
Manual Metodos DeLab FAO
61
1. INTRODUCCIÓN www.fao.org/docrep/ field/003/AB489S/AB489S03.htm El desarrollo de la acuacultura en sistemas semi intensivos e intensivos ha forzado el desarrollo de patrones de alimentación cada vez más eficientes, basados en los requerimientos nutricionales de cada una de las especies a cultivar. Para que el animal presente un crecimiento adecuado en el menor tiempo y más bajo costo posibles, el alimento suministrado debe poseer características nutricionales óptimas, por lo cual en la industria de alimentación de peces, como en las demás especies animales, la elaboración, los ingredientes y el almacenamiento de los alimentos procesados, deben de estar sometidos a estrictos controles de calidad. En la acuacultura semi-intensiva e intensiva, la calidad del alimento está considerada como uno de los factores de mayor influencia en el éxito de un cultivo, ya que los costos del alimento y la alimentación, representan por lo general entre el 50 y 70% de los costos de operación en un sistema de este tipo. La calidad de los alimentos va a depender en primer término de la calidad de los ingredientes utilizados en la preparación del alimento balanceado, así como también del tipo de procesamiento a que se sometan antes y durante la elaboración del alimento. Adicionalmente, el cuidado que se tenga para almacenar tanto ingredientes como alimento terminado, influirá notablemente en sus propiedades nutritivas. Los alimentos balanceados son preparados con base en los requerimientos nutricionales de cada especie y aún cuando la dieta se formula para satisfacerlos, no siempre contiene los niveles de nutrientes calculados una vez preparado, debido a que el proceso usado en su elaboración puede alterar significativamente su valor nutricional; por ejemplo, el calor puede dañar algunos nutrientes y/o puede hacerlos más disponibles eliminando los tóxicos termolábiles, mientras que por otro lado la molienda puede afectar la digestibilidad de proteínas y carbohidratos (Harris, 1980). La calidad del alimento también se modifica después de pasar cierto tiempo en el almacén, donde además de sufrir cambios en el valor nutricional, se pueden presentar alteraciones en otras características como son el color, la textura, el sabor y el olor. Por otra parte existe el grave riesgo de contaminarse con los desechos de roedores, insectos y micoorganismos que aceleran aún más su deterioro (Limborg, 1979; Chow et al., 1980; Smith and Moss, 1985). Teniendo en consideración todos estos factores, resalta la importancia de tener un control de calidad de los ingredientes alimenticios y del producto terminado, con el fin de asegurar que la dieta posea los niveles nutricionales mínimos requeridos, ya que nos proporciona la composición exacta del material y el nivel de sustancias tóxicas normalmente presentes. Este control se inicia con el análisis de los ingredientes con que se elaborará el alimento y finaliza con la certificación de los niveles de nutrientes en el alimento preparado, así como su adecuado almacenaje (Frazer, 1967; Harris, 1980). Adicionalmente, es claro que solo es posible formular una dieta completa, si se conoce con precisión al menos la composición proximal de los ingredientes con que se elaborará, sin embargo, Tacon (1979) recomienda que el valor nutricional de un material usado en la formulación de alimentos, debe evaluarse con base en: 1
-
Upload
mizukikami -
Category
Documents
-
view
125 -
download
0
Transcript of Manual Metodos DeLab FAO

1. INTRODUCCIÓN www.fao.org/docrep/ field/003/AB489S/AB489S03.htm
El desarrollo de la acuacultura en sistemas semi intensivos e intensivos ha forzado el desarrollo de patrones de alimentación cada vez más eficientes, basados en los requerimientos nutricionales de cada una de las especies a cultivar. Para que el animal presente un crecimiento adecuado en el menor tiempo y más bajo costo posibles, el alimento suministrado debe poseer características nutricionales óptimas, por lo cual en la industria de alimentación de peces, como en las demás especies animales, la elaboración, los ingredientes y el almacenamiento de los alimentos procesados, deben de estar sometidos a estrictos controles de calidad. En la acuacultura semi-intensiva e intensiva, la calidad del alimento está considerada como uno de los factores de mayor influencia en el éxito de un cultivo, ya que los costos del alimento y la alimentación, representan por lo general entre el 50 y 70% de los costos de operación en un sistema de este tipo. La calidad de los alimentos va a depender en primer término de la calidad de los ingredientes utilizados en la preparación del alimento balanceado, así como también del tipo de procesamiento a que se sometan antes y durante la elaboración del alimento. Adicionalmente, el cuidado que se tenga para almacenar tanto ingredientes como alimento terminado, influirá notablemente en sus propiedades nutritivas. Los alimentos balanceados son preparados con base en los requerimientos nutricionales de cada especie y aún cuando la dieta se formula para satisfacerlos, no siempre contiene los niveles de nutrientes calculados una vez preparado, debido a que el proceso usado en su elaboración puede alterar significativamente su valor nutricional; por ejemplo, el calor puede dañar algunos nutrientes y/o puede hacerlos más disponibles eliminando los tóxicos termolábiles, mientras que por otro lado la molienda puede afectar la digestibilidad de proteínas y carbohidratos (Harris, 1980). La calidad del alimento también se modifica después de pasar cierto tiempo en el almacén, donde además de sufrir cambios en el valor nutricional, se pueden presentar alteraciones en otras características como son el color, la textura, el sabor y el olor. Por otra parte existe el grave riesgo de contaminarse con los desechos de roedores, insectos y micoorganismos que aceleran aún más su deterioro (Limborg, 1979; Chow et al., 1980; Smith and Moss, 1985). Teniendo en consideración todos estos factores, resalta la importancia de tener un control de calidad de los ingredientes alimenticios y del producto terminado, con el fin de asegurar que la dieta posea los niveles nutricionales mínimos requeridos, ya que nos proporciona la composición exacta del material y el nivel de sustancias tóxicas normalmente presentes. Este control se inicia con el análisis de los ingredientes con que se elaborará el alimento y finaliza con la certificación de los niveles de nutrientes en el alimento preparado, así como su adecuado almacenaje (Frazer, 1967; Harris, 1980). Adicionalmente, es claro que solo es posible formular una dieta completa, si se conoce con precisión al menos la composición proximal de los ingredientes con que se elaborará, sin embargo, Tacon (1979) recomienda que el valor nutricional de un material usado en la formulación de alimentos, debe evaluarse con base en:
1

a. Su contenido de Nitrógeno proteico y no proteico. b. La composición de aceites y ácidos grasos. c. El contenido de fibra cruda y carbohidratos solubles d. Su contenido de minerales y vitaminas. e. La presencia de microbios. f. La presencia de compuestos orgánicos tóxicos. g. La variabilidad de la composición química.
De manera ideal cualquier ingrediente debería pasar por los puntos de control señalados, sin embargo, si por limitaciones económicas o de equipo no es posible, se debe entonces procurar cubrir al menos aquellos análisis que garanticen una calidad mínima deseable y una seguridad de que los animales no presentarán reacciones adversas al alimento. Aún cuando el control de calidad es un tema que merece tratarse exhaustivamente, este manual está enfocado particularmente a los métodos básicos de análisis químicos necesarios para una adecuada evaluación de ingredientes y dietas elaboradas, con el objeto de ponerlos a la disposición de la mayoría de acuacultores y personas interesadas en este tipo de evaluaciones que posean conocimientos básicos de química. Los métodos propuestos han sido recopilados de diferentes fuentes y la mayoría son considerados como estándares de norma. Con fines prácticos los análisis se han agrupado en tres secciones; en la primera se incluyen los métodos considerados dentro del análisis proximal, para determinar el contenido de humedad, proteína cruda, lípidos crudos, fibra cruda, ceniza y extracto libre de nitrógeno; este análisis permite conocer los niveles porcentuales de cada nutriente en el material pero no indica nada acerca de la calidad de los nutrientes, para lo cual se requieren otros análisis complementarios, algunos de los cuales se incluyen en este manual en la segunda sección donde se consideran como análisis especiales. En un tercer apartado se incluyen las técnicas más sencillas para la detección de algunos de los tóxicos y antinutrientes más comunes, ya que su presencia puede afectar negativamente la eficiencia de utilización de los alimentos, e inclusive, ser causa de mortalidad. Consideramos que este capítulo es particularmente importante cuando se incluye en los alimentos subproductos agroindustriales como harina de soya, algodón, o nuevas fuentes proteicas, particularmente de origen vegetal. Antes de realizar alguna técnica analítica, es importante considerar las siguientes recomendaciones:
a. Asegurarse de que cuenta con los reactivos y equipo necesarios para efectuar la determinación deseada, ya que una vez iniciado un análisis, por lo general no es posible suspenderlo temporalmente en tanto se consiguen los faltantes.
b. Efectúe cada análisis por TRIPLICADO. Es más exacto trabajar con valores promedio que con un sólo dato; si los resultados entre las réplicas varían notablemente, es preferible repetir todo el análisis.
c. Pese la muestra con la mayor aproximación posible. Tres o cuatro decimales nos dan una mayor exactitud en el cálculo. Siempre utilice una balanza analítica.
d. Después de pasar la muestra al recipiente donde se realizará el análisis, pese nuevamente el contenedor a fin de ajustar el peso final. La
2

exactitud en el peso es fundamental para obtener un buen resultado, ya que la mayor parte de las técnicas son gravimétricas.
e. Nunca modifique el tamaño de muestra, los tiempos en las diferentes etapas o el tipo de reactivos, a menos de que se hagan pruebas comparativas que garanticen la precisión de los resultados. Recuerde que los análisis normalmente se basan en métodos estándar aceptados o exigidos por las autoridades reguladoras.
f. Utilice siempre los equipos y accesorios de seguridad requeridos, como son campanas de extracción, máscaras y batas.
g. Recuerde que estará manejando substancias tóxicas o peligrosas para el organismo, por ello extreme las precauciones requeridas según el caso y no omita ninguna.
2. PREPARACION DE LA MUESTRA PARA ANALISIS
La muestra utilizada en el análisis debe ser representativa del total del lote de material, por lo cual se debe de aplicar la metodología apropiada para la toma de muestras. Se recomienda la siguiente rutina para tener una buena representatividad:
a. En lotes a granel menores de 10 ton tomar dos muestras por cada tonelada.
b. En lotes a granel mayores de 10 ton tomar una muestra por tonelada. c. Para materiales encostalados, para 1 a 10 costales tomar muestras de
cada uno; con más de 10 costales muestrear un 10% del total al azar.
Las muestras se deberán tomar de diferentes puntos para que el material sea representativo del total del lote; posteriormente se mezclan perfectamente y se dividen en sublotes de 1–2 kg, se colocan en recipientes herméticos y se almacenan de manera apropiada hasta su análisis. Para cada material se debe llevar un registro para conocer el tipo de proceso al que ha estado sujeto previamente (subproductos industriales), su origen (vegetal, animal, mineral, fármaco) y la parte usada como alimento (principalmente si ha estado sometido a un proceso que impida su reconocimiento). Estos datos son importantes particularmente cuando se están usando productos agrícolas, ya que éstos pueden variar su composición dependiendo de la variedad cultivada, las condiciones de cultivo o la época de cosecha; pueden también contener residuos de pesticidas o estar contaminados por mohos y en el caso de subproductos animales, presentar contaminación por antibióticos y hormonas (Frazer, 1967; Harris, 1980).
2.1. Preparación de la muestra.
Para que un material pueda ser utilizado en el laboratorio de análisis deberá ser preparado de manera apropiada, esto con el fin de que los resultados obtenidos sean representativos del total y puedan ser utilizados de manera confiable para la formulación del alimento o para la valoración del mismo, para lo cual se hacen las siguientes recomendaciones:
3

a. La cantidad de material debe ser adecuada para realizar todos los
análisis necesarios; debe ser una muestra homogénea y representativa.
b. El manejo de la muestra debe ser cuidadoso para evitar cualquier cambio o contaminación.
c. La muestra deberá molerse finamente, tamizarse y mezclarse homogéneamente. Esta operación debe hacerse rápidamente y con la mínima exposición al medio ambiente. Evite su sobrecalentamiento durante el molido, por lo cual materiales sensibles al calor deberán ser molidos a mano. Antes de usar el molino asegúrese de que está perfectamente limpio.
d. Si la muestra contiene mucha humedad y la preparación del material no puede hacerse sin cambios significativos en ésta, determine la humedad antes y después de la preparación.
e. Se recomienda un examen físico macro y microscópico para detectar la presencia de materiales contaminantes.
f. Mezcle la muestra perfectamente y divídala en dos partes iguales. De ser necesario haga un molido preliminar para facilitar esta operación. Almacene una de las partes en un frasco hermético, limpio y seco; la otra parte será usada en los análisis y su tamaño deberá ser adecuado para la totalidad de las pruebas requeridas.
g. Al menos que el método de análisis indique lo contrario, los materiales serán molidos de inmediato y pasados por una malla de 1 mm2; mezcle perfectamente la muestra tamizada y almacénela en un recipiente hermético. Antes de tomar material para cada análisis mézclese nuevamente.
h. Al menos que se señale lo contrario, las muestras húmedas deberán secarse para su molido y tamizado, siguiendo las indicaciones del punto anterior.
i. Las muestras líquidas y semilíquidas deberán conservarse en frascos tapados y mezclarse perfectamente antes de su análisis.
j. Los materiales deberán conservarse en refrigeración o a temperaturas que eviten cambios en su composición. Muestras para análisis de vitaminas u otras substancias sensibles a la luz se colocarán en recipientes de vidrio color ámbar.
3. ANALISIS PROXIMAL. Los análisis comprendidos dentro de este grupo, también conocido como análisis proximales Weende, se aplican en primer lugar a los materiales que se usarán para formular una dieta como fuente de proteína o de energía y a los alimentos terminados, como un control para verificar que cumplan con las especificaciones o requerimientos establecidos durante la formulación. Estos análisis nos indicarán el contenido de humedad, proteína cruda (nitrógeno total), fibra cruda, lípidos crudos, ceniza y extracto libre de nitrógeno en la muestra. Una descripción más amplia de estos análisis se puede encontrar en Osborne y Voogt (1978), MAFF (1982) y AOAC (1984).
3.1. Humedad.
4

Durante el balanceo de la ración, es fundamental conocer el contenido de agua en cada uno de los elementos que la compondrán; así mismo, es necesario vigilar la humedad en el alimento preparado, ya que niveles superiores al 8% favorecen la presencia de insectos y arriba del 14%, existe el riesgo de contaminación por hongos y bacterias (Cockerell et al., 1971). El método se basa en el secado de una muestra en un horno y su determinación por diferencia de peso entre el material seco y húmedo. Aparatos
• Horno de secado. • Desecadores.
Procedimiento
1. Pese alrededor de 5–10 g de la muestra previamente molida. 2. Coloque la muestra en un horno a 105°C por un mínimo de 12 h. 3. Deje enfriar la muestra en un desecador. 4. Pese nuevamente cuidando de que el material no este expuesto al
medio ambiente.
Cálculos Contenido de humedad (%) = 100(((B-A) - (C-A))/(B-A)) Donde: A = Peso de la charolilla seca y limpia (g) B = Peso de la charolilla + muestra húmeda (g) C = Peso de la charolilla + muestra seca (g)
FIGURA 1 Determinación del contenido de humedad en ingredientes alimenticios.
5

3.2. Proteína cruda.
Por su costo es este el nutriente más importante en la dieta en una operación comercial; su adecuada evaluación permite controlar la calidad de los insumos proteicos que están siendo adquiridos o del alimento que se está suministrando. Su análisis se efectúa mediante el método de Kjeldahl, mismo que evalúa el contenido de nitrógeno total en la muestra, después de ser digerida con ácido sulfúrico en presencia de un catalizador de mercurio o selenio. a) Método simple propuesto por Chow et al. (1980) Reactivos
• Oxido de mercurio, grado reactivo. • Sulfato de potasio o sulfato de sodio anhidro, grado reactivo. • Acido sulfúrico (98%), libre de Nitrógeno. • Parafina. • Solución de hidróxido de sodio al 40%; disolver 400 g de hidróxido de
sodio en agua y diluir a 1,000 ml. • Solución de sulfato de sodio al 4%. • Solución indicadora de ácido bórico; agregue 5 ml de una solución con
0.1% de rojo de metilo y 0.2% de verde de bromocresol a un litro de solución saturada de ácido bórico.
• Solución estándar de ácido clorhídrico 0.1N.
Materiales y Equipo
• Unidad de digestión y destilación Kjeldahl. • Matraces Kjeldahl de 500 ml. • Matraces Erlenmayer de 250 ml. • Perlas de ebullición.
Procedimiento
1. Pese con precisión de miligramos 1g de muestra y colóquelo en el matraz Kjeldahl; agréguele 10g de sulfato de potasio, 0.7g de óxido de mercurio y 20 ml de ácido sulfúrico concentrado.
2. Coloque el matraz en el digestor en un ángulo inclinado y caliente a ebullición hasta que la solución se vea clara, continúe calentando por media hora más. Si se produce mucha espuma, adiciónele un poco de parafina.
3. Deje enfriar; durante el enfriamiento adicione poco a poco alrededor de 90 ml de agua destilada y desionizada. Ya frío agregue 25 ml de solución de sulfato de sodio y mezcle.
4. Agregue una perla de ebullición y 80 ml de la solución de hidróxido de sodio al 40% manteniendo inclinado el matraz. Se formarán dos capas.
5. Conecte rápidamente el matraz a la unidad de destilación, caliente y colecte 50 ml del destilado conteniendo el amonio en 50 ml de solución indicadora.
6. Al terminar de destilar, remueva el matraz receptor, enjuague la punta del condensador y titule con la solución estándar de ácido clorhídrico.
6
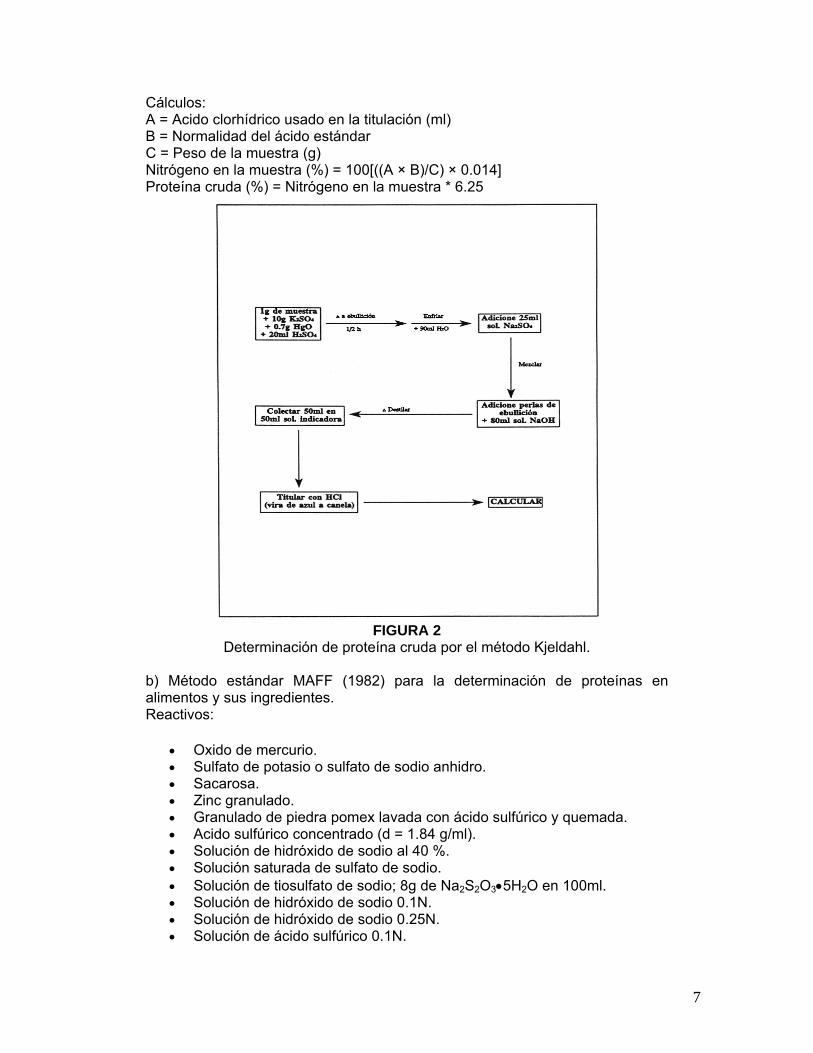
Cálculos: A = Acido clorhídrico usado en la titulación (ml) B = Normalidad del ácido estándar C = Peso de la muestra (g) Nitrógeno en la muestra (%) = 100[((A × B)/C) × 0.014] Proteína cruda (%) = Nitrógeno en la muestra * 6.25
FIGURA 2
Determinación de proteína cruda por el método Kjeldahl.
b) Método estándar MAFF (1982) para la determinación de proteínas en alimentos y sus ingredientes. Reactivos:
• Oxido de mercurio. • Sulfato de potasio o sulfato de sodio anhidro. • Sacarosa. • Zinc granulado. • Granulado de piedra pomex lavada con ácido sulfúrico y quemada. • Acido sulfúrico concentrado (d = 1.84 g/ml). • Solución de hidróxido de sodio al 40 %. • Solución saturada de sulfato de sodio. • Solución de tiosulfato de sodio; 8g de Na2S2O3•5H2O en 100ml. • Solución de hidróxido de sodio 0.1N. • Solución de hidróxido de sodio 0.25N. • Solución de ácido sulfúrico 0.1N.
7

• Solución indicadora de rojo de metilo; disuelva 0.3 g de rojo de metilo en 100 ml de etanol (95–96 % V/V).
• Solución indicadora rojo de metilo-azul de metileno; (a) disuelva 0.2g de rojo de metilo en 100ml de etanol (95–96 % V/V) y (b) disuelva 0.1g de azul de metileno en 100ml de etanol (95–96 % V/V), mezcle un volumen de (a) con uno de (b).
Materiales y Equipo
• Unidad de digestión y destilación Kjeldahl. • Matraces Kjeldahl.
Procedimiento
1. Pese 1g de muestra con aproximación de miligramos y pásela a un matraz Kjeldahl; adicione 10g de sulfato de potasio o sulfato de sodio, 0.6 – 0.7g de óxido de mercurio, 25 ml de ácido sulfúrico y unos pocos granos de piedra pomex.
2. Caliente el matraz moderadamente al principio, agitando ocasionalmente hasta que la materia este carbonizada y las burbujas hayan desaparecido, luego aumente la temperatura y permita que se establezca una ebullición suave. Evite que las paredes del matraz se sobrecalienten para que no se le peguen partículas orgánicas.
3. Cuando la solución se vea clara y sin color, continúe la ebullición por 2 horas más y luego permita que se enfríe. Si después de la digestión y del enfriamiento se cristaliza la solución repita el análisis; si sigue ocurriendo la cristalización repita el análisis usando una mayor cantidad de ácido sulfúrico.
4. Adicione con cuidado al matraz 250–350ml de agua destilada, mezclando el contenido al mismo tiempo; deje enfriar y agréguele unas lentejas de Zinc.
5. Transfiera 25 ml de solución de ácido sulfúrico 0.1 ó 0.5N al matraz de colecta del aparato de destilación, de acuerdo con el valor esperado de Nitrógeno en la muestra, así como unas cuantas gotas de indicador de rojo de metilo.
6. Tomando precauciones para evitar pérdida de amonio, adicione cuidadosamente a la muestra 100 ml de solución de hidróxido de sodio y luego 10 ml de solución de sulfato de sodio o 25 ml de solución de tiosulfato de sodio. Mezcle bien y conecte inmediatamente al aparato de destilación.
7. Caliente el matraz de tal manera que se destilen alrededor de 150 ml del líquido en 30 min. Al finalizar, mida con papel indicador el pH del destilado resultante y si es alcalino continúe con la destilación, la cual se suspenderá cuando el pH aparezca neutro. Durante este proceso agite ocasionalmente el contenido del matraz. Si el destilado se torna alcalino, la determinación deberá ser abandonada y el análisis repetido con los ajustes apropiados.
8. En el matraz de colecta titule el exceso de ácido sulfúrico con hidróxido de sodio 0.1 ó 0.25N, de acuerdo con la normalidad del ácido empleado, al punto final del indicador de rojo de metilo o rojo de metilo-azul de
8

metileno.
9. Corra un blanco de reactivos usando 1g de sacarosa en lugar de la muestra, para usarlo en el cálculo de los resultados.
Cálculos
a. Determine el H2SO4 consumido. 1 ml de ácido ≡ 1.4mg de Nitrógeno. b. Calcule el porcentaje de Nitrógeno en la muestra y conviértalo a
porcentaje de proteína multiplicando el resultado por 6.25. c. Si se sospecha de la presencia de Nitrógeno amoniacal o nitratos en la
muestra, deberán ser evaluados para restarse del Nitrógeno total. Exceptuando los alimentos para rumiantes, se deberá evaluar el contenido de Nitrógeno no proteico y también substraerse del Nitrógeno total.
FIGURA 3 Método estándar MAFF para la determinación de proteína cruda
3.3. Lípidos crudos
En este método, las grasas de la muestra son extraídas con éter de petróleo y evaluadas como porcentaje del peso después de evaporar el solvente. Reactivos, Materiales y Equipo
9

- Eter de petróleo, punto de ebullición 40–60°C.
• Aparato de extracción Soxhlet. • Horno de laboratorio ajustado a 105°C. • Desecador. • Dedales de extracción.
Procedimiento
1. Saque del horno los matraces de extracción sin tocarlos con los dedos, enfríelos en un desecador y péselos con aproximación de miligramos.
2. Pese en un dedal de extracción manejado con pinzas, de 3 a 5g de la muestra seca con aproximación de miligramos y colóquelo en la unidad de extracción. Conecte al extractor el matraz con éter de petróleo a 2/3 del volumen total.
4. Lleve a ebullición y ajuste el calentamiento de tal manera que se obtengan alrededor de 10 reflujos por hora. La duración de la extracción dependerá de la cantidad de lípidos en la muestra; para materiales muy grasosos será de 6 horas.
5. Al término, evapore el éter por destilación o con rotovapor. Coloque el matraz en el horno durante hora y media para eliminar el éter. Enfríe los matraces en un desecador y péselos con aproximación de miligramos. La muestra desengrasada puede usarse para la determinación de fibra cruda.
Cálculos A = Peso del matraz limpio y seco (g) B = Peso del matraz con grasa (g) C = Peso de la muestra (g) Contenido de lípidos crudos (%) = 100((B - A)/C)
10

FIGURA 4
Determinación de lípidos por el método de Soxhlet
3.4. Fibra cruda
Este método permite determinar el contenido de fibra en la muestra, después de ser digerida con soluciones de ácido sulfúrico e hidróxido de sodio y calcinado el residuo. La diferencia de pesos después de la calcinación nos indica la cantidad de fibra presente. Reactivos
• Solución de ácido sulfúrico 0.255N. • Solución de hidróxido de sodio 0.313N, libre de carbonato de sodio. • Antiespumante (ej. alcohol octil o silicona). • Alcohol etílico al 95% (V/V). • Eter de petróleo. • Solución de ácido clorhídrico al 1% (V/V).
Materiales y equipo.
• Matraz de bola fondo plano, 600 ml, cuello esmerilado. • Unidad de condensación para el matraz. • Matraz Kitazato de un litro. • Embudo Buchner. • Crisol de filtración.
11

• Conos de hule. • Papel filtro Whatman No. 541. • Pizeta de 500 ml. • Desecador. • Horno de laboratorio. • Mufla.
Método
1. Pese con aproximación de miligramos de 2 a 3 gramos de la muestra desengrasada y seca. Colóquela en el matraz y adicione 200ml de la solución de ácido sulfúrico en ebullición.
3. Coloque el condensador y lleve a ebullición en un minuto; de ser necesario adiciónele antiespumante. Déjelo hervir exactamente por 30 min, manteniendo constante el volumen con agua destilada y moviendo periódicamente el matraz para remover las partículas adheridas a las paredes.
4. Instale el embudo Buchner con el papel filtro y precaliéntelo con agua hirviendo. Simultáneamente y al término del tiempo de ebullición, retire el matraz, déjelo reposar por un minuto y filtre cuidadosamente usando succión; la filtración se debe realizar en menos de 10 min. Lave el papel filtro con agua hirviendo.
5. Transfiera el residuo al matraz con ayuda de una pizeta conteniendo 200ml de solución de NaOH en ebullición y deje hervir por 30 min como en paso 2.
6. Precaliente el crisol de filtración con agua hirviendo y filtre cuidadosamente después de dejar reposar el hidrolizado por 1 min.
7. Lave el residuo con agua hirviendo, con la solución de HCI y nuevamente con agua hirviendo, para terminar con tres lavados con éter de petróleo. Coloque el crisol en el horno a 105°C por 12 horas y enfríe en desecador.
8. Pese rápidamente los crisoles con el residuo (no los manipule) y colóquelos en la mufla a 550°C por 3 horas, déjelos enfriar en un desecador y péselos nuevamente.
Cálculos A = Peso del crisol con el residuo seco (g) B = Peso del crisol con la ceniza (g) C = Peso de la muestra (g) Contenido de fibra cruda (%)= 100((A - B)/C) Recomendaciones Uno de los problemas más frecuentes durante la evaluación de la fibra cruda es la oclusión de los filtros, por lo que en algunos casos se recomienda sustituir el papel (paso 4 del método) por una pieza de tela de algodón. Para evitar la saturación del crisol de filtración (paso 6) colóquelo ligeramente inclinado y agregue muy lentamente el material a filtrar, de manera que gradualmente se vaya cubriendo la superficie filtrante. Con el uso los crisoles de filtración tienden a taparse. Para su limpieza
12

calcínelos a 500 °C y hágales pasar agua en sentido inverso. Cuando se han tapado con partículas minerales, prepare una solución que contenga 20% KOH, 5% de Na3PO4 y 0.5% de EDTA sal sódica, caliéntela y hágala pasar por el crisol en sentido inverso. Este tratamiento erosiona al filtro de vidrio.
FIGURA 5
Determinación proximal de fibra cruda
3.5. Cenizas
El método aquí presentado se emplea para determinar el contenido de ceniza en los alimentos o sus ingredientes mediante la calcinación. Se considera como el contenido de minerales totales o material inorgánico en la muestra. Materiales y equipo.
• Crisoles de porcelana. • Mufla. • Desecador.
Procedimiento
1. En un crisol de porcelana que previamente se calcinó y se llevo a peso constante, coloque de 2.5 a 5g de muestra seca.
2. Coloque el crisol en una mufla y calcínelo a 550 °C por 12 horas, deje enfriar y páselo a un desecador.
3. Cuidadosamente pese nuevamente el crisol conteniendo la ceniza.
13

Cálculos A = Peso del crisol con muestra (g) B = Peso del crisol con ceniza (g) C = Peso de la muestra (g) Contenido de ceniza (%)= 100((A - B)/C)
FIGURA 6
Determinación del contenido de ceniza en ingredientes alimenticios.
3.6. Extracto Libre de Nitrógeno (ELN)
Dentro de este concepto se agrupan todos los nutrientes no evaluados con los métodos señalados anteriormente dentro del análisis proximal, constituido principalmente por carbohidratos digeribles, así como también vitaminas y demás compuestos orgánicos solubles no nitrogenados; debido a que se obtiene como la resultante de restar a 100 los porcientos calculados para cada nutriente, los errores cometidos en su respectiva evaluación repercutirán en el cómputo final. Cálculo Extracto Libre de Nitrógeno (%) = 100-(A+B+C+D+E) Donde: A = Contenido de humedad (%) B = Contenido de proteína cruda (%) C = Contenido de lípidos crudos (%) D = Contenido de fibra cruda (%) E = Contenido de ceniza (%)
14

3.7. Correcciones
Debido a que los análisis normalmente se hacen con muestras preparadas para tal fin, es necesario realizar ciertas correcciones en los resultados para que reflejen el contenido real de nutrientes en el material en las condiciones en que se usará. a) Humedad Si los análisis se efectuaron en base seca (BS), esto es material deshidratado, es necesario corregir el resultado para expresarlo en base húmeda (BH), tal como se encuentra en el alimento o material para su elaboración, mediante la siguiente expresión: A = Contenido de nutriente (%/BS) B = Contenido de humedad de el material (%) Contenido de nutriente (%/BH) = (A × ((100 - B)/100)) b) Lípidos Cuando se usa material desengrasado, por ejemplo en el análisis de fibra cruda, se aplica una expresión similar a fin de obtener un valor representativo de la muestra: A = Contenido de fibra (desengrasada, %) B = Contenido de lípidos en el material (%) Contenido de fibra ajustado (%) = (A × ((100 - B)/100))
4. ANALISIS ESPECIALES Entre estas técnicas se incluye algunos métodos aplicables a la determinación de la calidad de los ingredientes y los productos terminados, con base en su contenido de micronutrientes, tales como fibra digerible, minerales, vitaminas, actividad enzimática, etc.; así mismo, se presenta el método más ampliamente utilizado para la cuantificación de óxido de cromo durante la evaluación de la digestibilidad de las dietas.
4.1. Determinación de Fibra por Detergente Acido.
Este método permite tener una aproximación del grado de digestibilidad de las fibras en el alimento. La muestra es digerida por medio de cetil-trimetil-amonio en ácido sulfúrico y el residuo es considerado como la fibra no digerible. Reactivos
• Solución de detergente ácido al 1 % p/v. A un litro de agua agregar poco a poco 56 ml de ácido sulfúrico concentrado, lleve la solución a un volumen final de 2 1 con agua y agregue 20g de Cetil-trimetil-amonio.
• Antiespumante Dekalin (decahidronaftaleno). • Acetona.
Materiales y Equipo
• Horno • Matraces erlenmayer de 500 ml. • Mantilla de calentamiento.
15

• Condensador de dedo frío. • Crisoles de filtración, porosidad 1.
Procedimiento
1. Muela la muestra en un molino de martillos para que pase la malla de 1 mm.
2. Tome una submuestra y séquela durante la noche en un horno a 105 °C. 3. Enfríe la muestra en un desecador. 4. Pese con aproximación de miligramos un gramo de la muestra seca y
colóquela en un matraz erlenmayer de 500 ml. 5. Adicione 100 ml de la solución de detergente ácido y 2 ml del
antiespumante. 6. Coloque el matraz en la mantilla con el condensador instalado. 7. Lleve rápidamente a ebullición (3 a 5 minutos) y continúe el
calentamiento por 2 horas. 8. Filtre el contenido del matraz por gravedad en un crisol previamente
pesado. 9. Enjuague el matraz con agua destilada caliente y filtre. 10. Enjuague el contenido del crisol con 300 ml de agua destilada caliente.
Use succión débil (por vacío). 11. Enjuague el residuo con acetona y seque. 12. Coloque el crisol en el horno a 105 °C durante 12 horas. 13. Enfríe dentro de un desecador la muestra e inmediatamente después
determine el peso.
Cálculos Contenido de Fibra (%) = 100 (W2/W1) Donde W1 = Peso de muestra (g). W2 = Peso del residuo (g).
FIGURA 7 Determinación del contenido de fibra por el método de detergente ácido.
16

4.2. Determinación de Fibra por Detergente Neutro.
Este método es útil para la determinación de fibras vegetales en alimentos. Aparentemente tiene la capacidad de separar los componentes nutricionales solubles de aquellos que no son totalmente aprovechables o que dependen de la fermentación biológica para su aprovechamiento. El método tiene limitaciones en su precisión cuando los valores de proteína son muy altos y los valores de fibra son bajos. Reactivos
• Solución detergente neutra. Agregue a un litro de agua destilada 30 mg de sulfato lauril sódico USP, 18.61 g de etilendiamino tetra acetato disódico dihidrogenado dehidratado, 6.81 g de borato de sodio decahidrato y 4.56 g del reactivo fosfato ácido disódico anhidro. Agítese hasta disolver y mantenga el pH entre 6.9 y 7.1.
• Amilasa (Sigma A 1278) 2g en 90ml H2O mas 10 ml de etilenglicol. • Acetona grado reactivo. • Sulfito de sodio anhídro. Se usa únicamente en análisis de subproductos
animales.
Materiales y Equipo
• Equipo de reflujo. Cualquier equipo estándar adecuado para el análisis de fibra.
• Crisoles de filtración en vidrio. Use crisoles de tipo alto, con porosidad gruesa y placa de filtración de 40mm de diámetro con capacidad para 40 – 50 ml de líquido.
• Cuando los crisoles se obstruyen después de un uso continuo se prepara una solución limpiadora de 20% KOH, 5% de Na3PO4 y 0.5% de EDTA de sodio que se hace pasar caliente en sentido opuesto a través del crisol. Se debe de evitar el uso continuo de la solución pues tiende a erosionar el vidrio.
Procedimiento
1. Pese aproximadamente 1 g de muestra molida para pasar el tamiz de 1 mm y deposítela en un matraz de fondo redondo para iniciar el reflujo.
2. Agregue en orden 100 ml de detergente neutro a temperatura ambiente y 2 ml de amilasa por muestra.
3. Caliente para que la solución hierva en 5 a 10 minutos; en el momento de iniciar la ebullición reduzca la temperatura para evitar la formación de espuma. Ajuste la temperatura para que la solución hierva suavemente y mantenga en reflujo por 60 minutos a partir del instante en que empieza a hervir.
4. Agite el matraz para suspender y decante la muestra en un crisol previamente pesado y preparado para succión al vacío. Use poco vacío al principio incrementándolo a medida que lo vaya requiriendo. Pase toda la muestra al crisol utilizando un mínimo de agua caliente (80 °C) para el lavado del matraz. Elimine el vacío, afloje con cuidado la capa de muestra en el fondo del crisol sin removerla y agregue agua caliente,
17

repitiendo el lavado varias veces. Finalmente lave dos veces con acetona sin remover la muestra del filtro y seque con vacío.
5. Seque los crisoles a 105 °C durante 12 h y péselos en caliente (esta operación no debe durar más de 30 seg). Si no se puede pesar de inmediato, enfríe los crisoles en un desecador que contenga como desecante pentóxido de fósforo (P2O5).
6. El residuo de fibra recuperado se registra en términos de paredes celulares.
7. Calcule el contenido celular (material soluble) substrayendo este valor de 100.
NOTA: En el caso de granos y subproductos de molinos es posible que se forme un material gelatinoso derivado de los almidones y la proteína, el cual obstruye el filtro e impide el lavado. Para mejorar la filtración haga pasar aire en sentido inverso en el crisol. Así mismo al evite que la muestra se adhiera al fondo del matraz durante el inicio del calentamiento hasta la ebullición, calentando rápidamente con agitación constante. Cálculos
a. Paredes celulares (%) en “base seca” o “como se ofrece”. 100 ((peso del crisol + paredes celulares) - (peso del crisol))/peso de la muestra.
b. El porciento del contenido celular se calcula substrayendo de 100 el % de paredes celulares.
% contenido celular = 100 - % paredes celulares.Van Soest, P.J. and R.H. Wine, 1967, J. Assoc. Official Anal. Chem., 50:50.
FIGURA 8
Determinación del contendido de fibra por el método de detergente neutro
18

4.3. Ceniza insoluble en Acido Clorhídrico
Este método determina el contenido de substancias minerales insolubles en ácido clorhídrico contenidos en los alimentos; de acuerdo con el contenido mineral de la muestra se aplica ya sea el método A, para alimentos con alto contenido de materia orgánica, o el método B, recomendable para alimentos con un alto contenido de minerales, incluyéndose aquellos cuyo contenido de ceniza insoluble es mayor a 1 % al evaluarse previamente con el método A. Reactivos
• Solución HCI 3N. • Solución A de ácido tricloroacético al 20%, 20 g en 100 ml. • Solución B de ácido tricloroacético al 1 %, 1 g en 100 ml.
Materiales y Equipo
• Placa de calentamiento. • Mufla eléctrica con termostato. • Crisoles de calcinación: Platino o aleación de platino y oro (10% Pt, 90%
Au); Rectangulares (60 × 40 × 25 mm) o circulares (60–75 mm de diámetro con 20 – 25 mm de alto).
Método A Calcine la muestra siguiendo el método de determinación de ceniza. Transfiera el residuo a un vaso de precipitado de 250 – 400 ml usando 75 ml de ácido clorhídrico 3N y evapore a sequedad, continúe calentando durante una hora más para deshidratar cualquier sílice presente. Enfríe y agregue 75 ml de solución de HCI 3N y caliente a ebullición suavemente por 15 minutos. Filtre la solución en caliente a través de un papel filtro libre de ceniza y lave el residuo con agua caliente hasta que el filtrado deje de ser ácido. Seque el papel filtro conteniendo el residuo y calcine a 550–700 °C en un crisol de platino prepesado, enfríe en un desecador y posteriormente pese.
FIGURA 9
Determinación de ceniza insoluble en ácido clorhídrico. Método A.
19

Método B Pese alrededor de 5 g de muestra con una aproximación de 0.001 g y transfiérala a un vaso de precipitado de 250 – 400 ml. Adicione 25 ml de agua destilada y 25 ml de solución de ácido clorhídrico 3N. Mezcle con cuidado y espere hasta que ya no se liberen gases. Agregue otros 50 ml de ácido clorhídrico y espere hasta que cese la liberación de gases y coloque el vaso en un baño maría a ebullición durante 30 minutos, con el fin de hidrolizar los almidones presentes. Filtre la solución cuando todavía este caliente con papel filtro libre de cenizas, posteriormente lave el filtro con 50 ml de agua tibia hasta que el filtrado deje de ser ácido (ver nota). Coloque el papel filtro conteniendo el residuo en un crisol de platino previamente pesado, seque y calcine la muestra a 550 – 700 °C. Transfiera las cenizas a un vaso de precipitado usando 75 ml de ácido clorhídrico 3N y continúe el análisis como en el método anterior a partir de la etapa en la que se calienta la muestra suavemente por 15 minutos. NOTA: Si se hace difícil filtrar, repita el análisis reemplazando los 50 ml de ácido clorhídrico 3N con 50 ml de la solución A de ácido tricloroacético y enjuague el filtro con una solución tibia de la solución B de ácido tricloroacético. Cálculos Calcule el peso de residuo y exprese el resultado como porciento (%) de la muestra.
FIGURA 10
Determinación de ceniza insoluble en ácido clorhídrico. Método B.
20

4.4. Carbohidratos Totales Disponibles (Clegg-Anthrone).
Este método determina la cantidad de carbohidratos totales, basándose en su contenido de almidones hidrolizables y azúcares solubles. Reactivos
• Solución de ácido perclórico al 52 %. 279ml de ácido perclórico (grado específico 1.70) en 100 ml de agua destilada; deje enfriar antes de usar.
• Solución de ácido sulfúrico. 760ml de H2SO4 (grado específico 1.84) en 330ml de agua destilada; deje enfriar antes de usar.
• Reactivo Anthrone. Prepare suficiente reactivo Anthrone preparando una solución de ácido sulfúrico al 0.1 % con el fin de usarla el mismo día.
• Solución estándar de glucosa. Disuelva 100mg de glucosa en 100ml de agua.
• Solución estándar de glucosa diluida. Diluya 10ml del estándar de glucosa a 100 ml de agua destilada (1ml = 0.1mg de glucosa).
Materiales y Equipo
• Espectrofotómetro. • Papel filtro Wathman no. 542 o Schleicher y Schill no. 150.
Procedimiento Extracción:
1. Pese con aproximación de 0.001g 1.0g de muestra seca ó 2.5g de muestra húmeda conteniendo aproximadamente de 60 a 300 mg de carbohidratos totales disponibles.
2. Transfiera cuantitativamente a una probeta graduada de 100 ml con tapón.
3. Adicione 10 ml de agua y agite con una varilla de vidrio para dispersar la muestra.
4. Adicione 13 ml de la solución de ácido perclórico. Agite constantemente con la varilla de vidrio durante 20 minutos.
5. Enjuague la varilla con agua destilada y lleve el volumen a 100 ml. Mezcle y filtre a un matraz volumétrico de 250 ml.
6. Enjuague la probeta graduada con agua destilada y adicione al matraz volumétrico. Afore el matraz con agua destilada y agite.
Determinación:
1. Diluya 10 ml del extracto a 100 ml con agua destilada. Con una pipeta pase a un tubo de ensaye 1 ml del filtrado diluido.
2. Tome con la pipeta dos muestras de 1 ml de agua destilada que servirán como blancos por duplicado y coloque cada uno de ellos en un tubo de ensaye.
3. Tome dos blancos duplicados de 1 ml usando la solución de glucosa diluida.
4. Agregue rápidamente a todos los tubos 5ml de reactivo de anthrone recién preparado. Tape los tubos y mezcle vigorosamente. Colóquelos
21
en un baño maría y caliente durante 12 minutos.
5. Enfríe rápidamente a temperatura ambiente. Transfiera la solución a celdas para espectrofotómetro de 1 cm. El color verde es estable sólo por 2 horas.
6. Lea la absorbancia a 630 nm contra el blanco.
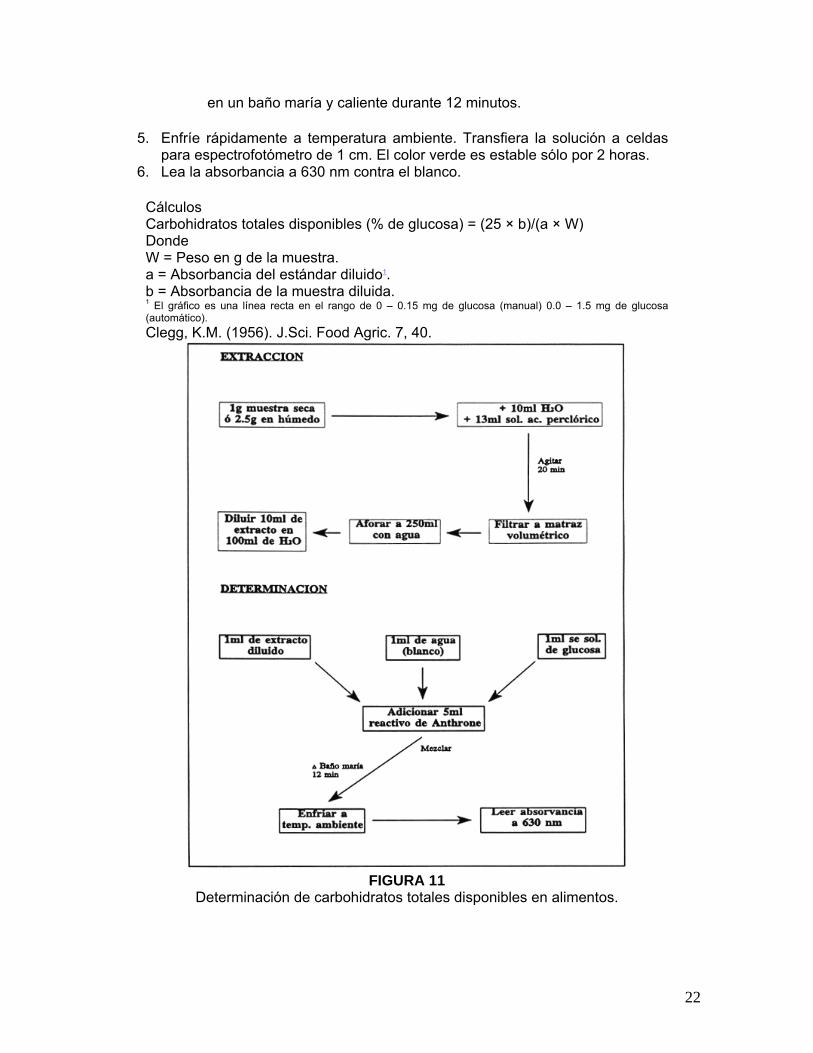
Cálculos Carbohidratos totales disponibles (% de glucosa) = (25 × b)/(a × W) Donde W = Peso en g de la muestra. a = Absorbancia del estándar diluido1. b = Absorbancia de la muestra diluida. 1 El gráfico es una línea recta en el rango de 0 – 0.15 mg de glucosa (manual) 0.0 – 1.5 mg de glucosa (automático). Clegg, K.M. (1956). J.Sci. Food Agric. 7, 40.
FIGURA 11
Determinación de carbohidratos totales disponibles en alimentos.
22

4.5. Nitrógeno Proteico y No Proteico
Esta técnica permite reconocer la porción de nitrógeno de origen proteico y no proteico de los materiales analizados. Puede aplicarse a todo tipo de materiales. Reactivos
• Acetato de cobre monohidratado en solución al 3 % p/v. • Sulfato de aluminio y potasio (2 H2O) en solución al 10% p/v. • Antiespumante de silicona.
Materiales y Equipo
• Matraces kjeldhal de 800 ml. • Embudos buchner de 12 cm de diámetro. • Matraces kitazato. • Papel filtro Whatman no. 541 ó Schleicher y Schill no. 1505, de 18 cm.
Procedimiento Pese 2 g de muestra con más de 25 % de proteína cruda, 1 g para rangos de 25 – 50 % de proteína y 0.5 g para materiales con más de 50 % de proteína cruda. Transfierala a un matraz kjeldahl y agregue aproximadamente 50 ml de agua destilada, unos pocos gránulos de antiebullente y una o dos gotas del antiespumante. Caliente a ebullición durante 30 minutos (teniendo cuidado de que no se seque). Cuando la muestra esté todavía caliente agregue 2 ml de la solución de sulfato de aluminio y potasio, mezcle perfectamente y caliente a ebullición, adicione 50 ml de la solución de acetato de cobre mezclando perfectamente bien y deje enfriar. Filtre usando vacío. Lave el matraz y el precipitado con 50 ml de agua destilada fría. Para evaluar el nitrógeno proteico analice el residuo por medio de la marcha de nitrógeno total de kjeldahl. El nitrógeno no proteico se analiza por medio de la misma técnica a partir del líquido filtrado. Los resultados respectivos de los sólidos y líquidos por medio de kjeldahl ofrecerán directamente los datos de nitrógeno proteico (NP) y no proteico (NNP) respectivamente.
FIGURA 12 Determinación del contenido de nitrógeno proteico y no proteico en alimentos.
23

4.6. Determinación de Proteínas por el Método de Lowry.
El método más preciso para la determinación de concentraciones proteicas es probablemente el método de hidrólisis ácida. La mayor parte de los otros métodos son sensibles a la composición de aminoácidos de las proteínas y no pueden ser obtenidas concentraciones absolutas. El procedimiento de Lowry no es la excepción, pero es sensiblemente constante de proteína a proteína, lo cual ha hecho que sea ampliamente usado y que las determinaciones sean una alternativa completamente aceptable con un rigor absoluto en todas las determinaciones en casi todas las circunstancias en donde se involucren mezclas de proteínas o extractos crudos. Reactivos
• Reactivo de formación compleja. Prepárese inmediatamente antes de usarse, mezclando las soluciones A, B y C en proporción 100:1:1 respectivamente.
• Solución A: 2% (P/V) Na2CO3 en agua destilada. • Solución B: 1% (P/V) CuSO4.5H2O en agua destilada. • Solución C: 2% (P/V) Tartrato de Sodio y Potasio en agua destilada. • Solución de Hidróxido de sodio 2N. • Reactivo de Folín (disponible comercialmente): úsese en concentración
21N. • Estándares: Use una solución madre de proteína estándar (por ejem.
fracción V de albúmina de suero bovino) conteniendo 4 mg / ml de proteína en agua destilada, almacenada a –20 °C. Prepare los estándares diluyendo la solución madre con agua destilada tal como se indica en la siguiente tabla:
PREPARACION DE ESTANDARES DE PROTEINA Solución madre (μl) 0 1.25 2.50 6.25 12.50 25.00 62.50 125.0 250.0 Agua (μl) 500 499 498 494 488 475 438 475 250 Concentración de proteína (μg/ml) 0 10 20 50 100 200 500 1000 2000 Procedimiento
1. A 0.1 ml de muestra o sol. estándar, adiciónele 0.1 ml de sol. NaOH 2N. Caliente a 100 °C durante 10 min a baño maría hirviendo para hidrolizar la muestra.
2. Deje enfriar a temperatura ambiente y agréguele 1 ml del reactivo de formación de complejo recién preparado y deje reposar a temperatura ambiente por 10 min.
3. Adicione 0.1 ml de reactivo de Folín, agite con un mezclador de vórtice y deje reposar a temperatura ambiente durante 30–60 min (no exceda este tiempo).
4. Lea la absorbancia a 750 nm si la concentración de la proteína fuera menor a 500 μg/ml, ó 550 nm si fuese entre 100 y 2000 μg/ml.
5. Trace una curva estándar de absorbancia como función de concentración inicial de proteína y úsela para determinar el contenido de proteína en la muestra.
24

NOTA
a. Si la muestra se encuentra como precipitado, disuélvalo en NaOH 2N e hidrolice como en el paso 1. Use alícuotas de 0.2 ml del hidrolizado para el paso 2.
b. Todas las células u otras muestras complejas pueden requerir un pretratamiento como el descrito por Burton (1984) para DNA.
c. Mezcle rápidamente en cuanto el reactivo de Folín sea adicionado; esto es importante para obtener una adecuada reproductibilidad.
d. Se requiere un grupo de estándares para cada ensayo, preferentemente por triplicado o duplicado.
FIGURA 13
Determinación de proteína en alimentos por el método de Lowry
4.7. Determinación de Urea
Este método permite cuantificar el contenido de urea en ingredientes alimenticios. Reactivos
• Carbón activado • Solución Carrez I: disuelva 21.9 g de acetato de zinc dehidratado en
agua, agregue 3 ml de ácido acético glacial y diluya a 100 ml con agua destilada.
• Solución Carrez II: disuelva 10.6 g de ferrocianuro de potasio en 100 ml de agua destilada.
25

• Acido clorhídrico 0.02N. • Solución de acetato de sodio: disuelva 136 g de acetato de sodio
trihidratado en 1000 ml de agua. • Solución de 4-dimetilaminobenzaldehído: disuelva 1.6 g de 4-
dimetilaminozenzaldehído (4-DMAB) en 100 ml de etanol al 96% y adicione 10 ml de ácido clorhídrico (d = 1.18 g/ml).
• Solución estándar de urea: disuelva 0.1 g de urea en 100 ml de agua.
Materiales y Equipo
• Agitador rotatorio • Espectrofotómetro con celdas de 10 mm.
Procedimiento Pese aproximadamente 2 g de muestra con una precisión de 0.001 g, o una cantidad que se espera contenga de 50 a 200 mg de urea y colóquela en un matraz volumétrico de 500 ml. Agregue 150 ml de ácido clorhídrico 0.02N, agite por 30 min y adicione 10 ml de la solución de acetato de sodio y mezcle. Agregue 1 g de carbón activado, agite perfectamente bien y deje reposar la mezcla por 15 min. Adicione 5 ml de la solución Carrez I, seguido por 5 ml de la solución Carrez II, mezclando perfectamente bien entre las adiciones. Afore con agua destilada y mezcle bien. Filtre una parte con un papel filtro seco en un vaso de precipitado de 250 ml limpio y seco. Determinación Transfiera 10 ml del filtrado a un tubo de ensaye con tapón esmerilado, adicione 10 ml de la solución 4-DMAB, mezcle y deje reposar por 15 min. Mida la absorbancia de la solución a 435 nm en una celda de 10 mm, contra una solución de referencia preparada con los reactivos. Curva de Calibración Diluya 5, 10, 20, 30 y 40 ml de solución de urea a 100 ml de agua. Transfiera 10 ml de cada solución a tubos de ensaye con tapón esmerilado y adicione 10 ml de la solución 4-DMAB a cada uno. Mezcle y deje reposar por 15 minutos, mida la absorbancia como se señaló anteriormente contra una solución de referencia preparada con 10 ml de solución 4-DMAB y 10 ml de agua, haga una gráfica relacionando las absorbancia con la cantidad de urea presente. Expresión de Resultados Determine la cantidad de urea en la muestra por referencia con la curva de calibración elaborada. Exprese los resultados como por ciento de la muestra. % Urea × 0.4665 = % N urea. NOTA: Si la muestra está muy coloreada, la cantidad de carbón activado deberá ser incrementada arriba de 5 g. La solución final después del filtrado deberá ser incolora.
26

FIGURA 14
Determinación del contenido de urea en ingredientes alimenticios.
4.8. Acido Urico.
A través de este método es posible determinar el contenido de ácido úrico y sus sales tanto en gallinaza como en alimentos e ingredientes que los constituyen. Reactivos
• Solución de hidróxido de sodio. Disuelva 50 g de NaOH en 50 ml de agua, mezcle bien y almacene la solución en un envase de plástico.
• Solución neutra de formaldehido.
a. Verifique la concentración de la solución disponible, para lo cual mezcle 30 ml de ésta con 50ml de solución de NaOH IN y 25ml de solución de Peróxido de hidrógeno (20 volúmenes). Caliente en baño de vapor hasta que no haya más efervescencia. Enfríe y titule con NaCl IN usando indicador de fenoftaleína. Realice una titulación de blanco usando 3 ml de agua en lugar del formaldehído y calcule la concentración como sigue:
1ml de la solución de NaOH IN = 0.0300 g de formaldehído.
Conc. de formaldehído (g/100 ml) = ((B-T) × ((0.0300 × 100)/3)
Donde B = Titulación del blanco. T = Titulación de la muestra.
b. Prepare una solución neutra que contenga 17.5g de formaldehído con 250ml de agua y 500ml de etanol. Ajuste el pH a 7.0 con una solución de
27

NaOH 0.1N. Diluya a 1000ml con agua, mezcle y ajuste nuevamente el pH de ser necesario.
• Solución buffer de succinato. Disuelva con calentamiento 29.5g de ácido succínico en 750ml de agua y 20ml de la solución de NaOH. Deje enfriar y adicione una cantidad adecuada de solución de formol que contenga 17.5g de formaldehído, mezcle bien y ajuste el pH a 6.0 con la solución de NaOH. Diluya a 1000ml con agua destilada, mezcle y ajuste el pH si es necesario.
• Solución de tiosulfato de sodio. Disuelva 25g de Na2S2O3.5H2O en 1000ml de agua destilada.
• Solución de lactato de plata. Disuelva con calentamiento 3g de lactato de plata en 50ml de agua destilada y 1ml de ácido láctico. Diluya a 100ml con agua, filtre y almacene en un frasco obscuro. No lo exponga a la luz directa.
• Solución de magnesio amoniacal. Disuelva 8.75g de MgSO4.7H2O y 17.5g de NH4CL en 50ml de agua. Agregue 30ml de NH4OH (d = 0.88g/ml), mezcle bien y diluya a 100ml con agua.
• Reactivo de Benedict y Hitchcock. Mezcle 35ml de solución de lactato de plata con 15ml de solución de magnesio amoniacal. Adicione 50ml de hidróxido de amonio (d = 0.88 g/ml) mézclese bien. Prepárese inmediatamente antes de usarse.
• Solución estándar de ácido úrico. Pese 250 mg de ácido úrico con una precisión de 0.1mg y transfiéralo a un matraz de fondo redondo acoplado a un condensador de reflujo. Adicione 100ml de la solución de formaldehído etílico y caliente a reflujo en un baño de vapor por 30min agitando frecuentemente. Enfríe y transfiera a un matraz volumétrico de 250ml, lavando el matraz redondo con sol. de formaldehído etílico, afore con esta solución y mezcle. 1 ml contiene 1 mg de ácido úrico.
• Eter de petróleo, punto de ebullición 40 – 60 °C.
Materiales y Equipo
• Epectrofotómetro con celdas de cuarzo de 10 mm. • Tubos de cristal para percolación, con aproximadamente 240 mm de
longitud, 18 mm de diámetro interno, y en la parte inferior aproximadamente 120mm de longitud y 8 mm de diámetro interno.
Extracción del ácido úrico:
1. De la gallinaza: Pese con una aproximación de 0.001 g cerca de 0.4g de gallinaza seca y colóquela en un matraz de fondo redondo de 150ml. Adicione 60ml de solución neutra de formaldehído, conecte a un condensador de reflujo y caliente en baño de vapor por una hora. Enfríe y filtre a través de un crisol (Porosidad 4) a un matraz volumétrico de 100ml. Enjuague el matraz 3 veces consecutivas con porciones de 10ml de solución de formaldehído etílico, pasando cada porción por el crisol de filtración al matraz volumétrico. Afore con la misma solución y agite.
2. De los alimentos: Pese 4 a 5g de muestra con una aproximación de 0.001 g y desengrasela por extracción con éter de petróleo. Transfiera
28

cuantitativamente la muestra desengrasada a un matraz de fondo redondo y remueva el solvente residual con una corriente suave de aire. Continúe el análisis como previamente se señaló, iniciando con la adición de 60 ml de solución de formaldehído etílico.
Determinación Transfiera con una pipeta de 20 ml de extracto de la muestra prepesado, según los métodos descritos, a un tubo de centrífuga de 50 ml. Adicione 10 ml del reactivo de Benedict y Hitchcock, mezcle bien y déjelos reposar en la oscuridad por una hora. Centrifuge a 2000 rpm por 15 min. Retire el sobrenadante y deje escurrir por 10 min. Cuidadosamente limpie cualquier líquido remanente sin perturbar el precipitado y adicione 20 ml de solución de tiosulfato de sodio. Disuelva el precipitado, agitando con una varilla de vidrio delgada. Con una pipeta transfiera 5 ml de esta solución a un matraz volumétrico de 200 ml conteniendo 40 ml de solución buffer de succinato, afore con agua destilada y mezcle bien. Mida la absorbancia de la solución a 294 nm en celdas de sílice de 10 mm comparada contra una solución preparada mezclando 5 ml de solución de tiosulfato de sodio con 40 ml de solución buffer de succinato aforada a 200 ml con agua destilada. Determine la cantidad de ácido úrico mediante una curva de calibración. Curva de Calibración: En una serie de tubos de centrífuga de 50 ml de capacidad transfiera con una pipeta 2, 4, 6, 8, 10 y 12 ml de solución estándar de ácido úrico (equivalentes a una cantidad similar de ácido úrico en mg) y llévelos a 20 ml con la solución de formaldehído etílico. Adicione a cada tubo 10 ml de reactivo de Benedict y Hitchcock, mezcle bien y déjelo reposar en la oscuridad por una hora. Continúe el método como en la determinación a partir del centrifugado. Mida la absorbancia de la solución y trace la curva de calibración graficando absorbancia (y) contra la cantidad correspondiente de ácido úrico en mg (x). Expresión de Resultados El contenido de N del ácido úrico como % de la muestra está dado por la fórmula: % N = A/(6 × W) Donde A = mg de ácido úrico (en la alícuota del extracto de la muestra) determinado por la medición fotométrica. W = peso de la muestra en g.
29
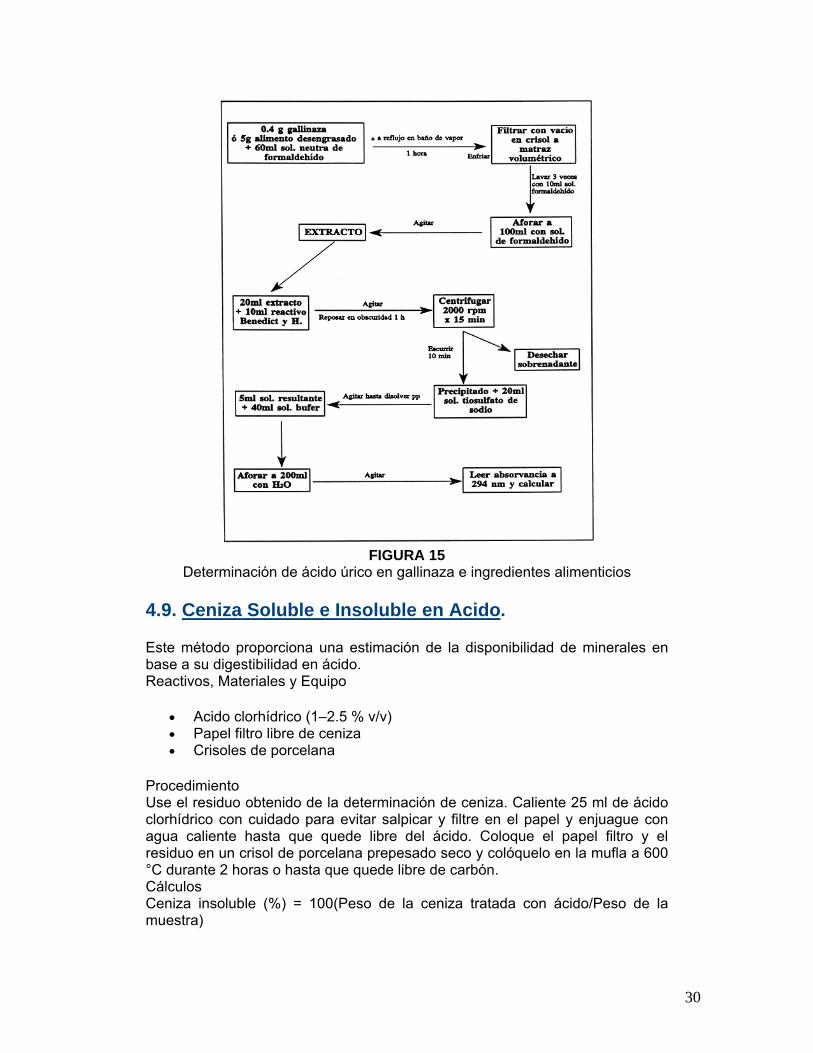
FIGURA 15
Determinación de ácido úrico en gallinaza e ingredientes alimenticios
4.9. Ceniza Soluble e Insoluble en Acido.
Este método proporciona una estimación de la disponibilidad de minerales en base a su digestibilidad en ácido. Reactivos, Materiales y Equipo
• Acido clorhídrico (1–2.5 % v/v) • Papel filtro libre de ceniza • Crisoles de porcelana
Procedimiento Use el residuo obtenido de la determinación de ceniza. Caliente 25 ml de ácido clorhídrico con cuidado para evitar salpicar y filtre en el papel y enjuague con agua caliente hasta que quede libre del ácido. Coloque el papel filtro y el residuo en un crisol de porcelana prepesado seco y colóquelo en la mufla a 600 °C durante 2 horas o hasta que quede libre de carbón. Cálculos Ceniza insoluble (%) = 100(Peso de la ceniza tratada con ácido/Peso de la muestra)
30

FIGURA 16
Determinación de ceniza soluble e insoluble en ácido clorhídrico
4.10. Determinación de Calcio en el Alimento.
La evaluación de calcio en la dieta reviste una gran importancia ya que un desbalance con el fósforo u otros minerales generará un bajo crecimiento. Reactivos
• Acido clorhídrico (1 – 3 %) • Acido nítrico 70% • Hidróxido de amonio (1:1 v/v) • Indicador de rojo de metilo (1 g en 200 ml de etanol) • Solución de oxalato de amonio 4.2 % • • Acido sulfúrico 98 % • Solución estándar de permanganato de potasio 0.05N
Materiales y Equipo
• crisoles de porcelana • matraces volumétricos de 250 ml • vasos de precipitado de 250 ml • Papel filtro para análisis cuantitativos libre de cenizas
Procedimiento Calcine en crisol de porcelana 2.5g de muestra finamente molida. Adicione 40 ml de HCI y unas gotas de HNO3 al residuo, caliente el crisol hasta ebullición, enfríe y transfiera a un matraz volumétrico de 250ml, afore y mezcle. Pase a un vaso de precipitado 100ml de sol. para cereales o alimentos con cereales ó 25ml para alimentos con minerales. Diluya a 100ml y adicione 2 gotas de rojo de metilo. Adicione NH4OH gota a gota hasta que vire a pardo anaranjado,
31

luego adicione 2 gotas de HCl para dar un color rosa. Diluya con 50ml de agua, hierva y adicione con agitación 10ml de sol 4.2% de oxalato de amonio. Ajuste el pH con ácido para regresar al color rosa si es necesario. Déje reposar, filtre y lave el precipitado con la solución de NH4OH (1.5%). Coloque el papel filtro con el precipitado en un vaso, adicione una mezcla de 125ml de agua y 5ml de H2SO4, caliente a 70°C y titule con la solución de permanganato y calcule: Ca (%)= 0.1((ml Sol. Permanganato / Peso de la Muestra) × (Alicuota usada en ml/250)
FIGURA 17
Determinación de calcio en alimento e ingredientes alimenticios.
4.11. Determinación de Fósforo
Al igual que el calcio, este mineral es indispensable para el buen desarrollo de los organismos cultivados, debiendo estar presente en las dietas en cantidades suficientes para satisfacer las necesidades metabólicas de los organismos. Este elemento en proteínas de origen vegetal puede estar formando parte de fitatos, por lo cual su disponibilidad puede estar limitada, por lo que se recomienda una determinación de ácido fítico previa al uso de tales materiales. Reactivos
• Reactivo de Molibdato. Disuelva 40 g de molibdato de amonio 4.H2O en 400 ml de agua caliente y enfríe. Disuelva 2 g de metavanadato de amonio en 250 ml de agua caliente, enfríe y adicione 450 ml de ácido perclórico al 70%. Gradualmente adicione la sol. de molibdato a la sol. de vanadato con agitación y diluya a 2 1.
• Estándar de Fósforo. Prepare una solución stock disolviendo 8.788 g de Ortofosfato dihidrógeno potasio en agua y llévelo a 1 1. Prepare la solución de trabajo diluyendo la sol. stock 1 en 20 (Conc. de trabajo 0.1 mg P//ml).
Materiales y Equipo
32

• Espectrofotómetro para leer a 400 nm • Matraces graduados de 100 ml
Procedimiento
1. Pase una alícuota como en la determinación de Ca a un matraz de 100 ml y adicione 20 ml del reactivo de molibdovanato. Afórelo, mezcle y deje reposar por 10 min.
2. Transfiera alícuotas del estándar de trabajo conteniendo 0.5, 0.8, 1.0 y 1.5 mg de P en matraces de 100 ml y trátelos como al anterior.
3. Lea la muestra a 400 nm ajustando el estándar de 0.5 mg a 100% de transmisión.
4. Determine mg de fósforo en la muestra usando una curva estándar.
FIGURA 18
Determinación de fósoforo en dietas e ingredientes alimenticios.
4.12. Determinación de Cloruro de Sodio
Este método permite determinar la cantidad de sal presente en harina de pescado y otros ingredientes. Reactivos
• Solución estándar 0.1N de Nitrato de Plata • Solución estándar 0.1N de Tiocianato de Amonio
33

• Indicador Férrico - Solución acuosa saturada • Solución de Permanganato de Potasio 6 % p/v • Solución de Urea 5 % p/v • Acetona grado analítico • Acido Nítrico concentrado
Procedimiento
1. Pese 2 g de muestra en un matraz erlenmayer de 250 ml, humedezca la muestra con 20 ml de agua, adicione con pipeta 15 ml de la solución de nitrato de plata y mezcle bien.
2. Adicione 20 ml de Acido Nítrico concentrado y 10 ml de la solución de Permanganato de Potasio y mezcle. Caliente continuamente la mezcla hasta que el líquido se aclare y desaparezca los vapores nitrosos, enfríe.
3. Adicione 10 ml de Solución de Urea y deje reposar por 10 min. 4. Adicione 10 ml de acetona y 5 ml de indicador férrico y titule el exceso
de nitrato de plata con la solución de Tiocianato hasta el punto final rojo-café.
Cálculos Calcule resultados como NaCl %NaCI= (15.00 - ml 0.1N NH4CNS × 0.585)/g de muestra
FIGURA 19
Determinación de cloruro de sodio en harina de pescado y otros ingredientes
4.13. Determinación de Potasio
Reactivos
34

• Acido Clorhídrico concentrado. • Solución estándar de Potasio: Para preparar solución stock (500 ppm),
disuelva 0.477 g de Cloruro de Potasio y llévelo a 500 ml con agua destilada. Para preparar el estándar de trabajo (10 ppm), diluya 1:50.
• Aceite de oliva (puro)
Materiales y Equipo
• Crisoles de Sílice. • Fotómetro de flama • Mufla
Procedimiento
1. Seque 2 g de muestra en un crisol de sílice a 100°C para eliminar la humedad. Adicione unas cuantas gotas de aceite de oliva y caliente sobre la flama hasta que deje de producir flama.
2. Calcine a 500°C en la mufla por 24 h, enfríe y adicione 2 ml de HCI concentrado para disolver el residuo.
3. Llévelo a 100 ml con agua destilada, tome 1 ml de la solución y haga una nueva dilución a 100 ml con agua destilada.
4. Ajuste el fotómetro de flama para que de una lectura de 100 con el estándar de 10 ppm y lea la solución muestra.
5. Si la lectura no se encuentra entre 50 y 100 haga una dilución al momento para dar una lectura apropiada.
FIGURA 20
Determinación del contenido de potasio en ingredientes alimenticios.
4.14. Cuantificación de óxido de cromo en heces y alimentos (Furukawa y Tsukahara, 1966)
35

El óxido de cromo es el marcador más ampliamente utilizado durante la evaluación de la digestibilidad en dietas experimentales para peces. El método aquí presentado es una modificación del propuesto por Furukawa y Tsukahara, a fin de manejar micromuestras durante la determinación del contenido de óxido de cromo en dietas y heces. Reactivos
• Acido nítrico concentrado, gr. • Acido perclórico, g.r.
Materiales y equipo
• Espectrofotómetro • Digestor kjeldahl para tubos de 100 ml • Matraces kjeldahl de 100 ml • Matraces volumétricos de 25 ml
Procedimiento Muela finamente el alimento y las heces, a las cuales se les habrá eliminado previamente escamas y cualquier otra materia extraña; manténgalos a sequedad. Pese con precisión de 0.0001g de 50 a 100mg de muestra, colóquela en un matraz kjeldahl de 100 ml y pese nuevamente la charolilla para ajustar peso de la muestra. Adicione 5ml de HNO3 y ponga a digerir en ebullición suave por un mínimo de 30 min hasta que desaparezcan los vapores amarillentos. En caso de que disminuya notablemente la cantidad de líquido y continúe habiendo vapores nitrosos, adicione otros 5ml de ácido nítrico y siga digiriendo. Al término la solución debe ser clara, de color verdoso y no debe desprender vapores ocres. Deje enfriar. Ya fría la solución, agregar cuidadosamente resbalando por las paredes del matraz 3ml de ácido perclórico. Realizar la adición dentro de una campana de extracción y con mucho cuidado, ya que en caso de una digestión incompleta se puede presentar una reacción explosiva. Coloque nuevamente el matraz en el digestor y continúe la ebullición hasta que la solución vire de verde a amarillo limón; apague el digestor y deje enfriar. Ya frío se debe formar un anillo rojizo en el borde del líquido; en caso de no formarse o si el líquido se torna verde nuevamente, volver a digerir hasta que el cambio sea permanente. Pase el líquido frío a un matraz volumétrico de 25 ml, enjuagando el matraz kjeldahl varias veces con agua destilada y afore. Ajuste a O el espectrofotómetro con un blanco de reactivos y leer a 350 mm. El blanco se prepara simultáneo a la muestras usando solamente los ácidos y agua destilada. Cálculos a) Calcule la cantidad de óxido de cromo (mg) presente en la muestra: X = ((Y - 0.0032)/0.2089)/4 Donde Y = absorbancia 0.0032 y 0.2089 son constantes b) Calcule el % de óxido de cromo en la muestra: O.C.% = 100(X/A)
36

Donde X = peso del óxido de cromo A = peso de la muestra
FIGURA 21
Determinación de óxido de cromo en alimentos y heces.
4.15. Método de Carpenter para la determinación de Lisina Disponible (Tejada, 1985)
El método permite determinar la cantidad de lisina libre que reacciona con el l- fluorodinitro benceno. Reactivos
• Solución madre de Mono E N dinitrofenil - lisina HCI (p. m. 366.77, 39.85 % lisina) Disolver 314 mg en 250 ml de HCl concentrado. Se disuelve lentamente, de manera que hay que iniciar su preparación un día antes.
• Solución estándar diluida de DPN-lisina. Diluya 10 ml de la solución madre en 100 ml de agua destilada para formar el estándar. Hecho lo anterior, 2 ml de la solución contienen alrededor de 0.1 mg de lisina y disuelto a 100 ml da una absorción neta de alrededor de 0–4 a 435 nm en celdilla de 1 cm.
• Solución de 1 - fluoro 2,4 dinitrobenceno (FDNB). Manéjese con guantes de plástico desechables. Sí el FDNB está sólido colóquese en balo de
37

agua caliente para que se funda, tome con una pipeta automática aproximadamente 0.4 ml y disuelva en 15 ml de alcohol etílico por muestra. Debe prepararse diariamente.
• Cloruro de metoxicarbonil (cloroformato de metilo). El MCC es inestable por lo que se debe almacenar en refrigeración. Manéjese con cuidado.
• Solución de bicarbonato de sodio al 8% en agua destilada. • Eter etílico libre de peróxidos. • Solución de fenoftaleína, 500 mg/l de alcohol etílico al 60%. • Solución de hidróxido de sodio al 12%. Se usa para incrementar el pH a
8.5 durante la reacción del MCC. Debe almacenarse en frasco de plástico. Tener a la mano un gotero con HCI 1M para corregir cualquier exceso.
• Buffer de bicarbonato de sodio - carbonato, pH 8.5. Disuelva 19.5g de NaHCO3 y 1g de Na2CO3 en 250 ml de agua destilada; ajuste el pH si es necesario. Almacenar en un frasco bien lleno y bien cerrado para evitar pérdida de CO2
• Acido clorhídrico concentrado
Materiales y Equipo
• Tamiz de 0.5 mm • Matraces de fondo redondo • Matraz volumétrico de 250 ml • Baño maría • Rotoevaporador • Aparato de reflujo • Papel filtro Whatman 541 • Tubos de ensaye con tapón • Perlas de vidrio • Espectrofotómetro
Procedimiento Muela la muestra para que pase el tamiz de 0.5 mm. La cantidad de muestra a utilizar deberá contener alrededor de 12mg de lisina disponible, lo que equivale a 0.3 – 2g de la muestra. Pásela a un matraz de fondo redondo. Agregue 2–3 perlas de vidrio y 10ml de NaHCO3, agitar suavemente para que la muestra no se adhiera a las paredes. Adicione 15 ml de FDNB y agite suavemente durante 2 horas. No permita que la muestra se quede en las paredes. Evapore el etanol pero no el agua; en un baño de agua hirviendo el matraz debe perder aproximadamente 12.5g. Enfríe y adicione 30ml de HCI 8.1M para neutralizar al NaHCO3. Sumando el agua presente, se tendrá un volumen de 40ml y una concentración de ácido de 6M. Reflujar suavemente durante 16 horas. Desconecte el matraz y lave el refrigerante con un poco de agua. Filtre en caliente a un matraz volumétrico de 250 ml. Enjuague el matraz y el filtro varias veces con agua destilada y colecte en el matraz volumétrico; afore en frío con agua destilada. En ocasiones se forma un precipitado de dinitrofenol que se puede extraer con pipeta una vez precipitado, o se elimina con el éter. Colocar 2 ml del filtrado en dos tubos con tapón, etiquetándolos A y B. Agregar
38

al tubo B 5 ml de éter y agita. Extraiga el éter (capa superior) con una pipeta automática. Coloque el tubo en un baño a 90 °C hasta que se elimine todo el éter y enfríe. Agregue una gota de fenoftaleína al tubo, adicione gota a gota la solución de NaOH hasta que aparezca un ligero color rosado; añada 2ml de buffer de carbonato y agregue dentro de una campana 0.5 ml de metilcloroformato. Tapar el tubo y agitar vigorosamente, liberando la presión con cuidado. Después de 5 ó 10 min, adicione gota a gota 0.75 ml de HCI concentrado agitando para evitar la formación de espuma. Se extrae cuatro veces la solución con éter como se describió anteriormente. Elimine el éter residual en baño maría, enfríe y lleve el volumen 10 ml con agua destilada. Durante las pausas de las manipulaciones del tubo B, extraiga tres veces con éter el tubo A; elimine el éter residual y lleve a 10 ml con HCl. Lea la absorvancia de ambos tubos a 435 nm en el espectrofotómetro contra agua destilada. La lectura del tubo menos la del tubo B (blanco) es la absorvancia atribuible al DNP - lisina. Absorbancia del estándar DPN - Lisina Coloque 2 ml de DNP-L diluido en dos tubos A y B siguiendo el procedimiento ya indicado-Practicar hasta que se tenga confianza en las manipulaciones. La absorbancia neta se usará para calcular las muestras problema. El blanco B tiene generalmente una absorbancia de 0.01; si el valor es mucho mayor revisar el procedimiento, especialmente el frasco de servicio, el pH del buffer de carbonato, la longitud de onda a que se está leyendo y el éter libre de peróxidos. Precauciones con alimentos de origen vegetal. En alimentos de origen vegetal en ocasiones no se logra una hidrólisis total a las 16 horas y como un tiempo más largo se descompone el DNP-L, debe hacerse una segunda digestión con materiales desconocidos. Para lograr esto, después de la digestión se filtra, se lava y se procesa el filtrado como se señaló. El residuo se coloca en un matraz de fondo redondo con HCI 6 M y algunas perlas de vidrio (se pueden juntar los residuos de las réplicas); el volumen del ácido clorhídrico debe ser menor a 30 ml. Se deja en reflujo por 16 horas, se filtra en caliente y se lleva a 100 ml según el procedimiento ya descrito. Si el resultado es muy pequeño se elimina, o de lo contrario, se hacen las correcciones necesarias para el material. Para determinar la pérdida de DNP-L se debe calcular un factor de corrección, el cual varía con el material. Para su cálculo, consultar a: Makade, M.L. y Liener, I.E., 1969. Anal. Biochem., 27:271. Cálculos C = (Ws × Au × V × 100 × 100)/Wu × As × a (cp) Donde C= g de lisina/16g de N Ws= Peso del estándar expresado como mg de Lis en 2 ml; 0.1 sí se prepara como se indica. Wu= Peso de la muestra en mg As= Absorción neta del estándar Au= Absorción neta del desconocido V= Volumen del hidrolizado filtrado. Se recomienda 250 ml (menos por la segunda digestión). a= Alícuota del filtrado. Se recomiendan 2 ml. CP= Proteína cruda, 6.25 × g N/100g material.
39

Para expresar el resultado como g lisina/Kg de PC, cambiar un 100 por 10 en la fórmula. NOTA: El material de vidrio puede tomar un tinte amarillento debido al dinitrofenol. Los hidrolizados y soluciones de DNP-Lisina deben protegerse de la luz especialmente a pH alcalino.
FIGURA 22
Determinación de lisina disponible en ingredientes alimenticios.
4.16. Análisis de Melazas
Los peces, según su especie, presentan una capacidad diferencial para utilizar carbohidratos en su dieta, por lo cual se considera importante contar con un método rápido para la determinación de azúcares totales disponibles. Reactivos:
• Solución de Fehling (modificada por Soxhlet): a. Disuelva 34.639g de Sulfato de Cobre 5H2O en agua destilada,
afore a 500ml, filtre. b. Disuelva 173 g de tartrato de sodio y potasio 4.H2O y 50 g de
Hidróxido de Sodio en agua destilada, diluya a 500 ml, deje reposar por 2 días y filtre a través de asbesto preparado.
• Estándar de Azúcar invertida. Prepare una solución stock adicionando 5ml de HCL (Sp. g 1.18) a 9.5g de sacarosa en solución y diluya a 100ml con agua destilada. Después de almacenar por 2 días a temperatura ambiente, afore a 1000 ml con agua destilada. Prepare soluciones de trabajo que contengan 5mg/ml, para lo cual mida con una pipeta volumétrica una alícuota de 100 ml de la solución stock. Vacíe a un matraz volumétrico de 200 ml y neutralice con una solución de NaOH al 20%, usando fenoftaleína como indicador, lleve a volumen y mezcle.
• Acido Clorhídrico concentrado (Sp.g 1.18). • Acido Clorhídrico (0.5N). • Hidróxido de Sodio, Solución 20%.
40
• Indicador de Fenoftaleína (1% en etanol). • Indicador de azul de metileno (1% en agua).
Materiales y Equipo
• Calentador eléctrico • Matraces erlenmayer de 500 ml
Procedimiento Disuelva 8g de melaza y llévela a 500ml con agua destilada (filtre). Hidrolice 100 ml del filtrado con 5ml de HCI (Sp. g 1.18) y deje reposar por 24 h. Neutralice con NaOH al 20% usando fenoftaleína como indicador y diluya a 200 ml. Estandarización de la Solución Soxhlet: Mida 10ml de la solución Soxhlet (a) y (b) con una pipeta volumétrica y vacíe a un matraz erlenmayer, mezcle y agregue 30ml de agua. Adicione con una bureta un volumen de estándar de trabajo casi suficiente para reducir el Cobre en la solución Soxhlet. Llévelo a ebullición y déjelo hervir por 2 min., adicione 4 gotas de azul de metileno y rápidamente complete la titulación mientras está hirviendo, hasta que se obtenga un color naranja brillante. Repita varias veces y determine el volumen de solución requerido para reducir completamente 20 ml de solución Soxhlet. Titulación de Muestra: Primero realice una titulación aproximada; pase a un matraz 10ml de las soluciones (a) y (b), adicione alícuotas de 10ml de la solución muestra. Añada 40 ml de agua y llévelo a ebullición. Si persiste el color azul, titule con una solución estándar de trabajo y calcule el contenido aproximado de azúcar en la muestra. Para determinar con precisión el contenido de azúcar, pase a un matraz 10 ml de las soluciones Soxhlet (a) y (b); adicione una alícuota de la solución muestra. El volumen de muestra usado dependerá del contenido de azúcar en la muestra. Adicione agua como se indica en la tabla, mezcle y hierva. Durante la ebullición, agregue estándar de trabajo con una bureta hasta que la titulación sea casi completa. Añada azul de metileno y complete la titulación.
VOLUMEN DE MUESTRA USADO EN LA TITULACION SOXHLET (AOAC, 1970)
ml de H2O ml de muestra g de muestra en alícuota Total como azúcar invertida (%) 40 10 0.08 73 35 15 0.12 82-58 30 20 0.16 61-41 25 25 0.20 49-35 20 30 0.24 49-29 Calcule el % de azúcar (como invertida) = (F - M) × I × 100/w Donde: F= Volumen de estándar necesario para reducir 20 ml de Solución Soxhlet. M= Volumen de solución estándar de azúcar requerido para completar la titulación I= Peso de la azúcar invertida en 1 ml de Standard de trabajo. W= Peso de la muestra en la alícuota usada.
41

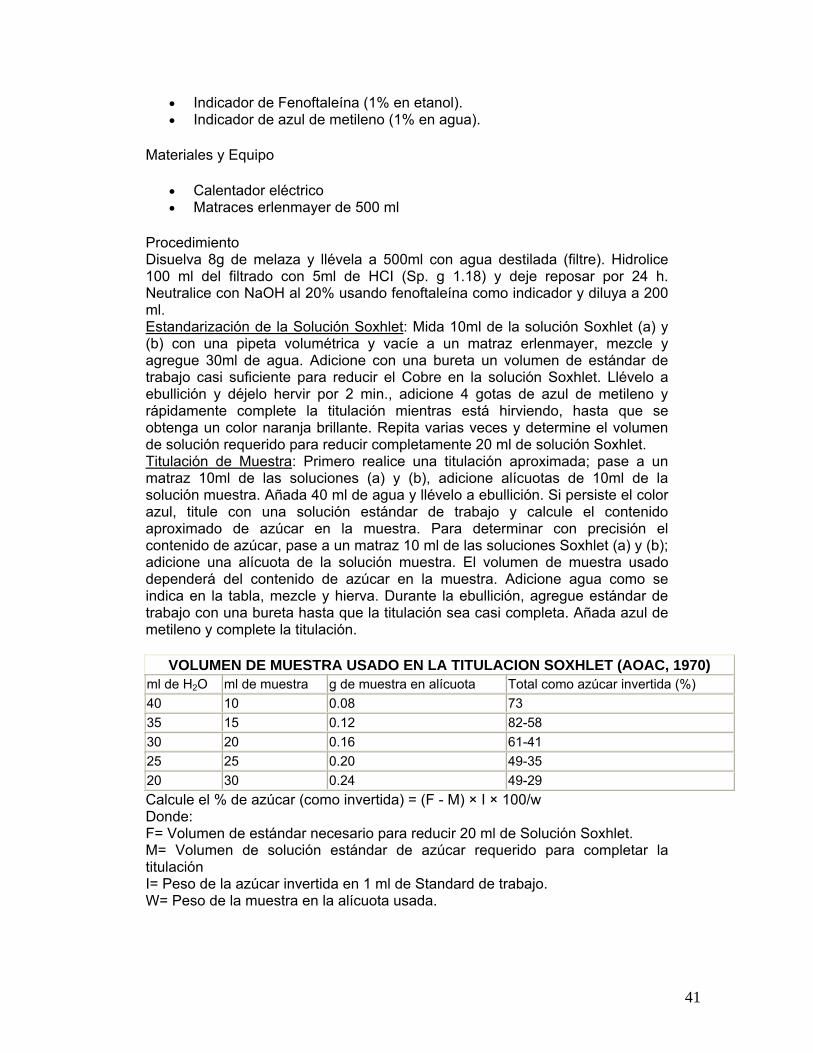
FIGURA 23
Determinación de azúcares totales disponibles en melaza
5. ANTIMETABOLITOS Y TOXINAS EN ALIMENTOS El valor nutricional de un alimento depende en gran medida de la calidad de los ingredientes utilizados, la cual se mide en función de su capacidad de promover el crecimiento y mantener en buen estado a los organismos. En este sentido, el origen y/o el tratamiento previo a que se someten los materiales influirá sobre su calidad, en función de la disponibilidad de nutrientes para usarse en las funciones metabólicas del animal, o también, el contenido de factores tóxicos endógenos característicos de cada material, particularmente aquellos de origen vegetal, que funcionan como antinutrientes afectando negativamente al organismo. En este sentido, existen varias técnicas que permiten conocer la disponibilidad de un nutriente determinado, principalmente aminoácidos, así como también, determinar el contenido de sustancias tóxicas a fin de eliminarlas o desechar el material. En este documento se incluyen los métodos para análisis de los antinutrientes más comunes encontrados en las proteínas vegetales.
5.1. Método Rápido Potenciométrico para Medir Actividad Ureásica en Harina de Soya
42

Este método determina la ureasa residual en harina de soya y sus derivados de acuerdo al método Caskey-Knapp (1944) modificado por AACC (1969) y Quím. Leticia Comar (Nutrimentos del Sureste, Mérida, Yucatán, México). El resultado esta dado en unidades de pH proporcionales a la actividad ureásica. Los valores aceptables oscilan entre 0.05 y 0.5; valores menores indican sobrecocimiento y mayores falta de cocimiento. Reactivos
• Solución bufer de fosfatos 0.05M. Disuelva 3.403g de fosfato de potasio monobásico g.r. (KH2PO4) en 100ml de agua destilada recién hervida. Disuelva 4.355g de fosfato de potasio dibásico g.r. (K2HPO4) en 100ml de agua destilada. Combine ambas soluciones, adicione 10ml de indicador rojo de fenol al 0.1% y afore a 1000ml con agua destilada. Ajuste el pH a 7.0 con un ácido o base fuerte antes de usarse. En refrigeración tiene una vida útil de 90 días.
• Solución bufer de urea. Disuelva 15g de urea g.r. en 500ml de sol. bufer de fosfatos. Ajuste el pH a 7.0 como en el caso anterior.
• Indicador rojo de fenol al 0.1%. Disuelva 0.1g de rojo fenol en 15ml de sol. de hidróxido de sodio 0.02N y afore a 100ml.
Materiales y Equipo
• Baño maría con agitación, precisión ± 0.5°C. • Medidor de pH con electrodo de vidrio y calomel adecuado para medir
muestras de 5ml, precisión de ± 0.02 unidades de pH y compensación de temperatura.
• Tubos de ensaye, 20 × 150mm con tapón de hule • Vasos de precipitado de 10ml
Procedimiento
1. Pese 0.2g de harina de soya finamente molida (sin sobrecalentar) por duplicado y colocar en tubos de ensaye (A y B); adicione al tubo A 10ml de la solución bufer de urea. Tape, agite e incube en baño maría por 30 min a 30 ± 0.5°C.
2. Después de 5 min, adicione al tubo B 10ml de sol. bufer de fosfatos, tape, agite y coloque en el baño maría.
3. Agitar cada uno de los tubos cada 5 minutos (seis agitaciones en 30 min).
4. Retire el tubo A del baño maría, decante el sobrenadante en un vaso de pp de 10ml y mida el pH dentro de un período menor a 3 min. Repita el procedimiento con el tubo B 5 minutos después del A.
5. Calcule el índice de actividad ureásica como unidades de pH resultantes de la diferencia de las lecturas del tubo A menos el tubo B y determine la calidad de la muestra según la tabla de ejemplo incluida:
IAU= pH tubo A (muestra + bufer urea) - pH tubo B (muestra + bufer fosfatos)
43

Grado de cocimiento Unidades de pH pH tubo A pH tubo B Color Cruda 1.9 8.80 6.90 Rojo Subcocida 0.80 7.60 6.80 Rosa Cocido adecuado 0.30 7.10 6.80 Rosa Cocido adecuado 0.08 6.98 6.90 Rosa tenue Sobrecocida 0.03 6.93 6.90 Ambar
FIGURA 24
Determinación de actividad ureásica en harina de soya y sus derivados.
5.2. Gosipol Libre en Harina de Semilla de Algodón
Este antinutriente es característico de la harina de algodón, siendo necesario determinar su contenido debido a sus efectos adversos sobre los organismos alimentados con la semilla, aun cuando los peces aparentemente resisten mayores concentraciones del alcaloide debido al elevado contenido proteico de su dieta. En este documento se describen dos métodos, el primero para harinas normales y el segundo para harinas que han sido tratadas químicamente y contienen dianilinogosipol. Reactivos:
• Acetona acuosa, 7 partes de acetona en 3 partes de agua destilada (v/v).
• Solución de Acetona acuosa - Anilina. En 700 ml de acetona y 300 ml de agua, adicione 0.5ml de anilina purificada. Prepare diariamente.
• Solución acuosa de alcohol isopropílico, 8 partes de alcohol isopropílico + 2 partes de agua destilada (v/v).
• Anilina purificada. Destile anilina grado reactivo sobre una pequeña cantidad de polvo de zinc, descartando el primero y último 10% del destilado. Almacene refrigerada en un frasco ámbar de vidrio. Estable por varios meses.
44

• Solución estándar de Gosipol: a. Disuelva 25 mg de gosipol puro en acetona libre de anilina y
transfiera a un matraz aforado de 250 ml, usando 100 ml de acetona. Adicione 75 ml de agua destilada, afore con acetona y mezcle.
b. Tome 50 ml de la solución (a), adicione 100 ml de acetona pura, 60 ml de agua destilada, mezcle y diluya a 250 ml con acetona pura. La solución (b) contiene 0.02 mg de gosipol/ml y es estable por 24 h en oscuridad.
Materiales y Equipo
• Agitador mecánico. • Espectrofotómetro • Matraces erlenmayer de 250 ml • Matraces Volumétricos de 25 y 250 ml • Baño maría
Procedimiento Muela la muestra para que pase la malla de 1 mm, cuidando de no sobrecalentarla. Tome aproximadamente 1 g de la muestra y agréguele 25 ml de acetona pura. Agite por unos minutos, filtre y divida el filtrado en dos. A una porción, adiciónele una lenteja de Hidróxido de Sodio y caliente en baño maría por unos minutos. Un extracto ligeramente amarillo que no cambia de color con el Hidróxido indica que la harina no ha sido tratada, proceda con el método 1. Si se obtiene un color rojo-anaranjado profundo, indica la presencia de dianilinogosispol y requiere del método 2. Método 1 Pese 0.5–1 g de muestra, dependiendo de la cantidad de gosipol esperada, en un matraz erlenmayer y adicione 2 ó 3 perlas de vidrio. Agregue 50 ml de solución de acetona acuosa, tape el matraz y agite por una hora. Filtre, deseche los primeros mililitros de filtrado y luego tome dos alícuotas iguales (2–10 ml, dependiendo del contenido de gosipol esperado) y páselas a matraces volumétricos de 25 ml. Diluya una de las alícuotas, afore a volumen con alcohol isopropílico acuoso (solución a), mientras la otra alícuota (solución b). Adiciónele 2 ml de anilina purificada, caliente en baño maría (90–100°C) por 30 min. junto con un blanco conteniendo 2 ml de anilina y un volumen de solución acuosa de acetona igual a la alícuota. Retire la solución b y el blanco, adicione suficiente alcohol isopropílico acuoso para efectuar solución homogénea, y enfría a temperatura ambiente en un baño con agua. Afore con alcohol isopropílico acuoso. Lea las muestras a una absorbancia de 400 nm en el espectrofotómetro. Ajuste el instrumento a 0 absorbancia con alcohol isopropílico acuoso y determine la absorbancia de la solución (a) y el blanco de reactivo. Si el blanco está abajo de 0.022 de absorbancia continúe con el análisis, de lo contrario repita el análisis usando anilina recién destilada. Determine la absorvancia de la solución (b) con el blanco de reactivo ajuste a 0 absorvancia. Calcule la absorvancia corregida de la alícuota: absorbancia de la solución (b) menos absorbancia de la solución (a). Determine los mg de gosipol libre presentes en la solución de la muestra usando la curva de calibración. Método 2.
45

Pese 1 g de muestra en un matraz erlenmayer, agregue 50 ml de acetona acuosa, agite y filtre como el anterior. Agregue alícuotas duplicadas del filtrado (de 2 a 5 ml, dependiendo de la cantidad esperada de gosipol libre) a matraces volumétricos de 25 ml. Afore una de las alícuotas (solución a) con el alcohol isopropílico acuoso y déjelo reposar 30 minutos antes de leerlo en el espectrofotómetro. Afore la otra alícuota (solución b) como en el procedimiento (1), determine las absorbancia de las soluciones a y b como el procedimiento anterior y calcule el contenido aparente de gosipol de ambas soluciones usando la curva de calibración. Preparación de la Curva de Calibración. Tome alícuotas duplicadas de 1, 2, 3, 4, 5, 7, 8 y 10 ml de estándar de gosipol 0.02 mg/ml y colóquelas en matraces volumétricos de 25 ml. Diluya una serie (solución A) aforando con alcohol isopropílico acuoso y determine la absorbancia como en el anterior. A la otra serie (solución b) adiciónele 2 ml de anilina purificada y proceda como anteriormente. Prepare un blanco de reactivos usando 2 ml de anilina y 10 ml de acetona acuosa, calentada junto con el estándar. Determine absorbancia como en el procedimiento (1) y calcule la densidad óptica corregida para cada solución estándar: Absorbancia Corregida = (Abs. solución b - Abs. solución a) Trace la curva estándar, graficando absorbancia corregida contra concentración de gosipol en 25 ml. Calule % de gosipol libre en harinas normales. Gosipol libre % = 5G/WV Donde: G= Lectura en la Gráfica W= Peso de la muestra V= Volumen de la alícuota usada. Para harinas químicamente tratadas. Gosipol libre % = 5(B - A)/WV Donde: A = mg Gosipol libre aparente en alícuota (a) B= mg Gosipol libre aparente en alícuota (b)
FIGURA 25
Determinación de gosipol libre en harinas de semilla de algodón.
46

5.3. Determinación de Tioglucósidos
El método descrito proporciona el contenido aproximado de tioglucósidos pero no determina tioglucósidos e isocianatos individuales. Reactivos y Aparatos
• Solución Cloruro de Bario 5% • Matraces volumétricos de 600 ml • Baño de vapor
Procedimiento
1. A 10 g de harina (desengrasada por extracción Soxhlet) adiciónele 250 ml de agua destilada, hidrolice a 54 °C por 1 h y caliente a ebullición por 2 h., manteniendo el volumen constante.
2. Filtre, reteniendo el filtrado y lave el residuo tres veces con 50 ml de agua caliente, agregando el líquido al filtrado inicial y afore a 600 ml.
3. Precipite el sulfato de bario por calentamiento y adicionando un exceso de solución de Cloruro de bario. Déjelo en un baño de vapor por algunas horas y filtre.
4. Calcine en una mufla y pese el precipitado.
Calcule el contenido aproximado de tioglucósido como: % Tiogl = (M. wt. tioglucósido × Peso de BaSO4) × 100/M.wt. BaSO4 × Peso Muestra
FIGURA 26
Determinación de tioglucósidos en ingredientes alimenticios.
5.4. Análisis de Aflatoxinas
47

El método que se incluye es adecuado para materiales como harina de cacahuate, harina de copra y harina de palma kernel; para detalles del método, así como para procedimientos alternativos, refiérase a Methods of Aflatoxin Analysis por B.D. Jones (1972), Report No. G70, Tropical Products Institute, London. Reactivos:
• Cloroformo (G.R.) • Eter etílico (G.R.) • Mezcla cloroformo/metanol (95/5 v/v) • Celita, tierra de diatomeas • Gel de sílica “G” (Merck) • Estándares Cualitativos. Permiten distinguir marcas de aflatoxina de
otras manchas fluorescentes que puedan estar presentes. Se puede usar con este propósito harina de cacahuate conteniendo aflatoxina B obtenible en el Inst. de Productos Tropicales de Londres.
Materiales y Equipo
• Placas para cromatografía de capa fina 20 × 20 cm. • Lámparas UV pico de emisión a 365 • Botellas de boca ancha de 250 ml • Micropipetas • Agitador • Rotoevaporador
Procedimiento
1. Pese 10 mg del material en un frasco de boca ancha y mézclelo con 10 ml de agua (si se usa material con alto nivel de grasa, será necesaria una extracción previa con soxhlet). Adicione 100 ml de cloroformo y agite por 30 min.
2. Filtre el extracto a través de la celita, tome 20 ml del filtrado y llévelo a 25 ml (solución a). Tome otros 20 ml del filtrado y concéntrelos a 5 ml (solución b).
3. Prepare placas de cromatografía agitando 100 g de gel de sílica “G” con 220 ml de agua por 20 min y aplicando la mezcla a las placas con el aparato apropiado, con un grosor de 509μ. Déjela reposar por 1 hora y seque a 100°C.
4. Coloque gotas de 10 y 20 μl de solución b y 15 y 10μl de solución a en la placa, junto con una gota de estándar cualitativo, en una línea a 2 cm del fondo de la placa y al menos a 2 cm de cada lado. Realice la aplicación bajo luz tenue.
5. Revele la placa en éter etílico a una altura de 12 cm, déjela secar en luz tenue y luego vuélvala a revelar en mezcla de cloroformo/metanol hasta una altura de 10 cm desde la línea basal.
6. Examine la placa en un cuarto obscuro a 30 cm de la fuente de UV. La presencia de una mancha azul fluorescente a Rf 0.5–0.55 indica aflatoxina B (asegúrese de que la mancha del estándar se encuentre
48

también en ese rango). la presencia de una segunda mancha a Rf 0.45 – 0.5 indica aflatoxina G.
La toxicidad de una muestra puede ser clasificada en términos de aflatoxinas B y G de acuerdo con la tabla siguiente:
TABLA COMPARATIVA PARA DETERMINAR TOXICIDAD DE AFLATOXINAS B Y G Conc. de Aflatoxina (mg/kg)
Volumen aplicado (ml) No fluoresencia c/fluoresencia Toxicidad de fluorescencia observada
5 (sol. A) <1000 >1000 Muy alta 10 (sol. A) <500 500–1000 Alta 10 (sol. B) <100 100–500 Media 20 (sol. B) <50 50–100 Baja Tomado de Harris, 1980
FIGURA 27
Determinación cromatográfica de aflatoxinas en ingredientes alimenticios.
5.5. Método Colorimétrico para la Determinación de Mimosina (Matsumoto y Sherman, 1951).
La mimosina es un aminoácido libre muy común en algunas leguminosas, incluyendo a Leucaena leucocephala y L. glauca, consideradas excelentes fuentes de proteína para la alimentación animal. Su presencia limita el uso de las hojas y semillas en la alimentación de monogástricos, ya que afecta la función de la tiroides, provocando reducción del crecimiento. Reactivos
• Solución de Mimosina, 0.1% en HCl 0.1N (mimosina de L. glauca recristalizada)
• Solución de Cloruro férrico al 0.5% en HCl 0.1N • Carbón Activado. • Acido Clorhídrico 0.1N.
49

Materiales y Equipo
• Colorímetro (usar espectrofotómetro 535 nm) Klett-Summerson con Filtro No. 54.
• Matraz Kitazato. • Tubos de ensaye. • Crisol de Filtración. • Vasos de precipitado de 250 ml. • Vasos de precipitado de 150 ml. • Matraz Volumétrico de 250 ml. • Matraz Volumétrico de 100 ml.
Procedimiento Extracción: Coloque 1.25 g de hoja seca de leucaena en un Vaso de precipitado de 250 ml y digiera con 100 ml de HCl 0.1N por hora con agitación constante, deje enfriar y transfiera todo a un matraz volumétrico de 250 ml, agite bien y deje reposar hasta que los sólidos se asienten en el fondo. Clarificación: Transfiera 10 ml de sobrenadante a un V.P. de 150 ml con 30 mg de Carbón. Agregue agua para llevar el volumen a 25 ml, cúbralo con un vidrio de reloj y caliente a ebullición por 15 min., deje enfriar y filtre con vacío en el crisol de filtración; el filtrado es colectado en un tubo colocado en el matraz kitazato. Enjuague el vaso de precipitado y el material en el crisol con 10 ml de HCl 0.1N dividido en tres porciones. Enjuague finalmente el crisol con 5 pequeñas porciones de agua. Determinación Colorimétrica: Transfiera el líquido obtenido a un matraz volumétrico de 100 ml, agregue 4 ml de solución de cloruro férrico al 0.5% en HCl 0.1N y afore con agua. El pH deberá ser entre 1.5 y 2.5. Lea en el Colorímetro y calcule la cantidad de mimosina en una curva de calibración. Curva de Calibración: Disuelva 0.1046 g de mimosina pura en HCl 0.1N y afore a 100 ml con el ácido. La solución contiene 0.1% de mimosina. Coloque 1 ml de solución de mimosina en un matraz volumétrico de 100 ml y adicione 10 ml de HCl 0.1N y 4 ml de la solución de cloruro férrico al 0.5% y afore con agua. Lea la intensidad del color en el Colorímetro (espectro 535 nm). Use agua destilada como referencia y repita el procedimiento usando 2,3,4 y 5 ml de solución de mimosina. Se obtiene una línea al graficar la lectura del Colorímetro contra la concentración.
50

FIGURA 28
Determinación colorimétrica de mimosina.
5.6. Método Colorimétrico para la Determinación de Canavanina (Archibald, 1946)
Las semillas y hojas de leguminosas y en particular las de canavalia (Canavalia ensiformis) y sesbania (Sesbania spp.) contienen cantidades variables del aminoácido libre canavanina, el cual es tóxico para animales monogástricos. El método permite determinar la cantidad del antinutriente en ingredientes alimenticios. Reactivos
• Buffer fosfato 1 M, pH 7.2. Mezcle 34.5 g de NaH2PO4.H2O con 130.6g de K2PO4 y ajuste el volumen a 1 litro
• Solución de nitroprusiato de sodio al 2%. Se debe usar solución recién preparada. Es estable por una semana cuando se almacena en obscuridad a 0–4°C.
• Peróxido de hidrógeno (30% H2O2) • Solución de carbonato de potasio al 20% • Solución stock de canavanina. 2.5mg de canavanina/ml. Almacenar en
hielo seco • Solución estándar de canavanina. Lleve 1 ml de la solución stock a 10
ml con agua • Reactivo de acuoprusiato. Mezcle 0.5ml de la solución de carbonato al
20% y 0.4 ml de H2O2 con 10ml de la solución de nitroprusiato. Deje reposar por 30 min a temperatura ambiente (20–25°C). Deberá prepararse diariamente. El uso de cantidades mayores de peróxido disminuye el color obtenido con la canavanina.
Materiales y Equipo
51

• Tubos de ensaye • Colorímetro • Espectrofotómetro
Procedimiento
1. Ponga en un tubo de ensaye una muestra de 1 ml de extracto de la muestra, conteniendo 0–0.25 mg de canavanina, se le adicionan 0.5 ml de buffer fosfato y 0.5 ml de reactivo de acuoprusiato. Maneje de la misma manera blancos conteniendo 0.2, 0.5, 0.8, y 1 ml de solución madre diluida y llevados a 1 ml con agua, así como también un blanco de reactivos con 1 ml de agua, tratados con el fosfato y acuoprusiato.
2. Mezcle vigorosamente y guarde los tubos en un gabinete obscuro; se desarrollará una coloración rojiza en presencia de canavanina.
3. Después de 2 horas lea en un colorímetro o espectrofotómetro. En este último, ajuste a una longitud de onda de 520 nm y el blanco a una densidad óptica de cero. Puede usar cubetas del espectrofotómetro para desarrollar el color; si usa cubetas de más de 2 ml, puede adicionarles hasta 10 ml de agua después de las 2 horas de incubación para desarrollo del color.
Cálculos Se grafica la densidad óptica de cada estándar contra su cantidad correspondiente de canavanina y el contenido de canavanina de las muestras se calcula a partir de la curva.
FIGURA 29
Determinación colorimétrica de canavanina en ingredientes alimenticios.
52

5.7. Determinación de Acido Cianhídrico
El ácido cianhídrico se encuentra de manera natural en diferentes materiales como son la semilla de lino, harina de yuca y algunas semillas de leguminosas. Este método permite determinar el ácido libre o en forma de glucósido, para lo cual, la muestra se suspende en agua, se libera el ácido mediante enzimas y se separa por destilación para colectarse en una solución de nitrato de plata acidificada. Reactivos
• Antiespumante (Silicona) • Acido nítrico (d= 1.42g/ml) • Solución de amonio: Prepare diluyendo un volumen de hidróxido de
amonio (d=0.88 g/ml) en dos volúmenes de agua. • Sulfato férrico amoniacal, solución saturada • Suspensión de almendras dulces. Triture 20 almendras dulces
blanqueadas en 100 ml de agua a 37–40 °C. Asegúrese de que no existe ácido cianhídrico en 10 ml de la suspensión usando papel de picrato de sodio o corriendo un blanco testigo como se describe más adelante
• Solución de acetato de sodio, neutral a la fenoftaleína: 10g de acetato de sodio anhidro en 100 ml de agua destilada.
• Solución 0.02N de tiocianato de amonio • Solución 0.02N de nitrato de plata.
Materiales y Equipo
• Estufa regulada a 37–38 °C • Aparato para destilación de vapor • Matraz de fondo plano de 1000 ml con tapón esmerilado • Baño de aceite • Bureta graduada a 0.05 ml
Procedimiento Pese con exactitud de 0.005 g aproximadamente 20 g de la muestra preparada, colóquela en un matraz de fondo plano de 1000 ml, adicione 50 ml de agua y 10 ml de suspensión de almendras dulces. Tape el matraz y póngalo en la estufa durante 16 horas a 37–38 °C. Enfríe a temperatura ambiente, adicione 80 ml de agua, 10 ml de solución de acetato de sodio y una gota de antiespumante. Conecte el matraz al aparato de destilación y colóquelo en el baño de aceite previamente ajustado a una temperatura ligeramente superior a 100 °C; destile 200–300 ml de líquido pasando una corriente de vapor a través del matraz y calentando suavemente el baño de vapor. Colecte el destilado en un matraz erlenmayer protegido de la luz conteniendo exactamente 50ml de solución de nitrato de plata 0.02N y 1ml de ácido nítrico. Asegúrese de que la extensión del condensador esté sumergida en la solución de nitrato de plata. Transfiera el contenido del matraz erlenmayer a un matraz aforado de 500 ml y llévelo al volumen con agua, mezcle y filtre. Remueva 250 ml del filtrado, adicione aproximadamente 1 ml de sulfato férrico amoniacal y titule el exceso de nitrato de plata con la solución de tiocianato de amonio. Si se desea, se
53

puede correr un blanco siguiendo el mismo procedimiento con 10ml de suspensión de almendras dulces omitiendo la muestra. Expresión de resultados Si el blanco indica que la solución de nitrato de plata ha sido consumida, reste su valor del volumen consumido por la destilación de la muestra. 1 ml de 0.02N AgNO3 ≡ 0.54mg de HCN. Exprese el resultado como porcentaje de la muestra. NOTA: Si la muestra (ej. frijoles) contiene una elevada cantidad de azufre, se formará un precipitado negro de sulfato de plata, el cual se filtra junto con el depósito de cianuro de plata. La formación de este precipitado consume solución de nitrato de plata y su efecto debe ser eliminado para el cálculo de contenido de HCN, para lo cual, trate el depósito sobre el papel filtro con 50ml de amoníaco a fin de disolver el cianuro de plata. Lave el residuo en amoníaco diluido y determine su contenido de plata. Convierta el valor a ml de solución 0.02N de solución de nitrato de plata y résteselo al volumen de solución de nitrato de plata consumida por el destilado de la muestra.
FIGURA 30
Determinación de ácido cianhídrico en ingredientes alimenticios.
5.8 Taninos en Plantas y Forrajes. (Krishna y Ranjhan, 1980)
Los taninos son glucósidos de polipéptidos solubles en agua presentes en muchas plantas, con un sabor agrio astringente, confiere sabor y olor indeseable a los alimentos. Afectan la utilización de las proteínas debido a que ligan a la lisina, haciéndola indisponible para animales monogástricos. Reactivos
54

• Solución 0.1N de ácido oxálico. 1 ml = 0.006235 g de ácido quercitánico ó 0.0008g 02 absorbido
• Solución de permanganato de potasio. Disuelva 1.333g de KMnO4 en un litro de agua y estandarice contra el ácido oxálico.
• Solución índigo. Disuelva 6g de indigotin disulfonato de sodio en 500ml de agua mediante calentamiento, enfría y adicione 50ml de ácido sulfúrico. Diluya a 1 litro y filtre.
Procedimiento
1. Extraiga 2g de muestra durante 20 horas con éter anhidro. Hierva el residuo por 2 horas con 300ml de agua, enfríe, diluya a 500 ml y filtre.
2. Mida 25 ml de la infusión dentro de una cápsula de porcelana de 2 litros, adicione 20ml de la solución de índigo y 750ml de agua. Adicione la solución estándar de permanganato, 1 ml a la vez, hasta que cambie el color azul a verde, luego agregue otras gotas hasta que se torne de color amarillo dorado.
3. De manera similar, titule una mezcla de 20ml de solución de índigo y 750ml de agua. Multiplique la diferencia entre las titulaciones por el factor deseado para obtener ácido quercitánico u oxígeno absorbido.
55

FIGURA 31
Método simple para la determinación de taninos en ingredientes alimenticios
5.9. Método Simple para la Determinación de Acido Fítico (Wheeler y Ferrel, 1971).
El ácido fítico se puede encontrar en algunas semillas como ajonjolí, trigo, arroz, avena, sorgo, cebada, etc. Su presencia puede provocar deficiencias de fósforo en monogástricos por ligar y formar fitatos indigeribles que hacen indisponible ese elemento; afecta también la disponibilidad de calcio, magnesio o el fierro con los que forma el complejo fitina. Reactivos
• Solución de ácido tricloroacético (ATA) al 3% • Solución concentrada de FeCl3 • Solución de sulfato de sodio al 3% en ATA • Solución 1.5N de hidróxido de sodio • Solución 3.2N de HNO3
56

• Solución 1.5M de KSCN • Solución FeCl3 - ATA que contenga 2 mg de Fe2 en sol 3% ATA
Materiales y equipo
• Matraz erlenmayer de 125 ml • Tubos cónicos para centrífuga • Matraz volumétrico de 100 ml • Baño maría • Agitador mecánico • Espectrofotómetro • Centrífuga de laboratorio • Embudos • Papel filtro Whatman 2
Procedimiento
1. Coloque en un matraz erlemayer de 125 ml una muestra finamente molida (40 mallas), estimada para contener de 5 a 50 mg de fitato-P. Extraiga durante 30 min con 50 ml de solución al 3% de ATA bajo agitación constante.
2. Centrifuge la suspensión y transfiera una alícuota de 10 ml del sobrenadante a un tubo de centrífuga cónico. Agréguele 4 ml de la solución de FeCl3 preparada para contener 2 mg de Fe2 soplando rápidamente la pipeta.
3. Caliente durante 45 min el tubo con la muestra en un baño maría a 90–100°C. Si el sobrenadante no está claro después de 30 min adicione 1 ó 2 gotas de la sol. al 3% de sulfato de sodio en ATA y continúe con el calentamiento. Centrifuge por 10–15 min y decante con cuidado el sobrenadante. Lave dos veces el precipitado con 20–25 ml de solución de ATA al 3% dispersándolo bien y calentándolo en agua a ebullición por 5–10 min y centrifuge. Finalmente repita una vez el lavado con agua.
4. Disperse el precipitado en un poco de agua destilada, adicione 3 ml de la solución 1.5N de NaOH y mezcle. Lleve el volumen a aproximadamente 30 ml con agua destilada y caliente en agua a ebullición durante 30 minutos. Filtre en caliente (cuantitativamente) con un papel de retención moderada (Whatman 2 ó SyS 597).
5. Lave el precipitado con 60–70 ml de agua destilada caliente y deseche el filtrado. Si se desea hacer determinación de fósforo se puede usar el filtrado. Disuelva el precipitado en el papel con 40 ml de la solución 3.2N de HNO3 pasándolo a un matraz volumétrico de 100 ml. Lave varias veces el papel con agua destilada colectándola en el matraz. Enfríe la muestra a temperatura ambiente y afore con agua destilada.
6. Transfiera una alícuota de 5 ml a un matraz volumétrico de 100 ml y diluya a aproximadamente 70 ml. Agregue 20 ml de la solución 1.5M de KSCN y afore con agua destilada. Lea de inmediato (dentro de un lapso de 1 min) en el espectrofotómetro a 480 nm.
7. Corra un blanco de reactivos con cada muestra. Calcule el contenido de fierro a partir de un estándar de Fe(NO3)3 corrido al mismo tiempo o mediante una curva estándar preparada con anterioridad.
57

Cálculos Calcule el fitato-P a partir de los resultados de fierro, asumiendo una tasa molecular de 4:6 fierro:fósforo
FIGURA 32
Método simple para la determinación de ácido fítico en ingredientes alimenticios.
5.10. Determinación de Saponinas en Vegetales y Alimentos (Fenwick y Oakenfull, 1983)
Las saponinas se pueden encontrar en una gran variedad de plantas incluyendo la alfalfa, soya y semillas de leguminosas, habiéndose demostrado un efecto negativo sobre el crecimiento de monogástricos. Es un antinutriente estable al calor. Reactivos
• Acetona G.R. • Metanol • Solución n-butanol-metanol-amoníaco concentrado (3.5:1:2.5) • Solución estándar de saponina purificada en metanol • Solución de ácido sulfúrico en metanol (100 ml por litro)
Materiales y equipo
• Extractor soxhlet • Rotoevaporador • Densitómetro con graficador • Planímetro • Desecador
58

• Placa de gel de sílica para cromatografía (Kieselgel 60 F-254 Merck)
Procedimiento Muela finamente la muestra y séquela a peso constante. Pese alrededor de 40 g y colóquela a reflujo con acetona en el extractor soxhlet por al menos 24 horas para remover lípidos, pigmentos, etc. Cambie el solvente por metanol y continúe la extracción por al menos otras 24 horas. Deje enfriar el extracto metanólico y llévelo a 250 ml con metanol. En este punto hay una modificación al método propuesta por Miriam Monforte (CICY, Mérida, Yucatán, México). En lugar de llevarlo a 250 ml como sugiere el método original, es más recomendable concentrar la muestra, lo que permite cuantificar cantidades muy pequeñas de saponina, para lo cual, pase el extracto metanólico a un rotoevaporador y concentre a sequedad. Resuspenda el residuo con un mínimo de metanol y páselo a un vial previamente pesado; si no se va a usar de inmediato, seque con nitrógeno y guárdelo en desecador. Pese el vial con la muestra seca y calcule el peso del residuo. Para utilizar la muestra, resuspenda con una cantidad conocida de metanol (alrededor de 1 ml). Cromatografía cualitativa en capa finaColoque puntos del extracto sobre una placa de gel de sílica y revélelos con una solución n-butanol-etanol-amoníaco concentrado (3.5:1:2.5). Se pueden aplicar tres diferentes métodos para identificar las manchas de saponina: (1) aspersíon con ácido acético-anisaldeido; (2) tratamiento con nitrato de plata seguido de hidróxido de sodio y (3) tratamiento con una suspensión de eritrocitos en buffer isotónico. Para mayores detalles consulte a Fenwick y Oakenfull (1981). Cromatografía cuantitativa en capa finaColoque series de puntos con volúmenes conocidos del extracto y de una solución estándar de saponina sobre las placas de cromatografía. Los puntos deben ser colocados de tal manera que cada punto de extracto vaya al lado de los de estándar de saponina. Revele las placas de la misma manera que en el apartado anterior y las manchas deben ser visualizadas mediante la aspersíon de una solución de ácido sulfúrico en metanol, seguida de calentamiento a 110 °C por 30 min. Mida la intensidad de las manchas de saponina por medio de un densitómetro y calcule las áreas de los picos en el graficador con un planímetro. los resultados son expresados como la relación (R) de las áreas de los picos de la muestra desconocida con respecto a la del estándar. Se ha determinado que la concentración de saponina en la muestra (en μg) es proporcional al cuadrado de las áreas relativas (R2). Grafique R2 contra el volumen de la gota de extracto metanólico colocada en la placa. La pendiente de la línea (calculada por el método de cuadrados mínimos), divido entre la pendiente de una línea obtenida de una curva patrón, dará la concentración de la saponina en el extracto y de esa manera, el contenido de saponina en la muestra. El método puede tener un error experimental acumulado de ±20%.
59

FIGURA 33 Determinación de saponinas en ingredientes alimenticios
6. REFERENCIAS AOAC, 1984. Official Methods for Analysis of the Association of Official
Analytical Chemists, 14th edition. Arlington, VA, 1141 pp. Archibald, R.M., 1946. Colorimetric determination of canavanine. J.Biol.Chem.,
165: 169–178. Chow, K.W., Rumsey, G.L. and Woldroup, P.W., 1980. Linear programming in
fish diet formulation. In: Fish feed technology,UNDP/FAO/ADCO/REP/80/11, 395 pp.
Cockerell, I., Francis, B. and Halliday, D., 1971. Changes in nutritive value of concentrate feedingstuffs during storage. Tropical Products Institute, London, U.K.
F.A.O., 1980. Fish feed technology. ADCP/REP/80/11. 395 pp. F.A.O., 1983. Fish feeds and feeding in developing countries - An interim report
on the ADCP Feedo Development Programme. ADCP/REP/83/18. 97 pp. Fenwick, D.E. and Oakenfull, D., 1981. Saponin content of soya beans and
some commercial soya bean products. J. Sci. Food Agric., 32:273–278. Frazer, A.C., 1967. Health problems in quality control: Chemical aspects. In: S.
M. Herschdoerfer (Editor), Quality Control in the Food Industry. Academic Press, London, U. K., 385 pp.
Furukawa, H. and Tsukahara, H., 1966. On the acid digestion method for the determination of chromic oxide as an index substance in the study of digestibility of fish fed. Bull. Jpn. Soc. Sci. Fish., 32(6):502–508.
60

Caskey, D. D., Jr. and Knapp, F. C., 1944. Method for detecting inadequately heathed soybean oil meal. Ind. Eng. Chem. Anal., 16:640.
Harris, L. E., 1980. Feedstuffs. pp. 111–170. Kent-Jones, D. W. and Amos, A. J. 1957. Modern Cereal Chemistry, fifth edition.
The Northern Publishing Co., LTD, Liverpool, England. pp. 59–60. Krishna, G. and Ranjhan, S.K., 1980. Laboratory manual for nutrition research.
Vikas Publishing House PVT LTD, New Delhi, India. 134 pp. Limborg, C. L., 1979. Industrial production of ready to use feeds for mass
rearing of fish larvae. Proc. World Symp. on Finfish Nutrition and Fish Feed Technology, Hamburg 20–23 june, 1978. Vol. II.
Lowry, O. H., Rosebrough, N. J., Farr, A. L. and Randall, R. J., 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275
MAFF, 1982. The feeding stuffs (sampling and Analysis) regulations 1982. Minister of Agriculture, Fisheries and Food. Her Majesty's Stationery Office, London U.K., 99 pp.
Matsumoto, H. and Sherman, G., 1951. A rapid colorimetric method for the determination of mimosine. Arch. Biochem. Biophys., 33: 195–200.
New, B. B., 1987. Feed and feeding of fish and shrimp. A manual on the preparation and presentation of compound feeds for shrimp and fish in aquaculture. FAO, ADCP/REP/87/26, 275 pp.
Osborne, D. R. and Voogt, P. 1978. The analysis of nutrients in foods. Academic Press, London, U.K., 240-pp.
Smith, J. E. and Moss, M. O. 1985. Micotoxins, formation, analysis and significance. John Wiley & Sons. 148 pp.
Tacon, A. G. J., 1979. The nutritional evaluation of animal and food processing wastes. 3rd International Conference on Effluent Treatment in the Biochemical Industries. London, 7th november 1979.
Tacon, A. G. J. and Jackson, A. J., 1985. Utilization of conventional and unconventional protein sources in practical fish feeds. In: C. B. Cowey, A. M. Mackie and J. G. Bell (Eds.), Nutrition and Feeding in Fish. Academic Press, London. pp. 119–145.
Tejada, de H. I., 1985. Manual de laboratorio para análisis de ingredientes utilizados en la alimentación animal. Patronato de Apoyo a la Investigación by Experimentación Pecuaria de México, D. F., 387 pp.
Wheeler, E. L. and Ferrel, R. E., 1971. A method for phytic acid determination in wheat and wheat fractions. Cereal Chem., 48:312–320.
Williams, D. R., 1987. Animal feeding stuffs legislation of the U. K. A concise guide Butterworths, London, U. K., 135 pp.
61


















