Tumores y lesiones pseudotumorales del hueso
31
Tumores y lesiones pseudotumorales del hueso. Lameiro Frida Paulina
-
Upload
frida-lameiro -
Category
Health & Medicine
-
view
105 -
download
3
Transcript of Tumores y lesiones pseudotumorales del hueso
Tumores y lesiones pseudotumorales del hueso.
Lameiro Frida Paulina

Se clasifican según la célula normal o el tipo de tejido del que proceden o características clínico-patológicas distintivas. Tumores formadores de hueso Osteoma
Osteoma osteoide y osteoblastomaOsteosarcoma
Tumores formadores de cartílago
OsteocondromaCondromaCondroblastomaFibroma condromixoideCondrosarcoma
Tumores fibrosos y fibroóseos Defecto cortical fibroso y fibroma no osificanteDisplasia fibrosaVariantes de fibrosarcoma
otros Sarcoma de EwingTumor de celulas gigantesQuiste óseo aneurismático

Más frecuentes: productores de matriz y fibrosos.Más frecuentes benignos: osteocondroma y defecto cortical fibroso.
Los tumores benignos son mas frecuentes que los malignos, y aparecen sobre todo en las primeras tres décadas de la vida.
- Predilección por los huesos largos.
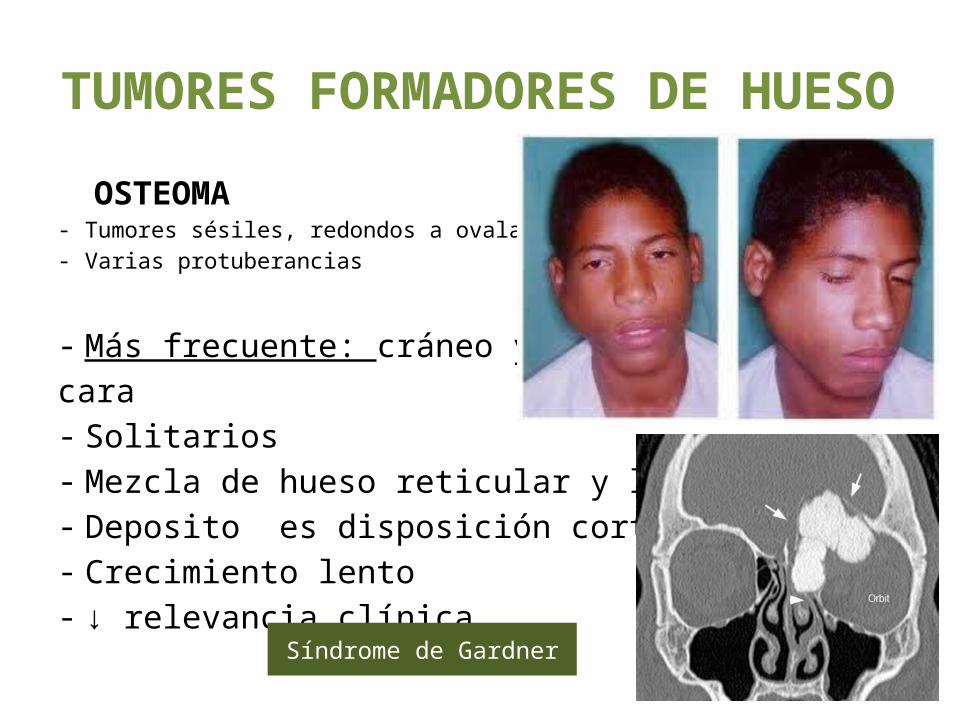
TUMORES FORMADORES DE HUESO
OSTEOMA- Tumores sésiles, redondos a ovalados- Varias protuberancias
- Más frecuente: cráneo y cara - Solitarios- Mezcla de hueso reticular y laminar.- Deposito es disposición cortical- Crecimiento lento- ↓ relevancia clínica.Síndrome de Gardner
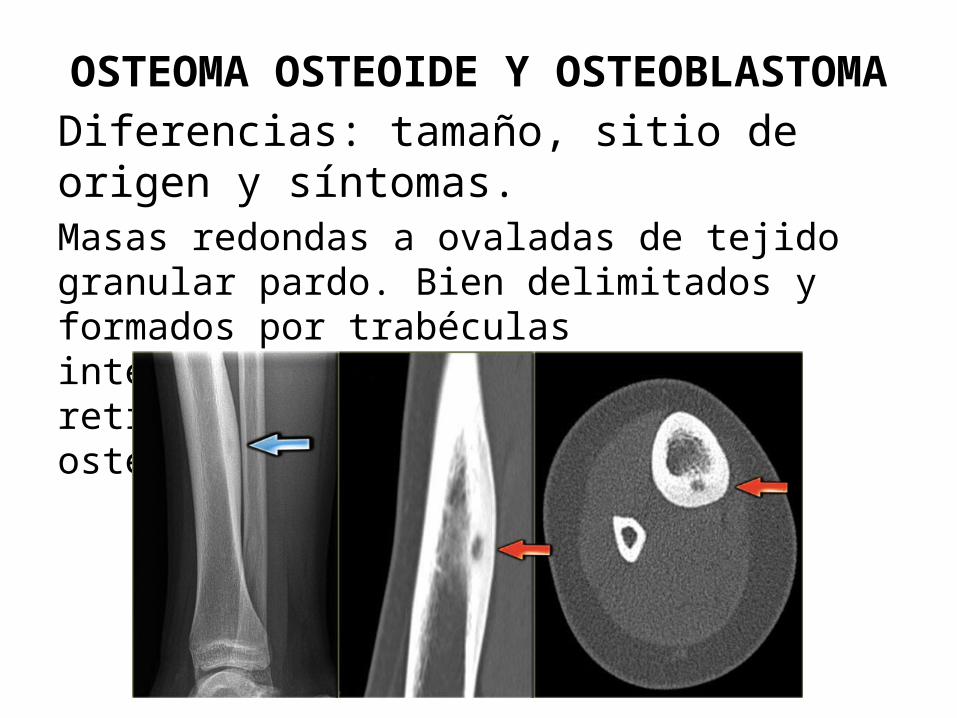
OSTEOMA OSTEOIDE Y OSTEOBLASTOMADiferencias: tamaño, sitio de origen y síntomas.Masas redondas a ovaladas de tejido granular pardo. Bien delimitados y formados por trabéculas interconectadas al azar en hueso reticular. Borde prominente de osteoblastos.
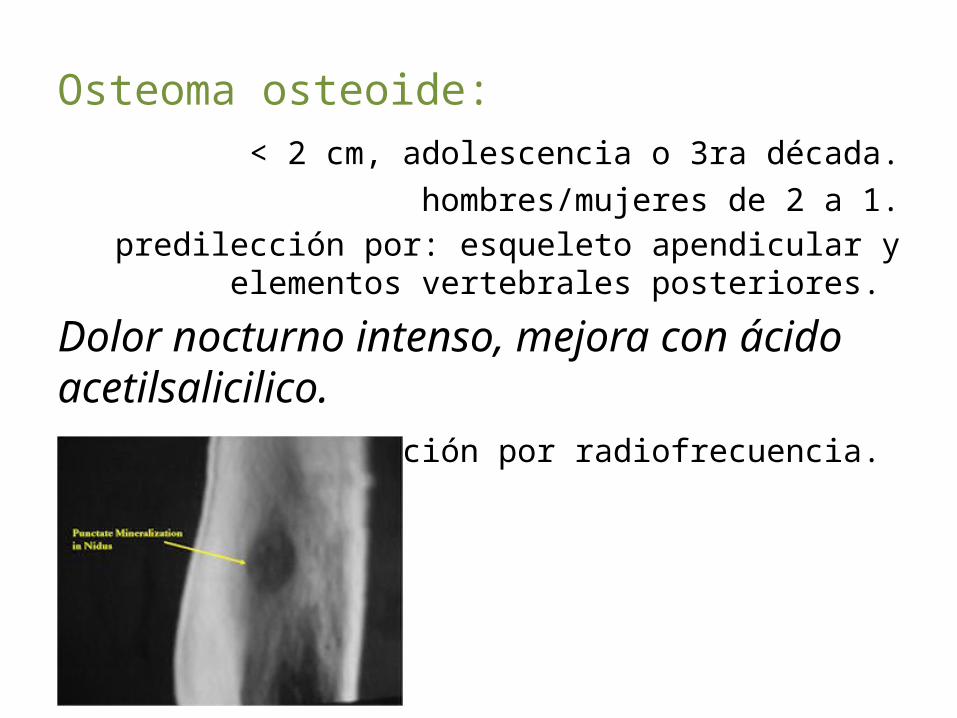
Osteoma osteoide: < 2 cm, adolescencia o 3ra década.
hombres/mujeres de 2 a 1.predilección por: esqueleto apendicular y elementos
vertebrales posteriores.
Dolor nocturno intenso, mejora con ácido acetilsalicilico.
Tx. Ablación por radiofrecuencia.
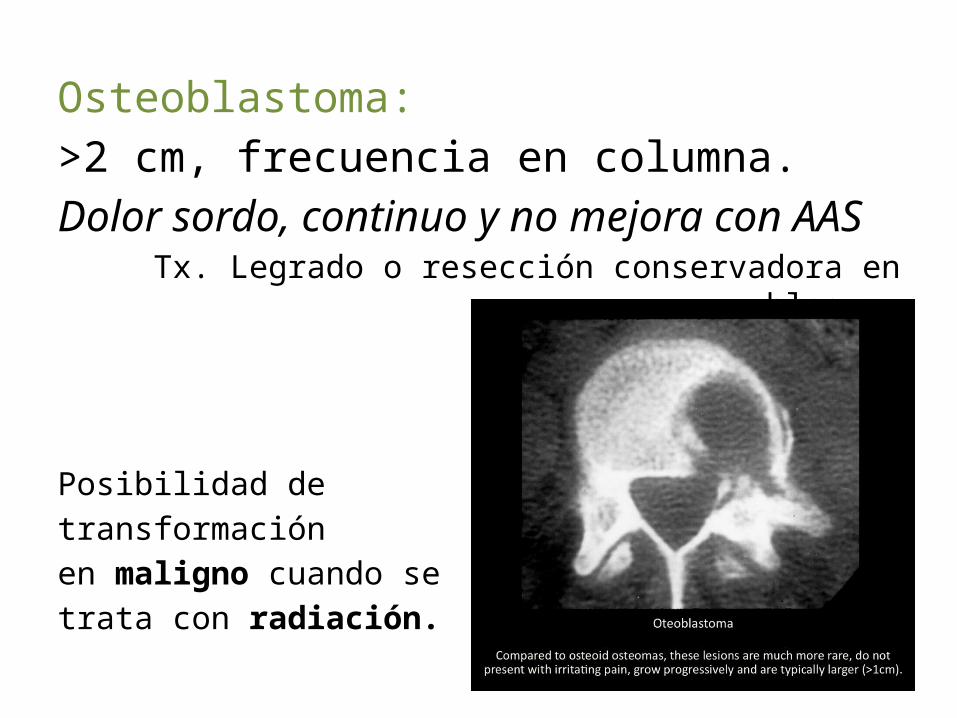
Osteoblastoma:>2 cm, frecuencia en columna.Dolor sordo, continuo y no mejora con AAS
Tx. Legrado o resección conservadora en bloque.
Posibilidad de transformación en maligno cuando se trata con radiación.
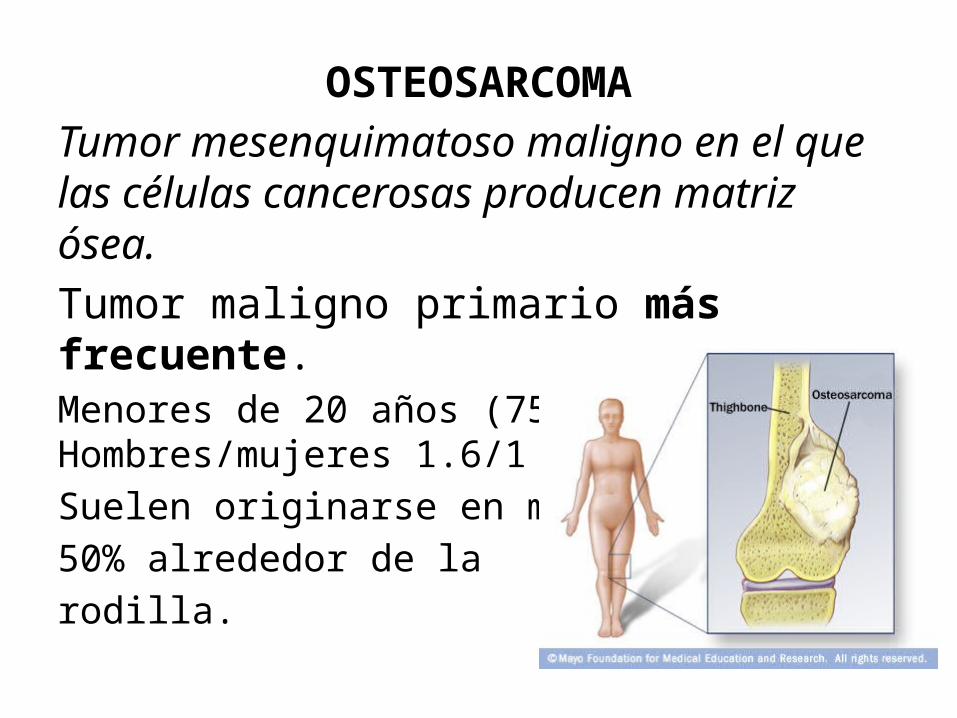
OSTEOSARCOMATumor mesenquimatoso maligno en el que las células cancerosas producen matriz ósea. Tumor maligno primario más frecuente.Menores de 20 años (75%). Hombres/mujeres 1.6/1Suelen originarse en metafisis. 50% alrededor de larodilla.
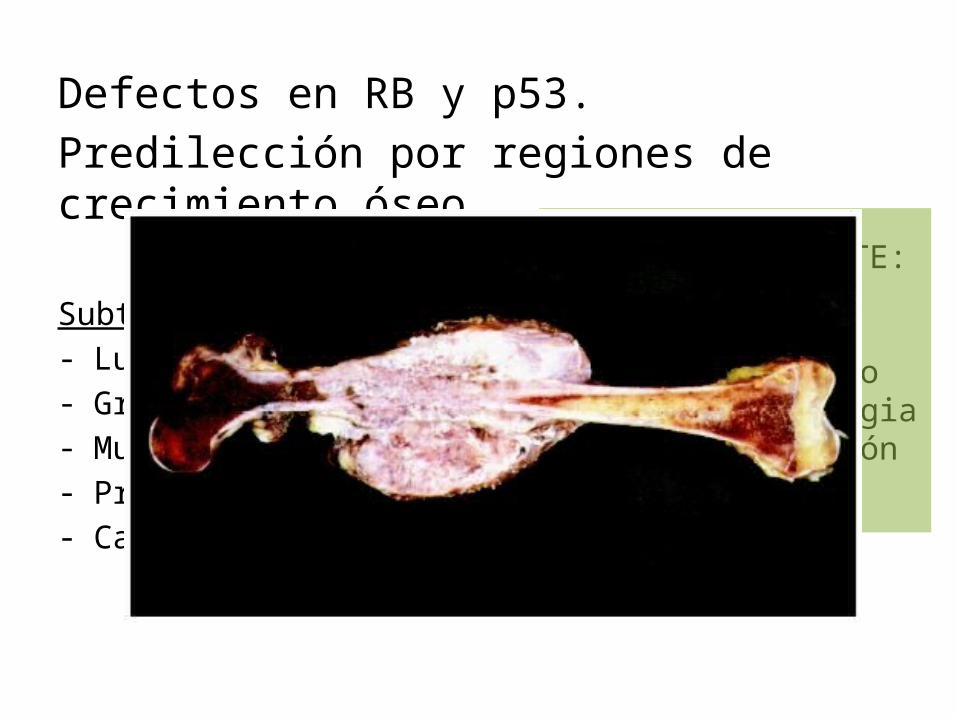
Defectos en RB y p53.Predilección por regiones de crecimiento óseo.
Subtipos:- Lugar de origen- Grado de diferenciación- Multicentricidad- Primario o secundario- Características histológicas
MACROSCÓPICAMENTE:Voluminosos granulares,
blanco-grisáceoZonas de hemorragia y de
degeneración quística.
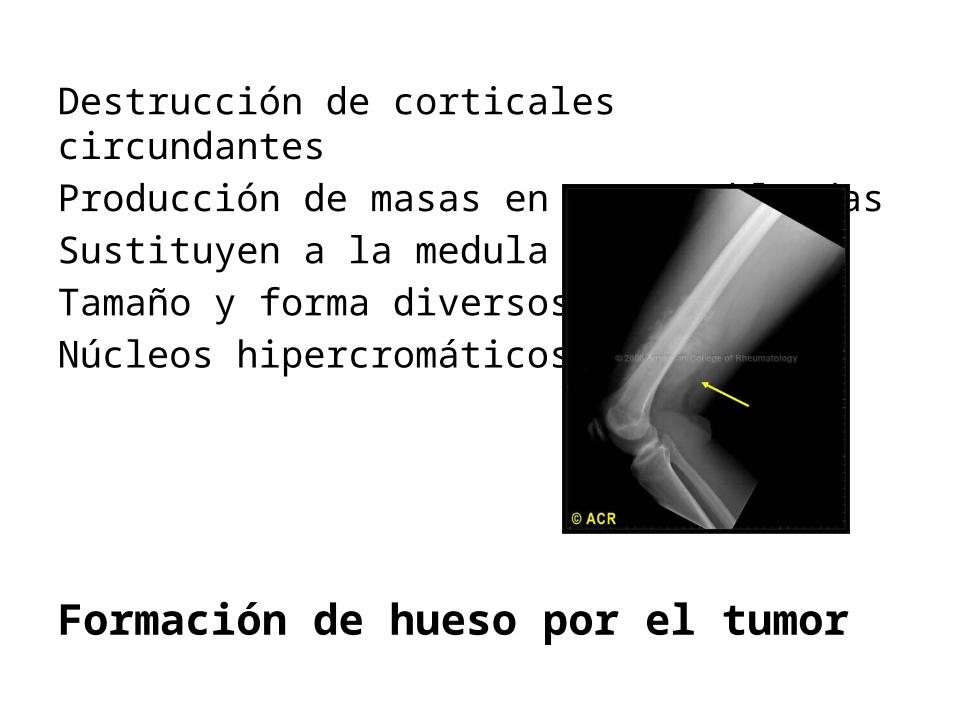
Destrucción de corticales circundantesProducción de masas en partes blandasSustituyen a la medula óseaTamaño y forma diversos Núcleos hipercromáticos
Formación de hueso por el tumor

Evolución clínica: Masas dolorosas de crecimiento progresivo.Primer síntoma fractura ósea.Triángulo de Codman: sombra triangular entre la cortical y los extremos elevados del periostio.
10-20% metástasis pulmonar.90% de fallecidos metástasis en: pulmón, hueso, cerebro y otros órganos.
Supervivencia a 5 años:Sin metástasis -> 60-70%Con metástasis -> 20%
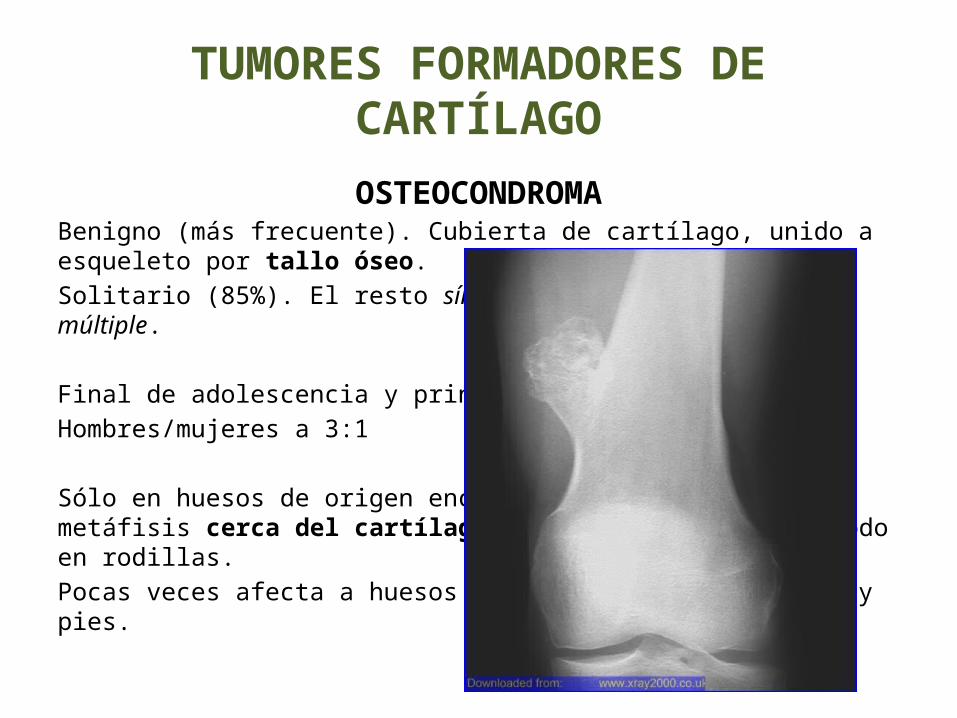
TUMORES FORMADORES DE CARTÍLAGO
OSTEOCONDROMABenigno (más frecuente). Cubierta de cartílago, unido a esqueleto por tallo óseo.Solitario (85%). El resto síndrome de exóstosis hereditaria múltiple. Final de adolescencia y principio de edad adulta.Hombres/mujeres a 3:1
Sólo en huesos de origen encondral y asienta en la metáfisis cerca del cartílago de crecimiento, sobre todo en rodillas. Pocas veces afecta a huesos tubulares cortos de manos y pies.
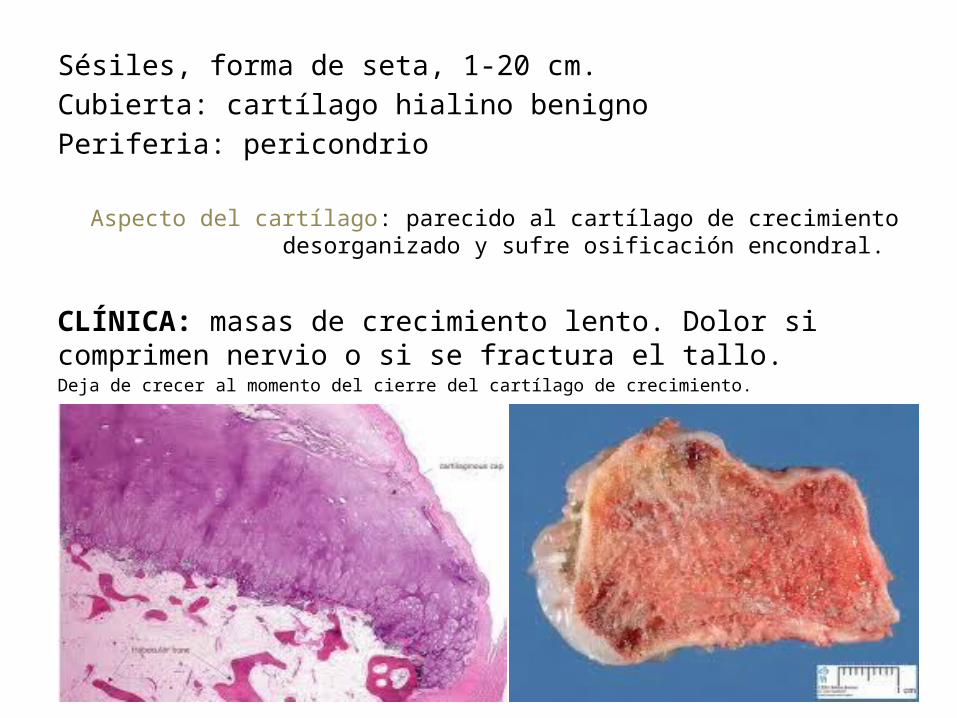
Sésiles, forma de seta, 1-20 cm.Cubierta: cartílago hialino benignoPeriferia: pericondrio
Aspecto del cartílago: parecido al cartílago de crecimiento desorganizado y sufre osificación encondral.
CLÍNICA: masas de crecimiento lento. Dolor si comprimen nervio o si se fractura el tallo. Deja de crecer al momento del cierre del cartílago de crecimiento.
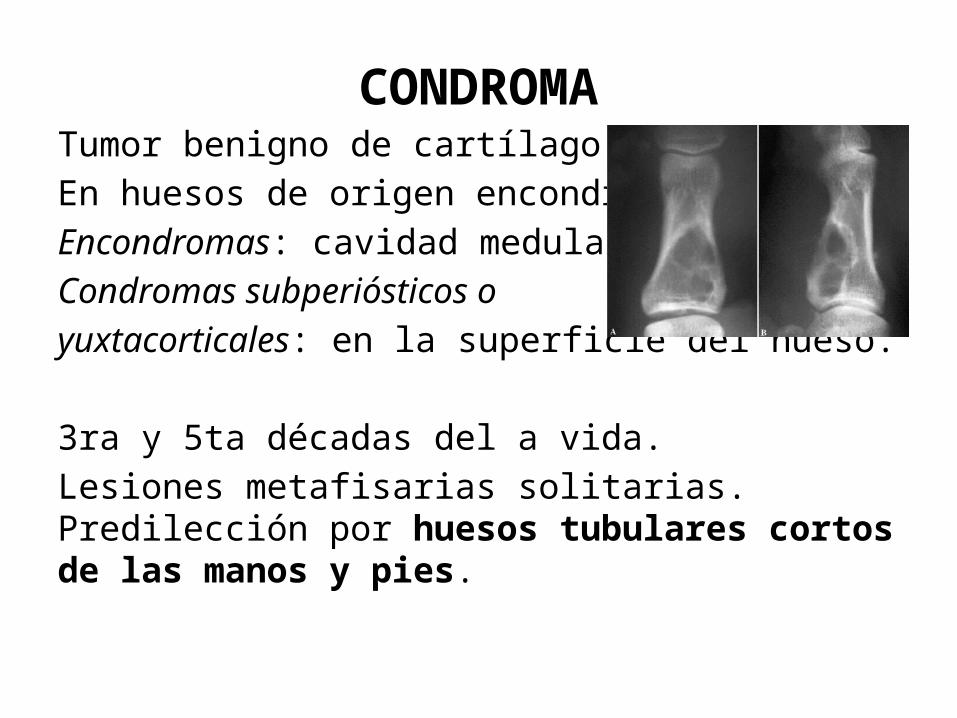
CONDROMATumor benigno de cartílago hialino.En huesos de origen encondral.Encondromas: cavidad medularCondromas subperiósticos o yuxtacorticales: en la superficie del hueso.
3ra y 5ta décadas del a vida.Lesiones metafisarias solitarias. Predilección por huesos tubulares cortos de las manos y pies.
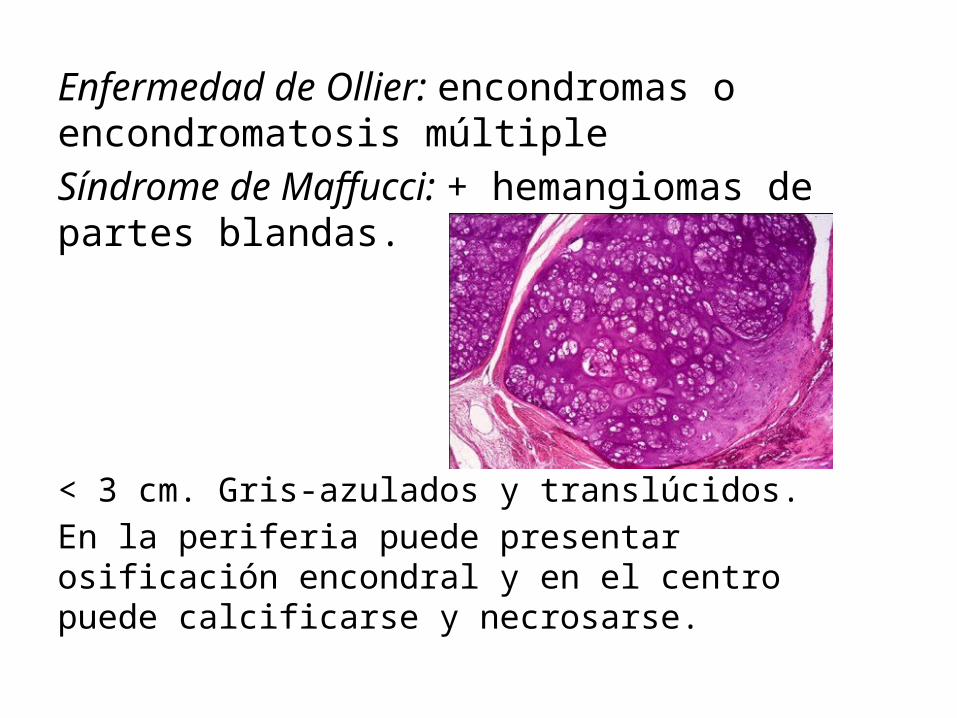
Enfermedad de Ollier: encondromas o encondromatosis múltipleSíndrome de Maffucci: + hemangiomas de partes blandas.
< 3 cm. Gris-azulados y translúcidos. En la periferia puede presentar osificación encondral y en el centro puede calcificarse y necrosarse.
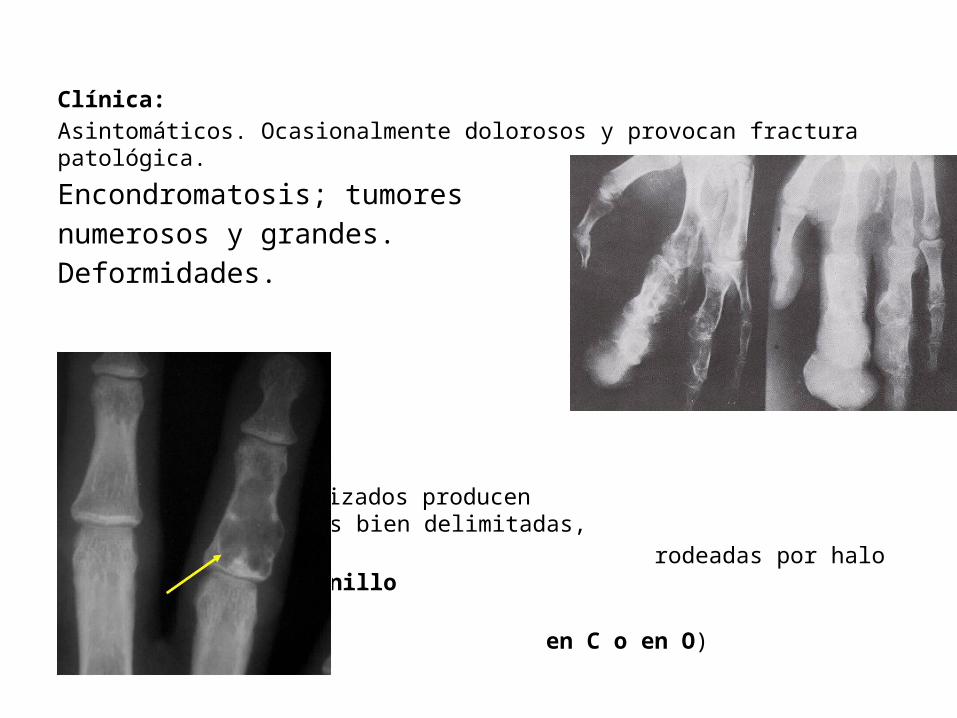
Clínica:Asintomáticos. Ocasionalmente dolorosos y provocan fractura patológica.
Encondromatosis; tumores numerosos y grandes. Deformidades.
Nodulos no mineralizados producen transparencias ovales bien delimitadas, rodeadas por halo delgado (signo del anillo en C o en O)
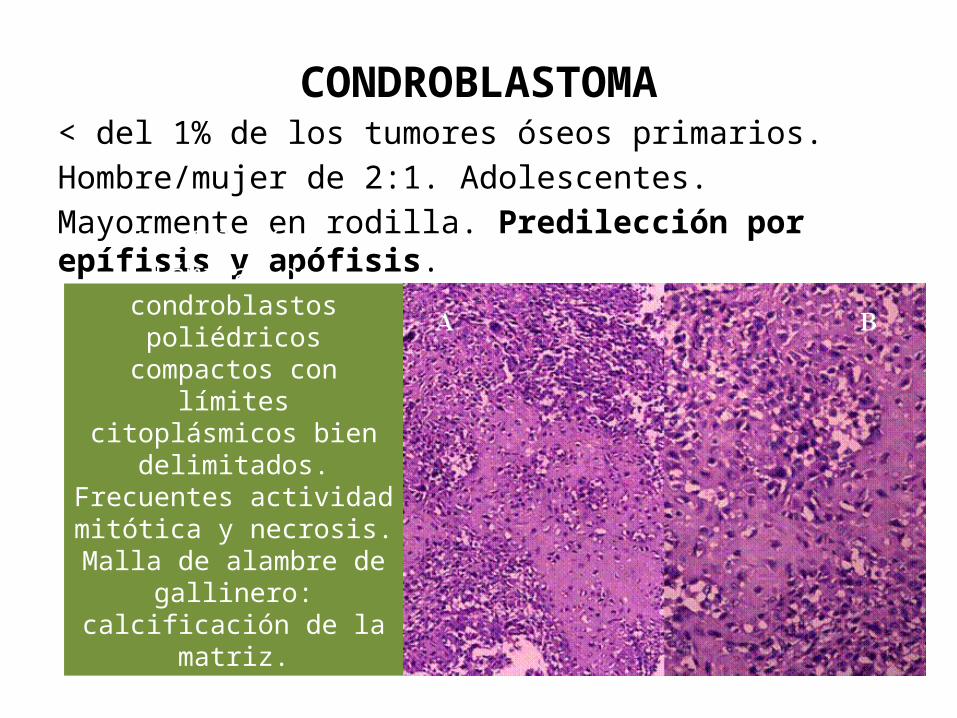
CONDROBLASTOMA< del 1% de los tumores óseos primarios. Hombre/mujer de 2:1. Adolescentes. Mayormente en rodilla. Predilección por epífisis y apófisis.
Morfología: Láminas de condroblastos poliédricos compactos con límites citoplásmicos bien
delimitados.Frecuentes actividad mitótica
y necrosis. Malla de alambre de
gallinero: calcificación de la matriz.
HAY CELULAS GIGANTES DE TIPO OSTEOCLASTO
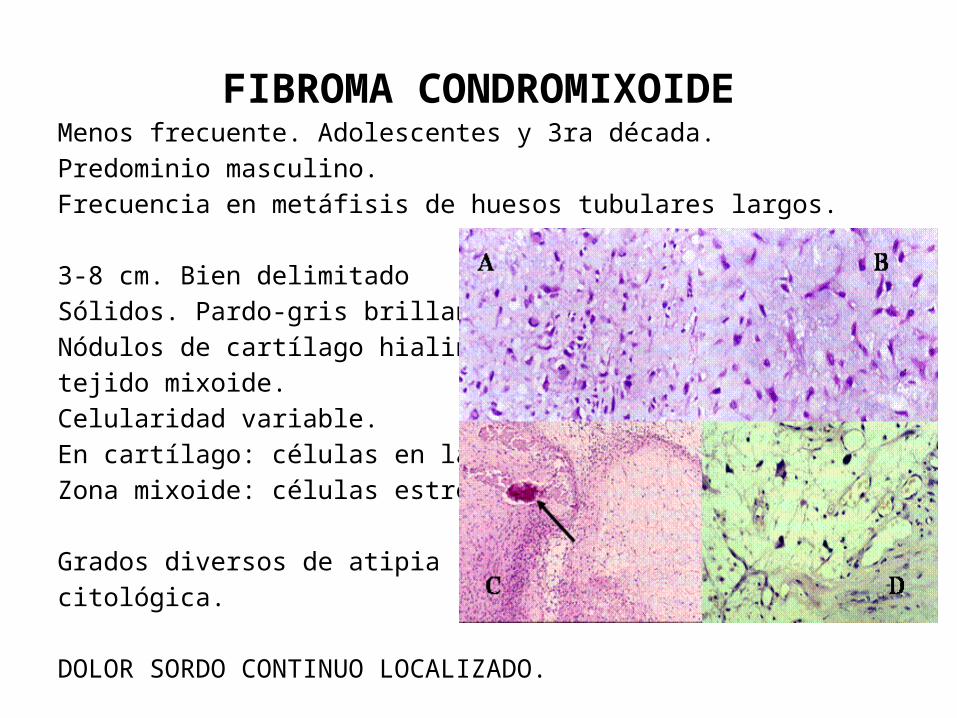
FIBROMA CONDROMIXOIDEMenos frecuente. Adolescentes y 3ra década.Predominio masculino.Frecuencia en metáfisis de huesos tubulares largos.
3-8 cm. Bien delimitadoSólidos. Pardo-gris brillanteNódulos de cartílago hialino y tejido mixoide.Celularidad variable. En cartílago: células en lagunas.Zona mixoide: células estrelladas.
Grados diversos de atipia citológica.
DOLOR SORDO CONTINUO LOCALIZADO.
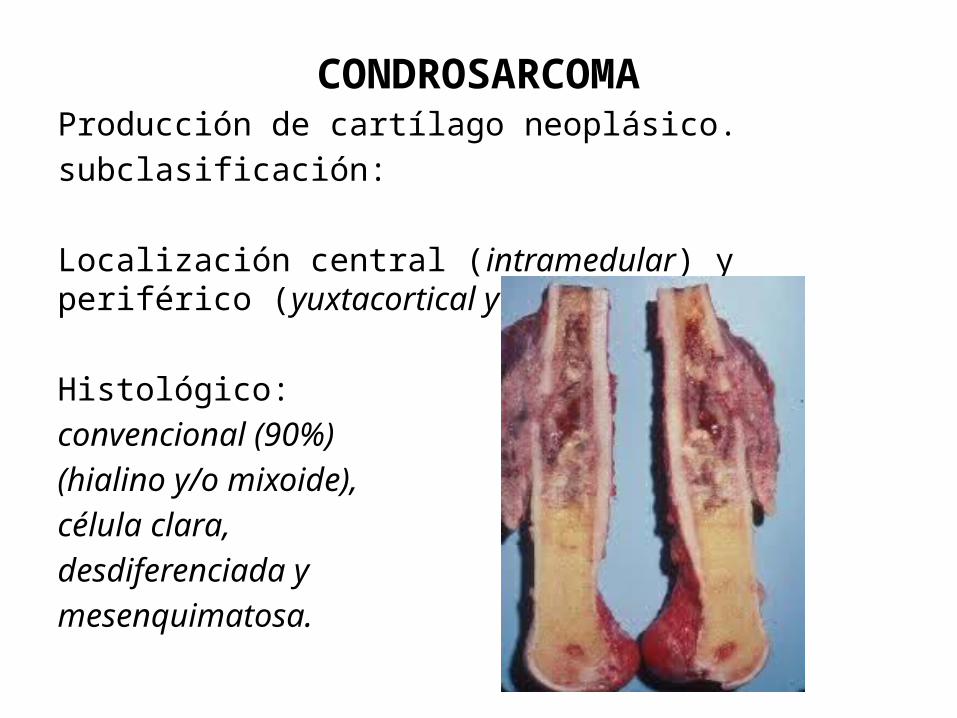
CONDROSARCOMAProducción de cartílago neoplásico.subclasificación:
Localización central (intramedular) y periférico (yuxtacortical y superficial).
Histológico: convencional (90%)(hialino y/o mixoide), célula clara, desdiferenciada y mesenquimatosa.
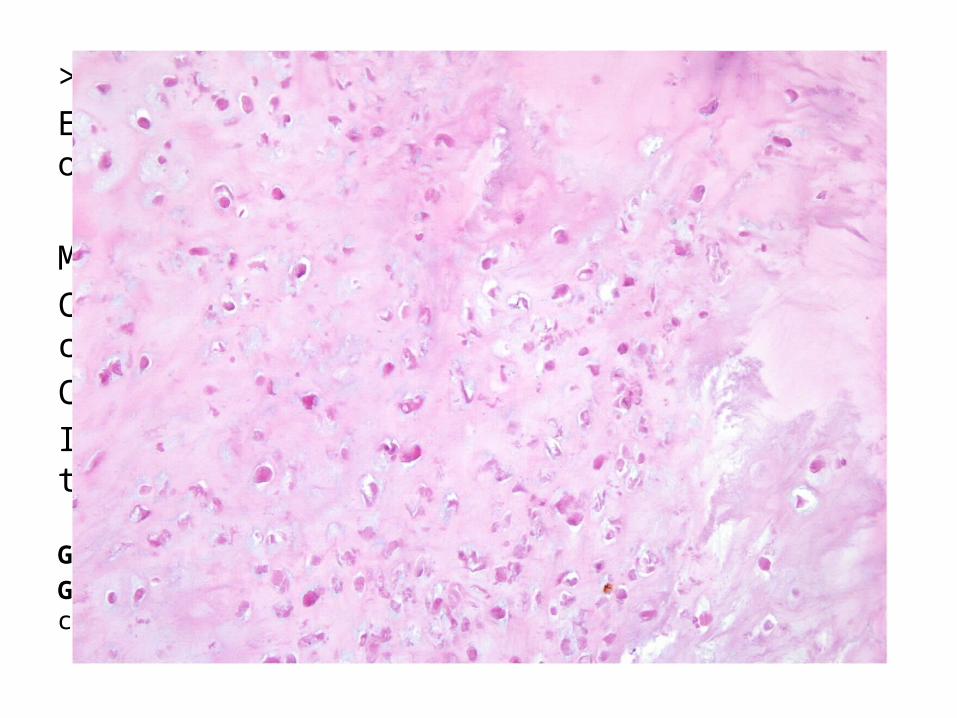
>40 años. El 15% se origina sobre un encondroma u osteocondroma previo.
Morfología: Calcificaciones punteadas y necrosis central.Crecimiento hacia partes blandas.Infiltración a medula ósea y rodea trabéculas.
Grado 1: celularidad escasa. Osificación encondral.Grado 3: hipercelularidad, pleomorfismo, extremo con células gigantes y mitosis.

Frecuencia en: pelvis, hombro y costillas.No suele afectar a la región distal de las extremidades.Masas dolorosas con crecimiento progresivo.
La mayoría corresponde a grado 1 y 2.
Supervivencia a 5 años: Grado 1 – 90%Grado 2 – 81%Grado 3 – 43%
Mayores de 10 cm son mas agresivos.Metástasis: pulmón y hueso.
Tx. Extirpación quirúrgica.

TUMORES FIBROSOS Y FIBROÓSEOS
DEFECTO CORTICAL FIBROSO Y FIBROMA NO OSIFICANTE
Muy frecuentes. 30-50% en menores de 2 años.Excéntricos en la metáfisis distal del fémur y proximal de la tibia. Pequeños (0.5 cm). Entre 5-6cm son fibromas no osificantes. Asintomáticos, desaparecen de modo espontaneo.
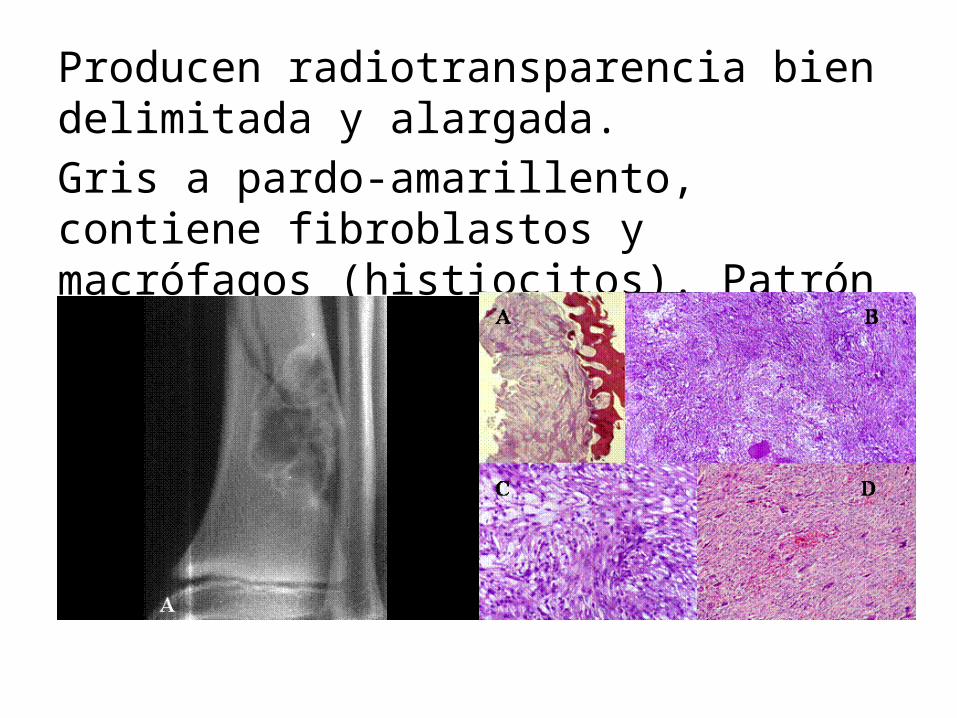
Producen radiotransparencia bien delimitada y alargada. Gris a pardo-amarillento, contiene fibroblastos y macrófagos (histiocitos). Patrón arremolinado.

DISPLASIA FIBROSAVinculado con una detención localizada del desarrollo. No hay maduración de las estructuras.
Durante el crecimiento.Lesiones circunscritas, intramedulares y de tamaño muy diverso.
Formas de trabéculas como caracteres chinos. Degeneración quística, hemorragia y macrófagos espumosos.

1) Afectación de un solo hueso (monostótica):70%. Frecuencia fémur, tibia, costillas, maxilares, calora y húmero.
Asintomática. Puede haber deformidad. Radiológicamente aspecto en vidrio esmerilado.
2) Afectación de múltiples huesos (poliostótica):27%. Fémur, cráneo, tibias, húmero, peroné, radio, cúbito,
mandíbula y vertebras.
Afectación craneofacial 50% de los casos.Deformidades marcadas, incapacitantes.

3) Enfermedad poliostótica asociada a pigmentación cutánea con manchas café con leche (costa de Maine) y anomalías endocrinas.
Pubertad precoz. Síndrome de McCune-Albright3%. Precocidad sexual, hipertiroidismo, adenomas hipofisarios, hiperplasia suprarrenal primaria. Más frecuente en las niñas
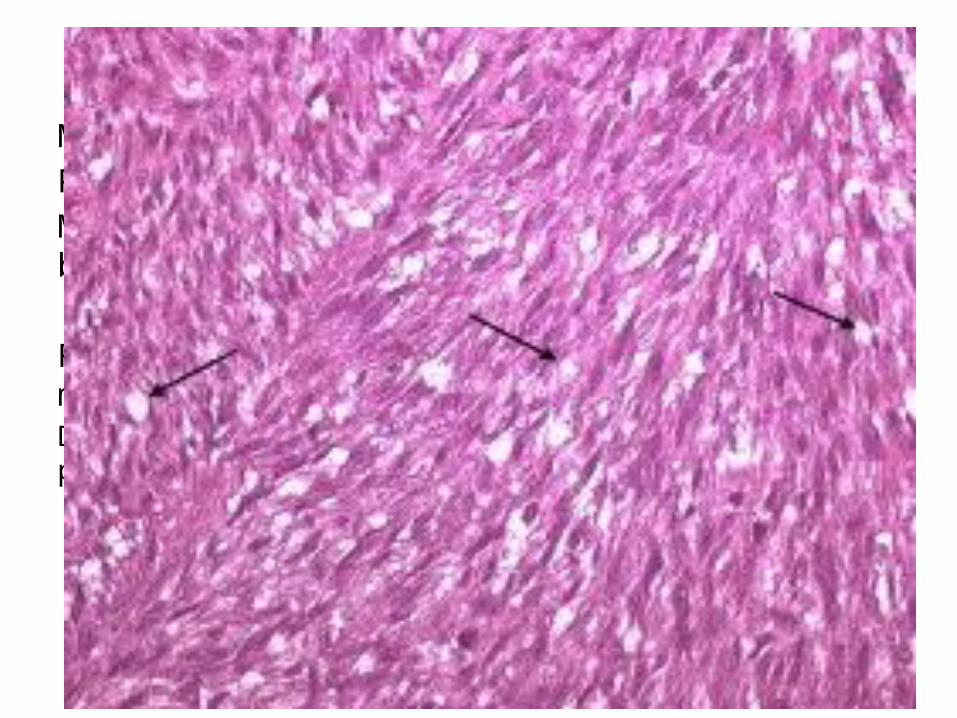
VARIANTES DE FIBROSARCOMAMediana o avanzada edad.Primarios.Masas grandes hemorrágicas de color pardo-blanquecino que destruyen el hueso.
Formados por fibroblastos citológicamente malignos. Diferenciación determina la cantidad de colágeno producido y el grado de atipia citológica.
Masa dolorosa que aumenta de tamaño.En la metáfisis de los huesos largos y en los huesos planos de la pelvis.
Fractura patológica.
OTROS TUMORES
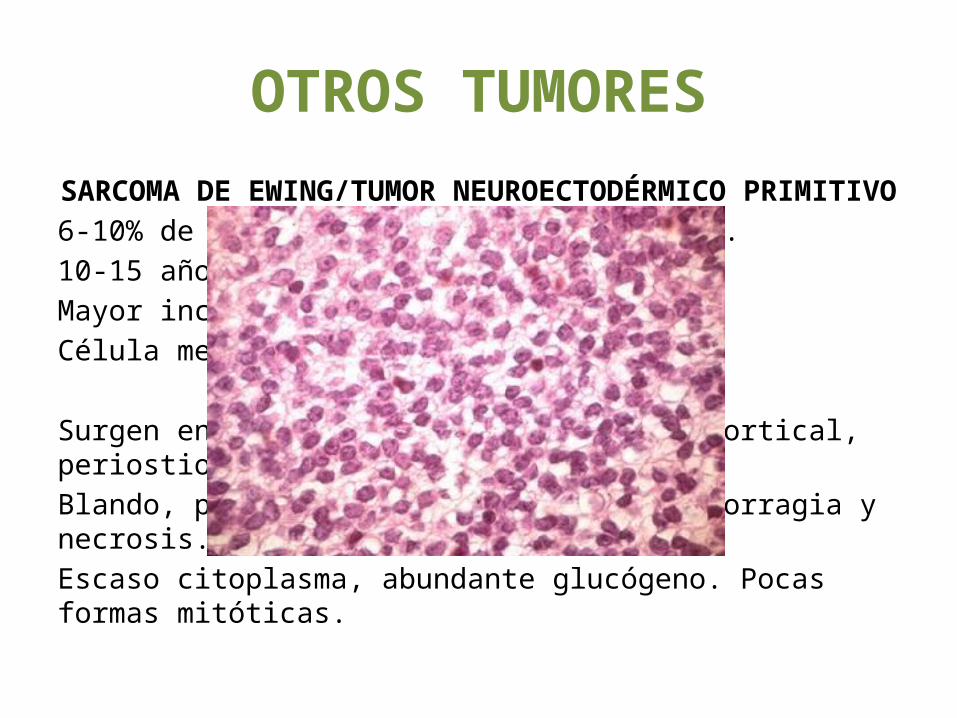
SARCOMA DE EWING/TUMOR NEUROECTODÉRMICO PRIMITIVO6-10% de los tumores malignos primarios.10-15 años.Mayor incidencia en niños. Raza blanca.Célula mesenquimatosa multipotente.
Surgen en la cavidad medular, invaden cortical, periostio y partes blandas. Blando, pardo-blanquecino, zonas de hemorragia y necrosis.Escaso citoplasma, abundante glucógeno. Pocas formas mitóticas.

Diáfisis de los huesos tubulares largos. Masas que aumentan de tamaño, dolorosas. Dolor, edema y calor en región afectada.Signos sistémicos.Reacción perióstica, piel de cebolla75% supervivencia en 5 años.
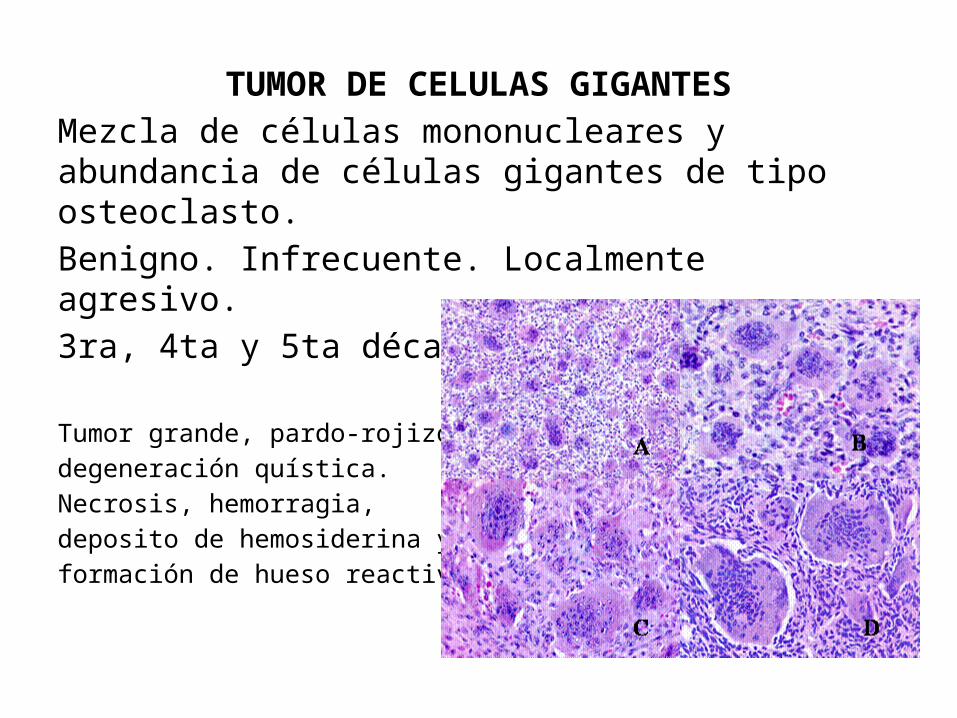
TUMOR DE CELULAS GIGANTESMezcla de células mononucleares y abundancia de células gigantes de tipo osteoclasto.Benigno. Infrecuente. Localmente agresivo.3ra, 4ta y 5ta década de la vida.
Tumor grande, pardo-rojizo, degeneración quística.Necrosis, hemorragia, deposito de hemosiderina y formación de hueso reactivo.
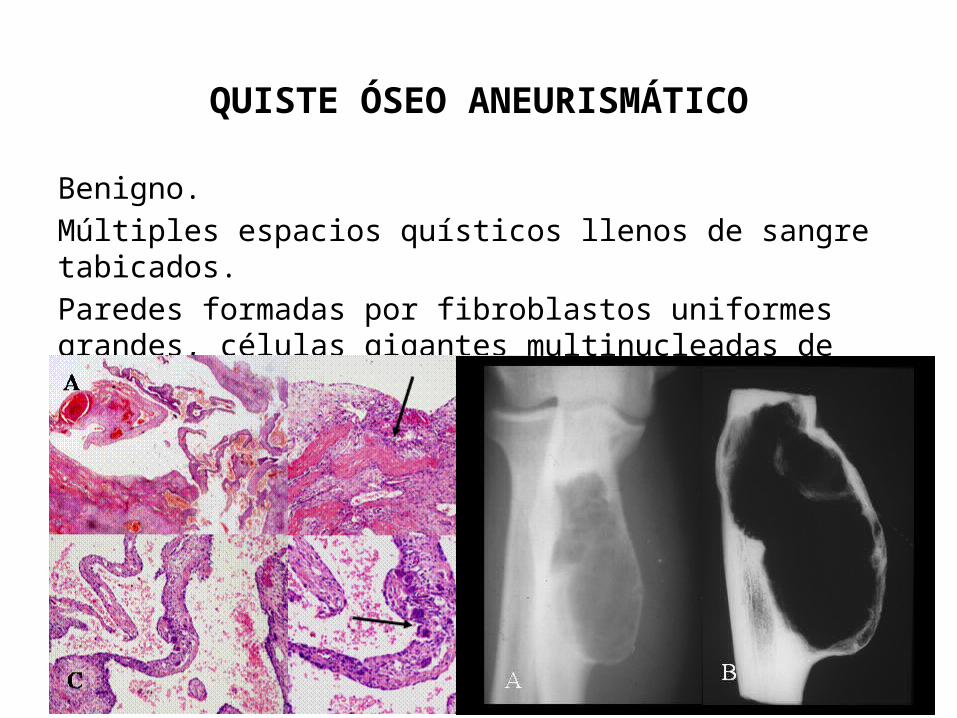
QUISTE ÓSEO ANEURISMÁTICO
Benigno.Múltiples espacios quísticos llenos de sangre tabicados.Paredes formadas por fibroblastos uniformes grandes, células gigantes multinucleadas de tipo osteoclastos y hueso reticular reactivo.
Frecuencia en las 2 primeras décadas de la vida.Huesos largos y elementos vertebrales posteriores.Dolor y tumefacción.Lesión excéntrica con bordes nítidos.