
Manual de Guías de Laboratorios y Procedimientos Agosto 2013
Upload
azv-fentanesCategory
view
41download
2description

UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO
FACULTAD DE ESTUDIOSSUPERIORES CUAUTITLÁN
DEPARTAMENTO DE CIENCIAS BIOLÓGICAS
SECCIÓN CIENCIAS DE LA SALUD HUMANA
MANUAL BÁSICO DE LABORATORIO DE INMUNOBIOLOGÍA
BIOQUÍMICA DIAGNÓSTICA
Coordinador de Edición y Revisión: Ladislao Palomar Morales
Edición y Revisión: Fonseca Coronado Salvador, Martínez Sosa Ángel Germán, Palomar Morales Ladislao, Vega López Marco Antonio, Romero Rojas Andrés, González Ballesteros Eric
AGOSTO 2015

2
INDICE
Página Programa de la asignatura 3
Calendario de prácticas 6
Reglamento general para los laboratorios 7
1 Seguridad en el laboratorio de inmunobiología 8
2 Morfología de las células del sistema inmunitario 14
3 Técnicas de separación y purificación de células del sistema inmunitario 18
4 Fagocitosis 26
5 Pruebas de aglutinación, precipitación y turbidimetría 32
6 Modelos experimentales y vías de inmunización 36
7 Citometría de flujo 41
8 Ensayo Inmuno Enzimático (ELISA) 47
9 Anatomía del sistema inmunitario 53
10 Histología e inmunohistoquímica 56
11 Electroforesis en gel de poliacrilamida 64
12 Inmunoelectrotransferencia 71

3
Programa de la Asignatura

4

5

6
UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO FACULTAD DE ESTUDIOS SUPERIORES CUAUTITLÁN
DEPARTAMENTO DE CIENCIAS BIOLÓGICAS SECCIÓN CIENCIAS DE LA SALUD HUMANA
CALENDARIZACIÓN CÓDIGO: FPE-CB-DEX-01-02
N° Revisión: 03 ASIGNATURA (1638) Inmunobiología GRUPO(S) 1601, 1652 SEMESTRE 2016-I CARRERA BQD
SEM
AN
A
FECHA (SEMANAL)
NÚMERO Y/O NOMBRE DE LA PRÁCTICA, PROYECTO Y/O ACTIVIDAD
CUMPLIÓ OBSERVACIONES
SI NO
1 13 y 14/08/2015 Presentación del curso y organización de equipos
2 20 y21/08/2015 Seguridad en el laboratorio de inmunobiología
3 27 y 28/08/2015 Morfología de las células del sistema inmunitario
4 03 y 04/09/2015 Técnicas de separación y purificación de células del sistema inmunitario
5 10 y 11/09/2015 Fagocitosis
6 17 y 18/09/2015 Pruebas de aglutinación, precipitación y turbidimetría y primera inmunización
7 24 y 25/09/2015 EXAMEN, Citometría de flujo y segunda inmunización
8 01 y 02/10/2015 TEORÍA tercera inmunización
9 08 y 09/10/2015 ELISA
10 15 y 16/10/2015 Anatomía del Sistema Inmunitario
11 22 y 23/10/2015 Histología e Inmunohistoquímica
12 29 al 30/10/2015 Electroforesis en gel de poliacrilamida, Inmunoelectrotransferencia
13 05 y 06/11/2015 SEMINARIOS
14 12 y 13/11/2015 SEMINARIOS
15 19 y 20/11/2015 EXAMEN
16 26 y 27/11/2015
SEM
FECHA (SEMANAL) EXAMEN CUMPLIÓ OBSERVACIONES SI NO
7 24 y 25/09/2015 Primer examen de laboratorio
15 19 y 20/11/2015 Segundo examen de laboratorio*

UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO FACULTAD DE ESTUDIOS SUPERIORES CUAUTITLÁN
DEPARTAMENTO DE CIENCIAS BIOLÓGICAS
REGLAMENTO GENERAL PARA LOS LABORATORIOS Fecha de revisión: 30/06/2014
No revisión: 03
1) Este reglamento aplicará para personal académico, alumnos y laboratoristas.
2) Para todo trabajo realizado en el laboratorio deberá utilizarse bata blanca con manga larga y zapatos cerrados.
3) La tolerancia para el inicio de la sesión de laboratorio será hasta de 10 minutos a partir de la hora señalada.
4) Por seguridad, no deben cerrarse las puertas del laboratorio con llave durante las prácticas.
5) En todo momento deberá mostrarse una conducta adecuada en el área de trabajo.
6) Queda prohibido en los laboratorios: a) Tirar basura fuera del cesto. b) Ingerir alimentos y/o bebidas. c) Fumar. d) Recibir visitas. e) La entrada a los inter-laboratorios a toda persona ajena a los mismos. f) Realizar reuniones o convivios en los laboratorios. g) Salir del laboratorio en el horario asignado para la sesión experimental. h) Sentarse sobre las mesas de trabajo. i) Mover el mobiliario de su lugar. j) Utilizar las gavetas para guardar material que no corresponda a la asignatura.
7) Los residuos peligrosos deben depositarse en los contenedores destinados para tal fin, entendiendo por residuo peligroso: elementos, sustancias, compuestos, desechos o mezclas de ellos que en cualquier estado físico representan un riesgo para el ambiente, la salud o los recursos naturales, por sus características corrosivas, reactivas, explosivas, tóxicas, inflamables o biológico-infecciosas (Art. 3º de la Ley General del Equilibrio y Protección del Ambiente).
8) Dentro del laboratorio no se permite el uso de teléfonos celulares, reproductores de sonido o cualquier medio electrónico de entretenimiento. El uso de las computadoras portátiles queda restringido a temáticas relacionadas con la asignatura.
9) El acceso al laboratorio se permitirá únicamente cuando esté presente uno de los profesores del grupo.
10) El uso del laboratorio para trabajo extraordinario, deberá programarse con el profesor responsable en un horario que no interfiera con aquel destinado para el desarrollo de las prácticas.
11) Para solicitar material y equipo, es requisito indispensable que el alumno llene debidamente el vale de material (FPE-CB-DEX-01-09) y lo entregue a la persona responsable, dejando como depósito la credencial vigente de la UNAM.
12) El alumno deberá revisar el material y/o equipo al momento de recibirlo indicando cualquier anomalía (faltante o material dañado) y será devuelto en las condiciones en que se recibió, de no hacerlo, se hará acreedor a las sanciones establecidas en cada laboratorio.
13) Es obligación de todos mantener limpio y ordenado el lugar de trabajo y todo el laboratorio.

PRÁCTICA 1
SEGURIDAD EN EL LABORATORIO DE INMUNOBIOLOGÍA
QFB Ladislao Palomar Morales
OBJETIVO Introducir las medidas de seguridad, químicas y biológicas, en el laboratorio de
Inmunobiología para que el alumno las ponga en práctica y pueda minimizar los riesgos de
trabajo.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO Es fundamental que el alumno conozca las medidas mínimas de seguridad, química y biológica,
que deben seguirse en un laboratorio donde se manejan reactivos químicos y muestras
biológicas, por esto se plantea en el programa de la asignatura como primera práctica del curso.
INTRODUCCIÓN Existe un riesgo evidente de contaminación al que está expuesto el personal de salud
(profesionales, estudiantes, investigadores, etc., personal de limpieza), al tratar con pacientes
infectados o potencialmente infectados, en especial cuando el personal de salud, está en
contacto con sangre o hemoderivados, con agujas, jeringas e instrumental contaminado,
pudiendo adquirir infecciones como el HIV, Hepatitis B, C, etc., así como el riesgo de toxicidad,
cuando en su trabajo emplea substancias químicas, ya sea como reactivos, o como productos
de desinfección, los cuales pueden dañar también el medio ambiente.
Según la NOM-018-STPS-2000 se definen como: peligro a la capacidad intrínseca de una
sustancia química para generar un daño y riesgo, a la probabilidad de que una sustancia
química peligrosa afecte la salud de los trabajadores o dañe el centro de trabajo. Es necesario
ampliar estas definiciones para ser aplicadas a y microorganismos y. productos de origen
biológico.

9
Aunque existe una gran variedad de sistemas para la identificación de riesgos los más usados a
nivel nacional e internacional son dos. Ambos sistemas son muy amigables por el uso de
colores y números para identificar los riesgos, y por tal razón son los recomendados por la
NOM-018-STPS-2000.
A mediados del siglo XX, la Agencia Nacional de Protección al Fuego (NFPA, por sus siglas en
inglés) diseña el Código 704 para la identificación de riesgos de las sustancias químicas.
Rombo (o diamante) del Sistema NFPA
Algunos años más tarde la Asociación Nacional de productores de Pinturas y Recubrimientos
(NPCA, por sus siglas en inglés) desarrolla un sistema alterno para la identificación de
materiales peligrosos. Hay que mencionar que tienen ligeras diferencias en su aplicación,
debido a su origen.
Rectángulo HMIS
Las sustancias químicas son producidas en una región determinada y posteriormente se
trasladan al lugar donde se van a utilizar, para esto se deben seguir en México, y en el mundo,

10
las recomendaciones de la ONU para el transporte de materiales peligrosos, estas
recomendaciones surgen por la movilidad de estos materiales después de la Segunda Guerra
Mundial. En México se debe aplicar la NOM-003-SCT-2008, que obliga a los transportistas a
identificar mediante un código el tipo de material que se está transportando, utilizando colores y
números. Existe un sistema alterno llamado HazChem, utilizado en Europa.
Gas inflamable
Sólido inflamable
Misceláneos
Sistema HazChem Paneles del Sistema de Identificación para el Transporte de
Materiales Peligrosos
Una vez en el lugar donde se utilizaran las sustancias químicas, deben ser almacenadas en
base a sus características químicas, y no en orden alfabético. Aunque existen varios sistemas
para almacenamiento, los más amigables son aquellos que utilizan, simultáneamente, colores,
números y pictogramas. Estos sistemas toman el nombre de las empresas productoras que los
crearon, y todos ellos fueron creados a principios de la década de los 80’s del siglo pasado.
Sistema Baker Saf-T-Date
Sistema ChemAlert
Sistema Winkler
Los colores asignados en base al tipo de riesgo, en estos tres sistemas, son los siguientes:
Tipo de riesgo Baker Saf-T-Data ChemAlert Winkler Salud Azul Azul Azul
Inflamabilidad Rojo Rojo Rojo Reactividad Amarillo Amarillo Amarillo
Contacto Blanco Blanco Blanco General (riesgo menor o igual a 2 en cualquiera de los tipos) Verde Gris Verde
Incompatibles con otras del mismo grupo
Color asignado a rayas diagonales NO EXISTE Color asignado y la
leyenda SEPARADO

11
La Organización Mundial de la Salud (OMS) publicó la primera edición del Manual de
bioseguridad en el laboratorio en 1983. En ella se alentaba a los países a aceptar y aplicar
conceptos básicos en materia de seguridad biológica y a elaborar códigos nacionales de
prácticas para la manipulación sin riesgo de microorganismos patógenos en los laboratorios que
se encuentran dentro de sus fronteras nacionales. Desde 1983, muchos países han seguido la
orientación especializada que se ofrece en el manual para elaborar esos códigos de prácticas.
La OMS establece que la clasificación por grupos de riesgo se utilizará exclusivamente para el trabajo de laboratorio. Y hace referencia a los peligros relativos que entrañan los
microorganismos infecciosos, clasificados por grupos de riesgo (grupos de riesgo 1, 2, 3 y 4).
Clasificación de los microorganismos infecciosos por grupos de riesgo Grupo de riesgo 1(riesgo individual y poblacional escaso o nulo) Microorganismos que tienen pocas probabilidades de provocar enfermedades en el ser humano o los animales.
Grupo de riesgo 2(riesgo individual moderado, riesgo poblacional bajo) Agentes patógenos que pueden provocar enfermedades humanas o animales pero que tienen pocas probabilidades de entrañar un riesgo grave para el personal de laboratorio, la población, el ganado o el medio ambiente. La exposición en el laboratorio puede provocar una infección grave, pero existen medidas preventivas y terapéuticas eficaces y el riesgo de propagación es limitado.
Grupo de riesgo 3(riesgo individual elevado, riesgo poblacional bajo) Agentes patógenos que suelen provocar enfermedades humanas o animales graves, pero que de ordinario no se propagan de un individuo a otro. Existen medidas preventivas y terapéuticas eficaces.
Grupo de riesgo 4(riesgo individual y poblacional elevado) Agentes patógenos que suelen provocar enfermedades graves en el ser humano o los animales y que se transmiten fácilmente de un individuo a otro, directa o indirectamente. Normalmente no existen medidas preventivas y terapéuticas eficaces.
En el siguiente cuadro se relacionan, no se equiparan, los grupos de riesgo con el nivel de
bioseguridad de los laboratorios destinados al trabajo con microorganismos de cada uno de
esos grupos.

12
Relación de los grupos de riesgo con los niveles de bioseguridad, las prácticas y el equipo
GRUPO DE RIESGO
NIVEL DE BIOSEGURIDAD
TIPO DE LABORATORIO
PRÁCTICAS DE LABORATORIO
EQUIPO DE SEGURIDAD
1 Básico Nivel 1
Enseñanza básica, investigación
TMA Ninguno; trabajo en mesa de laboratorio al descubierto
2 Básico Nivel 2
Servicios de atención primaria; diagnóstico, investigación
TMA y ropa protectora; señal de riesgo biológico
Trabajo en mesa al descubierto y CSB para posibles aerosoles
3 Contención Nivel 3
Diagnóstico especial, investigación
Prácticas de nivel 2 más ropa especial, acceso controlado y flujo direccional del aire
CSB además de otros medios de contención primaria para todas las actividades
4 Contención
máxima Nivel 4
Unidades de patógenos peligrosos
Prácticas de nivel 3 más cámara de entrada con cierre hermético, salida con ducha y eliminación especial de residuos
CSB de clase III o trajes presurizados junto con CSB de clase II, autoclave de doble puerta (a través de la pared), aire filtrado
En todo laboratorio escolar, o de diagnóstico clínico, se generan residuos de diferentes tipos,
cada uno de los cuales requiere diferentes condiciones para su recolección, traslado,
almacenamiento y disposición definitiva. De acuerdo con las NOM-052-SEMARNAT-2005 y
NOM-087-ECOL-SSA1-2002 los residuos deben separarse de acuerdo con el código CRETIB
(acrónimo de Corrosivo, Reactivo, Explosivo, Toxico, Inflamable y Biológico infeccioso).
Cada residuo debe segregarse de los otros y ser depositado en contenedores adecuados, sin
mezclarse entre sí. En nuestra facultad debemos seguir las indicaciones proporcionadas en la
“Instrucción de trabajo específica para la identificación clasificación y eliminación de residuos
peligrosos”, elaborada por profesores del Departamento de Ciencias Biológicas, que engloba
las normas anteriores.
Cada contenedor de residuos químicos debe etiquetarse de la siguiente forma:
Laboratorio que lo generó. Tipo de residuos según la clave CRETIB, y de ser posible el
pictograma correspondiente. Composición aproximada de la mezcla. Fecha de inicio y término de la recolección. Generador del residuo

13
Los residuos biológicos deben ser segregados de acuerdo con la NOM-087-ECOL-SSA1-2002 de la siguiente forma:
TIPO DE RESIDUOS ESTADO FISICO ENVASE COLOR
Sangre y derivados Líquido Recipiente hermético Rojo
Cultivos y cepas de agentes infecciosos Sólido Bolsa de
polietileno Rojo
Patológicos Sólidos Bolsa de
polietileno Amarillo
Líquidos Recipiente hermético Amarillo
Residuos no anatómicos
Sólidos Bolsa de polietileno Rojo
Líquidos Recipientes herméticos Rojo
Objetos punzocortantes Sólidos Recipiente rígido de polipropileno
Rojo
DISCUSIÓN Discutir la importancia de conocer, implementar, y trabajar con las normas de seguridad
adecuadas al laboratorio de esta asignatura.
BIBLIOGRAFÍA 1. Funes Espinoza Fátima, Panozo Meneces Adela y Cardozo Salinas Teresa. (2005). Bioseguridad y
Seguridad Química en Laboratorio. [En línea] http://www.swisscontact.bo/sw_files/mvhvmxjnomq.pdf obtenido el 12 de junio de 2011.
2. NOM-018-STPS-2000. Sistema para la identificación y comunicación de peligros y riesgos por sustancias químicas peligrosas en los centros de trabajo. [En línea] http://132.248.50.5/CLS/INFORMACIONPARACONSULTA/DERRAMESDEQUIMICOS/NOM-018-STPS-2000.pdf, consultada el 12 de junio de 2011.
3. NOM-087-ECOL-SSA1-2002. Protección ambiental- Salud ambiental-Residuos peligrosos biológico-Infecciosos-Clasificación y especificaciones de manejo. [En línea] http://www.semarnat.gob.mx/leyesynormas/Normas%20Oficiales%20Mexicanas%20vigentes/NOM-087-ECOL-SSA1-2002.pdf, obtenida el 12 de junio de 2011.
4. NOM-052-SEMARNAT-2005. Que establece las características, el procedimiento de identificación, clasificación y los listados de los residuos peligrosos. [En línea] http://www.semarnat.gob.mx/leyesynormas/Normas%20Oficiales%20Mexicanas%20vigentes/NOM%20052_23_JUN_2006.pdf, consultada el 12 de junio de 2011.
5. NOM-003-SCT-2008, Características de las etiquetas de envases y embalajes, destinadas al transporte de substancias, materiales y residuos peligrosos. [En línea] http://www.sct.gob.mx/fileadmin/normatividad/transporte_terrestre/43.pdf, obtenida el 12 de junio de 2011.
6. Organización Mundial de la Salud. (2005). Manual de Bioseguridad en el laboratorio. [En línea] http://whqlibdoc.who.int/publications/2005/9243546503_spa.pdf, obtenido el 12 de junio de 2011.

14
PRÁCTICA 2
MORFOLOGÍA DE LAS CÉLULAS DEL SISTEMA INMUNITARIO
QFB Ladislao Palomar Morales
OBJETIVOS Conocer y aplicar la metodología y cálculos para el conteo de células en cámara de
Neubauer.
Identificar microscópicamente a las células del sistema inmune presentes en circulación y
conocer sus principales características fenotípicas.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la unidad 2 se abordan los Mecanismos Inespecíficos y parcialmente específicos de Defensa
(MID), en el punto 2.5 se deben describir el fenotipo y la función de las células que participan en
los MID Y MPE.
INTRODUCCIÓN El conteo de leucocitos se refiere a la cantidad total
de leucocitos en sangre, contados utilizando la
cámara de Neubauer; La cámara de Neubauer tiene
dibujado en el centro un gran cuadrado, subdividido
en nueve cuadros más pequeños. Los cuatro
cuadrados de las esquinas se utilizan para el
recuento leucocitario y, a su vez, están subdivididos
en dieciséis cuadrados terciarios.
El cuadro central es el de cuenta de eritrocitos.

15
La Cuenta Diferencial consiste en evaluar la proporción y la morfología de los diferentes tipos
de leucocitos que hay en sangre periférica. El examen de una extensión de sangre, es parte
importante de la evaluación de la serie blanca, la fiabilidad de la información obtenida depende
principalmente de lo bien hechas y bien teñidas que estén las extensiones, las cuales son
sistemáticamente examinadas.
MATERIAL Placa de 96 pozos Cámara de Neubauer Microscopio Juego de micropipetas y puntas Portaobjetos Tubo Capilar Algodón Piano Contador
REACTIVOS Solución de Turk EDTA Colorante de Wright Aceite de Inmersión Amortiguador de fosfatos con pH 6.6 Agua destilada
DIAGRAMA DE FLUJO
R1: Aguja, al contenedor de punzocortantes; Algodón, basura municipal; tubo con sangre a contenedor con cloro R2: Residuos de Wright, Contenedor especial
Sangre con anticoagulante
Recuento de leucocitos
Cuenta diferencial
Diluir 1/20 con líquido de Turk
Llenar la cámara de Neubauer
Contar a 10X
Teñir con Wright
Contar a 100X
R1
R2

16
METODOLOGÍA Tomar una muestra de sangre con sistema Vacutainer®, utilizando EDTA como anticoagulante.
1. Conteo de leucocitos En un pozo de una microplaca, colocar 5 L de sangre completa y adicionar 95 L de líquido de
Turk, mezclar suavemente. Tomar 10 L de la sangre diluida y llenar un lado de la cámara de
Neubauer. Dejar reposar durante 1 minuto.
Colocar la cámara de Neubauer en el microscopio y buscar la cuadricula con el objetivo de 10X.
Realizar el conteo de leucocitos con el objetivo de 40X, en forma de culebra. Reportar el valor
en: leucocitos/mm3 y leucocitos/mL.
2. Cuenta diferencial de la serie blanca Colocar una gota del tubo capilar en el extremo de un portaobjetos. Extenderla con otro
portaobjetos con un movimiento suave, haciendo la extensión de sangre lo más delgada y fina
que se pueda. Secar al aire rápidamente. Cubrir con colorante de Wright durante 5 a 8 minutos.
Agregar el amortiguador, sin tirar el colorante, hasta que se forme una capa metálica que cubra
el frotis y dejarlo reposar de 5 a 8 minutos. Enjuagar con agua destilada y secar.
Colocar el frotis en el microscopio y enfocar con el objetivo de 40X, colocar una gota de aceite
de inmersión y realizar la cuenta con el objetivo 100X, en forma de culebra, contando un mínimo
de 200 células.

17
RESULTADOS Valores absolutos y relativos de los leucocitos.
DISCUSIÓN Discuta los valores obtenidos contra los valores de referencia y las implicaciones que tienen un
aumento y/o una disminución de estas células del sistema inmune.
BIBLIOGRAFÍA 1. Bernard, Henry John. (1993). Diagnóstico y Tratamiento Clínico por el Laboratorio, 9ª edición, Ed.
Salvat Medicina, México, DF. 1509 pp. 2. Velez A, Hernan. (1992). Fundamentos de Medicina Hematología. 4ª edición. Editorial Corporación
para Investigaciones Biológicas. Medellín, Colombia. 395 pp. 3. Williams, William J. (1979). Hematología. 1ª edición. Salvat Editores, S.A. Barcelona, España.1503
pp.

18
PRÁCTICA 3
TÉCNICAS DE SEPARACIÓN Y PURIFICACIÓN DE CÉLULAS DEL SISTEMA INMUNITARIO
Dr. Salvador Fonseca Coronado y QFB Ladislao Palomar Morales
OBJETIVO Y COMPETENCIAS Al finalizar el procedimiento experimental es deseable que el alumno haya adquirido las
siguientes competencias:
Conocer las principales técnicas de separación y purificación de células a partir de sangre
periférica.
Realizar el procedimiento de estratificación de sangre sobre Ficoll-Hypaque y la obtención
de las células mononucleares.
Conocer y aplicar los principios de purificación de células por el método de perlas
superparamagnéticas acopladas a anticuerpos específicos.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la unidad 3 se describen los mecanismos específicos de defensa (MED) y específicamente
en el apartado 3.1 se hace mención del fenotipo y función de las células de los MED y en el 3.9
de la estructura y función de los receptores celulares, en esta práctica se lleva a cabo un
procedimiento para la separación de células mononucleares, entre las que se encuentran
células de la inmunidad innata, y un procedimiento para la purificación específica de células NK
las cuales son células fundamentales en los MID por su función citotóxica.
INTRODUCCIÓN En inmunología, al igual que en la mayoría de las ciencias de la vida, para describir un
fenómeno particular, es necesario aislarlo del conjunto de variables con los que coexiste.

19
En la actualidad, casi todos los estudios con células del sistema inmune requieren su
aislamiento y purificación, bien para describir funciones específicas, o para el estudio delos
efectos de otras células, agentes o moléculas sobre ellas; el conocimiento de todas las
funciones descritas de las células hasta el momento, ha sido posible gracias a que se cuenta
con técnicas para la purificación de una estirpe en particular, a partir de un grupo heterogéneo
de estas.
Las células inmunes se pueden separar por varios métodos
Métodos físicos
Aislamiento por centrifugación en gradiente de densidad (Ficoll-Hypaque): Con esta técnica
podemos obtener de forma separada las células mononucleares (linfocitos y monocitos)
delos granulocitos.
Separación por adhesión selectiva:
Los monocitos se adhieren a superficies de vidrio o plástico y se separan de los
linfocitos.
Los linfocitos B se fijan a columnas o jeringas en lana de nylon o algodón (se obtienen
poblaciones separadas de LB y LT).
Estudio de marcadores y antígenos de membrana (diferencia poblaciones y
subpoblaciones de células). Las técnicas más empleadas son:
Citometría de flujo con FACS (separador celular activado por fluorescencia)
Formación de rosetas por linfocitos B y T
Separación por sus antígenos de membrana:
Separación magnética
Separación por afinidad en columnas
MATERIAL Y EQUIPO Sistema Vacutainer® para obtención de sangre con anticoagulante Cámara de Neubauer. Microscopio. Tubos cónicos de 15 mL Tubos de vidrio 13x100 Algodón (Torundas) Contador de células. Micropipetas de 10 y 100 μL. con puntas Pipetas de 5 y 10 mL y Pipetas Pasteur
REACTIVOS Ficoll-hypaque SSF o PBS Colorante de Wright Aceite de Inmersión Amortiguador de fosfatos con pH 6.6 Solución de Turk

20
Centrífuga clínica Balanza de dos platos Portaobjetos
Opcional Equipo magnético para purificación de células
Opcional Medio RPMI 1640 suplementado Mezcla de anticuerpos monoclonales contra marcadores de identidad acoplados a biotina Anticuerpos monoclonales anti-biotina acoplados a perlas magnéticas Amortiguador de purificación: PBS albúmina- EDTA
DIAGRAMA DE FLUJO
R1: Aguja, al contenedor de punzocortantes; Algodón, basura municipal; tubo con sangre a contenedor con cloro R2 y R3:Palangana con cloro R4:Residuos de Wrigth, Contenedor especial
METODOLOGÍA 1. Obtener dos tubos de sangre, 5 mL en cada uno, con anticoagulante (heparina de
preferencia).
Obtener 2 tubos de sangre periférica
Centrifugar un tubo
Estratificar el contenido del otro tubo sobre Ficoll-hipaque
Obtener la capa de Leucocitos
Centrifugar y obtener la capa de mononucleares
Resuspender en 1 mL
Preparar un frotis, teñir con Wright y observar morfología
Contar en cámara de Neubauer
R1
R3
R4
R2

21
Obtención de Buffy Coat 2. Centrifugar, uno de los tubos, a 700 g
durante 5 minutos.
3. Con pipeta Pasteur extraer y desechar el
plasma.
4. Con pipeta Pasteur obtener la capa de
Leucocitos (1 mm aproximadamente) y
colocar en un tubo de ensaye.
5. Completar a 1 mL con SSF o PBS.
6. Realizar el conteo de las células
obtenidas en la cámara de Neubauer.
7. Preparar un frotis con la suspensión y
teñir con Wright. Observar morfología a
100X.
Separación por gradiente de densidad en Ficoll-Hypaque. 8. Colocar 2 mL de Ficoll-hypaque en un tubo de ensaye de 13x100, estratificar lentamente la
sangre de un tubo sobre la superficie del Ficoll con ayuda de una pipeta Pasteur, de tal
manera que se formen dos fases.
9. Centrifugar a 300 g por 15 minutos.
10. Como se observa en la figura se forman cuatro
estratos diferentes introducir una pipeta Pasteur
hasta el estrato de células mononucleares (PBMC
del inglés: Peripheral blood Mononuclear Cells) y
extraer el anillo en forma circular evitando extraer la
menor cantidad de Ficoll posible.
11. Colocar las PBMC en otro tubo, adicionar 5 mL de
PBS y centrifugar 5 min a 300 g, repetir el lavado
una vez más. Resuspender en 1 mL de PBS
12. Prepara un frotis con la suspensión de PBMC y teñir con Wright; Observar la morfología a
100X.
13. Realizar el conteo de las células obtenidas en la cámara de Neubauer.

22
RESULTADOS Indicar la cantidad de células/mL de suspensión y morfología de las células obtenidas por cada
método.
DISCUSIÓN El alumno debe comprender y describir detalladamente el fundamento de la purificación de
células por selección negativa con perlas magnéticas.
REFERENCIAS Current Protocols in Immunology. (1997) A.3F.1-A.3F.2 Common Immunologic Techniques. Supplement 21, John Wiley & Sons, Inc. DOI: 10.1002/0471142735.ima03fs21 URL´s http://onlinelibrary.wiley.com/doi/10.1002/0471142735.ima03fs21/pdf (Libre acceso dentro de la UNAM). www.miltenyibiotec.com/download/datasheets_en/253/MiltenyiBiotec_DataSheet_CD4+-T-Cell-Isolation-Kit-II,-human_130-091-155.pdf

23
Opcional: Purificación de células por perlas superparamagnéticas (MACS) DIAGRAMA DE FLUJO

24
Importancia: La purificación de una población celular en particular, nos permite evaluar una gran variedad de funciones específicas y de respuestas a diferentes estímulos entre ellos por supuesto, los efectos de un determinado fármaco.
1. De acuerdo al número de células contadas, ajuste una solución de 1x107 células en 40 L
de buffer y adicione la cantidad de cocktail de anticuerpos anti-CD indicado en el instructivo
del producto. (La finalidad de esto es familiarizar al estudiante con insertos en idioma
inglés).
2. Incube 10 minutos en hielo
3. Adicione 10 μL de anticuerpos anti biotina acoplados a perlas magnéticas mezcle y deje en
incubación por 15 minutos.
4. Mientras esta en incubación la suspensión, coloque una columna LS sobre él magneto y
lave tres ocasiones por adición de 3 mL de PBS-albúmina.
5. Adicione la suspensión en la columna y colecte el eluato en un tubo cónico, lave en tres
ocasiones por adición de 2 mL de PBS-albúmina colectando el eluato en el mismo tubo
(esta fracción es rica en células purificadas).
6. Separe la columna del magneto y adicione en tres ocasiones 3 mL de buffer para sacar de
la columna las células marcadas (Esta fracción es rica en las poblaciones no purificadas).
7. Realice el conteo de ambas fracciones y calcule la eficacia del proceso, compare su
resultado experimental con el teórico esperado de acuerdo a la población purificada.

25
APÉNDICE Preparación de medio RPMI 1640 Dependiendo del fabricante, el medio puede venir en varias presentaciones, hay presentaciones
de 100 mL listas para su uso, sobres con polvo para disolver en un litro y frascos con polvo para
preparar 10 litros, en este último caso se pesa la cantidad adecuada para los mL que se desee
preparar. El polvo debe ser disuelto en agua tridestilada y desionizada o en agua milliQ.
Para preparar un litro de medio: Disolver la cantidad de polvo indicada en aproximadamente 970 mL de agua; agregar 2 gramos
de bicarbonato de sodio en polvo (grado cultivo celular) y 1 gramo de HEPES; adicionar 10 mL
de L-glutamina 100 mM y 10 mL de antibiótico-antimicótico (penicilina/estreptomicina-
Fungizona); ajustar el pH a 7 con NaOH 0.1 N y aforar a 1 litro. Filtrar en una unidad de
esterilización de 0.22 μm y etiquetar como medio base. El medio para cultivo celular se
suplementa por adición de 10 % de suero fetal bovino estéril.
Preparación de solución amortiguadora de fosfatos (PBS) pH 7.2 Pesar 8 g de NaCl, 0.2 g de KCl, 0.16 g de KH2PO4 y 0.79 g de Na2HPO4 anhidro, disolver en
agua desionizada y ajustar el pH a 7.2 con NaOH o HCl según sea el caso.
Esterilizar por filtración y almacenar a 4 °C.
Preparación de solución amortiguadora de separación para MACS. Preparar 1 litro de PBS 1X y adicionar 0.5 % de albúmina sérica bovina y EDTA 2 mM

26
PRÁCTICA 4
FAGOCITOSIS
QFB Ladislao Palomar Morales
OBJETIVO Evaluar la función fagocítica en células de sangre periférica humana para determinar:
a) Porcentaje de células con capacidad fagocítica
b) Porcentaje de células con capacidad de producir radicales intermediarios del oxígeno
c) Número de levaduras, en promedio, que fagocita una célula
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO Mecanismos inespecíficos y parcialmente específicos de defensa (MID), Unidad 2, punto 2.7
INTRODUCCIÓN En la fagocitosis (del griego -phagos, 'el que come', kytos, 'célula') algunas células rodean con
su membrana citoplasmática a una sustancia extracelular (un sólido generalmente) y la
introducen al interior celular.
Células fagocíticas En los animales superiores, la fagocitosis es una función de células especializadas (fagocitos
profesionales) del sistema inmune capaces de remover cuerpos extraños y combatir infecciones
como primera línea de defensa natural. Varias células ejercen funciones fagocíticas: Neutrófilos,
monocitos y macrófagos.
Los macrófagos son células fagocíticas de gran tamaño presentes en la
mayoría de los tejidos y cavidades, proceden de los monocitos que migran
desde la sangre hacia los tejidos. Algunos permanecen en los tejidos
durante años y otros circulan por los tejidos linfoides secundarios. También
pueden actuar como células presentadoras de antígenos.

27
Su cinética de diferenciación es la siguiente: monocito (en sangre),
Histiocito (células de Kupffer, osteoclastos, células de la microglia, células
del mesangio, etc.), macrófagos inflamatorios y finalmente los macrófagos
clásicamente (M1/CAM) y alternativamente activados (M2/AAM).
Los macrófagos expresan receptores de membrana para numerosos antígenos bacterianos, por
ejemplo: receptor para lipopolisacárido (CD14), receptores CD11b/CD18, receptores para
manosa, y receptor para glúcidos entre otros. Los macrófagos participan en gran medida de la
respuesta inmune innata a infecciones gracias a sus receptores "scavengers", o barredores,
que poseen una especificidad a ligandos muy amplia como: lipoproteínas, proteínas, poli y
oligonucleótidos, polisacáridos aniónicos, fosfolípidos y otras moléculas.
Los neutrófilos son los leucocitos más abundantes (>70 %). Su
tamaño es de 10-20 m de diámetro y se clasifican como granulocitos
debido a sus gránulos citoplasmáticos de lisozimas y de lactoferrina.
Pasan menos de 48 horas en la circulación antes de migrar a los
tejidos, debido a la influencia de los estímulos quimiotácticos. Es en
ellos donde ejercen su acción fagocítica y eventualmente mueren.
Los monocitos son células circulares cuyo diámetro oscila entre 15 a 30 μm
y poseen una alta relación núcleo/citoplasma. Se originan en la médula
ósea y constituyen cerca del 5 % del total de leucocitos de la sangre, donde
permanecen sólo unos tres días. Después atraviesan las paredes de las
vénulas y capilares (diapédesis) donde la circulación es lenta. Una vez en
los órganos, se transforman en macrófagos, lo que se refleja en el aumento
de su capacidad fagocítica, de la síntesis de proteínas, el número de
lisosomas y la cantidad de aparato de Golgi, microtúbulos y microfilamentos.
Estos últimos se relacionan con la formación de seudópodos, responsables
del movimiento de los macrófagos.
Reconocimiento y unión de partículas por fagocitos A pesar de que la fagocitosis de partículas fue demostrada por Haeckel en 1862 y que los
macrófagos y micrófagos fueron descritos por Metchnikoff en 1892, el rol de los factores séricos
que intervienen en este proceso fue puesto en evidencia hasta 1904 cuando Wright y Douglas
determinaron que dichas sustancias eran fundamentales para una óptima ingestión.

28
Etapas de la fagocitosis 1- Quimiotáxis Es la etapa de movilización y reclutamiento de células fagocíticas por medio de interacciones
celulares con la zona o tejido lesionado. El fagocito se adhiere a la superficie del endotelio
previamente activado por citocinas, a través de uniones moleculares de baja afinidad entre
receptores del fagocito y selectinas presentes en el endotelio.
En un punto específico, determinado por la presencia y activación de quimiocinas, los fagocitos
movilizados establecen interacciones intercelulares de gran afinidad con el endotelio por medio
de integrinas y otros ligandos endoteliales. En especial las moléculas endoteliales selectina-E,
ICAM-1,2,3; VCAM-1 se adhieren a ligandos específicos sobre los fagocitos, entre ellos CLA,
LFA-1 y Mac-1.
Los fagocitos atraídos por gradientes de concentración de las quimiocinas, atraviesan entonces
el endotelio vascular hacia el foco de infección patógena.
2- Opsonización La opsonización se consigue recubriendo las partículas con anticuerpos específicos de la clase
IgG, con o sin complemento. Debido a que los fagocitos tienen receptores de membrana para el
fragmento Fc de IgG, reconocen a estas partículas recubiertas por los anticuerpos. La IgM no
tiene capacidad de opsonizar, pero su unión a las partículas induce la activación del sistema de
complemento. Esto lleva a que el componente C3b se deposite sobre las partículas, las cuales
son reconocidas por los receptores CR1, CR3 y CR4, de los fagocitos para el fragmento C3b.
A pesar que el reconocimiento de partículas recubiertas con IgG y/o C3b vía los receptores Fc
y C3b, es el principal mecanismo de ingestión de partículas extrañas, llamado “fagocitosis
inmune”, existen otros factores que median o ejercen su influencia sobre este proceso.
3- Adherencia Receptores específicos sobre la membrana de los fagocitos permiten la adherencia sobre los
microorganismos, ya sea a productos microbianos específicos o sobre opsoninas del sistema
inmune del hospedador.
Algunos receptores de membrana presentes en las células fagocíticas son: receptor de manosa,
receptor para el complejo LPS-LBP (LBP: proteína de unión al LPS) que está formado por el
TLR4 y el CD14, receptores para los fragmentos Fc de los anticuerpos opsonizantes IgG2 e
IgG3.

29
4- Ingestión La unión a receptores de adherencia promueve señales de comunicación intracelular que
resultan en la invaginación de la membrana del fagocito rodeando al receptor y su ligando
patogénico. Al rodear por completo al complejo receptor-molécula, la membrana se une en sus
extremos y libera al interior de la célula un fagosoma. Esto puede ocurrir en más de un punto de
la membrana celular.
5- Digestión Los fagocitos cuentan con variados mecanismos microbicidas, los cuales se activan al
fusionarse el fagosoma con un lisosoma intracelular. Las enzimas del lisosoma se liberan dentro
del fagolisosoma recién formado actuando sobre su contenido. Otros componentes tóxicos
usados en la digestión de microorganismos son los intermediarios reactivos del O2 y el óxido
nítrico.
La Autofagia es un proceso del fagocito en el cual su retículo endoplásmico crea un
autofagosoma en su misma mitocondria liberando gránulos para la destrucción de la misma
mitocondria y así liberar los gránulos contenidos de la mitocondria que son los gránulos
secundarios específicos.
MATERIAL 1 Tubo vacutainer® con heparina 1 pipeta Pasteur 2 portaobjetos Baño María Charola para Tinción Microscopio óptico Contador de Células
REACTIVOS Suspensión de levaduras en SSF Nitroazul de Tetrazolio al 0.1 % en SSF (NBT) Colorante de Wright Solución amortiguadora pH 5.5 a 7.2 Aceite de inmersión

30
DIAGRAMA DE FLUJO
R1: Aguja, al contenedor de punzocortante; Algodón, basura municipal R2: Sangre con levaduras, Contenedor con cloro R3: Residuos de Wright, Recipiente para resíduos de wrigth METODOLOGIA 1. Obtener 3 mL de Sangre periférica con Heparina.
2. Agregar 1 mL de levaduras y 1 ml nitroazul de tetrazolio al tubo vacutainer® y mezclar
suavemente.
3. Incubar, en baño María, durante 30 minutos a 37 °C, mezclando ocasionalmente.
Obtención de sangre periférica
Incubar a 37 °C, 30 min
Suspensión de levaduras
Realizar un frotis
Teñir con Wrigth
Contar a 100X
R1
R3
R2

31
4. Mezclar suavemente, tomar una gota de la sangre y extender sobre un portaobjetos.
5. Teñir como si fuera un frotis para conteo diferencial.
6. Observar a 100X, contando por lo menos 200 células, contando las levaduras fagocitadas
por cada célula.
RESULTADOS Expresar los resultados como:
%푑푒푓푎푔표푐푖푡표푠푖푠 =푐é푙푢푙푎푠푞푢푒푒푠푡á푛푓푎푔표푐푖푡푎푛푑표
푓푎푔표푐푖푡표푠푡표푡푎푙푒푠(100)
퐼푛푑푖푐푒푑푒푓푎푔표푐푖푡표푠푖푠 =퐿푒푣푎푑푢푟푎푠푓푎푔표푐푖푡푎푑푎푠
푐é푙푢푙푎푠푞푢푒푒푠푡á푛푓푎푔표푐푖푡푎푛푑표
DISCUSIÓN Discuta la importancia de la fagocitosis como un mecanismo inespecífico y parcialmente
específico de defensa.
BIBLIOGRAFÍA 1. Aguilar Torrenta, Faviola. (2001). Manual del Curso Practico de Inmunología, 1ª edición, Editorial
ENCB IPN, México DF, 204 pp. 2. Gradwohl, RBH; Sonnenwirth, AC; Jarett, L; Zlochevsky Landes, D. (1986). Métodos y Diagnósticos
del Laboratorio Clínico, Volumen I, 8ª edición, Editorial Panamericana. Buenos Aires, Argentina. 1110 pp.
3. Palomar Morales, Ladislao. (1988). Fagocitosis en cerdos alimentados con diferentes niveles de aflatoxina B1. Tesis UNAM. FES Cuautitlán.
4. Roitt, Ivan. (2000). Inmunología, 5ª edición. Harcourt ediciones. Madrid, España. 423 pp.

32
PRÁCTICA 5
AGLUTINACIÓN, PRECIPITACIÓN Y TURBIDIMETRÍA
QFB Ladislao Palomar Morales
OBJETIVOS Que el estudiante comprenda las bases teóricas de la reacción de aglutinación, los
diferentes factores que contribuyen a la reacción, y sus aplicaciones clínicas para el
diagnóstico de algunas patologías.
Determinar la presencia de inmunoglobulinas (Ig’s) séricas llevando a cabo la prueba de
precipitación de sulfito de sodio.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la unidad 3, Mecanismos específicos de defensa, se describen los conceptos de antígeno,
antigenicidad e inmunoglobulinas. En la parte del contenido práctico se hace mención de las
reacciones antígeno-cognato.
INTRODUCCIÓN Jules Bordet propuso que la aglutinación tomaba lugar en dos fases: a) la combinación
específica del anticuerpo y el antígeno y b) la agregación visible de las partículas. Ambas fases
son mediadas por la atracción específica entre el anticuerpo y el antígeno.
A principios del siglo XX, Karl Landsteiner, menciona que en los eritrocitos deben existir
aglutinógenos (hoy llamados antígenos) que reaccionaban con las aglutininas del suero (hoy
llamados anticuerpos) formando agregados de gran tamaño visibles a simple vista.

33
Las reacciones de aglutinación y de precipitación son la base de la mayor parte de las técnicas
inmunológicas. Su principio se basa en la reacción antígeno-anticuerpo. Para comprender lo
anterior es necesario recordar qué es un antígeno y un anticuerpo.
Antígeno es una sustancia de alto peso molecular, con cierta rigidez estructural y que tiene la
particularidad de ser parcialmente “metabolizado” por células especializadas llamadas
macrófagos, por lo tanto es capaz de generar una respuesta inmune en un organismo que la
detecte como un agente extraño. Mientras que un anticuerpo es una glicoproteína, producida
por linfocitos B activados, llamados células plasmáticas, como respuesta a la presencia de un
antígeno en el organismo, a su vez los anticuerpos pueden ser producidos por líneas celulares
in vitro, como es el caso de la producción de anticuerpos monoclonales. Dichos anticuerpos
llamados también inmunoglobulinas, se presentan en cinco clases principales, IgG, IgM, IgA,
IgD e IgE, que se diferencian entre sí por sus características físicas, químicas y biológicas.
La capacidad inmunogénica de distintas partículas, es en orden decreciente la siguiente:
proteínas, glicoproteínas, polisacáridos, lípidos y azúcares. Las técnicas de aglutinación son
sólo semicuantitativas y algo más difíciles. La aglutinación de los antígenos nativos insolubles o
de las partículas recubiertas por el antígeno puede evaluarse a simple vista con o sin la ayuda
del microscopio. Entre las ventajas de las reacciones de aglutinación están su alto grado de
sensibilidad y la enorme variedad de substancias identificables a través del uso de partículas
que están recubiertas por antígeno o por anticuerpo.
Según Coombs existen 3 requerimientos principales en las pruebas de aglutinación:
1. Disponibilidad de una suspensión estable de células o de partículas.
2. Presencia de uno o más antígenos cercanos a la superficie.
3. Conocimiento de que los anticuerpos "incompletos" o no aglutinables no son
localizables sin modificación (reacciones antiglobulina).
Existen algunas variantes de la técnica de aglutinación, entre las cuales podemos mencionar:
Aglutinación Directa: Anticuerpo soluble y antígeno presente en una célula o partícula
insoluble.
Aglutinación Indirecta: Anticuerpo unido a una célula o partícula insoluble y antígeno
soluble.

34
Inhibición de la Aglutinación: Reacción en dos pasos; primero se ponen a reaccionar
el anticuerpo y el antígeno (ambos en forma soluble) y en una segunda etapa se agrega
uno de ellos unido a una célula o partícula insoluble.
Floculación: El complejo antígeno-anticuerpo no es tan grande como en las reacciones
de aglutinación, y en ocasiones el complejo puede ser inestable.
MATERIAL REACTIVOS Placas de cristal o plástico de fondo negro y/o fondo transparente 5 tubos de ensaye 13X100 Centrifuga Balanza de dos platos Juego de micropipetas y puntas
Equipos de diagnóstico para VDRL, Factor Reumatoide, Reacciones Febriles y/o grupos sanguíneos Solución de sulfito de sodio al 14, 16 y 18 %
DIAGRAMA DE FLUJO
R1: Aguja: Contenedor de punzocortantes, Algodón: contenedor municipal, Paquete eritrocitario:
Contenedor con hipoclorito de sodio. R2 y R3: Contenedor con hipoclorito de sodio. METODOLOGÍA 1. Extraer, con el sistema Vacutainer®, un tubo de sangre periférica, para obtener suero o
plasma.
2. Separar el suero o el plasma y colocarlo en un tubo de ensaye.
TURBIDIMETRÍA 3. En un tubo de ensaye colocar 1.9 mL de sulfato de amonio al 14 %; otro al 16% y uno más
al 18%. Agregar 0.1 mL de suero o plasma a cada tubo. Mezclar y dejar en reposo 30 min.
Obtener sangre periférica
Separar plasma o suero
Mezclar antígeno y suero en una placa
1.9 mL de sulfato de sodio, agregar 0.1 mL de suero
R1
R2
R3

35
PRUEBAS DE AGLUTINACIÓN 4. En una placa: colocar una gota del antígeno unido a látex, en una de las divisiones. Se
usarán tantas divisiones como antígenos se tengan. Agregar a cada división una gota del
suero o plasma y mezclar con la punta de un palillo. La lectura de la aglutinación debe
realizarse antes de dos minutos.
RESULTADOS Si hay turbidez en los tres tubos de sulfato de amonio, se debe reportar una concentración de Ig’s mayor o igual a 15 mg/mL de suero.
Si hay turbidez en los tubos de sulfato de amonio al 16 y 18 %, se debe reportar una concentración de Ig’s mayor entre 5 y 15 mg/mL de suero.
Si hay turbidez solo en el tubo de sulfato de amonio al 18 %, se debe reportar una concentración de Ig’s menor o igual a 5 mg/mL de suero.
Para las reacciones de aglutinación se reportara positivo (+) o negativo (-). CONCLUSIONES BIBLIOGRAFÍA 1. Aguilar-García, Vicente. (2004). VI. Reacciones de aglutinación. Gac Méd Méx Vol. 140, Suplemento
No. 3. Pp: S50-S52. 2. Martínez Romero, Aurora y Ortega Sánchez, José Luis. (2011). Manual de Laboratorio de
Inmunología Básica y Clínica. Universidad Autónoma de Chapingo y Revista electrónica de Veterinaria [En línea] http://www.veterinaria.org/revistas/redvet/n040411/041108.pdf, obtenido el 26 de agosto de 2013.
3. Morales de Ramírez, Ana Margarita Paz y Gaitán Fernández, Isabel Cristina. (2011). Manual de procedimientos de laboratorio: INMUNOLOGÍA. Universidad Mariano Galvez. Guatemala.

36
PRÁCTICA 6
MODELOS EXPERIMENTALES Y VÍAS DE INMUNIZACIÓN
Dr. Salvador Fonseca Coronado
OBJETIVOS Y COMPETENCIAS Al finalizar el procedimiento experimental es deseable que el alumno haya adquirido las
siguientes competencias:
Conocer la importancia de los modelos de experimentación en el ámbito de la investigación
básica, clínica y diagnóstica en inmunología.
Entender los procedimientos y manejar de manera adecuada y ética a los ratones sujetos de
experimentación en el curso.
Conocer las vías de inoculación de antígenos y adquirir la habilidad de realizar la
inoculación de antígenos por vía intraperitoneal.
Comprender la importancia del uso de adyuvantes en la activación de los MID y los MED.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la unidad 3 se abordan los mecanismos específicos de defensa (MED), en esta práctica se
lleva a cabo una introducción a la forma en que se da lugar a la activación de los MED mediante
la inoculación (exposición) a agentes inmunógenicos y a la importancia de los adyuvantes como
activadores de los MID.
Manejo de animales de laboratorio e identificación de las principales vías de inoculación en ratón. Importancia: Un profesional del área debe adquirir la competencia de conocer y manejar adecuadamente modelos experimentales y animales de experimentación.

37
INTRODUCCIÓN Un modelo experimental es un sistema que nos permite imitar un fenómeno en condiciones
controladas con la finalidad de describirlo lo más detalladamente posible. Casi todo lo que
damos por sentado del conocimiento en inmunología se ha estudiado y descrito a partir de
modelos experimentales tanto in vitro como in vivo.
Las líneas celulares y los cultivos primarios constituyen el principal modelo in vitro, en tanto que
el estudio en ratones es, por mucho, el modelo in vivo que más aportaciones a dado en el
estudio de la inmunología.
Con respecto a los modelos in vitro, en esta práctica, el alumno adquirirá los conocimientos
para el establecimiento de un cultivo primario y el mantenimiento de una línea celular. En este
punto el alumno ya ha trabajado modelos experimentales in vivo en asignaturas como
farmacología, ahora se abordara el modelo en ratón para conocer y analizar uno de los
fenómenos más relevantes en el campo: la reacción antígeno-anticuerpo.
El reconocimiento Ag-Ab es una reacción de complementariedad, se efectúa a través de
múltiples enlaces no covalentes entre una parte del antígeno (denominada epítopo) y los
aminoácidos del sitio de unión del anticuerpo. La reacción se caracteriza por su especificidad,
espontaneidad y reversibilidad.
Especificidad: Capacidad del anticuerpo de unirse al antígeno que lo estimuló. La unión dada
por la especificidad es muy precisa y permite distinguir entre grupos químicos con diferencias
mínimas a pesar de su similitud; además, permite la detención de un sólo antígeno en cuestión.
Espontaneidad: La reacción Ag-Ab no requiere energía adicional para efectuarse.
Reversibilidad: Dado que la reacción se debe a fuerzas no covalentes, es reversible y, en
consecuencia, se ve afectada por factores como la temperatura, la proporción de Ag-Ab, el pH y
la fuerza iónica.
En este primer acercamiento se conocerán las principales vías de inoculación de antígenos en
ratón y las implicaciones inmunológicas dependientes de cada vía, en particular la activación de
linfocitos B con la consiguiente producción de anticuerpos, los cuales serán posteriormente
obtenidos y utilizados en las pruebas de ELISA y Western blot.

38
MATERIAL Y EQUIPO Jeringas de 1 y 3 mL Cánulas de alimentación oral para neonatos
REACTIVOS Ácido pícrico Adyuvante completo de Freund Adyuvante incompleto de Freund Solución inyectable Albúmina Sérica Bovina Gama Globulinas Humanas
DIAGRAMA DE FLUJO
R1: Aguja (contenedor rígido para punzocortantes), Jeringas (bolsa roja)
Repartición de ratones y aprendizaje de
manipulación
Demostración de las diferentes vías de inoculación para animales de laboratorio:
Oral, Subcutánea, Intraperitoneal
Formación de grupos de experimentación e inoculación
intraperitoneal
Lote 1 100 L de solución
inyectable/ratón
Lote 2 20 g de IgG purificadas
en 100L de solución inyectable/ratón
Lote 3 20 g de IgG
purificadas en 100L de adyuvante/ratón
Mantener a los ratones e inmunizar nuevamente a los 7, 14 y 21 días.
El día 28 se obtiene muestra de sangre y se sacrifican los ratones
R1 R1

39
METODOLOGÍA Calendario de actividades de esta práctica
Actividad Día Manejo e inmunización de ratones I puntos 1 al 5 y (6, 7 u 8)
17 ó 18 de septiembre/2015
Inmunización de ratones II puntos 6, 7 o 9
24 ó 25 de septiembre/2015
Inmunización de ratones III puntos 6, 7 o 9
01 ó 02 de octubre/2015
Sacrificio y muestreo Práctica Anatomía del Sistema Inmune
15 ó 16 de octubre/2015
1. A cada equipo se le proporcionará un ratón y será responsable de su cuidado durante el
tiempo de desarrollo del proceso experimental.
2. Se hará una demostración del manejo adecuado de los ratones y los cuidados y riesgos de
su manipulación.
3. Se hará una demostración de las diferentes rutas de administración de sustancias a los
ratones (oral, intramuscular, intraperitoneal, subcutánea) y cada alumno seleccionará una
ruta para practicar evitando al máximo maltratos innecesarios de los animales.
4. Se enseñara a los alumnos las técnicas de marcaje, la selección e importancia estadística
de los grupos de experimentación, y se llevará a cabo la primera inoculación en ratones, con
las inmunoglobulinas humanas purificadas y cuantificadas en las prácticas anteriores (las
cuales se comportarán ahora como inmunógenos).
5. Se formarán tres grupos de experimentación de 6 ratones cada uno, los alumnos llevarán a
cabo el marcaje de sus animales.
6. Al grupo 1 se le administrarán 100 µL de solución inyectable a cada ratón por vía
intraperitoneal.
7. Al grupo 2 se le administrarán 20 µg de Ig´s en 100 µL de solución inyectable a cada ratón
por vía intraperitoneal.
8. Al grupo 3 se le administrarán 20 µg de Ig´s en 100 µL del adyuvante seleccionado a cada
ratón por vía intraperitoneal.
9. Se administrará la misma dosis a cada grupo a los 7, 14 y 21 días, solo se sustituirá el
adyuvante completo por adyuvante incompleto.

40
10. Los animales serán mantenidos en condiciones adecuadas con agua y alimento ad libitum
durante todo el periodo de experimentación.
11. El día 28 se tomara una muestra de sangre y se sacrificaran los animales, para utilizarlos en
la Práctica de Anatomía del Sistema Inmune,
Discusión El alumno en base a la información previa obtenida, discutirá las implicaciones de la
administración de un determinado antígeno por cada una de las vías analizadas y que órganos
y células se ven implicadas en cada caso.
Los alumnos tendrán una mesa de discusión acerca de la importancia de los adyuvantes.
Referencias 1. Delgado, N; Revuelta, M. (1993). Guía Práctica para el Manejo de Animales de Laboratorio. UNAM.
Facultad de estudios Superiores Cuautitlán. México, D.F. 115 p. 2. Donovan, J. C. and Brown, P. (2007). Care and Handling of Laboratory Animals. Current Protocols in
Immunology. 76:1.0.1–1.0.4. DOI: 10.1002/0471142735.im0100s76 3. Shevach, E. M. (2010). Animal Models for Infectious Diseases. Current Protocols in Immunology.
91:19.0.1–19.0.4. DOI: 10.1002/0471142735.im1900s91

41
PRÁCTICA 7
CITOMETRÍA DE FLUJO
Dr. Salvador Fonseca Coronado OBJETIVO Comprender los fundamentos de la Citometría de flujo y sus aplicaciones en la
determinación de marcadores celulares en investigación básica, clínica y diagnóstica.
RELACIÓN CON LOS CONTENIDOS TEORICOS DEL PROGRAMA Se relaciona con los Mecanismos Específicos de Defensa, con la estructura y función de los
receptores celulares para el antígeno, con la estructura y función de inmunoglobulinas y con la
interacción antígeno-anticuerpo.
FUNDAMENTOS La citometría de flujo (CF) es un método que permite el análisis de múltiples características
celulares como su morfología, la presencia de moléculas de superficie, intracelulares y
secretadas, así como su dinámica de expresión, tanto de células eucariotas como procariotas.
El equipo utilizado para estos fines es el citofluorómetro de flujo (denominado FACS, del inglés:
Fluorescence Activated Cell Sorter), el cual permite la detección de los parámetros de tamaño,
granularidad (o complejidad de la célula) y la emisión de fluorescencia.
El principio básico de la CF (Figura 1) consiste en obtener una suspensión celular (las células
se pueden marcar con anticuerpos u otras proteínas acopladas a fluorocromos capaces de
unirse en la superficie o intracelularmente a las moléculas a evaluar) y hacerlas pasar de forma
individual a través de un capilar, a una cámara donde incide sobre ellas un rayo láser. El haz de
luz láser cuando atraviesa una célula sufre una dispersión que puede ser evidenciada en un
fotodetector situado enfrente de la fuente emisora, como una disminución de la luz incidente. El
tiempo que dure el corte en la llegada de fotones al detector será proporcional al diámetro, y por
tanto al volumen o tamaño celular. Este estudio de interrupción frontal del haz láser se
denomina como Forward Scatter (FSC). Además del detector de luz situado frente al haz láser,

42
existe otro grupo óptico, que detecta la luz dispersada por las células y que se coloca formando
un ángulo de 90º con el haz láser. Esta luz es descompuesta, mediante los filtros apropiados
para poder estudiar tanto la dispersión de luz de la misma longitud de onda del láser como la
emitida por los fluorocromos. La mayor o menor dispersión de la luz y por tanto la mayor o
menor detección de luz en éste detector, será proporcional a la complejidad de la superficie
celular y a las estructuras y organelos celulares, denominándose Side Scatter (SSC); con estos
parámetros, el equipo amplifica las señales eléctricas y las convierte, mediante un sistema de
tarjetas electrónicas, en un gráfico en dos dimensiones que permite la separación de las
diferentes poblaciones celulares de acuerdo a su tamaño y granularidad (Figura 2).
Figura 1. Esquema de los sistemas de detección del citómetro de flujo.
Como se observa en la figura 1, existen una serie de detectores incorporados en el citómetro
que permiten detectar estas señales de fluorescencia. El marcaje de toda una amplia gama de
moléculas, permite que la CF sea una herramienta poderosa para realizar la caracterización de
múltiples funciones celulares.

43
Figura 2. Diagrama de tamaño vs granularidad (DOT-PLOT) de una muestra de sangre periférica donde se muestra la capacidad de separación de las diferentes poblaciones celulares mediante los sistemas detectores FSC y SSC.
Como se mencionó, las células pueden ser marcadas con fluorocromos libres o acoplados a
moléculas con afinidad por el componente celular que se desee evaluar, las moléculas más
utilizadas para este fin son los anticuerpos (debido a su especificidad), mediante los cuales es
posible analizar marcadores de la superficie celular (CD4, CD8 y todos los marcadores de
identidad); marcadores intracelulares (citocinas, caspasas, cinasas, proteínas virales, etc.) y
marcadores nucleares (DNA, RNA).
Los fluorocromos son una serie de compuestos químicos que tienen la capacidad de absorber
la luz del rayo láser en forma de fotones y emitirlos con menor energía (mayor longitud de
onda), si estos compuestos se encuentran acoplados a anticuerpos contra una molécula en
particular, la intensidad de la emisión será directamente proporcional a la concentración o
cantidad de la citada molécula. En la figura 3 se muestra el espectro de emisión de algunos
fluorocromos utilizados en CF.

44
Figura 3. Principales fluorocromos utilizados en CF, se muestran los excitados por un rayo láser de argón (488 nm) y de helio-Neón (630 nm). FITC: Isotiocianato de fluoresceína, PE: Ficoeritrina, ECD: Electron Coupled Dye
A partir de la grafica FSC vs SSC se pueden generar regiones para realizar el análisis de
poblaciones en particular, por ejemplo, en la figura, 1 se señala en rojo la región
correspondiente a los linfocitos, si se marcó esta suspensión con anticuerpos acoplados a
fluorocromos contra las subpoblaciones de linfocitos cooperadores (CD4-PE) y citotóxicos
(CD8-FITC), estos se pueden visualizar en un gráfico denominado histograma en el que se
grafica la intensidad del fluorocromo en cuestión contra el número de eventos (células) positivos
a dicha molécula (Figura 4).
Figura 4. Histogramas de la región correspondiente a los linfocitos donde se observan las poblaciones positivas a CD8 y a CD4
45
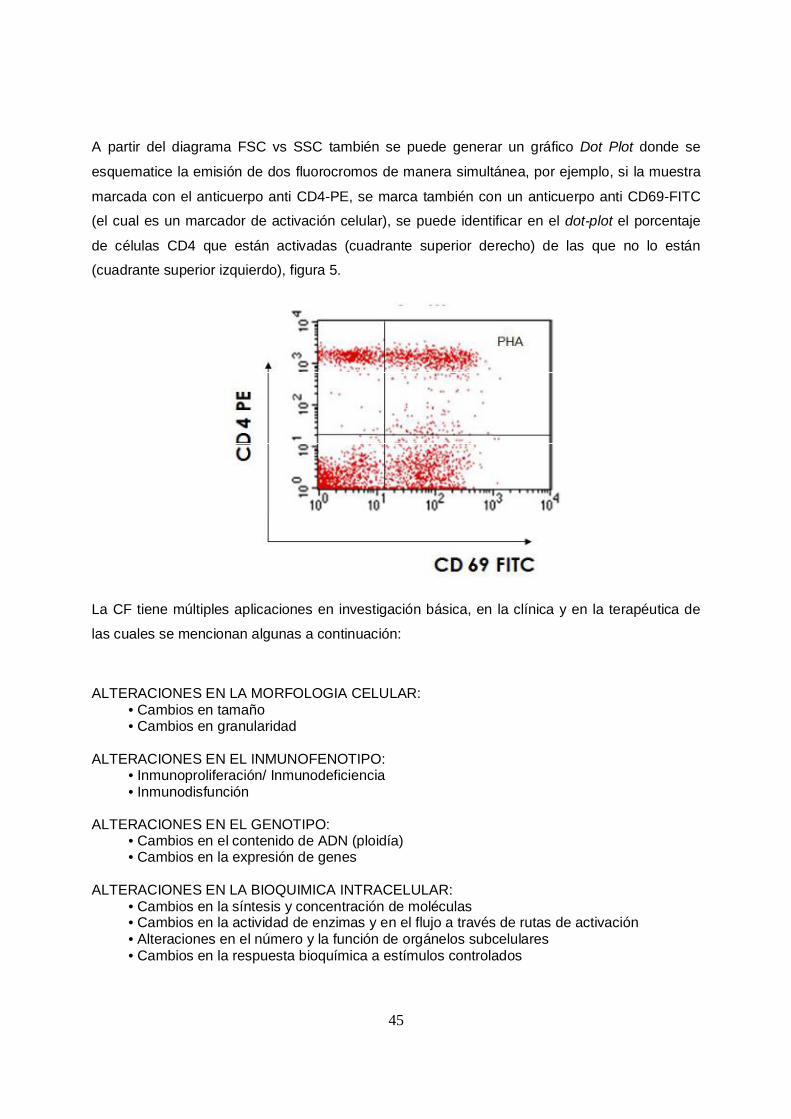
A partir del diagrama FSC vs SSC también se puede generar un gráfico Dot Plot donde se
esquematice la emisión de dos fluorocromos de manera simultánea, por ejemplo, si la muestra
marcada con el anticuerpo anti CD4-PE, se marca también con un anticuerpo anti CD69-FITC
(el cual es un marcador de activación celular), se puede identificar en el dot-plot el porcentaje
de células CD4 que están activadas (cuadrante superior derecho) de las que no lo están
(cuadrante superior izquierdo), figura 5.
La CF tiene múltiples aplicaciones en investigación básica, en la clínica y en la terapéutica de
las cuales se mencionan algunas a continuación:
ALTERACIONES EN LA MORFOLOGIA CELULAR:
• Cambios en tamaño • Cambios en granularidad
ALTERACIONES EN EL INMUNOFENOTIPO:
• Inmunoproliferación/ Inmunodeficiencia • Inmunodisfunción
ALTERACIONES EN EL GENOTIPO:
• Cambios en el contenido de ADN (ploidía) • Cambios en la expresión de genes
ALTERACIONES EN LA BIOQUIMICA INTRACELULAR:
• Cambios en la síntesis y concentración de moléculas • Cambios en la actividad de enzimas y en el flujo a través de rutas de activación • Alteraciones en el número y la función de orgánelos subcelulares • Cambios en la respuesta bioquímica a estímulos controlados

46
DETECCION DE CELULAS INFRECUENTES (“CELULAS RARAS”) • Detección de células tumorales circulantes • Detección de células fetales en sangre materna • Detección de células activadas o antígeno-específicas circulantes
PARAMETROS IMPLICADOS DIRECTAMENTE EN EL PROCESO PATOLOGICO:
• Análisis de parámetros específicos de la patología SELECCION DE CELULAS EN LA TERAPIA CELULAR:
• Análisis de progenitores en el transplante autólogo • Pruebas cruzadas (“cross-match”) en transplantes heterólogos • Detección y cuantificación de leucocitos residuales en hemopreparados
ANALISIS DE LA ACCION TERAPEUTICA A NIVEL CELULAR:
• Cambios en parámetros estructurales • Cambios en parámetros funcionales
ANALISIS DE LA ACCION TERAPEUTICA A NIVEL DEL PACIENTE:
• Detección de recidivas y enfermedad mínima residual • Establecimiento de patrones pronósticos de éxito terapéutico
DETECCION Y ANALISIS DE RESISTENCIA A LA TERAPIA:
• Análisis de la captación y retención de fármacos (fenotipo MDR) • Análisis del metabolismo de fármacos
CARACTERIZACION DE LOS MECANISMOS DE MUERTE CELULAR:
• Identificación y análisis de células apoptóticas • Identificación de células necróticas
Discusión Discuta con sus compañeros otras aplicaciones de la citometría de flujo como la cuantificación
de citocinas con perlas fluorescentes, estudios del ciclo celular, purificación de subpoblaciones celulares, identificación de vías de señalización, etc.
REFERENCIAS 1. Shevach, Ethan M. (2009). Immunofluorescence and Cell Sorting. Current Protocols in Immunology
5.0.1-5.0.3, Published online November 2009 in Wiley Interscience (www.interscience.wiley.com). DOI: 10.1002/0471142735.im0500s87
2. Darzynkiewicz, Z., Robinson, J.P., and Crissman, H.A. (eds.) (1994). Flow Cytometry, 2nd ed. Methods Cell Biol.: 41 & 42. Academic Press, San Diego.
3. Mason, D.W. and Williams, A.F. (1980). The kinetics of antibody binding to membrane antigens in solution and at the cell surface. Biochem. J.187:1-9.
4. Shapiro, H. M. (1988). Practical Flow Cytometry, 2nd ed. Wiley-Liss, New York.

47
PRÁCTICA 8
ENSAYO INMUNO ENZIMÁTICO (ELISA)
Dr. Marco Antonio Vega López
OBJETIVO Determinar la presencia de anticuerpos en muestras biológicas, para el diagnóstico indirecto
de patologías como el sarampión a través del inmunoensayo enzimático ELISA.
INTRODUCCIÓN Los inmunoanálisis ligados a enzimas son técnicas en las cuales uno de los reactantes, el
anticuerpo o el antígeno, se fija en un soporte sólido, casi siempre una placa de plástico, antes
de su interacción con la muestra del paciente y el reactante revelador de la reacción que, por lo
general, es un anticuerpo conjugado a una enzima que actúa sobre un substrato-cromógeno,
que indica y detecta la presencia del reactante a analizar virando a un color.
Las pruebas de ELISA o EIA, (enzyme-linked-immuno-sorbent-assay), se basan en dos
fenómenos biológicos importantes:
1 La especificidad de los anticuerpos (Ab). 2 La amplificación de la señal generada a través de la acción de una enzima.
La sencillez metodológica del ELISA y sus reducidos requerimientos de equipos especiales,
sumada a la especificidad de los Ab monoclonales (MAb), hace que sean sencillos, baratos y de
sensibilidad aceptable para su uso en el laboratorio, por encima de los basados en la utilización
de un trazador radiactivo, (radioinmunoanálisis –RIA-), un trazador emisor de luz
(quimioluminiscencia) o un trazador fluorescente (fluorografía).
Clasificación de las pruebas inmuno enzimáticas: Las pruebas inmuno enzimáticas pueden clasificarse en:
a) Homogéneas. b) Heterogéneas.

48
Las primeras se realizan exclusivamente en fase liquida y con los reactivos añadidos en forma
simultánea. Se emplean para aparatos automatizados y no son de uso común. Las segundas
emplean un soporte sólido para inmovilizar a uno de los inmunorreactantes y, a partir de ello, se
realiza la reacción específica de detección. Estas últimas son las de mayor uso en el
laboratorio.
En las pruebas heterogéneas, existen muchas variantes, dependiendo del tipo de analito que se
busque. Las pruebas más comunes son las de detección indirecta, donde se busca la presencia
de anticuerpos (Ab) en los pacientes. En ellas se fija el Ag de interés (de un virus o bacteria) en
la placa y sobre él se aplica la muestra (suero, saliva, líquido cefalorraquídeo, etc.). Los
anticuerpos específicos en la muestra (anticuerpo primario) se fijarán en el Ag y se harán
evidentes usando otro anticuerpo (secundario) que reconozca al primero y que está conjugado
con una enzima. El Ab secundario reaccionará con el sustrato, que debe ser incoloro y soluble y
éste, al ser degradado, deberá convertirse en un producto colorido y soluble, cuya
concentración se medirá por espectrofotometría.
También existe la variante de “sándwich”, donde se fija en la placa un anticuerpo primario (Ab
de captura), específico para el analito que se busca (por ejemplo citocinas), se coloca la
muestra y el analito capturado por el Ab primario se identifica mediante otro Ab que está
conjugado con una enzima y que también reacciona con el analito, pero en otro epítopo. El
revelado se hace como ya se explicó.
Finalmente, existe la variante competitiva, donde se trata de cuantificar la cantidad de analito de
interés. En este caso se usa una variante de la técnica de “sándwich”, donde al pozo con el Ab
de captura se añade tanto la muestra como una cantidad conocida del analito marcado con un
fluorocromo o una enzima. Esto establecerá una competencia entre el analito de la muestra y el
que nosotros añadimos en cantidades conocidas. En este caso, si la muestra no contiene al
analito, nuestro conjugado se unirá por completo al Ab de captura y tendremos la máxima señal.
Si la muestra contiene cantidades altas del analito, éste competirá con el conjugado que
añadimos y evitará que se fije al Ab de captura, de esta manera, entre más concentración de
analito tenga nuestra muestra, menos señal tendremos porque no se fijará el conjugado.
Haciendo curvas de concentración puede cuantificarse la cantidad de analito en la muestra.

49
Diagrama con los componentes principales de las pruebas de ELISA. Se esquematiza la prueba de “sándwich”.
Sistemas alternativos de reconocimiento. Es posible aumentar la sensibilidad de la técnica usando conjugados con otras proteínas
distintas a los Abs. Uno de esos sistemas involucra a la estreptavidina que es una proteína
producida por estreptococos y ciertas levaduras y que tiene elevada afinidad por la biotina
(Ka = ). Debido a que la biotina puede conjugarse fácilmente a distintas proteínas (entre
ellas, Ab y enzimas) por unión covalente, sin afectar su actividad biológica, se han desarrollado
métodos inmunoenzimáticos basados en la interacción estreptavidina/biotina.
Proteína A. Es una proteína de la pared de Staphylococus aureus (cepa Cowan I) que tiene alta afinidad
(Ka = 8 110 M ) por el Fc de algunos isotipos de las Ig. Tiene 4 sitios de unión pero usualmente
sólo reaccionan dos. Estas propiedades permitieron el desarrollo de diversos ELISA con
proteína A.
En la presente práctica se realizará un ELISA indirecto, que consiste en buscar Ab contra Ag
conocidos, los pozos se recubren con el Ag, después se lavan y se tratan con proteína
bloqueadora irrelevante y luego se incuban con en el suero problema. Los Ab, retenidos por el
Ag se detectan por adición de un segundo Ab conjugado a una enzima (conjugado), que
después se revela adicionando el substrato de la enzima y un cromógeno. El resultado de la
reacción es la formación de un producto coloreado soluble, se realiza para diagnosticar
sarampión.
15 110 M

50
MATERIAL 1 gradilla con 10 tubos de ensayo de 12x75 1 placa con micropozos 1 Juego de micropipetas 1 Pipeta multicanal (8 ó 12 puntas) Cubetas para micropipetas Puntas para micropipetas Estufa de incubación
REACTIVOS IgG humana purificada por Salting Out Suero de ratón inmunizado con IgG humana Leche descremada al 5 % en PBS Solución de lavado (PBS-Tween 20) Conjugado de cabra anti-IgG de Ratón Sustrato para peroxidasa Solución de paro (HCl 2 N)
DIAGRAMA DE FLUJO
R1: Sobrenadantes y residuos de los lavados al contenedor con agua clorada
Placa de micropozos
R1
IgG Humana diluida en amortiguador de carbonatos
Incubar
Lavar
Incubar
Solución bloqueadora
Lavar
Suero de ratón diluido Incubar
Lavar
Conjugado
Incubar
Lavar
Sustrato/cromógeno Incubar
Leer absorbancia
Sustrato/cromógeno

51
METODOLOGÍA Búsqueda de Ab de ratón contra -Globulina Humana, IgG) Los pasos 1 al 6 se realizan previamente en otra sesión 1. Sensibilizar una placa de micropozos con 100 μL de IgG humana (50 μg/pozo) en
amortiguador de carbonatos pH 9.6, incubar durante 1 hora a 37 °C (o toda la noche a 4 °C).
2. Vaciar el contenido en recipiente con cloro, sacudir contra papel absorbente.
3. Agregar 300 μL de la solución de lavado, vaciar el contenido al recipiente con cloro (lavar
tres veces).
4. Agregar 200 μL de caseína (leche descremada) al 5 % en PBS-Tween, incubar durante
1 hora a 37 °C (o toda la noche a 4 °C).
5. Vaciar el contenido en un recipiente con cloro, sacudir contra papel absorbente.
6. Agregar 300 μL de la solución de lavado, vaciar el contenido al recipiente con cloro (tres
veces). Dejar secar, cubrir con papel parafilm hasta el momento de su uso y guardar en
refrigeración.
El día de la práctica 7. Realizar una serie de diluciones dobles, empezando por 1/10 de suero de ratón (inmunizado
con IgG); el volumen final de cada dilución debe ser de 100 L.
8. Colocar 100 L de los sueros diluidos en cada pozo. Cubrir los pozos con papel parafilm,
agitar suavemente 15 segundos. Incubar 40 minutos a temperatura ambiente.
9. Vaciar el contenido en recipiente con cloro, sacudir contra papel absorbente.
10. Agregar 300 μL de la solución de lavado (PBS-Tween), vaciar el contenido al recipiente con
cloro (lavar tres veces).
11. Agregar 100 μL del conjugado (anti IgG de ratón, producido en cabra, unido a peroxidasa)
diluido 1/10 000 con solución bloqueadora a cada pozo. Agitar suavemente 15 segundos,
incubar 30 minutos a temperatura ambiente.
12. Vaciar el contenido en recipiente con cloro, sacudir contra papel absorbente.

52
13. Agregar 300 μL de la solución de lavado, vaciar el contenido al recipiente con cloro (tres
veces).
14. Agregar 50 μL de solución reveladora (diaminobencidina/peróxido de hidrógeno) a cada
pozo. Agitar suavemente 15 segundos. Incubar 10 minutos en obscuridad.
15. Agregar 50 μL de la solución de paro a cada pozo.
16. Una coloración amarilla indica prueba positiva y negativa si es incolora.
RESULTADOS Si se cuenta con un lector de ELISA el resultado (+) se asignará a la lectura mayor del valor de
la densidad óptica por sobre los controles, en caso de no contar con el lector de ELISA se hará
la lectura de manera visual.
BIBLIOGRAFÍA 1. Aguilar Torrenta, Faviola. (2001). Manual del Curso Practico de Inmunología, 1ª edición, Editorial
ENCB IPN, México DF, 204 pp. 2. Margini, Ricardo, Anibal. (1996). Inmunología e Inmunoquímica. 5ª edición. Editorial Panamericana.
Buenos Aires, Argentina. 976 pp. 3. Rojas Espinosa, Oscar. (2001). Inmunología (de memoria), 2ª edición, Editorial Panamericana.
México, DF. 374 pp. 4. Campbell, MA. (2002). ELISA (Enzyme-Linked ImmunoSorbant Assay). [En línea]
http://www.bio.davidson.edu/courses/genomics/method/ELISA.html consultado el 30 de enero 2014. 5. The Biology Project Home. (2000). Introduction to ELISA Activity. [En línea]
http://www.biology.arizona.edu/immunology/activities/elisa/technique.html consultado el 30 de enero 2014.

53
PRÁCTICA 9
ANATOMÍA DEL SISTEMA INMUNITARIO
MVZ Ángel Germán Martínez Sosa
OBJETIVOS Que el estudiante identifique in situ los órganos linfáticos visibles del ratón (ganglios
linfáticos axilares, inguinales y mesentéricos; timo, bazo y placas de Peyer) haciendo
énfasis en su posición anatómica, apariencia, color y textura.
Que logren la separación de células de un órgano linfoide primario (timo) y uno secundario
(bazo y placas de Peyer) y su identificación.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la unidad Mecanismos específicos de defensa, los órganos primarios y secundarios del
sistema inmune y la circulación de células del sistema inmune son analizados; y a su vez
complementados con el desarrollo experimental de esta práctica.
INTRODUCCIÓN El Sistema Inmune está formado por células, tejidos y órganos con un origen embriológico común: el mesodermo; está constituido por alrededor de 1011 linfocitos B y 1018 linfocitos T
con diferentes receptores para el antígeno, que realizan una función de patrullaje o vigilancia de
antígenos procedentes tanto de nuestro medio ambiente interno como externo.
Los órganos linfáticos primarios, centrales o independientes son el Timo para los linfocitos T
y la Bursa de Fabricio, dependiendo de la especie animal, para los linfocitos B; su función
primordial es la maduración de las células, con la eliminación por apoptosis de las células
autorreactivas.
Los órganos linfáticos secundarios, periféricos o antígeno-dependientes son los ganglios
linfáticos que constituyen cadenas ganglionares en diversas partes del cuerpo, el bazo y las
placas de Peyer; su función primordial es filtrar la linfa formada durante los procesos

54
inflamatorios en los tejidos circunvecinos; en su interior se activa y desarrolla la respuesta
inmune y mediante los vasos linfáticos eferentes egresan los linfocitos B y T activados/memoria
para posteriormente ingresar a la circulación sanguínea sistémica.
Los órganos linfáticos terciarios son aquellos que alojan a las células de memoria y en los
procesos inflamatorios crónicos son los sitios ectópicos donde se organizan estructuras linfoides
in situ.
A nivel celular es importante tener presente que cada estirpe celular, así como sus diferentes
estadios funcionales se caracterizan por la expresión de un fenotipo o marcadores celulares
específicos; los cuales debemos conocer para su identificación y purificación que pueden
realizarse tanto por de citometría de flujo, como por purificación por perlas magnéticas MACS
(del inglés: Magnetic Antibody Cell Sorter).
MATERIAL Equipo de disección 1 gradilla con 10 tubos de ensayo de 13x100 3 pipetas Pasteur 3 coladeras y 3 émbolos de jeringa 2 cajas Petri y 3 portaobjetos 2 pipetas de 2 mL Microscopio
REACTIVOS 100 mL de SSF o PBS pH 7.4 Tinción de Giemsa
DIAGRAMA DE FLUJO
R1: Los cadáveres de ratón y órganos, envolverlos en papel y depositarlos en bolsa amarilla para
congelarlos hasta el momento de su incineración. R2: Todos los residuos líquidos depositarlos en el contenedor que contiene agua con cloro. R3:Contenedor para residuos de Giemsa o Wright.
Sacrificar un ratón por dislocación cervical
Incidir en la piel para localizar e identificar los ganglios linfáticos inguinales y axilares
Obtener bazo, timo y placas de Peyer
Macerar y lavar las células obtenidas
Resuspender en 1 mL de PBS
Realizar frotis y teñir con Giemsa
Observar a 100X para realizar conteo diferencial
R1
R2

55
METODOLOGÍA 1. Sacrificar un ratón por alumno, por dislocación
cervical.
2. Cortar la piel sin interesar órganos internos, desde
la laringe hasta la pelvis, para localizar e
identificar los ganglios linfáticos inguinales y
axilares.
3. Realizar la identificación y disección de los
siguientes órganos, bazo, timo y placas de Peyer,
colocándolos en una caja de Petri con un poco de
SSF.
4. Macerar individualmente los órganos anteriores
en una coladera, con el extremo plano del émbolo
de una jeringa, agregar 1 ó 2 mL de SSF.
5. Lavar, centrifugando 2 veces con SSF.
6. Resuspender las células en 1 mL de SSF.
7. Realizar un frotis con cada una de las poblaciones
celulares obtenidas y teñir con Giemsa o Wright.
RESULTADOS Describir el aspecto macroscópico de los ganglios y órganos linfáticos del ratón-
Reportar lo observado en los frotis de cada suspensión celular.
BIBLIOGRAFÍA 1. Goldsby, RA.; Kindt, TJ.; Osborne, BA. & Kuby, J. (2004). Inmunobiología. Quinta edición Mc Graw
Hill, Iztapalapa, México D.F.; 665 pp. 2. Roitt, I. (2000). Inmunología. Quinta edición Harcourt Editions; Madrid, España. 423 pp.

56
PRÁCTICA 10
HISTOLOGÍA E INMUNOCITOQUÍMICA
M. en C. Erik González Ballesteros
OBJETIVO Conocer y comprender los fundamentos y metodología básicos de las técnicas
inmunocitoquímicas e inmunohistoquímicas, así como sus aplicaciones en la inmunología
básica, clínica y diagnóstica. RELACIÓN CON LOS CONTENIDOS TEORICOS DEL PROGRAMA Morfología y fisiología de los órganos y tejidos del sistema inmune e interacción antígeno
anticuerpo.
INTRODUCCIÓN Se conocen como técnicas inmunocitoquímicas e
inmunohistoquímicas a aquellas metodologías que
permiten la detección y localización de antígenos
determinados en preparaciones citológicas e histológicas,
esto mediante el uso de anticuerpos específicos y
sistemas de detección conjugados principalmente con
moléculas fluorescentes o enzimas, y que en mayor o
menor grado permiten la observación simultanea de las
características morfológicas celulares y tisulares
utilizando varios tipos de microscopía.
La inmunohistoquímica comienza a utilizarse en la
década de 1940 con técnicas de inmunofluorescencia en
cortes de tejidos congelados y es hasta la década de
1990 que adquiere mayor relevancia sobre todo por su
utilidad en patología quirúrgica para la clasificación y
diagnóstico de neoplasias, gracias a una serie de
Tabla 1. Historia de la inmunohistoquímica

57
avances técnicos que de manera acumulada permitieron mejorar diversos aspectos de la
metodología.La posibilidad de conjugar anticuerpos con enzimas como la peroxidasa de rábano
y su empleo en conjunto con diferentes sustratos cromogénicos, permitieron la localización de
los antígenos al poder observar las preparaciones en el microscopio oṕtico compuesto
convencional, en técnicas comúnmente conocidas como de inmunoperoxidasa. Otros de estos
avances técnicos fueron la posibilidad de detectar antígenos en muestras fijadas con formalina
y embebidas en parafina, así como la capacidad de incrementar la sensibilidad de las técnicas
empleando marcajes indirectos que emplean anticuerpos secundarios, sistemas de detección
con avidina o estreptavidina - biotina, y más recientemente uso de sistemas de amplificación
basados en polímeros con los que se logra una elevada sensibilidad. El desarrollo de la
tecnología para producir anticuerpos monoclonales impacto de manera similar a lo observado
con la mayoría de las técnicas inmunológicas: incrementando la especificidad y la
reproducibilidad de las técnicas en las cuales son empleados estos reactivos.
FUNDAMENTOS TECNICOS Origen, selección y preparación de la muestra. El origen de las células y tejidos pueden ser muestras de órganos obtenidas postmortem,
biopsias obtenidas en procedimientos quirúrgicos incluyendo la obtención en el transoperatorio
o en cirugías ambulatorias, improntas de tejidos o mucosas, aspirados de órganos
parenquimatosos, frotis de líquidos corporales, preparaciones obtenidas por citocentrifugación a
partir de líquidos con baja concentración celular o a partir de cultivos celulares. Las muestras
empleadas en técnicas de inmunohistoquímica o inmunocitoquímica en la mayoría de los casos
se encuentran fijadas mediante el uso de agentes químicos como formaldehído y etanol con el
fin de preservar la morfología celular y tisular, pero con el inconveniente de la probable pérdida
de las características antigénicas de las células de la muestra. A pesar de los problemas que
representa la perdida y necesaria recuperación de antígenos la formalina amortiguada con pH
neutro continúa representando la mejor opción para conservar las características morfológicas
del tejido y la retención de moléculas de bajo peso molecular que podrían perderse en los
procesos subsecuentes. En casos donde no se conozca el resultado sobre la antigenicidad
después de la fijación de la muestra, se podrían emplear preparaciones hechas a partir de
cortes de tejido congelado en las cuales la morfología puede alterarse en diferentes grados pero
garantizando el reconocimiento de los antígenos de interés con los anticuerpos disponibles. En
la actualidad muchos de los reactivos que se encuentran disponibles en el mercado son
obtenidos con metodologías que garantizan la utilidad de los mismos en muestras fijadas y
embebidas en parafina.

58
Con el fin de lograr un resultado óptimo en
las técnicas inmunocitoquímicas e
inmunohistoquímicas es necesario realizar
un proceso de recuperación de antígenos
con el fin de que los anticuerpos puedan
reconocer los determinantes antigénicos
conformacionales que resulten alterados
por la acción del agente fijador, ya sea
que actúe coagulando y precipitando
(etanol y metanol) o formando ligamientos
cruzados (formaldehído y glutaraldehído)
en las proteínas celulares.
Tabla 2. Esquema de estandarización para recuperación de
antígenos
Existen múltiples métodos para la recuperación de antígenos que principalmente se basan en
aplicación de calor, pH o acción de enzimas proteolíticas para lograr revertir el efecto del agente
fijador particularmente el ligamiento cruzado provocado por el formaldehído.Las condiciones
particulares de estos dos factores para cada caso dependerán de las características de la
muestra, el método de fijación empleado, objetivo de la tinción y antígeno en estudio. Se
colocan las preparaciones en agua destilada o diversas soluciones con pH ácido o básico y se
calientan temperaturas alrededor a los 100 ºC dado que las proteínas fijadas resisten más altas
temperaturas que las proteínas nativas e incluso se puede emplear un horno de microondas
convencional para calentar la muestra, aunque se debe estandarizar la metodología en cada
laboratorio.
Proceso de tinción y reactivos utilizados. Dependiendo de las células o tejidos utilizados se deberán realizar uno o varios pasos de
bloqueo con los cuales se busca reducir al máximo el fondo por uniones inespecíficas en la
tinción. En algunos casos se buscará bloquear sitios de unión inespecíficos empleando
soluciones con proteínas ajenas al sistema, esto es no relacionadas con el antígeno o con los
reactivos de detección, por ejemplo soluciones de leche descremada o suero normal de la
especie en las cual se generó el anticuerpo secundario.
En otros casos se deberá bloquear la actividad endógena de enzimas en el tejido como es el
caso de peroxidasas o la presencia de biotina que podría interferir con el paso de detección en
caso de utilizar el sistema avidina o estreptavidina con biotina.

59
Después de cada uno de los pasos que requieran
incubación con alguno de los reactivos se deberán
realizar lavados adecuados comúnmente con soluciones
isotónicas balanceadas y neutras como alguna de las
diferentes formulaciones de PBS. Una vez que se tiene
la muestra bloqueada comúnmente se utiliza un
anticuerpo primario monoclonal o policlonal específico
para los antígenos que se busca detectar y dependiendo
de la disponibilidad de reactivos o si de las
características y objetivo de la tinción, se busque
incrementar la sensibilidad o la especificidad de la
técnica. A pesar de que el emplear técnicas indirectas
puede incrementar el fondo de la tinción, en muchos
casos es necesario incrementar la sensibilidad para
conseguir la detección del antígeno en cuestión.
También cabe mencionar que muchas de las técnicas
tienen como objetivo la detección de anticuerpos
específicos en el suero de los individuos, como sucede
en fenómenos de autoinmunidad. En cualquiera de los
casos se deben estandarizar los periodos y temperatura
de incubación con cada uno de los reactivos, así como
realizar la titulación de los mismos con el fin de
determinar la concentración óptima obtener en la tinción
la máxima señal específica y el menor fondo
inespecífico. En todos los casos en la incubación se
debe evitar que la muestra se deshidrate mediante el
empleo de cámaras húmedas, ya que si la muestra se
seca en cualquiera de los pasos de incubación se
incrementará el pegado inespecífico y con ello el fondo
de la tinción.
Figura 1. Sistemas de detección en
inmunohistoquímica

60
La incubación con los reactivos para detección sigue los mismos principios que los
mencionados para los anticuerpos primarios, con la diferencia que en la mayoría o
prácticamente todos los casos los anticuerpos empleados son de tipo policlonal. También
pueden emplearse avidina o estreptavidina como sistema de detección cuando el anticuerpo
primario se encuentra conjugado con biotina. Existen sistemas de detección que utilizan
moléculas con las cuáles se permite que exista un mayor número de anticuerpos conjugados
que detecten los antígenos, como ejemplo se tiene a moléculas de dextrana con múltiples
anticuerpos unidos a ella y permiten la amplificación de la señal, sobretodo cuando los
antígenos a detectar se encuentran en baja cantidad en la muestra. En todos los casos estos
reactivos secundarios estarán conjugados con moléculas que permitan la detección del
antígeno en la muestra directamente o mediante la adición posterior de una solución reveladora
que contendrá por ejemplo el sustrato y una sustancia cromogénica en caso de que el sistema
de detección dependa de la acción de una o varias enzimas en el caso de la tinción múltiple. Es
importante en este paso que la totalidad de la muestra este inmersa o cubierta con la dilución
del conjugado.
Después de lavar adecuadamente las preparaciones para eliminar al máximo el exceso de
conjugado si la técnica es inmunoenzimática se procede a agregar la solución reveladora que
contiene el sustrato y el cromógeno específicos para la enzima del sistema de detección.
Existen varias sustancias cromogénicas utilizadas en las tinciones inmunohistoquímicas que
pueden ser utilizadas con las enzimas más comúnmente utilizadas como peroxidasa de rábano,
fosfatasa alcalina y glucosa oxidasa, y que tienen diferentes características de color y
solubilidad, lo que permite implementar técnicas de tinción doble con la selección adecuada de
reactivos para detectar dos antígenos simultáneamente en la misma preparación. Es importante
la estandarización de los tiempos necesarios para el desarrollo de la coloración óptima de la
tinción, frecuentemente esto se realiza directamente con la observación de las laminillas en el
microscopio. También se pueden realizar tinciones dobles empleando conjugados fluorescentes
en las cuales se detectan los antígenos diferentes empleando fluorocromos que se exciten con
luz de la misma o diferente longitud de onda y que emitan luz con diferente longitud de onda,
para realizar el análisis de microscopia de fluorescencia e incluso por microscopia confocal para
determinar colocalización.
La preparación finalmente debe ser contrateñida en las técnicas que permitan la observación
simultanea de los detalles morfológicos de las células o tejidos estudiados, como sucede en

61
técnicas enzimáticas, en las cuales comúnmente se utiliza hematoxilina para evidenciar los
núcleos de las células y en este caso también debe estandarizarse el tiempo óptimo para la
contratinción o tinción de contraste. En esta caso se debe considerar el solvente en el cual se
encuentra preparado el colorante para no eliminar de la muestra el cromógeno en caso de que
el producto colorido sea soluble en dicho solvente, por ello en muchos casos se prefiere evitar
tinciones con hematoxilina en alcohol. Otra consideración en este tipo de técnicas es el interés
en poder conservar las preparaciones mediante montaje permanente, para lo cual también se
debe tomar en cuenta las características de solubilidad en alcohol del producto colorido. Como
en toda prueba o técnica para diagnóstico o investigación es importante el contemplar controles
positivos, negativos y otros que sean necesarios para dar soporte a los resultados obtenidos.
Figura 2. Tinción inmunohistoquímica doble. Se
muestra la tinción secuencial con peroxidasa de rábano y fosfatasa alcalina de un corte de linfonodo para la
detección de cadenas ligeras y .
Tabla 3: Cromógenos utilizados en inmunohistoquímica.

62
USOS DE LAS TÉCNICAS INMUNOCITOQUÍMICAS E INMUNOHISTOQUÍMICAS Las técnicas de inmunocitoquímica e inmunohistoquímica tienen diversas aplicaciones en
diferentes campos de las ciencias biomédicas sin limitarse a uno de ellos en particular tanto en
estudios in vitro como in vivo. En inmunología tiene aplicaciones en inmunología básica
utilizándose en una gran variedad de estudios morfológicos y fisiológicos de células, tejidos y
órganos del sistema inmune; inducción y activación de la respuesta inmune, ontogenia y
filogenia del sistema inmune o en estudios de inmunología comparada; en inmunología clínica
en estudios de inmunopatología como en hipersensibilidades, autoinmunidad e
inmunodeficiencias; en inmunología diagnóstica para el diagnóstico de enfermedades
infecciosas virales, bacterianas, micóticas y parasitarias. En biología celular y molecular para
estudios de morfología y fisiología celular como aquellos que involucran el citoesqueleto,
marcadores celulares, ciclo celular, etc. Una de las áreas con mayor desarrollo en el campo de
la inmunocitoquímica e inmunohistoquímica es la patología, en particular en estudios
oncológicos que permiten el diagnóstico, clasificación y determinación del grado de malignidad
de neoplasias a partir de biopsias o aspirados de tejido neoplásico o de linfonodos regionales
con el fin de determinar metástasis a partir del tumor primario, detección de las alteraciones a
nivel genético asociadas con diferentes neoplasias para lo cual pueden complementarse con
técnicas moleculares como hibridación in situ, detección de marcadores de linaje o
diferenciación celular.
DESPARAFINIZADO Y TINCIÓN DE DE MUESTRAS EMBEBIDAS EN PARAFINA POR INMUNOHISTOQUÍMICA (Técnica empleada en CINVESTAV) Todas las incubaciones se hacen en cámara húmeda y debe evitarse que la muestra se seque
una vez desparafinizada.
1. La laminilla deberá haber estado a 40 °C toda la noche en horno para adherir bien el tejido
al vidrio. 2. La laminilla se sumerge en xilol por 15’ y se repite la operación en otro baño de xilol. 3. Se procede a rehidratar la muestra 2 veces en alcohol absoluto. 4. Se bloquea la peroxidasa endógena en metanol absoluto adicionado con 2 % de peróxido
de hidrógeno, por 45’. 5. El tejido se lava con PBS tres veces (3’ c/u) y se pone suero normal de cabra (frasco 1) por
45’ en cámara húmeda. 6. Se decanta el suero y se pone el anticuerpo monoclonal (MAb) a la concentración óptima.
Se incuba 2 hrs en cámara húmeda. 7. Se lava con PBS, tres veces y se incuba con el anticuerpo secundario (cabra anti-ratón
biotinilado, frasco 2), 45’ en cámara húmeda.

63
8. Se vuelve a lavar con PBS, tres veces, y se incuba con estreptavidina-peroxidasa (frasco 3) por 45’ en cámara húmeda.
9. Se lava 3 veces con PBS y se añade el substrato (DAB), vigilando la reacción bajo microscopio y deteniéndola sumergiendo la laminilla en PBS o agua.
10. Se contratiñe con hematoxilina de Harris por unos segundos, se azulea con agua de la llave y, de ser necesario, se destiñe con alcohol ácido.
11. La laminilla se deshidrata con alcoholes seriados (70-100 %) y en xilol dos veces. 12. Se monta en resina sintética y se deja secar 12 hrs. Con esta técnica las células que fueron reconocidas por el anticuerpo, y que posteriormente son reveladas con DAB se tiñen de color café y la hematoxilina nos sirve como color de contraste. METODOLOGÍA Se observaran al microscopio diferentes cortes histológicos de órganos y/o tejidos teñidos como se describió en la técnica. BIBLIOGRAFÍA 1. Dabbs DJ. (2010). Diagnostic Immunohistochemistry.. Theranostics and Genomic Applications, 3rd
edition. Saunders Elsevier, USA. 2. Hofman FM & Taylo CR. (2013). Immunohistochemistry. En Current Protocols in Immunology 21.4.21-
21.4.26, DOI: 10.1002/0471142735.im2104s103, John Wiley & Sons, Inc. USA 3. Taylor, CR. & Rudbeck L. (editors). (2013). Immunochemical Staining Methods Handbook, 6rd edition.
Dako, Agilent Technologies. USA.

64
PRÁCTICA 11
ELECTROFORESIS
Dr. Marco Antonio Vega López y QFB Ladislao Palomar Morales
OBJETIVO Realizar la separación de una mezcla de proteínas sometiéndolas a un campo eléctrico
utilizando como soporte un gel de poliacrilamida.
RELACIÓN CON LOS CONTENIDOS TEORICOS DEL PROGRAMA En la Unidad tres se estudian los Mecanismos Específicos de Defensa, abordándose en el
punto 3.3 y 3.10 los conceptos de Antígeno e inmunoglobulinas, respectivamente, ambos de
naturaleza proteínica principalmente. Planteándose además como contenido práctico la
separación y purificación de anticuerpos.
INTRODUCCIÓN La electroforesis puede definirse como un tamizado molecular a través de un gel de corrimiento.
En algunos geles, como los de agarosa, los poros no son homogéneos y son suficientemente
grandes como para no impedir la migración de la mayoría de las moléculas proteínicas, por lo
que aquí la separación dependerá fundamentalmente de su carga neta. En los geles de
poliacrilamida, modificando las condiciones de la polimerización, pueden producirse poros de
diferentes diámetros, por lo que, para un gel de determinado poro, el tamaño molecular y la
carga neta serán los factores determinantes de la separación de las moléculas de una mezcla.
Los geles de poliacrilamida son el resultado de la polimerización, de la acrilamida
2 2(CH CH CO NH ) y de su entrecruzamiento con la N-N-metilen bisacrilamida
2 2 2(CH CH CO NH CH NH CO CH CH ) . El poro del gel formado dependerá de las
concentraciones relativas de ambos reactivos durante la polimerización. La polimerización de la
acrilamida necesita de un iniciador del proceso. Los más comúnmente usados son el persulfato

65
de amonio y la riboflavina. Se añade, además, como catalizador N,N,N',N'-tetrametil
etilendiamina (TEMED). El persulfato de amonio-TEMED cataliza la formación de radicales
libres, lo que inicia la polimerización. La base libre TEMED retarda la polimerización.
La electroforesis en geles de poliacrilamida se realiza en condiciones fijas de pH, conteniendo
dodecilsulfato de sodio (SDS),para eliminar la carga intrínseca del péptido ante el exceso de
carga negativa del SDS; como consecuencia, la carga de todas las moléculas será
prácticamente idéntica, por lo que su desplazamiento en un campo eléctrico, en un gel de
determinada porosidad, dependerá exclusivamente de su tamaño molecular. Por lo que
comparando su movilidad con sustancias de peso molecular conocido, que se corren
simultáneamente, se determina el peso molecular relativo de las proteínas a analizar.
La electroforesis en gel de poliacrilamida se desarrolla usando sistema amortiguadores
continuos o discontinuos. En el primer caso, los iones que constituyen la solución
amortiguadora durante todo el recorrido de la muestra (gel separador) son los mismos y el pH
es constante. En algunas situaciones sólo puede variar la concentración iónica en los vasos de
contención o tanques. Cuando se usan estos sistemas, las muestras a analizar se colocan
directamente en el gel separador. En los sistemas discontinuos, la solución amortiguadora de
los geles y el de los tanques de los electrodos son diferentes. Normalmente el pH de ambas
soluciones amortiguadoras es diferente. Las muestras a analizar se colocan en un gel de poro
grueso (gel concentrador), que se ha hecho polimerizar sobre el gel separador (pH 8.8). La
solución amortiguadora del recipiente que contiene el cátodo es Tris-glicina con pH 8.3.
Si la muestra ha sido reducida con 2-mercaptoetanol (u otro reductor como el ditiotreitol), los
enlaces disulfuro intra e intercatenarios se disocian, la estructura cuaternaria se pierde y las
subunidades se separan como cadenas polipeptídicas individuales.
TINCIÓN CON AZUL DE COOMASSIE Una vez terminado el corrimiento electroforético, el gel se coloca en azul de Coomassie, para
teñir las proteínas corridas. Este colorante permite detectar de cantidades hasta a 0.1 g de
cualquier proteína en una banda fina. Dado que el colorante es preparado con ácido acético, la
fijación de las proteínas y su coloración ocurren al mismo tiempo.

66
MATERIAL Cámara de electroforesis con accesorios Fuente de poder 2 vasos de precipitados de 50 mL 2 vasos de precipitados de 200 mL 1 vaso de precipitados de 500 mL 1 matraz Erlenmeyer 1 pipeta Pasteur y propipeta 1 triángulo de porcelana 1 tripie Tela de asbesto Mechero Tubos Eppendorf Micropipetas y puntas para micropipetas Gradilla de unicel para colocar muestras Baño María Recipiente hermético Agitador Embudo y triángulo de porcelana Transluminador Regla
REACTIVOS Solución de monómeros TEMED SDS al 10% Persulfato de amonio al 10% Amortiguador TRIS-HCl pH = 6.8 y pH = 8.8. Agar noble al 1% Agua destilada Solución reguladora de corrimiento Solución reguladora de muestra Marcadores de peso molecular Muestras a separar Solución de azul de Coomassie Soluciones decolorantes I y II Carbón activado
*Opcional 2-mercaptoetanol, urea 6 M o enzimas
DIAGRAMA DE FLUJO
R1:El polímero de acrilamida aunque no es un residuo peligroso, se debe incinerar R2:La solución de corrimiento del tanque inferior puede reutilizarse dos o tres veces; la del tanque
superior deberá neutralizarse antes de ser desechada al drenaje
Preparación de las
muestras
Preparación de la cámara
Colocar las muestras en la cámara
Correr la electroforesis
Teñir el gel Determinar el Rf de las diferentes proteínas y
calcular sus pesos moleculares
R2
R3
R1

67
R3: Los decolorantes I y II deben filtrarse con carbón activado para su recuperación y posterior reutilización; En caso de desecharse deben guardarse en contenedores destinados para ello. El papel filtro debe ser incinerado.
METODOLOGÍA 5. Ensamblar los cristales de la cámara de electroforesis, sellar tres lados con agar y dejar
secar.
6. Preparar el gel separador, a la concentración deseada (ver tabla en el anexo), el persulfato
de amonio y el TEMED, se agregan a la mezcla justo antes de verterla en la cámara; verter
el gel hasta una altura de aproximadamente 75% de la cámara, cubrir con 150 µl de
isopropanol, dejar gelificar y desechar el isopropanol.
7. Preparación del gel concentrador, en forma semejante al gel separador, y verter en la
cámara; colocar el peine para formar los carriles.
8. En tubos Eppendorf colocar de 20 a 30 µg de la muestra a separar y agregar el mismo
volumen de la solución reguladora de muestra; hervir 3 minutos en baño María.
9. Llenar los tanques superior e inferior con la solución de corrimiento.
10. Colocar las muestras y marcadores de peso molecular, en los carriles, con micropipeta.
11. Conectar los cables de la corriente eléctrica en los polos correspondientes, prender la
fuente de poder a 100 volts, dejar correr hasta la separación total de los antígenos.
12. Una vez terminada la electroforesis desmontar la cámara y retirar el gel; teñir el gel con azul
de Coomassie.
13. Desteñir el gel con el decolorante I (ácido acético, metanol y agua), se deben observar
bandas de antígeno reveladas y finalmente colocar en decolorante II.
14. Colocar en el transluminador y medir la distancia recorrida por el frente del disolvente y
cada una de las muestras, para calcular el Rf de cada proteína y de los marcadores de
peso molecular.
distancia recorrida por la muestradistancia del frente de corrimiento
Rf
68
Ejemplo de bandas de Proteínas en una electroforesis
RESULTADOS Grafica de Rf vs. Log del peso molecular de los marcadores de peso molecular.
Tabla de Rf y peso molecular de las proteínas de las muestras.
PRECAUCIONES
1. La cámara de electroforesis debe quedar bien ensamblada y bien sellada con el agar.
2. Manejar con guantes la acrilamida y bisacrilamida, ya que son carcinógenos. 3. Preparar correctamente los geles, ya que pueden fragmentarse, no gelificar o no
separar adecuadamente las proteínas. 4. Cuidar el proceso de decoloración, para evitar decolorar totalmente las proteínas
DISCUSIÓN Discutir la importancia de la electroforesis para la separación, purificación e identificación de
antígenos y anticuerpos.
BIBLIOGRAFÍA 1. Cultek. (2006). Soluciones Western Blot. [En línea]
http://www.cultek.com/inf/otros/soluciones/CULTEK-TecnicaWB.pdf, obtenida el 31 de junio de 2011. 2. Laemmli, V. K. (1970). Cleavage of Structural Proteins During the Assembly of the Head
Bacteriophage t. Nature. 227: 680-685. 3. Margini, Ricardo Anibal. (1996). Inmunología e Inmunoquímica. 5ª edición. Editorial Panamericana.
Buenos Aires, Argentina. p. 976. 4. Towbin. H. Stahehelin, T, Gordon. (1979). Electrophoretic Transfer of Proteins from Polyacrylamide
Gels to Nitrocellulose-Sheets; procedure and some applications, proc, Nat, Acad. Sci, USA, 76 (9): 4350-4354.

69
APÉNDICE: PREPARACIÓN DE REACTIVOS Tris/HCl 1.5 M pH 8.8
Pesar 18.15 g de Tris Disolver el Tris en 50 mL de agua destilada Añadir gota a gota HCl concentrado para ajustar el pH a 8.8 Completar con agua destilada hasta el volumen final (100 mL)
Tris/HCl 1 M pH 6.8
Pesar 12.1 g de Tris Disolver el Tris en 50 mL de agua destilada Añadir gota a gota HCl concentrado para ajustar el pH a 6.8 Completar con agua destilada hasta el volumen final (100 mL)
SDS 10 % (p/v)
Pesar 10 g de SDS Añadir agua destilada hasta completar el volumen de 100 mL
Glicerol 50 % (v/v)
Tomar un volumen de 50 mL de glicerol al 100 % Añadir 50 mL de agua destilada
Azul de bromofenol 1 % (p/v)
Pesar 100 mg de azul de bromofenol Completar con agua destilada hasta 10 mL y mezclar hasta disolver Filtrar la solución en papel Whatman No 1 para eliminar el colorante no disuelto
Acrilamida 30%
La solución de acrilamida al 30 % (p/v) es una mezcla de acrilamida (29.2 g) y bisacrilamida (0.8 g). Pesar ambos reactivos y añadir agua destilada hasta 100 mL Filtrar en papel de filtro Whatman No1
Persulfato de amonio al 10 % (p/v)
Pesar 0.5 g de sulfato de amonio Disolver en 5 mL de agua destilada
Solución Amortiguadora de Corrimiento
Pesar 3 g de Tris (25 mM) Pesar 14.4 g de glicina (192 mM) Pesar 1 g de SDS (0.1 %) Añadir agua destilada hasta completar 1 L NOTA: El pH debería ser de aproximadamente 8.3. Se puede preparar una solución 10 veces más concentrada y diluir el volumen necesario en el momento del corrimiento. Esta solución puede almacenarse indefinidamente a temperatura ambiente.
Solución Reguladora de muestra 2X
0.24 mL de Tris/HCl 1M pH 6.8 2 mL de Glicerol 50 % 0.8 mL de SDS 10 % 0.5 mL de 2-mercaptoetanol* Opcional 0.5 mL de azul de bromofenol 0.96 mL de agua destilada

70
CANTIDADES PARA LA PREPARACIÓN DE GELES POLIACRILAMIDA.
Gel Separador Cantidad a preparar 6% 5 mL 10mL 15 mL 20 mL 25 mL 30 mL 40 mL 50 mL
H2O 2.7 5.3 8.0 10.6 13.3 15.9 21.1 26.5 30 % Acrilamida Mix 1.0 2.0 3.0 4.0 5.0 6.0 8.0 10.0 1.5 M Tris (pH 8.8) 1.3 2.5 3.8 5.0 6.3 7.5 10.0 12.5 10 % SDS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 10 % APS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 TEMED 0.004 0.008 0.012 0.016 0.02 0.024 0.032 0.04
8% 5 mL 10mL 15 mL 20 mL 25 mL 30 mL 40 mL 50 mL H2O 2.3 4.6 7.0 9.3 11.6 13.9 18.6 23.2 30 % Acrilamida Mix 1.3 2.7 4.0 5.3 6.7 8.0 10.7 13.4 1.5 M Tris (pH 8.8) 1.3 2.5 3.8 5.0 6.3 7.5 10.0 12.5 10 % SDS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 10 % APS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 TEMED 0.003 0.006 0.009 0.012 0.015 0.018 0.024 0.03
10% 5 mL 10mL 15 mL 20 mL 25 mL 30 mL 40 mL 50 mL H2O 2.0 4.0 5.9 7.9 9.9 11.9 15.8 20.0 30 % Acrilamida Mix 1.7 3.3 5.0 6.7 8.3 10.0 13.3 16.6 1.5 M Tris (pH 8.8) 1.3 2.5 3.8 5.0 6.3 7.5 10.0 12.5 10 % SDS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 10 % APS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 TEMED 0.002 0.004 0.006 0.008 0.01 0.012 0.016 0.02
12% 5 mL 10mL 15 mL 20 mL 25 mL 30 mL 40 mL 50 mL H2O 1.7 3.3 5.0 6.6 8.3 9.9 13.2 16.4 30 % Acrilamida Mix 2.0 4.0 6.0 8.0 10.0 12.0 14.0 20.0 1.5 M Tris (pH 8.8) 1.3 2.5 3.8 5.0 6.3 7.5 10.0 12.5 10 % SDS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 10 % APS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 TEMED 0.002 0.004 0.006 0.008 0.01 0.012 0.016 0.02
15% 5 mL 10mL 15 mL 20 mL 25 mL 30 mL 40 mL 50 mL H2O 1.2 2.3 3.5 4.6 5.7 6.9 9.2 11.4 30 % Acrilamida Mix 2.5 5.0 7.5 10.0 12.5 15.0 20.0 25.0 1.5 M Tris (pH 8.8) 1.3 2.5 3.8 5.0 6.3 7.5 10.0 12.5 10 % SDS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 10 % APS 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5 TEMED 0.002 0.004 0.006 0.008 0.01 0.012 0.016 0.02 Gel Concentrador 1 mL 2 mL 3 mL 4 mL 5 mL 6 mL 8 mL 10 mL H2O 0.68 1.4 2.1 2.7 3.4 4.1 5.5 6.8 30 % Acrilamida Mix 0.17 0.33 0.5 0.67 0.83 1.0 1.3 1.7 1.0 M Tris (pH 6.8) 0.13 0.25 0.38 0.5 0.63 0.75 1.0 1.25 10 % SDS 0.01 0.02 0.03 0.04 0.05 0.06 0.08 0.1 10 % APS 0.01 0.02 0.03 0.04 0.05 0.06 0.08 0.1 TEMED 0.001 0.002 0.003 0.004 0.005 0.006 0.008 0.01

71
PRÁCTICA 12
INMUNOELECTROTRANSFERENCIA
QFB Ladislao Palomar Morales
OBJETIVO Realizar la transferencia de proteínas separadas electroforéticamente de un gel de
poliacrilamida hacia una membrana, de tal manera que se conserve el perfil electroforético.
Realizar el reconocimiento de proteínas, separadas por electroforesis y transferidas a un
soporte sólido (por ejemplo: papel de nitrocelulosa), por medio de anticuerpos y su posterior
revelado con el uso de conjugados, sustratos y cromógenos.
RELACIÓN CON EL CONTENIDO DEL PROGRAMA TEÓRICO En la Unidad 3 se estudian los Mecanismos Específicos de Defensa (MED); en los puntos 3.3
antígenos y antigenicidad, 3.10 Estructura y función de inmunoglobulinas y 3.11 reacción
antígeno cognato, planteándose además como práctica de laboratorio la conjugación y
manipulación de inmunoglobulinas.
INTRODUCCIÓN La inmunoelectrotransferencia (o Western blot) se realiza en tres etapas:
1. Electroforesis 2. Transferencia 3. Revelado
La primera de ellas, la electroforesis, es semejante a la realizada la clase pasada. La
electroforesis difiere en que sólo se forman dos carriles en el gel; uno pequeño para los
marcadores de peso molecular y otro grande para la muestra.
La segunda de ellas, la transferencia de proteínas o botín supone la inmovilización de las
proteínas sobre membranas sintéticas, seguido de la detección empleando sistemas
especialmente diseñados para la tinción de blogs.

72
El método para proteínas más potente es el denominado Western blot en el que las proteínas
son separadas en primer lugar mediante electroforesis en geles de poliacrilamida en
condiciones desnaturalizantes Y posteriormente se transfieren a una membrana, mediante la
aplicación de un campo eléctrico perpendicular al gel (electroblotting). Una vez completada esta
operación las proteínas de interés son reveladas por el agregado de un anticuerpo específico.
El nombre Western (oeste, occidental) le fue dado por el investigador W. Neal Brúñete
(Analytical Biochemistry, 112:195-203, 1981) como un juego de palabras por los nombres
Southern y Northern de las técnicas que se usan con el mismo fin pero que son aplicadas a los
ácidos nucleicos. Luego que Edwin Southern diseñara la técnica que lleva su nombre para
detectar ADN, por oposición se denominó Northern a la técnica aplicada al ARN. Finalmente
como una broma, la técnica de inmunoblotting (inmunotransferencia) para proteínas fue
denominada Western
El electroblotting es el método donde las proteínas son transferidas desde el gel a la membrana
electroforéticamente. El procedimiento se inicia apilando sucesivamente sobre una esponja
plana papel de filtro empapado en buffer de transferencia, el gel, la membrana en contacto
directo con el gel, más papel de filtro y finalmente una esponja plana. Este conjunto se recoge
entre dos capas de plástico perforado (cartucho) y se introduce en una cámara en la que se
encuentra una solución amortiguadora de transferencia y dos electrodos planos (diseñados para
conseguir un campo uniforme en toda la superficie del gel). Se dispone de forma que el gel
quede hacia el ánodo (-) y la membrana hacia el cátodo (+) (figura 1). La carga neta de las
proteínas en el caso de los geles de SDS-PAGE es negativa debida a la carga del SDS.
Figura 1. Armado del cartucho de transferencia

73
La electroforesis se realiza a 8-10 V/cm (de separación entre electrodos), que suele
corresponder a 0.3 a 2 Ampere, de 1 a 4 horas. La velocidad de transferencia es inversamente
proporcional al tamaño de la proteína. El proceso provoca un fuerte calentamiento de la
solución por lo que se recomienda refrigerar el sistema y recircular el amortiguador.
Una vez realizada la transferencia la membrana se puede analizar inmediatamente o bien
conservarla en frío (2 a 8 ºC) durante meses.
La tercera etapa consta de tres reacciones sobre la membrana, las primeras dos de ellas son
reacciones inmunológicas, cuya velocidad puede ser modificada por factores como: La
concentración de los reactivos, la temperatura, el pH, etc.
En la primera reacción el antígeno pegado al papel de nitrocelulosa interacciona con su
anticuerpo específico:
Ag + Ab AgAb
Una segunda reacción ocurre entre este complejo y un anticuerpo ligado a una molécula para
hacer visible la reacción:
AgAb + AbM AgAbAbM
Y finalmente esta molécula reacciona para hacer visible la reacción.
Esta molécula puede ser una enzima, un flourocromo, o un isótopo radiactivo: En el caso de la enzima se agrega el sustrato y un cromógeno; En el caso de un flourocromo se hace incidir luz de determinada longitud de onda.
Figura 2. Esquema de las reacciones en el papel de nitrocelulosa

74
Parte importante de esta técnica es la eliminación de las moléculas que no han reaccionado,
formando enlaces permanentes (covalentes), mediante el uso de tensoactivos suaves como el
tween 20.
DIAGRAMA DE FLUJO
R1: Residuos de electroforesis, ver práctica anterior. R2: Amortiguador de transferencia, puede ser reutilizado dos o tres veces, el residuo final se coloca en un
contenedor adecuado. R3: La solución de rojo de Ponceau, puede ser reutilizada varias veces. R3a: El agua de los lavados del rojo de Ponceau, se puede tirar al drenaje. R4, R5, R6, R7 y R8: Se colocan en la palangana con cloro, antes de desecharlos al drenaje.
Cortar el papel de nitrocelulosa en tiras de3 a 5 mm
Colocar el número de tiras necesarias en los carriles
Agregar el suero problema e incubar. Lavar
Agregar el conjugado e incubar
Agregar el sustrato y cromógeno Detener la reacción
Diluir el suero de ratón con solución
de bloqueo
Diluir el conjugado con solución de
bloqueo
Preparar el sustrato/cromógeno
Electroforesis de IgG Humana
Hidratar membrana de nitrocelulosa, papel filtro y
esponjas en amortiguador de transferencia
Formar el “cartucho” para transferir
Transferir
Revisar la transferencia con rojo de Ponceau
Eliminar el colorante con agua destilada
Bloquear la membrana con Leche descremada
R1 R2
R3
R5
R6
R7
R8
R4
R3

75
MATERIAL Cámara de electroforesis con accesorios Cámara de transferencia con accesorios Micropipetas y puntas para micropipetas Papel de nitrocelulosa Fuente de poder 2 vasos de precipitados de 50 mL 2 vasos de precipitados de 200 mL 1 vaso de precipitados de 500 mL 1 matraz Erlenmeyer 1 pipeta Pasteur y propipeta 1 triángulo de porcelana 1 tripie Tela de asbesto Mechero Tubos Eppendorf Gradilla de unicel para colocar muestras Baño María Pipeta. Pinzas y Bisturí. Regla Placa de pozos para tiras de papel de nitrocelulosa Agitador
REACTIVOS Solución de monómeros TEMED SDS al 10% Persulfato de amonio al 10% Amortiguador TRIS-HCl pH = 6.8 y pH = 8.8. Muestras a separar (IgG humana purificada) Marcadores de peso molecular Agar noble al 1 % Agua destilada Solución reguladora de corrimiento Solución reguladora de muestra Amortiguador de transferencia Tiras de papel de nitrocelulosa Rojo de Ponceau Solución bloqueadora Sueros problema Conjugado (anticuerpo-enzima) Solución de lavado: PBS-tween 20 Sustrato para la enzima y cromógeno
*Opcional 2-mercaptoetanol, urea 6 M o enzimas
METODOLOGÍA 1. Una vez terminada la electroforesis, desmontar el gel y colocar en solución reguladora de
transferencia
2. Hidratar la membrana de nitrocelulosa durante 10 segundos en metanol, aclarar en agua
destilada y poner en la solución reguladora de transferencia.
3. Hidratar en la solución reguladora de transferencia las esponjas y el papel filtro.
4. Llenar con la solución amortiguadora de transferencia la cámara de transferencia hasta el
85 %.
5. Colocar en la posición adecuada en función de los electrodos, una esponja, un papel de
filtro y el gel PAGE-SDS.
6. Sobre el gel perfectamente plano, colocar cuidadosamente la nitrocelulosa, y alisar varias
veces MUY CUIDADOSAMENTE con un tubo de ensayo para eliminar las burbujas de aire.
7. Cubrir de nuevo con papel de filtro y esponja. Colocar el cartucho en la cámara de
transferencia; Transferir a 125 mA, por 1 hora.

76
8. Teñir el papel de nitrocelulosa con rojo de Ponceau, para comprobar si se realizó la
transferencia.
9. Decolorar con agua destilada, incubar con la solución bloqueadora durante 2 Hr a
temperatura ambiente o toda la noche a 4 °C.
10. Cortar el papel de nitrocelulosa en tiras de 3 a 5 mm de ancho.
11. Colocar las tiras de papel de nitrocelulosa en los carriles de la placa.
12. Incubar las tiras de papel, con 1.5 mL de los sueros problema diluidos 1/100 con solución
bloqueadora, e incubar 20 minutos a temperatura ambiente; decantar el sobrenadante.
13. Lavar 5 veces, con la solución de lavado, por 1 minuto cada vez.
14. Incubar las tiras de papel, con 1.5 mL del conjugado diluido 1/10 000 en solución
bloqueadora, por 20 minutos a temperatura ambiente; decantar el sobrenadante.
15. Lavar 5 veces con la solución de lavado, por 1 minuto cada vez.
16. Incubar las tiras de papel, con 1.5 mL de la mezcla sustrato/cromógeno hasta la aparición
de las bandas.
17. Agregar 0.5 mL de la solución de paro; decantar el sobrenadante.
18. Secar las tiras al aire.
RESULTADOS Indicar el PM de las proteínas del extracto antigénico que son reconocidas por cada uno de los
sueros problemas.
BIBLIOGRAFÍA 1. CulteK. (2006). Soluciones Western Blot. [En línea]
http://www.cultek.com/inf/otros/soluciones/CULTEK-TecnicaWB.pdf, obtenida el 31 de junio de 2011. 2. Laemmli, V. K. (1970). Cleavage of Structural Proteins During the Assembly of the Head
Bacteriophage Nature . 227: 680-685 pp. 3. Margini, Ricardo (Anibal). 1996 Inmunología e Inmunoquímica. 5ª edición. Editorial Panamericana.
Buenos Aires, Argentina. 976 pp. 4. Towbin. H. Stahehelin, T, Gordon. (1979). Electrophoretic Transfer of Proteins from Polyacrylamide
Gels to Nirtocellulose-Sheets; procedure and some applications, proc Nat Acad Sci, USA, 76 (9): 4350-4354 pp.

77
ANEXO PREPARACIÓN DE REACTIVOS Reactivos para la electroforesis Ver práctica anterior Amortiguador de transferencia (según Laemmli) Tris -HCL 50 mM, glicina 0.196 M, pH 8,3 con 20 % de metanol añadido Solución de rojo de Ponceau Rojo Ponceau S al 0.5 % y ácido tricloroacético al 5 % en agua destilada. Solución Bloqueadora Debe ser una proteína inerte al sistema inmune, las más usadas son albúmina, caseína o gelatina. Albúmina bovina al 1 % (caseína al 1% o gelatina al 1 %) en PBS pH 7.2; no utilizar azida de sodio (inhibe la peroxidasa). Para algunos procedimientos se puede agregar Tween 20 al 0.05 %. Sueros y anticuerpos Todos los sueros y anticuerpos deben ser diluidos en solución bloqueadora. Solución de lavado PBS pH 7.2 con Tween-20 al 0.05%. Solución de paro Ácido sulfúrico 2N. Solución de sustrato cromógeno (para peroxidasa) Para 60 mL, disolver 30 mg de 4-Cloro-1-naftol en 10 mL de metanol (proteger de la luz). Agregar 50 mL del amortiguador Tris pH 7.5 y 25 μL de peróxido de hidrógeno concentrado (35 %).


















