
Cáncer de tiroides
39
CÁNCER DE TIROIDES • Sebastián Acosta • David Domínguez • Damián Vargas
-
Upload
comediante-xxx-bromas-con-alto-contenido-sexual -
Category
Documents
-
view
666 -
download
2
Transcript of Cáncer de tiroides

CÁNCER DE TIROIDES • Sebastián Acosta •David Domínguez •Damián Vargas
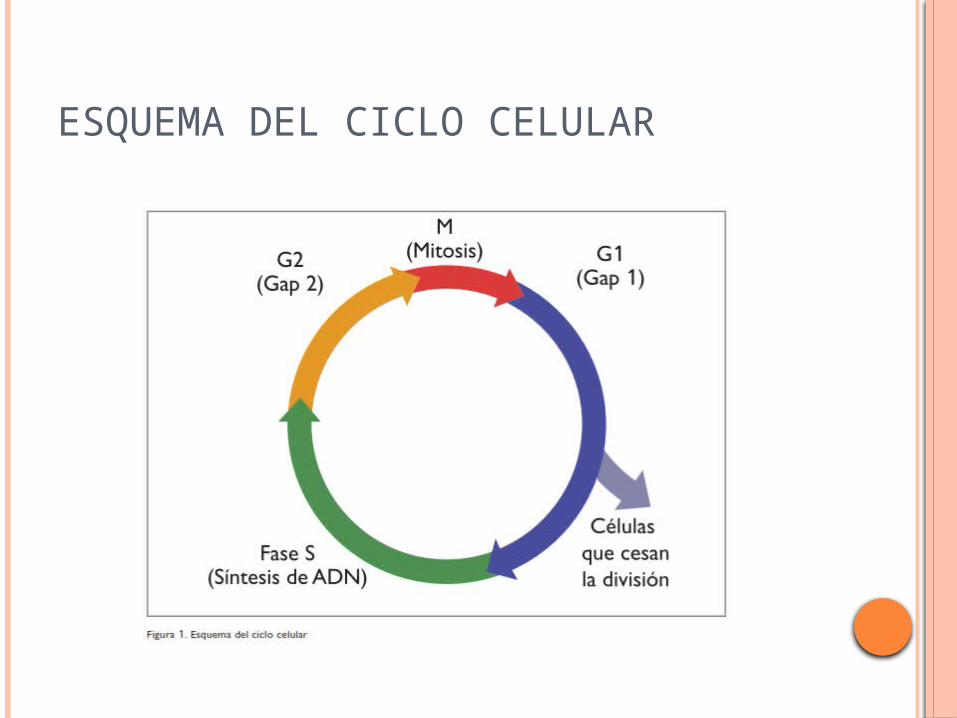
ESQUEMA DEL CICLO CELULAR

La célula, en una primera fase, está en quiescencia (G0) y para pasar a G1 se requiere la fosforilación de la proteína del retinoblastoma Rb.
La misma está en reposo asociada a la proteína E2F y cuando es fosforilada se separan y ésta última activa el ciclo celular. Sucesivamente la células va pasando por los estadíos S (síntesis de ADN), G2 y M (mitosis).
En cada una de estas etapas participan una serie de ciclinas que son fosforiladas por las quinasas de ciclinas.
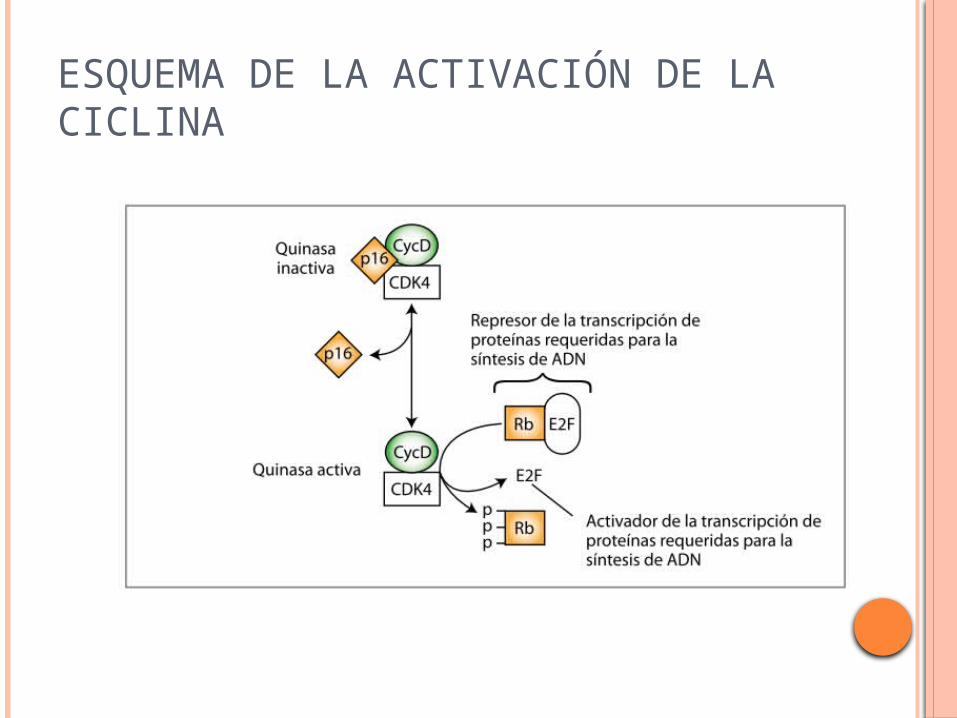
ESQUEMA DE LA ACTIVACIÓN DE LA CICLINA

P53 Otra proteína importante en estos procesos es la denominada
p53. Se trata de un factor antiproliferativo que también participa del proceso de reparación del ADN. Cuando por alguna causa, sea radiación o compuestos químicos, se daña el ADN, se pone en función la maquinaria necesaria para reparar el ADN. El p53 detiene el ciclo celular a la espera de la finalización del proceso reparador. Cuando esto sucede la célula puede seguir tres vías alternativas:
a) si el ADN fue reparado correctamente todo vuelve a la normalidad y puede reiniciarse el ciclo celular
b) la reparación por unión de genes que normalmente no están unidos, generando un oncogen o alterando la actividad de un factor antiproliferativo
c) el daño es inviable de reparar y p53 envía a la célula a la muerte celular programada o apoptosis.

FACTORES IMPLICADOS EN REGULACIÓN TIROIDEA

CAUSAS CÁNCER TIROIDEO

TSH
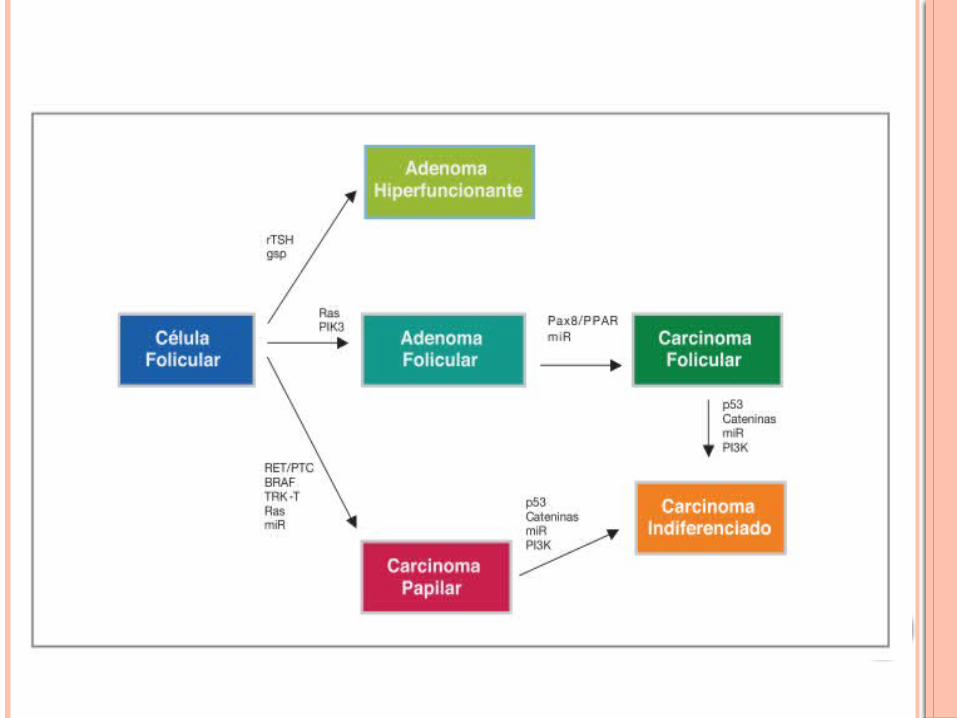
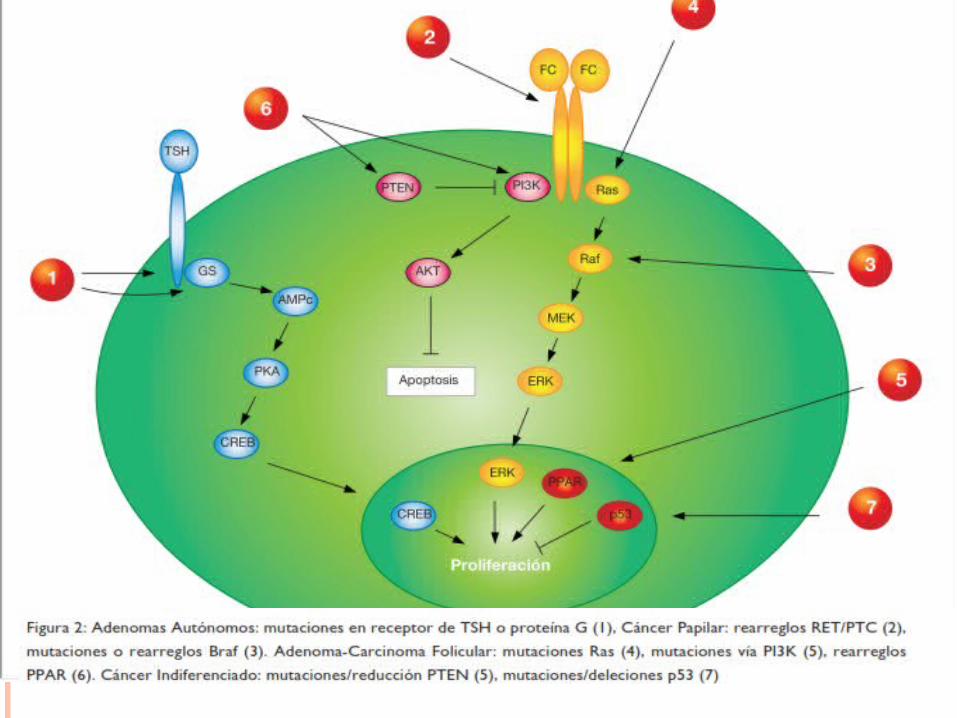
La TSH es el principal regulador del crecimiento en condiciones normales, pero también a su vez de la función tiroidea. La TSH actúa principalmente a través del estímulo de la proteína Gs (en lo que res- pecta al crecimiento) activando la adenilato ciclasa con la consiguiente formación de AMPc .En la mayoría de los adenomas hiperfuncionantes, ocurre una mutación en el receptor de TSH, por las cuales adquiere ganancia de función estimulando constitutivamente la formación de AMPc. De no ocurrir mutaciones en otros genes, este proceso no implica una desdiferenciación celular. Por lo tanto es raro encontrar mutaciones en el receptor de TSH en tumores malignos.

En lo que respecta a la variante folicular (CFT) (10-20% de los tumores de tiroides) los oncogenes principales involucrados son:
Ras:
Se lo encuentra alterado en aproximadamente 20-50% de los CF. Las proteínas RAS son GTPasas (su- bunidad de proteína G) activadas por factores de crecimiento con la consecuente activación de la MAP quinasa (MAPK) y otras vías de señalización como la PI3K/AKT. Cesado el estímulo, la proteína se inac- tiva. Diversas mutaciones le confieren ganancia de función favoreciendo la proliferación celular y desdi- ferenciando la célula. En menor proporción también se han encontrado mutaciones en la variante papilar.

Rearreglos Pax8-PPARγ:Ocurren por fusión del promotor del gen Pax8 con la mayor parte de la región codificante para el re- ceptor nuclear PPARγ (Peroxisome Proliferator-Activated Receptor Gamma). Este rearreglo da como resultado una sobre expresión del PPAR γ. Las consecuencias de esta sobre expresión no están muy claras pero se sabe de la desregulación de varios genes involucrados, entre otros, en la proliferación celular. Estos rearreglos se los encuentran en aproximadamente 35% de los CF.
microARNs (miARNs):Básicamente, los miARNs son ARN pequeños, una veintena de nucleótidos aproximadamente, con una secuencia complementaria a la de un ARN mensajero determinado, pudiendo interferir por ello con la síntesis de la proteína respectiva. Se ha visto que también pueden activar el proceso de traducción. En el cáncer de tiroides, tanto en la variante folicular como papilar se ha encontrado una desregulación en la expresión de diferentes miARNs.

En lo que respecta a la variante papilar (CPT) (80%) podemos encontrar:
RET:
Las alteraciones del gen RET corresponden a rearreglos conocidos como RET/PTC. Estos rearreglos se producen por fusión del dominio quinasa del RET (el gen RET codifica para un receptor con activi- dad tirosina quinasa. Normalmente no se expresa en cantidades importantes en células foliculares, pero sí en células C) con porciones de diferentes genes. Los rearreglos más comunes son el RET/PTC1 y el RET/PTC3. Estas fusiones activan la cascada RAS-RAF-MAPK. Se los encuentra en aproximadamente un 20% de los CP.
BRAF:
Codifica para una serina-treonina quinasa perteneciente a la familia de las proteínas RAF. Se lo encuen- tra mutado en un 45% aproximadamente de los CP. La presencia de un BRAF mutado correlaciona con una mayor agresividad del tumor.En el cáncer indiferenciado (CIT) o pobremente diferenciado se han encontrado además de alteracio- nes en RAS y BRAF, en el gen supresor de tumores p53, PTEN-PI3K/AKT y en β-cateninas.

p53: Codifica para una proteína que induce la detención del ciclo celular
y/o apoptosis. Se han encontrado alteraciones en esta proteína en la mayoría de los CIT.
PTEN: codifica para una enzima con actividad de fosfatasa que regula
negativamente la vía PI3K/AKT la cual pro- mueve la sobrevida celular. Se ha encontrado mutaciones o reducción de PTEN y mutaciones o ampli- ficación en la vía PI3K/AKT en CFT, CPT y CIT principalmente.
β-cateninas: son proteínas que interaccionan con ciclinas D, myc y otras proteínas
involucradas en el ciclo celular. Se observó que en tumores anaplásicos presentan mutaciones, no así en tumores benignos.

NUEVOS TRATAMIENTOS

Alteración de diferentes proteínas lleva al desarrollo de diferentes clases de cáncer de tiroides.
La caracterización de estas proteínas lleva al aparecimiento de nuevas terapias.
Algunas de estas están dirigidas al bloqueo de vías de señalización, inhibición de laexpresión de genes, destrucción tumoral, etc.

INHIBIDORES DE LAS VÍAS DE SEÑALIZACIÓN
Inhibición de receptores tirosinas quinasas
Variabilidad en la expresión de los genes para estos.
Algunos asociados a la angiogénesis. Mayoría de inhibidores intervienen en la unión
de ATP dentro del domino catalítico.

INHIBIDORES DE RET
Vandenatib: inhibición de RET y VEGF, inhibe el crecimiento de tumores.
Sorafenib: inhibidor de RET y oncoprotíenas, utilizado en terapias contra carcinoma de hígado y riñón. Experimentalmente comprobado que interviene en el control de tumores tiroides indiferenciados.

INHIBIDORES DEL RECEPTOR PARA EGF
Gefinitib: Inhibición de tumores tiroides en animales.
No utilizado ya que en los paciente sometidos a esta terapia no mostraron respuesta tumoral.

INHIBIDORES DE LA ANGIOGÉNESIS
Empleo de anticuerpos monoclonales contra VEGF y fosbretabulina, redujeron el crecimiento de implantes tumorales en modelos animales.
Fosbretabulina: efectiva en el tratamiento, única desventaja es el tiempo de duración del mismo que alcanza los nueve años.

Existen muchas desventajas en la utilización de estos fármacos.
Baja eficacia Hepatotoxicidad Hemoptisis Fatiga Neutropenia Diarrea

INHIBIDORES DE LA ACCIÓN DEL RAS
Fenilacetato y manumicina: Inhiben el crecimiento de tumores tiroideos en los humanos.

INHIBIDORES DE LAS PROTEÍNAS HSP90
La inhibición de esta proteína disminuye el crecimiento de varias líneas celulares tumorales.

INHIBICIÓN DE PROTEASAS
Inhibición reversible
Evita la proteólisis prevista que puede afectar a múltiples cascadas de señalización en la célula.

AGENTES DESMETILANTES
La metilación del ADN reprime la expresión de genes.
La hipótesis que la metilación en genes supresores de tumores resulta en una contribución al desarrollo del cáncer ha llevado a realizar estudios de fase II en pacientes con tumores que no responden a la terapia con iodo empleando agentes demetilantes.

INHIBIDORES DE DESACETILASAS DE HISTONAS
Se ha observado, en cultivos celulares, que la presencia de inhibidores de desacetilasas resulta en una acumulación de histonas acetiladas induciendo diferenciación o apoptosis en células transformadas.
Estos compuestos posiblemente tienen acción antitumoral y pueden llegar a sinérgica el efecto de la radioterapia y la quimioterapia.

Se han comenzado estudios de fase II en 16 pacientes con cáncer diferenciado de tiroides y tres con cáncer medular de tiroides (CMT) los mismos que fueron tratados con vorinostat oral, sin combinación con otra terapia.
Ninguno de los pacientes tuvo una respuesta completa o parcial.
Las razones de la finalización de la terapia se debieron al progreso de la enfermedad o a efectos adversos (fatiga, deshidratación, ataxia, neumonía, trombocitopenia severa).

INHIBIDORES DE LA CICLOOXIGENASA 2 (COX-2) La activación de esta proteína inhibe la
apoptosis aumentando la angiogénesis.
Se ha demostrado que esta proteína se encuentra aumentada en tumores de tiroides benignos y malignos.
Se lo ha utilizado para el tratamiento del cáncer de tiroides aunque los datos disponibles no muestran una buena respuesta de este compuesto.

TALIDOMIDA Y LENALIDOMIDA
Estos compuestos son potentes antiinflamatorios, antiangiogénicos e inmunomoduladores.
Su mecanismo de acción no está bien comprendido aunque se ha observado un aumento de la relación linfocitos T CD8/CD4 por disminución de los linfocitos T colaboradores circulantes.
En estudios realizados se ha demostrado que un 23.5 % de pacientes responden de manera correcta a este tratamiento

IROFULVENO (HMAF, MGI-114, IROF)
El irofulveno se une al ADN de la célula bloqueando el crecimiento de la misma.
Se ha demostrado que en la clínica puede ser utilizado este tratamiento en combinación con otros, ya que se ha demostrado una reducción en el tamaño de la masa tumoral en Pctes. con cáncer diferenciado de tiroides.

ROSIGLITAZONA
El PPARγ es un factor de transcripción perteneciente a la familia de receptores nucleares de esteroides y puede ser considerado como un gen supresor de tumores, aunque su rol en la tumorigenesis no está totalmente aclarado.
La rosiglitazona es un agonista del PPARγ por el cual disminuye la resistencia a la insulina en el músculo y en el tejido adiposo e inhibe la gluconeogénesis hepática y usada por lo tanto para el tratamiento de la diabetes.

BEXAROTENO
El bexaroteno activa los receptores retinoides (vitamina A) los cuales son responsables de controlar la división celular.
En trabajos “in vitro” se demostró que los retinoides aumentaron la expresión del ARN mensajero del NIS y la consecuente captación de iodo por parte de líneas celulares cancerosas.

TERAPIA GÉNICA
La introducción del transportador de iodo (NIS) en células que han perdido la capacidad de concentrar al halógeno permitiría el tratamiento con una terapia convencional utilizando Iodo (Esta estrategia se está tratando de implementar para otro tipo de tumores tales como pulmón, próstata, etc.).

Otro tipo de terapia génica empleada fue la de inducir la expresión de la enzima viral timidina quinasa mediante inyección intratumoral del vector retroviral.
La presencia de esta enzima permite la activación de la droga antiviral, ganciclovir, resultando en la ruptura de las dobles cadenas del ADN con la consiguiente muerte celular.

ARN DE INTERFERENCIA (ARNI)
Los ARNi se han convertido en una herramienta de laboratorio para silenciar genes y estudiar la función de estos.
En la actualidad se los ha propuesto para el tratamiento y prevención de enfermedades.

CONCLUSIONES Es interesante analizar la discrepancia respecto
a los resultados exitosos obtenidos previamente con animales de experimentación y la menor efectividad conseguida en los estudios de fase II en pacientes.
La mayoría de los compuestos utilizados tienen amplios efectos actuando en la mayoría de los tejidos con la consecuente toxicidad aparejada.
En los estudios de experimentación con animales no se miden los efectos adversos, tan sólo una reducción en el tamaño del tumor implantado.

En lo que respecta a las terapias génicas in vivo la dificultad es, que no se puede tener control sobre el número de copias de un gen o RNAi inyectado dentro de una célula utilizando vectores virales, liposomales, etc.
Por último no hay, para cada uno de los productos ensayados, muchos estudios sobre posible resistencia a los mismos.

BIBLIOGRAFÍA Ain KB, Lee C & Williams KD 2007 Phase II trial of
thalidomide for therapy of radioiodine-unresponsive and rapidly progressive thyroid carcinomas. Thyroid 17 663–670.
Ain KB, Lee C, Holbrook K, Dziba JM & Williams KD. 2008. Phase II study of lenalidomide in distantly metastatic, rapidly progressive, and radioiodineunresponsive thyroid carcinomas: preliminary results. Journal of Clinical Oncology 26 Supplement abstract 6027.
Barzon L, Pacenti M, Taccaliti A, Franchin E, Bruglia M, Boscaro M, Palù G. 2005. A pilot study of combined suicide/cytokine gene therapy in two patients with end-stage anaplastic thyroid carcinoma. J Clin Endocrinol Metab., 90: 2831-4.

Castellone M.D., Carlomagno F, Salvatore G, Santoro M.Receptor Tyrosine Kinase Inhibitors in Thyroid cancer. 2008.Best Practice & Research Clinical Endocrinology & Metabolism, 22: 1023-1038.
• Cohen E., Needles B., Cullen K., Wong S., Wade J., Ivy S., Villaflor V., Seiwert T., Nichols K., Vokes E.2008a. Phase 2 study of sunitinib in refractory thyroid cancer.Journal of Clinical Oncology 26 Supplement abstract 6025.
• Cohen E.E., Rosen L.S., Vokes E.E., Kies M.S., Forastiere A.A., Worden F.P., Kane M.A., Sherman E.,Kim S., Bycott P.2008b. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. Journal of Clinical Oncology, 26: 4708-4713.


















