







Idiomas
Páginas
Jurídico


Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
CAPÍTULO I: INTRODUCCIÓN Y DEFINICIÓN DE TÉRMINOS
1.1 INTRODUCCIÓN
La Termodinámica es la ciencia que estudia todas las transformaciones o conversiones de unas
formas de energía en otras.
La Termodinámica cubre muchas áreas de la ingeniería, pero el análisis de un amplio tipo de
sistemas se lleva a cabo usando sólo cuatro principios básicos.
Ejemplos de sistemas a estudiar son:
• Plantas de potencia de vapor de agua.
• Máquinas de combustión interna.
• Células de combustión.
• Plantas de potencia geotérmicas.
• Plantas de energía solar.
• Ciclos frigoríficos y bombas de calor.
1.2 CONCEPTOS Y DEFINICIONES
Sistema: región restringida, no necesariamente de volumen constante o fija en el espacio, en
donde se puede estudiar la transferencia y transmisión de masa y energía. Todo sistema tiene
límites que pueden ser reales o imaginarios.
Tipos de sistemas: Cerrado: región de masa constante, a través de sus límites sólo se transfiere energía. Se
denomina “masa de control”.
Abierto: se transfiere masa y energía. Se denomina “volumen de control”. Sus límites deben ser
permeables o imaginarios y se llaman “superficie de control”.
Aislado: no hay transferencia de materia ni energía con el medio exterior ( por ejemplo el
Universo).
Tipos de límites de los sistemas: Adiabáticos: no pueden ser atravesados por el calor
Diatérmicos: permiten la transferencia de calor.
Rígidos: no permiten el cambio de volumen
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 1

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
Permeables: permiten el paso de cualquier sustancia.
Semipermeables: permiten el paso de determinadas sustancias.
Fase: cantidad de materia homogénea en composición química y estructura física. Puede estar
compuesta de una sustancia pura o varios componentes.
Sistema homogéneo: contiene una fase.
Sistema heterogéneo: consta de dos o más fases.
Propiedad: cualquier característica evaluable de un sistema, cuyo valor depende de las
condiciones de éste. Las propiedades de un sistema definen su “estado”.
Las “propiedades termodinámicas” son aquellas relacionadas con la energía y definen el
“estado termodinámico”.
Ejemplos: masa, presión, temperatura, volumen, energía interna, entropía, etc.
Propiedades extensivas: dependen de la masa del sistema (volumen, todas las energías)
∑=
=N
1iixX
Propiedades intensivas: son independientes de la masa del sistema (presión, temperatura,
viscosidad, altura).
Concepto de estado: Si fuera posible conocer masa, velocidad, posición, etc. de cada una de
las partículas que componen un sistema, se conoce el estado microscópico y esto determina las
propiedades del sistema.
Cuando no se tiene un conocimiento tan detallado como el requerido para determinar el estado
microscópico del sistema, la Termodinámica considera las propiedades del sistema, las cuales,
una vez conocidas determinan el estado macroscópico del sistema.
TODAS LAS PROPIEDADES EL ESTADO MACROSCÓPICO
DEL SISTEMA ⇒ DEL SISTEMA
ESTÁN FIJADAS QUEDA FIJADO
En un sistema existe un gran número de propiedades, pero cuando se determinan sólo algunas
de ellas, el resto de las propiedades quedan automáticamente fijadas. De hecho, cuando se
considera un sistema simple como por ejemplo una cierta cantidad de una sustancia de
composición fija, al fijar el valor de dos propiedades se fija el valor de todo el resto. Por lo tanto
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 2

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
en un sistema simple tenemos dos propiedades independientes que se llaman “variables
independientes”. Si se fija el valor de estas dos propiedades, el valor del resto de las
propiedades del sistema quedan fijadas y estas propiedades son las denominadas “variables
dependientes”. El estado termodinámico de un sistema simple queda determinado cuando se
fija el valor de las dos variables independientes y en este caso se pueden elegir dos
propiedades cualesquiera y la elección será cuestión de conveniencia. En un sistema simple
podemos elegir como variables independientes P (presión) y T (temperatura) ya que son las
más fáciles de medir y si consideramos el volumen V como variable dependiente de P y T,
entonces:
V=V (P, T) (1.1)
La relación matemática entre V, P y T se denomina “ecuación de estado del sistema”.
b
c
T2
P
T1
d
V
a
T
P1
P2
Figura 1.1: Representación esquemática de los estados de existencia de una cantidad de gas
en el espacio V-T-P.
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 3

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
En un diagrama tridimensional (V, P, T) los puntos que representan estados de equilibrio de un
sistema caen en una superficie. Esto se muestra en la Figura 1.1, en la cual se puede apreciar
que fijando dos de las tres variables se fija el valor de la tercera. Consideremos un proceso que
lleva al sistema de “a” a “c”, este proceso causa un cambio en el volumen del sistema igual a
∆V=Vc-Va
Es obvio que ese proceso se puede llevar a cabo por distintos caminos en la superficie P-V-T,
dos de los cuales, a→b→c y a→d→c, se muestran en la Figura 1.1. Consideremos el camino
a→b→c, el cambio de volumen será
∆V=Vc-Va=(Vb-Va)+(Vc-Vb)
el proceso a→b ocurre a temperatura constante T1 y b→c ocurre a presión constante P2.
( ) dPPVVV
1
2
1 T
P
Pab ∫
∂∂
=− y ( ) dTTVVV
2
2
1 P
T
Tbc ∫
∂∂
=−
Por lo tanto
dTTVdP
PVV
2
2
11
2
1 P
T
TT
P
P∫∫
∂∂
+
∂∂
=∆ (1.2)
Consideremos el camino a→d→c
( ) dTTVVV
1
2
1 P
T
Tad ∫
∂∂
=− y ( ) dPPVVV
2
2
1 T
P
Pdc ∫
∂∂
=−
y otra vez
dPPVdT
TVV
2
2
11
2
1 T
P
PP
T
T∫∫
∂∂
+
∂∂
=∆ (1.3)
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 4

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
Las ecuaciones (1.2) y (1.3) son idénticas y son la representación física de lo que se obtiene
cuando la diferencial completa de la ecuación (1.1)
dTTVdP
PVdV
PT
∂∂
+
∂∂
= (1.4)
se integra entre los límites P2, T2 y P1, T1.
El valor del cambio de volumen de a hasta c sólo depende de los valores del volumen en a y c y
es independiente del camino tomado por el sistema entre estos puntos. Esto es consecuencia
del hecho de que el volumen es una “función de estado” y la ecuación anterior es el diferencial
exacto del volumen V.
1.3 EQUILIBRIO SIMPLE
Estado de equilibrio: un sistema está en equilibrio cuando no tiene tendencia por sí mismo a
cambiar de estado y a variar, por tanto, sus propiedades.
Equilibrio termodinámico: en el sistema no hay flujo de energía, materia, carga, etc.,
permaneciendo ellas y la composición, constantes en el interior.
Cuando se produce la variación de una o varias propiedades del sistema se da “un cambio de
estado” y el sistema experimenta “un proceso”.
Proceso cíclico: el sistema a través de una serie de cambios de estado finalmente vuelve a su
estado inicial.
Proceso cuasiestático: se verifica a través de sucesivos estados de equilibrio. Realmente no
existe, es ideal o teórico.
Proceso no estático: no cumple las condiciones anteriores. Son procesos de igualación.
Proceso reversible: es un proceso cuasiestático, se vuelve al estado inicial por los mismos
estados intermedios. No queda efecto residual ni en el sistema ni en el medio exterior.
Proceso irreversible: proceso real, hay degradación de energía y generación de entropía. Se
pueden llevar a cabo a través de cambios no estáticos o cambios cuasiestáticos con efectos
disipativos.
En la Figura 1.1 el estado de un sistema es tal que sólo puede ocurrir en la superficie en el
espacio P-V-T, para cualquier valor de temperatura y presión, el sistema está en equilibrio sólo
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 5

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
cuando tiene un único valor de volumen el cual corresponde a esos particulares valores de
presión y temperatura.
Un sistema particularmente simple se muestra en la Figura 1.2. Este consiste en una
determinada cantidad de gas contenida en un cilindro provisto de un pistón movible. El sistema
se encuentra en equilibrio cuando:
g a s
Figura1.2: Representación esquemática de
un sistema simple.
1.La presión ejercida sobre el gas por el
pistón iguala a la presión ejercida por el gas
sobre el pistón.
2. La temperatura del gas iguala a la
temperatura del medio exterior (siempre y
cuando el cilindro sea conductor del calor.
El estado del gas queda por lo tanto fijado
como el resultado de establecer un balance
entre la tendencia de los agentes externos a
provocar un cambio de estado del sistema y
la tendencia del sistema a resistirse a dicho
cambio.
Al fijar la presión a P1 y la temperatura en T1 se define el estado del sistema y por lo tanto el
valor del volumen queda fijado en V1. Si se produce una disminución del peso W, la presión
ejercida por el pistón sobre el gas decrece a P2,
1.4. ECUACIÓN DE ESTADO DE UN GAS IDEAL
La relación presión volumen de un gas a temperatura constante fue determinada
experimentalmente en 1660 por Robert Boyle, quien encontró que a temperatura constante,
P ∝V1
ESta expresión es conocida como la Ley de Boyle. De manera similar, una relación entre el
volumen y la temperatura de un gas a presión constante fue determinada experimentalmente
por Jacques Charles en 1787. Esta relación, conocida como la Ley de Charles, dice que a
presión constante
V α T
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 6

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
Figura 1.3.: (a) Variación con la presión del volumen de 1 mol de gas ideal a 300 y 100 K. (b)
Variación con la temperatura del volumen de 1 mol de gas ideal a 1, 2 y 5 atm.
7

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
En 1802 Joseph Luis Gay – Lussac observó que el coeficiente térmico de lo que entonces se
llamaban “gases permanentes” era constante. El coeficiente de expansión térmica α, se define
como el incremento con la temperatura, a presión constante, del volumen de un gas a 0 ºC,
Po TV
V1
∂∂
=α
donde Vo es el volumen del gas a 0 ºC. Gay – Lussac obtuvo un valor de 267
1 =α , Regnault
mediante experimentación más refinada obtuvo, en 1847, un valor de 273
1 =α .
Posteriormente se encontró que la exactitud con que las leyes de Boyle y charles describen el
comportamiento de diferentes gases varía de un gas a otro. Las leyes de Boyle y Charles
representan mejor los gases con menor punto de ebullición y cuando la presión del gas es
menor. Es por esto que se definió un gas ideal como un gas hipotético que cumple con las leyes
de Boyle y Charles a todas las presiones y temperaturas y tiene, además, un coeficiente de
expansión térmica igual a 273.16
1 =α
La existencia de un coeficiente de expansión téemica con valor finito fija un límite en la
contracción térmica del gas ideal. Como α es igual a 1/273.16, luego el decrecimiento en el
volumen del gas por grado de disminución de la temperatura es 1/273,16 ºC del volumen del
gas a 0 ºC. Por lo tanto, a -273,16 ºC el volumen del gas es cero y por lo tanto el límite en el
descenso de la temperatura. –273.16 ºC, es el cero absoluto de temperatura. Esto define una
escala absoluta de temperatura, llamada la escala de temperatura del gas ideal, la cual está
relacionada con la escala Celsius por la ecuación.
T (grados absolutos) = T (ºC) + 273,16
Combinación de las leyes de Boyle y Charles
Boyle:
Po. V (T, Po) = P. V (T, P)
Charles:
( ) ( )T,PTVT,P
TV
oooo
o =
donde Po: presión estándar (1atm)
To: temperatura estándar (0 ºC o 273.16 grados absolutos)
8

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
V : volumen a la temperatura T y presión P.
.cteT
V.PTV.P
o
oo == (1.5)
De la hipótesis de Avogadro, el volumen por mol de los gases a 0 ºC y 1atm de presión
(llamadas temperatura y presión estándar) es: 22,414 l
mol K.l .atm082054.0
molK 273,16l 22,414 x atm 1
TV.Po
oo ==
Esta constante denominada R, la constante de los gases, y como puede aplicarse a todos los
gases es una constante universal, la ecuación (1.5) puede entonces escribirse como:
P . V = R . T (1.6)
que es la ecuación de estado para 1 mol de gas ideal. La ecuación (1.6) también es conocida
como la ley de los gases ideales.
1.5. UNIDADES DE ENERGÍA Y TRABAJO
La unidad “l x atm” que aparece en las unidades de R es un término de energía. El trabajo es
una forma ordenada de energía y sus unidades son fuerza x distancia. Presión es fuerza por
unidad de área, por lo tanto, trabajo y energía pueden tenr las dimensiones de presión x área x
distancia o presión x volumen. La unidad de energía en el S.I. es el “joule”, que e el trabajo
hecho por una fuerza de 1 newton que recorre una distancia de 1 metro. Litros x atmósferas
puede ser convertido a joules de la siguiente forma:
1 atm = 101.325 mewtons/ metros2
multiplicando ambos lados por litros (10-3 m3) da:
1 litro . atm = 101.325 newton . metro
= 101.325 joules
y por lo tanto
R = 0.082057 litro . atm / grado . mol
= 8.3144 joules / grado . mol
9

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
1.6. PROPIEDADES EXTENSIVAS E INTENSIVAS
Las propiedades extensivas tienen un valor que depende del tamaño del sistema y las
propiedades intensivas son independientes del tamaño de sistema. El volumen es una
propiedad extensiva, y la temperatura y la presión son propiedades intensivas. El valor de una
propiedad extensiva, expresado por unidad de masa del sistema, tiene las características de
una propiedad intensiva, por ejemplo el volumen por unidad de masa ( volumen específico) o el
volumen por mol (volumen molar) son propiedades cuyos valores son independientes del
tamaño del sistema. Para un sistema con n moles de un gas ideal, la ecuación de estado es
P . V’ = n R T
donde V’ es el volumen del sistema. Por mol de sistema, la ecuación de estado es
P . V = R T
donde V, el volumen molar del gas es igual a V’/ n.
1.7. DIAGRAMAS DE FASE Y COMPONENTES TERMODINÁMICOS
De las muchas formas de representar gráficamente los estados de existencia en equilibrio de un
sistema los diagramas de fases son los más convenientes. La complejidad de un diagrama de
fases está determinada en principio por el número de componentes que tiene el sistema, estos
componentes son especies químicas de composición fija. Los componentes más simples son
elementos químicos y compuestos estequiométricos. Los sistemas primero se categorizan por
el número de componentes que contienen, por ejemplo, sistemas de un componente (unarios),
de dos componentes (binarios), de tres componentes (ternarios), de cuatro componentes
(cuaternarios), etc. El diagrama de fases de un sistema de un componente es la representación
bidimensional de la dependencia del estado de equilibrio del sistema con dos variables
independientes. Temperatura y presión son las dos variables independientes elegidas
normalmente. La figura 1.4 muetsra una representación esquemética de una parte del diagrama
para H2O. Las lineas llenas de la figura 1.4 dividen al diagrama en tres áreas designadas sólido,
líquido y vapor. Si una cantidad de H2O pura está a una temperatura y presión que están
representadas por un punto dentro del área AOB, el estado de equilibrio para el agua es líquido.
De manera similar, dentro de las áreas COA y COB los estados de equilibrio son,
respectivamente, sólido y vapor. Si el estado de existencia cae en una línea, por ejemplo, en la
10

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO I
línea AO, H2O líquida y sólida coexisten en equilibrio una con otra, y el equilibrio será con dos
fases.
Figura 1.4: Representación esquemática de parte del diagrama de fases para H2O.
Una fase se define como una porción finita de volumen de un sistema dentro de la cual las
propiedades son constantes, por ejemplo no experimenta ningún cambio abrupto al pasar de un
punto a otro del volumen. Dentro de las áreas de una sola fase en el diagrama de fases, se dice
que el sistema es homogéneo. El sistema es heterogéneo cuando contiene dos o más fases
11

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 12
CAPÍTULO II: LA PRIMERA LEY DE LA TERMODINÁMICA
2.1. INTRODUCCIÓN
La energía cinética se conserva en un sistema sin fricción en el que se produce la interacción
de cuerpos rígidos elásticos. La colisión entre 2 cuerpos ocasiona la transferencia de Ec de un
cuerpo a otro y la Ec total del sistema no cambia producto de la colisión.
Si el sistema cinético se encuentra bajo la influencia de campo gravitatorio, la suma de las
energías cinética y potencial de los cuerpos es constante; cambios en la posición de los
cuerpos en el campo gravitatorio, sumados a cambios en las velocidades de los cuerpos, no
alteran la energía dinámica total del sistema. Como resultado de las posibles interacciones, la
energía cinética puede ser convertida en energía potencial y viceversa, pero la suma de ambas
permanece constante.
Si el sistema tiene fricción, cuando se producen colisiones o interacciones entre los cuerpos, la
energía dinámica total disminuye y se produce calor. Es por lo tanto razonable esperar que
exista una relación entre la energía dinámica disipada y el calor producido por efecto de la
fricción.
El establecimiento de esta relación constituye la base para el desarrollo del método
termodinámico. El desarrollo del método termodinámico desde sus comienzos hasta nuestros
días fue alcanzado como el resultado de la invención de funciones termodinámicas de estado
convenientes. En este capítulo se introducen las dos primeras, la energía interna U y la entalpía
H.
2.2. RELACIÓN ENTRE CALOR Y TRABAJO
La relación entre calor y trabajo fue sugerida por primera vez en 1798 por Count Runford, quien
durante el taladrado de un cañón en el Arsenal de Munich, observó que el calor producido
durante el taladrado de un metal era proporcional al trabajo realizado durante el taladrado.
Esta observación era novedosa y hasta ese momento el calor era considerado como un fluido invisible llamado calórico que residía entre las partículas constituyentes del sistema. En la
Teoría calórica la temperatura de una sustancia queda determinada por la cantidad de fluido
calórico que ella contiene y cuando se pone en contacto dos cuerpos a diferente temperatura,

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 13
éstos alcanzan una temperatura común intermedia como resultado del flujo de fluido calórico
entre ellos. El equilibrio térmico se alcanzaba cuando la presión del fluido calórico era igual en
ambos cuerpos. La observación de Rumford que la producción de calor acompaña a la
realización de trabajo fue explicada por la teoría calórica diciendo que se debía al hecho que la
cantidad de calórico que puede ser contenido por un cuerpo, por unidad de masa del cuerpo,
depende de la masa del cuerpo. Pequeñas piezas de metal (virutas producidas por el taladrado)
contienen menos calórico por unidad de masa que la que tenía la masa original del metal
grande, y por lo tanto reduciendo la masa original a un número de pequeños trozos, el calórico
era entregado como calor sensible. Rumford luego demostró que si utilizaba un taladro grueso
que producía poca viruta, la misma producción de calor acompañaba a igual realización de
trabajo. La teoría calórica “explicó” la producción de calor en este caso como debida a la acción
del aire sobre la superficie del metal durante la realización del trabajo.
La teoría calórica fue finalmente desacreditada en 1799 cuando Humprey Davy derritió 2
bloques de hielo frotando uno contra otro en vacío. En este caso el calor latente necesario para
fundir el hielo que provisto por el trabajo mecánico realizado al frotar los bloques.
Desde 1840 en adelante la relación entre calor y trabajo fue puesta en bases firmemente
cuantitativas como resultado de una serie de experimentos realizados por James Joule. Joule
realizó experimentos en los cuales el trabajo era realizado sobre una cierta cantidad de agua
contenida adiabáticamente y medía el aumento de temperatura del agua. Joule observó que
existía proporcionalidad directa entre el trabajo realizado y el aumento de temperatura sin
importar el medio empleado en la producción del trabajo. Métodos de producción de trabajo
usados por Joule incluían
1. Rueda rotatoria inmersa en el agua
2. Motor eléctrico haciendo pasar una corriente a través de una bobina inmersa en el agua
3. Comprimiendo un cilindro de gas inmerso en el agua
4. Frotando dos bloques de metal inmersos en el agua
Esta proporcionalidad introdujo la noción de “equivalente mecánico del calor” y con el propósito
de definirlo es necesario definir una unidad de calor. Esta unidad es la Caloría que es la
cantidad de calor necesaria para subir la temperatura de 1g de agua de 14,5 a 15,5 ºC. Sobre la
base de esta definición Joule determinó el valor del equivalente mecánico del calor que era
0.241 calorías por joule. El valor aceptado actualmente es 0.2389 calorías por joule, que
redondeado a 0.239 calorías por joule define la caloría termodinámica, la cual fue hasta la
introducción en 1960 del S.I, la unidad tradicional de energía utilizada en termoquímica.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 14
2.3. ENERGÍA INTERNA Y LA PRIMERA LEY DE LA TERMODINÁMICA
Los experimentos de Joule dieron lugar al siguiente enunciado. “ El cambio en un cuerpo dentro
de un recipiente adiabático desde un estado inicial a uno final involucra la misma cantidad de
trabajo sin importar de que forma se llevó a cabo el proceso”. Este enunciado es una
formulación preliminar de la 1º Ley de la Termodinámica y desde este punto de vista es
necesario introducir una función que dependa únicamente del estado interno del sistema. Esta
función es U energía interna. Esta función se entiende mejor por medio de comparación con
conceptos más familiares. Cuando un cuerpo de masa m es movido en un campo gravitatorio
desde la altura h1 hasta la altura h2, el trabajo W hecho en el cuerpo está dado por
W = fuerza x distancia
W = mg x (h2 – h1)
= mgh2 – mgh1
= Ep2 – Ep1
Así como la energía potencial de un cuerpo de masa m depende solamente de la posición del
cuerpo en el campo gravitatorio, se aprecia que el trabajo hecho en el cuerpo depende sólo de
su estado inicial y final y es independiente del camino tomado por el cuerpo entre las dos
posiciones, por ejemplo, entre los dos estados. De manera similar la aplicación de una fuerza f
a un cuerpo de masa m provoca la aceleración del cuerpo de acuerdo a la Ley de Newton
dtdv.ma.mf ==
donde a = dv/dt es la aceleración
El trabajo realizado en el cuerpo es por lo tanto obtenido integrando
dl .fdw =
donde l es la distancia
dv v. m. dv .dtdl.mdl
dtdv.mdw ===
Integrando da
21
22 v.m
21v.m
21w −=
= energía cinética del cuerpo a la velocidad v2 (estado 2) – energía cinética del cuerpo a la
velocidad v1 (estado 1)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 15
Por lo tanto, otra vez el trabajo realizado sobre un cuerpo es la diferencia entre los valores de
una función de estado del cuerpo y es independiente del camino tomado por el cuerpo entre los
dos estados.
En el caso del trabajo realizado sobre un cuerpo contenido adiabáticamente con Ec y Ep
constantes, la función que describe adecuadamente el estado del cuerpo, o el cambio en el
estado del cuerpo es la energía interna U. Por lo tanto, el trabajo hecho por o sobre el cuerpo
contenido adiabáticamente es igual al cambio en energía interna del cuerpo, es decir, la
diferencia entre los valores de U en el estado final e inicial. En el caso del trabajo es una
convención asignar signo negativo al trabajo realizado sobre el cuerpo y signo positivo al trabajo
realizado por el cuerpo. Esta convención surge porque cuando un gas se expande y por lo tanto
realiza trabajo contra una presión externa, la integral ∫2
1dVP , que es el trabajo realizado, es una
cantidad positiva. Por lo tanto para un proceso adiabático en el cual el trabajo w se hace sobre
el cuerpo y como resultado del cual su estado se mueve de A a B,
w = - (UB - UA)
Si el trabajo se realiza sobre el sistema, UB > UA y si el trabajo lo realiza el sistema UB < UA
En los experimentos de Joule el cambio en el estado del agua contenida adiabáticamente se
midió como un incremento de temperatura del agua. El mismo aumento de temperatura y por lo
tanto el mismo cambio de estado se puede obtener poniendo en contacto el agua con una
fuente de calor y consiguiendo que el calor q fluya hacia el agua. Por convención se asigna
signo negativo al calor que fluye fuera del cuerpo (proceso exotérmico) y signo positivo al calor
que se entrega al cuerpo (proceso endotérmico). entonces,
q = (UB - UA)
Por lo tanto cuando el calor fluye hacia el cuerpo, q es una cantidad positiva y UB > UA mientras
que si el calor fluye fuera del cuerpo UB < UA y q es una cantidad negativa.
Es ahora de interés considerar el cambio en la energía interna de un sistema que
simultáneamente realiza trabajo y absorbe calor. Consideremos un cuerpo, inicialmente en el
estado A, el cual realiza trabajo w, absorbe calor q, y, como consecuencia de esto, se mueve al
estado B. La absorción de calor q incrementa la energía interna del cuerpo en una cantidad q, y
la realización de trabajo por parte del cuerpo disminuye la energía interna del cuerpo. Por lo
tanto, el cambio total en energía interna, ∆U, es
∆U = UB - UA = q - w (2.1)
Este es el enunciado de la 1º Ley de la Termodinámica.
Para un cambio de estado infinitesimal, la ecuación (2.1) puede ser escrita como un diferencial

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 16
dU = δq - δw (2.2)
El término de la izquierda de la ecuación (2.2) da el valor del incremento en una propiedad
existente del sistema, mientras que el lado derecho no tiene interpretación. Como U es una
función de estado, la integración de dU entre dos estados es independiente del camino seguido
por el sistema entre esos dos estados. Este no es el caso cuando δq y δw son integrados. Los
efectos del calor y trabajo, los cuales involucran energía en tránsito, dependen del camino
seguido entre los dos estados, como consecuencia de esto δq y δw no pueden ser evaluados
sino se conocen el camino. Esto se ilustra en la Figura 2.1, en esta figura el valor U2-U1, es
independiente del camino tomado entre los estados 1 (P1, V1) y 2 (P2, V2). Sin embargo, el
trabajo hecho por el sistema, el cual está dado por la integral ∫ ∫=δ2
1
2
1PdVw y que es por lo tanto,
el área bajo la curva entre V2 y V1 puede variar de manera importante según que camino se
tome.
Figura 2.1.: Tres caminos para el cambio de estado de una cantidad fija de gas que va de 1 a 2.
En la figura 2.1 el trabajo realizado en el proceso 1→2 por el camino c es menor que el
realizado por el camino b, el que a su vez es menor que el realizado por a. De la ecuación 2.1
se desprende que la integral de δq también debe depender del camino, y en el proceso 1→2 se
absorbe más calor por el camino a que por el b que a su vez es mayor que por el camino c. En
la ecuación (2.2) el uso del símbolo “d” indica un diferencial de una función o propiedad de

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 17
estado cuya integral es independiente del camino, el uso de símbolo “δ” indica un elemento
diferencial de una cantidad que no e función de estado. En la ecuación (2.1) la suma algebraica
de dos cantidades, ninguna de las cuales es independiente del camino, da una cantidad que si
es independiente del camino.
En el caso de un proceso cíclico el cual retorna al sistema a su estado inicial, por ejemplo el
proceso 1→2→1 en al figura 2.1, el cambio en U como resultado de ese proceso es cero.
( ) ( ) 0UUUUdUdUU 211
212
2
1=−+−=+=∆ ∫∫
El que la integral cíclica 0 dU =∫ es una propiedad de las funciones de estado.
En los experimentos de Joule donde (U2–U1)=-w, el proceso era adiabático (q=0), y por lo tanto
es camino del proceso estaba especificado.
Como U es una función de estado, para un sistema simple que consiste en una cantidad dada
de una sustancia de composición fija, el valor de U queda fijado una vez que dos propiedades
cualesquiera (las variables independientes) se fijan. Si temperatura y volumen se eligen como
variable independientes, luego
U = U (U,T)
El diferencial exacto en términos de las derivadas parciales da
dTTUdV
VUdU
VT
∂∂
+
∂∂
=
Como el estado de un sistema queda fijado cuando las dos variables independientes se fijan, es
de interés examinar aquellos procesos en los cuales el valor de una de las variables
independientes se mantiene constante y a la otra se le permite variar. De esta manera
examinaremos procesos en los cuales la temperatura T se mantiene constante (procesos
isotérmicos), o la presión P se mantiene constante (procesos isobáricos), o el volumen V se
mantiene constante (procesos isocóricos). También examinaremos los procesos adiabáticos en
los cuales q=0.
2.4. PROCESOS A VOLUMEN CONSTANTE
Si el volumen de un sistema se mantiene constante durante un proceso, el sistema no realiza
trabajo ∫ = )0PdV( , y por la 1º Ley, ecuación 2.2,
dU = δqv (2.3)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 18
donde el subíndice v indica volumen constante. Integrando la ecuación (2.3)
∆U = qv
el incremento o disminución de la energía interna del sistema es igual, respectivamente, al calor
absorbido o cedido por el sistema durante el proceso.
2.5. PROCESOS A PRESIÓN CONSTANTE Y LA ENTALPÍA H
Si la presión se mantiene constante durante un proceso que lleva al sistema del estado 1 al
estado 2, el trabajo realizado por el sistema estará dado por
( )122
1
2
1VVPdVPPdVw −=== ∫ ∫
por la 1º ley:
U2 – U1= qp - P (V2 - V1)
donde el subíndice p indica presión constante. Reordenando tenemos
( ) ( ) p1122 qPV,UPVU =+−+
y como la expresión (U+PV) contiene sólo funciones de estado, toda la expresión es una
función de estado llamada entalpía H;
H = U + PV (2.4)
Por lo tanto, para un proceso a presión constante,
H2 – H1 = ∆H = qp (2.5)
El cambio de entalpía durante un proceso a presión constante es igual al calor absorbido o
cedido por el sistema durante el proceso.
2.6. CAPACIDAD CALORÍFICA
Antes de discutir los procesos isotérmicos y adiabáticos, es conveniente introducir el concepto
de capacidad calorífica. La capacidad calorífica, C, de un sistema es la relación entre el q
entregado o cedido por el sistema y el cambio de temperatura del mismo. Por lo tanto
TqC∆
=
o si el cambio de temperatura es muy pequeño

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 19
dTq C δ
=
El concepto de capacidad calorífica se usa sólo cuando la adición o sustracción de calor del
sistema produce un cambio de temperatura, este concepto no se utiliza cuando se producen
cambios de fase. Por ejemplo, si el sistema es una mezcla de hielo y agua a 1 atm de presión y
0 ºC de temperatura, la adición de calo simplemente funde algo de hielo y no produce cambio
en la temperatura. En este caso la capacidad calorífica, de acuerdo a su definición, sería
infinita.
Nótese que si el sistema está en un estado 1 y la absorción de cierta cantidad de calor
incrementa su temperatura de T1 a T2, decir que la temperatura final es T2 es insuficiente para
determinar el estado final del sistema. Esto es porque el sistema tiene dos variables
independientes, y por lo tanto otra variable además de la temperatura debe especificarse a fin
de definir el estado del sistema. Esta segunda variable independiente puede variar de una
forma determinada o mantenerse constante durante el cambio. La última posibilidad es la más
práctica y por eso la adición de calor a un sistema para producir un cambio de temperatura se
considera normalmente a presión o volumen constante. De esta manera el camino del proceso
queda especificado y el estado final del sistema se conoce.
Así la capacidad calorífica a volumen constante Cv y la capacidad calorífica a presión constante
Cp son definidas como
vv dT
q C
δ= capacidad calorífica a volumen constante
pp dT
q C
δ= capacidad calorífica a presión constante
Por lo tanto de las ecuaciones (2.3) y (2.5)
dTCdUodtdU
dTq C v
vvv =
=
δ= (2.6)
dTCdHodTdH
dTq C p
ppp =
=
δ= (2.7)
La capacidad calorífica al ser dependiente del tamaño del sistema es una propiedad extensiva.
Sin embargo, normalmente es conveniente usar la capacidad calorífica por unidad de cantidad
de sistema. Luego el calor específico de un sistema es la capacidad calorífica por gramo a P
constante y la capacidad calorífica molar es la capacidad calorífica por mol a presión o volumen
constante. Entonces para un sistema que contiene n moles,
n cp = Cp y n cv = Cv

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 20
donde cp y cv son valores molares.
Es de esperar que, para cualquier sustancia, cp sea mayor que cv. Si se requiere que la
temperatura de un sistema se incremente en una cierta cantidad, entonces, si el proceso es
llevado a cabo a volumen cte., todo el calor entregado sólo se ocupa en aumentar T. Sin
embargo, si el proceso es llevado a cabo a presión cte., el calor entregado además es utilizado
para proveer el trabajo necesario para expandir el sistema a presión cte.
Este trabajo de expansión en contra de la presión cte. por grado de incremento de temperatura
se calcula como:
pTVPó
dTdV.P
∂∂
y sería de esperar que
pvp T
VPcc
∂∂
=−
La diferencia entre cp y cv se calcula de la siguiente manera
pppp T
VPTU
THc
∂∂
+
∂∂
=
∂∂
=
y
vv T
Uc
∂∂
=
entonces
vppvp T
UTVP
TUcc
∂∂
−
∂∂
+
∂∂
=−
pero
dTTUdV
VUdU
vT
∂∂
+
∂∂
=
y por lo tanto
pTvp TV
VU
TU
TU
∂∂
∂∂
+
∂∂
=
∂∂
entonces
vpvpTvp T
UTVP
TU
TV
VUcc
∂∂
−
∂∂
+
∂∂
+
∂∂
∂∂
=−

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 21
∂∂
+
∂∂
=−Tp
vp VUP
TVcc (2.8)
Las dos expresiones para cp-cv difieren en el término
pT TV.
VU
∂∂
∂∂
Joule trató de evaluar el término TV
U
∂∂ para los gases, mediante un experimento que consistía
en un recipiente de Cu lleno con un gas a cierta presión, conectado a otro similar pero vacío.
Ambos recipientes estaban sumergidos en agua contenida adiabáticamente, se abría la
conexión entre ambos recipientes y se deja expandir libremente el gas dentro del recipiente
vacío. Después de la expansión Joule no pudo detectar ningún cambio en la temperatura del
sistema. Como éste estaba contenido adiabáticamente y no se realiza trabajo, de acuerdo a la
1º Ley,
∆U = 0
y entonces
0dTTUdV
VUdU
vT=
∂∂
+
∂∂
=
Por lo tanto, como dT = 0 (medio experimentalmente) y dV ≠ 0, luego el término TV
U
∂∂ debe
ser cero. Joule concluyó: “La U de un gas es función sólo de la temperatura e independiente de
V y P”. Consecuentemente, para un gas
pvp T
VPcc
∂∂
=−
Sin embargo, en un experimento realizado por Joule y Thomson, en el cual un gas de volumen
molar V1 contenido adiabáticamente a una presión P1, se hace pasar a través de un diafragma
poroso hasta una presión P2 y un volumen molar V2, se observa cambio en la temperatura, lo
que muestra que para los gases reales 0VU
T≠
∂∂ .
De todas maneras, si 0VU
T=
∂∂
de la ecuación (2.8)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 22
Pvp T
VPcc
∂∂
=−
y como para un mol de gas ideal P. V.= RT, luego
RPxPRcc vp ==−
La razón por la cual Joule no observó un aumento en la temperatura en el experimento original,
fue porque el recipiente de Cu y el agua tienen una capacidad calorífica mucho más grande que
el gas, y por lo tanto los pequeños cambios de calor que se producen en el gas fueron
absorbidos por el recipiente y el agua. El cambio que se produce en la temperatura queda por
debajo del límite de medida.
En la ecuación (2.8) el término
pTV.P
∂∂
representa el trabajo hecho por el sistema por grado de cambio en la temperatura, para
expandir el gas en contra de la presión externa cte. P que actúa sobre el sistema. El otro
término en la ecuación (2.8),
PT TV
VU
∂∂
∂∂
representa el trabajo hecho por grado de cambio de la temperatura, para expandir el gas en
contra de las fuerzas cohesivas internas que actúan entre las partículas constituyentes del
sistema. Como se verá en el Capítulo VIII, los gases ideales están constituidos por partículas no
interactuantes, y entonces los átomos de un gas ideal pueden ser separados uno de otro sin
realizar trabajo. Entonces para un gas ideal, el término anterior y también el término
TVU
∂∂
son cero. En los gases reales la contribución de la presión interna es mucho más pequeña en
magnitud que la contribución de la presión externa, pero en líquidos y sólidos, en los cuales las
fuerzas de cohesión internas son considerables, el trabajo hecho al expandir el sistema en
contra de la presión externa es insignificante en comparación con el trabajo hecho contra la
presión interna. Por lo tanto, para líquidos y sólidos el término
TVU
∂∂
es muy grande.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 23
2.7. PROCESOS ADIABÁTICOS REVERSIBLES
Durante un proceso reversible en el cual el estado del gas cambia, el estado del gas nunca deja
la superficie de equilibrio mostrada en la Figura 1.1. Consecuentemente, durante un proceso
reversible, el gas pasa a través de continuos estados de equilibrio y el trabajo w está dado por
la integral ∫2
1PdV solamente si el proceso es llevado a cabo reversiblemente. En un proceso
adiabático q = 0 y por lo tanto de la 1º ley dU = -δw.
Consideremos un sistema de 1 mol de gas ideal. De la ecuación (2.6)
dU = cv dT
y para procesos adiabáticos reversibles
δw = P dV
Entonces
cv dT = -P dV
Como el sistema es 1 mol de gas ideal, luego VT.RP = y entonces
VRTdVdTCv −= reordenando
VdVR
TdTCv −=
Integrando entre los estados 1 y 2 da
=
2
1
1
2v V
Vnl RTTln.C
o R
2
1C
1
2VV
TT v
=
o
vcR
2
1
1
2VV
TT
=
Para un gas ideal se ha mostrado que cp – cv = R. Entonces cp/cv = R/cv; y si cp/cv = γ, luego
R/cv= γ - 1, y entonces 1
2
1
1
2VV
TT
−γ
−
De la ecuación de estado de un gas ideal

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 24
1
2
1
11
22
1
2VV
V.PV.P
TT
−γ
==
Por lo tanto γ
=
2
1
1
2VV
PP
y entonces
.ctePVVPVP 1122 === γγγ
Esta es la relación entre P y V para un gas ideal que realiza un proceso adiabático reversible.
2.8. PRESIÓN ISOTÉRMICA REVERSIBLE O CAMBIOS DE VOLUMEN EN UN GAS IDEAL
De la 1º Ley
w -q dU δδ=
y como dT = 0 (proceso isotérmico), luego dU = 0. Por lo tanto dVV
RTP.dV wq ==δ=δ por
mol de gas.
Integrando entre los estados 1 y 2 da
=
==
2
1
1
2PPnl RT
VVnl RTqw (2.10)
Para un gas ideal un proceso isotérmico es un proceso de U = cte. en el cual el trabajo
realizado por el sistema es igual al calor absorbido por el sistema y ambos están dados por la
ecuación (2.10).
Un proceso isotérmico reversible y uno adiabático reversible se muestran en un diagrama P-V
en la Figura 2.2 en la cual se puede apreciar que para una dada disminución de presión, el
trabajo hecho por el proceso isotérmico (el cual es igual al área bajo la curva) es mayor que el
hecho en el proceso adiabático. Esta diferencia se debe al hecho que durante el proceso
isotérmico el calor es absorbido por el sistema a fin de mantener la temperatura constante,
mientras que durante el proceso adiabático no hay absorción de calor por parte del sistema.
Durante la expansión isotérmica la energía interna del gas permanece constante y durante la
expansión adiabática la energía interna disminuye en una cantidad igual al trabajo realizado.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 25
Figura 2.2.: Comparación entre una expansión isotérmica reversible y una expansión adiabática
reversible realizadas por un gas ideal entre una presión inicial de 20 atm y una presión final de 4
atm.
2.9. RESUMEN
1. La relación entre w y q se determina introduciendo la función termodinámica energía
interna, U, que es una función de estado y por lo tanto ∆U depende sólo de los estados final e
inicial y es independiente del camino seguido por el proceso. La relación entre el cambio de
energía interna, el trabajo realizado y el calor absorbido por mol de sistema de composición fija
que se mueve de un estado a otro está dado por wqU −=∆ o para un incremento
w-q dU δδ= . Esta relación se denomina 1ºLey de la Termodinámica.
2. Las integrales de δq y δw sólo se pueden obtener si se conoce el camino tomado por el
sistema al moverse de un estado a otro. Procesos que son convenientes considerar incluyen
a. Proceso a volumen constante en el cual ∫δw = ∫PdV = 0
b. Proceso a presión constante en el cual ∫δw = P∫dV = P∆V
c. Proceso a temperatura constante en el cual ∆U = 0 y w = q
d. Proceso adiabático en el cual q = 0

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 26
3. Para un proceso a volumen constante como w = 0, luego ∆U = q. La definición de la
capacidad calorífica molar a volumen constante como vv
v TU
dtq c
∂∂
=
δ= (esta es una
cantidad medible experimentalmente) facilita la determinación del cambio en U resultante de un
proceso a volumen constante como ∆U = dTc2
1v∫ .
4. La consideración de un proceso a presión constante se facilita por la introducción de la
función termodinámica H, la entalpía, definida como H = U + P.V. Como la expresión sólo
contiene funciones de estado H también será una función de estado.
( ) pp qVPVPqVPUH =∆+∆−=∆+∆=∆
dT.CHTH
dTq C
2
1p
ppp ∫∆⇒
∂∂
=
δ=
5. Para un gas ideal U sólo función de la temperatura y Cp - Cv = R
6. Proceso adiabático reversible de un gas ideal
vp
CC dondecteV.P =γ=γ
durante una expansión adiabática q = 0
wdU δ=⇒
7. Durante un proceso isotérmico U = cte.
2
1
1
2PPnl RT
VVnl RTwq0dU ===⇒=
8. Sólo ∆U y ∆H pueden ser medidos, los valores absolutos de H y U no pueden ser
determinados.
2.10. EJEMPLO NUMÉRICO
10 l de un gas ideal monoatómico a 25ºC y 10 atm. de presión se expanden hasta una presión
final de 1 atm. La capacidad calorífica a vol. cte. es Cv = 3/2 R y es independiente de la
temperatura.
Calcular el w hecho y el q absorbido y el cambio en U y h para el gas si el proceso es:
a) isotérmico y b) adiabático.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 27
Habiendo determinado el estado final después de una expansión adiabática, verificar que el
cambio en U es independiente del camino entre los estados inicial y final, considerando que los
procesos fueron:
1) Proceso isotérmico + proceso o V = cte.
2) Proceso a V = cte. + proceso isotérmico
3) Proceso isotérmico + proceso a P = cte.
4) Proceso a V = cte. + proceso a P = cte.
5) Proceso P = cte. + proceso a V = cte.
Lo primero es calcular el tamaño del sistema.
T. R.
V.P.n T R. n. V.P =⇒=
moles089,4K298x
Kmollatm.0,08206
l10 atm. 10n ==
a) Expansión isotérmica reversible (a → b)
l 100tm a 1
tm a 10 x l 10P
V.PVcteV.Pb
aab ===⇒=
VaVb nl nRT
VdVnRTdV.Pwq0U
b
a
b
a====⇒=∆ ∫ ∫
KJ 328,2310
100 nl .K 298 xmolK J 8,3144 x mol089,4wq ===
q = W = 23,328 KJ
Para un gas ideal H es sólo función de T
( ) 0baH =→∆
( ) ( ) ( )( )
( ) 0baHTTTT nR nRTa - RTb n
V.PV.PV.PV.PbaUbaH
abab
aabbaabb
=→∆
=−==
−=−+→∆=→∆
b) Expansión reversible adiabática (a → b)
351
cvRV.PV.PcteV.P ccaa =+=γ=⇒= γγγ

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 28
( ) 53
351
c
aac atm1
l10.atm10P
V.PV
=
=
γγ
l81,39VC =
molKlatm08206,0xmol089,4
l81,39xatm1R.nV.PTy cc
c ==
Tc = 118.6 K
Proceso adiabático ⇒ q = 0
( ) ( )ac
c
avv TTncdTncwcaU −==−=→∆ ∫
( )K2986.118xKmol
J3144.8 . 23 . mol089,4 −=
( ) KJ 9,149- caU =→∆
( ) ( ) ( )aacc V.PVPcaUcaH −+→∆=→∆
( )
KJ098.6KJ149,9atml
J32.101.l10.atm100l81.39.atm1KJ149,9
−−=
−+−=
( ) KJ247,15caH −=→∆
1.- T = cte + V = cte (a→ e → c)
( ) ( ) ( )0w0VqceU0eaU v =⇒=∆=→∆=→∆
( ) dTncqceUc
evv ∫==→∆
( )KJ 149,9
K2986,118.K mol
J3144,8.23mol089.4
−=
−=
( ) ( ) ( ) KJ 149,9ccUeaUcaU −=→∆+→∆→→∆
2.- V = cte + T = cte (a →d → c)
( )
( )
( ) KJ 149,9daU
K298-118,6 . K mol
J 8,3144 . 23 . mol089,4dTnc
0w0VqdaUd
av
v
−=→∆
==
=⇒=∆=→∆
∫

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 29
( ) .cteTporque0cdU ==→∆
( ) KJ 149,9caU −=→∆
3.- T = cte + P = cte (a → b → c)
( ) cteT porque0baU ==→∆
( ) ( )bcbcbp VVPwPcomoPwqcbU −=⇒=−=→∆
( )l.atm
J32,101 . l10081,39 . atm 1w −=
KJ098,6w −=
( )∫ −==c
bbcp TTR
25.nncpdTq
K )2986,118(K mol
J3144,8 .25. mol089,4qp −=
( ) ( )
( ) ( ) ( )cbUbaUcaUKJ 150.9
UKJ 248,15q
KJ 098,6KJ 248,15cb
p
→∆+→∆=→∆−=
∆
−=
−−−=→
( ) 150,9caU −=→∆
4.- V = cte + P = cte (a → f → c)
( )
( )
K 8.29
l mo Kl . atm08206,0 x mol089,4
l10 x atm1nR
P.PT
TTncdTnc
0w0UqfaU
fff
afv
f
av
v
===
−==
=⇒=∆=→∆
∫
( ) ( )( )
( ) ( )K2988.29PdTncwqcfU
KJ 677,13faU
K2988.29 . K mol
J3144.8 . 23 . mol089,4U
f
c
fpp
fa
−−=−=→∆
−=→∆
−=∆
∫
→

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO II
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 30
( ) ( )
( ) ( )
KJ020,3wlatm
J32,101.l1081,39atm1VVPw
KJ547,7q
K8,296,118.molK
J3144,8.25.mol089,4TTncq
fcf
p
fcpp
=
−=−=
=
−=−=
( ) ( )( ) ( ) ( )
( ) KJ150,9UKJ527,4KJ677,13UUU
KJ527,4KJ020,3547,7U
ca
cffaca
cf
−=∆
+−=∆+∆=∆
=−=∆
→
→→→
→
5.- P = cte + V = cte (a → g → c)
( ) ( )aga
g
appga VVPdTncwqU −−=−=∆ ∫→
Kmoll.atm08206,0x.mol089,4
l81,39xatm10nR
V.PT gg
g ==
( ) ( ) ( )( )
( ) KJ155,9UKJ433,54278,45
UUU
ca
cggaca
−=∆−=
∆+∆=∆
→
→→→

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 31
CAPITULO III: LA 2º LEY DE LA TERMODINÁMICA
3.1. INTRODUCCIÓN
Cuando un sistema cambia de estado hay un cambio en la energía interna del sistema, que sólo
depende de los estados inicial y final y es la suma algebraica de los efectos de trabajo y calor.
La pregunta que surge es: ¿Qué magnitudes pueden tener los efectos de calor y trabajo y qué
criterios gobiernan esas magnitudes?. Dos casos extremos pueden suceder: w = 0 ó q = 0 y en
esos casos ∆U = q ó ∆U = w. Pero si w ≠ 0 y q ≠ 0, ¿existe una cantidad máxima de trabajo que
el sistema puede hacer durante el cambio de estado?
La respuesta a esto requiere conocer la naturaleza del proceso.
Podemos identificar 2 clases de procesos: reversibles e irreversibles, y para estudiarlos
introduciremos el concepto de “Entropía”. Este será introducido desde dos puntos de vista: 1º
para cuantificar el grado de irreversibilidad de los procesos y 2º como resultado de la
examinación de las propiedades de las máquinas térmicas operadas de forma reversible, ellas
naturalmente desarrollan una cantidad que tiene todas las propiedades de una función de
estado y esa función es la entropía.
3.2. PROCESOS NATURALES O ESPONTÁNEOS
Un sistema dejado libre tiene 2 opciones:
1. Permanecer en el estado en que se encuentra.
2. Moverse por su propia iniciativa hacia otro estado.
Si el sistema está en equilibrio con el medio exterior ⇒ 1º opción.
Si el sistema no está en equilibrio con el medio exterior ⇒ 2º opción.
El estado de equilibrio es como el estado de descanso y una vez alcanzado el sistema no se
moverá a menos que se ejerza una acción externa.
El nuevo sistema (sistema original + ext.) se moverá hacia el estado de equilibrio del sistema
combinado.
Un proceso que involucra el movimiento espontáneo de un sistema desde un estado de no
equilibrio se llama “Proceso natural o espontáneo”

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 32
Un proceso espontáneo no puede revertirse sin la aplicación de una acción externa, se dice que
es un “Proceso irreversible”
Natural ≡ Espontáneo ≡ Irreversible
En ambos casos el proceso inverso nunca puede ocurrir espontáneamente.
En sistemas simples es fácil predecir el estado de equilibrio sin conocer los criterios de
equilibrio. Por ejemplo si el estado inicial de sistema es de dos cuerpos a distinta temperatura,
cuando los cuerpos son puestos en contacto , el proceso espontáneo que ocurre es el flujo de
calor desde el cuerpo más caliente al más frío y el equilibrio se alcanza cuando los cuerpos
tienen una temperatura común uniforme.
T1 T2
En sistemas más complicados se deben establecer primero los criterios que gobiernan el
equilibrio antes de que se hagan los cálculos para conocer el estado de equilibrio.
La determinación del estado de equilibrio es de suma importancia en Termodinámica.
A + B = C + D
Para esa reacción química permitirá conocer en que dirección procederá la reacción
espontánea desde el estado inicial.
El estado inicial puede ser una mezcla de A, B, C, y D, conocido el estado de equilibrio
sabremos si la reacción procederá de izquierda a derecha o viceversa y cuanto se extenderá
hasta lograr el equilibrio.
Si el sistema realiza un proceso espontáneo que involucra la producción de w y Q, mientras
más cerca esté del equilibrio menos capacidad tendrá el sistema para realizar futuros cambios
espontáneos. Finalmente cuando el sistema alcanza el equilibrio su capacidad de realizar w es
nula.
Proceso espontáneo ⇒ sistema se degrada
La energía disponible para realizar w se convierte en energía térmica (Q) y en esa forma no
está disponible para propósitos externos.
Proceso espontáneo: Q (-) . W (+) A → B UA > UB
2TT 21 +
2TT 21 +

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 33
3.3. ENTROPÍA Y CUANTIFICACIÓN DE LA IRREVERSIBILIDAD.
Los dos tipos de procesos espontáneos son:
1. Conversión de W en Q (degradación de energía mecánica a U)
2. Flujo de Q a través de un gradiente de temperatura.
Proceso irreversible: El sistema se degrada a través del proceso. Esa degradación varía de un
proceso a otro y esto sugiere que debe existir una medida cuantitativa de la degradación o
grado de irreversibilidad de un proceso.
Lewis y Randalll consideran 3 procesos:
1. El depósito esta a T2. Se deja caer el peso W,
se realiza w y el q producido entra al depósito.
2. El depósito a T2 se pone en contacto con otro a
T1 < T2 el mismo q fluye del depósito a T2 al a T1.
3. El depósito está a T1, W cae. Produce w y entra
q.
Figura 3.1.
Los 3 procesos son espontáneos y por lo tanto irreversibles ⇒ se produce degradación.
Como el proceso (3) es la suma de los procesos (1) y (2), la degradación producida en él debe
ser mayor
Proceso (3) es más irreversible que (1) y (2).
Magnitud del q producido o el flujo de q. Importantes para definir
T a la cual el q es producido o ∆T que produce el flujo una escala de irreversibilidad.
w

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 34
Proceso (3): q / T1
Proceso (1): q / T2
q / T1 > q / T2 ⇒ Proceso (3) más irreversible que Proceso (1)
La cantidad q / T es una medida del grado de irreversibilidad del proceso.
q /T es el incremento de entropía S que ocurre como resultado del proceso.
Proceso espontáneo que causa absorción de T/qScteT a q =∆∴= (3.1)
3.4. PROCESOS REVERSIBLES
Como el grado de irreversibilidad es variable, el proceso se puede realizar de manera de que
éste se minimice.
El límite es un proceso en el que el grado de irreversibilidad sea cero y por lo tanto no ocurra
degradación y esto es un “Proceso reversible”
En este caso el concepto de espontaneidad, o sea ir de un estado de no equilibrio, a uno de
equilibrio, no es aplicable.
Es un proceso reversible el sistema nunca está lejos del equilibrio, todos los puntos que recorre
entre el estado inicial y el final son estados de equilibrio.
Este camino es imaginario, pero el proceso puede llevarse a cabo de manera que sea
virtualmente reversible.
La fuerza impulsora es infinitesimal ⇒ el sistema nunca está más allá de una distancia
infinitesimal del equilibrio.
Si en cualquier punto del camino la fuerza externa se quita, el proceso se detiene o si la
dirección de la fuerza externa se invierte la dirección del proceso se revierte.
3.5. ILUSTRACIÓN DE PROCESOS REVERSIBLES E IRREVERSIBLES
Consideremos un sistema con agua y vapor de agua a una temperatura uniforme T contenidos
en un cilindro cerrado mediante un pistón sin fricción, dejemos que el cilindro sea puesto en

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 35
contacto térmico con un baño térmico que está también a temperatura T constante, el sistema
se muestra en la Figura 3.2.
El vapor de agua en el cilindro ejerce una
presión PH2O (T), que es la presión de vapor
saturada de agua a la temperatura T.
El sistema se encuentra en equilibrio cuando
la presión externa que actúa sobre el pistón,
Pext iguala a la presión interna actuante sobre
el pistón, PH2O (T), y cuando la temperatura
del agua y el vapor de agua en el cilindro es
igual a la temperatura constante T del baño
térmico.
Figura 3.2
Si la P ext. disminuye repentinamente en un valor finito ∆P, el desbalance de presiones, (Pext -
∆P) ≠ Pint., hará que el cilindro suba rápidamente y se produzca la expansión del vapor de H2O.
Esta expansión hará decrecer la presión del vapor de agua PH2O a un valor menor que la de
saturación y por lo tanto se producirá la evaporación espontánea de H2O para restablecer el
equilibrio.
La evaporación espontánea es endotérmica y por lo tanto disminuye la temperatura del H2O,
esto crea un ∆T entre el baño térmico y el cilindro y por lo tanto se establece un flujo de q
espontáneo desde el baño térmico al cilindro para restablecer el equilibrio térmico.
Si cuando 1 mol de H2O se ha avaporado la Pext aumenta a su valor original, la evaporación de
H2O y el flujo de calor cesan y se establece el equilibrio nuevamente. El w hecho por el sistema
durante este proceso es:
w = (Pext - ∆P) . V
V: volumen molar del vapor de H2O a PH2O
(T) y la temperatura T.
Baño térmico T
H2O
PH2O (T) vapor de H2O
Pext.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 36
Si por el contrario la Pext. aumenta en ∆P ⇒ (Pext + ∆P) ≠ Pint el pistón baja provocando la
compresión del vapor de H2O. Esta compresión del vapor de agua hará decrecer la PH2O
a un
valor inferior al de saturación lo que provocará la condensación espontánea exotérmica con el
consiguiente aumento de la temperatura en el cilindro. Debido a esto se establecerá un ∆T entre
el cilindro y el baño térmico y un flujo de q espontáneo entre el cilindro y el baño.
Si cuando 1 mol de H2O se ha condensado la Pext. retorna a su valor original, se restablece el
equilibrio. El w hecho por el sistema durante este proceso es: W = (Pext + SP) . V
Veamos otra vez el proceso de evaporación. Sí Pext disminuye una cantidad infinitesimal δP el
infinitesimal desbalance de presiones (Pext – δP) ≠ Pint, hace que el pistón suba lentamente
provocando una lenta expansión del vapor, la PH2O
disminuye a un valor infinitesimalmente
menor que el de saturación y comienza la espontánea evaporación del H2O que al ser
endotérmica establece un gradiente infinitesimal de temperatura entre el baño térmico y el
cilindro y se crea un flujo de calor entre el baño y el cilindro.
Cuanto más pequeño sea δP, más lento serás el proceso, menor el grado de insaturación del
vapor de H2O y menor el gradiente de temperatura. Mientras más lentamente se lleve a cabo el
proceso, más oportunidad habrá de que la evaporación y el flujo de q sean procesos de
equilibrio.
Sí después de la evaporación de 1mol de H2O, la Pext aumenta a su valor original, se
restablecerá el equilibrio y el trabajo hecho por el sistema será w=(Pext-δP).V. Sí la Pext se
aumenta un δP, el trabajo hecho para condensar 1 mol de vapor de H2O será: w = (Pext + δP).V.
El cambio permanente en el proceso cíclico es el w hecho por el sistema menos el w hecho
sobre el sistema. Mientras menor sea δP, más similares serán los dos términos de trabajo. En el
límite serán iguales y el proceso cíclico se habrá llevado a cabo reversiblemente. Cuando se
aproxima a una completa reversibilidad el proceso se vuelve infinitamente lento.
3.6. ENTROPÍA Y CALOR REVERSIBLE
Consideremos sólo el proceso de evaporación de 1 mol de H2O. El trabajo realizado por el
sistema durante la evaporación de 1 mol de agua será máximo cuando el proceso sea
reversible: wmáx = Pext . V

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 37
Cualquier proceso de evaporación irreversible realiza menos trabajo, w = (Pext. - ∆P) V.
El cambio en el valor de U para el sistema como resultado de la evaporación de 1 mol de H2O
es independiente de si el proceso es llevado a cabo de manera reversible o irreversible.
De la 1º Ley, la máxima cantidad de calor que entra al cilindro desde el baño térmico, si el
proceso es reversible, será qrev = ∆U + wmáx. Sí el proceso es irreversible, se transfiere menos
calor q = ∆U + w.
La diferencia (wmáx – w) es la energía mecánica que ha sido degradada a energía térmica en el
cilindro como resultado de la naturaleza irreversible del proceso.
El calor producido por degradación está dado por
(qrev – q) = (wmáx. – w)
Si el proceso de evaporación es reversible, qrev deja el baño y entra al cilindro a la temperatura
T. El cambio de entropía en el baño será:
∆Sbaño = T
qrev− ( - porque el q deja el baño)
El cambio de entropía en el cilindro es:
∆SH2O + vapor = T
qrev ( + porque el q entra al cilindro)
El cambio total de entropía en el sistema combinado como resultado de un proceso reversible
será por lo tanto.
∆Stotal = ∆Sbaño + ∆SH2O + vapor = 0
Por lo tanto para un proceso reversible el cambio de entropía es cero y no se produce
degradación durante el proceso.
Si el proceso de evaporación es irreversible, el calor q (q < qrev) deja el baño térmico y entra al
cilindro a la temperatura T. El cambio en la entropía del baño térmico es
∆Sbaño = -Tq cambio de S en el baño
El calor total producido en el cilindro será el calor q proveniente del baño más el calor producido
por la degradación del w debido a la naturaleza irreversible del proceso.
(wmáx – w) = (qrev – q)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 38
El cambio en la S del cilindro será:
∆SH2O + vapor = T
qT
qqTq revrev =
−+
El cambio total de S en el sistema como resultado de un proceso irreversible será:
∆Stotal = ∆Sbaño + ∆SH2O + vapor = Tqq
Tq
Tq revrev −
=+−
Como qrev > q el cambio de S es (+) ⇒ se ha creado entropía debido a un proceso irreversible.
( )irr
rev ST
qq∆=
− es una medida de la degradación producida como resultado del proceso.
Para un proceso de evaporación, independientemente del grado de irreversibilidad
∆SH2O + vapor irrSTq
∆+= (3.2)
En un proceso de condensación reversible
∆SH2O + vapor T
qrev−=
∆Sbaño T
qrev=
y ∆Stotal = 0 y por lo tanto no hay creación de entropía.
Para un proceso de condensación irreversible
∆SH2O + vapor = ( )Tqq
Tq rev−+−
∆Sbaño = Tq
∆Stotal= ( )
Tqq
Tq
Tqq
Tq revrev −
=+
−+−
Como q > qrev se ha creado S como resultado del proceso ⇒ ∆Sirr y el cambio de entropía en el
agua más vapor de agua está dado por

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 39
∆SH2O + vapor irrSTq
∆+−= (3.3)
Lo importante de las ecuaciones (3.2) y (3.3)
∆S vapor + H2O T
qSTq rev
irr =∆+=
∆S vapor + H2O TqS
Tq rev
irr−
=∆+−=
es que la diferencia de S entre el estado inicial y final es independiente de si el proceso es
reversible o irreversible, o sea independiente del camino, lo que indica que la S es una función
de estado. Para un cambio de estado entre A y B
irrAB STqSSS ∆+=−=∆ (3.4a)
Tqrev= (3.4b)
3.7. PROCESOS DE COMPRENSIÓN ISOTÉRMICA REVERSIBLE DE UN GAS IDEAL.
Consideremos la compresión isotérmica reversible de 1 mol de gas ideal, que realiza este
proceso pasando de un estado inicial (VA, T) a un estado final (VB, T). El gas se pone en
contacto con un baño térmico a la temperatura T y es comprimido lentamente de manera que la
Pext. es sólo infinitesimalmente mayor que la Pinst. del gas y Pinst. = RT / Vinst.
El estado del gas cae siempre en una sección de T = cte. en la superficie P – V – T y por lo
tanto el gas pasa a través de continuos estados de equilibrio al ir desde el estado (VA, T) al
estado (VB, T) , esto implica que el proceso es reversible, no hay degradación y no se crea
entropía. La entropía simplemente es transferida del gas al baño térmico.
Como el proceso es isotérmico ∆U = 0 y por lo tanto el w hecho en el gas es igual al q que sale
del gas:
wmáx = qrev
donde
=== ∫∫
A
BV
V
V
Vmáx V
Vln RTV
RTdVdV.PwB
A
B
A

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 40
Como VB < VA, wmáx es una cantidad negativa, en concordancia con que el trabajo es hecho en
el gas. La transferencia de calor desde el gas al baño térmico causa un cambio en la entropía
del gas
===∆
A
Bmáxrevgas V
Vln RT
wT
qS esta cantidad también es negativa.
Consecuentemente, como no hay cambio en la entropía total del sistema durante la
compresión reversible, el cambio de en el baño está dado por
=∆−=∆
B
Agasbaño V
Vln RSS
como el proceso es reversible AStotal=0
Como VB < VA, el resultado de la comprensión reversible es una disminución de la entropía del
gas y un aumento (de igual magnitud) de la entropía del baño térmico.
3.8. EXPANSIÓN ADIABÁTICA REVERSIBLE DE UN GAS IDEAL
Consideremos 1 mol de gas ideal que se expande adiabáticamente desde el estado (PA, TA) al
estado (PB, TB).
La expansión debe llevarse a cabo muy lentamente para que el proceso sea reversible y el
estado del gas caiga siempre en la superficie P – V – T.
Como el proceso es adiabático q = 0 y por lo tanto P Vγ = cte.
Como el proceso es reversible no hay degradación y por ser adiabático no hay flujo del calor.
El cambio de entropía en el gas es cero, un proceso reversible adiabático es un proceso
“isoentrópico”.
Durante este proceso ∆U = - w máx
Si la expansión ocurre irreversiblemente y ocurre degradación, como el gas está contenido
adiabáticamente el calor producido por lo degradación permanece en el gas y la temperatura
final será mayor que TB.
Estado final de una expansión Estado final de una expansión

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 41
adiabática irreversible adiabática reversible
La S producida en el gas debido al proceso irreversible es la diferencia en entropía entre el
estado final e inicial. El estado final estará determinado por el grado de irreversibilidad del
proceso.
Para un cambio de presión PA → PB, mientras mayor sea el grado de irreversibilidad del
proceso, mayor será el calor producido en el gas debido a la degradación, la temperatura final y
la energía interna y el aumento de entropía.
Por lo tanto, durante una expansión irreversible el w hecho por el gas, iguala la disminución de
U del gas, pero esa disminución es menor que en la expansión reversible de PA a PB debido a la
aparición de calor en el gas como resultado de la degradación.
3.9. RESUMEN
1. Cuando un sistema realiza un proceso irreversible o espontáneo, la entropía del sistema
aumenta.
2. Cuando un sistema realiza un proceso reversible, no se crea entropía, la entropía sólo se
transfiere de una parte a otra del sistema.
3. La entropía es una función de estado.
3.10. PROPIEDADES DE LAS MÁQUINAS TÉRMICAS
Una máquina térmica es un dispositivo que convierte calor en trabajo.
q2 q1
w
Figura 3.3
Depósito de calor de
alta temperatura t2 Depósito de calor de
baja temperatura t1
Máquina
térmica

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 42
Vapor sobrecalentado se pasa desde el depósito de alta temperatura a los cilindros donde se
realiza trabajo de expansión contra los pistones. Como resultado de esa expansión la
temperatura del vapor disminuye y al final de la carrera del pistón el vapor es liberado a la
atmósfera que es el depósito de baja temperatura. Una rueda retorna el pistón a su posición
original, se completa el ciclo y se prepara para una nueva carrera del pistón.
La eficiencia de la máquina térmica será
Eficiencia = 2q
wingresado calor
obtenido trabajo=
Los factores que gobiernan la eficiencia de este proceso fueron explicados en 1824 por Sadi
Carnot, quien consideró el proceso de la figura.
P
A → B: se transfiere reversiblemente q2
desde el depósito de alta temperatura a una
sustancia termodinámica, como resultado de
esto la sustancia se expande isotérmica y
reversiblemente de A + B y realiza w, = área
Abba.
B → C: la sustancia termodinámica realiza
una expansión adiabática de B a c, como
resultado de esto la temperatura cae a t1, y
realiza w2 = BCcb.
Figura 3.4
C → D: se transfiere q1 isotérmica y reversiblemente desde la sustancia termodinámica hacia el
depósito de baja temperatura t1. El trabajo w3 = DCcd es realizado sobre la sustancia que se
mueve de C a D.
D → A: la sustancia se comprime reversible y adiabáticamente desde D a A, la temperatura
sube de t1 a t2 y w4 = Adda es realizado sobre la sustancia.
Durante el proceso cíclico, la sustancia ha realizado trabajo.
w = w1 + w2 – w3 – w4 = área ABCD y ha absorbido calor q = q2 – q1
Para un proceso cíclico ∆U = 0 y por la 1º Ley
A B
D
C
V a b c d
t2
t1

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 43
q = w ⇒ q2 – q1 = w
y la eficiencia de un proceso cíclico (ciclo de Camot) está dada por
Eficiencia = 2
12
2 qqq
qw −
=
Consideremos una segunda máquina que trabaje con una sustancia distinta, entre las
temperaturas t1 y t2, y que sea más eficiente.
Mayor eficiencia se puede obtener de dos formas.
1. q2 sale del depósito a t2 y se realiza más trabajo w’ (w’ > w), entonces se libera menos calor
1q′ a deposito de bajo temperatura t1 ( 1q′ < q1).
2. El mismo w se obtiene absorbiendo menos calor 2q′ del depósito a t2 ( 2q′ < q2), entonces
menos calor pasa al depósito a t1 2q′ < q1
Consideremos ahora que la 2º máquina está moviéndose en sentido directo y la 1º en sentido
reverso (actuando como una bomba de calor):
Entonces de (1) para la 2º máquina
w’ = q2 - 1q′
y para la 1º máquina
-w = - q2 + q1
la suma de los dos procesos es: (w’ – w) = (q1 - 1q′ ) esto es una cantidad de w (w’ – w) se ha
obtenido de una cantidad de q (q1 - 1q′ ) sin que otro cambio ocurra. Si bien esto no contraviene
la 1º Ley de la Termodinámica es contrario a la experiencia humana ya que este proceso
corresponde al movimiento perpetúo de 2º especie.
De (2) para la 2º máquina moviéndose en sentido directo w = 2q′ - 1q′ , para la 1º máquina
12 qqw +−=− y la suma de los dos procesos
( ) ( ) qqqqq 1122 =′−=−′
una cantidad de calor q a una temperatura se ha convertido en q a mayor temperatura sin que
ningún otro cambio ocurra. Esto corresponde a flujo espontáneo de calor en contra de un
gradiente de temperatura y esto es más contrario a la experiencia humana que el caso anterior.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 44
La discusión anterior da lugar a una formulación preliminar de la 2º Ley de la Termodinámica.
1. Principio de Thomsem: es imposible por medio de un proceso cíclico tomar calor de un
depósito y convertirlo en trabajo sin que en la misma operación se transfiera calor a un depósito
frío.
2. Principio de Clausius: es imposible transferir calor de un depósito frío a uno caliente sin
en la misma operación convertir cierta cantidad de trabajo en calor.
3.11. LA ESCALA TERMODINÁMICA DE TEMPERATURA.
Lo visto en el punto anterior sugiere que todos los ciclos reversibles de Carnot operando entre
las mismas temperaturas superior e inferior deben tener la misma eficiencia, es decir la máxima
posible. Esta eficiencia máxima es independiente de la sustancia de trabajo y sólo es función de
las temperaturas de trabajo t1 y t2.
Eficiencia = ( )2
121
2
12qq1t,tf
qqq
−=′=− o ( )21
2
1 t,tfqq
=
Consideremos los ciclos de Carnot
mostrados en la Figura 3.5
ciclo entre t, y t2 + ciclo entre t2 y t3 =
ciclo entre t, y t3
( )212
1 t,tfqq
=
( )323
2 t,tfqq
=
( )313
1 t,tfqq
=
Figura 3.5
entonces ( )( ) ( )21
2
1
32
31
2
3
3
1 t,tfqq
t,tft,tf
xqq
===
P
V
t1
t2
t3

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 45
Como ( )21 t,tf es independiente de t3, luego ( )31 t,tf y ( )32 t,tf pueden ser de la forma
( ) ( ) ( )3131 tFtFt,tf = y ( ) ( ) ( )3232 tFtFt,tf = , por ejemplo la función eficiencia ( )21 t,tf es el
cociente de una función de t1 sola y t2 sola.
Por lo tanto ( )( )2
1
2
1tFtF
= Kelvin llamó a las funciones T1 y T2 simplemente con lo que la
eficiencia del ciclo de Carnot es:
Eficiencia = 2
12
2
12T
TTq
qq −=
− (3.5)
y la Eficiencia =1 para T1 =0
Esta escala es idéntica a la discutida en el Capítulo 1 para los gases ideales. Esto puede ser
demostrado considerando que 1 mol de gas ideal es la sustancia de trabajo en un ciclo de
Carnot. Considerando la figura 3.4
A – B: expansión isotérmica reversible a T2
===∆
A
B212 V
VlnRTwq0U
B – C: expansión adiabática reversible
dTCUw0q1
2
T
Tv2 ∫−=∆−==
C – D: comprensión isotérmica reversible a T1
===∆
C
D131 V
VlnRTwq0U
D – A: comprensión adiabática reversible
dTCUw0q2
1
T
Tv4 ∫−=∆−==
Trabajo total hecho por el gas: w = w1 + w2 + w3 + w4
dTCVVlnRTdTC
VVlnRT
2
1
1
2
T
Tv
C
A1
T
Tv
A
B2 ∫∫ −
+−
=

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 46
( )
−=∴=
∆ A
B12
D
CBVVln.TTRw
VV
VV
Calor absorbido del depósito caliente
=
A
B22 V
VlnRTq
Eficiencia = 2
12
2 TTT
qw −
=
3.12. LA SEGUNDA LEY DE LA TERMODINÁMICA
La ecuación 2
12
2
12T
TTq
qq −=
− se puede escribir como: 0Tq
Tq
1
1
2
2 =− (3.6)
Un proceso cíclico puede ser dividido en un gran número de ciclos de Carnot, como lo muestra
la Figura 3.6.
En el ciclo ABA, el trabajo hecho por el sistema será el área encerrada por el ciclo, que se
puede aproximar por un número de ciclos de Carnot, de la ecuación 0Tq=∑ donde el calor
que entra al sistema es (+) y el deja el sistema (-).

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 47
Si hacemos los ciclos de Carnot cada vez más pequeños podemos hacer que coincida con el
camino ABA y en el límite la sumatoria será reemplazada por una integral cerrada o cíclica.
0Tq=
δ∫
esto indica que la integral es un diferencial exacto de una función de estado del sistema.
Esta nueva función de estado es la entropía S y se define como: TqdS δ
=
Para el ciclo ABA
( ) ( ) 0SSSSdSdS0dS BBB
B=−+−=+== ∆∆
∆
∆∫∫ ∫
recordar que q es el incremento de color “reversible”
TqdS revδ
=
La 2º Ley de la Termodinámica puede ser expresada como:
1. La entropía S definida por la ecuación T
qdS revδ= es una función de estado.
2. La entropía de un sistema con límites adiabáticos nunca puede decrecer, aumenta en
procesos irreversibles y permanece constante en procesos reversibles.
> 0 proceso irreversible
De 2 0dSi ≥∑
= 0 proceso reversible
∑ =i
irrdSdSi grado de irreversibilidad del proceso
3.13. TRABAJO MÁXIMO
Para un cambio de estado de A a B, la 1ºLey da: UB –UA = q-w, pero no las magnitudes de q y
w, q y w varían dependiendo del grado de irreversibilidad del proceso.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 48
La 2º Ley establece un límite al máximo trabajo que se puede obtener del sistema y a la
cantidad de calor que puede absorber.
Para un cambio de estado infinitesimal
d Ssistema = irrdSTq+
δ
y de la 1º Ley
wdUq sistema δ+=δ
irrsistema
sistema dST
wdUdS +δ+
=∴
.sistsistwirrsist.sist dUTdSóTdSdUTdSw −≤δ−−=δ (3.11)
Sí T = cte. (es igual a la temperatura del depósito caliente) integrando entre los estados A y B
( ) ( )ABAB UUSSTW −−−≤
U y S son funciones de estado, luego w no puede ser mayor que una cierta cantidad wmdx, que
es el trabajo obtenido cuando el proceso se lleva a cabo de manera reversible y ese
corresponde a la máxima absorción de calor qrev.
( ) ( )ABABmáx UUSSTw −−−=
Como la entropía es una función de estado, cuando se realice cualquier cambio de estado de A
a B, el cambio en la entropía del sistema es el mismo independientemente que el proceso sea
reversible o irreversible. La discusión anterior indica que es el efecto del calor es que es
diferente en los dos casos, por ejemplo, si el proceso involucra la absorción de calor y es
llevado a cabo reversiblemente, el calor absorbido qrev es mayor que el calor que se habría
absorbido si el proceso hubiese sido conducido de manera irreversible. Como puede apreciarse,
cuando 1 mol de gas ideal es expandido isotérmica y reversiblemente desde el estado A al B, el
calor
=
A
BVVlnRTq es reversiblemente transferido desde el baño térmico al gas y el
incremento en la entropía del gas SB-SA es igual a R ln (VB/VA). La entropía en el baño térmico
disminuye en una cantidad igual y por lo tanto no hay creación de entropía ∆Sirr = 0. Sin

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 49
embargo, si 1 mol de gas ideal se expande libremente de PA a PB, como el gas no realiza
trabajo no transferencia de calor del baño térmico al gas y no hay cambio de entropía en el
baño térmico. Como la entropía es una función de estado , el valor de SB-SA es independiente
del camino seguido en el proceso y por lo tanto la entropía creada, ∆Sirr, iguala a SB-SA=R ln
(VB/VA). Esta entropía es creada como resultado de la degradación del trabajo que podría haber
realizado el gas si la expansión no hubiese sido en contra de una presión externa igual a cero.
El trabajo degradado es
= wmáx-w
= wmáx-0
=wmáx
=qrev
Por lo tanto la expansión libre representa el límite de irreversiubilidad en el cual todo el trabajo
potencial se degrada a calor y ese calor hace que se incremente la entropía del gas. Por lo
tanto, para la expansión isotérmica de 1 mol de gas ideal desde el estado A al estado B, el valor
de ∆Sirr puede variar entre cero y R ln (VB/VA) dependiendo del grado de irreversibilidad del
proceso.
3.14. ENTROPÍA Y EL CRITERIO DE EQUILIBRIO
Al principio de este capítulo se estableció que un sistema dejado solo podía quedarse en ese
estado, si se encontraba en equilibrio, o moverse espontáneamente hacia otro estado, si el
sistema inicialmente estaba fuera del equilibrio. Este proceso espontáneo es, por definición,
irreversible y durante el movimiento del sistema desde su estado inicial de no equilibrio a su
estado final de equilibrio la S del sistema aumenta. Cuando se alcanza el equilibrio la entropía
tiene su máximo valor, por lo tanto la S puede usarse como criterio para la determinación del
equilibrio.
Para un sistema aislado con U y V = cte., el equilibrio se alcanza cuando la entropía del sistema
es máxima.
reacción química A + B = C + D adiabática
U = cte.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 50
Comenzando con A y B la reacción irá hacia la derecha y con C y D hacia la izquierda. En
ambos casos la reacción seguirás hasta alcanzar Smdx.
3.15. COMBINACIÓN DE LA 1º Y 2º LEY
Para un cambio incremental en el estado de un sistema cerrado, la 1º Ley da:
wqdU δ−δ=
si el proceso ocurre reversiblemente, la 2º Ley dice:
PdVw
TdSqoTqdS
=δ
=δδ
=
Combinando las 2 leyes queda la siguiente ecuación
dV.PTdSdU −= (1)
esto es sólo aplicable si:
1. El sistema es cerrado (no intercambia materia con el medio exterior)
S
(A+B) (C+D) Composición
Equilibrio

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO III
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 51
2. El W debido al cambio de volumen, es la única forma de w realizada por el sistema.
La ecuación (1) relaciona la variable dependiente U con las variables independientes δ y V
U = U (S, V)
Su diferencial total es dVVUdS
SUdU
SV
∂∂
+
∂∂
= (2)
Comparando (1) y (2) Sv V
UPySUT
∂∂
−=
∂∂
=
Consideremos ahora S como variable dependiente y U y V como independientes.
( )V,USS =
dVVSdU
USdS
UV
∂∂
+
∂∂
= (3)
la ecuación (1) se puede escribir
TPdV
TdUdS += (4)
comparando (3) y (4)
TP
VSyT1
US
UV=
∂∂
=
∂∂
El equilibrio se alcanza cuando
En un sistema de U = cte y V = cte cuando la entropía S se maximiza.
En un sistema de S = cte y V = cte. cuando la energía interna se minimiza.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 52
CAPÍTULO IV: FUNCIONES AUXILIARES
4.1. FUNCIONES AUXILIARES
Combinando 1º y 2º Ley ⇒ dU = TdS – P.dV
Relación entre variable dependiente U y variable independientes S y V, para un sistema
cerrado, de independientes S y V, para un sistema cerrado, de composición fija y el cambio
de volumen contra la presión externa es el único tipo de w que realiza (trabajo P-V). S y V
no son la mejor elección para variables independientes, S es difícil de medir y controlar.
La ecuación (1) no puede aplicarse a sistemas que presenten cambios composicionales y
realicen otro tipo de w diferente del trabajo P – V.
Aunque la ecuación (1) representa la base de la Termodinámica, para que esta expresión
sea útil es necesario crear funciones termodinámicas auxiliares, las cuales como variables
dependientes están relacionadas en forma simple a variables independientes más
convenientes. Así aparecen las funciones termodinámicas.
A: energía libre de Helmholtz o función trabajo.
A = U – TS (4.1)
G: energía libre de Gibbs
G = U + PV – TS = H – TS (4.2) 4.2. ENTALPÍA H
Sistema cerrado realizando un cambio de estado a P = cte., del estado 1 al 2 la 1º Ley dice:
( )12p12 VVPqUU −−=−
de la cual ( ) ( ) p1122 qPVUPVU =+−+
p12 qHHH =−=∆∴
en un proceso a P = cte., ∆H es igual al calor que entra sale del sistema. Sólo aplicable a
sistemas que realizan trabajo P – V.
4.3. ENERGÍA LIBRE DE HELMHOLTZ (A) A = U –TS
Cambio de estado de 1 a 2
( ) ( ) ( )11221212 STSTUUAA −−−=−
si es un sistema cerrado ( ) wqUU 12 −=−
( ) ( )112212 STSTwqAA −−−=−∴

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 53
si el proceso es isotérmico ⇒ T1 T2 = T (temperatura del depósito que da o recibe calor
durante el proceso), de la 2º Ley: ( ) ( ) wAASSTq 1212 −≤−⇒−≤
( ) wSTAA irr12 −=∆+− (4.3)
para un proceso a T = cte reversible ( )12 AA − =- wmáx para un proceso a T =
cte y V = cte ( )12 AA − + T∆Sirr = 0 (4.4)
0TdSd irr =+∆ en equilibrio 0dA0dSirr =⇒=
criterio de equilibrio para un sistema a T y V = cte.
consideremos n átomos de una sustancia que se encuentran como fase sólida cristalina y
fase vapor, ambas fases están contenidas en un recipiente de volumen constante que está
inmerso en un depósito de calor a T = cte.
∆ = U – TS en el equilibrio debe ser mínima y se logro con bajos valores de U y altos
de S.
los 2 estados extremos para el sistema son:
1. n átomos en fase sólida
2. n átomos en fase vapor
Consideremos 1, en la fase sólida los átomos son mantenidos juntos por las fuerzas
interatómicas, sise saca un átomo de la superficie del cristal y se coloca en el vacío encima
del cristal (eje vacío será ocupado por la base vapor) el w deberá hacerse en contra de los
esfuerzos de atracción que existen entre el cristal y el átomo removido. Pero que esta
separación sea a T = cte., la energía necesaria debe provenir de calor el cual fluye del
depósito de calor hacia el sistema. Ese flujo de q que entra al sistema aumenta la energía
interna y la entropía del sistema.
A medida que el proceso de evaporación continúa (+átomos en la fase vapor) U y S
continúan aumentando. Cuando los n átomos estén en fase vapor la U y S de un sistema
con V = cte y T = cte., tentrán sus máximos valores.
U S
nv nv
Como la contribución de la entropía, -TS, depende de la temperatura y la de la energía
interna es independiente de la temperatura, la influencia de la entropía aumenta cuando
aumenta la temperatura.
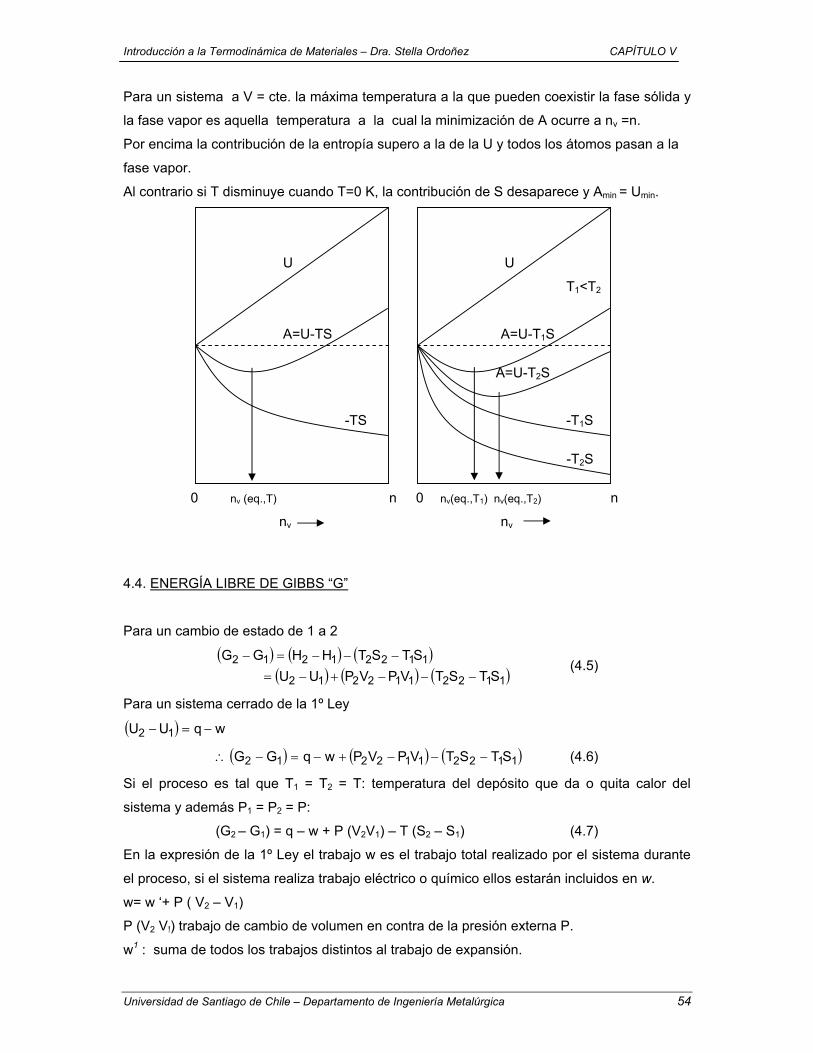
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 54
Para un sistema a V = cte. la máxima temperatura a la que pueden coexistir la fase sólida y
la fase vapor es aquella temperatura a la cual la minimización de A ocurre a nv =n.
Por encima la contribución de la entropía supero a la de la U y todos los átomos pasan a la
fase vapor.
Al contrario si T disminuye cuando T=0 K, la contribución de S desaparece y Amin = Umin.
U U
T1<T2
A=U-TS A=U-T1S
A=U-T2S
-TS -T1S
-T2S
0 nv (eq.,T) n 0 nv(eq.,T1) nv(eq.,T2) n
nv nv
4.4. ENERGÍA LIBRE DE GIBBS “G”
Para un cambio de estado de 1 a 2
( ) ( ) ( )( ) ( ) ( )1122112212
11221212STSTVPVPUU
STSTHHGG−−−+−=
−−−=− (4.5)
Para un sistema cerrado de la 1º Ley
( ) wqUU 12 −=−
( ) ( ) ( )1122112212 STSTVPVPwqGG −−−+−=−∴ (4.6)
Si el proceso es tal que T1 = T2 = T: temperatura del depósito que da o quita calor del
sistema y además P1 = P2 = P:
(G2 – G1) = q – w + P (V2V1) – T (S2 – S1) (4.7)
En la expresión de la 1º Ley el trabajo w es el trabajo total realizado por el sistema durante
el proceso, si el sistema realiza trabajo eléctrico o químico ellos estarán incluidos en w.
w= w ‘+ P ( V2 – V1)
P (V2 V!) trabajo de cambio de volumen en contra de la presión externa P.
w1 : suma de todos los trabajos distintos al trabajo de expansión.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 55
(G2 – G1) = q – w`- T (S2 – S1)
de la 2º Ley ( )12 SSTq −≤
( )12 GG'w −−≤∴
( ) irr12 STGG'w ∆+−=− (4.8)
En el caso de un proceso isotérmico e isobárico durante el que no se desarrolla otro trabajo
que no sea el de expansión (w`= 0)
(G2 –G1) + T∆Sirr = 0 (4.9)
Esto indica que un proceso sólo puede producirse espontáneamente (esto es con
producción de S) si la energía libre disminuye.
Como la condición para equilibrio termodinámico es que dSirr = 0 ⇒ que par un proceso
isotérmico e isobárico la condición de equilibrio está definida como:
dG = 0
Por lo tanto en procesos a T = cte y P = cte., la energía libre de Gibbs G sólo puede
decrecer o permanecer constante y el equilibrio se alcanza cuando G es mínima para los
valores fijados de P y T.
Para un sistema cerrado
dU = TdS – PdV
H = U + PV ⇒ dH = TdS + VdP (4.10)
A = U – TS ⇒ dA = -SdT – PdV (4.11)
G = H – TS ⇒ dG = -SdT + VdP (4.12) 4.6. VARIACIÓN DE LA COMPOSICIÓN Y TAMAÑO DEL SISTEMA
Toda la discusión anterior está restringida a sistemas cerrados, en cuyo caso el sistema
tiene dos variables independientes que cuando se fijan, fijan el estado del sistema.
Sin embargo, si el tamaño y/o composición varían durante el proceso, 2 variables no son
suficientes para fijar el estado del sistema.
Ejemplo: para un proceso a T y P = cte. el equilibrio se alcanza cuando G es mínima. Si la
composición del sistema varía porque ocurre una reacción química, la minimización de G
ocurrirá cuando, siendo T y P = cte., el sistema tenga una determinada composición. Como
G es una propiedad extensiva, depende del tamaño del sistema, es necesario que el número
de moles de todas las especie sean especificados.
G será por tanto función de T,P y el número de moles de todas las especies presentes en el
sistema.
G = G (T,P, ni, nj, nk,….) (4.13)
ni, nj, nk……número de moles de las especies i, j, k,……..

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 56
.....dnnGdn
nGdP
PGdT
TGdG j
....n,n,P,Tji
....n,n,P,Ti,.....n,n,T,....n,n,Pkikjjiji
+
∂∂
+
∂∂
+
∂∂
+
∂∂
= (4.14)
Si el número de moles permanece cte. durante el proceso
VdPSdTdG +−=
de donde VPGyS
TG
....n,n,T....n,n,P jiji
=
∂∂
−=
∂∂
i....n,P,T
ki
1i idn
nGVdPSdTdG
j
∑=
=
∂∂
++−= (4.15)
4.7. POTENCIAL QUÍMICO
El término ,.....j,n,P,Tin
G
∂∂ se denomina potencial químico de la especie i y se designa como µi
i,.....j,n,P,Tin
Gµ=
∂∂ (4.16)
El potencial químico de una especie i en una fase homogénea (µi) se define como el
incremento de G del sistema (la fase homogénea) para una adición infinitesimal de la
especie i, con la condición que la adición se haga a T,P y número de moles de las otras
especies constantes.
Si el sistema es muy grande de manera que la adición de 1 mol de i, a T y P = cte., no
introduce un cambio composicional medible ui es la cantidad mediante la cual la capacidad
del sistema para hacer trabajo, distinto del de expansión, se ve incrementada por mol de i
añadido a T, P y composición constante.
∑µ++=k
1iidnVdPSdTdG (4.17)
Esta ecuación se puede aplicar a sistemas abiertos que intercambian materia o calor con el
medio exterior y a sistemas cerrados en los que se produce cambio de composición.
∑
∂∂
+−=k
1i
...n,V,Sidn
nUPdVTdSdU
j
(4.18)
i,...n,P,S
k
1 idn
nHVdPTdSdH
j
∑
∂∂
++= (4.19)
i,...n,V,T
k
1 idn
nAPdVSdTdA
j
∑
∂∂
++−= (4.20)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 57
Observando las ecuaciones (4.16), (4.18), (4.19) y (4.20) se puede apreciar que
jjjj n,V,Tin,P,Sin,V,Sii
n,P,Ti nA
nH
nUu
nG
∂∂
=
∂∂
=
∂∂
=≡
∂∂ (4.21)
∑µ+−= dniPdVTdSdU i (4.22)
∑µ++= dniVdPTdSdH i (4.23)
∑µ+−−= dniPdVSdTdA i (4.24)
∑µ++= dniVdPSdTdG i (4.25)
4.8. RELACIONES TERMODINÁMICAS
Si la composición es constante, de las ecuaciones (4.22) a (4.25) se deduce que:
PV SH
SUT
∂∂
=
∂∂
= (4.26)
TV VA
VUP
∂∂
−=
∂∂
−= (4.27)
TS PG
PHV
∂∂
=
∂∂
= (4.28)
PV TG
TAS
∂∂
−=
∂∂
−= (4.29)
4.9. RELACIONES DE MAXWELL
Si Z es una función de estado y “x” e “y” son elegidas como variables independientes para
un sistema cerrado de composición fija, luego
Z = Z (x, y)
La diferencial de esa función es
dyyZdx
XZdZ
XY
∂∂
+
∂∂
=
Si la derivada parcial Yx
Z
∂∂ es por sí misma una función de x e y, puede expresarse como:
( ) ( ))y,xMyZyy,xL
xZ
XY=
∂∂
=
∂∂ , luego

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 58
MdyLdxdZ +=
Por lo tanto
XXY yL
xZ
y
∂∂
=
∂∂
∂∂
y
YYX xM
yZ
x
∂∂
=
∂∂
∂∂
Pero, como Z es una función de estado, el cambio en Z es independiente del orden de
diferenciación
Y
2
YXXY xdyZ
yZ
xxZ
y
∂∂
=
∂∂
∂∂
=
∂∂
∂∂
por lo tanto
YX XM
yL
∂∂
=
∂∂ (4.30)
Aplicando esto a las ecuaciones (3.12) y (4.10) a (4.12)
dU = TdS – PdV dA = - SdT – PdV
dH = TdS + VdP dG = -SdT + Vd P
Se obtienen las relaciones de Maxwell
VS SP
VT
∂∂
−=
∂∂ (4.31)
PS SV
PT
∂∂
=
∂∂ (4.32)
VT TP
VS
∂∂
=
∂∂ (4.33)
PT TV
PS
∂∂
−=
∂∂ (4.34)
Para un sistema cerrado y con composición fija, la ecuación (3.12) da
dU = TdS – PdV
luego
PVST
VU
TT−
∂∂
=
∂∂
usando las relaciones de Maxwell, ecuación (4.33)
PTPT
VU
VT−
∂∂
=
∂∂

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 59
ecuación de estado que relaciona la U de un sistema cerrado de componente con
cantidades medibles experimentales T, P y V.
Si el sistema es un gas ideal VR
TPRTV.P
V=
∂∂
⇒=
y P = RT / V ⇒ (∂U / ∂V)T = 0
esto indica que la energía interna de un gas ideal es independiente del volumen del gas.
Similarmente para un sistema cerrado de composición fija: dH = Tds + VdP
VTVT
PH
TV
PSyV
PST
PH
PT
PTTT
+
∂∂
−=
∂∂
∴
∂∂
=
∂∂
+
∂∂
=
∂∂
Ecuación de estado que relaciona la variación de entalpía con la variación de cantidades
medibles experimentalmente T,P y P
Si el sistema es una cantidad fija de un gas ideal
0PHP/RTyV
PR
TVRTV.P
TP=
∂∂
⇒==
∂∂
⇒=
esto indica que la entalpía de un gas ideal es independiente de su presión.
4.10. FÓRMULA DE TRANSFORMACIÓN
Sean X, Y y 2 3 propiedades de estado de un sistema cerrado y de composición fija.
( )YZ Z
xdyyxdxZ,yxx
∂∂
+
∂∂
=⇒=
Para un cambio de estado incremental a x = cte
dZZXdy
yx
YZ
∂∂
−=
∂∂
1XZ
ZY
YX
ZX
ZY
YX
YXZYXZ−=
∂∂
∂∂
∂∂
∴
∂∂
−=
∂∂
∂∂
4.11. LA ECUACIÓN DE GIBBS-HELMHOLTZ
La ecuación (4.2) da
G=H-TS
y la ecuación (4.12)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO V
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 60
STG
P−=
∂∂
Por lo tanto a presión y composición constante
G=H-T
dTdG
o
GdT=HdT+TdG
Dividiendo por T2 y rearreglando
22 THdT
TGdTTdG
−=−
ecuación que comparada con la identidad d(x/y) = (ydx-xdy)/y2, muestra que
2TH
dT)T/G(d
−= (4.36)
Esta ecuación se conoce como la ecuación de Gibbs-Helmholtz y es aplicable a sistemas
cerrados de composición fija que realizan procesos a presión constante. Para un cambio de
estado a presión constante en un sistema cerrado de composición fija, la ecuación (4.36) da
la relación del cambio de G con el cambio de H como
2TH
dT)T/G(d ∆
−=∆ (4.36a)
Esta ecuación es muy usada en termodinámica experimental, ya que permite obtener ∆H , el
calor de reacción, a partir de medidas de la variación de ∆G con la temperatura, o viceversa.
La utilidad de esta ecuación será desarrollada con más profundidad en el Capítulo 10.
La correspondiente relación entre A y U se obtiene de la siguiente manera: la ecuación (4.1)
da:
A=U-TS
A=U-TVT
A
∂∂
Y manipulando de manera similar a la anterior
2TU
dT)T/A(d
−= (4.37)
Esta ecuación es sólo aplicable a sistemas cerrados de composición fija que realizan
procesos a volumen constante y por lo tanto para un cambio de estado en esas condiciones:
2TU
dT)T/A(d ∆
−=∆ (4.37a)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 61
CAPITULO V: CAPACIDAD CALORÍFICA
5.1. INTRODUCCIÓN
Las ecuaciones (2.6) y (2.7) definen dos capacidades caloríficas, a volumen y presión constante
Volumen constante V
v TUc
∂∂
=
Presión constante P
P THc
∂∂
=
Si E es una propiedad extensiva, E’ será el valor de la propiedad para un sistema que contiene
n átomos.
E`= n E
Por lo tanto
dU’ = CvdT = ncvdT
dH’ = CpdT = n cpdT
Integrando esta última ecuación
( ) ( ) ∫=−=∆2
1
T
Tp12 dTcP,THP,THH
se puede apreciar que para conocer la dependencia de la entalpía con la temperatura es
necesario conocer la dependencia con la temperatura de cp. Para conocer la dependencia de U
con la temperatura necesitamos conocer como varía cv con la temperatura.
5.2. CÁLCULOS TEÓRICOS DE LA CAPACIDAD CALORÍFICA
Dulong y Petit introducen una regla empírica: la capacidad calorífica molar de todos los
elementos sólidos es igual a 3R (24.9 J/K)
Posteriores determinaciones experimentales mostraron que la capacidad calorífica de varios
elementos siempre aumentaba con la temperatura y esos valores tendían a ser mucho menores
que 3R a baja temperatura. Además las capacidades caloríficas pueden ser mayores que 3R a
temperaturas superiores a la temperatura ambiente.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 62
Einstein realizó cálculos de la capacidad calorífica de elementos sólidos utilizando la teoría
cuántica.
( )2T
T2E
v1C
CT
R3cE
E
−
θ=
θ
θ
donde θE es la temperatura característica de Einstein cuando T/θE aumenta CV → 3R de
acuerdo a Dulong y Petit y cuando T → 0, CV → 0 de acuerdo con las observaciones
experimentales.
Pb
Cu Si
Diamante
0
25
0 300 Temperatura (K)
c v (J
/K)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 63
Otros cálculos los realizó Debye
( )KTDhxdx
e1
e.xTR9cT
D
o2K
x43
Dv =
−
θ
= ∫θ
−
−
donde θ0 es la temperatura característica de Debye ajusta mejor que la Einstein a datos
experimentales a bajas temperaturas.
5.3. REPRESENTACIONES EMPÍRICAS DE LAS CAPACIDADES CALORÍFICAS
Las variaciones de cp (de una sustancia) con la temperatura medidas experimentalmente,
generalmente ajustan a una expresión del tipo:
2P cTbTaC −++=
esta expresión es aplicable sólo al rango de temperatura en que cp fue medido.
Ejemplo: α-Ca estable entre 737K y Tm y β-Ca estable entre 737K y 273K
kmol/JT10.5.10T10.4.323.6CCa
kmol/JT10.9.132.22cpCa253
P
3
−−
−
++=−β
+=−α

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 64
Elemento Cp (joules/degree mole) Rango Temperatura, K
Al (sólido) 20.7 + 12.4 x 10-3T 298 –Tm
Al (liquido) 29.3 Tm - 1273
Au (sólido) 23.7 + 5.19 x 10-3T 298 - Tm
Au (liquido) 29.3 Tm - 1600
Cu (sólido) 22.6 + 6.28 x 10-3 T 298 - Tm
Cu (líquido) 31.4 Tm - 1600
Fe (α) 17.5 + 24.8 x 10-3T 273 - 1033
Fe (β) 38 1033 - 1181
Fe (γ) 7.70 + 19.5 x 10-3 T 1181 - 1674
Fe (δ) 43.9 1674 - Tm
Fe (liquido) 41.8 Tm - 1873
C (diamante) 9.12 + 13.2 x 10-3 T – 6.19 x 105 T-2 298 - 1200
C (grafito) 17.2 + 4.27 x 103 T – 8.79 x 105 T-2 298 - 2300
O2 (gas) 30.0 + 4.18 x 103 T – 1.7 x 105 T-2 298 - 3000
5.4. ENTALPÍA EN FUNCIÓN DE LA TEMPERATURA Y COMPOSICIÓN Para un sistema cerrado de composición fija que realiza un cambio de estado desde 1 (T1, P)
hasta 2 (T2, P)
( ) ( ) ∫=−=∆2
1
T
Tp1122 dTCP,THP,THH
∆H es el área bajo la curva cp-T entre los límites T1 y T2, además sabemos que ∆H = qp el cual
es el calor necesario para aumentar la temperatura de 1 mol del sistema desde T1 a T2 a P= cte.
Par un sistema en el que se produce una reacción química A + B = AB:
∆H (T,P) = HAB (T,P) – HA (T,P) – HB (T,P)
∆H es la diferencia entre la entalpía de los productos del proceso (estado 2) y la entalpía de los
reactivos (estado 1) y esta es la Ley de Hess
si ∆H > 0 proceso endotérmico
si ∆H < 0 proceso exotérmico
Los cambios de entalpía resultantes de cambios de temperatura y/o composición pueden
representarse en un diagrama entalpía – temperatura.
Proceso que involucra cambio de fase A (3) = A (2)
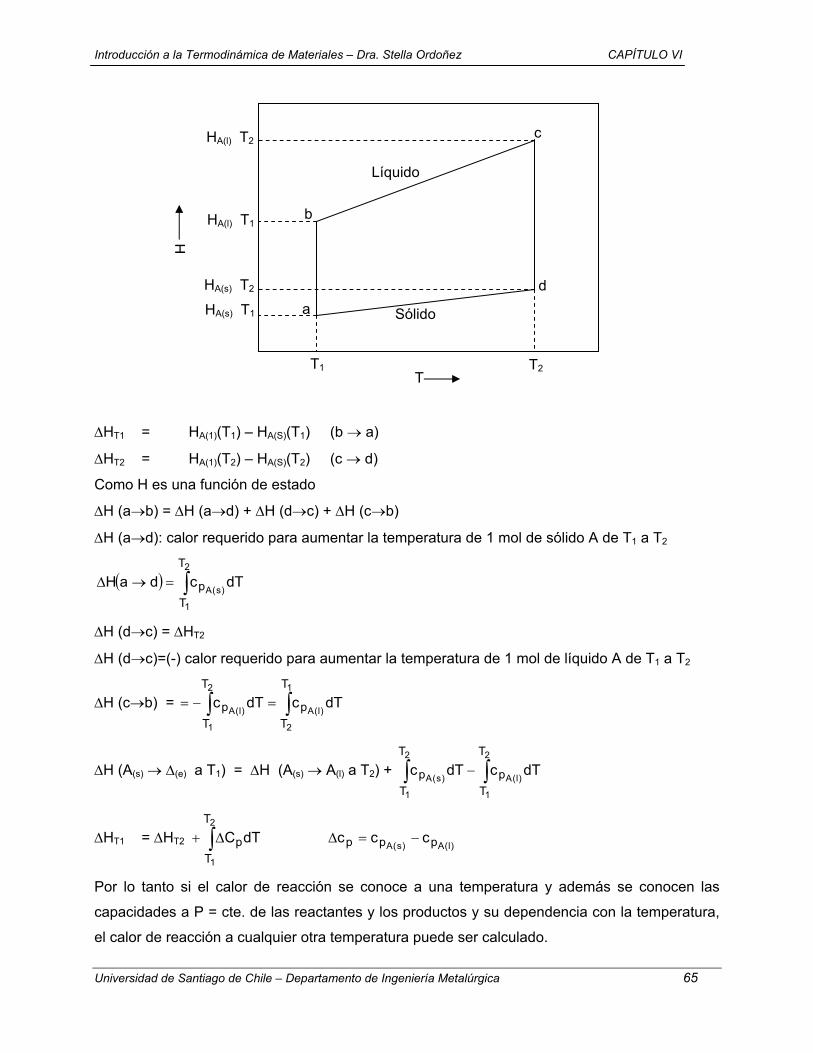
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 65
∆HT1 = HA(1)(T1) – HA(S)(T1) (b → a)
∆HT2 = HA(1)(T2) – HA(S)(T2) (c → d)
Como H es una función de estado
∆H (a→b) = ∆H (a→d) + ∆H (d→c) + ∆H (c→b)
∆H (a→d): calor requerido para aumentar la temperatura de 1 mol de sólido A de T1 a T2
( ) dTcdaH2
1
)s(A
T
Tp∫=→∆
∆H (d→c) = ∆HT2
∆H (d→c)=(-) calor requerido para aumentar la temperatura de 1 mol de líquido A de T1 a T2
∆H (c→b) = dTcdTc1
2
)l(A
2
1
)l(A
T
Tp
T
Tp ∫∫ =−=
∆H (A(s) → ∆(e) a T1) = ∆H (A(s) → A(l) a T2) + dTcdTc2
1
)l(A
2
1
)s(A
T
Tp
T
Tp ∫∫ −
∆HT1 = ∆HT2 ∫ −=∆∆+2
1
)l(A)s(A
T
Tpppp cccdTC
Por lo tanto si el calor de reacción se conoce a una temperatura y además se conocen las
capacidades a P = cte. de las reactantes y los productos y su dependencia con la temperatura,
el calor de reacción a cualquier otra temperatura puede ser calculado.
Líquido
a
b
c
d
Sólido
T1 T2 T
HA(l) T2
HA(l) T1
HA(s) T2
HA(s) T1
H

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 66
Si ∆cp = 0 ∆HT1 = ∆HT2, el calor de reacción es independiente de la temperatura.
En la figura pendiente de la línea (bc) que es (∂H/∂T)p para el líquido A, por definición es cp(l)
∴(bc) será una línea recta solamente si cp es independiente de la temperatura.
Como el valor absoluto de H para cualquier estado es desconocido, se asigna el valor cero de H
de sustancias elementales o su estado estable, eso es, a 298 K (25ºC).
La H de un compuesto a 298 K es igual al calor de formación del compuesto desde sus
elementos a 298 K.
( ) ( ) ( ) K298aMOO21M sg2s =+
∆H298= HMO(s)298 – HM(s)298 - 1/2 HO2(g)298
por convención HM,298 y H02,298 = 0
∆H298 = HMO,298
La variación de los colores de reacción (o calores de formación) con la temperatura a P=cte.
puede ser representada en un diagrama H – T.
Ejemplo: PbOO21Pb 2 =+
( )
( )
( )
( )
( ) K3000K298mol/JT10.18.40.30O,c
TK298mol/JT10.8.269.37,c
K1200Tmol/JT10.1.34.32,c
TK298mol/JT10.75.96.23,c
mol/J219000H
23g2p
0Pb,m3
sPbOp
PbO,m3
ePbp
Pb,m3
sPbp
298PbO
−+=
−+=
−−=
−+=
−=
−−
−
−
−
Calor latente de fusión de 1Pb ∆Hm, Pb = 4810 J/mol a
Tm = 600 K
Tm,Pb0 = 1159 K
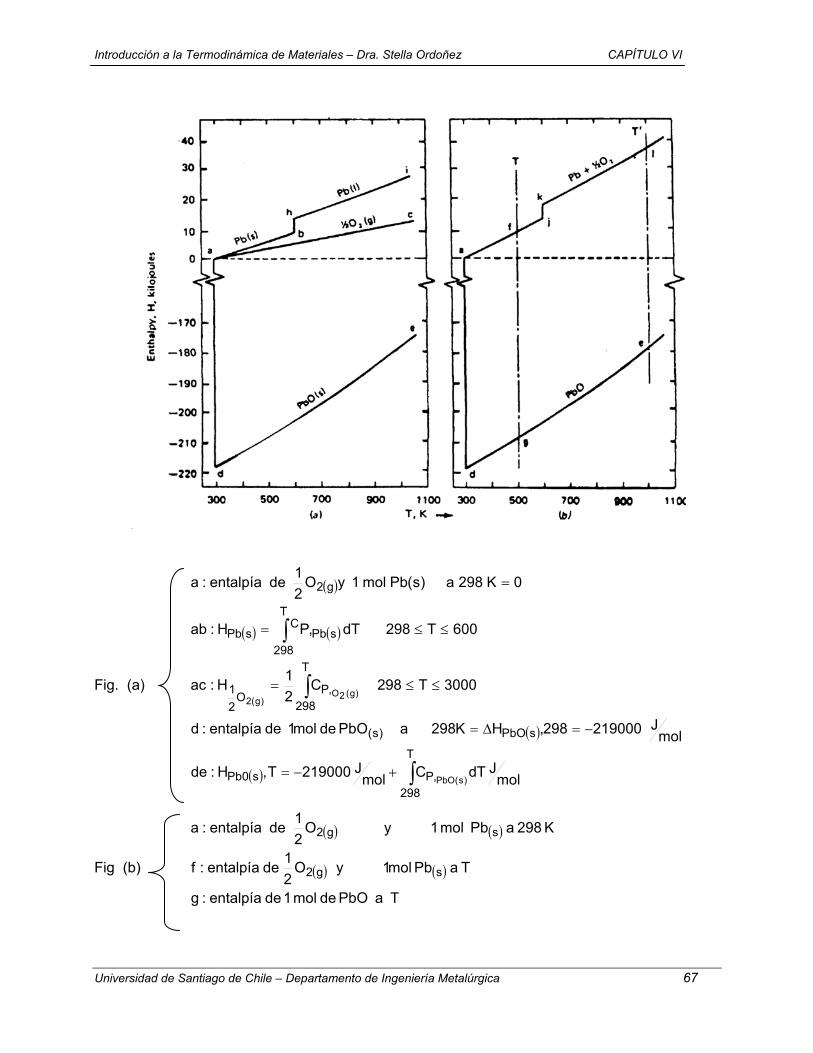
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 67
Fig. (a)
( )
( ) ( )
( )
( ) molJdTCmol
J219000T,H:de
molJ 219000298,HK298aPbO de mol1 de entalpía:d
3000T298C21H:ac
600T298dT,PH:ab
0K298a)s(Pbmol1 yO21 de entalpía:a
T
298,Ps0Pb
sPbO)s(
T
298,PO
21
T
298sPb
CsPb
g2
)s(PbO
)g(2O)g(2
∫
∫
∫
+−=
−=∆=
≤≤=
≤≤=
=
Fig (b)
( ) ( )
( ) ( )
T a PbO de mol1 de entalpía:g
T a Pb mol1yO21 de entalpía:f
K 298 a Pb mol1yO21 de entalpía:a
sg2
sg2

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 68
∆HPb0,298 = ∆H (a → f) + ∆H (f → g) + ∆H (g → d)
( ) ( ) ( )dT,CHdT,CC21 298
TsPbOPT,PbO
T
298sPbPgO,P 2 ∫∫ +∆+
+=
∫ ∆+∆=∆T
298p298T dTCHH
donde ( ) ( ) ( )gO,PsPb,PsPbO,P 2C
21CCCp −−=∆
( )
( )
( ) ( )
−−−+−−−=∆
+−+−=∆
++−=∆
−+−+−+=∆
−
−−
−−
−−−−
∫
2981
T110.85.0298T
210.96,14298T7.0219000H
dTT10.85.0T10.96.147.0219000H
T10.85.0T10.96.147.0C
T10.7.1T10.18.40.3021T10.75.96.23T810.269.37C
5223
T
T
298
253T
253p
25333P
Expresión válida para el rango 298 K a 600K
( ) ( ) ( )
( ) ( ) ( ) dTC21CCH
dTC21CCHH
1
Pb,m
2
2
T
TgO,PlPb,PsPbO,PPb,m
Pb,Tm
298gO,PsPb,PsPbO,P298T
∫
∫
−−+∆−
−−+∆=∆ ′

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 69
Variación con la temperatura del cambio en H para la reacción Pb + ½ O2 = PbO
1
2
3
4
1. ∆H298 = -219000 J
2. ( ) ( ) ( ) dTC21CC
Pb,m
2
T
298gO,PsPb,PsPbO,P∫
−−
3. ∆Hm,Pb
4. ( ) ( ) ( ) dTC21CC
T
600gO,PsPb,PsPbo,P 2∫
−−
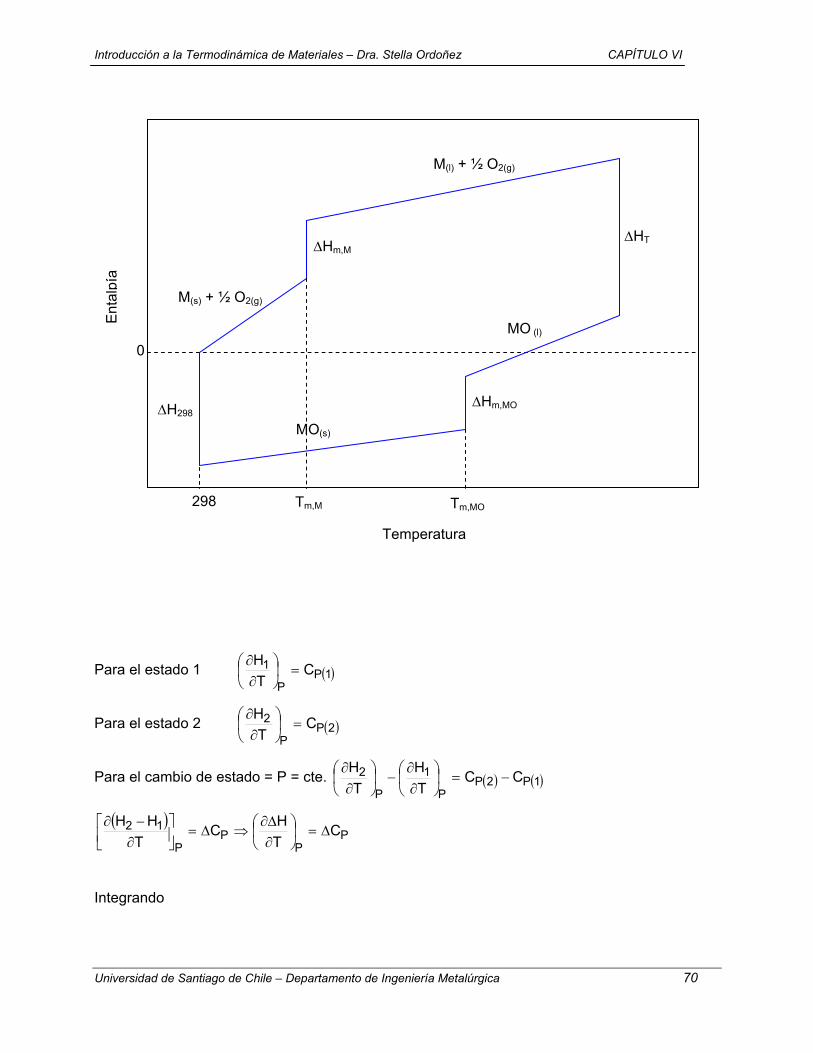
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 70
Para el estado 1 ( )1PP
1 CTH
=
∂∂
Para el estado 2 ( )2PP
2 CT
H=
∂∂
Para el cambio de estado = P = cte. ( ) ( )1P2PP
1
P
2 CCTH
TH
−=
∂∂
−
∂∂
( )P
PP
P
12 CTHC
THH
∆=
∂∆∂
⇒∆=
∂−∂
Integrando
∆Hm,MO
MO (l)
∆HT
M(l) + ½ O2(g)
∆Hm,M
M(s) + ½ O2(g)
∆H298
MO(s)
298 Tm,M Tm,MO
Temperatura
Enta
lpía
0

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 71
∫ ∆=∆−∆2
1
12
T
TPTT dTCHH Ley de Kirchhoff
5.5. DEPENDENCIA DE LA S CON LA TEMPERATURA Y 3º LEY DE LA TERMODINÁMICA
2º Ley, para sistema cerrado que realiza un proceso reversible.
TqdS δ
=
si el proceso es a P = cte.
TdTc
TH
TqdS P
PP=
∂=
δ=
por lo tanto si la temperatura de un sistema cerrado de composición fija aumento de T1 a T2 a P
= cte., el incremento de entropía por mol de sistema, ∆S, será.
( ) ( ) TlndcT
dTcP,TSP,TSS2
1
2
1
T
Tp
T
Tp12 ∫∫ ==−=∆
Para este cambio de estado, ∆S se obtiene como el área bajo la curva de:
1. cP/T vs. T entre T1 y T2
2. cP vs. ln T entre ln T1 y ln T2
Generalmente, ST , la entropía por mol de sistema a cualquier temperatura T está desde por:
∫+=T
oPT TlndcSoS
Donde So es la S del sistema por mol a 0 K
La consideración del valor numérico de So lleva al enunciado de la 3º Ley.
Nernst postuló que, para una reacción química entre sólidos puros o líquidos puros.
K0TaoTHy
TG
PP=≈
∂∆∂
∂∆∂
Para un cambio de estado (por ejemplo reacción química) a T= cte., la ecuación G = H –TS da
∆GT = ∆HT - T∆ST
y el postulado de Nernst era que ∆G para la reacción varía con T como se muestra en la figura.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 72
A cualquier temperatura T la pendiente de la línea es -∆ST y la intersección de la tangente a la
línea en T = 0 es ∆HT. A medida que T se aproxima a cero, la pendiente de la línea tiende a
cero.
T → 0 ⇒ ∆S → 0 y ∆cP → 0
STST
TH
TG
PPP∆−
∂∆∂
=
∂∆∂
=
∂∆∂
VdPSdTdGSTG
P+−=∆−=
∂∆∂
VdPTdSdHCTST
TH
PPP
+=∆=
∂∆∂
=
∂∆∂
como K0TaTHy
TG
PP=→=
∂∆∂
∂∆∂
⇒ ∆S y∆cp → 0 cuando T → 0
El teorema de Nernst dice:
pendiente 0 a T = 0 K
∆G
0 K T

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 73
“Para todas las reacciones que involucren sustancias en estado condensado, ∆S es cero a T =
0 K”
Para la reacción general
A + B = ∆B
∆S = SAB – SA – SB = o cuando T = 0 K
si SA = 0 y SB = 0 ⇒ SAB = 0
Planck completó el teorema de Nernst, enunciando que:
“La entropía de cualquier sustancia homogénea, que está en completo equilibrio interno puede
ser tomada como cero a T = 0 K”.
1. Vidrios (líq. Superenfriado): desorden atómico S ≠ 0 a T = 0 K, el valor de S depende del
grado de desorden.
2. Soluciones: mezcla de átomo, ion o moléculas, entropía de mezcla. La aleatoriedad atómica
de una mezcla determinada su grado de orden. El grado del equilibrio es función de la
temperatura y aumenta cuando T disminuye. El mantenimiento de este equilibrio depende de
la capacidad de las partículas de cambios, su posición en la mezcla y esta capacidad
decrece con la temperatura lo que hace difícil mantener el equilibrio interno.
Soluciones sólidas S ≠ 0 a T = 0 K
3. Algunos elementos químicos puros son mezcla de isótopos (Cl35- Cl35, Cl-35 Cl37, Cl37- Cl37)
existirá una entropía de mezcla y en estricto rigor S ≠ 0 a T = 0 K.
4. A cualquier temperatura los sólidos cristalinos puros contienen un número de defectos en
equilibrio (vacancias, intersticiales). Para que el equilibrio interno se mantenga el número de
defectos debe disminuir con la temperatura, pero como la difusividad también disminuye se
puede tener una concentración de defectos de no – equilibrio en el cristal sólido y por lo tanto
S ≠ 0 a T = 0 K.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 74
5.6. VERIFICACIÓN EXPERIMENTAL DE LA 3º LEY
Transformación de fase α → β, donde α y β son dos formas alotrópicas de un elemento.
a Ttrans y P = 1atom α y β están en equilibrio
∆SIV = ∆SI + ∆SII + ∆SIII
a T = 0 K por la 3º Ley ∆SIV = 0
∆SII = - (∆SI + ∆SIII)
donde ( ) T lndcStransT
0PI ∫ α=∆
( ) T lndcS
THS
0
TPIII
trans
transII
trans
∫ β=∆
∆=∆
∆SII: cambio de entropía de la 3ª Ley
Ejemplo: fase monoclínica estable sobre 368.5 K
0 K
∆SII
∆SI ∆SIII
∆SIV
T trans.
α β

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 75
Fase romboédrica estable bajo 368.5 K
( )
( ) KJ 8.37T lndcS
KJ 86.36T nldcS
0
5.368mpIII
5.368
0rp1
−==∆
==∆
∫
∫
( ) ( )KJ09.1S
KJ94.0KJ8.3786.36SS
II
IIII=∆
=−−=∆+∆−
Como la diferencia es menor que el error experimental, esto se toma como una verificación
experimental de la 3ª Ley.
Asumiendo So = 0, la entropía absoluta de cualquier sustancia pura será
KJT ln d cST
0PT ∫=
Las entropías molares normalmente están tabuladas a 298 K
KJT ln d cS298
0P298 ∫=

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 76
Elemento S298 (J/K) ∆Hm (J) Tm ∆Sm (J/K)
Al
Au
Cu
Fe
Pb
C (grafito)
C (diamante)
O2
28.3
47.36
33.3
27.2
64.9
5.694
2.44
205
10500
12800
13000
13800
4810
932
1336
1356
1808
600
11.3
9.58
8.59
7.63
8.02

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 77
KJT ln d cSST
298P298T ∫+=
cp = a + bT + cT-2
KJ298
1T1c
21)298T(b
298T ln aSS 22298T
−−−+
+=
Por encima de Tm
KJT ln d CST ln d CSST
T(l)Pm
T
298(s)P298T
m
m
∫∫ +∆++=
donde ∆Sm es la entropía de fusión = m
mTH∆
Graficando ∆Hm vs. Tm para un gran número de metales se tiene que ∆Hm/Tm cae en las
cercanías de 9 J/K. Esta correlación se conoce como la regla de Richards
m
mTH∆ = ∆Sm ∼ 8 –16 J/K
Similarmente graficando ∆Hv (calor molar de evaporación) vs. Tb (temperatura de ebullición) los
puntos caen cercanos a una línea recta de pendiente 88 J/K, esta correlación se conoce como
la regla de Trouton.
b
vTH∆ = ∆Sb ∼ 88 J/K

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 78

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 79
5.7. ENTALPÍA Y ENTROPÍA COMO FUNCIÓN DE P
Para un sistema cerrado de composición fija que realiza un cambio de presión a T= cte.
dPPHdH
T
∂∂
=
dH = TdS + VdP
VPST
PH
T+
∂∂
=
∂∂
de acuerdo a las relaciones de Maxwell
PT TV
PS
∂∂
−=
∂∂
VTVT
PH
PT+
∂∂
=
∂∂
Como el coeficiente de expansión térmica PT
VV1
∂∂
=α
)T1(VVVTPH
Tα−=+α−=
∂∂
Para un cambio de estado de (P1, T) a (P2, T)
∫ α−=−=∆2
1
P
P12 dP)T1(V)T,P(H)T,P(HH
Sistema cerrado de composición fija que realiza un cambio de presión a T = cte.
dPPSdS
T
∂∂
=
PT TV
PS
∂∂
−=
∂∂ y como
PTV
V1
∂∂
=α
por lo tanto VPS
Tα−=
∂∂
para un cambio de estado de (P1, T) a (P2, T)
∫ α−=−=∆2
1
P
P12 dPV)T,P(S)T,P(SS

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 80
para un gas ideal con α = 1/T
−=
−=−=∆ ∫
2
1
1
2P
PVVlnR
PPlnRPlndRS
2
1
En el caso de Fe a 298 K un aumento de presión de 1 a 100 atm, disminuye la entropía molar
en 0,0022 J/K y para Al 0,007 J/K. Estas disminuciones se pueden obtener bajando las
temperaturas de Fe y Al en 0,27 y 0,09 grados respectivamente para 1 atm de presión.
Esto confirma que las entalpías y entropías de las fases condensadas son relativamente
insensibles a los cambios de presión.
Para sistemas cerrados de composición fija que realiza cambios de temperatura y presión.
∫∫ α−+=−=∆2
1
2
1
P
P
T
Tp1122 dP)T1(VdTc)T,P(H)T,P(HH
∫∫ α+=−=∆2
1
2
1
P
P
T
Tp1122 dPVTlndc)T,P(S)T,P(SS
se requiere conocer la dependencia de cp con la temperatura y de V y α con la presión. Sin
embargo, para fases condensadas consideradas en rangos pequeños de presión, ambas
dependencias con la presión pueden ser usualmente ignoradas.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 81
CAPITULO VI: EQUILIBRIO DE FASES EN UN SISTEMA DE UN COMPONENTE
6.1. INTRODUCCIÖN
Propiedades intensivas: T, P, µ, son medidas de distintas clases de potencial
T: medida del potencial o intensidad del calor en un sistema. La T es una medida de la
tendencia del calor a dejar el sistema.
T1 < T2 ⇒ ∆T
Existe un gradiente que produce una fuerza impulsora para el flujo de calor a través de ese
gradiente desde la T mayor a la menor. Se produce un flujo espontáneo de calor hasta que el
gradiente potencial se elimina.
P: medida de la tendencia al movimiento masivo
P1 > P2 ⇒ ∆P
Si F1 se expande, P1 disminuye y F2 se contrae, P2 aumenta. El equilibrio se alcanza cuando
∆P=0.
µ: medida de la tendencia de las especies i a dejar la fase. Es una medida de la presión química
ejercida por i en la fase. Si µi es distinto en diferentes fases del sistema que están a igual T y P,
la especie i tenderá a ir de la fase en la que tiene mayor µi a la que tiene menor µi. La existencia
de un gradiente de potencial químico es la fuerza impulsora para la difusión química.
6.2. VARIACIÓN DE G CON LA T A P=CTE.
A 1 atm y 0ºC, hielo y agua están en equilibrio y G’ del sistema es mínima. Si se añade calor al
sistema de manera que algo de hielo se derrita a 0ºC y 1 atm, el equilibrio no se perturba y G’
permanece constante.
T1 T2
F1 F2

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 82
Si añadiendo calor derretimos 1 mol de hielo
H2O(s) = H2O(l) a 1 atm y 273 K
∆G = 0GG )s(OH)l(OH 22=−
444 3444 21molareslibresenergías
)s(OH)l(OH 22GG = (1)
Para un sistema que contiene )l(OH)s(OH 22nyn , La energía libre del sistema será:
)l(OH)l(OH)s(OH)s(OH 2222GnGnG +=′ (2)
a 0ºC y 1 atm )s(OH)l(OH 22GG = , por lo tanto el valor de G’ es independiente de las
proporciones de fase líquida y sólida presente.
Esto ocurre porque en el equilibrio, la tendencia del H2O a dejar la fase líquida es igual a la
tendencia a del H2O a dejar la fase sólida.
∑µ++−= iidnVdPSdTdG
Integrando esa expresión para T y P=cte.
∑µ=′i
iinG
para agua + hielo
)l(OH)l(OH)s(OH)s(OH 2222nnG µ+µ=′ (3)
comparando (2) y (3) OHOH 22G=µ en general ii G=µ , esto es el potencial químico de una
especie en un estado en particular iguala a la energía libre por mol de esa especie en ese
estado.
iP,Tin
Gµ=
∂
′∂
Para un sistema de un componente, el potencial químico de la especie i es igual al incremento
en el valor de G’ para el sistema como resultado de la adición de 1 mol de i a T y P=cte.
iG µ=′∆
y como iGG =′∆
iiG µ=

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 83
H2O + hielo a 1 atm y T> 0ºC, sistema inestable y el hielo se derrite espontáneamente. Este
proceso hace disminuir la energía libre del sistema y el equilibrio se alcanza cuando todo el
hielo se ha derretido.
H2O(s) = H2O(l) a 1 atm y T> 273 K
∆G = 0GG )s(OH)l(OH 22<−
)s(OH)l(OH 22GG <
para T< 273 K
)s(OH)l(OH 22GG >
Las variaciones de )s(OH)l(OH 22GyG con la temperatura a P=1 atm pueden graficarse
pendiente STG
P−=
∂∂ S
TG
P∆−=
∂∆∂
Tc
TS
TG P
PP2
2−=
∂∂
−=
∂
∂
Como la pendiente de la figura de la derecha es negativa, indica que para todas las
temperaturas
)s(OH)l(OH 22SS >
lo cual es lógico ya que la fase líquida es más desordenada que la fase sólida.
G = H – TS
Líquido
Sólido
Tm
G
T
P = cte.
Tm
P = cte.
+
0
-
T
∆G (s
→l)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 84
)l()l()l( TSHG −= )s()s()s( TSHG −=
para la reacción sólido → líquido
)ls()ls()ls( STHG →→→ ∆−∆=∆
en el equilibrio 0G )ls( =∆ → y eso ocurre a Tm
)ls(m)ls( STH →→ ∆=∆ (4)
-10
-8
-6
-4
-2
0
2
4
250 275 300 325 350Temperatura
Enta
lpía
8
12
16
20
24
28
250 275 300 325 350Temperatura
TS
H2O(l)
H2O(s)
Fig. 3a Fig. 3b
)ls(m HH →∆=∆ = 6008 J a 273 K
K/J38c
K/J44.75c
K/J77.44S
K/J08.70S
)s(OHP
)l(OHP
298),s(OH
298),l(OH
2
2
2
2
=
=
=
=
∫ −==T
298)l(PT),l( K/J)298T(44.75dTcH
∫ ∫ −+−−=+∆−=273
298
T
273)s(Pm)l(PT),s( K/J)273T(386008)298273(44.75dTcHdTcH
La separación vertical de las dos líneas a la temperatura T es igual a T),ls(H →∆
H2O(l)
H2O(s)
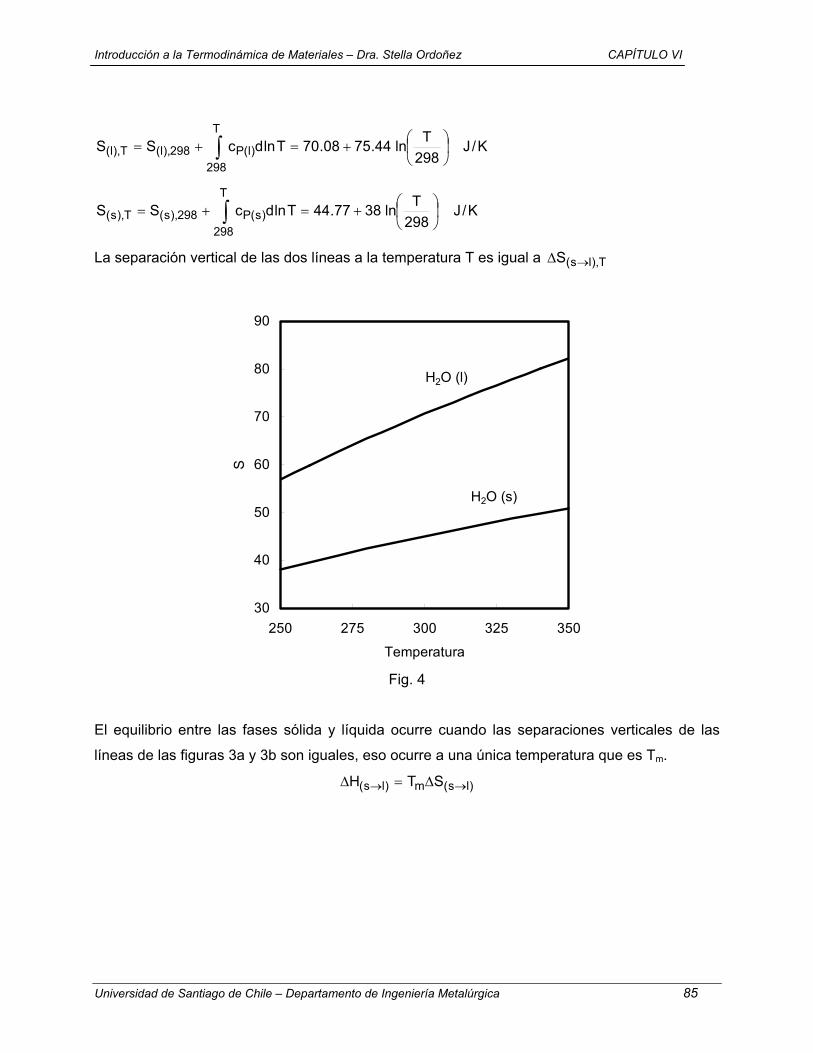
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 85
∫
+=+=T
298)l(P298),l(T),l( K/J
298Tln44.7508.70TlndcSS
∫
+=+=T
298)s(P298),s(T),s( K/J
298Tln3877.44TlndcSS
La separación vertical de las dos líneas a la temperatura T es igual a T),ls(S →∆
30
40
50
60
70
80
90
250 275 300 325 350Temperatura
S
H2O (l)
H2O (s)
Fig. 4
El equilibrio entre las fases sólida y líquida ocurre cuando las separaciones verticales de las
líneas de las figuras 3a y 3b son iguales, eso ocurre a una única temperatura que es Tm.
)ls(m)ls( STH →→ ∆=∆

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 86
-2
0
2
4
6
8
10
12
250 275 300 325 350Temperatura
∆G, T
∆S, ∆
H (K
J)
Fig. 5
∆G (s→l) = 0 a T=Tm=273 K para P=1atm
El equilibrio entre dos fases ocurre como resultado de un compromiso entre consideraciones
entálpicas y entrópicas. La minimización de G’ requiere que H’ sea pequeña y S’ grande. La
figura 3a muestra que siempre H(l) > H(s), entonces desde el punto de vista de la entalpía y sin
ninguna otra consideración la fase más estable es la sólida. Desde el punto de vista de la
entropía, como S(l) > S(s) la fase líquida será las más estable. Sin embargo, como la contribución
de la entropía es dependiente de la temperatura (TS), por encima de Tm la contribución de S
supera a H y por de bajo de Tm es a la inversa.
∆H (s→l)
∆G (s→l)
T∆S (s→l)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 87
6.3. VARIACIÓN DE G CON LA PRESIÓN A T=CTE.
H2O + hielo a 0ºC y 1 atm, si la presión aumenta a T=cte., de acuerdo al principio de Le
Chatelier el sistema tenderá a anular ese aumento de presión disminuyendo el volumen. A 0ºC
el hielo tiene un volumen molar mayor que el agua líquida, por lo tanto el cambio producido por
un aumento de presión es el derretimiento del hielo.
)l(T
)l( VP
G=
∂
∂ )s(
T
)s( VP
G=
∂
∂ )ls(
T
)ls( VP
G→
→ ∆=
∂
∆∂
6.4. G COMO FUNCIÓN DE T Y P
ES posible mantener el equilibrio entre las fases líquida y sólida variando T yP simultáneamente
de manera que ∆G (s→l) = 0
)s()l( GG =
y para cualquier cambio infinitesimal en T yP
)s()l( dGdG =
dPVdTSdG )l()l()l( +=
dPVdTSdG )s()s()s( +=
dPVdTSdPVdTS )s()s()l()l( +−=+−
1 atm
líquido
sólido
T = 0ºC
P →
G →

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 88
)sl(
)sl(
)l()s(
)l()s(
eq VS
VVSS
dTdP
→
→
∆
∆=
−
−=
En el equilibrio ∆G=0 ⇒ ∆H=T∆S
VTH
dTdP
eq ∆∆
=
ecuación de Clapeyron (5)
En el caso de hielo-agua ∆V es negativo y en cualquier sistema ∆H(s→l) es positivo, por lo tanto
eqdTdP
es negativo, es decir cuando la presión aumenta la Tm disminuye. En sistemas
normales eqdT
dP
es positivo, o sea la Tm aumenta con la presión.
Los estados de las fases líquida y sólida se pueden representar en un diagrama G-T-P, en esta
figura cada fase está representada por una superficie y la línea que une las intersecciones de
las superficies determina los valores de P yT que permiten el equilibrio de las dos fases.

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 89
6.5. EQUILIBRIO ENTRE LA FASE VAPOR Y LA FASE LÍQUIDA
Si se aplica la ecuación de Clapeyron al equilibrio sólido-vapor, ∆V es el cambio en el volumen
molar que acompaña la sublimación y ∆H el calor latente de sublimación.
∆V = Vvapor – V(s)
Vvapor >> V(s) ⇒ ∆V = Vvapor
veq TVH
dTdP ∆
=
si el vapor en equilibrio con la fase sólida se comporta idealmente, PV=RT, por lo tanto
dTRT
HPlnddTRT
HPdP
RTHP
dTdP
222eq
∆=⇒
∆=⇒
∆=
si ∆H es independiente de la temperatura
constanteRT
HPln +∆
−=
Como hay equilibrio la P a cualquier temperatura será la presión de vapor saturada ejercida por
la fase condensada a la temperatura T. La presión aumenta exponencialmente con la
temperatura.
Si ∆cP para la sublimación no es cero, pero es independiente de la temperatura
( ) ( ) Tcc298H298TcHH pp298p298T ∆+∆−∆=−∆+∆=∆
reemplazando en la ecuación (6) e integrando
.cteTlnRc
T1
RHc298Pln P298P +
∆+
∆−∆=
que es de la forma CTlnBTAPln ++=
Representación gráfica del equilibrio de fases en un sistema de un componente
En el caso de equilibrio líquido-vapor, el punto de ebullición normal se define como la
temperatura a la cual la presión de vapor de saturación ejercida por el líquido es 1 atm.
Conociendo las capacidades caloríficas del líquido y el vapor, el calor de evaporación ∆Hevap,T y
la temperatura de ebullición se pueden determinar de las relaciones presión de vapor-
temperatura de una sustancia particular. Para el caso del agua:

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 90
]2500298
253)v(OH,P K/JT10x33.0T10x7.1030c
2−− ++=
]373273)l(OH,P K/J44.75c
2=
K/JT10x33.0T10x7.1044.45c 253)vl(OH,P 2
−−→ ++−=∆
∆Hevap=41090 J/mol a 373 K
( )
( ) ( )
T110x33.0T10x35.5T44.4558872
3731
T110x33.0373T10x35.5373T44.4541090
dTcHH
523
5223
T
373vlP373,evapT,evap
−+−=
−−−+−−=
∆+∆=∆
−
−
→∫
dTRT
HPlnd 2
evap∆= R=8.3144 J/mol K
.cteRT2
10x33.0R
T10x35.5R
Tln44.45RT
58872Pln 2
53+++−−=
−
a la temperatura de ebullición 373 K, p=1 y la constante de integración es 51.092.
303.2092.51
RT303.2x210x33.0
R303.2T10x35.5
RTlog44.45
RT303.258872)atm(Plog 2
53+++−−=
−
185.22T862T10x279.0Tlog465.5
T3075)atm(Plog 2
3 +++−−= − (9)
relación presión de vapor saturada-temperatura para agua en el rango de temperatura 273 a
373 K. La curva que ajusta las medidas experimentales de la presión de vapor es de la forma
732.19Tlog65.4T
2900)atm(Plog +−−= (10)

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 91
Esta figura representa las ecuaciones 9 y 10, y se puede apreciar que son indistinguibles. En la
figura la pendiente de la línea a cualquier T es -∆Hevap,T/4.575.
- Línea AOA’
representación gráfica de
veq TVH
dTdP ∆
=
que expresa
la relación P-T para el
equilibrio sólido-líquido. “m”
es el punto de fusión y la
pendiente de la línea es
m
mVT
H∆∆ .
- Línea BOB’
representación de las
ecuaciones 7 u 8. Es la línea
del equilibrio líquido-vapor.
La línea BOB’ pasa por el
punto de ebullición “b” e
intersecta a la línea AOA’ en
P (a
tm)
bSólido Líquido
Vapor
m
O
AB
C
C’
A’
B’
1
0.006
0º
0 0075ºC
100º
Fig. 10

Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 92
el punto triple. El punto triple es el estado representado por valores invariables de P y T a los
cuales sólido, líquido y vapor coexisten en equilibrio.
- Línea COC’
representa el equilibrio sólido-vapor. Si al superficie G-T-P para la fase vapor se incluyera en la
figura 7, intersectaría con la superficie de estado sólido a lo largo de una línea e intersectaría
con la superficie de estado líquido a lo largo de otra línea. Las proyecciones de esas líneas en
el plano basal P-T producen la figura 10.
Introducción a la Termodinámica de Materiales – Dra. Stella Ordoñez CAPÍTULO VI
Universidad de Santiago de Chile – Departamento de Ingeniería Metalúrgica 93
Diagrama de fases del azufre
Diagrama de fases del agua Diagrama de fases del carbono
Top Related