Osteogenesis imperfeta
70
Osteogénesis imperfecta QBB. Jorge Hernández Bello Guadalajara, Jalisco, México. 2014
-
Upload
jorge-hdz -
Category
Health & Medicine
-
view
359 -
download
2
Transcript of Osteogenesis imperfeta
Osteogénesis imperfecta
QBB. Jorge Hernández Bello
Guadalajara, Jalisco, México. 2014
Carvajal AM, 2007
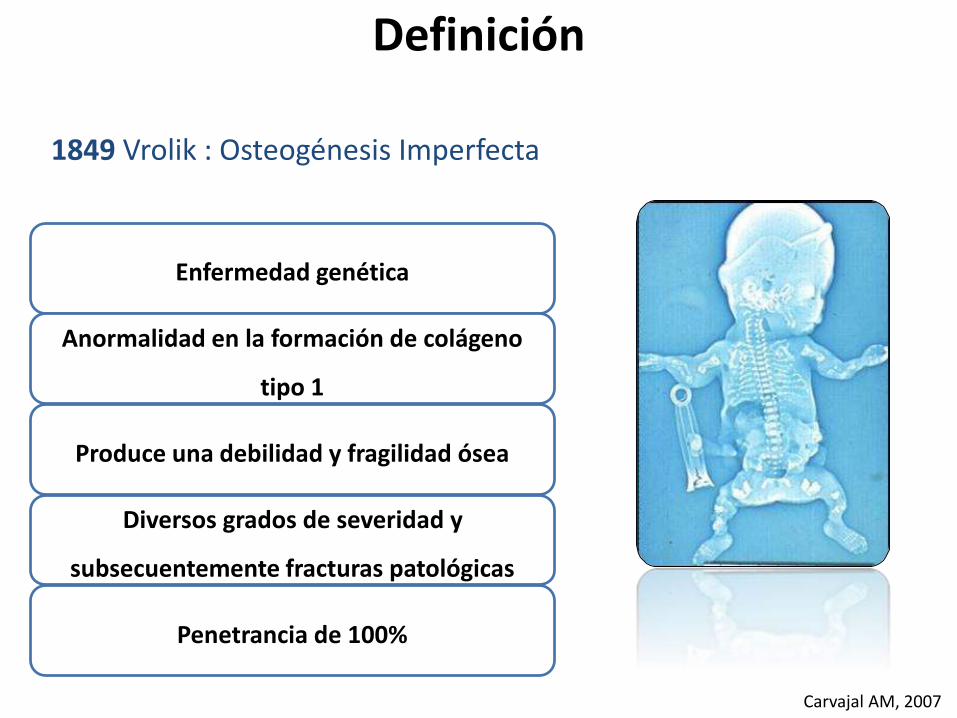
Enfermedad genética
Anormalidad en la formación de colágeno
tipo 1
Produce una debilidad y fragilidad ósea
Diversos grados de severidad y
subsecuentemente fracturas patológicas
Penetrancia de 100%
Definición
1849 Vrolik : Osteogénesis Imperfecta

Enfermedad de Lobstein
Enfermedad de Porak y Durante
Osteopsatirosis
Conocida comúnmente como
“Enfermedad de los huesos frágiles o de cristal“
Orphanet
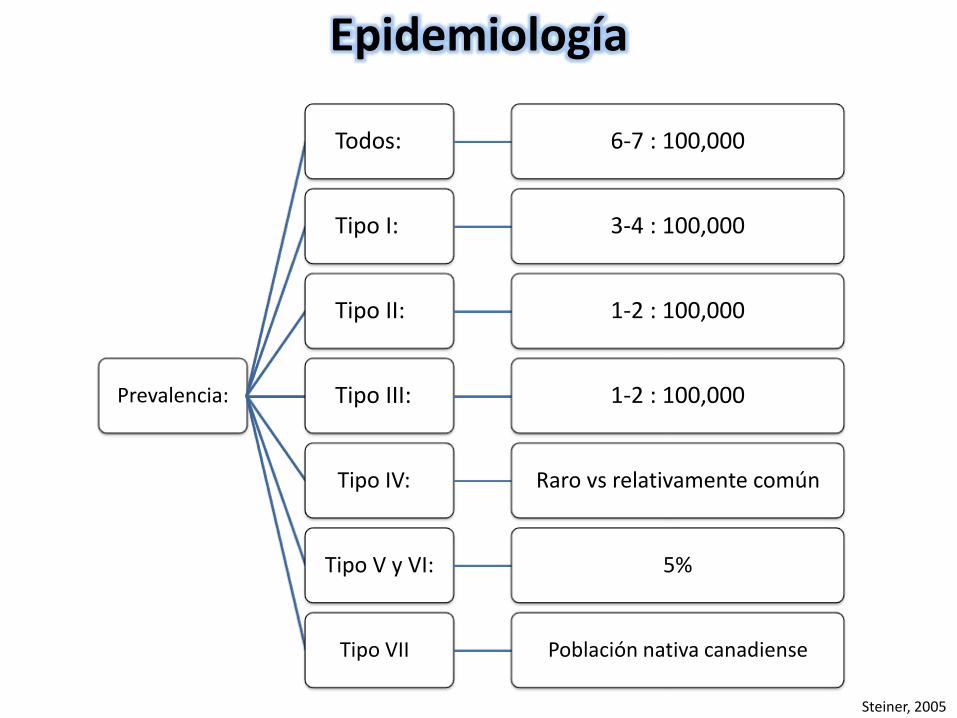
Prevalencia:
Todos: 6-7 : 100,000
Tipo I: 3-4 : 100,000
Tipo II: 1-2 : 100,000
Tipo III: 1-2 : 100,000
Tipo IV: Raro vs relativamente común
Tipo V y VI: 5%
Tipo VII Población nativa canadiense
Epidemiología
Steiner, 2005

Causada por mutaciones
Colágeno tipo 1 (90%)
• COL1A1 en 17q21.33
• COL1A2 en 7q21.3
10 % (Enzimas, “AR”)
Hidroxilasas de Prolina de COL A1: (CRTAP, LEPRE1 y PPIB)
Chaperonas : codificada por FKBP10 y SERPINH1 (plegamiento y secreción)
SERPINF1: un factor secretable que interacciona con la M.E
TMEM38B: Canal de Ca2+
Etiología
Chio y cols, 2007; Gutierrez et al., 2013
•60% de los casos de OI leve son mutaciones de novo•100% de los casos con OI letal o severa son mutaciones de novo
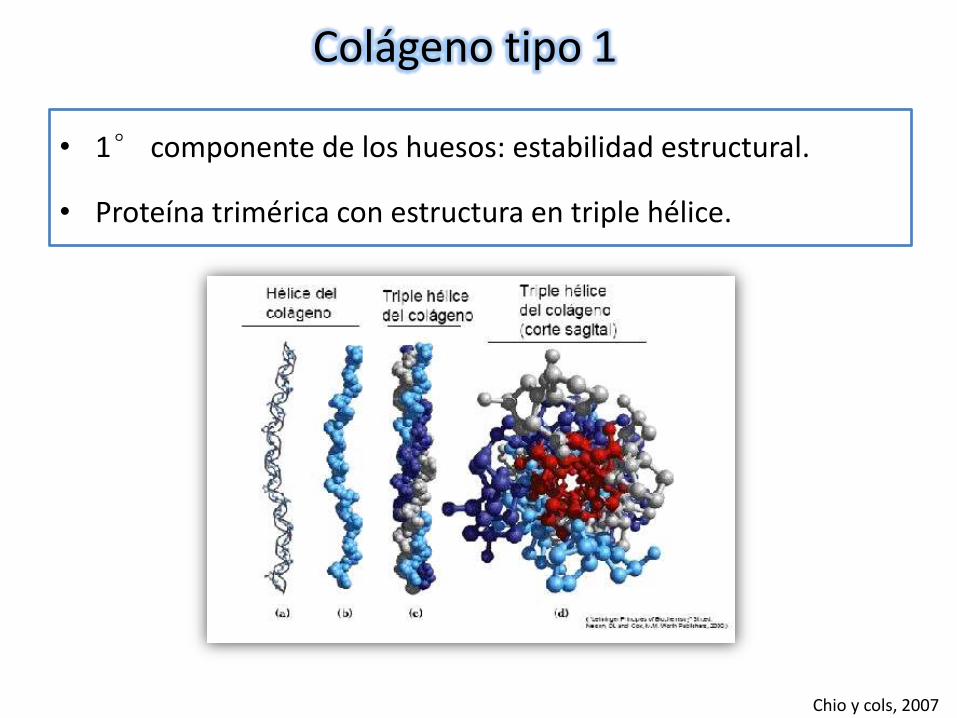
Colágeno tipo 1
• 1° componente de los huesos: estabilidad estructural.
• Proteína trimérica con estructura en triple hélice.
Chio y cols, 2007
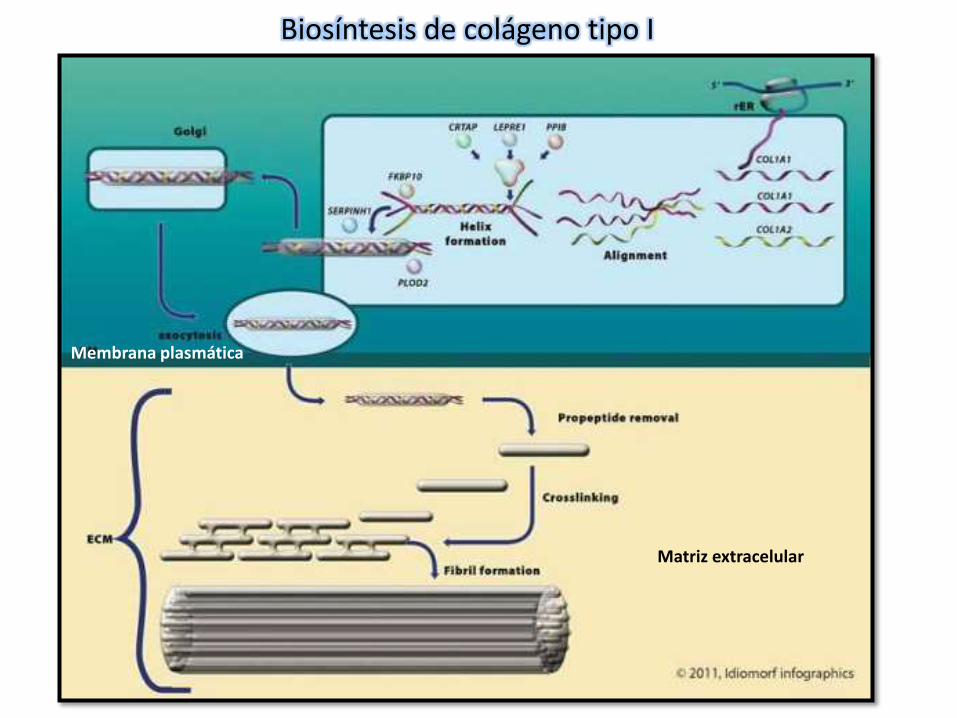
Matriz extracelular
Biosíntesis de colágeno tipo I
Membrana plasmática
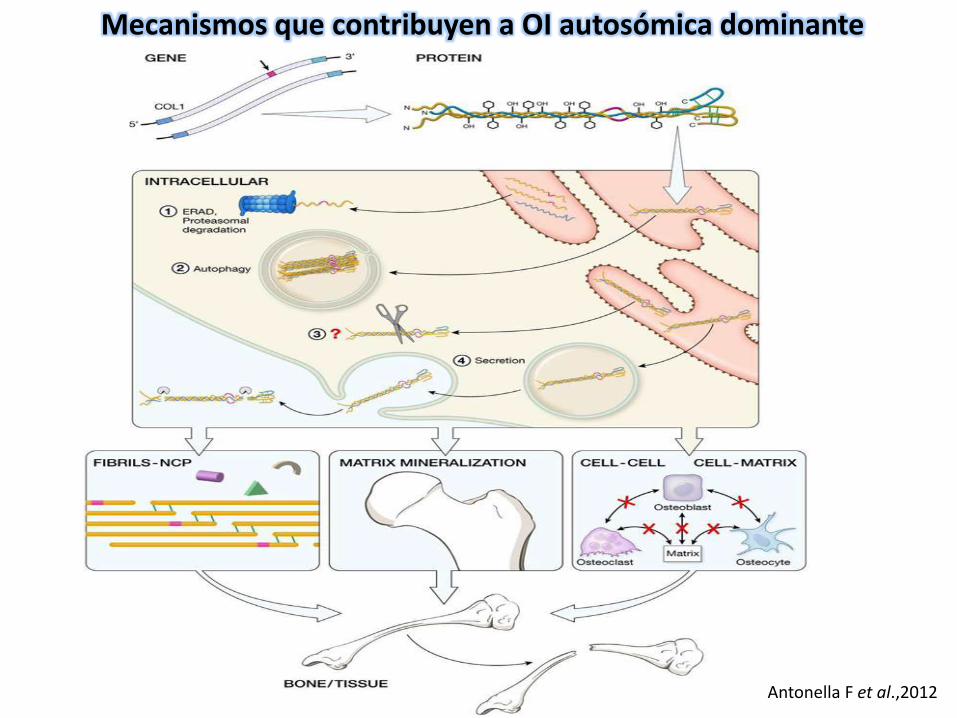
Antonella F et al.,2012
Mecanismos que contribuyen a OI autosómica dominante
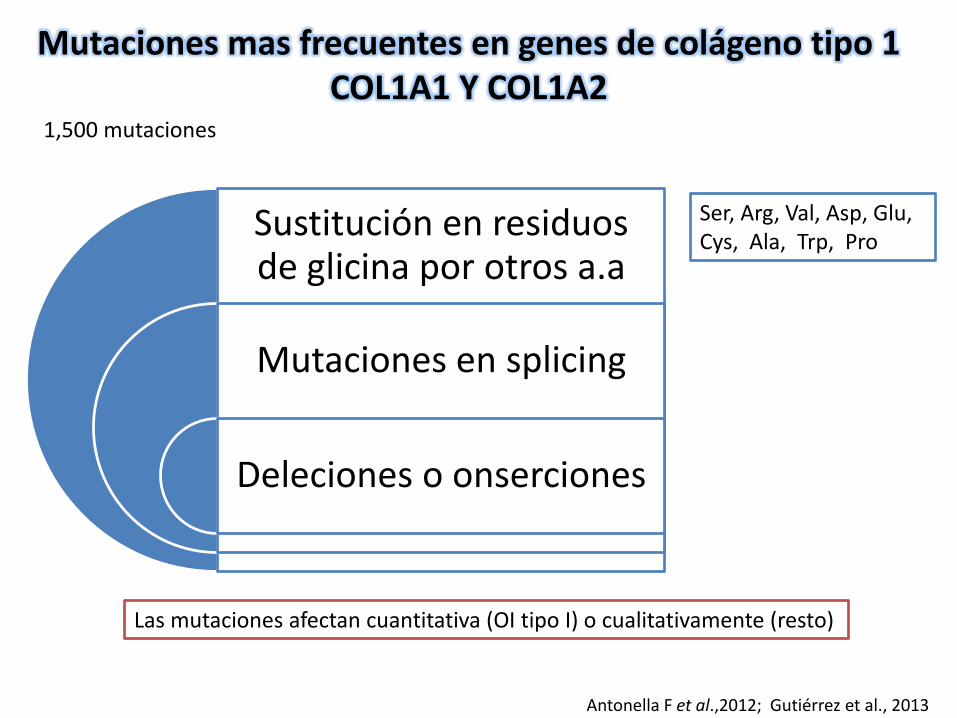
Mutaciones mas frecuentes en genes de colágeno tipo 1COL1A1 Y COL1A2
Sustitución en residuos de glicina por otros a.a
Mutaciones en splicing
Deleciones o onserciones
Ser, Arg, Val, Asp, Glu, Cys, Ala, Trp, Pro
Antonella F et al.,2012; Gutiérrez et al., 2013
Las mutaciones afectan cuantitativa (OI tipo I) o cualitativamente (resto)
1,500 mutaciones
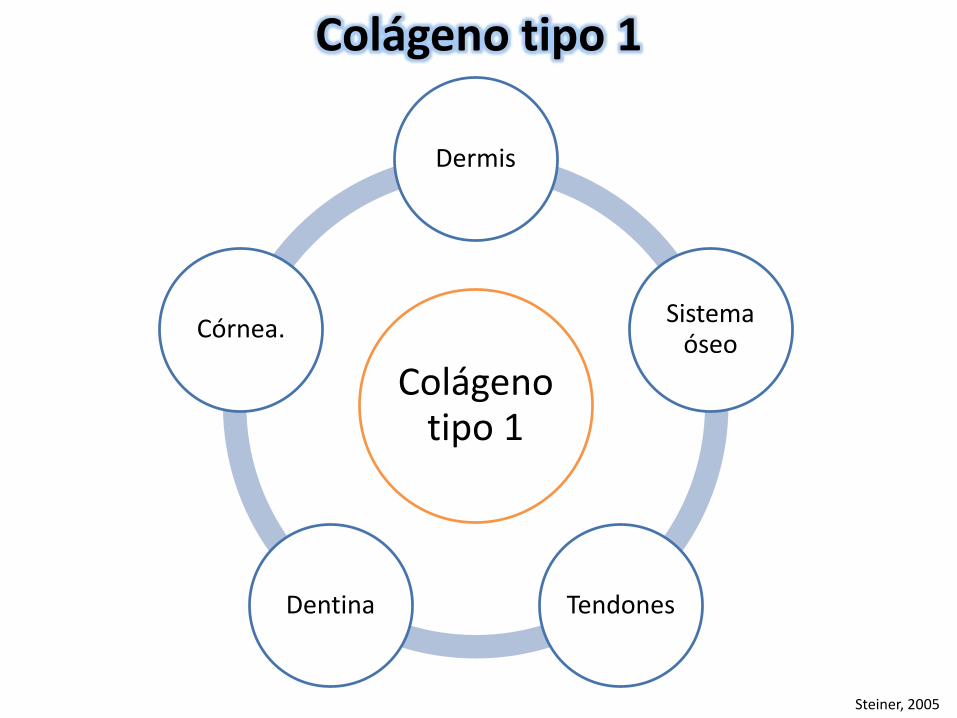
Colágeno tipo 1
Dermis
Sistema óseo
TendonesDentina
Córnea.
Colágeno tipo 1
Steiner, 2005
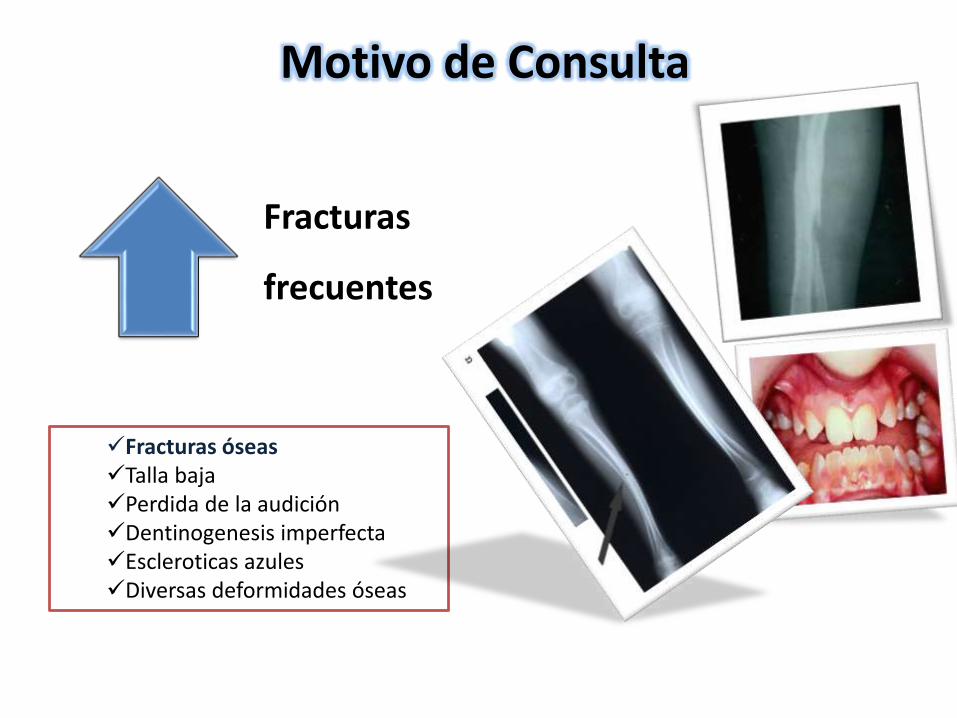
Fracturas
frecuentes
Motivo de Consulta
Fracturas óseasTalla bajaPerdida de la audición Dentinogenesis imperfectaEscleroticas azulesDiversas deformidades óseas
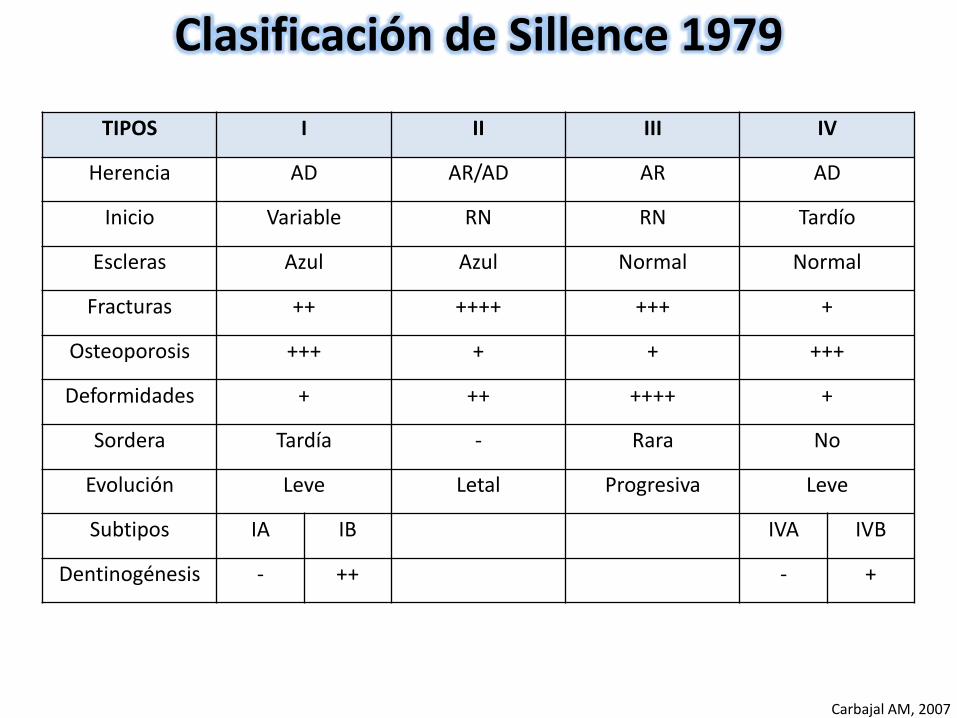
TIPOS I II III IV
Herencia AD AR/AD AR AD
Inicio Variable RN RN Tardío
Escleras Azul Azul Normal Normal
Fracturas ++ ++++ +++ +
Osteoporosis +++ + + +++
Deformidades + ++ ++++ +
Sordera Tardía - Rara No
Evolución Leve Letal Progresiva Leve
Subtipos IA IB IVA IVB
Dentinogénesis - ++ - +
Clasificación de Sillence 1979
Carbajal AM, 2007
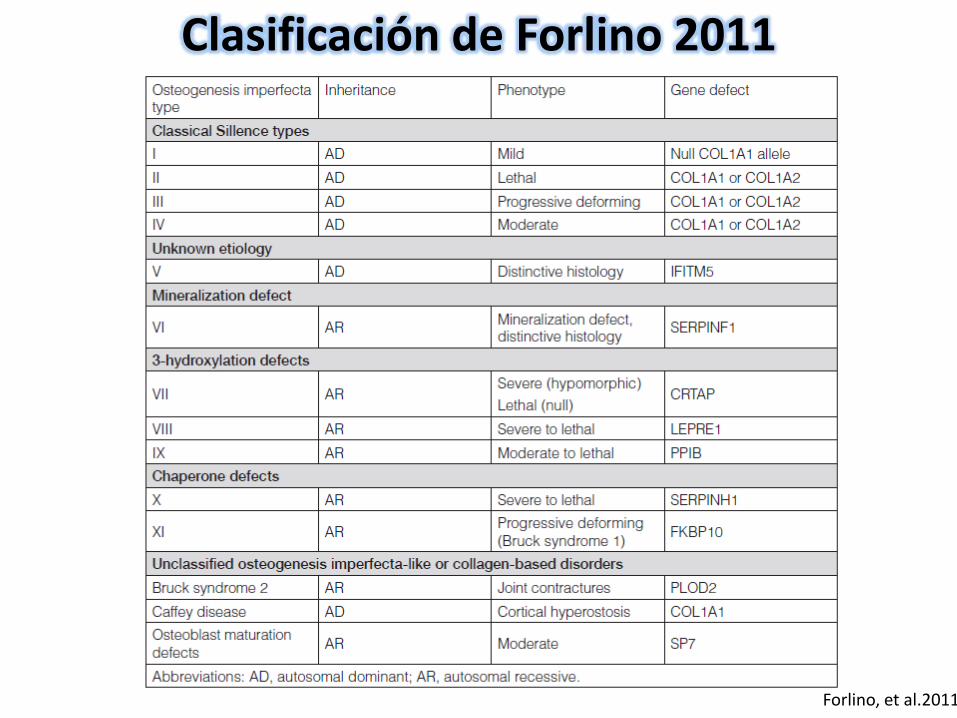
Forlino, et al.2011
Clasificación de Forlino 2011
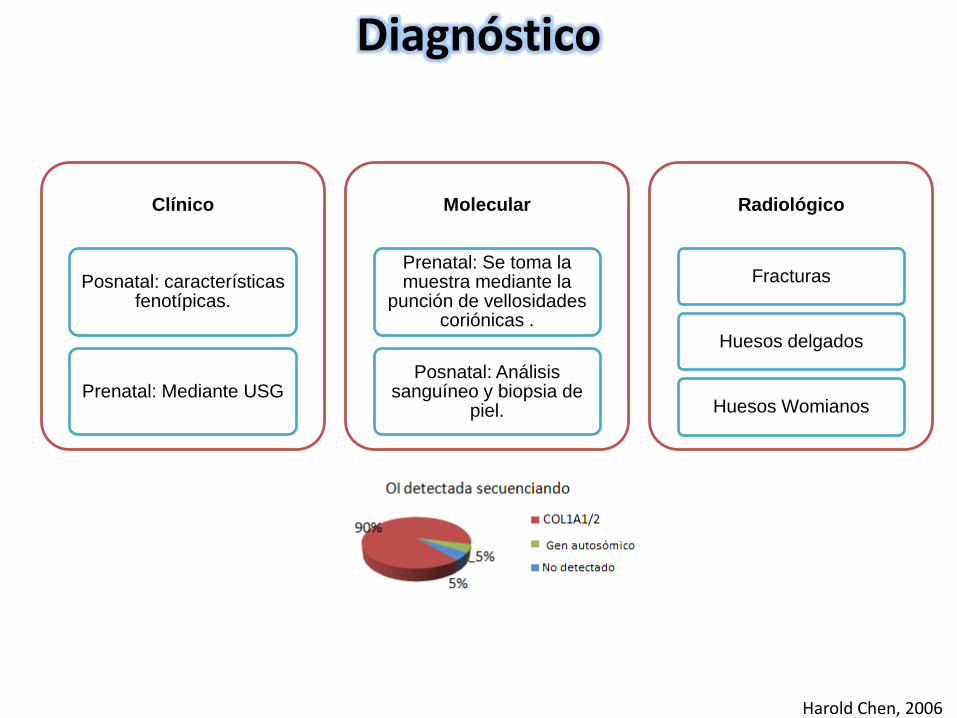
Clínico
Posnatal: características fenotípicas.
Prenatal: Mediante USG
Molecular
Prenatal: Se toma la muestra mediante la
punción de vellosidades coriónicas .
Posnatal: Análisis sanguíneo y biopsia de
piel.
Radiológico
Fracturas
Huesos delgados
Huesos Womianos
Diagnóstico
Harold Chen, 2006
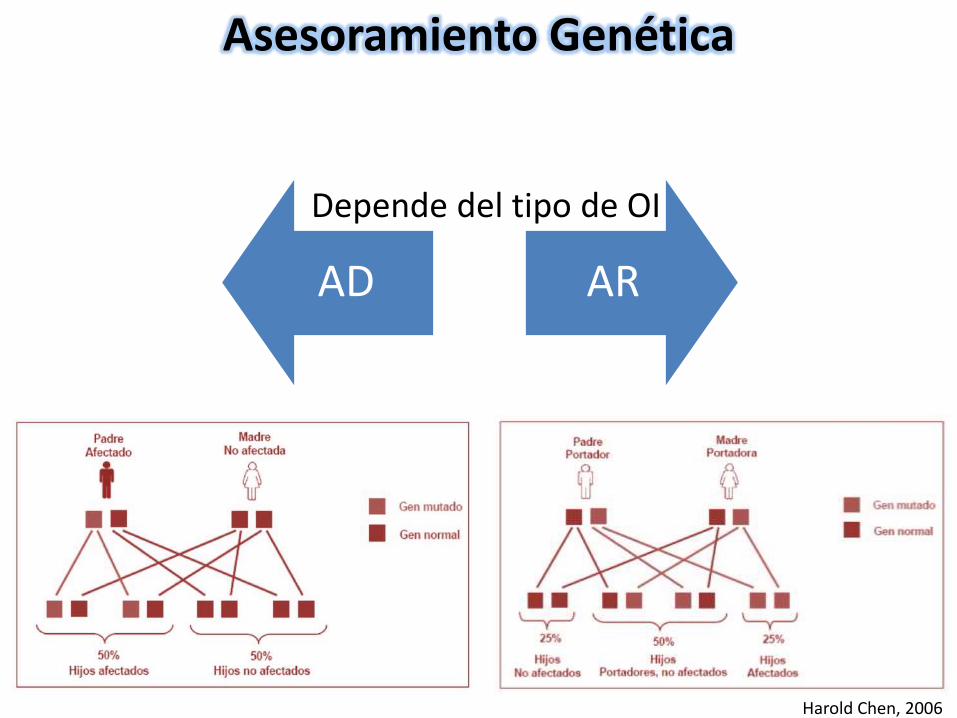
Asesoramiento Genética
AD AR
Depende del tipo de OI
Harold Chen, 2006

Osteogénesis imperfecta tipo I(escleróticas azules y herencia autosómica dominante)
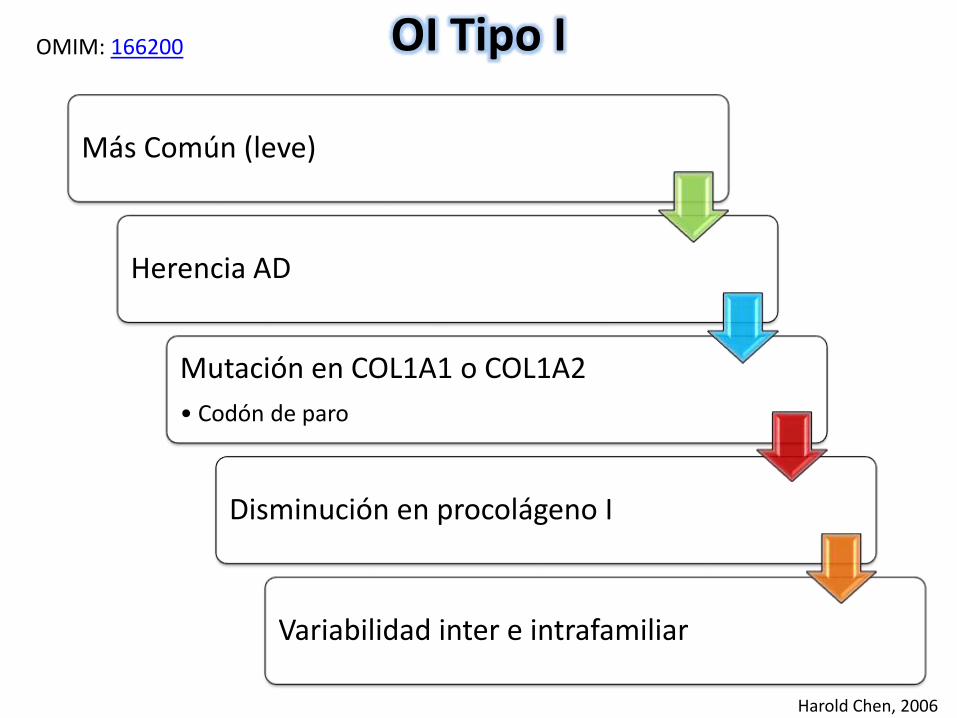
OI Tipo I
Más Común (leve)
Herencia AD
Mutación en COL1A1 o COL1A2
• Codón de paro
Disminución en procolágeno I
Variabilidad inter e intrafamiliar
Harold Chen, 2006
OMIM: 166200
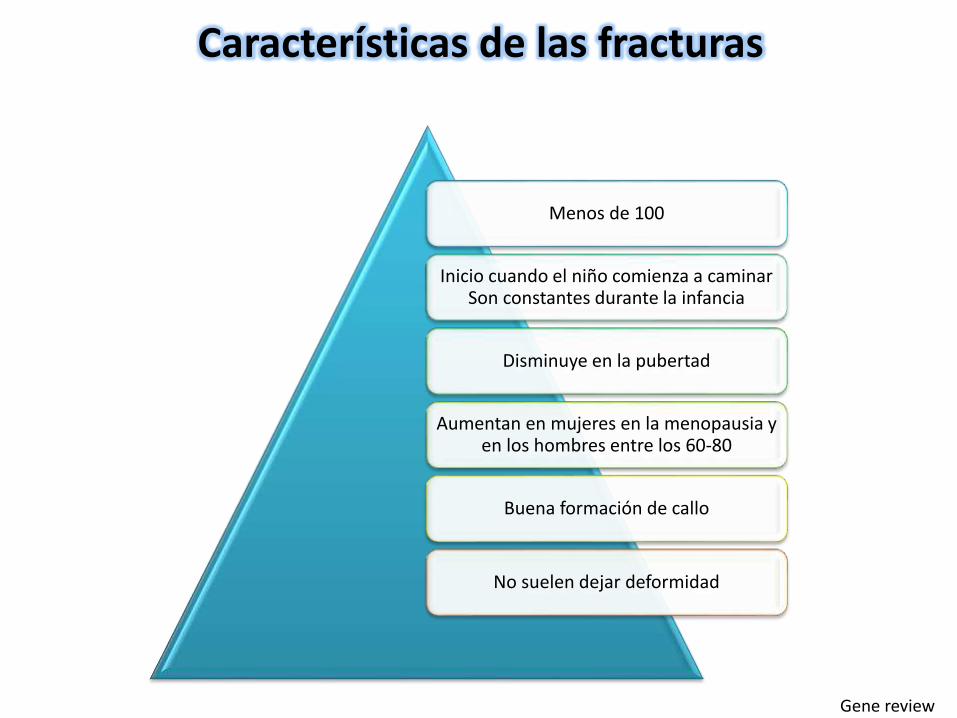
Menos de 100
Inicio cuando el niño comienza a caminar Son constantes durante la infancia
Disminuye en la pubertad
Aumentan en mujeres en la menopausia y en los hombres entre los 60-80
Buena formación de callo
No suelen dejar deformidad
Características de las fracturas
Gene review
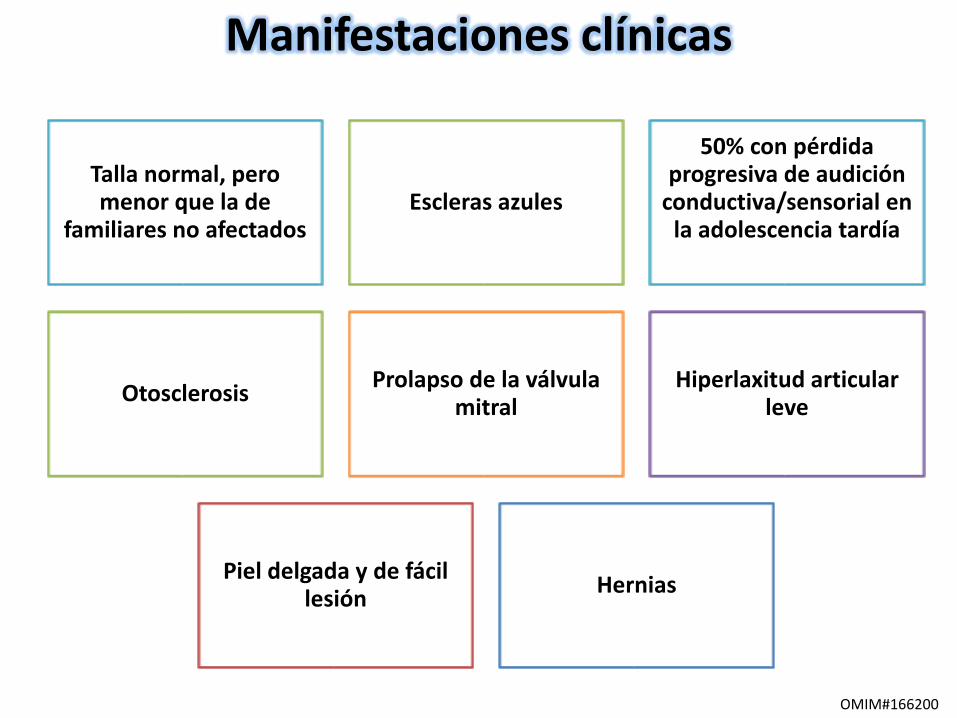
Manifestaciones clínicas
Talla normal, pero menor que la de
familiares no afectadosEscleras azules
50% con pérdida progresiva de audición
conductiva/sensorial en la adolescencia tardía
Otosclerosis Prolapso de la válvula
mitralHiperlaxitud articular
leve
Piel delgada y de fácil lesión
Hernias
OMIM#166200
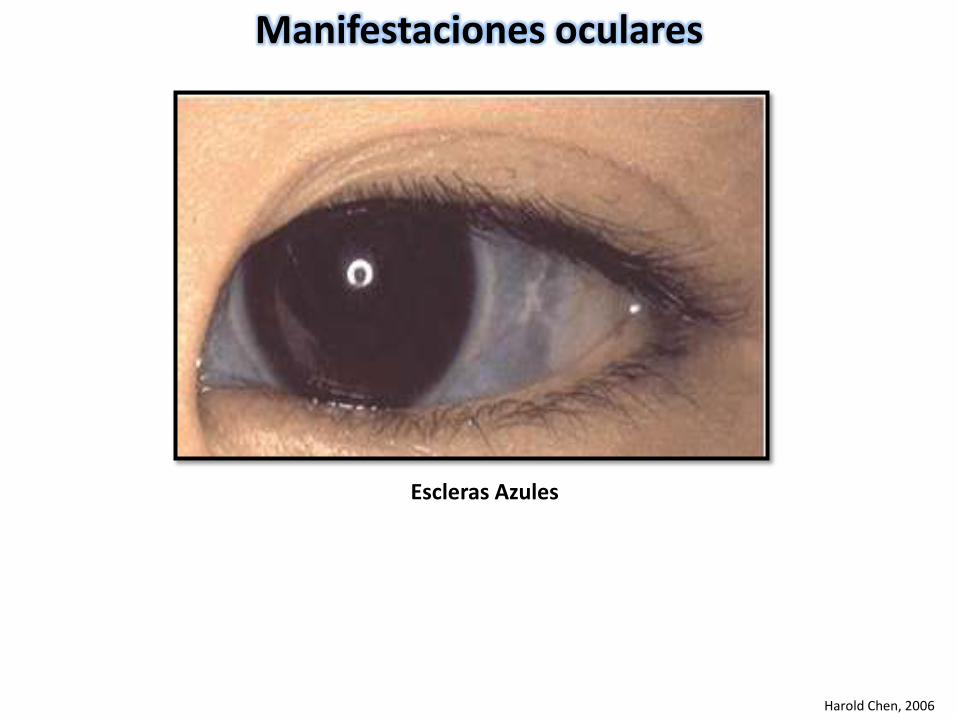
Manifestaciones oculares
Harold Chen, 2006
Escleras Azules
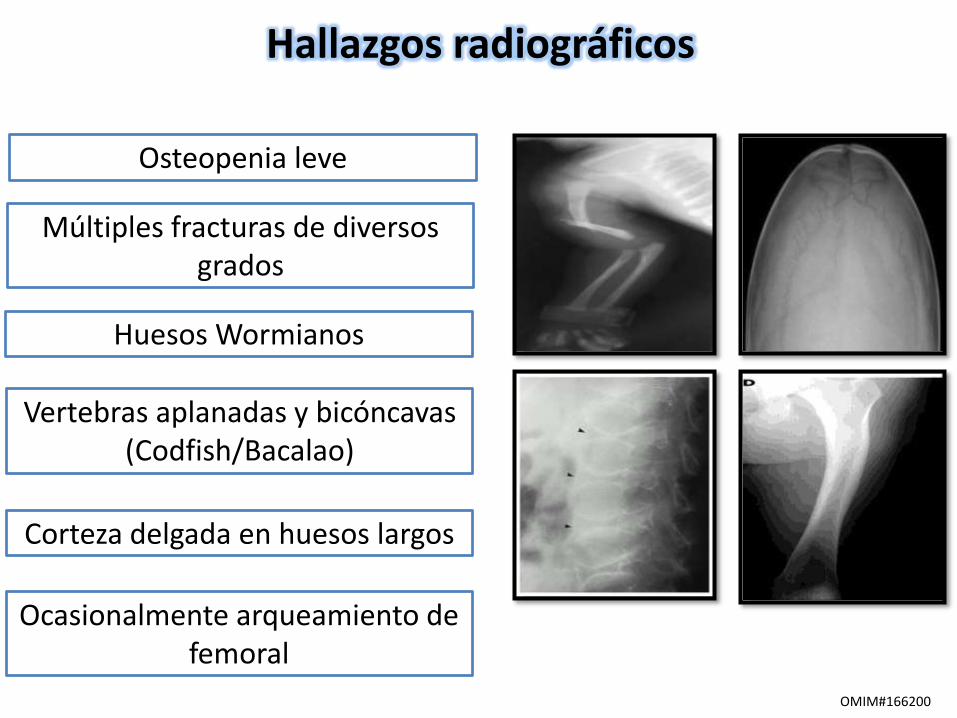
Hallazgos radiográficos
Múltiples fracturas de diversos grados
Osteopenia leve
Huesos Wormianos
Vertebras aplanadas y bicóncavas (Codfish/Bacalao)
Corteza delgada en huesos largos
Ocasionalmente arqueamiento de femoral
OMIM#166200
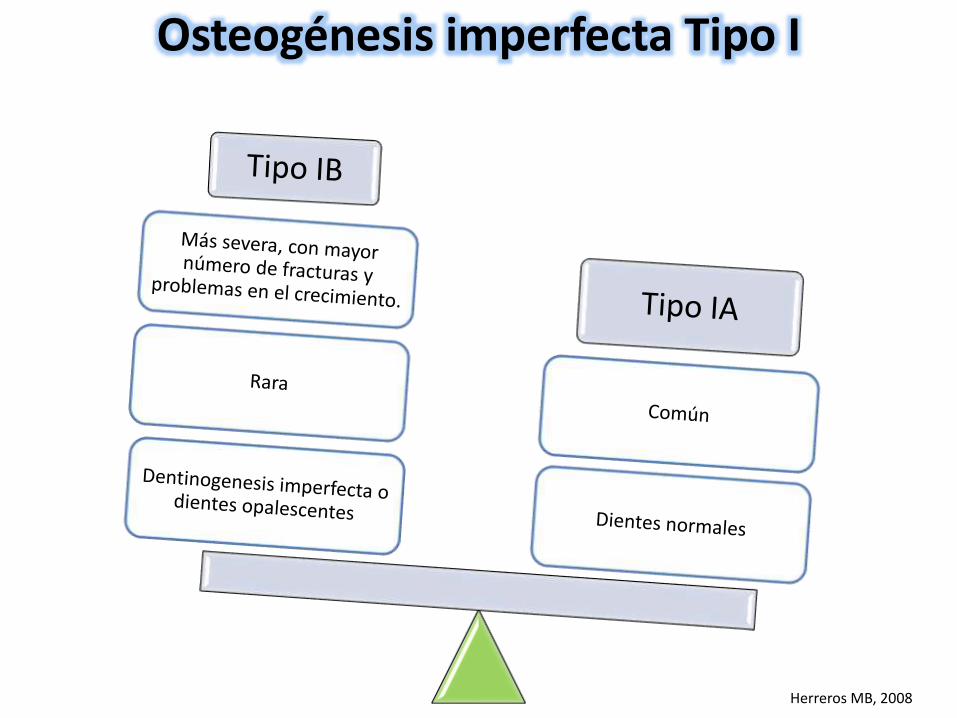
Osteogénesis imperfecta Tipo I
Herreros MB, 2008
Buen Pronóstico
SubdiagnósticoConfundido con maltrato
infantil

OSTEOGÉNESIS IMPERFECTA TIPO II

Es la forma más grave de la enfermedad, siendo mortal en el período prenatal.
OI Tipo II
OMIM: 166210
Se produce por nuevas mutaciones dominantes esporádicasMenor porcentaje: mosaicismo germinal
OTROS NOMBRES: OSTEOGÉNESIS IMPERFECTA CONGÉNICA, PERINATAL MORTAL
OMIM: 166210
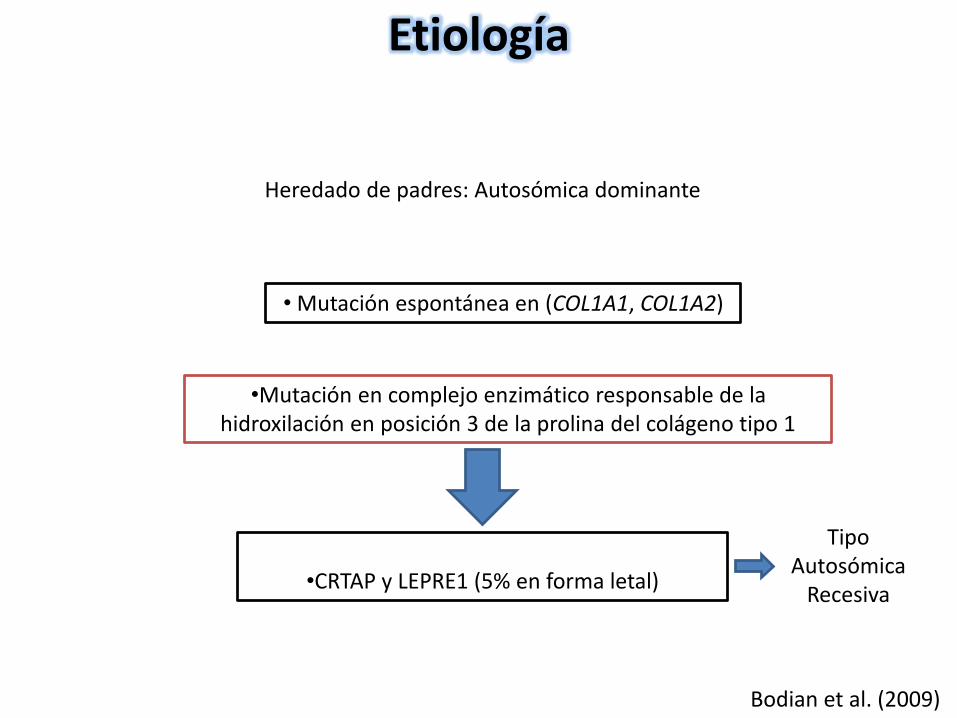
Heredado de padres: Autosómica dominante
•CRTAP y LEPRE1 (5% en forma letal)
Etiología
Bodian et al. (2009)
• Mutación espontánea en (COL1A1, COL1A2)
•Mutación en complejo enzimático responsable de la hidroxilación en posición 3 de la prolina del colágeno tipo 1
Tipo Autosómica
Recesiva
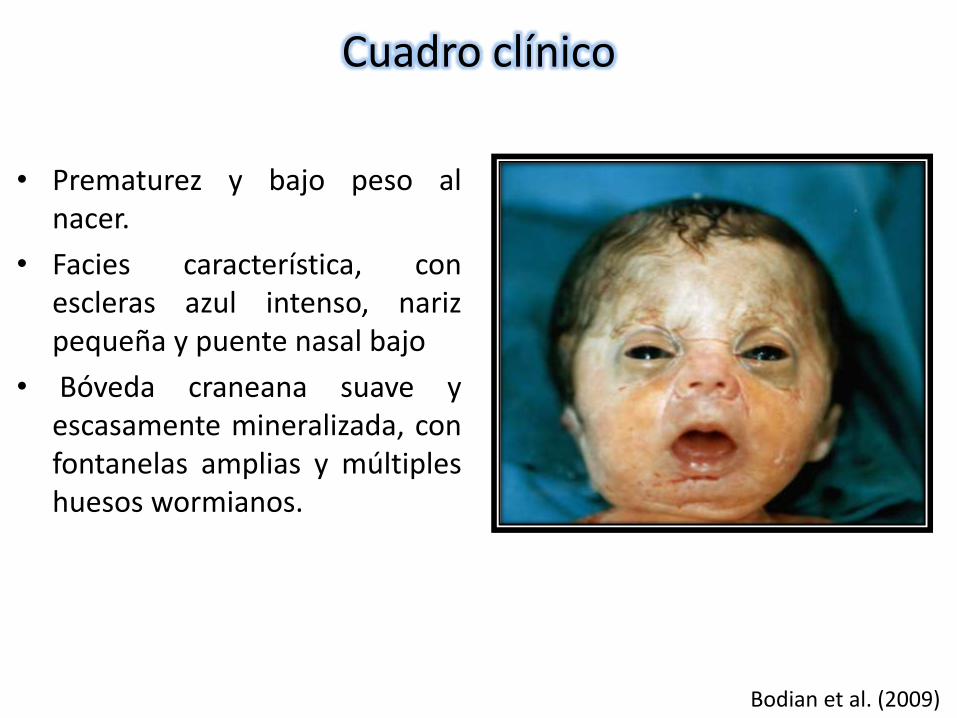
Cuadro clínico
• Prematurez y bajo peso alnacer.
• Facies característica, conescleras azul intenso, narizpequeña y puente nasal bajo
• Bóveda craneana suave yescasamente mineralizada, confontanelas amplias y múltipleshuesos wormianos.
Bodian et al. (2009)
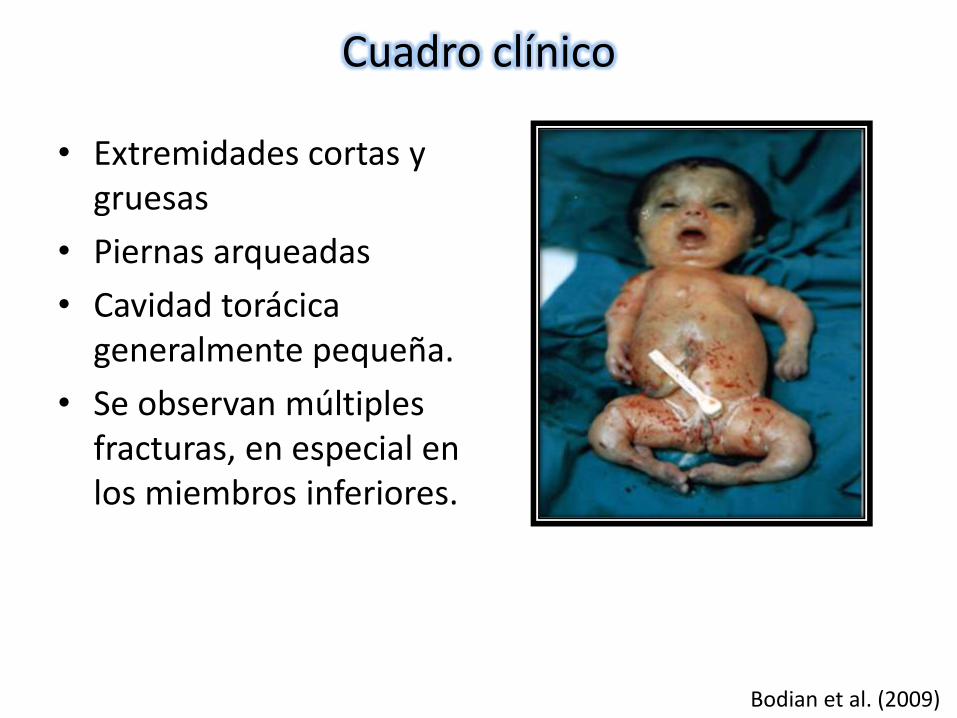
• Extremidades cortas y gruesas
• Piernas arqueadas
• Cavidad torácica generalmente pequeña.
• Se observan múltiples fracturas, en especial en los miembros inferiores.
Bodian et al. (2009)
Cuadro clínico

OMIM: 166210
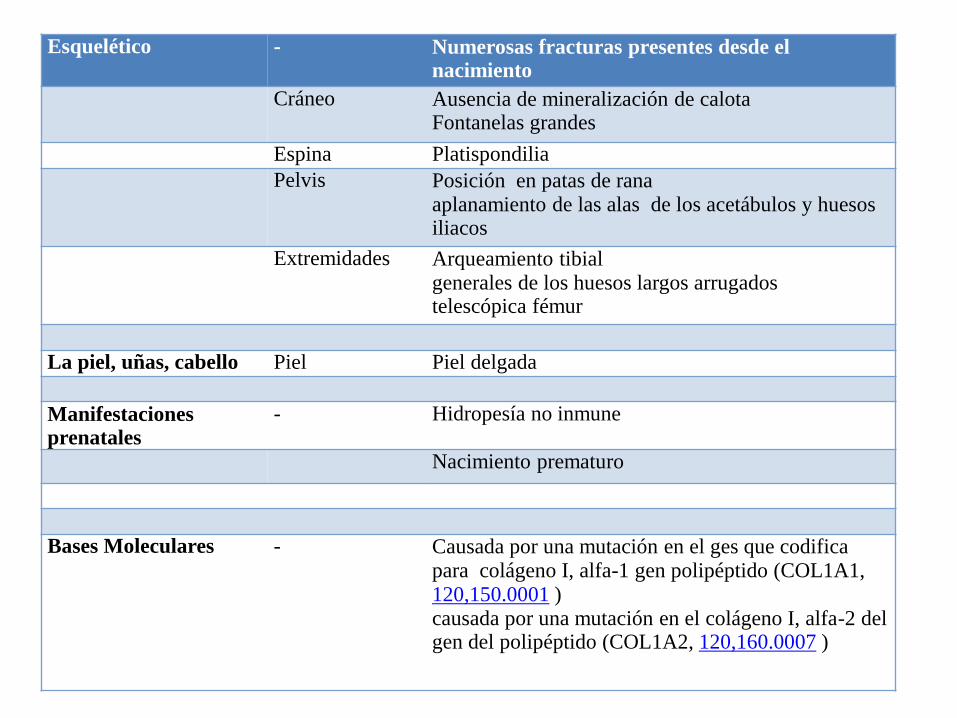
Esquelético - Numerosas fracturas presentes desde el nacimiento
Cráneo Ausencia de mineralización de calotaFontanelas grandes
Espina Platispondilia
Pelvis Posición en patas de ranaaplanamiento de las alas de los acetábulos y huesos iliacos
Extremidades Arqueamiento tibial generales de los huesos largos arrugados telescópica fémur
La piel, uñas, cabello Piel Piel delgada
Manifestaciones prenatales
- Hidropesía no inmune
Nacimiento prematuro
Bases Moleculares - Causada por una mutación en el ges que codifica para colágeno I, alfa-1 gen polipéptido (COL1A1, 120,150.0001 ) causada por una mutación en el colágeno I, alfa-2 del gen del polipéptido (COL1A2, 120,160.0007 )
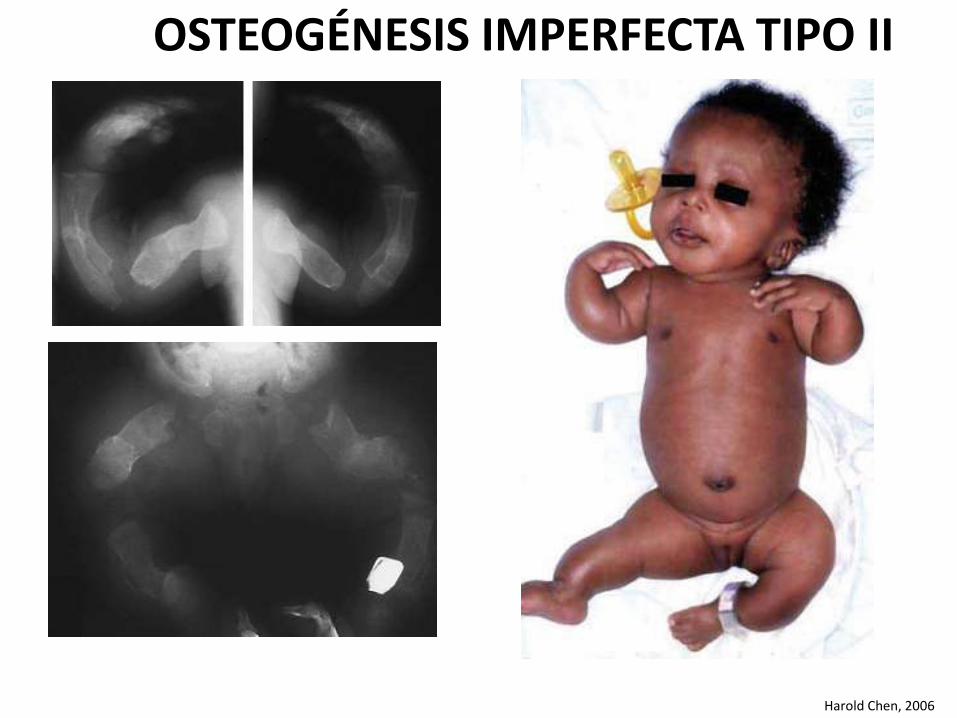
OSTEOGÉNESIS IMPERFECTA TIPO II
Harold Chen, 2006
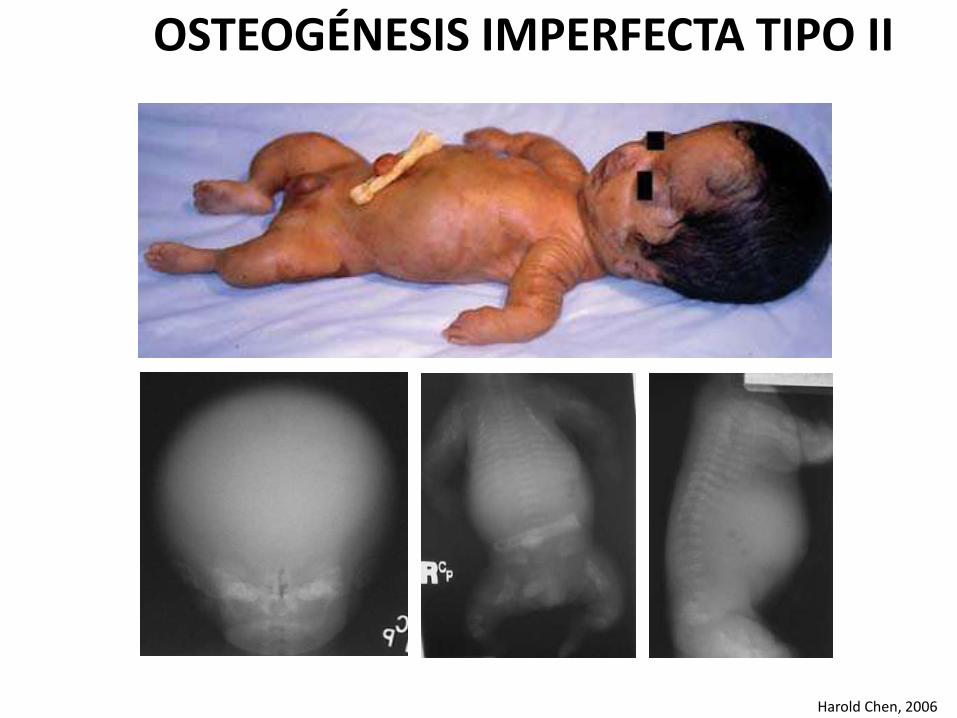
Harold Chen, 2006
OSTEOGÉNESIS IMPERFECTA TIPO II
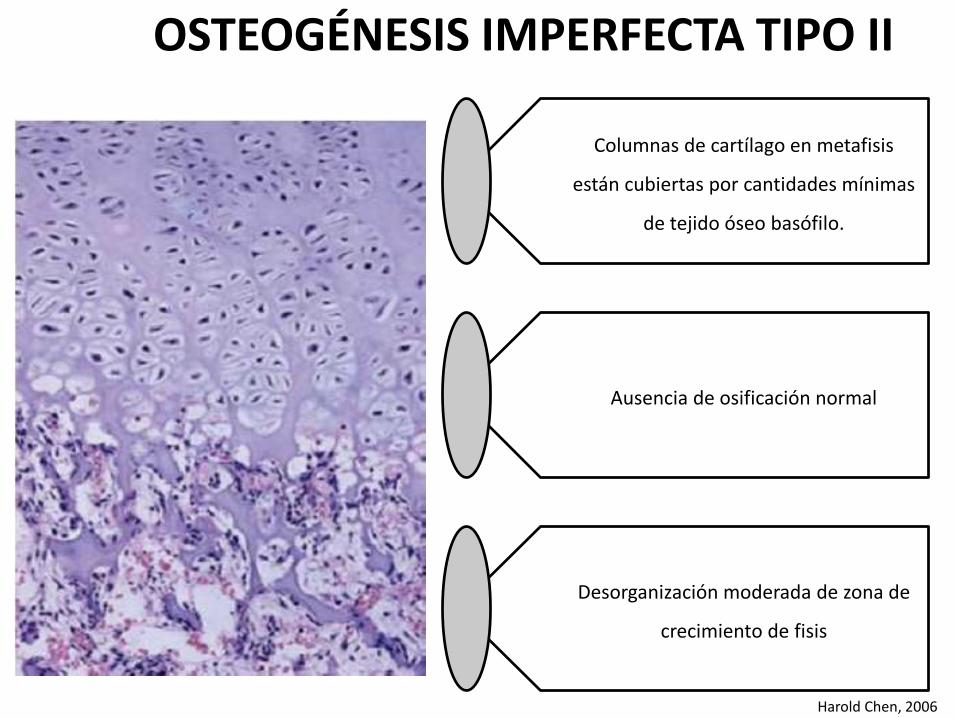
Columnas de cartílago en metafisis
están cubiertas por cantidades mínimas
de tejido óseo basófilo.
Ausencia de osificación normal
Desorganización moderada de zona de
crecimiento de fisis
Harold Chen, 2006
OSTEOGÉNESIS IMPERFECTA TIPO II
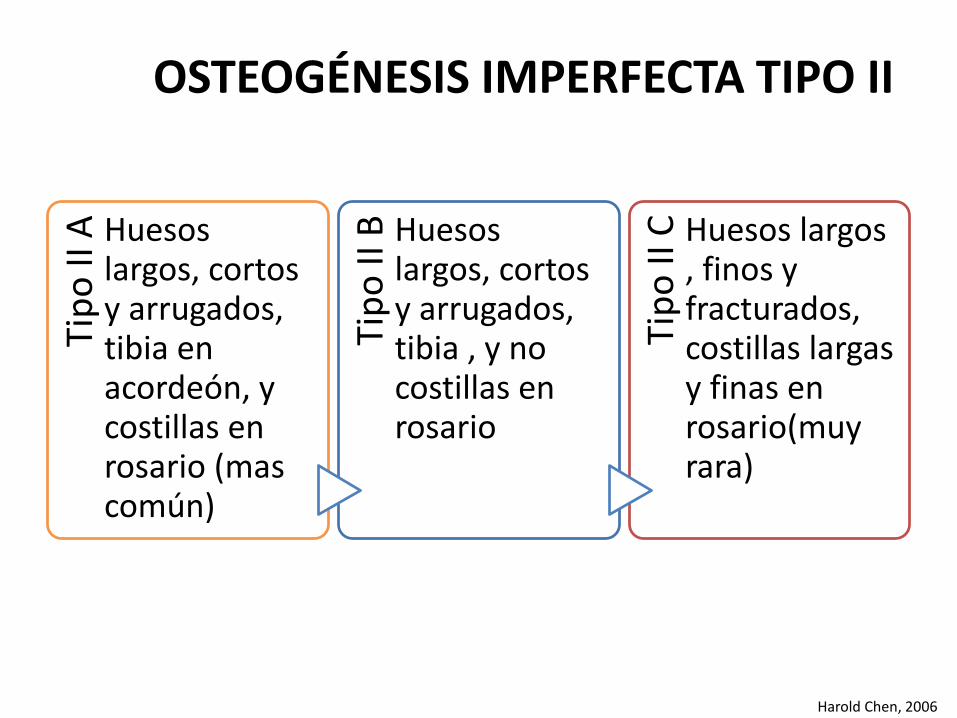
Tip
o II
A Huesos largos, cortos y arrugados, tibia en acordeón, y costillas en rosario (mas común)
Tip
o II
B Huesos largos, cortos y arrugados, tibia , y no costillas en rosario
Tip
o II
C Huesos largos , finos y fracturados, costillas largas y finas en rosario(muy rara)
Harold Chen, 2006
OSTEOGÉNESIS IMPERFECTA TIPO II
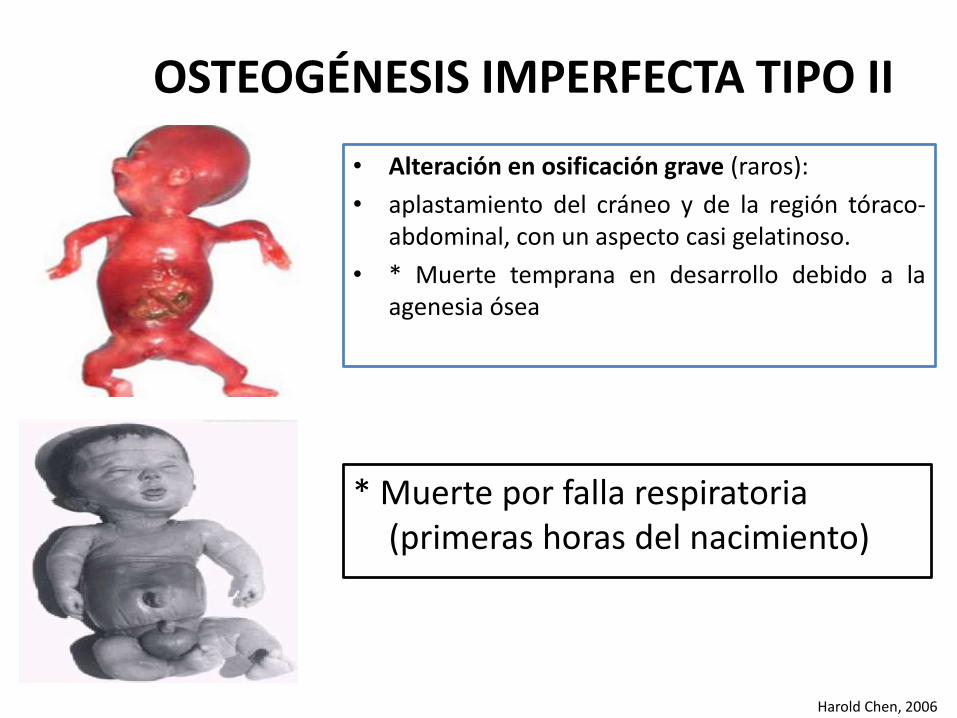
• Alteración en osificación grave (raros):
• aplastamiento del cráneo y de la región tóraco-abdominal, con un aspecto casi gelatinoso.
• * Muerte temprana en desarrollo debido a laagenesia ósea
Harold Chen, 2006
OSTEOGÉNESIS IMPERFECTA TIPO II
* Muerte por falla respiratoria(primeras horas del nacimiento)

OSTEOGÉNESIS IMPERFECTA TIPO III(Deformante)

7q21.3 Osteogenesisimperfecta tipo III
COL1A2
17q21.33 Osteogenesisimperfecta tipo III
COL1A1
“Deformidad progresiva y esclera normal”
Autosómica dominante (Común)
Autosómica recesiva (infrecuente)
Exón 49 en COL1A2
OMIM #259420, Faqeih et al. (2009)
OMIM: 259420
OSTEOGÉNESIS IMPERFECTA TIPO III
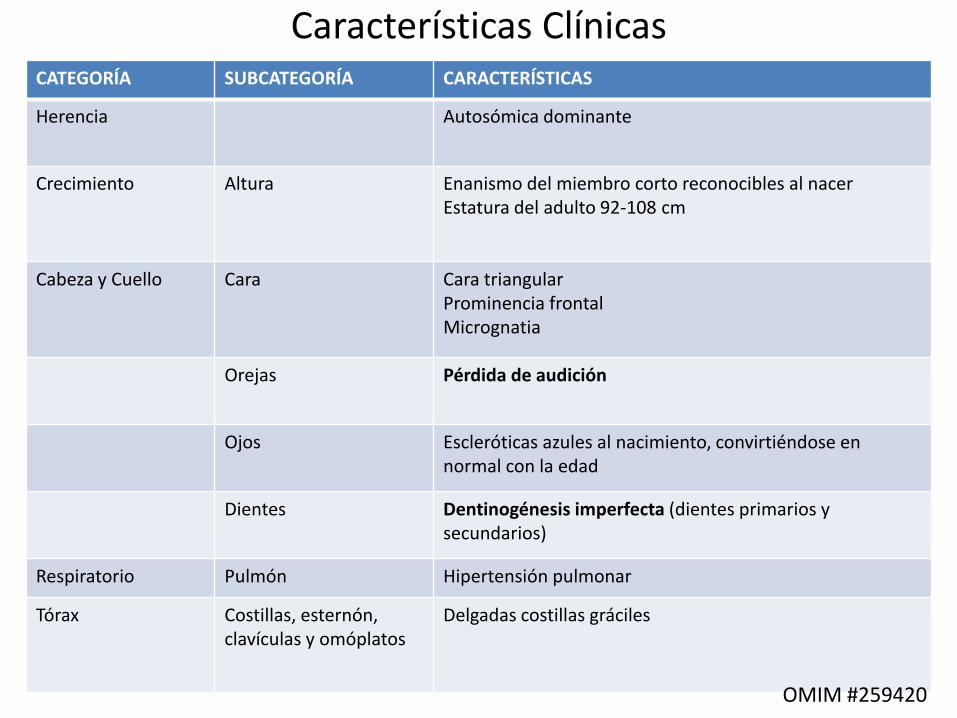
Características ClínicasCATEGORÍA SUBCATEGORÍA CARACTERÍSTICAS
Herencia Autosómica dominante
Crecimiento Altura Enanismo del miembro corto reconocibles al nacer Estatura del adulto 92-108 cm
Cabeza y Cuello Cara Cara triangular Prominencia frontal Micrognatia
Orejas Pérdida de audición
Ojos Escleróticas azules al nacimiento, convirtiéndose en normal con la edad
Dientes Dentinogénesis imperfecta (dientes primarios y secundarios)
Respiratorio Pulmón Hipertensión pulmonar
Tórax Costillas, esternón, clavículas y omóplatos
Delgadas costillas gráciles
OMIM #259420
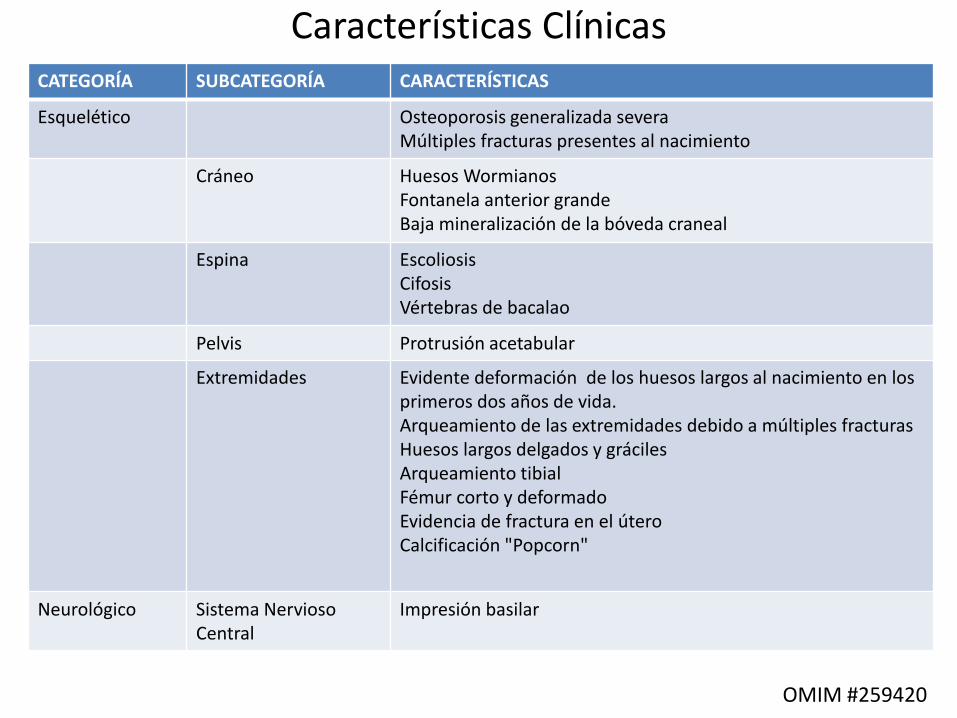
CATEGORÍA SUBCATEGORÍA CARACTERÍSTICAS
Esquelético Osteoporosis generalizada severaMúltiples fracturas presentes al nacimiento
Cráneo Huesos WormianosFontanela anterior grande Baja mineralización de la bóveda craneal
Espina Escoliosis Cifosis Vértebras de bacalao
Pelvis Protrusión acetabular
Extremidades Evidente deformación de los huesos largos al nacimiento en los primeros dos años de vida. Arqueamiento de las extremidades debido a múltiples fracturas Huesos largos delgados y gráciles Arqueamiento tibialFémur corto y deformadoEvidencia de fractura en el útero Calcificación "Popcorn"
Neurológico Sistema Nervioso Central
Impresión basilar
OMIM #259420
Características Clínicas
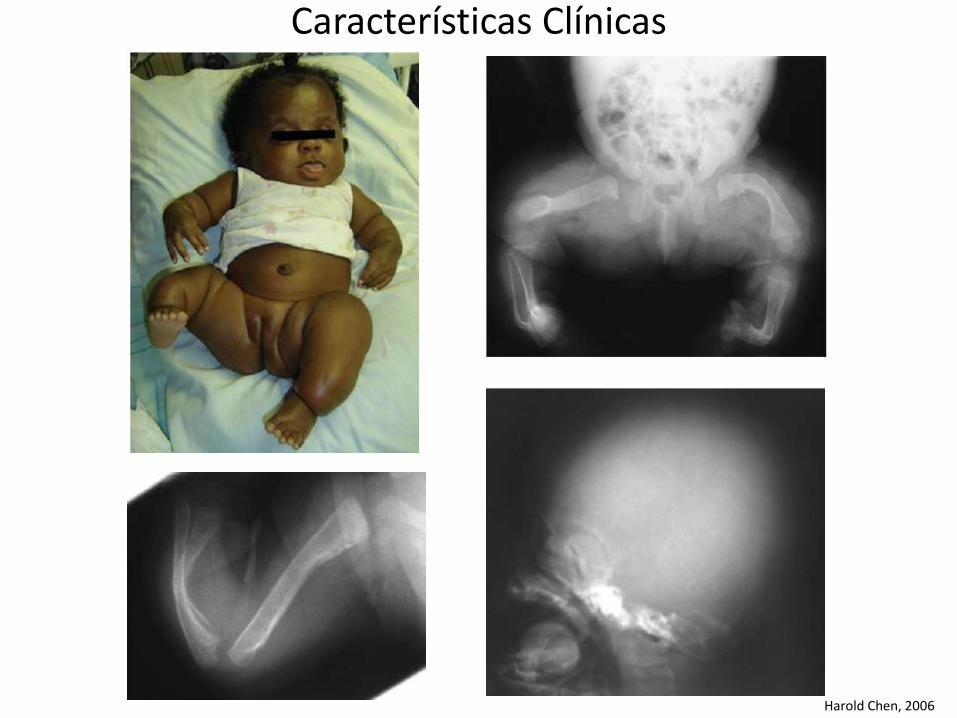
Harold Chen, 2006
Características Clínicas
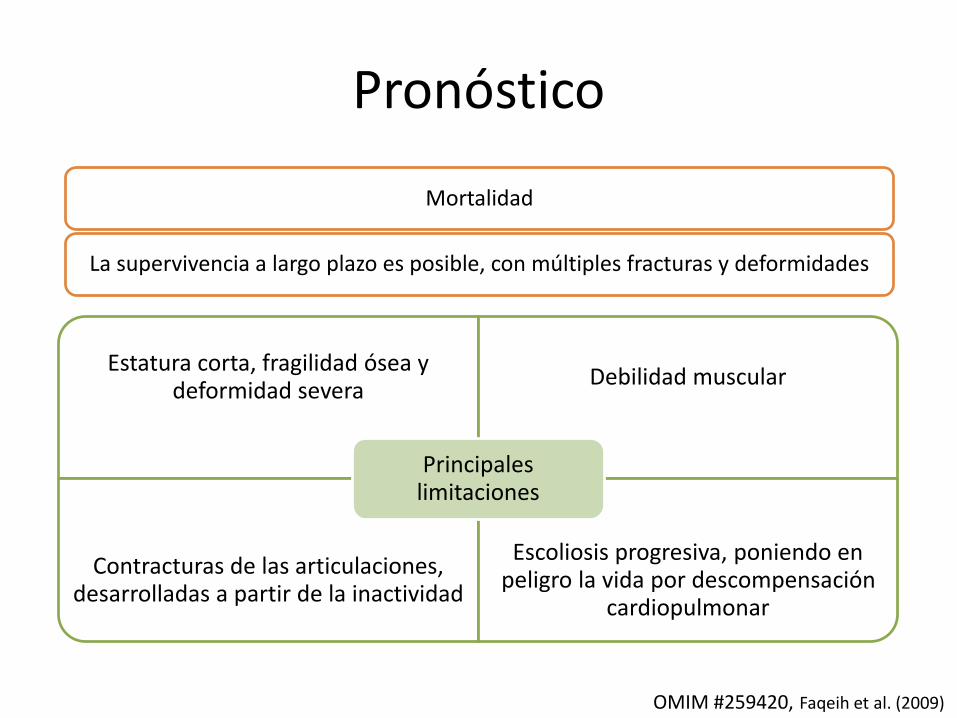
Pronóstico
Mortalidad
La supervivencia a largo plazo es posible, con múltiples fracturas y deformidades
Estatura corta, fragilidad ósea y deformidad severa
Debilidad muscular
Contracturas de las articulaciones, desarrolladas a partir de la inactividad
Escoliosis progresiva, poniendo en peligro la vida por descompensación
cardiopulmonar
Principales limitaciones
OMIM #259420, Faqeih et al. (2009)

OSTEOGÉNESIS IMPERFECTA TIPO IV(Escleróticas blancas)

Tipo moderado de osteogénesis imperfecta
Hernández y cols. 2007 ; POSSUM 2012; Onphanet rare disease
Es la forma más variable de OI
La OI tipo IV da cuenta de un tercio de todos los casos diagnosticados con OI
Osteogénesis imperfecta tipo IV
OMIM: 166220
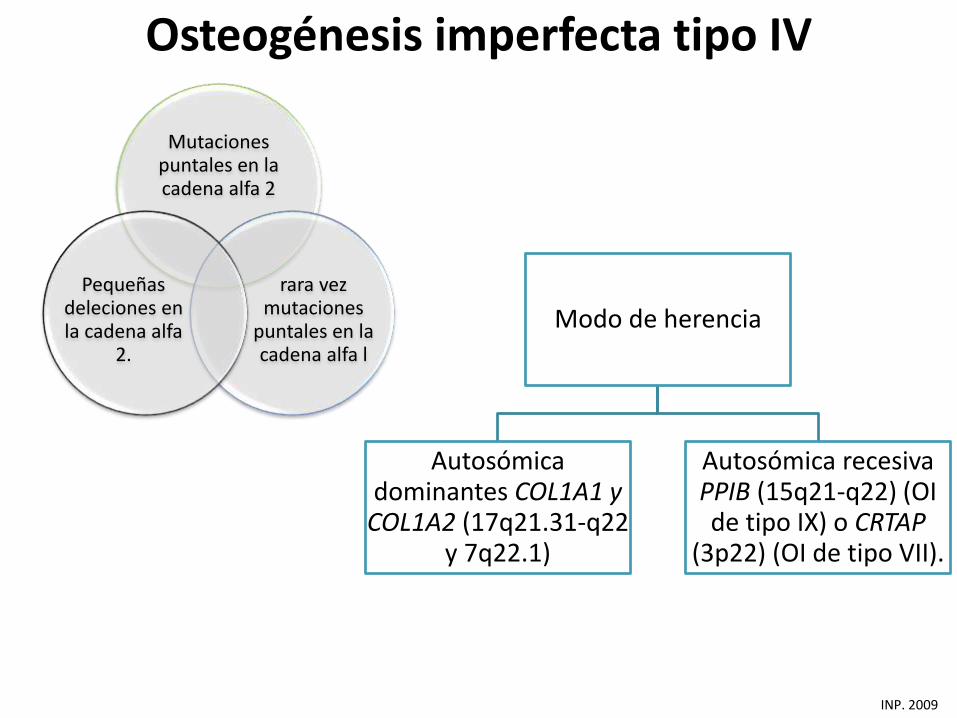
Mutaciones puntales en la cadena alfa 2
rara vez mutaciones
puntales en la cadena alfa l
Pequeñas deleciones en la cadena alfa
2.
INP. 2009
Modo de herencia
Autosómica dominantes COL1A1 y
COL1A2 (17q21.31-q22 y 7q22.1)
Autosómica recesiva PPIB (15q21-q22) (OI de tipo IX) o CRTAP
(3p22) (OI de tipo VII).
Osteogénesis imperfecta tipo IV
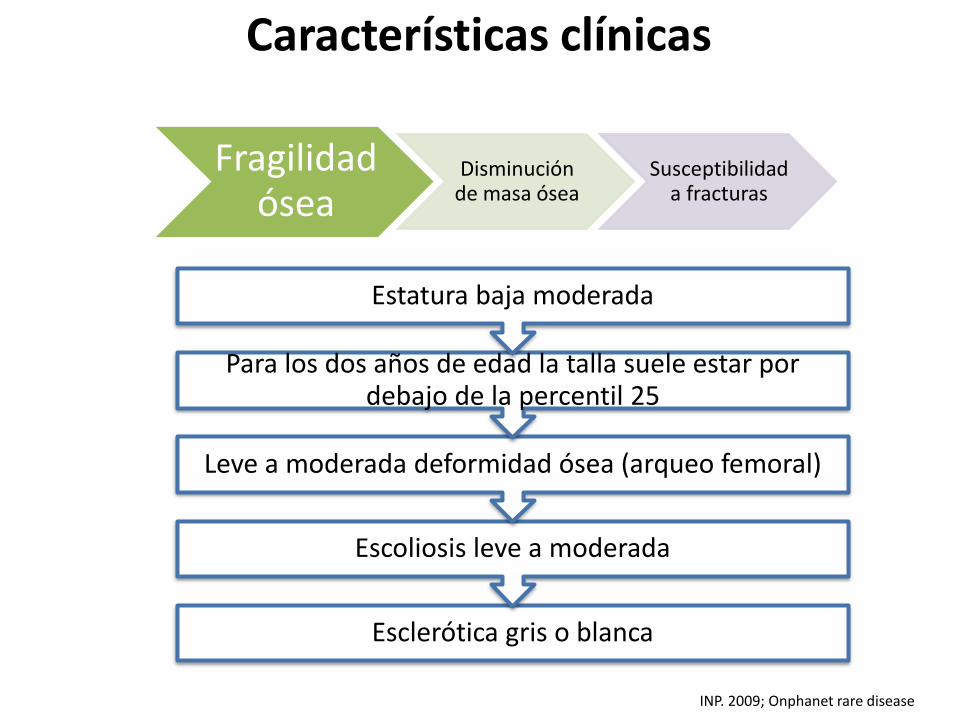
Esclerótica gris o blanca
Escoliosis leve a moderada
Leve a moderada deformidad ósea (arqueo femoral)
Para los dos años de edad la talla suele estar por debajo de la percentil 25
Estatura baja moderada
Fragilidad ósea
Disminución de masa ósea
Susceptibilidad a fracturas
INP. 2009; Onphanet rare disease
Características clínicas

• Disminuye en la pubertad
• Aumenta en las edades mayores
• Especialmente en mujeres postmenopausicas
La frecuencia de las fracturas
• Se ha subdividido este tipo en las formas A y B
• Ambas formas tienen escleras normalesDentinogénesis
imperfecta
La perdida de la audición afecta a menos de la
mitad de los individuos
POSSUM 2012; INP. 2009; Hernández y cols. 2007
Las escleróticas pueden ser azules en la infancia. Tipo IV-A con dientes normales. Tipo IV-B, con dentinogénesis imperfecta.
Características clínicas
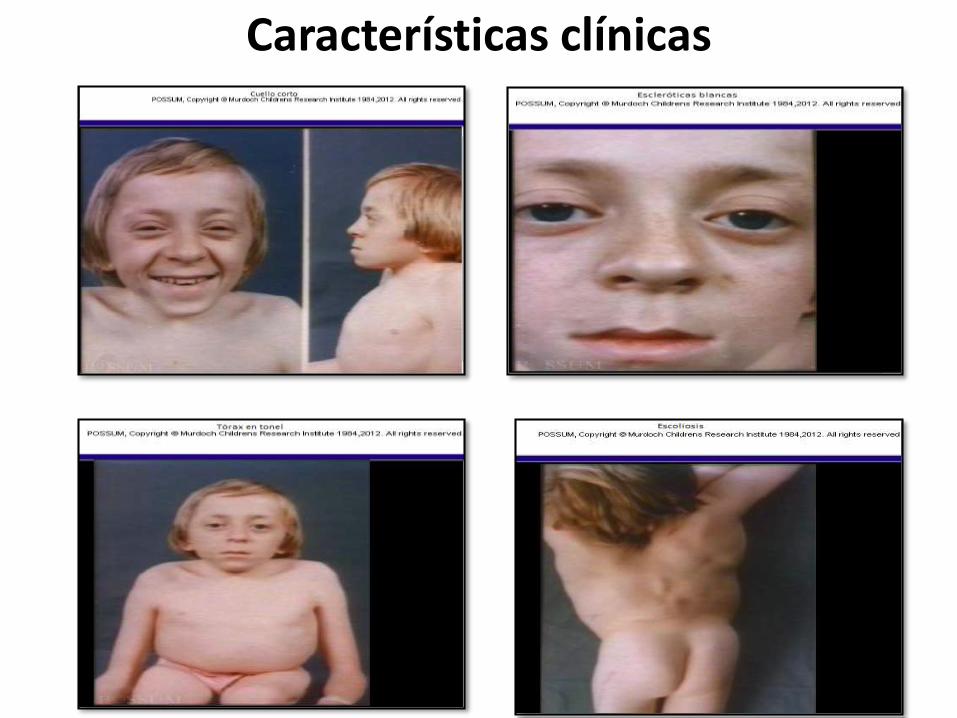
Características clínicas
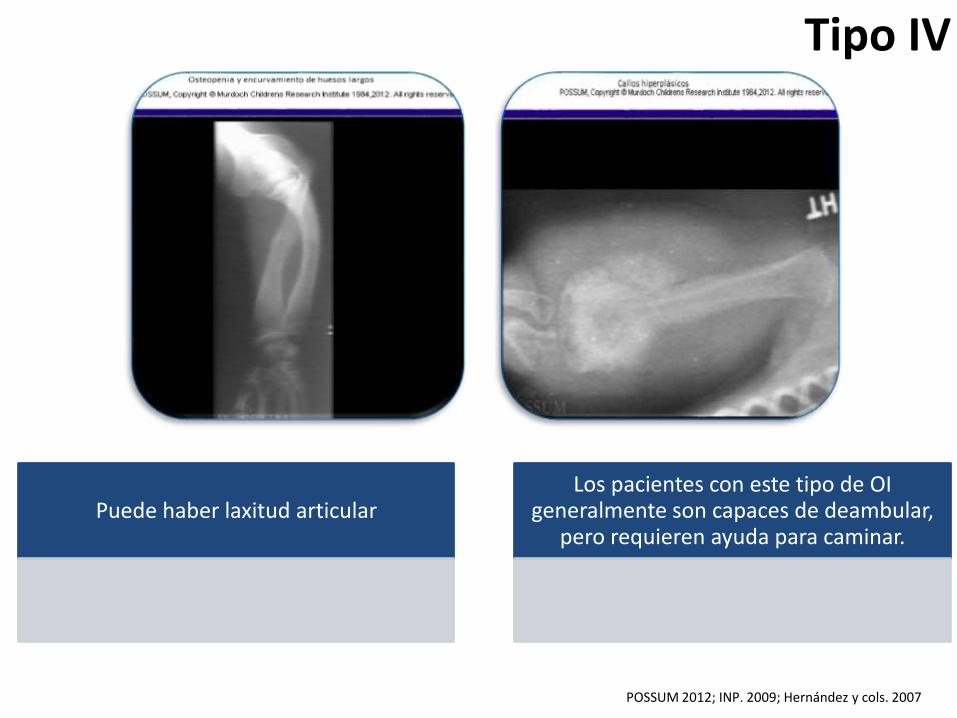
Puede haber laxitud articularLos pacientes con este tipo de OI
generalmente son capaces de deambular, pero requieren ayuda para caminar.
POSSUM 2012; INP. 2009; Hernández y cols. 2007
Tipo IV

Osteogénesis imperfecta tipo VII
La OI tipo VII es una forma autosómica recesiva de grave a letal
OMIM 610682
Gen CRTAP
http://omim.org/entry/605497
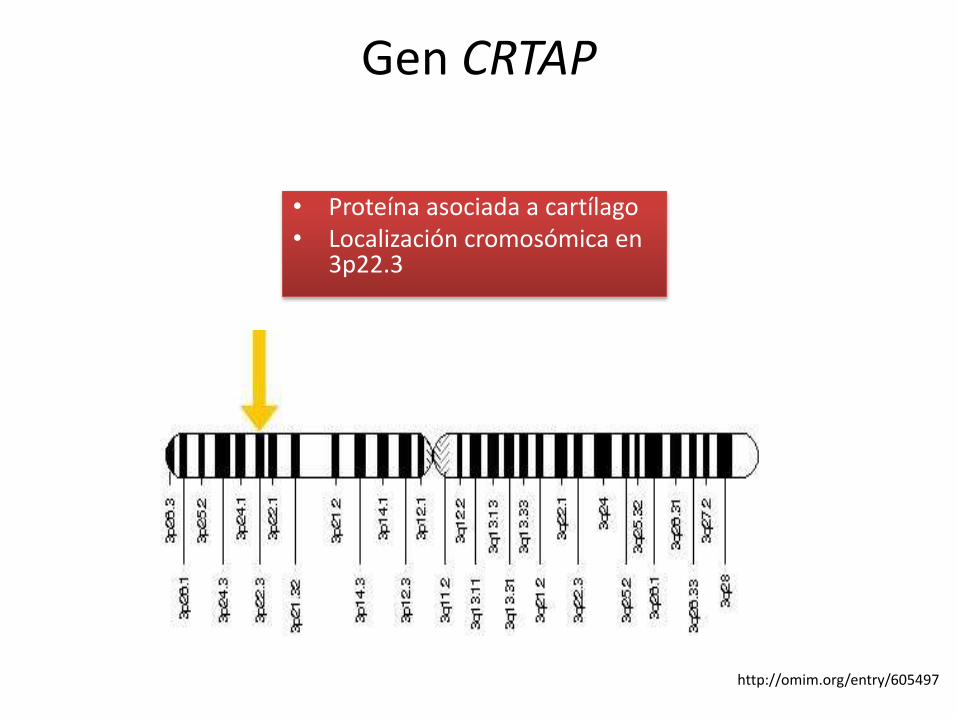
Gen CRTAP
• Proteína asociada a cartílago• Localización cromosómica en
3p22.3
http://omim.org/entry/605497
Función normal del gen CRTAP
CRTAP Proteína asociada a cartílago
La función específica de esta proteína no se conoce , pero se propone que desempeña un papel importante en el desarrollo
normal del hueso .
Fratzl-Zelman et al., 2010

Osteogénesis imperfecta tipo VIII
Cabral et al. 2007 describió una forma de OIautosómica recesiva, que se designan OI tipo VIII.
OMIM 610915Se caracteriza por la esclerótica blanca, deficiencia de
crecimiento, baja mineralización esquelética ymetáfisis bulbosas.
Gen LEPRE1
Genetic Home Reference
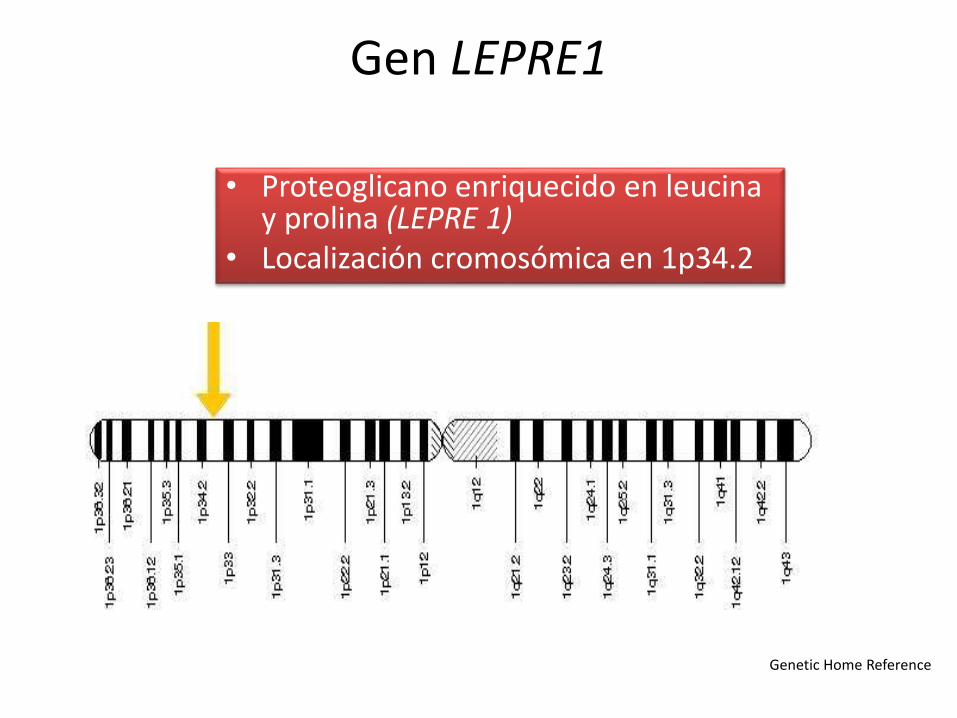
Gen LEPRE1
• Proteoglicano enriquecido en leucina y prolina (LEPRE 1)
• Localización cromosómica en 1p34.2
Genetic Home Reference

Función normal del gen LEPRE 1
LEPRE 1 Prolil-3 hidroxilasa 1
Funciones adicionales, por ejemplo esta enzima puede jugar un papel en las interacciones entre ciertos tipos de células y la matriz extracelular que las rodea, también puede actuar como un supresor tumoral , evitando que las células crezcan y se dividan demasiado rápido o de forma incontrolada.
Moul et al., 2013
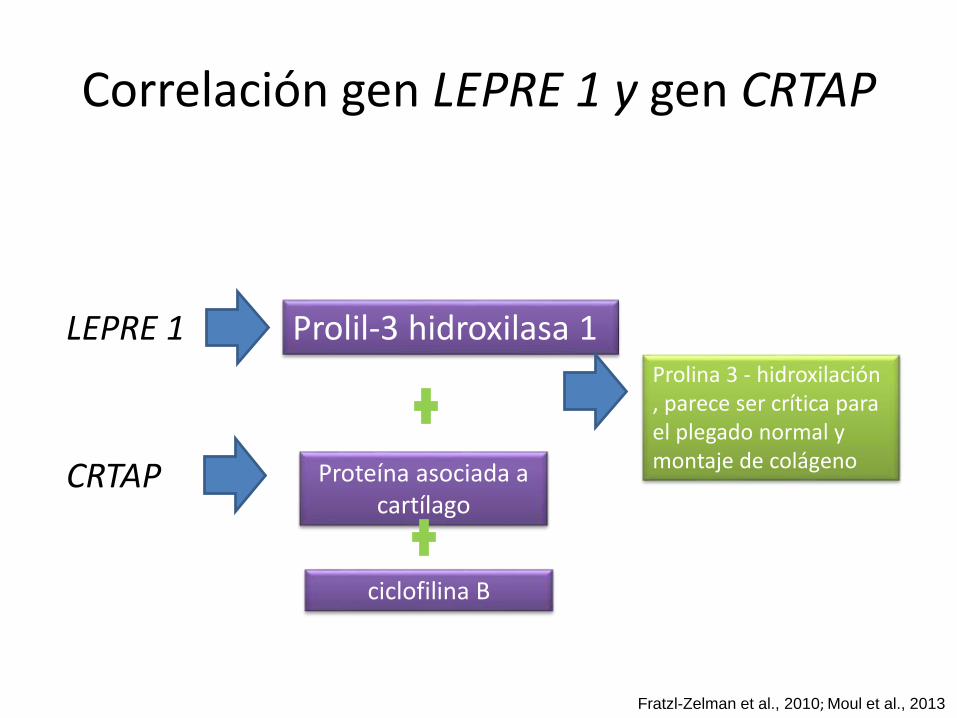
Correlación gen LEPRE 1 y gen CRTAP
LEPRE 1 Prolil-3 hidroxilasa 1
Proteína asociada a cartílago
Prolina 3 - hidroxilación, parece ser crítica para el plegado normal y montaje de colágeno
CRTAP
ciclofilina B
Fratzl-Zelman et al., 2010; Moul et al., 2013
Fratzl-Zelman et al., 2010; Moul et al., 2013

Mutaciones en el gen CRTAP• Al menos cinco mutaciones en el gen CRTAP son responsables
de OI tipo VII.
• Varias de estas mutaciones evitan que las células produzcanproteína asociada a cartílago.
• Sin esta proteína, los huesos y otros tejidos conectivos no seforman adecuadamente, dando lugar a una forma muy gravede la enfermedad.
• Otra mutación en el gen CRTAP reduce en gran medida lacantidad de proteína producida cartílago asociada, queinterrumpe la formación normal de colágeno. Este cambiogenético provoca signos y síntomas de la osteogénesisimperfecta menos graves
Fratzl-Zelman et al., 2010
Baldridge et al., 2008
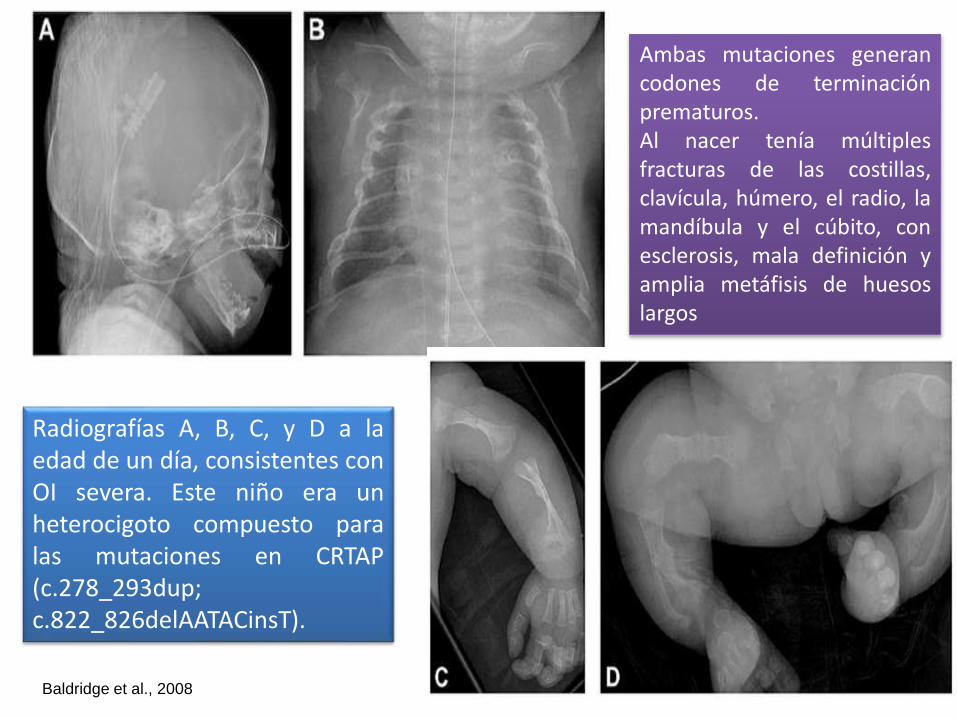
Radiografías A, B, C, y D a laedad de un día, consistentes conOI severa. Este niño era unheterocigoto compuesto paralas mutaciones en CRTAP(c.278_293dup;c.822_826delAATACinsT).
Ambas mutaciones generancodones de terminaciónprematuros.Al nacer tenía múltiplesfracturas de las costillas,clavícula, húmero, el radio, lamandíbula y el cúbito, conesclerosis, mala definición yamplia metáfisis de huesoslargos
Baldridge et al., 2008
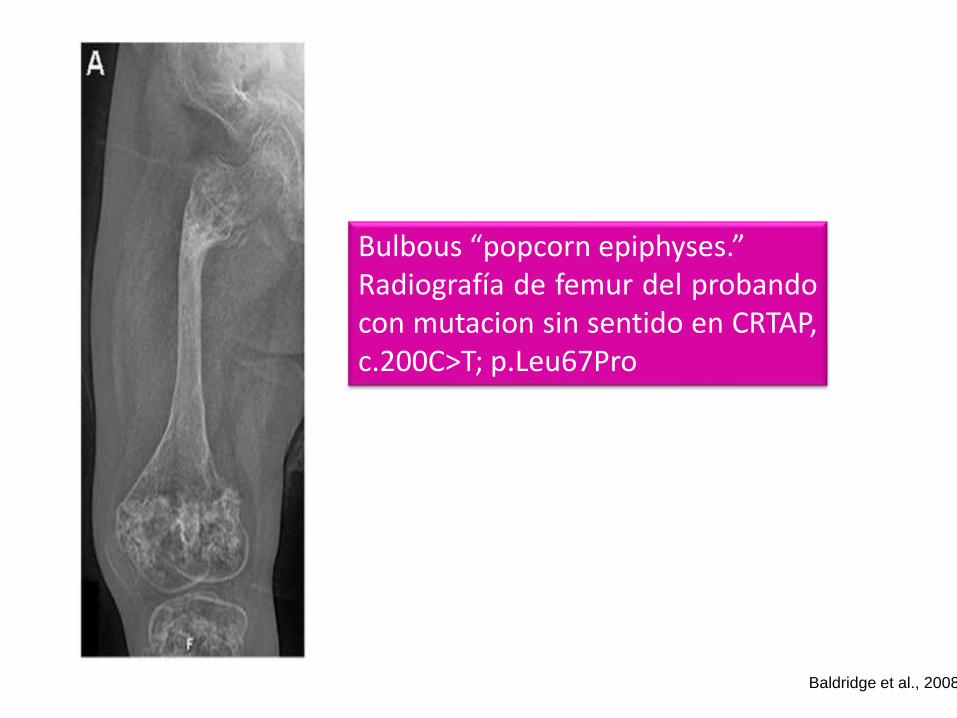
Bulbous “popcorn epiphyses.”Radiografía de femur del probandocon mutacion sin sentido en CRTAP,c.200C>T; p.Leu67Pro
Baldridge et al., 2008

Mutaciones en el gen LEPRE1• Al menos cuatro mutaciones en el gen LEPRE1 se han identificado
en OI tipo VIII.
• Estas mutaciones impiden que las células produzcan Leprecanfuncional.
• Sin esta enzima, ciertas formas de colágeno no se modifican através de prolina 3-hidroxilación.
• Las moléculas de colágeno alteradas son incorrectamenteplegadas, y algunos de los colágenos anormales son secretados apartir de células más lentamente de lo usual.
• Estos defectos de colágeno se debilitan los tejidos conectivos,resultando en un crecimiento extremadamente lento y, huesosfrágiles delgadas que pueden fracturar antes del nacimiento.
Moul et al., 2013
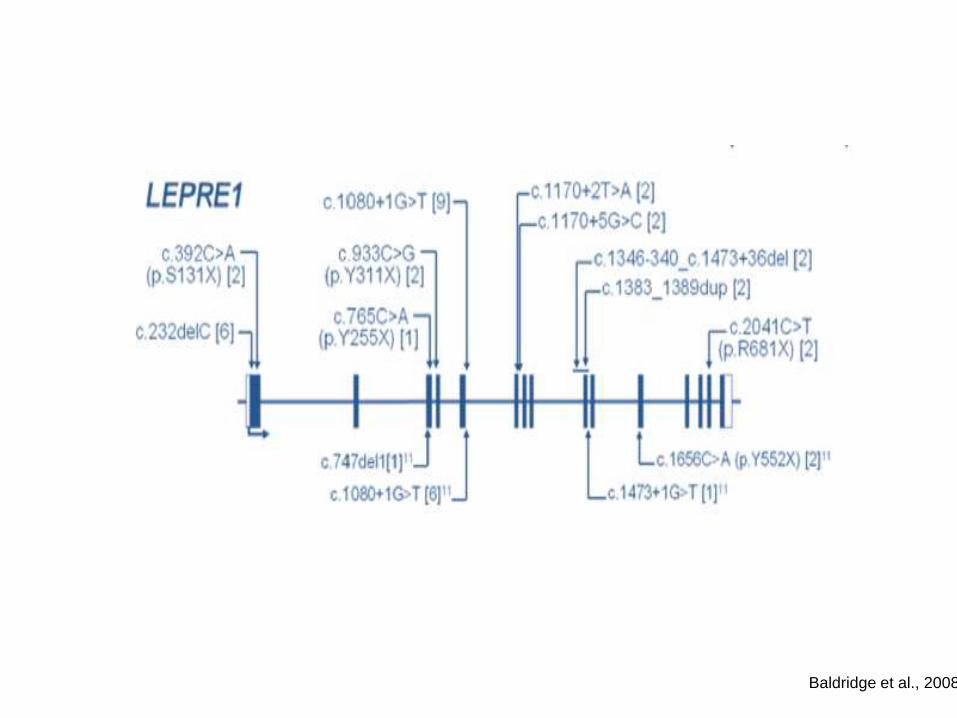
Baldridge et al., 2008
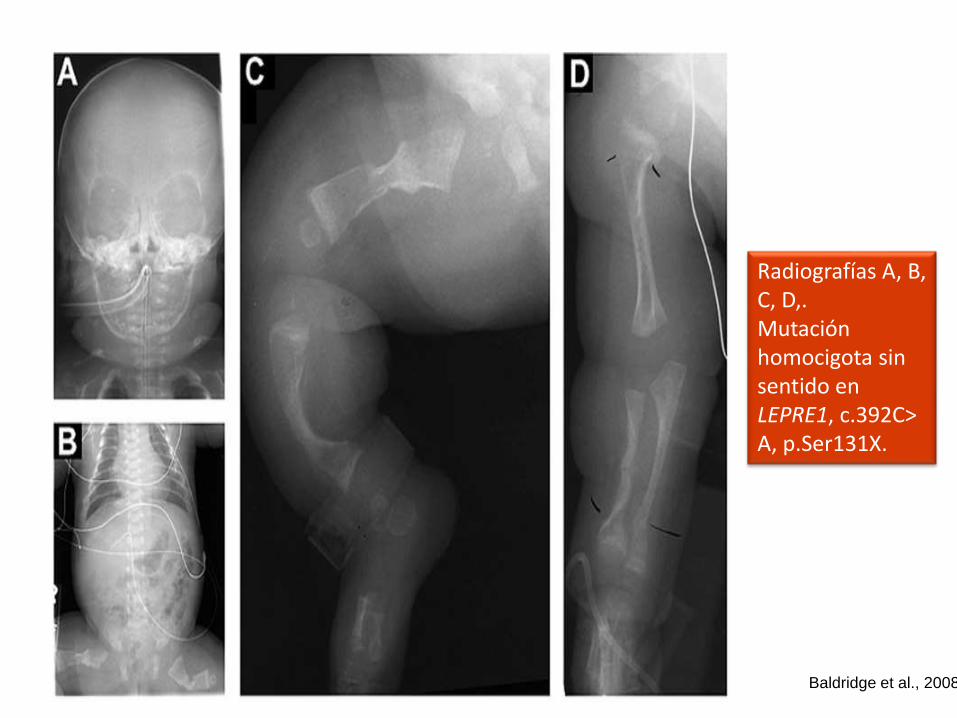
Radiografías A, B, C, D,.Mutación homocigota sin sentido en LEPRE1, c.392C> A, p.Ser131X.
Baldridge et al., 2008

Clínica OI tipo VII Y VIII
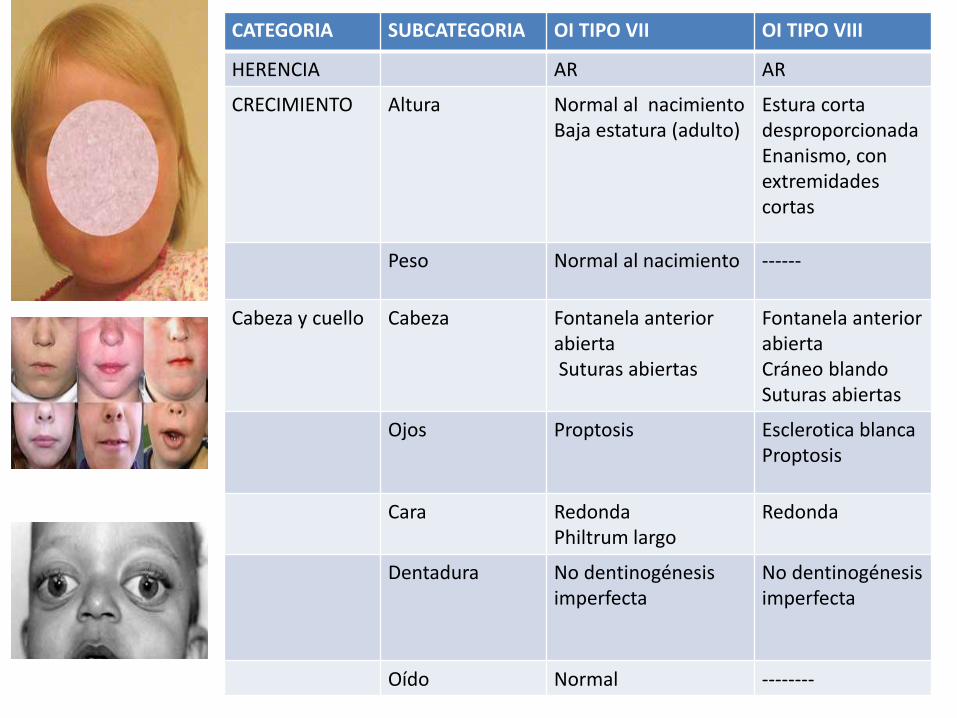
CATEGORIA SUBCATEGORIA OI TIPO VII OI TIPO VIII
HERENCIA AR AR
CRECIMIENTO Altura Normal al nacimientoBaja estatura (adulto)
Estura corta desproporcionadaEnanismo, con extremidades cortas
Peso Normal al nacimiento ------
Cabeza y cuello Cabeza Fontanela anterior abierta Suturas abiertas
Fontanela anterior abierta Cráneo blando Suturas abiertas
Ojos Proptosis Esclerotica blanca Proptosis
Cara RedondaPhiltrum largo
Redonda
Dentadura No dentinogénesisimperfecta
No dentinogénesisimperfecta
Oído Normal --------
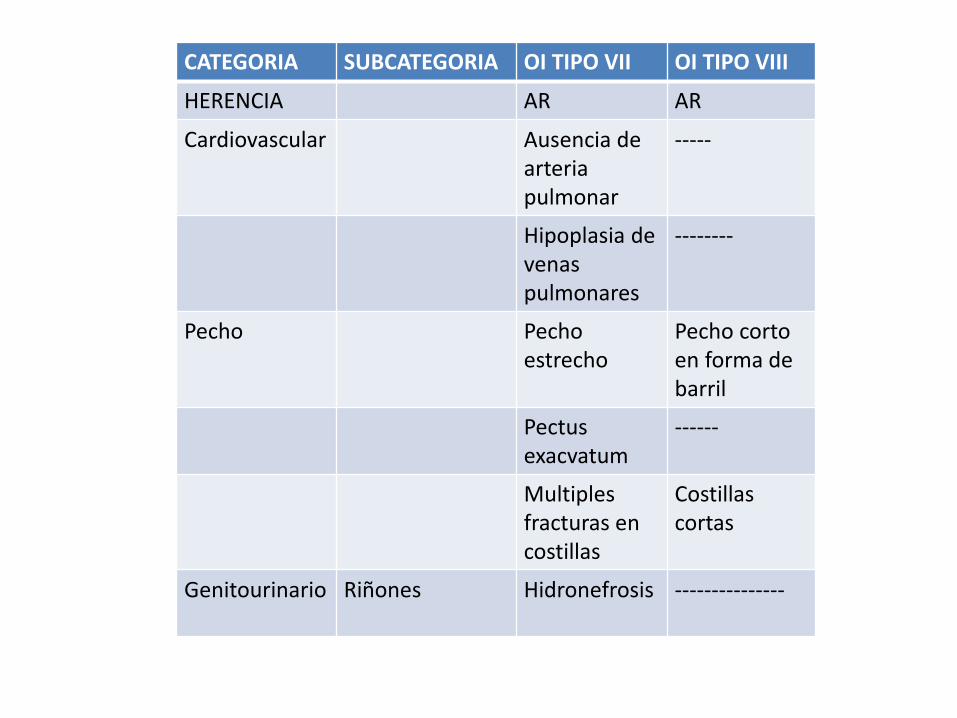
CATEGORIA SUBCATEGORIA OI TIPO VII OI TIPO VIII
HERENCIA AR AR
Cardiovascular Ausencia de arteria pulmonar
-----
Hipoplasia de venaspulmonares
--------
Pecho Pecho estrecho
Pecho corto en forma de barril
Pectusexacvatum
------
Multiplesfracturas en costillas
Costillas cortas
Genitourinario Riñones Hidronefrosis ---------------
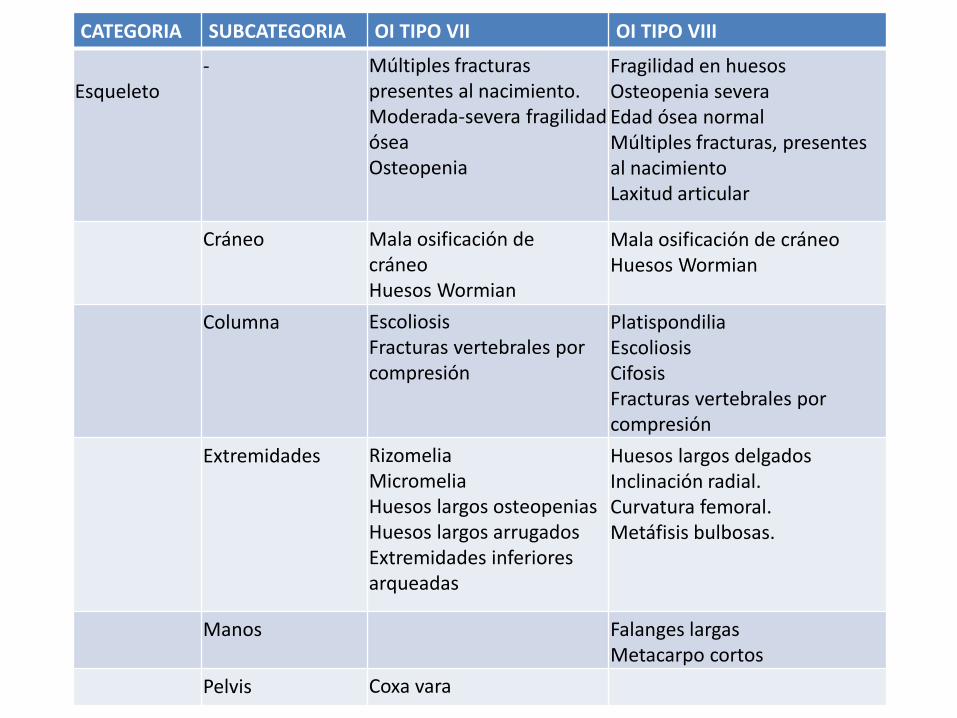
CATEGORIA SUBCATEGORIA OI TIPO VII OI TIPO VIII
Esqueleto- Múltiples fracturas
presentes al nacimiento. Moderada-severa fragilidad ósea Osteopenia
Fragilidad en huesos Osteopenia severa Edad ósea normal Múltiples fracturas, presentes al nacimientoLaxitud articular
Cráneo Mala osificación de cráneo Huesos Wormian
Mala osificación de cráneo Huesos Wormian
Columna EscoliosisFracturas vertebrales por compresión
PlatispondiliaEscoliosisCifosis Fracturas vertebrales por compresión
Extremidades RizomeliaMicromeliaHuesos largos osteopenias Huesos largos arrugados Extremidades inferiores arqueadas
Huesos largos delgados Inclinación radial. Curvatura femoral. Metáfisis bulbosas.
Manos Falanges largasMetacarpo cortos
Pelvis Coxa vara
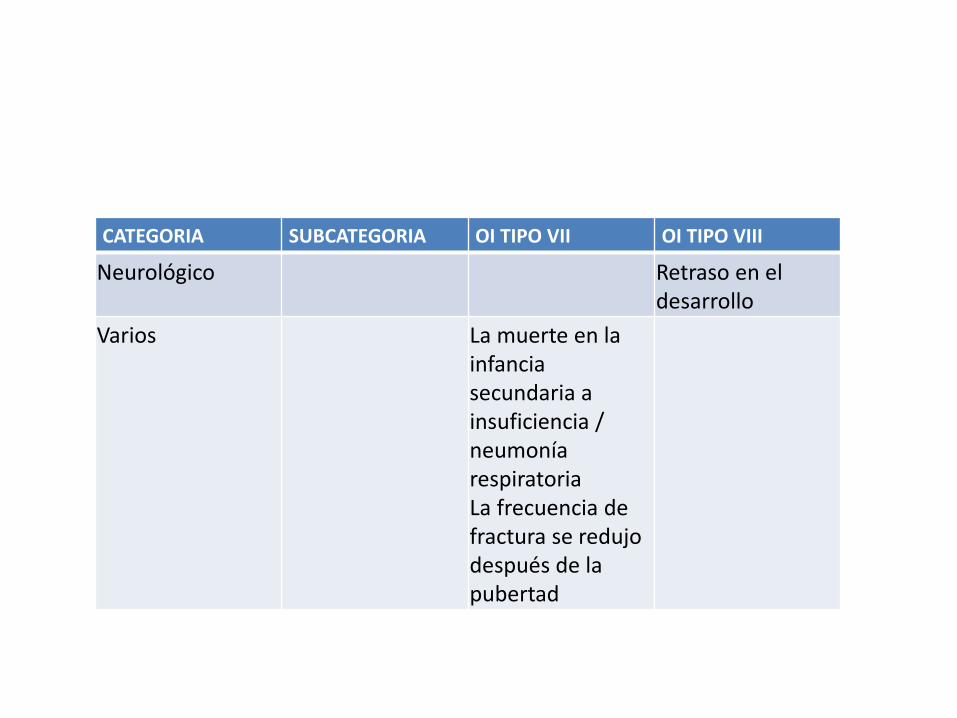
CATEGORIA SUBCATEGORIA OI TIPO VII OI TIPO VIII
Neurológico Retraso en el desarrollo
Varios La muerte en la infancia secundaria a insuficiencia / neumonía respiratoria La frecuencia de fractura se redujo después de la pubertad
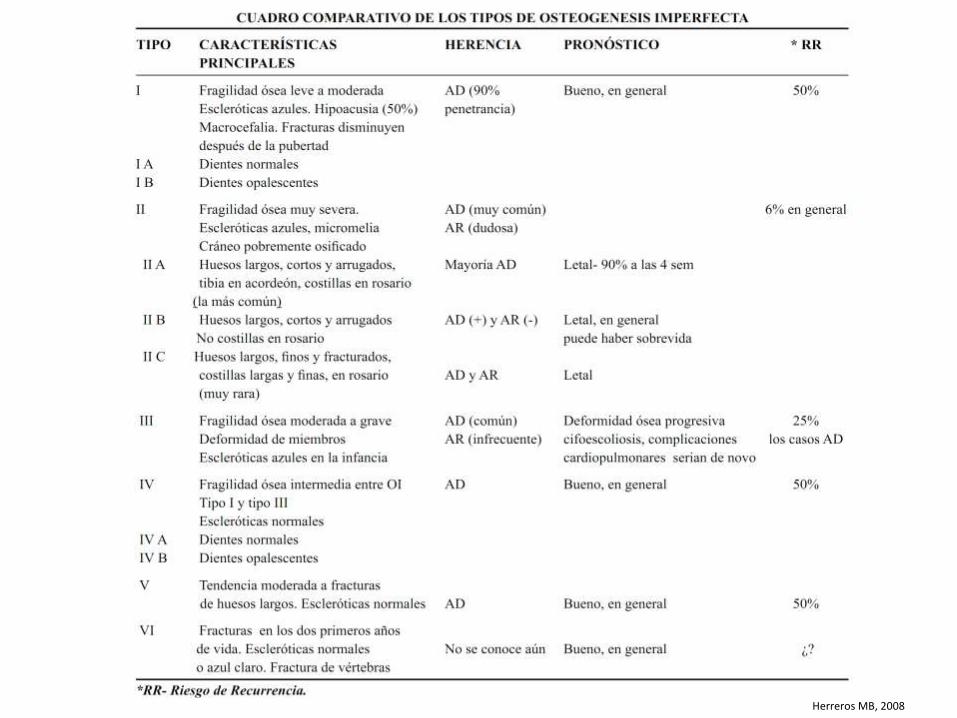
Herreros MB, 2008

www.pediatria.gob.mx Ultima modificación Viernes, 10de abril del 2009.
www.orpha.net Última actualización: Marzo 2010
Data bases POSSUM 20122
Herreros MB, Franco R, Ascurra M. Osteogenesisimperfecta. Pediatr. 2008. 35(1): 33-37
Barnes AM, Chang W, Morello R, y cols. Deficiency ofcartilage-associated protein in recessive lethalosteogenesis imperfecta. N Engl J Med. 2006 Dec28;355(26):2757-64.
Carvajal AM, Iturriaga SR. Osteogénesis Imperfecta.Rev Med de Costa Rica y Centroamérica. 2007;LXIV(580):161-165
Robert D Steiner, MD, Melanie G Pepin, MS, CGC,and Peter H Byers, MD,Osteogenesis Imperfecta BrittleBone Disease, OI. Gene Review. 2005
Harold Chen, Atlas of Genetic Diagnosis andCounseling, 2006, pp762-772
Choi JH, Shin YL, Yoo HW. Short-term Efficacy of
Monthly Pamidronate Infusion in Patients with
Osteogenesis Imperfecta J Korean Med Sci 2007;
22: 209-12
Hernández García Iván, Fernández Martín Mirely,
León Pérez Silvia, y cols. Osteogénesis imperfecta:
mosaicismo germinal o evidencia de heterogeneidad
genética. Presentación de una familia y revisión
bibliográfica. Rev Cubana Pediatr. 2007.79(3)
http://www.uth.tmc.edu/GeneWise/ShortTallLoose/08.html
Bibliografía


















