
Anesthésie et Hémoglobinopathies · • Les différent formes de thalassémie sont classifiées...
52
La thalassémie et Anesthésie Dre. Margaret Haig Hôpital Ste Justine
Transcript of Anesthésie et Hémoglobinopathies · • Les différent formes de thalassémie sont classifiées...

La thalassémie et Anesthésie Dre. Margaret Haig
Hôpital Ste Justine

Plan• Structure d’hémoglobine
• Génétique de thalassémie
• Pathophysiologie des maladies
• Organes atteintes
• Soins globaux
• Grossesse et hémoglobinopathie
• Management péri opératoire

Structure de l’hémoglobine• Hb adulte Hb A est fait de 4 chaines polypeptides
• 2 chaines α
globine
• 2 chaines de β
globine
• (α₂/β₂)

Thalassémies• Group de maladies autosomales récessives
• Caractérisés par une diminution ou une absence de
production d’une des globines (α
ou β
)
• Les différent formes de thalassémie sont classifiées selon la
chaine impliquée

Thalassémie• Un défaut de la production de la chaîne β
s’appelle β‐
thalassémie
• Un défaut de la chaîne α
s’appelle α‐thalassémie

Thalassémie• Thalassa = mer (Grec)
• Haima = sang

Populations à risque? • β‐thalassémie est répandue autour de la Méditerrané, en Asie
central, sud, et sud‐est et le sud de Chine
• α‐
thalassémie est plus fréquent en Asie sud –est, en Afrique,
et aux Indes
• Mondialement, 1.5% des êtres humains sont porteurs du
gène pour β‐thalassémie
• et 5% sont porteurs des gènes pour α‐thalassémie

Prévalence au Canada• Laboratoire à
Hamilton,
Ontario
• 800 échantillons pour anémie
microcytaire
• 664 échantillons de bonne
qualité
• 24.5 % porteurs de
βthalassémie majeure
• 10 % α
thalassémie (risque
d’anasarque fœtal
• 54% une forme de
thalassémie

Alpha‐thalassémie• Individus normales ont 4 copies du gène pour la chaine α
2 de
chaque parent (α,α/α,α)
• En plus il y a• 3 catégories d’anormalité:
• 1.Délétion du gène• 2.Mutation qui diminue la production
• 3.α‐globine anormale qui est aussi produit en quantité
réduit

Le trait d’α thalassémie• Les individus qui on 1 ou 2 gènes pour α
globine qui sont
fonctionnelles sont asymptomatiques (_,_,α,α
ou _,α,_,α)
• Espérance de vie N
• Porteurs silencieux de maladie grave
• Génétique très compliqué
plus que 128 différent défauts
moléculaire qui causent α
thalassémie

• 4 gènes pour α
globine donc multiples différent combinaisons
sont possible

• Homozygote pour absence totale de chaine α
• Génotype _,_ /_,_
• Le fétus ne peut pas produire HbF (α2 γ2)

Alphathalassémie
Produisent Hb Bart’s
(ϒ₄)Hb Bart’s: ne
transport pas d’O₂
Une anémie profonde
et hydrops fétalis
Chine 0.23% des
naissances

α thalassémie: Hb H• Dysfonction de 3/4 des gènes pour α
globine
• _,_, _,α• Excédant de ϒ
chaines in utéro cause une haute concentration
d’Hb Bart’s à la naissance mais à
cause de la présence de
• HbF (α2 ϒ
2) le bébé
n’a pas l’air malade à la naissance
• plus tard quand la production d’HbF ↓
Hb H (β₄)
• Le NN développe une anémie hémolytique et une hépato
splénomégalie

Maladie d’Hb H• Produisent < 30% de la quantité
N d’α
globine
• Anémiques
• HbH (β4) 0.8‐
40%
• Splénomégalie
• Jaunisse

Maladie d’Hb H• Sujet aux ulcères des jambes
• Cholélithiasis
• Déficience en acide folique
• Épisodes de hémolyse accélérée 2⁰
aux infections ou
médicaments

β thalassémie• L’individu normal a 1 copie du gène pour le β
globine sur
chaque chromosome 11
• Plus que 200 mutations de ces gènes ont étaient identifiées
avec des conséquences cliniques variables

Le trait de β thalassémie = thalassémie mineure• 1 gène normal et 1 anormal (le trait de β
thal)
• Généralement en santé
sauf pour une légère anémie
microcytaire qui est souvent mal diagnostiqué
comme anémie
ferriprive

β thalassémie majeure• 2 gènes anormales pour la chaine β
• Normalement le nombre de chaines α
doit être égale au
nombre de chaines β
• Les chaines α
en surplus font un précipité
dans les précurseurs
des globules rouges
• Ce qui cause du dommage au membrane du GR et une
hémolyse avant même que les globules rouges matures sont
produites

Beta thalassémie

Β thalassémie majeure• Érythropoïèse très, très inefficace
• 2 phénomènes
• la mort des précurseurs des GR (apoptose) dans la moelle
• La destruction précoce des GR circulants qui sont enlevés par la rate ou hémolysés directement ( survie seulement de 4‐8
jours)

Beta thal• Ce défaut d’hématopoïèse amène tous les effets secondaires
• 1. anémie
• 2. expansion de la moelle osseuse
• 3.hépatosplénomégalie
• 4. déformation des os
• 5.hypermétabolisme
• 6.accumulation de fer →dommages aux organes

anémie

Hypertrophie de la moelle

Hépatosplénomégalie
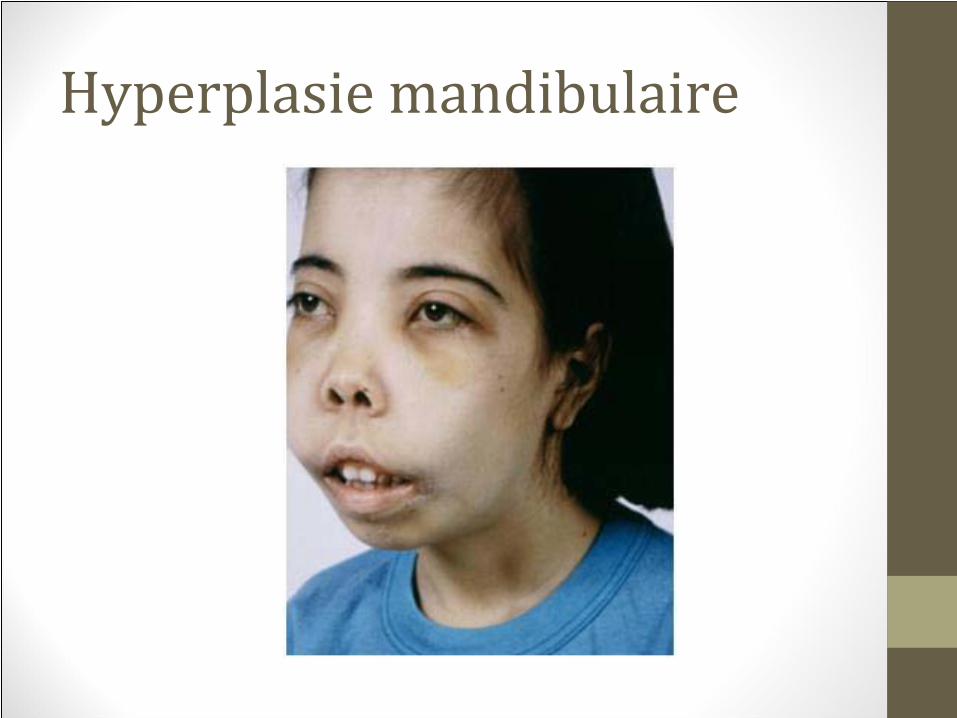
Hyperplasie mandibulaire

Beta thalassémie• 50% des cas de β
thalassémie sévère en Asie sud‐est, sont
causés par la gène pour β
thalassémie d’un parent et Hb E de
l’autre
• HbE est une autre forme anormale de la chaine β

Beta thalassémie inter‐média• Le nom vient de l’ère de l’électrophorèse d’Hb et la possibilité
d’identifier les mutations d’ADN
• Le phénotype de β
thalassémie intermédia était décrit
• Une anémie qui débutait plus tardivement, et qui était moins
sévère que β
thalassémie majeure
• La cause de cette forme moins sévère n’est pas encore
totalement élucidé
( HbF, présence d’autre gène qui modifie
le fonctionnement du gène pour la chaine β, alpha
thalassémie …)

Β thalassémie intermédiaire• L’anémie moins sévère
• ont moins besoin de transfusions (pendant les infections,
pendant la grossesse, périodes de croissance augmenté)
• Séquelles d’érythropoïèse augmentés plus sévères
•• Même si non transfusMême si non transfuséé, l, l’’absorption de fer alimentaire est absorption de fer alimentaire est
augmentaugmentééee
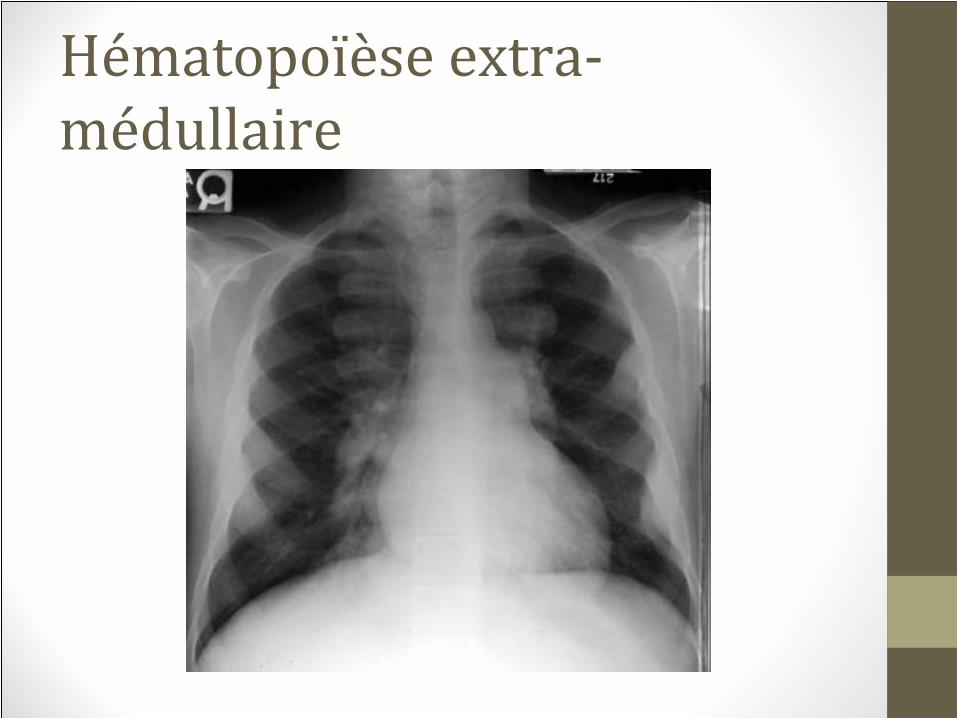
Hématopoïèse extra‐médullaire


Érythropoïèse extra médullaire• Tous les sites du corps peuvent être impliqués
• Incidence de 20% chez les malades avec β
thalassémie
intermédiaire
• vs 1% chez les gens avec βTM qui sont régulièrement
transfusés

Érythropoïèse extra médullaire• Para spinale peut présenter comme pseudotumeur
• Symptômes neurologiques (compression)
• Traitement:
• Transfusions• Radiation à
faible dose
• Laminectomie
• (risque de saignement massif)

Le diagnostique de thal.Anémie qui est: 1. microcytaire MCV <70fL
2.hypo chromique
et
3. ne répond pas à un supplément de fer
4. la ferritine est normale

Électrophorèse d’Hb• β
thalassémie majeure
• Hb A très bas ou absente
• Hb F élevée
• HbA2 élevée

Traitement de βthalassémie• Transfusions régulière à maintenir une Hb au dessus de 95g/L
• Chélation de fer
• Transfusions sans chélation →
mortalité
entre l’âge de 10 à 25
ans 2⁰
à la toxicité
cardiaque de fer
• Transfusions + chélation = espérance de vie 40 ans+

Surcharge en fer
• Chaque culot ajout ≈
200 mg de fer au corps
• Sans traitement le corps perd difficilement ce fer
• Cause des dommages aux glandes endocriniennes
(hypothalamus, gland pituitaire, les gonades, le thyroïde, le
parathyroïde et le pancréas
• L’effet est le retard de croissance, la puberté
retardée ou
absente et la diabète

Surcharge en fer• La défaillance cardiaque 2⁰
à
l’accumulation de fer dans le
myocarde est la cause le plus importante de mortalité
chez les
malades avec βthalassémie majeure
• Le concentration de fer dans le foie peut être suivi avec des biopsies hépatique
• Malheureusement la corrélation entre l’accumulation
hépatique et cardiaque n’est pas bonne
• Les biopsies cardiaques sont un peut trop invasives• Une méthode de suivie avec MRI a été
développée

Cardiaque IRM• Le niveau de fer dans le cœur est inversement proportionnel
au niveau de T₂*
• Les valeurs < 20 ms correspondent à fer cardiaque et peut
prédire le développement de la défaillance cardiaque
• Aux Royaume‐
Unis, depuis que IRM T2* a été
introduit il y a
une réduction de mortalité
pour thalassémie de 60%

Chélation de fer• Après 10 – 20 transfusions de globules rouges, la ferritine
sérique est saturée
• il faut aider le corps à excréter le fer en surplus
• 3 chélateurs qui sont utilisés maintenant:
• 1.Deferoxamine ( SC 8‐12 h /nuit, 5‐7 nuits par semaine)
• Sauve des vies mais l’observance est mauvaise
• Deferiprone po tid• Deferasirox (le nouveau) une dose de 20 mg/kg /jour
↑35/mg/kg/jour

Deferiprone• Avantage po• Complication majeure →agranulocytose
• 0.2 par 100 par années‐patient
• Neutropénie est plus fréquente• 2.1 par 100 années patient

Deferasirox• Effets secondaires GI
• Nausée, vomissement, douleur abdominal et diarrhée→15.8%
des enfants

Traitement• Hydroxy‐urée peut aider
• Greffe de moelle osseuse est la seule possibilité
de guérison
• Une sœur ou un frère HLA identique donne le meilleur
chance
• Survie globale jusqu’à
90% a été
rapporté
• Succès est augmenté
si le malade est plus jeune au moment
de la greffe

Grossesse et thalassémie

Grossesse et thalassémie• Malade avec le trait de β
thalassémie peut avoir un chute
d’Hb
et être incapable d’augmenter la production assez pour
les besoins de la grossesse
• Supplément en acide folique est nécessaire
• Jusqu’à
récemment les femmes avec β
thalassémie étaient
infertile
• Mais, intervention avec hormones et chélation agressive peut
maintenir la fertilité

Grossesse et β thal• Les données à propos des mères et fétus avec βthal
sont
rarissime
• La majorité
des femmes vivent dans les pays pauvres
• Hypogonadisme hypgonadotrophique
est présente en 60%
des femmes avec βthal
majeure
• La majorités des données sont tirées des séries de cas

• Arythmie et défaillance cardiaque sont des problèmes
importante chez les femmes qui commencent leur grossesse
avec sidérose cardiaque
• Les rapports plus récents chez les femmes sans problème
cardiaque et traitée avec chélation agressive avant le début
de la grossesse sont plutôt favorable.
• Diabète gestationnelle est fréquente
• La grossesse augmente les besoins transfusionnelle

Grossesse et β thal majeure• Origa
et al (Hematologica
2010)
• Étude multicentrique (Italie)
• 46 femmes avec βthal
majeure et 11 femmes avec βthal
inter.
• 91% des grossesses →45 bébés simples, 5 paires de jumeaux
et 1 triplet
• RCIU et accouchement pré‐terme était augmentait (40%)

Anésthésie
• Très peut d’information
• Ils vient en SO pour séquelles de la maladie eg. Fractures
pathologiques, chx
esthétique, splénectomie,
cholécystectomie…
• Si thalassémie majeure avec transfusions sous optimale ou
thalassémie intermédiaire la malformation du visage peut
causer une intubation difficile
• Comme AF possibilité
d’anticorps Cross‐match étendu

Qu’elle sorte d’anesthésie?• Une bonne• Évaluation des organes atteints
• Contrôle de la diabète péri‐op•• réticent à considérer une technique neuraxiale
si il y à la
possibilité
d’érythropoïèse para spinale ?
• thrombo‐prophylaxie

Références
• Update on Thalassemia: Clinical
Care and Complications ,
Melody J. Cunningham, Hematol
Oncol
Clin N Am 24
(2010)215‐227
• Β‐thalassémie: Medical
and Surgical
Considerations
in
Managing
Facial Deformities: Case Report and Review
of the
Literature, Park N, et al , J Oral Maxillofac
Surg70:e284‐e289,
2012
• ACOG technical
bulletin Hemoglobinopathies
in pregnancy, Int
J Gyne
Obstet, no 220‐February
1996, pp184‐194
• Cardiovascular
magnetic
resonance
T2 for tissue iron
assessment
in the heart, Taigang
He, Quant Imaging Med Surg
2014;4(5):407‐412

Références• Baby on board: what you need to know about pregnancy
in
the hemoglobinopathies, Hematology
Am Soc Hematol
Educ
Program. 2012;2012:208‐14.
• Guidelines for the Standard Monitoring of Patients with
Thalassemia: Report of the Thalassemia
Longitudinal Cohort,
Tubman VN et al, J Pediatr
Hematol
Oncol, 2015,37(3); e162‐
169
















