USO SEGURO DE LA TERAPIA ONCOLÓGICA: INTERCAMBIABILIDAD DE...
73
USO SEGURO DE LA TERAPIA ONCOLÓGICA: INTERCAMBIABILIDAD DE MEDICAMENTOS Dra. Carmen Juliana Pino. MD. Esp. Farmacóloga Clínica. MSc. (c) en Docencia de la Educación Superior. Profesora Asistente Universidad El Bosque. Medico Especializado Programa de Farmacovigilancia del INVIMA, Dirección de Medicamentos y productos Biológicos.
Transcript of USO SEGURO DE LA TERAPIA ONCOLÓGICA: INTERCAMBIABILIDAD DE...

USO SEGURO DE LA TERAPIA
ONCOLÓGICA:
INTERCAMBIABILIDAD DE
MEDICAMENTOS
Dra. Carmen Juliana Pino. MD. Esp. Farmacóloga Clínica.
MSc. (c) en Docencia de la Educación Superior. Profesora Asistente Universidad El Bosque.
Medico Especializado Programa de Farmacovigilancia del
INVIMA, Dirección de Medicamentos y productos Biológicos.

TEMARIO
FASES DEL DESARROLLO DE
MEDICAMENTOS.
PARALELO MEDICAMENTOS SÍNTESIS
QUÍMICA Y MEDICAMENTOS BIOLOGICOS.
INTERCAMBIABILIDAD.
INTERCAMBIABILIDAD -MEDICAMENTOS
ONCOLOGICOS –BIOSIMILARES Y
SEGURIDAD DEL PACIENTE.

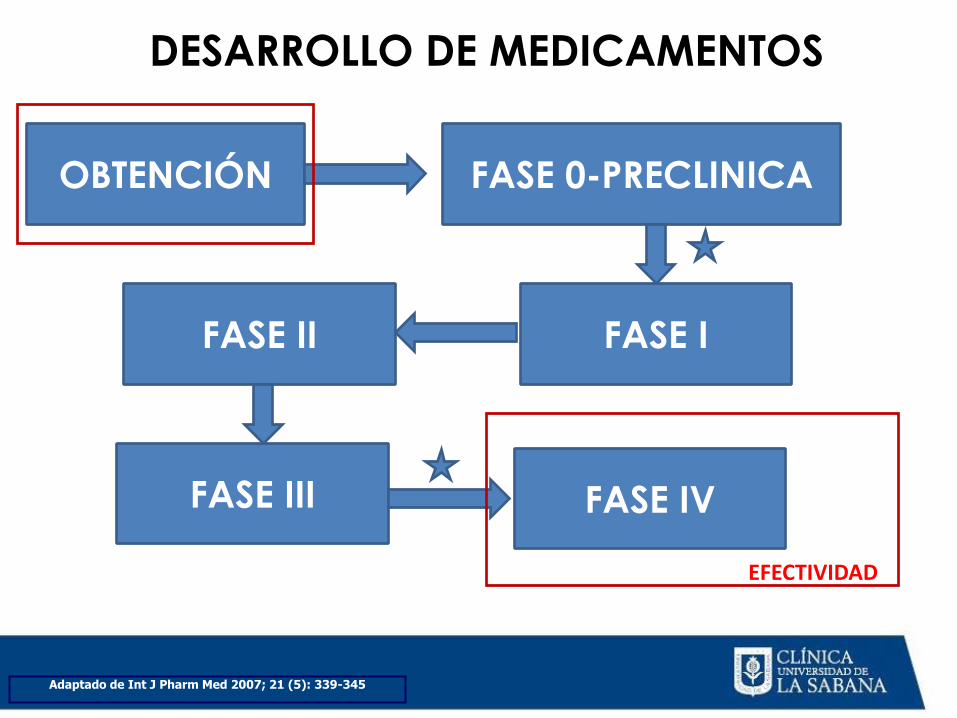
FASES DEL DESARROLLO DE MEDICAMENTOS
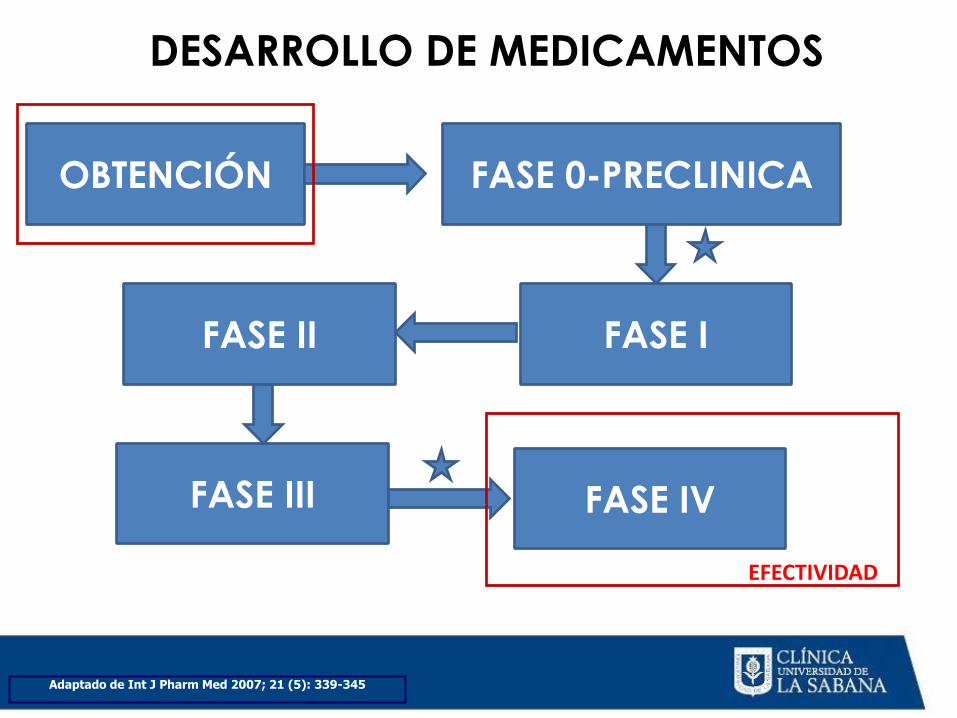
DESARROLLO DE MEDICAMENTOS
Adaptado de Int J Pharm Med 2007; 21 (5): 339-345
OBTENCIÓN FASE 0-PRECLINICA
FASE I FASE II
FASE III FASE IV
EFECTIVIDAD

Métodos más utilizados:
a) Extracción y purificación de un principio
activo presente en un producto natural.
b) Modificación química de una molécula ya
conocida.
c) Síntesis química de una nueva molécula.
d) Diseño racional de fármacos basados en la
relación estructura–actividad.
e) Utilización de la Biotecnología.
OBTENCIÓN
BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana

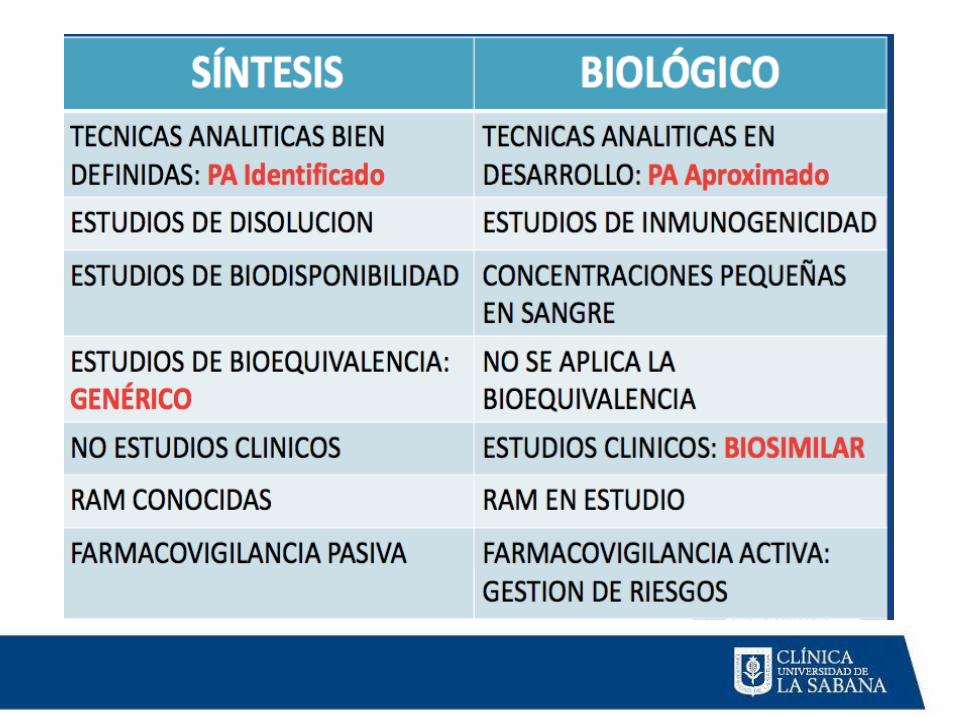
SINTESIS QUÍMICA BIOLÓGICOS
Síntesis de medicamentos cuyo
principio activo una estructura
química.
Semisintesis.
Se obtienen a partir de organismos vivos o de
sus tejidos, fluidos, órganos.
Virus - bacterias - hongos - células animales o
vegetales.
Los medicamentos de origen biológico se
producen mediante procesos más sencillos
sin uso de información genética.
Subconjunto denominado biotecnológicos: los biotecnológicos usan información
genética y tecnologías especiales para que
las células actúen como fábrica de
sustancias para luego convertirlas en
medicamentos. manipulación genética
técnicas de ADN reocmbinante.
Aunque los principios generales de producción son semejantes, las características de
calidad, seguridad y eficacia de los medicamentos de origen biológico dependen
del material biológico de origen, la complejidad de su estructura y los procesos
tecnológicos de su obtención.

MEDICAMENTOS DE SINTESIS
QUIMICA
7

8

9

10
Relaciones espaciales
• Define intervalos para
distancias y ángulos
• Relación estructura
actividad.
1.Janet B. Clarke, Mechanisms of ADRs to Biologics, Springer‐Verlag Berlin Heidelberg, 2010 2.EJHP Practice, Volume 14, 2008
11

12
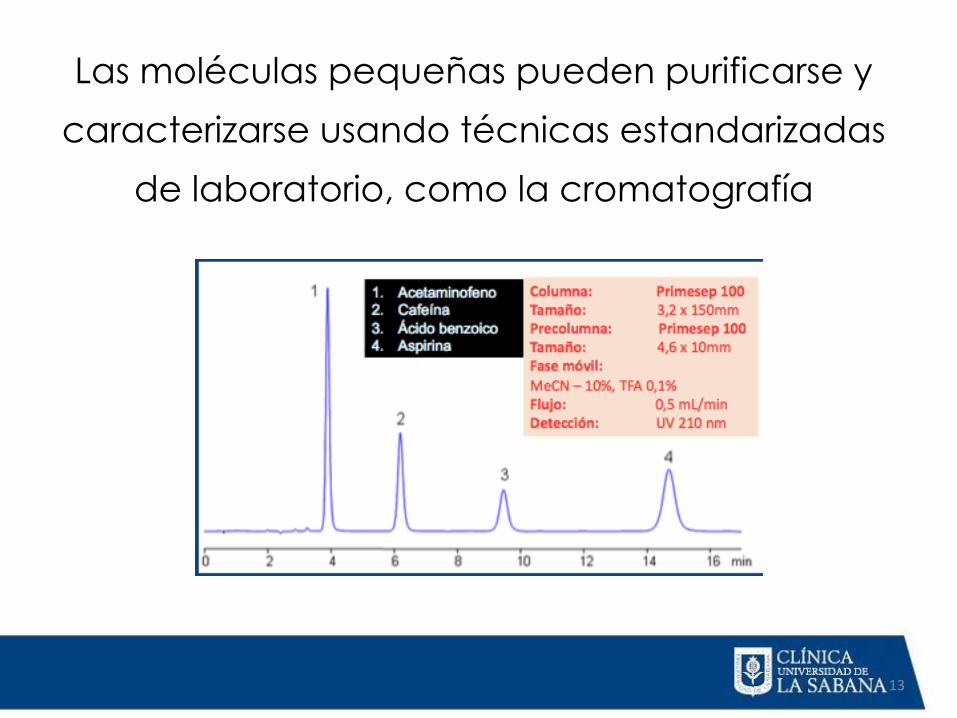
Los medicamentos de molécula
pequeña,
como la aspirina, se sintetizan a través de
procesos químicos definidos
13
Las moléculas pequeñas pueden purificarse y
caracterizarse usando técnicas estandarizadas
de laboratorio, como la cromatografía

MEDICAMENTOS BIOLÓGICOS
14
15
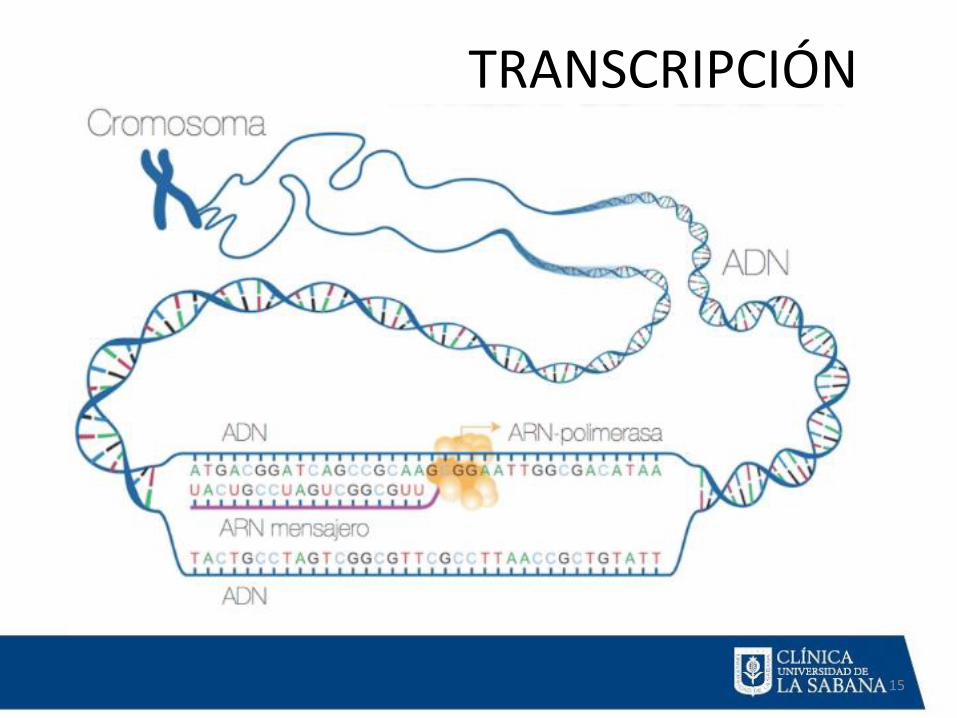
TRANSCRIPCIÓN
16
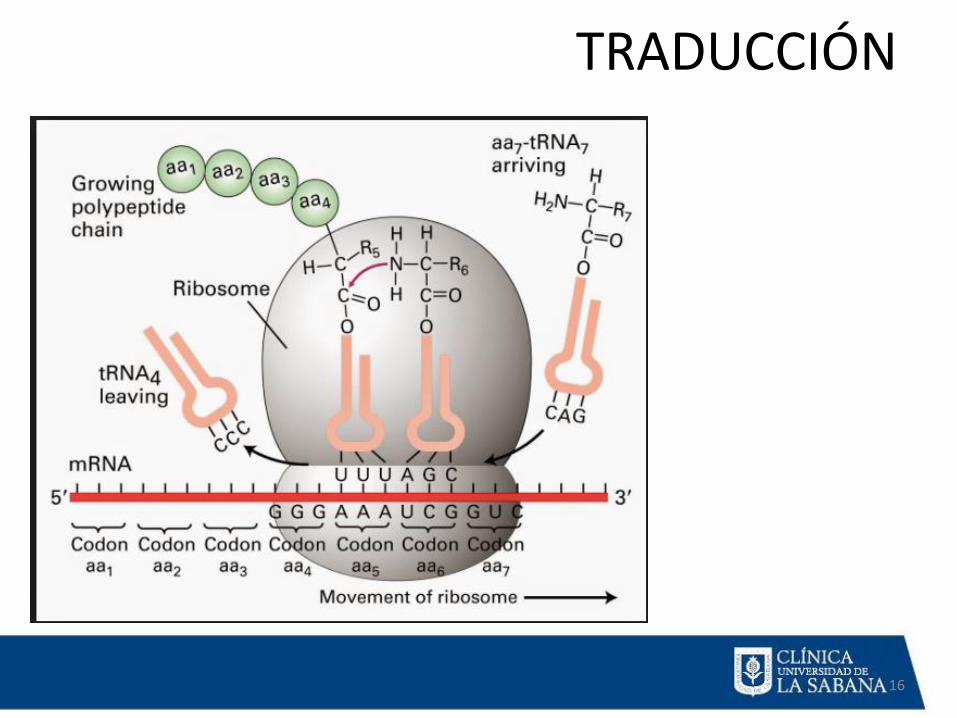
TRADUCCIÓN

17
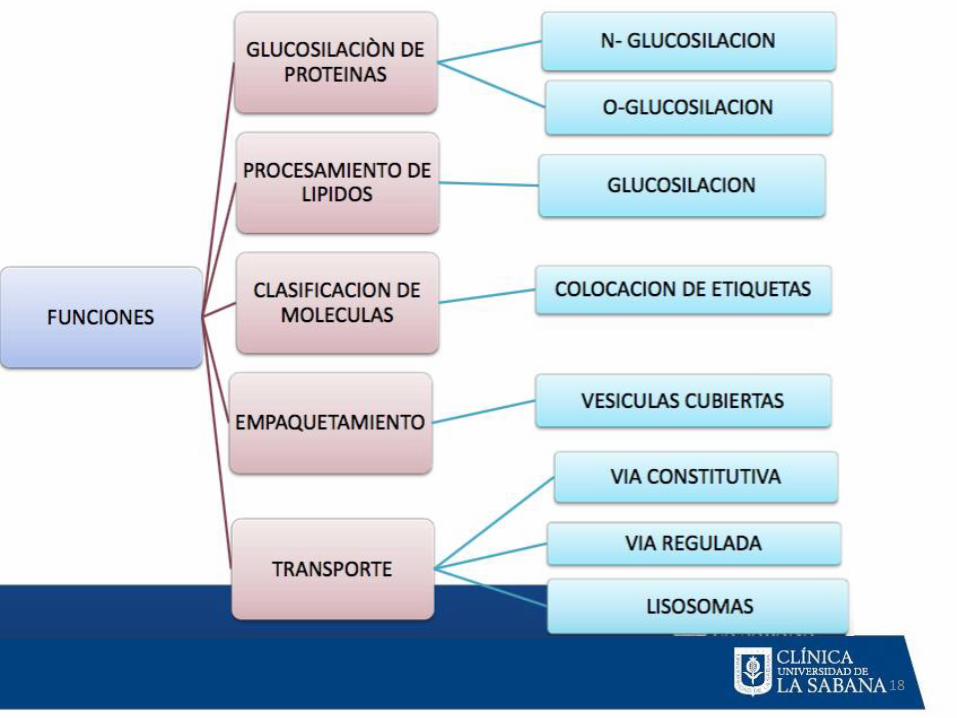
MODIFICACIONES POST TRADUCCIONALES
18

19 http://www.faba.org.ar/fabainforma/363/acta01.html

PARALELO ENTRE MEDICAMENTOS DE SÍNTESIS QUÍMICA Y MEDICAMENTOS
BIOLÓGICOS.
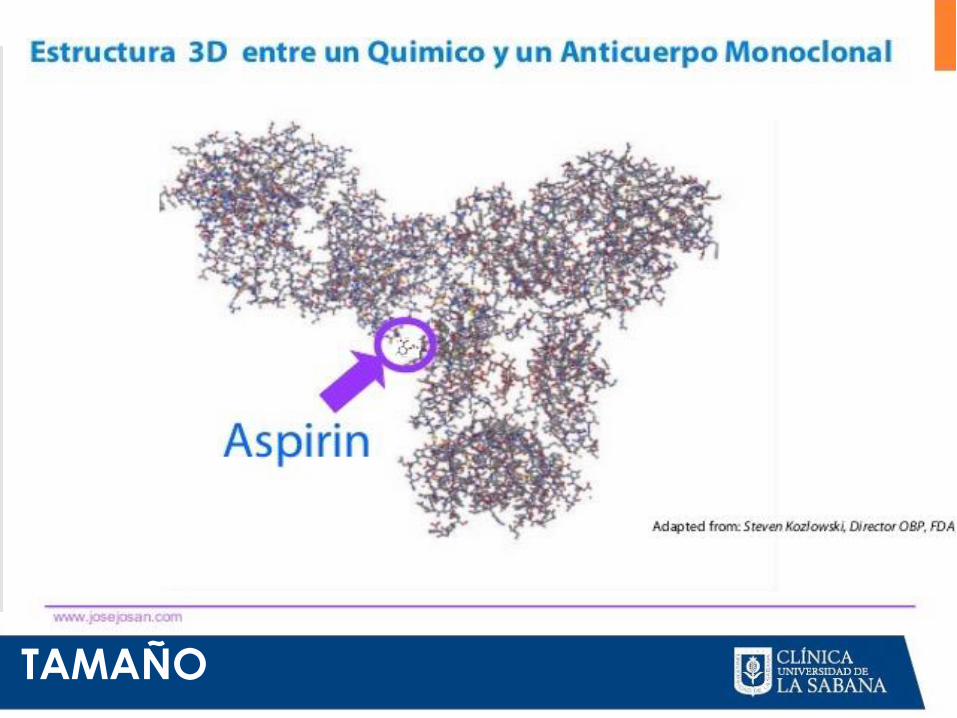
TAMAÑO

22

MEDICAMENTOS DE SINTESIS QUIMICA
•Estructura relativamente sencilla
•Fácil de reproducir
•PM: entre 100-1000 D(<1000 átomos)
•Estabilidad química : adecuada
•Fabricación: sencilla .
MEDICAMENTOS BIOLOGICOS
•Estructura muy compleja
•Difícil de reproducir
•PM: entre 10.000-300.000 D(5000-20000
átomos)
•Estabilidad química: inestables, lábiles a
cambios de pH, temperatura humedad.
•Fabricacion: compleja , sensibles a
pequeños cambios

ESTRUCTURA BIOLÓGICOS
Son macromoléculas– se afecta la
farmacocinética
Fácilmente degradables – proteasas
Estructura química muy inestable
Estructira tridimensional compleja
EFICACIA SEGURIDAD

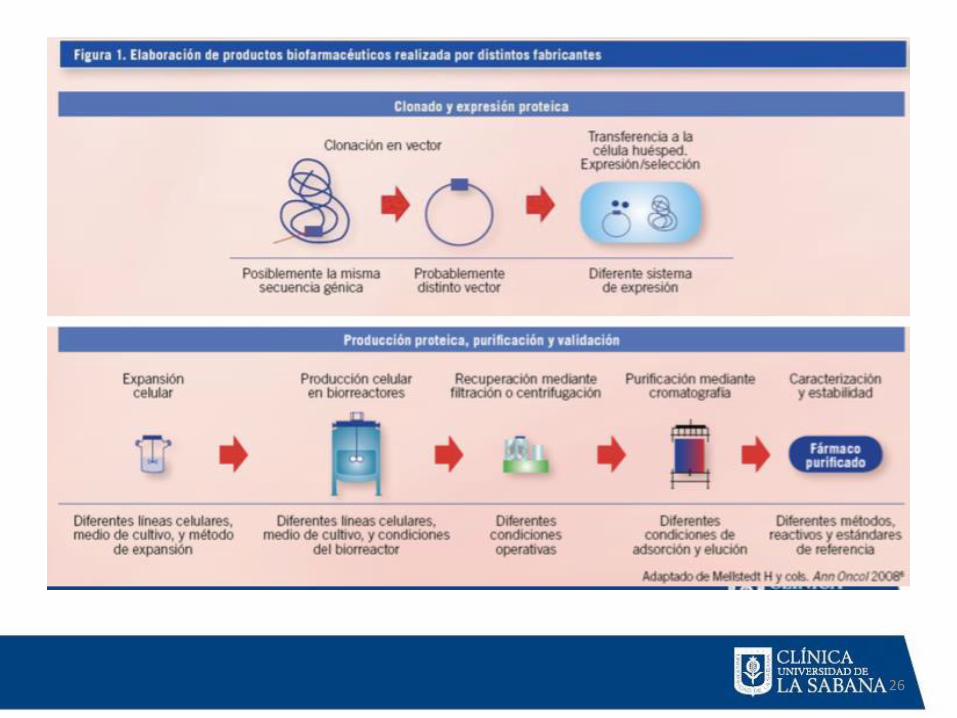
PROCESO DE PRODUCCION BIOLÓGICOS
• Tipo celular.
• Medio de cultivo.
• Método de purificación.
• Cambios estructurales de la proteína.
• Aprox. 2000 pruebas de calidad de
producción comparado con 200 de
moléculas pequeñas.
26

27 International Federation of Pharmaceutical Manufacturers & Associations

28

29

30
Experiencia en fabricación estéril (controles de etapas críticas y etapas intermedias)
• Estabilidad de los productos terminados
• Lixiviados (es decir, lixiviación de los componentes de las
agujas/ tapones de goma, etc.)
• Sistema de cierre del envase /acondicionamiento
• Control de excipientes (es decir, especificaciones,
procedimientos analíticos, justificación de especificaciones, excipientes de origen animal o humano,
excipientes originales, etc.)
DIFERENCIAS

INMUNOGENICIDAD
Muy baja en los medicamentos de
síntesis química.
Alto riesgo en medicamentos
biológicos, proteínas son potentes
antígenos.
Perfil de seguridad.

32
DESARROLLO DE MEDICAMENTOS
Adaptado de Int J Pharm Med 2007; 21 (5): 339-345
OBTENCIÓN FASE 0-PRECLINICA
FASE I FASE II
FASE III FASE IV
EFECTIVIDAD

FASE 0 - PRECLINICA
BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
1. FASE QUÍMICA:
Caracterización fisicoquímica
Desarrollo farmacéutico
2. FASE BIOLÓGICA:
Farmacología básica
Toxicología

AL FINALIZAR FASE 0 - PRECLINICA
BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
IND (“Investigational New Drug”)/NFI
DCI (Nombre Genérico)
Tiempo de PATENTE
Autorización para iniciar estudios Clínicos

BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
FASE I • Biofarmacéutica:
Cmax, tmax, ABC
Biodisponibilidad
Formas farm. y Vías adm.
• Farmacocinética:
Ke, Vd, t1/2
Unión a PP
Efectos de los alimentos
• Farmacodinámica:
Tolerabilidad
Reacciones adversas
Efectos sobre signos vitales
Interacciones farmacológicas

BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
FASE II
• Relación Dosis-Efecto • Rango de dosis • Ventana terapéutica • Esquema posológico (Do, ) • Tolerabilidad • Reacciones adversas

BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
FASE III
EFICACIA ECCs multicéntricos,
multinacionales,
comparativos “cabeza a cabeza”
Ampliación de formas farm. y vías de adm.

BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
NDA (“New Drug Aplication”)/SNF
REGISTRO SANITARIO
COMERCIALIZACIÓN : Nombre de marca
(comercial)
AL FINALIZAR LA FASE III

BUSTAMANTE ROJAS, CARLOS. FASES DEL DESARROLLO DE UN NUEVO MEDICAMENTO Químico y Médico Farmacoepidemiólogo M.Sc. Profesor Titular de Farmacología Clínica Universidad de La Sabana
FASE IV
• EFECTIVIDAD
• Farmacoepidemiología:
• (Epidemiología del medicamento: “Ciencia que estudia los
EFECTOS [benéficos y perjudiciales] y las CONDICIONES DE USO
de los medicamentos en grandes grupos de población” /B.Strom)

INTERCAMBIABILIDAD

Medicamento INNOVADOR
“Es aquel que recibe primero la autorización para
su comercialización sobre la base de la
documentación relativa a la eficacia , inocuidad
y calidad ”
OMS, serie de informes técnicos 863:134
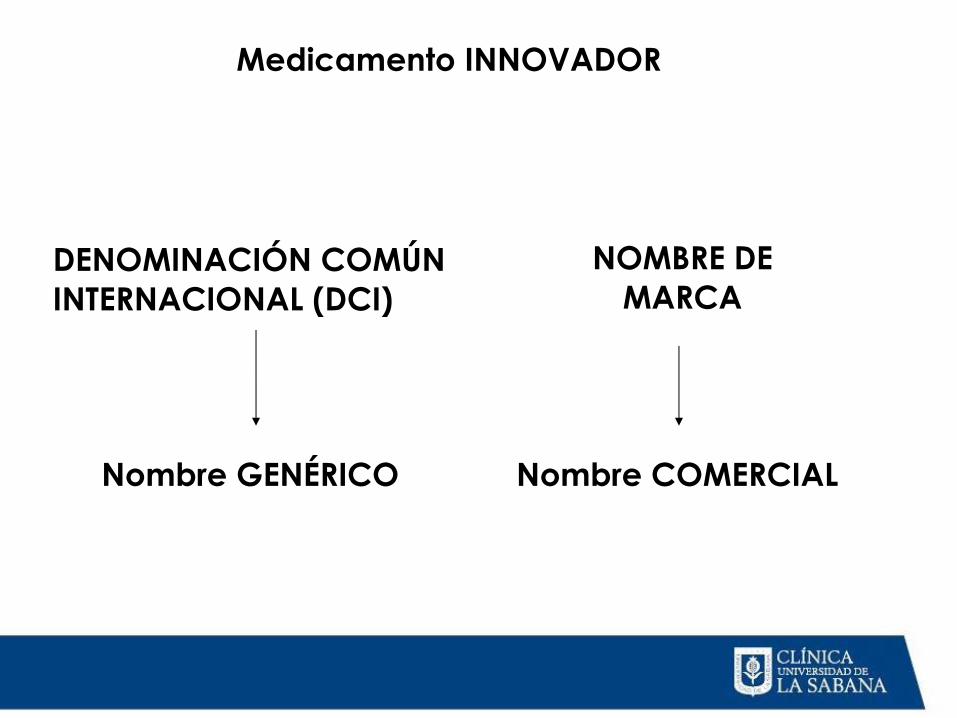
Medicamento INNOVADOR
DENOMINACIÓN COMÚN INTERNACIONAL (DCI)
NOMBRE DE MARCA
Nombre GENÉRICO Nombre COMERCIAL

Equivalencia Terapéutica
Se considera que dos medicamentos son Equivalentes Terapéuticos
si cumplen con los siguientes criterios:
1. Son Equivalentes Farmacéuticos o Alternativas Farmacéuticas.
2. Se espera que tengan esencialmente el mismo perfil de eficacia y
seguridad cuando se administran a pacientes a las mismas dosis
molares, por la misma vía y en las indicaciones aprobadas de uso.
“Multisource (generic) pharmaceutical products: guidelines on
registration requirements to establish interchangeability”. Fortieth Report, WHO Technical Report Series, No. 937, Annex 7
Geneva, 2006

Medicamento GENÉRICO (producto farmacéutico de fuentes múltiples)
“ Son equivalentes farmacéuticos o alternativas
farmacéuticas que pueden o no ser equivalentes
desde el punto de vista terapéutico.
Los productos farmacéuticos de fuentes múltiples que
sean equivalentes terapéuticos , se consideran
intercambiables”
OMS, serie de informes técnicos 863:134

47
MEDICAMENTO GENERICO INTERCAMBIABLE
Es aquel producto de síntesis química que
contiene el mismo principio activo del
medicamento innovador y que es
terapéuticamente intercambiable porque ha
demostrado mediante estudios
debidamente estandarizados de
equivalencia terapéutica correspondientes,
similar seguridad y eficacia terapéutica con
relación al medicamento de referencia.

48
¿Cómo se puede demostrar la
Equivalencia Terapéutica?

49
1. Estudios de Bioequivalencia
2. Estudios Farmacocinéticos
3. Estudios Farmacodinámicos
4. Estudios Clínicos
5. Estudios in-vitro
“ Multisource (generic) pharmaceutical products: guidelines on
registration requirements to establish interchangeability” . Fortieth Report, WHO Technical Report Series, No. 937, Annex 7, Geneva,
2006

50
BIOEQUIVALENCIA
“ Dos productos farmacéuticos son bioequivalentes
si además de ser equivalentes farmacéuticos o
alternativas farmacéuticas, tienen una Biodisponibilidad
Comparativa tan similar, que puede esperarse que
sus efectos sean esencialmente los mismos”
(FDA, EMEA, INVIMA-Resolución 1400 del
Ministerio de Salud)

51
Estudios de BIODISPONIBILIDAD
y BIOEQUIVALENCIA
NORMAS INTERNACIONALES
• Resolución 1400 del Ministerio de Salud
(24 de agosto de 2001)
NORMAS NACIONALES
• ICH: Normas de GCP. 1997
• FDA: Guidance for industry. October 2000
1st revision: march 2003
• EMEA: Note for guidance on the investigation of
Bioavailability and Bioequivalence. London,2001
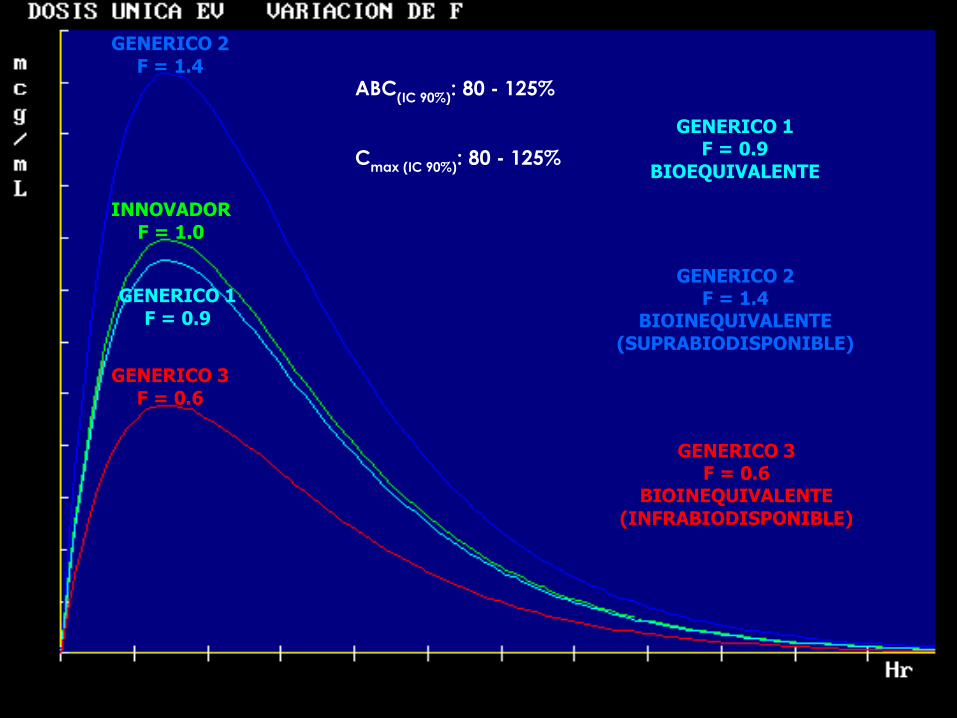
52
GENERICO 2
F = 1.4
INNOVADOR
F = 1.0
GENERICO 1
F = 0.9
GENERICO 3
F = 0.6
GENERICO 1 F = 0.9
BIOEQUIVALENTE
GENERICO 2 F = 1.4
BIOINEQUIVALENTE
(SUPRABIODISPONIBLE)
GENERICO 3 F = 0.6
BIOINEQUIVALENTE
(INFRABIODISPONIBLE)
ABC(IC 90%)
: 80 - 125%
Cmax (IC 90%)
: 80 - 125%

53
Sustitución Genérica de la prescripción
(Intercambiabilidad de medicamentos)
• Ética
• Segura
• Efectiva

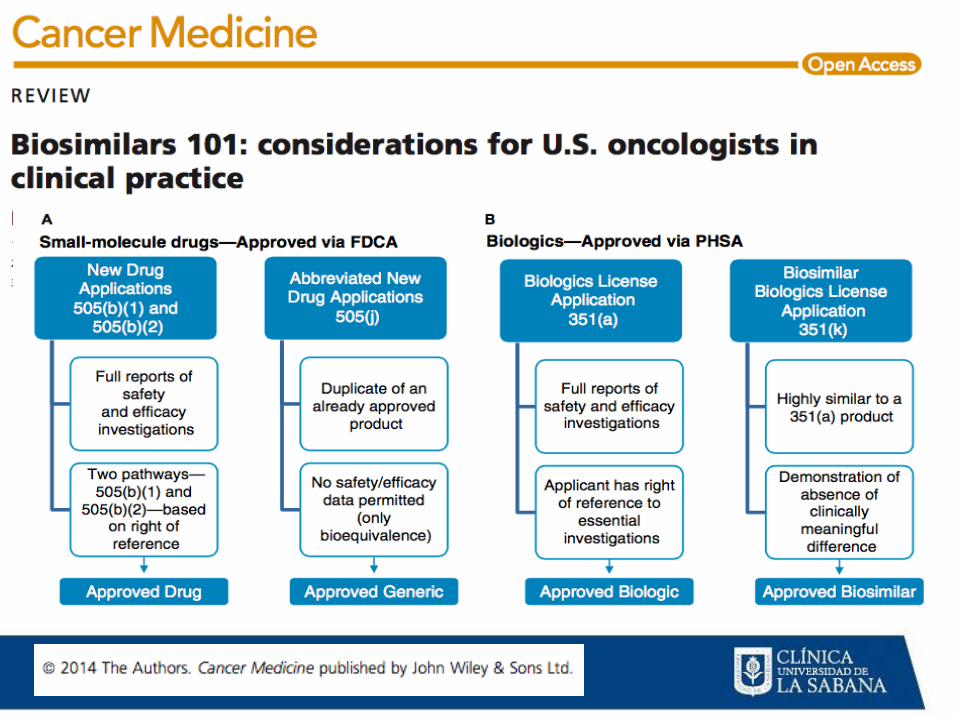
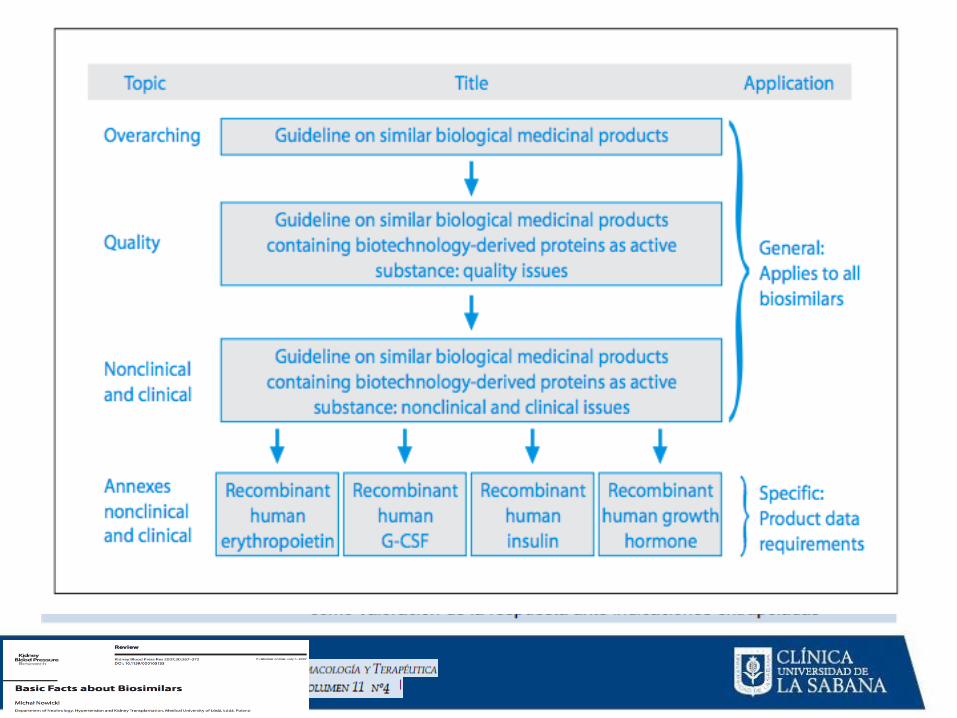
MEDICAMENTOS BIOLÓGICOS ONCOLÓGICOS

APROBACIÓN
•Ruta expediente completo
Ruta biogenéricos-biosimilares:
Ruta de compatibilidad
Ruta abreviada de compatibilidad
Pruebas de inmunogenicidad


58

59

¿SE PUEDE HABLAR DE INTERCAMBIABILIDAD?
REGULACION -BIOSIMILARES -MEDICAMENTOS BIOLOGICOS
BIOSIMILARES Y MEDICAMENTOS ONCOLOGICOS
SEGURIDAD DEL PACIENTE

Salud pública.
Costos en salud.
Eficiencia.





La guía propuesta se elaboró tomando como referencia documentos de la Agencia de Estados Unidos (FDA), expedida en
agosto de 2014 y la Agencia Europea (EMA), expedida en diciembre de 2007, que se presentan a continuación:
• “Guidance for Industry: Immunogenicity Assesment for
Therapeutic Protein Products” del Center for Drug Evaluation and
Research (CDER), el Center for Biologics Evaluation and Research
(CBER) de la agencia sanitaria de Estados Unidos, Food And Drug Administration –FDA-, adscrita al U.S. Department of Health and
Human Services.
• "Guideline on Immunogenicity Assessment of Biotechnology-
Derived Therapeutic Proteins" del Committee for Medicinal Products for Human Use (CHMP) de la agencia sanitaria europea, European Medicines Agency -EMA- (Documento Ref.
EMEA/CHMP/BMWP/14327/2006)
academia e investigadores experiencia regulatoria y con experiencia en la
industria