
Reporte BLAST
16
UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO FACULTAD DE ESTUDIOS SUPERIORES CUAUTITLÁN DEPARTAMENTOS DE CIENCIAS BIOLÓGICAS SECCIÓN BIOQUÍMICA Y FARMACOLOGÍA HUMANA ALUMNO: López Toledo Gustavo GRUPO: 2001 CARRERA: Bioquímica Diagnóstica PROFESORA: M. en C. Maritere Domínguez Rojas
-
Upload
gustavo-lopez-toledo -
Category
Documents
-
view
84 -
download
1
Transcript of Reporte BLAST

UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICOFACULTAD DE ESTUDIOS SUPERIORES
CUAUTITLÁNDEPARTAMENTOS DE CIENCIAS BIOLÓGICAS
SECCIÓN BIOQUÍMICA Y FARMACOLOGÍA HUMANA
ALUMNO: López Toledo Gustavo
GRUPO: 2001
CARRERA: Bioquímica Diagnóstica
PROFESORA: M. en C. Maritere Domínguez Rojas
Cuautitlán Izcalli, Abril de 2013

En la bioinformática , Basic Local Alignment Search Tool o BLAST , es un algoritmo para comparar información primaria de la secuencia biológica, tales como los aminoácidos secuencias de diferentes proteínas o los nucleótidos de las secuencias de ADN .Una búsqueda BLAST permite a un investigador comparar una secuencia de consulta con una biblioteca o base de datos de secuencias, e identificar secuencias de la biblioteca que se asemejan a la secuencia de consulta por encima de un cierto umbral.
Los diferentes tipos de explosiones están disponibles de acuerdo a las secuencias de la consulta. Por ejemplo, tras el descubrimiento de un gen previamente desconocido en el ratón , un científico suele realizar una búsqueda BLAST de la genoma humano para ver si los seres humanos tienen un gen similar; BLAST identifican secuencias en el genoma humano que se asemejan a los genes del ratón basado en la similitud de secuencia.
BLAST se puede utilizar para varios fines. Esto incluye la identificación de especies, dominios, posicionamiento filogenia establece, mapeo de ADN y la comparación.
La identificación de especies:
Con el uso de BLAST, puede posiblemente identificar correctamente una especie y / o encontrar especies homólogas. Esto puede ser útil, por ejemplo, cuando se está trabajando con una secuencia de ADN de una especie desconocida.
Localización de dominios
Cuando se trabaja con una secuencia de la proteína que puede ingresar en él BLAST, para localizar dominios conocidos dentro de la secuencia de interés.
El establecimiento de la filogenia
Con los resultados recibidos a través de BLAST puede crear un árbol filogenético utilizando el BLAST página web. Filogenias basadas en BLAST solo son menos fiables que otras construidas específicamente filogenética computacional métodos, por lo que sólo se debe confiar en los análisis de "primer paso" filogenéticos.
Secuenciar el ADN
Cuando se trabaja con una especie conocida, y que mira a la secuencia de un gen en un lugar desconocido, BLAST puede comparar la posición cromosómica de la secuencia de interés, a las secuencias correspondientes en la base de datos (s).
Comparación
Cuando se trabaja con genes, BLAST puede localizar genes comunes en dos especies relacionadas, y se puede utilizar para asignar anotaciones de un organismo a otro.

Programas de BLAST Hay un número de variaciones del programa BLAST para comparar el ácido nucleíco o secuencias de
proteínas de consulta con ácido nucleíco o bases de datos de secuencias de proteínas. Si es necesario, los programas de traducir secuencias de ácidos nucléicos en los seis marcos de lectura posibles para compararlas con las secuencias de proteínas.
La selección apropiada de un programa BLAST para una búsqueda dada está influenciada por los siguientes tres factores 1) la naturaleza de la consulta, 2) el objeto de la búsqueda, y 3) la base de datos destinados como el objetivo de la búsqueda y su disponibilidad. Las siguientes tablas proporcionan recomendaciones sobre cómo hacer esta selección.
Programa de Selección para consultas de nucleótidos
Longitud ¹ Base de datos Propósito Programa Explicación
20 pb o más 28 pb o más para megablast
Nucleótidos
Identificar la secuencia de la consulta
megablast contigua , megablasto blastn Más
información ...
Encontrar secuencias similares a la secuencia de consulta
megablast contigua o blastnMás
información ...
Encontrar secuencia similar desde el archivo de
seguimiento
Traza megablast o megablast contigua Rastro
Más información ...
Encuentra proteínas similares a la consulta traducido en una base de datos traducida
BLAST Traducido (tblastx)Más
información ...
PéptidoEncuentra proteínas similares a la consulta traducido en una base de datos de proteínas
BLAST Traducido (blastx)Más
información ...
7 a 20 pb NucleótidosEncontrar sitios de unión o
mapa contiguos motivos cortosBúsqueda de torneos cortos, casi
exactasMás
información ...
NOTA: ¹ El corte es sólo una recomendación. Para consultas cortas, es más probable que obtenga los partidos si la opción "Buscar torneos cortos, casi exactas" de la página se utiliza. Una discusión detallada se encuentra en la sección 4 a continuación. Con el ajuste predeterminado, el menor consulta inequívoca se puede utilizar es 11 para blastn y 28 para MEGABLAST.
Programa de Selección para consultas de proteínas
Longitud ¹ Base de datos Propósito Programa
http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=ProgSelectionGuide#mega
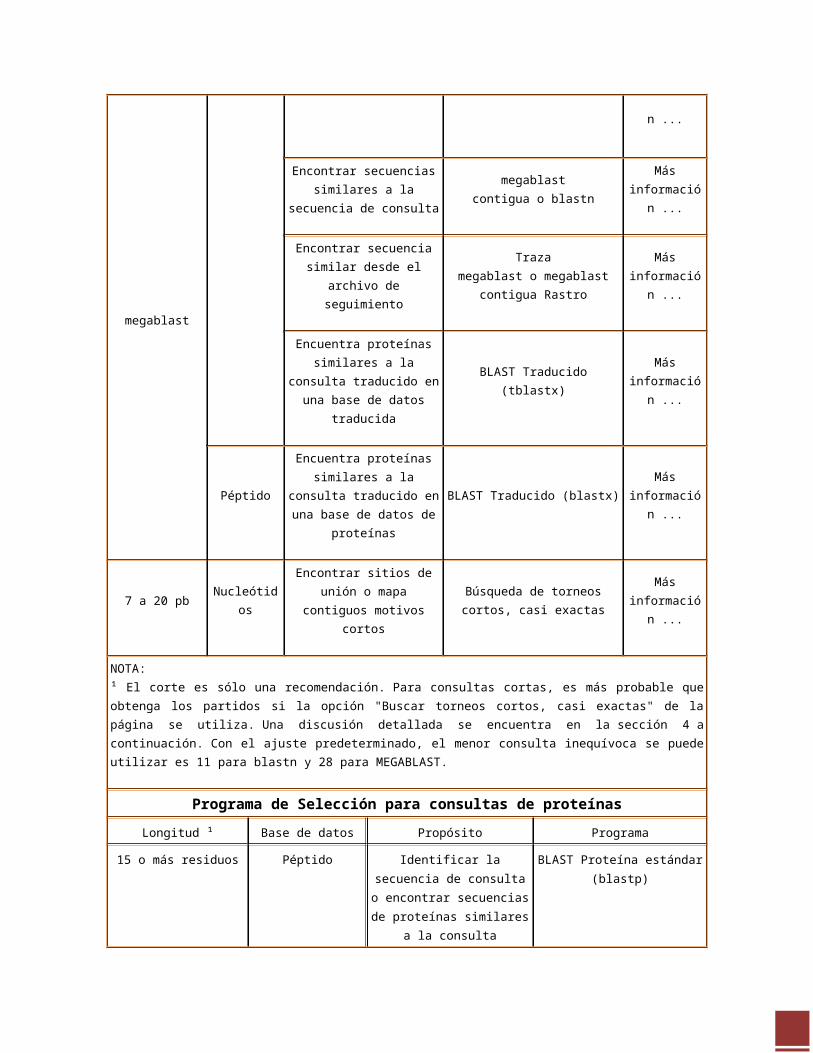
15 o más residuos
Péptido
Identificar la secuencia de
consulta o encontrar
secuencias de proteínas
similares a la consulta
BLAST Proteína estándar
(blastp)
Encuentra los miembros de
una familia de proteínas o
construir una costumbre
posición específica matriz de
puntuación
PSI-BLAST
Encuentra proteínas similares
a la consulta en torno a un
patrón dado
PHI-BLAST
Encuentre los dominios
conservados de la consultaCD-búsqueda (RPS-BLAST)
Buscar dominios conservados
en la consulta e identificar
otras proteínas con
arquitecturas de dominio
similares
Conservada arquitectura de
dominio de recuperación
Herramienta (CDART)
Nucleótidos
Encuentra proteínas similares
en una base de datos de
nucleótidos traducida
BLAST Traducido (tblastn)
5-15 residuos Péptido Buscar por motivos peptídicosBúsqueda de torneos cortos,
casi exactas
Nota:
¹ El corte es sólo una recomendación. Para consultas cortas, es más probable que obtenga los partidos si la
opción "Buscar torneos cortos, casi exactas" de la página se utiliza.
INICIO DE LA BUSQUEDA EN BLASTn
Enter query sequence. Es el espacio designado para introducir la secuencia del usuario. Choose search sets. Permite elegir los parámetros a seguir en la búsqueda de secuencias
para realizar el alineamiento Program Selection. Permite elegir el programa que llevará a cabo el alineamiento. logorithm parameter. Permite modificar los algoritmos que regirán la búsqueda.
En la ventana inicial se puede apreciar un menú principal en donde se pueden encontrar las siguientes opciones:
Recent results. Permite acceder a los resultados obtenidos de forma reciente, cada búsqueda en BLAST proporciona un número ID de búsqueda, éste podrá ser indicado con la finalidad de encontrar el resultado de interés, sin la necesidad de revisar los resultados uno por
Saves strategies. Permite seleccionar parámetros determinados previamente utilizados y guardados por el usuario.
Help. Permite acceder a manuales y guías de uso para las diferentes herramientas que componen el sistema

Home. Esta pestaña permitirá visualizar la ventana de inicio de BLAST. En esta opción se tendrá acceso a una breve descripción de BLAST; además de proporcionar un acceso directo a todas las herramientas disponibles en BLAST.
Compara una secuencia problema de nucleótidos contra una base de datos de secuencias de
Compara una secuencia problema de aminoácidos contra una base de datos de secuencias de proteínas.
Compara una secuencia problema de nucleótidos traducida en sus seis posibles marcos de lectura contra una base de secuencias de proteínas.
Compara una secuencia problema de aminoácidos contra toda la base de datos de nucleótidos traducida en sus seis posibles marcos de lectura.
Compara las seis traducciones en sus marcos de lectura de la secuencia problema de nucleótidos, contra las seis traducciones en sus marcos de lectura
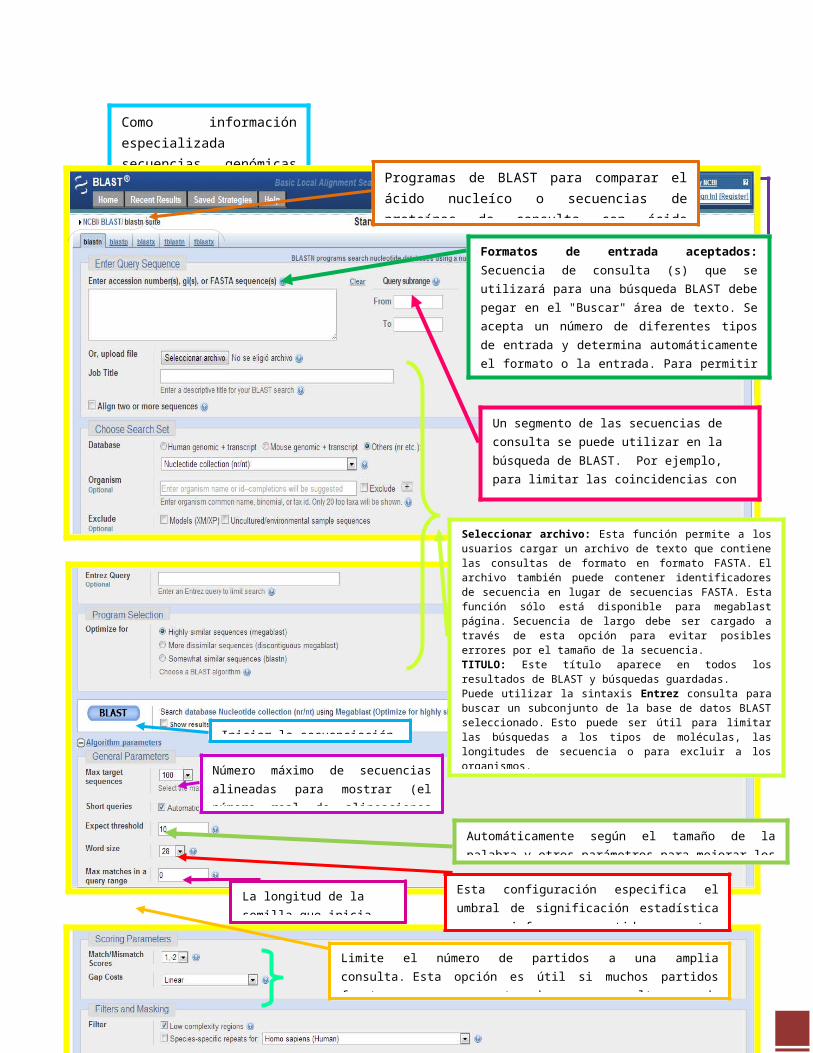
Como información especializada secuencias genómicas y otras se pondrán a disposición del público, NCBI BLAST crea páginas especializadas para esas secuencias.
Secuencias de Genomas de BLAST
Programas de BLAST para comparar el ácido nucleíco o secuencias de proteínas de consulta con ácido nucleíco o bases de datos de secuencias de proteínas.
Formatos de entrada aceptados: Secuencia de consulta (s) que se utilizará para una búsqueda BLAST debe pegar en el "Buscar" área de texto. Se acepta un número de diferentes tipos de entrada y determina automáticamente el formato o la entrada. Para permitir esta función, no son necesarias ciertas convenciones con respecto a la entrada de los identificadores (por ejemplo, adhesiones o gi, FASTA, Secuencias Bare.).

Descripción gráfica
Un segmento de las secuencias de consulta se puede utilizar en la búsqueda de BLAST. Por ejemplo, para limitar las coincidencias con la región de 24 a 200 de la secuencia de consulta, deberá introducir 24 en el campo "De" y 200 en el campo
Seleccionar archivo: Esta función permite a los usuarios cargar un archivo de texto que contiene las consultas de formato en formato FASTA. El archivo también puede contener identificadores de secuencia en lugar de secuencias FASTA. Esta función sólo está disponible para megablast página. Secuencia de largo debe ser cargado a través de esta opción para evitar posibles errores por el tamaño de la secuencia. TITULO: Este título aparece en todos los resultados de BLAST y búsquedas guardadas. Puede utilizar la sintaxis Entrez consulta para buscar un subconjunto de la base de datos BLAST seleccionado. Esto puede ser útil para limitar las búsquedas a los tipos de moléculas, las longitudes de secuencia o para excluir a los organismos. Megablast está destinado a comparar una consulta para secuencias estrechamente relacionadas y funciona mejor si el porcentaje de identidad de destino es 95% o más, pero es muy rápido. Megablast contiguo utiliza una semilla inicial que ignora algunas bases (permitiendo desajustes) y está destinada a comparaciones entre especies. BlastN es lento, pero permite una palabra de tamaño hasta siete bases.
Iniciar la secuenciación con BLAST
Número máximo de secuencias alineadas para mostrar (el número real de alineaciones puede ser mayor que esta).
Automáticamente según el tamaño de la palabra y otros parámetros para mejorar los resultados de las consultas cortas.
Esta configuración especifica el umbral de significación estadística para informar partidos contra secuencias de bases de datos.
La longitud de la semilla que inicia una alineación
Limite el número de partidos a una amplia consulta. Esta opción es útil si muchos partidos fuertes con una parte de una consulta, puede prevenir que BLAST presente los partidos más débiles a otra parte de la consulta.
Muchas búsquedas de nucleótidos utiliza un sistema de puntuación simple que consiste en una "recompensa" por un partido y una "pena" por una falta de coincidencia. El aumento de los costos Gap resultará en alineaciones que disminuyen el número de huecos introducidos.
Regiones de baja complejidad filtrar, Específicas para cada especie.
Inicio de comparación de la secuencia en BLAST estableciendo los criterios en los algoritmos.
Una visión general de las secuencias de la base de datos alineados con la secuencia de consulta. La puntuación de cada alineación se indica mediante uno de los cinco colores diferentes, que divide el rango de puntuaciones en cinco grupos. Múltiples segmentos de alineamientos de la secuencia misma base de datos están conectados por una línea gris delgada. Pasar el ratón sobre una secuencia de coincidencias hace que la definición y la puntuación que se muestra en la ventana en la parte superior, al hacer clic en una secuencia de golpe llevan al usuario a las alineaciones correspondientes.
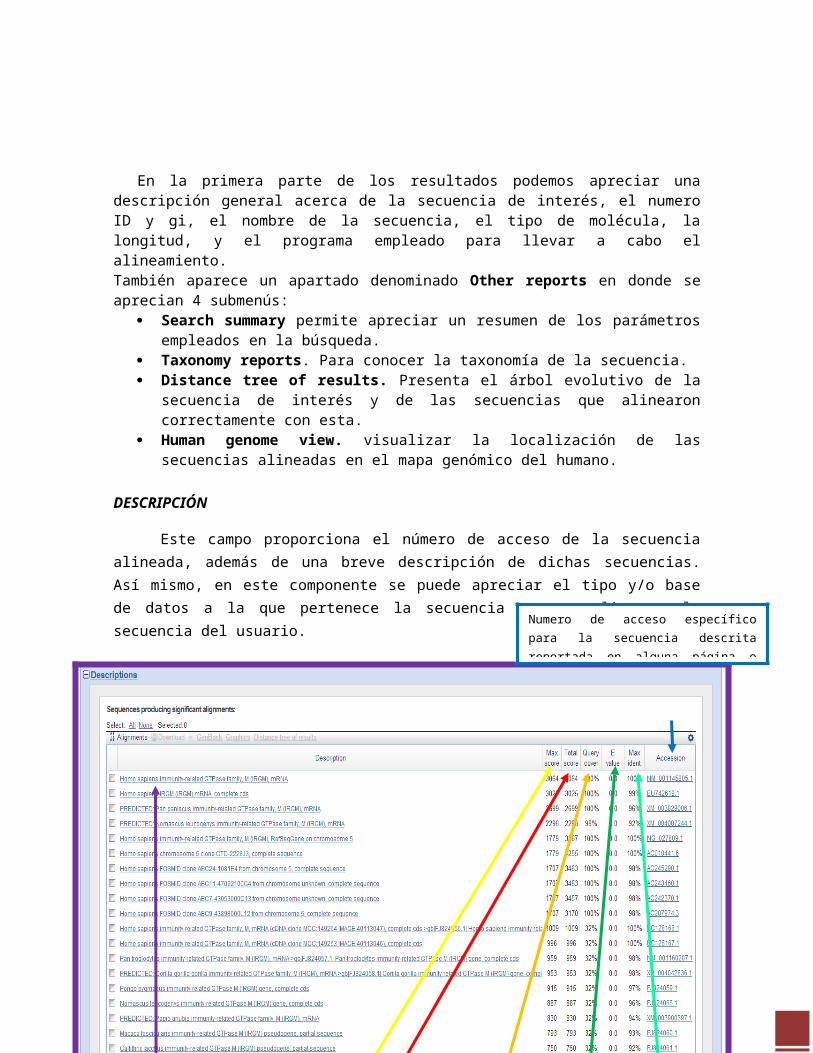
En la primera parte de los resultados podemos apreciar una descripción general acerca de la secuencia de interés, el numero ID y gi, el nombre de la secuencia, el tipo de molécula, la longitud, y el programa empleado para llevar a cabo el alineamiento. También aparece un apartado denominado Other reports en donde se aprecian 4 submenús:
Search summary permite apreciar un resumen de los parámetros empleados en la búsqueda.
Taxonomy reports. Para conocer la taxonomía de la secuencia. Distance tree of results. Presenta el árbol evolutivo de la secuencia de interés y de las
secuencias que alinearon correctamente con esta. Human genome view. visualizar la localización de las secuencias alineadas en el mapa
genómico del humano.
DESCRIPCIÓN
Muestra un resumen gráfico de las secuencias alineadas. El resumen cuenta con un código de colores que varía según el puntaje que reciba cada alineamiento; para el mejor alineamiento se encuentra el color rojo, de rosa se colocan los fragmentos de la secuencia alineada que cuenta con algunas no concordancias en la similitud, el verde muestra alineamientos poco confiables y los colores azul y negro son indicativos de alineamientos con una similitud muy pobre. Cada línea representa una secuencia para una especie en
Este gen codifica un miembro de la p47 inmunidad relacionada GTPasa de la familia. La proteína codificada puede desempeñar un papel en la respuesta inmune innata mediante la regulación de la formación en respuesta a la autofagia

Este campo proporciona el número de acceso de la secuencia alineada, además de una breve descripción de dichas secuencias. Así mismo, en este componente se puede apreciar el tipo y/o base de datos a la que pertenece la secuencia que se alineo a la secuencia del usuario.
ALINEACIONES
Max score. Muestra la puntuación máxima obtenida por un segmento alineado de la secuencia.
Total score. Muestra una sumatoria de la puntuación obtenida por todos los fragmentos que alinearon de la secuencia.
Query coverage. Indica el porcentaje total de la secuencia que alineo correctamente con respecto a la secuencia del
E-value. Indica el valor o procedimiento matemático para calcular la probabilidad de que un alineamiento particular sea producto únicamente del azar y no se base en homología sabiendo que mientras menor sea este, mayor será la similitud entre las secuencias.
Max ident. Muestra el porcentaje de identidad de la secuencia alineada con respecto a la secuencia de interés.
Numero de acceso específico para la secuencia descrita reportada en alguna página o liga de interés científico llámese NCBI o EBI.
La secuencia comparada en BLAST corresponde a la GTPasa de la familia IRGM en una molécula de RNA de la especie Homo sapiens, de las 1659 pb que conformar la secuencia se alineo en un 100% y presenta un 0.0% E-value esto nos dice que si es su homologo más que una secuencia al azar.
Esta opción restringe secuencias de la base de datos al número especificado para el que la alta puntuación segmento de pares (HSPs) son reportados. Páginas diferentes tienen diferentes configuraciones por defecto. Si el número de secuencias de bases de datos satisface el umbral de significación estadística se presentan los informes, sólo los partidos que posean la mayor importancia estadística son reportados.
Nos muestra en un formato FASTA de manera textual, los resultados del alineamiento, mostrando una descripción general tanto de la secuencia alineada como la que tiene similitud con la misma, así como algunos porcentajes respecto a los gaps, el porcentaje de las bases identificadas. Así como un apartado que nos permite elegir el orden en que se muestran los alineamientos.
Nombre, número de acceso y descripción de la secuencia perteneciente a la base de datos, longitud de la secuencia 1659 pb.
Valores estadísticos, resultados tras el alineamiento con 100% de similitud y al poseer 0.0% expect comprueba la homología de la secuencia con BLAST.
Alineamiento textual entre secuencias.
Los resultados obtenidos pueden ser descargados directamente.
Se puede acceder a información de la secuencia en GenBank y observar formato gráfico en la misma página.
LinkOuts de bases de datos. Al habilitar esta opción proporciona enlaces de referencias cruzadas de los BLAST hits a las entradas que se encuentran en otras bases de datos especializadas de NCBI. Si una secuencia de bases de datos coincide con la consulta y también se incluye en Gene, UniGene, estructura o base de datos GEO, usted será capaz de seguir el enlace al GIF icono marcado enlaces para obtener información adicional para que los hit.records en esos recursos.
= Enlace a Gene = Enlace a UniGene
= Enlace a GEO = Enlace a la estructura

Aplicando la búsqueda de secuencias homologas en BLASTn para la proteína ECSIT en homo sapiens con el menor e-value posible se obtienen los siguientes resultados.
gi|7407498|gb|AAF62100.1|AF243044_1 ECSIT [Homo sapiens] MSWVQATLLARGLCRAWGGTCGAALTGTSISQVPRRLPRGLHCSAAAHSSEQSLVPSPPEPRQRPTKALV PFEDLFGQAPGGERDKASFLQTVQKFAEHSVRKRGHIDFIYLALRKMREYGVERDLAVYNQLLNIFPKEV FRPRNIIQRIFVHYPRQQECGIAVLEQMENHGVMPNKETEFLLIQIFGRKSYPMLKLVRLKLWFPRFMNV NPFPVPRDLPQDPVELAMFGLRHMEPDLSARVTIYQVPLPKDSTGAADPPQPHIVGIQSPDQQAALACHN PARPVFVEGPFSLWLRNKCVYYHILRADLLPPEEREVEETPEEWNLYYPMQLDLEYVRSGWDNYEFDINE VEEGPVFAMCMAGAHDQATMAKWIQGLQETNPTLAQIPVVFRLAGSTRELQTSSAGLEEPPLPEDHQEED DNLQRQQQGQS
Se encontraron alrededor de 50 homologías que correspondían a la secuencia basados en un e-value del 0% pero estas presentaban diferente identidad en las secuencias pero afirmando que corresponden a la misma proteína, aunque en diferentes especies. Como podemos apreciar los aprox 50 homólogos seleccionados tienen un e-value de 0 lo que nos indica, que las secuencias son muy similares entre sí.

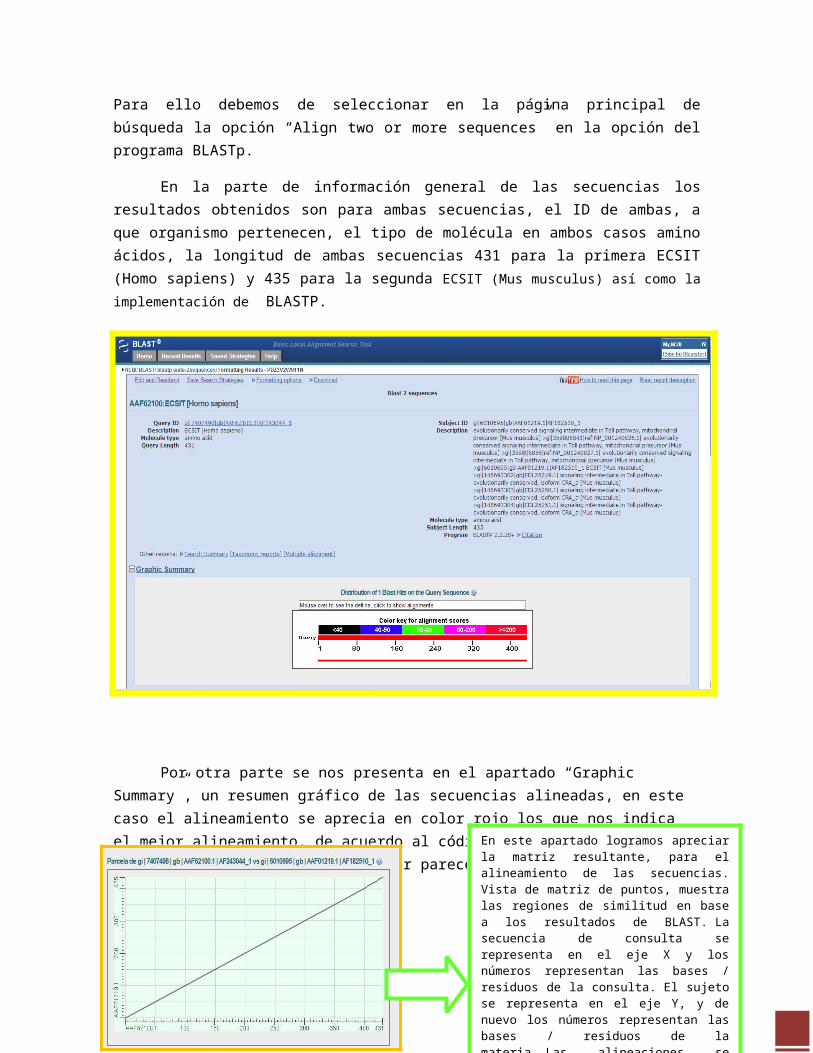
Se procedió a comparar ambas secuencias proteicas y analizar los resultados.Para ello debemos de seleccionar en la página principal de búsqueda la opción “Align two or more sequences” en la opción del programa BLASTp.
En la parte de información general de las secuencias los resultados obtenidos son para ambas secuencias, el ID de ambas, a que organismo pertenecen, el tipo de molécula en ambos casos amino ácidos, la longitud de ambas secuencias 431 para la primera ECSIT (Homo sapiens) y 435 para la segunda ECSIT (Mus musculus) así como la implementación de BLASTP.
Por otra parte se nos presenta en el apartado “Graphic Summary”, un resumen gráfico de las secuencias alineadas, en este caso el alineamiento se aprecia en color rojo los que nos indica el mejor alineamiento, de acuerdo al código de colores, es decir ambas secuencias son homologas por parecerse entre sí. En este apartado logramos apreciar la matriz
resultante, para el alineamiento de las secuencias. Vista de matriz de puntos, muestra las regiones de similitud en base a los resultados de BLAST. La secuencia de consulta se representa en el eje X y los números representan las bases / residuos de la consulta. El sujeto se representa en el eje Y, y de nuevo los números representan las bases / residuos de la materia. Las alineaciones se muestran en el diagrama como líneas. Relación de partidos entre las líneas de proteínas aquellas que están inclinadas desde la parte inferior izquierda a la esquina superior derecha, menos partidos las líneas que están inclinadas desde la parte superior izquierda a la inferior derecha. El número de líneas que se muestran en la trama es el mismo que el número de alineaciones que han encontrado los

En la sección de “descriptions” se muestra el numero de acceso de la secuencia alineada con la secuencia de interés, su descripción y el valor Max Score de 627 que corresponde a la puntuación máxima obtenida por el segmento alineado de la secuencia, el valor de Total score que se refiere a la puntuación total de todos los fragmentos alineados que también es de 627, el porcentaje de alineamiento correcto fue de un 100% y el valor de e-value de 0 lo que nos indica una mayor similitud entre ambas secuencias al ser menor que 1 , pero la identidad de la secuencia alineada con respecto a la secuencia de interés fue de 73% es decir que no toda la secuencia de aa se parece entre la especie Homo sapiens y la Mus mulusculus.
En este apartado podemos apreciar el resultado del alineamiento de manera textual, se nos proporciona otra vez la descripción general de la secuencia alineada, respecto a la secuencia de interés, así como la identidad entre aminoácidos en este caso se identificaron 318 de 435 lo que equivale al 73% de identidad, además de que hay zonas de deleción o inserción dentro de la secuencia, ya que se señala la existencia de 4 gaps correspondiente al 1% de toda la secuencia alineada, proporciona visualmente los tipos de aa que se modifican entre las secuencias de comparación de las especies en humano y rata.
C O M E N T A R I O
BLAST, es una herramienta bioinformatica altamente funcional que permite encontrar regiones similares entre secuencias biológicas. Normalmente BLAST es usado para encontrar probables genes homólogos. Por lo general, cuando una nueva secuencia es obtenida, se usa BLAST para compararla con otras secuencias que han sido previamente caracterizadas, para así poder inferir su función. BLAST es la herramienta más usada para la anotación y predicción funcional de genes o secuencias proteicas. Muchas variantes han sido creadas para resolver algunos problemas específicos de búsqueda.
Se ha convertido en la herramienta predilecta para llevar a cabo el alineamiento de grandes bases de datos de genomas. Debido que aborda un problema fundamental basado en la heurística y su algoritmo que es mucho más rápido que el cálculo de una alineación óptima. Se hace énfasis en la velocidad vital para el algoritmo práctico sobre las enormes bases de datos genómicos disponibles actualmente, aunque los algoritmos siguientes pueden ser aún más rápidos.
Es así como se convierte en un medio de gran utilidad para las investigaciones de genéticas, biología molecular, medicina y con el paso de los años BLAST se consolida como la herramienta más poderosa y actualizada para llevar a cabo alineamientos entre una secuencia particular con respecto a otra, pudiendo incluso realizar un alineamiento con el total de las secuencias presentes en las bases de datos a nivel mundial.


















