
OBTENCION Y CARACTERIZACION DE LA ENZIMA FRUCTOSA-1,6 ...
91
Profesor Patrocinante Dr. Juan Carlos Slebe Instituto de Bioquímica Facultad de Ciencias OBTENCION Y CARACTERIZACION DE LA ENZIMA FRUCTOSA-1,6-BISFOSFATASA DE RIÑON DE CERDO MUTADA EN EL RESIDUO LISINA 274 A TRIPTÓFANO Tesis de Grado presentada como parte de los requisitos para optar al grado de Licenciado en Bioquímica y Título Profesional de Bioquímico LUCIANO ANDRÉS RIVERA MARCOS VALDIVIA – CHILE 2013
Transcript of OBTENCION Y CARACTERIZACION DE LA ENZIMA FRUCTOSA-1,6 ...

Profesor Patrocinante Dr. Juan Carlos Slebe Instituto de Bioquímica Facultad de Ciencias
OBTENCION Y CARACTERIZACION DE LA ENZIMA FRUCTOSA-1,6-BISFOSFATASA DE RIÑON DE CERDO MUTADA EN EL RESIDUO LISINA 274 A TRIPTÓFANO
Tesis de Grado presentada como parte de los requisitos para optar al grado de Licenciado en Bioquímica y Título Profesional de Bioquímico
LUCIANO ANDRÉS RIVERA MARCOS
VALDIVIA – CHILE 2013

Dedicada a mi esposa, hijas y madre, que gracias a su apoyo y sacrificio me ha permitido cerrar
este ciclo.

Agradecimientos
Les doy mis más sinceros agradecimientos al Dr. Juan Carlos Slebe y al Dr. Alejandro
Yañez, que me permitieron ser parte de su equipo de trabajo y apoyaron durante esta etapa de mi
formación.
Gracias a todos los tesistas y estudiantes que participaron junto a mi y de modo conjunto
logramos crecer profesionalmente, de modo especial al Dr. Joel Asenjo quién fue un guía en los
momentos más complejos, a Francisco Montero quien estuvo siempre junto a mi como el gran
amigo que es y a Stephan Schott por su ayuda en bioinformatica.

i
I. Índice.
a. Índice de contenidos
Página
I. Índice i
a. Índice de Contenidos i
b. Índice de Figuras iv
c. Índice de Tablas vi
II. Abreviaturas vii
1. Resumen ix
1.1. Summary x
2. Introducción 1
2.1. Generalidades del metabolismo de la glucosa 1
2.2. Características cinéticas de la fructosa-1,6-bisfosfatasa. 4
2.3. Características estructurales de la fructosa-1,6-bisfosfatasa. 6
2.4. Estudio de la relación entre la estructura y función de la fructosa-1,6-
bisfosfatasa.
9
2.5. Objetivos. 11
2.5.1. Objetivo General. 11
2.5.2. Objetivos Específicos. 11
3. Materiales y Métodos. 12
3.1. Materiales. 12
3.1.1. Reactivos. 12

ii
3.1.2. Equipos. 13
3.2. Métodos. 14
3.2.1. Mutagénesis Sitio dirigida. 14
3.2.2. Preparación y transformación de células competentes. 16
3.2.3. Análisis de los plasmidios obtenidos. 18
3.2.4. Expresión y purificación de proteína recombinante. 19
3.2.5. Determinación de la actividad enzimática. 23
3.2.6. Determinación de las constantes cinéticas. 24
3.2.7. Determinación de la constante de activación por Magnesio. 26
3.2.8. Determinación de la constante de inhibición por AMP. 26
3.2.9. Determinación de la constante de inhibición por fructosa-2,6-
bifosfato.
27
3.2.10. Determinación de los cambios en la emisión de fluorescencia. 27
4. Resultados. 29
4.1. Obtención de la mutante de FBPasa de riñón de cerdo, Lys274Trp. 29
4.1.1. Mutagénesis sitio dirigida. 29
4.1.2. Análisis de los plasmidios obtenidos. 29
4.1.3. Secuenciación de los pADNs. 31
4.2. Expresión y purificación de la mutante de FBPasa Lys274Trp. 33
4.2.1. Expresión. 33
4.2.2. Purificación. 33
4.3. Caracterización Cinética. 37
4.3.1. Curva de saturación por sustrato. 37

iii
4.3.2. Curva de activación por Mg2+. 39
4.3.3. Curva de inhibición por AMP. 39
4.3.4. Curva de inhibición por fructosa-2,6-bisfosfato. 42
4.4. Caracterización de las propiedades de fluorescencia de las mutantes de
FBPasa.
45
4.4.1. Unión de fructosa1,6-bisfosfato a la mutante FBPasaLys274Trp. 47
4.4.2. Unión de fructosa-2,6-bisfosfato a la mutante FBPasa Lys274Trp. 47
4.4.3. Unión de Mg2+ a la mutante FBPasa Lys274Trp. 50
4.4.4. Unión de AMP a la mutante FBPasa Lys274Trp. 50
5. Discusión. 54
6. Bibliografía 68

iv
b. Índice de Figuras
Figura Título Página
1 Esquema resumido que representa las tres reacciones irreversibles de la
glicólisis, al lado derecho y de la gluconeogénesis, al lado izquierdo.
3
2 Estructura cuaternaria del tetrámero de FBPasa de riñón de cerdo, mostrado
en diagrama de cintas.
7
3 Diagrama de cintas de la subunidad C1 del tetrámero de FBPasa. 8
4 Mecanismo de expresión inducido por IPTG en una cepa bacteriana BL21
DE3 transformada con el vector pET 15b.
22
5 Análisis de restricción y PCR de los pADNs obtenidos desde las colonias de
bacterias transformadas.
33
6 Alineamiento del producto de la secuenciación contra la secuencia tipo
silvestre de FBPasa de riñón de cerdo.
35
7 Perfil parcial de elusión de la mutante Lys274Trp desde una columna de
sefarosa-níquel.
38
8 Purificación de la mutante de FBPasa Lys274Trp. 39
9 Efecto de la concentración del sustrato Fru-1,6-P2 sobre la actividad de la
fructosas-1,6-bisfofatasas recombinante y mutante.
41
10 Efecto de la concentración de Mg2+ sobre la actividad de las enzimas
silvestre y mutante de FBPasa.
43
11 Efecto de la concentración de AMP+ sobre la actividad de las enzimas
silvestre y mutante de FBPasa.
44

v
12 Efecto de la concentración de Fru-2,6-P2 sobre la actividad de las enzimas
silvestre y mutante de FBPasa.
46
13 Espectros de emisión de fluorescencia de las FBPasas recombinantes
silvestre y mutante Lys274Trp.
50
14 Efecto de la concentración de Fru-1.6-P2 en la emisión de fluorescencia de
la mutante FBPasa Lys274Trp.
52
15 Efecto de la concentración de Fru-2.6-P2 en la emisión de fluorescencia de
la mutante FBPasa Lys274Trp.
53
16 Efecto de la concentración del catión bivalente magnesio en la emisión de
fluorescencia de la mutante FBPasa Lys274Trp.
55
17 Efecto en la intensidad de la fluorescencia en la enzima Lys274Trp mediado
por la unión de AMP
56
18 Ampliación del dominio de unión de Fru-2,6-P2 67
19 Superposición de los esquemas de cintas, ampliado el dominio de unión de
Fru-2,6-P2 de la FBPasa tipo silvestre y la mutada computacionalmente.
70

vi
c. Índice de Tablas
Tabla Título Página
I Parámetros de preparación de la reacción de mutagénesis para 1 tubo 15
II Parámetros de las temperaturas y tiempos utilizados en el termociclador para
la reacción de mutagénesis.
16
III Parámetros utilizados en la preparación de la mezcla de digestión por
endonucleasas.
19
IV Parámetros de las temperaturas y tiempos utilizados en el termociclador para
la reacción de PCR
20
V Parámetros de tiempo y volumen de la cromatografía, configurados en el
equipo.
25
VI Purificación de la mutante de FBPasa Lys274Trp. 37
VII Resumen de las constantes cinéticas de la enzima tipo silvestre y mutante. 48
VIII Resumen de las constantes de disociación del complejo FBPasa Lys274Trp-
ligando y de los coeficientes de Hill para las interacciones
58

vii
II. Abreviaturas
AMP adenosina 5’-monofosfato.
ATP adenosina trifosfato.
EtBr bromuro de etidio.
BSA albúmina sérica de bovino.
cDNA ácido desoxiribonucleico complementario
DAB diaminobencidina.
RPMI medio de cultivo Roswell park memorial institute.
dNTPs deoxinucleotidos trifosfato.
EDTA ácido etilendiamonotetraacético.
FBPasa fructosa-1,6-bisfosfatasa.
Fru-1,6-P2 fructosa-1,6-bisfosfato.
Fru-2,6-P2 fructosa-2,6-bisfosfato.
Fru-6-P fructosa-6-fosfato.
kb kilobase(s).
kDA kilodalton(s).
pb pares de base(s).
PCR reacción de la DNA polimerasa en cadena.
RNA ácido ribonucleico.
SDS lauril sulfato de sodio.

viii
TEMED N,N,N’,N’- tetrametiletilendiamina.
Tris tris[hidroximetil]aminometano.
U unidad enzimática.

ix
1. Resumen
La enzima gluneogénica fructosa-1,6-bifosfatasa, la cual en presencia de cationes
bivalentes cataliza la hidrólisis de fructosa-1,6-bifosfato a fructosa-6-fosfato más un fosfato
inorgánico, es irreversible en condiciones fisiológicas. Esta enzima es inhibida por AMP y
fructosa-2,6-bifosfato de modo alostérico y competitivo respectivamente y ambas presentan un
efector sinérgico en la inhibición y además presenta una inhibición por exceso de sustrato.
Debido a la importancia que tiene esta enzima para la regulación del proceso gluconeogénico, se
han realizado numerosos estudios sobre la relación estructura y función de la FBPasa y es así
como se decide generar y caracterizar la enzima mutante de FBPasa Lys274Trp, principalmente
con la intención de dilucidar los posibles efectos que se producen en el putativo sitio activo
visualizados por las variaciones en la fluorescencia que sean mediadas por la unión de los
distintos efectores descritos para la enzima. Los resultados cinéticos muestran un aumento de
aproximadamente 6 veces en la Km, 12 veces en la I50 para fructosa-2,6-bifosfato, 2 veces en la
Ka para Mg2+, una perdida en la cooperatividad para la unión de AMP y un aumento en la
concentración de sustrato necesaria para producir la inhibición por exceso de sustrato. Con
respecto a los ensayos de unión el principal efecto detectado corresponde a la unión de AMP. De
este modo se concluye que el residuo Lys274 tiene un efecto en la estabilización de la unión del
sustrato y el inhibidor fructosa-2,6-bifosfato y además se sugiere que este residuo puede estar
involucrado en la transducción de señal al sitio activo mediado por la unión de AMP.

x
1.1 Summary
The gluconegenic enzyme fructose-1,6-bisphosphate, catalyzes the hydrolysis of fructose-
1,6-bisphosphate, in presence of bivalent cations, to fructose-6-phosphate and inorganic
phosphate, which is irreversible in physiologic conditions. This enzyme is inhibited by AMP and
fructose-2,6-bisphosphate, the first in allosteric way and the second in competitive way and both
do a synergic effect, additionally, the excess of sustrate present a inhibitory effect. Because this
enzyme is very important for the regulation of the gluconeogenic process, has been a target for a
lot of structure-function studies, for this reason we chose to generate and characterize the mutant
of FBPase Lys274Trp, primarily intended to elucidate the possible effects that occur in the
putative active site displayed by changes in fluorescence, mediated through binding of various
effectors described for the enzyme. Kinetic results show an increase of about 6 times the Km, 12
times the I50 for fructose-2,6-bisphosphate, 2-fold for Mg2+ Ka, a loss in cooperativity to AMP
binding and increased the concentration of substrate needed to produce the inhibition for excess
substrate. With respect to binding assays, the main change detected it was for the effect of AMP.
Thus we conclude that the residue Lys274 has a stabilizing effect on the binding of substrate and
inhibitor fructose-2,6-bisphosphate, but the effect of AMP suggested that may be involved in
signal transduction to active site mediated for the binding of this inhibitor.

1
2. Introducción.
2.1. Generalidades del metabolismo de la glucosa
Por distintos motivos la glucosa ocupa una posición central en el metabolismo pues en la
mayoría de las células de organismos superiores es la principal fuente de energía. En breve, su
importancia radica en:
- Aportar energía a la célula en forma de ATP, cuando esta se oxida a piruvato por medio
de la vía glicolítica, y posteriormente el piruvato puede aportar aún más energía al
incorporarse al ciclo de Krebs (Nelson & Cox 2005).
- Ser almacenada como glucógeno, y frente a hipoglicemias o carencias energéticas la
degradación de este polímero es el primer modo de normalizarlas (Nelson & Cox 2005).
- Aporta poder reductor a la célula en forma de NADPH, al oxidarse a ribosa-5-fosfato en
la vía de las pentosas fosfato (Nelson & Cox 2005).
La mantención de la glicemia en valores relativamente constantes es de vital importancia,
considerando que una hipoglicemia prolongada puede ser catastrófica para los distintos tipos
celulares que ocupan la glucosa como principal o única fuente de energía, por ejemplo:
eritrocitos, células del sistema nervioso, testicular y cerebral. Por esto mismo, se debe
proporcionar a la célula un suministro constante de este azúcar, reconociéndose tres formas en
que la glucosa puede ser aportada: alimentación, la obtenida por glicogenólisis y
gluconeogénesis (Pilkis & Granner 1992).
La vía gluconeogénica es un proceso anabólico que ante el estimulo de bajos niveles
sanguíneos de glucosa, que en condiciones normales solo está dado por ayunos prolongados, es

2
capaz de utilizar moléculas no glucídicas de bajo peso molecular, tales como lactato, piruvato,
glicerol y aminoácidos, para generar glucosa (Pilkis et al. 1988).
La regulación de esta vía tiene una trascendental importancia, porque al estar en un
estrecho equilibrio con la glicólisis, y al ser la mayoría de sus reacciones reversibles y comunes
para ambas vías, se podrían generar ciclos fútiles y el consecuente desaprovechamiento de
energía. La unidireccionalidad de estas vías esta asegurada por tres reacciones prácticamente
irreversibles denominadas “etapas claves”, las cuales se esquematizan en la figura 1.
Estas vías metabólicas poseen un sistema de control muy sensible a los requerimientos
celulares. El reciclaje de sustratos y productos de estas reacciones opuestas puede ser controlado
por la velocidad y dirección del flujo neto; por una serie de metabolitos en todos los organismos,
y por varias hormonas en animales. Además, este control opera por varios medios, los que
incluyen cambios en la concentración de las enzimas involucradas, modificación covalente de
estas enzimas y la interacción con efectores positivos y negativos (Pilkis et al., 1988).

3
Figura 1. Esquema resumido que representa las tres reacciones irreversibles de la glicólisis,
al lado derecho y de la gluconeogénesis, al lado izquierdo. (Pilkis & Granner 1992) Glucose
corresponde a glucosa, Glu-6-P es glucosa-6-fosfato, Fru-6-P es fructosa-6-fosfato, Fru-1,6-P2 es
fructosa-1,6-bisfosfato, PEP es fosfoenolpiruvato, PYR es piruvato. Las enzimas representadas
en la vía gluconeogénica son: Glu-6-Pase que es glucosa-6-fosfatasa, Fru-1,6-P2 que es fructosa-
1,6-bisfosfatasa y PEPCK que corresponde a la carboxiquinasa de fosfoenolpiruvato. Las
enzimas mostradas en la vía glicolítica son: GK que es glucoquinasa, 6PF-1-K que es fructosa-6-
fosfato-1-quinasa y PK que corresponde a piruvato quinasa. Es importante destacar que este
esquema no muestra la totalidad de las etapas para la conversión de PEP a Fru-1,6P2, solo por
motivos de resumen.

4
2.2. Características cinéticas de la fructosa-1,6-bisfosfatasa.
La enzima fructosa-1,6-bisfosfatasa (D-fructosa-1,6-bisfosfato-1-fosfohidrolasa, EC.
3.1.3.11; FBPasa), participa en la vía gluconeogénica y en presencia de cationes bivalentes como
Mg2+ cataliza la hidrólisis, que en condiciones fisiológicas es irreversible, de fructosa-1,6-
bisfosfato, generando como productos fructosa-6-fosfato más fosfato inorgánico (Taketa &
Pogell 1965).
Frente a estímulos glicolíticos la vía gluconeogénica es rápidamente desfavorecida (Pilkis
et al. 1988), y una de las principales etapas de regulación es la catalizada por FBPasa, para lo
cual esta enzima es potentemente inhibida por dos moléculas: adenosina monofosfato (AMP), de
modo alostérico y por fructosa-2,6-bisfofato (Fru-2,6-P2), de manera competitiva (McGrane et al.
1983). Además, existe un fuerte sinergismo en la inhibición al unirse ambas moléculas a la
enzima, lo que se piensa que es el principal mecanismo de control del flujo de la vía
gluconeogénica (Van Schaftingen 1987).
El mecanismo propuesto para la inhibición por AMP consiste en que al unirse el
nucleótido a la enzima el lazo, compuesto por los residuos de aminoácidos 52 al 72, se
desengancha de su posición sobre el dominio de unión del sustrato, con lo cual se desestabiliza la
unión de los metales bivalentes, con la consiguiente pérdida en la actividad (Choe et al. 1998;
Choe et al. 2000; Nelson et al. 2000b). Cabe mencionar que a bajas concentraciones de sustrato
esta enzima muestra una típica grafica hiperbólica de saturación por sustrato, pero a
concentraciones más altas (> 50µM) aparece inhibición por exceso de sustrato. Se observa una
disminución en un 50% en la actividad enzimática, respecto a la velocidad máxima experimental
observada a una concentración 1mM de Fru-1,6-P2. Es importante destacar que la inhibición por

5
exceso de sustrato se desplaza a concentraciones mayores del sustrato cuando se aumenta la
concentración del catión monovalente K+ (Slebe et al. 1985).
La fructosa-2,6-bisfosfato, es un análogo estructural del sustrato e inhibidor de la
reacción, pero el mecanismo de inhibición es aún tema de controversia. Los grupos de trabajo han
enfrentado el problema de distintas maneras, encontrando distintas respuestas, sin que ninguna
sea completamente satisfactoria o verdadera. Una de las posiciones es que la inhibición es de tipo
competitiva respecto al sustrato (Pilkis et al. 1981b). No obstante, la interpretación que el Fru-
2,6-P2 se une a los mismos determinantes de unión que el Fru-1,6-P2, no es compatible con los
resultados de nuestro laboratorio. Estos estudios muestran diferentes efectos, por ejemplo que la
modificación química de la Cys-128 afecta las interacciones de los azucares con la enzima de
distinta manera (Reyes et al., 1993). Por este motivo, propusimos que: la enzima tiene un dominio
de unión de fructosa-bisfosfato, que puede acomodar tanto al sustrato como al inhibidor, pero en
el que cada ligando ocupa distintos determinantes de unión, y como consecuencia tiene distintos
efectos. Por otro lado, hay autores que postulan que la inhibición por Fru-2,6-P2 es debido tanto a
la unión al sitio catalítico como a un sitio alostérico (Meek et al., 1984). Asimismo, una buena
indicación que Fru-2,6-P2 se asocia a la enzima en un sitio diferente al catalítico, son los
resultados que muestran que Fru-2,6-P2 induce cooperatividad en las curvas de saturación por el
sustrato (Hers and Van Schaftingen 1982; Reyes et al., 1993), y que la modificación de la FBPasa
de riñón de cerdo con N-etilmaleimida genera un derivado de la enzima insensible a la inhibición
por Fru-2,6-P2, pero que mantiene su afinidad al sustrato (Reyes et al, 1987 y 1993).

6
2.3. Características estructurales de la fructosa-1,6-bisfosfatasa.
La FBPasa de riñón de cerdo es un homotetrámero y cada una de sus subunidades tiene
337 aminoácidos con una masa aproximada de 36,5 KDa. (Marcus et al. 1982; Williams &
Kantrowitz 1992). Por convención a la subunidad ubicada en el vértice superior derecho del
tetrámero se ha denominado C1, continuando con C2, C3 y C4 siguiendo el sentido de las
manecillas del reloj (Figura 2).
Estudios de difracción de rayos X sobre la enzima cristalizada han demostrado que esta
posee dos estados conformacionales, uno de alta actividad (estado R) y uno de baja actividad
(estado T), donde la transición del estado R al estado T involucra una rotación de 15°-17º del
dímero superior (C1-C2) con respecto al dímero inferior (C3-C4), además, implica una
reorganización del dominio de unión de AMP con una rotación de 1,9º con respecto del dominio
de unión de fructosa-1,6-bisfosfato, los cuales se encuentran separados por aproximadamente 28
Å. (Ke et al. 1991; Villeret et al. 1995). En adición, se ha visto que el sitio activo se encuentra
ubicado adyacente a las interfaces C1-C2 y C3-C4 y el sitio de unión a AMP se encuentra
adyacente a las interfaces C1-C4 y C2-C3. En la subunidad la secuencia de 337 residuos de
aminoácidos se ordena de manera tal que forman una estructura hexaédrica (Figura 3). En detalle,
la estructura terciaria de la enzima está formada por un ordenamiento alternado de conjuntos de α
hélices y conjuntos de hojas β plegada.

7
Figura 2 Estructura cuaternaria del tetrámero de FBPasa de riñón de cerdo, mostrado en
diagrama de cintas (Ke et al. 1991). Se aprecia en azul la subunidad C1, en rojo la subunidad
C2, en naranjo la subunidad C3 y en gris la subunidad C4. En verde se resalta la posición de la
Lys274.

8
Figura 3 Diagrama de cintas de la subunidad C1 del tetrámero de FBPasa, resaltando la
posición relativa de sus efectores (Ke et al. 1990). En verde se representa la fructosa-6-fosfato, en
amarillo el AMP y en negro señalizado con una flecha azul el Mg2+.

9
2.4. Estudio de la relación entre la estructura y función de la fructosa-1,6-bisfosfatasa.
Existen numerosos métodos para analizar la relación entre la estructura y la función de
una proteína; en este trabajo y con la intención de variar lo menos posible las propiedades
cinéticas y estructurales de la enzima el método utilizado fue la introducción de un residuo de
triptófano en su estructura primaria.
La fluorescencia intrínseca de las proteínas se debe a la presencia de aminoácidos
aromáticos en su estructura primaria, principalmente triptófano y en menor medida a la presencia
de fenilalanina y/o tirosina. El triptófano como sonda fluorescente reportera es una herramienta
muy útil, ya que nos permite observar en las proteínas distintos tipos de interacciones y cambios
conformacionales, siempre y cuando estos modifiquen el entorno de este aminoácido. La
exposición de este aminoácido a condiciones polares provocan un corrimiento al rojo en su
máximo en la emisión de fluorescencia y en condiciones de menor polaridad se provoca un
corrimiento al azul, y también se puede afectar la intensidad de la emisión de fluorescencia
cuando se acercan o alejan grupos apagadores (Lakowicz 2006; Royer 2006).
La FBPasa de riñón de cerdo en su estructura primaria no posee triptófanos (Marcus et al.
1982; Williams & Kantrowitz 1992). Por lo tanto, la mutación de un residuo a Trp coloca una
sonda fluorescente única en una región específica de la proteína, supuestamente responsable de la
catálisis y su regulación. Por este motivo, y por lo anteriormente descrito, se han generado y
caracterizado distintas mutantes fluorescentes de la enzima FBPasa (Hodgson et al. 1998; Nelson
et al. 2000b), y específicamente en nuestro laboratorio: Phe6Trp, Phe16Trp, Phe89Trp,
Phe219Trp y Phe232Trp (Asenjo 2000; Maureira 2005; Ludwig et al. 2007). No obstante, no
existe información sobre la incorporación de un triptófano en Lys-274, residuo del sitio activo
que se ha sugerido a partir de la estructura cristalina como importante en la interacción con el

10
grupo fosforilo en posición 6 del Fru-1,6-P2 (Ke, H. et al., 1989). Para estudiar cómo influye la
unión de distintos efectores en el dominio del sitio activo y específicamente conocer más acerca
del mecanismo de inhibición de la enzima por fructosa-2,6-bisfosfato y por altas concentraciones
de sustrato, se decidió reemplazar el residuo de lisina en posición 274 por un residuo de
triptófano.
En base a los antecedentes previamente expuestos se postula la siguiente hipótesis:
“La mutación Lys274Trp en la fructosa-1,6-bisfosfatasa de riñón de cerdo afectará las
interacciones del sustrato fructosa-1,6-bisfosfato y del inhibidor fructosa-2,6-bisfosfato con
la enzima de distinta manera”.

11
2.5. Objetivos.
Para demostrar esta hipótesis se propusieron los siguientes objetivos.
2.5.1. Objetivo General.
Estudiar la relación entre la estructura y función en la enzima gluconeogénica FBPasa,
mediante el uso de mutagénesis sitio dirigida.
2.5.2. Objetivos Específicos.
I. Obtener la mutante de FBPasa por medio de mutagénesis sitio dirigida.
II. Caracterizar cinéticamente la mutante con los distintos activadores e inhibidores.
III. Estudiar la unión de AMP, Fru 1,6-P2 y Fru-2,6-P2 a la mutante de FBPasa,
utilizando las propiedades de fluorescencia de la enzima. Se analizará si la unión de
la unión de cada ligando a la enzima provoca cambios conformacionales, los cuales
serán registrados como cambios en la emisión de fluorescencia del triptófano
introducido en la enzima.

12
3. Materiales y Métodos.
3.1 Materiales.
3.1.1 Reactivos.
Agar, agarosa, ampicilina, tris[hidroximetil]aminometano (Tris-base), fructosa-1,6-bisfosfato
(Fru-1,6-P2), fructosa-2,6-bisfosfato (Fru-2,6-P2), nicotinamida adenina dinucleótido (NAD+),
adenina monofosfato (AMP), ácido etilendiaminotetraacético (EDTA), laureldodecilsulfato de
sodio (SDS), azul de bromofenol, isopropil-thio-β-D-galactosido (IPTG) y las enzimas auxiliares
glucosa-6-fosfato deshidrogenasa y fosfoglucosa isomerasa fueron suministrados por Sigma
Chemicals Co (USA). Hidróxido de sodio, isopropanol, etanol, ácido acético, N,N,N',N'-
tetrametiletilendiamina (Temed) y metanol fueron provistos por Merck Darmstadt, Alemania.
Los productos obtenidos por invitrogen fueron medio LB Broth base en polvo (Lennox L Broth
base), agar en polvo, agarosa ultra pura, acrilamida, bisacrilamida (N,N'- metilenbisacrilamida),
bromuro de etidio, Cepas de E. coli JM 109 y BL21 (DE3) se compraron a promega y novagen
respectivamente, endonucleasas de restricción, marcadores de masa molecular de DNA (1 kb y
100 kb), de proteínas y preteñido fueron adquiridos en BioLabs, oligonucléotidos sintetizados
requeridos como partidores para la mutagénesis sitio dirigida de FBPasa y para la reacción de
PCR se sintetizaron en Bioschile.
Partidores mutagénesis: Lys274Trp Fw 5'- AAG AAA AGC CCC AAA GGA TGG TTA AGA
CTG CTA TAC GAA -3', Lys274Trp Rv 5'- TTC GTA TAG CAG TCT TAA CCA TCC TTT
GGG GCT TTT CTT -3'

13
Partidores para PCR: FBPasa-N Fw 5’- TTC ATA TGA CGG CTA CCA GGC GG -3’, FBPasa-
C Rev 5’- TTG GAT CCT CAC CTT GGC TGC TTC TG -3’.
La determinación de la concentración de proteínas se realizó mediante el sistema “Bio-
Rad Protein Assay” de Bio-Rad, que es una variación del método de Bradford (Bradford 1976),
los plásmidos se aislaron y purificaron utilizando el sistema de purificación de DNA plasmidial
“E.Z.N.A. Plasmid Miniprep kit”. Todos los reactivos utilizados fueron de grado analítico o
superior.
3.1.2 Equipos.
Balanza analítica Shimadzu (Libror AEX-120 G), espectrofotómetro Shimadzu (UV -150-
02), espectrofluorímetro Perkin-Elmer LS-50B, pHmetro Radiometer Copenhagen (PHM 83
autocal pH meter), estufa 37ºC Bluem (dry type bacteriological incubator), balanza Shimadzu
(Libror EB-3200 S), termociclador MJ Research (Mini Cycler), centrífuga Fisher Scientific
(Micro V), micropipetas Gilson, horno microondas Somela (E 70 TF-7), fuente de poder Life
Technologies (model 500), agitador termorregulado lab-line instruments (Lab-Line Orbit
Environ-Shaker), centrífuga Eppendorf (5417 R), centrifuga Sorvall (RC 5C), baño
termorregulado Kottermann y vortex Fisher (Genie2). Espectrofotómetro de arreglo de diodo
Hewlett Packard modelo 8453, columnas y sistema de cromatografía FPLC Äcta Prime plus de
Amersham-Pharmacia.

14
3.2 Métodos.
3.2.1 Mutagénesis Sitio dirigida.
La mutagénesis se realizó utilizando el kit “QuikChange® Site-Directed Mutagenesis Kit”
provisto por Stratagene, que genera las mutaciones por medio de una reacción de PCR, según lo
descrito en la tabla I y II, que gracias a la ADN polimerasa Pfu turbo, replica con alta fidelidad y
completamente ambas hebras del templado utilizado. En este trabajo se usó el vector pET15b con
el inserto de FBPasa de riñón de cerdo tipo silvestre. Para incluir la mutación, se requiere que los
partidores sean específicos y que solo difieran en su complementariedad con la secuencia
codificante de FBPasa por el codón que codificaba para el aminoácido que se desea mutar. Es
importante mencionar que este kit incluye un paso de degradación del pADN templado por la
acción de la enzima Dpn I, ya que esta solo corta de modo específico los sitios Dam metilados,
que solo poseen los plasmidios templados, considerando que estos fueron amplificados en la cepa
bacteriana de E. coli JM109 que es Dam metilasa positiva, de este modo, se favorece la
probabilidad de trasformar bacterias solo con los pADN mutados. Es importante indicar que la
digestión con Dpn I se realizó inmediatamente después del término del PCR, agregando 1 µL de
la endonucleasa directamente sobre los productos y se incubo por 1h a 37°C.
Los partidores fueron diseñados según las recomendaciones del fabricante, al igual que las
condiciones de la reacción de mutagénesis.

15
Tabla I Parámetros de preparación de la reacción de mutagénesis para 1 tubo
Mezcla de Reacción 1X (µL)
Buffer 10X 5
ADN Templado 1
Partidor Sentido 3
Partidor Anti-Sentido 3
dNTP’s 1
H2O 36
Pfu turbo 1
Volumen Total 50

16
Tabla II Parámetros de las temperaturas y tiempos utilizados en el termociclador para la reacción
de mutagénesis.
Programa Termociclador Temperatura (°C) Tiempo (Min)
Denaturación inicial 95 0,5
Denaturación 95 0,5
Alineamiento 58 1
Extensión 68 12
Ciclos 16 -

17
3.2.2 Preparación y transformación de células competentes.
Las células competentes se prepararon por el método del cloruro de calcio, el cual consiste
en:
1. Realizar un precultivo inoculando 50 mL de medio LB fresco y sin antibiótico con una
colonia o alícuota de células E. coli. Incubar a 37ºC en agitación constante a 250 r.p.m. por
toda la noche.
2. Inocular 320mL de medio LB con 3,2mL del precultivo. Incubar en las mismas condiciones
antes descritas hasta obtener una OD590 igual a 0,4.
3. Luego se divide el cultivo en tubos Falcon de 50mL esterilizados, y mantenidos a baja
temperatura, centrifugar a 4 ºC a 4500r.p.m. por 7min.
4. Resuspender suavemente el pellet en 10 mL de Cl2Ca frío, centrifugar a 4ºC por 7 min a 4500
r.p.m.
5. Resuspender suavemente el pellet en 10 mL de Cl2Ca frío, mantener las células a 4ºC por 30
min. centrifugando a 4ºC y 4500 r.p.m. por 5 min.
6. Resuspender suavemente el pellet en 2 mL de Cl2Ca frío. Dividiendo la suspensión en tubos
eppendorf de 0.6 mL, agregando 105 µL. Guardar inmediatamente a –80ºC.

18
La transformación de las células competentes se realizó del siguiente modo:
1. Se toma una alícuota congelada a -80°C y se deja reposar en hielo por 5 min.
2. Se agrega 1µL del pADN sobre las bacterias, o el volumen suficiente para lograr una masa de
50 ng, y se deja reposar en hielo por 1h.
3. Luego se incuba a 42 °C por 90 s.
4. En un tubo Falcon de 15 mL con 1 ml de medio LB fresco se le agrega todo el contenido
bacteriano. Se deja incubar en un agitador por 1h a 37 °C y 250 r.p.m.
5. En una placa con medio de cultivo LB-agar y el respectivo antibiótico se agregan 200 µL de
la suspensión bacteriana. (Cuando se transformaron las bacterias con el pADN mutado, se
centrifugó a 4500 r.p.m. por 5 min y el pellet se resuspendió en 200 µL de medio LB para
luego inocular la placa con la totalidad del volumen). Las placas se incubaron en una estufa
por 12h a 37 °C.
3.2.3 Análisis de los plasmidios obtenidos.
Se analizaron los plasmidios con dos técnicas: restricción por endonucleasas y PCR.
Las restricciones se realizaron siguiendo las instrucciones del fabricante de la enzima,
según lo indicado en la Tabla III.
La reacción de PCR se realizó según el protocolo indicado en la tabla IV, utilizando la
enzima Taq ADN polimerasa.

19
Tabla III Parámetros utilizados en la preparación de la mezcla de digestión por endonucleasas.
Mezcla de restricción 1X (µL)
pADN 4
Buffer 10X 1
H2O 4,4
Enzima de Restricción 0,6
Volumen Total 10
Tiempo de acción 1h
Temperatura de acción 37 °C
20
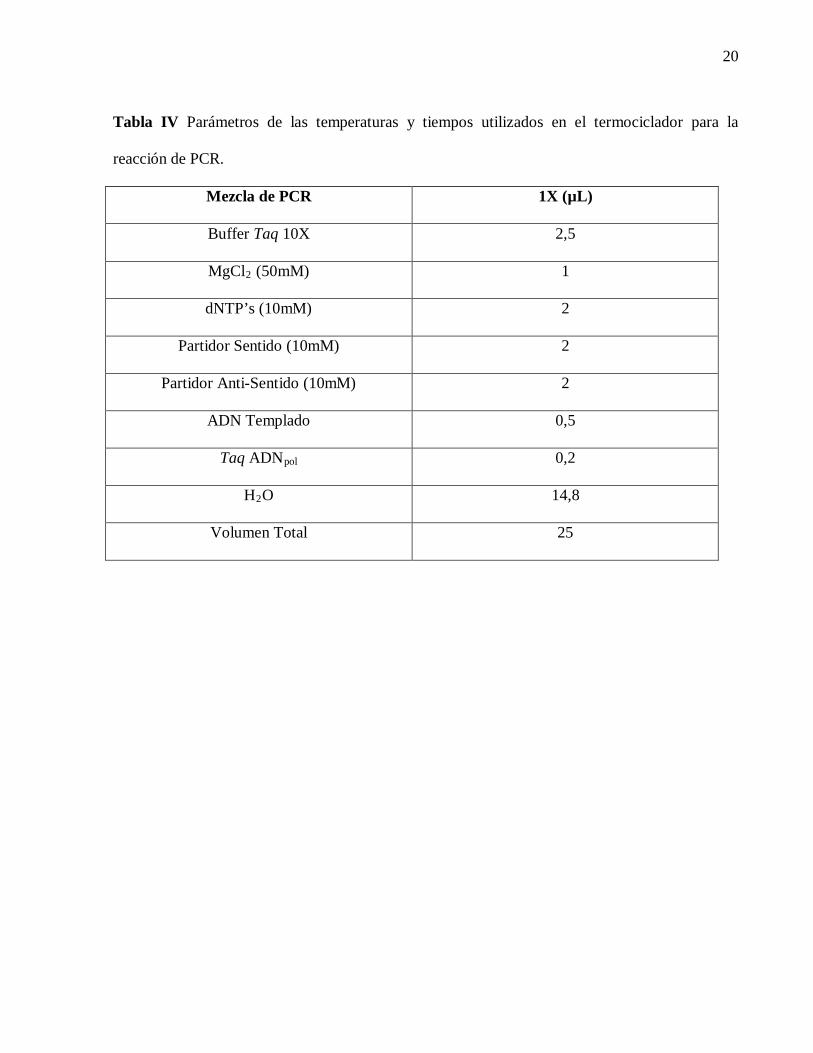
Tabla IV Parámetros de las temperaturas y tiempos utilizados en el termociclador para la
reacción de PCR.
Mezcla de PCR 1X (µL)
Buffer Taq 10X 2,5
MgCl2 (50mM) 1
dNTP’s (10mM) 2
Partidor Sentido (10mM) 2
Partidor Anti-Sentido (10mM) 2
ADN Templado 0,5
Taq ADNpol 0,2
H2O 14,8
Volumen Total 25

21
3.2.4 Expresión y purificación de proteína recombinante.
Para expresar la proteína recombinante de FBPasa se utilizó el sistema pET adquirido
desde Novagen.
El sistema pET nos permite obtener una gran cantidad de proteína recombinante con la
ventaja de ser relativamente sencillo purificarla al agregar una cola de histidinas a la proteína
recombinante.
El sistema elegido para expresar proteínas recombinantes en esta tesis es el que utiliza
como vector el plasmidio pET 15b, porque permite una rápida y fácil purificación, por medio de
una columna de sefarosa níquel, de la proteína expresada al incluirle una cola de 6 histidinas al
extremo amino, sin afectar significativamente las propiedades cinéticas o fluorescentes, según lo
demostrado en nuestro laboratorio.
La cepa bacteriana de expresión es la BL21 DE3, la cual contiene en su genoma el gen
que codifica para la T7 ARNpol y para el represor lac, el que es expresado constitutivamente
regulando la expresión de la T7 ARNpol, por lo que evita posibles autoinducciones.
El detalle del método de expresión se muestra en la figura 4 extraída del manual del
sistema pET.

22
Figura 4 Mecanismo de expresión inducido por IPTG en una cepa bacteriana BL21 DE3
transformada con el vector pET 15b. El represor lac es expresado constitutivamente desde el
gen lac I, presente en la cepa bacteriana y en el vector. Así el represor se une a las regiones
operadoras impidiendo la transcripción de la T7 ARNpol y de la proteína objetivo. Al momento de
agregar IPTG, este se une al represor lac liberando ambas regiones promotoras, con lo cual
comienza la transcripción de la T7 ARNpol, esta a su vez se une a su respectiva región promotora
en el vector, dando inicio a la transcripción de la proteína objetivo.

23
La expresión se realizó según el siguiente protocolo, basado principalmente en las
recomendaciones del fabricante y experiencias empíricas de nuestro laboratorio.
1. Pre-cultivo. Se inoculan 50 ml de medio LB más ampicilina (125 µg/mL) y glucosa 1%, para
evitar la autoinducción, incubando por toda la nocha a 37 ºC con una agitación de 250 r.p.m.
2. Crecimiento. El precultivo se centrifuga a 4500 r.p.m. por 5 min a 4ºC eliminando el
sobrenadante y resuspendiendo el sedimento en 50 mL de medio LB fresco. Se inoculan 6
matraces que contienen 500 ml de medio LB con 5 mL del precultivo resuspendido. Luego se
incuba con una agitación constante de 250 r.p.m. a 37ºC, hasta obtener una D.O.600 de 0,6-0,8
U.A.
3. Inducción de la expresión. Una vez finalizada la incubación los matraces son retirados del
agitador y se incuban en hielo hasta disminuir la temperatura a 20ºC. Luego se agrega el
inductor IPTG a una concentración 0,4 mM final y se deja incubar por 4 h con una agitación
constante de 250 r.p.m. a 30ºC.
4. Lisis bacteriana. Finalizada la inducción se centrifugan las bacterias 7000 r.p.m. por 7 min a
4ºC y el sedimento bacteriano se resuspende en Tris-HCl 20 mM, pH 7,5; EDTA 0,1 mM;
EGTA 0,1 mM; NaCl 500 mM; Triton X-100 0,1% y PMSF 4 mM. Luego se dejo lisando a 4
°C por 4 horas con una suave agitación constante, posteriormente, se sometió a esta
suspensión a 5 ciclos de sonicasión con pulsos de 1 s por 1 min. Previo al paso
cromatográfico se realizó un corte con calor incubando la suspensión bacteriana a 55ºC por 5
min, posteriormente la fase soluble fue recuperada por centrifugación a 10.000 r.p.m. por 30
min.

24
5. Purificación. La purificación de las proteínas recombinantes fue realizada con el sistema de
cromatografía FPLC Äcta Prime plus, cargando el sobrenadante del corte con calor en una
columna de 15cm con la resina Sefarosa-níquel “His-binding” y el equipo se configuró de
acuerdo a lo indicado en la tabla V.
6. Concentración. Con las fracciones eluídas que tuvieron mayor actividad se generó un pool,
el cual se precipitó con (NH4)2SO4 al 70% de saturación, y se dejó a 4 °C con agitación
constante por 30 min. Luego se centrifugo a 10000 r.p.m. por 15 min, resuspendiendo el
precipitado en la solución tampón Tris-HCl 20 mM, pH 7,5; EDTA 0,1 mM, para finalmente
dializarla realizando 4 cambios de 250 mL con la misma solución tampón en una bolsa de
diálisis (Dialyzer tubing) de tamaño de poro de 4,8 Å, eliminando las proteínas y sales de un
tamaño menor a 30 kDa. Así se logra concentrar la proteína aproximadamente 10 veces, la
cual se guardo a –20ºC en alícuotas de 0,5 y 1 mL.

25
Tabla V Parámetros de tiempo y volumen de la cromatografía, configurados en el equipo.
Intervalo
(mL) Buffer
Flujo
(mL/min)
Volumen fracción
colectada (mL)
Posición
llave
0,0
Unión
2,0 0,0 Lavado
14,0 2,0 0,0 Carga
15,0 2,0 8,0 Inyección
165,0 Lavado
2,0 8,0 Carga
265,0 2,0 8,0 Carga
265,1 Elusión
2,0 4,0 Carga
345,0 2,0 4,0 Carga

26
3.2.5 Determinación de la actividad enzimática.
Los productos generados en la reacción catalizada por la FBPasa no son fácilmente
detectables ni menos cuantificables por métodos espectroscópicos, por lo cual, la determinación
de la actividad FBPasica es realizada mediante una serie de reacciones acopladas que producen
como producto β-NADH el cual es detectado al medir la absorbancia a 340 nm. El siguiente
esquema muestra las reacciones acopladas.
La mezcla de reacción contiene: Tris-HCl 50 mM pH 7,5; EDTA 0,1 mM; β-NAD+ 0,3
mM; MgSO4 5,0 mM; 1,2 U de fosfoglucosa isomerasa (PGI) y 1,2 U de glucosa-6-fosfato
deshidrogenasa (GPD), en un volumen final de 500 µL. La cantidad a agregar de sustrato o de los
inhibidores dependió del ensayo a realizar. Para comenzar la reacción se agregaban 0,4 µg de la
enzima. Todos los ensayos fueron medidos a 30°C, salvo que se indique lo contrario.
D-Fructosa-6-fosfato + Pi FBPasa D-Fructosa-1,6-bisfosfato
D-Glucosa-6-fosfato PGI D-Fructosa-6-fosfato
6-Fosfo-D-gluconato + β-NADH GPD Glucosa-6-fosfato + β-NAD+

27
3.2.6 Determinación de las constantes cinéticas.
La curva de saturación por sustrato se realizó graficando la actividad específica versus la
concentración de sustrato. Para la determinación de las constantes se realizó una regresión no
lineal de los datos obtenidos con la siguiente ecuación (Cárcamo et al. 2000).
Donde:
υ = Velocidad inicial;
Vm = Velocidad máxima;
[S] = Concentración de sustrato;
Km = Constante de Michaelis;
KSS = Constante de disociación del complejo ESS;
β = Factor que afecta la kcat a altas concentraciones de sustrato;

28
Esta ecuación se deriva de la suposición de que la enzima es un dímero de dímeros, que
frente a la unión de una segunda molécula de sustrato se agrega un factor que disminuye la
constante de formación del producto, lo que explica la inhibición por exceso de sustrato.
A continuación se esquematiza el mecanismo propuesto.
Donde:
E = Enzima; S = Sustrato; P = Producto; ES = Complejo enzima-sustrato;
ESS = Complejo enzima-sustrato-sustrato; k2 = Constante de deformación;
KS = Constante de disociación del complejo ES;
KSS = Constante de disociación del complejo ESS;
β = Factor que determina la máxima actividad para muy altas [S];
E + S
ESS
KS k2
KSS
E + P
E + P
βk2
ES + S

29
3.2.7 Determinación de la constante de activación por Magnesio.
Para la determinación de la constante activación por magnesio se midió la actividad
específica a concentraciones crecientes del catión (0-5mM), manteniendo constante la
concentración de sustrato y los demás parámetros del análisis. Los datos obtenidos se ajustaron a
la siguiente ecuación.
Donde Vmax corresponde a la máxima actividad medida y V a la actividad medida a una
concentración definida de Mg2+, n es la constante de Hill o índice de cooperatividad y Ka
corresponde a la concentración de Mg2+ necesaria para alcanzar el 50% de la actividad máxima.
3.2.8 Determinación de la constante de inhibición por AMP.
Para construir la curva de inhibición por AMP se agregaron concentraciones crecientes
del inhibidor y se graficó la actividad relativa versus la concentración de AMP. La determinación
de las constantes cinéticas y curva de tendencia se utilizó una ecuación derivada de la constante
de equilibrio de la unión del inhibidor a la enzima (Taketa & Pogell 1965).
Su forma logarítmica se muestra a continuación:
Donde Vmax corresponde a la actividad sin el inhibidor y V es la actividad a una
determinada concentración del inhibidor, I50 es la concentración de AMP necesaria para
alcanzaron un 50% de la actividad máxima, y n es el número aparente de moléculas del inhibidor
unidas a una sola molécula de enzima.
Log [Vmax - V] . [V]
n Log Ka n Log [Mg2+] = +
+ Log [Vmax - V] . [V]
Log I50 n Log [AMP] =

30
3.2.9 Determinación de la constante de inhibición por fructosa-2,6-bifosfato.
La curva de inhibición por Fru-2,6-P2 se construyo graficando las actividades medidas
normalizadas versus la concentración del inhibidor. Las constantes se determinaron al ajustar los
datos a la siguiente ecuación.
Donde Vmax corresponde a la actividad sin el inhibidor y V es la actividad a una
determinada concentración del inhibidor, I50 es la concentración de Fru-2,6-P2 necesaria para
alcanzar un 50% de la actividad máxima, y n es el índice de cooperatividad o coeficiente de Hill.
3.2.10 Determinación de los cambios en la emisión de fluorescencia.
La determinación se hizo en un espectrofluorímetro Perkin-Elmer LS-50B acoplado a un
computador. Para medir los espectros de emisión se preparó una solución de la enzima a 60
µg/mL en tampón Tris-HCl 50 mM pH 7,5; EDTA 0,1 mM, en una cubeta de cuarzo con camino
óptico de 1 cm, a una temperatura de 15 °C y agitación constante.
Los parámetros de medición del equipo fueron: longitud de onda de excitación 295 nm,
ranuras de excitación y emisión 7 nm y se hizo un barrido de detección desde 310 hasta 390 nm.
Para determinar las influencias de los distintos ligandos sobre la enzima mutante se
agregaron volúmenes de ligando a concentraciones crecientes sobre la solución que contenía la
enzima, con un tiempo de incubación de 60 s antes de medir la fluorescencia. Las mediciones
fueron corregidas por los respectivos factores de dilución y la posible fluorescencia del ligando
(efecto de filtro interno). Además se midió la fluorescencia de la enzima en ausencia del ligando,
Log [Vmax - V] . [V]
n Log I50 n Log [Fru-2,6-P2] = +

31
o sea, se realiza una titulación en iguales condiciones a la del ligando, pero en vez de este se
agrega agua a la enzima.
El análisis de los datos se realizó graficando ΔF/F0 versus la concentración del ligando y
los datos experimentales se ajustaron, mediante una regresión no lineal, a la ecuación de Hill, que
se muestra a continuación.
Donde ΔF es la diferencia entre la fluorescencia a una determinada concentración de ligando y la
fluorescencia sin el ligando (F0), Kd es la constante de disociación, L es la concentración del
ligando y n el coeficiente de Hill.

32
4. Resultados.
4.1. Obtención de la mutante de FBPasa de riñón de cerdo Lys274Trp.
4.1.1 Mutagénesis sitio dirigida.
Para efectuar las mutagénesis se utilizó el Kit QuickChangeTM site-directed de Stratagene,
según lo descrito en Materiales y métodos. Como templado de la reacción de mutagénesis se
utilizó el plasmidio pET 15b, el que contenía el segmento codificante de la FBPasa.
4.1.2 Análisis de los plasmidios obtenidos.
Con el fin de amplificar los plasmidios para analizar la presencia de las mutaciones, se
transformaron bacterias competentes con los plasmidios obtenidos desde las mutagénesis, por el
método de cloruro de calcio. Desde las colonias obtenidas se aislaron y purificaron los plasmidios
utilizando el kit E.Z.N.A. Plasmid Miniprep, obteniendo una concentración de aproximadamente
de 100 ng/µl. Los plasmidios fueron analizados por un ensayo de restricción con la enzima Pst I y
además por un PCR utilizando partidores externos y específicos para FBPasa renal de cerdo. En
la figura 5, panel A se muestran los resultados para el PCR, donde se ve claramente un
amplificado de 1000 pb correspondiente a la FBPasa mutada y en el panel B de la figura 5 se
muestra el análisis de restricción, en el cual se pueden visualizar las dos bandas esperadas por el
corte con esta enzima, aproximadamente 5200 pb y 1500 pb. Con esto se concluye que los
plasmidios tienen el inserto y por lo tanto fueron enviados a secuenciar.

33
Figura 5. Análisis de restricción y PCR de los pADNs obtenidos desde las colonias de
bacterias transformadas. Gel de agarosa al 1% teñido con bromuro de etidio. Panel A
corresponde al ensayo de PCR, carril 1 estándar de ADN de 1Kb (BioLabs), carriles 2 y 3:
control negativo y positivo respectivamente, carriles 4-9: pADNs obtenidos de las colonias 1-6,
transformadas con el vector pET15b con el inserto de FBPasa que contiene la mutación
Lys274Trp. Panel B corresponde al análisis de restricción con la enzima Pst I, carril 1 estándar de
ADN de 1Kb (BioLabs), carril 2 control negativo, carriles 3-8: pADNs obtenidos de las colonias
1-6, transformadas con el vector Pet15b con el inserto de FBPasa que contiene la mutación
Lys274Trp.
A 1 2 3 4 5 6 7 8 9
B 1 2 3 4 5 6 7 8
1000 pb
1500 pb
5000 pb

34
4.1.3 Secuenciación de los pADNs.
Los plasmidios se enviaron a secuenciar a Macrogen Inc. en Seúl Corea, donde se realizó
la secuenciación mediante el método de “single primer extension”. Se utilizaron como partidores
para la secuenciación los complementarios a la secuencias T7 promotora (partidor sentido) y
terminadora (partidor anti-sentido), contenidas en el vector pET15b río arriba y río abajo
respectivamente del sitio de múltiple clonamiento. Estos partidores fueron provistos por la
empresa secuenciadora.
Los análisis de los resultados de la secuenciación, vistos mediante el alineamiento
contra la secuencia de ADN codificante de la FBPasa renal de cerdo descrita por Marcus F. et al
1982, mostraron la presencia de la mutación esperada, y además, no se aprecian mutaciones en
otros sitios. En la Figura 6, se muestra una porción de los alineamientos de la secuencia silvestre
de la FBPasa renal de cerdo contra la mutante de FBPasa renal de cerdo Lys274Trp.

35
Figura 6. Alineamiento del producto de la secuenciación contra la secuencia tipo silvestre de
FBPasa de riñón de cerdo. Solo se muestra una porción de los alineamientos entre la secuencia
codificante de la FBPasa renal de cerdo tipo silvestre con la mutante Lys274Trp. En la secuencia
superior corresponde a la enzima silvestre y la inferior a la mutante.
WT 787 CAGAGGAAGAAGTTTCCCCCAGACAACTCAGCCCCCTACGGGGCCAGGTACGTGGGCTCC 846 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Mut_s 371 CAGAGGAAGAAGTTTCCCCCAGACAACTCAGCCCCCTACGGGGCCAGGTACGTGGGCTCC 312 WT 847 ATGGTGGCCGATGTCCACCGCACGCTGGTCTATGGAGGGATCTTTATGTACCCAGCAAAC 906 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Mut_s 311 ATGGTGGCCGATGTCCACCGCACGCTGGTCTATGGAGGGATCTTTATGTACCCAGCAAAC 252 K274 WT 907 AAGAAAAGCCCCAAAGGAAAGTTAAGACTGCTATACGAATGTAACCCGATGGCCTATGTC 966 |||||||||||||||||| |||||||||||||||||||||||||||||||||||||||| Mut_s 251 AAGAAAAGCCCCAAAGGATGGTTAAGACTGCTATACGAATGTAACCCGATGGCCTATGTC 192 WT 967 ATGGAGAAGGCAGGAGGACTGGCCACCACTGGGAAGGAAGCTGTGCTGGACATCGTTCCC 1026 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Mut_s 191 ATGGAGAAGGCAGGAGGACTGGCCACCACTGGGAAGGAAGCTGTGCTGGACATCGTTCCC 132 Wt 1027 ACTGACATCCACCAGAGGGCGCCAATCATCTTGGGGTCTCCTGAAGACGTGACTGAGCTC 1086 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Mut_s 131 ACTGACATCCACCAGAGGGCGCCAATCATCTTGGGGTCTCCTGAAGACGTGACTGAGCTC 72

36
4.2. Expresión y purificación de la mutante de FBPasa Lys274Trp.
4.2.1 Expresión.
Esta se realizó transformando bacterias BL21 DE3 que contenían el vector pRP4, con los
plasmidios obtenidos de la mutagénesis, estas fueron seleccionadas por crecimiento en placas de
LB-agar con ampicilina 125 µg/ml y kanamicina 40 µg/ml. Posteriormente y por medio de mini
expresiones de proteína recombinante se seleccionaron las colonias que fueran capaz de expresar
la proteína en cantidad suficiente.
Con la colonia que expresaba mejor la FBPasa se realizó la expresión inducida por IPTG,
desde un cultivo de 3L de medio LB con aproximadamente una densidad óptica de 0,8 U.A. de
bacterias.
4.2.2 Purificación.
La Purificación se realizó en varias etapas, según lo descrito en Materiales y métodos,
logrando una masa de aproximadamente 8 mg de proteína purificada disueltos en 10 ml de
tampón Tris-HCl 20 mM pH 7,5 y EDTA 0,1 mM, la cual tenía una actividad específica de 16
U/mg a 30 µM de sustrato (Tabla VI).
El cromatograma representado en la figura 7, muestra una porción de la cromatografía,
correspondiente a la etapa de elusión, observándose que la mayor actividad se encuentra en las
fracciones que tienen el máximo de absorbancia. Las otras fracciones colectadas muestran una
actividad específica igual a 0.
En la figura 8, se muestra un SDS-PAGE en la cual se cargaron alícuotas de distintas
etapas de la purificación de la enzima mutante Lys274Trp.

37
Tabla VI: Purificación de la mutante de FBPasa Lys274Trp.
Fracción Volumen
(ml)
Actividad 1
(UT)
Proteína
(mg/ml)
Actividad
Especifica
(U/mg)
Rendimiento
%
Sobrenadante
Lisado 2 70.0 195 2.00 1.40 100
Pool de
eluídos3 28.0 160 0.45 12.7 82.1
Corte4
(NH4)2SO4
10.0 128 0.80 16.0 65.6
1 La actividad enzimática fue medida a 30 µM de sustrato y a 30° C.
2 Previo a cargar en la columna de sefarosa-níquel.
3 Fracciones mezcladas del eluído con mayor actividad.
4 Etapa final posterior a la diálisis.

38
Figura 7. Perfil parcial de elusión de la mutante Lys274Trp desde una columna de sefarosa-
níquel. Solo se muestra la porción del cromatograma correspondiente a la etapa de elusión, por
fines de resumen y debido a que ninguna de las otras fracciones colectadas presentó actividad. En
el eje izquierdo se muestra la mAu que corresponde a las mili unidades de absorbancia a 280 nm
entregadas por el equipo cromatógrafo, así la línea verde representa la absorbancia de la fracción
eluída y el eje derecho se muestra la actividad específica, medida a 30 µM de sustrato, de cada
fracción colectada graficado como una línea roja. Mayores antecedentes en Materiales y
Métodos.
0
50
100
150
200
250
300
0 10 20 30 40 50 60 70 80
Volumen (ml)
mA
u
0
5
10
15
20
A.E
.

39
Figura 8. Purificación de la mutante de FBPasa Lys274Trp. SDS-PAGE al 12% teñido con
azul de coomasie, en los carriles 3-9 se cargo 10 µg de proteína. En el carril 1 se cargaron 2 µg de
proteína purificada, FBPasa Phe219Trp, como control positivo. Carril 2 corresponde a un
estándar de proteínas sin preteñir de amplio marcaje (BioLabs). Carril 3, lisado bacteriano
después de las 4 horas de inducción con IPTG. Carriles 4-5, alícuotas colectadas después de la
sonicación y corte con calor respectivamente. Carriles 6-7, sobrenadante y pellet obtenido
posterior al paso de centrifugación respectivamente. Carril 8, pool de fracciones con mayor
actividad, colectadas de la elusión cromatografía de sefarosa-níquel. Carril 9, Proteína purificada
posterior a la precipitación con sulfato de amonio y diálisis.
1 2 3 4 5 6 7 8 9

40
4.3 Caracterización Cinética.
Los ensayos para la determinación de las constantes cinéticas se realizaron según lo
descrito en Materiales y métodos; para los análisis de inhibición y activación todas las reacciones
fueron realizadas a una concentración de 30 µM de sustrato y a 30° C.
4.3.1 Curva de saturación por sustrato.
En la Figura 9 se muestran los gráficos de actividad enzimática en función de la
concentración de sustrato de la mutante, comparada con la enzima FBPasa tipo silvestre. Se
midió la actividad enzimática a distintas concentraciones de sustrato, desde 0 hasta 3000µM y
las constantes se determinaron mediante una regresión no lineal a la ecuación descrita por
Cárcamo et al. 2000. La curva descrita para la saturación por sustrato en la FBPasa es de tipo
hiperbólica hasta una concentración de 30-40 µM, para luego decaer, en un fenómeno llamado
inhibición parcial por exceso de sustrato. Ambas curvas tienen formas similares, pero la enzima
mutante FBPasa Lys274Trp requiere una concentración de sustrato aproximada de 300 µM para
alcanzar el máximo de actividad. Esta mutante, además, muestra un leve aumento en los valores
de Km obtenidos. El aumento en el valor de Km es aproximadamente 6 veces respecto a la enzima
nativa, para la FBPasa Lys274Trp. También se aprecia una disminución tanto en el valor de la
constante catalítica (kcat) como en la actividad específica máxima para la mutante, lo cual,
probablemente está dado por la inclusión del triptófano en el putativo sitio activo impidiendo la
correcta catálisis, posiblemente porque este no estabilice la unión del sustrato al tener mayor
volumen además de distinta carga que la lisina.

41
Figura 9 Efecto de la concentración del sustrato Fru-1,6-P2 sobre la actividad de la
fructosas-1,6-bisfofatasas recombinante y mutante. La medición de la velocidad inicial se
realizó en amortiguador Tris-HCl 50 mM, pH 7,5; EDTA 0,1 mM; MgSO4 5,0 mM; a las
concentraciones de sustrato que se indican. El gráfico semi-logarítmico muestra las curvas de
saturación por sustrato de las enzimas FBPasa wt (azul) y FBPasa Lys274Trp (rojo). Todas las
enzimas tienen una cola de histidina pues para su expresión se utilizó el sistema vector pET15b+.
Los puntos representan los datos experimentales, mientras que las líneas continuas corresponden
a las curvas predichas de acuerdo a la ecuación descrita por Segel (1975) para la inhibición
parcial por exceso de sustrato. Detalles del procedimiento ver en Material y Métodos.
0
5
10
15
20
25
30
0,1 1 10 100 1000 10000
Act
ivid
ad E
spec
ífic
a(u
mol
/ m
g x
min
)
Fru-1.6-P2 (µM)

42
4.3.2 Curva de activación por Mg2+.
La hidrólisis de Fru-1,6-P2 requiere necesariamente la presencia de Mg2+, pues este metal
se posiciona estratégicamente cerca del fosfato 1 del azúcar, permitiendo la ruptura del enlace
fosfodiéster. Se determinó la activación de la reacción de hidrólisis, a concentraciones crecientes
de magnesio (0-5 mM) en la enzima silvestre y la mutante de FBPasa. En la figura 10, se muestra
el efecto de la concentración de Mg2+ sobre la actividad de las diferentes FBPasas quedando de
manifiesto la activación por el metal bivalentes. La constante de activación Ka para la enzima
mutante Lys274Trp aumenta su valor en aproximadamente 2 veces, resultado congruente con los
descritos para la mutante de hígado de cerdo Lys274Leu (Shyur et al., 1995) y de riñón de cerdo
Lys274Ala (Asenjo, 2008). Desafortunadamente, no existen datos para la enzima mutante de
hígado de rata Lys274Ala (El Magharabi et al., 1992).
4.3.3 Curva de inhibición por AMP.
La inhibición alostérica por AMP de la FBPasa es no competitiva respecto al sustrato y la
unión a su sitio provoca una serie de cambios conformacionales, descritos como la transición R a
T. Se estudió el efecto de la concentración del inhibidor alostérico AMP (0-100µM) sobre la
actividad de la enzima silvestre y la mutante de FBPasa. Las curvas de inhibición se muestran en
la figura 11. Se observa que la mutante FBPasa Lys274Trp, mantiene prácticamente igual el de
I50 pero pierde la cooperatividad para AMP, comparado con los valores obtenidos para la enzima
silvestre.

43
0
25
50
75
100
0 1 2 3 4 5
Act
ivid
ad R
elat
iva
%
Mg2+ (mM)
Figura 10. Efecto de la concentración de Mg2+ sobre la actividad de las enzimas silvestre y
mutante de FBPasa. La medición de la velocidad inicial se realizó en amortiguador Tris-HCl 50
mM, pH 7,5; EDTA 0,1 mM; y Fru-1,6-P2 30 µM, a las concentraciones de MgSO4 que se
indican. El gráfico muestra las curvas de saturación por Mg2+ de las enzimas FBPasa tipo
silvestre (azul), FBPasa Lys274Trp (rojo). Todas las enzimas tienen una cola de histidina pues
para su expresión se utilizó el sistema vector pET15b+. Los puntos corresponden a los datos
experimentales y las líneas continuas corresponden a la curva de tendencia ajustada a la ecuación
de Hill. Detalles del procedimiento ver en material y métodos.

44
0
25
50
75
100
0,1 1 10 100AMP (µM)
Act
ivid
ad R
elat
iva
%
Figura 11. Efecto de la concentración de AMP sobre la actividad de las enzimas silvestre y
mutante de FBPasa. La medición de la velocidad inicial se realizó en amortiguador Tris-HCl 50
mM, pH 7,5; EDTA 0,1 mM; MgSO4 5,0 mM y Fru-1,6-P2 30 µM, a las concentraciones de
AMP que se indican. El gráfico muestra las curvas de inhibición de las enzimas FBPasa tipo
silvestre (azul) y FBPasa Lys274Trp (rojo). Las dos enzimas tienen una cola de histidina pues
para su expresión se utilizó el sistema vector pET15b+. Los puntos corresponden a los datos
experimentales y las líneas continuas corresponden a la curva de tendencia ajustada a la ecuación
de Hill modificada por Taketa y Pogell (1965). Detalles del procedimiento ver en Material y
métodos.

45
4.3.4 Curva de inhibición por fructosa-2,6-bisfosfato.
El tipo de inhibición por Fru-2,6-P2, es tema de controversia. Se describe como un
inhibidor competitivo, pero también como uno de tipo alostérico, lo que depende del tipo de
abordaje experimental usado. Por esta razón, fue atrayente utilizar la mutante FBPasa Lys274Trp
para estudiar el mecanismo por el cual el azúcar bisfosfato inhibe la actividad de la enzima. Las
curvas de inhibición de la actividad de la enzima silvestre y de la enzima mutante Lys274Trp se
muestran en la figura 12. Las curvas de inhibición muestran comportamientos muy diferentes, los
que se reflejan en los valores de I50 para Fru-2,6-P2. Destaca que en la mutante Lys274Trp el
valor de I50 (concentración de Fru-2,6-P2 requerida para disminuir a la mitad la velocidad de la
reacción) aumenta 12 veces, con respecto al valor de I50 de la FBPasa silvestre.

46
Figura 12. Efecto de la concentración de Fru-2,6-P2 sobre la actividad de las enzimas
silvestre y mutante de FBPasa. La medición de la velocidad inicial se realizó en amortiguador
Tris-HCl 50 mM, pH 7,5; EDTA 0,1 mM; MgSO4 5,0 mM y Fru-1,6-P2 30 µM, a las
concentraciones de Fru-2,6-P2 que se indican. El gráfico muestra las curvas de inhibición de las
enzimas FBPasa tipo silvestre (azul) y FBPasa Lys274Trp (rojo). Las dos enzimas tienen una
cola de histidina pues para su expresión se utilizó el sistema vector pET15b+. Los puntos
corresponden a los datos experimentales y las líneas continuas corresponden a la curva de
tendencia ajustada a la ecuación de Hill, de acuerdo a Taketa y Pogell (1965). Detalles del
procedimiento ver en Material y métodos.
0
25
50
75
100
0,1 1 10 100
Act
ivid
ad R
elat
iva
%
Fru-2,6-P2 (μM)

47
Los parámetros cinéticos para las diferentes enzimas fueron obtenidos de los datos de las
figuras 9, 10, 11 y 12. Todas las enzimas son de riñón de cerdo recombinante expresadas
utilizando el sistema vector pET15b+. Las mediciones de la velocidad inicial se realizaron a 30°C
en amortiguador Tris-HCl 50 mM, pH 7,5; EDTA 0,1 mM; MgSO4 5,0 mM y Fru-1,6-P2 30 µM,
en el caso que no se indique otro parámetro. Km corresponde a la constante de Michaelis y
Menten y kcat a la constante catalítica. I50 corresponde a la concentración de Fru-2,6-P2 ó AMP a
la cual se inhibe en un 50% la actividad y nH es el coeficiente de Hill. Ka corresponde a la
concentración de Mg2+ a la cual se alcanza el 50% de la actividad máxima, y sus constantes son
resumidas en la tabla VII. Para más detalles ver en Material y métodos.

48
Tabla VII. Resumen de las constantes cinéticas de la enzima tipo silvestre y mutante.
Enzima kcat s-1
Km
µM
I50 Fru-2,6-P2
µM
I50 AMP
µM
nh AMP
ka Mg2+
µM
nh Mg2+
Tipo Silvestre 19,7±0,1 4,8±0,6 1,0±0,2 7,1±0,1 2,1±0,1 280±1 3,2±0,1
Lys274Trp 12,4±0,2 27,9±0,8 12,3±0,5 8,7±0,2 1,2±0,1 530±8 2,0±0,2

49
4.4 Caracterización de las propiedades de fluorescencia de las mutantes de FBPasa.
Se conoce que el triptófano usado como sonda entrega valiosa información dinámica y
estructural acerca del microambiente en el que se encuentra. La ausencia de triptófanos en la
estructura primaria de la FBPasa (Marcus et al., 1980) le otorgan la propiedad que tenga una muy
baja emisión de fluorescencia al excitar a 295nm, por lo cual la introducción de un flouróforo
intrínseco permite develar posibles cambios conformacionales que ocurren en la enzima,
mediados por sus distintos efectores. En la figura 13 se muestran los espectros de emisión de
fluorescencia de las enzimas silvestre y mutante Lys274Trp. La baja emisión de fluorescencia de
la FBPasa wt concuerda con la noción que la enzima de riñón de cerdo no posee residuos
triptófanos. La FBPasa Lys274Trp presentó un máximo de emisión alrededor de 350 nm,
indicando que el residuo de triptófano se encuentra localizado en un ambiente polar. Para la
determinación de las constantes de disociación, los cambios en la fluorescencia registrados se
corrigieron y se ajustaron a la ecuación de Hill, según lo descrito en Materiales y métodos. Todos
los datos de fluorescencia mostrados en adelante corresponden a la mutante de FBPasa
Lys274Trp.

50
λ (nm)
300 320 340 360 380 400
Fluo
resc
enci
a U
.A.
0
100
200
300
400
Figura 13. Espectros de emisión de fluorescencia de las FBPasas recombinantes silvestre y
mutante Lys274Trp La concentración de cada enzima fue de 60 µg/ml en solución
amortiguadora Tris-HCl 50 mM, pH 7,5, EDTA 0,1 mM. La longitud de onda de excitación fue
de 295 nm. El gráfico muestra los espectros de emisión de fluorescencia de las enzimas FBPasa
tipo silvestre (línea azul) y a la mutante Lys274Trp (línea roja), en el rango de 310 a 390 nm.
Para más detalles ver Material y métodos.

51
4.4.1 Unión de fructosa-1,6-bisfosfato a la mutante FBPasaLys274Trp.
Utilizando las propiedades de fluorescencia de la mutante Lys274Trp, se procedió a la
determinación de las constantes de unión realizando titulaciones con el sustrato. En las
titulaciones se encontró que en la FBPasa mutante no hubo corrimiento del máximo de emisión
de fluorescencia, indicando que no se alteró el microambiente donde se encuentra el residuo
triptófano. Los datos obtenidos para la unión del sustrato a la enzima mutante ajustan a una
hipérbola equilátera (Figura 14). Cabe indicar que la unión del sustrato a la enzima produce un
aumento en la eficiencia cuántica, que se representa como una disminución en la razón ΔF/F0,
siendo ΔF la diferencia entre la emisión de fluorescencia de la enzima sin ligando (F0) y el de la
enzima en presencia de ligando. El cambio en la emisión de fluorescencia ocurre a bajas
concentraciones de sustrato (< 40µM), observándose un cambio de aproximadamente 2%. El
valor de la constante de disociación del complejo enzima-sustrato, Kd, es de 17,1 µM, con un
valor de coeficiente de Hill para el sustrato cercano a 1,0, lo que concuerda con la noción que no
hay cooperatividad en la unión del sustrato a bajas concentraciones.
4.4.2 Unión de fructosa-2,6-bisfosfato a la mutante FBPasa Lys274Trp.
Se realizaron ensayos para determinar el efecto de la concentración del inhibidor Fru-2,6-
P2 en la emisión de fluorescencia de la mutante FBPasa Lys274Trp, en las condiciones ya
descritas, encontrándose que no hay variación significativa en la intensidad de fluorescencia
(Figura 15).

52
Fru-1,6-P2(µM)
0 200 400 600 800 1000
∆F/F
0
-0,20
-0,15
-0,10
-0,05
0,00
Figura 14. Efecto de la concentración de Fru-1.6-P2 en la emisión de fluorescencia de la
mutante FBPasa Lys274Trp La concentración de enzima fue de 60 µg/ml en solución
amortiguadora Tris-HCl 50 mM, pH 7,5, EDTA 0,1 mM, a las concentraciones de sustrato que se
indican, incubando por 1 min en cada adición de sustrato. La longitud de onda de excitación fue
de 295 nm a 15°C. Los puntos corresponden a los datos obtenidos experimentalmente y la línea
continua a la curva de tendencia ajustada a la ecuación de Hill. En el gráfico pequeño en la
esquina inferior derecha se muestra una ampliación del eje Y para destacar la tendencia.

53
Fru-2,6-P2 (µM)
0 2 4 6 8 10 12 14 16 18
∆F/F
0
-0,10
-0,05
0,00
0,05
0,10
Figura 15. Efecto de la concentración de Fru-2.6-P2 en la emisión de fluorescencia de la
mutante FBPasa Lys274Trp. La concentración de enzima fue de 60 µg/ml en solución
amortiguadora Tris-HCl 10 mM, pH 7,5, EDTA 0,1 mM, a las concentraciones de inhibidor que
se indican, incubando por 1 min en cada adición de Fru-2.6-P2. La longitud de onda de excitación
fue de 295 nm a 15°C. Los puntos corresponden a los datos obtenidos experimentalmente y la
línea continua a la curva de tendencia ajustada a la ecuación de Hill modificada por Taketa y
Pogell (1965).

54
4.4.3 Unión de Mg2+ a la mutante FBPasa Lys274Trp.
Se realizaron ensayos para determinar el efecto de la concentración del catión bivalente
magnesio en la emisión de fluorescencia de la mutante FBPasa Lys274Trp, en las condiciones ya
descritas, encontrándose que sólo hay una variación en la intensidad de fluorescencia menor a 1%
(Figura 16), valor que no es sorpresivo considerando que no se ha descrito que Mg2+ interaccione
con el residuo 274. No obstante, la baja variación en la intensidad de fluorescencia, existe una
clara tendencia a la disminución, y los valores de Ka. y nH muestran similitud con los obtenidos
cinéticamente.
4.4.4 Unión de AMP a la mutante FBPasa Lys274Trp.
Los mayores cambios en la intensidad de emisión de fluorescencia registrados
corresponden a la unión de AMP a la FBPasa Lys274Trp. Los datos obtenidos para la unión del
nucleótido a la enzima mutante se ajustan a una hipérbola equilátera, con una variación
aproximada a un 20% (Figura 17). Si bien el dominio de unión de este efector está ubicado
aproximadamente a 30Ǻ del sitio activo, la unión de AMP a la enzima genera numerosos
cambios en la estructura cuaternaria que podría este residuo ser capaz de evaluar, entre los cuales
destaca el desenganchamiento del sitio activo del lazo que comprende los residuos de
aminoácidos 52-72. Probablemente, este cambio conformacional inducido por AMP y que
provoca la desestabilización del sitio activo, sea capaz de generar el cambio de intensidad de
fluorescencia observado cuando se une el nucleótido.

55
Mg2+ (mM)
0 1 2 3 4 5
∆F/F
0
-0,20
-0,15
-0,10
-0,05
0,00
Figura 16. Efecto de la concentración del catión bivalente magnesio en la emisión de
fluorescencia de la mutante FBPasa Lys274Trp La concentración de enzima fue de 60 µg/ml
en solución amortiguadora Tris-HCl 10 mM, pH 7,5, EDTA 0,1 mM, a las concentraciones de
Mg2+ que se indican, incubando por 1 min en cada adición del catión bivalente. La longitud de
onda de excitación fue de 295 nm a 25°C. Los puntos corresponden a los datos obtenidos
experimentalmente y la línea continua a la curva de tendencia ajustada a la ecuación de Hill. En
el gráfico pequeño en la esquina inferior derecha se muestra una ampliación del eje Y para
destacar la tendencia.

56
AMP (µM)
0 20 40 60 80 100
∆F/F
0
0,00
0,05
0,10
0,15
0,20
Figura 17. Efecto en la intensidad de la fluorescencia en la enzima Lys274Trp mediado por
la unión de AMP. Los puntos corresponden a los datos obtenidos experimentalmente y la línea
continua a la curva de tendencia ajustada a la ecuación de Hill. La concentración de enzima fue
de 60 µg/ml en solución amortiguadora Tris-HCl 10 mM, pH 7,5, EDTA 0,1 mM, a las
concentraciones de AMP que se indican, incubando por 1 min en cada adición del catión
bivalente. La longitud de onda de excitación fue de 295 nm a 25°C. Los puntos corresponden a
los datos obtenidos experimentalmente y la línea continua a la curva de tendencia ajustada a la
ecuación de Hill.

57
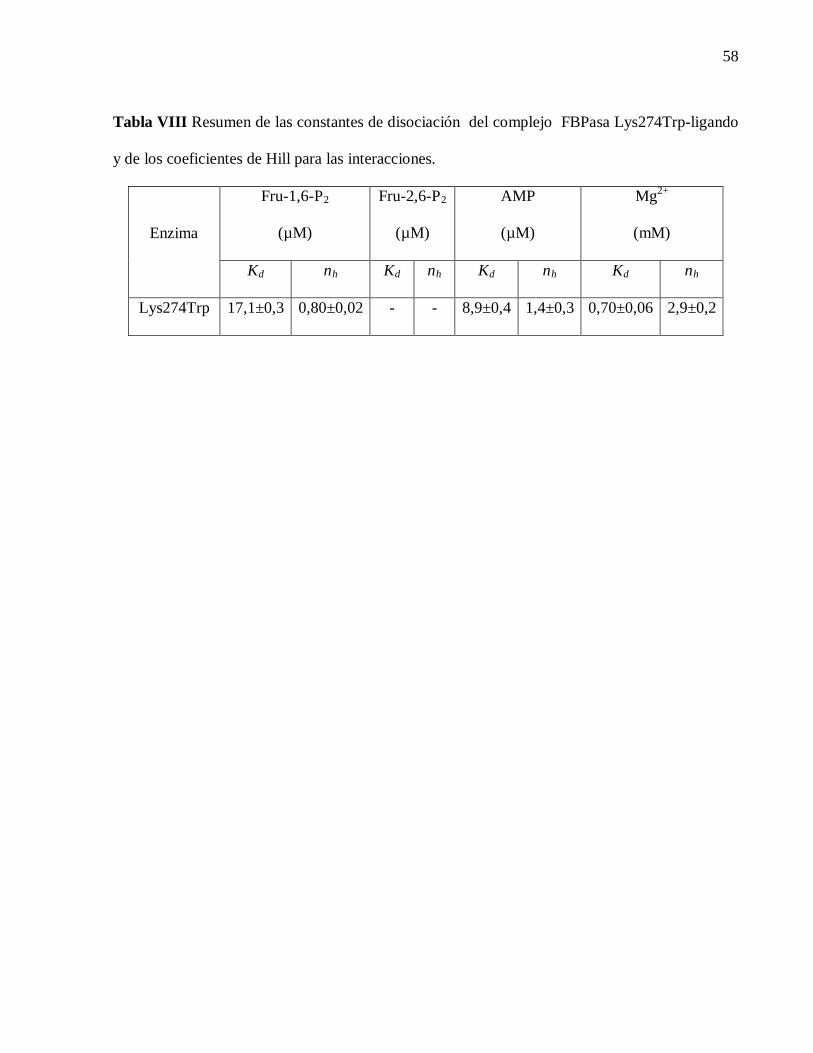
Kd es la constante de disociación para la formación del complejo enzima-ligando y nH
corresponde al coeficiente de Hill para la unión del respectivo ligando. Los valores de Kd y nH
para los diferentes ligandos fueron obtenidos de los datos de las figuras 14, 16, 17 y 18, fueron
calculados aplicando la ecuación descrita en el punto 3.2.10 de Material y métodos y resumidos
en la Tabla VIII. En general los principales cambios en la intensidad de la fluorescencia son
ocurridos de modo negativo, a excepción del AMP que ocurre de modo positivo. Este hecho
implica que la mayoría de los efectores produce un apagamiento del fluoroforo, probablemente
por el acercamiento de grupos apagadores, a diferencia del AMP que genera un aumento en la
intensidad de la fluorescencia.
58
Tabla VIII Resumen de las constantes de disociación del complejo FBPasa Lys274Trp-ligando
y de los coeficientes de Hill para las interacciones.
Enzima
Fru-1,6-P2
(µM)
Fru-2,6-P2
(µM)
AMP
(µM)
Mg2+
(mM)
Kd nh Kd nh Kd nh Kd nh
Lys274Trp 17,1±0,3 0,80±0,02 - - 8,9±0,4 1,4±0,3 0,70±0,06 2,9±0,2

59
5. Discusión.
La enzima fructosa-1,6-bisfosfatasa, por catalizar una etapa clave en la regulación del
flujo de la vía gluconeogénica, ha sido un blanco muy atractivo para realizar estudios
estructurales y cinéticos, con la intención principal de encontrar posibles drogas capaces de
inhibir su actividad y así disminuir la producción endógena de glucosa en estados diabéticos (van
Poelje et al. 2007; Dang et al. 2008), con lo cual se reducirían los efectos nocivos producidos por
los excesos de este azúcar (Qian et al. 2008). Los esfuerzos en este sentido se han centrado en el
estudio del efecto que tienen sobre la actividad catalítica análogos del inhibidor alostérico AMP
(van Poelje et al. 2007; Dang et al. 2008). No obstante, se conoce que sustratos o análogos de
sustrato que se unen al sitio activo son generalmente más efectivos que inhibidores que se unen a
otros sitios de la enzima. Entonces, análogos de Fru-1,6-P2 o Fru-2,6-P2, es probable que sean
ligandos de la enzima más adecuados para ser usados como drogas, que análogos del inhibidor
AMP, porque se espera que sean más específicos.
Con el propósito de aportar más información sobre el mecanismo de inhibición de la
enzima por fructosa-2,6-bisfosfato y por su sustrato Fru-1,6-P2, sobre todo con respecto a las
posibles diferencias entre los determinantes de unión del sustrato y del inhibidor Fru-2,6-P2, en
este trabajo la mutación de Lys274 a triptófano permitió colocar una sonda fluorescente única en
el putativo sitio activo de la enzima, generándose la mutante FBPasa Lys274Trp. Los resultados
apoyan la idea que la inhibición de la enzima por sustrato se debe a interacciones entre las
subunidades, de modo que la unión de las dos primeras moléculas de sustrato induce menor
afinidad y actividad catalítica en los otros dos sitios de unión. Además, muestran que Lys274 es
crucial para la unión de los azúcares bisfosfatos y catálisis, y demuestran que la espectroscopia de

60
fluorescencia es de primera importancia en el sondeo de la dinámica de cambios
conformacionales en la FBPasa. La mutagénesis del residuo Lys274 fue corroborada por la
secuenciación de los plásmidos que tenían el inserto de la FBPasa. La caracterización cinética de
la mutante Lys274Trp con respecto a la curva de saturación por sustrato mostró un claro
desplazamiento a la derecha, con un cambio en la concentración de sustrato a la cual la enzima se
inhibe por exceso de este (disminución de la inhibición de la enzima por sustrato). La enzima tipo
silvestre se encuentra inhibida sobre un 60% a una concentración de sustrato de 100µM, en
cambio la mutante logra su máxima actividad sobre 300µM. Este cambio fue acompañado por
una considerable baja en la actividad catalítica, lo que se refleja en una disminución alrededor de
la mitad en el valor de la kcat y en el aumento de seis veces en el valor de la Km, resultados que se
explican por la mutación del sitio activo.
Estos resultados sugieren que la mutación afecta la capacidad de la enzima para estabilizar
la unión del sustrato, debido principalmente a que el triptófano no sería capaz de interaccionar
con el sustrato del mismo modo a lo descrito para la lisina en esta posición; estabilización del
oxígeno en el anillo de furanosa y el grupo fosfato en posición 6 (Hines et al. 2007). Entonces, el
residuo lisina en posición 274 juega un importante papel en la unión del sustrato.
Al comparar las curvas de saturación de la mutante Lys274Trp con los datos obtenidos
para la mutante Lys274Ala (el-Maghrabi et al. 1992; Asenjo 2008), se observa que esta última
variante también presenta una disminución tanto en la afinidad de la enzima por el sustrato (Km
aumenta entre 6 y 40 veces respecto a la enzima silvestre) (el-Maghrabi et al. 1992; Asenjo
2008), como en la inhibición por exceso de sustrato (Asenjo 2008). Sin embargo, es notable que
la mutante Lys274Ala muestre, por el contrario a la enzima silvestre, un marcado aumento en el
valor de la constante catalítica (Asenjo 2008). Por otra parte, el residuo leucina en la posición 274

61
(FBPasa Lys274Leu) hace aumentar en 27 veces el valor de Km, sin afectar la constante catalítica
(Shyur et al., 1995). Los datos disponibles indican claramente que el triptófano también genera
un impedimento estérico en la unión del sustrato.
Por lo tanto, se puede postular que el residuo lisina en posición 274 tendría un importante
efecto estabilizador del sustrato, con un cierto grado de impedimento estérico, lo que se refleja en
su eficiencia catalítica, medida por el valor de la constante de especificidad (kcat / Km = 18 x 107
M-1s-1). Asimismo, este residuo estaría implicado en la capacidad que tiene el sustrato de inducir
el cambio conformacional que genera los sitios de baja afinidad para Fru-1,6-P2, mecanismo que
explica la inhibición por exceso de sustrato (Asenjo, 2008). En cambio, el residuo alanina en esta
posición (Lys274Ala) facilitaría el ingreso e interacción del sustrato al centro activo (bajo
impedimento estérico), pero con una baja estabilización del sustrato, lo que se refleja en un
aumento en los valores de kcat (29,2 s-1) y Km (32 µM), generándose una enzima con menor
eficiencia catalítica (kcat / Km = 9,1 x 107 M-1s-1), respecto de la enzima silvestre. Esta variante de
la FBPasa, a la vez, presenta una marcada disminución en la inhibición por exceso de sustrato. El
residuo triptófano en posición 274 tendría un mayor impedimento estérico y un menor efecto
estabilizador del sustrato (kcat = 12,4 s-1; Km = 27,9 µM), con una menor eficiencia catalítica (kcat
/ Km = 4,4 x107 M-1s-1). Congruente con este análisis, la sustitución de lisina por leucina en la
posición 274 (Shyur et al., 1995), genera una mutante que muestra un gran aumento en el valor
de Km (94 µM) y una leve disminución en el valor de la constante catalítica, mostrando una
eficiencia catalítica aún menor (kcat / Km = 1,3 x 107 M-1s-1). Digno de interés es el cambio de
tirosina por fenilalanina en la posición 244 (Shyur et al., 1995; Asenjo, 2008; Asenjo et al.,
2013), pues siendo un residuo que se también se encuentra en el sitio activo e interacciona con el
oxígeno del grupo fosfato en posición 6, muestra curvas de saturación y parámetros cinéticos

62
similares a los obtenidos con la mutante Lys274Trp (Asenjo, 2008; Asenjo et al., 2013); la
variante Tyr244Phe muestra un valor de constante de especificidad de 3,4 x107 M-1s-1.
Es interesante que cationes monovalentes como el ion potasio, a concentraciones
fisiológicas, producen también una disminución en la afinidad de la enzima por el sustrato y un
aumento en su actividad molecular (aumentan los valores de Km y kcat), efectos acompañados por
una marcada disminución de la inhibición por exceso de sustrato (Slebe et al., 1985). Luego, es
plausible pensar que el ion potasio induce un cambio conformacional en la enzima, generando un
sitio activo con una geometría tal que facilitaría el ingreso e interacción del sustrato (bajo
impedimento estérico), pero con una buena estabilización del Fru-1,6-P2. Dado que la unión de
cationes monovalentes altera el correcto funcionamiento del lazo comprendido entre los residuos
52-72 (Choe et al., 2003) es probable que la mutante Lys274Trp genere un efecto análogo sobre
el funcionamiento de este lazo. Por otra parte, no se puede descartar que el efecto del triptófano
en posición 274 podría perturbar el lazo 190, que comprende los residuos 187-195, y/o un
conjunto de cadenas laterales, entre las que destaca Ala54, que estabilizan la conformación
desenganchada del lazo 52-72 (estado T), pues mutaciones Ala54Leu, Glu97Ala, Asp118Ala o
Asp121Ala muestran una drástica disminución en el valor de kcat (Nelson et al., 2004; Iancu et
al., 2005). Sin embargo, esta explicación es menos plausible pues junto a la disminución en el
número de recambio las mutantes muestran una disminución en el valor de la constante de
activación de la enzima por Mg2+, efecto opuesto a lo observado en la FBPasa Lys274Trp, Más
aun, en el caso de la FBPasa Ala54Leu (Iancu et al. 2005) también se observa un aumento de 50
veces en el valor de IC50-AMP (concentración de AMP que causa 50% de inhibición).
La hidrólisis de Fru-1,6-P2 requiere la presencia de Mg2+, pues este metal se posiciona
estratégicamente cerca del fosfato 1 del azúcar, permitiendo la ruptura del enlace fosfodiester. Al

63
observar las curvas de activación por magnesio se puede apreciar que la mutación Lys274Trp
aumenta aproximadamente al doble la constante de activación manteniendo el efecto cooperativo
del metal. El aumento al doble de la constante de activación está de acuerdo con los resultados
descritos para las enzimas mutantes FBPasa Lys274Ala, FBPasa Tyr244Phe y FBPasa
Lys274Leu (Asenjo, 2008; Shyur, et al., 1995). Se sabe que Mg2+ estabiliza el correcto
“enganche” del lazo comprendido entre los residuos 52-72 al sitio activo (conformación del lazo
enganchado), posicionando la cadena lateral de un residuo catalítico esencial, Asp74 (Nelson et
al. 2000 a; b). Más aun, al interaccionar el lazo dinámico 52-72 con el sitio activo proporciona
una cadena lateral (Asp68) para la coordinación del metal en el sitio 3 y, a su vez, orienta las
cadenas laterales (Asp74 y Glu97) esenciales para la catálisis. Subunidades con el lazo
desenganchado tienen una afinidad reducida por los iones metálicos esenciales, y como
consecuencia, pueden verse afectadas con respecto a la catálisis. Entonces, nuestros resultados
sugieren que la posición del triptófano probablemente desestabiliza en parte la conformación
enganchada del lazo 52-72.
Se sabe que Ala54 está en el centro de un conjunto crucial de cadenas laterales
hidrofóbicas, y que este conjunto de interacciones se forma sólo cuando el lazo 52-72 está en la
conformación desenganchada (estado T) (Iancu, et al., 2005). La introducción en esta posición de
una cadena lateral de mayor volumen (Leu), rompe el conjunto de interacciones hidrofóbicas y
desestabiliza la conformación desenganchada del lazo 52-72, desplazando el equilibrio de la
población de moléculas de FBPasa hacia el estado R (enganchado). No es sorprendente, entonces,
que la mutante Ala54Leu muestre una importante disminución en el valor de la constante de
activación para Mg2+ (aumento de afinidad de la FBPasa por el metal) (Iancu et al., 2005). Sin
embargo, también muestra una disminución a la mitad en el valor de la constante catalítica, sin

64
afectar significativamente la afinidad de la enzima por la Fru-1,6-P2. Este efecto se reflejaría en
una rotación en 3º del dímero C1C2 respecto al dímero C3C4, estabilizando los metales en su
sitio de unión. Por lo tanto, es plausible pensar que la introducción de Trp en la posición 274
podría perturbar este giro y, a su vez, la conformación enganchada/desordenada del lazo 52-72.
Cabe recordar que este lazo puede estar en el estado R, activo, en dos conformaciones
(enganchado y desordenado), mientras el estado T favorece una única conformación
(desenganchada), la cual no puede estabilizar la asociación del catión divalente al sitio activo
(Choe et al., 2003; Nelson et. al., 2004). Así, la conformación enganchada se requiere para la
asociación con alta afinidad de los cationes bivalentes y para la estabilización del estado de
transición, mientras la conformación desordenada facilitaría la liberación de producto y
asociación del sustrato. Más aun, la presencia de Mg2+ en el sito 3 se correlaciona con la
aparición de una estructura ordenada para el segmento 61-69 del lazo 52-72; este segmento se
encuentra inmediatamente sobre el sitio activo (Choe, et .al, 2003). Así, el lazo 52-72 puede estar
en su conformación enganchada, mientras los residuos 61-69 oscilan entre una conformación que
estabiliza Mg2+ en el sitio 3 y otras conformaciones que permiten el intercambio de ligando entre
el sitio activo y el solvente. La movilidad del segmento sería importante en el recambio catalítico
(kcat) en la enzima al estado R con un lazo enganchado. Debido a que mutaciones en este
segmento ocasionan un aumento en la constante de activación para Mg2+ y una disminución en el
valor de kcat (Choe, et al., 2003), una explicación plausible para el efecto observado al incorporar
Trp en posición 274 sería una perturbación en la movilidad del segmento 61-69.
La inhibición por fructosa-2,6-bisfosfato en la mutante Lys274Trp disminuye
aproximadamente 12 veces, en comparación a la enzima tipo silvestre, sugiriendo que este
residuo es importante para la unión y efecto de este inhibidor. Es importante destacar que en

65
trabajos realizados con la enzima de hígado de rata, se describe el residuo Lys274 como de
trascendental importancia para la inhibición por Fru-2,6-P2, en base a que el cambio de este
residuo a alanina aumenta en 1.000 veces el valor de I50 para Fru-2,6-P2 (el-Maghrabi et al.
1992). Esta dramática disminución en la afinidad de la mutante por el Fru-2,6-P2 no se observó
en la enzima de riñón de cerdo, lo que probablemente refleja que en esta enzima la cadena lateral
de lisina interacciona con la misma fuerza con ambos azúcares bisfosfatos, mientras que en la de
hígado de rata el grupo ε-amino de la lisina 274 interacciona con mayor fuerza con Fru-2,6-P2
que con el sustrato. No obstante, en ambos casos, el residuo Lys274 está directamente implicado
en la unión y estabilización de ambos azúcares a la enzima. Estos datos son coincidentes con los
estudios de difracción de rayos X, los cuales postulan que la Lys274 es capaz de estabilizar el
grupo fosfato en la posición 6 y al anillo de furanosa, tanto del inhibidor como del sustrato (Ke et
al. 1991; Liang et al. 1992; Lu et al. 1996). Por otro lado, la sustitución del residuo Lys274 por
leucina en la enzima de hígado de porcino (Shyur et al. 1995), además de aumentar el valor de
Km en 27 veces, aumenta casi en 2.000 veces el valor de I50 para Fru-2,6-P2, lo que llevó a los
autores a plantear que este residuo lisina permite a la enzima discriminar entre el sustrato y el
inhibidor. Sin embargo, los resultados de nuestro laboratorio (esta tesis; mutante Lys274Ala), que
muestran un aumento de seis veces en la afinidad por Fru-1,6-P2 y de doce a dieciocho veces en
la afinidad por Fru-2,6-P2, no apoyan esta idea. Más bien, pareciera ser que la disminución en la
afinidad de la enzima por el Fru-2,6-P2 depende de la naturaleza de la cadena lateral que se
introduce en la posición 274; sin embargo, no se puede descartar que en la enzima de hígado de
rata el péptido carboxilo terminal también tenga un papel importante. Por otra parte, la pérdida de
Asp121 (FBPasa Asp121Ala) elimina la unión del metal al sitio 1 (Nelson, et al., 2003). Este es
el único sitio para el Mg2+ que hace de puente entre los dominios de AMP y Fru-1,6-P2 en una

66
subunidad, por lo que la pérdida del metal del sitio 1 causa una orientación errónea del dominio
de Fru-1,6-P2, respecto al dominio de AMP, con la consiguiente disminución en la afinidad por
Fru-2,6-P2, implicando un mecanismo de acoplamiento entre el sitio de unión de Fru-2,6-P2 y el
sitio 1 de unión del metal. Entonces, es perfectamente posible suponer que el residuo Trp274
puede afectar la unión del metal al sitio 1. Sin embargo, no se puede descartar que tenga un
efecto sobre la unión de Mg2+ a los sitios 2 y 3, pues el metal bivalente en estos sitios de unión
coordina el oxígeno distal de Pi. El análisis de las imágenes obtenidas desde los datos
cristalográficos muestra que el grupo amino, cargado positivamente, de la cadena lateral del
residuo Lys274 es capaz de interactuar con el grupo fosfato en posición 6 y 2 de la Fru-2,6-P2,
así como con el oxígeno del anillo de furanosa, considerando que la distancia entre el nitrógeno
de este grupo y los átomos anteriormente mencionados están entre 3 y 4Å (Figura 18).

67
Figura 18. Ampliación del dominio de unión de Fru-2,6-P2. Destacando con una flecha negra
el residuo Lys274, con una roja y verde los grupos fosfato en posición 6 y 2 de la Fru-2,6-P2
respectivamente. Los átomos de carbono se muestran como esferas color celeste al igual que la
estructura secundaria de la proteína, en rojo se aprecian los átomos de oxígeno, en dorado los
átomos de fósforo, en verde los de Mg2+ y en azul los de nitrógeno. El cuadro pequeño muestra
una subunidad de la FBPasa, desde la cual se obtuvo la ampliación, destacando en azul el lazo
comprendido entre los residuos 52 a 72. Código PDB: 2QVU (Hines et al. 2007).

68
Los resultados de la inhibición por AMP sugieren que este residuo no afecta
significativamente la afinidad de la enzima por el nucleótido, pues los valores de I50 para AMP
de la mutante y la enzima tipo silvestre son similares. No obstante, el principal efecto visualizado
es la perdida de la cooperatividad entre los sitios de unión de AMP, la cual podría deberse a un
efecto del residuo de triptófano en la posición 274 sobre la transición del lazo 52-72 entre las
conformaciones enganchada, desordenada y desenganchada. Considerando que este lazo es
esencial para que la inhibición por AMP sea alostérica (Nelson et al. 2000a), podemos suponer
que el triptófano introducido en la posición 274 afecta la correcta transducción de la señal al
unirse AMP. En este sentido, un mecanismo secuencial permite explicar la inhibición cooperativa
de la enzima por AMP (Iancu et al., 2005), el cual predice que existe un acoplamiento entre la
mitad superior e inferior del tetrámero, implicando las subunidades C1 y C4. La primera
molécula de AMP puede unirse a cualquier subunidad con la misma afinidad, causando el
movimiento de la hélice H2 en esa subunidad. Si la unión ocurre en la subunidad C1,
interacciones entre la hélice H2 de esta subunidad y el lazo 190 de la subunidad C4 se debilitan,
de manera que la segunda molécula de AMP se une con mayor afinidad a la subunidad C4, pues
requeriría una menor energía para el movimiento de la hélice H2 en esa subunidad. Cabe recordar
que, para explicar el efecto de triptófano en posición 274 sobre la disminución en eficiencia
catalítica de la enzima, propusimos que Trp274 podría perturbar el lazo 190. Por otra parte, el
modelo secuencial también considera los cambios conformacionales en el lazo dinámico
(residuos 52-72). Interesantemente, todas las mutaciones descritas de Lys42, Arg49, Lys50,
Ile190, Gly191 y Glu192 y modificación química de Arg49 y Lys50, residuos que son parte de la
hélice H2 o del lazo 190, eliminan la cooperatividad para AMP (Lu et al., 1996; Shyur et al.,

69
1996; Lu et al., 1997; Ludwig et al., 1999; Cárcamo et al., 2000), en forma similar al efecto
causado por Trp274.
En relación a los ensayos de unión de los efectores de la enzima, registrados por los
cambios en la emisión de fluorescencia, se encontró que el máximo en la intensidad de la
fluorescencia no mostró variación debido a la unión de ningún efector, manteniéndose
aproximadamente en 350nm, lo cual indica que ningún efector produce cambios significativos en
la polaridad del microambiente alrededor del residuo de triptófano introducido, y además, este
valor indica que se encuentra bastante expuesto al solvente o a condiciones polares (Royer 2006).
Los cambios que se lograron observar fueron en la intensidad de la fluorescencia, probablemente
por el acercamiento o alejamiento de grupos apagadores del triptófano.
De modo adicional, es importante destacar que la posición relativa del grupo funcional del
triptófano con respecto a su carbono alfa no sigue el mismo plano, a diferencia de la lisina, como
se visualiza en la figura 19. Esta diferencia tiene como posible efecto que la orientación del grupo
funcional del triptófano este dirigida hacia una posición distinta a la de unión del sustrato y Fru-
2,6-P2, además, que la distancia entre el carbono alfa y el átomo más distante es menor para el
triptófano.
Por lo tanto, es plausible sugerir que la posición relativa de la cadena lateral del residuo de
triptófano tiene una orientación y proximidad diferente a la de la cadena lateral de lisina, sobre
todo al considerar la flexibilidad del residuo Lys274 (Giroux et al. 1994).

70
Figura 19. Superposición de los esquemas de cintas, ampliado el dominio de unión de
Fru-2,6-P2 de la FBPasa tipo silvestre y la mutada computacionalmente (Cod. PDB 3FBP).
Destacando con una flecha amarillo la posición relativa del triptófano en posición 274, una flecha
roja la posición de la lisina 274 y en una flecha verde la Fru-2,6-P2.

71
La mutante Lys274Trp detecta la unión de Mg2+ y Fru-1,6-P2 por un leve incremento en la
emisión de fluorescencia, aproximadamente entre 1-2%, pero esta emisión de fluorescencia no
fue afectada por la unión del Fru-2,6-P2, resultados concordantes con el microambiente polar y el
alto grado de exposición del residuo 274. Sin embargo, debido al diferente efecto producido por
la unión de los azúcares bisfosfatos, es posible sugerir que la posición del sustrato es más cercana
al Trp274 que el inhibidor, así como que ambas moléculas podrían poseer distintos determinantes
de unión.
El mayor cambio en la intensidad de la fluorescencia fue observado por la unión de AMP,
aproximadamente un 20% de disminución con respecto a la enzima sin el ligando. Esta
significativa disminución en la emisión de fluorescencia, comparada al leve o nulo efecto
generado por la unión del sustrato y Fru-2,6-P2, puede implicar que la posición relativa del
residuo de triptófano es capaz de detectar el efecto de la unión de AMP mediado por a) la
transición entre las conformaciones enganchada, desordenada y desenganchada del lazo
comprendido por los residuos 52 -72; b) otros cambios en la estructura cuaternaria, como por
ejemplo el giro del dominio del sitio activo con respecto al dominio de unión de AMP (Liang et
al. 1993); y c) cambios diferentes en la conformación del lazo comprendido entre los residuos
262-275, por la unión de Fru-2,6-P2 y AMP.
Aunque si bien el efecto de la unión de AMP en la estructura cuaternaria de la enzima de
riñón de cerdo es similar al de la unión de Fru-2,6-P2 (Hines et al 2007; Nelson et al 2000b), lo
que descartaría la hipótesis descrita en a), las acciones de AMP en el sitio alostérico
(aproximadamente a 28 Å del centro catalítico) y de Fru-2,6-P2 en el sitio activo desplazan el lazo
dinámico (52-72) enganchado por distintos mecanismos (Hines et al 2007), con lo cual es posible
que el Trp274 sea capaz de diferenciar los mecanismos mediados por cada inhibidor.

72
Hemos sugerido que el lazo comprendido entre los residuos 262-275 podría estar
implicado en la detección de la unión de AMP por Trp274, ya que este lazo adopta diferentes
conformaciones dependiendo del complejo FBPasa-ligando formado. Sin embargo, esta hipótesis
parece menos plausible pues se ha demostrado que la introducción de Trp en la posición de
Phe219 permite detectar la unión del sustrato y de Fru-2,6-P2, por un gran aumento en la emisión
de fluorescencia, pero no es capaz de detectar la unión de AMP (Asenjo, 2008). El residuo
Phe219 está próximo al sitio activo, posicionado al extremo del lazo L10.
Como conclusión, el residuo Lys274 tiene importancia en la estabilización de la unión del
sustrato y el inhibidor Fru-2,6-P2 de distinto modo, aunque este no sea el principal residuo
participante en la unión de estos dos efectores, pero los datos sugieren fuertemente que este
residuo puede ser uno de los principales involucrados en la transducción de señal, al sitio activo,
mediada por la unión de AMP.

73
6. Bibliografía
Asenjo, J. (2000). Obtención y caracterización de las mutantes 219 y 232 de fenilalanina a
triptófano (Phe219Trp y Phe232Trp) de la fructosa-1,6-bisfosfatasa de riñón de cerdo.
Tesis de Bioquímico. Escuela de Bioquímica. Facultad de Ciencias. Universidad Austral
de Chile.
Asenjo, J. (2008). Asimetría en la fructosa-1,6-bisfosfatasa. Análisis del mecanísmo de
inhibición por fructosa-2,6-bisfosfato y por altas concentraciones de fructosa-1,6-
bisfosfato, mediante el uso de enzimas híbridas con subunidades sensibles y
desensibilizadas al efector. Tesis de Doctorado en Ciencias, mención biología celular y
molecular. Escuela de Graduados. Facultad de Ciencias. Universidad Austral de Chile.
Bradford, MM (1976). "A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding." Anal Biochem 72:
248-54.
Carcamo, JG, Yanez AJ, Ludwig HC, Leon O, Pinto RO, Reyes AM and Slebe JC (2000). "The
C1-C2 interface residue lysine 50 of pig kidney fructose-1, 6-bisphosphatase has a crucial
role in the cooperative signal transmission of the AMP inhibition." Eur J Biochem 267(8):
2242-51.
Choe, JY, Fromm HJ, and Honzatko RB (2000). "Crystal structures of fructose 1,6-
bisphosphatase: mechanism of catalysis and allosteric inhibition revealed in product
complexes." Biochemistry 39(29): 8565-74.
Choe, JY, Poland BW, Fromm HJ, and Honzatko RB (1998). "Role of a dynamic loop in cation
activation and allosteric regulation of recombinant porcine fructose-1,6-bisphosphatase."
Biochemistry 37(33): 11441-50.

74
Choe JY, Nelson SW, Fromm HJ, Honzatko RB (2003). "Interaction of Tl+ with product
complexes of fructose-1,6-bisphosphatase." J Biol Chem. 2003 May 2;278(18):16008-14.
Dang, Q, Kasibhatla SR, Jiang T, Fan K, Liu Y, Taplin F, Schulz W, Cashion DK, Reddy KR,
van Poelje PD, Fujitaki JM, Potter SC and Erion MD (2008). "Discovery of phosphonic
diamide prodrugs and their use for the oral delivery of a series of fructose 1,6-
bisphosphatase inhibitors." J Med Chem 51(14): 4331-9.
el-Maghrabi, MR, Austin LR, Correia JJ and Pilkis SJ (1992). "Lysine 274 is essential for
fructose 2,6-bisphosphate inhibition of fructose-1,6-bisphosphatase." J Biol Chem
267(10): 6526-30.
Giroux, E, Williams MK. and Kantrowitz ER (1994). "Shared active sites of fructose-1,6-
bisphosphatase. Arginine 243 mediates substrate binding and fructose 2,6-bisphosphate
inhibition." J Biol Chem 269(50): 31404-9.
Hers, HG. and Van Schaftingen E (1982). Fructose-2,6-bisphosphate two years after its
discovery. Biochem. J. 206: 1-12.
Hines, JK, Chen X, Nix JC, Fromm HJ and Honzatko RB (2007). "Structures of mammalian and
bacterial fructose-1,6-bisphosphatase reveal the basis for synergism in AMP/fructose 2,6-
bisphosphate inhibition." J Biol Chem 282(49): 36121-31.
Hodgson, RJ, Jia Z and Plaxton WC (1998). "A fluorescence study of ligand-induced
conformational changes in cytosolic fructose-1,6-bisphosphatase from germinating castor
oil seeds." Biochim Biophys Acta 1388(2): 285-94.
Ke H, Thorpe CM, Seaton BA, Marcus F, y Lipscomb WN. (1989). Molecular structure of
fructose-1,6-bisphosphatase at 2.8-A resolution. Proc Natl Acad Sci U S A 86: 1475-
1479.

75
Ke, HM, Liang JY, Zhang YP and Lipscomb WN (1991). "Conformational transition of fructose-
1,6-bisphosphatase: structure comparison between the AMP complex (T form) and the
fructose 6-phosphate complex (R form)." Biochemistry 30(18): 4412-20.
Ke, HM, Zhang YP and Lipscomb WN (1990). "Crystal structure of fructose-1,6-bisphosphatase
complexed with fructose 6-phosphate, AMP, and magnesium." Proc Natl Acad Sci U S A
87(14): 5243-7.
Lakowicz, JR (2006). Principles of Fluorescence Spectroscopy, 3° Edición.
Liang, JY, Huang S, Zhang Y, Ke H and Lipscomb WN (1992). "Crystal structure of the neutral
form of fructose 1,6-bisphosphatase complexed with regulatory inhibitor fructose 2,6-
bisphosphate at 2.6-A resolution." Proc Natl Acad Sci U S A 89(6): 2404-8.
Liang, J, Zhang YY, Huang S and Lipscomb WN (1993). "Allosteric transition of fructose-1,6-
bisphosphatase." Proc Natl Acad Sci U S A 90(6): 2132-6.
Lu G, Giroux EL, Kantrowitz ER. (1997). "Importance of the dimer-dimer interface for allosteric
signal transduction and AMP cooperativity of pig kidney fructose-1,6-bisphosphatase.
Site-specific mutagenesis studies of Glu-192 and Asp-187 residues on the 190's loop". J
Biol Chem. 1997 Feb 21;272(8):5076-81.
Lu, G, Stec B, Giroux EL. and Kantrowitz ER (1996). "Evidence for an active T-state pig kidney
fructose 1,6-bisphosphatase: interface residue Lys-42 is important for allosteric inhibition
and AMP cooperativity." Protein Sci 5(11): 2333-42.
Ludwig, HC, Pardo FN, Asenjo JL, Maureira MA, Yanez AJ and Slebe JC (2007). "Unraveling
multistate unfolding of pig kidney fructose-1,6-bisphosphatase using single tryptophan
mutants." Febs J 274(20): 5337-49.

76
Ludwig HC, Herrera R, Reyes AM, Hubert E, Slebe JC. (1999), "Suppression of kinetic AMP
cooperativity of fructose-1,6-bisphosphatase by carbamoylation of lysine 50". J Protein
Chem. 1999 Jul;18(5):533-45.
Meek DW and Nimmo HG.(1984) Effects of phosphorylation on the kinetic properties of rat liver
fructose-1,6-bisphosphatase. Biochem J 222: 125-130.
Marcus, F, Edelstein I, Reardon I and Heinrikson RL (1982). "Complete amino acid sequence of
pig kidney fructose-1,6-bisphosphatase." Proc Natl Acad Sci U S A 79(23): 7161-5.
Maureira, MA (2005). Obtención y Caracterización de la Mutante Sitio Eespecífica Phe16Trp de
la Fructosa-1,6-Bisfosfatasa de Riñón de Cerdo. Tesis de Bioquímico. Escuela de
Bioquímica. Facultad de Ciencias. Universidad Austral de Chile.
McGrane, MM, El-Maghrabi MR and Pilkis SJ (1983). "The interaction of fructose 2,6-
bisphosphate and AMP with rat hepatic fructose 1,6-bisphosphatase." J Biol Chem
258(17): 10445-54.
Nelson, DL and Cox MM (2005). Lehninger, Principios de Bioquímica, 4° Edición, Capitulo 14
y 15.
Nelson, SW, Choe JY, Honzatko RB and Fromm HJ (2000a). "Mutations in the hinge of a
dynamic loop broadly influence functional properties of fructose-1,6-bisphosphatase." J
Biol Chem 275(39): 29986-92.
Nelson, SW, Iancu CV, Choe JY, Honzatko RB and Fromm HJ (2000b). "Tryptophan
fluorescence reveals the conformational state of a dynamic loop in recombinant porcine
fructose-1,6-bisphosphatase." Biochemistry 39(36): 11100-6.
Nelson SW, Honzatko RB, Fromm HJ. (2004) "Origin of cooperativity in the activation of
fructose-1,6-bisphosphatase by Mg2+. J Biol Chem. 2004 Apr 30;279(18):18481-7,

77
Pilkis SJ, El-Maghrabi MR, Pilkis J and Claus T (1981). “Inhibition of fructose-1,6-
bisphosphatase. J. Biol. Chem. 256: 3619-3622.
Pilkis, SJ, el-Maghrabi MR and Claus TH (1988). "Hormonal regulation of hepatic
gluconeogenesis and glycolysis." Annu Rev Biochem 57: 755-83.
Pilkis, SJ, El-Maghrabi MR, Pilkis J and Claus T (1981). "Inhibition of fructose-1,6-
bisphosphatase by fructose 2,6-bisphosphate." J Biol Chem 256(8): 3619-22.
Pilkis, SJ and Granner DK (1992). "Molecular physiology of the regulation of hepatic
gluconeogenesis and glycolysis." Annu Rev Physiol 54: 885-909.
Qian, Y, Feldman E, Pennathur S, Kretzler M and Brosius FC, 3rd (2008). "From fibrosis to
sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy." Diabetes 57(6):
1439-45.
Reyes AM, Burgos ME, Hubert E, y Slebe JC.(1987). Selective thiol group modification renders
fructose-1,6-bisphosphatase insensitive to fructosa-2,6-bisphosphate inhibition. J Biol
Chem 262: 8451-8454, 1987.
Reyes AM, Bravo N, Ludwig H, Iriarte A, y Slebe JC. (1993). Modification of Cys-128 of pig
kidney fructose 1,6-bisphosphatase with different thiol reagents: size dependent effect on
the substrate and fructose-2,6-bisphosphate interaction. J Protein Chem 12: 159-168.
Royer, C. A. (2006). "Probing protein folding and conformational transitions with fluorescence."
Chem Rev 106(5): 1769-84.
Segel, I. (1975). Enzime Kinetics.
Shyur LF, Aleshin AE, Honzatko RB, Fromm HJ. (1996). "Biochemical properties of mutant and
wild-type fructose-1,6-bisphosphatases are consistent with the coupling of intra- and

78
intersubunit conformational changes in the T- and R-state transition". J Biol Chem. 1996
Dec 27;271(52):33301-7.
Shyur, LF, Zhang R and Fromm HJ (1995). "Site-directed mutagenesis of the substrate binding
site of porcine fructose-1,6-bisphosphatase." Arch Biochem Biophys 319(1): 123-7.
Slebe, JC, Reyes A and Hubert E (1985). "Activation of fructose-1,6-bisphosphatases by
monovalent cations and its relationship with a fructose-2,6-bisphosphate allosteric site."
Arch Biol Med Exp (Santiago) 18(3-4): 309-15.
Taketa, K and Pogell BM (1965). "Allosteric Inhibition of Rat Liver Fructose 1,6-Diphosphatase
by Adenosine 5'-Monophosphate." J Biol Chem 240: 651-62.
van Poelje, PD, Dang Q and Erion MD (2007). "Discovery of fructose-1,6-bisphosphatase
inhibitors for the treatment of type 2 diabetes." Curr Opin Drug Discov Devel 10(4): 430-
7.
Van Schaftingen, E (1987). "Fructose 2,6-bisphosphate." Adv Enzymol Relat Areas Mol Biol 59:
315-95.
Villeret, V, Huang S, Zhang Y and Lipscomb WN (1995). "Structural aspects of the allosteric
inhibition of fructose-1,6-bisphosphatase by AMP: the binding of both the substrate
analogue 2,5-anhydro-D-glucitol 1,6-bisphosphate and catalytic metal ions monitored by
X-ray crystallography." Biochemistry 34(13): 4307-15.
Williams, MK and Kantrowitz ER (1992). "Isolation and sequence analysis of the cDNA for pig
kidney fructose 1,6-bisphosphatase." Proc Natl Acad Sci U S A 89(7): 3080-2.


















