
NOM 240 SSA 1-2012. Instalación y operación de la ... · NOM-240-SSA1-2012 ARTÍCULO 38 Las...
35
Mayo 2013 NOM 240 SSA 1-2012. Instalación y operación de la Tecnovigilancia Q. María del Carmen Becerril Martínez
Transcript of NOM 240 SSA 1-2012. Instalación y operación de la ... · NOM-240-SSA1-2012 ARTÍCULO 38 Las...

Mayo 2013
NOM 240 SSA 1-2012. Instalación y
operación de la Tecnovigilancia
Q. María del Carmen Becerril Martínez

El gobierno tiene la
responsabildad de garantizar la
seguridad calidad y eficacia de
los Dispositivos Médicos,
usados en los servicios de salud
y por los pacientes.

Para garantizar la seguridad, eficacia, calidad y funcionalidad de los Dispositivos Médicos a través de:
Garantía de información: El diseño del dispositivo y los métodos de producción deben
documentarse (por ejemplo, Archivo de Documentación Técnica)
Garantía de seguridad:
El dispositivo no debe involucrar un riesgo para el paciente, usuario o tercero (por ejemplo, cumplimiento con las normas de seguridad).

Garantía de desempeño: El dispositivo debe lograr las características de desempeño
descritas por el fabricante (por ejemplo, de acuerdo con los datos clínicos y la evaluación).
Garantía de calidad: Los dispositivos fabricados en serie deben proveer las mismas
características de seguridad y desempeño que el tipo de dispositivo original (por ejemplo, la organización debe tener implementado un sistema de aseguramiento de calidad).
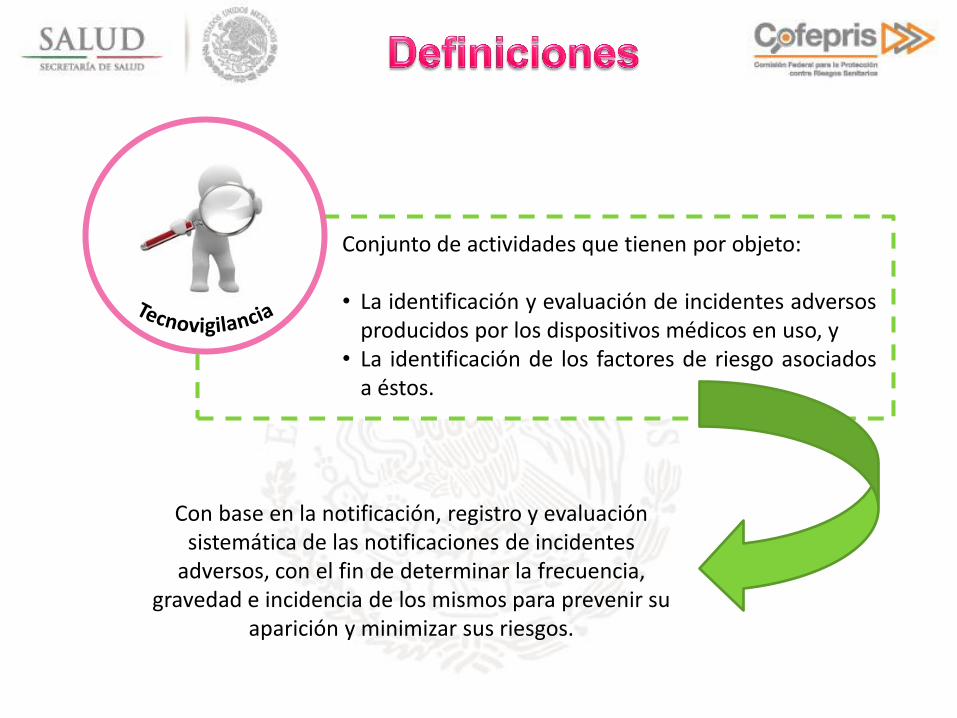
Con base en la notificación, registro y evaluación sistemática de las notificaciones de incidentes
adversos, con el fin de determinar la frecuencia, gravedad e incidencia de los mismos para prevenir su
aparición y minimizar sus riesgos.
Conjunto de actividades que tienen por objeto: • La identificación y evaluación de incidentes adversos
producidos por los dispositivos médicos en uso, y • La identificación de los factores de riesgo asociados
a éstos.

A la sustancia, mezcla de sustancias, material, aparato o instrumento empleado solo o en
combinación en el diagnóstico, monitoreo o prevención de enfermedades en humanos o
auxiliares en el tratamiento de las mismas y de la discapacidad, así como los empleados en el
reemplazo, corrección, restauración o modificación de la anatomía o procesos fisiológicos humanos.
Prótesis
Material quirúrgico

Aparatos, accesorios e instrumental para uso específico destinados a la atención médica, quirúrgica o a procedimientos de exploración, diagnóstico, tratamiento y rehabilitación de pacientes, así como aquellos para efectuar actividades de investigación biomédica.
Equipo médico
Aquellos dispositivos destinados a sustituir o complementar una función, un órgano, o un tejido del cuerpo humano.
Prótesis, órtesis y ayudas funcionales
Todos los insumos incluyendo antígenos, anticuerpos calibradores, verificadores o controles, reactivos, equipos de reactivos, medios de cultivo y de contraste y cualquier otro similar que pueda utilizarse como auxiliar de otros procedimientos clínicos o paraclínicos.
Agentes de diagnóstico

Todas las sustancias o materiales empleados para la atención de la Salud dental.
Insumos de uso odontológico
Los dispositivos o materiales que adicionados o no de antisépticos o germicidas se utilizan en la práctica quirúrgica o en el tratamiento de las soluciones de continuidad, lesiones de la piel o sus anexos.
Material quirúrgico y de curación
Los materiales y sustancias que se apliquen en la superficie de la piel o cavidades corporales y que tengan acción farmacológica o preventiva.”
Productos higiénicos
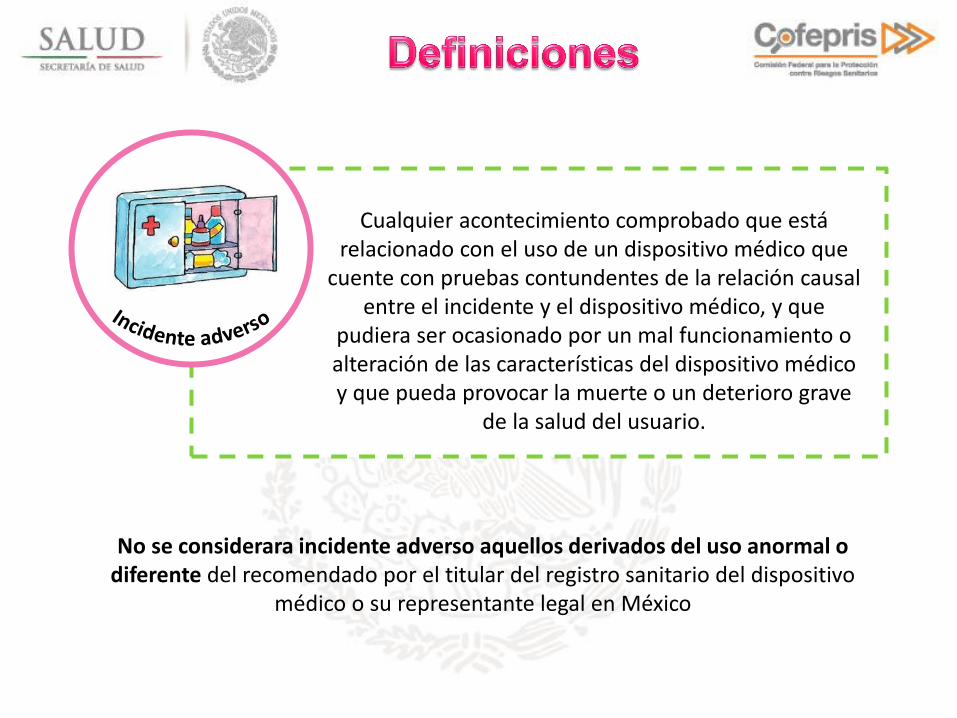
Cualquier acontecimiento comprobado que está relacionado con el uso de un dispositivo médico que
cuente con pruebas contundentes de la relación causal entre el incidente y el dispositivo médico, y que
pudiera ser ocasionado por un mal funcionamiento o alteración de las características del dispositivo médico y que pueda provocar la muerte o un deterioro grave
de la salud del usuario.
No se considerara incidente adverso aquellos derivados del uso anormal o diferente del recomendado por el titular del registro sanitario del dispositivo
médico o su representante legal en México
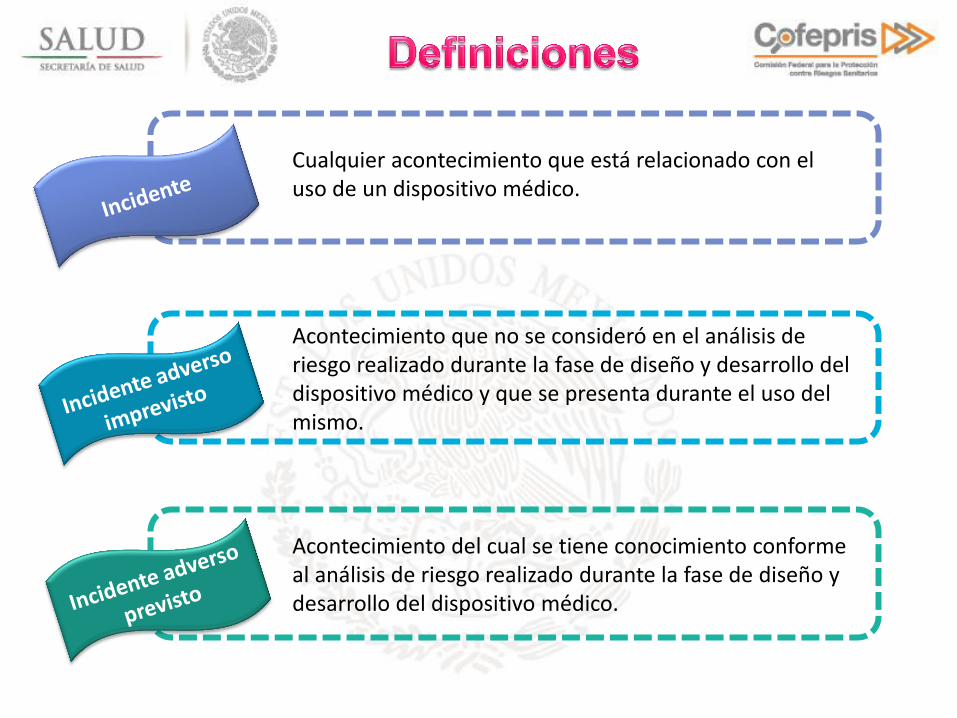
Cualquier acontecimiento que está relacionado con el uso de un dispositivo médico.
Acontecimiento del cual se tiene conocimiento conforme al análisis de riesgo realizado durante la fase de diseño y desarrollo del dispositivo médico.
Acontecimiento que no se consideró en el análisis de riesgo realizado durante la fase de diseño y desarrollo del dispositivo médico y que se presenta durante el uso del mismo.

Propósito final del dispositivo médico, conforme a las instrucciones de uso e
información suministrada por el fabricante.
Acción u omisión que conduce a un resultado diferente del
previsto por el fabricante o al esperado por el usuario. El error
de uso incluye descuidos, equivocaciones y todo uso
indebido que se pueda prever.

RIS
NOM-240-SSA1-2012
ARTÍCULO 38 Las reacciones adversas de los medicamentos u otros insumos que se presenten durante la comercialización o uso de éstos, las notificadas por los profesionales de la Salud, las publicadas en la literatura científica y las reportadas por los organismos sanitarios internacionales, deberán hacerse del conocimiento inmediato de la Secretaría por el titular del registro, por los distribuidores o comercializadores de los insumos.

Instalación y operación de la Tecnovigilancia. (Vigilancia de la seguridad de los dispositivos médicos)
Establece los lineamientos sobre los cuales se deben realizar las actividades de vigilancia de la seguridad de los dispositivos médicos (tecnovigilancia) con la finalidad de: • Garantizar la protección de la salud y seguridad de los productos • Evitar la repetición de incidentes adversos, mediante la
recolección, evaluación y difusión de la información sobre los citados incidentes, sanciones y medidas correctivas adoptadas.
Publicada en el DOF el 30 de octubre de 2012.

Esta norma permite unificar criterios de aplicación a nivel nacional, pretendiendo así establecer perfiles
de seguridad, a través de la participación y comunicación activa entre cada uno de los
integrantes y la autoridad sanitaria, para la práctica médica nacional en procesos fisiológicos humanos.
Prótesis
Material quirúrgico

Se utilizará la misma infraestructura de la Farmacovigilancia ampliando su alcance a los
dispositivos médicos.

Centro Nacional
Centros Estatales
Centros Institucionales
Titulares de registro sanitario
Establecimientos dedicados a la venta y suministro
de insumos para la salud
Instituciones del Sistema nacional de salud
Centros de investigación
Usuarios y pacientes

Notificación inicial de incidentes adversos involucrados con Dispositivos médicos con registro sanitario en México.
Reportes de seguimiento y final de incidentes adversos que incluyan las acciones preventivas y correctivas llevadas a cabo en el territorio nacional.
Informe de Tecnovigilancia que se genere como parte del proceso de prórroga de registros sanitarios

Centro Nacional de Farmacovigilancia
Pacientes
Médico
CE y CI
IQF
Unidades de Investigación
Clínica

TITULAR, FABRICANTE, DISTRIBUIDOR, COMERCIALIZADOR
PROFESIONALES DE LA SALUD PACIENTES Ó USUARIOS
FORMATOS DE NOTIFICACIONES DE INCIDENTES ADVERSOS DE DISPOSITIVOS MEDICOS

TITULAR, FABRICANTE, DISTRIBUIDOR, COMERCIALIZADOR
FORMATOS DE NOTIFICACIONES DE INCIDENTES ADVERSOS DE DISPOSITIVOS MEDICOS

PROFESIONALES DE LA SALUD
FORMATOS DE NOTIFICACIONES DE INCIDENTES ADVERSOS DE DISPOSITIVOS MEDICOS

PACIENTES Ó USUARIOS
FORMATOS DE NOTIFICACIONES DE INCIDENTES ADVERSOS DE DISPOSITIVOS MEDICOS
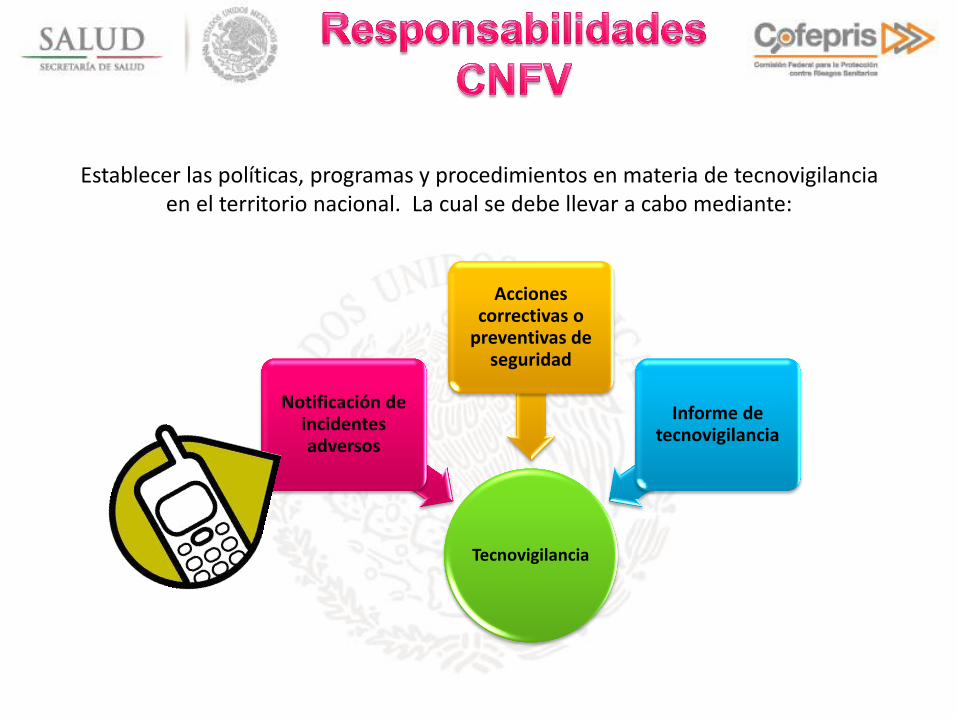
Tecnovigilancia
Notificación de incidentes adversos
Acciones correctivas o
preventivas de seguridad
Informe de tecnovigilancia
Establecer las políticas, programas y procedimientos en materia de tecnovigilancia en el territorio nacional. La cual se debe llevar a cabo mediante:

Contar con un líder de proyecto que será el mismo de Farmacovigilancia
Recibir y enviar la notificación inicial de los
incidentes al CNFV y turnar una copia de la misma al
titular del registro o representante legal en
México para su seguimiento.
Contar con un Comité de
Tecnovigilancia

Reportar a los centros de Tecnovigilancia o al CNFV
todo incidente adverso relacionado con
dispositivos médicos.
Asignar una persona responsable de llevar a cabo la vigilancia de los dispositivos médicos.

Fomentar, registrar y recopilar notificaciones de incidentes adversos
Dar continuidad a las acciones que la SSA
determine incluyendo aquellas en coordinación
con autoridades sanitarias extranjeras
Desarrollar y mantener actualizados PNO´s para:
•Recibir, registrar e investigar informes de incidentes adversos.
•Validar datos verificando fuentes.
•Detectar posible duplicidad.
•Conservar todos los datos del informe por cinco años o en función de la vida útil del producto

Garantizar la confidencialidad de la
identidad de los informantes, así como la
integridad de almacenamiento y
transmisión de datos.
A solicitud del CNFV, estimar la frecuencia del
incidente adverso e investigar el posible
factor de riesgo.
Informar al CNFV:
•Los incidentes adversos dentro del tiempo establecido.
•Sobre la implementación de las acciones requeridas.
•Sobre los plazos estipulados por la autoridad competente del país donde se presenten los incidentes adversos en el extranjero con productos que se comercialicen en México.

Realizar notificación inicial, reporte de seguimiento o
reporte final de los incidentes adversos
Proveer al personal asignado de información, entrenamiento y
capacitación en el área de Tecnovigilancia
Realizar el informe de Tecnovigilancia
quinquenalmente, como parte del proceso de
prórroga (renovación) o modificación del registro
sanitario

Notificación inicial • Datos de quien presenta la
notificación: Nombre Institución Dirección Número de teléfono y fax Fecha de la notificación. • Datos del fabricante y distribuidor: Nombre Dirección • Identificación del paciente: Nombre Edad Sexo
• Lugar del incidente
• Descripción del incidente
• Fecha del incidente
• Nombre comercial del dispositivo
• Tipo de dispositivo médico
• Modelo o número de catálogo
• Número de serie o de lote
• Accesorios o dispositivo médicos asociados
• Versión del software (si aplica)
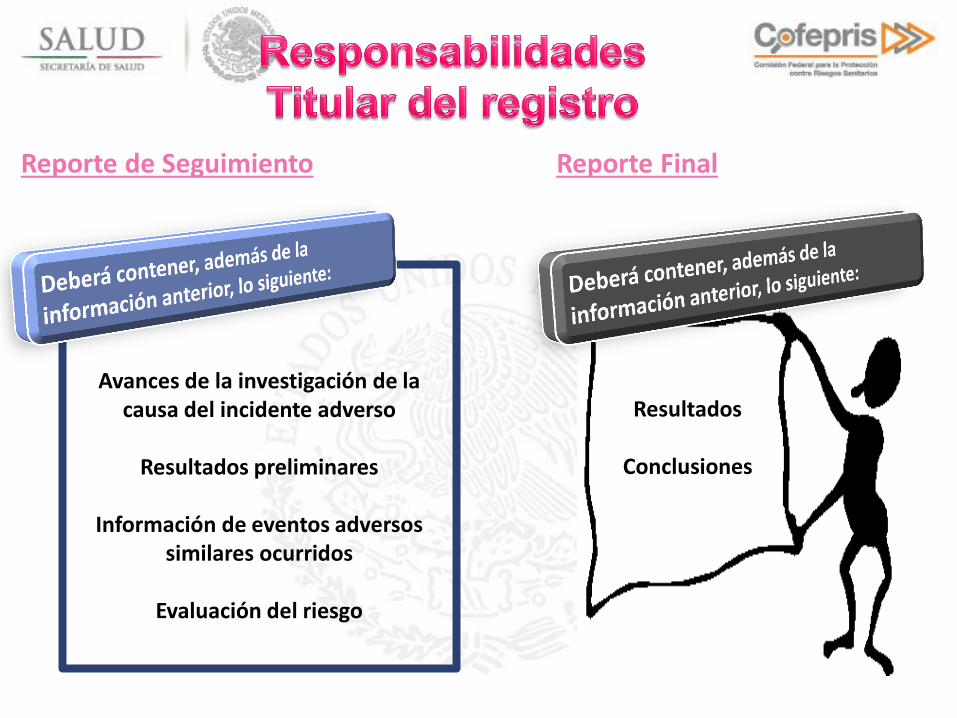
Reporte de Seguimiento
Avances de la investigación de la causa del incidente adverso
Resultados preliminares
Información de eventos adversos
similares ocurridos
Evaluación del riesgo
Reporte Final
Resultados
Conclusiones

Notificar al CNFV los incidentes adversos que se presenten durante el estudio, en los
tiempos establecidos
Colaborar con las Unidades de Tecnovigilancia de los Centros
Estatales e Institucionales

TIEMPOS DE NOTIFICACIÓN
1 El plazo para presentar al
CNFV el reporte de seguimiento y final será de
6 meses máximo, dependiendo de la
gravedad.
Otros incidentes

La autoridad debe recibir las notificaciones y los reportes por parte de
los fabricantes e iniciar sus propias investigaciones, y monitorear el
progreso de la investigación del fabricante.
El monitoreo puede incluir la solicitud de nuevos reportes de
seguimiento de parte del fabricante sobre:
• Cuántos dispositivos están involucrados y dónde han sido
comercializados.
• La situación actual del dispositivo
• Cuánto tiempo llevan los dispositivos en el mercado
• Detalles de los cambios realizados en el diseño

Describe los criterios que se deben utilizar para detectar
un aumento significativo de la tasa de incidentes, brinda
orientación, incluye:
Notificación de tendencias relativas a los incidentes.
Análisis de tendencias en relación con los incidentes
Ejemplo de análisis estadístico de tendencias y de
aumento significativo.

GRACIAS
Q. María del Carmen Becerril Martínez
Directora Ejecutiva de Farmacopea y Farmacovigilancia
Mayo 2013
Somos COFEPRIS,
somos ARN

















