
Proteinas l
19
SEMINARIO DE PROTEINAS HEVER ALEJO VALVERDE LIZETH VALVERDE LOPEZ
-
Upload
hever-alejito -
Category
Health & Medicine
-
view
1.042 -
download
0
Transcript of Proteinas l

SEMINARIO DE PROTEINAS
HEVER ALEJO VALVERDE
LIZETH VALVERDE LOPEZ
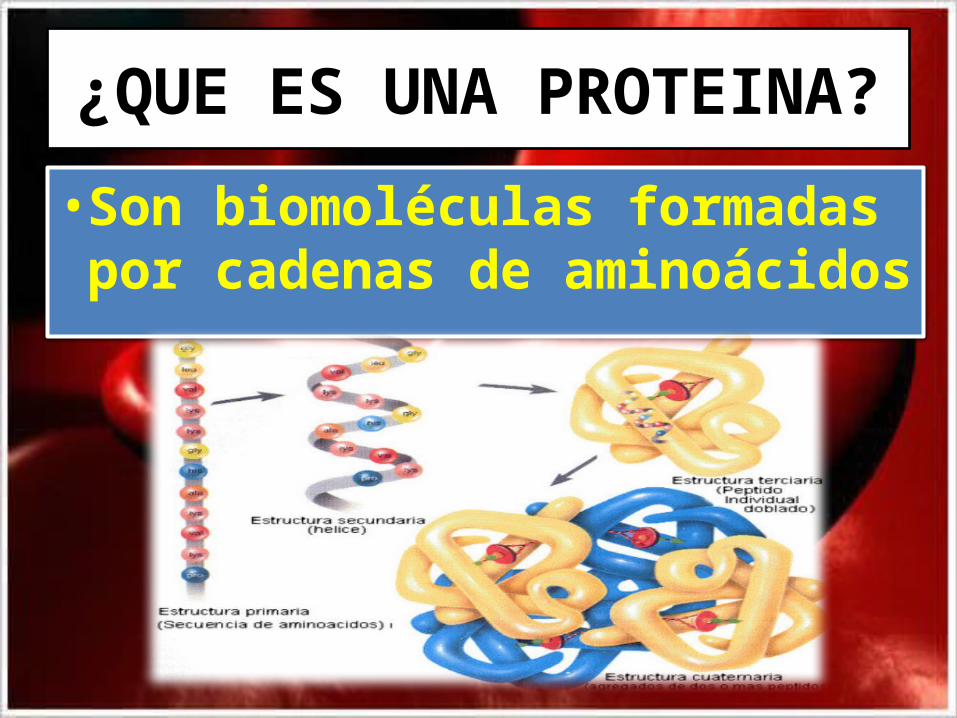
¿QUE ES UNA PROTEINA?
• Son biomoléculas formadas por cadenas de aminoácidos
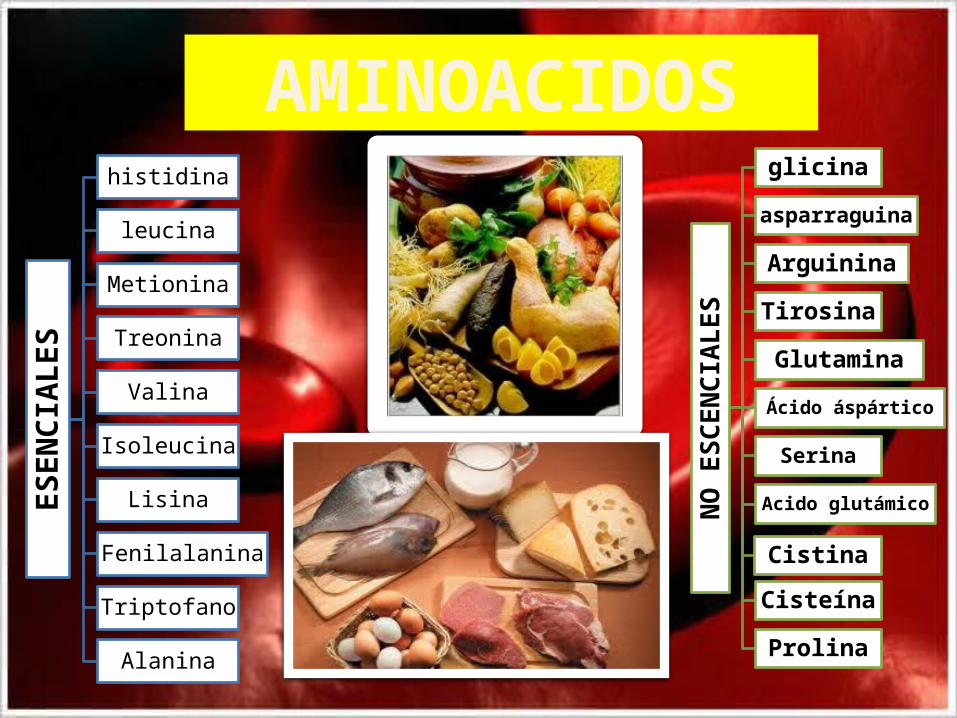
ES
EN
CIA
LES
histidina
leucina
Metionina
Treonina
Valina
Isoleucina
Lisina
Fenilalanina
Triptofano
Alanina
AMINOACIDOS
NO
ES
CEN
CIA
LES
glicina
asparraguina
Arguinina
Tirosina
Glutamina
Ácido áspártico
Serina
Acido glutámico
Cistina
Cisteína
Prolina
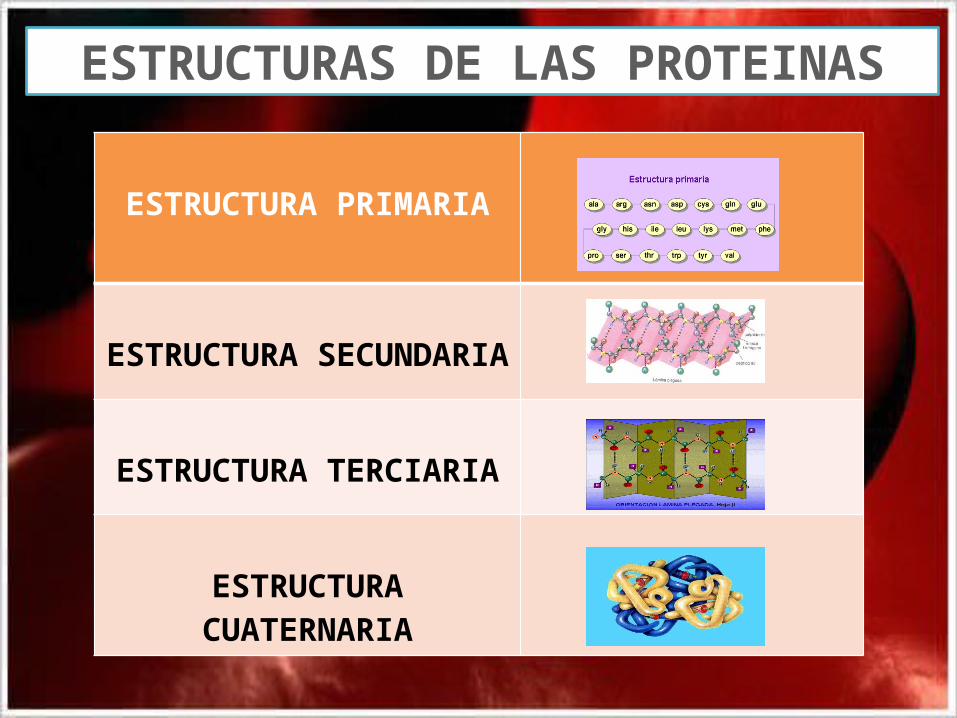
ESTRUCTURA PRIMARIA
ESTRUCTURA SECUNDARIA
ESTRUCTURA TERCIARIA
ESTRUCTURA CUATERNARIA
ESTRUCTURAS DE LAS PROTEINAS

FUNCIONESFunción Estructural
Función Enzimática
Función Hormonal
Función Reguladora
Función Homeostática
Función de Transporte(Hemoglobina)
Función Contráctil
Función de Reserva

¿Qué es la Anemia? ¿Cuáles son los Síntomas de la Anemia?
¿Qué causa la Anemia? Tratamiento de la Anemia
ANEMIA

Existen varios tipos de anemia, cada uno con una causa y tratamiento específico, incluyendo los siguientes: • Anemia ferropénica. • Anemia megaloblástica (perniciosa). • Anemia por deficiencia de folato. • Anemia hemolítica. • Anemia drepanocítica o de células
falciformes. • Anemia de Cooley (beta talasemia). • Anemia alfa talasemia • Anemia aplásica. • Anemia crónica.
TIPOS DE ANEMIA

• La talasemia es un grupo de anemias hemolíticas hereditarias en las que existe disminución de la síntesis de una o más de las cadenas polipeptídicas de la hemoglobina. Hay varios tipos genéticos con cuadros clínicos que van desde anomalías hematológicas difícilmente detectables hasta anemia severa y fatal.
Herencia mendeliana autosómica recesiva: dos mutaciones de línea germinal (una de cada uno de los padres) para desarrollar la enfermedad; igualmente transmitida por hombres y mujeres
TALASEMIA

Estructura de la proteína Hemoglobina.
• La Hemoglobina (Hb) es una hemoproteína de estructura cuaternaria, constituida por cuatro cadenas polipeptídicas denominadas alfa(a), beta (b), gamma (g) y delta (d); las cuales difieren en la secuencia de aminoácidos que la conforman. Las cadenas polipeptídicas a tienen 141 aminoácidos en su estructura primaria, mientras que las , b, g d contienen 146 aminoácidos(Fig.1).
• Cada una de las cuatro cadenas polipeptídicas de la Hb, contiene un grupo prostético Hem, que es un tetrapirrol cíclico formado por un átomo de Hierro y un anillo de porfirina. Este grupo Hem posee una estructura planar con propiedades apolares que lo hacen insoluble en agua (fig.2).

• En la parte central de este grupo Hem se encuentra un átomo de Hierro en estado de oxidación ferroso (+2) que puede formar 5 ó 6 enlaces de coordinación dependiendo de la unión del oxígeno a la Hb (oxiHb, desoxiHb), cuatro de estos enlaces se forman entre las cadenas polipeptídicas, mientras que los otros se realizan con la histidina distal y el oxígeno . Debido a que cada cadena posee un grupo prostético Hem, hay 4 átomos de Hierro en cada molécula de Hb, pudiendo así transportar un total de 4 moléculas de oxígeno cada molécula de hemoglobina (Fig.3).
• Debido al carácter apolar y a su superficie planar, el grupo Hem, se localizará entre las estructuras terciarias de las cadenas polipeptídicas en una especie de “bolsillo” hidrofóbico, que se formará en cada una de ellas.
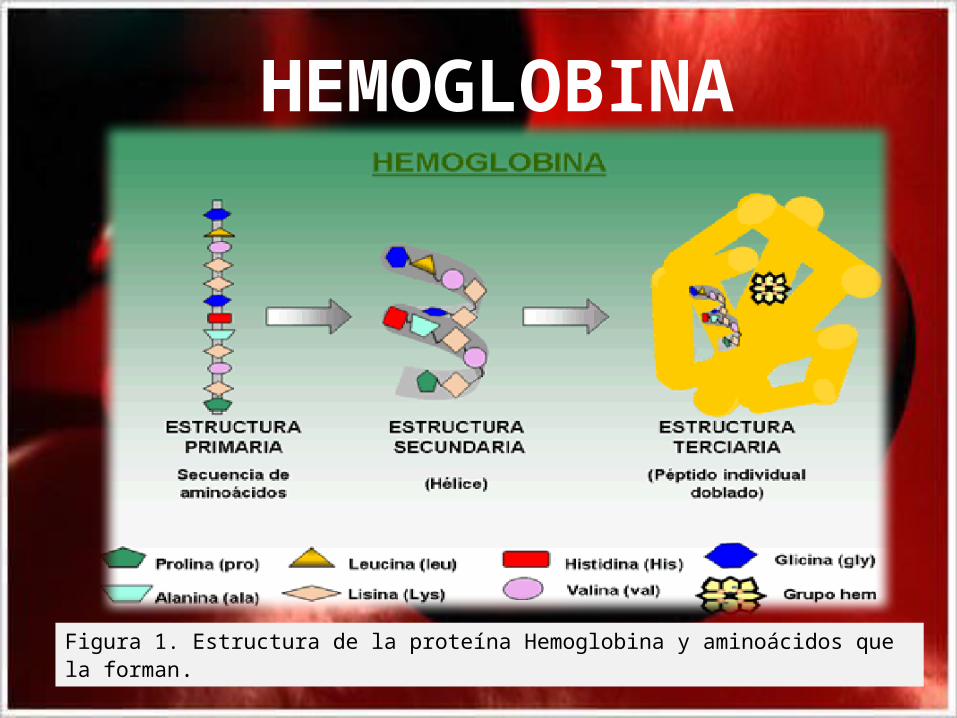
Figura 1. Estructura de la proteína Hemoglobina y aminoácidos que la forman.
HEMOGLOBINA

Alteraciones de la proteína Hb.
• Todos los genes funcionales de la Hb comparten una estructura general que consiste en 3 exones (secuencias codificadoras) y dos intrones o sectores interpuestos (secuencias que no se traducen). La causa mas común de hemoglobinopatías es la mutación puntual , es decir, la sustitución de un nucleótido de DNA por otro, lo que modifica el código genético y puede inducir un cambio en una aminoácido de la proteína resultante.
• Este es el caso de la Anemia Falciforme, donde un cambio en el codón GAC normal, a GTG produce un cambio de un aminoácido por otro(Fig.4), resultando la Hemoglobina S.

• La Hemoglobina S, se debe a una alteración del gen que codifica a la cadena b de la hemoglobina. De las cuatro cadenas polipeptídicas que forman la hemoglobina (dos a y dos b), las a son idénticas a la de la hemoglobina normal (HbA), pero existe una alteración en las cadenas b. Estas tienen un aminoácido valina en el lugar que normalmente ocuparía un ácido glutámico, en el codón numero 6 de la subunidad polipeptídica b, en el cromosoma 11.
• El reemplazo de estos aminoácidos crean un punto de contacto hidrofóbico adhesivo en la posición 6 de la cadena β globina normal. Estos puntos de adherencia provocan que las moléculas de desoxihemoglobina S se asocien anormalmente entre ellas, formando largos agregados fibrosos propios de la enfermedad, causa de que la Hb S al desoxigenarse, pasa a un estado insoluble, provocando la vaso oclusión de vasos sanguíneos(Fig.5). Esta es la razón de la deformación de los eritrocitos en forma de hoz (Fig.6).

Figura 5. Sustitución de aminoácidos (Glu porVal), que provocan la no solubilidad de la HbSal desoxigenarse, dándole al glóbulo rojo una estructura rígida y en forma de hoz que por consecuente causa la vaso oclusión de capilaressanguíneos.
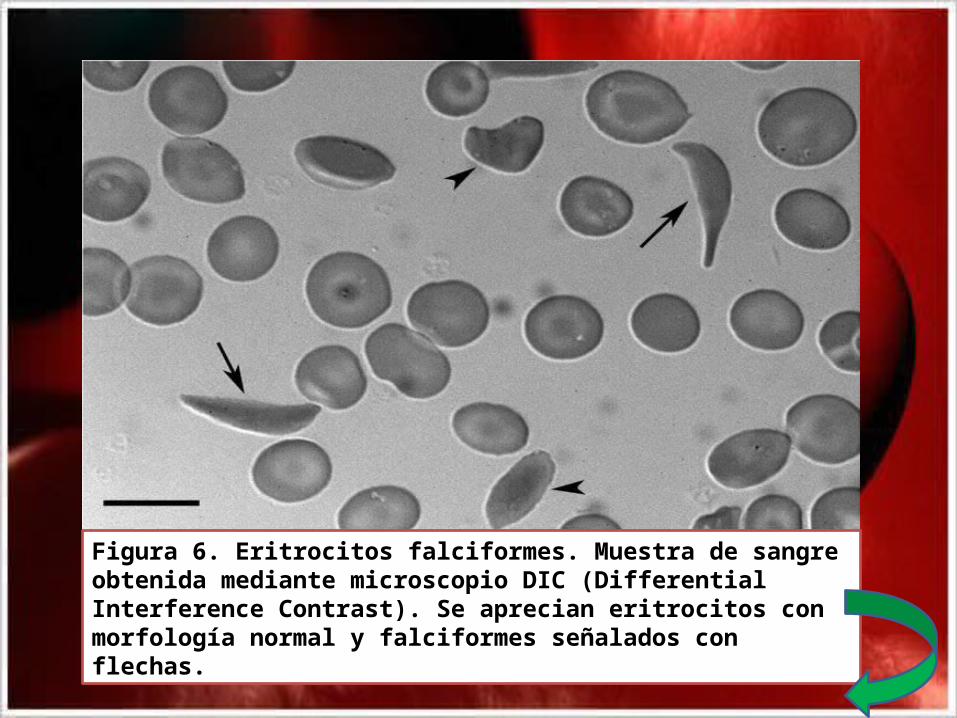
Figura 6. Eritrocitos falciformes. Muestra de sangre obtenida mediante microscopio DIC (Differential Interference Contrast). Se aprecian eritrocitos con morfología normal y falciformes señalados con flechas.

Anemia Falciforme.
• La anemia falciforme es una patología genética, en la que los eritrocitos de un individuo presentan una estructura alargada y en forma de hoz, conformación considerada anómala, debido a que la morfología regular de un hematíe es oval, bicóncava y aplanada.
• La concentración de glóbulos rojos en la sangre es mucho menor al rango considerado normal (4,5 a 5 millones/ml), debido a que la vida media de los eritrocitos falciformes (10 a 20 días) es mucho menor a la de los glóbulos rojos normales (120 días). Es por ello que luego de cumplir su función se degradan fácilmente produciendo anemia.
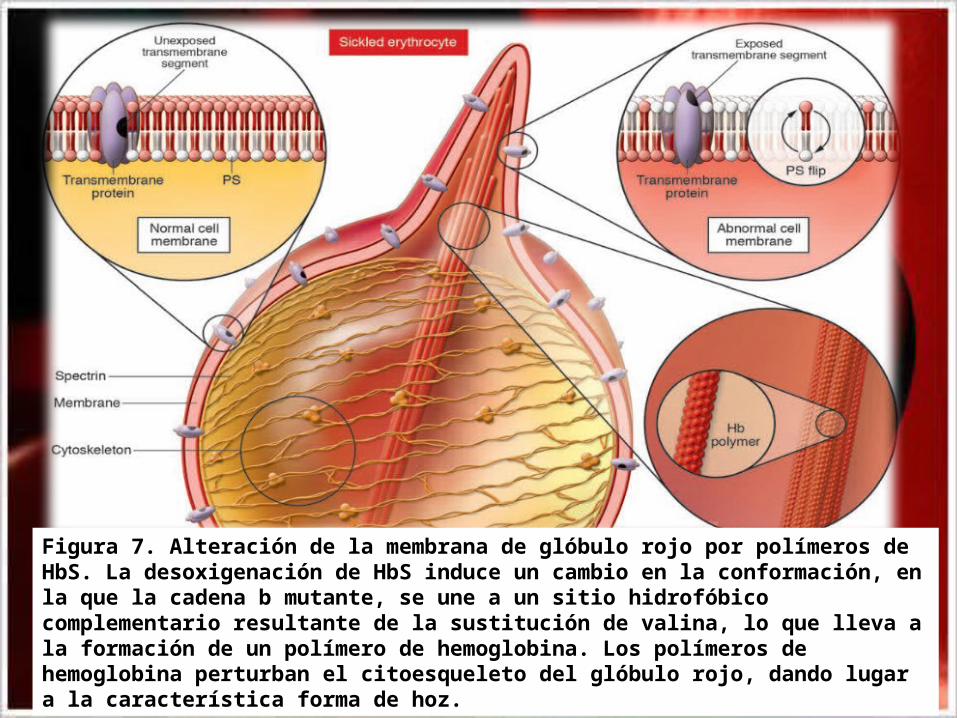
Figura 7. Alteración de la membrana de glóbulo rojo por polímeros de HbS. La desoxigenación de HbS induce un cambio en la conformación, en la que la cadena b mutante, se une a un sitio hidrofóbico complementario resultante de la sustitución de valina, lo que lleva a la formación de un polímero de hemoglobina. Los polímeros de hemoglobina perturban el citoesqueleto del glóbulo rojo, dando lugar a la característica forma de hoz.

Figura 8. Vaso oclusión de células Falciformes. Los glóbulos rojos falciformes inducen la expresión de una respuesta inflamatoria y mediadora de la coagulación, lo que lleva a la activación del endotelio vascular. Los glóbulos rojos falciformes también pueden estimular directamente al endotelio por las células de adhesión. Al activarse el endotelio se produce la captura de neutrófilos y glóbulos rojos falciformes lo que produce episodios de vaso oclusiones vasculares que se producen los capilares mas pequeños.

• Recién nacido -----------------13,5 a 19,5 gr/dl
• A los 3 meses -------------------9,5 a 12,5 gr/dl
• Al año de edad ---------------------11 a 13 gr/dl
• Entre los 3 y 5 años--- -----------12 a 14 gr/dl
• De los 5 a los 15 años----------1,5 a 15 gr/dl
• Hombre adulto-----------------------13 a 16 gr/dl
• Mujer adulta ----------------------1,5 a 14,5 gr/dl
VALORES NORMALES DE HEMATOCRITO


















