







Idiomas
Páginas
Jurídico


ESCUELA: BIOQUIMICA Y FARMACIA
NOMBRE:
TECNOLOGÍA FARMACÉUTICA
FECHA:
B.F. Geovanni López
ABRIL – AGOSTO 2008

FARMACOCINETICA:lo que el organismo hace sobre el
fármaco:
Dosis-Concentración
FARMACODINAMICAlo que el fármaco hace en el organismo
Concentración-Efecto

FARMACOCINÉTICA
Para que un fármaco pueda actuar, debe:
Llegar al lugar de acción (superar barreras biológicas)
Llegar al receptor Alcanzar la concentración

Para alcanzar la concentración efectiva: penetrarse en el organismo
absorción para llegar al plasma distribuirse por los tejidos
distribución Pero tan pronto como penetra
eliminación en el organismo es sometido
Dos mecanismos
vías naturales (orina, bilis, saliva, etc..)
metabolismo o biotransformación enzimática
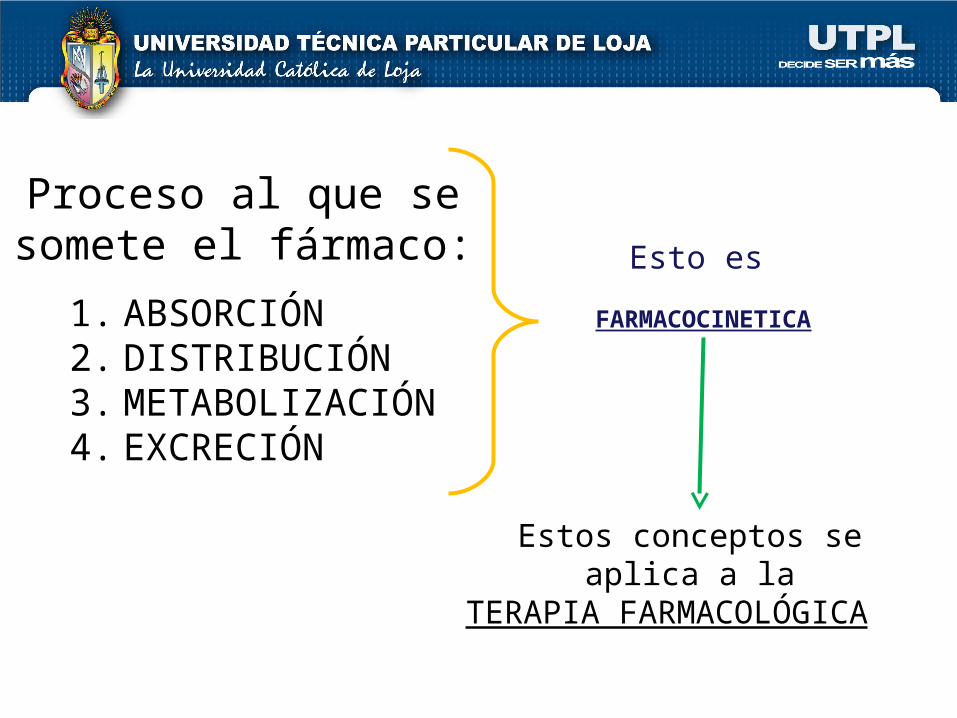
Proceso al que se somete el fármaco:
1. ABSORCIÓN2. DISTRIBUCIÓN 3. METABOLIZACIÓN4. EXCRECIÓN
Esto es
FARMACOCINETICA
Estos conceptos se aplica a la
TERAPIA FARMACOLÓGICA
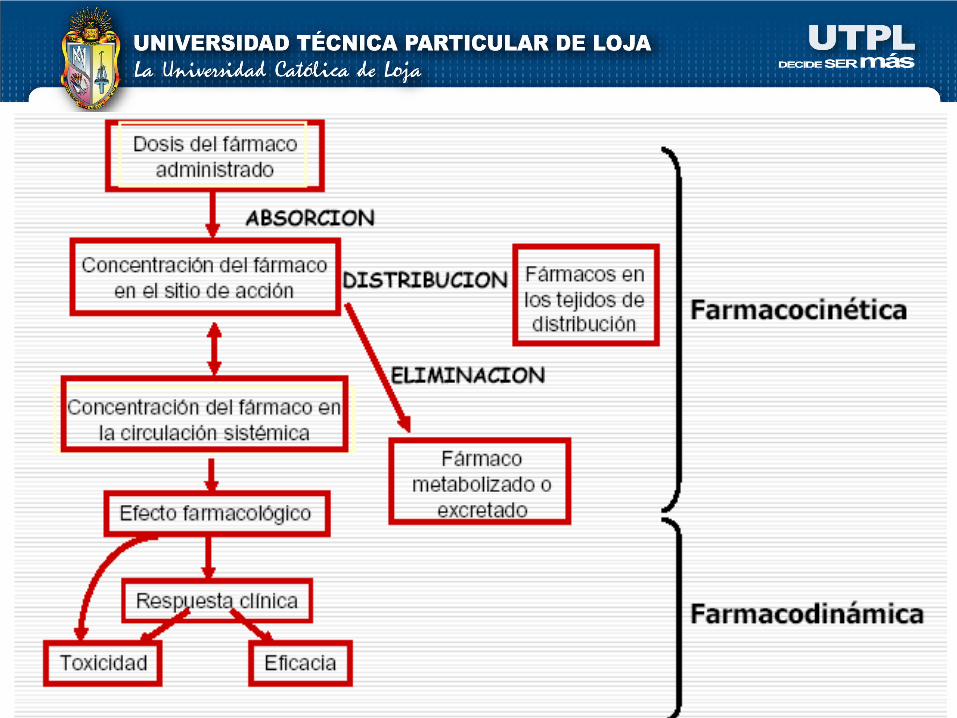

“PROCESO LADMER”
L : LIBERACIÓN
A : ABSORCIÓN
D : DISTRIBUCIÓN
M : METABOLISMO
E : ELIMINACIÓN
R : RESPUESTA

“PROCESO LADMER”
LIBERACIÓNConsiste en pasar el principio activo
de la forma farmacéutica al lugar donde se absorbe
Se efectúa bajo la influencia del medio biológico y de las
condiciones mecánicas del lugar de administración.

ABSORCIÓN
Comprende:Liberación del fármaco Índice de
disoluciónEliminación presistémica
Proceso:Fármaco va desde:Sitio o vía de administración Circulación
sanguínea
ATRAVEZANDO MEMBRANAS
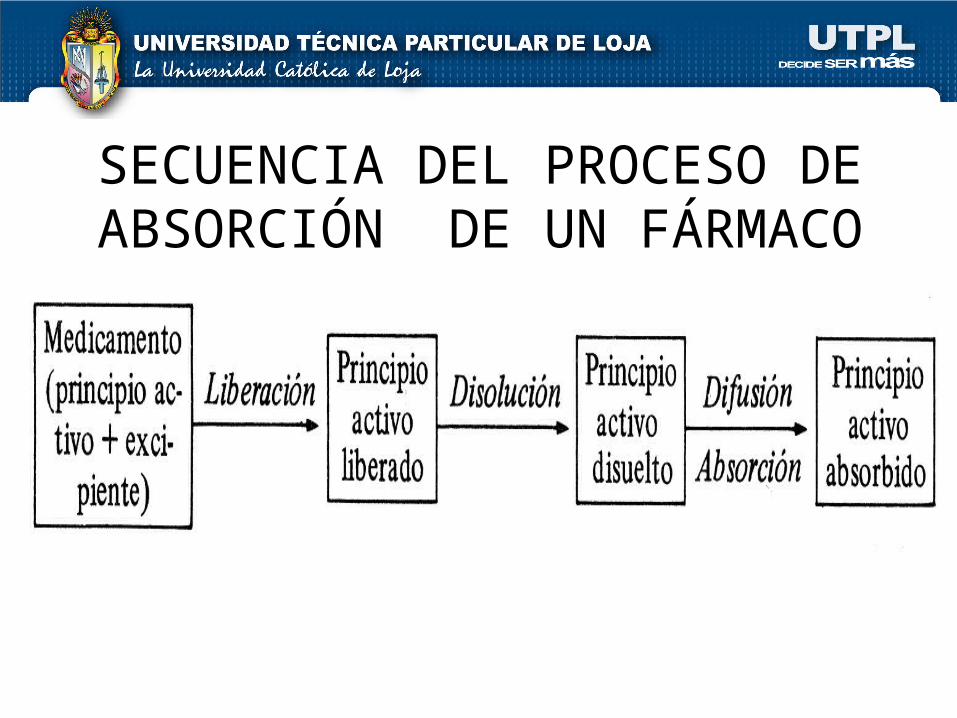
SECUENCIA DEL PROCESO DE ABSORCIÓN DE UN FÁRMACO

Los factores importantes:Dependientes del fármaco
Dosis Propiedades fisicoquímicas, particularmente la estereoquímica y
solubilidad del fármaco Tamaño de partícula y área superficial Forma farmacéutica y sistema de liberación
Dependientes del sitio orgánico de absorción. Ruta y sitio de administración Tiempo de contacto con la superficie de absorción Área de la superficie de absorción PH del sitio de absorción Integridad de la membrana Vascularización del sitio de absorción.
FACTORES QUE MODULAN LA ABSROCIÓN

FACTORES DEPENDIENTES DEL FÁRMACO
Gradiente de concentración: concentración del fármaco al lado externo de la membrana celular.
Grado de ionización: la fracción no ionizada del fármaco es liposoluble y difunde con facilidad, en tanto que la fracción ionizada es hidrosoluble y sólo difunde con dificultad a través de las membranas celulares.

FACTORES DEPENDIENTES DEL FÁRMACO
Liposolubilidad: mientras más liposoluble es una droga mas fácilmente se absorbe viceversa.
Tamaño de molécula: mientras más pequeño es el tamaño de la molécula del fármaco, su absorción es más rápida e inversamente, será más lenta cuando mayor sea el diámetro molecular.

FACTORES DEPENDIENTES DEL SITIO DE ABSORCIÓN
SUPERFICIE: La absorción es tanto más rápida, cuanto mayor y más prolongado es el contacto del fármaco en la superficie de absorción.
RIEGO SANGUÍNEO: a mayor irrigación sanguínea mayor absorción

FACTORES DEPENDIENTES DEL SITIO DE ABSORCIÓN
Ph del medio: el valor del pH en el que se encuentra un fármaco incide en el grado de ionización, en efecto los ácidos débiles se ionizan más a valores altos de pH, y las bases débiles se ionizan a valores bajos de pH.
Tiempo de contacto: a mayor tiempo de contacto con la superficie mayor absorción.

El proceso de absorción se realiza por varios mecanismos, entre los cuales están:
1. Difusión pasiva (para la mayoría de fármacos).
2. Transporte activo3. Filtración a través de poros, para
moléculas pequeñas, 4. Pinocitosis.- para macromoléculas.

FACTORES QUE MODIFICAN LA ABSORCIÓN POR LAS DIFERENTES VÍAS DE
ADMINISTRACIÓN

Vía sublingual:La absorción es mediante la mucosa
sublingual.Sirve para evitar el efecto de primer paso
(hepático)Es más rápida que la oral.
Ejemplo:
la nitroglicerina usada en el tratamiento de angina de pecho se absorbe con facilidad
y a gran velocidad por la mucosa oral.

2.-Vía oral:
Es la más común, cómoda y barata.El sujeto está consciente. No se puede administrar cuando existen
vómitos. El fármaco se va a absorber:Si es ácido estómago Si es básico intestino delgado. La mayoría de fármacos son ácidos y bases
débiles.

El efecto tarda en aparecer Es la de más lenta absorción Sabor desagradable Metabolización al paso por el
hígado antes de distribuirse Sublingual una variedad de esta
vía, la ventaja es que elude la travesía hepática

Factores que influyen en la absorción del fármaco por vía
oral pueden ser: Los alimentos.-disminuyen la
absorción del fármaco. Ejemplo: Cimetidina ingerida con alimentos se
absorbe lentamenteTetraciclina con desayuno no se absorbeRiboflavina y griseofulvina.- el alimento
favorece la absorción pH del estómago .-Si las moléculas
del fármaco se destruyen se administra por otra vía o se recubre con una cubierta entérica.

Factores que influyen en la absorción del fármaco por vía
oral pueden ser: El tubo digestivo ofrece:
Superficie absorbente de 70 m2 Gran irrigación intestinal (mas profusa que el
estómago)Rango de pH de 5 a 8 en las diferentes porciones
del intestino
Y la prolongada permanencia de la droga favorece la absorción de ácidos y bases débiles. Ejemplo barbitúricos, salicilatos, morfina, quinina, fedrina.

Factores que influyen en la absorción del fármaco por vía
oral pueden ser: Efecto de primer paso: El fármaco en el tubo digestivo antes de pasar
a la circulación sistémica: Las venas del tubo digestivo llegan al hígado
por vena porta, metabolizándose ciertas sustancias en determinadas proporciones, pasando después a la circulación sistémica.
Algunas sufren una metabolización importante.
A mayor vaciamiento gástrico la absorción intestinal se hace a mayor velocidad.

3.-Vía rectal: El fármaco se absorbe en la mucosa
rectal.Absorción rápida a través de la mucosa colónica pasa a las venas hemorroidales inferiores y por esta a las vena hipogástrica para ganar la circulación general sin pasar por el hígado.
Es errática.- ya que los fármacos se absorben mal y de forma irregular.

VÍA RECTAL
Formas de supositorios y enemas Las formas líquidas se van a absorber
mejor que las sólidas. Evita las sustancias irritantes y mal
sabor por vía oral y problemas de deglución. Se aplica a pacientes con vómito

4.-Vía intramuscular:Normalmente el músculo va a estar muy bien
vascularizado (permitir una gran absorción) Es muy rápida (más que la oral). Se utiliza porque no se puede absorber por la
mucosa.Los problemas que conlleva esta vía son los
siguientes: va a requerir un instrumental estéril, la técnica es dolorosa, pueden producirse lesiones e infecciones.
Siempre existe el riesgo de administración intravenosa errónea

Factores que influyen en la absorción del fármaco por vía
intramuscular pueden ser: La solubilidad.- las drogas:
Las liposolubles se absorben con mayor facilidad. Las hidrosolubles se absorben mientras menor sea el tamaño de la molécula.
Las suspensiones o preparaciones coloidales se absorben más lentamente que las soluciones acuosas.
La absorción depende de la superficie y el riego sanguíneo en la zona.

6.-Vía intravenosa:Administración de fármacos a la corriente
sanguínea. Es muy rápida.-es la vía de elección en las
urgencias.Permite administrar grandes cantidades de
líquido y obtener concentraciones plasmáticas altas y precisas.
Los problemas son: Requiere instrumental, esterilización, aumenta
los efectos indeseables, posibilidad de infecciones, posibilidad de embolias vasculares y cuadros alérgicos.

7.-Vía cutánea:
A través de la piel. Normalmente hay que disolver la sustancia para facilitar su paso.
Es difícil atravesar las capas de piel. Esto hace que la vía cutánea quede para el tratamiento de patología superficial.
Esta vía esta en relación directa con la solubilidad en lípidos, así ocurre con la Vitamina D, las hormonas esteroides y anestésicos

7.-Vía génito-urinaria:
La vagina tiene una mucosa que se deja penetrar por sustancias liposolubles como los esteroides sexuales, más difícilmente por fármacos hidrosolubles, de ahí el objeto primordial de esta vía es producir efectos locales.

VIAS DE ADMINISTRACIÓN DE MEDICAMENTOS
1. Vías enterales: oral y rectal2. Vías parenterales: intradérmica,
subcutánea, intramuscular, intravenosa, intraarterial, intratecal
3. Vía inhalatoria4. Vía tópica

VÍA INTRADÉRMICA Soluciones isotónicas inferior a un
volumen de 1 ml se inyectan en la epidermis

VÍA INTRAMUSCULAR Vía de absorción rápida ya que tiene
mayor irrigación Útil en pacientes que no degluten,
inconscientes
VÍA INTRARTERIAL• Permite tener efectos en un área
determinada, en forma instantánea y en altas concentraciones, pues la droga no se diluye en todo el volumen plasmático.

VÍA INTRAVENOSA
Produce efectos inmediatos ya que la droga es entregada directo al torrente sanguíeno
Alcanza niveles plasmáticos en pocos segundos
Conlleva a mayor riesgo de infecciones virales, bacteriana

VÍA INHALATORIA
La cantidad de sangre que atraviesa por la superficie de los alvéolos en unidad de tiempo es bastante grande, por lo mismo la velocidad de absorción por esta vía es mayor que por el intestino, los músculos o la piel
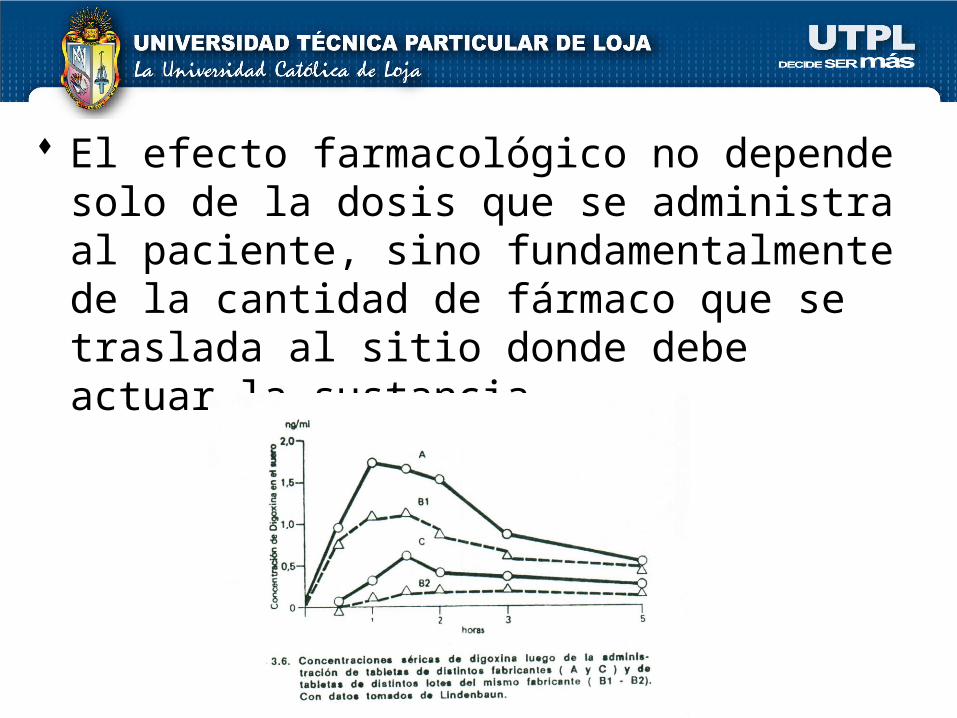
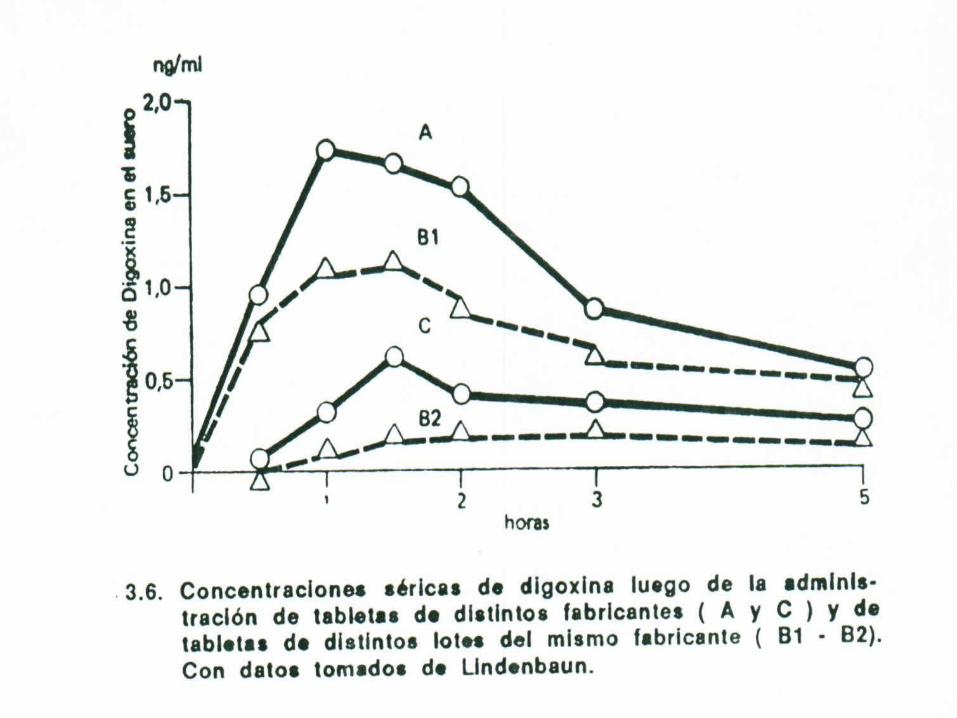
El efecto farmacológico no depende solo de la dosis que se administra al paciente, sino fundamentalmente de la cantidad de fármaco que se traslada al sitio donde debe actuar la sustancia
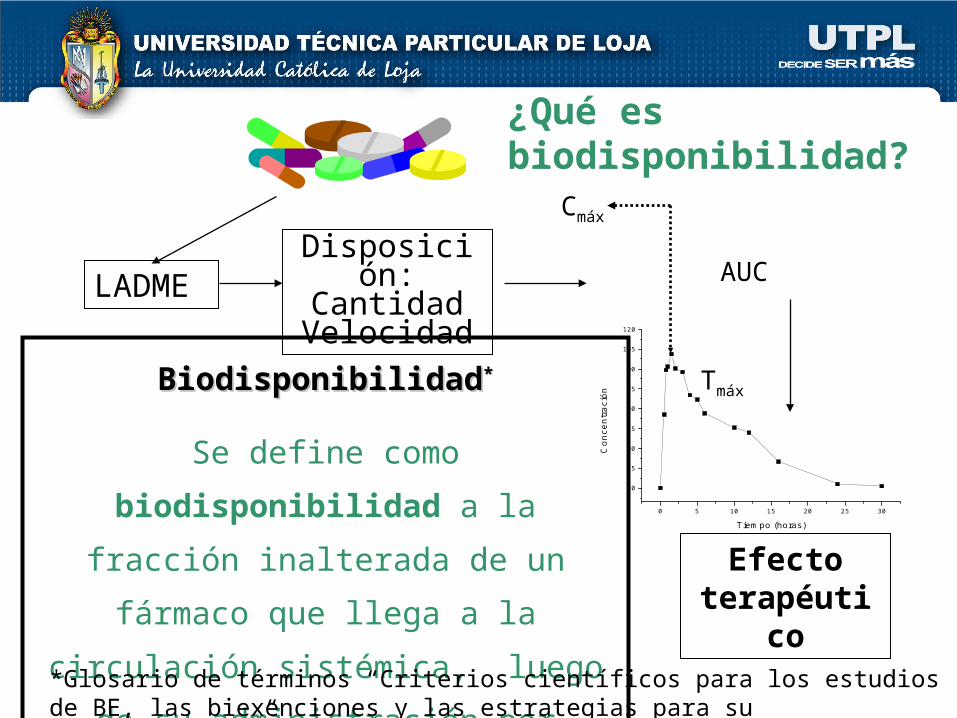
AUCLADMEDisposición:
Cantidad Velocidad
Efecto terapéutico
BiodisponibilidadBiodisponibilidad**
Se define como biodisponibilidad a la
fracción inalterada de un fármaco que
llega a la circulación sistémica, luego de
su administración por cualquier vía.
0 5 10 15 20 25 30
0
15
30
45
60
75
90
105
120
Conce
ntr
aci
ón
Tiempo (horas)
Cmáx
Tmáx
¿Qué es biodisponibilidad?
*Glosario de términos “Criterios científicos para los estudios de BE, las biexenciones y las estrategias para su implementación” Rep. Dom. Marzo 2005

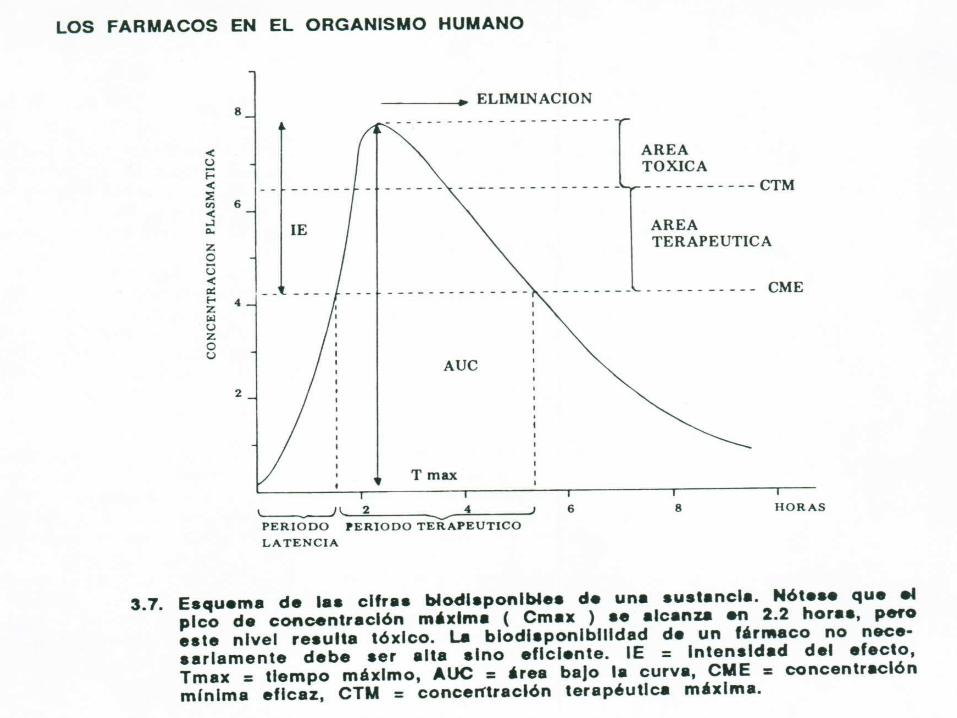
La máxima biodisponibilidad se puede alcanzar cuando es administrado por vía
intravenosa:tiene un valor de 1 (£ 100%) ya que por esta vía el fármaco no sufre el efecto de
primer paso
BIODISPONIBILIDAD
Otras vías que requieran absorción tienen valores de biodisponibilidad iguales o
menores a 1 (£ 100%) debido principalmente a dos fenómenos: absorción incompleta o
efecto de primer paso

BIODISPONIBILIDAD El efecto de primer paso consiste en
una degradación que sufre el fármaco por metabolización en la misma pared intestinal al ser absorbido (testosterona), en la sangre de la circulación portal y más a menudo a su primer paso por el hígado, antes de alcanzar la circulación sistémica (propranolol).

BIODISPONIBILIDADEl concepto de biodisponibilidad
comprende dos aspectos: la intensidad de la absorción, medida
como el área bajo la curva (AUC) la velocidad de absorción,
representada por la concentración plasmática máxima alcanzada (Cmax) y el tiempo requerido para alcanzar dicha concentración máxima (Tmax).

Equivalentes farmacéuticos:
Son dos medicamentos que contienen el mismo principio activo en igual cantidad y forma farmacéutica. Y
presentan perfiles de concentración plasmática versus tiempo (área bajo
la curva, Cmax y Tmax) estadísticamente iguales

BioequivalenciaBioequivalencia**
Significa la ausencia de una diferencia significativa en la
tasa y el grado en que el principio activo o la fracción
activa en equivalentes farmacéuticos o alternativas
farmacéuticas está disponible en el sitio de acción del
fármaco cuando se administra a la misma dosis molar
en condiciones similares en un estudio apropiadamente
diseñado.
*Glosario de términos “Criterios científicos para los estudios de BE, las biexenciones y las estrategias para su implementación” Rep. Dom. Marzo 2005

Por ejemplo, dos fabricantes diferentes producen tabletas con 500 mg de amoxicilina; estos dos medicamentos son equivalentes farmacéuticos, pero este solo hecho no nos garantiza que su efectividad clínica sea igual, pues por definición los excipientes pueden ser diferentes (en calidad y cantidad) y también la técnica de fabricación empleada (dureza de las tabletas, friabilidad) que pueden llevar a patrones de liberación y de absorción diferentes.

FACTORES QUE INFLUYEN EN LA BIODISPONIBILIDAD
1. Dependientes del Fármaco: Caracteres físico químicos: liposolubilidad,
tiempo de desintegración de la fórmula farmacéutica, tiempo de disolución, excipientes
Porcentaje de transformación tanto a nivel intestinal como hepático. Ejemplo levodopa metabolizado a nivel intestinal y propanolol en el hígado
Forma farmacéutica: cimetidina tiempo de vida muy corto

FACTORES QUE INFLUYEN EN LA BIODISPONIBILIDAD
1. Dependientes del paciente:a) pH de los líquidos orgánicos, motilidad y perfusión gastrointestinal, función hepática, velocidad de vaciado gástrico.b) Presencia de otros productos en el tubo digestivoc) Caracteres genéticosd) Factores sicológicos

FACTORES QUE INFLUYEN EN LA BIODISPONIBILIDAD
FACTORES TECNOLÓGICOS.
Más importante que los factores fisiológicos, por ser previsibles y rectificables, los cuales pueden afectar profundamente la disponibilidad biológica de los fármacos.
Los coadyuvantes o excipientes.- contribuyen
a retardar en mayor o menor grado la liberación del fármaco desde una forma farmacéutica sólida según la naturaleza y cantidad con que se empleen en la formulación.

FACTORES QUE INFLUYEN EN LA BIODISPONIBILIDAD
Los factores tecnológicos propiamente dichos: los métodos de granulación, el tamaño del granulado, la fuerza de compresión, etc.
Influencia en la velocidad de disolución de los fármacos en los fluidos del tracto
gastrointestinal.

TRANSPORTE Y DISTRIBUCIÓN DE
FÁRMACOS

TRANSPORTEEl objetivo :Llegar a los receptores en su sitio de
acción.Y La eliminación y excreción.En el plasma hay tres posibilidades
para la circulación de las drogas:1. Ligada a proteínas2. Unida a hematíes3. Disueltas en el agua plasmática

TRANSPORTEEntre las proteínas están: Las albúminas son las más importantes, en
esta se han descritos dos puntos de fijación:Punto I llamado punto Warfarina Punto II que transporta clorotiazida,
indometacina, fenilbutazona La hemoglobina Las lipoproteínas Las globulinas como la glucoproteína
(bases débiles se unen )

Esta fijación proteica es específica para cada fármaco y no todo el fármaco se liga:
se mantienen
en equilibrio.
La fracción libre es la que produce el efecto farmacológico ó el efecto farmacodinámico
La fracción ligada determina el tiempo de acción de una droga.
EN EL PLASMA

Mientras la droga no se libera de las albúminas, no puede ser metabolizada ni eliminada por el riñón.
Varias drogas pueden competir entre ellas por su fijación proteica y esto va a depender del grado afinidad.
Varias drogas pueden competir por su fijación proteica con sustancias fisiológicas como la bilirrubina.

DISTRIBUCIÓNEs el conjunto de los fenómenos que rigen
el reparto del principio activo en el organismo:
en el plasma, fluidos intersticiales y agua intracelular hasta obtener un equilibrio
M O D E L O M O N O C O M P A R T I M E N T A L
D istribu ción instantán ea
A ntes D esp u es
Es el transporte de sustancias desde una parte del cuerpo a otra

DISTRIBUCIÓNLos factores importantes son:En el organismo:
Flujo de sangre en órganos y tejidos Permeabilidad de las membranas Diferencias de PH entre plasma y tejidos
De acuerdo a las propiedades del fármaco: Tamaño molecular Unión a eritrocitos y proteínas del plasma y
tejidos Solubilidad y propiedades físico-químicas Lipofilicidad Afinidad.

DISTRIBUCIÓN
Los factores que afectan la distribución son:1. Unión a las proteínas plasmáticas, como la
albúmina, lipoproteínas y glucoproteínas plasmáticas.
2. Unión a constituyentes celulares de carácter lipídico o proteínico.
3. Naturaleza química del fármaco4. Interacciones con otros fármacos, metabolitos
y sustancias endógenas.

VOLUMEN DE DISTRIBUCIÓN El volumen de líquidos en el cuál
debe distribuirse la droga para alcanzar una concentración
semejante a la de la sangre en un momento dado.
esangrelaendrogadeiónConcentraccuerpoelendrogadeCantidad
Vd

Volumen plasmático (70 Kg es de 3 L) x ejemplo si tenemos una droga que es una macromolécula que no atraviesa el endotelio capilar y se fija fuertemente a proteínas plasmáticas el volumen máximo en el que puede disolverse será de 3 litros.
Volumen de líquido extracelular (12 L) ejemplo si tenemos una droga que es liposoluble y vence el endotelio capilar, esta difundirá hasta el líquido extravascular y el volumen máximo en el que puede disolverse será de 12 litros.
Volumen de agua corporal en el plasma total 42 L y si un fármaco es capaz de atravesar todas las barreras biológicas el volumen máximo en el que puede disolverse será de 42 litros

BIOTRANSFORMACIÓN
Es el proceso por el cuál la droga introducida al organismo sufre un
proceso de transformación química (metabolismo) por acción de
enzimas que cambian su comportamiento farmacológico
original, sea cuantitativo, cualitativo o ambos.

La acción que tienen los tejidos frente a los fármacos pueden ser:
1. Tejidos indiferentes: no modifican al fármaco
2. Activos: cambia el comportamiento y la estructura química del fármaco por la acción de enzimas metabolitos
3. Susceptibles: el tejido cambia su funcionamiento
4. Emuntorios: tejidos que expulsan las drogas activas o sus metabolitos fuera del organismo

METABOLIZACIÓN
Proceso de transformación de un medicamento liposoluble en hidrosoluble.
Son las distintas transformaciones químicas que ocurren en el organismo encaminadas, sobre todo, a reducir la liposolubilidad y la actividad biológica de los fármacos.

VÍAS QUÍMICAS DE LA BIOTRANSFORMACIÓN
1.Fase I: Reacciones no sintéticas Formación de centros de conjugación en la molécula del medicamento (oxidación, reducción e hidrólisis).
2. Fase II: Reacciones de Conjugación con un sustrato endógeno.

REACCIONES FASE ILas enzimas que participan en esta reacción
de biotransformación están ubicadas en los microsomas hepático (hígado).
Este metabolismo de la molécula conduce a:
1. Inactivación del fármaco2. Conversión de un producto
Farmacológicamente inerte en activo PRODROGA
3. Conversión de un producto activo en otro activo o tóxico
Copi pg 14 gooman y gilman y pg 37 samaniego

REACCIONES FASE IISe realizan fundamentalmente en el hígado
y en menor extensión en el riñón mediante la participación de enzimas transferasas.
Esta reacción es con sustancias endógenas representadas por moléculas como el ácido glucorónico, glicina, metilo, etc.
Las moléculas o fármacos que se van a biotransformar deben poseer centros de conjugación

Hay 6 reacciones de conjugación ó 6 agentes que utiliza el aparato metabólico humano para las reacciones de síntesis:
1. Ácido glucorónico se forman los glucorónidos que sirven para inactivar alcoholes fenoles y ácidos aromáticos.
2. Aminoácidos glicina y glutamina se combinan con ácidos aromáticos y heterocíclicos
3. Acetilación participación de acetil transferasas incorpora grupos carboxilo
4. Metilación participación de metiltransferasa
5. Sulfatación incorpora sulfatos6. Metiltiolación Pg 39 sama

METABOLIZACIÓN
Los aspectos importantes en la Biotransformación son:
Efecto del Primer Paso (Eliminación presistémica). Fracción que se elimina durante la primera exposición a la pared del intestino o el hígado debido a la presencia de Enzimas microsomales a este nivel.
Inducción enzimático Inhibición enzimática

EXCRECION
Proceso en que los medicamentos se eliminan del organismo, ya sea inalterados o en forma de metabolitos inactivos y quizá productos tóxicos
Las vías de eliminación son:1. Excreción renal, que es la más importante
2. Excreción biliar e intestinal (heces)
3. Excreción pulmonar (aire exhalado)
4. Excreción por otras vías: sudor, saliva, lágrimas, leche materna, cabellos y piel. La excreción dérmica es a través de la piel y glándulas sudoríparas.

EXCRECION
El principal órgano excretor es el riñón.
Existen tres tipos de procesos implicados en la excreción renal de un fármaco o metabolito que son: Filtración glomerular Secreción tubular Reabsorción tubular.
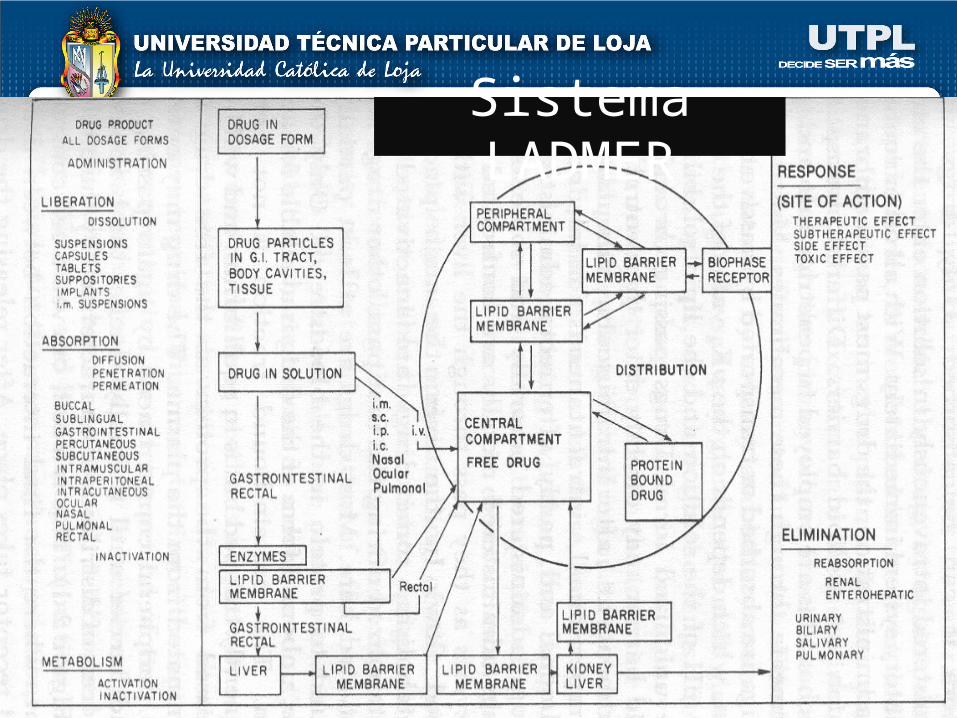
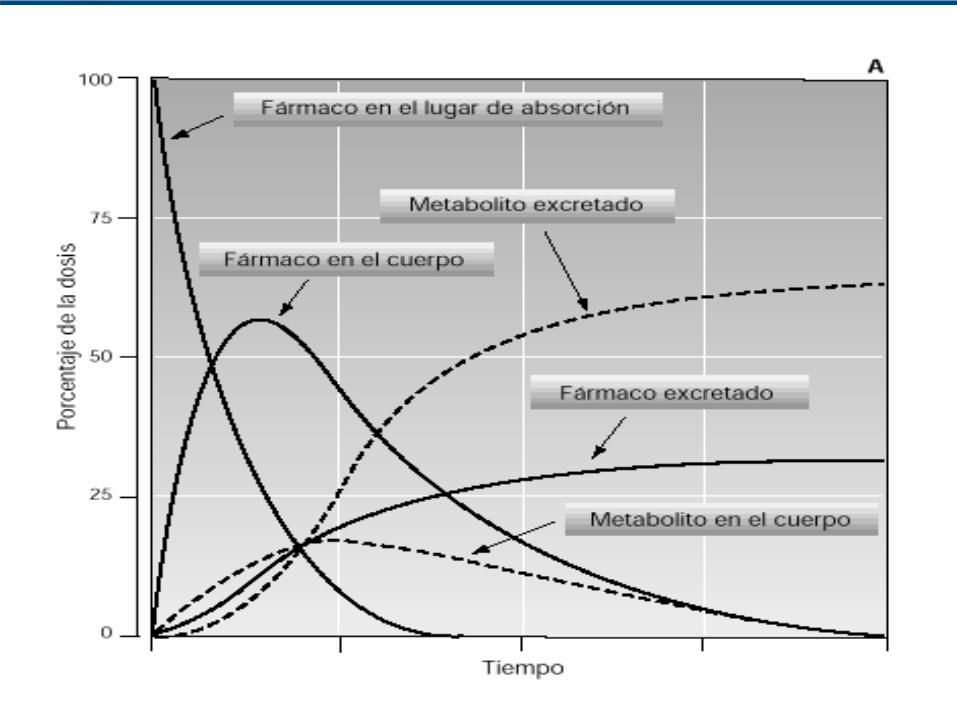
Sistema LADMER
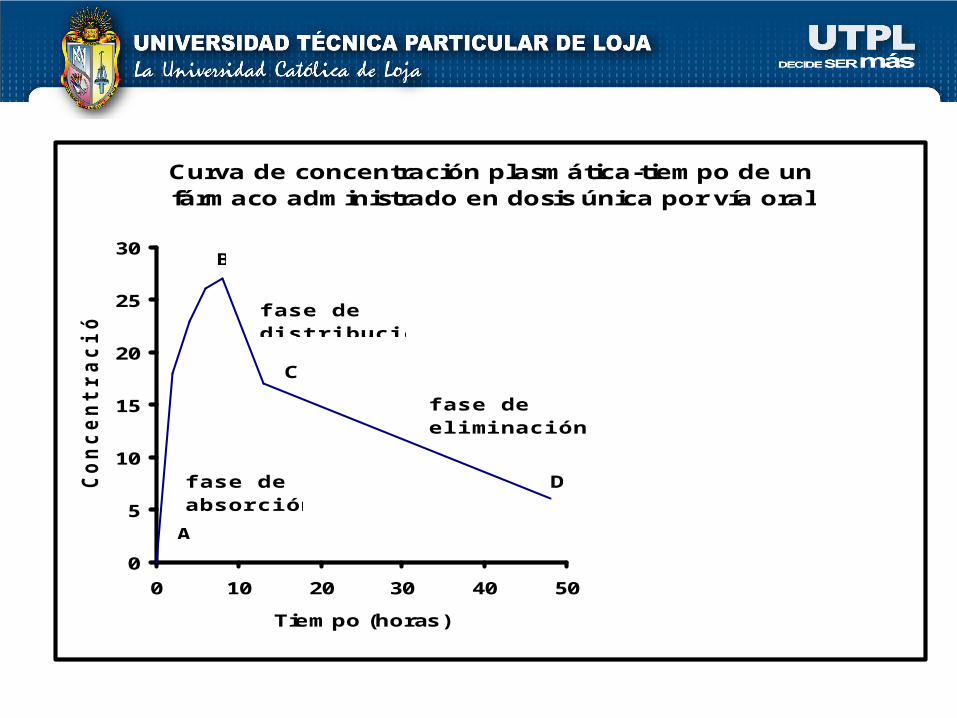
Curva de concentración plasmática-tiempo de un fármaco administrado en dosis única por vía oral
0
5
10
15
20
25
30
0 10 20 30 40 50
Tiempo (horas)
Co
ncen
tració
n (
log
)
fase dedistribución
fase deabsorción
fase deeliminación
A
B
C
D
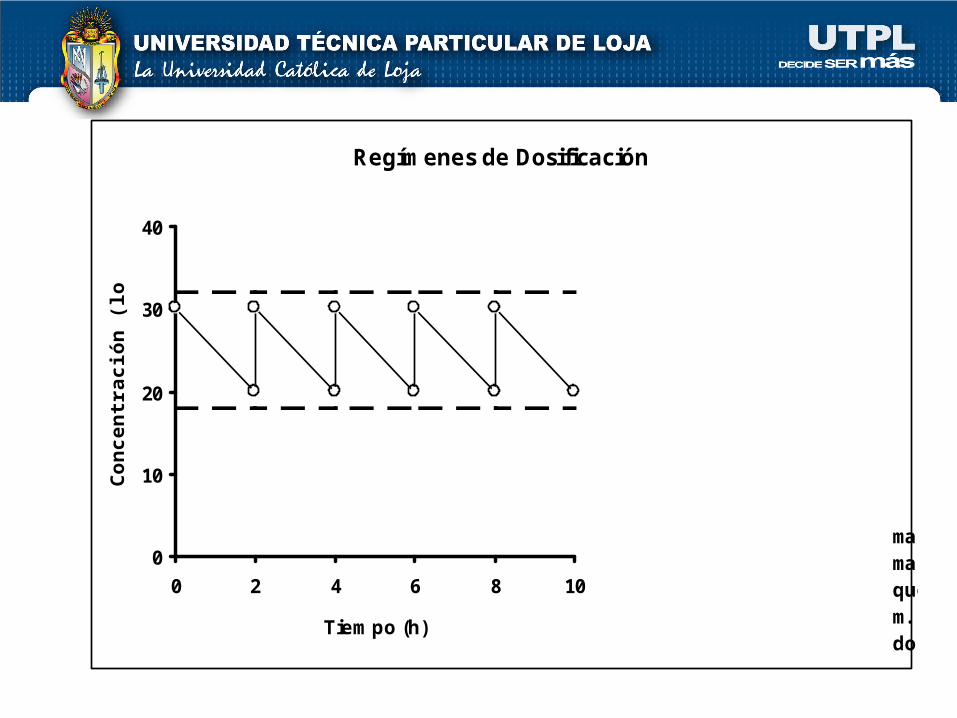
Regímenes de Dosificación
0
10
20
30
40
0 2 4 6 8 10
Tiempo (h)
Co
nce
ntr
ació
n (
log
)
Cmáx
Cmín
D*
DMARGEN TERAPEUTICO
Simbología:Cmáx = Conc. máximaCmín = Conc. mínimaD* = Dosis de ataqueD = Dosis mantenim.T = Intervalo de dosif.
T