
Proyecto Genoma Humano - bioinformatica.uab.esbioinformatica.uab.es/base/documents\masterGP\Proyecto...
20
1 Proyecto Genoma Humano 2. El paisaje genómico global 1 Contenido en CG 2 Islas CpG 3 Tasas de recombinación 4 Secuencias repetidas 5 Contenido en genes 6 El Proyecto ENCODE Referencias: Lander et al. 2001, Nature 409:860- 891. The Encode Project Consortium 2007, Nature 447: 799-816. Range: 31-65% (20-kb windows) Mean: 41% SD: 5.2%
Transcript of Proyecto Genoma Humano - bioinformatica.uab.esbioinformatica.uab.es/base/documents\masterGP\Proyecto...

1
Proyecto Genoma Humano2. El paisaje genómico global
1 Contenido en CG2 Islas CpG3 Tasas de recombinación4 Secuencias repetidas5 Contenido en genes6 El Proyecto ENCODE
Referencias: Lander et al. 2001, Nature 409:860-891. The Encode Project Consortium 2007, Nature 447: 799-816.
Range: 31-65% (20-kb windows)
Mean: 41%
SD: 5.2%
2
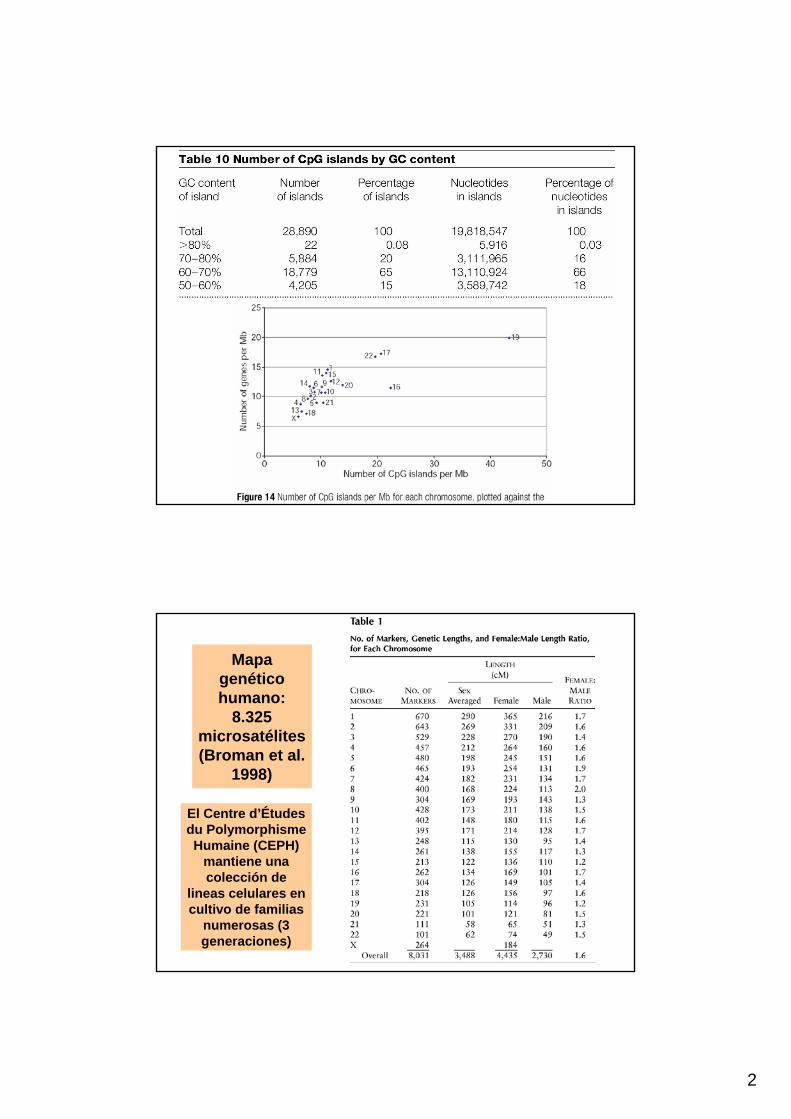
Mapagenético humano:
8.325 microsatélites(Broman et al.
1998)
El Centre d’Étudesdu PolymorphismeHumaine (CEPH)
mantiene una colección de
lineas celulares en cultivo de familias
numerosas (3 generaciones)

3

4
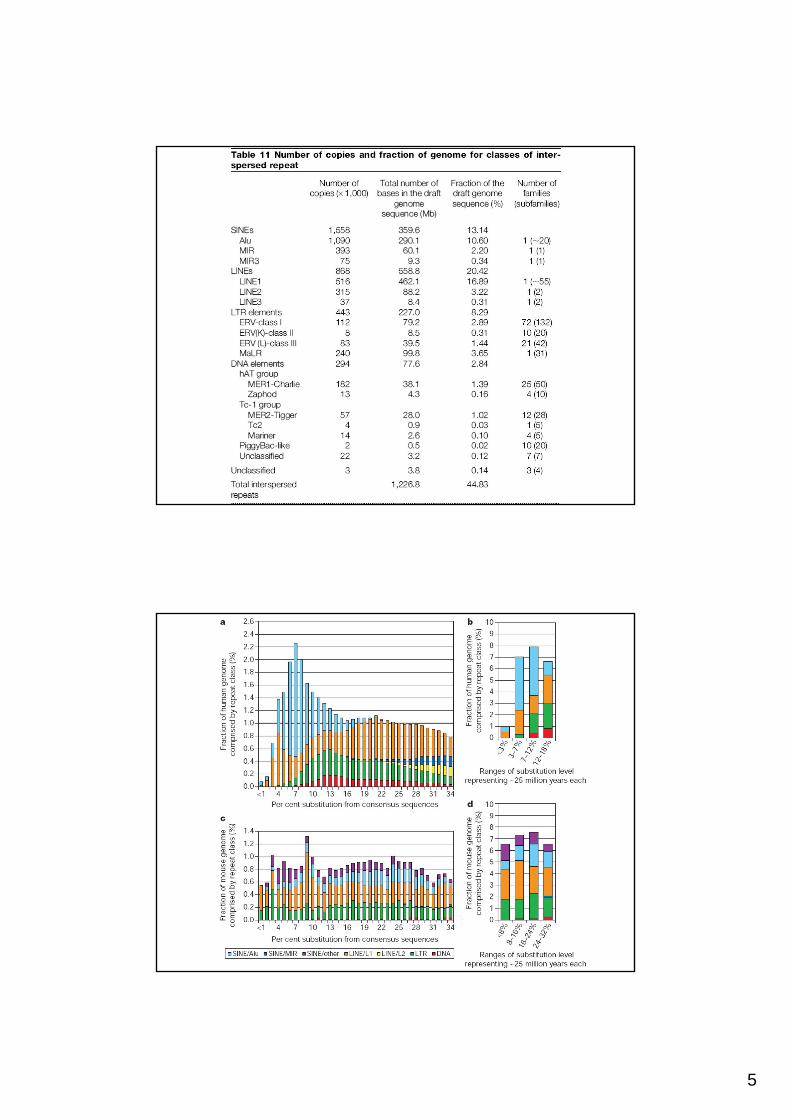
DNA repetitivo en el genoma humanoDNA repetitivo en el genoma humano
i. Repeticiones derivadas de elementos transponibles (repeticiones dispersas)
ii. Pseudogenes procesados (copias inactivas de genes celulares producto de retrotranscripción)
iii. Microsatélites o repeticiones de secuencia simple como (A)n, (CA)n o (CGG)n
iv. Duplicaciones segmentales (10-300 kb)
v. Grandes bloques de secuencias altamente repetidas en tándem (centrómeros y telómeros)
Elementos transponibles en el genoma humano
80-3000 bp
100-300 bp
5

6
Contribución de los elementos transponibles al genoma de diversas especies
Elementos H. sapiens D. melanogaster C. elegans A. thaliana
LINE/SINE 33.40% 0.70% 0.40% 0.50%
LTR 8.10% 1.50% 0.00% 4.80%
DNA 2.80% 0.70% 5.30% 5.10%
Total 44.40% 3.10% 6.50% 10.50%
Mutaciones causadas por la inserción de elementos transponibles
• En Drosophila, ~50% de las mutacionesmorfológicas clásicas (por ejemplo, white) están causadas por la inserción de elementos transponibles.
• En el raton, 10% de las mutacionesespóntáneas están causadas portransposiciones.
• En humanos, 1/600 de las mutacionesespontáneas están causadas por la inserciónde elementos transponibles.

7
DistribuciDistribucióón de los elementosn de los elementos
• La región con más alta densidad de TEs (89%) esuna región de 525 kb en el cromosoma Xp11. Contiene un segmento de 200 kb con una densidadde TEs de 98%.

8

9

10
Genes que cifran RNA no Genes que cifran RNA no codificantecodificante en el genoma humano.en el genoma humano.
Genes deRNA
Número esperado
Número Observado
Genes relacionados (pseudogenes, fragmentos,
parálogos)
tRNA 1.310 497 324
18S rRNA 150-200 0 40
5.8S rRNA 150-200 1 11
28S rRNA 150-200 0 181
5S rRNA 200-300 4 520
snoRNA 97 84 645
snRNA (U1-U12) ?? 78 1542
7SL RNA 4 3 773
Características de los genes humanos que codifican proteínas(10.272 mRNAS en la base de datos RefSeq alineados con el borrador del
genoma, Lander et al. 2001)
Característica Mediana Promedio Tamaño muestra
Tamaño exones 122 bp 145 bp 43.317
Número exones 7 8.8 3.501
Tamaño intrones 1.023 bp 3.365 bp 27.238
3’ UTR 400 bp 770 bp 689 (crom. 22)
5’ UTR 240 300 463 (crom. 22)
Secuencia codificadora 1.100 bp 1.340 bp 1.804
(CDS) 367 aa 447 aa
Extensión genómica 14 kb 27 kb 1.804

11
Comparación de los genes humanos con los de Caenorhabditis y Drosophila
Correlación entre la densidad génica y el contenido en CG

12
•Programas informáticos diseñados para la predicción ab initio de exones e intrones.
• Similaridad de la secuencia del DNA o de la proteína con la de otros organismos (por ej. el ratón o la rata).
• Evidencia directa de transcripción proporcionada por mRNAs o ETS.
Estrategias para la identificaciEstrategias para la identificacióón de genesn de genes
Genes detectados por el Proyecto Genoma Humano Genes detectados por el Proyecto Genoma Humano (IGI/IPI ver 1)(IGI/IPI ver 1)
MétodoNúmero
de genesLongitud
promedio (aa)
Genes conocidos (RefSeq/SwissProt/TrEMBL)
14.882 469
Ensembl (Genscan + similaridad con prot, EST y mRNA de cualquier organismo) + Genie
4.057 443
Ensembl 12.839 187
Total 31.778 352

13
Evaluación de IGI/IPI• IGI contains 19/31 (61%) novel genes in the human
genome.• 81% of 15.294 mouse cDNAS showed similarity to the
human genome seq whereas only 69% showed similarity to IGI suggesting a sensitivity 69/81=85%.
• 477 IGI gene predictions were compared to 539 confirmed genes and 133 pseudogenes on chr. 22. 43 (9%) hit 34 pseudogenes (fragmentation rate=1.2). 63 (13%) did not overlap any current annotation.
• Assuming a rate of overprediction of 20% and a rate of fragmentation of 1.4, the IGI would be estimated to contain 24.500 actual human genes.
• Assuming that the gene predictions contains only 60% of previously unknown human genes, the total number of genes in the human genome would be 31.000.
Estimas del nEstimas del núúmero de genes existentes en el genoma humano (2001)mero de genes existentes en el genoma humano (2001)
Método Estima Autores
Cinética de reasociación 40.000 Lewin 1980
Tamaño del genoma/tamaño del gen 100.000 W. Gilbert (Lewin 1990)
Número de islas CpG 70.000-80.000
Antequera and Bird 1993
Número de EST 35.000-120.000
Ewing and Green 2000; Liang et al. 2000
Asociación de EST e islas CpG 142.000 Dickson 1999
Comparación del genoma humano con el de Tetraodon nigroviridis
30.000 Roest Crollius et al. 2000
Extrapolación de los cromosomas 21 y 22
30.500-35.000
Dunham 2000
Análisis inicial del genoma humano 32.000 Lander et al. 2001
Análisis inicial del genoma humano 38.000 Venter et al. 2001

14
La paradoja del valor NLa paradoja del valor N¿Puede la complejidad del proteoma humano explicar la complejidad fenotípica de los vertebrados?
Jean Michael Claverie (Science, 2001) ha definido la complejidad de un organismo como el número de transcriptomas distintos que teóricamente es capaz de generar.
Si cada gen puede tener dos estados (ON y OFF), un genoma con N genes puede dar lugar a 2N diferentes transcriptomas. Por lo tanto el aumento de complejidad que se puede conseguir al pasar de 20.000 a 30.000 genes es igual a
2 30.000/2 20.000 = 2 10.000 = 10 3.000
una cantidad astronómica (mayor que el número de átomos en el universo).
Genoma Humano (versión 21-Oct-2004)• Se han conseguido secuenciar 2.850 Mb (99%
de la eucromatina).• La tasa de error es 1/100.000 bases (Q = 50).• Se ha reducido el número de “gaps” (huecos)
de ~150.000 a sólo 341.• De ellos, 33 (total ~198 Mb) en la
heterocromatina y 308 (total ~28 Mb) en la eucromatina.
• Tamaño total: 2.850 + 198 + 28 = 3.080 Mb.• Número de genes: 20.000-25.000 (19.600 genes
conocidos + 2.200 predicciones).• Pseudogenes: ~20.000.

15
El proyecto ENCODE• El proyecto ENCODE (Encyclopedia od
DNA elements) pretende identificar TODOS los elementos funcionales de la secuencia del genoma humano.
• Fase piloto. Análisis detallado de 44 regiones discretas repartidas por todo el genoma que suman ~30 Mb (~1%).
• Fase de desarrollo tecnológico.• Fase de producción. Aplicación de las
técnicas desarrolladas en la fase anterior al restante 99% del genoma.

16
El proyecto ENCODE
Resultados iniciales del proyectoENCODE (Junio 2007)
• La mayor parte del genoma humano se transcribe.• Se han identificado muchos transcritos nuevos que
no codifican proteínas.• Se han identificado numerosos sitios de inicio de la
transcripción nuevos. • Se estimado que ~ 5% de las bases están
evolutivamente constreñidas en los mamíferos. Unatercera parte de ellas codifican proteínas. Un 40% no están anotadas.
• Hay una correlación general entre regiones bajoconstreñimiento evolutivo y regiones funcionales(experimentalmente determinadas) pero no muy alta. Algunos elementos funcionales parecen no estarconstreñidos.

17
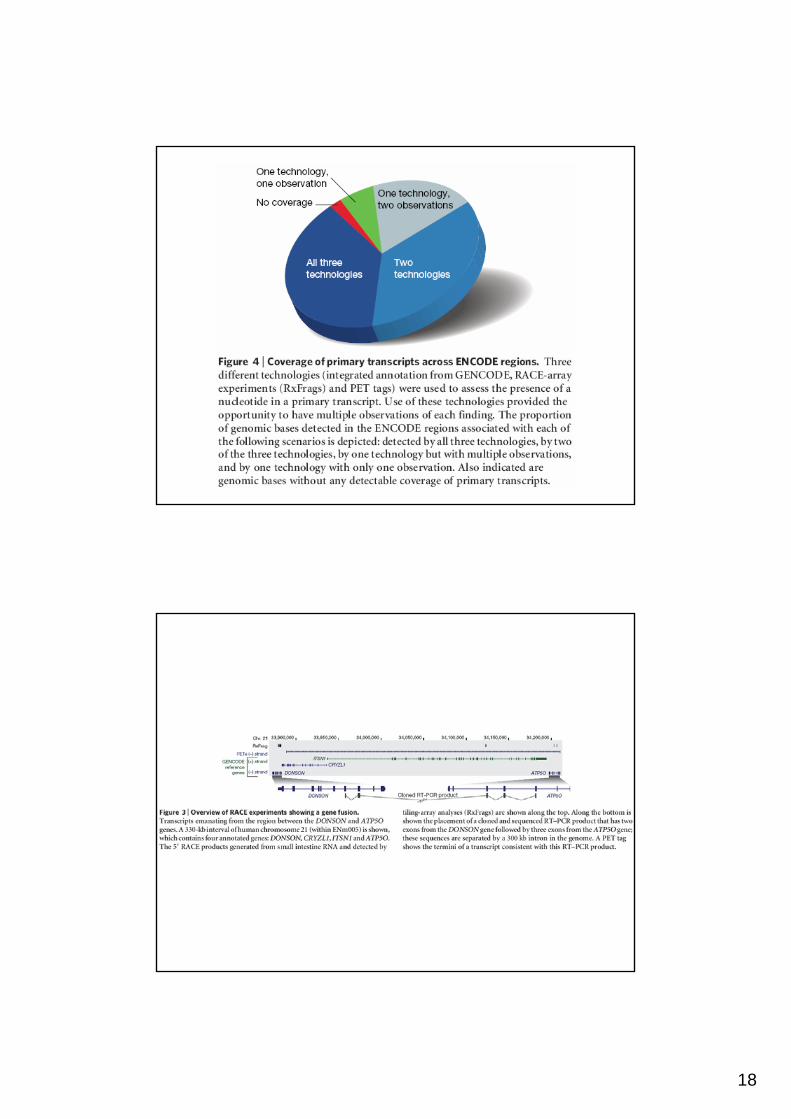
GENCODE = Integrated annotation of existinf cDNA and proteinresources to define transcrips.
TxFrag = Fragment of a transcript; a genomic region found to be present in a transcript by an unbiased tiling-array essay.
CODE and PET tags = short sequences from the 5’ end or containingboth the 5’ and 3’ ends of a transcript.
18

19

20


















