Tecnología Analítica de Proceso - … · Escalamiento de la síntesis de APIs ... directa con los...
76
8324 332.95 7452.0 1793.00 1784.0 9325.0 93.00 93.99% 8371.00 1793.00 4003.0 92.1% 43.2% 24.3% 21.980% 92.1% 56.1% 64.2% 92.1% 18.4% 478.00 563.00 Tecnología Analítica de Proceso Sección Especial: APIs, Excipientes y Manufactura Seguimiento del avance en el análisis de datos Más: Escalamiento de la síntesis de APIs Cómo lograr la estereoselectividad de los APIs POSTURA OFICIAL: Manejo del análisis de impurezas elementales Volumen 10, Número 5 Investigación Arbitrada • Simulación numérica del recubrimiento de tabletas • Métodos de medición de carbón para validación de la limpieza
Transcript of Tecnología Analítica de Proceso - … · Escalamiento de la síntesis de APIs ... directa con los...

8324
332.95
7452.0
1793.00
1784.0
9325.0
1793.00
93.99%
8371.00
1793.00
4003
.0
92.1%43.2%
24.3%
21.980%92.1%
56.1%
64.2%
92.1%
18.4%
478.00
563.00
Tecnología Analíticade Proceso
Sección Especial: APIs, Excipientes y Manufactura
Seguimiento del avance enel análisis de datos
Más:Escalamiento de la síntesis de APIs
Cómo lograr la estereoselectividad de los APIs
POSTURA OFICIAL: Manejo del análisis de impurezas elementales
Volumen 10, Número 5
Investigación Arbitrada• Simulación numérica del recubrimiento de tabletas
• Métodos de medición de carbón para validación de la limpieza

ACG Pam Mexico.pdf 1 05/09/12 11:42
Marque en la tarjeta de servicio al lector el No. 2

Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
5

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 20122
NOVIEMBRE / DICIEMBRE 2012 VOLUMEN 10, NÚMERO 5
En el terreno de juego
Investigación Arbitrada
10 Reporte desde: JapónJane WanDespués de una serie de reformas gu-bernamentales que hacen un llamado a los participantes tanto domésticos como extranjeros, el mercado farmacéutico japonés está haciendo su reaparición. 12 Atención: Descubrimiento Los premios a la investigación de PhRMA se concentran en la enfermedad de Alzheimer
CIENCIAS FARMACÉUTICAS Y NOVEDADES TECNOLÓGICAS
Pharmaceutical Technology en Español, proporciona información importante, confiable, y oportuna sobre todos los aspectos relacionados con Desarrollo e Investigación Aplicada; y con lasTecnologías de Proceso, Fabricación, Formulación, y Empaque para la Industria Farmacéutica Convencional y la de Biotecnología.
AspectosFORO TÉCNICO
INGREDIENTES FARMACÉUTICOS
ENTREGA DE FÁRMACOS
POSTURA OFICIAL
MÉTODOS DE LIMPIEZA
RECUBRIMIENTO DE TABLETAS
POSTURA OFICIAL
5 Liofilizado: ¿Está la tecnología avanzando lo suficientemente rápido?Foro Técnico moderado por Stephanie Sutton Los expertos discuten las mejores prácticas para desarrollar un proceso de liofilización basado en la QbD.
36 Escalamiento en la síntesis de APIsPatricia Van Arnum Las estrategias se centran en la manera de optimizar las condiciones y operabilidad del proceso. 61 Logro de la estereoselectividad en APIs e intermedios farmacéuticosPatricia Van Arnum La tecnología quiral está creciendo en importancia conforme la industria y la academia avanzan en nuevos esquemas para lograr la enantioselectividad.
55 Futura innovación en la entrega de fármacosMesa redonda de la industria con representantes de Aptalis, Sistemas de Entrega de Fármacos 3M y la Universidad de Nuevo México.
14 GMPs para validación de métodos en el desarrollo inicial (Parte II)Donald Chambers, Gary Guo, Brent Kleintop, Henrik Rasmussen, Steve Deegan, Ste-ven Nowak, Kristin Patterson, John Spicuzza, Michael Szulc, Karla Tombaugh, Mark D. Trone, Zhanna Yuabova Los representantes del Consorcio IQ exploran los esquemas de la industria para la aplicación de las GMPs en el desarrollo inicial.
42 Métodos de medición de carbón para la validación de limpieza Robert Clifford y Minako TankaLos autores comparan la combustión directa con los métodos de enjuague y muestreo con hisopo.
50 Simulación numérica del recubrimiento de tabletas Gregor Toschkoff, Daniele Suzzi, Siegfried Adam, y Johannes KhinastLos autores investigan el proceso de recubrimiento de tabletas utilizando una combinación de diferentes técnicas de simulación.
63 Análisis de impurezas elementales Alan CrossEl autor discute la manera de manejar los cambios farmacopeicos pendientes.
8324
332.95
7452.0
1793.00
1784.0
9325.0
1793.00
93.99%
8371.00
1793.00
4003
.0
92.1%43.2%
24.3%
21.980%92.1%
56.1%
64.2%
92.1%
18.4%
478.00
563.00
46 Tecnología Analítica de Proceso: seguimiento del avance en el análisis de datosLos expertos de la industria comparten las perspectivas sobre la instrumentación analítica, los métodos y el análisis de datos.
Ilust
raci
ón
por
Dan
War
d

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 3
40 Una revisión estadística del ICH Q10 Sistema de Calidad FarmacéuticaLynn D. TorbeckLa aplicación de las recomendaciones del ICH Q10 al análisis estadístico puede ayudar a prevenir las recuperaciones de producto.
39 Cambios pequeños, gran impacto en el sistemaSimon Chalk y Steve JonesUn vistazo al reemplazo del elastómero mejora a veces las operaciones.
58 Expedidores con temperatura controladaHallie ForcinioLas herramientas de empacado y monitoreo protegen a los farmacéuticos sensibles a la temperatura.
SOLUCIONES ESTADÍSTICAS
BIOFORO
FORO DE EMPAQUE
CONTENIDO
Pharmaceutical Technology es selectivamente extraida o indexada en:Biological Sciences Database (Cambridge Scientific Abstracts)Biotechnology and Bioengineering Database (Cambridge Scientific Abstracts)Business and Management Practices (RDSI)Chemical Abstracts (CAS)Current Packaging AbstractsDECHEMADerwent Biotechnology Abstracts (Derwent Information, Ltd.)Excerpta Medica (Elsevier)International Pharmaceutical Abstracts (ASHP)Science Citation Index (Thomson)Pharmaceutical Technology está orgullosa de ser miembro asociado de DCAT, IPEC y PDA.
ColumnasSección Especial: APIs, Excipientes y ManufacturaQUÍMICA DEL ESTADO SÓLIDO
RECUBRIMIENTO DE TABLETAS
EXCIPIENTES
CADENA DE SUMINISTRO
BIOFARMACÉUTICOS
22 Desarrollo de la forma sólida óptimaDavid Igo y Stephen Carino
24 Logrando la comprensión y el control del proceso en el recubrimiento pelicularEric Van Ness, Beverly Schad, Thomas Riley y Brian Cheng
29 Excipientes en el enmascaramiento del saborSesión de preguntas y respuestas con BASF
31 Cambio a las inspecciones basadas en el riesgoPatricia Van Arnum
34 Ponderación del acceso y la costeabilidadKenneth I. Kaitin y Joshua P. Cohen
Secciones
69 ¿Qué hay de nuevo?
69 Calendario de eventos
70 Cápsulas Farmacéuticas
71 Directorio Clasificado
72 Índice de anunciantes

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 20124
James P. AgallocoPresident, Agalloco & Associates
Larry L. Augsburger, PhDProfessor, Department of Pharmaceutics, University of Maryland
David H. Bergstrom, PhDCOO, NovaDel Pharma Inc.
Phil BormanQbD Lead & Data Management & Analysis Manager GlaxoSmithKline
Rory BudihandojoDirector, Quality Systems Audit, Boehringer-Ingelheim Shanghai Pharmaceuticals Co. (China)
Todd L. CecilVice-PresidentCompendial ScienceUnited States Pharmacopeia
Metin Çelik, PhDPresident, Pharmaceutical Technologies International (PTI)
Zak T. Chowhan, PhDConsultant, Pharmaceutical Development
Suggy S. Chrai, PhDPresident and CEO,Chrai Associates, Inc.
Roger Dabbah, PhDPrincipal Consultant, Tri-Intersect Solutions
Tim FreemanManaging Director, FreemanTechnology
Sanjay Garg, PhDProfessor, Pharmaceutical Sciences, University of South Australia
R. Gary Hollenbeck, PhDChief Scientific Officer, UPM Pharmaceuticals
Ruey-ching (Richard) Hwang, PhDSenior Director, Pharmaceutical Sciences,Pfizer Global R&D
Mansoor A. Khan, PhDDirector, FDA/CDER/DPQR
Russell E. MadsenPresident, The Williamsburg Group, LLC
Heidi M. Mansour, PhDAssistant Professor,College of Pharmacy, University of Kentucky
Jim MillerPresident, PharmSource Information Services Bio/Pharmaceutical Outsourcing Report
Colin Minchom, PhDVice President Particle DesignHovione
Christine Moore, PhDDeputy Director for Science and Policy, Office of New Drug Quality Assessment, CDER, FDA
R. Christian Moreton, PhDVice-President, Pharmaceutical Sciences, Finnbrit Consulting
Fernando J. Muzzio, PhDDirector, NSF Engineering Research Center on Structured Organic Particulate Systems, Dept. of Chemical and Biochemical Engineering, Rutgers University
Moheb M. Nasr, PhDVice-President, CMC Regulatory Strategy, Global Regulatory Affairs, GlaxoSmithKline
Garnet E. Peck, PhDProfessor Emeritus of Industrial Pharmacy, Purdue University
James Polli, PhDProfessor, School of Pharmacy, University of Maryland
Wendy Saffell-ClemmerDirector, Research, BioPharma Solutions
Gurvinder Singh Rekhi, PhDDirector,Research and Development, Elan Drug Delivery Inc.
Susan J. SchnieppPharmaceutical Consultant, Schniepp & Associates, LLC
David R. SchonekerDirector of Global Regulatory Affairs, Colorcon
Eric B. Sheinin, PhDPresident, Sheinin and Associates
Charles A. Signorino, PhDCEO, Emerson Resources, Inc.
Aloka SrinivasanPrincipal Consultant, PAREXEL International
Heinz Sucker, PhDProfessor Emeritus,Pharmaceutical Institute, University of Bern
Scott Sutton, PhDMicrobiology Network
Lynn D. TorbeckStatistician, PharmStat Consulting
Pharmaceutical Technology en Español V.10 No.5 Noviembre - Diciembre de 2012. Publicación Bimestral, editada por Revistas para la Industria, S.A. de C.V. Editor Res-ponsable: Ma. Antonieta Guerrero Paz. No. de Certificado de Reserva otorgado por el instituto nacional de derecho de Autor 04-2011-010610533100-102 No. de Certificado de licitud de Titulo 12699. No. de Certificado solicitud de contenido 10271. Domicilio de la publícacion: Av. Insurgen-tes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, México, D.F. Impreso en: Polymasters de México, S.A. de C.V. Distribuida por Revistas para la Industria, S.A. de C.V. Av. Insurgentes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, México, D.F.
Toda la información y conceptos que aquí aparecen son responsabilidad exclusiva de cada uno de los autores y firmas comerciales.
Esta prohibida y será castigada la reproducción total o parcial de cualquiera de los materiales que aquí aparecen.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 5
Foro Técnico: LioFiLización
Secado por congelamiento: ¿Está avanzando suficientemente rápido la tecnología?Foro técnico moderado por Stephanie Sutton
Para saber más acerca de las mejores prácticas para desarrollar un proceso de liofilización, incluso cuando se utiliza un enfoque de calidad por diseño (QbD), Pharmaceutical Technology platicó con Henning Gieseler, líder de grupo, Grupo de Enfoque para Secado por Congelamiento, en la División de Farmacia en la Universidad de Erlangen-Nuremberg; Yves Mayeresse, director, Centro de Manufactura de Llenado de Excelencia y Operaciones de Secado por Congelamiento, en GSK Biológicos; Steven Nail, científico principal en Baxter Pharmaceutical Solutions; Trevor Page, director técnico de grupo, y Manfred Steiner, gerente de ventas del área, ambos en GEA Pharma Systems; y Michael J. Pikal, profesor de farmacia en la Escuela de Farmacia, Universidad de Connecticut.
ESTRATEGIAS DE QBDPharmTech: ¿Qué tipos de estrategias únicas y conocimiento del producto se requieren cuando se utiliza un enfo-que de QbD para la liofilización?
Gieseler (Universidad de Erlan-gen-Nuremberg): Necesitamos encon-trar una interpretación más profunda de los experimentos realizados en di-ferentes escalas del equipo. El secado por congelamiento exitoso requiere una profunda comprensión de los atribu-tos relacionados tanto con el producto como con el proceso, así como las co-rrespondientes herramientas analíticas usadas durante el desarrollo del produc-to y del proceso para medirlos repre-sentativamente. Cuando buscamos las características de calidad finales desea-das de un producto secado por conge-lamiento, el término ‘calidad’ está, en primera instancia, no relacionado con la estabilidad de un API, sino que apun-ta a otras características, tales como la elegancia de la torta, el tiempo de re-constitución, el contenido de humedad y otros parámetros. Un vial con una torta colapsada se rechaza de manera rutinaria del lote durante la inspección
óptica, aún cuando la estabilidad del API puede ser perfectamente aceptable desde un punto de vista farmacéutico. La inspección óptica es una de las pri-meras pruebas a ser realizadas en un producto secado por congelamiento, no la estabilidad del API.
El vínculo que conecta entre ‘atri-butos de calidad’ y ‘atributos del pro-ducto/proceso’ está con frecuencia fundamentado en la conducta fisicoquí-mica de la formulación, la cual es una función de la temperatura y el tiempo. Las propiedades fisicoquímicas, tales como la temperatura crítica de formu-lación (la temperatura de transición ví-trea del soluto concentrado por conge-lamiento [Tg’] para productos amorfos o la temperatura eutéctica [Teu] para materiales cristalinos) son parámetros importantes que deben ser determina-dos antes del desarrollo del ciclo. En-tonces, el objetivo es controlar la tem-peratura del producto en la interfase de sublimación del hielo por debajo de esta temperatura crítica durante el ciclo para evitar la movilidad elevada en el siste-ma y los cambios morfológicos, tales como encogimiento, colapso y fundido. En la industria, la calorimetría diferen-cial de barrido (DSC) ha sido usada por décadas para evaluar la huella térmica de un material. La DSC es una pode-rosa herramienta, aunque no es perfec-tamente representativa de la situación real del secado por congelamiento de un producto. Un procedimiento más representativo es la determinación de la temperatura de colapso (Tc) mediante microscopía de secado por congelación (FDM). La configuración técnica de un experimento de FDM es actualmente la mejor manera de simular el secado por congelamiento a microescala, aunque todavía presenta obstáculos en la inter-pretación de los datos.
Teniendo estas temperaturas críticas en mente, el secado por congelamiento demanda un control confiable y repre-sentativo de las temperaturas del pro-ducto en la interfase de sublimación del hielo durante el secado primario para obtener un producto de alta calidad. Muchas herramientas PAT disponibles comercialmente (p.ej., medición ma-nométrica de temperatura, TDLAS, y otras) ayudan durante la etapa de desa-

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 20126
rrollo a determinar las temperaturas de interfase del producto, aunque dichas herramientas con frecuencia no pueden ser usadas en un entorno de produc-ción. Como resultado, el obstáculo más grande y el reto para el futuro cuando se establezca un concepto confiable de QbD para el secado por congelamiento es determinar los parámetros críticos (relevantes) del producto y el proceso que sean también escalables.
Mayeresse (GSK Biológicos): La liofilización ha evolucionado mucho durante los pasados 20 años. Hace años, el desarrollo de la liofilización se apo-yaba principalmente en las habilidades de los científicos que aprendían a través del ensayo y error. Hoy día, existen he-rramientas analíticas para ayudar al de-sarrollo del proceso de secado por con-gelamiento. Por ejemplo, aparatos como un criomicroscopio permiten la determi-nación de la temperatura de transición vítrea, la cual se utiliza para determinar la temperatura y presión durante la fase de secado primario de un ciclo de seca-do por congelamiento. Para un enfoque de QbD, es muy fácil definir en qué paso se aplicará cada herramiento y cuál será su resultado en el proceso.
Actualmente, el desarrollo de un nuevo proceso es más sistemático, lo cual le da a los desarrolladores más tiempo para concentrarse en la especi-ficidad del producto.
Nail (Baxter Pharmaceutical So-lutions): En Baxter, el enfoque por QbD para el desarrollo y optimización del ciclo de secado por congelamiento se apoya fuertemente en una tecnolo-gía analítica de proceso llamada espec-trometría de absorción láser con diodo afinable (TDLAS). Esta es una tecno-logía de infrarrojo cercano que mide la velocidad de flujo instantánea de la masa del vapor de agua desde la cámara del secador por congelamiento hasta el condensador. También usamos métodos bastante estándar para la caracterización de la formulación, tales como análisis térmico a baja temperatura y microsco-pía de secado por congelamiento, para determinar el límite superior de tempe-ratura del producto durante el secado primario. Utilizamos un método gráfico para el espacio de diseño que incorpo-ra limitaciones colocadas en el proceso
Foro Técnico: LioFiLización
que se basan tanto en las características del producto como en la capacidad del equipo de secado por congelamiento. El TDLAS facilita la medición del coe-ficiente de transferencia de calor del vial como una función de la presión, la medición de la resistencia de la capa de producto seco al flujo del vapor de agua, y el índice máximo de sublimación so-portado por el equipo como una función de la presión. Todos estos son necesa-rios para construir el espacio de diseño.
Page/Steiner (GEA Pharma Sys-tems): En cualquier proceso de QbD, es importante definir primero el desem-peño requerido del producto terminado. En otras palabras, ¿cuáles son los atri-butos de calidad críticos del producto? Para un producto secado por congela-miento estos atributos son típicamente cosas tales como tiempo de reconstitu-ción, apariencia, merma, colapso, via-bilidad del producto y vida de anaquel.
El siguiente paso es utilizar méto-dos analíticos para determinar la con-ducta del producto durante el proceso de congelamiento y secado. Una técni-ca de evaluación de riesgo, tal como el análisis en modo de fallo y el análisis de efectos, puede determinar cuáles fac-tores en el proceso pueden impactar la calidad del producto final.
La base de la QbD es asegurar el nivel de conocimiento con respecto al producto y cómo varía la calidad del producto con los cambios en la materia prima o la variabilidad en las condicio-nes del proceso asegura que el proceso es totalmente capaz de producto un pro-ducto que cumple las especificaciones.
Pikal (Universidad de Connecti-cut): Casi todos los productos liofiliza-dos deben ser estériles, lo cual impone un atributo de calidad crítico que no es relevante para los productos orales. También, mientras que la estabilidad con frecuencia es un problema con los productos orales, es casi siempre un problema con un producto liofilizado; de otra forma, ¿Por qué liofilizar? Adi-cionalmente, el Diseño de Experimen-tos (DOE) es con frecuencia una parte crítica de la QbD. Aunque la QbD pue-de ser útil para el diseño de formulacio-nes y procesos para productos liofiliza-dos, no es útil en el diseño de la etapa de secado primario de la liofilización. Esto
es resultado del hecho de que la física del secado primario está bien entendida. Diseñar procesos con base en la física es mejor y más eficiente que el diseño de éstos basado en la estadística.
ESPACIO DE DISEÑOPharmTech: ¿Qué factores clave de-ben considerarse cuando se determi-na un espacio de diseño para un pro-ducto liofilizado?
Gieseler (Universidad de Erlan-gen-Nuremberg): El espacio de dise-ño debe estar definido por la formula-ción crítica y los factores del proceso. Considerando la formulación, dichos factores podrían incluir la temperatura de formulación crítica (es decir, la tem-peratura de colapso), el contenido de humedad, los parámetros de estabilidad del API, la apariencia y los parámetros morfológicos (p.ej., superficie especí-fica de la estructura interior de la torta que influye, entre otras cosas, el tiempo de reconstitución). La mayoría de los científicos, sin embargo, se enfocan en el espacio de diseño del proceso, o más precisamente en el espacio de diseño del secado primario. Aquí, el factor más importante es la temperatura de interfa-se del producto, la cual, a su vez, es re-sultados de la temperatura del anaquel aplicada, la presión de la cámara y las características de transferencia de calor y de masa del sistema de contenedor aplicado.
Los estudios recientes han sugeri-do que la determinación del espacio de diseño del secado primario solo, parece insuficiente para obtener un cuadro re-presentativo de la conducta del produc-to durante el proceso. Al menos, el paso del congelamiento debe considerarse también ya que éste determina la dis-tribución del tamaño de poro y, por lo tanto, afecta la resistencia del flujo de masa durante el secado primario. Ade-más, el paso de congelamiento puede causar inestabilidad del API debido a que ocurre concentración por congela-miento o desnaturalización (proteínas) superficial inducida por hielo/agua. Una morfología del producto que se ha formado en diferentes temperaturas de nucleación durante el paso de conge-lamiento también puede proporcionar un diferente grado de estabilidad para

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 7
la estructura de la torta durante el se-cado primario. Por ejemplo, tempera-turas de nucleación más tibias forman poros más grandes. Algunas tortas del producto han demostrado una firmeza estructural mayor durante la fase de su-blimación cuando estuvieron presentes poros más grandes. La morfología del producto formada durante la fase de congelamiento incluso influye el des-empeño del secado secundario del pro-ducto farmacéutico.
El obstáculo más grande es deter-minar representativamente el espacio de diseño de la formulación y el pro-ceso. Aunque el espacio de diseño del proceso típicamente está definido en el equipo a escala de laboratorio, dicha in-formación debe escalarse después a la manufactura. El reto es que el espacio de diseño del proceso originalmente de-finido podría no cuadrar perfectamente con el espacio de diseño del proceso en la manufactura.
Mayeresse (GSK Biológicos): Los factores clave para determinar un pro-ceso de secado por congelamiento son la temperatura de los anaqueles, la pre-sión en la cámara y el tiempo. El valor de estos parámetros está influido por el equipo, lo que significa que el espacio de diseño debe ser tan grande como sea posible. Para los parámetros de salida del proceso, los factores clave son la elegancia de la torta, el contenido de humedad y la potencia. Dependiendo del producto, pueden añadirse algunos parámetros específicos. Por ejemplo, si el ingrediente activo es propenso a la oxidación, puede desarrollarse una prueba específica.
Nail (Baxter Pharmaceutical So-lutions): Los factores clave son el lími-te superior de temperatura del producto durante el secado primario (ya sea una temperatura de colapso o una tempera-tura de fusión eutéctica) y la capacidad del equipo. Además de esto, necesita-mos conocer la relación entre las varia-bles que controlamos, como es la tem-peratura de anaquel y la presión de la cámara, y la variable en la que estamos más interesados, la cual es la tempera-tura del producto. Esto se hace utilizan-do ecuaciones bien establecidas para la transferencia de calor y masa junto con el coeficiente de transferencia de calor
del vial y la resistencia de la capa de producto seco al flujo de vapor de agua.
En mi compañía, hemos dirigido la mayoría de nuestra atención al desarro-llo del espacio de diseño para el secado primario, porque éste es generalmente la parte del proceso que consume la mayor parte de tiempo, y está gene-ralmente asociado con el mayor riesgo para la calidad del producto. También necesitamos dirigir nuestra atención al congelamiento y a las fases de secado secundario del ciclo.
Page/Steiner (GEA Pharma Sys-tems): El espacio de diseño define las condiciones aceptables del proceso que han mostrado dar como resultado un producto dentro de especificaciones. Con frecuencia, el concepto se consi-dera en términos del rango permitido de ajuste de los parámetros críticos del proceso. Sin embargo, también es útil utilizarlo para considerar el rango de condiciones del proceso que ocurren naturalmente dentro de un liofilizador.
El cambio principal de paradigma que se presenta actualmente dentro del mundo de la liofilización es admitir que cada contenedor tiene su propio proce-so individual, el cual está determinado por factores influyentes tales como la posición en el anaquel o las fuentes de nucleación. Esto aplica para todas las clases de contenedores, incluyendo via-les, jeringas o charolas.
Pikal (Universidad de Connecti-cut): Normalmente existen tres tipos de restricciones. Primero, quieres restrin-gir la temperatura del producto durante el secado primario a un valor menor que alguna temperatura máxima permitida, la cual con frecuencia (aunque no siem-pre) es la temperatura de colapso. La selección de la combinación apropiada de temperatura de anaquel y de presión de la cámara asegurará que se cumpla este objetivo, pero el proceso también debe al menos cerrarse hasta el mínimo tiempo que sea posible para lograr la mejor eficiencia del proceso. Segundo, el tiempo pasado en el secado primario necesita ser lo suficientemente largo de manera que todo el producto esté vacío de hielo antes de que la temperatura de anaquel se incremente para el secado secundario. El incremento prematuro de la temperatura de anaquel puede cau-
sar colapso del producto.Finalmente, el proceso necesita co-
rrerse a una velocidad de sublimación que esté dentro de las capacidades de transferencia de masa y calor para el sistema. Correrlo bajo condiciones que sean excesivamente agresivas puede, por ejemplo, resultar en un flujo asfixia-do, lo que significa pérdida del control de presión de la cámara y quizás lleva-ría a la pérdida del lote completo.
PharmTech: De acuerdo a las guías del ICH, la determinación del “bor-de del fallo” es no esencial cuando se establece el espacio de diseño pero puede ser informativo. ¿Qué tanta consideración se le debe dar a la de-terminación del borde del fallo en el desarrollo del proceso de liofilización y porqué?
Gieseler (Universidad de Erlan-gen-Nuremberg): En mi opinión, el ‘borde del fallo’ es importante tanto para conocer como para comprender la ciencia del secado por congelación. Aunque los procesos o las formulacio-nes no deben ser diseñados en el ‘bor-de’, no se puede estimar un ‘margen de seguridad’ apropiado que sea requerido. En los casos en los que el ‘borde del fa-llo’ no ha sido investigado, un margen de seguridad podría ser demasiado con-servador, o definirse sobre una base de ensayo y error. De manera más impor-tante, para algunos parámetros críticos del proceso o del producto, no existen las condiciones del ‘borde del fallo’, lo cual entonces es muy relevante. Por ejemplo, un producto que puede ser procesado en secado primario a tem-peraturas de anaquel my por arriba del ambiente, el parámetro limitante ya no es el producto, sino el diseño del equi-po. Nuevamente, debemos trabajar con un margen de seguridad en el espacio de diseño establecido, pero necesitamos establecer racionalmente el margen de seguridad con base en el conocimiento del ‘borde del fallo’.
Mayeresse (GSK Biológicos): Es interesante conocer dónde está el borde del fallo incluso si es un dato no esen-cial ya que proporciona conocimiento acerca de la robustez total de la formula-ción. En un esquema de QbD, el alcance del espacio de diseño viene del análisis

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 20128
de riesgo que utilizase para determinar el margen necesario. Imaginémonos que para la temperatura de anaquel de-finimos un rango de 5°C alrededor del blanco. Para alguna formulación, 5°C está cerca del borde del fallo, pero para otras tenemos cinco grados más. A par-tir de este valor, pueden clasificarse las diferentes formulaciones en términos de robustez contra el colapso.
Nail (Baxter Pharmaceutical So-lutions): Nosotros le damos a esto mu-cha consideración para el desarrollo de productos secados por congelamiento. Nuestra comprensión de la idea de un espacio de diseño es conocer todas las combinaciones de, por ejemplo tempe-ratura de anaquel y presión de la cáma-ra, que den como resultado un producto farmacéuticamente aceptable. Nos gus-ta que este espacio de diseño sea tan grande como sea posible, de manera que las fronteras del espacio de diseño sean el límite superior de temperatura del producto durante el secado prima-rio (es decir, el borde del fallo del pro-ducto) y la capacidad de equipo, la cual es el borde de fallo del equipo. Por lo tanto, pensamos que el ‘borde del fallo’ es un componente clave en el desarrollo del espacio de diseño.
Page/Steiner (GEA Pharma Sys-tems): Es importante el valor de cono-cer donde puede fallar el proceso. La determinación del espacio de diseño depende del nivel de confianza en la ve-locidad de cambio de los parámetros re-levantes en la región entre el límite del espacio de diseño y el borde del fallo. Si un proceso es muy pronosticable y lineal, entonces puede ser razonable-mente pronosticado el riesgo de fallo. Sin embargo, en un proceso de secado por congelamiento, el impacto de la condición del proceso sobre la calidad del producto puede ser no lineal y la predicción de la proximidad al borde del fallo ser menos fácilmente definida. En este caso, puede ser mejor explorar explícitamente el borde del fallo.
Pikal (Universidad de Connec-ticut): En general, concuerdo con esta filosofía. Sin embargo, sostengo que con respecto al impacto del colapso, es aconsejable secar por congelamiento un producto por arriba de la temperatura de colapso para observar el impacto sobre
Foro Técnico: LioFiLización
la calidad del producto. La razón es que las temperaturas de colapso se determi-nan utilizando técnicas que no siempre pronostican cuantitativamente el colap-so en un producto que se está secando por congelamiento en un vial. Algunas veces se puede secar por congelamien-to a 5°C o más sobre la temperatura de colapso, medida por microscopía de secado por congelamiento sin colapso observable en el vial. Existen razones teóricas y varias observaciones que pro-porcionan documentación sobre este enunciado. Sin embargo, incluso si el colapso ocurre en el vial, existe la cues-tión de si hay o no algún otro atributo de calidad crítico comprometido. Con frecuencia, la respuesta es no, y algunas veces la calidad del producto (estabili-dad) es mejor en un producto colapsado. La explicación de esta información es el conocimiento sobre la evaluación del riesgo de colapso. La medida del riesgo es realmente el producto de la proba-bilidad del evento y la severidad de la ocurrencia del evento. La corrida sobre la temperatura del colapso aborda la ‘se-veridad’ de la ocurrencia del evento.
MANEJO DEL CONOCIMIENTOPharmTech: La guía del ICH Q8R2 Desarrollo Farmacéutico establece, “Los cambios en la formulación y procesos de manufactura durante el desarrollo y el manejo del ciclo de vida deben verse como oportunidades de adquirir conocimiento adicional y mayor soporte para el establecimien-to del espacio de diseño.” ¿Cómo pue-de este principio ser aplicado al desa-rrollo de un proceso de liofilización?
Gieseler (Universidad de Erlan-gen-Nuremberg: Uno de los mejores métodos para adquirir conocimiento adicional acerca del producto y del pro-ceso es implementar protocolos para análisis de robustez durante el desa-rrollo del ciclo en el laboratorio. Aquí, uno podría utilizar la composición de la formulación final deseada y la receta para el secado por congelamiento de-seado (optimizado) y después desarro-llar protocolos que desafíen el ciclo y la formulación. La temperatura de anaquel es elevada en pasos pre-definidos para obtener temperaturas mayores de pro-ducto durante el secado primario. Puede
aplicarse el mismo principio al punto de ajuste de presión de la cámara. Después del ciclo, los atributos de calidad del producto son inspeccionados y correla-cionados con los atributos observados de desempeño del proceso, tales como velocidades de flujo de masa, perfiles de temperatura del producto y resistencia del producto. De esta manera es posible simular, por ejemplo, el impacto de una situación de pérdida del control de pre-sión en la cámara de secado y su impac-to sobre la apariencia del producto, el contenido de humedad, y otros factores relevantes. Una pérdida del control de presión en la cámara dará como resulta-do temperaturas de producto incremen-tadas que a su vez pueden causar defec-tos cosméticos o de otra índole.
El mismo concepto de análisis de robustez puede aplicarse a la etapa de formulación. Un cambio en la formula-ción de excipientes o incluso un simple intercambio de excipientes, típicamente resulta en un cambio significativo del desempeño de la formulación, y por lo tanto del espacio de diseño. El uso de métodos estadísticos, como el DoE, ayu-da durante el desarrollo de la formula-ción a identificar factores relevantes. En general los DoEs son mucho más valio-sos durante la etapa de formulación que durante el desarrollo del proceso debido al número de experimentos requeridos. En general, la obtención de conocimien-to adicional es ciertamente el deseo de muchos científicos de formulación o de ingenieros de proceso, pero los estrictos tiempos en el trabajo rutinario del día a día generalmente no dan suficiente tiem-po para adentrarse más en la ciencia.
Mayeresse (GSK Biológicos): El papel de estabilizadores tales como azúcar, buffer, polímeros y surfactantes es ahora mejor entendido. No obstante, dependiendo de la complejidad de las moléculas, pueden darse algunas sor-presas durante el desarrollo que nece-sitarán corregirse. El conocimiento que surge de esto será recordado cuando se desarrollen nuevas moléculas. Es nece-sario detectar todas las especificidades de la molécula durante la primera etapa del desarrollo porque en esta etapa pue-de corregirse fácilmente sin impactar los resultados de los estudios clínicos. Los cambios de formulación en la últi-

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 9
ma etapa de un proyecto pueden ser un problema mayor.
Nail (Baxter Pharmaceutical So-lutions): No consideramos los cambios en la formulación en el contexto del ma-nejo del ciclo de vida; esto es, una vez que se ha establecido la formulación, no hacemos cambios. Sin embargo, du-rante el desarrollo del producto, hacer cambios sistemáticos de la composición es parte del desarrollo del espacio de di-seño en la formulación. Esto podría ser una gráfica de Tg’ contra pH, por ejem-plo. La idea es obtener un buen sentido de cómo los cambios en la composición afectan la conducta del producto. Para el examen de estabilidad, generalmente nos apoyamos en el análisis a corto pla-zo bajo condiciones de estrés.
Page/Steiner (GEA Pharma Sys-tems): En el pasado, las técnicas de control de calidad tendían a considerar y registrar sólo datos binarios de los atri-butos, tales como pasa no pasa/bueno o malo. Este enfoque está frecuentemente enraizado en conceptos antiguos de con-
trol de calidad por inspección del pro-ducto final. Pero si sólo se registran atri-butos de pasa/no pasa, se pierde mucha información útil acerca del parámetro que se está midiendo. Mediante la de-terminación y el registro de mediciones reales es posible aplicar simples técni-cas de control estadístico de proceso que pueden caracterizar la capacidad del proceso y dar advertencias de variabili-dad no pronosticada mucho antes de que el proceso llegue a un límite de especi-ficación. Por ejemplo, la medición de la merma dentro de un rango aceptable puede ser un indicador útil de que el proceso se está moviendo cada vez más cerca a un punto en donde puede ocurrir un nivel de colapso inaceptable.
Pikal (Universidad de Connecti-cut): Me gustaría pensar que hay ejem-plos reales de la industria implementan-do este asesoramiento y encontrando una respuesta entusiasta de la FDA. Sin embargo, no tengo ningún conocimiento directo de tales ejemplos. Sé, no obstan-te, que muchos procesos se corren más
allá de lo óptimo. Si éstos fueran optimi-zados o al menos mejorados utilizando buenas prácticas de secado por congela-miento existente, resultaría en procesos más cortos con absolutamente ningún incremento en el riesgo de pérdida de los atributos de calidad del producto. Ciertamente, en algunos casos, el ries-go de pérdida de los atributos de calidad del producto sería menos en el proceso rediseñado.
En el área de formulación, sabemos ahora formular al menos algunas proteí-nas de tal manera que se obtiene una es-tabilidad muy superior, con la cantidad de agregación desarrollándose durante el almacenamiento a temperatura am-biente en dos años siendo mucho me-nor que la agregación que se desarrolla durante dos años en almacenamiento refrigerado. Sin embargo, no he visto ejemplos de una compañía que refor-mule para reducir el nivel de agregados, en tanto que la ‘formulación actual’ cumple los requerimientos mínimos del producto. PT
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
7

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201210
Ima
ge
So
ur
ce
/ge
tt
y I
ma
ge
S
Reporte desde:
JapónJane Wan
Después de una serie de reformas guber-namentales que hacen un llamado a los participantes domésticos y extranjeros, el mercado farmacéutico japonés está ha-ciendo una reaparición. En marzo de 2012, Pfizer Japón, una subsidiaria de Pfizer EEUU, estableció una División de Enfermedades Raras con el objetivo de convertirse en un líder global en el tratamiento de enfermedades raras. El Instituto Genómico de Beijing (BGI), China en el Distrito de Yantian de Shenzhen, formó el BGI Ja-pón en Kobe, en septiembre de 2011, para incrementar su rango de socios y para realizar investigación conjunta con compañías japonesas. La Corporación Golden Biotechnology en Taipei, Taiwán, estableció una subsidiaria con base en Tokio en abril de 2011 para hacer salir sus suplementos de salud patentados. Y la lista continúa.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 11
El bien establecido mercado farmacéutico japonés conti-núa atrayendo un extenso interés e inversión extranjera. Alan Thomas, director de planeación de negocios y análisis en IMS Japón K.K., dice: “Un incremento en las enfermedades crónicas, tales como las relacionadas con la diabetes y las cardiovasculares, y el número de pacientes tratados dentro de estas enfermedades continúa viendo un acceso expandido para los farmacéuticos. Adicionalmente, un incremento en las áreas de enfermedad relacionadas con especialidades, tales como oncología y osteoporosis, conduce el crecimiento con un mayor uso de biológicos y farmacéuticos de especialidad. Con estas áreas de enfermedad que emergen y se expanden, el número de opciones innovadoras de tratamiento también está aumentando.”
La economía estancada de Japón y el envejecimiento de la población han acicateado al gobierno a rediseñar las políticas de salud que favorecen la entrada de empresas extranjeras. La agencia ha dado pasos para acortar el proceso de aprobación de fármacos y facilitar el acceso sencillo a menores opcio-nes de tratamiento. La mediana de aprobación de fármacos ha caído desde 22 hasta 15 meses en los últimos dos años; y el tiempo de aprobación para productos bajo prioridad de revisión ha caído desde 15 a 9 meses.
El gobierno está considerando un sistema de “uso com-pasivo” que permita que los pacientes seriamente enfermos usen fármacos que todavía no se han aprobado para uso en Japón. Este programa está pensado para pacientes que no han respondido a tratamientos estándar y en donde no se dispone de opciones domésticas. El gobierno también está buscando permitir a las aseguradoras absorber algunos de los costos en los que incurren los pacientes bajo este sistema. Japón ha de-dicado tiempo a examinar los estudios clínicos del sector y valorarlos también, y bajo las nuevas iniciativas, la cada vez mayor aceptación y uso de datos de estudios clínicos globales ha reducido los costos y acelerado los tiempos. El llamado
“triángulo clínico” comprendido por China, Corea y Japón, también ayuda a reducir el tiempo de desarrollo en Japón de-bido a los datos adicionales del estudio de que se dispone.
Adicionalmente, la extensión de la “prima para el desarrollo de nuevos fármacos y eliminación del uso fuera de las indica-ciones” es aplicable a las modificaciones de precio del Seguro Nacional de Salud (INH) establecidas en abril de 2010. Bajo este sistema, los fabricantes están estimulados para desarro-llar nuevos fármacos y proporcionar información adicional para los existentes cuando los productos son elegibles para una modificación de precios menor del NIH.
Recientemente, el gobierno cambió su enfoque de los fár-macos genéricos con el objetivo de incrementar la participa-ción en el mercado a 30% en el 2013 en un intento por hacer frente al sobrecargado sistema de salud del país. Para desva-necer la percepción común del público de que los fármacos genéricos son inferiores, los fármacos genéricos y sus ingre-dientes activos se colocan bajo un riguroso control de cali-dad. Se requiere que los fabricantes de genéricos suministren todas las potencias y formas farmacéuticas de la versión de marca. El Ministerio de Salud, Trabajo y Bienestar (MHLW) también requiere que los fabricantes suministren potencias de la misma forma farmacéutica que las de los productos de marca y que las versiones genéricas sean una pareja perfecta de los productos de marca.
Ranjith Gopinathan, gerente de programa de ciencias de la vida y prácticas de salud en Frost & Sullivan, agrega, “Fuer-temente respaldado por el gobierno, el mercado de genéricos está estimulado por iniciativas tales como procedimientos de registro relajados y proveyendo incentivos a los doctores que los prescriben por encima de los fármacos de marca.”
De manera interesante, la presencia extranjera ha redise-ñado las estrategias de negocios de los participantes domésti-cos. Jamie Davies, jefe de farmacéuticos y salud en Business Monitor International, dice, “Típicamente, las compañías farmacéuticas japonesas son conservadoras por naturaleza y se enfocan principalmente en el mercado doméstico y tie-nen exposición limitada en estados menos desarrollados. Sin embargo, el efecto dual de la expiración de la patente y la reducida productividad en investigación los ha forzado a ver cada vez más allá de sus fronteras para el crecimiento de las
Marque en la tarjeta de servicio al lector el No. 9

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201212
ventas. Como resultado, varios están buscando en mercados emergentes para generar nuevo crecimiento.”
La economía estancada de Japón y el envejecimiento de la población han ins-tado a establecer políticas que favore-cen la entrada de empresas extranjeras.
Las compañías domésticas han empezado a forjar fuertes vínculos con empresas internacionales y mercados extranjeros. Todas las empresas líderes de Japón obtienen alrededor del 40% de sus utilidades de los mercados extranjeros, principal-mente de EEUU, aunque la exposición a los mercados emer-gentes está aumentando, agrega Davies. En octubre de 2010, Takeda Pharmaceutical en Tokio anunció planes para formar alianzas con las compañías de la India para vender sus fárma-cos patentados y para ofrecer servicios básicos del negocio.
La búsqueda de M&A también ha cobrado impulso en Japón con la adquisición de Takeda de Nycomed, con base en Zurich, Suiza, por $13,700 mdd en septiembre de 2011. Takeda Farmacéutica Brasil firmó un convenio para adquirir Multilab Indústria e Comércio de Productos Farmacéuticos, con base en Río Grande do Sul, Brasil, a finales del segundo trimestre de 2012.
Desde luego, existen excepciones. Recientemente, Eisai, con base en Tokio, ha cambiado su enfoque de vuelta a Asia Oriental, citando el enorme potencial de la región, especial-mente ahora que el mercado japonés se está volviendo atrac-tivo nuevamente gracias a las iniciativas gubernamentales. De manera similar, Sawai Pharmaceutical en Osaka se está concentrando en el mercad japonés y explorando sociedades con compañías farmacéuticas más grandes para expandir su rango terapéutico.
Thomas agrega, “Desde la perspectiva de un portafolio, las compañías farmacéuticas japonesas pondrán su enfoque en los farmacéuticos de especialidad, oncología y platafor-mas biológicas. Esta actividad de adquisición ganará impulso como estrategia para la entrada al mercado y expansión del portafolio.” Aunque las empresas domésticas controlan el 66% de la participación del mercado farmacéutico japonés, el mercado sigue siendo atractivo para los grandes jugadores extranjeros por su menor riesgo y exposición en comparación con otros mercados. De hecho, una fuente de Bloomberg in-dica que las compañías prominentes lo están haciendo muy bien. Pfizer aumentó las ventas japonesas a $7,300 mdd el año pasado y las cifras de GlaxoSmithKline fueron hasta de 28% en el mismo año. Las compañías de gran escala están bien posicionadas debido a sus fuertes proyectos de última etapa y sus agresivos objetivos de lanzamiento para continuar creciendo los siguientes dos o tres años. Por otro lado, los jugadores a menor escala sin presencia en Japón pueden tener una opción para obtener licencias o asociarse para mantener el crecimiento.
Jane Wan es escritora independiente con base en Singapur
El 12 de septiembre de 2012, la asociación de Investigación Farmacéutica y Fabricantes de América (PhRMA) es co-anfitrión de un nuevo programa de premios nacionales en el Newseum, en Washington, DC, para honrar a los individuos y organizaciones que han contribuido significativamente para el avance del cuidado del paciente y la innovación médica en los Estados Unidos. Los nuevos premios anuales, llamados los Premios de Investigación y Esperanza, están reemplazan-do al Premio del Descubrimiento de PhRMA y este año, se concentrará en la lucha contra la Enfermedad de Alzheimer. La Fundación de Alzheimer de América, la Alianza Nacional para el Cuidado y la Asociación Nacional para el Cuidado en Casa y Hospicios son co-anfitriones del evento del 2012. Pharmaceutical Technology es un copartícipe de medios para los premios, los cuales se darán en las siguientes categorías, de acuerdo al PhRMA:
•El Premio de Investigación y Esperanza para la Investiga-ción Académica en el Alzheimer: Presentado a los indivi-duos o grupos de trabajo de la comunidad académica por la investigación excepcional en la búsqueda de una cura de la enfermedad de Alzheimer.•El Premio de Investigación y Esperanza para la Investiga-ción de la Industria Biofarmacéutica en el Alzheimer: Pre-sentado a individuos o grupos de trabajo de una compañía biofarmacéutica por la investigación excepcional en la bús-queda de una cura de la enfermedad de Alzheimer.•El Premio de Investigación y Esperanza para la Defensa del Paciente: Este premio se presenta para un individuo u organización que haya tenido un impacto significativo en el respaldo y defensa de temas legislativos o regulatorios de política de salud. Su trabajo ha incrementado la conciencia, y el financiamiento para la investigación o el acceso al cui-dado. El premio reconoce el involucramiento, el compromi-so y el logro para la lucha contra el Alzheimer.•El Premio de Investigación y Esperanza – Campeón Vo-luntario: este premio se le presenta a un individuo que haya afectado significativamente las vidas de los pacientes y/o de los cuidadores de una manera caritativa o humanitaria. El Campeón Voluntario reconoce a los individuos y cuidadores que se entregan sin egoísmos para ayudar a otros que están en necesidad.
“Esta ceremonia de premiación celebra la evolución de la investigación y el progreso hecho en la batalla contra dicha enfermedad tan compleja y devastadora. Aún así, a pesar de los enormes retos que los científicos biofarmacéuticos conti-núan enfrentando para luchar contra el Alzheimer, los logros que estaremos reconociendo nos dan esperanzas,” dijo John J. Castellani, presidente y CEO de PhRMA. “Esta es también una oportunidad para reconocer mejor a aquéllos individuos que han dedicado su vida a ayudar a pacientes que sufren de esta enfermedad tan debilitante.”Los ganadores del premio a la ciencia están siendo seleccio-nados por un comité especial de la Fundación de PhRMA. Los premios de defensa están siendo seleccionados por un comité formado por PhRMA y representantes del coanfitrión. Las entrevistas con los ganadores aparecerán en el número del Pharmaceutical Technology de diciembre de 2012. Para mayor información, visite el sitio web de Premios de PhRMA http://www.phrma.org/AWARDS.
–Angie Drakulick
Atención: DescubrimientoLos premios de investigación de PhRMA se centran en la enfermedad de Alzheimer

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 13Marque en la tarjeta de servicio al lector el No. 17

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201214
Los autores, parte del Consorcio Internacional sobre Innovación y Calidad en Desarrollo Farmacéutico, IQ Consortium), exploran y definen los esquemas y prácticas comunes de la industria cuando se aplican las GMPs en el desarrollo inicial. Un grupo de trabajo del consorcio tiene como objetivo desarrollar una serie de recomendaciones que pueden ayudar a la industria a identificar oportunidades para mejorar el tiempo para llegar a los primeros estudios en humanos y reducir los costos del desarrollo, manteniendo mientras tanto los estándares de calidad requeridos y garantizando la seguridad del paciente. Este artículo es el segundo de una serie y se enfoca a la validación del método en la primera etapa del desarrollo.
Donald Chambers está en ciencias analíticas en Merck Research Laboratories, Gary Guo está en IyD analítico en Amgen, Brent Kleintop* está en desarrollo analítico y bioanalítico en Bristol-Myers Squibb Co., Henrik Rasmussen está en desarrollo analítico en Vertex Pharmaceuticals, Steve Deegan está en Operaciones de Aseguramiento de Calidad en Abbott, Steven Nowak está en IyD Analítico NCE, IyD Farmacéutico Global en Abbott, Kristin Patterson está en IyD de mercados emergentes en GlaxoSmithKline, John Spicuzza está en desarrollo analítico en Baxter, Michael Szuluc está en desarrollo analítico en Bioden Idec, Karla Tombaugh está en IyD/Calidad de Comercialización, División de Manufactura de Merck, en Merck & Co., Mark D. Trone está en desarrollo analítico, moléculas pequeñas, en Millennium Pharmaceuticals, y Zhanna Yuabova está en desarrollo analítico, EEUU, en Boehringer Ingelheim Pharmaceuticals.
*A quien debe dirigirse la correspondencia.
El Consorcio Internacional sobre Innovación y Cali-dad en Desarrollo Farmacéutico (IQ) se formó en el 2010 como una asociación de más de 25 compañías farmacéuticas y de biotecnología con la misión de
lograr avances en estándares basados en la ciencia y mane-jados científicamente y en regulaciones para medicamentos en todo el mundo. En la edición pasada del Pharmaceutical Technology, se presentó un artículo el cual describe un pano-rama general de las recomendaciones consolidadas de IQs de las Buenas Prácticas de Manufactura (GMPs) en el grupo de trabajo (WG) para el Desarrollo Inicial (1). El centro de esta WG de IQ ha sido desarrollar esquemas recomendados sobre cómo aplicar las GMPs en las actividades de desarrollo del CMC en su fase inicial que abarcan desde la Fase I hasta la Fase IIa. Una premisa clave de las GMPs en el WG de Desa-rrollo Inicial es que las guías existentes de las GMPs para el desarrollo inicial son vagas y que una claridad mejor en la de-finición de las expectativas de las GMPs produciría un avance en la innovación en el desarrollo farmacéutico de moléculas pequeñas mejorando los tiempos del ciclo y reduciendo los costos, manteniendo mientras tanto la calidad apropiada del producto y garantizando la seguridad del paciente.
Una consecuencia de la ausencia de claridad que rodea las expectativas de las GMP en la fase inicial es que se ha variado la interpretación y aplicación de las guías existentes de las GMP a través de la industria, dependiendo de la propia cultura de la compañía individual y de la tolerancia al riesgo. Los debates internos dentro de una compañía han resultado con frecuencia en la aplicación inapropiada de las interpre-taciones conservadoras de “un tamaño único para todo” que se apoyan en las guías de la Conferencia Internacional de Armonización (ICH) que son más apropiadas para produc-tos farmacéuticos que se aproximan al punto de la solicitud de autorización para la comercialización. En muchos casos, la aplicación errónea de estas expectativas comerciales GMP del ICH durante el desarrollo clínico inicial no distinguen las diferencias en requerimientos entre el desarrollo inicial y el desarrollo en la última etapa (Fase IIb y más allá). Un ob-jetivo clave de este WG del IQ, por lo tanto, ha sido definir colectivamente en el desarrollo inicial –dentro de las prácticas industriales aceptables- algunas expectativas GMP que per-miten tener una flexibilidad apropiada que sea consistente con las guías y estatutos regulatorios existentes (2).
Postura oficial: GMPs en la fase inicial
GMPs para la validación de métodos en el desarrollo inicialPerspectiva de la Industria (Parte II) Donald Chambers, Gary Guo, Brent Kleintop, Henrik Rasmussen, Steve Deegan, Steven Nowak, Kristin Patterson, John Spicuzza, Michael Szuluc, Karla Tombaugh, Mark D. Trone, Zhanna Yuabova

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 15
Según se señaló en el artículo de introducción anterior, los esfuerzos de las GMPs en el WG de Desarrollo Inicial se han concentrado en las siguientes cuatro áreas de actividades: validación de métodos analíticos, especificaciones, manu-factura de productos farmacéuticos y estabilidad. El alcance inicial de estos esfuerzos ha estado limitado al desarrollo de fármacos de molécula pequeña el cual soporta los estudios clínicos Primero en Humanos (FIH) hasta la Fase IIa (Prueba de Concepto). Dentro de esta revista, se estarán publicando en los próximos meses una serie de artículos que describen un esquema recomendado para la aplicación de las GMPs en cada una de estas áreas. En la edición de este mes, los autores abogan por un enfoque de ciclo de vida para la validación de métodos, el cual es iterativo por naturaleza con el fin de alinearse con la evolución del proceso de manufactura y la expansión del espacio de conocimiento del producto.
Desde 2004 no se ha publicado una perspectiva colectiva de la industria farmacéutica sobre la validación del método analítico con respecto a la fase de desarrollo (3). La génesis del artículo del 2004 se presentó durante una serie de talleres patrocinados por el Grupo Técnico Analítico de Investigación Farmacéutica y Fabricantes de América (PhRMA) en sep-
tiembre de 2003. El artículo al que se hace referencia, resu-mió las recomendaciones para un enfoque en fases para la va-lidación del método para sustancias farmacéuticas de pequeña molécula y productos farmacéuticos en el desarrollo clínico inicial. Aunque se han publicado otras pocas revisiones so-bre las prácticas de validación del método (4), este artículo proporciona una perspectiva de la industria actual, con una amplia base, sobre los enfoques apropiados de validación del método durante las primeras fases del desarrollo del producto farmacéutico.
Esta amplia evaluación de la industria de la validación de métodos también reveló la necesidad de diferenciar cla-ramente el contexto de los términos de “validación” y “cali-ficación”. La calificación del método está basada en el tipo, propósito que se persigue, y comprensión científica del tipo de método en uso durante la experiencia del desarrollo inicial. Aunque no se usen para la liberación GMP de los materiales clínicos, los métodos calificados son métodos experimentales confiables que pueden ser usados para el trabajo de caracte-rización, como es el caso de los estándares de referencia y la predicción científica de la vida de anaquel.
También se presente una perspectiva sobre algunos re-
Tabla I: Resumen del enfoque propuesto para la validación de métodos para el desarrollo inicial y posterior.Tipo de método analítico
Identificación EnsayoImpurezas
(cuantitativa)Impurezas
(prueba límite)Pruebas físicas
Parámetro de validaciónPrimera fase
Última fase
Primera fase
Última fase
Primera fase
Última fase
Primera fase
Última fase
Primera fase
Última fase
especificidad
componente mayor + + + + + + + + + +
Degradación forzada + + + +
Impurezas conocidas/excipientes
+ + + + +
exactitud +1 +1 +3 +
Precisión
repetibilidad + + +3 + + +
Precisión intermedia + + +
reproducibilidad + +
Sensibilidad
Límite de cuantificación +3 +
Límite de detección + + +
Linealidad
componente principal +2 + +2 + +2 +
Impurezas + +3 +
rango + +
robustez del método
estabilidad de la solución
+ + + + + + + +
Variación del parámetro del método
+ + + + +
Nota: 1 se refiere al análisis del producto farmacéutico solamente. 2 es la linealidad derivada utilizando menos estándares que la validación de la última fase. 3 se determina utilizando el aPI como sustituto.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201216
cientes desafíos y estrategias de los métodos analíticos, como los métodos de impureza genotóxica, el uso de métodos ge-néricos, y métodos usados para el análisis de materiales de toxicología o muestras de estabilidad para determinar las con-diciones de almacenamiento etiquetadas, períodos de reanáli-sis y vida de anaquel de los APIs y productos farmacéuticos. El enfoque a la validación del método descrita en el presente se basa en lo que se consideran las mejores prácticas vigentes usadas por organizaciones de desarrollo que participan en el consorcio IQ. Adicionalmente, este enfoque contiene algunos aspectos los cuales representan nuevos esquemas científi-camente sólidos y apropiados que les podrían facilitar a los científicos de desarrollo para ser más eficientes sin compro-meter la calidad del producto o la seguridad del paciente. Se presentan estas mejores prácticas aceptables dirigidas cien-tíficamente para proveer orientación y un hito para los gru-pos de trabajo en colaboración de los científicos analíticos, colegas regulatorios y expertos en cumplimiento que están desarrollando estándares de prácticas a ser usadas durante las primeras etapas del desarrollo farmacéutico. Los puntos de vista expresados en este artículo se basan en la experiencia industrial acumulada de los miembros del grupo de trabajo IQ y no refleja la política oficial de sus respectivas compañías.
Parámetros del método en la primera fase que requieren validaciónEn el desarrollo inicial, uno de los principales propósitos de los métodos analíticos es determinar la potencia de los APIs y los productos farmacéuticos para asegurar que se entregue la dosis correcta en la clínica. Los métodos deben ser también indicadores de estabilidad, capaz de identificar impurezas y degradantes, y permitir la caracterización de los atributos cla-ve, tales como la liberación del fármaco, la uniformidad del contenido, y las propiedades relacionadas con la forma. Estos métodos son necesarios para asegura que los lotes tengan un perfil de seguridad consistente y acumular el conocimiento de los parámetros clave del proceso con el fin de controlar y asegurar una manufactura y biodisponibilidad consistente en la clínica. En las últimas etapas del desarrollo del fármaco, cuando los procesos están asegurados y necesitan ser trans-feridos a instalaciones de manufactura en todo el mundo, los métodos necesitan ser rentables, operacionalmente viables y adecuadamente robustos de manera que los métodos se com-porten consistentemente sin importar dónde son ejecutados. En la consideración del propósito de los métodos en el desa-rrollo inicial contra el posterior, los autores hablan en favor de que no es necesaria la misma cantidad de experimentos rigu-rosos y extensos de validación del método, según se describe en el ICH Q2 Validación Analítica, para los métodos usados para soportar el desarrollo del fármaco en la etapa inicial (5). Este enfoque es consistente con el ICH Q7 Buenas Prácticas de Manufactura, el cual defiende el uso de controles de la-boratorio científicamente sólidos (más que validados) para el API en los estudios clínicos (6). Adicionalmente, un proyecto de guía de la FDA sobre procedimientos y métodos analíti-
Postura oficial: GMPs en la fase inicial
cos está en favor de que la cantidad de información necesaria sobre la validación de procedimientos y métodos analíticos variará con la fase de la investigación (7).
La perspectiva de IQ con respecto a cuáles parámetros del método deben ser validados tanto para los métodos en prime-ra etapa como en última etapa se resume en la Tabla I. En esta tabla, los métodos de identificación se consideran aquéllos
que discriminan el analito de interés de los compuestos con estructuras similares (o disimilares) o de una mezcla de otros compuestos para asegurar la identidad. Esta categoría inclu-ye, aunque no está limitada a métodos de identificación que utilizan cromatografía de líquidos de alta resolución (HPLC), espectroscopía de infrarrojo con transformada de Fourier (FTIR), y Espectroscopía Raman. Los métodos de ensayo se utilizan para cuantificar el componente principal de interés. Esta categoría incluye, pero no está limitada al ensayo del fármaco, uniformidad de contenido, ensayo de contra-ión, ensayo de conservadores, y mediciones de disolución. Los métodos de impurezas se utilizan para la determinación de impurezas y degradantes e incluyen métodos para impurezas orgánicas, impurezas inorgánicas, productos de degradación y total de volátiles. Para diferenciar más esta categoría de métodos, se proveen recomendaciones por separado para los métodos cuantitativos y de prueba límite, los cuales miden las impurezas. La categoría de las “pruebas físicas” en la Tabla I puede incluir tamaño de partícula, distribución de la gota, patrón de rocío, rotación óptica y metodologías tales como Difracción con Rayos X y Espectroscopía Raman. Aunque se dan recomendaciones representativas de parámetros potencia-les a considerar para la validación de estas pruebas físicas, los parámetros específicos a ser evaluados probablemente difie-ran para cada tipo de prueba.
Cuando se compara el enfoque de validación del método señalado para el desarrollo inicial contra los estudios de vali-dación del método realizados para soportar los sometimientos del NDA y el control de los productos comerciales, los pará-metros que involucran los estudios inter-laboratorio (es decir, precisión intermedia, reproducibilidad y robustez) no se reali-zan típicamente durante el desarrollo en etapa inicial. Los es-tudios inter-laboratorio pueden ser reemplazados por evalua-ciones apropiadas de transferencia del método y verificados mediante los requisitos de aptitud del sistema que garanticen que el método se comporta según lo proyectado a través de los laboratorios. Debido a los cambios en las rutas de síntesis
No son necesarios experimentos rigurosos y extensos de validación de métodos para los métodos usados en el soporte del desarrollo del fármaco en la primera etapa.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 17
y las formulaciones, las impurezas y los productos de degra-dación formados pueden cambiar durante el desarrollo. En consecuencia, las sustancias relacionadas son con frecuenta determinadas utilizando el porcentaje de área, asumiendo que los factores de respuesta relativos son similares a los del API. Si se utiliza el mismo supuesto para realizar los análisis y en la evaluación y calificación de las impurezas toxicológicas, cualquier corrección posterior al nivel de impurezas es auto-correctiva y por lo tanto mitiga el riesgo de que los sujetos estén expuestos a impurezas no calificadas. Como resultado, típicamente no se realizan estudios extensos para demostrar el balance de masas durante el desarrollo inicial.
Además de un número menor de parámetros que se eva-lúan en el desarrollo preclínico e inicial, también es típico re-ducir el alcance de la evaluación de cada parámetro y el uso de criterios de aceptación más amplios para demostrar la aptitud de un método. Dentro del desarrollo inicial, el enfoque para la validación o calificación también difiere en lo que se está analizando, con expectativas más estrictas para métodos que soportan las especificaciones de liberación y de estabilidad clínica, que para los métodos cuyo objetivo es adquirir cono-cimiento de los procesos (es decir, análisis en proceso, y así sucesivamente). Sigue una evaluación de los requerimientos para los métodos de liberación y de estabilidad clínica. Las definiciones de cada parámetro están en las guías del ICH y no serán repetidas en el presente (5). La evaluación defendida permite un régimen de análisis reducido apropiado. Aunque IQ defiende el hecho de realizar la validación de los métodos de liberación y estabilidad como se presentan aquí, los deta-lles se presentan como un esquema general, con la compren-sión de que el número de replicados y criterios de aceptación pueden diferir sobre la base de caso por caso. Como tal, el siguiente esquema no pretende ofrecer una guía completa.
Especificidad. La especificidad provee típicamente el mayor desafío en los métodos de la fase inicial ya que cada componente a ser medido debe medirse como una sola en-
tidad química. Este reto es también cierto para los últimos métodos, pero se amplifica durante los métodos de la primera fase para el ensayo y las impurezas en que:
• El conocimiento químico con respecto a las sustancias relacionadas está limitado.• Con frecuencia hay un mayor número de sustancias relacionadas que en las rutas sintéticas comerciales.• Las sustancias relacionadas que necesitan ser cuantifi- cadas pueden diferir significativamente de lote a lote conforme la síntesis cambia y se introducen nuevas formulaciones.
Un enfoque común para demostrar la especificidad para el ensayo y el análisis de impurezas se basa en llevar a cabo descomposición forzada y experimentos de compatibilidad de excipientes para generar potenciales productos de degrada-ción y para desarrollar un método que separe los potencia-les productos de degradación, las impurezas del proceso, los excipientes del producto farmacéutico (cuando aplique), y el API. Notablemente, los requerimientos son menos estrictos para los métodos en donde las impurezas no están cuantifi-cadas tales como los métodos del ensayo o de la disolución. En estos casos, la especificidad se requiere sólo para el API.
Exactitud. Para los métodos usados en el desarrollo ini-cial, la exactitud generalmente es evaluada aunque típica-mente con menos replicados que los que se tendrían para un
Aunque los datos necesitan estar documentados, no se requieren reportes detallados del método y la validación para asegurar el cumplimiento en el desarrollo inicial.
Headquaters:SPAMI Via Molinella, 17 35017 Piombino Dese (PD) Italywww.optrel-ltd.com | www.stevanatogroup.com - [email protected]
Local Representative:PACK & PROCESSTels. 52 (55) 5536-0490 y 52 (55) [email protected]. Insurgentes Sur 605, Desp. 1106-B, Col. Nápoles, México, D.F. C.P. 03810
Tecnología de inspección Optrel:- Maquinaria automática y semi-automatica- Para frascos, ampolletas, cartuchos y jeringas- Para contenedores vacíos y llenos (Líquidos, polvos y liofilizados)- Sistema de detección de derrames disponible- Soluciones de maquinas individuales, en línea y combos

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201218
método destinado a soportar los estudios clínicos en última etapa. Para determinar el API en el producto farmacéutico, pueden realizarse experimentos de adición de placebo por triplicado en el 100% de la concentración nominal y deter-minar los recobros. Son aceptables recobros de 95 – 105% para métodos del producto farmacéutico (con especificacio-nes de 90 – 110% del etiquetado). Se requieren criterios de aceptación de la validación más cerrados para los productos farmacéuticos con especificaciones más cerradas. Para las impurezas, la exactitud puede ser evaluada utilizando el API como un sustituto asumiendo que el sustituto es indicador del comportamiento de todas las impurezas, incluyendo el mis-mo factor de respuesta. La exactitud puede ser realizada en el límite de la especificación (o el umbral de reporte) mediante la adición por triplicado. Los recobros de 80 – 120% se consi-deran por lo general aceptables, aunque dependerán del nivel de concentración de la impureza. Para las pruebas donde las mediciones se hacen en diferentes concentraciones (contra una concentración nominal), como el análisis de disolución, puede ser necesario evaluar la exactitud en más de un nivel.
Precisión. Para los métodos de la fase inicial, sólo se exa-mina la repetibilidad de la inyección y el análisis. La des-viación estándar relativa (DER) del % de área se determina típicamente con 5 replicados. La repetibilidad es determinada en el 100% de la concentración nominal para el API, siendo las impurezas evaluadas en el umbral del reporte utilizando el API como sustituto. El criterio de aceptación de 1% de DER (repetibilidad de la inyección) o de 2% de DER (repetibilidad del análisis) para el API son señalados con frecuencia. Para las impurezas, son aceptables límites de precisión más eleva-dos (p.ej., 10-20%) y deben considerar el nivel de la impureza que se está midiendo (repetibilidad de la inyección y del aná-lisis). Para las pruebas en las que las mediciones se hacen en diferentes concentraciones (contra una concentración nomi-nal), tales como el análisis de disolución, puede ser necesario evaluar la repetibilidad en más de un nivel.
Límite de detección y límite de cuantificación. Es ne-cesaria una evaluación de la sensibilidad para determinar el nivel al cual pueden observarse las impurezas. Utilizando el API como sustituto, puede hacerse una evaluación “práctica” demostrando que la señal de una muestra preparada en el um-bral del reporte produce una relación de señal a ruido de más de 10. Puede determinarse un límite de cuantificación a partir de esta evaluación, calculando la concentración que sería re-querida para producir una relación señal a ruido de 10:1. De manera similar, puede calcularse un límite de detección como la concentración que produciría una relación de señal a ruido de 3:1. Sin embargo, se hace énfasis en que el “límite de cuan-tificación práctico” al cual se verifica que el nivel más bajo de interés (umbral de reporte) proporciona una señal al menos 10 veces la del ruido y por lo tanto puede ser cuantificada, es de importancia primordial.
Linealidad. La linealidad puede determinarse de curvas de calibración de 3 puntos en concentraciones de la prueba de 70, 100 y 130% del nominal (API) para el ensayo o de 3
Postura oficial: GMPs en la fase inicial
puntos que vayan desde el umbral de reporte hasta 130% del límite de especificación para impurezas. El API se usa como el analito sustituto para las impurezas. Para ambos análisis, puede establecerse un criterio de validación como R2 > 0.995. Para las pruebas donde son necesarias las mediciones sobre rangos de concentración más amplios, tales como el análisis de disolución, puede examinarse un rango lineal más amplio utilizando una calibración de 3 puntos.
Rango. Al igual que para los métodos de la última fase, el rango se infiere de los estudios de exactitud, precisión y linealidad.
Robustez. El análisis completo de robustez no se reali-za durante el desarrollo inicial. No obstante, debe realizarse una evaluación de la estabilidad de la solución para demostrar el período de vida viable de los estándares y las muestras. Específicamente, las soluciones deben considerarse estables cuando se cumplen las siguientes condiciones:
• El ensayo del API cambia en no más de 2%• No se observa ninguna nueva impureza mayor que el umbral de reporte.• Las impurezas en el umbral de reporte cambian en no más de 30%; las impurezas en los niveles entre el umbral de reporte y el límite de la especificación cambian en no más de 20% y las impurezas en o por arriba del límite de especificación cambian en no más de 15%. De manera notable, si la validación se realiza concurren-
temente con el análisis de la muestra como una aptitud del sistema extendida, debe evaluarse la estabilidad de la solución por separado. Esta evaluación se realiza típicamente como parte del desarrollo del método.
Métodos en fase inicial que requieren validaciónDurante las discusiones que se sostuvieron para desarro-
llar este esquema para la validación de métodos en la fase inicial, fue evidente que el contexto de los términos “valida-ción” y “calificación” no era usado universalmente dentro de todas las compañías del IQ. Para facilitar un entendimiento común de este enfoque, los autores se referirán por lo tanto a “métodos validados” como aquéllos métodos que se compor-tan como se esperaba cuando se someten a la serie de pruebas analíticas descritas en este esquema. Los “métodos califica-dos” se considera que son métodos analíticos que se someten a estudios menos estrictos para demostrar que son científica-mente válidos para el uso que se pretende. En las siguientes secciones, los autores recomiendan qué tipos de métodos tí-picamente empleados en el desarrollo inicial requieren ya sea validación o calificación.
Métodos para análisis de liberación y para soportar la manufactura GMP. En el desarrollo inicial, se utilizan es-pecificaciones para controlar la calidad de los APIs y de los productos farmacéuticos. La consideración de las especifica-ciones le pone gran énfasis a la seguridad del paciente ya que el conocimiento del API o del proceso del producto farmacéu-tico está limitado debido al bajo número de lotes producidos en esta etapa del desarrollo. Las especificaciones típicamente

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 19
contienen un número de diferentes pruebas analíticas que de-ben realizarse para asegurar la calidad del API o del producto farmacéutico. Los atributos típicos del material, tales como la apariencia, la potencia, la pureza, la identidad, la unifor-midad, los solventes residuales, el contenido de agua, y las impurezas orgánicas/inorgánicas, se analizan contra criterios de aceptación establecidos. Los métodos específicos para el API y el producto farmacéutico para potencia, impurezas, uniformidad y otros deben ser validados según se describió anteriormente y que demuestren que son adecuados para el uso que se pretende en el desarrollo de la fase inicial antes de la liberación. Si se utilizan métodos farmacopeicos para ana-lizar contra una especificación (p.ej., FTIR para identificación y titulación por Karl Fischer [KF] para contenido de agua), éstos deben ser evaluados y/o calificados para que sean aptos para analizar el API o el producto farmacéutico antes de usar-lo sin validación. Los materiales usados en la manufactura de la sustancia farmacéutica y del producto farmacéutico GMP usados para los estudios clínicos en la fase inicial, para los cuales no están detalladas las especificaciones en un some-timiento regulatorio (p.ej., penúltimos, materiales de inicio, intermedios aislados, reactivos y excipientes) sólo necesitan estar calificados para su uso proyectado. La transferencia de métodos es menos rigurosa en esta etapa inicial de desarrollo y puede lograrse utilizando experimentos de covalidación o evaluaciones simplificadas.
Según se mencionó, la calificación del método se diferen-cia con frecuencia de la validación del método. Los experi-mentos para demostrar la calificación del método se basan en el propósito proyectado del método, la comprensión científica del método adquirida durante el desarrollo del mismo y el tipo de método. Este es un paso importante para asegurar que pueden generarse reproduciblemente datos confiables para nuevos fármacos en investigación en las etapas iniciales del desarrollo. Los métodos calificados no deben usarse para la liberación del API o del producto farmacéutico contra especi-ficaciones y estudios de estabilidad concurrentes. Sin embar-go, puede hacerse la caracterización del material de referencia con métodos calificados.
La generación del conocimiento del proceso en el desarro-llo inicial está evolucionando rápidamente. Numerosas mues-tras son analizadas durante el desarrollo inicial para adquirir conocimiento del producto en diversas etapas del proceso. Los resultados de estas muestras son sólo para información y los métodos usados para este tipo de análisis no se requiere que sean validados o calificados. Sin embargo, para asegu-rar la exactitud del conocimiento que se está generado, debe usarse un juicio científico válido para garantizar la aptitud de cualquier método analítico usado para propósitos informati-vos solamente.
Métodos “genéricos” o “generales. Una estrategia analí-tica común empleada con frecuencia en el desarrollo inicial es el uso de métodos genéricos o generales que se ajustan al pro-ceso para una prueba específica a través de múltiples productos (p.ej., cromatografía de gases para solventes residuales). Estos
métodos deben ser validados si se utilizan para probar con-tra una especificación establecida. El esquema sugerido para validar estos métodos en el desarrollo inicial se lleva a cabo típicamente en dos etapas. La etapa 1 involucra validar los pa-rámetros que son comunes para cada producto con el cual pue-de usarse el método. La linealidad de las soluciones estándar y la repetibilidad de la inyección pertenecen a esta etapa. La etapa 2 de la validación involucra la identificación de los pará-metros que son específicos para el producto individual, como la exactitud. La especificidad puede ser demostrada en la etapa 1 para atributos no relacionados con el producto y en la etapa 2 para atributos relacionados con el producto. La etapa 1 de va-lidación se presenta previa al análisis GMP. Este enfoque para la validación de métodos que se ‘ajustan al propósito’ puede aportar eficiencia para el desarrollo del fármaco conservando recursos en las primeras fases del desarrollo y puede asegurar la confiabilidad de la aplicación proyectada del método.
Métodos para GTI y calificación toxicológica del lote. La necesidad de métodos analíticos para demostrar el control de impurezas genotóxicas (GTI) se ha desarrollado reciente-mente debido a las expectativas y lineamientos provistos por las autoridades regulatorias (8, 9). Con frecuencia, estos mé-todos requieren alta sensibilidad con límites de cuantificación en el rango de partes por millón (ppm). Aunque los niveles de control para los GTIs (conocidos como umbral de problema toxicológico) es menos estricto para los estudios clínicos ini-ciales (p.ej., ingestión del paciente < 50 µg/día para estudios clínicos de <30 días contra 1.5 µg/día para estudios clínicos más prolongados), las autoridades regulatorias esperan que el control de las GTI sea demostrado durante el desarrollo ini-cial. Dependiendo de cuándo un GTI es generado potencial-mente durante una síntesis del API, las GTIs pueden estar lis-tadas en las especificaciones. La validación de estos métodos es nuevamente dependiente del uso proyectado del método. Los métodos usados para la evaluación pueden ser califica-dos a menos que sean usados para analizar contra una espe-cificación como parte de la liberación clínica. La calificación del método también se considera apropiada si el método está destinado para caracterización o liberación de los artículos de prueba para un estudio de toxicología.
Métodos para estabilidad de los APIs y productos far-macéuticos. Los lotes del API y del producto farmacéutico se exponen típicamente a condiciones de estrés acelerado y se analizan en intervalos programados para evaluar si se ha presentado alguna degradación. La vida de anaquel del API o del producto farmacéutico –esto es, el período de tiempo de almacenamiento en una condición específica dentro del cual la sustancia farmacéutica y el producto farmacéutico cumplen sus especificaciones establecidas, se basa en los datos analíti-cos generados a partir de estos estudios. Para esta aplicación, los métodos analíticos necesitan ser indicadores de estabili-dad (p.ej., capaces de detectar y cuantificar los degradantes) para garantizar la calidad, seguridad y eficacia de una sus-tancia farmacéutica y producto farmacéutico. Con frecuencia, los métodos analíticos para realizar pruebas de estabilidad son

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201220
los mismos métodos usados para analizar contra una especifi-cación para el análisis de liberación; estos métodos deben ser validados. Sin embargo, si se realizan pruebas adicionales que no están incluidas en la especificación establecida, pueden es-tar calificadas para su uso proyectado, en lugar de validarlas.
Métodos de análisis en proceso. El análisis en proceso (IPT) durante la manufactura de la sustancia farmacéutica y el producto farmacéutico puede hacerse en línea, dentro de la línea o fuera de la línea. Los resultados generados del IPT son usados para monitorear procesos que involucran el térmi-no de la reacción, la remoción de solventes, la remoción de impurezas y la uniformidad de contenido de la mezcla. Los parámetros de manufactura pueden ajustarse con base en los resultados del IPT. Los métodos de IPT son con frecuencia muy limitados en su alcance. En el desarrollo inicial, el be-neficio primario de llevar a cabo IPTs es la generación del conocimiento del proceso y no como un control o especifica-ción. Como resultado, aún cuando el IPT es esencial para la fabricación de la sustancia farmacéutica y del producto far-macéutico, la calificación de un método IPT es apropiada en el desarrollo en la fase inicial.
Documentación y otros requerimientos. El alcance de la documentación y las prácticas asociadas en el desarrollo ini-cial debe estar alineado con el nivel apropiado de validación del método según se discutió antes. En este artículo, los auto-res proveen una perspectiva sobre el nivel de documentación apropiado, el protocolo y la generación de criterios de acep-tación, la calificación de los instrumentos y la supervisión de la unidad de aseguramiento de calidad para la validación y calificación del método en la fase inicial. El enfoque les da a los científicos de desarrollo la flexibilidad para adaptarse efi-cientemente al entorno dinámico típico dentro del desarrollo farmacéutico en la fase inicial, asegurando mientras tanto la seguridad del paciente y la integridad científica del proceso de validación.
Con respecto a la documentación, la perspectiva de IQ es que los datos crudos que se generan durante la validación del método en la primera fase deben ser generados y mantenidos en un formato de datos compatible con el almacenamiento. La integridad de los datos crudos debe controlarse de tal ma-nera que puedan recuperarse para abordar futuras cuestiones técnicas y relacionadas con el cumplimiento. La documenta-ción apropiada de los datos y los experimentos de validación también debe considerarse como un aspecto importante de la validación en la fase inicial. La disponibilidad de los sistemas de libreta electrónica (ELN) ha proporcionado una alternativa viable, más eficiente a la del uso de libretas de papel tradi-cionales. En el desarrollo de las políticas para implementar ELNs, el objetivo no debe ser que todas las prácticas de do-cumentación usadas con libretas de papel sean replicadas. Más bien, el ELN debe poseer los suficientes controles para el uso proyectado de los datos. En muchos casos, los sistemas electrónicos tales como las ELNs transformarán el proceso de trabajo y los controles que proporciona se lograrán de manera completamente nueva en comparación con el sistema anticua-do al cual están reemplazando.
Postura oficial: GMPs en la fase inicial
Aunque los datos necesitan estar documentados como se describe anteriormente, es posición de los autores que no se requieran reportes formales detallados del método y la vali-dación para asegurar el cumplimiento en el desarrollo inicial. Necesitan estar en vigor los controles adecuados para asegu-rar que los parámetros del método, usados para ejecutar los métodos validados son equivalentes a los parámetros usados durante la validación. La generación de breves reportes su-marios del método y la validación se requieren sólo cuando son necesarios para completar los requisitos del sometimiento regulatorio o para tratar solicitudes o cuestiones de las auto-ridades de salud. Los resúmenes de validación no son reque-ridos para presentar todos los datos de validación, sino más bien un resumen de los estudios pertinentes suficientes para demostrar que el método está validado para cumplir los re-querimientos de su uso proyectado. Una vez que los reportes son generados y aprobados internamente, debe disponerse de procedimientos aprobados de control de cambios y seguirse para mantener un estado apropiado de control sobre la ejecu-ción del método y la disponibilidad del reporte.
Aunque la perspectiva de los autores es que necesita exis-tir un plan de validación para la validación de métodos para la fase inicial, la organización analítica podría considerar di-ferentes mecanismos para llenar esta necesidad. Por ejemplo, las guías internas o las mejores prácticas de documentación pueden detallar suficientemente los requerimientos de vali-dación de manera que no necesita generarse un plan de vali-dación por separado para cada método. En ausencia de dicha guía o procedimiento, podría documentarse un plan de valida-ción en una bitácora de laboratorio o ELN la cual incluya una breve descripción de los elementos de validación y los pro-cedimientos a ser evaluados. Los planes de validación deben garantizar que el método será apropiado para el uso a que se destina. El uso de estrictos criterios de validación dentro del plan de validación debe estar limitado en estas etapas iniciales del desarrollo. Los estudios de validación para los métodos de desarrollo inicial pueden ser realizados en instrumentos que sirven al propósito que están calibrados y mantenidos, aunque no necesariamente calificados o bajo estrictos estándares de control de cambios.
El papel del sistema de calidad farmacéutica y la supervi-sión de las prácticas de validación del método en fase inicial y la documentación es otra área a ser considerada. En la indus-tria farmacéutica, la gestión de calidad está supervisada por una “Unidad de Calidad” que califique y supervise activida-des en las áreas de materiales GMP tales como los controles de laboratorio. En la práctica, el tamaño y complejidad de la Unidad de Calidad que supervisa la manufactura GMP varía con base en el tamaño del fabricante y la etapa del desarrollo del fármaco. No obstante, los aspectos básicos de un sistema de calidad deben estar en funciones. En el desarrollo inicial, la postura de IQ es que, como los procesos de manufactura del API y del producto farmacéutico están evolucionando, los métodos analíticos no requieren todavía una validación completa como se indica en el ICH Q2. Similarmente, el sis-

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 21
tema de calidad implementado durante las fases iniciales po-dría considerar que los métodos analíticos en evolución son intrínsecos al trabajo que se está realizando para desarrollar los procesos finales del API y del producto farmacéutico y podrían permitir flexibilidad para implementar fácilmente los cambios al método durante el desarrollo inicial. Por ejemplo, la Unidad de Calidad debería delegar la supervisión para la aprobación del plan de validación, el control de cambios, la aprobación de desviaciones y reportes a los departamentos analíticos antes de la finalización y realización de la completa validación ICH Q2 de los métodos analíticos. Este enfoque sería consistente con el Capítulo 19 del ICH Q7A. Sin embar-go, los departamentos analíticos deben asegurar que los estu-dios de validación en fase inicial sean realizados por personal calificado con supervisión, quienes les den seguimiento a los procedimientos departamentales aprobados. Claramente, los acuerdos entre las Unidades de Calidad y los departamentos analíticos para implementar un sistema apropiado, estratégi-co, de supervisión de calidad basada en fases, aportarían mu-chos beneficios dentro de la industria.
ConclusionesDentro de este artículo, los representantes de IQ han presen-tado una perspectiva de la industria sobre los requerimientos y consideraciones apropiados para la validación de métodos analíticos en la fase inicial. Se ha presentado un esquema sugerido de experimentos aceptables que garanticen procedi-mientos analíticos, desarrollados para respaldar la producción del API y del producto farmacéutico para materiales clínicos en la fase inicial que sean adecuados para su uso proyecta-do. Adicionalmente, los autores han proporcionado una pos-tura sobre los enfoques por fases para otros aspectos de la validación de métodos tales como requerimientos de docu-mentación, generación de planes de validación de métodos, criterios de validación y el involucramiento estratégico de la supervisión de la unidad de calidad. Cuando se aplica apro-piadamente, este enfoque puede ayudar a asegurar que las or-ganizaciones de desarrollo analítico proporcionen controles analíticos apropiados para los procesos del API y del produc-to farmacéutico, los cuales servirán al objetivo último que es la seguridad del paciente. Aunque el alcance de los experi-mentos de validación de métodos en fase inicial es menor que el empleado en las últimas etapas del desarrollo, vemos que cualquier riesgo relacionado con este enfoque no se materiali-
zará, especialmente cuando se considera la calidad global y el esquema de seguridad usado por las compañías farmacéuticas para los estudios clínicos en fase inicial.
Es deseo de los autores que la aportación de dicho es-quema a la validación de métodos en etapa inicial, junto con los esquemas señalados en esta serie de artículos de GMP en fases iniciales, servirá como un trampolín para estimular las discusiones sobre estos esquemas dentro de la industria y con las autoridades de salud de todo el mundo. Para estimular más diálogo, este grupo de trabajo de IQ está planeando realizar un taller en el futuro cercano para promover un debate y dis-cusión robustos sobre estos esquemas recomendados para las GMPs en el desarrollo inicial. Estas discusiones permitirán, idealmente, un mejor alineamiento entre desarrollo de IyD, Calidad y organizaciones regulatorias de CMC a través de la industria farmacéutica, y, más importante, con las autoridades regulatorias del mundo. La concordancia entre la industria y las autoridades sanitarias con respecto a las prácticas acepta-bles para aplicar las GMPs en las fases iniciales del desarrollo de fármacos serán claramente beneficiosas para los científicos de desarrollo farmacéutico del CMC y permitirá un enfoque más ágil y flexible para abordar el entorno dinámico típico de las primeras fases del desarrollo clínico, garantizando al mis-mo tiempo controles apropiados para garantizar la seguridad del paciente durante el desarrollo inicial.
Referencias1. A. Eylath at al., Pharm. Technol. 36 (5) 54–58 (2012).2. FDA, Guidance for Industry: cGMP for Phase 1 Investigational
Drugs (July 2008 FDA). 3. S.P. Boudreau et al., Pharm. Technol. 28 (11) 54–66 (2004).4. M. Bloch, “Validation During Drug Product Development –
Considerations as a Function of the Stage of Drug Development,” Method Validation in Pharmaceutical Analysis, a Guide to Best Practice, Eds. J. Ermer, J.H. Miller (Wiley, 2005), pp. 243–264.
5. CH, Q2 (R1) Validation of Analytical Procedures: Text and Me-thodology (Nov. 2005).
6. ICH, Q7A Good Manufacturing Practice Guidelines for Active Phar-maceutical Ingredients (Aug. 2001).
7. FDA, Draft Guidance for Industry: Analytical Procedures and Me-thod Validation, Chemistry, Manufacturing and Controls Docu-mentation (Aug. 2000).
8. EMA, Guideline on the Limits of Genotoxic Impurities (June 2006).9. FDA, Draft Guidance for Industry: Genotoxic and Carcinogenic
Impurities in Drug Substances and Products: Recommended Appro-aches (2008). PT
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486
Fax: 52 (55) 5659-8879E-mail: [email protected]
Y APROVECHE NUESTRAPROMOCIÓN ESPECIAL
CONTRATE ANTES DEL 7 DE FEBRERO

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201222
La evaluación de compuestos dirigidos para usarse en una formulación oral sólida convencional, co-múnmente emplean estrategias de modificación del fármaco que están enfocadas en mejorar la so-
lubilidad del mismo. Esto se basa en el reconocimiento de que el fármaco debe estar en un estado de solución para ser absorbido a través del tracto gastrointestinal (GI). Maximizar la cantidad de fármaco que se disuelve y permanece disuelto conforme viaja a través del tracto GI proporciona una mayor probabilidad de que sean maximizadas la absorción y la bio-disponibilidad del fármaco.
Las primeras fases del desarrollo están con frecuencia acompañadas de esfuerzos para establecer el perfil de las pro-piedades fisicoquímicas del fármaco e intenta medir su velo-cidad de disolución y la conducta de solubilidad para modelar el desempeño in vivo del fármaco (1). Se han utilizado herra-mientas predictivas para desarrollar un entendimiento gene-ral de estos atributos (2, 3). Las mediciones empíricas (p.ej., solubilidad cinética y velocidad de disolución intrínseca), sin embargo, continúan siendo un pilar. Los datos que revelan solubilidad/velocidad de disolución pobre o sub-óptima no necesariamente evitan el avance de un fármaco. Sin embargo, una estrategia de avance con frecuencia necesitará explorar un modo de entrega alternativa o la modificación de la sus-tancia farmacéutica como un medio de mejorar la solubilidad y la biodisponibilidad. Lo segundo es a menudo el esquema más sencillo y rentable. Las opciones de que se dispone para considerar requieren la comprensión de cómo se comporta la molécula (p.ej., solubilidad y precipitación) en diferentes re-giones del tracto GI. Algunos conocimientos sobre los niveles de dosis de la formulación que se pretende también pueden ser valiosos en la evaluación de las opciones potenciales.
Cuando se combina con el conocimiento del pKa del com-
puesto y su solubilidad intrínseca, se vuelve por lo general más claro si una sal puede ser usada para permitirse una me-jora en las propiedades que pueden llevar a una mejora en la biodisponibilidad (4). Para moléculas que tienen el potencial de formar sales estables, los estudios de selección de la sal son típicamente el esquema más efectivo en la medición de un amplio rango de ácidos, bases y condiciones de cristalización. La selección también puede lograrse en una manera de mate-rial eficiente, una consideración siempre importante durante el desarrollo inicial.
Puede requerirse la modificación de la forma sólida de una sustancia farmacéutica para los compuestos farmacéuticos que muestran escasa solubilidad y biodisponibilidad. Los autores describen una plataforma de tecnología de formas sólidas, utilizada para optimizar la selección de la sal, la identificación de la co-cristalización y la modificación, o el desarrollo de una forma libre.
Desarrollo de la forma sólida óptimaDavid Igo y Stephen Carino
sección esPecial: aPis, exciPientes y Manufactura
Química DeL esTaDo sóLiDo
ca
taLe
Nt
Ph
ar
ma
So
Lut
IoN
S
David Igo*, PhD, es director y Stephen Carino, PhD, es científico principal, ambos en Optiform Technologies, Catalent Pharma Solutions, 160 Pharma Drive, Morrisville, NC, [email protected].
*A quien debe dirigirse la correspondencia continúa en la pág. 28

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 23
sección esPecial: aPis, exciPientes y Manufactura
DuraLac®.Lactosa anhidra ahora disponible. De Meggle.
– Perfectamente adecuada para la compresión directa y formulaciones de granulación seca
p p– Excipiente de elección para formulaciones con p pprincipios activos sensibles a la humedad
– Excelente rendimiento de dureza a baja fuerza de compactación
– Humedad estable debido a la relativamente baja higroscopicidad
DuraLac® de MEGGLE: Lactosa anhidra de calidad conocida.
Inmediatamente eficaz, MEGGLE también ofrece Inmedi tlactosa anhidra para una amplia variedad dep ¡ paplicaciones farmacéuticas. ¡Optimice su for-
pmulación! Los beneficios hablan por sí solos.Y por DuraLac®:
MEXALC, S.A. DE [email protected]. (52 55) 5616 0668; 5616 1651
MEGGLE HEAD OFFICE:[email protected].: +49 [0]807 173 476
Marque en la tarjeta de servicio al lector el No. 16

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201224
recubrimienTo De TabLeTas
sección esPecial: aPis, exciPientes y Manufactura
El recubrimiento de tabletas involucra la selección de un color y brillo específicos que se usan para distinguir un producto y construyen la identidad de la marca para ese producto. Además de la cuestión del color y el brillo, el recubrimiento debe tener las propiedades de deslizamiento de superficie apropiadas y adherirse adecuadamente a la tableta. El proceso de recubrimiento y los materiales usados en el sistema de recubrimiento pueden optimizarse utilizando un esquema de calidad por diseño (QbD) para cumplir estos requisitos. Los autores describen un estudio de QbD que se realizó para optimizar un sistema de recubrimiento.
Los atributos de calidad críticos (CQAs) del recubri-miento de las tabletas son el color, el deslizamiento, el brillo y la adhesión. Un estudio de calidad por diseño (QbD) que utilice un diseño estadístico y el
análisis de los datos, pueden identificar los parámetros críti-cos del proceso (CPPs) necesarios para obtener los deseados CQAs.
Los CPPs incluyen tanto la composición del material (es decir, la fórmula) como los parámetros en el proceso de recu-brimiento. En este estudio, se utilizó el despliegue de la fun-ción de calidad (QFD) para identificar los efectos esperados de la composición del material sobre los CQAs. El QFD pue-de ser usado para minimizar el número de experimentos, lo cual limita el error experimental. Con base en el QFD que se muestra en la Tabla I, la cantidad de dióxido de titanio (TiO2
) y un colorante rojo fueron identificados como los CQAs para el color, y se determinó que no era necesario un diseño de la mezcla para probar el efecto de la composición del mate-rial sobre el color. En lugar de esto, se seleccionó un factorial completo de tres factores con un punto central para el efecto de color como un diseño de experimento (DOE) práctico.
Se estudiaron las composiciones del sistema de recubri-miento. El nivel de colorante rojo (Rojo 40 Lacas, 12-14% concentrado) y el nivel de TiO
2 se variaron para establecer
el espacio de diseño para el efecto de color. Los niveles del triglicérido de cadena media (MCT) se ajustaron para man-tener el balance de masas. La Tabla II lista la temperatura y las variables del proceso de velocidad de la aspersión usadas para probar cada una de las nueve composiciones del sistema de recubrimiento.
Un lote de placebo de 1200 g fue recubierto con cada uno de los nueve sistemas de recubrimiento. Después del recu-brimiento por aspersión, se colectaron las muestras con un aumento de peso de 3 y 5% y se hicieron girar en el bombo de recubrimiento durante 5 minutos más. El porcentaje de au-mento de peso se basó en el consumo teórico de la solución de recubrimiento y no se consideró para la eficiencia del re-cubrimiento. Se llevaron a cabo un total de 45 experimentos de recubrimiento y se colectaron 90 muestras. Las muestras se analizaron, con cinco replicados, para el color, el brillo, el deslizamiento y la adhesión.
Calidad del colorSe utilizó la colorimetría para medir el matiz del color. El valor L* es el espacio de color blanco/negro. El valor a* es el espa-
Eric Van Ness* es gerente de soporte técnico, Beverly Schad es gerente técnica de ventas, Thomas Riley es gerente de formulaciones y Brian Cheng es científico de formulaciones, todos ellos en Sensient Pharmaceutical Coating Systems, Saint Louis, MO, tel. 314.286.7135, [email protected]
*A quien debe dirigirse la correspondencia
Logrando la comprensión y control del proceso en el recubrimiento pelicularUna estrategia de calidad por diseño Eric Van Ness, Beverly Schad, Thomas Riley y Brian Cheng
Foto
gr
aF
IaB
aS
Ica
/Ve
tta
/ge
tt
y Im
ag
eS

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 25
sección esPecial: aPis, exciPientes y Manufactura
cio rojo/verde y el valor b* es el espacio azul/amarillo. Los valores objetivo L*, a* y b* se listan en la Tabla III. La di-ferencia total de color (∆E*) se calcula a partir de las diferencias entre los valores L*, a* y b* y el objetivo, tomando la raíz cuadrada de la suma de las diferencias al cuadrado. Las especificaciones para los valores L*, a* y b* se definieron con base en una ∆E* < 2.0.
El total de las 450 muestras se ana-lizaron por regresión para detectar los factores que tenían un valor L* signi-ficativo. El análisis de los datos reveló que los seis factores (es decir, el nivel de dióxido de titanio, el colorante rojo, y el MCT en el sistema de recubrimien-to, y el porcentaje de aumento de peso, la temperatura de desfogue del recubri-miento y la velocidad de aspersión) te-nían un efecto significativo sobre el va-lor L*. El modelo de regresión tuvo una R2 (ajustada) de 92.10% y un valor p de la carencia de ajuste de 0.000. El efecto relativo de las variables se muestra en la Figura 1. El análisis de los datos reveló que el TiO
2 y el colorante rojo tuvieron
el efecto más significativo; tanto la tem-peratura de desfogue como la velocidad de aspersión también tuvieron un efecto significativo sobre L*.
Se llevó a cabo un análisis de regre-sión similar para identificar los factores que pueden tener un efecto significativo sobre el valor a*. El análisis de los da-tos reveló que los mismos seis factores tuvieron un efecto significativo sobre el valor de a*. El modelo de regresión tuvo una R2 (ajustada) de 91.57% y un valor p de carencia de ajuste de 0.000. El efecto relativo de las variables se muestra en la Figura 2. El análisis de datos reveló que los niveles de TiO
2 y
del colorante rojo tuvieron el efecto más significativo; tanto la temperatura de desfogue como la velocidad de as-persión también tuvieron un efecto sig-nificativo sobre el valor de a*.
Finalmente, se realizó un análisis de regresión similar para identificar los factores que pueden tener un efec-to significativo sobre el valor de b*. El análisis de datos reveló sólo cuatro fac-tores (es decir, los niveles de TiO
2 y de
colorante rojo, porcentaje de aumento de peso y temperatura de desfogue del recubrimiento) tuvieron un efecto sig-nificativo sobre el valor b*. El modelo de regresión tuvo una R2 (ajustada)
Tabla I: Despliegue de la función de calidad para los efectos del material sobre las propiedades de recubrimiento (Spectrablend II Pink, Sensient Pharmaceutical Coating Systems).
MaterialEfecto sobre el color
Efecto sobre el brillo
Efecto sobre el deslizamiento
Efecto sobre la adhesión
Dióxido de titanio --- --- - 0
Lacas rojo 40 +++ 0 0 0
triglicéridos de cadena media (mct)
0 +++ +++ 0
Los símbolos indican el efecto relativo: altamente negativo (---), ligeramente negativo (-), neutro (0), positivo (+), y altamente positivo (+++).
Tabla III: Valores de colorimetría blanco(Spectrablend II Pink, Sensient Pharmaceutical Coating Systems).
L* a* b* ∆E*
Objetivo 77.43 27.09 7.56 0
Límite inferior 77 25.5 6.5 2.0
Límite superior 78.4 28.5 8.5 2.0
L* es el espacio blanco/negro, a* es el espacio rojo/verde, b* es el espacio azul/amarillo, Δe* es la diferencia de color total.
Tabla II: Condiciones de recubrimiento experimental.Temperatura (°C) Velocidad de atomizado (g/min)
40 30
40 10
50 30
50 10
45 20
Lacas rojo 40 (%)
1.65
0 10 20 30 40 50 60
Efecto estandarizado
Térm
ino
Dióxido detitanio (%)
Temperaturade desfogue (°C)
Triglicéridos decadena media (%)
Aumentode peso (%)
Velocidad deatomizado (g/min)
Figura 1: Diagrama de Pareto de los efectos estandarizados de las variables experimentales en el valor L*, el cual describe el espacio de color blanco/negro, Alfa = 0.10.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201226
de 86.67% y un valor p de carencia de ajuste de 0.000. El efecto relativo de las variables se muestra en la Figura 3. El análisis de datos reveló que los niveles de TiO
2 y del colorante rojo tuvieron el
efecto más significativo; tanto la tempe-ratura de desfogue como la velocidad de aspersión también tuvieron un efecto significativo sobre el valor de b*.
Se calculó una optimización de los factores dirigidos para el valor L*, a*, y b* utilizando análisis multirregresional. El material óptimo y los factores de pro-ceso se destacan en rojo en la Figura 4, la cual muestra que el nivel de coloran-te tuvo un efecto más grande sobre el matiz del color en comparación con los
sección esPecial: aPis, exciPientes y Manufactura
Figura 2: Diagrama de Pareto de los efectos estandarizados de las variables experimentales en el valor a*, el cual describe el espacio de color rojo/verde,Alfa = 0.10.
Figura 3: Diagrama de Pareto de los efectos estandarizados de las variables experimentales en el valor b*, el cual describe el espacio de color azul/amarillo, Alfa = 0.10.
1.65
Térm
ino
0 10 20 30 40 50 60Efecto estandarizado
Lacasrojo 40 (%)
Dióxido detitanio (%)
Temperaturade desfogue (°C)
Aumentode peso (%)
1.65
Térm
ino
0 10 20 30 40Efecto estandarizado
Lacas rojo 40 (%)
Dióxidode titanio (%)
Temperaturade desfogue (°C)
Aumentode peso (%)
efectos del proceso (p.ej., temperatura de desfogue y velocidad de aspersión). Para la solución optimizada en 3% de aumento de peso, los valores pronos-ticados de L*, a* y b* fueron 75.86, 32.21 y 9.07, respectivamente. La ∆E* pronosticada fue de 0.0, lo cual es una combinación perfecta. También se cal-culó una especificación del proceso del TiO
2 y del colorante rojo con base en
los límites inferior y superior de ambos ingredientes. El área blanca de la gráfi-ca en la Figura 4 es el nivel permisible del ingrediente de color que cumpliría la ∆E* < 2.0. Puede determinarse una diferente especificación del color utili-zando el mismo espacio de diseño.
Calidad del brilloEl brillo es una medida de la proporción de luz que tiene un reflejo especular desde la superficie. Una superficie lisa tiene un alto brillo, mientras que una superficie rugosa tiene menos, debido a que la luz reflejada es difusa. Los altos porcentajes de sólidos en la mezcla seca pueden hacer una superficie rugosa que puede afectar el brillo. Las condiciones del proceso, tales como la temperatu-ra de recubrimiento y la velocidad de atomización, también pueden tener un efecto significativo sobre la reología de la superficie de la película, lo cual pue-de afectar el brillo. Se utilizó un medi-dor de brillo novo-curve para medir el brillo del recubrimiento pelicular en la tableta con 60° de reflexión de la luz.
El análisis de regresión mostró un modelo débil con R2 (ajustada) de 40.45% aún cuando el valor p de ca-rencia de ajuste fue de 0.000. Un bajo R2 (ajustado) significa que otros fac-tores significativos no fueron tomados en cuenta o que el proceso no está en control. Un modelo débil resultó en una baja previsibilidad. Sin embargo, se sabe que el brillo puede ser inducido por el peso del lote y el tiempo de gira-do del bombo después de completar el atomizado.
El resumen estadístico de los datos del brillo mostró un amplio rango de re-sultados del brillo. Se utilizó el análisis de variables (ANOVA) para deducir el efecto principal. Con base en el resulta-do del ANOVA, los altos niveles de TiO2
y de colorante rojo tuvieron un efecto negativo en el brillo. El alto porcentaje de aumento de peso tuvo un efecto po-sitivo en el brillo. La alta temperatura y la alta velocidad de atomizado tuvieron un efecto positivo en el brillo pero no tuvieron efecto arriba de una tempera-tura de desfogue de 45°C y una velo-cidad de atomización de 20 g/min. Se espera que el alto por ciento de sólidos cause mayor rugosidad, la cual puede afectar la reflexión de la luz. El nivel de MCT no mostró un efecto significativo sobre el brillo. El requerimiento para la igualación de color restringe los ni-veles de TiO2
y colorante rojo, pero se recomienda una temperatura mínima de desfogue del recubrimiento de 45°C y una velocidad de aspersión de 20 g/min para lograr el brillo máximo.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 27
sección esPecial: aPis, exciPientes y Manufactura
Figura 4: Gráfica de contornos de mediciones de calorimetría (valores L*, a* y b*) en relación con las concentraciones del colorante rojo y del dióxido de titanio en la formulación. Las variables que se mantuvieron constantes fueron el aumento de peso (3%), concentración del triglicérido de cadena media (7.0%), temperatura de desfogue (40°C) y velocidad del atomizador (10 g/min).
Dióxido de titanio
Col
oran
te ro
jo
1.00%
0.90%
0.80%
0.70%
0.60%
0.50%10.00% 12.00% 14.00% 16.00% 18.00% 20.00%
L*7576
a*31.833
b*8.59.5
L* (espacio blanco/negro)a* (espacio rojo/verde)b* (espacio azul/amarillo)
Calidad del deslizamientoEl deslizamiento (es decir, la fricción con la superficie) del recubrimiento pe-licular puede ser medido utilizando un analizador de textura. Se midieron cin-co tabletas para cada experimento (es decir, un total de 2250 muestras) para obtener una justificación estadística, y los datos se analizaron por regresión. La R2 (ajustada) de 68.80% fue mar-ginalmente aceptable. El análisis de regresión indicó que la velocidad de aspersión, la temperatura de desfogue, y el nivel de MCT también tienen un efecto significativo sobre el desliza-miento. El modelo de regresión pronos-ticó que podía alcanzarse una fuerza de deslizamiento mínima de 27.56 g con la composición de material y condicio-nes de proceso recomendadas. Como la composición del material debe mante-nerse constante para igualar el color ob-jetivo, la respuesta óptima se ajustó uti-lizando el aumento de peso y los niveles de dióxido de titanio y colorante rojo.
Calidad de la adhesiónLa calidad de la adhesión se determinó midiendo la fuerza de tensión en una tableta. Se colocaba una tableta en una pieza de cinta adhesiva por los dos la-dos, adherida a la parte superior de la plataforma plana del analizador de tex-
tura. Otro pedazo de cinta se colocaba plana a la parte de abajo de una sonda cilíndrica de acero inoxidable, de 25 mm, se comprimía a una fuerza de 800 g sobre las tabletas durante 10 s, des-pués se separaba a 1 mm/s. El análisis de regresión tuvo una R2 (ajustada) de 59.04%, lo que sugirió factores no to-mados en cuenta o error experimental alto. La distribución de los datos para los datos de adhesión fue una distri-bución no Gaussiana, la cual se ajustó como una distribución de Weibull de 3 parámetros. La probabilidad de fallo basada en el espacio de diseño se esti-mó que es baja (0.94%), lo cual indicó que la adhesión no era sensible a los factores del material (es decir, TiO
2,
colorante rojo y niveles de MCT) o fac-tores de proceso (es decir, temperatura de desfogue y velocidad de atomizado).
Hoja de ruta de QbDUn ejercicio típico de QbD es detallar el diagrama de flujo de proceso y construir el mapeo del proceso para identificar las variables clave de entrada del proceso (KPIVs) y las variables clave de salida del proceso (KPOVs). La importan-cia relativa de las KPIVs y las KPOVs fue evaluada con una matriz de causa y efecto (C&E). Las KPOVs con las ma-yores puntuaciones (p.ej., en el percen-
til superior 20o.) fueron consideradas CQAs para las cuales necesitaban de-finirse las especificaciones del proceso. El resultado de la matriz C&E mostró que la uniformidad del mezclado antes y después de la adición del MCT era crítica. La uniformidad del mezclado antes de la adición del MCT determinó la uniformidad del mezclado después de la adición del MCT. Por lo tanto, sólo hubo un CQA en el proceso de mezclado. La sensibilidad de la detec-ción necesaria para determinar la uni-formidad del mezclado se basó en la de-tección visual obteniendo un retiro de la mezcla. Se recomienda detección visual bajo aumento, la cual puede además asegurar la calidad de la uniformidad. De la matriz C&E, los KPOVs con altas puntuaciones (es decir, carga de materia prima y verificación de uniformidad de mezclado de la muestra) fueron defini-das como los CPPs. Es importante con-trolar los CPPs para alcanzar la calidad deseada. Se llevó a cabo un análisis del efecto en modo de fallo (FMEA) con base en los CPPs identificados.
QbD del recubrimientoCon base en la evaluación del FMEA, se generó el número de prioridad del ries-go (RPN), y la carga de la materia prima tuvo un alto RPN. Se desarrollaron pla-nes de acción correctiva para controlar el procedimiento de carga de la materia prima y asegurar la exactitud y ejecu-ción del procedimiento de producción. Similarmente, el paso de retiro también fue un CPP. El RPN no fue tan severo en comparación con el paso de carga de la materia prima y también se propuso un procedimiento de control del proce-so. La evaluación del C&E indicó que un Spectrablend II rosa completamen-te disperso en agua era un CQA. Este CQA podía ser controlado mediante examen visual. Todos los KPIVs fueron CPPs. El proceso de recubrimiento tuvo seis variables de proceso (es decir, tem-peratura de recubrimiento, velocidad de atomizado, velocidad del bombo, pre-sión de atomización, patrón de AIR, y flujo de aire).
Efecto relativo del recubrimientoLa evaluación de la C&E sugirió que la eficiencia del recubrimiento, la eficien-cia del secado, y la calidad del recubri-

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201228
miento eran los CQAs. La eficiencia del secado es una observación visual durante el recubrimiento y depende de la experiencia del operador. La calidad del recubrimiento puede ser medida cuantitativamente por colorimetría y otras pruebas reológicas. La evaluación de C&E sugirió que el total de los seis KVIPs tenían un efecto significativo. Se llevó a cabo una evaluación de FMEA.
FMEA para el proceso de recubrimientoLa carga de las tabletas, el flujo de aire,
sección esPecial: aPis, exciPientes y Manufactura
la temperatura de recubrimiento, la ve-locidad de aspersión y la velocidad del bombo necesitaban ser controladas. La cantidad de carga de las tabletas debía ser maximizada de acuerdo al diseño del bombo de recubrimiento. El flujo de aire estaba correlacionado con la temperatura de recubrimiento y debía ajustarse a la configuración óptima para estabilizar la temperatura de re-cubrimiento. La velocidad del bombo debía ajustarse para tener un flujo ideal sin excesivas volteretas que causaran astilladuras de las tabletas o del recu-brimiento. El efecto de la temperatura
de recubrimiento y de la velocidad de atomización estuvo más sujeto al mate-rial de recubrimiento según se describió previamente.
ConclusiónLos atributos de calidad críticos, tales como el color, el matiz, el brillo, el deslizamiento y la adhesión, pueden controlarse mediante el material apro-piado y la especificación del proceso. Un enfoque de QbD es una herramienta práctica para comprender y controlar la calidad y el proceso. PT
sección esPecial: aPis, exciPientes y Manufactura
Los estudios de selección de sal realizados utilizando una plataforma de tecnología de formas sólidas (Optiform, Cata-lent Pharma Solutions) permiten que sean usados múltiples elementos diversos en una operación de alto rendimiento, la cual a su vez maximiza la probabilidad de producir y descu-brir sales cristalinas. Los elementos de diversidad importantes a ser considerados cuando se hace la selección de las sales y disponibles con la plataforma Optiform incluyen estequiome-tría ácido-base, el método de dosificación de fármaco (p.ej., dosificación del fármaco como un sólido o una solución), el método de dosificación de contra-ión (p.ej., como sólido o como solución), la selección del solvente de cristalización y el modo de cristalización (p.ej., enfriamiento y adición de antisolvente). La evaluación inicial de la solubilidad del compuesto original, así como la posibilidad de generar sa-les cristalinas de clorhidrato o sodio en una pequeña serie de experimentos ofrece información de los solventes poten-ciales que pueden ser usados y proporcionan una guía sobre el método de cristalización apropiado. El mezclado efectivo combinado con un formato de sal/vial que permite que las muestras individuales sean seleccionadas del arreglo, aisladas y caracterizadas utilizando aislamiento y análisis de alto ren-dimiento proporcionan una metodología de selección rentable en material y tiempo.
Para las moléculas que son ya sea no ionizables o que no forman sales estables, los co-cristales con frecuencia se consideran un medio de entregar un material cristalino que tiene el potencial de mejorar la biodisponibilidad en compara-ción con la forma libre del compuesto. Similar a la selección de la sal, la identificación de un co-cristal adecuado requiere la combinación del API con una cantidad estequiométrica de un segundo compuesto que sea tanto un sólido a temperatura ambiente y generalmente considerado como seguro (GRAS). Estas combinaciones pueden ser efectivamente construidas utilizando la tecnología de plataforma Optiform. Las combi-naciones estequiométricas están sometidas a estudios de ma-durez basados en pastas acuosas, molido con gotas de solven-te, y/o experimentos basados en solución como un medio de promover la formación de co-cristales. Los experimentos de madurez y basados en solución pueden estar soportados, en
su totalidad por la plataforma Optiform. El molido con gotas de solvente de alto rendimiento, no obstante, requiere el uso de equipo de molido especializado. Las técnicas de molido de alto rendimiento se han asociado con la plataforma Optiform para mostrar la eficiencia de la combinación en la realización de estudios de co-cristalización (5).
Si no puede obtenerse con éxito ninguna modificación de sales ni de co-cristales, entonces puede modificarse la forma libre del compuesto para impartir un cambio en la solubili-dad y velocidad de disolución y, en correspondencia, en la biodisponibilidad del compuesto. Esta modificación puede lo-grarse mediante la reducción del tamaño para incrementar el área de superficie a través de diversos medios, incluyendo el molido mecánico bajo condiciones ambientales o criostáticas. La conversión del estado cristalino altamente ordenado a una forma sólida que tiene un orden bajo (es decir, escasamente cristalina) o de largo rango negligible (es decir, amorfa) en un esquema generalmente considerado para mejorar la biodispo-nibilidad. Esta opción puede ser explorada realizando un ran-go de estudios de cristalización en la forma libre de la sustan-cia farmacéutica, típicamente empleando experimentos que se prestan por sí mismos a producir formas menos ordenadas o altamente energéticas (p.ej., métodos de desupersaturación rápida). Otras técnicas que pueden producir materiales amor-fos incluyen liofilización, secado por aspersión y/o evapora-ción rápida del solvente. En casi todos los casos, se requiere del conocimiento de la solubilidad del fármaco para permitir un estudio bien diseñado. La plataforma de tecnología Opti-form utiliza la información de solubilidad para crear diseños experimentales y ejecutar estos diseños de manera paralela.
Referencias1. L. F. Huang and W.G. Tong. Adv. Drug Delivery Rev. 56 (3), 321–
334 (2004).2. C.A. Lipinski et al., Adv. Drug Delivery Rev. 46 (1–3), 3-26 (2000). 3. S. R. Carino, D.C. Sperry, and M. Hawley, J. Pharm. Sci. 99 (9),
3923–3930 (2010). 4. W.Q. Tong and G. Whitesell, Pharm. Dev. Technol. 3 (2), 215–223
(1998). 5. S. Bysouth, J.A. Bis, and D.H. Igo, Int. J. of Pharm. 11 (1–2), 169–171
(2011). PT
continuación de la pág. 22

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 29
sección esPecial: aPis, exciPientes y Manufacturasección esPecial: aPis, exciPientes y Manufactura
excipienTes
El enmascaramiento de sabor de formas farmacéuticas sólidas y fármacos líquidos es un reto para los fabricantes farmacéuticos. La mayoría de los APIs son desagradables o con un sabor áspero, lo que puede ocasionar el incumplimiento del paciente. Este reto afecta a todos los grupos de edad, pero es específicamente problemático para pacientes pediátricos. La implementación de programas para el enmascaramiento del sabor dentro del proceso de manufactura del fármaco es crucial para evitar pérdidas debidas al incumplimiento. Los fabricantes farmacéuticos se enfrentan con retos en la gestión del ciclo de vida, el control de costos, las regulaciones globales y la protección de patentes. En una sesión de preguntas y respuestas, Pharmaceutical Technology examina la selección de excipientes y la funcionalidad en las aplicaciones de enmascaramiento del sabor.
En un webcast, Pharmaceutical Technology examinó el desarrollo de la formulación en la gestión del ci-clo de vida del producto, incluyendo formulaciones especializadas como las formulaciones pediátricas,
y los asuntos técnicos relacionados en la selección y funcio-nalidad de los excipientes, incluyendo el enmascaramiento de sabor y la protección de humedad, para desarrollar un producto oralmente sabroso (1). Participando en el webcast estuvieron presentes: Avinash Thombre, PhD, becario de in-vestigación en ciencias farmacéuticas con Pfizer Investiga-ción y Desarrollo Global, quien discutió la gestión del ciclo de vida y las opciones de nuevas formas farmacéuticas; Ka-ren C. Thompson, PhD, investigadora senior distinguida, en ciencias farmacéuticas en Merck & Co., quien ahondó en las formulaciones pediátricas y formas farmacéuticas relaciona-das; y Nigel Langley, PhD, MBA, Jefe de ventas técnicas en Norteamérica, Pharma Ingredients & Services, BASF, quien discutió las nuevas soluciones de excipientes para enmasca-ramiento del sabor. Para una perspectiva sobre la selección de excipientes y la funcionalidad en aplicaciones para el enmas-caramiento del sabor, Patricia Van Arnum, editora ejecutiva de Pharmaceutical Technology y moderadora del webcast discutió además estos temas con Langley, de BASF.
El diálogo se inició después del lanzamiento por parte de BASF de un excipiente, Kollicoat Smartseal 30 D, una disper-sión acuosa de un polímero formador de película con aplica-ciones para enmascaramiento de sabor y barrera de humedad para formas farmacéuticas sólidas. El producto es un nuevo polímero de recubrimiento y se suministra como una disper-sión en agua al 30%. Desde una perspectiva de característica del producto, el excipiente es altamente impermeable al vapor de agua, lo cual ayuda a preservar la potencia de ingredientes activos sensibles. El polímero es estable en saliva y específi-camente soluble en jugo gástrico. Estas propiedades permiten la protección efectiva de un sabor desagradable en la boca del paciente y la rápida liberación e inicio de la acción del ingre-diente activo en el estómago.
En octubre de 2011, BASF y Colorcon formaron una co-laboración para desarrollar futuros sistemas de recubrimiento pelicular utilizando el Kollicoat Smartseal 30 D de BASF y un nuevo aditivo preformulado de Colorcon. Colorcon de-sarrolló el sistema de aditivo preformulado para usar con el Kollicoat Smartseal 30 D para permitir la preparación y apli-cación eficiente de este polímero en aplicaciones de enmasca-ramiento del sabor. El aditivo preformulado reduce el número de materiales a ser dispensados en 50% y reduce el tiempo de preparación en casi 40%, de acuerdo a un comunicado de prensa de Colorcon, en octubre 21, 2011.
Excipientes en el enmascaramiento del saborSesión de preguntas y respuestas moderada por Patricia Van Arnum
Para el webcast editorial, “Enmascaramiento de Sabor en el Desarrollo de Formulaciones” (Tastemasking in Formulation Development), ver http://www.pharmtech.com/fds, o la sección de webcast en www.PharmTech.com/webcasts.
La
ur
eN
NIc
oLe
/ge
tt
y Im
ag
eS

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201230
sección esPecial: aPis, exciPientes y Manufactura
Propiedades del excipiente en el enmascaramiento del saborPharmTech: ¿Qué características necesita tener un excipien-te para proveer el enmascaramiento del sabor?
Langley: El material tiene que ser estable a pH 6.8 a 7.2, de manera que no perciba nada del fármaco amargo liberado en la saliva. Además, el fármaco tiene que, al entrar al estóma-go, ser liberado rápidamente a un pH bajo. Estamos buscando básicamente un polímero entérico invertido para proporcionar esta funcionalidad. Con respecto al Kollicoat Smartseal 30 D, específicamente, el polímero es insoluble en un valor de pH básico y neutro y tiene muy baja permeabilidad al vapor, lo cual está caracterizado por la estructura –principalmente, los dos grupos etilo que están unidos a la porción del nitrógeno y al propio polímero es el copolímero de metil metacrilatro die-tilaminoetil metacrilato. Es muy insoluble en valores de pH neutros así como en pH básico. Forma una sal en valores de pH por debajo de 5.5, de manera que el polímero se disuelve rápidamente en el estómago liberando el fármaco.
Aplicaciones, desempeño y ejemplosPharmTech: ¿Cuáles son las aplicaciones para este excipien-te, cómo se utiliza para enmascarar el sabor, y cuáles son al-gunos ejemplos?
Langley: Tiene una amplia aplicabilidad, no sólo en re-cubrimientos de tabletas, sino también en recubrimientos de pellets, partículas y cristales. Es útil para tabletas que se des-integran oralmente, para el enmascaramiento de sabor de los dispersables orales, y también en los procesos de granulación.
La recomendación de los niveles de recubrimiento de ta-bleta para proporcionar enmascaramiento del sabor está entre 2-5 mg/cm2, un poquito más es necesario para la protección de la humedad en 5-20 mg/cm2. Kollicoat Smartseal 30 D tie-ne muy baja permeabilidad al vapor de agua. Como resultado, no se plastifica con agua. De esta manera, en el recubrimiento, es necesario adicionar un plastificante con el fin de reducir la temperatura mínima de formación de película (-57°C) a un nivel aceptable. Para este propósito se recomiendan el trietil citrato y el tributil citrato. Adicionalmente, un antioxidante, de preferencia el hidroxi tolueno butilado (BHT, 1-2.5% con base en el peso del polímero) también se requiere en la formu-lación del recubrimiento.
Un buen polímero para enmascarar el sabor no muestra ninguna liberación del fármaco a pH 6.8 a 7.2. Y esto es para lo que se ha diseñado Kollicoat Smartseal 30 D. Comparemos diferentes niveles de recubrimiento y la cantidad de liberación del fármaco con relación a los núcleos sin recubrir para el fármaco amargo, clorhidrato de quinina. En niveles de recu-brimiento entre 4 mg/cm2 y 5 mg/cm2, no hay liberación del fármaco a pH 6.8 a 7.2 durante 60 min. Cuando reduces el pH a 1.2, verás la liberación completa del fármaco, y ésta es una situación ideal para el enmascaramiento del sabor.PharmTech: ¿Cuáles son otras aplicaciones del producto?
Langley: Smartseal 30 D también es muy adecuado para recubrimiento de pellets y cristales. Creemos que estas aplica-ciones se incrementan en importancia debido al surgimiento de formas farmacéuticas innovadoras, tales como las table-
tas oralmente dispersables. Por ejemplo, puede enmascararse efectivamente el sabor de los pellets de cafeína en un nivel de recubrimiento de 5 mg/cm2. Los cristales de acetaminofén son efectivamente enmascarados en 30% de aumento de peso donde no hay liberación del fármaco a pH 6.8 – 7.2 durante 120 min. Se logra algo de enmascaramiento incluso con un aumento de peso tan bajo como 7.5%. En cada ejemplo, el recubrimiento del pellet y del cristal puede visualizarse inclu-yendo un pigmento en la formulación. Kollicoat Smartseal 30 D tiene un beneficio adicional ya que provee brillo.PharmTech: ¿Cómo evalúa el desempeño del enmascara-miento y cuáles son las características clave para lograr el enmascaramiento de sabor?
Langley: Aunque es bueno mostrar los datos de disolu-ción, nosotros necesitamos ver en la realidad si se está lo-grando un efecto de enmascaramiento del sabor en las pro-pias tabletas cuando se ponen en la boca. Utilizar un grupo profesional para enmascaramiento del sabor es obviamente importante ya que ellos están entrenados para hacer precisa-mente este ejercicio.
En nuestro análisis, BASF comparó el Kollicoat Smartseal 30 D con el copolímero disponible comercialmente de metil acrilato butilado y productos de hidroxipropil metilcelulosa. En diferentes niveles de recubrimiento, los panelistas die-ron sus evaluaciones del sabor amargo, Por ejemplo, con el Kollicoat Smartseal 30 D, a 5 mg/cm2, todos los panelistas reportaron una completa ausencia de sabor amargo para el clorhidrato de quinina en comparación con los otros materia-les probados. Por lo tanto, el Kollicoat Smartseal 30 D es un polímero muy efectivo de enmascaramiento del sabor en ese nivel de recubrimiento.
En suma, el Kollicoat Smartseal 30 D tiene una permeabi-lidad al vapor de agua extremadamente baja, proporcionan-do de esta forma la oportunidad de protección de humedad. Muestra una rápida disolución en medio ácido por debajo de pH 5.5 y una fuerte resistencia a pH neutro. Es, por lo tanto, considerado como altamente adecuado como recubrimiento tanto para enmascaramiento de sabor como para protección de la humedad. Otra ventaja es que es un recubrimiento con base acuosa. No se requieren solventes. Tiene muy bajo olor e incluso más importante, no es pegajoso, lo cual puede ser una ventaja real, especialmente en una escala de manufactura. Permite la preparación rápida y simple de la suspensión de recubrimiento. Tiene baja viscosidad y es fácil de atomizar en concentraciones altas de sólidos. Es efectivo en niveles de recubrimiento relativamente bajos. El Kollicoat Smartseal 30 D ha sido diseñado específicamente para mostrar exactamente todas aquéllas características para dar la solución que el mer-cado ha estado buscando. El resultado es el enmascaramiento del sabor y la protección a la humedad en combinación con la liberación instantánea y un recubrimiento pelicular fácil y económico.
Referencia1. “Tastemasking in Formulation Development” Webcast, Pharm.
Technol. Sept. 2011, http://pharmtech.com/fds, accessed Aug. 20, 2012. PT

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 31
caDena De suminisTro
sección esPecial: aPis, exciPientes y Manufacturasección esPecial: aPis, exciPientes y Manufactura
La aprobación del Acta de Cuotas de Usuarios de Fármacos Genéricos (GDUFA) a principios de este año agregó requisitos para mejorar e intensificar las actividades regulatorias para el beneficio de los proveedores globales de fármacos genéricos y de los usuarios de fármacos genéricos en EEUU en intercambio de la aportación de la industria de genéricos global a la FDA con financiamiento para las cuotas de usuarios. La autora describe las provisiones clave de la GDUFA en lo que se refiere a la cadena de suministros global, incluyendo la paridad de inspecciones entre sitios domésticos y extranjeros para formas farmacéuticas terminadas y APIs de fármacos genéricos.
Cambiando a las inspecciones basadas en el riesgoAnálisis del Acta de Cuotas de Usuarios de Fármacos Genéricos Patricia Van Arnum
cr
ea
tIV
e c
ro
P/g
et
ty
Ima
ge
S El 1o. de octubre de 2012, el Acta de Cuotas de Usua-rios de Fármacos Genéricos (GDUFA), la cual se convirtió en ley por el Presidente Barack Obama el 9 de julio de 2012, entró en vigor y proporciona
cambios importantes para los fabricantes de fármacos ge-néricos y los productores de APIs genéricos. La ley impone cuotas de usuarios de fármacos genéricos, pagables por los productores de fármacos terminados y APIs genéricos, para aportar un financiamiento adicional a la FDA para mejorar el proceso de revisión regulatoria, incluyendo las inspecciones a las instalaciones de manufactura de fármacos, para los fár-macos genéricos. Estas medidas, que están soportadas por las industrias de fármacos genéricos y especialidades químicas, pretenden acelerar la entrega y asegurar la calidad de los fár-macos genéricos.
Cuotas de usuarios de fármacos genéricosBajo la GDUFA, la FDA recibirá $299 mdd anualmente (ajus-tado a la inflación) durante la autorización de cinco años del GDUFA desde el 1o. de octubre de 2013 hasta septiembre 30, 2017 (es decir, año fiscal [FY] 2013 hasta el año fiscal 2017) (2). Las cuotas de usuarios requeridas por la GDUFA son
• Una cuota de una sola vez para las solicitudes originales de nuevos fármacos abreviados (ANDAs) pendientes al 1o. de octubre de 2012 (también conocidas como solici-tudes atrasadas)
• Cuotas anuales para instalaciones de APIs y formas far-macéuticas finales
• Cuotas para nuevos ANDAs y suplementos de aprobación previa (PASs)
• Una cuota de una sola vez para los expedientes maestros de fármaco (DMFs) (1).
Setenta por ciento de las cuotas del GDUFA se deriva-rán de las cuotas anuales para instalaciones que producen o están pendientes de revisión para producir APIs o formas farmacéuticas terminadas para una solicitud de fármaco ge-nérico y aproximadamente el 30% de las cuotas de usuario se derivarán de las cuotas de solicitud de una vez (cuotas de DMF y cuotas de ANDA y PAS). Las cuotas generales fueron estructuradas para que reflejen el respectivo peso en la ca-dena de suministro, de manera que el 80% de las cuotas son pagadas por fabricantes de formas farmacéuticas terminadas y el 20% por los fabricantes del API. En el primer año del programa, $50 mdd del financiamiento total por la cuota del usuario GDUFA será generada por la cuota de atraso de una sola vez para los ANDAs pendientes (excepto por los ANDAs

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201232
sección esPecial: aPis, exciPientes y Manufactura
que están pendientes pero han recibido aprobación tentativa) el 1o. de octubre de 2012. En general, se estima que la FDA recibirá el financiamiento a través de aproximadamente 750 ANDAs por año sometidos electrónicamente, 750 PASs, 350 DMFs nuevamente referenciados por año y 2000 instalacio-nes asociadas con los ANDAs (2).
Objetivos principales del programaCon el financiamiento adicional proporcionado por las cuotas de usuarios de fármacos genéricos bajo el GDUFA, la FDA está obligada a cumplir ciertos objetivos de desempeño con respec-to a su revisión regulatoria de fármacos genéricos. El GDUFA destaca objetivos en cinco áreas principales del programa.
• Métrica de las solicitudes: a finales del año fiscal 2017, la FDA revisará y dictaminará el 90% de los ANDAs electrónicos completos en un lapso de 10 meses después de la fecha de sometimiento. Ciertas solicitudes enmen-dadas pueden tener diferente métrica.
• Métrica de los atrasos: la FDA revisará y dictaminará el 90% de todos los ANDAs, enmiendas a los ANDAs y PASs del ANDA del estatus actual de revisión (ya sea electrónicos, en papel, o híbridos) pendientes al 1o. de octubre de 2012, para finales del año fiscal 2017.
• Métrica de inspecciones cGMP: la FDA llevará a cabo inspecciones de vigilancia cGMP bianuales ajustadas al riesgo de fabricantes de APIs genéricos y de formas far-macéuticas terminadas, con el objeto de lograr la paridad de la frecuencia de inspecciones entre las empresas ex-tranjeras y domésticas en el año fiscal 2017.
• Mejoras de eficiencia: la FDA implementará varias me-joras de eficiencia, las cuales incluyen esfuerzos por parte de la industria y la FDA para poblar y mantener bases de datos que sean necesarias para las instalaciones, evaluación de cuotas, eficiencia y otras mejoras. La FDA también hará mejoras basadas en el DMF e inspección así como crear una base de datos de registros vigente de manufactura química y controles (CMC) para ayudar a la eficiencia de las revisiones e inspecciones.
• Ciencia regulatoria: la FDA continuará o empezará di-versas iniciativas regulatorias-científicas. Algunos temas clave son la continuación de las iniciativas de calidad por diseño para los fármacos genéricos, incluyendo el desa-rrollo de recomendaciones basadas en la ciencia para el desarrollo de producto, materia prima, API y controles en proceso, y el manejo del ciclo de vida de formas far-macéuticas complejas (p.ej., productos farmacéuticos inhalados oralmente y formas farmacéuticas de libera-ción modificada). Otro objetivo clave es desarrollar un conocimiento basado en el riesgo de potenciales impac-tos adversos a la calidad del producto farmacéutico que resulten a raíz de cambios en la manufactura y controles del API. Este esfuerzo ayudará en la capacidad para pro-nosticar el potencial impacto que los cambios en la ma-nufactura tendrán sobre la calidad del producto, de ma-nera que los fabricantes puedan dirigir las evaluaciones y controles en áreas de alto riesgo y que los reguladores puedan concentrar sus revisiones en estas áreas (2).
Respuesta de la industriaLa respuesta de la industria al GDUFA ha sido en gran medida positiva con los grupos, tales como la Asociación Farmacéutica de Genéricos (GPhA), el Grupo de Especialidades Químicas Europeas (EFCG), y la Fuerza de Tarea de Farmacéuticos a
Granel (BPTF), una filial de la Sociedad de Fabricantes Quí-micos y Afiliados (SOCMA), respaldando el pase del GDUFA y ofreciendo su participación a lo largo del proceso legislativo.
“La legislación para cuotas de usuario histórica –la legis-lación farmacéutica más importante desde el Acta de Hatch-Waxman en 1984- le dará a la FDA recursos adicionales y asegurará que todos los participantes en el sistema de fárma-cos genéricos de EEUU, ya sea localizados en EEUU o en el extranjero, cumplan con nuestros estrictos estándares de calidad del país,” dijo Ralph G. Neas, presidente y CEO de GPhA, en un comunicado de prensa de GPhA del 9 de julio de 2012. Se espera que las nuevas cuotas reduzcan el tiem-po promedio de revisión para la mayoría de aplicaciones de fármacos genéricos en casi dos años –de más de 30 meses actualmente a 10 meses para el final del quinto año del pro-grama, de acuerdo al GPhA, y proporcionará mayor financia-miento para las inspecciones de instalaciones del fabricante de fármacos genéricos.
Para los productores de especialidades químicas y APIs, una de las provisiones clave del GDUFA es la adopción de un proceso de inspección basado en el riesgo para lograr pa-ridad en la inspección de instalaciones entre los fabricantes de EEUU y los extranjeros, un punto realizado por la Comi-sionada de la FDA Margaret Hamburg a principios de este año (3). “Bajo el GDUFA, la FDA también garantizará que los fabricantes –extranjeros o domésticos- que participan en el mercado de fármacos genéricos de EEUU se sometan al mismo, consistente, estándar de alta calidad,” dijo Hamburg en la Reunión Anual del GPhA el 23 de febrero de 2012 (3). “Realizaremos inspecciones de vigilancia bianuales ajustadas al riesgo de todos los fabricantes de APIs y formas terminadas de genéricos –con el objetivo de lograr paridad de la frecuen-cia de inspección entre empresas extranjeras y domésticas para el año cinco. Teniendo los recursos para realizar estas inspecciones regulares se nivelará el campo de juego para los fabricantes extranjeros y domésticos...” (3).
“Bajo la GDUFA, la FDA también garan-tizará que los fabricantes –extranjeros y domésticos- que participan en el merca-do de fármacos genéricos en EEUU se apeguen al mismo y consistente están-dar de alta calidad.” – Comisionada de la FDA, Margaret Hamburg

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 33
sección esPecial: aPis, exciPientes y Manufactura
La industria de especialidades químicas concuerda. “La frecuencia de inspección obligatoria por el Congreso de cada dos años ha sido muy difícil de lograr debido a los recursos insuficientes dentro de la FDA y a la rápida globalización de la manufactura de fármacos genéricos,” dice el Director Ejecutivo de BPTF, John DiLoreto. “Mientras que las instala-ciones de EEUU han sido inspeccionadas aproximadamente cada 2.5 años, muchas instalaciones extranjeras nunca han sido inspeccionadas. La implementación exitosa el GDUFA dará como resultado la identificación y el registro de todas las instalaciones de producción de fármacos,” explica. “Previa-mente, la FDA incluso tuvo dificultades determinando cuán-tas instalaciones estaban produciendo fármacos genéricos que entraban a la cadena de suministros cada día,” dijo. Para el final de los primeros cinco años de la GDUFA, se espera que todas las instalaciones extranjeras y domésticas sean inspec-cionadas a la misma tasa nominal de cada dos años. “Esto nivelará el campo de juego para los productores extranjeros y domésticos y establecerá un balance en la calidad del fármaco asegurando que todos los fabricantes estén implementando las cGMPs,” dice DiLoreto.
Bajo la GDUFA, la FDA llevará a cabo inspecciones de vigilancia de cGMPs semestrales, ajustadas al riesgo, de fa-bricantes de APIs genéricos y de formas farmacéuticas termi-nadas genéricas, con el objeto de lograr paridad de la frecuen-cia de inspección entre las empresas extranjeras y domésticas en el año fiscal 2017. La FDA priorizará las inspecciones de establecimientos asociados con los ANDAs que de otra forma serían aprobables o elegibles para una aprobación tentativa excepto por una inspección excepcional , así como los esta-blecimientos asociados con ANDAs que no han sido inspec-cionados previamente. En circunstancias apropiadas, la FDA puede apoyarse en una inspección de vigilancia de rutina en lugar de una inspección específica de la solicitud. General-mente, la FDA se apoya en una inspección previa de un sitio del producto terminado que se presentó dentro de un lapso de dos años de la evaluación cGMP para una solicitud pendiente, tres años para un sitio del API o un laboratorio de análisis de control y cuatro años para un sitio sólo de acondicionado (2). Existen excepciones a esta práctica general, las cuales están habitualmente relacionadas con la naturaleza del fármaco que está siendo procesado o de la complejidad de las operacio-nes asociadas del proceso. Bajo la GDUFA, la FDA pretende continuar la práctica de utilizar una evaluación basada en el riesgo para determinar el período de tiempo desde la última inspección, guiada por un ciclo de dos años para los sitios de producto terminado y un ciclo de tres años para los sitios del API con consideración del tipo de producto terminado o API en la solicitud. Para tomar decisiones acerca de las solicitudes pendientes para las cuales la FDA no tiene información de inspección actual dentro del período de tiempo indicado, la FDA puede utilizar la información de la inspección previa y/o utilizar información de inspección de otra autoridad regulato-ria, según convenga (2).
TODO LO QUE USTED NECESITA EN UN FTIR:
LA SIGUIENTE GENERACIÓN EN ESPECTROFOTOMETROS FTIRALPHA LA SIGUIENTE GENERACIÓN EN
Sobre un cuaderno del laboratorio, el ALPHA es un pequeño es-pectrómetro de FT-IR que jugará una gran parte en su rutina diaria. Set-up Plug & play , software fácil de usar, combinado con los módulos accesorios Quick-Snap aseguran el funcionamiento y confi abilidad del FTIR BRUKER de gran alcance para usted.
Contáctenos para más detalles: www.brukeroptics.com
Bruker Mexicana S.A. de C.V.
Concepción Beistegui No. 1402Col. Narvarte PonienteC.P. 03020, D.F., MéxicoTel: +52 (55) 2455 3143Fax: +52 (55) 5601 1189E-mail: [email protected]
Innovation with IntegrityF T- IR
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
3
“Cambiando a las inspecciones basadas en el riesgo”continúa en la pág. 35

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201234
sección esPecial: aPis, exciPientes y Manufactura
El fallecido Dr. Louis Lasagna, padre visionario del campo de la farmacología clínica y fundador del Cen-tro Universitario Tufts para el Estudio del Desarrollo de Fármacos (CSDD), acostumbraba referirse a la
“cruel ironía” de desarrollar fármacos avanzados que salvan la vida que no pueden pagar los pacientes. En muchos aspectos, este dilema se está jugando a través de muchos mercados farma-céuticos mayores alrededor del mundo.
Los mercados abiertos, que permiten estrategias de precios más flexibles, ofrecen un incentivo significativo a las compañías de fármacos para buscar la aprobación en esa región, lo que lleva a un mayor número de medicamentos que alcanzan el mercado a pesar del hecho de que algunos de estos medicamentos pue-den ser incosteables para algunos pacientes. En el otro extremo, los mercados cerrados, que regulan más estrechamente los pre-cios farmacéuticos y los reembolsos, pueden llevar a que menos medicamentos nuevos lleguen al mercado. Estos productos, sin embargo, pueden tener precios más costeables y, por lo tanto, ser accesibles para un mayor número de pacientes.
La cuestión básica para los estrategas de la salud y los legis-ladores es la siguiente: ¿Quiere más fármacos, sabiendo que al-gunos pacientes que los necesitan no pueden costearlos, o quiere menos fármacos, los cuales son costeables para todos?
Para explorar este tema, CSDD Tufts realizó un estudio so-bre el acceso a fármacos para el cáncer en los Estados Unidos y la Unión Europea y revisó los precios y las restricciones de reembolso en cada región. Los hallazgos del estudio, los cua-les fueron encabezados por un co-autor de este artículo, Joshua Cohen, fueron publicados en una reciente publicación del Tufts CSDD Impact Report (1).
Brevemente, el estudio encontró que en la década pasada, la FDA aprobó más fármacos para el cáncer que su contraparte Eu-ropea, la EMA, y para los fármacos aprobados en ambos merca-dos, la FDA los aprobó más rápido. Adicionalmente, los terceros pagadores en EEUU, tanto privados como públicos, aprobaron un mayor porcentaje de fármacos comercializados para reem-
Kenneth I. Kaitin*, PhD, es profesor y director y Joshua P. Cohen, PhD, es profesor asistente, ambos en el Centro Tufts para el Estudio de Desarrollo de Fármacos, Escuela de Medicina Tufts, [email protected].
*A quien debe dirigirse la correspondencia.
punTo De VisTa: bioFarmacéuTicos
Ponderación del acceso y la costeabilidadKenneth I. Kaitin y Joshua P. Cohen
Los legisladores deben balancear los problemas fundamentales que involucran el acceso a los medicamentos y los precios.
bolso, aunque con una participación paciente-costo mucho más elevada que en Europa. En contraste, mientras que se aprobaron menos fármacos para el cáncer en Europa, los precios de estos fármacos fueron, en promedio 9% menores que en EEUU.
Aunque el estudio no evaluó si el mejor acceso a los fármacos para cáncer más nuevos llevaba a mejores resultados de salud, los resultados destacan un desafío clave para los legisladores. Dado que la lista anual de nuevos fármacos para el cáncer relativamen-te costosos se está incrementando, el reembolso continuará sien-do una preocupación significativa en todos los sistemas de salud.
A este respecto, los legisladores pueden aprovechar extrayendo cuidadosamente las lecciones de las experiencias, tanto positi-vas como negativas, de sistemas que han integrado evaluaciones económicas dentro del proceso de toma de decisiones de la pres-cripción y el reembolso.
Desde luego, esta conclusión plantea las preguntas: ¿Por qué se aprobaron más fármacos nuevos para el cáncer en EEUU que en Europa, y entre los 29 fármacos comunes para ambas re-giones, por qué fueron todos ellos aprobados y comercializados primero en EEUU? La respuesta es que las aprobaciones son, en parte, una función de la política regulatoria (p.ej., acceso a la re-visión prioritaria y procedimientos de aprobación acelerados) así como a características institucionales, tales como si un entorno es más conducente a la comercialización de los productos. Los EEUU tuvieron menos controles de precios y tienen menos ins-titucionalización de las evaluaciones de rentabilidad como una posible barrera para el acceso al mercado. En consecuencia, es más probable que las compañías farmacéuticas busquen la apro-bación y comercialicen sus productos primero en EEUU.
Si el siguiente objetivo político se presupone –esto es, maxi-mizar el agregado de los resultados de salud sujetos a una restric-ción del presupuesto- entonces el escenario Europeo de otorgar menos acceso a medicamentos que se considera que ofrecen me-
¿Por qué se aprobaron más fármacos nuevos para el cáncer en EEUU que en Europa?

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 35
“Las inspecciones basadas en el riesgo tendrán un efecto positivo en la FDA y los fabricantes de fármacos genéricos,” dice DiLoreto. “En la administración más eficiente de los recursos, la FDA podrá concentrarse en aquéllos fabricantes que tienen la mayor dificultad en la implementación y mante-nimiento de programas de producción de fármacos de calidad. Las instalaciones de manufactura que nunca han sido inspec-cionadas se considerarán de alto riesgo, y los fabricantes con una historia de programas activos de calidad exitosos serán considerados de menor riesgo,” explica.
Carla Vozone, directora de desarrollo de negocios en Ho-vione y representante de la industria del EFCG durante el proceso de negociación para el GDUFA, también apunta al beneficio del GDUFA en “corregir el desajuste que existió en el programa de inspección y la cadena de suministro real.” Aunque aproximadamente el 80% de APIs para los fármacos comercializados en EEU/U provienen de India o China, las inspecciones de las instalaciones de manufactura de fármacos en estos países no son proporcionales para estos niveles, ex-plica Vozone. “La paridad en las inspecciones es crítica para garantizar la seguridad de los medicamentos genéricos”, dice. Ella también señala el valor en las provisiones del GDUFA que proveen un sistema de inspecciones de vigilancia ajusta-das al riesgo para priorizar inspecciones de instalaciones de alto riesgo.
También de significancia es la mayor autoridad para la FDA para tratar con fármacos de baja calidad y falsificados. “Antes del GDUFA, la FDA tenía poca autoridad para evitar que los fármacos de baja calidad y falsificados entraran a la cadena de suministro,” explica DiLoreto. A partir del 1o. de octubre de 2012, la FDA tiene la autoridad para considerar mal etiquetados a cualquier fármaco no producido en una ins-
nos valor (es decir, menos redituables) puede tener sentido. Este escenario, sin embargo, es una perspectiva social o del pagador y no puede representar los mejores intereses de los pacientes in-dividuales, quienes lo más probable es que no les importe mucho el promedio o los datos agregados. Para ellos, tener más acceso inmediato ofrece esperanza, una oportunidad para extender su vida o una mejora en la calidad de vida.
“Cambiando a las inspecciones basadas en el riesgo”continuación de la pág. 33
talación de manufactura registrada e inspeccionada. Todos los fármacos considerados mal etiquetados pueden ser destruidos en lugar de devolverlos de manera que no se permita que en-tren a la cadena de suministro a través de diferentes canales de la misma, explica.
Como la barra de calidad para fármacos genéricos y APIs se eleva bajo el GDUFA, la cadena de suministros para fárma-cos genéricos también puede afectarse, señala Vozone. “Los fabricantes de genéricos tendrán que hacer una evaluación de riesgo de sus cadenas de suministro,” dice. Los asuntos tales como el incremento del suministro secundario o las evalua-ciones posteriores de los proveedores existentes, entrarán en juego. Ella también señala que la cantidad de las cuotas de usuarios, las cuales todavía se están determinando, obligarán a los productores a evaluar sus estructuras de costo para el suministro de un API genérico dado, y como resultado, cier-tos productores pueden salir del mercado para ese producto o sector. Estas cuestiones serán importantes para tomarse en consideración en las estrategias de suministro para los fabri-cantes de fármacos genéricos.
Referencia1. FDA, “Agency Information Collection Activities; Proposed Co-
llection; Comment Request; Generic Drug User Fee Cover Sheet; Form FDA 3794,” Federal Register 77 (144), 43844–43846 (2012).
2. FDA, Human Generic Drug Performance Goals and Procedures Fiscal Years 2013 through 2017 (Rockville, MD), July 7, 2012, www.fda.gov/downloads/ForIndustry/UserFees/GenericDrugUserFees/UCM282505.pdf, accessed Aug. 15, 2012.
3. M. Hamburg, “Challenges and Opportunities for the Generic Drug Industry Remarks,” presented at the GPhA Annual Meeting, Or-lando, FL, Feb. 23, 2012. PT
Puede haber una compensación entre el acceso y el precio: mayor acceso a costa de precios más elevados, posiblemente in-costeables para los pacientes. Es una “cruel ironía”.
Referencia1. K.I. Kaitin, Ed., “US Offers Patients Faster, Greater Access to Can-
cer Drugs than Europe”, Tufts CSDD Impact Report 14 (4), 1–4 (2012). PT
EN LA REVISTA MÁS LEIDA POR LA INDUSTRIA FARMACÉUTICA
CONTRATE SU PAUTA PUBLICITARIAANTES DEL 14 DE DICIEMBREINICIE 2013 SU PRESENCIA
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486 Fax: 52 (55) 5659-8879E-mail: [email protected]

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201236
Patricia Van Arnum es editora ejecutiva de Pharmaceutical Technology, USA, 485 route one South, edif. F, Primer Piso, Iselin, NJ 08830 tel. 732.346.3072, [email protected]
ingreDienTes FarmacéuTicos: apis y excipienTes
Escalamiento de la Síntesis de APIsPatricia Van Arnum
Las estrategias se centran en lasmaneras de optimizar las condiciones del proceso y la operatividad.
Los químicos de proceso en-frentan el reto de desarrollar rutas de manufactura comer-cial rentable y eficiente para
APIs. Ellos se encuentran con los retos de incrementar el rendimiento del pro-ducto, alcanzar mayor estereoselecti-vidad y regioselectividad, y de mejorar las condiciones del proceso, tales como la temperatura y la presión, todo como una manera de producir un compuesto farmacéutico de alta calidad de forma se-gura. En este esfuerzo pueden usarse una variedad de herramientas, incluyendo la aplicación de enfoques químico-ecológi-cos, como la biocatálisis, el reemplazo de solventes y la química de flujo conti-nuo, para ayudar a lograr transformacio-nes químicas más eficientes bajo condi-ciones de reacción mejoradas.
Ruta biocatalítica para la simvastatinaEn junio de 2012, Codexis, una compa-ñía que se especializa en biocatálisis, y Yi Tang, profesor en el Departamento de Ingeniería Química y Biomolecular en la Universidad de California en los Ángeles (UCLA), fueron reconocidos con el “Premio 2012 de Vías de Síntesis Ecológicas” como parte de los Premios de los Retos Químicos Ecológicos Pre-sidenciales de la Agencia de Protección Ambiental (EPA) por el desarrollo de una ruta biocatalítica para hacer sim-vastatina, el ingrediente activo del fár-maco anticolesterol Zocor de Merck & Co., que hoy día ya perdió la patente (1). Codexis y Tang ganaron el premio este año después de haber sometido previamente el nuevo proceso en 2010 y 2011 (2-4).
Para la ruta de simvastatina, Co-dexis obtuvo la licencia para la tecno-logía de Tang. Las rutas de síntesis pre-vias para la simvastatina involucraban la conversión de lovastatina a simvas-tatina agregando un grupo metilo que se requería para la protección y después la
ingreDienTes FarmacéuTicos: apis y excipienTes
desprotección de otras funcionalidades en la molécula de lovastatina en una sín-tesis multipasos. En la primera ruta, la lovastatina fue hidrolizada al triol, mo-nacolina J, seguido de protección con sililación selectiva, esterificación con cloruro de dimetil butiril y desprotec-ción. La segunda ruta involucró la pro-tección de las funcionalidades del ácido carboxílico y del alcohol, metilando el carbón C2’ con yoduro de metilo y desprotegiendo el producto. Estas rutas fueron ineficientes porque produjeron menos de 70% de rendimiento global y fueron concentradas en masa debido a la protección y desprotección (1-4).
La ruta desarrollada por Tang y su grupo eludieron la protección y despro-tección y resultó en una mayor econo-mía de átomos, desperdicio reducido, y en general en menos condiciones de re-acción peligrosas. Primero, clonaron el LovD, una acetiltransferasa natural de Aspergillus terreus que está involucrada en la síntesis de lovastatina y que puede aceptar donadores de acilo no natura-les. Reconociendo que el LovD puede ser un tipo de simvastatina sintasa y un punto de inicio para crear un nuevo pro-ceso biocatalítico, ellos evolucionaron la enzima hacia la utilidad comercial (1-5). Codexis adquirió la licencia de la tecnología de Tang y optimizó la manu-factura de simvastatina.
El biocatalizador LovD transfirió selectivamente la cadena lateral 2-me-tilbutiril a la sal sódica o de amonio del alcohol C8 de monacolin J. El donador del acilo, dimetilbutiril-S-metil-mer-captopropionato (DMB-SMMP), es efi-ciente para la reacción catalizada por el LovD, es más seguro que las alternati-vas tradicionales y se prepara en un solo paso a partir de precursores económi-cos, de acuerdo al reporte sumario del premio (1). Codexis adquirió la licencia de este proceso de la UCLA y posterior-mente optimizó la enzima y el proceso químico para la manufactura comercial. Codexis llevó a cabo nueva iteraciones de la evolución in vitro, creando 216 bibliotecas y eligiendo 61,779 variantes para desarrollar una variante de LovD con actividad mejorada, estabilidad en proceso y tolerancia a la inhibición

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 37
del producto (1). La enzima mejorada aproximadamente 1000 veces y el nue-vo proceso empujaron la reacción para terminar en una alta carga de sustrato y minimizó las cantidades del donador acilo y de los solventes para extracción y separación del producto. En la nue-va ruta, la lovastatina es hidrolizada y convertida a la sal de amonio soluble en agua del monacolin J (1-5).
Según se especificó en el reporte sumario del Premio al Reto Quími-co Ecológico Presidencial, la variante evolucionada genéticamente de LovD acetiltransferasa de Escherichia coli utiliza DMB-SMMP del donador acilo para hacer la sal de amonio insoluble en agua de simvastatina. El único co-producto de la síntesis de simvastatina es el ácido metil 3-mercaptopropiónico, el cual es reciclado. El rendimiento fi-nal de la sal de amonio de simvastatina es mayor de 97% en una carga de 75 g/L de monacolin J. Esto evita el uso de químicos peligrosos, incluyendo el cloruro de tert-butil dimetil silano, yo-duro de metilo, y n-butil litio. Se han fabricado más de 10 toneladas métricas de simvastatina utilizando este nuevo proceso (1).
Ruta biocatalítica para atorvastatinaPfizer también empleó un enfoque más amigable ambientalmente en el desarro-llo de una ruta biocatalítica para hacer atorvastatina, el ingrediente activo del Lipitor, y sometió el proceso a conside-ración de los Premios del Reto Químico Ecológico Presidencial de la EPA (1). El nuevo proceso incorporó una enzima con base de agua 2-deoxirribosa-5-fos-fato aldolasa (DERA) al inicio de la ruta para hacer un lactol a partir de un amino aldehído (es decir, 3-ftalimidopropio-naldehído; PPA) y acetaldehído (1).
La síntesis eliminó el uso de por-ciones de cianuro o azida para introdu-cir nitrógeno debido a que éste ya está presente en el lactol. En contraste con la síntesis original, la enzima DERA establece ambos estereocentros con alta selectividad en agua a temperatura am-
biente. La conversión del lactol resul-tante en isopropil acetónido atorvasta-tina (IAA) involucró sólo cuatro pasos químicos de alto rendimiento (oxida-ción, esterificación, desprotección, Paal Knorr). El producto IAA fue convertido a atorvastatina (1). La nueva síntesis eliminó el paso previo de hidrogena-ción a alta presión con sus catalizadores metálicos asociados. También evita el n-butil litio pirofórico y su gas butano de desecho asociado. La FDA aprobó el nuevo proceso de manufactura en abril de 2010, y Pfizer fabricó lotes de validación a escala comercial en 2011 y actualmente está transitando a la ma-nufactura comercial a escala completa, de acuerdo al reporte de los Premios del Reto Químico Ecológico Presidencial de la EPA (1).
Química de Grignard de Eli LillyLa reacción de Grignard es una reac-ción bien establecida en química orgá-nica, pero plantea algunos retos en es-calamiento a escala comercial; activa-ción fuertemente exotérmica y pasos de reacción; reacciones heterogéneas con problemas potenciales en la suspensión y mezclado de la mezcla de reacción; y riesgos operacionales planteados por solventes etéreos como dietil éter (1). Eli Lilly desarrolló una química Grig-nard más segura utilizando un reactor con agitación continua (CSTR) que permite la formación continua de reac-tivos de Grignard con operaciones de acoplamiento continuo y apagado, un proceso del cual la compañía sometió su ingreso a los Premios del Reto Quí-mico Ecológico Presidencial de la EPA (1). De acuerdo al ingreso, el enfoque del CSTR mitigó los riesgos operando volúmenes de reacción pequeños, reali-zó activación metálica sólo una vez por cada campaña y utilizó 2-metiltetrahi-drofurano como reactivo de Grignard y solvente de reacción, resultando en producto con una quimio- y estereose-lectividad mejoradas. Con relación al procesado del lote, el enfoque continuo permitió el control del estado estacio-nario y reducciones generales de hasta
43% en magnesio, 10% en la estequio-metría del reactivo Grignard, y 30% en la intensidad de la masa del proceso (1).
Solventes mejorados para hacer diaril aldiminasLas iminas son intermedios usados en muchas síntesis farmacéuticas. Las dia-ril aldiminas, por ejemplo, se usan en la síntesis del fármaco anticáncer Taxol (paclitaxel) y el fármaco anticolesterol Zetia (ezetimibe) (1). La síntesis tradi-cional de las diaril aldiminas con fre-cuencia requieren solventes peligrosos e incluyen pasos con mucha carga de energía y de reflujo de muchas horas. Aunque algunas síntesis de imina usan solventes o condiciones más benignas, aún así requieren largos tiempos de re-acción, recristalización u otros proce-dimientos ambientalmente más rudos. Jacqueline Bennett, profesora en el Departamento de Química y Bioquími-ca, Universidad Estatal de Nueva York (SUNY) Oneonta y la Fundación de Investigación SUNY, desarrollaron un proceso que utilizó etil L-lactato como solvente para sintetizar iminas. El pro-ceso se sometió al Premio del Reto Quí-mico Ecológico Presidencial de la EPA EEUU 2012 (1). De acuerdo al reporte resumido para el premio, el método fue eficiente bajo condiciones ambientales, requirió menos solvente que otros mé-todos, tuvo una mediana de rendimiento de más de 92%, y tuvo una mediana de tiempo de reacción de menos de 10 min (1). Las iminas resultantes fueron gene-ralmente lo suficientemente puras sin recristalización ya que la polaridad del etil L-lactato fue modulada agregando agua. Los materiales de inicio permane-cieron disueltos, pero la imina se crista-lizó de la solución conforme se formaba (1). Aunque los métodos tradicionales con frecuencia hacen avanzar las reac-ciones removiendo agua, este método dirigió la reacción haciendo avanzar el producto a través de la cristalización, de acuerdo al reporte sumario (1). Ben-nett y su grupo de investigación han sintetizado casi 200 iminas utilizando este método y obtuvieron una patente EEUU para el proceso (1, 6).

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201238
Etileno en la síntesis deespecialidades químicasOtra entrada interesante para los Pre-mios del Reto Químico Ecológico Pre-sidencial de la EPA involucró el uso de etileno en la síntesis de especialidades químicas desarrollada por T. V. Ra-janBabu, profesor en el Departamen-to de Química, de la Universidad del Estado de Ohio (1). De acuerdo con el reporte sumerio de los premios, los métodos prácticos que utilizan fuentes de carbón como materiales de inicio para formar enlaces enantioselectivos carbón-carbón no son comunes. Una re-acción ampliamente aplicable que uti-liza etileno para instalar grupos vinilo enantioméricamente, como la desarro-llada por RajanBabu, podría tener un impacto significativo en la síntesis de especialidades médicas.
RajanBabu y su equipo de trabajo desarrollaron protocolos altamente ca-talíticos (relación sustrato-catalizador hasta de 7,412:1) para la codimeriza-ción casi cuantitativa (rendimientos aislados de más de 99%) y altamente selectiva (aproximadamente 100% de regioselectividad; relaciones enan-tioméricas de más de 99:1) de etileno y varios vinilarenos funcionalizados, 1,3-dienos y alquenos tensionados. Es-tas reacciones se dieron bajo condicio-nes suaves (-52° a 25°C; 1 atmósfera de etileno) para producir intermedios, tales como 3-arilbutenos, los cuales pueden ser transformados a fármacos anti-infla-matorios no esteroidales (FAINEs) en dos pasos. Estas reacciones consumen materiales de inicio, no dejando pro-
ductos colaterales. Los éxitos incluyen síntesis altamente enantioselectivas de FAINEs comunes, tales como ibuprofe-no, naproxeno, flurbiprofeno y fenopro-feno, de los correspondientes estirenos y etilenos (1).
Los 1,3-dienos cíclicos y acíclicos también pasaron por una adición eficien-te enantioselectiva de etileno. La síntesis de varios 1-vinilcicloalquenos y 1-susti-tuido-1,3-butadienos alcanzó rendimien-tos hasta de 99% (1). El enfoque tam-bién ha sido aplicado a otras clases de compuestos biológicamente relevantes, incluyendo bisabolanos, herbindoles, tri-quentrinas, esteroides D-ring 20S- ó 20R derivados, (-)-desoxieserolina, pseudop-terosina A-F, G-J y K-L agliconas y he-lioporinas (1).
Eliminadores de impurezasLos químicos, intermedios y reactivos asó como subproductos de procesos sintéticos pueden tener propiedades tóxicas y estar presentes como impure-zas en niveles bajos en un API o formu-lación final del fármaco. La detección y remoción de estas impurezas es de vital importancia para los químicos de pro-ceso, particularmente en el caso de im-purezas genotóxicas. Una de las impu-rezas potencialmente genotóxicas con base en alertas estructurales es la acro-leína, un aldehído a, b-insaturado que se utiliza como bloque de construcción en la producción de farmacéuticos (7).
Los investigadores de MIP Tech-nologies, una subsidiaria de Biotage, y la Universidad Dortmund en Alema-nia reportaron recientemente sobre un
ingreDienTes FarmacéuTicos: apis y excipienTes
esquema para la remoción selectiva de acroleína de los APIs utilizando iodixa-nol como modelo de API. Se analizó el comportamiento eliminador de acroleí-na de los eliminadores de aldehídos con base de poliestireno y sílica en medios orgánicos en presencia del API iodixa-nol. Se probaron varios eliminadores, y las resinas que mostraron la eficiencia y selectividad de unión más alta fueron evaluadas después. La eliminación más efectiva y selectiva se obtuvo con po-liestireno-amina, la cual removió hasta 97.8% de acroleína y sólo 2.0% de io-dixanol dentro de los 20 min utilizan-do un procedimiento de extracción en modo de lote (7).
Referencia1. EPA, “The Presidential Green Che-
mistry Challenge Awards Program: Summary of 2012 Award Entries and Recipients” (Washington, DC, 2012).
2. P. Van Arnum, Pharm. Technol. 35 (9), 54–58 (2011).
3. EPA, “The Presidential Green Che-mistry Challenge Awards Program: Summary of 2010 Award Entries and Recipients” (Washington, DC, 2010).
4. EPA, “The Presidential Green Che-mistry Challenge Awards Program: Summary of 2011 Award Entries and Recipients” (Washington, DC, 2011).
5. Y. Tang et al., Science 326 (5952), 589–592 (2009).
6. J. Bennett, Green Synthesis of Aryl Al-dimines Using Ethyl Lactate, US Patent Application 20110196174 (Aug. 11, 2011).
7. R. Kecili et al., Org. Proc. Res. Dev. 16 (6), 1225–1229 (2012). PT
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486
Fax: 52 (55) 5659-8879E-mail: [email protected]
CONTRATE ANTES DEL 7 DE FEBREROY APROVECHE NUESTRA
PROMOCIÓN ESPECIAL

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 39
Ima
ge
N:
Sto
cK
By
te
/ge
tt
y Im
ag
eS
BIOFORO
Pequeños cambios, gran impacto en el sistemaSimon Chalk y Steve Jones
El efecto mariposa es un fenó-meno muy citado en donde un pequeño cambio en un sistema puede tener un impacto signi-
ficativo sobre el estado global del siste-ma. De manera similar, los elastómeros relativamente no costosos pueden con-tribuir desproporcionadamente al costo de correr una operación de manufactu-ra biofarmacéutica. Los elastómeros y los plásticos juegan un papel vital en la operación de una planta de bioproce-so, formando empaques, arillos (“o”-rings), y diafragmas muy adentro de la estructura del equipo de proceso. Su función es evitar fugas y separar fluidos que no deben entrar nunca en contacto. Estos materiales, parecidos al hule, son útiles porque son flexibles, elásticos y pueden asegurar sellos herméticos entre las superficies metálicas duras.
Con el tiempo, y con la severa tem-peratura, los químicos y los ciclos de presión a los que están sujetos, estos materiales pueden volverse quebradi-zos y deformados y fallar. Necesitan cambiarse bastante antes de que exista un riesgo de falla, cuya consecuencia podría ser un producto contaminado o una peligrosa grieta de un sistema. Mu-chas plantas biofarmacéuticas tienen una gran base instalada de válvulas, por ejemplo, quizás 5000 o más. Cada una necesita ser mantenida correctamente para evitar problemas. Aunque el costo de la falla es elevado, el costo del cam-bio también es elevado. Se estima que hasta 50% de la actividad de manteni-miento es consumida por el cambio de
Un vistazo al reemplazo del elastómero muestra a veces cómo el conocimiento de la industria mejora las operaciones y el costo.
Simon Chalk ([email protected]) y Steve Jones son ambos directores del Grupo BioPhorum Operations
partes blandas. Sume esto a los tiempos muertos de la planta y hay un claro ob-jetivo para revisar el ahorro de costos. De manera que ¿Cuál es el alcance para la mejora? ¿Puede desafiarse la práctica actual?
La estrategia actualmente aceptada y común para el cambio del elastómero es temporal de base (es decir, hay una frecuencia fija, quizás bianual o anual para el mantenimiento programado para reemplazar el componente). Aunque esta estrategia es aceptable, no toma en cuenta las condiciones a las que ha es-tado sujeto el elastómero. En los casos en los que el componente se ha usado ligeramente, puede reemplazarse aún cuando el uso continuado sería perfec-tamente aceptable. En el otro extremo del espectro, el uso severo podría tener un riesgo de fallo del elastómero antes de haber alcanzado su período fijo de tiempo.
Varios líderes de ingeniería en operaciones biofarmacéuticas se están cuestionando esta metodología. Ellos se están conduciendo por una búsque-da implacable de la excelencia opera-cional y por maneras más efectivas de trabajar. Así como hay ahorros en el costo, también está la idea de que sus talentosos ingenieros podrían estar me-jor empleados trabajando en proyectos técnicos de mayor valor agregado en lugar del mantenimiento de rutina. Uno de dichos ingenieros adquirió el hábito de colectar las partes suaves desechadas e inspeccionarlas visualmente. Su cu-riosidad y disgusto por el desperdicio lo llevó a preguntarse si existía una mejor manera de sistematizar el reemplazo de estos artículos de manera que fueran usados durante más tiempo sin el riesgo
de una falla en la operación. Su involu-cramiento con un grupo de evaluación comparativa con la industria y las dis-cusiones con sus pares que pensaban de manera similar, demostraron que sí existían mejores prácticas. Este cono-cimiento lo impulsó a implementar una nueva manera de trabajar, lo que llevó a significativos ahorros.
Siguiendo estrategias simples cien-tíficas y basadas en el riesgo, algunas compañías están ahora extendiendo la vida de los elastómeros en tres, cuatro o cinco veces. Los anteriores ciclos de mantenimiento basados en el tiempo han sido reemplazados con ciclos basa-dos en las condiciones en donde se ana-liza cuidadosamente el uso y el desgas-te de los componentes y se gradúa, de manera que la vida de los componentes puede pronosticarse con exactitud. Los factores que afectan el uso y el desgas-te, tales como el número de ciclos de limpieza, las temperaturas y los quími-cos usados, son registrados y proporcio-nan una base racional para el análisis y posteriormente, la medición.
Los datos operacionales que mues-tran variaciones de los resultados pro-nosticados son además fuentes de cono-cimiento profundo, que arrojan luz so-bre los factores desconocidos llevando a la reducción de la variabilidad y a ma-yores niveles de confianza para pronos-ticar la condición del componente. Uno de dichos análisis de causa raíz reveló que el correcto o incorrecto ensambla-do de las válvulas del diafragma puede contribuir significativamente al desem-
“Pequeños cambios, gran impacto en el sistema”continúa en la pág. 41

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201240
Revisión estadística del ICH Q10 Sistema de Calidad FarmacéuticaLynn D. Torbeck
La aplicación de las recomendaciones del ICH Q10 al análisis estadístico puede llevar a evitar recuperaciones de producto del mercado.
SOLUCIONES ESTADÍSTICAS
La guía de la Conferencia Inter-nacional para la Armonización ICH Q10, Sistema de Calidad Farmacéutica, y sus dos guías
acompañantes Q8 Desarrollo Farma-céutico y Q9 Manejo del Riesgo de Ca-lidad, han sido fácilmente aceptadas si bien no completamente implementadas por la industria farmacéutica en los pasa-dos pocos años (1-3). Las discusiones de las implicaciones estadísticas de la Q8 y Q9 se han dado desde que las guías se armonizaron (4). Sin embargo, poco se ha dicho acerca del contenido estadístico del modelo Q10, probablemente debido a que se percibió que estaba enfocado solamente en la gestión del sistema de calidad. Existen muchas recomendacio-nes del Q10 que afectan los problemas estadísticos que enfrente la industria far-macéutica, no obstante, la guía establece que no está “destinada a crear ninguna nueva expectativa más allá de los reque-rimientos regulatorios actuales” (1).
Aunque explícitamente en la Q10 no se requiere ninguna nueva estadística o plan de muestreo, no hace falta decir que los requerimientos regulatorios actuales son, de hecho, obligatorios. Adicional-mente, las cGMPs continúan mejorando con el tiempo y de acuerdo a la Q10, “La
Ph
oto
DIS
c/g
et
ty
Ima
ge
S
Lynn D. Torbeck es estadístico en PharmStat consulting, 2000 Dempster, evanston, IL 60202, tel. 847.424.1314, [email protected], www.PharmStat.com.
implementación de la ICH Q10 a lo lar-go del ciclo de vida del producto debe facilitar la innovación y la mejora con-tinua y fortalecer el vínculo entre el de-sarrollo farmacéutico y las actividades de manufactura” (1). Este vínculo debe incluir los resultados de experimentos diseñados estadísticamente y el análisis estadístico y de riesgo relacionado.
Aunque explícitamente no requiere estos enfoques, el ICH claramente im-plica que las compañías necesitan ser proactivas cuando se trata de progra-mas de acción correctiva y preventiva (CAPA). En el entorno de hoy, no es suficiente ser reactivo sólo cuando se presentan problemas. El departamento de Calidad debe buscar rutinariamente los problemas potenciales y evitarlos antes de que resulten en rechazos o recuperación de productos del merca-do. Por ejemplo, el Q10 señala que las compañías deben “Establecer y mante-ner un estado de control para desarro-llar sistemas efectivos de monitoreo y control para el desempeño del proceso y la calidad del producto, proporcionan-do así la seguridad de aptitud continua y capacidad de los procesos” (1).
Teniendo el control sobre el propio producto y proceso no es una nueva ex-pectativa, aunque todavía existe confu-sión de lo que un “estado de control” apropiado significa (4). No es suficiente pedir un estado de control; la industria debe proveer y definir modificadores adicionales. Existen varias maneras en las cuales un proceso puede estar en un estado de control o, al contrario, en un “estado de fuera de control”.
Un proceso puede estar en control, por ejemplo, para finanzas y contabili-dad, para el cumplimiento regulatorio y para el control organizacional y geren-cial. Se supone que estas formas de con-trol están habitualmente en funciones. Existen otros dos estados de control que están relacionados con las estadísticas: la ingeniería y la estadística.
Se dice que un proceso está en un estado de control de ingeniería cuando el proceso puede ser cambiado y ajus-tado utilizando los mandos de control y/o ajustando los parámetros críticos del proceso (variables independientes) que afectan las respuestas dependientes (5). Cuando está en control, el producto siempre cumple sus especificaciones in-cluso si es inconsistente y errático. Las gráficas de tiempo con líneas de espe-cificación se utilizan para monitorear el proceso. Se dice que un proceso está fuera de control de ingeniería cuando no cumple sus especificaciones.
Se dice que un proceso está en es-tado de control estadístico cuando el proceso ha sido diseñado, desarrollado y ajustado para producir producto que, aunque todavía contiene algo de variabi-lidad en los atributos de calidad críticos (variables dependientes), es pronosti-cable de esa variabilidad con el tiem-po. Las cartas de control estadístico se utilizan para monitorear el proceso. Se dice que un proceso está fuera de control estadístico cuando falla uno o más de las ocho reglas de las cartas de control Wes-tern Electric (6). Como señala el Q10, “El sistema de calidad farmacéutica debe incluir los siguientes elementos,

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 41
monitoreo del desempeño del proceso y de la calidad del producto, acciones co-rrectivas y preventivas, gestión del cam-bio y revisión de la gestión” (1).
El monitoreo de la calidad del pro-ducto puede ser interpretado como la investigación de las tendencias de los atributos de calidad críticos. Nueva-mente, el CAPA proactivo es preferi-do al CAPA reactivo. Como destaca el Q10: “Defender la mejora continua” (1). Esta mejora continua debe incluir la reducción proactiva de la variabilidad.
También está recomendado en la Q10: “...un acuerdo por escrito entre el que da el contrato y el que acepta el contrato.” Este acuerdo debe incluir los límites de calidad aceptables (AQL) y los límites de cantidad limitada (LQ) para los planes del muestreo de entrada así como los métodos de la especifica-ción habitual y los criterios de acepta-ción. Los datos colectados a la entrada y en el proceso pueden usarse para deter-minar el cumplimiento con un acuerdo contractual. De acuerdo a la Q10, “A lo largo del ciclo de vida del producto, las compañías están alentadas a evaluar las oportunidades para las estrategias in-novadoras para mejorar la calidad del producto” (1).
Existen muchas formas, estadísti-camente, para alcanzar este objetivo. Estudios de tendencia, experimentos diseñados, reducción de la variabilidad y espacio de diseño son sólo algunas
de las herramientas que pueden usarse para hacer mejoras en el proceso.
Muchos de los términos en el ICH Q10 implican las tendencias de los pa-rámetros críticos y los atributos. Es un hecho que esto debe hacerse. La Q10 establece: “Un sistema de monitoreo efectivo proporciona la seguridad de la capacidad continuada de los procesos y los controles para producir un producto de la calidad deseada y para identificar áreas para la mejora continua” (1).
La capacidad del proceso se mide comparando la variabilidad del produc-to/proceso con el ancho del rango de la especificación. Esta comparación puede lograrse mejor utilizando intervalos de tolerancia estadística debido a que to-man en cuenta el tamaño de la mues-tra, conde el Cpk y Ppk no lo hacen. De acuerdo a la Q10, “Identifica fuentes de variación que afectan el desempeño del proceso y la calidad del producto para la actividad potencial de mejora con-tinua para reducir o controlar la varia-ción” (1).
Algunos de los programas Seis Sig-ma han adquirido una pobre reputación en ciertos círculos debido al enfoque limitado de ahorrar dinero a diferencia de darle igual consideración a la mejo-ra de calidad y a la reducción de varia-ción. En opinión del autor, la gerencia necesita darle igual atención y recursos a ambos. Según lo demanda el Q10, “Los cambios propuestos deben ser
evaluados por grupos de expertos que contribuyan con la experiencia y el co-nocimiento apropiados de las áreas rele-vantes (p.ej., Desarrollo Farmacéutico, Manufactura, Calidad, Asuntos Regula-torios y Médicos), para asegurar que el cambio está técnicamente justificado” (1). El departamento de estadística de la compañía o un estadístico deben ser incluidos en el grupo.
Muchos enunciados en la ICH Q10 tienen importantes implicaciones para el uso correcto y consistente de la esta-dística en la implementación cotidiana de los sistemas de calidad farmacéuti-ca. Abordando estas recomendaciones armonizadas proactivamente y en con-texto puede ayudar a fortalecer el sis-tema de calidad y de esta forma reducir rechazos y recuperaciones de producto del mercado.
Referencias y notas1. ICH, Q10 Pharmaceutical Quality Sys-
tem (2008).2. ICH, Q8 Pharmaceutical Development
(2009).3. ICH, Q9 Quality Risk Management
(2005).4. L. Torbeck, Pharm. Technol. 35 (10)
46–47 (2011).5. Note: Other definitions of Engineering
Control exist in other industries.6. Note: It is common practice to use only
one to three of the eight Western Elec-tric rules for a given control chart. It is counterproductive to use more than three rules at a time. PT
peño de las partes suaves. Se descubrió que la lubricación correcta de los per-nos de fijación y el torque adecuado por ejemplo, son factores que contribuyen a la vida de los diafragmas.
La cuestión de la conformidad con la especificación fue otra rica área ob-jetivo, con la ausencia de estándares claros y métodos de prueba no existen-tes o inconsistentes en la industria. Los proveedores de elastómeros tienen un largo camino que recorrer para cumplir las necesidades del ambiente biofarma-céutico. El desempeño ha sido históri-
“Pequeños cambios, gran impacto en el sistema”continuación de la pág. 39
camente problema del cliente. Siendo la ausencia de control alrededor de los cambios una asunto particular de pre-ocupación en donde la cadena de sumi-nistro de los proveedores y los provee-dores de los proveedores no se maneja rigurosamente.
El mismo grupo de la industria que comparte las mejores prácticas está avanzando hoy en la causa proponiendo estándares centrados en el cliente que cubran secuencias de pruebas genéri-cas, criterios de inspección visual y me-jor control de cambios. Con los acuer-
dos de los diversos participantes, estos estándares se escribirán en códigos glo-balmente reconocidos que preparen el escenario para un mejor cumplimiento de la industria.
En este ejemplo de una dirección para las mejores prácticas en la manu-factura biofarmacéutica, uno puede tra-zar una línea directa desde un ingeniero que examina las partes desarmadas de una válvula de mariposa hasta un nuevo sistema en la industria de estándares y niveles de desempeño de calidad, no ex-perimentados anteriormente. PT

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201242
La validación de limpieza proporciona la seguridad de que la cantidad de sustancias residuales colectadas de las superficies del equipo están dentro de los límites permisibles, ayudando a asegurar el control de calidad y la seguridad en las instalaciones de manufactura farmacéutica. Se compararon tres diferentes métodos de validación de limpieza para medir el carbón en muestras residuales de diversas sustancias farmacéuticas.
Robert Clifford*, PhD, es gerente de la unidad industrial de negocios en Shimadzu Scientific Instruments, 7102 Riverwood Drive, Columbia, MD 21046, tel. 800.447.1227, [email protected]. Minako Tanaka es científico en el Centro de Desarrollo de Aplicaciones de Shimadzu, Kyoto, Japón, [email protected]
*A quien debe dirigirse la correspondencia.
Sometido: Noviembre 17, 2011; Aceptado: Marzo 12, 2012.
Los desafíos de llevar a cabo la validación de la lim-pieza están documentados en la literatura. R. Baffi et al., por ejemplo, describieron los diversos retos analíticos que surgen en la validación de los pro-
cedimientos de limpieza para productos biofarmacéuticos producidos mediante ADN recombinante, en el cual un am-plio rango de potenciales componentes celulares residuales y niveles traza de detergentes deben ser cuantificados (1). M.A. Strege et al. describieron el análisis de carbón orgánico total (TOC) de muestras de hisopo para la validación de limpie-za de equipo de bioproceso de fermentación y discutieron la exactitud, los límites de detección, el límite de cuantificación, la linealidad y la precisión (2). K.M. Jenkins et al. compara-ron las ventajas y desventajas de múltiples métodos para va-lidación de limpieza, incluyendo la cromatografía de líquidos de alta resolución (HPLC), la cromatografía en capa delgada (CCD), la espectrometría, el TOC y la conductividad (3). A. J. Holmes et al. describieron el método TOC para la medición de aspirina residual en aluminio, acero inoxidable, acero al carbón pintado, y Plexiglas (4). Los dos últimos autores des-criben el desafío con hisopo señalado en la guía de la FDA para inspecciones de superficies de limpieza.
Para la validación de limpieza utilizando un analizador TOC, se dispone de los siguientes tipos de métodos de muestreo:
• muestreo del enjuague• muestreo con hisopo con extracción acuosa• muestreo con hisopo con combustión directa
Estos métodos fueron comparados utilizando un analiza-dor de carbón orgánico total (TOC-LCPH, Shimadzu) para medir productos farmacéuticos residuales y sus sustancias constituyentes.
Preparación de las muestras de residuoLas muestras de residuo se prepararon aplicando varios tipos de productos farmacéuticos y sus constituyentes a recipien-tes de acero inoxidable. Se evaluaron compuestos con niveles variables de solubilidad en agua (es decir, soluble, insoluble y muy insoluble) para determinar cómo se comportaba cada método. Las sustancias solubles en agua se disolvieron en agua y las sustancias insolubles en agua se disolvieron en etanol o acetona, según se muestra en la Tabla I. Las concen-traciones de la solución se ajustaron a 2,000 mgC/L (es decir, concentración de carbón de 2000 mg/L).
Métodos de liMPieza
Métodos de medición del carbón para validación de limpiezaComparación de la combustión directa con los métodos de enjuague y muestreo con hisopo Robert Clifford y Minako Tanka
toc
-Lc
Ph
, S
hIm
aD
zu

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 43
Se estimaron los contenidos de carbón de ácido tranexámi-co (C
8H
15NO
2), cafeína anhidra (C
8H
10N
4O
2), isopropilantipiri-
na (C14
H18
N2O), y nifedipina (C
17H
18N
2O
6) mediante la fórmula
molecular. Los contenidos de carbón del ungüento Gentashin (antibiótico aminoglicósido) y el ungüento Rinderon (corticos-teroide) fueron determinados con el analizador TOC agregando muestras de los ungüentos directamente en una unidad de com-bustión de muestras sólidas (SSM-5000A, Shimadzu) ya que la fórmula molecular para estos compuestos es desconocida.
Cada muestra de residuo consistió en un área de 5 cm2 sobre la superficie de un recipiente al cual se aplicó y se secó 100 µL de cada solución. Por lo tanto, había 200 µg de carbón en la muestra en cada sitio de aplicación.
toD
aS
La
S F
Igu
ra
S S
oN
co
rt
eS
Ia D
e L
oS
au
tor
eS
Tabla I: Sustancias usadas para las mediciones del residuo.
Nombre de la sustancia Solubilidad en aguaSolvente usado en la preparación de la solución
Ácido tranexámico Soluble Agua
Cafeína anhidra Soluble Agua
Isopropilantipirina Insoluble Etanol
Nifedipina Insoluble Acetona
Ungüento Gentashin Muy insoluble Etanol
Ungüento Rinderon Muy insoluble Acetona
Tabla II: Mediciones utilizando el muestreo con enjuagueNombre de la sustancia
Concentración TOC (mgC/L)
Tasa de recobro
Coeficiente de variación (%)
Blanco 0.030 - -
Ácido tranexámico 2.14 105 1.26
Cafeína anhidra 2.19 108 1.86
Isopropilantipirina 2.20 109 1.97
Nifedipina 2.17 107 1.97
Ungüento Gentashin 0.117 4.35 16.73
Ungüento Rinderon 0.333 15.2 58.60
Figura 1: Concentraciones de carbón orgánico total (TOC) para (a) ácido tranexámico, (b) isopropilantipirina y (c) Gentashin ungüento utilizando muestreo con enjuague.
40
30
20
10
-4
(a) (b)
0 5 10 15 20
Time [min]
Inj. No. Conc. Result CV Conc.
123
2.1082.1602.147
NPOC:2.138mg/L 1.26Inj. No. Conc. Result CV Conc.
123
2.2502.1722.178
NPOC:2.200mg/L 1.97
Sig
nal
[m
V]
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
(c)
Inj. No. Conc. Result CV Conc.
123
0.13960.10560.1059
NPOC:0.1170mg/L 16.73
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
Método de muestreo con enjuagueEn el muestreo con enjuague, el agua del enjuague final de la limpieza de una unidad del equipo de producción se utiliza como la muestra para la medición TOC. Este método es ade-cuado para sistemas que no pueden desarmarse fácilmente, como el equipo de limpieza en el sitio (CIP) y la tubería es-trecha. El muestreo se considera difícil si los residuos no son solubles en agua.
Para evaluar el recobro de las diversas sustancias utilizan-do este método, se agitaron 100 mL de agua pura durante 15 min en el recipiente de acero inoxidable que contenía un par-che de muestra seca. La medición TOC se realizó en la solu-ción de enjuague utilizando un analizador TOC (TOC-LCPH, Shimadzu) con un catalizador de alta sensibilidad. El análisis del TOC fue mediante el método de acidificar y salpicar. La curva de calibración fue una curva de 2 puntos utilizando 0-3 mgC/L de solución acuosa de ftalato ácido de potasio. Se uti-lizó un volumen de inyección de 500 µL. Debido al contenido de carbón en cada una de las muestras de medición del resi-duo era de 200 µg, la concentración TOC teórica (es decir, si todo el carbón se disolviera en el agua de enjuague) sería de 2 mgC/L. La Figura 1 muestra las concentraciones TOC me-didas para muestras solubles en agua representativas (a, ácido tranexámico), muestras insolubles en agua (b, isopropilanti-pirina) y ungüentos insolubles en agua (c, Gentashin ungüen-to). Las otras muestras (es decir, cafeína anhidra, nifedipina y Rinderon ungüento) tienen perfiles similares a las muestras con la solubilidad correspondiente.
Para el blanco, la medición se realizó de la misma manera utilizando agua en un recipiente de acero inoxidable sin la muestra seca aplicada a su superficie. La concentración medi-da del blanco se restó de cada concentración TOC y se dividió entre el valor teórico de 2 mgC/L (es decir, la concentración teórica si toda la muestra se disolviera en el agua) para deter-minar la tasa de recobro, según se muestra en la Ecuación 1.
Ec. 1
Todas las muestras se corrieron por triplicado, y los valo-res del coeficiente de variación (CV) se muestran en la Tabla II junto con las concentraciones TOC y las tasas de recobro.
El ácido tranexámico soluble en agua y la cafeína anhidra insoluble en agua tuvieron altas tasas de recobro, según lo es-

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201244
Tabla III: Mediciones utilizando el muestreo con hisopo con extracción con agua Nombre de la sustancia
Concentración TOC (mgC/L)
Tasa de recobro
Coeficiente de variación (%)
Blanco 0.059 - -
Ácido tranexámico 2.19 107 0.94
Cafeína anhidra 2.23 109 0.91
Isopropilantipirina 1.90 92.2 1.34
Nifedipina 1.86 89.9 0.36
Ungüento Gentashin
0.093 1.70 20.15
Ungüento Rinderon 0.208 7.45 10.72
Figura 2: Concentraciones de carbón orgánico total (TOC) para (a) ácido tranexámico, (b) isopropilantipirina y (c) Gentashin ungüento utilizando muestreo con hisopo con extracción con agua.
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
Inj. No. Conc. Result CV Conc.
123
2.2182.1792.186
NPOC:2.194mg/L 0.94
Inj. No. Conc. Result CV Conc.
123
1.9321.8901.886
NPOC:1.902mg/L 1.34
(a) (b)
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
Inj. No. Conc. Result CV Conc.
123
0.11330.075830.09115
NPOC:0.09342mg/L 20.15
(c)
Métodos de liMPieza
perado. Además, la isopropilantipirina y la nifedipina, insolu-bles en agua, tuvieron altas tasas de recobro. Sin embargo, las tasas de recobro de Gentashin ungüento y Rinderon ungüento fueron ambas bajas, en menos de 20%. En consecuencia, el método de enjuague de para TOC, aunque es aceptable para algunas sustancias es inadecuado para ungüentos y otras sus-tancias similares.
Método de muestreo con hisopo con extracción con aguaEl muestreo con hisopo con extracción con agua consiste en limpiar la superficie interna del aparato de producción con un material fibroso, extrayendo el material adherido con agua y realizando la medición del TOC de la solución del extracto. Como el residuo se retira físicamente con el hisopo de un área fija de la superficie, la eficiencia del muestreo es alta. Los re-siduos que son insolubles en agua, sin embargo, son difíciles de extraer con agua. En consecuencia, la evaluación de resi-duos insolubles en agua con este método puede presentar difi-cultades similares a las del método de muestreo con enjuague.
Para evaluar el recobro de las diversas sustancias utilizan-do el muestreo con hisopo con extracción con agua, la mues-tra aplicada al recipiente de acero inoxidable se limpió con una pieza de 5 cm2 de material fibroso, la cual fue colocada en un tarro de vidrio conteniendo 100 mL de agua pura. El material fibroso de la pieza (Texwipe Alpha 10 lavado en agua
pura y secado) consiste en poliéster, de manera que se extrae muy poco material orgánico del propio paño. El residuo se extrajo agitando durante 1 h y la medición del TOC se llevó a cabo utilizando el mismo equipo y condiciones usadas para el método de muestreo con enjuague. Se corrieron tres replica-dos de cada muestra. Al igual que en el método de muestreo con enjuague, como el contenido de carbón en cada una de las muestras para medición del residuo es de 200 µg, la concen-tración del TOC (es decir, el TOC teórico) en la solución de extracción sería de 2 mgC/L si toda la muestra fuera limpiada. Los datos representativos se muestran en la Figura 2. Para el blanco, la medición se llevó a cabo de la misma manera, limpiando el recipiente de acero inoxidable, el cual no tenía ninguna muestra aplicada antes de realizar la extracción. La tasa de recobro se determinó utilizando la Ecuación 1. Los resultados se muestran en la Tabla III.
El ácido tranexámico y la cafeína anhidra, solubles en agua, tuvieron altas tasas de recobro, según era de esperar. Adi-cionalmente, la isopropilantipirina y la nifedipina, insolubles en agua, tuvieron altas tasas de recobro de aproximadamente 90%. Sin embargo, las tasas de recobro del Gentashin ungüen-to y del Rinderon ungüento fueron ambas bajas, de menos del 10%. Estos resultados muestran que el método de enjuague con extracción con agua para el TOC es confiable y exacto para algunas sustancias, pero inadecuado para ungüentos y quizás otras sustancias, debido a las bajas tasas de recobro.
Método de muestreo con hisopo con combustión directaEl muestreo con hisopo con combustión directa consiste en limpiar la superficie interna del aparato de producción con una pieza de material de hisopo con filtro de cuarzo-papel, y reali-zando después la medición utilizando un sistema de medición de carbón con combustión directa. El material de hisopo con el residuo adherido se mide directamente (es decir, sin extraer primero con agua) en un analizador TOC utilizando una uni-dad o módulo de combustión de muestras sólidas (SSM).
Para evaluar la tasa de recobro de los diferentes tipos de sustancias utilizando este método, el material de hisopo de pa-pel (papel de vidrio de cuarzo QR-100 Advantec de 45 mm, tratado con calor a 600°C durante 15 min) se utilizó para limpiar la muestra adherida al recipiente de acero inoxidable y colocado después en a nave de muestras, la cual se colo-
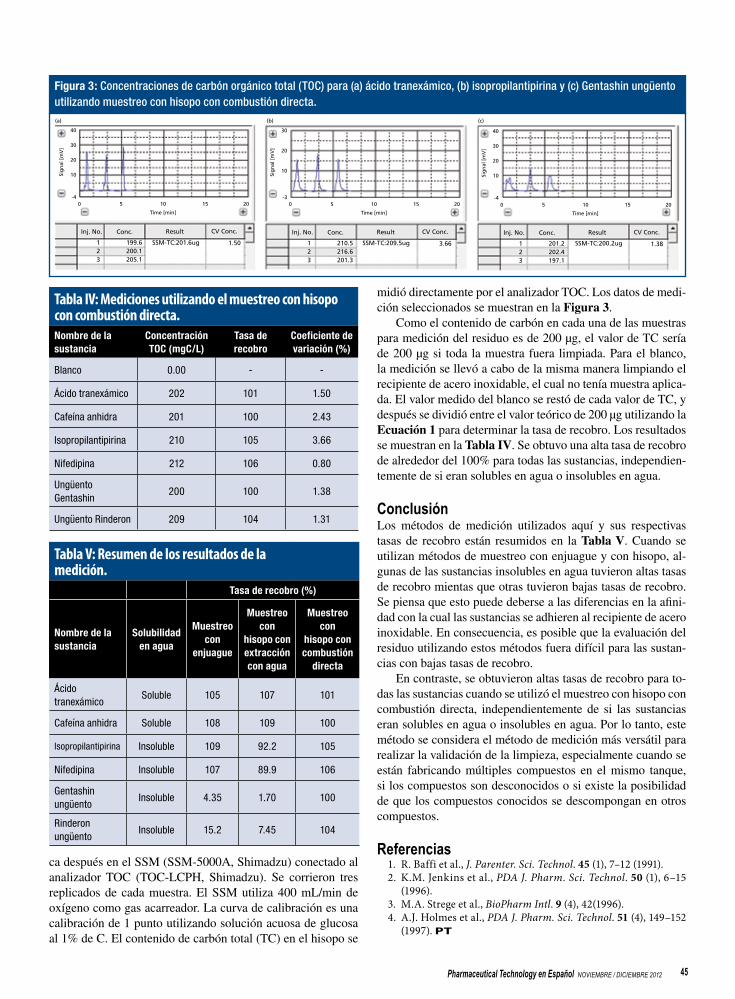
Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 45
ca después en el SSM (SSM-5000A, Shimadzu) conectado al analizador TOC (TOC-LCPH, Shimadzu). Se corrieron tres replicados de cada muestra. El SSM utiliza 400 mL/min de oxígeno como gas acarreador. La curva de calibración es una calibración de 1 punto utilizando solución acuosa de glucosa al 1% de C. El contenido de carbón total (TC) en el hisopo se
Figura 3: Concentraciones de carbón orgánico total (TOC) para (a) ácido tranexámico, (b) isopropilantipirina y (c) Gentashin ungüento utilizando muestreo con hisopo con combustión directa.
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
30
20
10
-30 5 10 15 20
Time [min]
Sig
nal
[m
V]
Inj. No. Conc. Result CV Conc.
123
199.6200.1205.1
SSM-TC:201.6ug 1.50
Inj. No. Conc. Result CV Conc.
123
210.5216.6201.3
SSM-TC:209.5ug 3.66
(a) (b)
40
30
20
10
-40 5 10 15 20
Time [min]
Sig
nal
[m
V]
Inj. No. Conc. Result CV Conc.
123
201.2202.4197.1
SSM-TC:200.2ug 1.38
(c)
Tabla IV: Mediciones utilizando el muestreo con hisopo con combustión directa.Nombre de la sustancia
Concentración TOC (mgC/L)
Tasa de recobro
Coeficiente de variación (%)
Blanco 0.00 - -
Ácido tranexámico 202 101 1.50
Cafeína anhidra 201 100 2.43
Isopropilantipirina 210 105 3.66
Nifedipina 212 106 0.80
Ungüento Gentashin
200 100 1.38
Ungüento Rinderon 209 104 1.31
Tabla V: Resumen de los resultados de la medición.
Tasa de recobro (%)
Nombre de la sustancia
Solubilidad en agua
Muestreo con
enjuague
Muestreo con
hisopo con extracción con agua
Muestreo con
hisopo con combustión
directa
Ácido tranexámico
Soluble 105 107 101
Cafeína anhidra Soluble 108 109 100
Isopropilantipirina Insoluble 109 92.2 105
Nifedipina Insoluble 107 89.9 106
Gentashin ungüento
Insoluble 4.35 1.70 100
Rinderon ungüento
Insoluble 15.2 7.45 104
midió directamente por el analizador TOC. Los datos de medi-ción seleccionados se muestran en la Figura 3.
Como el contenido de carbón en cada una de las muestras para medición del residuo es de 200 µg, el valor de TC sería de 200 µg si toda la muestra fuera limpiada. Para el blanco, la medición se llevó a cabo de la misma manera limpiando el recipiente de acero inoxidable, el cual no tenía muestra aplica-da. El valor medido del blanco se restó de cada valor de TC, y después se dividió entre el valor teórico de 200 µg utilizando la Ecuación 1 para determinar la tasa de recobro. Los resultados se muestran en la Tabla IV. Se obtuvo una alta tasa de recobro de alrededor del 100% para todas las sustancias, independien-temente de si eran solubles en agua o insolubles en agua.
ConclusiónLos métodos de medición utilizados aquí y sus respectivas tasas de recobro están resumidos en la Tabla V. Cuando se utilizan métodos de muestreo con enjuague y con hisopo, al-gunas de las sustancias insolubles en agua tuvieron altas tasas de recobro mientas que otras tuvieron bajas tasas de recobro. Se piensa que esto puede deberse a las diferencias en la afini-dad con la cual las sustancias se adhieren al recipiente de acero inoxidable. En consecuencia, es posible que la evaluación del residuo utilizando estos métodos fuera difícil para las sustan-cias con bajas tasas de recobro.
En contraste, se obtuvieron altas tasas de recobro para to-das las sustancias cuando se utilizó el muestreo con hisopo con combustión directa, independientemente de si las sustancias eran solubles en agua o insolubles en agua. Por lo tanto, este método se considera el método de medición más versátil para realizar la validación de la limpieza, especialmente cuando se están fabricando múltiples compuestos en el mismo tanque, si los compuestos son desconocidos o si existe la posibilidad de que los compuestos conocidos se descompongan en otros compuestos.
Referencias1. R. Baffi et al., J. Parenter. Sci. Technol. 45 (1), 7–12 (1991).2. K.M. Jenkins et al., PDA J. Pharm. Sci. Technol. 50 (1), 6–15
(1996).3. M.A. Strege et al., BioPharm Intl. 9 (4), 42(1996).4. A.J. Holmes et al., PDA J. Pharm. Sci. Technol. 51 (4), 149–152
(1997). PT

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201246
8324
332.957452.0
1793.00
1784.0
9325.0
1793.00
93.99%
8371.00
1793.00
4003
.0
92.1%43.2%
24.3%
21.980%92.1%
56.1%
64.2%
92.1%
18.4%
478.00
563.00
Tecnología Analítica de Proceso: seguimiento del avance en el análisis de datosMesa redonda de la industria moderada por Patricia Van Arnum
Los expertos de la industria comparten perspectivas sobre instrumentación analítica, métodos y análisis de datos.
primera pLana: TecnoLogía anaLíTica
Cuando la FDA anunció por primera vez en 2002 una nueva iniciativa, Buenas Prácticas de Manufactura
Farmacéutica Vigentes (CGMPs) para el Siglo XXI, y posteriormente publicó su reporte, cGMPs Farmacéuticas para el Siglo XXI – Un enfoque basado en el riesgo, en el 2004, puso en movimiento un esfuerzo para mejorar la calidad del producto y modernizar la manufactura farmacéutica a través de un enfoque ba-sado en la ciencia y en el riesgo bajo los principios de la calidad por diseño (QbD) (1). Este esfuerzo fue además motivado por la emisión de la guía
sobre tecnología analítica de proceso (PAT) en 2004 para facilitar la introduc-ción de nuevas tecnologías que mejora-rían la comprensión del proceso y apo-yarían en la identificación y control de puntos críticos en un proceso (2). Estas tecnologías incluyen: dispositivos de medición apropiados, los cuales pueden colocarse, en, dentro y sobre la línea; herramientas estadísticas y de tecnolo-gía de la información; y un esquema de sistemas científicos para el análisis de los datos para controlar los procesos y asegurar la producción de materiales en proceso y productos finales de la cali-dad deseada (3).
Así que, ¿Qué tan lejos ha llegado la industria en el avance del PAT y dón-de puede residir la innovación futura? Pharmaceutical Technology realizó una mesa redonda con la industria para ob-tener perspectiva sobre los avances en instrumentación analítica y desarrollo de métodos. Los participantes fueron: Tim Freeman, director general de Free-man Technology y presidente del Grupo de Enfoque de Tecnología Analítica de Proceso de la Asociación Americana de Científicos Farmacéuticos; Kevin Au-miller, gerente de producto TOC en GE Analytical Instruments; Andy Salamon, científico senior del personal y defensor de clientes en PerkinElmer; Chris Heil, especialista de producto, analizadores Antaris NIR en Thermo Fisher Scienti-fic; y Scott Samojla, director senior de sistemas de proceso PATROL en Waters.
Avances en PATPharmTech: ¿Qué identificaría usted como el avance más significativo en el PAT usado en la industria farmacéutica durante los últimos cinco años?
Freeman (Freeman Technology): Antes de enfocarme en tecnologías espe-cíficas, me gustaría destacar lo que pien-so que es un cambio importante en la percepción del PAT durante los últimos cinco años. Introducido inicialmente por los reguladores en el 2004 para respaldar la eficiencia en el desarrollo, la manu-factura y el aseguramiento de calidad, el PAT ha, en los años en que ha inter-venido, demostrado progresivamente su valor potencial. La experiencia colectiva e individual ha llevado a una aprecia-ción muy amplia del papel integral que jugará el PAT en el logro de nuevos ni-veles de eficiencia de manufactura y de calidad de producto, y como resultado, la captación del PAT se ha incrementado ahora significativamente, de manera más notable por la colecta de información necesaria para soportar el desarrollo de procesos más eficientes.
Este enfoque sobre colecta de in-formación y monitoreo del proceso se refleja en las tecnologías que han avanzado más durante los últimos cin-co años. El uso de sistemas dentro de la línea o en línea con un registro de rastreo demostrado se ha incrementado considerablemente pero de la misma

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 47
forma también la aplicación de métodos en línea que proporcionan información única y conocimiento.
Los sistemas dentro y en línea apor-tan valor al permitir el rastreo en tiempo real de la conducta del proceso y de las propiedades cambiantes de los materia-les en proceso. Hoy día, la industria far-macéutica está haciendo mayor uso de algunas técnicas en línea tradicionales, tales como la medición del pH, y tam-bién de tecnologías en tiempo real más recientes, tales como el dimensiona-miento de partículas y, desde luego, los métodos espectroscópicos tales como el NIR. En la plaza de la medición en lí-nea, yo reconocería la imagenología de partículas y los sistemas de caracteriza-ción de polvos en volumen, como ejem-plos de tecnología que son realmente un avance del conocimiento. Mediante la aportación de la información necesa-ria para racionalizar la conducta de la partícula y del polvo, estas tecnologías soportan el logro de un mejor desempe-ño del procesamiento de polvos, el cual es vital para la mayor eficiencia de la manufactura.
Heil (Thermo Fisher Scientific): La espectroscopía NIR ha madurado en una técnica común para el análisis PAT a través del ciclo de vida de manufac-tura farmacéutica completa, desde la identificación de la materia prima hasta granulación y secado hasta el mezclado y la producción de tabletas. La llegada de los analizadores NIR que se ajustan al propósito y para la solución total ha desbloqueado el potencial comple-ta de la espectroscopía NIR como una tecnología analítica de proceso para el monitoreo y el control de los procesos de producción farmacéutica. Nosotros hemos atestiguado el desarrollo de ana-lizadores dedicados PAT NIR durante los últimos cinco años, los cuales ofre-cen soluciones de análisis de proceso totales. La evolución en el diseño fue atestiguada no sólo en los analizadores NIR sino también en el software, los accesorios y las sondas requeridas para una solución de análisis total. La inte-gración del software y la comunicación del proceso también son aspectos clava de un paquete total del analizador.
La experiencia ha demostrado que la faceta más desafiante y con frecuen-
cia más importante para un análisis de proceso exitoso es el muestreo repre-sentativo. La interfaz del analizador con el punto de muestreo es crítica ya que ésta es donde el espectrómetro NIR cuestiona la muestra para el análisis. Un ejemplo perfecto es el reto para ob-tener una muestra representativa en una sonda instalada en un secador de lecho fluido cuando el producto se está fluidi-ficando y el nivel de humedad dicta si el producto cubrirá o ensuciará la ventana de la sonda. Los avances en el diseño de sondas de fibra óptica han llevado a sondas que se auto-limpian con aire de purga o mecanismos de retracción para evitar que la sonda se ensucie. Adicio-nalmente, se han diseñado sondas que usan diseños en forma de copas, de ventanas con vista lateral, puntas en ángulo, y curvadas, para asegurar que las muestras representativas estén en contacto con la ventana de la sonda. Estos avances en los analizadores NIR, el software y los accesorios han promo-vido la aplicación del PAT para la opti-mización del proceso de producción, la reducción en los costos de producción y la calidad mejorada del producto a tra-vés de mediciones oportunas de atribu-tos de calidad típicos.
Samojla (Waters): La necesidad de un conocimiento científicamente mejorado del proceso de manufactura ha sido y continuará siendo la clave para el éxito futuro en la industria far-macéutica. La llegada de cromatografía de líquidos de ultra-resolución propor-cionó el poder no sólo para cumplir es-
tas necesidades dentro del laboratorio, sino que permite el uso cromatográfico en línea para el monitoreo y control del proceso. Los beneficios de este enfoque reducen la variabilidad del proceso y el riesgo agregando mientras tanto auto-matización e información de la que no se disponía anteriormente en línea.
Aumiller (GE Analytical Instru-ments): Un buen ejemplo del avance del PAT ha sido asociado con la limpie-za y liberación del equipo. Los sistemas de limpieza en el lugar (CIP), los cuales han sido usados históricamente en el espacio biofarmacéutico, han ganado fuerza dentro de las plantas farmacéu-ticas tradicionales y las organizacio-nes de manufactura por contrato. Esto ha sido promovido por la actividad sin precedentes de fusiones y adquisiciones en los últimos tres a cinco años. Con un enfoque de la industria sobre la capa-cidad cada vez mayor de las plantas y la reducción de los costos de operación globales, las plantas están adquiriendo más productos, lo cual a su vez, gene-ra la necesidad de cambios rápidos en el equipo. La implementación del CIP para reemplazar la limpieza manual ha proporcionado oportunidades significa-tivas para la eficiencia en esta área.
Al ofrecer los beneficios de los ci-clos de limpieza automatizados, pro-nosticables y repetibles, los sistemas CIP satisfacen los objetivos principales del PAT construyendo la calidad den-tro del proceso y proporcionando el aseguramiento en tiempo real de la ca-lidad. Los atributos críticos de calidad
Participantes de la mesa redonda de la industria, de izquierda a derecha: Tim Freeman, director general de Freeman Technology y presidente del Grupo de Enfoque de Tecnología Analítica de Proceso de AAPS; Kevin Aumiller, gerente de producto TOC en GE Analytical Instruments; Andy Salamon, científico senior del personal y defensor de clientes en PerkinElmer; Scott Samojla, director senior de sistemas de proceso PATROL en Waters; y Chris Heil (no aparece), especialista de producto, analizadores Antaris NIR en Thermo Fisher Scientific

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201248
y desempeño asociados con la limpieza pueden ser monitoreados y controlados continuamente. Por ejemplo, la tempe-ratura y la concentración de las solu-ciones de limpieza pueden ser monito-readas con sondas de conductividad en línea y sensores de temperatura. La agi-tación de los vasos puede ser controla-da activamente con medidores de flujo y transductores de presión. Con el uso de sistemas de control lógico del pro-ceso (PLC), las secuencias pueden ser cronometradas para dar una exposición consistente a los agentes de limpieza y al agua de enjuague.
Una vez que la limpieza está com-pleta, puede realizarse el muestreo in-directo del agua de enjuague final en casi tiempo real con los analizadores de carbón orgánico total (TOC) en línea y los sensores de conductividad en línea. Las muestras de hisopo directo también pueden ser analizadas con instrumen-tación TOC en línea. Los instrumentos TOC proveen detección del produc-to residual y otras especies no iónicas presentes en el proceso. Los sensores de conductividad pueden detectar la presencia de contaminantes iónicos, ta-les como los agentes de limpieza en el agua de enjuague. La combinación de métodos de prueba provee retroalimen-tación oportuna para liberar el equipo para uso.
El futuro del PATPharmTech: Viendo a cinco años en el futuro, ¿Cómo ves el PAT evolucionan-do en la industria farmacéutica?
Freeman (Freeman Technology): Aunque el uso del PAT para colecta de información y monitoreo del pro-ceso, particularmente en el desarrollo, ha crecido sustancialmente durante los últimos cinco años, hay todavía manera de avanzar en términos de la optimiza-ción de su aplicación a través del ciclo completo de manufactura y su utiliza-ción como mecanismo para el control de proceso. En los próximos cinco años, yo espero que el PAT penetre más a la manufactura comercial conforme la confianza en su uso y las capacidades de expandan.
La automatización del control de proceso es una tendencia continua y
primera pLana: TecnoLogía anaLíTica
continuará para motivar a los provee-dores a llevar nuevas tecnologías en línea. Por otro lado, la experiencia de otros sectores sugeriría que la sofistica-da aplicación de las técnicas más rele-vantes en línea es un esquema extrema-damente complementario y productivo. Por lo tanto, parece que probablemente el enfoque debe y seguirá estando en las tecnologías que verdaderamente producen en términos de información relevante y sea en línea o dentro de la línea. Los parámetros, tales como la morfología de las partículas, y la fluidez del polvo, la compresibilidad, la fuerza de cizallamiento y el área de superficie seguirán siendo todos probablemente pertinentes para los procesos de polvo.
Heil (Thermo Fisher Scientific): En la industria farmacéutica, el PAT está bien adoptado entre las compañías far-macéuticas más grandes, especialmente en los países más desarrollados. En los próximos cinco años, veo la evolución del PAT en compañías farmacéuticas más pequeñas, en organizaciones de manufactura por contrato, en fabricantes de suplementos dietéticos, y en compa-ñías de países menos desarrollados. Las compañías y la industria en estas regio-nes han visto el progreso que las com-pañías farmacéuticas más grandes han hecho y ven el valor de las herramientas del PAT, tales como la espectroscopía NIR, para el monitoreo y control de sus procesos para mejorar la calidad y redu-cir los costos de producción.
Los avances en análisis de datos y manejo de datos son críticos para el éxito futuro del PAT. El manejo de la gran cantidad de datos multivariados generados en tiempo real, de un proceso de producción, de manera que puedan ser usados para ajustes del proceso en tiempo real o en el análisis de produc-ción fuera de la línea es un reto. Duran-te los últimos diez años, se ha adquirido conocimiento significativo sobre cómo implementar apropiadamente la instru-mentación del proceso para el análisis en línea.
Para muchas compañías, el pun-to de arranque para implementar las herramientas del PAT fue cambiar las técnicas de espectroscopía del labo-ratorio a la línea. El valor del PAT se
construye alrededor del análisis en tiempo real de los atributos críticos de calidad y desempeño para mejorar la calidad del proceso. Para un proceso farmacéutico, esto significa combinar los datos de múltiples fuentes para ob-tener un cuadro completo de la calidad global del material crudo, en proceso y de los productos finales. Este enfoque requiere sistemas avanzados de manejo de datos capaces de recibir información de varios formatos de múltiples analiza-dores y sistemas de automatización de la producción. Tomando como ejemplo el manejo de datos requerido cuando se aplica el PAT al proceso farmacéutico de extrusión con fundido caliente. Los datos en tiempo real de múltiples for-matos y fuentes de un analizador NIR, la sonda de temperatura del barril, el sensor de presión, la velocidad de la es-piral del extrusor, y las velocidades de alimentación de los ingredientes nece-sitarían archivarse en una ubicación co-mún para el tiempo real, el análisis de datos multivariados así como el análisis del proceso posterior fuera de la línea.
Con los avances futuros en el análi-sis de datos y el software de manejo de datos, las compañías podrán enfocar sus esfuerzos del PAT en la optimización de su muestreo en el punto de análisis, mo-delos quimiométricos, y en herramien-tas de análisis multivariado permitién-doles así aplicar el PAT a procesos de producción más desafiantes.
Samojla (Waters): El análisis cro-matográfico en tiempo real mejorará cada vez más los procesos de manufac-tura –desarrollo, transferencia, monito-reo y control. Integrando estas mejoras dentro de un modelo de acceso de in-formación unificado, capaz de analíti-cas de muy alta velocidad se obtendrá un manejo de decisiones mejorad, el análisis predictivo y el análisis amplio –sin el papel. Esta integración puede ser lograda mediante el despliegue de la analítica en tiempo real, eliminando mientras tanto los retrasos en tiempo, la analítica, y la variabilidad del proceso
“Tecnología Analítica de Proceso: seguimiento del avance en el análisis de datos”continúa en la pág. 54

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 49
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
6

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201250
Existe la necesidad de un conocimiento más profundo de los principios subyacentes del proceso farmacéutico de recubrimiento de tabletas en un bombo. Los autores investigan el proceso de recubrimiento de tabletas utilizando una combinación de diferentes técnicas de simulación, incluyendo el método de elemento discreto y los métodos de dinámica de fluidos computacionales multifase. Los resultados pueden ser usados para optimizar el tiempo de residencia en la zona de aspersión, la formación de película o el secado del atomizado.
Gregor Toschkoff, Daniele Suzzi, PhD y Siegfried Adam, PhD, son investigadores en el Centro de Investigación para Ingeniería Farmacéutica (RCPE) en Graz, Austria. Johannes Khinast*, PhD, es director del RCPE y jefe del Instituto para Ingeniería de Procesos y Partículas, Universidad Graz de Tecnología, Inffeldgasse 21/a/II 8010 Graz, Austria, tel. +43 316 873 7978, [email protected].
*A quien debe dirigirse la correspondencia.
Sometido: Abril 15, 2011; Aceptado: Enero 16, 2012.
1In silico es una expresión usada para indicar que algo se realiza por computadora o a través de una simulación en computadora; es el
equivalente a los términos in vivo ó in vitro usados en biología.
Los bombos de recubrimiento son ampliamente usa-dos en la industria farmacéutica para producir ta-bletas con recubrimiento pelicular. La o las capas de recubrimiento alrededor del núcleo de las table-
tas puede servir a muchos propósitos, tales como enmascara-do del sabor, color, control de liberación del API del núcleo de la tableta, aplicación de un API adicional, o protección del núcleo de la tableta de las influencias ambientales. En este proceso, una boquilla atomizadora colocada por arriba de las tabletas rocía la solución de recubrimiento sobre las tabletas, las cuales son mezcladas por el bombo rotatorio. El aire ca-liente se fuerza a través del sistema para mejorar el secado del recubrimiento. De esta forma, cada tableta recibe una serie de recubrimientos parciales (ver Figura 1). Un tema crucial es la uniformidad del recubrimiento, la cual incluye la uniformidad inter-tableta (es decir, variación del peso del recubrimiento de una tableta a la otra) y la uniformidad intra-tableta (es decir, variación del espesor del recubrimiento y calidad en la su-perficie de una sola tableta) (1-3). La no homogeneidad en el espesor del recubrimiento, por ejemplo, podría llevar a varia-ciones significativas en el contenido del API o en la velocidad de liberación. En muchos casos, una sola tableta que falla el análisis puede ocasionar el rechazo del lote completo.
Los parámetros que influyen el desempeño del recubri-miento con bombo pueden dividirse en dos grupos. El pri-mer grupo incluye parámetros que pueden ser ajustados por el operador (p.ej., velocidad de rotación del bombo, nivel de llenado, velocidad de atomización, o temperatura del aire de secado). El segundo grupo son parámetros que no pueden ajustarse directamente, pero tienen una influencia directa en la calidad del producto (p.ej., extensión de la zona de ato-mizado, eficiencia del mezclado, patrón de flujo de aire, y calidad del atomizado de recubrimiento) (4).
Aunque el recubrimiento con bombo es usado común-mente en la industria farmacéutica, han habido relativamente pocas investigaciones científicas reportadas en la literatura. Además, el diseño del proceso se ha basado mayormente en el ensayo y error y en la experiencia del operador. Reciente-mente, además del trabajo experimental, los avances en las simulaciones computacionales se han convertido en una im-portante herramienta para dichas investigaciones (5-8).
El principal objetivo de este trabajo es mostrar cómo una combinación de diferentes técnicas puede ayudar a propor-
recubriMiento de tabletas
Simulación numérica del recubrimiento de tabletasPrimeros pasos hacia un espacio de diseño in-silico1 Gregor Toschkoff, Daniele Suzzi, Siegfried Adam y Johannes Khinast
ch
rIS
ro
ge
rS
/ge
tt
y Im
ag
eS

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 51
cionar un análisis amplio, in silico (es decir, vía una simulación en computa-dora) del proceso completo de recubri-miento de la tableta. Se identificaron los siguientes aspectos del proceso de recubrimiento pelicular (ver Figura 2):
• Simulación del mezclado del lecho de tabletas utilizando el método del elemento discreto (DEM)
• Análisis numérico detallado de la interacción entre las gotas del ato-mizado y la superficie de la tableta por medio de métodos avanzados de dinámica de fluidos computa-cional (CFD) multifase
• Análisis computacional del flujo interior del recubridor en térmi-nos de aire de secado, atomizado y efectos de la pared utilizando CFD.
El trabajo tiene como objetivo pro-veer una comprensión más profunda de:
• El proceso de mezclado de las tabletas en un bombo de recubri-miento industrial, el cual permite la optimización del tiempo de re-sidencia de la tableta en la zona de atomizado
• La conducta local de las gotas del atomizado que impactan sobre una sola tableta, lo que lleva a una com-prensión más amplia de los meca-nismos de formación de la película
• La interacción entre el atomizado y el aire de secado del recubri-dor, permitiendo así un análisis detallado de la eficiencia de de-posición del material de recu-brimiento sobre el lecho de las tabletas y la información de los problemas operativos como el sobre-atomizado, el taponado de los filtros o el curado incomple-to del recubrimiento de la tableta.
Método de simulación computacionalLas simulaciones DEM de partículas no esféricas en diferentes geometrías del recubridor fueron usadas para es-tudiar el flujo de las partículas dentro del recubridor y el tiempo de residencia de las tabletas en la zona de atomiza-ción (EDEM software v2.3, Soluciones DEM). Los atributos del material de una muestra de tableta placebo fueron cuantificados experimentalmente como control. Las simulaciones DEM consi-deran las fuerzas del cuerpo y las fuer-
zas de contacto tangenciales sobre las tabletas. Las ecuaciones de Newton son resueltas para calcular la velocidad y la velocidad rotacional de la tableta. Los modelos de contacto apropiados, que se aplican en los puntos de contacto de las tabletas, explican las colisiones tableta-tableta y tableta-pared. En este estudio se usó un modelo de contacto basado en la teoría de Hertz-Mindlin.
Se hicieron análisis del proceso de recubrimiento local y del flujo del aire y del atomizado en la cámara de recu-brimiento utilizando simulaciones mul-
tifase CFD (FIRE v2009, AVL List). El software se utilizó para estudiar la interacción entre el atomizado de re-cubrimiento y las tabletas, tanto local-mente (es decir, para una sola tableta) como globalmente (es decir, calculando el flujo de aire en el recubridor com-pleto). Para este fin, se simularon las gotas del atomizado con una técnica de Euler-Lagrange para el método de gotas discretas (DDM). Se toma en cuenta la evaporación del solvente con el fin de estimar los efectos del flujo del aire de secado. Un modelo bidimensional que
Atomizado
HumectaciónGota
Particula
Ciclo derecubrimiento Partícula
recubierta
Análisis por SEM deuna película de tableta
(a) Simulación del mezclado de la tableta
(b) Simulación detallada del atomizado/tableta
(c) Simulación del flujo del recubridor
Figura 1: En el proceso de recubrimiento, cada tableta recibe recubrimientos parciales, lo cual lleva a una capa homogénea de recubrimiento. La foto de microscopía electrónica de barrido (SEM) muestra una sección transversal de la capa de recubrimiento sobre el núcleo de la tableta.
Figura 2: Representación esquemática que muestra diferentes regímenes del proceso de recubrimiento con un esquema de simulación apropiado para cada uno. Para (a) y (c), los colores denotan velocidad desde lenta (azul) hasta rápida (roja). En (b), el color rojo representa el espesor del recubrimiento; la tableta a la derecha en el cuadro inferior tiene un recubrimiento más grueso.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201252
incorpora sub-modelos para la fuerza de cizallamiento interfacial, la evapora-ción de la película y la transferencia de calor entre la película, la pared sólida y el aire, describe la película de recubri-miento no seca.
recubriMiento de tabletas
Investigación DEM del movi-miento de la tabletaLa Figura 3 muestra una simulación DEM para un recubridor continuo (Driaconti, DRIAM Anlagenbau). Las tabletas se colorean de acuerdo a su ve-
Tiempo de residencia fraccional
Relación longitud a altura Nivel de llenado
0.038
0.036
0.034
0.032
0.03
0.028
0.026
0.024
0.022
0.021
1.21.4
1.61.8
0.060.055
0.05
0.0450.04
2
Figura 3: Simulaciones DEM en un recubridor de ciclo continuo (DriaConti, Driam). El color de la tableta indica la velocidad desde lento (azul) a rápido (rojo).
Velocidad
1.25
1
0.75
0.5
0.25
0
Figura 4: Tiempo de residencia fraccional promedio en la zona de atomizado como función de la relación longitud a altura de la forma de la tableta (redonda, biconvexa y ovalada en orden ascendente) y niveles de llenado de la cubierta.
locidad, con la más rápida cerca de la superficie, en la parte superior del recu-bridor (región roja). La boquilla de ato-mizado se posiciona típicamente en esta región, ya que las tabletas deben mover-se a través de la zona del atomizado tan rápido como sea posible para eliminar la sobre-humectación.
El tiempo de residencia en la zona de atomizado es un parámetro impor-tante. Aunque el tiempo de residencia es difícil de determinar a partir de los experimentos, es posible fácilmente de los datos de simulación del DEM. La Figura 4 muestra el tiempo de residen-cia fraccional (es decir, el tiempo de residencia con relación al tiempo tota) para un tiempo de simulación de 60 s (9). Tanto la forma de la tableta (cuan-tificada como relación de longitud a al-tura) como el nivel de llenado (relación del volumen de la tableta con respecto al volumen total del bombo) influyen en el tiempo que una sola tableta pasa ex-puesta al atomizado. Para minimizar la variabilidad del recubrimiento o el tiem-po requerido para el proceso de recubri-miento, el tiempo de residencia fraccio-nal debe ser tan alto como sea posible.
En la Figura 4, los niveles de llena-do más bajos generalmente se compor-tan mejor que los niveles altos, como es de esperar. La dependencia del tiempo de residencia sobre el nivel de llenado, sin embargo, no es lineal. Las tabletas esféricas y ovaladas muestran depen-dencia similar del nivel de llenado. Las tabletas biconvexas, por otro lado, muestran una conducta cualitativamen-te diferente; éstas están menos afecta-das por el nivel de llenado y, por lo tan-to, superan a las otras formas en niveles de llenado bajos.
Formación de la películaLas simulaciones CFD de Euler-La-grange muestran cómo los diferentes parámetros físicos del atomizado de recubrimiento afectan el proceso de re-cubrimiento en una sola tableta. La con-ducta de deposición de las gotas sobre la superficie de la tableta se analiza en términos del espesor de la película y su homogeneidad en la tableta. Las simu-laciones estiman los efectos de la forma de la tableta, el diámetro de la gota, la temperatura del aire, la velocidad de

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 53
Contenido de agua de la gota del atomizado
Boquilla delatomizador
Masa de atomizado perdida / %
(a)
(b)
(c)
B1
2
34 5
8
6
4
2
0
0.8
0.6
0.4
0.2
0DER del espesor de la película
(a)
(b)
t = 0.1 s t = 0.2 s
100 80 60 40 20 0
Espesor pared-película (µm)
t = 0.3 s t = 0.4 s t = 0.5 s
Temperatura del aire / K Presión de atomización / bar
0.016
0.0155
0.015
0.0145
0.014
0.0135
0.013
0.0125355
345
335
350
340
330 1
2
3
1.5
2.5
atomizado del recubrimiento y el contenido de agua de la so-lución de recubrimiento en la formación de la película. Los datos de simulación permiten el análisis de importantes carac-terísticas del proceso tales como la conducta de la película sin secar sobre una tableta o la uniformidad inter-tabletas de la película de la tableta, medida como desviación estándar rela-tiva (DER), como una función de los parámetros del proceso (ver Figura 5). Los valores pequeños de la DER se traducen en alta uniformidad. La gráfica de superficie en la Figura 5 muestra que los resultados más bajos de la presión de atomi-zado resultan en mayor uniformidad, pero la temperatura del aire tiene poca influencia.
Proceso de atomizado en un recubridor industrialSe utilizaron simulaciones CFD multifase para estudiar el flu-jo del aire y de la gota dentro de un dispositivo de bombo de recubrimiento. Este trabajo simuló los efectos de la posición de la pistola de atomización y el ángulo de inyección sobre las pérdidas del atomizado y la eficiencia del recubrimiento.
La Figura 6a muestra el atomizado dentro del recubridor después de 0.5 s. El color y el tamaño de la gota son pro-porcionales al contenido de agua. Las grandes gotas cercanas a la boquilla del atomizador tuvieron un contenido de agua cercano a la fracción inicial de agua atomizada de 0.8. Las gotas pequeñas al lado derecho estuvieron casi parcialmente
o completamente secas. Una fracción del atomizado se mo-vió hacia el lecho de las tabletas pero entonces fue arrastrada hacia arriba, impactando así el cilindro del aire de entrada/salida. Las gotas secas siguieron el flujo del aire de secado y lo dejaron después de un tiempo de residencia variado. La Figura 6b muestra que la cantidad de pérdida del atomizado varía para las diferentes posiciones de la boquilla del atomiza-dor. Las posiciones 1 y 3 se desempeñaron significativamente mejor que las otras, incluyendo la colocación de la boquilla estándar (B).
Figura 5: (a) En un esquema de la técnica de simulación (izquierda), los colores indican el contenido de vapor de agua que va desde los bajo (azul) hasta lo alto (rojo oscuro). En el tiempo (t), la evolución de la película sobre las tabletas con diferentes formas (derecha), el espesor se muestra en tonos grises. (b) En una gráfica de superficie de la uniformidad del recubrimiento intra-tableta para la forma biconvexa, los valores más pequeños significan mejor uniformidad.
Figura 6: (a) Flujo de aire y su efecto sobre el movimiento de la gota del atomizado. La dirección del flujo9 se muestra con las flechas grises, el diámetro de la gota está dibujado a escala y el color de las gotas indica el contenido de agua (es decir, las gotas azules son partículas ya secas); (b) Efectos de diferentes posiciones y ángulos de la pistola de atomizado sobre la cantidad de pérdida del atomizado (definida como la cantidad relativa de masa que ha abandonado el sistema sin contacto con las tabletas).

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201254
ConclusiónLas técnicas avanzadas de simulación numérica pueden ayudar en el desarrollo y optimización de los procesos far-macéuticos de recubrimiento de tabletas. La investigación del desempeño del mezclado y la resultante distribución del tiempo de residencia de diferentes equipos de recubrimiento puede usarse para incrementar la homogeneidad intra-tableta del recubrimiento. Se usaron simulaciones numéricas de la interacción del aire de secado, el atomizado y el lecho de las tabletas para examinar el desempeño del recubrimiento tanto local como global. Los resultados computacionales mostraron cómo los parámetros del proceso influyen en la calidad de la película y en la uniformidad del recubrimiento. Una regla empírica es colocar la boquilla a alrededor de un tercio del lecho de la tableta, medido a lo largo de la superficie del le-cho empezando por la parte superior. El estudio de diferentes preparaciones de la boquilla del atomizado demostró que en términos de pérdida del atomizado, seguir esta regla dio los mejores resultados. Sin embargo, apuntando la boquilla lige-ramente lejos de la entrada de aire se pudo reducir la cantidad de pérdida del atomizado. Finalmente, se cuantificó la varia-bilidad del recubrimiento inter-tableta utilizando el método DEM para determinar los efectos del flujo de tabletas dentro del recubridor en términos de tiempo de residencia bajo el
recubriMiento de tabletas
asociada con el análisis tradicional fue-ra de línea.
Aumiller (GE Analytical Instru-ments): En los siguientes cinco años, la adopción del PAT probablemente será muy evolucionaria por naturaleza. Los procesos existentes serán cambia-dos cada vez más para proporcionar una mejor eficiencia. Es improbable que una tecnología destructiva sea fácil-mente adoptada y que reemplace total-mente un proceso existente.
Dentro del contexto de los sistemas de limpieza, probablemente veremos el desarrollo de mejores herramientas y analíticas que permitan un control más óptimo del proceso. Como se indicó en un principio, muchos procesos de limpieza actualmente hacen uso de la conductividad y de las mediciones del TOC como verificación de la limpieza del equipo. Este análisis es realizado con más frecuencia en el laboratorio pero también puede llevarse a cabo en línea para acelerar el tiempo para el re-sultado. Con el uso de instrumentación
robusta y de rápida respuesta en línea, los ciclos de CIP podrían ser diseñados para correr hasta que los contaminantes caigan por debajo de un umbral predefi-nido en lugar de correrlos por períodos de tiempo fijo. Con la inmediata retroa-limentación provista por la conductivi-dad y los monitores de TOC integra-dos, los procesos podrían volverse más rápidos y requerir menores costos de servicio para lograr la misma o mejor calidad. Esta transición requerirá el uso de instrumentación que pueda detectar confiablemente los contaminantes de interés dentro de la compleja matriz de un proceso de limpieza.
Salamon (PerkinElmer): Para considerar los avances potenciales en la instrumentación analítica, yo vería otros avances significativos en el desarrollo farmacéutico y no sólo en los avances del PAT. La nanotecnología en la entre-ga de fármacos es el mayor avance en los últimos cinco años y será el avance más grande en los futuros cinco años. Actualmente no existen prácticas y pro-
cesos de manufactura desarrollados que se haya demostrado su escalamiento para producir consistentemente, a gran escala, nanofarmacéuticos. En el labo-ratorio de desarrollo, la mejora más sig-nificativa es la partícula simple-plasma acoplado por inducción-espectrometría de masas (SP-ICP-MS) de nanomate-riales. Este no es un análisis dentro de la línea o en línea, sino que es un avance significativo en las pruebas analíticas.
Referencias1. FDA, Pharmaceutical cGMPs for the 21st
Century—Risk-Based Approach: Final Report (Rockville, MD, 2004).
2. FDA, Guidance for Industry: PAT—A Framework for Innovative Pharma-ceutical Development, Manufacturing and Quality Assurance (Rockville, MD, 2004).
3. FDA, Progress Report on Process Analytical Technology, www.fda.gov/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswer-sonCurrentGoodManufacturingPrac-ticescGMPforDrugs/ucm072006.htm, accessed June 18, 2012. PT
“Tecnología Analítica de Proceso: seguimiento del avance en el análisis de datos”continuación de la pág. 48
atomizado.Además de ayudar a lograr una comprensión mecanísti-
ca del proceso de recubrimiento, los datos computacionales pueden ser colectados en espacios de diseño in silico, en los cuales la correlación entre los parámetros críticos del proceso y los atributos de calidad críticos proporcionan un mejor co-nocimiento del diseño, optimización y operación de los dispo-sitivos industriales para recubrimiento.
Referencias1. A. Kalbag and C. Wassgren, Chem. Eng. Sci. 64 (11), 2705–2717
(2009).2. D. Suzzi, S. Radl, and J.G. Khinast, Chem. Eng. Sci. 65 (21),
5699–5715 (2010).3. B. Freireich and C. Wassgren, Chem. Eng. Sci. 65 (3), 1117–1124
(2010).4. S. Tobiska and P. Kleinebudde, Intl. J. Pharmaceutics 224 (1–2),
141–149 (2001).5. L. Ho et al., J. Control. Rel., 119 (3), 253–261 (2007).6. W.R. Ketterhagen, M.T. am Ende, and B.C. Hancock, J. Pharm.
Sci. 98 (2), 442–470 (2009).7. A. Alexander, T. Shinbrot, and F.J. Muzzio, Powd. Tech. 126 (2),
174–190 (2002).8. G. Toschkoff et al., AIChE J. 58 (2), 399–411 (2012).9. D. Suzzi et al., Chem. Eng. Sci. 69 (1), 107–121 (2012). PT

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 55
reporTe especiaL: enTrega De Fármacos
Innovación futura en la entrega de fármacos Mesa redonda de la industria moderada por Patricia Van Arnum
Los avances en la entrega dirigida de fármacos y los perfiles de liberación personalizados son objetivos clave.
¿Cuál será la innovación futu-ra en la entrega de fármacos? Para adquirir una perspectiva, Pharmaceutical Technology le
preguntó a líderes expertos sus puntos de vista. En la mesa redonda de la in-dustria participaron Anthony Recupero, PhD, director senior de otorgamiento de licencias, Aptalis Pharmaceutical Tech-nologies; Linda A. Felton, pHd, direc-tora del Departamento de Ciencias Far-macéuticas y profesora asociada de far-macia en el Colegio de Farmacia de la Universidad de Nuevo México, Centro de Servicios de Salud, y miembro del Comité Directivo del Grupo de Enfoque de Liberación Modificada de la Asocia-ción Americana de Científicos Farma-céuticos; y Louise Righton, gerente de marketing global de la división en 3M Sistemas de Entrega de Fármacos.PharmTech: Viendo a futuro por los próximos 5 a 10 años, ¿En qué áreas ve la oportunidad de la mayor innovación en la entrega de fármacos? ¿Cómo pue-den los factores, tales como el aumento del desarrollo de especialidades en me-dicamentos dirigidos a poblaciones de pacientes y medicamentos personaliza-
dos, afectan las estrategias en la entrega de fármacos o influyen en la tecnología de entrega de fármacos específicos?
Recupero (Aptalis Pharmaceuti-cal Technologies): En los próximos 5 a 10 años, vemos una gran oportunidad para la innovación en la entrega de fár-macos diseñando nuevas formulaciones dirigidas a perfiles farmacocinéticos (FC) particulares, necesarios para las correspondientes poblaciones de pa-cientes que están desatendidas por las formulaciones actuales de fármacos en el mercado. Bien sea que estos fár-macos utilicen formulaciones orales o parenterales, el enfoque clave estará en la liberación de fármacos personaliza-dos que proporcionen un nuevo perfil FC que lleve a una mayor eficacia y/o a riesgos de seguridad reducidos para poblaciones específicas de pacientes dentro de las áreas terapéuticas parti-culares. Estos pacientes pueden estar identificados por características tales como edad, co-morbilidades, o una variedad de biomarcadores potenciales que indiquen una respuesta favorable al fármaco de interés, si se administra bajo las condiciones apropiadas. El sistema nervioso central es un área terapéutica general en donde estas estrategias pro-bablemente sean benéficas significa-tivamente, dada la variabilidad en las respuestas del paciente basadas en una variedad de factores personales.
Los perfiles requeridos persona-lizados de liberación del fármaco ne-cesitarán el uso de múltiples tipos de tecnologías, incluyendo la liberación extendida, la liberación sostenida y la liberación en pulsos de tiempo. Mucha de la innovación vendrá del desarrollo de un mejor conocimiento del paciente más que de la identificación de nuevas moléculas.
Desde una perspectiva del negocio, será clave tener tecnologías patentadas disponibles con fuertes estados de pa-tente detrás de ellas para estas aplica-ciones. Esto le permitirá a las compa-ñías desarrollar vías protegidas para la exclusividad comercial de manera que puedan estar protegidas no sólo por las patentes de tecnología y formulación, sino también por nuevas patentes de uso que sean el resultado de nuevos hallaz-gos clínicos basado en la habilitación

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201256
reporTe especiaL: enTrega De Fármacos
La entrega de fármacos dirigidos es particularmente importante en la entrega de terapias anticáncer para bajar la dosificación, reducir los efectos colaterales y maximizar la entrega de la sustancia farmacéutica a los sitios del tumor. La reciente investigación en nanolipogeles y encapsulación basada en lípidos termo-responsivos ayudan al avance de las estrategias en este campo.
Nanolipogeles como vehículos de entrega de fármacosLos investigadores en la Universidad de Yale desarrollaron recientemente nanolipogeles como una nueva tecnología de transporte de fármacos para entregar terapias anti-cáncer. Los nanolipogeles son esferas biodegradables, huecas, a escala nano, que son capaces de portar moléculas químicamente diversas, de acuerdo a un comunicado de prensa el 15 de julio de 2012 por la Fundación Nacional de Ciencias (NSF), la cual está aportando financiamiento para la investigación. Los nanolipogeles contenían factor de crecimiento transformado (TGF-β) e interleucina-2 (IL-2).
Puedes imaginarte el tumor y su microentorno como un castillo y un foso,” dijo Tarek Fahmy, el profesor de ingeniería de la Universidad de Yale y co-autor de un reciente artículo que detalla la investigación, en el comunicado de prensa de la NSF (1). “Los ‘castillos’ son tumores cancerosos que han evolucionado a una estructura altamente inteligente –las células tumorales y la vasculatura. El ‘foso’ es el sistema de defensa del cáncer, el cual incluye el TGF-β. Nuestra estrategia es ‘secar’ ese foso y neutralizar el TGF-β. Hacemos esto utilizando el inhibidor que se libera de los nanolipogeles. El inhibidor efectivamente detiene la capacidad del tumor para impedir una respuesta inmune.”
Específicamente, los investigadores desarrollaron geles poliméricos liposomales a nanoescala (es decir, nanolipogeles) de ciclodextrinas acomplejadas con fármaco y polímeros biodegradables que encapsulan citocinas para entregar pequeños inhibidores moleculares hidrofóbicos y citocinas de proteína solubles en agua. El nanolipogel que libera el inhibidor del TGF-β y el IL-2 retrasaron significativamente el crecimiento del tumor, incrementaron la sobrevivencia de los ratones portadores de tumores e incrementó la actividad de las células asesinas naturales y de CD8 activado intratumoral+ infiltración de células T (1).
“Se puede imaginar la citocina como una manera de obtener refuerzos para cruzar el foso seco y entrar al castillo y mandar una señal para que lleguen más fuerzas,” dijo Fahmy en el comunicado de la NSF. “En este caso, los refuerzos son las células T, la ‘armada’ anti-invasores del cuerpo. Logrando ambos objetivos del tratamiento de una sola vez, el cuerpo tiene mayor oportunidad de derrotar al cáncer,” dijo.
Un aspecto importante de los nanolipogeles es su capacidad para “empacar” dos tipos de moléculas
completamente diferentes, -proteínas grandes, solubles en agua, como la IL-2 y moléculas hidrofóbicas más pequeñas, tales como el inhibidor de TGF-β- en un solo vehículo de entrega, de acuerdo al comunicado de la NSF. La cubierta exterior de cada nanolipogel está hecha de un lípido sintético, biodegradable, que se degrada de manera controlada, puede encapsular un complejo fármaco-andamiaje, y es fácil de formar en una coraza esférica. Cada coraza rodea una matriz hecha de polímeros biodegradables, biocompatibles que están impregnados con las moléculas del inhibidor TGF-β. Estas esferas, casi completas se ponen en una solución que contiene IL-2, que queda atrapado dentro del andamiaje, un proceso llamado carga remota, de acuerdo al comunicado de la NSF. El resultado es un vehículo de entrega de fármacos a nanoescala que es lo suficientemente pequeño para viajar a través de la corriente sanguínea pero lo suficientemente grande para ser atrapado para entregarse en los sitios del tumor.
Nanopartículas súper-paramagnéticasLos investigadores en la Universidad de Sidney en Australia han desarrollado nanopartículas encerradas en grasa como posible tratamiento para el cáncer de pulmón. El grupo diseñó partículas dirigibles, inhalables que pueden atacar tumores pero dejan a las células sanas sin daño, reduciendo así los efectos colaterales del tratamiento de cáncer, de acuerdo a un comunicado de prensa de la Universidad de Sidney el 27 de julio de 2012. Las partículas consisten en un fármaco encerrado en un lípido que puede ser activado utilizando un campo magnético. Los investigadores incorporaron budenosida y nanopartículas de óxido de fierro superparamagnético (SPIONs) dentro de partículas de lípido utilizando una emulsificación de aceite en agua (2). Se investigaron el tamaño de partícula, la composición química, el grado de respuesta al campo magnético, la termosensibilidad y el desempeño de la inhalación in vitro (2).
“Cuando se exponen a un campo magnético, las nanopartículas super-paramagnéticas encerradas vibran, fundiendo la grasa y liberando el fármaco,” dijo el líder de la investigación Wojciech Chrzanowski y profesor de farmacia en la Universidad de Sidney, en el comunicado de prensa. “El sistema que hemos desarrollado aborda uno de los problemas más importantes relacionados con os efectos colaterales de las terapias para el cáncer, tales como interacciones indeseadas con tejidos sanos. Como el fármaco está escondido en una estructura de lípidos hasta que alcanza el sitio objetivo, las células sanas están protegidas,” dijo. “Podemos activar la liberación del fármaco debido a que nuestra formulación es termosensible. El estímulo externo, en nuestro caso el campo electromagnético, induce el aumento de la temperatura local dentro de las partículas, con lo cual se activa la liberación del fármaco. Bajo el
microscopio, el campo electromagnético externo parece agitar las nanopartículas súper-paramagnéticas en la formulación, produciendo calor que después abre las partículas, liberando el fármaco en el sitio del tumor. Como la formulación incluye partículas súper-paramagnéticas, también podemos guiar las partículas a sitios específicos utilizando magnetos,” explicó.
Los investigadores encontraron que las partículas de diámetro promedio de 2-4 µm con budesonida y SPIONs dentro de la matriz del lípido respondían a un campo magnético con una extracción del 100% a una distancia de 5 mm. Las formulaciones demostraron tener velocidad acelerada de liberación del fármaco a temperaturas hipertérmicas (45°C) de liberación controlada. El polvo seco para inhalación producido tenía una fracción inhalable de partículas finas de 30%. El sistema de lípidos tenía características termosensibles, adecuadas para la entrega controlada, y el fármaco modelo y el sistema de lípidos cargados con el SPION era magnéticamente activo y movible utilizando simples magnetos permanentes. En general, el sistema demostró tener aplicación como una terapia de inhalación dirigida y controlada (2).
La investigación tiene potencial no sólo para formulaciones inhalables, sino también para otras aplicaciones de entrega de fármacos. “Nuestro sistema tiene gran potencial no sólo como formulación inhalable sino para un rango amplio de aplicaciones donde la focalización y la estimulación o activación externa de liberación del fármaco es crítica,” dijo el profesor asociado de la Universidad de Sydney, Paul Young, recientemente designado como jefe de tecnología respiratoria en el Instituto Woolcock de Investigación Médica, en el comunicado. “La característica importante del sistema es su capacidad para ‘aprovechar’ la sensibilidad a la temperatura de la formulación, de manera que la liberación del fármaco puede lograrse a diferentes temperaturas. Esto es particularmente importante para la entrega multi-fármacos en donde diferentes compuestos activos podrían ser liberados en diferentes momentos.”
Fuentes
Perspectivas de entrega de fármacos: Nanotecnología
1. T. Fahmy et al., “Combination Delivery of TGF-β inhibitor and IL-2 by Nanoscale Liposomal Poly-meric Gels Enhances Tumor Immunotherapy,” Nature Materials, online, DOI:10.1038/nmat3355, July 15, 2012. 2. W. Chrzanowski et al., “Magnetized Thermo Responsive Lipid Vehicles for Targeted and Con-trolled Lung Drug Delivery,” Pharm. Res., online, DOI: 10.1007/s11095-012-0774-9, May 15, 2012.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 57
del sistema de entrega. Esto le permi-tirá a las compañías y a los inversores maximizar su retorno de la inversión y estimular más innovación en la entrega de fármacos.
Felton (Universidad de Nuevo México): La próxima década continua-rá siendo una época excitante para ser un científico de formulaciones confor-me la industria se aleje de innovaciones incrementales y fármacos ‘me-too’1 en favor de avances más significativos. Cuando miro el futuro de la farmacia, veo muchas oportunidades de mejora en la entrega de fármacos y seguridad del paciente. Con una mejor compren-sión de las enfermedades y de la ana-tomía y fisiología, espero ver que se desarrollen sistemas de entrega de fár-macos más dirigidos. Estos sistemas incluirán sitios objetivo específicos en el cuerpo así como químicos dirigidos a células específicas en el cuerpo. Ac-tualmente se está invirtiendo una gran cantidad de esfuerzo en la focalización a nivel celular para mejorar los resulta-dos terapéuticos y reducir las toxicida-des sistémicas. Estas nuevas entidades químicas, sin embargo, probablemente creen retos de entrega adicionales para los científicos de formulación. Dichos retos probablemente serán resueltos uti-lizando estrategias innovadoras.
Un significativo esfuerzo de inves-tigación está actualmente enfocado en
la identificación de nuevas indicaciones para los fármacos existentes. Dicho re-direccionamiento contribuirá además a la necesidad de rutas de entrega al-ternativas así como a la liberación de fármacos dirigidos. Espero ver mejoras en la entrega de vacunas, que incluyan tecnologías sin uso de aguja, entrega mediante la ruta de inhalación y esta-bilidad mejorada del producto. Existen también oportunidades para terapias mejoradas en el manejo del dolor cró-nico, donde la liberación prematura del fármaco (descarga de dosis) está mini-mizada. Dichos sistemas también redu-cirían el potencial de abuso. Se espera el desarrollo e implementación adicio-nal de tecnologías anti-falsificación y esto también contribuye a la seguridad del paciente, especialmente conforme el mercado global continúe creciendo.
El envejecimiento de la población también creará oportunidades para el desarrollo de sistemas nuevos y nove-dosos de entrega de fármacos. Además de las mejoras en las tecnologías de enmascaramiento del sabor y rutas de entrega alternativas, existe una crecien-te necesidad de productos combinados para reducir las dosis diarias totales de los medicamentos. Aunque todavía existe mucha discusión acerca de la medicina personalizada, la creación de productos combinados únicos para pa-cientes individuales tienen el potencial
de impactar enormemente el apego del paciente a los regímenes terapéuticos, mejorar el manejo del estado de la en-fermedad y mejorar la calidad de vida y podría ser fácilmente implementado en farmacias comunitarias.
Participantes de la mesa redonda de la industria de izquierda a derecha: Anthony Recupero, PhD, director senior, otorgamiento de licencias, Aptalis Pharmaceutical Technologies; Louise Righton, gerente de marketing divisional global, para 3M Sistemas de Entrega de Fármacos. Foto no mostrada: Linda A. Felton, PhD, directora del Departamento de Ciencias Farmacéuticas y profesora asociada de farmacia, Colegio de Farmacia de la Universidad de Nuevo México, Centro de Ciencias de la Salud, y miembro del Comité Directivo del Grupo de Enfoque de Liberación Modificada de la Asociación Americana de Científicos Farmacéuticos.
Las microesferas o nanoesferas que entregan fármacos a áreas focalizadas del cuerpo y sólo las liberan ahí podrían ayudar a superar el problema encontrado con frecuencia con las terapias anticáncer que entregan fármaco no sólo a los sitios del tumor sino también a células sanas. Un método desarrollado por los investigadores en el Instituto Max Planck de Coloides e Interfases en Postdam-Golm, Alemania, hace posible producir dichas esferas en un amplio rango de tamaños y equiparlas con diferentes funciones.
El método de fabricación de las microesferas es importante en la encapsulación y liberación de un fármaco. El investigador desarrolló una simple técnica para producir microesferas porosas de carbonato de calcio como plantillas para la producción de pelotas huecas tridimensionales como microesferas para la entrega de fármacos, de acuerdo a un comunicado de prensa del Instituto Max Planck, del 2 de julio de 2012. Específicamente, los investigadores mostraron que las esferas coloidales híbridas tridimensionales con funciones integradas y propiedades colectivas pueden fabricarse utilizando nano-objetos inorgánicos comunes como bloques de construcción con encapsulación de polielectrolitos a través de una estrategia de plantilla simple, la cual abre la estrategia de
fabricación como un enfoque para el desarrollo de materiales híbridos con múltiples funciones y propiedades colectivas (1).
Los investigadores usaron microesferas de carbonato de calcio y llenaron los poros de las esferas de carbonato de calcio con nanopartículas, rodeando las esferas de carbonato de calcio llenas con una red de largas cadenas de proteína o hilos de polímero, y después disolvieron la plantilla de carbonato de calcio utilizando ácido, de acuerdo con el comunicado. Las nanopartículas se organizaron a sí mismas en una esfera porosa que fue encerrada en la red de proteína. “Podemos combinar fácilmente sustancias para formar una unidad multifuncional y adaptar sus propiedades químicas y físicas para la función requerida,” dijo Helmuth Möhwald, profesor del Instituto Max Planck de Coloides e Interfases, en el comunicado. La red de proteína no sólo cubre la esfera hueca, sino que también es biocompatible y puede contener sustancias bioquímicas señalizadoras que envían las esferas directamente a su objetivo en el cuerpo.
Fuente 1. X. Yan, J. Li, and H. Möhwald, “Templating Assembly of Multifunctional Hybrid Col-
loidal Spheres,” Advanced Materials, online, DOI: 10.1002/adma.201200408, Apr. 23, 2012.
Perspectivas de entrega de fármacos: Microesferas de carbonato de calcio
1Los fármacos ‘me-too’ (yo también) son fármacos que estructuralmente son muy similares a los fármacos ya conocidos, con sólo diferencias menores.
“Innovación futura en la entrega de fármacos ”continúa en la pág. 66

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201258
FORO DE EMPAQUE
tetr
a Im
ag
eS
/ge
tt
y Im
ag
eS
Expedidores con temperatura controladaHallie Forcinio
Herramientas de empaque y monitoreo que protegen los fármacos sensibles a la temperatura
Los fabricantes farmacéuticos con productos sensibles a la temperatura disfrutan de una variedad cada vez mayor de
empaque protector, monitoreo ambiental y opciones para rastreo del embarque.
Empaque protectorLos expedidores aislados con paneles de aislamiento al vacío y refrigerantes basados en materiales con cambio de fase han capturado la participación en el mercado de las estructuras tradicio-nales de poliestireno expandido (EPS) y poliuretano y de los empaques de gel o hielo seco. Los expedidores de alto desempeño ofrecen tapas de compre-sión controlada, paneles de aislamien-to al vacío, y material con cambio de fase a +5°C que provee una transición fundido-congelado precisa y consisten-te para el control de temperatura. Los expedidores pesan aproximadamente 50% menos que los expedidores tradi-cionales aislados y reducen los costos de distribución. El diseño agiliza el pro-ceso de preacondicionado y mantiene cargas útiles en 2-8°C durante 140 h en condiciones ambientales extremada-mente calientes o frías, se dice que 30%
Hallie Forcinio es editora del Foro de empaque del Pharmaceutical Technology, 4708 morningside Drive, cleveland, oh 44109, tel. 216.351.5824, fax 216.351.5684, [email protected].
más prolongado que con otras opciones. Las etiquetas termocrómicas cambian de color para proporcionar una verifica-ción visual de que los expedidores es-tán listos para empacarse (expedidores ORCA 2-8°C, Intelsius).
Muchos diseños de empaque pro-tector son de un solo uso, pero los es-fuerzos para elegir opciones más sus-tentables están estimulando el interés en los diseños reutilizables. Una op-ción, disponible en una configuración de un solo uso o reutilizable, protege la carga entera de la tarima y mantiene 15-30°C hasta por 120 h. La configura-ción de un solo uso utiliza una tarima de madera y una cubierta externa de cartón corrugado, mientras que la unidad reuti-lizable utiliza plástico para la tarima y la cobertura exterior (Pallet Transporter CRT, Cryopak).
Los diseños reutilizables se han vuelto tan populares que un proveedor ha expandido su centro de servicio re-gional para renovar sus expedidores con temperatura controlada. Los servicios incluyen inspecciones de calidad y man-tenimiento, reemplazo de componentes, sanitización y limpieza vía un proceso de descontaminación con ultravioleta a ser certificado pronto. La compañía también realiza labores de equipamien-to y/o empacado, preacondicionado de los expedidores y proporciona rastreo, informes personalizados y funciones de análisis certificado. Su último diseño reutilizable reemplaza las cajas exterio-res de papel kraft corrugado o plástico estándar con un contenedor exterior de polietileno de alta densidad que tiene
un período de vida proyectado de dos o tres años. El contenedor exterior rígido contiene seis paneles con aislamiento al vacío que pueden ser retirados y re-emplazados independientemente para reducir los costos de mantenimiento. Inicialmente disponible en tamaños de 10, 12 y 16 L, el diseño también me-jora el comportamiento térmico hasta en 15% contra las construcciones con cajas corrugadas (Credo DuraCUBE, Minnesota Thermal Science).
Las unidades precalificadas repre-sentan otra tendencia. Las opciones incluyen un sistema con temperatura controlada capaz de mantener 2-8°C en condiciones invernales. Diseñado para soportar el frío extremo de las carrete-ras canadienses, la unidad protege su contenido en condiciones ambientales de -25°C durante más de 72 h (Time-SaverCN72 Extreme Cold PreQualified Shipper, Cryopak).
Para aplicaciones menos estrictas, otro diseño precalificado mantiene em-
Estaremos viendo más...•Expedidores aislados con pan-
eles aislados al vacío
•Uso de materiales con cambio de fase para mantener el rango de temperatura
•Expedidores reutilizables
•Expedidores precalificados
• Integración de expedidores con monitoreo ambiental y rastreo

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 59
barques pequeños a 20 – 24°C en con-diciones ambientales calientes o frías hasta por 24 h. El expedidor ofrece espacio suficiente de carga hasta para cuatro viales de 20 mL (TimeSaver 24 CRT Mini shipper, Cryopak).
Una familia de expedidores preca-lificados y validados acomoda tamaños de embarque que van desde una sola jeringa hasta una tarima completa. La mayoría de los diseños combinan el EPS o el aislamiento con espuma de po-lietileno de baja densidad con refrige-rantes para mantener las temperaturas deseadas durante 60-120 hrs (InTemp Pallet Solutions e InTemp Temperature Control Packaging, Inmark). Un nuevo laboratorio interno valida el empaque con temperatura controlada.
Otra familia de expedidores preca-lificados ha añadido una opción de 5 L de tamaño a las de 11 y 36 L. El dise-ño reutilizable provee protección hasta por 120 h en temperaturas refrigeradas, congeladas y de hielo seco que depende del cambio de fase del material usado para la unidad preacondicionada de re-gulación térmica (expedidores intelli-Therm, Intelligent Thermal Solutions).
Monitoreo del embarqueLos registradores de datos rastrean las condiciones durante el embarque. Una segunda generación, unidad parecida a una etiqueta con sensor de temperatura integrado y punto de conexión USB se fija fácilmente al empaque (ver Figura 1). El sensor de temperatura se calibra durante la manufactura a los estándares establecido por el Instituto Nacional de Estándares y Tecnología (NIST) de EEUU y es exacto hasta ±0.5°C a lo largo de su rango de operación de -30 a 60°C. Durante el tránsito, toma una lectura de superficie del objeto al cual está adherida. Los diodos emisores de luz en la etiqueta destellan si ocurre una excursión de la temperatura. Una vez en su destino, un punto de conexión USB integrado permite que la etiqueta sea co-nectada directamente a la computadora, eliminando así la necesidad de un lector o software especializado para bajar los datos de la cadena fría. La etiqueta ge-nera automáticamente un archivo PDF que contiene el tiempo completo y la
historia de la temperatura, incluyendo gráficas y datos sumarios. Las nuevas características incluyen un archivo in-crustado, de valores separados por co-mas (CSV), que elimina la necesidad de software de terceros para generar el ar-chivo CSV. El archivo incrustado CSV lista los datos de hora y temperatura en incrementos de cinco minutos. El archi-vo CSV puede ser copiado y salvado a otra ubicación de acuerdo a los procedi-mientos estándar de operación (PNOs) del usuario. Los datos del archivo pue-den ser manipulados utilizando otros programas de acuerdo con los PNOs de la compañía (Etiqueta de Monitoreo de Temperatura BIOmed XpressPDF, Pak-Sense).
La misma forma compacta y tecno-logía USB se utiliza para registradores de datos de un solo uso introducidos en INTERPHEX 2012. Las unidades pue-den ordenarse precalibradas y prepro-gramadas para rangos de temperatura específicos (Registrados de Cadena Fría CCL100, Vaisala).
La protección completa de la ca-dena fría también se apoya en senso-res que monitorean las áreas de alma-cenamiento tales como cuartos fríos, refrigeradores y congeladores. Los registradores de temperatura inalám-bricos que usan baterías proporcionan alertas en tiempo real si las condiciones se desvían más allá de los parámetros establecidos. Algunos modelos también monitorean la humedad o la presión así como la temperatura, Los intervalos de muestreo pueden ocurrir tan frecuente-mente como una vez por segundo con una exactitud de ±0.1°C. Las unidades también pueden monitorear autoclaves, túneles de esterilización y otras áreas con temperatura controlada. El soft-ware patentado automatiza las pruebas de validación, garantiza la seguridad de los datos y genera una variedad de re-portes (Sistema de Validación y Mapeo Térmico Inalámbrico XpertLog, Lives International Corp.).
Un sistema basado en identificación con radiofrecuencia (RFID) consiste en una tarjeta inteligente RFID, capaz de monitorear la hora y la temperatu-ra, un lector de RFID con escáner in-tegrado para código de barras opcional,
Figura 1: La etiqueta se fija al empaque para monitorear las condiciones del tránsito (Etiqueta de Monitoreo de Temperatura BIOmed XpressPDF, PakSense)
y el Internet (ver Figura 2). La tarjeta inteligente RFID se coloca en la tari-ma, la caja individual, u otra ubicación designada. Para iniciar el proceso de monitoreo, el lector escanea el código de barras de la tarima. Las temperaturas se registran en intervalos específicos. Cuando el embarque llega a su destino, se leen las etiquetas (tags) para determi-nar las fluctuaciones de temperatura que hayan ocurrido. Los resultados se trans-miten vía el USB o Wi-Fi a una página dedicada en Internet, y pueden enviarse correos electrónicos o textos de alerta a los socios de la cadena de suministros. Las tarjetas inteligentes también pue-den ser devueltas por correo para bajar los datos, de los cuales se dispone en línea dentro de las 48 horas. Las etique-tas pueden ser reiniciadas en cualquier momento para registrar nuevos segmen-tos. Por ejemplo, conforme son recibi-das las tarimas, las etiquetas continúan registrando el tiempo y la temperatura en el camión, en el muelle y en el alma-cén. A las etiquetas se les puede incluso asignar nuevo propietario y tiempo y parámetros de temperatura conforme el embarque se mueve a través de la cade-na de suministro (proceso de monitoreo de tiempo/temp, cadena fría, basado en la web Temp-TRIP, TempTRIP).
La tecnología, que se originó en Australia, también hace uso de tecno-logía de RFID activa, GPS y programa de rastreo basado en Internet para moni-torear productos en tránsito, almacén o refrigerador. El sistema, ya en uso en la

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201260
industria de alimentos, colecta datos y proporciona alertas en tiempo real cuan-do las temperaturas se desvían fuera de los parámetros requeridos (hardware y software Fresh InTransit, Fresh InSide, Fresh Mobile, CoolTrax EEUU).
El nuevo software permite el acceso a datos colectados mediante un sistema de monitoreo de temperatura o cargado por los socios de la cadena de suminis-tros, como los embarcadores, los recep-tores y los transportistas. El programa archiva instantáneamente y automáti-camente los datos de los embarques de
FORO DE EMPAQUE
Figura 3: Un contenedor de carga aérea protege y monitorea los productos farmacéuticos y biotecnológicos sensibles a la temperatura (PharmaPort 360, fabricado por Cool Containers y disponibles a través de servicio UPS Temperature True, UPS).
Figura 2: Los datos colectados de los sensores del empaque se usan para graficar la ruta de la cadena fría y las banderas rojas muestran cuando la carga experimenta temperaturas fuera de especificaciones (proceso de monitoreo de tiempo/temp, cadena fría, basado en la web Temp-TRIP, TempTRIP).
perecederos entrantes en el momento de la llegada. La transmisión inalámbrica a un servidor seguro elimina la necesidad de descargar registradores individuales (Sistema de Monitoreo de Temperatura Automático, Cargo Data).
Para la recepción de los empleados portuarios, un dispositivo portátil pro-porciona acceso inmediato a los datos de temperatura, lo cual elimina la ne-cesidad de esperar la descarga antes de decidir aceptar o rechazar un embarque. La unidad también transmite datos para el almacenamiento permanente (recep-tor portátil Express, Cargo Data).
Enfoque de sistemasUn innovador contenedor para carga aé-rea combina la protección al producto y el monitoreo para farmacéuticos, vacu-nas y biológicos sensibles a la tempe-ratura (ver Figura 3). Completamente validado y probado para confirmar el cumplimiento con los estándares de la industria, el contenedor está alimentado por una batería recargable y se calienta o enfría según sea necesario para man-tener el rango de temperatura apropiado durante más de 100 h, independiente-mente de la temperatura ambiente. Los sensores integrados monitorean la con-dición del embarque y la ubicación GPS y transmiten los datos en tiempo cerca-
no al real a través de una red estándar de celular global. Si se disparan las alertas, indicando un riesgo potencial, el trans-portista puede intervenir con planes de contingencia pre-establecidos para res-catar un embarque en peligro y evitar pérdida del producto. Mientras está en el modo de tránsito, el contenedor no consume energía ni emite calor externo, vapor o gases permitiendo así que vue-le en posiciones de transporte aéreo en cubierta alta o baja. Esto ofrece más op-ciones de enrutamiento, especialmente en carriles de transporte restringidos. El contenedor también tiene la aproba-ción de la Administración de Aviación Federal para volar en transporte aéreo estrecho y ancho, lo que permite un alto grado de flexibilidad en selecciones de vuelo y abre el acceso a mercados glo-balmente (PharmaPort 360, fabricado por Cool Containers y disponible a tra-vés de servicio UPS Temperature True, UPS).
La protección integrada del pro-ducto y el monitoreo también pueden incluir opciones, tales como nitrógeno líquido precargado, contenedor de va-por seco que mantiene -150°C hasta por 10 días. Con 10 días de tiempo de per-manencia, el expedidor soporta com-plejos itinerarios internacionales y eli-mina el re-enfriamiento requerido con los embarques con hielo seco. Como la unidad se entrega lista para empacar y recogidas para su renovación o recicla-je después de descargar en su destino, los embarcadores no necesitan comprar, inventariar o manejar cajas aisladas y hielo seco (Deep Frozen Shipping So-lution, FedEx).
Si es necesario, un registrador de datos de RFID, un sensor GPS y una plataforma basada e la web monitorean la temperatura, la humedad, la presión barométrica, la exposición a la luz y la ubicación; proporciona visibilidad en tiempo casi real para los embarques y envía alertas si se exceden los paráme-tros pre-establecidos de la ruta. El re-gistrador de datos/unidad GPS reusable también está disponible por una cuota mensual (dispositivo y servicio de in-formación SenseAware, FedEx). PT
Recursos para el manejo de la cadena fría• ISPE Guía de Buenas Prácticas:
Manejo de la Cadena Fría www.ISPE.org
•Grupo de Aseguramiento de Temperatura www.Linkedin.com
•Cadena Fría IQ, subgrupo de Farma IQ, www.Linkedin.com
•Foro Global de Manejo de Cadena Fría y Temperatura, Sept 24-28, www.iqpc.com
•Grupo de Interés de la Cadena Fría Farmacéutica, www.pda.org

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 61
Patricia Van Arnum es editora ejecutiva del Pharmaceutical Technology, USA, 485 route one South, edif. F, Primer Piso, Iselin, NJ 08830; tel. 732.346.3072, [email protected] twitter@PharmtechVarnum
IngredIentes FarmacéutIcos: apIs y excIpIentes
Cumplimiento de los desafíos en la síntesis asimétricaPatricia Van Arnum
La tecnología quiral es importante en la síntesis farmacéutica conforme la industria y la academia avanzan a esquemas novedosos para lograr la enantioselectividad.
La química quiral juega un pa-pel significativo en el desa-rrollo de intermedios farma-céuticos y APIs y como tal,
los avances en la síntesis asimétrica son de valor para las compañías farmacéuti-cas. Los investigadores de la academia y de la industria continúan desarrollan-do nuevas rutas para lograr la enantiose-lectividad deseada. Algunas estrategias recientes incluyen una ruta más eficien-te para las prostaglandinas, una ruta biocatalítica para hacer un intermedio clave en la producción de boceprevir, el API del Victrelis de Merck & Co., y un catalizador bifuncional derivado de ligandos BINOL para la producción bromolactonizaciones altamente enan-tioselectivas de ácidos carboxílicos no saturados.
Evaluando el mercadoSe espera que el mercado global para la tecnología quiral, incluso de aplicacio-nes en farmacéuticos, muestre un creci-miento moderado durante los próximos cinco años. El mercado de la tecnolo-gía quiral global está valorado en casi $5,300 mdd en 2011, de acuerdo a un análisis reciente de BCC Research. Se espera que este mercado se incremente en una tasa compuesta de crecimien-to anual (CAGR) de 6.5% del 2011 al 2016 y se aproximará a los $7,200 mdd a finales del período previsto. Los pro-ductos de síntesis quiral comprenden la mayoría del mercado de tecnología qui-ral en 2010, con una participación del 80.0% (es decir, $3,900 mdd de utili-dades). Este segmento del mercado fue estimado en $4,200 mdd en 2011 y se espera que sea de $5,700 mdd para el 2016, un CAGR de cinco años de 6.4%, de acuerdo a BCC. El mercado de aná-lisis quiral fue valuado en $785.7 mdd en 2010 y creció a $839.4 mdd en 2011. Está proyectado que este mercado al-cance casi $1,100 mdd para el 2016, un CAGR de 5.8% sobre el período de cin-co años, de acuerdo a BCC.
Una mejor ruta para las prostaglandinasLos investigadores en la Universidad de Bristol, en Inglaterra, reportaron recien-temente sobre un método mejorado para hacer prostaglandinas, químicos pareci-dos a hormonas que tienen aplicaciones farmacéuticas. El análogo de prosta-glandina lanoprost, el cual se utiliza para tratar el glaucoma y la hiperten-sión ocular, es una prostaglandina bien conocida. Es el ingrediente activo del Xalatan de Pfizer, el cual generó ventas en 2011 de $1,250 mdd; la patente para el fármaco expiró en 2011 (1).
Debido a la actividad biológica de las prostaglandinas, pero a la dificul-tar en sintetizarlas, las estrategias para mejores rutas de síntesis para las pros-taglandinas es un área de investigación activa. Por ejemplo, la síntesis actual del latanoprost requiere 20 pasos y utiliza la metodología y estrategia desarrolla-da por E.J. Corey, ganador del Premio Nobel de 1990 en Química, de acuerdo a un comunicado de prensa de la Universi-dad de Bristol del 15 de agosto de 2012.
Los investigadores de la Universi-dad de Bristol reportaron la síntesis de la prostaglandina PGF2a
, la cual se apo-ya en el uso de un organocatalizador, una pequeña molécula orgánica, para catalizar un paso clave en el proceso, el cual produce altos niveles de este-reoquímica relativa y absoluta y menos pasos, de acuerdo al comunicado de prensa de la universidad y al reciente artículo que detalla la investigación (2). El nuevo proceso utiliza una nueva des-conexión que les permitió a los inves-tigadores completar la síntesis en sólo siete pasos, de acuerdo al comunicado de prensa. El paso clave es una reacción aldólica en cascada del succinaldehído que utiliza organocatálisis de prolina para crear un enal bicíclico en un paso con un exceso enantiomérico de 98%. Este enal bicíclico intermedio está com-pletamente listo con la funcionalidad apropiada para la unión de los grupos restantes (1). La ruta para el enal bicí-clico es importante para una ruta más eficiente y potencialmente rentable pero también sirve como base para examinar estructuras químicas relacionadas de los análogos de prostaglandinas (1, 2).
“A pesar de las largas síntesis y del enorme esfuerzo resultante que se re-quiere para la preparación de estas mo-léculas, éstas son todavía usadas en la

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201262
clínica debido a su importante actividad biológica”, dijo Varinder K. Aggarwal, profesor en la Escuela de Química, Uni-versidad de Bristol, en el comunicado universitario. “Poder hacer complejos farmacéuticos en un número menor de pasos y, por lo tanto, más efectivamen-te, significaría que muchas más personas podrían ser tratadas por el mismo costo.”
Biocatalizadores en acciónLos investigadores de la Escuela de Química e Ingeniería Biomolecular en el Instituto Georgia de Tecnología y Merck & Co. recientemente desarro-llaron una amina deshidrogenasa para la síntesis de aminas quirales. Especí-ficamente, los investigadores alteraron con éxito una leucina deshidrogenasa a través de ingeniería de proteínas a una amina deshidrogenasa enantioselectiva. En lugar del a-ceto ácido del tipo sil-vestre, la nueva amina deshidrogenasa aceptó la cetona análoga metil isobutil cetona, la cual intercambió el grupo carboxi por un grupo metilo para produ-cir (R)-1,3-dimetilbutilamina quiral (3).
A principios de este año, Codexis, una compañía de biocatalizadores, re-portó sobre su trabajo con Merck & Co. para el desarrollo de un método de producción basado en enzimas para un intermedio clave en la producción de bo-ceprevir, el API del Victrelis de Merck, un fármaco para tratar la hepatitis C. Las compañías reportaron sobre un proceso quimioenzimático para la manufactura del intermedio de prolina bicíclica del boceprevir, basado en desimetrización catalizada por amina oxidasa. La carac-terística estructural clave en el bocepre-vir es la porción de prolina bicíclica, la cual durante las etapas de desarrollo, era producida mediante una resolución clá-sica. Conforme el candidato a fármaco avanzó, Codexis y Schering-Plough (hoy Merck) desarrollaron conjuntamente una síntesis asimétrica quimioenzimática donde la reacción neta fue una reacción oxidativa de Strecker. La parte clave de la secuencia de reacción es una desime-
IngredIentes FarmacéutIcos: apIs y excIpIentestrización oxidativa enzimática de un sus-trato de amina proquiral (4, 5).
Según Codexis, el nuevo método in-crementó el rendimiento del intermedio químico 150% sobre el proceso anterior. También redujo el uso de materia pri-ma en 60%, el uso de agua en 61% y el desperdicio general del proceso en 63%. Codexis utilizó su tecnología patentada
de evolución dirigida CodeEvolver para desarrollar la enzima a la medida para uso en la manufactura a escala comercial del intermedio del boceprevir (5).
Codexis se había asociado inicial-mente con Merck & Co. para otra ruta biocatalítica. Las compañías desarrolla-ron una síntesis asimétrica biocatalítica de aminas quirales a partir de cetonas en la manufactura de la sitagliptina, el API del fármaco anti-diabetes de Merck, Januvia (5). En mayo de 2012, Merck amplió su pacto para la biocatálisis con Codexis por otros tres años hasta el 2015. El acuerdo inicial fue anunciado en abril de 2007.
N-heterociclos enantioselectivosLos compuestos heterocíclicos son
importantes en las aplicaciones farma-céuticas y los investigadores del Institu-to de Tecnología de California (Caltech) reportaron recientemente sobre el avan-ce en la síntesis de dichos compuestos. Ellos reportaron sobre su trabajo en la construcción enantioselectiva de N-heterociclos cuaternarios mediante la alquilación alílica decarboxilativa, ca-talizada con paladio, de lactamas (5, 6).
“Pensamos que va a ser una reac-ción altamente posible, no sólo para la preparación de complejos productos na-turales, sino también por hacer sustan-cias farmacéuticas que incluyen compo-nentes que anteriormente representaban un gran reto para hacerlos,” dijo Brian Stoltz, profesor de química en Caltech, en un comunicado de prensa universi-tario del 13 de enero de 2012. “Esto ha hecho que de repente sean muy fáciles de hacer y debe permitir que los quími-
cos de medicamentos accedan a niveles de complejidad a los que anteriormente no podían acceder.”
Específicamente, los investigadores reportaron sobre la alquilación alílica decarboxilativa catalizada con paladio, altamente enantioselectiva, de lactamas, para formar pirrolidinonas 3,3-disusti-tuidas, piperidinonas, caprolactamas y lactamas estructuralmente relacionadas. Los investigadores afirmaron que la sín-tesis proporciona un nuevo enfoque para la síntesis asimétrica de tales estructuras, un desarrollo importante dada la preva-lencia de N-heterociclos cuaternarios en los alcaloides biológicamente activos y en los agentes farmacéuticos. Los inves-tigadores reportaron que la catálisis pro-porcionaba lactamas cuaternarias enan-tiopuras que interceptan los intermedios sintéticos previamente usados en la sín-tesis de los alcaloides de Aspidosperma, quebrachamina y rhazilinam, pero que anteriormente eran producidos median-te esquemas quirales auxiliares o como mezclas racémicas (5, 6).
Otros enfoques catalíticosEl BINOL y sus derivados son clases de ligandos ampliamente usados en síntesis asimétrica, tales como las re-acciones de Diels-Alder, la adición de carbonilo, y las reducciones (7). Los investigadores en la Universidad de Texas, en Austin, desarrollaron recien-temente un catalizador bifuncional de-rivado del BINOL para producir bro-molactonizaciones altamente selectivas de ácidos carboxílicos no saturados (8, 9). Específicamente, el catalizador pro-mueve bromolactonizaciones altamente enantioselectivas de ácidos 4- y 5-aril-4-pentenoico aunque también cataliza las bromolactonizaciones altamente enantioselectivas de ácidos 5-alquil-4(Z)-pentenoicos. Los investigadores afirman que estas reacciones represen-tan las primeras bromolactonizaciones catalíticas de ácidos olefínicos alquil-sustituidos que procedían por medio de ciclizaciones en modo 5-exo para dar lactonas en las cuales se forman nue-vos enlaces carbón-bromo en un centro
Los Investigadores del Tecnológico de Georgia y Merck, desarrollaron una amina deshidrogenasa para la síntesis de aminas quirales.
“Cumplimiento de los desafíos en la síntesis asimétrica”continúa en la pág. 66

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 63
Do
rLI
Ng
KIN
De
rS
Ley
/ge
tt
y Im
ag
eS
La Farmacopea de los Estados Unidos (USP) ha modificado sus límites de impurezas elementales y los capítulos de procedimiento con la implementación establecida para mayo del 2014. El autor explica la necesidad de estas modificaciones y proporciona un vistazo a algunas de las técnicas propuestas por la USP para la detección e identificación de impurezas elementales.
Alan Cross es científico en RSSL, Centro de Ciencias Reading, Campus Whiteknights, Pepper Lane, Reading, Berks, RG6 6LA, RY. tel. +44 (0)118 918 4129, [email protected].
Las modificaciones a la Farmacopea de EEUU Ca-pítulo General <231> Metales Pesados han sido debatidas y propuestas durante más de una década, y se sabe desde hace tiempo que los métodos ac-
tuales son altamente subjetivos y probablemente inexactos, al menos para ciertos metales. El camino para la reforma ha sido algo balbuceante, pero después de un largo período de revisiones y comentarios, el 1o. de diciembre de 2012, el Ca-pítulo General <232> Impurezas Elementales – Límites y el Capítulo <233> Impurezas Elementales – Procedimientos se-rán publicados en el segundo suplemento de la Farmacopea de EEUU 35 – Formulario Nacional 30 (USP-NF).
El 1o. de mayo de 2014, cuando la USP 37 – NF 32 se vuelva oficial, todas las referencias al Capítulo <231> dejarán de existir, y se requerirá la conformidad al Capítulo <232> y al Capítulo <233> dentro de las Noticias Generales. La acep-tación de estos capítulos abrirá la puerta para que los labora-torios utilicen un rango más amplio de métodos para analizar los contaminantes de metales pesados. Desde luego, estos métodos todavía necesitarán ser validados, y aún puede haber espacio para el debate acerca de cuáles métodos son mejores para cualquier situación dada, pero al menos los métodos du-dosos del Capítulo <231> dejarán de estar disponibles para los medicamentos comercializados en Estados Unidos. (Por el momento, los métodos comparables usados en las farma-copeas europea y japonesa continuarán estando disponibles). Este artículo aborda la necesidad de nuevos requisitos com-pendiales, con un enfoque en la detección e identificación de las impurezas elementales.
Necesidad del cambioEl análisis para metales pesados es actualmente una de las ideas más establecidas, contenidas dentro de las farmacopeas nacionales en todo el mundo. De hecho, la USP ha incluido un prueba general para metales pesados desde 1905 en el oc-tavo volumen de la farmacopea, la cual utilizaba precipitación con sulfuro para detectar antimonio, arsénico, cadmio, cobre, fierro, plomo y zinc. Como sucede, el propósito de la prueba tenía más que ver con la prevención del etiquetado incorrecto que con la prevención de la contaminación, ya que las sales de metales pesados eran usadas con frecuencia en la terapia y uno tenía que conocer cuáles sales estaban presentes en un tratamiento. La necesidad de detectar contaminación residual se estableció en 1942, con la introducción del volumen XII de la USP, en la cual se incluyó en la prueba un estándar que contenía plomo. El objetivo era detectar los residuos de meta-
Postura oficial: iMPurezas eleMentales
Análisis de impurezas elementalesCómo manejar los cambios farmacopeicos que se avecinan Alan Cross

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201264
les pesados potencialmente venenosos, tales como el plomo y el cobre, ya que estos metales eran ampliamente usados en el equipo de producción de esa época. Curiosamente, los metales como el fierro, el cromo y el níquel no se revelaban con la prueba (1). Por último, es la falta de aplicabilidad de una prue-ba “estándar” (tal como la definida por el Capítulo <231>) la que ha llevado a su caída y la necesidad de mayor flexibilidad.
Conocimiento de la industria de los contaminantes metálicos comunes. Las impurezas metálicas son correcta-mente una causa de preocupación en los productos farma-céuticos y existen muchos medios mediante los cuales puede contaminarse un producto. Existen muchas impurezas inorgá-nicas que son agregadas deliberadamente a los procesos far-macéuticos (p.ej., catalizadores). Existen otras impurezas que pueden surgir como contaminantes no detectados provenien-tes de materiales de inicio o reactivos, o que vienen del pro-pio proceso (p.ej., lixiviación de las tuberías y otro equipo). Entonces, por supuesto, hay iones metálicos que se presentan naturalmente dentro de la planta o fuentes minerales que son usadas para producir los ingredientes activos de los farmacéu-ticos y las medicinas herbales.
Independientemente de cómo pueden llegar los metales a un producto, o de la certificación previa de estos metales, los productores farmacéuticos deben llevar a cabo pruebas para demostrar la ausencia de impurezas antes de usar los materia-les en un producto farmacéutico.
Capítulo General <232>: Nuevo límitesEl Capítulo General de la USP <232> Impurezas Elementales – Límites, establece los niveles aceptables de 15 elementos en los productos farmacéuticos finales. Estos límites han sido evaluados de los datos toxicológicos y se expresan en térmi-nos de un límite de exposición diaria permitida (DPE). El DPE también toma en consideración la ruta de administración (p.ej., oral, parenteral o inhalable) con fármacos oralmente administrados que tienen un límite permitido más elevado que los productos farmacéuticos parenterales o inhalables. Cuando se sabe que los elementos de la lista están presentes o tienen el potencial de estar presentes, entonces debe evaluarse el cumplimiento con las especificaciones. Los 15 elementos abordados en el Capítulo <232> se basan en el proyecto de guía en pre-Etapa 2 del Grupo de Trabajo de Impurezas Ele-mentales de la Conferencia Internacional de Armonización (ICH), Q3D (2).
El Capítulo <232> cubre el arsénico, cadmio, mercurio y plomo –todos estos elementos que son considerados omni-presentes y por lo tanto deben evaluarse en todos los casos. Adicionalmente, el capítulo cubre el iridio, osmio, paladio, platino, rodio, rutenio, cromo, molibdeno y níquel. El segun-do grupo de elementos puede estar presente en los productos como resultado de la adición deliberada, por ejemplo, en la forma de catalizadores o a través de interacciones con compo-nentes metálicos a lo largo del proceso de manufactura.
Como la guía Q3D del ICH aún se está revisando y proba-blemente se expanda para cubrir más elementos, se ha decidido que se dará una revisión del Capítulo <232> después de que ha-yan terminado las deliberaciones sobre las guías Q3D del ICH. En esta etapa, el alcance del Capítulo <232> podría expandirse
Postura oficial: iMPurezas eleMentalespara cubrir más elementos, o puede incorporarse un capítulo de información para cubrir los elementos de baja toxicidad.
La desactualización de las pruebas antiguasEs justo señalar que los métodos del Capítulo General <231> de la USP fueron desarrollados antes de la introducción de modernos instrumentos analíticos. Estos métodos fueron fá-cilmente transferibles de un laboratorio al otro y no requirie-ron de instrumentos sofisticados o de experiencia especiali-zada. Por lo tanto, un miembro competente del personal de laboratorio podría realizar las mismas técnicas con relativa facilidad. El problema era que los métodos por sí mismos eran defectuosos, sin importar qué tan competente es el analista.
Por ejemplo, los métodos del Capítulo <231> involucran un examen visual subjetivo y la comparación de la solución de la muestra con un estándar de plomo. Similar al método de 1905, los métodos compendiales utilizaban una reacción para formar el sulfuro de cualquier ión metálico presente y el contenido total de metales se reportaba contra la respuesta del estándar de plomo con una prueba límite.
La validez de esta comparación se apoyaba en varios su-puestos, todos los cuales pueden ser cuestionados. Por ejem-plo, el método compendial asumía que cada uno de los meta-les pesados en la matriz de la muestra reaccionaría de igual manera al plomo para formar las especies de sulfuro. Este supuesto se aplicó a pesar de que se sabe que muchos sulfuros son insolubles y a pesar de que se sabe que algunos elementos tienen un sulfuro de color mucho más intenso que el estándar de plomo contra el cual se está evaluando. De manera similar, el método compendial asumió que la cinética de la reacción para el sulfuro de plomo sería muy similar a la de los otros sulfuros metálicos y que la cinética de reacción no se afectaba mucho por la matriz de la muestra. Un supuesto final mayor e inseguro era que el paso de calentamiento y/o carbonización del método no tendría impacto sobre los metales volátiles (3).
Se han realizado trabajos que sugieren que el recobro de mercurio puede ser tan pequeño como 2% utilizando el método compendial <231>, lo cual introduce claramente un error masivo en el resultado final (2). Otros laboratorios han reportado un recobro escaso similar de metales tales como el estaño, selenio y antimonio. Estos ejemplos no son en modo alguno las únicas razones para retar la validez, aplicabilidad y confiabilidad de los métodos compendiales. De hecho, con los años se han agregado capítulos adicionales a la USP para el control de metales específicos y otras impurezas inorgánicas. Significativamente entre estas adiciones han sido el Capítulo de la USP <730> Espectroquímica de Plasma, el cual le dio a los laboratorios la oportunidad de usar técnicas tales como el plasma inductivamente acoplado ya sea con espectrometría de masas o con espectroscopía de emisión atómica (ICP-MS e ICP-AES).
La ventaja de los métodos ICP es que pueden proporcionar la detección y cuantificación específicas para cada uno de los elementos especificados en el Capítulo <232>. La subjetividad de la comparación semicuantitativa que es requerida por los métodos compendiales se elimina con el ICP. Las técnicas de ICP son también más rápidas en la mayoría de los casos, requi-riendo un tamaño de muestra más pequeño y dando un mejor

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 65
límite de detección para todos los elementos de interés. El mé-todo de preparación de la muestra para el ICP, por ejemplo, es menos probable que lleve a la pérdida de elementos volátiles.
Capítulo <233>: Nuevas TécnicasEl Capítulo General de la USP <233> Impurezas Elementales – Procedimientos establece las condiciones generales para el análisis, que cubre la preparación, el análisis y los parámetros para la validación. Los métodos de preparación referidos para lo anterior son en forma pura, solución acuosa directa, solu-ción orgánica directa y solución indirecta.
Las muestras puras están en tal estado que pueden usarse sin más preparación. Las soluciones más comúnmente usadas necesitarán ser preparadas antes del análisis y el más simple de estos procedimientos es la preparación de una solución di-recta siempre que un producto se disuelva o se diluya con agua/ácido diluido o con un solvente orgánico para dar una solución para el análisis.
En muchos casos, puede ser deseable tratar la muestra descomponiendo cualquier material orgánico contenido den-tro de ésta; dicho paso típicamente reduce el efecto de la ma-triz que podría de otra forma dar lugar a resultados falsos po-sitivos/negativos. Si se prepara una muestra de esta forma, en-tonces se hace referencia a ésta como una solución indirecta. Estas soluciones son preparadas generalmente utilizando un digestor de microondas. En esta técnica, se pesa una pequeña cantidad de muestra en un recipiente y se le agrega ácido. El recipiente se sella y se coloca dentro del microondas. En el microondas, la muestra se calienta a temperaturas de hasta 250°C y presiones hasta de 55 bar. Bajo estas condiciones, la matriz de la muestra se destruye efectivamente y los átomos metálicos son liberados en la solución. Después de enfriar la muestra, se lleva ésta a un volumen adecuado con agua lista para el análisis.
KP-MS e ICP-AES. Según se señaló anteriormente, el Capítulo <233> establece dos procedimientos para el análisis, el ICP-MS y el ICP-AES. El segundo también se conoce a ve-ces como ICP-OES, que significa espectrometría de emisión óptica. En esta técnica, la solución de la muestra se alimenta dentro de un plasma de argón, el cual tiene una temperatura de aproximadamente 10,000 °C. La matriz de la muestra se destruye bajo estas condiciones y se liberan los átomos indi-viduales. Estos átomos son después excitados a un estado de energía más alto. Conforme los átomos excitados se enfrían, vuelven a un “estado fundamental”. El proceso libera energía en forma de luz, cuya longitud de onda es específica para un elemento particular. Cuando esta luz cae en un detector, pue-de ser cuantificada y puede evaluarse la cantidad del analito.
El ICP-MS es el segundo procedimiento especificado en el Capítulo <233>. Esta técnica también usa un plasma, pero con esta técnica, el plasma se utiliza para ionizar los átomos metálicos los cuales son después alimentados dentro de un cuadrupolo el cual separa los iones de acuerdo a su relación masa a carga. Después de la separación, los iones caen en un detector y la muestra puede ser cuantificada.
Diferenciando las nuevas técnicas. Tanto el ICP-AES como el ICP-MS pueden analizar varios elementos simul-táneamente. Como resultado, el rendimiento de la muestra
puede ser muy rápido, típicamente 2-3 minutos por muestra. En general, es justo decir que la instrumentación ICP-AES es más económica que la ICP-MS, aunque ambos instrumentos tienen costos de corrida relativamente altos debido al consu-mo de argón en el plasma. La diferencia clave entre los instru-mentos es el límite de detección. El ICP-MS típicamente tiene límites de detección 100 – 10,000 veces más bajos que los del ICP-AES. Ambas técnicas son capaces de analizar hasta los niveles requeridos por la USP, pero el ICP-MS puede ofrecer un límite de detección mucho más bajo. El Capítulo <233> establece que para ambas técnicas, pueden tomarse pasos para remover interferencias de la matriz. Para el ICP-AES, estas interferencias pueden presentarse por longitudes de onda que se traslapan. En este caso, pueden usarse para el análisis lon-gitudes de onda alternativas. También, muchos fabricantes de instrumentos tienen técnicas de corrección integradas en el software de operación.
En el caso del ICP-MS, las fuentes de interferencias de la matriz provienen del hecho de que las diferentes especies pueden tener la misma relación masa/carga. Por ejemplo, el cloruro de argón aparece en la misma masa que el arsénico, dando resultados falsos positivos. Para remover estas interfe-rencias, muchos fabricantes de instrumentos utilizan celdas especiales dentro del instrumento que pueden agregar gases a los iones y mitigar las interferencias.
Métodos alternativosPueden usarse otras técnicas en el análisis de impurezas ele-mentales, pero cada una debe estar validada para garantizar que es adecuada y capaz de detectar los analitos en el nivel requerido. A continuación están unas pocas opciones.
Espectrometría de absorción atómica con flama (FAAS). Esta técnica simple y relativamente barata tiene límites de detección relativamente altos, especialmente para elementos tales como el mercurio y el arsénico. El FAAS sólo puede analizar un elemento a la vez.
Espectrometría de absorción atómica con generación de vapor (VG-AAS). Esta técnica involucra una reacción quími-ca para liberar metales en forma de hidruros gaseosos. Tiene mejores límites de detección en comparación con el FAAS pero sólo puede usarse para arsénico, bismuto, germanio, plo-mo, antimonio, selenio, estaño, y telurio. Sólo puede anali-zarse un elemento a la vez. Se usan reactivos para generar el hidruro, generando por lo tanto un mayor costo que el AAS tradicional.
Espectrometría de absorción atómica en Horno de Gra-fito (GFAAS). En esta técnica, se calienta lentamente una pe-queña cantidad de muestra para secar primero y luego incinerar la muestra. Posteriormente, la temperatura se eleva muy rápi-damente para volatilizar el metal de interés. Esta técnica tiene muy buena sensibilidad y puede usarse para ver niveles muy bajos de analito, similares a los que se logran con el ICP-AES, aunque es propenso a interferencias químicas que afectan los resultados. También el análisis es lento y puede ser costoso.
La caída del Capítulo <231> significa que las técnicas modernas como las referidas anteriormente, y otras, se con-vertirán ahora en más comunes, y los resultados de la vieja química húmeda dejarán de ser válidos.

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201266
ConclusiónUno sólo puede simpatizar con los científicos de la USP que tienen responsabilidad para los métodos farmacopeicos es-tándar que involucran metales pesados. De las más de 4000 monografías en la USP-NF, existen aproximadamente 1000 que especifican un límite de metales pesados, ya sea en una sustancia farmacéutica, un excipiente o un producto farma-céutico (4). Diciembre de 2012 marca el inicio del fin para el Capítulo <231> y la introducción de los Capítulos <232> y <233>. Para mayo de 2014, el <231> dejará de existir, y para entonces, los procedimientos validados necesitan estar en su lugar para cubrir el retiro del Capítulo <231>. Los 18 meses entre estas fechas pueden parecer un tiempo largo, pero con-siderando el número de monografías existentes que contienen la prueba límite de metales pesados, este marco de tiempo
Postura oficial: iMPurezas eleMentales
parece muy corto. En general, los cambios en la USP, aun-que desalentadores, pueden llevar a mejoras para la industria, incluyendo la mejor protección del público a través de medi-camentos analizados efectivamente. Los fabricantes tendrán la tranquilidad de que están proveyendo productos limpios y seguros al mercado.
Referencias1. O. Pedersoen, Pharmaceutical Chemical Analysis: Methods for
Identification and Limit Tests (Taylor & Francis, 2006).2. N. Lewen et al., J. of Pharm. and Biomedical Anal. 35 (4) 739–
752 (2004). 3. ICH, Q3D Impurities: Guideline for Metal Impurities, Final
Concept Paper (2009).4. D.R. Abernethy, Chief Science Officer, USP, presentation online
at www.usp.org. PT
Righton (3M Sistemas de Entre-ga de Fármacos): La consideración de la experiencia del usuario y la in-terfaz con los dispositivos de entrega de fármacos está a la alta y continuará teniendo un impacto significativo en el éxito de un producto. A lo largo de la próxima década, las compañías conti-nuarán buscando maneras eficientes y amigables con el usuario para entregar los fármacos, lo que crea la preferen-
“Innovación futura en la entrega de fármacos ”continuación de la pág. 57
“Cumplimiento de los desafíos en la síntesis asimétrica”continuación de la pág. 62
cia del paciente, cumpliendo mientras tanto las necesidades de los proveedo-res de salud y de las agencias regula-torias. En este mercado manejado cada vez más por el paciente, las compañías están observando que la integración de aspectos amigables para el paciente, ta-les como la adición de un contador de dosis para dispositivos de inhalación de dosis medidas, puede no sólo servir como un bien en el proceso regulatorio,
sino que también puede construir la pre-ferencia y lealtad del paciente. Además, la cada vez mayor conveniencia del paciente encontrada en los dispositivos de entrega de fármacos sin aguja puede ofrecerle a las compañías una importan-te herramienta del manejo del ciclo de vida que puede diferenciar su producto y proveer una ventaja competitiva. PT
estereogénico con alta enantioselecti-vidad. Los investigadores también re-portaron lo que dicen que es la primera desimetrización catalítica de un ácido dienoico proquiral por bromolactoniza-ción enantioselectiva (8, 9).
Referencias1. B. Halford, Chem. & Eng. News 90 (34),
9 (2012).2. G. Coulthard, W. Erb, and K. Ag-
garwal, “Stereocontrolled Organo-catalytic Synthesis of Prostagladin PGF2α in Seven Steps,” Nature, online DOI10.1038/nature11411, Aug. 15, 2012.
3. M.J. Abrahamson, Angew. Chem. Int. Ed. Engl. 51 (16), 3969–3972 (2012).
4. T. Li et al., J. Am. Chem. Soc. 134 (14), 6467–6472 (2012).
5. P. Van Arnum, Pharm. Technol. 36 (5), 56–60 (2012).
6. B.M. Stoltz et al., Nature Chem. 4 (2) 130–133 (2012).
7. W. Sommer and D. Weibel, Aldrich ChemFiles 8.2 (56), 2008.
8. D.H. Paull, J. Am. Chem. Soc., 134 (27), pp 11128–11131 (2012).
9. C. Drahl, Chem. & Eng. News 90 (28), 29 (2012). PT
EN LA REVISTA MÁS LEIDA POR LA INDUSTRIA FARMACÉUTICA
CONTRATE SU PAUTA PUBLICITARIAANTES DEL 14 DE DICIEMBREINICIE 2013 SU PRESENCIA
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486 Fax: 52 (55) 5659-8879E-mail: [email protected]

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 67
Inaugura nueva planta de Macrosoluciones
Constituida desde hace más de 15 años, “Grupo Neolpharma” a la vanguardia en la producción de pro-ductos farmacéuticos, medicamentos para neuropsi-quiatría, síndrome metabólico, oncología, algología, entre otras; inauguró el pasado 29 de octubre su nueva planta de Macro Soluciones ubicada en el norte de la Ciudad de México en la zona industrial de Vallejo; donde bajo un ambiente ameno y agradable se reunió el gremio farmacéutico para ser testigos del nacimiento de su nuevo proyecto “Parenterales”.
El objetivo de esta nueva planta de PARENTERA-LES, es manufacturar macrosoluciones de gran volu-men en presentaciones de plástico de 250 a 1000 ml, así como también soluciones orales de 500 ml para la
recuperación electrolítica y funcionalidad metabólica. La producción se realizará en equipos y áreas con tec-nología de punta y sistemas de alta seguridad biológica que cumplen las exigencias y regulaciones más estric-tas tanto nacionales como internacionales.
La capacidad de producción proyectada será de 15,000,000 de unidades bajo la más estricta normas de calidad, a través de un proceso “BLOW, FILL & SEAL”, e incorporando Equipos de la más alta tecno-logía mundial en una planta totalmente automatizada de forma robotizada. Cabe mencionar que las resinas utilizadas en la producción de las botellas son únicas en su tipo en toda América Latina.
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486
Fax: 52 (55) 5659-8879E-mail: [email protected]
Y APROVECHE NUESTRAPROMOCIÓN ESPECIAL
CONTRATE ANTES DEL 7 DE FEBRERO

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201268
El Presidente Ejecutivo de “Grupo Neolpharma”, Lic. Efrén
Ocampo López, demostrando su calidad humana, con una
sonrisa saluda a quienes va encontrando en el camino hacia
el podio, nos pide antes de iniciar que nos pongamos de pie
y cantemos el Himno Nacional Mexicano y al terminar; con un
rotundo “Viva México” agradeció a los visitantes.
Estuvo acompañado en el podio por el Dr. Dagoberto
Cortés Cervantes, Presidente de ANAFAM, Dr. René Asomoza
Palacio, Director General de Investigación y Estudios Avan-
zados de CINVESTAV, Mtro. Mikel Andoni Arriola Peñalosa,
Comisionado Federal de Protección contra Riesgos Sanitarios
COFEPRIS, Ing. Eric Hágsater Gartenberg, Presidente de CA-
NIFARMA, Lic. José Antonio González Fernández, Ex Secreta-
rio de Salud, Alejandro Carbajal González, Diputado Federal.
Para la construcción de esta planta, se necesitó una inversión cercana a los 450 MDP, y se conto con la par-ticipación de expertos de Inglaterra, Italia, Alemania, Estados Unidos y de México.
Con este nuevo proyecto “Grupo Neolpharma” re-afirma su compromiso al estrechar vínculos con insti-tuciones académicas y centros de Investigación, tales como el IPN, UNAM, CINVESTAV, UAM; promoviendo la investigación y el Desarrollo Tecnológico, y para im-pulsar a los investigadores. En conmemoración a la inauguración Grupo Neolpharma ha diseñado el “Pre-mio Neolpharma en Biotecnología y Nanotecnología” y el CINVESTAV será la sede de dicho premio en 2013.
En su participación; el Presidente de ANAFAM, el director del CIVESTAV y el Comisionado de COFEPRIS, destacaron el compromiso de Neolpharma con Méxi-
co, al instituir el premio en Biotecnología y Nanotecno-logía, la inversión de esta nueva planta y su contribu-ción a la economía del país al comprometerse a innovar la producción de medicamentos utilizando tecnología de punta.
El reto para “Neolpharma” será lograr un crecimien-to anual entre un 12% y 14%, crecer la exportación en América Latina un 10%, y exportar al mercado Nor-teamericano; ya que sus condiciones regulatorias son muy estrictas. Además por supuesto el seguir con su compromiso de desarrollo sustentable que se verá reflejado en su contribución a la comunidad. Hoy día cuenta con una planta de Hidroponia, áreas verdes, una granja y una planta de tratamiento de agua resi-dual, agua que próximamente se utilizará para el riego de áreas verdes.
En hora buena Grupo Neolpharma!

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 69
El lugar para los amantes de sus mascotas
Recibe consejos e información para el cuidado de tu mascota y Consulta a Expertos que responderán a tus principales inquietudes sobre:
•
Comportamiento / Entrenamiento•
Alimentación•
Higiene•
Control de plagas•
Enfermedades / padecimientos comunes •
y másComparte experiencias con otros dueños de mascotasConoce a otras mascotas como la tuyaEncuentra pareja para tu mascotaCompra regalos para tu mascota
Síguenos en...
CHAT MASCOTAS
CALENDARIO DE EVENTOS
Cuando usted contacte a alguno de los organizadores de éstos eventos, por favor mencione que vio su evento en
NOVIEMBRE 12 – 14Curso taller “PROY-NOM-241-SSA1-2011 Buenas Prácticas de Fabricación para Fabricantes de Dispositivo. Lugar: Hotel Sevilla Palace, Ciudad de México, Teléfonos: 52 (55) 47530632, E-mail: [email protected], Página Web: www.globalsystemsandenterprisesolutions.com
13 - 14Pharmaceutical Supply Chain Conference. Lugar: Bethesda, MD USA, Página Web: www.pda.org/GlobalEventCalendarandRegistration/PDAFDA-Pharmaceutical-Supply-Chain-Conference-.aspx
15 – 16Curso Taller “Liderazgo en GMP bajo la NOM-059-SSA1-2006” Good Manufacturing Practice for the Pharma. Lugar: Hotel Sevilla Palace, Ciudad de México. Teléfonos: 52 (55) 47530632, E-mail: [email protected], Página Web: www.globalsystemsandenterprisesolutions.com
19 - 22 EMBALLAGE 2012 - Packaging exhibition. Lugar: Paris Nord Villepinte, France. Página Web: en.emballageweb.com/
26 – 28The Annual World Drug Manufacturing Summit sources and presents leading global case studies from Pfizer, GSK, AstraZeneca, Baxter, Bayer and many more. Lugar: Dusseldorf, Germany, Página Web: www.wdmsummit.com/
28 – 29Pharma Integrates 2012: Where do you see partnerships strategically? Lugar: London, UK. Riverbank Park Plaza Hotel. Página Web: www.lifesciencesindex.com/pharma-integrates-2012-agenda
promueva su evento aquí CONTRATACIONES (55) 5659-8880
30 NOVIEMBRE – 3 DICIEMBRE ‘12CPhI India. Lugar: Bombay Exhibition Centre - Western Express Highway, Goregaon, Mumbai, India. Página Web: www.cphi-india.com
DICIEMBRE3-4 Pharmaceutical Partnering Meeting Pharma Venue. Lugar: Barcelona (España). Página Web: www.pharmavenue.com/registration.php
3 – 4 PDA FDA Vaccines Conference. Lugar: Bethesda, MD USA. Página Web: www.pda.org/GlobalEventCalendarandRegistration/PDAFDA-Vaccines- Conference.aspx
7 – 8 5th Annual Contract Manufacturing. Lugar: London, UK. Página Web: www.visiongain.com/Conference/311/5th-Annual-Contract-Manufacturing
ENERO 201328 – 29Pre-Filled Syringes. Lugar: London, UK. Página Web: www.smi-online.co.uk/events/overview.asp?is=4&ref=4023

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201270
PRUEBAS DE INTEGRIDAD DE FILTROS HEPA Y HERMETICIDAD EN SELLOS DE MARCOS.
Con esta prueba se determina la presencia o ausencia de fugas en los filtros y la estanqueidad entre el marco de alojamiento y el filtro.
PRUEBAS DE LAMINARIDAD DE FLUJO DE AIRE.Con esta prueba se determina la laminaridad del aire detectando posibles turbulen-cias que provoquen contaminación interna.
CONTEO DE PARTÍCULAS VIABLES PARA CLASIFICACIÓN DE ÁREA.Con esta prueba se determina la cantidad las partículas vivas en el ambiente.
CONTEO DE PARTÍCULAS NO VIABLES PARA CLASIFICACIÓN DE ÁREA.Con esta prueba se lleva a cabo la monitorización de las partículas volátiles sus-pendidas en el aire.
VELOCIDADES DE FLUJO Y CAMBIO DE AIRE POR HORA. Con esta prueba se verifica la cantidad, velocidad y renovación del aire en las áreas.
MONITOREO DE TEMPERATURA Y HUMEDAD. Con esta prueba se verifican los rangos de temperatura y humedad en las diferen-tes zonas acorde a normatividad.
PRESIONES DIFERENCIALES ENTRE CUARTOS.Con esta prueba se determina el sentido de flujo de aire y la presión existente entre cuartos.
PRUEBA DE RECUPERACIÓN.Con esta prueba se determina el tiempo necesario sistema (HVAC) en recuperar sus condiciones iniciales.
CALIFICACIÓN DE EQUIPOS Y SISTEMAS CRÍTICOS.Equipos y Líneas de Producción, Sistemas de Agua Purificada y Agua Grado Inyecta-ble (WFI), Aire Comprimido, Sistema de Nitrógeno, Sistemas Computarizados.
ASESORIA ESPECIALIZADA.De Validación, Calificación y Proyectos.
Estamos seguros que con nuestra experiencia en la Validación / Calificación, podremos encontrar la mejor relación costo – beneficio que satisfaga sus necesidades.
www.eolis.com.mx [email protected] www.comsaemte.com Tel. +52 55 3640
Eolis cuenta con una larga experiencia en la planificación, construcción y mantenimiento de instalaciones y Sistemas completos de cuartos limpios para la Industria Farmacéutica, Alimenticia, Cosmética, Veterinaria e Industrial en general.Sabemos que una de las partes principales para tener resultados satisfactorios es la obtención de ambientes controlados con características particulares como: partículas, temperatura, hu-medad, diferenciales de presión.
EOLIS, ofrece la calificación de instalaciones, sistemas y equi-pos para todo tipo de Industria regulada por normas naciona-les e internacionales, incluyendo:
70
CALIFICACION Y VALIDACION DE EQUIPOS, SISTEMAS CRITICOS, Y SISTEMAS COMPUTARIZADOS

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 2012 71
MAQUINARIA DE ACONDICIONAMIENTO / LLENADORAS Y TAPONADORAS
MAQUINARIA DE ACONDICIONAMIENTO / BLISTERAS Y ENCARTONADORAS
Directorio Clasificado
CONSTRUCCIÓN Y SISTEMAS CRÍTICOS / SISTEMAS DE AIRE ACONDICIONADO EQUIPO DE PROCESO / AISLADORES
MAQUINARIA DE ACONDICIONAMIENTO / LLENADORAS Y TAPONADORAS
MAQUINARIA DE ACONDICIONAMIENTO / AGRUPADORAS Y ENVOLVEDORAS

Pharmaceutical Technology en Español NOVIEMBRE / DICIEMBRE 201272
Cuando usted contacte a alguno de estos anunciantes, por favor mencione que vió su anuncio en
ÍND
ICE
DE
AN
UN
CIA
NT
ES
PRO
DU
CTO
/SERVIC
IOEM
PRESA
No. PÁ
GIN
AN
O. PA
RA TA
RJETA D
E SER
VICIO
AL LEC
TOR
Accesorios para Producción y M
antenimiento / Sistem
as de Pesado y Surtido
Cápsulas de Gelatina Blanda
Construcción y Sistemas Críticos / Cuartos Am
bientales
Construcción y Sistemas Críticos / Sistem
as de Aire Acondicionado
Equipo de Proceso / Aisladores
Equipo de Proceso / Tableteadoras y Encapsuladras
Ingredientes Activos / Excipientes y Vehículos
Inst. y Equipo para Inspección. Control y Monitoreo de Procesos
Instrumentos y Equipos Analíticos
Instrumentos y Equipos Analíticos - Biotecnología
Maquinaria de Acondicionam
iento / Agrupadoras y Envolvedoras
Maquinaria de Acondicionam
iento / Blisteras y Encartonadoras
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Maquinaria de Acondicionam
iento y Embalaje
Materiales y Accesorios para Laboratorio / Purificadores de Agua para Laboratorio
Sistemas de Aire Acondicionado
Sistemas de Aire Acondicionado: Equipos, Filtros, Etc
Varios / Chat mascotas
AERSA
División M
aquinaria
CATALEN
T
GRU
PO ID
EA
CEDIN
SA
COM
ECER - PACK & PRO
CESS
ACG -pam
- PACK & PRO
CESS
MEXA
LC, S.A D
E C.V.
STEVAN
ATO G
ROU
P
BRUKER M
EXICAN
A, S.A
. DE C.V.
SARTO
RIUS
POLY PACK - PACK &
PROCESS
HEIN
O ILSEM
AN
N - PACK &
PROCESS
MA
R - PACK & PRO
CESS
ROTA
- PACK & PRO
CESS
PACK FEEDER - PACK &
PROCESS
VEOLIA
WATER SYSTEM
S MEXICO
S.A. D
E C.V.
EOLIS A
MERICA
LATINA
S.A. D
E C.V.
CALEFACIÓ
N Y VEN
TILACIÓN
S.A. D
E C.V.
PETFACEBOO
K
9
4ta. Forros
13
7171
2da. Forros
2317331717171716949
3ra. Forros
11
69
7 1
17
11
10
2
16
8 3 5
1213
15
14
18
6 4919
ww
w.aersa.net / E-m
ail: maquinaria@
aersa.net
ww
w.catalent.com
/ Email: consultas@
catalent.com
ww
w.grupoidea.com
.mx / e-m
ail: [email protected]
.mx
ww
w.cedinsa.com
.mx / Em
ail: info@cedinsam
ex.net
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.m
exalc.com.m
x / E-mail: tklabe@
mexalc.com
ww
w.optrel-ltd.com
/ ww
w.stevanatogroup.com
- spami@
stevanatogroup.com
ww
w.brukeroptics.com
/ Email: info@
bruker.com.m
x
ww
w.sartorius.com
/ E-mail: sartorius-stedim
@sartom
ex.com.m
x
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.purelabflex.com
/ Email: elga.m
exico@veoliaw
ater.com
ww
w.eolis.com
.mx y w
ww
.comsaem
te.com
ww
w.cyvsa.com
/ Email: salud@
cyvsa.com
ww
w.petfacebook.com
.mx
DIR
ECC
IÓN
WEB
/ CO
RR
EO ELEC
TRÓ
NIC
O

Marque en la tarjeta de servicio al lector el No. 4

Marque en la tarjeta de servicio al lector el No. 1