
SIGNOS Y SÍNTOMAS DE LOS TUMORES...
92
1 SIGNOS Y SÍNTOMAS DE LOS TUMORES CEREBRALES PRIMARIOS PREVIOS AL DIAGNÓSTICO, EN PACIENTES ENTRE LOS 6 MESES Y 17 AÑOS EN EL INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT EVIDENCIADOS ENTRE ENERO DE 2008 A NOVIEMBRE DE 2011 JOHANNA ANDREA JIMÉNEZ JIMÉNEZ JULIET ROCÍO PICHÓN RODRÍGUEZ UNIVERSIDAD DE CIENCIAS APLICADAS Y AMBIENTALES FACULTAD DE CIENCIAS DE LA SALUD MEDICINA HUMANA BOGOTÁ 2012
Transcript of SIGNOS Y SÍNTOMAS DE LOS TUMORES...

1
SIGNOS Y SÍNTOMAS DE LOS TUMORES
CEREBRALES PRIMARIOS PREVIOS AL DIAGNÓSTICO, EN PACIENTES
ENTRE LOS 6 MESES Y 17 AÑOS EN EL INSTITUTO DE ORTOPEDIA
INFANTIL ROOSEVELT EVIDENCIADOS ENTRE ENERO DE 2008 A
NOVIEMBRE DE 2011
JOHANNA ANDREA JIMÉNEZ JIMÉNEZ
JULIET ROCÍO PICHÓN RODRÍGUEZ
UNIVERSIDAD DE CIENCIAS APLICADAS Y AMBIENTALES
FACULTAD DE CIENCIAS DE LA SALUD
MEDICINA HUMANA
BOGOTÁ
2012

2
SIGNOS Y SÍNTOMAS DE LOS TUMORES CEREBRALES PRIMARIOS
PREVIOS AL DIAGNÓSTICO, EN PACIENTES ENTRE LOS 6 MESES Y 17
AÑOS EN EL INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT
EVIDENCIADOS ENTRE ENERO DE 2008 A NOVIEMBRE DE 2011
Trabajo de grado presentado como requisito para obtener título de Médico cirujano
JOHANNA ANDREA JIMÉNEZ JIMÉNEZ
JULIET ROCÍO PICHÓN RODRÍGUEZ
Asesor Científico
Dra. Vanessa Gonzales Vargas
Médico Residente de Neurocirugía
UNIVERSIDAD DE CIENCIAS APLICADAS Y AMBIENTALES
FACULTAD DE CIENCIAS DE LA SALUD
MEDICINA HUMANA
BOGOTÁ
2012

3
Nota de Aceptación:
________________________________
________________________________
________________________________
________________________________
________________________________
_________________________________
Firma del Presidente del Jurado
_________________________________
Firma del Jurado
_________________________________
Firma del Jurado

4
DEDICATORIA
Queremos dedicarle este trabajo a nuestros padres, porque creyeron en nosotras, dándonos ejemplo digno de superación y entrega, porque en gran parte gracias a ellos hoy podemos ver alcanzada nuestra meta. Gracias porque siempre estuvieron impulsándonos en los momentos más difíciles de esta laboriosa carrera. Este maravilloso logro es por ustedes, por lo que valen, porque admiramos su fortaleza y por lo que han hecho de nosotras. Y a las personas que de algún modo nos fomentaron el deseo de superación y el
anhelo de triunfo en la vida. Mil palabras no bastarían para agradecerles su apoyo,
su comprensión y sus consejos en los momentos difíciles.
A todos, esperamos no defraudarlos y contar siempre con su valioso apoyo,
sincero e incondicional.

5
AGRADECIMIENTOS
Queremos agradecerle la culminación de este trabajo a Dios que nos ha dado la
vida y fortaleza, pero sobre todo la sabiduría para hacer las cosas de manera
correcta.
Y una vez más agradecer a nuestros padres, hermanos (as) y demás familiares
por ser la luz guía de nuestros pasos y decisiones, porque para nosotras son el
motor que nos impulsa cada día a ser mejores.
Agradecer a la Dra. Eugenia Espinosa (Neuropediatra), por su paciencia y
colaboración, a la Dra. Martha Rocha (Pediatra) por su amabilidad y paciencia en
realizar las correcciones de nuestro trabajo, a la Dra. Karen (Epidemióloga) por
que sin su ayuda incondicional todo habría sido más complicado y al Dr. Germán
Molano (Medico y docente de investigación / metodología) por acompañarnos en
todo el proceso hasta su culminación, a ellos muchas gracias.
Y a todas las personas que directa o indirectamente colaboraron para concluir esta
etapa de formación, a esos compañeros que siempre tuvieron una palabra de
aliento, aquellos que nos vieron caer, se rieron y al final nos tendieron la mano,
gracias.
Gracias a todos y cada uno de los que lean y han leído este trabajo porque, por
ese simple hecho, ya forman parte de él.
Gracias a todos por depositar su confianza en nosotras, por el buen ejemplo y
porque, tal vez sin que lo sepan, nos han inspirado para ser cada día mejores: a
todos gracias por su amistad y apoyo.

6
TABLA DE CONTENIDO Pág.
INTRODUCCIÓN ………………………………………………………………….…….10 JUSTIFICACIÓN ………………………………………………………………….……..11 1. PROBLEMA Y PREGUNTA DE INVESTIGACIÓN……………………….……....12 2. OBJETIVOS…………………………………………………………………………...13 2.1 Objetivo general …………………………………………………………………….13 2.2 Objetivos específicos …………………………………………………….………..13 3. MARCO TEÓRICO …………………………………………………………………..14 3.1 Tumor cerebral primario ……………………………………………………………14 3.2 Epidemiologia ……………………………………………………………………….15 3.3 Factores de riesgo ………………………………………………………………….15 3.4 CLASIFICACIÓN TUMORES CEREBRALES PRIMARIOS …………..……....16 3.4.1 Astrocitomas ………………………………………………………………………18 3.4.1.1 Astrocitoma grado I o de bajo grado …………………………………………18 3.4.1.2 Astrocitoma difuso ………………………………………………….…………..18 3.4.1.3 Astrocitoma anaplasico …………………………………………….………….18 3.4.1.4 Astrocitoma IV de alto grado o glioblastoma multiforme……….…………..19 3.4.1.4.1 Glioblastoma de células gigantes…………………………….………….....19 3.4.2 Oligodendrogliomas ……………………………………………………………...19 3.4.3 Ependimarios ……………………………………………………………………..19 3.4.3.1 Subependimoma (Grado I) ……………………………………………………20 3.4.3.2 Ependimoma mixopapilar (Grado I) ………………………………………….20 3.4.3.3 Ependimoma grado II……………………………………………………….….20 3.4.3.3.1 Ependimoma celular …………………………………………………….......20 3.4.3.3.2 Ependimoma papilar ……………………………………………..............…20 3.4.3.3.3 Ependimoma de células claras ………………………………………….....20 3.4.3.3.4 Ependimoma tanicitico ………………………………………………………20 3.4.3.3.5 Ependimoma anaplasico (Grado III) ……………………………………….20 3.4.4 Tumores neurales, mixtos, ganglionares ………………………………………21 3.4.4.1 Gangliocitoma…………………………………………………………..……….21 3.4.4.2 Ganglioma desmoplásico infantil ……………………………………………..21 3.4.4.3 Tumores Neuroepiteliales disembrioplasicos ……………………………….21 3.4.4.4 Neurocitoma central…………………………………………………………….21 3.4.4.5 Neuroblastoma olfatorio ……………………………………………………….22 3.4.5 Gliomas mixtos ……………………………………………………………………22 3.4.6 Plexos coroideos ……………………………………………………………….…22 3.4.6.1 Papiloma del plexo coroideo …………………………………………....…….22 3.4.6.2 Carcinoma del plexo coroideo ………………………………………….……..22 3.4.7 Tumores pineales ………………………………………………………………...22 3.4.7.1 Pineocitoma, pineoblastoma y tumores mixtos/ transicionales ……………22 3.4.8 Tumores embrionarios……………………………………………………………23

7
Pág.
3.4.8.1 Meduloepitelioma…………………………………………………….………... 23 3.4.8.2 Neuroblastoma………………….……………………………………………... 23 3.4.8.3 Ependimoblastoma…………………………………………………………..... 23 3.5 SÍNTOMAS SEGÚN LA LOCALIZACIÓN DEL TUMOR CEREBRAL PRIMARIO………………………………………………………………………………. 24 3.5.1 Lóbulo frontal………………………………………………………………………24 3.5.2 Lóbulo temporal……………………………………………………………………24 3.5.3 Lóbulo parietal……………………………………………………………………..24 3.5.4 Síntomas cerebelosos ……………………………………………………………24 3.5.5. Lóbulo occipital……………………………………………………………………25 3.6 SIGNOS Y SÍNTOMAS DE LOS TUMORES CEREBRALES PRIMARIO …...25 3.6.1 ALTERACIONES EN LA MARCHA …………………………………………….25 3.6.1.1 Macha hemiparetica …………………………………………………………...26 3.6.1.2 Marcha atáxica ………………………………………………………………….26 3.6.2 Rigidez nucal ……………………………………………………………………...27 3.6.3 Cefalea …………………………………………………………………………….27 3.6.4 Apraxias ……………………………………………………………………………29 3.6.5 Agnosias …………………………………………………………………………..30 3.6.6 Nistagmus …………………………………………………………………………30 3.6.7 Hipertensión endocraneana ……………………………………………………..31 3.6.7.1 Diagnostico clínico ……………………………………………………………..32 3.6.7.2 Diagnostico radiológico ………………………………………………………..37 3.6.7.3 Monitorización presión intracraneal …………………………………………..37 3.6.7.4 Saturación yugular de oxigeno …………………………………………….….37 3.6.7.5 Electroencefalograma ……………………………………………………….…38 3.6.7.6 Eco-doppler trasncraneal ………………………………………………….…..38 3.6.7.7 Presión tisular de oxigeno………………………………………………….…..38 3.6.7.8 Tratamiento hipertensión endocraneana ……………………………….……38 3.6.7.8.1 Estabilización inicial ………………………………………………………….38 3.6.7.8.2 Optimización flujo sanguíneo cerebral …………………………………....39 3.6.7.8.3 Pseudoanalgesia…………………………………………..……………….. 39 3.6.7.8.4 Hematológico …………………………………………………………………40 3.6.7.8.5 Profilaxis anticonvulsiva ………………………………………….………....40 3.6.7.8.6 Evitar fiebre …………………………………………………………………...40 3.6.7.8.7 Profilaxis antibiótica ………………………………………………………….40 3.6.7.8.8 Hidroelectrolitico……………………………………………..……………... 40 3.6.7.8.9 Esteroides …………………………………………………………………….41 3.6.8 Nauseas y émesis ………………………………………………………………..41 3.6.9 Vértigo ……………………………………………………………………………..41 3.6.10 Somnolencia……………………………………………………..……………….41

8
Pág.
3.6.11 Dificultad para la marcha ……………………………………………………….41 3.6.12 Irritabilidad…………………………………………………………………….….42 3.6.13 Alteración comportamiento psicomotor………………………………………..42 3.6.14 Hidrocefalia y aumento perímetro cefálico ……………………………….….42 3.6.15 Letargia ……………………………………………………………………….….43 3.6.16 Papiledema ……………………………………………………………………...43 3.6.17 Ataxia ……………………………………………………………………………..43 3.6.17.1 Ataxia cebelosa …………………………………………………………….…43 3.6.17.2 Ataxia sensitiva …………………………………………………………..……43 3.6.17.3 Ataxia aguda de origen tumoral ………………………………………..……43 3.6.18 Lipotimia ……………………………………………………………………….…44 3.6.19 Disminucion en la fuerza muscular …………………………………………...44 3.6.20 Microcefalia ……………………………………………………………………..45 3.6.21 Convulsiones…………………………………………………………………….45 3.6.22 Otros……………………………………………………………………………...48 3.7 DIAGNOSTICO ……………………………………………………………………..49 3.7.1 Historia clínica……………………………………………………………………..49 3.7.2 Examen físico……………………………………………………………………...49 3.7.3 Paraclinicos………………………………………………………………………..49 3.8 TRATAMIENTO …………………………………………………………………….49 3.8.1 Cirugía ……………………………………………………………………………..50 3.8.2 Radioterapia ………………………………………………………………………51 3.8.2.1 Indicacion radioterapia…………………………………………………..…..…51 3.8.2.2 Radioterapia externa convensional…………………………………………...51 3.8.2.3 Radioterapia externa estereotaxica…………………………………………...51 3.8.2.4 Radioterapia intersticial y braquiterapia…………………………………...…52 3.8.2.5 Terapia con haces de partículas………………………………………………52 3.8.2.6 Radioterapia externa hiperfraccionada…………………………………..…...52 3.8.2.7 Radioterapia intensiva ,modulada ……………………………………………52 3.8.3 Quimioterapia ……………………………………………………………………..53 4 METODOLOGIA ………………………………………………………………………54 4.1 Diseño de la investigacion …………………………………………………………54 4.2 Poblacion estudiada…………………………………………………………………55 4,2,1 Poblacion diana……………………………………………………………………55 4.2.2 Criterios selección de la muestra………………………………………………..55 4.2.2.1 Criterios inclusión ………………………………………………………………55 4.2.2.2 Criterios exclusión ……………………………………………………………..55 4.2.3 Poblacion estudiada ……………………………………………………………...55 4.2.4 Tamaño d ela población …………………………………………………………55 4.3 Recolección de la información e instrumento ……………………………….…..56 4.5 Calidad del dato, control de sesgos y error ………………………………….…..56

9
Pág.
4.6 Procesamiento estadístico de la información y plan de análisis de la información ………………………………………………………………………………56 5. RESULTADOS INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT …...…57 5.1 Discusión instituto de ortopedia infantil Roosevelt …………………………......77 5.2 Conclusiones instituto de ortopedia infantil Roosevelt ………………………...80 5.3. Recomendaciones………………………………………………………………….81 ANEXOS ………………………………………………………………………………....82 Anexo A Instrumento recolección de datos …………………………………………..83 Anexo B Discusión Instituto Ortopedia Roosvelt………………………………….….84 Lista de Graficas …………………………………………………………………….…..85 Lista de Tablas ……………………………………………………………….……….…86 Lista de Anexos…………………………………………………………………………..87 REFERENCIAS ………………………………………………………………………....88

Bogotá; Mayo/2012
SIGNOS Y SÍNTOMAS DE LOS TUMORES CEREBRALES PRIMARIOS PREVIOS AL DIAGNÓSTICO, EN PACIENTES ENTRE LOS 6 MESES Y 17 AÑOS EN EL INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT EVIDENCIADOS ENTRE ENERO DE 2008 A
NOVIEMBRE DE 20111
Johanna Jimenez2
Juliet Pichón3 2012
RESUMEN
Los tumores cerebrales en niños y lactantes son la segunda forma más común de cáncer después de la leucemia. El meduloblastoma es el tumor más frecuente seguido por los astrocitomas cerebelosos constituyendo los tumores del SNC propios de la infancia. Se identificaron los principales signos y síntomas de los tumores cerebrales primarios, previos al diagnóstico, en pacientes entre los 6 meses y 17 años del Instituto de Ortopedia Infantil Roosevelt entre enero de 2008 a noviembre de 2011. Los datos analizados en este proyecto se tomaron a partir de la revisión de historias clínicas de los pacientes que acuden al servicio de pediatría, neuropediatría y neurooncología de esta institución que cumplen con los criterios de inclusión; la información recolectada fue ingresada a una base de datos en excel y posteriormente se realizo el análisis estadístico con SPSS. Los resultados: En total se conto con 68 casos que cumplían con criterios de inclusión, se identificó que los signos de los tumores cerebrales primarios más frecuentes son: Convulsiones (61.8%), seguido de hidrocefalia (47.1%), aumento de perímetro cefálico (38.2%), hiporeflexia (30.9%), disminución de la fuerza muscular (20.6%), alteración de la marcha (17.6%), disartria (7.4%), hipotonía (7.4%), hemiplejia (8.8%) y hemiparesia (14.7%) y dentro de los síntomas los más frecuentes son: Emesis se encontró en el (47%) siendo el mas representativo, seguido de cefalea en un (46%), irritabilidad (43%), alteraciones del desarrollo psicomotor (38%), somnolencia (24%), náuseas (22%), vértigo (12%), retraso de crecimiento y alteraciones de comportamiento cada uno con un (10%) y dificultad para la marcha (1.2%). Este trabajo de investigación es solo un capítulo del consolidado del trabajo de investigación Signos y Síntomas de los tumores cerebrales primarios previos al diagnóstico, en pacientes entre los 6 meses y 17 años en el Instituto de Ortopedia Infantil Roosevelt y el Hospital Occidental de Kennedy evidenciados entre Enero de 2008 a Noviembre de 2011.
Palabras Clave: tumores cerebrales primarios, signos, síntomas, localización anatómica. Clasificación histológica.
1 Trabajó de grado en modalidad investigación. 2 Estudiante de medicina X semestre. 3 Estudiante de medicina X semestre.

10
INTRODUCCIÓN
Se conoce como tumor cerebral primario al grupo celular anormal que tiene como
origen el tejido neuronal, meninges, nervios o glándulas que ocupan la región
intracraneal. Este tipo de alteración puede afectar directamente las células
neuronales causando la destrucción o inflamación de estas, llevando así al
aumento de la presión intracraneal, afectando otras partes del cerebro.
Los tumores cerebrales primarios, a través de los años, han representado un
porcentaje importante de las muertes a nivel mundial con tendencia al aumento;
siendo por estudios estadísticos los gliomas los que conforman más del 80% de
los tumores cerebrales, siendo estos más prevalente en la población caucásica
(1).
De acuerdo con las estadísticas proporcionadas por el Departamento de
Neurocirugía de la Universidad del Valle (Guía de Manejo: Tumores. Cali. 2009), la
incidencia de los tumores cerebrales es de 10 x 100.000 habitantes al año, de los
cuales el 60% son de etiología maligna, resaltando que los tumores cerebrales,
corresponden a la segunda causa de muerte en niños, siendo más frecuentes en
hombres que en mujeres a razón de 6.5%:4.5% por 100.000 habitantes al año.
Por la Fundación Anna Vázquez. Tumores Cerebrales (Primera Parte). Bogotá.
2008.(2), expone como en niños menores de 15 años, del 15 - 20% de los tumores
cerebrales son intracraneales, siendo los más prevalentes en la infancia los
meduloblastomas, astrocitomas y ependimomas.
Esta investigación descriptiva retrospectiva, serie de casos; tiene como propósito
dar una orientación precisa sobre los signos y los síntomas que presentan los
pacientes con tumores cerebrales primarios, antes de ser diagnosticados y
confirmados por exámenes de laboratorio e imagenológicos; logrando la sospecha
temprana para el correcto diagnostico y oportuno inicio de tratamiento para
mejorar el estado de salud y calidad de vida de estos pacientes.

11
JUSTIFICACIÓN
Se busca obtener beneficio a nivel salud en el mejoramiento de la calidad de vida
en niños entre los 6 meses y 17 años de edad quienes desarrollan una serie de
trastornos fisiológicos, que a lo largo de su evolución se expresan en un tumor
cerebral primario; así como repercusiones graves a nivel social, disminuyendo las
tasas de mortalidad a nivel nacional, en donde la incidencia de los tumores
cerebrales en los niños ocupan el segundo lugar en frecuencia (22%),
representando el 1% de las muertes en el mundo (3).
Al identificar los signos y síntomas que presentan estos pacientes entre los 6
meses y 17 años previos al diagnóstico de tumor cerebral primario en el Instituto
de Ortopedia Infantil Roosevelt y el Hospital Occidente de Kennedy evidenciados
entre enero de 2008 a noviembre de 2011, obteniendo una anamnesis completa y
oportuna, nos hará sospechar de la presencia de un tumor cerebral primario,
facilitando así el diagnostico temprano y el tratamiento oportuno, evitando la
evolución de la enfermedad y aparición de secuelas.
Con lo anterior se desea mejorar no solo la capacidad de diagnóstico del médico y
el nivel de calidad de atención hospitalaria, sino también mejorar la calidad de vida
y la supervivencia en años de estos pacientes, además de fortalecer las relaciones
psicoafectivas y familiares, como medio de apoyo para el contribuir al
mejoramiento del estado físico y mental de estos pacientes.

12
1. PROBLEMA Y PREGUNTA DE INVESTIGACIÓN
En los lactantes y niños pequeños, los tumores cerebrales son la segunda forma
más común de cáncer después de la leucemia. La distribución por sexos muestra
un discreto predominio de los tumores benignos en mujeres, mientras que los
malignos y las cifras globales son mayores en varones. Las tasas de mortalidad de
algunas estadísticas con tendencia ascendente serán de 6,5 por 100 mil
habitantes/año para varones y 4,5 para mujeres. (3)
Sólo el 1,5% de todos los cánceres se observa en niños, pero con una alta
incidencia de neoplasias del SNC siendo la segunda causa más importante de
cáncer. El meduloblastoma es el tumor más frecuente en niños y conjuntamente
con los astrocitomas cerebelosos constituyen los tumores propios de la infancia.
(3)
El cuadro clínico del tumor cerebral primario, depende de la edad, el género, la
localización anatómica, la clasificación histológica, los signos y síntomas, que en
la mayoría de casos son muy inespecíficos; y a su vez son comunes en otro tipo
de patología presentes en la edad pediátrica; lo que dificulta un diagnostico
temprano. Todo esto motiva a la realización de un estudio en donde por clínica a
través de los signos y síntomas se contribuya a la sospecha del tumor, realizando
un diagnostico temprano que se verá reflejado en el mejoramiento de la calidad de
vida de estos pacientes, evitando la aparición de secuelas, y al mismo tiempo
mejorado el criterio de los médicos al momento de diagnosticar esta patología,
realizándola de forma oportuna para así lograr una intervención terapéutica
temprana e individualizada para cada paciente.
Con el propósito de concientizar a los profesionales de la salud sobre la sospecha
temprana del tumor cerebral primario se formula la siguiente pregunta de
investigación:
¿Cuáles son los signos y síntomas que presentan los pacientes entre
los 6 meses y 17 años, previos al diagnóstico de tumor cerebral
primario, en el Instituto Infantil de Ortopedia Roosevelt y el Hospital
Occidente de Kennedy evidenciados entre enero de 2008 a noviembre
de 2011?

13
OBJETIVOS
2.1 OBJETIVO GENERAL
Identificar los principales signos y síntomas de los tumores cerebrales primarios,
previos al diagnóstico, en pacientes entre los 6 meses y 17 años del Instituto
Infantil de Ortopedia Roosevelt y el Hospital Occidente de Kennedy evidenciados
entre enero de 2008 a noviembre de 2011.
2.2 OBJETIVOS ESPECÍFICOS
Caracterizar la población estudiada de acuerdo con las variables demográficas y clinicopatológicas de los tumores cerebrales primarios, previos al diagnóstico, en pacientes entre los 6 meses y 17 años del Instituto Infantil de Ortopedia Roosevelt y el Hospital Occidente de Kennedy evidenciados entre enero de 2008 a noviembre de 2011.
Tipificar los tumores cerebrales primarios de acuerdo a su clasificación histológica y localización en pacientes entre los 6 meses y 17 años del Instituto Infantil de Ortopedia Roosevelt y el Hospital Occidente de Kennedy evidenciados entre enero de 2008 a noviembre de 2011.
Establecer las relaciones entre la localización, signos y síntomas de los tumores cerebrales primarios, previos al diagnóstico, en pacientes entre los 6 meses y 17 años del Instituto Infantil de Ortopedia Roosevelt y el Hospital Occidente de Kennedy evidenciados entre enero de 2008 a noviembre de 2011.
Cumplir los objetivos citados anteriormente en el análisis único de cada hospital.

14
2. MARCO TEÓRICO
Los tumores cerebrales primarios son la entidad de tumor solido más frecuente en
la infancia, teniendo dentro de los tipos más comunes según el grupo etario los
astrocitomas que son quistes benignos de crecimiento lento que se presentan en
niños en edades entre los 5 y los 8 años; los gliomas ocurren casi exclusivamente
en niños con edad aproximada de 6 años que expresa su clínica cuando su
tamaño es bastante significativo; los ependimomas representan entre un 8 y 10%
de los tumores cerebrales pediátricos, los cuales se encuentran localizados en los
ventrículos cerebrales ocasionando la obstrucción de los mismos y por ende del
flujo de líquido cefalorraquídeo y por último los meduloblastomas que son el tipo
de tumor cerebral primario más frecuente en la Infancia, se presenta en niños
menores de 10 años con una edad promedio de 5 años, siendo más común en
niños que en niñas. Los síntomas suelen ser inespecíficos dependiendo no solo de
la localización del tumor sino también de la edad del niño que empeoran
gradualmente y/o pueden ocurrir de una manera muy rápida.
Según los registros del Instituto Nacional de Cancerología (INC), la tasa de
incidencia de tumores del SNC es 2 a 5 de cada 100.000 niños con una incidencia
que ha ido incrementándose anualmente en 1% durante los últimos 20 años.
3.1 TUMOR CEREBRAL PRIMARIO
Dr. Niño J. y el Dr. Barrientos C. (4) define un tumor cerebral primario como un
crecimiento desordenado de las células cerebrales que generan lesiones
infiltrativas que se van expandiendo hasta el parénquima logrando obstruir las
fronteras normales de la sustancia gris y sustancia blanca.
Las cuales pueden ser de tipo benigno o maligno dependiendo de las
características histológicas; en el caso de los tumores benignos se encuentran
con bordes bien delimitados, no invasivos, con poca probabilidad de reaparición
después del tratamiento quirúrgico; en cuanto a los malignos se caracterizan
por tener bordes indefinidos, de comportamiento metastásico y tienden a
reaparecer después del tratamiento quirúrgico.
3.2 EPIDEMIOLOGÍA

15
Los Tumores Cerebrales Primarios representan entre el 80 y 95 % las
neoplasias a nivel cerebral, con una incidencia del 21% de las neoplasias
presentes en niños. Según el Instituto nacional de cancerología (5) la tasa de
incidencia de tumores cerebrales primarios es de 4 por cada 100.000 personas
para los hombres y de 3 por cada 100.000 personas en mujeres menores
de 14 años de edad. Representando entre las neoplasias la segunda causa
de muerte en la infancia después de las leucemias y segunda causa de
mortalidad después de los accidentes de tránsito. La distribución es similar para
ambos sexos, predominando los tumores Infratentoriales (55%) sobre los
Supratentoriales (45%), excepto en lactantes, en los que predominan los
supratentorial. De todas formas, cada grupo de edad, dentro de la población
pediátrica, manifiesta una preferencia para ciertos tipos tumorales. En Europa y
Norteamérica, predominan el astrocitoma cerebeloso y el meduloblastoma. En
África y Japón, hay una mayor incidencia de craneofaringiomas y tumores de la
región pineal. En cambio, el ependimoma es más frecuente en la India que en
cualquier otro país.
3.3 FACTORES DE RIESGO
Se han identificado algunos factores de riesgo asociados que contribuyen a la
aparición de los tumores cerebrales primarios obteniendo:
Factores Genético-Hereditarios: Neurofibromatosis o enfermedad de Von Recklinhausen (Neurinomas del acústico y gran variedad de gliomas), Esclerosis Tuberosa, Enfermedad de Von Hipel-Lindau, Síndrome de Sturge-Weber, y un 16% de pacientes con historia Familiar de cáncer.
Factores Ambientales: Radiaciones, Traumatismos, Factores ocupacionales, Factores infecciosos.
Factores relacionados con radiaciones: Asociación entre el riesgo de aparición de glioma y meningioma por antecedente de exposición radiológica repetida.
Factores relacionados con Traumatismos: Traumatismos encefalocraneanos graves.
Factores de Inmunosupresión: SIDA o inmunosuprimidos (Linfoma Cerebral Primario).

16
3.4 CLASIFICACIÓN DE TUMORES CEREBRALES PRIMARIOS
Según la Organización Mundial de la Salud a nivel Histológico los Tumores
Cerebrales Primarios se dividen en:
TUMORES ASTROCÍTICOS:
Astrocitoma de células gigantes subependimario G I Astrocitoma pilocítico G I Astrocitoma pilomixoides G II Astrocitoma difuso G II Xantoastrocitoma pleomórfico G II Astrocitoma anaplásico G III Glioblastoma G IV Glioblastoma de células gigantes G IV Gliosarcoma G IV
TUMORES OLIGODENDROGLIALES: Oligodendroglioma G II Oligodendroglioma anaplásico G III
TUMORES OLIGOASTROCÍTICOS: Oligoastrocitoma G II Oligoastrocitoma anaplásico G III
TUMORES EPENDIMARIOS: Subependimoma G I Ependimoma mixopapilar G I Ependimoma G II Ependimoma anaplásico G III
TUMORES DE PLEXOS COROIDES: Papiloma de plexos coroides G I Papiloma de plexos coroides atípico G II Carcinoma de plexos coroides G III OTROS TUMORES NEUROEPITELIALES:
Glioma angiocéntrico G I Glioma coroide de tercer ventrículo G II TUMORES NEURO-GLIALES MIXTOS Y NEURONALES:
Gangliocitoma G I Ganglioglioma G I

17
Ganglioglioma anaplásico G III Astrocitoma desmoplásico infantil y ganglioglioma G I Tumor neuroepitelial disembrioplásico G I Neurocitoma central y neurocitoma extraventricular G II Liponeurocitoma cerebelar G I Paraganglioma de cordón espinal G I Tumor glioneural papilar G I Tumor glioneural formador de rosetas del IV ventrículo G I
TUMORES PINEALES: Pineocitoma G I Tumor de parénquima pineal de diferenciación intermedia G I Pineoblastoma G IV Tumor papilar de región pineal G III o IV
TUMORES EMBRIONARIOS: Meduloblastoma G IV Tumor neuroectodérmico primitivo del sistema nervioso central G IV Tumor rabdoide/teratoide atípico G IV
TUMORES DE NERVIOS CRANEALES Y PARAESPINALES:
Schwanoma G I Neurofibroma G I Perineurioma G I Tumor maligno de la vaina nerviosa periférica G II, III o IV
TUMORES MENÍNGEOS:
Meningioma G I Meningioma atípico G II Meningioma anaplásico G III Hemangiopericitoma G II Hemangiopericitoma anaplásico G III Hemangioblastoma G I
TUMORES DE LA REGIÓN SELLAR: Craneofaringioma G I Tumor de células granulares de la neurohipófisis G I Pituicitoma G I Oncocitoma de células alargadas de la adenohipófisis G I
LINFOMAS Y NEOPLASIAS HEMATOPOYÉTICAS: Linfoma maligno G IV Plasmocitoma G IV Sarcoma Granulocítico G IV

18
TUMORES DE CÉLULAS GERMINALES:
Germinoma G IV Carcinoma embrionario G IV Tumor de saco vitelino G IV Coriocarcinoma G IV Teratoma: Maduro G 0 Inmaduro G IV Teratoma con transformación maligna G IV Tumor de células germinales mixto G IV
TUMORES METASTÁSICOS
Clasificación 2007 de Histológica los tumores del Sistema Nervioso Central de la
Organización Mundial de la Salud
3.4.1 Astrocitomas. Son tumores primarios del SNC y se definen como tumores
del tejido neuroepitelial en su variedad de tumores astrocíticos. La Organización
Mundial de la Salud clasifica los astrocitomas cerebrales según sus características
histológicas, capacidad de invasión y progresión, de la siguiente forma:
3.4.1.1 Grado I o astrocitoma de bajo grado. Constituyen un grupo heterogéneo
de tumores curables quirúrgicamente como es el caso del astrocitoma pilocítico
que es un tumor benigno debido a que no invade tejido vecino al cerebro. (6) En
general los tumores grado I son de crecimiento lento y son comúnmente
diagnosticado en niños entre edades de 15-19 años.
3.4.1.2 Grado II o astrocitoma difuso. Son tumores de crecimiento lento con una
evolución crónica, tienen poca capacidad para invadir estructuras cerebrales. La
principal sintomatología de este tipo de tumores es la crisis epiléptica.
Encontramos como representantes: Astrocitoma fibrilar, Protoplasmático y
Gemistocítico.
3.4.1.3 Grado III o astrocitoma anaplásico. Constituyen el 4% de los tumores
cerebrales primarios. Este tipo de tumores tiene un crecimiento rápido e invaden
tejidos vecinos sanos.
3.4.1.4 Grado IV o astrocitoma de alto grado o glioblastoma multiforme. “Es el
tumor mas maligno. Se clasifican en: Primarios y Secundarios que se presentan
más en pacientes jóvenes” (7). Gliosarcoma: “Según la Organización Mundial de

19
la Salud se clasifica con un grado tumoral IV. Principalmente afecta regiones
Supratentoriales” (8).
3.4.1.4.1 Glioblastoma de células gigantes. “Se caracterizan histológicamente
por células gigantes multinucleadas. Las manifestaciones clínicas se relacionan
con su localización y tamaño” (9). “Los tumores astrocíticos mas frecuentes son el
astrocitoma anaplásico y el Glioblastoma multiforme en donde su presentación
clínica más frecuente se establece con el defecto motor y alteraciones de la esfera
psíquica” (10).
3.4.2 Oligodendrogliomas. Corresponden entre el 4 y 7% de todos los tumores
primarios del SNC. La frecuencia de este tumor en pacientes pediátricos se
encuentra entre el 1 y el 2% de todas las neoplasias primarias del SNC. El signo
inicial más frecuente es la epilepsia, seguida de cefalea, alteración de funciones
mentales y vértigo-náusea que en los niños es el segundo en importancia (11).
3.4.3 Ependimarios. Según la clasificación más reciente de la Organización
Mundial de la Salud y el Instituto Nacional del Cáncer de los Institutos Nacionales
de la Salud de EE.UU. (12), sobre los tumores cerebrales primarios; éste tipo de
tumor lo clasifican en 4 subtipos principales.
3.4.3.1 Subependimoma (Grado I según OMS). Considerada una neoplasia
maligna de crecimiento lento que se une a la pared ventricular componiéndose de
conglomerados de células tumorales gliales en una matriz fibrilar.
3.4.3.2 Ependimoma Mixopapilar (Grado I según OMS). Surge casi
exclusivamente del cono medular terminal, la cauda equina y el filo terminal de la
médula espinal, el cual se caracteriza por células tumorales organizadas de forma
papilar alrededor de núcleos estrómicos mixoides vascularizados.
3.4.3.3 Ependimoma (Grado II según OMS). Se origina en las paredes del
ventrículo o del canal espinal, componiéndose de células neoplásicas
ependimarias. A la vez se subdivide en:
3.4.3.3.1 Ependimoma Celular. Subtipo más común; habitualmente muestra una
celularidad significativa sin aumento en la actividad mitótica.
3.4.3.3.2 Ependimoma Papilar. Forma superficies lineales de tipo epitelial a lo
largo de las exposiciones del líquido cefalorraquídeo.

20
3.4.3.3.3 Ependimoma de Células Claras. Presenta una apariencia
oligodendroglial con halos perinucleares; esta variante se localiza preferentemente
en el compartimiento supratentorial del cerebro.
3.4.3.3.4 Ependimoma Tanicítico. Se localiza con mayor frecuencia en la
columna vertebral; las células tumorales se disponen en fascículos de ancho y
densidad celular variables que se entrelazan precariamente.
3.4.3.3.5 Ependimoma Anaplásico (Grado III según OMS). Muestra un aumento
de celularidad y un aumento de la actividad mitótica que, con frecuencia, se
relacionan con proliferación microvascular y necrosis pseudoempalizada.
En las investigaciones realizadas por el Instituto Nacional del Cáncer de los
Institutos Nacionales de la Salud de EE.UU., se concluye (12): Que los
subependimomas y los ependimomas mixopapilares habitualmente se consideran
tumores diferentes que los ependimomas de grado II y grado III. En los
ependimomas de grado II y grado III, la relación entre las características
histológicas y la supervivencia varió entre estudios, aunque en la mayoría de los
estudios y meta análisis amplios recientes, se demostró que el grado histológico
es un factor pronóstico independiente de la supervivencia sin complicaciones. En
un estudio de una sola institución se indica que los pacientes con ependimomas
de células claras pueden tener un riesgo más alto de fracaso del tratamiento que
los pacientes con otras formas de ependimomas de grado II; sin embargo, se
necesita una confirmación en un grupo más numeroso de pacientes no
seleccionados. Los ependimoblastomas, que por lo general se comportan más
como meduloblastomas o tumores cerebrales neuroectodérmicos, se consideran
entidades separadas de los ependimomas y ahora se clasifican con los tumores
embrionarios.
3.4.4 Tumores neuronales y mixtos ganglionares.
3.4.4.1 Gangliocitoma. El gangliocitoma central según el autor J. Escalona (13)
se define como un tipo de tumor que se desarrolla a expensas de células
nerviosas adultas, con una ubicación anatómica a nivel de los hemisferios
cerebrales especialmente afectando los lóbulos temporales, ganglios basales e
infundíbulo. Su prevalencia es mayor entre jóvenes que van desde los 10
años y 30 años. Afectando en mayor proporción a género femenino.

21
3.4.4.2 Ganglioma Desmoplásico Infantil. El ganglioma desmoplásico infantil
(GDI) según el autor J. Escalona (13) se puede resumir como el tumor más
frecuente en los neonatos y niños menores de 2 años, presentando un
mayor pico de incidencia a los 4 meses afectando a el género masculino, su
localización anatómica es a nivel de la parte anterior del cerebro es decir el
lóbulo frontal, parietal y temporal. Macroscópicamente se va observar un
tumor firme, con bordes mal delimitados y van a tener una gran adherencia a
las meninges, es un tumor que no va a producir necrosis, ni hemorragias.
Tiene una sobrevida muy alta en los pacientes que la padecen, su recibida es
de 10% en paciente que han sido tratados mediante resecciones quirúrgicas.
3.4.4.3 Tumores neuroepiteliales disembrioplásicos. Basados en la
información suministrada por los autores Pérez J. Resendiz M. Aguirre D (14); El
ganglioma desmoplásico infantil tiene una prevalencia entre los tumores del
sistema nervioso central entre el 1 y 2 %. su incidencia se presenta entre los
20 a 39 años de edad con una relación entre hombres y mujeres de 3 :1, su
ubicación anatomía es a nivel supratentorial e intracortical. Macroscópicamente
se aprecia como una neoplasia nodular bien delimitada. Compuesta por material
mucoso de baja malignidad.
3.4.4.4 Neurocitoma Central. Como lo define Aguirre (14) y Rodríguez (15) el
neurocitoma central es un tumor de naturaleza neural y glioneuronal, se
desarrolla durante la embriogénesis, su localización a nivel anatómico es
supratentorial , interventricular y en niños a nivel de la línea media. Tiene una
evolución lenta y poca expansiva. su prevalencia está entre los 2 a 4 años
de edad, con una afectación para ambos sexos equitativos.
3.4.4.5 Neuroblastoma Olfatorio. Es un tumor maligno de aparición
infrecuente, supone el 3% de todas las neoplasias intranasales, es de etiología
desconocida, se ubica a nivel del epitelio olfatorio de la placa cribiforme a
nivel del septum nasal, se extiende a nivel del cráneo e intracraneal. Su
aparición se da entre los 10 a 20 años de edad, afectando en la misma
proporción hombres y mujeres.(16)
3.4.5 Gliomas mixtos. Es un crecimiento anormal de las células gliales, que
conforman el Sistema Nervioso Central como astrocitos, células ependimales y/o
oligodendrocitos, entre otras; que dependiendo el compromiso del componente
oligodendroglial se definirán como: Oligodendroglioma (90-100% de compromiso
glial), Oligoastrocitoma (inferior al 90%). (17)

22
3.4.6 Plexos coroideos.
3.4.6.1 Papiloma del Plexo Coroideo. Considerada una neoplasia benigna
originada a partir del epitelio cuboidal de los plexos coroideos en el
neuroectodermo; tomando la forma de uno de ellos hipertrófico, contribuyendo a la
formación de líquido cefalorraquídeo. En los niños se forma en los ventrículos
laterales, produciendo aumento de la presión intracraneal, alteración neurológica,
cefalea y ataxia. (18)
3.4.6.2 Carcinoma del Plexo Coroideo. Es una neoplasia maligna de rara
frecuencia que representa entre el 20% y el 30% de los tumores de los plexos
coroideos; se presenta con mayor frecuencia en niños a nivel de ventrículos
laterales. Éste tipo de tumor presenta “características invasivas, áreas de necrosis,
focos de hemorragia y en su mayor parte pierden el patrón papilar, que se ve
sustituido por nidos y áreas difusas de células anaplásicas con marcado
pleomorfismo y actividad mitótica” (19).
3.4.7 Tumores pineales.
3.4.7.1 Pineocitoma, Pineoblastoma y Tumores Mixtos/Transicionales. Son
una serie de neoplasias, originadas en el parénquima pineal, produciendo una
hipertrofia invasiva de las mismas sobre la Glándula Pineal; Siendo la más
agresiva la forma de pineoblastoma, ya que éste se ubica a nivel del neuroeje,
produciendo hidrocefalia obstructiva o Síndrome de Parinaud. (20)
3.4.8 Tumores embrionarios.
3.4.8.1 Meduloepitelioma. El meduloblastoma o meduloepitelioma es el tumor
de mayor prevalencia a nivel embrionario. Aguirre (14) lo define como un
tumor infiltrativo de alta capacidad de invasión debido a que se disemina a
través del liquido cefalorraquídeo por medio de las leptomeninges y cavidades
ventriculares supratentoriales sobre el epitelio ependimario. En los niños se
ubica a nivel del vermis cerebeloso y ápex del cuarto ventrículo. Afecta a
niños menores de 16 años con un pico de aparición a los 7 años afectando
principalmente al género masculino.
3.4.8.2 Neuroblastoma. El Neuroblastoma es uno de los tumores sólidos
malignos más frecuentes en los niños, se origina en la cresta neural durante la

23
embriogénesis y aparece sobre la cadena ganglionar simpática. Es el más
frecuente en lactantes. Su diagnostico se hace antes de los 10 años en un
95% y antes de los 5 años en un 90% . Es un tumor que tiene una evolución
y comportamiento muy variable, es por esto que su sobrevida es incierta, su
pronóstico se ve muy relacionado con la edad y factores de riesgo asociados.
3.4.8.3 Ependimoblastoma. Según Aguirre (14) se trata de un tumor poco
frecuente que se desarrolla en neonatos y en niños menores de 2 años,
afectando a ambos géneros, su localización es a nivel ventricular y
supratentorial aunque puede aparecer en medula espinal y leptomeninges
además de tener una gran incidencia de metástasis extracerebral.
3.5 SÍNTOMAS SEGÚN LA LOCALIZACIÓN DEL TUMOR CEREBRAL PRIMARIO La sintomatología que experimentan los pacientes con tumores cerebrales
primarios varía según la localización anatómica como Carlos Uribe lo describe:
3.5.1 Lóbulo frontal. Las crisis características de la región frontal posterior son la
oculocefalogiras y crisis adversivas secundarias a compromiso de la región
precentral. Cuando existe compromiso del lóbulo frontal dominante se presentan
alteraciones del lenguaje como afasia de tipo motora. Además cambios
psicológicos característicos de la lesión frontal como depresión aunque no tan
frecuente o excitación donde se encuentra euforia, irritabilidad. También se
encuentran trastornos de la marcha (ataxia) y alteraciones en la diadococinesia.
(21)
3.5.2 Lóbulo temporal. Son frecuentes crisis epilépticas de tipo uncinadas en las
cuales se perciben olores desagradables, obsesiones persistentes. También se
asocia a alteraciones de memoria y labilidad emocional, trastornos del lenguaje,
tipo afasia receptiva. (21)
3.5.3 Lóbulo parietal. Por la vecindad con el área motora, las hemiparesias
contra laterales al inicio flácidas y luego espásticas es la manifestación más
frecuente continuado por alteraciones sensoriales, trastornos práxicos y de

24
lenguaje. Alteración de coordinación motora que se ponen en manifiesto con
movimientos finos como escritura y costura. (21)
3.5.4 Síntomas cerebelosos. Según la topografía se consideran dos síndromes
según sintomatología: los tumores del a línea media que comprometen el lóbulo
floculo-nodular, más frecuentes en niños como el meduloblastoma que se
manifiesta por trastornos del equilibrio como ataxia del tronco y movimientos
oculares tumores del hemisferio causan alteraciones de la coordinación motora de
las extremidades que se manifiesta por hipotonía, adiadococinesia, disartria y
temblor de intensión. (21)
3.5.5 Lóbulo occipital. Poco frecuentes, su principal síntoma es dolor occipital
con irradiación a región cervical y compromisos del tracto motor y sensitivo. (21)
3.6 SIGNOS Y SÍNTOMAS DE LOS TUMORES CEREBRALES PRIMARIOS
La forma más común de presentación de los tumores cerebrales primarios, es un
déficit neurológico progresivo (68%), frecuentemente parésia (45%), seguido por
cefalea (54%) y crisis epilépticas (26%). Los tumores del sistema nervioso central
pueden cursar durante un periodo de tiempo en forma asintomática, siendo en
ocasiones de inicio insidioso, lento y progresivo o bien con manifestaciones
clínicas de inicio súbito. La forma de presentación de los tumores cerebrales es
muy variada y depende de su localización, tipo y velocidad de crecimiento, así
como de la edad del niño. En general, hay dos formas distintas de presentación:
Síntomas y signos de hipertensión intracraneal
Signos neurológicos focales. (22)
“El primer síntoma son los cambios de personalidad. En el niño, semanas o meses
antes de ser diagnosticado el tumor, se puede presentar, irritable, hiperactivo u
olvidadizo, y/o puede disminuir su rendimiento escolar” (23).
Con frecuencia los tumores presentan síntomas de mayor importancia son:
3.6.1 Alteraciones en la marcha. La presencia de alteraciones en la marcha, bien
condicionadas por una ataxia o por una hemiplejia, orientan a la localización del
proceso neoplásico.
La ataxia la encontramos en las lesiones cerebelosas, que se ubicaran en el
vermix si son tronculares (ataxia línea media) y en los hemisferios cerebelosos si

25
son lateralizadas (acompañadas de dismetría, disdiadococinesia, temblor
intencional, hipotonía muscular o nistagmo ocular).
En ocasiones también las lesiones localizadas en el lóbulo frontal pueden producir
una inestabilidad de la marcha que puede simular una ataxia típica de las lesiones
de fosa posterior.
Si el tumor está localizado en áreas motoras de los hemisferios cerebrales, se
manifestaran clínicamente como una hemiparesia o hemiplejia contralateral
espástica. (21)
3.6.1.1 Marcha hemiparética. Este tipo de marcha se origina por una lesión de la
vía piramidal. El paciente camina lentamente, apoyando el peso del cuerpo sobre
el miembro no afectado, desplazando el parético en arco («marcha del segador»),
al tiempo que el brazo afectado permanece pegado al cuerpo en semiflexión. En la
exploración se constatan, en el hemilado parético los correspondientes signos de
afectación piramidal: aumento del tono muscular (espasticidad), hiperreflexia,
signo de Babinski, clonus, etc. El diagnóstico del tipo de marcha se realiza
simplemente con la exploración física. La valoración de los antecedentes (historia
clínica), junto con los estudios neurorradiológicos sirven para determinar la
etiología y establecer la morfología de la lesión cerebral subyacente. El paciente
con este trastorno puede beneficiarse de la fisioterapia y de la atención por parte
del traumatólogo-ortopeda (ortesis). (24)
3.6.1.2 Marcha atáxica. La ataxia se define como una alteración de la
coordinación de los movimientos voluntarios y del equilibrio. Este tipo de marcha
se origina por alteraciones que pueden asentar a diversos niveles: cerebelo, vías
cerebelo-vestíbulo-espinales, cordones posteriores. En los procesos cerebelosos
en los que la afectación reside en un hemisferio, el paciente se desvía hacia el
lado de la lesión; cuando realiza la marcha hacia adelante y atrás de manera
alternativa, hace la «marcha en estrella». En las lesiones del arquicerebelo camina
con aumento de la base de sustentación, con el hemicuerpo superior hacia atrás y
los brazos separados del cuerpo. Las situaciones responsables de una marcha
atáxica son muy diversas incluyendo procesos expansivos cerebelosos,
enfermedades desmielinizantes (esclerosis en placas), enfermedades infecciosas
(cerebelitis) y parainfecciosas (síndrome de Guillain-Barré), degeneraciones
espinocerebelosas, el raro síndrome de Kinsbourne (ataxia, mioclonus y opsoclus,
a veces en relación con tumores, como neuroblastoma), intoxicaciones,
enfermedades metabólicas de expresión intermitente, etc. El tratamiento es el de
la situación responsable. (24)

26
3.6.2 Rigidez nucal. “Puede ser la manifestación de diversas patologías, como
puede ser una herniación cerebelosa a través del foramen magnum, o sangrado
en el espacio subaracnoideo” (21).
3.6.3 Cefalea. La cefalea es uno de los síntomas más frecuentes, suele ser de
carácter intermitente y su intensidad varia de moderada a severa, y es progresiva,
pero puede faltar en tumores infiltrativos con crecimiento lento. Aunque
típicamente se describe como de predominio nocturno y/o matutino, despertando
al paciente por la noche y agravándose con las maniobras de Valsalva o con los
cambios de posición de la cabeza, estas características solo están presentes en
aproximadamente una cuarta parte de los casos.
Se debe a la tracción de estructuras sensibles intracraneales (meninges, senos
venosos, arterias, nervios craneales), al efecto masa y a la hidrocefalia. La cefalea
en la mayoría de los casos tiene características inespecíficas pero con cierta
frecuencia (en un tercio de los casos) puede indicar la localización del tumor en un
hemisferio y puede ser unilateral con características de migraña en un porcentaje
pequeño de pacientes (entre un 7 y un 9%). Así un tumor localizado en las fosas
craneales anterior y media podría producir típicamente una cefalea frontotemporal
por un tumor supratentorial que comprimiese estructuras inervadas por pares
craneales, mientras que los localizados en la fosa posterior darían clínica en la
región occipital y nucal, por afectación de pares craneales bajos o bien raíces
cervicales. Si está producida por hipertensión intracraneal la cefalea es frontal u
occipital sin relación con la localización del proceso neoplásico. En cuanto al
tamaño tumoral, no hay estudios concluyentes a cerca de su relación con la
intensidad de la cefalea. (21)
“La prevalencia de cefalea primaria varía de forma considerable entre diversos
estudios, pero se estima que no menos de un 5-10% de niños sufren migraña”
(25).
De forma general, puede considerarse que existen dos grandes grupos de
cefaleas según su causa: primarias, sin trastorno definido causante; y secundarias
o sintomáticas, directamente causadas por un trastorno definido. Todas ellas se
clasifican en función de una serie de criterios propuestos por la International
Headache Society en el año 2004; hoy en día, esta clasificación se ha impuesto y
difundido tanto en edad pediátrica como adulta (International Classification of
Headache Disorders, 2nd edition: ICHD-II). En la ICHD-II, se desglosan todas las
causas posibles de cefalea secundaria en función del origen del trastorno
(traumático, vascular, etc.), así como los criterios para los diferentes tipos de
cefalea. (25)

27
La anamnesis proporciona la orientación principal de la cefalea y entre ellos datos
que tienen máxima importancia en la evaluación, como los enumera Cancho
Candela R (25):
Tiempo de evolución
Hora de aparición: La cefalea por hipertensión intracraneal (HTIC) es,
recuentemente, nocturna o matutina, justo al despertar. La migraña puede ocurrir en cualquier hora del día, aunque es algo más frecuente por la mañana. La cefalea tensional es, predominantemente, vespertina.
Duración: La mayoría de las migrañas duran más de 30 minutos y menos de 3-4 horas; la cefalea hemicraneal paroxística son breves (minutos, algunas horas). La cefalea continua es sospechosa.
Frecuencia: La cefalea episódica es más probable que tenga un origen
primario. La progresividad en la intensidad y/o frecuencia y la presencia de síntomas intercríticos es sospechosa de ser sintomática.
Agente desencadenante: Alimentos, sueño escaso o menstruación. La
cefalea que empeora con el ejercicio o con maniobras de aumento de presión intracraneal es sospechosa de ser sintomática.
Ubicación: La migraña pediátrica es, al menos en la mitad de pacientes,
frontal bilateral, y la cefalea tensional suele ser holocraneal y difusa. La cefalea unilateral fija y la selectivamente occipital pueden ser sospechosas de ser sintomáticas.
Carácter: La cefalea pulsátil es, característicamente, migrañosa. La cefalea
descrita como opresiva es inespecífica y puede corresponder a cualquier tipo de cefalea, primaria o secundaria.
Pródromos: Debe preguntarse de forma orientada acerca de síntomas
previos de tipo sensitivo motriz, visual, etc. La sintomatología vegetativa (náuseas, vómitos, mareo) en la mayoría de episodios es orientativa de migraña. Los vómitos de la HTIC no ocurren en el contexto de un episodio de cefalea, sino de forma independiente y repetida en un periodo prolongado de tiempo, y sin náuseas.
Interfiere con las actividades diarias.

28
Si la historia clínica y el patrón de la cefalea son claros y el examen físico del niño
es normal, no es necesario recurrir a otros exámenes. Se efectuaron estudios por
imágenes en niños con patrones de cefaleas atípicas, con síntomas sistémicos
(p.ej., pérdida de peso, fatiga), con anormalidades en el examen neurológico o
porque los padres estaban preocupados por la posibilidad de un “tumor cerebral”.
Solamente el 3.8% presento una anormalidad en la tomografía de cráneo o en la
resonancia magnética y esta normalidad no explicaba la causa de cefalea. (26)
“La observación de los padres, de los maestros y el uso de “diarios de cefaleas”
suelen ser una fuente de datos importantes en la historia clínica para arribar a
diagnósticos más precisos” (26).
3.6.4 Apraxias. Se definen como una incapacidad de ejecutar actos motores a
pesar de conservar la motilidad elemental y tener el propósito de hacerlo.
Geschwind las definió como la perdida de habilidades motoras adquiridas, gracias
a la memoria procedural. Las principales son las apraxias constructivas, ideatoria,
ideomotora, del vestir y oral. También se han descrito apraxias de la mirada, de
los parpados, de la marcha.
Las apraxias constructivas se observan espacialmente en pacientes con lesiones
parietales (o subcorticales) derechas o izquierdas.
En la apraxia ideatoria existe una desorganización de las etapas de un acto motor
complejo, por ejemplo abrir una botella. Servir un vaso y beber, cepillarse los
dientes o fumar, y los movimientos son poco precisos o se contaminan por
perseveración entre ellos. Heilman y cols. Han propuesto que la representación
del movimiento o engramas motores visuokinestésicos que subyacen a estas
habilidades se almacenan en el lóbulo parietal izquierdo y que su destrucción
causa no solo una apraxia sino que también una agnosia de la pantomima.
Cuando estos engramas se desconectan de las áreas motoras puede existir una
apraxia sin agnosia.
La apraxia del vestir puede observarse en lesiones parietotemporales derechas (o
bilaterales, como en la enfermedad de Alzheimer). El paciente no sabe colocarse
las prendas de vestir, o se las coloca en un orden anormal. La apraxia oral se
refiere a la incapacidad de hacer gestos voluntarios con los labios o con la lengua
y suele asociarse a afasias y a disartria cortical. (27)
3.6.5 Agnosias. Las agnosias son trastornos en el reconocimiento que no se
explican por defectos sensoriales ni por un defecto cognitivo general del paciente.
En las agnosias auditivas el sujeto escucha ruidos pero no sabe identificarlos. En
la agnosia táctil o estereognosia el sujeto conserva la sensibilidad elemental pero
no logra identificar los objetos que se le ponen en la mano.

29
La agnosia visual para los objetos puede ser aperceptible o asociativa. En la
primera existe una percepción fragmentaria, una incapacidad para construir un
todo visual en base a lo percibido. En la agnosia visual asociativa existe una
desconexión entre la información visual, que puede ser adecuada, y las áreas del
lenguaje o la memoria, y el sujeto puede parear objetos o dibujos que no logra
denominar. La agnosia visual aperceptiva suele verse en lesiones del territorio
cerebral posterior izquierdo; la agnosia asociativa requiere habitualmente lesiones
bilaterales. (27)
3.6.6 Nistagmus. Son oscilaciones rítmicas que se superponen a los movimientos
normales de los ojos. Se trata de un problema de estática ocular y los movimientos
son congruentes, conjugados, involuntarios y rítmicos. El tono oculomotor está
regulado por estímulos laberinticos, propioceptivos, visuales, cerebelosos, sistema
extrapiramidal y sustancia reticular. El desequilibrio de estos provoca la aparición
del nistagmus. Los nistagmus de origen cerebral son de comienzo lento. El
nistagmus espontaneo es bastante constante cuando existe tumor del IV
ventrículo” (29).
En todo nistagmus existe un movimiento de oscilación con dos fases y podemos
dividirlos:
Según su velocidad:
Pendular: Las dos fases tienen la misma velocidad, duración y amplitud.
Resorte: tiene una fase lenta y una fase rápida.
En saltamontes (N. de Frenkel): En este tipo existen dos fases rápidas con un intervalo de quietud.
Según la dirección:
Simples: Los movimientos son en un solo plano
Compuestas: Movimientos de más de un plano (oblicuos)
Según la amplitud:
Leves, moderados, severos.
Según la frecuencia:
Lento, mediano y rápido. (28)
Se clasifica en:
Nistagmus unidireccional: En trastornos vestibulares agudos; fase lenta hacia el oído afectado, y fase rápida en contra.

30
Nistagmus multidireccionales: Más común en tóxicos o enfermedades de
tallo o fosa posterior.
Nistagmus de fijación: Solo aparece en la fijación ocular. Es típico de la lesión en SNC. La fijación ocular tiende a abolir el nistagmus periférico.
Nistagmus vertical: Particularmente cuando es persistente, es
característico de vermis cerebeloso o tallo. (29)
3.6.7 Hipertensión endocraneana. Algunos signos y síntomas de los tumores
cerebrales se encuentran relacionados al grado de obstrucción del drenaje del
líquido cefalorraquídeo, aumento en la producción o bien disminución de la
absorción que produce elevación de la presión endocraneal. “La hipertensión
endocraneana es la causa más frecuente de morbimortalidad en niños con
patología neuroquirúrgica” (31).
Este cuadro puede manifestarse de manera aguda o crónica:
Agudo: En menores de 1 año, el incremento inesperado del perímetro
cefálico, separación de suturas, alteraciones del estado de alerta (somnolencia, irritabilidad) disminución en la ingesta. En preescolares, escolares y adolescentes puede manifestarse con cefalea y vómito generalmente matutino que se exacerba con maniobras de Valsava y disminuye en el transcurso del día. El vómito puede ser en proyectil, irritabilidad, letargia, edema de papila, discromatopsia (pérdida de la visión de colores), escotomas centrales y la parésia del VI par.
Crónico o Intermitente: Irritabilidad, letargia, vómito, atrofia de papila o pérdida progresiva de la visión, cambios conductuales, de personalidad, del rendimiento académico, anorexia y pérdida o ganancia ponderal. (30)
La asociación Española de pediatría (31) diferencia tres conjuntos de síntomas y
signos: Triada de inicio: cefalea, vómitos y edema de papila. Progresión clínica,
con disminución del nivel de conciencia por:
Disminución de la presión de perfusión cerebral y disminución del flujo sanguíneo cerebral.
Lesión de la formación reticular del tronco cerebral.
Fenómenos de enclavamiento. Se producen al desplazarse la masa cerebral por el aumento de la presión intracraneal (PIC). Los signos clínicos serán diferentes según la herniación sea central o a través del tentorio.
Si la herniación es central: Aparecen signos de disfunción neurológica
descendentes y progresivos. Con estupor inicial que progresa a coma,

31
alteraciones en el proceso respiratorio, pupilas inicialmente mióticas que pasan a
ser midriáticas no reactivas al progresar la herniación. Y posturas de decorticación
(rigidez y flexión de los brazos sobre el tórax, puños cerrados y piernas
extendidas), descerebración (extensión rígida de los brazos con rotación interna y
piernas, inclinación de los dedos de los pies hacia abajo y arqueo hacia atrás de la
cabeza) y finalmente flacidez. Aparecen los signos constitutivos de la triada de
Cushing (bradicardia, hipertensión arterial y respiración irregular). Por otra parte si
es una herniación uncal se produce una afectación del III par (midriasis unilateral
al hemisferio cerebral dañado) y hemiparesia (por lo general contralateral al
hemisferio cerebral dañado).
Para el diagnostico de la Hipertensión intracraneal, la asociación Española de
pediatría (31) describe siete situaciones diagnosticas:
3.6.7.1 Diagnostico Clínico. Aparecen signos de afectación general,
caracterizados por la disminución progresiva del nivel de conciencia. Es muy
importante destacar que esta disminución ocurre siempre siguiendo los siguientes
niveles, de mejor a peor:
Bradipsiquia (lentitud intelectual y de ejecución). Desorientación témporo-espacial.
Estupor. El paciente tiene tendencia a quedar dormido. El paciente entra en situación de hiperactividad elemental intelectual y psíquica, sin responder al entorno.
Coma. El paciente está inmóvil. Sólo responde ante estímulos externos.
Muerte cerebral.
La escala de Glasgow es un instrumento útil para valorar la disfunción cerebral y
sobre todo permite el seguimiento evolutivo. (31)
Como describe el Dr. Conrad Stephens K (32) los signos y los síntomas de la
Hipertensión intracraneal son variables dependiendo de la edad del paciente y de
la velocidad de instauración del proceso y separa este síndrome en:
Síndrome de hipertensión intracraneana subagudo o progresivo
Síndrome de hipertensión intracraneana agudo
Síndrome de hipertensión intracraneana del lactante

32
Síndrome de hipertensión intracraneana subagudo o progresivo. Cuando el
agente causal es un proceso expansivo que crece en forma gradual. Ej.: Tumor
cerebral
La manifestación más frecuente es la cefalea que es progresiva y que va
aumentando de intensidad a través del tiempo. Cada vez que nos enfrentemos a
un paciente que tiene cefalea y que es claramente progresiva, debe alertarnos
ante la posibilidad de un proceso expansivo. Por el contrario, una cefalea de
carácter recurrente, con largos periodos de remisión, y con episodios de distinta
localización e intensidad, es muy probable que sea por síndrome de hipertensión
intracraneana (SHI).
La cefalea tiene un horario matutino, el paciente se despierta con cefalea y se
alivia luego de levantarse. Este fenómeno se explica porque al dormir, en posición
horizontal, la presión sanguínea del seno longitudinal superior es mayor, lo que
dificulta el retorno venoso y la reabsorción de liquido cefalorraquídeo (LCR). Una
vez que la persona se levanta, la presión del seno disminuye, mejora el retorno
venoso y facilita la reabsorción de líquido cefalorraquídeo, con lo que se alivia la
presión intracraneal, y la cefalea disminuye.
La intensidad de la cefalea aumenta con las maniobras que provocan aumento de
presión del LCR, como son el toser, pujar, hacer movimientos bruscos. Por esta
razón las personas con síndrome de hipertensión intracraneana se mueven con
cautela, hablan en voz baja, monótona, cara menos expresiva, se paran y caminan
lento. Es frecuente que al toser se lleven la mano a la cabeza. Asociado pueden
tener emesis y nauseas, que no alivian la cefalea.
Generalmente la cefalea es holocránea, en ocasiones pueden ser de predominio
frontal y otras de predominio occipital. Algunas veces el lugar donde apareció una
cefalea progresiva, puede indicar la ubicación del proceso expansivo y esto es
debido a que el tumor se encuentra cerca de la duramadre y pude irritar las
terminaciones nerviosas que posee la dura madre en la cercanía de los vasos
meníngeos, causar dolor pero cuando la cefalea es debida a síndrome de
hipertensión intracraneana, se debe principalmente al estiramiento y compresión
de las arterias del cerebro y de las meninges en la base del cráneo y se expresa
como holocránea, de predominio frontal si es supratentorial, y a veces occipital, si
es un proceso de la fosa posterior.
El examen de fondo de ojos: si la presión intracraneal aumenta, también aumenta
la presión de LCR que roda al nervio óptico, lo que dificulta el retorno venoso de
las venas de la retina que se dilatan y pierden el latido venoso que normalmente

33
es apreciable en la mayor parte de las personas. Si persiste la hipertensión
intracraneal se congestiona la papila; se pone hipertérmica y sus bordes se hacen
menos precisos, se difuminan. Posteriormente aparece edema de la papila con
levantamiento y finalmente aparecen hemorragias peripapilares y exudados
fibrinosos. Si no se trata la causa del problema, la compresión del nervio se hace
intolerable y la persona comienza a perder la visión hasta quedar ciega, por atrofia
del nervio óptico.
Alteración de la conciencia también puede ser una manifestación de SHI y
habitualmente es debida a la deformación y malfuncionamiento de las estructuras
de la línea media del encéfalo (hipotálamo, formación reticular, tálamo) cuando el
proceso expansivo las empuja hacia el lado contrario. Generalmente cuando se
produce la hernia subfacial el compromiso de conciencia (obnulación, confusión),
aumenta drásticamente y el paciente cae en sopor. Posteriormente la hernia
transtentorial descendente puede comprimir el III par y aparecer parálisis, primero
con dilatación de la pupila, luego ptósis palpebral y finalmente parálisis de todos
los músculos inervados por el motor ocular común. Si la hernia continua creciendo
puede comprimir el mesencéfalo y el paciente cae en coma mesencefálico y
decimos que ha habido “enclavamiento”, pues esta hernia estrangula
progresivamente las estructuras neuronales y se producen hemorragias por
obstrucción venosa que agravan mas la condición del paciente.
Manifestaciones neurovegetativas: es frecuente que un paciente con síndrome de
hipertensión intracraneana presente taquicardia, diaforesis, hipertensión arterial,
esto como expresión de estrés pero también un intento del organismo de mejorar
la presión de perfusión cerebral. Posteriormente cuando las herniaciones causan
alteraciones del funcionamiento del mesencéfalo y el tronco cerebral, aparecen
alteraciones del ritmo respiratorio pro con una respiración rápida y profunda que
puede causar alcalosis. Luego se presenta una respiración de Sheyne-Stockes
(aumento progresivo de la amplitud y frecuencia, luego descenso hasta hacer una
pausa de varios segundos, para repetir el ciclo). Finalmente cuando la alteración
del tronco se extiende hacia abajo se presenta una respiración atáxica.
Cuando la hipertensión intracraneal es muy intensa, o de rápida progresión puede
presentarse bradicardia e hipertensión arterial severa y se debería a un aumento
en la presión arterial para mejorar la perfusión cerebral, pero al mismo tiempo,
causa reflejo inhibitorio de la frecuencia cardiaca por estimulación del seno
carotideo del cuello.

34
Finalmente es necesario recalcar que cualquier manifestación neurológica con un
perfil evolutivo claramente progresivo debe hacer pensar en que el agente causal
más probable es un proceso expansivo.
Síndrome de hipertensión intracraneana Agudo. Aparece cuando la
enfermedad causante del síndrome es de instalación aguda, como: una
hemorragia subaracnoidea, obstrucción aguda de la circulación del líquido
cefalorraquídeo en los ventrículos, por un cisticerco intraventricular por ejemplo,
hemorragia secundaria a un traumatismo, una encefalitis viral grave, obstrucción
aguda de un seno venoso importante, etc.
Se inicia con cefalea, muchas veces de aparición brusca y acompañada de
emesis. Puede rápidamente aparecer alteración del estado de conciencia, que
progresa en pocas horas o días hacia el coma y la muerte, si no es debidamente
tratado o si la causa no es curable. De igual modo, puede presentar alteración de
los nervios craneales por compromiso directo de la lesión o secundaria a la
herniación. En estos casos no es infrecuente la aparición de convulsiones y de las
manifestaciones vegetativas.
En el examen del fondo de ojo no es esperable que haya edema de papila, pues el
aumento brusco de la presión intracraneal causará dilatación de las venas,
desaparición del latido venoso e incluso hemorragias perivenosas, pero para que
aparezca edema se precisa de un aumento sostenido de la presión intracraneal a
lo largo de unas dos o tres semanas.
Síndrome de hipertensión intracraneana del lactante. En los recién nacidos y
en los lactantes si la presión intracraneal aumenta por un proceso expansivo, o por
hidrocefalia, la fontanela se abomba y se pone tensa, el latido se hace menos
evidente y las oscilaciones respiratorias desaparecen aun poniendo vertical al
infante. Además las suturas se separan progresivamente de modo que el
perímetro del cráneo aumenta anormalmente y el niño presenta macrocefalia.
Normalmente la fontanela se cierra a los 18 meses de vida pero en estos casos
puede permanecer abierta mayor tiempo. Este crecimiento anormal de la cabeza
hace que el aumento de la presión intracraneal nunca sea muy grande y por eso
no se produce edema de papila en los niños pequeños. Pero la circulación venosa
intracraneal es obstaculizada y por eso es muy notoria la circulación colateral
extracraneal; las venas del cuello cabelludo se dilatan y se hacen muy evidentes.
Si la causa de esta situación no es corregida se producirá un retardo cada vez
más grave del desarrollo psicomotor del pequeño.

35
La craneosinostosis es una condición en la que las suturas se consolidan en una
forma anormalmente precoz, y como consecuencia el cráneo crece en forma
defectuosa y adopta formas diversas y características según la o las suturas
comprometidas. Hay varias de estas alteraciones en las que la cavidad craneal no
se expande acorde con el desarrollo del cerebro y este va quedando aprisionado,
comprimido por un cráneo rígido y se produce hipertensión intracraneal por
craneosinestosis. En este caso en vez de macrocefalia, hay microcefalia, la
fontanela se cierra precozmente y el niño presenta edema en las papilas.
Es importante destacar que si se presenta SHI en niños preescolares y escolares
con un proceso expansivo de evolución progresivo lento, el cráneo a esta edad
aun es bastante plástico y también puede presentar macrocefalia, por lo cual
siempre debe tenerse presente como un signo sospechoso de hipertensión
intracraneal, examinar bien el fondo de ojo que frecuentemente estará claramente
alterado si hay un tumor cerebral. (32)
Continuando con el diagnostico de la Hipertensión intracraneal, la asociación
Española de pediatría (31) también describe:
3.6.7.2 Diagnostico Radiológico. La radiografía de cráneo. En situaciones de
hipertensión intracraneal crónica, se pueden ver las impresiones digitiformes de
las circunvoluciones cerebrales sobre la tabla interna. En los niños es fácil apreciar
la separación o diástasis de suturas aún no cerradas.
La tomografía axial computarizada (TAC) y la resonancia nuclear magnética
(RNM). Visualizan los procesos expansivos ocasionantes de la hipertensión
intracraneal, así como la existencia de desviaciones de línea media y los
fenómenos de enclavamiento del parénquima cerebral a nivel del tentorio o de las
amígdalas cerebelosas en el agujero magno.
3.6.7.3 Monitorización presión intracraneal (PIC). Permite el diagnóstico de
hipertensión intracraneal. Técnica invasiva, con riegos de sangrado e infección; se
debe realizar en una Unidad de Cuidados Intensivos. Permite la evaluación de las
medidas terapéuticas y el cálculo de la presión de perfusión cerebral.
3.6.7.4 Saturación yugular de oxígeno (SjO2). Catéter en el bulbo de la yugular
para obtener medidas de la saturación venosa. Los valores normales oscilan entre
el 55% y el 75%. En situaciones de isquemia aumenta la extracción de oxígeno
por las células y la SjO2 será menor de 55% y, en los casos de hiperemia,

36
aumentará. La utilidad de esta técnica radica en que nos informa si debemos
aplicar medidas que disminuyan el flujo sanguíneo cerebral en casos de hiperemia
(hiperventilación, barbitúricos), o bien, medidas que aumenten el flujo sanguíneo
cerebral y la presión de perfusión cerebral (expansión con salino hipertónico,
inotrópicos).
3.6.7.5 Electroencefalograma (EEG). En el paciente crítico se puede realizar
monitorización continua, pudiendo detectar de forma precoz crisis o bien simetrías
que correspondan a lesiones focales. Es aconsejable su utilización para la
valoración del paciente en tratamiento con barbitúricos.
3.6.7.6 Eco-doppler transcraneal. Posibilita la medición del flujo sanguíneo
cerebral, permite detectar alteraciones secundarias a hipertensión intracraneal
(disminución de la velocidad media y sobre todo de la diastólica). Es posible
identificar hiperemia cerebral, permitiendo así valorar el uso de hiperventilación
agresiva, siempre en combinación con la medición de la SjO2.
3.6.7.7 Presión tisular de oxígeno (PtiO2). A través de un electrodo se mide la
presión del oxigeno disuelto en el intersticio cerebral. Dado que el oxígeno difunde
libremente, una disminución de la PtiO2 indicaría aumento de la extracción por las
células o disminución del flujo sanguíneo cerebral (de forma análoga a la SjO2).
Los valores normales son 25-30 mmHg, correspondiendo valores <15 a hipoxia
cerebral.
3.6.7.8 Tratamiento de la hipertensión endocraneal. Para el tratamiento la
asociación Española de pediatría (31) tiene como objetivos:
PPC de 40-60 mmHg (dependiendo de la edad) en niños con traumatismo craneal e hipertensión intracraneal.
PIC < 20 mmHg.
Saturación venosa yugular entre 55-75%.
Eco-doppler sin hiperalfujo o isquemia.
3.6.7.8.1 Estabilización inicial. Asegurar la estabilidad respiratoria y
hemodinámica del paciente. Es útil seguir el “ABC” de las normas de reanimación
cardiopulmonar.
A. Vía aérea: Asegurar la permeabilidad de la vía aérea. Aspirar
secreciones y sangre o cuerpos extraños de la boca si los hubiera. Las indicaciones de intubación endotraqueal son:

37
Incapacidad de mantener la vía aérea permeable (vómitos, traumatismo facial).
Glasgow > 9. Hipoxemia pese a aporte de oxígeno al 100%. Inestabilidad hemodinámica.
B. Ventilación: El objetivo es normoventilar y evitar la hipoxemia. La
saturación de oxígeno debe mantenerse por encima de 95%. La PaCO2 debe mantenerse alrededor de 35-40 mmHg. El paciente en ventilación mecánica se debe programar una frecuencia y volumen corriente adecuados a su edad y peso. La presión positiva al final de la espiración (PEEP) debe ser de 3-5 cm. de agua, suficiente para asegurar la oxigenación sin dificultar el retorno venoso al aumentar la presión intratorácica.
C. Circulatorio: Debe evitarse la hipotensión. En caso de hipotensión se realiza una expansión con 10-20 ml/kg en 20 minutos de Suero Salino Fisiológico (SSF). La canalización de una vía venosa central que permite medida la presión venosa central facilita la pronta detección de cuadros de hipovolemia, y permiten evaluar la respuesta las expansiones.
Varios estudios indican que el uso de suero salino hipertónico (3 al 6%) es más
eficaz que el salino en la estabilización del paciente además de disminuir la PIC.
Los pacientes tratados con salino hipertónico presentan menor requerimiento de
volumen, mejor respuesta hemodinámica, disminuyendo el número de
complicaciones y mejorando el pronóstico. Si la respuesta no es adecuada o
precisa expansión de volemia en más de tres ocasiones, se inicia soporte
inotrópico en perfusión continua. La dopamina es la primera opción, a dosis de 5 a
20 mcg/kg/min. Algunos autores prefieren el uso de noradrenalina dado que
dopamina y adrenalina aumentan el consumo cerebral de oxígeno.
3.6.7.8.2 Optimización del flujo sanguíneo cerebral. Mantener la cabeza
centrada en línea media y elevada 30º facilita el flujo venoso y el drenaje del
líquido cefalorraquídeo. Limitar la presión positiva al final de la espiración (PEEP)
a 5 cm de agua suele ser recomendado para que no limite el retorno venoso, aun
cuando estudio experimentales en animales han demostrado que la obstrucción
del flujo se produce con valores mucho más latos, de alrededor de 15 cm. de
agua.
3.6.7.8.3 Sedoanalgesia. La agitación y el dolor incrementan dos o tres veces el
gasto metabólico cerebral, lo que puede aumentar el flujo sanguíneo cerebral y la

38
presión intracraneal. Una adecuada sedoanalgesia puede ayudar al control de la
hipertensión intracraneal, al disminuir la demanda de oxígeno. El sedante más
utilizado es el midazolam en perfusión intravenosa a dosis inicial de 0,1- 0,2
mg/kg/h. Como analgésico se utiliza un opioide de vida corta, generalmente
fentanilo en perfusión a 1-3 mcg/kg/h; el cloruro mórfico o el remifentanilo son
otras opciones. Está contraindicado el uso de ketamina por que incrementa el
consumo cerebral de oxígeno y puede aumentar la presión intracraneal. El
propofol no se recomienda por que puede producir acidosis metabólica en niños.
Los relajantes musculares son utilizados en el tratamiento de la hipertensión
intracraneal en base a estudios que demuestran una reducción de la presión
intracraneal por varios mecanismos entre los que se encuentran la reducción en la
presión intratorácica, la mejora del flujo venoso y el mejor acoplamiento a la
ventilación mecánica. El agente más utilizado es el vecuronio (0,1mg/kg/h).
3.6.7.8.4 Hematológico. Se debe corregir la anemia para asegurar un correcto
aporte de oxígeno al tejido cerebral.
3.6.7.8.5 Profilaxis anticonvulsiva. Se recomienda la profilaxis con fenitoína en
los TCE graves durante la primera semana. Dosis de choque 20 mg/kg, dosis de
mantenimiento 5 mg/kg/día.
En el caso de aparecer crisis convulsivas deben tratarse agresivamente con
diacepam, midazolam, o fenitoína.
3.6.7.8.6 Evitar la fiebre. Dado que produce aumento de la lesión secundaria,
aumento de la las demandas metabólicas, y además favorece la aparición de
convulsiones.
3.6.7.8.7 Profilaxis antibiótica. No se recomienda de forma rutinaria. En casos de
traumatismos con fractura de la base de cráneo se administrará amoxicilina-
clavulanico a dosis de 100 mg/kg/día.
3.6.7.8.8 Hidroelectrolítico. Debe evitarse la hipoglucemia y la hiperglucemia, el
aporte hídrico debe realizarse con SSF, aportando las necesidades basales, y la
glucosa necesaria para mantener la glucemia entre 100 y 120 mg/dl. Sumamente
importante es el control de natremia y osmolaridad. Tanto la diabetes insípida
central como la secreción inadecuada de ADH son complicaciones que pueden
empeorar el pronóstico.

39
3.6.7.8.9 Esteroides. Indicados en la hipertensión intracraneana secundaria al
edema vasogénico que acompaña a los tumores. El régimen más empleado es la
dexametasona 0,15 mg/kg/dosis cada 6 horas. No existe evidencia de que
mejoren la morbimortalidad en el resto de casos de hipertensión intracraneana.
3.6.8 Nauseas y emesis. Manuel González refiere “Las nauseas y el vomito en
los tumores cerebrales primarios tienen una prevalencia del 30% en los pacientes
en el momento del diagnostico, se presentan en forma brusca (proyectil)
relacionados con la posición corporal” (33). Asociada a cefaleas matutina, más
frecuentes en la fosa posterior secundario a compresión de los centros eméticos.
En neoplasias infantiles puede aparecer como único y primer síntoma.
3.6.9 Vértigo- Según Suarez (34) el vértigo relacionado con tumores cerebrales
primarios a nivel infratentorial ocasionan vértigo por afectación del cerebelo o del
tronco encefálico, mientras los supratentorial en pocas ocasiones se manifiestan
con vértigo, los pacientes los van a describir más como mareo e inestabilidad
posicional.
3.6.10 Somnolencia. Como lo describe González (33) y Díaz (35) síntoma
referido por el paciente o sus familiares como perdida de la energía, tendencia al
sueño. Se asocia a tumores de localización frontal, del cuerpo calloso y
temporal. Se produce alteración de las fibras de asociación de sustancia blanca y
el aumento de la hipertensión endocreneana, evoluciona a estupor y coma.
3.6.11 Dificultad para la marcha. Las alteraciones de la marcha son dadas
principalmente por ataxia o hemiplejia como Reyes Olivero lo describió:
Ataxia se encuentra en lesiones cerebelosas que se encuentran en el vermix si
son tronculares y en los hemisferios cerebelosos si sin lateralizadas. En ocasiones
los tumores de ubicación a nivel frontal pueden producir una inestabilidad de la
marcha que puede disimular una ataxia típica de fosa posterior. Si el tumor está
localizado en áreas motoras de los hemisferios cerebrales se manifiesta como
hemiparesia y hemiplejia contralateral espástica. (36)
3.6.12 Irritabilidad. Como lo refiere Aguirre este síntoma “Se presenta en
pacientes con grandes tumores a nivel frontal y temporal secundario a
hipertensión endocraneana. Se asocia a cambios de personalidad. Uno de los
primeros síntomas en lactantes” (37).

40
3.6.13 Alteraciones del comportamiento y psicomotor. Aguirre (37) y la
asociación Española de pediatría (30) manifiesta que los tumores que invaden de
manera directa el diencéfalo en etapas tempranas se presentan con alteraciones
en el comportamiento principalmente en cambios de personalidad, emocional,
hábitos, depresiones, agresividad y rendimiento escolar que en muchas ocasiones
se convierte en el motivo de consulta expresado por los familiares
3.6.14 Hidrocefalia y aumento de perimetro cefálico. La hidrocefalia se
caracteriza por el acumulo de LCR en la cavidad craneal, específicamente en los
ventrículos cerebrales. “Este acúmulo de líquido aumenta la presión en el interior
de la cavidad intracraneal y comprime el cerebro lesionándolo, a veces de forma
irreversible” (38). Como dice Álvarez (39) la hidrocefalia está asociada a una
obstrucción en la circulación del líquido cefalorraquídeo, que puede ser de dos
tipos:
Congénita: Es debida a malformaciones cerebrales durante la gestación,
provocadas por causas ambientales durante el desarrollo del feto o por predisposición genética, que impiden la circulación del líquido cefalorraquídeo.
Adquirida: Debida a lesiones o enfermedades cerebrales que se dan en el
momento del nacimiento o después, las cuales impiden la circulación o la reabsorción del líquido cefalorraquídeo. Ejemplo: tumores cerebrales, hemorragias intracraneales, o infecciones como meningitis.
A propósito de las Hidrocefalias tumorales, Álvarez dice: Los tumores
intraventriculares son los más frecuentes en los niños, y los supratentoriales son
los que más se asocian a hidrocefalia. Los de la fosa posterior, son
especialmente frecuentes y es en estos donde más frecuentemente se presenta la
hidrocefalia de origen tumoral. También se describe una hidrocefalia que a veces
se presenta después de algunas intervenciones sobre tumores cerebrales y deben
considerarse como inflamatorias pos hemorrágicas.
El diagnostico se hace por medio imágenes diagnosticas (Resonancia magnética
nuclear, tomografía axial computarizada) pero hay que tener en cuenta signos
como el aumento progresivo del perímetro cefálico en los niños cuyas suturas aun
no están consolidadas, y el abombamiento de las fontanelas. El tratamiento a
este tipo de enfermedad, va dirigido a la causa principal que provoca el acumulo
del liquido cefalorraquídeo. (39)

41
3.6.15 Letargia. Síntoma característico de enfermedades nerviosas, infecciosas o
tóxicas, caracterizado por un estado de somnolencia profunda y prolongada (41).
En pacientes con tumores cerebrales suele deberse a la presencia de aumento de
la presión intracraneal, consecuente con la hidrocefalia.
3.6.16 Papiledema. Como dice Rosaralis S (42) El papiledema hace referencia al
edema pasivo del disco, asociado con aumento de la presión intracraneal. La
mayoría de los casos es diagnóstico de un tumor cerebral, lo que frecuentemente
va acompañado de náuseas, cefalea y diplopía horizontal directa por lesión de los
VI nervios craneales. Los tumores infratentoriales son los que comúnmente
dependen de la localización, tipo, velocidad de crecimiento, estado de la pupila,
entre otros. “Para que se produzca papiledema debe existir un aumento de la
presión del líquido cefalorraquídeo por encima de 200 mm de agua (H2O), cuyo
valor normal fluctúa entre 100 y 180 mm H2O. En niños muy pequeños con
hipertensión endocraneana por grandes hidrocefalias congénitas, no se produce
papiledema, debido a la extensibilidad ósea del cráneo” (42).
3.6.17 Ataxia. Como dice Mateos Beato Fernando (43) La ataxia es un signo
neurológico que indica una alteración de la coordinación motora voluntaria y del
control postural. Describe varios tipos de ataxia:
3.6.17.1 Ataxia Cerebelosa. Suele acompañarse de otros signos de disfunción
del cerebelo. Cuando la lesión se localiza en línea media se afecta la estática y la
marcha; cuando es hemisférica se presenta el temblor, dismetría e hipotonía, del
mismo lado de la lesión.
3.6.17.2 Ataxia sensitiva. Se refiere a la pérdida del estímulo propio de los
miembros inferiores, la cual genera en el sujeto la pérdida del conocimiento de su
posición en el espacio, de sus movimientos, la contracción muscular y de los
detalles del terreno por donde se desplaza.
3.6.17.3 Ataxia aguda de origen tumoral. Aunque los tumores de fosa posterior
suelen cursar con una ataxia lentamente progresiva, su comienzo agudo es
relativamente frecuente, “debido a un brusco aumento de tamaño por hemorragia
intratumoral o a hidrocefalia. La ataxia se asocia a cefaleas, vómitos y edema de
papila, nistagmos y afectación de pares craneales. Un TAC debe ser siempre la
primera prueba diagnóstica” (43).

42
3.6.18 Lipotimia. Se define como una pérdida de conciencia incompleta, brusca,
breve y transitoria, asociada a una pérdida del tono postural. Etiología: cardíaca,
disfunción circulatoria (sistémica y cerebral), neurológica, metabólica, psicógena y
tóxica. El factor desencadenante final es un descenso crítico en la perfusión
cerebral, o bien una alteración en la composición química de la propia sangre. El
tipo más común es el síncope vasovagal o neurocardiogénico (44).
El diagnóstico se fundamenta en una anamnesis detallada y en una exploración
física completa, la prueba de la mesa basculante, que permite confirmar el
diagnóstico de la variante más frecuente (síncope vasovagal). El tratamiento va de
acuerdo a la etiológica. Cuando el síncope vasovagal tiene una presentación
fugaz, recurrente o con componente convulsivo, debe considerarse el tratamiento
farmacológico. (44)
3.6.19 Disminución fuerza muscular. Los pacientes que padecen afecciones
cerebrales, presentan consecuentemente disminución en la fuerza muscular hasta
llegar a la atrofia muscular debido a la hipertensión endocraneana que genera
compresión de masa encefálica y cordones nerviosos que alteran el correcto
funcionamiento del cuerpo; suele presentarse de manera progresiva y va de
acuerdo al grupo muscular afectado.
3.6.20 Microcefalia. Como dice García Peñas J y Romero Andújar F (45) La
presencia de un perímetro craneal menor de dos desviaciones estándar por
debajo de la media, lo que suele indicar la presencia de microencefalia.
Un 90% de las microcefalias se asocian a retraso mental, salvo en los casos de
microcefalias de origen familiar que pueden tener una inteligencia normal. En el
origen de las microcefalias se implican factores diversos, incluyendo anomalías del
desarrollo cerebral y daño cerebral adquirido de origen diverso (prenatal, perinatal
o postnatal), los cuales se asocian a diversos grados de atrofia cerebral en la
tomografía computarizada (TAC) craneal y en las imágenes de resonancia
magnética (IRM) cerebral. Dentro de la etiopatogenia: La microcefalia primaria se
refiere a un cerebro es pequeño y que no completó su normal desarrollo
embrionario por factores genéticos, cromosómicos y malformativos o por factores
ambientales. La microcefalia secundaria el cerebro completó un desarrollo
embrionario normal, pero luego sufre un daño difuso y se alteró su crecimiento
evolutivo.
3.6.21 Convulsiones. Definida como una alteración repentina en la actividad
eléctrica cortical, manifestada como una alteración en el estado de conciencia ó la

43
aparición de sintomatología motora, sensitiva o conductual. En sus estadios más
avanzados se presenta como un trastorno intermitente del sistema nervioso,
causado por una alteración en la conducción del impulso nervioso caracterizada
por sus recurrencias durante meses o años denominado Epilepsia. (46)
“Las convulsiones se pueden presentar con sintomatología inicial o durante la evolución del tumor cerebral, el paciente puede presentar una exploración neurológica inicial normal, refiriendo solo cambios comportamentales aislados. Es una alteración súbita en la actividad eléctrica cortical, una descarga neuronal hipersincrónica, que se manifiesta clínicamente por alteración de la conciencia ó por aparición de sintomatología motora, sensitiva o conductual” (47).
Clasificación de epilepsias y síndromes convulsivos Según su forma de presentación, las crisis pueden ser:
A. Generalizadas: inician en ambos hemisferios. Pueden ser convulsivas ó no.
B. Parciales: se puede discernir un inicio focal. Pueden ser simples (sin pérdida de conocimiento) ó complejas (con pérdida de conocimiento). Secundariamente generalizadas: son crisis parciales que evolucionan a generalizadas, más frecuentemente tónico-clónicas.
Causas de sindromes convulsivos
“En el paciente HIV positivo la patología infecciosa debe ser tenida en cuenta como una de las más frecuentes, así como las neoplasias intracraneales. En los pacientes HIV positivos deben tenerse en cuenta las siguientes patologías: toxoplasmosis cerebral, chagoma cerebral, criptococosis meníngea, abscesos cerebrales, tuberculoma, infoma primario, otros” (47)
Clínica Convulsión tónico-clónico generalizada – Gran mal
El paciente puede sentir que se aproxima una crisis al percibir uno de
diversos fenómenos subjetivos. Durante algunas horas se puede sentir
apático, deprimido, irritable o en éxtasis. La aparición de una ó más
sacudidas mioclónicas del tronco ó las extremidades al despertar puede
sugerir la ocurrencia de una crisis convulsiva más tarde. Otros pródromos
pueden ser dolores abdominales, palidez ó enrojecimiento de la cara y
cefalea pulsátil, entre otros signos Generalmente dura unos cuantos
segundos, puede constituir toda la crisis o progresar a pérdida del
conocimiento y crisis convulsiva generalizada. (47)
Fase clónica: Comienza con un temblor generalizado leve, que
corresponde en realidad a la relajación repetitiva de la contracción tónica.

44
Se inicia con una frecuencia de 8 por segundo, cambia a 4 por segundo y rápidamente cede el camino a espasmos flexores violentos breves que se producen a descargas rítmicas y agitan a todo el cuerpo. La cara toma un color violáceo y contorsionado por una serie de gesticulaciones.
Ausencia – pequeño mal
Son notables por su brevedad y escasez de actividad motora. Para el
testigo parece un momento de abstracción de la mente. El ataque
sobreviene sin advertencia, consiste en la interrupción de la conciencia que
recibe el nombre de ausencia. El paciente clava la mirada fija y deja de
hablar o de responder. Sólo el 10 % de los pacientes queda totalmente
inmóvil durante el ataque; en los restantes se observa una descarga breve
de movimientos clónicos finos de los párpados, los músculos faciales o de
los dedos de las manos o movimientos asincrónicos de ambos brazos a un
ritmo de 3 por segundo. Como regla éstos pacientes no caen al piso,
pueden incluso continuar con movimientos tan complejos como caminar o
andar en bicicleta. Después de 2 a 10 segundos restablece el contacto
completo y readopta su actividad previa a la crisis.(47)
Crisis Jacksoniana o motora focal
Las convulsiones motoras focales se atribuyen a lesión del lóbulo frontal opuesto. Su manifestación más frecuente es el giro forzado de la cabeza y de los ojos hacia el lado opuesto al foco irritativo, que se acompaña a menudo de contracción tónica del tronco y extremidades de ese lado. La convulsión motora jacksoniana se inicia con una contracción tónica de los dedos de una mano o un lado de la cara o pié; se transforman en movimientos clónicos de ésas partes de manera análoga a lo que ocurre en las crisis T-C generalizadas
Tratamiento de los estados convulsivos
Drogas anticonvulsivantes – mecanismo de acción Fármaco Mecanismo de
acción Acido valproico Prolonga el período inactivo de los canales de sodio
Inhibición del receptor GABA Bloquea los canales T de calcio
Benzodiacepinas Prolonga el período inactivo de los canales de sodio
Aumenta el tono GABAérgico (unión reversible) Fenobarbital Idem que BZD
pero con unión irreversible a los receptores GABA Carbamacepina Prolonga
el período inactivo de los canales de sodio Fenitoína Prolonga el período
inactivo de los canales de sodio Vigabatrina Inhibición de GABA t específica
e irreversible Etosuximida Bloquea los canales T de calcio Lamotrigina
Disminuye la liberación de neurotransmisores excitatorios Felbamato /

45
Gabapentina Mecanismo de acción aún no conocido Tanto la
carbamacepina como la fenitoína son estabilizantes de membrana. Elección
de la droga según el tipo de crisis Tipo de crisis Primera elección Segunda
elección Crisis parciales CarbamacepinaFenitoina Acido valproico
Fenobarbital Primidona Benzodiacepinas Crisis tónico-clónicas
generalizadas Fenobarbital Fenitoína carbamacepina Acido valproico
Primidona Ausencias Acido valproico etosuxinida Benzodiacepinas Crisis
mioclónicas Acido valproico benzodiacepinas Etosuxinida * las
benzodiacepinas incluyen: clobazam, nitracepam, clonazepam.(47)
Criterios de internación de un paciente con convulsiones
1. Status epiléptico 2. Crisis reiteradas 3. Existencia de foco neurológico tras crisis generalizada 4. Crisis secundaria a otros procesos (infecciones, alteraciones
hidroelectrolíticas, lesiones ocupantes de espacio)
Estado Epiléptico
Convulsiones continuas por lo menos durante 5 minutos o dos o más crisis entre las cuales exista una recuperación incompleta de la conciencia.
Causas: abandono de la medicación, toxicidad por drogas, infecciones del
SNC tumor cerebral, traumatismo encéfalocraneano (TEC) epilepsia refractaria stroke trastornos metabólicos encefalopatía anóxica encefalopatía metabólica supresión o intoxicacion etílica otras
Manifestaciones neurológicas del mal epiléptico Inicio del cuadro: usualmente comienza con convulsiones tónico-clónicas, tónicas ó clónicas clínicamente obvias que afectan las extremidades. Las manifestaciones pueden ser sutiles, algunos presentan sólo movimientos de pequeña amplitud en el rostro, manos, pies o nistagmus. Clínicamente inaparente: ciertos pacientes no presentan actividad motora evidente y la detección de las convulsiones ininterrumpidas requiere del registro electroencefalográfico Estado epiléptico mioclónico: consiste en breves movimientos restringidos a una ó más partes del cuerpo, puede no haber pérdida de conocimiento. Se ve después de anoxia prolongada.

46
3.6.22 Otros. Puede dar también signos de tractos largos como el piramidal
(debilidad, hipertonía, hiperreflexia y reflejos anormales), sensitivos (superficial o
profunda), extrapiramidal (corea, atetosis, distonía).
En general la población pediátrica presenta:
Cambios en la personalidad y el comportamiento.
Alteración en la concentración.
Aumento del sueño.
Pérdida de memoria.
Problemas con el razonamiento. Los bebés pueden tener los siguientes signos físicos:
Fontanelas abultadas
Suturas separadas
3.7 DIAGNÓSTICO
Al momento de realizar el diagnostico debemos tener en cuenta varios aspectos
como:
3.7.1 Historia clínica. Donde es preciso valorar antecedentes de importancia que
predispongan a la aparición del tumor, además valorar los síntomas que presente
el paciente, considerado en el apartado 3.6 previo.
3.7.2 Examen físico. Evaluación neurológica de pares craneanos, alteración de
conciencia, déficit sensibilidad, motilidad, lenguaje, signos vitales entre ellos la
tensión arterial, fundoscopia y reflejos.
3.7.3 Paraclínicos. Radiografía de cráneo anteroposterior y lateral
(Abombamiento de fontanelas, calcificación), Tomografía axial computarizada,
Resonancia magnética (Sitio - localización del tumor), gammagrafías cerebrales,
angiografías (vascular).
3.8 TRATAMIENTO
A propósito del tratamiento de los tumores cerebrales, el Dr. García Jose y el Dr. Benavides Manual proponen: El paciente con un tumor cerebral debe ser sometido

47
a una terapia específica, principalmente con cirugía con o sin radioterapia y/o quimioterapia, y a un tratamiento para el alivio de los síntomas. El tratamiento de soporte o de alivio de síntomas se basará principalmente en el uso de esteroides, sobre todo, dexametasona, y de fármacos para evitar las crisis convulsivas o antiepilépticos. Los esteroides pretenden reducir el edema circundante y como consecuencia de ello, los efectos compresivos sobre el tejido sano. Si el paciente ha presentado alguna crisis convulsiva, los antiepilépticos reducirán el riesgo de aparición de nuevas crisis. Las dosis de todos estos fármacos dependerán de los síntomas que presente el paciente. (48) Las herramientas terapéuticas en los tumores cerebrales son:
Cirugía
Radioterapia
Quimioterapia 3.8.1 Cirugía. Es la principal forma de tratamiento en la mayoría de los tumores cerebrales e incluso puede ser suficiente para controlar la enfermedad en aquellos pacientes con tumores benignos. La cirugía se lleva a cabo para alcanzar varios objetivos:
Obtener tejido tumoral para realizar un diagnóstico histológico preciso.
Realizar una resección intentando conservar la máxima función a la vez que se extirpa la mayor cantidad posible de tejido tumoral. Con este planteamiento se consigue descomprimir los tejidos sanos por lo que el paciente mejora en su funcionalidad y se le proporciona un mejor pronóstico y calidad de vida.
En diferentes estudios se ha podido demostrar una relación entre la resección realizada y el pronóstico de los pacientes con varios tipos de tumores cerebrales, incluyendo las metástasis cerebrales y los gliomas de bajo y alto grado. Pero, en ocasiones, el tumor está cercano a zonas que rigen funciones críticas para el paciente y la resección no es posible. En estos casos la realización de una biopsia puede permitir el diagnóstico del tipo de tumor y de este modo planificar el tratamiento más adecuado. (48)
La cirugía es en todos los casos de gran complejidad para preservar la mayor funcionalidad al paciente. Con este objetivo el neurocirujano se ayudará de técnicas radiológicas como la tomografía axial computarizada y la resonancia magnética craneal para localizar con la mayor exactitud el tumor. Hoy en día otras técnicas permiten precisar al máximo la localización de las zonas críticas para el paciente y sus relaciones con el tumor: La estereoataxia (o sistema de localización del tumor mediante un sistema geométrico para localizar el punto más adecuado para la biopsia), los sistemas de neuronavegación (instrumentos informáticos que fusionan las imágenes radiológicas durante la intervención para una mejor localización del tumor), el mapeo funcional cortical intraoperatorio (que

48
permite localizar áreas elocuentes y evitar su daño durante la intervención), la microcirugía, la ecografía intraoperatoria, etc. (48) Una resonancia tras la intervención permitirá identificar la existencia o no de este tejido tumoral, que tendrá que ser sometido a seguimiento y/o tratado con radioterapia y/o quimioterapia. La cirugía lleva asociada un aumento del riesgo de complicaciones como empeoramiento de los déficits neurológicos, convulsiones, infecciones, hemorragias, pérdida de líquido cefalorraquídeo y otras de carácter más general como pueden ser episodios trombóticos, infarto, hemorragia digestiva, etc. Aunque la mortalidad y el riesgo de complicaciones han mejorado en los últimos años con los avances técnicos, se evaluará en cada paciente el balance riesgo/beneficio para plantear la mejor de las opciones de tratamiento. (48) 3.8.2 Radioterapia. Es una forma de tratamiento que consiste en el envío de partículas ionizadas de alta energía contra las células del tumor que tienen como objetivo producir daños en su material genético; este daño facilitará su muerte. La radioterapia puede dañar al tejido neurovascular que rodea al tumor; para minimizar este efecto se limita la dosis máxima que recibirán y se realiza un fraccionamiento diario de la dosis total a recibir durante varios días o semanas. (48) 3.8.2.1 Indicaciones de la Radioterapia. La radioterapia tiene un papel fundamental en aquellos pacientes con metástasis cerebrales, con gliomas de alto grado y con tumores irresecables. En aquellos con tumores de bajo grado y con pronóstico muy favorable se evaluará el balance entre el riesgo de toxicidad tardía y el beneficio esperable con el tratamiento. La radioterapia puede aplicarse mediante distintas técnicas; cada una de ellas puede tener diferentes indicaciones, según la extensión de la irradiación, la dosis que se necesita alcanzar para el control de la enfermedad y la toxicidad esperable. (48) 3.8.2.2 Radioterapia externa convencional. Utiliza distintos haces de irradiación externa para irradiar el volumen tumoral y minimizar la cantidad de tejido sano circundante irradiado, y con ello, la toxicidad. El tratamiento es administrado, según la dosis a alcanzar, durante varios días o semanas, recibiendo dosis diarias iguales. Durante el tratamiento el paciente es inmovilizado con una máscara para asegurar los puntos de irradiación durante todo el tratamiento. (48) 3.8.2.3 Radioterapia externa estereoatáxica. Es una irradiación externa en la
que el volumen de irradiación es pequeño, sobre él se alcanzan dosis más altas en una sola sesión y se determina mediante un sistema de ejes de coordenadas colocado en el paciente, que permite localizar con precisión el punto de

49
irradiación. Se precisa la utilización de una maquinaria específica como puede ser el acelerador lineal, o un gammaknife (una unidad de cobalto modificada para un tratamiento multiplanar) o un ciberknife (con sistema robótico que permite delimitar mejor el área de irradiación). Puede ser útil en pacientes con metástasis cerebrales y en algunos casos, de meningiomas, gliomas y neurinomas, aunque de forma más controvertida. (48) 3.8.2.4 Radioterapia intersticial o braquiterapia. Consiste en la implantación dentro del tejido tumoral de catéteres que son fuentes de irradiación (por ejemplo agujas de iridio 192, etc.) que alcanzan dosis terapéuticas alrededor de ellos y se evita la irradiación del tejido sano a distancia. Estas técnicas son altamente especializadas y en este momento está por definir el beneficio de su uso en el tratamiento de los tumores cerebrales. (48) 3.8.2.5 Terapia con haces de partículas. Se utilizan principalmente unas
partículas llamadas protones o neutrones en centros muy especializados, puesto que es preciso un ciclotrón para generarlas. La principal ventaja de estas técnicas es la posibilidad de circunscribir de forma más precisa el área de irradiación. Aunque se han utilizado para algunos tipos especiales de tumores, están por definir sus indicaciones y sus ventajas frente a las formas de tratamiento convencional. (48) 3.8.2.6 Radioterapia externa hiperfraccionada. Es una forma de radioterapia
externa en la que con un mayor número de fracciones y dosis por día, se quiere aumentar la capacidad de destrucción del tejido tumoral. Puede ser estereoatáxica, si se quiere irradiar un pequeño volumen. (48) 3.8.2.7 Radioterapia de intensidad modulada. Es un sistema por el cual se consigue dar dosis más altas en unas áreas del tumor y dosis más bajas en otras áreas; el objetivo es alcanzar las dosis adecuadas en todos los tejidos tumorales, con la menor toxicidad de los tejidos circundantes. Está por definir sus indicaciones y sus ventajas en casos seleccionados frente a la radioterapia externa convencional. (48) Es importante recalcar que la radioterapia puede tener efectos secundarios que serán diferentes dependiendo del momento en el que se produzcan:
Agudos: Aparecen horas o días tras el inicio del tratamiento y son transitorios; consisten generalmente en cefalea o empeoramiento de los déficits neurológicos. Estos efectos son producidos por aumento del edema asociado al tumor y pueden ser tratados con esteroides. (48)
Toxicidades diferidas tempranas: Pueden aparecer desde seis semanas
hasta seis meses después de finalizar la radioterapia. Son producidas por

50
un daño neurológico reversible y causan empeoramiento de los déficits neurológicos que se trata también con esteroides. Clínicamente es indistinguible de los cambios que se producen en el paciente cuando se produce un empeoramiento temprano del tumor, pero una respuesta favorable a los esteroides hará pensar en que el cuadro fue producido por la toxicidad de la radioterapia. (48)
Toxicidades tardías: Pueden aparecer años después de finalizar la
radioterapia y se producen por destrucción del tejido cerebral (radionecrosis); los síntomas no siempre mejoran con los esteroides y en ocasiones el tratamiento es quirúrgico. Otro efecto indeseable tardío es el daño cognitivo que puede oscilar entre un ligero daño neurológico y una auténtica demencia. Un efecto secundario tardío muy poco frecuente es la aparición de tumores radioinducidos. Dependiendo de la localización del área a irradiar se puede producir pérdida de agudeza visual y déficits hormonales. (48)
3.8.3 Quimioterapia. Consiste en la administración de medicamentos que tienen como objetivo la destrucción de la célula tumoral por diferentes vías (oral, intravenosa o locorregional). El tratamiento quimioterápico de los tumores cerebrales presenta dos importantes dificultades:
La existencia de la barrera biológica hematoencefálica, que protege al sistema nervioso central de la llegada de sustancias tóxicas por la sangre.
La resistencia biológica de estos tumores al tratamiento, que impide que mejoren con la mayoría de los fármacos quimioterápicos.
En los tumores cerebrales primarios de la infancia el tratamiento quimioterápico es fundamental, por la mayor sensibilidad que muestran frente a los adultos y para evitar los efectos tóxicos de la radioterapia. Los gliomas son los tumores primarios más frecuentes. En aquellos tumores de alto grado (astrocitoma anaplásico y glioblastoma) la quimioterapia basada en nitrosoureas (carmustina, lomustina, etc.), que han demostrado un beneficio limitado y controvertido tanto en supervivencia como en calidad de vida. Otros gliomas de alto grado, como lo son los oligodendrogliomas anaplásicos han demostrado ser tumores más quimiosensibles que los astrocitomas. De este modo se han demostrado tasas altas de respuesta con esquemas con lomustina, procarbacina y vincristina (PCV) en oligodendrogliomas que reaparecen tras los tratamientos iniciales. Su uso tras la cirugía y la radioterapia puede aumentar la supervivencia libre de enfermedad del paciente (periodo de tiempo en el que el

51
paciente no progresa), aunque no la supervivencia global. Por ello, teniendo en cuenta los factores pronósticos del paciente, se deberá evaluar su indicación. Los tumores embrionarios, como el meduloblastoma, son poco frecuentes en los adultos y por ello la información científica es limitada. En estos tumores se ha demostrado actividad de la quimioterapia tras otras líneas de tratamiento. Su uso como tratamiento complementario a la cirugía y a la radioterapia se podrá considerar en pacientes de alto riesgo de recaída de la enfermedad. La quimioterapia es el tratamiento de elección en la mayoría de los pacientes con linfomas primarios del sistema nervioso central, tras la confirmación biópsica del diagnóstico. Son tumores muy sensibles a la radioterapia, aunque su toxicidad ha hecho que se limiten las indicaciones. La quimioterapia presenta efectos secundarios variables según los medicamentos empleados. Dada la eficacia limitada de estos fármacos y el estado funcional en que se encuentran algunos de estos pacientes, se deberá evaluar de forma individualizada la relación riesgo/beneficio de estos tratamientos para establecer la indicación. (48)
3. METODOLOGÍA
4.1 DISEÑO DE LA INVESTIGACIÓN
Se propone una investigación de tipo descriptivo retrospectiva, serie de casos;
que tiene el objeto de estudiar los signos y síntomas de los tumores cerebrales
primarios en pacientes entre los 6 meses y 17 años en el Instituto de Ortopedia
Infantil Roosevelt y el Hospital Occidente de Kennedy evidenciados entre enero de
2008 a noviembre de 2011, relacionando de una manera descriptiva a fin de
extraer generalizaciones significativas que contribuyan al conocimiento del cuadro
clínico antes del diagnóstico.
Los datos analizados en este proyecto se tomaran a partir de la revisión de
historias clínicas, tomando como referencia variables como la edad, genero,
motivos por lo cual consultan los pacientes y por ultimo tanto la revisión por
sistemas como el examen físico, todo esto con el fin de tener claridad de esta
patología antes de ser diagnosticada como tal.

52
4.2 POBLACIÓN ESTUDIADA
4.2.1 Población Diana. Pacientes que acuden al servicio de neuropediatría y
neurooncología del Instituto de Ortopedia Infantil Roosevelt y el Hospital Occidente
de Kennedy evidenciados entre enero de 2008 y noviembre de 2011.
4.2.2 Criterios de Selección de la Muestra.
4.2.2.1 Criterios de Inclusión
Pacientes con edades entre 6 meses y los 17 años.
Pacientes que asistan al servicio de neuropediatría y neurooncología.
Pacientes en estudio de tumor cerebral primario en el Hospital Occidente de Kennedy e Instituto de Ortopedia Infantil Roosevelt durante el periodo de estudio.
Pacientes con diagnóstico de tumor cerebral primario en el Hospital Occidente de Kennedy e Instituto de Ortopedia Infantil Roosevelt durante el periodo de estudio.
Pacientes con información completa en la historia clínica.
Pacientes con mas de dos localizaciones de tumor cerebral primario. 4.2.2.2 Criterios de Exclusión
Pacientes con diagnóstico de tumor Cerebral Metastásico.
Pacientes con información incompleta en la historia clínica.
4.2.3 Población estudiada. Se realizara un muestreo por conveniencia, para
incluir en el estudio todos los pacientes que cumplan los criterios de selección.
4.2.4 Tamaño de la Población. La muestra será conformada por todos los
pacientes que se encuentren dentro de los criterios de selección previamente
establecidos.
4.3 RECOLECCIÓN DE LA INFORMACIÓN E INSTRUMENTO
Esta es una investigación de tipo descriptiva de serie de casos el cual por medio
de un instrumento de recolección de datos (Ver anexo - A), que tiene el objeto de
estudiar los signos y síntomas de los tumores cerebrales primarios en pacientes
entre los 6 meses y 17 años en el Instituto de Ortopedia Infantil Roosevelt y el
Hospital Occidente de Kennedy evidenciados entre enero de 2008 a noviembre de
2011, relacionando de una manera descriptiva a fin de extraer generalizaciones

53
significativas que contribuyan al conocimiento del cuadro clínico antes del
diagnóstico.
En este sentido, la metodología de esta investigación se divide en 4 fases, que
son:
Fase 1: Revisión bibliográfica previa acerca de los signos y síntomas de Tumores
Cerebrales Primarios en pacientes entre los 6 meses y 17 años.
Fase 2: Indagación de historias clínicas del Instituto de Ortopedia Infantil
Roosevelt y el Hospital Occidental de Kennedy evidenciados entre enero de 2008
a noviembre de 2011 que permitan la identificación de variables como la edad,
motivo de consulta, signos y síntomas de la enfermedad previos al diagnóstico.
Fase 3: Análisis de signos, síntomas y condiciones previas al diagnóstico de
tumores cerebrales primarios en pacientes entre los 6 meses y 17 años del
Instituto de Ortopedia Infantil Roosevelt y el Hospital Occidente de Kennedy
evidenciados entre enero de 2008 a noviembre de 2011; teniendo en cuenta los
criterios de inclusión y exclusión para la investigación descriptiva.
Fase 4: Conclusiones para próximas investigaciones previas al diagnóstico de
tumores cerebrales primarios en pacientes entre los 6 meses y 17 años del
Instituto de Ortopedia Infantil Roosevelt y el Hospital Occidente de Kennedy
evidenciados entre enero de 2008 a noviembre de 2011.

54
4.4 TABLA DE VARIABLES

55
4.5 CALIDAD DEL DATO, CONTROL DE SESGOS Y ERROR
SESGO CONTROL
De selección
La población tomada para el estudio es amplia, pero al
momento de seleccionar la muestra, se debe especificar
exactamente los criterios que lo incluyan dentro del estudio.
Se toma como base la clasificación de tumor cerebral de la
Organización Mundial de la Salud (OMS) para así poderlos
incluir en el estudio.
De Información
Se recolecta de forma estricta la información de bases de
datos de los sitios u hospitales donde se realiza la
investigación.
Se obtendrá la información del estudio por revisión de las
historias clínicas de estos pacientes.
Habrá un tutor guía quien verifique que la información
obtenida sea estrictamente la necesaria para el estudio.
Tabla 3: Representantes del presente trabajo de investigación. 2011
ERROR CONTROL
Del Observador
Revisión previa de información sobre tumores cerebrales
primarios.
Tener en cuenta los criterios especificados que se deben
valorar de la historia clínica del paciente.
Del Observado Imposible determinar.
Del Instrumento
La revisión de las historias clínicas se debe realizar en
compañía de un tutor capacitado en este tema para la
correcta recolección de los datos.
Tabla 4: Representantes del presente trabajo de investigación. 2011

56
4.6 PROCESAMIENTO ESTADÍSTICO DE LA INFORMACIÓN Y PLAN DE
ANÁLISIS DE LA INFORMACIÓN
Se realizará una descripción de las variables demográficas y clínicas de los
pacientes seleccionados en el estudio; en las variables cualitativas se utilizarán
distribuciones de frecuencias y distribuciones porcentuales y en las cuantitativas
se utilizarán medidas de tendencia central (promedio, mediana y moda) y medidas
de dispersión o variabilidad utilizando el rango y desviación estándar. La
información recolectada será ingresada a una base de datos en Excel, la cual
posteriormente será trasladada a un paquete estadístico para el análisis de los
datos en el programa Statistical Package for the Social Sciences (SPSS).
Se realizó análisis de tipo univariado obteniendo el comportamiento de las
mismas; para calcular la normalidad de las variables se utilizo la prueba de Fisher.
Se estableció un nivel de significancia de P ≤ 0,05 e intervalos de confianza del
95%. También se utilizo la prueba estadística de Shapiro que se usa en muestras
menores de 200 pacientes, donde se pudo evidenciar que el comportamiento de
las variables.

57
5. RESULTADOS INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT
Se capturaron un total de 68 historias clínicas en el Instituto de Ortopedia Infantil Roosevelt, posterior a la realización de los criterios de inclusión y exclusión. Este trabajo de investigación es solo un capítulo del consolidado del trabajo de investigación Signos y Síntomas de los tumores cerebrales primarios previos al diagnóstico, en pacientes entre los 6 meses y 17 años en el Instituto de Ortopedia Infantil Roosevelt y el Hospital Occidental de Kennedy evidenciados entre Enero de 2008 a Noviembre de 2011.
Gráfica 11. Distribución por género de los tumores cerebrales primarios en el
Instituto Ortopedia Infantil Roosevelt
GENERO
n %
hombre 21 30,9
mujer 47 69,1
Total 68 100,0
Como se puede observar en la gráfica - 11, la relación mujer: hombre observada fue de 2,24 pacientes del sexo femenino por cada paciente masculino.
31%
69%
hombre
mujer
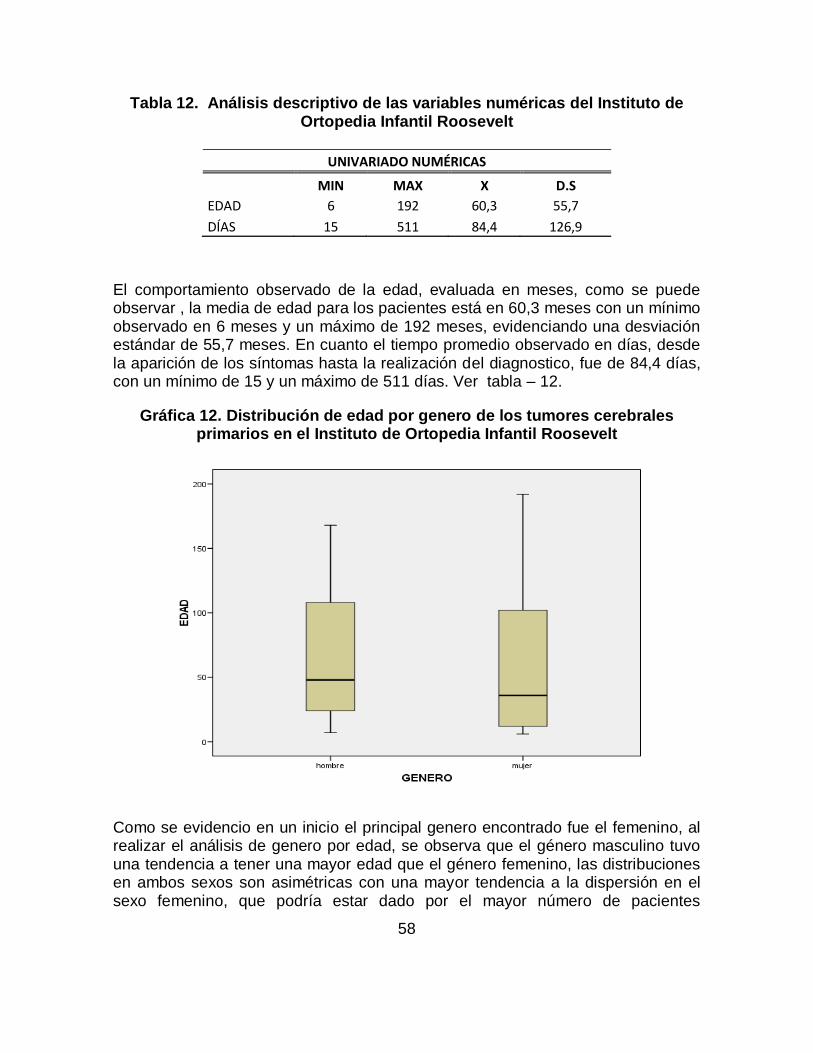
58
Tabla 12. Análisis descriptivo de las variables numéricas del Instituto de Ortopedia Infantil Roosevelt
UNIVARIADO NUMÉRICAS
MIN MAX X D.S
EDAD 6 192 60,3 55,7
DÍAS 15 511 84,4 126,9
El comportamiento observado de la edad, evaluada en meses, como se puede observar , la media de edad para los pacientes está en 60,3 meses con un mínimo observado en 6 meses y un máximo de 192 meses, evidenciando una desviación estándar de 55,7 meses. En cuanto el tiempo promedio observado en días, desde la aparición de los síntomas hasta la realización del diagnostico, fue de 84,4 días, con un mínimo de 15 y un máximo de 511 días. Ver tabla – 12.
Gráfica 12. Distribución de edad por genero de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
Como se evidencio en un inicio el principal genero encontrado fue el femenino, al realizar el análisis de genero por edad, se observa que el género masculino tuvo una tendencia a tener una mayor edad que el género femenino, las distribuciones en ambos sexos son asimétricas con una mayor tendencia a la dispersión en el sexo femenino, que podría estar dado por el mayor número de pacientes

59
presentes en este grupo, la mediana está más cercana en ambos grupos hacia el primer cuartil lo que indica un predominio de pacientes en menor edad de esta escala. Ver gráfica -12.
Gráfica 13. Distribución de días transcurridos desde el inicio de los síntomas a la realización del diagnostico de tumor cerebral primario por genero en el
Instituto de Ortopedia Infantil Roosevelt
En la gráfica – 13, se muestra la distribución de los días transcurridos desde el inicio de los síntomas hasta la realización del diagnostico de tumor cerebral primario por género, la cual muestra una distribución más simétrica en ambos géneros pero con mayor tendencia a un comportamiento más estable y con menos días transcurridos desde el inicio de los síntomas hasta la realización del diagnostico en el género femenino, aunque con mayor número de pacientes con comportamientos atípicos y duración del diagnostico muy prolongadas, así mismo la mediana de comportamiento muestra una mayor duración del diagnostico en el género masculino.
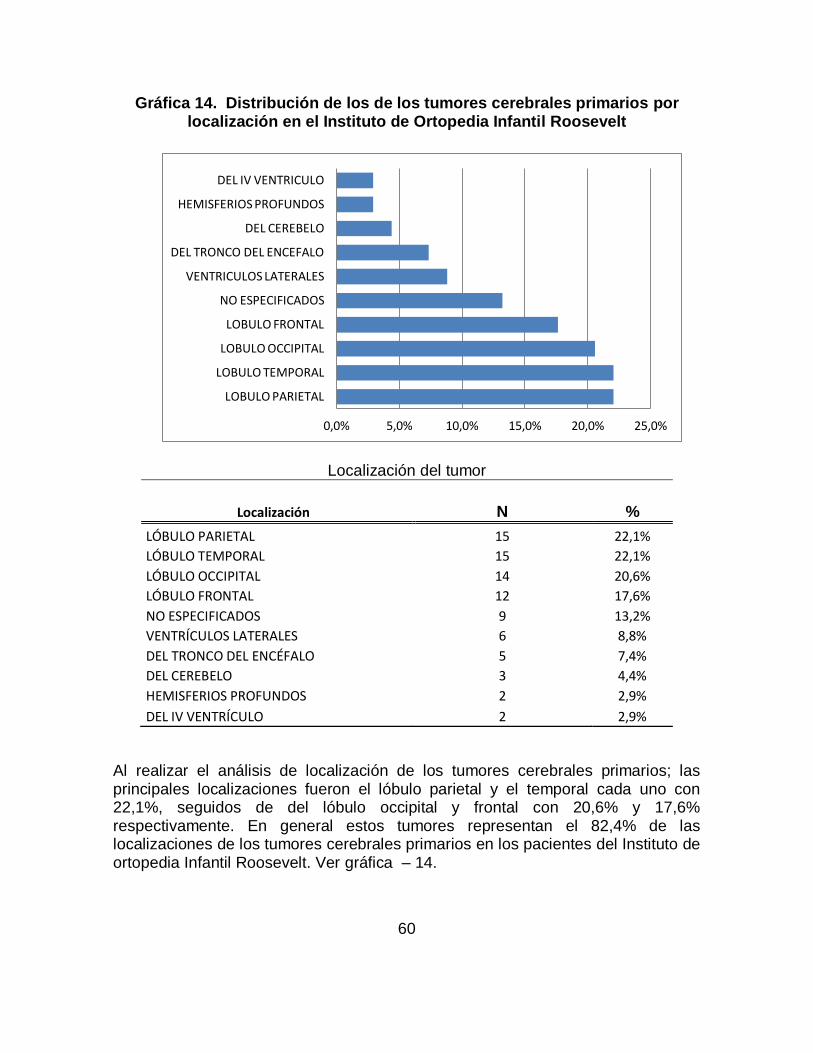
60
Gráfica 14. Distribución de los de los tumores cerebrales primarios por localización en el Instituto de Ortopedia Infantil Roosevelt
Localización del tumor
Localización N %
LÓBULO PARIETAL 15 22,1%
LÓBULO TEMPORAL 15 22,1%
LÓBULO OCCIPITAL 14 20,6%
LÓBULO FRONTAL 12 17,6%
NO ESPECIFICADOS 9 13,2%
VENTRÍCULOS LATERALES 6 8,8%
DEL TRONCO DEL ENCÉFALO 5 7,4%
DEL CEREBELO 3 4,4%
HEMISFERIOS PROFUNDOS 2 2,9%
DEL IV VENTRÍCULO 2 2,9%
Al realizar el análisis de localización de los tumores cerebrales primarios; las principales localizaciones fueron el lóbulo parietal y el temporal cada uno con 22,1%, seguidos de del lóbulo occipital y frontal con 20,6% y 17,6% respectivamente. En general estos tumores representan el 82,4% de las localizaciones de los tumores cerebrales primarios en los pacientes del Instituto de ortopedia Infantil Roosevelt. Ver gráfica – 14.
0,0% 5,0% 10,0% 15,0% 20,0% 25,0%
LOBULO PARIETAL
LOBULO TEMPORAL
LOBULO OCCIPITAL
LOBULO FRONTAL
NO ESPECIFICADOS
VENTRICULOS LATERALES
DEL TRONCO DEL ENCEFALO
DEL CEREBELO
HEMISFERIOS PROFUNDOS
DEL IV VENTRICULO

61
Gráfica 15. Localización de los de los tumores cerebrales primarios agrupada en el Instituto de Ortopedia Infantil Roosevelt
Localización agrupada
N %
Supratentorial 51 75,0
Infratentorial 8 11,8
no especificado 8 11,8
infra y supra 1 1,5
Total 68 100,0
Posterior a este análisis inicial y evidenciando que gran parte de los tumores cerebrales primarios se localizaban en los cuatro lóbulos cerebrales se decidió hacer el análisis de la distribución de los tumores, a través de la clasificación internacional para agrupación del sistema nervioso central de lesiones como supratentorial e infratentorial y como se puede evidenciar en la como se evidencia en la gráfica – 15, el 75% de los tumores tienen una localización supratentorial mostrándolos como los más frecuentes en este caso, en tanto la infratentorial y la no especificada se ubican en segundo lugar con un 11,8% de los casos cada uno.
supratentorial infrantentorial no especificado infra y supra
Series1 51 8 8 1
0
10
20
30
40
50
60

62
Gráfica 16. Clasificación histopatológica de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
Clasificación del Tumor
n %
QUISTE 34 50,0%
ASTROCITOMA 14 20,6%
GLIOMA 7 10,3%
MEDULOBLASTOMA 6 8,8%
EPENDIMOMA 1 1,5%
GANGLIOGLIOMA 1 1,5%
NEUROCITOMA 1 1,5%
NO ESPECIFICADOS 1 1,5%
En cuanto a la clasificación de los tumores cerebrales primarios se realizó a través de la histopatología de los mismos, donde se evidencia que los quistes son los más frecuentes representando un 50% del total de los tumores cerebrales primarios encontrados, seguidos del astrocitoma con un 20,6%, glioma 10,3%, meduloblastoma 8,8% y el resto de los tumores como los ependimomas, ganglioma, neurocitoma y los no especificados se presentaron en solo uno de los pacientes lo que representa un 1,5% de la clasificación. Ver gráfica – 16.
QUISTE ASTROCITO
MA GLIOMA
MEDULOBLASTOMA
EPENDIMOMA
GANGLIOGLIOMA
NEUROCITOMA
NO ESPECIFICAD
OS
Series1 50,0% 20,6% 10,3% 8,8% 1,5% 1,5% 1,5% 1,5%
0,0%
10,0%
20,0%
30,0%
40,0%
50,0%
60,0%
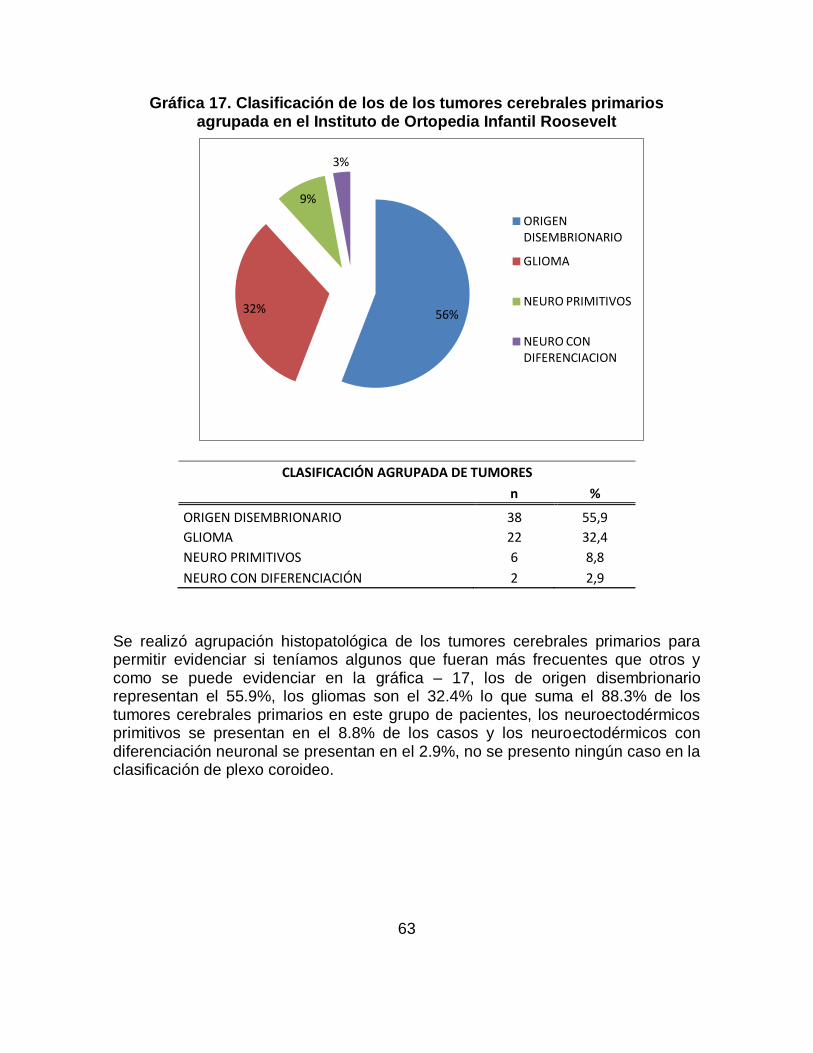
63
Gráfica 17. Clasificación de los de los tumores cerebrales primarios agrupada en el Instituto de Ortopedia Infantil Roosevelt
CLASIFICACIÓN AGRUPADA DE TUMORES
n %
ORIGEN DISEMBRIONARIO 38 55,9
GLIOMA 22 32,4
NEURO PRIMITIVOS 6 8,8
NEURO CON DIFERENCIACIÓN 2 2,9
Se realizó agrupación histopatológica de los tumores cerebrales primarios para permitir evidenciar si teníamos algunos que fueran más frecuentes que otros y como se puede evidenciar en la gráfica – 17, los de origen disembrionario representan el 55.9%, los gliomas son el 32.4% lo que suma el 88.3% de los tumores cerebrales primarios en este grupo de pacientes, los neuroectodérmicos primitivos se presentan en el 8.8% de los casos y los neuroectodérmicos con diferenciación neuronal se presentan en el 2.9%, no se presento ningún caso en la clasificación de plexo coroideo.
56% 32%
9%
3%
ORIGEN DISEMBRIONARIO
GLIOMA
NEURO PRIMITIVOS
NEURO CON DIFERENCIACION

64
Gráfica 18. Signos neurológicos de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
Signos Neurológicos
N %
CONVULSIONES 42 61,8% HIDROCEFALIA 32 47,1% AUMENTO PERIMETRO CEFÁLICO 26 38,2% HIPOREFLEXIA 21 30,9% DISMINUCIÓN FUERZA MUSCULAR 14 20,6% ALTERACIÓN MARCHA 12 17,6% HEMIPARESIA 10 14,7% HEMIPLEJIA 6 8,8% DISARTRIA 5 7,4% HIPOTONÍA 5 7,4% LIPOTIMIA 4 5,9% HIPERREFLEXIA 4 5,9% LETARGIA 3 4,4% RIGIDEZ NUCAL 3 4,4% NISTAGMUS 3 4,4% AFASIA SENSITIVA 2 2,9% PARÁLISIS FACIAL 2 2,9% PAPILEDEMA 2 2,9% MICROCEFALIA 2 2,9% AFASIA MOTORA 1 1,5% ATAXIA 1 1,5% APRAXIA 1 1,5%
61,8%
47,1%
38,2%
30,9%
20,6%
17,6%
14,7%
8,8%
7,4%
7,4%
CONVULSIONES
HIDROCEFALIA
AUMENTO PERIMETRO CEFALICO
HIPOREFLEXIA
DISMINUCION FUERZA MUSCULAR
ALTERACION MARCHA
HEMIPARESIA
HEMIPLEJIA
DISARTRIA
HIPOTONIA

65
En cuanto a los signos relacionados con los tumores cerebrales primarios se analizaron un total de 22 signos los cuales en donde se evidencian los 10 principales signos entre los que se encuentran convulsiones como el principal signo representado en un 61.8%, seguido de hidrocefalia 47.1%, aumento del perímetro cefálico 38.2%, hiporeflexia 30.9%, disminución de la fuerza muscular 20.6%, alteración de la marcha 17.6%, hemiparesia 14.7%, hemiplejia 8.8%, disartria e hipotonía 7.4% cada uno respectivamente. Así mismo se muestran los signos que menos frecuencia de presentación tuvieron en los pacientes fueron: Afasia motora, ataxia y apraxia, seguidos de afasia sensitiva, parálisis facial, papiledema, microcefalia, nistagmus, rigidez nucal, letargia, hiperreflexia y lipotimia. Ver gráfica - 18.
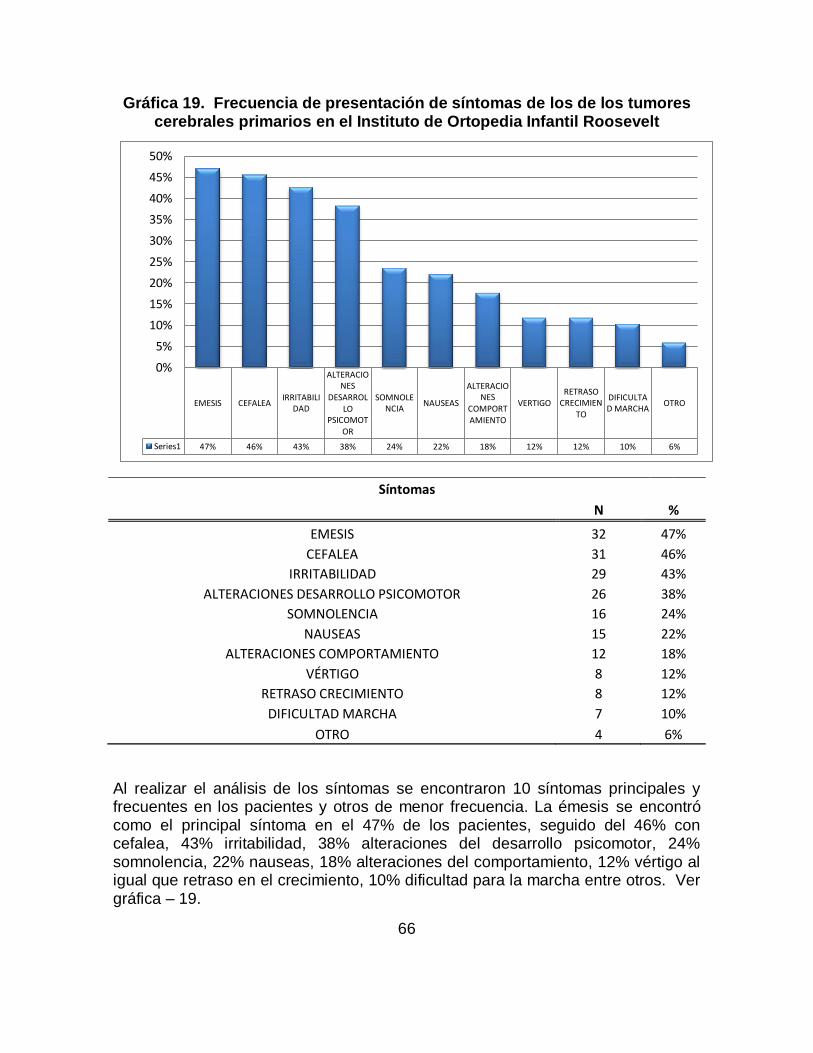
66
Gráfica 19. Frecuencia de presentación de síntomas de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
Síntomas
N %
EMESIS 32 47%
CEFALEA 31 46%
IRRITABILIDAD 29 43%
ALTERACIONES DESARROLLO PSICOMOTOR 26 38%
SOMNOLENCIA 16 24%
NAUSEAS 15 22%
ALTERACIONES COMPORTAMIENTO 12 18%
VÉRTIGO 8 12%
RETRASO CRECIMIENTO 8 12%
DIFICULTAD MARCHA 7 10%
OTRO 4 6%
Al realizar el análisis de los síntomas se encontraron 10 síntomas principales y frecuentes en los pacientes y otros de menor frecuencia. La émesis se encontró como el principal síntoma en el 47% de los pacientes, seguido del 46% con cefalea, 43% irritabilidad, 38% alteraciones del desarrollo psicomotor, 24% somnolencia, 22% nauseas, 18% alteraciones del comportamiento, 12% vértigo al igual que retraso en el crecimiento, 10% dificultad para la marcha entre otros. Ver gráfica – 19.
EMESIS CEFALEA IRRITABILI
DAD
ALTERACIONES
DESARROLLO
PSICOMOTOR
SOMNOLENCIA
NAUSEAS
ALTERACIONES
COMPORTAMIENTO
VERTIGO RETRASO
CRECIMIENTO
DIFICULTAD MARCHA
OTRO
Series1 47% 46% 43% 38% 24% 22% 18% 12% 12% 10% 6%
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%

67
Luego del análisis descriptivo se pudo evidenciar algunos signos y síntomas más frecuentes, se continuo, con la realización de un análisis de normalidad, con ayuda de la prueba estadística de Shapiro que se usa en muestras menores de 200 pacientes, donde se pudo evidenciar que el comportamiento de las variables fue no normal, y al tratarse de un análisis de una variable numérica con variables categóricas, se realizara una prueba no paramétrica U de Mann Whitney, el análisis bivariado de la edad se realizara con los signos más frecuentes, ya que son los que podrían mostrar alguna relación o significancia estadística.
Tabla 13. Análisis bivariado edad y signos neurológicos de los tumores cerebrales primarios más frecuentes en el Instituto de Ortopedia Infantil
Roosevelt
IC 95%
N X D.S. P Inf. Sup.
CONVULSIONES si 42 55,2 51,9 0,3497 -40,894 14,678
no 26 68,3 61,6
HIDROCEFALIA si 32 36,1 41,8 0,0005 -70,394 -20,745
no 36 81,7 58,2
HIPOREFLEXIA si 21 62,7 55,0 0,8131 -25,913 14,730
no 47 59,2 56,6
AUMENTO PERIMETRO CEFÁLICO si 26 28,5 32,4 0,0001 -76,362 12,500
no 42 79,9 58,3
DISMINUCIÓN FUERZA MUSCULAR si 14 81,6 54,4 0,1087 -6,115 59,814
no 54 54,7 55,2
ALTERACIÓN MARCHA si 12 89,1 56,0 0,0474 0,408 69,616
no 56 54,1 54,2
HEMIPARESIA si 10 115,9 63,3 0,0004 30,373 100,117
no 58 50,7 48,8
En la tabla – 13, se evidenciaron 3 valores de P estadísticamente significativos (P ≤ 0,05), lo que demuestra una posible asociación entre edad e hidrocefalia, aumento de perímetro cefálico y hemiparesia; al realizar el análisis con los intervalos de confianza se mostró que no existe asociación entre la edad y el aumento del perímetro cefálico en pacientes con tumor cerebral primario pero que la hidrocefalia se presenta en pacientes de menor edad y tiende a disminuir a medida que el paciente crece y en el caso de la hemiparesia a medida que aumenta la edad aumenta la presentación de hemiparesia.
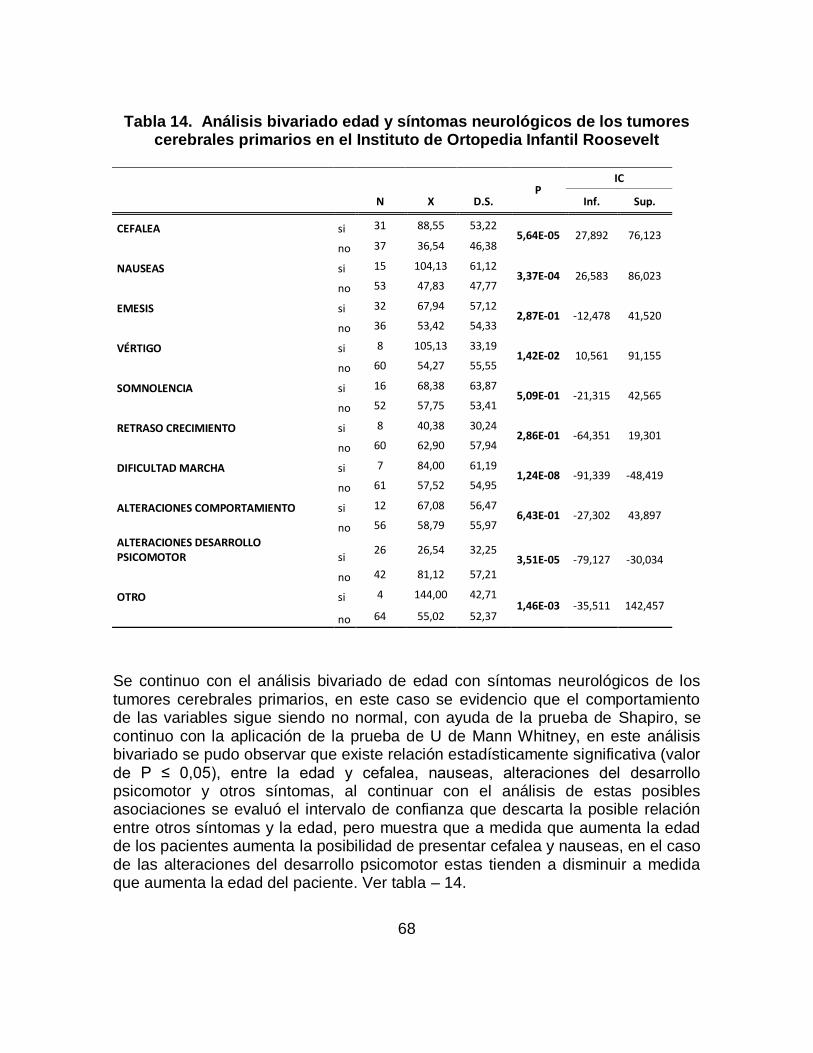
68
Tabla 14. Análisis bivariado edad y síntomas neurológicos de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
P
IC
N X D.S. Inf. Sup.
CEFALEA si 31 88,55 53,22 5,64E-05 27,892 76,123
no 37 36,54 46,38
NAUSEAS si 15 104,13 61,12 3,37E-04 26,583 86,023
no 53 47,83 47,77
EMESIS si 32 67,94 57,12 2,87E-01 -12,478 41,520
no 36 53,42 54,33
VÉRTIGO si 8 105,13 33,19 1,42E-02 10,561 91,155
no 60 54,27 55,55
SOMNOLENCIA si 16 68,38 63,87 5,09E-01 -21,315 42,565
no 52 57,75 53,41
RETRASO CRECIMIENTO si 8 40,38 30,24 2,86E-01 -64,351 19,301
no 60 62,90 57,94
DIFICULTAD MARCHA si 7 84,00 61,19 1,24E-08 -91,339 -48,419
no 61 57,52 54,95
ALTERACIONES COMPORTAMIENTO si 12 67,08 56,47 6,43E-01 -27,302 43,897
no 56 58,79 55,97
ALTERACIONES DESARROLLO PSICOMOTOR si
26 26,54 32,25 3,51E-05 -79,127 -30,034
no 42 81,12 57,21
OTRO si 4 144,00 42,71 1,46E-03 -35,511 142,457
no 64 55,02 52,37
Se continuo con el análisis bivariado de edad con síntomas neurológicos de los tumores cerebrales primarios, en este caso se evidencio que el comportamiento de las variables sigue siendo no normal, con ayuda de la prueba de Shapiro, se continuo con la aplicación de la prueba de U de Mann Whitney, en este análisis bivariado se pudo observar que existe relación estadísticamente significativa (valor de P ≤ 0,05), entre la edad y cefalea, nauseas, alteraciones del desarrollo psicomotor y otros síntomas, al continuar con el análisis de estas posibles asociaciones se evaluó el intervalo de confianza que descarta la posible relación entre otros síntomas y la edad, pero muestra que a medida que aumenta la edad de los pacientes aumenta la posibilidad de presentar cefalea y nauseas, en el caso de las alteraciones del desarrollo psicomotor estas tienden a disminuir a medida que aumenta la edad del paciente. Ver tabla – 14.

69
Tabla 15. Análisis bivariado género y signos neurológicos de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
Se continuo el análisis bivariado con el análisis de genero por signos neurológicos como se muestra en la tabla – 13, con la prueba estadística de Fisher por el comportamiento no normal de las variables, se evidencio que existe asociación estadísticamente significativa (P≤0,05), entre el género y la disartria, es decir, que ser del género masculino aumenta la posibilidad de presentar disartria en 2,9 veces más que si se pertenece al género femenino; en el caso de la rigidez nucal que también mostro asociación estadísticamente significativa, la posibilidad de
GENERO
Hombre mujer
n % n % P OR
IC 95%
inf Sup
DISARTRIA si 4 19,05 1 2,13 0.029 2,965 1.63 5,389
no 17 80,95 46 97,87
AFASIA MOTORA si 1 4,76 0 0,00 0,309 3,300 2,300 4,830
no 20 95,24 47 100
AFASIA SENSITIVA si 0 0,00 2 4,26 0,475 1,400 1,200 1,790
no 21 100 45 95,74
CONVULSIONES si 10 47,62 32 68,09 0,176 0,560 0,270 1,130
no 11 52,38 15 31,91
ALTERACION MARCHA si 6 28,57 6 12,77 0,168 1,860 0,910 3,800
no 15 71,43 41 87,23
PARALISIS FACIAL si 1 4,76 1 2,13 0,520 1,650 0,390 6,920
no 20 95,24 46 97,87
HEMIPLEJIA si 4 19,05 2 4,26 0,068 2,430 1,213 4,816
no 17 80,95 45 95,74
AUMENTO PERIMETRO CEFALICO si 4 19,05 22 46,81 0,034 0,380 0,144 1,101
no 17 80,95 25 53,19
LETARGIA si 1 4,76 2 4,26 0,636 1,080 0,210 5,500
no 20 95,24 45 95,74
PAPILEDEMA si 1 4,76 1 2,13 0,523 1,650 0,394 6,918
no 20 95,24 46 97,87
ATAXIA si 0 0,00 1 2,13 0,691 1,457 1,200 1,700
no 21 100 46 97,87
LIPOTIMIA si 3 14,29 1 2,13 0,080 2,660 1,300 3,230
no 18 85,71 46 97,87
DISMINUCION FUERZA MUSCULAR si 5 23,81 9 19,15 0,740 1,200 0,500 2,700
no 16 76,19 38 80,85
MICROCEFALIA si 1 4,76 1 2,13 0,525 1,600 0,390 6,900
no 20 95,24 46 97,87
HIDROCEFALIA si 6 28,57 26 55,32 0,065 0,450 0,199 1,020
no 15 71,43 21 44,68
RIGIDEZ NUCAL si 3 14,29 0 0,00 0,027 3,600 2,400 5,300
no 18 85,71 47 100
HIPERREFLEXIA si 2 9,52 2 4,26 0,580 1,680 0,380 4,800
no 19 90,48 45 95,74
HIPOREFLEXIA si 8 38,10 13 27,66 0,408 1,300 0,670 2,800
no 13 61,90 34 72,34
HEMIPARESIA si 5 23,81 5 10,64 0,269 1,810 0,850 3,820
no 16 76,19 42 89,36

70
presentar rigidez nucal en el sexo masculino es 3,6 veces más que en el sexo femenino. El resto de signos no evidencio ningún tipo de asociación con el género del paciente. Ver tabla – 15.
Tabla 16. Análisis bivariado genero por síntomas neurológicos de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
GENERO
hombre Mujer P OR
IC 95%
n % n % Inf. Sup.
CEFALEA si 9 42,86 22 46,81 0,762 0,852 0,302 2,404
no 12 57,14 25 53,19
NAUSEAS si 5 23,81 10 21,28 0,816 1,156 0,340 3,929 no 16 76,19 37 78,72 EMESIS si 10 47,62 22 46,81
0,915 1,033 0,369 2,895 no 11 52,38 25 53,19 VÉRTIGO si 4 19,05 4 8,51
0,213 2,529 0,567 11,283 no 17 80,95 43 91,49 SOMNOLENCIA si 5 23,81 11 23,40
0,971 1,023 0,305 3,430 no 16 76,19 36 76,60
RETRASO CRECIMIENTO si 2 9,52 6 12,77 0,469 1,792 0,364 8,829
no 19 90,48 41 87,23
DIFICULTAD MARCHA si 3 14,29 4 8,51 0,469 1,792 0,364 8,829 no 18 85,71 43 91,49
IRRITABILIDAD si 6 28,57 23 48,94 0,117 0,417 0,138 1,262 no 15 71,43 24 51,06
ALTERACIONES COMPORTAMIENTO si 4 19,05 8 17,02 0,84 1,147 0,304 4,331 no 17 80,95 39 82,98
ALTERACIONES DESARROLLO PSICOMOTOR si 7 33,33 19 40,43 0,577 0,737 0,251 2,166
no 14 66,67 28 66,67 OTRO si 1 4,76 3 6,38
0,793 0,733 0,072 7,493 no 20 95,24 44 93,62
Se realizó el análisis de normalidad para género y síntomas neurológicos con la prueba estadística de Fisher por el comportamiento no normal de las variables, los síntomas no mostraron asociación estadísticamente significativa con el género (P ≥ 0,05). Ver tabla – 16.

71
Tabla 17. Análisis bivariado localización de los tumores cerebrales primarios por signos neurológicos en el Instituto de Ortopedia Infantil
Roosevelt
Se realizó el análisis de normalidad para localización y signos neurológicos de los tumores cerebrales primarios con la prueba estadística de Fisher por el comportamiento no normal de las variables, evidenciando asociación estadísticamente significativa entre localización y afasia sensitiva, hidrocefalia e hipotonía; los demás signos no mostraron asociación estadísticamente significativa (P ≥ 0,05). Ver tabla – 17.
LOCALIZACION
supratentorial infratentorial no especificado P
n % n % n %
AFASIA SENSITIVA si 2 3,9 0 0,0 0 0,0
0,023 no 49 96,1 8 100,0 8 100,0
CONVULSIONES si 34 66,7 4 50,0 3 37,5
0,055 no 17 33,3 4 50,0 5 62,5
ALTERACION MARCHA si 7 13,7 1 12,5 4 50,0
0,876 no 44 86,3 7 87,5 4 50,0
PARALISIS FACIAL si 1 2,0 0 0,0 1 12,5
0,308 no 50 98,0 8 100,0 7 87,5
HEMIPLEJIA si 5 9,8 0 0,0 1 12,5
0,083 no 46 90,2 8 100,0 7 87,5
AUMENTO PERIMETRO CEFALICO si 22 43,1 3 37,5 1 12,5
0,391 no 29 59,0 5 62,0 7 87,5
LETARGIA si 2 3,9 1 12,5 0 0,0
0,640 no 49 96,1 7 87,5 8 100,0
PAPILEDEMA si 0 0,0 1 12,5 1 12,5
0,785 no 51 100,0 7 87,5 7 87,5
ATAXIA si 0 0,0 1 12,5 0 0,0
0,336 no 51 100,0 7 87,5 8 100,0
LIPOTIMIA si 1 2,0 3 37,5 0 0,0
0,640 no 50 98,0 5 62,5 8 100,0
DISMINUCION FUERZA MUSCULAR si 10 19,6 1 12,5 3 37,5
0,082 no 41 80,0 7 87,0 5 62,0
MICROCEFALIA si 2 3,9 0 0,0 0 0,0
0,055 no 49 96,1 8 100,0 8 100,0
HIDROCEFALIA si 27 52,9 3 37,5 2 25,0
0,001 no 24 47,1 5 62,5 6 75,0
RIGIDEZ NUCAL si 2 3,9 1 12,5 0 0,0
0,571 no 49 96,1 7 87,5 8 100,0
HIPERREFLEXIA si 2 3,9 1 12,5 1 12,5
0,876 no 49 96,1 7 87,5 7 87,5
HIPOREFLEXIA si 15 29,4 1 12,5 4 50,0
0,327 no 36 70,6 7 87,5 4 50,0
HEMIPARESIA si 5 9,8 0 0,0 4 50,0
0,640 no 46 90,2 8 100,0 4 50,0
APRAXIA si 1 2,0 0 0,0 0 0,0
0,641 no 50 98,0 8 100,0 8 100,0
NISTAGMUS si 1 2,0 1 12,5 1 12,5 0,177

72
Tabla 18. Análisis bivariado localización por síntomas neurológicos de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil
Roosevelt
LOCALIZACIÓN
P
Supratentorial Infratentorial
No especificado
Supra e Infra
N % n % n % n %
CEFALEA si 21 41,18 6 75 3 37,5 1 100
0,204 no 30 58,82 2 2,94 5 7,35 0 0
NAUSEAS si 9 17,65 3 4,41 3 4,41 0 0 0,380
no 42 82,35 5 62,5 5 62,5 1 100
EMESIS si 25 49,02 4 50 3 37,5 0 0 0,732
no 26 50,98 4 50 5 62,5 1 100
VÉRTIGO si 5 9,80 2 25 1 12,5 0 0 0,642
no 46 90,20 6 75 7 87,5 1 100
SOMNOLENCIA si 10 19,61 3 37,5 2 25 1 100 0,207
no 41 80,39 5 62,5 6 75 0 0
RETRASO CRECIMIENTO si 7 13,73 0 0 1 12,5 0 0 0,707
no 44 86,27 8 100 7 87,5 1 100
DIFICULTAD MARCHA si 5 9,80 0 0 2 25 0 0 0,404
no 46 90,20 8 100 6 75 1 100
IRRITABILIDAD si 26 50,98 2 25 1 12,5 0 0 0,103
no 25 49,02 6 75 7 87,5 1 100
ALTERACIONES COMPORTAMIENTO si 8 15,69 2 25 2 25 0 0 0,815
no 43 84,31 6 75 6 75 1 100 ALTERACIONES DESARROLLO PSICOMOTOR si 21 41,18 2 25 3 37,5 0 0 0,705
no 30 58,82 6 75 5 62,5 1 100
OTRO si 3 5,88 0 0 1 12,5 0 0 0,754
no 48 94,12 8 100 7 87,5 1 100
Como se puede evidenciar en la tabla – 18. Con la prueba estadística de Fisher, la localización al igual que en el caso de los signos a realizar el análisis por género, no muestra ningún tipo de asociación estadística, a pesar que para darle mayor soporte de análisis se realizó agrupación de esta localización, por separado no mostro tampoco ningún tipo de asociación.

73
Tabla 19. Análisis bivariado clasificación de los de los tumores cerebrales primarios y signos neurológicos en el Instituto de Ortopedia Infantil
Roosevelt
Continuando con la evaluación bivariada de las variables se procedió a realizar el análisis de la clasificación de los tumores cerebrales primarios tanto de forma simple como agrupada pero por facilidades de presentación y al no evidenciar diferencias con la forma no agrupada se toma la presentación agrupada, se logró evidenciar asociación estadísticamente significativa entre la clasificación del tumor
CLASIFICACION TUMORES
glioma
neuro con diferenciación
Neuroprimitivo
plexo coroideo P
n % n % n % N %
DISARTRIA si 0 0,0 0 0,0 2 33,3 3 8,82 0,082
no 22 100,0 2 100,0 4 66,7 31 91,18
AFASIA MOTORA si 0 0,0 0 0,0 0 0,0 1 2,94 0,908
no 22 100,0 2 100,0 6 100,0 33 97,06
AFASIA SENSITIVA si 1 4,5 0 0,0 0 0,0 1 2,94 0,967
no 21 95,5 2 100,0 6 100,0 33 97,06
CONVULSIONES si 15 68,2 1 50,0 2 33,3 23 0,254
no 7 31,8 1 50,0 4 66,7 11 32,35
ALTERACION MARCHA si 3 13,6 0 0,0 4 66,7 5 14,71 0,020
no 19 86,4 2 100,0 2 33,3 29 85,29
PARALISIS FACIAL si 1 4,5 0 0,0 0 0,0 1 2,94 0,967
no 21 95,5 2 100,0 6 100,0 33 97,06
HEMIPLEJIA si 1 4,5 0 0,0 0 0,0 5 14,71 0,537
no 21 95,5 2 100,0 6 100,0 29 85,29
AUMENTO PERIMETRO CEFALICO
si 8 36,4 1 50,0 1 16,7 14 41,18 0,792
no 14 63,6 1 50,0 5 83,3 20 58,82
LETARGIA si 1 4,5 0 0,0 1 16,7 1 2,94 0,629
no 21 95,5 2 100,0 5 83,3 33 97,06
PAPILEDEMA si 0 0,0 0 0,0 0 0,0 1 2,94 0,102
no 22 100,0 2 100,0 6 100,0 33 97,06
ATAXIA si 0 0,0 0 0,0 0 0,0 0 0,00 0,003
no 22 100,0 2 100,0 6 100,0 34 100,00
LIPOTIMIA si 1 4,5 0 0,0 1 16,7 1 2,94 0,328
no 21 95,5 2 100,0 5 83,3 33 97,06
DISMINUCION FUERZA MUSCULAR
si 6 27,3 0 0,0 3 50,0 5 14,71 0,195
no 16 72,7 2 100,0 3 50,0 29 85,29
MICROCEFALIA si 1 4,5 0 0,0 0 0,0 1 2,94 0,967
no 21 95,5 2 100,0 6 100,0 33 97,06
HIDROCEFALIA si 10 45,5 2 100,0 0 0,0 17 50,00 0,006
no 12 54,5 0 0,0 6 100,0 17 50,00
RIGIDEZ NUCAL si 1 4,5 0 0,0 0 0,0 1 2,94 0,335
no 21 95,5 2 100,0 6 100,0 33 97,06
HIPERREFLEXIA si 1 4,5 1 50,0 0 0,0 1 2,94 0,031
no 21 95,5 1 50,0 6 100,0 33 97,06
HIPOREFLEXIA si 6 27,3 0 0,0 3 50,0 11 32,35 0,707
no 16 72,7 2 100,0 3 50,0 23 67,65
HEMIPARESIA si 3 13,6 1 50,0 2 33,3 4 11,76 0,332

74
cerebral primario y alteraciones de la marcha, así como con ataxia, hidrocefalia, hiperreflexia y nistagmus con un valor de P ≤ 0,05. Ver tabla – 19.
Tabla 20. Análisis bivariado clasificación de los tumores cerebrales
primarios y síntomas neurológicos en el Instituto de Ortopedia Infantil Roosevelt
Al realizar el análisis bivariado de la clasificación de los tumores cerebrales primarios junto con los síntomas neurológicos, no se evidencia ningún tipo de asociación estadísticamente significativa (P≥0,05). Ver tabla – 20.
CLASIFICACION TUMORES
glioma
nuero con diferenciación
neuroprimitivo
plexo coroideo
otros P
n % n % n % n % n %
CEFALEA si 11
50,00 1 50 3 50,00 14 41,18 2 50 0,970
no 11
50,00 1 50 3 50,00 20 58,82 2 50
NAUSEAS si 4 18,18 1 50 1 16,67 6 17,65 3 75 0,080
no 18
81,82 1 50 5 83,33 28 82,35 1 25
EMESIS si 12
54,55 1 50 2 33,33 15 44,12 2 50 0,896
no 10
45,45 1 50 4 66,67 19 55,88 2 50
VERTIGO si 3 13,64 1 50 1 16,67 3 8,82 0 0 0,427
no 19
86,36 1 50 5 83,33 31 91,18 4 100
SOMNOLENCIA si 7 31,82 0 0 2 33,33 6 17,65 1 25 0,656
no 15
68,18 2 100 4 66,67 28 82,35 3 75
RETRASO CRECIMIENTO si 4 18,18 0 0 0 0,00 3 8,82 1 25 0,575
no 18
81,82 2 100 6 100,00 31 91,18 3 75
DIFICULTAD MARCHA si 2 9,09 0 0 1 16,67 4 11,76 0 0 0,900
no 20
90,91 2 100 5 83,33 30 88,24 4 100
IRRITABILIDAD si 12
54,55 1 50 1 16,67 13 38,24 2 6,90
0,504
no 10
45,45 1 50 5 83,33 21 61,76 2 50
ALTERACIONES COMPORTAMIENTO si
2 9,09 0 0 2 33,33 7 20,59 1 25 0,574
no 20
90,91 2 100 4 66,67 27 79,41 3 75
ALTERACIONES DESARROLLO PSICOMOTOR si
6 27,27 1 50 1 16,67 18 52,94 0 0 0,091
no 16
72,73 1 50 5 83,33 16 47,06 4 100
OTRO si 2 9,09 0 0 0 0,00 2 5,88 0 0 0,885
no 20
90,91 2 100 6 100 32 94,12 4 100

75
Tabla 21. Análisis bivariado edad agrupada y signos neurológicos de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
EDAD AGRUPADA
0 A 1 AÑO 2 A 5 AÑOS 5 AÑOS O MAS P
n % n % n %
CONVULSIONES si 16 59,3 15 83,3 11 47,8
0,064 no 11 40,7 3 16,7 12 52,2
HIDROCEFALIA si 20 74,1 7 38,9 5 21,7
0,001 no 7 25,9 11 61,1 18 78,3
AUMENTO PERIMETRO CEFÁLICO si 19 70,4 4 22,2 3 13,0
0,000 no 8 29,6 14 77,8 20 87,0
HIPORREFLEXIA si 6 22,2 9 50,0 6 26,1
0,118 no 21 77,8 9 50,0 17 73,9
DISMINUCIÓN FUERZA MUSCULAR si 2 7,4 4 22,2 8 34,8
0,057 no 4 92,6 14 77,8 15 65,2
ALTERACIÓN MARCHA si 0 0 5 27,8 7 30,4
0,008 no 27 100 13 72,2 16 69,6
HEMIPARESIA si 1 3,7 2 11,1 7 30,4
0,026 no 26 96,3 16 88,9 16 69,6
HEMIPLEJIA si 1 3,7 3 16,7 2 8,7
0,324 no 26 96,3 15 83,3 21 91,3
DISARTRIA si 0 0 1 5,6 4 17,4
0,060 no 27 100 17 94,4 19 82,6
HIPOTONÍA si 3 11,1 1 5,6 1 4,3
0,622 no 24 88,9 17 94,4 22 95,7
Luego se procedió a realizar el análisis bivariado por edad agrupada con el estadístico de Fisher para pruebas no paramétricas pues el comportamiento de las variables fue no normal, en este análisis se puede evidenciar asociación estadísticamente significativa entre la edad e hidrocefalia, aumento de perímetro cefálico, alteración de la marcha y hemiparesia, el resto de los signos no presento ningún tipo de asociación estadística. Ver tabla – 21.

76
Tabla 22. Análisis bivariado edad agrupada y síntomas neurológicos de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt
EDAD AGRUPADA
0 A 1 AÑO 2 A 5 AÑOS 5 AÑOS O MAS P
n % n % N %
EMESIS si 10 37 9 50,0 13 56,5
0,372 no 17 63 9 50,0 10 43,5
CEFALEA si 4 14,8 9 50,0 18 78,3
0,000 no 23 85,2 9 50,0 5 21,7
NAUSEAS si 2 7,4 3 16,7 10 43,5
0,007 no 25 92,6 15 83,3 13 56,5
VÉRTIGO si 0 0 1 5,6 7 30,4
0,002 no 27 100 17 94,4 16 69,6
IRRITABILIDAD si 21 77,8 8 44,4 0 0,0
0,000 no 6 22,2 10 55,6 23 100,0
SOMNOLENCIA si 6 22,2 5 27,8 5 21,7
0,884 no 21 77,8 13 72,2 18 78,3
RETRASO CRECIMIENTO si 3 11,1 4 22,2 1 4,3
0,209 no 24 88,9 14 77,8 22 95,7
DIFICULTAD MARCHA si 1 3,7 2 11,1 2 17,4
0,281 no 26 96,3 16 88,9 16 82,6
ALTERACIONES DESARROLLO PSICOMOTOR
si 18 66,7 6 33,3 2 8,7 0,000
no 9 33,3 12 66,7 21 91,3
ALTERACIONES COMPORTAMIENTO si 4 14,8 3 16,7 5 21,7
0,808 no 23 85,2 15 83,3 18 78,3
Se realizó el análisis bivariado por edad agrupada con el estadístico de Fisher para pruebas no paramétricas pues el comportamiento de las variables fue no normal, en este análisis se puede evidenciar asociación estadísticamente significativa entre la edad y cefalea, nauseas, vértigo, irritabilidad y alteraciones del desarrollo psicomotor, el resto de los síntomas no presento ningún tipo de asociación estadística. Ver tabla – 22.

77
5.1 DISCUSIÓN INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT
Se capturaron un total de 68 pacientes en el Instituto de Ortopedia Infantil Roosevelt posterior a la realización de criterios de inclusión y exclusión; obteniendo que la relación entre mujer: hombre fue 1:2.24; según Taquechel y colaboradores “Tumores del sistema nervioso central en la infancia” se encontró un ligero predominio del género femenino con respecto al masculino en una proporción de 1.1:1. Con respecto a la edad evaluada en meses se encontró que la media de edad para los pacientes está en 60,3 meses con un mínimo observado en 6 meses y un máximo de 192 meses. En cuanto el tiempo promedio observado en días, desde la aparición de los síntomas hasta la realización del diagnóstico fue de 84,4 días, con un mínimo de 15 y un máximo de 511 días, comparado con el estudio de Vázquez R. y colaboradores “Síntomas y signos iniciales de los tumores pediátricos” en donde el tiempo medio para llegar al diagnóstico fue de 172.6 días con una mediana de 30 días (50).
De acuerdo a la localización de los tumores cerebrales primarios las principales son los encontrados en el lóbulo parietal y el temporal cada uno con 22,1%, seguidos de del lóbulo occipital y frontal con 20,6% y 17,6% respectivamente, donde el 75% son supratentoriales siendo estos los más frecuentes; los infratentoriales y los de localización no especificada se ubican en segundo lugar con un 11,8% de los casos cada uno. La clasificación histológica de los tumores cerebrales primarios muestra como principal representante el astrocitoma con un 20,6%, glioma 10,3%, meduloblastoma 8,8%, donde se muestra que los de origen disembrionario representan el 55.9%, los gliomas son el 32.4%, los neuroectodérmicos primitivos se presentan en el 8.8% y los neuroectodérmicos con diferenciación neuronal se presentan en el 2.9% de los casos estudiados. Según Ramos – Clason y colaboradores, el tipo predominante fue el meningioma con 32.1%, seguido de glioblastomas con 18.5% y astrocitomas con un 8.5% (51). De acuerdo el estudio realizado por Suarez Ay colaboradores, el 62,5% de los tumores tuvieron localización supratentorial, y el 29,7% estaban ubicados en el espacio infratentorial (53). Se analizaron un total de 22 signos en la población del Instituto de Ortopedia Infantil Roosevelt en el cual se encuentro que las convulsiones el principal signo representado en un 61.8%, seguido de hidrocefalia 47.1%, aumento del perímetro cefálico 38.2%, hiporeflexia 30.9%, disminución de la fuerza muscular 20.6%, alteración de la marcha 17.6%, hemiparesia 14.7%, hemiplejia 8.8%, disartria e hipotonía 7.4% cada uno respectivamente; en cuanto al total de los síntomas analizados la emesis se encontró como el principal en un 47% de los pacientes, seguido de cefalea con 46%, irritabilidad 43%, alteraciones del desarrollo

78
psicomotor 38%, somnolencia 24%, náuseas 22%, alteraciones del comportamiento 18%, vértigo al igual que retraso en el crecimiento 12%, dificultad para la marcha 10%, entre otros. Según el estudio realizado por Suarez A y colaboradores, los síntomas más frecuentes fueron cefalea (52%), vómito (42%). La combinación de cefalea y vómitos fue encontrada en el 34,3%; la convulsión, en el 27%; la alteración en la marcha, en el 27%; y el estrabismo, en el 21%. Los signos más comunes detectados fueron la cabeza ladeada (28%), la separación de suturas del cráneo (28%), la ataxia (17%), el aumento del polígono de sustentación (14%) y el compromiso de pares craneales (8%) (53). Se considero la relación existente entre la edad de los pacientes y los signos neurológicos que estos presentan, evidenciando la relación significativa con hidrocefalia, aumento de perímetro cefálico y hemiparesia, demostrando que la hidrocefalia se presenta en pacientes de menor edad y tiende a disminuir a medida que el paciente crece y en el caso de la hemiparesia a medida que aumenta la edad aumenta la presentación de esta; mientras que al realizar el análisis de la edad en cuanto a los síntomas, se evidencia una relación significativa con la cefalea, las nauseas y las alteraciones del desarrollo psicomotor, observando que a medida que aumenta la edad de los pacientes aumenta la posibilidad de presentar cefalea y nauseas, en el caso de las alteraciones del desarrollo psicomotor estas tienden a disminuir a medida que aumenta la edad del paciente. En el estudio realizado por Vázquez R y colaboradores, en niños menores de 2 años los síntomas mas prevalentes son somnolencia y aumento del perímetro cefálico en un 25%, náuseas y emesis, letargia, convulsiones y cambios de humor 17% respectivamente, los niños de 2 a 5 años presentaron fundamentalmente nauseas y vómitos 55%, cefalea 36%, seguido de alteración en el hábito alimentario, y tortícolis 18%, los niños de 5 a 16 años presentaron cefalea en un 39%, náuseas y vómitos 35%, alteraciones visuales en un 13% y mareos inespecíficos en un 20% (50).
El análisis bivariado entre el genero y los signos demostró que existe una asociación estadísticamente significativa (P≤0,05), entre el género y la disartria, es decir, que ser del género masculino aumenta la posibilidad de presentar disartria en 2,9 veces más que si se pertenece al género femenino; en el caso de la rigidez nucal que también mostro asociación estadísticamente significativa, la posibilidad de presentar rigidez nucal en el género masculino es 3,6 veces más que en el género femenino. Pero en el análisis con respecto a los síntomas no existió una relación significativa con ninguno de los síntomas planteados en la investigación.
En la observación de la relación entre la localización de los tumores cerebrales primarios y los signos, se demuestra que es estadísticamente significativa con la afasia sensitiva, la hidrocefalia e hipotonía; con respecto a los síntomas se evidencio que no muestra ningún tipo de asociación estadística, a pesar que para darle mayor soporte de análisis se realizó agrupación de esta localización y por separado no mostro tampoco ningún tipo de asociación. El análisis bivariado con

79
la clasificación histológica de los tumores cerebrales primarios y los signos logró evidenciar asociación estadísticamente significativa con las alteraciones de la marcha, ataxia, hidrocefalia, hiperreflexia y nistagmus (P ≤ 0,05), pero con respecto a los síntomas no se demostró una relación estadísticamente significativa.

80
5.2 CONCLUSIONES INSTITUTO DE ORTOPEDIA INFANTIL ROOSEVELT
En la elaboración del estudio retrospectivo serie de casos en el Instituto de Ortopedia Infantil Roosevelt sobre signos y síntomas previos al diagnostico de tumor cerebral primario en pacientes entre los 6 meses y 17 años de edad; en el periodo comprendido entre los años 2008 a 2011 se puede concluir:
Los síntomas referidos por los pacientes y/o familiares reportados fueron la emesis en primer lugar, continuando con cefalea, irritabilidad, alteraciones del desarrollo psicomotor, somnolencia y los signos entre los que se encuentran convulsiones como el principal signo, seguido de hidrocefalia, aumento del perímetro cefálico.
Respecto a la localización se evidencio que los tumores cerebrales primarios se ubican con mayor frecuencia a nivel supratentorial principalmente en el lóbulo parietal y temporal y en menor proporción en lóbulo occipital y frontal.
En cuanto a la clasificación histológica de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt, se muestra que los de origen disembrionario se encuentran en mayor porcentaje, seguido de los gliomas.
En la relación entre la localización de los tumores cerebrales primarios y los signos, se demuestra que es estadísticamente significativa con la afasia sensitiva, la hidrocefalia e hipotonía; con respecto a los síntomas no muestra ningún tipo de asociación estadística.
El análisis bivariado con la clasificación histológica de los tumores cerebrales primarios y los signos se evidencia asociación con las alteraciones de la marcha, ataxia, hidrocefalia, hiperreflexia y nistagmus, pero con respecto a los síntomas no hay relación significativa.
Finalmente el Instituto de Ortopedia Infantil Roosevelt, abrió sus puertas y facilitó
la recolección de los respectivos datos para el presente estudio en donde se pudo
observar que la información plasmada en las historias clínicas por el especialista y
medico general, se encontraban incompletas, en cuanto a la ausencia del reporte
de patología y la clasificación histológica del tumor, dificultó la recolección de los
datos.

81
5.3 RECOMENDACIONES
El tumor cerebral primario es una patología poco frecuente a nivel de nuestra
población, lo cual conlleva a que muchas veces los profesionales de la salud
principalmente el médico general; quien es el primero en tener contacto con esta
población en centros de atención primaria, no tengan en cuenta un conjunto de
signos y síntomas que los lleven a sospechar de la presencia de un tumor cerebral
primario. Esto hace que sea una patología de difícil diagnostico.
Nuestra recomendación es incentivar al personal médico general para que realicen
una sospecha diagnostica basado en la realización de una historia clínica
detallada e interpretando los síntomas y correlacionándolos con los signos
encontrados en el momento de realizar un examen físico completo; de esta forma
mejoramos el tiempo transcurrido desde el inicio de los síntomas hasta el
momento en que se diagnostica, ofreciéndole al paciente un diagnostico
oportuno, que resultaría en disminución de las complicaciones y reducción de la
aparición de secuelas y como mayor objetivo mejorar la calidad de vida de estos
pacientes.
Para concluir insistimos en la importancia de identificar tempranamente los signos
y los síntomas que alertan sobre la posible sospecha de aparición de un tumor
cerebral, a fin de lograr un diagnostico precoz y tratamiento oportuno, mejorando
la calidad de vida de estos pacientes y prolongando la supervivencia de los
mismos.

82
ANEXOS

83
ANEXO A. INSTRUMENTO DE RECOLECCIÓN DE DATOS

84
ANEXO B. DISCUSIÓN INSTITUTO ORTOPEDIA ROOSEVELT
DIS
CU
SIÓ
NIN
ST
ITU
TO
DE
OR
TO
PE
DIA
INF
AN
TIL
RO
OS
EV
ELT
NO
. DE
CA
SO
S E
ST
UD
IAD
OS
#
68
VA
RIA
BL
EIN
ST
ITU
TO
OT
RA
S IN
VE
ST
IGA
CIO
NE
S
AN
TE
CE
DE
NT
ER
ES
UL
TA
DO
Cla
sif
ica
ció
n h
isto
lóg
ica d
el
tum
or ce
reb
ral p
rim
ari
o
Astr
ocito
ma
20
,6%
,g
liom
a10
,3%
,
med
ulo
bla
sto
ma
8,8
%,
de
orig
en
dis
em
bri
on
ario
rep
rese
nta
ne
l55
.9%
,
gliom
as
32
.4%
,lo
sn
eu
roe
cto
dé
rmic
os
8.8
%
ne
uro
ecto
dé
rmic
os
co
nd
ife
ren
cia
ció
nn
eu
ron
al
se
pre
se
nta
ne
ne
l2
.9%
Ra
mo
s E
, T
uñ
ón
M,
Riv
as F
. C
art
ag
en
a 2
00
1-
20
06
.
Me
nin
gio
ma 3
2.1
%, G
lio
bla
sto
ma 1
8.5
%
astr
ocito
ma
s 8
.5%
Lo
ca
liza
ció
n d
el t
um
or
75
% s
up
rate
nto
ria
les in
fra
ten
tori
ale
s 1
1,8
%
loca
liza
ció
n n
o e
sp
ecif
icad
a 1
1,8
%
Su
are
z a
. C
aste
lla
no
s M
. eta
l. 2
01
16
2,5
% s
up
rate
nto
ria
l, 2
9,7
% in
fra
ten
tori
al
Sig
no
s
Co
nvu
lsio
ne
s 6
1.8
% H
idro
ce
falia
47
.1%
Au
me
nto
de
l pe
rím
etr
o c
efá
lico
38
.2%
Hip
ore
fle
xia
30
.9%
Dis
min
ució
n d
e la
fu
erz
a
mu
scu
lar 2
0.6
% A
lte
ració
n d
e la
ma
rch
a 1
7.6
%
He
mip
are
sia
14
.7%
He
mip
lejia
8.8
%
Dis
art
ria
e h
ipo
ton
ía 7
.4%
Su
are
z a
. C
aste
lla
no
s M
. eta
l. 2
01
1C
ab
eza
lad
ea
da
(2
8%
)
Se
pa
ració
n d
e s
utu
ras d
el c
rán
eo
(2
8%
)
Ata
xia
(1
7%
)
Au
me
nto
de
l po
líg
on
o d
e s
uste
nta
ció
n (1
4%
)
Co
mp
rom
iso
de
pa
res c
ran
ea
les (8
%)
Sín
tom
as
47
% e
me
sis
46
% c
efa
lea
43
% ir
rita
bilid
ad
38
% a
lte
racio
ne
s d
el d
esa
rro
llo
psic
om
oto
r 2
4%
so
mn
ole
ncia
22
% n
au
se
as
18
% a
lte
racio
ne
s d
el c
om
po
rta
mie
nto
12
% v
ért
igo
Y r
etr
aso
en
el cre
cim
ien
to
10
% d
ific
ulta
d p
ara
la m
arc
ha
Su
are
z a
. C
aste
lla
no
s M
. eta
l. 2
01
1C
efa
lea
(5
2%
)
Vó
mito
(4
2%
)
La
co
mb
ina
ció
n d
e c
efa
lea
y v
óm
ito
s fu
e
en
co
ntr
ad
a e
n e
l 34
,3%
; la
co
nvu
lsió
n 2
7%
La
alte
ració
n e
n la
ma
rch
a 2
7%
Estr
ab
ism
o 2
1%
.
Ed
ad
–sín
tom
as
Au
me
nta
la e
da
d y
au
me
nta
la p
osib
ilid
ad
de
pre
se
nta
r ce
fale
a y
na
use
as.
las a
lte
racio
ne
s d
el d
esa
rro
llo
psic
om
oto
r
tie
nd
en
a d
ism
inu
ir a
me
did
a q
ue
au
me
nta
la
ed
ad
de
l pa
cie
nte
Vá
sq
ue
z R
, M
art
íne
z A
, L
lore
nte
O.
20
08
< 2
añ
os lo
s s
ínto
ma
s m
ás p
reva
len
tes s
on
so
mn
ole
ncia
y a
um
en
to d
el p
erí
me
tro
ce
fálic
o
en
un
25
%, n
áu
se
as y
em
esis
, le
targ
ia,
co
nvu
lsio
ne
s y
ca
mb
ios d
e h
um
or 1
7%
resp
ectiva
me
nte
,
2 a
5 a
ño
s: n
au
se
as y
vó
mito
s 5
5%
, ce
fale
a
36
%, a
lte
ració
n e
n e
l há
bito
alim
en
tari
o, y
tort
íco
lis 1
8%
5 a
16
añ
os: c
efa
lea
en
un
39
%, n
áu
se
as y
vó
mito
s 3
5%
, alte
racio
ne
s v
isu
ale
s e
n u
n 1
3%
y m
are
os in
esp
ecíf
ico
s e
n u
n 2
0%

85
LISTA DE GRAFICAS
Pág.
Gráfica 11. Distribución por género de los tumores cerebrales primarios en el Instituto Ortopedia Infantil Roosevelt…………………………………………………..57
Gráfica 12. Distribución de edad por género de los tumores cerebrales primarios
en el Instituto de Ortopedia Infantil Roosevelt……………………………………..…58
Gráfica 13. Distribución de días transcurridos desde el inicio de los síntomas a la realización del diagnostico de tumor cerebral primario por genero en el Instituto de Ortopedia Infantil Roosevelt………………………………………………………….…59
Gráfica 14. Distribución de los de los tumores cerebrales primarios por localización en el Instituto de Ortopedia Infantil Roosevelt………………………….60
Gráfica 15. Localización de los de los tumores cerebrales primarios agrupada en el Instituto de Ortopedia Infantil Roosevelt…………………………………………....61
Gráfica 16. Clasificación histopatológica de los de los tumores cerebrales
primarios en el Instituto de Ortopedia Infantil Roosevelt………………………….…62
Gráfica 17. Clasificación de los de los tumores cerebrales primarios agrupada en el Instituto de Ortopedia Infantil Roosevelt……………………………………………63
Gráfica 18. Signos neurológicos de los de los tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt………………………………………………64
Gráfica 19. Frecuencia de presentación de síntomas de los de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt…………….…66

86
LISTA DE TABLAS Pág.
Tabla 12. Análisis descriptivo de las variables numéricas del Instituto de
Ortopedia Infantil Roosevelt…………………………………………………………….58
Tabla 13. Análisis bivariado edad y signos neurológicos de los tumores cerebrales
primarios más frecuentes en el Instituto de Ortopedia Infantil Roosevelt…………67
Tabla 14. Análisis bivariado edad y síntomas neurológicos de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt…………….…68
Tabla 15. Análisis bivariado género y signos neurológicos de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt……………….69
Tabla 16. Análisis bivariado genero por síntomas neurológicos de los de los
tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt…….70
Tabla 17. Análisis bivariado localización de los tumores cerebrales primarios por
signos neurológicos en el Instituto de Ortopedia Infantil Roosevelt……………….71
Tabla 18. Análisis bivariado localización por síntomas neurológicos de los de los
tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt.......72
Tabla 19. Análisis bivariado clasificación de los de los tumores cerebrales
primarios y signos neurológicos en el Instituto de Ortopedia Infantil Roosevelt...73
Tabla 20. Análisis bivariado clasificación de los tumores cerebrales primarios y
síntomas neurológicos en el Instituto de Ortopedia Infantil Roosevelt.................74
Tabla 21. Análisis bivariado edad agrupada y signos neurológicos de los tumores
cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt……………...75
Tabla 22. Análisis bivariado edad agrupada y síntomas neurológicos de los
tumores cerebrales primarios en el Instituto de Ortopedia Infantil Roosevelt…...76

87
LISTA DE ANEXOS
Pág.
ANEXO A. INSTRUMENTO DE RECOLECCIÓN DE DATOS…………………..83
ANEXO B. DISCUSIÓN INSTITUTO ORTOPEDIA ROOSEVELT……………..…84

88
REFERENCIAS
1. Universitam, 2010 Mayo 18, Disponible en: URL:
http://universitam.com/academicos/?p=2334
2. Fundación Anna Vázquez. Tumores Cerebrales (Primera Parte).Bogotá. octubre 27 de
2008. URL: http://fundacionannavazquez.wordpress.com/2007/10/09/meduloblastoma-
tumor-neuroectodermico-periferico-pnet/
3. Beyer A. Jimenez P., tumores del sistema nervioso central, diciembre 2003, capitulo 13,
URL: http// http://www.med.ufro.cl/Recursos/neurologia/doc/c13.pdf
4. Dr. Niño J. y el Dr. Barrientos C. Tumores Cerebrales. Volumen 1. Lima, Perù.
UNMSM. 2008.
5. Martínez M., García A., Garaizar C. Tumores Cerebrales Infantiles: Diagnóstico y
Semiología Neurológica. Asociación Española de Pediatría (en línea) 2008.
6. Valenzuela R. Gliomas de bajo grado: Viejas controversias, nuevas evidencias. Rev.
chile. neuro-psiquiatria. [revista en la Internet]. 2005 . Disponible en:
http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0717-
92272005000300007&lng=es. doi: 10.4067/S0717-92272005000300007.
7. Nogués P, Aguas J, Pallarés J. Glioblastoma cerebeloso: Caso clínico. Oncología
(Barcelona) [revista en la Internet]. 2006 . Disponible en:
http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0378-
48352006000300006&lng=es. doi: 10.4321/S0378-48352006000300006.
8. Melo J, Roberto T, Souza A, Reis C, Almeida M. Infantile gliosarcoma. Arq. Neuro-
Psiquiatr. [serial on the Internet]. 2008. Available from:
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-
282X2008000100022&lng=en. doi: 10.1590/S0004-282X2008000100022.
9. Alvarez L, López S, Caldera A. Glioblastoma de células gigantes: Reporte de un caso.
Gac. Méd. Méx [revista en la Internet]. 2004 . Disponible en:
http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0016-
38132004000300016&lng=es.
10. Pérez L, Rodríguez E, Figueredo R, Barroso E. Astrocitoma anaplásico y glioblastoma multiforme: Factores que influyen en la supervivencia. Rev Cubana Cir [revista en la Internet]. 2001. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-74932001000200001&lng=es
11. Moreno S, Vanegas A, Bramasco A, García C, Terrazo J, Tena. Oligodendrogliomas en
el Instituto Nacional de Neurología y Neurocirugía: comportamiento biológico en una
población definida. Arch. Neurociencias. (Mexico., D.F.) [revista en la Internet]. 2005.
Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0187-
47052005000300004&lng=es.
12. Instituto Nacional del Cáncer de los Institutos Nacionales de la Salud de EE.UU.
Tumores Ependimarios. Versión para profesionales de la Salud. Informe del Instituto
Nacional de Càncer de los Institutos Nacionales de la Salud. EE.UU. 2011.
13. Escalona J. tumores del sistema nervioso central. Madrid: complutense; 2006
14. Aguirre S. tumores cerebrales tomo 1 .Mexico. Ed. Panamericana. 2008.

89
15. Ceferina F. Rodriguez V. Alert J. Suarez A. Neurocitoma. 2010;49 (4) : 395 -401.
Disponible en www.scielo.Sld.cu/pdf7med/
16. Guitierrez L. estesioneuroblastoma revisión Oncológica. 2005; 28 (7): 351 – 36
disponible en http://scielo.isciii.es/pdf/onco/v28n7/07.pdf
17. Arràzola M., Comuñas F., Conde B., Urculo E. y Dìaz F,. Oligodendrogliomas: el
problema de los Tumores Mixtos. Volumen 10. N.5, pp. 398-405. Sociedad Española
de Neurocirugìa. España. 2009.
18. Martínez M., Weil B., Herrero A., Papiloma y carcinoma de plexos coroideos en la
edad pediátrica. Volumen 49, Nº.4, pàgs. 279-286. España. Dialnet. 2007.
19. Sociedad Española de Neuroraquis. Tumor de Plexo Coroideo. España. Sociedad
Española de Neuroraquis. 2008.
20. Rostion C. Jaurequi L. Braussain V. Neuroblastoma: forma de presentación y
probabilidad de resección quirúrgica {en línea}. 2005; 2 (2: 16 – 20 disponible en
http://www.revistapediatria.cl/vol2num2/pdf/11_neuroblastoma.pdf.
21. Reyes F, Lema M. Gliomas del Encéfalo. Santiago de Compostela: Instituto
Universitario de Ciencias Neurológicas Pedro Barrié de la Maza; 2007.
22. Cancho R. “Cefalea en el Niño”. Pediatría Integral 2011; XV(9): 868-875.
23. Micheli F, Nogués M, Asconapé J, Fernández M, Biller J. Tratado de Neurología
Clínica. Madrid: Editorial Médica Panamericana S.A.; 2002.
24. Palencia R. “Trastornos de la marcha. Protocolo diagnóstico”. Neuropediatría. 2000;
40: 97-99.
25. Cancho R. “Cefalea en el Niño”. Pediatría Integral 2011; XV (9): 868-875.
26. Micheli F, Nogués M, Asconapé J, Fernández M, Biller J. Tratado de Neurología
Clínica. Madrid: Editorial Médica Panamericana S.A.; 2002.
27. Nogales J, Donoso A, Verdugo R. Tratado de Neurología Clínica. Santiago de Chile:
Editorial Universitaria; 2005.
28. Caviedes S, Collado J, Gomez A. Oftalmoligia II. España: Universidad de
Cantabria;1991.
29. Uribe C, Arana A, Lorenzana P. Fundamentos de Medicina Neurología. Bogotá:
Corporación para Investigaciones Biológicas; 2005.
30. Centro Nacional de Excelencia Tecnológica en Salud. Diagnóstico, Tratamiento inicial
y Prevención de los Tumores Cerebrales Infantiles en el Primer y Segundo Nivel de
Atención. México: Secretaria de Salud; 2008. Disponible en:
http://www.cvsp.cucs.udg.mx/guias/TODAS/IMSS_264_10_ASTROCITOMA_Y_ME
DULOBLASTOMA/IMSS_264_10_EyR.pdf
31. Arjona D, Borrego R, Huidobro B, Fernández B, Verdú A. Asociación Española de
Pediatría. Protocolos Diagnóstico Terapéuticos de la AEP: Neurología Pediátrica:
Hipertensión intracraneal. Hospital Virgen de la Salud, Toledo; 2008.
32. Conrad S. Síndrome de Hipertensión Intracraneal. 2003. Disponible en:
http://www.med.ufro.cl/Recursos/neurologia/doc/c8.pdf
33. González M, Ordoñez A, Feliu J. tratado de medicina paliativa. Buenos Aires: ed
panamericana 2.ed; 2007.
34. Suarez C, Gil J, Marco J. tratado de otorrinolaringología y cirugía de cabeza y cuello.
Buenos Aires: ed. panamericana; 2da ed., tomo II; 2007

90
35. Díaz E, García J. Oncología Clínica Básica. Madrid: Aron editores; 2000
36. Universidad Santiago de Compostela. Gliomas del encéfalo. Santiago de Compostela.
USC. 2007
37. Aguirre m, Sotelo J. Tumores cerebrales. México D.C: Ed. panamericana. 2008
38. Elke B. La Hidrocefalia. Centro Especial de Educación Especial. 2009. Disponible en:
http://compartir-educacionespecial.org/pdf/9_La_hidrocefalia.pdf
39. Álvarez Prof. Prat Prof. Hidrocefalias. Asignatura de Neurocirugía Curso 2010. 2010.
Disponible en:
http://centros.uv.es/web/departamentos/D40/data/informacion/E125/PDF930.pdf
40. Rosaralis S. Oftalmología pediátrica. Capítulo 20. Enfermedades del nervio óptico y vía
visual. 2010. Disponible en: http://gsdl.bvs.sld.cu/cgi-bin/library?e=d-000-00---
0oftalmol--00-0 oftalmol--0prompt-10---4----0-0-0-0l-0-1-es-50---20-about--4-00031-
001-1-0utfZz-8-00--0-0l--11-es-50---20-home---00-3-1-00-0-0-11-1-0utfZz-8-
00&a=d&c=oftalmol&cl=CL1&d=HASH53a0ed82a8f1b49caba721.28.2.1
41. Grupo Océano. Diccionario de Medicina. MMVI Editorial Océano. Milanesat 21-
23.Barcelona España. 2007
42. Rosaralis S. Oftalmología pediátrica. Capítulo 20. Enfermedades del nervio óptico y vía
visual. 2010. Disponible en: http://gsdl.bvs.sld.cu/cgi-bin/library?e=d-000-00---
0oftalmol--00-0 oftalmol--0prompt-10---4----0-0-0-0l-0-1-es-50---20-about--4-00031-
001-1-0utfZz-8-00--0-0l--11-es-50---20-home---00-3-1-00-0-0-11-1-0utfZz-8-
00&a=d&c=oftalmol&cl=CL1&d=HASH53a0ed82a8f1b49caba721.28.2.1
43. Mateos F. Ataxia Aguda. 2009. Disponible en:
http://www.sld.cu/galerias/pdf/sitios/rehabilitacion-temprana/ataxia_aguda.pdf
44. Sánchez J, Rodríguez A. Sincopes y Mareos en Niños y Adolescentes. Pediatría
Integral. 2008; XII(8): 757-776. Disponible en:
http://www.sepeap.org/imagenes/secciones/Image/_USER_/Sincopes_mareos_ninos_ad
olescentes.pdf
45. García J. Alteraciones del Perímetro Craneal: Microcefalia y Macrocefalia.
Pediatría Integral. 2003; VII(8): 587-600. Disponible en:
http://www.sepeap.org/imagenes/secciones/Image/_USER_/Perimetro_craneal_microce
falia_macrocefalia_alteraciones(1).pdf
46. Herranz, J, Argumosa A.. “Convulsiones Agudas”. Neuropediatría, Hospital
Universitario M. de Valdecilla, Santander.
47. Sicca M. Libros Virtuales Intramed. Convulsiones. Intramed. Disponible en :
http://www.intramed.net/sitios/librovirtual1/pdf/librovirtual1_47.pdf

91
48. García J, Benavides M. Tratamiento tumores cerebrales. Geino Grupo Español de
Investigación en Neurooncología [en línea], [Revisado el 13 mayo de 2012]. Disponible
en: http://www.geino.es/tumores_cerebrales/tumores_cerebrales_tratamiento.html
49. Chater G, Aristizabal G, Aristizabal j. características demográficas y patológicas de
tumores del sistema nervioso central estudiados en la clínica del bosque. Acta
neurológica Colombiana.2011; vol 27 nª2:107-113
50. Vasquez R, Martinez A, Llorente O. signos y síntomas iniciales de los tumores
cerebrales pediátricos. revista neurológica. 2008; vol23:1-7
51. Ramos E, Tuñon M, Rivas F. Tumores primarios del sistema nervioso central en
Cartagena 2001-2006. revista de salud pública; vol2: 257-267.
52. Taquechel o, Corral N. tumores del sistema nervioso central en la infancia.2006; 7:30-
34
53. Suarez a. Castellanos M. etal. Aspectos clínicos y demora para el diagnóstico en niños
con tumores del sistema nervioso central en el instituto nacional de cancerología en
Colombia.2011;vol15: 1-8


















