
03 Alteraciones Del Crecimiento
14
PED 3. ALTERACIONES DEL CRECIMIENTO. Clasificación y estudio clínico de las alteraciones del crecimiento más frecuentes. El niño con talla baja: Orientación diagnóstica y actitud terapéutica. Principales cuadros clínicos de hipercrecimiento. La talla es una variable continua que sigue una distribución normal. Se acepta como límite de la normalidad el intervalo situado entre 2 DS (desviaciones estándar), que se corresponden con percentiles 3 y 97. Estos límites son orientativos y deben considerarse inicialmente patológicos por encima o debajo de 3DS; valores entre 2 y 3 DS son casos límite que deben evaluarse cuidadosamente, valorar la velocidad de crecimiento (incrementos de altura en intervalos regulares de tiempo), y correlación con talla media de los padres (mejor índice para estimar potencial genético de crecimiento individual). Si la velocidad de crecimiento se encuentra entre los percentiles 25 y 75, la talla se mantendrá en el mismo nivel; si se sitúa entre los percentiles 10 y 25 ó 75 y 90, probablemente el crecimiento y la talla finales se desviarán de los límites normales; si es inferior al percentil 10 o superior a 90, es hipo o hipercrecimiento. 1.ETIOPATOGENIA DE LAS ALTERACIONES DEL CRECIMIENTO: Ruptura del equilibrio entre el conjunto de los factores genéticos y ambientales que regulan la multiplicación y el crecimiento celular. Se puede hablar de 4 grupos etiopatogénicos:Hipo/hipercrecimietos por alteración de los factores determinantes Ídem con los factores permisivos Ídem con los factores reguladores Ídem con los factores realizadores 1.1.Hipocrecimientos. Es útil dividirlos en dos grupos: Variantes normales: el patrón de crecimiento de estos niños se sitúan en los límites extremos del rango de variación normal y la talla baja no se acompaña de ninguna otra alteración. Diferenciamos 2 formas clínicas evolutivas distintas.
-
Upload
kerly-madeleine -
Category
Documents
-
view
12 -
download
5
Transcript of 03 Alteraciones Del Crecimiento

PED 3. ALTERACIONES DEL CRECIMIENTO. Clasificación y estudio clínico de las alteraciones del crecimiento más
frecuentes. El niño con talla baja: Orientación diagnóstica y actitud terapéutica. Principales cuadros clínicos de hipercrecimiento.
La talla es una variable continua que sigue una distribución normal. Se acepta como límite de la normalidad el intervalo situado entre 2 DS (desviaciones estándar), que se corresponden con percentiles 3 y 97. Estos límites son orientativos y deben considerarse inicialmente patológicos por encima o debajo de 3DS; valores entre 2 y 3 DS son casos límite que deben evaluarse cuidadosamente, valorar la velocidad de crecimiento (incrementos de altura en intervalos regulares de tiempo), y correlación con talla media de los padres (mejor índice para estimar potencial genético de crecimiento individual).Si la velocidad de crecimiento se encuentra entre los percentiles 25 y 75, la talla se mantendrá en el mismo nivel; si se sitúa entre los percentiles 10 y 25 ó 75 y 90, probablemente el crecimiento y la talla finales se desviarán de los límites normales; si es inferior al percentil 10 o superior a 90, es hipo o hipercrecimiento.1.ETIOPATOGENIA DE LAS ALTERACIONES DEL CRECIMIENTO:Ruptura del equilibrio entre el conjunto de los factores genéticos y ambientales que regulan la multiplicación y el crecimiento celular. Se puede hablar de 4 grupos etiopatogénicos:Hipo/hipercrecimietos por alteración de los factores determinantes Ídem con los factores permisivos Ídem con los factores reguladores Ídem con los factores realizadores1.1.Hipocrecimientos. Es útil dividirlos en dos grupos:Variantes normales: el patrón de crecimiento de estos niños se sitúan en los límites extremos del rango de variación normal y la talla baja no se acompaña de ninguna otra alteración. Diferenciamos 2 formas clínicas evolutivas distintas.1-Talla baja familiar: muy frecuente en clínica. Al nacimiento tienen peso y talla prácticamente normales, y a lo largo de la infancia se mantienen ligeramente por debajo de la media (entre -2 y -3 DS). La maduración ósea no está retrasada y la talla final se sitúa por debajo del percentil 3.Habitualmente se encuentran entre los familiares próximos individuos con talla baja, ya que este hipocrecimiento se transmite, como la talla normal, como un carácter poligénico. Criterios diagnósticos: Historia familiar positiva Talla baja para la edad cronológica, pero normal relación con talla media de los padres. velocidad de crecimiento estable Ausencia en retraso de maduración ósea y de alteraciones orgánicas.La patogenia no se conoce bien, pero los ensayos terapéuticos con GH biosintética no modificaron la talla final. Se realizaron ensayos con GH sintética, rhGH, pero la talla final no se vio modificada. También se pensó en que existía una resistencia del cartílago a la acción de la GH, pero no se ha demostrado que el acortamiento de la talla se deba a esto.La talla final debe ser considerada como una variación fisiológica del patrón de crecimiento de origen genético.

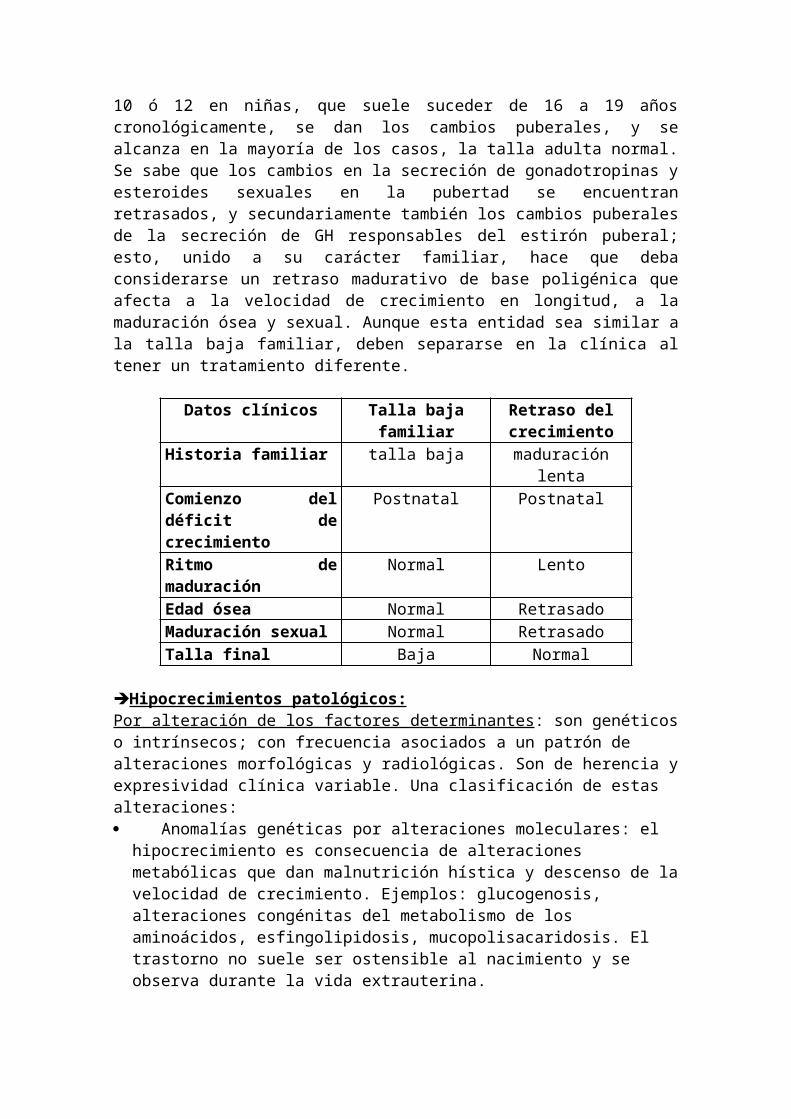
2- Retraso constitucional del crecimiento y de la pubertad: es en realidad una situación fisiológica que transitoriamente simula una alteración definitiva del crecimiento; tiene un patrón de crecimiento retardado, un peso y talla normales al nacer, pero a partir de los 6-12 meses enlentecen el ritmo de crecimiento y de maduración ósea situándose en los percentiles inferiores; a partir de los 4 ó 5 años crecen a ritmo normal y se sitúan en uno de los canales normales o ligeramente por debajo para su edad cronológica, pero con talla y velocidad adelantados para la edad ósea, que suele estar retrasado de 2 a 3 años. A los 12 ó 13 años, no se produce aceleración del crecimiento de la pubertad, ni se inicia maduración sexual ni avanza el desarrollo esquelético, y la talla se desvía claramente de los valores normales para la edad cronológica, finalmente, al alcanzar la edad ósea los 12 ó 13 años en los niños, los 10 ó 12 en niñas, que suele suceder de 16 a 19 años cronológicamente, se dan los cambios puberales, y se alcanza en la mayoría de los casos, la talla adulta normal.Se sabe que los cambios en la secreción de gonadotropinas y esteroides sexuales en la pubertad se encuentran retrasados, y secundariamente también los cambios puberales de la secreción de GH responsables del estirón puberal; esto, unido a su carácter familiar, hace que deba considerarse un retraso madurativo de base poligénica que afecta a la velocidad de crecimiento en longitud, a la maduración ósea y sexual. Aunque esta entidad sea similar a la talla baja familiar, deben separarse en la clínica al tener un tratamiento diferente.
Datos clínicos Talla baja familiar Retraso del crecimiento
Historia familiar talla baja maduración lentaComienzo del déficit de crecimiento
Postnatal Postnatal
Ritmo de maduración Normal LentoEdad ósea Normal RetrasadoMaduración sexual Normal RetrasadoTalla final Baja Normal
Hipocrecimientos patológicos:Por alteración de los factores determinantes: son genéticos o intrínsecos; con frecuencia asociados a un patrón de alteraciones morfológicas y radiológicas. Son de herencia y expresividad clínica variable. Una clasificación de estas alteraciones: Anomalías genéticas por alteraciones moleculares: el hipocrecimiento es
consecuencia de alteraciones metabólicas que dan malnutrición hística y descenso de la velocidad de crecimiento. Ejemplos: glucogenosis, alteraciones congénitas del metabolismo de los aminoácidos, esfingolipidosis, mucopolisacaridosis. El trastorno no suele ser ostensible al nacimiento y se observa durante la vida extrauterina.
Alteraciones cromosómicas: casi todas la cromosopatías, exceptuando Klinefelter y XYY, cursan con dismorfia, deficiencia mental e hipocrecimiento, destacando el síndrome de Down y el de Turner, así como en otras traslocaciones y anomalías estructurales.
Exposición del embrión a radiaciones, sustancias radiomiméticas, tóxicos o agentes infecciosos: hipocrecimiento severo, como en la exposición a alcohol. Se suele manifestar con hipocrecimiento severo, microcefalia, microftalmía y retraso mental. Pueden asociarse malformaciones genitales y de las extremidades.
Otros síndromes dismórficos de etiología no aclarada: con alteración morfológica de la cara, que les confiere gran parecido entre sí. Se asocian otras malformaciones como envejecimiento prematuro, alteraciones cutáneas y deformidades de las extremidades.
Hipocrecimiento por alteración de los factores permisivos: son por alteraciones sistémicas o del medio interno; generalmente son de moderada intensidad, postnatales, armónicos, edad ósea y velocidad normal, y no presentan dismorfias llamativas; si bien, la gravedad depende de la fecha de comienzo y duración de la enfermedad que la origina: son siempre muy graves en las etapas iniciales del desarrollo cuando el incremento de masa y la dinámica morfogenética tienen un ritmo muy acelerado.os mecanismos fisiopatológicos que intervienen son: Malnutrición : con efecto directo sobre el metabolismo celular, y además repercute negativamente sobre la regulación paracrina del crecimiento; hay un estado reversible de resistencia a GH y una bajada de receptores, con alteración del proceso de transducción de la señal y de la respuesta intracelular; esto origina una disminución de IGF-1 acompañada de descenso de sus proteínas transportadoras con descenso de IGFBP-3 y aumento de IGFBP-1; se trata de un mecanismo adaptativo a la malnutrición, frenando el crecimiento y favoreciendo el catabolismo proteico, redistribuyendo los nutrientes a los órganos más importantes.Los nutrientes esenciales se clasifican en tipo I, cuya repercusión sobre el crecimiento es tardía e inconstante porque cumplen funciones específicas en determinadas vías metabólicas y su carencia sólo afecta a éstas; mientras que los tipo II, su déficit conduce a la detención del crecimiento, ya que forma parte de vías metabólicas esenciales de todas las vías del organismo. A este grupo de nutrientes pertenecen el Zn, P, Mg y algunos aminoácidos esenciales. El aporte mínimo de energía es de 18-20 kcal/kg/día pero además es necesario el aporte de cantidades mínimas y equilibradas de nutrientes esenciales. Incremento de los requerimientos : como ocurre en las infecciones, asma bronquial, y la afección respiratoria grave de la fibrosis quística. El incremento produce un desequilibrio entre los ingresos y las necesidades (que aumentan en los ejemplos citados) Modificación de la biodisponibilidad de los factores de crecimiento : como en insuficiencia renal crónica, con reducción del aclaramiento de las proteínas transportadoras de la IGF-1, con descenso de la IGF-1 libre, que es la realmente activa (ésta filtra y se pierde al no reabsorberse. La que está unida a IGFBP1 e IGFBP3 no se pierde pero es la forma inactiva de la hormona) Efecto desfavorable de algunos tratamientos farmacológicos : como con corticoesteroides en el tratamiento del asma, en la enfermedad inflamatoria intestinal o en la artritis crónica juvenil. La administración aguda de corticosteroides aumenta la producción de GH pero la administración disminuye la síntesis de GH y de IGF-I, alterando la síntesis de matriz extracelular por los condorcitos en crecimiento.Hipocrecimientos por alteración de los factores reguladores: son hipocrecimientos neuroendocrinos; por alteración de secreción de uno o varios neurotransmisores u hormonas, lo que da una disminución de la velocidad de crecimiento y retraso de maduración ósea, con hipocrecimiento intenso, comienzo postnatal, armónico, comienza con crecimiento normal, con deterioro progresivo de la talla.La alteración puede consistir en descenso de secreción de hormonas que favorecen el crecimiento (GH, tiroxina, insulina), exceso de hormonas con acción catabólica (corticoesteroides) o modificaciones del ritmo de secreción de los factores que regulan

velocidad de crecimiento, morfogénesis y maduración. El fallo se puede producir en cualquier punto: SNChipotálamo-hipófisisglándulas periféricasórgano diana.El patrón de crecimiento suele ser característico: Crecimiento normal durante un periodo de tiempo variable y después disminuye la velocidad de crecimiento y se deteriora progresivamente la talla. Los cuadros más importantes se ven en el capítulo de Endocrinología. Ver figura 1.Hipocrecimientos por alteración de los factores realizadores: el responsable fundamental es el esqueleto, y dentro de él, el cartílago fisario o de crecimiento. Se caracterizan por ser disarmónicos, de comienzo prenatal, con alteraciones radiológicas y maduración normal. Los mecanismos patogénicos pueden reducirse a 2:I.- displasias: alteración genética que perturba la capacidad de multiplicación y crecimiento del condrocito o su respuesta a estímulos.II.- distrofias: lesión “secundaria” por insulto tóxico, carencial, infecioso, o de otro tipo, de un cartílago de crecimiento genéticamente normal.
Existen muchas clasificaciones de estas patologías, siendo la más útil la de Bierich, que utiliza criterios morfológicos, y los divide según los segmentos corporales afectados en:
Hipocrecimientos de miembros cortosCon acortamiento proximal (RIZOMÉLICOS): Ejemplo: Acondroplasiacon acortamiento medial y ocasionalmente distal (MESOMÉLICOS)
como por ejemplo las displasias mesomélicas (muy infrecuentes)Hipocrecimientos del tronco: acortamiento del tronco que hace que los
miembros parezcan mucho más largos. Existe una platispondilia (descenso de la altura de las vértebras) que se asocia frecuentemente a deformidad de uno o varios cuerpos vertebrales. Hipocrecimientos con afectación del tronco y de los miembros: Talla baja y deformidades, como en la displasia espóndilo-epifisaria. Hipocrecimientos con alteración del crecimiento como síntoma secundario e inconstante, como en las distrofias óseas, fragilidades óseas constitutivas u osteopatías condensantes.2.METÓDICA DE ESTUDIO DE LOS HIPOCRECIMIENTOSPara encasillar al niño primero en retraso patológico o variante normal, basta con valorar su patrón de crecimiento por medio de la historia clínica, exploración física y patrón de crecimiento.2.1.Evaluación clínica:a)Historia clínica: en la historia familiar es importante conocer la talla adulta y la edad en que alcanzaron la madurez sexual los familiares próximos, así como presencia de alteraciones metabólicas, enfermedades hereditarias o retrasos del crecimiento. Se debe precisar la edad de comienzo del hipocrecimiento. Anamnesis cuidadosa encaminada a descubrir patología durante el embarazo, el parto o la presencia de síntomas reveladores de los distintos sistemas orgánicos: dieta, apetito, tolerancia digestiva, procesos infecciosos, enfermedades renales, respiratorias, cianosis u otros síntomas de enfermedades cardiovasculares y el tipo de medicamentos dados. También fijarse en maduración de los distintos sistemas orgánicos y la adquisición de las principales funciones: dentición, lenguaje, desarrollo estático, motor, maduración intelectual y psicoafectiva.b) Exploración clínica: estudio antropométrico (peso, talla, relaciones segmentarias, desarrollo sexual y estado de nutrición), añadido a la exploración normal del niño

(encaminada a buscar pequeñas anomalías que se asocien a los cuadros de hipocrecimiento)c) Valoración del patrón de crecimiento: valorar velocidad de crecimiento y talla, corregida en función de talla media de los padres. Las posibilidades son:1. Talla se encuentra dentro de los límites de variación fisiológica (> de P3 o de -2 DS),
realizar predicción de talla adulta, con revisión anual para comprobar velocidad de crecimiento normal y que el niño sigue la senda prevista.
2. Talla entre -2 y -3 DS: Si la velocidad de crecimiento ha sido > P10 en último año para la edad cronológica, actuar igual que en el caso anterior; pero si la velocidad de crecimiento es baja, se realizará una exploración completa. Si se desconoce la velocidad de crecimiento se controlará a los 6-8 meses: si la velocidad es normal, se harán las revisiones anuales como en el caso primero.
3. Si talla inferior en más de 3 DS a la media o velocidad de crecimiento < a P 10: estudio completo.
2.2.Orientación diagnóstica inicial: es un “diagnóstico provisional” que nos permitirá orientar el diagnóstico final y las pruebas que se realizarán al niño (las necesarias). Valorando: gravedad de hipocrecimiento, comienzo pre o postnatal, armónico o disarmónico, existencia o no de anomalías asociadas, relación entre edad de crecimiento, edad cronológica y edad ósea, y la velocidad de crecimiento.2.3.Pruebas diagnósticas complementarias: Dirigidas a hallar el diagnóstico etiopatológico final.1. De laboratorio : análisis de orina (pH, densidad, albúmina, sedimento citológico,
estudio de la capacidad de concentración y urocultivo), de sangre (hemograma, VSG, pH, gases, ionograma, urea, fosfatasa alcalina, colesterol, proteínas, Zn y Fe) o heces (aspecto, consistencia, peso, grasa total en 24h, presencia de parásitos).
2. Estudio radiológico : radiografía de carpo y mano izda. para determinar edad ósea, y radiografía lateral de cráneo, para valorar silla turca, calcificaciones o anomalías. Se complementa con un TAC si hay sospecha de alteración hipotálamo-hipofisaria.3. Determinaciones hormonales : función tiroidea (T3, T4 y TSH); GH: como ésta se libera de forma episódica, hay que recurrir a estímulos farmacológicos, lo cual tampoco es fisiológico, y por tanto, hay otras técnicas como secreción espontánea durante 24 h, y respuesta a inyección de GHRH, que determina si el problema es hipotalámico o hipofisario; también se pueden determinar las proteínas transportadoras (GHBP, IGFBP-3 e IGFBP-1) y la IGF-1 e IGF-2.4. Exploraciones especiales : como determinación de otras hormonas, cariotipo, estudio radiológico del esqueleto o biopsias.3.ACTITUD TERAPÉUTICA.3.1.Variantes normales:I) Talla baja familiar: no se ha podido demostrar que el tratamiento de rhGH (GH biosintética) modifique la talla definitiva.II) Retraso constitucional del crecimiento y de la pubertad: en la mayoría de los casos no necesita tratamiento: el crecimiento se acelera al final de la pubertad y la talla es normal. Sobretodo en varones, la talla baja y el retraso en la aparición de los caracteres sexuales secundarios puede crear problemas graves de adaptación psicosocial; por ello, en estos casos, la oxandrolona (derivado androgénico con menos capacidad virilizante que la testosterona) produce una aceleración de la velocidad de crecimiento, sin modificar la relación entre talla y maduración ósea, y sin efectos secundarios. La dosis es de 0,0625mg/kg/día durante 3 meses.b) Cuadros patológicos:

I) Hipocrecimientos por alteración de los factores determinantes: en la actualidad sin tratamiento eficaz (sólo se puede realizar cirugía ortopédica en casos aislados), a excepción del S. Turner y sus variantes, que pueden beneficiarse de un tratamiento hormonal (con oxandrolona 0,0625mg/kg/día y/o con hGH a la dosis de 1U(0,375mg)/kg/semana distribuída en 7 dosis)II) Hipocrecimientos por alteración de los factores permisivos: la única terapéutica eficaz es el tratamiento de la enfermedad primaria, que debe hacerse lo antes posible, en el periodo en que la alteración de crecimiento es aún reversible. El tratamiento hormonal no está justificado (excepto algunos casos de niños con insuficiencia renal)III) Hipocrecimiento por alteración de los factores reguladores: son los más accesibles al tratamiento; los “hipopituitarismos” deben tratarse con GH tan pronto como se diagnostiquen, porque la talla final guarda una estrecha relación con la precocidad con que se inicie el tratamiento; en los déficit múltiples hay que asociar las demás hormonas deficitarias, y hacer un control de la función tiroidea, ya que un % de las formas aisladas, al año de tratamiento, desarrollan un cuadro de hipotiroidismo, y será necesario añadir T4 al tratamiento con GH. La dosis recomendada de GH es 0.5-0,7U/kg/semana. Otra posibilidad será dar GHRH cuando el déficit se encuentre en el hipotálamo, que suele suceder en un 60-70% de los pacientes.El hipotiroidismo es una causa frecuente de talla baja, que puede ser la única manifestación, junto al retraso de maduración ósea de las formas leves de enfermedad: su tratamiento es la L-tiroxina.En el síndrome de Cushing iatrogénico, el tratamiento es la supresión o reducción de la dosis esteroidea; cuando está originado por un tumor suprarrenal o un tumor ectópico secretor de ACTH, la conducta terapéutica es la extirpación; en las situaciones de hiperplasia suprarrenal bilateral, el defecto primario es un microadenoma o tumor hipofisario, y su tratamiento será quirúrgico.En los niños con “pubertad precoz”, los cuales muestran, aparte de la maduración sexual acelerada, una aceleración del crecimiento en longitud y una maduración ósea aún más acelerada, que conduce en una 2ª fase a una disociación entre la evolución de la maduración ósea (que sigue acelerándose) y el crecimiento estatural (que se estabiliza). Finalmente, las epífisis se sueldan y la talla final queda reducida; por eso, en estos niños se han introducido para su tratamiento análogos de LHRH, que pueden mejorar el pronóstico de talla final.IV) Hipocrecimientos por alteración de los factores realizadores: También llamadas osteocondrodisplasias. Se han beneficiado por técnicas de alargamiento quirúrgico de los miembros, que corrige la talla y la proporcionalidad de los distintos segmentos corporales; sus indicaciones serán tallas bajas disarmónicas, dismetrías que superen 3 ó 4 cm, y excepcionalmente las tallas bajas proporcionadas en las que el paciente tenga estatura muy baja que le plantee dificultades de integración social. En la actualidad este procedimiento quirúrgico es capaz de alargar la talla hasta 20-24cm. Se trata de un proceso largo (1 a 3 años) que debe estar muy bien planificado.4.HIPERCRECIMIENTOS. CLASIFICACIÓN Y ESTUDIO CLÍNICO.Es un problema mucho menos frecuente; además, factores socioculturales han contribuido a que la talla alta sea considerada un índice de buena salud, por lo que es excepcional que los padres del niño acudan a consulta por esto, a no ser que sea un gigantismo dismórfico. Aparte de esto encontramos cuadros patológicos en los que la talla alta son una manifestación de enfermedades graves, síndromes complejos que

asocian crecimiento excesivo, visceromegalia, tendencia a neoplasias malignas o enfermedades sistémicas.4.1. Variantes normales:1-Talla alta familiar: determinado poligénicamente, y representa el límite superior de la distribución normal de la talla. Tienen talla alta en toda la infancia, con ritmo de maduración normal, finalizan su crecimiento a edad habitual y alcanzan una estatura superior a +2 DS por encima de la media; en la historia familiar ocurre lo mismo, y la talla media de los padres suele ser superior al promedio. Clínicamente son niños altos, sin alteraciones morfológicas asociadas, proporcionados, y las únicas manifestaciones patológicas son las derivadas de sus dificultades para la adaptación social de algunos niños. Esto justifica los ensayos terapéuticos para acelerar la maduración ósea y limitar la talla adulta. En las niñas se han usado estrógenos, y en los niños, testosterona, consiguiéndose reducciones en la talla de entre 3,2 y 4,6cm, si bien, faltan aún controles suficientes, y el tratamiento debe preservarse para situaciones con afectación psicológica grave.2-Maduración acelerada: se presenta en límite superior de la variación normal, pero para la maduración, no para la talla. Cuadro familiar caracterizado por velocidad de crecimiento por encima de la media, acompañada de aceleración de maduración ósea y desarrollo puberal, y el crecimiento finaliza a edad temprana: la talla adulta es normal.4.2. Hipercrecimientos patológicos:Hipercrecimiento por alteración de los factores determinantes: son hipercrecimientos dismórficos, en los que la talla adulta se asocia a anomalía morfológicas y a un elevado riesgo de padecer neoplasias de distinta localización. Distinguimos: Anomalías cromosómicas: lo habitual es que curse con hipocrecimiento, pero existen 3 anomalías numéricas de los cromosomas sexuales (aneuploidías 47XXY, 47XYY y 48XXYY) con talla alta y hábitos eunucoides. Sólo una alteración de los autosomas cursa con hipercrecimiento: la trisomía de los brazos cortos del cromosoma 20. Otros síndromes dismórficos: determinados genéticamente, acompañados de anomalías fenotípicas, esqueléticas o viscerales, como el S. de Beckwitt-Wiedemann (cursa con macrosomía, macroglosia, onfalocele, y visceromegalia), el de Weaver ( que cursa con aceleración de la maduración ósea), y el síndrome de Berdinelli o lipodistrofia congénita (que cursa con hipertrofia muscular, hematomegalia y retraso mental).Hipercrecimientos por alteración de los factores permisivos: como el organismo posee un control adaptativo del aporte energético a las necesidades celulares en cada momento, es excepcional que se produzca un crecimiento excesivo por este motivo, a no ser que existan anomalías del S.N. o del aparato circulatorio, que altere la distribución de los nutrientes y secundariamente la regulación del índice mitótico. El crecimiento excesivo suele afectar a una parte del cuerpo dando lugar a gigantismos parciales (hiperplasias regionales) con escasa o nula repercusión sobre la talla; con frecuencia se acompañan de angiomas y otras anomalías vasculares, manchas cutáneas y alteraciones neurológicas; los más importantes son la hemihipertrofia congénita, el síndrome de Klippel-Trenaunay-Weber ,el linfedema congénito, el síndrome de Mafucci, el síndrome de Sturge-Weber y la neurofibromatosis de Recklinhausen.Hipercrecimiento por alteración de los factores reguladores: son los hipercrecimientos neuroendocrinos: los cuadros más importantes son: Gigantismo hipofisario: casi siempre por adenoma secretor de GH (raras veces se debe a hiperplasia de la hipófisis). Hipercrecimiento armónico y poco a poco se van haciendo ostensibles los rasgos acromegaloides (se alargan la nariz y mentón, y los pies

y manos adquieren un tamaño excesivo), la silla turca suele estar agrandada, y es frecuente, que por compresión de las zonas vecinas, se origine un déficit de las restantes hormonas hipofisarias, apareciendo síntomas de hipofunción; hay hiperglucemia resistente a insulina. El diagnóstico se basa en el cuadro clínico, ver anomalías en RX/TAC y la hiperglucemia resistente a insulina. Gigantismo cerebral o síndrome de Sotos: de etiología desconocida, con talla alta, dismorfia facial y retraso mental. Se inicia intraútero y al nacer son ya grandes, con talla y peso superior al P97. Ritmo de crecimiento acelerado con maduración ósea avanzada. Se diagnostica por datos somatométricos, y el fenotipo característico: cráneo dolicocéfalo, frente prominente, hendidura palpebral inclinada de forma antimongoloide, paladar ojival y con frecuencia asimetría facial. El hipercrecimiento podría deberse a anomalías del córtex o del hipotálamo, ya que el ventrículo cerebral suele estar dilatado. Hipertiroidismo: es poco frecuente, y su repercusión sobre el crecimiento es menos importante que el resto de la clínica. Se caracteriza por aceleración de la maduración ósea junto a déficit de peso en relación con la talla. Clínica de hipertiroidismo. Síndrome de hiperandrogenismo: hipersecreción de hormonas con actividad androgénica, que origina una aceleración del crecimiento en longitud y de la maduración ósea, además de cambios en la esfera sexual, como virilización de la niña y macrogenitosomía en el varón. La etiología más frecuente es la hiperplasia suprarrenal congénita, neoplasias suprarrenales, el tumor de cél. Intersticiales en el niño y el arrenoblastoma en la niña.Existe siempre una acción más intensa sobre la maduración ósea que sobre el crecimiento en longitud, por lo que aunque en un examen único el paciente tenga una talla superior a la correspondiente a su edad, al edad esquelética siempre por delante de la estatural, el periodo de crecimiento se acorta y la talla final es baja; se trata por tanto de un hipocrecimiento que pasa por un periodo transitorio de crecimiento acelerado.Hipercrecimientos por alteración de los factores realizadores: se trata de enfermedades hereditarias del tejido conjuntivo y derivados de él, que intervienen como órganos efectores en la realización del patrón de crecimiento. Los cuadros más importantes son: Síndromes de aracnodactilia: son hasta 6 síndromes distintos todos ellos con fenotipo o hábito marfanoide: talla alta, facies alargada, miembros gráciles y dedos largo y finos. Destacan la homocistinuria y los síndromes de Marfán y de Achard. Displasia epifisaria hemimélica: sólamente esta displasia cursa con hipercrecimiento con gigantismo parcial. La alteración patológica más importante es un crecimiento exce-sivo y fragmentación de la epífisis distal del fémur y de la tibia en un miembro inferior.


















