
BLAST: Búsqueda de homologías - VHIR · • Para cuantificar la similaridad entre dos secuencias,...
40
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 1 BLAST: Búsqueda de homologías
Transcript of BLAST: Búsqueda de homologías - VHIR · • Para cuantificar la similaridad entre dos secuencias,...

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 1
BLAST: Búsqueda de homologías

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 2
Outline
- Motivación- Alineamiento por parejas
- Sistemas de puntuación- Matrices de substitución (PAM, BLOSUM)
- BLAST (Alineamiento contra BBDD)- Ejemplo Básico- Cómo funciona?- E-values, Bit-scores- Donde cortar?

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 3
Predicción de la función de un proteína/gen
• Secuencia problemaQueremos averiguar sus posibles propiedades
• La evolución es un proceso conservativoCambian los residuos en una secuencia pero se conservan las
propiedades bioquímicas y los procesos fisiológicos
• Si somos capaces de encontrar secuencias homólogas a la secuencia problema podemos inferir que ésta “debe de tener” propiedades similares a las de la secuencia conocida
• La búsqueda (el hallazgo, de hecho) de secuencias homólogas puede ser una vía para predecir la función de una proteína o un gen.

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 4
Homología vs Similaridad
• Homología– Descendencia de un ancestro común
– Medida cualitativa• Dos secuencias son homólogas o no lo son
– Podemos usar una medida de similaridad para inferir homología
• Similaridad– Medida cuantitativa para determinar el grado de relación
entre dos secuencias
– Se basa en la combinación de• Identidad: grado en que dos secuencias de AA o nucleótidos son
invariantes
• Conservación: cambios en una posición de una secuencia de AA que mantienen las propiedades químicas de la secuencia

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 5
Ejemplo: similaridad
Queremos medir el grado de similaridad de dos secuencias
Es necesario definir un criterio (sistema de puntuación) que evalúe esta similaridad
Ejemplo:
- Match=1- Mismatch=0
S= A T G C A G TT= A T A A G Tp(s,t) 1 1 0 0 0 0 Σ= 2

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 6
Alineamiento de secuencias
• Para destacar las regiones similares entre las moléculas, probablemente la herramienta más utilizada en bioinformática es el alineamiento de secuencias
• IDEA: Comparar dos (pairwise) o más (multiple) secuencias buscando los caracteres o patrones que aparezcan en el mismo orden en las secuencias
• Distinguiremos entre alineamientos– Globales: Alineamiento de secuencias completas– Locales: Alineamiento de subsecuencias

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 7
Infiriendo homología
• Si, al alinear dos secuencias, se obtiene una puntuación “elevada” se puede inferir que ambas secuencias son posibles homólogos
• Esto sugiere que dada una secuencia, para encontrar secuencias homólogas en una base de datos, tan sólo hace falta alinear ésta con todas las de la BD
• Las secuencias con mayor puntuación serán posibles homólogas de la secuencia problema

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 8
Sistemas de puntuación
• Para cuantificar la similaridad entre dos secuencias, S y T, definimos sistemas de puntuaciones de forma que para cada alineamiento se pueda calcular un número tal que, a mayor valor, mayor sea su significación (biológica)
• Pueden ser esquemas sencillos Coincidencia: S[i]=T[i] 1
No coincidencia: S[i]≠T[i] 0
Inserción de espacios: Gap -1
• O bien sistemas más complejos basados en afinidades químicas o en frecuencias de emparejamiento observadas

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 9
Puntuación de un alineamiento
• Una vez establecido un sistema de puntuación la puntuación de una pareja de caracteres s, talineados se define como p(s,t)
• La puntuación de un alineamiento (score) entre S i T
• Un alineamiento es óptimo si su puntuación es la más grande posible
score S ,T =∑∀ i
pS [i ] , T [i ]

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 10
Ejemplo: Alineamiento de secuencias
S= A T G C A G TT= A T A A G Tp(s,t) 1 1 0 0 0 0 Σ= 2
S= A T G C A G TT= A T A A ▬ G Tp(s,t) 1 1 0 0 -1 1 1 Σ= 3
S= A T G C A G TT= A T ▬ A A G Tp(s,t) 1 1 -1 0 1 1 1 Σ= 4
- Match=1- Mismatch=0- Gap=-1
- El alineamiento de las secuencias puede aumentar la puntuación
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 11
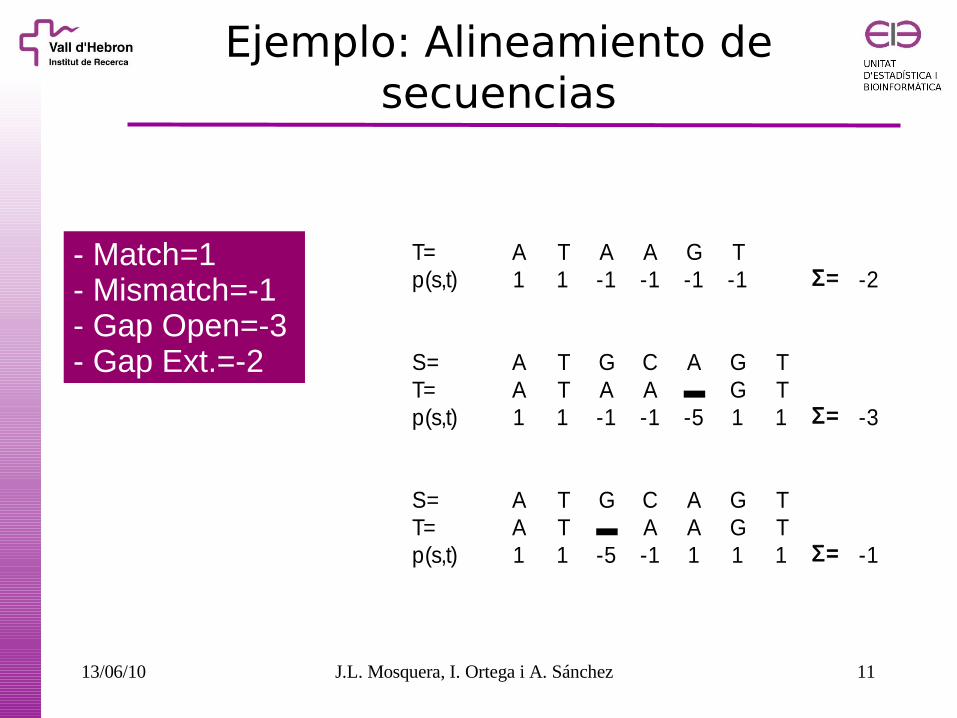
Ejemplo: Alineamiento de secuencias
T= A T A A G Tp(s,t) 1 1 -1 -1 -1 -1 Σ= -2
S= A T G C A G TT= A T A A ▬ G Tp(s,t) 1 1 -1 -1 -5 1 1 Σ= -3
S= A T G C A G TT= A T ▬ A A G Tp(s,t) 1 1 -5 -1 1 1 1 Σ= -1
- Match=1- Mismatch=-1- Gap Open=-3- Gap Ext.=-2

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 12
El sentido de las puntuaciones
• Ambos alineamientos puntúan igual. Sin embargo,– conserva residuos comunes (A, S, T)
– conserva residuos menos habituales (W, Y)
• El sistema de puntuar los emparejamientos entre AA debería reflejar su relación química y biológica
– Residuos similares/distintos deberían puntuar alto/bajo porque cambiar uno por otro afectará poco/mucho la función de la proteína
- Match=1- Mismatch=0- Gap=-1
S= T T Y G A P P W C ST= − T G Y A P P P W Sp(s,t) -1 1 0 0 1 1 1 0 0 1 Σ= 4
S= T T Y G A P P W C ST= T G Y A P P P W S −p(s,t) 1 0 1 0 0 1 1 1 0 -1 Σ= 4

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 13
Matrices de puntuación o de substitución
• Tabla que contiene las puntuaciones que asignamos a cada pareja posible de caracteres
• Sirve para las coincidencias y las no coincidencias
• El término “substitución” refleja que lo que se pretende al puntuar un emparejamiento es valorar el coste evolutivo de cambiar un residuo por otro
actaccagttcatttgatacttctcaaa
taccattaccgtgttaactgaaaggacttaaagact
Secuencia 1
Secuencia 2
Matriz identidad
P(i,i)=1, P (i,j)=0
o alguna variante P(i,i)=0.9,
P (i,j)=-0.1
A G C T
A 1 0 0 0
G 0 1 0 0
C 0 0 1 0
T 0 0 0 1
Match: 1Mismatch: 0Score = 5
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 14
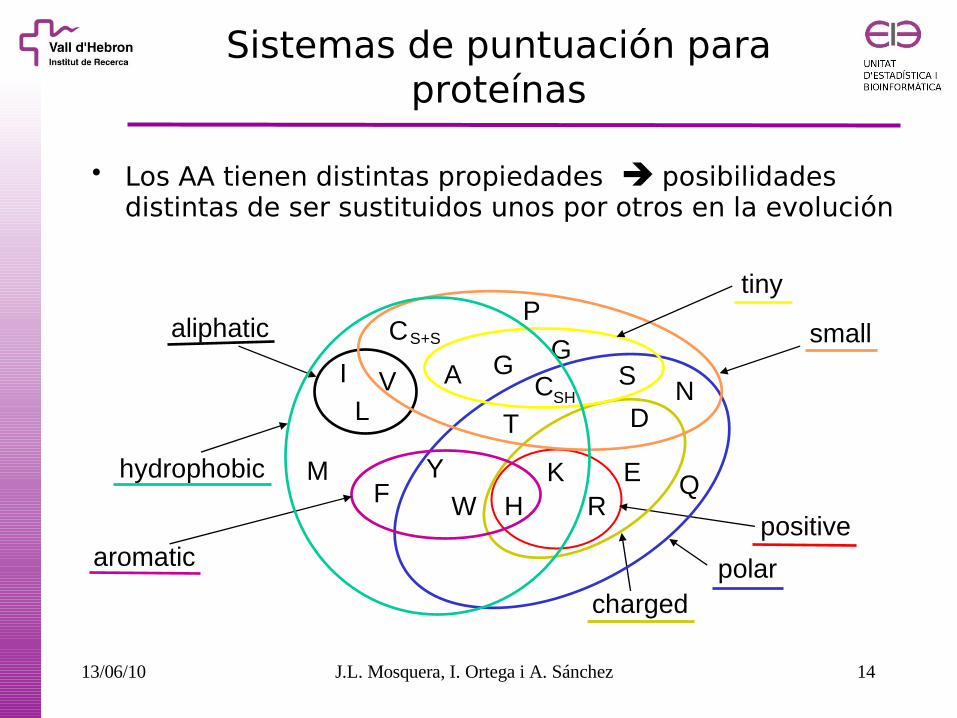
Sistemas de puntuación para proteínas
Los AA tienen distintas propiedades posibilidades distintas de ser sustituidos unos por otros en la evolución
CP
GGAVI
L
MF
Y
W HK
RE Q
DN
S
T
CSH
S+S
positive
chargedpolar
aliphatic
aromatic
small
tiny
hydrophobic

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 15
Substituciones de aminoácidos
• Ciertas substituciones de AA son muy comunes en proteínas homólogas. Otras no lo son en absoluto
• Esto puede interpretarse como que– Las primeras mantienen la función de la proteína
– Las segundas afectan negativamente a su función
• Las sustituciones “inusuales” tendrán menor grado de aceptación por por parte de la selección natural
• Para poder hacer alineamientos que reflejen el proceso evolutivo que ha llevado a cambiar una secuencia por otra es preciso disponer de estimaciones de la frecuencia con que se produce cada cambio o sustitución
• Para responder a esta necesidad se crearon las matrices de sustitución.

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 16
Familias de matrices de substitución
• No hay una matriz única que se pueda usar siempre
• Según la familia de proteínas y el grado de similitud esperado se usa una u otra
• Las más utilizadas PAM y BLOSUM– PAM: Point Accepted Mutation Matrix (Dayhoff et al, 1978)
• Derivadas de alineamientos globales de secuencias próximas
• A mayor número mayor distancia evolutiva
• PAM40 (similares) PAM250 (distantes)
– BLOSUM: BLOcks of amino acid SUbstitution Matrix (Henikoff & Henikoff, 1992)
• Derivadas de alineamientos locales de secuencias distantes
• A mayor número mayor proximidad evolutiva
• BLOSUM90 BLOSUM45 (El número representa porcentaje de identidad)

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 17
PAM250
A R N D C Q E G H I L K M F P S T W Y V B Z
A 2 -2 0 0 -2 0 0 1 -1 -1 -2 -1 -1 -3 1 1 1 -6 -3 0 2 1 R -2 6 0 -1 -4 1 -1 -3 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2 1 2 N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -3 0 1 0 -4 -2 -2 4 3 D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -6 -1 0 0 -7 -4 -2 5 4 C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2 -3 -4 Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 -1 -1 -5 -4 -2 3 5 E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2 4 5 G 1 -3 0 1 -3 -1 0 5 -2 -3 -4 -2 -3 -5 0 1 0 -7 -5 -1 2 1 H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2 3 3 I -1 -2 -2 -2 -2 -2 -2 -3 -2 5 2 -2 2 1 -2 -1 0 -5 -1 4 -1 -1 L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -3 -3 -2 -2 -1 2 -2 -1 K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2 2 2 M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2 -1 0 F -3 -4 -3 -6 -4 -5 -5 -5 -2 1 2 -5 0 9 -5 -3 -3 0 7 -1 -3 -4 P 1 0 0 -1 -3 0 -1 0 0 -2 -3 -1 -2 -5 6 1 0 -6 -5 -1 1 1 S 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1 2 1 T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3 -5 -3 0 2 1 W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17 0 -6 -4 -4 Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2 -2 -3 V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4 0 0 B 2 1 4 5 -3 3 4 2 3 -1 -2 2 -1 -3 1 2 2 -4 -2 0 6 5 Z 1 2 3 4 -4 5 5 1 3 -1 -1 2 0 -4 1 1 1 -4 -3 0 5 6
PAM250 = 250 mutaciones por 100 residuos
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 18
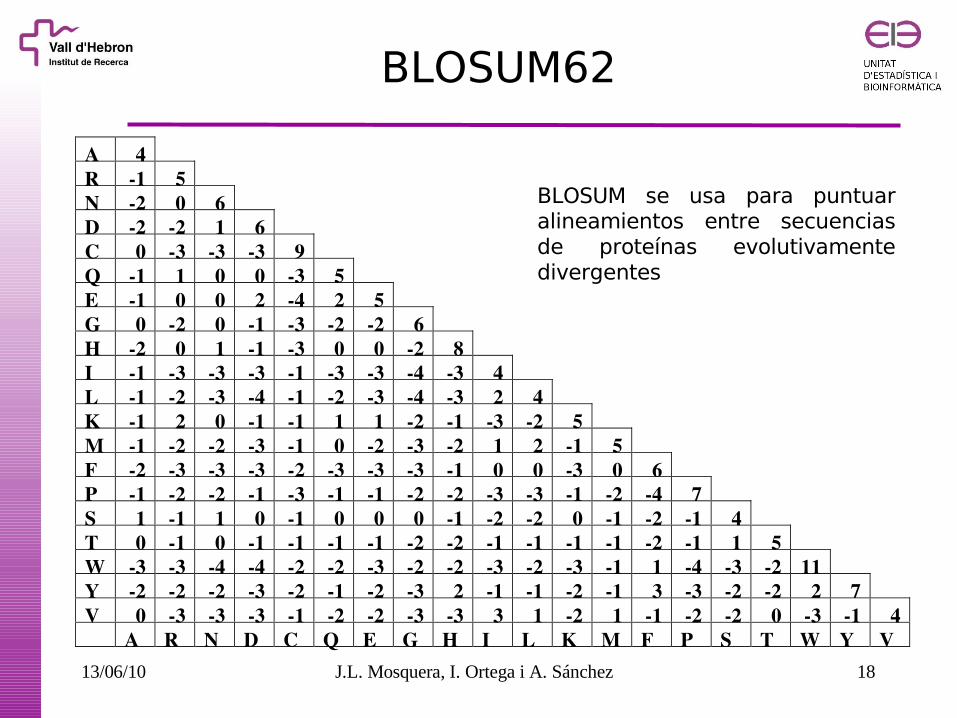
BLOSUM62
A 4 R 1 5 N 2 0 6 D 2 2 1 6 C 0 3 3 3 9 Q 1 1 0 0 3 5 E 1 0 0 2 4 2 5 G 0 2 0 1 3 2 2 6 H 2 0 1 1 3 0 0 2 8 I 1 3 3 3 1 3 3 4 3 4 L 1 2 3 4 1 2 3 4 3 2 4 K 1 2 0 1 1 1 1 2 1 3 2 5 M 1 2 2 3 1 0 2 3 2 1 2 1 5 F 2 3 3 3 2 3 3 3 1 0 0 3 0 6 P 1 2 2 1 3 1 1 2 2 3 3 1 2 4 7 S 1 1 1 0 1 0 0 0 1 2 2 0 1 2 1 4 T 0 1 0 1 1 1 1 2 2 1 1 1 1 2 1 1 5 W 3 3 4 4 2 2 3 2 2 3 2 3 1 1 4 3 2 11 Y 2 2 2 3 2 1 2 3 2 1 1 2 1 3 3 2 2 2 7 V 0 3 3 3 1 2 2 3 3 3 1 2 1 1 2 2 0 3 1 4 A R N D C Q E G H I L K M F P S T W Y V
BLOSUM se usa para puntuar alineamientos entre secuencias de proteínas evolutivamente divergentes
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 19
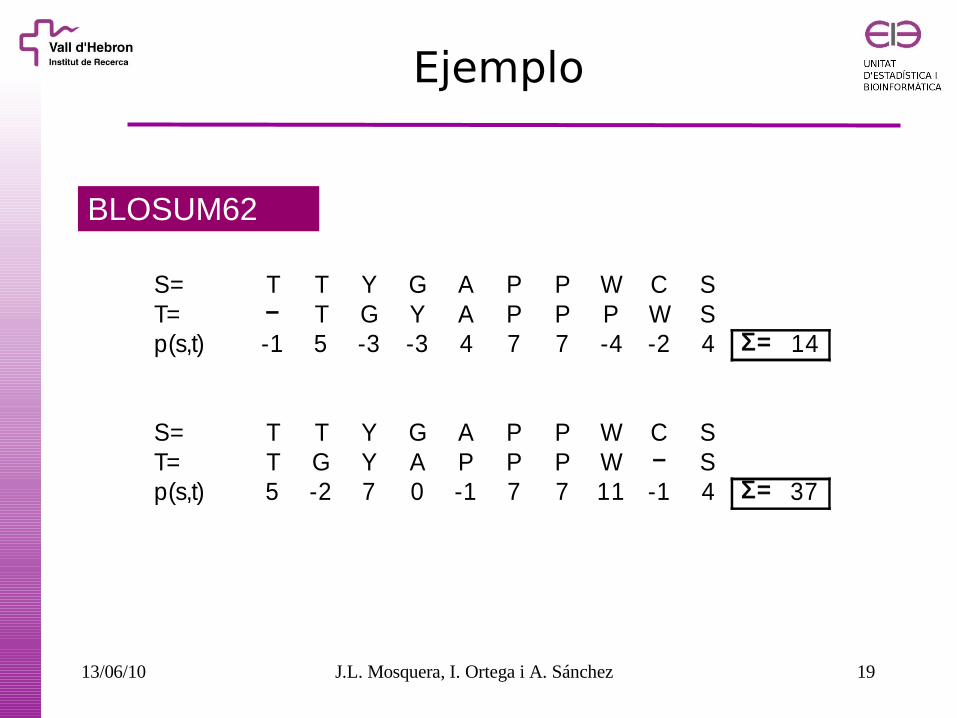
Ejemplo
BLOSUM62
S= T T Y G A P P W C ST= − T G Y A P P P W Sp(s,t) -1 5 -3 -3 4 7 7 -4 -2 4 Σ= 14
S= T T Y G A P P W C ST= T G Y A P P P W − Sp(s,t) 5 -2 7 0 -1 7 7 11 -1 4 Σ= 37

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 20
Consejos para elegir una matriz de substitución
• Generalmente, la matrices BLOSUM funcionan mejor que las PAM para búsquedas de similaridad local
• Cuando comparamos– proteínas cercanas deberíamos usar
• PAM bajas o• BLOSUM altas
– proteínas distantes sería mas conveniente• PAM altas o• BLOSUM bajas
• Para búsquedas en BBDD sin información previa es bastante común el uso de una BLOSUM62 (o PAM70)

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 21
Alineamiento contra BBDD
• Supongamos que buscamos secuencias homólogas a nuestra secuencia problema
• Una estrategia posible es hacer alineamientos contra una base de datos de secuencias
• Una manera de obtener el alineamiento óptimo seria– Construir todos los alineamientos posibles
– Calcular la puntuación de cada uno
– Mirar que alineamiento tiene el valor más grande (OJO! puede haber más de uno)
– PROBLEMA: El número de alineamientos posibles es muy alto Demasiado lento!!!

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 22
Alineamiento contra BBDD
• Una alternativa a la búsqueda exhaustiva es la programación dinámica (PD)
• Los dos algoritmos más conocidos són– Needleman-Wunsch (J. Mol Biol., 1970) para alineamientos globales
– Smith-Waterman (J. Mol Biol., 1981), una variante para alineamientos locales
• Sirven para alinear tanto DNA como proteínas
• Retornan los alineamientos con la máxima puntuación posible para una matriz de substitución y un coste de gaps dados
• El alineamiento obtenido no tiene necesariamente un significado biológico

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 23
Alineamiento contra BBDD
• Una forma razonable de comparar una secuencia con las de una base de datos es mediante alineamientos locales
• El algoritmo de Smith-Waterman obtiene un alineamiento local óptimo dado un sistema de puntuaciones
• Demansiado lento para buscar contra una BBDD
• Alternativa: Los métodos heurísticos (BLAST o FastA)

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 24
BLAST
• BLAST: Basic Local Alignment Search Tool (Alschultz, NAR 1997)– Rápido– Preciso – Accesible vía web
Haz clic aquí para verlo
– PROBLEMA: Al ser un algoritmo heurístico puede obviar alineamientos óptimos
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 25
BLAST en NCBI
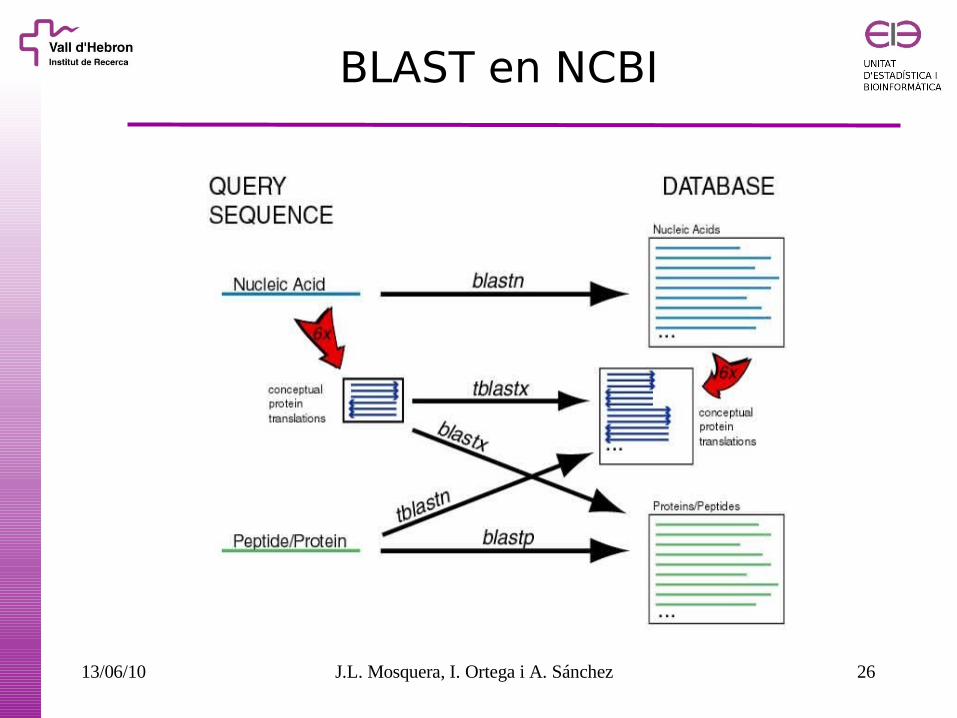
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 26
BLAST en NCBI

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 27
Ejemplo
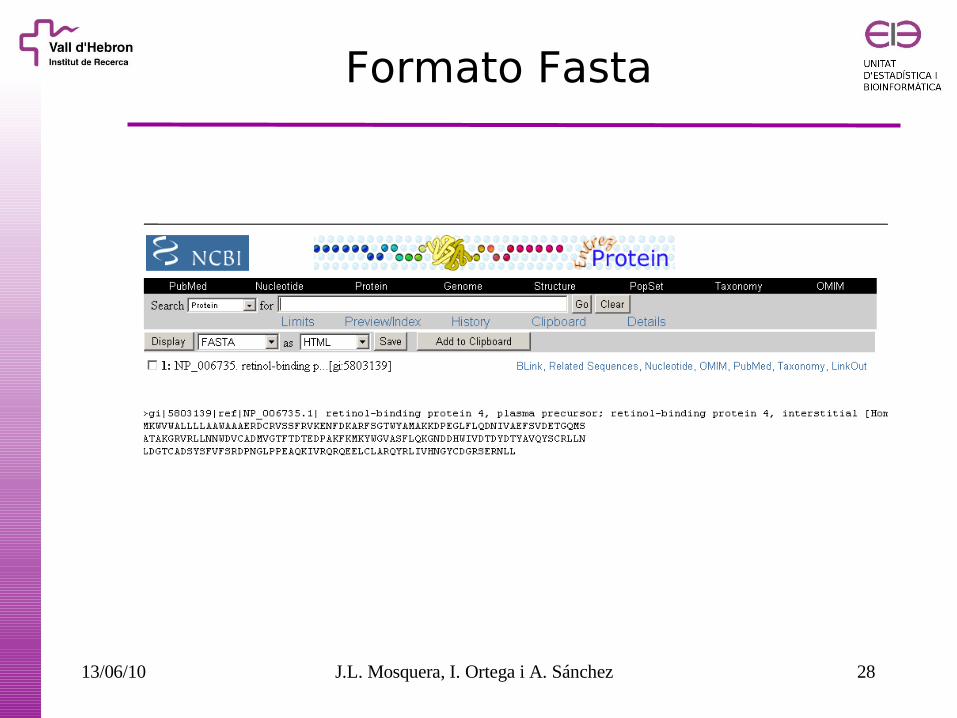
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 28
Formato Fasta

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 29
Ejemplo

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 30
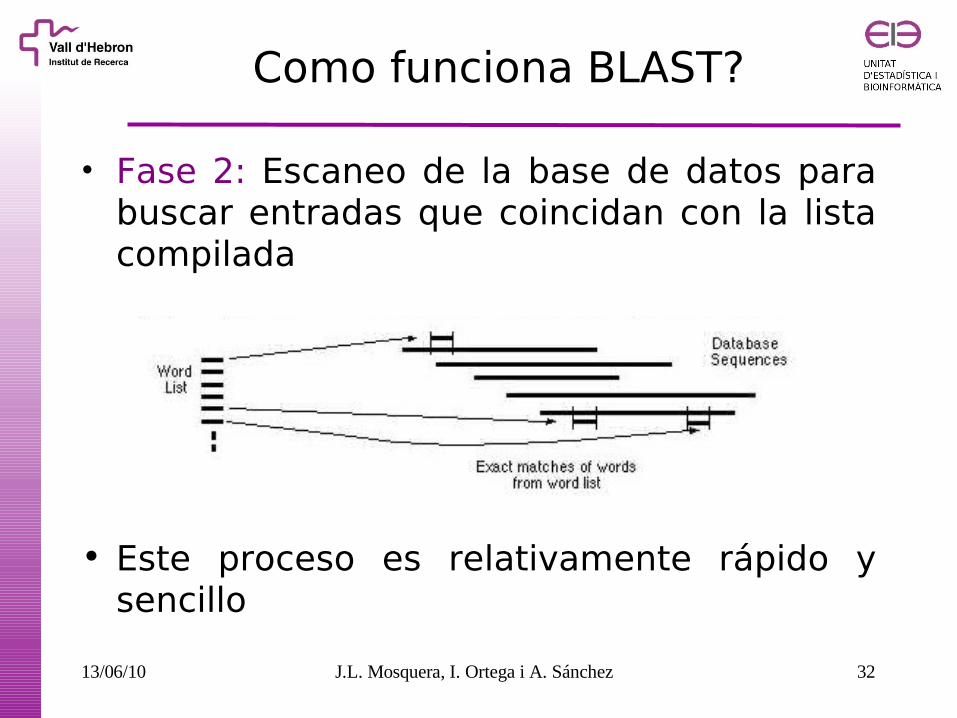
Como funciona BLAST?
• Fase 1: compila una lista de palabras (de tamaño w=3) que generen una puntuación superior a un umbral mínimo HSP (High-scoring Segment Pairs)

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 31
Como funciona BLAST?
Ejemplo: Búsqueda para “human RBP”:
FSG SGT GTW TWY WAY
…FS GTW YA… Lista de palabras (w=3) YSG TGT ATW SWY WFA
FTG SVT GSW TWF WYA
Neighborhood word hits > HST
GTW 6,5,11 22
GSW 6,1,11 18
ATW 0,5,11 16
NTW 0,5,11 16
GTY 6,5,2 13
...
Neighborhood word hits < HST
GNW 10
GAW 9
...
HSP = 11
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 32
Como funciona BLAST?
• Fase 2: Escaneo de la base de datos para buscar entradas que coincidan con la lista compilada
• Este proceso es relativamente rápido y sencillo

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 33
Como funciona BLAST?
• Fase 3: Para cada palabra “hit”, extiende el alineamiento en ambas direcciones para encontrar alineamientos cuya puntuación sea mayor que un cierto “cutoff”
• La extensión cesa si – la puntuación
• desciende por debajo del “cutoff”,
• llega a 0, o
– Se acaba la secuencia
KENFDKARFSGTWYAMAKKDPEG RBP (query)
MKGLDIQKVAGTWYSLAMAASD lactoglobulin (hit)
Hit!extender extender
13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 34
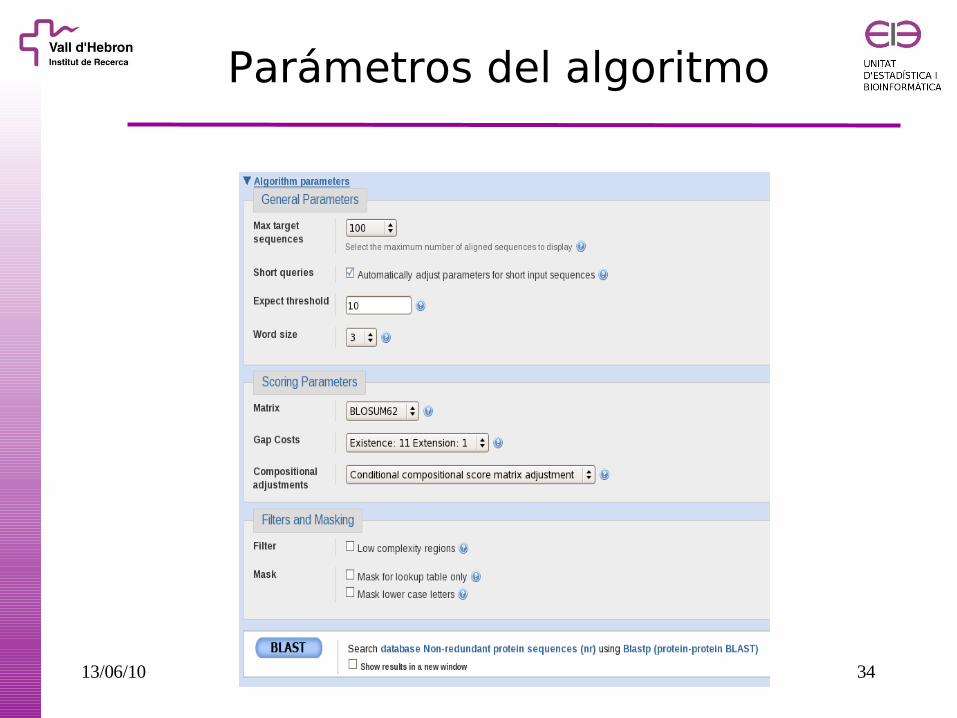
Parámetros del algoritmo

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 35
Criterios de selección
• Dado que los programas de búsqueda heurística tan sólo encuentran coincidencias aproximadas conviene poder cuantificar cuan aproximadas son
• En el BLAST saremos los estadísticos
– Bit-score– E-value

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 36
Bit-score
• El valor de las puntuaciones obtenidas por un emparejamiento carecen de sentido si no se tiene en cuenta– el tamaño de la base de datos, y– el sistema de puntuación
• Los Bit-score normalizan las puntuaciones para independizarlas de ambos factores de forma que podamos compararlas
S'=S−ln K
ln 2

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 37
E-value
• Dada una secuencia que ha obtenido una puntuación S, E-value (o Expectation value) es el número esperado de puntuaciones iguales o superiores a las de dicha secuencia atribuibles al azar
• Un E-value de 10 para una coincidencia significa que, en una base de datos de secuencias aleatorias del mismo tamaño en la que se ha realizado la búsqueda, se podría esperar encontrar hasta 10 coincidencias con la misma puntuación o similar
• El E-value es la medida de corte más utilizada en las búsquedas en bases de datos. Sólo se informa de las coincidencias que superan un nivel mínimo
• El E-value oscila entre 0 y cualquier valor
E=K mne− S

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 38
Donde cortar?
• Valores bajos de E se pueden interpretar como un p-valor (probabilidad de encontrar por azar una secuencia con la misma puntuación o superior)
• Si queremos la seguridad de que las secuencias que encontramos sean realmente homologas (más especificidad)
– E-values pequeños (p.e. E=0.05, 0.1),
– Bit-scores altos, y
– Porcentajes de identidad altos
• Si nos interesa explorar y priorizamos no perder información por delante de la seguridad (más sensibilidad)
– E-values más relajados (p.e. E=1, 10),
– Bit-scores normales,
– Porcentajes de identidad medios

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 39
Existe homologia?
>gb|AAA60147.1| placental protein 14 [Homo sapiens]Length=162 Score = 33.9 bits (76), Expect = 0.34 Identities = 24/107 (22%), Positives = 46/107 (42%), Gaps = 11/107 (10%) Query 28 RVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWD- 86 + K++ + + +GTW++MA + L + A V T + +L+ W+ Sbjct 5 QTKQDLELPKLAGTWHSMAMA-TNNISLMATLKAPLRVHITSLLPTPEDNLEIVLHRWEN 63Query 87 -VCADMVGTFTDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTY 132 C + T +P KFK+ Y ++ ++DTDYD +Sbjct 64 NSCVEKKVLGEKTGNPKKFKINYTVA--------NEATLLDTDYDNF 102
Ejemplo: RBP4 y PAEP
• Bit-score bajo, E-value 0.34, 22% identidad (“zona gris”)
• Pero son, en efecto, homologas. Se puede comprobar con una búsqueda BLAST con PAEP como secuencia “query”, y se encuentran muchas lipocalinas.

13/06/10 J.L. Mosquera, I. Ortega i A. Sánchez 40
Porque usar BLAST?
• BLAST searching is fundamental to understanding the relatedness of any favorite query sequence to other known proteins or DNA sequences
• Applications include– identifying orthologs and paralogs– discovering new genes or proteins– discovering variants of genes or proteins– investigating expressed sequence tags (ESTs)– exploring protein structure and function– …


















