
AGUA LÍQUIDA SOBREENFRIADA: UN ESTUDIO DE … · Presentado ante la Facultad de Matemática,...
97
AGUA LÍQUIDA SOBREENFRIADA: UN ESTUDIO DE SIMULACIÓN CON DINÁMICA MOLECULAR por Aleida Josefina Bermúdez Di Lorenzo Presentado ante la Facultad de Matemática, Astronomía y Física como parte de los requerimientos para la obtención del grado de Doctor en Física de la UNIVERSIDAD NACIONAL DE CÓRDOBA, en el marco del convenio de beca FUNDAYACUCHO-VENEZUELA Octubre de 2014 FaMAF-UNC Directores: Dr. Marcelo Andrés Carignano, Dr. Rodolfo Guillermo Pereyra
Transcript of AGUA LÍQUIDA SOBREENFRIADA: UN ESTUDIO DE … · Presentado ante la Facultad de Matemática,...

AGUA LÍQUIDA SOBREENFRIADA: UN ESTUDIO
DE SIMULACIÓN CON DINÁMICA MOLECULAR
por Aleida Josefina Bermúdez Di Lorenzo
Presentado ante la Facultad de Matemática, Astronomía y Física como parte de los requerimientos para la obtención del grado de Doctor en Física
de la UNIVERSIDAD NACIONAL DE CÓRDOBA, en el marco del convenio de beca FUNDAYACUCHO-VENEZUELA
Octubre de 2014
FaMAF-UNC
Directores: Dr. Marcelo Andrés Carignano, Dr. Rodolfo Guillermo Pereyra

Dedicado a mi amado pueblo venezolano revolucionario en su lucha por la integración latinoamericana, en particular Argentina-Venezuela.

iii
AGRADECIMIENTOS
Las primeras líneas son especialmente para agradecer al Estado venezolano por la beca de Doctorado, instrumento del pueblo y para el pueblo. La beca me fue asignada a través de la Fundación Gran Mariscal de Ayacucho (FUNDAYACUCHO), después de haber cumplido con los requisitos necesarios para ser merecedora de la misma. Las siguientes líneas son para el Dr. Ávila Edgardo, miembro del grupo de Física de la Atmósfera de la Facultad de Matemática, Astronomía y Física (FaMAF), por su celeridad para llevar a cabo los trámites administrativos necesarios para mi ingreso al doctorado. También a la Dra. Castellano Nesvit y su familia; de ellos recuerdo mucho y nunca olvidaré cuando me abrieron las puertas de su casa para vivir unas cuantas semanas en su hogar. Sus hijos Pilar, Milagros y Felipe junto a su madre me trataron de manera muy especial. Las puertas de mi casa en Venezuela siempre estarán abiertas para todos Ustedes. Luego de conectarme con algunos miembros del grupo de Física de la atmósfera me encuentro con el Dr. Pereyra Rodolfo, co-directo de tesis. Al Dr. Rodolfo (conocido como “Rolo”), el cual me sigue resultando todo un personaje por su espontaneidad para decir las cosas y su sencillez, le debo un substancial agradecimiento. A ti “Rolo” te doy especialmente las gracias por todo tu apoyo; tu persona ha sido esencial para desarrollar este trabajo de tesis. Te debo unas cuantas botellas de Ron venezolano que sé te gustan y las disfrutas mucho al lado de tu esposa.
El siguiente es el Dr. Carignano Marcelo, director de tesis. A ti Marcelo te agradezco tus invaluables aportes a nivel académico. Me hubiese gustado haber tenido más contacto personal contigo, estoy segura que tus valiosas lecciones hubiesen resultados en muchos más frutos. Soy más del aprendizaje presencial que a distancia.
Las siguientes líneas son para manifestar mi más profundo agradecimiento con todo mi amor a mis amigos Adriana Pérez (“la Chesa”) y Fernando Nollas (“el Ferchu”). Sin el amor de Ustedes no hubiese aguantado situaciones desesperantes que viví en este largo camino. A ti “Ferchu” no me alcanzará la vida a nivel académico para compensar todas las horas de tu tiempo que dedicaste a explicarme conocimientos en física de los cuales yo desconocía o simplemente no recordaba; mi vida espiritual siempre estará conectada a la tuya, nuestro lazo es uno de los más hermosos y poderosos que puedo recordar en las distintas semillas de amistad que he formado en mi vida; me quedo corta de palabras para manifestar el profundo amor que siento por vos. A ti “Chesa” con tu manera de ser tan espontánea has abierto puertas que desconocía y que no se cerrarán nunca más; tu amistad es un regalo que siempre atesoraré; nuestras vidas están conectadas por un profundo lazo de amistad, de esos que son “para toda la vida”. A tu lado he desarrollado una de las labores más maravillosas de mi vida, el proyecto de “yoga y musicoterapia para adultos mayores”. Este proyecto surgió como una propuesta para cumplir actividades sociales exigidas por la beca; el proyecto ha sido puesto en práctica en distintas instituciones geriátricas y los resultados han sido maravillosos. Las actividades con los abuelos al igual que las presentaciones realizadas con la banda “sin fronteras”, han sido parte de las extraordinarias panaceas de este largo transitar.
Para finalizar y por eso más importante, mi familia. Han pasado casi cuatro años desde que nos vimos en Venezuela. A ti madre amada, mi madre siempre mi madre, has sido pilar fundamental en todo los logros de mi vida, y en mis “malos ratos” siempre

iv
has estado para apoyarme y levantarme como nadie más sabe hacerlo. A mis hermanas, “Tama”, “Fira” y a mi sobrino Andrés, gracias por todo. Las conversas por teléfono, las risas, alguna que otras lágrimas, han sido empujes para terminar esta etapa de mi vida y demuestra que la distancia es solo una ilusión, pues siempre están conmigo y son referencias para cerrar ciclos y que la lucha sigue no importe lo que pase. Todo mi amor y entrega para Ustedes, ese amor sin límites que damos, cada quien a su modo y en donde estemos, en todos los cambios y transformaciones revolucionarias que llevamos a cabo en nuestra maravillosa Venezuela y en el Nuestro Americano.
GRACIAS A CADA UNO DE USTEDES Y A TODOS LOS QUE COLABORARON EN EL DESARROLLO DE ESTE TRABAJO.
Haciendo remisión a las palabras del Che Guevara “Hasta la victoria siempre” y como dijo Hugo Chávez Frías “Nosotros no andamos chillando, no. Los verdaderos combatientes no chillan; combaten y pujan. Nosotros pujamos, nosotros lloramos, sí, pero sobre todo amamos inmensamente lo que hacemos. Es el amor que reina en nuestros corazones, en nuestras acciones, en el cada día que batallamos. No queremos tregua”. Chávez ante la primera Asamblea Nacional del Polo Patriótico, 1999.

v
Clasificación: 42.68.Ge Effects of clouds and water; ice crystal phenomena 82.20.Db Transition state and statistical theories of rate constants 82.20.Pm Rate constants, reaction cross sections, and activation energies 31.15.xv Molecular dynamics and other numerical methods. Palabras claves: agua sobre-enfriada, nucleación de hielo, tasa de nucleación, modelos de agua, dinámica molecular.

vi
RESUMEN
Se investigó el comportamiento de algunas propiedades del agua líquida sobre-enfriada y se evaluó la naturaleza de la nucleación heterogénea de hielo mediante simulación de dinámica molecular. La mayoría de los sistemas fueron representados por un bulk de agua y todas las simulaciones se hicieron a temperatura, presión y número de moléculas constantes, con modelos rígidos de distintas arquitecturas de construcción. Las propiedades investigadas fueron: la temperatura de máxima densidad (TMD), el coeficiente de difusión (D), la temperatura de fusión (Tf), la estructura tetragonal del agua mediante el tiempo de vida promedio de los enlaces puentes de hidrógeno (��̅(�)) y el parámetro de orden tetraédrico (q), la capacidad calorífica isobárica (CP) y el tamaño del embrión de hielo crítico (N) en nucleación homogénea.
Los resultados mostraron que el agua sobre-enfriada presenta una tendencia clara a formar enlaces puentes de hidrógeno (Hb) más estables que el agua a temperaturas mayores a 273 K. Esta tendencia fue apreciada en mayor medida en las simulaciones realizadas con modelos construidos con puntos de carga que asemejan a los pares de electrones no enlazantes del átomo de oxígeno (LP). También se encontró que las CP calculadas en función de una temperatura escaleada exhibieron un pico justo donde los resultados experimentales muestran una divergencia aparente, divergencia que se adjudica a una posible transición de fase del agua líquida sobre-enfriada. El tamaño de N necesario para la nucleación homogénea fue calculado por primera vez en sistemas tipo bulk; al comparar los resultados de las simulaciones con los experimentales se obtuvo una muy buena correspondencia entre ellos, particularmente en aquellos modelos con LP.
Por otra parte, en uno de los estudios se llevó a cabo una reparametrización de uno de los modelos de agua, mediante la utilización de metodologías sencillas. Los resultados mostraron que la representación de algunas de las propiedades investigadas por el modelo reparametrizado no fueron mejores que las representadas por el modelo original. En el estudio sobre la naturaleza de la nucleación heterogénea se encontró que la nucleación es estocástica. Este estudio se considera el primero en describir a nivel de los nanómetros, la estadística de la nucleación heterogénea del agua sobre-enfriada.
Por último, el presente trabajo de tesis ha sido motivo de diversas presentaciones en la Asociación Física de Argentina (AFA) desde el año 2011. Los trabajos llevan por títulos “Reparametrización del modelo de molécula de agua TIP5P-E” (SUF-AFA-2011), “Relación entre los enlaces tipo puentes de hidrógeno, orden tetraédrico y movilidad molecular en distintos modelos de agua” (AFA-2012) y “Cálculo del tamaño del núcleo crítico en la nucleación homogénea de hielo” (AFA-2013). Así mismo, la tesis ha arrojado las siguientes publicaciones “On the relation between hydrogen bonds, tetrahedral order and molecular mobility in model water” (Chem. Phys. Lett., 538: 35-38, 2012) y “The water supercooled regime as described by four common water models” (J. Chem. Phys. 139024506-1, 2013). Actualmente, se encuentra en proceso la tercera publicación: “Stochastic vs Singular hypothesis in heterogeneous nucleation of ice by molecular dynamics”.

vii
ABSTRACT
The behavior of some properties of the supercooled water was investigated and
the nature of the heterogeneous ice nucleation was evaluated by Molecular Dynamics simulation. Most systems were represented by a bulk water and all simulations were made to constant temperature, pressure and number of molecules for several rigid water models. The properties investigated were: the temperature of maximum density (TMD), the diffusion coefficient (D), the melting temperature (Tm), the water tetragonal structure by an average lifetime of hydrogen bonds ((��̅(�)) and tetrahedral order parameter (q), isobaric heat capacity (CP) and critical embryo size (N) in homogeneous nucleation.
The results showed that supercooled water have to form a clear stable hydrogen bonds trend (Hb). This trend was appreciated more in the simulations with models includes specific sites resembling the water lone pairs (LP). We also found that CP calculated based on a rescaled temperature display a peak just where the experimental results show an apparent divergence, divergence that is allocated to a possible phase transition supercooled liquid water. N was estimated for the first time in bulk systems; by comparing simulation with experimental results a good agreement between them was obtained, particularly in models with LP.
Moreover, in one study we did a simple reparametrization of one water model. The results showed that the representation of some of the properties investigated by the reparameterized model were no better than those represented by the original model. Moreover, it was found that the nature of the heterogeneous nucleation is stochastic; this study is considered the first to describe the nanometer level, the statistics of the heterogeneous ice nucleation of supercooled water.
Finally, this thesis has been the subject of several presentations in Argentina Physics Association (AFA) since 2011. It has yielded the following publications "On the relation Between hydrogen bonds, tetrahedral molecular mobility and order in model water" (Chem Phys Lett, 538: 35-38, 2012) and "The supercooled water regime as Described by four common water models "(J. Chem. Phys. 139024506-1, 2013). The third publication "Stochastic Singular vs hypothesis in heterogeneous nucleation of ice by molecular dynamics", is in process.

viii
ÍNDICE GENERAL
INTRODUCCIÓN 1
CAPÍTULO I: AGUA LÍQUIDA SOBREENFRIADA 5
I.1. ANOMALÍAS 8
I.2. INTERPRETACIONES DEL COMPORTAMIENTO ANÓMALO 11
I.3. NUCLEACIÓN DE HIELO 13
I.3.1. Teoría Clásica de la Nucleación (T.C.N.) 13
I.3.2. Determinación Experimental de la Tasa de Nucleación 15 I.3.3. Estudios de Nucleación Homogénea mediante Simulaciones
Computacionales
17 CAPÍTULO II: DINÁMICA MOLECULAR 20
II.1. PRINCIPIOS TEÓRICOS 21
II.2. TÉCNICAS 23
II.3. PROPIEDADES MACROSCÓPICAS 26
II.4. MODELOS DE AGUA 27 CAPÍTULO III: ESTUDIO DEL COMPORTAMIENTO DE ALGUNAS PROPIEDADES DEL AGUA LÍQUIDA REPRESENTADA POR DISTINTOS MODELOS
32
III.1. ANÁLISIS DE DISTINTOS MODELOS DE AGUA EN EL RÉGIMEN SOBRE-ENFRIADO METAESTABLE. REPARAMETRIZACIÓN DEL MODELO TIP5P-Ew 32
III.2. ESTRUCTURA Y DINÁMICA DE LOS ENLACES PUENTES DE HIDRÓGENO
39
III.2.1. Tiempo de Vida Promedio de los Enlaces Puentes de Hidrógeno 40
III.2.2. Orden Tetraédrico 41
III.2.3. Relación entre el Orden Tetraédrico y la Dinámica de los Hb 42
CAPÍTULO IV: NUCLEACIÓN HOMOGÉNEA DE HIELO. CONJETURA SOBRE LA CRISTALIZACIÓN DEL AGUA EN CONDICIONES NO MAN´S LAND EN MODELOS ATOMÍSTICOS
46
IV.1. ESTIMACIÓN DE LA ENERGÍA DE ACTIVACIÓN 48
IV.2. CÁLCULO DE LA ENTALPÍA DE CRISTALIZACIÓN (∆Ηc) 50
IV.3. ESTIMACIÓN DEL TAMAÑO DEL EMBRIÓN CRÍTICO 54
CAPÍTULO V: NUCLEACIÓN HETEROGÉNEA DE HIELO 58
V.1. PREPARACIÓN DE LOS SISTEMAS 59
V.2. MONITOREO DEL PROCESO DE NUCLEACIÓN 60
V.3. ESTADÍSTICA DE LA NUCLEACIÓN 62

ix
CONCLUSIONES 67
APÉNDICE 1 70
1.1. DEDUCCIÓN MATEMÁTICA DE GAMMA (γ) 70 1.2. DEPENDENCIA DE LAS PROPIEDADES D (T= 255 K) Y ρ (T= 255
K) CON LA VARIACIÓN INDEPENDIENTE (NO SIMULTÁNEA) DE LOS PARÁMETROS σ, ε Y q DEL TIP5P-Ew
71
APÉNDICE 2 73 2.1. CÁLCULO DEL TIEMPO DE VIDA DE LOS ENLACES PUENTES
DE HIDRÓGENO
73 2.2. CÁLCULO DEL ORDEN TETRAÉDRICO (q) 75
REFERENCIAS BIBLIOGRÁFICAS 77
LISTA DE SÍMBOLOS 87

1 Introducción
INTRODUCCIÓN
Para el filósofo Tales de Mileto (624-547 A.C.) el agua es el principio de todas las cosas. Dedujo tal convicción, según Aristóteles, “de la constatación de que el sustento de todas las cosas es húmeda”,
de las simientes y semillas “que poseen una naturaleza húmeda y por consiguiente la
desecación total provoca la muerte puesto que la vida está ligada a la humedad y la humedad
proviene del agua”.
El agua es la sustancia de la vida. Desde los inicios de la ciencia, los filósofos presocráticos la consideraban uno de los cuatro elementos del universo. Hoy en día es bien conocido que toda la evolución biológica en nuestro planeta tiene al agua como sustancia esencial; la misma está involucrada de manera directa o indirecta en todos los procesos evolutivos terrestres. Esta situación le adjudica al agua una posición imprescindible en la vida de cualquier especie. Tal importancia ha sido motivo de que enormes esfuerzos científicos se vengan realizando para obtener un entendimiento completo de sus propiedades. Si bien el avance en esta línea ha sido muy importante, aún quedan muchos interrogantes por responder.
La molécula de agua está constituida por apenas tres átomos ligeros: dos de hidrógeno y uno de oxígeno. Esta aparente simplicidad contrasta con la enorme cantidad de propiedades anómalas que presenta el agua, como sustancia. Se llaman propiedades anómalas, porque al compararlas con otros hidruros del grupo del oxígeno, el agua presenta comportamientos marcadamente distintos. Entre las propiedades anómalas, cabe mencionar, por su importancia, la temperatura de máxima densidad (TMD), la aparente divergencia de la compresibilidad isotérmica (KT) y de la capacidad calorífica isobárica (CP), el aumento de la difusividad (D) con la densidad, entre otras. Tales anomalías han despertado y despiertan gran interés en la comunidad científica, en particular, cuando el agua se encuentra en estado líquido sobre-enfriado.
El agua tiene la particularidad que, a la presión de 1 atm, se puede evitar su cristalización a temperaturas menores que su temperatura de fusión; por debajo de la temperatura de fusión el agua líquida presenta diferentes particularidades. Presenta la característica de que se puede sobre-enfriar con relativa facilidad bajo condiciones experimentales bien controladas; se ha alcanzado una temperatura límite de sobre-

2 Introducción
enfriamiento de aproximadamente 231 K [2], debajo de la cual cristaliza espontáneamente para formar hielo hexagonal (Ih). Esta característica ha dado lugar a numerosos estudios en esta fase metaestable conocida como la región del agua sobre-enfriada metaestable, comprendida entre la temperatura de nucleación espontánea (231 K [2]) y la temperatura de fusión (273 K [2]). Por otro lado, si la temperatura del agua en estado líquido es reducida cuasi-instantáneamente a temperaturas, aproximadamente, menores a 130 K el sistema queda en un estado vítreo conocido como agua vítrea o hielo amorfo [2, 6]. Además, también presenta otro comportamiento anómalo entre los 150K y la temperatura de nucleación espontánea, en cuyo caso, experimentalmente no se puede obtener un estado amorfo del agua líquida, siempre se forma hielo hexagonal independientemente de la velocidad de enfriamiento. Esta región de temperaturas es conocida como la zona no man's land [2, 6] y la ausencia del estado amorfo en esta zona es una notable característica del agua líquida sobre-enfriada aun no explicada.
La simulación por computadoras es una herramienta útil para comprender el comportamiento macroscópico de las sustancias a partir de parámetros moleculares. La Dinámica Molecular (D.M.) es una técnica computacional utilizada para estudiar las propiedades dinámicas y termodinámicas de las sustancias a partir de trayectorias atomísticas. Si bien la D.M. es ampliamente usada para todo tipo de sistemas, la literatura muestra que la mayoría de los trabajos publicados basados en esta técnica incluyen al agua; la razón de esto es porque la D.M. es una herramienta fundamental utilizada en estudios biológicos que involucran proteínas, lípidos, membranas, etc. Este hecho resultó en el desarrollo de métodos, modelos y programas muy eficientes para el tratamiento de sistemas acuosos en condiciones de temperatura y presión estándares para la vida. En este sentido, los avances que la comunidad de biofísica computacional logró en términos de modelos y métodos de D.M. simplificaron la tarea a quienes estudian al agua en condiciones diferentes a las estándares.
La dinámica molecular se basa en la integración de las ecuaciones clásicas de movimiento de un sistema de moléculas representado de manera simplificada. Por ejemplo, la molécula de agua se representa como un objeto rígido constituido por tres partículas, o sitios, ubicadas en las posiciones correspondientes a las de sus átomos. La interacción entre dos moléculas de agua se representa, típicamente, por una suma de interacciones Lennard-Jones y Coulómbicas. La energía potencial total del sistema se calcula sumando las interacciones de todos los pares de moléculas. Al usar este esquema simplificado, se logra transformar un problema cuántico, el cual involucra núcleos y electrones de todo el sistema, en un problema clásico que involucra objetos rígidos interactuando con una forma funcional simple. La arquitectura de la molécula junto con la forma funcional y los parámetros de las interacciones son las características que conforman el campo de fuerza o modelo. La literatura reporta diversos modelos de agua, los cuales se han venido desarrollando desde 1933; su amplia diversidad abarca modelos rígidos y flexibles, en donde ambos pueden ser polarizables y no polarizables.
A pesar de los enormes esfuerzos que se han realizado para entender el comportamiento del agua, aún queda un largo trayecto por transitar. En los últimos años se han desarrollado numerosas investigaciones de fenómenos relacionados con agua sobre-enfriada, todas ellas con el propósito de entender o corroborar alguna de sus propiedades. Varias investigaciones han orientado sus esfuerzos en estudiar problemas relacionados con la existencia de un segundo punto crítico a muy bajas temperaturas [2, 6, 133, 134], con los mecanismos involucrados en la nucleación de hielo en el régimen sobre-enfriado [1, 2, 68, 88, 100, 115, 133], con la transformación estructural del agua

3 Introducción
líquida en condiciones correspondientes a la región no man's land [2, 117, 122, 123, 134], etc. Algunos de los estudios, realizados mediante simulaciones computacionales, tienen en común que fueron llevados a cabo utilizando un solo tipo de modelo de agua; otros han simulado los sistemas a temperatura constante o en cortos rangos de T; y otros se han abocado al desarrollo de nuevos modelos de agua o a la reparametrización de los existentes con el propósito de calcular y evaluar ciertas propiedades del agua.
En base a lo expuesto, la presente tesis se enfoca en estudiar el comportamiento de algunas propiedades del agua líquida en un amplio rango de temperatura, con especial interés en el régimen sobre-enfriado metaestable, mediante simulaciones de dinámica molecular utilizando distintos modelos de agua atomísticos, rígidos y no polarizables. En particular:
� Se analizan diferentes modelos en su capacidad para representar el régimen
sobre-enfriado metaestable. � Se lleva a cabo una reparametrización de uno de los modelos de agua a T = 255
K. � Se investiga la dependencia con la temperatura de la estructura tetragonal del
agua y su relación con la vida media de los enlaces puentes de hidrógeno. � Se estudia la nucleación homogénea de hielo y se presenta una conjetura sobre la
cristalización del agua en hielo hexagonal en condiciones no man’s land. � Se propone un re-escaleo de la temperatura en el régimen sobre-enfriado para
obtener una mejor evaluación de la calidad de diferentes modelos. � Se estudia la naturaleza de la nucleación heterogénea a partir de agua sobre-
enfriada.

5
Capítulo I: Agua Líquida Sobre-enfriada
CAPÍTULO I
AGUA LÍQUIDA SOBRE-ENFRIADA
El agua es una sustancia que está presente en todos los fenómenos de la naturaleza. Es la única, en condiciones ambientales de presión y temperatura, que se encuentra en las tres fases estables: sólido, líquido y gaseoso. A diferentes condiciones de temperatura y presión, los estados sólido y líquido tienen diferentes formas. Para el estado sólido, se han caracterizado experimentalmente los hielos estables: Ih II, III, V, VI, VII, VIII, X y XI; y los metaestables: Ic IV, IX, XII, XIII, XIV y XV [4, 5, 16]
Los hielos Ih e Ic, se encuentran a presión atmosférica, teniendo el Ih mayor presencia en la atmósfera. Se obtiene enfriando agua líquida entre ~ 190 K y la temperatura de fusión Tf = 273 K; tiene una estructura hexagonal y una red poco empaquetada (figura I.1), lo que le proporciona una densidad (ρIh) menor que la fase líquida. Presenta una variante conocida como hielo cúbico (Ic), de estructura tipo diamante, de red poco compacta y con una densidad ρIc muy parecida a la del Ih (ρIc = 0.93 g.cm-3; dato corregido a 273 K para comparación directa con la ρIh = 0.92 g.cm-3 [7]). Aunque el Ih es la forma cristalina predominante del agua que se encuentra en la tierra, la cristalización del agua líquida en Ic se observa bajo condiciones naturales en la supertropósfera y estratósfera [64, 115]. A nivel de laboratorio, el Ic se ha obtenido mediante la congelación de agua líquida en materiales porosos, por deposición de vapor, por calentamiento a altas presiones de hielos cristalinos y amorfos, etc. [4, 7, 16, 64].
El agua se mantiene en estado líquido bajo distintas condiciones de temperatura
T y presión P. A presiones elevadas y a temperaturas por encima de su punto de ebullición (373 K), se obtiene agua sobre-calentada hasta las condiciones críticas de temperatura (Tc = 647 K) y presión (Pc = 220.64 atm), en cuyo caso las fases líquida y vapor son indistinguibles.
A la presión de 1 atm y en distintos dominios de temperatura, el agua líquida presenta diferentes formas; las mismas se ilustran esquemáticamente en la figura I.2. Entre su punto de fusión Tf y ebullición Te, el agua se mantiene como un líquido estable. Por debajo de su punto de Tf, se ha logrado enfriar sin cristalizar hasta ~ 231 K (~ -42°C) [2]; de esta forma se obtiene agua líquida sobre-enfriada en estado metaestable. La temperatura límite de sobre-enfriamiento en la región metaestable, denominada temperatura de nucleación homogénea (TN), se ha alcanzado experimentalmente con gotitas de agua altamente pura y diámetros entre 1-10 µm [6].
Figura I.1. Estructura cristalina del hielo Ih vista desde una de las caras prismáticas.

6
Capítulo I: Agua Líquida Sobre-enfriada
Debajo de la TN el agua cristaliza espontáneamente para formar hielo hexagonal (Ih); la formación de hielo Ih por debajo de TN es independiente de la velocidad de enfriamiento [2, 6].
Cuando la temperatura del agua líquida es reducida cuasi-instantáneamente
(velocidades de enfriamiento ≥ 107 K/s) a T < 130 K, el sistema queda en un estado vítreo conocido como agua vítrea o hielo amorfo. Si el agua vítrea se calienta a temperaturas entre130 K ≤ T < 150 K, se obtiene agua ultra viscosa que cristaliza a Ic a una temperatura Tx ~ 150 K [2, 3, 6]. La T = 130 K es la temperatura de transición vítrea Tg [6]. La región entre TN y Tx se conoce como no man´s land; en ella, el agua líquida no se puede observar a escalas de tiempo experimentales ya que su cristalización en ese rango es inevitable [2, 6]. Del agua vítrea se distinguen dos estados, el hielo amorfo de alta densidad, HDA (del acrónimo en inglés high-density amorphous ice) y el hielo amorfo de baja densidad, LDA (del acrónimo low-density amorphous ice). El LDA se obtiene enfriando rápidamente el agua líquida desde T > Tf hasta T < Tg a P = 1 atm; el HDA se obtiene fundiendo cristales de hielo a altas presiones (> 2000 atm) a T < Tg [3].
El agua líquida presenta propiedades excepcionales que evidencian un comportamiento diferente (anómalo) al de otros líquidos formados por moléculas de tamaño y características similares. Estas anomalías son responsables de una gran cantidad de fenómenos y algunas de ellas tienden a ser más pronunciadas en el régimen sobre-enfriado [6]. Se argumenta que el responsable de estas anomalías es el enlace puente de hidrógeno (Hb), el cual se forma cuando la parte positiva de una molécula de agua (un átomo de hidrógeno) se une a la parte negativa (átomo de oxígeno) de otra molécula análoga, originándose fuerzas electrostáticas intermoleculares. La presencia de densidades de cargas positiva y negativa en una molécula de agua es porque, a pesar que el enlace oxígeno-hidrógeno presenta compartición de electrones típico de un enlace covalente, al mismo tiempo se produce un desplazamiento de la nube electrónica del hidrógeno hacia el oxígeno; de esta forma, los átomos asumen cargas eléctricas opuestas. La fuerza del enlace puente hidrógeno es aproximadamente 20 kJ/mol, considerablemente más fuerte que una interacción de tipo van der Waals, ∼ 1kJ/mol, pero significativamente más débil que un enlace covalente, ∼ 400 kJ/mol [6].
En el agua líquida, cada molécula forma, en promedio, cuatro Hb con las moléculas vecinas [5]; es decir, dos moléculas de agua tienen enlaces de hidrógeno
Tem
pera
tura
[K]
Figura I.2. Esquema de diferentes dominios de temperatura del agua líquida a la presión de 1 atm. Tf, TN y Tg son las temperaturas de fusión, nucleación y transición vítrea, respectivamente [22].
373
250
200
150
100
273
235
150
130?
Tf
TN
Tx
Tg
agua estable
agua sobreenfriada (metaestable)
“no man´s land”
agua ultraviscosa
agua vítrea

7
Capítulo I: Agua Líquida Sobre-enfriada
aceptores con la molécula central, y los hidrógenos de la molécula central forman enlaces dadores, los cuales apuntan a los pares de electrones libres del átomo de oxígeno de las otras dos moléculas de agua (figura I.3).
La influencia del enlace puente de hidrógeno en el agua se aprecia comparando las temperaturas de fusión y de ebullición del agua con otras moléculas similares. Los hidruros de nitrógeno (NH3) y flúor (HF), caracterizados por formar puentes de hidrógeno, presentan temperaturas de ebullición y fusión menores a las del agua (Tf = 195 K y Te = 240 K para el NH3; Tf = 190 K y Te= 293 para el HF [4]). Las Tf y Te son mucho menores en aquellos hidratos que no forman puentes de hidrógeno. Por lo tanto, el elevado punto de fusión y ebullición del agua son alguna de las tantas anomalías que presenta el agua líquida. También se pueden mencionar otras anomalías tales como: su elevada constante dieléctrica (ε = 78,5) [1], lo que convierte al agua en un poderoso solvente; el incremento, en el régimen sobre-enfriado, de la compresibilidad isotérmica KT cuando la temperatura disminuye; una densidad ρ mayor que su fase sólida Ih, exhibiendo un máximo de 1g/cm3 a la presión de 1atm y una temperatura de máxima densidad TMD = 277 K (4°C) [6]; el aumento de la difusividad con la densidad; la disminución, a presión atmosférica, de la capacidad calorífica isobárica CP cuando T aumenta, exhibiendo un mínimo débil en T ∼ 308 K [6]; etc. En la figura I.4 se ilustran, cualitativamente, los fuertes contrastes de algunas de las propiedades entre el agua y los líquidos “simples”; el comportamiento de las mismas se vuelve más dicotómico en el rango sobre-enfriado. La línea punteada y las flechas en la figura I.4 corresponden, respectivamente, a la temperatura de fusión y las temperaturas donde se produce el máximo o el mínimo de algunas propiedades.
Figura I.3. Asociación de moléculas de agua a través de enlaces puentes de hidrógeno (líneas punteadas). Las esferas rojas y blancas representan los átomos de oxígeno e hidrógeno, respectivamente.
Figura I.4. Comparación esquemática, a presión constante, de la dependencia de la densidad ρ, la compresibilidad isotérmica KT y la capacidad calorífica isobárica CP para el agua y un líquido simple.
Figura extraída de ref. [6].
agua
Líquido simple
277 K
ρρρρ
T
KT
319K
Líquido simple
agua
T
308 K Líquido simple
agua
CP
T Tf Tf Tf

8
Capítulo I: Agua Líquida Sobre-enfriada
Las funciones respuestas de la CP y la KT están asociadas con una correspondiente fluctuación. La CP es proporcional a las fluctuaciones entrópicas experimentadas por N partículas del sistema a presión constante; mientras que KT es una medida de las fluctuaciones volumétricas para un N constante. En muchos líquidos, cuando la temperatura disminuye las fluctuaciones volumétricas y entrópicas se hacen cada vez menores y están positivamente correlacionadas, es decir, un aumento del volumen V implica un aumento de la entropía S. En el agua, dichas fluctuaciones son marcadas en el régimen sobre-enfriado y a T < TMD están anti-correlacionadas (una disminución de V implica un aumento de la S). La mencionada anti-correlación es una consecuencia de la formación de una estructura abierta que van formando los enlaces puentes de hidrógeno del agua a medida que T disminuye, por lo que, un detrimento en la entropía orientacional está acompañada con un aumento del volumen [6].
La profundización de algunos aspectos acerca del agua líquida sobre-enfriada se describen en distintas secciones. La sección I.1 está dedicada a las anomalías, la I.2 a las interpretaciones que se han propuesto para explicar su comportamiento anómalo, y la sección I.3 resume las bases teóricas de uno de los procesos que ocurren en agua líquida sobre-enfriada, la nucleación de hielo.
I.1 ANOMALÍAS.
� Capacidad Calorífica Isobárica. La capacidad calorífica isobárica CP es una medida de la fluctuación entrópica
⟨(∆�)⟩ y por ende, debe disminuir con la temperatura. Sin embargo, la CP del agua tiene un comportamiento distinto, incrementa cuando T disminuye y exhibe una pronunciada pendiente negativa por debajo de 273 K y a presión atmosférica [6, 7]. Los experimentos de Anisimov et. al. [1972] [109] hasta los 266 K (sobre-enfriamiento moderado), ya habían mostrado un incremento anómalo de la CP. Angell et. al. [1982] [43] amplían el rango de mediciones reduciendo T hasta 236 K y demuestran que la CP sigue aumentando; sus resultados sugieren una divergencia a temperaturas menores a las medidas. Este comportamiento también se aprecia en los estudios de Bertollini et. al [1985] [110], Tombari et. al. [1999] [44], Archer D. y Carter R. [2000] [45]. En la figura I.5 se muestran los valores de la CP medidos por los últimos cuatro autores mencionados.

9
Capítulo I: Agua Líquida Sobre-enfriada
Las simulaciones atomísticas de agua sobre-enfriada muestran un comportamiento algo distinto a los resultados experimentales, pero no necesariamente en contradicción. La CP del agua presenta un máximo en el régimen sobre-enfriado y la cristalización homogénea o espontánea del agua no se observa. Por ejemplo, Kumar y Stanley [2011] [46] encuentran que el máximo de la capacidad calorífica isobárica está en T ~ 250 K. Longinotti et. al. [2011] [47] reportan un máximo de esta propiedad a 247 K. Los estudios de Pi et. al. [2009] [111] y Vega et. al [2010] [108] no reportan la temperatura del máximo de la CP, pero la tendencia cualitativa de esta propiedad es semejante a la experimental en el rango de temperatura que los autores evaluaron, 240 K<T < 290 K. Por otra parte, es importante mencionar que todas las simulaciones fueron realizadas con distintos modelos de agua, lo que trae como consecuencia que los resultados pueden verse afectados por el modelo elegido.
� Densidad. El incremento de la densidad (ρ) con la disminución de la temperatura es el
comportamiento natural de muchos líquidos; sin embargo y como se indicó antes, la densidad del agua disminuye con la temperatura siendo más pronunciado este efecto para T < 273 K. Resultados experimentales de la densidad como función de la temperatura a presión atmosférica se reproduce en la figura I.6 para T > 235 [6]. Por otra parte, diferentes estudios a nivel atomístico han reportado los valores de ρ, a P = 1 atm, en un amplio rango de temperatura. Por ejemplo, Pi et. al. [2009] [111] analizan el rango de temperaturas comprendido entre 150 K y 375 K y encuentran que las ρ calculadas están, cualitativa y cuantitativamente, en buena correspondencia con los datos experimentales. Sciortino et. al. [1996] [112] y Qvist et. al. [2011] [113] obtienen comportamientos cualitativos de la ρ parecidos a los experimentales en el rango de temperatura entre ~ 200 K y ~ 300 K; el valor de la TMD que obtienen es de 240 K, muy alejada de la TMD experimental. Los autores Dick y Madura [2005] [37] refieren, al buen acuerdo cualitativo y cuantitativo que existe entre los valores calculados y experimentales de ρ, para un rango de 235 K < T < ~ 345 K. Al igual que en la CP y
Figura I.5. Capacidad calorífica isobárica CP vs T del agua sobre-enfriada. Mediciones experimentales realizadas por distintos autores a presión atmosférica. Figura extraída de ref. [107].
T [K]
CPP PP [k
J.kg
-1.K
-1]

10
Capítulo I: Agua Líquida Sobre-enfriada
como se explicará más adelante, los resultados de las simulaciones dependen del modelo utilizado.
� Coeficiente de Difusión. El comportamiento anómalo del coeficiente de difusión del agua D, se refleja en
la marcada dependencia que este presenta para temperaturas menores a 273 K y presión atmosférica. Tal dependencia, ha sido explicada mediante el argumento que los Hb del agua líquida en el rango de T sobre-enfriada adquieren una estructura tipo red más definida. La figura I.7 muestra los resultados experimentales reportados por Price et. al. [2000] [7] para agua pura y para un sistema de moléculas de agua disueltas en nitrometano. Los autores concluyen que, la suave disminución de D con T del sistema agua-nitrometano en comparación con el agua pura, se debe a la ausencia de Hb.
Figura I.6. Dependencia con T de la densidad del agua (ρ) medida a presión atmosférica. Figura extraída de ref. [6].
233 253 273 293 0.97
0.98
0.99
1
T [K]
ρρ ρρ [g
.cm
-3]
1000/T [K -1] 3.0 3.2 3.4 3.6 3.8 4.0 4.2 4.4
-23
-22
-21
-20
-19
-18
LnD
[m
2 .s-1]
294 K 273 K 250 K
FiguraI.7. Dependencia con T del coeficiente de difusión del agua (D). Las mediciones son a P = 1 atm. Figura extraída de ref. [7].

11
Capítulo I: Agua Líquida Sobre-enfriada
Los resultados de las simulaciones de agua líquida sobre-enfriada, para el cálculo de D, muestran distintos comportamientos. Por ejemplo, Molinero y Moore [2009] [114] obtienen, para un rango de 222 K < T < 363 K, D más altos y menos sensible con T que los D experimentales. Longinotti et. al. [2011] [47] señalan que el coeficiente de difusión calculado disminuye más rápido con la temperatura que el medido; sus resultados abarcan un rango de T entre 232 K y 294 K. Los D calculados por Pi et. al. [2009] [111] presentan buena correspondencia cualitativa y cuantitativa con los experimentos. Las simulaciones realizadas por Qvist et. al. [2011] [113] entre T ~ 200 K y T ~ 300 K sobreestiman los valores de D. Hans et. al. [2004] [33] obtienen resultados excelentes entre T = 242.5 K y T = 298 K. Nuevamente, el modelo utilizado en las simulaciones afecta los resultados.
I.2 INTERPRETACIONES DEL COMPORTAMIENTO ANÓMALO.
La aparente divergencia de las funciones respuestas (figura I.4) se interpretan en términos de varios escenarios teóricos: conjetura del límite de estabilidad, libre singularidad y la transición de fase líquida-líquida [6]. El último escenario, mejor conocido como hipótesis del segundo punto crítico líquido-líquido LLCP (del acrónimo en inglés liquid-liquid critical point), ha sido ampliamente avalado en las últimas dos décadas por estudios experimentales y de simulaciones computacionales. El escenario del LLCP fue propuesto en 1992 por Poole et. al. [116] como un resultado de extensas simulaciones. Años más tarde, Mishima et. al. [1998] [3] proveyeron soporte experimental a tal interpretación.
La hipótesis LLCP propone la existencia de un segundo punto crítico a una presión mayor a 1 atm y a una temperatura cercana al límite superior de la zona no man’s land. En esta zona el agua líquida se separa en dos fases distintas (inmiscibles), una líquida de baja densidad LDL (del acrónimo en inglés low-density liquid) y una líquida de alta densidad HDL (high-density liquid). Como se ha mencionado en parágrafos anteriores, el acceso experimental a la zona no man´s land es inviable (en estado líquido), por lo que solo existe evidencia indirecta del LLCP a partir de datos obtenidos de los estados amorfos del agua vítrea (LDA y HDA) a muy bajas temperaturas (T < Tx), y de datos del agua líquida obtenidos a temperaturas más altas (T > TN) y a presiones mayores a 1atm.
Los estudios experimentales de Mishima et. al. [1998] [3], Koza et. al. [2003] [117], Nelmes et. al. [2006] [118], etc., muestran que, a presiones entre 1000 y 2000 atm, las fases LDA y HDA se inter-convierten mediante una transición marcada y reversible a T < Tx. Dicha transición, la interpretan como una manifestación estructural de la transición de fase entre las dos formas del agua líquida, LDL y HDL; es decir, LDA y HDA son simplemente las correspondientes formas vítreas del agua LDL y HDL, respectivamente. Los distintos autores también señalan que, el agua cerca del segundo punto crítico es una mezcla fluctuante de moléculas (las estructuras locales son las dos fases LDL y HDL), por lo que tales fluctuaciones son las que influencian las propiedades del agua líquida y generan el comportamiento anómalo en la misma.
En la figura I.8 se muestran las relaciones entre las fases agua líquida L, LDL, HDL, LDA y HDA. El punto C´ denota el segundo punto crítico, el cual se ubica a una Pc ́~ 1000 atm y a una Tc ́~ 220 K [2, 3, 6, 46]; F es la línea de coexistencia a lo largo

12
Capítulo I: Agua Líquida Sobre-enfriada
de la cual las fases sólidas de baja y alta densidad (LDL y HDL) coexisten. Esta línea de coexistencia termina en el punto C´. Las curvas L y H son los límites de metaestabilidad de las fases HDA y LDA, respectivamente.
Los estudios de simulaciones, desde hace más de una década, han mostrado que el agua a bajas temperaturas presenta una inmiscibilidad líquido-líquido [119, 120, 121]. Diversos trabajos realizados por Moore et. al. [2009, 2010, 2010, 2009] [100, 1140, 122, 123], mediante simulaciones que abarcan un amplio rango de temperatura (entre 100 K y 350 K), han permitido profundizar sobre el comportamiento del agua en la zona no man´s land. Trabajos recientes, como los de Liu et. al. [2012] [133] cuyas simulaciones las realizaron en condiciones sobre-enfriadas (T = 228.6 K y 235 K a P = 2.2 kbar), muestran evidencia directa de una transición de primer orden entre la fase líquida de baja densidad (LDL con ρ ~ 0.9 g.cm-3) y la de alta densidad (HDL con ρ ~ 1.15 g.cm-3). A pesar de estos resultados, aún se presentan controversias sobre la existencia del segundo punto crítico. Una interpretación diferente ha sido expuesta por Limmer y Chandler [2011] [134], quienes concluyeron que la transición que ocurre es líquido-sólido y no líquido-líquido.
Por otra parte, dentro del escenario LLCP, la línea de coexistencia entre LDL y HDL a P < Pc´ y T > Tc ́ es llamada Widom Line, la cual se expresa en términos de presión y temperatura como Tw(P). La línea Tw(P) es la prolongación de la línea de coexistencia F de la figura I.8; se inicia en el punto donde se produce la divergencia de las funciones respuestas termodinámicas en el plano P - T (P ~ 1 atm y T ~ 235 K) y finaliza en el segundo punto crítico [46, 123]. De esta forma, la línea Tw(P) se relaciona con las anomalías del agua.
El cambio de fases líquido-líquido también se ha relacionado con cantidades dinámicas. Diferentes estudios experimentales han investigado los procesos de relajación en el agua sobre-enfriada en un amplio rango de temperatura y presión; han sugerido, que el agua exhibe una transición dinámica no-Arrhenius (líquido frágil) hacia
TN
Tem
pera
tura
[°C
]
1000 2000 3000 Presión [atm]
Tem
pera
tura
[K]
Figura I.8. Representación de las relaciones de fases entre el agua líquida: L, LDL, HDL, LDA y HDA . Figura extraída de ref. [3].

13
Capítulo I: Agua Líquida Sobre-enfriada
una de tipo Arrhenius (líquido fuerte) a P < Pc ,́ y cambios dinámicos no ocurren por encima del segundo punto crítico [6, 46]. En la figura I.9 se esquematiza un hipotético diagrama de fase en el cual se representa la línea Widom y la transición líquido-líquido HDL y LDL.
Por otra parte, distintos estudios han argumentado que el agua sobre-enfriada a la presión de 1atm presenta una transición fuerte – frágil representada por dos formas funcionales distintas, Arrhenius y Vogel-Tammann-Fulcher (VTF), respectivamente [6, 46]. Tales formas funcionales son frecuentemente usadas para analizar la transición frágil a fuerte en líquidos que forman, al igual que el agua, estructuras tipo red; esto ocurre en la temperatura del máximo del CP [124].
I.3. NUCLEACIÓN DE HIELO.
I.3.1. Teoría Clásica de la Nucleación (T.C.N.)
La congelación del agua es uno de los tantos fenómenos fundamentales que ocurren en la naturaleza, y a pesar que a nivel experimental y teórico se pueden encontrar diversos trabajos sobre la termodinámica y la cinética del proceso de nucleación de agua líquida sobre-enfriada metaestable [1, 2, 57, 75, 81, 91,101, 102], aún hay una amplia disertación sobre este tema.
Las bases teóricas del proceso de nucleación de hielo son dadas por la Teoría Clásica de Nucleación (T.C.N.) [1]. La misma plantea que la creación de la fase sólida del agua desde su estado metaestable sobre-enfriado, se produce a través de la formación aleatoria de “embriones de hielo”. Estos embriones tienen fluctuaciones continúas en sus tamaños debido a la incorporación o desprendimiento de moléculas. Si tales fluctuaciones generan un embrión con tamaño suficientemente grande como para que el crecimiento del mismo sea más probable que su desaparición, se dice que el embrión ha alcanzado el tamaño crítico que conlleva a la nucleación de la fase hielo.
La nucleación de hielo puede ocurrir espontáneamente en el seno del agua líquida pura sobre-enfriada, o puede ser inducida por un ente extraño. Estas situaciones se conocen como nucleación homogénea y nucleación heterogénea, respectivamente [80, 93].
Pc´
HDL (frágil)
LDL (fuerte)
Tc ́
Arrhenius a no-Arrhenius
Figura I.9. Representación esquemática del diagrama de fase planteado por la hipótesis LLCP.

14
Capítulo I: Agua Líquida Sobre-enfriada
Para el caso de la nucleación homogénea, la energía libre de Gibbs de la formación aleatoria de un embrión de hielo esférico se calcula mediante la ecuación ec.I.1.
∆�� �(�) = −���(�) − ��(�)� ∗ 4���33v(�)+ 4������(�) ec.I.1
donde µs y µl (µs > µl) son los potenciales químicos de las fases sólida y líquida, respectivamente; v es el volumen molecular del hielo; T la temperatura absoluta en Kelvin; re es el radio del embrión de hielo y σsl es la tensión interfacial sólida-líquida.
El primer término, del lado derecho de la igualdad, corresponde al cambio volumétrico de la energía libre de formación del embrión (∆���); el segundo término toma en cuenta el cambio de la energía libre superficial debido a la formación de la nueva interface (agua líquida sobre-enfriada metaestable/hielo). Dependiendo del tamaño de re habrá un punto donde ∆�� � alcanzará un máximo correspondiente al radio crítico re
* del embrión (∆�� �∗ ). El comportamiento cualitativo de la energía libre de Gibbs como función del radio se muestra en la figura I.10.
La probabilidad de formar un embrión crítico �∗ en un sistema compuesto de N moléculas por unidad de volumen está relacionada con la energía libre de Gibbs de la formación del embrión crítico (∆�� �∗ ) según ec.I.2.
�∗ = !exp &− ∆'()*∗ (+),-+
. ec.I.2
La formación del embrión crítico de hielo y su posterior crecimiento se encuentran limitados por el flujo (difusión) de moléculas de agua entre la interface agua líquida sobre-enfriada metaestable/hielo. Por lo tanto, el sistema debe vencer una barrera de energía libre de activación debida a la difusión de moléculas de agua hacia el embrión de hielo (∆/∗). La probabilidad de esta difusión (pd) por unidad de tiempo se expresa como en ec.I.3.
Figura I.10. Cambio de energía libre de Gibbs (∆Ghom) como función del radio del embrión. Las líneas punteadas corresponden a la energía superficial y volumétrica. La línea gruesa es la energía
total. Después que ∆Ghom alcanza el máximo en ��∗, la nucleación se da espontáneamente.
re re*
∆∆∆∆0123∗
∗
superficial
volumétrica

15
Capítulo I: Agua Líquida Sobre-enfriada
4 = ,-+� exp &− ∆g∗(+)
,-+. ec.I.3
donde h es la constante de Planck [1]. Una vez que el embrión de hielo alcanza el tamaño crítico, este crece rápidamente a expensa de las moléculas de agua líquida circundantes.
El producto de las ec.I.2 y ec.I.3 conduce a la tasa de nucleación homogénea (probabilidad de formación de embriones crítico de hielo por unidad de tiempo y volumen) según ec.I.4. Por lo tanto, la tasa de nucleación incluye parámetros cinéticos y termodinámicos; los cinéticos están relacionados con el flujo de moléculas sobre la superficie del núcleo que favorecen su crecimiento, y los termodinámicos con la energía libre de formación de embriones de hielo.
6� �(�) = ,-+� exp &− ∆g∗(+)
,-+. . !exp &− ∆'()*∗ (+)
,-+. ec.I.4
La T.C.N. también se aplica a la nucleación heterogénea de agua líquida sobre-enfriada metaestable, y la ec.I.4. sigue siendo válida mientras se considere que la formación de embriones de hielo es aleatoria. La diferencia radica en que la energía libre de formación del embrión crítico se expresa como ∆���8∗ = ∆�� �∗ 9, en donde f satisface 0 ≤ f ≤ 1 y es conocido como el factor de potencia catalítica. Este factor cuantifica la eficiencia que tiene un sustrato o núcleo de hielo IN (del acrónimo en inglés Ice Nucleus) para llevar a cabo la nucleación heterogénea [84]. Como puede observarse por los valores de f, la nucleación heterogénea siempre favorece la formación de hielo. Un f = 0 corresponde a un IN ideal, mientras que f = 1 es un sustrato totalmente ineficiente (nucleación homogénea). El valor de f depende del ángulo de contacto θ formado entre el IN y el embrión de hielo (figura I.11.); a su vez, el ángulo está vinculado con la composición química, tamaño, área superficial, morfología y estructura cristalográfica del IN [1, 82, 85].
I.3.2. Determinación Experimental de la Tasa de Nucleación.
La determinación experimental de J homogéneo y heterogéneo de agua líquida sobre-enfriada metaestable, en su mayoría se realiza mediante estudios estadísticos de congelación de gotas [54-56, 87]. Si la congelación de gotas se lleva a cabo a temperatura constante en un intervalo de tiempo, la tasa de nucleación se determina mediante una distribución de Poisson según ec.I.5 [1, 56, 86, 135]. En este caso, al igual que en la T.C.N., se asume que los embriones de hielo en gotas de agua sobre-enfriada, tienen la misma probabilidad de alcanzar el tamaño crítico, debido a las fluctuaciones
embrión de hielo θθθθ
sustrato (IN )
Agua sobre-enfriada
Figura I.11. Representación del ángulo de contacto θ entre el IN y el embrión de hielo en un entorno de agua líquida sobre-enfriada.

16
Capítulo I: Agua Líquida Sobre-enfriada
aleatorias de desprendimiento e incorporación que experimentan las moléculas de agua sobre los embriones. Por lo tanto la nucleación es estocástica y dependiente del tiempo.
,(�) = :(;.8�<,! > exp��j.�� ec.I.5
donde pk(t) es la probabilidad que k embriones de radio crítico se formen en un tiempo t dentro de una gota; y j es la tasa de nucleación extensiva (j=J*V; V es el volumen de la gota) que resulta ser la probabilidad de formación de embriones de hielo con radio crítico por unidad de tiempo, en esa gota.
La medición directa del número k de embriones que se forman en una gota de volumen V a un tiempo t, no es posible realizarlo. Sin embargo, lo que se mide es el número de gotas que permanecen líquidas a un tiempo t, Nt. El cociente entre éste número y el número inicial de gotas, Nt/N0, permite calcular j mediante la ecuación I.6.
ln &BCBD. � �j� ec.I.6
La figura I.12. muestra, a modo de ejemplo, el ln (Nt/N0) vs t de mediciones experimentales de nucleación homogénea de agua sobre-enfriada realizadas por Krämer et. al. [1999] [53] a la temperatura de 236 K. Según ec.I.6, la pendiente de la curva es la tasa de nucleación j.
Los estudios realizados por Huang et.al. [1995] [52], Krämer et.al. [1999] [53],
Stöckel et.al. [2005] [54], Kabath et.al. [2006] [55] y Baumgärtel y Zimmermann [2011] [56], han reportado Jhom = 1, 30, 4, 8.13 y 2.1 (x105 cm-3s-1), respectivamente, a T = 237 K. Tales resultados muestran las importantes discrepancias de Jhom entre diferentes autores, siendo de hasta un orden de magnitud entre resultados obtenidos a la misma temperatura. Estas discrepancias se pueden atribuir a los distintos diseños experimentales utilizados por los autores.
La determinación experimental de Jhet ha conllevado, desde hace décadas, a que exista un debate intenso en cuanto a si la nucleación heterogénea desde agua líquida sobre-enfriada se da por un proceso estocástico y dependiente del tiempo (hipótesis
Figura I.12. Relación de gotas no congeladas de radio ~ 60µm como función del tiempo a T = 236 K. La pendiente del ajuste lineal es la j homogénea. Figura copiada explícitamente de ref. [53].

17
Capítulo I: Agua Líquida Sobre-enfriada
estocástica), o si tiene lugar, de forma instantánea, sobre los “sitios activos” presentes en las superficies de los IN inmersos en las gotas de agua sobre-enfriada metaestable (hipótesis singular o determinística e independiente del tiempo) [1, 89, 91, 99, 135]. El comportamiento de la nucleación heterogénea estocástica es similar a la mostrada en la figura I.12. El comportamiento de la nucleación heterogénea singular se ejemplifica en la figura I.13. La figura muestra las mediciones experimentales realizadas por Niedermeier et. al. [2010] [136]. Los autores midieron, en función de la temperatura, la fracción de gotas que congelaron a distintos tiempos de experimentación (cuadrados anaranjados para un t = 1.6 s, cuadrados azules para un t = 10 s). El experimento lo realizaron en gotas que tuvieron inmersas partículas minerales de distintas composiciones y estructuras cristalinas.
Se puede observar en la figura I.13 que a distintos tiempos, la fracción de gotas congeladas no cambia, indicando que la nucleación heterogénea en el estudio de Niedermeier et. al. [2010] [136] soporta la hipótesis singular. En el Capítulo V de la presente tesis se ampliará sobre ambas hipótesis.
I.3.3. Estudios de Nucleación Homogénea mediante Simulaciones Computacionales.
Los estudios de nucleación homogénea de hielo desde agua líquida sobre-enfriada mediante simulaciones computacionales, se han visto limitados por la actual capacidad de cómputo. La nucleación homogénea usualmente no se observa a pesar de los enormes tiempos de simulación que se emplean (del orden de los microsegundos, lo que se traduce en varios meses de simulación para una única trayectoria atomística); solo en dos casos se observó nucleación homogénea. A saber, los estudios realizados por Matsumoto et. al. [2002] [62] y por Vrbka y Jungwirth [2006] [63].
Matsumoto et. al. [2002] [62] realizaron simulaciones a 230 K en un sistema tipo bulk constituido por 512 moléculas de agua. De las seis simulaciones realizadas, solo vieron nucleación en una de ellas. Al evaluar el cambio temporal de la energía potencial promedio del sistema calculado para cada simulación, los autores identificaron (en aquella simulación donde se produjo la nucleación del agua líquida sobre-enfriada)
Figura I.13. Dependencia con T, de la fracción de gotas de agua (con partículas minerales inmersas) que congelaron a distintos tiempos de experimentación. Cuadrados anaranjados son los datos obtenidos a t =
1.6 s. Los cuadrados azules son los datos obtenidos a t =10 s. La línea punteada señala la T donde la nucleación homogénea se produce. Figura modificada desde su fuente original [136].

18
Capítulo I: Agua Líquida Sobre-enfriada
cuatro zonas características del proceso de nucleación. En la figura I.14 se muestra un gráfico de la energía potencial vs tiempo de simulación; la región de color rojo (zona 1) corresponde a agua líquida sobre-enfriada, las regiones verde y azul (zonas 2 y 3) indican el proceso de nucleación y la región violeta (zona 4) a la formación total del hielo. Hasta la fecha este evento fortuito de nucleación no ha sido posible reproducirlo, ni siquiera por los mismos autores.
Vrbka y Jungwirth [2006] [63] estudiaron la nucleación homogénea de hielo a
250 K, mediante simulaciones en sistemas acuosos tipo slabs (estructuras bidimensionales generalmente utilizadas para evaluar procesos que se llevan a cabo en la interface) y constituidos con regiones bulk. Encontraron que la nucleación homogénea de hielo ocurre tanto en la zona bulk como en la región ubicada debajo de la capa cuasi-líquida (sub-superficie). En esta última es donde hubo mayor ocurrencia del evento de nucleación (figura I.15). Los autores señalan que las razones físicas para estos resultados se deben a un mejor ajuste del cambio del volumen cuando el agua congela en la sub-superficie que en el bulk, a la presencia de un campo eléctrico (0.1 - 1 V/Å) en la interface aire/agua producido por la orientación de las moléculas de agua de la superficie, y al hecho que la difusión del agua en la capa interfacial es ligeramente mayor que en la región bulk.
Figura I.14. Cambio temporal de la energía potencial. (1) período largo de reposo (rojo); (2) período de lenta disminución de la energía (verde); (3) período de rápida disminución de la energía (azul oscuro); (4) período de completa cristalización (violeta). La columna de color azul claro indica el
período en donde se produce la nucleación. Figura copiada explícitamente de la ref. [62].
t [ns]
Ene
rgía
Pot
enci
al [k
J m
ol-1
]

19
Capítulo I: Agua Líquida Sobre-enfriada
Las simulaciones para estudiar la nucleación heterogénea están enmarcadas principalmente en caracterizar distintos tipos de sustratos. Yokoyama y Hagiwara [2003] [65] elucidaron las interacciones entre un sustrato tipo hielo rodeado de agua líquida sobre-enfriada; encontraron que las moléculas de agua sobre-enfriadas más cercanas al sustrato se congelan rápidamente (~100 ps). Okawa et. al. [2006] [66] investigaron la nucleación heterogénea de agua sobre dos sustratos de platino, uno de superficie lisa y el otro de superficie rugosa; esta última la construyeron removiendo alguno de los átomos de la superficie lisa. Los resultados de este estudio mostraron que solo en el sustrato con superficie rugosa se produce la nucleación heterogénea de hielo.
Las investigaciones mencionadas evidencian los enormes esfuerzos que se vienen realizando para entender las propiedades del agua, en especial en la región sobre-enfriada donde las anomalías del agua se manifiestan marcadamente. Aún queda un largo trayecto por transitar; estudios experimentales, teóricos y de simulación computacional están muy lejos de explicar las anomalías observadas. Se siguen buscando respuestas para comprender fenómenos que se producen en una sustancia tan compleja y extraordinaria como el agua.
Figura I.15. Snapshots de la nucleación homogénea de sistemas slabs con regiones bulk de: a) 384 moléculas de agua. b) 576 moléculas de agua. Las regiones sombreadas corresponden a la formación del
embrión de hielo en la sub-superficie y en el bulk. Figura copiada explícitamente de ref. [63].
a) b)

20 Capítulo II: Dinámica Molecular
CAPÍTULO II
DINÁMICA MOLECULAR
El desarrollo y la rápida evolución de las computadoras han permitido que surjan numerosos métodos computacionales que son empleados para la resolución de problemas científicos. Entre los distintos métodos, la Dinámica Molecular (D.M.) es una de las principales herramientas utilizada en el estudio de sistemas complejos constituidos por muchas partículas, y en especial, en aquellos sistemas que se encuentran en fase líquida [9]. Las primeras conclusiones relevantes sobre el comportamiento de líquidos empleando esta técnica se remonta a los 50´s [13]. Años más tardes, destacados avances se obtienen con las primeras simulaciones en argón líquido, en agua líquida y en un sistema biológico (proteína tripsina pancreática bovina) [13]. Desde entonces hasta hoy en día, y debido a la influencia fundamental del agua en los procesos biológicos, ha habido un amplio desarrollo de métodos y modelos de D.M. que han permitido caracterizar sistemas acuosos no solo en condiciones ambientales de temperatura y presión, sino también en condiciones diferentes [9, 13].
La dinámica molecular es una técnica que permite predecir la evolución temporal de un sistema molecular. La información que se obtiene a nivel microscópico son las posiciones y las velocidades de cada partícula en cada instante de tiempo. A partir de este conocimiento microscópico de la dinámica del sistema se calculan los valores de diferentes propiedades macroscópicas estáticas y dinámicas.
En dinámica molecular clásica se trata a las moléculas que conforman un sistema, como un conjunto de centros de interacción ubicados en diferentes posiciones (la posición de la molécula usualmente se define según las posiciones de sus átomos), que interactúan mediante potenciales simples. En la mayoría de las aplicaciones la energía potencial total del sistema (V) se calcula mediante el enfoque de aproximación aditiva de pares, el cual consiste en sumar todas las interacciones entre todos los pares presentes en el sistema. La interacción entre dos partículas puede ser afectada por la presencia de una tercera, una cuarta o más; por lo tanto, la interacción, por ejemplo, entre tres partículas A, B y C no se obtiene estrictamente como la suma de las energías de interacción entre pares: V(A,B,C) ≠ V(A,B) + V(A,C) + V(B,C). Sin embargo, como la contribución de interacciones entre tres cuerpos es en general muy pequeña, especialmente en condiciones bulk, habitualmente no se considera [13].
La elección del tipo de potencial intermolecular juega un papel central en simulaciones de D.M. Las características de este potencial relacionadas con la arquitectura de las moléculas, la forma funcional del potencial y los parámetros de interacción se pueden obtener mediante cálculos mecánico-cuánticos. El conjunto de tales características se conoce como Campos de Fuerzas (del acrónimo en inglés Force Field) y se emplean para tratar los sistemas complejos que se simulan con D.M. En la actualidad existen diferentes campos de fuerzas que se usan en distintos paquetes (software) de D.M. En el caso del agua, desde hace décadas, se vienen desarrollando diversos campos de fuerzas o modelos de agua con el fin de obtener una descripción física del agua, la cual permita reproducir sus propiedades [17] y por ende lograr comprender el origen de las diversas anomalías. En dichos modelos, la interacción entre

21 Capítulo II: Dinámica Molecular
dos moléculas de agua se representa típicamente por una suma de interacciones Lennard-Jones y Coulómbicas.
Los estudios llevados a cabo en la presente tesis se realizaron con distintos modelos de agua, bajo condiciones de temperatura, presión y número de moléculas constantes. Todas las simulaciones se llevaron a cabo con el paquete de cálculo Gromacs v.4.5.5 [50, 51]. Los sistemas evaluados, en su mayoría, fueron representados por un bulk de agua en el régimen sobre-enfriado metaestable. Desde el punto de vista de la simulación, estas condiciones pueden ser consideradas de equilibrio ya que la barrera de energía libre para acceder al verdadero equilibrio (hielo hexagonal) del sistema es relativamente alta y por lo tanto el sistema permanece en estado líquido durante toda la simulación. Los sistemas utilizados en las simulaciones realizadas y cuyos resultados se presentan en el Capítulo V, se tratan de sistemas en el régimen sobre-enfriado pero no en condiciones de equilibrio, los mismos están compuestos inicialmente por un sustrato o núcleo de hielo (IN) inmerso en agua sobre-enfriada. La evolución del sistema lleva esta mezcla a un estado de equilibrio, que puede ser hielo o agua dependiendo de las condiciones.
A continuación se comentan algunos aspectos relacionados con los fundamentos teóricos de la D.M. En la siguiente sección se mencionan ciertas técnicas que se deben emplear cuando se realizan simulaciones en sistemas líquidos tipo bulk y de tamaños considerablemente pequeños (~ 10000 moléculas) en comparación con los reales (~ 1023 moléculas). La última sección describe algunas de las propiedades macroscópicas que se pueden calcular con D.M.
II.1 PRINCIPIOS TEÓRICOS.
La dinámica molecular asume que la dinámica de N partículas (átomos o moléculas) que conforman un sistema está gobernada por las ecuaciones clásicas de movimiento de Newton (ec.II.1):
( )i
Nii
iiN r
rrVtam
dt
rdmrr r
rrr
rrrr
∂∂−=== ),...,(
),...,( 12
2
1iF ec.II.1
FEFFFG es la fuerza actuando sobre el i-esimo átomo debido a las interacciones con los otros
N-1 átomos; HGI la aceleración, irr
la posición y mi la masa del i-esimo átomo; JKJLGM es el
gradiente de la energía potencial. N��GO, … , �GB� describe la energía potencial total del sistema.
Una simulación con D.M. consta básicamente de cuatro etapas [9]: 1. Establecer los parámetros de la simulación: temperatura, presión, densidad, paso
de integración (∆t), número de pasos a integrar (tiempo total de simulación), etc. El tiempo real de la simulación sería ∆t * número de pasos a integrar.
2. Asignar posiciones y velocidades iniciales a las moléculas del sistema. Las velocidades iniciales se escogen aleatoriamente a partir de una distribución de Maxwell-Boltzmann. Esta distribución gaussiana de velocidad, da la probabilidad p de que una molécula i tenga una velocidad vi en la dirección i(x,y,z) a una temperatura T predefinida:

22 Capítulo II: Dinámica Molecular
�RI� � S& �MT,-+. �U &�MVMW,-+. ec.II.2
Las velocidades se ajustan de forma tal que el momento lineal del sistema sea cero, a fin de mantener la energía del sistema constante.
3. Realizar una corrida al sistema, con las posiciones y velocidades iniciales, para calcular las fuerzas sobre todas las moléculas e ir integrando las ecuaciones de movimiento. Esta integración se realiza para cada ∆t (que debe ser suficientemente pequeño) durante el tiempo total de simulación. Típicamente ∆t = 1fs, un orden de magnitud menor que los movimientos más rápidos que ocurren en el sistema (vibraciones de enlaces que involucran átomos de hidrógeno, ~10 fs). En cada paso de integración se almacenan datos de posición y velocidad de cada molécula para su posterior análisis.
4. Analizar las trayectorias de cada molécula para determinar las propiedades dinámicas y termodinámicas del sistema.
La aplicación de un método numérico discreto es necesaria para integrar las
ecuaciones de movimiento, explícitamente a partir del cálculo de las fuerzas FFFGI, de las cuales se determinan las nuevas posiciones rGI de la molécula para un cierto ∆t (ec.II.1). Se han desarrollado distintos algoritmos para integrar las ecuaciones de movimiento [10, 15]: el método de Euler, el de Verlet, el salto de rana, el de velocidad de Verlet, el de Beeman y el de Gaer, entre otros. A continuación se describe brevemente el algoritmo salto de rana ya que por defecto es usado por el Gromacs y por ende, empleado en la presente tesis.
El salto de rana usa las posiciones �GI y aceleraciones HGI a un tiempo t, y las
velocidades RGI a un tiempo � � O∆� . Las relaciones son:
�GI�� + ∆�� � �GI��� + RGI &� + O∆�.∆� ec.II.3
RGI &� + O∆�. � RGI &� � O∆�. + HGI���∆� ec.II.4
Las velocidades de las moléculas se incluyen explícitamente pero a un tiempo distinto al de las posiciones; es decir, las posiciones se calculan en intervalos enteros y las velocidades en la mitad de ellos. Las velocidades a un tiempo t se calculan mediante la siguiente aproximación (promedio):
RGI��� � VFGM�8X∆8/�ZVFGM�8Z∆8/� ec.II.5
La simplicidad de este algoritmo y el cálculo preciso de las posiciones conducen a que a menudo, ésta sea la primera opción para la simulación molecular. La precisión tiene que ver con la magnitud del error. El salto de rana incluye un error de ∆t2 para la posición.

23 Capítulo II: Dinámica Molecular
II.2 TÉCNICAS.
Las simulaciones con D.M. se realizan para un número pequeño de moléculas (~ 10000 partículas) en comparación a sistemas reales que contienen del orden de 1023 moléculas. Tal limitación es debido a que el tiempo requerido para evaluar las interacciones entre moléculas es proporcional a N [9, 13, 50, 51], lo cual incrementa enormemente el tiempo de cálculo. Bajo estas condiciones, en las simulaciones se deben emplear técnicas especiales para que un sistema pequeño cumpla ciertas características, de tal forma que se asemeje al comportamiento de uno real.
� Condiciones de contorno periódicas. Toda simulación de un sistema de partículas se debe realizar en un determinado
volumen. Tal confinamiento genera que las moléculas cercanas a las paredes del recipiente presenten efectos de superficie y por ende tienen energía y propiedades distintas a las ubicadas en el seno del sistema. Para reducir dichos efectos y con el objetivo de obtener propiedades bulk, en el cálculo se imponen condiciones de contorno periódicas (pbc) [12, 14, 20, 50, 51]. Las pbc consisten en rodear la caja o celda básica (celda de simulación) de réplicas de ella misma (figura II.1). En la práctica, la caja de simulación no tiene que ser necesariamente cúbica. La periodicidad de las condiciones de contorno genera dos cosas: 1. la partícula al dejar la celda central de simulación a través de una cara particular del volumen, inmediatamente entra a la celda central por la cara contraria, haciendo que el número de partículas de la celda de simulación y por ende la del sistema total se conserve. 2. una partícula determinada (por ejemplo la partícula 1 en la figura II.1) interacciona con todas las demás, es decir, con las que se encuentran en la celda de simulación y en las réplicas, generando múltiples interacciones que conlleva a elevados tiempos de simulaciones y costos computacionales; para evitar esto, se consideran solo las interacciones de la partícula con las otras partículas más cercanas; por ejemplo, las interacciones que sólo se toman en cuenta entre la partícula 1 y las demás (figura II.1) son aquellas que se ejercen dentro del cuadrado punteado, mientras que las interacciones con las partículas más lejanas (fuera del cuadrado punteado) se desprecian. Este criterio se conoce como convención de mínima imagen [50, 51].
Por último, la replicación del sistema, a nivel práctico, no presenta problemas técnicos. El almacenamiento de las coordenadas de todas las réplicas es innecesario, solo son importantes las de la celda central de simulación.
Figura II.1 : Ilustración en dos dimensiones de las condiciones de contorno periódicas, convención
de mínima imagen y radio de corte. Las partículas pueden entrar y salir a través de cuatro
1
2 3
4
5
24 Capítulo II: Dinámica Molecular
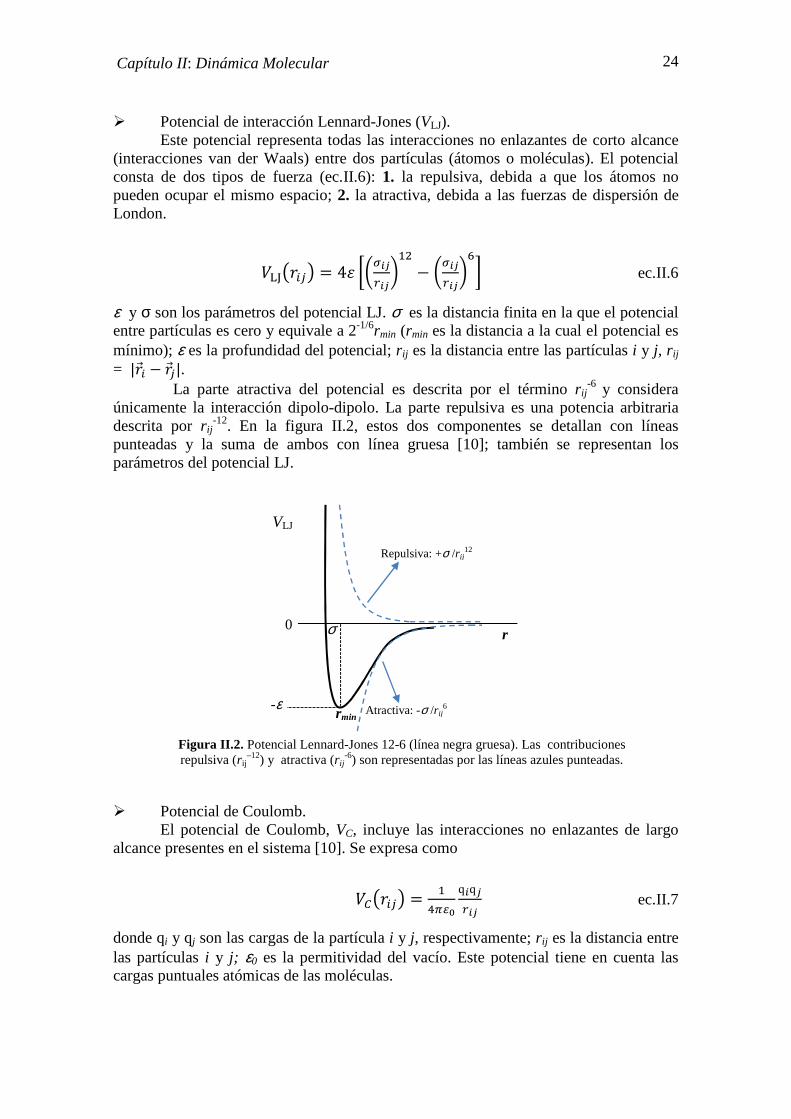
� Potencial de interacción Lennard-Jones (VLJ). Este potencial representa todas las interacciones no enlazantes de corto alcance
(interacciones van der Waals) entre dos partículas (átomos o moléculas). El potencial consta de dos tipos de fuerza (ec.II.6): 1. la repulsiva, debida a que los átomos no pueden ocupar el mismo espacio; 2. la atractiva, debida a las fuerzas de dispersión de London.
N[\]�I;^ � 4_ `abMcLMcdO � abMcLMcd
ef ec.II.6
ε y σ son los parámetros del potencial LJ. σ es la distancia finita en la que el potencial entre partículas es cero y equivale a 2-1/6rmin (rmin es la distancia a la cual el potencial es mínimo); ε es la profundidad del potencial; r ij es la distancia entre las partículas i y j, rij = |�GI � �G;|. La parte atractiva del potencial es descrita por el término r ij
-6 y considera únicamente la interacción dipolo-dipolo. La parte repulsiva es una potencia arbitraria descrita por r ij
-12. En la figura II.2, estos dos componentes se detallan con líneas punteadas y la suma de ambos con línea gruesa [10]; también se representan los parámetros del potencial LJ.
� Potencial de Coulomb. El potencial de Coulomb, VC, incluye las interacciones no enlazantes de largo
alcance presentes en el sistema [10]. Se expresa como
Nh]�I;^ � OiTjDqMqcLMc ec.II.7
donde qi y qj son las cargas de la partícula i y j, respectivamente; r ij es la distancia entre las partículas i y j; ε0 es la permitividad del vacío. Este potencial tiene en cuenta las cargas puntuales atómicas de las moléculas.
σ
VLJ
0
-ε
Repulsiva: +σ /r ij12
Atractiva: -σ /r ij6
Figura II.2. Potencial Lennard-Jones 12-6 (línea negra gruesa). Las contribuciones repulsiva (r ij
–12) y atractiva (r ij-6) son representadas por las líneas azules punteadas.
r
rmin

25 Capítulo II: Dinámica Molecular
� Truncamiento del Potencial. Esta técnica se utiliza para minimizar el número de interacciones no enlazantes
presentes en el sistema; con ello se reduce el costo computacional causado por las pbc impuestas al sistema. Cuando se trunca el potencial solo se consideran las fuerzas ejercidas por los vecinos más cercanos a cada partícula. Para tal fin, se establece un radio de corte (cutoff), rc, en donde las interacciones más allá de él se desprecien. En otras palabras, para r ij > rc el V(r ij) = 0. En la práctica se suele usar un rc = 2.5σ [12, 14].
Los resultados de las simulaciones dependen mucho del valor asignado a rc puesto que un truncamiento simple del potencial puede provocar resultados erróneos. La mayor incidencia del rc se presenta en las interacciones Coulómbicas, su efecto no es despreciable incluso si este es lo suficientemente grande; esta situación es contraria a la presentada con las interacciones dispersivas (LJ), en las cuales el efecto de rc es casi inapreciable. Tomando en consideración lo anterior, en la literatura se encuentran diferentes tratamientos para la parte del potencial que involucra las interacciones de largo alcance (el potencial de Coulomb) [21, 22, 23, 50, 51, 105]. El más usado en D.M. es el método sumas de Ewald [23] y su variante PME (acrónimo de Particle Mesh Ewald) [22, 23].
La energía total de Coulomb VC para un sistema periódico de N partículas situadas en una celda cúbica genérica de lados Lx, Ly y Lz y sus infinitas réplicas o imágenes se expresa como:
Nh]�I;^ � O∑ ∑ ∑ ∑ ∑ qMqciTjDLMc�m�B;BImnmomp ec.II.8
donde r ij(L) es la distancia entre las cargas eléctricas. La ec.II.8 tiene la característica que su convergencia es lenta. En 1921 Ewald P.
propone una nueva técnica para el cálculo de Vc, conocida como el método de sumas de Ewald [21, 50, 51]. Esta técnica consiste en transformar la ec.II.8 en una suma de tres términos, dos de ellos rápidamente convergentes y uno constante; su expresión es:
N � NL�q� + NL�r + Nr88� ec.II.9
Vreal es el potencial debido a las sumas de las interacciones electrostáticas en el espacio real; Vrec en el espacio recíproco o de Fourier; y el término constante Vctte el cual anula las interacciones de las partículas consigo mismas consideradas en Vreal y Vrec.
Las sumas de Ewald es un método poco práctico cuando se simulan sistemas grandes (requerimiento de cálculo es N3/2). Con el fin de mejorar la eficiencia computacional, Darden T. et. al. (1993) [22] desarrollaron el algoritmo PME (Particle Mesh Ewald). La técnica PME está basado en las sumas de Ewald con un tiempo de cálculo proporcional a N log N [22, 23, 24].
� Termostato de Nosé-Hoover. Los termostatos permiten mantener constante la temperatura de un sistema. En
simulaciones se utilizan algoritmos cuya aplicación implica, que las ecuaciones de Newton para las partículas de un sistema se modifican de tal forma que el intercambio energético mantenga constante la temperatura. Desde el punto de vista mecánico estadístico, la imposición de una temperatura a un sistema se hace a través de colocar a

26 Capítulo II: Dinámica Molecular
este en contacto con un baño térmico externo (acoplamiento) [9, 11, 50, 51]. Hoy en día existen diversos termostatos [25, 26, 27, 106]. Brevemente se describirá el de Nosé-Hoover ya que fue utilizado en la presente tesis.
Nosé S. en 1984 demostró que la temperatura de un sistema se puede controlar mediante la modificación del Hamiltoniano de las ecuaciones de movimiento [27]. Para ello, Nosé agrega una nueva coordenada s al Lagrangiano del sistema, la cual representa virtualmente a un termostato. Hoover W. (1985) [28] optimiza el Hamiltoniano de Nosé con el fin de mantener constante el paso de integración (∆t) de las ecuaciones de movimiento (requisito para el cálculo adecuado de las propiedades termodinámicas), contrario a lo que ocurría con el Hamiltoniano de Nosé. El termostato de Nosé-Hoover reescalea las velocidades en cada ∆t en función de la diferencia de temperatura entre el baño térmico y el sistema. El factor de escalamiento incluye una constante de tiempo que permite, según el requerimiento del usuario, un acoplamiento débil o fuerte. El valor de la constante de tiempo suele elegirse de 0.01 ps para acelerar la aproximación del sistema al equilibrio. Una vez que está equilibrado, la constante de tiempo se incrementa a valores mayores (0.5 ps) para minimizar el efecto del termostato sobre la dinámica del sistema.
� Barostato de Parinello-Raphman. Los barostatos permiten mantener constante la presión de un sistema. En
simulaciones se emplean algoritmos en donde las ecuaciones de Newton para las partículas se deben modificar de tal forma que la fluctuación en el volumen mantenga constante la presión. El sistema se acopla a un baño de presión para mantener constante a esta variable dentro del volumen de la celda de simulación [9, 50, 51]. Los baróstatos más utilizados en dinámica molecular son el de Berendsen [26] y el de Parinello- Raphman [29].
En el barostato de Parinello-Raphman tanto las coordenadas de las partículas que conforman el sistema como las de la caja de simulación son escaladas en cada paso de integración; y al igual que el termostato de Nosé-Hoover, el factor de escalamiento incluye una constante de tiempo que la elige el usuario. En muchos casos se recomienda combinar el termostato de Nosé-Hoover con este baróstato, ya que funciona muy bien cuando el sistema está cerca del equilibrio o en el equilibrio.
II.3. PROPIEDADES MACROSCÓPICAS.
La información microscópica del espacio multidimensional o espacio de fase (conjunto de posiciones y velocidades de las moléculas para cada tiempo de simulación) obtenida con la D.M. se debe trasladar al nivel macroscópico para estudiar propiedades observables tales como temperatura, presión, capacidad calorífica, coeficiente de difusión, densidad, etc. Para ello, la dinámica molecular promedia en el tiempo las trayectorias de las diferentes partículas que constituyen el sistema. La forma de construir el promedio temporal es:
⟨s⟩8��t Lq� � Ou∑ s�∆��u8v8�w ec.II.10

27 Capítulo II: Dinámica Molecular
donde A es el número de etapas o el número total de pasos de integración de la simulación (adimensional), teq el tiempo de la simulación donde el sistema ha alcanzado el estado de equilibrio y B(∆t) es el valor de la propiedad B en el paso de integración ∆t. En resumen, la D.M. permite calcular observables en condiciones de equilibrio y a su vez apreciar la evolución temporal de algunos fenómenos que se encuentran ligeramente fuera del equilibrio, como son los procesos de nucleación o de crecimiento de cristales.
A continuación, se mencionan algunas de las propiedades macroscópicas que se calculan [8, 9, 13, 50, 51]. Los valores promedios de las energías potencial (V) y cinética (K) se obtienen a través de:
N � ⟨N⟩ � Ou∑ ∑ N]�GI;^BIvOu8v8�w ec.II.11
x � ⟨x⟩ � Ou∑ ∑ OBIvOu8v8�w yIRGI�∆�� ec.II.12
Las propiedades estructurales estáticas de un sistema se describen a través de la función de distribución radial g(r), también llamada función de correlación de pares., Ésta mide la probabilidad de encontrar la partícula j a una distancia r =|�G| de la partícula de referencia i. El algoritmo consiste en determinar el número de partículas (n) que se encuentran a una distancia �G y �G + ∆�G de la partícula i. Matemáticamente se expresa:
g�r� � VB ⟨z�L,LX∆L�⟩iTLW∆L ec.II.13
La propiedad dinámica “coeficiente de difusión” de la i-ésima partícula Di, se puede calcular utilizando
6|I� � lim8→�⟨|�GI(�) − �GI(0)|⟩ ec.II.14
donde ⟨|�GI(�) − �GI(0)|⟩ representa la desviación cuadrática media de la posición de la partícula i respecto a su posición inicial �GI(0). Esta ecuación es válida para tiempos largos de simulación [13].
La capacidad calorífica isobárica, CP, se puede determinar a partir de las fluctuaciones de la entalpía H de la forma �P = ]⟨HW⟩Z⟨H⟩W^
,-+W; o mediante una diferenciación directa de una función que ajusta los
datos entalpía-temperatura. Ambos métodos proporcionan resultados similares; sin embargo, el segundo método se utiliza con mayor frecuencia puesto que reduce el ruido en los resultados y por lo tanto permite una determinación más precisa de la CP [47].
II.4. MODELOS DE AGUA.
El cálculo de las propiedades promedios de un sistema mediante simulaciones de D.M. requiere de un modelo que defina las interacciones entre las moléculas. Como la sustancia de estudio en esta tesis es el agua, la descripción de tales interacciones se

28 Capítulo II: Dinámica Molecular
realiza mediante modelos de agua. Desde los primeros trabajos de simulaciones, se encuentra un amplio espectro de modelos enfocados en mejorar la descripción física del agua. La intención de los mismos es reproducir las propiedades del agua en un amplio rango de condiciones termodinámicas y garantizar la transferibilidad de la mencionada descripción a varios escenarios (soluciones, interfaces, clusters, etc.).
Los modelos de agua, según la manera de representarla, se clasifican en tres tipos [10, 18]: los rígidos, que son los más simples porque cada molécula de agua mantiene una geometría fija; los flexibles, que incluyen vibraciones intramoleculares; y tanto para los modelos rígidos y flexibles, se le pueden incluir explícitamente las polarizabilidades atómicas. La utilización de un modelo dado depende básicamente de dos factores: la precisión y la eficiencia computacional; este último ha tenido un peso importante en las simulaciones computacionales, lo que ha traído como consecuencia que la mayoría de los potenciales de agua desarrollados sean empíricos.
El desarrollo de la presente tesis se realiza con modelos rígidos y no polarizables. Indiscutiblemente, el agua no es una molécula rígida con tres, cuatro, cinco, o más cargas puntuales estratégicamente colocadas; sin embargo, la intencionalidad de estos modelos simples es tratar de reproducir la característica más importante del agua en sus fases condensadas: el enlace puente de hidrógeno.
La plataforma general de los modelos empíricos es representar el enlace puente de hidrógeno como una competencia entre una energía potencial intermolecular atractiva, bien aproximada por interacciones electrostática clásicas (potencial de Coulomb), y una energía potencial intermolecular repulsiva representado por un potencial Lennard-Jones centrado sobre el átomo de oxígeno [17, 18]. Los parámetros de carga y de repulsión son ajustados para reproducir algunas propiedades del estado gaseoso (momento dipolar, energía de interacción entre dímeros, etc.) o algunas del estado líquido en condiciones de temperatura y presión estándar (calor de vaporización, densidad, etc.) mediante cálculos con D.M. o Monte Carlo. El mencionado ajuste, conocido como parametrización, consiste en ir modificando los parámetros hasta alcanzar un nivel adecuado de concordancia entre los valores calculados y experimentales de distintas características del agua. La distribución de carga de la molécula de agua se modela mediante cargas positivas ubicadas sobre los átomos de hidrógeno, y la negativa, dependiendo del modelo, se puede localizar sobre el átomo de oxígeno o en varios sitios denominados ficticios ubicados en el plano de la estructura molecular, o fuera de este.
Los modelos empíricos, rígidos y no polarizables utilizan potenciales de pares efectivos para obtener la energía potencial total del sistema. Consideran interacciones electrostáticas entre tres y seis sitios y requieren de menor esfuerzo computacional que los flexibles y los polarizables. A continuación, se describen las características generales de los modelos usados en la presente tesis y se enfatizan las más relevantes.
Los modelos SPC y SPC/E [30] presentan una distribución de carga eléctrica representada por tres cargas puntuales, la positiva se distribuye entre los hidrógenos y se balancea por una carga negativa apropiada localizada sobre el átomo de oxígeno (-qO = 2qH). Estos modelos no utilizan los valores experimentales de distancia (dOH) ni ángulo (∠H-O-H), sino dO-H = 1 Å y ∠H-O-H = 109.5°. La diferencia entre ellos radica que el SPC/E incluye una corrección llamada auto-polarización. La corrección toma en cuenta el trabajo necesario para polarizar una molécula con momento dipolar, de la fase gaseosa a la fase líquida [69].

29 Capítulo II: Dinámica Molecular
Los modelos TIPXP fueron propuestos por Jorgensen et. al. [1983, 2000] [31, 35] y consideran que los valores dO-H y ∠H-O-H son los experimentales: 0.9578 Å y 105.46 °, respectivamente. Entre ellos, se puede mencionar el TIP3P, que al igual que el SPC/E, está construido con tres cargas puntuales localizadas sobre los átomos de la molécula de agua. Además de la diferencia geométrica con el SPC/E, los parámetros potenciales del TIP3P fueron obtenidos ajustando solo la densidad del líquido al valor experimental en condiciones de temperatura y presión ambiente (298 K y 1 bar). El SPC/E no solo emplea como parámetro de ajuste la densidad, sino también la entalpía de vaporización del agua (corregida por la llamada corrección de auto-polarización) [69].
El TIP4P incluye cuatro sitios de interacción. La carga negativa está localizada en un centro M (sitio ficticio) a una distancia dO-M= 0.015nm [18] del átomo de oxígeno a lo largo de la bisectriz del ∠H-O-H, y la carga positiva se distribuye igualmente entre los átomos de hidrógeno. Esta parametrización se hace ajustando la densidad a T y P ambiente y la entalpía de vaporización a T ambiente. El TIP4P presenta variantes tales como el TIP4P-Ew [33], el TIP4P-2005 [103] y el TIP4P-Ice [34]. En todos ellos los parámetros han sido ajustados para reproducir las propiedades en la que el modelo original fallaba. Por ejemplo, para el caso del TIP4P-Ew y TIP4P-2005 se ajustaron los parámetros para reproducir las densidades isobáricas a presión ambiente; además, en el caso del TIP4P-2005 también se ajustaron un par de propiedades relacionadas con la densidad y la estabilidad de estructuras polimórficas de hielo. Por otro lado, en el TIP4P-Ice los ajustes se realizaron para reproducir la temperatura de fusión experimental. La Ew del TIP4P-Ew hace referencia a las interacciones electrostáticas de largo alcance tratadas mediante sumas de Ewald; esto es, sus parámetros potenciales fueron optimizados con dichas sumas para reproducir las densidades y entalpías de vaporización experimentales a diferentes T. A diferencia del TIP4P (método cut-off para tratar las interacciones electrostáticas), el TIP4P-Ew reproduce mejor la densidad isobárica y el coeficiente de difusión del agua.
El TIP5P es otro miembro de la familia de los TIP construido con cinco cargas puntuales. Fue propuesto en el año 2000 por Mahoney y Jorgensen [2000] [35], cuya carga negativa se divide entre dos cargas parciales ubicadas en posiciones ficticias L denominadas par solitario (LP), el cual representa uno de los pares de electrones no enlazantes del oxígeno. Un modelo similar a este, muy utilizado en los 70, es el ST2 [104], pionero en simulaciones computacionales; a diferencia del TIP5P, el ST2 es incapaz de reproducir el máximo de densidad y el punto de fusión. El TIP5P ha sido parametrizado para ser usado considerando interacciones de largo alcance truncadas a un cut-off = 9 Å; y se ha mostrado que reproduce muy bien muchas de las propiedades del agua líquida, incluyendo la contante dieléctrica, la constante de difusión y el máximo de la densidad. Sin embargo, debido a los efectos de condiciones de contorno, las propiedades del modelo TIP5P son dependientes del tamaño del sistema y se modifican notablemente cuando el tamaño del mismo es cambiado, por ejemplo, de 512 a 216 moléculas para un cut-off = 8Å [36]. También, su imprecisión incrementa considerablemente cuando es usado con otros métodos para tratar las interacciones de largo alcance y comparado con el TIP4P, el TIP5P muestra mayor dependencia sobre el tamaño de los sistemas [36]. Empero, cuando se usa Ewald para reparametrizar el TIP5P, la dependencia con el tamaño del sistema se elimina. De esta forma se obtiene el modelo TIP5P-Ew de Rick [2004] [36], el cual reproduce con exactitud las propiedades

30 Capítulo II: Dinámica Molecular
termodinámicas, dieléctricas y dinámicas en un rango de temperatura y presión, tal como lo hacía el original TIP5P.
El modelo Six-Sites está construido con seis sitios de interacción. Fue desarrollado por Nada et. al. en el 2003 [19] con la intención de simular hielo y agua cerca del punto de fusión. La carga positiva se distribuye entre los hidrógenos, y la negativa entre tres posiciones ficticias (un LP como en el TIP5P) y en un centro M localizado sobre la bisectriz del ángulo H-O-H (como en el TIP4P). A diferencia de los modelos TIP, su interacción Lennard-Jones no sólo actúa sobre el átomo de oxígeno sino sobre los hidrógenos. Los valores de dH-O y ∠H-O-H provienen de los promedios experimentales de la fase vapor e hielo.
La figura II.3 muestra la representación de las estructuras de los distintos modelos de agua previamente descritos; la tabla II.1 reúne los valores de sus parámetros: σ y ε del potencial Lennard-Jones, y del parámetro q del potencial de coulomb para el átomo de oxígeno o de los sitios ficticios (centro M y LP).
Tabla II.1. Parámetros de los modelos de agua SPC/E, TIP3P, TIP4P, TIP4P-Ew, TIP4P-2005,
TIP5P, TIP5P-Ew, Six-Sites. PARÁMETROS
MODELO σ
[nm] ε
[kJ mol-1] q1 [e]
q2 [e]
d1 [Å]
d2 [Å] θ° φφφφ°
SPC/E[30] 0.3166 0.650 +0.4238 -0.8476 1.0000 - 109.47 -
TIP3P [31] 0.315061 0.6364 +0.4170 -0.8340 0.9572 - 104.52 -
TIP4P [32] 0.315365 0.6480 +0.5200 -1.0400 0.9572 0.15 104.52 52.26
TIP4P-Ew [33] 0.316435 0.680946 +0.52422 -1.0484 0.9572 0.125 104.52 52.26
TIP4P-2005 [103] 0.31589 0.7749 +0.5564 -1.1128 0.9572 0.1546 104.52 52.26
TIP5P [35] 0.312000 0.6694 +0.2410 -0.2410 0.9572 0.70 104.52 109.47
TIP5P-Ew[36] 3.097 0.7448 +0.2410 -0.2410 0.9572 0.70 104.52 109.47
Six-Sites [19] 3.115OO 0.673HH
0.715OO 0.115HH
+0.477 -0.044L -0.866M
0.980 0.8892L 0.2300M
108.00 111.00
Figura II. 3. Representación esquemática de diferentes modelos de agua. En a) Los de tres puntos; b) Los de cuatro puntos; c) Los de cinco puntos; d) El Six-Sites. Las esferas blancas,
rojas y amarillas simbolizan los átomos de hidrógeno, oxígeno y los sitios ficticios (el centro M y los L para el LP), respectivamente. La representación del Six-Sites fue extraída explícitamente de
ref. 29. q1 es la carga del H; q2 es la carga del O o del M o del L, según el modelo de agua.
q1 q1 q1
H H
d1
θθθθ
M d2
O
φφφφ
b) a)
H H
d1
θθθθ
O
c)
H H
d1
θθθθ
d2
L L
O
d)
q1
q2
q2 q1 q1
q2 q2 φφφφ
φφφφ
θθθθ

31 Capítulo II: Dinámica Molecular
Los modelos descritos tienen en común que el momento dipolar siempre es mayor que el momento dipolar de la molécula de agua en fase gas (µ = 1.885 D [17]), lo que permite compensar la falta de polarizabilidad en los mismos. Por otra parte, cuando se utilizan determinadas propiedades para llevar a cabo el proceso de parametrización de un modelo específico, las demás propiedades siempre sufren modificaciones, unas en mayor medida que otras [10, 17, 18, 69].

32 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
CAPÍTULO III
ESTUDIO DEL COMPORTAMIENTO DE ALGUNAS PROPIEDADES
DEL AGUA LÍQUIDA REPRESENTADAS POR DISTINTOS MODELO S
III.1. ANÁLISIS DE DISTINTOS MODELOS DE AGUA EN EL RÉGIMEN
SOBRE-ENFRIADO METAESTABLE. REPARAMETRIZACIÓN DEL MODELO TIP5P-Ew.
La presente sección muestra la metodología, los resultados y análisis de diferentes modelos de agua en su capacidad para representarla en el régimen sobre-enfriado metaestable. Este fue el primer estudio desarrollado en la presente tesis y consistió en evaluar la habilidad de los modelos para reproducir propiedades tales como: el coeficiente de difusión D, la densidad de masa ρ y la temperatura de fusión Tf. La variable principal de control para el monitoreo de los modelos fue D; su elección radicó en que la misma permite caracterizar la dinámica de las moléculas de agua, comportamiento vinculado con la cinética y por ende con la tasa de nucleación de hielo.
Los modelos seleccionados fueron: TIP3P [31], TIP4P [32], TIP4P-Ew [33], TIP5P [35] y TIP5P-Ew [36], motivado por el amplio uso que se les ha dado en estudios vinculados con la biología, las ciencias atmosféricas, la industria de alimentos, etc [17, 36, 37, 40, 41, 69]. Como estos modelos representan la molécula de agua con distintas arquitecturas, ofrecieron una forma de evaluar la mejor representación del agua en el régimen de interés. En particular, cómo se relaciona la arquitectura de la molécula con la dependencia de D y ρ con la temperatura. La estructura de los modelos se mostró en la figura II.3 del Capítulo II.
Las simulaciones fueron realizadas con una caja cúbica que contuvo 512 moléculas de agua a presión de 1 atm y en el rango de temperatura 235 K ≤ T ≤ 300 K. El rango de temperaturas se cubrió con pasos de 5K. Cada modelo fue simulado durante un tiempo total de 10 ns. La dependencia con la temperatura de D y ρ obtenida con cada modelo, y los resultados experimentales correspondientes [ref. 38 para D; ref. 36 para ρ], se presentan en la figura III.1. El coeficiente se difusión se calculó a partir del desplazamiento cuadrático medio de las moléculas de agua, como se explicó en la sección II.3 (ver la ec.II.14 del Capítulo II).
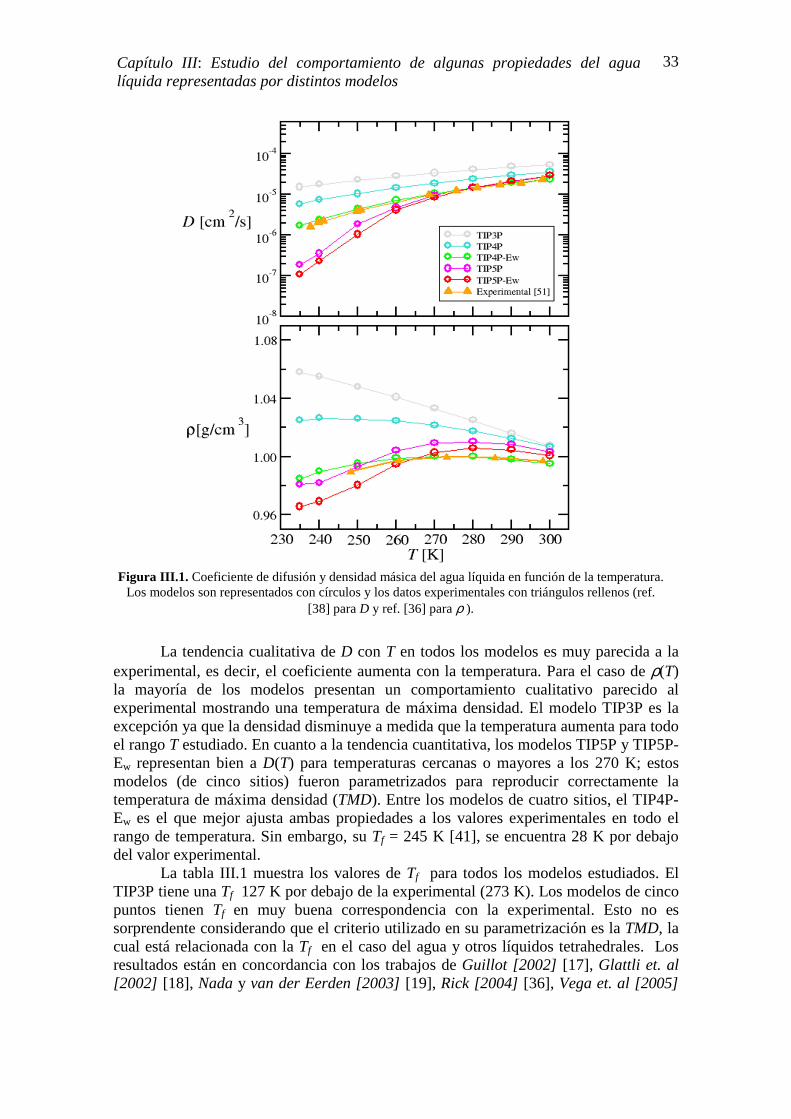
33 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
La tendencia cualitativa de D con T en todos los modelos es muy parecida a la
experimental, es decir, el coeficiente aumenta con la temperatura. Para el caso de ρ(T) la mayoría de los modelos presentan un comportamiento cualitativo parecido al experimental mostrando una temperatura de máxima densidad. El modelo TIP3P es la excepción ya que la densidad disminuye a medida que la temperatura aumenta para todo el rango T estudiado. En cuanto a la tendencia cuantitativa, los modelos TIP5P y TIP5P-Ew representan bien a D(T) para temperaturas cercanas o mayores a los 270 K; estos modelos (de cinco sitios) fueron parametrizados para reproducir correctamente la temperatura de máxima densidad (TMD). Entre los modelos de cuatro sitios, el TIP4P-Ew es el que mejor ajusta ambas propiedades a los valores experimentales en todo el rango de temperatura. Sin embargo, su Tf = 245 K [41], se encuentra 28 K por debajo del valor experimental.
La tabla III.1 muestra los valores de Tf para todos los modelos estudiados. El TIP3P tiene una Tf 127 K por debajo de la experimental (273 K). Los modelos de cinco puntos tienen Tf en muy buena correspondencia con la experimental. Esto no es sorprendente considerando que el criterio utilizado en su parametrización es la TMD, la cual está relacionada con la Tf en el caso del agua y otros líquidos tetrahedrales. Los resultados están en concordancia con los trabajos de Guillot [2002] [17], Glattli et. al [2002] [18], Nada y van der Eerden [2003] [19], Rick [2004] [36], Vega et. al [2005]
Figura III.1. Coeficiente de difusión y densidad másica del agua líquida en función de la temperatura. Los modelos son representados con círculos y los datos experimentales con triángulos rellenos (ref.
[38] para D y ref. [36] para ρ ).

34 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
[40], Fernández et. al [2006] [41], Vega y Abascal [2011] [68] y otros, en relación a que no hay modelo que reproduzca simultáneamente D y ρ en un rango extenso de temperatura. En consecuencia, aún se siguen abocando considerables esfuerzos para desarrollar un modelo que represente al agua de manera satisfactoria en estado líquido, ya sea a temperaturas mayores que Tf o en el régimen sobre-enfriado metaestable.
Tabla III.1. Temperatura de fusión (Tf) de distintos modelos de agua y experimental.
Modelo TIP3P TIP4P TIP4P-Ew TIP5P TIP5P-Ew Experimental
Tf [K] 146 [40] 232 [40] 245 [41] 270 [40] 271 [41] 273.15 [40]
El análisis anterior muestra que los modelos TIP de cinco puntos describen bien la TMD y la temperatura de fusión del agua sobre-enfriada; sin embargo, no logran representar de manera satisfactoria al coeficiente de difusión a T < 270 K. Con el propósito de mejorar la representación del D(T) en el régimen sobre-enfriado sin afectar significativamente la ρ(T) y la Tf, se llevó a cabo una modificación del campo de fuerza (force field) del modelo TIP5P-Ew. La elección de este modelo sobre el TIP5P fue debido a que sus propiedades termodinámicas, dieléctricas y dinámicas son independientes del tamaño del sistema [36] (ver sección II.4 Capítulo II). El cambio del force field (reparametrización) del TIP5P-Ew consistió en variar los parámetros σ y ε del potencial LJ del átomo de oxígeno y la carga q del potencial de Coulomb de los átomos hidrógeno y LP. De tal variación se obtuvieron nuevos parámetros, σ´, ε´ y q´, los cuales se utilizaron para sustituir los no primados σ, ε y q en el modelo TIP5P-Ew.
Los σ´, ε´ y q´ se formularon como factores multiplicativos de los originales σ, ε, q; a saber:
σ´ = fσ σ ec.III.1
ε´= fε ε ec.III.2
q´= fq q ec.III.3
La variación independiente de los factores fσ, fε y fq es poco práctica; por tal motivo, se buscaron condiciones para relacionar los tres factores de tal forma que sólo uno de ellos fuese independiente y los otros dos dependieran de este. Se eligió a fq como el factor independiente y se derivaron las expresiones matemáticas para las funciones fσ(fq) y fε(fq). Tales expresiones se muestran en las ecuaciones ec.III.4 y ec.II.5. Una descripción detallada sobre las deducciones de estas funciones se presenta en el Apéndice 1 sección 1.1.
fb � O�W��qW���fqW�����)�� XO� ec.III.4
f� � ai�qW�OZfqW��eTj)bj + 1d ec.III.5

35 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
La reparametrización del TIP5P-Ew consistió en: 1. Modificar fq entre 1.0001 y 1.0005 en intervalos de 0.0001. Fuera de este rango
los resultados se alejaron considerablemente de los experimentales. 2. Calcular los otros dos factores fσ y fε. 3. Calcular σ´, ε´ y q´. 4. Sustituir en el original TIP5P-Ew los parámetros q, σ, ε por los primados q´, σ´,
ε´. 5. Simular sistemas de 512 moléculas de agua líquida a T = 255 K con cada modelo
TIP5P-Ew modificado. T = 255 K se consideró representativa del rango sobre-enfriado metaestable por ubicarse en el medio del mismo. Cada simulación tuvo una duración de 10ns.
6. Calcular el coeficiente de difusión y la densidad con cada modelo TIP5P-Ew modificado.
7. Validar las propiedades calculadas. Se estableció la condición que la diferencia entre ellas y las experimentales fuese ≤ 5% (condición de validación). La tabla III.2 reúne los valores de fσ, fε, fq, q´, σ´, ε´ y de las propiedades
calculadas. Las mismas son cotejadas con los datos experimentales [38 para el D; 36 para ρ] y con el original TIP5P-Ew (fσ = fε = fq = 1.0000).
Tabla III.2. Valores de fσ, fε, fq, q´, σ´, ε´ y de las propiedades D y ρ del agua líquida calculadas a
T = 255K para cada modificación realizada al TIP5P-Ew. Los D y ρ son cotejados con los experimentales [ref. 38 para el D; ref. 36 para ρ] y el original TIP5P-Ew.
fq fσσσσ fεεεε q´ [e] σσσσ´ [nm] εεεε´ [kJ.mol-1] D[x 10-5cm2/s] ρρρρ [g/cm3]
1.0000 1.0000 1.0000 -0.2410000 0.3097000 0.7448000 0.225 0.988
1.0001 1.0123 0.8632 -0.2410241 0.3135206 0.6428979 0.201 0.963
1.0002 1.0258 0.7364 -0.2410482 0.3176980 0.5484797 0.271 0.945
1.0003 1.0407 0.6197 -0.2410723 0.3222999 0.4615478 0.273 0.923
1.0004 1.0572 0.5130 -0.2410964 0.3274131 0.3821041 0.298 0.903
1.0005 1.0757 0.4164 -0.2411205 0.3331553 0.3101516 0.404 0.882
Experimental 0.469 0.993
Los resultados muestran que para cada conjunto de valores q´, σ´, ε´, el coeficiente de difusión del agua (D) incrementa con fq, comportamiento que se manifiesta de manera contraria para la densidad. Con respecto a la condición de validación, la misma fue cumplida para la ρ con fq = 1.0001 y 1.0002; en el caso de D, la condición de validación no se cumplió para ningún fq. Lo anterior refleja que la reparametrización elegida del TIP5P-Ew mediante la relación conjunta de los parámetros Lennard-Jones (σ, ε ) y Coulomb (q), no logra mejorar la representación del agua en el régimen sobre-enfriado metaestable.
En base a lo obtenido, se decidió realizar otro tipo de reparametrización al TIP5P-Ew. La misma consistió en modificar sólo σ. Los fundamentos para esta modificación tiene su justificación en un análisis previo sobre la dependencia y la

36 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
sensibilidad de las propiedades D(T= 255 K) y ρ(T = 255 K) del agua con la variación independiente (no simultánea) de σ, ε y q. Se encontró que el coeficiente de difusión del agua es altamente sensible a la variación de σ. Parte de los resultados de estas pruebas se muestran en el Apéndice 1 sección 1.2.
La nueva reparametrización del TIP5P-Ew consistió en calcular σ´ multiplicando a σ por un factor entre 1.001 y 1.005 en intervalos de 0.001, y simulando un sistema de 512 moléculas de agua líquida a T = 255 K durante 10ns. La validación de D y ρ se realizó con el mismo criterio que antes (diferencia con datos experimentales < 5%).
La tabla III.3 muestra los valores de D (255 K) y ρ (255 K) para cada barrido de σ´ aplicada al TIP5P-Ew. Los datos de las propiedades son cotejados con los experimentales y con el original TIP5P-Ew.
Tabla III.3. Propiedades D y ρ del agua líquida calculadas a T = 255 K con el modelo TIP5P-Ew para cada barrido de σ´. Los valores son cotejados con los experimentales [ref. 38 para el D; ref. 36
para ρ] y el original TIP5P-Ew.
σσσσ [nm] Factor de Incremento σσσσ´ [nm] D [x 10-5cm2/s] ρρρρ [g/cm3]
0.3097000
1.001 0,3100097 0.291 0.963
1.002 0,3103194 0.331 0.961
1.003 0,3106291 0.441 0.960
1.004 0,3109388 0.504 0.959
1.005 0,3112485 0.638 0.958
1.000 (original) 0.3097000 0.225 0.988
Experimental 0.469 0.993
Los resultados indican que todos los valores de σ´ cumplen con la condición de validación para la ρ; para el D, los factores de incremento 1.003 y 1.004 cumplen la validación, obteniéndose mayor correspondencia con el factor de incremento 1.003. A esta modificación del TIP5P-Ew se denominó modelo TIP5P-EwR. Se completó el rango sobre-enfriado 235 ≤ T ≤ 270 K con simulaciones de 10 ns para cada temperatura. En dicho rango, los valores de D(T) y ρ(T) calculados con los modelos TIP5P-EwR y TIP5P-Ew se representan en la figura III.2 y se cotejan con los datos experimentales.

37 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
Las distintas propiedades del agua líquida sobre-enfriada calculadas con el
modelo reparametrizado TIP5P-EwR, presentan la misma tendencia cualitativa que la experimentación. Cuantitativamente, el modelo TIP5P-EwR ajusta el coeficiente de difusión mejor que el original TIP5P-Ew, garantizando una excelente reproducibilidad de la dinámica de las moléculas de agua sobre-enfriada. La densidad fue ligeramente subestimada por el TIP5P-EwR, por lo que en este modelo, el líquido presenta menor densidad e interacciones intermoleculares más débiles que en el TIP5P-Ew.
La otra propiedad a valorar fue la temperatura de fusión. La Tf se estimó mediante simulaciones realizadas entre 260 K y 273 K en sistemas agua-hielo constituidos de 1016 moléculas (508 moléculas de agua líquida y 508 de hielo). Los sistemas se prepararon mediante el método de coexistencia directa [42]. Este método consiste en colocar en contacto ambas fases del agua y dejar que el sistema llegue a su fase más estable a la temperatura escogida. En consecuencia, la Tf se define como la temperatura comprendida entre la temperatura más alta donde el sistema permanece congelado y la temperatura más baja a la cual el sistema ha fundido.
El valor de la Tf se monitoreó mediante las gráficas energía potencial (Ep) vs tiempo de simulación. La condición impuesta fue que, el TIP5P-EwR no experimentara cambios mayores o menores de 5 K con respecto a la Tf del original TIP5P-Ew (271 K). La Tf del nuevo modelo fue ~ 262 K, valor que está a 9 K y 11 K por debajo del TIP5P-Ew y del experimental, respectivamente. Los cambios estructurales y la energía potencial promedio en función del tiempo (Ep(t)) para la Tf del TIP5P-EwR se muestran en las figuras III.3 y III.4, correspondientemente.
Figura III.2 . Propiedades D(T) y ρ(T) del agua líquida calculadas para los modelos TIP5P-Ew, TIP5P-EwR cotejados con valores experimentales [ref. 38 para el D; ref. 36 para ρ].
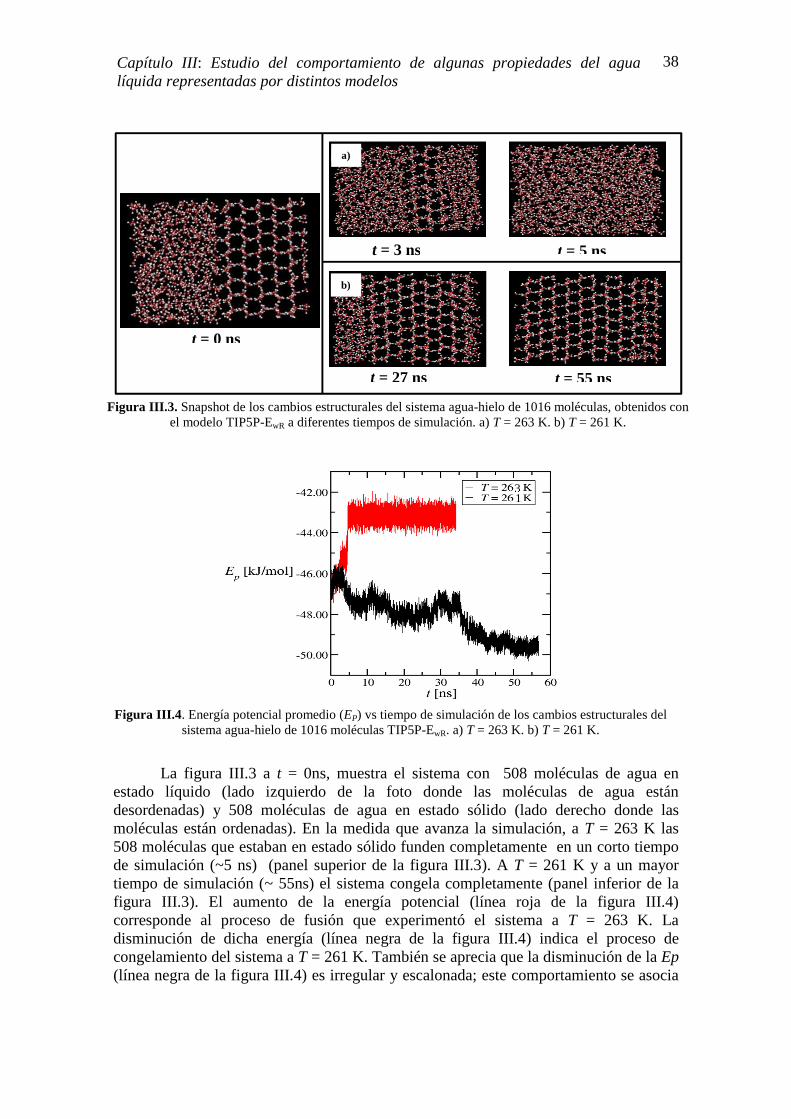
38 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
La figura III.3 a t = 0ns, muestra el sistema con 508 moléculas de agua en
estado líquido (lado izquierdo de la foto donde las moléculas de agua están desordenadas) y 508 moléculas de agua en estado sólido (lado derecho donde las moléculas están ordenadas). En la medida que avanza la simulación, a T = 263 K las 508 moléculas que estaban en estado sólido funden completamente en un corto tiempo de simulación (~5 ns) (panel superior de la figura III.3). A T = 261 K y a un mayor tiempo de simulación (~ 55ns) el sistema congela completamente (panel inferior de la figura III.3). El aumento de la energía potencial (línea roja de la figura III.4) corresponde al proceso de fusión que experimentó el sistema a T = 263 K. La disminución de dicha energía (línea negra de la figura III.4) indica el proceso de congelamiento del sistema a T = 261 K. También se aprecia que la disminución de la Ep (línea negra de la figura III.4) es irregular y escalonada; este comportamiento se asocia
Figura III.4 . Energía potencial promedio (EP) vs tiempo de simulación de los cambios estructurales del sistema agua-hielo de 1016 moléculas TIP5P-EwR. a) T = 263 K. b) T = 261 K.
3 1
t = 3 ns
t = 0 ns
t = 27 ns t = 55 ns
a)
b)
t = 5 ns
Figura III.3. Snapshot de los cambios estructurales del sistema agua-hielo de 1016 moléculas, obtenidos con el modelo TIP5P-EwR a diferentes tiempos de simulación. a) T = 263 K. b) T = 261 K.

39 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
al crecimiento de hielo debido a la incorporación paulatina que realizan las moléculas de agua líquida sobre-enfriada a la estructura de hielo.
Por último, el análisis de la reparametrización del TIP5P-Ew muestra que cambiar los parámetros Lennard-Jones y las cargas de los LP y H para que las simulaciones ajustaran lo mejor posible algunas propiedades experimentales del agua en el régimen sobre-enfriado metaestable, siempre generó alguna discrepancia con la experimentación. En un trabajo realizado por Glattli et. al [2002] [18] el cual consistió en reparametrizar el modelo SPC a 298.15 K, los autores concluyeron que hay gran dificultad en mejorar simultáneamente una gama de propiedades cuando se modifican diversos parámetros en el modelo (incorporación de sitios van der Waals a los núcleos de H, incremento de las cargas parciales de los átomos de O y H, geometría). Además, remarcan que de los dieciocho modelos obtenidos solo uno exhibió mejor acuerdo con los datos experimentales.
Lo mostrado y analizado permite concluir que: • Las reparametrizaciones establecen metodologías sencillas para la modificación
de algunos parámetros del force field en modelos de agua atomísticos. Sin embargo no se logró mejorar el TIP5P-Ew en su representación del agua líquida sobre-enfriada metaestable.
• El original TIP5P-Ew sigue presentando para las tres propiedades evaluadas mejor correlación con los valores experimentales.
• El estudio permitió analizar la dependencia y la sensibilidad de las propiedades D y ρ del agua con la variación independiente (no simultánea) de σ, ε y q. Se encontró que el coeficiente de difusión del agua es altamente sensible a la variación del σ del potencial LJ.
Los detalles computacionales implementados en las simulaciones fueron: • Condiciones de contorno periódicas. • Paso de integración de las ecuaciones de movimiento (∆t) igual a 1 fs. • Un cut-off esférico de 0.9 nm para considerar las interacciones Lennard-Jones e
interacciones electrostáticas de corto alcance. • La técnica PME (Particle Mesh Ewald) para considerar las interacciones
electrostáticas en los modelos TIP4P-Ew, TIP5P-Ew. • Presión de trabajo igual a 1 atm. • Tiempo de relajación del baróstato Parinello-Raphman (τb) igual a 0.1 ps.
Tiempo de relajación del termostato Nose-Hoover (τT) igual a 0.5 ps con compresibilidad de 4.5 x 10-5 bar.
• Tiempo total de cada simulación para el posterior cálculo de las propiedades, fue igual a 10 ns. La propiedades se calcularon en los últimos 2ns de simulación (el sistema estaba equilibrado).
III.2. ESTRUCTURA Y DINÁMICA DE LOS ENLACES PUENTES DE HIDRÓGENO.
La caracterización estructural y dinámica de los enlaces puentes de hidrógeno (Hb) establecidos entre las moléculas de agua, tiene gran interés por el papel que ellos

40 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
juegan en el comportamiento del agua condensada. Las anomalías del agua líquida, las cuales son más pronunciadas en el régimen sobre-enfriado, están conectadas con la influencia microscópica que ejercen los Hb sobre sus propiedades dinámicas y termodinámicas. Además, la formación de hielo es un reflejo directo de la relación entre la ganancia energética de los Hb permanentes y la pérdida entrópica correspondiente [4, 16, 78, 79].
La literatura reporta diferentes estudios, experimentales y de simulaciones, relacionados con diversos aspectos de los Hb: la formación de distintos poliedros (redes), la conectividad entre redes, tiempos de relajación, tiempos de vida medio, etc. [72, 78, 125, 126, 127, 128]. Algunos de los estudios tienen en común, que la temperatura de trabajo siempre es de 298 K; y los de simulaciones los han desarrollado implementando un solo tipo de modelo de agua.
La presente sección remite al segundo estudio desarrollado en la tesis. El estudio consistió en investigar la dependencia con la temperatura de la dinámica de los Hb empleando los modelos SPC/E, TIP4P-Ew, TIP5P-Ew y Six-Sites. Se analizaron los efectos que ocasionan las diferentes arquitecturas de construcción de los modelos en el patrón de formación y ruptura de los Hb. La dinámica de los enlaces se evaluó relacionando el tiempo de vida de los Hb, el orden tetraédrico y el coeficiente de difusión del agua. La representación de los modelos se mostró en la figura II.3 y los parámetros geométricos en la tabla II.1 (ver sección II.4 Capítulo II). III.2.1. Tiempo de Vida Promedio de los Enlaces Puentes de Hidrógeno.
El tiempo de vida promedio de los enlaces puentes de hidrógeno presentes en el sistema,tH(t), se calculó implementando un programa (desarrollo propio) que contabilizó todos los Hb que permanecieron formados (antes de romperse) a un tiempo t de simulación promediado por el número total de moléculas del sistema. Una descripción detallada del cálculo de ��̅��� se presenta en el Apéndice 2 sección 2.1.
Los sistemas simulados estuvieron constituidos de 512 moléculas de agua para los modelos SPC/E, TIP4P-Ew y TIP5P-Ew, y de 1536 moléculas para el Six-Sites; el rango de temperatura estudiado fue 230 K ≤ T ≤ 300 K. La figura III.5. (a), muestra la dependencia detH (t) con el tiempo de simulación para el TIP5P-Ew a 230 K (color negro), 250 K (color rojo), 270 K (color verde) y 300 K (color azul). Se aprecia quetH (t) incrementa a medida que la temperatura disminuye. La figura III.5 (b) describe, para todos los modelos de agua, la dependencia con T del promedio temporal detH (t) en los últimos 5ns de simulación (<tH>); para el TIP5P-Ew a T = 230 K, <tH> es el valor indicado por la línea horizontal amarilla de la figura III.5. (a).

41 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
Los resultados indican que a medida que la temperatura disminuye las moléculas del sistema experimentan una cinética más lenta. Los <tH> son muy similares para los cuatro modelos a temperaturas entre 270 K y 300 K; a T < 270 K, <tH> incrementa considerablemente siendo más pronunciado su aumento para el TIP5P-Ew y el Six-Sites. A 230 K el <tH> para estos modelos, construidos con pares de electrones explícitos, resultó ser un orden de magnitud mayor que los modelos sin LP.
III.2.2. Orden Tetraédrico.
El factor de orden tetraédrico, denominado q, mide el nivel de arreglo tetraédrico de una molécula k con sus cuatro vecinas más cercanas [70]. Para q(k) = 1 se tiene un orden tetraédrico perfecto; y q(k) = 0 implica un orden molecular aleatorio. El cálculo de q en cada simulación (todos los modelos y para todas las T), se realizó con un programa (desarrollo propio) que contabiliza los seis posibles ángulos entre la molécula central k y sus cuatro vecinas más cercanas. Para estimar la densidad de probabilidad del parámetro de orden tetraédrico, P(q), el programa realiza un histograma normalizado de la cantidad de moléculas (en todo el sistema y en los 5ns de simulación) que tienen un q específico (entre 0 y 1). Una explicación más amplia de q y P(q) se presenta en el Apéndice 2 sección 2.2.
Los resultados de la P(q) del agua líquida obtenidos para los cuatro modelos y temperaturas de 230 K, 250 K, 270 K y 300K se muestran en la figura III.6. A 300 K las curvas son notablemente similares para todos los modelos y están caracterizadas por una distribución bimodal [70, 71]. Esta bimodalidad ha sido presentada y discutida en trabajos que han realizado el cálculo del índice de estructura local (LSI) [72]; la misma se ha usado para soportar la idea que el agua líquida consiste de una mezcla de moléculas en dos estados estructurales diferentes, uno de baja densidad en donde las moléculas están bien estructuras y coordinadas de una forma altamente tetraédrica con sus cuatro moléculas de agua vecinas, y otro de alta densidad en cuyo caso las moléculas se encuentran desestructuradas [73, 74]. Cuando la temperatura disminuye, el
Figura III.5. (a) Tiempo de vida promedio instantáneo de todos los Hb del agua líquida (��������) vs tiempo de simulación obtenido en el TIP5P-Ew. Negro, rojo, verde y azul corresponden a las T = 230 K, 250 K, 270 K y 300 K, respectivamente. La línea horizontal representa el tiempo promedio de los Hb, <tH>. (b) <tH> vs
T calculado con el Six-Sites (negro), TIP5P-Ew (rojo), TIP4P-Ew (verde) y SPC/E (azul).
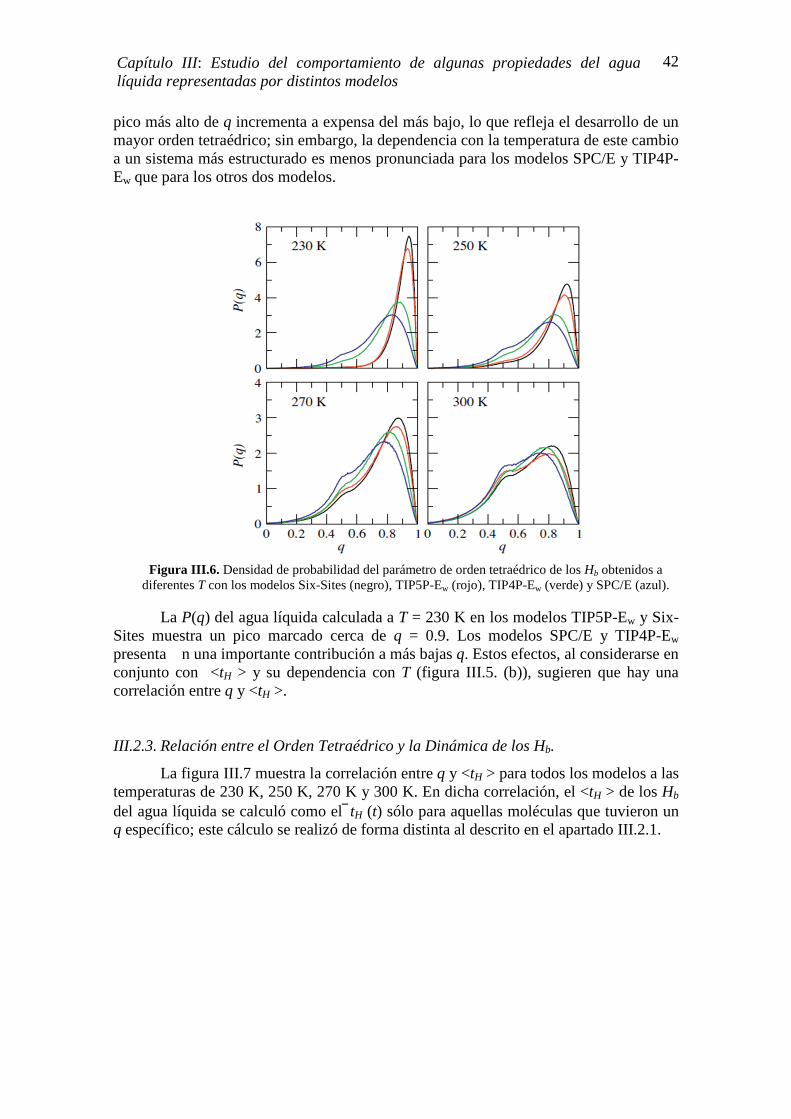
42 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
pico más alto de q incrementa a expensa del más bajo, lo que refleja el desarrollo de un mayor orden tetraédrico; sin embargo, la dependencia con la temperatura de este cambio a un sistema más estructurado es menos pronunciada para los modelos SPC/E y TIP4P-Ew que para los otros dos modelos.
La P(q) del agua líquida calculada a T = 230 K en los modelos TIP5P-Ew y Six-
Sites muestra un pico marcado cerca de q = 0.9. Los modelos SPC/E y TIP4P-Ew presenta n una importante contribución a más bajas q. Estos efectos, al considerarse en conjunto con <tH > y su dependencia con T (figura III.5. (b)), sugieren que hay una correlación entre q y <tH >.
III.2.3. Relación entre el Orden Tetraédrico y la Dinámica de los Hb.
La figura III.7 muestra la correlación entre q y <tH > para todos los modelos a las temperaturas de 230 K, 250 K, 270 K y 300 K. En dicha correlación, el <tH > de los Hb del agua líquida se calculó como eltH (t) sólo para aquellas moléculas que tuvieron un q específico; este cálculo se realizó de forma distinta al descrito en el apartado III.2.1.
Figura III.6. Densidad de probabilidad del parámetro de orden tetraédrico de los Hb obtenidos a diferentes T con los modelos Six-Sites (negro), TIP5P-Ew (rojo), TIP4P-Ew (verde) y SPC/E (azul).
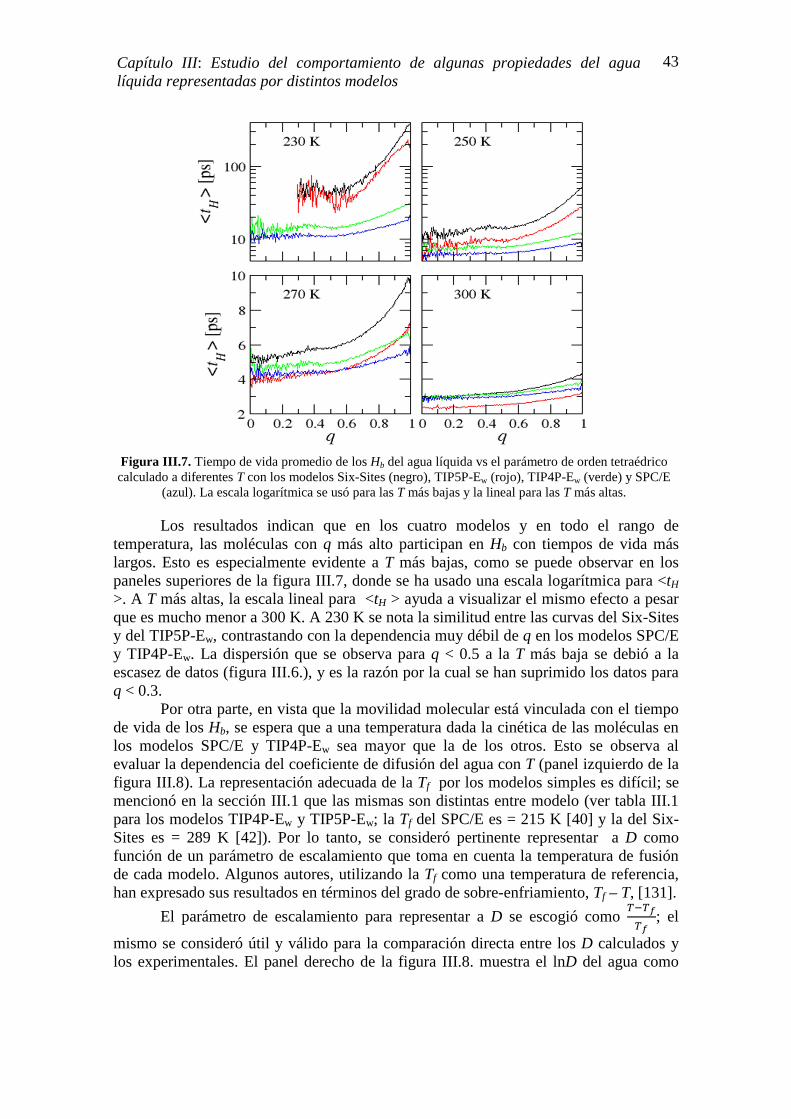
43 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
Los resultados indican que en los cuatro modelos y en todo el rango de
temperatura, las moléculas con q más alto participan en Hb con tiempos de vida más largos. Esto es especialmente evidente a T más bajas, como se puede observar en los paneles superiores de la figura III.7, donde se ha usado una escala logarítmica para <tH >. A T más altas, la escala lineal para <tH > ayuda a visualizar el mismo efecto a pesar que es mucho menor a 300 K. A 230 K se nota la similitud entre las curvas del Six-Sites y del TIP5P-Ew, contrastando con la dependencia muy débil de q en los modelos SPC/E y TIP4P-Ew. La dispersión que se observa para q < 0.5 a la T más baja se debió a la escasez de datos (figura III.6.), y es la razón por la cual se han suprimido los datos para q < 0.3.
Por otra parte, en vista que la movilidad molecular está vinculada con el tiempo de vida de los Hb, se espera que a una temperatura dada la cinética de las moléculas en los modelos SPC/E y TIP4P-Ew sea mayor que la de los otros. Esto se observa al evaluar la dependencia del coeficiente de difusión del agua con T (panel izquierdo de la figura III.8). La representación adecuada de la Tf por los modelos simples es difícil; se mencionó en la sección III.1 que las mismas son distintas entre modelo (ver tabla III.1 para los modelos TIP4P-Ew y TIP5P-Ew; la Tf del SPC/E es = 215 K [40] y la del Six-Sites es = 289 K [42]). Por lo tanto, se consideró pertinente representar a D como función de un parámetro de escalamiento que toma en cuenta la temperatura de fusión de cada modelo. Algunos autores, utilizando la Tf como una temperatura de referencia, han expresado sus resultados en términos del grado de sobre-enfriamiento, Tf – T, [131].
El parámetro de escalamiento para representar a D se escogió como +Z+�+� ; el
mismo se consideró útil y válido para la comparación directa entre los D calculados y los experimentales. El panel derecho de la figura III.8. muestra el lnD del agua como
Figura III.7. Tiempo de vida promedio de los Hb del agua líquida vs el parámetro de orden tetraédrico calculado a diferentes T con los modelos Six-Sites (negro), TIP5P-Ew (rojo), TIP4P-Ew (verde) y SPC/E
(azul). La escala logarítmica se usó para las T más bajas y la lineal para las T más altas.

44 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
función de la temperatura re-escaleada. Se dilucida que los modelos con pares de electrones libres presentan la mejor aproximación.
La figura III.8 también dilucida que el D(T) calculado con el TIP5P-Ew tiene una dependencia en T más pronunciada que los valores experimentales. En vista que el TIP5P-Ew presenta mejor aproximación a la temperatura de fusión del hielo, este modelo podría ser el que mejor represente al agua en el régimen sobre-enfriado metaestable.
Lo mostrado y analizado en esta sección III.2 permite concluir que: • Las distintas arquitecturas de construcción de los modelos estudiados
permitieron comparar sus diversos patrones de formación y ruptura de los Hb. • Los modelos construidos con LP tienen una clara tendencia a formar enlaces
puentes de hidrógenos más estables y con tiempos de vida más largos que los modelos sin pares de electrones.
• Los modelos de agua TIP5P-Ew y el Six-Sites tienen un efecto claro para favorecer considerablemente la estructura tetraédrica del agua a T < 0°C.
• Un escalamiento de la temperatura fue necesario para comparar las predicciones del coeficiente de difusión del agua en los diferentes modelos.
• El Six-Sites y el TIP5P-Ew presentaron un D en mejor correspondencia con los valores experimentales cuando D se representa en función de la temperatura re-escaleada. De los dos, el TIP5P-Ew podría representar mejor al agua en el régimen sobre-enfriado metaestable, tiene una Tf en mejor correspondencia con la experimental. Los detalles computacionales se describen como:
• Simulaciones realizadas con condiciones de contorno periódicas. • Steptime de integración de las ecuaciones de movimiento (∆t) de 1 fs. • Cut-off esférico de 0.9 nm para considerar las interacciones Lennard-Jones e
interacciones electrostáticas de corto alcance.
Figura III.8 . Panel izquierdo: D(T) del agua (círculos abiertos) calculado con los modelos Six-Sites (negro), TIP5P-Ew (rojo), TIP4P-Ew (verde) y SPC/E (azul). Las líneas punteadas verticales representan
las Tf de los modelos. La Tf del SPC/E cae fuera del rango de T graficado. El D experimental es representado con símbolos anaranjados [67] y violeta [38]. La línea marrón vertical indica la Tf
experimental. Panel derecho: son los mismos datos pero en función del parámetro de escalamiento de T.

45 Capítulo III: Estudio del comportamiento de algunas propiedades del agua líquida representadas por distintos modelos
• Interacciones electrostáticas de largo alcance fueron consideradas a través de la técnica PME para los modelos TIP4P-Ew y TIP5P-Ew; mientras que para el Six-Sites, se empleó un cut-off de 0.9 nm.
• Presión de trabajo: 1 atm. La compresibilidad fue = 4.5 x 10-5 bar. • Tiempo de relajación del baróstato Parinello-Raphman (τb): 0.1 ps. Tiempo de
relajación del termostato Nose-Hoover (τT): 0.5 ps.

46 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
CAPÍTULO IV
NUCLEACIÓN HOMOGÉNEA DE HIELO. CONJETURA SOBRE LA CRISTALIZACIÓN DEL AGUA EN CONDICIONES NO MAN´S LAND
EN MODELOS ATOMÍSTICOS
Las transformaciones cinéticas del agua líquida en el régimen sobre-enfriado metaestable vía nucleación homogénea, han ameritado enormes esfuerzos tanto a nivel experimental como teórico [2, 47, 58, 75, 76]. Muchos estudios sobre mecanismo de la nucleación homogénea desde agua líquida sobre-enfriada, se han centrado en la determinación de la tasa de nucleación. La T.C.N. (ver Capítulo I sección I.3.1.) plantea que el valor de la tasa de nucleación homogénea Jhom (T) se puede calcular si se conocen los valores de la energía libre de Gibbs de la formación de embriones críticos (∆�� �∗ ���) y los valores de la energía libre de activación para la difusión de moléculas en la interface líquida/sólida (∆/∗���). Los trabajos teóricos se han enfocado en plantear ecuaciones que permiten estimar ∆�� �∗ ���; para tal estimación se usan datos experimentales como: presiones de vapor de equilibrio del hielo y del agua líquida sobre-enfriada, coeficiente de difusión del agua líquida, etc. Como ∆�� �∗ ��� es la máxima barrera de energía que un embrión, ya alcanzado su tamaño crítico, debe vencer, tanto la energía libre como la velocidad de nucleación Jhom dependen del tamaño del embrión crítico.
La determinación directa del tamaño del embrión crítico homogéneo no ha sido posible con las simulaciones moleculares, debido a la imposibilidad que tienen los modelos atomísticos de representar la cristalización espontánea de agua (ni siquiera logran representar la cristalizan en la zona no man´s land). En la literatura, se encuentran solo dos trabajos de simulaciones en donde se logra nucleación espontánea de agua sobre-enfriada metaestable con modelos atomísticos: Matsumoto et. al. [2002] [62] y Vrbka y Jungwirth [2006] [63] (ver Capítulo I apartado I.3.3). Utilizando un modelo distinto a los atomísticos (el mW, del acrónimo en inglés model of water), Moore E. y Molinero V. [2010, 2011] [100, 129], observaron nucleación homogénea en simulaciones suficientemente largas en la zona no man´s land.
Por otra parte, de la literatura revisada, solo un trabajo a nivel experimental reporta mediciones directas del tamaño del núcleo crítico. En este estudio, Liu. et. al. 2007 [61] usaron un método para determinar la temperatura de cristalización de microemulsiones de agua en aceite con y sin agente nucleante (heptacosanol). Los autores determinaron una relación directa entre el tamaño del embrión y la temperatura de cristalización. En las condiciones de nucleación homogénea (sin heptacosanol), encontraron que el embrión crítico contiene entre 70 y 280 moléculas de agua en un rango de T entre 225 y 232 K. Se presume que la escases de trabajos experimentales sobre la determinación del embrión de hielo crítico en agua pura sobre-enfriada, se debe a las limitaciones que presentan los diseños experimentales para cuantificar el tamaño de un embrión microscópico. En cuanto a las simulaciones numéricas, Pereyra et al. 2011 [68] estudiaron la estabilidad de nanocolumnas de hielo en agua líquida sobre-enfriada; los autores determinaron el radio mínimo necesario para el crecimiento de las nanocolumnas. El tamaño del embrión crítico para partículas 3D fue deducido a partir de resultados de simulaciones 2D y la ecuación de Gibbs-Thomson.

47 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
Este capítulo de la tesis (tercer estudio) está dedicado principalmente al cálculo del tamaño del embrión de hielo crítico en el rango de 235 K ≤ T ≤ 273 K. Para ello, se utilizaron argumentos termodinámicos y se tomó como base la hipótesis planteada por Baumgärtel y Zimmermann (2011) [56]. Estos autores propusieron que la energía necesaria para la cristalización de agua sobre-enfriada (energía de activación Ea) es igual al cambio de entalpía entre las fases líquida y cristalina de las moléculas que conforman el embrión crítico. A saber:
Ea =N ∆Hc ec.IV.1
donde N es el número de moléculas que conforman el embrión crítico y ∆Hc es la entalpía de cristalización para una molécula de agua. Para la estimación final de N, Baumgärtel y Zimmermann 2011 [56] usaron para ∆Hc una expresión polinómica de cuarto orden planteada por Pruppacher y Klett [1997] [1]. Para la implementación de la ec.IV.1 se realizó lo siguiente: 1. Se estimaron los valores de Ea a partir de un ajuste matemático aplicado a la tasa de nucleación homogénea calculada teóricamente por Pruppacher y Klett [1997] [1] a la presión de 1 atm. Estos autores realizaron una amplia valoración de Jhom (T) para el agua sobre-enfriada, aplicando la T.C.N. y un conjunto de datos experimentales obtenidos de evaluar la dependencia con T de algunos parámetros característicos (calor latente de fusión, densidad del agua, densidad del hielo, tensión superficial del agua, presiones de vapor de equilibrio del hielo y del agua líquida sobre-enfriada, coeficiente de difusión del agua líquida, etc.). En la tabla IV.1 se reúnen los valores de Jhom (T) los cuales forman el conjunto más completo. La Jhom experimental no fue posible implementarla ya que solo se ha determinado en cortos rangos de temperatura (2 o 3 K). 2. Se calcularon los valores de ∆Hc a partir de los resultados obtenidos por las simulaciones computacionales.
Tabla IV.1: Tasa de nucleación homogénea de agua líquida sobre-enfriada calculadas por kPruppacher y Klett [1997] [39] a la presión de 1 atm.
T [°C] T [K] Jhom [cm−−−−3s−−−−1]
-29.00 244.15 46×10−11 -30.00 243.15 43×10−8 -31.00 242.15 15×10−5 -32.00 241.15 65×10−3 -33.00 240.15 15×100 -34.00 239.15 30×102 -35.00 238.15 20×104 -36.00 237.15 10×106 -37.00 236.15 30×107 -38.00 235.15 50×108 -39.00 234.15 90×109 -40.00 233.15 10×1011 -41.00 232.15 20×1012 -42.00 231.15 10×1014 -43.00 230.15 50×1015 -44.00 229.15 20×1017

48 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
IV.1. ESTIMACIÓN DE LA ENERGÍA DE ACTIVACIÓN.
Los valores de Jhom (T) se ajustaron con la ecuación de Vogel-Tammann-Fulcher (VTF) [6, 47, 48, 57-60] para temperaturas mayores a una T dada, para la cual algunas propiedades físicas del agua sobre-enfriada reflejan la transición de fase HDL-LDL; y un ajuste de tipo Arrhenius para temperaturas menores a esta T de transición. Tales ajustes tuvieron sus bases en el hecho de que, al ser Jhom(T) una propiedad estrechamente vinculada con la formación de embriones cristalinos y por ende con la cinética de las moléculas de agua, su comportamiento debe mostrar una dependencia Arrhenius o VTF a una temperatura de transición. A pesar que la hipótesis del segundo punto crítico líquido-líquido LLCP (ver Capítulo I sección I.2) no define inexorablemente la temperatura de nucleación espontánea, ni explica por qué no se puede observar un estado vítreo inmediatamente debajo de la temperatura Widom Line, la LLCP permite definir un punto de referencia o de transición a temperaturas bajas.
Los experimentos en el régimen sobre-enfriado metaestable son difíciles de realizar: diferentes mediciones conducen a diferentes temperaturas para aquellas propiedades físicas que reflejan la transición de fase HDL-LDL. Por ejemplo, Mallamace et. al. [2006] [124] muestran que el coeficiente de expansión térmica del agua confinada en sílica mesoporosa a P = 1 atm, (negativo) presenta un máximo a los 238 K. Estos mismos autores, usando RMN para medir el cambio químico protónico (δ), encuentran un máximo en -T(∂lnδ/∂T)P a casi la misma temperatura; esta cantidad se comporta similar al CP y por ende es otra manifestación de una transición de fase entre los líquidos de alta y baja densidad. Maruyama et. al. [2003, 2004] [130], a través de mediciones, encontraron el máximo de la capacidad calorífica a 227 K para agua confinada en poros de sílica gel. Por otra parte, se ha reportado que el bulk de agua a presión atmosférica congela espontáneamente a 235 K [43]. Considerando la dispersión que existe en estos valores experimentales de T, se decidió usar una temperatura Widom Line Tw (P = 1atm) igual a 235 K.
La figura IV.1 muestra los ajustes VTF (línea azul) y Arrhenius (línea roja) de la Jhom(T), y el valor experimental a 237 K (Jhom = 2.1 x 105 cm-3s-1) obtenido por Baumgärtel y Zimmermann [2011] [56].
Figura IV.1. Tasa de nucleación homogénea del agua calculada por Pruppacher [39] (círculos). Las líneas representan los ajustes VTF (azul) y Arrhenius (rojo).

49 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
Las ecuaciones para los distintos ajustes de Jhom(T) se expresan en la ec.IV.2 para Arrhenius y ec.IV.3 para VTF; de ellas, se obtuvieron las distintas cantidades para la energía de activación (Ea). De la ec.IV.2 se calculó una Ea igual a -2.56 x 10-18 J y de la ec.IV.3, los valores de ajuste para los tres parámetros: A = 106.844, B = 2177.49 K y T0 = 261.074 K. Con estos datos y siguiendo lo planteado por Ediger et al. 1996 [58], se definió una energía de activación aparente Ea(T) expresada por la ec.IV.4.
ln 6 = Z��,-++ ln � ec.IV.2
ln 6 = �+Z+D
+ � ec.IV.3
�q(�) = −�� 4 �� �&OZ�D
� .W = − ,-�
&OZ�D� .
W ec.IV.4
Los valores de Ea y Ea (T) se reúnen en la tabla IV.2 y representan en la figura IV.2.
Tabla IV.2. Energía de activación de la nucleación homogénea de agua calculada mediante un ajuste VTF y Arrhenius a la Jhom (ec.IV.4) (ec.IV.2).
T [K] Ea [ x 10-18 J] Tipo de Ajuste Matemático
245.00 -6.93
244.15 -6.25
VTF
243.15 -5.53 242.15 -4.92 241.15 -4.40 240.15 -3.96 239.15 -3.58 238.15 -3.24 237.15 -2.95 236.15 -2-70 235.15 -2.47 234.15
-2,56
Arrhenius
233.15
232.15
231.15
230.15
229.15
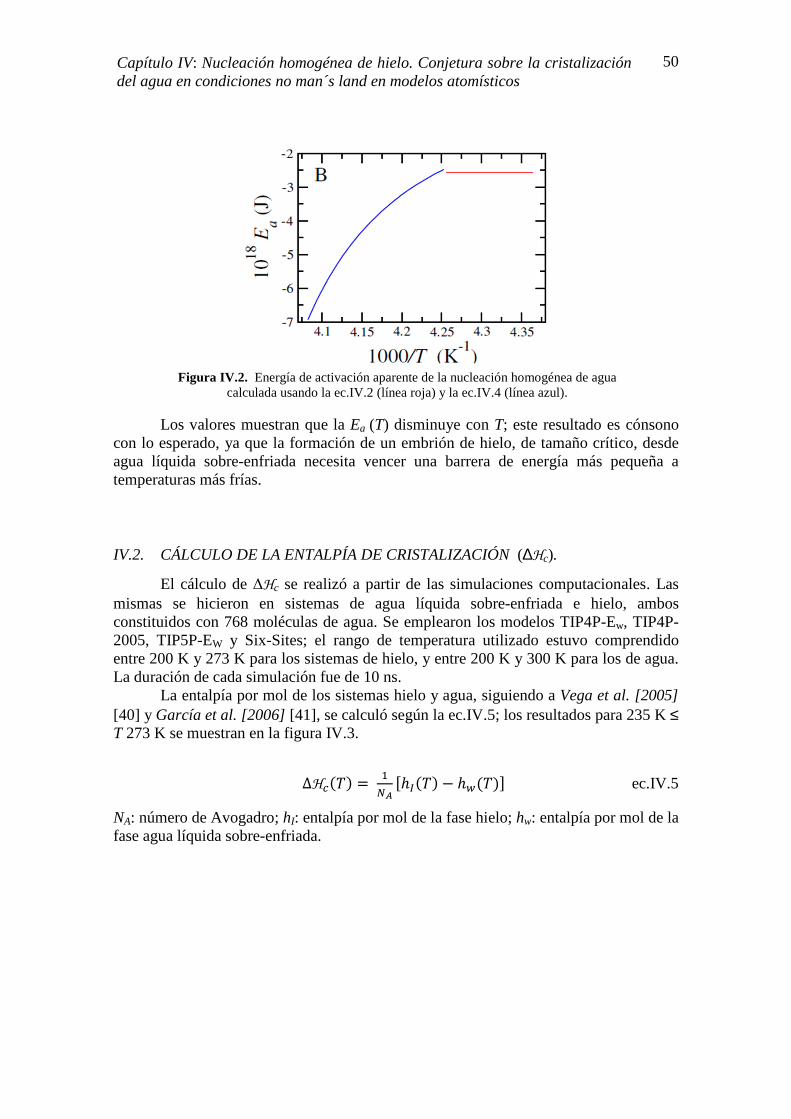
50 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
Los valores muestran que la Ea (T) disminuye con T; este resultado es cónsono con lo esperado, ya que la formación de un embrión de hielo, de tamaño crítico, desde agua líquida sobre-enfriada necesita vencer una barrera de energía más pequeña a temperaturas más frías.
IV.2. CÁLCULO DE LA ENTALPÍA DE CRISTALIZACIÓN (∆Hc).
El cálculo de ∆Hc se realizó a partir de las simulaciones computacionales. Las mismas se hicieron en sistemas de agua líquida sobre-enfriada e hielo, ambos constituidos con 768 moléculas de agua. Se emplearon los modelos TIP4P-Ew, TIP4P-2005, TIP5P-EW y Six-Sites; el rango de temperatura utilizado estuvo comprendido entre 200 K y 273 K para los sistemas de hielo, y entre 200 K y 300 K para los de agua. La duración de cada simulación fue de 10 ns.
La entalpía por mol de los sistemas hielo y agua, siguiendo a Vega et al. [2005] [40] y García et al. [2006] [41], se calculó según la ec.IV.5; los resultados para 235 K ≤ T 273 K se muestran en la figura IV.3.
∆Hr��� � OB� �� ��� � �¡���� ec.IV.5
NA: número de Avogadro; hI: entalpía por mol de la fase hielo; hw: entalpía por mol de la fase agua líquida sobre-enfriada.
Figura IV.2. Energía de activación aparente de la nucleación homogénea de agua calculada usando la ec.IV.2 (línea roja) y la ec.IV.4 (línea azul).

51 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
Los resultados exhiben una marcada discrepancia cuantitativa entre los ∆Hc(T)
estimados con los modelos de cuatro puntos (TIP4P-Ew y TIP4P-2005) y los modelos de cinco y seis puntos. Este comportamiento se puede adjudicar al hecho que los modelos TIP5P-Ew y Six-Sites están construidos con sitios explícitos que asemejan a los pares de electrones libres del agua. Por otra parte, se ha mencionado en varias oportunidades que la temperatura de fusión de los modelos es distinta entre ellos (Capítulo III), provocando que la comparación de los resultados experimentales con las simulaciones sean pocos significativos. Dado que el régimen sobre-enfriado implica un amplio rango de temperatura, en el presente estudio se decidió utilizar una variable de re-escaleo diferente a la planteada en la sección III.2 del Capítulo III. Esta nueva variable denominada τ considerara dos límites de temperatura.
• Re-escaleo de la Temperatura en el Régimen Sobre-enfriado.
El re-escaleo de la temperatura se formuló de la siguiente forma τ � +Z+£+�Z+£. En
el caso del τ experimental se asumió temperaturas iguales a 235 K y 273.15 K, correspondientes al máximo de la capacidad calorífica isobárica (�hP
= Tw) y a la temperatura de fusión del agua (Tf), respectivamente. En el caso de las simulaciones, el τ de cada modelo se calculó con sus respectivas Tf (ver tabla IV.1 para l los modelos TIP4P-Ew y TIP5P-Ew; la Tf del TIP4P-2005 es igual a 252 K [103] y la del Six-Sites igual a 289 K [42]).
La �hP del agua líquida en el régimen sobre-enfriado, se determinó usando el
método de diferenciación directa de una función de ajuste de los datos calculados de la entalpía (H) en función de la temperatura. La función de ajuste se muestra en la ec.IV.6
Figura IV.3. ∆Hc(T) por molécula de agua estimada con los modelos TIP4P-Ew (círculos verdes), TIP4P-2005 (círculos azules), TIP5P-Ew (círculos rojos) y Six-Sites (círculos negros).
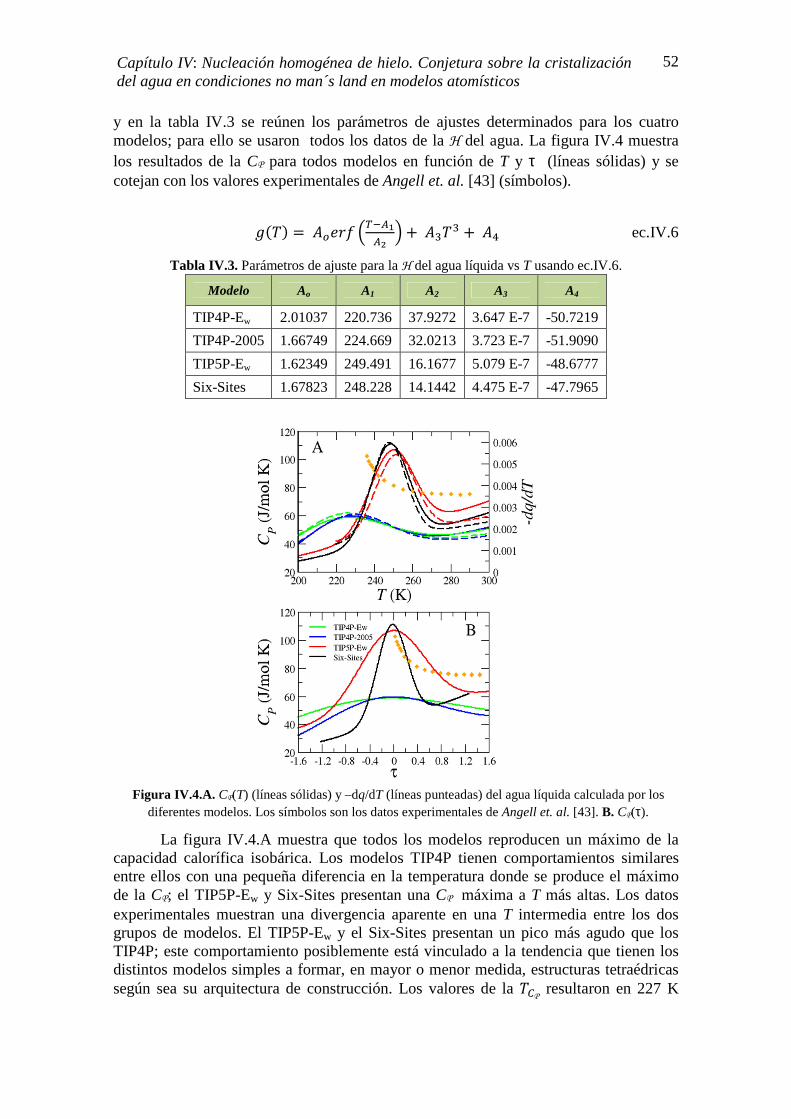
52 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
y en la tabla IV.3 se reúnen los parámetros de ajustes determinados para los cuatro modelos; para ello se usaron todos los datos de la H del agua. La figura IV.4 muestra los resultados de la CP para todos modelos en función de T y τ (líneas sólidas) y se cotejan con los valores experimentales de Angell et. al. [43] (símbolos).
/��� � � ��9 &+Zu�uW . +���� +�i ec.IV.6
Tabla IV.3. Parámetros de ajuste para la H del agua líquida vs T usando ec.IV.6.
Modelo Ao A1 A2 A3 A4
TIP4P-Ew 2.01037 220.736 37.9272 3.647 E-7 -50.7219
TIP4P-2005 1.66749 224.669 32.0213 3.723 E-7 -51.9090
TIP5P-Ew 1.62349 249.491 16.1677 5.079 E-7 -48.6777
Six-Sites 1.67823 248.228 14.1442 4.475 E-7 -47.7965
La figura IV.4.A muestra que todos los modelos reproducen un máximo de la
capacidad calorífica isobárica. Los modelos TIP4P tienen comportamientos similares entre ellos con una pequeña diferencia en la temperatura donde se produce el máximo de la CP; el TIP5P-Ew y Six-Sites presentan una CP máxima a T más altas. Los datos experimentales muestran una divergencia aparente en una T intermedia entre los dos grupos de modelos. El TIP5P-Ew y el Six-Sites presentan un pico más agudo que los TIP4P; este comportamiento posiblemente está vinculado a la tendencia que tienen los distintos modelos simples a formar, en mayor o menor medida, estructuras tetraédricas según sea su arquitectura de construcción. Los valores de la �hP
resultaron en 227 K
Figura IV.4.A. CP(T) (líneas sólidas) y –dq/dT (líneas punteadas) del agua líquida calculada por los diferentes modelos. Los símbolos son los datos experimentales de Angell et. al. [43]. B. CP(τ).
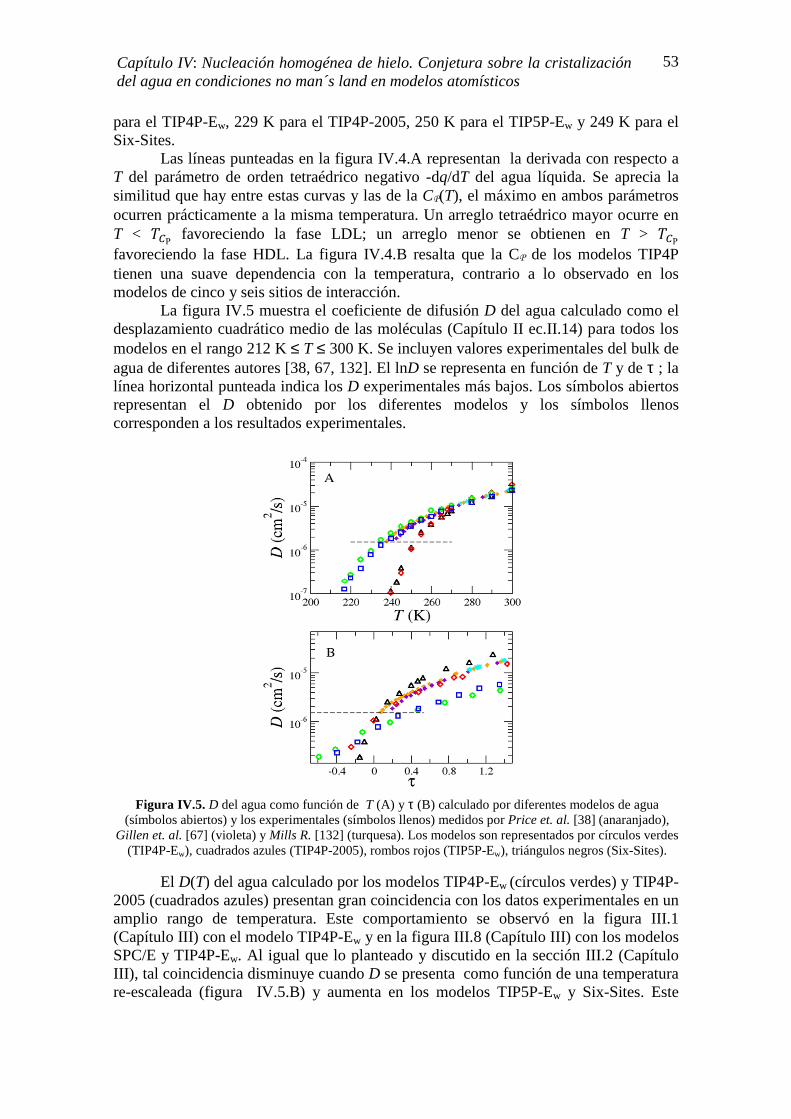
53 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
para el TIP4P-Ew, 229 K para el TIP4P-2005, 250 K para el TIP5P-Ew y 249 K para el Six-Sites.
Las líneas punteadas en la figura IV.4.A representan la derivada con respecto a T del parámetro de orden tetraédrico negativo -dq/dT del agua líquida. Se aprecia la similitud que hay entre estas curvas y las de la CP(T), el máximo en ambos parámetros ocurren prácticamente a la misma temperatura. Un arreglo tetraédrico mayor ocurre en T < �hP favoreciendo la fase LDL; un arreglo menor se obtienen en T > �hP favoreciendo la fase HDL. La figura IV.4.B resalta que la CP de los modelos TIP4P tienen una suave dependencia con la temperatura, contrario a lo observado en los modelos de cinco y seis sitios de interacción.
La figura IV.5 muestra el coeficiente de difusión D del agua calculado como el desplazamiento cuadrático medio de las moléculas (Capítulo II ec.II.14) para todos los modelos en el rango 212 K ≤ T ≤ 300 K. Se incluyen valores experimentales del bulk de agua de diferentes autores [38, 67, 132]. El lnD se representa en función de T y de τ ; la línea horizontal punteada indica los D experimentales más bajos. Los símbolos abiertos representan el D obtenido por los diferentes modelos y los símbolos llenos corresponden a los resultados experimentales.
El D(T) del agua calculado por los modelos TIP4P-Ew (círculos verdes) y TIP4P-2005 (cuadrados azules) presentan gran coincidencia con los datos experimentales en un amplio rango de temperatura. Este comportamiento se observó en la figura III.1 (Capítulo III) con el modelo TIP4P-Ew y en la figura III.8 (Capítulo III) con los modelos SPC/E y TIP4P-Ew. Al igual que lo planteado y discutido en la sección III.2 (Capítulo III), tal coincidencia disminuye cuando D se presenta como función de una temperatura re-escaleada (figura IV.5.B) y aumenta en los modelos TIP5P-Ew y Six-Sites. Este
Figura IV.5. D del agua como función de T (A) y τ (B) calculado por diferentes modelos de agua (símbolos abiertos) y los experimentales (símbolos llenos) medidos por Price et. al. [38] (anaranjado),
Gillen et. al. [67] (violeta) y Mills R. [132] (turquesa). Los modelos son representados por círculos verdes (TIP4P-Ew), cuadrados azules (TIP4P-2005), rombos rojos (TIP5P-Ew), triángulos negros (Six-Sites).

54 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
hecho se adjudica a que la Tf de los modelos de cuatro puntos están muy lejos de la experimental; al ser muy bajas, Tf TIP4P-Ew = 244 K, Tf TIP4P-2005 = 252 K, los TIP4P representan el agua, en la mayor parte del rango sobre-enfriado experimental, en su fase líquida estable y no en su correspondiente estado metaestable sobre-enfriado. Contrario a lo que ocurre con el Six-Sites y en particular con el TIP5P-Ew, cuya Tf está muy próxima a la experimental.
La línea punteada que se muestra en la figura IV.5, correspondiente a los D experimentales más bajos. Cuando se compara con los resultados de las simulaciones, se observa que la misma se produce a temperaturas cercanas a la �hP
de cada modelo.
IV.3. ESTIMACIÓN DEL TAMAÑO DEL EMBRIÓN CRÍTICO.
El tamaño del embrión crítico de hielo en función de la temperatura re-escaleada N(τ) fue estimado con la ec.IV.1; se utilizaron los valores de Ea, Ea(T) y ∆Hc(τ). A la entalpía de cristalización se le aplicó un ajuste lineal (∆Hc(τ) = aτ +b) en el rango VTF del Jhom (entre ∼ 235.15 y 245.00 K, correspondiente a un τ entre ∼ 0.01 y 0.26). Las constantes a y b del ajuste se reportan en la tabla IV.4. La figura IV.6 muestra los mismos valores de ∆Hc de la figura IV.3 en función de τ y la línea sólida es el ajuste lineal aplicado a ∆Hc(τ). Tabla IV.4. Constantes del ajuste lineal del ∆Hc(τ)para el rango de τ [0.01-0.26]. Modelos de agua
TIP4P-Ew, TIP4P-2005, TIP5P-Ew y Six-Sites. TIP4P-Ew TIP4P-2005 TIP5P-Ew Six-Sites
a [×10−23 J] −145 ± 7 -197,49 ± 2 −404 ± 20 −691 ± 30
b [×10−21 J] −6.07 ± 0.01 -6,30 ± 0.01 −8.87 ± 0.04 −8.20 ± 0.06
Figura IV.6. Entalpía de cristalización por molécula de agua en función de τ (∆Hc(τ)) estimada por los modelos TIP4P-Ew (verde), TIP4P-2005 (azul), TIP5P-Ew (rojo) y Six-Sites (negro). Las líneas rectas
son el ajuste lineal de ∆Hc(τ) para el rango 0.01 ≤ τ ≤ 0.26.

55 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
El número de moléculas N que conforman al embrión de hielo crítico estimado
para los distintos modelos se muestran en la tabla IV.5 y figura IV.7. En la figura, las líneas corresponden a los distintos modelos (verde TIP4P-Ew, azul TIP4P-2005, roja TIP5P-Ew, negra Six-Sites) y el rombo es el valor calculado por Baumgärtel y Zimmermann [2011] [56] (N = 358 moléculas a T = 237 K).
Tabla IV.5. Tamaño del embrión de hielo crítico como función de la temperatura escaleda N(τ)
estimado para distintos modelos de agua.
T [K] ττττ N [N° moléculas]
TIP4P-Ew TIP4P-2005 TIP5P-Ew Six-Sites
245.00 0.26 1010 960 664 670
244.15 0.24 927 882 612 620
243.15 0.21 820 782 545 556
242.15 0.19 758 725 506 520
241.15 0.16 678 650 456 473
240.15 0.14 632 606 427 445
239.15 0.11 570 549 388 409
238.15 0.08 518 499 355 379
237.15 0.06 486 470 335 361
236.15 0.03 444 431 309 337
235.40 0.01 419 407 293 323
La figura muestra que N(τ) incrementa con la temperatura. Este resultado es consistente con el hecho de que la probabilidad de formación de un embrión de hielo desde agua líquida sobre-enfriada metaestable, con tamaño mucho mayor que el del embrión crítico, cae notablemente cuando N(τ) aumenta; comportamiento que explica la rápida anulación del proceso de nucleación homogénea de hielo a temperaturas de unos pocos grados por encima de la �hP
. También se encuentra que los N(τ) estimados por los modelos sin LP son significativamente mayores que los correspondientes a los estimados para el Six-Sites y TIP5P-Ew, y presentan una dependencia mucho más
Figura IV .7. Tamaño del embrión de hielo crítico en agua líquida sobre-enfriada como función de τ estimado por los distintos modelos. Las líneas representan la predicción de los diferentes modelos, y el diamante es el valor calculado por Baumgärtel y Zimmermann [2012] [56].

56 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
pronunciada con la temperatura. Los N(τ) estimados por los modelos TIP5P-Ew y Six-Sites están en buena correspondencia con el valor experimental. Una vez más este hecho contribuye a reafirmar que los modelos construidos con pares de electrones explícitos representan mejor al comportamiento del agua sobre-enfriada. De ellos, es el TIP5P-Ew quien presenta coeficientes de difusión en mejor correspondencia con los experimentales cuando la escala de temperatura es corregida por las temperaturas que definen los límites del régimen sobre-enfriado.
En base a lo discutido en este Capítulo IV se pueden señalar las siguientes
conclusiones: • El uso de la variable τ representa una nueva forma de comparar las predicciones
de diferentes modelos de agua que ayuda a revelar sus ventajas y deficiencias. • El tamaño del embrión crítico N(τ) fue calculado por primera vez en sistemas
tipo bulk en T > �hP.
• N(τ) fue calculado de forma indirecta debido a la imposibilidad que presentan los modelos atomísticos para la cristalización espontánea del agua sobre-enfriada. En este sentido, se plantea una conjetura sobre tal imposibilidad, especialmente en condiciones no man´s land (T < �hP
). Está claro que el parámetro de orden tetraédrico q del sistema incrementa monotónicamente cuando la temperatura disminuye (sección III.2 Capítulo III). El máximo de la capacidad calorífica isobárica ocurre a temperaturas donde las fluctuaciones tetraédricas son máximas; a T < �hP
, las fluctuaciones decaen por lo que se debería esperar cristalización. Las simulaciones realizadas por Moore E. y Molinero V. [100, 129] con el modelo mW (representa a la molécula de agua como una sola partícula con interacciones de corto alcance que imitan a los enlaces puentes de hidrógeno; la separación entre partículas es dada por el ángulo), logran cristalizar. Para una velocidad alta de enfriamiento, el sobre-enfriado mW se transforma del estado HDL al estado LDL cuando la T disminuye; a una velocidad baja de enfriamiento, el líquido sobre-enfriado mW cristaliza espontáneamente. Se considera que la razón para ello, es debido al suave potencial repulsivo que utiliza el modelo mW en comparación al r-12 de los modelos atomísticos; por lo tanto, el mW es capaz de rotar hacia una estructura organizada y por ende cristalizar a T < �hP
. Una manera en que podrían los modelos atomísticos incorporar una repulsión más suave es permitiendo la transferencia de protón entre moléculas.
• El modelo TIP5P-Ew fue seleccionado en vez del Six-Sites para el estudio de la nucleación heterogénea. El desarrollo de dicho estudio se expone en el siguiente Capítulo. Los detalles computaciones se presentan a continuación.
• Las moléculas de los sistemas de hielo y agua líquida estuvieron confinadas en un paralelepípedo y en una caja cúbica, respectivamente, con condiciones de contorno periódicas. Los sistemas hielos fueron optimizados con las reglas de Bernal-Fowler usando el algoritmo propuesto por por Buch and Sadlej 1998 [49].

57 Capítulo IV: Nucleación homogénea de hielo. Conjetura sobre la cristalización del agua en condiciones no man´s land en modelos atomísticos
• La temperatura y la presión de los sistemas fueron controlados usando el termostato Nose-Hoover con una constante temporal de 0.1 ps, y el baróstato de Parinello-Raphman con una constante temporal de 0.5 ps.
• La compresibilidad fue de 4.5 x 10-5 bar-1 para los sistemas de agua y de 1.0 x 10-7 bar-1 para los sistemas de hielo.
• El cut-off esférico se impuso igual a 0.9 nm para las interacciones Lennard-Jones e interacciones electrostáticas de corto alcance.
• Las interacciones electrostáticas de largo alcance fueron consideradas a través de la técnica PME para los modelos TIP4P-Ew y TIP5P-Ew; para el Six-Sites se empleó un cut-off de 0.9 nm.
• El paquete de cálculo empleado fue Gromacs v.4.5.5 [50, 51]. • El número de simulaciones fue aproximadamente de 140, cada una con duración
de 10ns. • Las distintas propiedades fueron calculadas considerando los últimos 5 ns del
paso de simulación. Los primeros 5ns fueron suficientes para garantizar la relajación del sistema.

58
Capítulo V: Nucleación heterogénea de hielo.
CAPÍTULO V
NUCLEACIÓN HETEROGÉNEA DE HIELO.
La nucleación heterogénea de hielo es propiciada por la presencia de partículas o sustratos sólidos e insolubles en agua. Estos sustratos, denominados núcleos de hielo (IN), actúan como catalizadores en el proceso de nucleación; son catalizadores porque disminuyen la energía de activación en el proceso de formación de “embriones de hielo”. La presencia de IN provoca que la nucleación heterogénea se produzca en mayor medida que la homogénea [80-83].
La T.C.N., como se señaló en el Capítulo I sección I.3, refiere a que la energía libre de formación del embrión crítico de hielo es igual a ∆���8∗ � ∆�� �∗ 9 (f es el factor de potencia catalítica y cuantifica la habilidad que tiene un IN para llevar a cabo la nucleación heterogénea [84]); el valor de f está vinculado con las distintas características que presentan los IN [1, 82, 85].
Por otra parte, también se mencionó en la sección I.3.2, que la visión estadística de la nucleación heterogénea de agua líquida sobre-enfriada metaestable presenta dos hipótesis conceptuales opuestas. Una de ellas es la “hipótesis estocástica”, la cual se ejemplifica en los trabajos de Bigg [1953, 1955] [94-96], Carte [1956, 1959] [97, 98] y Dufour y Defay [1963] [99]. Todos ellos señalaron, que si bien es cierto que el proceso aleatorio de nucleación de hielo desde agua sobre-enfriada aumenta debido a la presencia de los IN inmersos en las gotas, estos no alteran la naturaleza estocástica de la formación de los embriones de hielo. Recientes estudios experimentales soportan esta visión [91, 137, 138, 139]. Estos estudios refieren que a temperatura constante, en una población de gotas de agua sobre-enfriada de tamaños similares, donde cada gota contiene inmerso IN de tamaños y composiciones químicas muy parecidas, todas las gotas tienen la misma oportunidad de congelar en un período de tiempo dado.
La otra hipótesis, en principio propuesta por Levine [1950] [89], plantea que el congelamiento de una población de gotas de agua sobre-enfriada con IN inmersos, no es dependiente del tiempo sino que se produce de manera instantánea por los “sitios activos” presentes en las superficies de los IN. Estos sitios pueden ser de origen físico (grietas, poros, etc.) y/o de origen químico (las superficies se encuentran impregnadas de compuestos químicos insolubles), los cuales presentan características particulares (singulares) de congelamiento; es decir, la nucleación es determinística [1, 90, 91, 135, 139]. Diferentes estudios asumieron, que cada IN inmerso en una gota de agua sobre-enfriada presenta una temperatura característica (Ts < 273 K), en la cual la nucleación/congelación de la gota se iniciará; por lo tanto, la nucleación/congelación de la gota es propiciada por aquellos IN con Ts más alta [139]. Los IN con tamaños y características superficiales diferentes nuclean a Ts diferentes, en caso contrario (IN iguales), nuclean todos a la misma Ts [135]. Si la temperatura de sobre-enfriamiento permanece constante, el número de gotas no congeladas no cambiará con el tiempo (solo permanecen congeladas aquellas gotas cuyos IN tuvieron sitios activos a la Ts más alta); en otras palabras, nuevos eventos de nucleación no ocurrirán si las condiciones ambientales son iguales; en este sentido, el tiempo no es un factor importante [92]. Autores como Möhler et al. [2006] [83], Connolly et al. [2009] [100] han mostrado que la tasa de nucleación heterogénea cumple con la hipótesis singular.

59
Capítulo V: Nucleación heterogénea de hielo.
El desarrollo de este Capítulo V correspondiente al cuarto y último estudio llevado a cabo en la presente tesis, se enmarcó dentro de la hipótesis estocástica de un IN específico inmerso en un bulk de agua líquida sobre-enfriada. Con ello, esta investigación es la primera que describe a nivel de los nanómetros, la estadística de la nucleación heterogénea del agua sobre-enfriada a través de simulaciones de D.M.
La naturaleza estocástica y dependiente del tiempo, plantea que la tasa de nucleación heterogénea se mide como la probabilidad por unidad de tiempo que un embrión de hielo, en ciertas condiciones, alcanza el tamaño crítico. Esta probabilidad es descrita por una distribución de Poisson de la forma mostrada en la ec.I.6 (Capítulo I sección I.3). Para los propósitos del presente estudio, la ec.I.6. será referida como la ec.V.1 en cuyo caso la tasa de nucleación heterogénea extensiva se representa como jM.
ln &BCBD. � �¤¥� ec.V.1
donde Nt es el número de gotas que permanecen líquidas a un tiempo t y N0 es el número inicial de gotas de agua líquida.
Los sistemas simulados estuvieron compuestos de agua sobre-enfriada y un núcleo cristalino insoluble; el IN fue construido con las mismas moléculas y características estructurales del hielo Ih (los mejores nucleadores de hielo son IN con estructuras cristalinas muy parecidas al Ih [66, 83, 88]). La tasa de nucleación fue calculada para distintos tamaños de IN con la ec.V.1. A continuación se describen los procedimientos, las técnicas, los resultados y análisis del estudio.
V.1. PREPARACIÓN DE LOS SISTEMAS.
Los sistemas se constituyeron con 768 moléculas de agua TIP5P-Ew; M de ellas conformaron un núcleo sólido INM y el resto se dispusieron en estado líquido. Las posiciones de las moléculas del INM fueron parcialmente restringidas con la técnica position-restraints. Esta técnica mantiene las moléculas confinadas localmente sin impedir una agitación térmica realista de las mismas, de modo que el núcleo permanece sólido a lo largo de toda la simulación.
Las configuraciones iniciales de los sistemas se prepararon de la siguiente forma: 1. Un sistema de 768 moléculas de agua congelada (hielo Ih), fue simulado a
temperatura constante de 250 K a la presión constante de 1 atm durante 10ns. Las dimensiones de la caja de simulación fueron apropiadas para que el volumen del hielo fuese correctamente reproducido con condiciones de contorno periódicas. El volumen final de la caja fue igual a xyz = (2.636×3.043×2.959) nm3.
2. A partir del estado final de la simulación anterior se prepararon seis sistemas. Cada sistema estuvo conformado con un solo núcleo de hielo constituido con una cantidad específica de moléculas M (M = 48, 56, 64, 72, 80 y 90). Solo las posiciones moleculares de estos núcleos fueron restringidas. Por ejemplo, de las 768 moléculas de hielo, las posiciones de 678 moléculas no se restringieron cuando el INM fue igual a 90. Los núcleos fueron paralelepípedos con caras planas paralelas a los planos de la caja de simulación (figura V.1).

60
Capítulo V: Nucleación heterogénea de hielo.
3. A volumen constante, los seis sistemas fueron agitados a la temperatura constante de 400 K por un tiempo suficientemente largo (10 ns), de modo que el hielo no restringido se fundiera completamente.
4. De cada sistema (constituido con un particular INM) se extrajeron al azar catorce estados de los últimos 2 ns de la trayectoria de las simulaciones realizadas en el punto 3; tales estados fueron denominados “réplicas”. Estas 14 réplicas tuvieron posiciones y velocidades diferentes para las moléculas y se utilizaron como configuraciones iniciales para realizar las simulaciones de nucleación. Se aseguró la mayor descoordinación entre las réplicas, eligiendo estados no más cercanos que 100 ps. Para desarrollar el estudio estadístico de la nucleación, cada una de las réplicas
fueron simuladas a 250 K hasta obtener congelación total del sistema o hasta que el tiempo de simulación alcanzó los 200 ns. Dicha T corresponde al máximo de la capacidad calorífica isobárica (�h¦) estimada para el TIP5P-Ew (ver sección IV.2 Capítulo IV). El modelo TIP5P-Ew fue escogido por las razones presentadas y discutidas en las secciones previas.
V.2. MONITOREO DEL PROCESO DE NUCLEACIÓN.
El monitoreo cualitativo del proceso de nucleación en los sistemas de agua sobre-enfriada con el núcleo cristalino insoluble inmerso en ella, se realizó mediante las gráficas energía potencial vs tiempo de simulación, Ep(t). La figura V.2.a muestra la EP(t) para una réplica particular del sistema con IN64 (núcleo de 64 moléculas de agua restringidas con position-restraints). Comparando la figura con los resultados obtenidos en el estudio de nucleación de Matsumoto et. al. [2002] [62] (ver Capítulo I sección I.3.3), se distinguen zonas con estados bien marcados que caracterizan al proceso de nucleación.
b. c. a.
Figura V.1. Paralelepípedo que representa a la estructura del núcleo de hielo para el caso M = 64, visto desde el plano basal en dirección z (a.) y los planos prismáticos en las direcciones x e y (b. y
c., respectivamente). Esferas rojas y blancas representan los átomos de oxígeno e hidrógeno, respectivamente, y el rectángulo la caja de simulación.

61
Capítulo V: Nucleación heterogénea de hielo.
La zona 1 (color negro) de la figura V.2, se encuentra ubicada en el intervalo temporal 0 ns < t < 125 ns y está caracterizada por una Ep(t) relativamente constante y es la zona en donde el agua líquida no presenta cambios físicos; el ruido que se observa en esta zona se debe a los movimientos moleculares generados por la agitación térmica. La zona 2 (color azul) correspondiente al intervalo temporal 125 ns < t < 140 ns, está definida por una Ep(t) que disminuye rápidamente en el tiempo de simulación; este comportamiento revela el comienzo del proceso de nucleación seguido de la congelación del agua. La zona 3 (color violeta) ubicada entre 140 ns < t < 200 ns y caracterizada con una Ep(t) constante con un valor promedio menor que el correspondiente al de la zona 1, refleja la formación completa de la fase hielo. En la base de la figura V.2. se representa el cambio estructural de cada zona.
El monitoreo cuantitativo del proceso de nucleación, en los distintos sistemas, se realizó identificando “el tiempo de nucleación tN” a través de un nuevo parámetro. Este parámetro, dependiente del tiempo y denotado como ni (t), contabiliza el número de moléculas que permanecen confinadas en un volumen de 0.001 nm3 durante un período
Figura V.2. Energía potencial (a) y número de moléculas en “estado hielo” (b) vs tiempo de simulación a T = 250 K para una de las réplicas del sistema de agua líquida sobre-enfriada con IN64. Regiones de
diferentes colores se explican en el texto. La intercepción entre la zona 1 y zona 2 indica el tiempo de nucleación tN. Las tres fotos son propias de cada zona (izquierda, central y derecha corresponde a la zona
1, 2 y 3, respectivamente). Las esferas rojas y blancas, respectivamente, representan a los átomos de oxígeno e hidrógeno del IN. Los segmentos rojos son los enlaces de hidrógeno del agua líquida.

62
Capítulo V: Nucleación heterogénea de hielo.
de 1ns desde un tiempo t. es decir, ni (t) representa el número de moléculas involucradas en el estado sólido presentes en el sistema. La implementación de ni(t) permitió determinar tN con mayor exactitud, ya que ni(t) presenta menor ruido que Ep(t), tal como se muestra en la figura V.2.b. Para una de las 14 réplicas del sistema con IN64, tN resulta igual a 125 ns.
V.3. ESTADÍSTICA DE LA NUCLEACIÓN.
Las réplicas de los sistemas de agua sobre-enfriada constituidos con los IN de 64, 72, 80 y 90 moléculas congelaron completamente a diferentes tiempos tN. Estos resultados hicieron posible monitorear el número de réplicas no congeladas (Nt), calcular el cociente Nt/N0 (N0 =14) y estimar la tasa de nucleación para cada tamaño de núcleo (jM). N0 =14 representa el número total de réplicas a t = 0, es decir, según ec.V.1, son las gotas iniciales de agua líquida (14 gotas), cada una con un específico INM. En el sistema con IN igual a 56 moléculas, se obtuvo nucleación en sólo una de las réplicas y ningún evento de nucleación con IN48.
La figura V.3 muestra el ln (Nt/14) vs tN para los sistemas de agua sobre-enfriada constituidos con IN64 (puntos negros), IN72 (puntos rojos), IN80 (puntos verdes), IN90 (puntos azules). El ajuste lineal de los valores mediante ec.V.1 se representa con líneas rectas. Se aprecia que la dispersión de los datos, atribuido al escaso número de réplicas, es mayor para los IN de 64 y 72 moléculas.
También se observa que jM decrece con el tamaño del IN. Los valores de la velocidad de nucleación para cada tamaño de IN y el coeficiente de correlación lineal (R2) se reúnen en la tabla V.1.
Figura V.3. ln (Nt/14) vs tN para sistemas de agua sobre-enfriada a T = 250 K constituidos con el IN64 (negro), el IN72 (rojo), el IN80 (verde) y el IN90 (azul). Las líneas rectas representan el ajuste lineal de
los datos.
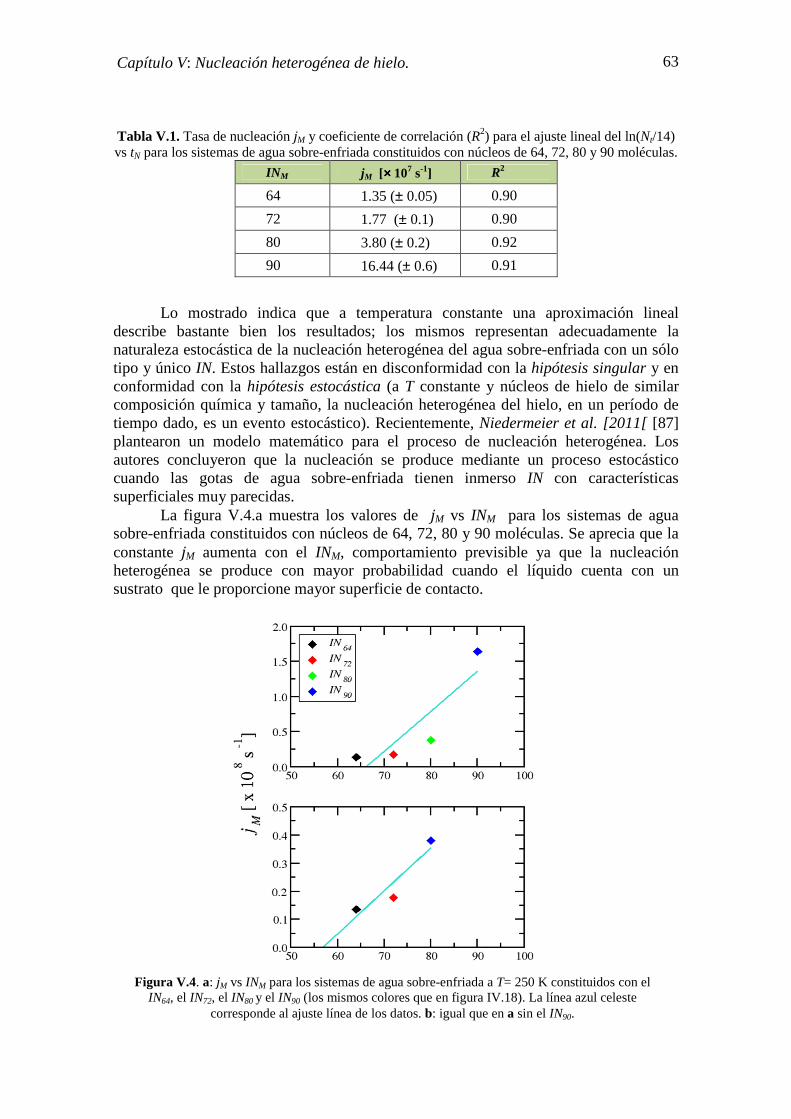
63
Capítulo V: Nucleación heterogénea de hielo.
Tabla V.1. Tasa de nucleación jM y coeficiente de correlación (R2) para el ajuste lineal del ln(Nt/14) vs tN para los sistemas de agua sobre-enfriada constituidos con núcleos de 64, 72, 80 y 90 moléculas.
INM jM [×××× 107 s-1] R2
64 1.35 (± 0.05) 0.90
72 1.77 (± 0.1) 0.90
80 3.80 (± 0.2) 0.92
90 16.44 (± 0.6) 0.91
Lo mostrado indica que a temperatura constante una aproximación lineal
describe bastante bien los resultados; los mismos representan adecuadamente la naturaleza estocástica de la nucleación heterogénea del agua sobre-enfriada con un sólo tipo y único IN. Estos hallazgos están en disconformidad con la hipótesis singular y en conformidad con la hipótesis estocástica (a T constante y núcleos de hielo de similar composición química y tamaño, la nucleación heterogénea del hielo, en un período de tiempo dado, es un evento estocástico). Recientemente, Niedermeier et al. [2011[ [87] plantearon un modelo matemático para el proceso de nucleación heterogénea. Los autores concluyeron que la nucleación se produce mediante un proceso estocástico cuando las gotas de agua sobre-enfriada tienen inmerso IN con características superficiales muy parecidas.
La figura V.4.a muestra los valores de jM vs INM para los sistemas de agua sobre-enfriada constituidos con núcleos de 64, 72, 80 y 90 moléculas. Se aprecia que la constante jM aumenta con el INM, comportamiento previsible ya que la nucleación heterogénea se produce con mayor probabilidad cuando el líquido cuenta con un sustrato que le proporcione mayor superficie de contacto.
Figura V.4. a: jM vs INM para los sistemas de agua sobre-enfriada a T= 250 K constituidos con el
IN64, el IN72, el IN80 y el IN90 (los mismos colores que en figura IV.18). La línea azul celeste corresponde al ajuste línea de los datos. b: igual que en a sin el IN90.

64
Capítulo V: Nucleación heterogénea de hielo.
La estimación del tamaño del núcleo mínimo se realizó sobre la conjetura que si el número de moléculas de los IN disminuyen lo suficiente, la velocidad de nucleación sería cero para algún valor INM mínimo. En efecto, se observó que para INM = 48 no hubo nucleación. En base a esto, se consideró que un ajuste lineal de jM vs INM permitiría estimar el número mínimo de moléculas que pueden iniciar nucleación de hielo en sistemas que presenten las mismas características de los simulados. La línea azul celeste de la figura V.4.a. representa el ajuste lineal para los cuatro casos.
Una extrapolación de la línea azul a jM = 0 permitió estimar INM mínimo igual a 66 moléculas. Sin embargo, este número no es consistente con el hecho que se obtuvo nucleación en todas las réplicas de los sistemas de agua sobre-enfriada constituidos con el IN64; y es menos consistente con el evento de nucleación observado en el sistema constituido con el IN56. En este sentido, se consideró que la fuente de esta inconsistencia se debe al valor de j90, notablemente más alto que el de los demás. Es pertinente recordar que las simulaciones se llevaron a cabo en un sistema constituido con un total de 768 moléculas con condiciones de contorno periódicas (pbc), por lo que sustratos de grandes tamaños promueven a que los indeseados efectos de bordes sean mayores. En este sentido, se discierne que las imágenes de un IN grande (en comparación con el tamaño total del sistema) pueden favorecer artificialmente el proceso de nucleación.
En base a lo anterior, se realizó el ajuste lineal excluyendo los resultados de los sistemas con IN90 (figura V.4.b). El INM mínimo para este caso fue ~ de 57 moléculas, valor que está en concordancia con los resultados obtenidos en los sistemas con INM = 48 y 56 moléculas. En la sección de nucleación heterogénea del trabajo experimental de Liu et al. [2007] [61], los autores reportan que el tamaño mínimo del embrión de hielo es de 70 moléculas de agua a T = 240.85 K.
Otro respaldo en cuanto a la estimación del INM mínimo descartando los resultados del IN90, proviene de las gráficas ni(t). En la figura V.5 se representa ni (t) de una de las 14 réplicas de los sistemas sobre-enfriados constituidos con IN64, IN72, IN80 y IN90; estas representación es característica de todas las simulaciones. Indudablemente el sistema con IN90 presenta un comportamiento diferente a los otros; el aumento de ni(t) es irregular y escalonado en comparación a lo rápido y sin pausa del ni(t) de los otros INM. Así mismo, las EP(t) de los sistemas con IN64, IN72 e IN80 (no mostradas) tuvieron características similares a las presentadas y discutidas en la sección V.2. Además, los tN para los sistemas con IN90 fueron tan pequeños en relación al tiempo total de simulación, que el proceso de crecimiento de hielo tuvo poca demora.
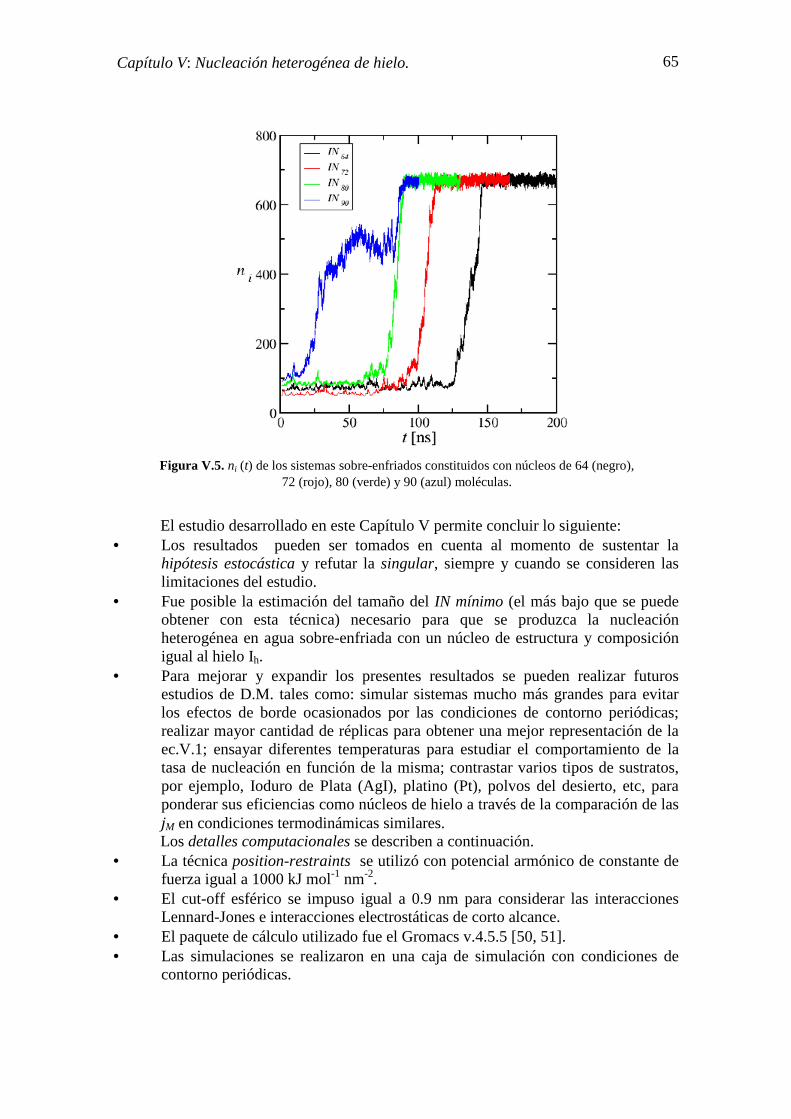
65
Capítulo V: Nucleación heterogénea de hielo.
El estudio desarrollado en este Capítulo V permite concluir lo siguiente: • Los resultados pueden ser tomados en cuenta al momento de sustentar la
hipótesis estocástica y refutar la singular, siempre y cuando se consideren las limitaciones del estudio.
• Fue posible la estimación del tamaño del IN mínimo (el más bajo que se puede obtener con esta técnica) necesario para que se produzca la nucleación heterogénea en agua sobre-enfriada con un núcleo de estructura y composición igual al hielo Ih.
• Para mejorar y expandir los presentes resultados se pueden realizar futuros estudios de D.M. tales como: simular sistemas mucho más grandes para evitar los efectos de borde ocasionados por las condiciones de contorno periódicas; realizar mayor cantidad de réplicas para obtener una mejor representación de la ec.V.1; ensayar diferentes temperaturas para estudiar el comportamiento de la tasa de nucleación en función de la misma; contrastar varios tipos de sustratos, por ejemplo, Ioduro de Plata (AgI), platino (Pt), polvos del desierto, etc, para ponderar sus eficiencias como núcleos de hielo a través de la comparación de las jM en condiciones termodinámicas similares. Los detalles computacionales se describen a continuación.
• La técnica position-restraints se utilizó con potencial armónico de constante de fuerza igual a 1000 kJ mol-1 nm-2.
• El cut-off esférico se impuso igual a 0.9 nm para considerar las interacciones Lennard-Jones e interacciones electrostáticas de corto alcance.
• El paquete de cálculo utilizado fue el Gromacs v.4.5.5 [50, 51]. • Las simulaciones se realizaron en una caja de simulación con condiciones de
contorno periódicas.
Figura V.5. ni (t) de los sistemas sobre-enfriados constituidos con núcleos de 64 (negro), 72 (rojo), 80 (verde) y 90 (azul) moléculas.

66
Capítulo V: Nucleación heterogénea de hielo.
• La temperatura y la presión del sistema se controlaron usando el termostato de Nose-Hoover y el barostato de Parrinello-Rahman, con tiempos de relajación de 0.5ps y 0.1ps, respectivamente; y la compresibilidad fue de 4.5×10−5 bar-1.
• Las interacciones electrostáticas de largo alcance se consideraron con la técnica PME.
• El número de sistemas simulados para evaluar el proceso de nucleación, fue de cincuenta y seis; con los recursos computacionales disponibles, cada sistema tardó en nuclear/congelar ~ cinco días.

67 Conclusiones
CONCLUSIONES
El presente Capítulo reúne de manera articulada las conclusiones arrojadas por los resultados obtenidos en los diversos estudios de simulación de agua líquida, resultados que fueron mostrados y discutidos en los Capítulos III, IV y V. En los primeros tres estudios se investigó el comportamiento de algunas propiedades del agua líquida tales como: la TMD, el D, la Tf, el ��̅���, el q, la CP y N, con particular interés en el régimen sobre-enfriado. El último estudio se enfocó en evaluar la naturaleza de la nucleación heterogénea de hielo.
A continuación se enumeran las principales conclusiones: 1. Los procedimientos diseñados para modificar el campo de fuerza (force field)
del modelo TIP5P-Ew en sistemas de agua líquida, permitieron plantear metodologías sencillas para llevar a cabo reparametrizaciones de modelos de agua.
2. En el rango sobre-enfriado metaestable, el modelo reparametrizado TIP5P-EwR logró ajustar el D(T) mejor que el original TIP5P-Ew, subestimó ligeramente la ρ(T) y presentó un marcado detrimento en la Tf (~ 262 K). Por lo tanto, el original TIP5P-Ew siguió representando mejor las tres propiedades evaluadas.
3. No fue posible obtener un mejor modelo mediante cambios sistemáticos de los parámetros de los potenciales LJ y Coulomb. Esta imposibilidad se debió al alto costo de tiempo y trabajo que se requiere, así como a la poca eficacia de las metodologías planteadas para las reparametrizaciones.
4. La exploración en sistemas de agua líquida a T = 255 K sobre la sensibilidad de
las propiedades D y ρ con los parámetros σ, ε y q del TIP5P-Ew, permitió observar que D es altamente sensibilidad a la variación del parámetro σ del potencial LJ. Dicha exploración fue previa a la obtención del reparametrizado TIP5P-EwR.
5. El cálculo del tiempo de vida promedio de los enlaces puentes de hidrógeno tH
(t) y el parámetro de orden tetraédrico q, mostró que el agua sobre-enfriada presenta una tendencia clara a formar enlaces puentes de hidrógeno (Hb) más estables que el agua a temperaturas mayores a 273 K. Esta tendencia fue mayor en las simulaciones realizadas con modelos construidos con puntos de carga que asemejan a los pares de electrones no enlazantes del átomo de oxígeno (LP), es decir, en los modelos TIP5P-Ew y Six-Sites. Estos modelos tuvieron un efecto claro para favorecer considerablemente la estructura tetraédrica del agua a T < 270 K. Tal situación es cónsona con la estructura tipo red más definida que adquieren los Hb del agua líquida en el rango de T sobre-enfriada [6, 7].
6. En los sistemas de agua líquida se realizaron distintos escalamientos de la temperatura; los mismos fueron necesarios para comparar las predicciones de los

68 Conclusiones
diferentes modelos. Los escalamientos de T fueron planteados como +Z+�+� y
§ � +Z+£+�Z+£, los cuales ayudaron a revelar las ventajas y deficiencias de los
modelos utilizados. Cuando se evaluó el D(+Z+�+� ) y el D(τ), los modelos TIP5P-
Ew y Six-Sites presentaron mejor correspondencia con los valores experimentales en comparación a los modelos sin LP (SPC/E, TIP4P-Ew y TIP4P-2005.
7. Las capacidades caloríficas a presión constante calculadas en función de una temperatura escaleada, CP(τ), exhibieron un pico justo donde la experimental muestra una divergencia aparente, divergencia que se adjudica a una transición de fase HDL-LDL del agua líquida sobre-enfriada (hipótesis LLCP). Las CP (τ) para los modelos TIP5P-Ew y el Six-Sites no solo exhibieron un pico más agudo sino que estuvieron en mejor correspondencia con la experimentación, contrario a lo obtenido en los modelos sin LP (TIP4P-Ew y TIP4P-2005).
8. El tamaño crítico del embrión de hielo necesario para la nucleación homogénea N(τ) fue calculado por primera vez en sistemas tipo bulk de agua líquida y en un amplio rango de temperatura sobre-enfriada. Los modelos construidos con LP (TIP5P-Ew y Six-Sites) estuvieron en muy buena correspondencia con la experimentación.
9. De lo concluido en los puntos anteriores se reafirma que los modelos construidos con puntos de carga que asemejan a los pares de electrones no enlazantes del átomo de oxígeno del agua (LP), representan mejor las propiedades evaluadas. Tal situación es contraria a lo obtenido con los modelos sin LP; algunos de ellos ni siquiera lograron representar la tendencia cualitativa de algunas propiedades.
10. El N(τ) se calculó de forma indirecta debido a la imposibilidad que presentan los
modelos atomísticos para la cristalización espontánea del agua sobre-enfriada. Debido a esta situación, se presenta una conjetura sobre tal imposibilidad, especialmente en condiciones no man´s land (T < �hP
). Se considera que tal situación se debe al fuerte potencial repulsivo que utilizan los modelos atomísticos, impidiendo la rotación de las moléculas de agua hacia una estructura más organizada y por consiguiente a su posterior cristalización.
11. Se realizó el primer estudio sobre la estadística de la nucleación heterogénea en agua sobre-enfriada. En base a las conclusiones previamente señaladas, un modelo construido con LP fue escogido para llevar a cabo las simulaciones. De los modelos estudiados, el TIP5P-Ew fue seleccionado sobre el Six-Sites. Tal selección fue motivada porque la Tf del TIP5P-Ew está en mejor correspondencia con la Tf experimental.
El estudio arrojó que la naturaleza de la nucleación heterogénea del agua sobre-enfriada con un tipo y único IN (construido con las mismas moléculas y características estructurales del hielo Ih) es estocástica. En este sentido, la

69 Conclusiones
hipótesis estocástica se sustenta y la hipótesis singular se refuta, considerando, indudablemente, las limitaciones del estudio.
12. Se estimó el tamaño mínimo del núcleo de hielo (IN mínimo) necesario para
llevar a cabo la nucleación heterogénea en agua sobre-enfriada con un núcleo de estructura y composición igual al hielo Ih. Con base en lo anterior, es pertinente realizar las siguientes recomendaciones:
� Intentar modificar la parte repulsiva del potencial LJ de los modelos atomísticos hacia un potencial más suave mediante la transferencia protónica entre moléculas. Este punto se vincula con la conjetura planteada sobre la ausencia de nucleación en los modelos atomísticos incluso a temperaturas muy bajas (T < �hP
). � Mejorar y expandir los resultados obtenidos en el estudio de la nucleación
heterogénea. Sería conveniente realizar futuros estudios de D.M. orientados a simular sistemas más grandes, ensayar otras temperaturas y/o contrastar distintos sustratos.
Por último, es importante mencionar que las publicaciones arrojadas por el
desarrollo de la presente tesis: “On the relation between hydrogen bonds, tetrahedral order and molecular mobility in model water” (Chem. Phys. Lett., 538: 35-38, 2012) y “The water supercooled regime as described by four common water models” (J. Chem. Phys. 139024506-1, 2013), están vinculadas con los resultados mostrados en los Capítulos III (sección III.2) y IV. Los resultados del Capítulo V se encuentran en proceso de ser publicados bajo el título “Stochastic vs Singular hypothesis in heterogeneous nucleation of ice by molecular dynamics”.

70 Apéndice 1
APÉNDICE 1
1.1. DEDUCCIÓN MATEMÁTICA DE GAMMA (γ).
Los criterios tomados en cuenta para relacionar los factores fσ, fε y fq entre sí fueron: • La modificación sólo del término atractivo del potencial LJ (línea roja punteada de la figura 1.1) mientras el término repulsivo permaneció igual. Esta elección estuvo basada en el hecho de querer mantener la topología o estructura de la molécula de agua inalterable, mientras la energía de las fuerzas de dispersión de London se modificaban. Se planteó que el término atractivo del nuevo modelo (parámetros primados) respecto al modelo original (parámetros no primados), cambiase en un factor γ según ec.1.1; y el término repulsivo primado en función del no primado permaneciera igual según ec.1.2. 4_´�´e � 4_�eγ ec.1.1
4_´�´O � 4_�O ec.1.2
Reemplazando σ´ = fσ σ y ε´= fε ε , se obtiene:
f��fb��e � γ ec.1.3
f��fb��O � 1 ec.1.4
Las ecuaciones de los factores fσ y fε en función de γ se obtienen relacionando estas dos expresiones; a saber: fb � O
√γ� ec.1.5
fj �γ ec.1.6
• La vinculación del parámetro q del potencial de Coulomb con el potencial LJ. Para ello se estableció la aproximación de que a una distancia ro las fuerzas entre las moléculas no cambiasen entre el modelo primado y no primado, es decir: «GCoul�� � +
σ
VLJ
0
término atractivo
Figura 1.1. Potencial Lennard-Jones 12-6 (línea negra gruesa). La modificación del término atractivo es representada por la línea roja punteada.
r
término repulsivo

71 Apéndice 1
«GLJ�� � � «G´Coul�� � + «G´LJ�� �. La sustitución de cada una de las fuerzas por sus expresiones correspondientes resulta en:
� ±WiTjDL)W + 4_ &Ob�WL)�� . � 4_ &eb�L)² . � � ±´WiTjDL)W + 4_´ &Ob´�WL)�� . � 4_´ &eb´�L)² . ec.1.7
Las ecuaciones 1.1 y 1.2 se reemplazan en 1.7 para obtener:
� ±WiTjDL)W � 4_ &eb�L)² . � � ±´WiTjDL)W � 4_ &eb�L)² . γ ec.1.8
Eligiendo ro = 3σ, la expresión queda:
� ±W�eTjDbW � ³j´�b � � ±´W�eTjDbW � ³j´�b γ ec.1.9
El factor γ se despeja de la ec.1.9 para dar:
µ � i��eT�)b �q � q´� + 1 ec.1.10
Y sustituyendo q´= fq ⋅ q, se obtiene γ en función de los parámetros correspondientes al modelo original y en función del factor independiente fq:
γ � i�qW�OZfqW��eTjDbj + 1 ec.1.11
Al reemplazar γ en las ecuaciones 1.5 y 1.6, los factores fσ y fε se expresan como:
fb � 1�243q�1 � fq�96�_ �_ + 1�
f� � ¸243q�1 � fq�96�_ �_ + 1¹
1.2. DEPENDENCIA DE LAS PROPIEDADES D (T= 255 K) y ρ (T= 255 K) CON
LA VARIACIÓN INDEPENDIENTE (NO SIMULTÁNEA) DE LOS PARÁMETROS σ, ε Y q DEL TIP5P-Ew.
La siguiente tabla muestra parte de los resultados obtenidos cuando se modificaron de manera independiente los parámetros σ, ε y q del modelo original
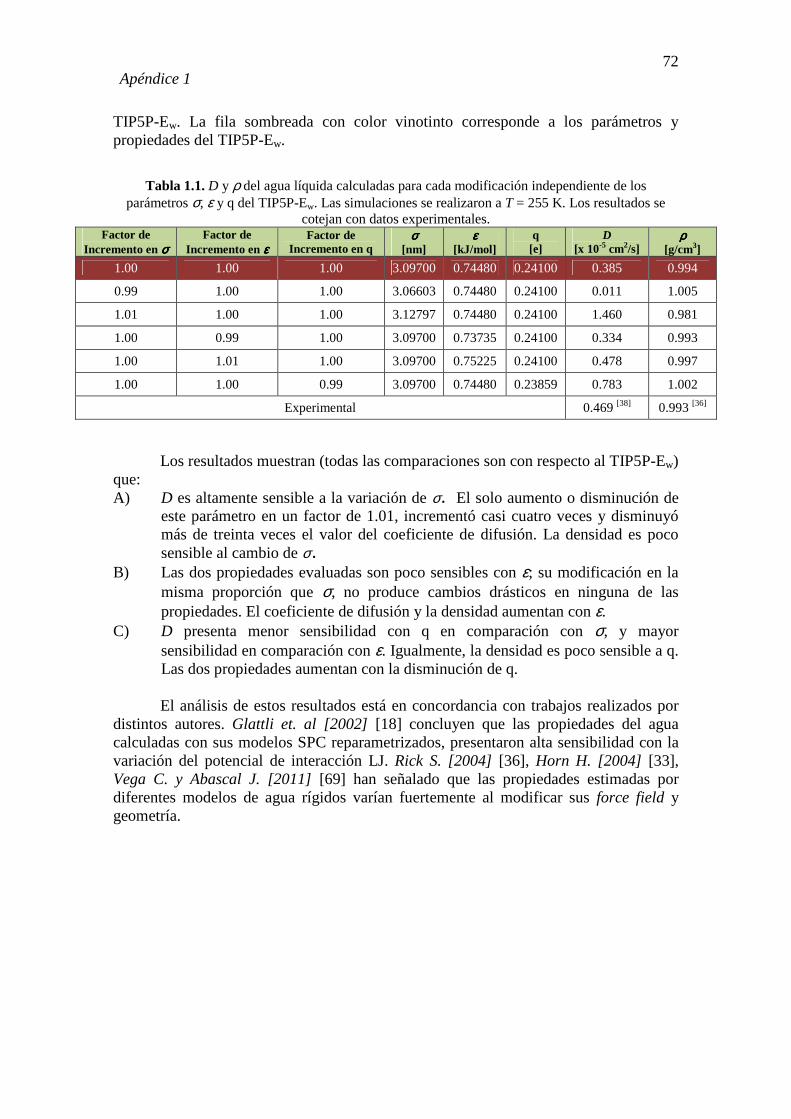
72 Apéndice 1
TIP5P-Ew. La fila sombreada con color vinotinto corresponde a los parámetros y propiedades del TIP5P-Ew.
Tabla 1.1. D y ρ del agua líquida calculadas para cada modificación independiente de los parámetros σ, ε y q del TIP5P-Ew. Las simulaciones se realizaron a T = 255 K. Los resultados se
cotejan con datos experimentales. Factor de
Incremento en σσσσ Factor de
Incremento en εεεε Factor de
Incremento en q σσσσ
[nm] εεεε
[kJ/mol] q
[e] D
[x 10-5 cm2/s] ρρρρ
[g/cm3]
1.00 1.00 1.00 3.09700 0.74480 0.24100 0.385 0.994
0.99 1.00 1.00 3.06603 0.74480 0.24100 0.011 1.005
1.01 1.00 1.00 3.12797 0.74480 0.24100 1.460 0.981
1.00 0.99 1.00 3.09700 0.73735 0.24100 0.334 0.993
1.00 1.01 1.00 3.09700 0.75225 0.24100 0.478 0.997
1.00 1.00 0.99 3.09700 0.74480 0.23859 0.783 1.002
Experimental 0.469 [38] 0.993 [36]
Los resultados muestran (todas las comparaciones son con respecto al TIP5P-Ew) que: A) D es altamente sensible a la variación de σ. El solo aumento o disminución de
este parámetro en un factor de 1.01, incrementó casi cuatro veces y disminuyó más de treinta veces el valor del coeficiente de difusión. La densidad es poco sensible al cambio de σ.
B) Las dos propiedades evaluadas son poco sensibles con ε; su modificación en la misma proporción que σ, no produce cambios drásticos en ninguna de las propiedades. El coeficiente de difusión y la densidad aumentan con ε.
C) D presenta menor sensibilidad con q en comparación con σ, y mayor sensibilidad en comparación con ε. Igualmente, la densidad es poco sensible a q. Las dos propiedades aumentan con la disminución de q. El análisis de estos resultados está en concordancia con trabajos realizados por
distintos autores. Glattli et. al [2002] [18] concluyen que las propiedades del agua calculadas con sus modelos SPC reparametrizados, presentaron alta sensibilidad con la variación del potencial de interacción LJ. Rick S. [2004] [36], Horn H. [2004] [33], Vega C. y Abascal J. [2011] [69] han señalado que las propiedades estimadas por diferentes modelos de agua rígidos varían fuertemente al modificar sus force field y geometría.

73 Apéndice 2
APÉNDICE 2
2.1. CÁLCULO DEL TIEMPO DE VIDA DE LOS ENLACES PUENTES DE HIDRÓGENO.
Un programa fue desarrollado para calcular el tiempo de vida promedio de los enlaces puentes de hidrógeno. El programa determina cuando dos moléculas de agua, dada sus posiciones, están unidas a través de un enlace puente hidrogeno (Hb). El criterio usado para esta unión, fue que la distancia entre los oxígenos (O) y el ángulo O⋅⋅⋅O-H estuviesen dentro de un cierto rango de tolerancia. Los cut-off elegidos fueron 30° para el ángulo y para la distancia, el primer mínimo O-O de la función distribución radial (g(rO-O)) calculada en los sistemas de agua líquida por los distintos modelos. Los valores de g(rO-O) en función de T se reúnen en la tabla 2.1; como ejemplo, en la figura 2.1 está representada la forma de g(rO-O) obtenida por el TIP5P-Ew a la T = 250 K.
Tabla 2.1. Valores del primer mínimo de la función radial oxígeno-oxígeno (g(rO-O)) en función de T, calculados en sistemas de agua líquida por los modelos: SPC/E, TIP4P-Ew, TIP5P-Ew y Six-Sites.
SPC/E TIP4P-Ew TIP5P-Ew Six-Sites
T [K] g(rO-O) [nm] g(rO-O) [nm] g(rO-O) [nm] g(rO-O) [nm]
230 0.3181 0.3227 0.3271 0.3334
240 0.3228 0.3239 0.3289 0.3340
250 0.3232 0.3245 0.3303 0.3344
260 0.3255 0.3259 0.3314 0.3355
270 0.3281 0.3278 0.3329 0.3375
280 0.3298 0.3295 0.3349 0.3399
290 0.3311 0.3308 0.3381 0.3421
300 0.3340 0.3310 0.3427 0.3426
El Gromacs por defecto establece un paso de integración ∆t = 1fs (utilizado en toda la tesis); sin embargo, permite grabar las posiciones de las moléculas a tiempos
Figura 2.1. Función de distribución radial oxígeno-oxígeno (g(rO-O)) calculada por el TIP5P-Ew en un sistema de agua líquida a la temperatura de 250 K.

74 Apéndice 2
mayores mediante la elección de distintas frecuencias de grabado (fg). El tiempo real entre cada captura dependerá de la frecuencia con la cual se graba.
El programa cuenta el número de capturas en la cual un Hb está presente (serán referidos como pasos). Para ello considera que: 1. Se ha formado un nuevo Hb cuando aparece en una captura determinada y no en
la anterior. 2. Un Hb contabiliza un paso más de vida cuando existe en una captura
determinada y existía en la anterior. 3. Se ha cortado un Hb en una captura determinada cuando existía en una previa y
no en ésta.
El tiempo de vida de los Hb se cuantifica mediante la frecuencia de grabado, una vez dado el número de pasos donde los mismos estuvieron presentes. La frecuencia de grabado elegida para todas las simulaciones (todas las T y todos los modelos) fue = 1ps-
1, la cual se estableció después de haber realizado pruebas con diferentes valores (1000ps-1, 100ps-1, 10ps-1, 1ps-1 y 0.1ps-1). Se encontró que todas las fg, excepto fg = 0.1ps-1, fueron mayores a la frecuencia típica de ruptura del enlace puente de hidrógeno; sin embargo, la elección de fg = 1ps-1 fue porque el tiempo de grabado y el consumo de recurso computacional es menor.
El tiempo de vida individual de cada enlace en cada paso no es posible establecerlo por razones técnicas. Empero, se establece como dato de salida del programa, un tiempo de vida promedio de todos los Hb existentes en cada paso (tH (t)). La fórmula detH (t) es: ��̅��� � ∑tMº»Bz ec.2.1
pi es la cantidad de pasos del i-esimo enlace que se ha mantenido presente hasta esa captura; ¼ es la cantidad de enlaces existentes en esa captura; ! es la cantidad de moléculas del sistemas.
La esquematización del cálculo de tH (t) para un número pequeño de enlaces en un sistema de 512 moléculas de agua se muestra en la tabla 2.2. Cada columna representa una captura particular y cada fila representa el i-esimo Hb. Se observa que en la primera columna (captura 1) existen tres enlaces (enlace 1, enlace 2, enlace 3).Como en esta captura no hay información sobre el número de pasos previos donde han estado presente los tres enlaces, esta se contabiliza asignándole el 0 a pi de cada enlace, es decir, enlace 1, enlace 2, enlace 3 tienen p1=0, p2=0 y p3=0. Esta falta de información se subsana realizando simulaciones suficientemente largas lo que garantiza la inexistencia de los primeros enlaces (todos los Hb iniciales se han roto). Para esta parte del estudio de tesis las simulaciones tuvieron una duración de 10ns.
La columna dos, correspondiente a la captura 2, no solo están presentes los tres enlaces de la captura 1 sino que aparece un nuevo Hb (enlace 4). Esta captura se contabiliza adjudicándole el número 1 a p1, p2 y p3 y el 0 a p4. En la captura 3 (columna tres), los enlaces 1, 2 y 4 todavía permanecen, el enlace 3 deja de existir (representado con la letra X) y un nuevo Hb aparece (enlace 5); en este caso, se asignan los números 2, 2, 1 y 0 a p1, p2, p4 y p5, respectivamente. Y Así sucesivamente.

75 Apéndice 2
Tabla 2.2. Esquematización de los tiempos de vida de los enlaces puentes de hidrógeno tH y ��̅��� para un sistema de 512 moléculas de agua líquida. captura 1 captura 2 captura 3 captura 4 captura 5
enlace 1 p1= 0 p1 = 1 p1 = 2 p1 = 3 p1 = 4
enlace 2 p2 = 0 p2 = 1 p2 = 2 p2 = X
enlace 3 p3 = 0 p3 = 1 p3 = X
enlace 4 p4 = 0 p4 = 1 p4 = 2 p4 = X
enlace 5 p5 = 0 p5 = 1 p5 = X
enlace 6
p6 = 0 p6 = 1
∑½¾ 0 3 5 6 5
¿ 3 4 4 4 2
tH (t) [ps] 0
1psZO ∙ 512 ∙ 3 �
0.0000 ps
31psZO ∙ 512 ∙ 4 �
0.0015 ps
51psZO ∙ 512 ∙ 4 �
0.0024 ps
61psZO ∙ 512 ∙ 4 �
0.0029 ps
51psZO ∙ 512 ∙ 2 �
0.0049 ps
2.2. CÁLCULO DEL ORDEN TETRAÉDRICO (q).
El parámetro de orden local tetraédrico mide el nivel de arreglo tetraédrico de una molécula k con sus cuatro vecinas más cercanas. Viene expresado por la siguiente ecuación [70]:
Ã��� � 1 � �³∑ ∑ &cosψI,; �
O�.i
;vIXO�IvO ec.2.2
ψikj es el ángulo entre puentes de hidrógeno que forma la molécula central k con sus moléculas vecinas i y j (ver figura 2.2.). En otras palabras, q es un factor que se calcula a partir de los seis diferentes ángulos posibles entre la molécula central k y sus cuatro primeras vecinas. Toma valores en el rango -3 ≤ q ≤ 1. Si una molécula es localizada en el centro de un tetraedro regular cuyos vértices son ocupados por sus cuatro vecinos más cercanos entonces cos(ψikj) = -1/3. Un orden tetraédrico perfecto implica un q(k) = 1. Si el arreglo entre moléculas es aleatorio los ángulos asociados con la molécula

76 Apéndice 2
central k son independientes y q(k) = 0.
El cálculo de q se realizó con un programa que contabiliza el ángulo ψikj que presenta cada molécula central k con sus cuatro vecinas más cercanas. El programa calcula la densidad de probabilidad de q, P(q), mediante un histograma normalizado de la cantidad de moléculas (en todo el sistema y en los 5ns de simulación) que tienen un q específico (entre 0 y 1). El intervalo de clase fue igual a 100.
Figura 2.2. Identificación del ángulo ψikj que forma la molécula central k y sus vecinas i, j más cercanas.

77 Referencias bibliográficas
REFERENCIAS BIBLIOGRÁFICAS
[1] Pruppacher H. R. and Klett J. D. Microphysics of clouds and precipitation. 2nd ed., Kluwer Academic Publishers, 1997.
[2] Koop T. Homogeneous ice nucleation in water and aqueous solutions. Z. Phys.
Chem. 218: 1231–1258, 2004. [3] Mishima O. and Stanley H. E. The relationship between liquid, supercooled and
glassy water. Nature 396: 329-335, 1998. [4] Petrenko V. F. Structure of ordinary ice Ih. Part I: Ideal structure of ice. Special
Report 93-25. US Army Corps of Engineers, 1993. [5] Aragones J.L. Simulación del equilibrio de fases del agua: cristales plásticos,
constantes dieléctricas y disoluciones. Tesis Doctoral. Universidad Complutense de Madrid, 2013.
[6] Debenedetti P. G. Supercooled and glassy water. J. Phys.: Condens. Matter. 15:
R1669-R1726, 2003. [7] Water models. On-line en http://www1.lsbu.ac.uk/water/models.html. [8] Dr. Muñoz Francisco. Temas QFC-T2, QFC-T3, QFC-T4, QFC-T5, QFC-
T6,QFC-T7. Departamento de físico-química. Universidad de las Islas Baleares, 2007. On-line en: http://www.uib.es/depart/dqu/dqf/paco/docencia/.
[9] Frenkel D. and Smit B. Understanding molecular simulation. Academic Press,
London, 2002. [10] Leach A. R. Molecular Modelling. 2nd ed. Pearson Education, 2001. [11] Dr. Cannas Sergio. Curso de Termodinámica y Mecánica Estadística II. Facultad
de Matemática, Astronomía y Física de la Universidad Nacional de Córdoba, 2010. On-line en: http://www.famaf.unc.edu.ar/~cannas/notas.html
[12] Valderrama A. Estudio mecanicocuántico, Docking y dinámica molecular de
tioazúcares como inhibidores de la proteína fucosidasa. Tesis Doctoral. Universidad de Granada. 2009.
[13] Allen M. P. and Tildesley D.J. Computer simulation of liquids. The Ipswich Book
Co Ltd, London, 1991. [14] Heermann D. W. Computer simulation methods. 2nd ed. Springer-Verlag, 1990.

78 Referencias bibliográficas
[15] López J. G y Moyano G. El método de trayectorias clásicas. Curso de Postgrado sobre teoría de la dinámica de reacciones química. Universidad de Antioquia, Colombia, 2009.
[16] Petrenko, V. y Whitworth, R. Physics of Ice. Oxford University press, 1998. [17] Guillot B. A reappraisal of what we have learnt during three decades of computer
simulations on water. J. Mol. Liq., 101: 219–260, 2002. [18] Glättli A., Daura X. and van Gunsteren W. Derivation of an improved simple
point charge model for liquid water: SPC/A and SPC/L. J. Chem. Phys., 116: 9811-9828, 2002.
[19] Nada H. and van der Eerden J. P. M. An intermolecular potential model for the
simulation of ice and water near the melting point: A Six-Sites model of H2O. J. Chem. Phys., 118: 7401-7413, 2003.
[20] Rapaport D. C. The art of molecular dynamics simulation. 2nd ed. Cambridge
University Press, 2004. [21] Toukmaji A. Y. and. Board J. A. Ewald summation techniques in perspective: A
survey. Comp. Phys. Comm., 95: 73-92, 1996. [22] Darden T., York D. and Pedersen L. Particle mesh Ewald: An N-log(N) method
for Ewald sums in large systems. J. Chem. Phys., 98: 10089-10092, 1993. [23] Essmann U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. and Pedersen, L.
G. A smooth particle mesh Ewald potential. J. Chem. Phys., 103: 8577-8592, 1995.
[24] Toukmaji A. and Board J. A. Technical Report No. 95-004, Duke University
(unpublished). [25] Andersen H. Molecular dynamics simulations at constant pressure and/or
temperature. J. Phys. Chem., 72: 2384-2393, 1980. [26] Berendsen H., Postma, J., DiNola A. and Haak J. Molecular dynamics with
coupling to an external bath. J. Chem. Phys., 81: 3684 -3690, 1984. [27] Nosé S. A molecular dynamics method for simulations in the canonical ensemble.
Mol. Phys., 52: 255 - 268, 1984. [28] Hoover W. G. Canonical dynamics: equilibrium phase-space distributions. Phys.
Rev. A., 31: 1695 - 1697, 1985. [29] Parinello M. and Raphman, A. Polymorphic transitions in single crystals: A new
molecular dynamics method. J. Appl. Phys., 52: 7182 - 7190, 1981.

79 Referencias bibliográficas
[30] Berendsen H., Grigera J. and Straatsma T. The missing term in effective pair potentials. J. Phys. Chem., 91: 6269-6271, 1987.
[31] Jorgensen W., Chandrasekhar J., Madura J., Impey R. and Klein M. Comparison
of simple potential functions for simulating liquid water, J. Chem. Phys., 79: 926-935, 1983.
[32] Jorgensen W. and Madura J. Temperature and size dependence for Monte Carlo
simulations of TIP4P water. Mol. Phys., 56: 1381-1392, 1985. [33] Horn H., Swope W., Pitera J., Madura J., Dick T., Hura G. and Head-Gordon T.
Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew, J. Chem. Phys., 120: 9665 – 9678, 2004.
[34] Abascal L., Sanz E., Fernández R. and Vega C. A potential model for the study of
ices and amorphous water: TIP4P/Ice, J. Chem. Phys., 122: 234511, 2005. [35] Mahoney M. and Jorgensen W. A five-site model for liquid water and the
reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys., 112: 8910-8922, 2000.
[36] Rick S. A reoptimization of the five-site water potential (TIP5P) for use with
Ewald sums. J. Chem. Phys., 120: 6085 - 6093, 2004. [37] Dick T. and Madura J. A review of the TIP4P, TIP4P-Ew, TIP5P, and TIP5P-E
water models. Annu. Rep. Comp. Chem., 1: 59-74, 2005. [38] Price W., Ide H. and Arata Y. Self-diffusion of supercooled water to 238 K using
PGSE NMR diffusion measurements. J. Phys. Chem. A., 103: 448-450, 1999. [39] Pruppacher, H.R. A new look at homogeneous ice nucleation in supercooled water
drops. J. Atmos. Sci., 52: 1924-1933, 1994. [40] Vega C., Sanz E. and Abascal J. The melting temperature of the most common
models of water. J. Chem. Phys., 122: 114507, 2005. [41] Fernández R., Abascal J. and Vega C. The melting point of ice Ih for common
water models calculated from direct coexistence of the solid-liquid interface. J. Chem. Phys., 124: 144506, 2006.
[42] Abascal J., Fernandez R., Vega C. and Carignano M. The melting temperature of
the six site potential model of water. J. Chem. Phys., 125: 166101, 2006. [43] Angell C., Oguni M. and Sichina W. Heat capacity of water at extremes of
supercooling and superheating. J. Phys. Chem., 86: 998-1002, 1982. [44] Tombari E., Ferrari C. and Salvetti G. Heat capacity anomaly in a large sample of
supercooled water. Chem. Phys. Lett., 300: 749-751, 1999.

80 Referencias bibliográficas
[45] Archer D. and Carter R. Thermodynamic properties of the NaCl + H2O System. Heat Capacities of H2O and NaCl(aq) in cold-stable and supercooled states. J. Phys. Chem. B, 104: 8563-8584, 2000.
[46] Kumar P., Buldyrev S., Becker S., Poole P., Starr F. and Stanley H. Relation
between the Widom line and the breakdown of the Stokes–Einstein relation in supercooled water. PNAS, 104: 9575-9579, 2007.
[47] Longinotti M., Carignano M., Szleifer I. and Corti, H. Anomalies in supercooled
NaCl aqueous solutions: A microscopic perspective. J. Chem. Phys., 134: 244510, 2011.
[48] Faraone A., Liu L., Mou C.-Y., Yen C.-W. and. Chen. S.-H. Fragile-to-strong
liquid transition in deeply supercooled confined water. J. Chem. Phys., 121: 10843-10846, 2004.
[49] Buch V., Sandler P. and Sadlej J. Simulations of H2O solid, liquid, and clusters,
with an emphasis on ferroelectric ordering transition in hexagonal ice. J. Phys. Chem. B, 102: 8641-8653, 1998.
[50] Van der Spoel D., Lindahl E., Hess B., Groenhof G., Mark, A. and Berendsen, H. Gromacs. J. Comp. Chem., 26: 1701-1718, 2005.
[51] Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs. J. Chem. Theory
Comp., 4: 435-447, 2008. [52] Huang J. and Bartell L. Kinetics of homogeneous nucleation in the freezing of
large water clusters. J. Phys. Chem., 99: 3924-3931, 1995. [53] Krämer B., Hübner O., Vortisch H., Wöste L., Leisner T., Schwell M., Rühl E.,
Baumgärtel H. Homogeneous nucleation rates of supercooled water measured in single levitated microdroplets. J. Chem. Phys., 111: 6521-6527, 1999.
[54] Stöckel P., Weidinger I., Baumgärtel H. and Leisner T. Rates of homogeneous ice
nucleation in levitated H2O and D2O droplets. J. Phys. Chem. A, 109: 2540-2546, 2005.
[55] Kabath P., Stöckel P., Lindinger A., Baumgärtel H. The nucleation of ice in
supercooled D2O and H2O. J. Mol. Liq., 125: 204-211, 2006. [56] Baumgärtel H. and Zimmermann H. The homogeneous nucleation in supercooled
water. An examination using statistics and irreversible thermodynamics. J. Mol. Liq., 164: 178–186, 2011.
[57] Jeffery C. and Austin P. H. Homogeneous nucleation of supercooled water:
Results from a new equation of state. J. Geophys. Res., 102: 25269-25279, 1997. [58] Ediger M., Angell C. and Nagel S. Supercooled liquids and glasses. J. Phys.
Chem., 100: 13200-13212, 1996.

81 Referencias bibliográficas
[59] Angell, C. A. Glass formation and glass transition in supercooled liquids, with
insights from study of related phenomena in crystals. J. Non-Cryst. Solids, 354: 4703-4712, 2008.
[60] Angell C. Heat capacity and entropy functions in strong and fragile glass-
formers, relative to those of disordering crystalline materials. Extraído del libro Glassy, Amorphous and Nano-Crystalline Materials, Pag. 21-40. Springer 2011.
[61] Liu J., Nhicholson C. and Cooper S. Direct measurement of critical nucleus size
in confined volumes. Langmuir, 23: 7286-7292, 2007. [62] Matsumoto M., Saito S. and Ohmine I. Molecular dynamics simulation of the ice
nucleation and growth process leading to water freezing. Nature, 416: 409 – 413, 2002.
[63] Vrbka L. and Jungwirth P. Homogeneous freezing of water starts in the
subsurface. J. Phys. Chem. B., 110: 18126-18129, 2006. [64] Svishchev I. and Kusalik P. Electrofreezing of liquid water: A Microscopic
Perspective J. Am. Chem. Soc.,118: 649-654, 1996. [65] Yokoyama T. and Hagiwara Y. Molecular dynamics simulation for the mixture of
water and an ice nucleus. Mol. Sim., 29: 235-248, 2003. [66] Okawa S., Saito A. and Matsui T. Nucleation of supercooled water on solid
surfaces. Journal of Refrigeration, 29: 134-141, 2006. [67] Gillen K., Douglass D. and Hoch J. Self-diffusion in liquid water to −31°C. J.
Chem. Phys., 57: 5117 - 5119, 1972. [68] Pereyra R, Szleifer I. and Carignano M. Temperature dependence of ice critical
nucleus size J. Chem. Phys., 135: 034508, 2011. [69] Vega C. and Abascal J. Simulating water with rigid non-polarizable models: a
general perspective. Phys. Chem. Chem. Phys., 13: 19663-19688, 2011. [70] Errington J. and Debenedetti P. Relationship between structural order and the
anomalies of liquid water. Nature, 409: 318-321, 2001. [71] Kumar P., Buldyrev S. and Stanley H. A tetrahedral entropy for water. Proc. Natl.
Acad. Sci. USA, 106: 22130-22134, 2009. [72] Shiratani E. and Sasai M. Grow and collapse of structural patterns in the hydrogen
bond network in liquid water. J. Chem. Phys., 104: 7671-7680, 1996. [73] Appignanesi G., Rodriguez J. and Sciortino F. Evidence of a two-state picture for
supercooled water and its connections with glassy dynamics. Eur. Phys. J. E, 29: 305-310, 2009.

82 Referencias bibliográficas
[74] Malaspina D., Rodriguez J., Appignanesi G. and Sciortino F. Identifying a causal
link between structure and dynamics in supercooled water. Eur. Lett., 88: 16003-16003, 2009.
[75] Earle M., Kuhn T., Khalizov A. and Sloan J. Volume nucleation rates for
homogeneous freezing in supercooled water microdroplets: results from a combined experimental and modelling approach. Atmos. Chem. Phys., 10: 7945-7961, 2010.
[76] Hagen D., Rodney A. and James K. Homogeneous condensation-Freezing
nucleation rate messurements for small water droplets in an expansion cloud chamber. J. Atmos. Sci., 38: 1236-1243, (1981).
[77] Gunton J. Homogeneous Nucleation. J. Statistic. Phys., 95: 903-923, 1999. [78] Starr F., Nielsen J. and Stanley H. Fast and slow dynamics of hydrogen bonds in
liquid water. Phys. Rev. E, 62: 2294-2297, 1999. [79] Rapaport D. Hydrogen bonds in water. Mol. Phys., 50: 1151-1162, 1983. [80] Vali G. Ice nucleation-A review. Plenary lecture presented at the 14th
International Conference on Nucleation and Atmospheric Aerosols. Extraído del libro Appeared in Nucleation and Atmospheric Aerosols 1996, pp.271-279, Pergamon Press 1996.
[81] Wood S, Baker M. and Swanson B. Instrument for studies of homogeneous and
heterogeneous ice nucleation in free-falling supercooled water droplets. Rev. Sci. Instrum., 73: 3988-3996, 2002.
[82] Hoose C. and Möhler O. Heterogeneous ice nucleation on atmospheric aerosols: a
review of results from laboratory experiments. Atmos. Chem. Phys., 12: 9817- 9854, 2012.
[83] Möhler O., Field P., Connolly P., Benz1 S., Saathoff H., Schnaiter M., Wagner
R., Cotton R., Krämer M., Mangold A., and Heymsfield A. Efficiency of the deposition mode ice nucleation on mineral dust particles. Atmos. Chem. Phys., 6: 3007-3021, 2006.
[84] Wilson P., Heneghan A. and Haymet A. Ice nucleation in nature: supercooling
point (SCP) measurements and the role of heterogeneous nucleation. Cryobiology, 46: 88-98, 2003.
[85] Mason B. The Physics of Clouds. Oxford University Press, 1971. [86] Vali G. Freezing rate due to heterogeneous nucleation. J. Atmos. Sci., 51: 1843-
1856, 1994.

83 Referencias bibliográficas
[87] Niedermeier D., Shaw R., Hartmann S., Wex H., Clauss T., Voigtländer J. and Stratmann F. Heterogeneous ice nucleation: exploring the transition from stochastic to singular freezing behavior. Atmos. Chem. Phys., 11: 8767-8775, 2011.
[88] DeMott, P.J. Quantitative descriptions of ice formation mechanisms of silver
iodide-type aerosols. Atmos. Res., 38: 63-99, 1995. [89] Levine J. Statistical explanation of spontaneous freezing of water droplets, NACA
Tech. Notes N° 2234, 1950. [90] Fletcher N. Active sites and ice crystal nucleation J. Atmos. Sci., 26: 1266-1271,
1969. [91] Vali G. Repeatability and randomness in heterogeneous freezing nucleation.
Atmos. Chem. Phys., 8: 5017-5031, 2008. [92] Chen J., Hazra A. and Levin Z. Parameterizing ice nucleation rates using contact
angle and activation energy derived from laboratory data. Atmos. Chem. Phys., 8: 7431-7449, 2008.
[93] Vali, G. Nucleation terminology, J. Aerosol Sci., 16: 575-576, 1985. [94] Bigg E. The supercooling of water, Proc. Phys. Soc. B: 66, 688-694, 1953a. [95] Bigg E. The formation of atmospheric ice crystals by the freezing of droplets.
Quart. J. Royal Meteorol. Soc., 79: 510-519, 1953b. [96] Bigg E. Ice-crystal counts and the freezing of water drops. Quart. J. Royal
Meteorol. Soc., 81: 478-479, 1955. [97] Carte A. The freezing of water droplets. Proc. Phys. Soc. B, 69: 1028-1037, 1956. [98] Carte A. Probability of freezing. Proc. Phys. Soc. B, 73: 324, 1959. [99] Connolly P., Möhler O., Field P., Saathoff H., Burgess R., Choularton T. and
Gallagher M. Studies of heterogeneous freezing by three different desert dust samples. Atmos. Chem.Phys., 9: 2805-2824, 2009.
[100] Moore E. and Molinero V. Ice crystallization in water’s “no-man’s land”. J.
Chem. Phys., 132: 244504, 2010. [101] Bartell. L. A. Analyses of nucleation rates from molecular dynamics simulations.
J. Phys. Chem. 106: 10893-10897, 2002. [102] Jung S., Tiwari M., Doan N. and Poulikakos D. Mechanism of supercooled
droplet freezing on surfaces. Nat. Commun, 3: 615 doi: 10.1038/ncomms1630, 2012.

84 Referencias bibliográficas
[103] Abascal J. and Vega C., A general purpose model for the condensed phases of water: TIP4P-2005. J. Chem. Phys., 123: 234505, 2005.
[104] Rahman A. y Stillinger F. Molecular dynamics study of liquid water. J. Chem.
Phys., 55: 3336-3359, 1971. [105] van Buuren A., Marrink S. and Berendsen H. A molecular dynamics study of the
decane/water interface. J. Phys. Chem., 97: 9206-9212, 1993. [106] Stoyanov S. and Groot R. From molecular dynamics to hydrodynamics-a novel
Galilean invariant thermostat. J. Chem. Phys., 08: 114112, 2005. [107] Holten V, Bertrand C., Anisimov M. and Sengers J. Thermodynamics of
supercooled water. J. Chem. Phys., 136: 094507, 2012. [108] Abascal J. and Vega C. Widom line and the liquid-liquid critical point for the
tip4p/2005 water model. J. Chem. Phys., 133: 234502, 2010. [109] Anisimov M., Voronel A., Zaugol´nikova N. and Ovodov G., JETP Lett 15: 317,
1972. [110] Bertolini D., Cassettari M. and Salvetti G. Anomalies in the “latent heat” of
solidification of supercooled water Chem. Phys. Lett., 199: 553-555, 1985. [111] Pi H., Aragones J., Vega C., Noya E., Abascal J., Gonzalez M. Anomalies in
water as obtained from computer simulations of the TIP4P/2005 model: density maxima, and density, isothermal compressibility and heat capacity minima. Mol. Physics, 107: 365–374, 2009.
[112] Sciortino F., Gallo P., Tartaglia P. and Chen S. Supercooled water and the kinetic
glass transition. Phys. Rev. E, 54: 6331 – 6343, 1996. [113] Qvist J., Schober H. and Halle B.. Structural dynamics of supercooled water from
quasielastic neutron scattering and molecular simulations. J. Chem. Phys., 134: 144508, 2011.
[114] Molinero V. and Moore E. Water modeled as an intermediate element between
carbon and silicon. J. Phys. Chem. B, 113: 4008-4016, 2009. [115] Murray B., Knopf D. and Bertram A. The formation of cubic ice under conditions
relevant to Earth´s atmosphere. Nature, 434: 202-205, 2005. [116] Poole P., Sciortino F., Essmann U. and Stanley H. Phase-behavior of metastable
water. Nature, 360: 324-328, 1992 [117] Koza M., Schober H., Fischer H., Hansen T. and Fujara F. Kinetics of the high- to
low-density amorphous water transition. J. Phys.: Condens. Matter, 15: 321-332, 2003.

85 Referencias bibliográficas
[118] Nelmes R., Loveday J., Strässle T., Bull C., Guthrie M., Hamel G. and Klotz S.
Annealed high-density amorphous ice under pressure Nat. Phys., 2: 414 - 418 , 2006.
[119] Harrington S, Zhang R., Poole P., Sciortino F. y Stanley H. Liquid-liquid phase
transition: Evidence from simulation. Phys. Rev. Lett., 78: 2409-2412, 1997. [120] Mahoney M. and Jorgensen W. A five-site model for liquid water and the
reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys., 112: 8910-8922, 2000.
[121] Scala A., Starr F.W. La Nave E, Stanley H. E. y Sciortino F. Free energy surface
of supercooled water. Phys. Rev. E, 62: 8016-8020, 2000. [122] Moore E., de la Llave E., Welke K., Scherlis D. and Molinero V. Freezing,
melting and structure of ice in a hydrophilic nanopore. Phys. Chem. Chem. Phys., 12: 4124 - 4134, 2010.
[123] Moore E. and Molinero V. Growing correlation length in supercooled water. J.
Chem. Phys., 130: 244505-244512, 2009. [124] Mallamace F., Broccio M., Corsaro C., Faraone A., Wanderlingh U., Liu L., Mou
C. and Chen S., The fragile-to-strong dynamic crossover transition in confined water: nuclear magnetic resonance results. J. Chem. Phys., 124: 161102, 2006.
[125] Stillinger H. Frank. Water revisited. SCIENCE, 209: 451-457, 1980 [126] Sciortino F., Poole H., Stanley E. and Havlin S. Lifetime of the bond network and
gel-like anomalies in supercooled water. Phys. Rev. Lett., 64: 1686-1689, 1990. [127] Gale M., Gallot G. and Lascoux N. Frequency-dependent vibrational population
relaxation time of the OH stretching mode in liquid water. Chem. Phys. Lett., 311: 123-125, 1999.
[128] Fecko J., Eaves D., Lparo J., Tokmakoff A. and Geissler L.P. Ultrafast hydrogen-
bond dynamics in the infrared spectroscopy of water. SCIENCE, 301: 1698-1702, 2003.
[129] Moore E. and Molinero V. Structural transformation in supercooled water controls
the crystallization rate of ice. Nature, 479: 506-508, 2011. [130] Maruyama S., Wakabayashi K. and Oguni M. Thermal properties of supercooled
water confined within silica gel pores. 3rd International Symposium on Slow Dynamics in Complex Systems. Sendai, JAPAN, NOV 03-08, 2004.
[131] Shevchuk R., Prada-Gracia D. and Rao F. Water structure-forming capabilities are
temperature shifted for different models. J. Phys. Chem. B, 116: 7538-7543, 2012.

86 Referencias bibliográficas
[132] Mills R. Self-diffusion in normal and heavy water in the range 1 - 45. deg. J.
Phys. Chem., 77: 685-688, 1973. [133] Liu Y., Palmer J., Panagiotopoulos A. and Debenedetti P. Liquid-liquid transition
in ST2 water. J. Chem. Phys., 137: 214505, 2012. [134] Limmer D. and Chandler D. The putative liquid-liquid transition is a liquid-solid
transition in atomistic models of water. J. Chem. Phys., 135: 134503, 2011. [135] Hellmuth, O., Khvorostyanov, V., Curry, J., Shchekin, A., Schmelzer, J., Feistel,
R., Djikaev, Y. and Baidakov, V. Nucleation Theory and Applications. Schmelzer J. and Hellmuth O. editors. Dubna, 2013.
[136] Niedermeier, D., Hartmann, S., Shaw, R. A., Covert, D., Mentel, T. F., Schneider,
J., Poulain, L., Reitz, P., Spindler, C., Clauss, T., Kiselev, A., Hallbauer, E., Wex, H., Mildenberger, K., and Stratmann, F. Heterogeneous freezing of droplets with immersed mineral dust particles measurements and parameterization, Atmos. Chem. Phys., 10: 3601–3614, 2010.
[137] Shaw, R. Durant, A. and Mi, Y. Heterogeneous surface crystallization observed in
undercooled water. J. Phys. Chem. B., 109: 9865-9868, 2005. [138] Zobrist, B. Koop, T. Luo, B. Marcolli, C. and Peter, T. Heterogeneous ice
nucleation rate coefficient of water droplets coated by a nonadecanol Monolayer. J. Phys. Chem. C., 111: 2149-2155, 2007.
[138] Wright, T. and Petters, M, The role of time in heterogeneous freezing nucleation.
J. Geophys. Res: Atmos., 118: 1–13, doi:10.1002/jgrd.50365, 2013.

87 Lista de símbolos
LISTA DE SÍMBOLOS
∠: ángulo de enlace. ε: parámetro que representa la profundidad del potencial Lennard-Jones. σ: parámetro del potencial Lennard-Jones. ε: constante dieléctrica. ρ: densidad de masa. µ: momento dipolar del agua en fase gaseosa. τ: temperatura re-escaleda en el régimen sobre-enfriado. ε0: permitividad del vacío. ∆g*: energía de activación de la difusión de moléculas de agua líquida sobre-
enfriada metaestable a través de la interface líquido/sólido. ∆Ghet: cambio de energía libre de Gibbs para la formación de embriones de hielo en
nucleación heterogénea. ∆Ghom: cambio de energía libre de Gibbs para la formación de embriones de hielo en
nucleación homogénea. ρIc: densidad de masa del hielo cúbico. ρIh: densidad de masa del hielo hexagonal. µl(T): potencial químico de la fase líquida. µs(T): potencial químico de la fase sólida. σsl: tensión interfacial sólida-líquida. ∆t: paso de integración de las ecuaciones de movimiento. HGI: vector aceleración de la i-esima partícula. RGI: vector velocidad de la molécula i. �hP
: temperatura calculada en algunos modelos de agua donde ocurre el máximo de la capacidad calorífica isobárica CP. �∗: probabilidad para formar un embrión crítico de hielo.
∆Hc: entalpía de cristalización para una molécula de agua. C´: segundo punto crítico (hipótesis LLCP). CP: capacidad calorífica isobárica. d: distancia de enlace. D: coeficiente de difusión del agua. Ea: energía de activación. Ep: energía potencial configuracional. fσ: factor multiplicativo del parámetro σ del potencial LJ. fε: factor multiplicativo del parámetro ε del potencial LJ. fq: factor multiplicativo del parámetro q del potencial de Coulomb. f: factor de potencia catalítica. fg: frecuencia de grabado.
FEFFFG: vector fuerza actuando sobre la i-esima partícula debido a las interacciones con las otras N-1 partículas.
g(r): función de distribución radial. h: constante de Planck.

88 Lista de símbolos
H: entalpía. Hb: enlaces puentes de hidrógeno. hI: entalpía por mol de la fase hielo. hw: entalpía por mol de la fase agua líquida sobre-enfriada. Ic: hielo cúbico. Ih: hielo hexagonal IN: sustrato o núcleo de hielo en nucleación heterogénea. INM: sustrato o núcleo de hielo constituido de M moléculas. j: tasa de nucleación extensiva. J: tasa de nucleación intensiva. Jhom: tasa de nucleación homogénea intensiva. jM: tasa de nucleación heterogénea correspondiente a un sistema de agua sobre-
enfriada con un IN constituido de M moléculas. K: energía cinética. kB: constante de Boltzman. KT: compresibilidad isotérmica. LP: sitios ficticios que asemejan los pares de electrones libres del oxígeno en los
modelos de agua. M: centro de carga M de los modelos de agua de cuatro puntos. M: número de moléculas de conformaron el IN. mi: masa de la i-esima partícula. N: número de partículas que constituyen un sistema. En el estudio de nucleación
homogénea (Capítulo IV) se hizo referencia a N como el tamaño crítico del embrión de hielo.
N0: número de gotas que permanecen líquidas a un tiempo t = 0. NA: número de Avogadro. Nt: número de gotas que permanecen líquidas a un tiempo t. ¼: cantidad de enlaces Hb presentes en una captura. P(q): densidad de probabilidad del parámetro de orden tetraédrico q. P: presión. Pc: presión crítica. Pc :́ presión crítica del segundo punto crítico (hipótesis LLCP). pd: probabilidad de difusión de moléculas de agua líquida sobre-enfriada
metaestable en la interface líquido/sólido. q: carga de Coulomb. q: parámetro de orden tetraédrico.
irr
: vector posición de la i-esima partícula.
rc: radio cutoff. re
*: radio crítico del embrión de hielo. r ij: distancia entre las partículas i y j. rmin: distancia a la cual el potencial de LJ es mínimo. T: temperatura Tc: temperatura crítica. Tc :́ temperatura crítica del segundo punto crítico (hipótesis LLCP). Te: temperatura de ebullición. Tf: temperatura de fusión. Tg: temperatura de transición vítrea.

89 Lista de símbolos
TN: temperatura de nucleación. Tx: temperatura donde el agua ultra viscosa cristaliza a Ic. Tw(P): temperatura de la widom line. t: tiempo. ��̅���: tiempo de vida promedio de los enlaces puentes de hidrógeno. <tH>: ��̅��� promediado en los últimos 5ns de simulación. tN: tiempo de nucleación. v: volumen por molécula. V: energía potencial. V: volumen. VC: potencial de Coulomb. vi: velocidad de la i-esima partícula. VLJ: potencial de Lennard-Jones.


















