
R E S U M E N · se depositó sobre los nanotubos en una estructura cristalina en fase beta...
149
Transcript of R E S U M E N · se depositó sobre los nanotubos en una estructura cristalina en fase beta...


R E S U M E N
Se prepararon materiales de electrodo de composición dual basados en nanotubos de
dióxido de titanio (TiO2-Nts) y óxidos metálicos (PbO2 o SnO2) mediante la anodización
de placas de titanio (de 1 x 1 cm2 de área por lado) en un baño electrolítico compuesto
de glicerol:agua (1.3:1), Na2SO4 0.2 M y NaF 0.5% (m/v); bajo la aplicación de un
potential de 30 V durante 4 h y usando una placa de circonio de 4 cm2 de área como
contraelectrodo. Despúes de la anodización, los TiO2-Nts se trataron térmicamente a
500°C durante 90 minutos usando una rampa de calentamiento de 5°C/min. Después del
tratamiento térmico de los nanotubos de TiO2, se depositó PbO2 o SnO2 en el arreglo de
nanotubos. Ambos tipos de electrodos de composición dual se caracterizaron por SEM,
EDS, XRD, Espectroscopia de Reflectancia Difusa y, Voltametría Lineal y Cíclica.
En el caso de PbO2, la electrodeposición anódica de dióxido de plomo a partir de
Pb(NO3)2 en HNO3 0.1M a una densidad de corriente de 50 mA/cm2 se realizó sobre el
arreglo de nanotubos a diferentes tiempos de depósito. Estos tiempos de deposición se
variaron con el fin de alcanzar la mayor dispersión del óxido sobre los nanotubos, y este
objetivo se alcanzó a los 5-10 s de deposición.
En el caso de SnO2, se eligió un método de rociado pirolítico artesanal como el método
de deposición, mediante el cual se realizaron sobre los nanotubos, cinco depósitos de
una solución precursora que contenía SnCl2 y Sb2O3 en una solución de metanol: HCl
(5:1) a una temperatura de 550-600°C.
Los análisis de MEB demostraron que el SnO2 se dispersó adecuadamente sobre los
nanotubos La morfología del depósito de óxido de estaño varió dependiendo de la
concentración de los materiales iniciales en la solución precursora. Debido a una baja
foto-respuesta de estos electrodos, sólo se realizaron electrocatálisis con estos
electrodos en oscuridad. Con estos electrodos, una solución ácida de rojo de metilo de
15 ppm se decoloró en un 80% en 1.5 h y la oxidación de una solución ácida de 50 ppm
de p-nitrofenol llegó a un 75% en 3.5 h. De la muestra final de esta reacción, se
detectaron compuestos orgánicos como hidroquinona, benzoquinona y ácidos alifáticos,
Sin embargo, este depósito de SnO2 mostró muy baja estabilidad, ya que, el depósito
perdía adherencia después de 4-8 h de uso. Asimismo, en la polarización acelerada en

medio de ácido sulfúrico sobre este electrodo, el potencial se mantiene constante durante
23 horas, pero se observa desprendimiento del depósito y el arreglo de nanotubos
desaparece según lo muestra la MEB; lo cual indica menor estabilidad de estos
electrodos con respecto a sus análogos de PbO2.
PbO2 se depositó sobre los nanotubos en una estructura cristalina en fase beta
(tetragonal). Esta estructura se desarrolla conforme avanza el tiempo de depósito hasta
formar una estructura microscópica parecida a una coliflor, la cual se reproduce sobre el
arreglo de nanotubos. De la voltametria cíclica, se observó que se alcanza la mayor área
superficial con el depósito de dióxido de plomo de 5 s (Ti/TiO2-Nts::PbO2 5s). Por lo tanto,
se eligió este electrodo para llevar a cabo oxidaciones electrocatalíticas y/o
fotoelectrocatalíticas de soluciones de rojo de metilo y p-nitrofenol. Sin embargo, se
observó un comportamiento catalítico diferente para los electrodos hechos a base de una
placa de titanio que provenía de un lote diferente de fabricación. Por lo tanto, los
electrodos se clasificaron como electrodos de primera y segunda generación. Esta
clasificación se basa en el desempeño del electrodo. Básicamente, los electrodos de la
primera generación mostraron un buen desempeño electrocatalítico y
fotoelectrocatalítico. Es decir, una solución ácida de rojo de metilo de 15 ppm se decoloró
completamente en 90 minutos de electrocatálisis bajo condiciones de oscuridad y el
mismo tipo de solución se decoloró completamente en 30 minutos de electrocatálisis (a
una densidad de corriente de 30 mA/cm2) bajo iluminación con una lámpara de longitud
de onda de 365 nm. Por otro lado, los electrodos de la segunda generación sólo
presentaron un buen desempeño electrocatalítico, pero un bajo o incluso nulo
desempeño fotoelectrocatalítico. Para estos electrodos, la electrooxidación de la solución
de rojo de metilo en oscuridad logra su decoloración completa en 30 minutos. Asimismo,
la oxidación completa de p-nitrofenol (50 ppm) con estos electrodos se logra en 3.5 h de
reacción, siendo los ácidos alifáticos como maleico, málico y oxálico los productos
remanentes de la electrooxidación. La estabilidad de este material se probó mediante
polarización acelerada en ácido sulfúrico 1 M, a j=100 mA/cm2, y se observó que el
potencial aplicado se mantiene constante durante al menos 29 horas. Asimismo, la MEB
del electrodo al final de la polarización, muestra ligeros cambios en el depósito de PbO2,
mientras que, el arreglo de nanotubos se mantiene intacto.

A B S T R A C T
Electrode materials of dual composition based on titanium dioxide nanotubes (TiO2-Nts)
and metal oxides (PbO2 or SnO2) were prepared by anodization of titanium plates (1 x 1
cm2 of area by side). Anodization was held at an applied potential of 30 V, in an electrolytic
bath composed of glycerol:water (1.3:1), 0.2 M Na2SO4, and 0.5% (m/v) NaF. A 4 cm2
zirconium plate was used as the counter electrode. The reaction was stopped after 4
hours. TiO2-Nts were annealed at 500°C during 90 minutes using a heating ramp of
5°C/min. Once nanotubes were annealed, electrodes of titania nanotubes and PbO2 or
SnO2 were prepared by deposition of these metal oxides on the nanotube array. Both
types of composite electrode materials were characterized by SEM, EDS, XRD, Diffuse
Reflectance Spectroscopy and, Linear and Cyclic Voltammetry.
In the case of PbO2, electrodeposition of lead dioxide from a 0.25 M Pb(NO3)2 in 0.1 M
HNO3 at current density of 50 mA/cm2 was performed on the nanotube array during
different deposition times. This deposition times varied in order to achieve the highest
dispersion of the oxide over the nanotubes, and this objective was achieved at 5-10 s of
deposition.
In the case of SnO2, deposition from spray-pyrolisis was chosen as the deposition
method, in which five depositions from a solution containing SnCl2 and Sb2O3 in a
methanol: HCl (5:1) solution were made on the nanotubes array at 550-600°C.
The electrodes based on TiO2-Nts and SnO2 (Ti/TiO2-Nts/SnO2) were well dispersed over
the nanotubes as it was shown through SEM analyzes. SnO2 deposits morphology varied
depending on the concentration of the starting materials in the precursor solution. Due to
a low photo-response with these electrodes, only electrocatalysis under darkness was
performed on them. With these electrodes, a 15 ppm methyl red solution was almost fully
bleached (80% bleaching) in 1.5 h; and p-nitrophenol oxidation reached a 75% oxidation
in 3.5 h; being organic compounds such as hydroquinone, benzoquinone and aliphatic
acids, the remaining products of its electrooxidation. However, this SnO2 deposit showed
very low stability since it was lost after 4 to 8 h of use. Also, accelerated polarization
experiments proved these electrodes have a lower stability compared to their analogue
Ti/TiO2-Nts::PbO2 electrodes, since SnO2 deposit is completely lost after 23 hours of

anodic polarization and SEM analyses showed the nanotube array is also lost although
the potential remained constant during the whole experiment.
On its side, lead dioxide deposited on TiO2-Nts in a β-PbO2 crystalline structure
(tetragonal). This structure develops with deposition time to form a cauliflower-like
microscopic structure of the lead dioxide dispersed over the nanotubes array. From cyclic
voltammetry, it was observed that the highest surface area is achieved when PbO2
deposits within five seconds (Ti/TiO2-Nts::PbO2 5s). Therefore, this electrode was chosen
to perform electrocatalytic and/or photoelectrocatalytic oxidations of methyl red and p-
nitrophenol. However, a different catalytic behavior was observed for electrodes made
from a titanium plate coming from a different fabrication lot. Therefore, electrodes were
classified as first-generation electrodes and second-generation electrodes. This
classification is based on the electrode performance. Basically, electrodes from the first
generation presented both, electrocatalytic and photoelectrocatalytic, behavior. That is, a
15 ppm methyl red acidic aqueous solution is fully bleached in 90 minutes of
electrocatalysis under darkness and the same type of solution is fully bleached in 30
minutes of photoelectrocatalysis (at current density of 30 mA/cm2) under illumination with
a 365 nm wavelength source. On the other side, electrodes from the second generation
only presented electrocatalytic behavior, but very low or none photoelectrocatalytic
activity was observed. For these electrodes, the methyl red solution is fully bleached in
30 minutes of electrocatalysis under dark conditions. Also, p-nitrophenol oxidation was
performed on this electrode and a complete oxidation of these biorefractory compound
(50 ppm initial concentration) was achieved after 3.5 h of reaction, being aliphatic acids
such as maleic, malic and oxalic acids, the main remaining products of its
electrooxidation. Stability of this material was tested with accelerated polarization in 1 M
sulphuric acid, and at j=100 mA/cm2, and it was observed that the applied potential was
kept constant for at least 29 hours and there were slight changes in the PbO2 deposit
while the nanotubes array remained intact as it was shown by SEM.

Este trabajo fue realizado en el Centro de
Investigación y Desarrollo Tecnológico en
Electroquímica (CIDETEQ), bajo la dirección de
Dr. Yunny Meas-Vong
Dr. Marco A. Quiroz Alfaro

AGRADECIMIENTOS
Doy gracias a Dios por haberme dado la fortaleza para llegar a la conclusión de mi trabajo
doctoral. Así como también, a las amables personas de CIDETEQ: compañeros,
académicos y administrativos, a los cuales siempre deberé su paciencia, apoyo y trato
cordial. A mis amigos, jefes y compañeros de trabajo por su soporte y porras. Y por
supuesto a mi familia, mi hermosa familia, quienes supieron esperar, animarme y
reanimarme en los momentos difíciles de esta travesía.
De mi trabajo de investigación, me llevo, más que la comprensión de un tema: una visión
diferente de la ciencia y cómo se construye el saber. He aprendido que falta mucho por
conocer y que, los retos del quehacer científico son aún vastos y dignos de afrontarse; y
que, mientras exista ese deleite por buscar la explicación de un fenómeno, la
investigación merece la pena el tiempo y esfuerzo invertidos.
He aprendido que en la investigación, como en la vida, vale más la humildad que la
soberbia; pues es preciso poder reconocer los momentos en los que nuestra propia
ignorancia, falta de habilidad, o poca percepción, nos limitan. En este punto no tengo
palabras para agradecer a los doctores Marco Quiroz y Yunny Meas, sin quienes,
indudablemente, no habría podido reconocer esos momentos.

Para Ana, Samuel y Héctor

ÍNDICE DE CONTENIDO
Pág.
CAPÍTULO I: INTRODUCCIÓN 1
1. Antecedentes 1
2. Marco Teórico 7
2.1. Nanotubos de dióxido de titanio en la eliminación de contaminantes
orgánicos acuosos
7
2.1.1. Fotocatálisis sobre nanotubos de TiO2 8
2.1.2. Fotoelectrocatálisis sobre Nts-TiO2 9
2.2. Aspectos importantes en la formación de nanotubos de dióxido de
titanio por anodización
10
2.2.1. Mecanismo de formación de nanotubos por anodización 11
2.2.2. Fases cristalinas del TiO2 14
2.2.3. Variables que afectan el crecimiento de los nanotubos 16
2.2.3.1 Voltaje aplicado 16
2.2.3.2. Viscosidad 17
2.2.3.3. Efecto del pH 17
2.2.3.4. Tiempo de anodizado 18
2.2.3.5. Temperatura 18
2.2.3.6. Efecto de la concentración de fluoruro 18
2.2.3.7. Difusión: factor que controla la velocidad de crecimiento
del tubo
19
2.3. Depósitos de Dióxido de Plomo 21
2.3.1. Modificaciones en la construcción de electrodos de PbO2 22
2.3.2. Modificaciones en el sustrato 22
2.3.3. Modificaciones electrónico-estructurales 23
2.3.4. Electrodepósito de dióxido de plomo 23
2.3.5. Fases y estructura del PbO2 24
2.3.6. Conductividad de PbO2 25
2.4. Depósitos de SnO2 26
2.4.1. Características físicas y químicas del SnO2 y sus variantes
dopadas
26

2.4.2. Métodos de preparación de depósitos de SnO2 27
2.4.2.1. Deposición por rociado pirolítico 28
2.4.2.2. Modalidades de los sistemas de rociado pirolítico 29
2.4.2.3. Ventajas del rociado pirolítico 30
3. Justificación 31
4. Hipótesis 32
5. Objetivo General 33
6. Objetivos Específicos 33
CAPÍTULO II: PREPARACIÓN Y CARACTERIZACIÓN DE NANOTUBOS DE
DIÓXIDO DE TITANIO
34
1. Resumen 34
2. Anodizaciones preliminares 35
3. Metodología de preparación de nanotubos: Metodología A 42
4. Resultados y Discusión 43
4.1. Rugosidad de la placa como un factor determinante en la formación y
crecimiento de nanotubos
46
4.1.1. Formación de PP debido a la rugosidad a microescala del Ti
metálico.
48
4.1.2. Transitorios de corriente en la formación de nanotubos a partir de
placas de titanio diferentes
54
5. Conclusiones 56
CAPÍTULO III. ELECTRODEPOSICIÓN DE DIÓXIDO DE PLOMO SOBRE
NANOTUBOS DE DIÓXIDO DE TITANIO
58
1. Resumen 58
2. Electrodepósitos preliminares 58
3. Metodología de electrodeposición de PbO2 60
4. Resultados y Discusión 60
4.1. Medida del área superficial real de los electrodos de Nts- TiO2 y Ti/Nts-
TiO2::PbO2
68
4.2. Estabilidad electroquímica de los electrodos de Ti/Nts-TiO2::PbO2 5s 71
5. Conclusiones 73

CAPÍTULO IV. DEPOSICIÓN DE DIÓXIDO DE ESTAÑO SOBRE
NANOTUBOS DE TiO2
74
1. Resumen 74
2. Metodología deposición de SnO2 74
2.1. Detalles del sistema de rociado pirolítico (spray-pyrolysis) 74
2.2. Preparación de la solución precursora 76
2.3. Rociado pirolítico de la solución precursora sobre las placas de Nts-
TiO2
77
3. Resultados y Discusión 77
3.1 Efecto en la morfología del depósito de SnO2 debido a la variación en la
concentración de la solución precursora
3.2. Estabilidad electroquímica de los electrodos de Ti/Nts-TiO2/SnO2
79
83
4. Conclusiones 85
CAPÍTULO V. ELECTROCATÁLISIS Y FOTOELECTROCATÁLISIS DE
COMPUESTOS ORGÁNICOS CON ELECTRODOS DE Ti/Nts-TiO2::PbO2
86
1. Resumen 86
2. Metodología 87
2.1 Precisiones sobre la preparación de nanotubos de segunda generación,
2- Ti/Nts-TiO2
87
2.2. Evaluación de fotoactividad 87
2.3. Decoloración de rojo de metilo 87
2.4. Oxidación de p-nitrofenol 88
2.5. Análisis cromatográfico por HPLC 88
3. Resultados y Discusión 88
3.1 Electrocatálisis y fotoelectrocatálisis de rojo de metilo sobre electrodos
de Ti/Nts-TiO2::PbO2 (5, 10 y 15 s) de la primera generación
88
3.1.1 Evaluación de fotoactividad 89
3.1.2 Decoloración de rojo de metilo 92
3.2 Electrocatálisis y fotoelectrocatálisis con electrodos de la segunda
generación.
94
3.2.1. Evaluación de fotoactividad 95
3.2.2. Electrocatálisis y fotoelectrocatálisis de rojo de metilo 96
3.2.2.1. Electrocatálisis de rojo de metilo 97

3.2.3 Electrocatálisis de p-nitrofenol . 100
3.2.3.1 Análisis de HPLC de intermediarios de reacción de la
oxidación de PNF.
102
3.2.3.2 Ruta de oxidación de p-nitrofenol propuesta 106
4. Conclusiones 108
CAPÍTULO VI. ELECTROCATÁLISIS DE COMPUESTOS ORGÁNICOS
MEDIANTE ELECTRODOS DE 2-Ti/Nts-TiO2/SnO2
110
1. Resumen 110
2. Metodología 110
3. Resultados y Discusión 111
3.1. Electrocatálisis de rojo de metilo 111
3.2. Electrocatálisis de p-nitrofenol 112
3.3. Estabilidad de los depósitos de SnO2 115
4. Conclusiones 117
CONCLUSIONES GENERALES 119
REFERENCIAS 121
ANEXO: Lista de Equipos 128

ÍNDICE DE TABLAS
Capítulo II Pág.
Tabla II.1. Resumen de las primeras condiciones de anodización. 35
Capítulo III
Tabla III.1. Área real y factor de rugosidad de los electrodos de Nts-TiO2
y Ti/Nts-TiO2::PbO2. 70
Tabla III.2. Razones de electroactividad (REs) para los electrodos de Nts- TiO2
y Ti/Nts-TiO2::PbO2. 70
Capítulo IV
Tabla IV.1. Análisis elemental del depósito de SnO2 obtenido por EDS. 78
Capítulo V
Tabla V.1. Valores de DQO en diferentes puntos de color durante la electrooxidación
de rojo de metilo, a) color rosa inicial, b) color rosa más claro, c) color amarillo
claro y d) solución transparente. 96
Tabla V.2. Constantes cinéticas de la decoloración de RM en, 2-Ti/Nts-TiO2,
2-Ti/Nts-TiO2::PbO2 5 s 2-Ti/Nts-TiO2::PbO2 15 s y Pb/PbO2. 98
Tabla V.3. Constantes cinéticas de la oxidación de PNF en 2-Ti/Nts-TiO2::PbO2 5 s,
2-Ti/Nts-TiO2, y Pb/PbO2. 99
Capítulo VI
Tabla VI.1. Constantes cinéticas de pseudo-primer orden para la decoloración
de rojo de metilo con electrodos 2-Ti/Nts-TiO2/SnO2. 100

ÍNDICE DE FIGURAS
Pág.
Capítulo I
Figura I.1. Probables rutas de oxidación que pueden seguir los ánodos. 2
Figura I.2. Nanotubos de TiO2 vistos por microscopía electrónica de barrido. 4
Figura I.3. Esquema de formación de un electrodo de NTs-TiO2/SnO2. 5
Figura I.4. Representación de la anodización de Ti, a) en ausencia de fluoruros
(lo que produce capas de óxido), y b) en presencia de fluoruros (lo que produce
crecimiento del tubo). 11
Figura I.5. Etapas de formación de la capa de nanotubos. 13
Figura I.6. a) Esquema de la unidad periódica entre ondulaciones, b) Transitorio
de corriente durante la formación de nanotubos, c) Imagen de MEB de nanotubos
con las ondulaciones periódicas. 14
Figura I.7. Estructuras de rutilo y anatasa del TiO2. 15
Figura I.8. Empaquetamiento cristalino de las fases anatasa y rutilo. 15
Figura I.9. Transitorios de corriente registrados durante anodización de Ti en
electrolito de NH4F 0.27 M y glicerol:agua (50:50) en condiciones de agitación
(10 minutos) y sin agitación. 20
Figura I.10. Perfiles de concentración de las especies dentro y fuera del área de
difusión de los tubos con y sin agitación. 21
Figura I.11. Empaquetamiento del oxígeno octaédrico en dióxido de plomo en
α y β-PbO2, también se muestran las distancias plomo-oxígeno y oxígeno-oxígeno. 25
Figura I.12. Comparación de la estructura de las superficies de (a) SnO(001) y (b)
SnO2(101). Las similitudes entre las dos orientaciones cristalinas se han usado para
explicar la transformación de películas de SnO(001) a películas de SnO2(101). 27
Figura I.13. Componentes del sistema de rociado pirolítico. 29

Capítulo II
Figura II.1. Nanotubos de las reacciones 7 (izquierda) y 8 (derecha), recubiertos
por PP y fracturados para la reacción 8. 38
Figura II.2. Nanotubos de reacción 9, se observan recubiertos por una menor
cantidad de sal. 39
Figura II.3. Nanotubos de la reacción 13 (izquierda), se observa con un poco
de PP sobre la superficie. Nanotubos de la reacción 14 (derecha), se observan
libres de PP. 39
Figura II.4. Reacción 17, el depósito de nanotubos se fracturó y se pudo
visualizar la longitud de los mismos (aproximadamente 3000 nm). La reacción
duró 4 h. 40
Figura II.5. a) Electrodo de la reacción 28 se resquebrajó debajo del vástago.
b)Electrodo de la reacción 29, se resquebrajó de la parte lateral cercana al vástago.
c)Electrodo de la reacción 30, se resquebrajó en más de la mitad de la superficie
del electrodo, en la parte cercana al vástago. 41
Figura II.6. Reacción 31, los nanotubos se fracturaron. La reacción duró 6 horas. 41
Figura II.7. Análisis de EDS del precipitado formado sobre los nanotubos de Ti,
del que se puede apreciar diferentes elementos provenientes del medio de reacción
como F, Na y Si. 42
Figura II.8. Micrografías de nanotubos producidos por la metodología A. 44
Figura II.9. Análisis de EDS de nanotubos producidos por la metodología A. 44
Figura II.10. Espectro de Rayos X de Nts-TiO2 sobre titanio después de tratamiento
térmico a 500°C durante 1.5 h, donde se observa la formación de la fase anatasa
del TiO2. 45
Figura II.11. Voltamograma de nanotubos de TiO2 en medio de H2SO4 0.5 M. 45
Figura II.12. Espectro de absorción (Kubelka-Munk) de Nts-TiO2. El espectro fue
obtenido mediante la aplicación directa de la herramienta matemática, que
proporciona el espectrofotómetro (Cary 50), sobre el espectro de reflectancia
difusa obtenido por el mismo equipo. 46

Figura II.13. a) Ti metálico sin ningún tratamiento, placa UNK. b) Ti tratado con HCl
(18%), c) Ti tratado con HCl, anodizado en medio electrolítico de la metodología A
(no hay nanotubos). 47
Figura II.14. a) Placa de Ti (UNK) sin pretratamiento, b) Placa de Ti (MKB) sin
pretratamiento c) Nanotubos con PP, provenientes de placa de Ti (MKB) sin
pretratamiento. 49
Figura II.15. Transitorio de corriente del anodizado de placas de Ti pulido y
sin pulir. 50
Figura II.16. Sección transversal de nanotubos crecidos sobre una placa de Ti
sin pulir (izquierda) y una placa de titanio pulida (derecha). 51
Figura II.17. a) Placa de Ti (MKB) donde se observa un rayado pronunciado
del Ti metálico, b) Nanotubos crecidos sobre placa MKB cuya longitud es mayor
en las crestas, c) Doblado de nanotubos en zonas elevadas de la superficie. 52
Figura II.18. a) Placa de Titanio AME sin pulir. b) Nanotubos producidos a partir de
placa AME. c) Nanotubos a mayores aumentos. 53
Figura II.19. Nanotubos producidos a partir de placa de titanio tratada con ácido
oxálico. 54
Figura II.20. Izquierda: Transitorio de corriente que muestra las primeras etapas de
formación de nanotubos. Derecha: Imágenes de MEB de cortes transversales de
dichas etapas iniciales. 55
Figura II.21. Transitorios de corriente en el anodizado de tres placas de titanio con
rugosidad inicial diferente. 56
Capítulo III
Figura III.1. a) Depósito masivo de PbO2 sobre Ti/Nts-TiO2, b) Otro depósito masivo de
PbO2, donde se observa la reproducibilidad del depósito cristalino, c) Depósito de
PbO2 sobre Pb. 59
Figura III.2. a) Imagen de MEB del electrodo de Ti/Nts-TiO2::PbO2 a los 5 s de
depósito, b) Imagen de MEB del electrodo de Ti/Nts-TiO2::PbO2 a los 10 s de depósito,
c) Imagen de MEB del electrodo de Ti/Nts-TiO2::PbO2 a los 15 s de depósito,
d) Imagen aumentada de los depósitos incipientes de PbO2.. 61

Figura III.3. Imagen aumentada de MEB de depósitos de PbO2 en forma de coliflores,
b) Análisis de EDS de la zona de nanotubos, c) Análisis de EDS de la zona de PbO2
(coliflores). 62
Figura III.4. a) Imagen de MEB del fondo de los nanotubos, llenos de PbO2, se
observan como puntos blancos; b) Análisis elemental por EDS del fondo de los
nanotubos, c) Imagen ampliada del fondo de los nanotubos, d) Punto blanco del fondo
de los nanotubos analizado por EDS, e) Espectro de EDS del punto blanco. 64
Figura III.5. Espectro de DRX del depósito de PbO2 sobre nanotubos de TiO2 que
muestra que β-PbO2 es su estructura cristalina predominante. 65
Figura III.6. Espectro de absorción (Kubelka-Munk) de Ti/Nts-TiO2/PbO2 5s. 66
Figura III.7. a) Voltamograma de Ti/Nts-TiO2, b) Voltamograma de
Ti/Nts-TiO2::PbO2 5 s, c) Voltamograma Ti/TiO2-Nts/PbO2 15 s, y
d) Voltamograma Pb/PbO2. Todos los voltamogramas se obtuvieron en
0.5 M H2SO4, a una velocidad de barrido de 100mV/s. 67
Figura III.8. Voltamogramas lineales de Ti/Nts-TiO2 y Ti/Nts-TiO2::PbO2 en
H2SO4 0.5 M en oscuridad, vel. barrido de 100 mV/s. El intercepto en el eje X
(línea gris claro) es el sobrepotencial de evolución de oxígeno para cada
electrodo: 1.21 V (15 s), 1.28 V (10 s) y 1.32 V (5 s). Esta forma de obtener el
sobrepotencial de oxígeno, se infirió en base al artículo de José L. Nava (et al). 68
Figura III.9. Gráfica de corriente(I) vs. Velocidad de barrido para la obtención de la
capacitancia. Inserto: Voltamogramas cíclicos de Nts-TiO2 a diferentes
velocidades de barrido (10, 20, 40, 60, 80, 100, 150 y 200 mV/s). 69
Figura III.10. Representación esquemática del crecimiento de PbO2 dentro de los
nanotubos a: a) 5 s, b) 10 s y c) 15 s. 71
Figura III.11. Evolución del potencial bajo condiciones de Polarización Anódica en
Nts-TiO2::PbO2 (5s). 75
Figura III.12. Imágenes de MEB de electrodos de Ti/Nts-TiO2::PbO2 5s
a) antes y b) después de polarización anódica 72

Capítulo IV
Figura IV.1. Componentes del sistema de rociado pirolítico construido en el
laboratorio. 75
Figura IV.2. Distancia entre la punta del aerógrafo (rociador) y el sustrato
(~11cm). 76
Figura IV.3. Imágenes SEM correspondientes a (a) depósito con una rociada, se
observan estructuras de SnO2 (conglomerados) cortas, (b) depósito con cinco
rociadas, se observan conglomerados de SnO2 alargados y entrelazados.
(c) Depósito con una rociada, se observan algunos conglomerados, (d) Depósito
con 5 rociadas, se observa la distribución de mayor número de conglomerados.
Las imágenes a y b, se encuentran a una escala de 5 µm con el fin de observar
el crecimiento de los conglomerados; y las imágenes c y d, se encuentran a
escala de 50 µm con el fin de observar la dispersión y aumento numérico de los
conglomerados. 78
Figura IV.4. Depósito de SnO2 proveniente de la solución precursora ES2, donde
se observa un carácter granular del depósito. 80
Figura IV.5. Análisis de EDS del electrodo ES2 en una zona del depósito de
SnO2. 80
Figura IV.6. Espectro de absorción (Kubelka-Munk) del electrodo de
2-Ti/Nts-TiO2/SnO2 (ES1). 81
Figura IV.7. Espectro de absorción (Kubelka-Munk) del electrodo de
2-Ti/Nts-TiO2/SnO2 (ES2). 82
Figura IV.8. Difractograma de electrodo ES2, donde se muestra la formación de
SnO2 sobre los nanotubos de TiO2. La asignación de los picos de difracción se
basó en los artículos de Tosun y Kumar. 83
Figura IV.9. Evolución del potencial bajo condiciones de Polarización Anódica
en Nts-TiO2/SnO2. 84
Figura IV.10. Imágenes de MEB de electrodos de Ti/Nts-TiO2/SnO2
a) antes y b) después de polarización anódica. 84

Capítulo V
Figura V.1. Densidad de fotocorriente (cuadrados azules) y eficiencia de
fotoconversión con respecto a Eapp (círculos rojos). 90
Figura V.2a. Densidad de fotocorriente para Ti/Nts-TiO2 (círculos morados), Ti/Nts-
TiO2::PbO2 10s (círculos rojos), Ti/Nts-TiO2/ PbO2 15s (triángulos verdes),
Ti/Nts-TiO2/ PbO2 5s (rombos azules). 91
Figura V.2b. Densidad de fotocorriente para Ti/Nts-TiO2 (círculos morados), Ti/Nts-
TiO2::PbO2 10s (círculos rojos), Ti/Nts-TiO2::PbO2 15s (triángulos verdes), Ti/Nts-
TiO2::PbO2 5s (rombos azules). Figura tomada de M. Cerro-Lopez (et al). 91
Figura V.3. Se muestra la decoloración de rojo de metilo por parte de los diferentes
electrodos: electrocatálisis enTi/ Nts-TiO2 (cuadrados rojos vacíos),
fotoelectrocatálisis en Ti/Nts-TiO2 (cuadrados rojos llenos), electrocatálisis en
Pb/PbO2 (círculos negros), electrocatálisis en Ti/Nts-TiO2::PbO2 5s (triángulos
azules), fotoelectrocatálisis en Ti/Nts-TiO2::PbO2 5s (triángulos azules llenos).
Figura tomada de M. Cerro-Lopez (et al). 92
Figura V.4. Imágenes de MEB de depósitos de PbO2 de a) 5 s para electrodos de
segunda generación y, b) de 10s para electrodos de primera generación. 94
Figura V.5a. Curvas I-V de electrodos de segunda generación en H2SO4 0.5M, bajo
radiación de 365 nm a una I=4W/cm2, velocidad de barrido de 5 mV/s. 95
Figura V.5b. Eficiencia de fotoconversión de electrodos de segunda generación en
H2SO4 0.5M, bajo radiación de 365 nm a una I=4W/cm2, velocidad de barrido
de 5 mV/s. 96
Figura V.6. Electrocatálisis y fotoelectrocatálisis de RM sobre electrodos 2-Nts-
TiO2::PbO2 5 s (de segunda generación). 96
Figura V.7. Decoloración de rojo de metilo en H2SO4 0.5 M sobre electrodos
2-Ti/Nts-TiO2 (círculos grises), 2-Ti/Nts-TiO2::PbO2 5 s (círculos azules),
2-Nts-TiO2::PbO2 15 s (círculos amarillos) y Pb/PbO2 (círculos naranjas) a
j=30 mAcm-2, Co= 15 ppm. 97
Figura V.8. Decoloración de rojo de metilo en H2SO4 0.5 M sobre el electrodo
2-Ti/Nts-TiO2::PbO2 5s , Co= 15 ppm, j=30 mAcm-2. 98

Figura V.9. Detección por HPLC de productos de oxidación de RM en la muestra
final de reacción donde se han identificado ácidos maleico y oxálico mediante el
método cromatográfico: Columna PL-Hiplex H; Fase Móvil : H2SO4 4mM acuoso,
Vel. de flujo: 0.4 mL/min. T=35°C; λ=210 nm. 99
Figura V.10. Curvas cinéticas de la electrooxidación de PNF en H2SO4 0.5 M sobre
electrodos de 2-Ti/Nts-TiO2::PbO2 5 s, 2Ti/Nts-TiO2, y Pb/PbO2, j=30 mA/cm2,
Co=50 ppm. 101
Figura V.11. Cromatogramas de la muestras de oxidación de PNF sobre el
electrodo 2-Ti/Nts-TiO2 a diferentes tiempos de reacción para la identificación
de intermediarios aromáticos mediante el método cromatográfico: Columna
Inertsil ODS 3; Fase Móvil: Acetonitrilo: Agua: Ac. Acético, 49:49:2; Vel.
de flujo: 0.8 mL/min; T=25°C; λ= 288 nm. 102
Figura V.12. Cromatograma de la muestra final de oxidación de PNF sobre el
electrodo 2-Ti/Nts-TiO2. Columna PL-Hiplex H; Fase Móvil : H2SO4 4mM acuoso,
Vel. de flujo: 0.4 mL/min. T=35°C; λ=210 nm. 103
Figura V.13. Cromatogramas de la muestras de oxidación de PNF sobre el electrodo
2-Ti/Nts-TiO2::PbO2 a diferentes tiempos de reacción para identificación de
intermediarios aromáticos mediante el método cromatográfico: Columna Inertsil
ODS 3; Fase Móvil: Acetonitrilo: Agua: Ac. Acético, 49:49:2; Vel. de
flujo: 0.8 mL/min; T=25°C; λ= 288 nm. 104
Figura V.14. Cromatogramas de la muestras de oxidación de PNF sobre el electrodo
2-Ti/Nts-TiO2::PbO2 a diferentes tiempos de reacción para identificación de ácidos
alifáticos. Método cromatográfico: Columna Allure®Organic Acids, Fase Móvil:
Fosfato de potasio 0.1M a pH 2.5, Vel. flujo: 0.5 mL/min, λ=226 nm. 105
Figura V.15. Esquema de ruta de reacción general. 106
Figura V.16. Ruta general de degradación para compuestos aromáticos oxidados
electroquímicamente en electrodos de PbO2. 107
Figura V.17. Ruta de degradación de p-nitrofenol en medio de H2SO4 0.5 M,
j=30 mA/cm2 para los electrodos de 2-Ti/Nts-TiO2 y 2-Ti/Nts-TiO2::PbO2. 108

Capítulo VI
Figura VI.1. Decoloración de rojo de metilo frente a electrodos de 2-Ti/Nts-TiO2/SnO2,
cobertura parcial y total, y electrodo de 2-Ti/Nts-TiO2 (para fines de comparación). 111
Figura VI.2. Electrooxidación de PNF en medio ácido frente a electrodos de 2Ti/Nts-
TiO2/SnO2.Se incluye la oxidación de PNF con electrodos 2-Ti/Nts-TiO2, para fines
de comparación. 112
Figura VI.3. Análisis por HPLC de intermediarios aromáticos de la oxidación de PNF
con electrodos 2Ti/Nts-TiO2/SnO2. El pico en 1.7 corresponde al frente del
solvente. 113
Figura VI.4. Análisis por HPLC de ácidos alifáticos en muestra final de reacción
de oxidación de PNF con 2-Ti/Nts-TiO2/SnO2 (cobertura total). 114
Figura VI.5. Imágenes de MEB de los depósitos de SnO2 y nanotubos de TiO2,
a) antes de reacción, b) y c) después de reacción. 116
Figura VI.6. Imágenes de MEB donde se observa en a) el depósito original de SnO2
que cubre totalmente a los nanotubos y en b) que el mismo depósito se ha
desprendido de los nanotubos y éstos han perdido su forma tubular. 117

1
CAPÍTULO I: INTRODUCCIÓN
1. Antecedentes
Dentro de los procesos avanzados de oxidación (PAOs) para el tratamiento de aguas
residuales, la oxidación electroquímica se ha situado como uno de los procesos con
mayores ventajas, entre las que se encuentran: versatilidad, compatibilidad ambiental, y
rentabilidad para la degradación de diversos agentes orgánicos contaminantes.1
Dentro de la degradación por oxidación electroquímica es de particular interés el
mecanismo por el cual se llevará a cabo la oxidación, influenciado principalmente por el
material de electrodo. En general, el mecanismo de oxidación electroquímica involucra,
en un primer paso, la descarga de agua sobre la superficie del electrodo; este paso
genera radicales hidroxilo (*OH) que, dependiendo de la naturaleza química del electrodo
llevarán a cabo, ya sea, la mineralización del compuesto orgánico hasta CO2, o bien, una
oxidación parcial que produce derivados oxidados de los compuestos orgánicos
biorrefractarios (compuestos no degradables por tratamiento biológico convencional)
iniciales.2-4 Entre los compuestos biodegradables se hallan los constituyentes de
desechos industriales provenientes de refinerías de petróleo, de industrias químicas y de
polímeros, plantas de coque; y de desechos agrícolas de pesticidas.5
Los electrodos que se han usado para este propósito son óxidos metálicos (MOx) y el
diamante dopado con boro (BDD). De acuerdo al mecanismo por el cual estos materiales
llevan a cabo la oxidación, éstos se clasifican en activos e inactivos. Los electrodos
activos son aquéllos cuya superficie interactúa fuertemente con los radicales hidroxilo, lo
que puede conducir a la oxidación del ánodo dando lugar a un óxido metálico de mayor
estado de oxidación (MOx+1). Este óxido lleva a cabo la oxidación del compuesto orgánico
(R). Por su parte, los electrodos inactivos, no reaccionan con los radicales libres (*OH),
por lo que, éstos últimos se hallan disponibles para atacar a las especies orgánicas,
Figura 1.6-7

2
Figura I.1. Probables rutas de oxidación que pueden seguir los ánodos.7
La elección de un determinado material de electrodo para la oxidación anódica depende
fundamentalmente de tres factores: a) estabilidad química y electroquímica, b) alta
conductividad eléctrica y, c) alta hidrofobicidad. Entre éstos materiales se encuentran las
películas sintéticas de diamante dopado con boro (BDD) en sustratos de Ti o Si, 8-9 que
han demostrado alta eficiencia electrocatalítica; y los óxidos metálicos como el IrO2,4,10
RuO2,11 PbO2, 7,12-14 SnO2, 8-9,12 SnO2-SbO5 (sobre sustrato de Ti) y sus derivados
dopados, con resultados variados.15-17 PbO2 y SnO2, así como el BDD son electrodos
inactivos con alto sobrepotencial de oxígeno; por su parte IrO2 y RuO2 son electrodos
activos con bajo sobrepotencial de oxígeno. 6-9
Con el fin de mejorar el desempeño de los electrodos, se han investigado algunas
modificaciones en su preparación, entre las que resaltan: el dopaje, la adición de
polímeros y modificaciones al sustrato. La literatura reporta dopajes de SnO2 con Sb, Bi
y F, y de PbO2 con cationes metálicos (Bi, Co, Ce, Fe) que inducen modificaciones
electrónico-estructurales y, por lo tanto, cambios en la actividad catalítica. Por su parte,
algunas resinas poliméricas (p.ej. resina de flúor)18 se han añadido para aumentar la
hidrofobicidad de la superficie de electrodo, disminuyendo así, la interacción con los

3
radicales hidroxilo; lo que, deja a los radicales hidroxilo disponibles para reaccionar con
el compuesto orgánico en solución. Asimismo, la membrana de Nafión ® también se ha
usado con el fin de acumular especies químicas con carga. 19 En lo que se refiere al
sustrato, generalmente Ti, éste se ha tratado por medios físicos y/o químicos con el fin
de aumentar el área superficial.
El titanio como sustrato se ha preferido por encima de otros metales (Ta, Zr) por su
dureza, conductividad, alta estabilidad química, adherencia y bajo costo.20 En la
preparación de ánodos de óxidos metálicos donde la superficie del sustrato se impregna
con una sal del metal para posteriormente, en presencia de aire, descomponerse
térmicamente y producir el óxido, se observó que el Ti formaba una capa aislante, y por
lo tanto, indeseable de TiO2 a temperaturas mayores a 550°C.20 Sin embargo, si el TiO2
se mezclaba con óxidos de Ir o Ru, éste contribuía a aumentar la vida útil del electrodo,
aunque disminuía la actividad catalítica del electrodo.21
El dióxido de titanio, en sí mismo, es un material fotocatalítico de importantes
aplicaciones como la descomposición de microorganismos, la desactivación de células
cancerosas, la degradación de sustancias de olor desagradable, la descomposición
fotoasistida de agua, en la construcción de celdas solares fotosensibilizadas (DSSC) y
sensores de gases, así como en la eliminación de contaminantes ambientales del agua
y aire, por mencionar algunas.22-23 El uso del TiO2 como material de electrodo se había
limitado a la formación de fotoánodos de películas delgadas del óxido que, presentaban
baja actividad catalítica. Con el objeto de mejorar dicha actividad se ha probado el dopaje
con iones metálicos, la depositación de metales pesados, la disminución del tamaño de
partícula y, actualmente, la formación de nanotubos.24-25 Los nanotubos de TiO2 han
mostrado ser una estructura estable física y químicamente, provistos de una gran área
superficial.26 Para su preparación se han investigado diferentes métodos entre los que
podemos mencionar a: el método sol-gel, congelación-secado, electrodepositación,
depositación sonoquímica y tratamientos químicos de partículas de TiO2, los cuales ven
limitado su uso como electrodos de trabajo debido a que no están soportados.27
La respuesta a la falta de adherencia de los nanotubos sintetizados por los métodos
anteriormente mencionados se encuentra en la preparación de nanotubos mediante la
oxidación anódica de placas de titanio a potenciales que varían entre 20 V y 60 V, en un
medio acuoso que, además de un electrolito soporte, contiene ácido fluorhídrico y/o

4
alguna sal de flúor. Los nanotubos de TiO2 así obtenidos son estructuras tipo carrizo,
cuyos diámetros van de 30 a 200 nm y de longitudes que varían de los 300 nm a los 250
µm, fuertemente adheridas al soporte metálico.28 Los patrones de difracción de rayos X
muestran que el TiO2 obtenido de la anodización es amorfo por lo que, para pasar a la
fase anatasa, debe tratarme térmicamente a temperaturas superiores a los 450°C.
Anatasa es la fase cristalina que se desea favorecer debido a que presenta un ancho de
banda adecuado para evitar la recombinación de los portadores de carga y una masa
electrónica efectiva más pequeña, lo que se traduce en una mayor movilidad de los
portadores de carga. Figura 2. 24,27
a) Vista frontal b) Vista lateral
Figura I.2. Nanotubos de TiO2 vistos por microscopía electrónica de barrido. 27
Xie Quan y colaboradores,24 han reportado la actividad fotocatalítica y
fotoelectrocatalítica de los nanotubos de TiO2 para la oxidación de pentaclorofenol
(PCP). Sus estudios muestran una mineralización mayor de PCP (70 %) con
fotoelectrocatálisis por nanotubos de TiO2 que con película de TiO2 (50%). Los mismos
autores comparan la fotocatálisis y la fotoelectrocatálisis por estos nanotubos, siendo
esta última la de mayor eficiencia, pues degrada al PCP en un 98%, mientras que, la
fotocatálisis sólo mineraliza el 70%.
Debido a la estabilidad natural que estos nanotubos presentan y a la posibilidad de
proveer un sustrato con gran área superficial, algunas investigaciones recientes se han
enfocado en la preparación de electrodos inactivos de óxidos metálicos sobre una base
de nanotubos de TiO2.29 El fundamento de estas preparaciones es el uso de este arreglo

5
de nanotubos de TiO2 como una especie de pozos múltiples susceptibles de ser
llenados con el óxido metálico, Figura 3.
Figura I.3. Esquema de formación de un electrodo de NTs-TiO2/SnO2.30
De esta manera, Zhao y colaboradores,30 han preparado electrodos de dióxido de estaño
(dopado con antimonio) implantado en nanotubos de TiO2 (NTs-TiO2/SnO2). Estos
electrodos se probaron frente a la electrooxidación de ácido benzoico y mostraron una
mejora sobresaliente en su desempeño; sobre todo en el tiempo de vida útil que aumentó
de 3 h para un electrodo de SnO2 convencional a 36 h para un electrodo de NTs-
TiO2/SnO2. En esta misma línea, Zhao y Zhang desarrollaron sobre un sustrato de
nanotubos, electrodos de PbO2 modificados mediante resinas poliméricas a base de flúor
para aumentar la hidrofobicidad y aumentar su actividad electrocatalítica al disminuir la
interacción de los radicales *OH con el óxido. La estabilidad de estos electrodos, así
como su actividad catalítica es incluso comparable con la del diamante dopado con boro.
31
Otras investigaciones enfocadas en las aplicaciones fotocatalíticas y
fotoelectrocatalíticas apuestan por la fabricación de materiales combinados donde los
nanotubos de TiO2 se combinan con nanopartículas de plata, oro o, platino, para
incrementar su foto-respuesta debido a que el depósito de estas partículas facilita la
separación electrón-hueco (e--h+) y promueve la transferencia electrónica interfacial , lo
cual incrementa la reactividad superficial.32 Otros óxidos como ZnFe2O433 o Cu2O,34 por
mencionar algunos, se han también depositado sobre los nanotubos de TiO2 con la
finalidad de que estos materiales asistan a los nanotubos en la absorción de luz visible;
ya que, poseen anchos de banda menores a la fase anatasa del TiO2. En este mismo
sentido se han realizado modificaciones electrónico-estructurales al TiO2 a través del

6
dopaje con N, C o Si, de manera que la energía del ancho de banda del TiO2 disminuya
para que la captura de luz se dé en la región visible.35
Es evidente que la relativa facilidad de preparación de los nanotubos de TiO2 y sus
propiedades únicas los hacen un material semiconductor ampliamente estudiado ya sea
desde el punto de vista del control y optimización de las variables que afectan su
formación y crecimiento, así como en las modificaciones que pueden sufrir para mejorar
su desempeño en alguna aplicación determinada, que en el caso del presente proyecto
es la oxidación electrocatalítica y fotoelectrocatalítica de compuestos orgánicos de
interés ambiental.

7
2. Marco Teórico
2.1. Nanotubos de dióxido de titanio en la eliminación de contaminantes orgánicos
acuosos
Como ya se ha mencionado, una de las nanoestructuras de dióxido de titanio con un alto
potencial de aplicación en los procesos avanzados de oxidación, como la fotocatálisis y
la fotoelectrocatálisis, son los nanotubos de dióxido de titanio (Nts-TiO2). Los Nts-TiO2
sintetizados a partir de la anodización de placas de titanio han despertado gran interés,
ya que pueden mejorar el rendimiento en aplicaciones conocidas del dióxido de titanio,
como la fotoelectrólisis del agua, fotocatálisis, las heterouniones en celdas solares,
sensores de gases y purificación del medio ambiente.36-38 La mejora en el rendimiento
en estas aplicaciones radica en que los Nts-TiO2 proporcionan un arreglo nanotubular
altamente ordenado con una gran superficie específica. La estructura altamente
ordenada puede mejorar el transporte de electrones fotogenerados en la película de
TiO2, ya que proporciona un canal eléctrico unidireccional y reduce los límites de grano
en la fotocatálisis y en aplicaciones fotoelectroquímicas. Por su parte, la gran superficie
puede permitir la captación más eficiente de luz.39-40
Como material para la oxidación electrocatalítica de colorantes tipo azo, los Nts-TiO2 han
mostrado una nula o muy baja capacidad para oxidarlos. Sin embargo, cuando la
oxidación electroquímica se combina con radiación UV, la degradación se vuelve más
eficaz; como en el caso de la decoloración de naranja de metilo, la cual, según Zhang y
colaboradores, alcanzó una eficiencia de degradación de 56.3% para un tiempo de
irradiación de 90 minutos con una concentración de colorante inicial de 2 x 10-4 M.40
Asimismo, en electrocatálisis, los nanotubos de TiO2 se han utilizado como sustrato para
soportar materiales electroactivos, como los metales nobles (Ag, Au, Pd, Pt y Ru)41-43 u
óxidos metálicos.29-30,44 Como se mencionó anteriormente, los Nts-TiO2, constituyen un
material de gran interés en electroquímica debido a sus propiedades semiconductoras,
la capacidad de inserción de huéspedes y su estabilidad a largo plazo. Estas
características hacen a los Nts-TiO2 muy adecuados para contener otros materiales,
pues proporcionan gran área superficial y una estructura químicamente estable que
puede ser utilizada en varias ocasiones.43 De tal manera que, los Nts-TiO2 se han usado
como sustrato sobre el cual se han dispersado nanopartículas de oro de 3 nm, las cuales

8
han demostrado ser más eficaces para la reducción de oxígeno que las nanopartículas
de oro dispersadas en una película compacta de TiO2. Asimismo, el oro y otros metales
nobles como la plata, el paladio, el platino y el rutenio se han depositado en los Nts-TiO2
con el fin de mejorar las propiedades fotocatalíticas del TiO2.41-42
2.1.1. Fotocatálisis sobre nanotubos deTiO2
En los últimos años, el TiO2 coloidal y en partículas se ha utilizado para fotodegradar
contaminantes tanto en fases líquidas y gaseosas. Sin embargo, estos sistemas
suspendidos deben enfrentarse a tres problemas técnicos inherentes: a) separación o
filtración después de la reacción, b) agregación de partículas y c) los problemas
asociados a sistemas de flujo continuo. Se han reportado métodos para preparar TiO2
sobre un soporte sólido, pero la eficiencia del sistema inmovilizado es mucho menor que
la del sistema suspendido, debido a la reducción de la superficie de área activa en el
catalizador inmovilizado. Por consiguiente, los Nts-TiO2 pueden superar este
inconveniente debido a su alta superficie específica. 45 A este respecto, se evaluó la
actividad fotocatalítica de los Nts-TiO2 hacia la decoloración de naranja metilo y se
comparó con la de una película de nanopartículas TiO2. En este estudio, los autores
reportan una decoloración más eficiente sobre el arreglo de nanotubos que en la película
de nanopartículas. Esta eficiencia más alta se atribuye a una separación más efectiva
entre los pares electrón-hueco fotogenerados y una mayor área superficial expuesta en
la estructura de nanotubos.45
Se ha observado que la actividad fotocatalítica del TiO2 se mejora mediante el depósito
de partículas de metales nobles, ya que, esto aumenta la separación electrón-hueco y
se promueven procesos de transferencia electrónica. Se ha corroborado que los
nanotubos recubiertos con nanopartículas de oro y plata forman uniones Schottky locales
que presentan un gradiente de potencial más alto que en la interfase TiO2/electrolito. 42
La fotocatálisis también puede mejorarse si se utilizan dos metales simultáneamente
para modificar el TiO2, como se investigó para una película co-modificada de nanotubos
de TiO2 con Au-Pd (Au-Pd-TiO2). Las actividades, tanto del TiO2 descubierto como de
Au-Pd-TiO2 se compararon con la degradación del malatión. El estudio mostró que se
eliminó el 73.8% de malatión en presencia de TiO2 descubierto, pero se logró una
eliminación del 98.2% cuando se utilizó Au-Pd-TiO2 en lugar de TiO2 descubierto.43

9
Como puede apreciarse, una de las principales tendencias en el uso ambiental de los
nanotubos de TiO2 es combinarlo con otro material, metal o semiconductor, con el fin de
mejorar sus propiedades fotocatalíticas o para obtener un material dual que presente
propiedades de los materiales constitutivos. En algunos casos, el arreglo de nanotubos
de TiO2 sólo desempeña el papel de sustrato con una gran área superficial, buena
resistencia mecánica y una mayor capacidad de adsorción para soportar un óxido
metálico altamente electroactivo como PbO2 o, SnO2 dopado con Sb. 29-30,46. PbO2 y
SnO2 son conocidos por tener un alto sobrepotencial de evolución de oxígeno que los
hace más eficientes para electrooxidación de contaminantes orgánicos. Estos óxidos han
sido soportados en cerámicas, Ti y otros materiales de soporte adecuados para
aplicaciones ambientales debido a su bajo costo, buena resistencia a la corrosión y alta
conductividad, pero la inestabilidad mecánica de sus recubrimientos es una desventaja
importante que debe ser superada. Para lograr ese objetivo, se han realizado varias
modificaciones a sus métodos de síntesis. La modificación del sustrato es una de ellas;
y dentro de esta modificación, el uso de Nts-TiO2 proporciona una estructura donde los
materiales electroactivos pueden penetrar y ser combinados con más firmeza, lo que
aumenta la estabilidad de los electrodos.29-30,46
2.1.2. Fotoelectrocatálisis sobre Nts-TiO2
La idea de combinar TiO2 con otros óxidos para obtener electrodos que presenten ambas
propiedades, tanto fotocataliticas y electrocataliticas no es nueva.47 Sin embargo, la
posibilidad de utilizar los Nts-TiO2 preparados anódicamente en placas de Ti, que sirvan
tanto de soporte como de material fotoactivo, hace que sea factible crecer o implantar
óxidos electroactivos para crear un material de electrodo con propiedades
fotoelectrocataliticas. De este modo, la preparación de electrodos de este tipo, ha
recibido gran atención recientemente.
El uso de Nts-TiO2 descubiertos como fotoelectrocatalizadores ha sido reportado en la
decoloración de pigmentos tipo azo, como es el caso de naranja de metilo o naranja
ácido 7. En el caso del naranja de metilo, las actividades fotoelectrocatalíticas (PEC) y
fotocataliticas (PC) del electrodo de nanotubos se compararon en experimentos de
decoloración donde se aplicó un potencial de 0.5V e iluminación UV (365 nm). Una
remoción de 99.06% se alcanzó bajo condiciones PEC mientras que sólo el 21.5% del
naranja de metilo se eliminó por el proceso de PC para un tiempo total de 90 minutos.40

10
Para el naranja ácido 7, la remoción total (decoloración) fue alcanzada después de 45
min de descomposición fotoelectrocatalitica bajo un potencial aplicado de 1.0 V vs.
Ag/AgCl.48 De acuerdo con este autor,48 la aplicación de un potencial electroquímico
ayuda a controlar el doblado de las bandas que puede resultar en una separación más
eficiente de los portadores de carga.
Una forma de mejorar las propiedades fotoelectrocataliticas de los Nts-TiO2 consiste en
acoplarlos con otros materiales como Sb dopado, SnO2, SiO2, Bi2O3, Fe2O3, ZnO y CdS.
29-30,44, 49-50 Así, por ejemplo, un fotocatalizador como Bi2O3 que es capaz de oxidar el
agua bajo irradiación de luz visible, se puede crecer sobre un electrodo de arreglo de
nanotubos de TiO2 y el electrodo resultante de la combinación de ambos óxidos presenta
una mayor actividad catalítica hacia la degradación del 2,4-diclorofenol que los
electrodos de los materiales individuales de Bi2O3 ó TiO2. La mayor actividad catalítica,
radica en que un electrodo combinado como Bi2O3/TiO2, presentará alta actividad
fotocatalítica tanto en las regiones de UV y del visible, ya que, Bi2O3 es fotoactivo bajo
luz visible y TiO2 es activo bajo luz UV.44
2.2. Aspectos importantes en la formación de nanotubos de dióxido de titanio
por anodización
Como se ha mencionado, el dióxido de titanio es un material con diversas propiedades
físicas y químicas que encuentran numerosas aplicaciones. En la actualidad, se busca
mejorar dichas propiedades al sintetizar este material en forma de nanoestructuras, entre
las que se pueden enumerar a: nanopartículas, nanovarillas, nanalambres y nanotubos.51
Entre éstos, los nanotubos de TiO2 encuentran aplicaciones importantes en: a) las DSSC,
b) en sensores de gases altamente sensibles a hidrógeno, c) como sustratos de
catalizadores para la electrooxidación de metanol, d) sustratos para el crecimiento de
hidroxiapatita en aplicaciones biomédicas, e) aplicaciones fotocatalíticas,
fotoelectrolíticas, y fotovoltaicas; y, f) en la liberación de fármacos y filtración de
biofluidos.52-53 El desempeño de los nanotubos depende de las diferentes características
que presentan entre las que se puede destacar el diámetro, el ancho de pared, su
longitud y su grado de ordenamiento.

11
2.2.1. Mecanismo de formación de nanotubos por anodización
La formación de los nanotubos de dióxido de titanio por anodización, en presencia de
fluoruro, está gobernada por las reacciones en competencia de: a) la formación del óxido
y b) de su disolución como complejos de fluoruro.
Ti + 2H2O TiO2 + 4e- + 4H+ (1)
TiO2 + 6F- + 4H+ [TiF6]2- + 2H2O (2)
El mecanismo de formación de nanotubos por anodización de una placa de titanio
involucra los siguientes pasos: 1) Se forma una capa de óxido en la superficie metálica
de Ti por la reacción con O2- y OH- en el ánodo; después, estos aniones difunden dentro
de la capa de óxido y continúan reaccionando con el metal que se encuentra debajo; 2)
El Ti4+ presente en la interface metal/óxido migra a la interface metal/electrolito bajo la
influencia del campo eléctrico, 3) Se inicia la disolución en la interface óxido/electrolito
debido a que el enlace Ti-O se debilita bajo la acción del campo eléctrico aplicado y esto
causa la disolución. 4) Debido a la presencia del anión fluoruro en los electrolitos usados
en el anodizado o bien, debido a algunos ácidos, la disolución química es un fenómeno
determinante en el crecimiento del nanotubo,54-55 Figura 4.
Figura I.4. Representación de la anodización de Ti, a) en ausencia de fluoruros (lo que produce
capas de óxido), y b) en presencia de fluoruros (lo que produce crecimiento del tubo).56

12
Cao y colaboradores, 55 sugieren que el proceso de crecimiento del nanotubo es un
modelo periódico, ya que en un transitorio de corriente, la corriente muestra oscilaciones
con una amplitud asimétrica; estas oscilaciones se deben a la oxidación y disolución que
ocurren de manera periódica. Las micrografías de la microscopía electrónica de barrido
(MEB) muestran que las ondulaciones que rodean a los nanotubos, se conectan entre sí
y existen en un plano perpendicular al tubo. Las ondulaciones conectadas entre sí forman
una capa, la distancia entre dos ondulaciones es la misma que el grosor de la capa del
fondo, lo que Cao explica de la siguiente manera:
1) La primera capa de óxido se forma en el estado inicial de la anodización, lo que
requiere un alto potencial, la capa formada es rugosa y con rupturas distribuidas
aleatoriamente en diversos sitios. En estos sitios se inicia la disolución de TiO2
que deriva en la formación de pozos. Figura 5a.
2) Al incrementarse el tamaño de los pozos, el electrolito tiene la oportunidad de
infiltrarse en la interfase de la capa de óxido/metal y se forma una segunda capa
de óxido, la cual se rompe nuevamente por acción del campo eléctrico. Debido a
que, la capa de óxido se forma y se rompe una y otra vez, es que ocurren las
oscilaciones de corriente observadas en los transitorios de corriente. Los poros se
forman debido a la prolongación longitudinal y a la expansión lateral de los pozos
con el incremento en el número de las capas de óxido, Figura 5b y 5c.
3) Con el incremento en el tamaño del poro, el área superficial interna aumenta y la
tensión superficial que ocasiona el encogimiento del poro también aumenta. La
cercanía entre poros expulsa al óxido entre ellos, lo que conduce a la separación
entre poros y a la eventual formación del tubo, Figura 5d.

13
Figura I.5. Etapas de formación de la capa de nanotubos. 56
4) El tamaño del poro que se incrementa en las últimas capas demora el incremento
del poro en la capa anterior, por lo que, la separación de poros en las capas
subsecuentes ocurre después. De ahí que, algo del óxido entre capas adyacente
quede remanente, lo que se traduce en la formación de las ondulaciones de las
paredes de los nanotubos. La figura 6, muestra la unidad periódica entre dos
ondulaciones y la capa-barrera formada entre los fondos conectados, lo que
explica la formación de la estructura tubular como resultado del avance periódico
de la capa-barrera por el sustrato de Ti, lo cual se manifiesta en las oscilaciones
de corriente, figura 6b.

14
Figura I.6. a) Esquema de la unidad periódica entre ondulaciones, b) Transitorio de corriente
durante la formación de nanotubos, c) Imagen de MEB de nanotubos con las ondulaciones
periódicas.55
2.2.2. Fases cristalinas del TiO2
El dióxido de titanio recién preparado es amorfo y debe recocerse a más de 250°C para
cristalizar en la fase cristalina que se desea de acuerdo a las posibles aplicaciones del
material; ya que, por ejemplo, la fase anatasa es adecuada para el dióxido de titanio en
DSSC y la fase rutilo, se prefiere en sensores de gases.
La titania o dióxido de titanio (TiO2) puede presentarse en cuatro polimorfos naturales:
brookita, anatasa y rutilo, así como también una fase amorfa; y existen, al menos tres
polimorfos que se han producido sintéticamente.57 Brookita es la fase más rara, estable
sólo a muy bajas temperaturas, y rutilo es la única fase termodinámicamente estable en
el material en el bulto. Por su parte, anatasa es la fase comúnmente hallada en
nanocristales. A este respecto, Zhang y Banfield señalan que anatasa es más estable
que rutilo cuando el tamaño de partícula decrece por debajo de los 14 nm.58
Ambas, anatasa y rutilo se conforman de cadenas de octaedros distorsionados de TiO6,
pero difieren en el grado de distorsión de cada octaedro y en el ensamble de las cadenas.
El octaedro de anatasa se halla más distorsionado y su simetría es octaédrica elongada,
D2d; mientras que, rutilo presenta mayor simetría, Oh.
Las distancias Ti-Ti en la anatasa son mayores que en el rutilo y las distancias Ti-O son
menores. A su vez, en el rutilo cada octaedro está en contacto con otros diez vecinos
formando una cadena lineal. Por otro lado, anatasa está en contacto con ocho vecinos
formando una cadena en forma de zigzag. Estas diferencias en sus estructuras de red

15
les confieren diferentes densidades de masa y estructuras electrónicas de bandas, 59-60
Figuras 7 y 8.
Figura I.7. Estructuras de rutilo y anatasa del TiO2.60
Figura I.8. Empaquetamiento cristalino de las fases anatasa y rutilo.61
Para algunas aplicaciones como en dispositivos de separación de cargas, tales como
celdas solares sensibilizadas por pigmentos y en fotocatálisis, anatasa es la fase
cristalina que se prefiere favorecer en el TiO2, lo cual se debe a que presenta un ancho
de banda prohibida más amplio y una masa efectiva electrónica más pequeña, lo cual le
permite una movilidad mayor de los portadores de carga 59,62 Por otro lado, se prefiere el
uso de la fase rutilo en capas dieléctricas y en sensores de gases.55 Zihan Poh y
colaboradores,63 concuerdan en que cristales incipientes de anatasa, rutilo y titanio se
forman bajo el calentamiento a temperaturas mayores a los 300°C. La cristalización de

16
anatasa comienza entre los 250-280°C, y el calentamiento posterior hasta 400°C
fomenta la cristalización de esta fase, lo que incrementa su concentración. El
calentamiento a temperaturas entre 550° y 700°C favorece el crecimiento de anatasa
pero también de rutilo, este último se favorece a partir de 600°C. El calentamiento a más
de 650°C provoca el colapso de los nanotubos lo que favorece su descomposición y,
por ende, el retorno de la fase amorfa.54, 64 Es importante tomar en cuenta que el
calentamiento puede acarrear la reducción de los poros y del área superficial, así como
también una reducción de la longitud de los nanotubos.55
2.2.3. Variables que afectan el crecimiento de los nanotubos
La eficiencia de las estructuras nanotubulares en las aplicaciones antes mencionadas
estará determinada por su diámetro, longitud, y su superficie más expuesta. El
crecimiento en longitud de los nanotubos es una característica deseada debido a que
esto incrementa la razón de aspecto (aspect ratio) de los nanotubos. Sin embargo, a
mayor crecimiento, se presenta el doblado de los nanotubos en la capa superficial de
los mismos, lo que afecta el transporte del excitón (par electrón-hueco), la dinámica de
recombinación y la penetración de materiales de interés. Por otro lado, aún cuando los
nanotubos no presenten este doblado, los pozos de sellado y decapado sobre la
superficie incidirán desfavorablemente en su desempeño y restringirán sus aplicaciones.
Se ha planteado, por lo tanto, la necesidad de controlar las condiciones de anodización
para lograr la morfología deseada.65
Wang y colaboradores,65 señalan que la temperatura, el voltaje aplicado, el tiempo de
anodización, la concentración de flúor, y otras variables, alteran la velocidad de
anodizado y la velocidad de grabado químico. Por lo tanto, es necesario conocer su
influencia para controlar el crecimiento del nanotubo.
2.2.3.1. Voltaje aplicado
El voltaje aplicado tiene una clara influencia en el diámetro de los tubos. En general se
observa una dependencia lineal del diámetro y el grosor de la capa de nanotubos con el
potencial aplicado. 52, 56. Aunque es importante notar que este comportamiento puede
mostrar desviaciones a ciertos potenciales dependiendo del medio de anodizado. Macak
y colaboradores,52 en un medio de glicerol:agua (1:1), indican que potenciales de 2V
producen diámetros de 20 nm, mientras que potenciales de 40 V producen tubos de 300

17
nm de diámetro, asimismo, reportan que la dependencia del diámetro con el potencial
aplicado es prácticamente lineal, mientras que la dependencia del grosor de la capa con
el potencial es lineal hasta un voltaje de 20 V, a partir del cual el grosor de la capa se
mantiene más o menos constante, efecto que puede atribuirse a un adelgazamiento de
las paredes del tubo debido a la disolución química del óxido. En contraste, Wang reporta
que voltajes que van de 10 a 100 V producen capas de nanotubos que van de los 20 a
los 170 nm de diámetro y cuya longitud va de los 0.1 µm a los 6 µm en electrolito no
acuoso de etilenglicol con 2% (v/v) de HF y 0.5 % (p/p) de NH4F.65
Existe también un efecto importante a considerar: el grosor de la capa de óxido en el
fondo de los nanotubos (capa de barrera), el cual también se incrementa con el voltaje.
Esta barrera de óxido se incrementa a razón de 2-2.5 nm por voltio aplicado. 56
2.2.3.2. Viscosidad
La viscosidad es otra variable que influye en la morfología de los nanotubos. En general,
los electrolitos viscosos contribuyen a la formación de tubos más largos y más lisos en
comparación con los electrolitos a base de agua. Además, se ha demostrado que en
electrolitos a base de glicerol la eficiencia de corriente para la formación de nanotubos
es casi del 100%.52
Otro aspecto, que reafirma el control por difusión en la etapa de crecimiento se puede
deducir de las curvas de polarización y de corriente-tiempo para experimentos en agua,
glicerol y etilenglicol en presencia de NH4F, ya que, éstas coinciden con una disminución
en la densidad de corriente para los medios no acuosos. Esto corrobora la dependencia
del coeficiente de difusión de la viscosidad bajo el modelo de Stokes-Einstein,
D=kBT/6πηa,
Donde kB es la constante de Boltzmann, T la temperatura absoluta, n la viscosidad
dinámica, y a, el radio del cuerpo esférico. 52
2.2.3.3. Efecto del pH
El pH juega un papel clave en la longitud de los tubos, en la rugosidad de sus paredes y
en la eficiencia de corriente. Lo que ocurre es que bajo condiciones limitadas por difusión,
la acidificación local en la punta del poro adquiere un papel determinante en su
crecimiento. La acidificación es el resultado de la oxidación de titanio (Ti0 Ti4+) y de la

18
hidrólisis (ver ecuación 1); asimismo, la velocidad de disolución química de TiO2 se
incrementa en soluciones ácidas que contienen fluoruro. Ambas, la acidificación local y
la disolución química disminuyen en electrolitos viscosos, lo que se traduce en la
elongación de los tubos.
La lisura de la pared del tubo se origina por el amortiguado de picos locales en el flujo de
las especies de reacción dentro de los tubos, es decir, minimiza las diferencias locales
de los valores de pH; como resultado, no existen oscilaciones detectables de corriente y
los tubos crecen libres de ondulaciones.52
2.2.3.4. Tiempo de anodizado
Dependiendo del medio de anodizado, los nanotubos pueden crecer en mayor o menor
grado. En el caso de electrolitos a base de agua, una extensión en el tiempo de
anodizado tiene un efecto menor; ya que, en electrolitos acuosos, el crecimiento del tubo
se ve limitado por la disolución del óxido a pH´s ácidos locales. Por otra parte, si el
electrolito es a base de glicerol, la longitud del tubo se incrementa linealmente hasta
llegar a un equilibrio. 65
2.2.3.5. Temperatura
La temperatura controla el diámetro y la longitud de los nanotubos, se ha observado que,
a mayor temperatura, mayor es el diámetro y la longitud de los tubos. Macak y
Schmuki,52 encontraron que en medio de glicerol esto es cierto para temperaturas de 0,
20 y 40°C, pero temperaturas mayores favorecen el doblado indeseado de los tubos,
asimismo, a estas temperaturas ocurre la separación de la capa de nanotubos del
sustrato.
La gran dependencia del crecimiento de los nanotubos de la temperatura y la viscosidad
indican que el factor limitante en su crecimiento es la difusión de los reactantes a la punta
del poro o de los productos que se alejan de ella al dirigirse al seno de la solución.52
2.2.3.6. Efecto de la concentración de fluoruro
Diversos autores señalan que la concentración de fluoruros es un factor clave en la
electroquímica de los nanotubos de TiO2. Se ha observado que a mayor concentración
de fluoruro, se obtienen mayores densidades de corriente de las curvas de polarización.
Las curvas de polarización típicas de la anodización de Ti para un sistema glicerol/agua

19
muestran en la primera etapa (justo en el inicio de la anodización), que las densidades
de corriente se elevan durante el barrido hasta alcanzar un primer máximo, en esta etapa
se forma la capa de óxido. Este incremento en corriente es más pronunciado en
presencia de fluoruros lo que puede atribuirse a dos factores: existe un transporte
asistido de F- de campo rápido a través de la capa de óxido que compite con el transporte
de O2-, o bien existe una porosidad nanoscópica en el óxido que crece en los electrolitos
con fluoruro. El grosor de la capa de óxido en ese preciso momento, es proporcional al
potencial aplicado. Después de este máximo, la corriente continúa su incremento, sin
embargo, sólo en la presencia de fluoruro se nota claramente la formación de un
segundo pico máximo. Este pico se ve acompañado por la formación de poros que se
forman aleatoriamente. Después de este segundo pico, la corriente decae debido al
traslape de dos efectos: por un lado, a tiempos muy cortos de anodización, la capa
compacta de óxido alcanza un grosor de estado estacionario y por otro, a tiempos más
largos, los efectos de difusión dentro de los tubos se vuelven dominantes en el control
de la velocidad.56 En este artículo, Macak y colaboradores describen las etapas de
crecimiento de los nanotubos. En la primera etapa, se forma una capa delgada y
compacta de TiO2, la cual contiene sitios de ruptura estadísticamente distribuidos, es
decir, puntos donde ocurre la disolución acelerada. Despúes de 1-3 minutos estos poros
de nucleación son evidentes en casi toda la superficie y aún se visualizan zonas de la
capa inicial de óxido. Después de 3-10 minutos, la distribución de poros tiene una
apariencia aleatoria y entonces comienza una disolución más pronunciada en el fondo
de los poros. En esta etapa, no quedan indicios de la capa inicial de TiO2. Es después
de los 30 minutos de anodización que, se comienza a formar la estructura nanotubular y
su grosor ya ha alcanzado los 500 nm. Si se anodiza por más tiempo, se favorece el
desarrollo de la capa de nanotubos auto-organizada y su crecimiento en grosor. Este
crecimiento continúa hasta que se alcanza un estado estacionario.56
2.2.3.7. Difusión: factor que controla la velocidad de crecimiento del tubo
En la formación de nanotubos el tratamiento por anodizado se lleva a cabo,
generalmente, sin agitación de la solución electrolítica debido a que se ha encontrado
que esto conduce a un crecimiento más homogéneo de los tubos sobre la superficie. En
un experimento de corriente-tiempo, se observa que al iniciar la agitación, la corriente se
incrementa significativamente y que al cesar la agitación, la corriente regresa a su nivel
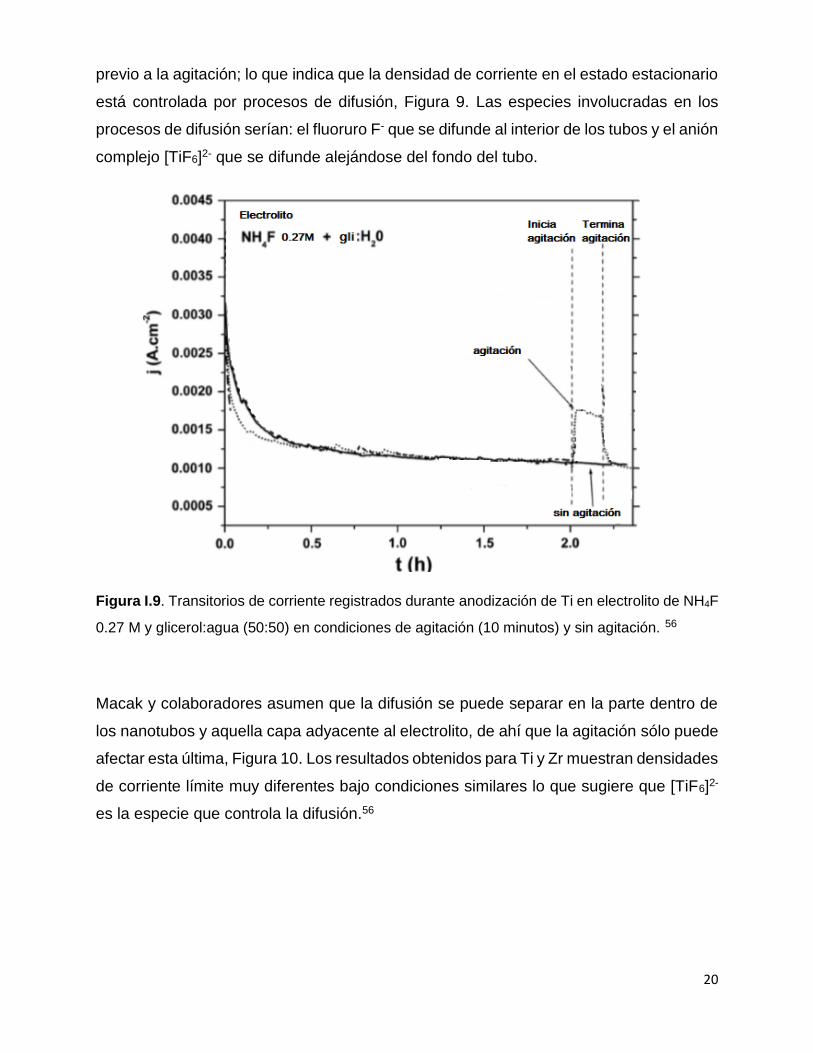
20
previo a la agitación; lo que indica que la densidad de corriente en el estado estacionario
está controlada por procesos de difusión, Figura 9. Las especies involucradas en los
procesos de difusión serían: el fluoruro F- que se difunde al interior de los tubos y el anión
complejo [TiF6]2- que se difunde alejándose del fondo del tubo.
Figura I.9. Transitorios de corriente registrados durante anodización de Ti en electrolito de NH4F
0.27 M y glicerol:agua (50:50) en condiciones de agitación (10 minutos) y sin agitación. 56
Macak y colaboradores asumen que la difusión se puede separar en la parte dentro de
los nanotubos y aquella capa adyacente al electrolito, de ahí que la agitación sólo puede
afectar esta última, Figura 10. Los resultados obtenidos para Ti y Zr muestran densidades
de corriente límite muy diferentes bajo condiciones similares lo que sugiere que [TiF6]2-
es la especie que controla la difusión.56

21
Figura I.10. Perfiles de concentración de las especies dentro y fuera del área de difusión de los
tubos con y sin agitación.56
2.3. Depósitos de Dióxido de Plomo
El dióxido de plomo es un material de especial interés en electroquímica debido que es
un material de bajo costo, alta conductividad electrónica, buena estabilidad bajo
condiciones controladas, resistencia al ataque de iones, y alto sobrepotencial para la
evolución de oxígeno en medio acuoso (1.9 V/she). 66. Algunas de sus aplicaciones más
importantes son en la generación de ozono, oxidación y reacciones de transferencia de
oxígeno, así como en la degradación oxidativa de contaminantes orgánicos, tales como
fenol, dimetilsulfóxido, glucosa y ácido cloranílico.67-68
Por lo general, se le deposita sobre sustratos metálicos como Ti,69 Au,70 y Pt,71 con el
objeto de mejorar sus propiedades mecánicas y catalíticas.72 Asimismo, sus
electrodepósitos en numerosos sustratos cerámicos y metálicos son de gran interés
debido a la facilidad de obtención de PbO2 a partir de las sales de nitrato o acetato de
plomo. Diversos autores señalan que las características del depósito obtenido dependen
de varios parámetros: tipo de substrato (generalmente titanio) y su pretratamiento,

22
composición del baño, temperatura, agitación, densidad de corriente anódica, entre
otros.73
Sin embargo, a pesar de las numerosas ventajas que ofrecen los electrodos de dióxido
de plomo, éstos presentan dos desventajas que limitan su periodo de servicio: la
liberación de Pb a la solución de trabajo y su predisposición a la corrosión bajo
condiciones de alta densidad de corriente impuesta. Asimismo, cuando se deposita PbO2
sobre algún sustrato, puede presentar problemas de resistencia mecánica y de
adherencia como ha sido el caso de los sustratos de titanio. 74
2.3.1. Modificaciones en la construcción de electrodos de PbO2
Para mejorar la estabilidad del óxido y su actividad catalítica, se han probado diversas
modificaciones en la construcción de sus electrodos, entre las que destacan: variaciones
al sustrato donde se crece la capa de PbO2,75 modificaciones electrónico-estructurales a
base de dopajes con iones fluoruro76 o metálicos en el depósito de PbO2,77-78 cambios
en la hidrofobicidad de la superficie, e incluso, el uso de nanotubos de titania como
sustrato.31, 77
2.3.2. Modificaciones en el sustrato
En el depósito de PbO2 se han usado como sustratos metales como Ti, Pt, Ta y Nb; y
materiales a base de carbono como grafito y diamante dopado con boro (BDD). Entre
éstos, titanio se presenta como un material de soporte adecuado debido a su resistencia
su dureza, conductividad, alta estabilidad química, adherencia y bajo costo.31
En el pretratamiento al sustrato de titanio, con el objeto de eliminar óxido de titanio, grasa
y otros contaminantes, se puede incluir: sandblasting, desengrasado alcalino seguido por
grabado químico en ácido oxálico en ebullición o ácido clorhídrico. Después de estos
procesos, el titanio presenta una superficie limpia y rugosa, que es capaz de adherirse
fuertemente al dióxido de plomo. También se puede depositar sobre esta superficie,
alguna intercapa que tiene como finalidad mejorar la conductividad, estabilidad y
actividad del electrodo, entre estas intercapas se puede mencionar al Pt, SnO2, Sb2O5,
TiO2/Ta2O5 y TiO2/RuO2, así como multintercapas aplicadas antes del catalizador.42-44.
Entre estas intercapas se comienza a incluir al dióxido de plomo en fase alfa, ya que ésta
reduce el stress y evita que el electrolito penetre hasta el sustrato.31,79

23
2.3.3. Modificaciones electrónico-estructurales
Para aumentar la actividad catalítica del electrodo de PbO2 se han incluido en su
preparación especies dopantes como iones hierro, cobalto, bismuto, cobre, cerio, y
fluoruro, entre otros. El objeto del dopaje tiene como finalidad aumentar, dirigir, mejorar
las propiedades del óxido resultante para un proceso electrocatalítico particular.
En el caso de cationes como bismuto y hierro, se ha demostrado que su inclusión
favorece la actividad en reacciones de transferencia anódica de oxígeno en sustratos
orgánicos e inorgánicos, así como en la evolución de ozono.80 El uso de cobalto aumenta
la rugosidad de composites de PbO2 + Co3O4 y de PbO2 + CoOx, y la actividad inicial
para reacciones de evolución de oxígeno también se ve incrementada, aunque decae
después de algunos días probablemente debido a la disolución de Pb, lo que deriva en
una pérdida de resistencia mecánica. 81-82
2.3.4. Electrodepósito de dióxido de plomo
El dióxido de plomo puede ser electrogenerado a partir de soluciones de Pb2+, ácidas o
básicas. La electrodeposición de PbO2 a partir de soluciones que contienen iones nitrato
es ampliamente utilizada y la formación del óxido involucra el siguiente mecanismo de
reacción:71, 81
H2O OHads + H+ + e (1)
Pb2+ + OHads Pb(OH)2+ (2)
Pb(OH)2+ + H2O PbO2 + 3H+ + e (3)
En el primer paso se forma, electroquímicamente, OH quimisorbido, seguido por la
interacción de éste con plomo (II) en solución para formar especies solubles que
probablemente contienen Pb(III); éste se oxida electroquímicamente para formar PbO2.
Velichenko y colaboradores,71 proponen que el último paso de transferencia electrónica
y la difusión de Pb2+ controlan la formación del óxido. La transferencia electrónica
controla la velocidad de formación cuando se trabaja en un intervalo bajo de

24
sobrepotencial; por el contrario, a intervalos más altos de potencial, la difusión de Pb2+
limita dicha velocidad.
El PbO2 electrodepositado se presenta en dos fases cristalinas α y β; y, dependiendo de
las condiciones de deposición, se puede favorecer una fase sobre la otra. La fase
tetragonal β-PbO2 se obtiene predominantemente de soluciones ácidas y, bajo distintas
condiciones de densidad de corriente se puede favorecer la fase α. Al respecto,
Velichenko hace notar que la literatura puede ser contradictoria, por lo que, deben
especificarse las condiciones experimentales bajo las cuales se obtiene una fase u otra.
En este sentido, el autor reportó que en condiciones de intervalos bajos de
sobrepotencial (1.3-1.55 V) la velocidad de reacción puede estar limitada por el paso 1 ó
3. Si el potencial es mayor a 1.55V, el control difusional prevalece. La electrodeposición
de PbO2 a partir de solución 0.1 M de Pb(NO3)2 en HNO3 0.1M genera PbO2 en fase β y
un poco del óxido en su fase α. La cantidad de ésta última se incrementa si se incrementa
el potencial cuando predomina el control cinético, mientras que disminuye cuando el
incremento ocurre bajo control difusional.
Es importante conocer la naturaleza exacta del PbO2 electrodepositado, ya que, sus
fases tienen diferentes propiedades. El α-PbO2 tiene una movilidad electrónica menor y,
por lo tanto, una mayor concentración electrónica. Esto se manifiesta en la conductividad
del α-PbO2, la cual es de un orden de magnitud menor que la del β-PbO2. A su vez, la
fase α tiene una estructura más compacta lo que implica una menor actividad
electroquímica; esto, por ejemplo, en el caso de las baterías disminuye la capacidad de
almacenamiento de energía por unidad de peso pero favorece un ciclo de vida más largo.
α-PbO2 también proporciona tiempos más largos de operación (>2500 h) como electrodo
en la generación de ozono.66
2.3.5. Fases y estructura del PbO2
Las fases α y β del dióxido de plomo son bien conocidas; así, α-PbO2 tiene la estructura
ortorrómbica de la columbita (grupo espacial Pbcn, Vh) y β-PbO2 tiene la estructura
tetragonal del rutilo (grupo espacial P4/mnm, D4h). En ambas fases, el ion Plomo (IV)
está en el centro de un octaedro distorsionado pero se diferencian en la manera en que
estos octaedros se encuentran empacados. Los octaedros en el β-PbO2 forman cadenas

25
lineales mientras que en el arreglo ortorrómbico del α-PbO2 forman cadenas con forma
de zig-zag, 83 Figura 11.
Figura I.11. Empaquetamiento del oxígeno octaédrico en dióxido de plomo en α y β-PbO2,
también se muestran las distancias plomo-oxígeno y oxígeno-oxígeno.83
Estas fases son fácilmente diferenciables por difracción de rayos X y ninguna de las dos
es completamente estequiométrica, lo que les confiere su alta conductividad. Rüetschi 84
sugiere que la interpretación cristalográfica de la no estequiometría de PbO2 indica que
la subred aniónica de la estructura está completa y que un empaquetamiento muy denso
de O2- excluye la presencia de H2O u OH interscicial, la desviación de la estequiometría
se debe a un desorden en la subred catiónica. El modelo de Rüetschi propone que el
PbO2 contiene vacancias catiónicas las cuales se agrupan en capas que actúan como
interfaces entre regiones cristalográficas ordenadas. Estas capas pueden hospedar
cationes de iones Pb (II). De acuerdo a este modelo, la composición de PbO2 se escribe
como (Pb4+) 1-x-y(Pb2+)y(O2-)2-4x-2y(OH-)4x+2y, donde x representa la fracción de la vacancia
catiónica y, y la fracción de iones Pb2+ presentes con respecto al número total de sitios
catiónicos presentes.84
2.3.6. Conductividad de PbO2
De acuerdo a Mindt 85 las conductividades de α-PbO2 y β-PbO2 son cercanas a 103 ohm-
1cm-1 y 104 ohm-1cm-1 respectivamente, así también, los estudios del efecto Hall

26
confirman que la conducción se debe al movimiento de electrones. Estos electrones
libres podrían provenir de la no estequiometría mencionada, de la incorporación de
protones y de impurezas dopantes.
La conductividad de β -PbO2 es comparable a metales como Ti o Hg y superior a la de
la mayoría de las formas alotrópicas del carbono. Para varios autores, el β-PbO2 es un
conductor metálico. La naturaleza metálica de PbO2, de acuerdo a Payne y
colaboradores se debe a la ocupación de estados de la banda de conducción arriba del
nivel de Fermi del PbO2 estequiométrico, debida a defectos de la vacancia de oxígeno.86
2.4. Depósitos de SnO2
El dióxido de estaño (SnO2) es un material semiconductor (tipo n) transparente,
denominado así porque presenta una alta transmitancia óptica, con energía de banda
prohibida de 3.6-3.8eV.87-89. El dióxido de estaño presenta una adecuada combinación
de propiedades químicas, electrónicas y ópticas, que lo hacen útil como material para
sensores de gas y de biomoléculas,90 conductor a alta temperatura, electrodo
transparente para películas delgadas, catalizador y en dispositivos optoelectrónicos, con
gran estabilidad química y mecánica.91 El SnO2 es muy estable, adherente y
biocompatible92 estas características además de su morfología, tamaño de grano y
cristalinidad lo hacen un material de electrodo de gran interés para su uso en
electrocatálisis.93-96
2.4.1. Características físicas y químicas del SnO2 y sus variantes dopadas
El óxido de estaño posee una valencia dual que rige su comportamiento superficial. La
valencia dual facilita la transformación reversible de la composición superficial, la cual
puede variar de cationes superficiales Sn4+ a cationes reducidos Sn2+ dependiendo del
potencial químico de oxígeno en el sistema. Esta valencia dual refleja la existencia de
dos óxidos principales de estaño: el óxido estánico (SnO2) y el óxido estanoso (SnO). En
el caso del SnO, su band gap no se encuentra bien definido y se conoce que se encuentra
en el intervalo de 2.5-3 eV, mientras que el band gap de SnO2 generalmente se acepta
como 3.6 eV. El óxido estanoso tiene la estructura conocida como litargirio mientras que
el óxido estánico posee una estructura tipo rutilo (tetragonal) y a presiones altas se forma
una estructura ortorrómbica ligeramente más densa. Termodinámicamente, el óxido
estánico es la forma más estable del óxido de estaño. La oxidación de SnO a SnO2 ocurre

27
fácilmente porque existe una gran similitud entre la matriz del plano (0 0 1) de SnO y el
plano (101) del SnO2, la cual parece formarse por la introducción de una capa de oxígeno
al plano (001) del SnO, 97 Figura 12.
Figura I.12. Comparación de la estructura de las superficies de (a) SnO(001) y (b) SnO2(101).
Las similitudes entre las dos orientaciones cristalinas se han usado para explicar la
transformación de películas de SnO(001) a películas de SnO2(101).97
En la mayoría de sus aplicaciones, el SnO2 rara vez se encuentra puro, pues,
generalmente se modifica por el dopaje y otros aditivos. La conductividad del SnO2 se
puede aumentar mediante la introducción controlada de un átomo dopante en la red
cristalina. Algunos dopantes empleados son el arsénico, el cloro, el fosforo; sin embargo
los elementos más utilizados para aumentar la conductividad del SnO2, a temperatura
ambiente han sido el indio, el antimonio y el fluor.88, 98-99
Los electrodos de SnO2 dopados se han utilizado en la eliminación y transformación
electroquímica de compuestos orgánicos e inorgánicos, al igual que en la detección de
gases tóxicos. El SnO2 ha mostrado resultados favorables en la oxidación electroquímica
de compuestos aromáticos.95
2.4.2. Métodos de preparación de depósitos de SnO2
Entre los métodos de síntesis de los polvos cerámicos de SnO2, puro o dopado, estos
incluyen: sol-gel, coprecipitación de oxalatos o hidróxidos, precursor polimérico,
reacciones en estado sólido a través de la descomposición de carbonatos, deposición en
fase vapor y la oxidación de SnO (α) a SnO2 por crecimiento epitaxial, deposición por haz

28
de iones, pulverización catódica (sputtering), síntesis hidrotermal y solvotermal, método
del citrato amorfo, autocombustión y rociado pirolítico (spray-pyrolysis).100-102
2.4.2.1. Deposición por rociado pirolítico
Entre las técnicas mencionadas anteriormente, la técnica de rociado pirolítico ha probado
ser simple, reproducible y de bajo costo, así como adecuada para varias aplicaciones.
Ya que es una técnica versátil para la deposición de óxidos metálicos. Además, la
disposición experimental simple, alta tasa de crecimiento y la capacidad de producción
en masa de los revestimientos de gran área, la hace útil para procesos industriales
(fabricación del vidrio) así como en la fabricación de celdas solares. Lo que es más, esta
técnica abre la posibilidad de controlar la morfología de la película y el tamaño de
partícula en una escala nanométrica. 100
La técnica de rociado pirolítico es muy socorrida en la investigación de materiales para
preparar películas tanto delgadas como gruesas, revestimientos cerámicos y, polvos. A
diferencia de muchas otras técnicas de deposición de películas, esta técnica involucra
un procesamiento muy simple y relativamente rentable ofreciendo así, un método fácil
para preparar películas de cualquier composición, que no requiere una alta calidad de
los sustratos o productos químicos.103
En general, el rociado pirolítico es un proceso en el cual, se atomiza una solución
precursora, la cual reacciona a cierta temperatura, para generar la nucleación y
crecimiento de partículas sólidas. Lo cual puede involucrar procesos en los que las gotas
reaccionan sin evaporarse completamente, produciendo una partícula por gota o, bien,
procesos donde las gotas se evaporan completamente y las partículas comienzan la
nucleación desde la fase gaseosa. 104
La deposición efectiva del SnO2 por rociado pirolítico puede ser controlada por cinco
parámetros: temperatura del ambiente gaseoso, distancia entre la punta del atomizador
y el sustrato, el radio de la gota, concentración de la solución y el flujo de la solución.100
El esquema básico de un sistema de spray-pyrolysis consiste en un atomizador o
nebulizador, un gas acarreador, una solución precursora, un sustrato, y una fuente de
calentamiento (horno, placa caliente, llama) 103-104 y, dependiendo del método de
atomización se añadirán otros componentes al sistema como un compresor de aire en el

29
caso del método de atomización pneumático; o un sonicador en el caso de atomización
asistida por ultrasonido, por mencionar algunos.104 En la figura 13, se puede apreciar
algunos de estos componentes básicos.
Figura I.13. Componentes del sistema de rociado pirolítico.105
2.4.2.2. Modalidades de los sistemas de rociado pirolítico
El rociado pirolítico puede incluir diferentes formas de energía para inducir la formación
de las partículas como aquéllas que proveen un calentamiento local rápido como una
llama, plasma o láser; o bien, un calentamiento externo controlable mediante un horno.
Dentro de los métodos de rociado pirolítico podemos distinguir los siguientes: a) rociado
pirolítico en un reactor tubular, b) rociado pirolítico mediante la utilización de un reactor
de vapor de llama, c) combustión de emulsión y, d) rociado pirolítico de llama. Su
clasificación se basa en cómo se transfiere la energía térmica al precursor, cómo se
libera el precursor a la posición de reacción, y las características del producto final.106
En el método de rociado pirolítico en un reactor tubular, la solución se nebuliza dentro de
un reactor de pared caliente a temperaturas bien definidas. Las gotas del aerosol se
someten a evaporación, concentración del soluto dentro de la gota, secado, termólisis de
las partículas precipitadas y, finalmente, sinterización para densificar las partículas.106

30
En el método de rociado pirolítico de vapor de llama, la energía es aportada por una
llama. El precursor es atomizado directamente hacia la llama, reaccionando
endotérmicamente y enfriándose posteriormente. Por su parte, en el método de la
combustión de emulsión, se coloca la solución precursora en un aceite emulsionado, se
atomiza y se somete a ignición.106
El método de rociado pirolítico de llama consiste en un generador de gotas, un
pulverizador de llama, un reactor de cuarzo, un colector de partículas y una bomba de
vacío. Utiliza un nebulizador ultrasónico que genera gotas y las gotas generadas son
arrastradas hacia una llama de difusión de alta temperatura por un gas portador. El
secado, la evaporación, la descomposición, la cristalización y la disolución de la gota se
realizan durante la difusión de la llama. Se utilizan propano (combustible) y oxigeno
(oxidante) para generar la difusión de la llama. Este método permite ajustar la velocidad
de producción, la temperatura de la llama, el tiempo de residencia de las partículas en la
llama y el crecimiento de las partículas.106
2.4.2.3. Ventajas del rociado pirolítico
Las ventajas del método de rociado pirolítico frente a otros métodos de síntesis son las
siguientes: formación de partículas de tamaño nanométrico, alta pureza, partículas muy
homogéneas con composición estequiométrica, distribución estrecha de tamaño de
partícula, control sencillo de la morfología en función de la naturaleza del precursor y de
las condiciones experimentales, obtención de partículas densas, compactas o huecas,
geometría esférica, facilidad para la síntesis de materiales basados en multicomponentes
de alta homogeneidad composicional, alta velocidad de producción, producción continua
del material y no requiere un sistema de ultra alto vacío.
Este método de síntesis, para la obtención del dióxido de estaño, tiene un efecto
importante sobre el tamaño de partícula, la morfología de la misma y sus propiedades
semiconductoras.

31
3. Justificación
La investigación en el desarrollo de nuevos materiales de electrodo cuya capacidad de
oxidación de contaminantes orgánicos sea equiparable, o mejor, a los ya existentes y,
que además, sean estables y de bajo costo, es un área de la electroquímica aplicada de
gran interés hoy en día.
Los materiales de electrodo en este sentido deberán oxidar al compuesto a un grado tal
que los productos de oxidación sean biodegradables (ácidos alifáticos); o bien, oxidar al
compuesto hasta su eliminación completa mediante conversión a dióxido de carbono y
agua. Dentro de estos materiales, los óxidos metálicos de PbO2 y SnO2, son ampliamente
utilizados y los esfuerzos por mejorar sus propiedades físicas y catalíticas, son objeto de
numerosas publicaciones en las que el dopaje, o las modificaciones al sustrato,
generalmente titanio, son una vía para el logro de estos objetivos. En general, se apuesta
por un material cuyo desempeño iguale al del material preferido por antonomasia: el
diamante dopado con boro (BDD), material cuya eficiencia se ha explicado en términos
de: su alto sobrepotencial de oxígeno, capacidad de producción de radicales hidroxilo,
su alta hidrofobicidad y gran estabilidad. El desarrollo de un material con tales
características, pero de menor costo, es aún uno de los mayores retos en la actualidad.
En décadas recientes, el dióxido de titanio ha sido de especial interés en la
descontaminación del agua debido a que, sus propiedades fotocatalíticas encuentran
aplicación en la desinfección de agua y en la degradación de contaminantes orgánicos
acuosos. El dióxido de titanio es un semiconductor tipo n, estable en diversos medios,
de bajo costo y no tóxico. Parte del enfoque en la investigación actual sobre este material
se basa en la formación de nanoestructuras de TiO2 que representen una mejora en las
propiedades inherentes al material.
En los últimos años, los nanotubos de TiO2 han demostrado ser ventajosos en la
preparación de materiales de electrodo para la fotoelectrocatálisis o la electrocatálisis.
En la fotoelectrocatálisis, los nanotubos de dióxido de titanio han demostrado tener un
desempeño sobresaliente en la oxidación de compuestos biorrefractarios como el
pentaclorofenol, 4-clorofenol y algunos pigmentos tipo azo. En este sentido, la formación
de composites de nanotubos de TiO2 y otros óxidos como ZnFe2O4, Cu2O ó Bi2O3 permite
a estos materiales la absorción de luz visible debido a los anchos de banda prohibida

32
más pequeños de estos óxidos con respecto al ancho de banda de TiO2 (3.2 eV). Otra
modificación a los nanotubos para lograr la absorción de luz en el intervalo de radiación
visible es mediante el dopaje con elementos como N, C, Si, y metales de transición.
Otra tendencia en el uso de nanotubos de TiO2 como materiales de electrodo es en su
uso como soporte de materiales electrocatalíticos como nanopartículas de Ag, Au, Pd y
Pt, así como SnO2 y PbO2, los cuales son dispersados sobre el arreglo nanotubular
incrementando su área superficial y, por ende, su actividad catalítica. En el caso de los
óxidos metálicos de estaño y plomo, se han preparado electrodos en los que estos
materiales recubren completamente al arreglo de nanotubos, siendo la actividad
electrocatalítica de estos materiales la que se ha explotado con resultados prometedores.
Deben resaltarse algunas características de los nanotubos de dióxido de titanio que los
hacen aptos para el desarrollo de materiales de electrodo en la oxidación electro y
fotoelectrocatalítica: su gran área superficial, las propiedades fotoanódicas del TiO2, la
mayor movilidad electrónica a través de sus paredes, la relativa facilidad de preparación
y su costo accesible.
Por lo anterior, este proyecto de investigación se ha planteado la posibilidad de preparar
materiales de electrodo duales a base de nanotubos de TiO2 combinados con los óxidos
metálicos de SnO2 ó PbO2. En estos electrodos duales se busca que el material
fotocatalítico (TiO2) se halle expuesto a la superficie al igual que el material
electrocatalítico (SnO2 o PbO2); de manera que, ambos materiales puedan actuar
combinando sus propiedades para tener un efecto sinérgico en la actividad
electrocatalítica y/o fotoelectrocatalítica del material dual.
4. Hipótesis
Se puede obtener electrodos de composición dual basados en nanotubos de dióxido de
titanio (Nts-TiO2) y óxidos metálicos (PbO2 ó SnO2) con un efecto de sinergia en sus
propiedades electrocatalíticas y fotoelectrocatalíticas.

33
5. Objetivo General
Depositar partículas de MO2 (M = Pb, Sn) sobre una interfase reticulada de nanotubos
de TiO2 (Nts-TiO2); de manera que, el material fotocatalítico (TiO2) se halle expuesto a la
superficie al igual que el material electrocatalítico (SnO2 o PbO2). y fortalecer así, las
propiedades electrocatalíticas y/o fotoelectrocatalíticas del material dual (Nts-TiO2/SnO2
o Nts-TiO2/PbO2).
6. Objetivos Específicos
1. Establecer un procedimiento electroquímico para producir interfases de
nanotubos de TiO2 sobre sustratos de Ti.
2. Establecer condiciones químicas y/o electroquímicas para el depósito de MO2 (M
= Pb, Sn) sobre la estructura de nanotubos de TiO2.
3. Determinar la composición química y las características morfológicas y
estructurales de los electrodos de Nts-TiO2/MO2, y su estabilidad.
4. Determinar las propiedades electro y fotoelectrocatalíticas de los electrodos de
Nts-TiO2/MO2
5. Proponer una ruta de oxidación del compuesto orgánico modelo empleado en la
catálisis sobre estos electrodos.

34
CAPÍTULO II: PREPARACIÓN Y CARACTERIZACIÓN DE NANOTUBOS DE
DIÓXIDO DE TITANIO
1. Resumen
En la preparación de nanotubos de TiO2 se llevaron a cabo diversas metodologías. En
una etapa inicial se siguieron métodos de preparación reportados en la literatura con el
fin de obtener el crecimiento efectivo de los nanotubos (anodizaciones preliminares). De
estos primeros experimentos se observó que la morfología y rugosidad de la placa de
titanio influye fuertemente en el crecimiento de nanotubos y en la aparición de un
precipitado (PP) indeseado sobre los nanotubos así formados. De igual manera, la
composición de la solución electrolítica en cuanto al porcentaje de agua es un factor a
considerar en la aparición de dicho precipitado. En esta primera etapa se obtuvo la
composición óptima de la solución de anodizado, el voltaje de anodización y la geometría
de la celda necesarios para el crecimiento de la capa de nanotubos libres de precipitado.
Estas condiciones constituyen la denominada metodología A, la cual sufrió
modificaciones debido al uso de placas diferentes de titanio, las cuales poseían
morfologías y rugosidades iniciales diferentes, por lo que, se implementaron algunos
pretratamientos tanto físicos como químicos para lograr una superficie de titanio
favorable al crecimiento. A lo largo de este capítulo, se discute el efecto del
pretratamiento del sustrato en la morfología del arreglo nanotubular resultante del
anodizado, así como algunos aspectos inherentes al titanio como sustrato de nanotubos
de TiO2 que ya se han estudiado por otros investigadores. Esta discusión se origina en
el hecho de que en el transcurso de esta investigación se observaron diferencias
significativas en el desempeño de los nanotubos debidas al cambio de placa de titanio.
Las diferencias en el desempeño de los nanotubos se describen en los capítulos 3 y 5.
En este capítulo, también se incluye la caracterización fisicoquímica de los nanotubos
(Nts), la cual se llevó a cabo mediante microscopía electrónica de barrido (MEB),
espectroscopía de dispersión de energía de rayos X (EDS), difracción de rayos X (XRD),
voltametría cíclica (VC) y espectroscopía UV-vis de reflectancia difusa (ERD).

35
2. Anodizaciones preliminares
Como se mencionó con anterioridad, en el inicio de esta investigación se realizaron
diversos experimentos basados en lo reportado en la literatura, de los cuales se obtuvo
una metodología única; la cual, debido a que se fueron utilizando placas nuevas de
titanio, provenientes de diferentes lotes de fábrica, se modificó con respecto al
tratamiento inicial de la superficie de titanio. Las placas de titanio utilizadas fueron, en
general, compradas de Aldrich con número CAS: 7440-32-6. El espesor fue de 0.127 mm
en la mayoría de los casos y la pureza de 99.7%.
Las primeras placas de titanio se trataron por diferentes metodologías sugeridas por la
literatura, entre ellas, a) lijado con lija de 600 seguido de calentamiento por 10 minutos
con HCl al 18%(v/v), b) Lijado con lija de 600, c) Sin lijado. A cualquiera de estos
tratamientos le siguió una limpieza por sonicación en uno o más de los siguientes
solventes como acetona, etanol y metanol.
Aunado al tratamiento de la placa se probaron diferentes medios electrolíticos y voltajes
de anodizado hasta encontrar las condiciones que darían como resultado el crecimiento
eficaz de los nanotubos de TiO2 (NTs-TiO2). Las condiciones específicas de estos
experimentos se resumen en la Tabla 1, se incluye en la tabla los resultados de los
tratamientos de la placa y de las diferentes condiciones de anodización. La tabla nos
dirige también hacia las micrografías MEB que ilustran los nanotubos obtenidos,
obviando aquellos experimentos en los que no se obtuvieron nanotubos.
Tabla II.1. Resumen de las primeras condiciones de anodización.
No. de
reacción
Solución de
anodizado
Pretratamiento de la
placa de titanio
Condiciones de
reacción
Observaciones
1,2 50 ml de
solución NH4F
0.8%
Na2SO4 1.6%
PEG 400 10%
Placa de Ti. Lijada, sonicada
en acetona por 10 minutos y
calentada en HCl al 18% por
10 minutos
Contraelectrodo (CA)
de platino en barra o
placa. Voltaje aplicado
(VA) de 15 V. Tiempo
de anodizado (TA) 3 y 7
h, respectivamente.
La separación entre el
ánodo y el cátodo es
de 1.3 cm y de 1 cm
para la reacción 2.
NO HAY
NANOTUBOS
3,4 Mismas
condiciones
anteriores
Mismas condiciones
anteriores
CA=Barra de Platino. 3
h de reacción a 20 y 30
V respectivamente.
NO HAY
NANOTUBOS
36
5 50 ml de
solución
Glicerina:Agua
(2:1), Na2SO4
0.2 M y
NaF0.5% (p/v)
Mismas condiciones
anteriores
CA=Barra de Platino. 3
h de reacción a 30 V.
NO HAY
NANOTUBOS
6 10 ml de
solución NH4F
0.8%
Na2SO4 1.6%
PEG 400 10%
Pretratada con calentamiento
en HCl por 10 minutos
CA=Placa de Pt , 3 h de
reacción a 30 V.
NO HAY
NANOTUBOS
7,8 10 ml de
solución
Glicerina:Agua
(2:1), Na2SO4
0.2 M y NaF
0.5% (p/v)
Placa de Ti, 1 X 1.5 cm2,
espesor de 0.25 mm.
Pretratada con lijado y
sonicación por 10 min con
etanol y por 10 minutos con
acetona, sin calentamiento
con HCl
CA=Barra de Pt, VA=30
V a 3 h y 6 h de
reacción
respectivamente.
SI HAY
NANOTUBOS, pero
se observan
recubiertos por un
precipitado (PP) en
gran parte de la
superficie y también
se han fracturado en
el caso del No. 8, ver
Fig. 1.
9 Mismas
condiciones
anteriores
Pretratada con sonicación
por 10 min en etanol y por 10
minutos en acetona, sin lijado
y sin calentamiento con HCl
CA=Barra de Pt, VA=30
V a 3 h y 6 h de
reacción
respectivamente.
SI HAY
NANOTUBOS. Se
observa menos
superficie recubierta
por el PP. Ver Fig.2
10 Mismas
condiciones
anteriores
Pretratada con sonicación
por 10 min en etanol y por 10
minutos en acetona, con
lijado y sin calentamiento con
HCl
CA=Barra de Pt, VA=30
V a 6 h de reacción.
SI HAY
NANOTUBOS. Se
observa menos
superficie recubierta
por el PP.
11 50 ml de
solución
Glicerina:Agua
(2:1), Na2SO4
0.2 M y
NaF0.5% (p/v).
Pretratada con sonicación
por 10 min con etanol y por
10 minutos con acetona, sin
lijado y sin calentamiento con
HCl
CA=Barra de Grafito
VA=30 V a 3 h de
reacción.
SI HAY
NANOTUBOS. Se
observa menos
superficie recubierta
por la sal que en las
reacciones 9 y 10.

37
12 10 ml de
solución
Glicerina:Agua
(2:1), Na2SO4
0.2 M y NaF
0.5% (p/v).
Mismas condiciones
anteriores
CA=Barra de Pt, VA=30
V a 3 h de reacción.
SI HAY
NANOTUBOS. Se
observa recubierta
por el PP.
13 50 ml de
solución
Glicerina:Agua
(2:1), Na2SO4
0.2 M y
NaF0.5% (p/v).
Celda grande,
agitador de 2
cm de largo.
Pretratada con sonicación
por 10 min con etanol y por
10 minutos con acetona, sin
lijado y sin calentamiento con
HCl
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 4 h
de reacción.
SI HAY
NANOTUBOS. Se
observa recubierta
por el PP. Fig. 3a
14,15 Mismas
condiciones
anteriores
Solución nueva.
Mismas condiciones
anteriores
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 4 h
de reacción.
SI HAY
NANOTUBOS. Ya no
hay recubrimiento con
PP. Se usó glicerol
Merck al 87%. Fig. 3b
16 50 ml de
solución de
Na2SO4 1 M y
0.5%(m/m) NaF
Mismas condiciones
anteriores
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 2 h
de reacción.
NO HAY
NANOTUBOS
17-27 50 ml de
solución
Glicerol:Agua
(1,3:1),Na2SO4
0.2 M y
NaF0.5% (p/v).
Celda grande,
agitador de 2
cm de largo.
Placa de Ti, 1.0 X 1.0 cm2,
espesor de 0.127 mm.
Pretratada con sonicación
por 10 min con etanol y por
10 minutos con acetona, sin
lijado.
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 3 h,
4 h y 6 h de reacción.
SI HAY
NANOTUBOS, pero
hay formación del PP
y en algunos,
fracturas. Los
nanotubos se
observan
desordenados, vista
lateral.Fig. 4.
28-30 Mismas
condiciones
anteriores
Mismas condiciones
anteriores
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 3 h
de reacción.
Se usó glicerina
marca Merck. Se
reusó el mismo medio
en la 29 y en la 30. La
separación entre
electrodos es de 1.3
cm. Fig.5

38
31 Mismo medio
de anodizado
pero con
agitador de 1
cm de largo.
Mismas condiciones
anteriores
CA=Placa de Zr, A=3 x
4 cm2, VA=30 V a 6 h
de reacción.
Se reusó el medio de
la reacción 29 y 30.
La distancia entre
electrodos es 1cm. Se
ven recubiertos por
PP y con fracturas,
Fig. 6
Figura II.1. Nanotubos de las reacciones 7 (izquierda) y 8 (derecha), recubiertos por PP y
fracturados para la reacción 8.

39
Figura II.2. Nanotubos de reacción 9, se observan recubiertos por una menor cantidad de sal
Figura II.3. Nanotubos de la reacción 13 (izquierda), se observa con un poco de PP sobre la
superficie. Nanotubos de la reacción 14 (derecha), se observan libres de PP..

40
Figura II.4. Reacción 17, el depósito de nanotubos se fracturó y se pudo visualizar la longitud de
los mismos (aproximadamente 3000 nm). La reacción duró 4 h.
(a) (b)

41
(c)
Figura II.5. a) Electrodo de la reacción 28 se resquebrajó debajo del vástago. b)Electrodo de la
reacción 29, se resquebrajó de la parte lateral cercana al vástago. c)Electrodo de la reacción 30,
se resquebrajó en más de la mitad de la superficie del electrodo, en la parte cercana al vástago.
Figura II.6. Reacción 31, los nanotubos se fracturaron. La reacción duró 6 horas.
De los resultados anteriores se infieren algunos aspectos importantes a considerar en la
preparación de los nanotubos: a) la rugosidad inicial de la placa juega un papel
determinante en la formación de los nanotubos, b) la composición del medio electrolítico,
sobre todo en lo que se refiere a la proporción de agua afecta el crecimiento de los
nanotubos y la formación del PP se pudo evitar si se maneja la adecuada proporción de
agua, y c) la agitación moderada es conveniente para evitar la formación del PP. Dicho

42
PP, en su análisis de EDS está conformado por una mezcla de elementos provenientes
del TiO2 amorfo y del medio de reacción, incluyendo silicio, por lo que, se determina evitar
el uso de material de vidrio para el anodizado, Figura 7.
Element Weight% Atomic%
C K 4.62 8.71
O K 37.92 53.68
F K 12.60 15.02
Na K 2.46 2.43
Si K 0.34 0.28
Ti K 42.06 19.89
Totals 100.00
Figura II.7. Análisis de EDS del precipitado formado sobre los nanotubos de Ti, del que se puede
apreciar diferentes elementos provenientes del medio de reacción como F, Na y Si.
3. Metodología de preparación de nanotubos: Metodología A
Las observaciones anteriores permitieron obtener la metodología denominada A, la cual
se describe en los siguientes pasos.
1. Se corta una placa de titanio de 1 cm x 1 cm (Aldrich Cat. No. 348791-13G, No.
CAS: 7440-32-6). El espesor de la placa puede ser de 0.125 o 0.27 mm. Se deja
un vástago de 1 a 1.5 cm.
2. Si la placa presenta baja rugosidad se puede dejar sin lijar o, lijar con lija de 3
micras hasta conseguir mayor lisura de la placa, después se talla con papel filtro y
se sonica con acetona, etanol y metanol por 10 minutos en cada solvente.
3. Se anodiza la placa de Ti con una fuente de poder a 30 V bajo agitación moderada
(agitador magnético de 1 cm de largo) en 20-30 ml de un medio de glicerol:agua
(1.3:1) conteniendo Na2SO4 0.2 M y NaF 0.5% (p/v). Se usa como contraelectrodo
un placa de zirconio de 3.0 x 4.0 cm2 separada a una distancia de
aproximadamente 1 cm del electrodo de trabajo (Ti). La anodización dura 4 h.
Como celda de trabajo se utiliza un vaso de polipropileno de 100 ml, marca
Nalgene.

43
4. Una vez anodizada, la placa se lava con agua desionizada y se seca bajo corriente
de nitrógeno.
5. El electrodo así preparado se trata térmicamente en horno tubular bajo atmósfera
ambiental con una rampa de temperatura de 5°C/min hasta llegar a los 500°C; se
mantiene a esta temperatura por 1.5 h y después se enfría con rampa de
temperatura de 5°C/min, de acuerdo a la literatura.
6. La solución de anodizado no debe reusarse, ya que, se observa en todos los casos
que los nanotubos formados se separan fácilmente de la placa de titanio.
4. Resultados y Discusión
Las placas de titanio anodizadas y tratadas térmicamente siguiendo la metodología A,
fueron caracterizadas por microscopía electrónica de barrido (MEB), espectroscopía de
dispersión de energía de rayos X (EDS), difracción de rayos X (XRD), voltametría cíclica
(VC) y espectroscopia UV-vis de reflectancia difusa (ERD). La MEB revela que los
nanotubos formados presentan diamétros internos que varían de 110 a 150 nm y
diámetros externos de 140 a 180 nm, y ancho de pared de 10-30 nm, ver Figura 8. Los
análisis de EDS muestran que el oxígeno y titanio del óxido se hallan en una relación de
54 % oxígeno y 26% titanio, lo que indica la formación de TiO2, Figura 9. Difracción de
rayos X corroboró que, después del tratamiento térmico (annealing), los nanotubos
presentan la fase cristalina anatasa, Figura 10. La VC de los electrodos de Nts-TiO2 en
H2SO4 0.5 M a una ν= 100 mV/s muestra un voltamograma típico para la fase anatasa
de nanotubos de TiO2, con un pico catódico a un potencial de 0.3 V (vs. Hg/Hg2SO4) que
corresponde a la reducción de Ti4+ combinada con la inserción de H+, y un pico anódico
a 0.022 V correspondiente a la reacción inversa,107 Figura 11.
Ti4+O2 + H+ + e- ↔ Ti3+O(OH) Ec. 1

44
Figura II.8. Micrografías de nanotubos producidos por la metodología A.
Element Weight% Atomic%
C K 10.46 20.44
O K 36.50 53.56
Ti K 53.04 25.99
Totals 100.00
Figura II.9. Análisis de EDS de nanotubos producidos por la metodología A.

45
Figura II.10. Espectro de Rayos X de Nts-TiO2 sobre titanio después del tratamiento térmico a
500°C durante 1.5 h, donde se observa la formación de la fase anatasa del TiO2.
Figura II.11. Voltamograma de nanotubos de TiO2 en medio de H2SO4 0.5 M
0
200
400
600
800
1000
1200
20 30 40 50 60 70 80 90 100 110 120
Cu
en
tas
2θ
A: Anatasa
AA
T: Titanio
T
T
T
T
A
-0.0001
-0.00008
-0.00006
-0.00004
-0.00002
0
0.00002
0.00004
0.00006
0.00008
-0.2 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
I(A
)
E(V) vs. Hg/Hg2SO4
v=100mV/s

46
Por su parte, los espectros de reflectancia difusa (ERD) modificados bajo la función de
Kubelka-Munk para obtener la absorbancia de los Nts-TiO2, muestran que los nanotubos
obtenidos presentan una longitud de onda de corte promedio29,108-109 (tomado de al
menos tres muestras) de λ= 380 nm, lo que indica un ancho de banda, Eg=1240 eV/λ,
característico de la fase anatasa del TiO2 de 3.2 eV, Figura 12.
Figura II.12. Espectro de absorción (Kubelka-Munk) de Nts-TiO2. El espectro fue obtenido
mediante la aplicación directa de la herramienta matemática, que proporciona el
espectrofotómetro (Cary 50), sobre el espectro de reflectancia difusa obtenido por el mismo
equipo.
4.1. Rugosidad de la placa como un factor determinante en la formación y
crecimiento de nanotubos
De las anodizaciones preliminares, se tenía conocimiento de que la rugosidad inicial de
la placa era un factor determinante en la formación de nanotubos, como lo demostró el
hecho de que en ningún medio electrolítico se formaron nanotubos cuando la placa de
Ti fue tratada con ácido clorhídrico (18%), lo cual provoca un cambio radical en la
morfología inicial de la superficie, pero la nueva morfología no es favorable a la formación
de nanotubos; ya que, como se puede observar en la figura 13, el tratamiento con ácido
en la placa UNK (Fig.13a) genera una superficie altamente escarpada (Fig. 13b) que al
ser anodizada, sólo favorece el crecimiento del óxido (TiO2) en un patrón repetido de la

47
morfología anterior al anodizado (Fig.13c). De lo anterior fue claro que se debía tener
especial cuidado en el estado superficial de la placa de Ti para obtener, en primera
instancia, los nanotubos.
(a) (b)
(c)
Figura II.13. a) Ti metálico sin ningún tratamiento, placa UNK. b) Ti tratado con HCl (18%), c) Ti
tratado con HCl, anodizado en medio electrolítico de la metodología A (no hay nanotubos).

48
4.1.1. Formación de PP debido a la rugosidad a microescala del Ti metálico.
A lo largo de este proyecto, las placas de Ti que se usaron provinieron de tres lotes
diferentes de titanio. La primera placa (15x15cm2), de la cual se obtuvieron los primeros
nanotubos y se hicieron las primeras pruebas de desempeño de nanotubos, correspondió
a una placa que se tenía con anterioridad en el laboratorio y de la cual se desconoce el
número de lote, y que en lo sucesivo, se denominará UNK. Por otro lado, las dos placas
que le sucedieron correspondieron a los lotes No. MKBD6581V y MKBG1575 de la marca
Aldrich, a estas placas se les denominará MKB. La MEB de estas placas MKB revela que
la rugosidad de éstas es similar para ambas placas, sin embargo, difiere de la placa UNK,
Fig. 14a y 14b. De la figura 14, es importante puntualizar que las placas MKB presentan
un rayado en toda su superficie, mientras que las placas UNK, no presentan tal rayado.
La ausencia de este rayado explica porqué al utilizar la metodología A, sin ningún
pretratamiento del titanio más que la sonicación en diferentes solventes, las placas UNK
producen nanotubos libres de PP, ver figura 8; mientras que las placas MKB producirán
nanotubos altamente recubiertos por PP, estos recubrimientos de PP se producirán en
las zonas más elevadas (crestas) de la superficie del Ti metálico, Figura 14.
(a) (b)

49
(c)
Figura II.14. a) Placa de Ti (UNK) sin pretratamiento, b) Placa de Ti (MKB) sin pretratamiento c)
Nanotubos con PP, provenientes de placa de Ti (MKB) sin pretratamiento.
De lo anterior, se establece que para evitar la formación del PP se realice un lijado que
permita eliminar las zonas más elevadas del Ti metálico, lo cual evita la formación del
PP pero aún favorece el crecimiento de los nanotubos. G.R. Dale y colaboradores,110
observaron que los nanotubos crecen de manera preferencial en los valles o depresiones
de la superficie de titanio. Dale (et al) sugiere que las variaciones en la topología inicial
del titanio derivan en variaciones en las densidades de corriente en regiones específicas
sobre la superficie, lo que conducirá a una distribución variante de la cobertura de
nanotubos. Los mismos autores concluyen que un pulido mecánico es conveniente para
disminuir la rugosidad a microescala y aumentar la rugosidad a nanoescala lo cual
favorece el crecimiento de los tubos.

50
Por su parte, con respecto del pulido mecánico, Lan Zhang y Yong Han,111 señalan que
no existe diferencia, mostrada por los análisis de difracción de rayos X, en los
componentes de las fases del TiO2 al utilizar una placa sin pulir de una placa pulida;
asimismo, sus análisis de MEB no muestran diferencias en la morfología superficial de
los nanotubos formados en ambas placas. La única diferencia observada reside en el
hecho de que al comparar los transitorios de corriente al llevar a cabo el proceso de
anodizado, los autores encuentran que el conocido decremento en corriente asociado a
la formación de la capa de óxido inicial es menor en la placa pulida que en la placa sin
pulir, Figura 15.
Figura II.15. Transitorio de corriente del anodizado de placas de Ti pulido y sin pulir.3

51
El seguimiento de MEB que los autores hacen del crecimiento del nanotubo, demuestra
que esto tiene un efecto en el grosor de la capa de nanotubos, la cual crece más sobre
la placa pulida (25µm) que sobre la placa sin pulir (15 µm).
Figura II.16. Sección transversal de nanotubos crecidos sobre una placa de Ti sin pulir
(izquierda) y una placa de titanio pulida (derecha).111
Del presente trabajo realizado con el anodizado de placas de titanio, se observa que la
rugosidad inicial del Ti se mantiene en la película de nanotubos, es decir, las zonas
elevadas (crestas) y de depresión (valles) de la morfología original del Ti serán zonas
donde habrá nanotubos más largos (crestas) y nanotubos más cortos (valles),
respectivamente. Incluso la orientación del nanotubo podría variar, ya sea si éste ha
crecido en una cresta o en un valle, pues en las zonas elevadas, puede observarse que
los nanotubos se llegan a doblar ligeramente, Figura 17.
(a) (b)

52
(c)
Figura II.17. a) Placa de Ti (MKB) donde se observa un rayado pronunciado del Ti metálico, b)
Nanotubos crecidos sobre placa MKB cuya longitud es mayor en las crestas, c) Doblado de
nanotubos en zonas elevadas de la superficie.
Por otro lado las placas que no presentan este rayado tan pronunciado, como la que se
obtuvo recientemente de la marca American Elements (AME), presentan nanotubos más
uniformes en longitud y más ordenados, Figura 18. El análisis mediante MEB del estado
inicial de las placas de Ti y de su influencia en la formación de nanotubos sustenta el
hecho de que la rugosidad de las mismas afecta el crecimiento de los tubos, por lo que,
su longitud y orientación varía a lo largo de la superficie del electrodo, lo cual podría
afectar el depósito de otros materiales sobre el arreglo de nanotubos.

53
Figura II.18. a) Placa de Titanio AME sin pulir. b) Nanotubos producidos a partir de placa AME.
c) Nanotubos a mayores aumentos.
En este sentido cabe mencionar que incluso el devastado químico con ácido oxálico,112
seguido de un pulido mecánico para eliminar el óxido, trae como consecuencia un
ordenamiento diferente del arreglo de nanotubos, Figura 19.

54
Figura II.19. Nanotubos producidos a partir de placa de titanio tratada con ácido oxálico.
4.1.2. Transitorios de corriente en la formación de nanotubos a partir de placas de
titanio diferentes
En transitorios de corriente registrados durante la anodización de placas de titanio, se
han identificado tres zonas relacionadas con diferentes etapas de la formación de
nanotubos: a) en el estado inicial de la anodización (I), se manifiesta un decremento
abrupto en la corriente, como se presentaría en el caso de anodización libre de fluoruro,
debido a la formación de una capa compacta de óxido, b) en el estado II, se registra un
incremento de corriente debido a la formación de poros a nanoescala que penetran la
capa inicial de óxido (la corriente se incrementa como resultado del incremento en el
área reactiva) y c) en el estado III, la corriente desciende nuevamente al formarse una
capa regular de nanoporos o nanotubos. Después de que ocurre la auto-organización, el
crecimiento del tubo continúa a densidades de corriente constantes, 113-114 Figura 20.

55
Figura II.20. Izquierda: Transitorio de corriente que muestra las primeras etapas de formación
de nanotubos. Derecha: Imágenes de MEB de cortes transversales de dichas etapas iniciales.
113
Con el fin de corroborar la posible influencia del estado superficial inicial de la placa de
titanio en la topología final de los nanotubos observada por MEB, se obtuvieron
transitorios de corriente en tres tipos de placas: a) Placa AME: Placa de American
Elements sin lijar; b) Placa MKBL: Placa MKB lijada y; c) Placa MKBSL: Placa MKB sin
lijar. En estas anodizaciones la placa de titanio se anodizó de acuerdo a la metodología
A, con la diferencia de que en estos experimentos se excluyó la agitación. De la figura
21, se puede apreciar que en los tres casos los transitorios de corriente varían entre sí,
lo que indica que el mecanismo de formación de nanotubos en sus tres etapas es
diferente. Por un lado, el grosor de la capa inicial del óxido podría ser diferente en los
tres casos debido a las diferencias en el descenso inicial de la corriente (zona I), siendo
las placas AME y MKBL las que sufren el mayor decremento, lo que indica un mayor
grosor de la capa inicial del óxido; sin embargo, estas placas difieren en que el pequeño
incremento en corriente (zona II), asociado al primer devastado químico por parte del ión
F- es casi imperceptible para las placas AME y un poco más notorio para las placas
MKBL. Por su parte, las placas MKBSL, manifiestan un menor decremento en la corriente
inicial pero el incremento en corriente en la zona II es más pronunciado que en las placas
AME y MKBL.

56
Figura II.21. Transitorios de corriente en el anodizado de tres placas de titanio con rugosidad
inicial diferente.
5. Conclusiones
Se logró desarrollar una metodología reproducible (metodología A) para la formación de
nanotubos de TiO2 por anodización electroquímica de una placa de titanio. La
metodología involucra tres pasos importantes: 1) Pretratamiento y/o limpieza de la placa
de Ti, 2) Anodización a 30 V en celda de dos electrodos, 3) Tratamiento térmico a 500°C
para pasar a la fase anatasa. En la preparación de todas las placas utilizadas en esta
investigación, la metodología no sufrió modificaciones en los pasos 2 y 3, pero sí en el
paso 1, debido a la compra de nuevas placas de titanio con rugosidad inicial diferente.
El lijado de las placas se realizó con la finalidad principal de evitar la formación del PP.
De la MEB, no es posible establecer diferencias significativas en la topología final de los
nanotubos formados a partir de la placa lijada y de la placa sin lijar, lo cual concuerda
con lo reportado por Zhang y Han. Es observable que, el lijado disminuye la altura de las
crestas en el titanio metálico, pero preserva la topología base del mismo, la cual se
reproduce en el arreglo de nanotubos. Lo que se observa es que la topología de la
película de nanotubos entre las placas del mismo origen (MKB), con y sin lijado, es
1
10
100
1000
0 200 400 600 800 1000 1200 1400 1600
I, u
A
t(s)
Placa MKBL
Placa AME
Placa MKBSL
Zona I
Zona II
Zona III

57
similar, sin embargo, el grosor de la película podría ser diferente de acuerdo al
comportamiento diferenciado de los transitorios de corriente. Asimismo, la placa de
diferente origen (AME) muestra una película de nanotubos con un arreglo de los mismos
más uniforme, y un transitorio de corriente también diferente, lo cual confirma el supuesto
de que las diferencias entre placas de titanio traen diferencias en el desarrollo de los
nanotubos y el resultado final no debe ignorarse, ya que, podría tener consecuencias
importantes en las propiedades de los mismos, como se discutirá en el capítulo 5 con
respecto a los electrodos denominados de segunda generación que se describen en él.
Es importante tomar en cuenta que, aunque la composición de las placas sea titanio de
alta pureza, las variaciones en el proceso de deformado para su obtención así como la
disposición de los planos cristalinos del metal afecta el crecimiento de los nanotubos. Al
respecto Silvia Leonardi (et al), 115 advierte que existe una interdependencia entre la
orientación de los granos del sustrato y el crecimiento de los nanotubos de TiO2. Leonardi
incluso identifica algunas orientaciones que son favorables o desvaforables al
crecimiento, como por ejemplo, la orientación Ti (111) es un sitio preferencial de
formación de nanotubos, mientras que Ti (010) sólo genera TiO2 como una película de
óxido compacta.
Dado que estas condiciones iniciales del sustrato producen morfologías diferentes de
los nanotubos, el desempeño de los mismos puede modificarse. Al respecto, A. G.
Kontos y colaboradores,116 mostraron que las diferencias morfológicas tanto externas:
forma y tamaño de las bocas, espacio entre nanotubos, como internas: longitud y
rugosidad de las paredes (ondas de las paredes), influyen en las propiedades
fotocatalíticas y de superhidrofilicidad. En su estudio, Kontos encuentra que los
nanotubos con longitud de 10 µm, cuya estructura es más regular y más compacta que
la de los nanotubos de longitudes menores a 1 µm, presentan una mayor actividad
fotocatalítica, así como una mayor hidrofilicidad.

58
CAPÍTULO III: ELECTRODEPOSICIÓN DE DIÓXIDO DE PLOMO SOBRE
NANOTUBOS DE DIÓXIDO DE TITANIO
1. Resumen
Se prepararon electrodos de nanotubos de dióxido de titanio con depósitos de dióxido de
plomo (Ti/Nts-TiO2::PbO2). Con el objetivo de tener un material dual, el PbO2 se
electrodepositó a partir de una solución de Pb(NO3)2, durante 5, 10 y 15 segundos, bajo
el régimen de corriente controlada con el fin de definir las condiciones que lograran una
adecuada dispersión del material electrocatalítico (PbO2) pero que dejaran al descubierto
al material fotocatalítico (TiO2). Los electrodos de Ti/Nts-TiO2::PbO2, se caracterizaron
mediante MEB, EDS, DRX, VC y ERD. La VC basada en la metodología de Trasatti
permitió la obtención del factor de rugosidad de los electrodos de Ti/Nts-TiO2 y de Ti/Nts-
TiO2::PbO2. Se observó que el electrodo de nanotubos de dióxido de titanio con dióxido
de plomo depositado durante cinco segundos (Ti/Nts-TiO2::PbO2 5s) tiene el mayor factor
de rugosidad, así como también la mayor eficiencia de fotoconversión obtenida a partir
de las curvas I-V de los materiales bajo luz de onda larga de 365 nm, resultados que se
discuten en el capítulo 5.
2. Electrodepósitos preliminares
Los primeros electrodepósitos se hicieron a partir de la solución acuosa de Pb(NO3)2, a
un potencial de 1.826 V, a temperatura ambiente y durante 35 minutos; en un sistema de
tres electrodos, donde el ánodo es la placa de Ti/Nts-TiO2, el cátodo es una barra de
grafito y se usó un electrodo de sulfato mercuroso como referencia. A los 5 minutos de
iniciada la reacción se observó un depósito negro uniforme correspondiente a PbO2 que
creció conforme avanzó la reacción hasta que ésta se detuvo a los 35 minutos. La
morfología de la superficie de este óxido se observa altamente cristalina (ver figura 1a),
con la forma tetragonal característica de la fase beta de PbO2. Esta reacción se replicó
y sólo se observaron diferencias en el tamaño de grano del depósito (figura 1b). Estos
depósitos se compararon con el depósito tradicional de PbO2 sobre Pb y las micrografías
muestran una mayor cristalinidad en el caso de los depósitos de PbO2 sobre nanotubos
de TiO2 (ver figura 1c).

59
Figura III.1. a) Depósito masivo de PbO2 sobre Ti/Nts-TiO2, b) Otro depósito masivo de PbO2,
donde se observa la reproducibilidad del depósito cristalino, c) Depósito de PbO2 sobre Pb.
De lo anterior se observó que debían cuidarse los tiempos de depósito y que el PbO2 se
electrodepositaba en los primeros segundos de reacción. Así también, se decidió probar
otras concentraciones iniciales de Pb(NO3)2 en HNO3 0.1M y utilizar densidad de
corriente de 50 mA/cm2. Se encontró que la concentración de 0.25 M era adecuada para

60
el depósito de PbO2 a tiempos cortos de reacción, por lo que, a esta concentración se
hicieron los depósitos posteriores de PbO2 a tres diferentes tiempos: 5, 10 y 15 s.
3. Metodología de electrodeposición de PbO2
De los experimentos preliminares se estableció la siguiente metodología de
electrodeposición. Se monta un sistema de tres electrodos donde el electrodo de trabajo
es la placa de Ti/Nts-TiO2 de 1 cm2 de área (un lado ha sido recubierto con silicón y cinta
Teflón) ó 2 cm2 (ningún lado sin recubrir), el electrodo auxiliar es una placa de zirconio
de 2 x 2 cm2, y el electrodo de referencia es de sulfato mercuroso. La solución electrolítica
es una solución de Pb(NO3)2 0.25 M en HNO3 0.1M y la densidad de corriente utilizada
es 50 mA/cm2. Durante la electrodeposición, el sistema se mantiene en agitación
moderada. Los tiempos de depósito utilizados fueron 5, 10 y 15 s.
4. Resultados y Discusión
La morfología de los electrodos así preparados se caracterizó por MEB. De las
micrografías de los electrodos a los diferentes tiempos de reacción se puede observar
un mayor grado de recubrimiento por parte de PbO2, el cual se observa como un depósito
brillante, conforme se incrementa el tiempo de reacción, Figura 2a-c. Las imágenes
aumentadas de estos depósitos permiten distinguir diferentes grados de crecimiento del
dióxido de plomo sobre las bocas de los tubos. Los crecimientos incipientes se observan
como puntos brillantes mientras que los crecimientos de mayor tamaño presentan la
forma de coliflores (Figura 2d). El tamaño y cantidad de dichas coliflores aumenta
conforme aumenta el tiempo de depósito.

61
(a) (b)
(c) (d)
Figura III.2.a) Imagen de MEB del electrodo de Ti/Nts-TiO2::PbO2 a los 5 s de depósito, b)
Imagen de MEB del electrodo de Ti/Nts-TiO2::PbO2 a los 10 s de depósito, c) Imagen de MEB
del electrodo de Ti/Nts-TiO2::PbO2 a los 15 s de depósito, d) Imagen aumentada de los
depósitos incipientes de PbO2.
Los análisis de EDS de estos electrodos mostraron que PbO2 se encuentra dispersado
en toda la superficie exterior de nanotubos y su concentración aumenta, en la zona de
las coliflores, Figura 3.

62
(a)
(b) (c)
Figura III.3. Imagen aumentada de MEB de depósitos de PbO2 en forma de coliflores, b) Análisis
de EDS de la zona de nanotubos, c) Análisis de EDS de la zona de PbO2 (coliflores).
Asimismo, se obtuvieron micrografías del fondo de los nanotubos (de electrodos que se
fracturaron accidentalmente) con depósito de PbO2 que, demuestran que el PbO2 crece
desde el fondo de los nanotubos, llenándolos hasta que eventualmente emerge de las
bocas de los mismos, formando los depósitos de PbO2 en forma de coliflor. Los análisis
de EDS del fondo de los tubos, demuestran que PbO2 se encuentra en el fondo de los
mismos, Figura 4.
EDS DE LA ZONA DE NANOTUBOS
Element Weight% Atomic%
O K 26.07 67.93
Ti K 25.69 22.36
Pb L 48.25 9.71
Totals 100.00
EDS DE LA ZONA DE PbO2 (coliflores)
Element Weight% Atomic%
O K 18.52 69.41
Ti K 7.28 9.11
Pb L 74.20 21.48
Totals 100.00

63
(a) (b)
(c)
Element Weight% Atomic%
O K 41.78 76.54
Ti K 32.36 19.80
Pb M 25.86 3.66
Totals 100.00

64
(d) (e)
Figura III.4.a) Imagen de MEB del fondo de los nanotubos, llenos de PbO2, se observan como
puntos blancos; b) Análisis elemental por EDS del fondo de los nanotubos, c) Imagen ampliada
del fondo de los nanotubos, d) Punto blanco del fondo de los nanotubos analizado por EDS, e)
Espectro de EDS del punto blanco.

65
Asimismo, los análisis de DRX, muestran que estos electrodos se hallan compuestos de
TiO2 en su fase anatasa, y PbO2 en su fase beta, Figura 5.
Figura III.5. Espectro de DRX del depósito de PbO2 sobre nanotubos de TiO2 que muestra que
β-PbO2 es su estructura cristalina predominante.
Por su parte, los espectros UV- vis de reflectancia difusa, modificados a través de la
función de Kubelka-Munk, de los electrodos de Ti/Nts-TiO2::PbO2 5s, mostraron un ligero
desplazamiento hipsocrómico de la longitud de onda de corte; la cual, en promedio (de
al menos tres muestras), es de 370 nm, lo cual da un ancho de banda de 3.35 eV, Figura
6.
0
20
40
60
80
100
120
140
160
180
200
20 30 40 50 60 70 80
Cu
en
tas
2θ
A: Anatasa
A
A
A
T: Titanio
T
T
T
P: β-PbO2
P
T
T
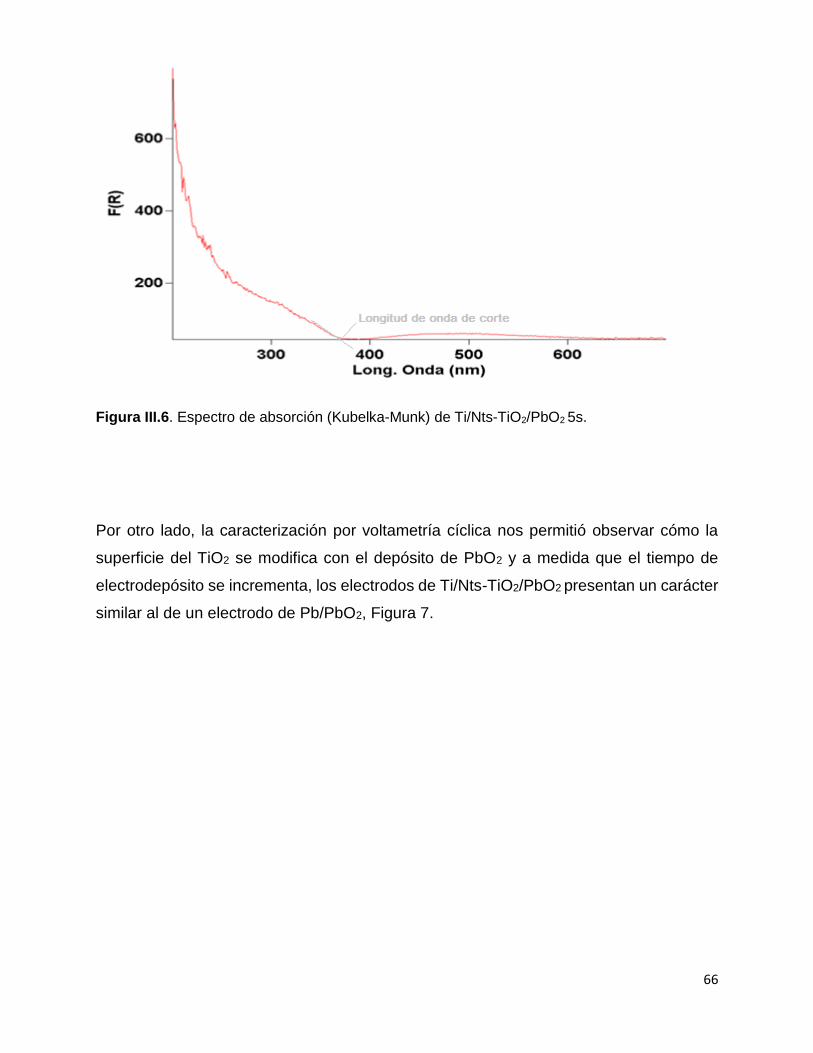
66
Figura III.6. Espectro de absorción (Kubelka-Munk) de Ti/Nts-TiO2/PbO2 5s.
Por otro lado, la caracterización por voltametría cíclica nos permitió observar cómo la
superficie del TiO2 se modifica con el depósito de PbO2 y a medida que el tiempo de
electrodepósito se incrementa, los electrodos de Ti/Nts-TiO2/PbO2 presentan un carácter
similar al de un electrodo de Pb/PbO2, Figura 7.

67
Figura III.7. a) Voltamograma de Ti/Nts-TiO2, b) Voltamograma de Ti/Nts-TiO2::PbO2 5 s, c)
Voltamograma Ti/TiO2-Nts/PbO2 15 s, y d) Voltamograma Pb/PbO2. Todos los voltamogramas
se obtuvieron en 0.5 M H2SO4, a una velocidad de barrido de 100mV/s.
Asimismo, los voltamogramas lineales de estos electrodos en medio de H2SO4 0.5 M
muestran un ligero aumento de los sobrepotenciales de oxígeno conforme disminuye el
tiempo de depósito. Figura 8.

68
Figura III.8. Voltamogramas lineales de Ti/Nts-TiO2 y Ti/Nts-TiO2::PbO2 en H2SO4 0.5 M en
oscuridad, vel. barrido de 100 mV/s. El intercepto en el eje X (línea gris claro) es el sobrepotencial
de evolución de oxígeno para cada electrodo: 1.21 V (15 s), 1.28 V (10 s) y 1.32 V (5 s). Esta
forma de obtener el sobrepotencial de oxígeno, se infirió en base al artículo de José L. Nava (et
al).117
4.1. Medida del área superficial real de los electrodos de Nts- TiO2 y Ti/Nts-
TiO2::PbO2
De algunas pruebas preliminares hechas con ácido cloranílico con los electrodos de
Ti/Nts-TiO2::PbO2, se observa que la decoloración de ácido cloranílico es más rápida en
los electrodos de Ti/Nts-TiO2::PbO2 5s. Lo que sugiere una mayor área superficial en
este electrodo. De ahí que, con la finalidad de mostrar que el dióxido de plomo
incrementa su área superficial electrocatalítica inversamente con el tiempo de depósito,
se realizaron estudios de voltametría cíclica en los electrodos de Ti/Nts-TiO2 y Ti/Nts-
TiO2::PbO2, basados en la metodología sugerida por Trasatti.118 En base a esta
metodología, se obtuvo la capacitancia de la doble capa eléctrica de los electrodos
mencionados en un intervalo muy pequeño de potencial (50-100mV) a diferentes

69
velocidades de barrido. La gráfica de la corriente (medida en el medio del intervalo de
potencial) contra la velocidad de barrido da una línea recta. La pendiente de esta línea
recta corresponde a la capacitancia diferencial de la interface:
C= dQ/dE= Idt/dE= I/(dE/dt) Ec. 1
Para obtener el área superficial, esta capacitancia debe compararse con un valor de
referencia, C*= 60 μFcm-2, el cual se considera el área superficial real unitaria de un
óxido, independientemente de su naturaleza.
A= C/C* Ec. 2.
De acuerdo a esta metodología, se obtuvieron las capacitancias de la doble capa para
los diferentes electrodos, Figura 9.
Figura III.9. Gráfica de corriente(I) vs. Velocidad de barrido para la obtención de la capacitancia. Inserto: Voltamogramas cíclicos de Nts-TiO2 a diferentes velocidades de barrido (10, 20, 40, 60, 80, 100, 150 y 200 mV/s)
y = 1E-05x + 4E-07R² = 0.9946
0.00E+00
5.00E-07
1.00E-06
1.50E-06
2.00E-06
2.50E-06
3.00E-06
0 0.05 0.1 0.15 0.2 0.25
I, A
Velocidad de barrido, V/s

70
De estos voltamogramas cíclicos, se obtuvo la capacitancia y el factor de rugosidad para
cada electrodo (fr= Área real/Área geométrica), considerando para el cálculo de éste, que
el área geométrica de los electrodos fue de 1 cm2, Tabla 1.
Tabla III.1. Área real y factor de rugosidad de los electrodos de Nts-TiO2 y Ti/Nts-
TiO2::PbO2
Tiempo de depósito de
PbO2 sobre Ti/Nts-TiO2
Área real, cm2 Factor de rugosidad
0 s 0.167 0.167
5 s 23.333 23.333
10 s 1.333 1.333
15 s 0.333 0.333
Se definió una razón de electroactividad como RE=CPbO2/CTiO2. La Tabla 2 muestra las
REs para los electrodos de Nts-TiO2 y Ti/Nts-TiO2::PbO2. Se puede observar de la Tabla
2 que la RE para el depósito de 5 s es más de 15 veces mayor que la RE del depósito
de 10 s.
Tabla III.2. Razones de electroactividad (REs) para los electrodos de Nts- TiO2 y Ti/Nts-
TiO2::PbO2
Tiempo de depósito de PbO2 sobre Ti/Nts-TiO2 Razón de electroactividad, RE
5 140
10 8
15 2
Lo anterior corrobora lo observado en la MEB con respecto al hecho de que PbO2
comience su crecimiento desde el fondo de los nanotubos y continúe creciendo sobre la
superficie de los nanotubos, cubriendo sus paredes y conforme crece, eventualmente
llene los tubos y salga de las bocas de los mismos para formar las coliflores, Figura 10.

71
a b c
Figura III.10. Representación esquemática del crecimiento de PbO2 dentro de los nanotubos a:
a) 5 s, b) 10 s y c) 15 s.
4.2. Estabilidad de los electrodos de Ti/Nts-TiO2::PbO2 5s
Como se sabe, la evolución del potencial de celda con respecto al tiempo, en un sistema
electroquímico en el que se controla la corriente, da cuenta de la estabilidad del electrodo
de trabajo y, por lo tanto, de su tiempo de servicio. Por lo que, se hicieron mediciones de
polarización anódica del electrodo de Ti/Nts-TiO2::PbO2 5s, en H2SO4 0.1 M, a densidad
de corriente de 100 mA/cm2 durante 29 horas, Figura 11. Por su parte, la superficie de
este electrodo, antes y después de polarización anódica presenta ligeros cambios en
cuanto a la morfología del depósito de PbO2 y el sustrato de nanotubos conserva su
integridad, Figura 12.

72
Figura III.11. a) Evolución del potencial bajo condiciones de Polarización Anódica en Nts-
TiO2::PbO2 (5s).
a b
Figura III.12. Imágenes de MEB de electrodos de Ti/Nts-TiO2::PbO2 5s a) antes y b) después
de polarización anódica
0
0.5
1
1.5
2
2.5
3
0 5 10 15 20 25 30 35
Po
ten
cial
(V
)
t (h)

73
5. Conclusiones
Se prepararon electrodos de composición dual formados por depósitos de dióxido de
plomo (PbO2) dentro de nanotubos de dióxido de titanio. El PbO2 se depositó vía
electroquímica en condiciones galvanostáticas bajo un control manual del tiempo de
depósito del dióxido de plomo (5, 10 o 15s). Se observó que a menores tiempos de
deposición se logra la mayor dispersión de PbO2 sobre los nanotubos de TiO2; los cuales
proveen un sustrato con una gran área superficial. Lo anterior se manifiesta en la mayor
área superficial expuesta para el electrodo con el menor tiempo de depósito; lo que
concuerda con el mecanismo de crecimiento del PbO2 sobre los nanotubos, en el cual,
bajo las condiciones de densidad de corriente utilizadas, se propone que el crecimiento
de PbO2 inicie en el fondo de los nanotubos y de ahí se extienda hacia sus paredes hasta
que los llene y eventualmente, emerja de los mismos.
La caracterización morfológica por SEM muestra que estos electrodos se hallan
compuestos por un arreglo nanotubular de TiO2 en fase anatasa y PbO2 en fase beta
altamente dispersado el cual se desarrolla dentro de los nanotubos y conforme avanza
el tiempo de depósito forma estructuras con forma de coliflores; las cuales crecen en
tamaño y número al incrementarse el tiempo de depósito. La voltametría cíclica en medio
ácido muestra que al mayor tiempo de depósito de PbO2 los electrodos tienen un
comportamiento similar al de un electrodo de Pb/PbO2.
De los estudios de espectrofotometría de reflectancia difusa se calculó un ancho de
banda óptico de 3.35 eV, el cual representa un ligero desplazamiento hipsocrómico con
respecto al espectro de absorción (Kubelka-Munk) de nanotubos de TiO2. Por otra parte,
estos electrodos manifiestan ser estables bajo condiciones de polarización anódica, ya
que el potencial aplicado permanece constante durante el experimento y la MEB muestra
ligeros cambios en el depósito de PbO2; mientras que el arreglo de nanotubos conserva
su integridad.

74
CAPÍTULO IV: DEPOSICIÓN DE DIÓXIDO DE ESTAÑO SOBRE
NANOTUBOS DE TiO2
1. Resumen
Se prepararon electrodos de dióxido de estaño sobre los nanotubos de dióxido de titanio,
a través de la deposición de dióxido de estaño sobre la superficie de nanotubos de
dióxido de estaño por un método de rociado pirolítico (spray-pyrolisis) artesanal. El
método involucró el uso de una solución precursora de cloruro de estaño a la cual se le
añadió trióxido de antimonio con el fin de dopar el SnO2, sin embargo la caracterización
fisicoquímica no mostró evidencia de que se haya dopado al óxido de estaño, ya que no
se detectó antimonio por EDS. El rociado de esta solución se hizo a través de un
aerógrafo cuyo orificio, de diámetro ajustable, permitió el rociado de finas gotas de la
solución precursora sobre las placas de nanotubos de TiO2 (Ti/Nts-TiO2) que habían sido
calentadas a 600°C durante 10 minutos. El rociado se realizó hasta cinco veces con el
fin de aumentar el grosor del depósito de SnO2, entre intervalos de cinco minutos. Las
variaciones en la concentración de SnCl2 de la solución precursora y en el número de
rociadas condujeron a diferencias apreciables en la morfología del depósito.
2. Metodología de deposición de SnO2
La metodología de preparación de los electrodos de nanotubos de dióxido de titanio con
depósitos de SnO2 (Ti/Nts-TiO2/SnO2) se llevó a cabo por el método de rociado pirolítico
artesanal, en el que la solución precursora del dióxido de estaño se roció sobre el sustrato
de nanotubos de dióxido de titanio a alta temperatura (calentado previamente a 600°C)
y se sometió a un proceso de calcinación a 550-600°C para formar el dióxido de estaño.
Los detalles de la metodología se describen a continuación.
2.1 Detalles del sistema de rociado pirolítico (spray-pyrolysis)
El esquema experimental establecido del sistema de rociado pirolítico se construyó en el
laboratorio. En la Figura 1 se observa este sistema, el cual consta de un aerógrafo que
hace la función de la pistola rociadora del sistema (Figura 1a); el aerógrafo cuenta con
una boquilla de rociado que se puede manipular para ajustar el diámetro de salida de la
solución. La solución está contenida en un frasco de vidrio, Figura 1a. En la Figura 1b,

75
se observa el sustrato caliente (placa de Nts-TiO2). El sistema construido también consta
de una unidad de control para la temperatura (horno), Figura 1c, y un compresor de aire
con un regulador de presión y una fuente de alimentación, Figura 1d.
(a) (b)
(c) (d)
Figura IV.1. Componentes del sistema de rociado pirolítico construido en el laboratorio.
Se controlaron también condiciones para realizar el rociado (spray), como el ajuste en el
diámetro de la boquilla del aerógrafo. Una vez ajustado el diámetro, ésta se fijó a una
distancia de 10 cm del sustrato caliente, Figura 2. A esta distancia, la sustancia
precursora se roció sobre el sustrato (que es la zona de alta temperatura) en forma de

76
gotas de tamaño micrométrico, que se dispersaron en un área de 5 cm2. El aire
comprimido es el gas portador de la solución dopante, el caudal de salida de dicho gas
se controla con la aguja del aerógrafo y la presión con el compresor de aire,
proporcionando una velocidad de flujo volumétrico de 2.33x10-8 m3/s.
Para conseguir una deposición uniforme, tanto el sustrato como el sistema se mantienen
estacionarios, lo que cambia, es la cantidad de rociadas que se depositan sobre el
sustrato caliente o la concentración de la solución precursora.
Figura IV.2. Distancia entre la punta del aerógrafo (rociador) y el sustrato (~11cm).
2.2. Preparación de la solución precursora
Se prepara la solución precursora de dióxido de estaño mediante la disolución de cloruro
de estaño dihidratado (SnCl2.H2O) en 5mL de metanol (MeOH), con agitación vigorosa.
Después de una completa disolución, con el fin de dopar al SnO2, se adiciona Sb2O3
previamente disuelto en 1ml de HCl concentrado. La agitación se mantiene constante
por un par de minutos más para así obtener una mezcla homogénea, viscosa, de color
ligeramente amarillo.

77
2.3. Rociado pirolítico de la solución precursora sobre las placas de Nts-TiO2
Para realizar los depósitos por rociado pirolítico sobre las placas de Nts-TiO2 se siguieron
los siguientes pasos: a) la placa con Nts-TiO2 se introduce a un horno, previamente
calentado a 600°C, por 10 minutos; b) se retira la placa del horno y se realiza la
deposición mediante rociado de la solución precursora; c) hecho esto, la placa con Nts-
TiO2 se mete de nuevo al horno por 5 minutos; d) se retira de nuevo la placa del horno y
se vuelve a rociar, hasta que se alcanza el número de rociadas deseadas, repitiendo los
pasos b) y c). Cuando se completa el número de rociadas, e) la placa se deja en el horno
por 10 minutos más y para finalizar; f) la placa se deja enfriar a temperatura ambiente.
De esta manera se obtienen los electrodos de nanotubos de dióxido de titanio
combinados con dióxido de estaño (Nts-TiO2/SnO2).
3. Resultados y Discusión
Se obtuvieron dos tipos de electrodos debido a que en su preparación se utilizaron 2
concentraciones diferentes del SnCl2 en la solución precursora (2.4 M y 1.8 M). En
adelante, se hará referencia a estas soluciones como S1 y S2, respectivamente. La
solución S1 contenía Sb2O3 0.057 M y la solución S2, Sb2O3 0.028 M. El número de
rociadas se mantuvo en 5, debido a que se observó, para la solución S1, que con este
número de rociadas se logra una mayor cobertura del SnO2 sobre el arreglo nanotubular.
Las observaciones por MEB de los cambios morfológicos y de grado de cobertura con
el número de rociadas mostraron que los depósitos de SnO2 sobre Nts-TiO2
(independientemente del número de rociadas) adoptan una morfología de partículas
granulares, las cuales se concatenan una sobre otra para formar una estructura similar
a un rosario; en adelante, se hará referencia a estas estructuras como conglomerados
de SnO2. Con el aumento en el número de rociadas, el número de estos conglomerados
se incrementa sobre la superficie del sustrato de nanotubos; así también, el número de
partículas que los conforman, se incrementa dando lugar a depósitos (rosarios) más
alargados de SnO2. Cabe aclarar que debido a que, pueden existir variaciones al hacer
el rociado manual, pudieran existir zonas con poco depósito aún en electrodos con varias
deposiciones. Sin embargo, la tendencia general es hacia el aumento en el tamaño y
número de los conglomerados de SnO2 sobre la superficie de nanotubos, Figura 3.

78
(a) (b)
(c) (d)
Figura IV.3. Imágenes SEM correspondientes a (a) depósito con una rociada, se observan
estructuras de SnO2 (conglomerados) cortas, (b) depósito con cinco rociadas, se observan
conglomerados de SnO2 alargados y entrelazados. (c) Depósito con una rociada, se observan
algunos conglomerados, (d) Depósito con 5 rociadas, se observa la distribución de mayor número
de conglomerados. Las imágenes a y b, se encuentran a una escala de 5 µm con el fin de
observar el crecimiento de los conglomerados; y las imágenes c y d, se encuentran a escala de
50 µm con el fin de observar la dispersión y aumento numérico de los conglomerados .

79
Los análisis de EDS de estos electrodos muestran que SnO2 se deposita tanto en la zona
donde aparentemente sólo hay nanotubos como en la zona de conglomerados y el
porcentaje atómico de estaño se halla entre 1.5 a 2.5% para un electrodo con 5 rociadas
que es el número máximo de rociadas elegido. En adelante, los electrodos preparados
con esta solución precursora y con cinco deposiciones se denominarán ES1.
Tabla IV.1. Análisis elemental del depósito de SnO2 obtenido por EDS.
Elemento % Peso % Atómico
C 3.24 6.80
O 43.31 68.30
Ti 43.08 22.70
Sn 10.37 2.20
3.1. Efecto en la morfología del depósito de SnO2 debido a la variación en la
concentración de la solución precursora
La caracterización por MEB muestra depósitos morfológicamente diferentes debido a las
diferencias de concentración de la sal precursora: los electrodos ES1, preparados a partir
de la solución precursora S1; y los ES2, preparados a partir de la solución S2. De la
MEB, se observó que, mientras el SnO2 en el electrodo ES1 es un depósito que deja
expuestos a los nanotubos, en los electrodos ES2, el depósito de SnO2 es granular
(tamaño de grano de aproximadamente 500 nm) y cubre completamente la capa de
nanotubos.

80
Figura IV.4. Depósito de SnO2 proveniente de la solución precursora ES2, donde se observa un
carácter granular del depósito.
El análisis de EDS de los electrodos ES2, preparados a partir de la solución S2, revela
que el % atómico de Sn varía de 13 a 17% entre diferentes zonas del análisis, Figura 4.
Element Weight% Atomic%
O K 38.45 77.77
Cl K 1.23 1.13
Ti K 11.54 7.80
Sn L 48.78 13.30
Totals 100.00
Figura IV.5. Análisis de EDS del electrodo ES2
en una zona del depósito de SnO2
Por su parte, los espectros UV- vis de reflectancia difusa, modificados a través de la
función de Kubelka-Munk, de los electrodos de Ti/Nts-TiO2/SnO2, mostraron diferencias
en los dos tipos de electrodos. Por su parte, los electrodos ES1 muestran un espectro de
características similares a las de un espectro de nanotubos de TiO2, con una longitud de
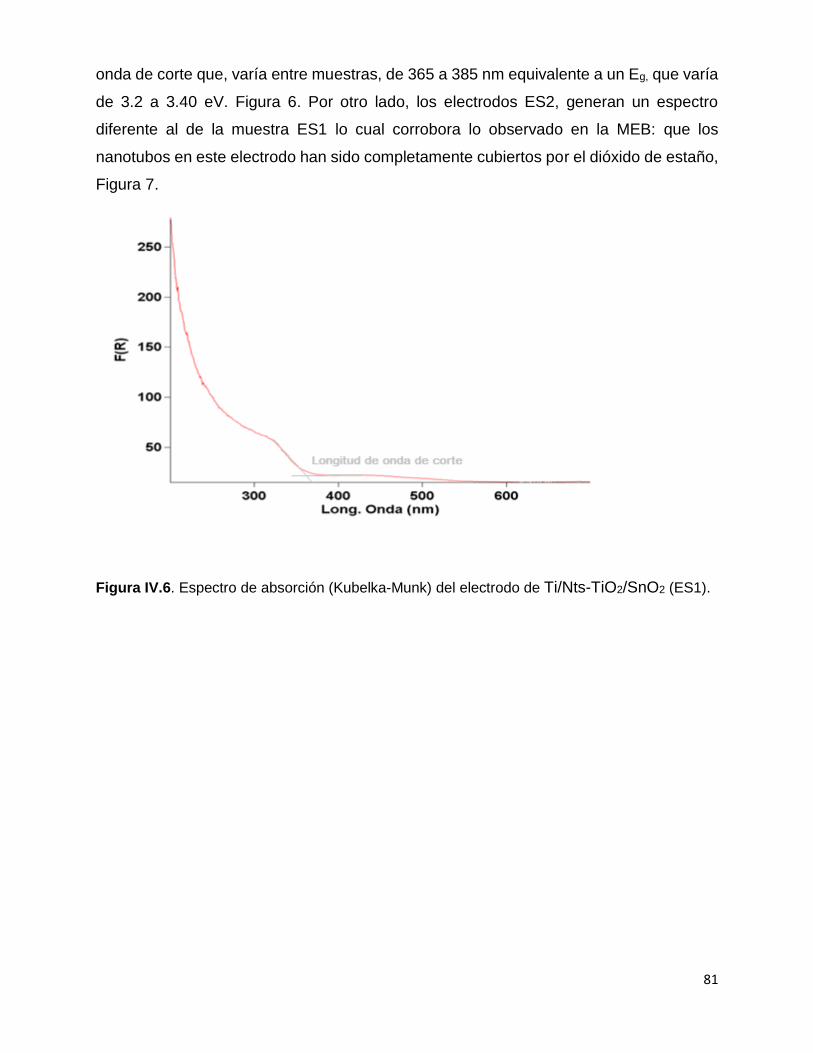
81
onda de corte que, varía entre muestras, de 365 a 385 nm equivalente a un Eg, que varía
de 3.2 a 3.40 eV. Figura 6. Por otro lado, los electrodos ES2, generan un espectro
diferente al de la muestra ES1 lo cual corrobora lo observado en la MEB: que los
nanotubos en este electrodo han sido completamente cubiertos por el dióxido de estaño,
Figura 7.
Figura IV.6. Espectro de absorción (Kubelka-Munk) del electrodo de Ti/Nts-TiO2/SnO2 (ES1).

82
Figura IV.7. Espectro de absorción (Kubelka-Munk) del electrodo de Ti/Nts-TiO2/SnO2 (ES2).

83
Por su parte, los análisis de Difracción de Rayos X para el electrodo ES2, corroboran
que la identidad del depósito corresponde a dióxido de estaño, Figura 8.
Figura IV.8. Difractograma de electrodo ES2, donde se muestra la formación de SnO2 sobre los
nanotubos de TiO2. La asignación de los picos de difracción se basó en los artículos de Tosun119
y Kumar.120
3.2. Estabilidad electroquímica de los electrodos de Ti/Nts-TiO2/SnO2
Con el fin de evaluar la estabilidad del electrodo de Ti/Nts-TiO2/SnO2, éste se sometió a
mediciones de polarización anódica en medio de H2SO4 0.1 M, a densidad de corriente
de 100 mA/cm2 durante 23 horas, Figura 9. El potencial se mantuvo constante durante
este tiempo pero a las 23 horas se observó el desprendimiento del depósito. La MEB de
este electrodo corroboró la desaparición del depósito de SnO2 y de la capa de nanotubos.
Figura 10.
2θ
Cu
enta
s
SnO
2
SnO
2
TiO
2
TiO
2
Ti
Ti

84
Figura IV.9. Evolución del potencial bajo condiciones de Polarización Anódica en Nts-
TiO2/SnO2.
(a) (b)
Figura IV.10. Imágenes de MEB de electrodos de Ti/Nts-TiO2/SnO2 a) antes y b) después de
polarización anódica.
0
0.5
1
1.5
2
2.5
3
0.0 5.0 10.0 15.0 20.0 25.0
Po
ten
cial
(V
)
t (h)

85
4. Conclusiones
Se depositó SnO2 sobre nanotubos de dióxido de titanio mediante la técnica de rociado
pirolítico. Mediante DRX, se comprobó que SnO2 crece sobre los nanotubos a partir de
la calcinación de SnCl2. En el rociado pirolítico se usó un aerógrafo cuyo diámetro de
boquilla se fijó de manera que se obtuvieran gotas muy finas de la solución precursora.
El diámetro de boquilla se mantuvo constante y se varió el número de rociadas o bien, la
concentración de la solución precursora.
De la MEB se observó que en los depósitos de la solución precursora de mayor
concentración se obtienen estructuras de SnO2 en forma de rosario (conglomerados), los
cuales aumentan su tamaño y número conforme aumenta el número de rociadas. En
estos depósitos, los nanotubos se encuentran descubiertos. Por otro lado, la reducción
en la concentración de cloruro de estaño de la solución precursora, conduce a la
deposición de una película continua de SnO2 que cubre completamente a los nanotubos,
como lo muestra la MEB.
La estabilidad de estos electrodos es menor en comparación con la estabilidad mostrada
por los electrodos de Ti/Nts-TiO2::PbO2 5s, ya que la polarización acelerada conlleva al
desprendimiento del depósito de dióxido de estaño y a la pérdida de la estructura
nanotubular del dióxido de titanio.

86
CAPÍTULO V. ELECTROCATÁLISIS Y FOTOELECTROCATÁLISIS DE
COMPUESTOS ORGÁNICOS MEDIANTE ELECTRODOS DE Ti/Nts-
TiO2::PbO2
1. Resumen
Los electrodos de Ti/Nts-TiO2::PbO2, preparados y caracterizados según lo descrito en
el capítulo 3, se probaron frente a la oxidación electroquímica de los siguientes
compuestos orgánicos: ácido cloranílico, rojo de metilo y p-nitrofenol. En todos los casos,
la electrooxidación se realizó bajo condiciones de corriente controlada (30 mAcm-2). La
electrooxidación de ácido cloranílico y de rojo de metilo se probó en condiciones de
oscuridad y de iluminación con luz ultravioleta de onda larga (365 nm), con electrodos
denominados de primera generación debido a que el arreglo nanotubular se hizo sobre
placas de Ti denominadas UNK en el capítulo 2 (párrafo 4.1.1). La decoloración de la
solución de ácido cloranílico se usó para obtener las primeras evidencias del
comportamiento de estos electrodos. Sin embargo, este compuesto se descartó al
comprobarse que sufría fotólisis bajo irradiación de luz a 365 nm. Para la investigación,
el ácido cloranílico se sustituyó por rojo de metilo, compuesto que demostró ser estable
frente a la iluminación y cuya decoloración en solución se pudo seguir mediante
espectrofotometría UV-vis a 525 nm. Se comparó la electrooxidación, a corriente
controlada, de rojo de metilo entre los electrodos, de primera generación: Ti/Nts-
TiO2::PbO2 5s, Ti/Nts-TiO2::PbO2 10s y Ti/Nts-TiO2::PbO2 15s, en condiciones de
oscuridad e iluminación. De estos experimentos, se observó que, bajo iluminación, el
electrodo Ti/Nts-TiO2::PbO2 5s producía la mayor decoloración de la solución del
compuesto orgánico en el menor tiempo de electrólisis. Este comportamiento se explicó
en términos de una mayor fotoeficiencia presentada por este electrodo.
Por otro lado, con los electrodos de segunda generación, preparados a partir de la placa
de Ti rotulada como MKB, en el capítulo 2 (párrafo 4.1.1); se llevó a cabo la oxidación
electrocatalítica de rojo de metilo y p-nitrofenol. Cabe destacar que la razón para esta
clasificación de electrodos, como de primera y segunda generación, es la diferencia en
desempeño fotoelectroquímico observado: los de primera generación presentaron una
elevada actividad fotoelectrocatalítica, mientras que los de segunda generación

87
presentaron una nula o muy baja fotoactividad. Sin embargo, la oxidación
electrocatalítica de rojo de metilo y p-nitrofenol sobre estos electrodos, aun siendo en
oscuridad, es notable. Por lo que se presentan, en este capítulo, el comportamiento
cinético de la oxidación de los compuestos rojo de metilo y p-nitrofenol, obtenido
mediante análisis por 1) espectrofotometría UV-vis, así como por 2) cromatografía de
líquidos (HPLC) de los productos de oxidación sobre estos electrodos de segunda
generación. En ambos casos se logró el 100% de oxidación.
2. Metodología
2.1. Precisiones sobre la preparación de nanotubos de segunda generación, 2-
Ti/Nts-TiO2
Las placas MKBSL sólo recibieron como tratamiento previo al anodizado, sonicación por
10 minutos con cada uno de los solventes siguientes: acetona, etanol, metanol. Se
enjuagaron con agua tridestilada y se secaron en atmósfera de nitrógeno; a continuación,
se sometieron a anodización. Después de la anodización, estos electrodos presentan el
precipitado indeseado PP debido a la rugosidad inicial de la placa de Ti. Para eliminar
dicho precipitado, las placas recién anodizadas, se sonicaron en acetona por 10-15 s.
Una vez eliminado el exceso de PP, los nanotubos recién sintetizados se sometieron a
tratamiento térmico (annealing).
2.2. Evaluación de fotoactividad
Se obtuvieron curvas I-V en un sistema de tres electrodos donde el electrodo de trabajo
correspondió al electrodo de Ti/Nts-TiO2::PbO2, el electrodo auxiliar fue una varilla de Ti
y el electrodo de referencia fue de sulfato mercuroso. Estas curvas se realizaron en
medio acuoso de H2SO4 0.5 M para los electrodos de Ti/Nts-TiO2 y de Ti/Nts-TiO2::PbO2
preparados a 5, 10 y 15 s. La velocidad de barrido utilizada fue de 5 mV/s. Las curvas I-
V en iluminación se realizaron irradiando sobre el sistema electroquímico con una
lámpara UV de 365 nm. La lámpara tenía una intensidad de 1.4 W/cm2 a una distancia
de 14 cm.
2.3. Decoloración de rojo de metilo
Se comprobó la actividad electro y fotoelectrocatalítica de los electrodos preparados
frente a la decoloración de rojo de metilo. Se utilizó una solución de rojo de metilo de 15

88
o 25 ppm en H2SO4 0.5 M (50 mL). La decoloración se siguió mediante análisis de
espectrometría UV-vis a longitud de onda de 525 nm. Las alícuotas de análisis fueron de
0.3 mL. Tanto las electrocatálisis como las fotoelectrocatálisis se llevaron a cabo a una
densidad de corriente de 30 mA/cm2, utilizando un sistema de tres electrodos donde el
contraelectrodo fue una varilla de titanio o un espiral de platino y el electrodo de
referencia fue de sulfato mercuroso, bajo agitación constante. Las fotoelectrocatálisis se
irradiaron con lámpara de 365 nm de intensidad de 4 W/cm2 a una distancia de 7 cm.
2.4. Oxidación de p-nitrofenol
La oxidación electrocatalítica de p-nitrofenol se llevó a cabo con una solución de 50 ppm
en H2SO4 0.5 M (50 mL). Se utilizó el sistema de tres electrodos mencionado
anteriormente así como la j=30mA/cm2. La oxidación se siguió espectrofotométricamente
a una longitud de onda de análisis de 290 nm. Esta reacción se siguió también mediante
HPLC en las condiciones que a continuación se indican.
2.5. Análisis cromatográfico por HPLC
El análisis por HPLC para la identificación de compuestos aromáticos se realizó con el
siguiente método. Columna cromatográfica Inertsil ODS 3, 5µm, 4.6 x 150 mm; Fase
Móvil: Acetonitrilo: Agua: Ac. Acético, 49:49:2; Velocidad de flujo: 0.8 mL/min; T=25°C;
Longitud de onda de detección: 288 nm. Mientras que, el análisis por HPLC para la
identificación de ácidos alifáticos se llevó a cabo por dos métodos diferentes: a) con una
columna cromatográfica PL-Hiplex H 8µm, 300 x 7.7mm; Fase Móvil: H2SO4 4mM
acuoso, Velocidad de flujo: 0.4 mL/min. T=35°C; Longitud de onda de detección: 210 nm;
b) con una columna Allure®Organic Acids, 5 µm, 4.6 mm x 250 mm, Fase Móvil: Fosfato
de potasio 0.1M a pH 2.5, Velocidad de flujo: 0.5 mL/min, Longitud de onda de detección:
226 nm.
3.1. Electrocatálisis y fotoelectrocatálisis de rojo de metilo sobre electrodos de
Ti/Nts-TiO2::PbO2 (5, 10 y 15 s) de la primera generación
Los electrodos de la primera generación se prepararon a partir de las placas de titanio
denominadas UNK sin lijar de acuerdo a la metodología A (Cap. 2). Después del
tratamiento térmico, se depositó PbO2 en los nanotubos como se indica en el capítulo 3.

89
En base a la caracterización de los electrodos de Ti/Nts-TiO2::PbO2 (5, 10 y 15 s), en la
que se puede observar que a menor tiempo de depósito se tiene una menor cobertura
de PbO2 sobre los nanotubos, pero una mayor dispersión del mismo, se llevaron a cabo
los siguientes experimentos: a) evaluación de fotoactividad de los tres tipos de electrodos
(5, 10 y 15 s), b) una vez que se ha corroborado con qué electrodo se logra la mayor
fotoeficiencia, se llevaron a cabo experimentos de decoloración de rojo de metilo. La
decoloración de rojo de metilo se siguió mediante espectroscopía UV-vis con dicho
electrodo y con electrodos de Ti/Nts-TiO2.
3.1.1. Evaluación de fotoactividad
Se obtuvieron curvas I-V en un sistema de tres electrodos donde el electrodo de trabajo
correspondió al electrodo de Ti/Nts-TiO2/PbO2, el electrodo auxiliar fue una varilla de
grafito y el electrodo de referencia fue de sulfato mercuroso. Estas curvas de
fotocorriente se realizaron en medio acuoso de H2SO4 0.5 M para los electrodos de
Ti/Nts-TiO2 y de Ti/Nts-TiO2::PbO2 preparados a 5, 10 y 15 s. La velocidad de barrido
utilizada fue de 5 mV/s. Las curvas I-V en iluminación se llevaron a cabo irradiando sobre
el sistema electroquímico con una lámpara UV de 365 nm. La lámpara tenía una
intensidad de 1.4 W/cm2 a una distancia del electrodo de trabajo de 14 cm.
De las curvas I-V se obtuvo la eficiencia de fotoconversión, η, de acuerdo a la ecuación
propuesta por Shahed U. M. Khan121 donde % η eff( photo) = [ jp(E°rev – |Eapp|)x100/(Io)]. De
acuerdo a esta ecuación, % η para Ti/Nts-TiO2 fue de 6.4%, similar a lo reportado en la
literatura, ver Figura 1.

90
Figura V.1. Densidad de fotocorriente (cuadrados azules) y eficiencia de fotoconversión con respecto a Eapp (círculos rojos). Figura tomada de M. Cerro-Lopez (et al).122
Se obtuvieron curvas I-V para los Ti/Nts-TiO2::PbO2 y se realizó el cálculo de eficiencia
de fotoconversión con el cual se pudo comparar la eficiencia presentada por los
electrodos, siendo electrodo de Ti/Nts-TiO2::PbO2 5 s, el que mostró la mayor eficiencia
(11%), ver Figura 2.
Eapp (V vs. ENH)
Efic
ien
cia
de
foto
con
vers
ión
(%
)
Den
sid
ad d
e fo
toco
rrie
nte
(m
A/c
m2
)
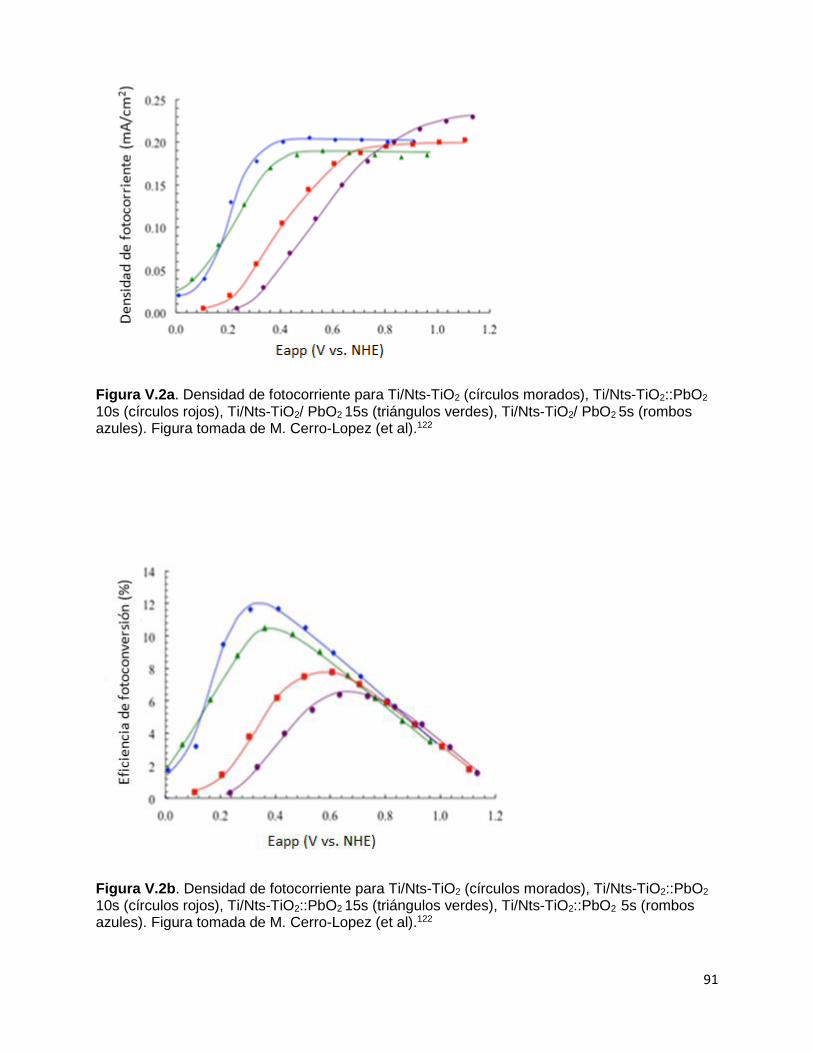
91
Figura V.2a. Densidad de fotocorriente para Ti/Nts-TiO2 (círculos morados), Ti/Nts-TiO2::PbO2 10s (círculos rojos), Ti/Nts-TiO2/ PbO2 15s (triángulos verdes), Ti/Nts-TiO2/ PbO2 5s (rombos azules). Figura tomada de M. Cerro-Lopez (et al).122
Figura V.2b. Densidad de fotocorriente para Ti/Nts-TiO2 (círculos morados), Ti/Nts-TiO2::PbO2 10s (círculos rojos), Ti/Nts-TiO2::PbO2 15s (triángulos verdes), Ti/Nts-TiO2::PbO2 5s (rombos azules). Figura tomada de M. Cerro-Lopez (et al).122

92
3.1.2. Decoloración de rojo de metilo
Las gráficas comparativas para la decoloración de rojo de metilo, Figura 3, muestran que
el electrodo de Ti/Nts-TiO2::PbO2 5s decolora completamente a la solución en los
primeros 30 minutos de reacción mientras que en la electrocatálisis para el mismo
electrodo la decoloración completa se alcanza en 110 minutos. El electrodo decolora
completamente al RM, pero el tiempo al que se alcanza esta decoloración es casi 4 veces
menor para la reacción en iluminación. Por otro lado, la fotoelectrocatálisis con
nanotubos descubiertos, apenas logra un porcentaje de decoloración del 30% a los130
min de reacción, lo cual indica que existe un efecto sinérgico entre PbO2 y los Nts-TiO2;
mediante el cual se logra una decoloración mayor a la que se esperaría de la suma de
los procesos individuales (fotoelectrocatálisis sobre nanotubos y electrocatálisis sobre
Ti/Nts-TiO2::PbO2 5s). En la misma gráfica se puede observar, para fines de
comparación, la decoloración electrocatalítica sobre un electrodo tradicional de Pb/PbO2,
la cual es de un 65% a los 130 min de reacción.
Figura V.3. Se muestra la decoloración de rojo de metilo por parte de los diferentes electrodos: electrocatálisis enTi/ Nts-TiO2 (cuadrados rojos vacíos), fotoelectrocatálisis en Ti/Nts-TiO2 (cuadrados rojos llenos), electrocatálisis en Pb/PbO2 (círculos negros), electrocatálisis en Ti/Nts-TiO2::PbO2 5s (triángulos azules), fotoelectrocatálisis en Ti/Nts-TiO2::PbO2 5s (triángulos azules llenos). Figura tomada de M. Cerro-Lopez (et al).122

93
El efecto sinérgico observado para los electrodos de Ti/Nts-TiO2::PbO2 5s se explica de
la siguiente manera. El sistema PbO2::TiO2::electrolito tiene dos interfaces importantes:
PbO2::TiO2 y TiO2::electrolito. En cada uno se crea un gradiente de potencial, el cual
estimula la transferencia electrónica por dos portadores de carga diferentes. En el primer
caso, cuando PbO2 se forma dentro de los nanotubos de TiO2, las diferencias en
conductividad producen un gradiente de potencial en la heterounión; el cual es capaz de
mejorar la transferencia electrónica a través de la interface TiO2::PbO2, incrementando
la eficiencia de fotoconversión. En el segundo caso, el gradiente de potencial generado
por la unión TiO2::electrolito estimula la transferencia de huecos a la interface. Cuando
se aplica un potencial externo bajo la radiación UV, el gradiente de potencial se
incrementa a tal grado que produce una separación completa de los portadores de carga
(condición de agotamiento). En esta condición, los electrones fotogenerados son
transportados a través de la interface PbO2::TiO2 y de ahí hacia el cátodo siguiendo la
secuencia:
VB(TiO2) CB(TiO2) LEO(PbO2) (TiO2) en el bulto (Ti)sustrato (C)Cátodo
Donde VB y CB representan las bandas de valencia y conducción de TiO2, LEO el orbital
de menor energía de PbO2 y el resto la representación típica para TiO2, Ti y electrodos
auxiliares de carbono.
Por otro lado, la ocurrencia de esta transferencia electrónica asistida por el contacto
cercano entre TiO2 y PbO2 también produce un flujo opuesto importante de huecos hacia
la interface TiO2::electrolito, donde se involucran en la oxidación de agua para formar
radicales libres .OH o la oxidación directa del colorante RM, dependiendo del potencial
de oxidación fijado por la densidad de corriente aplicada. Adicionalmente, debe notarse
que los ánodos de PbO2 exhiben propiedades electrocatalíticas adecuadas para la
oxidación de moléculas orgánicas debido a las interacciones entre los sitios de Pb(IV) y
los grupos activos del contaminante orgánico, en este caso RM.121 Este mecanismo,
favorece la oxidación rápida de contaminantes orgánicos en solución, como se ha
reportado previamente por Bonfatti,123 Martínez-Huitle 124 y Ferro.125

94
3.2. Electrocatálisis y fotoelectrocatálisis de rojo de metilo con electrodos de la
segunda generación
Los electrodos de la segunda generación, a los cuales se les denominará 2Ti/Nts-
TiO2::PbO2, el 2 significa de segunda generación, presentaron dos características
distintas de los electrodos de la primera generación. Una de ellas, relacionada con una
baja actividad fotoelectrocatalítica y, la otra, relacionada con una mayor cobertura de
PbO2 sobre los nanotubos como mostró la MEB de estos electrodos. Es decir, los
depósitos de PbO2 de 5 s en las placas de la segunda generación se parecía más a los
depósitos de 10 s o de 15 s de la primera generación, Figura 4.
(a) (b)
Figura V.4. Imágenes de MEB de depósitos de PbO2 de a) 5 s para electrodos de segunda generación y, b) de 10s para electrodos de primera generación.
Los nanotubos de estos electrodos (2-Ti/Nts-TiO2), se prepararon partir de las placas de
titanio MKB sin lijar (MKBSL). Cabe mencionar que, al comienzo de las pruebas
catalíticas, lo nanotubos se prepararon a partir de placas MKB lijadas, pero los
experimentos con los electrodos de segunda generación provenientes de estas placas
mostraron baja actividad tanto electrocatalítica como fotoelectrocatalítica, por lo que, no
se presentan dichos resultados.
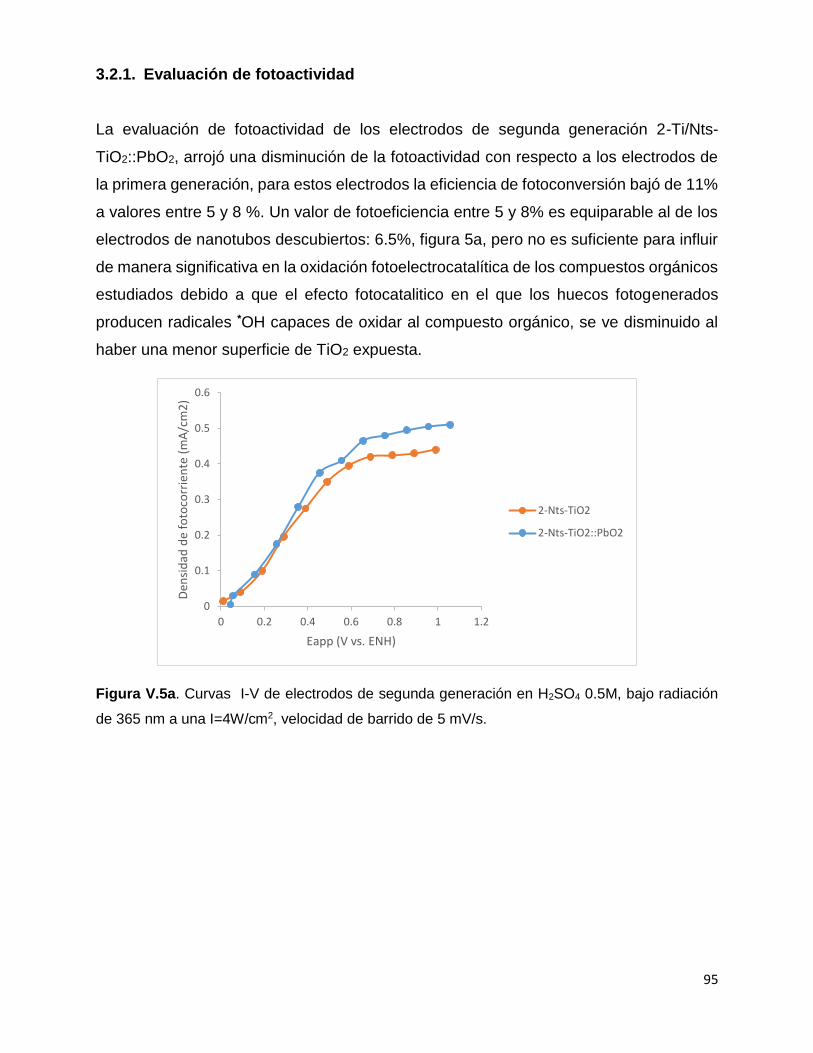
95
3.2.1. Evaluación de fotoactividad
La evaluación de fotoactividad de los electrodos de segunda generación 2-Ti/Nts-
TiO2::PbO2, arrojó una disminución de la fotoactividad con respecto a los electrodos de
la primera generación, para estos electrodos la eficiencia de fotoconversión bajó de 11%
a valores entre 5 y 8 %. Un valor de fotoeficiencia entre 5 y 8% es equiparable al de los
electrodos de nanotubos descubiertos: 6.5%, figura 5a, pero no es suficiente para influir
de manera significativa en la oxidación fotoelectrocatalítica de los compuestos orgánicos
estudiados debido a que el efecto fotocatalitico en el que los huecos fotogenerados
producen radicales *OH capaces de oxidar al compuesto orgánico, se ve disminuido al
haber una menor superficie de TiO2 expuesta.
Figura V.5a. Curvas I-V de electrodos de segunda generación en H2SO4 0.5M, bajo radiación
de 365 nm a una I=4W/cm2, velocidad de barrido de 5 mV/s.
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.2 0.4 0.6 0.8 1 1.2
Den
sid
ad d
e fo
toco
rrie
nte
(m
A/c
m2
)
Eapp (V vs. ENH)
2-Nts-TiO2
2-Nts-TiO2::PbO2

96
Figura V.5b. Eficiencia de fotoconversión de electrodos de segunda generación en H2SO4 0.5M,
bajo radiación de 365 nm a una I=4W/cm2, velocidad de barrido de 5 mV/s.
3.2.2. Electrocatálisis y fotoelectrocatálisis de rojo de metilo
En los experimentos de electrocatálisis y fotoelectrocatálisis se observó una actividad
catalítica similar para la oxidación de RM entre ambas pruebas, y algunas veces, incluso
inferior; contrario a lo que había sucedido para los electrodos de primera generación,
Figura 6.
Figura V.6. Electrocatálisis y fotoelectrocatálisis de RM sobre electrodos 2-Nts-TiO2::PbO2 5 s
(de segunda generación).
0
1
2
3
4
5
6
7
8
0 0.2 0.4 0.6 0.8 1 1.2
Efic
ien
cia
de
foto
co
nve
rsió
n (
%)
Eapp (V vs. ENH)
2-Nts-TiO2
2-Nts-TiO2::PbO2 5s
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
0 20 40 60 80
Co
nce
ntr
ació
n r
elat
iva,
Ci/
Co
t (min)
Fotoelectrocatálisis deRM
Electrocatálisis de RM

97
3.2.2.1. Electrocatálisis de rojo de metilo
Debido a la baja actividad fotoelectrocatalítica de estos nuevos electrodos, se decidió
estudiar únicamente el comportamiento electrocatalítico frente a RM, para lo cual, una
solución de 15 ppm de rojo de metilo se oxidó a una densidad de corriente de 30 mA/cm2
con los siguientes electrodos 2-Ti/Nts-TiO2, 2-Ti/Nts-TiO2::PbO2 5 s, 2-Ti/Nts-TiO2::PbO2
15 s y Pb/PbO2 para fines de comparación. Como se puede apreciar de la Figura 7, la
decoloración completa de rojo de metilo se logra a los 30 minutos de reacción para el
electrodo 2-Ti/Nts-TiO2::PbO2 5 s. Visualmente, la decoloración de rojo de metilo pasa
por 4 puntos principales: a) color rosa inicial, b) color rosa más claro, c) color amarillo y
d) solución transparente, Figura 8. La demanda química de oxígeno (DQO) en estos
puntos, muestra que los valores de DQO no cambian prácticamente de los puntos a a b,
pero éstos sí cambian drásticamente de 23 ppm O2 a valores indetectables (debajo de 4
ppm O2 según lo indica el método Merck utilizado) al final de la reacción, puntos c a d,
Tabla 1. Cabe mencionar que la DQO teórica para RM es de 34 ppm, pero diferentes
soluciones de RM de 15 ppm de concentración inicial dieron una DQO experimental entre
25-30 ppm.
Figura V.7. Decoloración de rojo de metilo en H2SO4 0.5 M sobre electrodos electrodos 2-
Ti/Nts-TiO2 (círculos grises), 2-Ti/Nts-TiO2::PbO2 5 s (círculos azules), 2-Nts-TiO2::PbO2 15 s
(círculos amarillos) y Pb/PbO2 (círculos naranjas) a j=30 mAcm-2, Co= 15 ppm
0
0.2
0.4
0.6
0.8
1
1.2
0 50 100 150 200 250
Co
nce
ntr
ació
n r
elat
iva,
Ci/
Co
Tiempo de electrólisis (min)

98
Figura V.8. Decoloración de rojo de metilo en H2SO4 0.5 M sobre el electrodo 2-Ti/Nts-
TiO2::PbO2 5s , Co= 15 ppm, j=30 mAcm-2
Tabla V.1. Valores de DQO en diferentes puntos de color durante la electrooxidación
de rojo de metilo, a) color rosa inicial, b) color rosa más claro, c) color amarillo claro y
d) solución transparente
Punto de color DQO, O2 ppm
a 25
b 23
c y d Por debajo del límite de detección (4 ppm)
Este comportamiento puede ser explicado en los siguientes términos: la decoloración de
rojo de metilo inicia con la ruptura del enlace azo, lo cual podría explicar la decoloración
observada en la primera etapa. Dicha ruptura conlleva a la formación de intermediarios
aromáticos cuya concentración es responsable de que la DQO no cambie en los puntos
a y b. Sin embargo, conforme la reacción avanza, tales intermediarios pueden
convertirse a ácidos carboxílicos y dióxido de carbono, lo cual disminuye los valores de
DQO. El análisis por HPLC de los productos finales de reacción, mostró que aunque se
logra una decoloración total y la DQO disminuye a valores por debajo del límite de
0
0.2
0.4
0.6
0.8
1
1.2
0 5 10 15 20 25 30 35
Rel
ativ
e co
nce
ntr
acio
n (
Ci/
Co
)
Electrolysis time(min)
a
b
c d

99
detección, (4 ppm O2) la electrooxidación no alcanza la mineralización completa del
compuesto; ya que, se detectaron en la muestra final de reacción: ácido oxálico, maleico
y, ácidos no identificados, Figura 9.
Figura V.9. Detección por HPLC de productos de oxidación de RM en la muestra final de reacción donde se han identificado ácidos maleico y oxálico mediante el método cromatográfico: Columna PL-Hiplex H; Fase Móvil : H2SO4 4mM acuoso, Vel. de flujo: 0.4 mL/min. T=35°C; λ=210 nm.
Con el fin de obtener información de la cinética de decoloración por parte de tales
electrodos, y poder comparar su comportamiento catalítico, de las curvas cinéticas
mostradas en la Figura 7, se obtuvieron las constantes cinéticas para la decoloración de
RM en los electrodos de 2-Ti/Nts-TiO2, y Pb/PbO2. Para el ajuste de la curvas cinéticas
se partió de la cinética propuesta por Panizza y Cerisola126, quienes indican que, en el
caso de electrodos con altos sobrepotenciales de oxígeno (como PbO2 o BDD), los
Aci
do
Oxá
lico

100
compuestos orgánicos son mineralizados en la superficie del ánodo por los radicales
hidroxilo electrogenerados por la descarga de agua, Ec. 1, lo cuales reaccionan con el
rojo de metilo para formar dióxido de carbono, Ec. 2.
H2O *OH + H+ + e- Ec. 1
C15H15N3O2 + 86 *OH 15CO2 + 3NO3- + 3H+ + 49H2O Ec. 2
De la ecuación anterior, la ley de velocidad propuesta sería
−𝑑[𝑅𝑀]
𝑑𝑡= 𝑘[𝑅𝑀][𝑂𝐻] Ec.3
Sin embargo, el radical hidroxilo (*OH) es una especie muy reactiva que, no sólo se
acumula en solución, si no que su concentración asume un valor de estado estacionario
durante el proceso, por lo que, la Ec. 3 puede reescribirse como una ecuación de pseudo-
primer orden, Ec. 4; donde kapp= k[*OH].127
−𝑑[𝑅𝑀]
𝑑𝑡= 𝑘𝑎𝑝𝑝[𝑅𝑀] Ec. 4
En general, las curvas cinéticas se ajustaron a una cinética de pseudo-primer orden. Sin
embargo, el electrodo 2-Ti/Nts-TiO2::PbO2 5 s, no mostró un comportamiento cinético
ajustable a algún orden de reacción. Las constantes cinéticas obtenidas se muestran en
la Tabla 2.
Tabla V.2. Constantes cinéticas de la decoloración de RM en, 2-Ti/Nts-TiO2, 2-Ti/Nts-
TiO2::PbO2 5 s 2-Ti/Nts-TiO2::PbO2 15 s y Pb/PbO2.
Electrodo k (min-1)
2-Ti/Nts-TiO2 0.0011
2-Ti/Nts-TiO2::PbO2 15 s 0.0121
Pb/PbO2 0.0147
2-Ti/Nts-TiO2::PbO2 5 s Sin ajuste
3.2.3. Electrocatálisis de p-nitrofenol
Se realizaron electrooxidaciones de p-nitrofenol (PNF) con los electrodos de 2-Ti/Nts-
TiO2::PbO2 5 s, 2-Ti/Nts-TiO2, y Pb/PbO2. Cada electrodo presentó un comportamiento
cinético análogo al observado para la decoloración de rojo de metilo. Es decir, la

101
oxidación del p-nitrofenol con 2-Ti/Nts-TiO2 fue incompleta, del 13%, a las 3.5 h de
reacción; mientras que para el electrodo 2-Ti/Nts-TiO2::PbO2 5s, el porcentaje de
oxidación de PNF fue del 100% en el mismo tiempo de reacción (3.5 h), Figura 10. Por
otro lado, las curvas cinéticas de los electrodos 2-Ti/Nts-TiO2, y Pb/PbO2 se ajustaron a
una cinética de pseudo-primer orden cuyas constantes se muestran en la Tabla 3. Por
otro lado, el electrodo 2-Ti/Nts-TiO2::PbO2 5s, no se ajustó a ningún orden de reacción,
lo cual es consistente con lo observado para este electrodo en rojo de metilo.
Figura V.10. Curvas cinéticas de la electrooxidación de PNF en H2SO4 0.5 M sobre electrodos
de 2-Ti/Nts-TiO2::PbO2 5 s, 2Ti/Nts-TiO2, y Pb/PbO2, j=30 mA/cm2, Co=50 ppm.
Tabla V.3. Constantes cinéticas de la oxidación de PNF en 2-Ti/Nts-TiO2::PbO2 5 s, 2-
Ti/Nts-TiO2, y Pb/PbO2.
Electrodo k (min-1)
2-Ti/Nts-TiO2 0.0006
Pb/PbO2 0.0131
2-Ti/Nts-TiO2::PbO2 5s Sin ajuste
0
0.2
0.4
0.6
0.8
1
1.2
0 50 100 150 200 250
Co
nce
ntr
ació
n r
elat
iva,
Ci/
Co
Tiempo de electrólisis (min)
2Nts-TiO2::PbO2 5 s
Pb/PbO2
2-Nts-TiO2

102
3.2.3.1. Análisis de HPLC de intermediarios de reacción de la oxidación de PNF
De los cromatogramas de los intermediarios aromáticos de la oxidación de PNF en
electrodo 2-Ti/Nts-TiO2 se observaron cuatro picos analíticos correspondientes a: PNF
(TR=4.30 min), hidroquinona (RT=2.35 min), benzoquinona (RT=3.30 min) y un pico no
identificado a RT=2.77 min. La figura 11, muestra los cromatogramas obtenidos a tres
diferentes tiempos de reacción de la oxidación de PNF sobre nanotubos descubiertos.
De la figura 11 se puede observar cómo la oxidación de PNF genera los intermediarios
aromáticos como hidroquinona y benzoquinona, los cuales aumentan conforme avanza
la reacción. En la misma figura, se aprecia que PNF apenas decrece.
Figura V.11. Cromatogramas de la muestras de oxidación de PNF sobre el electrodo 2-Ti/Nts-TiO2 a diferentes tiempos de reacción para la identificación de intermediarios aromáticos mediante el método cromatográfico: Columna Inertsil ODS 3; Fase Móvil: Acetonitrilo: Agua: Ac. Acético, 49:49:2; Vel. de flujo: 0.8 mL/min; T=25°C; λ= 288 nm.
Para esta reacción se analizó por HPLC la formación de ácidos sólo en la muestra final
de reacción, el cromatograma obtenido muestra la formación incipiente del ácido
maleico a TR= 14.95 min, Figura 12.
t=0
t=60 min
t=225 min
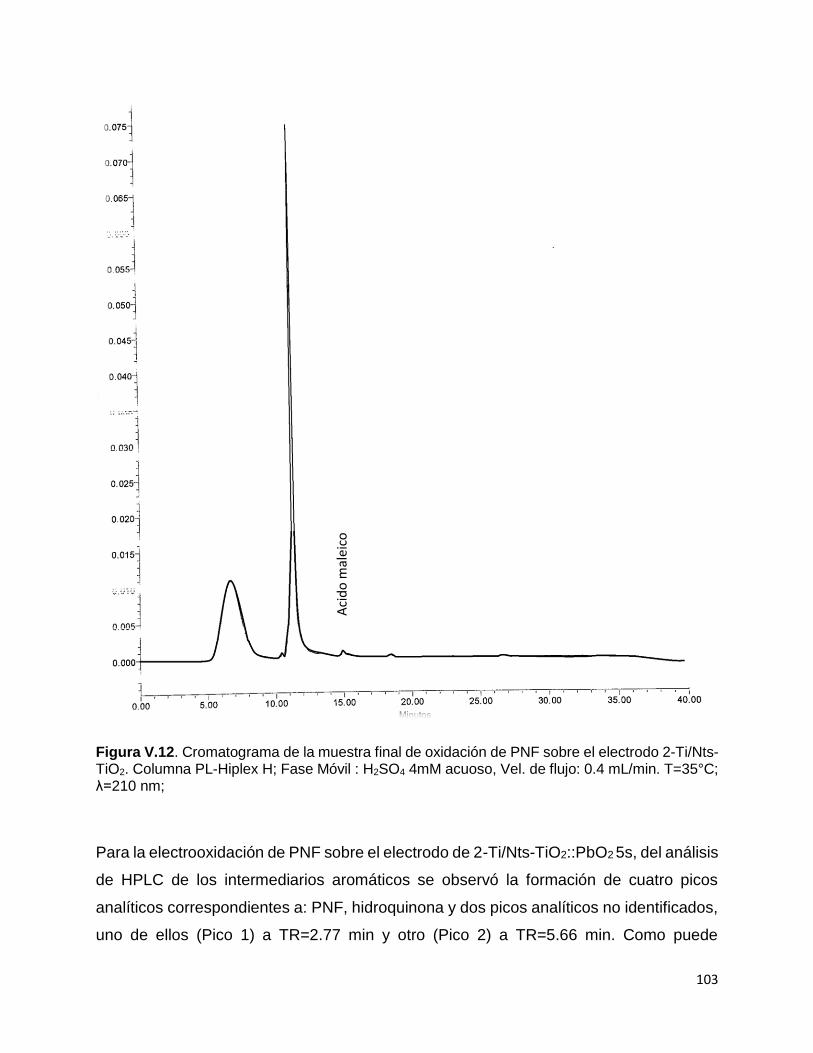
103
Figura V.12. Cromatograma de la muestra final de oxidación de PNF sobre el electrodo 2-Ti/Nts-TiO2. Columna PL-Hiplex H; Fase Móvil : H2SO4 4mM acuoso, Vel. de flujo: 0.4 mL/min. T=35°C; λ=210 nm;
Para la electrooxidación de PNF sobre el electrodo de 2-Ti/Nts-TiO2::PbO2 5s, del análisis
de HPLC de los intermediarios aromáticos se observó la formación de cuatro picos
analíticos correspondientes a: PNF, hidroquinona y dos picos analíticos no identificados,
uno de ellos (Pico 1) a TR=2.77 min y otro (Pico 2) a TR=5.66 min. Como puede
Aci
do
mal
eico

104
observarse de los cromatogramas de la Figura 13, a diferencia de lo observado para el
electrodo 2-Ti/Nts-TiO2, estos compuestos experimentan un aumento en los primeros 90
minutos y después una disminución hasta su eventual desaparición a las 3.5 h de
reacción. Como puede apreciarse del cromatograma del t= 210 min (Figura 13) sólo
queda un pico muy pequeño de PNF cuya área se relaciona con una concentración de
~0.5 ppm. Cabe destacar que se analizó catecol (TR 2.62), 4-nitrocatecol (TR 3.20) y
resorcinol (TR 2.56). Puede observarse, de los tiempos de retención, que ninguno de
estos intermediarios polihidroxilados se detectó en las muestras de reacción.
Figura V.13. Cromatogramas de la muestras de oxidación de PNF sobre el electrodo 2-Ti/Nts-
TiO2::PbO2 a diferentes tiempos de reacción para identificación de intermediarios aromáticos mediante el método cromatográfico: Columna Inertsil ODS 3; Fase Móvil: Acetonitrilo: Agua: Ac. Acético, 49:49:2; Vel. de flujo: 0.8 mL/min; T=25°C; λ= 288 nm.
Por otro lado, el análisis de HPLC de los ácido alifáticos producidos en la misma reacción
indica la formación de varios picos analíticos correspondiente a ácidos, de los cuales se
identificaron ácidos oxálico (RT=6.5 min), maleico (RT=10.2min) y málico (10.9min), con
el método cromatográfico de la columna Allure®Organic Acids. En los cromatogramas
de la Figura 14, se puede observar que la cantidad de ácido oxálico crece durante el
t=0
t=90 min
t=150 min
t=210 min
Hid
roq
uin
on
a
PNF

105
tiempo que dura la reacción, nunca disminuye, mientras que para los ácidos maleico y
málico, se nota un incremento en los primeros 150 minutos de reacción y después un
decremento debido a su oxidación a ácido oxálico y, eventualmente, a dióxido de
carbono, lo cual se comprobó a través del análisis de Carbón Orgánico Total (COT) de
la muestra final de esta reacción, el cual arrojó un valor de 17.8 mg/L lo cual refleja la
mineralización parcial de la solución inicial de p-nitrofenol (50 ppm), cuyo COT era 28.2
mg/L (COT Teórico= 26 ppm).
Figura V.14. Cromatogramas de la muestras de oxidación de PNF sobre el electrodo 2-Ti/Nts-
TiO2::PbO2 a diferentes tiempos de reacción para identificación de ácidos alifáticos. Método
cromatográfico: Columna Allure®Organic Acids, Fase Móvil: Fosfato de potasio 0.1M a pH 2.5,
Vel. flujo: 0.5 mL/min, λ=226 nm.
Aci
do
mal
eico
Aci
do
mál
ico
Aci
do
oxá
lico
t= 0 min
t= 90 min
t=150 min
t=210 min

106
3.2.3.2. Ruta de oxidación de p-nitrofenol propuesta
De acuerdo al mecanismo de reacción general propuesto por Torres y colaboradores,128
los fenoles p-sustituidos como el p-nitrofenol pueden sufrir mineralización directa o bien
indirecta a través de la formación de intermediarios aromáticos que experimentan
rupturas para formar ácidos alifáticos, los cuales formarán dióxido de carbono, Figura 15.
Figura V.15. Esquema de ruta de reacción general127
Por su parte, Zhou y colaboradores,129 a través de estudios de HPLC, señalan que para
electrodos de PbO2, la oxidación de algunos compuestos fenólicos p-sustituidos , ocurre
principalmente vía el mecanismo propuesto en la figura 16.

107
Figura V.16. Ruta general de degradación para compuestos aromáticos oxidados
electroquímicamente en electrodos de PbO2.128
De estas premisas y del análisis de intermediarios de HPLC realizado. Se propone la
siguiente ruta de degradación de la cual se ha descartado la mineralización directa, pues
se encontraron ácidos, principalmente maleico y oxálico a los diferentes tiempos de
reacción, Figura 17.

108
Figura V.17. Ruta de degradación de p-nitrofenol en medio de H2SO4 0.5 M, j=30 mA/cm2 para
los electrodos de 2-Ti/Nts-TiO2 y 2-Ti/Nts-TiO2::PbO2.
4. Conclusiones
Se detectaron diferencias importantes en el desempeño electrocatalítico y
fotoelectrocatalítico de los electrodos de dióxido de plomo soportados en nanotubos de
dióxido de titanio debido al uso de placas de titanio de diferente procedencia. Lo anterior
se atribuye a los antecedentes de fabricación de las placas de titanio, sobre todo en lo
que respecta al deformado y acabado; lo cual, como señala la literatura, influye en la
forma en que crecen los nanotubos, y por ende, su comportamiento catalítico.
Por otra parte, se logró la mineralización parcial de p-nitrofenol mediante el uso de
electrodos de PbO2 soportados en nanotubos de TiO2 bajo condiciones galvanostáticas.
La eliminación de p-nitrofenol fue de casi 100% después de 3.5 h. Los compuestos
remanentes de la electrooxidación son ácidos alifáticos de cadena corta susceptibles de
biodegradación posterior.

109
Las curvas cinéticas de la decoloración de rojo de metilo y la oxidación de p-nitrofenol,
muestran que los electrodos de nanotubos solos, dióxido de plomo e incluso del depósito
de dióxido de plomo de 15 segundos sobre nanotubos, muestran una cinética de pseudo-
primer orden, mientras que los electrodos de nanotubos con dióxido de plomo depositado
durante 5 segundos no pudieron ajustarse a algún orden de reacción, lo cual sugiere una
influencia notable del dióxido de titanio en el mecanismo de reacción que podría
atribuirse a procesos de adsorción sobre la superficie de mismo.130-131

110
CAPITULO 6: ELECTROCATÁLISIS DE COMPUESTOS ORGÁNICOS
MEDIANTE ELECTRODOS DE 2-Ti/Nts-TiO2/SnO2
1. Resumen
Los electrodos de 2-Ti/Nts-TiO2/SnO2, de segunda generación (el número 2 inicial indica
segunda generación), fueron preparados y caracterizados según lo descrito en el capítulo
4. De estos electrodos, tanto ES1 (cobertural parcial de nanotubos) como ES2 (cobertura
total de nanotubos), presentaron una baja eficiencia de fotoconversión; y, en el caso de
los electrodos ES2, completamente cubiertos por SnO2, presentaron también una
respuesta alta en corriente bajo condiciones de oscuridad. Por lo anterior, sólo se
realizaron oxidaciones electrocatalíticas de rojo de metilo y p-nitrofenol. La decoloración
de rojo de metilo se llevó a cabo frente a electrodos ES1 y ES2, mientras que la
electrooxidación de p-nitrofenol se realizó únicamente con electrodos ES2. La
decoloración de rojo de metilo con el electrodo ES1, se logró en un porcentaje del 100%
en un tiempo de 3 h, mientras que con el electrodo ES2 se observa una decoloración del
90% en aproximadamente el mismo tiempo.
La oxidación de p-nitrofenol se realizó de manera parcial con el electrodo ES2, con el
cual se logró una oxidación del 75% en 3.5 h. Los análisis de HPLC, mostraron que al
tiempo final de reacción se encuentran presentes en solución: PNF, hidroquinona,
benzoquina, y ácidos alifáticos. Ambos electrodos presentaron baja estabilidad, ya que,
el depósito se pierde después de 4-8 h de reacción.
2. Metodología
Para el estudio del comportamiento electrocatalítico de estos electrodos se siguió la
metodología descrita en el capítulo 5. En general, las electrooxidaciones se llevaron a
cabo bajo condiciones galvanostáticas (j=30 mA/cm2) y la oxidación de siguió
espectrofotométricamente a la longitud de onda de análisis del compuesto orgánico
estudiado. Sólo para el electrodo ES2, la formación de intermediarios, aromáticos y de
ácidos alifáticos, provenientes de la oxidación de p-nitrofenol también se siguió mediante
HPLC, bajo las mismas condiciones analíticas descritas en el capítulo 5.

111
3. Resultados y Discusión
3.1. Electrocatálisis de rojo de metilo
La electrocatálisis de una solución de rojo de metilo (15 ppm) en H2SO4 0.5 M fue seguida
espectrofotométricamente a λ=520 nm, frente a los electrodos ES1 y ES2. La oxidación
de RM con ambos electrodos no muestra diferencias significativas en cuanto al
porcentaje de decoloración alcanzado a un determinado tiempo, Figura 1. En ambos
casos, un alto porcentaje de decoloración (mayor o igual al 90%) ocurre en
aproximadamente 3.0 h. La decoloración de rojo de metilo en el electrodo de cobertura
total se ajusta a una cinética de pseudo-primer orden; mientras que, el ajuste para la
decoloración de RM con el electrodo de cobertura parcial no puede atribuirse a una
cinética de primer orden dado que R2 de la regresión lineal es bajo (0.95). Sin embargo,
se realizó el ajuste para obtener las constantes de velocidad de estas reacciones con
fines de comparación, ver Tabla 1. Lo anterior corrobora lo observado para los electrodos
de dióxido de plomo: que los materiales con cobertura parcial no presentan cinéticas de
orden similar al de los electrodos donde el material electrocatalítico expuesto
corresponde a un solo tipo de óxido, sea TiO2, SnO2 o PbO2.
Figura VI.1.Decoloración de rojo de metilo frente a electrodos de 2-Ti/Nts-TiO2/SnO2, cobertura
parcial y total, y electrodo de 2-Ti/Nts-TiO2 (para fines de comparación).
0
0.2
0.4
0.6
0.8
1
1.2
0 50 100 150 200
Co
nce
ntr
ació
n r
elat
iva
(Ci/
Co
)
Tiempo de electrólisis (min)
2-Ti/TiO2/SnO2cobertura total
2-Ti/TiO2/SnO2cobertura parcial
2-Ti/TiO2-Nts

112
Tabla VI.1. Constantes cinéticas de pseudo-primer orden para la decoloración de rojo de
metilo con electrodos 2-Ti/Nts-TiO2/SnO2.
Electrodo k(min-1) R2
2-Ti/Nts-TiO2/SnO2
cobertura parcial (ES1)
0.023 0.95
2-Ti/Nts-TiO2/SnO2
cobertura total (ES2)
0.016 0.98
3.2. Electrocatálisis de p-nitrofenol
La electrooxidación de una solución de 50 ppm de PNF en medio de H2SO4 0.5 M a
densidad de corriente de 30 mA/cm2, se realizó frente a electrodos de cobertura total
(ES2). Bajo estas condiciones, la oxidación de p-nitrofenol se siguió mediante
espectrofotometría UV-vis a λ=290 nm. El análisis espectrofotométrico muestra que se
el PNF se oxida de manera parcial, resultando en un 85% de oxidación en 3.5 h, Figura
2. La curva se ajusta a una cinética de pseudo-primer orden. La constante de pseudo-
primer orden para esta reacción es 0.0096 min-1 con un valor de R2 de 0.99.
Figura VI.2. Electrooxidación de PNF en medio ácido frente a electrodos de 2Ti/Nts-
TiO2/SnO2.Se incluye la oxidación de PNF con electrodos 2-Ti/Nts-TiO2, para fines de
comparación.
0
0.2
0.4
0.6
0.8
1
1.2
0 50 100 150 200 250
Co
nce
ntr
ació
n r
elat
iva
(Ci/
Co
)
Tiempo de electrólisis (min)
2-Ti/Nts-TiO2/SnO2cobertura total
2-Ti/TiO2-Nts

113
El análisis de HPLC de los intermediarios producidos en esta reacción indica que la
oxidación inicia con la formación de intermediarios aromáticos como hidroquinona
(TR=2.3), benzoquinona (TR=3.3) y otro compuesto aromático no identificado (TR=2.7),
Figura 3. Como puede observarse del cromatograma de la figura 3, el PNF disminuye
hasta desaparecer completamente en el tiempo final de reacción, sin embargo,
hidroquinona permanece en la muestra final de reacción lo que indicaría que, en realidad,
la oxidación de p-nitrofenol fue completa pero su conversión hacia ácidos carboxílicos
no.
Figura VI.3. Análisis por HPLC de intermediarios aromáticos de la oxidación de PNF con
electrodos 2Ti/Nts-TiO2/SnO2. El pico en 1.7 corresponde al frente del solvente.

114
En lo que se refiere a la formación de ácidos alifáticos, sólo se analizó por HPLC la
muestra final de reacción. La cromatografía muestra la formación de ácidos oxálico y
maleico, Figura 4.
Figura VI.4. Análisis por HPLC de ácidos alifáticos en muestra final de reacción de oxidación de
PNF con 2-Ti/Nts-TiO2/SnO2 (cobertura total).

115
3.3. Estabilidad de los depósitos de SnO2
Se observa que después de 4-8 horas de reacción, los electrodos de 2Ti/Nts-TiO2/SnO2
(cobertura parcial) pierden actividad. Por lo que se analizó mediante MEB el cambio en
tales electrodos después de ser usados. La MEB muestra que se pierde el depósito de
SnO2, Figura 5a y 5b. Asimismo, la morfología del depósito de SnO2 pierde su
característica granular y tiende hacia un depósito amorfo, lo cual también ocurre con
algunas zonas de los nanotubos, Figura 5c.
(a) (b)

116
(c)
Figura VI.5. Imágenes de MEB de los depósitos de SnO2 y nanotubos de TiO2, a) antes de
reacción, b) y c) después de reacción.
En cuanto a los electrodos de 2-Ti/Nts-TiO2/SnO2 cobertura total, éstos también pierden
su actividad después de unas horas de uso. La microscopía electrónica de barrido
muestra que en estos electrodos, el depósito SnO2 también se desprende, y la estructura
nanotubular desaparece, Figura 6.

117
(a) (b)
Figura VI.6. Imágenes de MEB donde se observa en a) el depósito original de SnO2 que cubre
totalmente a los nanotubos y en b) que el mismo depósito se ha desprendido de los nanotubos y
éstos han perdido su forma tubular.
4. Conclusiones
Se probó la actividad catalítica de electrodos preparados a base de nanotubos de TiO2 y
dióxido de estaño frente a la decoloración de rojo de metilo y p-nitrofenol. Se observa
que, en general, con estos electrodos es factible alcanzar la decoloración completa de
rojo de metilo en un tiempo 3 h aproximadamente; asimismo, la oxidación completa de
p-nitrofenol también se alcanza en este tiempo aproximadamente. Sin embargo, la
oxidación de p-nitrofenol se dirige mayoritariamente a la formación de hidroquinona. De
las curvas cinéticas, se observa que la actividad de los electrodos de 2-Ti/Nts-TiO2/SnO2
es mayor a la actividad de los nanotubos descubiertos (2-Ti/Nts-TiO2), pues la constante
cinética para las oxidaciones con los electrodos de 2-Ti/Nts-TiO2/SnO2 es 16-20 veces
mayor que con los electrodos de 2-Ti/Nts-TiO2.
Por otro lado, de acuerdo a los análisis de HPLC en los que se detectan intermediarios
aromático (hidroquinona y benzoquinona) y ácidos alifáticos (ácido oxálico y maleico), se
propone la misma ruta de degradación de p-nitrofenol propuesta en el capítulo 5, la cual
involucra la formación de hidroquinona y benzoquinona y la formación de ácidos maleico
y oxálico a partir de ésta última.

118
Sin embargo, la estabilidad de estos electrodos es baja, ya que se observa, que los
electrodos pierden actividad después de unas horas de uso, lo que se explica debido a
cambios en la morfología del dióxido de estaño original, así como también a una pérdida
del óxido de estaño depositado de acuerdo a lo observado en la MEB. Lo anterior podría
atribuirse a una escasa penetración del SnO2 en los nanotubos debido a la volatilización
de la solución precursora sobre la superficie.

119
CONCLUSIONES GENERALES
Se prepararon electrodos de composición dual a base de óxidos metálicos
electrocatalíticos (PbO2 o SnO2) y nanotubos de dióxido de titanio, con el fin de observar
un efecto sinérgico entre el óxido metálico y el dióxido de titanio nanotubular en la
actividad electrocatalítica y/o fotoelectrocatalítica del nuevo material. De manera que, las
propiedades físicas y químicas del material dual superaran a las de los materiales
individuales.
En el caso de los electrodos de dióxido de plomo y nanotubos de TiO2 se demostró que,
la actividad electrocatalítica de estos electrodos frente a la oxidación en medio ácido de
dos tipos de compuestos orgánicos, un colorante (rojo de metilo) y un compuesto
biorrefractario modelo (p-nitrofenol), se incrementa sustancialmente cuando se usan
estos electrodos como ánodos. El aumento en la actividad electrocatalítica se atribuyó
principalmente a un aumento en el área superficial del dióxido de plomo. Con estos
electrodos, la oxidación de p-nitrofenol llegó a la conversión total a ácidos alifáticos y a
una mineralización parcial del 40%, mediante una ruta de oxidación parcial.
En lo que respecta a la actividad fotoelectrocatalítica de estos electrodos, se demostró
que existe un efecto sinérgico entre el dióxido de plomo y los nanotubos de TiO2 (de
primera generación), ya que se observó un incremento de casi el doble en la eficiencia
de fotoconversión en los electrodos con depósito de PbO2 de 5 s con respecto a la
eficiencia de fotoconversión de los nanotubos descubiertos. Este incremento de
eficiencia de fotoconversión es notorio al someter a los electrodos a la fotoelectrocatálisis
de rojo de metilo, con la cual se logra la decoloración total del rojo de metilo en el menor
tiempo.
En lo que se refiere a los electrodos de composición dual de dióxido de estaño y
nanotubos de TiO2, se demostró que, sobre estos electrodos, la oxidación
electrocatalítica de p-nitrofenol no es efectiva para su conversión a ácidos alifáticos.
Pues, aunque espectrofotométricamente, la oxidación total del p-nitrofenol se observa en
un tiempo aproximado de 3-3.5 h, su oxidación (seguida por HPLC) produce
mayoritariamente hidroquinona; lo cual implica una baja destoxificación de dicha
solución.

120
Por otro lado, los electrodos a base de Nts-TiO2 y PbO2 mostraron mayor estabilidad
frente a la oxidación en medio ácido que sus análogos de SnO2, los cuales se
desprenden de los nanotubos después de algunas horas de reacción. Lo que implica el
reto de mejorar la estabilidad de estos electrodos mediante la introducción de materiales
dopantes o variaciones en el método de depósito.
Finalmente, es muy importante tomar en cuenta las características iniciales de la placa
de titanio a partir de la cual se crecen los nanotubos de TiO2; ya que, como se ha
planteado en los capítulos 2 y 5, el crecimiento de los nanotubos se afecta notablemente
por diferencias en la superficie de titanio, diferencias que pueden ser estructurales y/o
morfológicas. Estas diferencias deberán considerarse cuidadosamente cuando se
investiga el desempeño de los nanotubos de TiO2.

121
REFERENCIAS
1. D. Gandini, E. Mahè, P.A. Michaud, W. Haenni, A. Perret, Ch. Comninellis. J. Appl.
Electrochem., 30 [12], 1345 (2000).
2. A. Da Pozzo, C. Merli, I. Sirés, J. Garrido, M. Rodríguez, E. Brillas. Environmental
Chemistry Letters. 3, 7 (2005).
3. K. Jüttner, U. Galla, H. Schmieder. Electrochim. Acta. 45, 2575 (2000).
4. A. M. Polcaro, S. Palmas, S. Dernini. Current Topics in Electrochemistry. 4, 137
(1997).
5. A. Vlyssides, E. M. Barampouti, S. Mai. Environ,Sci. Technol, 38, 6125 (2004).
6. B. Marselli, J. García-Gómez, P. Michaud, M. A. Rodrigo, Ch. Comninellis. J.
Electrochem. Soc. 150 [3] D79 (2003).
7. F. Bonfatti, S. Ferro, F. Lavezzo, M. Malacarne, G. Lodi, A. De Battisti,. J.
Electrochem. Soc., 146 [6], 2175 (1999).
8. V. Fisher, D. Gandini, S. Laufer, E. Blank, Ch. Comninellis. Electrochim. Acta. 44
[2-3], 521 (1998).
9. M. Fryda, D. Herrmann, L. Schafer, C.-P. Klages, A. Perrer, W. Haenni, Ch.
Comninellis, D. Gandini. New Diamond and Frontier Carbon Technol., 9 [3], 229-
240 (1999).
10. Ch. Comninellis. Electrochim. Acta. 39 [11-12], 1857 (1994).
11. A. De Battisti,, G. Battaglin, A. Benedetti, J. Kristof, J. Liszi. Chimia. 49, 17 (1995).
12. A. M. Polcaro, S. Palmas, F. Renoldi, M. Mascia. J. Appl. Electrochem. 29 [2], 147
(1999).
13. T. N. Belhadj, A. Savall. J. Electrochem. Soc., 145 [10], 3427 (1998).
14. J. Feng, L. Houk L., D. C. Johnson, S. N. Lowery, J. J. Carey. J. Electrochem. Soc.
142 [11], 3626 (1995).
15. G. Foty, D. Gandini, Ch. Comninellis. Curr. Top. Electrochem., 5, 71 (1997).
16. Ch. Comninellis, A. De Battisti. J. Chim. Phys., 93 [4], 673 (1996).
17. Ch. Comninellis, C. Pulgarin. J. Appl. Electrochem., 23 [2], 108 (1993).
18. M. Zhou, Q. Dai, L. Lei, C. Ma, D. Wang. Environ,Sci. Technol, 39, 363 (2005).
19. C.A, Martínez-Huitle, M. Cerro, S. Ferro. A. de Battisti, M.A. Quiroz. Can. J. Anal.
Sci. Spect., 52 [6], 335 (2007).

122
20. B. Correa-Lozano, Ch. Comninellis, A. De Battisti, J. Appl. Electrochem, 26, 683
(1996).
21. Ch. Comninellis, G.P. Vercesi, J. Appl. Electrochem. 21, 335 (1991).
22. J.L.G. Fierro (editor), Metal Oxides: Chemistry and Applications, CRC Taylor &
Francis, Berkely, California (2006).
23. Y.K. Lai, L. Sun, C. Chen, C.G. Nie, J.Zuo, C.J. Lin. Appl.Surf. Sci. 252 1101
(2005).
24. X. Quan, S. Yang, X. Ruan, H. Zhao. Environ. Sci. Technol. 39, 3770 (2005).
25. B. E. Hayden, D. V. Malevich, D. Pletcher. Electrochem. Commun. 3 390 (2001).
26. J. Wang, L. Zhao, V. S-Y. Lin. Z. Lin. J. Matter. Chem, 19 3682 (2009).
27. S. Li, G. Zhang, D. Guo, L. Yu, W. Zhang, J. Phys. Chem. C. 113, 12759 (2009).
28. J. Kunze, L. Müller, J. M. Macak, P. Greil, P. Schmuki, F. A. Muller. Electrochim.
Acta 53, 6995 (2008).
29. P. Li, G. Zhao, X. Cui, Y. Zhang, Y. Tang. J. Phys. Chem. C, 113, 2375 (2009).
30. G. Zhao, X. Cui, M. Liu, P. Li, Y. Zhang, T. Cao, H. Li, Y, Lei, L. Liu, D. Li.
Environ. Sci. Technol. 43, 1480 (2009).
31. G. Zhao; Y. Zhang. Environ. Sci. Technol., 44, 1754 (2010).
32. B. K. Vijayan, N. M. Dimitrijevic, J. Wu, K. A. Gray.J. Phys. Chem. C. 114, 21262
(2010).
33. Y. Hou, X. Li, Q. Zhao, X. Quan, G. Chen. Environ. Sci. Technol. 44, 5098 (2010).
34. Y. Hou, X. Li , X. Zou, X. Quan, G. Chen, Environ. Sci. Technol. 43, 858 (2009).
35. A. Ghicov, J. M. Macak, H.Tsuchiya, J. Kunze, V. Haublein, S. Kleber, P. Schmuki.
Chem. Phys. Lett, 419, 426 (2006).
36. P. Kamat. J. Phys. Chem. C, 116, 11849 (2012).
37. H. Zhang, X. Quan, S. Chen, H. Zhao, Environ. Sci. Technol. 40, 6104 (2006).
38. W. Sun, Y. Yu, H. Pan, X. Gao, Q. Chen, L. Peng. J. Am. Chem. Soc., 130, 1124
(2008).
39. Z. Liu, X. Zhang, S. Nishimoto, M. Jing, D. Tryk, T. Murakami, A. Fujishima. J.
Phys. Chem. C, 112, 253 (2008).
40. Z. Zhang, Y. Yuan, G. Shi, Y. Fang, L. Liang, H. Ding, L. Jin. Environ. Sci.
Technol., 41, 6259 (2007).
41. J. Macak, F. Schmidt-Stein, P. Schmuki. Electrochem. Commun. 9, 1783 (2007).

123
42. I. Paramasivam, J. Macak, P. Schmuki. Electrochem. Commun. 10, 71 (2008).
43. H. Yu, X. Wang, H. Sun, M. Huo. J. Hazard. Mater. 184, 753 (2010).
44. X. Zhao, H. Liu, J. Qu. Appl. Surf. Sci. 257, 4621 (2011).
45. H. Zhuang, C. Lin, Y. Lai, L. Sun, J. Li. Environ. Sci. Technol., 41, 4735 (2007).
46. Y. Jiang, Z. Hu, M. Zhou, L. Zhou, B. Xi. Sep. Purif. Technol., 128, 67 (2014).
47. J. Li, L. Zhen, L. Li, G. Shi, Y. Xian, L. Jin, Electroanal. 18, 2251 (2006).
48. M. Zlamal, J. Macak, P. Schmuki, J. Krýsa. Electrochem. Commun., 9, 2822
(2007).
49. T. Jeon, W. Choi, H. Park. J. Phys. Chem. C, 115, 7134 (2011).
50. Y. Zhang, G. Zhao, Y. Lei, P. Li, M. Li, Y. Jin, B. Lv. ChemPhysChem. 11, 3491
(2010).
51. X. Chen, S.S. Mao. Chem. Rev. 107, 2891 (2007).
52. J. M. Macak, P. Schmuki, Electrochim. Acta, 52, 1258 (2006).
53. Y. Li, X. Yu, Q. Yang. Journal of Sensors, Volume 2009, pag.1-19 (2009).
Disponible en http://dx.doi.org/10.1155/2009/402174
54. J.M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer, P. Schmuki.
Curr. Opin. Solid St. M., 11, 3 (2007).
55. C. Cao, G. Zhang, X. Song, Z.Sun. Nanoscale Res. Lett., 6, 64 (2011).
56. J.M. Macak, H. Hildebrand, U. Marten-Jahns, P. Schmuki. J. Electroanal.Chem.,
621, 254 (2008).
57. J.F. Banfield y D.R. Veblen. Am. Mineral., 77, 545 (1992).
58. H. Zhang, J. F. Banfield. J. Mater. Chem. 8 [9], 2073 (1998).
59. K. M. Reddy, S. V. Manoramaa, A. R.Reddy. Materials Physics and Chemistry. 78,
239 (2002).
60. A. L. Linsbigler, G. Lu, J.T. Yates, Jr. Chem. Rev., 95, 735 (1996).
61. N. Satoh, T. Nakashima, K. Yamamoto en Scientific Reports, Publicado el 7 de
Junio de 2013, 3:1959, Doi: 10.1038/srep01959. Disponible en
www.nature.com/scientificreports.
62. S.B. Aldabergenova, A. Ghicov, S. Albu, J.M. Macak, P. Schmuki. Journal of Non-
Crystalline Solids, 354, 2190 (2008).
63. Z. Poh, K. Vasilev, K. Kant, R. Emamali Sabzi, D. Losic.(Ian Wark Research
Institute, University of South Australia, SA 5095, Australia).
64. C. A. Grimes. J. Mater. Chem. 17, 1451 (2007).

124
65. D. Wang, Y. Liu, B. Yu, F. Zhou, W. Liu. Chem. Mater, 21, 1198 (2009).
66. R. Inguanta, S. Piazza, C.Sunseri. J. Electrochem. Soc, 155 [12], K205 (2008).
67. A.J. Saterlay, S. J. Wilkins, K. B. Holt, J. S. Foord; R. G. Compton, F. Marken. J.
Electrochem. Soc., 148 [2], E66 (2001).
68. C. A. Martinez-Huitle, M. A. Quiroz, C. Comninellis, S. Ferro, A. De Battisti.
Electrochim. Acta. 50, 949 (2004).
69. M.Ueda, A. Watanabe, T. Kameyama, Y. Matsumoto, M. Sekimoto y T.
Shimamune, J. Appl. Electrochem., 25, 817 (1995).
70. A. B. Velichenko, D. V. Girenko, F.I. Danilov, J. Electroanal. Chem. 405, 127
(1996).
71. A.B. Velichenko, R. Amadelli, A. Benedetti, D.V. Girenko, S.V. Kovalyov and F.I.
Danilov, J. Electrochem. Soc., 149 C445 (2002).
72. J. González-García, J. Iniesta, E. Expósito, V. García-García, V. Montiel and A.
Aldaz, Thin Solid Films, 352, 49, (1999).
73. N. Vatistas, S. Cristofaro. Electrochem. Commun. 2, 334 (2000).
74. F. Hine, M. Yasuda, T. Iida, Y. Ogata and K. Hara. Electrochim. Acta, 29, 1447
(1984).
75. Y. Mohd, D. Pletcher. Electrochim. Acta. 52, 786 (2006).
76. Y. Cong, Z. Wu. J. Phys. Chem. C. 111, 3442 (2007).
77. Y. Liu, H. Liu. Electrochim. Acta, 53, 5077 (2008).
78. A.B. Velichenko, R. Amadelli, E.A. Baranova, D.V. Girenko, F.I. Danilov. J.
Electroanal. Chem. 527, 56 (2002).
79. Yun-Hai, Wang, Qing-Yun, Chen, Li Guo, Xiang-Lin Li (2012). Anodic Materials
with High Energy Efficiency for Electrochemical Oxidation of Toxic Organics in
Waste Water, Industrial Waste, Prof. Kuan-Yeow Show.(Ed.), ISBN: 978-953-51-
0253-3, InTech, Disponible en:
http://www.intechopen.com/books/industrialwaste/anodic-materials-with-high-
energy-efficiency-for-electrochemical-oxidation-of-toxic-organics-in-wast
80. A.B. Velichenko, R. Amadelli, G.L. Zucchini, D.V. Girenko, F.I. Danilov.
Electrochim. Acta 45 4341 (2000).
81. S. Cattarin, I. Frateur, P. Guerriero, M. Musiani. Electrochim. Acta. 45, 2279
(2000).
82. S. Cattarin, P. Guerrero, M. Musiani. Electrochim. Acta 46, 4229 (2001).

125
83. J. P. Carr y N. A. Hampson. Chem. Rev., 72[6] 679 (1972).
84. P. Rüetschi. J. Electrochem. Soc. 139, 1347 (1992).
85. W. Mindt. J. Electrochem. Soc. 116, 1076 (1969).
86. X. Li, D. Pletcher, F. C. Walsh. Chem. Soc. Rev. 40, 3879 (2011).
87. K. Anandan, V. Rajendran. Journal of Non-Oxide Glasses. 2[2], 83 (2010).
88. K. Kolentsov, L. Yourukova, A. Zheliaskova, A. Rachkova. Bulg. J. Phys. 31, 87
(2004).
89. Muhammad Akhyar Farrukh, Heng Boon Teck, Rohana Adnana. Turk. J. Chem
34[4], 537 (2010).
90. D. Chu, Y. Masuda, T. Ohji, K. Kato. Chem. Eng. J., 168, 955 (2011).
91. A. A. Mosquera Lozano, J. Arana Varela, J. E. Rodríguez Páez, Rev.fac.ing.univ.
Antioquia, Medellín. Jan./Mar. 39, 33 (2007).
92. Ali Hassanzadeh, Behrang Moazzes, Hossein Haghgooie, Mohesen Nasseri, Mir
Maqsood Golzan, Hassan Sedghi, Cent. Eur. J. Chem. 6[4], 651 (2008).
93. B. Correa-Lozano, Ch. Comninellis. A. de. Battisti. J. Appl. Electrochem, 26, 683-
688, (1996).
94. A. M. Polcaro, S. Palmas. S. Dernini. Curr. Top. Electrochem.. 4. 137 (1997).
95. P. Li, G. Zhao, X. Cui, Y. Zhang, Y. Tang. J. Phys. Chem. C. 113, 2375 (2009).
96. S. Chai, G. Zhao, P. Li, Y. Lei, Y. Zhang, D. Li. J. Phys. Chem. C, 115, 18261
(2011).
97. M. Batzil, U. Diebold, Prog. Surf. Sci.79, 47 (2005).
98. R. Rivera, F. Marcillo, W. Chamba, P. Puchaicela, A. Stashans. SnO2 Physical and
Chemical Properties due to the Impurity Doping. Proceedings of the International
Multiconference of Engineers and Computer Scientists. Vol II.IMECS 2013. March
13-15, 2013. Hong Kong (2013).
99. A.R. Babar, S.S. Shinde, A. V. Moholkar. C. H. Bhosale, J. H. Kim and K. Y.
Rajpure. Journal of Semiconductors. Vol. 32. No. 5 (2011).
100. G. E. Patil, D. D. Kajale, D. N. Chavan, N. K. Pawar, P. T. Ahire, S. D.
Shinde, V. B. Gaikwad and G. H. Jain, Bull. Mater. Sci. Indian Academy of
Sciences., 34[1], 1 (2011).
101. Mahesh Bhagwat, Pallavi Shah, Veda Ramaswamy. Mater. Lett. 57, 1604
(2003).

126
102. C. E. Ararat-Ibarguen, A. Montenegro y J. E. Rodríguez-Páez. Quim. Nova,
30[7], 1578 (2007).
103. D. Perednis, L. J. Gauckler, Journal of Electroceramics, 14, 103 (2005)
104. Hongwang Zhang, Aerosol Spray Pyrolysis and Solution Phase Synthesis
of Nanostructures. ProQuest Information and Learning Company, Ann arbor, MI,
2008.
105. J. Hirunlabh, S. Suthateeranet, K. Kirtikara and Ralph D. Pynn. Thammasat
Int. J. Sc. Tech., 3[2], 10 (1998).
106. A.I. Bárcena Millán. Síntesis y Caracterización de Partículas
Nanoestructuradas del sistema Gd2‐xEuxO3 con un porcentaje atómico de 1% en
europio y del sistema Gd2O3. Tesis de Licenciatura. Universidad Carlos III de
Madrid. Escuela Politécnica Superior, Abril 2010. 109 pp.
107. A. Ghicov, H. Tsuchiya, R.Hahn, J. M. Macak, A. G. Muñoz, P. Schmuki.
Electrochem. Commun. 8, 528 (2006).
108. A.B. Murphy, Sol. Energ. Mat. Sol.C., 91, 1326 (2007).
109. Shimadzu Application News, Spectrophotometric Analysis No.A428,
Measurements of Band Gap in Compound Semiconductors - Band Gap
Determination from Diffuse Reflectance Spectra.
110. G.R. Dale, J.W. J. Hamilton, P.S. M. Dunlop, P.Lemoine, J.A. Byrne..
Journal of Nanoscience and Nanotechnology. 9, 4215 (2009).
111. L. Zhang, Y. Han. Nanotechnology 21, 8 (2010).
112. S. Cheng, K. Chan. Electrochem. Solid. St., 7[3] D4 (2004).
113. P. Roy, S. Berger, P. Schmuki. Angew. Chem. Int. Ed. 50, 2904 (2011).
114. S. H. Kang, J. Kim, H.S. Kim, Y. Sung, J. Ind. Eng. Chem. 14, 52 (2008).
115. S. Leonardi, A. Li Bassi, V. Ruso, F. Di Fonzo, O. Paschos, T. M. Murray,
H. Efstathiadis, J. Kunze. J. Phys. Chem. C. 116, 384 (2012).
116. A. G. Kontos, A. I. Kontos, D. S. Tsoukleris, V. Likodimos, J. Kunze, P.
Schmuki, and P. Falaras. Nanotechnology, 20, 045603 (2009).
117. J.L. Nava, M.A. Quiroz, C.A. Martínez-Huitle, J. Mex. Chem. Soc. 52[4],
249 (2008).
118. S. Trasatti, O.A. Petri. Real surface Area Measurements in
Electrochemistry. Pure & Appl. Chem. 63[5], 711 (1991).

127
119. B. S. Tosun, R. K. Feist, A. Gunawan, K. A. Mkhoyan, S. A. Campbell, E. S.
Aydill. Thin Solid Films, 520, 255 (2012).
120. R. Kumar, I. Verma, N. Verma. International Journal of Electronics and
Computer Science Engineering, 1[2] 1422, ISSN- 2277-1956. Disponible en línea
en www.ijecse.org
121. Shahed U.M Khan, Science, 297, 2243 (2002).
122. M. Cerro-Lopez, Y. Meas-Vong, M.A. Méndez-Rojas, C.A. Martínez-Huitle, M.A. Quiroz. Appl.Cat. B: Environ., 144, 174 (2014).
123. F. Bonfatti, S. Ferro, F. Lavezzo, M. Malacarne, G. Lodi, A. De Battisti, J. Electrochem. Soc. 147, 592 (2000).
124. C.A. Martinez-Huitle, S. Ferro, A. De Battisti, Electrochim. Acta 49, 4027 (2004).
125. S. Ferro, C.A. Martínez-Huitle, A. De Battisti, J. Appl. Electrochem. 40,1779 (2010).
126. M. Panizza y G. Cerisola. Ind. Eng. Chem. Res. 47, 6816 (2008).
127. E. Guivarch, S.Trevin, C. Lahitte, M. A. Oturan. Environ. Chem Lett 1, 38
(2003).
128. R. A. Torres, W.Torres, P. Peringer, C. Pulgarin, Chemosphere 50, 97
(2003).
129. M. Zhou, Q. Dai, L. Lei, C. Ma , y D. Wang. Environ. Sci . Technol . 39,
363 (2005).
130. C. Wu, J. Chern, Ind. Eng. Chem. Res., 45, 6450 (2006).
131. N. Vatistas, J Appl. Electrochem. 40,1743 (2010).

128
ANEXO
Lista de Equipos
1. Potenciostato-Galvanostato, modelo PJT24-1 (Tacussel Electronique; Lyon,
Francia)
2. Potenciostato de Barrido modelo 362, (EG&G Princeton Applied Research, Oak
Ridge, TN) equipado con un graficador Graphtec X-Y modelo WX2400
(Yokohama, Japón)
3. Potenciostato-Galvanostato AUTOLAB, modelo PGSTAT 30 (Utretch, Holanda)
4. Fuente de poder, GW Instek modelo GPC-3030D (Chino, CA, EUA)
5. Microscopio electrónico de barrido, TESCAN modelo VEGA LSU (Kohoutovice,
República Checa) con voltaje máximo de aceleración de 30 KV acoplado a un
Detector de Análisis Elemental de Espectroscopía de Dispersión de Energía de
Rayos X (EDAX), Oxford Instruments Ltd (Oxfordshire, Inglaterra)
6. Espectrofotómetro UV-vis Varian modelo Cary 50 (Palo Alto, CA, EUA) con esfera
de integración para espectrofotometría de reflectancia difusa.
7. Espectrofotómetro UV-vis HACH (Loveland, CO, EUA)
8. Cromatógrafo de Líquidos de Alta Resolución, Waters modelo 1515 (Milford, MA,
EUA)
9. Difractómetro de Rayos X, Bruker Axs D8 Advance (Billerica, MA, EUA) , con
fuente de radiación Cu Kα.(1.54Å)
10. Lámpara de longitud de onda larga de 100 W, Blak-Ray modelo B 100AP (Upland,
CA, EUA) para los experimentos 4W/cm2 de intensidad.
11. Lámpara de longitud de onda larga de 6 W, Spectroline ENF-260C 6W (Westbury,
NY, EUA) para los experimentos de 1.4 mW/cm2 de intensidad.


















