Ministerio de Desarrollo Social y Medio AmbienteMinisterio de Desarrollo Social y Medio Ambiente...
149
Ministerio de Desarrollo Social y Medio Ambiente Secretaría de Desarrollo Sustentable y Política Ambiental Manual de tecnologías de medición de concentración de gases y material particulado en chimeneas y atmósfera Dr. Jaime A. Moragues Programa Desarrollo Institucional Ambiental Control de Contaminación Industrial Coordinador a cargo: Lic. Patricia Galán El PRODIA fue financiado por el Tesoro Nacional y los préstamos Nº 768 /OC - AR y 907 / SF - AR del BID
Transcript of Ministerio de Desarrollo Social y Medio AmbienteMinisterio de Desarrollo Social y Medio Ambiente...

Ministerio de Desarrollo Socialy Medio Ambiente
Secretaría de Desarrollo Sustentable y Política Ambiental
Manual de tecnologías de medición
de concentración de gases y material
particulado en chimeneas y atmósfera
Dr. Jaime A. Moragues
Programa Desarrollo Institucional AmbientalControl de Contaminación Industrial
Coordinador a cargo: Lic. Patricia Galán
El PRODIA fue financiado por el Tesoro Nacionaly los préstamos Nº 768 /OC - AR y 907 / SF - AR del BID

ii
I N D I C E
1. Características más Importantes de los Contaminantes y sus Efectos Sobre el Medio
Ambiente....................................................................................................................1
1.1 Introducción....................................................................................................................1
1.2 Contaminantes fotoquímicos...........................................................................................2
1.2.1 Hidrocarburos no metano (HCNM)............................................................................2
1.2.2 Oxidos de nitrógeno (NO y NO2)...............................................................................4
1.2.3 Oxidantes fotoquímicos. Ozono (O3).........................................................................5
1.3 Dióxido de azufre (SO2).................................................................................................6
1.4 Monóxido de carbono (CO)............................................................................................7
1.5 Material particulado en suspensión (MP).......................................................................8
2. Procesos Involucrados en la Contaminación del Aire. Definiciones..........................11
2.1 Fuente de Emisión.........................................................................................................11
2.2 Propagación de los Contaminantes...............................................................................12
2.3 Contaminación del Aire................................................................................................12
3. Mediciones. Unidades. Presentación de Datos...........................................................15
3.1 Mediciones....................................................................................................................15
3.2 Unidades de Medición..................................................................................................15
Tabla 3.2-I : Unidades de Medición de Contaminantes...........................................15
3.3 Presentación de Datos. Calidad del Aire.......................................................................16
Tabla 3.3-I. Efectos Sobre la Salud de los Distintos Estados de Contaminación.....17
Tabla 3.3-II Puntos de Quiebre de IEC...................................................................18
3.4 Relación entre la concentración expresada en fracción de volumen (ppm) y en masa por
unidad de volumen (µg/m3)................................................................................18
Tabla 3.4-I : Unidades............................................................................................19
4. Normas de Calidad de Aire...........................................................................................20
Tabla 4 -I: Normas de Calidad de Aire de la EPA...................................................22
Tabla 4-II: Normas de control de calidad de Aire....................................................23

iii
5. Métodos de Medición y Análisis. Instrumentación......................................................24
5.1 Introducción..................................................................................................................24
5.2 Clasificación de los métodos de medición....................................................................24
5.3 Criterios de selección de equipos de medición.............................................................27
5.4 Mediciones de Calidad de Aire en Atmósfera..............................................................29
5.4.1 Sistema de toma de muestra.....................................................................................29
5.4.1.1 Cabeza de toma de muestra......................................................................................29
i) Contaminantes gaseosos....................................................................................30
Figura 5.4.1–1: Cabeza muestreador para toma de muestra de contaminantes
gaseosos....................................................................................................................30
ii) Material Particulado..........................................................................................32
Toma de muestra de material particulado total........................................................32
Tabla 5.4.1 –I : Dimensionamiento del dispositivo de entrada al sistema de
muestreo...................................................................................................................33
Figura 5.4.1 -2: Cabeza del muestreador para toma de muestra de material
particulado................................................................................................................33
Toma de muestra de material particulado con φ < 10 µm........................................34
1 Muestreador de gran volumen...........................................................................34
1.1 Separación por impacto....................................................................................34
Figura 5.4.1 –3: Toma de muestra partículas con φ < 10 µm con separador por
impacto.....................................................................................................................35
1.2 Separación ciclónica.........................................................................................35
Figura 5.4.1-4: Toma de muestra partículas con φ < 10 µm con separador
ciclónico...................................................................................................................36
Muestreador Dicotómico o Dichot...........................................................................37
Figura 5.4.1-5 : Cabeza de entrada de muestreador Dicotómico para toma de muestra de
Partículas con φ < 10µm........................................................................38
Figura 5.4.1–6 : Principio de separador secundario de partículas con φ < 2,5 µ con
muestreador Dicotómico..........................................................................................38

iv
5.4.1.2 Tubo de entrada y de distribución de muestra (múltiple).........................................39
5.4.2 Equipo de calibración para mediciones automáticas................................................39
5.4.3 Descripción de los métodos de medición.................................................................41
5.4.3.1 Métodos de referencia..............................................................................................41
1. Hidrocarburos no metano (HCNM)...................................................................42
Método de cromatografía gaseosa - detector de ionización de llama.......................42
2. Óxidos de nitrógeno (NOx)................................................................................44
Método de Quimioluminiscencia en fase gaseosa. (NO, NO2)................................44
Figura 5.4.3-1: Determinación de NO y NO2 por método de Quimioluminiscencia
...............................................................................................45
Método de Griess-Saltzman modificado para determinación de NO2......................45
3. Ozono (O3)........................................................................................................46
Método de Quimioluminiscencia.............................................................................46
4. Dióxido de azufre (SO2)....................................................................................47
Método espectrofotométrico con Thorin..................................................................47
Método espectrofotométrico del tetracloromercurato/ pararosanilina......................48
5. Monóxido de carbono (CO)..............................................................................49
Método de cromatografía gaseosa............................................................................49
Método de fotometría infrarroja no dispersiva.........................................................50
6. Material particulado (MP).................................................................................51
Método de gran volumen. Material particulado en suspensión total (MPT)............51
Figura 5.4.3-2: Monitor de gran volumen (hi-vol) para material particulado..........52
Material particulado con φ<10 µm. PM10...............................................................53
Determinación de un índice de humo negro.............................................................54
7. Plomo (Pb).........................................................................................................55
Método de espectrometría de absorción atómica.....................................................55
5.4.3.2 Métodos equivalentes de medición..........................................................................56
1. Óxidos de nitrógeno (NOx) ..............................................................................57

v
Método de espectroscopia de absorción óptica diferencial......................................57
Figura 5.4.3-3: Espectroscopía de absorción óptica diferencial ...………………...58
2. Ozono (O3).........................................................................................................59
Método de fotometría (absorción) ultravioleta.........................................................59
Figura 5.4.3-4: Método de fotometría ultravioleta para la medición de ozono........59
Método de espectroscopia de absorción óptica diferencial......................................60
3. Dióxido de azufre (SO2)....................................................................................60
Fluorescencia Molecular..........................................................................................60
Figura 5.4.3-5: Método de medición de SO2 por fluorescencia molecular................61
Variación de la conductividad..................................................................................61
Método de espectroscopia de absorción óptica diferencial......................................62
4. Monóxido de carbono (CO)..............................................................................63
Variación de la conductividad..................................................................................63
5. Material particulado (MP).................................................................................63
Microbalanza Electrónica.........................................................................................63
Vibración de un filtro (microbalance oscilante).......................................................64
Atenuación (o absorción) de rayos beta...................................................................64
Absorción de luz (sensibilidad óptica)....................................................................64
5.4.4 Casilla de la estación de monitoreo..........................................................................65
5.5 Mediciones de Concentración de Gases y Material Particulado en Chimenea.............68
5.5.1 Toma de muestras de gases y material particulado. Determinación de velocidad dentro del
conducto y el caudal volumétrico............................................................68
Toma de muestra para la determinación automática de concentración de gases......68
Figura 5.5.1-1: Ejemplo de un sistema de toma de muestra y equipamiento para
acondicionamiento de la misma...............................................................................70
Método del centroide para toma de muestras y determinación de velocidad para fuentes
estacionarias.................................................................................................71
Figura 5.5.1-2: Muestra de la selección de puntos de toma de muestra por el método del
centroide en la sección transversal de la chimenea............................................72

vi
Determinación de velocidad y caudal volumétrico del gas dentro de la chimenea (Tubo
Pitot tipo S)....................................................................................................73
Figura V.5.1-3 Conjunto de tubo Pitot tipo S y manómetro....................................74
Figura 5.5.1-4: Propiedades constructivas de un tubo Pitot tipo S...........................74
Métodos para la toma de muestra en gas de escape.................................................75
5.5.2 Métodos de determinación de concentración de contaminantes en chimenea.........76
5.5.2.1 Métodos de análisis externo.....................................................................................76
1. Dióxido de azufre..............................................................................................76
Método Thorin. /Peróxido de hidrógeno/ Perclorato de bario/.................................76
Figura 5.5.2-1: Tren de toma de muestra para la determinación de concentración de SO2
77
Método de precipitación y titulación con Arsenazo III............................................78
Método de absorción infrarrojo no dispersivo..........................................................79
Método de electrólisis a un potencial controlado.....................................................80
Figura 5.5.2-2: Componentes de un analizador de electrólisis a un potencial
controlado.................................................................................................................81
2. Niebla de ácido sulfúrico (H2SO4)....................................................................82
Método de titulación con bario-Thorin.....................................................................82
3. Óxidos de nitrógeno (NOx)................................................................................83
Método fenol disulfónico (PDS)..............................................................................83
Figura 5.5.2-3: Tren de toma de muestra para la determinación de NOx................83
Método de cromatografía iónica...............................................................................84
Método de espectrometría ultravioleta.....................................................................85
Método de Quimioluminiscencia.............................................................................86
Método de electrólisis a un potencial controlado.....................................................87
4. Monóxido de carbón (CO).................................................................................88
Método de analizador infrarrojo no dispersivo.........................................................89
5. Material particulado...........................................................................................89
Método gravimétrico manual...................................................................................89
Figura 5.5.2-4: Tren de toma de muestra de material particulado con filtro fuera de la
chimenea...............................................................................................................90
Método de filtrado en chimenea...............................................................................91

vii
Figura 5.5.2-5: Se muestra el área bloqueada por la sonda y el portafiltro dentro de la
chimenea para la toma de material particulado....................................................91
Figura 5.5.2-6: Tren de extracción de muestra de material particulado con el filtro dentro
de la chimenea...............................................................................................92
6. Plomo inorgánico (Pb)......................................................................................93
Método de espectrometría de absorción atómica.....................................................93
Método polarográfico...............................................................................................94
Figura 5.5.2-7: Toma de muestra y portafiltro para análisis de concentración de
plomo........................................................................................................................95
7. Azufre total en forma reducida (ATFR) Formado por el conjunto de compuestos:
Acido sulfídrico (S2H), Metil mercaptano (CH3SH), Dimetil sulfuro ([CH3]2S), Disulfuro
de Dimetilo [(CH3)2S2]...........................................................96
Método de separación por cromatografía gaseosa y detección con fotometría de
llama.........................................................................................................................96
Método de conversión a SO2 y análisis por Thorin. /Peróxido de hidrógeno/ Perclorato de
bario/...................................................................................................97
Método de conversión a SO2 y análisis por cromatografía gaseosa y detección fotométrica
de llama.................................................................................................98
8. Compuestos orgánicos gaseosos........................................................................99
Método de cromatografía gaseosa............................................................................99
Método de analizador infrarrojo no dispersivo.......................................................100
Método de determinación de gases orgánicos totales excepto metano, como
carbono...................................................................................................................100
18. Fenoles (C6H5OH)...........................................................................................102
Método de absorciometría con 4-aminoantipirina..................................................102
Método de absorciometría ultravioleta...................................................................102
Método de cromatografía gaseosa con detector de llama de hidrógeno ionizante.103
19. Formaldehído (HCHO)...................................................................................104
Método absorciometría AHMT (4-amino-3-hydrazino-5-mercapto-1,2,4-triazol)104
Método absorciometría con ácido cromotrópico Acido 4,5-dihidroxi-2,7-naftalen
disulfónico..............................................................................................................104
Método de cromatografía gaseosa con detector de llama de hidrógeno ionizante.105
Método de cromatografía líquida con detector de espectrometría ultravioleta......106
5.5.2.2 Métodos de medición automática...........................................................................107

viii
1. Dióxido de azufre (SO2)..................................................................................107
Característica de comportamiento de métodos de medición automática............107
Figura 5.5.2-8: Ejemplo de método extractivo y no extractivo..............................109
2. Óxidos de nitrógeno (NOx)..............................................................................110
Método de celda electroquímica.............................................................................110
3. Dióxido de azufre (SO2) y monóxido de nitrógeno (NO)...............................111
Método de absorción ultravioleta...........................................................................111
Figura 5.5.2-9: Método automático de medición de concentración de NO y SO2 por
absorción ultravioleta.............................................................................................112
4. Dióxido de azufre (SO2), óxidos de nitrógeno (NOx), óxidos de carbono (COx), ácido
clorhídrico (HCl), ácido fluorhídrico (HF), amoníaco (NH3).......................113
Método continuo de espectroscopia de absorción óptica diferencial.....................113
Figura 5.5.2-10: Disposición del sistema de medición en chimenea con el sistema de
medición continua de espectroscopia de absorción óptica diferencial..............114
5. Monóxido de carbono (CO).............................................................................115
Método de absorción infrarrojo y correlación de filtros.........................................115
Figura 5.5.2-11: Esquema del equipo para medición por el método de absorción infrarrojo
y correlación de filtros............................................................................116
6. Material particulado.........................................................................................117
Método de opacimetría...........................................................................................117
5.6 Descripción de Técnicas de Análisis..........................................................................118
1 Espectrometría de absorción atómica..............................................................118
Tabla 5.6-I Temperatura de llama para diferentes mezcla....................................119
Figura 5.6-2: Espectrofotómetro de llama típico con un sólo haz..........................120
2 Cromatografía (Gaseosa y Líquida)................................................................121
Figura 5.6-3: Representación esquemática de un cromatógrafo gaseoso...............123
3 Cromatografía iónica.......................................................................................123
4 Detector de ionización de llama.....................................................................124
Figura 5.6-4: Detector de ionización de llama.......................................................124
5 Detector de fotometría de llama......................................................................125
6 Procedimientos luminiscentes.........................................................................125
6.1 Fluorescencia molecular.................................................................................125
Figura 5.6-5 Esquema de un fluorímetro.............................................................126

ix
6.2 Método de Quimioluminiscencia...................................................................127
7 Espectrometría de absorción ultravioleta........................................................127
Figura 5.6-6 : Ejemplo del método de medición por quemiluminiscencia.............128
Figura 5.6-7: Esquema de un típico espectrofotómetro manual de doble haz........129
8 Espectrometría infrarroja (dispersiva y no dispersiva)....................................129
8.1 Absorción radiación infrarrojo no dispersivo................................................131
8.2 Correlación filtro de gas..............................................................................131
Figura 5.6-8: Absorción de radiación infrarroja no dispersivo.............................132
Figura 5.6-9: Método de correlación filtro de gas..................................................132
9 Métodos potenciométricos...............................................................................133
9.1 Método de electrodo selectivo de iones o electrodo específico (denominado también
electrodo a membrana selectiva de iones)................................................133
Figura 5.6-10: Sistema de electrodos típicos para medir el pH..............................135
Figura 5.6-11: Electrodo de membrana líquida sensible a M2+ (catión cuya actividad se
determina).........................................................................................................135
9.2 Titulación potenciométrica.........................................................................136
10 Electrólisis a un potencial controlado (Método coulombimétrico).................136
Figura 5.6-12: Sistema para la electrólisis a un potencial de cátodo controlado. El
contacto C se ajusta para mantener el potencial del cátodo al valor deseado........137
Figura 5.6-13: Esquema de aparato para electrólisis a un potencial controlado...137
11 Método polarográfico....... ..............................................................................138
Figura 5.6-14 : Electrodo de gota de mercurio......................................................138
12 Método de absorción de rayos beta. ...............................................................139
Figura 5.6-15: Método de absorción beta para determinar concentración de material
particulado. ........................................... ..................................................139
13 Método de vibración de una filtro cargado con material particulado (microbalanza
oscilante) ........................................... ............................................140

1
1. CARACTERÍSTICAS MÁS IMPORTANTES DE LOS
CONTAMINANTES Y SUS EFECTOS SOBRE EL MEDIO
AMBIENTE.
1.1 Introducción
Es reconocida la influencia que la contaminación atmosférica tiene sobre el deterioro de la
salud humana, especialmente en las grandes ciudades, llegando en algunos casos a
episodios críticos donde se observan síntomas respiratorios de tipo irritativo, fenómenos
respiratorios de tipo obstructivo y una mortalidad más elevada que los promedios
normales. Se ha establecido la estrecha relación de la contaminación atmosférica con
alteraciones de la función respiratoria, con el transporte de oxígeno en el cuerpo, con
enfermedades respiratorias cardiovasculares y dermatológicas y con la presencia de
diversos y numeroso casos de cáncer.
Existen diversos factores a tener en cuenta para analizar los efectos sobre la salud, entre los
que podemos citar.
Intensidad y composición de los contaminantes: No es lo mismo estar expuesto, aún en
períodos cortos, a concentraciones altas de contaminantes cuyo valores medios en un
período mayor sean aceptables. Las variaciones de la intensidad de la concentración
depende no sólo de la emisión sino también de las condiciones meteorológicas y el relieve
geográfico. Asimismo, el tipo de contaminante es muy importante por sus diversos efectos,
por lo que los períodos de exposición cambia según aquellos
El horario: Las concentraciones varían en general con respecto a la hora del día por
variaciones en las emisiones y las condiciones meteorológicas locales. Para un flujo de
emisión constante, la concentración de los contaminantes a nivel del suelo dependerá de las
lluvias y de los vientos
Los contaminantes que se producen directamente en algún proceso natural o debido a la
actividad humana se denominan contaminantes primarios, o precursores. Si el tiempo
de residencia de estos en la atmósfera es suficiente, pueden participar en reacciones
químicas y transformarse en otras sustancias contaminantes denominadas contaminantes

2
secundarios. En algunos casos los contaminantes primarios no son dañinos, por ejemplo el
NO, y si lo son los secundarios, en este ejemplo el NO2.
La mayoría de los residuos volátiles generados por el hombre sólo ascienden unos pocos
centenares de metros en la atmósfera. El aire en esta zona está en contacto con la
superficie terrestre y su movimiento afectado por la rugosidad de aquella. Por esta razón
se producen turbulencias que generan una mezcla constante de los componentes
atmosféricos. A esta zona más baja de la atmósfera se la denomina capa de mezcla.
Del conjunto de contaminantes atmosféricos que pueden afectar de una u otra forma el
medio ambiente, solamente se miden para su control un grupo de ellos que se consideran
fundamentales. Los limitaremos por lo tanto al estudio de los siguientes contaminantes.
a) Contaminantes fotoquímicos
a1) Hidrocarburos no metano (HCNM); contaminante primario.
a2) Oxidos de nitrógeno, (NO) contaminante primario, y (NO2) contaminante
secundario.
a3) Oxidantes fotoquímicos. Ozono (O3); contaminante secundario.
b) Dióxido de azufre (SO2); contaminante primario.
c) Monóxido de carbono (CO); contaminante primario.
d) Material particulado en suspensión con diámetro menor de 10 µm (MP10);
contaminante primario y secundario. En casos particulares se mide, además,
específicamente plomo (Pb).
Es de destacar que al analizar los efectos de los contaminantes mencionados, es importante
considerar las interacciones entre ellos y la influencia que la presencia de uno tiene sobre
otros.
1.2 Contaminantes fotoquímicos
1.2.1 Hidrocarburos no metano (HCNM)
El metano no se considera un contaminante importante por que no reaccionar
apreciablemente para formar compuestos dañinos. La característica más importante de los

3
demás hidrocarburos desde el punto de vista de la contaminación, es su habilidad para
reaccionar con otros compuestos produciendo contaminantes dañinos secundarios.
El análisis del aire por contaminantes individuales de hidrocarburos no metano (HCNM)
revela la presencia de numerosos compuestos diferentes por lo que es muy difícil de
especificar un grupo de características definidas de “hidrocarburos”. Para nombrar algunos,
dentro de los HC emitidos al aire por lo procesos de combustión, se encuentran los HC
alifáticos o aquellos que sólo tienen enlaces simples y las olefinas o hidrocarburos con uno
o más enlaces dobles, además de otras sustancias orgánicas como aldehídos, cetonas, e
hidrocarburos aromáticos policíclicos. Los métodos de referencia para la determinación de
hidrocarburos definen como de interés a los compuestos que pasan a través de un filtro con
una porosidad de 3 a 5 µm y que produce una señal en un detector de llama ionizante (1).
Un sumario de las reacciones complejas por lo cual aparecen oxidantes a partir de HCNM
es el siguiente. El punto de partida es el dióxido de nitrógeno (NO2), un producto formado
por oxidación del monóxido de nitrógeno (NO) el cual es producido durante la combustión.
En ausencia de hidrocarburos, el NO2 es disociado por la luz solar produciendo NO y un
átomo de oxigeno. Este último se combina con oxigeno molecular para dar ozono (O3) el
cual no se acumula puesto que se combina con el NO para producir nuevamente NO2. El
proceso es continuo y se establece una concentración de equilibrio de cada especie. La
concentración de oxidante fotoquímico aumenta cuando el estado de régimen del ciclo
fotolítico del NO2 es roto por los HCNM, que reaccionan con agentes oxidantes como
radicales OH u oxígeno atómico para dar otras especies radicales, que reaccionan a su vez
con el NO y desbalancean el ciclo. La concentración de ozono aumenta al encontrar menor
cantidad de NO libre para reaccionar. Una de las especies químicas que se origina en la
reacción de los HCNM con el NO es el nitrato de peroxiacetilo (PAN), compuesto
relativamente estable y de elevado poder oxidante. Es el responsable de la típica irritación
en los ojos en situaciones de “smog” (2) intenso.
En general los HCNM no presentan efectos nocivos sobre la salud al nivel de
concentraciones que se encuentran en el aire. Sin embargo, los hidrocarburos que tienen
(1) Lawrence Berkeley Laboratory (LBL), 1973: Instrumentation for Environmental Monitoring. NSF GrantNo. AG-271, Environmental Instrumentation Laboratory, Berkeley, CA.
(2) El término smog proviene de las palabras inglesas “smoke” (humo) y “fog” (niebla) y se emplea paradescribir el fenómeno en la ciudad debido al estancamiento de las masas de aire producido por lainversión térmica que incrementa los niveles de residuos.

4
incorporado oxígeno a su estructura molecular, como en el caso de aldehídos, cetonas y
algunos ácidos orgánicos sustituidos, por lo general son perjudiciales al ser humano.
Las sustancias aromáticas como el benceno tienen alto poder cancerígeno, así como otras
sustancias cíclicas con anillos bencénicos.
1.2.2 Oxidos de nitrógeno (NO y NO2)
El monóxido de nitrógeno (NO) es formado en el quemado de combustibles fósiles por
oxidación del nitrógeno del aire. No hay evidencia que el NO sea dañino para la salud en
las concentraciones normalmente encontradas en la atmósfera. La importancia de conocer
su concentración en el aire ambiente, y su relación con la calidad de este, se debe a que
frecuentemente el NO es oxidado formando dióxido de nitrógeno (NO2). El NO2 no sólo
es tóxico, sino también corrosivo y altamente oxidante. Pequeña cantidad de NO2,
usualmente menor que décimas por ciento, son formadas directamente durante la
combustión a temperatura elevada. Menos del 10 % del NO2 se forma por oxidación
directa del NO en el corto intervalo entre la eyección de NO como un producto de la
combustión y el tiempo en que este diluye su concentración debajo de 1 ppm. La mayoría
del NO2 que se encuentra en la atmósfera resulta por la oxidación del NO en presencia de
luz solar, ozono e hidrocarburos. Otros óxidos del nitrógeno no son considerados por su
baja concentración en la atmósfera y por no reaccionar fotoquímicamente.
Antes de la salida del sol las concentraciones de NO y NO2 permanecen relativamente
constante. Cuando aumenta la actividad urbana de las 6 a las 8 horas de la mañana, la
concentración de contaminantes primarios, CO y NO, aumenta dramáticamente. Luego,
por aumento de la radiación solar ultravioleta, aumenta la concentración de NO2 a partir
del NO. Cuando la concentración de NO cae a niveles muy bajos (menor del 0,1 ppm) los
oxidantes fotoquímicos comienzan acumularse y alcanzan un pico al mediodía. El aumento
del trafico de automotores por la tarde causa un nuevo aumento en la concentración de NO.
Aún en la ausencia de luz solar, se continua formando el NO2 a partir del NO por acción
del ozono, hasta que el aporte del O3 se agota.
Al igual que el SO2, el dióxido de nitrógeno es otro de los contaminantes gaseoso que
poseen carácter ácido. Su acción se manifiesta mediante la descomposición del NO2 con la
humedad presente en el sistema respiratorio, transformándose en ácido nítrico (HNO3) y
nitroso (HNO2). Dado que el NO2 no es muy soluble en agua pasa a través de la tráquea y

5
bronquios, relativamente secos, alcanzando el área húmeda de los pulmones (los alvéolos)
donde forma los ácidos mencionados, ambos irritantes y corrosivos para la cubierta
mucosa de los pulmones.
1.2.3 Oxidantes fotoquímicos. Ozono (O3)
Los oxidantes son definidos como las sustancias atmosféricas que reaccionan
específicamente oxidando. El más abundante de esos oxidantes es el ozono (O3). Por esta
razón, el termino oxidante y ozono son frecuentemente usados en forma indistinta. El
ozono no es directamente emitido a la atmósfera, sino que es un contaminante secundario
formado por una variedad de reactivos atmosférico. El ozono interacción con el ambiente
más que cualquier otro contaminante ambiental. Reacciona con otros contaminantes, con
vegetales, etc. y es fácilmente destruido en esas reacciones. La reacción del ozono con el
NO causa que la concentración de O3 en la cercanía de autopistas o caminos de gran
circulación de vehículos sea mucho menor que el que se encuentra en otros lugares.
La formación de oxidantes esta afectada por la intensidad y duración de la luz solar, la
temperatura, y los procesos de emisión y dilución que afectan la concentración en la
atmósfera de los otros participantes en las reacciones fotoquímicas. La relación entre las
emisiones primarias de NO e HCNM y la subsiguiente formación de ozono atmosférico es
difícil de cuantificar. La lenta formación y el transporte de contaminantes secundarios
tienden a producir una gran separación, espacial y temporalmente, entre las principales
fuentes de emisión y las áreas de elevada concentración de contaminantes oxidantes.
Existen pocas fuentes de producción primaria de ozono, usualmente vinculadas con
descarga eléctrica. En general, no se observa contribución importante en la concentración
urbana, excepto en las vecindades inmediata de donde se produce. El ozono puede también
crecer en la superficie por el proveniente de la estratosfera donde es formado por foto
disociación de oxígeno y recombinación a ozono. La acumulación de ozono ha sido
frecuentemente observada dentro de la capa de inversión sobre áreas urbanas y rurales.
Procesos de convección pueden traer esas capas elevadas a la altura de suelo.
Aparte de otros contaminantes, el índice de peligrosidad de una atmósfera urbana se
acostumbra a medir en función de los niveles de ozono presente. Existe una cierta
variación de los niveles de ozono en la atmósfera urbana durante el día, que refleja la
actividad propia de la ciudad. Los niveles de ozono comienzan a aumentar por la mañana,

6
en el inicio de la actividad urbana, alcanzándose las concentraciones máximas cerca del
mediodía, un par de horas después de que se registren los niveles más altos de NO2 y de
hidrocarburos en la atmósfera. A partir del medio día la concentración de ozono disminuye
a causa de ciertas reacciones químicas, como la transformación de NO a NO2 y
determinados hidrocarburos a aldehídos, en las que el ozono manifiesta su gran poder
oxidante. Los niveles más bajos de ozono se observan durante la noche, puesto que
reacciona con el NO2 presente en la atmósfera dando radicales NO3 muy reactivos. Estos
radicales oxidan el NO a NO2 y se combinan con otros NO2 para dar N2O5, inestable, que
reacciona con el vapor de agua dando ácido nítrico, responsable de la acidez de las nieblas
matutinas urbanas.
El ozono afecta a la salud humana a través de la alta reactividad que muestra en presencia
de compuestos orgánicos con doble enlace, como por ejemplo proteínas y elementos
constitutivos de las células. Esto conduce a transformaciones químicas que pueden dar
lugar a mutaciones a nivel celular, cuando esta sustancia penetra al organismo a través de
las vías respiratorias. Es uno de los contaminantes de mayor estudio en el campo
epidemiológico por sus efectos nocivos sobre la salud humana.
1.3 Dióxido de azufre (SO2)
El dióxido de azufre (SO2) se genera por oxidación del azufre contenido en los
combustibles al quemarse estos. Actualmente su nivel tiende a bajar dado que se exigen
combustibles con bajo contenido de azufre. Como el SO2 es soluble en agua, interacciona
física y químicamente con la humedad ambiente. Reacciona fotoquímica y catalíticamente
con otros componentes de la atmósfera. La cinética de las reacciones son complejas,
algunas de las cuales no son entendidas. El SO2 puede ser catalíticamente oxidado a SO3 en
presencia de óxidos de nitrógeno. El SO3 luego convierte óxidos básico a sulfato. La tasa
de oxidación de SO2 a sulfatos varía de 0,17 % / hora a 50 % / hora, dependiendo de la
humedad relativa y la presencia y concentración relativa de otros contaminantes. La tasa es
típicamente más rápida en zonas urbanas. El SO2 puede ser foto oxidado a ácido sulfúrico
(H2SO4) en aerosol en la presencia de vapor de agua y más rápidamente cuando está
presente hidrocarburos, dióxido de nitrógeno o compuestos de hierro o de manganeso.
Debido a las interacciones complejas del SO2 con otros contaminantes es muy difícil
determinar tiempo de residencia y vida media. En general la vida media va de una hora a

7
varios días. La vida media más corta es característica del SO2 urbano donde existen
muchos otros contaminantes. Un decaimiento exponencial con una vida media de tres
horas es típica de una ciudad. Se ha encontrado que el océano es el mayor sumidero de SO2
El dióxido de azufre, por su carácter ácido, tiene efectos irritativos sobre las vías
respiratorias, creando problemas de bronquitis obstructiva. Al encontrarse en presencia de
partículas en suspensión, la sinergía producida por ambas sustancias aumenta su
agresividad. Su solubilidad en agua y posible transformación a ácido sulfúrico, así como su
propio carácter ácido, permiten que origine problemas puntuales en regiones de alta
sensibilidad del sistema respiratorio.
La temperatura en un punto dado tiene un pequeño efecto directo sobre la concentración de
SO2. Sólo gradientes de temperatura, principalmente en el sentido vertical, tienen alguna
influencia. Sin embargo, la temperatura ambiente puede influenciar la emisión de SO2 (en
realidad su producción), sobre todo cuando se quema combustibles para calefacción.
También si la generación de electricidad es fundamentalmente térmica, los requerimientos
de refrigeración en verano influye en la emisión de SO2.
1.4 Monóxido de carbono (CO)
El monóxido de carbono (CO) se produce por la oxidación incompleta del carbono en el
proceso de combustión. En general esto ocurre en los automotores, dado que en las
industrias y en las centrales térmicas se controla que la oxidación sea total, generando CO2
pues es la forma de obtener el mayor rendimiento térmico de los hidrocarburos, o sea hay
un aspecto económico por medio; el CO2 no es dañino para el ser humano, si bien produce
otros efectos como el invernadero. La producción de CO2 es inevitable, dado que es
inherente al proceso mismo de oxidación del combustible, pero la generación de CO puede
ser reducida al máximo con un control adecuado de la combustión.
El monóxido de carbono actúa por asociación con la hemoglobina de la sangre, formando
carboxihemoglobina, reduciendo ostensiblemente la oxigenación debido a que el CO es
210 veces más reactivo que el oxígeno con la hemoglobina. Por lo tanto se observa una
disminución en el transporte de oxígeno por la sangre hacia las células del cuerpo humano.
Cuando la concentración de CO supera las 120 ppm, se puede producir pérdida de reflejos,
dolores de cabeza, nauseas, vómitos, y si persiste puede llevar a la muerte.

8
1.5 Material particulado en suspensión (MP)
El material particulado en suspensión (MP) como un contaminante del aire incluye una
amplia clase de sustancias líquidas o sólidas con una variedad de propiedades físicas y
químicas. Una característica importante es su tamaño, dado que partículas grandes no son
colectadas por el sistema respiratorio del ser humano por lo que no son consideradas
dañinas a la salud. Las partículas con diámetro aerodinámico menor o igual a 10 µm,
usualmente mencionada como MP10, pueden penetrar las vías respiratorias y llegar a los
pulmones, depositándose en las paredes alveolares.
A causa de su irregularidad en forma, densidad, composición y estructura, el material
particulado es caracterizado convenientemente por su diámetro aerodinámico
equivalente. Partículas teniendo la misma velocidad de caída son definidas como teniendo
el mismo diámetro aerodinámico equivalente, el cual por conveniencia se especifica como
el diámetro de una esfera con densidad unidad que tiene esa velocidad de caída. Cuando
son considerados los efectos de las partículas sobre la visibilidad o la dispersión de la luz,
puede ser necesario emplear una definición más relacionada con el tamaño físico real de
las mismas.
El principal daño a la salud del material particulado es por su deposición en el sistema
respiratorio. Los aerosoles atmosféricos que contienen material con diámetro hasta 10 µm
varían en distribución de tamaño y composición química. Se puede considerar tres
tamaños:
a) Las partículas más pequeñas, con diámetro < 0,1 µm, tienen vida corta y frecuentemente
se observan como una clase distinta cerca de la fuente de combustión; se denominan
modo núcleo. El modo núcleo pequeño (Aitken) crece rápidamente por coagulación en
la clase superior.
b) Las partícula de tamaño medio (diámetro de 0,1 a 2,5 µm) son formados principalmente
por coagulación y condensación de vapor sobre las partículas modo núcleo.
c) Las partículas más grandes de modo tamaño grueso (diámetro >2,5 µm) generalmente
forman la mayoría de la masa e incluye partículas formadas por procesos
antropogénicos y partículas de superficie.
Las dos primeras se denominan partículas finas y las mayores partículas gruesas. Las
partícula finas resultan principalmente de procesos de combustión, incluyendo la

9
condensación y transformación atmosférica de gases de escape para formar MP. Procesos
mecánicos y erosión del viento producen partículas gruesas.
Las partículas finas típicamente consisten de sulfatos, nitratos, carbonatos orgánicos,
amonio y plomo, mientras que las partículas gruesas están constituidas típicamente de
óxidos de silicio, hierro, aluminio, sal del mar, partículas de cubiertas de automotores, y
partículas de plantas.
Las fuentes naturales de emisión de partículas son pulverización del mar, incendios,
emanaciones biogenéticas, y volcanes. La mayoría de las emisiones producidas por el
hombre son fugas desde rutas o calles (pavimentadas o no), actividades de construcción,
agricultura, actividades mineras e industrial. La mitad del material particulado urbano está
formado por negro de grafito procedente de la combustión de carburantes fósiles,
principalmente en automotores, sobretodo los que funcionan con motor Diesel. También
contribuyen a su formación los calefactores domiciliarios, las centrales térmicas y las
industrias que operan con fuel oil o carbón.
Como mencionamos, las MP10 pueden penetrar las vías respiratorias y llegar a los
pulmones. La distinta solubilidad de las partículas, según su carga química, determinará su
transferencia a la sangre. La deposición de partículas en el sistema respiratorio depende de
tres fuerzas física:
a) Fuerzas inerciales: Son las causales de deposición en la nasofaringe. La inercia es muy
importante en los grandes conductos del sistema respiratorio, especialmente cuando se
requiere respiración rápida forzada. Su importancia decrece mientras más adentro del
sistema respiratorio se encuentren las partículas.
b) Sedimentación gravitacional: Es proporcional a la velocidad de deposición de la
partícula y al tiempo disponible para sedimentar. Como la velocidad decrece en los
conductos estrechos del sistema, el efecto gravitacional se ve aumentado.
c) Difusión: En el caso de partículas finas, la fuerza más importante es la de difusión y
conduce a una sedimentación o depósito en las paredes de los ductos finos del sistema,
como es el espacio alveolar. Esta fuerza es una magnitud significativa para partículas de
diámetro superior a 0,5 µm.
La acidez inherente a las partículas urbanas puede provocar la irritación de las membranas
mucosa y conducir a una constricción bronquial. La función irritante de las partículas no es

10
función sólo de la naturaleza de las mismas, sino también de la facilidad de absorber o
adsorber otras sustancias en la superficie de ellas, que en ciertas ocasiones da lugar al
denominado sinergismo (3). Un ejemplo típico se observa cuando las partículas se
encuentran en presencia de SO2 en el aire. Otro ejemplo lo presenta los hidrocarburos
aromáticos policíclicos (HAP) que en algunos casos no son agentes mutagénicos, pero se
comportan como tales cuando están en presencia de otros HAP.
Otras sustancias que están presentes en el material particulado son el plomo, arsénico,
cadmio, mercurio, ácido sulfúrico y sulfatos. El plomo, emitido a causa de la combustión
de gasolina que incorporan como aditivo tetraetilplomo u otros compuestos orgánicos de
plomo, es una de las partículas metálicas que más presente se encuentra en la atmósfera
urbana. El plomo en el interior del organismo interfiere en el proceso de maduración de las
células rojas de la sangre, así como también induce la excreción, a través de la orina, de
porfirinas, que son sustancias precursoras de la hemoglobina. Se acumula en los huesos y
tejidos, produciendo alteraciones nerviosas y reducción de la función renal. Por esa razón
se suele medir su concentración en aire por separado.
Las partículas sólidas en suspensión actúan de agentes de condensación del vapor de agua
presente en la atmósfera. Por ello, el material particulado puede participar en procesos
químicos que ocurren en la atmósfera urbana, actuando incluso de catalizadores. Por
ejemplo, los óxidos de azufre y nitrógeno se transforman rápidamente a ácidos sulfúrico y
nítrico, respectivamente, en la superficie de las partículas, las cuales actúan de catalizador
del proceso. El material particulado favorece así la formación de nieblas ácidas que
acostumbran a estar presente en los núcleos urbanos muy contaminados.
Actualmente la concentración de MP10 es empleada como indicador de calidad de aire
ambiente, en reemplazo del material particulado en suspensión total que se empleaba
anteriormente.
(3) Efecto sinérgico es el fenómeno que presentan algunas sustancias que al encontrarse en presencia de otrasincrementan su agresividad frente al medio que los rodea.

11
2. PROCESOS INVOLUCRADOS EN LA CONTAMINACIÓN DEL
AIRE. DEFINICIONES
Existe una relación causal entre las fuentes de emisión, la propagación de los
contaminantes y la contaminación del aire. A fin de cuantificar cualquier relación entre
ellos, se definen los conceptos básicos pertinentes. En términos generales se seguirán las
definiciones dadas en la Organización Internacional de Normalización (ISO) (4).
2.1 Fuente de Emisión
Atmósfera:
La masa total de aire que circunda la tierra.
Emisión:
Es la transferencia o descarga de sustancias contaminantes del aire desde la fuente a la
atmósfera libre. El punto o lo superficie donde se efectúa la descarga se denomina
“fuente”. Este término se utiliza para describir la descarga y el caudal de esa descarga.
Concentración de la emisión:
Concentración de contaminantes del aire en una emisión en sus puntos de descarga.
Flujo de emisión:
Caudal de emisión por unidad de área de la superficie apropiada de una fuente emisora.
Caudal de emisión:
Masa de contaminante transferida a la atmósfera por unidad de tiempo.
Factor de emisión:
Expresión de la razón del caudal en que se emite un contaminante del aire como resultado
de un actividad, respecto del caudal de esa actividad. Por ejemplo: los kilogramos de
dióxido de azufre emitidos por tonelada de acero producido.
(4) International Organization for Standardization ISO Standard Compendium – Environment Air Quality,Edition 1994. ISO 4225:1994. Air Quality – General Aspects – Vocabulary.

12
Norma de emisión:
Caudal de emisión especificado que tiene un estado legal. Se define frecuentemente en
forma estadística fijando un límite al caudal de emisión. Se especifica en el caso de
concentración el nivel de dilución u opacidad de referencia.
Altura eficaz de chimenea:
Altura utilizada con la finalidad de calcular la dispersión de los gases emitidos por una
chimenea y que difiere de la altura real de esa chimenea en una cantidad que depende de
factores tales como la velocidad de salida, los efectos de flotación y la velocidad del
viento; puede ser afectada por la topografía.
Muestreo isocinético:
Método de toma de muestra de material particulado o de metales en suspensión en una
corriente de gas para determinar su concentración, de tal modo que la velocidad de
muestreo (velocidad y dirección del gas entrando a la tobera o conducto de toma de
muestra) sea la misma que la de la corriente gaseosa en el punto de muestreo. Para ello es
necesario medir la velocidad del gas.
2.2 Propagación de los Contaminantes
Transmisión:
Describe fenómenos colectivos que afectan los contaminantes del aire en la atmósfera libre
entre la fuente y el receptor. Son efectos combinados de transporte y reacciones
atmosféricas sobre aquellos; incluyen todos los efectos de dinámica física como dilución
del contaminante con aire, así como las reacciones físicas y químicas que pueden ocurrir.
2.3 Contaminación del Aire
Aire ambiente:
Aire exterior al cual pueden estar expuestos personas, plantas, animales y materiales.
Calidad del aire ambiente:
Estado del aire ambiente según lo indique su grado de contaminación.

13
Normas de calidad del aire ambiente:
Calidad del aire ambiente especificada, que posee un estado legal, frecuentemente definida
en forma estadística por la fijación de un límite en la concentración de un contaminante del
aire respecto de un período promedio especificado.
Concentración de fondo natural:
Concentración de una especie dada en una masa de aire prístina en la cual las emisiones
antropogénicas (5) son despreciables.
Contaminante del aire:
Cualquier sustancia emitida a la atmósfera, por una actividad humana o por un proceso
natural, que afecte al ser humano o al medio ambiente.
Contaminación del aire:
La presencia habitual, en la atmósfera, de sustancias resultantes de la actividad humana o
de procesos naturales, en concentración suficiente, durante un tiempo suficiente y en
circunstancias tales como para afectar el confort, la salud o el bienestar de personas, o el
medio ambiente.
Materia en suspensión:
Toda materia particulada que queda en la atmósfera o en una corriente de gas de chimenea
durante largos períodos debido a que el tamaño de las partículas es demasiado pequeño
para tener una velocidad de caída apreciable.
Contaminante primario:
Contaminante del aire emitido directamente por una fuente.
Contaminante secundario:
Contaminante que puede ser producido en la atmósfera por procesos físicos o químicos, a
partir de contaminantes primarios u otras sustancias presentes como resultado de emisiones
de fuentes estacionarias o móviles.
Concentración a nivel de suelo.
Cantidad de sólido, líquido o materia gaseosa por unidad de volumen de aire,
generalmente medida a una altura especificada.
(5) Generada por el hombre

14
Inmisión:
Es la transferencia de contaminantes del aire desde la atmósfera libre a un receptor tal
como un ser humano, planta o edificio. La suma de las inmisiones en un intervalo de
tiempo da la dosis de inmisión, o sea la cantidad total de contaminantes del aire admitido,
aspirado, absorbido o ingerido por parte del receptor. De acuerdo a esta definición,
inmisión es tasa, medida o proporción de masa, u otra propiedad cuantificable determinada
por unidad de intervalo de tiempo, la cual debe ser medida en lo posible en el receptor.
Esto lleva a que se debe conocerse la inmisión de un gran número de receptores diferentes.
Un estudio de la contaminación del aire debe ser diseñada para medir la inmisión en
receptores y los efectos posibles. Uno puede introducir un “receptor virtual” con superficie
unidad y propiedades unidades y estudiar, para cada receptor, la posible inmisión como
una función de espacio y tiempo. Un receptor virtual puede ser simulado por un sistema de
medición especial o tener una correlación definida con una concentración a nivel de suelo.
No tiene el mismo significado que concentración a nivel del suelo, pero tiene significado
opuesto a emisión. Sin embargo, en muchas oportunidades se emplea el término inmisión
en el mismo sentido que el de concentración a nivel del suelo.
Dosis de inmisión:
Integral del caudal de inmisión en el receptor durante un período de exposición.
Flujo de inmisión:
Caudal de inmisión por unidad de área de la superficie del receptor.
Caudal de inmisión:
Masa de contaminantes transferida al receptor por unidad de tiempo.
Monitoreo:
1. En un sentido amplio, este término designa las mediciones repetidas destinadas a
seguir la evolución de un parámetro durante un intervalo de tiempo.
2. En un sentido más restrictivo se aplica a la medida regular de niveles de
contaminantes respecto de una norma, o para evaluar la eficacia de un sistema de
regulación y de control.

15
3. MEDICIONES. UNIDADES. PRESENTACIÓN DE DATOS
3.1 Mediciones
Las mediciones de los gases contaminantes en atmósfera se hacen según el tipo de sistema
o método de medición. Para métodos discontinuos, generalmente manuales, se mide un
tiempo dado, normalmente de 1 a 24 horas, según el contaminante y la norma
correspondiente. Para métodos continuos o automáticos se mide en forma continua,
registrando los datos cada minuto. Para informar los valores se calculan las
concentraciones en promedio por minuto, y a partir de éstos se calculan los promedios
horarios. Con ellos se calculan los promedios móviles (6) dentro de las 24 horas, según el
contaminante, informándose el valor máximo.
3.2 Unidades de Medición
Las unidades de medición, según referencia ISO (7), son dadas en la Tabla 3.2-I. Se
diferencian en contaminantes gaseosos y material particulado en suspensión.
Tabla 3.2-I : Unidades de Medición de Contaminantes
Cantidad Unidad Símbolo
Gases y Vapores
Fracción de volumen o de masa de los
principales constituyentes (por ejemplo
nitrógeno, oxígeno, dióxido de carbono) en aire
Porciento (en volumen)
Porciento (en masa)
%
%
Fracción en volumen de gases contaminantes.
Indica en volumen las partes de contaminante
contenida en un millón de partes de atmósfera,
siempre considerando a la temperatura de 25 °C
y 760 mm de presión.
Parte por millón (10-6) ppm
(6) Los promedios se realizan tomando el número de horas consideradas, desplazando en una hora el tiempode inicio (por ejemplo, para el CO se calculan los promedios móviles de 8 h. en forma corrida).
(7) ISO 4226:1993(E) International Standard. Air Quality - General aspect - Units of measurement.

16
Cantidad Unidad Símbolo
Gases y Vapores
Concentración en masa de contaminantes
gaseosos (a)
Miligramo por metro
cúbico
Microgramo por metro
cúbico
Nanogramo por metro
cúbico
mg/m3
µg/m3
ng/m3
Material Particulado
Concentración en masa de material particulado
en suspensión
Miligramo por metro
cúbico
Microgramo por metro
cúbico
Nanogramo por metro
cúbico
mg/m3
µg/m3
ng/m3
[a] Cuando las concentraciones son expresadas en términos de masa por unidad de volumen, sedeben dar la temperatura y la presión, así como la humedad.
En el caso de contaminantes gaseosos, la relación entre la concentración expresada en
µg/m3 y ppm es dada en la Sección 3.4
3.3 Presentación de Datos. Calidad del Aire
Para la contaminación del aire atmosférico o calidad del aire, la información de base que se
transmite es el denominado Indice Estándar de Contaminación (IEC), Indice Normalizado
de Contaminantes (INC), o Indice Diario de Contaminación del Aire (ICA). Según la
norma EPA (8) se lo conoce como Pollutants Standad Index (PSI),.
La estructura de este índice incluye los cinco contaminantes para los cuales la EPA ha
establecido estándares (primarios y secundarios): O3, NO2, SO2, CO, MP. Se calcula para
(8) EPA 40 CFR, Part 58, Appendix G.

17
cada contaminante un índice a partir de una función lineal fragmentada que transforma la
concentración ambiente a una misma escala numérica que va de 0 a 500 teniendo en cuenta
el efecto que pueden causar sobre la salud de la población, siendo 100 el correspondiente a
la concentración de los estándares primarios, o sea que el estado de concentración de algún
contaminante ha superado el límite aceptable de contaminación y 500 el de niveles de
perjuicios significativos. Para los valores de 200, 300 y 400 sus efectos son
aproximadamente normalizados usando los puntos de quiebre correspondientes a alerta,
alarma, y emergencia. En la Tabla 3.3-I se describen, para cada nivel de contaminación,
los efectos sobre la salud humana.
Tabla 3.3-I. Efectos Sobre la Salud de los Distintos Estados de Contaminación
ESTADO DE
CONTAMINACIÓNIEC EFECTO SOBRE LA SALUD
Bajo o Bueno < 50
Medio o Moderado 50-100
Alto o Insalubre 101-200 Agravamiento de síntomas en personassusceptibles, síntomas de irritación en poblaciónenferma.
Crítico o Muy insalubre 201-300 Agravamiento significativo de síntomas ydisminución de tolerancia a los ejercicios físicos enancianos y personas con problemas de corazón.Amplio espectro de síntomas en la poblaciónenferma.
Peligroso 301-400 Prematura aparición de síntomas junto con unasignificativa disminución de la tolerancia a losejercicios físicos en la población enferma.
Muy Peligroso > 400 Muerte prematura en personas enfermas y ancianos.Personas sanas experimentan síntomas que afectansu actividad normal
En la Tabla 3.3-II se resumen los puntos de quiebre en unidades métricas y en ppm.
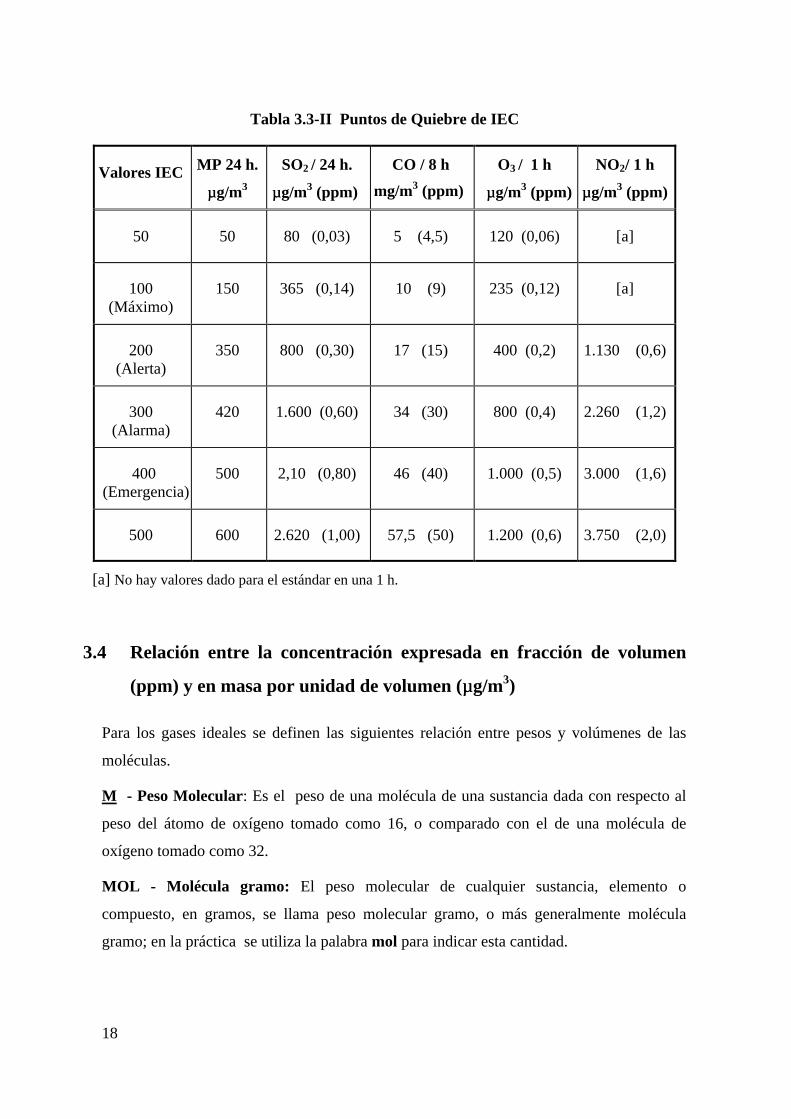
18
Tabla 3.3-II Puntos de Quiebre de IEC
Valores IEC MP 24 h.
µµg/m3
SO2 / 24 h.
µµg/m3 (ppm)
CO / 8 h
mg/m3 (ppm)
O3 / 1 h
µµg/m3 (ppm)
NO2/ 1 h
µµg/m3 (ppm)
50 50 80 (0,03) 5 (4,5) 120 (0,06) [a]
100(Máximo)
150 365 (0,14) 10 (9) 235 (0,12) [a]
200(Alerta)
350 800 (0,30) 17 (15) 400 (0,2) 1.130 (0,6)
300(Alarma)
420 1.600 (0,60) 34 (30) 800 (0,4) 2.260 (1,2)
400 (Emergencia)
500 2,10 (0,80) 46 (40) 1.000 (0,5) 3.000 (1,6)
500 600 2.620 (1,00) 57,5 (50) 1.200 (0,6) 3.750 (2,0)
[a] No hay valores dado para el estándar en una 1 h.
3.4 Relación entre la concentración expresada en fracción de volumen
(ppm) y en masa por unidad de volumen (µµg/m3)
Para los gases ideales se definen las siguientes relación entre pesos y volúmenes de las
moléculas.
M - Peso Molecular: Es el peso de una molécula de una sustancia dada con respecto al
peso del átomo de oxígeno tomado como 16, o comparado con el de una molécula de
oxígeno tomado como 32.
MOL - Molécula gramo: El peso molecular de cualquier sustancia, elemento o
compuesto, en gramos, se llama peso molecular gramo, o más generalmente molécula
gramo; en la práctica se utiliza la palabra mol para indicar esta cantidad.

19
VGI (0 °°C, 1 atm)
Volumen ocupado por 1 mol de un gas ideal a 0°°C (273,16 °°K), 1 atm. (760 mm de Hg,
101,325 kPa) : 22,414 litros (0,022414 m3)
VGI (25 °°C, 1 atm)
Volumen ocupado por 1 mol de un gas ideal a 25°°C (298,16 °°K), 1 atm (760 mm de
Hg, 101,325 kPa) : 24,467 litros (0,024467 m3).
En función de las definiciones dadas se determinan las siguientes relaciones
Fracción en volumen de gases contaminantes (ppm) y
Concentración en masa de por unidad de volumen (µµg/m3)
y ppm . (M/VGI) = x µg/m3
x µg/m3 . (VGI/M) = y ppm
Ejemplo
(x µg de sustancia / 1 m3 de aire) x (0,022414 m3/ M 106 µg) = (z 10-6 m3 de sustancia) / 1
m3 de aire = (z m 3 de sustancia) / (1 millón de m3 de aire) = parte por millón en volumen
Las unidades mencionadas de presión y temperatura se expresan en la Tabla 3.4-I en
función del SISTEMA INTERNACIONAL DE UNIDADES
Tabla 3.4-I : Unidades
CANTIDAD UNIDAD SIMBOLO RELACIÓN
Temperatura Kelvin (termodinámica)
Grado Celsius
K
° C
1 K
273,15 K
Presión Pascal
Milímetro de mercurio
Torricelli
Atmósfera
Pa
Mm Hg
Torr
Atm
1 Pa
133,3224 Pa
133,3224 Pa
101.325 Pa

20
4. NORMAS DE CALIDAD DE AIRE
La calidad del aire se define no en función del aire por si mismo, sino que para que éste, en
razón de las concentraciones de los contaminantes que contenga, no represente un riesgo
para el hombre o para la flora o los suelos. Se define (9) la calidad que debe tener el aire
respecto a un contaminante dado, o a la inversa, la forma en que puede ser usado el aire, en
cuanto componente del medio ambiente, como receptor de las descargas de un
contaminante dado que generan determinadas actividades naturales o productivas. La
fijación de un estándar de calidad ambiental debe estar basada en un fundamento de
carácter científico, lo que obliga a la realización de los estudios pertinentes, que
establezcan las asociaciones o correlaciones relevantes entre contaminantes y los efectos
que se quieren evitar. Dadas las limitaciones económicas que tienen países como la
Argentina para realizar un estudio de esa naturaleza, es razonable adoptar estándares de
otros países que han efectuado tales estudios de respaldo, como EE.UU. de Norteamérica,
pero adaptándolos en lo pertinente a la realidad nacional.
En EE.UU. se definen dos tipo de estándares de calidad de aire
Estándar de calidad de aire primario.
Define niveles de calidad de aire el cual la entidad responsable juzga que es el necesario,
con un adecuado margen de seguridad, para proteger la salud de la población.
Los estándares primarios tienen por objetivo proteger la vida y salud de los seres humanos,
con un margen razonable de seguridad.
Estándar de calidad de aire secundario.
Define niveles de calidad de aire el cual la entidad responsable juzga que es el necesario
para proteger el bienestar de la población de un efecto adverso conocido o esperado
(previsto) de un contaminante. Los estándares secundarios están destinados a proteger,
conservar o preservar la vida y salud de los seres vivos no humanos (flora y fauna), la
renovabilidad de los recursos de los cuales tales especies dependen y la sustentabilidad de
las funciones de los ecosistemas de que forman parte, también con un grado razonable de
(9) Se sigue las definiciones dadas por EPA. Environmental Protection Agency, Title 40: Code of FederalRegulations (40 CFR), Part 50 - National primary and secondary air quality standards (Pt.50), Appendix(APP.)

21
seguridad. Asimismo son funcionales para la protección de monumentos, visibilidad,
paisajes u otros aspectos del medio ambiente.
En la Tabla 4 -I se resumen las normas de calidad de aire dadas por EPA y en la Tabla 4 -
II las normas de calidad de aire ambiental nacional, vigentes en la Argentina, así como las
dadas por la EPA como comparación.
Todas la mediciones de calidad de aire son corregidas para referirlas a 25 ° de temperatura
y a presión de 760 milímetro de Hg (1.013,2 milibars) ( 101,3 kPa).
Hidrocarburos no metano HCNM
Aunque los HCNM no son considerados a nivel ambiente como peligrosos para la salud,
algunos estudios muestran que una concentración promedio de 0,24 ppm entre las horas 6
y 9 en la mañana, puede producir una concentración promedio horario máxima de ozono
de 0,1 ppm. Para guardar los niveles de oxidantes debajo de estos niveles, los valores guía
para HCNM fueron elegido para que el máximo de 0,24 ppm para la concentración
promedio de 6 a 9 en la mañana no sea excedido más que una vez por año. Sin embargo,
esos número guía son sólo aplicables en áreas donde están también presentes los otros
precursores necesarios para producir violaciones del estándar de oxidantes.
Recientemente, el efecto conjunto de producción de ozono de los hidrocarburos y los
óxidos de nitrógeno han sido considerados en la formulación de estrategias de control del
ozono urbano.
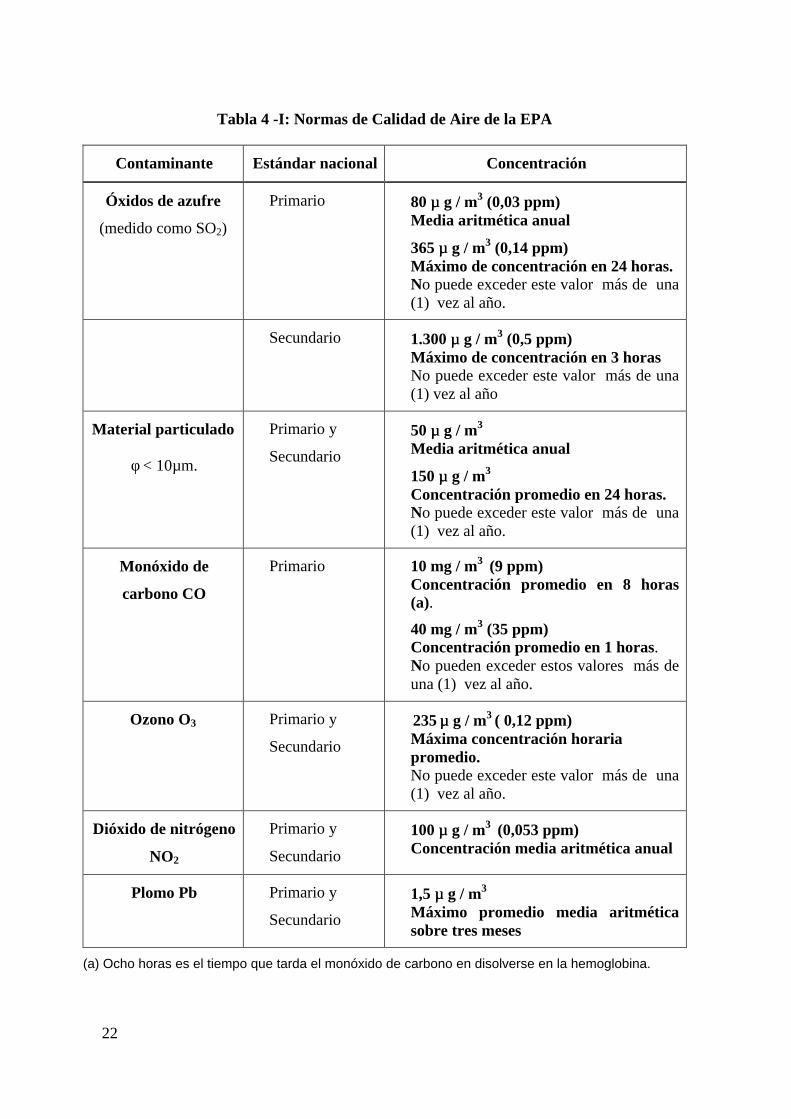
22
Tabla 4 -I: Normas de Calidad de Aire de la EPA
Contaminante Estándar nacional Concentración
Óxidos de azufre
(medido como SO2)
Primario 80 µµ g / m3 (0,03 ppm)Media aritmética anual
365 µµ g / m3 (0,14 ppm)Máximo de concentración en 24 horas.No puede exceder este valor más de una(1) vez al año.
Secundario 1.300 µµ g / m3 (0,5 ppm)Máximo de concentración en 3 horasNo puede exceder este valor más de una(1) vez al año
Material particulado
φ < 10µm.
Primario y
Secundario
50 µµ g / m3
Media aritmética anual
150 µµ g / m3
Concentración promedio en 24 horas.No puede exceder este valor más de una(1) vez al año.
Monóxido de
carbono CO
Primario 10 mg / m3 (9 ppm)Concentración promedio en 8 horas(a).
40 mg / m3 (35 ppm)Concentración promedio en 1 horas.No pueden exceder estos valores más deuna (1) vez al año.
Ozono O3 Primario y
Secundario
235 µµ g / m3 ( 0,12 ppm)
Máxima concentración horariapromedio.No puede exceder este valor más de una(1) vez al año.
Dióxido de nitrógeno
NO2
Primario y
Secundario
100 µµ g / m3 (0,053 ppm)Concentración media aritmética anual
Plomo Pb Primario y
Secundario
1,5 µµ g / m3
Máximo promedio media aritméticasobre tres meses
(a) Ocho horas es el tiempo que tarda el monóxido de carbono en disolverse en la hemoglobina.
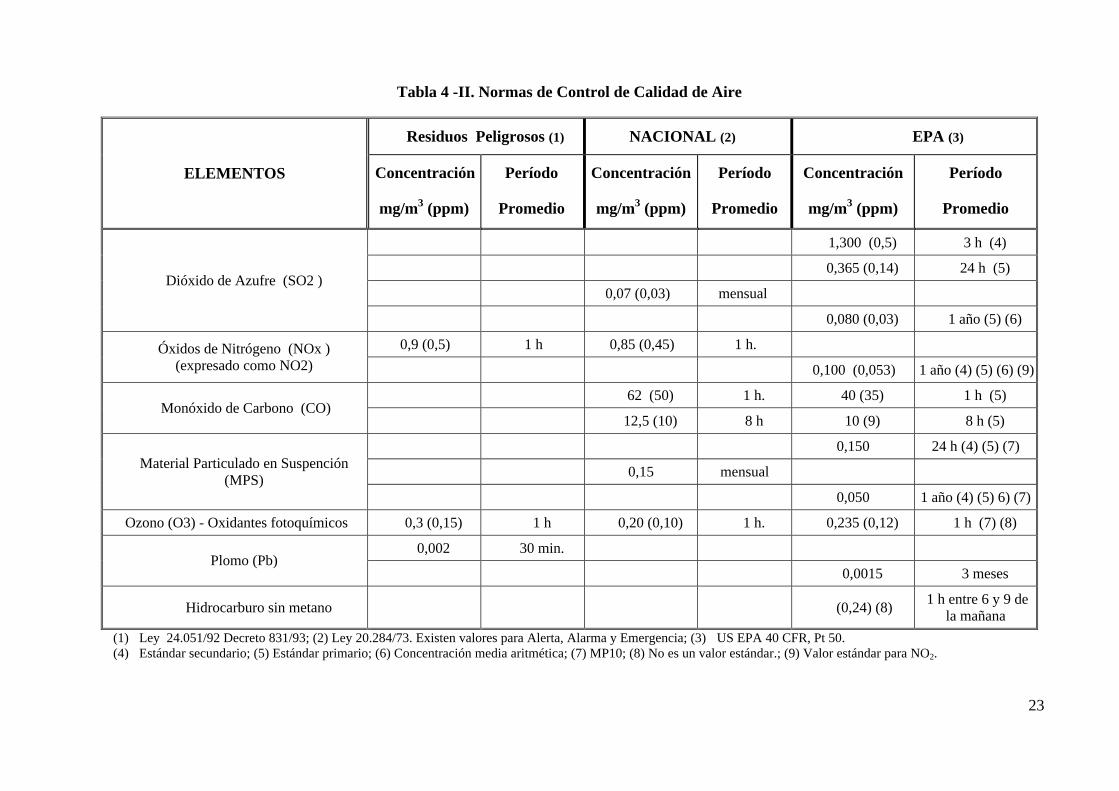
23
Tabla 4 -II. Normas de Control de Calidad de Aire
Residuos Peligrosos (1) NACIONAL (2) EPA (3)
ELEMENTOS Concentración
mg/m3 (ppm)
Período
Promedio
Concentración
mg/m3 (ppm)
Período
Promedio
Concentración
mg/m3 (ppm)
Período
Promedio
1,300 (0,5) 3 h (4)
0,365 (0,14) 24 h (5)
0,07 (0,03) mensualDióxido de Azufre (SO2 )
0,080 (0,03) 1 año (5) (6)
0,9 (0,5) 1 h 0,85 (0,45) 1 h.Óxidos de Nitrógeno (NOx )(expresado como NO2) 0,100 (0,053) 1 año (4) (5) (6) (9)
62 (50) 1 h. 40 (35) 1 h (5) Monóxido de Carbono (CO)
12,5 (10) 8 h 10 (9) 8 h (5)
0,150 24 h (4) (5) (7)
0,15 mensualMaterial Particulado en Suspención
(MPS)0,050 1 año (4) (5) 6) (7)
Ozono (O3) - Oxidantes fotoquímicos 0,3 (0,15) 1 h 0,20 (0,10) 1 h. 0,235 (0,12) 1 h (7) (8)
0,002 30 min.Plomo (Pb)
0,0015 3 meses
Hidrocarburo sin metano (0,24) (8) 1 h entre 6 y 9 de
la mañana
(1) Ley 24.051/92 Decreto 831/93; (2) Ley 20.284/73. Existen valores para Alerta, Alarma y Emergencia; (3) US EPA 40 CFR, Pt 50.(4) Estándar secundario; (5) Estándar primario; (6) Concentración media aritmética; (7) MP10; (8) No es un valor estándar.; (9) Valor estándar para NO2.

24
5. MÉTODOS DE MEDICIÓN Y ANÁLISIS. INSTRUMENTACIÓN.
5.1 Introducción
En el presente Capítulo se resumen los métodos de medición de concentración de gases
contaminantes tanto en atmósfera, como en chimenea de una fuente fija de emisión. Se
presenta en forma muy sucinta la aplicación del método y las posibles interferencias, el
principio en el cual se basa el mismo y los límites de concentración para los cuales es
válido.
En la Sección 5.4 se describen los métodos de medición de calidad de aire atmosférico y en
la Sección 5.5 los correspondientes a las mediciones en chimenea. Dado que muchos de
los principios de medición son similares para diferentes elementos, tanto sea para
mediciones en atmósfera como en chimenea, como por ejemplo ionización de llama,
absorción ultravioleta, etc., el detalle del funcionamiento de los mismos se dan en la
Sección 5.6

25
5.2 Clasificación de los métodos de medición
Los métodos de medición de calidad de aire ambiente y de concentración en chimenea se
pueden clasificar de diferentes maneras. Cada una de ellas destacan las características
fundamentales que los diferencia.
Ì Según su forma de operación
Métodos
discontinuos
Son, en términos generales, métodos manuales paralos cuales la toma de muestra en el lugar y el análisisen el laboratorio son dos pasos separados. Puede habermediciones discontinuas que pueden realizarse conequipos automáticos tanto en la toma de muestra comoen el análisis de laboratorio.
Métodos
continuos o
automáticos
Típicamente involucran equipamientos automáticos enun lugar fijo que realiza ambos procesos, toma demuestra y análisis. Estos métodos son fundamentalescuando existen regulaciones que determinan nivelesde pre-alerta y diferentes grados de alerta.
Ì Según el método utilizado
Químicos-físicos El contaminante sufre una transformación química y elproducto de la reacción se determina por una técnicaanalítica apropiada.
Físicos Se mide una propiedad física, o su variación, paracada contaminante en forma selectiva, sin que seproduzcan cambios en la composición de la muestrade aire.

26
Ì Según el volumen espacial sobre el que se promedia la medición.
Métodos
puntuales
La muestra de aire se toma en forma continua en unpunto determinado y se hace pasar esta por el detector.La mayoría de los equipos actualmente disponibles sebasan en este concepto. Los sensores necesitan unacantidad finita de aire contaminado antes que puedanresponder y un intervalo de tiempo finito antes quepueda ser observado un valor de la concentración. Senecesita un equipo para cada contaminante.
Métodos zonales Miden la concentración promedio de loscontaminantes directamente en una sección de laatmósfera, a través de la perdida de intensidad de unhaz de luz por absorción molecular de loscontaminantes. Se pueden medir simultáneamentetodos los gases que tengan valores suficientementediferentes de longitud de onda de absorción. Mide a lolargo de una zona relativamente grande, mínimo de100 m por problema de sensibilidad, y el método esaplicable hasta espesores de atmósfera, o lo que esequivalente distancia entre emisor y receptor, de1.500 metros. No sirve para material particulado.
Ì Según sean considerados métodos de referencia o equivalentes
Métodos de
referencia
Son dados para las mediciones de cada uno de losgases contaminantes más importantes y se losconsidera de referencia para determinar elcumplimiento de las normas. Generalmente sonmétodos manuales, pocos automatizados y querequieren la colección de muestra por un tiempodiscreto relativamente largo.
Métodos
equivalentes
Se han desarrollado equipos de medición continua oautomáticos que permiten obtener datos en formaprácticamente continua (intervalo de medición de unminuto) y conectados “on line” con el centro deinformación, lo cual es sumamente útil para detectaraccidentes y actuar en forma inmediata. Deben pasaruna prueba que determine que son adecuados frente alos métodos de referencia, la cual es realizada porinstituciones autorizadas. Han sido desarrollado solopara los contaminantes más importantes.

27
5.3 Criterios de selección de equipos de medición
Cuando se seleccionan monitores para un estudio de calidad de aire se debe tener en cuenta
el objetivo del programa a fin de evaluar la economía relativa de los distintos sistemas
considerados. Por ejemplo, si el estudio es realizado para determinar tendencias de
contaminantes de aire en intervalos de tiempos largos, no hay razón de seleccionar
monitores que den salidas continuas si hay otros disponibles. Para ese fin, con los
monitores de salida continua, se debe seleccionar algún método para promediar los
resultados obtenidos y esto aumenta los costos. Debe ponerse especial atención a las
rutinas de calibración y mantenimiento de todos los equipos para asegurar una operación
continua tan extensa como sea posible.
En términos generales hay ciertos parámetros técnicos y operacionales que deben ser
considerados al evaluar la aptitud y utilidad de una técnica analítica o un equipo de
medición.
Parámetros técnicos
Ø Selectividad, indica el grado de independencia de interferencias del método.
Ø Especificidad: indica el grado de interferencias en la determinación.
Ø Límite de detección,
Ø Sensibilidad: Tasa o amplitud del cambio de la lectura del instrumento con
respecto a los cambios de los valores característicos de la calidad del aire.
Ø Exactitud: Grado de acuerdo o semejanza entre el valor real o verdadero y el
valor medio o medido. Depende tanto de la especificidad del método como de la
exactitud de la calibración; esta última dependen de la disponibilidad de
estándares primarios y de la forma como es calibrado el equipo. Denota en que
manera están ausentes errores por predisposición o sesgo o por azar.
Ø Precisión, o reproducibilidad de las medidas: grado de acuerdo o semejanza entre
los resultados una serie de mediciones aplicando un método bajo condiciones
predescriptas y el valor medio de las observaciones.

28
Ø Facilidad de calibración del instrumento: Está asociada a la disponibilidad de
gases de calibración en el mercado (estándares primarios) y a su aplicación en el
sistema de muestreo, así como a la necesidad de la frecuencia de su empleo.
Ø Disponibilidad de gases de calibración. Gases primarios o secundarios.
Ø Volumen de gas necesario para la determinación: Depende del
comportamiento de las concentraciones de sustancias a medir.
Ø Tiempo de respuesta del instrumento: Corresponde al tiempo necesario para
que el monitor responda a una señal dada, o sea el período transcurrido desde la
entrada del contaminante al instrumento de medición hasta la emisión del valor de
la medición. Se suele distinguir dos partes: a) tiempo de retraso, aquel en que se
alcanza el 10 % del cambio final en el instrumento de lectura, b) tiempo de
crecimiento o caída, durante el cual se pasa del 10 % al 90 % del cambio final en
el instrumento de lectura.
Parámetros operacionales
Ø Disponibilidad de los sensores.
Ø Resolución espacial.
Ø Mantenimiento.
Ø Porcentaje de intervalo de tiempo fuera de operación.
Ø Equipamiento adicional necesario.
Ø Mano de obra especializada requerida para operación y mantenimiento.

29
5.4 MEDICIONES DE CALIDAD DE AIRE EN ATMOSFERA
5.4.1 Sistema de toma de muestra
El sistema de toma de muestra consiste del equipamiento entre el punto de entrada del aire
de muestreo hasta el punto de entrada al instrumento de medición.
Componentes
del sistema
de toma de
muestra
• Cabeza de toma de muestra
• Tubo de entrada
• Tubo de distribución de muestra (múltiple) desde el tubo deentrada a los instrumentos de medición individuales.
• Bomba o ventilador.
Dado que dentro de la casilla de medición se tiene una temperatura controlada,
normalmente existe un gradiente de temperatura con el exterior y en ambientes altamente
húmedo existe condensaciones. Por ello las condiciones de transporte por el tubo de
entrada y el múltiple debe representar las mismas que tiene el aire en el exterior. Para ello
ambos van recubierto con aislante térmico, para evitar transferencia de calor, y elementos
de precalentamiento. Por lo general se eleva la temperatura de la muestra entre 25 y 35 °C.
El material del tubo de toma de muestra y el múltiple puede ser vidrio, acero inoxidable o
teflón para que no haya reacción de los contaminantes con las paredes de estos. Por
economía se construyen comúnmente de vidrio. Se incluye una bomba para la toma de
muestra.
5.4.1.1 Cabeza de toma de muestra
La cabeza de toma de muestra difiere según se midan gases o material particulado. Para
cada caso se discuten los diseños más empleados.

30
i) Contaminantes gaseosos
La cabeza del sistema de toma de muestra debe ser diseñada como un pre-separador de
partículas grandes, por ejemplo mayores de 50 µm, basado en un sistema inercial de
cambio brusco de dirección (las partículas más pesadas tienden a seguir en la dirección
original), como se indica en la Figura 5.4.1 -1.
Figura 5.4.1–1: Cabeza muestreador para toma de muestra de contaminantes gaseosos
Dimensión de la
cabeza del sistema
de toma de muestra
Función del flujo
de la muestra y la
eficiencia de
separación del
material
particulado
D=√ [72 VaL / π(bP-bL) g. dac2 ] + da
2 = √ [4,25 V / (106.d2
ac)] + da2
• D: diámetro interno del capuchón de la entrada delseparador en cm.
• V : Caudal del aire en cm3/s• da : diámetro exterior del tubo de muestreo en cm.• aL : viscosidad dinámica del aire (1,82 10-4 g/cm .s a 20
°C)• bp : peso específico de las partículas (1 g/ cm3)• bL : peso específico del aire (1,20 .10-3 g/cm3 a 20 °C y
1,103 mb)• g : constante gravitacional (981 cm/s2)• dac : diámetro aerodinámico de las partículas
(se considera aquí 0,005 cm = 50 µm)• Valores típicos : D = 14 cm ; da = 4 cm.

31
Algunas
características
• La cabeza del muestreador debe ser de acero inoxidable, sinfiltros o grillas protectoras. El borde de la boca de aspiracióndebe ser redondeada.
• El borde superior del tubo de entrada debe esta al menos un (1)metro sobre el techo de la estación y 0,5 m debajo de la toma demuestra para las mediciones de material particulado.
• El tubo de entrada de muestra debe ser construido de vidrio-borosilicato y debe tener un diámetro nominal entre 2 y 4 cm.El múltiple debe ser construida de material inerte (por eje.PTFE, vidrio, acero inoxidable).
• El caudal que permita el sistema de entrada debe ser al menos10 veces mayor que el consumo de gas de muestra de todos losanalizadores.
• El tiempo de retención de la muestra dentro del sistema (desdeel punto de entrada al sistema hasta el último punto de salida) nodebe exceder 10 segundos.
• Los instrumentos de medición deben estar conectados a la líneade entrada de muestra de acuerdo a la actividad de la sustancia,en el orden siguiente: O3; b) NO,NO2; c) SO2; d) H2; e) CmHn; f) CO..
• El sistema de entrada de muestra, incluyendo las líneas demuestra, debe ser limpiada, por lo menos, cada seis meses.

32
ii) Material Particulado
Hay diferentes tipos de cabeza del sistemas de toma de muestra según se quiera sólo
permitir el paso de material particulado de un tamaño máximo (por ejemplo PM1; PM2,5;
PM10) o si se quiere hacer toma de muestra de todo el material particulado en suspensión.
Toma de muestra de material particulado total
Principio
del
método
La cabeza del sistema de muestreo es diseñada para colectar elmaterial particulado total. Está dimensionado de acuerdo al caudalde aire (v) como se indica en la Tabla 5.4.1-I.En la Figura 5.4.1-2 se muestra un esquema del mismo
Algunas
características
• El extremo superior de la cabeza del sistema de muestreo debesobresalir verticalmente del techo de la estación al menos en1,5 m. La longitud del sistema de muestreo debe ser menor de3 m.
• La cabeza, o toma de muestra, del muestreador debe ser hechade acero inoxidable.
• La sonda de muestreo debe llegar verticalmente al instrumentode medición a fin de evitar curvas o cambios de sección. Debeser hecha de material resistente a la corrosión.
• Debe asegurarse que la temperatura del sistema de muestreoeste arriba del punto de rocío.
• De acuerdo al sitio y a los contaminantes el sistema demuestreo debe ser limpiado regularmente, con períodos nomenor de seis meses.

33
Tabla 5.4.1 –I : Dimensionamiento del dispositivo de entrada al sistema de muestreo
Símbolo V=1-1,3 m3/h V= 2,5-3 m3/h Comentarios
D 100 120 Diámetro del protector de lluvia en mm.
a 10 10 Altura del protector de lluvia, en mm
b 15 15 Distancia entre el protector de lluvia y lasuperficie de entrada en mm.
s 1 1 Espesor del plato superior mm
c 3 3,5 Altura del como deflector en mm
h 5 8 Ancho del canal anular en mm
d1 55 75 Diámetro del plato superior
d2 12 14 Diámetro de la base cono deflector
d3 20 27 Diámetro superior del tubo de succión en mm
d4 12 14 Diámetro inferior del tubo de succión en mm
t 12 12 Profundidad de la parte cilíndrica del tubo desucción en mm.
α 73N 7° Ángulo de la parte cónica del tubo de succión
l 43,5 72,5 Altura del tubo de succión
Figura 5.4.1 -2: Cabeza del muestreador para toma de muestra de material particulado

34
Toma de muestra de material particulado con φφ < 10 µµm
Hay dos tipo de equipos de toma de muestra para la medición de material particulado con
diámetro menor de 10 µm: a) el denominado muestreador de gran volumen,
b) el muestreador dicotómico (10) o Dichot.
1 Muestreador de gran volumen
Componentes
básicos
• Cabeza de toma de muestra que transmite solamente laspartículas con diámetros menor de 10 µ. Normalmente haydisponible dos tipo de diseño de cabeza de toma de muestra,que se diferencian por el método de discriminación de laspartículas: a) por impacto
b) ciclónico
• Sistema de control capaz de mantener un flujo constantedentro de las especificaciones de diseño de la cabeza. Haydos tipos de sistemas de control de flujo: i) de masa,
ii) volumétrico.
1.1 Separación por impacto
Principio
del
método
El diseño simétrico de la cabeza de toma de muestra aseguraque sea independiente de la dirección del viento. El aireambiente pasa a través de seis toberas de aceleración hacia laprimera cámara de impacto. Luego pasa a través de 16 toberasde aceleración a la segunda cámara de impacto. Las toberastienen un diámetro crítico a fin de proveer el cambio necesariode velocidad para producir el correcto fraccionamiento portamaño de las partículas dentro de la cámara de impacto. Elaire finalmente sale hacia la cámara de filtros a través de nuevetubos de venteo. La velocidad del aire es crítica; un valor dediseño típico es de 1,13 m3/minuto. En la Figura 5.4.1-3 semuestra un esquema del separador.
(10) Se denomina al método de clasificación en que las divisiones sólo tienen dos partes

35
Figura 5.4.1 –3: Toma de muestra partículas con φφ < 10µµm con separador por impacto
1.2 Separación ciclónica
Principio
del
método
Se imparte a las partículas una componente angular de lavelocidad a través de una serie de alabes uniformementeespaciados. Al pasar luego la muestra por el tubo de colección,las partículas más pesadas, expulsadas hacia afuera, sonretenidas en la superficie del mismo, cubierta por unabsorbedor a fin de evitar rebotes. La muestra pasa luego a untubo intermedio donde la trayectoria es alterada en direcciónascendente y finalmente otro tubo modifica el sentido del flujohacia abajo permitiendo que las partículas sean recogidas sobreun filtro. El control de la velocidad de las partículas es críticopara mantener el correcto punto de corte del tamaño de lasmismas; los valores típicos de diseños son similares alseparador por impacto. En la Figura 5.4.1 - 4 se muestra unesquema del separador.
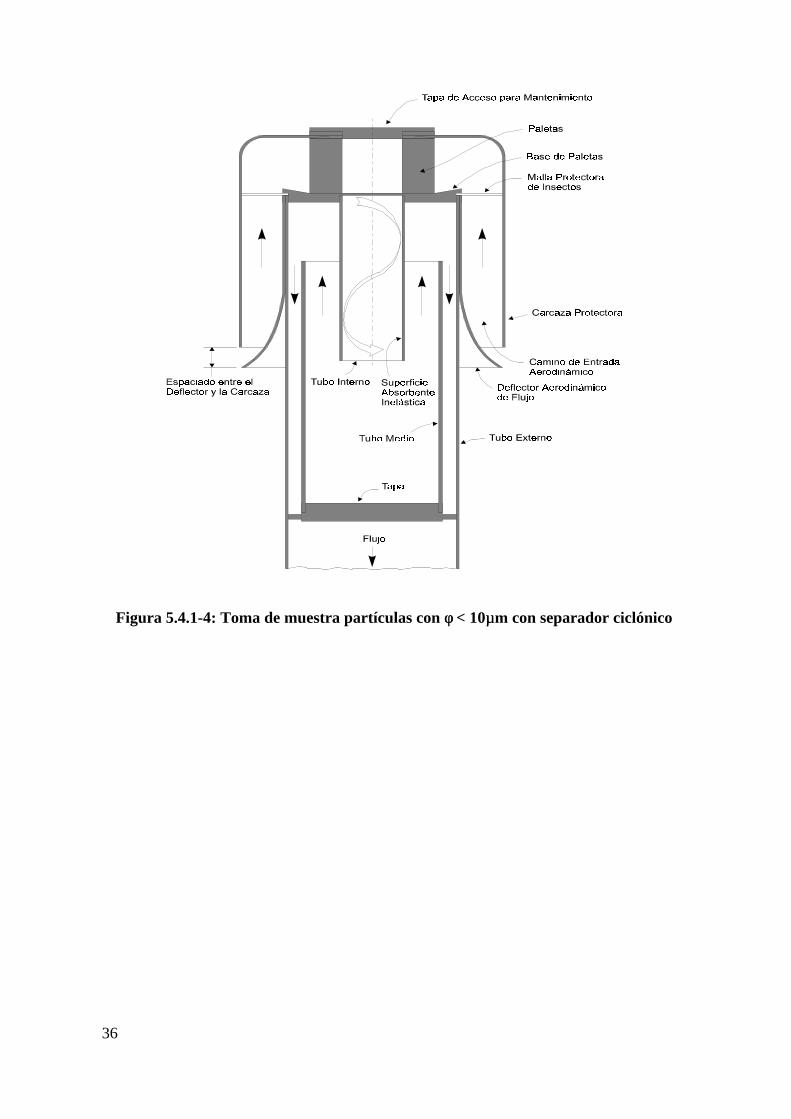
36
Figura 5.4.1-4: Toma de muestra partículas con φφ < 10µµm con separador ciclónico

37
Muestreador Dicotómico o Dichot
Principio
del
método
Colecta partículas de dos tamaños: menores que 10 µm y mayoresde 2,5 µm (denominadas gruesas) y menores de 2,5 µm(denominadas finas). La concentración es la suma de lasfracciones gruesas y finas. El sistema dicotómico de toma demuestra más común tiene un caudal de entrada de 16,7 l/min. Laspartículas con diámetro mayor de 10 µm son separadosinercialmente. En la Figura 5.4.1-5 se muestra un esquema deldiseño de la cabeza de entrada del sistema que separa laspartículas con φ > 10 µ. El aire conteniendo el resto del materialparticulado es forzado a través de un acelerador de tipo boquilla opitón pasando luego a un impactador virtual donde se expande,dividiéndose el flujo de aire. La partículas finas hacen un girobrusco hacia afuera para seguir la velocidad elevada del flujoinicial, pasando a través del filtro de partículas finas. A causa desu mayor inercia, las partículas gruesas continúan su recorrido porel tubo receptor del impactador virtual y son colectados en el filtrode partículas gruesas. Una pequeña porción de partículas finas sontambién colectados en el filtro de partículas gruesas por lo que hayque realizar una corrección en el cálculo. En la Figura 5.4.1-6 semuestra un esquema del conjunto donde se observa la segundaparte del separador.

38
Figura 5.4.1-5 : Cabeza de entrada de muestreador Dicotómico para toma de muestra de
Partículas con φφ < 10µµm
Figura 5.4.1–6 : Principio de separador secundario de partículas con φφ < 2,5 µµ con
muestreador Dicotómico.

39
5.4.1.2 Tubo de entrada y de distribución de muestra (múltiple)
Se destacan algunas características que deben ser consideradas para los tubos de entrada y
distribución de muestra hasta los instrumentos de medición individuales.
• La entrada de toma de muestra debe estar protegida de la lluvia y de los insectos. Si se
utiliza un prefiltrado, deberá elegirse y mantenerse (limpieza regular) de forma que su
influencia sobre la concentración de los gases a medir sea mínima.
• La línea de toma de muestra entre el punto de muestreo y el instrumento debería ser lo
más corta posible. El tiempo de tránsito de las muestras de volúmenes de gas en la
línea de toma de muestra no debería pasar de 10 segundos.
• El tubo de entrada y la línea de toma de muestra o múltiple (tubería y conexiones)
deberá estar realizada a partir de materiales inertes (por ejemplo, vidrio, teflón o acero
inoxidable) que no modifiquen la concentración de los gases a medir.
• La línea de toma de muestra debe ser hermética y el flujo controlarse regularmente.
• Debe evitarse la condensación en la línea de toma de muestra para lo cual,
eventualmente, debe ser calefaccionado.
• La línea de toma de muestra deberá limpiarse regularmente teniendo en cuenta las
condiciones locales
• Las emisiones de gases de los instrumentos de medición y las emisiones procedentes
del sistema de calibración no deberá afectar a la toma de muestra
• Las instalaciones anexas (acondicionador del aire y dispositivos de transmisión de
datos) no deberán afectar al muestreo en la cabeza de toma de muestra.
5.4.2 Equipo de calibración para mediciones automáticas
Para todos las mediciones se debe disponer de un de equipo de calibración que, en general,
consta de un calibrador multigas y un generador de aire cero. Permite realizar la
calibración del fondo de escala (o rango) y comprobación de puntos intermedios de los
analizadores de O3, CO, SO2, NOx, hidrocarburos, etc. por el método de dilución de gases
de referencia en aire cero. El calibrador debe dar la proporción de dilución específica

40
requerida, para concentraciones de los gases contaminantes considerados. Se entiende por
gas de referencia el que contiene una concentración bien determinada de un gas en
particular, dentro de un gas portador. El gas más comúnmente usado como portador es el
nitrógeno (N2). El aire cero se emplea también para la calibración del punto cero de los
equipos. Se entiende por aire cero a un aire libre de los contaminantes a calibrar; puede ser
directamente N2 en grado ultrapuro. Por ejemplo, si se quiere calibrar el punto cero del
analizador de CO, el aire introducido debe tener una pureza de 2 ppb de CO como
máximo; valores similares se deben cumplir para los otros contaminantes..
El equipo de calibración incluye a) un generador de aire cero, b) un medidor de flujo de
aire, b) un equipo de calibración multigas automático y programable, que proporciona
concentraciones requeridas de los contaminantes cuyas concentraciones se van a medir.
También contienen un generador de ozono para la calibración del analizador de ozono. El
calibrador aporta los niveles requeridos de los gases contaminantes considerados,
comprobación de precisión y de nivel 1 del rango, así como multipuntos de calibración de
aquellos. Se debe introduce por teclado en cada equipo el valor que debe alcanzar el punto
fondo de escala y el punto cero, (la norma EPA no acepta la calibración remota). La
calibración de los equipos se deben hacer cada 15 días.
En lo posible se debe disponer de tubos de gases de referencia en cada estación. Tienen
una duración típica de 2 años. Las concentraciones requeridas par ser usados por el
método de dilución dinámica (calibración dinámica y multipunto de los equipos
analizadores de gases) son: CO = 5.000 ppm/N2; SO2 = 50 ppm/N2, NO = 50 ppm/N2. Los
mismos se encuentran dentro de un mismo cilindro. Se debe tener presente que, por normas
internacionales, estos gases no pueden ser traídos por avión, por lo que se debe prever
tiempo de viaje si se hace su compra en el extranjero. También puede usarse un método de
cilindros múltiples, donde se utilizan varios cilindros que contienen gas patrón, con una
concentración específica para cada punto de la calibración, cuyas concentraciones estén
dentro del rango de medición del analizador. Se debe disponer de dispositivos de control
(ajuste y regulación de la velocidad de flujo) y medidor de flujo. Para el caso del método
de dilución dinámica, el control debe ser regulado con una exactitud de ± 1 % y la
medición se debe hacer con una exactitud de ± 2 % del valor real de flujo.

41
5.4.3 Descripción de los métodos de medición
Se han clasificados los métodos de medición en dos grandes grupos:
i) Métodos de referencia: Para aquellos contaminantes normalmente considerados en el
análisis de calidad de aire, dióxido de azufre, óxidos de nitrógeno, monóxido de
carbono, ozono, hidrocarburos totales no metano, material particulado en suspensión y
plomo.
ii) Métodos equivalentes: Los equipos de medición automática o continua de los gases
mencionados anteriormente se basan o en los métodos de referencia o en métodos
equivalentes para los cuales se establecen las condiciones de prueba, el procedimiento
y la interferencia admitida con otros componentes para ser aprobados, especificándose
los parámetros que deben medirse y los valores a lo que deben responder para ser
considerados métodos automáticos equivalentes..
5.4.3.1 Métodos de referencia
Se describen los métodos de referencia para los contaminantes normalmente considerados
en el análisis de calidad de aire, dióxido de azufre, óxidos de nitrógeno, monóxido de
carbono, ozono, hidrocarburos totales no metano (CH4), material particulado en suspensión
y plomo (Pb). Se indica en cada caso la norma ISO - International Organization for
Standardization, y/o EPA - Environmental Protection Agency; US, basadas en el principio
descripto (11).
(11) La información en detalle de las normas ISO se puede obtener de: International Organization forStandardization, ISO Standard Compendium – Environment Air Quality. Edition 1994.La correspondientes a las normas EPA se puede obtener de : Environmental Protection Agency, 40CFR, Part 50.

42
1. Hidrocarburos no metano (HCNM)
Método de cromatografía gaseosa - detector de ionización de llama.
EPA 40 CFR Pt. 50 App. E.
Aplicación e
interferencias
El método se aplica a mediciones semicontinuas de hidrocarburoscorregida por metano (CH4).
Principio
del
método
Se envían volúmenes medidos de aire en forma semicontinua (4 a12 veces por hora) a un detector de ionización por llama dehidrógeno (Ver Sección 5.6, Apartado 4) para medir el contenidode hidrocarburo total (HCT).Una alícuota de la misma muestra de aire es introducida en unacolumna (“stripper”) (Ver Sección 5.6, Apartado 2) la cualremueve el agua, el dióxido de carbono y otros hidrocarburossalvo metano (CH4). El monóxido de carbono y el metano sonseparados en una columna cromatográfica. El metano es eluidoprimero y pasado sin cambios a través de un reductor catalítico aldetector de ionización de llama. El monóxido de carbono es eluidoen un tubo reductor catalítico donde es reducido a metano antes depasar a través del detector de ionización de llama.Las concentraciones de hidrocarburos corregidas por metano sondeterminadas sustrayendo el valor de metano del valor dehidrocarburo total.
Modos
de
operación
Es posible realizar dos modos de operación:
1) Modo cromatográfico espectral: Se realiza un análisiscromatográfico completo mostrando una salida continua delos detectores de cada muestra inyectada.
2) Modo barográfico o normal: El sistema es programado parair al cero automático y extenderse para mostrar los anchosde banda seleccionados del cromatógrafo. La altura del picoes usada como la medida del cromatógrafo.

43
Intervalo
de
aplicación
• Aplicaciones normales
Concentración de hidrocarburos totales: 0 – 13,1 mg/ m3 (0 - 20 ppm) de carbono (como metano).Concentración de metano: 0 - 6,55 mg/m3 (0 - 10 ppm).
• Para aplicaciones especiales están disponibles intervalosmenores
Concentración de hidrocarburos totales: 0 -1,31 mg/m3 (0 – 2 ppm) de carbón (como metano)Concentración de metano: 0-1,31 mg/m3 (0-2 ppm).
SensibilidadPara el límite superior del intervalo la sensibilidad es:
Para hidrocarburos totales: 0,065 mg/m3 (0,1 ppm) decarbón (como metano)
Para metano 0,33 mg/m3 (0,05 ppm).

44
2. Óxidos de nitrógeno (NOx)
Método de Quimioluminiscencia en fase gaseosa. (NO, NO2)
ISO - 7996/85. EPA 40 CFR Pt. 50 App. F
Aplicación e
interferencias
El método permite determinar la concentración de monóxido denitrógeno (NO) y dióxido de nitrógeno (NO2)
Principio
del
método
Determinación de NO. Se hace pasar la muestra de aire por unfiltro a flujo constante e incidir en la cámara de reacción delanalizador donde se mezcla con un exceso de ozono. El NO y elozono da NO2 excitado que al decaer emite luz (reacciónquimioluminiscente [Ver Sección 5.6, Apartado 6.2]). Se filtra laradiación emitida, proporcional a la cantidad de NO presente, conun filtro óptico selectivo y se convierte la radiación filtrada en unaseñal eléctrica por medio de un tubo fotomultiplicador, midiendola intensidad de luz a longitudes de onda mayores de 600nanometro (El pico intenso de radiación quimioluminiscente es auna longitud de 1,2 µm).
Determinación de NO2. La muestra se pasa por un convertidor(horno a 400 ° C constante) para reduce el NO2 a monóxido antesde hacerla entrar a la cámara de reacción. La señal eléctricaobtenida en este caso es proporcional a la cantidad total de óxidosde nitrógeno presente en la muestra. La cantidad de dióxido denitrógeno se obtiene por diferencia entre este valor y el obtenidoen la medición de NO.
Las mediciones de NO y NO+NO2 pueden hacerse con sistemadual, o cíclicamente con el mismo sistema teniendo en cuenta quelos tiempos del ciclo no exceda un minuto.
El volumen total se determina midiendo el caudal y el tiempo decolección.
Intervalo
de
aplicación
Concentración de monóxido de nitrógeno: hasta 12,5 mg/m3
Concentración de dióxido de nitrógeno: hasta 19 mg/m3
A 25 °C de temperatura y 101,3 kPa de presión.

45
Figura 5.4.3-1: Determinación de NO y NO2 por método de Quimioluminiscencia .
Método de Griess-Saltzman modificado para determinación de NO2
ISO - 6768/85
Aplicación e
interferencias
El método permite determinar la concentración de dióxido denitrógeno (NO2)
Principio
del
método
Se absorbe el dióxido de nitrógeno presente en la muestrahaciéndola pasar a través de una reactivo que forma azo colorante(grupo –N=N-), resultando la formación de un color rosado en 15minutos. Se determina la absorbancia de la solución para unalongitud de onda entre 540 y 550 nm, con un espectrofotómetroapropiado (o colorímetro), evaluando la concentración másica dedióxido de nitrógeno por medio de una calibración gráfica conmezcla de gases de calibración. De acuerdo a la disponibilidad deequipos en el laboratorio puede ser conveniente usar, para pruebasde rutina, soluciones de nitrito de sodio.El tiempo de muestreo puede ser de 10 minutos a 2 horas. Debidoal tiempo de estabilidad de la muestra, no debe pasar más de 8horas desde la toma de la misma y su análisis.
Intervalo de
aplicación
Concentración de dióxido de nitrógeno de 0,010 a 20 mg/m3.

46
3. Ozono (O3)
Método de Quimioluminiscencia.
ISO - 10313/93. EPA 40 CFR Pt. 50 App.D
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deozono (O3). El material particulado puede producir puede causardiferentes perturbaciones en la medición y debe ser previamentefiltrado. El óxido de nitrógeno (NO) puede reaccionar con elozono por lo que el tiempo de duración en el cual el aire ambientedebe permanece en la línea de toma de muestra debe sersuficientemente corto para mantener este efecto al mínimo.
Principio
del
método
La muestra de aire es pasada continuamente, a flujo constante, através de un filtro de partículas antes de hacerlo incidir sobre elanalizador de quimioluminiscencia (Ver Sección 5.6, Apartado6.2). Luego fluye a una cámara de reacción donde se mezcla conun exceso de etileno (C2H4) . El ozono y el etileno reaccionaninstantáneamente emitiendo luz en la región visible con unmáximo en la longitud de onda alrededor de 400 nm. Laintensidad de la luz emitida es proporcional a la concentración deozono en la muestra de aire y es medida con tubosfotomultiplicadores. El voltaje resultante es amplificado ycalibrado en términos de la concentración de ozono ambiente. Lafotometría ultravioleta es especificada como método primario decalibración a causa de su probada exactitud y especificidad paraozono. El uso de otros estándares requiere previa calibraciónfrente al método primario.Existe una norma (12) para determinar cuando el número de díaspor año calendario esperado con concentración promedio horariamáxima superior a 0,12 ppm (235 µg/m3) es igual o menor a uno.
Intervalo
de
aplicación
Concentración másica de ozono de 2 µg/m3 (0,001 ppm[V/V]) a10 mg/m3 (5 ppm [V/V]) en las condiciones de referencia de298 K y 101,3 kPa.
(12) EPA 40 CFR Pt. 50 App. H

47
4. Dióxido de azufre (SO2)
Método espectrofotométrico con Thorin.
ISO - 4221/80
Aplicación e
interferencias
Se aplica a la determinación de la concentración de dióxido deazufre (SO2). Puede haber interferencia con amoníaco (NH3) ysulfuro de hidrógeno (SH2) si están presente en concentracionesmuy elevadas. Para eliminar interferencia con materialparticulado se filtra la muestra del aire.
Principio
del
método
El dióxido de azufre presente en la muestra de aire es absorbido yoxidado haciéndolo pasar a través de una solución acidificada deperóxido de hidrogeno (H2O2) de pH definido (entre 4 y 4,5 conácido perclórico [HClO4]), durante un período de tiempodeterminado, generando ácido sulfúrico (SO4H2). Los ionessulfatos son precipitados por formación de sulfato de bario(SO4Ba) a través de una reacción con perclorato de bario(Ba[ClO4]2) en exceso; se determina la cantidad de iones de barioen la solución por complejamiento con Thorin y midiendo elcomplejo coloreado espectrométricamente en la longitud de ondade 520 nm. La diferencia entre la cantidad inicial y final de ionesde bario corresponde a la concentración de iones sulfato en lasolución de absorción, y por lo tanto a la cantidad de dióxido deazufre que fue oxidado. La absorbancia varia en forma inversa conla concentración de iones sulfato presentes en la solución deabsorción.Para un período de muestreo de 24 horas, el volumen de soluciónabsorbente debe ser de 50 ml, mientras para un período de 48horas debe ser de 150 ml.
Intervalo
de
aplicación
Concentración másica de dióxido de azufre de 3,5 a 150 µ g/m3,asumiendo un volumen de muestra de aire de 2 m3 y un volumende solución de muestra de 50 ml.

48
Método espectrofotométrico del tetracloromercurato/ pararosanilina
ISO - 6767/90. EPA 40 CFR Pt. 50 App.A
Aplicación e
interferencias
Se aplica a la determinación de la concentración de dióxido deazufre (SO2).
Principio
del
método
El dióxido de azufre presente en la muestra de aire es absorbido enuna solución 0,04 M de tetracloro mercurato de sodio[Na2(HgCl4)] (o de potasio) (TCM), resultando la formación de uncomplejo diclorosulfito-mercurato. El CO2 presente en la corrientede aire reacciona con la solución TCM formando un compuestoestable de monocloro-sulfonato mercurato. Una vez formado, estecompuesto es resistente a la oxidación y es estable en la presenciade oxidantes fuertes como el ozono y óxidos de nitrógeno. Sedeben destruir los iones nitrito formados en la solución detetracloro mercurato de sodio (Na2[HgCl4]) por la presencia deóxidos de nitrógeno en la muestra, adicionado solución de ácidosulfámico (NH3O3S). Se realiza la conversión del complejo TCMen ácido pararosanilina metilsulfónico intensamente coloreado devioleta, adicionando solución de formaldehído (CH2O) y soluciónde pararosanilina (C19H19N3O) acidificada con ácido clorhídrico(ClH). Se determina la absorbancia de la muestra a una longitudde onda de 550 nm usando un espectrofotómetro (o colorímetro),y calculando la concentración másica de dióxido de azufre usandouna calibración gráfica con mezcla de gases de calibración. Elvolumen total de muestra de aire corregida a 25 °C y 101,3 kPa, sedetermina midiendo el caudal y el tiempo de toma de muestra. Eltiempo de toma de muestra es de 30 a 60 minutos. Si el períodode muestreo es mayor de 60 minutos, hasta 24 horas, o si se esperaconcentraciones de dióxido de azufre mayores que las indicadas(hasta 2.000 µg/m3), debe tomarse precauciones para que laconcentración de dióxido de azufre en la solución de absorción noexceda de 0,25 a 2,5 µg por mililitro de solución absorbente.
Intervalo
de
aplicación
Límite inferior de detección de SO2 : 0,75 µg en 10 ml de TCM.Esto representa una concentración de 25 µg / m3 (0,01 ppm) enuna muestra de aire de 30 litros (muestreo de tiempo corto) y unaconcentración de 13 µg / m3
(0,005 ppm) en una muestra de 288litros (muestreo tiempo largo).Límite superior de detección de SO2: 34 µg en 25 ml desolución final. Este límite superior del intervalo de análisisrepresenta concentraciones de 1.130 µg / m3 (0,43 ppm) en unamuestra de aire de 30 litros y concentración de 590 µg / m3 (0,23ppm) en muestra de aire de 288 litros. Para concentración entre

49
80-200 µg SO2 / m3 la desviación estándar es del orden de ± 10 %.
Concentraciones más elevadas pueden medirse colectandovolúmenes menores de aire, o aumentando el volumen de soluciónabsorbente, o diluyendo una porción apropiada de la muestracolectada con solución absorbente antes del análisis.
5. Monóxido de carbono (CO)
Método de cromatografía gaseosa.
ISO - 8186/89
Aplicación e
interferencias
Se aplica a la determinación de la concentración de monóxido decarbono (CO). El método está libre de cualquier interferencia.
Principio
del
método
Un volumen fijo de muestra de aire se hace pasar por una columnacromatográfica (ver Sección 5.6, Apartado 2) para separaciónefectiva de monóxido de carbono de los otros gases contenidos enla muestra. Se reduce el CO separado a metano (CH4) haciendopasar gas arrastrado por hidrógeno por un catalizador de níquelcalentado. El metano resultante se pasa a través de un detector dellama ionizante. La señal de salida es proporcional a la cantidad deCO presente en la muestra.
Intervalo
de
aplicación
Concentración de CO1) de 0 a 1 mg/m3
2) de 0 a 25 mg/m3
3) puede ser aplicado también para concentraciones hasta1.000 mg/m3 en aire
Valores para 25 °C de temperatura y 101,3 kPa de presión.

50
Método de fotometría infrarroja no dispersiva
EPA 40 CFR Pt. 50 App.C
Aplicación e
interferencias
Se aplica a la determinación de la concentración de monóxido decarbono (CO).
Principio
del
método
La radiación infrarroja se hace pasar a través de un celdaconteniendo el gas de muestra que se desea analizar, y laabsorción cuantitativa de energía por el CO es medida por undetector apropiado en un fotómetro no dispersivo (Ver Sección5.6, Apartado 8.1). El fotómetro es sensibilizado a CO empleandogas CO en un filtro en el camino óptico, con lo cual se limita lamedición de absorción sólo a uno o más de las longitudes de ondapara las cuales se produce una fuerte absorción por parte del CO.Se puede también usar filtros para limitar la sensibilidad delfotómetro a una banda angosta de interés. Se hace pasar laradiación infrarroja alternativamente por el filtro con CO,produciendo un haz de referencia, y con otro gas, por ejemplo N2,que es transparente a la radiación infrarroja de interés,generándose el haz de medición, que luego es absorbido por elCO de la muestra (esta variante se conoce también con el nombrede método de correlación de filtros). Ver Sección 5.6 con eldetalle de ambos métodos y los esquemas correspondientes.La absorción medida es convertida en un señal eléctrica que esrelacionada con la concentración de CO.

51
6. Material particulado (MP)
Método de gran volumen. Material particulado en suspensión total
(MPT)
EPA 40 CFR Pt. 50 App.B
Hasta que se desarrollaron suficiente datos de base para medir material particulado con
φ<10 µm (MP10), la mayor información disponible para indicar la naturaleza de la
concentración de material particulado estaba basada en mediciones de material particulado
en suspensión total (MPT) realizadas con monitores de gran volumen (hi-vol).
Actualmente se siguen empleando estos equipos en muchas mediciones por lo que
describiremos su principio de funcionamiento.
Aplicación e
interferencias
El método se aplica a la determinación de concentración de materialparticulado total.
Principio
del método
El proceso de medición no es destructivo y el tamaño de la muestracolectada es usualmente adecuada para realizar otros análisisquímicos.Se hace pasar una cantidad medida de aire ambiente a través de unfiltro de fibra de vidrio durante 24 horas, período nominal demuestreo, a una velocidad relativamente alta (caudal de ≈ 1,1 a1,7 m3/min). El caudal del equipo de muestreo y la geometría delprotector o cubierta favorece la colección de partículas, avelocidades del aire entre 1,3 y 4,5 m/seg (3 a 10 mph), condiámetro máximos entre 25 y 50 µm (diámetro aerodinámico),dependiendo de la dirección del viento. Los filtros sonespecificados para tener una eficiencia de colección mínima del99 % para partículas de 0,3 µm.El filtro es pesado (después de equilibrar la humedad porcalentamiento) antes y después de la colección de materialparticulado, cuya masa se obtiene por diferencia. El volumen deaire recogido es determinado a través de la medición del caudal yel tiempo de toma de muestra, corrigiéndolo para las condicionesestándar de 25 °C y 101,3 kPa. La concentración de materialparticulado en aire ambiente se obtiene por cociente entre la masade material pesada y el volumen de aire total recogido.

52
Intervalo
de
aplicación
Concentración de material particulado: 2 a 750 µg / m3
normal.El límite superior está determinado por el punto al cual el equipode muestreo no puede mantener el caudal específico debido alaumento de la caída de presión del filtro cargado.El límite inferior está dado por la sensibilidad de la balanza.
Variabilidad
del
Método
La variabilidad en la medición de concentración no es más del 10% de día a día teniendo en cuenta la velocidad del viento. Bajoidénticas condiciones meteorológicas un coeficiente típico devariabilidad es de 3 a 5 %. Un problema más significativo es laformación de masa artificial por reacción de gases ácidos con elmaterial colectado en el filtro durante la colección de muestras de24 horas. Una estimación de 6 a 7 µg/m3 puede ser adicionado auna medición de 24 horas por este efecto. Errores tambiénpueden ocurrir por pérdida de partículas volátiles, deposiciónantes y después de la toma de muestra, reacción de gas despuésdel muestreo, y manipuleo del filtro.
Figura 5.4.3-2: Monitor de gran volumen (hi-vol) para material particulado

53
Material particulado con φφ<<10 µµm. PM10
EPA 40 CFR Pt. 50 App.J
Aplicación e
interferencias
El método se aplica a la determinación de concentración dematerial particulado con diámetro menor de 10 µm.
Principio
del
método
El método de referencia está diseñado para medir la porción dematerial particulado en suspención en la atmósfera que puededepositarse en la región torácica del sistema de respiraciónhumano. Es aplicable a la medición de concentración másica dematerial particulado con diámetro aerodinámico menor o igual a10 micrómetro en aire ambiente por un período de 24 horas. Tieneuna eficiencia de colección del 50 % para partículas con 10 µm.Ser define como corte del tamaño de partícula de una muestras deMP como el diámetro al cual la eficiencia de colección es menorque 50 % para todas la partículas mayores, y se indica como D50= 10 µm.La muestra de aire es llevada a caudal constante a un equipo detoma de muestra con una entrada de forma especial donde elmaterial particulado en suspención es separado inercialmente enuno o más fracciones de tamaño dentro de cada intervaloconsiderado. Cada fracción de tamaño en el intervaloconsiderado es colectada sobre filtros separados duranteperíodos específico de muestreo.
Cada filtro es pesado, después de secado, antes y después derecoger las muestras y de su diferencia se obtiene el peso delmaterial particulado con diámetro menor de 10 micrómetro. Elvolumen de aire se determina midiendo el caudal y el tiempo detoma de muestra, corrigiéndolo a condiciones estándares.
Intervalo
de
aplicación
Concentraciones de masa de partículas con diámetro menorde 10 micrometro : al menos de 300 µg/m3 durante 24 horasoperando dentro de los límites de caudal establecidos.

54
Determinación de un índice de humo negro.
ISO - 9835/93
Aplicación e
interferencias
El método se aplica a la medición del índice de humo negro.Se entiende por humo negro al formado por material particuladoen suspensión altamente absorbente de la luz en el aire ambiente.La mayor contribución al humo negro son la partículas de hollín(partículas conteniendo carbón en su forma elemental).
Principio
del
método
El método se basa sobre el efecto de mancha producido al pasarla muestra de aire por un papel de filtro. Se mide la reflectanciade la mancha producida. El método no mide la concentraciónmásica de partículas directamente.
Intervalo
de
aplicación
Es aplicable a la medición de índice de humo negro en el intervalode 6 a 375.

55
7. Plomo (Pb)
Método de espectrometría de absorción atómica.
ISO - 9855/93 . EPA 40 CFR Pt. 50 App.G.
Aplicación e
interferencias
Se aplica a la determinación de concentración de plomo (Pb)particulado.
Principio
del
método
El material particulado en suspensión en el aire ambiente escolectado sobre un filtro de fibra de vidrio durante 24 horasusando un equipo de muestreo de gran volumen. El análisis delas muestra puede realizarse en forma individual o colectando lasmuestras diarias durante un mes o un trimestre. El Plomocontenido en el material particulado es extraído disolviendo conácido nítrico (NO3H), calentando la muestra para facilitar lareacción, o usando una mezcla de ácido nítrico (NO3H) yclorhídrico (ClH), facilitando la reacción con ultrasonido. Elcontenido de plomo de la muestra es luego analizado porespectrometría de absorción atómica usando una llama de aire-acetileno, en la líneas de absorción del plomo de 283,3 o 217,0nm (Ver Sección 5.6, Apartado 1).
La extracción con mezcla de ácidos nítrico y clorhídrico conultrasonido, puede disolver otros metales .
Intervalo
de
aplicación
El intervalo típico de aplicación del método es de 0,07 a 7,5 µgPb / m3 , suponiendo límite superior de intervalo de análisis de15 µg / ml y un volumen de aire de 2.400 m3.
El método es aplicable en los casos en que las partículas deplomo colectadas en el filtro sea mayor a 1 µg, si ladeterminación final se realiza con un espectrómetro de absorciónatómica con quemador de llama. Cantidades menores de 1 µgpueden realizarse si la determinación final se realiza con unespectrómetro de absorción atómica con quemador horno degrafito; se debe validar en este caso los límites de detección.

56
5.4.3.2 Métodos equivalentes de medición
Existen diferentes clasificación de los equipo de medición automática o continua, según las
normas del país que los certifica. Se ha seguido la normativa (13) de la Agencia de
Protección Ambiental (EPA) de los EE. UU. de Norteamérica como guía. La norma EPA
40 CFR, Part 53 - Subpart B (14), especifica los parámetros que deben medirse y los
valores a lo que deben responder para ser considerados métodos automáticos equivalentes.
Establece las condiciones de prueba, el procedimiento y la interferencia admitida con otros
componentes.
Hay equipos que se basan en los métodos de referencia antes descriptos. Otros emplean
diferentes principios de medición, denominados equivalente. Se resumen los principios de
funcionamiento de los métodos equivalente de equipos automáticos de medición de
concentraciones de gases y material particulado en aire ambiente.
(13) Identificación de los equipos de medición continua
RFEQNSOCLPMPSAMesAñoNúmero
Método de ReferenciaMétodo EquivalenteÓxido de NitrógenoDióxido de AzufreOzonoMonóxido de CarbonoPlomoAnalizadores automáticos de Material ParticuladoTomadores de muestra de Material ParticuladoSistema AutomáticoMes de autorizaciónAño de autorizaciónSecuencia cronológica de autorización
(14) EPA (Environmental Protection Agency), 40 CFR (Title 40: Code of Federal Regulations), Part 53 -Ambient air monitoring reference and equivalent methods, Subpart B - Procedures for testing performancecharacteristics of automated methods for SO2, CO, O3 and NO2 “

57
1. Óxidos de nitrógeno (NOx) .
Método de espectroscopia de absorción óptica diferencial.
Aplicación e
interferencias
Se aplica a la determinación de concentración de monóxido ydióxido de nitrógeno (NO y NO2).
Principio
del
método
Basado en la ley de absorción de Beer-Lambert que establece larelación entre la cantidad de luz absorbida y el número demoléculas en el camino que atraviesa la luz. Se emplea una fuenteespecial, lámpara de xenón a alta presión que emite luz delongitudes de onda en el espectro visible, ultravioleta, e infrarrojo.El haz de luz atraviesa un camino de longitud determinada dondese produce la absorción molecular. La luz es luego capturada porun receptor y conducida por fibras óptica hasta el analizador, unespectrómetro de elevada calidad. Usando un programa decomputo se puede evaluar y analizar la perdida de luz debido a laabsorción molecular que se produjo en el camino recorrido, y enfunción de dicha longitud obtener la concentración por unidad devolumen.Dado los bajos niveles de concentración en atmósfera, el sistemaes sensible para recorrido de la luz como mínimo de 300 m. Sepuede acortar esta longitud introduciendo reflexión del haz con loque se lo obliga a pasar tres veces por el mismo camino, con loque se puede llegar a valores de 100 m. La distancia máxima esde 1.500 m.
Intervalo
de
aplicación
NO2: 1 a 1000 µg/m3. Longitud recomendada 300 a 800 m
NO : 2 a 2000 µg/m3. Longitud recomendada 100 a 200 m
Otras
aplicaciones
del
Método
Dado que cada tipo de molécula tiene propiedades únicas delespectro de absorción, es posible identificar y determinar laconcentración de diferentes gases simultáneamente con un sóloequipo a lo largo de un dado recorrido de luz. Hasta el momentoEPA tiene homologado el equipo solamente para SO2, NO2, y O3.
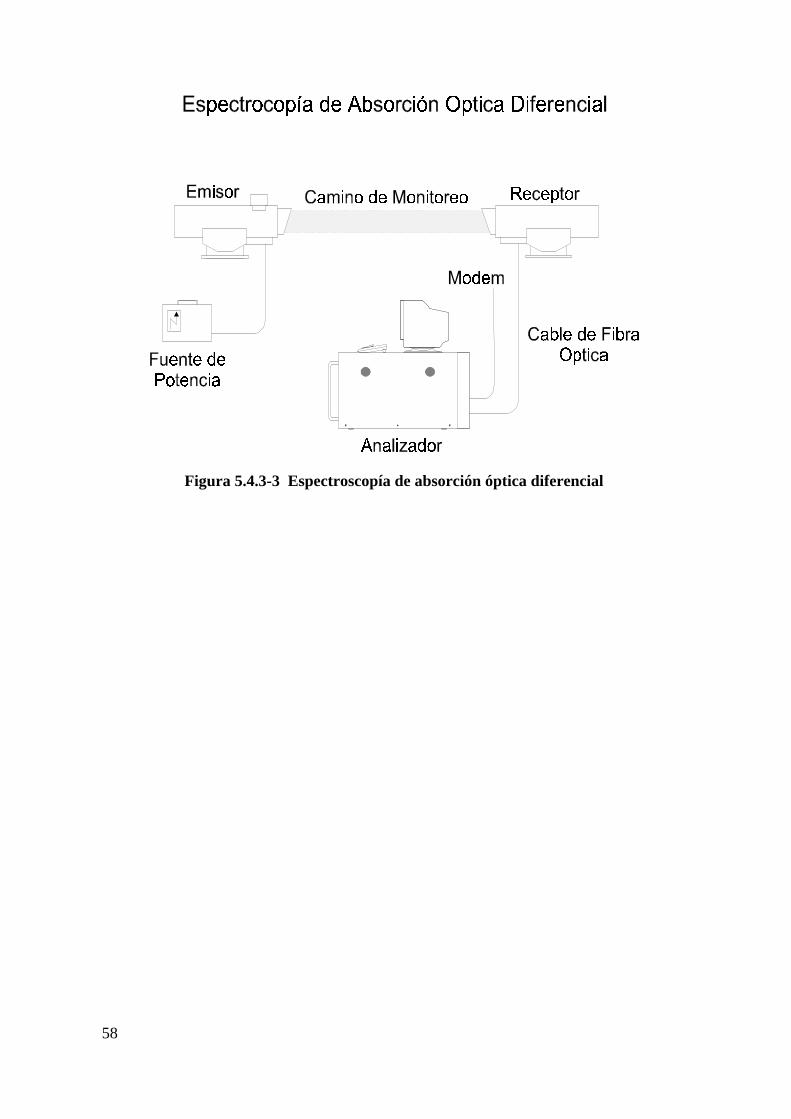
58
Figura 5.4.3-3 Espectroscopía de absorción óptica diferencial

59
2. Ozono (O3).
Método de fotometría (absorción) ultravioleta
Aplicación e
interferencias
Se aplica a la determinación de concentración de ozono (O3).
Principio
del
método
La concentración de ozono es determinada por atenuación de laradiación ultravioleta de 254 nm de longitud de onda al pasar porla celda de muestreo, llena con el aire en análisis (Ver Sección5.6, Apartado 7). Se emplean dos celdas, una con el gas de lamuestra a analizar y otra con aire de referencia (el mismo aire aanalizar se trata con un conversor selectivo que elimina sólo elozono, por ejemplo, el ozono se convierte catalíticamente aoxígeno). Los detectores miden la intensidad de la luz transmitidaa través de cada celda. Hay otros compuestos que absorben laradiación ultravioleta de 254 nm, tal como SO2, aromáticos, etc.,que es corregida con la comparación con la celda de referenciadonde se elimina el ozono. El instrumento determina laconcentración de ozono computando la atenuación de la luz en lasdos celdas. Hay un control continuo de cambios de temperatura ypresión para corregir los cambios registrados de esos parámetrosen el ambiente.
Intervalo
de
aplicación
El intervalo de concentración para el cual se aplica es, envolumen, de 0 a 0,1 ppm.
Figura 5.4.3-4: Método de fotometría ultravioleta para la medición de ozono

60
Método de espectroscopia de absorción óptica diferencial.
Aplicación e
interferencias
Se aplica a la determinación de concentración de ozono (O3)
Principio
del
método
Ver método equivalente en determinación de NO y NO2
Intervalo
de
aplicación
3 a 1.000 µg/m3. Longitud recomendada 300 a 500 m
3. Dióxido de azufre (SO2)
Fluorescencia Molecular
Aplicación e
interferencias
Se aplica a la determinación de concentración de dióxido deazufre (SO2).
Principio
del
método
Se excita las moléculas de SO2 con radiación ultravioleta en elintervalo de longitud de onda de 230 a 190 nm, las cuales luegodecaen a su nivel energético fundamental, emitiendo una radiaciónfluorescente característica (Ver Sección 5.6, Apartado 6.1). Laintensidad de la radiación emitida es directamente proporcional ala concentración del dióxido de azufre.

61
Figura 5.4.3-5: Método de medición de SO2 por fluorescencia molecular.
Variación de la conductividad
Aplicación e
interferencias
Se aplica a la determinación de concentración de dióxido deazufre (SO2).
Principio
del
método
La muestra de aire se pone en contacto con una solución deperóxido de hidrógeno (H2O2) acidificada con ácido sulfúrico(SO4H2); el dióxido de azufre es absorbido y oxidado formandoácido sulfúrico (SO4H2). Esto aumenta la conductividad eléctricade la solución reactiva sirviendo como una indicación de laconcentración de SO2. Se mide alternativamente la conductividadde la solución reactiva antes del pasaje de la muestra de aire ydespués, y la diferencia es proporcional a la concentración deSO2. Este método es aplicable cuando las mediciones no sonafectadas por otros gases que se disuelven en el absorbente yproducen cambios en la conductividad, tales como cloro (Cl),amoníaco (NH3) y dióxido de carbono (CO2).
Se identifican dos tipos de mediciones:Mediciones acumulativas: Las mediciones registran laconcentración de SO2 correspondiente al incremento de laconductividad del absorbente al pasar una cantidad fija demuestra de aire durante un tiempo predeterminado.. Mediciones instantáneas: Las mediciones registrancontinuamente la concentración de SO2 midiendo el cambio de laconductividad del absorbente con un flujo de caudal constante delaire de muestra.

62
Método de espectroscopia de absorción óptica diferencial.
Aplicación e
interferencias
Se aplica a la determinación de concentración de dióxido deazufre (SO2)
Principio
del
método
Ver método equivalente en determinación de NO y NO2
Intervalo
de
aplicación
1 a 2.000 µg/m3. Longitud recomendada 300 a 500 m

63
4. Monóxido de carbono (CO).
Variación de la conductividad
Aplicación e
interferencias
Se aplica a la determinación de concentración de monóxido decarbono (CO).
Principio
del
método
El CO contenido en la muestra de aire es oxidado a CO2 a 150 °Cen un horno de combustión conteniendo iodine pentonxide. ElCO2 es absorbido en una solución de hidróxido de sodiocambiando la conductividad. Se mide la conductividad antes ydespués de ser absorbido el CO2.
5. Material particulado (MP)
Todos los métodos emplean un separador de tamaño a la entrada del sistema de toma de
muestra.
Microbalanza Electrónica
Aplicación e
interferencias
Se aplica a la determinación de concentración de materialparticulado con diámetro < 10 µm.
Principio
del
método
Se colecta las MP10 sobre un pequeño filtro conectado a través deuna interface a un traductor de masa inercial que permite pesar elfiltro en forma casi continua a medida que se acumula el materialparticulado. El filtro es cambiado manualmente cuando la cargadel material particulado sobrepasa el límite de tolerancia deldispositivo.

64
Vibración de un filtro (microbalance oscilante)
Aplicación e
interferencias
Se aplica a la determinación de concentración de materialparticulado con diámetro < 10 µm.
Principio
del
método
La muestra de aire pasa a través de un filtro el cual es parte de unsistema que vibra a su resonancia característica (Ver Sección 5.6,Apartado 13). El material particulado colectado sobre el filtroaumenta la masa vibrante y por lo tanto decrece la frecuencia deoscilación en forma proporcional. La concentración de materialparticulado es calculado a partir de una calibración que relacionala frecuencia de vibración y la cantidad de material particulado,teniendo en cuenta el volumen de muestra de aire.
Atenuación (o absorción) de rayos beta
Aplicación e
interferencias
Se aplica a la determinación de concentración de materialparticulado total o con diámetro < 10 µm, según el tipo deseparador que se coloca en la entrada.
Principio
del
método
La radiación emitida por una fuente de rayos β pasa por un filtrode fibra de vidrio limpio, generalmente en forma de cinta (Versección 5.6, Apartado 12). Un volumen conocido de muestra deaire se hace pasar por el filtro y en la mancha se mide laatenuación de rayos beta por absorción en la muestra.Periódicamente se corre el filtro. La relación entre ambasmediciones dan la cantidad de MP depositado. La fuente de rayos-β es usualmente un isótopo de C14.
Absorción de luz (sensibilidad óptica)
Aplicación e
interferencias
Se aplica a la determinación de concentración de materialparticulado total o con diámetro < 10 µm, según el tipo deseparador que se coloca en la entrada.
Principio
del
método
Un haz de luz pasa por un filtro de fibra de vidrio limpio,generalmente en forma de cinta. Un volumen conocido demuestra de aire se hace pasar por el filtro y en la mancha se midela transmisión de luz para determinar la densidad óptica ocoeficiente de niebla u opacidad. Periódicamente se corre el filtro.La relación entre ambas mediciones dan la cantidad de MPdepositado.

65
5.4.4 Casilla de la estación de monitoreo
Los equipos de medición de concentración de gases en atmósfera deben ser colocados
dentro de una casilla, para las cuales es necesario especificar una serie de condiciones y
seguir pautas generales para su diseño. Es común el empleo de casillas para las estaciones
de monitoreo prefabricadas, pero por un problema de costo se debe analizar la posibilidad
de su construcción por métodos convencionales. Una ventaja relativa de las casillas
prefabricadas es que pueden ser movidas de sitio en caso de decidir un cambio del lugar de
toma de muestra.
Se describen algunas especificaciones a tener en cuenta, tanto en la construcción de la
estación cuanto en el diseño del sistema de operación y control de la misma. Lo referente
al sistema de toma de muestra y transporte del aire hasta los equipos de medición se
describe en la Sección 5.4.1.
Tamaño Debe tener entre 4 - 3 m de largo x 3 – 2 m de ancho y2,5 m de alto.
Especificación de
estabilidad
• Carga del techo > 750 N/m2
• El techo debe ser suficientemente fuerte como paracaminar sobre el.
• Carga del piso > 300 N/m2.
Aislación del calor Número de conductividad térmica media < 2 W/m2 .K(el valor recomendado es de 1,2 W/m2 .K).
Materiales Los materiales de construcción y los accesorios nodeben ser inflamables o al menos de difícil ignición.
Protección de rayos La estación de medición, el sistema de muestreo y laestación meteorológica deben estar protegidas frente ala caída de rayos y el exceso de voltaje. Se debe preverla colocación de un pararrayos, con la correspondientepuesta a tierra.

66
Aire acondicionado • La humedad relativa interna normal debe estar entre30 y 60 %. Esto es importante pues con humedadarriba del 60 % será difícil tener mediciones reales deSO2 dado que tiende a reaccionar en condicionesmuy húmedas.
• La temperatura interior debe estar dentro delintervalo de +15 °C y +30 °C. No debe variar mas de± 5K
• La estación debe ponerse automáticamente fuera deoperación si la temperatura interior supera los 35 °C.
• El flujo de aire desde el acondicionar de aire debe serfiltrado antes de su emisión dentro de la estación.
• El aire de acondicionamiento debe ser descargadohacia afuera de la estación de forma tal que noinfluya en la toma de muestra de aire.
Equipamiento
eléctrico
Se debe colocar cuatro circuitos diferentes y separados,cada uno de los cuales debe tener protección para elpersonal y circuito de emergencia, para:
a) Procesamiento de datos y comunicación
b) Sistema de muestreo y medición,
c) Acondicionador de aire,
d) Ventilación y luz
Cuando el circuito de emergencia para los circuitos a) yc) es activado, la ventilación y la luz debe permaneceren operación.
Tableros, soportes
y sistema de
adquisición de
datos
• Tablero general con interruptores termomagnéticosy un regulador de voltaje,
• Lugar para fijar cilindros de gases de referencia parala calibración del rango o fondo de escala y cero ocalibración multipunto de los analizadores,
• Mesa de trabajo con el equipo de adquisición dedatos (dataloggers) basado en una microcomputadora (por ejemplo PC Pentium de 75MHertz, disco duro de 1 Gigabyte, monitor color,software, tarjeta de adquisición de datos, módulosmultipuertas en serie para interconectarla con cadauna de las salidas de los equipos de medición).

67
Señales del estado
del sistema
Las funciones esenciales de las estaciones de medicióndeben ser comprobadas por señales de estadotransmitidas telemétricamente, por ejemplo
• Funcionamiento del sistema de aire acondicionado,temperatura interior.
• La puerta de entrada debe estar equipada con uncontacto que provea una señal telemétrica indicandosu estado.
• Flujo de gas en le sistema de muestreo.
Tratamiento del
gas de descarga
Cuando son usados cilindros de gas presurizados, el gasde calibración en exceso debe ser descargado a través deun bypass, a fin de evitar sobrepresión cuando el sistemaes conmutado de medición a calibración. Todo el gas enexceso y de descarga debe ser emitidos en forma seguradesde la estación de medición.En los casos de un defecto del sistema o una falla depotencia, la descarga incontrolada de los gases decalibración y operación debe ser prevenida, pordispositivos de seguridad, tanto como sea posible.
Estación
meteorológica
En principio debería incluirse una estaciónmeteorológica mínima que permita interpretar ypredecir la dispersión de contaminantes; por ejemplo amayor radiación solar, menor humedad, modificándosela posible reacción fotoquímica de compuestoshidrocarburos para generar ozono.Una estación meteorológica para este servicio debeconstar de sensores de: velocidad y dirección del viento,humedad relativa, temperatura, radiación solar, presiónbarométrica. Se necesita una torre de 10 m de alturapara las mediciones de velocidad y dirección del vientoy la conexión a la casilla de toma de datos.

68
5.5 MEDICIONES DE CONCENTRACIÓN DE GASES Y MATERIAL
PARTICULADO EN CHIMENEA
5.5.1 Toma de muestras de gases y material particulado.
Determinación de velocidad dentro del conducto y el caudal
volumétrico.
Se describe el alcance y las limitaciones de las distintas normas, dado que si bien en
general coinciden, en cada una se especifican particularidades que deben tenerse en cuenta
y enriquecen la información.
Toma de muestra para la determinación automática de concentración de
gases.
ISO - 10396 / 93.
Aplicación y
limitaciones
El método estándar especifica procedimientos y equipamiento quepermite, dentro de ciertos límites, tomar muestras representativaspara la determinación automática de concentraciones de gases enefluentes gaseosos por chimenea. La aplicación se limita a ladeterminación de oxígeno (O2), dióxido de carbono (CO2),monóxido de carbono (CO), monóxido de nitrógeno (NO) ydióxido de nitrógeno (NO2) (15). Aunque solo se mencionabrevemente, medición detallada de la velocidad del gas esrequerida para determinar la intensidad de flujo másico.La norma abarca el muestreo de gas para métodos extractivos y noextractivos. En el primer caso, la muestra de gas es acondicionadapara remover aerosoles, material particulado y otras sustanciasque pueden producir interferencia, antes de llevar la misma alinstrumento de medición. En el segundo caso, como lasmediciones son in-situ, no hay acondicionamiento de la misma,salvo algún filtrado.
Existen algunos procesos de combustión y situaciones que puedenlimitar la aplicabilidad del método, entre las que se pueden citar:a) Componentes corrosivos o altamente reactivos.b) Vacío elevado; flujo de gas a presión o temperatura elevada.
(15) Las condiciones de toma de muestra isocinética está dada en la norma ISO 9096/92 específica para la
determinación de material particulado.

69
c) Gases húmedos, que se encuentran en el punto de saturación opróximo y pueden contener gotas de agua.
d) Fluctuaciones en velocidad, temperatura o concentracióndebido a variaciones incontroladas en los procesos.
e) Estratificación del gas debido a la falta de mezcla de los gasesen la chimenea.
f) Mediciones realizadas usando dispositivos de controlambiental.
g) Bajo nivel de concentración de gases.
Principio
del
método
1. Ubicación del lugar de toma de muestra. Generalmente laconcentración a través de una sección de la chimenea esuniforme debido a la difusión y mezcla turbulenta del gas.Para ello el lugar de toma de muestra debe estar posicionado aun tercio de la mitad de camino de la chimenea, lejos de loscambios de dirección.
2. Se debe medir la temperatura y velocidad en el punto de tomade muestra.
3. La línea de extracción de muestra no debe estar compuestapor materiales que tengan propiedades de absorción de gasesque pueden afectar el tiempo de respuesta de la sección demedición. Asimismo, no debe haber corrosión o reaccionescon componentes de la mezcla.
4. Debe haber un apropiado calentamiento, secado y prueba depérdidas, según los casos.

70
Figura 5.5.1-1: Ejemplo de un sistema de toma de muestra y equipamiento para
acondicionamiento de la misma.

71
Método del centroide para toma de muestras y determinación de
velocidad para fuentes estacionarias.
EPA – 40 CFR, Pt. 60, App. A, Meth. 1 / 94
Aplicación y
limitaciones
El método es aplicable en la extracción de muestras de gasesfluyendo por tubos, chimeneas y conductos. No puede ser usadocuando:1) El flujo es ciclónico o en forma de remolinos,2) La chimenea tiene menos de 0,30 m de diámetro, ó un área de
la sección horizontal de la chimenea menor de 0,071 m2,3) El sitio de medición está ubicado a menos de dos diámetro de
la chimenea “arriba” (en el sentido del flujo) y menos demedio diámetro “abajo” (en el sentido del flujo) de un puntode perturbación del flujo.
Los requerimientos de este método deben ser tenidos en cuentaantes de construir una nueva chimenea a fin de instalaradecuadamente la toma de muestras.
Principio
del
método
Para lograr una medición representativa de las emisionescontaminantes y/o del caudal o flujo volumétrico total de unafuente estacionaria, se selecciona un sitio de toma de muestra omedición donde la corriente del efluente está fluyendo en unadirección conocida y se divide la sección de las chimenea en áreasiguales. Se localiza luego un punto en el centroide de cada una delas áreas predeterminadas donde se harán la toma de muestra.
Selección del sitio de medición. La toma de muestra o lamedición de velocidad se realiza en un sitio localizado al menosocho (8) diámetros de chimenea “arriba” (en el sentido del flujo)y dos (2) diámetros “abajo” (en el sentido del flujo) de cualquierperturbación tal como una curva, una expansión o contracción, dela chimenea o una llama visible. Si la sección de la chimenea esrectangular, el diámetro equivalente se calcula comoD=2LW/(L+W) donde L = longitud y W = ancho de la sección.Cuando no se pueden cumplir estas condiciones se dispone de unprocedimiento alternativo para determinar la aceptabilidad de lalocalización de toma de muestra, donde se debe medir el ángulode inclinación o pendiente del flujo de gas y el ángulo dedesviación lateral ambos respecto a una línea paralela al eje de lachimenea, en 40 o más puntos .
Determinación del número de puntos en la sección transversal.Cuando se cumplen las condiciones de selección del sitio demedición antes dadas, el número mínimo de puntos de toma demuestra en forma transversal son:1) Para chimeneas con diámetro (o diámetro equivalente) mayor

72
de 0,61 m, doce (12) puntos.2) Para chimeneas con diámetro circular entre 0,30 y 0,61 m,
ocho (8) puntos.3) Para Chimeneas rectangulares con diámetro equivalente entre
0,30 y 0,61 m, nueve (9) puntos.Cuando no se cumplen las condiciones de 8 y 2 diámetros, elnúmero mínimo de puntos es determinado a través de un grupo defiguras, según se mida material particulado o velocidad o caudal(pero no material particulado).Asimismo, se determina la disposición de los puntos de toma demuestra en la sección transversal (ver Figura V.5.1-2).
Figura 5.5.1-2: Muestra de la selección de puntos de toma de muestra por el método del
centroide en la sección transversal de la chimenea

73
Determinación de velocidad y caudal volumétrico del gas dentro de la
chimenea (Tubo Pitot tipo S).
EPA – 40 CFR, Pt. 60, App. A, Meth. 2 / 94
Aplicación y
limitaciones
El método se aplica para la medición de velocidad promedio delflujo del gas y para cuantificar su caudal.Este procedimiento no se aplica a lugares de mediciones que nocumplan el criterio del Método EPA – 40 CFR, Pt. 60, App. A,Meth. 1 . Tampoco puede aplicarse para medición directa de flujosde gas ciclónicos o en forma de remolinos. Cuando existancondiciones no aceptables se debe usar métodos alternativos.
Principio
del
método
La velocidad promedio del gas en la chimenea es determinadadesde la densidad del gas y desde mediciones de la velocidadpromedio con un tubo Pitot (16) tipo S (Stausscheibe o tipoinvertido). Se puede emplear un tubo de Pitot estándar.En la Figura V.5.1-3 se muestra un esquema del tubo Pitot tipo Sjunto con el manómetro y en la Figura V.5.1-4 se muestranalgunas propiedades constructivas del mismo. El tubo Pitot debeser metálico (por ejemplo de acero inoxidable), el diámetroexterno (Dt) entre 0,48 y 0,95 centímetro. Se mide la temperaturadel fluido a la entrada del tubo de Pitot.
(16) Tubo de Pitot. Tubo en forma de L para le medición de velocidad de una corriente de líquido o gas. La boca
de la rama horizontal se coloca frente a la corriente y contra ella, y el fluido, empujado por la corriente, se
eleva cierta altura en la rama vertical. Esta elevación depende de la velocidad de acuerdo a la expresión
V = c √(2gH), donde H es la elevación del fluido en el tubo vertical, g la aceleración de la gravedad, c una
constante que se determina previamente con un medidor patrón y V la velocidad. Acoplando un tubo de Pitot
con otro piezométrico, que indica la presión estática, puede hallarse por diferencia la presión dinámica,
debida a la velocidad. Se suele combinar ambos con las dos ramas de un manómetro en U. Esto puede
lograrse también con el tubo Pitot tipo S..
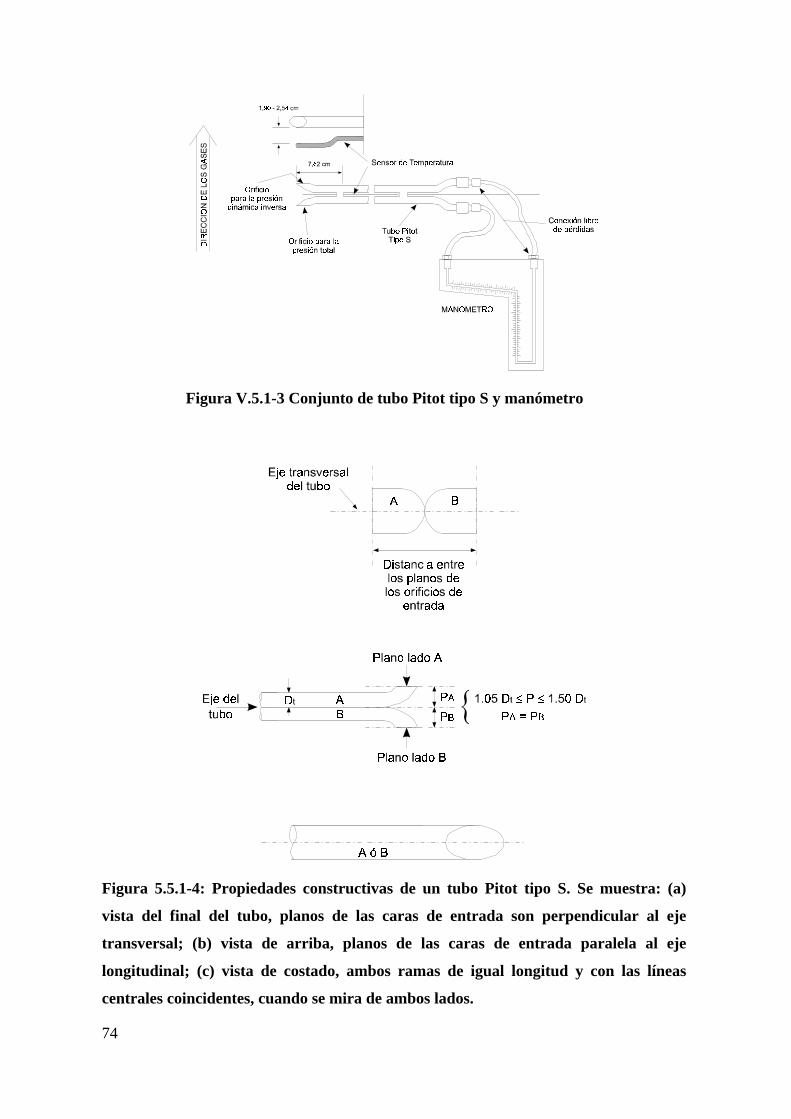
74
Figura V.5.1-3 Conjunto de tubo Pitot tipo S y manómetro
Figura 5.5.1-4: Propiedades constructivas de un tubo Pitot tipo S. Se muestra: (a)
vista del final del tubo, planos de las caras de entrada son perpendicular al eje
transversal; (b) vista de arriba, planos de las caras de entrada paralela al eje
longitudinal; (c) vista de costado, ambos ramas de igual longitud y con las líneas
centrales coincidentes, cuando se mira de ambos lados.

75
Métodos para la toma de muestra en gas de escape.
JIS - K-0095 / 88
Aplicación e
interferencias
Especifica el método de toma de muestra para análisis de gasescomponentes de los gases de salida por chimeneas, conductos,etc. productos del quemado de combustibles u otros componentesen centrales, industrias, en cualquier clase de procesosproductivos u otros.

76
5.5.2 Métodos de determinación de concentración de contaminantes en
chimenea
5.5.2.1 Métodos de análisis externo
1. Dióxido de azufre.
Método Thorin. /Peróxido de hidrógeno/ Perclorato de bario/.
EPA – 40 CFR, Pt. 60, App. A, Meth. 6/94;
ISO - 7934 / 89.
Aplicación e
interferencias
Es aplicable a la determinación de concentración de dióxido deazufre (SO2), con presencias despreciables de trióxidos de azufre(SO3) y ácido sulfúrico (SO4H2). Posible interferencia sonamoníaco (NH3) libre, cationes solubles en agua y fluoruros. Loscationes y fluoruros son removidos previamente filtrando con lanade vidrio y haciendo burbujear la muestra en isopropanol (CH3-CH.OH-CH3). El amoníaco (NH3) libre interfiere reaccionandocon el SO2, formado iones de sulfito (SO3
-) que reacciona con elindicador. Para eliminar la interferencia se calienta la probetatoma muestra a 275 °C y se coloca un filtro de alta eficiencia.
Principio
del
método
El método se basa en la absorción del gas de muestra en unasolución de peróxido de hidrógeno (H2O2) dentro de un períodoespecífico, resultando la formación de ácido sulfúrico (SO4H2). Seajusta el pH de la solución a 3,5 con solución de hidróxido desodio (NaOH) o solución de ácido perclórico (HClO4), según serequiera. Se determina la concentración másica de iones sulfatopresentes en la solución por titulación con solución de percloratode bario (Ba[ClO4]2) usando Thorin (17) como indicador; de ellose calcula la concentración de dióxido de azufre. Para eliminar elmaterial particulado se coloca un filtro antes de la soluciónabsorbente.El tiempo de toma de muestra normal es de 30 min.
(17) Thorin, también conocido como Thoron o Thoronol, es una sal de sodio del ácido 4-{(2-arsonophenyl)-azo}-3-hydroxy-naphthalene-disulfonic.

77
Intervalo
de
aplicación
El límite mínimo de detección del método se ha determinado en3,4 mg de SO2 por m3
de aire seco en condiciones estándar (18). Sibien no se a establecido un límite superior, las pruebas hanmostrado que pueden determinarse eficientementeconcentraciones tan elevadas como 80.000 mg/m3 cuando serecoge usando dos pequeños tubos de burbujeo, cada unoconteniendo 15 mililitros de peróxido de hidrógeno (H2O2) al 3 %,a una velocidad de 1 litro por minuto durante 20 minutos.Basado sobre un cálculo teórico, el límite superior de detección enuna muestra de 20 litros es alrededor de 93.300 mg/m3 .
Figura 5.5.2-1: Tren de toma de muestra para la determinación de concentración de
SO2
(18) Presión y temperatura estándar absoluta de 760 mm Hg (101,3 kPa) y 293 °K según norma EPA. La
norma ISO especifica 273,1 °K .

78
Método de precipitación y titulación con Arsenazo III
K-0103/88
Aplicación e
interferencias
El método permite la determinación de concentración de óxidosde azufre (SO2 + SO3) en la muestra de gas.
Principio
del
método
Los óxidos de azufre de la muestra son absorbidos por el peróxidode hidrógeno (H2O2) formando ácido sulfúrico (SO4H2). Con elagregado de 2-propanol (ó alcohol isopropílico; CH3-CHOH-CH3)y ácido acético (CH3-COOH), se titula la solución de acetato debario ([CH3-COO]2Ba) usando arsenazo III como indicador. Elácido sulfúrico (SO4H2) reacciona con el acetato de bario ([CH3-COO]2Ba) formando precipitado de sulfato de bario (BaSO4). Eneste proceso los iones de Ba en exceso reaccionan con As III yforman un complejo de color azul. La concentración de óxidos deazufre se calcula a partir de la cantidad de acetato de bario ([CH3-COO]2Ba) titulado.
Intervalo
de
aplicación
Para concentraciones de SOx de 140 a 700 ppm en la muestra degas.

79
Método de absorción infrarrojo no dispersivo
EPA-40 CFR, Pt. 60, App. A, Meth. 6C / 94
Aplicación e
interferencias
El método se aplica a la determinación de concentración dedióxido de azufre (SO2) en la muestra de gas.
Principio
del
método
Una muestra de gas es extraída en forma permanente de lachimenea y una porción de la misma es transportada a uninstrumento analizador de gases para la determinación de laconcentración de SO2 usando un analizador infrarrojo nodispersivo (Ver Sección 5.6, Apartado 8). El SO2 absorberadiación infrarroja a 7.300 nm de longitud de onda. Se aplicacuando el efecto de coexistencia de humedad, CO2 ehidrocarburos es despreciable o removible. Para tiempo deextracción de muestra de 1 h, se deben hacer mediciones cada 1min. o un mínimo de 30 mediciones. Para tiempo de extracción demuestra mayor, las mediciones se deben realizar cada 2 min. o unmínimo de 96 mediciones.
Intervalo
de
aplicación
Para concentraciones de SOx de 0 a 500 ppm en la muestra degas.
El intervalo analítico está determinado por el diseño del
instrumento. Una porción del mismo es seleccionado eligiendo el
valor de fondo de escala del sistema de monitoreo; esto se realiza
de forma tal que la concentración del gas contaminante
equivalente a la emisión estándar no sea menor que el 30 % del
fondo de escala.
Sensibilidad Límite mínimo detectable depende del intervalo analítico, la señal
a fondo de escala del instrumento y la relación señal ruido del
sistema de medición. Para un sistema bien diseñado, el mínimo
detectable será menor que el 2 % del fondo de escala.

80
Método de electrólisis a un potencial controlado
JIS - K-7981/84
Aplicación e
interferencias
Se aplica a la determinación de la concentración de dióxido deazufre (SO2) cuando el efecto de coexistencia con ácido sulfídrico(SH2), dióxido de nitrógeno (NO2), hidrocarburos y ozono (O3) esdespreciable o removible.
Principio
del
método
Una muestra de gas es introducida en una solución electrolítica através de una membrana permeable. El SO2 que difunde en elelectrolito es electrolizado a un potencial constante. Laconcentración de SO2 es calculada midiendo la corrienteelectrolítica entre los electrodos.La membrana permeable prevé la salida y evaporación delelectrolito y también reduce el efecto de componentes que puedanproducir interferencia. El electrodo de trabajo es de platino (Pt),oro (Au) o paladium (Pd) y el de medición, que le da al primero elpotencial de oxidación que es necesario para la electrólisis apotencial controlado, debe ser elegido para hacer la mejorcombinación con aquel, y puede ser de platino, níquel ocompuesto de níquel, plomo (Pb) o compuesto de plomo. Elelectrolito es una solución aproximadamente ½ mol / l soluciónde ácido sulfúrico (SO4H2). (Ver Sección 5.6, Apartado 10)
Intervalo
de
aplicación
Para concentraciones de SOx de 0 a 2000 ppm en la muestra degas.

81
Figura 5.5.2-2: Componentes de un analizador de electrólisis a un potencial controlado

82
2. Niebla de ácido sulfúrico (H2SO4)
Método de titulación con bario-Thorin
EPA-40 CFR, Pt. 60, App. A, Meth. 8 / 94
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deniebla de ácido sulfúrico (SO4H2) (incluyendo trióxido de azufre(SO3), y en la ausencia de otro material particulado) y dióxido deazufre (SO2) emitidos por fuentes estacionarias. Posible agentesde interferencia del método son fluoruros, amoníaco (NH3) libre, yanilina dimetílica (ó dimetilanilina C6H5N(CH3)2).
Principio
del
método
Se extrae una muestra de gas isocinéticamente de la chimenea. Laniebla de ácido sulfúrico (SO4H2) (incluyendo trióxido de azufre)y el dióxido de azufre son separados y la concentración de ambasfracciones son determinadas separadamente por el método detitulación con bario-Thorin.La velocidad de toma de muestra no debe ser mayor a 0,030 m3
por minuto.
Intervalo
de
aplicación
El límite menor de detección es de 0,05 mg/m3 para trióxido deazufre y 1,2 mg/m3 para dióxido de azufre. No se ha establecidoun límite superior, pero en base a cálculos teóricos, el límitesuperior de concentración de dióxido de azufre, para 200 ml desolución de 3 % de peróxido de hidrógeno (H2O2), es de 12.500mg/m3.

83
3. Óxidos de nitrógeno (NOx)
Método fenol disulfónico (PDS).
EPA – 40 CFR, Pt. 60, App. A, Mth. 7 / 94
JIS-K-0104/84
Aplicación e
interferencias
Se aplica a la determinación de los óxidos de nitrógeno enconjunto (NO+NO2).
Principio
del
método
El NO es oxidado a NO2 por: a) el oxígeno del aire (dejandotranscurrir el menos 16 horas), b) adicionando oxígeno a lamuestra , c) adicionado ozono a la muestra.Los óxidos de nitrógeno contenidos en la muestra de gas, enpresencia de oxígeno, son convertidos en ion nítrico (NO3
-) porabsorción en una solución de ácido sulfúrico (H2SO4) y peróxidode hidrógeno (H2O2) [agua oxigenada]. La solución así obtenidase hace reaccionar con ácido fenol disulfónico[C6H3OH(SO3H)2] y la concentración de óxidos de nitrógeno,medida como NO2, se determina midiendo la absorbancia dellíquido con un espectrofotómetro (absorbancia a 410 nm)previamente calibrado con muestra patrón.
Intervalo
de
aplicación
EPA: Para concentraciones de 2 a 400 mg NOx (como NO2 ) pormetro cúbico seco estándar (20 °C (293 °K) y 760 mm Hg), sintener que diluir la muestra.JIS: Para concentraciones de óxidos de nitrógeno en la muestrade 10 a 4.900 ppm en volumen, según el tamaño de la muestra .
Figura 5.5.2-3: Tren de toma de muestra para la determinación de NOx

84
Método de cromatografía iónica.
EPA – 40 CFR, Pt. 60, App. A, Mth. 7A / 94
Aplicación e
interferencias
Se aplica a la determinación de los óxidos de nitrógeno enconjunto (NO+NO2).
Principio
del
método
El NO es oxidado a NO2 por: a) el oxígeno del aire (dejandotranscurrir el menos 16 horas), b) adicionando oxígeno a lamuestra , c) adicionado ozono a la muestra.Los óxidos de nitrógeno contenidos en la muestra de gas, enpresencia de oxígeno, son convertidos en ion nítrico (NO3
-) porabsorción en una solución de ácido sulfúrico (H2SO4) y peróxidode hidrógeno (H2O2) [agua oxigenada]. Los nitrato (NO3-) sonmedidos con un cromatógrafo iónico. El cromatógrafo debetener una columna capaz de separa iones de nitrato de iones desulfato u otros iones presentes y otra columna supresora deaniones (Ver Sección 5.6, Apartado 3).
Intervalo
de
aplicación
El intervalo analítico de aplicación es de 125 a 1.250 mg NOx
/m3 como NO2 (65 a 655 ppm); concentraciones mayorespueden se analizadas diluyendo la muestra. El límite más bajode detección es de 19 mg/m3 (10 ppm), pero puede variarfuertemente entre instrumentos.

85
Método de espectrometría ultravioleta.
EPA – 40 CFR, Pt. 60, App. A, Mth. 7B / 94
Aplicación e
interferencias
Se aplica a la determinación de los óxidos de nitrógeno enconjunto (NO+NO2). Se emplea para la medición de óxidos denitrógeno emitidos en plantas de ácido nítrico (HNO3).
Principio
del
método
El NO es oxidado a NO2 por: a) el oxígeno del aire (dejandotranscurrir el menos 16 horas), b) adicionando oxígeno a lamuestra , c) adicionado ozono a la muestra.Los óxidos de nitrógeno contenidos en la muestra de gas, enpresencia de oxígeno, son convertidos en ion nítrico (NO3
-) porabsorción en una solución de ácido sulfúrico (H2SO4) y peróxidode hidrógeno (H2O2). Los óxidos de nitrógeno, excepto óxidonitroso, son medidos por absorción de la luz ultravioleta en lalongitud de onda de 210 nm. (Ver Sección 5.6, Apartado 7).
Intervalo
de
aplicación
Para concentraciones de NOx (como NO2) entre 57 y 1.500mg/m3 estándar (30 a 786 ppm).

86
Método de Quimioluminiscencia
EPA – 40 CFR, Pt. 60, App. A, Mth. 7 E / 94
JIS – B- 7982 / 84
Aplicación e
interferencias
Se aplica para la determinación de monóxido y dióxido denitrógeno (NO y NO2) y del conjunto de óxidos de nitrógeno(NOx).
Principio
del
método
Determinación de NO. Se hace incidir la muestra de aire en lacámara de reacción del analizador donde se mezcla con un excesode ozono (O3). El NO y el ozono da NO2 excitado que al decaeremite luz. Se filtra la radiación emitida (la intensidadquimioluminiscente) (Ver Sección 5.6, Apartado 6.2),proporcional a la cantidad de NO presente, con un filtro ópticoselectivo y se convierte la radiación filtrada en una señal eléctricapor medio de un tubo fotomultiplicador, midiendo la intensidad deluz a longitudes de onda entre 590 y 875 nanometro.Determinación de NO2. La muestra se pasa por un convertidor(por eje. horno a 400 °C constante) para reduce el NO2 amonóxido antes de hacerla entrar a la cámara de reacción. Laseñal eléctrica obtenida en este caso es proporcional a la cantidadtotal de óxidos de nitrógeno presente en la muestra. La cantidad dedióxido de nitrógeno se obtiene por diferencia entre este valor y elobtenido en la medición de NO.El volumen total se determina midiendo el caudal y el tiempo decolección.
Intervalo
de
aplicación
Se aplica para la determinación de monóxido de nitrógeno yóxidos de nitrógeno en el intervalo de concentraciones de 0 a2.000 ppm.
El intervalo analítico está determinado por el diseño delinstrumento. Una porción del mismo es seleccionado eligiendo elvalor de fondo de escala del sistema de monitoreo; esto se realizade forma tal que la concentración del gas contaminanteequivalente a la emisión estándar no sea menor que el 30 % delfondo de escala.
Sensibilidad Límite mínimo detectable depende del intervalo analítico, la señala fondo de escala del instrumento y la relación señal ruido delsistema de medición. Para un sistema bien diseñado, el mínimodetectable será menor que el 2 % del fondo de escala.

87
Método de electrólisis a un potencial controlado
JIS-B-7982/84
Aplicación e
interferencias
El método mide monóxido de nitrógeno (NO). El dióxido denitrógeno (NO2) se mide convirtiéndolo en monóxido.
Principio
del
método
Una muestra de gas se introduce en una celda electrolítica através de una membrana permeable al NO, éste se oxida porelectrólisis y genera una corriente electrolítica proporcional a suconcentración (Ver Sección 5.6, Apartado 10). La fuente depotencia debe proveer a los electrodos un voltaje continuoconstante, un potencial controlado. Las concentraciones de NO yNO2 son calculadas midiendo la corriente de electrólisis.
Intervalo
de
aplicación
Se aplica a la determinación de monóxido (NO) y óxidos denitrógeno (NO+NO2) en el intervalo de 0 a 2.000 ppm

88
4. Monóxido de carbón (CO)
Método de analizador infrarrojo no dispersivo
EPA – 40-CFR. Pt. 60, App.A, Meth 10/94
Aplicación e
interferencias
Se aplica a la determinación de concentración de monóxido decarbono (CO). Cualquier sustancia que tenga una fuerteabsorción en la zona de energía del infrarrojo interferirá enalguna manera; en particular H2O y CO2. Por eje., las razones dediscriminación de H2O y CO2 son 3,5 % de agua para 7 ppm COy 10 % de anhídrido carbónico para 10 ppm de CO,respectivamente para dispositivo midiendo en el intervalo de1.500 a 3.000 ppm. Para dispositivos midiendo en el intervalo de0 a 100 ppm, las razones de interferencias pueden ser 3,5 % H2Opara 25 ppm CO y 10 % CO2 para 50 ppm CO. El uso de trampade “silica gel” y “ascarite” puede aliviar los mayores problemasde interferencia.
Principio
del
método
Se extrae una muestra de gas y se analiza el contenido de COutilizando un analizador infrarrojo no dispersivo (AIND) tipoLuft o equivalente (Ver Sección 5.6, Apartado 8.1). Se empleaun tubo de Pitot, tipo S, junto con la toma de muestra para que lavelocidad de muestreo pueda ser regulada proporcional a lavelocidad del gas en la chimenea.
Intervalo
de
aplicación
De 0 a 1.000 ppm. La mínima concentración detectable es de 20ppm en el intervalo mencionado.
Precisión
y
exactitud
La precisión de la mayoría de los AIND es ≈ ± 2 % del fondo deescala.
La exactitud de la mayoría de los AIND es ≈ ± 5 %del fondo deescala después de la calibración

89
5. MATERIAL PARTICULADO
Método gravimétrico manual.
ISO - 9096 / 92.
EPA-40 CFR, Pt60, App.A, Meth.5 /94
JIS - Z-8808/92
Aplicación e
interferencias
Es un método de referencia para determinación de materialparticulado, que incluye todo material que condense arriba de latemperatura de filtrado, que puede ser empleado para calibrarmétodos continuos. La cantidad de material particulado es funciónde la temperatura y la presión, por lo que estas deben se medidas.
Principio
del método
La muestra de gas debe ser extraída isocinéticamente del conductopor donde sale el gas, haciéndola incidir sobre un filtro de fibra devidrio manteniendo la temperatura del conducto de extracción ydel filtro en el intervalo de 120±4 °C y una temperatura que seaespecificada para una aplicación particular; el contenido en elfiltro es luego secado. Se determina el peso de materialparticulado por diferencia con el peso del filtro previamentetarado y se calcula la concentración midiendo el volumen del gasde muestra extraído. Para asegurar que las muestras soncaracterísticas, se deben sacar las mismas de un número deposiciones preseleccionadas en la sección transversal delconducto.La posición y el número de los puntos de muestreo en conductosde secciones circulares y rectangulares y el cuidado y uso detubos Pitot para determinar la velocidad promedio del gas en elducto están dados en las normas correspondientes. Como ejemplose puede mencionar para una chimenea circular con diámetroentre 0,35 y 0,7 m se deben medir a lo largo de dos diámetros untotal de 5 puntos; para diámetro mayor de 2 m se deben medir 17puntos a lo largo de dos diámetros perpendiculares.El tiempo de toma de muestra no debe ser inferior a 3 minutos afin de minimizar los errores de ajuste del flujo.
Intervalo
de
aplicación
El método puede usarse para determinar concentraciones que vande 5 mg/m3 a 1x 104 mg m3. Para concentraciones debajo de 5mg/m3 , la inexactitud del método es mayor que ± 10%.

90
Figura 5.5.2-4: Tren de toma de muestra de material particulado con filtro fuera de la
chimenea

91
Método de filtrado en chimenea
EPA-40 CFR, Pt60, App.A, Meth. 17 / 94
Aplicación e
interferencias
Cuando la concentración del material particulado, dentro delintervalo normal de asociado con una fuente específica, es sabidoque es independiente de la temperatura, es deseable eliminar lossistemas de calefacción y tomar la muestra directamente a latemperatura de la chimenea. No es aplicable en chimeneas dondeel gas contengan gotas de líquido o esté saturado de vapor deagua. Tampoco debe usarse si la sección del tomador de muestraes mayor al 5 % del área de la sección de chimenea.
Principio
del
método
La muestra de gas es extraída isocinéticamente y el materialparticulado colectada sobre un filtro dentro de la chimenea a latemperatura de esta. La masa de material es determinadagravimétricamente después de secar la muestra. El filtro debe serrejilla de fibra de vidrio o un dedal de fibra, sin ligazón orgánico.
Intervalo
de
aplicación
Similar al método gravimétrico manual.
Figura 5.5.2-5: Se muestra el área bloqueada por la sonda y el portafiltro dentro de la
chimenea para la toma de material particulado.

92
Figura 5.5.2-6: Tren de extracción de muestra de material particulado con el filtro
dentro de la chimenea

93
6. Plomo Inorgánico (Pb)
Método de espectrometría de absorción atómica
EPA-40 CFR, Pt60, App.A, Meth. 12 / 94
Aplicación e
interferencias
Determinación de plomo (Pb) gaseoso y particulado.Concentración elevada de Cu puede interferir con el análisis de Pben la longitud de onda de 217,0 nm. Esta interferencia puedeeliminarse analizando la muestra a 283,3 nm.El análisis de Pb por absorción atómica es sensitivo a lacomposición química y a las propiedades físicas (viscosidad, pH)de la muestra, el denominado efecto matriz, que puede interferircon la medición.
Principio
del
método
La emisión de plomo gaseoso y particulado son extraídaisocinéticamente desde la fuente y colectada en un filtro (elparticulado) y en ácido nítrico (HNO3) diluido en un burbujeador(la parte gaseosa). Las muestras colectadas son digeridas ensolución de ácido nítrico y el nitrato de plomo analizado porespectrometría de absorción atómica usando una llama deacetileno/aire a longitudes de onda de 217,0 o 283,3 nm (VerSección 5.6, Apartado 1). El espectrómetro se calibra conmuestras patrones.
Intervalo
de
aplicación
Para una precisión analítica de ±10 por ciento, el límite más bajodel intervalo es de 100 µg. El límite superior puede serconsiderablemente extenso por dilución.
Precisión
y
exactitud
Sensibilidad típica para un cambio de 1 % en absorbancia (0,0044unidades de absorbancia) son 0,2 y 0,5 µg Pb/ml para las líneas de217,0 y 283,3, respectivamente.La precisión, medida por el coeficiente de variación, va de 0,2 a9,5 % relativa a la concentración de una medición media. En losanálisis de prueba realizados el intervalo de concentracionesfueron de 0,61 a 123,3 mg de Pb por m3 de gas.

94
Método polarográfico
JIS – K – 0097 / 97
Aplicación e
interferencias
Se aplica a la determinación de plomo (Pb) como materialparticulado contenido en la muestra de aire.
Principio
del
método
La muestra extraída isocinéticamente es pasada por un filtrodonde son retenidos los elementos. El tipo de filtro empleadodepende de la temperatura del gas en la chimenea. Debajo de 120°C se emplea papel de filtro de celulosa, entre 120 y 500 °C papelde filtro de fibra de vidrio y entre 500 y 1000 °C papel de filtro defibra de cuarzo.Según el contenido de sustancias orgánicas en la muestra, se trataesta para su análisis. Si no contiene sustancias orgánicas, se tratacon ácido nítrico (HNO3); si contiene pequeñas cantidades, setrata con mezcla de ácidos nítrico e clorhídrico (HCl) o ácidonítrico y peróxido de hidrógeno (H2O2); con gran cantidad desustancias orgánicas y carbón libre, se realiza incineración debajode 200 °C y tratamiento con ácido clorhídrico (HCl) y peróxido dehidrógeno (H2O2).Para la determinación de la concentración de plomo, se toma unpolarograma de ion Pb en solución de ácido nítrico (HNO3), en elintervalo de 0,2 –0,6 V. Luego se mide la altura de onda ocurridaa un potencial de media onda (potencial de pico) de Pb (alrededorde 0,44 V) y se determina la cantidad de plomo a través de lacurva de calibración previamente realizada (Ver Sección 5.6,Apartado 11).
Intervalo
de
aplicación
El limite de sensibilidad del método es más de 0,05 mg de Pbpara polarografía de corriente continua, más de 0,025 mg parapolarografía de corriente alterna y más de 0,005 mg parapolarografía de onda trapezoidal. La desviación estándar de larepetición es de 10 a 3 %.
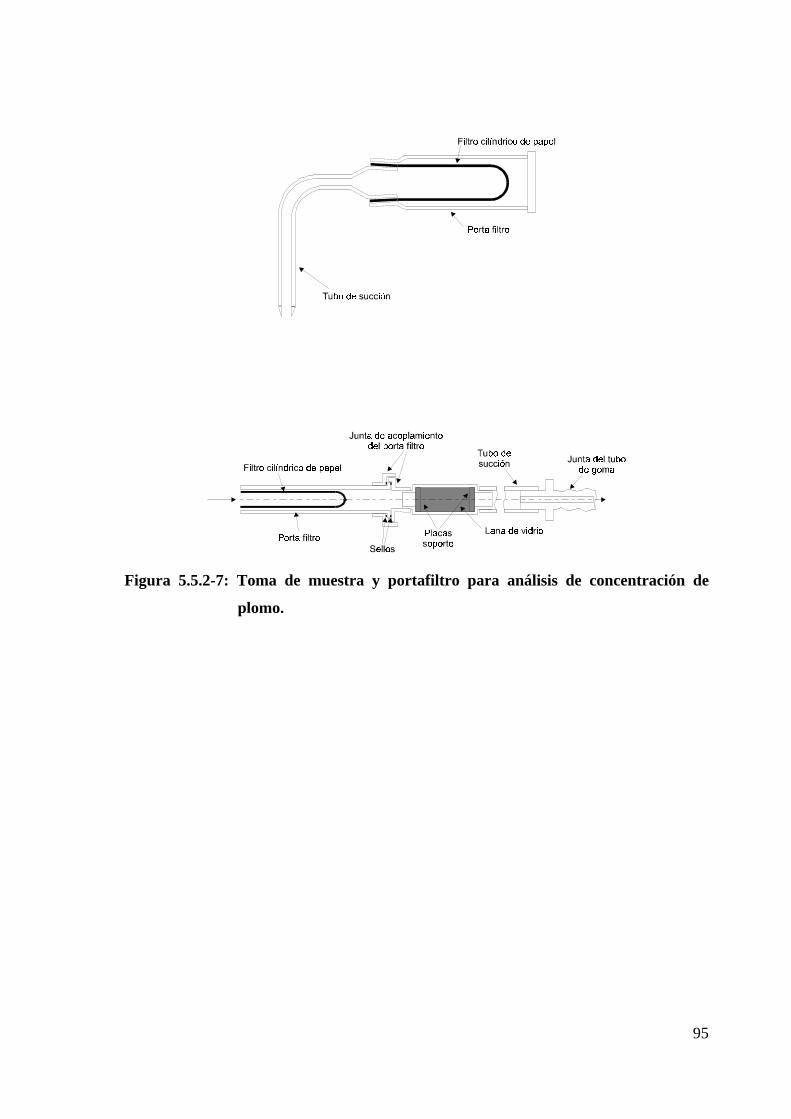
95
Figura 5.5.2-7: Toma de muestra y portafiltro para análisis de concentración de
plomo.

96
7. Azufre total en forma reducida (Atfr)
formado por el conjunto de compuestos: Acido Sulfídrico (S2h), Metil
mercaptano (Ch3sh), Dimetil sulfuro ([Ch3]2s), Disulfuro de Dimetilo
[(Ch3)2s2].
Método de separación por cromatografía gaseosa y detección con
fotometría de llama
EPA – 40 CFR, Pt. 60, App. A, Mth. 16 /94
Aplicación e
interferencias
Se aplica a la determinación de concentración de ácido sulfídrico(H2S), metil mercaptano (CH3SH), dimetil sulfuro ([CH3]2S) ydisulfuro de dimetilo [(CH3)2S2], conjunto conocidocolectivamente como “azufre total en forma reducida” (ATFR).Es un método semi - continuo.Hay diversos efectos de interferencia producidos por: humedadcondensada, CO y CO2, material particulado y SO2.
Principio
del
método
Una muestra de gas es extraída desde la fuente de emisión y unaalícuota es analizada separando los componentes concromatografía en fase gaseosa (Ver Sección 5.6, Apartado 2),separados adecuadamente mediante procesos de evaporación, ydetectados con fotometría de llama (llama de hidrógeno de bajatemperatura que calienta el S y se observa la luz emitida confotómetro de filtro en la longitud de 394 nm) (Ver Sección 5.6,Apartado 5) .
Intervalo
de
aplicación
El intervalo analítico varía con el tamaño de la muestra.Típicamente, este puede extenderse desde 0,1 a 100 ppm usandotamaños de muestra de 10 a 0,1 ml.
Sensibilidad Usando un tamaño de muestra de 10 ml la mínima concentracióndetectable es aproximadamente de 50 ppb.

97
Método de conversión a SO2 y análisis por Thorin. /Peróxido de
hidrógeno/ Perclorato de bario/.
EPA – 40 CFR, Pt. 60, App. A, Mth. 16A /94
Aplicación e
interferencias
Se aplica a la determinación de concentración de ácido sulfídrico(H2S), metil mercaptano (CH3SH), dimetil sulfuro ([CH3]2S) ydisulfuro de dimetilo [(CH3)2S2], conjunto conocidocolectivamente como “azufre total en forma reducida” (ATFR).Hay interferencias con sulfuro de carbonilo (se oxida a SO2) ymaterial particulado, primariamente carbonato de calcio queincrementa el pH del burbujeador con citrato produciéndose laabsorción de H2S. Además, el carbonato de calcio en la soluciónde peróxido de hidrógeno (H2O2) produce precipitación de ionessulfato.La muestra de gas debe tener por lo menos 1 % de oxígeno paraproducir la oxidación completa del conjunto de compuestosATFR a dióxido de azufre.
Principio
del
método
La muestra de gas es extraída de la chimenea y se hace pasar porun burbujeador con solución reguladora de pH (buffer) decitrato(19) a fin de separar selectivamente el SO2. El conjunto decompuestos ATFR son oxidados térmicamente a SO2, colectandoéste sobre peróxido de hidrógeno (H2O2) como sulfato ydeterminando la concentración másica de iones sulfato portitulación con solución de perclorato de bario (Ba[ClO4]2)usando Thorin como indicador.
Intervalo
de
aplicación
El menor límite de medición de concentración es de 0,1 ppm deSO2 cuando el caudal de muestreo es de 2 litros/min. durante 3 hy de 0,3 ppm para el mismo caudal durante 1 h. El límitesuperior de concentración excede los niveles de concentracióndel conjunto de compuestos ATFR que generalmente seencuentra en los gases emitidos por chimenea.
(19) Ácido cítrico (CH2-HOC-CH2)-(COOH)3 - -citrato de sodio.

98
Método de conversión a SO2 y análisis por cromatografía gaseosa y
detección fotométrica de llama
EPA – 40 CFR, Pt. 60, App. A, Mth. 16B /94
Aplicación e
interferencias
Se aplica a la determinación de concentración de ácido sulfídrico(H2S), metil mercaptano (CH3SH), dimetil sulfuro ([CH3]2S) ydisulfuro de dimetilo [(CH3)2S2], conjunto conocidocolectivamente como “azufre total en forma reducida” (ATFR).La muestra de gas debe tener por lo menos 1 % de oxígeno paraproducir la oxidación completa del conjunto de compuestosATFR a dióxido de azufre. Produce interferencia sulfuro decarbonilo (COS), material particulado (sobre todo carbonato decalcio [CO3Ca]), CO, y CO2.
Principio
del
método
La muestra de gas es extraída de la chimenea y se hace pasarpor un burbujeador con solución reguladora de pH (buffer) decitrato a fin de separar selectivamente el SO2. El conjunto decompuestos ATFR son oxidados térmicamente a SO2. Este esanalizado por cromatografía gaseosa (Ver Sección 5.6, Apartado2) realizando la detección fotométrico de llama (llama dehidrógeno de baja temperatura que calienta el S y se observa laluz emitida con fotómetro de filtro en la longitud de 394 nm)(Ver Sección 5.6, Apartado 5).
Intervalo
de
aplicación
Usando un tamaño de muestra de 1 ml para el cromatógrafogaseosos, el límite máximo de determinación de concentraciónes de 10 ppm. Este límite se expande por dilución del gasmuestra antes del análisis. Para fuentes con niveles de emisiónentre 10 y 100 ppm, los intervalos de medición pueden ser mejorextendidos reduciendo el tamaño de la muestra.

99
8. Compuestos orgánicos gaseosos
Método de cromatografía gaseosa.
EPA – 40 CFR, Pt. 60, App. A, Meth. 18 / 94
Aplicación e
interferencias
El método se aplica al análisis de aproximadamente 90 porciento del total de compuestos orgánicos emitidos por laindustria. No incluye técnicas para identificar y medir trazas decompuestos orgánicos, tales como los que se encuentran en elaire de los edificios o determinación de pérdidas.El método no determina compuestos que: 1) son polímeros(moléculas de elevado peso), 2) son polimerizados antes delanálisis, 3) tienen una presión de vapor muy baja en la chimenea.
Principio
del
método
Los principales compuestos orgánicos de una mezcla de gasesson separados por cromatografía gaseosa (Ver Sección 5.6,Apartado 2) y cuantificados individualmente por el método dellama ionizante, fotoionización, captura electrónica u otroprincipio apropiado de detección.Los tiempos de retención de cada compuesto por separado soncomparados con aquellos de compuestos conocidos bajoidénticas condiciones. Por lo tanto, el análisis confirma laidentidad y concentración aproximada de las emisionesorgánicas. Con esta información, el analista prepara o compramezclas de estándares disponibles comercialmente para calibrarel cromatógrafo gaseoso bajo condiciones idénticas a las de lasmuestras. Se determina, además, la necesidad de diluir lasmuestras para evitar la saturación del detector, de filtrar el gaspara eliminar el material particulado y prevenir la condensaciónde humedad.
Intervalo
de
aplicación
El valor más bajo del intervalo de aplicación del método estádeterminado por el sistema de toma de muestra; se puede usarabsorbentes para concentrar la muestra y bajar así el límite dedetección debajo de 1 ppm. El límite superior está gobernado porla saturación del detector del cromatógrafo gaseoso o lasobrecarga de la columna; se puede extender diluyendo lamuestra con un gas inerte. El límite superior también puede sergobernado por condensación de compuestos de elevado punto deebullición.

100
Método de analizador infrarrojo no dispersivo
EPA – 40 CFR, Pt. 60, App. A, Meth. 25B / 94
Aplicación e
interferencias
Se aplica a la medición de concentración gaseosa orgánica total devapores consistentes primariamente en alcanos (hidrocarburosparafínicos). (Otros compuestos orgánicos pueden ser medidosusando el procedimiento general de este método, utilizando el gasde calibración apropiado y un analizador ubicado en la banda deabsorción correspondiente). La concentración es expresada entérminos de propano (u otro gas apropiado de calibración) o entérminos de carbón.
Principio
del
método
La muestra de gas es obtenida a través de una línea calentada deextracción de muestra y pasada, si es necesaria, a través de unfiltro de fibra de vidrio, hacia un analizador infrarrojo nodispersivo (Ver Sección 5.6, Apartado 8.1). Los resultados sonexpresados como concentración de volumen equivalente del gasde calibración o como carbono equivalente.
Método de determinación de gases orgánicos totales excepto metano,como carbonoEPA – 40 CFR, Pt. 60, App. A, Meth. 25 / 94
Aplicación e
interferencias
El método se aplica a la medición de compuestos orgánicosvolátiles, así como gases orgánicos totales sin metano (CH4),expresados como carbón. El material particulado orgánico puedeinterferir con el análisis por lo que se requiere un filtrado previo.
Principio
del
método
Una muestra es extraída de la chimenea a una tasa constante através de un filtro calentado y una trampa enfriada decondensación, por medio de la succión con un tanque evacuado detoma de muestra. El contenido orgánico de la fracción condensadaen la trampa es oxidada a CO2 y colectado cuantitativamente elefluente en un frasco o tanque evacuado; luego una porción de CO2
es convertido a CH4 y medido con un analizador con detector dellama ionizante (DLLI) (Ver Sección 5.6, Apartado 4). La fracciónde compuestos orgánicos de la muestra que queda en el tanqueevacuado es medido inyectando una porción a la columna de uncromatógrafo gaseoso para separar los gases orgánicos sin metano(GOSM) del CO, CO2 y CH4. Los gases orgánicos sin metano sonoxidados a CO2 , reducidos a CH4 y medidos con DLLI. De estamanera se elimina la respuesta variable del DLLI asociados condiferentes tipos de compuestos orgánicos.

101
Cuando está presente CO2 y vapor de agua en la chimenea, puedenproducir una influencia positiva en la muestra, dependiendo ésta dela concentración de los mismos. Como una guía general, si elproducto de la concentración de CO2, como por ciento en volumen,por la concentración de vapor es menor que 100, la influencia esdespreciable.La medición directa del efluente con un DLLI puede ser apropiadapara una caracterización previa del flujo de gas y conocer si larespuesta del detector es adecuada para los compuestos orgánicosde la muestra. El método de DLLI puede ser aplicado para ladeterminación de la concentración másica de la estructuramolecular total de la emisión orgánica bajo las siguientescondiciones: 1) Cuando se conoce la existencia de sólo uncompuesto, 2) Cuando los compuestos orgánicos consisten sólo dehidrógeno y carbón, 3) Cuando los porcentajes relativos de loscompuestos son conocidos o pueden ser determinados y sonconocidas las respuestas de DLLI a los compuestos, 4) Cuandoexiste una mezcla consistente de los compuestos antes y despuésdel control de emisión y sólo las concentraciones relativas puedenser determinadas, 5) Cuando el método DLLI puede ser calibradocon masas estándares de los compuestos emitidos .En situaciones donde es requerido un análisis cualitativo/cuantitativo del afluente, se puede aplicar un sistema decromatografía gaseosa con detector de llama ionizante. Sinembargo, para emisiones con una gran cantidad de compuestosorgánicos, el tiempo y el gasto con este método pueden ser muygrande.
Intervalo
de
aplicación
El mínimo detectable con el método es de 50 ppm como carbón.

102
9. Fenoles (C6H5OH)
Método de absorciometría con 4-aminoantipirina
JIS - K-0086 / 83
Aplicación e
interferencias
Se aplica a la determinación de concentración de fenoles(C6H5OH). Cuando hay presente gases oxidantes como cloro,bromo, etc., y gases reductores como dióxido de azufre, sulfurode hidrógeno, etc., se generan errores negativos.
Principio
del
método
El gas de muestra es absorbido en solución de hidróxido desodio (NaOH). Después de ajustar el pH a 10±0,2, se adicionansoluciones de 4-aminoantipirina y de ferrocianuro de potasio([Fe(CN)6]K4), en ese orden, se mide la absorbancia en 510 nmde la solución roja obtenidas y se calcula la concentración defenoles.
Intervalo
de
aplicación
Concentración de muestra al cual es aplicado el método es de:1 a 20 ppm cuando la muestra de gas es de 10 l..
Método de absorciometría ultravioleta.
JIS - K-0086 / 83
Aplicación e
interferencias
Se aplica a la determinación de concentración de fenoles(C6H5OH). Se genera errores positivos si hay presentecompuestos que tengan espectro de absorción con longitud deonda entre 220 y 350 nm tales como carburos hidrogenadosaromáticos.
Principio
del
método
Los gases de muestra son absorbidos en agua para colectar losfenoles. La absorbancia de radiación ultravioleta de la soluciónes medida cerca de la longitud de onda de 270 nm con unespectrofotómetro y con ello se calcula la concentración de losfenoles. (Ver Sección 5.6, Apartado 7).
Intervalo
de
aplicación
Concentración de fenoles en la muestra al cual es aplicado elmétodo es de 1 a 50 ppm cuando la muestra de gas es de 10 l.

103
Método de cromatografía gaseosa con detector de llama de hidrógeno
ionizante.
JIS - K-0086 / 83
Aplicación e
interferencias
Se aplica a la determinación de concentración de fenoles(C6H5OH).
Principio
del
método
Los fenoles contenidos en el gas de muestra son absorbidos ensolución de hidróxido de sodio (NaOH), se acidifica la misma yse extraen con acetato de etilo [(CH3-COO)C2H5]. Con uncromatógrafo de gases con detector de llama de hidrógenoionizante se determina la concentración de fenoles (Ver Sección5.6, Apartados 2 y 4).
Intervalo
de
aplicación
Concentración de fenoles en la muestra al cual es aplicado elmétodo es de 1 a 300 ppm cuando la muestra de gas es de 10 l ,extraída con solvente y determinada a 150 °C usando carburohidrógeno saturado en fase líquida y de 10 a 2000 ppm cuandola muestra de gas es tomada por 1 a 3 ml como gas.
.

104
10. Formaldehído (HCHO)
Método absorciometría AHMT (4-amino-3-hydrazino-5-mercapto-1,2,4-
triazol)
JIS - K-0303 / 93
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deformaldehido (HCHO) en muestra de gas. La coexistencia deiones sulfito (SO3
-) y nitrito (NO2-) no son impedimento para el
método. Otros aldehidos presentes no son obstáculos si laconcentración es hasta alrededor del doble de la del formaldehido.
Principio
del
método
El formaldehído de la muestra de gas es absorbido en una soluciónde ácido bórico haciendo pasar el gas con un caudal de 1 l / min..Se debe medir el volumen de muestra de gas, la temperatura y lapresión. La muestra luego se alcaliniza con hidróxido de sodio(NaOH), se le agrega solución AHMT para colorarlo y soluciónperiodato de potasio (KIO4). Se coloca una porción de la soluciónen la celda de absorción y se mide su absorbancia en la longitudde onda alrededor de 550 nm, usando agua como solución dereferencia. El equipo se calibra con soluciones patrones deformaldehido.
Intervalo
de
aplicación
Concentración de muestra al cual es aplicado el método es de 0,3a 50 ppm cuando es tomada 20 l de la muestra de gas para lasconcentraciones bajas y 2 l para las concentraciones altas.
Método absorciometría con ácido cromotrópico
Acido 4,5-dihidroxi-2,7-naftalen disulfónico
JIS - K-0303 / 93
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deformaldehido (HCHO) en muestra de gas. La coexistencia deotros aldehidos dan un ligero error positivo y la de etanol,alcoholes de moléculas grande, hidrocarburos aromáticos,olefinas (hidrocarburos saturados) y ciclohexanos dan un ligeroerror negativo. La coexistencia de fenol con concentración hasta8 veces la del formaldehido da errores negativos de 10 a 20 %.

105
Principio
del
método
El formaldehido de la muestra de gas es absorbido en unasolución de bisulfito de sodio. A la solución de muestra se leagrega ácido cromotrópico para colorarlo, se acidifica con ácidosulfúrico (SO4H2) y se coloca una muestra en la celda deabsorción. Se mide su absorbancia con la longitud de ondaalrededor de 580 nm, usando agua como solución de referencia.El equipo se calibra con muestras patrones de formaldehido.
Intervalo
de
aplicación
El intervalo de concentración de muestra al cual es aplicado elmétodo es de 0,4 a 60 ppm cuando es tomada 20 l de la muestrade gas para las concentraciones bajas y 2 l para lasconcentraciones altas.
Método de cromatografía gaseosa con detector de llama de hidrógeno
ionizante
JIS - K-0303 / 93
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deformaldehido (HCHO) en muestra de gas.
Principio
del
método
El formaldehido de la muestra de gas es absorbido en unasolución de 2,4-dinitrofenilhidrazona, se extrae con cloroformo(CHCl3), se lo introduce en el cromatógrafo gaseoso y sedetermina la concentración con un detector de llama dehidrógeno ionizante previamente calibrado con muestraspatrones (Ver Sección 5.6, Apartados 2 y 4).
Intervalo
de
aplicación
Concentración de muestra al cual es aplicado el método es de 0,1a 100 ppm cuando es tomada 10 l de la muestra de gas.

106
Método de cromatografía líquida con detector de espectrometría
ultravioleta
JIS - K-0303 / 93
Aplicación e
interferencias
El método es aplicable a la determinación de concentración deformaldehido (HCHO) en muestra de gas.
Principio
del
método
El gas de muestra se pasa por un cartucho con el colectorimpregnado con solución 2,4-dinitrofenilhidrazona para colectarel formaldehído, levigar(20) con acetonitrilo (CH3-CN),introduciéndose luego la muestra en un cromatógrafo líquido decaracterísticas elevadas. Las mediciones se realizan con undetector de espectrometría ultravioleta (Ver Sección 5.6,Apartado 7). Las condiciones típicas de medición son:temperatura de la columna 40 a 65 °C, caudal del líquido, 0,7 a1,2 ml/min., longitud de onda del detector, 350 a 360 nm.
Intervalo
de
aplicación
Concentración de muestra al cual es aplicado el método es de:0,05 a 50 ppm cuando es tomada 1 l de la muestra de gas.
(20) Disolver y desunir en líquido una materia en polvo para separar las partes más tenues de las más gruesa

107
5.5.2.2 Métodos de medición automática
1. Dióxido de azufre (SO2)
Característica de comportamiento de métodos de medición automática.
ISO - 7935/92.
Campo de
aplicación
Se especifican características de sistemas de medición automática(SMA) para la medición continua de concentración de dióxido deazufre (SO2).Se aplica a métodos extractivos y no extractivos (ó “in-situ” ómétodos a través de la chimenea).
Interferencias Para ambos métodos las principales sustancias contenidas en elgas que producen interferencia con la determinación de SO2 son:dióxido de carbono (CO2), monóxido de carbono (CO), monóxidode nitrógeno (NO), agua (H2O) y en pequeñas concentraciones,dióxido de nitrógeno (NO2) y amoníaco (NH3). Si el vapor deagua no se extrae del gas, puede haber también interferencia conácido clorhídrico (HCl) y grupo iónico HN. El efecto deinterferencia es ± 2 %. Una concentración típica de estassustancias en una planta de combustión es CO2=275 g/m3;CO=100mg/m3; NO=400 mg/m3; resto N2.
Algunas
características
Se define un parámetro de “comportamiento integral” de un SMAparticular como la diferencia entre pares de valores medidos deconcentración de SO2 usando un método SMA dado y un métodomanual ISO. Es una medida de la exactitud del método yconforma un límite superior del mismo.
No es posible determinar la desviación estándar de un SMA bajocondiciones de trabajo repetibles dado que:a) Las mezclas de gases de calibración disponibles
comercialmente conteniendo SO2 no tienen todas laspropiedades de los gases de chimenea ni cubren todas lasposible influencias.
b) La concentración de masa de SO2 en el gas de chimeneausualmente varía con el tiempo.
c) No es posible mantener las propiedades de los gases dechimenea presentes cuando los mismos son transferidos a unrecipiente.
Tiempo de respuesta < 200 s, asumiendo un tiempo de integraciónde 30 min.

108
Intervalo
de
aplicación
Los intervalos de concentraciones de SO2 en el total de gas para elcual son aplicables las especificaciones son entre 0 a 100 mg/m3 y0 a 8 g/m3, variando los mismos según sea el proceso que generalos gases.
Límite de detección 2 %.
1. Métodos extractivos
Partes
constitutivas
1) extractor de muestra,
2) línea de transporte de muestra,
3) sistema de acondicionamiento del gas,
4) analizador.
Ver Figura 5.5.2-8
El diseño del extractor de muestra y el sistema deacondicionamiento del gas depende de las característicasfisicoquímicas del efluente (composición de la fase gaseosa,concentración de partículas, temperatura, punto de rocío, etc.) y elprincipio del analizador utilizado. En general la línea contienefiltros para la supresión de material particulado y de humedad,dado que ambos pueden alterar las mediciones. Para evitarpérdidas de SO2 y lecturas inconsistentes, la línea esfrecuentemente calentada.
Métodos de
detección
comúnmente
usado
1) Absorción, usando radiación infrarroja o ultravioleta
2) Fluorescencia, usando radiación ultravioleta,
3) Interferometría
4) Conductometría
2. Métodos no extractivos, in-situ, o de medición a través de la chimenea.
Se usa un dispositivo óptico el cual es posesionado directamente en el conducto por
donde fluyen los gases. Consiste en dos módulos, uno emisor de la radiación, el otro el
receptor de la misma; esta pasa a través del gas que contiene el dióxido de azufre. Ver
Figura 5.5.2-8 . La instalación de los dos módulos, en relación a la chimenea, depende
del aparato usado.
Cuando se comparan los resultados del método no - extractivo con el extractivo seco, es
necesario conocer el contenido de agua en el gas para corregir.

109
Figura 5.5.2-8: En la parte superior se muestra un ejemplo de componentes del
método extractivo. En la parte inferior se muestra un ejemplo de componentes de
un método no extractivo

110
2. Óxidos de nitrógeno (NOx)
Método de celda electroquímica
Aplicación e
interferencias
Se determina la concentración de óxidos de nitrógeno (NOx) engases de chimenea en forma continua.
Principio
del
método
Se introduce en la chimenea una celda electrolítica que puedeoperar a alta temperatura (hasta 260 °C). Está separada del gasque fluye por la chimenea por una membrana permeable, a travésde la cual penetra el gas para análisis, que produce la reacciónelectroquímica. Esta reacción genera una corriente eléctrica que esfunción de la concentración de NOx presente.
Intervalo
de
aplicación
Se puede disponer varios intervalos de aplicación:
De 0 a 100 ppm, con exactitud de ± 3 ppm.
De 0 a 3.000 ppm, con exactitud de ± 10 ppm.
De 3.000 a 30.000 ppm, con exactitud de ± 10 % de la lectura

111
3. Dióxido de azufre (SO2) y monóxido de nitrógeno (NO)
Método de absorción ultravioleta
Aplicación e
interferencias
Determinación de la concentración de dióxido de azufre (SO2) ymonóxido de nitrógeno (NO).
Principio
del
método
Se mide la absorción de la radiación ultravioleta en las bandasespectrales correspondientes al SO2 (218,5 nm) y al NO(226,5 nm) (Ver Sección 5.6, Apartado 7). Se introduce dentro dela chimenea una cavidad cuyas paredes están formadas pormaterial cerámico poroso, a través de las cuales difunden losgases, haciendo a su vez de filtro del material particulado.Para el análisis se emplea la segunda derivada de la banda deabsorción angosta de cada contaminante.Una fuente de deuterio se emplea como emisor de radiaciónultravioleta, la cual se envía a la cavidad y después de atravesarlos gases es reflejada retornando el haz de medición a la unidad deprocesamiento; allí un sintonizador monocromático separa la luzen bandas espectrales discretas. Un sistema de ranuras de salidasobre el monocromador permite que lleguen al detector doslongitudes de onda separadas correspondientes a los gases enanálisis. La luz es convertida por un tubo fotomultiplicador en unaseñal eléctrica.
Intervalo
de
aplicación
SO2 : de 25 a 75.000 ppm
NO : de 75 a 75.000 ppm.
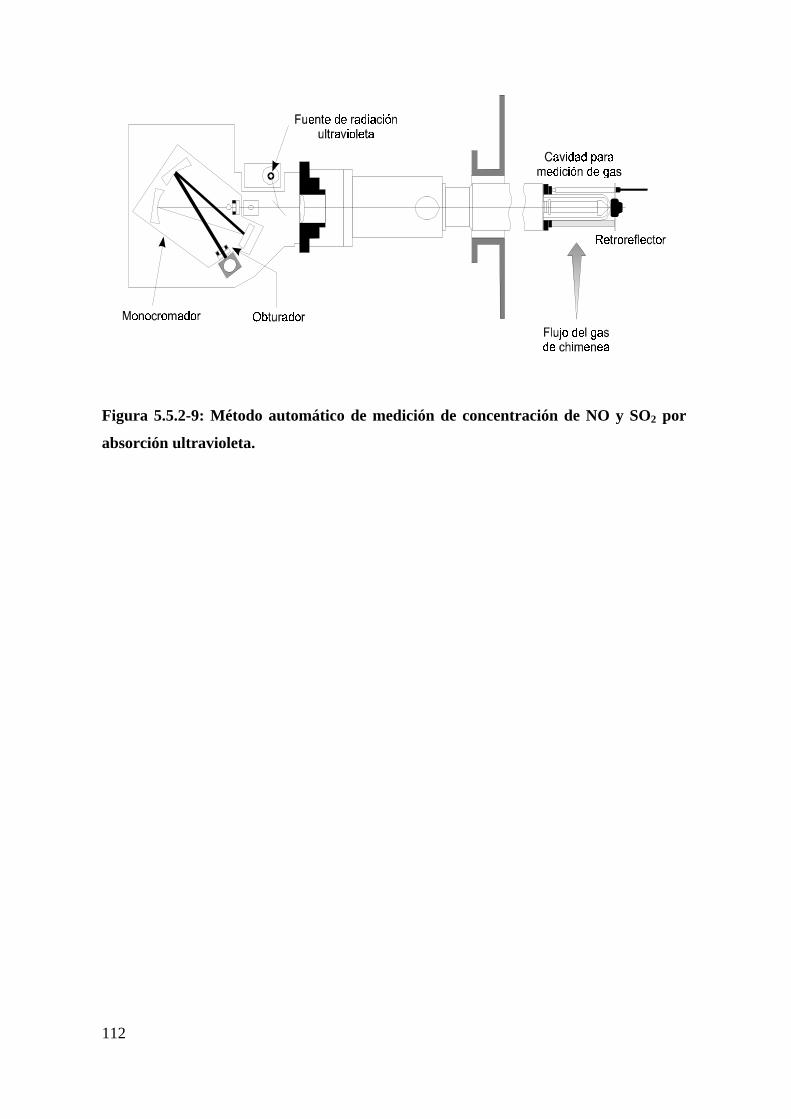
112
Figura 5.5.2-9: Método automático de medición de concentración de NO y SO2 por
absorción ultravioleta.

113
4. Dióxido de azufre (SO2), óxidos de nitrógeno (NOx), óxidos de carbono
(COx), ácido clorhídrico (HCl), ácido fluorhídrico (HF), amoníaco
(NH3).
Método continuo de espectroscopia de absorción óptica diferencial.
Aplicación e
interferencias
Se aplica a la determinación de concentración de dióxido deazufre (SO2); monóxido y dióxido de nitrógeno (NO y NO2);monóxido y dióxido de carbono (CO y CO2), ácido clorhídrico(HCl); ácido fluorhídrico (HF); amoníaco (NH3).
Principio
del
método
Basado en la ley de absorción de Beer-Lambert que establece larelación entre la cantidad de luz absorbida y el número demoléculas en el camino que atraviesa la luz. Se emplea una fuenteespecial, lámpara de xenón a alta presión que emite luz delongitudes de onda en el espectro visible, ultravioleta, e infrarrojo.El haz de luz atraviesa un camino de longitud determinada dondese produce la absorción molecular. La luz es luego capturada porun receptor y conducida por fibras óptica hasta el analizador, unespectrómetro de elevada calidad. Usando un programa decomputo se puede evaluar y analizar la perdida de luz debido a laabsorción molecular que se produjo en el camino recorrido, y enfunción de dicha longitud obtener la concentración por unidad devolumen. Dado que cada tipo de molécula tiene propiedadesúnicas del espectro de absorción, es posible identificar ydeterminar la concentración de diferentes gases simultáneamentecon un sólo equipo a lo largo de un dado recorrido de luz. Elcamino de medición recomendado es de 1 a 5 m. En la Figura5.5.2-10 se muestra un esquema del sistema.
Intervalo
de
aplicación
Los valores dados son para un camino de medición de 1 m. Paracaminos mayores, el límite máximo es proporcionalmente menor.
SO2 : 1 a 5.000 mg/m3.NO2: 1 a 2.000 mg/m3; 1-10.000 mg/m3, según el analizadorNO : 1 a 2.000 mg/m3. Máxima concentración de SO2 5 g/m3x mCO : 3 a 10.000 mg/m3
CO2 : 0,1 a 100 % vol.HCl : 10 a 10.000 mg/m3. Camino de medición 5 m, tiempo demedición 30 s.HF : 5 a 1.000 mg/m3
NH3 : 0,5 a 1.000 mg/m3. Máxima concentración de SO2
500 g/m3x m.

114
Figura 5.5.2-10: Disposición del sistema de medición en chimenea con el sistema de
medición continua de espectroscopia de absorción óptica diferencial.

115
5. Monóxido de carbono (CO)
Método de absorción infrarrojo y correlación de filtros
Aplicación e
interferencias
El método se aplica a la determinación continua de monóxido decarbono (CO)
Principio
del
método
El método empleado se basa en la absorción de la radiacióninfrarroja por el método de correlación de filtros (Ver Sección 5.6,Apartado 8.2). Se emite el haz de radiación infrarroja a través deuna rueda con gases de referencia (CO y N respectivamente),creando una relación entre la absorción de los gases de referenciaindependiente del contenido de gases en la chimenea. El haz sehace pasar luego a través del gas fluyendo por la chimenea dondese produce una absorción adicional en el monóxido de carbonopresente, modificando la relación antes mencionada. Esta esmedida en el receptor usando filtros específicos para caracterizarla longitud de onda de absorción del CO y un detector infrarrojo.Los cambios son electrónicamente procesados dando laconcentración de CO en ppm o mg/m3. El método se puede aplicara chimeneas con diámetro entre 0,5 a 10 m.
Intervalo
de
aplicación
Varios intervalos entre 0 y 5.000 ppm (6.250 mg/m3).
Exactitud, ± 2 % del valor de fondo de escala
La temperatura del gas puede ser hasta 370 °C

116
1. Figura 5.5.2-11: Esquema del equipo para medición por el método de absorción
infrarrojo y correlación de filtros

117
6. Material particulado
Método de opacimetría
Aplicación e
interferencias
Se aplica a la determinación de concentración de materialparticulado. Es un método relativo, no da valores absolutos.
Principio
del
método
Se mide la atenuación (o la transmisión) de un haz de luz al pasara través de un flujo de gas conteniendo material particulado ensuspensión. El método se basa en el principio que parte de la luzes absorbida por las partículas o deflectadas por refracción enestas. Mayor es la cantidad de material particulado en el flujo degas, mayor es la perdida de radiación y menor la transmisión. Latransmisión (T) se expresa como la relación entre la intensidad dela luz emitida (Io) y la intensidad de la luz recibida (I) :T= I/Io . El haz de luz generado por una lámpara incandescente esemitido en línea recta a través del lente objetivo y posteriormentede la chimenea (y por lo tanto del gas) hacia el reflector. El hazde luz retorna usando el mismo camino (atravesando nuevamentela chimenea) hacia el receptor. Tanto el haz de referencia como eltransmitido generan en el receptor un voltaje que se proporcional ala cantidad de luz recibida. Su relación es proporcional a latransmisión. El sistema permite medir en chimeneas condiámetros hasta 10 m.
Intervalo
de
aplicación
El intervalo de transmisión es de 10 % al 100 % de opacidad

118
5.6 DESCRIPCIÓN DE TECNICAS DE ANÁLISIS (21)
En la presente Sección se describen únicamente algunas de las técnicas de análisis que son
mencionadas en las Secciones 5.4 y 5.5.
1 Espectrometría de absorción atómica
La espectroscopia atómica se basa en la absorción y emisión por parte de átomos o iones.
Los espectros atómicos se obtienen mediante un adecuado tratamiento térmico que
convierte los componentes de una muestra en átomos o iones elementales gaseosos.
El proceso de la espectrometría de absorción atómica puede ser dividido en dos etapas:
1. La atomización, o sea la producción de átomos libres desde la muestra.
La llama de combustión provee un medio de convertir las soluciones en análisis en
átomos libres en fase vapor. Se emplea un nebulizador neumático para transformar la
solución de muestra en una niebla o aerosol, constituida por pequeñas gotas de líquido,
la cual se introduce en un quemador donde, por efecto del calor, se generan átomos
libres. Se debe seleccionar la fuente de llama adecuada para proveer la temperatura
requerida según el un análisis que se realice. La máxima temperatura está dada por el
tipo de combustible y de oxidante y el valor exacto de temperatura está dado por la
relación entre ellos. El intervalo de temperatura de atomización característico según el
tipo de llama empleada es de 1700 a 3150 °C. En la Tabla 5.6-I se dan ejemplos de
temperatura de llama para diferentes mezcla de combustibles y oxidantes.
2. La absorción de radiación de una fuente externa por los átomos generados con la
llama de combustión.
Los métodos analíticos que se basan en la absorción atómica son muy específicos
debido a que las líneas de absorción son bastante estrechas (de 0,002 a 0,005 nm). La
fuente de emisión de radiación produce radiación con ancho de banda aún más
(21) Bibliografía utilizada: Análisis Instrumental. D.A.Skoog y J.J.Leary. Editorial McGraw-Hill. 1994.Instrumental Methods of Analysis. H.H.Willard, L.L.Merritt Jr, J.A.Dean,F.A.Settle Jr. Wadsworth Publishing Company. 1988.
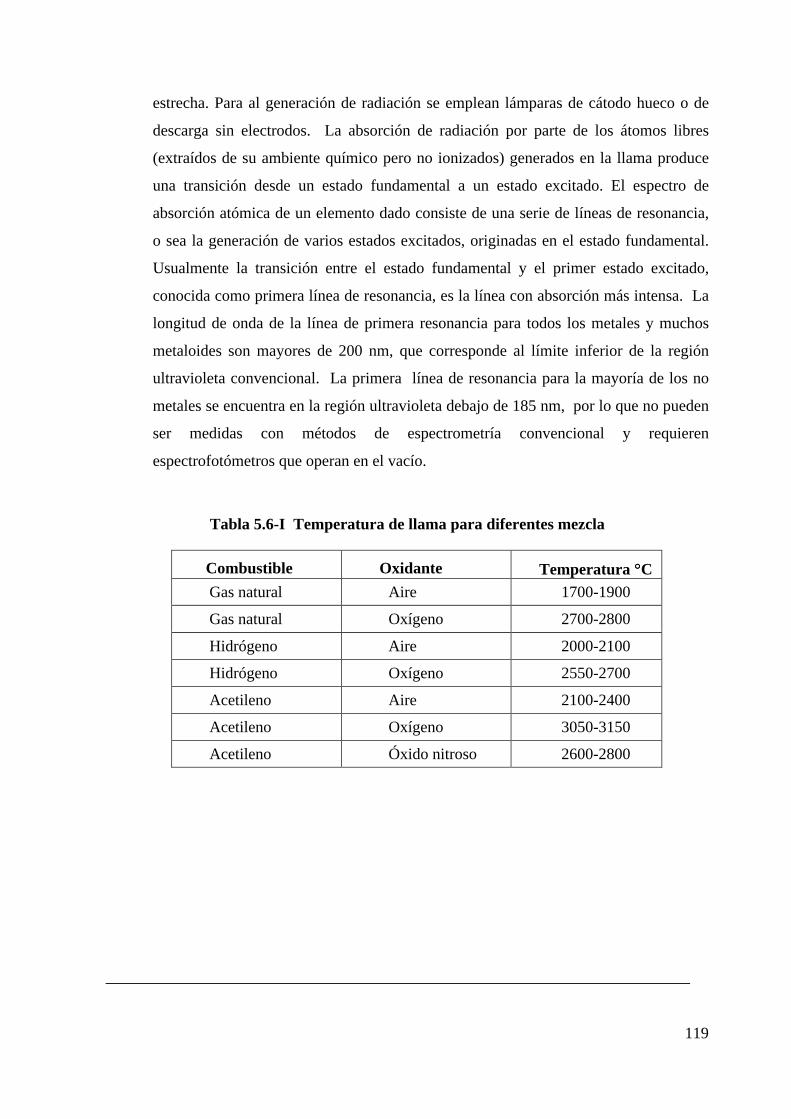
119
estrecha. Para al generación de radiación se emplean lámparas de cátodo hueco o de
descarga sin electrodos. La absorción de radiación por parte de los átomos libres
(extraídos de su ambiente químico pero no ionizados) generados en la llama produce
una transición desde un estado fundamental a un estado excitado. El espectro de
absorción atómica de un elemento dado consiste de una serie de líneas de resonancia,
o sea la generación de varios estados excitados, originadas en el estado fundamental.
Usualmente la transición entre el estado fundamental y el primer estado excitado,
conocida como primera línea de resonancia, es la línea con absorción más intensa. La
longitud de onda de la línea de primera resonancia para todos los metales y muchos
metaloides son mayores de 200 nm, que corresponde al límite inferior de la región
ultravioleta convencional. La primera línea de resonancia para la mayoría de los no
metales se encuentra en la región ultravioleta debajo de 185 nm, por lo que no pueden
ser medidas con métodos de espectrometría convencional y requieren
espectrofotómetros que operan en el vacío.
Tabla 5.6-I Temperatura de llama para diferentes mezcla
Combustible Oxidante Temperatura °°CGas natural Aire 1700-1900
Gas natural Oxígeno 2700-2800
Hidrógeno Aire 2000-2100
Hidrógeno Oxígeno 2550-2700
Acetileno Aire 2100-2400
Acetileno Oxígeno 3050-3150
Acetileno Óxido nitroso 2600-2800

120
Figura 5.6-1: Ejemplo de nebulizador
Los gases generados en la llama forman un conjunto de átomos libres no excitados
capaces de absorber radiación, a la longitud de onda de la línea de resonancia, emitida por
una fuente externa. La radiación no absorbida pasa a través de un monocromador que aísla
la línea resonante y luego llega a un fotodetector que mide la potencia de la radiación
transmitida. La absorción es determinada por diferencia de potencia de la línea resonante
en presencia y ausencia de los átomos generados en la llama.
Figura 5.6-2: Espectrofotómetro de llama típico con un sólo haz.

121
2 Cromatografía (Gaseosa y Líquida)
El hecho que distingue la cromatografía gaseosa o líquida de otros métodos físicos o
químicos de separación es que dos fases mutuamente inmisibles son puestas en contacto;
una fase es estacionaria y la otra móvil. Las dos fases se eligen de forma tal que los
componentes de la muestra que se quiere analizar se distribuyen de modo distinto entre la
fase móvil y la estacionaria. Una muestra introducida en la fase móvil es llevada a través
de una columna (múltiple) conteniendo una fase estacionaria distribuida. Especies en la
muestra es sometida a repetidas interacciones (particiones) entre la fase móvil y la
estacionaria. Al final del proceso emergen componentes separados en función de la
diferencia de interacción con la fase estacionaria. El componente menos retardado emerge
primero. El componente más fuertemente retenido emerge último. La partición entre las
fases explota diferencia en las propiedades física y/o químicas de los componentes de la
muestra.
La columna de separación es el corazón de la cromatografía. Provee versatilidad en los
tipos de análisis que pueden realizarse. Esta versatilidad, debido a la amplia elección de
materiales para las fases estacionaria y móvil, hace posible separa moléculas que difieren
levemente en sus propiedades físicas y químicas.
Hablando en términos generales la distribución de un soluto(22) entre las dos fases resulta
del balance de fuerzas entre las moléculas del soluto y las moléculas de cada fase. Ello
refleja la atracción relativa o repulsión que las moléculas o iones de las fases competitivas
muestran para el soluto o entre ellas mismas. Esas fuerzas pueden ser polares en su
naturaleza, apareciendo desde momentos dipolares inducidos o permanentes, y ello puede
ser debido a fuerzas dispersas de London. La polaridad relativa de los solventes es
manifestada en su constante dieléctrica.
La fase móvil puede ser un gas o un líquido, mientras que la fase estacionaria puede ser
solamente líquido o sólido. Cuando la fase móvil es un gas, se denomina cromatografía
gaseosa y cuando la fase móvil es un líquido, cromatografía líquida.
(22) Soluto: en las disoluciones, el cuerpo que se disuelve.

122
Cromatografía gaseosa
Dentro de la cromatografía gaseosa la combinación de fases más empleada es la de gas (fase
móvil) – líquido (fase estacionaria).
Las columnas pueden ser de relleno o capilares.
a) Las columnas de relleno se fabrican con tubo de vidrio, metal (acero inoxidable, cobre o
aluminio) o de teflón, con una longitud de 2 a 3 m y un diámetro de 2 a 4 mm. Estos
tubos se empaquetan densamente con un material de relleno sólido, finamente dividido
y homogéneo, que se cubre con una delgada capa (0,05 a 1 µm) de la fase estacionaria
líquida.
b) Las columnas capilares son de dos tipo: 1) tipo capilares de pared recubierta, donde la
pared interior del tubo capilar está cubierta de una fina capa de fase estacionaria; 2) tipo
capilares con soporte recubierto, donde la superficie interna está revestida de una fina
capa (30 µm) de material soporte, tal como tierra de diatomeas; tienen varias veces la
fase estacionaria que una de pared cubierta y por lo tanto mayor capacidad.
Los componentes separados deben luego ser detectados; en el caso de cromatografía
gaseosa la versatilidad y especificidad de los detectores y la posibilidad de combinar el
método con un espectrómetro de masa o un espectrofotómetro de infrarrojo incrementa la
utilidad de este método.
El detector de ionización de llama es uno de los más extensamente utilizado en
cromatografía gaseosa (ver Apartado 4) y también el de fotometría de llama (ver
Apartado 5).

123
Figura 5.6-3: Representación esquemática de un cromatógrafo gaseoso
3 Cromatografía iónica
Es un tipo de cromatografía líquida (ver Apartado 2) cuyo nombre fue acuñado para
describir el análisis de iones disueltos por cromatografía de intercambio iónico, difiriendo
en la naturaleza de la resina de intercambio iónico. Los procesos de intercambio iónico se
basan en los equilibrios de intercambio entre los iones de una disolución y los iones del
mismo signo que están en la superficie de un sólido de elevada masa molecular y
esencialmente insoluble.
La columna consiste de un corazón de polímero neutro de alrededor de 10 µm de diámetro.
Dependiendo de si será usado para la separación de iones o cationes, el corazón es
ligeramente sulfonado o aminado, lo cual lleva en el primer caso a la formación de una
fina cascara superficial exterior de ácido sulfónico (-SO3-H*), un ácido fuerte, o en algunos
casos ácido carboxílico (-COO-H*), y en el segundo caso a la formación de una fina
cáscara exterior de grupos de amina primaria (-NH3+(OH-), o también grupos de amina
cuaternaria (-N(CH3)3+HO-). Esto produce la tradicional resina de intercambio iónico. El
hecho nuevo es la introducción de espuma polimérica, aminada o sulfonada y con 100 a

124
300 nm de diámetro, las cuales son vinculadas electrostáticamente sobre la superficie del
corazón del polímero.
4 Detector de ionización de llama
La técnica de medición por ionización de llama se basa en la medición de los iones de una
muestra de gas determinado que son formados durante la combustión de hidrógeno. En una
cámara de ionización, la nube de iones formados es extraída aplicando un campo eléctrico,
vía electrodos, produciendo una corriente eléctrica. Esta corriente es aproximadamente
proporcional al caudal de masa del gas, proporcional a su concentración. Es necesario
realizar una calibración con muestra patrón.
El detector de ionización de llama consiste de una cámara de combustión (ver Figura 5.6-
4). A través de una boquilla o tobera se introduce hidrógeno puro (combustible), y por un
tubo que rodea la tobera aire atmosférico (oxidante) para la combustión. La llama de
hidrógeno (2.660 °C) produce una pequeña densidad de iones (valor cero) en la ausencia
de la muestra de gas. Los electrodos necesarios para la extracción de la nube de iones son
dispuestos cerca de la llama. La misma tobera puede ser usada como uno de los electrodos.
Se emplea un amplificador sensible a corriente continua para lograr una señal de amplitud
adecuada. Por una entrada adicional se introduce en la tobera el gas de muestra. Para
mediciones continuas se debe mantener constante la temperatura y el caudal de la muestra
de gas.
Figura 5.6-4: Detector de ionización de llama

125
5 Detector de fotometría de llama.
Se trata de un detector selectivo que se emplea fundamentalmente para los compuestos que
contiene azufre (S) y fósforo (P). La muestra pasa a la cámara de combustión donde a
través de una boquilla o tobera se introduce hidrógeno puro (combustible), y por un tubo
que rodea la tobera aire atmosférico (oxidante) para la combustión y se genera una llama
de hidrógeno de baja temperatura. La llama calienta los compuestos de azufre y fósforo y
convierte parte del fósforo en una especie HPO que emite bandas de radiación con
longitudes de onda alrededor de 510 y 526 nm y el azufre se convierte en S2, emitiendo en
una banda centrada en 394 nm. Las bandas se separan empleando filtros adecuados y se
mide sus intensidades con fotómetros.
6 Procedimientos luminiscentes
La luminiscencia es el término aplicado al caso en que las moléculas en análisis son
excitadas dando una especie cuyo espectro de emisión suministra información para el
análisis cualitativo y cuantitativo. Dentro de estos procedimientos se encuentran la
fluorescencia molecular y la quimioluminiscencia molecular.
6.1 Fluorescencia molecular
En la fluorescencia molecular fotones de radiación electromagnética son absorbidos por
moléculas, alcanzando estas un estado excitado y retornando luego a su estado
fundamental con la emisión de radiación; la transición energética en esta caso no genera un
cambio de spin de los electrones. A causa de que los niveles de vibración de ambos
estados, fundamental y excitado, son similares, el espectro de fluorescencia es una imagen
especular del espectro de absorción. El tiempo de vida de un estado excitado simple es de
10 –9 a 10 –6 segundos y el tiempo de vida fluorescente se encuentra dentro de este
intervalo.
Es un método muy usado por su elevada sensibilidad y especificidad. La elevada
sensibilidad resulta de la diferencia en longitud de onda entre la radiación excitante y la
fluorescente. La elevada especificidad proviene de la dependencia de dos espectros, el de
excitación y el de emisión, y la posibilidad de medir la vida media del estado fluorescente.

126
Para los fluorímetros de filtro, como fuente de radiación se emplea la lámpara de mercurio
a baja presión que produce 8 líneas muy intensas entre 254 y 773 nm. Las líneas
individuales se pueden aislar con los filtros de interferencia o absorbancia adecuados. Para
espectros donde se requiere una fuentes de radiación continua, normalmente se emplea una
lámpara de arco de xenón a elevada presión que emite un espectro continuo de 300 a 1300
nm.
Como detectores se emplean tubos fotomultiplicadores dado que la señal es de baja
intensidad. También se emplean detectores de diodo alineados.
Figura 5.6-5 Esquema de un fluorímetro

127
6.2 Método de Quimioluminiscencia
La quimioluminiscencia se produce cuando una reacción química da una especie
electrónicamente excitada, la cual emite radiación para volver a su estado fundamental.
La reacción más sencilla para producir quimioluminiscencia se puede formular como
A + B → C* + D
C* → C + hν
Donde C* representa el estado excitado de la especie C y hν la radiación emitida.
El aire que contiene el contaminante cuya concentración se quiere determinar se mezcla en
el reactor con un exceso de un reactivo dado. En general este suele ser ozono. La reacción
produce otro gas en un estado excitado que al decaer emite radiación. La intensidad de
esta radiación quimiolumiscencia es proporcional a la concentración del contaminante si el
gas auxiliar necesario para producir la reacción está presente en exceso. La
instrumentación es muy simple y en general se compone de un recipiente de reacción
adecuado y un tubo fotomultiplicador.
El método es relativo y debe ser calibrado con muestras de concentración conocida.
7 Espectrometría de absorción ultravioleta
La absorción de radiación ultravioleta visible está en el intervalo de longitud de onda de
180 a 780 nm. La absorbancia molecular de la radiación ultravioleta es directamente
proporcional a la longitud de la trayectoria del haz a través de la muestra y a la
concentración de la especie absorbente. Dado que la solución de la muestra que contiene el
elemento absorbente debe colocarse en alguna clase de recipiente transparente o cubeta, se
produce en este perdidas de radiación por reflexión y dispersión, por lo que la potencia del
haz transmitido se debe comparar con la de una cubeta idéntica que solo contenga el
disolvente.

128
El instrumento para medir la absorción de la radiación ultravioleta consta de: 1) fuente de
radiación ultravioleta, 2) selectores de longitud de onda, 3) recipiente de muestra, 4)
detectores de radiación.
Figura 5.6-6 : Ejemplo del método de medición por quemiluminiscencia
1) Fuente de radiación ultravioleta : Se necesita una fuente continua cuya potencia no
varíe bruscamente en un intervalo considerable de longitud de onda ; se consideran:
a) Lámpara de deuterio e hidrógeno: La excitación eléctrica de deuterio o hidrógeno a
baja presión, produce un espectro continuo en la región ultravioleta de 160 a 375
nm.
b) Lámpara de arco de xenón: Se produce una intensa radiación al pasar una corriente
a través de una atmósfera de xenón. El espectro continuo es entre 250 y 600 nm.
c) Lámpara de vapor de mercurio con filtros que aíslan la línea de emisión de 254
nm.
2) Recipiente de muestra: Debe ser realizado de un material a través del cual pase la
radiación. El cuarzo o sílice fundida permite el paso de radiación por debajo de 350
nm. En la región arriba de 350 nm se puede usar vidrios de silicatos.

129
3) Detectores de radiación: La mayoría de los espectrofotómetros utilizan tubos
fotomultiplicadores.
Figura 5.6-7: Esquema de un típico espectrofotómetro manual de doble haz.
8 Espectrometría infrarroja (dispersiva y no dispersiva)
La región Infrarroja del espectro electromagnético se extiende entre 0,7 y 500 µm. El
intervalo más usado es en la región media de 2,5 a 50 µm. La radiación infrarroja no es
suficientemente energética como para producir las transiciones electrónicas que se dan
cuando se trata de radiaciones ultravioleta y visible. La absorción de radiación infrarroja se
limita así en gran parte a especies moleculares para las cuales existen pequeñas diferencias
de energía entre los distintos estados de vibraciones y rotaciones. El método se basa en la
absorción selectiva de la radiación infrarroja por lo distintos componentes de una muestra.
Al entrar en interacción con la radiación infrarroja, las moléculas absorben porciones de la
misma a longitudes de onda específicas. La multiplicidad de vibraciones que ocurren
simultáneamente producen un espectro de absorción altamente complejo que es
unívocamente característico de los grupos funcionales que integran la molécula y la
configuración que incluye a la molécula misma. Luego cuando se interpreta un espectro es
posible establecer que están presente cierto grupos funcionales, cierto compuestos de
interés, y determinar su concentración por comparación. Parte de la energía de la radiación
infrarroja es absorbida por el compuesto en análisis en forma proporcional a su

130
concentración; en una célula de referencia no existe dicha absorción y por diferencia se
puede determinar la concentración.
La instrumentación usada para el estudio de espectrometría infrarroja se divide en dos
grupos:
a) Instrumentos dispersivos: Usan un prisma o retículo para seleccionar o aislar la
longitud de onda deseada.
b) Instrumentos no dispersivos: usan filtros de interferencia, fuentes de láser sintonizable
o un interferómetro (espectrometría infrarroja por transformada de Fourier), para
aislar la longitud de onda deseada.
En la región infrarroja se usan espejos de primera superficie pues el vidrio o cuarzo usado
en las lentes son opacos a esa radiación.
Se necesita una fuente de radiación infrarroja continua. Consisten en un sólido inerte que
se calienta eléctricamente a una temperatura entre 1.500 y 2.200 K. La máxima intensidad
radiante se produce entre 2 a 1,7 µm. Se pueden mencionar : a) emisor de Nernst,
constituido por óxidos de tierras raras, b) fuente global, constituido de varilla de carburo
de silicio, c) filamento incandescente, constituido de un filamento de alambre de níquel
cobre, d) arco de mercurio, especialmente para infrarrojo lejano (λ> 50 µm), e) lámpara de
filamento de tungsteno, ideal para infrarrojo cercano (2,5 a 0,78 µm). f) fuente láser de
dióxido de carbono, que produce una banda en el intervalo de 11 a 9 µm.
Los detectores usados para longitudes de onda arriba de 1,2 µm se clasifican en dos
grupos:
a) Detectores térmicos: la radiación infrarroja produce un calentamiento que altera alguna
propiedad física del detector (por eje. expansión de un sólido, gas o fluido, resistencia
eléctrica, termocupla, termopila, piroeléctrico).
b) Detectores de fotones: usa el efecto cuántico de la radiación infrarroja para cambiar la
propiedad eléctrica de un semiconductor (detector fotovoltaico o fotoconductor).
Las moléculas heteroatómicas como CO y CO2 poseen un espectro de absorción
característica típica en el intervalo de radiación infrarrojo. La radiación infrarroja se hace
pasar a través de un celda conteniendo el gas de muestra que se desea analizar, y la

131
absorción cuantitativa de energía por el CO es medida por un detector apropiado en un
fotómetro no dispersivo.
Para los casos de contaminantes con espectros de absorción muy similares, puede haber
interferencias. En ese caso, se puede utilizar una celda en serie con una muestra del gas que
no se quiere medir y puede interferir que elimina la parte del espectro correspondiente.
Dependiendo del método de almacenaje de muestra, se distingue entre los métodos de
Absorción radiación infrarrojo no dispersivo y Correlación filtro de gas.
8.1 Absorción radiación infrarrojo no dispersivo
Se utiliza un doble haz de radiación infrarroja (ver Figura 5.6-8), que pasan a través de dos
celdas; una es la de medición llena con el gas de muestra y la otra, de referencia, llena por
ejemplo de N2. En la primera celda se produce la absorción en el componente que se desea
medir, mientras que en la de referencia no hay absorción. Una rueda interruptora
(“chopper”) permite que el haz llegue alternativamente al detector. El método usa el
receptor de luz como almacenaje. El detector consiste en dos comportamientos separados
por un diagrama a los que llega la radiación no absorbida en las celdas, llenos con el
mismo tipo de gas que se analiza. Este absorbe la energía radiante del haz (mayor en la
parte que recibe el haz que pasó por la celda de referencia), aumentando su temperatura y
ejerciendo una presión sobre el diafragma La modulación producida por la rueda
interruptora (“chopper”) producen variación periódica de presión en las cámaras. Estas
señales son detectadas y convertidas en señal eléctrica, o por un capacitor de membrana
(el diafragma forma parte del capacitor y la variación de posición da lugar una
modificación de su capacidad) o con un detector de micro flujo, los cuales miden el flujo
de presión compensado entre las dos cámaras receptoras.
8.2 Correlación filtro de gas
En el método de correlación filtro de gas (ver Figura5.6-9) se usa un sólo haz de radiación
infrarroja que pasa por la celda de medición.. Para limitar la sensibilidad del fotómetro a una
banda angosta de interés se hace pasar la radiación infrarroja alternativamente por el filtro con
CO, produciendo un haz de referencia, y con otro gas, por ejemplo N2, que es transparente a la
radiación infrarroja de interés, generándose el haz de medición, que luego es absorbido por el
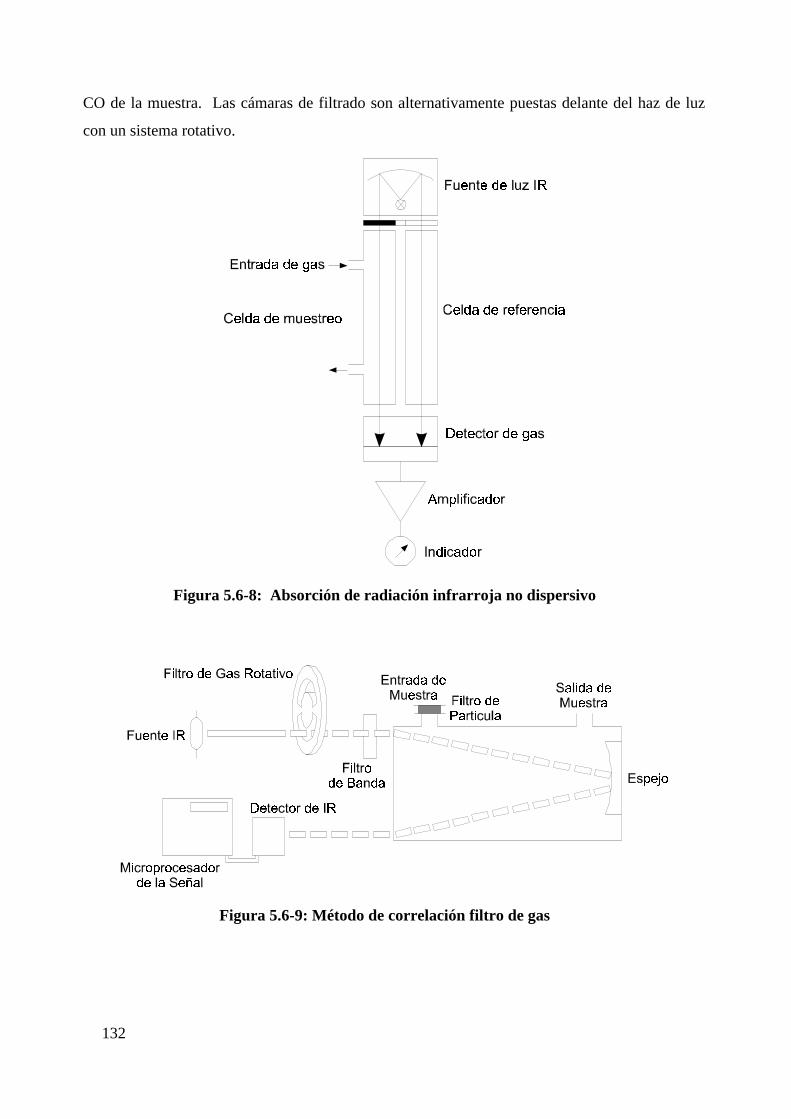
132
CO de la muestra. Las cámaras de filtrado son alternativamente puestas delante del haz de luz
con un sistema rotativo.
Figura 5.6-8: Absorción de radiación infrarroja no dispersivo
Figura 5.6-9: Método de correlación filtro de gas

133
9 Métodos potenciométricos
Los denominados métodos potenciométricos están basados en la medición del potencial de
las celdas electroquímicas empleadas cuando no se producen corrientes apreciables. Se
emplea para la detección de puntos finales en métodos volumétricos de análisis. También
incluyen los métodos en los que las concentraciones de los iones se obtienen directamente
del potencial de un electrodo de membrana selectiva de iones.
El equipamiento necesario para el análisis por métodos potenciométricos es simple e
incluye un electrodo de referencia, un electrodo indicador (que puede ser metálico o de
membrana) y un dispositivo para medir potencial.
9.1 Método de electrodo selectivo de iones o electrodo específico (denominado también
electrodo a membrana selectiva de iones)
Desde el punto de vista analítico el método de electrodo selectivo de iones es una
herramienta de medición ideal por su habilidad de monitorear selectivamente la actividad
de ciertos iones en solución en forma continua y no destructiva. El sistema está compuesto
de un electrodo de referencia, una solución interna y una membrana (electrodo indicador o
de trabajo, cuya respuesta depende de la concentración del elemento o compuesto en
estudio) a través de la cual pasa selectivamente los iones cuya concentración se desea
determinar. Si ocurre una separación de cargas entre iones en una interface, se genera una
diferencia de potencial a través de la interface. El potencial de un electrodo selectivo de
iones está compuesto de dos o más contribuciones discretas que aparecen de los varios
procesos en las interfaces y en el volumen o masa del material de la membrana activa. El
problema es encontrar una interface cuya composición favorezca selectivamente un tipo
de iones sobre otros. En una solución diluida, la actividad de los iones es usualmente
cercano a la concentración de iones. El mecanismo general por el que se produce en estos
montajes un potencial selectivo de iones es independiente de la naturaleza de la membrana
y es completamente diferente del mecanismo que origina el potencial en los electrodos
indicadores metálicos. En estos últimos el potencial se produce debido a la tendencia a que
tenga lugar una reacción de oxidación/reducción en la superficie del electrodo. En los
electrodos de membrana, por el contrario, el potencial observado es una especie de

134
potencial de unión que se forma en la membrana que separa la disolución del elemento o
compuesto en estudio y la disolución de referencia.
Los electrodo de referencia deben ser reversibles, presentar un potencial que sea constante
con el tiempo, volver al potencial original después de haber estado sometido a corrientes
pequeñas y presentar poca histéresis con los ciclos de temperatura Algunos electrodos de
referencia son: a) electrodos de calomelanos, que consisten en mercurio en contacto con
una disolución saturada de cloruro de mercurio y que también contiene una concentración
conocida de cloruro de potasio b) electrodos de plata – cloruro de plata (Ag-AgCl), que
consiste en un electrodo de plata sumergido en una disolución de cloruro de potasio (KCl)
que ha sido saturada de cloruro de plata.
Los electrodos de membrana se llaman también electrodos de iones debido a la alta
selectividad de la mayoría de estos dispositivos. Hay dos tipos de electrodos de membrana
selectivos de iones:
1. Electrodo de membrana cristalina
a) Cristal único. Eje. LaF3 para F-.
b) Policristalina o mezcla de cristales.
Eje. Ag2S para S2- y Ag+
2. Electrodos de membrana no cristalina
a) Vidrio Eje. Vidrios de silicatos para Na+ y H+
b) Líquido Eje. Intercambiadores de iones líquidos para Ca2+ y portadores
neutros para K+.
c) Líquido inmovilizado en un polímero rígido.
Eje. Matriz de cloruro de polivinilo para Ca2+ y NO3-.

135
Figura 5.6-10: Sistema de electrodos típicos para medir el pH
Figura 5.6-11: Electrodo de membrana líquida sensible a M2+ (catión cuya actividad se
determina)

136
9.2 Titulación potenciométrica
El potencial de un electrodo indicador adecuado se puede utilizar para establecer el punto
de equilibrio de una valorización. Se mide la variación en la fuerza electromotriz (fem)
inducida por la adición de un titulador a la muestra. El método se puede aplicar a cualquier
reacción de titulación para la cual un electrodo indicador es obtenible para seguir la
actividad de al menos una de las sustancias comprometidas. La medida de potencial es
usada para detectar un cambio rápido de actividad en el punto equivalente de la titulación.
10 Electrólisis a un potencial controlado (Método coulombimétrico)
El método de electrólisis a un potencial controlado es un método coulombimétrico,
denominado también electrólisis a potencial de cátodo (o de ánodo) controlado o
electrólisis potenciométrica. Se basa en la oxidación o reducción electródica de un soluto.
Los métodos electrolíticos están caracterizados por una gran relación de área de electrodo a
volumen de solución y condiciones de transferencia de masa tan efectiva como sea posible.
Están caracterizados por corrientes grandes y escala de tiempo de los experimentos de
minutos o mayores.
El potencial del electrodo de trabajo es la variable básica que controla el grado de
cumplimiento de un proceso de electrólisis. La técnica de un potencial controlado es
usualmente la más deseada para electrólisis exhaustivas. Para ello se utiliza un tercer
electrodo, o electrodo de referencia, cuyo potencial en la disolución es conocido y
constante. El potencial entre el electrodo de trabajo y el de referencia puede ajustarse a un
valor que proporcione el potencial deseado en el cátodo (o en el ánodo) con respecto al
electrodo de referencia.
El equipo empleado se representa en la Figura 5.6-12. Los electrodos estables deben ser
cuidadosamente posicionados para minimizar efectos de resistencias no compensados. El
electrodo auxiliar es posicionado para proveer una distribución de corriente regularmente
uniforme a través de la superficie del electrodo de trabajo. Algunas veces el electrodo
auxiliar es puesto en un comportamiento separado aislado del comportamiento del
electrodo de trabajo.

137
Figura 5.6-12: Sistema para la electrólisis a un potencial de cátodo controlado. El
contacto C se ajusta para mantener el potencial del cátodo al valor deseado.
Figura 5.6-13: Esquema de aparato para electrólisis a un potencial controlado.

138
11 Método polarográfico
El método polarográfico pertenece al grupo de voltaperometría que engloba un conjunto
de métodos electroanalíticos en los que la información sobre el elemento o compuesto en
estudio es obtenido midiendo la intensidad de corriente en función del potencial aplicado
en condiciones tales que se favorece la polarización de un electrodo indicador, o de trabajo.
Se basa en la medida de una intensidad de corriente que se desarrolla en condiciones de
polarización total de concentración. Se aplica una señal de excitación que es un potencial
variable a una celda electroquímica que contiene un microelectrodo. La señal de excitación
provoca una respuesta de intensidad de corriente característica. En el caso del método
polarográfico el electrodo de trabajo es un electrodo de gotas de mercurio (ver Figura 5.6-
14); además las intensidades límites polarográficas están controladas sólo por la difusión.
La intensidad de corriente de este tipo de celda experimenta fluctuaciones que corresponde
en frecuencia a la velocidad de goteo. Cuando una gota se desprende del capilar, la
intensidad de corriente cae a cero; entonces aumenta rápidamente cuando el área del
electrodo crece debido a la mayor superficie en la que la difusión puede tener lugar. La
intensidad de corriente promedio es la hipotética intensidad de corriente constante.
El método polarográfico se utiliza mucho para el análisis de sustancias inorgánicas. La
mayoría de los cationes metálicos, por ejemplo, se reducen en el electrodo de gotas de
mercurio. Incluso los metales alcalinos y alcalinotérreos se reducen, a condición de que el
electrolito soporte no reaccione a los elevados potenciales requeridos.
Figura 5.6-14 : Electrodo de gota de mercurio

139
12 Método de absorción de rayos beta
La cantidad de material particulado medido con un sistema de absorción de rayos beta
determina la atenuación gradual cuando estos rayos pasan a través de un filtro donde son
retenido aquellos.
Como emisor de rayos beta se emplea normalmente una fuente radioactiva de actividad
apropiada (por eje. isótopos de carbón 14 o kriptón 85) y como detector un contador
Geiger Müller o una cámara de ionización. Para compensar por la gradual reducción de la
radioactividad sobre un período de tiempo, y la consiguiente variación de la disminución
de la radiación debido al material particulado, se hacen en cada caso mediciones de la
absorción sin y con depósito y se comparan ambas mediciones. Generalmente se en
emplea un método de compensación de doble haz como se muestras en la Figura 5.6-15.
La señal que proviene del haz que pasa por la muestra es positiva y la del haz de control
negativa; ambas se suman. Cuando no hay material particulado en el filtro, la suma de las
señales se regulan para dar un voltaje cero. El sistema permite una medición de la
concentración de material particulado en tiempo real.
Figura 5.6-15: Método de absorción beta para determinar concentración de material
particulado.

140
13 Método de vibración de una filtro cargado con material particulado (microbalanza
oscilante)
Para la medición de material particulado en suspensión por el método de microbalanza
oscilante, se hace pasar una muestra de aire a través de un filtro el cual es parte de un
sistema vibrante u oscilante con su resonancia característica. El material particulado
colectado sobre el filtro aumenta la masa vibrante y por lo tanto decrece la frecuencia de
resonancia. Un sistema electrónico monitorea continuamente la frecuencia de resonancia.
La concentración de material particulado en suspensión se calcula por una calibración entre
la frecuencia de resonancia y la cantidad de material depositado, teniendo en cuenta el
volumen medido de gas de muestra.