
Aproximación diagnóstica de la Enfermedad Pulmonar ...
31
Aproximación diagnóstica de la Enfermedad Pulmonar Intersticial Difusa Dr. Matías Florenzano Valdés
Transcript of Aproximación diagnóstica de la Enfermedad Pulmonar ...

Aproximación diagnóstica de la Enfermedad Pulmonar Intersticial DifusaDr. Matías Florenzano Valdés

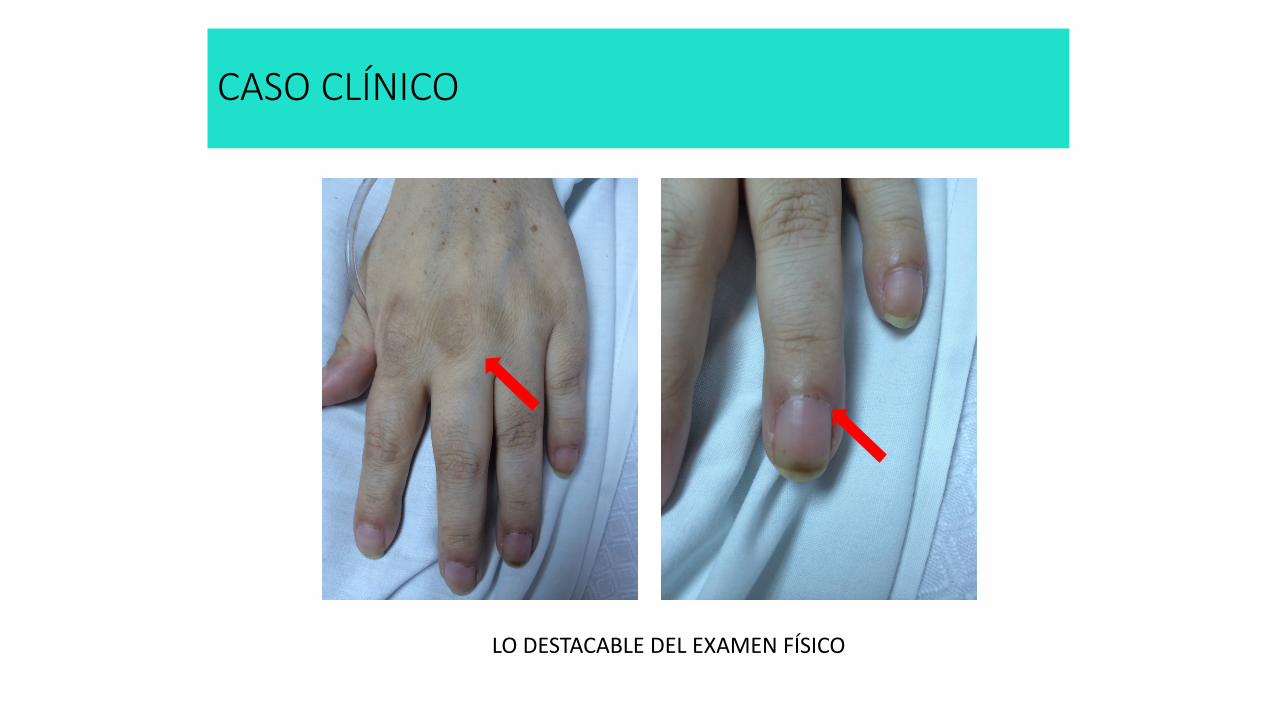
CASO CLÍNICO
• Mujer 45 años, casada, 2 hijos. G2P2A0• Procedente de Graneros• Dueña de casa• 6 meses de disnea progresiva y fiebre• 2 hospitalizaciones y varios tratamientos antibióticos sin
respuesta• Derivada a INT

CASO CLÍNICO
CVF 50% DLCO 35% C6M Sat 92 – 78%
CASO CLÍNICO
LO DESTACABLE DEL EXAMEN FÍSICO

CASO CLÍNICO
• ANA –• ENA –• FR –• CCP –

Caso clínico
M.-P. Debray et al. / European Journal of Radiology 84 (2015) 516–523 521
Fig. 2. (a) Axial transverse CT image of mid-lung zones in a 57-year-old womanpositive for anti-PL12 antibody. It shows small foci of consolidations superim-posed on ground-glass opacities and reticulations, with relative subpleural sparing,which is consistent with a non-specific interstitial pneumonia–organizing pneu-monia (NSIP–OP) CT pattern. (b) Same patient 16 months later. Consolidations haveresolved whereas ground-glass opacities persist and traction bronchiectasis haveincreased.
anti-Jo1–positive patients, and 22 months (range 13–54 months)for anti-PL12–positive patients (p = 0.067). Because they were fewin number, anti-PL7 positive patients were not compared to otherpatients.
4. Discussion
We report here a large imaging series of AS-ILD and the firstdetailed follow-up of CT features. We found CT features weremainly suggestive of NSIP or OP at presentation, isolated or incombination with a high prevalence of consolidations. Whereasconsolidations most often resolved, follow-up also revealed a pro-gression towards fibrosis in some patients.
In order to provide a global and concise description of CT find-ings, which can improve clinical management, especially sincehistopathological data are usually lacking for such patients, CTabnormalities were described by four distinct CT patterns. In fact,surgical lung biopsies are not usually performed in AS-ILD cases,just as they are not performed for other connective tissue diseasesassociated ILD either. We observed a very good interobserver agree-ment for the CT patterns, which can be partly attributed to therestriction to only four pattern categories.
The most frequent CT findings at presentation were GGO, retic-ulations and traction bronchiectasis, consistent with a NSIP CT
Fig. 3. (a) Axial transverse CT image of lower lung zones in a 78-year-old womanpositive for anti-Jo1 antibody. It shows ground-glass opacities in a peripheral sub-pleural distribution with superimposed reticulations and traction bronchiectasis,suggestive of a non-specific interstitial pneumonia (NSIP) CT pattern. (b) Samepatient, one year later. A dramatic improvement of traction bronchiectasis hasoccurred with marked decrease in overall extent of abnormalities.
pattern [10,16], which is in phase with most previous reports ofAS-ILD [4,6,17,18]. CT features were consistent with OP or mixedNSIP–OP patterns in most of the remaining cases, in view of thehigh prevalence of consolidations, present in 45% of cases. A highprevalence of consolidations has been previously reported, bothin patients with anti-Jo1 antibody [3,5] and patients with myositis[19], which is not the case for other connective-tissue-disease asso-ciated ILD [20]. Therefore, we believe searching for the presenceof anti-synthetase antibody should be included in the diagnosticwork-up of ILD when showing OP or mixed NSIP–OP patterns onCT. NSIP–OP is not usually described as a distinct CT pattern ininterstitial pneumonias [21,22], although a combination of differ-ent features is well recognized in idiopathic or connective-tissuedisease-associated ILD [23–25]. Here we chose to individualizeNSIP–OP as a distinct pattern in order to highlight the high fre-quency of consolidations. Furthermore, this association has beenidentified in series with lung biopsies performed in patients withAS [5,14,26]. In our series, the majority of cases with OP or NSIP–OPCT pattern for which histology was available had biopsy-confirmedOP. However, because of the small number of CT-pathologic-correlations in our series, we cannot state definitive conclusionson the underlying pathologic features for AS ILD patients and CTpatterns cannot be transposed into histopathological patterns.
Despite the decrease in consolidations, 81% of patients showedpersistent signs of ILD on follow-up CT. Disease extent over timewas variable, showing an increase in 35% of cases, a result simi-lar to the ILD progression rate reported in a series of 31 anti-Jo1
M.-P. Debray et al. / European Journal of Radiology 84 (2015) 516–523 521
Fig. 2. (a) Axial transverse CT image of mid-lung zones in a 57-year-old womanpositive for anti-PL12 antibody. It shows small foci of consolidations superim-posed on ground-glass opacities and reticulations, with relative subpleural sparing,which is consistent with a non-specific interstitial pneumonia–organizing pneu-monia (NSIP–OP) CT pattern. (b) Same patient 16 months later. Consolidations haveresolved whereas ground-glass opacities persist and traction bronchiectasis haveincreased.
anti-Jo1–positive patients, and 22 months (range 13–54 months)for anti-PL12–positive patients (p = 0.067). Because they were fewin number, anti-PL7 positive patients were not compared to otherpatients.
4. Discussion
We report here a large imaging series of AS-ILD and the firstdetailed follow-up of CT features. We found CT features weremainly suggestive of NSIP or OP at presentation, isolated or incombination with a high prevalence of consolidations. Whereasconsolidations most often resolved, follow-up also revealed a pro-gression towards fibrosis in some patients.
In order to provide a global and concise description of CT find-ings, which can improve clinical management, especially sincehistopathological data are usually lacking for such patients, CTabnormalities were described by four distinct CT patterns. In fact,surgical lung biopsies are not usually performed in AS-ILD cases,just as they are not performed for other connective tissue diseasesassociated ILD either. We observed a very good interobserver agree-ment for the CT patterns, which can be partly attributed to therestriction to only four pattern categories.
The most frequent CT findings at presentation were GGO, retic-ulations and traction bronchiectasis, consistent with a NSIP CT
Fig. 3. (a) Axial transverse CT image of lower lung zones in a 78-year-old womanpositive for anti-Jo1 antibody. It shows ground-glass opacities in a peripheral sub-pleural distribution with superimposed reticulations and traction bronchiectasis,suggestive of a non-specific interstitial pneumonia (NSIP) CT pattern. (b) Samepatient, one year later. A dramatic improvement of traction bronchiectasis hasoccurred with marked decrease in overall extent of abnormalities.
pattern [10,16], which is in phase with most previous reports ofAS-ILD [4,6,17,18]. CT features were consistent with OP or mixedNSIP–OP patterns in most of the remaining cases, in view of thehigh prevalence of consolidations, present in 45% of cases. A highprevalence of consolidations has been previously reported, bothin patients with anti-Jo1 antibody [3,5] and patients with myositis[19], which is not the case for other connective-tissue-disease asso-ciated ILD [20]. Therefore, we believe searching for the presenceof anti-synthetase antibody should be included in the diagnosticwork-up of ILD when showing OP or mixed NSIP–OP patterns onCT. NSIP–OP is not usually described as a distinct CT pattern ininterstitial pneumonias [21,22], although a combination of differ-ent features is well recognized in idiopathic or connective-tissuedisease-associated ILD [23–25]. Here we chose to individualizeNSIP–OP as a distinct pattern in order to highlight the high fre-quency of consolidations. Furthermore, this association has beenidentified in series with lung biopsies performed in patients withAS [5,14,26]. In our series, the majority of cases with OP or NSIP–OPCT pattern for which histology was available had biopsy-confirmedOP. However, because of the small number of CT-pathologic-correlations in our series, we cannot state definitive conclusionson the underlying pathologic features for AS ILD patients and CTpatterns cannot be transposed into histopathological patterns.
Despite the decrease in consolidations, 81% of patients showedpersistent signs of ILD on follow-up CT. Disease extent over timewas variable, showing an increase in 35% of cases, a result simi-lar to the ILD progression rate reported in a series of 31 anti-Jo1
Ciclofosamida iv 6 mesesMicofenolato 2,5 g/día 1a
CVF 80% DLCO 55% C6M Sat 92 – 88%

Contexto• Grupo heterogéneo de enfermedades que comprometen el intersticio
pulmonar con grados variables de inflamación y fibrosis• Originalmente descritas y clasificadas en base a elementos
histopatológicos*• Actualmente el rol fundamental de tomografía computada permite su
diagnóstico, sin necesidad de biopsia en muchos casos**• La evaluación multidisciplinaria es el estándar de cuidado de pacientes
con EPI
* Katzenstein AA. Surgical pathology of non-neoplastic lung disease. Can J Surg 1997;40:394** Müller NL. Idiopathic interstitial pneumonias: high-resolution CT and histologic findings. Radiographics 1997;17:1016–22

Enfermedades Pulmonares Intersticiales: más de 200!!
287
idiopática lo hará como una forma crónica. Esta forma de clasi-
ficación es bastante útil para acotar el espectro de diagnósticos
etiológicos y por lo general se puede complementar paralela-
mente a la clasificación etiológica, antes arriba mencionada.
ESTUDIO DIAGNÓSTICO
Como en todas las patologías, el apoyo del laboratorio es una
herramienta de gran ayuda en el estudio de enfermedades
pulmonares. El laboratorio debiera incluir, pruebas hemato-
lógicas como hemograma, velocidad de sedimentación, perfil
bioquímico y pruebas reumatológicas como anticuerpos anti-
nucleares, factor reumatoideo y otras específicas en caso
de sospecha de mesenquimopatia. Ante la sospecha de una
neumonitis por hipersensibilidad también es muy útil solicitar
precipitinas específicas para aves como catas o paloma y cuando
el antecedente lo amerite, pecipitinas para hongos, como
Aspergilus.
Una vez establecida la sospecha de una EPD, el examen que no
debe faltar para confirmar la enfermedad es la radiografía de
tórax. La imagen radiológica dará la definición del compromiso
difuso caracterizado por extensión de varios lóbulos, segmentos
o pulmón completo ya sea en forma uni o bilateral. Al mismo
tiempo podremos evaluar en forma indirecta tamaño de los
pulmones, predominio espacial del compromiso, presencia de
neumotórax o derrame pleural, así como también tamaño de la
silueta cardiaca asociado a elementos radiológicos de conges-
tión pulmonar. Sin embargo la interpretación más fina y precisa
de una EPD debe ser complementada con la tomografía axial
computada (TAC) de tórax con cortes de alta resolución.
Tomograf�a axial computada de t�rax de alta resoluci�nTAC de tórax es el examen radiológico que definirá la presencia
más detallada de alteraciones intersticiales, compromiso
alveolar, vía aérea o alteraciones vasculares y también explo-
ración del mediastino. Las imágenes tomográficas de pulmón
pueden establecer patrones radiológicos que orientan a
un determinado grupo de enfermedades que lo poseen. La
técnica tomográfica está dada por imágenes de alta resolución
(Tabla 1). La interpretación radiológica debiera incluir informa-
ción previa para el radiólogo otorgada por el médico internista
o especialista, con el fin de precisar mejor la alternativa diag-
nóstica (6).
La neumonía intersticial usual (UIP) constituye un diagnóstico
radiológico que deriva de los hallazgos de la anatomía pato-
lógica. Se trata de elementos de fibrosis caracterizados por
opacidades reticulares de predominio basal, compromiso de
ubicación subpleural, presencia de panal y bronquiectasias por
tracción. La interpretación radiológica a cargo del radiólogo
de tórax podrá acotar los diagnósticos de EPD según el patrón
UIP, Usual interstitial pneumonia, NSIP: Nonspecific interstitial pneumonia, DIP: Desquamative interstitial pneumonia, RB-ILD: Respiratory bronchiolitis and interstitial lung diseases, COP: Cryptogenic organizing pneumonia, AIP: Acute interstitial pneumonia, LIP: Limphocytic interstitial pneumonia. (Modificado de referencia 4).
UIP
Fibrosis pulmonar idiop�tica (55%)
Neumon�a intersticial idiop�ticas
No UIP
NSIPDIPRB-ILDCOPAIPLIP
Sarcoidosis
Granulomatosa
Proteinosis alveolar
Linfangioleiomiomatosis
Fibroelastosis pleuropulmonar
Raras
ETIOLOGÍA CONOCIDA ETIOLOGÍA DESCONOCIDA
Neumonitis por hipersensibilidad
Neumoconiosis
Da�o pulmonar inducido por drogas
Mesenquimopatias
Causas infecciosas
FIGURA 1. CLASIFICACIÓN DE LAS ENFERMEDADES PULMONARES DIFUSAS
[APROXIMACIÓN DIAGNÓSTICA A LAS ENFERMEDADES PULMONARES DIFUSAS - Dr. Alfredo Jalilie E.]Document downloaded from http://www.elsevier.es, day 08/07/2017. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited.Document downloaded from http://www.elsevier.es, day 08/07/2017. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited.
Jalilie A. Rev Med Clin Condes 2015; 26(3) 285-291]

Am J Respir Crit Care Med 2013;188:733–748
American Thoracic Society Documents
An Official American Thoracic Society/EuropeanRespiratory Society Statement: Update of theInternational Multidisciplinary Classification of theIdiopathic Interstitial PneumoniasWilliam D. Travis, Ulrich Costabel, David M. Hansell, Talmadge E. King, Jr., David A. Lynch, Andrew G. Nicholson,Christopher J. Ryerson, Jay H. Ryu, Moises Selman, Athol U. Wells, Jurgen Behr, Demosthenes Bouros,Kevin K. Brown, Thomas V. Colby, Harold R. Collard, Carlos Robalo Cordeiro, Vincent Cottin, Bruno Crestani,Marjolein Drent, Rosalind F. Dudden, Jim Egan, Kevin Flaherty, Cory Hogaboam, Yoshikazu Inoue, Takeshi Johkoh,Dong Soon Kim, Masanori Kitaichi, James Loyd, Fernando J. Martinez, Jeffrey Myers, Shandra Protzko,Ganesh Raghu, Luca Richeldi, Nicola Sverzellati, Jeffrey Swigris, and Dominique Valeyre; on behalf of the ATS/ERSCommittee on Idiopathic Interstitial Pneumonias
THIS OFFICIAL STATEMENT OF THE AMERICAN THORACIC SOCIETY (ATS) AND THE EUROPEAN RESPIRATORY SOCIETY (ERS) WAS
APPROVED BY THE ATS BOARD OF DIRECTORS, JUNE 2013, AND BY THE ERS STEERING COMMITTEE, MARCH 2013
CONTENTS
Executive SummaryIntroductionMethodsSummary of Major Revisions of the IIP ClassificationGeneral Progress in IIPs since 2002
Multidisciplinary ApproachObserver Agreement in Diagnosis of IIP
Important Differential Diagnostic ConsiderationsHypersensitivity PneumonitisCollagen Vascular DiseaseFamilial Interstitial PneumoniaCoexisting Patterns
Progress in Specific IIPs since 2002Chronic Fibrosing IIPsSmoking-related IIPsAcute or Subacute IIPs
Rare IIPsIdiopathic Lymphoid Interstitial PneumoniaIdiopathic Pleuroparenchymal Fibroelastosis
Rare Histologic PatternsAcute Fibrinous and Organizing PneumoniaBronchiolocentric Patterns of Interstitial Pneumonia
Unclassifiable IIPClinical Classification of Disease BehaviorBiomarkers
Background: In 2002 the American Thoracic Society/European Res-piratory Society (ATS/ERS) classification of idiopathic interstitialpneumonias (IIPs)definedseven specificentities, andprovided stan-dardizedterminologyanddiagnosticcriteria. Inaddition, thehistorical“gold standard” of histologic diagnosis was replaced by amultidiscipli-nary approach. Since2002manypublicationshaveprovidednew infor-mation about IIPs.
Purpose: The objective of this statement is to update the 2002 ATS/ERS classification of IIPs.Methods: An international multidisciplinary panel was formed anddeveloped key questions that were addressed through a review ofthe literature published between 2000 and 2011.Results: Substantial progress has beenmade in IIPs since thepreviousclassification. Nonspecific interstitial pneumonia is now better de-fined. Respiratory bronchiolitis–interstitial lung disease is now com-monly diagnosed without surgical biopsy. The clinical course of idi-opathic pulmonary fibrosis and nonspecific interstitial pneumonia isrecognized to be heterogeneous. Acute exacerbation of IIPs is nowwell defined. A substantial percentage of patients with IIP are diffi-cult to classify, often due to mixed patterns of lung injury. A classifi-cation based on observed disease behavior is proposed for patientswho are difficult to classify or for entities with heterogeneity in clin-ical course. A group of rare entities, including pleuroparenchymalfibroelastosis and rare histologic patterns, is introduced. The rapidlyevolving field of molecular markers is reviewed with the intent ofpromoting additional investigations that may help in determiningdiagnosis, and potentially prognosis and treatment.Conclusions: This update is a supplement to the previous 2002 IIPclassification document. It outlines advances in the past decadeand potential areas for future investigation.
Keywords: idiopathic interstitial pneumonia; usual interstitial pneumo-nia; nonspecific interstitial pneumonia; respiratory bronchiolitis; desqua-mative interstitial pneumonia; cryptogenic organizing pneumonia; acuteinterstitial pneumonia; lymphoid interstitial pneumonia; pleuroparenchy-mal fibroelastosis; acute fibrinous and organizing pneumonia
EXECUTIVE SUMMARY
There are several specific areas that are given special attention inthis revision of the 2002 American Thoracic Society/EuropeanRespiratory Society idiopathic interstitial pneumonia (IIP) state-ment.
1. Idiopathic nonspecific interstitial pneumonia (NSIP) isnow accepted as a specific clinicopathologic entity. It hasbecome evident that clinical progression is highly het-erogeneous, with several studies suggesting that a subsetof patients demonstrate progression to end-stage fibro-sis; criteria to define this group at the time of diagnosiswould be helpful.
This document has an online supplement, which is accessible from this issue’stable of contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 188, Iss. 6, pp 733–748, Sep 15, 2013Copyright ª 2013 by the American Thoracic SocietyDOI: 10.1164/rccm.201308-1483STInternet address: www.atsjournals.org
2. New information has accumulated on smoking-related inter-stitial lung disease, including patients with combined emphy-sema and interstitial fibrosis. In clinical practice, respiratorybronchiolitis–interstitial lung disease is increasingly diag-nosed without surgical lung biopsy in smokers on the basisof clinical and imaging features (ground-glass opacities andcentrilobular nodules) and bronchoalveolar lavage (smok-er’s macrophages and absence of lymphocytosis).
3. The natural progression of idiopathic pulmonary fibrosis(IPF) is acknowledged to be heterogeneous with somepatients remaining stable for prolonged periods, othersshowing more rapid steady progression, and still otherssuccumbing to acute exacerbation.
4. Acute exacerbation is better defined and recognized tooccur in chronic fibrosing IIPs (IPF and NSIP).
5. Some patients with IIP are difficult to classify, oftenbecause of mixed patterns of lung injury.
6. It is recognized that there is a need to provide a clinicalalgorithm for classifying and managing IIP cases. This isparticularly applicable when no biopsy is available andhigh-resolution computed tomography is not diagnostic.
7. Pleuroparenchymal fibroelastosis is recognized as a specificrare entity, usually idiopathic. Other less well-defined his-tologic patterns, such as bronchiolocentric inflammationand fibrosis, are also included.
8. A rapidly emerging field of molecular markers holdspromise for improving diagnostic approaches. Thesemarkers may also be useful in predicting prognosis andresponse to different therapies. Incorporation of geneticand molecular studies may revolutionize the approachto diagnosis and classification of the IIPs.
INTRODUCTION
The objective of this statement is to update the 2002 AmericanThoracicSociety/EuropeanRespiratorySociety (ATS/ERS)clas-sification of idiopathic interstitial pneumonias (IIPs) (1). Focus isplaced on describing changes to previously described clinical enti-ties, describing new clinical entities, and describing new histologicpatterns. This update is not intended as a stand-alone documentand should be used as a supplement to the original 2002 IIP classi-fication. In 2002, the ATS/ERS IIP classification (1) defined sevendisease categories, and proposed standardized terminology anddiagnostic criteria. In addition, the historical “gold standard” ofhistologic diagnosis was replaced by a “dynamic integrated ap-proach” using multidisciplinary discussion (MDD). The 2002 IIPclassification was used in 75% (157 of 208) of all clinical publica-tions on the topic of IIPs between 2004 and 2011. The new infor-mation from these publications is incorporated in this update.
METHODS
This project was performed under supervision by the ATSDocu-ments Development and Implementation Committee in collab-oration with the ERS (Table E1 in the online supplement). Aninternational multidisciplinary panel was assembled. The panelconsisted of 34 experts in interstitial lung diseases (19 pulmonol-ogists, 4 radiologists, 5 pathologists, 2 experts in evidence-basedmedicine, and 4 molecular biologists). Several meetings wereheld by members of the international multidisciplinary panel(Table E2), who disclosed conflicts of interest, which were vettedaccording to ATS and ERS policies.
Key questions were developed that the committee believedimportant for the classification of IIPs (see APPENDIX 1 in the
online supplement). A literature search was performed to iden-tify new publications that pertained to these key questions,assisted by two librarians experienced in literature searches forpulmonary diseases. Literature retrieved from Medline searchesbetween 2000 and 2011 was used to produce this statement.
The committee was divided into subgroups assigned to spe-cific sections of the document. These subgroups reviewed the rel-evant literature and produced the first draft of their respectivesections. These sections were compiled by the committee chairand a complete first draft was edited by the writing subcommit-tee. This document was reviewed and edited by all committeemembers before final review by the writing subcommittee.The revised document was approved by all authors.
SUMMARY OF MAJOR REVISIONS OF THEIIP CLASSIFICATION
In the revision of the IIP classification, the main entities are pre-served (Table 1). However, there are several important changes.First, cryptogenic fibrosing alveolitis is removed, leaving idiopathicpulmonary fibrosis (IPF) as the sole clinical term for this diagnosis.Second, idiopathic nonspecific interstitial pneumonia (NSIP) is nowaccepted as a distinct clinical entity with removal of the term “pro-visional” (2). Third, major IIPs are distinguished from rare IIPs andunclassifiable cases. Fourth, rare histologic patterns of acute fibri-nous and organizing pneumonia (AFOP) and interstitial pneumo-nias with a bronchiolocentric distribution are recognized. Fifth, themajor IIPs are grouped into chronic fibrosing (IPF and NSIP; Fig-ures 1 and 2), smoking-related (respiratory bronchiolitis–interstitiallung disease [RB-ILD] and desquamative interstitial pneumonia[DIP]; Figure 3), and acute/subacute IIPs (cryptogenic organizingpneumonia [COP] and acute interstitial pneumonia [AIP]; Figure 4and Table 2). Sixth, a clinical disease behavior classification is pro-posed. Last, molecular and genetic features are reviewed.
GENERAL PROGRESS IN IIPS SINCE 2002
Multidisciplinary Approach
The process of achieving amultidisciplinary diagnosis in a patientwith IIP is dynamic, requiring close communication between cli-nician, radiologist, and when appropriate, pathologist (1). Clin-ical data (presentation, exposures, smoking status, associated
TABLE 1. REVISED AMERICAN THORACIC SOCIETY/EUROPEANRESPIRATORY SOCIETY CLASSIFICATION OF IDIOPATHICINTERSTITIAL PNEUMONIAS: MULTIDISCIPLINARY DIAGNOSES
Major idiopathic interstitial pneumoniasIdiopathic pulmonary fibrosisIdiopathic nonspecific interstitial pneumoniaRespiratory bronchiolitis–interstitial lung diseaseDesquamative interstitial pneumoniaCryptogenic organizing pneumoniaAcute interstitial pneumonia
Rare idiopathic interstitial pneumoniasIdiopathic lymphoid interstitial pneumoniaIdiopathic pleuroparenchymal fibroelastosis
Unclassifiable idiopathic interstitial pneumonias*
*Causes of unclassifiable idiopathic interstitial pneumonia include (1) inade-quate clinical, radiologic, or pathologic data and (2) major discordance betweenclinical, radiologic, and pathologic findings that may occur in the following situa-tions: (a) previous therapy resulting in substantial alteration of radiologic or histo-logic findings (e.g., biopsy of desquamative interstitial pneumonia after steroidtherapy, which shows only residual nonspecific interstitial pneumonia [153]);(b) new entity, or unusual variant of recognized entity, not adequately character-ized by the current American Thoracic Society/European Respiratory Society classi-fication (e.g., variant of organizing pneumonia with supervening fibrosis) (79); and(c) multiple high-resolution computed tomography and/or pathologic patterns thatmay be encountered in patients with idiopathic interstitial pneumonia.
734 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 188 2013

American Thoracic Society Documents
An Official American Thoracic Society/EuropeanRespiratory Society Statement: Update of theInternational Multidisciplinary Classification of theIdiopathic Interstitial PneumoniasWilliam D. Travis, Ulrich Costabel, David M. Hansell, Talmadge E. King, Jr., David A. Lynch, Andrew G. Nicholson,Christopher J. Ryerson, Jay H. Ryu, Moises Selman, Athol U. Wells, Jurgen Behr, Demosthenes Bouros,Kevin K. Brown, Thomas V. Colby, Harold R. Collard, Carlos Robalo Cordeiro, Vincent Cottin, Bruno Crestani,Marjolein Drent, Rosalind F. Dudden, Jim Egan, Kevin Flaherty, Cory Hogaboam, Yoshikazu Inoue, Takeshi Johkoh,Dong Soon Kim, Masanori Kitaichi, James Loyd, Fernando J. Martinez, Jeffrey Myers, Shandra Protzko,Ganesh Raghu, Luca Richeldi, Nicola Sverzellati, Jeffrey Swigris, and Dominique Valeyre; on behalf of the ATS/ERSCommittee on Idiopathic Interstitial Pneumonias
THIS OFFICIAL STATEMENT OF THE AMERICAN THORACIC SOCIETY (ATS) AND THE EUROPEAN RESPIRATORY SOCIETY (ERS) WAS
APPROVED BY THE ATS BOARD OF DIRECTORS, JUNE 2013, AND BY THE ERS STEERING COMMITTEE, MARCH 2013
CONTENTS
Executive SummaryIntroductionMethodsSummary of Major Revisions of the IIP ClassificationGeneral Progress in IIPs since 2002
Multidisciplinary ApproachObserver Agreement in Diagnosis of IIP
Important Differential Diagnostic ConsiderationsHypersensitivity PneumonitisCollagen Vascular DiseaseFamilial Interstitial PneumoniaCoexisting Patterns
Progress in Specific IIPs since 2002Chronic Fibrosing IIPsSmoking-related IIPsAcute or Subacute IIPs
Rare IIPsIdiopathic Lymphoid Interstitial PneumoniaIdiopathic Pleuroparenchymal Fibroelastosis
Rare Histologic PatternsAcute Fibrinous and Organizing PneumoniaBronchiolocentric Patterns of Interstitial Pneumonia
Unclassifiable IIPClinical Classification of Disease BehaviorBiomarkers
Background: In 2002 the American Thoracic Society/European Res-piratory Society (ATS/ERS) classification of idiopathic interstitialpneumonias (IIPs)definedseven specificentities, andprovided stan-dardizedterminologyanddiagnosticcriteria. Inaddition, thehistorical“gold standard” of histologic diagnosis was replaced by amultidiscipli-nary approach. Since2002manypublicationshaveprovidednew infor-mation about IIPs.
Purpose: The objective of this statement is to update the 2002 ATS/ERS classification of IIPs.Methods: An international multidisciplinary panel was formed anddeveloped key questions that were addressed through a review ofthe literature published between 2000 and 2011.Results: Substantial progress has beenmade in IIPs since thepreviousclassification. Nonspecific interstitial pneumonia is now better de-fined. Respiratory bronchiolitis–interstitial lung disease is now com-monly diagnosed without surgical biopsy. The clinical course of idi-opathic pulmonary fibrosis and nonspecific interstitial pneumonia isrecognized to be heterogeneous. Acute exacerbation of IIPs is nowwell defined. A substantial percentage of patients with IIP are diffi-cult to classify, often due to mixed patterns of lung injury. A classifi-cation based on observed disease behavior is proposed for patientswho are difficult to classify or for entities with heterogeneity in clin-ical course. A group of rare entities, including pleuroparenchymalfibroelastosis and rare histologic patterns, is introduced. The rapidlyevolving field of molecular markers is reviewed with the intent ofpromoting additional investigations that may help in determiningdiagnosis, and potentially prognosis and treatment.Conclusions: This update is a supplement to the previous 2002 IIPclassification document. It outlines advances in the past decadeand potential areas for future investigation.
Keywords: idiopathic interstitial pneumonia; usual interstitial pneumo-nia; nonspecific interstitial pneumonia; respiratory bronchiolitis; desqua-mative interstitial pneumonia; cryptogenic organizing pneumonia; acuteinterstitial pneumonia; lymphoid interstitial pneumonia; pleuroparenchy-mal fibroelastosis; acute fibrinous and organizing pneumonia
EXECUTIVE SUMMARY
There are several specific areas that are given special attention inthis revision of the 2002 American Thoracic Society/EuropeanRespiratory Society idiopathic interstitial pneumonia (IIP) state-ment.
1. Idiopathic nonspecific interstitial pneumonia (NSIP) isnow accepted as a specific clinicopathologic entity. It hasbecome evident that clinical progression is highly het-erogeneous, with several studies suggesting that a subsetof patients demonstrate progression to end-stage fibro-sis; criteria to define this group at the time of diagnosiswould be helpful.
This document has an online supplement, which is accessible from this issue’stable of contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 188, Iss. 6, pp 733–748, Sep 15, 2013Copyright ª 2013 by the American Thoracic SocietyDOI: 10.1164/rccm.201308-1483STInternet address: www.atsjournals.org
Coexisting Patterns
Most patients with a chronic IIP can be given a single clinical–radiologic–pathologic diagnosis. However, multiple pathologicand/or HRCT patterns may be found in the same patient. Dif-ferent patterns may be seen in a single biopsy or in biopsiesfrom multiple sites (e.g., usual interstitial pneumonia [UIP] inone lobe and NSIP in another) (39), or when pathologic andHRCT patterns differ. In smokers, multiple HRCT and histo-logic features may coexist including Langerhans’ cell histiocy-tosis, respiratory bronchiolitis (RB), desquamative interstitialpneumonia (DIP), pulmonary fibrosis (UIP or NSIP), and em-physema (40–42). Combined pulmonary fibrosis and emphy-sema (CPFE) is an example of coexisting patterns. CPFEcomprises a heterogeneous population of patients, not believedto represent a distinctive IIP. Patients with CPFE have increased
risk of developing pulmonary hypertension, which portends poorprognosis (43–46). When coexisting patterns occur, MDD maydetermine the clinical significance of individual patterns (4, 47, 48).
PROGRESS IN SPECIFIC IIPS SINCE 2002
Chronic Fibrosing IIPs
Idiopathic pulmonary fibrosis. An updated evidence-based guide-line for the diagnosis and management of IPF was recently pub-lished (8). A new diagnostic algorithm and schema for correlatinghistologic and radiologic findings in patients with suspected IPFwas provided in this guideline (8). New aspects of this algorithmincluded criteria for three levels of certainty for patterns of UIPbased on HRCT findings (UIP, possible UIP, and inconsistent withUIP) and four levels of certainty for pathologic diagnosis (UIP,
Figure 4. Cryptogenic organizingpneumonia. Computed to-mography (CT) features with(A) peripheral consolidationand air bronchograms, (B)bronchocentric distribution,(C) perilobular pattern show-ing focal right lower lobe con-solidation, with more centralground-glass opacity, corre-sponding to the reversedhalo sign, and (D) bandlikeconsolidation.
TABLE 2. CATEGORIZATION OF MAJOR IDIOPATHIC INTERSTITIAL PNEUMONIAS
Category Clinical–Radiologic–Pathologic DiagnosesAssociated Radiologic and/or
Pathologic–Morphologic Patterns
Chronic fibrosing IP Idiopathic pulmonary fibrosis Usual interstitial pneumoniaIdiopathic nonspecific interstitial pneumonia Nonspecific interstitial pneumonia
Smoking-related IP* Respiratory bronchiolitis-interstitial lung disease Respiratory bronchiolitisDesquamative interstitial pneumonia Desquamative interstitial pneumonia
Acute/subacute IP Cryptogenic organizing pneumonia Organizing pneumoniaAcute interstitial pneumonia Diffuse alveolar damage
Definition of abbreviation: IP ¼ interstitial pneumonia.*Desquamative interstitial pneumonia can occasionally occur in nonsmokers.
American Thoracic Society Documents 737
Am J Respir Crit Care Med 2013;188:733-748

RARE IIPS
The category of rare IIPs has been created to include idiopathiclymphoid interstitial pneumonia (LIP) and idiopathic pleuropar-enchymal fibroelastosis (PPFE). In addition, several rare histo-logic patterns of ILD have been described including AFOP anda group of bronchiolocentric patterns, although current data donot support these as distinct IIPs.
Idiopathic Lymphoid Interstitial Pneumonia
Most cases of LIP are associated with other conditions, althoughidiopathic cases still rarely occur (111). Idiopathic LIP hastherefore been moved to the category of rare IIPs. The clinical,imaging, and histopathologic criteria for LIP proposed in 2002remain unchanged, apart from recognition that some cases showstriking cyst formation on HRCT (111, 112). Both the 2002 IIPclassification and the ATS NSIP project demonstrated thatmany of the cases previously diagnosed as LIP are now consid-ered cellular NSIP (1, 2). Consequently, few cases of idiopathicLIP have been published since 2002 (111).
Idiopathic Pleuroparenchymal Fibroelastosis
PPFE is a rare condition that consists of fibrosis involving thepleura and subpleural lung parenchyma, predominantly in theupper lobes. HRCT shows dense subpleural consolidation withtraction bronchiectasis, architectural distortion, and upper lobevolume loss (Figures 7A and 7B) (113). The fibrosis is elastotic,and intraalveolar fibrosis is present (Figures 8A and 8B) (113–117). It presents in adults with a median age of 57 years and hasno sex predilection (113). Approximately half of patients haveexperienced recurrent infections. Pneumothorax is common. Aminority has familial interstitial lung disease and nonspecific auto-antibodies. Histologically, biopsies may show mild changes ofPPFE or other patterns such as UIP. Disease progression occursin 60% of patients with death from disease in 40% (113, 118).
RARE HISTOLOGIC PATTERNS
Rare histologic interstitial pneumonia patterns have been de-scribed and these were not included as new IIP entities becauseof questions concerning whether they are variants of existing IIPsor exist only in association with other conditions such as HP orCVD. When encountered histologically, these terms may be ofvalue in provisionally classifying biopsy features before MDD.
Acute Fibrinous and Organizing Pneumonia
AFOP was first reported in 17 patients with acute respiratoryfailure and initially regarded to represent a possible new IIP(119). The principal HRCT findings are bilateral basal opacitiesand areas of consolidation (Figure 9A). The dominant histo-logic pattern is intraalveolar fibrin deposition and associatedorganizing pneumonia (Figures 9C and 9D). Classical hyalinemembranes of DAD are absent. AFOP may represent a histo-logic pattern that can occur in the clinical spectrum of DAD andOP or it may reflect a tissue sampling issue. AFOP may beidiopathic or associated with CVD (120), hypersensitivity pneu-monitis (121), or drug reaction (122). As this pattern can beseen in eosinophilic pneumonia, this diagnosis should be ex-cluded by absence of tissue and peripheral eosinophilia.
Bronchiolocentric Patterns of Interstitial Pneumonia
Several small retrospective series have recently described bron-chiolocentric fibroinflammatory changes (123–126). Of these,two studies in particular suggest these cases may be an IIPcentered on airways (123, 125), although imaging findingswere not well characterized and in one series there were
environmental or occupational exposures in most cases (123).One study described cases of peribronchiolar metaplasia-ILD,which probably represent a form of small airway disease. TheHRCTs in these cases were either normal or showed air trap-ping (124).
UNCLASSIFIABLE IIP
The 2002 ATS/ERS classification proposed an “unclassifiable”category of IIP, acknowledging that a final diagnosis may not beachieved, even after lengthy MDD (1). Examples of circum-stances in which a case cannot be satisfactorily classified aresummarized in Table 1 (127). Cases that are “unclassifiable”in terms of overlap of histologic patterns often prove to berelated to CVD (e.g., interstitial pneumonia and follicular bron-chiolitis in a patient with rheumatoid arthritis) or drug induced,rather than being idiopathic on MDD. If ILD is difficult, orimpossible, to classify, management should be based on themost probable diagnosis after MDD and consideration of theexpected disease behavior (as described below).
CLINICAL CLASSIFICATION OF DISEASE BEHAVIOR
Patterns of disease behavior in diffuse lung disorders and relatedtreatment approaches can be broadly subdivided as shown inTable 3. This approach is most useful in unclassifiable casesand for some IIPs, such as NSIP, that can be associated withall five patterns of disease behavior. This disease behavior clas-sification is complementary to the IIP classification and shouldnot be used as a justification for delaying SLB. Such delaysincrease the risk of surgical complications and may result in
TABLE 3. IDIOPATHIC INTERSTITIAL PNEUMONIAS:CLASSIFICATION ACCORDING TO DISEASE BEHAVIOR*
Clinical Behavior Treatment Goal Monitoring Strategy
Reversible and self-limited (e.g., manycases of RB-ILD)
Remove possible cause Short-term (3- to 6-mo)observation to confirmdisease regression
Reversible disease withrisk of progression(e.g., cellular NSIPand some fibroticNSIP, DIP, COP)
Initially achieve responseand then rationalizelonger term therapy
Short-term observationto confirm treatmentresponse. Long-termobservation to ensurethat gains arepreserved
Stable with residualdisease (e.g., somefibrotic NSIP)
Maintain status Long-term observation toassess disease course
Progressive, irreversibledisease with potentialfor stabilization (e.g.,some fibrotic NSIP)
Stabilize Long-term observation toassess disease course
Progressive, irreversibledisease despite therapy(e.g., IPF, some fibroticNSIP)
Slow progression Long-term observation toassess disease courseand need for transplantor effective palliation
Definition of abbreviations: COP ¼ cryptogenic organizing pneumonia; DIP ¼desquamative interstitial pneumonia; HRCT ¼ high-resolution computed tomog-raphy; IPF ¼ idiopathic pulmonary fibrosis; NSIP ¼ nonspecific interstitial pneu-monia; RB-ILD ¼ respiratory bronchiolitis–interstitial pneumonia.
* The distinctions in Table 3 are made by assimilating several factors: (1) Aconfident multidisciplinary diagnosis that often identifies the expected patternof disease behavior (e.g., IPF). However, in other idiopathic interstitial pneumo-nias (e.g., NSIP) more than one pattern of behavior is possible; (2) disease sever-ity, based on lung function and/or HRCT. In severe NSIP (154) a progressiveirreversible course is frequent; (3) evaluation of potentially reversible and irrevers-ible features based on review of the HRCT and biopsy if available; and (4) short-term disease behavior. Disease behavior classification must be refined over timein individual patients considering longitudinal changes in disease severity.
American Thoracic Society Documents 741
American Thoracic Society Documents
An Official American Thoracic Society/EuropeanRespiratory Society Statement: Update of theInternational Multidisciplinary Classification of theIdiopathic Interstitial PneumoniasWilliam D. Travis, Ulrich Costabel, David M. Hansell, Talmadge E. King, Jr., David A. Lynch, Andrew G. Nicholson,Christopher J. Ryerson, Jay H. Ryu, Moises Selman, Athol U. Wells, Jurgen Behr, Demosthenes Bouros,Kevin K. Brown, Thomas V. Colby, Harold R. Collard, Carlos Robalo Cordeiro, Vincent Cottin, Bruno Crestani,Marjolein Drent, Rosalind F. Dudden, Jim Egan, Kevin Flaherty, Cory Hogaboam, Yoshikazu Inoue, Takeshi Johkoh,Dong Soon Kim, Masanori Kitaichi, James Loyd, Fernando J. Martinez, Jeffrey Myers, Shandra Protzko,Ganesh Raghu, Luca Richeldi, Nicola Sverzellati, Jeffrey Swigris, and Dominique Valeyre; on behalf of the ATS/ERSCommittee on Idiopathic Interstitial Pneumonias
THIS OFFICIAL STATEMENT OF THE AMERICAN THORACIC SOCIETY (ATS) AND THE EUROPEAN RESPIRATORY SOCIETY (ERS) WAS
APPROVED BY THE ATS BOARD OF DIRECTORS, JUNE 2013, AND BY THE ERS STEERING COMMITTEE, MARCH 2013
CONTENTS
Executive SummaryIntroductionMethodsSummary of Major Revisions of the IIP ClassificationGeneral Progress in IIPs since 2002
Multidisciplinary ApproachObserver Agreement in Diagnosis of IIP
Important Differential Diagnostic ConsiderationsHypersensitivity PneumonitisCollagen Vascular DiseaseFamilial Interstitial PneumoniaCoexisting Patterns
Progress in Specific IIPs since 2002Chronic Fibrosing IIPsSmoking-related IIPsAcute or Subacute IIPs
Rare IIPsIdiopathic Lymphoid Interstitial PneumoniaIdiopathic Pleuroparenchymal Fibroelastosis
Rare Histologic PatternsAcute Fibrinous and Organizing PneumoniaBronchiolocentric Patterns of Interstitial Pneumonia
Unclassifiable IIPClinical Classification of Disease BehaviorBiomarkers
Background: In 2002 the American Thoracic Society/European Res-piratory Society (ATS/ERS) classification of idiopathic interstitialpneumonias (IIPs)definedseven specificentities, andprovided stan-dardizedterminologyanddiagnosticcriteria. Inaddition, thehistorical“gold standard” of histologic diagnosis was replaced by amultidiscipli-nary approach. Since2002manypublicationshaveprovidednew infor-mation about IIPs.
Purpose: The objective of this statement is to update the 2002 ATS/ERS classification of IIPs.Methods: An international multidisciplinary panel was formed anddeveloped key questions that were addressed through a review ofthe literature published between 2000 and 2011.Results: Substantial progress has beenmade in IIPs since thepreviousclassification. Nonspecific interstitial pneumonia is now better de-fined. Respiratory bronchiolitis–interstitial lung disease is now com-monly diagnosed without surgical biopsy. The clinical course of idi-opathic pulmonary fibrosis and nonspecific interstitial pneumonia isrecognized to be heterogeneous. Acute exacerbation of IIPs is nowwell defined. A substantial percentage of patients with IIP are diffi-cult to classify, often due to mixed patterns of lung injury. A classifi-cation based on observed disease behavior is proposed for patientswho are difficult to classify or for entities with heterogeneity in clin-ical course. A group of rare entities, including pleuroparenchymalfibroelastosis and rare histologic patterns, is introduced. The rapidlyevolving field of molecular markers is reviewed with the intent ofpromoting additional investigations that may help in determiningdiagnosis, and potentially prognosis and treatment.Conclusions: This update is a supplement to the previous 2002 IIPclassification document. It outlines advances in the past decadeand potential areas for future investigation.
Keywords: idiopathic interstitial pneumonia; usual interstitial pneumo-nia; nonspecific interstitial pneumonia; respiratory bronchiolitis; desqua-mative interstitial pneumonia; cryptogenic organizing pneumonia; acuteinterstitial pneumonia; lymphoid interstitial pneumonia; pleuroparenchy-mal fibroelastosis; acute fibrinous and organizing pneumonia
EXECUTIVE SUMMARY
There are several specific areas that are given special attention inthis revision of the 2002 American Thoracic Society/EuropeanRespiratory Society idiopathic interstitial pneumonia (IIP) state-ment.
1. Idiopathic nonspecific interstitial pneumonia (NSIP) isnow accepted as a specific clinicopathologic entity. It hasbecome evident that clinical progression is highly het-erogeneous, with several studies suggesting that a subsetof patients demonstrate progression to end-stage fibro-sis; criteria to define this group at the time of diagnosiswould be helpful.
This document has an online supplement, which is accessible from this issue’stable of contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 188, Iss. 6, pp 733–748, Sep 15, 2013Copyright ª 2013 by the American Thoracic SocietyDOI: 10.1164/rccm.201308-1483STInternet address: www.atsjournals.org
Am J Respir Crit Care Med 2013;188:733–748
diseases, lung function, laboratory findings) and radiologic find-ings are essential for multidisciplinary diagnosis.
The multidisciplinary approach does not lessen the impor-tance of lung biopsy in the diagnosis of IIPs; rather, it definesthe settings where biopsy is more informative than high-
resolution computed tomography (HRCT) and those where bi-opsy is not needed. Also, once a pathologist has recognized a histo-logic pattern (e.g.,NSIPor organizingpneumonia [OP]), the clinicianshould reconsider potential causes (e.g., hypersensitivity pneumonitis[HP], collagen vascular disease [CVD], and drug exposure).
Figure 1. Atypicalusual intersti-tial pneumonia/idiopathic pul-monary fibrosis. Computedtomography (CT) features: (A)CT image of atypical usual in-terstitialpneumonia(UIP).Proneaxial CT through the lung basesin a patient with histologicallyproven UIP shows predominantlyperibronchovascular ground-glass/reticular opacities with tractionbronchiectasis. Under the ATS/ERS/JRS/ALAT Statement(8), thiswould be classified as inconsistentwith UIP. Histologic features: (B)Thebiopsy showspatchy subpleu-ral dense fibrosis with honeycombchange adjacent to preservedlung. (C) Dense scarring fibrosisis present with a small nodularfibroblastic focus.
4C/FPO
Figure 2. Nonspecific intersti-tial pneumonia. Computed to-mography (CT) features: (A)Axial and (B) coronal CT recon-structions show confluent bi-lateral lower lobe ground-glass opacities with markedtraction bronchiectasis andlower lobe volume loss. Theperibronchovascular predomi-nance with subpleural sparingis well shown on the axial im-age. (C and D) Histologic fea-tures: Lung biopsy shows diffusealveolar wall thickening by uni-form fibrosis. The alveolar archi-tecture is preserved and nohoneycombing or fibroblasticfoci are seen. Interstitial inflam-mation is mild.
4C/FPO
American Thoracic Society Documents 735

Herramientas diagnósticas utilizadas en EPI• Historia y examen físico• Imágenes de tórax• Pruebas de función pulmonar• Análisis serológico• Lavado broncoalveolar• Biopsia pulmonar• Discusión multidisciplinaria

Historia y examen físico en EPI• Cuestionarios estandarizados• EPI autoinmunes más frecuentes en mujeres jóvenes• FPI en hombres mayores, ex-fumadores• Exposición ambiental a antígenos en Neumonitis por Hipersensibilidad y
Neumoconiosis (silicosis, asbestosis)• Compromiso multisistémico en ETC y Sarcoidosis
Adegunsoye A, Ryerson CJ. Diagnostic Classification of Interstitial Lung Disease in Clinical Practice. Clin Chest Med 2021;42:251-261

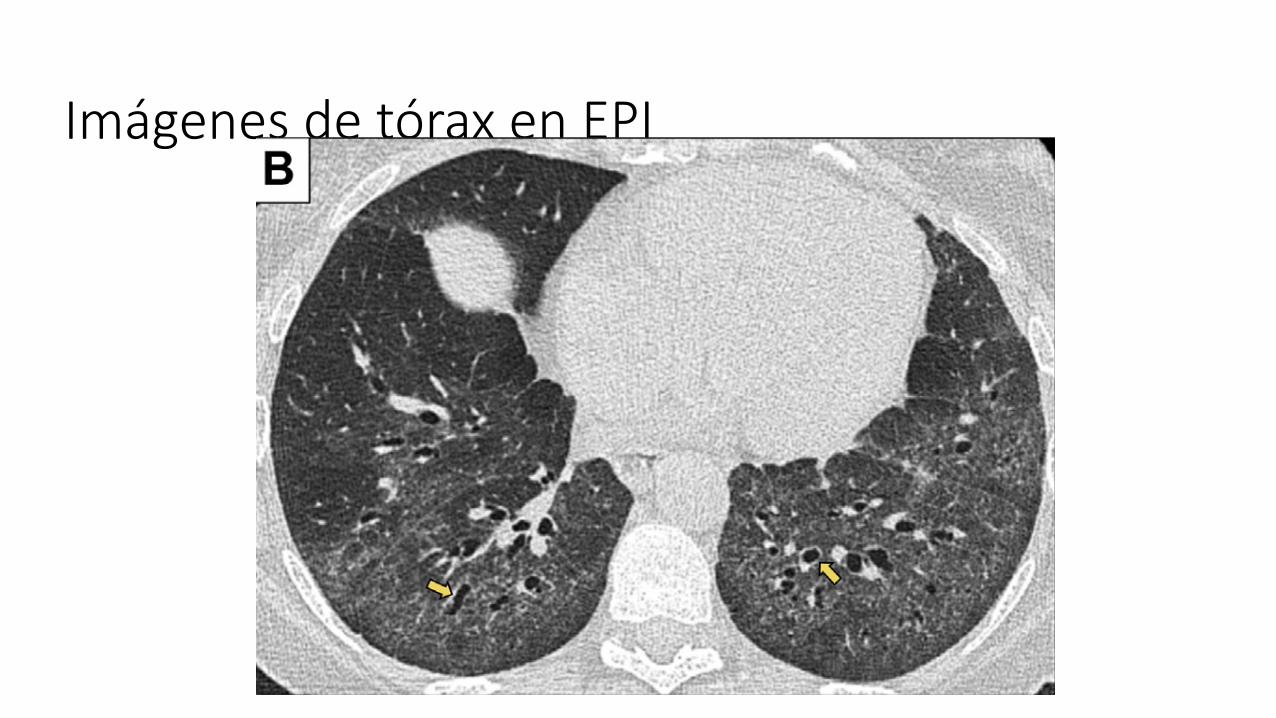
Imágenes de tórax en EPI:
such as computerized quantitative algorithms mayeventually be useful to help eliminate the interob-server variability that currently exists, particularlyamong nonexperts.32,33 This approach also holdsappeal for its potential to improve precision inassessing ILD severity and estimatingprognosis.32–34
Pulmonary Function Testing
Although pulmonary function tests are typically un-helpful in distinguishing ILD subtypes, these are acritical component of assessing disease severityand disease behavior.35,36 Longitudinal monitoringof forced vital capacity (FVC) and diffusing capac-ity of the lung for carbon monoxide (DLCO) is
valuable for monitoring therapeutic response,quantifying disease progression, and identifyingpatients with high mortality risk. Even in patientswithout a defined ILD, the subtle presence of inter-stitial lung abnormalities on chest HRCT is associ-ated with an increased rate of pulmonary functiondecline,37 indicating the importance of monitoringpatients with subtle abnormalities on chest imag-ing. In some patients, disease behavior providesuseful information that impacts the diagnostic like-lihood. For example, long-term stability decreasesthe probability of IPF, whereas ongoing rapidworsening may argue against non-IPF etiologies,particularly if a potential offending cause hasbeen removed (eg, a potentially offending medica-tion or environmental exposure).
Fig. 2. Representative axial CT images of common ILD patterns. (A) A typical UIP pattern characterized by sub-pleural and basal predominant honeycombing (blue triangle), reticulation (red asterisks), and traction bronchi-ectasis (yellow arrows). (B) A typical nonspecific interstitial pneumonia (NSIP) pattern with peripheral andlower lung predominant abnormalities, including the classic feature of subpleural sparing that is seen in approx-imately 25% of patients with NSIP and helps distinguish from UIP when present. In this case, the presence ofbronchiectasis (yellow arrows) indicates the presence of “microfibrosis” that is represented by ground glass onthis image. (C) A typical pattern of fibrotic HP including the 3-density sign that is indicated by the presence ofwell-demarcated areas of high attenuation or ground glass (green circle), low attention (blue rectangle), andnormal attenuation. There is diffuse abnormality in both the craniocaudal and axial planes, with presence ofreticulation (red asterisks) and traction bronchiectasis (yellow arrows). (D) An indeterminate pattern of fibrosischaracterized by mild peripheral reticulation and ground-glass abnormality without any defining feature. Thesuggestion of well-demarcated subpleural sparing (red lightning bolts) is an image processing artifact anddoes not suggest NSIP.
Adegunsoye & Ryerson254
2 24 2 2 ( E 1 2 1 C AE ) 4 ) 2 E C 2E /2 2 2D C2 A . A A 2 A 2 ) E A C , 0 4 4 C24

Imágenes de tórax en EPI
the benefits of establishing a secure diagnosisof IPF; therefore, the final decision regardingwhether or not to pursue a biopsy must betailored to the clinical situation of theindividual patient. Multiple biopsies shouldbe obtained from two to three lobes, becausethe histologic patterns on SLB specimensobtained from different segments can bediscordant (e.g., coexisting UIP pattern andfibrotic NSIP pattern from different lobes).
Methods for processing SLBs arevariable and require careful handling ofsamples to avoid iatrogenic mechanicalatelectasis and use of inflation techniques topreserve normal lung architecture. Specialstains may be used in some patients,including iron stains to identify asbestosbodies in patients with incriminatingexposure histories and elastic tissue stainsfor patients in whom vascular abnormalitiesdiffer from the secondary changescommon in the UIP pattern. Connectivetissue stains may also have value indistinguishing patterns of fibrosis but are oflimited incremental value compared withbiopsies processed with high-quality routinestaining techniques like hematoxylinand eosin.
Histopathology Features of theUIP PatternThe histopathologic hallmark and chiefdiagnostic criterion of UIP is a lowmagnification appearance of patchy densefibrosis that 1) is causing remodeling of lungarchitecture, 2) often results in honeycombchange, and 3) alternates with areas of less-affected parenchyma (Figure 7). Thesehistopathologic changes typically affect thesubpleural and paraseptal parenchyma mostseverely. Inflammation is usually mild andconsists of a patchy interstitial infiltrate oflymphocytes and plasma cells associatedwith hyperplasia of type 2 pneumocytes andbronchiolar epithelium. The fibrotic zonesare composed mainly of dense collagen,although scattered convex subepithelialfoci of proliferating fibroblasts andmyofibroblasts (so-called fibroblast foci)are a consistent finding. Microscopichoneycombing is characterized by cysticfibrotic airspaces that are frequently linedby bronchiolar epithelium and filled withmucus and inflammatory cells. Smoothmuscle metaplasia in the interstitium iscommonly seen in areas of fibrosis andhoneycombing. A definitive pathologic
diagnosis of the UIP pattern can be madewhen all of the above features are present,particularly when honeycombing ispresent. However, even in the absence ofhoneycombing, a definite diagnosis of aUIP pattern can still be made if all of theother typical features are present.
Key histologic features can be helpfulin excluding alternate diagnoses, such ashypersensitivity pneumonitis (e.g.,bronchiolocentric distribution withlymphocyte-rich bronchiolitis, extensiveperibronchiolar metaplasia, poorlyformed nonnecrotizing granulomasin peribronchiolar interstitium), acuteexacerbation of IPF or acute interstitialpneumonia (i.e., hyaline membranes),cicatricial variants of cryptogenic organizingpneumonia with fibrosis (prominentorganizing pneumonia), pneumoconiosis(e.g., asbestos bodies, prominent dustmacules and/or silicotic nodules),sarcoidosis (prominent well-formednonnecrotizing granulomas in a lymphaticdistribution), smoking-related interstitialfibrosis (extensive respiratory bronchiolitisand exquisitely subpleural and/orperibronchiolar paucicellular densely
Figure 1. High-resolution computed tomography (CT) images demonstrating a usual interstitial pneumonia pattern. (A–C) Transverse CT section and (D)coronal reconstruction illustrating the presence of honeycombing with subpleural and basal predominance. Note the concurrent presence of mild ground-glass opacity. (E) Magnified view of the left lower lobe showing typical characteristics of honeycombing, consisting of clustered cystic airspaces with well-defined walls and variable diameters, seen in single or multiple layers (arrows).
AMERICAN THORACIC SOCIETY DOCUMENTS
American Thoracic Society Documents e51

Imágenes de tórax en EPI
the benefits of establishing a secure diagnosisof IPF; therefore, the final decision regardingwhether or not to pursue a biopsy must betailored to the clinical situation of theindividual patient. Multiple biopsies shouldbe obtained from two to three lobes, becausethe histologic patterns on SLB specimensobtained from different segments can bediscordant (e.g., coexisting UIP pattern andfibrotic NSIP pattern from different lobes).
Methods for processing SLBs arevariable and require careful handling ofsamples to avoid iatrogenic mechanicalatelectasis and use of inflation techniques topreserve normal lung architecture. Specialstains may be used in some patients,including iron stains to identify asbestosbodies in patients with incriminatingexposure histories and elastic tissue stainsfor patients in whom vascular abnormalitiesdiffer from the secondary changescommon in the UIP pattern. Connectivetissue stains may also have value indistinguishing patterns of fibrosis but are oflimited incremental value compared withbiopsies processed with high-quality routinestaining techniques like hematoxylinand eosin.
Histopathology Features of theUIP PatternThe histopathologic hallmark and chiefdiagnostic criterion of UIP is a lowmagnification appearance of patchy densefibrosis that 1) is causing remodeling of lungarchitecture, 2) often results in honeycombchange, and 3) alternates with areas of less-affected parenchyma (Figure 7). Thesehistopathologic changes typically affect thesubpleural and paraseptal parenchyma mostseverely. Inflammation is usually mild andconsists of a patchy interstitial infiltrate oflymphocytes and plasma cells associatedwith hyperplasia of type 2 pneumocytes andbronchiolar epithelium. The fibrotic zonesare composed mainly of dense collagen,although scattered convex subepithelialfoci of proliferating fibroblasts andmyofibroblasts (so-called fibroblast foci)are a consistent finding. Microscopichoneycombing is characterized by cysticfibrotic airspaces that are frequently linedby bronchiolar epithelium and filled withmucus and inflammatory cells. Smoothmuscle metaplasia in the interstitium iscommonly seen in areas of fibrosis andhoneycombing. A definitive pathologic
diagnosis of the UIP pattern can be madewhen all of the above features are present,particularly when honeycombing ispresent. However, even in the absence ofhoneycombing, a definite diagnosis of aUIP pattern can still be made if all of theother typical features are present.
Key histologic features can be helpfulin excluding alternate diagnoses, such ashypersensitivity pneumonitis (e.g.,bronchiolocentric distribution withlymphocyte-rich bronchiolitis, extensiveperibronchiolar metaplasia, poorlyformed nonnecrotizing granulomasin peribronchiolar interstitium), acuteexacerbation of IPF or acute interstitialpneumonia (i.e., hyaline membranes),cicatricial variants of cryptogenic organizingpneumonia with fibrosis (prominentorganizing pneumonia), pneumoconiosis(e.g., asbestos bodies, prominent dustmacules and/or silicotic nodules),sarcoidosis (prominent well-formednonnecrotizing granulomas in a lymphaticdistribution), smoking-related interstitialfibrosis (extensive respiratory bronchiolitisand exquisitely subpleural and/orperibronchiolar paucicellular densely
Figure 1. High-resolution computed tomography (CT) images demonstrating a usual interstitial pneumonia pattern. (A–C) Transverse CT section and (D)coronal reconstruction illustrating the presence of honeycombing with subpleural and basal predominance. Note the concurrent presence of mild ground-glass opacity. (E) Magnified view of the left lower lobe showing typical characteristics of honeycombing, consisting of clustered cystic airspaces with well-defined walls and variable diameters, seen in single or multiple layers (arrows).
AMERICAN THORACIC SOCIETY DOCUMENTS
American Thoracic Society Documents e51
to the 2011 guidelines are highly likely tohave histopathologic UIP, despite the absenceof radiologic honeycombing (77). Specifically,an HRCT pattern of possible UIP withperipheral traction bronchiectasis orbronchiolectasis in the correct clinical settinglikely represents histopathologic UIP onbiopsy (65, 78–80). Therefore, subpleural,basal-predominant reticular abnormalitieswith peripheral traction bronchiectasis orbronchiolectasis should be regarded as“probable UIP.” As with a UIP pattern,ground-glass opacification may be presentin probable UIP, but it is not a dominantfeature. Many patients with an HRCT patternof probable UIP will be determined to haveIPF once other factors such as histopathologyare considered.
Indeterminate for UIP pattern. It is nowrecognized that atypical HRCT featuresfrequently (i.e., about 30%) accompany ahistopathologic pattern of UIP/IPF (81).Therefore, the category “indeterminate forUIP pattern” should be assigned when HRCT
demonstrates features of fibrosis but does notmeet UIP or probable UIP criteria and doesnot explicitly suggest an alternative diagnosis.This category includes a subset of patientswith very limited subpleural ground-glassopacification or reticulation without obviousCT features of fibrosis, for whom there is asuspicion that early UIP or probable UIP ispresent. In such cases, it should be confirmedwith prone inspiratory views that thesubpleural opacities do not representdependent atelectasis (Figure E2).
Alternative diagnosis. In some cases offibrotic lung disease, there is clinical suspicionof IPF, but the HRCT pattern suggests analternative diagnosis. Examples includebronchocentric fibrosis in the upper lobes orprofuse mosaic attenuation that suggesthypersensitivity pneumonitis, posterior fibroticretraction of the hila in sarcoidosis, or extensiveground-glass opacification with subpleuralsparing in fibrotic nonspecific interstitialpneumonia (NSIP). Occasionally, the HRCTpresentation may be that of a UIP, probable
UIP, or indeterminate for UIP pattern, butancillary findings suggest an alternativediagnosis. In such situations, an alternativediagnosis to IPF should be reconsidered.
CT findings in the presence of an acuteexacerbation. Patients with an acuteexacerbation of IPF have bilateral ground-glass opacification with or withoutconsolidation on a background of lungfibrosis (Figure 6). In the absence of aprevious HRCT study, bilateral ground-glass opacity and/or consolidation on abackground of a UIP pattern is highlysuggestive of an acute exacerbation and canbe used to confirm an underlying IPFdiagnosis in the appropriate clinical context.
SLB TechniqueVideo-assisted thoracoscopic surgery is thepreferred approach to SLB for patients whocan tolerate single-lung ventilation, ratherthan open thoracotomy. In patients withsevere physiologic impairment or substantialcomorbidity, the risks of SLB may outweigh
Table 4. High-Resolution Computed Tomography Scanning Patterns
UIP Probable UIP Indeterminate for UIP Alternative Diagnosis
Subpleural and basalpredominant; distribution isoften heterogeneous*
Subpleural and basalpredominant;distribution is oftenheterogeneous
Subpleural and basal predominant Findings suggestive of anotherdiagnosis, including:
Honeycombing with or withoutperipheral tractionbronchiectasis orbronchiolectasis†
Reticular pattern with peripheraltraction bronchiectasis orbronchiolectasis
Subtle reticulation; may have mildGGO or distortion (“early UIPpattern”)
d CT features:
May have mild GGO
CT features and/or distribution oflung fibrosis that do not suggestany specific etiology (“trulyindeterminate for UIP”)
∘ Cysts∘ Marked mosaicattenuation
∘ Predominant GGO∘ Profuse micronodules∘ Centrilobular nodules∘ Nodules∘ Consolidation
d Predominant distribution:∘ Peribronchovascular∘ Perilymphatic∘ Upper or mid-lung
d Other:∘ Pleural plaques (considerasbestosis)
∘ Dilated esophagus(consider CTD)
∘ Distal clavicular erosions(consider RA)
∘ Extensive lymph nodeenlargement (considerother etiologies)
∘ Pleural effusions, pleuralthickening (considerCTD/drugs)
Definition of abbreviations: CT = computed tomography; CTD = connective tissue disease; GGO= ground-glass opacities; RA = rheumatoid arthritis;UIP = usual interstitial pneumonia.*Variants of distribution: occasionally diffuse, may be asymmetrical.†Superimposed CT features: mild GGO, reticular pattern, pulmonary ossification.
AMERICAN THORACIC SOCIETY DOCUMENTS
e50 American Journal of Respiratory and Critical Care Medicine Volume 198 Number 5 | September 1 2018
Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice GuidelineAm J Respir Crit Care Med 2018;198; e44–e68
Imágenes de tórax en EPI
such as computerized quantitative algorithms mayeventually be useful to help eliminate the interob-server variability that currently exists, particularlyamong nonexperts.32,33 This approach also holdsappeal for its potential to improve precision inassessing ILD severity and estimatingprognosis.32–34
Pulmonary Function Testing
Although pulmonary function tests are typically un-helpful in distinguishing ILD subtypes, these are acritical component of assessing disease severityand disease behavior.35,36 Longitudinal monitoringof forced vital capacity (FVC) and diffusing capac-ity of the lung for carbon monoxide (DLCO) is
valuable for monitoring therapeutic response,quantifying disease progression, and identifyingpatients with high mortality risk. Even in patientswithout a defined ILD, the subtle presence of inter-stitial lung abnormalities on chest HRCT is associ-ated with an increased rate of pulmonary functiondecline,37 indicating the importance of monitoringpatients with subtle abnormalities on chest imag-ing. In some patients, disease behavior providesuseful information that impacts the diagnostic like-lihood. For example, long-term stability decreasesthe probability of IPF, whereas ongoing rapidworsening may argue against non-IPF etiologies,particularly if a potential offending cause hasbeen removed (eg, a potentially offending medica-tion or environmental exposure).
Fig. 2. Representative axial CT images of common ILD patterns. (A) A typical UIP pattern characterized by sub-pleural and basal predominant honeycombing (blue triangle), reticulation (red asterisks), and traction bronchi-ectasis (yellow arrows). (B) A typical nonspecific interstitial pneumonia (NSIP) pattern with peripheral andlower lung predominant abnormalities, including the classic feature of subpleural sparing that is seen in approx-imately 25% of patients with NSIP and helps distinguish from UIP when present. In this case, the presence ofbronchiectasis (yellow arrows) indicates the presence of “microfibrosis” that is represented by ground glass onthis image. (C) A typical pattern of fibrotic HP including the 3-density sign that is indicated by the presence ofwell-demarcated areas of high attenuation or ground glass (green circle), low attention (blue rectangle), andnormal attenuation. There is diffuse abnormality in both the craniocaudal and axial planes, with presence ofreticulation (red asterisks) and traction bronchiectasis (yellow arrows). (D) An indeterminate pattern of fibrosischaracterized by mild peripheral reticulation and ground-glass abnormality without any defining feature. Thesuggestion of well-demarcated subpleural sparing (red lightning bolts) is an image processing artifact anddoes not suggest NSIP.
Adegunsoye & Ryerson254
2 24 2 2 ( E 1 2 1 C AE ) 4 ) 2 E C 2E /2 2 2D C2 A . A A 2 A 2 ) E A C , 0 4 4 C24

Imágenes de tórax en EPI
such as computerized quantitative algorithms mayeventually be useful to help eliminate the interob-server variability that currently exists, particularlyamong nonexperts.32,33 This approach also holdsappeal for its potential to improve precision inassessing ILD severity and estimatingprognosis.32–34
Pulmonary Function Testing
Although pulmonary function tests are typically un-helpful in distinguishing ILD subtypes, these are acritical component of assessing disease severityand disease behavior.35,36 Longitudinal monitoringof forced vital capacity (FVC) and diffusing capac-ity of the lung for carbon monoxide (DLCO) is
valuable for monitoring therapeutic response,quantifying disease progression, and identifyingpatients with high mortality risk. Even in patientswithout a defined ILD, the subtle presence of inter-stitial lung abnormalities on chest HRCT is associ-ated with an increased rate of pulmonary functiondecline,37 indicating the importance of monitoringpatients with subtle abnormalities on chest imag-ing. In some patients, disease behavior providesuseful information that impacts the diagnostic like-lihood. For example, long-term stability decreasesthe probability of IPF, whereas ongoing rapidworsening may argue against non-IPF etiologies,particularly if a potential offending cause hasbeen removed (eg, a potentially offending medica-tion or environmental exposure).
Fig. 2. Representative axial CT images of common ILD patterns. (A) A typical UIP pattern characterized by sub-pleural and basal predominant honeycombing (blue triangle), reticulation (red asterisks), and traction bronchi-ectasis (yellow arrows). (B) A typical nonspecific interstitial pneumonia (NSIP) pattern with peripheral andlower lung predominant abnormalities, including the classic feature of subpleural sparing that is seen in approx-imately 25% of patients with NSIP and helps distinguish from UIP when present. In this case, the presence ofbronchiectasis (yellow arrows) indicates the presence of “microfibrosis” that is represented by ground glass onthis image. (C) A typical pattern of fibrotic HP including the 3-density sign that is indicated by the presence ofwell-demarcated areas of high attenuation or ground glass (green circle), low attention (blue rectangle), andnormal attenuation. There is diffuse abnormality in both the craniocaudal and axial planes, with presence ofreticulation (red asterisks) and traction bronchiectasis (yellow arrows). (D) An indeterminate pattern of fibrosischaracterized by mild peripheral reticulation and ground-glass abnormality without any defining feature. Thesuggestion of well-demarcated subpleural sparing (red lightning bolts) is an image processing artifact anddoes not suggest NSIP.
Adegunsoye & Ryerson254
2 24 2 2 ( E 1 2 1 C AE ) 4 ) 2 E C 2E /2 2 2D C2 A . A A 2 A 2 ) E A C , 0 4 4 C24

Imágenes de tórax en EPI
such as computerized quantitative algorithms mayeventually be useful to help eliminate the interob-server variability that currently exists, particularlyamong nonexperts.32,33 This approach also holdsappeal for its potential to improve precision inassessing ILD severity and estimatingprognosis.32–34
Pulmonary Function Testing
Although pulmonary function tests are typically un-helpful in distinguishing ILD subtypes, these are acritical component of assessing disease severityand disease behavior.35,36 Longitudinal monitoringof forced vital capacity (FVC) and diffusing capac-ity of the lung for carbon monoxide (DLCO) is
valuable for monitoring therapeutic response,quantifying disease progression, and identifyingpatients with high mortality risk. Even in patientswithout a defined ILD, the subtle presence of inter-stitial lung abnormalities on chest HRCT is associ-ated with an increased rate of pulmonary functiondecline,37 indicating the importance of monitoringpatients with subtle abnormalities on chest imag-ing. In some patients, disease behavior providesuseful information that impacts the diagnostic like-lihood. For example, long-term stability decreasesthe probability of IPF, whereas ongoing rapidworsening may argue against non-IPF etiologies,particularly if a potential offending cause hasbeen removed (eg, a potentially offending medica-tion or environmental exposure).
Fig. 2. Representative axial CT images of common ILD patterns. (A) A typical UIP pattern characterized by sub-pleural and basal predominant honeycombing (blue triangle), reticulation (red asterisks), and traction bronchi-ectasis (yellow arrows). (B) A typical nonspecific interstitial pneumonia (NSIP) pattern with peripheral andlower lung predominant abnormalities, including the classic feature of subpleural sparing that is seen in approx-imately 25% of patients with NSIP and helps distinguish from UIP when present. In this case, the presence ofbronchiectasis (yellow arrows) indicates the presence of “microfibrosis” that is represented by ground glass onthis image. (C) A typical pattern of fibrotic HP including the 3-density sign that is indicated by the presence ofwell-demarcated areas of high attenuation or ground glass (green circle), low attention (blue rectangle), andnormal attenuation. There is diffuse abnormality in both the craniocaudal and axial planes, with presence ofreticulation (red asterisks) and traction bronchiectasis (yellow arrows). (D) An indeterminate pattern of fibrosischaracterized by mild peripheral reticulation and ground-glass abnormality without any defining feature. Thesuggestion of well-demarcated subpleural sparing (red lightning bolts) is an image processing artifact anddoes not suggest NSIP.
Adegunsoye & Ryerson254
2 24 2 2 ( E 1 2 1 C AE ) 4 ) 2 E C 2E /2 2 2D C2 A . A A 2 A 2 ) E A C , 0 4 4 C24

Signos sugerentes de EPI-ETC

Pruebas de función pulmonar en EPI• Espirometría restrictiva (CVF < 80%, VEF/CVF normal o
aumentada)• Ojo con normalización de CVF en FPI/Enfisema• DLCO disminuída (+ sensible)• Caminata de 6 minutos desaturación > 4%• Seguimiento longitudinal fundamental en pronóstico y
respuesta a terapia
Am J Respir Crit Care Med 2013;188:733–748

Análisis serológico en EPI• PCR, VHS• Tamizaje autoinmune: ANA, FR, CCP, ANCA• Estudio autoinmune dirigido: ENA, Panel de miositis,
Panel de esclerodermia, ANA 23 • Tamizaje Panel Antígenos de Hipersensibilidad (IgG)
(aviares, fúngicos, etc)
Am J Respir Crit Care Med 2013;188:733–748

An official European Respiratory Society/American Thoracic Society researchstatement: interstitial pneumonia withautoimmune features
Aryeh Fischer1,17,18, Katerina M. Antoniou2, Kevin K. Brown3, Jacques Cadranel4,Tamera J. Corte5,18, Roland M. du Bois6, Joyce S. Lee7,18, Kevin O. Leslie8,David A. Lynch9, Eric L. Matteson10, Marta Mosca11, Imre Noth12,Luca Richeldi13, Mary E. Strek12,18, Jeffrey J. Swigris3,18, Athol U. Wells14,Sterling G. West15, Harold R. Collard7,18,19 and Vincent Cottin16,18,19, on behalf ofthe “ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD”
Affiliations: 1Dept of Medicine, University of Colorado School of Medicine, Denver, CO, USA. 2ThoracicMedicine, University of Crete, Heraklion, Greece. 3Dept of Medicine, National Jewish Health, Denver, CO, USA.4Pneumologie, Hopital Tenon, Paris, France. 5The Aldred Hospital, Melbourne, Australia. 6Interstitial LungDisease Unit, Dept of Occupational Medicine, Royal Brompton Hospital, London, UK. 7Medicine, University ofCalifornia San Francisco, San Francisco, CA, USA. 8Pathology, Mayo Clinic, Scottsdale, AZ, USA. 9Dept ofRadiology, National Jewish Health, Denver, CO, USA. 10Division of Rheumatology, Mayo College of Medicine,Rochester, MN, USA. 11University of Pisa, Pisa, Italy. 12Medicine, University of Chicago, Chicago, IL, USA.13Southampton General Hospital, Southampton, UK. 14Interstitial Lung Disease Unit, Royal BromptonHospital, London, UK. 15University of Colorado School of Medicine, Aurora, CO, USA. 16Service dePneumologie, Hopital L. Pradel, Lyon, France. 17Task force chair. 18Members of the writing group of the taskforce. 19Task force vice-chairs, and contributed equally to this manuscript.
Correspondence: Aryeh Fischer, 1775 Aurora Court, P.O. Box 6511, Mail Stop B-115, Aurora, CO 80045, USA.E-mail: [email protected]
ABSTRACT Many patients with an idiopathic interstitial pneumonia (IIP) have clinical features that suggestan underlying autoimmune process but do not meet established criteria for a connective tissue disease (CTD).Researchers have proposed differing criteria and terms to describe these patients, and lack of consensus overnomenclature and classification limits the ability to conduct prospective studies of a uniform cohort.
The “European Respiratory Society/American Thoracic Society Task Force on Undifferentiated Forms ofConnective Tissue Disease-associated Interstitial Lung Disease” was formed to create consensus regardingthe nomenclature and classification criteria for patients with IIP and features of autoimmunity.
The task force proposes the term “interstitial pneumonia with autoimmune features” (IPAF) and offersclassification criteria organised around the presence of a combination of features from three domains: a clinicaldomain consisting of specific extra-thoracic features, a serologic domain consisting of specific autoantibodies, anda morphologic domain consisting of specific chest imaging, histopathologic or pulmonary physiologic features.
A designation of IPAF should be used to identify individuals with IIP and features suggestive of, but notdefinitive for, a CTD. With IPAF, a sound platform has been provided from which to launch the requisitefuture research investigations of a more uniform cohort.
@ERSpublicationsERS/ATS task force provides nomenclature and classification criteria for patients with IIP andautoimmune features http://ow.ly/O7qao
Copyright ©ERS 2015
Received: Jan 27 2015 | Accepted after revision: May 11 2015 | First published online: July 09 2015
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
Support statement: Support for this task force was provided by the European Respiratory Society and the AmericanThoracic Society. Funding information for this article has been deposited with FundRef.
976 Eur Respir J 2015; 46: 976–987 | DOI: 10.1183/13993003.00150-2015
ERS/ATS TASK FORCEINTERSTITIAL LUNG DISEASE
An official ERS/ ATS research statement: interstitial pneumonia with autoimmune features. ERJ 2015

Lavado broncoalveolar (FBC) en EPI• Fundamental ante sospecha de infección o neoplasia• Útil al encontrar linfocitosis en LBA en NHS o
sarcoidosis, eosinofilos en neumonía eosinofílica o hemático progresivo en hemorragia alveolar difusa
• No informativo en otros escenarios de EPI
Am J Respir Crit Care Med 2013;188:733–748

Lavado broncoalveolar (FBC) en EPI• Fundamental ante sospecha de infección o neoplasia• Útil al encontrar linfocitosis en LBA en NHS o
sarcoidosis, eosinofilos en neumonía eosinofílica o hemático progresivo en hemorragia alveolar difusa
• No informativo en otros escenarios de EPI
Am J Respir Crit Care Med 2013;188:733–748

Biopsia pulmonar en EPI• Histología por biopsia pulmonar quirúrgica (VATS)
siempre aporta información diagnóstica y pronóstica• Mortalidad a 30 días post BPQ: 1.7%• Se eleva a 15-20% cuando se realiza en contexto
agudo por admisión respiratoria
In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States: 2000 to 2011. Am J Respir Crit Care Med 2016;193:1161–7 Surgical lung biopsy for the diagnosis of interstitial lung disease in England: 1997-2008. Eur Respir J 2016; 48:1453–61

Biopsia pulmonar en EPI• No justificada en:• Patrón definitivo o probable de NIU en TC tórax• EPI-ETC establecida • NH con exposición conocida, linfocitosis en LBA y
patrón tomográfico característico

28Arch Pathol Lab Med 2016-0233-RA
Review Article
Transbronchial Cryobiopsy in Diffuse Lung Disease
Update for the Pathologist
Thomas V. Colby, MD; Sara Tomassetti, MD; Alberto Cavazza, MD; Alessandra Dubini, MD; Venerino Poletti, MD
! Context.—Transbronchial cryobiopsy has recently beenproposed as an alternative to surgical biopsy in thediagnosis of diffuse lung disease.
Objective.—To familiarize pathologists with transbron-chial cryobiopsy, including what it is, how it is performed,how it compares to other techniques of lung biopsy indiffuse lung disease, what are the technical issues relatingto it, what the complications are, how cryobiopsies shouldbe interpreted, and the clinical usefulness of cryobiopsy.
Data Sources.—All the available literature on cryo-biopsy in diffuse lung disease through May 2016, primarilyin the last 5 years, was reviewed, and some unpublisheddata known to the authors were included.
Conclusions.—Cryobiopsies are considerably larger than
forceps biopsies and allow pattern recognition approach-ing that of a surgical lung biopsy in many cases. Artifactsassociated with cryobiopsy are minimal. In comparisonwith surgical lung biopsies, the diagnosis rate with cryo-biopsies is lower, in the neighborhood of 80%, versushigher than 90% for surgical lung biopsies. Cryobiopsy isproposed as an alternative to surgical lung biopsy and atechnique that may appreciably decrease the number ofpatients who require surgical lung biopsy for diagnosis.This is important because the mortality from cryobiopsy isvery small (0.1% to date) compared with surgical lungbiopsy (1.7% for elective procedures and considerablyhigher for nonelective procedures).
(Arch Pathol Lab Med. doi: 10.5858/arpa.2016-0233-RA)
Transbronchial cryobiopsies are now used at manycenters around the world to assess patients with diffuse
lung disease,1–13 and this has implications for pathologistswho will encounter them.
WHAT IS A TRANSBRONCHIAL LUNG CRYOBIOPSY?
Put simply, a cryoprobe is inserted into the lung, cooledbelow freezing, and then retrieved with the lung tissue thathas frozen onto it. This tissue is thawed in saline and thentransferred to formalin and processed routinely for histo-logic evaluation.
Background
Cryoprobes have been used bronchoscopically for morethan 40 years,1,14 initially for the debulking of endobronchiallesions. Because more tissue can be retrieved than withbronchoscopic forceps biopsies, cryoprobe-retrieved speci-
mens are larger and have also proven more effective thanforceps biopsy specimens for retrieving sufficient tissue forhistologic diagnosis (and immunohistochemistry as needed)of tumors biopsied endobronchially.14,15 In a prospectivemulticenter trial of nearly 600 patients among 8 centers,cryobiopsy proved superior to forceps biopsy in providing adiagnosis for an endobronchial tumor: 95% versus 85% (268of 282 patients versus 239 of 281 patients), and the safetyprofile for cryobiopsy was similar to that of forceps biopsy.16
Transbronchial cryobiopsy has recently been used in thesetting of diffuse lung disease,1–13 a setting in whichtransbronchial forceps biopsies have had only limitedusefulness. As stated in the 2009 report by Babiak et al1:‘‘transbronchial cryobiopsy is a novel technique, which allowsto obtain large biopsy samples of lung parenchyma thatexceed the size and quality of forceps biopsy samples.’’ Sincethe initial studies, there have been a large number ofadditional reports from around the world describing thistechnique in diffuse lung disease, primarily interstitialpneumonias1–13 but also in other settings, including lungtransplant sampling17,18 and airway disease19; the safety profileis acceptable and the diagnostic usefulness much greater thanforceps biopsies.4,20–22
HOW IS TRANSBRONCHIAL CRYOBIOPSYPERFORMED?
Transbronchial cryobiopsy is performed with a rigid orflexible bronchoscope, with or without intubation.23 Usually,under fluoroscopic guidance a cryoprobe (1.9 or 2.4 mm indiameter) is inserted through the operating channel of theflexible bronchoscope into the region of interest (typically~1.0 cm from the pleura), and it is cooled for a short period
Accepted for publication July 13, 2016.From the Department of Pathology, Mayo Clinic Arizona,
Scottsdale (Dr Colby); the Departments of Diseases of the Thorax(Drs Tomassetti and Poletti) and Pathology (Dr Dubini), G.B.Morgagni Hospital, Forli, Italy (Dr Tomassetti); the Department ofPathology, Arcispedale S Maria Nouva, Istituti di Ricovero e Cura aCarattere Scientifico, Reggio Emilia, Italy (Dr Cavazza); and theDepartment of Respiratory Diseases and Allergy, Aarhus UniversityHospital, Aarhus, Denmark (Dr Poletti).
The authors have no relevant financial interest in the products orcompanies described in this article.
Presented in part at the Third Princeton Integrated PathologySymposium; May 14, 2016; Plainsboro, New Jersey.
Reprints: Thomas V. Colby, MD, Mayo Clinic Arizona, 13400 EShea Blvd, Scottsdale, AZ 85259 (email: [email protected]).
Arch Pathol Lab Med Transbronchial Cryobiopsy in Diffuse Lung Disease—Colby et al 1
of time (5–6 seconds), at which point the cryoprobe iswithdrawn with tissue stuck/frozen to it; the tissue is thawedand removed from the probe in a saline bath, and then it istransferred to formalin and processed for routine histology.Multiple biopsies can be obtained in the same sitting.
As has been emphasized recently,23 there is a clear needfor procedural standardization for this recently introducedtechnique. Procedural issues include: type and degree ofsedation, intubation versus no intubation, types of ventila-
Figure 1. Performing cryobiopsies. The cryoprobe is introducedthrough a rigid bronchoscope by the bronchoscopist (A). The shadedregion at the top shows the cryoprobe inserted into the operativechannel of the flexible bronchoscope. With fluoroscopic guidance theprobe is inserted into the periphery of the lung and cooled (B). Oncethe specimens are retrieved on the cryoprobe, they are thawed in salineand typically put directly into formalin. Some gross examples ofcryobiopsies are shown in C.
Figure 2. Twelve consecutive unselected cryobiopsies from the G.B.Morgagni Hospital in Forli, Italy. It is apparent that most of these are atleast 0.5 cm in greatest dimension and that multiple specimens can betaken at one sitting. Specimens that are sufficiently large enough can bedivided in two once they are fixed in formalin.
Figure 3. Low-power photomicrograph of a cryobiopsy showingpristine lung tissue without any crush artifact. A small airway cutlongitudinally shows some sloughing of epithelium. Such a biopsywould be considered nondiagnostic/normal. Other specimens from thiscase are shown in Figure 14 (hematoxylin-eosin, approximate originalmagnification 360).
2 Arch Pathol Lab Med Transbronchial Cryobiopsy in Diffuse Lung Disease—Colby et al
of time (5–6 seconds), at which point the cryoprobe iswithdrawn with tissue stuck/frozen to it; the tissue is thawedand removed from the probe in a saline bath, and then it istransferred to formalin and processed for routine histology.Multiple biopsies can be obtained in the same sitting.
As has been emphasized recently,23 there is a clear needfor procedural standardization for this recently introducedtechnique. Procedural issues include: type and degree ofsedation, intubation versus no intubation, types of ventila-
Figure 1. Performing cryobiopsies. The cryoprobe is introducedthrough a rigid bronchoscope by the bronchoscopist (A). The shadedregion at the top shows the cryoprobe inserted into the operativechannel of the flexible bronchoscope. With fluoroscopic guidance theprobe is inserted into the periphery of the lung and cooled (B). Oncethe specimens are retrieved on the cryoprobe, they are thawed in salineand typically put directly into formalin. Some gross examples ofcryobiopsies are shown in C.
Figure 2. Twelve consecutive unselected cryobiopsies from the G.B.Morgagni Hospital in Forli, Italy. It is apparent that most of these are atleast 0.5 cm in greatest dimension and that multiple specimens can betaken at one sitting. Specimens that are sufficiently large enough can bedivided in two once they are fixed in formalin.
Figure 3. Low-power photomicrograph of a cryobiopsy showingpristine lung tissue without any crush artifact. A small airway cutlongitudinally shows some sloughing of epithelium. Such a biopsywould be considered nondiagnostic/normal. Other specimens from thiscase are shown in Figure 14 (hematoxylin-eosin, approximate originalmagnification 360).
2 Arch Pathol Lab Med Transbronchial Cryobiopsy in Diffuse Lung Disease—Colby et al
of time (5–6 seconds), at which point the cryoprobe iswithdrawn with tissue stuck/frozen to it; the tissue is thawedand removed from the probe in a saline bath, and then it istransferred to formalin and processed for routine histology.Multiple biopsies can be obtained in the same sitting.
As has been emphasized recently,23 there is a clear needfor procedural standardization for this recently introducedtechnique. Procedural issues include: type and degree ofsedation, intubation versus no intubation, types of ventila-
Figure 1. Performing cryobiopsies. The cryoprobe is introducedthrough a rigid bronchoscope by the bronchoscopist (A). The shadedregion at the top shows the cryoprobe inserted into the operativechannel of the flexible bronchoscope. With fluoroscopic guidance theprobe is inserted into the periphery of the lung and cooled (B). Oncethe specimens are retrieved on the cryoprobe, they are thawed in salineand typically put directly into formalin. Some gross examples ofcryobiopsies are shown in C.
Figure 2. Twelve consecutive unselected cryobiopsies from the G.B.Morgagni Hospital in Forli, Italy. It is apparent that most of these are atleast 0.5 cm in greatest dimension and that multiple specimens can betaken at one sitting. Specimens that are sufficiently large enough can bedivided in two once they are fixed in formalin.
Figure 3. Low-power photomicrograph of a cryobiopsy showingpristine lung tissue without any crush artifact. A small airway cutlongitudinally shows some sloughing of epithelium. Such a biopsywould be considered nondiagnostic/normal. Other specimens from thiscase are shown in Figure 14 (hematoxylin-eosin, approximate originalmagnification 360).
2 Arch Pathol Lab Med Transbronchial Cryobiopsy in Diffuse Lung Disease—Colby et al
of time (5–6 seconds), at which point the cryoprobe iswithdrawn with tissue stuck/frozen to it; the tissue is thawedand removed from the probe in a saline bath, and then it istransferred to formalin and processed for routine histology.Multiple biopsies can be obtained in the same sitting.
As has been emphasized recently,23 there is a clear needfor procedural standardization for this recently introducedtechnique. Procedural issues include: type and degree ofsedation, intubation versus no intubation, types of ventila-
Figure 1. Performing cryobiopsies. The cryoprobe is introducedthrough a rigid bronchoscope by the bronchoscopist (A). The shadedregion at the top shows the cryoprobe inserted into the operativechannel of the flexible bronchoscope. With fluoroscopic guidance theprobe is inserted into the periphery of the lung and cooled (B). Oncethe specimens are retrieved on the cryoprobe, they are thawed in salineand typically put directly into formalin. Some gross examples ofcryobiopsies are shown in C.
Figure 2. Twelve consecutive unselected cryobiopsies from the G.B.Morgagni Hospital in Forli, Italy. It is apparent that most of these are atleast 0.5 cm in greatest dimension and that multiple specimens can betaken at one sitting. Specimens that are sufficiently large enough can bedivided in two once they are fixed in formalin.
Figure 3. Low-power photomicrograph of a cryobiopsy showingpristine lung tissue without any crush artifact. A small airway cutlongitudinally shows some sloughing of epithelium. Such a biopsywould be considered nondiagnostic/normal. Other specimens from thiscase are shown in Figure 14 (hematoxylin-eosin, approximate originalmagnification 360).
2 Arch Pathol Lab Med Transbronchial Cryobiopsy in Diffuse Lung Disease—Colby et al

Am J Respir Crit Care Med 2016;193:745–752
• Corte transversal• 117 pacientes con EPID no NIU• 58 CBTB; 59 Bp Qx• Confianza diagnóstica FPI
• CBTB 29 a 63%• Bp Qx 30 a 65%
• Kappa Bp Qx: 0.93• Kappa CBTB: 0.96
Neumólog@Reumatólog@
Higienista ambiental
Radiólog@
Patólog@Paciente con EPI
Discusión multidisciplinaria en EPI

Tratamiento de EPI: inflamación vs fibrosis
Inflamación
Fibrosis
• Corticioides• Azatioprina• Micofenolato• Anticalcineurínicos• Ciclofosfamida• Rituximab• Tocilizumab• Otros biológicos
• Pirfenidona• Nintedanib• Nuevos antifibróticos• Trasplante pulmonar


















