
Polimiositis asociada a lupus profundo. Argent. Dermatol. 57 7-10, 2007.pdf · das a LP, entre...
4
Polimiositis asociada a lupus profundo Mario A. Marini\o Saponaro^, Carina I. Simionato^, Gabriel Moriega^, Fernando Ariel'*, José G. Casas^ y Andrea Bosaleh® RESUMEN: El lupus profundo (LP) es una variedad clínica inusual del lupus eritematoso (LE). En algunas ocasiones se lo ha asociado con otras enfermedades sistemicas severas, como la esclerodermia y la dermatomiositis. Presentamos un paciente de 52 años de edad, masculino, con diagnóstico de polimiositis (PM) primaria asociada a placas y nodulos indurados en tórax, miembros superiores e inferio- res, compatibles clínica e histopatológicamente con LP. En algunas de las manifestaciones cutáneas aparecieron con posterioridad lesiones de lupus eritematoso discoide (LED) e imáge- nes histológicas de enfermedad de Degos símil. La inusual presentación de esta asociación y sus manifestaciones clínicas proteiformes motivan la publicación de este caso. SUMMARY: Lupus profundus is an unusual variety of lupus erythematosus. It is sometimes associated with others severe systemic disease, like scleroderma and dermatomyositis. We present a man of 52 years oíd, who developed primary polymyositis with indurated pla- ques and nodules in chest, arms and legs, related with PL. Later, in a few cutaneous manifesta- tion, he developed lesions of discoid lupus erythematosus and Degos símil disease in the histo- pathology. We present this case because of the unusual presentation of this association and their va- riety of the manifestation in this patients. Arch. Argent. Dermatol. 57:7-10, 2007 INTRODUCCION El lupus profundo (LP), publicado por primera vez por Kaposi' en 1883 y denominado en 1956 por Arnold^ como "lupus eritematoso profundo de Kaposi-Irgang", re- presenta una variante rara de LE (1-3%). La paniculltis puede ser la única expresión de la enfermedad o aso- ciarse con otras manifestaciones cutáneas y/o sistemi- cas en pacientes con lupus eritematoso crónico o sisté- mico o subagudo^ ^ Aparece preferentemente en la edad adulta y puede desencadenarse por traumas o infecciones^. Se publicaron enfermedades inmunológicas asocia- das a LP, entre ellas la dermatomiositis y la escleroder- mia^ ^-^ Sin embargo, su asociación con polimiositis pri- maria es extremadamente inusual. Hospital Británico de Buenos Aires. Perdrle! 74. Buenos Aires . Argentina. ' Profesor Titular de Dermatología. Facultad de Medicina. Univer- sidad de Buenos Aires. Jefe del Servicio de Dermatología. ^ Médicos Dermatólogos. Servicio de Dermatología. ^ Médico concurrente de la carrera de especialistas en dermatolo- gía del Hospital Británico. " Médico Reumatólogo. Servicio de Reumatología.. ^ Profesor Titular de Patología de la Facultad de Medicina. Univer- sidad de Buenos Aires. Jefe del Servicio de Anatomía Patológica. ^ Fellowship del Servicio de Patología. Presentamos un'paciente con diagnóstico clínico e histopatológico de LP y polimiositis primaria. CASO CLINICO Paciente de 52 años de edad, sexo masculino, sin an- tecedentes personales ni familiares de importancia. En octubre del 2004, luego de un traumatismo, comen- zó con una placa eritematosa, indurada y dolorosa en el hombro izquierdo, que se fue extendiendo hacia el brazo homolateral (Fig. 1). Luego aparecieron lesiones similares Fig. 1: Placa indurada, eritematosa, en brazo izquierdo. Recibido: 24-8-2006. Aceptado para publicación: 7-12-2006. 7
Transcript of Polimiositis asociada a lupus profundo. Argent. Dermatol. 57 7-10, 2007.pdf · das a LP, entre...

Polimiositis asociada a lupus profundo Mario A. Marini\o Saponaro^, Carina I. Simionato^, Gabriel Moriega^,
Fernando Ariel'*, José G. Casas^ y Andrea Bosaleh®
RESUMEN: El lupus profundo (LP) es una variedad clínica inusual del lupus eritematoso (LE). En algunas ocasiones se lo ha asociado con otras enfermedades sistemicas severas, como la esclerodermia y la dermatomiositis.
Presentamos un paciente de 52 años de edad, masculino, con diagnóstico de polimiositis (PM) primaria asociada a placas y nodulos indurados en tórax, miembros superiores e inferiores, compatibles clínica e histopatológicamente con LP. En algunas de las manifestaciones cutáneas aparecieron con posterioridad lesiones de lupus eritematoso discoide (LED) e imágenes histológicas de enfermedad de Degos símil.
La inusual presentación de esta asociación y sus manifestaciones clínicas proteiformes motivan la publicación de este caso.
SUMMARY: Lupus profundus is an unusual variety of lupus erythematosus. It is sometimes associated with others severe systemic disease, like scleroderma and dermatomyositis.
We present a man of 52 years oíd, who developed primary polymyositis with indurated plaques and nodules in chest, arms and legs, related with PL. Later, in a few cutaneous manifesta-tion, he developed lesions of discoid lupus erythematosus and Degos símil disease in the histo-pathology.
We present this case because of the unusual presentation of this association and their variety of the manifestation in this patients.
Arch. Argent. Dermatol. 57:7-10, 2007
INTRODUCCION
El lupus profundo (LP), publicado por primera vez por Kaposi' en 1883 y denominado en 1956 por Arnold^ como "lupus eritematoso profundo de Kaposi-Irgang", representa una variante rara de LE (1-3%). La paniculltis puede ser la única expresión de la enfermedad o asociarse con otras manifestaciones cutáneas y/o sistemicas en pacientes con lupus eritematoso crónico o sisté-mico o subagudo^ ^
Aparece preferentemente en la edad adulta y puede desencadenarse por traumas o infecciones^.
Se publicaron enfermedades inmunológicas asociadas a LP, entre ellas la dermatomiositis y la esclerodermia^ ̂ -̂ Sin embargo, su asociación con polimiositis primaria es extremadamente inusual.
Hospital Británico de Buenos Aires. Perdrle! 74. Buenos Aires . Argentina. ' Profesor Titular de Dermatología. Facultad de Medicina. Universidad de Buenos Aires. Jefe del Servicio de Dermatología. ^ Médicos Dermatólogos. Servicio de Dermatología. ^ Médico concurrente de la carrera de especialistas en dermatología del Hospital Británico. " Médico Reumatólogo. Servicio de Reumatología.. ^ Profesor Titular de Patología de la Facultad de Medicina. Universidad de Buenos Aires. Jefe del Servicio de Anatomía Patológica. ^ Fellowship del Servicio de Patología.
Presentamos un'paciente con diagnóstico clínico e histopatológico de LP y polimiositis primaria.
CASO CLINICO
Paciente de 52 años de edad, sexo masculino, sin antecedentes personales ni familiares de importancia.
En octubre del 2004, luego de un traumatismo, comenzó con una placa eritematosa, indurada y dolorosa en el hombro izquierdo, que se fue extendiendo hacia el brazo homolateral (Fig. 1). Luego aparecieron lesiones similares
Fig. 1: Placa indurada, eritematosa, en brazo izquierdo.
Recibido: 24-8-2006. Aceptado para publicación: 7-12-2006. 7
Mario A. Marini y colaboradores
en parrilla costal derecha y raíz de miembro inferior derecho. Presentó además síntomas generales como astenia, adinamia, disfagia progresiva a sólidos y debilidad proximal de los cuatro miembros, motivo por el cual fue internado en sala general del Hospital Británico para estudio y tratamiento.
Ante la sospecha de un proceso inflamatorio autoinmu-ne muscular se solicitaron enzimas musculares, electromio-grama, resonancia magnética nuclear (RMN), biopsia muscular y de piel y otros exámenes complementarios.
El laboratorio general y el colagenograma con complemento se encontraban dentro de los límites normales.
Las enzimas musculares fueron aumentando: láctico de-hidrogenasa-LDH: 616 Ul/L (25-100 U/L), creatina fosfo-quinasa-CPK: 1953 Ul/L en la primera determinación y 3229 Ul/L en la segunda (25-90 Ul/L en varones), aldolasa: 15,8 Ul/L en la primera determinación y 369 Ul/L en la segunda (0,6 Ul/L).
El electromiograma, la RMN y la biopsia muscular demostraron un proceso miopático compatible con polimiositis. Los estudios complementarios serológicos y por imágenes descartaron que tal miopatía fuese secundaria.
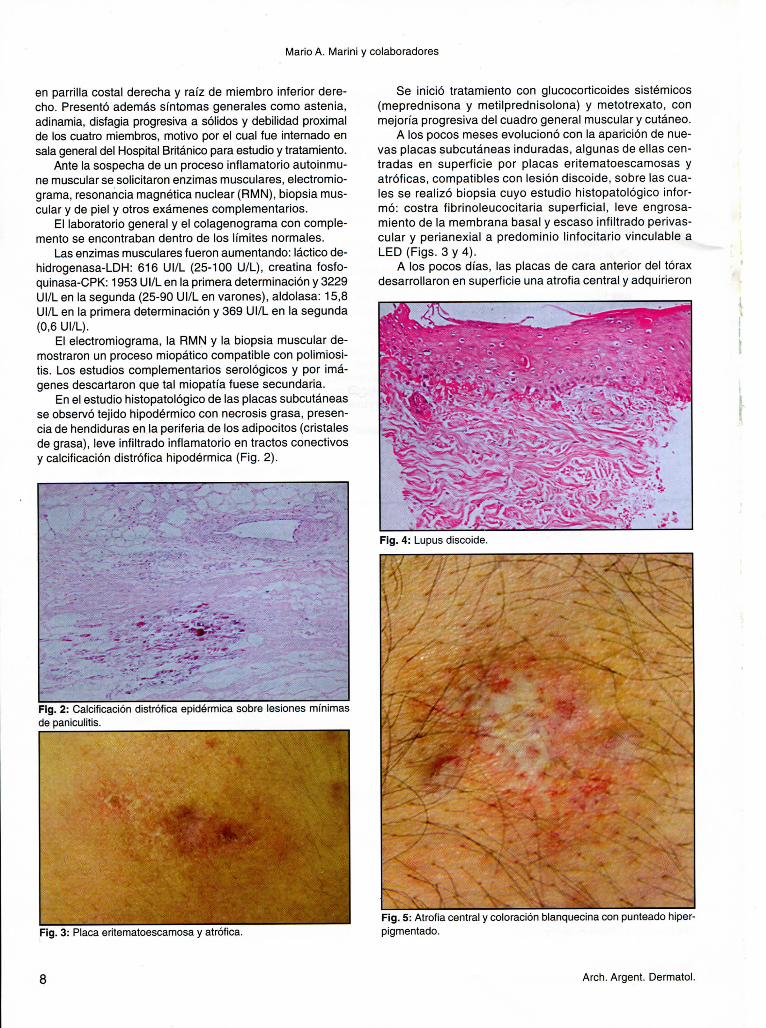
En el estudio histopatológico de las placas subcutáneas se observó tejido hipodérmico con necrosis grasa, presencia de hendiduras en la periferia de los adipocitos (cristales de grasa), leve infiltrado inflamatorio en tractos conectivos y calcificación distrófica hipodérmica (Fig. 2).
Fig. 2: Calcificación distrófica epidérmica sobre lesiones mínimas de paniculitis.
Fig. 3: Placa eritematoescamosa y atrófica. •
Se inició tratamiento con glucocorticoides sistémicos (meprednisona y metilprednisolona) y metotrexato, con mejoría progresiva del cuadro general muscular y cutáneo.
A los pocos meses evolucionó con la aparición de nuevas placas subcutáneas induradas, algunas de ellas centradas en superficie por placas eritematoescamosas y atróficas, compatibles con lesión discoide, sobre las cuales se realizó biopsia cuyo estudio histopatológico informó: costra fibrinoleucocitaria superficial, leve engrosa-miento de la membrana basal y escaso infiltrado perivas-cular y perianexial a predominio linfocitario vinculable a LED (Figs. 3 y 4).
A los pocos días, las placas de cara anterior del tórax desarrollaron en superficie una atrofia central y adquirieron
Fig. 4: Lupus discoide.
Fig. 5: Atrofia central y coloración blanquecina con punteado hiper-pigmentado.
8 Arch. Argent. Dermatol.

Polimiositis asociada a lupus profundo
Fig. 6: Paniculitis lúpica, con imágenes vasculares oclusivas y atro-fia-hialinización dérmica enfermedad de Degos-símll.
una coloración blanquecina con punteado hiperpigmenta-do, por lo cual se realizó una nueva biopsia, cuyo informe fue: paniculitis lúpica que coexiste con imágenes vasculares oclusivas y atrofia con hialinización dérmica enfermedad de Degos símil (Figs. 5 y 6).
Ante estas nuevas lesiones se pidió anticuerpos anti-cardiolipina y anticoagulante lúpico, que fueron negativos.
Se comenzó tratamiento coadyuvante con hidroxicloro-quina a dosis creciente de 200 a 400 mg/día con control laboratorial y oftalmológico. Las lesiones cutáneas y la sin-tomatología mejoraron parcialmente, sin aparición de nuevas placas y con persistencia de las calcificaciones en las lesiones más antiguas.
DISCUSION
El LP es un proceso crónico, recurrente, que puede presentar recaídas periódicas, así como largos períodos de remisiones y en general tiene buen pronóstico^ Algunos autores utilizan los términos LP y paniculitis lúpica como sinónimos; sin embargo, otros utilizan el nombre paniculitis lúpica para designar a aquellos pacientes que no presentan cambios cutáneos suprayacentes de LE^.
Aparece preferentemente en la edad adulta y raramente en niños menores de 13 años. Se hian publicado casos familiares^". El 80% de los pacientes son del sexo femenino.
Al examen clínico se caracteriza por placas y nodulos subcutáneos, indurados, bien delimitados y dolorosos. Suele comprometer tronco, cara y parte proximal de las extremidades.
La piel suprayacente a las lesiones de LP puede presentar lesiones de LED, con atrofia, eritema y escamas, lo cual aporta sustancialmente al diagnóstico clínico, o puede ser normal, firmemente adherida al tejido celular subcutáneo o más raramente ulcerada^". No son infrecuentes las calcificaciones en las placas de LP, prefe
rentemente en las lesiones de mayor evolución'. Massone^ y Martens^^ revisaron las características
clínicas y laboratoriales de 9 y 40 pacientes respectivamente con paniculitis lúpica y hallaron que un 22 a 33% de ellos reunían los criterios completos del American Co-llege of Rheumatology (ACR) para LES. Los anticuerpos antinucleares (ANA) son positivos en un 65 % de los pacientes con LP, habitualmente a títulos bajos''^2^^. Otras alteraciones que pueden observarse son: leuco-penia, hipocomplementemia, factor reumatoideo positivo, VDRL reactiva y aumento de la eritrosedimentación^^.
La histopatología demuestra una paniculitis mixta con necrosis hialina de la grasa, infiltrado linfohistiocitario focal con formación de nodulos linfoides a nivel lobuli-llar, acompañado de alteraciones septales como engro-samiento, fibrosis e infiltrado linfocitario. Pueden verse hallazgos dermoepidérmicos habituales de cualquier tipo de LE como ser: atrofia epidérmica, degeneración hidrópica en la unión dermoepidérmica, depósitos de mu-cina en la dermis y de calcio en el tejido adiposo^'.
Los hallazgos histopatológicos encontrados en la tercera biopsia, correspondientes a un área cicatrizal blanquecina, que mostraba un sector triangular hipocelular bien definido, con el vértice inferior ubicado en la profundidad y coronado con una arteria trombosada, símil enfermedad de Degos, nos llevó a investigar la presencia de anticuerpos antifosfolipídicos, que fueron negativos.
Sabemos que la papulosis atrofiante se relaciona con enfermedades autoinmunes como la dermatomiositis, la artritis reumatoidea y, especialmente, el LES. Para algunos autores sería la expresión de distintos procesos patológicos y posiblemente el resultado final común a injurias vasculares, como las que ocurren en el LES^"-^^. Con respecto a la PM, constituye uno de los 2 tipos más comunes de miopatías inflamatorias idiopáticas. En ocasiones puede aparecer concurrentemente con enfermedades del colágeno y/o malignidad.
Los corticoides son la primera línea terapéutica para la PM, con dosis decrecientes luego del control inicial de la enfermedad. El metotrexato suele ser útil y necesario^^.
Por otro lado, coincidentemente, el manejo del paciente con LP comparte ciertas similitudes con la terapéutica de la P M . Los inmunosupresores sistémicos como los glucocorticoides son particularmente útiles al inicio de la terapia en pacientes con lesiones extensas, con inflamación, siendo retirados gradualmente. Sin embargo, los antimaláricos son los de elección para el tratamiento del LP, hidroxicloroquina, cloroquina, a dosis de 200-400 mg/día y 250-500 mg/día, respectivamente. Otros tratamientos coadyuvantes incluyen quinacrina, metotrexato, ciclosporina A, ciclofosfami-da, azatioprina, dapsona y talidomida, con respuestas variables'^»''.
El monitoreo de los datos de laboratorio y el examen
Tomo 57 n M , Enero-Febrero 2007 9

Mario A. Marini y colaboradores
oftalmológico periódico es mandatario en los pacientes que necesitan la terapéutica con antimaláricos por períodos prolongados.
Se han publicado casos de calcinosis cutánea tratados con diferentes fármacos, entre los que destacamos el diltiazem, probenecid, bajas dosis de warfarina, corticoides intralesionales y colchicina^
Presentamos este paciente por lo inusual de la asociación de LP con PM y resaltamos su proteiforme presentación clínica y el interesante hallazgo histopatológico tipo papulosis atrofiante de Degos.
BIBLIOGRAFIA
1. Kaposi, M.: Patliologie und therapie der hantkrankheiten in vorlesung für pral<tische aerzte und studierende. Wien Ur-ban & Schawarzenberg 1883; 642.
2. Arnoid, H.L. Jr.: Lupus erythematosus profundus: commen-tary and report of four more cases. Arch Dermatol 1956; 73: 15-33.
3. Requena, L.; Sánctnez Yus, E.: Panniculitis. Part II. Mostiy lobular panniculitis. J Am Acad Dermatol 2001; 45: 325-361.
4. Caproni, M.; Palleschi, G.M.; Papi, C ; Fabbri, P; Discoid lupus erythematosus lesions developed on lupus erythematosus profundus nodules. Int J Dermatol 1995; 34: 357-359.
5. Balian, M.C.; Retamar, R.; Abeldaño, A.; Harris, P.; Paoloni, G. ; Kien, C ; Azcune, R.; Chouela, E.: Lupus eritematoso: formas infrecuentes. Dermatol Argent 1997; 3: 327-335.
6. Yoo, J.Y.; Jo, S.J.;Cho, K.H.: Lupus panniculitis with combined features of dermatomyositis resulting in severe lipoatrophy. J Dermatol 2004; 31: 552-555.
7. Massone, C ; Kodama, K.; Saimhofer, W.; Abe, R.; Shimizu, H. ; Parodi, A.; KerI, H.; Cerroni, L.: Lupus erythematosus panniculitis (lupus profundus): clinical, histopathological, and molecular analysis of nine cases. J Cut Pathol 2005; 32: 396-404.
8. Stork, J . ; Vosmik, F.: Lupus erythematosus panniculitis with morphea-like lesions. Clin Exp Dermatol 1994;19: 79-82.
9. Morgan, K.; Callen, J . : Calcifying lupus panniculitis ¡n a pa-tient with subacute cutaneous lupus erythematosus: response
to diltiazem and chioroquine. J Rheumatol 2001; 28: 2129-2132.
10. RoweII, N.; Goodfieid, M.: The connective tissue disease. En: Champion, R.H.; Burton, J.L.; Burns, D.; Breathnach, S.M. edits.: Rook/Wiikinson/Ebling Textbook of Dermatology; 6th edition. 1998; Chapter58; págs. 2451-2452.
11. Bondi, E.; Margolis, D.; Lazarus, G.: Disorders of subcutane-ous tissue. Lupus Panniculitis. En: Freedberg, I.; Eisen, A.; Wolff, K.; Austen, K.F; Goldsmith, L.A.; Katz, S.I.: Fitzpatrick Dermatology in general medicine; 5th edition. McGraw-Hill. Págs. 1281-1282.
12. Martens, P.; Moder, K.; Iftikhar, A.: Lupus panniculitis: clinical perspectives from a case series. J Rheumatol 1999; 26: 68-72.
13. Grossberg, E.; Scherschun, L.; Fivenson, D.P.: Lupus profundus: not a benign disease. Lupus 2001; 10: 514-516.
14. Marini, M.A.; Casas, J.G.; Lagodín, C ; Acuña, K.; Dahbar, M.; Casas, G.: Papulosis atrofiante maligna. Presentación de un caso fatal. Act Terap Dermatol 2005; 28: 44-49.
15. Black, M.M.; Hudson, P.M.: Atrophie Planche lesions closely resembling malignant atrophie papulosis (Degos' disease) in systemic lupus erythematosus. B J Dermatol 1976; 95: 649-652.
16. Grilli, R.; Soriano, M.L.; Izquierdo, M.J.; Fariña, M.C.; Martín, L.; Manzarbeitia, F.; Requena, L.: Panniculitis mimicking lupus erythematosus profundus: a new histopathologic finding in malignant atrophie papulosis (Degos disease). Am J Dermatopathol 1999; 21: 365-368.
17. Cario, J.R.: El tratamiento de las miopatías inflamatorias. 18. Hae-Shin Chung; Seung-Kyung Hann: Lupus panniculitis
treated by a combination therapy of hydroxychioroquine and quinacrine. J Dermatol 1997; 24: 569.572.
19. Saeki, Y; Ohshima, S.; Kurimoto, I.; Miura, H.; Suemura. M.: Maintaining remission of lupus erythematosus profundus (LEP) with cyclosporin A. Lupus 2000; 9: 390-392.
Dirección postal M.A. Marini José Bonifacio 634 1424 Buenos Aires [email protected] •
10 Arch. Argent. Dermatol.


















