







Idiomas
Páginas
Jurídico


UNIVERSIDAD NACIONAL AUTÓNOMA DE NICARAGUA
UNAN-León
FACULTAD DE CIENCIASY TECNOLOGÍA DEPARTAMENTO DE QUÍMICA
“VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA
CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO”
TESIS PARA OPTAR AL TÍTULO DE LICENCIADO EN QUÍMICA PRESENTADA POR: Br. FERNANDO GALEANO CARRION CATEDRÁTICO GUÍA: MSc. FABIO JOSÉ PALLAVICCINI NARVÁEZ Lic. MANUEL ANTONIO VANEGAS CARVAJAL
LEÓN, NICARAGUA, AGOSTO DEL 2009

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
INDICE I. RESUMEN, 1 II. INTRODUCCIÓN, 2 III. OBJETIVOS, 3 III.1 Objetivo General, 3 III.2 Objetivos Específicos, 3 IV. MARCO TEÓRICO, 4 IV.1 Acetaminofén, 4 IV.2 Tamadol HCl, 6 IV.3 Cromatografía Líquida de Alta Eficiencia (HPLC), 7 IV.3.1 Causas que afectan un análisis por HPLC, 9 IV.3.2 Razones por las cuales se utiliza HPLC, 11 IV.3.3 Aplicaciones de HPLC, 11 IV.3.4 Limitaciones, 11 IV.3.5 Campos de uso de HPLC, 11 IV.3.6 Diagrama básico de un sistema de HPLC, 12 IV.4 Validación de Métodos Analíticos, 18 IV.4.1 Jerarquía de la metodología, 18 IV.4.2 Proceso de validación, 20 IV.4.3 Validación general, 20 IV.4.4 Validación para uso específico, 22 IV.4.5 Proceso general para la metodología, 23 IV.4.6 El método de adición patrón, 24 IV.4.6.1 Adición patrón, 25 IV.4.7 Criterios de Eficiencia, 25 IV.4.7.1 Precisión, 26 IV.4.7.2 Exactitud o Bias (sesgo), 26 IV.4.7.3 Límite de detección, 26 IV.4.7.4 Límite de cuantificación, 27 IV.4.7.5 Linealidad, 28 IV.4.7.6 Rango, 28 IV.4.7.7 Selectividad, 28 IV.4.7.8 Sensibilidad, 28 IV.5 Aproximación ISO para el cálculo de la incertidumbre, 29 V. PARTE EXPERIMENTAL, 31 V.1 Equipos y Materiales, 31 V.2 Reactivos, 32 V.3 Procedimientos, 33 V.3.1 Condiciones cromatográficas, 33 V.3.2 Preparación de la fase móvil, 33 V.3.3 Preparación de la curva de calibración normal de acetaminofén y tramadol, 33 V.3.4 Preparación de la muestra, 34 V.3.5 Selectividad del método, 34 V.3.6 Influencia del placebo en la curva de calibración normal, 34 V.3.7 Idoneidad del Sistema, 35 V.3.8 Linealidad del sistema y del método cromatográfico, 35
V.3.9 Precisión del sistema y del método cromatográfico, 36 V.3.10 Exactitud del método, 37 V.3.11 Efecto de matriz, 38 V.3.12 Límite de detección y límite de cuantificación, 38 V.3.13 Repetibilidad de las curvas de calibración normal, 39 V.3.14 Aplicación del método y estimación de la incertidumbre asociada a la cuantificación, 39 VI. RESULTADOS Y DISCUSIÓN, 40 VI.1 Selectividad, 40 VI.2 Influencia del placebo, 43 VI.3 Idoneidad del sistema, 45 VI.4 Linealidad del sistema y linealidad del método, 46 VI.5 Precisión del sistema y precisión del método, 50 VI.6 Exactitud del método, 52 VI.7 Efecto de matriz y exactitud del método, 56 VI.8 Límite de detección y límite de cuantificación, 59 VI.9 Repetibilidad de las curvas de calibración normal, 61 VI.10 Aplicación del método y estimación de la incertidumbre asociada a la cuantificación de acetaminofén y tramadol en tabletas de ingeril compuesto, 63 VI.10.1 Modelo matemático, 63 VI.10.2 Identificación de las fuentes de incertidumbre, 63 VI.10.3 Cuantificación de los componentes de incertidumbre, 64 VI.10.4 Cálculo de los coeficientes de sensibilidad, 65 VI.10.5 Estimación de la incertidumbre combinada, 67 VI.10.6 Estimación de lo incertidumbre expandida, 67 VI.10.7 Cuantificación y estimación de la incertidumbre asociada a la cuantificación de tramadol en tabletas de ingeril compuesto, 68 VII. CONCLUSIONES, 70 VIII. RECOMENDACIONES, 72 IX. BIBLIOGRAFÍA, 73 XANEXOS, 76 X.1 Datos completos de la curva de calibración normal sin placebo, 76 X.2 Datos completos de la curva de calibración normal cargada con placebo, 77 X.3 Flujograma del proceso analítico, 78

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
1
I. RESUMEN En el presente trabajo se llevo a cabo la validación de un método analítico por HPLC para
la cuantificación de acetaminofen y tramadol en tabletas de Ingeril Compuesto, la cual es
una nueva formulación de los laboratorios CEGUEL, en donde se llevaron a cabo todas las
actividades de adecuación y optimización de las condiciones cromatográficas para de esta
manera lograr el desarrollo de una metodología adecuada para la cuantificación de
acetaminofen y tramadol los cuales son los principios activos de la tableta de Ingeril
Compuesto.
Con el propósito de realizar la validación de esta nueva metodología primeramente se
evaluó la selectividad del método y se analizó la influencia que ejerce el placebo en la
curva de calibración normal, los resultados mostraron que el método es selectivo para los
propósitos previstos y que el placebo no provoca ningún efecto en la curva de calibración
normal, seguidamente se realizó el estudio de la idoneidad del sistema observándose que el
método cumple con los criterios de aceptación propuestos por la USP, posteriormente se
evaluaron los parámetros de validación como: Linealidad del sistema y del método para los
cuales se obtuvieron valores cercanos a la unidad ≥ 0.999, Precisión del sistema y del
método obteniéndose valores de %RSD < 1% es decir tiene muy buena precisión. La
exactitud del método se evaluó por medio del porcentaje de recuperación (%R), los cuales
fueron de 103.20 ± 1.50 y de 105.37 ± 2.01 para acetaminofen y tramadol respectivamente
es decir el método es exacto, se llevo a cabo el estudio del efecto de matriz relacionándolo
con la exactitud del método aquí se encontró que no hay efecto de matriz y se confirmó la
exactitud del método, luego se determinaron el límite de detección obteniéndose valores de
1.80 μg/mL y 0.22 μg/mL para acetaminofen y tramadol respectivamente y el límite de
cuantificación 4.47 μg/mL y 0.68 μg/mL para acetaminofen y tramadol respectivamente,
seguidamente se evaluó la repetibilidad de calidad de la curva de calibración normal.
Posteriormente se estimó la incertidumbre asociada a la cuantificación de acetaminofén y
tramadol en tabletas de Ingeril Compuesto, y finalmente se realizó la aplicación del
método, para la cual se realizó la cuantificación de acetaminofén y tramadol en tabletas
formuladas de Ingeril Compuesto (proyecto piloto), encontrándose valores aceptables de
acuerdo a lo especificado en el empaque de la tableta.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
2
II. INTRODUCCIÓN
El Acetaminofén es un medicamento usado como analgésico y antipirético en pacientes
sensibles a la aspirina y que experimentan otras reacciones adversas a la aspirina. Raras
veces induce efectos adversos y suele ser bien tolerado por los pacientes sensibles a la
aspirina [1]. Por otro lado el Tramadol es un medicamento que calma el dolor y actúa
reduciendo los efectos de las endorfinas (moléculas que intervienen en la transmisión del
dolor) que se encuentran en el cerebro y en la columna vertebral [2]. La tableta de Ingeril
compuesto es una nueva formulación realizada en los Laboratorios CEGUEL, la cual es un
comprimido que contiene como principios activos estas dos sustancias “Acetaminofen y
Tramadol”. En la industria farmacéutica, el desarrollo de métodos de análisis de los
productos farmacéuticos, son necesarios para asegurar y cumplir diversos parámetros de
calidad, seguridad y eficacia de sus productos formulados. Estos métodos están basados en
las farmacopeas internacionales y son adecuados a normas internas propias del laboratorio
para asegurar el control analítico de un producto farmacéutico.
Debido a que en los laboratorios CEGEL se ha llevado a cabo el desarrollo y optimización
de las condiciones cromatográficas del método por HPLC para la cuantificación de
acetaminofen y tramadol en tabletas de Ingeril Compuesto resulta de suma importancia la
evaluación de los parámetros que definen el buen desempeño de este método para asegurar
tanto la calidad del producto elaborado así como también la salud de la toda la población.
En vista de que este es un método adaptado o desarrollado recientemente, no se encuentran
antecedentes de estudios realizados en Nicaragua, sin embargo se encuentran antecedentes
de estudios realizados respecto a la validación de un método de análisis cuantitativo de
acetaminofén por HPLC [1], por otro lado se han encontrados publicaciones internacionales
de estudios realizados pero por separados de acetaminofén [2] y tramadol en capsulas por
cromatografía de capa fina de alta eficiencia [3].
En el presente trabajo se llevo a cabo la validación del método por HPLC utilizado en los
laboratorios CEGUEL en el departamento de Control de Calidad para la determinación
simultánea de acetaminofen y tramadol en tabletas de Ingeril Compuesto.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
3
III. OBJETIVOS
III.1. Objetivo General ♦ Validar el método analítico por HPLC para la cuantificación simultánea de
acetaminofen y tramadol en tabletas de ingeril compuesto
III.2. Objetivos Específicos ♦ Evaluar la selectividad del método.
♦ Analizar la influencia del placebo en la curva de calibración normal.
♦ Evaluar la Idoneidad del Sistema.
♦ Evaluar la linealidad del sistema y del método cromatográfico.
♦ Realizar un estudio de la precisión del sistema y del método cromatográfico.
♦ Evaluar la exactitud del método.
♦ Estudiar el efecto de matriz.
♦ Determinar el límite de detección y el límite de cuantificación.
♦ Evaluar la repetibilidad de calidad de la curva de calibración normal.
♦ Aplicar el método estimar la incertidumbre asociada a la cuantificación.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
4
IV. MARCO TEÓRICO IV.1. Acetaminofén
El acetaminofén, es un fármaco con propiedades analgésicas y antipiréticas. Ejerce su
acción analgésica inhibiendo la síntesis de prostaglandina en el sistema nervioso central (en
el hipotálamo) y a través de una acción periférica al bloquear la generación del impulso
doloroso, esta acción periférica puede deberse también a la inhibición de la síntesis de
prostaglandinas o a la inhibición de otras sustancias que sensibilizan los receptores
dolorosos a la estimulación mecánica o química. Por otro lado ejerce su acción antipirética
al actuar sobre el hipotálamo en el centro regulador de la temperatura, produciendo
vasodilatación con aumento del flujo sanguíneo a través de la piel, con perdida de
sudoración y calor. Estas acciones se traducen en alivio sintomático del dolor y reducción
de la fiebre, respectivamente. No obstante, carece o tiene mínimos efectos antiinflamatorios
ya que es un inhibidor débil de la ciclooxigenasa en presencia de altas concentraciones de
peróxidos que aparecen en las lesiones inflamatorias, a diferencia de ello su efecto
antipirético puede explicarse por su capacidad de inhibir la ciclooxigensa en el encéfalo [2].
El acetaminofén se puede encontrar por sí solo e en combinación con otros medicamentos
para aliviar los síntomas de la gripe, resfriados, dolores de cabeza y osteoartritis. El tomar
demasiado acetaminofén puede causarle problemas hepáticos, daño a los riñones y anemia.
También se ha visto que puede causar los mismos problemas a los bebés. Por lo tanto, es
importante que no se ingiera más de la cantidad recomendada [4].
Figura No. 1. Estructura de la Acetaminofén

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
5
El Acetaminofén en la actualidad es uno de los analgésicos más utilizados al ser bastante
seguro y no interactuar con la gran mayoría de los medicamentos. Las dosis recomendadas
varían según el paciente; siendo las más usuales: 325 a 650 mg cada 4 ó 6 horas o bien
1000 mg cada 6 a 8 horas, sin exceder 4000 mg por día. En usos pediátricos se recomienda
administrar de 10 mg por cada kg de peso cada 4 horas o 15 mg por kg de peso por cada 6
horas [4].
El Acetaminofén por vía oral se absorbe en el tracto digestivo en un periodo que oscila
entre los 30 y los 90 minutos después de una dosis terapéutica, sin embargo, las
preparaciones líquidas pueden absorberse en un periodo de 20 minutos. Se estima que el
inicio de la actividad analgésica ocurre en aproximadamente 30 minutos, los alimentos
reducen la concentración máxima del acetaminofén en un 40 %, por lo cual no se
recomienda su administración con las comidas. Tiene una baja unión a proteínas
plasmáticas y su vida media de eliminación oscila entre 1 y 3 horas. Se metaboliza
principalmente en el hígado, menos del 5% se elimina en forma inalterada y la mayoría se
excreta por la orina en forma de metabolitos. Ninguna de las conjugaciones metabólicas
que sufre el acetaminofén son potencialmente tóxicas; sin embargo el metabolito N-Acetil-
p-benzoquinonimida (NAPQ1), que aunque posee una vida media corta de alrededor de
unos pocos nanosegundos, se une a la membrana celular hepática causando daño sobre la
bicapa lípidica sino es neutralizado por un antioxidante, como el sistema glutatión. A dosis
terapeuticas el acetaminofén es una excelente droga. Pero fuera de rango es peligrosa [4]

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
6
IV.2. Tramadol HCl El Tramadol es un analgésico de acción central de tipo opioide que alivia el dolor actuando
sobre células nerviosas específicas de la médula espinal y del cerebro.
Tras la administración oral de tramadol se absorbe más de un 90% de la dosis,
independientemente de la ingestión simultánea de alimentos. La biodisponibilidad es
aproximadamente del 70%. El tramadol surte efecto de primer paso, en aproximadamente
un 30% de la dosis administrada [5].
Tramadol posee una elevada afinidad tisular, siendo su unión a las proteínas plasmáticas del
20%. La concentración plasmática máxima se alcanza cinco horas después de la
administración.
Tramadol atraviesa las barreras hematoencefálica y placentaria y en unos porcentajes
mínimos (<0.2%) pasa a leche materna.
Independientemente del modo de administración, la semivida es aproximadamente de 6 h.
La metabolización de tramadol tiene lugar en el hígado. Sufre procesos de O-desmetilación
y N-desmetilación así como por la conjugación de los derivados O-desmetilados con ácido
glucurónico. Únicamente el O-desmetiltramadol es farmacológicamente activo. Existen
considerables diferencias cuantitativas interindividuales entre los demás metabolitos. Hasta
ahora se han identificado 11 metabolitos en la orina. Los estudios realizados en animales
han demostrado que O-desmetiltramadol es 2-4 veces más potente que la sustancia de
origen [5].
Tramadol y sus metabolitos se eliminan casi completamente por vía renal (90%).
El perfil farmacocinético de tramadol es lineal dentro del margen de dosificación
terapéutico. La relación entre las concentraciones séricas y el efecto analgésico es dosis-
dependiente [5].

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
7
Figura No. 2. Estructura del Clorhidrato de Tramadol
IV.3. Cromatografía Líquida de Alta Eficiencia (HPLC) La Cromatografía Líquida de Alta Eficiencia (HPLC) es una técnica de separación basada
en una fase estacionaria sólida y una fase móvil liquida. Las separaciones se logran por
procesos de partición, absorción o intercambio iónico según el tipo de fase estacionaria
empleada. Los compuestos que se van analizar en un disolvente adecuado y la mayoría de
las separaciones tiene lugar a temperatura ambiente, la mayoría de los fármacos aun siendo
no volátiles o térmicamente inestables, pueden separarse sin descomposición o sin
necesidad de hacer derivados volátiles [6].
El HPLC consta de un recipiente que contiene la fase móvil, una bomba para forzar el paso
de la fase móvil a través del sistema a alta presión, un inyector para introducir la muestra a
la fase móvil, una columna cromatográfica, un detector y un depósito de correlación de
datos (una computadora o un registrador). Las columnas consta de partículas de fase
estacionaria que permiten un intercambio rápido de los compuestos entre la fase móvil y la
fase estacionaria, reciben y reproducen las señales enviadas por el detector, las
computadoras se emplean para controlar las operaciones y los parámetros cromatográficos
y permiten largos periodos de operación sin necesidad de supervisión [6].

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
8
Los sistemas de bombeo de HPLC administran cantidades exactas de fase móvil desde los
recipientes hasta la columna mediante una tubería y uniones adecuadas para altas presiones,
constan de una o varias bombas reguladoras controladas por computadoras que se
programan para variar la relación entre los compuestos de la fase móvil en corridas
isocráticas controladas con mayor exactitud.
Los compuestos que se van a separar se inyectan en la fase móvil, usando jeringas o
inyectores de espiral. Algunos inyectores automáticos pueden programarse para controlar el
volumen de muestreo, el número de inyecciones y los ciclos de enjuague del espiral, el
intervalo entre las inyecciones y otras variables operativas [6].
Por lo general, las columnas empleadas para las separaciones analíticas tienen diámetros
internos de 2 mm a 5 mm; para la cromatografía preparativa se emplean columnas de
diámetros más grandes. Las columnas pueden calentarse para proporcionar separaciones
más eficaces, pero rara vez se las utiliza a temperaturas por encima de 60° C debido a la
potencial degradación de la fase estacionaria o a la volatilidad de la fase móvil [6].
El PH de la fase móvil, la temperatura, el tipo de ión, la concentración iónica y los
modificadores orgánicos influyen en el equilibrio; estas variables pueden ajustarse para
obtener el grado deseado de separación.
Muchos métodos farmacopéicos de HPLC requieren el uso de detectores, entre los más
usados tenemos:
- Detectores espectrofotometricos
- Detectores de longitud de onda múltiple
- Detectores de red de diodos
- Detectores de refractometría diferencial
- Detectores fluorométricos
- Detectores electroquímicos potenciométricos, voltamétricos o polarográficos
La composición de la fase móvil influye significativamente en el desempeño
cromatográfico y en la resolución de los compuestos de la mezcla que se esta

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
9
cromatografiando. Para el trabajo cuantitativo exacto deben emplearse reactivos de alta
pureza y disolventes orgánicos de grado HPLC. El agua de calidad adecuada debe de tener
una conductividad baja y una absorción UV baja [6].
Los reactivos utilizados con tipos especiales de detectores pueden requerir que se
establezcan tolerancias adicionales para especies que puedan interferir. La composición de
la fase móvil ejerce un efecto mucho mayor que la temperatura en el factor de capacidad,
k´.
IV.3.1 Causas que afectan un análisis por HPLC
Presión Alta
A causa de la Obstrucción de la Columna de HPLC o Guarda Columna por partículas, la
solución es: invertir la Columna y Enjuagar con solvente, teniendo la columna
desconectada del detector. Si esto no funciona reemplace el filtrado a la entrada de la
columna. Si la presión sigue alta reemplace la columna.
Asegurarse que todas las fases móviles se filtren apropiadamente antes que entren a la
bomba de HPLC. También filtre todas las muestras antes de inyectarlas [6].
Pérdida de la Resolución
A causa de la Obstrucción de la Columna de HPLC ó del Guarda Columna por partículas,
la solución es: es necesario filtrar todo antes que se introduzcan las fases móviles en el
sistema de HPLC [6].
Picos Hendidos
A causa de la Obstrucción de la Columna de HPLC o del Guarda Columna por partículas,
la solución es: si es necesario reemplace el fritado de la entrada o la columna, filtrar todo
antes que se introduzcan las fases móviles en el sistema de HPLC [6].

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
10
Variación en los Tiempos de Retención
A causa de aire atrapado en la bomba debido a gases disueltos en fase móvil, la solución
es: que se debe asegurar que la fase móvil esté propiamente y adecuadamente desgasificada [6].
Variaciones de la Línea Base
A causas de Burbujas del aire atrapados en la celda del detector debido a una mala
desgasificación de los solventes de la fase móvil, la solución es: asegurarse que todas las
fases móviles estén debidamente desgasificadas y considerar el uso de un restrictor de la
presión a toma de corriente del detector [6].
Línea Base con mucho Ruido
A causa de aire atrapado en celda del detector o en la bomba, la solución es enjuagar el
sistema y purgar la bomba de HPLC, la solución es: use Solventes desgasificados
adecuadamente para mantener constante la velocidad de flujo de la fase móvil del sistema [6].
Picos Falsos (Detectores Electroquímicos y de Fluorescencia)
A causa de Oxígeno Disuelto, la solución es: desgasificar adecuadamente las fases móviles
para reducir la concentración de oxígeno disuelto o agregar un sistema de filtración al vacío
en línea. Periódicamente chequear el nivel de oxígeno disuelto [6].
Baja ó Ninguna Presión
A causa de trabajar con bombas, sellos ó pistones expuestos por mucho tiempo a partículas
en suspensión en la fase móvil, la solución es: reemplazar los sellos o pistones, si es
necesario [6].

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
11
IV.3.2. Razones por las cuales se utiliza HPLC:
o Gran variedad de configuración operativa.
o Gran cantidad de diferentes sistemas de fase estacionaria.
o Gran numero de fases moviles y modificaciones de ella.
o Amplio campo de uso.
IV.3.3. Aplicaciones de HPLC:
o Para sustancias solubles en un solvente o una mezcla de solventes.
o Sustancias no solubles.
o Sustancias muy polares y/o ionicas.
o Sustancias de alto peso molecular
o Sustancias termolabiles.
IV.3.4. Limitaciones:
o Solubilidad de la sustancia (sustancias poco o no solubles)
o Detectabilidad de la sustancia.
IV.3.5. Campos de uso de HPLC:
o Control de pureza y calidad de producto
o Analisis de medicamentos
o Analisis de sustancias en matrices biologicas.
o Analisis de residuos de plaguicidas.
o Analisis de sustancias toxicas para el medio ambiente.
o Analisis de polimeros sinteticos.
o Separacion y limpieza de biopolímeros.
o Aislamientos de productos sensibles (HPLC preparativa).

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
12
IV.3.6. Diagrama Básico de un Sistema de HPLC
Una instalación HPLC está compuesta de varias unidades especializadas las cuales pueden ser encontradas como entidades separadas o ser integradas dentro de un marco de trabajo común. Un sistema de tuberías de diámetros internos muy pequeños (0.1 mm) asegura la circulación de la fase móvil entre los módulos. Estos tubos de transferencia están hechos de acero inoxidable o de PEEK (Polyether-etherketone), un polímero coloreado y flexible, capaz de resistir los solventes comunes bajo presiones altas (por encima de 350 bares).
Figura No. 3. Esquema de un instrumento HPLC modular.
Un sistema modular permite a los usuarios adaptar la instalación de acuerdo a las
aplicaciones que serán llevadas a cabo. El ensamble vertical de los diferentes módulos
permite una economía de espacio. Aquí el cromatógrafo HP 1200, comprende un auto
muestreador que permite la operación continua y una columna controlada
termostáticamente para mejorar la reproducibilidad de las separaciones.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
13
Eleccion del solvente [6]
Caracteristicas:
• Disponible comercialmente
• Precio
• Pureza y estabilidad. En la actualidad contamos con productos de calidad de pureza
cromatografica. Bajo contenido de impurezas.
• Disolver la muestra
• Miscible con otros solventes para formar mezclas utiles
• No degradar o disolver la fase estacionaria
• Tener baja viscosidad para reducir las caidas de presion
• Ser compatible con el detector utilizado. Transpariencia optica (cuando se usan
detectores UV).
Filtración y desgasificacion de solventes [6]
Los solventes para HPLC, pueden acumular partículas en suspensión que pueden ser
perjudicial a los componentes del sistema HPLC.
Las partículas pueden ocasionar costosos daños a la bomba HPLC, al guardar columnas, y
en general causar desgaste del sistema de HPLC. Los fabricantes de los instrumentos tienen
en cuenta este problema y recomiendan que se filtre y desgasifique los solventes HPLC
antes de usarlos.
En el instante que se abre una nueva botella de solvente para HPLC se expone el interior
del solvente a la atmósfera y empieza a acumular gases disueltos que se encuentran en la
atmósfera.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
14
El nitrógeno disuelto puede producir burbujas en la columna de HPLC y cuando el solvente
entra al detector produce picos falsos y desviaciones de la línea base. El dióxido de
Carbono disuelto algunas veces puede ser la causa de los cambios de pH en el sistema de
solvente.
Métodos de filtración de solventes en HPLC [6]
Hay tres metodos comunes que se utilizan hoy para la filtración previa de los solventes
en HPLC:
Filtro a la entrada del Solvente
Filtración al Vacio
Filtración en linea
Métodos de Desgasificación de Solventes en HPLC [6]
Existen cuatro métodos comunes usados para desgasificar solventes en HPLC previos a su
uso:
Sonificación
Burbujear Helio
Desgasificación electrónica en la línea del flujo
Desgasificación al Vacío en línea
Bombas [6]
Requisitos o aspectos más importantes que debe reunir una bomba o sistema de bombeo:
Debe producir presiones estables hasta 6000 psi.
Mantener el flujo libre de pulsaciones
General intervalos de caudales de flujo (0.1 a 10 mL/min.)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
15
Control y Reproducibilidad del flujo del solvente
Componentes de la bomba resistentes a la corrosion
Programación del Solvente [6]
Existen dos metodos de programación de solventes en HPLC:
Isocrático. Es un término que se utiliza para describir el proceso analítico mediante
el cual no se cambia la composición de la fase móvil.
Gradiente de elución. Es término que se utiliza para describir el proceso mediante el
cual se cambia la composición de la fase móvil.
Sistemas de inyección de muestra [6]
Estos sistemas han variados durante la historia del sistema de HPLC, en un principio se
utilizaba la inyección de la muestra con jeringas de alta presión, las cuales, ya están en
desuso. Hoy se utiliza el sistema de válvulas inyectoras.
Columnas y Fases Estacionaria [6]
Columnas:
El material para la fase estacionaria consiste en particulas esfericas.
Las columnas analiticas son normales de acero o de vidrio con longitudes de 250, 125, 100,
60 milimetros y un diámetro interno de 2, 3, 4 milimetros.
Columnas preparativas pueden tener longitud hasta de un metro con diámetro interno de
hasta 50 milimetros. Para el uso industrial las medidas pueden ser mayores.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
16
Fases estacionarias [6]
La siguiente tabla muestra las fases estacionarias empleadas comúnmente en cromatografía
de líquida de la modalidad invertida.
Tabla No. 1. Fases estacionarias empleadas comúnmente
Tipo de Cadena -R Tamaño de Partícula
Larga -C18 H37 5 – 10
Intermedia -C8 H17 5 - 10
Los soportes de fases enlazadas poseen grupos funcionales incorporados a la cadena
hidrocarbonada saturada que se enlazan químicamente a la superficie del soporte, este tipo
de cromatografía es la llamada cromatografía de fase inversa (invertida o reversa) que
involucra una fase estacionaria relativamente poco polar como cadenas de hidrocarburos de
8 a 18 carbonos unidas a los grupos silanos del soporte y se utilizan por lo general con fases
móviles muy polares, esta modalidad cromatografía es la de mayor interés e impacto en la
actualidad.
La cromatografía de fase inversa, la retención de un analito dado y la composición de fase
móvil constante puede variar por el cambio de la polaridad de la fase estacionaria
químicamente enlazada. La única manera de que el factor de capacidad del analito de
iguales valores para diferentes columnas es variando la polaridad de la fase móvil.
En fase inversa, los solutos son aludidos en orden de polaridad, siendo eludido primero los
componentes más polares. Se pueden variar el tiempo de retención cambiando la polaridad
de la fase estacionaria o más fácilmente cambiando la polaridad de la fase móvil.
La polaridad de la fase estacionaria esta en dependencia del grupo no polarizado, por
ejemplo C – 8 > C – 18 y del porcentaje del carbono presente.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
17
La retención de un soluto dado con composición de fase móvil constante aumenta con la
longitud de la cadena alquílica químicamente agregada.
En cromatografía liquida de fase reversa la hidrofobicidad de la fase estacionaria y la del
soluto es la propiedad más importante. La prueba de retención del soluto depende de la
hidrofobicidad de la fase estacionaria en eluyentes semejantes. Los componentes de la fase
móvil por lo tanto pueden ser cambiados para diferentes tipos de fases estacionarias con el
fin de obtener el máximo desarrollo.
Detección [6]
La eficiencia de un detector cromatografico depende de la relacion entre la cantidad fisica
medida y la composición del efluente, asi como tambien de las caracteristicas de la señal
transferida. En farmacia el detector mas utilizado es el de Ultravioleta.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
18
IV.4. Validación de los métodos analíticos La validación de los métodos analíticos es un tema de considerable interés. Documentos
tales como “Directrices ACS para la adquisición de datos y evaluación de la calidad de los
datos “(1), recomendó el uso de métodos validados. La promulgación de regulaciones
federales al medio ambiente requiere la inclusión de métodos de referencia validados. Las
organizaciones que redactan estándares invierten considerable tiempo en métodos de
pruebas colaborativas que ellos preparan validándolos en aplicaciones típicas y
determinando sus características de resolución. Sin embargo, frecuentemente surgen
preguntas acerca de la convivencia de los métodos y la valides de su uso en situaciones
especificas. Algunas de esas preguntas pueden ser debido a la diferencia en entender tanto
qué significa el proceso de validación.[ 7]
La validación del método es un requisito importante en la práctica del análisis químico.
Validación del método es el “proceso de establecer las características de desempeño y
limitaciones del método y la identificación de los aspectos influyentes que puedan cambiar
estas características así como hasta que punto se puede cambiar”.
La validación de un método es un proceso basado en la confirmación del desempeño o de
que el mismo es consistente con los requerimientos de su aplicación. El laboratorio de
servicio y su personal tiene una clara responsabilidad, la confianza del cliente,
proporcionado la respuesta correcta a la parte analítica del problema en otras palabras debe
demostrarse que los resultados son “adecuados para el propósito” para esto será suficiente
que cualquier decisión que se tome basada en el sea confiable.
De manera que, el desempeño del método debe ser valido, y de igual manera, deberá
estimarse la incertidumbre del resultado y analizar las muestras adecuadas. [7]
IV.4.1 Jerarquía de la metodología La jerarquía de la metodología procede de lo general a lo específico, puede considerarse
como sigue:
Técnica método procedimiento protocolo.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
19
Una técnica es principio científico que se ha encontrado utilidad para proporcionar
información composicional; la espectrofotometría es un ejemplo.
Un método es una adaptación distinta de una técnica para un propósito de medición
seleccionado.
Un procedimiento consiste en una guía escrita necesaria para utilizar el método.
Los métodos estándares desarrollados por la ASTM y la AOAC, en realidad son
procedimientos estandarizados. Por tanto algún nivel de sofisticación es supuesto para el
usuario de todo el procedimiento publicado; si es muy sofisticado, el usuario solo
contemplara aquel mínimo detalle necesario o viceversa. Sin embargo se debe de notar que
cualquier omisión en la descripción del paso crítico es una fuente potencial de variación o
tendencia (desviación) aún en las manos de un analista muy experto y hábil. Debido a la
flexibilidad intencionada o no intencionada proporcionada al analista, o debido a las
diferencia en las interpretación que es justo decir que las menores /mayores diferencias de
la aplicación se dan aún en los procedimientos definidos con mas precisión. Tales
diferencias frecuentemente ocurren para la variabilidad de Inter.- laboratorio observada en
muchas pruebas de laboratorio colaborativas.
Además en algún punto de desviación del procedimiento publicado resulta un nuevo
método que puede necesitar su propia validación.
El termino protocolo es el nombre mas especifico para un método. Un protocolo es un
conjunto de guías definitivas que deben ser seguidas sin excepción si los resultados
analíticos son aceptados para un propósito dado. Los protocolos pueden consistir de
procedimientos o métodos ya existentes o modificaciones de los mismos o ellos pueden ser
desarrollados para propósitos específicos.
Una cantidad de métodos, procedimientos, y protocolos basados en el mismo principio de
medición puede surgir para una determinación analítica dada. Usualmente ellos son
diferentemente redactados y pueden contener sutilezas o mayores diferencias en los detalles
técnicos.[8]
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
20
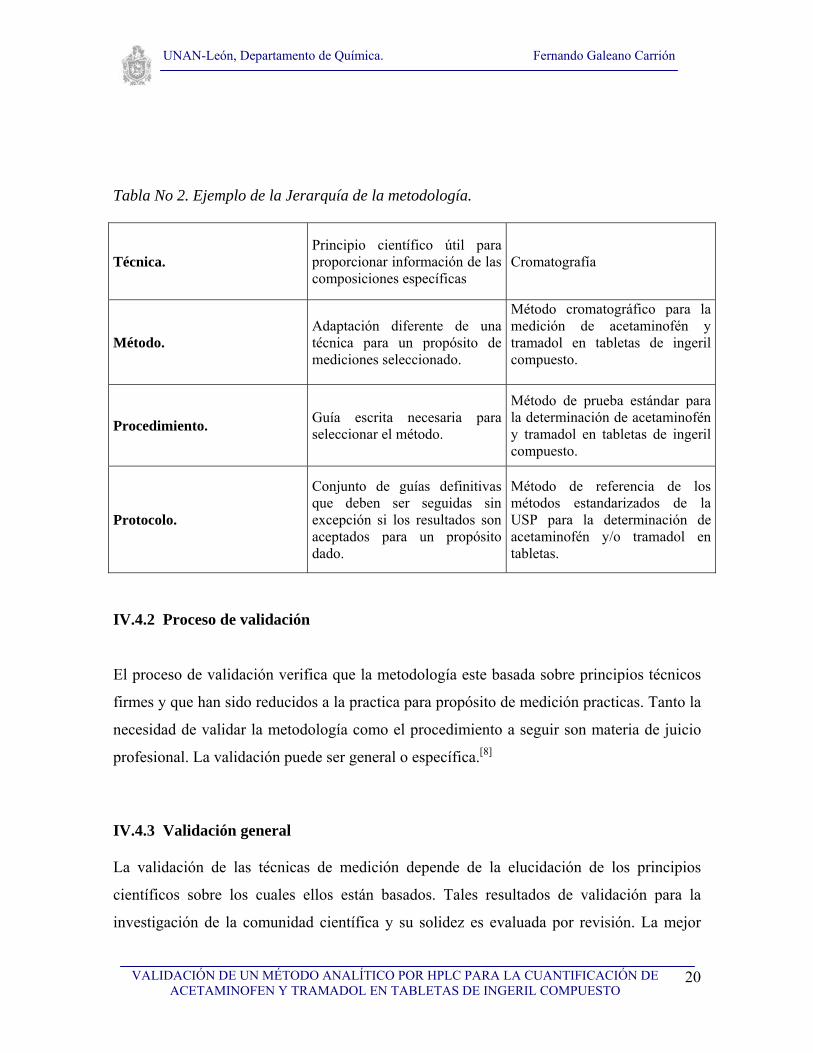
Tabla No 2. Ejemplo de la Jerarquía de la metodología.
Técnica. Principio científico útil para proporcionar información de las composiciones específicas
Cromatografía
Método. Adaptación diferente de una técnica para un propósito de mediciones seleccionado.
Método cromatográfico para la medición de acetaminofén y tramadol en tabletas de ingeril compuesto.
Procedimiento. Guía escrita necesaria para seleccionar el método.
Método de prueba estándar para la determinación de acetaminofén y tramadol en tabletas de ingeril compuesto.
Protocolo.
Conjunto de guías definitivas que deben ser seguidas sin excepción si los resultados son aceptados para un propósito dado.
Método de referencia de los métodos estandarizados de la USP para la determinación de acetaminofén y/o tramadol en tabletas.
IV.4.2 Proceso de validación El proceso de validación verifica que la metodología este basada sobre principios técnicos
firmes y que han sido reducidos a la practica para propósito de medición practicas. Tanto la
necesidad de validar la metodología como el procedimiento a seguir son materia de juicio
profesional. La validación puede ser general o específica.[8]
IV.4.3 Validación general La validación de las técnicas de medición depende de la elucidación de los principios
científicos sobre los cuales ellos están basados. Tales resultados de validación para la
investigación de la comunidad científica y su solidez es evaluada por revisión. La mejor

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
21
comprensión de los principios de medición puede extender su alcance y mejorar la calidad
de su uso.
Los métodos surgen como resultado de una investigación aplicada, típicamente por
individuos que frecuentemente involucran tanto el entendimiento comprensible de las
técnicas de medición como el alto grado de ingenuidad e innovación en su aplicación. La
comprobación de los métodos en situaciones prácticas típicas juegan un papel clave, tanto
en el proceso de desarrollo como en el de la validación.
Los procedimientos son desarrollados para el uso final de los métodos en situaciones
analíticas prácticas. El usuario de laboratorio comúnmente necesita mas detalle
experimentales que están contenidos en reportes de investigación publicados de un método
para utilizarlos en aplicaciones practicas. Frecuentemente cuando un método gana un uso
extensivo el usuario puede decidir que este necesite ser estandarizado.
Un proceso de revisión completo incluye pruebas colaborativas (ver figura No. 4) en los
cuales los materiales de prueba estable típicos son analizados para verificar la utilidad del
procedimiento y para identificar tanto la debilidad técnica como editoriales.
Figura No. 4. Prueba colaborativa.
Método candidato
Método de referencia de
exactitud conocida
Muestra de referencia Homogéneamente
estable
Composición desconocida
Composición conocida
Precisión e inclinación de la evaluaciónPrecisión de la evaluación

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
22
Si la composición de la muestra de referencia es conocida, su precisión y su desviación
tanto a nivel intra o ínter laboratorio pueden ser evaluadas. Si un método de exactitud
conocida está disponible, el ensayo colaborativo puede consistir de su comparación con el
método propuesto en cualquier caso tanto la precisión como la desviación pueden ser
evaluadas. Un protocolo es preescrito por Fiat de una organización que requiere un tipo
específico de medición. Presumiblemente resulta de una decisión inteligente basado en el
proceso de validación de la organización o de otros. Esto puede consistir de un ensayo
colaborativo extenso o la publicación de protocolo propuesto para comentario público.
Desafortunadamente el juicio científico ha estado invalidado ante la ausencia de protocolos
defectuosos e invalidados. Los protocolos que han sido especificados en un arreglo
contractual pueden escoger arbitrariamente o a través de un proceso de selección bien
concebido. La verificación de su validez para su uso especifico debe ser de primera
consideración.[8]
IV.4.4 Validación para uso especifico El uso final de la metodología analítica es producir información composicional acerca de
las muestras específicas necesarias para la solución de un problema. El proceso de
validación clásica se ilustra en la figura siguiente.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
23
IV.4.5 Proceso general para la metodología
Figura No 5. Evaluación / validación
Cuando las muestras de referencia están disponibles y es similar en todos los aspectos a las
muestras de prueba el proceso es muy simple: consiste en el análisis de un número
significativo de muestras de referencia y comparando los resultados que se esperan o
valores certificados. Antes o durante la práctica, el analista debe demostrar la habilidad del
estado del control estadístico del sistemas de medición para que los resultados puedan ser
realizados sobre otros, representativos a aquellos esperados cuando se utiliza en la
metodología de un sistema de medición. Cuando no se dispone de un material de referencia
apropiado se pueden usar otras técnicas. Una de ellas consiste de la comparación de los
resultados del método propuesto con aquellos de otros métodos conocidos sean aplicables y
Muestra fortificada con analito / Muestras de referencia
Método Independiente
Material Estándar de Referencia
Precisión
Método de exactitud conocida
Ensayo colaborativo
Bias
Método Evaluado validado
Mediciones Replicadas
Control de Calidad
Método Propuesto
Sustituta

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
24
confiables pero no útil en la situación real debido al costo indisponibilidad del personal o
equipo u otras razones.
Las muestras llamadas “spiked” (fortificada con el analíto) y “surrogated” pueden ser
usadas como muestras de referencia. Esta técnica es menos deseable y menos satisfactoria
debido a la dificultad en la preparación confiable de las muestras y debido a la artificialidad
añadida de materiales tales como “spikes” y “surrogates” que pueden exhibir diferentes
efectos de matrices a aquellas de las muestras naturales. La división de las muestras reales
de ensayos puede ser utilizada para evaluar la precisión de un método o procedimiento,
pero ellas no proporcionan información acerca de la presencia o magnitud de cualquier
“bias” (inclinación) de la medición.
Otra técnica es inferir la metodología apropiada de las mediciones sobre análogos, pero
diferentes orígenes. Se necesita un análisis crítico del analista para decidir la validez de la
interferencia.
En todos los casos las pruebas suficientes se pueden hacer para evaluar la metodología para
una variedad de matrices y rangos de composición esperada dentro del proceso de
medición, comúnmente debería incluir tres niveles de concentración, es decir, los extremos
y el rango medio de composición esperada. Las consideraciones estadísticas sugieren que al
menos seis grados de libertad (ordinariamente siete mediciones) deberían estar
involucrados en cada punto de medición. [8]
IV.4.6 El método de adición patrón Uno de los principales problemas en las determinaciones de los analitos en las muestras es
el efecto de matriz, la cual se define como las interferencias de los diferentes componentes
de la muestra en la determinación del componente principal. Esto significa que la señal
detectada será una respuesta no solamente del componente principal (o analito) sino debido
a otros componentes de la matriz. El efecto de matriz puede aumentar o disminuir la señal
verdadera, lo que trae como consecuencia un error sistemático en la determinación
analítica. Para minimizar las interferencias de la matriz, se ha implementado la curva de
calibración por el método de adición estándar.[7]

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
25
IV.4.6.1 Adición Patrón Para ganar seguridad en la utilización de las rectas de calibrado, o sea, de la correcta
similitud en el comportamiento de patrones y problemas, es aconsejable el método de la
adición estándar.
Las dudas suelen plantearse en los sistemas analíticos complejos o para las muestras con
matrices difícilmente reproducibles en los patrones, el caso de alimentos, muestras
biológicas o del medio ambiente, y evidentemente cuando concurren las dos circunstancias.
La duda surge al no disponer de muestras de la misma matriz pero sin contaminación del
metal, para utilizarla en la preparación de los patrones de calibración. Una solución es
preparar los patrones con la misma muestra. Para ello se toman varios volúmenes iguales de
la disolución a analizar, añadiendo a algunas de ellas cantidades perfectamente conocidas
de la especie química a determinar y todas las alícuotas se enrazan al mismo volumen. Con
ello se dispone de una serie de preparaciones que contienen, además de la cantidad
problema (no conocida), las cantidades adicionadas, incluyendo la adición cero y valores
que cubren el margen de linealidad. El método de las adiciones se apoya en dos premisas
no siempre válidas; una es suponer que en el sistema analítico los patrones adicionados se
comportan de la misma forma que lo hace la sustancia contenida en la muestra, pues no
podemos asegurar que correspondan a la misma especie o combinación química (estado de
oxidación o de complejación, etc.). Otra suposición es que la relación señal-concentración
mantiene la misma función lineal (concretamente la misma pendiente) fuera del margen
calibrado, pues se deduce la concentración por extrapolación. Si con el método de la
adición patrón se detecta interferencia, se recomienda modificar las condiciones
experimentales o utilizar modificaciones de matriz para eliminar o reducir al mínimo la
interferencia.[9]
IV.4.7 Criterios de eficiencia
Los criterios de eficiencia pueden clasificarse en primarios y secundarios. Los criterios
primarios son:

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
26
IV.4.7.1 Precisión
Es una medida de que tan cerca están los resultados entre sí y usualmente se expresa por
medio del valor de la desviación estándar, que describe la dispersión, o sea que, describe la
magnitud de los errores aleatorios. [9]
IV.4.7.2 Exactitud o Bias (sesgo)
Que mide la magnitud de los errores sistemáticos.
IV.4.7.3 Límite de detección
Que mide la cantidad mínima que puede diferenciarse de la señal de ruido de fondo. Es la
concentración del analito que produce una señal que es tres desviaciones estándar mayor
que la del blanco.
Uno de los puntos característicos de la recta de calibrado es la señal para la concentración
cero, que puede tener diferente significado si corresponde a la medida realizada sobre los
reactivos sin la muestra (prueba en blanco de los reactivos). O bien, se realiza sobre la
muestra sin alguno de los reactivos esenciales (prueba en blanco de la matriz de la
muestra). Las explicaciones que puedan darse a la desviación desde cero hacia valores
positivos o negativos de la señal para las pruebas en blanco, constituyen un buen índice del
grado de conocimiento que el científico posee del procedimiento analítico. [9]
Ambas señales de las pruebas en blanco suelen acumularse sobre las de la muestra, aunque
de manera diferente a las señales de los patrones, por lo que es más prudente mantener
estos valores en la representación de la recta de calibrado, y sustraer al resultado de
concentración del correspondiente a la prueba en blanco. La señal de la prueba en blanco
10 /29.3. bSbDL = Ec. (1)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
27
también presenta errores experimentales y, por lo tanto, una dispersión de valores que
afectan al límite de detección del procedimiento.
Así como la pendiente de la recta de calibrado es un índice de la sensibilidad del
procedimiento analítico, la precisión de esta pendiente y la de la ordenada en el origen
establecen el límite de detección, o sea la cantidad mínima de sustancia detectable
cuantitativamente. El valor del límite de detección se establece con criterios de
probabilidad de cometer error por asignar un valor a la señal de un blanco, o bien por
asignar el valor del blanco (no detectable) cuando en realidad la muestra contiene cantidad
significativa de sustancia. También aquí la estadística proporciona diversos modelos para
establecer estos límites, según el grado de seguridad que se desee alcanzar. En algunas
técnicas instrumentales se llama "ruido de fondo" a la dispersión de la señal del blanco,
estableciéndose como límite de detección la concentración que da una señal doble o triple
respecto a la señal de fondo.
Es incorrecto utilizar expresiones como "no detectable", "ausencia" o "0", como resultado
correspondiente a señales iguales a la prueba en blanco, siendo correcto decir que el
resultado de una señal no diferenciable de la prueba en blanco es "inferior al límite de
detección", cuyo valor se ha determinado previamente.[9]
Los criterios secundarios son los que tienen influencia en los primarios; estos son:
IV.4.7.4 Límite de cuantificación
Es la más baja concentración del analito que puede ser cuantificada con suficiente precisión
y exactitud. También es definido, convencionalmente, como la concentración de analito que
corresponde al blanco de la muestra, más 5, 6 ó 10 desvíos estándar de la media (del
blanco), también se le conoce como “límite de determinación”.
10 /10. bSbCL = Ec. (2)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
28
IV.4.7.5 Linealidad
Que describe el comportamiento entre la respuesta y la concentración a través del modelo
de calibración (una desviación del modelo representa un bias).
IV.4.7.6 Rango
Representa el intervalo (niveles inferior y superior del analito)en el cual la relación lineal u
otro modelo de calibración utilizado es correcta. [9]
IV.4.7.7 Selectividad
Este parámetro asegura que la señal medida no es influenciada por otras sustancias
presentes en la muestra y en caso contrario, garantiza la remoción de las mismas, otro
aspecto importante de la selectividad es si el analito puede existir en la muestra de una
forma, tal como enlazado o libre, inorgánico u órgano - metálico, o en diferentes estados de
oxidación. [9]
IV.4.7.8 Sensibilidad
Es el parámetro que mide la magnitud del cambio en la función de respuesta (o señal) con
la concentración y corresponde a la pendiente de la curva de respuesta. [17]

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
29
IV.5. Aproximación ISO para el cálculo de la Incertidumbre
Para llevar a cabo el cálculo de la incertidumbre comúnmente se divide el proceso de
medida químico en sus partes fundamentales, cada una de estas partes fundamentales se
subdivide a la vez en contribuciones más pequeñas, etc. Posteriormente, se calcula la
incertidumbre de cada una de las partes y se combinan para obtener la incertidumbre global
del proceso de medida química.[10]; [11]; [12]
Figura No. 6, Flujograma para el cálculo de la incertidumbre de un procedimiento según
la aproximación ISO.
Especificación
Aquí debe modelarse el proceso de medida. Es decir, debe establecerse cual es la relación
que existe entre el resultado analítico y los parámetros de los cuales depende.[10]; [13; [14]
Identificación
Una vez modelado el proceso de medida, deben identificarse todas las fuentes de
incertidumbre independientemente de la importancia que pueda tener cada una de ellas en
la incertidumbre final de los resultados. [10]; [11]; [12]
Especificación Modelado del proceso de medida
Identificación Identificación de las fuentes de incertidumbre
Cuantificación Cálculo de la incertidumbre estándar
Combinación Cálculo de la incertidumbre estándar combinada
Cálculo de la incertidumbre expandida

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
30
Combinación
Los componentes de incertidumbre individuales deben combinarse siguiendo la ley de
propagación del error. De esta forma, se obtiene la incertidumbre total estándar. [10]; [13; [14]
( ) ( ) ( )ij
n
i
n
ij ji
n
ii
ic X
Xc
XcXU
XcU cov.2.
1 1
1
1
2
∑ ∑∑= +=
−
= ∂∂
∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
= Ec. (3)
Donde “c” es el resultado obtenido con el procedimiento analítico, Xi es el parámetro “i”
del que depende el resultado, U(Xi) es la incertidumbre estándar de Xi y cov (Xij) es la
covarianza que hay entre los parámetros Xi y Xj.
Finalmente el cálculo de la incertidumbre expandida, U(c), proporciona un intervalo de
confianza donde se encuentra el valor verdadero con una determinada probabilidad. Esta
incertidumbre se obtiene multiplicando la incertidumbre estándar por un factor de cobertura
“k” . [10]; [11]; [12]
( ) ( )cukcU .= Ec. (4)
el factor de cobertura “k” depende de la probabilidad con la que queremos que se encuentre
el valor verdadero dentro del intervalo c ± U(c) así como de la distribución y de los grados
de libertad asociados a U(c). Normalmente, se utiliza el valor de k = 2. este valor asume
una distribución normal y una probabilidad aproximada del 95.45 % de contener el valor de
“t” tabulado de dos colas para el nivel de significación escogido y los grados de libertad
efectivos, veff, calculados con la aproximación de Welch-Satterthwaite. [10]; [11]; [12]
Ec. (5)
Donde vi es el número de grados de libertad asociados a U(xi).
( )( )∑
=
=n
i i
iffe
vxu
cuv
1
4
4

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
31
V. PARTE EXPERIMENTAL
V.1. Equipos y Materiales
Tabla No. 3. Equipos y materiales
Descripción Marca Modelo
Agitador ultrasónico. Fisher Scientific FS -30
Balanza analítica. Ohaus Explorer
Balones aforados. Pirex Clase A
Beaker. Pirex -
Bomba de vacío. Welch 2534B-01
Calculadora científica. - -
Columna. Agilent Zorbax SB-C8
Cromatógrafo líquido de alta eficiencia
(HPLC).
Varian ProStar. -
Espátulas - -
Filtro econofiltro. Agilent -
Filtro de membrana de Nylon. Agilent -
Jeringa. Agilent -
Kit de micro filtración. Agilent -
Mortero y Pilón - -
pH-metro Termo Orion 420 -
Pipetas Pirex Clase A
Probeta Pirex -
Viales de 2 mL Varian Varian

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
32
V.2. Reactivos Tabla No. 4. Reactivos
Descripción Calidad
Fosfato de Sodio Monobásico de Potasio
grado HPLC
Agua destilada
grado HPLC
Trietilamina (TEA)
(p.a.)
Metanol
grado HPLC
V.3. Contenido de la Tableta de Ingeril Compuesto de 650 mg V.3.1. Principios activos Acetaminofen DC 90 361.11 mg equivalente a 325 mg Acetaminofen Tramadol Clorhidrato 37.5 mg
V.3.2. Excipientes (Placebo) Almidón de maíz
Almidón pregelatinizado
Avicel pH-102
Cab-o-sil
Estereato de Magnesio
Vivastar-p

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
33
V.4. Procedimientos
V.4.1 Condiciones cromatográficas
• Equipo: Cromatógrafo liquido de alta Resolución
• Columna: Agilent Zorbax SB-C8
• Sistema: Binario, Fosfato Monobásico de Potasio (FMK)/Metanol (MeOH) en relación
(60/40)
• Longitud de onda: 270 nm
• Temperatura: ambiente (30 oC)
• Flujo: 1.2 mL/min
• Volumen de inyección: 20 µL
• Tiempo de Retención: 1.2 min (Acetaminofén) y 4.2 - 4.3 min. (Tramadol HCl)
aproximadamente
• Tiempo total de corrida: 5 min.
• Método de calculo: Estándar externo
V.4.2. Preparación de la Fase Móvil
Buffer PH 4.9
Desgasificar 300 mL Metanol. Pesar exactamente 3.4 g de Fosfato Monobásico de Potasio (FMK)
y disolver con 500 mL de Agua destilada, ajustar el PH con una o mas gotas de trietilamina (TEA)
y desgasificar.
Fase Móvil: Buffer PH 4.9 ajustado con trietilamina (TEA), proporción Fosfato Monobásico de
Potasio (FMK)/Metanol (MeOH) en realación 60/40
V.4.3. Preparación de la curva de calibración normal de acetaminofén y tramadol
Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80, 90, 100,
110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8
mg de Tramadol HCl, posteriormente aforar cada uno de los matraces con agua desionizada y
someterlos a una baño ultrasónico por 10 min. Luego tomar 10 mL de cada una de las soluciones
estándar y colocarlos en otros 5 matraces volumétricos de 100 mL y aforar con agua desionizada.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
34
V.4.4. Preparación de la muestra
Determinar el peso promedio de 20 tabletas de Ingeril Compuesto posteriormente molerlas hasta
polvo fino. Pesar 200 mg de muestra para obtener de acuerdo a la relación entre peso promedio y
la cantidad existente de activo, un equivalente 100 mg de acetaminofén y 11.5 mg de tramadol.
Luego transferir a un balón de 100 mL y aforar con agua desionizada. Someter a baño ultrasónico
por 30 min. Luego tomar 10 mL de la solución conteniendo la muestra, transferir a un balón de
100 mL y aforar con agua destilada.
Usar una jeringa con filtro 0.2 µm tomar 2 mL de cada solución y transferir a un vial de 2 mL
respectivamente.
V.4.5. Selectividad del método.
Realizar tres inyecciones del solvente utilizado para los análisis y tres inyecciones del
placebo utilizado en la formulación de las tabletas de ingeril compuesto, luego realizar tres
inyecciones de un estándar de acetaminofén y tres inyecciones de un estándar de tramadol
posteriormente realizar tres inyecciones de la muestra a analizar. En todos los casos
observar que no exista ninguna señal cormatográfica que coincida con los tiempos de
retención de los principios activos a estudiar es decir que no deben existir interferencias con
las señales de nuestro interés.
V.4.6. Influencia del placebo en la curva de calibración normal
Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80, 90, 100,
110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8
mg de Tramadol HCl, posteriormente aforar cada uno de los matraces con agua desionizada y
someterlos a una baño ultrasónico por 10 min. Luego tomar 10 mL de cada una de las soluciones
estándar y colocarlos en otros 5 matraces volumétricos de 100 mL y aforar con agua desionizada.
Posteriormente tomar otros 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada
uno 80, 90, 100, 110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3,
11.5, 12.6, 13.8 mg de Tramadol HCl, luego adicionar las cantidades necesarias del placebo
descrito en V.3.2 (equivalente al contenido en una tableta) posteriormente aforar cada uno de los

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
35
matraces con agua desionizada y someterlos a una baño ultrasónico por 10 min. Luego tomar 10
mL de cada una de las soluciones estándar conteniendo placebo y colocarlos en otros 5 matraces
volumétricos de 100 mL para posteriormente aforar con agua desionizada, de éstos se toman 20μL
y se inyectan al cromatógrafo.
Graficar en el mismo plano la curva de calibración normal sin contener placebo y la otra
conteniendo placebo (realizar esto con cada principio activo), luego comparar los resultados
gráficamente, las curvas no deberán cruzarse en ningún punto para que no exista ninguna
influencia del placebo en la curva de calibración normal.
V.4.7. Idoneidad del Sistema
Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80, 90, 100,
110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8
mg de Tramadol HCl, posteriormente aforar cada uno de los matraces con agua desionizada y
someterlos a una baño ultrasónico por 10 min. Luego tomar 10 mL de cada una de las soluciones
estándar y colocarlos en otros 5 matraces volumétricos de 100 mL y aforar con agua desionizada,
tomar 20 μL e inyectarlos al cromatógrafo.
Seguidamente calcular los parámetros de idoneidad del sistema los cuales deberán de
cumplir con los valores de aceptación sugeridos en la Farmacopea de Estados Unidos
(USP).
V.4.8. Linealidad del sistema y del método cromatográfico
Linealidad del sistema
Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80, 90, 100,
110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8
mg de Tramadol HCl, posteriormente aforar cada uno de los matraces con agua desionizada y
someterlos a una baño ultrasónico por 10 min. Luego tomar 10 mL de cada una de las soluciones
estándar y colocarlos en otros 5 matraces volumétricos de 100 mL y aforar con agua desionizada
tomar 20 μL e inyectarlos al cromatografo.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
36
Linealidad del método
Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80, 90, 100,
110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8
mg de Tramadol HCl, luego adicionar a cada uno 200 mg de una muestra (tableta), posteriormente
aforar cada uno de los matraces con agua desionizada y someterlos a una baño ultrasónico por 10
min. Luego tomar 10 mL de cada una de las soluciones estándar y colocarlos en otros 5 matraces
volumétricos de 100 mL para posteriormente aforar con agua desionizada, de éstos se toman 20μL
y se inyectan al cromatógrafo.
Realizar los gráficos independientes para la linealidad del sistema y del método,
posteriormente calcular los coeficientes de determinación los cuales deberán de ser
mayores o igual a 0.99.
V.4.9. Precisión del sistema y del método cromatográfico
Precisión del sistema
Realizar 5 inyecciones de tres niveles de concentración diferentes: uno bajo, un nivel
intermedio y un nivel alto. Para acetaminofén 80, 100, 120 μg/mL y para tramadol 9.2,
11.5, 13.8 μg/mL. Calcular el factor de respuesta y su desviación estándar relativa
expresada como porcentaje (%RSD) la cual deberá ser menor del 2 %.
Precisión del método
Realizar 5 inyecciones de tres niveles de concentración diferentes: uno bajo, un nivel
intermedio y un nivel alto. Para acetaminofén 80, 100, 120 μg/mL (dopando cada uno con
muestra “equivalente a 100 μg/mL de acetaminofén”) y para tramadol 9.2, 11.5, 13.8
μg/mL (dopando cada uno con muestra “equivalente a 11.5 μg/mL de tramadol”). Calcular
el factor de respuesta y su desviación estándar relativa expresada como porcentaje (%RSD)
la cual deberá ser menor del 2 %.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
37
V.4.10. Exactitud del método
La exactitud del método se evaluó de dos maneras, para las cuales se describe a
continuación el procedimiento.
1) Tomar 5 matraces volumétricos de 100 mL, limpios y secos, agregarles a cada uno 80,
90, 100, 110, 120 mg de Acetaminofén, seguidamente agregarles exactamente 9.2,
10.3, 11.5, 12.6, 13.8 mg de Tramadol HCl, posteriormente aforar cada uno de los
matraces con agua desionizada y someterlos a una baño ultrasónico por 10 min. Luego
tomar 10 mL de cada una de las soluciones estándar y colocarlos en otros 5 matraces
volumétricos de 100 mL y aforar con agua desionizada tomar 20 μL e inyectarlos al
cromatografo. Calcular el porcentaje de recuperación (%R) para cada nivel de
concentración de la curva dividiendo la concentración experimental entre la
concentración teórica multiplicado por 100, éste deberá estar entre 90% - 110 %.
2) Curva de calibración normal: Tomar 5 matraces volumétricos de 100 mL, limpios y
secos, agregarles a cada uno 80, 90, 100, 110, 120 mg de Acetaminofén, seguidamente
agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8 mg de Tramadol HCl,
posteriormente aforar cada uno de los matraces con agua desionizada y someterlos a
una baño ultrasónico por 10 min. Luego tomar 10 mL de cada una de las soluciones
estándar y colocarlos en otros 5 matraces volumétricos de 100 mL y aforar con agua
desionizada, tomar 20 μL e inyectarlos al cromatografo.
Curva de adición patrón: Tomar 5 matraces volumétricos de 100 mL, limpios y secos,
agregarles a cada uno 80, 90, 100, 110, 120 mg de Acetaminofén, seguidamente
agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8 mg de Tramadol HCl, luego
adicionar a cada uno 200 mg de una muestra (tableta), posteriormente aforar cada uno
de los matraces con agua desionizada y someterlos a una baño ultrasónico por 10 min.
Luego tomar 10 mL de cada una de las soluciones estándar y colocarlos en otros 5
matraces volumétricos de 100 mL para posteriormente aforar con agua desionizada, de
éstos se toman 20μL y se inyectan al cromatógrafo.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
38
Calcular el porcentaje de recuperación (%R) para acetaminofén y para tramadol
respectivamente, el %R se calcula dividiendo la pendiente de la curva de adición patrón
ente la pendiente de la curva de calibración normal, el %R deberá estar entre 90 % a 110 %.
V.4.11. Efecto de matriz
Curva de calibración normal: Tomar 5 matraces volumétricos de 100 mL, limpios y
secos, agregarles a cada uno 80, 90, 100, 110, 120 mg de Acetaminofén, seguidamente
agregarles exactamente 9.2, 10.3, 11.5, 12.6, 13.8 mg de Tramadol HCl, posteriormente
aforar cada uno de los matraces con agua desionizada y someterlos a una baño ultrasónico
por 10 min. Luego tomar 10 mL de cada una de las soluciones estándar y colocarlos en
otros 5 matraces volumétricos de 100 mL y aforar con agua desionizada, tomar 20 μL e
inyectarlos al cromatografo.
Curva de adición patrón: Tomar 5 matraces volumétricos de 100 mL, limpios y secos,
agregarles a cada uno 80, 90, 100, 110, 120 mg de Acetaminofén, seguidamente agregarles
exactamente 9.2, 10.3, 11.5, 12.6, 13.8 mg de Tramadol HCl, luego adicionar a cada uno
200 mg de una muestra (tableta), posteriormente aforar cada uno de los matraces con agua
desionizada y someterlos a una baño ultrasónico por 10 min. Luego tomar 10 mL de cada
una de las soluciones estándar y colocarlos en otros 5 matraces volumétricos de 100 mL
para posteriormente aforar con agua desionizada, de éstos se toman 20μL y se inyectan al
cromatógrafo.
Graficar las dos curvas (adición patrón y calibración normal) en un mismo plano, para
acetaminofén y para tramadol. Las curvas deberán ser paralelas entre si para que no exista
efecto de matriz.
V.4.12. Límite de detección y de cuantificación
Elaborar dos curvas de calibración normal a concentraciones diluidas cercanas a la señal
del ruido, una para Acetaminofén (0.5, 1.0, 1.5, 3.0, 5.0, 7.0, 9.0 μg/mL) y otra para
Tramadol (0.06, 0.12, 0.17, 0.35, 0.58, 0.81, 1.03 μg/mL). Calcular el LD y el LC
utilizando las ecuaciones 1 y 2.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
39
V.4.13. Repetiblidad de las curvas de calibración normal
Elaborar dos curvas de calibración normal, una para Acetaminofén y otra para Tramadol en
el rango de concentración de 80 a 120 μg/mL y de 9.2 a 13.8μg/mL respectivamente,
realizar una curva diariamente durante cinco días, posteriormente realizar el gráfico de
control para el intercepto y la pendiente de las curvas, todos los valores deberán estar
dentro de la zona de aceptación de la elipse.
V.4.14. Aplicación del método y estimar la incertidumbre asociada a la cuantificación
Las muestras a analizar se trataron de acuerdo al procedimiento V.4.4 para luego realizar
los cálculos correspondientes según el modelo matemático. Para evaluar la incertidumbre
primeramente se realizó la deducción del modelo matemático a partir del proceso de
medición analítico, posteriormente se identificaron las fuentes de incertidumbres que
contribuyen a la incertidumbre global del proceso de medición y se cuantificaron cada uno
de estos componentes, seguidamente se calcularon los coeficientes de sensibilidad para
cada parámetro del modelo matemático, a continuación se combinaron las incertidumbres y
se calculó la incertidumbre combinada la cual al multiplicarla por un factor de cobertura de
2 para un nivel de confianza de 95.45 % se obtuvo la incertidumbre expandida con la cual
se reportan nuestros resultados.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
40
VI. RESULTADOS Y DISCUSIÓN
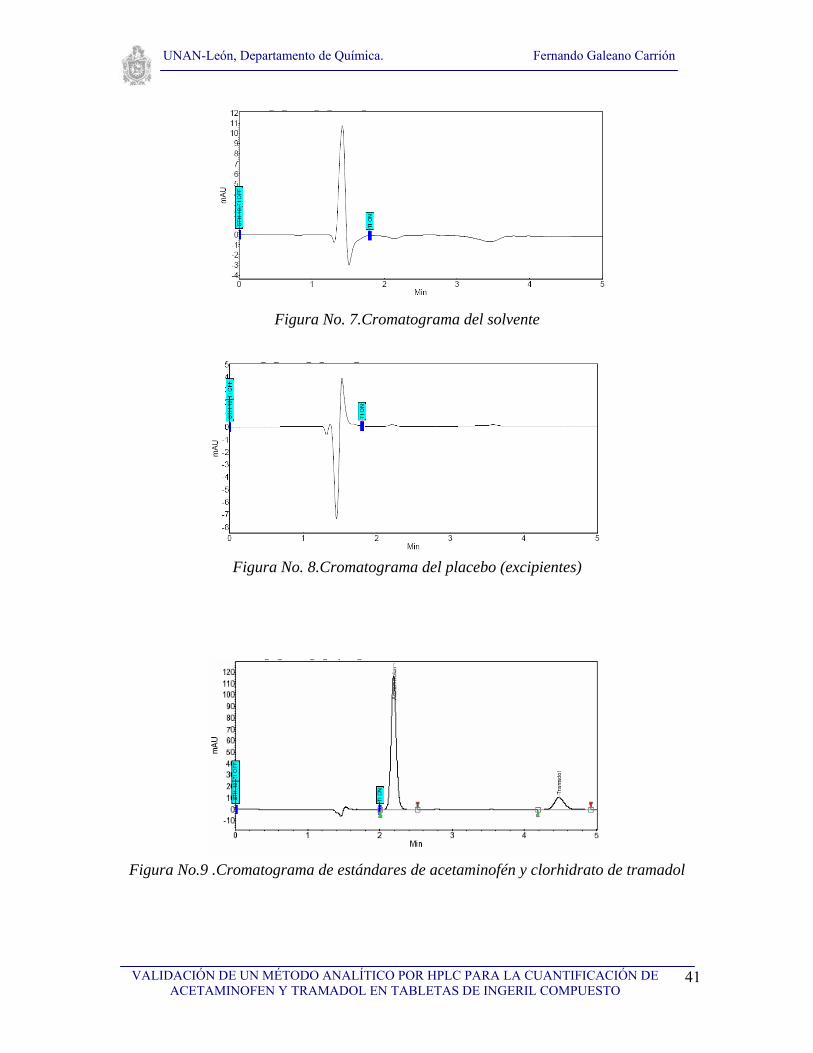
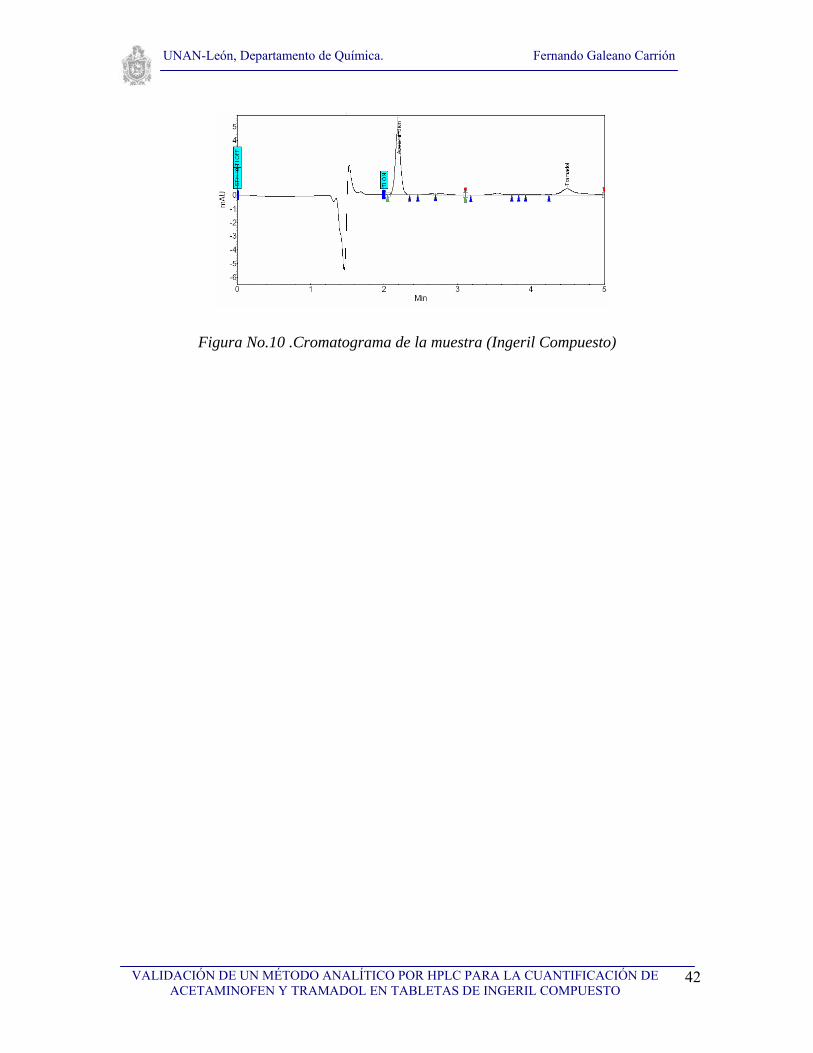
VI.1. Selectividad
Para realizar el estudio de selectividad del método se analizaron los cromatogramas de: el
solvente, los ingredientes placebos usados en la formulación de la tableta de ingeril
compuesto, así como los cromatogramas de estándares de los dos principios activos de la
tableta y de la muestra (tableta Ingeril Compuesto). Se realizaron tres inyecciones del
solvente y del placebo, tres inyecciones del estándar de acetaminofen y tres inyecciones del
estándar de tramadol así como tres inyecciones de la muestra a analizar. Primeramente se
analizó el solvente con el objetivo de observar que no hubieran interferencias con ninguno
de los picos cromatográficos de interés (fig. 7), seguidamente se analizó el cromatograma
correspondiente al placebo (fig. 8) con el propósito de demostrar que no existen
interferencias debida a los excipientes, tal y como se puede observar en la figura No. 8, no
existe ninguna interferencia, posteriormente se analizaron los cromatogramas de los
estándares de acetaminofen y de tramadol (fig. 9). Finalmente se analizaron los
cromatogramas de la muestra conteniendo acetaminofen y tramadol (Ingeril Compuesto)
para observar la resolución de los picos, como se puede observar en la figura No. 10, los
picos tienen muy buena resolución. Los tiempos de retención obtenidos fueron de 2.15 y de
4.47 para acetaminofen y tramadol respectivamente tal a como se puede observar en la
figura No. 10, el tiempo muerto fue de 1.45. Se logró observar una diferencia adecuada
entre los tiempos de retención, por otro lado no se observó ninguna señal cromatográfica
que pudiera interferir en la determinación o cuantificación de acetaminofen y tramadol en
la tableta de ingeril compuesto, por lo que podemos afirmar que el método es selectivo para
la determinación de los dos principios activos.
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
41
Figura No. 7.Cromatograma del solvente
Figura No. 8.Cromatograma del placebo (excipientes)
Figura No.9 .Cromatograma de estándares de acetaminofén y clorhidrato de tramadol
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
42
Figura No.10 .Cromatograma de la muestra (Ingeril Compuesto)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
43
VI.2. Influencia del placebo
Este estudio se realizó con el propósito de comprobar la influencia que ejerce el placebo
(interferencia de los excipientes) sobre la curva de calibración normal, ya que muchas
determinaciones se realizan mediante interpolación de la curva de calibración normal la
cual pudiera ser elaborada tomando o no en cuenta el placebo lo cual podría generar un
error en dicha determinación en caso de que éste provoque alguna influencia o algún efecto
en la curva de calibración normal.
Para llevar a cabo dicho estudio se elaboraron dos curvas de calibración normal, una sin
tomar en cuenta el placebo y la otra tomando en cuenta el mismo. Los resultados se
muestran a continuación en la siguiente tabla.
Tabla No. 5. Datos de las curvas de calibración normal con placebo y sin placebo.
Acetaminofen Tramadol
Áreas Áreas Concentración
μg/mL Con Placebo Sin Placebo
Concentración
μg/mL Con Placebo Sin Placebo
80 1492.78 1486.92 9.2 32.50 37.04
90 1725.32 1671.96 10.3 39.40 41.44
100 1948.88 1874.26 11.5 42.22 46.08
110 2084.9 2051.04 12.6 48.00 52.34
120 2284.28 2248.08 13.8 54.30 57.24
r2 0.9922 0.9997 r 2 0.9853 0.9957
A continuación se pueden observar los gráficos elaborados a partir de los datos mostrados
en la tabla anterior, tanto para los estándares de acetaminofen como para los estándares de
Tramadol con y sin placebo.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
44
Tramadol
30
35
40
45
50
55
60
8,5 9,5 10,5 11,5 12,5 13,5 14,5
µg/mL
Áre
as
Sin Placebo Con Placebo
Acetaminofen
1200
1400
1600
1800
2000
2200
2400
75 85 95 105 115 125
µg/mL
Áre
as
Sin Placebo Con Placebo
Gráfico No. 1. Gráfico de concentración Vs área para la Acetaminofen con y sin placebo
Gráfico No. 2. Gráfico de concentración Vs área para la Tramadol con y sin placebo
Como se puede observar en los gráficos anteriores no existe ningún efecto provocado por
los excipientes en la curva de calibración normal (estándares puros) ya que las rectas
obtenidas con y sin placebo son paralelas en ambos casos tanto para acetaminofen como
para tramadol. Por tanto la curva de calibración normal podrá ser elaborada tomando en
cuenta o no el placebo.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
45
VI.3. Idoneidad del Sistema
La determinación de la Idoneidad del Sistema es un conjunto de ensayos que se realizan
para comprobar que un sistema (analista, reactivos e instrumentos) es adecuado para una
metodología analítica determinada. Este estudio se realiza antes de que un sistema analítico
se utilice por primera vez, y cuando una columna o material de separación de otro
fabricante, sea utilizado por primera vez. Se considera como parte integral del
procedimiento de análisis y requisito previo a su realización.
Con el objetivo de realizar el estudio de la idoneidad del Sistema se elaboraron dos curvas
de calibración normal, una para Acetaminofen y otra para Tramadol, las curvas se
realizaron en el rango de concentración de 80 a 120 μg/mL y de 9.2 a 13.8μg/mL
respectivamente, se realizaron en total 5 puntos para cada curva y tres réplicas por cada
punto, los detalles de las curvas de calibración normal pueden ser observados en los anexos.
Posteriormente se calcularon el tiempo de retención (Tr), el factor de asimetría (T) y el
número de platos teóricos (N) tanto para la curva de acetaminofen como para la curva de
tramadol. A continuación se muestran los datos obtenidos para cada caso.
Tabla No. 6. Tabla de los parámetros de idoneidad para Acetaminofen
Tiempo de retención (Tr)
Factor de Asimetría (T)
Número de platos Teóricos (N)
Promedio 2.15 1.11 4377.98 Desviación Estándar 0.005 0.031 18.51
Criterio de Aceptación USP - ≤ 2 > 2000 Cumple Si/No - Sí Si
Tabla No. 7. Tabla de los parámetros de idoneidad para Tramadol
Tiempo de retención (Tr)
Factor de Asimetría (T)
Número de platos Teóricos (N)
Promedio 4.47 1.22 5108.73 Desviación Estándar 0.011 0.028 24.300
Criterio de Aceptación USP - ≤ 2 > 2000 Cumple Si/No - Sí Si

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
46
Los resultados de los parámetros de idoneidad obtenidos con las condiciones
cromatográficas establecidas fueron comparados con los valores de los parámetros de
aceptación reportados en la USP. Como se puede observar los resultados obtenidos
cumplen con los criterios de aceptación propuestos en la USP.
VI.4. Linealidad del Sistema y Linealidad del Método
Este estudio se realizó con el objetivo de encontrar el grado de linealidad que existe a lo
largo de un buen rango de aplicación, para comprobar la linealidad del sistema se realizó
una curva de calibración normal en un intervalo de concentración entre 80 – 120 μg/mL
para acetaminofen y de 9.2 – 13.8 μg/mL para Tramadol, los datos se muestran en la tabla
No. 8.
El valor del coeficiente de determinación (r2) obtenido fue: 0.9997 para acetaminofen y de
0.9990 para Tramadol, una linealidad ideal indica un valor de coeficiente de determinación
igual a 100 % (r2 = 1),
Tabla N. 8. Datos de las curvas de calibración normal para acetaminofen y tramadol
Acetaminofen Tramadol
Concentración
μg/mL Área
Concentración
μg/mL Área
80 1535.82 9.2 37.04 90 1735.86 10.3 41.44 100 1915.56 11.5 47.26 110 2119.28 12.6 52.34 120 2307.00 13.8 57.24 r 2 0.9997 r 2 0.9990
Como el valor de r2 obtenido experimentalmente es cercano a la unidad se puede decir que
existe una excelente linealidad, es decir existe una fuerte correlación entre las áreas
obtenidas como función de la concentración inyectada. A continuación se muestran los
gráficos obtenidos.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
47
Linealidad del Sistema(Acetaminofen)
1400
1600
1800
2000
2200
2400
75 80 85 90 95 100 105 110 115 120 125
µg/mL
Áre
as
Linealidad del Sistema(Tramadol)
30
35
40
45
50
55
60
9 9,5 10 10,5 11 11,5 12 12,5 13 13,5 14
µg/mL
Áre
as
Gráfico No. 3. Linealidad del Sistema para Acetaminofen
Gráfico No. 4. Linealidad del Sistema para Tramadol

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
48
A demás de evaluar la linealidad del Sistema a través de la curva de calibración normal se
evaluó la linealidad del Método por medio de una curva de adición patrón dentro del mismo
rango de concentraciones de (80-120 μg/mL para acetaminofen y de 9.2 a 13.8 μg/mL para
tramadol), los datos se muestran en la tabla No 9.
Tabla N. 9. Datos de las curvas de adición patrón para acetaminofen y tramadol
Acetaminofen Tramadol
Concentración
μg/mL Área
Concentración
μg/mL Área
80 1736.3 9.2 39.1 90 1933.1 10.3 45.4 100 2140.0 11.5 51.8 110 2353.0 12.6 57.6 120 2580.0 13.8 64.0 r 2 0.9993 r 2 0.9997
El valor del coeficiente de determinación (r2) obtenido fue: 0.9993 para acetaminofen y de
0.9997 para tramadol, como los valores de coeficientes de determinación obtenidos están
cercanos a la unidad (r2 = 1) es decir casi 100 %, entonces podemos afirmar que existe una
fuerte correlación entre la variable dependiente (Áreas) y la variable independiente
(Concentración) es decir el método tiene una excelente linealidad en el rango de aplicación
seleccionado tanto para acetaminofen como para tramadol. Las gráficas se muestran a
continuación

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
49
Linealidad del Método(Acetaminofen)
150017001900210023002500
75 80 85 90 95 100 105 110 115 120 125
µg/mL
Área
Linealidad del Método(Tramadol)
30,0
40,0
50,0
60,0
70,0
9 9,5 10 10,5 11 11,5 12 12,5 13 13,5 14
µg/mL
Área
Gráfico No. 5. Linealidad del Método para Acetaminofen
Gráfico No. 6. Linealidad del Método para Tramadol

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
50
VI.5. Precisión del Sistema y Precisión del Método
La precisión del sistema se llevo a cabo realizando cinco inyecciones de tres estándares a
tres niveles de concentraciones diferentes, 80, 100 y 120 para acetaminofen y de 9.2, 11.5 y
13.8 para tramadol, seguidamente se calculó el factor de respuesta (FR =
Área/Concentración) para cada estándar, posteriormente se calculó el promedio y la
desviación estándar correspondiente a cada estándar, para finalmente calcular el %RSD por
medio del cual se evalúa la precisión del sistema. Los resultados se muestran a
continuación en las siguientes tablas.
Tabla No. 10. Resultados del estudio de precisión del Sistema para Acetaminofen
Concentración (μg/mL) Réplicas 80 100 120 1 1483.3 1872.7 2245.6 2 1485.9 1874.3 2247.5 3 1486.8 1874.3 2247.6 4 1489.4 1876.1 2251.2 5 1489.2 1873.9 2248.5
18.5 18.7 18.7 18.6 18.7 18.7 18.6 18.7 18.7 18.6 18.8 18.8
FR
18.6 18.7 18.7 Promedio 18.6 18.7 18.7 Desv. Est. 0.03 0.012 0.017 %RSD 0.17 0.04 0.09
Tabla No. 11. Resultados del estudio de precisión del Sistema para Tramadol
Concentración (μg/mL) Réplicas 9.2 11.5 13.8 1 37.4 46.5 59.1 2 36.8 46.1 59.0 3 37.2 46.0 59.0 4 37.2 46.0 58.8 5 36.6 45.8 59.0
4.1 4.0 4.3 4.0 4.0 4.3 4.0 4.0 4.3 4.0 4.0 4.3
FR
4.0 4.0 4.3 Promedio 4.0 4.0 4.3 Desv. Est. 0.04 0.023 0.008 %RSD 0.89 0.56 0.19
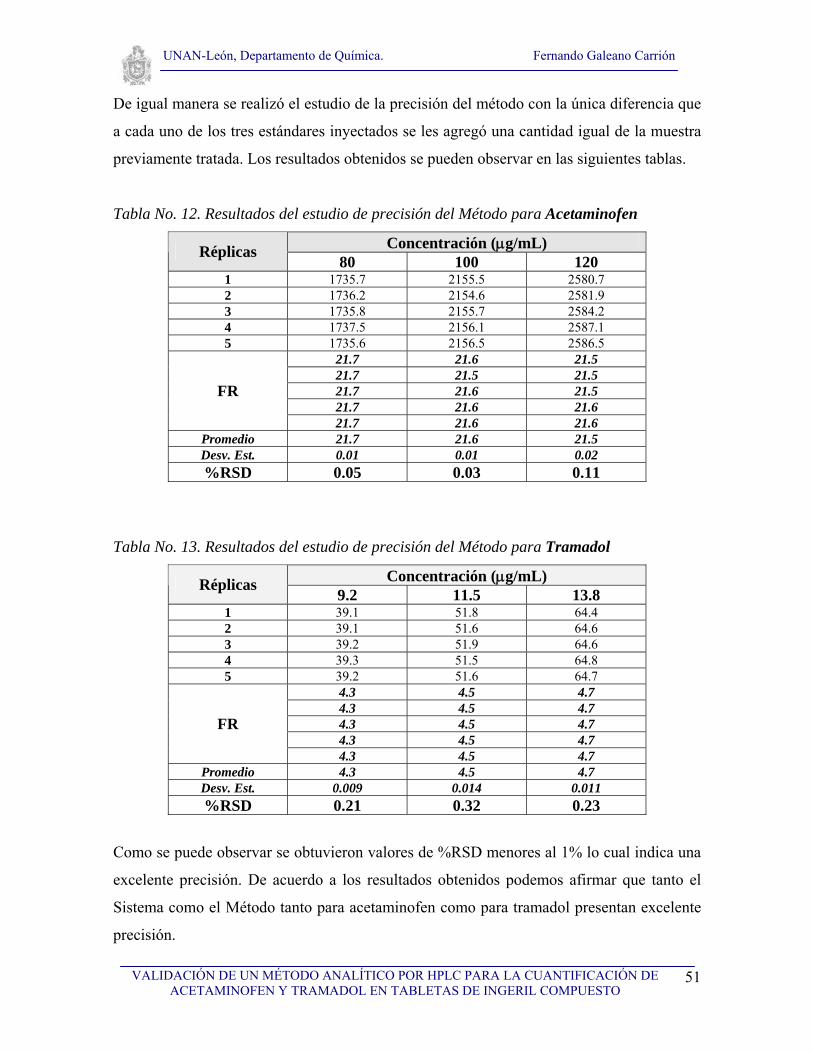
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
51
De igual manera se realizó el estudio de la precisión del método con la única diferencia que
a cada uno de los tres estándares inyectados se les agregó una cantidad igual de la muestra
previamente tratada. Los resultados obtenidos se pueden observar en las siguientes tablas.
Tabla No. 12. Resultados del estudio de precisión del Método para Acetaminofen
Concentración (μg/mL) Réplicas 80 100 120 1 1735.7 2155.5 2580.7 2 1736.2 2154.6 2581.9 3 1735.8 2155.7 2584.2 4 1737.5 2156.1 2587.1 5 1735.6 2156.5 2586.5
21.7 21.6 21.5 21.7 21.5 21.5 21.7 21.6 21.5 21.7 21.6 21.6
FR
21.7 21.6 21.6 Promedio 21.7 21.6 21.5 Desv. Est. 0.01 0.01 0.02 %RSD 0.05 0.03 0.11
Tabla No. 13. Resultados del estudio de precisión del Método para Tramadol
Concentración (μg/mL) Réplicas 9.2 11.5 13.8 1 39.1 51.8 64.4 2 39.1 51.6 64.6 3 39.2 51.9 64.6 4 39.3 51.5 64.8 5 39.2 51.6 64.7
4.3 4.5 4.7 4.3 4.5 4.7 4.3 4.5 4.7 4.3 4.5 4.7
FR
4.3 4.5 4.7 Promedio 4.3 4.5 4.7 Desv. Est. 0.009 0.014 0.011 %RSD 0.21 0.32 0.23
Como se puede observar se obtuvieron valores de %RSD menores al 1% lo cual indica una
excelente precisión. De acuerdo a los resultados obtenidos podemos afirmar que tanto el
Sistema como el Método tanto para acetaminofen como para tramadol presentan excelente
precisión.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
52
VI.6. Exactitud del Método
Para llevar a cabo el estudio de la exactitud del método se realizaron lecturas de cinco
estándares (cinco niveles de concentración) de concentraciones perfectamente conocidas
tanto de acetaminofen como de tramadol, realizándose cinco réplicas para cada una de
ellas, posteriormente se determinó el porcentaje de recuperación para evaluar la exactitud
del método. Al mismo tiempo se realizó el test de COCHRAN para evaluar si las varianzas
del %R de los cinco niveles de concentración son homogéneas es decir para evaluar la
precisión del %R. A continuación se muestran los resultados obtenidos.
Tabla No. 14. Resultados de la exactitud del método para Acetaminofen
Concentración Teórica (μg/mL) 80 90 100 110 120
Réplicas Concentración Recuperada (μg/mL) 1 80.23 93.73 104.28 113.82 124.27 2 80.63 93.74 104.63 113.64 124.11 3 80.63 93.54 104.12 114.06 123.9 4 80.32 93.55 104.51 113.97 123.91 5 80.7 93.95 104.46 113.52 124.22
Promedio 80.50 93.70 104.40 113.80 124.08 Desv. Estd 0.21 0.17 0.20 0.22 0.17
%R 100.63 104.11 104.40 103.46 103.40 Desv Est %R 0.26 0.19 0.20 0.20 0.14
IC %R 100.63 ± 0.26 104.11 ± 0.19 104.40 ± 0.20 103.46 ± 0.20 103.40 ± 0.14 Varianza %R 0.070 0.035 0.040 0.042 0.020
Suma de Var %R 0.207 Var máxima 0.070
Gcal 0.34
Gtab (α = 0.05, K =5, n = 5) 0.68
Como Gcal < Gtab (α = 0.05, K =5, n =
5), entonces No hay diferencias significativas entre las varianzas
del %R de los 5 niveles de concentración
Como se puede observar los resultados de %R obtenidos en los cinco niveles de
concentración tienen igual precisión, entonces el valor de %R a reportar será el promedio, a
continuación se puede observar el %R total o promedio con su desviación estándar los
cuales son usados para encontrar el intervalo de confianza del %R ( SX ± ) y la precisión
como expresada como %RSD.
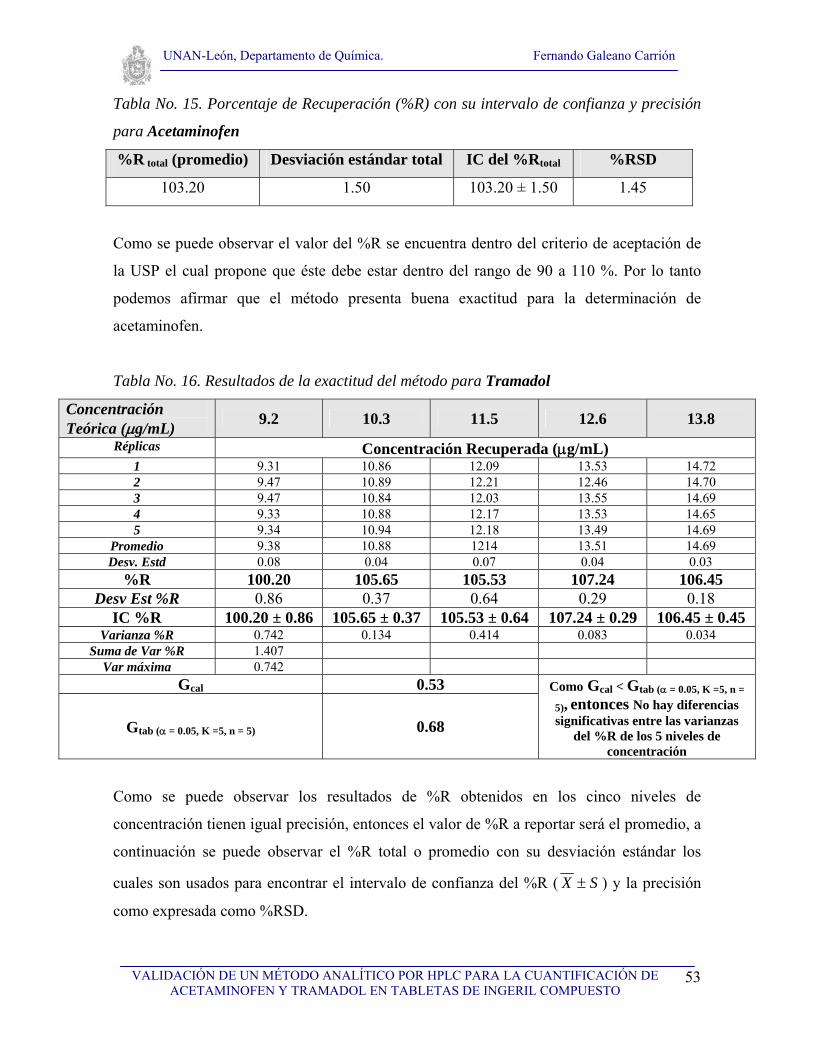
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
53
Tabla No. 15. Porcentaje de Recuperación (%R) con su intervalo de confianza y precisión
para Acetaminofen
%R total (promedio) Desviación estándar total IC del %Rtotal %RSD
103.20 1.50 103.20 ± 1.50 1.45
Como se puede observar el valor del %R se encuentra dentro del criterio de aceptación de
la USP el cual propone que éste debe estar dentro del rango de 90 a 110 %. Por lo tanto
podemos afirmar que el método presenta buena exactitud para la determinación de
acetaminofen.
Tabla No. 16. Resultados de la exactitud del método para Tramadol
Concentración Teórica (μg/mL) 9.2 10.3 11.5 12.6 13.8
Réplicas Concentración Recuperada (μg/mL) 1 9.31 10.86 12.09 13.53 14.72 2 9.47 10.89 12.21 12.46 14.70 3 9.47 10.84 12.03 13.55 14.69 4 9.33 10.88 12.17 13.53 14.65 5 9.34 10.94 12.18 13.49 14.69
Promedio 9.38 10.88 1214 13.51 14.69 Desv. Estd 0.08 0.04 0.07 0.04 0.03
%R 100.20 105.65 105.53 107.24 106.45 Desv Est %R 0.86 0.37 0.64 0.29 0.18
IC %R 100.20 ± 0.86 105.65 ± 0.37 105.53 ± 0.64 107.24 ± 0.29 106.45 ± 0.45 Varianza %R 0.742 0.134 0.414 0.083 0.034
Suma de Var %R 1.407 Var máxima 0.742
Gcal 0.53
Gtab (α = 0.05, K =5, n = 5) 0.68
Como Gcal < Gtab (α = 0.05, K =5, n =
5), entonces No hay diferencias significativas entre las varianzas
del %R de los 5 niveles de concentración
Como se puede observar los resultados de %R obtenidos en los cinco niveles de
concentración tienen igual precisión, entonces el valor de %R a reportar será el promedio, a
continuación se puede observar el %R total o promedio con su desviación estándar los
cuales son usados para encontrar el intervalo de confianza del %R ( SX ± ) y la precisión
como expresada como %RSD.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
54
Exactitud del MétodoAcetaminofen
75,0085,0095,00
105,00115,00125,00135,00
75 85 95 105 115 125
µg/mL(Teórica)
µg/m
L(E
xper
imen
tal)
Tabla No. 17. Porcentaje de Recuperación (%R) con su intervalo de confianza y precisión
para Tramadol
%R total (promedio) Desviación estándar total IC del %Rtotal %RSD
105.37 2.01 105.37 ± 2.01 1.90
Como se puede observar el valor del %R se encuentra dentro del criterio de aceptación de
la USP el cual propone que éste debe estar dentro del rango de 90 a 110 %. Por lo tanto
podemos afirmar que el método presenta buena exactitud para la determinación de
tramadol.
A continuación se muestran los gráficos elaborados de concentraciones teóricas Vs
concentraciones experimentales tanto para acetaminofen como para tramadol.
Gráfico No. 7. Gráfico de valuación de la exactitud del Método para Acetaminofen
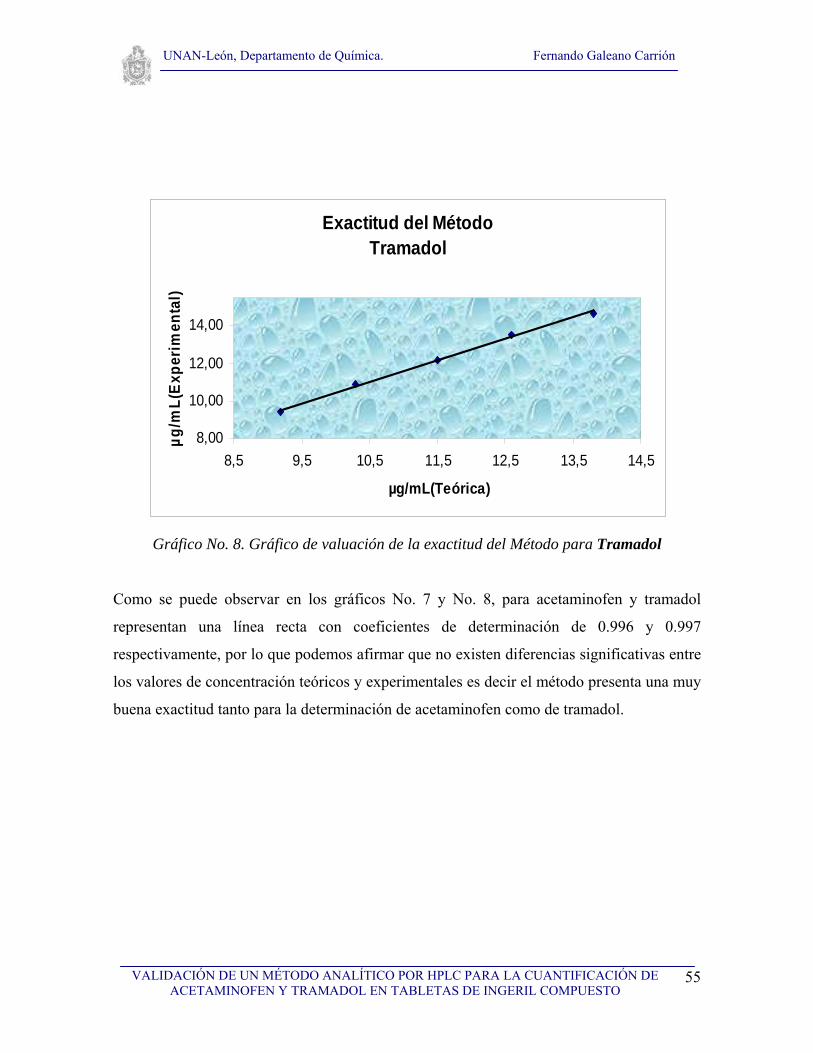
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
55
Exactitud del MétodoTramadol
8,00
10,00
12,00
14,00
8,5 9,5 10,5 11,5 12,5 13,5 14,5
µg/mL(Teórica)
µg/m
L(Ex
peri
men
tal)
Gráfico No. 8. Gráfico de valuación de la exactitud del Método para Tramadol
Como se puede observar en los gráficos No. 7 y No. 8, para acetaminofen y tramadol
representan una línea recta con coeficientes de determinación de 0.996 y 0.997
respectivamente, por lo que podemos afirmar que no existen diferencias significativas entre
los valores de concentración teóricos y experimentales es decir el método presenta una muy
buena exactitud tanto para la determinación de acetaminofen como de tramadol.
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
56
VI.7. Efecto de Matriz y Exactitud del Método
El efecto de matriz es uno de los principales problemas que se presenta a la hora de realizar
una medición analítica ya que los componentes que están presentes en la muestra podrían
aumentar o disminuir la señal analítica y obtener resultados erróneos de las lecturas. Para
evaluar el efecto de matriz en el resultado del análisis de acetaminofen y tramadol en las
muestras de tabletas de Ingeril Compuesto se elaboraron dos curvas, una de calibración
normal (Estándares puros) y otra de adición patrón, para observar dicho efecto se graficaron
ambas curvas en un mismo plano de coordenadas, a continuación se muestran los datos de
las curvas.
Tabla No. 18. Datos de las curvas de calibración normal para acetaminofen y tramadol
Acetaminofen Tramadol
Concentración
μg/mL Área
Concentración
μg/mL Área
80 1486.92 9.2 37.04 90 1671.96 10.3 41.44 100 1874.26 11.5 46.08 110 2051.04 12.6 52.34 120 2248.08 13.8 57.24 b1 19.014 b1 4.459
Sb1 0.194 Sb1 0.169
Tabla No. 19. Datos de las curvas de adición patrón para acetaminofen y tramadol
Acetaminofen Tramadol
Concentración
μg/mL Área
Concentración
μg/mL Área
80 1736.30 9.2 39.05 90 1933.10 10.3 46.28 100 2055.48 11.5 51.78 110 2375.25 12.6 57.58 120 2505.20 13.80 60.60 b1 19.799 b1 4.723
Sb1 1.615 Sb1 0.391

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
57
Acetaminofen
1000,00
1500,00
2000,00
2500,00
3000,00
70,00 90,00 110,00 130,00
µg/mL
Áre
as CAP
CCN
Tramadol
6,00
8,00
10,00
12,00
14,00
16,00
35,00 45,00 55,00 65,00
µg/mL
Áre
as CAP
CCN
Con los datos mostrados en las tablas No. 18 y No. 19 se elaboraron los gráficos para la
evaluación del efecto de matriz, para ello se graficaron en un mismo plano tanto la curva de
calibración normal como la curva de adición patrón.
Gráfico No. 9. Gráfico de valuación del efecto de matriz para Acetaminofen
Gráfico No. 10. Gráfico de valuación del efecto de matriz para Tramadol
Como se puede observar en los gráficos de evaluación del efecto de matriz se obtuvieron
rectas paralelas entre si para acetaminofen y tramadol respectivamente (no se cruzan en
ningún punto) por lo que podemos afirmar que no existe efecto de matriz (ni depresor ni
intensificador de la señal) en la determinación de acetaminofen y tramadol en tabletas de
Ingeril Compuesto.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
58
Con los datos que se muestran en las tablas No. 18 y No. 19, se calculó también en % de
Recuperación (%R) utilizando la siguiente ecuación:
100%)(1
)(1 xbb
RCCN
CAP=
Donde:
%R = es el porcentaje de recuperación
b1(CAP) = es la pendiente de la curva de adición patrón
b1(CCN) = es la pendiente de la curva de calibración normal
Sustituyendo los datos de las tablas 18 y 19 en la ecuación 1 se obtienen los siguientes
resultados:
Tabla No. 20. Resultados de %R calculado a partir de la curva de calibración normal y de
la curva de adición patrón para acetaminofen y tramadol
Acetaminofen Tramadol %R 104.13 105.91
Como se puede observar el valor del %R obtenido de esta forma coincide con los resultados
encontrados en el acápite VI.5 mostrados en las tablas No 15 y No 17, los cuales están
dentro de los límites de aceptación 90 a 110% por lo que de esta forma se confirma que el
método para determinar acetaminofen y tramadol en tabletas de Ingeril Compuesto presenta
muy buena exactitud.
Ec. (6)
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
59
VI.8. Límite de Detección y Límite de Cuantificación
Una de las ventajas de utilizar métodos instrumentales de análisis es que son capaces de
detectar y determinar cantidades de analito mucho más pequeñas que los métodos clásicos.
Uno de los principales problemas que se presentan cuando se trabaja a bajos niveles de
concentración se da a la hora de informar sobre la presencia o ausencia de un analito
determinado por esta razón es muy importante llevar a cabo la determinación tanto del
límite de detección como del límite de cuantificación, esto se realizó utilizando la
desviación estándar del intercepto y la pendiente de la curva de calibración normal
elaborada con estándares de acetaminofen y tramadol, las concentraciones utilizadas en la
curva de calibración normal se encuentra cercana a la señal del blanco. los datos se pueden
observar en la siguiente tabla.
Tabla No. 21. Datos de las curvas de calibración normal para acetaminofen y tramadol
utilizadas para calcular el LD y el LC
Acetaminofen Tramadol
Concentración
μg/mL Área
Concentración
μg/mL Área
0.50 10.53 0.06 0.50 1.00 21.40 0.12 0.43 1.48 32.67 0.17 1.10 3.00 66.93 0.35 2.03 5.00 68.20 0.58 1.80 7.00 152.60 0.81 3.93 9.00 219.70 1.03 5.43 b1 22.97 b1 4.83 Sb0 12.57 Sb1 0.33

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
60
Para el cálculo del límite de detección y el límite de cuantificación se usaron las siguientes
ecuaciones:
Donde Sb0 es la Desviación estándar del intercepto de la curva de calibración normal y b1 es
la pendiente de la curva de calibración normal.
Los resultados obtenidos se muestran a continuación, como se puede observar los valores
encontrados son muy pequeños.
Tabla No. 22. Resultados del LD y LC para acetaminofen y tramadol
Acetaminofen Tramadol
Límite de Detección (L.D.) 1.80 μg/mL 0.22 μg/mL
Límite de Cuantificación (L.C.) 4.47 μg/mL 0.68 μg/mL
1
029.3bxSbLD =
1
010bxSbLC =
Ec. (1)
Ec. (2)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
61
1 2 3 4 580 1561,9 1560,4 1556,5 1566,8 1564,590 1701,2 1709,8 1702,6 1702,2 1704100 1899,8 1903,3 1899,6 1893 1898,3110 2069,7 2067,4 2078,5 2070,6 2080,2120 2276,3 2269,9 2269,2 2281,2 2272,8bo 104,48 125,56 99,98 105,56 111,16b1 17,973 17,766 18,013 17,972 17,928
Acetaminofén
μg/mLDías
1 2 3 4 59,2 41,8 41,8 42,1 42,3 42,510,3 46,9 47,3 47,3 47,1 47,311,5 52,5 52,5 52,3 51,7 52,212,6 57,2 57 56,9 57 57,113,8 63,3 62,7 63 63,4 62,4bo -0,8804052 0,85167826 1,00193529 0,28070759 2,78881161b1 4,6359238 4,47807681 4,4702147 4,53129725 4,31282129
μg/mLDías
Tramadol
VI.9. Repetibilidad de la curva de calibración normal Para observar la repetibilidad en las curvas de calibración normal se realizó un gráfico de
control donde se presentan los límites de tolerancia de los parámetros de regresión
(intercepto y pendiente) de dichas curvas las cuales se elaboraron por cinco días
consecutivos, construyendo los límites de control de la elipse con los parámetros de la
curva del primer día. Estos datos se muestran en las siguientes tablas.
Tabla No. 23. Parámetros de regresión de las curvas de calibración normal para
acetaminofén, realizadas por cinco días consecutivos. Tabla No. 24. Parámetros de regresión de las curvas de calibración normal para tramadol,
realizadas por cinco días consecutivos.
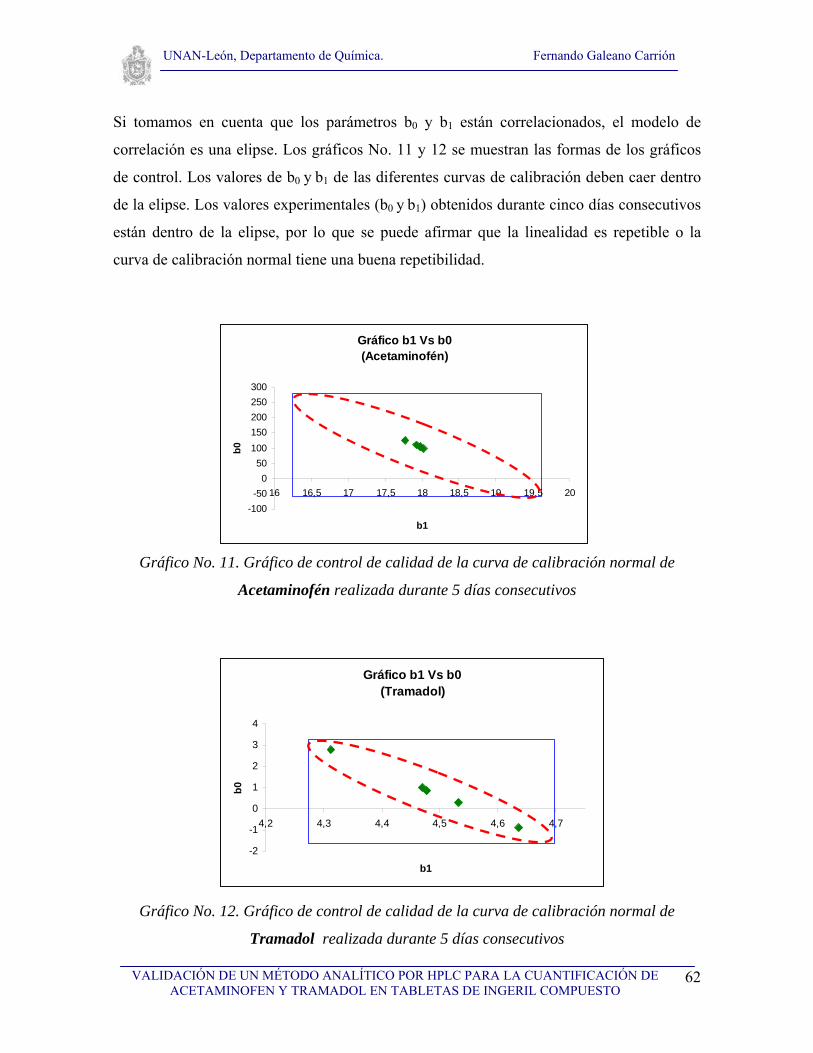
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
62
Si tomamos en cuenta que los parámetros b0 y b1 están correlacionados, el modelo de
correlación es una elipse. Los gráficos No. 11 y 12 se muestran las formas de los gráficos
de control. Los valores de b0 y b1 de las diferentes curvas de calibración deben caer dentro
de la elipse. Los valores experimentales (b0 y b1) obtenidos durante cinco días consecutivos
están dentro de la elipse, por lo que se puede afirmar que la linealidad es repetible o la
curva de calibración normal tiene una buena repetibilidad.
Gráfico No. 11. Gráfico de control de calidad de la curva de calibración normal de
Acetaminofén realizada durante 5 días consecutivos
Gráfico No. 12. Gráfico de control de calidad de la curva de calibración normal de
Tramadol realizada durante 5 días consecutivos
Gráfico b1 Vs b0(Acetaminofén)
-100-50
050
100150200250300
16 16,5 17 17,5 18 18,5 19 19,5 20
b1
b0
Gráfico b1 Vs b0(Tramadol)
-2
-1
0
1
2
3
4
4,2 4,3 4,4 4,5 4,6 4,7
b1
b0

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
63
VI.10. Aplicación del método y estimación de la incertidumbre asociada a la cuantificación de acetaminofén y tramadol en tabletas de Ingeril Compuesto
VI.10.1. Modelo matemático
A continuación se muestra el modelo matemático empleado para la cuantificación de los
principios activos (acetaminofén y tramadol) en tabletas de Ingeril compuesto, el
flujograma del proceso de cuantificación puede ser observado en los anexos.
Donde: Ax: es el área del principio activo obtenida experimentalmente.
b0: es el intercepto de la curva de calibración normal (CCN).
b1: es la pendiente de la curva de calibración normal (CCN).
V2: es el volumen de la alícuota tomada (10 mL).
V3: es el volumen de dilución final (100 mL).
W0: es el peso de la muestra a ser analizada.
W1: es el peso promedio de las tabletas de Ingeril compuesto
VI.10.2. Identificación de las fuentes de incertidumbre
Figura No.7. Identificación de las fuentes de incertidumbres asociadas a la determinación de los principios activos en la muestra de ingeril compuesto
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛ −=
1002
13
1
0
xxWVxWVx
bbAC x
x Ec. (7)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
64
VI.10.3. Cuantificación de los componentes de incertidumbre.
Incertidumbre debida a la lectura del área de la muestra (acetaminofén)
Incertidumbre debida a la lectura del área de la muestra (tramadol)
Incertidumbre del intercepto de la curva de calibración normal para acetaminofén
Incertidumbre de la pendiente de la curva de calibración normal para acetaminofén
Incertidumbre del intercepto de la curva de calibración normal para tramadol
Incertidumbre de la pendiente de la curva de calibración normal para tramadol
06.0506.0
511.0
222)(
2)(
min=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛=
n
U
n
UU métodoPsistemaP
A ofénaceta
27.0525.0
554.0
22)(
2)( =⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛=
n
U
n
UU métodoPsistemaP
Atramadol
75.8561.190
0===
nSbUb
09.0519.01
1===
nSbU b
59.0533.10
0===
nSb
U b
05.0511.01
1===
nSbU b
Ec. (9)
Ec. (8)
Ec. (11)
Ec. (10)
Ec. (12)
Ec. (13)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
65
Incertidumbre debida a la alícuota de 10 mL
01.032
101.2*3*10302.0
32**
3
4222
2
2=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛ Δ
+⎟⎠⎞
⎜⎝⎛=
−xtVUU TV
α
Incertidumbre debida a la dilución de 100 mL
05.032
101.2*3*100308.0
32**
3
4223
2
3=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛ Δ
+⎟⎠⎞
⎜⎝⎛=
−xtVUU TV
α
Incertidumbre debida a la pesada de la muestra (200 mg)
001.032
0001.02001.0
322
2222
0=⎟
⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=
UresUcalUW
Incertidumbre debida a la pesada de la tableta de Ingeril compuesto (650 mg)
001.032
0001.02001.0
322
2222
0=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛=
UresUcalUW
VI.10.4. Cálculo de los coeficientes de sensibilidad (Generalizado para acetaminofén y tramadol)
Coeficiente de sensibilidad par el área
Coeficiente de sensibilidad par el intercepto de la curva de calibración normal
( )( )10**
*
021
13
WVbWV
AC
Cx
xAX
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )10**
*
021
13
00 WVb
WVbC
C xb −=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
Ec. (14)
Ec. (15)
Ec. (16)
Ec. (17)
Ec. (18)
Ec. (19)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
66
Coeficiente de sensibilidad par la pendiente de la curva de calibración normal
Coeficiente de sensibilidad par la alícuota de 10 mL
Coeficiente de sensibilidad par la dilución a 100 mL
Coeficiente de sensibilidad par el peso de la muestra (200 mg)
Coeficiente de sensibilidad para el peso promedio de las tabletas (650 mg)
( )( )( )10**
*
022
1
130
11 WVb
WVbAbCC xx
b−
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10** 021
10
33 WVb
WbAVCC xx
V−
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10**
*
02
21
130
22 WVb
WVbAVC
C xxV
−−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10** 021
30
11 WVb
VbAWC
C xxW
−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10**
*2
021
130
00 WVb
WVbAWCC xx
W−
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
Ec. (20)
Ec. (21)
Ec. (22)
Ec. (23)
Ec. (24)

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
67
Ec. (25)
Ec. (26)
VI.10.5. Estimación de la incertidumbre combinada asociada al proceso de determinación de acetaminofén en ingeril compuesto
Aplicando la ley de propagación de la incertidumbre al modelo matemático (Ec. 7) se
obtiene la siguiente expresión la cual se aplica para el cálculo de la incertidumbre
combinada (La deducción completa de esta ecuación puede ser observada en los anexos).
La contribución de cada fuente de incertidumbre se obtiene finalmente multiplicando la
incertidumbre estándar con su coeficiente se sensibilidad, la incertidumbre combinada es la
suma cuadrática de las contribuciones individuales.
Sustituyendo los valores de los coeficientes de sensibilidad y los valores de las
incertidumbres de cada uno de los parámetros que contribuyen con la incertidumbre en la
ecuación 25, se obtiene el siguiente resultado.
05.2)( =XCUc
VI.10.6. Estimación de la incertidumbre expandida asociada al proceso de determinación de acetaminofén en ingeril compuesto
Tomando un factor de cobertura de 2, para un nivel de confianza del 95.45 %. La
incertidumbre expandida asociada a la cuantificación de acetaminofén en tableta de ingeril
compuesto estará dada por:
11.42 )()( ==XX CC xUcUe
( )22222222
,22222222
)( 1100332210101011002 WWWWVVVVbbbbbbbbbbAAC UCUCUCUCrUUCCUCUCUCUc
XXX+++++++=

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
68
Tabla No. 25. Resultado obtenido para la cuantificación de acetaminofén en tabletas de Ingeril compuesto
Tipo de muestra Analito Concentración Incertidumbre K = 2
Unidades
Ingeril compuesto (tableta) Acetaminofén 316.36 4.11 mg acetaminofén /
650 mg tableta
VI.10.7. Cuantificación y estimación de la incertidumbre expandida asociada al proceso de determinación de tramadol en ingeril compuesto
De igual manera que para el caso de la acetaminofén se introducen los valores
correspondientes a las áreas obtenidas correspondientes a la muestra y los valores de
intercepto y pendientes de la curva de calibración normal en el modelo matemático en el
cual se toman en cuenta los efectos de dilución y la relación de tramadol por tableta, para
cuantificar los mg de tramadol contenidos por tableta.
Al mismo tiempo se estimó la incertidumbre asociada a la cuantificación realizando los
mismos pasos descritos para el caso de la acetaminofén.
Los resultados se describen a continuación:
Tabla No. 26. Resultado obtenido para la cuantificación de Tramadol en tabletas de Ingeril compuesto
Tipo de muestra Analito Concentración Incertidumbre
K = 2 Unidades
Ingeril compuesto (tableta) Tramadol 37.76 1.28 mg tramadol / 650
mg tableta A continuación se muestran los resultados de una serie de determinaciones de acetaminofén y tramadol en tabletas de ingeril compuesto.
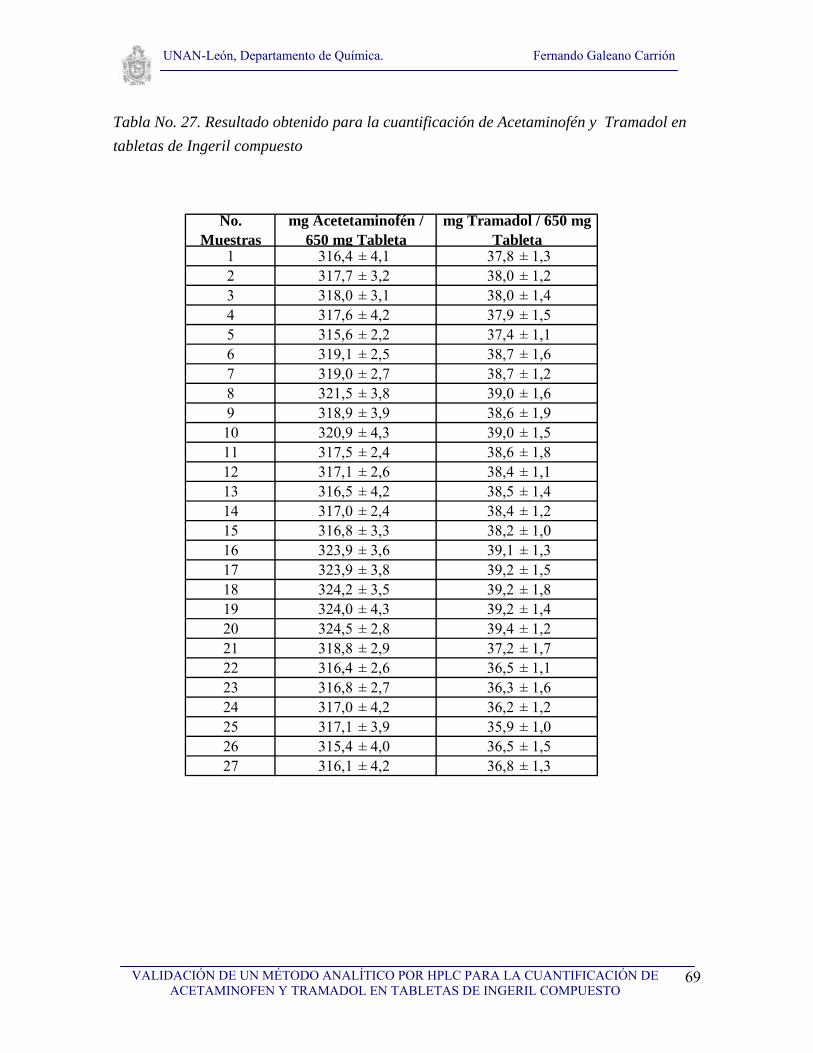
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
69
1 316,4 ± 4,1 37,8 ± 1,32 317,7 ± 3,2 38,0 ± 1,23 318,0 ± 3,1 38,0 ± 1,44 317,6 ± 4,2 37,9 ± 1,55 315,6 ± 2,2 37,4 ± 1,16 319,1 ± 2,5 38,7 ± 1,67 319,0 ± 2,7 38,7 ± 1,28 321,5 ± 3,8 39,0 ± 1,69 318,9 ± 3,9 38,6 ± 1,910 320,9 ± 4,3 39,0 ± 1,511 317,5 ± 2,4 38,6 ± 1,812 317,1 ± 2,6 38,4 ± 1,113 316,5 ± 4,2 38,5 ± 1,414 317,0 ± 2,4 38,4 ± 1,215 316,8 ± 3,3 38,2 ± 1,016 323,9 ± 3,6 39,1 ± 1,317 323,9 ± 3,8 39,2 ± 1,518 324,2 ± 3,5 39,2 ± 1,819 324,0 ± 4,3 39,2 ± 1,420 324,5 ± 2,8 39,4 ± 1,221 318,8 ± 2,9 37,2 ± 1,722 316,4 ± 2,6 36,5 ± 1,123 316,8 ± 2,7 36,3 ± 1,624 317,0 ± 4,2 36,2 ± 1,225 317,1 ± 3,9 35,9 ± 1,026 315,4 ± 4,0 36,5 ± 1,527 316,1 ± 4,2 36,8 ± 1,3
mg Acetetaminofén / 650 mg Tableta
mg Tramadol / 650 mg Tableta
No. Muestras
Tabla No. 27. Resultado obtenido para la cuantificación de Acetaminofén y Tramadol en tabletas de Ingeril compuesto

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
70
VII. CONCLUSIONES
Se demostró que el método es selectivo para la determinación de acetaminofen y tramadol
en tabletas de ingeril compuesto, no se observó ninguna interferencia de ningún tipo.
Se comprobó que el placebo no ejerce ningún efecto en la curva de calibración normal por
lo que ésta podrá ser elaborada tomando en cuenta o no el placebo.
Se logró comprobar la idoneidad del sistema obteniéndose parámetros aceptables es decir
que cumplen con los criterios de aceptación establecidos en la USP
Existe buena linealidad en un buen rango de aplicación, desde 80 μg/mL a 120 μg/mL para
acetaminofen y de 9.2 μg/mL a 13.8 μg/mL para tramadol obteniéndose valores de r2 ≥
0.999, tanto para el sistema como para el método.
El sistema y el método presentaron muy buena precisión para la determinación de
acetaminofen y tramadol en tabletas de Ingeril Compuesto, obteniéndose valores de %RSD
menores del 1%.
El método analítico presentó muy buena exactitud, este se evaluó de dos maneras una
mediante inyecciones de cinco niveles de concentración perfectamente conocidas para
luego compararlas con las concentración obtenida experimentalmente para luego calcular el
% de Recuperación, obteniéndose valores de %R de 103.20 ± 1.50 y %R 105.37 ± 2.01
para acetaminofen y tramadol respectivamente, y la otra manera fue calculando el %R por
medio de las pendientes de las curvas de calibración normal y adición patrón, obteniéndose
valores de %R de 104.13 y 105.915 % para acetaminofen y tramadol respectivamente.
Se demostró que no existe efecto de matriz en la cuantificación de acetaminofen y tramadol
por HPLC en tabletas de Ingeril Compuesto.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
71
Se lograron determinar el Límite de Detección obteniéndose valores de 1.80 μg/mL y 0.22
μg/mL para acetaminofen y tramadol respectivamente y el Límite de Cuantificación 4.47
μg/mL y 0.68 μg/mL para acetaminofen y tramadol respectivamente.
Las curvas de calibración normal presentaron una muy buena precisión, lo cual se demostró
a través de los gráficos de control para el intercepto y la pendiente, observándose que todos
los puntos están dentro de la zona de aceptación del gráfico (Elipse).
Se realizó la cuantificación de acetaminofén y tramadol, obteniéndose buenos resultados
tomando en cuenta que los pesos de las tabletas son muy variables (el peso debe estar entre
90 % – 110 %).
Se estimó la incertidumbre asociada a la cuantificación de acetaminofén y tramadol en
tabletas de ingeril compuesto, obteniéndose resultados aceptables para el % de error: 1 %
para acetaminofén y 3 % para tramadol.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
72
VIII RECOMENDACIONES
• Evaluar la exactitud del método utilizando un material de referencia certificado.
• Realizar el estudio de robustez de este método.
• Realizar el estudio de posibles interferencias debidas a los productos de degradación
de la muestra y del placebo.
• Realizar el estudio de la estabilidad de la muestra
• Realizar análisis de un mayor número de muestras

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
73
IX BIBLIOGRAFÍA
[1] Guillen Corrales Glenda Vanesa, Pavón Villanueva Tania Martina, Chamorro Bautista
Ronald José (Tutor), “Validación de un método de análisis cuantitativo de acetaminofén,
diclofenac sódico, dipirona, fluconazol, piroxicam, (materiaprima) por HPLC”. Tasis (Lic.
En farmacia y Química). Universidad Nacional Autónoma de Nicaragua, León 1999.
[2] A Adames, M. C. Caballero, A. Jonson y M. Puello. “Determinación de Acetaminofén
por HPLC por el Método de Patrón Externo”. Cromatografía de Gases y Líquida de Alta
Eficiencia. Maestría en Ciencias Químicas. Escuela de Química. Facultad de Ciencias
Naturales Exactas y Tecnología. Universidad de Panamá. Panamá 11 de Septiembre de
2008.
[3] Venkateshwarlu K, Reddy YN, Srisailam K, Rajkuermar V and Pai MG.
“Determinación de tramadol en capsulas por cormatografia de capa fina de alta eficiencia”.
Current Trenes in Biotechnology and pharmacy, Vol. 2 (3) 421 – 425 (2008).
[4] Acetaminofén y Tramadol. Consultado en Línea, Fecha de Acceso 21 de Julio del 2009.
Especialistas en Información de la Organización de Teratología (OTIS). Septiembre de
2005. Disponible en la URL:
http://www.otispregnancy.org/pdf/es_acetaminofen.pdf [5] Tramadol. Consultado en Línea, Ultima revisión 29 de mayo del 2009. Fecha de Acceso
22 de Julio del 2009. Disponible en Línea en la URL
http://es.winkipedia.org/wiki/Tramadol [6] Francis Rouessac and Annick Rouessac, Chemical Analysis, Modern Instrumentation
and Techniques, Second edition, John Wiley & Sons, Ltd., 2007.
[7] Jorge Chacón. “Curso teórico práctico en aspectos técnicos del control de calidad
interno del laboratorio de análisis”. CIRA/UNAN-Managua. 1999.
[8] Johk Taylor. AnalChem 55(1983) 600A-608A.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
74
[9] Gustavo Delgado. “Principios de estadística y técnicas de validación”. UNAN-León.
1999.
[10] Alicia Maroto Sánchez. “Incertidumbre en Métodos Analíticos de Rutina” Tesis
Doctoral, Universitar Rovira Virgili, Facultad de Química, Tarragona, 2002.
[11] G-ENAC-09. “Guía para la expresión de la incertidumbre en los ensayos
cuantitativos”. Rev. 1 julio de 2005.
[12] EURACHEM/CITAC Guide CG4. “Quantifying Uncertainty in Analytical
Measurement”. Second Edition. 2000.
[13] ISO 5725-3:1994(E). “Accurecy (trueness and precision) of meausurement
methods and results- Part 3: Intermediate mausures of precision of a standard
measurement methods”. First edition, 1994-12-15.
[14] Comisión Permanente de la Farmacopea de los Estados Unidos Mexicanos, Sexta
Edición, México, 1994.
[15] Cheng Chow Chan, Herman Lam, Y. C. Lee, Xue-Ming Zhang. “Analytical Method
Validation and Instrument Performance Verification”. Wiley-Interscience, a John Wiley &
sons, Inc., Publication. 2004.
[16] Robert A. Nash, Alfred H. Wachter. “Pharmaceutical Process Validation”. an
international third edition. Revised and expanded, Marcel Dekker, Inc. 2003
[17] Christopher M. Riley and Thomas W. Rosanske. “Development and Validation of
Analytical Methods”. Progress in Pharmaceutical and Biomedical Analysis. Volume 3.
1996.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
75
[18] Ludwig Huber, “Validation and Quantification in Analytical Laboratories”. Second
Edition. Agilent Thecnologies Waldbronn, Germany, Informa Healthcare USA, Inc. New
York. 2007.
[19] Douglas A. Skoog, F. James Holler, Timothy A. Nieman, “Principios de Análisis
Instrumental”. Quinta edición. McGraw-Hill/Interamericana de España S.A.. 2001.
[20] Reymond P. W. Scott. “Liquid Chomatography”, Chrom-Ed Book Series.
Library4science, LLC. 2003.
[21] David Harvey. “Modern Analytical Chemistry”. MacGraw-Hill Higher Education. 2000.

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
76
CONCENTRACION Acetaminofen
CONCENTRACION Tramadol Area/Acetaminofen Area/Tramadol
80 9,2 1483,3 32,780 9,2 1485,9 32,680 9,2 1486,8 32,380 9,2 1489,4 32,580 9,2 1489,2 32,490 10,3 1669,9 39,490 10,3 1669,7 39,490 10,3 1672,2 39,490 10,3 1674,1 39,490 10,3 1673,9 39,4100 11,5 1872,7 42,2100 11,5 1874,3 41,9100 11,5 1874,3 42,7100 11,5 1876,1 41,8100 11,5 1873,9 42,5110 12,6 2049,1 48,1110 12,6 2048,8 48110 12,6 2052 47,9110 12,6 2052,5 48110 12,6 2052,8 48120 13,8 2245,6 54,5120 13,8 2247,5 54,2120 13,8 2247,6 54,3120 13,8 2251,2 54,2120 13,8 2248,5 54,3
X. ANEXOS
X.1. Datos completes de la curva de calibración normal sin placebo
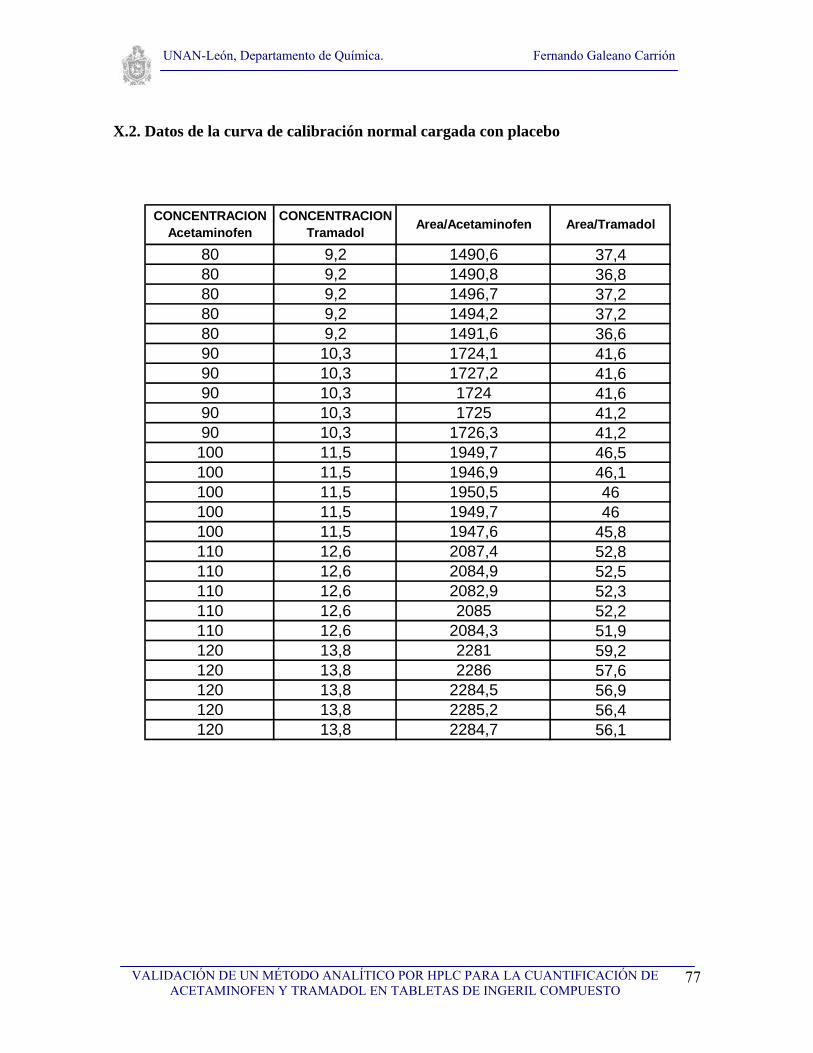
UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
77
80 9,2 1490,6 37,480 9,2 1490,8 36,880 9,2 1496,7 37,280 9,2 1494,2 37,280 9,2 1491,6 36,690 10,3 1724,1 41,690 10,3 1727,2 41,690 10,3 1724 41,690 10,3 1725 41,290 10,3 1726,3 41,2100 11,5 1949,7 46,5100 11,5 1946,9 46,1100 11,5 1950,5 46100 11,5 1949,7 46100 11,5 1947,6 45,8110 12,6 2087,4 52,8110 12,6 2084,9 52,5110 12,6 2082,9 52,3110 12,6 2085 52,2110 12,6 2084,3 51,9120 13,8 2281 59,2120 13,8 2286 57,6120 13,8 2284,5 56,9120 13,8 2285,2 56,4120 13,8 2284,7 56,1
CONCENTRACION Acetaminofen
CONCENTRACION Tramadol Area/Acetaminofen Area/Tramadol
X.2. Datos de la curva de calibración normal cargada con placebo

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
78
X.2. Flujograma del procedimiento analítico

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
79
X.3. Deducción de la ecuación para estimar la incertidumbre asociada a la determinación de acetaminofén y tramadol en tabletas de ingeril compuesto.
Modelo Matemático:
Aplicación de la Ley de Propagación de la Incertidumbre al Modelo Matemático se obtiene:
11
00
33
22
11
00
dWWC
dWWC
dVVC
dVVC
dbbC
dbbC
dAAC
dC xxxxxxx
x
xx ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
∑∑∑∑∑∑∑∑ ⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
= 11
00
33
22
11
00
dWWC
dWWC
dVVC
dVVC
dbbC
dbbC
dAAC
dC xxxxxxx
x
xx
( )2
11
00
33
22
11
00
2⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
= ∑∑∑∑∑∑∑∑ dWWC
dWWC
dVVC
dVVC
dbbC
dbbC
dAAC
dC xxxxxxx
x
xx
( ) ( ) ( ) ( )
( ) ( )∑∑
∑∑∑∑∑∑
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
21
2
1
20
2
0
23
2
3
22
2
2
2
11
00
22
2
dWWC
dWWC
dVVC
dVVC
dbbC
dbbC
dAAC
dC
xx
xxxxx
x
xx
( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( )∑∑∑∑
∑∑∑∑∑∑
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
21
2
1
20
2
0
23
2
3
22
2
2
101010
21
2
1
20
2
0
22
2 ,2
dWWC
dWWC
dVVC
dVVC
bbrdbdbbC
bC
dbbC
dbbC
dAAC
dC
xxxx
xxxxx
x
xx
⎟⎠⎞
⎜⎝⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛ −=
101
0
1
2
3
1
0
WW
VV
bbA
C xx

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
80
Correlación entre el intercepto y la pendiente.
Calculo de los coeficientes de Sensibilidad
Derivando parcialmente cada uno de los términos del modelo matemático se obtienen los coeficientes de sensibilidad, los cuales se muestran a continuación:
( )[ ] ( ) ( ) ( ) ( )
( ) ( ) ( ) ( )N
dWWC
dWWC
dVVC
dVVC
Nbbrdbdb
bC
bC
dbbC
dbbC
dAAC
dCN
xxxx
xxxxx
x
xx
1
1,21
21
2
1
20
2
0
23
2
3
22
2
2
101010
21
2
1
20
2
0
22
2
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+
⎥⎥⎦
⎤
⎢⎢⎣
⎡+⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
∑∑∑∑
∑∑∑∑∑∑
( ) ( )nXn
Xr
i
ibb ∑
∑−=2, 10
( )
22
1
22
0
22
3
22
2,
22
10
22
1
22
0
22
2
10
321010102
Wx
Wx
Vx
Vx
bbbbxx
bx
bx
Ax
xC
UWC
UWC
UVC
UVC
rUUbC
bC
UbC
UbC
UAC
UXX
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )10**
*
021
13
WVbWV
AC
Cx
xAX
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )10**
*
021
13
00 WVb
WVbC
C xb −=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
⎟⎠⎞
⎜⎝⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛ −=
101
0
1
2
3
1
0
WW
VV
bbA
C xx

UNAN-León, Departamento de Química. Fernando Galeano Carrión
VALIDACIÓN DE UN MÉTODO ANALÍTICO POR HPLC PARA LA CUANTIFICACIÓN DE ACETAMINOFEN Y TRAMADOL EN TABLETAS DE INGERIL COMPUESTO
81
( )( )
0
*
1
130
=
−=
dbdU
WVbAU x ( )
( )10**
10**
021
021
WVdbdU
WVbU
=
=
( )( )( )10**
*
022
1
130
11 WVb
WVbAbCC xx
b−
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )[ ] ( ) ( )( )[ ] ( )( )( )[ ]
( )( )[ ] ( )( )[ ]
( )( )[ ]( )10**
*10**
10****
10**10****0*10**
022
1302
021
02130
1
2021
02130021
1
1
1
1
WVbWVbA
WVbWVWVbA
bC
C
WVbWVWVbAWVb
bC
C
xxxb
xxb
−−=
−−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
−−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )[ ] ( )( )[ ] ( )( )[ ] ( )[ ]( )[ ]2021
021130130021
1 10**10******10**
1 WVbWVbdWVbAWVbAdWVb
bC
C xxxb
−−−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10** 021
10
33 WVb
WbAVCC xx
V−
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10**
*
02
21
130
22 WVb
WVbAVC
C xxV
−−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10** 021
30
11 WVb
VbAWC
C xxW
−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=
( )( )( )10**
*2
021
130
00 WVb
WVbAWCC xx
W−
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=
( )22222222
,222222222
1100332210101011002 WWWWVVVVbbbbbbbbbbAAC UCUCUCUCrUUCCUCUCUCU
XXX+++++++=
( )22222222
,22222222
1100332210101011002 WWWWVVVVbbbbbbbbbbAAC UCUCUCUCrUUCCUCUCUCU
XXX+++++++=
XX CC UUe 2= Incertidumbre expandida
Incertidumbre Combinada