscdb.hospitalclinic.org/media/upload/pdf//interdisc...CASO...

Coordinación
Dra. Irene Madrigal Bajo
Edición Esther Fernández Galán
Casos Clínicos Interdisciplinarios Servicio de Bioquímica y
Genética Molecular

Centre deDiagnòsticBiomèdic
Secuenciación del exoma:
nueva herramienta
diagnóstica en
la enfermedad mitocondrial
12 Diciembre 2017

3
Dr. Joan Anton Puig-Butillé
Especialista Sénior
Sección Genética Molecular Core Biologia Molecular
Laura Gort Mas
Especialista Sénior
Sección de Errores Congénitos del Metabolismo
Dra. Judith Garcia Villoria
Jefa de Sección
Sección de Errores Congénitos del Metabolismo

INTRODUCCIÓN DEFINICIÓN
Las enfermedades mitocondriales son un grupo heterogéneo de trastornos, caracterizados por defectos de
origen genético en el sistema de fosforilación oxidativa (OXPHOS), y que por tanto, tienen en común una
deficiencia en la producción de energía.
MANIFESTACIONES CLÍNICAS
Las mitocondrias están en todos los órganos y tejidos
y por tanto, las enfermedades mitocondriales tienen
manifestaciones multisistémicas muy variables. Se
verán afectados especialmente aquellos órganos con
necesidades energéticas elevadas como: cerebro,
musculo esquelético, corazón, hígado, riñón y retina.
La gran variabilidad en la presentación de la
enfermedad en los recién nacidos, juntamente con la
rápida evolución en algunos casos dificulta mucho su
diagnóstico.

5
CASO CLÍNICO
Tercera hija de padres no consanguíneos
Debut neonatal con acidosis láctica, hipoglucemia, cardiomiopatía, hipotonía y dificultad respiratoria
Exitus al los 13 días de vida por insuficiencia respiratoria
HISTORIA CLÍNICA
ANTECEDENTES FAMILIARES
1ª Hija: Retraso psicomotor, exitus por sepsis a los 15 meses . Sin diagnóstico
2ª Hija: Acidosis láctica, hiperamonemia, cardiomiopatía hipertrófica e hipertensión pulmonar, exitus a los 8 días y diagnosticada post-mortem de glucogenosis
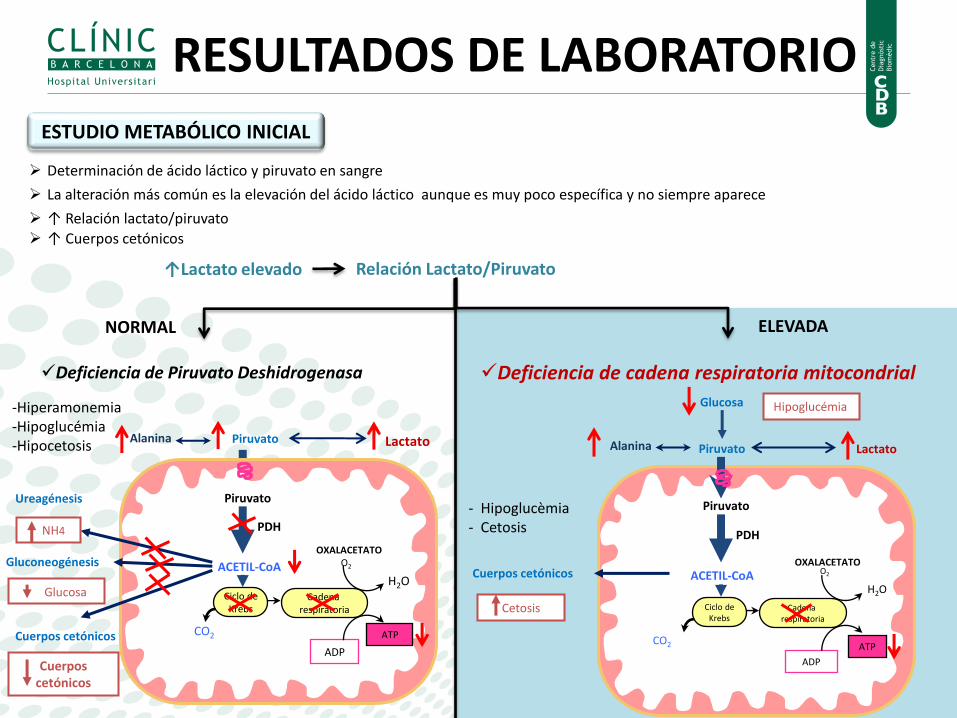
NORMAL ELEVADA
Relación Lactato/Piruvato
-Hiperamonemia -Hipoglucémia -Hipocetosis
Piruvato
Piruvato
ACETIL-CoA
Ciclo de Krebs
Cadena respiratoria
H2O
O2
ATP
ADP
CO2
PDH
OXALACETATO
Lactato Alanina
Cuerpos cetónicos
Ureagénesis
Gluconeogénesis
Glucosa
Cuerpos cetónicos
NH4
Piruvato
ACETIL-CoA
Piruvato
Ciclo de Krebs
Cadena respiratoria
H2O
O2
ATP
ADP
CO2
Cuerpos cetónicos
Hipoglucémia
Cetosis
PDH
OXALACETATO
Lactato
Glucosa
Alanina
Deficiencia de cadena respiratoria mitocondrial
- Hipoglucèmia - Cetosis
Deficiencia de Piruvato Deshidrogenasa
Determinación de ácido láctico y piruvato en sangre
La alteración más común es la elevación del ácido láctico aunque es muy poco específica y no siempre aparece
↑ Relación lactato/piruvato
↑ Cuerpos cetónicos
RESULTADOS DE LABORATORIO
ESTUDIO METABÓLICO INICIAL
↑Lactato elevado

ESTUDIOS BIOQUÍMICOS
• Plasma para valorar aminoácidos
• Orina para análisis de ácidos orgánicos
• Biopsia de músculo para estudio de la actividad de los complejos de la cadena respiratoria
mitocondrial.
AMINOÁCIDOS EN PLASMA
Perfil 46 aminoácidos. Diagnóstico de aminoacidopatias, más de 30 enfermedades.
RESULTADOS DE LABORATORIO

ÁCIDOS ORGÁNICOS EN ORINA
Estándar interno
3-Me-glutárico
3-Me-glutacónico cis y trans
Láctico
Paciente
Control
100 metabolitos en 43 minutos
Lactato + Alanina + 3-Metilglutárico + 3-Metilglutacónico:
ACIDURIA 3-METILGLUTACÓNICA
El estudio del perfil de ácidos orgánicos en orina se realiza mediante cromatografía de gases acoplada a espectrometría de masas (GC/MS). Este perfil puede estar alterado en más de 50 enfermedades, pero no todas son acidurias orgánicas. Los ácidos orgánicos son metabolitos clave en muchas vías metabólicas del metabolismo intermediario y por tanto su análisis nos aporta información sobre el estado de estas vías.
RESULTADOS DE LABORATORIO
Estándar interno Láctico 3-Me-glutárico
3-Me-glutacónico cis y trans

ACTIVIDAD CADENA RESPIRATORIA MITOCONDRIAL EN MÚSCULO
PACIENTE VALORES REFERÉNCIA (nmol/min/mg prot)
Complejo I + II 32 12-56
Complejo II 7 4-10
Complejo II + III 9 7-24
Complejo III 146 55-259
Complejo IV 208 59-170
Citrato sintasa 150 71-200
Se puede cuantificar mediante métodos espectrofotométricos que miden la actividad de los enzimas por separado y se puede estudiar en linfocitos o a partir de células en cultivo (fibroblastos). Estas pruebas pueden resultar muy útiles y guiar en el diagnóstico del paciente, pero muchas veces, como en este caso clínico, el resultado es normal, no se puede descartar una enfermedad mitocondrial y se debe continuar con estudios moleculares.
RESULTADOS DE LABORATORIO
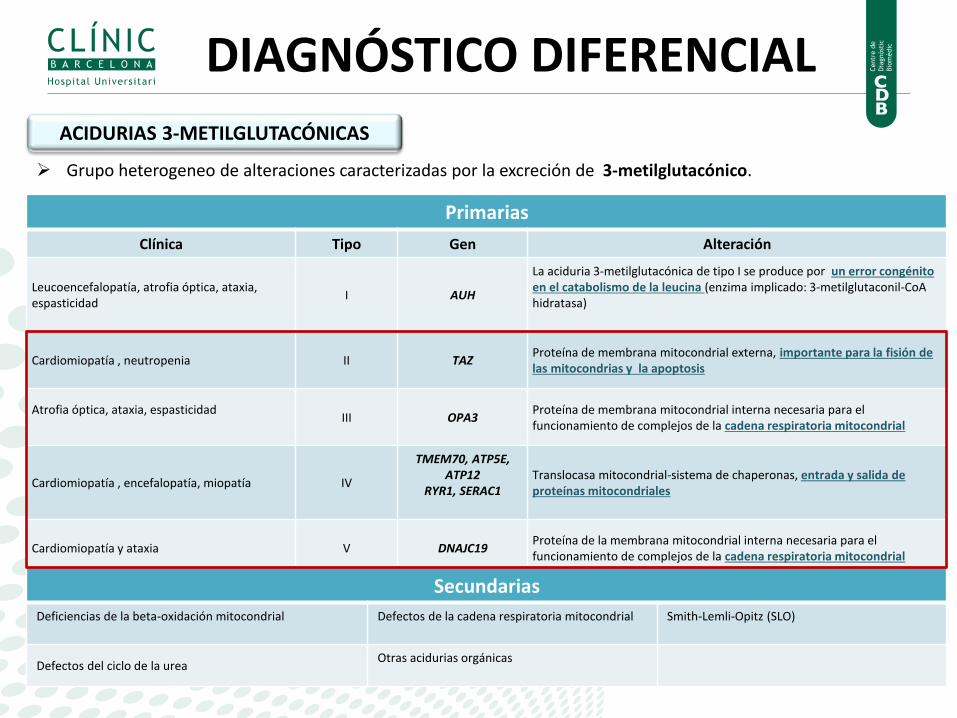
ACIDURIAS 3-METILGLUTACÓNICAS
Grupo heterogeneo de alteraciones caracterizadas por la excreción de 3-metilglutacónico.
Primarias
Clínica Tipo Gen Alteración
Leucoencefalopatía, atrofia óptica, ataxia, espasticidad
I AUH
La aciduria 3-metilglutacónica de tipo I se produce por un error congénito en el catabolismo de la leucina (enzima implicado: 3-metilglutaconil-CoA hidratasa)
Cardiomiopatía , neutropenia II TAZ Proteína de membrana mitocondrial externa, importante para la fisión de las mitocondrias y la apoptosis
Atrofia óptica, ataxia, espasticidad
III OPA3 Proteína de membrana mitocondrial interna necesaria para el funcionamiento de complejos de la cadena respiratoria mitocondrial
Cardiomiopatía , encefalopatía, miopatía IV
TMEM70, ATP5E, ATP12
RYR1, SERAC1
Translocasa mitocondrial-sistema de chaperonas, entrada y salida de proteínas mitocondriales
Cardiomiopatía y ataxia V DNAJC19 Proteína de la membrana mitocondrial interna necesaria para el funcionamiento de complejos de la cadena respiratoria mitocondrial
Secundarias
Deficiencias de la beta-oxidación mitocondrial
Defectos de la cadena respiratoria mitocondrial
Smith-Lemli-Opitz (SLO)
Defectos del ciclo de la urea Otras acidurias orgánicas
DIAGNÓSTICO DIFERENCIAL

Única posición Único gen Múltiples Genes
>5 Múltiples Genes
>100 Exoma
Genoma Completo
NGS NGS
NGS
NGS
NGS
(Exome Sequencing)
NGS
(Whole-Genome Secuencing)
qPCR/Sanger Sequencing
Sanger Sequencing
Alto
Bajo
Nú
mer
o d
e m
ues
tras
Screening de Variantes "Descubrimiento"
ESTUDIO MOLECULAR
Exoma Clínico: 4800 genes asociados a enfermedades
(12 Mb)
En este caso clínico el estudio molecular engloba más de 50 genes y se realiza mediante Next Generation Sequencing (NGS)
Decidir cuando se debe utilizar la PCR, secuenciación Sanger o estrategias de NGS, como la secuenciación del exoma o la secuenciación total del genoma, depende de una combinación de factores.
Sanger y la PCR son estrategias adecuadas cuando el numero de regiones diana es bajo (1-20), y los objetivos del estudio se limitan a la identificación de variantes conocidas. La secuenciación exómica y WGS son métodos excelentes para un análisis genético integral y el descubrimiento de variantes. La NGS dirigida ofrece una opción equilibrada entre estas dos estrategias.
Gráfico adaptado de Illumina Co.
RESULTADOS DE LABORATORIO

FRAGMENTACIÓN DNA (TAGMENTACIÓN)
PCR AMPLIFICACIÓN
gDNA
Muestra A (P1/N1)
P1 N1 P1 N2
Muestra B (P1/N2)
P1 N1
P1 N1
P1 N1
P1 N1 P1 N2
P1 N2
P1
N2
P1/N1 P1/N2
HIBRIDACIÓN
CAPTURA
SECUENCIACIÓN DE NUEVA GENERACIÓN (NGS)
GENERACIÓN DE LA LIBRERIA TRUSIGHT ONE (EXOMA CLÍNICO)

13 Metzker ML et al. Nature Reviews Genetics 11(1):31-46 (2010).
AMPLIFICACIÓN EN SUPERFICIE SÓLIDA
SECUENCIACIÓN POR SÍNTESIS TERMINACIÓN REVERSIBLE CÍCLICA (CRT)
Obtención de miles de copias del mismo fragmento localizadas en un área definida
La NGS requiere equipos de secuenciación masiva en paralelo más sofisticados que los utilizados para la secuenciación Sanger
SECUENCIACIÓN DE NUEVA GENERACIÓN (NGS)
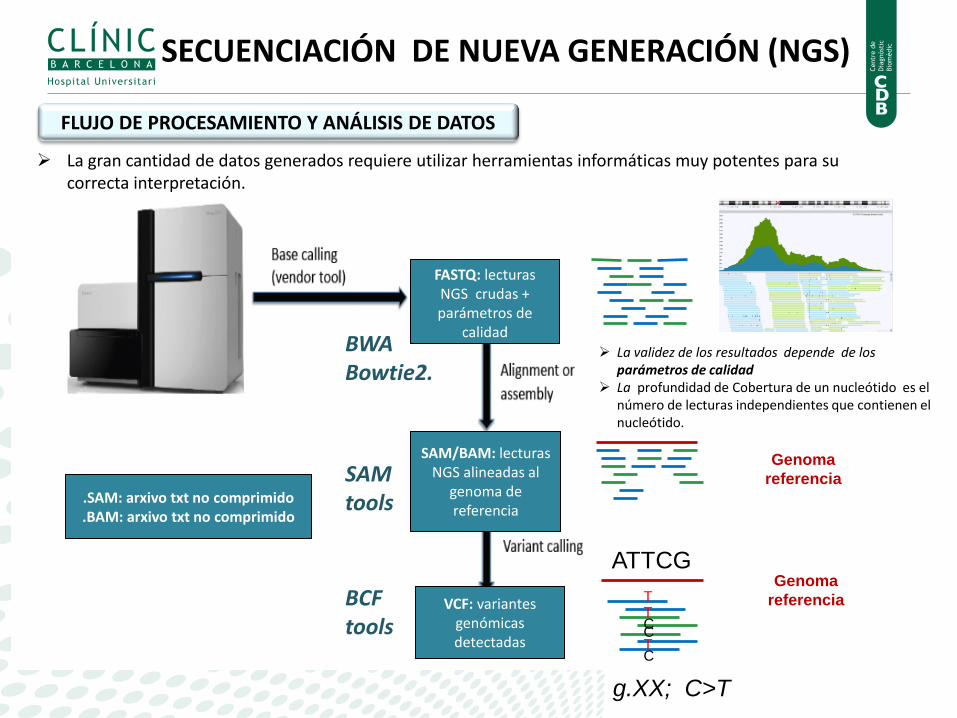
FASTQ: lecturas NGS crudas + parámetros de
calidad
SAM/BAM: lecturas NGS alineadas al
genoma de referencia
VCF: variantes genómicas detectadas
.SAM: arxivo txt no comprimido
.BAM: arxivo txt no comprimido
ATTCG
T T
T C
C C
g.XX; C>T
BWA Bowtie2.
SAM tools
BCF tools
Genoma
referencia
Genoma
referencia
La validez de los resultados depende de los parámetros de calidad
La profundidad de Cobertura de un nucleótido es el número de lecturas independientes que contienen el nucleótido.
La gran cantidad de datos generados requiere utilizar herramientas informáticas muy potentes para su correcta interpretación.
FLUJO DE PROCESAMIENTO Y ANÁLISIS DE DATOS
SECUENCIACIÓN DE NUEVA GENERACIÓN (NGS)
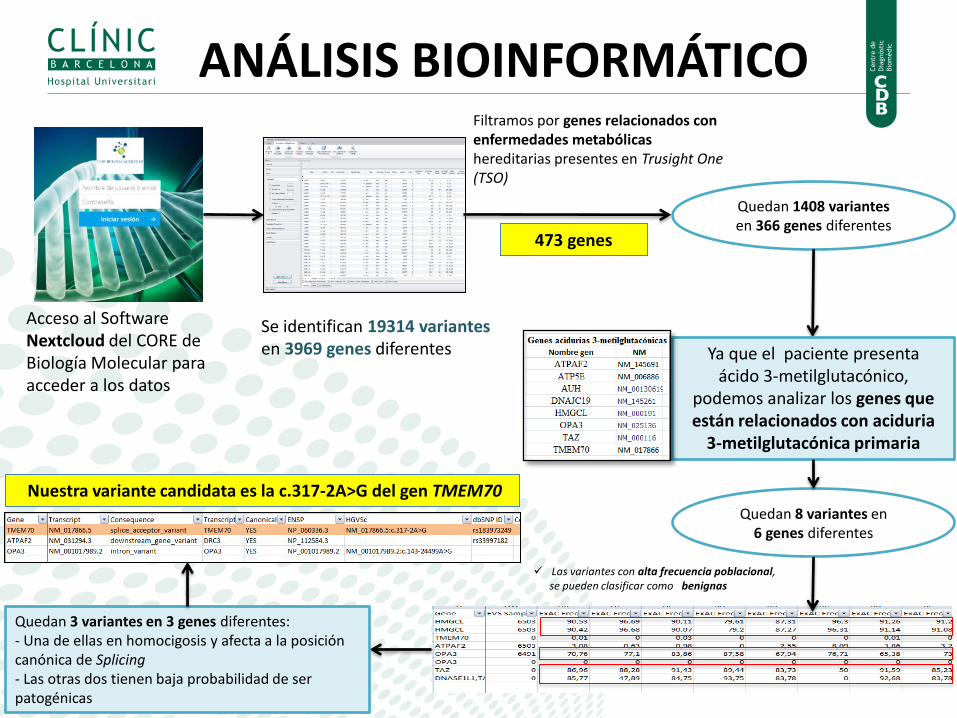
Acceso al Software Nextcloud del CORE de Biología Molecular para acceder a los datos
ANÁLISIS BIOINFORMÁTICO
Se identifican 19314 variantes en 3969 genes diferentes
Filtramos por genes relacionados con enfermedades metabólicas hereditarias presentes en Trusight One (TSO)
473 genes
Quedan 1408 variantes en 366 genes diferentes
Ya que el paciente presenta ácido 3-metilglutacónico,
podemos analizar los genes que están relacionados con aciduria
3-metilglutacónica primaria
Quedan 8 variantes en 6 genes diferentes
Quedan 3 variantes en 3 genes diferentes: - Una de ellas en homocigosis y afecta a la posición canónica de Splicing - Las otras dos tienen baja probabilidad de ser patogénicas
Las variantes con alta frecuencia poblacional, se pueden clasificar como benignas
Nuestra variante candidata es la c.317-2A>G del gen TMEM70

,
Se trata de una mutación descrita en Human Gene Mutation Database y muy prevalente en otros pacientes afectos.
Gen : TMEM70
Genotip : c.317-2A>G; c.317-2A>G;
DIAGNÓSTICO DEL PACIENTE
Acidúria 3-metilglutacónica por mutacions en TMEM70
ATP12 ATP5E TMEM70
Proteínas implicadas en la biosíntesis y ensamblaje del complejo V de la cadena respiratoria mitocondrial -Cardiomiopatía - Retraso psicomotor -Microcefalia -Hepatomegalia - Dismorfia facial -Hipotonía - Pueden ser exitus durante las primeras semanas de vida - Disfunción de la cadena respiratoria mitocondrial
c.317-2A>G (mutación prevalente)
RESOLUCIÓN DEL CASO CLÍNICO

BN-PAGE y Western blot mostraron una deficiencia en el ensamblaje del complejo V
Complejo II
Complejo V
BN-PAGE Western Blot
Control 1 Paciente Control 2 Paciente Control 1
BN-PAGE Actividad
Complejo II
Complejo V
Control 2
Estudios realizados por el Dr. Frederic Tort, Dra. Núria Bujan y Dra. Xènia Ferrer Proyecto FIS 16/01048
ESTUDIOS FUNCIONALES EN MÚSCULO DEL PACIENTE
RESOLUCIÓN DEL CASO CLÍNICO

Paneles de genes – Trusight One (Illumina)
> 4800 genes
Análisis bioinformático
Diagnóstico definitivo
Mutaciones patogénicas en gen candidato
Tipificación de la familia
Análisis de los padres y los familiares por secuenciación Sanger
Nuevo abordaje diagnóstico de las EMH

Estratificación clínica y bioquímica
Panel de genes – Trusight One Secuenciación Sanger
Diagnóstico definitivo
Mutaciones descritas
Diagnóstico definitivo
Predicciones bases de datos
Funcionalidad
Mutaciones no descritas
Diagnóstico definitivo
No mutaciones
Secuenciación Exoma
Análisis de los padres y familiares por secuenciación Sanger
Tipificación de la familia
Nuevo abordaje diagnóstico de las EMH

1. Los estudios bioquímicos orientan el diagnóstico y los genes que se tienen que estudiar
2 NGS es la tecnología de elección para el diagnóstico molecular de las enfermedades mitocondriales (>250 genes implicados), ya que permiten el estudio de grandes zonas del genoma en un solo análisis
3 La utilización de NGS puede cambiar los protocolos de diagnóstico:
Solo obtener biopsias de tejidos en caso de que las mutaciones sean de significado incierto para realizar estudios funcionales.
4 NGS puede disminuir el tiempo de diagnóstico y evitar pruebas invasivas
5 En ciertos casos la tecnología NGS puede identificar variantes no claramente patogénicas. Se necesitarán estudios complementarios para llegar al diagnóstico definitivo
4 El filtrado de los datos bioinformáticos es clave para obtener un buen resultado
20
CONCLUSIONES

Dra. Ribes Dr. Castellano Dr. Tort Dra. Ferrer Dra. Bujan
Patricia Alcalá Montserrat Fernández Ricard Isanta Sonia Moliner Laura Pacheco Sabine Richard Alba Roset














![Casos Clínicos Interdisciplinarioscdb.hospitalclinic.org/media/upload/pdf/...1. Caso índex p.[D561V] 2. Hermana portadora p.[D561V + =] 3. Hermana no portadora [= + =] 4. Control](https://static.fdocuments.ec/doc/165x107/604089d719f5a61d7530418e/casos-clnicos-in-1-caso-ndex-pd561v-2-hermana-portadora-pd561v-.jpg)



