







Idiomas
Páginas
Jurídico


TalasemiasPor: Tatiana Isabel Rodríguez Galán

Definición, historia y etiología
1. Cooley + Ley niños con anemia grave con esplenomegalia y alteraciones óseas ANEMIA DE COOLEY (cadena beta/talasemia mayor)
2. Talassa: mar, antepasados con origen Mediterráneo3. 1940: carácter genético
Grupo polifacético de trastornos heredados, causado por mutaciones genéticas que
disminuyen o anulan síntesis de una o más cadenas de globina
TalasemiaMayor
homocigoto
Talasemia menor
heterocigoto
Factor genéticos más
frecuente
Más gravesMortalidad y morbilidad elevadas

Un grupo de trastornos: Afección que disminuye la tasa de síntesis de uno
o más de las cadenas de globina produciendo un desequilibrios
Producción de hemoglobina defectuosa Deterioro de los eritrocitos o sus percusores
Se define su nombre por la cadena afectada:
Cadena alfa o beta de la
HbA
HbA;α2,ß2

Control genético de la síntesis de la hemoglobina
Hemoglobina normal: tetrámetro (dímero doble) Dos cadena tipo alfa Dos cadenas tipo beta
Producen 6 hemoglobinas
normales
Hb Gower-1Hb Gower-2Hb Portland
Embrionarias
HbF (2%)
Fetal
HbA (95-97%)
HbA2 (2-3%)
Adulta
Genes alfa y zeta Brazo corto cromosoma 16Genes beta Brazo corto cromosoma 11
Desplazamiento gamma-beta

Categorías de talasemias

Distribución geográfica
Α-talasemiaThailandia, China, Filipinas y asiáticos
B-talasemiaMediterráneo (Italia/Grecia)India, Pakistán y sudeste asiático
Baja de paludismoResistencia

Fisiopatología Desequilibrio de la síntesis de las cadenas de
hemoglobina
ß-talasemia
Falta de hemoglobina
Precursores eritroides
Eritrocitos pequeños e
hipocrómicos
Exceso de cadena de globina no pareadas
Daños a la membrana
Eritropoyesis ineficaz
Hemólisis prematura por
macrófagos
Asintomáticos hasta los 4-6
meses de vida
α-talasemia
Cadena alfa compartida en fetal y adulta
Un exceso de producción de otras cadenas
Feto: exceso producción de
gamma
Son solubles no
eritropoyesis ineficaz grave
Precipitan conforme eritrocito
envejece en la sangre
Forman cuerpo de inclusión
Eritrocitos: microcíticos e hipocrómicos
Ineficaz transporte de
oxígeno
Valores normales de hemoglobina en adultos
HbA 95-97%HbA2 2-3%HbF 2%

Defectos genéticos que causan talasemia
• Disminuyen cantidad de mRNA
Mutación puntual
• Altera la función promotora
Sustitución de bases
• Impide la síntesis del polipéptido de la globina completo y normal
Mutaciones de inserción o de deleción dentro de la región codificadora del mRNA
• Elimina uno o más gene y altera la regulación de los restantes
Grandes delaciones dentro de los grupos alfa o beta

Grupo de las ß-talasemias Más de 70 mutaciones asociadas Se dividió en tres síndromes clínicos, se
agrego uno nuevo1. B-talasemia menor (heterocigota): anemia
hemolítica leve, micrócítica e hipocrómica2. B-talasemia mayor (homocigota): anemia grave
que depende de transfusiones3. B-talasemia intermedia: síntomas de gravedad
intermedia entre los otros dos4. De portador silencioso
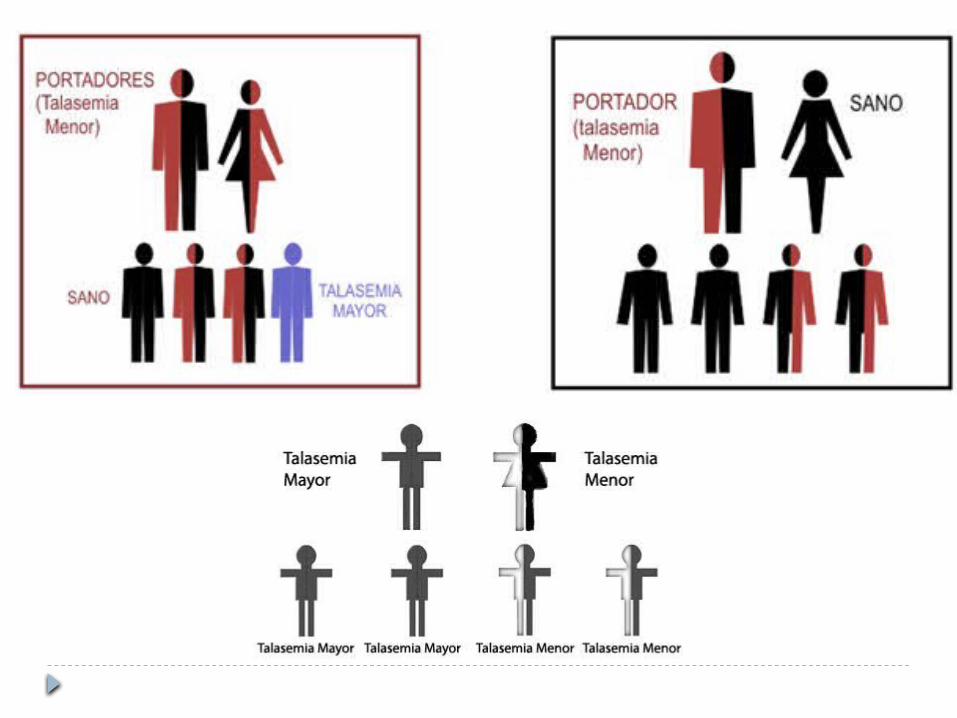

De portador silencioso Presentan diferentes mutaciones
heterogéneas beta Producen un pequeña disminución de la
cadena beta Proporción de cadena alfa/cadena beta casi
normal y ninguna anomalía hematológica
Niveles normales de HbA2 Microcitosis muy leve
Homocigotos: anemia microcítica hipocrómica moderada con hemoglobina 6-7 g/dl
HbF 10-15%

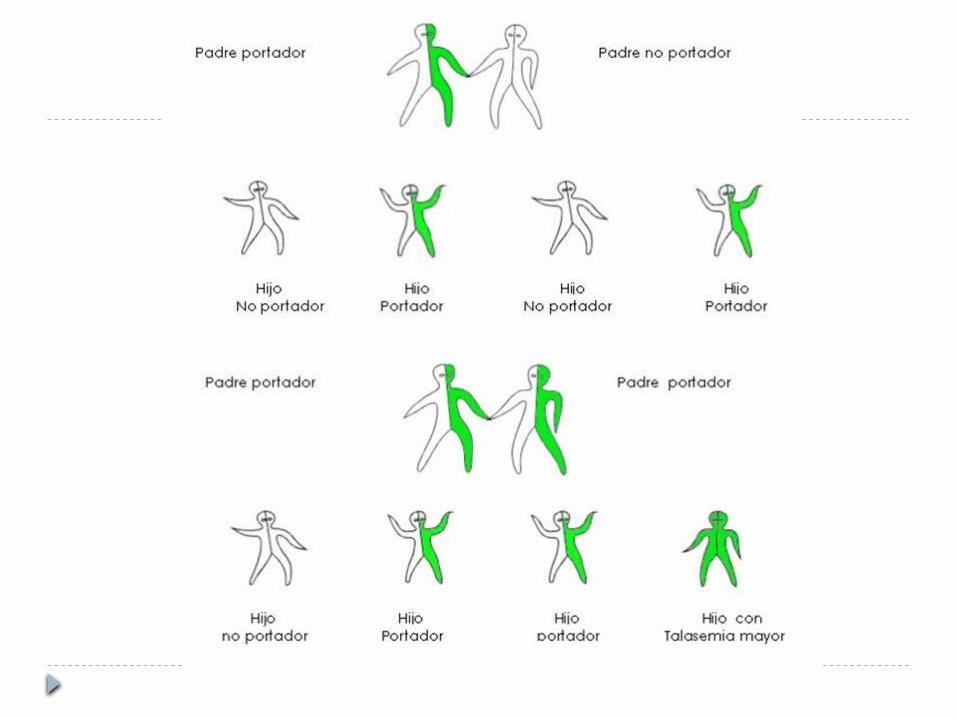
B-talasemia menor Consecuencia de mutaciones heterocigotos
que afectan la síntesis de la beta-globina Anemia hemolítica leve, ASINTOMATICA Un gen afectado disminuido o nulo pero el
otro normal Sangre periférica valores Hb: 10-13 g/dl Anemia microcítica hipocrómica Poiquilocitosis
Hiperplasia eritroide leve, eritropoyesis
ineficaz
Hepatomegalia y
esplenomegalia
Elevación de HbA2 y HbF

B-talasemia mayor Anemia grave detectada desde la infancia, en
el cambio gamma-beta Diagnóstico: 6 meses y 2 años Sin tratamiento con hipertransfusión:
hepatoesplenomegalia con ictericia y cambios óseos (agrandamiento de cavidad medular) + fracturas patologicos
Fascias típicas: prominencia frente, pómulos y maxilares superiores
Crecimiento físico y desarrollo retrasados.


B-talasemia mayor
Niveles de Hb muy bajos (3-4
g/dl)
Eritrocitos: hipocrómicos, poiquilocitos
extrema.
Presencia de ertitrocitos
agrandados y muy delgados
Reticulocitos bajos en
relación con grado de
hiperplasia
Muerte intramedular
de los precursores eritrocitarios
HbF (mayoría) con HbA2 algo aumentada y HbA nula o disminuida

B-talasemia mayor Nivel de ferretina: elevado Hipertransfusión: evitar anemia, eritropoyesis
y alteraciones óseas + NO megalia Causa de morbilidad: toxicidad en órganos
parenquimatosos por transfusiones carga de hierro y hemosiderosis transfusional
Administración de quelante (desferrioxamina) >4 años
Trasplante de médula ósea

Talasemia intermedia Describir pacientes que pueden mantener
nivel Hb ≥7 g/dl sin necesidad de transfusión Morfología de eritrocitos similar para
talasemia mayorHbF 2-100%
HbA2 7%HbA 180%
Evolución clínica variablePueden tener problemas de hierro aun sin transfusiones Eritropoyesis aceleradaComplicaciones cardiacas y endocrinas: 10-20 años después

Diagnóstico prenatal: ADN por biopsia de las vellosidades coriónicas o por amniocentesis
Persistencia hereditaria de
la hemoglobina
fetal
Hemoglobinas Lepore y Kenya
(globinas cruzadas)

α-talasemia Más frecuente: deleciones en los genes de la
alfa-globina Grado de producción = mutación especifica, #
genes afectados y si es gen a2 o a1α2: produ
ce 75%
de las caden
as
α1: solo
produce
25%
Portador silencioso
Rasgo de α-talasemia (menor)
Enfermedad por
hemoglobina H
Hidropesía fetal
(homocigoto)

Portador silencioso Deleción de un gen de alfa-globina, solo deja
tres funcionales (- α/ α α) Es casi normal, no se observan problemas
hematológicos Solamente por estudios genéticos

Rasgo de alfa-talasemia Causado por la forma homocigota (- α/- α) Muestra una anemia leve con eritrocitos con
microcitosis e hipocromía marcados

Enfermedad por hemoglobina H Presencia de un solo gen productor de cadena alfa (--/- α) Causada por combinaciones del gen Más frecuente en asiáticos Formara tetrámetros inestables de HbH Anemia hemolítica crónica leve a moderada Concentraciones de Hb: 7 a 10 g/dL y reticulocitos del 5
al 10% Medula ósea presenta hiperplasia eritroide y el bazo suele
estar agrandado Infecciones, embarazo, exposición a fármacos: crisis
hemolíticas Esplenectomía: beneficiosa Asociado a trastornos mieloproliferativos y
mielodisplásicos

Hidropesía fetal La alfa-talasemia homocigota (--/--) es
incompatible con la vida y se caracteriza por ausencia total
Lactante nace con hidropesía fetal: edema causado por acumulación de líquido seroso en tejidos fetales por anemia grave
Vive hasta tercer trimestre Nace pretérmino, muerto o muere poco
después Peligroso para la madre: toxemia y
hemorragias postparto graves Interrupción del emabarazo

Diagnóstico
Top Related