
ALTERACIONES TEGU.docx
34
PSORIASIS DEFINICIÓN La psoriasis es una enfermedad crónica, determinada genéticamente, caracterizada por sucesivos brotes de placas eritemato-descamativas en las superficies extensoras de la piel y cuero cabelludo, siendo la artropatía la única manifestación extracutánea ETIOLOGÍA Aunque la etiología es desconocida, existe una clara agregación familiar que apoya la implicación de factores genéticos. Además, factores ambientales conocidos como los traumatismos, diálisis, la hipocalcemia, algunas infecciones como la faringitis estreptocócica o el VIH, fármacos como el litio, betabloqueantes, antipalúdicos, interferón, interleucina 2, antidiabéticos orales, la retirada de corticoides sistémicos, algunos antiinflamatorios (indometacina) y, por último, las situaciones de estrés, pueden ser desencadenantes de la enfermedad. La psoriasis se produce como consecuencia de la interacción de 3 fenómenos que concurren simultáneamente: 1. aumento de la velocidad de crecimiento epidérmica; 2. proliferación venular postcapilar en las papilas dérmicas; y 3. Reacción inmune mediada por linfocitos T. La consecuencia de esta interacción es el fenómeno de la exudación cíclica papilar, que caracteriza microscópicamente a la enfermedad. CLÍNICA Es una enfermedad universal con una prevalencia de alrededor del 2% de la población europea, y con una incidencia de 150.000 casos nuevos/año. Puede debutar a cualquier edad, aunque cuando existen antecedentes familiares aparece de forma más temprana. Existen dos picos de incidencia: a los 16-22 años y a los 57-60 años, sin diferencia entre sexos, aunque las mujeres desarrollan antes la enfermedad. La lesión elemental es una placa eritematosa, de bordes bien definidos y superficie irregular descamativa, que tras el raspado metódico deja una superficie eritematosa con pequeños puntos sangrantes, “rocío hemorrágico de Auspitz”. Se disponen simétricamente
-
Upload
fanny-ayala -
Category
Documents
-
view
224 -
download
4
description
dermatologia
Transcript of ALTERACIONES TEGU.docx

PSORIASIS
DEFINICIÓN
La psoriasis es una enfermedad crónica, determinada genéticamente, caracterizada por sucesivos brotes de placas eritemato-descamativas en las superficies extensoras de la piel y cuero cabelludo, siendo la artropatía la única manifestación extracutánea
ETIOLOGÍA
Aunque la etiología es desconocida, existe una clara agregación familiar que apoya la implicación de factores genéticos. Además, factores ambientales conocidos como los traumatismos, diálisis, la hipocalcemia, algunas infecciones como la faringitis estreptocócica o el VIH, fármacos como el litio, betabloqueantes, antipalúdicos, interferón, interleucina
2, antidiabéticos orales, la retirada de corticoides sistémicos, algunos antiinflamatorios (indometacina) y, por último, las situaciones de estrés, pueden ser desencadenantes de la enfermedad. La psoriasis se produce como consecuencia de la interacción de 3 fenómenos que concurren simultáneamente: 1. aumento de la velocidad de crecimiento epidérmica; 2. proliferación venular postcapilar en las papilas dérmicas; y 3. Reacción inmune mediada por linfocitos T. La consecuencia de esta interacción es el fenómeno de la exudación cíclica papilar, que caracteriza microscópicamente a la enfermedad.
CLÍNICA
Es una enfermedad universal con una prevalencia de alrededor del 2% de la población europea, y con una incidencia de 150.000 casos nuevos/año. Puede debutar a cualquier edad, aunque cuando existen antecedentes familiares aparece de forma más temprana. Existen dos picos de incidencia: a los 16-22 años y a los 57-60 años, sin diferencia entre sexos, aunque las mujeres desarrollan antes la enfermedad. La lesión elemental es una placa eritematosa, de bordes bien definidos y superficie irregular descamativa, que tras el raspado metódico deja una superficie eritematosa con pequeños puntos sangrantes, “rocío hemorrágico de Auspitz”. Se disponen simétricamente en superficies extensoras y en cuero cabelludo. Hasta un tercio de los enfermos presentan lesiones en zonas previamente traumatizadas (fenómeno de Koebner o isomorfismo).
Se han descrito distintas formas clínicas:
1. Psoriasis vulgar: es la forma más frecuente. Las lesiones son crónicas y se localizan además de en los lugares ya mencionados, en la piel de abdomen y sacro.
2. Psoriasis en gotas: las lesiones son lenticulares o puntiformes, en tronco, generalmente en niños y adultos jóvenes, cursando en brotes. Tiene buen pronóstico y se ha relacionado con infección faringea estreptocócica previa.
3. Psoriasis invertida suele afectar grandes pliegues con predominio del eritema sobre la descamación
4. Psoriasis palmoplantar, con placas eritematodescamativas con fisuración.

5. Psoriasis ungueal: aparece hasta en un 35% en pies y un 50% en manos de enfermos con psoriasis. Es más frecuente en los casos de eritrodermia o artropatía asociada, aunque se puede presentar aislada. El espectro clínico va desde lesiones puntiformes en la lámina ungueal (pits) a onicodistrofia intensa con incluso pérdida ungueal, pasando por la característica “manchade aceite” o mancha marrón distal de la lámina.
6. Psoriasis pustulosa, que puede presentar varias formas:
6.1. Generalizada tipo von Zumbusch: Puede ser el debut de la enfermedad, asociada frecuentemente a artropatía, ser la evolución de una forma pustulosa localizada o estar desencadenada por fármacos (corticoides orales, litio, fenilbutazona), infecciones o embarazo (impétigo herpetiforme). De forma súbita, aparecen lesiones eritematosas confluentes que pueden evolucionar a eritrodermia sobre las que surgen brotes sucesivos de pústulas blanquecinas, anulares, agrupadas o dispersas. Asocia fiebre, malestar general, y leucocitosis con desviación a la izquierda. Suele respetar palmas y plantas, aunque es común la afectación ungueal.
6.2. Anular: puede ser generalizada o localizada. Son lesiones anulares con borde eritematoso y collarete descamativo interior con alguna pústula aislada; el centro es rosado con descamación grosera.
6.3. Localizadas, con 2 formas:
a) Pustulosis palmoplantar, de predominio en adultos y en el sexo femenino. No está clara su relación con la psoriasis. De hecho, sólo un 24% de estos enfermos tienen antecedentes de psoriasis;
b) Acrodermatitis continua de Hallopeau, caracterizada por pústulas en las falanges distales de los dedos (pulgares), que evolucionan dejando, al vaciarse, un superficie eritematosa brillante atrófica. Existe afectación ungüeal con onicodistrofia e incluso desaparición de la uña por daño de la matriz. Puede asociarse a otras lesiones a distancia de psoriasis vulgar o pustuloso e incluso generalizarse a una forma de von Zumbusch.
7. Eritrodermia psoriásica: eritrodermia exfoliativa no pruriginosa, con afectación del estado general. Las lesiones son de borde neto, con afectación ungueal importante. Suele aparecer en enfermos con psoriasis crónicas intensas.
8. Artropatía psoriásica: Esta artritis seronegativa afecta del 5 al 8% de los pacientes con psoriasis. En un 70% afecta de forma asimétrica a las pequeñas articulaciones de los dedos y en el resto aparece como poliartritis simétrica, espondilitis anquilosante y artritis mutilante. Ocurre en la cuarta y quinta década, y en un 50% de los casos de forma aguda. La psoriasis cutánea precede a la artropatía y la intensidad de la clínica articular es independiente de la afectación cutánea, aunque más grave en las formas pustulosas. Es muy frecuente la afectación ungueal, incluso como única manifestación cutánea.
HISTOLOGÍA

El patrón típico histopatológico de la psoriasis es el de la exudación cíclica papilar, por la que los neutrófilos y linfocitos salen de los capilares dilatados de las puntas de las papilas y permean las capas basales suprapapilares, produciendo atrofia suprapapilar, pústulas espongiformes de Kogoj, microabscesos de Pautrier, acantosis, papilomatosis regular y paraqueratosis confluente. El hallazgo de exocitosis neutrofílica en las puntas de las papilas o la presencia de capilares dilatados en las mismas, son dos claves importantes en el diagnóstico de esta enfermedad. En las formas especiales, como en la Psoriasis invertida, y en la eritrodérmica, la paraqueratosis es escasa, puesto que son formas poco descamativas, pero siempre se puede identificar la exocitosis neutrofílica suprapapilar. Lo mismo ocurre en otras formas como en la ungueal o en la gutata, donde la paraqueratosis es muy focal. En las formas pustulosas, a veces es necesario el uso de PAS para descartar una candidosis o de inmunofluorescencia directa para descartar un pénfigo IgA.
TRATAMIENTO
La elección del tratamiento se realiza en función de la extensión y localización de las lesiones de psoriasis, así como de los tratamientos previos y la edad del paciente. Entre los tratamientos tópicos destacan los corticoides, en cura abierta u oclusiva y los derivados de la vitamina D (tacalcitol y calcipotriol). Estos fármacos se utilizan conjuntamente con emolientes y breas o alquitranes, así como con queratolíticos como el ácido salicílico. La helioterapia, y su variante PUVA (psoraleno + radiación UVA) o radiación UVB de banda estrecha, es un tratamiento muy empleado, especialmente en las psoriasis eruptivas o en gotas.
Dentro de los tratamientos sistémicos, destaca el empleo de:
1) Retinoides (etretinato y acitretino) muy útiles en las formas pustulosas, eritrodérmicas y en la artropatía, solos o combinados con fototerapia (RePUVA);
2) Metotrexato, un potente antiproliferativo y antiinflamatorio, muy eficaz en las formas graves y, sobre todo, en la psoriasis con artropatía, aunque sus efectos secundarios agudos (afectación de médula ósea o fibrosis hepática) lo limitan; 3) Ciclosporina, agente inmunosupresor efectivo en un 70% de enfermos con psoriasis vulgar crónico intenso, aunque también se utiliza en casos de artropatía, psoriasis pustuloso generalizado o eritrodermia psoriásica.

ERITRODERMIAS DESCAMATIVAS
Las eritrodermias constiuyen un cuadro clínico multietiológico, a veces grave, que se presentan en cualquier momento de la vida. Se define como eritrodermia, cualquier estado inflamatorio de la piel que abarca a casi toda la totalidad del tegumento cutáneo.
Las eritrodermias infantiles son poco frecuentes con excepción de las eritrodermias atópicas y las eritrodermias hereditarias, ya que la mayoría suelen presentarse en sujetos de más de 45 años.
Las eritrodermias infantiles se pueden clasificar de varias maneras atendiendo a factores etiológicos, evolución (agudas o crónicas), patogenia (adquiridas o hereditarias), caracter primario o secundario de la enfermedad, etc.
Algunos autores anglosajones consideran las eritrodermias con variantes de los linfomas de células T, y por otra parte, no existe una frontera delimitada entre eritrodermia y eritema y muchas veces una eritrodermia es clasificada como eritema o vicerversa
Las eritrodermias hereditarias son las más frecuentes y se clasifican en tres grandes grupos:
Ictiosis o eritrodermias ictiosiformes asociados a trastornos de la queratinización
Eritrodermias asociadas a inmunodeficiencias
Eritrodermias asociadas a errores del metabolismo
Las eritrodermias también reciben el nombre de dermatitis exfoliativas. La eritrodermia exfoliativa primaria ha sido también descrita como el síndrome del hombre rojo (sindrome de l'homme rouge)

ICTIOSIS
Concepto
Las ictiosis son un grupo de enfermedades que producen escamas visibles en toda o gran parte de la superfi cie de la piel. Su nombre deriva de la palabra griega que signifi ca pez. Las ictiosis pueden ser debidas a anomalías hereditarias de la queratinización o a trastornos adquiridos, entre los que destacan las neoplasias. Las primeras son mucho más importantes en la edad pediátrica.
Existen diversas clasifi caciones de las ictiosis. la propuesta por Traupe, que divide las ictiosis por anomalías congénitas de la queratinización en “vulgares”, cuando las manifestaciones clínicas no son evidentes en el momento del nacimiento y “congénitas”, cuando las manifestaciones son ya visibles en el recién nacido. A su vez, estos dos grupos se subdividen según existan o no anomalías extracutáneas asociadas. Los trastornos metabólicos conocidos residen fundamentalmente en las proteínas estructurales epidérmicas o en los lípidos.
Clínica
El dato clínico común de las ictiosis hereditarias es la observación de un proceso descamativo, generalizado o al menos extenso, presente desde el nacimiento o que se inicia en los primeros meses de vida, prácticamente siempre antes de los dos años. En muchos casos existirán antecedentes familiares de la enfermedad o consanguinidad.
A partir de estos hallazgos compartidos por los diferentes tipos de ictiosis, cada uno de ellos agrega peculiaridades, tanto en sus manifestaciones cutáneas como en la posible participación sistémica. A continuación destacamos las principales características clínicas de las ictiosis más comunes. Ictiosis vulgar
Las lesiones suelen comenzar a ser detectables varios meses después del nacimiento, a veces tras el primer año de vida. Consisten en escamas fi nas y blanquecinas, que predominan en el tronco y la cara de extensión de los miembros.
La cara, el cuello y las flexuras corporales suelen estar respetadas. Muy a menudo se acompaña de hiperqueratosis folicular en las zonas de extensión de las extremidades, aumento de los pliegues e hiperqueratosis palmo-plantar. Suele mejorar en el verano y empeorar en el invierno.
La intensidad de la descamación es muy variable y en los casos leves muchas veces el diagnóstico es consecuencia de un hallazgo en una exploración rutinaria o por otro motivo. La ictiosis vulgar no produce manifestaciones extracutáneas, pero es una asociación frecuente en pacientes con dermatitis atópica.
Ictiosis laminares
Actualmente se admite que es un conjunto de enfermedades con distintas bases bioquímicas y manifestaciones clínicas. El niño puede tener inicialmente el aspecto de un bebé colodión . En las fases evolutivas iniciales suele haber un eritema como base del cuadro ictiosiforme , que en algunos casos

tiende a atenuarse o se hace imperceptible con el tiempo. La descamación es generalizada, con tamaño de escamas muy variable. El ectropión, el eclabión, la queratodermia palmoplantar y la alopecia son hallazgos frecuentes.
Las principales asociaciones descritas son talla baja y retraso mental.Eritrodermia ictiosiforme congénita ampollosa Aunque con menos frecuencia que las ictiosis laminares, puede presentarse como un bebé colodión. La mayoría de las veces el neonato está eritrodérmico y se observan áreas erosivas y denudadas; la observación de ampollas íntegras es posible, pero rara. La eritrodermia tiende a persistir, aunque se puede atenuar y las erosiones y las ampollas van disminuyendo con el tiempo, a la vez que aumenta la hiperqueratosis, sobre todo en las zonas flexurales, donde puede hacerse verrugosa. En conjunto hay una tendencia a la mejoría con la edad.
Eritrodermia ictiosiforme congénita ampollosa
Aunque con menos frecuencia que las ictiosis laminares, puede presentarse como un bebé colodión. La mayoría de las veces el neonato está eritrodérmico y se observan áreas erosivas y denudadas; la observación de ampollas íntegras es posible, pero rara. La eritrodermia tiende a persistir, aunque se puede atenuar y las erosiones y las ampollas van disminuyendo con el tiempo, a la vez que aumenta la hiperqueratosis, sobre todo en las zonas flexurales donde puede hacerse verrugosa. En conjunto hay una tendencia a la mejoría con la edad.
Bebé colodión
Es una forma de presentación de diversos tipos de ictiosis, sobre todo de las ictiosis laminares. Algunos casos pueden evolucionar hacia la curación. Los niños nacen con una piel eritematosa y con el aspecto de estar envueltos en celofán . Esta envoltura superficial tiende a agrietarse y más tarde a desprenderse en grandes láminas, tras lo cual se instauran las características clínicas del proceso de base. Es habitual que exista ectropión y eclabión y puede causar dificultad respiratoria por constricción torácica y abdominal.
Tratamiento
En la actualidad el tratamiento de las ictiosis es sobre todo sintomático, aunque cabe esperar que los avances en el conocimiento de su etiopatogenia lleven a permitir solucionar el defecto genético o sustituir las proteína defi citarias.

Medidas generales
Los pacientes con ictiosis, sobre todo los neonatos con formas graves, se benefician de un ambiente húmedo. En los recién nacidos con bebé colodión y otros tipos de ictiosis eritrodérmica es esencial controlar el balance hidroelectrolítico y la posibilidad de infecciones con puerta de entrada cutánea.
La higiene es esencial a lo largo de la vida de estos pacientes para evitar las sobreinfección y favorecer la eliminación de las escamas. Un gel de pH ácido es el producto más recomendable con este fi n.
Tratamientos tópicos
Las sustancias más empleadas son los emolientes y los queratolíticos. Muchas veces los productos de uso tópico comparten ambas propiedades. Tal es el caso de la urea, los alfahidroxiácidos, en especial el ácido láctico y el propilenglicol. Los aceites vegetales y minerales y la vaselina y la parafi na tienen una acción predominante como emoliente. El ácido salicílico es un excelente queratolítico, pero su uso en las ictiosis infantiles es poco aconsejable por el riesgo de absorción y toxicidad.
Los retinoides tópicos (ácido retinoico, retinal, isotretinoína, adapaleno, tazaroteno, etc.) tienen acción queratolítica y queratoplástica. Su principal limitación es debida a su acción irritante y elevado coste.
En algunos estudios se ha demostrado la eficacia de los derivados de la vitamina D, como el calcipotriol y el tacalcitol.
Tratamientos sistémicos
En la actualidad los retinoides orales son las únicas medicaciones utilizables en la mayoría de las ictiosis graves, en las que no resulta posible el control con tratamientos tópicos. La isotretinoína y el acitretino son los productos de este grupo comercializados en España.
Sus efectos adversos más constantes son la sequedad de la piel y las mucosas y debe controlarse el posible desarrollo de hiperlipemia, toxicidad hepática, miopatía, hipertensión intracraneal y calcifi caciones. En adolescentes y adultos debe recordarse que son medicaciones teratógenas

ALBINISMO
La palabra “albinismo” se refiere a un grupo de condiciones heredadas. Las personas con albinismo tienen muy poco ó quizás no tengan pigmento en sus ojos, piel ó pelo. Han heredado genes que no producen las cantidades correctas de un pigmento llamado melanina. El albinismo afecta a personas de todas las razas. La mayoría de niños con albinismo tienen padres con pelo y ojos normales típicos de su raza. Muchas veces las personas no reconocen que tienen albinismo.
Un mito común del albinismo es que los afectados tienen ojos rojos. En realidad, hay diferentes tipos de albinismo, y la cantidad de pigmento en los ojos varía. Aunque hay algunos individuos con ojos rojizos o violetas, la mayoría tienen ojos azules. Algunos tienen ojos castaños ó cafés.
Problemas Visuales
Las personas con albinismo siempre tienen problemas de visión y puede que tengan baja visión. Muchos son legalmente ciegos pero la mayoría usan su visión para leer y no usan el sistema Braille. Algunos tienen una visión lo bastante buena hasta para manejar un automóvil. Los problemas de visión resultan de desarrollos anormales de la retina y patrones anormales de conexiones de nervios entre el ojo y el cerebro. Son estos problemas visuales los que definen el diagnóstico del albinismo. Por eso, el principal examen para ver si uno tiene albinismo es simplemente un examen de la vista.
Tipos de Albinismo
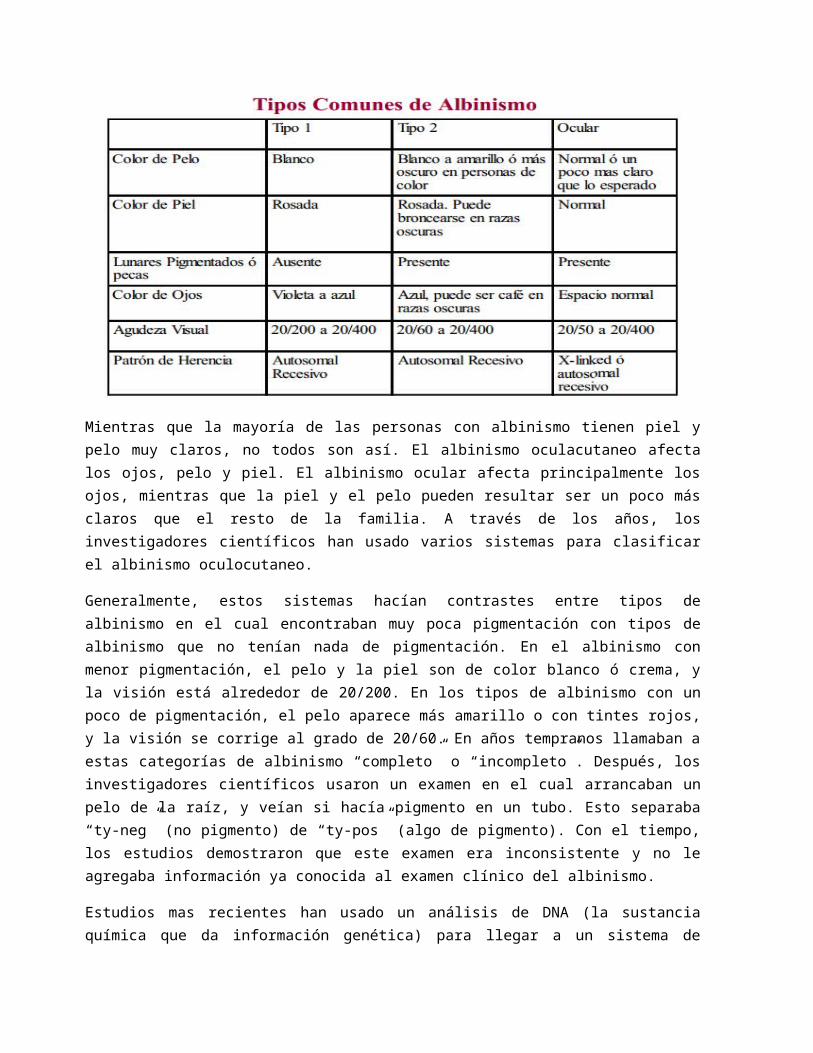
Mientras que la mayoría de las personas con albinismo tienen piel y pelo muy claros, no todos son así. El albinismo oculacutaneo afecta los ojos, pelo y piel. El albinismo ocular afecta principalmente los ojos, mientras que la piel y el pelo pueden resultar ser un poco más claros que el resto de la familia. A través de los años, los investigadores científicos han usado varios sistemas para clasificar el albinismo oculocutaneo.

Generalmente, estos sistemas hacían contrastes entre tipos de albinismo en el cual encontraban muy poca pigmentación con tipos de albinismo que no tenían nada de pigmentación. En el albinismo con menor pigmentación, el pelo y la piel son de color blanco ó crema, y la visión está alrededor de 20/200. En los tipos de albinismo con un poco de pigmentación, el pelo aparece más amarillo o con tintes rojos, y la visión se corrige al grado de 20/60. En años tempranos llamaban a estas categorías de albinismo “completo” o “incompleto”. Después, los investigadores científicos usaron un examen en el cual arrancaban un pelo de la raíz, y veían si hacía pigmento en un tubo. Esto separaba “ty-neg” (no pigmento) de “ty-pos” (algo de pigmento). Con el tiempo, los estudios demostraron que este examen era inconsistente y no le agregaba información ya conocida al examen clínico del albinismo.
Estudios mas recientes han usado un análisis de DNA (la sustancia química que da información genética) para llegar a un sistema de clasificación más confiable. El Albinismo Tipo 1 (también conocido por albinismo relacionado con la tiroxinaza) es el tipo que no tiene casi nada de pigmentación. El tipo 1 resulta de un defecto genético en una enzima llamada tiroxinaza (tyrosinase). Esta enzima le ayuda al cuerpo a cambiar el aminoácido tiroxina a pigmento. (Un aminoácido es un componente de proteína y viene de la proteína de la dieta). El Albinismo Tipo 2, el tipo con un poco de pigmento, resulta ser de un defecto en un gen diferente llamado el gen “P”.
Investigadores científicos han identificado a muchos otros genes que causan tipos de albinismo. En el síndrome Hermansky-Pudlack, un tipo de albinismo, pueden existir problemas sanguíneos, junto con enfermedades de los pulmones y de la digestión. Hermansky-Pudlak es una forma menos común del albinismo, pero debería ser motivo de sospecha si su niño muestra moretones ó sangra anormalmente.
Los Genes del Albinismo
Para casi todos los tipos de albinismo, los dos padres tienen que tener un gen para albinismo para tener un hijo con albinismo. Como el cuerpo tiene dos pares enteros de genes, una persona puede que se vea normal, pero puede contener los genes para el albinismo. Si una persona tiene un par de genes normales y un par de genes con albinismo, el ó ella tienen la información genética suficiente para hacer pigmento normal. El gen del albinismo es “recesivo” y no resultará en una persona con albinismo a menos que los dos pares de genes contengan albinismo y no hay copia del gen que tiene pigmento normal.
Cuando los dos padres tienen el gen pero ninguno de los dos tienen albinismo, existe una probabilidad de 25% en cada embarazo de que el bebé nazca con albinismo. Este tipo de herencia se llama “herencia recesiva autosomal” (autosomal recessive inheritance.)
Cada padre de un niño con albinismo oculacutaneo tiene que tener el gen. El padre y la madre juntos tienen que tener el gen para albinismo. Para parejas que no han tenido un hijo con albinismo, no hay un examen fácil para determinar si una persona tiene el gen del albinismo ó no. Investigadores científicos han analizado el DNA de personas con albinismo y han encontrado cambios que causan albinismo, pero estos cambios no son siempre en el mismo lugar, ni para un cierto tipo de albinismo. Por eso, los exámenes del gen pueden ser inconclusos.

HISTIOCITOSISConcepto
Entendemos por histiocitosis un grupo heterogéneo de enfermedades de causa desconocida que se caracterizan por la proliferación de células del sistema mononuclear fagocítico (SMF) (monocitos, macrófagos, células dendríticas) en diferentes órganos y sistemas. Dicha proliferación puede ser localizada (lesión afectando únicamente piel o una lesión aislada en hueso) o bien generalizada, afectando varios órganos o sistemas. Son enfermedades poco frecuentes, de predominio en la edad infantil, con gravedad muy diversa y con características biológicas limítrofes entre el campo de la Hematología, la Inmunología, la Oncología y la Dermatología.
En el término de histiocitosis se excluyen las enfermedades en las se produce secundariamente la proliferación histiocitaria, tales como la reacción granulomatosa en las infecciones crónicas, la enfermedad del injerto contra el huésped, el síndrome linfoproliferativo ligado al cromosoma X o las lipoidosis hereditarias, entre otras.
Clasificación
Esta diversidad clínica da lugar a múltiples cuadros, la mayoría de ellos con expresión cutánea precoz que permite orientar el diagnóstico, por lo que en la práctica con frecuencia es el dermatólogo quien las diagnostica, aunque el pediatra dirige la terapéutica, particular mente en los casos de compromiso sistémico.
Clásicamente la Sociedad Internacional del Histiocito (Histiocyte Society) las clasifica en tres grandes grupos:

1. Histiocitosis tipo I o histiocitosis de células de Langerhans. Enfermedad antiguamente conocida como histiocitosis X, y que ha recibido diversos epónimos: enfermedad de Abt-Letterer-Siwe, enfermedad de Hand- Schuller-Christian, Hashimoto-Pritzker, que refl ejan la variabilidad de su presentación.
Sin embargo, en la actualidad se prefi ere la denominación de histiocitosis de células de Langerhans y su catalogación en formas localizadas o sistémicas.
2. Histiocitosis tipo II o histiocitosis de células no Langerhans. Incluyen un grupo muy variado de enfermedades por proliferación de histiocitos cuyo fenotipo es diferente de la célula de Langerhans. Clínicamente se dividen en 3 grupos, las que afectan predominantemente la piel, otras que afectando la piel presentan una afectación sistémica predominante, y el tercer grupo de enfermedades que son principalmente extracutáneas. El prototipo del primer grupo es el xantogranuloma juvenil, y del tercer grupo la linfohistiocitosis hemofagocítica.
3. Histiocitosis tipo III o malignas. Más recientemente se han reclasificado según la célula que los produce
Histopatología
En la histiocitosis de células de Langerhans se halla un infi ltrado denso de células de Langerhans (núcleo reniforme, indentado, característico, y citoplasma denso). Dicho infi ltrado es epidermotropo en la piel o invasivo en otros órganos y sistemas, con inmunohistoquímica positiva para la proteína S-100 y el anticuerpo monoclonal CD1a. La microscopía electrónica permite observar en el citoplasma de dichas células los típicos cuerpos en raqueta o gránulos de Birbeck. La histología no distingue las formas agresivas de las localizadas, ya que el patrón es idéntico en ambas y por tanto sólo tiene un carácter diagnóstico pero no pronóstico.
En las xantohistiocitosis se halla un infiltrado de células espumosas y células gigantes multinucleadas de Touton, características de los xantomas. En realidad se trata de histiocitos cargados de grasa en diferentes estadíos de evolución. En fases iniciales de estos procesos se aprecia mayor componente inflamatorio (linfocitos, mastocitos). Las tinciones para grasas (oil-red, sudán) son positivas. En el grupo de histiocitosis no Langerhans autoinvolutivas el infiltrado es histiocitario, monomorfo, no invasivo y sin xantomización. Como en ambos grupos de cuadros clínicos probablemente la célula proliferante sea el macrófago se presentan positividades frente a anticuerpos monoclonales propios de dichas células: CD68, factor XIIIa, CD34 o MAC 387. En las formas autoin volutivas pueden hallarse los denominados cuerpos vermiformes en el citoplasma de estas células al microscopio electrónico, como signo, en general, de buen pronóstico.
En el síndrome hemofagocítico se observa la presencia de histiocitos con citoplasma vacuolado y el fenómeno característico de autocitofagia de hematíes en su interior . En la enfermedad de Rosai Dorfman se aprecia una proliferación de histiocitos benignos con el fenómeno característico de emperipolesis (englobamiento de linfocitos en el citoplasma). Dentro del grupo de las histiocitosis malignas, en el raro sarcoma histiocítico se halla un infi ltrado de macrófagos atípicos de gran tamaño con abundantes mitosis y áreas de necrosis. Estas células presentan una tinción difusa para esterasa. Afecta piel, ganglios, médula ósea y todos los demás órganos del SMF (hígado, bazo,…). Antiguamente se había confundido con el linfoma anaplásico de células grande CD30 positi vo, entidad con característica traslocación entre los cromosomas 2 y 5 (gen NPM-ALK), que es en realidad un linfoma no Hodgkin de alto grado y no una histiocitosis.
Tratamiento
El tratamiento de las Histiocitosis de células de Langerhans está protocolizado internacionalmente por la Sociedad Internacional del Histiocito Las formas cutáneas no invasivas precisan un control evolutivo exhaustivo para confirmar su autoinvolución. El seguimiento con conducta expectante se aplica a la mayoría de xantohistiocitosis, aunque es preciso excluir el compromiso sistémico o una posible asociación maligna. Dentro de las histiocitosis malignas los linfomas histiocitarios son enfermedades muy raras. Se tratan con poliquimioterapia siguiendo esquemas similares a los del linfoma no Hodgkin de alto grado.

EPIDERMÓLISIS AMPOLLOSA.
Definición
La epidermólisis ampollosa (EA) es un grupo heterogéneo de enfermedades hereditarias, formadoras de ampollas y vesículas hemorrágicas en piel y mucosas, con predominio en los sitios de presión o roce, y que aparecen de manera espontánea o como respuesta a traumatismos leves.
La enfermedad puede aparecer poco después del nacimiento o en edades más tardía Hasta el momento, se han descrito más de 26 subtipos clínicos con manifestaciones variables, desde leves alteraciones hasta procesos que llegan a ser fatales en corto tiempo. La clasificación más utilizada es la de tipo histológico y según la localización de las ampollas, se describen tres entidades: EA simple o intraepidérmica, la EA de unión y la EA distrófica o intradérmica. Éstas, son enfermedades que debe conocer el médico de primer contacto, pues el niño debe ser enviado lo más pronto posible al dermatólogo para el diagnóstico preciso y tratamiento oportunos.
Etiología
Diversos estudios han mostrado su origen genético. El subtipo simple se asocia a mutaciones en los genes que sintetizan las queratinas basales 5 y 14, localizadas en los cromosomas 14q y 12q respectivamente. Por otra parte, la tinción con anticuerpos en la EA de unión, ha revelado la falta de laminina 5 (también llamada kalinina o niceina), que a su vez se origina por la mutación en uno de los genes que codifican sus cadenas: α3 (LAMA3), β3 (LAMB3) y γ2 (LAMC2) y por último, en la EA distrófica se han encontrado mutaciones en el gen de la colágena tipo VII (COL7A1), localizado en el cromosoma 3p21 aunque también se describe una actividad incrementada de la colagenasa en cultivo de fibroblastos, la cual se conoce degrada la colágena.

Diagnóstico
El diagnóstico de exclusión se sospecha por la presencia de ampollas que aparecen en edad temprana, y que son secundarias a roce mínimo. Es indispensable realizar una historia clínica completa y detallada, con énfasis en: antecedentes familiares de enfermedades ampollosas, de abortos y óbitos, traumatismos obstétricos, la ingesta de medicamentos y de padecimientos infecciosos.
Es necesaria la toma de biopsia de piel, mediante la cual podremos localizar histológicamente el sitio de las ampollas; este procedimiento deberá incluir la piel circundante a la ampolla, teñirse con hematoxilina y eosina, y observarse con microscopio de luz. Sin embargo, el diagnóstico definitivo se realiza con microscopia electrónica que muestra la ultraestructura de la piel afectada.
El mapeo de antígenos por inmunofluorescencia directa es un método rápido para la clasificación de la EA y se basa en la incubación de la piel afectada con anticuerpos específicos y la posterior identificación del sitio de formación de la ampolla. Los anticuerpos contra membrana basal se utilizan para crear reacciones en la unión dermoepidérmica y así corroborar el diagnóstico. Esta técnica ayuda a diferenciar la EA de otras entidades como: el lupus eritematoso ampolloso, el penfigoide del recién nacido y otras.
Desde el punto de vista genético es muy importante determinar el patrón de transmisión, ya que la clasificación y pronóstico también dependen de si la herencia es autosómica dominante o recesiva.
En las formas simples e intraepidérmicas, se registra predominio del patrón autosómico dominante como en: las variantes de Weber-Cockayne, de Köebner, y de Dowling Meara; pero se puede encontrar también la transmisión de tipo recesivo como en la EA asociada a distrofia muscular6,9 (Figura 3).
En el grupo de EA de unión predomina la herencia autosómica recesiva: las variantes de Herlitz (gravis) y Mitis. En las de tipo distrófica e intradérmica existe tanto el patrón autosómico recesivo como el dominante, en el primero se encuentra la variante Hallopeau-Siemens, en el segundo encontramos las de Cockayne-Touraine, Pasini y la Transitoria del recién nacido
Tratamiento
No hay tratamiento curativo definitivo para estos pacientes, sin embargo, el tratamiento se debe enfocar a la prevención de traumas mecánicos y la curación de las ampollas.
Es necesario que el paciente presente una buena higiene corporal y dental. Las ampollas que se encuentran tensas o en sitios que comprometen el movimiento se pueden drenar, siempre y cuando no se desepitelicen para así proteger el área lesionada La estabilización del paciente debe de incluir aporte adecuado de líquidos y electrólitos, una dieta calórica y semilíquida, y el tratamiento y prevención de infecciones secundarias.
Son indispensables los vendajes protectores con vaselina u óxido de zinc y el enfriamiento de la piel, pueden llegar a ser necesarios los apósitos sustitutos de piel como el “Apligraft”, el “Epifast” y el “Ortec”. Para reducir la fricción en la piel puede aplicarse glutaraldehído tópico al 5% cada tercer día.

En algunos casos la difenilhidantoína a dosis de 3 mg/kg/día por vía oral y la ingestión de vitamina E a dosis altas disminuyen la formación de vesículas y permiten la rápida epitelización y cicatrización de las lesiones.
Es importante mencionar que el uso de corticosteroides está contraindicado en estos pacientes, al contrario de lo que ocurre en otras enfermedades ampollosas. Algunos de los medicamentos utilizados pero que no aportan beneficio, son los antimaláricos, retinoides, tetraciclinas, nitrato de plata y la ciclosporina. En caso de estenosis esofágica es necesario realizar dilataciones frecuentes, llegando a requerirse gastrostomía o transposición de colon.
En caso de fusión de los dedos es necesario realizar cirugía plástica para liberarlos y en las erosiones crónicas pueden ser útiles los injertos. Es importante estar alerta a la aparición temprana de carcinomas epidermoides cutáneos y realizar excisión de los tumores.
El tratamiento debe ser integral y multidisciplinario con valoración oftalmológica, urológica y gastroenterológica e incluir consejo genético y psicológico a la familia debido a la posibilidad de recurrencia de la enfermedad en otros de sus hijos.

PENFIGOIDE AMPOLLOSO
Definicion
El penfigoide ampolloso (PA) es una enfermedad autoinmune frecuentemente observada en pacientes de edad avanzada. Su diagnóstico se establece con la combinación de características clínicas, histológicas e inmunopatológicas. El PA se caracteriza por la presencia de ampollas tensas asentadas en piel normal o eritematosa que tienen predilección por las extremidades y frecuentemente la enfermedad tiende a generalizarse. En el estudio histológico se observa una ampolla subepidérmica, con predominio de eosinófilos. Los estudios de inmunofluorescencia (IF) directa de piel perilesional muestran depósitos lineales de IgG y/o C3 en la zona de la membrana basal (ZMB).
El término “penfigoide” es utilizado para un grupo de enfermedades que comparten características clínicas de vesículas y ampollas, que en la histopatología muestran una ampolla subepidérmica, con infiltrado inflamatorio rico en eosinófilos y en menor proporción neutrófilos, así como anticuerpos IgG tanto circulantes como unidos a piel, contra proteínas específicas de la zona de la membrana basal (ZMB). Hay tres tipos principales de penfigoide:
1) Penfigoide ampolloso (primariamente enfermedad cutánea).
2) Penfigoide de membranas mucosas (primariamente enfermedad de mucosas).
3) Penfigoide (herpes) gestacional (primariamente una enfermedad cutánea en mujeres embarazadas.
La enfermedad se considera una enfermedad ampollosa autoinmune. La base de la autoinmunidad del PA fue sugerida por Jordon et al (1967) con la identificación de los depósitos de IgG y componentes del complemento en la piel de los pacientes con PA El penfigoide ampolloso (PA) es la enfermedad ampollosa autoinmune más frecuente en el mundo occidental.
Etiologia
Las lesiones típicas son ampollas tensas y grandes, las cuales tienen predilección por las extremidades (cara interna de muslos y áreas flexurales de brazos y piernas), área inguinal, axilas, abdomen y cuello. Se involucra de forma menos común cara y piel cabelluda. El líquido de la ampolla es comúnmente claro, pero ocasionalmente puede ser hemorrágico.
Estas ampollas frecuentemente permanecen intactas y no se deforman con la presión, su ruptura conduce a la formación de erosiones que evolucionan a costras yocasionalmente pueden ser invadidas por bacterias patógenas. La dermatosis puede ser leve o intensamente pruriginosa o incluso ser asintomática. Pueden presentarse lesiones en mucosas la frecuencia varía del 8% al 50% y es la mucosa oral la más afectada. Dentro de otras membranas mucosas involucradas se incluyen faringe, uretra y conjuntiva; las lesiones tienden a ser leves y transitorias y son generalmente observadas en pacientes con enfermedad cutánea extensa.1

Existen múltiples variantes clínicas de la enfermedad: penfigoide dishidrosiforme, penfigoide vesicular, penfigoide nodular, penfigoide vulvar localizado de la infancia, penfigoide localizado pretibial y penfigoide vegetante.
Las características histológicas de la biopsia de una ampolla reciente son las mismas para el penfigoide ampolloso o gestacional e incluyen la presencia de ampolla subepidérmica con infiltración moderada a densa de eosinófilos y otras células inflamatorias dentro de la cavidad de la ampolla. No hay necrosis de la epidermis subyacente, excepto de la porción media del techo de la ampolla, donde la ampolla tiene más tiempo de aparición. En algunos casos, los eosinófilos pueden ser observados en una dirección en aproximación con la ZMB. Los linfocitos también se observan frecuentemente y, en casos raros, predominan los neutrófilos. El infiltrado inflamatorio usualmente es confinado a la dermis papilar y a la porción superficial de la dermis reticular. Puede haber también edema en dermis papilar. Los diagnósticos diferenciales histológicos incluyen casos de dermatitis herpetiforme, dermatosis lineal por IgA cuando presentan el número incrementado de esosinófilos. Contrariamente, de forma ocasional existen casos de PA que presentan un número incrementado de neutrófilos. En estas situaciones, la inmunofluorescencia (IF) directa es probablemente la forma más fácil de distinguirlos.8
Diagnostico
Los procedimientos diagnósticos convencionales para el PA, como la (IF) directa e indirecta, e inmunoblot(IB); se han complementado con el método sensible y específico de ELISA.
La (IF) es la investigación más notable para realizar el diagnóstico; la IF directa muestra depósitos de autoanticuerpos (IgG) en la unión dermo-epidérmica y la IF indirecta muestra autoanticuerpos circulantes directamente dirigidos contra las proteínas de la membrana basal que se localizan en el lado epidérmico de la piel en la técnica de SALT split.3
Una biopsia perilesional para la IF directa muestra depósitos lineales de IgG y/o C3 en la ZMB en cerca del 100% de los casos; se pueden observar adicionalmente otras inmunoglobulinas (IgM, IgA, IgD, o IgE) en la misma distribución.4-6,8 La IgG es la inmunoglobulina más comúnmente observada en un 60% a 90% de los casos en donde la IgG4 es la subclase predominante.
La IF indirecta puede ser positiva en más del 90% de los pacientes con PA. Los autoanticuerpos reaccionan con 2 componentes de los hemidesmosomas del epitelio estratificado: el antígeno del PA 230 (BP230) y el antígeno 180 (BP180). El antígeno del PA 230 ha sido implicado en la organización de los filamentos de queratina y es localizado en la placa de los hemidesmosomas de forma intracelular. El antígeno del PA 180, está involucrado en el anclaje del epitelio estratificado a la membrana basal subyacente, es una proteína transmembrana tipo II
Tratamiento
El PA es una enfermedad que resulta de una respuesta autoinmune anormal (autoanticuerpos) con una respuesta inflamatoria prominente (infiltrado celular). Las terapias del PA tienen como finalidad suprimir la inflamación y/o respuesta inmune. Antes de elegir la terapia, se deben considerar las variables relacionadas con la enfermedad (extensión del involucro y los síntomas) y con el paciente

(edad; otras enfermedades como diabetes, hipertensión). El objetivo del tratamiento es la cicatrización de las lesiones existentes y la prevención de nuevas lesiones. La aparición de una lesión no requiere del incremento de la dosis del tratamiento.
La mayoría de los pacientes con PA generalizado requieren de terapia sistémica. Los corticosteroides son los agentes sistémicos más comúnmente utilizados.
Los corticosteroides tienen tanto efectos antiinflamatorios como inmunosupresores que resultan en una disminución de los linfocitos circulantes, eosinófilos, monocitos y basófilos. Los neutrófilos circulantes aumentan porque existe una liberación incrementada de la médula ósea, una disminución en su remoción de la circulación y un aumento en su acumulación de la pared de los vasos.
La prednisona es el agente más frecuentemente utilizado y es suficiente como monoterapia en la mayoría de los casos. La dosis es de 0.5-1 mg/kg/día dependiendo de la severidad de la enfermedad. Se han utilizado los pulsos de corticosteroides con metilprednisolona intravenosa 1 mg diario por 3 días consecutivos para el control inicial de una enfermedad severa.
Se debe pensar en la utilización de otros agentes inmunosupresores sólo si la dosis de los corticosteroides no se puede reducir a niveles aceptables, presencia de efectos adversos y en los pacientes en quienes su enfermedad no responde completamente con la terapia con corticosteroides.
La azatioprina es el principal agente establecido, seguido del metotrexate. Otros inmunosupresores también frecuentemente utilizados son la ciclofosfamida, ciclosporina y micofenolato de mofetilo.

URTICARIA
DEFINICION
La urticaria constituye uno de los motivos de consulta más frecuentes en la práctica médica, la presentación aguda es extremadamente común, afectando posiblemente al 10-20% de la población en algún momento de su vida.
La urticaria se caracteriza por la aparición de elementos cutáneos, sobreelevados, de aspecto eritematoso y es característico la presencia de prurito. El angioedema puede considerarse como la misma manifestación que la urticaria pero su localización es distinta puesto que afecta la dermis profunda y al tejido subcutáneo, es precisamente su localización, la que hace que el angioedema no suela presentar el síntoma prurito, sin embargo suele acompañarse de una sensación de opresión.
El angioedema puede presentarse aislado o asociado a urticaria, aproximadamente en el 50% de los casos suelen coexistir, en el 40% suele presentarse la urticaria como fenómeno único y en el 10% de los casos angioedema aislado. Tanto la urticaria como el angioedema son entidades clínicas cuya etiología es variada y sus mecanismos etiopatogénicos también suelen ser múltiples. Suelen presentarse como episodios aislados de corta duración y la localización dependerá del tipo de manifestación, así como la urticaria puede afectar a toda la superficie cutánea el angioedema afecta al tejido subcutáneo siendo la región periorbitaria, los labios, lengua, genitales y zonas distales de extremidades, las más afectadas; a diferencia de la urticaria no suele ser pruriginoso.
La urticaria y el angioedema pueden presentarse como única manifestación de una reacción alérgica; puede que formen parte de una reacción generalizada como puede ser una reacción anafiláctica con shock y manifestaciones respiratorias o incluso como si se tratase de una manifestación más dentro de un complejo proceso sistémico. Inmunopatogenia
Desde el punto de vista inmunopatológico la urticaria y el angioedema son consecuencia de la liberación por parte de las células (mastocitos y basófilos) de mediadores preformados, siendo la histamina el más importante y sería el responsable de la expresión cutánea. Otros factores también son liberados a partir de los mastocitos, por ejemplo el Factor Activador de Plaquetas (PAF); su acción directa sobre las plaquetas hace que éstas liberen serotonina que podría ser uno de los responsables de la urticaria crónica. Otro mediador liberado de los mastocitos seria el Factor Quimiotáctico de los Eosinófilos el cual sería el responsable de la migración de estas células hacia el foco inflamatorio.
Un segundo grupo de mediadores van a ser sintetizados por las células o tejidos circundantes, por acción directa de los mediadores primarios; estos mediadores secundarios son de aparición más tardía y su acción se prolonga en el tiempo, son metabolitos del ácid araquidónico, leucotrienos C y D.
La activación del complemento da lugar a la producción de anafilatoxinas (C3a, C4a, C5a) y su acción directa sobre la superficie celular es capaz de liberar histamina. El factor C5a es el más activo sobre la permeabilidad vascular, y dado que su inhibición por parte del facto inhibidor de anafilotoxinas se produce más tardíamente, le hace actuar no sólo como favorecedor de la permeabilidad sino que

también como factor quimiotático de la células (eosinófilos, neutrófilos ....) que aparecen en el foco inflamatorio.
Etiología
Las causas más frecuentes de urticaria/ angioedema entre la población general son en primer lugar los medicamentos y con menor incidencia los alimentos, picaduras de insectos y determinados antígenos tanto inhalados, ingeridos o de contacto. Sin embargo, en el ámbito pediátrico son los alimentos los principales responsables de esta patología seguida de los fármacos y el resto de alérgenos; esta primacía de los alimentos se invierte en el tiempo a favor de los medicamentos.
Cuando la urticaria aguda o angioedema se presenta en relación con ciertos periodos estacionales debe considerarse que la etiología está en función del alérgeno prevalente en aquellos momentos y su mecanismo de acción puede ser tanto por vía inhalatoria, como por ingestión o incluso por contacto.
Algunos individuos con alergia al látex presentan reacciones de hipersensibilidad tras ingerir plátano, aguacate, castaña o kiwi. Diversas teorías se han sugerido para explicar la existencia de esta doble sensibilización látex-fruta.
Los estímulos físicos son causa frecuente de urticaria; con frecuencia un mínimo trauma es capaz de provocar urticaria (dermografismo), en otros individuos los responsables son estímulos del tipo frío, presión, ejercicio Otras etiologías pueden hallarse implicadas en la génesis de la urticaria: aditivos (salicilatos, ac. Benzóico, tartrazina), parásitos, enfermedades del tiroides, enfermedad sistémica (lupus, artritis reumatoide, hepatitis, carcinoma...). Cuando no han podido relacionarse con un agente específico se han querido involucrar factores psicosomáticos.
El rascado de la piel y la consiguiente aparición de urticaria (signo de Darier) nos orienta hacia una urticaria de tipo pigmentoso, de localización exclusivamente en piel.
Clasificación
No existe una unanimidad de criterio a la hora de clasificar los distintos tipos de urticarias, hay quien establece dos tipos de urticarias, aquellas que se presentan de manera brusca o inmediata ante el insulto de un agente bien sea específico o inespecífico, serían las urticarias agudas. En este tipo de urticarias puede detectarse en la mayoría de los casos un mecanismo inmunológico bien sea mediado por IgE, por activación de las distintas vías del complemento o por un mecanismo de tipo celular

Manifestaciones clínicas
El síntoma característico de la urticaria es el habón que puede aparecer en cualquier parte del cuerpo, puede ofrecer distintas formas más o menos variables de tamaño, con tendencia a confluir y suele acompañarse de prurito. La expresión de lesión de carácter puntiforme morbiliforme suele ser más frecuente en las urticarias colinérgicas. Una de las característica de la urticaria y/o angioedema es su carácter recidivante.
El llamado síndrome de alergia oral se caracteriza por afectación de la mucosa bucal y edema labial hasta posterior aparición de urticaria y/o angioedema sin descartar la posibilidad de que pueda aparecer síntomas bronquiales, nasales, conjuntivales o gastrointestinales y excepcionalmente shock anafiláctico. Puede ser atribuido en la mayoría de ocasiones a una hipersensibilidad de tipo inmediato (mediada por IgE) frente a frutas. Cuando la urticaria se mantiene por tiempo prolongado decimos que se trata de una urticaria crónica, este periodo se establece de forma aleatoria en 6 semanas. Dentro de las urticarias crónicas son las físicas las que ocupan el primer lugar.
Tratamiento
El diagnóstico entre urticaria aguda o crónica es básico dada la implicación terapéutica que conlleva. La presentación mas frecuente en Pediatría es la urticaria/angioedema agudo. Ante el hecho de hallarnos ante un angioedema recidivante es importante descartar el angioedema hereditario. En el caso de urticaria, angioedema crónico puede existir una remisión espontánea a los 3-4 años de su presentación

ACNÉ
El acné es una enfermedad inflamatoria de la unidad pilosebácea. Es una de las patologías dermatológicas más frecuentes, ya que afecta casi al 80% de los adolescentes entre los 13 y los 18 años. Representa el 25% de las consultas al dermatólogo y posiblemente este número sea aún mayor en la consulta del pediatra y médico de cabecera. Puede durar muchos años, dejar cicatrices persistentes y provocar efectos adversos importantes en el desarrollo psicológico del adolescente que la sufre. Su etiología es multifactorial y actualmente existen tratamientos muy efectivos para controlar cada uno de los factores patogénicos implicados. El conocimiento correcto de las bases fisiopatológico del acné permite efectuar una aproximación terapéutica adecuada que a menudo es definitiva en casos leves o moderados, y permit ayudar de forma correcta los más graves.
Patogenia
Se puede definir al acné como a una enfermedad inflamatoria de etiología multifactorial que afecta la unidad pilosebácea con la intervención del Propionibacterium acnes y otras bacterias. Su patogenia aún no queda del todo definida, pero el conocimiento de los distintos factores que intervienen en la misma ha permitido desarrollar nuevas medidas terapéuticas específicas.
Factores patogénicos
En la patogenia del acné es menester considerar cuatro factores básicos :
1. Aumento de la secreción sebácea.
2. Hiperqueratosis ductal con obstrucción del folículo pilosebáceo.
3. Colonización bacteriana por P. acnes.
4. Inflamación secundaria.
La lesión inicial, el microcomedón, es el resultado de la obstrucción de los folículos sebáceos por un exceso de sebo junto con células epiteliales descamadas procedentes de la pared folicular (hiperqueratosis ductal). Estos dos factores causan lesiones no inflamatorias como los comedones abiertos (puntos negros o barrillos) y los microquistes o comedones cerrados. Una bacteria anaerobia, el P. acnes, prolifera con facilidad en este ambiente y provoca la aparición de mediadores de la inflamación.
Tipo de lesión
Resulta fundamental diferenciar las lesiones inflamatorias de las no inflamatorias, así como también definir la lesión predominante
Lesiones no inflamatorias

Entre las lesiones no inflamatorias, los comedones cerrados o microquistes son el elemento más característico, y lo que define al acné. Por el contrario, los comedonesabiertos o barrillos no se encuentran siempre ni acostumbran a presentar cambios inflamatorios.
Su aspecto es debido a la compactación de células foliculares en el ducto y a la oxidación del sebo y la melanina, no a la suciedad. Es bueno recordar que el diagnóstico diferencial entre el acné auténtico o verdadero y las llamadas reacciones acneiformes se establece porque estas últimas suelen ser monomorfas y no presentan barrillos.
Lesiones inflamatorias
Las lesiones inflamatorias incluyen, pápulas, pústulas , nódulos, quistes y posteriormente cicatrices. Estas últimas no siempre van ligadas a una manipulación impulsiva de las lesiones ni tampoco a la gravedad del acné
Estadios de las lesiones
Para valorar el estadio inicial del acné y la posterior evolución del tratamiento instaurado, es muy importante señalar en la historia clínica la gravedad del cuadro estableciendo en cuál de los cinco grados de estadio se encuentran las lesiones , que, de un modo más práctico, se puede simplificar a tres: formas leves, moderadas y graves.
Localización y extensión de las lesiones
Casi todos los acnéicos presentan lesiones en la cara, y aproximadamente la mitad las presentan en la espalda y pecho. Sólo un 1% de ellos tiene lesiones severas en tronco sin afectación facial.
Tratamiento dependiendo del tipo de la lesión y la gravedad
Acné comedogénico no inflamatorio Un acné con barrillos en frente y en área paranasal, típico del inicio de la pubertad, puede ser tratado con tretinoína tópica una vez al día.

El ácido salicílico, el adapaleno y el ácido azelaico, así como el ácido glicólico, resultan opciones menos eficaces.
El tratamiento deberá mantenerse durante meses. Puede resultar aconsejable asociar un antibiótico tópico para evitar la aparición de lesiones pustulosas
Acné inflamatorio leve
Acné caracterizado por pápulas y pústulas con menos comedones y sólo en la cara. Se puede iniciar sólo con un antibiótico tópico (eritromicina) asociado a peróxido de benzoilo. Acné inflamatorio moderado Si existen más lesiones pustulosas y comedones se pueden combinar la tretinoína (predominio de comedones) o el peróxido de benzoilo (mayoría de lesiones pustulosas) con un antibiótico tópico y/o sistémico dependiendo de la severidad de las pústulas y de la afectación o no del tronco.



















