4-Estudios cos de La Bomba de Na
10
Review Mechanistic studies of sodium pump Larry D. Faller University of California at Los Angeles and Veterans Administration Greater Los Angeles Health Care System, Los Angeles, CA 90073, USA a r t i c l e i n f o Article history: Received 16 April 2008 and in revised form 22 May 2008 Available online 17 June 2008 Keywords: Na,K-ATPase Sodium pump Active ion transport Phosphoryl group transfer Conformational change Reaction mechanism a b s t r a c t Sodium pump was the first ion pump discovered. A member of the family of active transporters that cat- alyze adenosine 5 0 -triphosphate hydrolysis by forming a phosphorylated enzyme intermediate, sodium pump couples the energy released to unequal countertransport of sodium and potassium ions. The ion gradient generated by the pump is important for a variety of secondary physiological processes ranging from metabolite transport to electrical excitation of nerve and muscle. Selected experiments relating structure to function are reviewed. Ó 2008 Elsevier Inc. All rights reserved. The Royal Swedish Academy of Sciences spotlighted the vital role adenosine 5 0 -triphosphate (ATP) plays as the ‘‘energy cur- rency” of life by awarding the 1997 Nobel Prize in Chemistry for structural and mec hanistic studi es of ATP synthesis and for the first discovery of an ion pump [1]. Driven by a proton gradient across mitoc hondr ial membra nes, ATP synth ase (F 0 F 1 -ATPase) makes ‘‘about one half” of a resting person’s body-weight in ATP daily. Na,K-ATPase spends ‘‘a third” of the ATP minted to pump 3 Na + ions out of cells in exchange for 2 K + ions. Sodium pump is ubiqui- tous because of the need to regulate the volume of animal cells and organe lles bounded by flexible lipid bilayers . Loweri ng the intra- cellular Na + concentration helps to prevent cell lysis. The electro- chemical gradient generated by the unequal exchange of Na + for K + is used to drive a variety of fundamental physiological pro- cesses. The electrical gradient contributes to the resting membrane potent ial, thereb y affecting chann el-regulated electri cal stimu la- tion of nerve and muscle cells. The Na + gradient is the energy source for facilitated transport of ions (e.g. Na + /Ca 2+ exchange) and metabolites (e.g. Na + /glucose co-transport) by carriers. Pre- sumably because the pump is essential for cell viability, Na,K-ATP- ase has only been indirectly implicated in the etiology of diseases lik e ess ential hypertension and dia bete s. Sodium pump is the receptor for cardiac glycosides, like digitalis, used to treat patients with congestive heart failure. Only the structure of the water-soluble domain of ATP synthase, which catalyzes ATP hydrolysis (F 1 -ATPase), was solved. However, the ‘‘orange”-shaped oligomeric structure with three diprotomeric ‘‘segments” in different conformations and a central spindle [2] confirmed the elegant rotational mechanism that had been pro- posed for catalysis [3]. The first atomic -resol ution struct ure of an ion pump published in 2000 [4] showed the entire integral-mem- brane protein. However, the mechanism of catalysis-coupled vec- tori al tran sport was not obv iou s, becaus e the thr ee cyt osolic domains of monomeric Ca-ATPase were widely separated in the initial structure, and the Ca 2+ ions were embedded in the trans- membrane helices with no obvious pathway to either surface. Since publication of this initial ‘‘open” structure, a remarkable ser- ies of additional ‘‘snapshots” of intermediates in the calcium pump catalysis-transport cycle have been published by three laborato- ries, culminat ing in struc tures with the extrace llula r ‘‘gate” open [5,6] at the end of 2007. Simultaneously, the first pictures of so- dium pump [7] and a proton pump [8] were published, prompting an accompanying commentary entitled ‘‘Ion Pumps made Crystal Clear” [9]. The papers reporting the most recent still photographs of calcium pump [5,6] and the article on Ca-ATPase in this volume review the structural changes that mechanically couple catalysis to transport in detail. Relatively little has been written about the cat- alytic mechanism, or the multiple roles of water in catalysis and in structural changes. Therefore, the mechanism of phosphoryl group transfer is discussed with emphasis on a method that has only been used to follow the reaction catalyzed by mutants of sodium pump. Differences between ion binding sites in the sodium- and calcium-pump structure s, the rates of elementary steps in the reac- tion cycle, and studies of a specific inhibitor (ouabain) and of so- dium pump’s subunits are reviewed. Mechanism Na,K-ATPase and sodium pump are alternative names for the same integral-membrane protein that indicate its dual function 0003-9861/$ - see front matter Ó 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.abb.2008.05.017 E-mail address: [email protected] Archives of Biochemistry and Biophysics 476 (2008) 12–21 Contents lists available at ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi
-
Upload
bryanarriola -
Category
Documents
-
view
216 -
download
0
Transcript of 4-Estudios cos de La Bomba de Na

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 1/10
Review
Mechanistic studies of sodium pump
Larry D. Faller
University of California at Los Angeles and Veterans Administration Greater Los Angeles Health Care System, Los Angeles, CA 90073, USA
a r t i c l e i n f o
Article history:
Received 16 April 2008and in revised form 22 May 2008Available online 17 June 2008
Keywords:
Na,K-ATPaseSodium pumpActive ion transportPhosphoryl group transferConformational changeReaction mechanism
a b s t r a c t
Sodium pump was the first ion pump discovered. A member of the family of active transporters that cat-alyze adenosine 50-triphosphate hydrolysis by forming a phosphorylated enzyme intermediate, sodiumpump couples the energy released to unequal countertransport of sodium and potassium ions. The iongradient generated by the pump is important for a variety of secondary physiological processes ranging
from metabolite transport to electrical excitation of nerve and muscle. Selected experiments relatingstructure to function are reviewed.
Ó 2008 Elsevier Inc. All rights reserved.
The Royal Swedish Academy of Sciences spotlighted the vitalrole adenosine 50-triphosphate (ATP) plays as the ‘‘energy cur-rency” of life by awarding the 1997 Nobel Prize in Chemistry forstructural and mechanistic studies of ATP synthesis and for the first
discovery of an ion pump [1]. Driven by a proton gradient acrossmitochondrial membranes, ATP synthase (F0F1-ATPase) makes‘‘about one half” of a resting person’s body-weight in ATP daily.Na,K-ATPase spends ‘‘a third” of the ATP minted to pump 3 Na +
ions out of cells in exchange for 2 K+ ions. Sodium pump is ubiqui-tous because of the need to regulate the volume of animal cells andorganelles bounded by flexible lipid bilayers. Lowering the intra-cellular Na+ concentration helps to prevent cell lysis. The electro-chemical gradient generated by the unequal exchange of Na+ forK+ is used to drive a variety of fundamental physiological pro-cesses. The electrical gradient contributes to the resting membranepotential, thereby affecting channel-regulated electrical stimula-tion of nerve and muscle cells. The Na+ gradient is the energysource for facilitated transport of ions (e.g. Na+/Ca2+ exchange)
and metabolites (e.g. Na+/glucose co-transport) by carriers. Pre-sumably because the pump is essential for cell viability, Na,K-ATP-ase has only been indirectly implicated in the etiology of diseaseslike essential hypertension and diabetes. Sodium pump is thereceptor for cardiac glycosides, like digitalis, used to treat patientswith congestive heart failure.
Only the structure of the water-soluble domain of ATP synthase,which catalyzes ATP hydrolysis (F1-ATPase), was solved. However,the ‘‘orange”-shaped oligomeric structure with three diprotomeric‘‘segments” in different conformations and a central spindle [2]confirmed the elegant rotational mechanism that had been pro-
posed for catalysis [3]. The first atomic-resolution structure of anion pump published in 2000 [4] showed the entire integral-mem-brane protein. However, the mechanism of catalysis-coupled vec-torial transport was not obvious, because the three cytosolic
domains of monomeric Ca-ATPase were widely separated in theinitial structure, and the Ca2+ ions were embedded in the trans-membrane helices with no obvious pathway to either surface.Since publication of this initial ‘‘open” structure, a remarkable ser-ies of additional ‘‘snapshots” of intermediates in the calcium pumpcatalysis-transport cycle have been published by three laborato-ries, culminating in structures with the extracellular ‘‘gate” open[5,6] at the end of 2007. Simultaneously, the first pictures of so-dium pump [7] and a proton pump [8] were published, promptingan accompanying commentary entitled ‘‘Ion Pumps made CrystalClear” [9]. The papers reporting the most recent still photographsof calcium pump [5,6] and the article on Ca-ATPase in this volumereview the structural changes that mechanically couple catalysis totransport in detail. Relatively little has been written about the cat-
alytic mechanism, or the multiple roles of water in catalysis and instructural changes. Therefore, the mechanism of phosphoryl grouptransfer is discussed with emphasis on a method that has onlybeen used to follow the reaction catalyzed by mutants of sodiumpump. Differences between ion binding sites in the sodium- andcalcium-pump structures, the rates of elementary steps in the reac-tion cycle, and studies of a specific inhibitor (ouabain) and of so-dium pump’s subunits are reviewed.
Mechanism
Na,K-ATPase and sodium pump are alternative names for thesame integral-membrane protein that indicate its dual function
0003-9861/$ - see front matter Ó 2008 Elsevier Inc. All rights reserved.doi:10.1016/j.abb.2008.05.017
E-mail address: [email protected]
Archives of Biochemistry and Biophysics 476 (2008) 12–21
Contents lists available at ScienceDirect
Archives of Biochemistry and Biophysics
j o u r n a l h o m e p a g e : w w w . e l s e v i e r . c o m / l o c a t e / y a b b i

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 2/10
as both an enzyme and a transporter. Formation of a phosphory-lated enzyme intermediate catalyzes ATP hydrolysis. Therefore,the family of active transporters is named P-type. Ca-ATPase andthe gastric H,K-ATPase are other examples of P-type pumps. Theenergy released by hydrolyzing ATP is coupled to ion movementby a series of protein conformational changes that were first pro-posed for sodium pump [10,11]. The minimal steps shown sche-matically in Fig. 1 for sodium pump are binding of 3 Na+
intracellularly to a conformation of the enzyme (E1) that is phos-phorylated by ATP. The swung dash ($) is used to indicate a‘‘high-energy” bond. The Na+ ions are released into the extracellu-lar fluid by another conformation of the enzyme (E2) that binds 2external K+ and reacts with H2O releasing Pi into the cytosol. Thepump completes the cycle by changing back to the original confor-mation and releasing K+ ions into the cell. The transported ions areenclosed within the geometrical shapes used for the alternativeconformations to indicate that the ions are ‘‘occluded” at somesteps in the transport cycle. The gray ATP acts as an effector accel-erating the conformational change from E2 to E1. Alternating thephosphorylation and dephosphorylation steps with the Na+ andK+ transport steps assures coupling between catalysis and trans-port. Wedge shapes indicate the unequal Na+ and K+ gradients thatcontribute to the membrane potential.
In contrast to the rotary mechanism of catalysis, the Albers–Post scheme does not specify the nature of the conformationalchange. The minimum requirements for active transport are twoconformations with different chemical and vectorial specificity[12]. One conformation reacts only with nucleotide and binds theexported ion preferentially from the cell interior. The other confor-mation reacts only with inorganic phosphate and binds the counterion preferentially from the cell exterior. Calcium pump is pre-sumed to work by the same mechanism with Ca2+ or H+ insteadof Na+ or K+ acting as the switch between E1 and E2 conformations.Using substrate, transition state, and product analogs plus inhibi-tors to induce intermediate protein states, one or more snapshotsthat mimic most of the important intermediates in the calcium-
pump reaction cycle are now available as follows: holoenzyme
Ca2ÁE1 [4], substrate complex Ca2ÁE1ÁATP [13,14], transition statefor phosphoryl group transfer to the enzyme [5,15], high energyphosphoenzyme intermediate E1$P [6], low energy phosphoen-zyme intermediate E2–P [5,6], transition state for phosphorylgroup transfer to water [5,16], product complex E2ÁPi [17,18], effec-tor complex E2ÁATP [19,20], and apoenzyme E2 [21,22]. The onlyavailable Na,K-ATPase structure corresponds to the product com-plex E
2ÁP
i[7].
Evolution
Functional Na,K-ATPase is a heterodimer of two integral-mem-brane proteins. The larger a subunit ($106 kDa depending onsource) has four distinct domains. Only P-type pumps that count-ertransport K+ have b subunits. The three cytosolic domains of the a subunit catalyze phosphoryl group transfer. The nucleotide(N) domain binds ATP, and the phosphorylation (P) domain cata-lyzes transfer of the c-phosphoryl group to the enzyme. The thirdwater-soluble domain is called the actuator (A) domain, because itis required both for dephosphorylation of the enzyme and for cou-pling ATP hydrolysis to vectorial ion transport through the mem-brane (M) domain.
Only the evolutionary origin of the P-domain is known. New en-zymes originate by recruiting the chemistry evolved to catalyze anelementary reaction. Surprisingly, the P-domain of sodium pumpevolved from a family of bacterial enzymes [23], whose simplestmembers, such as L -2-haloacid dehalogenase, do not catalyze phos-phoryl group transfer, require Mg2+, or bind nucleotide. However,some more-evolved members of the haloacid dehalogenase(HAD) superfamily like phosphoserine phosphatase (PSP) do re-quire Mg2+ for catalysis of phosphoryl group transfer to an aspartylgroup on the enzyme and subsequent hydrolysis of the phosphoen-zyme intermediate [24]. Table 1 shows that three of the six signa-ture sequences for P-type pumps align with the three motifs thatdefine the HAD superfamily.
Phosphoryl group transfer
Enzymes commonly catalyze phosphoryl group transfer bynucleophilic attack on phosphorus forming a pentacoordinate tran-sition state that results in inversion of the stereochemistry aroundthe phosphorus atom [25]. The phosphotransferase activity of P-type pumps also appeared to involve nucleophilic attack for threereasons. First, the aspartyl oxygen of the enzyme is not exchangedby reacting with an isotopically labeled substrate [26]; second,vanadate in the +5 oxidation state with the trigonal bipyramidalgeometry of pentacoordinate phosphorus is a transition-state ana-log [27]; and third, the chirality of the c-phosphate of ATP does notchange, as predicted for hydrolysis catalyzed via a phosphoenzyme
intermediate in two sequential nucleophilic reaction steps thatfirst invert and then restore the stereochemistry around phospho-rus [28]. P-type pumps are reversible. The most impressive of thefour phosphoryl-group-transfer steps catalyzed is phosphorylationof the enzyme by inorganic phosphate. The probability of formingan acylphosphate from Pi nonenzymatically is only 1 in 40 million.
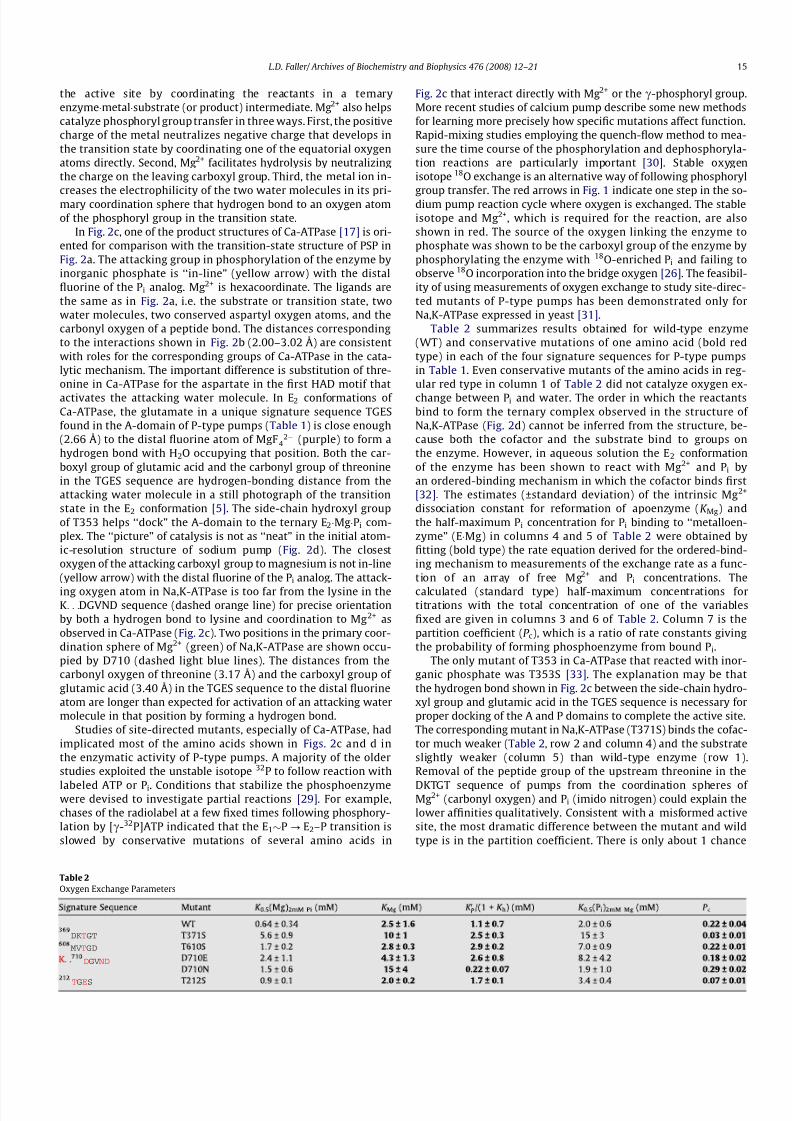
Fig. 2 compares the catalytic machinery of HAD superfamilymember PSP with the arrangement of the corresponding aminoacids in the phosphorylation sites of Ca-ATPase, as well as Na,K-ATPase, because the locations of water molecules are not shownin the initial release of the Na,K-ATPase structure. Fig. 2a is a viewof the PSP transition state in the hydrolysis reaction [24] tilted to-ward the viewer slightly to show the trigonal bipyramidal geome-try (dashed orange lines) of the transition state with the attacking
water molecule (orange) above and the leaving aspartate of the en-zyme below the plane formed by the phosphoryl group analog AlF 3
Fig. 1. Reaction cycle. The minimal steps in the catalysis-transport cycle of sodiumpump are shown schematically. Black arrows indicate the normal, clockwisedirection of the reaction sequence. Oxygen isotope (red) exchange may occur in thereversible step colored red. The cycle represents the sidedness, as well as repetition,of the reactions. For example, the 3 Na+ (blue) that displace 2 K+ (green)intracellularly are subsequently displaced by 2 K+ extracellularly, generating theinward Na+ (larger) and outward K+ (smaller) gradients indicated by wedge shapes.A circle and a square symbolize the limiting protein conformations (E 1 and E2,
respectively) with enclosed ions indicating occlusion. Different colors denote ATPacting as an effector (gray) or substrate (black).
L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21 13

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 3/10
(purple). The shortness of the axial distances from aluminum to thereactants water (2.16 Å) and aspartic acid (2.09 Å) favors an asso-
ciative mechanism in which catalysis occurs via a pentacoordinateintermediate. The first HAD motif extends from below the trigonalbipyramid along the right to near the top, where the downstreamaspartate positions and activates the nucleophile by hydrogenbonding to the attacking water molecule. The second HAD motif lies behind the trigonal bipyramid, and the third motif winds fromthe left side around to the front. For clarity, only the colored aminoacids in Table 1 are shown. Red in Table 1 indicates conserved ami-no acids or conservative substitutions. Orange is used to call atten-tion to amino acids in two positions in the first motif that are notconserved. They include the downstream aspartic acid crucial forreaction of the phosphoenzyme with H2O.
In Fig. 2b, the molecule is rotated toward the reader until theaxis of the bipyramid is perpendicular to the plane of the paper
with the attacking water molecule on top. The leaving carboxylgroup is omitted for clarity. Electrophiles facilitate phosphoryl
group transfer by neutralizing the excess negative charge thatdevelops on the oxygens in the transition state. The distances to
the fluorine atoms of the transition state analog in Fig. 2b predictthree hydrogen bonds, or ion–dipole interactions, indicated bydashed green lines with each of the oxygens of the phosphorylgroup in the transition state. One of the electrophiles is the diva-lent cation required for enzymatic activity. Mg2+ (green) coordi-nates one of the equatorial fluorine atoms directly. Watermolecules that also coordinate the transition-state analog fill twoadditional metal coordination sites. Three protein ligands completethe hexacoordinate geometry of Mg2+ (dashed light blue lines inFig. 2a). The side-chain carboxyl group of the upstream aspartatein the third HAD motif and the backbone carbonyl group of thedownstream aspartate in the first HAD motif that activates theattacking water molecule are shown below and to the right of Mg2+, respectively, in Fig. 2b. The third protein ligand is the oxygen
atom of the leaving carboxyl group that is not shown interactingwith AlF3 in Fig. 2a. Therefore, the divalent cation helps organize
Fig. 2. Comparison of haloacid dehalogenase (HAD) with P-type pumps in E2 conformation. (a) Transition-state structure of phosphoserine phosphatase (PSP) oriented withthe axis of the trigonal bipyramid (orange lines) in the plane of the paper (Protein Data Bank identifier 1L7N). Light blue lines show the hexacoordinate geometry of Mg2+
(green). Two of the ligands are water molecules (orange). The white labels identify the HAD signature motif in which the amino acids are found with only the amino acids thatparticipate directly in catalysis shown. (b) View of the PSP transition state rotated so that the axis of the bipyramid is perpendicular to the plane of the paper with theattacking water molecule on top and the leaving carboxyl group (bottom) omitted for clarity. The green lines show coordination of the phosphoryl group analog AlF 3 (purple)by amino acids in the protein, water, and Mg2+. (c) Product complex E2ÁPi of Ca-ATPase (1WPG). White and gray letters identify amino acids from signature sequences in the P-and A-domains, respectively. The yellow arrow indicates attack on the product analog MgF4
2À in purple. (d) Product complex of Na,K-ATPase (3B8E). The product analog isMgF4
2À. The orange line shows the distance between the attacking oxygen and the side-chain of K691. The blue lines show Mg 2+ coordination by both carboxylic oxygens of D714.
Table 1
Alignment with HAD
14 L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21
8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 4/10
the active site by coordinating the reactants in a ternaryenzymeÁmetalÁsubstrate (or product) intermediate. Mg2+ also helpscatalyze phosphoryl group transfer in three ways. First, the positivecharge of the metal neutralizes negative charge that develops inthe transition state by coordinating one of the equatorial oxygenatoms directly. Second, Mg2+ facilitates hydrolysis by neutralizingthe charge on the leaving carboxyl group. Third, the metal ion in-creases the electrophilicity of the two water molecules in its pri-mary coordination sphere that hydrogen bond to an oxygen atomof the phosphoryl group in the transition state.
In Fig. 2c, one of the product structures of Ca-ATPase [17] is ori-ented for comparison with the transition-state structure of PSP inFig. 2a. The attacking group in phosphorylation of the enzyme byinorganic phosphate is ‘‘in-line” (yellow arrow) with the distalfluorine of the Pi analog. Mg2+ is hexacoordinate. The ligands arethe same as in Fig. 2a, i.e. the substrate or transition state, twowater molecules, two conserved aspartyl oxygen atoms, and thecarbonyl oxygen of a peptide bond. The distances correspondingto the interactions shown in Fig. 2b (2.00–3.02 Å) are consistentwith roles for the corresponding groups of Ca-ATPase in the cata-lytic mechanism. The important difference is substitution of thre-onine in Ca-ATPase for the aspartate in the first HAD motif thatactivates the attacking water molecule. In E2 conformations of Ca-ATPase, the glutamate in a unique signature sequence TGESfound in the A-domain of P-type pumps (Table 1) is close enough(2.66 Å) to the distal fluorine atom of MgF 4
2À (purple) to form ahydrogen bond with H2O occupying that position. Both the car-boxyl group of glutamic acid and the carbonyl group of threoninein the TGES sequence are hydrogen-bonding distance from theattacking water molecule in a still photograph of the transitionstate in the E2 conformation [5]. The side-chain hydroxyl groupof T353 helps ‘‘dock” the A-domain to the ternary E2ÁMgÁPi com-plex. The ‘‘picture” of catalysis is not as ‘‘neat” in the initial atom-ic-resolution structure of sodium pump (Fig. 2d). The closestoxygen of the attacking carboxyl group to magnesium is not in-line(yellow arrow) with the distal fluorine of the Pi analog. The attack-
ing oxygen atom in Na,K-ATPase is too far from the lysine in theK. . .DGVND sequence (dashed orange line) for precise orientationby both a hydrogen bond to lysine and coordination to Mg2+ asobserved in Ca-ATPase (Fig. 2c). Two positions in the primary coor-dination sphere of Mg2+ (green) of Na,K-ATPase are shown occu-pied by D710 (dashed light blue lines). The distances from thecarbonyl oxygen of threonine (3.17 Å) and the carboxyl group of glutamic acid (3.40 Å) in the TGES sequence to the distal fluorineatom are longer than expected for activation of an attacking watermolecule in that position by forming a hydrogen bond.
Studies of site-directed mutants, especially of Ca-ATPase, hadimplicated most of the amino acids shown in Figs. 2c and d inthe enzymatic activity of P-type pumps. A majority of the olderstudies exploited the unstable isotope 32P to follow reaction with
labeled ATP or Pi. Conditions that stabilize the phosphoenzymewere devised to investigate partial reactions [29]. For example,chases of the radiolabel at a few fixed times following phosphory-lation by [c-32P]ATP indicated that the E1$P? E2–P transition isslowed by conservative mutations of several amino acids in
Fig. 2c that interact directly with Mg2+ or the c-phosphoryl group.More recent studies of calcium pump describe some new methodsfor learning more precisely how specific mutations affect function.Rapid-mixing studies employing the quench-flow method to mea-sure the time course of the phosphorylation and dephosphoryla-tion reactions are particularly important [30]. Stable oxygenisotope 18O exchange is an alternative way of following phosphorylgroup transfer. The red arrows in Fig. 1 indicate one step in the so-dium pump reaction cycle where oxygen is exchanged. The stableisotope and Mg2+, which is required for the reaction, are alsoshown in red. The source of the oxygen linking the enzyme tophosphate was shown to be the carboxyl group of the enzyme byphosphorylating the enzyme with 18O-enriched Pi and failing toobserve 18O incorporation into the bridge oxygen [26]. The feasibil-ity of using measurements of oxygen exchange to study site-direc-ted mutants of P-type pumps has been demonstrated only forNa,K-ATPase expressed in yeast [31].
Table 2 summarizes results obtained for wild-type enzyme(WT) and conservative mutations of one amino acid (bold redtype) in each of the four signature sequences for P-type pumpsin Table 1. Even conservative mutants of the amino acids in reg-ular red type in column 1 of Table 2 did not catalyze oxygen ex-change between Pi and water. The order in which the reactantsbind to form the ternary complex observed in the structure of Na,K-ATPase (Fig. 2d) cannot be inferred from the structure, be-cause both the cofactor and the substrate bind to groups onthe enzyme. However, in aqueous solution the E2 conformationof the enzyme has been shown to react with Mg2+ and Pi byan ordered-binding mechanism in which the cofactor binds first[32]. The estimates (±standard deviation) of the intrinsic Mg2+
dissociation constant for reformation of apoenzyme (K Mg) andthe half-maximum Pi concentration for Pi binding to ‘‘metalloen-zyme” (EÁMg) in columns 4 and 5 of Table 2 were obtained byfitting (bold type) the rate equation derived for the ordered-bind-ing mechanism to measurements of the exchange rate as a func-tion of an array of free Mg2+ and Pi concentrations. The
calculated (standard type) half-maximum concentrations fortitrations with the total concentration of one of the variablesfixed are given in columns 3 and 6 of Table 2. Column 7 is thepartition coefficient (P c), which is a ratio of rate constants givingthe probability of forming phosphoenzyme from bound Pi.
The only mutant of T353 in Ca-ATPase that reacted with inor-ganic phosphate was T353S [33]. The explanation may be thatthe hydrogen bond shown in Fig. 2c between the side-chain hydro-xyl group and glutamic acid in the TGES sequence is necessary forproper docking of the A and P domains to complete the active site.The corresponding mutant in Na,K-ATPase (T371S) binds the cofac-tor much weaker (Table 2, row 2 and column 4) and the substrateslightly weaker (column 5) than wild-type enzyme (row 1).Removal of the peptide group of the upstream threonine in the
DKTGT sequence of pumps from the coordination spheres of Mg2+ (carbonyl oxygen) and Pi (imido nitrogen) could explain thelower affinities qualitatively. Consistent with a misformed activesite, the most dramatic difference between the mutant and wildtype is in the partition coefficient. There is only about 1 chance
Table 2
Oxygen Exchange Parameters
TGES
MVTGD
DGVND
DKTGT
L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21 15

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 5/10
in 30 of the mutant forming phosphoenzyme from bound Pi com-pared with 1 chance in 4.5 for wild-type enzyme.
The other mutant with a P c significantly different from wildtype is T212S. Indirect measurements of vanadate binding ordephosphorylation obtained by following [c-32P]ATP hydrolysisor disappearance of phosphoenzyme formed from [c-32P]ATP,respectively, were interpreted to mean that the affinity of the E2
conformation for the transition-state analog is reduced 4-foldand dephosphorylation is slowed 5-fold by the mutation T212A[34]. The same mutant expressed in yeast did not catalyze measur-able 18O exchange. However, T212S could be studied as a functionof both reactant concentrations. No significant difference in eitherMg2+ or Pi binding between wild type (Table 2, row 1) and themutant (row 6) was detected. The lower probability of formingphosphoenzyme (column 7) indicates that at least one rate con-stant other than the rate constant for dephosphorylation is affectedby substituting serine for threonine, because P c does not containthe dephosphorylation rate constant. A reasonable explanation of the more dramatic effect of alanine than serine substitution onboth binding and phosphoryl group transfer is that the hydrogenbond shown in Fig. 2d between the side-chain hydroxyl group of threonine and the imido nitrogen of E214 is important for config-uring the TGES loop. However, the decrease in the probability of T212S forming phosphoenzyme suggests that one or both of theinteractions of the fluorine occupying the position of the leavinggroup in the phosphorylation reaction with the carbonyl oxygenof threonine and with the carboxyl group of glutamic acid becomesless favorable than in wild type. Activation of the attacking waterby the negatively charged carboxyl group of E214 is more impor-tant than hydrogen bonding to the carbonyl oxygen of T212, be-cause the mutant E214A was not expressed in COS-1 cells [34]and oxygen exchange could not be detected even when the mostconservative mutant E214D was expressed in yeast.
The only measurable effect of changing threonine in the MVTGDsignature sequence to serine (T610S) was lowered affinity of themetalloenzyme for Pi (Table 2, row 3 and column 5). Therefore,
the data quantitatively confirm a role in Pi binding predicted bythe distance from the side-chain hydroxyl group of T610 to theclosest fluorine atom of MgF4
2À in Fig. 2d (2.58 Å). Failure to detectany oxygen exchange by T610V was unexpected because multipleligands contribute to Pi binding and to stabilization of the transi-tion state in phosphoryl group transfer (Fig. 2b).
Single mutations of two amino acids in the K. . .DGVNDsequence of sodium pump also resulted in complete loss of ATPaseactivity, including the most conservative substitutions of argininefor lysine and glutamic acid for the downstream aspartic acid[35,36]. Mutants of the asparagine in both Na,K-ATPase [35] and
Ca-ATPase [37] catalyzed ATP hydrolysis, but the Ca-ATPase mu-tants did not form significant phosphoenzyme from [32P]Pi. Oxygenexchange was undetectable for even the most conservative muta-tions of the amino acids in the K. . .DGVND sequence of Na,K-ATP-ase indicated by standard red type in Table 1. However, conflictingdata and different interpretations of similar results have beenreported for mutants of the upstream aspartate in both the sodiumand calcium pumps by several laboratories. Small shifts comparedto wild-type Na,K-ATPase in the half-maximal Mg2+ concentrationfor Mg2+ and Pi (or vanadate)-dependent tritiated ouabain bindingto D710A were interpreted to mean that D710 does not bind Mg 2+
in the E2 conformation [35,38]. However, row 5 of Table 2 showsthat the K 0.5 values for Mg2+ and Pi may not differ dramaticallyfrom the wild-type values in row 1, if the mutation has compensat-ing effects on the dissociation constants for the cofactor and thesubstrate [39]. Reduced affinity for Mg2+, when the charge onD710 is neutralized, and increased affinity for P i are consistentwith ordered binding of the cofactor and substrate to form the ter-nary complex between the reactants shown in Fig. 2d. Retainingthe charge on D710 modestly decreased the estimated affinitiesof D710E for both Mg2+ and Pi by roughly the same amount (Table2, row 4). A study of the corresponding mutation in Ca-ATPase mayindicate the importance of water in the reaction mechanism. Amore dramatic shift in the wild-type half-maximal concentrationwhen D703E was titrated with Mg2+ at a fixed Pi concentration,than in the converse titration, was interpreted as evidence for acritical role of the upstream aspartate in metal binding [40]. How-ever, the measurements were made in an organic solvent–watermixture, where kinetic studies indicated the substrate is MgÁPi
[41]. Therefore, the half-maximum should occur at the same freeMgÁPi concentration for either choice of independent variable. Aspredicted, the free concentrations of MgÁPi calculated from theempirical half-maximal and fixed variable concentrations [40]and the dissociation constant (140 mM) of MgÁPi in the organic sol-vent–water mixture [41] for the titrations with Pi and Mg2+ are thesame (3 lM). Therefore, an alternative interpretation of the D703E
results is that the substrate changes from P i in water [42] to MgÁPi
in the organic solvent–water mixture. The organic solvent may dis-rupt second aqueous-sphere coordination of the metal by proteinligands like D707 (Fig. 3c) that favor sequential binding of the reac-tants in H2O.
Enzymes utilize binding energy to fix reactants on their surfaceand entropically lower the reaction barrier. The active site of E2
must be formed by an ‘‘induced fit” mechanism, because the pro-tein ligands that bind Mg2+ are more than twice as far apart inthe holoenzyme Ca2ÁE1 [4] as in the product structure E2ÁPi [17].A still photograph of the metalloenzyme (E2ÁMg) has not yet been
Fig. 3. Hypothetical formation of ternary EÁMgÁPi complex. (a) E: P-domain active site amino acids in Ca2ÁE1 (1SU4). (b) EÁMg: active site in effector complex E2ÁATP (2DQS)superimposed on Ca2ÁE1 (gray). (c) EÁMgÁPi: product complex (1WPG) superimposed on E 2ÁATP (gray).
16 L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 6/10
obtained. However, one snapshot of the E2 conformation with ATPbound at an effector site has Mg2+ at the active site [20]. Fig. 3shows how the catalytic site may be formed in the counter-clock-wise direction in Fig. 1 from sequential binding of Mg2+ and Pi.Fig. 3a is the holoenzyme Ca2ÁE1 [4]. In Fig. 3b, the correspondingamino acids of the effector complex E2ÁATP were superimposedon Ca2ÁE1 (gray) by fitting the backbone atoms of the seven b
strands in the P domain that form the Rossman fold. Three ligandsappear bound to Mg2+ (dashed green lines). Both aspartates in theDGVND sequence coordinate the metal ion directly. A water mole-cule is also in the primary coordination sphere. Nearly perfectoverlap of all three sequences of Ca2ÁE1 with the corresponding se-quences of E2ÁATP and only a slightly greater distance between theoxygen atoms coordinated to Mg2+ in Ca2ÁE1 than in E2ÁATP suggestthat the effector complex may be a good model for the P-domainresidues (white letters) of E2ÁMg that form the catalytic site. Thecarboxyl groups of D703 and D707 are nearly twice as far apartin E2 [21] as in E2ÁATP or Ca2ÁE1. Fig. 3c shows the product complexE2ÁPi superimposed on E2ÁATP (gray). Only the upstream asparticacid D703 coordinates the metal ion (green) directly. The DKT,K. . .D. . .ND, and TGE (gray letters) sequences collapse around theternary enzymeÁmetalÁproduct complex with the glutamic acidside chain rotated into position to bind Pi (Fig. 2c) or activate theattacking water molecule in the transition state (Fig. 2a). The TGsequence (white letters) is shifted to the left, so the side chain of threonine and the nitrogen of glycine can also assist in binding P i
and neutralizing the negative charge that develops on the oxygensin the transition state. The mechanism depicted in Fig. 3 canexplain larger effects than were predicted from the available struc-tures for mutating some of the amino acids denoted by regular redtype in Table 2. For example, complete loss of activity was not pre-dicted for mutations of the downstream aspartic acid in theDGVND sequence, because the only apparent contribution of D707 to the completed active site (Fig. 3c) is second-sphere coor-dination of both Mg2+ and Pi (dashed light blue lines). However,failure to bind Mg2+ (Fig. 3b) and subsequently Pi (Fig. 3c) could
explain why even the most conservative mutant of the correspond-ing amino acid in Na,K-ATPase (D721E) failed to catalyze oxygenexchange between Pi and water. In the normal clockwise directionindicated by unidirectional arrows in Fig. 1, the active site may beformed either from Ca2ÁE1 or E2ÁATP with the latter more likely atthe high concentrations of ATP present in cells [43]. In either case,replacing the TGES loop with the c-phosphate of ATP completesformation of the active site.
Mutations in the TGES sequences of both the sodium [34] andcalcium [44,45] pumps affect phosphorylation by inorganic phos-phate but not by ATP. Fig. 4 shows that in the substrate complexCa2ÁE1ÁATP, the TGES loop is replaced by the nucleotide. The aminoacids corresponding to the amino acids shown in Fig. 2b, the diva-lent cation, and the two water molecules are all close enough to
one of the oxygen atoms of the c-phosphate for roles in bindingthe substrate. They are also likely to catalyze transfer of the c-phosphate to the enzyme by stabilizing the transition state. Consis-tent with participation of the two water molecules in catalysis, therate of phosphoryl group transfer from ATP to the Ca-ATPase is re-duced 20-fold [46] in the organic solvent–water mixture in whichthe substrate for phosphorylation by Pi is MgÁPi [41]. A second Mg2+
ion coordinated to the a- and b-phosphates of ADP in snapshots of the transition state for phosphoryl group transfer to the enzyme[5,15] presumably facilitates loss of the leaving group. However,structural evidence that the substrate is MgÁATP is lacking, becauseMg2+ is not coordinated to AMPPCP in either structure of E1 withthe substrate analog bound [13,14]. The postulated difference inenzymatic specificity of the E1 and E2 conformations results from
movements of the N, P, and A domains that replace the nucleotidewith the TGES loop in the active site. The large tilt of the N domain
(60o), rotation of the A domain (90o), and inclination of the P
domain (30o) that bring nucleotide into the active site and replaceATP with the TGES loop are described and illustrated in the articleon Ca-ATPase in this volume. Counterintuitively, hydrolysis of ATPis not prevented by exclusion of water from the active site in the E 1
conformation. On the contrary, two water molecules are likely toplay important roles as hydrogen-bond donors in both the E 1 andE2 conformations. The attacking water molecule may be importedinto the active site of E2 along with the TGES loop.
Ion transport
Different vectorial specificity results from mechanical couplingbetween the movements of the cytosolic domains and changes in 6of the 10 transmembrane helices. One particularly long helix (M5)
spans the P domain as well as the membrane. The bending, tilting,translation relative to the plane of the membrane, and partialunwinding of the helices are pictured in the article on calciumpump in this issue. The transported ions are bound roughly a quar-ter of the way through the membrane from the cytosolic side byamino acids on helices M4–M6 in the mobile part of the M domainand M8.
The ion binding sites of the sodium and calcium pumps areremarkably similar. Six of the seven amino acids with side chainsthat bind the two Ca2+ ions in the E1 conformation of Ca-ATPaseare hypothesized to bind the congener of K+ (Rb+) in the E2 struc-ture of Na,K-ATPase [7]. The putative sodium pump ligands E327,S775, N776, E779, E804, E808, and Q923 include two conservativesubstitutions (aspartic acid for asparagine and glutamine for glu-
tamic acid). The distances from E808 (4.76 Å) and Q923 (6.67 Å)to the nearest Rb+ indicate second and third aqueous-sphere coor-dination, whereas the corresponding amino acids in Ca-ATPasebind Ca2+ directly. The ligand without a counterpart in calciumpump is S775. The seventh Ca-ATPase ligand is a threonine thatis conserved in Na,K-ATPase (T807) but distant from the monova-lent cation sites. Three carbonyl oxygens of the polypeptide chaincoordinate one of the Ca2+ ions in Ca-ATPase. The carbonyl oxygensof two valines (V322 and V325) occupy comparable positions inthe M4 helix of Na,K-ATPase and are roughly the same distanceas E327 from one of the Rb+ ions. Whether the observed differencesin ligation are sufficient to explain the different ion specificities of the two pumps is difficult to assess without knowing the hydrationnumbers of the monovalent cations in sodium pump. A proposed
third Na+
site [47] supported by site-directed mutagenesis [48] isnot occupied in the crystal structure of sodium pump. Residues
Fig. 4. Active site in E1 conformation. Structure of E1ÁATP (1VFP) in which thecatalytic site is completed by the polyphosphate chain of the substrate analogAMPPCP instead of the TGES sequence.
L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21 17

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 7/10
unique to sodium pump in a C-terminal extension also appear to beimportant for Na+ binding, because when the last five amino acidsKETYY were removed, the affinity for Na+ was reduced by morethan an order of magnitude [7].
The ions appear occluded in most snapshots of calcium pump.Only the two low energy phosphoenzyme (E2–P) structures [5,6]show the extracellular ‘‘gate” open with the distance betweenthe long extracellular loops L34 and L78 visibly lengthened inone [6]. Therefore, one change in chemical and vectorial specificityoccurs simultaneously in the E1$P? E2–P step. Triggered by ADPdissociation, the N domain disengages from the P domain, andthe A domain rotates into the space formerly occupied by thenucleotide. Some of the bonds formed between the TGES loopand the phosphorylated P domain are shown in Fig. 2c. The extra-cellular gate is closed in the E2–P? E2ÁP transition. The pathway tothe ion binding sites from the cell interior is not as obvious. Theintracellular gate is presumably open in the E2? Ca2ÁE1 transition,or the E2ÁATP? E1ÁATP transition at physiological concentrationsof ATP, and closes once the Ca2+ sites are filled. E309 on M4 is be-lieved to act as the intracellular gate by rotating in (E 1 conforma-tions) and out (E2 conformations) of the Ca2+ binding site. E327 isthought to play a similar role in Na,K-ATPase with evidence fromsite-directed mutagenesis that rotation of the side chain may becontrolled by van der Waals interactions with L97 [49].
Reaction rates
The large domain movements that change the enzyme specific-ity must be fast for tight coupling between catalysis and transport.Sodium pump has the advantage for kinetic studies of conforma-tional changes that both transported ions can be easily manipu-lated experimentally. In addition to establishing the rate of aconformational change, kinetic studies can reveal the mechanismof ligand binding to the protein and contributions to the energeticsof the reaction. The change from the sodium (Na3ÁE1) to potassium(K2ÁE2) conformation of sodium pump, which presumably includes
a tilting of the N domain and rotation of the A domain that movethe top of the N domain more than 50 Å [17], is diffusion controlled
[50]. The theoretical equation used to describe shielding of dimerformation from oppositely charged monomers fits the dependenceof the first-order rate constant for association of the cytosolic do-mains on ionic strength. Therefore, electrostatic interactions be-tween domains contribute to the driving force for the proteinrearrangement. The extrapolated value at zero ionic strengthexpressed as a second-order rate constant (1.1 Â 109 MÀ1 sÀ1)approaches the limit for a diffusion-controlled reaction. Dissocia-tion of the domains also depends on ionic strength. However, thedissociation rate is far below the diffusion limit, implying forma-tion of an intermediate encounter complex that is rate limiting inthe association reaction. Fig. 5 shows the activation entropies cal-culated from the temperature dependence of the domain associa-tion and dissociation rates. Without the increase in entropyexplained schematically in the figure by loss of structured waterfrom the protein surface, the units of the rate constants for forma-tion of the encounter complex would be dayÀ1 instead of sÀ1.Water increases the rate of the conformational change 105-fold.The reverse reaction in Fig. 5 is too slow (second half-time) forcompatibility with the rate of ATP hydrolysis (millisecond half-time). However, lower affinity ATP binding than required for acti-vation of hydrolysis increases the rate of the conformationalchange from E2 to E1 by nearly two orders of magnitude [51].Therefore, in vitro rate measurements anticipated the suggestionthat conformations without nucleotide bound are bypassed, andE1ÁATP is formed directly from E2ÁATP in vivo [43].
In contrast to calcium pump, which binds 2 Ca2+ cooperativelywith micromolar dissociation constants differing by more thanan order of magnitude [52], the E1 conformation of sodium pumpbinds 2 K+ statistically (or anticooperatively with millimolar disso-ciation constants serendipitously differing by the statistical factorfour) [53]. Na+ competes for the same sites on E1 with roughly10-fold greater affinity reversing the coupled conformationalchange between E1 and E2 [54]. Random binding to the sitesobserved in the crystal structure of Na,K-ATPase is hard to under-stand. A plausible explanation may be that sites closer to the cyto-
sol in E1 discriminate between Na+ and K+ based on their chargedensities, and the completely or partially dehydrated ions proceed
Fig. 5. Hypothetical role of water in the conformational change. The schematic shows how release of structured water from the protein surface lowers the energy barrier
accelerating the conformational change from E1 (1SU4) to E2. 1IWO is used to represent both the encounter complex Eec without and E2 with interdomain bonds (red circles)formed.
18 L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 8/10
to the occlusion sites. Consistent with the structural evidence foran open intracellular gate in E1 structures and an openextracellular gate only in E2–P, monovalent ions released fromthe occluded E2 conformation of sodium pump by ATP, whichaccelerates the conformational change to E1, are indistinguishable,but their release from the phosphoenzyme formed by reactionwith Pi is ordered [55]. Na+ release from the E2 conformation of so-dium pump measured electrically is also ordered [56]. The electri-cal signal results from ion movement through the membrane’selectrical field in an ‘‘access” channel, whose existence was con-firmed structurally by a recent snapshot of E 2–P [6]. Three tempo-rally resolved time constants were observed confirming theexistence of a third Na+ site. The estimate of the time required torelease the third Na+ (61 ls) in a sequential model was limitedby the time constant of the instrument used to perturb the mem-brane potential. The intermediate relaxation time corresponding torelease of the second Na+ was about 100 ls. The slowest relaxationtime was interpreted as release of the first Na+ ($1 ms) rate-lim-ited by the conformational change from E1$P to E2–P openingthe extracellular gate. Other methods for estimating the rate of the conformational change in phosphorylated enzyme are alsoindirect and require assumptions about coupling between thereaction observed and the conformational change, e.g. rapid-mix-ing experiments using extrinsic fluorophores to follow phosphory-lation by ATP [57]. Nevertheless, the half-times reported for theE1$P? E2–P transition generally fall in the range 1–20 ms.
Equilibrium constants for enzyme-catalyzed reactions are typi-cally close to unity [25]. From a comparison of the amount of phos-phoenzyme formed from Pi in K+ (E2–P) with the amount formedfrom ATP in Na+ (E1$P), the equilibrium constant between cova-lently and noncovalently bound Pi (K h) on E2 is also near unity[58]. Therefore, the intrinsic phosphate dissociation constant of bound Pi from metalloenzyme (K 0P ) is roughly twice the value incolumn 5 of Table 2, and the rate constants for the reactions inred in Fig. 1 can be estimated from the exchange rate and the ex-change parameters in columns 5 and 7. Remarkably, the estimated
relaxation time for phosphoenzyme formation and hydrolysis onthe surface of the protein by the molecular mechanism shown inFig. 2 is only 0.1 ms [31].
Ouabain
Ouabain is a specific inhibitor of Na,K-ATPase that binds to theprotein from the extracellular side. The high affinity of the enzymefor ouabain made the cardiac glycoside binding site one of the firsttargets for study by site-directed mutagenesis. However, a problemwith point mutations is that amino acids outside the targeted bind-ing site may affect binding because of concerted conformationalchanges throughout the pump. Random mutagenesis identified
amino acids in 4 of the 5 extracellular loops and 5 of the 10 trans-membrane helices that affected ouabain binding [59]. In a recentseries of papers, false positives were avoided by gross screeningwith chimeras of the sodium and gastric proton pumps. In the firstpaper [60], the ouabain binding site was localized by showing thata chimera of H,K-ATPase with the M3/M4 and M5/M6 hairpinsfrom Na,K-ATPase binds ouabain comparably to wild-type Na,K-ATPase. In the second paper [61], the functional chimera was mu-tated by changing the 21 amino acids in the M5/M6 hairpin of Na,K-ATPase that are different back to the amino acid in H,K-ATP-ase in groups and individually. The three individually mutatedamino acids that eliminated ouabain binding were then introducedinto chimeras containing only the M3/M4 hairpin from sodiumpump in various combinations to see if ouabain binding could be
restored. All three of the amino acids in the M5 helix were requiredwith the M3/M4 hairpin for high-affinity ouabain binding. In the
third paper [62], the same strategy of combining chimera construc-tion with site-directed mutagenesis was used to identify four ami-no acids in the M4 helix of sodium pump as important for ouabainbinding. Simultaneous introduction of E312, V314, I315, G319,F783, T797, and D804 into the gastric H,K-ATPase produced a mu-tant with the same affinity for ouabain as Na,K-ATPase. Finally, inthe fourth paper [63] a homology model of the ouabain binding sitewas refined by identifying three additional amino acids P778, L795,and C802 with apparent roles in ouabain binding that are con-served in the gastric H,K-ATPase. In the model, ouabain sits in apocket formed by M4 and the L12 and L56 loops close to the mono-valent cation binding sites. The side chains of D312 and T799 andthe imido nitrogens of T797 and V798 plus the side chains of Q111and N122, previously implicated in ouabain binding [64], appear tobind ouabain directly. Experiments designed to test the model mayreveal how ouabain inhibits sodium pump.
b Subunit
In contrast to Ca-ATPase and nearly all other P-type pumps,Na,K-ATPase and the other mammalian pump that countertran-
sports K+
the gastric H,K-ATPase are heterodimers (ab). Heavilyglycosylated, the protein ($37 kDa) is roughly a third the size of the a subunit. The b subunit of Na,K-ATPase spans the membraneonce and is required for activity. The b subunit has two main func-tions that have been extensively reviewed [65]. One function of theb subunit is to chaperone correct insertion of the newly synthe-sized protein into the membrane. There is also growing evidencethat the b subunit affects the intrinsic ion binding and transportproperties of the mature pump. One experiment illustrates anotherstructural feature of sodium pump. Different isoforms of both the aand b subunits are found in different tissues. When nine differentcombinatorial isozymes of three different a and b isoforms wereexpressed and assayed, significantly different results wereobtained [66]. For example, a1b1 had more than twice the apparentaffinity for external K+ exhibited by a
2b
2. Another experiment indi-
cates that the large, glycosylated, extracellular domain of the Na,K-ATPase b subunit, which contains six cysteines that can form disul-fide linkages, must be properly folded. When the disulfide bondswere partially reduced, occlusion of the K+ congener Rb+ wasroughly halved [67]. Unfortunately, only the transmembrane helixof the b subunit is resolved in the first structure of sodium pump.The b helix appears close enough to aM7 for predominately vander Waals interactions in the region between G848 and L856. How-ever, not enough amino acids in the b-subunit extracellular domaincould be traced for structural confirmation that the segment fromE63 to N159 is sufficient for interaction with 26 amino acids in theL78 loop including the conserved sequence SYGQ [68]. Electron-density maps indicate the rest of the glycosylated domain extendsunderneath the a subunit, and the cytosolic N-terminus of the b
subunit continues in the direction that the helix slopes wrappingaround the a subunit. Atomic resolution of the entire b subunitmay help explain why only mammalian P-type pumps that count-ertransport K+ are heterodimers.
c Subunit
A much smaller protein ($9 kDa) than either a or b was discov-ered 30 years ago in renal tissue and is referred to as the k subunitof Na,K-ATPase. The c subunit is not required for activity and wasrecently identified as a member of a family of transport regulatorsnamed for an invariant amino acid sequence FXYD in the signaturemotif [69]. So far, seven members of the FXYD family (numbered in
the order of sequencing) have been identified [70], and tissue-spe-cific association with Na,K-ATPase has been demonstrated for six
L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21 19

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 9/10
of them [71]. The N-terminus is located extracellularly with asingle helix spanning the membrane. Twenty-nine amino acidslocated in the helix beginning just after the signature sequenceof the c subunit (FXYD2) are resolved in the first structure of so-dium pump [7]. The c helix lies alongside M9 of the a subunit.The helices are farther apart than expected from mutagenesisexperiments implicating specific amino acids of M9 in the stabilityof the complex [72], but too close together to accommodate themutation of G41 to arginine implicated in hypomagnesaemia[73]. Some of the properties of the pump that may be affected bythe c subunit are the affinities for ATP [74], K+, and Na+ [75,76].The changes in half-maximal concentrations for ATPase activityor pump current caused by expressing c with ab are small. How-ever, there is precedent for regulation of P-type pumps by rela-tively small proteins that span the membrane once. For example,phospholamban (PLB) regulates the Ca-ATPase of cardiac sarco-plasmic reticulum [77]. PLB has little sequence homology withmembers of the FXYD family, is oppositely oriented with anintracellular N-terminus, and is predominantly a homopentamer.However, there are also striking similarities, especially with phos-pholemman (PLM) the family member (FXYD1) found in heart andskeletal muscle [78]. The folds of both PLB [79] and PLM [71] beginwith arginine-rich intracellular helices containing phosphorylationsites that are connected by a turn to apolar transmembrane helices.Association of monomeric PLB with Ca-ATPase inhibits the pump,and phosphorylation reverses the inhibition. Association withPLM may regulate sodium pump in a similar way. For example,ischemia-induced phosphorylation of PLM activates cardiacNa,K-ATPase [80].
Conclusion
Extraordinary progress toward understanding how biologicalpumps couple ATP hydrolysis to ion transport was made in the last7 years because structures of calcium pump showing the moleculardetails were solved. Partial resolution of the b and c subunits in the
firststructureofsodiumpumppublishedattheendof2007presagesanswers toquestionsabout theuniquepropertiesof sodiumpump inthe near future. For example, how do P-type pumps discriminateamong the ions transported? Why is the b subunit required onlyby K+ countertransporting ions? How does the c subunit modulateNa,K-ATPase activity? How does ouabain inhibit sodium pump?Some questions can never be answered by crystal structures. Forexample, how fast are the conformational changes? What is themechanism of cofactor, substrate, and transported-ion interactionwith thepump?Morequantitative approacheswillbe neededto teststructural hypotheses. This article has attempted to illustrate whyboth mechanistic and structural studies are required for deeperunderstanding of how biological pumps function.
References
[1] J.C. Skou, Biochimica et Biophysica Acta 23 (1957) 394–401.[2] J.P. Abrahams, A.G.W. Leslie, R. Lutter, J.E. Walker, Nature 370 (1994) 621–628.[3] P.D. Boyer, Biochimica et Biophysica Acta 1140 (1957) 215–250.[4] C. Toyoshima, M. Nakasako, H. Nomura, H. Ogawa, Nature 405 (2000) 647–655.[5] C. Toyoshima, Y. Norimatsu, S. Iwasawa, T. Tsuda, H. Ogawa, Proceedings of the
National Academy of Sciences of the United States of America 104 (2007)19831–19836.
[6] C. Olesen, M. Picard, A.-M.L. Winther, C. Gyrup, J.P. Morth, C. Oxvig, J.V. Møller,P. Nissen, Nature 450 (2007) 1036–1042.
[7] J.P. Morth, B.P. Pedersen, M.S. Toustrup-Jensen, T.L.-M. Sørensen, J. Petersen, J.P. Andersen, B. Vilsen, P. Nissen, Nature 450 (2007) 1043–1049.
[8] B.P. Pedersen, M.J. Buch-Pedersen, J.P. Morth, M.G. Palmgren, P. Nissen, Nature450 (2007) 1111–1114.
[9] D.C. Gadsby, Nature 450 (2007) 957–959.[10] R.W. Albers, Annual Review of Biochemistry 36 (1967) 727–756.[11] R.L. Post, L. Kume, T. Tobin, G. Orcutt, A.K. Sen, Journal of General Physiology
54 (1969) 306s–326s.[12] W.P. Jencks, Journal of Biological Chemistry 264 (1989) 18855–18858.
[13] T.L.-M. Sørensen, J.V. Møller, P. Nissen, Science 304 (2004) 1672–1675.[14] C. Toyoshima, T. Mizutani, Nature 430 (2004) 529–535.[15] T.L.-M. Sørensen, J.D. Clausen, A.-M.L. Jensen, B. Vilsen, J.V. Møller, J.P.
Andersen, P. Nissen, Journal of Biological Chemistry 279 (2004) 46355–46358.[16] C. Olesen, T.L.-M. Sørensen, R.C. Nielsen, J.V. Møller, P. Nissen, Science 306
(2004) 2251–2255.[17] C. Toyoshima, H. Nomura, T. Tsuda, Nature 432 (2004) 361–368.[18] K. Moncoq, D.A. Trieber, H.S. Young, Journal of Biological Chemistry 282 (2007)
9748–9757.[19] A.M. Jensen, T.L-M. Sørensen, C. Olesen, J.V. Møller, P. Nissen, European
Molecular Biology Organization Journal 25 (2006) 2305–2314.[20] G. Inesi, D. Lewis, H. Ma, A. Prasad, C. Toyoshima, Biochemistry 45 (2006)
13768–13769.[21] C. Toyoshima, H. Nomura, Nature 418 (2002) 605–611.[22] K. Obara, N. Miyashita, C. Xu, I. Toyoshima, Y. Sugita, G. Inesi, C. Toyoshima,
Proceedings of the National Academy of Sciences of the United States of America 102 (2005) 14489–14496.
[23] L. Aravind, M.Y. Galperin, E.V. Koonin, Trends in Biochemical Science 23 (1998)127–129.
[24] W. Wang, H.S. Cho, R. Kim, J. Jancarik, H. Yokota, H. H Nguyen, I.V. Grigoriev,D.E. Wemmer, S.-H. Kim, Journal of Molecular Biology 319 (2002) 421–431.
[25] J.R. Knowles, Annual Review of Biochemistry 49 (1980) 877–919.[26] A.S. Dahms, T. Kanazawa, P.D. Boyer, Journal of Biological Chemistry 248
(1973) 6502–6595.[27] L.C. Cantley Jr., L.G. Cantley, L. Josephson, Journal of Biological Chemistry 253
(1978) 7361–7368.[28] M.R. Webb, D.R. Trentham, Journal of Biological Chemistry 256 (1981) 4884–
4887.[29] D.M. Clarke, T.W. Loo, D.H. MacLennan, Journal of Biological Chemistry 265
(1990) 22223–22227.[30] T.L-M. Sørensen, Y. Dupont, B. Vilsen, J.P. Andersen, Journal of Biological
Chemistry 275 (2000) 5400–5408.[31] R.A. Farley, E. Heart, M. Kabalin, D. Putnam, K. Wang, V.N. Kasho, L.D. Faller,
Biochemistry 37 (1997) 941–951.[32] V.N. Kasho, M. Stengelin, I.N. Smirnova, L.D. Faller, Biochemistry 36 (1997)
8045–8052.[33] J.D. Clausen, D.B. McIntosh, D.G. Woolley, J.P. Andersen, Journal of Biological
Chemistry 276 (2001) 35741–35750.[34] M. Toustrup-Jensen, B. Vilsen, Journal of Biological Chemistry 278 (2003)
11402–11410.[35] P.A. Pedersen, J.R. Jorgensen, P.L. Jorgensen, Journal of Biological Chemistry
275 (2000) 37588–37595.[36] P. Su, G. Scheiner-Bobis, Biochemistry 43 (2004) 4731–4740.[37] J.D. Clausen, D.B. McIntosh, D.G. Woolley, A.N. Anthonisen, B. Vilsen, J.P.
Andersen, Journal of Biological Chemistry 281 (2006) 9471–9481.[38] P.L. Jorgensen, J.R. Jorgensen, P.A. Pedersen, Journal of Bioenergetics and
Biomembranes 33 (2001) 367–377.
[39] L.D. Faller, A.K. Nagy, D.J. Kane, R.A. Farley, Annals of the New York Academy of Science 986 (2003) 275–277.[40] D.B. McIntosh, J.D. Clausen, D.G. Woolley, D.H. MacLennan, B. Vilsen, J.P.
Andersen, Journal of Biological Chemistry 279 (2004) 32515–32523.[41] Ph.Champeil,F. Guillain,C. Vénien, M.P.Gingold,Biochemistry24 (1985)69–81.[42] A.K. Nagy, D.J. Kane, C.M. Tran, R.A. Farley, L.D. Faller, Journal of Biological
Chemistry 280 (2005) 7435–7443.[43] J.V. Møller, C. Olesen, A.-M.L. Jensen, P. Nissen, Journal of Bioenergetis and
Biomembranes 37 (2005) 359–364.[44] J.D. Clausen, B. Vilsen, D.B. McIntosh, A.P. Einholm, J.P. Andersen, Proceedings
of the National Academy of Sciences of the United States of America 101(2004) 2776–2781.
[45] A.N. Anthonisen, J.D. Clausen, J.P. Andersen, Journal of Biological Chemistry281 (2006) 31572–31582.
[46] H. Suzuki, T. Kanazawa, Journal of Biological Chemistry 271 (1996) 5481–5486.
[47] H. Ogawa, C. Toyoshima, Proceedings of the National Academy of Sciences of the United States of America 99 (2002) 15977–15982.
[48] C. Li, O. Capendeguy, K. Geering, J-D. Horisberger, Proceedings of the
National Academy of Sciences of the United States of America 102 (2005)12706–12711.
[49] A.J. Einholm, J.P. Andersen, B. Vilsen, Journal of Biological Chemistry 282(2007) 23854–23866.
[50] S.-H. Lin, L.D. Faller, Biochemistry 44 (2005) 1482–1494.[51] M. Steinberg, S.J.D. Karlish, Journal of Biological Chemistry 264 (1989) 2726–
2734.[52] G. Inesi, M. Kurzmack, C. Coan, D.E. Lewis, Journal of Biological Chemistry 256
(1980) 3025–3031.[53] I.N. Smirnova, S.-H. Lin, L.D. Faller, Biochemistry 34 (1995) 8657–8667.[54] S.J.D. Karlish, Journal of Bioenergetics and Biomembranes 12 (1980) 111–136.[55] B. Forbush, Journal of Biological Chemistry 262 (1987) 11116–11127.[56] M. Holmgren, J. Wagg, F. Bezanilla, R.F. Rakowski, P. De Weer, D.C. Gadsby,
Nature 403 (2000) 898–901.[57] D.J. Kane, K. Fendler, E. Grell, E. Bamberg, K. Taniguchi, J.P. Froehlich, R.J.
Clarke, Biochemistry 36 (1997) 13406–13420.[58] G. Berberian, L. Beauge, in: The Sodium Pump: Recent Developments, Society
of General Physiologists, vol. 44, Part 2, 1992, pp. 389–393.
[59] M.L. Croyle, A.L. Woo, J.B. Lingrel, European Journal of Biochemistry 248 (1997)488–495.
20 L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21

8/2/2019 4-Estudios cos de La Bomba de Na
http://slidepdf.com/reader/full/4-estudios-cos-de-la-bomba-de-na 10/10
[60] J.B. Koenderink, H.P.H. Hermsen, H.G.P. Swarts, P.H.G.M. Willems, J.J.H.H.M. DePont, Proceedings of the National Academy of Sciences of the United States of America 97 (2000) 11209–11214.
[61] L.Y. Qiu, J.B. Koenderink, H.G.P. Swarts, P.H.G.M. Willems, J.J.H.H.M. De Pont, Journal of Biological Chemistry 278 (2003) 47240–47244.
[62] L.Y. Qiu, E. Krieger, G. Schaftenaar, H.G.P. Swarts, P.H.G.M. Willems, J.J.H.H.M.De Pont, J.B. Koenderink, Journal of Biological Chemistry 280 (2005) 32349–32355.
[63] L.Y. Qiu, H.G.P. Swarts, E.C.M. Tonk, P.H.G.M. Willems, J.B. Koenderink, J.J.H.H.M. De Pont, Journal of Biological Chemistry 281 (2006) 13533–13539.
[64] E.M. Price, J.B. Lingrel, Biochemistry 27 (1988) 8400–8408.[65] K. Geering, Journal of Bioenergetics and Biomembranes 33 (2001) 425–438.[66] G. Crambert, U. Hasler, A.T. Beggah, C. Yu, N.N. Modyanov, J.-D. Horisberger, L.
Lelievre, K. Geering, Journal of Biological Chemistry 275 (2000) 1976–1986.[67] S. Lutsenko, J.H. Kaplan, Biochemistry 32 (1993) 6737–6743.[68] T.E. Colonna, L. Huynh, D.M. Fambrough, Journal of Biological Chemistry 272
(1997) 12366–12372.[69] K.J. Swaedner, E. Rael, Genomics 68 (2000) 41–45.[70] K. Geering, Journal of Bioenergetics and Biomembranes 37 (2005) 387–392.
[71] P.Teriete,C.M.Franzin,J.Choi,F.M.Marassi,Biochemistry46(2007)6774–6783.[72] C. Li, A. Grosdidier, G. Crambert, J.-D. Horisberger, O. Michielin, K. Geering,
Journal of Biological Chemistry 279 (2004) 38895–38902.[73] I.C. Meij, J.B. Koenderink, H. van Bokhoven, K.F.H. Assink, W.T. Groenestege,
J.J.H.H.M. De Pont, R.J.M. Bindels, L.A.H. Monnens, L.P.W.J. van den Heuvel,N.V.A.M. Knoers, Nature Genetics 26 (2000) 265–266.
[74] H.X. Pu, F. Cluzeaud, R. Goldshleger, S.J.D. Karlish, N. Farman, R. Blostein, Journal of Biological Chemistry 276 (2001) 20370–20378.
[75] E. Arystarkhova, R.K. Wetzel, N.K. Asinovski, K.J. Swaedner, Journal of Biological Chemistry 274 (1999) 33183–33185.
[76] P. Béguin, G. Crambert, S. Guennoun, H. Garty, J.-D. Horisberger, K. Geering,European Molecular Biology Journal 20 (2001) 3993–4002.
[77] H.K.B. Simmerman, L.R. Jones, Physiological Reviews 78 (1998) 921–947.[78] C.J. Palmer, B.T. Scott, L.R. Jones, Journal of Biological Chemistry 266 (1991)
11126–11130.[79] J. Zamoon, A. Mascioni, D.D. Thomas, G. Vegalia, Biophysical Journal 85 (2003)
2589–2598.[80] W. Fuller, P. Eaton, J.R. Bell, M.J. Shattock, Federation of American Societies for
Experimental Biology Journal 16 (2004) 197–199.
L.D. Faller/ Archives of Biochemistry and Biophysics 476 (2008) 12–21 21