
RM miniManual 1 - fcs.uner.edu.ar · asocia a infección por VHB ( 20-30% ), VHC (5%) y a leucemia...
32
Reumatología Pág. 1 Índice TEMA 1. ESTUDIO DE LAS ENFERMEDADES MUSCULOESQUELÉTICAS. ....................... 3 TEMA 2. VASCULITIS. ............................................................................................................. 3 2.1. Definición. ..................................................................................................................3 2.2. Poliarteritis nodosa (PAN).......................................................................................... 3 2.3. Poliangeítis microscópica (microPAN). ...................................................................... 4 2.4. Angeítis y granulomatosis alérgica (enfermedad de Churg-Strauss). ......................... 4 2.5. Granulomatosis de Wegener . ..................................................................................... 5 2.6. Arteritis temporal....................................................................................................... 5 2.7. Arteritis de Takayasu. ................................................................................................. 6 2.8. Púrpura de Schönlein-Henoch. .................................................................................. 6 2.9. Vasculitis predominantemente cutáneas (vasculitis por hipersensibilidad). ................ 7 2.10. Tromboangeítis obliterante (enfermedad de Buerger)............................................... 7 2.11. Síndrome de Behçet. .................................................................................................. 7 2.12. Crioglobulinemias. .....................................................................................................7 TEMA 3. ARTRITIS POR MICROCRISTALES.......................................................................... 8 3.1. Hiperuricemia y gota. ................................................................................................. 8 3.2. Artritis debida a depósito de cristales de calcio. ...................................................... 10 TEMA 4. LUPUS ERITEMATOSO SISTÉMICO...................................................................... 11 4.1. Epidemiología. ..........................................................................................................11 4.2. Etiopatogenia. ..........................................................................................................11 4.3. Manifestaciones clínicas. ...........................................................................................11 4.4. Diagnóstico. .............................................................................................................13 4.5. Síndrome antifosfolípido. .........................................................................................14 TEMA 5. ARTRITIS REUMATOIDE........................................................................................14 5.1. Definición. ................................................................................................................14 5.2. Etiopatogenia. ..........................................................................................................14 5.3. Manifestaciones clínicas. ...........................................................................................15 5.4. Diagnóstico. .............................................................................................................16 5.5. Tratamiento. .............................................................................................................16 5.6. Enfermedad de Still del adulto. ................................................................................ 17 TEMA 6. ESPONDILOARTROPATÍAS. SERONEGATIVAS. ................................................. 17 6.1. Espondilitis anquilosante (EA). .................................................................................17 6.2. Artritis reactiva (A Re)..............................................................................................20 6.3. Artropatía psoriásica. ...............................................................................................21 6.4. Artritis en la enfermedad inflamatoria intestinal. ..................................................... 21 TEMA 7. ENFERMEDADES METABÓLICAS ÓSEAS. ........................................................... 21 7.1. Osteoporosis............................................................................................................21 7.2. Raquitismo y osteomalacia. ......................................................................................23 7.3. Enfermedad ósea de Paget. ......................................................................................24
Transcript of RM miniManual 1 - fcs.uner.edu.ar · asocia a infección por VHB ( 20-30% ), VHC (5%) y a leucemia...

Reumatología
Pág. 1
ÍndiceTEMA 1. ESTUDIO DE LAS ENFERMEDADES MUSCULOESQUELÉTICAS. .......................3
TEMA 2. VASCULITIS. .............................................................................................................32.1. Definición. ..................................................................................................................32.2. Poliarteritis nodosa (PAN). .........................................................................................32.3. Poliangeítis microscópica (microPAN). ......................................................................42.4. Angeítis y granulomatosis alérgica (enfermedad de Churg-Strauss). .........................42.5. Granulomatosis de Wegener. .....................................................................................52.6. Arteritis temporal. ......................................................................................................52.7. Arteritis de Takayasu. .................................................................................................62.8. Púrpura de Schönlein-Henoch. ..................................................................................62.9. Vasculitis predominantemente cutáneas (vasculitis por hipersensibilidad). ................72.10. Tromboangeítis obliterante (enfermedad de Buerger). ..............................................72.11. Síndrome de Behçet. ..................................................................................................72.12. Crioglobulinemias. .....................................................................................................7
TEMA 3. ARTRITIS POR MICROCRISTALES. .........................................................................83.1. Hiperuricemia y gota. .................................................................................................83.2. Artritis debida a depósito de cristales de calcio. ......................................................10
TEMA 4. LUPUS ERITEMATOSO SISTÉMICO. .....................................................................11 4.1. Epidemiología. ..........................................................................................................114.2. Etiopatogenia. ..........................................................................................................114.3. Manifestaciones clínicas. ...........................................................................................114.4. Diagnóstico. .............................................................................................................134.5. Síndrome antifosfolípido. .........................................................................................14
TEMA 5. ARTRITIS REUMATOIDE. .......................................................................................145.1. Definición. ................................................................................................................145.2. Etiopatogenia. ..........................................................................................................145.3. Manifestaciones clínicas. ...........................................................................................155.4. Diagnóstico. .............................................................................................................165.5. Tratamiento. .............................................................................................................165.6. Enfermedad de Still del adulto. ................................................................................17
TEMA 6. ESPONDILOARTROPATÍAS. SERONEGATIVAS. .................................................176.1. Espondilitis anquilosante (EA). .................................................................................176.2. Artritis reactiva (A Re). .............................................................................................206.3. Artropatía psoriásica. ...............................................................................................216.4. Artritis en la enfermedad inflamatoria intestinal. .....................................................21
TEMA 7. ENFERMEDADES METABÓLICAS ÓSEAS. ...........................................................217.1. Osteoporosis. ...........................................................................................................217.2. Raquitismo y osteomalacia. ......................................................................................237.3. Enfermedad ósea de Paget. ......................................................................................24

miniMANUAL 1 CTO
Pág. 2
TEMA 8. ESCLEROSIS SISTÉMICA PROGRESIVA. ..............................................................258.1. Etiopatogenia. ..........................................................................................................258.2. Manifestaciones clinicopatológicas. ..........................................................................268.3. Datos de laboratorio. ...............................................................................................268.4. Diagnóstico. .............................................................................................................268.5. Evolución y pronóstico. ............................................................................................27
TEMA 9. ARTRITIS INFECCIOSAS. ......................................................................................279.1. Artritis no gonocócica. .............................................................................................279.2. Artritis gonocócica. .................................................................................................27
TEMA 10. AMILOIDOSIS. ........................................................................................................2810.1. Manifestaciones clínicas. ...........................................................................................2810.2. Diagnóstico, pronóstico y tratamiento. ....................................................................28
TEMA 11. SÍNDROME DE SJÖGREN. .....................................................................................2911.1. Manifestaciones clínicas. ..........................................................................................2911.2. Diagnóstico. .............................................................................................................2911.3. Tratamiento. .............................................................................................................29
TEMA 12. POLIMIOSITIS Y DERMATOMIOSITIS. .................................................................29
TEMA 13. ARTROSIS. ..............................................................................................................2913.1. Definición. ................................................................................................................2913.2. Manifestaciones clínicas. ...........................................................................................3013.3. Tratamiento. ...................................................................................................30
TEMA 14. OTRAS ARTROPATÍAS. .........................................................................................3014.1. Osteoartropatía hipertrófica (acropaquias). .............................................................3014.2. Fibromialgia. .............................................................................................................30
ANEXO. RESUMEN DE LAS PRINCIPALES ENFERMEDADES REUMATOLÓGICAS. ......31
Reumatología
Pág. 3
TEMA 1. ESTUDIO DE LAS ENFERMEDADES MUSCULOESQUELÉTICAS.
Los pacientes con síntomas musculoesqueléticos deben ser estu-diados mediante la historia clínica, la exploración física, las pruebas de laboratorio y pruebas de imagen.
Tabla 1. Dolor reumatológico
OCINÁCEMROLOD OIROTAMALFNIROLOD
osoperlenoceyunimsiD osoperlenocriunimsideleusoN
Tanto la historia clínica como la exploración aportan gran canti-dad de datos que sugieren el diagnóstico, el cual se suele confirmar mediante la realización de pruebas complementarias. Entre las pruebas complementarias utilizadas destacan:1. El análisis del líquido sinovial, que es esencial para filiar el ori-
gen de las monoatritis agudas, estando sólo contraindicado si el paciente presenta infección de las partes blandas adyacentes. (MIR 00-01, 76; MIR 99-00, 117; MIR 99-00, 121; MIR 96-97, 105; MIR 96-97F, 90, MIR 94-95, 181). También es de gran ayuda para el diagnóstico de las artritis crónicas (de >6 semanas de evolu-ción), en las que se puede requerir la realización de biopsias sinoviales.
2. Pruebas serológicas, como la determinación de niveles de complemento, ANAS (anticuerpos antinucleares) y factor reu-matoide.
El factor reumatoide se define como una inmunoglobulina tipo M , G, A ó E dirigida contra el fragmento Fc de la IgG. El factor reumatoide es mayoritariamente de clase IgM y suele detectarse realizando pruebas de aglutinación (Waaler Rose y en látex).
Los anticuerpos antinucleares aparecen en gran cantidad de enfermedades reumatológicas, siendo más frecuentes en el LES, la EMTC y la esclerodermia. Se detectan mediante la inmunofluores-cencia indirecta (IFI). (MIR 99-00,119 )
3. Pruebas de imagen, de las que las más utilizadas son:• Radiografía convencional, muy útil en el diagnóstico de la en-
fermedades articulares. Hay que recordar que gran cantidad de las enfermedades reumatológicas tienen un período de laten-cia radiológico de años, es decir, la enfermedad debe avanzar durante largos períodos de tiempo hasta conseguir producir alteraciones que sean visibles en la radiología simple.
• Ecografía, de elección en los quistes sinoviales, las alteraciones del manguito de los rotadores, las lesiones tendinosas y en la displasia congénita de cadera en menores de 3 meses.
• Gammagrafía isotópica, prueba muy sensible pero muy poco específica, de uso en la detección de metástasis óseas, en-fermedad de Paget y procesos inflamatorios e infecciosos.
• TAC, de gran utilidad en la patología por canal medular estrecho.
• RMN, más precisa que el TAC en las alteraciones de partes blandas y de elección en la zona de la rodilla.
TEMA 2. VASCULITIS.
2.1. Definición.
Las vasculitis son procesos caracterizados por la inflamación y lesión de los vasos sanguíneos. La inflamación llega a ocluir la luz vascular, por lo que los tejidos irrigados por dichos vasos sufren procesos isquémicos. Se puede alterar cualquier tipo de vaso y en cualquier localización, lo que origina diferentes tipos de manifestaciones clínicas .Se cree que la mayoría de las vasculitis son producidas por mecanismos inmunes.
2.2. Poliarteritis nodosa (PAN).
Se define como una vasculitis necrotizante multisistémica que afecta a las arterias musculares de pequeño y mediano calibre. Suele afectar a varones (4/1) de más de 50 años y característicamente se asocia a infección por VHB ( 20-30% ), VHC (5%) y a leucemia de células peludas o tricoleucemia. (MIR 96-97,114)
Las lesiones son parcheadas y se localizan en las zonas de bi-furcación y ramificación de las arterias. La aparición de dilataciones aneurismáticas de <1cm es muy característica de la PAN clásica.
En las fases agudas de la enfermedad se visualizan PMN infil-trando todas las capas de la pared vascular y zonas perivasculares, y se origina una proliferación de la íntima con fragmentación de la lámina elástica interna. En las fases subagudas y crónicas, el infiltra-do pasa a ser de células mononucleares y se produce una necrosis fibrinoide de los vasos que oblitera parcialmente la luz y origina trombosis e infarto de los tejidos irrigados por el vaso afectado. Con la curación, se deposita colágeno en la pared vascular, lo que provoca mayor oclusión del vaso.
CLÍNICA
Casi la mitad de los pacientes presentan signos y síntomas inespecíficos sistémicos como la pérdida de peso, la fiebre y el malestar general.
El riñón es el órgano más frecuentemente afectado (70%) y la manifestación más común es la hipertensión vasculorrenal (por isquemia de los glomérulos). A veces, el paciente puede debutar con hematuria y proteinuria, síndrome nefrítico, sd nefrótico o GNF extracapilar con oligoanuria, aunque la glomerulonefritis es excepcional en este síndrome. Si existe infección por VHB, es posible la aparición de GNF membranosa y mesangiocapilar.
Las alteraciones musculoesqueléticas son muy frecuentes, sobre todo las artralgias y mialgias, pudiendo llegar a padecer una artritis asimétrica.
Tabla 2. Análisis del líquido sinovial.
LAMRON OCINÁCEM OIROTAMALFNI OCITPÉS
ROLOC ollirama,etnerapsnarT.ojor-etnerapsnarT
ojor-olliramAollirama,oibruT ocapo,oibruT
DADISOCSIV atlA atlA ajaB ajabyuM
ASOCULG lamroN lamroN ajab-lamron ajabyuM
SALULÉC )lcunonom(3mm/002-0 )lcunonom(0003< )NMP(*00005-0003 )NMP(00005>
NMP %52 %03atsah %09-52 %09>
SANÍETORP lamroN lamroN otlA otlayuM
OCITCÁL lamroN lamroN otlA otlA
OTNEMELPMOC RA,SELneojaB retieRneotlA
SOLPMEJE omsitamuart,sisortrA,lfnisitirtra,atog,SEL,RA
sacitpéssitirtrasanuglaneseceva,acitpéssitirtrA
sairotamalfnisitirtra
sacinórcsenoiccefnisaloirartnocleroP.saluléc000.05salrarepussecevaedeupanilatsircorcimaly)retieR,RA(airotamalfnisitirtraaL*.)sognoh,alecurB,CBT(rolavetserarepusonsecevanedeup
miniMANUAL 1 CTO
Pág. 4
La afectación cutánea aparece en el 50% de los pacientes, siendo característica la púrpura palpable y produciéndose también una paniculitis de tipo septal.
Le sigue en frecuencia la alteración isquémica del SNP ( 50%), que se presenta como una mononeuritis múltiple con déficit sen-sitivomotor. (MIR 96-97,117)
Los vasos intestinales se afectan por esta vasculitis, siendo muy común la aparición de dolor abdominal, y en los casos más graves se puede producir isquemia intestinal. También es posible la aparición de apendicitis y colecistitis alitiásica. El hígado presenta un aumento de la fosfatasa alcalina, aunque no hay manifestaciones clínicas.
Los vasos bronquiales pueden llegar a afectarse, aunque típica-mente la PAN cursa sin alteración pulmonar. (MIR 03-04,12)
Otros órganos que presentan isquemia son el corazón (in-suficiencia cardíaca congestiva, y si la isquemia es más brusca, incluso infarto agudo de miocardio) y el aparato genitourinario (dolor testicular).
Tabla 3. Manifestaciones clínicas PAN.
� ����� ������ ���� �����������
laneR,laneraicneicifusnI
ATH07
acitéleuqseolucsuM,saíglaim,saiglartrA
sitirtra06-05
aenátuC elbaplaparupruP 05
ociréfireposoivren.SsitiruenonoM
elpitlúm05
ovitsegidobuT lanimodbaroloD 04
nózaroC sitidracirep,MAI,CCI 03
oiraniruotineG roloD 52
DIAGNÓSTICO.En la analítica se observa leucocitosis, trombocitosis y anemia nor-mocítica normocrómica (de procesos crónicos) con elevación de la VSG. Se detectan anticuerpos p-ANCA en el 20 % de los pacientes. (MIR 98-99, 85; MIR 99-00F, 96)
El diagnóstico se confirma mediante la biopsia de una zona afec-tada. La arteriografía sería eficaz para detectar los aneurismas.
TRATAMIENTO.Los corticoides en dosis altas (1mg/kg) son la base del tratamiento de la PAN. En algunas ocasiones se añaden inmunosupresores (de elección la ciclofosfamida a 2mg/kg/día.) para poder controlar la enfermedad con menores dosis de corticoides o detener la afecta-ción visceral (si ésta es extensa).
Si existe hepatitis B asociada, se añaden corticoides y antivi-rales.
La supervivencia es del 90% a los 5 años con el tratamiento inmunosupresor.
2.3. Poliangeítis microscópica (microPAN).
La PAN microscópica se caracteriza por presentar rasgos histo-lógicos típicos de la PAN clásica (vasculitis necrotizante), pero limitados a los vasos de pequeño calibre: arteriolas, capilares y vénulas. No suelen verse aneurismas. A diferencia de la PAN clásica, puede afectar a las arterias pulmonares (en forma de capilaritis pulmonar) provocando hemoptisis por hemorragia alveolar y en el riñón suele producir glomerulonefritis. Es la enfermedad más asociada con la presencia de p-ANCA (en el 50% de los casos).
2.4. Angeítis y granulomatosis alérgica (enfermedad de Churg-Strauss).
Se trata de una vasculitis necrotizante granulomatosa que afecta a arterias musculares de pequeño y mediano calibre, capilares, vénulas postcapilares y venas. En la pared vascular pueden observarse granu-lomas necrotizantes intra y extravasculares con infiltración tisular por eosinófilos. Se asocia con asma grave y eosinofilia periférica.
Se presenta en la edad media de la vida, siendo igual la preva-lencia en ambos sexos.
CLÍNICA.Como en todas las vasculitis , se observan signos inespecíficos como la fiebre y el malestar general.
La afectación pulmonar domina el cuadro, presentándose en forma de crisis asmáticas severas que radiológicamente se traducen en infiltrados transitorios no cavitados. Muchas veces existen previo al desarrollo de la vasculitis síntomas de alergia, como son la rinitis, la poliposis nasal o el asma. También se pro-duce afectación crónica de los senos paranasales en forma de sinusitis y poliposis.
Tabla 4. Diagnóstico diferencial clínico-patológico de las distintas vasculitis.
PAN CHURG-STRAUSS
VASCULITIS PORHIPERSENSIBILIDAD
VASCULITIS DE CÉLULAS GIGANTES
Eosinofiliatisular y periférica.Granulomas intra-y extra-vasculares.
• Segmentaria.
Bifurcaciones.
Lesiones en
distintos estadios.
Muy destructiva
(aneurismas).
•
•
•
•
•
VHB.
Tricoleucemia.p-ANCA.
Leucocitoclasia.Todas las lesiones
en un mismo estadio
VÍAS ALTAS+
PULMÓN+
RIÑÓN
Granulomasintra y
extravasculares(yuxtavasculares)
C-ANCA
Cefalea,claudicaciónmandibular y
ceguera
Granulomas con células gigantes.Afectación parcheada
RIÑÓN (AP)
Articulaciones(clínica)
PULMÓN••
•
Asma.Infiltrados
migratorios.Eosinofilia.
PIEL(púrpura palpable)
Schönlein-Henoch(púrpura, artralgias,
dolor abdominal,hematuria)
A. TEMPORAL A. TAKAYASU
VASCULITIS NECROTIZANTESSISTÉMICAS
Troncossupraórticos
Asimetría depulsos y TA
Arterias muscularesde pequeño y
mediano calibre
Arterias de pequeñoy mediano calibre,capilares, vénulas
postcapilares y venasCapilares y vénulas Arterias grandes
Arterias de pequeñoy mediano calibre,capilares, vénulas
postcapilares y venas
Ancianas occidentalescon
polimialgia reumática
Jóvenesasiáticas
ÓRGANOS
VASOS
ANATOMÍA
PATOLÓGICA
ASOCIACIONES
WEGENER
Behçet:similar AP pero Dx clínico
Buerger: varón joven, oriental yfumador, con claudicación en EE.
Microabscesos en los vasos
AP similar en ysíndrome de
Kawasakisuperposición

Reumatología
Pág. 5
La afectación cutánea aparece en forma de púrpura y nódulos subcutáneos en el 70% de los afectados.
Las alteraciones cardíacas representan la causa más importante de muerte y se detectan en el 30% de los pacientes.
También es frecuente encontrar mononeuritis múltiples y en el riñón produce una GNF necrotizante focal con o sin semilunas, junto con infiltrados intersticiales de eosinófilos (MIR 96-97, 227; MIR 98-99F, 95).
DIAGNÓSTICO
La presencia de asma, junto con elevación de la IgE y una eosinofi-lia de >1000/ml, ayuda a sugerir el diagnóstico. Se suelen detectar anticuerpos p-ANCA, que son poco específicos de la enfermedad. La confirmación se realiza mediante la realización de una biopsia, donde se objetivan los granulomas y la vasculitis.
TRATAMIENTO
Se administran corticoides en dosis altas, asociadas o no con ciclo-fosfamida. La supervivencia es del 60% a los 5 años
2.5. Granulomatosis de Wegener.
Es una vasculitis necrotizante granulomatosa de pequeño vaso, donde los granulomas son de localización intra o extravascular. Típicamente asocia afectación de las vías respiratorias superiores e inferiores, junto con glomerulonefritis. Afecta a personas en la edad media de la vida, con proporción 1/1 entre sexos. Es la vasculitis más frecuente de todas las que afectan al pulmón.
CLÍNICA
En el 96% de los pacientes existe alteración del tracto respiratorio superior y/o inferior. La sinusitis es el síntoma de debut más frecuente, representado clínicamente por dolor y supuración purulento-hemorrágica a través de la fosa nasal. Histológicamen-te existe inflamación, necrosis y formación de granulomas, con o sin vasculitis, llegando en ocasiones a producirse ulceraciones en la mucosa nasal. El tabique nasal se puede perforar, dando como resultado una nariz en silla de montar. A veces el proceso de cica-trización origina una estenosis subglótica que puede provocar una obstrucción respiratoria grave.
Tabla 5. Vasculitis de Wegener.
RENEGEWEDSITILUCSAV
acinórcsitisuniS-%89
seranomlupsenoicaretlA-%58
avisergorpetnemadipárFNG-%57
)..sitirelcseipe,sisotporp(seralucosenoicaretlA-%05
saenátucsenoicaretlA-%04
)elpitlúmsitiruenonom(sacigóloruentlA-%02
CNSsenoicaretlA-%8
La afectación pulmonar (85%) se caracteriza por tos, disnea, hemoptisis y dolor torácico. Radiológicamente se visualizan infil-trados nodulares bilaterales cavitados no migratorios que pueden alcanzar un gran tamaño.
En el 77% de los pacientes se afecta el riñón en forma de una GNF focal y segmentaria que evoluciona hacia una GNF rápida-mente progresiva con semilunas. La manifestación más común es la hematuria y la proteinuria, aunque es posible la aparición de un sd nefrítico, un sd nefrótico u oligoanuria. Excepcionalmente se origina una obstrucción en los uréteres por aparición de granulo-mas en su pared.
La afectación ocular se da en el 50% de los casos y varía des-de una simple conjuntivitis hasta proptosis (muy específica del Wegener) debido a la ocupación del espacio retroorbitario por granulomas.
Las lesiones cutáneas (46%) son las mismas que en otros tipos de vasculitis. En el 20 % de los pacientes hay manifestaciones del
SNP en forma de mononeuritis múltiple y el 8% presentan síntomas del SNC como neuropatía de los pares craneales. (MIR 04-05, 86; MIR 97-98, 232)
DIAGNÓSTICO.La bioquímica demuestra una gran elevación de la VSG, junto con leucocitosis, anemia, trombocitosis e hipergammaglobulinemia a expensas de la Ig A.
De gran utilidad son los anticuerpos c-ANCA tanto para el diagnóstico como para el seguimiento y evaluación de la actividad de la enfermedad. Son en un 95% específicos del Wegener y sus títulos son muy elevados cuando la afectación orgánica es severa (sobre todo en la afectación renal). Sin embargo, a pesar de tener un gran valor, no deben sustituir a la biopsia como confirmación diagnóstica. (MIR 94-95, 64, 187)
La biopsia se realiza de elección en el pulmón, puesto que en las vías aéreas superiores no se suele demostrar la vasculitis y en el riñón es difícil observar los característicos granulomas.
Se debe realizar diagnóstico diferencial entre la granuloma-tosis de Wegener y la granulomatosis linfomatoide. Este cuadro se engloba dentro de los trastornos inmunoproliferativos an-giocéntricos y cursa principalmente con infiltración destructiva del tracto respiratorio superior e inferior por linfocitos atípicos, células plasmáticas e histiocitos con mitosis anormales. Se pro-duce en varones de 50 años y característicamente se observan granulomas centrados en los vasos, a diferencia de en el Wegener. La radiología de tórax es superponible entre ambas enfermedades y también hay afectación renal, sólo que en la linfomatosis ésta no se expresa clínicamente. Suele derivar con el tiempo en un linfoma maligno y el tratamiento son los corticoides asociados o no a ciclofosfamida.
TRATAMIENTO.Es de elección la administración de ciclofosfamida en dosis de 2mg/kg/día durante al menos 1 año, que ha conseguido la remisión de la enfermedad en el 75% de los casos y gran mejoría en el 90% de los pacientes. También se ha demostrado útil en el manejo de las recidivas tras la suspensión del tratamiento. Se aconseja que durante los primeros meses la ciclofosfamida se asocie a corticoides en dosis altas, aunque estos no han demostrado mejorar el curso evolutivo de la enfermedad, sirviendo sólo para agilizar la mejoría de los síntomas.
En caso de objetivarse leucopenia, se deberá ajustar la ciclo-fosfamida hasta que los leucocitos asciendan a >3000/ml (>1500 neutrófilos). Entre sus efectos adversos cabe destacar la pancito-penia, la cistitis, el cáncer vesical, la mielodisplasia y la toxicidad gonadal. Si no es posible su uso por encontrarse contraindicado, se intentará una pauta con metotrexate o azatioprina asociado a corticoides (MIR 99-00F, 229, MIR 97-98, 225).
2.6. Arteritis temporal.
Es una panarteritis de vasos de mediano y gran calibre que cursa con infiltrados de células mononucleares y células gigantes en el interior de la pared vascular. Se observan granulomas en la biopsia. La afectación del vaso es parcheada o segmentaria y se observa proliferación de la íntima y degeneración de la lámina elástica. La arteria temporal en la más frecuentemente afectada.
Se observa en mujeres ancianas en una proporción 3/1 res-pecto a los varones y se asocia en el 50% de los casos a polimialgia reumática (rigidez, dolores sordos y mialgias en cinturas escapular y pelviana).
CLÍNICA.Debuta clásicamente como un cuadro de fiebre, anemia, VSG ele-vada y cefalea frontotemporal en un paciente anciano.
El síntoma más frecuente es la aparición de cefalea (65%) refractaria al tratamiento analgésico. Se puede asociar a una ar-teria temporal engrosada y dolorosa a la palpación que conserva inicialmente el pulso.
Otros síntomas característicos son la claudicación mandibular, la inflamación del cuero cabelludo o incluso la pérdida del gusto.
La complicación más grave de la arteritis temporal no tratada es la afectación ocular en forma de neuritis óptica isquémica por trombosis de la arteria central de la retina, que causa ceguera irre-

miniMANUAL 1 CTO
Pág. 6
versible unilateral, que puede convertirse en bilateral si el proceso sigue sin tratarse. (MIR 00-01, 81; MIR 95-96, 51)
Aunque ésta suele ser la clínica predominante, no hay que olvidar que otras partes del organismo pueden estar afectadas, siendo frecuentes los granulomas a nivel hepático que provocan un aumento de la fosfatasa alcalina (signo de ocupación hepática).
DIAGNÓSTICO.Se sospecha la enfermedad al hallarse cefalea, fiebre, anemia normo-normo y VSG elevada en un paciente, con o sin polimialgia reumá-tica. La confirmación se consigue a partir de muestras histológicas. Recuerda que el tratamiento con corticoides puede negativizar la biopsia, pero que es necesario iniciar cuanto antes el tratamiento para evitar las manifestaciones oculares. Además, la afectación es segmentaria, por lo que puede que las muestras recogidas sean de una zona de la arteria no afectada. (MIR 97-98, 229)
TRATAMIENTO.Es característica de esta enfermedad una excelente respuesta al tratamiento con corticoides sistémicos ( lo cual tiene utilidad diag-nóstica, cuando la biopsia no es concluyente).Se utilizan tanto para conseguir un alivio sintomático como para prevenir las complica-ciones oculares. Inicialmente se utiliza una dosis de 1mg/kg/día que se va disminuyendo hasta la dosis mínima eficaz para el control de los síntomas. El tratamiento debe de durar al menos 1-2 años para evitar la aparición de recaídas. La VSG es un parámetro de gran utilidad para monitorizar la eficacia del tratamiento, ya que indica la actividad inflamatoria.
RECUERDA
Si sólo existen síntomas de polimialgia reumática, el tratamiento son corticoides a dosis bajas (de 15-20 mg/día) (MIR 03-04, 11)
2.7. Arteritis de Takayasu.
También denominada enfermedad sin pulso o síndrome del arco aórtico. Se trata de una enfermedad inflamatoria y estenosante fundamentalmente de las arterias de gran calibre que muestra pre-dilección por los troncos supraaórticos. Histológicamente es una pa-narteritis granulomatosa con infiltrado inflamatorio mononuclear y células gigantes multinucleadas en la adventicia, proliferación de la íntima, fibrosis de la media y disgregación de la lámina elástica interna (igual que la arteritis de la arteria temporal). Esto produce un estrechamiento de la luz del vaso, con o sin trombosis. No existe necrosis fibrinoide.
Tabla 5. Arteritis de Takayasu.
AIRETRA EJATNECROP ACINÍLC
aivalcbuS 39,sozarbsolednóicacidualC
duanyaRonemónef
númocaditóraC 85,epocnís,selausivsonrotsarT
sutci,TIA
lanimodbaatroA 74,saesúan,lanimodbaroloD
sotimóv
selanersairetrA 83aicneicifusni,nóisnetrepiH
laner
ocitróazíaryodayaC 53 CCI,acitróaaicneicifusnI
selarbetreV 53 soeram,selausivsenoicaretlA
ocaílecejE 81,saesúan,lanimodbaroloD
sotimóv
roirepusacirétneseM 81,saesúan,lanimodbaroloD
sotimóv
sacaílI 71 sanreipsalednóicacidualC
seranomluP %04-01 aensid,ocipítaocicárotroloD
sairanoroC 01< MAI,ocicárotroloD
Es una enfermedad sistémica que aparece con más frecuencia en jóvenes asiáticas. Las arterias más afectadas son las subclavias, seguidas por la carótida común, la aorta abdominal y las arterias renales.
CLÍNICA.Cursa inicialmente con sintomatología general (fiebre, malestar, anorexia, y pérdida de peso). Posteriormente se presenta dolor por la lesión del vaso y síntomas secundarios a la isquemia en el territorio irrigado por el vaso afectado, siendo los más frecuentes los fenómenos isquémicos del SNC (que son la principal causa de muerte, junto a la insuficiencia cardíaca y el infarto de miocardio). (MIR 01-02, 50)
DIAGNÓSTICO.Debe sospecharse ante una mujer joven en la que se detecte asimetría de pulsos entre ambos brazos (por afectación asimétrica de ambas arterias subclavias) y soplos por obstrucción parcial de los vasos. La analítica es similar a la de otras vasculitis y el diagnóstico confirma-torio se realiza mediante arteriografía. Son datos característicos en ésta la irregularidad de las paredes vasculares, las estenosis, las dila-taciones postestenóticas, la formación de aneurismas, las oclusiones y aumento de la circulación colateral. No sirven las biopsias, por ser inaccesibles y porque la afectación es segmentaria.
TRATAMIENTO.Se debe intentar controlar la inflamación con corticoides en do-sis altas, para realizar posteriormente, con el paciente estable, angioplastias u otras técnicas quirúrgicas de reparación (endarte-rectomías y bypass). Si existe resistencia a los corticoides o no son suficientes, se puede usar metotrexato u otros inmunosupresores. La anticoagulación previene la trombosis y la oclusión completa de los vasos. La mortalidad es del 20% a los 5 años.
2.8. Púrpura de Schönlein-Henoch.
Es una vasculitis de pequeño vaso del tipo leucocitoclásica que se caracteriza por púrpura palpable no trombopénica (de distribu-ción en nalgas y miembros inferiores), artralgias, dolor abdominal y glomerulonefritis. Aunque se suele ver en varones de 4-7 años, también es posible en el adulto.
Se cree que el mecanismo patogénico es el depósito de inmu-nocomplejos (sobre todo formados por Ig A). La púrpura suele estar precedida por un cuadro de infección respiratoria, lo que apoya que los antígenos más frecuentemente implicados sean los de los gér-menes que producen estas infecciones, aunque también se asocia al uso de ciertos fármacos o alimentos.
CLÍNICA.Las manifestaciones más precoces son las cutáneas, en forma de púrpura palpable.
La mayoría de los pacientes (90%) presentan poliartralgias, llegando en algunos casos a presentar una verdadera poliartritis no erosiva migratoria.
En el 70% de los pacientes pediátricos hay síntomas gastroin-testinales, predominando el dolor abdominal cólico con nauseas, vómitos, diarrea o estreñimiento (producidos por edema de la pared intestinal secundario a la isquemia). Con frecuencia hay rectorragia causada por el daño en la mucosa.
La afectación renal es frecuente y cursa con hematuria con cilindros hemáticos y proteinuria. Más raramente hay evolución a síndrome nefrótico y a GN rápidamente progresiva (con semilu-nas). Histológicamente se visualiza una proliferación mesangial difusa o focal y una GN proliferativa segmentaria. Es característico el depósito mesangial de IgA y complemento. En los pocos casos en los que la enfermedad puede causar la muerte, ésta se debe al daño crónico renal. (MIR 04-05, 186; MIR 99-00, 127; MIR 99-00, 213, MIR 97-98F, 144; MIR 95-96F, 13)
DIAGNÓSTICO.Se apoya en la clínica y se confirma mediante la biopsia y la de-mostración de los inmunocomplejos en el tejido. En la analítica se observa leucocitosis con una cifra de plaquetas y de complemento sérico normal. Los niveles de IgA circulante están elevados en el 50% de los enfermos.

Reumatología
Pág. 7
TRATAMIENTO.No suele ser necesario porque la enfermedad es autolimitada, pre-sentando recidivas y remisiones durante un período de semanas o meses hasta su resolución espontánea. Se puede recurrir a los corticoides en dosis altas (1mg/kg/día) para disminuir el edema tisular, las artralgias y las molestias abdominales. En casos graves puede recurrirse a la plasmaféresis. (MIR 98-99, 183)
La mayoría de las veces el pronóstico es excelente, excepto en los casos que cursan de forma crónica o con sucesivos brotes.
2.9. Vasculitis predominantemente cutáneas (vasculitis por hipersensibilidad).
Se trata de las vasculitis más frecuentes y se caracterizan por la afectación cutánea y por su predilección por los vasos pequeños (capilares y vénulas). Su nombre se debe a que en su patogenia está implicada una reacción de hipersensibilidad tras la exposición a un antígeno ( fármacos, infecciones, neoplasias, etc.) . (MIR 02-03, 220), que lleva al depósito de inmunocomplejos en la piel y otros órganos como las articulaciones, el riñón o el sistema gastrointestinal .
La lesión característica se denomina leucocitoclasia, que defi-ne los restos nucleares procedentes de neutrófilos que infiltran las paredes de los vasos y zonas adyacentes durante las fases agudas. Se observa extravasación de hematíes a partir de los vasos lesionados, que clínicamente da lugar a una púrpura palpable. Como son en-fermedades que cursan generalmente en brotes, todas las lesiones están en el mismo estadio evolutivo.
CLÍNICA.Cursa con síntomas generales (fiebre, malestar, mialgias y anorexia) y púrpura palpable que se encuentra en las zonas declives, pudiendo ser pruriginosa e incluso muy dolorosa.
DIAGNÓSTICO
Se confirma mediante biopsia cutánea.
TRATAMIENTO.La mayoría de estas vasculitis se resuelven espontáneamente. También es posible realizar el tratamiento etiológico, asociando o no corticoides .
2.10. Tromboangeítis obliterante (enfermedad de Buerger).
Es una enfermedad vascular inflamatoria oclusiva caracterizada por la formación de trombosis en las arterias pequeñas y me-dianas de las zonas distales de las extremidades, que afectan de forma segmentaria a los vasos y se acompañan de un infiltrado de polimorfonucleares en todas las capas de la pared vascular, así como de pequeños microabscesos dentro de la estructura del trombo.
Se observa en varones jóvenes fumadores, estando este trastorno muy relacionado con el consumo de tabaco.
La tríada clínica típica es la claudicación de la extremidad afec-ta, junto con fenómeno de Raynaud y tromboflebitis superficiales migratorias.
DIAGNÓSTICO.Se realiza mediante la historia clínica y la exploración física. En la arteriografía, que es confirmatoria, se visualiza el afilamiento de los vasos distales y la presencia de vasos colaterales en las áreas ocluidas.
TRATAMIENTO.Es esencial abandonar el hábito tabáquico para evitar la progre-sión de la enfermedad. Si el vaso es lo suficientemente grande, será posible emplear técnicas de derivación quirúrgica. En caso contrario, la amputación será inevitable si se objetiva isquemia persistente grave.
2.11. Síndrome de Behçet.
El síndrome de Behcet es un proceso multiorgánico que se mani-fiesta por la aparición de úlceras bucales y genitales y por afectación ocular.
Histológicamente, la principal lesión es una vasculitis de pe-queño vaso con tendencia a la formación de trombos venosos. Su prevalencia es mayor entre los jóvenes varones de Japón y el Mediterráneo oriental y parece estar relacionado con el antígeno HLA –B5.
Tabla 6. Criterios diagnósticos de la enfermedad de Behçet.
1. Presencia de úlceras orales recurrentes (imprescindible) asociadas a 2 de los siguientes:
2. Úlceras genitales recurrentes.3. Lesión ocular (uveítis posterior o anterior).4. Lesiones cutáneas (eritema nodoso, foliculitis ...).5. Fenómeno de patergia positivo.
CLÍNICA.Es esencial la existencia de aftas orales (3 ó más episodios anuales) para el diagnóstico. Son úlceras dolorosas, superficiales o profundas, en cualquier localización en la mucosa oral. Curan sin dejar cicatriz en 1-2 semanas. (MIR 04-05, 148; MIR 97-98, 233; MIR 97-98F, 209). Las úlceras genitales (80%) se parecen a las orales, pero sí dejan cicatriz. En la piel se observan foliculitis o pseudofoliculitis (80%), eritema nodoso y exantema semejante al acné.
Las alteraciones oculares son la complicación más grave de la enfermedad, ya que evolucionan rápidamente a la ceguera. Sue-le ocurrir al inicio de la enfermedad y cursa en forma de uveítis posterior, uveitis anterior o neuritis óptica. Los pacientes afectos de Behçet poseen fenómeno de patergia positivo, es decir, la inyección intradérmica de suero salino provoca la aparición de pústulas en la piel (se produce también en el Sd de Sweet y en el pioderma gangrenoso). El 30-60% de los enfermos padecen artralgias, aunque puede llegar a originar una artritis de grandes articulaciones no erosiva.
En una cuarta parte de los pacientes se observan trombosis venosas superficiales y profundas (con el consecuente riesgo de TEP), y pueden llegar a afectarse las arterias, provocando aortitis y aneurismas y trombosis de arterias periféricas. (MIR 95-96F, 142),
La afectación del SNC por la vasculitis se traduce en hiper-tensión craneal benigna, meningoencefalitis aséptica, afectación piramidal y alteraciones psiquiátricas.
DIAGNÓSTICO.Es clínico y se requiere la conjunción de 3 de los criterios diagnós-ticos para diagnosticar la enfermedad, siendo imprescindible la existencia de aftas orales.
En la analítica se observa aumento de la VSG y de la PCR junto con leucocitosis. Pueden detectarse anticuerpos contra la mucosa oral humana.
TRATAMIENTO.Este síndrome se trata de forma sintomática y empírica.
Las úlceras mejoran con la aplicación tópica de corticoides, y en casos más graves se utiliza la talidomida, la colchicina y la pen-toxifilina. La uveítis posterior debe ser tratada enérgicamente y de forma precoz con ciclosporina (5-10mg/kg/día). La afectación del SNC requiere dosis altas de glucocorticoides, azatioprina o ciclos-porina. La artritis mejora con la colchicina y el interferón alfa y las tromboflebitis superficiales mejoran con el ácido acetilsalicílico. La gravedad del síndrome cede con el tiempo.
2.12. Crioglobulinemias.
Las crioglobulinas son inmunoglobulinas que precipitan a una temperatura de 4ºC y se disuelven por calentamiento. Según su etiología y el tipo de inmunoglobulinas que las formen, se clasifican en tres grupos.
Tanto el grupo II como el III pueden producir una crioglobuline-mia esencial. Puede asociarse a infección por el virus de la hepatitis C (en el 90% de los casos), a infecciones por hongos, bacterias o virus o a enfermedades malignas.
Es una enfermedad que afecta más a la mujer alrededor de la quinta década de la vida.
La manifestación más frecuente es la púrpura palpable (que indica la existencia de una vasculitis cutánea de pequeño vaso),

miniMANUAL 1 CTO
Pág. 8
seguido de manifestaciones musculoesqueléticas, fiebre y hepa-toesplenomegalia; a veces hay polineuropatía y adenopatías.
El riñón se afecta en la mitad de los casos. Cursa con hematuria y/o proteinuria que puede llegar a rango nefrótico. También puede debutar como síndrome nefrítico (glomerulonefritis con semilunas con oliguria).
DIAGNÓSTICO.Se demuestran mediante biopsia las lesiones cutáneas. En el riñón se puede visualizar una glomerulonefritis mesangial o mesangioca-pilar. Si hay semilunas, se tratará de un GN extracapilar tipo II.
En el plasma es necesario demostrar la presencia de crioglobu-linas y la disminución de los niveles séricos de complemento (por formación de inmunocomplejos). Será necesario investigar cuál es la causa de que se produzcan dichas inmunoglobulinas.
TRATAMIENTO.Se utilizan corticoides e inmunosupresores. A veces, la plasmafé-resis y el IFα producen mejoría de los síntomas.
TEMA 3. ARTRITIS POR MICROCRISTALES.
3.1. Hiperuricemia y gota.
El ácido úrico es el producto final de la degradación de las purinas. Se produce en los órganos que contienen xantín oxidasa (el intestino delgado y el hígado) y se elimina sobre todo por el riñón.
Las concentraciones de úrico son muy bajas en la infancia y van aumentando en los varones a partir de la pubertad y en las mujeres tras la menopausia (lo cual se cree que es debido a una mayor excre-ción de uratos en la época fértil). (MIR 98-99, 88)
Se define la hiperuricemia como la concentración plasmática >7mg/ dl. Su prevalencia en la población general es entre 2-13%. Sin embargo, el término gota engloba a las manifestaciones clínicas producidas por el depósito de cristales de urato monosódico en la cavidad articular o en otros tejidos. La prevalencia de la gota es del 1-4%.
Las causas de la hiperuricemia se pueden clasificar en tres grupos:
A. AUMENTO DE LA SÍNTESIS DE URATOS.Puede producirse por causas primarias o innatas, como fallos here-ditarios en las enzimas del catabolismo de las purinas. Entre ellas destacan (MIR 00-01, 64):1. Aumento de la actividad de la fosforribosilpirofosfato (PRPP)
sintetasa –Defecto ligado al cromosoma X.2. Déficit de la hipoxantina fosforribosiltransferasa(HPRT)-
Defecto ligado al cromosoma X. Puede presentarse de forma completa (Sd de Lesch-Nyhan) asociado a alteraciones neu-rológicas, o como un déficit parcial enzimático (Sd de Kelley Seegmiller).
Más frecuentes son las causas secundarias o adquiridas, en las que existe un aumento de la lisis celular, de la que se liberan las purinas que originarán el exceso de ácido úrico. Ejemplos son las enfermedades mielo-linfoproliferativas o el tratamiento de las mismas, los procesos hemolíticos, la policitemia vera, la psoriasis, la enfermedad de Paget, las glucogenosis de tipo III,V, VII, la rab-domiólisis, el ejercicio, el alcohol y la obesidad.
La dieta es una fuente exógena de purinas, pero no posee gran importancia para el control de la hiperuricemia (sólo puede dis-minuir 1 mg/dl la uricemia). NO se utiliza en el tratamiento de la
Tabla 7. Artritis por microcristales.
MICROSCOPIOPOLARIZACIÓN
BipiramidalRomboidal Aguja
MECÁNICO, suele tenermenos de 2000 cél.
Neutrófilos ymononucleares
ANCIANOS
MICROSCOPIOPOLARIZACIÓN
MICROSCOPIOPOLARIZACIÓN
Muy pequeños
PIROFOSFATOCÁLCICO (PPCD)
HIDROXIAPATITA (HA)OXALATO
CÁLCICO (OxCa)URATO
MONOSÓDICO (UMS)
Condrocalcinosissimétrica
INFLAMATORIOPredominio de
neutrófilos
MECÁNICOMononucleares
CalcificacionesDISTRÓFICAS yMETASTÁSICAS
RODILLA, MUÑECATOBILLO
, RODILLA, HOMBRO CUALQUIERA
Micr. electrónico, se tiñede rojo con
ALIZARINA ROJA
ANCIANOS con artrosis.Si <50a pensar en
alteración metabólicao hereditaria
ASINTOMÁTICOS
"PSEUDOGOTA"En ocasiones aguda:
Oxalosis 2ª a IRT endiálisis y vit. C.
Oxalosis 1ª en <20aVARÓN en la 5ª década
GOTA agudaSinovitis en
paciente con IRT
ASINTOMÁTICAArtritis aguda
Artropatía destructiva
CondrocalcinosisErosiones
Geodas
INFLAMATORIO,predominio de
neutrófilos
1ª METATARSOFALÁNGICA
No tiene Muy +++ Muy - - -(MIR 02-03, 227)
Débil +BIRREFRINGENCIA
LÍQUIDOSINOVIAL
RADIOLOGÍA
LOCALIZACIÓNmás frecuente
DIAGNÓSTICO
EDADmás frecuente
PRESENTACIÓNCLÍNICA
FORMA delCRISTAL

Reumatología
Pág. 9
gota, pero sí para el control de los factores de riesgo cardiovascular que se asocian a ésta.
B. DISMINUCIÓN DE LA EXCRECIÓN RENAL DE ÁCIDO ÚRICO.Este mecanismo causa más del 90% de las hiperuricemias. El ácido úrico es filtrado, reabsorbido y secretado en los túbulos renales, por lo que fallos en cualquiera de los tres pasos pueden disminuir la eliminación renal.1. Disminución de la filtración glomerular. Se origina por dis-
minución del volumen extracelular (diuréticos) o por causas parenquimatosas. Los diuréticos son actualmente la causa identificada más frecuente de hiperuricemia.
La insuficiencia renal se acompaña de gota en el 1% de los casos y la poliquistosis renal en el 30%. El tratamiento susti-tutivo mediante hemodiálisis también aumenta la prevalencia de hiperuricemia, así como la precipitación de otro tipo de cristales.
2. Aumento de la reabsorción renal. Los diuréticos y la diabetes insípida producen secundariamente disminución de la filtración y un aumento en la reabsorción distal de ácido úrico. (MIR 01-02, 76; MIR 00-01F, 83)
3. Disminución de la secreción. Otros ácidos compiten con el úrico en los túbulos por su secreción.• Ácidos endógenos: la acidosis láctica, la cetoacidosis diabé-
tica o alcohólica, la inanición .• Ácidos exógenos: la ingesta de salicilatos ( <2g/día), pirazi-
namida, etambutol, ácido nicotínico, levodopa o ciclospo-rina.
RECUERDA
Los salicilatos en dosis >2g/día tienen efecto uricosúrico.
Otras causas de hiperuricemia de probable origen renal son el hipotiroidismo, el hiperparatiroidismo, el hipoparatiroidismo, el saturnismo y la sarcoidosis.
C. MECANISMOS COMBINADOS.Son aquellas situaciones en las que se origina tanto un aumento de la producción como una disminución de la excreción.
Son el déficit de glucosa 6 fosfatasa, el déficit de fructosa 1 fosfato aldolasa y el alcohol ( ya que aumenta la síntesis de uratos y produce hiperlactacidemia que compite por la secreción renal).
MANIFESTACIONES CLÍNICAS.La hiperuricemia puede cursar de forma asintomática o provocar la aparición de complicaciones. Cuanto mayor sea la concentración plasmática, mayor será la posibilidad de desarrollar gota.
La hiperuricemia asintomática no debe tratarse. Pero sí hay que indagar sobre su posible causa y tratarla, si es posible. La hiperu-ricemia puede asociarse con hipertensión, hipercolesterolemia, diabetes mellitus y obesidad, que deberán controlarse. (MIR 99-00, 123; MIR 94-95, 34; MIR 95-96, 3)
Hiperuricemia asintomática Artritis gotosa aguda
Gota intercrítica
( )sucesivos ataques de gota
Gota tofácea crónica
Figura 1. Historia natural de la gota.
A. ARTRITIS GOTOSA.Es la primera manifestación clínica de la gota. Suele debutar como un ataque monoarticular, de predominio en la articulación metatarsofalángica del dedo gordo del pie (la clásica podagra). Se manifiesta de forma brusca, como una intensa inflamación articular (roja y dolorosa) acompañada de hiperestesia (no soporta ni el roce de la sábana).
Se han identificado factores desencadenantes, entre los que destacan las enfermedades médicas graves, las intervenciones quirúrgicas, el alcohol, las comidas copiosas, los traumatismos y sobre todo los medicamentos.
En algunas ocasiones la artritis puede ser poliarticular, lo cual sucede sobre todo en la mujeres.
RECUERDA
Que tanto los ascensos como los descensos (sobre todo estos) de la concentración plasmática de ácido úrico pueden producir un ataque de gota. Es por ello que en la fase aguda no se deben utilizar fármacos hipouricemiantes.
DIAGNÓSTICO.Se confirma mediante la visualización en el líquido sinovial de cristales de urato monosódico intracelulares (en el interior de los PMN) con birrefringencia negativa.
Tras un primer ataque de gota, se pueden producir recidivas, de-nominándose el período entre ataques artríticos “gota intercrítica.”
TRATAMIENTO.Un ataque de gota agudo se resuelve espontáneamente en 3-10 días, y en sólo 2, si se trata farmacológicamente.
Lo primero es tener en reposo la articulación. Si no existe con-traindicación a su uso (hemorragia digestiva o insuficiencia renal) los fármacos de elección son los AINES y la colchicina.
La colchicina ha sido el fármaco más usado en el paciente que no posee un diagnóstico de certeza. Tradicionalmente, la hiperu-ricemia, asociada a artritis monoarticular aguda y con respuesta al tratamiento con colchicina, daba el diagnóstico de gota, pero actualmente se conoce que otras artropatías por microcristales también responden a la colchicina. NO desciende la uricemia y su uso sigue siendo de elección en la profilaxis de nuevas crisis de gota (disminuye la inflamación subclínica).
Los AINES son el tratamiento de elección de la gota aguda. Aunque se utiliza la indometacina, todos los AINES tienen una eficacia similar .
En los casos de pacientes donde la colchicina y los AINEs están contraindicados, se pueden utilizar los glucocorticoides intraarticu-lares. Pero para ello es imprescindible que el diagnóstico haya sido confirmado (MIR 01-02, 81, MIR 96-97, 109; MIR 96-97F, 92; MIR 95-96F, 170, MIR 97-98F, 208, MIR 95-96F, 153).
B. GOTA TOFÁCEA.Tras dejar evolucionar la gota durante largo tiempo y sin tratamien-to, se producen los tofos, que son agregados de cristales de urato monosódico rodeados por una reacción granulomatosa (reacción a cuerpo extraño) con gran capacidad erosiva. Se localizan en los codos, el pabellón auricular, la 1ª articulación metatarsofalángica, la mano y en los tendones (sobre todo en el aquíleo). Si no se restablecen los niveles normales de ácido úrico, los tofos pueden crecer. Si por el contrario se normaliza la uricemia, con el tiempo los tofos llegan a desaparecer. En la radiografía simple se observa que la gota crónica produce una deformación y destrucción articular y la aparición de erosiones óseas con un borde esclerótico (borde resaltado).
TRATAMIENTO.Todos los pacientes que han padecido ataques de gota, nefrolitiasis o han desarrollado una gota tofácea crónica deben seguir un trata-miento hipouricemiante. La reducción del ácido úrico plasmático por debajo de su concentración de saturación facilita la disolución de los cristales y, por lo tanto, la desaparición de los tofos.
La aparición de nuevos ataques de gota no está relacionada con la uricemia, sino con la presencia de cristales de urato monosódico. Por ello, es posible sufrir ataques de gota aunque los niveles séricos de ácido úrico sean normales. Los niveles de ácido úrico pueden disminuirse por dos mecanismos:1. Inhibición de la producción de ácido úrico: ALOPURINOL. Este
fármaco inhibe a la xantin oxidasa y se utiliza en aquellos pa-cientes que:- Poseen excreción renal normal, con >800mg/día o >600mg/
día (si dieta sin purinas) o- Existen antecedentes de nefrolitiasis o- El paciente tiene insuficiencia renal o- Existen depósitos tofáceos o nefropatía por urato.
El alopurinol potencia la acción de la ciclofosfamida, azatioprina y 6 mercaptopurina. Además, es muy típico la aparición de lesio-

miniMANUAL 1 CTO
Pág. 10
nes cutáneas por vasculitis. Otros efectos son alopecia, molestias digestivas y la toxicidad hepática o renal.
2. Aumento de la excreción renal: de uso restringido en España. SULFINPIRAZONA, BENZOBROMARONA y PROBENECID. Están contraindicados si existe insuficiencia renal (clearance <80ml/minuto), nefrolitiasis (pueden desencadenarla) o si hay tofos. Limitado su uso por sus efectos secundarios si la excreción renal es de <600-800mg/día. Producen como efectos adversos hipersensibilidad, erupciones cutáneas o molestias digestivas.
Nunca se comienza a tomar fármacos hipouricemiantes en el curso de un ataque agudo, ya que al reducir la uricemia provocan una liberación de uratos de los cristales que puede agravar el cuadro. Por ello, siempre se debe añadir al tto. hipouricemiante colchicina durante los primeros meses, para evitar que se produzcan nuevos brotes.
C. NEFROLITIASIS.Los cálculos de ácido úrico pueden preceder a la artritis gotosa, siendo la primera manifestación de la gota en el 40% de los casos. El aumento de la excreción de ácido úrico en la orina es el factor que más influye en la aparición de la litiasis, pero parece que tam-bién existe relación con las concentraciones plasmáticas de uratos. Además, el ácido úrico puede actuar como nido para que el oxalato cálcico pueda precipitar.
TRATAMIENTO.Además del tratamiento hipouricemiante con alopurinol, se debe aumentar la ingesta hídrica hasta conseguir una diuresis >2 litros/día (lo que facilita la eliminación de los cálculos de urato). Se utiliza bicarbonato sódico o acetazolamida para aumentar la solubilidad del ácido úrico al alcalinizar la orina. Como alternativa es posible utilizar el citrato potásico, sobre todo si los cálculos tienen sales cálcicas.
D. NEFROPATÍA POR URATO.Es un síntoma tardío de la gota tofácea crónica. Se define como una nefropatía intersticial producida por el depósito de cristales de urato monosódico en el parénquima renal, rodeados por una reacción inflamatoria de células gigantes e infiltración de linfocitos. Como consecuencia de la inflamación se provoca fibrosis.
Clínicamente se observan desde casos asintomáticos hasta proteinuria, hipertensión e insuficiencia renal (según avanza la afectación). Se debe realizar DD con una posible intoxicación crónica por plomo.
MUY IMPORTATNTE
Esta es de las únicas nefropatías intersticiales que cursan con hipertensión arterial (recuerda que las alteraciones intersticiales tienden a perder sales y agua).
TRATAMIENTO.Tratamiento hipouricemiante con alopurinol.
E. NEFROPATÍA POR ÁCIDO ÚRICO.Causa reversible de insuficiencia renal aguda, originada por el de-pósito masivo de cristales de ácido úrico en los túbulos renales y los conductos colectores, lo que obstruye el flujo de orina. La causa más frecuente es el tratamiento con citotóxicos de las leucemias y los linfomas, que provocan una rápida destrucción de las células malignas y consiguiente sobreproducción de úrico. Clínicamente debuta como una IRA oligúrica acompañada de hematuria. El cociente ácido úrico/creatinina >1(este parámetro sólo sirve para sugerir una nefropatía por ácido úrico, siendo su valor escaso para evaluar la sobreproducción de urato).
TRATAMIENTO.Lo más adecuado es evitar la aparición de la misma, lo que ha de-mostrado aumentar la supervivencia. Se debe aumentar el flujo de orina mediante hidratación iv y furosemida, diluyéndose así el ácido úrico. Para aumentar la solubilidad del úrico, se alcaliniza la orina mediante bicarbonato sódico o acetazolamida. Se suele administrar
alopurinol para disminuir la producción de ácido úrico y que así disminuya la cantidad que llega hasta el riñón.
3.2. Artritis debida a depósito de cristales de calcio.
1. CRISTALES DE PIROFOSFATO CÁLCICO DIHIDRATADO( CONDROCALCINOSIS).
El depósito de cristales de pirofosfato cálcico dihidratado se observa frecuentemente en ancianos, ya que su prevalencia aumenta con la edad.• La forma etiológica más frecuente es la idiopática.• Las formas familiares (AD) son raras y debutan a los 30-50 años
con afectación poliarticular incapacitante.• Menos del 10% de las condrocalcinosis se asocian al depósito
de otros productos metabólicos, que favorecen el depósito de pirofosfato. Entre ellas se encuentran las 4 “H”: hemocromatosis, hipofosfatasia, hiperparatiroidismo, hipomagnesemia.Otras en-fermedades metabólicas son la gota tofácea crónica, la ocronosis y el hipotiroidismo. Según la enfermedad que se sospecha, se solicitan las correspondientes pruebas bioquímicas. (MIR 04-05, 85; MIR 95-96F, 144; MIR 99-00F, 97; MIR 95-96, 59; MIR 97-98, 231; MIR 96-97, 106).
Clínicamente puede ser asintomática o cursar con una artritis aguda, una artritis crónica o una combinación de ambas.
Figura 2. Depósitos de pirofosfato cálcico intraarticular.
A. ARTRITIS AGUDA (PSEUDOGOTA).Episodios autolimitados de artritis, de comienzo brusco o gradual, con intensos signos inflamatorios acompañados de fiebre, lo que la hace indistinguible de la gota. La rodilla es la articulación más frecuentemente afectada, seguida de la muñeca. Los desencade-nantes son los mismos que para la gota y el diagnóstico se basa en la visualización de cristales débilmente birrefringentes positivos en el líquido sinovial (MIR 00-01F, 78; MIR 98-99F, 99; MIR 97-98, 234; MIR 99-00F, 94; MIR 94-95, 178).
B. ARTRITIS CRÓNICA (ARTROPATÍA POR PIROFOSFATO).Se observa principalmente en mujeres de más de 65 años. Se trata de una artropatía degenerativa y bilateral caracterizada por dolor crónico, rigidez y limitación de la movilidad, sobre todo en rodillas y muñecas. Debe diferenciarse de la artrosis primaria, en la que la afectación es asimétrica, de menor gravedad y acompañada de escasa o nula inflamación. Además, la artrosis no suele afectar a las articulaciones MTCF, las muñecas, codos, hombros y tobillos. Radio-lógicamente se observan calcificaciones lineales y finas en el cartílago hialino( imagen de doble contorno), en el cartílago fibroso( de forma densa y punteada), en los meniscos y en el ligamento triangular del carpo (MIR 03-04, 19). También se localizan más raramente en otros ligamentos y tendones. El diagnóstico de confirmación también se consigue por el análisis del líquido sinovial con la demostración de los cristales, siendo de ayuda los hallazgos radiológicos.

Reumatología
Pág. 11
TRATAMIENTO.Está basado en la aspiración intraarticular, junto con la administra-ción de AINES o la inyección intraarticular de corticoides. De esta forma, se controla el brote agudo en unos 10 días.
En brotes sucesivos, el tratamiento es la colchicina en dosis bajas, que yugula las crisis agudas y previene nuevas crisis.
2. ENFERMEDAD POR DEPÓSITO DE HIDROXIAPATITA CÁLCICA (HA).
La hidroxiapatita es el principal mineral del hueso y de los dientes y el responsable de la mayoría de las calcificaciones de partes blandas en el organismo. Aunque la mayoría de las calcificacio-nes son idiopáticas, pueden estar asociadas a una lesión tisular (situación en la que se definen como calcificaciones distróficas), a enfermedades del tejido conectivo (LES, dermatomiositis, es-clerodermia), a enfermedades metabólicas (hiperparatiroidismo, hiperfosfatemia, intoxicación por vitamina D, síndrome leche –alcalinos o sd. de Burnett, IRC, diabetes mellitus, hemodiálisis) donde se denominan calcificaciones metastásicas ó a trastornos neurológicos.
CLÍNICA.El depósito predomina sobre todo en las bolsas y los tendones y dentro o alrededor de las articulaciones de la rodilla, hombro, ca-dera y dedos de las manos. Suelen ser asintomáticos, pero también pueden cursar como una artritis aguda, una artropatía crónica, bursitis o periartritis.• Artritis: similar al ataque de gota. Se produce en raras ocasiones
y se cree que es producida por la liberación de cristales.• Artropatía crónica: aproximadamente se han detectado cristales
de HA en la mitad de los afectos de artrosis y se cree que su pre-sencia se correlaciona con la gravedad radiológica de la artrosis y con la producción de episodios de sinovitis aguda (nódulos de Heberden calientes).
• Periartritis calcificante: el hombro es la localización más fre-cuente. Suele ser asintomática o puede acompañarse de dolor crónico que aumenta al contraer el tendón calcificado (visible en la radiografía simple).
• Artropatía destructiva: se observa en mujeres de más de 60 años, localizándose en el hombro (hombro de Milwaukee) y la rodilla. Se trata de una forma rara de artropatía rápidamente destructiva que se acompaña de debilidad y rotura de las estructuras de sostén de la articulación, lo que provoca una movilidad anormal y deformidad. Es muy frecuente que el manguito de los rotadores esté roto en la afectación del hombro.
DIAGNÓSTICO
Se confirma mediante la visualización de pequeños cristales (es necesario utilizar el microscopio electrónico) sin birrefringencia en el líquido sinovial (tipo no inflamatorio) o en los tejidos, que tiñen con alizarina roja y tinción de Wright. La radiología refleja calcificaciones intra o periarticulares algodonosas con posibilidad de hallar signos de erosión o hipertrofia en el hueso adyacente.
TRATAMIENTO.La inflamación aguda se controla con AINEs, colchicina o corticoi-des intraarticulares. En los casos avanzados, se recurre a cirugía ortopédica.
3. ENFERMEDAD POR DEPÓSITO DE CRISTALES DE OXALATO CÁLCICO.
EL oxalato es el producto final del catabolismo del ácido ascórbico y algunos aminoácidos.
La oxalosis 1ª es un raro trastorno hereditario, que cursa con hiperoxalemia, insuficiencia renal y muerte antes de los 20 años. Mucho más frecuente es la oxalosis 2º, observada en los pacientes con nefropatía terminal que se encuentran o no en tratamiento dializador de la misma y a los que antiguamente se les administraba grandes dosis de ácido ascórbico ( vitamina C). La falta de excreción renal del ácido ascórbico por el riñón enfermo o por la diálisis, favorecen que la oxalemia alcance su punto de sobresaturación y que este material empiece a depositarse en forma de cristales en el cartílago, el hueso, los vasos, la piel, el riñón y el corazón. Cuando los cristales se des-prenden originan un artritis aguda y tenosinovitis indistinguible de la producida por otros microcristales. Si los depósitos permanecen en
las articulaciones durante largo tiempo, provocan una proliferación sinovial y posterior destrucción de la articulación.
DIAGNÓSTICO.Se basa en la demostración de cristales extracelulares con fuerte birrefringencia positiva y forma bipiramidal en un líquido sinovial no inflamatorio. La radiología muestra imágenes de condrocalci-nosis .
TRATAMIENTO.El trasplante hepático es el tratamiento de la oxalosis 1º. En la oxalosis 2ª, la artropatía presenta una mínima mejoría con la administración de colchicina, AINEs o corticoides intraarticulares. También se ha pro-bado que el aumento de la frecuencia de las sesiones de hemodiálisis produce cierta mejoría. Actualmente se evita en estos pacientes el uso de suplementos de ácido ascórbico.
TEMA 4. LUPUS ERITEMATOSO SISTÉMICO.
4.1. Epidemiología.
Es una enfermedad autoinmune de causa desconocida en la que se produce una lesión tisular por el depósito anormal de inmuno-complejos y por la presencia de gran cantidad de autoanticuerpos contra las estructuras orgánicas.
Prevalece en mujeres en edad fértil, siendo más frecuente en la raza negra (y también más grave, por ser habitual la afectación renal) (MIR 97-98, 239).
4.2. Etiopatogenia.
Diversos factores etiológicos han sido involucrados. Entre ellos destacan:1. Asociación con factores genéticos, como determinados haploti-
pos de HLA ( el DR2 y el DR3) y déficits de factores del comple-mento (C1q, C2 y C4). En ciertas familias existe predisposición a padecer LES.
2. Las hormonas sexuales (estrogénicas) también parecen estar en relación con la patogenia de esta enfermedad. El LES es casi exclusivo del sexo femenino y se ha demostrado que el meta-bolismo de dichas hormonas está alterado en estas pacientes. Además, se conoce la capacidad de los estrógenos para inducir tolerancia inmunológica (lo que facilitaría la formación de au-toanticuerpos que no son destruidos por el sistema inmune y originan el daño histológico).
3. Factores ambientales como la radiación solar UVB-UVA y algu-nos fármacos, pueden inducir la aparición del LES.
4. Factores inmunológicos. Es sabido que estas pacientes tienen fallos en la regulación de la inmunidad y en la eliminación de inmunocomplejos, con una excesiva tolerancia frente a los autoanticuerpos y a la formación de inmunocomplejos.
Factores PatogénicosAlteración de la respuesta inmune
Alteración del equilibrio CD4+/CD8+
(Aumento de los Cd4 respecto de los CD8)
Pérdida de supresión de linfocitos B
(debido a la disminución de la función de los CD8)
Alta Producción de anticuerpos
Figura 3. Etiopatogenia del LES.
4.3. Manifestaciones clínicas.
El LES puede afectar prácticamente a cualquier órgano o sistema. Es frecuente que la paciente sufra exacerbaciones intercaladas entre períodos de inactividad de la enfermedad.• Manifestaciones generales inespecíficas: (95%) representadas
por febrícula, anorexia, astenia y pérdida de peso.• Manifestaciones musculoesqueléticas(95%): casi todos los pacien-
tes presentan artralgias y mialgias. También es posible la afectación en forma de poliartritis no erosiva migratoria en las articulaciones
miniMANUAL 1 CTO
Pág. 12
proximales de la mano y rodillas, excesivamente dolorosa para los signos inflamatorios. Es muy característica la artropatía de Jac-coud, consistente en desviación cubital en ráfaga y reductible de la muñeca junto con deformidad en cuello de cisne de los dedos.
En un pequeño número de pacientes es posible encontrar una verdadera miositis inflamatoria y el 15% sufre necrosis avascular. (MIR 97-98, 224; MIR 96-97F, 94)
• Alteraciones hematológicas(85%): en la mayoría de los pacientes con lupus activo se detecta anemia de procesos crónicos (nor-mocítica-normocrómica). En un 10% hay anemia hemolítica Coombs positiva que responde a dosis altas de corticoides.
La leucopenia y la linfopenia son frecuentes, pero no aumentan la susceptibilidad a las infecciones ni necesitan tratamiento. La trombocitopenia leve también es común, y si se asocia con anemia hemolítica autoinmune, conforma el denominado síndrome de Evans.
No hay que olvidar la existencia del anticoagulante lúpico, que origina trombosis de repetición.• Manifestaciones cutáneas(80%): la fotosensibilidad se presenta
en el 70% de los pacientes e indica la relación patogénica de los rayos UVB-UVA en el LES. Son frecuentes las aftas orales y nasales dolorosas. Según el tipo de lesión predominante, el lupus se subdivide en 3 grupos:1. Lesiones agudas: la más característica es la erupción malar
en alas de mariposa, una erupción eritematosa en el puente nasal y las mejillas que cura sin dejar cicatriz. También son frecuentes las erupciones exantemáticas maculopapulosas en zonas fotoexpuestas, que indican una exacerbación de la enfermedad.
2. Lesiones subagudas (LECS: lupus eritematoso cutáneo subagudo): pertenecen a un subgrupo de la enfermedad, donde dominan el cuadro las lesiones eritematosas en zo-nas fotoexpuestas. En dichas lesiones puede predominar la descamación (psoriasiformes) o la confluencia (polícíclico anular) y no dejan cicatriz al curar. Se asocia a fatiga y artritis, pero no hay afectación renal ni del SNC. Está relacionado con autoanticuerpos anti Ro y anti DNA monocatenario(ss). (MIR 04-05, 82, 142; MIR 00-01F, 150)
3. Lesiones crónicas o lupus discoide: es la forma más frecuente de presentación del lupus cutáneo y aparece en un 20% de los pacientes afectos de LES. Es muy rara la evolución del lupus cutáneo hacia la forma sistémica. Se manifiesta como lesiones atróficas y cicatriciales que producen pérdida de los anejos y desfiguraciones. Se localizan en el cuero cabelludo (alopecia irreversible), cara y resto de zonas expuestas al sol. Las lesiones son circulares con borde eritematoso elevado, descamación, taponamiento folicular y telangiectasias. Es posible la afecta-ción del panículo adiposo (lupus profundo)
• Alteraciones neurológicas (60%): cualquier región del siste-ma nervioso puede estar afectada.La disfunción cognitiva leve es la manifestación más frecuente y las convulsiones y la psicosis forman parte de los criterios diagnósticos del LES. Aunque el diagnóstico es clínico, la RMN puede detectar tanto lesiones agudas como crónicas. En el LCR existe aumento de las proteínas y las células, detectándose a veces bandas oligoclonales. Se cree que los mecanismos patogénicos de estas alteraciones son la vasculitis o el daño directo tisular por autoanticuerpos.
• Manifestaciones cardiopulmonares(60%): la pleuritis bilateral es la alteración pulmonar más frecuente. La causa más frecuente de infiltrados pulmonares es la infección, mucho más habitual que la neumonitis lúpica (que cursa de forma similar a una neumonía infecciosa).
En cambio, la hemorragia masiva alveolar por vasculitis de pe-queño vaso es la manifestación que más mortalidad ocasiona.
Respecto a las alteraciones cardíacas, la pericarditis es la más común de todas. Muy característica es la endocarditis de Libman-Sacks, que se visualiza mediante el ecocardiograma transesofágico y puede producir insuficiencia de válvulas iz-quierdas.
• Afectación renal(50%): prácticamente todos los pacientes con LES tienen alteraciones en la biopsia renal. Sin embargo, la nefropatía clínica sólo se objetiva en un 50% de los pacientes y su presencia oscurece el pronóstico de la enfermedad. Se correlaciona con los títulos de anticuerpos anti-ADNds y con el descenso marcado del complemento (C3, C4 y CH50).
Tabla 8. Hallazgos reversibles/irreversibles en la nefritis lúpica.
����������� �������������
raluremolgsisorceN sisorelcsE
selailetipesanulimeS sasorbifsanulimeS
soirotamalfnisodartlifnIselaicitsretni
lacitsretnisisorbiF
etnazitorcensitilucsaV ralubutaifortA
Las formas anatomopatológicas de GN lúpica se clasifican en 6 categorías y es frecuente la evolución de una a otra forma. En todas ellas la IFD demuestra depósitos compuestos por una mezcla va-riable de inmunoglobulinas (IgG, IgM e IgA) y complemento (C1q, C4 y C3) con un patrón granular. Esto hace creer que el mecanismo de lesión tisular sea el depósito de inmunocomplejos circulantes y la activación posterior del complemento.
Tabla 9. Formas de nefropatía lúpica.
������������� ����������������� ������ ���� �������������������!���
������ ���� �"��������# �� �����������"�����
$� lamroN laignasemnóicarefilorPralulecnóicarefilorP
lailetodneylaignasemairatnemgesylacof
ralulecnóicarefilorPlailetodneylaignasemnéibmatseceva(asufid)sanulimesnoclailetipe
osufidotneimasorgnE•.ralipacderapaled
.laignasemnóicarefilorP•
��%&�selaignasemsotisópeD
)sosacse(seralunargsotisópeD
selaignasem
selailetodnebussotisópeDsasayoignasemne
seralipac
selailetodnebussotisópeDsasayoignasemne
seralipac
selailetipebussotisópeD)sekips(
���$ — — —
.lacofsisorceN•.ediorbifsisorceN•
.seralulecsanulimeS•.erbmalaedsasA•
.socinílixotamehsopreuC•
—
������ lamronlanernóicnuF
etneucerfsámamroFsetneicapnesocitámotnisa
—neetneucerfsámamroF•.socitámotnissetneicap
.ocitsónorproepedaL•—
lamronlanernóicnuFlamronlanernóicnuF
)%57(laneraicneicifusnI
lamronlanernóicnuFlaicini
)NSorar(airunietorP airunietorP � )%03NS( airunietorP �� )%09NS( airunietorP �� )%09NS(
Reumatología
Pág. 13
Clase I: lesión glomerular lúpica mínima. Se caracteriza por la práctica normalidad clínica e histológica.• En el MO hay escasos o nulos cambios.• IF. Revela depósitos ocasionales en el mesangio exclusivamente.
Clínicamente el filtrado glomerular es normal.Puede haber hematuria, proteinuria.
Clase II: glomerulonefritis lúpica mesangial. Se objetiva pro-teinuria moderada y hematuria en casi la mitad de los pacientes y rara vez produce síndrome nefrótico con hipertensión arterial. El filtrado glomerular casi siempre es normal. Es la forma más frecuente en enfermos asintomáticos, con supervivencia mayor del 90% a los 5 años.• MO. Se visualiza esclerosis mesangial (tipo IIA) y/o proliferación
mesangial difusa (tipo IIB).• IF. Revela depósitos mesangiales de IgG, IgM Ig A y comple-
mento.
Clase III: glomerulonefritis lúpica proliferativa focal y segmen-taria (afectación de <50% de los glomérulos) Hay proteinuria en todos los pacientes y síndrome nefrótico en un tercio de ellos. El filtrado glomerular se altera en el 20-25% y es frecuente su evolución a GN proliferativa difusa.• MO. Proliferación celular mesangial focal y segmentaria asociada
con necrosis y aumento difuso de la matriz mesangial. Debe distinguirse de la GESF (forma de GN primaria) en la que hay esclerosis, pero no proliferación celular.
• IF. Hay depósitos subendoteliales en mesangio y asas capila-res.
Clase IV: glomerulonefritis lúpica proliferativa difusa(afectación de >50% de los glomérulos). Es la forma más frecuente en los enfer-mos sintomáticos, y la de peor pronóstico. Los pacientes presentan un sedimento urinario activo, proteinuria importante, hipertensión y alteración de la función renal en el 50% de los casos en el mo-mento del diagnóstico. Es de muy mal pronóstico, con evolución a insuficiencia renal terminal en un 20% de los pacientes a pesar del tratamiento.• MO. Existe proliferación celular difusa mesangial y endotelial
con amplia interposición de células mesangiales en la pared del capilar periférico. Hay necrosis fibrinoide, cuerpos hematoxilíni-cos y «asas de alambre» (capilares engrosados por el aumento de la membrana basal y la interposición del mesangio). Se pueden encontrar semilunas y diversos grados de lesión crónica.
• ME. Aparecen depósitos granulares de predominio subendotelial y mesangial en casi todas las asas capilares.
Clase V: glomerulonefritis lúpica membranosa. La mayoría (90%) presentan proteinuria en rango nefrótico. Aunque en prin-cipio la función renal es normal, con el paso de los años sufre un deterioro progresivo.• MO. Engrosamiento de la pared capilar. A veces existe prolifera-
ción mesangial (algo inusual en la GN membranosa idiopática o primaria).
• IFD y ME. Encontramos depósitos electrodensos de predominio subepitelial asociados con reacción de la membrana basal en forma de espigas (igual que en la GN membranosa idiopáti-ca).
Clase VI: glomerulonefritis esclerosante o terminal. Es el estadio final. Hay glomeruloesclerosis difusa, escasos depósitos y extensa afectación tubulointersticial.
La biopsia renal debe realizarse cuando se sospecha que su resultado puede variar el tratamiento.• Manifestaciones gastrointestinales (45%):predominan las naú-
seas, vómitos y la diarrea. La vasculitis intestinal es la manifesta-ción clínica más peligrosa, pero el LES también puede producir peritonitis aguda estéril, ascitis y pancreatitis. Las transaminasas están elevadas habitualmente (no implica lesión hepática), pero se suelen normalizar con el tiempo. (MIR 98-99F, 102)
• Alteraciones vasculares (15%): los anticuerposantifosfolípido y anticardiolipina suelen producir trombosis en vasos de cual-quier calibre. A largo plazo, el tratamiento esteroide produce una aterosclerosis acelerada.
4.4. Diagnóstico.
El diagnóstico es clínico y se establece al reunirse al menos 4 de los 11 criterios diagnósticos propuestos.
La presencia de autoanticuerpos es de gran utilidad tanto para el diagnóstico como por las asociaciones clínicas que éstos conllevan. (MIR 99-00F,241; MIR 02-03, 230; MIR 96-97F, 99; MIR 94-95, 184).
Analíticamente se suele observar una anemia de procesos crónicos, una elevacion de la VSG y un descenso de los niveles de complemento coincidiendo con los brotes de la enfermedad.
Puede detectarse FR y crioglobulinas, así como hipergammag-lobulinemia, a pesar de existir un característico déficit de IgA.
Tabla 10. Autoanticuerpos.
SOPREUCITNAOTUA AICNELAVERP NOICAICOSA
ANAne%4(%89
)largcalbop-
sD-ITNA %07
-SELedocifícepsEeddadivitcanocnóicalerroCedaicneserpydademrefneal
sitirfen
mS-ITNA %03aledsocifícepsesámsoL
dademrefne
PNR-ITNA %04 .duanyaRysonamedamedE
OR-ITNA %03
SEL,ovitagenANASELSEL,onaicnaSEL,odugabus
ticifédSEL,latanoenatnemuA.otnemelpmoc
sitirfenedogseir
AL-ITNA %51leeyunimsiD.º1nergöjSdS
sitirfenedogseir
ANOTSIHITNAne%59(%07
SEL)socamráf
SELledsocifícepseyuMsocamráfropodicudni
P-ITNA)selamosobirritna(
%02senoicatsefinaM
sisocisP(sacirtáiuqisporuen)nóiserpedy
Tabla 11. Criterios diagnósticos del LES.
• Eritema malar.• Lupus discoide.• Fotosensibilidad.• Úlceras orales o nasofaríngeas.• Artritis.• Serositis (pleuritis o pericarditis).• Enfermedad renal (proteinuria o cilindros celulares).• Enfermedad neurológica (psicosis o convulsiones).• Alteración hematológica.
- Leucopenia <4000/mm3.- Linfopenia <1500/mm3.- Trombopenia <100.000 plaquetas /mm3.- Anemia hemolítica.
• Trastorno inmunológico: anticuerpos antiDNAds, antiSm, anti fosfolípido o cualquier combinación de ellos.
• Anticuerpos antinucleares.
SITUACIONES ESPECIALES.1. Lupus inducido por fármacos. Fármacos como la procainamida y la hidralacina pueden oca-
sionar un síndrome parecido al LES. Las diferencias con la forma idiopática se reflejan en el cuadro. (MIR 96-97F, 87)
2. Lupus cutáneo subagudo. Se asocia a positividad en los ANA, antiRo y antiLa, así como
al haplotipo DR3. Son comunes los síntomas inespecíficos y musculoesqueléticos, pero no la afectación de órganos vitales.
3. Lupus y embarazo Es frecuente que las pacientes afectas de lupus sufran ame-
norrea. El 30% presenta abortos espontáneos, muerte fetal y prematuridad, que se suelen asociar a la existencia de un

miniMANUAL 1 CTO
Pág. 14
síndrome antifosfolípido o a nefritis activa. Se aconseja evitar la gestación hasta que la enfermedad se encuentre totalmente controlada.
Deben utilizarse con cuidado los fármacos empleados para el tratamiento del lupus. Se suele evitar el uso de los AINEs, la warfa-rina, los corticoides de larga vida media, la cloroquina y todos los inmunosupresores, a excepción de la ciclosporina. Los demás medi-camentos se deben administrar con precaución. (MIR 99-00, 42)
4. Lupus neonatal. Se produce en <5% de los recién nacidos de madres portadoras
de AntiRo y/o AntiLa. Suele manifestarse con la aparición de:- Lesiones cutáneas similares al lupus cutáneo subagudo.- Bloqueo cardíaco completo.- Alteraciones hematológicas. Los anticuerpos permanecen durante 6 meses, negativizándose
con el paso del tiempo. No hay mayor riesgo de padecer la enfer-medad en la edad adulta.
PRONÓSTICO.En la fase inicial del lupus, la mortalidad es debida a la propia actividad de la enfermedad. Destacan la afectación renal, las in-fecciones y la alteración del SNC como causas más frecuentes de fallecimiento.
Cuando el LES lleva años de evolución, la muerte es secundaria a la aterosclerosis producida por el tratamiento corticoideo y la inflamación persistente, que dañan el tejido endotelial y originan IAM s y ACVs.
Tabla 13. Tratamiento del lupus en función de su gravedad.(MIR 98-99, 86; MIR 94-95, 179; MIR 99-00F, 257)
,sitirtra(sevelsenoicatsefinaM)...sitisores,erbeif
sENIA + sisodasediocitrocsajab
saenátucsenoicatsefinaM.ralosnóicisopxealrativE•
socipótsediocitroC• +.aniuqorolcixordih
nóicatcefa(sevargsenoicatsefinaM)...,laner,acigóloruen
satlasisodasediocitroC +seroserpusonumni
4.5. Síndrome antifosfolípido.
Este síndrome aparece en la mayoría de los casos asociado al lupus eritematoso sistémico, aunque puede encontrarse en otras enferme-dades autoinmunes. En los casos en los que aparece aisladamente, el síndrome antifosfolípido se define como primario.
Patogénicamente se cree que los anticuerpos que se detectan en este síndrome se unen a los fosfolípidos de la membrana del endotelio y las plaquetas, produciendo las características trom-bosis.
La clínica está dominada por la aparición de trombosis ar-teriales y venosas, siendo más frecuentes las trombosis venosas profundas de las extremidades inferiores, que pueden originar secundariamente tromboembolismos pulmonares.
La otra característica importante del sd. AFL son los abortos espontáneos de repetición, sobre todo en el 2-3º trimestre. Se piensa que son provocados por las alteraciones en la circulación y trombosis de los vasos placentarios y fetales.
Pueden asociarse otro tipo de síntomas, como el livedo reticu-laris, la hipertensión pulmonar y alteraciones neurológicas tipo sd Guillain Barré. (MIR 97-98, 227; MIR 01-0, 51).
Tabla 14. Criterios diagnósticos del SAF.
Manifestaciones clínicas:• Trombosis arterial o venosa.• Abortos de repetición.
Alteraciones analíticas:• Anticuerpos anticardiolipina IgG o IgM.• Anticoagulante lúpico.
El diagnóstico se establece cuando se presente un criterio clínico acompañado de un criterio analítico.
DIAGNÓSTICO.Debe cumplirse al menos un criterio clínico y otro analítico. (MIR 98-99F, 97; MIR 97-98F; 213)
La presencia de anticuerpo antifosfolípido es imprescindicle para diagnosticar el síndrome. Se pueden detectar mediante prue-bas coagulométricas, reagínicas o inmunológicas:• Pruebas reagínicas (VDRL, RPR): es característica la positivi-
dad de las serologías sifilíticas, que se explica por una reacción cruzada de estos anticuerpos anticardiolipina con el material utilizado para realizar esta prueba.
• Pruebas coagulométricas (anticoagulante lúpico): aunque in vitro estos anticuerpos prolongan el TTPA aun añadiendo plasma fresco, in vivo producen trombosis.
• Prueba inmunológica: mediante la técnica de ELISA se detectan anticuerpos anticardiolipina IgG e IgM.
TRATAMIENTO (MIR 03-04, 13; MIR 02-03, 218). Se utiliza de elección la anticoagulación en rango alto, con INR 2.5-3. Si a pesar de estar anticoagulado, se presentan nuevos episodios trombóticos, será necesario incrementar la dosis de anticoagulación y añadir antiagregantes.
En el caso de gestación, se deberán seguir las siguientes pautas:• Si no aborto ni trombosis previas, solamente anticuerpos posi-
tivos: AAS o nada.• Si anticuerpos positivos y abortos previos: AAS + HBPM.
TEMA 5. ARTRITIS REUMATOIDE.
5.1. Definición.
Es la enfermedad inflamatoria articular crónica más frecuente en la población general ( 1%). Predomina en el sexo femenino (3/1) y la incidencia es mayor entre los 40-60 años.
5.2. Etiopatogenia.
Se considera que la AR se desarrolla en sujetos genéticamente pre-dispuestos por la acción de un factor ambiental .
Se relaciona en el 70% de los casos con el haplotipo DR4, lo que indica cierta predisposición genética.
DR4- asociación a la artritis reumatoide.DR3-asociación con toxicidad renal por sales de oro y penicila-
mina y toxicidad cutánea y hematológica por sales de oro.Histológicamente se observa una sinovitis crónica con hiper-
plasia e hipertrofia de las células sinoviales, acompañada por un infiltrado de monocitos y linfocitos T (sobre todo CD4 y en menor medida CD8 citotóxicos). Ambas poblaciones expresan el antígeno de activación inicial CD69.
En la membrana sinovial se forma factor reumatoide y otras inmunoglobulinas policlonales, que determinan la formación de inmunocomplejos in situ.
Tabla 12. Lupus inducido por fármacos.
AÍGOLOIMEDIPE ACINÍLC ACITÍLANA OICINI
SELserejumne1/9soña03-02
odipár/otnelrodaliteca,aenátuc,asores,ralucitra
CNS,lanersámol(sdANDitnA
mSitnA)ocifícepseotneL
ODICUDNISUPULotnelrodaliteca1/1,soña05
4RD,seralucitrasamotnís%59
sosoresysoenátucsamotnísANA
anotsihitnAogralnuedséupsed,ocsurB
otneimatartedopmeit

Reumatología
Pág. 15
Figura 4. Etiopatogenia de la artritis reumatoide.
5.3. Manifestaciones clínicas.
La AR es una poliartritis crónica bilateral, simétrica y erosiva que predomina en las pequeñas articulaciones de manos y pies.
En el 70% de los pacientes el inicio es insidioso, estando domi-nado por las manifestaciones generales inespecíficas y apareciendo posteriormente la sinovitis. En un porcentaje de los casos debuta de forma aguda como una poliartritis acompañada de fiebre, astenia, adenopatías y esplenomegalia. (MIR 97-98F, 201).
Figura 5. Frecuencia de afectación articular en la AR.
1. AFECTACIÓN ARTICULAR. Las articulaciones más frecuentemente afectadas son las IFP y las metacarpofalángicas, siendo raro la participación de las IFD ( lo que la diferencia de la artrosis) (MIR 95-96F, 146). La clínica está domi-nada por el dolor, la tumefacción y la disminución de la movilidad, causadas por la inflamación sinovial y el aumento del líquido sino-vial en la articulación, que distiende las estructuras y provoca una actitud en flexo (posición en la que el volumen articular es máximo).El dolor se intensifica a la presión y con el movimiento (típico de las inflamaciones) y suele acompañarse de calor. Característica-mente se asocia rigidez tras el reposo, de predominio matutino, y que requiere de al menos una hora para desaparecer. Este hallazgo se suele correlacionar con la actividad de la enfermedad (a mayor duración de la rigidez, más gravedad de la AR).
La extensión a las vainas tendinosas de la sinovitis produce tendinitis, que deteriora las estructuras llegando a producir roturas de los tendones. La inflamación e hipertrofia de la sinovial crea en ciertas localizaciones un compromiso de espacio con las demás estructuras, que típicamente se observa con la formación de un síndrome del túnel carpiano por atrapamiento del nervio mediano secundario a la tendosinovitis de los flexores de la mano. Si aparece dolor y tumefacción en el hueco poplíteo, puede deberse a la exten-
sión de la inflamación sinovial hacia la zona poplítea (quiste roto de Baker), cuadro que hay que diferenciar de una tromboflebitis y que se diagnostica mediante ecografía.
La afectación axial suele limitarse a la columna cervical y puede producir subluxación atloaxoidea (por laxitud del ligamento trans-verso del atlas) con dolor en el occipucio y rara vez compresión de la médula espinal. Tampoco suelen afectarse las articulaciones sacroilía-cas, a diferencia de las espondiloartropatías (MIR 97-98, 236).
Figura 6. Deformidades de las falanges en la AR.
Tabla 15. Deformidades en las falanges.
PFI DFI
ensicedolleuC nóisnetxerepiH nóixelF
lajo-nótobnE nóixelF nóisnetxE
ollitramedodeD nóisnetxE nóixelF
La persistencia del proceso inflamatorio favorece la destrucción articular y la aparición de deformidades. Las más características son la desviación en ráfaga cubital de los dedos y la desviación radial de la muñeca, los dedos en cuello de cisne, en botón o en ojal, en martillo. En pies, la lesión más característica es el hundimiento del antepié. (MIR 97-98, 228).
La incapacidad funcional deriva con el paso del tiempo a osteo-porosis y atrofia muscular.
Las articulaciones afectadas por la AR son más susceptibles a la infección, siendo el germen más frecuente el Staphylococcus aureus. Siempre que se produzca una monoartritis aguda en el curso de una AR, será necesario descartar la existencia de una artritis séptica la destrucción articular predispone a la invasión bacteriana).
2. MANIFESTACIONES EXTRAARTICULARES.A veces éstas son el signo principal de actividad de la enfermedad. Ha-bitualmente se relacionan con títulos altos de factor reumatoide.a) Los nódulos reumatoides (20-30% de pacientes con AR).Casi ex-
clusivo de pacientes seropositivos (FR positivos). Pueden aparecer en cualquier órgano, pero habitualmente se localizan en zonas sujetas a presión mecánica como la bolsa olecraniana y el codo, el tendón de Aquiles y el occipucio.
Son de consistencia firme, están adheridos a planos profundos, y suelen ser indoloros. El fenómeno inicial parece ser una vas-culitis focal (MIR 95-96, 54).
b) La vasculitis reumatoide. Es más frecuente en AR grave y se asocia con títulos altos de FR y disminución de los niveles sérico de complemento (por consumo periférico para la formación de inmunocomplejos). La forma más grave es la vasculitis necrotizante, que puede afectar a diversos órganos, como la piel (necrosis y ulceración), el sistema nervioso ( polineuropatía o mononeuritis múltiple) o el mesenterio (infarto visceral).
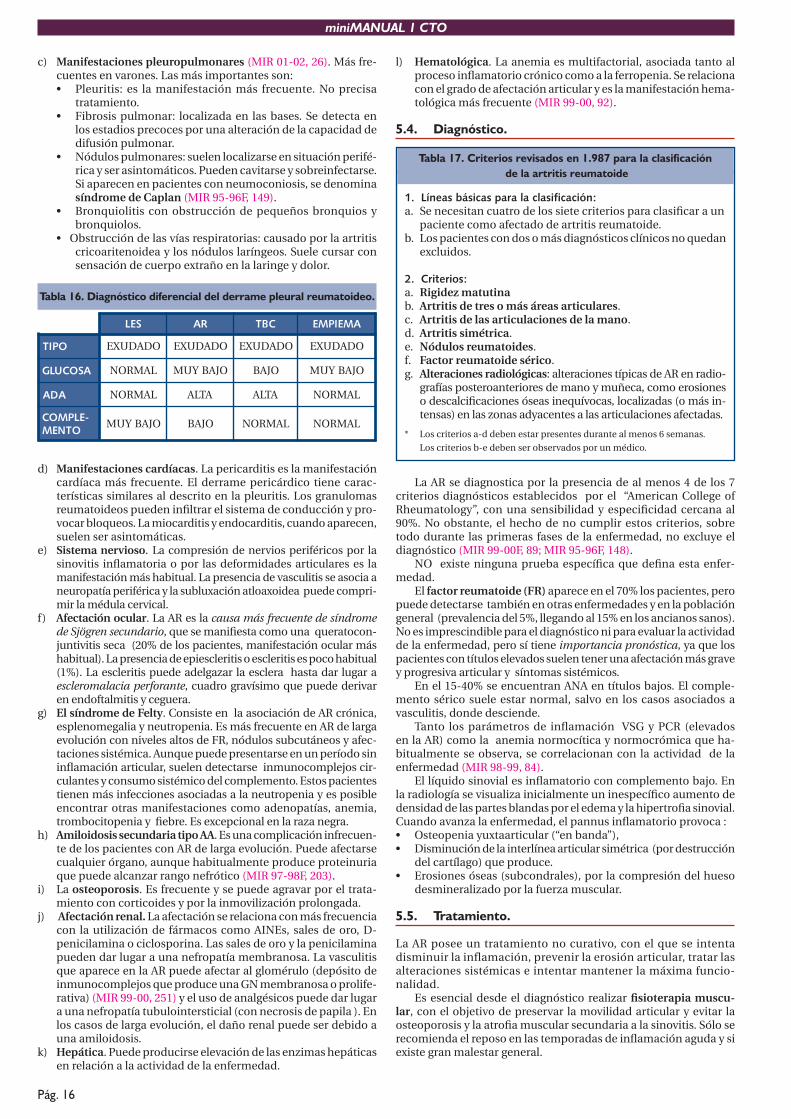
miniMANUAL 1 CTO
Pág. 16
c) Manifestaciones pleuropulmonares (MIR 01-02, 26). Más fre-cuentes en varones. Las más importantes son:• Pleuritis: es la manifestación más frecuente. No precisa
tratamiento.• Fibrosis pulmonar: localizada en las bases. Se detecta en
los estadios precoces por una alteración de la capacidad de difusión pulmonar.
• Nódulos pulmonares: suelen localizarse en situación perifé-rica y ser asintomáticos. Pueden cavitarse y sobreinfectarse. Si aparecen en pacientes con neumoconiosis, se denomina síndrome de Caplan (MIR 95-96F, 149).
• Bronquiolitis con obstrucción de pequeños bronquios y bronquiolos.
• Obstrucción de las vías respiratorias: causado por la artritis cricoaritenoidea y los nódulos laríngeos. Suele cursar con sensación de cuerpo extraño en la laringe y dolor.
Tabla 16. Diagnóstico diferencial del derrame pleural reumatoideo.
SEL RA CBT AMEIPME
OPIT ODADUXE ODADUXE ODADUXE ODADUXE
ASOCULG LAMRON OJABYUM OJAB OJABYUM
ADA LAMRON ATLA ATLA LAMRON
-ELPMOCOTNEM
OJABYUM OJAB LAMRON LAMRON
d) Manifestaciones cardíacas. La pericarditis es la manifestación cardíaca más frecuente. El derrame pericárdico tiene carac-terísticas similares al descrito en la pleuritis. Los granulomas reumatoideos pueden infiltrar el sistema de conducción y pro-vocar bloqueos. La miocarditis y endocarditis, cuando aparecen, suelen ser asintomáticas.
e) Sistema nervioso. La compresión de nervios periféricos por la sinovitis inflamatoria o por las deformidades articulares es la manifestación más habitual. La presencia de vasculitis se asocia a neuropatía periférica y la subluxación atloaxoidea puede compri-mir la médula cervical.
f) Afectación ocular. La AR es la causa más frecuente de síndrome de Sjögren secundario, que se manifiesta como una queratocon-juntivitis seca (20% de los pacientes, manifestación ocular más habitual). La presencia de epiescleritis o escleritis es poco habitual (1%). La escleritis puede adelgazar la esclera hasta dar lugar a escleromalacia perforante, cuadro gravísimo que puede derivar en endoftalmitis y ceguera.
g) El síndrome de Felty. Consiste en la asociación de AR crónica, esplenomegalia y neutropenia. Es más frecuente en AR de larga evolución con niveles altos de FR, nódulos subcutáneos y afec-taciones sistémica. Aunque puede presentarse en un período sin inflamación articular, suelen detectarse inmunocomplejos cir-culantes y consumo sistémico del complemento. Estos pacientes tienen más infecciones asociadas a la neutropenia y es posible encontrar otras manifestaciones como adenopatías, anemia, trombocitopenia y fiebre. Es excepcional en la raza negra.
h) Amiloidosis secundaria tipo AA. Es una complicación infrecuen-te de los pacientes con AR de larga evolución. Puede afectarse cualquier órgano, aunque habitualmente produce proteinuria que puede alcanzar rango nefrótico (MIR 97-98F, 203).
i) La osteoporosis. Es frecuente y se puede agravar por el trata-miento con corticoides y por la inmovilización prolongada.
j) Afectación renal. La afectación se relaciona con más frecuencia con la utilización de fármacos como AINEs, sales de oro, D-penicilamina o ciclosporina. Las sales de oro y la penicilamina pueden dar lugar a una nefropatía membranosa. La vasculitis que aparece en la AR puede afectar al glomérulo (depósito de inmunocomplejos que produce una GN membranosa o prolife-rativa) (MIR 99-00, 251) y el uso de analgésicos puede dar lugar a una nefropatía tubulointersticial (con necrosis de papila ). En los casos de larga evolución, el daño renal puede ser debido a una amiloidosis.
k) Hepática. Puede producirse elevación de las enzimas hepáticas en relación a la actividad de la enfermedad.
l) Hematológica. La anemia es multifactorial, asociada tanto al proceso inflamatorio crónico como a la ferropenia. Se relaciona con el grado de afectación articular y es la manifestación hema-tológica más frecuente (MIR 99-00, 92).
5.4. Diagnóstico.
Tabla 17. Criterios revisados en 1.987 para la clasificación de la artritis reumatoide
1. Líneas básicas para la clasificación: a. Se necesitan cuatro de los siete criterios para clasificar a un
paciente como afectado de artritis reumatoide.b. Los pacientes con dos o más diagnósticos clínicos no quedan
excluidos.
2. Criterios:a. Rigidez matutinab. Artritis de tres o más áreas articulares.c. Artritis de las articulaciones de la mano.d. Artritis simétrica.e. Nódulos reumatoides.f. Factor reumatoide sérico.g. Alteraciones radiológicas: alteraciones típicas de AR en radio-
grafías posteroanteriores de mano y muñeca, como erosiones o descalcificaciones óseas inequívocas, localizadas (o más in-tensas) en las zonas adyacentes a las articulaciones afectadas.
* Los criterios a-d deben estar presentes durante al menos 6 semanas.
Los criterios b-e deben ser observados por un médico.
La AR se diagnostica por la presencia de al menos 4 de los 7 criterios diagnósticos establecidos por el “American College of Rheumatology”, con una sensibilidad y especificidad cercana al 90%. No obstante, el hecho de no cumplir estos criterios, sobre todo durante las primeras fases de la enfermedad, no excluye el diagnóstico (MIR 99-00F, 89; MIR 95-96F, 148).
NO existe ninguna prueba específica que defina esta enfer-medad.
El factor reumatoide (FR) aparece en el 70% los pacientes, pero puede detectarse también en otras enfermedades y en la población general (prevalencia del 5%, llegando al 15% en los ancianos sanos). No es imprescindible para el diagnóstico ni para evaluar la actividad de la enfermedad, pero sí tiene importancia pronóstica, ya que los pacientes con títulos elevados suelen tener una afectación más grave y progresiva articular y síntomas sistémicos.
En el 15-40% se encuentran ANA en títulos bajos. El comple-mento sérico suele estar normal, salvo en los casos asociados a vasculitis, donde desciende.
Tanto los parámetros de inflamación VSG y PCR (elevados en la AR) como la anemia normocítica y normocrómica que ha-bitualmente se observa, se correlacionan con la actividad de la enfermedad (MIR 98-99, 84).
El líquido sinovial es inflamatorio con complemento bajo. En la radiología se visualiza inicialmente un inespecífico aumento de densidad de las partes blandas por el edema y la hipertrofia sinovial. Cuando avanza la enfermedad, el pannus inflamatorio provoca :• Osteopenia yuxtaarticular (“en banda”), • Disminución de la interlínea articular simétrica (por destrucción
del cartílago) que produce.• Erosiones óseas (subcondrales), por la compresión del hueso
desmineralizado por la fuerza muscular.
5.5. Tratamiento.
La AR posee un tratamiento no curativo, con el que se intenta disminuir la inflamación, prevenir la erosión articular, tratar las alteraciones sistémicas e intentar mantener la máxima funcio-nalidad.
Es esencial desde el diagnóstico realizar fisioterapia muscu-lar, con el objetivo de preservar la movilidad articular y evitar la osteoporosis y la atrofia muscular secundaria a la sinovitis. Sólo se recomienda el reposo en las temporadas de inflamación aguda y si existe gran malestar general.

Reumatología
Pág. 17
TRATAMIENTO FARMACOLÓGICO.a) Antiinflamatorios no esteroideos (AINE). Estos fármacos reali-
zan su acción analgésica y antiinflamatoria bloqueando, entre otros mecanismos, la producción de prostaglandinas por parte de la enzima ciclooxigenasa(COX1 o constitutiva y COX 2 o inducida). Aunque clásicamente se ha utilizado el AAS , se sabe que también se pueden utilizar otros AINEs. NO interfieren en la evolución a largo plazo de la enfermedad.
El efecto antiinflamatorio se observa desde el primer día de administración y desaparece rápidamente al suspenderlo.
Muchos de los efectos secundarios de estos fármacos se producen por la inhibición de la ciclooxigenasa 1, como son la irritación gástrica, hiperazoemia, disfunción plaquetaria, exacerbación de rinitis alérgica y asma. Es por ello que se ha aprobado el uso de los ICOX-2 (inhibidores selectivos de la COX-2 ) para minimizar estos efectos secundarios en pacientes ancianos que requieran dosis elevadas de AINES. Recuerda que actualmente existen estudios que indican el aumento de riesgo cardiovascular con el uso de estos fármacos, por lo que algunos ya han sido retirados del mercado preventivamente. Otros efectos, como la erupción cutánea o la depresión medular, no están relacionados con este mecanismo (MIR 96-97, 116).
b) Glucocorticoides. Las dosis bajas de esteroides por vía oral (por debajo de 7, 5 mg/día) ayudan de forma eficaz al control de los síntomas inflamatorios y recientemente se ha demostrado que pueden retrasar la aparición de las erosiones óseas. Los pulsos de corticoides en dosis altas se reservan para afectaciones vis-cerales graves.
c) Fármacos antirreumáticos modificadores de la enfermedad (FAME). Tienen la capacidad de producir una mejoría clínica y serológica (disminuyen los títulos de FR, de PCR y de VSG) y de retrasar la aparición de las alteraciones radiológicas. Se deben a empezar a utilizar sin demora desde que se confirma el diagnós-tico de AR. El metotrexate es actualmente el fármaco de elección. Su acción antiinflamatoria y analgésica es mínima, por lo que se requiere administrarlos junto con AINES. Los efectos beneficiosos suelen tardar semanas o meses en aparecer, y en el caso del me-trotexate, alcanzan su máximo a los 6 meses de tratamiento.
Este grupo comprende los antipalúdicos, la sulfasalazina y la auranofina, que son menos activos y menos tóxicos, por lo que se recomiendan en los pacientes con menor actividad, aunque los dos primeros actualmente están casi en desuso.
Cada uno de ellos produce una toxicidad considerable. La falta de respuesta o la aparición de toxicidad a algunos de estos agentes no impide que se produzca una respuesta beneficiosa frente a los demás (MIR 04-05, 79; MIR 02-03, 226; MIR 00-01, 79; MIR 97-98F, 205; MIR 96-97, 256; MIR 94-95, 182).
d) Inhibidores del TNF alfa. Tanto el etanercept (receptor TNF alfa tipo II unido a IgG1) como el infliximab (anticuerpo monoclonal quimérico frente a TNF alfa) controlan la sintomatología de la enfermedad en aquellos pacientes refractarios al tratamiento con fármacos modificadores de la enfermedad. Al interferir con el sistema inmune, en algunas ocasiones alteran sus funciones produciendo aumento en la incidencia de infecciones severas y aparición de anticuerpos anti-DNA, sin desarrollo concomitante de lupus .
AINES + Corticoides a dosis bajas
(hasta confirmación diagnóstica)
Añadir Metotrexate
Triple terapia (MTX + Hidroxiclorodiquina + Sulfasalacina)
o Leflunomida
(sólo o en combinación)
Si resultado insatisfactorio
añadir ANTI-TNF
Figura 7. Esquema terapéutico de la AR.
e) Tratamiento inmunosupresor. La azatioprina, la ciclofosfa-mida, la ciclosporina y la leflunomida han sido utilizadas en los pacientes con enfermedad severa que no responden a los fármacos modificadores de la enfermedad, o que presentan manifestaciones severas extraarticulares como vasculitis.
f) Cirugía. En las fases iniciales puede optarse por realizar sino-vectomías químicas para reducir la sintomatología y la progre-sión de la destrucción que el pannus inflamatorio produce en la articulación. También se puede utilizar la tenosinovectomía para evitar la rotura de los tendones y liberar estructuras com-primidas como en el síndrome del túnel carpiano.
En fases más avanzadas, cuando la articulación está irreversi-blemente destruida, es necesario realizar artroplastias (prefe-rentemente en rodilla y cadera).
5.6. Enfermedad de Still del adulto.
Es una forma de poliartritis asociada a manifestaciones sistémicas que afecta a adultos jóvenes con igual proporción en ambos sexos (MIR 03-04,23).
Se caracteriza por la aparición de fiebre alta en agujas, faringitis y rash eritematoso fugaz asalmonado. Suele acompañarse de mialgias, artralgias y oligo/poliartritis. El rash maculopapular suele coincidir con los picos febriles y puede dejar pigmentación residual.
Otras afecciones frecuentes son el derrame pleural, la pericar-ditis, la hepatoesplenomegalia y las adenopatías.
El diagnóstico es clínico, aunque se puede apoyar en pruebas serológicas, como FR negativo, ANA negativo con VSG elevado.
TEMA 6. ESPONDILOARTROPATÍAS. SERONEGATIVAS.
Las espondiloartropatías son un grupo de procesos que tienen ciertas manifestaciones clínicas en común, además de asociarse al alelo HLA-B27 (MIR 99-00, 116).
Tabla 18. Entidades incluidas dentro de la espondiloartritis.
• Espondilitis anquilosante.• Artropatía psoriásica.• Artritis reactiva.• Artritis de la enfermedad inflamatoria intestinal.• Espondilitis juvenil.• Espondiloartropatías indiferenciadas.• Síndrome SAPHO.
Tabla 19. Características de las espondiloartropatías.
• Ausencia de factor reumatoide.• Ausencia de nódulos reumatoides.• Artritis asimétrica de predominio en miembros inferiores,
mono u oligoartricular.• Sacroileitis y frecuente afectación axial.• Manifestaciones sistémicas características (mucocutáneas,
genitourinarias, intestinales y oculares).• Asociación a antígeno HLA B27.• Agregación familiar.• Presencia de entesitis.
6.1. Espondilitis anquilosante (EA).
La espondilitis anquilosante (EA) es una enfermedad inflamatoria crónica y sistémica que afecta predominantemente al esqueleto axial (de forma invariable a las articulaciones sacroilíacas), pero que también puede afectar a las articulaciones periféricas y a las estructuras yuxtaarticulares. Se la considera el prototipo de las espondiloartropatías.
EPIDEMIOLOGÍA.La enfermedad es más frecuente en varones (3/1) y suele comenzar entre los 15 y los 30 años.

miniMANUAL 1 CTO
Pág. 18
Se postula que la EA sigue un modelo de susceptibilidad genética poligénica. Es conocida la estrecha relación que la EA tiene con el antígeno de histocompatibilidad HLA B-27. El 90% de los pacientes presenta HLA B27, mientras que este antígeno sólo se encuentra en el 7% de la población general. (MIR 97-98, 237).
Se considera que la lesión fundamental que define a la EA es la entesitis, es decir, una inflamación de las zonas de inserción de los ligamentos y tendones en los huesos. Suele localizarse típicamente en la fascia plantar o en la inserción del tendón de Aquiles.
Figura 8. Clínica de la espondilitis anquilosante.
MANIFESTACIONES CLÍNICAS.La EA afecta principalmente al esqueleto axial. En el 80% de los casos el comienzo es insidioso, en forma de un dolor sordo bilateral en la zona lumbar y glútea, acompañado de rigidez lumbar matutina de varias horas de duración, que mejora con el ejercicio y empeora con el reposo (características inflamatorias). Es muy característico que el dolor despierte al enfermo de madrugada y que con el paso de los meses (debe durar al menos 3 meses para sugerir EA) se atenúe y desaparezca, reapareciendo en relación con las exacerbaciones de la enfermedad (MIR 97-98, 223). Cuando la enfermedad articular llega al grado de anquilosis (lo cual sólo ocurre en algunos casos de larga evolución donde no se ha seguido el tratamiento correcto), el dolor desaparece.
Pueden aparecer dolores extraarticulares producidos por la en-tesopatía en crestas ilíacas, trocánter mayor, tuberosidad isquiática, y sobre todo en los talones.
La afectación axial sigue un patrón ascendente. Tras iniciarse la enfermedad en la zona lumbar, afecta a la zona torácica, y en especial a las articulaciones costovertebrales y costoesternales. La consecuencia es una limitación de la expansión torácica.
La columna vertebral se rectifica al perderse la lordosis lum-bar y posteriormente se acentúa la cifosis torácica y el tórax se aplana. Tardíamente se afecta la columna cervical, curvándose
el cuello hacia delante. Así se crea la típica imagen del paciente espondilótico con gran cifosis de toda la espalda y con la cabeza en rotación lateral.
Se detectan artritis periféricas en el 25-35% de los pacientes. Las más frecuentemente afectadas son la cadera y los hombros (ar-ticulaciones de las cinturas), que aparecen en las fases precoces de la enfermedad. La artritis de la cadera suele ser bilateral y crónica, creando gran incapacidad. Esto suele suceder cuando la enfermedad comienza en la adolescencia y suele manifestarse en los primeros 10 años de evolución de la enfermedad.
La participación de otras articulaciones periféricas es mucho más rara y suelen seguir un patrón oligoarticular asimétrico no erosivo. (MIR 04-05, 81; MIR 99-00F, 90).
MANIFESTACIONES EXTRAARTICULARES.• Uveítis anterior aguda recidivante. Es la manifestación extraar-
ticular más habitual (25-30%) (MIR 95-96F, 145). Es más común en los pacientes HLA B27 +. Suele ser unilateral y dura más de 2-3 meses, curando sin dejar secuelas, aunque tiene una gran tendencia a recurrir, incluso en el ojo contralateral. A largo plazo producen sinequias pupilares y cataratas.
• Afectación cardiovascular. Lo más característico es la insufi-ciencia aórtica (10%) producida por la inflamación de la raíz aórtica, que suele producirse en espondilitis de larga evolución. También se puede encontrar bloqueos cardíacos completos por fibrosis del tejido de excitación-conducción .
• Manifestaciones pleuropulmonares. Son poco frecuentes y tar-díos. La más frecuente y característica es la aparición de fibrosis en los lóbulos superiores pulmonares, que se resuelve dejando cavitaciones que pueden ser colonizadas por Aspergillus, origi-nando un micetoma.
La restricción de la movilidad de la pared torácica produce una discreta disminución de la capacidad vital y un aumento de la capacidad residual funcional, pero los flujos aéreos suelen estar conservados y la función ventilatoria es normal.
• Manifestaciones neurológicas. La rigidez vertebral hace al en-fermo susceptible de traumatismos y fracturas vertebrales, que son la complicación más grave de la EA y se localizan principal-mente a nivel cervical (C5-C6, C6-C7). Otras alteraciones son la subluxación atloaxoidea por inestabilidad C1-C2 y raramente se observa un síndrome de cola de caballo secundaria a aracnoiditis crónica.
• Manifestaciones genitourinarias. Las más frecuentes son la prostatitis crónica y la nefropatía IgA. También se puede produ-cir proteinuria, deterioro de la función renal y necrosis papilar inducida por analgésicos.
• Alteraciones intestinales: existe una intensa asociación entre la EA y la enfermedad inflamatoria intestinal. La EII es un factor de riesgo para padecer EA independiente del HLA B27, aunque gran parte de los pacientes son HLA B27 positivos. Es muy frecuente (30-60%) la presencia de alteraciones inflamatorias histológicas en colon e íleon(como en el Crohn) asintomáticas.
• Raramente se produce amiloidosis secundaria (AA), en las formas de larga evolución.
EXPLORACIÓN FÍSICA.Inicialmente la exploración física sólo revela datos de inflamación. Suele observarse pérdida de la movilidad de la columna, limitación de los movimientos de flexoextensión y rotación lateral. La incapa-cidad funcional es desproporcionada para el grado de anquilosis, puesto que en gran parte es producida por la inflamación y la con-tractura muscular añadida. Existen ciertas maniobras exploratorias que indican el grado evolutivo de la EA:• Test de Schöber. Valora la limitación de la movilidad de la
columna lumbar. Se realiza con el paciente de pie, midiendo 10 cm por encima y 5 cm por debajo de la unión lumbosacra. Cuando la movilidad esté conservada, al realizar el paciente la flexión del tronco, la distancia entre las dos marcas establecida aumentará más de 5 cm (MIR 98-99F, 96).
• Expansión torácica. Se cuantifica midiendo la diferencia del perímetro torácico entre la inspiración y la espiración forzada. La expansión normal es superior a 5 cm.
• La sacroileítis puede demostrarse en la exploración mediante diferentes maniobras de provocación que desencadenan dolor.

Reumatología
Pág. 19
EXPLORACIONES COMPLEMENTARIAS.Ninguna tiene valor diagnóstico. Es habitual la anemia normocítica normocrómica de procesos crónicos y la hipergammaglobulinemia a expensas de Ig A (recuerda que la EA y la artritis psoriásica cursan con aumento de la Ig A y la AR y el LES con disminución). Los parámetros de actividad inflamatoria también se encuentran elevados:• La elevación de la VSG suele correlacionarse con la actividad
de la enfermedad articular periférica.• El aumento de la PCR sí se encuentra asociado a la actividad de
la enfermedad.Los niveles de complemento suelen estar normales o elevados.
El líquido articular posee características inflamatorias.
RADIOLOGÍA.Las alteraciones radiológicas son fundamentales para el diagnóstico de la EA(forman parte de sus criterios diagnósticos). Suele ser nece-sario un período mínimo de 5 años tras el inicio de la enfermedad para que se detecten alteraciones en las radiografías simples. Por eso, en fases muy precoces, puede tener utilidad el uso de la RMN, TC ó la gammagrafía ósea.
Habitualmente se demuestra la existencia de una sacroileítis bilateral y simétrica. Inicialmente la afectación se localiza en la parte ilíaca de la articulación sacroilíaca (puesto que aquí el hueso está más adelgazado y se erosiona con más facilidad).
Primero se observa borrosidad del borde cortical del hueso, que da paso a erosiones y esclerosis óseas. Según dichas erosiones se acen-túan, se observa un pseudoensanchamiento del espacio articular.
Tras la destrucción, se produce reactivamente formación de tejido fibroso y posteriormente sustitución por tejido óseo, lo que da lugar a la anquilosis (y desaparición del espacio articular).
Por lo tanto, la evolución de la afectación articular es: • Tejido de granulación en zonas ligamentarias y periostio.• Sinovitis y formación de pannus (que causan destrucción arti-
cular).• Borrosidad del hueso subcondral en las rx.• Bordes erosionados asimétricamente y esclerosis (pseudoen-
sanchamiento).• Anquilosis (regeneración fibrocartilaginosa formación ósea).• Desaparición de la ARTICULACIÓN.
Tejido de granulación en zonas
ligamentarias y periostio
Sinovitis y formación de Pannus (que causan destrucción articular)
Borrosidad del hueso subcondrial en las Rx
Bordes erosionados asimétricamente y esclerosis (pseudoensanchamiento)
Aquilosis
(regeneración fibrocartilaginosa)
Formación
ósea
Desaparición de
la articulación
Figura 9. Origen de las manifestaciones radiológicas en la EA.
Tabla 20. Clasificación de la sacroileítis.
Grado 0 Sacroilíacas normales.Grado I Pseudoensanchamiento del espacio articular.Grado II Estrechamiento del espacio articular, esclerosis y
erosiones.Grado III Formación de puentes óseos.Grado IV Anquilosis completa de la articulación.
En el esqueleto axial se produce una esclerosis ósea reactiva a la osteítis del borde anterior del cuerpo vertebral, seguida de erosión, que produce la característica cuadratura de las vértebras (“squaring”).
Los típicos sindesmofitos marginales son el producto de la osifi-cación de las capas superficiales del anillo fibroso del cartílago discal. Son puentes óseos entre las vértebras (a diferencia de los osteofitos o picos de loro de la artrosis, que no llegan a conectar los cuerpos).
La evolución progresiva ascendente de este proceso, junto con la anquilosis de las articulaciones interapofisarias da lugar a la característica imagen en caña de bambú (que confiere gran fragilidad a la columna y tendencia a la fractura vertebral, prin-cipalmente en la zona cervical). (MIR 01-02, 80; MIR 98-99, 89; MIR 96-97F, 95).
DIAGNÓSTICO.Para el diagnóstico de la EA se utilizan los criterios de Nueva York modificados de 1.984.
Tabla 21. Criterios diagnósticos de la EA.
Criterios clínicos.1. Limitación de la movilidad de la columna en los planos
frontal y sagital.2. Dolor lumbar de características inflamatorias.3. Limitación de la expansión torácica.
Criterios radiológicos.1. Sacroileítis bilateral grado II o superior.2. Sacroileítis unilateral grado III o IV.
El diagnóstico se establece cuando el paciente cumple el criterio radiológico y al menos un criterio clínico.
A pesar de que el 90% de los pacientes son HLA B27 positivos, la presencia de éste no es condición necesaria ni suficiente para el diagnóstico de la enfermedad.
DIAGNÓSTICO DIFERENCIAL.El principal diagnóstico diferencial se debe establecer con la hipe-rostosis anquilosante vertebral (enfermedad de Forestier). Esta enfermedad afecta a individuos de más de 60 años y está producida por la calcificación y osificación de los ligamentos paraespinosos, que forman la característica imagen en “cera derretida” en la parte anterior de los cuerpos vertebrales. Suele ser asintomática y no afecta a las sacroilíacas ni a las articulaciones interapofisarias. En ambas enfermedades se producen puentes óseos que pueden llegar a fusionar la columna por completo, pero éstos son más gruesos y exuberantes en la hiperostosis anquilosante.
EVOLUCIÓN Y PRONÓSTICO.La enfermedad tiene un curso lento, con exacerbaciones y sobre todo remisiones prolongadas. La anquilosis con graves deformi-dades y limitación de la movilidad sólo se observa en casos muy evolucionados. Los factores que se asocian a mal pronóstico son:• El comienzo precoz de la enfermedad (antes de los 16 años).• La afectación persistente de las articulaciones periféricas, fun-
damentalmente de la cadera.
TRATAMIENTO.No existe tratamiento definitivo para la EA. La finalidad es que el paciente siga un programa de ejercicios para mantener la movilidad y mejorar la capacidad funcional a la vez que se evitan las deformi-dades. Una de las actividades más beneficiosas es la natación.
El tratamiento antiinflamatorio se basa en el uso de AINEs (MIR 03-04, 16). Aunque la fenilbutazona es el fármaco más eficaz en el control sintomático de esta enfermedad, su uso se ha limitado a las formas especialmente graves por su toxicidad hematológica (anemia aplásica y agranulocitosis). Por lo tanto, la indometacina es el fármaco más utilizado.
La afectación periférica puede responder al uso de sulfasalaci-na o metrotexate, que también normalizan los datos analíticos de inflamación .Sin embargo, no sirven en el control de la enfermedad axial.
Los corticoides intraarticulares o intralesionales mejoran la entesopatía o la sinovitis persistente que no responda al trata-miento con AINEs. Incluso pueden ser de utilidad en el control de la sacroileítis, aunque para la administración intralesional en estos casos es necesario el control con TC (MIR 95-96F, 154).

miniMANUAL 1 CTO
Pág. 20
El tratamiento quirúrgico se utiliza en la artritis grave de la cadera, donde la artroplastia total alivia el dolor y la rigidez inva-lidantes.
6.2. Artritis reactiva (A Re).
La artritis reactiva (ARe) es una una artritis aguda no supurada que aparece como complicación de una infección localizada en otro lugar del organismo, preferentemente a nivel intestinal o genitourinario.
Se engloba dentro de las espondiloartropatías, que se caracte-rizan por artritis periférica, entesopatía, sacroileítis y se asocia a menudo al HLA B27.
Dentro de la artritis reactiva se encuadra una tríada con nombre propio, el síndrome de Reiter, que se define como la asociación de uretritis, conjuntivitis y artritis (MIR 94-95, 185).
ETIOLOGÍA Y EPIDEMIOLOGÍA.Se presenta habitualmente durante la 2º-3º década de la vida.
Mientras que en los casos de origen entérico la prevalencia entre ambos sexos es igual, aquellas artritis precedidas de un cuadro genitourinario son mucho más frecuentes en los varones (aunque esto se puede explicar teniendo en cuenta que la uretritis suele ser más sintomática que la cervicitis, y por ello es más fácil diagnosticarla).
Los principales gérmenes gastrointestinales que se relacionan con la aparición ARe son la Shigella flexneri, Salmonella, Yersinia y Campylobacter. En las formas de origen genitourinario, el agente asociado más frecuentemente suele ser la Chlamydia trachomatis y en segundo lugar el Ureoplasma urealiticum (MIR 97-98, 235).
El 60-80% de los pacientes son HLA B27 +. Dicha positividad implica un curso más crónico de la enfermedad y una mayor agresividad (MIR 94-95, 190) (MIR 00-01, 77).
En los pacientes VIH positivos, la ARe constituye la forma más frecuente de artritis. Los enfermos suelen ser HLA B27 positivos. La afectación suele ser más grave, especialmente a nivel cutáneo.
PATOGENIA.En la artritis reactiva se produce una interacción entre un factor externo (la infección) y un factor genético que regula la respuesta del huésped. Se cree que la presencia del HLA B27 es la responsa-ble del aumento de susceptibilidad a padecer artritis reactiva.
Se intentaría explicar la ARe mediante un mecanismo de mime-tismo molecular: los antígenos bacterianos y algunos componentes de la sinovial compartirían ciertas secuencias de aminoácidos. El HLA B27 se encargaría de presentar los antígenos bacterianos a los linfocitos T (sobre todo a los CD4), que producirían una respuesta inmune que lucharía contra las bacterias, y por reacción cruzada, dañaría las estructuras articulares (al producir sinovitis).
Todas las bacterias involucradas en el desarrollo de artritis reac-tivas son intracelulares y afectan característicamente a las mucosas. Hay autores que sugieren que existirían bacterias en el espacio sinovial de forma crónica, contra las que se produciría la respuesta inmune que daña la articulación. EL HLA B27 sería el encargado de presentar los antígenos bacterianos a los linfocitos.
MANIFESTACIONES CLÍNICAS.La artritis reactiva suele comenzar a manifestarse tras un período de latencia de 1 a 4 semanas desde la resolución de una diarrea infecciosa o tras un contacto sexual de riesgo. Hay que recordar que no siempre existen antecedentes de una infección previa.
Los pacientes habitualmente presentan síntomas generales inespecíficos. Lo más característico de la ARe es una artritis mono /oligoarticular aditiva y asimétrica, de predominio en los miembros inferiores. Suele comenzar de forma abrupta y es muy dolorosa. Se observa la existencia de derrame sinovial inflamatorio en muchos de los casos y los cultivos de líquido sinovial son siempre negativos.
Al igual que en la artritis psoriásica, se puede encontrar dacti-litis o “dedos en salchicha”, que consiste en la tumefacción difusa de un dedo de la mano o del pie, producida por afectación de la IFP e IFD.
Se puede encontrar dolores producidos por la entesitis, sobre todo en forma de talalgia (por afectación de la inserción en el cal-cáneo de la fascia plantar o del tendón aquíleo).
Es frecuente la presencia de dolor lumbar, que puede estar producido por la sacroileítis.
MANIFESTACIONES EXTRAARTICULARES.1) Urogenitales. Es frecuente que se observe uretritis y prostatitis en
los varones y cervicitis, cistitis o uretritis en las mujeres afectas. Tanto la uretritis como la diarrea pueden ser, además del síntoma propio de la infección desencadenante, una manifestación clínica de la fase “reactiva” de la enfermedad.
2) Lesiones mucocutáneas. Son muy frecuentes. Se suelen produ-cir aftas orales superficiales asintomáticas y lesiones ungueales (onicólisis, engrosamiento e hiperqueratosis).
En las artritis de origen urogenital, se observan las siguientes lesiones dérmicas:
A. La queratodermia blenorrágica : lesiones casi indistinguibles de las psoriasis pustulosa , pueden dominar el cuadro en el VIH. Son pústulas y costras hiperqueratósicas de localiza-ción palmoplantar.
B. La balanitis circinada: son vesículas que fácilmente se rom-pen, convirtiéndose en erosiones superficiales e indoloras localizadas en el glande.
3) Alteraciones oculares. Pueden ir desde una conjuntivitis leve (40% de los pacientes) hasta una uveítis anterior agresiva.
EVOLUCIÓN.En el 30-60% de los pacientes los síntomas persisten y hasta un 25% de los enfermos presentan recidivas.
El dolor crónico de los talones es uno de los síntomas más incapacitantes. Cuando la enfermedad presenta un curso crónico, se asemeja mucho a una EA, con asociación al HLA B27 y presencia de sacroileítis.
EXPLORACIONES COMPLEMENTARIAS.En la fase aguda, existe anemia y los parámetros de inflamación (reactantes de fase aguda) están elevados. El líquido sinovial es de tipo inflamatorio, con predominio de neutrófilos. Recuerda que en la ARe los cultivos de orina, sangre y articulares son negativos.
Inicialmente no se observan alteraciones en las radiografías, a excepción de una osteoporosis yuxtaarticular. En las formas avanzadas existen erosiones marginales y desaparición del es-pacio articular.
Como en todas las espondiloartropatías, la inflamación cróni-ca induce la formación de nuevo hueso de forma reactiva, siendo típico la formación del espolón calcáneo. Es posible encontrar datos de sacroileítis (20-30%) e incluso sindesmofitos.
DIAGNÓSTICO.El diagnóstico es clínico, ya que ningún dato analítico ni radiológico tiene valor diagnóstico. El antecedente de infección (si existe), junto con la distribución característica de la artropatía y los síntomas extraarticulares asociados, sirven para establecer el diagnóstico de ARe (MIR 02-03, 222).
Se debe establecer diagnóstico diferencial preferentemente con la artritis gonocócia y la psoriasis.1. Gonococia diseminada: no afecta al esqueleto axial, los
síntomas articulares se localizan tanto en las extremidades inferiores como en las superiores, es posible cultivar el gono-coco en la sangre, piel y la sinovial, las lesiones vesiculosas se encuentran por todo el cuerpo y responde al tratamiento antibiótico.
2. Artritis psoriásica: el comienzo es paulatino, afecta a miembros superiores y no se asocia a aftas orales, uretritis ni síntomas intestinales.
TRATAMIENTO.No existe tratamiento específico para la ARe.1. Los AINES mejoran la sintomatología de la artritis aguda. Se
utiliza la indometacina, reservándose la fenilbutazona para los casos refractarios (ya que tiene una alta toxicidad)
2. Los corticoides se pueden utilizar como en la EA de forma in-tralesional para calmar la sintomatología de las tendinitis y las entesitis, nunca de forma sistémica.
3. Según ciertos estudios, la sulfasacina es beneficiosa para el tratamiento de la ARe.

Reumatología
Pág. 21
4. Si el paciente es resistente a los AINEs y a la sulfasalacina, se ha probado con éxito la administración de inmunosupresores como el metotrexate y la azatioprina.
5. El tratamiento con tetraciclinas parece ser útil en las infecciones por Chlamydia , ya que parece que evita la aparición posterior de una artritis reactiva.
6. En los pacientes VIH positivos, el tratamiento con AZT (zidovu-dina) mejora notablemente las lesiones cutáneas.
6.3. Artropatía psoriásica.
EPIDEMIOLOGÍA.La artropatía psoriásica es una artritis inflamatoria crónica que afecta del 5 al 8% de los psoriáticos. Aunque la afectación cutánea suele preceder a la artritis, también es posible que ocurra de forma inversa.
La artropatía debuta entre la 3º y 4ª década y globalmente la proporción es de 1/1 entre ambos sexos. Los estudios en familias hacen pensar que existe una predisposición genética a padecer la enfermedad.
MANIFESTACIONES CLÍNICAS.Se aprecian diferentes patrones clínicos de la enfermedad. En todas ellas es muy frecuente la artritis de las IFD y la dactilitis o dedos en salchicha en manos y pies.
El 80% de los pacientes con artritis psoriásica tienen afectación ungueal , siendo típico el pitting o punteado, la queratosis subun-gueal y la onicólisis (MIR 01-02, 78; MIR 98-99F, 100; MIR 99-00F, 91; MIR 96-97F, 89).
EXPLORACIONES COMPLEMENTARIAS.Analíticamente se observa un aumento de los parámetros de actividad inflamatoria, junto con incremento de la IgA. Existe hiperuricemia debida al gran recambio celular que se produce a nivel cutáneo.
En la radiología destaca la desaparición del espacio articular y las erosiones. No hay osteoporosis (encontrándose en todo caso osteopenia en las primeras fases). Son datos típicos las erosiones en la IFD y la reabsorción de la falange distal. Imágenes propias de la artritis psoriásica son el “lápiz –copa” (reabsorción de un extremo y proliferación ósea del otro) y “lápiz-lápiz” (reabsorción de ambos extremos de la articulación)
Las alteraciones radiológicas en el esqueleto axial se presentan en forma de sacroileítis asimétrica y sindesmofitos, que son más asimétricos e irregulares que en la EA.
Tabla 22. Formas clínicas de la artritis psoriásica.
CERF OXES ALH ACINÍLC
SITIRTRAOGILOACIRTÉMISA
%05 M=V71B6wC
alneDFIalatcefAeroirepusdadimertxe
roirefni
SITIRTRAILOPACIRTÉMIS
%52 M 4RDecerapeS.ralucocatcefaoN
soludónnisorepRAala,sediotamuer
SITILIDNOPSE %52 V 72B
catcefaedañapmocaeSsitinidnet,aciréfirepcitraetneucerfsoneM.aelíuqaeneit%03lE.ralucocatcefa
selanitsetnilfnisenoisel
DFINÓICATCEFA %5 V.añualyDFIedcatcefA
amrofaanoiculovEralucitrailop/ogilo
AMROFETNALITUM
%5 V
,egnalafednóicrosbaeRoadalsiaamroF.TTM,PCM
cerF.seroiretnasalaº2sitíeliorcas
DIAGNÓSTICO.El diagnóstico es clínico, al encontrar la existencia de artritis y pso-riasis. Sin embargo, hay que recordar que las lesiones cutáneas de psoriasis pueden ser mínimas y ocultas (cuero cabelludo, pliegue interglúteo). (MIR 00-01F, 84)
Hay que pensar en artritis psoriásica cuando nos encontremos ante una artritis asimétrica, de predominio en miembros superiores, asociada a dactilitis, onicodistrofia y entesitis, con especial predilec-ción por las articulaciones IFD. La afectación de las articulaciones IFD se diferencia de la artrosis por las alteraciones radiológicas que se visualizan y por la ausencia de nódulos de Heberden.
TRATAMIENTO.El tratamiento se basa en 3 pilares:1. Fisioterapia para evitar la incapacidad funcional2. AINEs, que disminuyen el dolor y la inflamación (en su defecto,
se pueden utilizar corticoides en dosis bajas)3. Para evitar la progresión se añaden fármacos modificadores
de la enfermedad (FAME), al igual que en la AR. No sirven en la afectación axial. De elección se utiliza el metotrexato , que ha demostrado su eficacia tanto para el control en la artritis como en las lesiones cutáneas, pero que puede agravar la enfermedad en los pacientes VIH positivos.
Otros fármacos de posible uso son la sulfasalacina, la ciclospo-rina o la azatioprina. Aunque la cloroquina mejora la artropatía, puede empeorar notablemente la psoriasis.
6.4. Artritis en la enfermedad inflamatoria intestinal.
Los pacientes con enfermedad inflamatoria intestinal (colitis ulce-rosa o enfermedad de Crohn) pueden presentan síntomas de artritis periférica o de espondilitis-sacroileítis (MIR 98-99F, 98).
La artritis periférica aparece en el 25% de los casos, tras el debut de la enfermedad intestinal. Se produce con más frecuencia en los pacientes que tienen afectación de colon. La prevelencia es igual entre ambos sexos y el curso de los síntomas articulares es paralelo al de la afectación gastrointestinal.
Se trata de una poliartritis asimétrica migratoria no erosiva de grandes articulaciones de miembros inferiores. El tratamiento es el mismo que es de la afectación intestinal.
La espondilitis/sacroileítis se produce en el 20% de los pacientes con EII. Es más frecuente en varones y puede preceder a los síntomas gastrointestinales. El curso clínico es independiente de la actividad inflamatoria intestinal. Los síntomas son similares a los de la EA, así como los hallazgos radiológicos. Existe una asociación con el HLA B27 (50-75%). Recuerda que la uveitis y el pioderma gangre-noso también llevan un curso independiente del de la enfemedad intestinal.
TEMA 7. ENFERMEDADES METABÓLICAS ÓSEAS.
7.1. Osteoporosis.
La osteoporosis es la enfermedad metabólica ósea más frecuente. Se define como la reducción de la masa ósea o la presencia de una fractura por fragilidad. Otra forma de definirlo es como la densidad ósea menor de 2.5 desviaciones standard respecto a la de un adulto joven del mismo sexo ( puntuación T) . Se define como osteopenia a una densidad mineral inferior a una desviación standard.• Puntuación T: compara los resultados individuales con los de
una población joven de misma raza y género.• Puntuación Z: compara los resultados individuales con los de
una población de la misma edad, raza y género.
La masa ósea adquiere su pico máximo entre los 30-35 años, y a partir de aquí se produce un descenso progresivo por desequi-librio entre la formación y reabsorción, que hará perder entre un 20-30% de masa ósea a los varones y un 40-50% a las mujeres. Es por ello que la osteroporosis aumenta su frecuencia al aumentar la edad.
Se produce una disminución del grosor cortical y del número y grosor de las trabéculas del hueso esponjoso, lo que confiere una fragilidad ósea aumentada y un riesgo de fractura elevado.
ETIOPATOGENIA
Existe gran cantidad de factores que contribuyen a disminuir la masa ósea. Se puede clasificar la osteoporosis como primaria o secunda-ria:
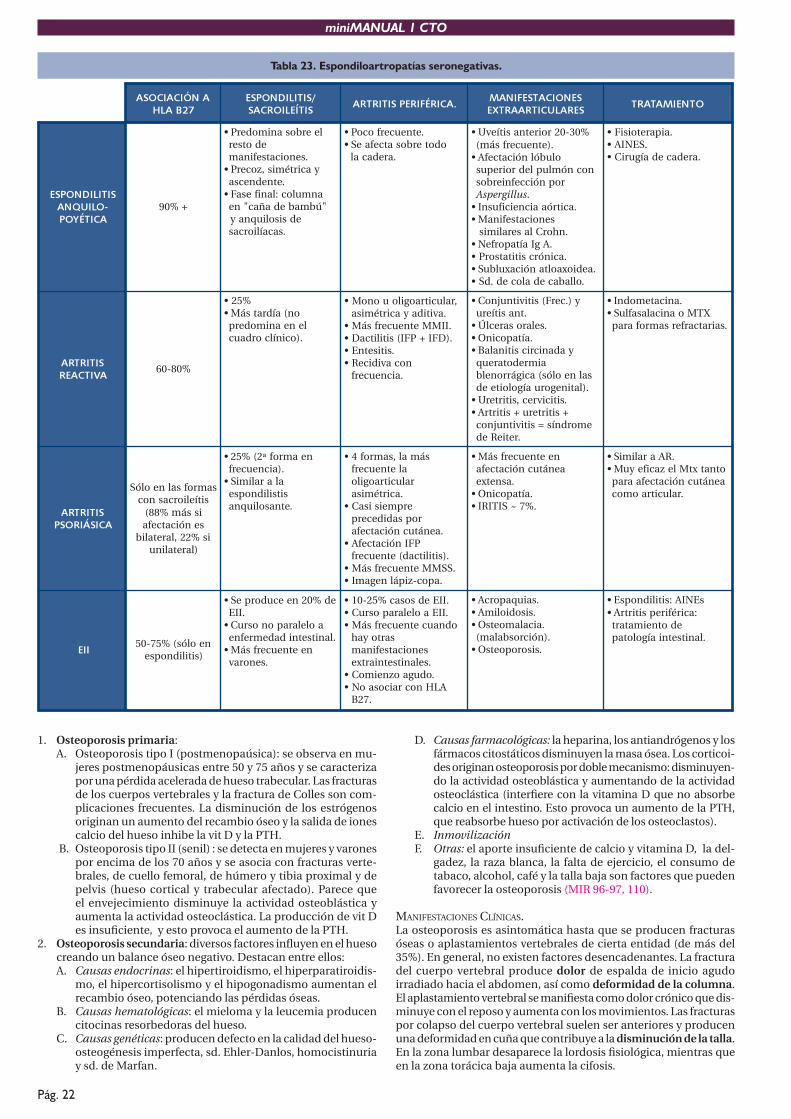
miniMANUAL 1 CTO
Pág. 22
1. Osteoporosis primaria: A. Osteoporosis tipo I (postmenopaúsica): se observa en mu-
jeres postmenopáusicas entre 50 y 75 años y se caracteriza por una pérdida acelerada de hueso trabecular. Las fracturas de los cuerpos vertebrales y la fractura de Colles son com-plicaciones frecuentes. La disminución de los estrógenos originan un aumento del recambio óseo y la salida de iones calcio del hueso inhibe la vit D y la PTH.
B. Osteoporosis tipo II (senil) : se detecta en mujeres y varones por encima de los 70 años y se asocia con fracturas verte-brales, de cuello femoral, de húmero y tibia proximal y de pelvis (hueso cortical y trabecular afectado). Parece que el envejecimiento disminuye la actividad osteoblástica y aumenta la actividad osteoclástica. La producción de vit D es insuficiente, y esto provoca el aumento de la PTH.
2. Osteoporosis secundaria: diversos factores influyen en el hueso creando un balance óseo negativo. Destacan entre ellos:A. Causas endocrinas: el hipertiroidismo, el hiperparatiroidis-
mo, el hipercortisolismo y el hipogonadismo aumentan el recambio óseo, potenciando las pérdidas óseas.
B. Causas hematológicas: el mieloma y la leucemia producen citocinas resorbedoras del hueso.
C. Causas genéticas: producen defecto en la calidad del hueso- osteogénesis imperfecta, sd. Ehler-Danlos, homocistinuria y sd. de Marfan.
D. Causas farmacológicas: la heparina, los antiandrógenos y los fármacos citostáticos disminuyen la masa ósea. Los corticoi-des originan osteoporosis por doble mecanismo: disminuyen-do la actividad osteoblástica y aumentando de la actividad osteoclástica (interfiere con la vitamina D que no absorbe calcio en el intestino. Esto provoca un aumento de la PTH, que reabsorbe hueso por activación de los osteoclastos).
E. InmovilizaciónF. Otras: el aporte insuficiente de calcio y vitamina D, la del-
gadez, la raza blanca, la falta de ejercicio, el consumo de tabaco, alcohol, café y la talla baja son factores que pueden favorecer la osteoporosis (MIR 96-97, 110).
MANIFESTACIONES CLÍNICAS.La osteoporosis es asintomática hasta que se producen fracturas óseas o aplastamientos vertebrales de cierta entidad (de más del 35%). En general, no existen factores desencadenantes. La fractura del cuerpo vertebral produce dolor de espalda de inicio agudo irradiado hacia el abdomen, así como deformidad de la columna. El aplastamiento vertebral se manifiesta como dolor crónico que dis-minuye con el reposo y aumenta con los movimientos. Las fracturas por colapso del cuerpo vertebral suelen ser anteriores y producen una deformidad en cuña que contribuye a la disminución de la talla. En la zona lumbar desaparece la lordosis fisiológica, mientras que en la zona torácica baja aumenta la cifosis.
Tabla 23. Espondiloartropatías seronegativas.
'����� ()*�+
, �-���.'�/ � �-0������ 1���2����/ �-��-� �'���- ���'�
���3��-��-4� - �-'���-�
�-���.'�/ �5���36'��-27�/
+%09
leerbosanimoderP •edotser
.senoicatsefinam yacirtémis,zocerP •
.etnednecsa anmuloc:lanifesaF •"úbmabedañac"ne
edsisoliuqnay.sacaíliorcas
.etneucerfocoP •odoterbosatcefaeS •
.aredacal
%03-02roiretnasitíevU •( sám .)etneucerf
olubólnóicatcefA •nocnómlupledroirepus
ropnóiccefnierbos sulligrepsA .
.acitróaaicneicifusnI •senoicatsefinaM •
.nhorClaseralimis .AgIaítaporfeN •
.acinórcsititatsorP•.aedioxaoltanóicaxulbuS •
.ollabacedaloced.dS•
.aiparetoisiF•.SENIA•
.aredacedaíguriC•
�-��-���-���
%08-06
%52•on(aídratsáM •
leneanimoderp .)ocinílcordauc
• ,ralucitraogilouonoM.avitidayacirtémisa
.IIMMetneucerfsáM•.)DFI+PFI(sitilitcaD•
.sitisetnE•nocavidiceR•
.aicneucerf
y).cerF(sitivitnujnoC •.tnasitíeru
.selarosareclÚ •.aítapocinO •
yadanicricsitinalaB •aimredotareuq
salneolós(acigárronelb .)latinegoruaígoloiteed
.siticivrec,sitirterU •+sitirteru+sitirtrA •
emordnís=sitivitnujnoc .retieRed
.anicatemodnI •XTMoanicalasafluS •
.sairatcarfersamrofarap
�-��-��� 8��� /
samrofsalneolóSsitíeliorcasnoc
%88( sám issenóicatcefa
is%22,laretalib)laretalinu
neamrofª2(%52 •.)aicneucerf
alaralimiS •sitsilidnopse
.etnasoliuqna
sámal,samrof4•aletneucerf
ralucitraogilo .acirtémisa
erpmeisisaC•ropsadidecerp
.aenátucnóicatcefa PFInóicatcefA•
.)sitilitcad(etneucerf .SSMMetneucerfsáM•
.apoc-zipálnegamI•
neetneucerfsáM •aenátucnóicatcefa
.asnetxe .aítapocinO •.%7~SITIRI •
.RAaralimiS •otnatxtMlezacifeyuM •aenátucnóicatcefaarap
.ralucitraomoc
���neolós(%57-05
)sitilidnopse
2neecudorpeS • 0 ed%.IIE
aolelaraponosruC •.lanitsetnidademrefne
neetneucerfsáM •.senorav
2-01• 5 .IIEedsosac%.IIEaolelaraposruC•
odnaucetneucerfsáM•sartoyah
senoicatsefinam .selanitsetniartxe .odugaozneimoC•
ALHnocraicosaoN•.72B
.saiuqaporcA •.sisodiolimA •
.aicalamoetsO •.)nóicrosbalam(
.sisoropoetsO •
sENIA:sitilidnopsE • • A :aciréfirepsitirtr
edotneimatart .lanitsetniaígolotap

Reumatología
Pág. 23
MANIFESTACIONES RADIOLÓGICAS.La disminución de la densidad ósea se puede manifestar radioló-gicamente como: acuñamiento anterior vertebral, biconcavidad o vértebra en pez (por disminución central del material óseo) y aplastamiento total o vértebra en galleta. Hay adelgazamiento de las corticales de los huesos tubulares.
DIAGNÓSTICO.La radiología no es de utilidad hasta que haya una disminución de la densidad ósea al menos del 30%. Es por ello que la densitometría es la técnica de elección, al determinar la masa ósea de cualquier localización de forma precisa. Además, la densitometría sirve tanto para el diagnóstico de la osteoporosis como para valorar la eficacia del tratamiento (MIR 00-01F, 77).
El patrón bioquímico de recambio óseo más habitual de la osteoporosis es el normal , aunque en un 20% de los casos de osteo-porosis tipo I se encuentra hipercalciuria por déficit de reabsorción tubular (MIR 95-96, 57).
Tabla 24. Diagnóstico diferencial de la osteoporosis.
AMOLEIMetnenopmoc,aimeclacrepih,adatnemuaGSV
aniroyoreusnelanolconom
AICALAMOETSOaledotnemuayaimerofsofopih,aimeclacopiH
namkliM-resooLedsaeníL.AF
SISENÉGOETSOATCEFREPMI
.aesódadisnedaledosufidosnecseDaesóaispoibropocitsóngaiD
º1HTPREPIHnocaimetafsofopih,aimeclacrepiH
airuiclacrepiheairutafsofrepih
SISATSÁTEMSAESÓ
ocrayolucídeplednóicatcefa,adatnemuaGSVroiretsop
TEGAPFNEynóicroseredsortemárapsolednóicavelE
aesónóicamrof
DIAGNÓSTICO DIFERENCIAL.El diagnóstico diferencial se debe realizar con aquellos procesos que produzcan pérdida de masa ósea y fracturas. La presencia de fracturas por encima de D4 debe hacer sospechar enfermedad tumoral maligna con infiltración del cuerpo vertebral.
TRATAMIENTO.1. Tratamiento de las fracturas osteoporóticas. Las fracturas de cadera generalmente requieren tratamiento
quirúrgico seguido de tratamiento rehabilitador. El aplasta-miento vertebral se trata mediante analgesia y actualmente se ha demostrado la eficacia de las inyecciones percutáneas de cemento artificial. El reposo prolongado no se aconseja, ya que la inmovilización aumenta la pérdida de masa ósea.
2. Tratamiento a largo plazo. Inicialmente se deben evitar los factores de riesgo y cubrir las nece-
sidades dietéticas de calcio y vitamina D, puesto que disminuyen la pérdida de masa ósea y suprimen el recambio óseo. El ejercicio también es beneficioso. Se indica el tratamiento profiláctico en los casos de osteopenia asociados a factores de riesgo.• Bifosfonatos: de primera elección. Alendronato, risedronato.
Son análogos del pirofosfato inorgánico y se caracterizan por ser potentes inhibidores de la resorción ósea. Sus efectos secundarios son principalmente gastrointestinales, como la esofagitis.
• Estrógenos. Sólo se utilizan en la osteoporosis postmeno-páusica acompañada de síntomas climatéricos.
• Modificadores selectivos de los receptores estrogénicos o SERM: raloxifeno. Alternativa a los bifosfonatos. Como efec-tos secundarios principales producen empeoramiento de los síntomas climatéricos y aumentan el riesgo de enfermedad tromboembólica. A diferencia de los estrógenos, disminuyen el riesgo de cáncer de mama. Reducen el recambio y la pér-dida de masa ósea, disminuyendo la incidencia de fracturas vertebrales.
• Calcitonina. Se utiliza por su efecto analgésico central y porque suprime la actividad de los osteoclastos. Los efectos secundarios y la falta de una gran mejora limitan su uso.
Osteoporosis tipo II: se emplean el calcio y la vitamina D. El fluoruro sódico es un fármaco que activa directamente los osteo-blastos y produce un aumento de masa ósea trabecular, aunque el hueso que crea no es de buena calidad.
Otros tratamientos utilizados son las tiacidas, para corregir la hipercalciuria que aparece en algunas osteoporosis, y los andróge-nos en las osteoporosis del varón con hipogonadismo.
7.2. Raquitismo y osteomalacia.
El raquitismo y la osteomalacia son trastornos que cursan con un defecto de la mineralización de la matriz orgánica del esqueleto. El raquitismo afecta al esqueleto en crecimiento y especialmente a las metáfisis, mientras que la osteomalacia es un trastorno del esqueleto del adulto. Para una mineralización normal del tejido osteoide se requiere una cantidad adecuada de calcio, fosfatos, así como una función metabólica normal. La alteración en cualquiera de los iones o del metabolismo origina estas enfermedades.
ETIOLOGÍA.Muchas son las causas que pueden derivar a osteomalacia o ra-quitismo:1. Déficit de vitamina D. Puede ser por:
- Aporte extrínseco insuficiente (ingestión inadecuada, expo-sición insuficiente a la luz solar).
- Trastornos gastrointestinales (malabsorción intestinal por enfermedad hepática que no produce la primera hidroxila-ción, insuficiencia pancreática que no produce las micelas necesarias para la absorción de la vit D liposoluble o enfer-medad del intestino delgado que por daño de la mucosa no absorbe el calcio ni la vitamina D).
- Trastornos del metabolismo de la vit D (adquiridos, como la insuficiencia renal, el sd. nefrótico, donde se pierde por orina la proteína transportadora de la vit D, o el tratamiento con anticonvulsionantes, que induce los enzimas hepáticas que catabolizan la vit D. Hay causas hereditarias, como el pseudodéficit o dependencia de vit D tipo I -provocado por déficit de la 25-OH hidroxilasa-, o la alteración del receptor de la vitamina D, que produce el raquitismo hereditario dependiente de vitamina D tipo II, típicamente asociado a alopecia).
2. Hipofosforemia crónica tanto por: - Déficit de aporte (abuso de antiácidos con aluminio que
quelan el fosfato e impiden su absorción).
Figura 11. Metabolismo de la vitamina D.
- Pérdida tubular de fosfato (raquitismo resistente a la vitami-na D ligado al cromosoma X, osteomalacia hipofosfatémica resistente a la vitamina D, de aparición en el adulto, acidosis tubular renal, ingesta de fármacos, síndrome de Fanconi).
RECUERDA
La sola reducción de fosfato produce osteomalacia, sin hiperPTH secundario.

miniMANUAL 1 CTO
Pág. 24
FISIOPATOLOGÍA. La figura 11 resume el metabolismo de la vitamina D y los cambios que aparecen en los pacientes con osteomalacia secundaria a su déficit.
Figura 12. Fisiopatología del raquitismo/osteomalacia.
MANIFESTACIONES CLÍNICAS.El raquitismo suele manifestarse de forma florida a partir del año de edad. Existe un retraso en el crecimiento y la formación de hueso no mineralizado (y por tanto frágil y muy deformable) da lugar a las deformidades óseas y a las fracturas. Las deformidades aparecen inicialmente en el cráneo y avanzan caudalmente. Apa-rece craneotabes (reblandecimiento óseo), junto con fontanelas amplias que tardan en cerrarse. Existe prominencia frontal (frente olímpica) y parietal (caput cuadratum) con aplastamiento occipi-tal (debido a la postura de decúbito). Hay retraso en la dentición. Los engrosamientos condrocostales (por producción de excesivo tejido osteoide que intenta compensar la falta de mineralización) producen el rosario raquítico. La deformidad de las últimas costi-llas en su inserción en el diafragma da lugar al surco de Harrison y el ensanchamiento de la base del tórax le confiere el aspecto de tórax alado. En las extremidades aparecen engrosamientos en las metáfisis, y como el hueso no mineralizado no posee suficiente resistencia como para soportar la carga, las piernas y los brazos se arquean. Se observa una pelvis en forma de naipe. La hipocalcemia que deriva del déficit de vit D puede provocar hipotonía e incluso tetania en casos extremos.
La osteomalacia se presenta de forma más insidiosa. La manifes-tación más característica es el dolor óseo, seguido por la debilidad muscular proximal (de la cintura escapular y de la cintura pelviana). Pueden producirse fracturas patológicas, entre las que destacan las pseudofracturas o líneas de Looser-Milkman, que son bandas radiotransparentes que cruzan de forma perpendicular la cortical (MIR95-96F, 3).
LABORATORIO. Ver tabla 26 en la página anterior.
RADIOLOGÍA.En el raquitismo, las alteraciones radiológicas son precoces. Se observan rarefacciones en el hueso y las metáfisis aparecen en-sanchadas en forma de copa de champán. La cortical está adel-gazada y la distancia entre metáfisis y epífisis está aumentada.
En la osteomalacia, la lesión más característica son las líneas de Looser-Milkman. Los cuerpos vertebrales suelen adoptar forma bicóncava, borrándose el patrón trabecular, y dando un aspecto de vidrio esmerilado.
DIAGNÓSTICO. El diagnóstico se establece por las alteraciones clínicas, bioquímicas y radiológicas. La confirmación se realiza mediante biopsia ósea, que muestra un tejido osteoide de grosor aumentado. El marcaje con tetraciclinas detecta un tiempo de desfase en la mineralización de más de 100 días.
TRATAMIENTO.Según la causa que produzca el raquitismo o la osteomalacia, se administran suplementos de calcio asociados o no a suplementos de vitamina D. Se prefiere el uso de la forma activa de la vitamina D (el calcitriol) para evitar las posibles alteraciones en el metabolismo de ésta que interfieran en la corrección del trastorno.
7.3. Enfermedad ósea de Paget.
La enfermedad de Paget es la 2ª osteopatía más frecuente en nuestro país (1.5% de los >55 años). Se presenta más en varones de más de 60 años y la prevalencia aumenta con la edad.
Respecto a su etiología se asocian factores genéticos y ambien-tales. En los osteoclastos se encuentran inclusiones citoplasmáticas que sugieren una infección por paramyxovirus (sarampión, virus respiratorio sincitial o del moquillo canino). Además, se demuestra una fuerte agrupación familiar, que apoyaría un patrón de heteroge-neidad genética (fallos distintos en los genes producen una misma enfermedad) (MIR 01-02, 81).
Fase Reabsortivaosteolítica
Fase Mixta Fase Osteoblástica
Resorción > Formación Resorción = Formación Formación > Resorción
Figura 13. Etapas evolutivas de la enfermedad de Paget.
FISIOPATOLOGÍA.Parece que el fallo primario del Paget reside en los osteoclastos, que son más numerosos y con mayor actividad. La enfermedad se caracteriza por una resorción excesiva de hueso por los os-teoclastos, seguida de un incremento de la síntesis ósea. En la fase inicial (fase osteoporótica o lítica) predomina la resorción ósea de un hueso muy vascularizado (balance óseo negativo), seguida de una fase mixta en la que la formación ósea se acopla a la resorción (balance óseo neutro). El hueso que se forma es de tipo laminar y se deposita de forma irregular ( se forma hueso muy rápidamente y no es hueso de calidad, es más frágil). En la última fase (osteoblástica o esclerótica) la formación de hueso supera a la velocidad de reabsorción, por lo que el balance óseo es positivo y se forma un hueso duro y poco vascularizado (poco deformable).
Se observa un aumento de producción de IL-6, citoquina que hace al hueso más sensible a la resorción provocada por la vit D en los focos de la enfermedad, que puede contribuir a la activación osteoclástica. Ver figura 14.
MANIFESTACIONES CLÍNICAS.El 60% de los pacientes se encuentran asintomáticos. El dolor óseo primario es la manifestación clínica más habitual, loca-lizándose en la cabeza, región facial o en la espalda. También son posibles las deformidades (por aumento de tamaño y por la tracción de los músculos sobre un hueso más deformable), la tumefacción en extremidades (por hipervascularización) y la dificultad para la marcha por dismetría de los miembros. Es típico que necesiten una talla mayor de sombrero o que los pantalones les queden largos por arqueamiento de las piernas. La hipoacusia se produce por una afectación directa de los huesecillos del oído interno o es secundaria a la compresión del VIII par en el orificio auditivo interno. El crecimiento de la base del cráneo (platibasia) comprime el troncoencéfalo, mientras que el engrosamiento de la columna dorsal produce paraplejía por compresión de la médula espinal.

Reumatología
Pág. 25
Figura 14. Fisiopatología de la enfermedad de Paget.
DATOS ANALÍTICOS.Destaca el aumento de los marcadores de formación ósea (fosfatasa alcalina, osteocalcina) y de reabsorción ósea ((hidroxiprolina, fos-fatasa ácida). Algunos pacientes con enfermedad inicial muy activa pueden presentar aumento de la calciuria, y rara vez hipercalcemia moderada (MIR 97-98F, 215; MIR 95-96, 53).
ALTERACIONES RADIOLÓGICAS.La pelvis es la estructura ósea más afectada, seguida del fémur. En la fase lítica de la enfermedad se observa en el cráneo la típica imagen de osteoporosis circunscrita y en los huesos largos lisis con avance en forma de V (tibia en sable).
En la fase mixta se visualiza un ensanchamiento irregular adya-cente a la zona de lisis, siguiendo un patrón tosco y estriado. En esta fase, el cráneo se ensancha y aumenta el grosor de la tabla externa. La tracción muscular y la fuerza gravitatoria son las que guían la dirección de las líneas de tensión del nuevo hueso formado, lo que da lugar a las deformidades. Es típico el engrosamiento del orificio pélvico superior y las “vértebras en marco “ al aumentar las estrías verticales y el refuerzo periférico.
En la fase esclerótica, el hueso presenta un aumento homogéneo de densidad. Así ocurre en la columna, formándose las “vértebras en marfil” (que se diferencian del Hodgkin en que la vértebra está aumentada de tamaño en el Paget).
COMPLICACIONES.1) Los huesos pagéticos están hipervascularizados, por prolifera-
ción de los vasos en la fase resortiva. Cuando se afecta más de 1/3 del esqueleto se puede producir una elevación del gasto cardíaco, aunque es rara la insuficiencia cardíaca.
2) En la fase destructiva son posibles las fracturas patológicas, ya que el hueso es demasiado débil. También es posible que se produzca una artropatía por vecindad (más frecuente coxofe-moral) y síndromes neurológicos compresivos
3) Aumenta la incidencia de cálculos urinarios por hipercalciuria.También es posible la hiperuricemia.
4) La complicación más grave es el sarcoma (1% de los pacientes). Suele localizarse en la metáfisis distal del fémur o en la tibia proximal. Se debe sospechar ante el aumento del dolor y de la tumefacción, junto con un aumento exagerado de los niveles de fosfatasa alcalina (MIR 96-97F, 101).
DIAGNÓSTICO.El diagnóstico se realiza por la elevación de los niveles de fos-fatasa alcalina o por la aparición de alteraciones radiológicas características. La gammagrafía con Tc99 indica la extensión de la enfermedad, ya que capta en aquellas zonas con aumento del metabolismo óseo.
TRATAMIENTO.Los enfermos asintomáticos no se tratan (MIR 04-05, 80). Los bi-fosfonatos son el fármaco de elección, reservándose la calcitonina
en perfusión iv para las compresiones medulares. Ambos fármacos actúan inhibiendo la acción de los osteoclastos.
Las indicaciones absolutas de tratamiento son los enfermos sin-tomáticos con dolor óseo persistente, compresión nerviosa, defor-midad ósea, insuficiencia cardíaca, hipercalcemia e hipercalciuria, fracturas óseas y preparación para cirugía ortopédica (al disminuir la resorción también disminuye la vascularización). Es necesaria la cirugía para aliviar la artropatía por vecindad. Las indicaciones rela-tivas son los pacientes asintomáticos con afectación de la base del cráneo y vertebral (por las posibles complicaciones neurológicas) y lesiones líticas en huesos de carga (MIR 99-00, 118).
TEMA 8. ESCLEROSIS SISTÉMICA PROGRESIVA.
La esclerosis sistémica progresiva (ESP) es una enfermedad gene-ralizada originada por el depósito excesivo de los componentes del tejido conjuntivo en los órganos, expresado en forma de fibrosis tisular. Afecta predominantemente a la piel. Suele aparecer en mujeres (3/1) entre los 30 y 50 años (en la edad fértil), siendo más frecuente y grave en la raza negra.
Dentro de la ESP se distinguen dos formas clínicas funda-mentales: • La ESP con afectación cutánea limitada, que puede presentarse
como síndrome de CREST (Calcinosis, fenómeno de Raynaud, alteraciones Esofágicas, eSclerodactilia, Telangiectasias). Es más frecuente en niños y mujeres jóvenes (MIR 95-96, 60).
• La ESP con afectación cutánea difusa, con afectación visceral importante.
La esclerosis puede afectar exclusivamente a la piel en la for-ma con afectación cutánea limitada. Dentro de la formas clínicas posibles, la más frecuente es la morfea en placas, que son placas nacaradas escleróticas localizadas en el tronco, no pruriginosas y con un halo adyacente violáceo (lilac ring). No se adhiere a los planos profundos y produce atrofia cutánea con pérdida de los anejos cutáneos. Otras formas son la morfea en gotas, morfea ge-neralizada y la morfea lineal (que se denomina en golpe de sable si se localiza en la cara).
Tabla 27. Diferencias entre afectación difusa y limitada.
asufiD adatimiL
nóicatcefAaenátuc
yarac,lamixorp,latsiD.ocnort
aatcefaoN.aracylatsiD.ocnort
duanyaRoiverpoñaleneazneimoC
senoicatsefinamsala.saenátuc
edsetnasoñaazneimoC.aenátucnóicatcefaal
aipocsoralipaC,sadatalidseralipacsasAedadidrépnocsasoutrot
.seralipacsasa
nissasaednóicataliDsasaedadidrép
seralipac
nóicatcefAlarecsiv
.ranomlupsisorbiF•.selanersisirC•
.airamirprailibsisorriC•.ranomlupnóisnetrepiH•
sopreucitnA1asaremosiopotitnA
.)07LCSitna(.oremórtnecitnA
osruC .ovisergorpetnemadipáR .ovisergorpetnematneL
8.1. Etiopatogenia.
Aunque es desconocida, se asocian factores genéticos y ambien-tales que producen cuadros similares (sílice, aceite de colza, clo-ruro de polivinilo, bleomicina) como causas de esta enfermedad. Se cree que la patogenia es debida a 3 mecanismos: anomalías inmunológicas, alteraciones vasculares y trastorno en la síntesis del colágeno.
Un agente lesivo frente al endotelio desencadenaría la activa-ción de diferentes tipos celulares, que sintetizarían factores pro-inflamatorios que exacerbarían el daño endotelial, manteniendo a los fibroblastos en una situación de activación permanente, produciendo colágeno de caraterísticas normales pero en canti-dad excesiva.

miniMANUAL 1 CTO
Pág. 26
Alteración del sistema inmuneInmunidad celular
Daño en el endotelio vascularEngrosamiento de la íntima, oclusión
de vasos
Plaquetas se adhieren al endotelioActivan la proliferación de los
fibroblastos
Producción anormal dañadode colágeno (excesiva)
Figura 15. Etiopatogenia de la esclerosis sistémica progresiva.
8.2. Manifestaciones clinicopatológicas.
1. Fenómeno de Raynaud (MIR 00-01F, 82; MIR 01-02, 82). Es el primer síntoma de la ES (95%) y expresa la regulación anormal del flujo sanguíneo como consecuencia de las lesiones vascu-lares. El stress y el frío son los factores desencadenantes más habituales. Recuerda la regla de PCR, que indica la cronología de la clínica: Palidez , Cianosis (con parestesias) y Rubor (asociado a dolor por reperfusión).
2. Alteraciones cutáneas. Es la manifestación más característica de la enfermedad. Aparece en todos los pacientes, excepto en la ESP sin esclerodermia. Se inicia en las zonas distales y avanza proximalmente. La rapidez de los cambios cutáneos se relaciona con la mayor gravedad de la afectación visceral. Se diferencian tres fases en la afectación cutánea:1. Fase edematosa (hinchazón, sobre todo en los dedos de las
manos).2. Fase indurativa (la piel aparece engrosada y tirante, adherida
al tejido celular subcutáneo).3. Fase atrófica (la epidermis se adelgaza).
Los pacientes suelen acudir al médico en la fase indurativa. En la cara es típico observar la pérdida de los pliegues lineales (las arrugas) que confiere inexpresividad al rostro, la microstomía (la piel engrosada y dura impide la apertura de la boca) y un aumento de los surcos peribucales verticales.
La piel pierde sus anejos debido a la infiltración colágena por debajo de la epidermis. Además se encuentra a tensión, lo que produce una limitación de la movilidad y ulceraciones en los ex-tremos de los dedos y en las prominencias óseas (donde la piel ya isquémica por la enfermedad puede ser presionada y sufrir una mayor hipoperfusión).
En las formas con afectación cutánea limitada es frecuente la calcinosis en forma de depósitos cálcicos localizados en tejido celular subcutáneo.
3. Alteraciones musculoesqueléticas. Pueden estar presentes desde el inicio de la ES en forma de rigidez, dolor articular e incluso poliartritis no erosiva. La infiltración colágena produce engrosamientos tendinosos, que dan lugar a crepitación y en la muñeca a un sd del túnel del carpo. En fases avanzadas, la sinovial se fibrosa, produciéndose contracturas en flexión por la limitación de la movilidad. Dicha limitación de la movilidad (por afectación de la piel y de las articulaciones) aboca a una atrofia muscular por desuso. Es típica la acroosteólisis o reabsorción de los penachos de las falanges distales.
4. Alteraciones gastrointestinales: es la manifestación visceral más frecuente. La afectación se produce por el adelgazamiento de la mucosa y atrofia de la capa muscular junto con depósito de colágeno en la submucosa, lámina propia y serosa. Puede afectarse cualquier zona del tracto intestinal y se caracteriza por la pérdida de peristalsis. • Esófago. La fibrosis impide la normal motilidad del esófago
en los 2/3 inferiores, perdiéndose la peristalsis y la función del esfínter esofágico inferior. Esto provoca la aparición de reflujo gastroesofágico, que evolutivamente termina produ-ciendo metaplasia de Barrett. Los pacientes con fenómeno de Raynaud aislado (sin conectivopatía) presentan también alteraciones en la motilidad esofágica. Puede haber retraso en el vaciamiento gástrico.
• Intestino delgado. La atonía originada por la fibrosis produce obstrucción intestinal (con distensión acompañante), íleo paralítico y malabsorción (ya que el estasis produce so-brecrecimiento bacteriano por fenómeno de asa ciega). La neumatosis cistoide es una complicación rara, consistente
en la aparición de quistes en la pared intestinal que pueden romperse y producir neumoperitoneo.
• Intestino grueso. La alteración de la motilidad se traduce en estreñimiento. La atrofia de la capa muscular produce divertículos de boca ancha. Menos frecuente es la aparición de incontinencia y prolapso rectal. Puede haber hemorragias por rotura de telangiectasias.
• Alteración hepática. No es común, excepto la cirrosis biliar pri-maria asociada a las formas de afectación cutánea limitada.
5. Afectación pulmonar. Segundo órgano más frecuentemente afectado y primera causa de muerte de la ES (MIR 03-04, 15). Se produce una fibrosis pulmonar basal difusa (por infiltración co-lágena en las tabiques alveolares) que se manifiesta por disnea de esfuerzo y tos seca. Presenta un patrón restrictivo en la espirometría (como las enfermedades puramente intersticiales) y típicamente hay disminución de la DLCO ( signo más precoz de afectación).
El daño en los pequeños vasos produce una oclusión de los mismos, que origina una hipertensión pulmonar. La afectación cutánea limitada puede asociarse a HTP primaria (en el 10% de los casos) (MIR 96-97F, 93).
Hay un riesgo aumentado de padecer cáncer de células alveo-lares (recuerda que las cicatrices pulmonares son un factor de riesgo, y la fibrosis es lo mismo que la cicatrización)
Se pueden producir también neumonías aspirativas (secunda-rias a la afectación esofágica).
6. Alteraciones cardíacas. Se asocian a mal pronóstico. Lo normal es encontrar una pericarditis asintomática con o sin derrame. También es posible la insuficiencia cardíaca, los bloqueos y las arritmias (por fibrosis en el sistema de excitación conducción) La alteración vascular propia de la enfermedad es la responsable de que se produzca angina de pecho por vasoespasmo, lo que da lugar a una alteración patológica característica denominada “necrosis en banda”.
7. Afectación renal. Hasta la aparición de los IECAs, era la 1ª causa de muerte. Es más frecuente en la forma de afectación difusa. La alteración vascular daña los vasos interlobulares y las arte-riolas aferentes y glomerulares (al igual que en la hipertensión maligna), produciendo una isquemia que activa al eje renina-angiotensina (que es contrarrestado mediante los IECAs) y da lugar a una crisis renal.
Las crisis renales suelen precederse por ciertos signos (pre-monitorios), como la anemia hemolítica microangiopática y el derrame pericárdico crónico. La clínica de las crisis renales es igual a la de la hipertensión arterial maligna (encefalopatía, cefalea, convulsiones, retinopatía e insuficiencia cardíaca). En el riñón produce hematuria y proteinuria, que se siguen de oliguria e insuficiencia renal (MIR 98-99, 87; MIR 97-98, 209).
8. Otras alteraciones. La fibrosis patológica puede afectar a otras partes del cuerpo, originando un síndrome seco, hipogonadis-mo, neuropatía periférica y neuralgia del trigémino.
Como enfermedad inmune que es, se puede asociar a otras autoinmunes (recuerda que el factor de riesgo más importante para sufrir una enfermedad autoinmune es padecer ya otra), un síndrome de Sjögren secundario o una tiroiditis autoinmune,
8.3. Datos de laboratorio.
Es habitual la elevación de la VSG.La anemia de trastornos crónicos es la más común, pero también es posible la anemia ferropénica por sangrado digestivo crónico, macrocítica por déficit de vitamina B12 o de ácido fólico secundario a sobrecrecimiento bacteriano o hemolítica microangiopática por la hipertensión maligna.
Casi todos los pacientes presentan ANAs (95%) con diferentes especificidades en función del cuadro clínico asociado.
Podemos encontrar también hipergammaglobulinemia, y en el 25% de los casos, factor reumatoide.
8.4. Diagnóstico.
Cuando la enfermedad alcanza una fase avanzada el diagnóstico es obvio. Sin embargo, para el diagnóstico precoz de la enfermedad, resultan muy útiles los hallazgos de la capilaroscopia, que estudia la microcirculación del lecho ungueal y demuestra la alteración endotelial típica de esta enfermedad. (MIR 94-95, 191).

Reumatología
Pág. 27
Tabla 28. Criterios para la clasificación de la ESP.
Criterio mayor.• Esclerodermia proximal (a las articulaciones MCF).
Criterios menores.• Esclerodactilia.• Fibrosis pulmonar bibasal.• Cicatrices puntiformes en los pulpejos de los dedos.
El diagnóstico se establece cuando se presenta un criterio mayor o dos criterios menores.
Los criterios diagnósticos se exponen en la tabla 28, aunque resultan ineficaces para detectar formas poco evolucionadas o incluso las formas con afectación cutánea limitada.
8.5. Evolución y pronóstico.
La evolución es más favorable para las formas con afectación cutá-nea limitada (supervivencia a 10 años 75%), exceptuando aquellos casos en que se produce hipertensión pulmonar primaria o cirrosis biliar primaria. El pronóstico es peor en las formas con afectación cutánea difusa (supervivencia a los 10 años 55%) con afectación visceral. (MIR 04-05, 84; MIR 99-00F, 98).
TEMA 9. ARTRITIS INFECCIOSAS.
Proceso infeccioso con tendencia a la destrucción articular se-cundaria a la colonización por un germen de la articulación. En el 90% de los casos se presenta como una monoartritis aguda. La vía de infección más frecuente es la hematógena, aunque también es posible la extensión desde una infección vecina o por inoculación externa.
9.1. Artritis no gonocócica.
Los pacientes afectos suelen tener factores predisponentes a la in-fección articular, como son la AR, el tratamiento con corticoides, la diabetes mellitus, las neoplasias malignas o la hemodiálisis. Las in-fecciones neuomocócicas son frecuentes en los pacientes alcohólicos, aquellos con déficit humoral o los que sufren hemoglobinopatías.
CLÍNICA.El 90% de los pacientes presentan una monoartritis aguda, siendo la rodilla la articulación más frecuentemente afectada, seguida por la
cadera. En los pacientes ADVP es muy característica la afectación de la columna, las articulaciones sacroilíacas o esternoclaviculares.
Se manifiesta por un dolor moderado en la articulación acompañado de derrame, espasmo muscular y limitación de los movimientos.
Tabla 30. Etiología de las artritis.
..� '����9
.aicnatcaLsuerua.S .
.sairetcaboretnE.BopurgsuccocotpertS
.soña5<eazneulfni.H .)nóicanucavonis(
suerua.S nóicanucavis( eazneulfni.H .)AopurgsuccocotpertS anucavis( eazneulfni.H .)
.soña04-51.ococonoG
.suerua.S
.soña04> .suerua.S
DIAGNÓSTICO.En la analítica se observan datos de infección (aumento de la VSG, PCR, anemia y leucocitosis). El diagnóstico confirmatorio se realiza mediante el análisis del líquido sinovial, de características infla-matorias. La tinción de Gram del líquido es positiva en el 75% de las infecciones por Staph. aureus y en el 30-50% de las infecciones por BGN. El cultivo sinovial es positivo en el 90% de los casos y los hemocultivos son positivos en el 50% de las infeccione por Staph. aureus.
TRATAMIENTO.Se administrará antibioticoterapia en función del organismo sos-pechado.
9.2. Artritis gonocócica.
La artritis gonocócica es la causa más frecuente de artritis séptica en los menores de 40 años. Se produce secundariamente a una bacterie-mia por infección gonocócica o a una colonización del tracto genital. Las mujeres están más predispuestas que los varones a padecer esta afección (sobre todo en la menstruación y el embarazo), ya que las infecciones gonocócicas suelen ser asintomáticas en ellas (por eso no se tratan y generan gonococemias). Las personas con déficit en los fac-tores C5-C9 del complemento también tienen mayor susceptibilidad a padecer gonococemia diseminada.
Tabla 29. Tratamiento de la ES.
��'0�� �-'���-�-
����:������������������
.ADIGÍRYARUDLEIPlarenegnE•.ralucitrairepsisoniclaC•
erbosysodedsoledsameysalnesareclÚ•.saesosaicnenimorp
etsoorcA• ol .soenátucsojenaedadidrépysisitsorciM• ímo .laicafnóiserpxeedadidrépya
• .)?(anirafraW• azeipmil,ovisulcoejadnev,odigírrotcetorP issBA,acinácem
.atcefnies• y ocioznebonimarapodicá,anicihcloc,animalicinep-D
.Eanimativ
����������������� zedilaP• → sisonaiC → robuR .)RCP:alger(• i,aniresnatek,oiclacledsatsinogatnA l sámtsorpo
.ramufon,sértseedosnecsed,oirflednóiccetorp
������������������������������
.acirtémissitirtrailoP•.onaipraclenútledemordníS•
• osENIA .)aíd/gm01(sediocitroc• .aiparetoisiF
���������������������������������
julferedsitigafosE• .o
,lanedouddadilitomopih(nóicrosbalaM•onairetcabotneimicercerbos ).
• .)nolocleddadilitomopih(otneimiñertsE
• sodicáitna,selarutsopysacitéteidsadideM y serotercesitna .
• itnA b .)setnetimretnisolcic(socitói
• .sevaussetnaxal,secehedserodadnalbA
����������������������• .laneraicneicifusnI• .ATH
• ysACEI d .)avisergorpRI(sisilái• rporp:serosnetopiH ona .olidixonimyanidinolc,lol
������������������������
• .sitidracoimysitidracireP• .CI• .ocidrácirepemarreD• .cte,soeuqolb,saimtirra:sortO
• .)ozalpogralasodacidniON(sediocitroC• .)ahcertseaicnaligiv(socitéruid,latigiD

miniMANUAL 1 CTO
Pág. 28
La artritis suele encuadrarse dentro de la enfermedad gonocó-cica diseminada. Esta se manifiesta por fiebre, erupción pustulosa hemorrágica en el tronco y extremidades y artritis migratoria con tenosinovitis de rodillas, manos, muñecas y pies.
DIAGNÓSTICO.Se confirma mediante el cultivo y la tinción de gram positiva del líquido sinovial. También es posible realizar hemocultivos y cultivos del tracto urogenital para confirmar la existencia de gonococo. La radiología sirve para el seguimiento del proceso, pero no para el diagnóstico inicial. La gammagrafía con Tc99 es un método muy sensible pero poco específico para localizar las zonas inflamadas. La ecografía visualiza los derrames y el TAC y la RMN sólo se usan en caso de sospecha de absceso u osteítis. TRATAMIENTO.Se requiere CEFTRIAXONA vía iv. En algunos casos es necesario el drenaje de la articulación.
Tabla 31. Tratamiento de las artritis.
nóiccefnI sámsenemréGselbaborp nóicceleedocitóibitnA
acitpéssitirtrA)sesem2<(
suerua.S ,sucoccotpertS ,
NGB
aíd/gK/gm001anilicaxolC001amixatofeC+
aíd/gK/gm
acitpéssitirtrA)soña3-sesem2(
eazneulfni.H ,suerua.S ,
sucoccotpertS
aíd/gK/gm001anilicaxolC001amixatofeC+
aíd/gK/gm
acitpéssitirtrA)soña3>(
suerua.S aíd/gK/gm001anilicaxolC
acicóconogsitirtrA eaeohrronog.N aíd/g1anoxairtfeC
PVDAnesitirtrA suerua.S NGB,+.v.ih8/g2anilicaxolCaíd/gm042anicimatneG
nesitirtrAsodimirpedonumni
sairetcaboretnEsanomoduesP
asonigureamarGsocoC
sovitisop
+.v.ih8/g2amidizatfeC.v.iaíd/g1anicakimA
nóiccefnIairotarepotsop
sucoccolyhpatS ,NCB
+.v.ih21/g1anicimocnaV.v.iaíd/g1anicakimA
atreibaarutcarF
marGsocoCsovitisopysoiborea
,soiboreanasoidirtsolc
ocináluvalc/anilicixomA
TEMA 10. AMILOIDOSIS.
La amiloidosis es un grupo de enfermedades en las que se deposita en el tejido extracelular una proteína de estructura fibrilar cuyas características histológicas son las que permiten su diagnóstico.
La sustancia amiloide tiene un componente constante o “p”, no fibrilar, que se parece a la proteína C reactiva y se origina en el hígado, y otro variable o fibrilar, que es quien proporciona las características tintoriales. Existen varias formas de amiloidosis:a. La amiloidosis 1ª (AL), idiopática o asociada al mieloma: es la
forma más frecuente. El componente fibrilar son las cadenas ligeras de las inmunoglobulinas (λ / κ = 2/1 ).
b. La amiloidosis 2ª (AA), que es la complicación de una enferme-dad inflamatoria crónica o de una infección.
Existen otras formas de amiloidosis, como las heredofamiliares, la asociada a la hemodiálisis (beta2microglobulina) o las localizadas. (MIR 01-02, 85; MIR 97-98F 17; MIR 01-02, 60; MIR 00-01F, 80; MIR 00-01F, 215).
10.1. Manifestaciones clínicas.
Las manifestaciones clínicas dependen de los órganos donde se ha depositado el amiloide. La proteinuria ( que se manifiesta en forma de edemas por disminución de la presión oncótica) es la primera manifestación de la amiloidosis AA y AL.1. Riñón. La afectación renal se produce en el 70-90% de los pacien-
tes. Es la primera causa de muerte en la amiloidosis secundaria (AA) y la segunda causa, después de la afectación cardíaca, en la amiloidosis primaria (AL). El material amiloide se deposita en el mesangio y en la pared capilar. Al dañar la membrana de filtración se produce proteinuria, que llega hasta rango nefrótico. Es muy frecuente la trombosis de la vena renal como compli-cación del síndrome nefrótico. La amiloidosis también puede manifestarse como daño tubular (síndrome de Fanconi, si altera el tcp, acidosis tubular renal y diabetes insípida nefrogénica si altera el tcd).
2. Corazón. Se observa en la amiloidosis primaria como una insuficiencia cardíaca tipo restrictivo (la infiltración impide la normal relajación del corazón en la diástole) que no responde al tratamiento. Si el depósito se realiza en el sistema de conduc-ción, se producen arritmias y bloqueos En la ecocardiografía se visualizan los depósitos fibrilares como hiperecogénicos, dando la imagen de “centelleo granular” .
3. Aparato digestivo. La infiltración del tubo digestivo también es habitual; el amiloide se deposita en la pared y produce obs-trucción de la luz, úlceras y dificulta la absorción (por lo que se pierden proteínas y el aumento de residuos origina diarreas). Las hemorragias son consecuencia de la fragilidad de los capilares. Es típica la macroglosia en la forma 1ª. La infiltración hepática cursa con hepatomegalia y aumento de las enzimas de la colestasis (MIR 96-97, 113).
4. Aparato locomotor. La forma AL asociada al mieloma es la que más frecuentemente afecta a las articulaciones produciendo ar-tritis, al depositarse el amiloide en el cartílago y en la membrana sinovial. Cuando se infiltran los músculos del hombro, puede observarse el signo del “almohadillado del hombro u hombro de jugador de rugby”.
5. Piel. La afectación cutánea es una de las manifestaciones más características de la amiloidosis primaria. Típicamente se obser-va la tríada formada por el sd de túnel carpiano, la macroglosia y las lesiones mucocutáneas. Las lesiones más frecuentes son las petequias y equimosis espontáneas (ya que el amiloide infiltra los capilares, haciéndose estos más frágiles) que producen el sd del ojo negro si se localizan alrededor de la órbita.
Las segundas en frecuencia son las pápulas céreas en la zona de la cara y cuello. Según qué estructura infiltre el amiloide se produce alopecia ( folículo piloso), cambios esclerodermiformes (difusamente), lesiones ampollosas, lesiones ungueales (matriz ungueal) o Sd de Sjögren (gl. exocrinas).
La amiloidosis puede afectar exclusivamente a la piel (forma locali-zada), dando lesiones pruriginosas de las cuales el liquen amiloideo es la más frecuente (lesiones verrucosas en placas en cara anterior de la piernas).También puede producir máculas y nódulos.
6. Sistema nervioso. Se afecta frecuentemente en las formas he-redofamiliares, manifestándose como neuropatías periféricas y alteraciones vegetativas. Recuerda que el sd de Down y la en-fermedad de Alzheimer se engloban dentro de las amiloidosis. El síndrome del túnel carpiano aparece especialmente en la amiloidosis primaria (AL) y en la asociada con la hemodiálisis crónica (β2 M).
7. Sistema endocrino. La infiltración del tejido de las glándulas endocrinas es habitualmente asintomática. Característicamente se visualiza amiloide en el cáncer medular de tiroides y en los islotes pancreáticos de la DM tipo 2 evolucionada.
8. Aparato respiratorio. El amiloide se deposita a cualquier nivel del árbol respiratorio, afectando a las vías inferiores en la ami-loidosis AL (en forma de un patrón restrictivo difuso, que se acompaña de disnea). Cuando se deposita en las vías respira-torias superiores, obstruye su luz, favoreciendo la infección.
10.2. Diagnóstico, pronóstico y tratamiento.
El diagnóstico específico se obtiene observando los depósitos de material amiloide hialino en el tejido.Las localizaciones más

Reumatología
Pág. 29
rentables para realizar la biopsia son el recto, el riñón y la grasa abdominal.
Característicamente el amiloide se tiñe con rojo Congo y muestra birrefringencia verda manzana a la luz polarizada. La hematoxilina –eosina muestra los depósitos de color rosado translúcido y fibri-lar. La forma 1ª se diferencia de la forma 2ª porque es resistente al permanganato potásico. Se tiñe de amarillo con yodo y de azul con el ácido sulfúrico.
La amiloidosis es una enfermedad lentamente progresiva que suele matar al paciente tras años de evolución. Las principales causas de muerte son la alteración renal y cardíaca.
El tratamiento se ha basado en el trasplante de hígado, riñón y en las amiloidosis por mieloma, se utilizan fármacos alquilantes (M2-Pr, melfalán –prednisona).
TEMA 11. SÍNDROME DE SJÖGREN.
El síndrome de Sjögren es una enfermedad autoinmune producida por la infiltración por linfocitos CD4 de las glándulas exocrinas del organismo. Es 9 veces más frecuente en las mujeres y suele debutar en la edad media de la vida.
Existe una forma primaria, que suele asociarse a un inicio más precoz, una mayor duración de la enfermedad y mayor afectación extraglandular.
La forma secundaria se asocia a otras enfermedades autoinmu-nes, siendo la AR la causa más frecuente de síndrome de Sjögren 2º.
Se cree que la patogenia está relacionada con un aumento de los linfocitos CD4 que activan a los linfocitos B, los cuales producen anticuerpos, entre ellos el Anti-Ro (60%) y Anti-La (50%), el FR (80%) y los ANAs. Se cree que existe cierta susceptibilidad genética , siendo más frecuente la enfermedad en los pacientes HLA DR3.
11.1. Manifestaciones clínicas.
1. La xerostomía o sequedad bucal es la manifestación más fre-cuente.
2. La xeroftalmía, por infiltración de las glándulas lacrimales, que origina una queratoconjuntivitis seca (manifestada por sensa-ción de cuerpo extraño en el ojo. (MIR 94-95, 259, 24).
Las manifestaciones extraglandulares son propias del Sjögren primario (30%) y son las que marcan el pronóstico. 1. Articulares. Es habitual la aparición de artralgias y en algu-
nos casos una poliartritis no erosiva.2. Pulmonares. Se suele producir una enfermedad pulmonar
intersticial difusa, de predominio en las bases, que suele ser asintomática.
3. Renales. Se puede producir daño en el glomérulo o en el intersticio. En el glomérulo se manifiesta como una GNF membranoproliferativa o membranosa. El intersticio es infiltrado por linfocitos (nefritis linfocitaria), lo que altera su función, dando lugar a anemia Fanconi, ATR y a DIN (hi-postenuria). También puede producirse nefrocalcinosis.
4. Vasculitis. Se produce una vasculitis leucocitoclástica o una vasculitis que afecta a vasos de pequeño y mediano calibre.
5. Neurológicas. Suele afectarse el SNP por la vasculitis, siendo posible también la aparición de síntomas del SNC .
6. Fenómeno de Raynaud en el 30% de los pacientes.
Suele presentarse adenopatías en el sd de Sjögren. En los pacien-tes con síndrome de Sjögren hay una mayor incidencia de linfomas no Hodgkin de células B y macroglobulinemia de Waldenström (suelen ser una manifestación de las fases avanzadas de la enfer-medad). La presencia de un linfoma debe ser sospechada cuando un paciente con Sjögren presente tumefacción parotídea de forma prolongada o reducción de los títulos de factor reumatoide. (MIR 94-95, 24; MIR 01-02, 83).
11.2. Diagnóstico.
El diagnóstico se realiza mediante la clínica y pruebas que indican hipofunción glandular. El diagnóstico confirmatorio se consigue gracias a las biopsias, en las que se observa la infiltración linfocitaria característica.
La xerostomía se diagnostica mediante sialometría, sialografía o gammagrafía. La biopsia de la glándula salivar menor posee una alta rentabilidad para confirmar la sequedad de boca.
La xeroftalmía se comprueba gracias al test de Schirmer tipo I (se coloca papel adsorbente en el párpado inferior. Según la longitud de papel empapado en un tiempo determinado, se conoce la cantidad de lágrima producida). La tinción de rosa Bengala permite observar ulceraciones corneales puntiformes, debidas al daño de la córnea por la falta de protección que proporciona la película lacrimal.
Tabla 32. Formas del síndrome de Sjögren.
º1NERGÖJS HIVROPNERGÖJS
.LOIMEDIPE aidemdade,rejum nevojnórav
CITNAOTUA aL-itnAyoR-itnA ON
CARTLIFNI 8DC 4DC
ALH 5RD 3RD
HIV OVITISOP OVITAGEN
11.3. Tratamiento.
No existe tratamiento específico. Se alivia los síntomas de xeros-tomía y xeroftalmía mediante colutorios de bromhexidina oral y lágrimas artificiales.
Las manifestaciones articulares pueden responder a la hidroxi-cloroquina. Los corticoides y los inmunosupresores se reservan para la afectación visceral grave.
TEMA 12. POLIMIOSITIS Y DERMATOMIOSITIS.
Las miopatías inflamatorias idiopáticas son enfermedades muscu-lares poco frecuentes que cursan con debilidad muscular, funda-mentalmente proximal y simétrica, y con inflamación no supurativa del músculo esquelético (MIR 96-97F, 97).
Clasificación de la Polimiositis-Dermatomiositis (Bohan y col).• Grupo I: polimiositis idiopática primaria.• Grupo II: dermatomiositis idiopática primaria.• Grupo III: dermatomiositis o polimiositis asociada a neoplasia: en
mayores de 50 años y ancianos. Asociados a tumores de mama, pulmón, ovario, gastrointestinales y mieloproliferativos.
• Grupo IV: dermatomiositis o polimiositis infantil asociada con vasculitis: afectación cutánea con signos de vasculitis sistémica y calcificación subcutánea
• Grupo V: polimiositis o dermatomiositis asociada con enferme-dad del colágeno.
DIAGNÓSTICO.Se requiere cuadro clínico típico elevación de CPK, EMG caracte-rístico, biopsia muscular positiva.
TRATAMIENTO.De elección los corticoides en dosis altas. Si no hay respuesta, se prueba con la azatioprina. Las manifestaciones cutáneas responden a la hidroxicloroquina.
TEMA 13. ARTROSIS.
13.1. Definición.
La artrosis es la enfermedad articular más frecuente y consiste en la insuficiencia de las articulaciones diartrodiales( móviles, con sinovial). Se puede distinguir una artrosis primaria ó idiopática y otra secundaria, asociada a traumatismos, anomalías del desarrollo, enfermedades metabólicas, endocrinas, depósito de cristales de calcio, enfermedades óseas o neuropática.
El factor de riesgo más importante para padecer artrosis es la edad (MIR 98-99F, 210). Otros factores asociados parecen ser la obesidad

miniMANUAL 1 CTO
Pág. 30
(que produce sobrecarga de las articulaciones), los traumatismos y el sobreuso de la articulación. También parece existir factores hereditarios implicados. La artrosis afecta a más del 50% de los sujetos con más de 65 años.
13.2. Manifestaciones clínicas.
Los síntomas característicos son el dolor de tipo mecánico, la rigidez de corta duración ( al contrario que en las enfermedades articulares inflamatorias), la crepitación y la limitación de la movilidad. En fases avanzadas puede producirse sinovitis por desgaste de la arti-culación y en los casos más evolucionados hasta deformidades.
En la artrosis primaria, los exámenes de laboratorio son normales y el líquido sinovial es de tipo no inflamatorio(MIR 00-01F, 81).
En la radiografía se observan los signos característicos de la artrosis: el pinzamiento articular asimétrico (en la AR es simétrico), la esclerosis subcondral, (en la AR hay erosiones), los osteofitos( en la AR se observa osteoporosis yuxtaarticular), las geodas y la defor-midad articular(MIR 96-97, 118).
FORMAS CLÍNICAS MÁS IMPORTANTES.Artrosis de IFD. Presenta herencia AD. Predomina en las mujeres. Se manifiesta con los nódulos de Heberden.
Artrosis de IFP. Produce los característicos nódulos de Bouchard.
Artrosis del pulgar o rizartrosis. Más frecuente en mujeres y asociado a los nódulos de Heberden. Provoca dificultad para hacer el gesto de la pinza (con el dedo índice y pulgar)
Artrosis coxofemoral. Es más frecuente en varones. Cursa con dolor inguinal acompañado de cojera y dificultad para levantarse o sentarse de una silla.
Artrosis de rodilla. Es la principal causa de discapacidad cró-nica en ancianos. Predomina en mujeres, con comienzo habitual-mente unilateral y con tendencia posterior a hacerse bilateral. Se presenta como dolor en la interlínea articular o en la cara anterior de la rodilla.
Artrosis vertebral. Puede afectar a cualquier nivel. Denomina-mos espondilosis a la enfermedad degenerativa de los discos junto con artrosis vertebral de las articulaciones interapofisarias. En la columna cervical produce cervicalgia y en los casos más graves puede llegar a comprimir la médula ósea. En la columna dorsal produce dolor a la rotación del tronco, y a nivel lumbar afecta a las últimas vértebras( L4-L5 –S1)
13.3. Tratamiento.
No existe un tratamiento definitivo, por lo que la finalidad es evitar la pérdida de movilidad y la aparición de dolor.A Se recomienda realizar ejercicios de tipo isométrico y medidas para
disminuir la carga sobre la articulación (como por ejemplo, dismi-nuir de peso para que la articulación soporte menos presión).
B. El dolor se controla mediante AINEs, estando aprobados los ICOX 2 para disminuir los efectos secundarios de los clásicos
AINEs.También son de utilidad las inyecciones intraarticulares de corticoides y de ácido hialurónico (estas últimas parecen proporcionar un alivio más duradero).
C. Si tras 6 meses de tratamiento médico correcto persiste una validez importante, se puede optar por la cirugía, sobre todo a nivel de la cadera y la rodilla (artroplastias de sustitución).
TEMA 14. OTRAS ARTROPATÍAS.
14.1. Osteoartropatía hipertrófica (acropaquias).
Se caracteriza por la deformidad de los dedos en palillo de tambor, periostitis y artritis. Aunque existe una forma primaria (herencia AD), lo más frecuente es la forma secundaria asociada a diferentes enfermedades, entre las que destacan las infeccio-nes pulmonares crónicas, la fibrosis quística, la sarcoidosis, la enfermedad inflamatoria intestinal y las neoplasias intestinales y torácicas.
14.2. Fibromialgia.
Es una enfermedad frecuente, cuya prevalencia aumenta con la edad, que se presenta en mujeres (relación 9/1) de más de 50 años.
Aunque no está clara la patogenia ni la etiología de este proceso, sí se han postulado diversos factores como los causantes de la fibromial-gia. Entre ellos destacan: los trastornos del sueño, principalmente de la fase 4; déficit de serotonina o de la hormona de crecimiento, que juegan un papel en la regulación del dolor, el sueño y la forma física; alteraciones psicológicas como la depresión, la ansiedad, la somati-zación y la hipocondría; alteraciones del SN autónomo ; disminución del umbral del dolor y de la resistencia muscular.
Hay algunas enfermedades psicosomáticas asociadas con la fibromialgia, que apoyan la relación entre esta enfermedad y ciertas alteraciones psiquiátricas.
CLÍNICA.Cursa con un dolor sordo generalizado junto con rigidez del tronco y de las cinturas escapular y pélvica. La rigidez es máxima por la mañana y va aliviándose a lo largo del día. Las pacientes presen-tan problemas de sueño (despertares precoces), debilidad y mala tolerancia al ejercicio.
En la exploración física se encuentran los característicos puntos gatillo, en los que se despierta dolor a la palpación. También pueden existir nódulos subcutáneos en esas mismas áreas. No hay signos inflamatorios y las pruebas complementarias son siempre normales. La enfermedad suele tener un curso crónico.
DIAGNÓSTICO.Se requiere una historia crónica de dolores generalizados y de-mostrarse la presencia de 11 de los 18 puntos dolorosos con una exploración articular y muscular normal.
Tabla 33. Características de las miositis.
SITISOIMOTAMRED SITISOIMILOP NÓISULCNISOPREUCAÍTAPOIM
H/VNÓICALER 2/1 2/1 1/3
TUBEDDADE sotluda-soñiN soña81edsáM soña05>
NÓICAZILACOL acirtémislamixorP acirtémislamixorP acirtémisa,latsidylamixorP
AÍNOFSID/AIGAFSID %02-01 %02-01 %06
SAENÁTUCSENOISEL )ocinómongotap(íS on on
KPCEDSELEVIN sodavelE sodavelE sodavelE
SOPREUCITNA )acisálcMD(2iMitnA %04-03 sovitageN
AISPOIBTfnil,4DC.ralucsavirepylaisimireP
ByTnil,sogafórcaM.laisimodnE
8DC,.8DC,Tfnil,sogafórcaM.laisimodnE
.sadaetebirsaloucaV.ediolimaotisópeD
atsahTORavresnoc(GME)sadaznavasesaf
ocitápoiM ocitápoiM ocitáporuen/ocitápoiM

Reumatología
Pág. 31
Tabla 34. Diagnóstico diferencial de la Fibromialgia.
AIGLAIMORBIF AIGLAIMILOPACITÁMUER SITISOIMILOP
DADE05-04rejuM
soñasoña05>rejuM
serejuMsenevój
ACINÍLCzedigiryroloD
ralucsumadazilareneg
neroloD.sarutnic
zedigiRralucsum
dadilibeDralucsum.lamixorp
ACITÍLANAKPCYGSV
LAMRON
KPC,ATLAGSV,LAMRON
.A.F,AIMENAATLA
KPC,ATLAGSVATLA
+GMEAISPOIB
RALUCSUMLAMRON LAMRON OCIGÓLOTAP
OTNEIMATART-ENIA
soviserpeditnAsediocitroCsajabsisod
sediocitroCsatlasisod
TRATAMIENTO.Es necesario explicar a la paciente las características de la enferme-dad y su curso crónico. Se utilizan los AINE como medida sintomá-tica. Fármacos muy útiles son los antidepresivos y los ansiolíticos.
ANEXO. RESUMEN DE LAS PRINCIPALES ENFERMEDADES REUMATOLÓGICAS.
Ver figura 16 en la página siguiente.
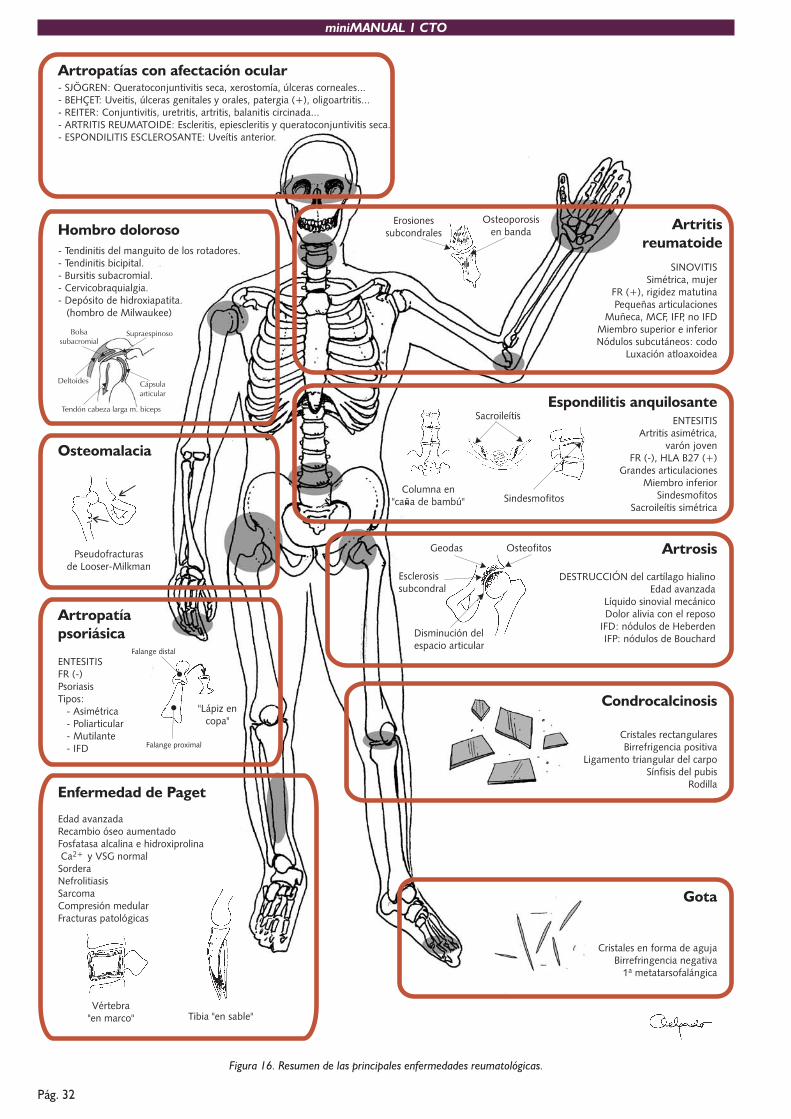
miniMANUAL 1 CTO
Pág. 32
- SJÖGREN: Queratoconjuntivitis seca, xerostomía, úlceras corneales...- BEHÇET: Uveitis, úlceras genitales y orales, patergia (+), oligoartritis...- REITER: Conjuntivitis, uretritis, artritis, balanitis circinada...- ARTRITIS REUMATOIDE: Escleritis, epiescleritis y queratoconjuntivitis seca.- ESPONDILITIS ESCLEROSANTE: Uveítis anterior.
- Tendinitis del manguito de los rotadores.- Tendinitis bicipital.- Bursitis subacromial.- Cervicobraquialgia.- Depósito de hidroxiapatita.
(hombro de Milwaukee)
Artropatías con afectación ocular
Hombro doloroso
Osteomalacia
Erosionessubcondrales
Artropatía
psoriásica
Gota
Condrocalcinosis
Artrosis
Espondilitis anquilosante
Artritis
reumatoide
Enfermedad de Paget
Pseudofracturasde Looser-Milkman
ENTESITISFR (-)PsoriasisTipos:
- Asimétrica- Poliarticular- Mutilante- IFD
Cristales en forma de agujaBirrefringencia negativa
1ª metatarsofalángica
Cristales rectangularesBirrefrigencia positiva
Ligamento triangular del carpoSínfisis del pubis
Rodilla
DESTRUCCIÓN del cartílago hialinoEdad avanzada
Líquido sinovial mecánicoDolor alivia con el reposo
IFD: nódulos de HeberdenIFP: nódulos de Bouchard
ENTESITISArtritis asimétrica,
varón jovenFR (-), HLA B27 (+)
Grandes articulacionesMiembro inferior
SindesmofitosSacroileítis simétrica
SINOVITISSimétrica, mujer
FR (+), rigidez matutinaPequeñas articulaciones
Muñeca, MCF, IFP, no IFDMiembro superior e inferiorNódulos subcutáneos: codo
Luxación atloaxoidea
Edad avanzadaRecambio óseo aumentadoFosfatasa alcalina e hidroxiprolinaCa y VSG normalSorderaNefrolitiasisSarcomaCompresión medularFracturas patológicas
2+
Falange distal
Falange proximal
"Lápiz encopa"
Tibia "en sable"
Osteoporosisen banda
Sacroileítis
SindesmofitosColumna en
"caña de bambú"
Geodas Osteofitos
Esclerosissubcondral
Disminución delespacio articular
Vértebra"en marco"
Deltoides
SupraespinosoBolsasubacromial
Tendón cabeza larga m. biceps
Cápsulaarticular
Figura 16. Resumen de las principales enfermedades reumatológicas.


















