
La Nocion de Substancia en La Filosofia Moderna (Descartes Spinoza y Leibniz)

80 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
NEUROPATOLOGÍA DE LAS DEMENCIAS*
Por EDMUNDO BETETA**
RESUMEN
Presentamos la revisión de los estudios de neuropatología en las demencias: Vascular,de Alzheimer, Asociadas con cuerpos de Lewy, Frontotemporal (Pick) y en enfermedad deHuntington. Los estudios incluyen los aspectos macroscópicos, microscópicos e histo-químicos que permiten la correlación clínico-patológica a la luz de los últimos estudiosbioquímicos genéticos.
ABSTRACT
A neuropathology and biochemistry review of the most important dementias ispresented. We study the macroscopic, microscopic and histochemistry pathology ofAlzheimer, Parkinson with Lewy´s bodies, frontotemporal (Pick´s disease) and Hun-tington´s disease. The present revisión will help the clinician to understand the clinic-pathological correlation based in Uptodate knowledge of biochemistry genetic.
Revista de Neuro-Psiquiatría 2004; 67: 80-105
PALABRAS-CLAVE : Neuropatología, histopatología, bioquímica, demencia, atrofia cerebral. KEY WORDS : Neuropathology, biochemistry, histopathology, dementia, brain atrophy.
* Ponencia en el Tercer Curso Internacional de Psicogeriatría “Actualización en Demencias”, Asociación Peruana dePsicogeriatría, Lima, julio de 2003.
** Profesor de Neurología, Clínica médica, sede Hospital Daniel Alcides Carrión, Departamento de Medicina, Facultad deMedicina, UNMSM. Jefe del Laboratorio de Psicofisiología, Facultad de Psicología, UNMSM.
Los modernos estudios de neuropa-tología incluyen las variantes de la biologíamolecular, por lo cual, se pueden relacionarlos cambios anatomo-patológicos con lapresencia de un substrato bioquímico anor-mal originado en un defecto genético. De
acuerdo a la descripción y los hallazgos deneuropatología es muy difícil establecer unademencia de localización cortical y otrasubcortical, de tal manera que esta diferen-ciación es fundamentalmente clínica apo-yada por los estudios de MRI.33
NEUROPATHOLOGY OF THE DEMENTIAS’

81NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
En esta presentación, revisaremos la pa-tología de las demencias genéticamente,determinadas (para diferenciarlas de la de-mencia vascular), las cuales, constituyen elsubstrato anormal cerebral de la mayoría delas demencias.
No revisaremos otras demencias des-critas en el texto de nomenclatura DSM IV,aunque alguna de ellas, comparte algunascaracterísticas de las genéticas (Ej. presen-cia de cuerpos de Lewy en la demencia trau-mática), tales como demencias de causasmédicas (nutricionales, metabólicas, anó-xicas, inflamatorias crónicas), infecciosas(sífilis, HIV, complejo demencial, encefalitispor virus o priones), traumáticas (demenciapugilística), toxicas (alcohol, drogas psi-coactivas) y las entidades que producen hi-drocefalia (meningoencefalitis, hemorragiasubcracnoidea, tumores del SNC cisticer-cosis cerebral, hematomas subdurales).
DEMENCIA VASCULAR (Fig.1)
Clásicamente, la demencia vascular estu-vo asociada a la hipertensión arterial, factorde riesgo que producía isquemia persistente,determinando desmielinización progresiva.En este proceso, la arterioloesclerosis, en laspartes más dístales del árbol vascular cere-bral, afecta esencialmente la substancia blan-ca hemisférica, tomando la denominación dedemencia multinfarto. En esta patología seincluía: 1) La encefalopatía subcortical cró-nica progresiva descrita por Binswanger en1894; 2) El estado lacunar o “etat lacunaire”o lesión cribiforme del cerebro, estudiado porPierre Marie en 1901; 3) La atrofia granularcortical o “etat granular” descrita por Pents-chew en 1933. En esta patología, la cortezacerebral aparecía “arrugada” por numerososmicro infartos simétricos, bilaterales loca-lizados en las zonas limítrofes de las arteriasde mayor calibre.3
Atrofia cerebral vascular. Corte vértico-frontal asimétrico retroquiasmático, mostrando atrofiacortical, dilatación ventricular y un área de infarto en el lóbulo parietal derecho (flecha).
FIGURA 1

82 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
Aunque los conceptos clásicos sirven debase al patólogo, las últimas revisiones des-tacan algunos hallazgos significativos. Enprimer lugar, la demencia vascular no soloafecta la substancia blanca, observándoseinfartos en el tálamo y núcleo caudado, deotro lado, existen procesos que causan des-mielinización multifocal o difusa del cere-bro, sin afectarse las arterias de pequeñocalibre, tales como la esclerosis múltiple,leucoencefalopatía multifocal progresiva,encefalitis por virus HIV, deficiencia devitamina B12, efectos a distancia de la radio-quimioterapia, encefalopatía postraumática,enfermedades de Alzheimer y CreutzfeldtJakob, estas últimas, genéticamente deter-minadas que presentaremos más adelante. Ensegundo lugar, la patología vascular arteriolarno, corresponde necesariamente a la hiper-tensión arterial o la Diabetes Mellitus (de-generación hialina concéntrica arteriolar),también se presenta en la angiopatía familiaramiloide, la arteriopatía cerebral autosomadominante con infartos subcorticales y leu-coencefalopatía (CADASIL).21, 25 La des-mielinización progresiva crónica que originademencia no es producida siempre por is-quemia, observándose degeneración hialinaconcéntrica en pacientes jóvenes no diabé-ticos, ni hipertensos.14
En la patología bioquímica, debe des-tacarse el diagnóstico entre la demencia deAlzheimer y la demencia vascular. En laprimera -como se describirá más adelante-existen hallazgos de inflamación (Cytoquinasy proteína acídica fibrilar glial), anomalíasdel stress oxidativo (Isoprostanos, 3-Nitro-tirosina) y del metabolismo lípido (Apoli-poproteína E, colesterol 24S-OH), destacandoel hallazgo de una reducción de sulfátido,90% en la substancia gris, 50% en la blanca y40% en el líquido cefaloraquídeo. El sul-fátido es un galactocerebrósido de los oligo-dendrocitos y constituye el 5% de los lípidosde la mielina, por lo cual, es un marcador de la
desmielinización activa. En la demenciavascular y otros desórdenes con disrupciónde la mielina (leucodistrofia meta cromática,esclerosis múltiple, infección por HIV-1 ymeningioma) se altera la degradación delsulfátido por lo que aparece aumentado en ellíquido cefalorraquídeo.15 Otro marcadorbioquímico diferencial, entre la demencia deAlzheimer y la demencia vascular, es elhallazgo de la elevación de nivel de cobresérico solo en primera, revelando la ho-meostasis perturbada del cobre (también zincy hierro) que se deposita en las placas seniles,los ovillos neurofibrilares y el líquido cefa-lorraquídeo.35
DEMENCIAS DE TIPO GENÉTICODemencia de Alzheimer ( Fig. 2,3,4)
La confirmación neuropatológica deldiagnóstico de enfermedad de Alzheimer, selogró por un grupo de neuropatólogos,que estudiaron, desde 1987, la correlaciónanatomo-clínica en 106 pacientes, proceden-tes de 18 centros médicos y los 24 neuro-patólogos del CERAD (Consorcio para esta-blecer un registro de enfermedad de Alz-heimer) estandarizaron un protocolo semi-cuantitativo para evaluar la frecuencia deplacas seniles, ovillos neurofibrilares y otroscambios sinápticos en relación con la evo-lución clínica de la demencia.26 Los cambioshistopatológicos incluyen: 1) Placas (seniles)neuríticas; 2) Ovillos neurofibrilares; 3) Pér-dida de sinapsis y neuronas; 4) Degeneracióngránulo vacuolar ; 5) Angiopatía amiloide y6) Placas AMY no amiloides asociadas a pre-sencia de mediadores inflamatorios.8, 32
Últimamente, sin embargo, lesiones is-quémicas subcorticales pueden causar de-nervación colinérgica cortical, observándoseen CADASIL (Arteriopatía cerebral auto-soma dominante con infartos subcorticales yleucoencefalopatía), el predominio de lesionesbioquímicas en la corteza parietal posterior,

83NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
FIGURA 2
FIGURA 3
Atrofia cerebral de Alzheimer. La cara superior-externa del cerebro presenta atrofia de lascircunvoluciones, agrandamiento de los surcos y espesamiento de la leptomeninge, predominante
en la región fronto-parietal izquierda.
Atrofia cerebral de Alzheimer. La cara interna del cerebro presenta atrofia de lascircunvoluciones predominante en la región fronto-parietal. La región está disminuida
mostrando fenestraciones.

84 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
FIGURA 4
Atrofia cerebral de Alzheimer. El corte vértico-frontal mesencefálico demuestra atrofiageneralizada, prevalente en el lóbulo parietal. Los vasos subcorticales están congestionados y la
substancia nigra está conservada.
frontal dorsal y occipital. Este modelo lesio-nal es exactamente opuesto a de la demenciade Alzheimer, en la cual, la mayor dener-vación colinérgica se observa en el lóbulotemporal.25
Estos hallazgos que serán discutidos,constituyen disturbios genéticos del meta-bolismo degradativo de las proteínas en elsistema ubiquitin-proteosoma, originandopéptidos olígomeros neurotóxicos y depó-sitos de proteína intracelular. La correlaciónclínico-patológica diferencial, está basada enanormalidades del péptido beta amiloide, dela proteína alfa sinucleína y de la proteína tauhiperfosforilada. Las 3 proteinopatías deter-minan (de acuerdo al gen de diferentes cro-mosomas), variantes clínico-patológicas dela enfermedad de Alzheimer, sin embargo, ladisfunción de la alfa sinucleína está com-prometida en la demencia de cuerpos deLewy de los pacientes con Alzheimer quedesarrollaran Parkinson, mientras que el
disturbio de la proteína tau (taupatías) secorrelaciona con la enfermedad de Pick odemencia frontemporal (3R), y la demenciacorticobasal, y la parálisis progresiva su-pranuclear (4R) y con la pre proteína (PrP)del prion responsable de la demencia deCreutzfeldt-Jacob y el síndrome de Gerts-man Straussler-Scheinker que presentanespongiosis y placas amiloides (Tabla I). Porúltimo, debe considerarse la repetición detrinucleotidos CAG en el cromosoma 4 quedetermina la Huntingtin, proteína anormalque produce degeneración de las interneuro-nas que secretan Gaba y Acetilcolina conincremento de la dopamina en el núcleocaudado, putamen y núcleo basal consti-tuyendo la enfermedad de Huntington ar-quetipo de la demencia subcortical (Tabla II).
En la tabla I puede observarse que elmecanismo patogénico de estos desórdenesestá basado en la agregación de polipéptidosque no son metabolizados adecuadamente

85NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
TABLA I
DEMENCIAS CON PATOLOGÍA DE FILAMENTOS TAU
1. Enfermedad de Alzheimer2. Demencia fronto temporal3. Degeneración córtico basal4. Parálisis supranuclear progresiva5. Enfermedad de Pick6. Enfermedad de Jakob – Creutzfeldt7. Enfermedad de Gerstmann – Sträussler – Scheinker8. Otras no descritas en este trabajo:
! Complejo demencial Parkinson – Esclerosis lateral amiotrófica! Pan encefalitis subaguda esclerosante! Demencia pugilística! Síndrome de Down ( después de 30 años)
por el sistema Ubiquitin proteosoma, resul-tando en monómeros solubles que evi-dencian neurotoxicidad, alterando la funcióncelular, apareciendo como inclusiones ca-racterísticas. Es interesante observar, queestas proteinopatías afectan químicamentediferentes poblaciones neuronales y circuitosbioquímicos, los cuales exhiben diferenciaregional de vulnerabilidad -volviendo a laclásica vulnerabilidad selectiva de losVOGT- resultando en diferentes manifes-taciones clínicas neuropsiquiátricas. De estemodo, las tres proteinopatías básicas seasocian en la demencia de Alzheimer y losfactores genéticos modificando la vulne-rabilidad regional cerebral determinan fe-notipos diagnósticos de alguna variante es-pecífica de enfermedad.5, 6, 9
El análisis histopatológico de la demen-cia de Alzheimer revela tres tipos de placasneuríticas que tienen relación con el progre-so de la enfermedad. Las placas difusas sinamiloide conteniendo proteína amiloide (A-Beta) inmunoreactiva, se observan en elcerebelo y en áreas asintomáticas de loshemisferios cerebrales en los primeros esta-díos de la enfermedad. Las placas neuríticas
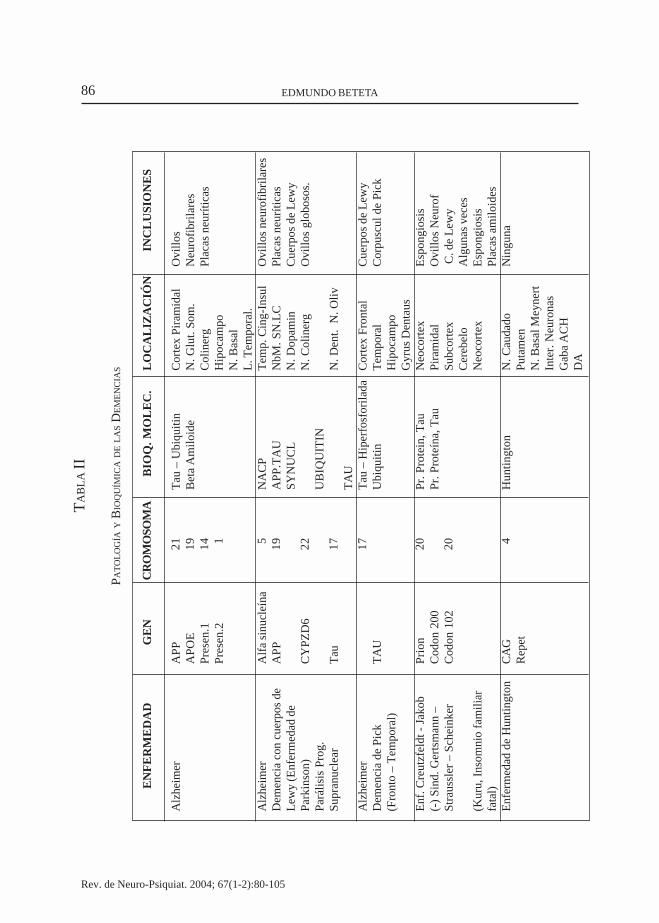
de 50-200 um de diámetro con amiloide,pares de filamentos helicoidales y organelasanormales asociados con astrocitos y mi-croglia reactiva dentro y, fuera de la placa, enfin, en los estadíos avanzados aparece eltercer tipo de placa conteniendo amiloidedenso aislado con marcada actividad infla-matoria ( Fig. 5).
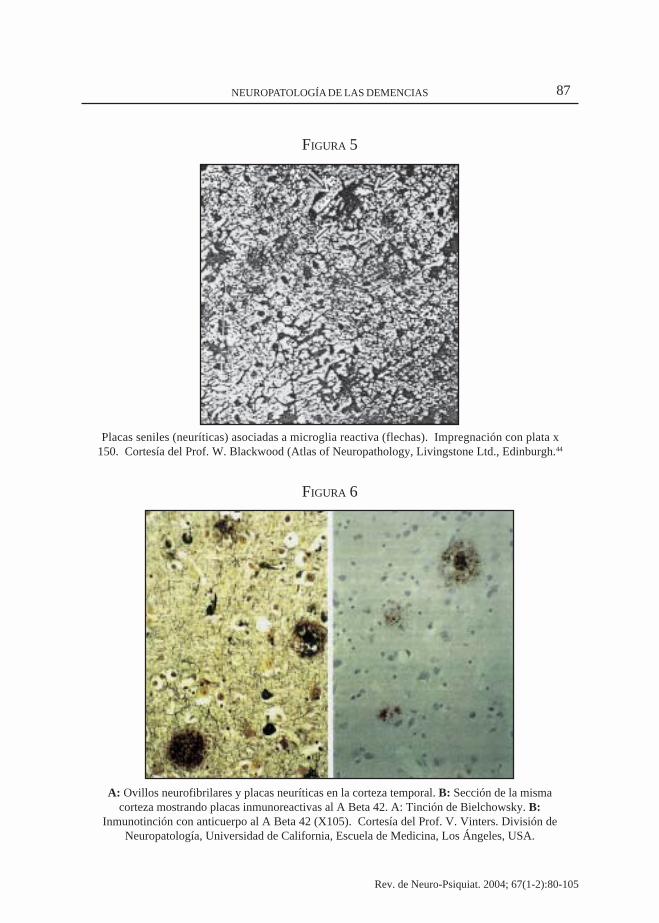
Los ovillos neurofibrilares (“Tangles”)consisten de filamentos espiralados en paresque ocupan el soma y las dentritas -nunca elaxon- que contienen proteínas tau fosforiladaanormal que representan el residuo insolublede las neuronas sometidas a la apoptosis.Aparecen primero, en las neuronas pirami-dales de la corteza entorinal, difunden en lasáreas límbicas y se extienden al neocortex.Hallazgos patológicos que se correlacionanclínicamente, primero con severas altera-ciones de la memoria reciente, defectosafectivo-instintivos y posteriormente congraves perturbaciones del lenguaje, praxia ygnosia (Fig. 6). Estos hallazgos -placas yovillos- corresponden al mecanismo de ladegeneración neurofibrilar; anormal hiper-fosforilación de la proteína tau que daña losmicrotúbulos y compromete el flujo axo-
86 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
EN
FER
ME
DA
D
Alz
heim
er
Alz
heim
erD
emen
cia
con
cuer
pos d
eLe
wy
(Enf
erm
edad
de
Park
inso
n)Pa
rális
is P
rog.
Supr
anuc
lear
Alz
heim
erD
emen
cia
de P
ick
(Fro
nto
– Te
mpo
ral)
Enf.
Cre
utzf
eldt
- Ja
kob
(-) S
ind.
Ger
tsm
ann
–St
raus
sler
– S
chei
nker
(Kur
u, In
som
nio
fam
iliar
fata
l)En
ferm
edad
de
Hun
tingt
on
GEN
APP
APO
EPr
esen
.1Pr
esen
.2
Alfa
sinu
cleí
naA
PP
CY
PZD
6
Tau
TAU
Prio
nC
odon
200
Cod
on 1
02
CAG
Rep
et
CR
OM
OSO
MA
21 19 14 1 5 19 22 17 17 20 20 4
BIO
Q. M
OL
EC
.
Tau
– U
biqu
itin
Bet
a A
milo
ide
NA
CP
APP
.TA
USY
NU
CL
UB
IQU
ITIN
TAU
Tau
– H
iper
fosf
orila
daU
biqu
itin
Pr. P
rote
in, T
auPr
. Pro
teín
a, T
au
Hun
tingt
on
LOC
ALI
ZAC
IÓN
Cor
tex
Pira
mid
alN
. Glu
t. So
m.
Col
iner
gH
ipoc
ampo
N. B
asal
L. T
empo
ral.
Tem
p. C
ing-
Insu
lN
bM. S
N.L
CN
. Dop
amin
N. C
olin
erg
N. D
ent.
N. O
liv
Cor
tex
Fron
tal
Tem
pora
lH
ipoc
ampo
Gyr
us D
enta
usN
eoco
rtex
Pira
mid
alSu
bcor
tex
Cer
ebel
oN
eoco
rtex
N. C
auda
doPu
tam
enN
. Bas
al M
eyne
rtIn
ter.
Neu
rona
sG
aba
AC
HD
A
INC
LU
SIO
NE
S
Ovi
llos
Neu
rofib
rilar
esPl
acas
neu
rític
as
Ovi
llos n
euro
fibril
ares
Plac
as n
eurít
icas
Cue
rpos
de
Lew
yO
villo
s gl
obos
os.
Cue
rpos
de
Lew
yC
orpu
scul
de
Pick
Espo
ngio
sis
Ovi
llos N
euro
f C
. de
Lew
yA
lgun
as v
eces
Espo
ngio
sis
Plac
as a
milo
ides
Nin
guna
TAB
LA II
PATO
LOG
ÍA Y
BIO
QU
ÍMIC
A D
E LA
S DEM
ENC
IAS
87NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
FIGURA 5
FIGURA 6
Placas seniles (neuríticas) asociadas a microglia reactiva (flechas). Impregnación con plata x150. Cortesía del Prof. W. Blackwood (Atlas of Neuropathology, Livingstone Ltd., Edinburgh.44
A: Ovillos neurofibrilares y placas neuríticas en la corteza temporal. B: Sección de la mismacorteza mostrando placas inmunoreactivas al A Beta 42. A: Tinción de Bielchowsky. B:
Inmunotinción con anticuerpo al A Beta 42 (X105). Cortesía del Prof. V. Vinters. División deNeuropatología, Universidad de California, Escuela de Medicina, Los Ángeles, USA.

88 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
plásmico, determinando fracaso de la con-ducción sináptica y muerte neuronal cuyadegradación “Ubiquitinada” caracteriza losovillos neurofibrilares.18
La degeneración gránulo vacuolar ocu-rre fundamentalmente en las neuronas pira-midales del hipocampo. Las vacuolas en elcitoplasma de las neuronas piramidales con-tienen densos gránulos que reaccionan fren-te a los anticuerpos antineurofilamentos yantiproteína tau, posteriormente, los pares defilamentos helicoidales de los ovillos de lasdendritas reaccionan a los anticuerpos anti-Ubiquitin. El material amiloide difunde enlos vasos sanguíneos corticales, leptome-
ninges y no se concentra en los sitios de lasplacas neuríticas (Fig. 7). Las placas deanticuerpos monoclonales (AMY) no de-positan amiloide pero contienen una pro-teína que es coexistente pero distinta de laproteína tau hiperfosforilada y coinciden conlas placas amiloides en las regiones ce-rebrales con patología Beta Amiloide. Porúltimo, la presencia de materiales insolublesdepositados crónicamente, asociados a lamuerte neuronal origina una inflamaciónadyacente o en el interior de la placa neurí-tica. La presencia de alfa1 antiquimotripsinay alfa 2 macro globulina, así como la mi-croglia activada inmunopositiva a interleu-quina -1 e interleuquina- 6, y el sistema com-
FIGURA 7
B: Degeneración granulovacuolar asociada a ovillos neurofibrilares en sección del Hipocampo.Tinción de Bielsehowsky (X 104). Cortesía del Prof. V. Vinters, División de Neuropatología,
Universidad de California. Escuela de Medicina, Los Ángeles, U.S.A.
89NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
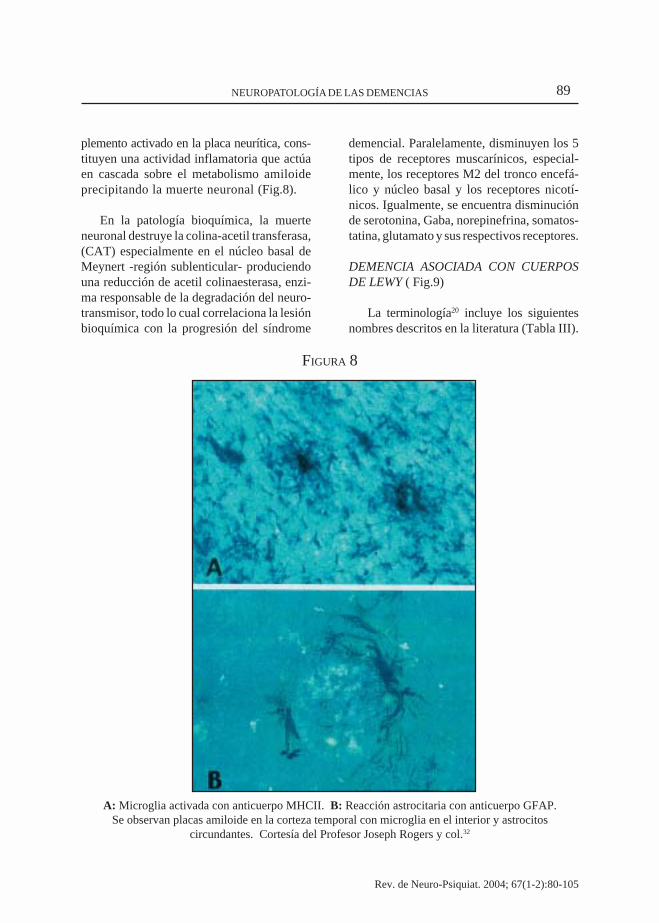
plemento activado en la placa neurítica, cons-tituyen una actividad inflamatoria que actúaen cascada sobre el metabolismo amiloideprecipitando la muerte neuronal (Fig.8).
En la patología bioquímica, la muerteneuronal destruye la colina-acetil transferasa,(CAT) especialmente en el núcleo basal deMeynert -región sublenticular- produciendouna reducción de acetil colinaesterasa, enzi-ma responsable de la degradación del neuro-transmisor, todo lo cual correlaciona la lesiónbioquímica con la progresión del síndrome
demencial. Paralelamente, disminuyen los 5tipos de receptores muscarínicos, especial-mente, los receptores M2 del tronco encefá-lico y núcleo basal y los receptores nicotí-nicos. Igualmente, se encuentra disminuciónde serotonina, Gaba, norepinefrina, somatos-tatina, glutamato y sus respectivos receptores.
DEMENCIA ASOCIADA CON CUERPOSDE LEWY ( Fig.9)
La terminología20 incluye los siguientesnombres descritos en la literatura (Tabla III).
A: Microglia activada con anticuerpo MHCII. B: Reacción astrocitaria con anticuerpo GFAP.Se observan placas amiloide en la corteza temporal con microglia en el interior y astrocitos
circundantes. Cortesía del Profesor Joseph Rogers y col.32
FIGURA 8
90 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
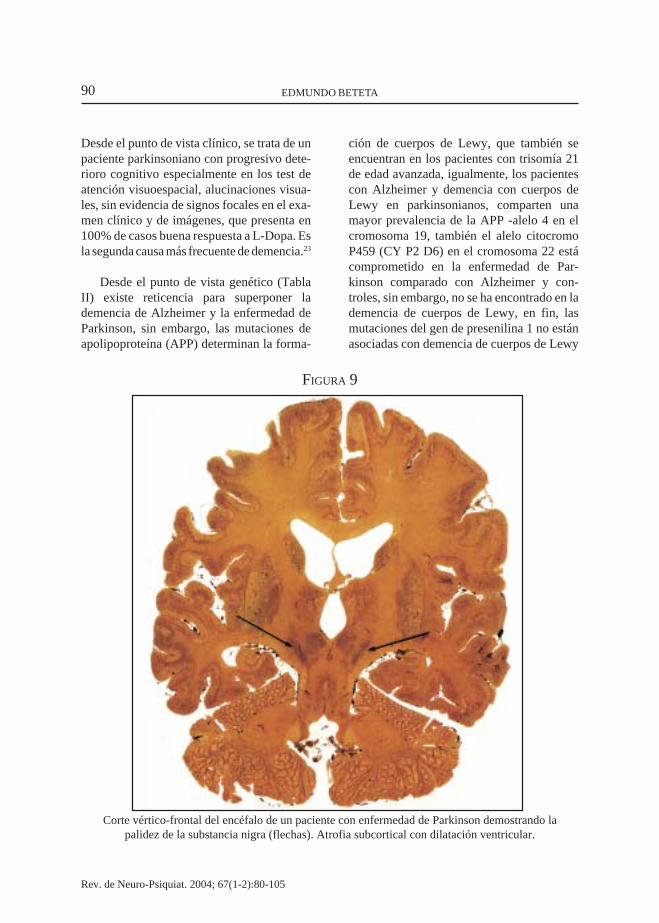
Desde el punto de vista clínico, se trata de unpaciente parkinsoniano con progresivo dete-rioro cognitivo especialmente en los test deatención visuoespacial, alucinaciones visua-les, sin evidencia de signos focales en el exa-men clínico y de imágenes, que presenta en100% de casos buena respuesta a L-Dopa. Esla segunda causa más frecuente de demencia.23
Desde el punto de vista genético (TablaII) existe reticencia para superponer lademencia de Alzheimer y la enfermedad deParkinson, sin embargo, las mutaciones deapolipoproteína (APP) determinan la forma-
ción de cuerpos de Lewy, que también seencuentran en los pacientes con trisomía 21de edad avanzada, igualmente, los pacientescon Alzheimer y demencia con cuerpos deLewy en parkinsonianos, comparten unamayor prevalencia de la APP -alelo 4 en elcromosoma 19, también el alelo citocromoP459 (CY P2 D6) en el cromosoma 22 estácomprometido en la enfermedad de Par-kinson comparado con Alzheimer y con-troles, sin embargo, no se ha encontrado en lademencia de cuerpos de Lewy, en fin, lasmutaciones del gen de presenilina 1 no estánasociadas con demencia de cuerpos de Lewy
Corte vértico-frontal del encéfalo de un paciente con enfermedad de Parkinson demostrando lapalidez de la substancia nigra (flechas). Atrofia subcortical con dilatación ventricular.
FIGURA 9

91NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
y, las mutaciones del gen de la alfa sinucleí-na en el cromosoma 4, rara vez producenenfermedad de Parkinson autosomo domi-nante y no se acompañan de demencia.23
Últimamente, se ha discutido el efecto neu-rotóxico de los cuerpos de Lewy al iden-tificarse el gen DJ-1 comprometido en ladegradación de la proteína en el núcleo. Deeste modo, el rol de los cuerpos de Lewy seríaneuroprotector, probablemente después defracasar el sistema proteosoma Ubiquitin.12
En la actualidad 4 genes se han ligado a laenfermedad de Parkinson; la Alfa sinucleína,el Parkin y últimamente, el DJ-1 y el UCH-L1. El Parkin causa con mayor frecuencia laforma autosoma recesiva de comienzo juve-nil, mientras el DJ-1 con similares carac-terísticas clínicas sólo ha sido detectado endos familias europeas consanguíneas.1, 7
La característica fundamental en pato-logía, es la presencia de los cuerpos de Lewy,inclusiones neuronales descritos por FrederichH. Lewy en 1912 en la substancia innomi-nada y en el núcleo motor dorsal del vago enpacientes portadores de Parkinson, tambiénse observan en la corteza límbica, amígdala,núcleo basal, substancia nigra, núcleo pe-dúnculo pontino, locus caeruleus, núcleo del
rafe, tálamo e hipotálamo. Se trata de in-clusiones protoplasmáticas esféricas, de 10 a30 um de diámetro, con una parte centralhialina eosinofílica y un halo periférico páli-do, ocasionalmente, tienen forma multicolory pueden ser más de uno en las neuronas(Fig.10 a, b, c). Ultraestructuralmente, exhibenfilamentos dispuestos radialmente asociadoscon material denso granular que contieneUbiquitin y Alfa sinucleína. Es interesantedestacar que predominan en las capas cor-ticales profundas, especialmente en la ínsula,cíngulo, corteza temporal y entorrinal, sinembargo, disminuyen en el cortex fronto-parietal y no se observan en el hipocampo.
De modo similar al cuadro patológico deAlzheimer, se observan las placas neuríticasen la región CA2 del hipocampo, la cortezalímbica y núcleos subcorticales, coincidiendocon el depósito de Beta Amiloide que tam-bién aparece en el neocortex, sin embargo, notienen inmunoreactividad al tau en pares defilamentos helicoidales. Los ovillos neuro-fibrilares, de acuerdo al criterio de CERAD26
aparecen en escasa cantidad en el neocortex,hallazgo, que marca la diferencia con lapatología de Alzheimer además de la falta deinmunoreactividad a la proteína tau en lasplacas seniles.30
TABLA III
TERMINOLOGÍA DE LA DEMENCIA ASOCIADA CON CUERPOS DE LEWY
1. Enfermedad cortical de cuerpos de Lewy2. Enfermedad difusa de cuerpos de Lewy3. Demencia de cuerpos de Lewy4. Demencia senil con cuerpos de Lewy5. Demencia de la enfermedad de Parkinson6. Enfermedad de Parkinson con enfermedad de Alzheimer7. Enfermedad de Alzheimer con enfermedad de Parkinson8. Enfermedad de Alzheimer con cuerpos de Lewy9. Enfermedad de cuerpos de Lewy variante de enfermedad de Alzheimer.

92 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
Neurona pigmentada conteniendo 2 cuerpos de Lewy en el Locus Caeruleus de un pacienteparkinsoniano (hematoxilina-eosina). Cortesía del Prof. Ian R.A. Mackenzie, Vancouver
General Hospital, Vancouver.41
Neuronas de la substancia nigra demostrando cuerpos de Lewy (hematoxilina-eosina). Cortesíadel Profesor Robert Perry, neuropatólogo del Newcastle General Hospital.30
FIGURA 10-A
FIGURA 10-B
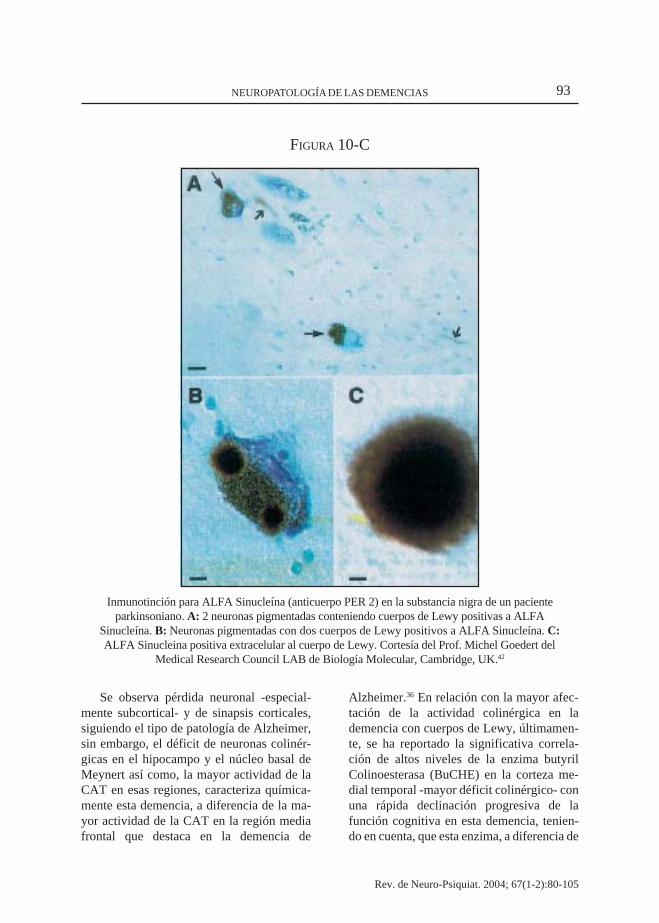
93NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
Se observa pérdida neuronal -especial-mente subcortical- y de sinapsis corticales,siguiendo el tipo de patología de Alzheimer,sin embargo, el déficit de neuronas colinér-gicas en el hipocampo y el núcleo basal deMeynert así como, la mayor actividad de laCAT en esas regiones, caracteriza química-mente esta demencia, a diferencia de la ma-yor actividad de la CAT en la región mediafrontal que destaca en la demencia de
Inmunotinción para ALFA Sinucleína (anticuerpo PER 2) en la substancia nigra de un pacienteparkinsoniano. A: 2 neuronas pigmentadas conteniendo cuerpos de Lewy positivas a ALFA
Sinucleína. B: Neuronas pigmentadas con dos cuerpos de Lewy positivos a ALFA Sinucleína. C:ALFA Sinucleina positiva extracelular al cuerpo de Lewy. Cortesía del Prof. Michel Goedert del
Medical Research Council LAB de Biología Molecular, Cambridge, UK.42
FIGURA 10-C
Alzheimer.36 En relación con la mayor afec-tación de la actividad colinérgica en lademencia con cuerpos de Lewy, últimamen-te, se ha reportado la significativa correla-ción de altos niveles de la enzima butyrilColinoesterasa (BuCHE) en la corteza me-dial temporal -mayor déficit colinérgico- conuna rápida declinación progresiva de lafunción cognitiva en esta demencia, tenien-do en cuenta, que esta enzima, a diferencia de

94 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
la AChE, actúa en sinapsis de la amígdala y elhipocampo, con actividad específica en lacélulas gliales.31 Por último, los estudios his-toquímicos en la demencia con cuerpos deLewy, han demostrado pérdida de las neu-ronas y sinapsis de dopamina en la substan-cia nigra y de Norepinefrina en el LocusCaeruleus. Además, el déficit de neuro-transmisores en esta sinucleopatía, se hasospechado que podría comprometer la fun-ción retiniana tratando de explicar las alu-cinaciones visuales en la demencia de cuer-pos de Lewy. A propósito de esta hipótesis, seha reportado un caso con alucinaciones vi-suales que presentó inclusiones pálidas en lacapa plexiforme externa, asociadas a una de-sorganización estructural de los conos y mo-dificación del modelo inmuno histoquímicode distribución de las sinucleínas en la retina,sin embargo, no se pudo demostrar en esasinclusiones los componentes inmunoquími-cos, de los cuerpos de Lewy.22
DEMENCIA FRONTOTEMPORAL (EN-FERMEDAD DE PICK) (Fig.11)
La primera descripción de Arnold Pick,distinguido neuropsiquiatra alemán en 1892,reunió 6 pacientes con afasia amnésica aso-ciada a una atrofia del lóbulo temporal.Después de la descripción clínico patológicode Alois Alzheimer en 1911, Pick conAltman, describieron en los cerebros de suspacientes las inclusiones argirofilas (cuerposde Pick) y las células acromáticas “hincha-das” (células de Pick), además, mencionaronla ausencia de placas seniles y ovillos neu-rofibrilares, destacando una patología focalcon preservación del hipocampo.
La terminología de la demencia frontotemporal y las variantes patológicas y bioquí-micas, han sido motivo de una encuestadiscutida en un seminario que reunió 30especialistas expertos en el tema. Los resul-tados seleccionaron las denominaciones:
Demencia frontotemporal para describir elsíndrome y degeneración frontotemporalpara el cuadro patológico. Estos términosclínicos patológicos son específicos en lataupatía ligada al cromosoma 17 distinguién-dose como se mencionó previamente formasdiferentes en la demencia frontotemporal (6iso formas de proteína tau derivadas de ungen aislado): 3 iso formas contienen tres re-peticiones (3R-T) de secuencia que com-promete los microtúbulus, corresponden a laenfermedad de Pick; 3 iso formas que pre-sentan una cuarta repetición codificada por elexon 10 (4R-T), en relación a la degene-ración corticobasal y la parálisis supranu-clear progresiva, por último, las 6 iso formasde tau que se reúnen para configurar lademencia de Alzheimer (Tabla IV)
La patología macroscópica destaca unaatrofia cerebral severa (750 gr. de peso) cir-cunscrita en la parte anterior de los lóbulosfrontal y temporal, predominante en el hemis-ferio cerebral izquierdo. Los ventrículos estándilatados y se puede observar la degeneracióndel cuerpo estriado, globus pallidus y la subs-tancia nigra. Microscópicamente, aparece unagran pérdida de las neuronas piramidalesgrandes, asociadas a gliosis y espongiosis dela corteza cerebral. Las “células de Pick”(Fig12) neuronas balonadas, débilmente ar-girofílicas, aparecen difusas y ultraestruc-turalmente exhiben material granulofilamen-toso, demostrando por inmunocitoquímicaanticuerpos a los neurofilamentos, fosfo-rilados y proteína TAU, en menor gradopositividad al Ubiquitin. También, exhibencon anticuerpos, reactividad a los filamentoshelicoidales en pares, estructura anormalresponsable de la formación de ovillos neu-rofibrilares. Los cuerpos de Pick son ne-gativos a la Alfa sinucleína, diferenciandoestas formaciones de los cuerpos de Lewy.
Las áreas límbicas, el hipocampo y laamígdala son las más vulnerables mostrando
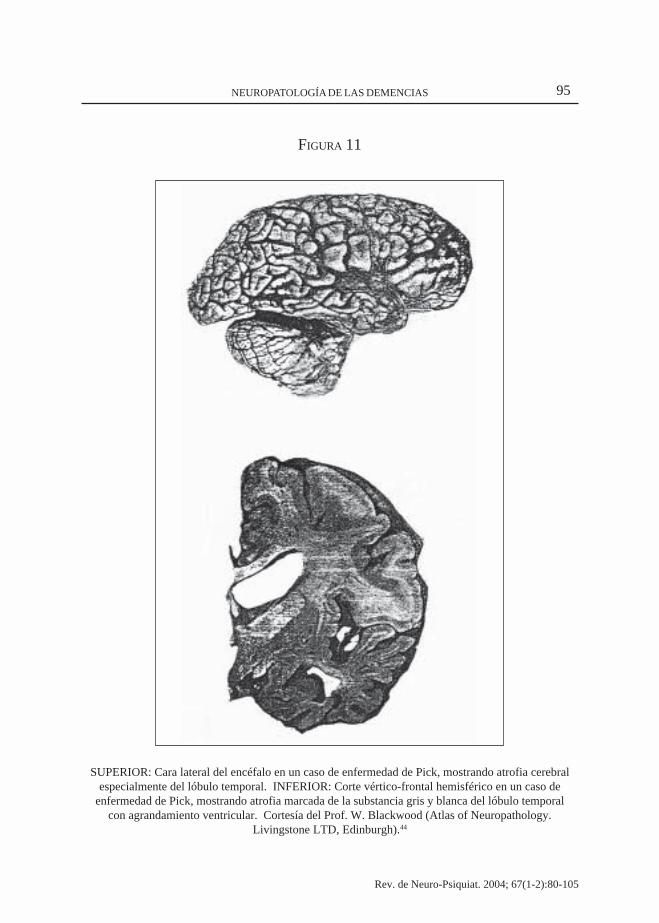
95NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
FIGURA 11
SUPERIOR: Cara lateral del encéfalo en un caso de enfermedad de Pick, mostrando atrofia cerebralespecialmente del lóbulo temporal. INFERIOR: Corte vértico-frontal hemisférico en un caso de
enfermedad de Pick, mostrando atrofia marcada de la substancia gris y blanca del lóbulo temporalcon agrandamiento ventricular. Cortesía del Prof. W. Blackwood (Atlas of Neuropathology.
Livingstone LTD, Edinburgh).44

96 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
TABLA IV
PATOLOGÍA BIOQUÍMICA DE LAS DEMENCIAS
severa atrofia, con numerosos cuerpos dePick en las neuronas piramidales y granu-losas, siguiendo la distribución de los ovi-llos neurofibrilares en Alzheimer, sin em-bargo, el núcleo basal de Meynert está rela-tivamente preservado en la demencia de Picky por ende no se alteran los marcadorescolinérgicos, más bien los serotoninérgicos.Los cuerpos de Pick aparecen en el cuerpoestriado, el globus pallidus, substancia nigra yen menor grado en el Locus Caeruleus (encomparación con la mayor densidad decuerpos de Lewy), núcleos del rafe dorsal,formación reticular bulbo protuberancial.Acompañando la pérdida neuronal, se ob-serva gliosis y degeneración axonal conmarcada afectación de la substancia blanca,por último, la microglia está incrementada, loque se puede demostrar, por anticuerpos quereaccionan a los antígenos (HLA-DR) delcomplejo de histocompatibilidad mayor-clase II, marcadores de la activación mi-croglial.
Los estudios cooperativos realizados porla Universidad de Western Ontario, London,Ontario, Canadá, y el Departamento de pato-logía genética de John Hospital University,24, 27
han presentado estudios estructurales ybioquímicos de la degeneración frontotem-poral, destacando la siguiente frecuencia deanormalidades: Microvacuolización en un60%; Status espongioso en 12%; neuronasedematosas en 32%; granos argirofílicos(taupatía 4R) en 16%; esclerosis hipocámpi-ca en 48%; inclusiones positivas a Ubiquitinen 72%, distribuidas en neuronas del gyrusdentato en 64% y en neuronas corticales en el60% de todos los casos. Por último, en laliteratura revisada de las variantes clínico-patológicas-genéticas de la degeneraciónfrontotemporal (Degeneración Córtico Ba-sal), debe mencionarse la asociación de lademencia frontotemporal con la esclerosislateral Amiotrófica. En el 72% de los casoscon demencia fronto-temporal se observaninclusiones citoplasmáticas Ubiquitinadas
ENFERMEDAD
Alzheimer
Demencia con cuerpos de Lewy.Parkinson
Demencia frontotemporal
Degeneración córtico basal.Parálisis supranuclear progresiva
Jakob - Creutzfeldt (PRIONES)
PATOLOGÍA
Placas euríticas (AB) ovillos neurofibrilares.(TAU) Cuerpos de Lewy (alfa-sinucleína)
Cuerpos de Lewy
Ovillos neurofibrilares. Célula de Pick (3RT)
Ovillos neurofibrilares (4RT)
Placas PrPOvillos neurofibrilaresCuerpos de Lewy

97NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
-tau negativas- en las motoreuronas espina-les.16 Se trata de familias afectadas con sín-drome de motoneurona espinal asociadas condemencia progresiva de tipo fronto-temporal28
que precede o desarrolla paulatinamente alfinal del síndrome neuromuscular (Tabla V).
Los estudios clínicos bioquímicos y deresonancia magnética, señalan diferenciasfundamentales entre la Demencia de Al-zheimer y la Demencia fronto-temporal. Laprimera, es más frecuente (90% de casos),afecta el sistema colinérgico de la formaciónHipocampo Temporo-insular, secundario a
la degeneración neuronal y la astrocitosisreactiva que incrementa los productos de laperoxidación lipídica en el tejido cerebral,39
tiene evolución lenta e insidiosa, predomi-nando la alteración de las funciones mnésicascognitivas. La segunda, es menos frecuente(10% de casos), afecta al sistema seroto-ninérgico del lóbulo temporal a la cortezafrontal, demostrando también degeneraciónneuronal y astrocitosis reactiva sin aumentarlos productos de la peroxidación lipídica;presenta una evolución maligna progresiva,17
predominando alteraciones de la personali-dad, interacción social, apatía, alternando
FIGURA 12
A: Cuerpos de Pick demostrados en 2 neuronas adyacentes. B: 2 neuronas balonadas (flechas),C: Cuerpos de Pick presentes en las células granulosas de la F. Dentata (flecha). D: Agregadosirregulares de proteína TAU y células gliales reactivas a TAU (flechas). Aumento 60x. Cortesía
del Prof. N. Tresser, Departamento de Patología, Universidad de Pennsylvania. Escuela de Medi-cina, Philadelphia, USA.43

98 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
TABLA V
con desinhibición, conductas estereotipadasy desarrollo de afasia fluente (mayor com-promiso temporal) o no fluente (frontal).
DEMENCIA EN LA ENFERMEDAD DEHUNTINGTON (Fig.13)
Los estudios de neuropatología en estaenfermedad autosoma dominante, repeticiónde nucleótidos CAG en el Cromosoma 4(tabla II ) siguen los trabajos de Vonsattel yCol.37, 29 En la primera evaluación clínico-patológica de 163 casos de enfermedad deHuntington,37 se confirmó el mayor compro-miso lesional del Striatum (N. Caudado +Putamen), proponiéndose una escala pro-gresiva de 5 grados en los hallazgos neuro-patológicos. El grado 0 refería ausencia deanormalidades, el grado 1 no exhibía anorma-lidades macroscópicas pero presentaba mo-derada pérdida neuronal y gliosis astrocita-ria en la región paraventricular del núcleocaudado y la región dorsal del putamen. Elgrado 2 presentaba evidencia macroscópicade atrofia de la cabeza del núcleo caudado,manteniendo su convexidad en el piso delventrículo lateral. Microscópicamente seobservó moderada pérdida de neuronas congliosis reactiva astrocitaria, específicamente
en la porción del núcleo caudado y regionesdorsales del putamen. El grado 3 demostrómacroscópicamente el bosquejo de la cabezadel núcleo caudado como una “línea recta” enel ventrículo lateral. La pérdida neuronal y lagliosis fue más difusa en el núcleo caudadosin comprometerse la región paracapsulares yventral. El grado 4 demostró el núcleo cau-dado medialmente cóncavo con apariencia de“arrugado” y atrofia del putamen y la cápsulainterna. La pérdida neuronal y la astrocitosisfibrilar fue muy severa y difusa en todo elnúcleo caudado y el putamen. En otros tra-bajos,29 los hallazgos patológicos se corre-lacionaron con la edad, grado de compromisofuncional y la duración de la enfermedad,determinándose que los mayores compro-misos neuropatológicos (grados 3 y 4) corres-pondieron a la edad de comienzo entre 4 y 19años (Forma Juvenil o adolescente) y la ma-yor alteración clínica referida a los problemasmotores, posturales, verbales y deglutorios.
En la última revisión neuropatológica sepresentan hallazgos importantes.38 El pesopromedio del cerebro post-morten es de1.067 gm. (normal 1.350 gm.) demostrán-dose atrofia de los lóbulos frontales en el80% de los casos de Huntington. Las seccio-
Variantes clínico-patológicas genéticas de ladegeneración fronto-temporal
Patología Genética Clínica
6 Isoformas Proteia Tau E. Alzheimer3 Isoformas (3R-T) E. Pick3 Isoformas (4R-T) Degeneración córtico basal
Parálisis supranuclearProgresiva
Ubiquitin positivo Demencia fronto-temporalInclusiones TAU negativas Esclerosis lateral amiotróficaCuerpos de Lewy
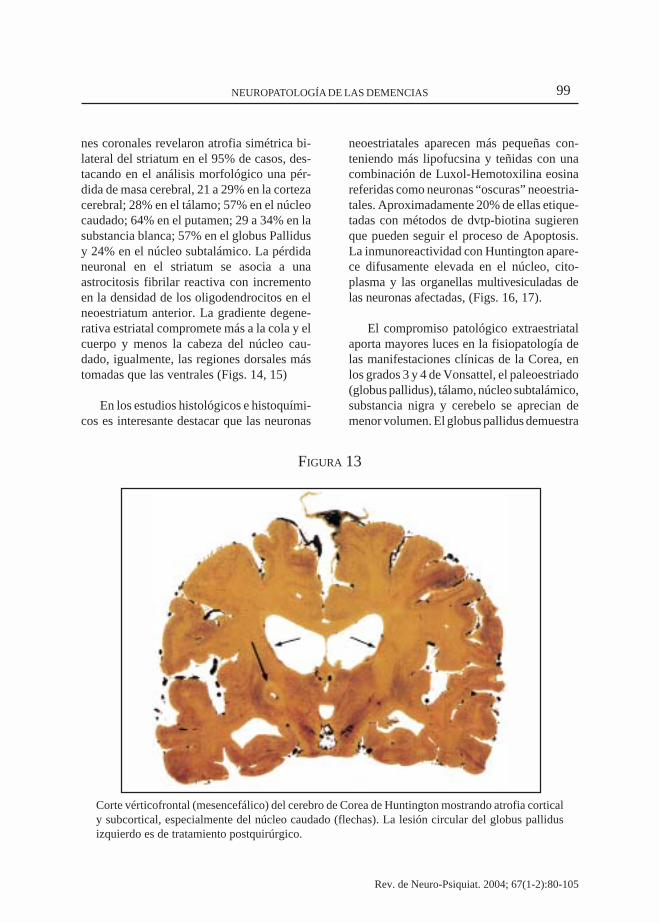
99NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
nes coronales revelaron atrofia simétrica bi-lateral del striatum en el 95% de casos, des-tacando en el análisis morfológico una pér-dida de masa cerebral, 21 a 29% en la cortezacerebral; 28% en el tálamo; 57% en el núcleocaudado; 64% en el putamen; 29 a 34% en lasubstancia blanca; 57% en el globus Pallidusy 24% en el núcleo subtalámico. La pérdidaneuronal en el striatum se asocia a unaastrocitosis fibrilar reactiva con incrementoen la densidad de los oligodendrocitos en elneoestriatum anterior. La gradiente degene-rativa estriatal compromete más a la cola y elcuerpo y menos la cabeza del núcleo cau-dado, igualmente, las regiones dorsales mástomadas que las ventrales (Figs. 14, 15)
En los estudios histológicos e histoquími-cos es interesante destacar que las neuronas
neoestriatales aparecen más pequeñas con-teniendo más lipofucsina y teñidas con unacombinación de Luxol-Hemotoxilina eosinareferidas como neuronas “oscuras” neoestria-tales. Aproximadamente 20% de ellas etique-tadas con métodos de dvtp-biotina sugierenque pueden seguir el proceso de Apoptosis.La inmunoreactividad con Huntington apare-ce difusamente elevada en el núcleo, cito-plasma y las organellas multivesiculadas delas neuronas afectadas, (Figs. 16, 17).
El compromiso patológico extraestriatalaporta mayores luces en la fisiopatología delas manifestaciones clínicas de la Corea, enlos grados 3 y 4 de Vonsattel, el paleoestriado(globus pallidus), tálamo, núcleo subtalámico,substancia nigra y cerebelo se aprecian demenor volumen. El globus pallidus demuestra
FIGURA 13
Corte vérticofrontal (mesencefálico) del cerebro de Corea de Huntington mostrando atrofia corticaly subcortical, especialmente del núcleo caudado (flechas). La lesión circular del globus pallidusizquierdo es de tratamiento postquirúrgico.
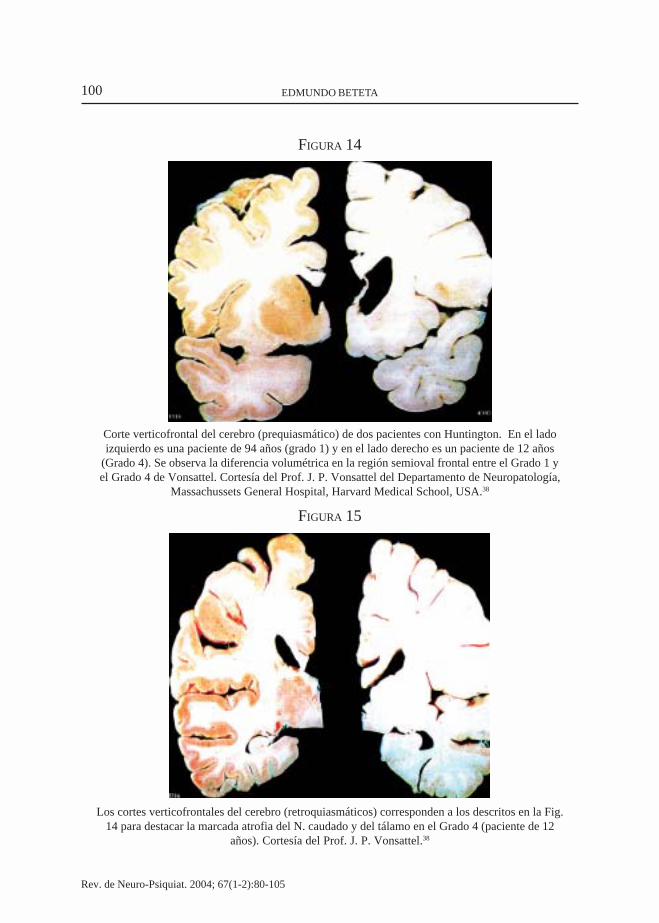
100 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
FIGURA 14
Corte verticofrontal del cerebro (prequiasmático) de dos pacientes con Huntington. En el ladoizquierdo es una paciente de 94 años (grado 1) y en el lado derecho es un paciente de 12 años
(Grado 4). Se observa la diferencia volumétrica en la región semioval frontal entre el Grado 1 yel Grado 4 de Vonsattel. Cortesía del Prof. J. P. Vonsattel del Departamento de Neuropatología,
Massachussets General Hospital, Harvard Medical School, USA.38
FIGURA 15
Los cortes verticofrontales del cerebro (retroquiasmáticos) corresponden a los descritos en la Fig.14 para destacar la marcada atrofia del N. caudado y del tálamo en el Grado 4 (paciente de 12
años). Cortesía del Prof. J. P. Vonsattel.38

101NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
Microfotografía de la pérdida neuronal y reacción astrocitaria en la cabeza del N. caudado(Grado 3). Colaboración Azul de Luxol) con Hematoxilina-eosina (312x). Cortesía del Prof. J.P.
Vonsattel.38
FIGURA 16
atrofia en el grado 4, específicamente en elsegmento externo con una disminución del40% de neuronas aparentemente por laspérdidas de las conexiones estriatales. Encuanto a la pérdida neuronal de la corteza,existe discrepancia en los estudios pato-lógicos10, 11, 13, 34, 40 indicando que el mayorcompromiso lesional apareció en las neuronaspiramidales grandes en las capas III, V y VI,sobre todo en el lóbulo occipital. En el tála-mo, substancia nigra y el núcleo subtalámico,también se ha observado pérdida neuronalvariando la astrocitosis reactiva, específica-mente en el grado 4. Por último, no hayconsenso en los hallazgos del cerebelo, así
Dunlap16 en sus 29 pacientes sólo encontróun caso con atrofia cerebelosa, pero Von-sattel29, 37, 38 en sus estudios de grado 3 y 4describió el cerebelo más pequeño sin atrofianeuronal en la corteza cerebelosa, excepto enlos casos agónicos o isquémicos-hipóxicos,similares a los encontrados en las neuronaspiramidales del sector de Sommer del hipo-campo (asta de Ammon) en el cerebro de epi-lépticos crónicos.2, 4
Al final de esta revisión, no se ocultanmuchas interrogantes, especialmente en lacorrelación de la clínica con la patología,aunque los avances de la metodología clínica

102 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
(tests de funciones mnésicas y funciones eje-cutivas) permiten un diagnóstico más certero.De otro lado, la patología estudiada en elmicroscopio óptico -aunque de gran valor- hasido mejorada por los estudios de la infraes-tructura y los progresos de la genética asocia-da a las investigaciones bioquímicas. De cual-quier modo, el trabajo presentado invita alclínico psiquiatra y neurólogo a continuar es-
Microfotografía del neoestriado demostrando las neuronas oscuras (“dark”), atróficas conalteraciones de la membrana, citoplasma granular oscuro y el núcleo con la cromatina
condensada (Grado 3). Coloración de azul de Luxol con hematoxilina-eosina (500x). Cortesíadel Prof. J.P. Vonsattel.38
FIGURA 17
tudiando las interrogantes en el diagnósticode las demencias.
A propósito de esta contribución, deborendir homenaje a mis maestros, profesoresEnrique Encinas y J. O. Trelles, con quienesaprendí incipientes bases de neuropatologíaque me sirvieron como base fundamental enel desarrollo de la neuropsiquiatría.

103NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
BIBLIOGRAFÍA
1. Abou-Sleiman P.M., Healy, D.G y Col.(2003): The role of Pathogenic DJ-1 Muta-tions in Parkinson´s Disease. Ann. Neurol.54:283-286.
2. Beteta E. (1961): El problema del asta delAmmon en la Epilepsia. Rev. Neurol. B.Aires, 19:1-23.
3. Beteta E. (1970): Demencias en vaculopa-tías. III Congreso Peruano de Neuropsiquia-tría, (22-27 nov.) Vol. 1:291-310). Lima.
4. Beteta E. (1974): ¿Tiene anatomía patológi-ca la Epilepsia? Rev. Neur. Psiq. 37:177-195.
5. Beteta E. (1999): Neurobiología y Neuro-genética. Rev. Per. Neurol. 5:75-79.
6. Beteta E. (2003): Neurogenética de lasfunciones cognitivas. Rev. Neur. Psiq.,66:335-343.
7. Bonifativ. Rizzu P. Van Baren, M.J. y Col(2003): Mutations in the DJ1 Gene asso-ciated Whith Autosomal recessive early-onset Parkinsonism. Science, 299: 256-259.
8. Cummings, J.L, Vinters H.V., Cole, G.M.,Khachaturin Z.C. (1998): Alzheimer´sDisease. Etiologies, Pathophysiology, Cog-nitive reserve, and Treatment opportunities,en “Current perspective in Alzheimer´s Di-sease”, Neurology 51 (suppl. 1), pags. 2-17.
9. Commines JL. (2003): Toward a molecularneuropsychiatry of Neurodegenerative Di-seases. Ann. Neurol. 54:147-154.
10. De la Monte SM. Vonsattel JP, RicharsonE.P. (1998): Monphonietric demostration
of Atrophic changes in the cerebral cortex,white matter and neoestriatum in Hun-tington´s disease. G. Neuropathol. ExpNeurol. 47:516-525.
11. Dunlap CB. (1927): Pathologic Changes inHuntingtons´s Chorea with special referenceto the Hábeas Striatum. Arch, Neurolog.Psychiat. 18:867-943.
12. Fahn, S. (2003): Clinical Trials Promotetreatment and Neuroprotection in Parkin-son Disease. Neurology Today AAN. 3,8:31-33.
13. Fornols LS., Jose C. (1073): Huntington´sChorea: A Pathological study en: BarbeauA., Chase TN, Paulson GW. Eds. Hun-tington´s Chorea-Advances in NeurologyVol. 1:453-470, Raven Press, New York.
14. Gijn Janvan (1998): Leukoaraisosis andvascular dementia en “Current Advance inthe managment of Stroke”, Neurology 51,Suppl. 3:35-85
15. Han, XM., Fagan. AM., Cheng H. y Col.(2003): Cerebro espinal fluid sulfatide isdecreased in subjects with incipient De-mentia. Ann. Neurol. 53:115-119.
16. Hirano A. (1996): Neuropathology 47.suppl. 2:563-566.
17. Hodges JR. Davies R., Xuereb, J. Krill, J.Halliday g. (2003): Survival in Frontotem-poral dementia. Neurology 61:349-354.
18. Iqbal, K. Grubde-Iqbal I (2000): Metabolichipotesis, mechanism and therapeutic tar-gets of Alzheimer neurofibrillary Dege-neration. Neuroscience News. 34:14-20.

104 EDMUNDO BETETA
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
19. Kertesz, A. Muñoz DG., Hillis A. (2003):Frontotemporal Dementia and Pick´s Di-sease; Preferred Terminology. Ann. Neurol.54. Suppl. 5:53-56.
20. Mackenzie, IRA (2000): Dementia withLewy Bodies Neuroscience News. 3, 4:28-32.
21. Markus, HS. Martin RJ. y Col (2002): Diag-nostic Strategies in Cadasil, Neurology,59:1134-1138.
22. Maurage, Claude-Alain y Col (2003): Reti-nal involvement in Dementia with LewyBodies: A clue to Hallucinations? Ann.Neurol. 54:542-547.
23. Mckeith, IG. y Col. (1996): Consensus Gui-delines for the clinical an Pathologic diag-nosis of Dementia with Lewy Bodies (DLB):Reports of the Consortium on DLB Inter-national workshop. Neurology 47:1113-1124.
24. Mckhann GM., Albert M., Grossman M. yCol. (2001): Clinical and Pathologicaldiagnosis of frontotemporal Dementia:report of the work group on frontotemporalDementia an Pick disease. Arch. Neurol.58: 1803-1809.
25. Mesulam M., Siddique J., Chen B., (2003):Cholinergic denervation in a pure multi-infarct state. Observation on Cadasil. Neu-rology, 60:1183-1185.
26. Mirra, Suzannes, Gearing Maria NashFlorence (1997): Neuropathologic asses-ment of Alzheimer Disease in “Consortumto establish a registry for AlzheimerDisease: the Cerad experience”, Neurology49, Suppl. 3:s14-s16.
27. Muñoz DG, Dickson DW y Col. (2003):The neuropathology and biochemistry offrontotemporal dementia, in “Frontotemporal
Dementia and Pick Disease”, ed. A. Kertesz,Ann. Neurol. 54; suppl 5:s24-s 28.
28. Murphy J., Henry R. (2003): Longitudinalanalisis of the anatomic. Neuropsycho-logical and Behavioral features of ALSDementia. Ann Neurol. 54: suppl 7. s52.
29. Myers RH., Vonsattel, JP, Stevens BA. yCol. (1988): Clinical and neuropathologicassesment of severity in Huntington´sdisease, Neurology, 38: 341- 347.
30. Perry R., McKeit I. & Perry E. (1998): De-mentia with lewy bodies: The second mostcommon cause of Dementia. NeurosciencieNews, 1, 3:28-35.
31. Perry R., McKeit I., Ballard C. (2003):Butyryl cholinoesterease and progresión ofcognitive deficits in Dementia with LewyBodies. Neurology 60:1852-1853.
32. Rogers J., Banyang Li, Fen Lue UN. y Col.(2002): Inflamatory mediators in the Al-zheimer’s disease Brain. NeuroscienceNews, 3, 4:38-45.
33. Silbert L. C., Quinn, J. F. y Col. (2003):Changes in premorbid brain volume predictAlzheimer´s disease pathology. Neurology61: 487-492.
34. Sotrel A, Myers RH (1990): Morphometricanálisis os prefrontal cortex in Huntington´sdisease (abstract), J. Neuropathol. Exp.Neurol., 49:346.
35. Squitt, R, Pasqualett, P, Casseta E. y Col.(2003): Elevation or serum Copper levelsdiscriminates Alzheimer disease from vas-cular dementia. Neurology, 60: 2013-2014.
36. Tirabosch, P. y Col. (2000): Cholinergicdysfunction in disease with Lewy Bodies.Neurology 54:407-411.

105NEUROPATOLOGÍA DE LAS DEMENCIAS
Rev. de Neuro-Psiquiat. 2004; 67(1-2):80-105
37. Vonsattel JP, Myers RH, Stevens TJ y Col(1985): Neuropathology of Huntington´sdisease. J. Neuropathol. Exp. Neurol.44:559-577.
38. Vonsattel JP. (2000): Neuropathology orHuntington´s disease. Neuroscience News.3, 2-3:45-53.
39. Yaoy, Zhukareya, Sung, S. y Col.(2003): Enhanced brain levels of 8, 12-150 -iPF2Alfa- VI differentiate AD from fron-totemporal dementia. Neurology 61:475-478.
40. Zalneraitis EL, Landis, DMD, RichardsonEP, Selkoe DJ. (1981): A comparison ofAstrocytic structure in cerebral cortex and
stratiatum huntington´s Disease (abstract)Neurolog 31, suppl. 1:151.
41. Mackenzie, I. (2000): Dementia with LewyBodies. Neuroscience News, 3,4:28-32.
42. Goedert, M. Takes, R. Spillantini, M.G.(1998): Alpha-Synuclein and the LewyBody, Neuroscience news, 1,3:47-52.
43. Shafig M. NEE, L. Tresser, N. Lee y V.M.Trokanowski, J.Q. Lippa, C.F. (2001): Frontotemporal dementia: Report of a familiarcase. Neurology, 56, 11, Suppl. 4:31-34.
44. Blackwood, Dodds and Sommerville (1964):Atlas of Neuropathology E.S. LivingstoneLtd. Edinburgh.


















