
TLC Parte6
7
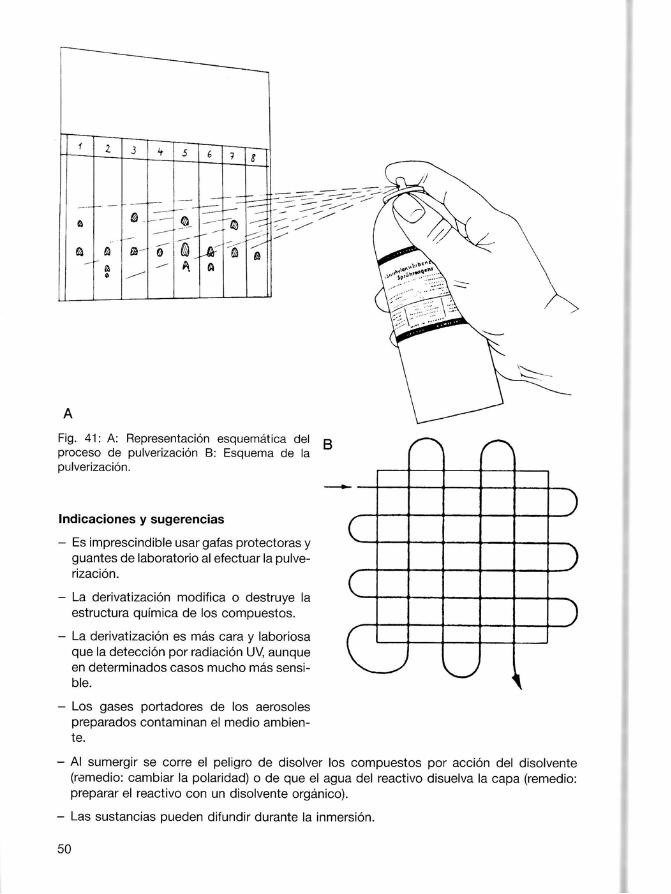
Las sustancias cuya propia fluorescencia puedeexcitarse por radiación UV se detectan preferentemente sobre capas sin indicador fluorescente. Se hacen visibles bajo radiación UV como zonas luminosas coloreadas y brillantes sobre fondooscuro. Ninguno de los dos procedimientos acostumbra a modificar ni destruir la estructura química de los compuestos detectados y son por ello los más adecuados para finespreparativos. 3.6.2 Detección por derivatización. Las reacciones de derivatización se utilizan cuando las fracciones individuales no reaccionan a laradiación UVocuando lasensibilidad de ladetección es insuficiente 124,5O1. Por principio, es irrelevante que el reactivo se utilice antes de la aplicación de la sustancia (derivatización precromatográfica) o después del desarrollo (derivatización postcromatográfica). La derivatización precromatográfica sirve no sólo para visualizar sinotambién para aumentar la selectividad delsistema de separación por los compuestos investigados o transformar los comouestos lábiles en estables. Laderivatización postcromatográfica sirve sobre todo para revelar lassustancias separadas o paraaumentar la sensibilidad de la detección. Usualmente los reactivos de detección se pulverizan sobre la placa o folio. Algunos de estos reactivos se venden ya preparados, p. ej. en forma de soluciones listaspara usar (p. ej. rodamina B,ninhidrina, ácido fosfomolíbdico). Lamayoría deben sinembargo prepararse enel laboratorio y pulverizarse mediante atomizadores adecuados (Fig. a0). Entre ellos se encueniran los atomizadores de laboratorio. acoolados a unaconducción de aire comprimido o nitrógeno, y las llamadas pistolas pulverizadoras quese componen de un depósito, un contenedor de gas portador y una cabeza pulverizadora (atención: por su contenido en CFCs- hidrocarburos fluoroclorados - nocivos para el medioambiente, las pistolas pulverizadoras y los botesde aerosol dejarán de utilizarse en un futuropróximo). La pulverización deberealizarse siempre bajounacampana de extracción con buen tiraje o mediante un dispositivo adecuado que se lleve la niebla de reactivo, generalmente tóxica o agresiva, y los vapores de disolvente. El cromatograma se coloca en posición veftical ligeramente inclinado. Cuando se ha formado una neblina uniforme se dirige el chorro del atomizador sobrela placa y se pulveriza de manera uniforme, generalmente hasta que se empieza a hacer transparente. Pulverizar en exceso puede producir la disolución o el arrastre de algúno de los compuestos de la placa (Fig. 41). Además de los dispositivos de pulverización se disponetambién de dispositivos de inmersión. La inmersión y extracción verticales así como el tiempode permanencia en la cubeta de inmersión (algunos segundos) pueden seleccionarse a voluntad y se realizan de manera automática 12, 511. Algún reactivopuede estar mezclado con la capa con lo que la derivatización por calentamiento se realiza después del desarrollo.También puede añadirse a través de la fase gas(p.ej. losgases nitrosos para loscompuestos aromáticos) o mezclarse con el eluyente (p. ej. la ninhidrina paralos aminoácidos) [52, 53]. A menudo loscromatogramas hande calentarse en unaestufa o sobre unaplaca calefactora después de añadir losreactivos de detección, para activar la reacción (p.ej.de 10a 15min. a 105-1 10 'C). lnmediatamente después delrevelado conviene marcar y rotular (alápiz) lasmanchas, yaque éstas pueden palidecer o cambiar de color. 49
-
Upload
quimionauta -
Category
Documents
-
view
218 -
download
8
description
Documento sobre cromatografía en capa fina.
Transcript of TLC Parte6

Las sustancias cuya propia fluorescencia puede excitarse por radiación UV se detectanpreferentemente sobre capas sin indicador fluorescente. Se hacen visibles bajo radiación UVcomo zonas luminosas coloreadas y brillantes sobre fondo oscuro.
Ninguno de los dos procedimientos acostumbra a modificar ni destruir la estructura químicade los compuestos detectados y son por ello los más adecuados para fines preparativos.
3.6.2 Detección por derivatización.
Las reacciones de derivatización se utilizan cuando las fracciones individuales no reaccionana la radiación UVo cuando la sensibilidad de la detección es insuficiente 124,5O1. Por principio,es irrelevante que el reactivo se utilice antes de la aplicación de la sustancia (derivatizaciónprecromatográfica) o después del desarrollo (derivatización postcromatográfica).
La derivatización precromatográfica sirve no sólo para visualizar sino también para aumentarla selectividad del sistema de separación por los compuestos investigados o transformar loscomouestos lábiles en estables.
Laderivatización postcromatográfica sirve sobre todo para revelar las sustancias separadas opara aumentar la sensibilidad de la detección.
Usualmente los reactivos de detección se pulverizan sobre la placa o folio. Algunos de estosreactivos se venden ya preparados, p. ej. en forma de soluciones listas para usar (p. ej.rodamina B, ninhidrina, ácido fosfomolíbdico). La mayoría deben sin embargo prepararse en ellaboratorio y pulverizarse mediante atomizadores adecuados (Fig. a0).
Entre ellos se encueniran los atomizadores de laboratorio. acoolados a una conducción deaire comprimido o nitrógeno, y las llamadas pistolas pulverizadoras que se componen de undepósito, un contenedor de gas portador y una cabeza pulverizadora (atención: por sucontenido en CFCs - hidrocarburos fluoroclorados - nocivos para el medio ambiente, laspistolas pulverizadoras y los botes de aerosol dejarán de utilizarse en un futuro próximo).
La pulverización debe realizarse siempre bajo una campana de extracción con buen tiraje omediante un dispositivo adecuado que se lleve la niebla de reactivo, generalmente tóxica oagresiva, y los vapores de disolvente. El cromatograma se coloca en posición vefticalligeramente inclinado. Cuando se ha formado una neblina uniforme se dirige el chorro delatomizador sobre la placa y se pulveriza de manera uniforme, generalmente hasta que seempieza a hacer transparente. Pulverizar en exceso puede producir la disolución o el arrastrede algúno de los compuestos de la placa (Fig. 41).
Además de los dispositivos de pulverización se dispone también de dispositivos deinmersión. La inmersión y extracción verticales así como el tiempo de permanencia en lacubeta de inmersión (algunos segundos) pueden seleccionarse a voluntad y se realizan demanera automática 12, 511.
Algún reactivo puede estar mezclado con la capa con lo que la derivatización porcalentamiento se realiza después del desarrollo.También puede añadirse a través de la fasegas (p.ej. los gases nitrosos para los compuestos aromáticos) o mezclarse con el eluyente (p.ej. la ninhidrina para los aminoácidos) [52, 53].
A menudo los cromatogramas han de calentarse en una estufa o sobre una placa calefactoradespués de añadir los reactivos de detección, para activar la reacción (p. ej. de 10 a 15 min. a105-1 10 'C).
lnmediatamente después del revelado conviene marcar y rotular (alápiz) las manchas, ya queéstas pueden palidecer o cambiar de color.
49
a 1-a-3l , - -
@
tw,{
@
'6
A
Fig. 41: A: Representación esquemática delproceso de pulverización B: Esquema de lapulverización.
lndicaciones y sugerencias
- Es imprescindible usar gafas protectoras yguantes de laboratorio al efectuar la pulve-rización.
- La derivatización modifica o destruye laestructura química de los compuestos.
- La derivatización es más cara y laboriosaque la detección por radiación UV, aunqueen determinados casos mucho más sensi-ble.
- Los gases portadores de los aerosolespreparados contaminan el medio ambien-
- Al sumergir se corre el peligro de disolver los compuestos por acción del disolvente(remedio: cambiar la polaridad) o de que el agua del reactivo disuelva la capa (remedio:preparar el reactivo con un disolvente orgánico).
- Las sustancias pueden difundir durante la inmersión.
50

- Los reactivos de detección se reparten de forma más homogénea por inmersiÓn que porpulverización. Los reactivos de inmersión contaminan menos al personal y al medioambiente que los de pulverización.
- Al calentar en una estufa debe ponerse una superfície metálica debajo de la placa para
conseguir un calentamiento homogéneo.
3.7 Evaluación
Tras finalizar un cromatograma de capa fina deben evaluarse los resultados. Existe una granvariedad de métodos adecuados a cada tipo de problemática (Fig. 42). Los más importantesse describen a continuación [54, 55].
3.7.1 Evaluación cualitativa.
Los cromatogramas de capa fina se realizan para identificar sustancias de una mezcla, pero
también para comprobar purezas o separar mezclas. Sirven especialmente para el control desíntesis o del transcurso de una reacción.
Semicuantitativa
lnd¡recta Directa (in situ)
- Recorridos- Colores/lntensidad- Comportamiento UV- Combinación o
acoplamiento conIR, EM, RMN, CG
- Comparación de laintensidad del color
- Comparación de laintensidad defluorescencia
- Comparación deltamaño de mancha
- ldem con una ser¡ede diluciones
- Fotometría- Gravimetría- Volumetría- Polarimetría- Polarographía- IR, EM, RMN- Media de isÓtoPos- Fosforescencia- Fluorescencia- EAA- Determinación
enzimática
- Dens¡tometría- Espectrofotometría- Medidas de centelleo- Radiometría
Fig. 42: Métodos de evaluación.
cl

El Rt (ver Cap.2.2) indica la posición del cromatograma en que se encuentra una sustancia. Esconveniente tratarlo como un valor meramente indicativo ya que son tantos los factores queintervienen durante la cromatografía que es muy difícil y laborioso obtener Rrs exactamentereproducibles.
Con fines de identificación es necesario relacionar los R,s de los compuestos analizados conlos de las sustancias patrón. Si coinciden es probable (pero no seguro) que se trate de lasmismas sustancias. La cerleza en la identificación sólo se consigue realizando, además de lacromatografÍa de capa fina, estudios espectroscópicos (p. ej. espectroscopía lR, RMN, demasas, o el acoplamientos de las mismas con la CCH.
3.7.2 Evaluación semicuantitativa.
La evaluación semicuantitativa se utiliza cuando hay que determinar si se está claramente porencima o por debajo de unos valores límite, o cuando bastan indicaciones aproximadas.
En cualquier caso se cromatografían junto a la muestra varias concentraciones de la sustanciade interés. La evaluación se realiza por comparación visual o por medición del diámetro o de lasuperficia de las manchas. La exactitud máxima se sitúa alrededor del + 10%.
3.7.3 Evaluación cuantitativa.
Parala evaluación cuantitativa se dispone de métodos indirectos y directos. En el primer casolas sustancias han de extraerse de la placa mediante un proceso de disolución para proseguirlos estudios. En el segundo la evaluación tiene lugar directamente sobre la placa (: ¡¡situ).
3.7.3. 1 Evaluación cuantitativa indirecta.
Una posibilidad de evaluación indirecta, poco impoftante actualmente, consiste en raspar laregión de la capa en que se encuentra la sustancia de interés, disolver la sustancia y, acontinuación, analizarla mediante un procedimiento analítico adecuado. Hay que tener encuenta que la extracción diluye la muestra y que generalmente hay que concentrarla de nuevo.También en este caso se recomienda cromatografiar patrones sobre la misma placa.
La elución de las zonas de placas de CCF en que hay sustancia puede hacerse tambiénautomáticamente (p. ej. con el Eluchrom CAMAG), sin necesidad de raspar la capa. Primero se
Fig. 43: Densitómetro de barrido (CAMAG TlC-Scanner ll) y camino óptico en medidasdensitométricas. L: fuente de luz, S : rendija, M : monocromador, A: objetivo de enfoque , PM :fotomultiplicador. C : cromatoorama.
52

raspa la capa en forma de anillo alrededor de las manchas. A continuación se posa lacabezade elución y se eluye. De esta forma se pueden eluir hasta seis manchas a la vez [56].
3.7.3.2 Evaluación cuantitativa directa.
En todos los métodos directos de evaluación cuantitativa es esencial aplicar los mismosvolúmenes de muestra y patrón sobre la misma placa. Las concentraciones deberían ser delmismo orden y los Rrs deberían estar comprendidos entre 0.3 y 0.7.
Los cromatogramas se registran pista por pista por densitometría en un espectrofotómetropara cromatogramas y se evalúan por comparación de las alturas de pico o de las superficiesde pico entre patrones y muestras. Las mediciones se realizan bajo luz visible o en la región UV- según las propiedades de las sustancias - generalmente a la longitud de onda en que lasustancia a determinar muestra un máximo de absorción o de fluorescencia (Fig. 43).
Debe evitarse la evaluación cuantitativa por "medida de la disminución de la fluorescencia".Las sustancias que producen una disminución de la fluorescencia cuando se utiliza unindicador fluorescente en el UV254 absorben en cualquier caso radiación UV así que es mejordeterrninarlas a la longitud de onda de su máximo de absorciÓn.
En general la absorción de luz se mide por reflexión (la radiación UVes absorbida por el vidrio oel gel de sílice y no atraviesa la placa). Las sustancias coloreadas o fluorescentes puedenmedirse también por transmisión, aunque normalmente no conlleva ninguna ventaja.
Referencias
11l R.E. Kaiser, GIT Fachz. Lab. 29, 268-269 (1985).
[2] H. Jork, W. Funk, W. Fischet H. Wimmer: Dünnschicht-Chromatographie - Reagenzienund Nachweismethoden,Vol. 1a, p. 6,VCH-Verlag Weinheim 1989.
t3l A.N. Stein, J. Chem. Ed. 53, 646 (1976).
[4] U.Wintermeyer: Die Wurzeln der Chromatographie, GIT-Verlag, Darmstadt 1989.
[5] E. Stahl (editor):Thin Layer Chromatography - A Laboratory Handbook, 2nd Edition,Springer-Verlag, Berlin 1 969.
16l J.G. Kirchner,Thin Layer Chromatography, John Wiley and Sons, New York 1978. (:
Techniques in Chemistry vol XIV).
[7] H. Halpaap, Kontakte (Merck), 1980 (3), 37.
[8] B. Fried, J. Sherma: Thin Layer Chromatography, Marcel Dekker, New York 1986' 4-7.
[9] F. Geiss: Fundamentals of Thin Layer Chromatography, Hüthig, Heidelberg 1987, 3-8.
t10l K. Randerath: Dünnschicht-Chromatographie,Verlag Chemie,Weinheim 1962.
t11l O. Mikes (Hrsg.), Laboratory Handbook of Chromatographic and Allied Methods, EllisHarwood Ltd. Chichester 1979, 472ss.
[12] P Pachaly: Dünnschichtchromatographie in derApotheke,WissenschaftlicheVerlagsge-sellschaft, Stuttgart 1 982.

[13] Ver ref. [8], pp. 9-19
[14i R.E. Kaiser en A. Zlatkis, R.E. Kaiser (editores): HPTLC; Journal of ChromatograpnyLibraryvol.9, pp.31ss., Elsevier, Amsterdam-Oxford-NewYork1977;vertambién ref. [9],pp. 73ss.
[15] Ver ref. [9], p .2.
[16] W. Christen: Dünnschichtchromatographie. Experimente für den naturwissenschaft-lichen Unterricht, GIT-Verlag, Darmstadt 1 975.
i17l H. Halpaap en R.E. Kaiser (Hrsg): Einführung in die Hochleistungs-Dünnschicht-Chromatographie HPDC, tfc-Vertag, Bad Dürkheim 1976, pp.232-233.
[18] Ref. l2l, p. 121
[19] Ref. [10], p. 16.
[20] S. Ebel, Nachr. aus Chemie,Technik & Labor 36, Sonderheft 1988.
[21] Folletos Merck, Mitteilungen zur Dünnschicht-Chromatographie l, 1993.
l22l Ver ref. [9], pp.185ss.
[23] Ver ref. [12], pp.16-18.
l24l W. Funk, Fresenius Z. Anal. Chem. 319,206-219 (1994).
[25j G. Mi ldau, H. Jork, Fresenius Z. Anat. Chem.318,302-304 (1984).
[26] Publicación Merck, Extrelut - Neues Verfahren zur Extraktion lipophiler Stoffe, 1g78.
l27l H. Halpaap, K.-F. Krebs, J. Chromatogr.142,823-853 (1977)
t28l H. Jork, W. Funk, W Fischer, H. Wimmer, J. Ptanar Chromatogr. 1,280-292, 1gBA.
[29] W. Funk, M. Schanze, U. Wenske, GtT-Supplement Chromatographie 3/83.
[30] R. Klaus, J. Chromatogr. 333,276-287 1985.
[31] w. Gótz, A. sachs, H. wimmer, Dünnschichtchromatographie, G. Fischer-Vertag,Stuttgaft-New York, 1 978.
[32] Ver ref .1121, cap. 3.5.
[33] Ver ref .l2l, p. 132.
[34] Catálogo CAMAG TLC 1987.
[35] R. Klaus, J. Chromatogr. 333,276-287 1985.
[36] D.C. Fenimore, C.J. Mezer, J. Chromatogr.186,555-561 1979;D.C. Fenimore, C.M. Davis,Anal. Chem. 53,252A-266A (1981)
[37] D.Volkmann, GIT Fachz. Lab.23,508-512, 1979.
[38] H. Jork, H. Wimmer: Quantitative Auswerlung von Dünnschicht-Chromatogrammen,Loseblattsammlung 1./3-3, GIT-Verlag, Darmstadt 1982.
[39] Ver ref. [5], pp. 86-105.
54

[40] Ver ref. [10], pp. 51-53.
[41] Ver ref. [8], pp. 93-94.
l42l D.Volkmann, GIT Supplement Chromatographie 5/84, 17 -22, 1984.
[43] R.E. Kaiser, J. High Resolut. Chromatogr., Chromatogr. Commun. 1,164-168, 1978.
1441 J.A. PerryT.H. Jupi l le, L.H. Glunz, Anal. Chem. 47,65A-744,1975.
[45] E. Stahl, Arch. Pharm., 292,411-416, 1959.
t46l K. Burger, G|T-Supplement Chromatographie 5/84, 29-31 (1984); D.E. Jaenchen,Internat ional Lab. March 1987, 66-71.
l47l Ver ref. [9], pp. 305ss.
[48] Ver ref. [34].
[49] Ver ref. [8] , pp. 100-115.
t50l F. Kreuzig, GIT Supplement Chromatographie, 1982, 46-49.
t51l W. Funk, M. Heiligenthal, GIT Supplement Chromatographie 5/84,49-51, 1984.
[52] J.C.Touchstone,T. Murawec, M. Kasparow,W.Wortmann, J. Chrom. Sci., 10, 490-493,1972.
l53l R. Bottler,T. Knuhr Fresenius Z. Anal. Chem.,302,286-289, 1986.
[54] Ver ref. [38], caps.l./1 y l./2.
[55] Ver ref. [9], pp. 420ss
[56] Folleto CAMAG, "CAMAG Eluchrom", 1986.
55


















