Linfomas curso facultad
60
Servicio de Hematología Dra. Susana Cerana
-
Upload
cursobianualmi -
Category
Documents
-
view
3.282 -
download
0
Transcript of Linfomas curso facultad

Servicio de Hematología Dra. Susana Cerana
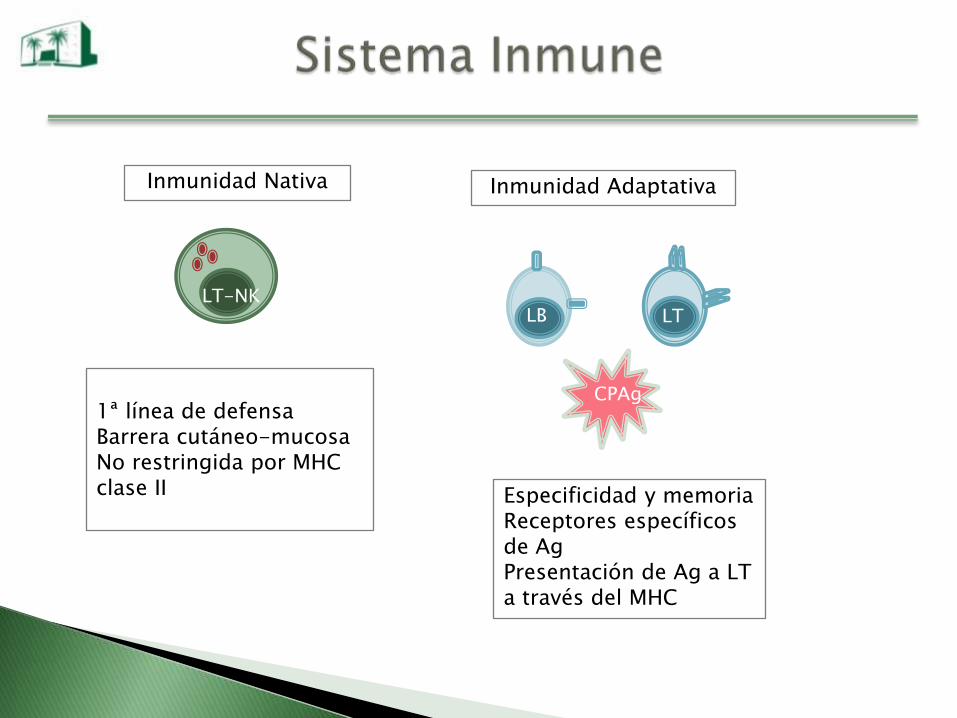
Inmunidad Nativa
LB LT
CPAg
LT-NK
1ª línea de defensaBarrera cutáneo-mucosaNo restringida por MHC clase II Especificidad y memoria
Receptores específicos de AgPresentación de Ag a LT a través del MHC
Inmunidad Adaptativa
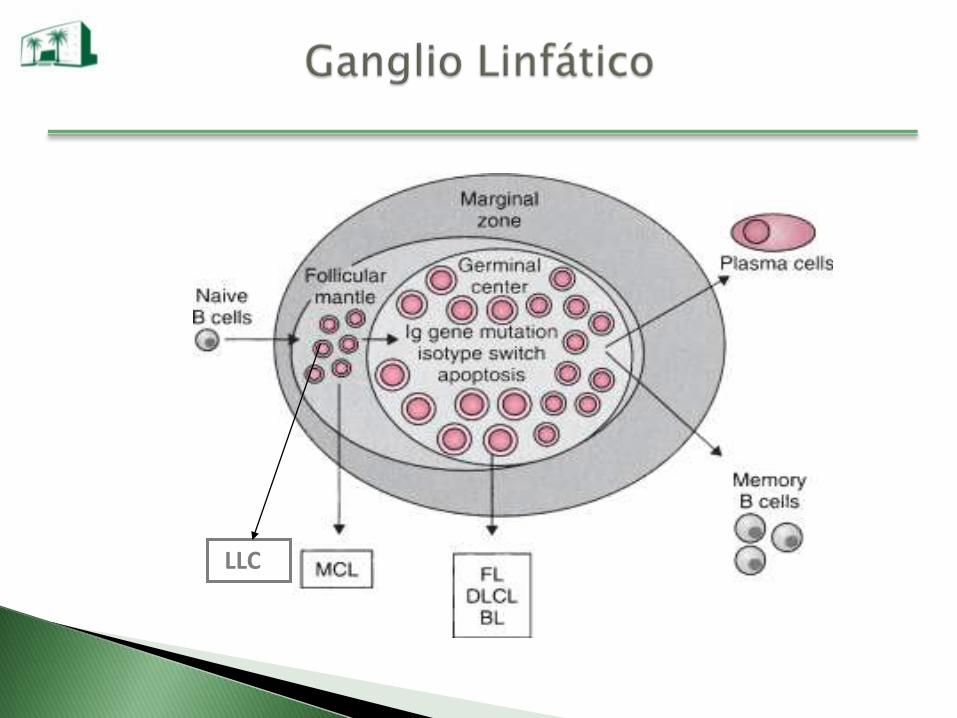
LLC

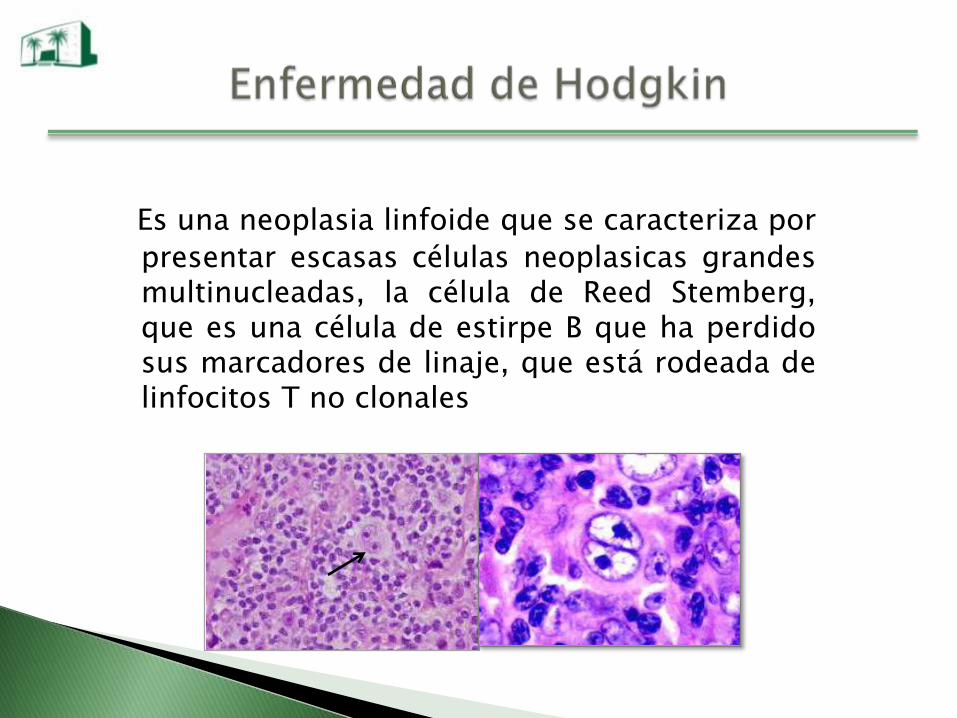
Es una neoplasia linfoide que se caracteriza por
presentar escasas células neoplasicas grandesmultinucleadas, la célula de Reed Stemberg,que es una célula de estirpe B que ha perdidosus marcadores de linaje, que está rodeada delinfocitos T no clonales

Adultos jóvenes. Edad media 30 años
2° pico más pequeño a los 50-55 años
Presentación nodal, generalmente cervical
Pueden presentar síntomas generales: fiebre, sudoración nocturna, pérdida de peso, prurito
Clasificación:
Hodgkin clásico CD30 y CD15 (+)
Predominio linfocítico nodular CD30 y CD15 (-) CD20 (+)

Examen Físico
T.A.C. de tórax abdomen y pelvis
Biopsia de médula ósea
Laboratorio
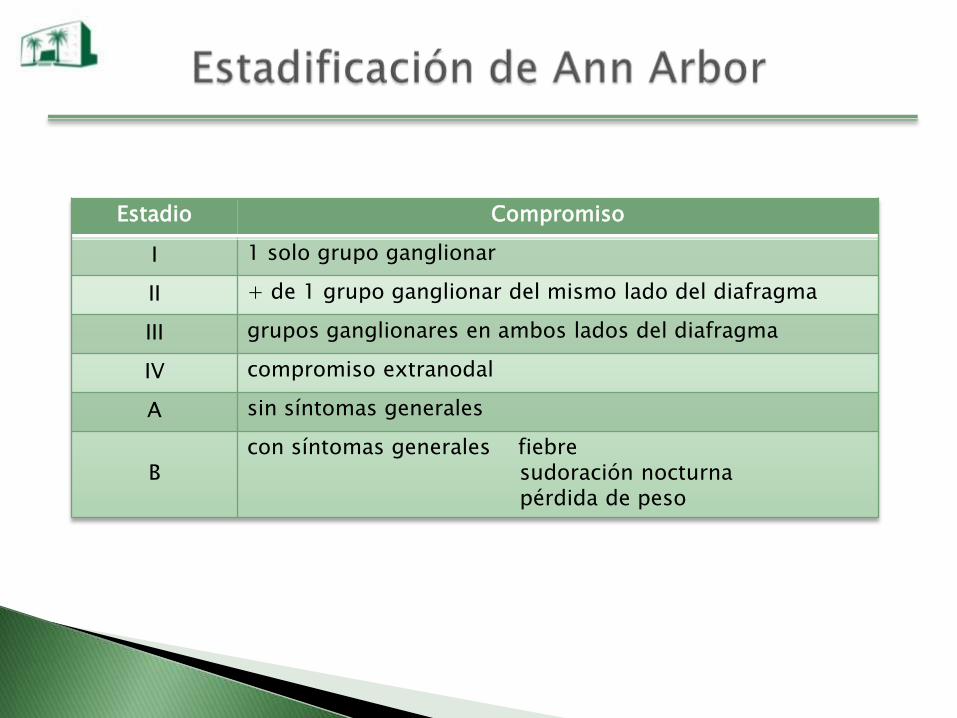
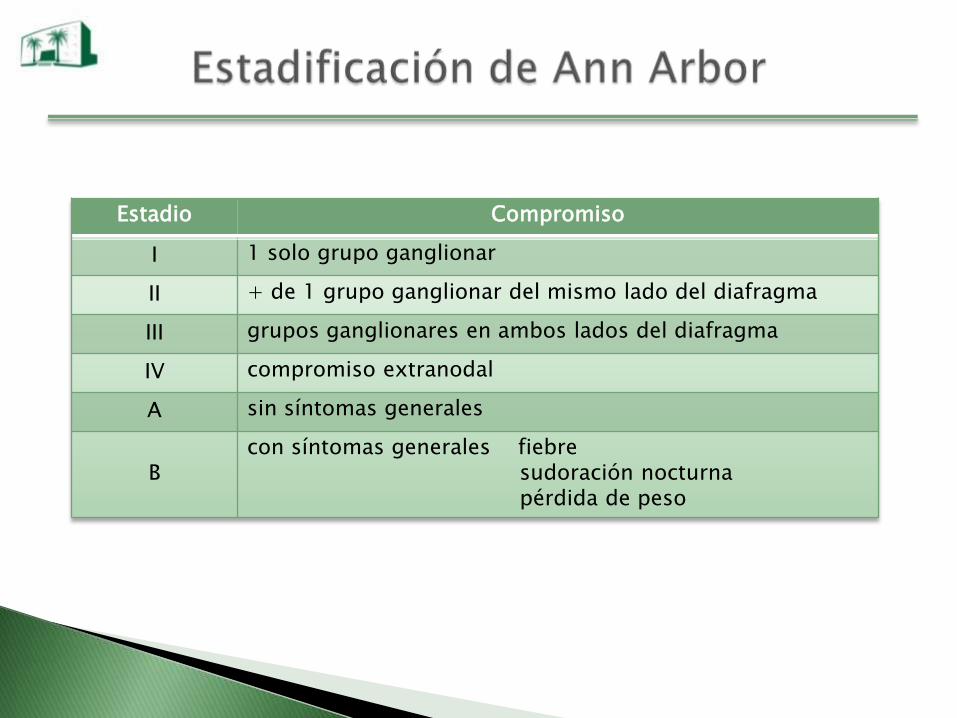
Estadio Compromiso
I 1 solo grupo ganglionar
II + de 1 grupo ganglionar del mismo lado del diafragma
III grupos ganglionares en ambos lados del diafragma
IV compromiso extranodal
A sin síntomas generales
Bcon síntomas generales fiebre
sudoración nocturnapérdida de peso

Estadío IV
Albúmina < 4g/dl
Hemoglobina < 10g/dl
Sexo masculino
Leucocitos > 15000/mm3
Linfocitos < 600/mm3
Eritrosedimentación aumentada
Edad >50 años

Estadíos tempranos
RadioterapiaQuimioterapia 2-4 ciclos (AVBD)
Estadíos avanzados
Quimioterapia (AVBD)Radioterapia en sitios con enfermedad voluminosa
Se debe evaluar respuesta con PET
Pacientes refractarios o recaídos
Quimioterapia de rescate y si responden consolidación con TAMO


Clasificación de Rappaport (1956)
Clasificación de Kiel (1974)
Working Formulation (1982)
Clasificación Real (1994)
Clasificación de la OMS (2001 – 2008)

Neoplasias de precursores linfoides
Leucemia - Linfoma Linfoblástico B
Leucemia - Linfoma Linfoblástico T
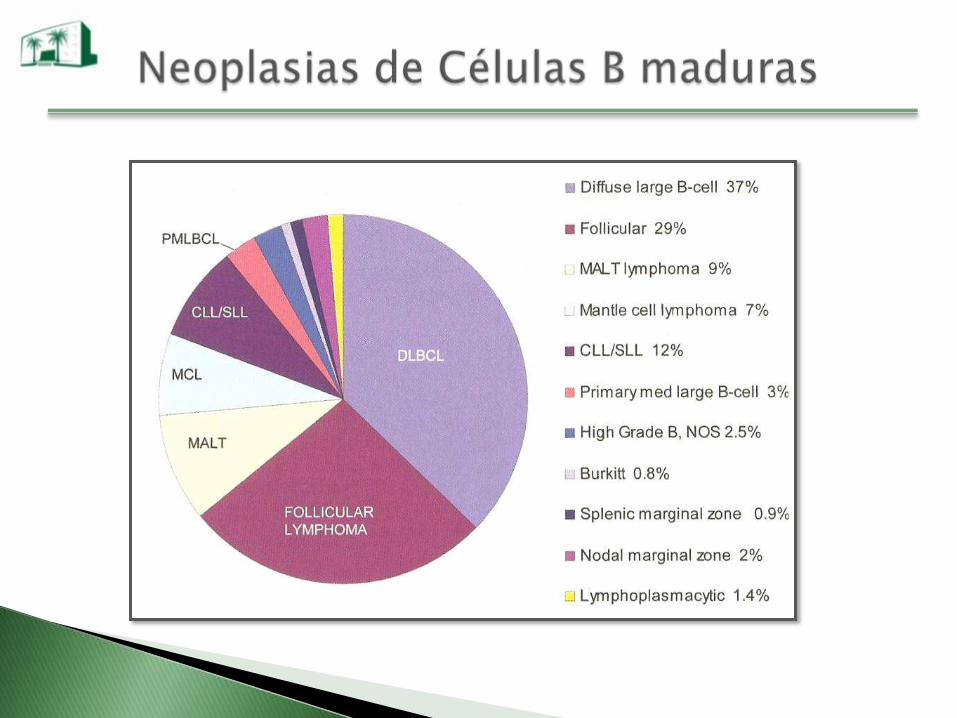
Neoplasias de Células B Maduras
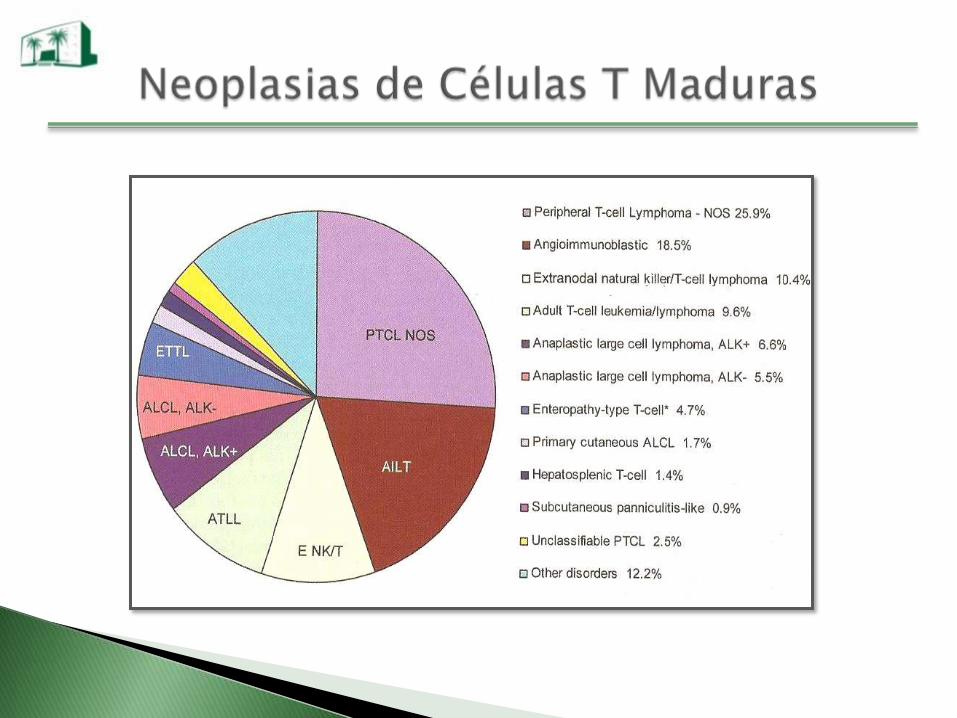
Neoplasias de Células T Maduras

LLC / SLL
Leucemia prolinfocítica B
Leucemia de células vellosas
Linfoma linfoplasmocítico
Linfoma del Manto
Linfoma Folicular
Linfoma de la zona marginal MALT
Nodal
Esplénico
Linfoma difuso de células grandes
Linfoma de Burkitt
Neoplasias de células plasmáticas Mieloma múltiple
Plasmocitoma solitario

Leucemia prolinfocítica T
Leucemia de linfocitos T grandes granulares
Leucemia NK agresiva
Linfoma extranodal NK/T tipo nasal
Linfoma T periférico no especificado
Linfoma T angioinmunoblástico
Leucemia/linfoma de células T del adulto (HTLV I)
Linfoma anaplásico de células grandes
Linfomas T cutáneos
Linfoma T intestinal tipo enteropatía
Linfoma T hepato-esplénico

Adenopatías
Hepatoesplenomegalia
Síntomas B pérdida de peso
sudoración profusa
fiebre
Compromiso extranodal

Las punciones ganglionares no son de utilidad
Se debe realizar siempre biopsia ganglionar
Se debe resecar en lo posible un ganglio entero
Morfología
Inmunotipificación IHQ o C.F.
Alteraciones genéticas
Marcadores B
MarcadoresT
CD 20CD19CD22Igs/c
CD7CD2CD3CD4CD8

Examen Físico
T.A.C. de tórax abdomen y pelvis
Biopsia de médula ósea
Laboratorio
Estadio Compromiso
I 1 solo grupo ganglionar
II + de 1 grupo ganglionar del mismo lado del diafragma
III grupos ganglionares en ambos lados del diafragma
IV compromiso extranodal
A sin síntomas generales
Bcon síntomas generales fiebre
sudoración nocturnapérdida de peso

Dependientes del linfoma
Estirpe T de peor pronóstico que BTipo histológico linfomas agresivos / linfomas indolentesIndice proliferativoMasa voluminosa (bulky disease)Síntomas BEstadioAlteraciones genéticas
Dependientes del pacienteEdadPerformance statusComorbilidades
Dependientes del tratamiento
Relación eficacia/toxicidad

Tipo HistológicoIndice
PronósticoParámetros
LDCG-B I.P.I.
Edad >60 Est. III - IV
P.S.> 2 LDH alta
N° Sitios Extranodales > 2
Linfoma Folicular F..L.I.P.I
Edad >60 Est. III - IV
Hb <12g/dl LDH alta
N° sitios Extranodales > 4
Linfoma del Manto M.I.P.I.Edad P.S.
LDH Cifra de G.B.
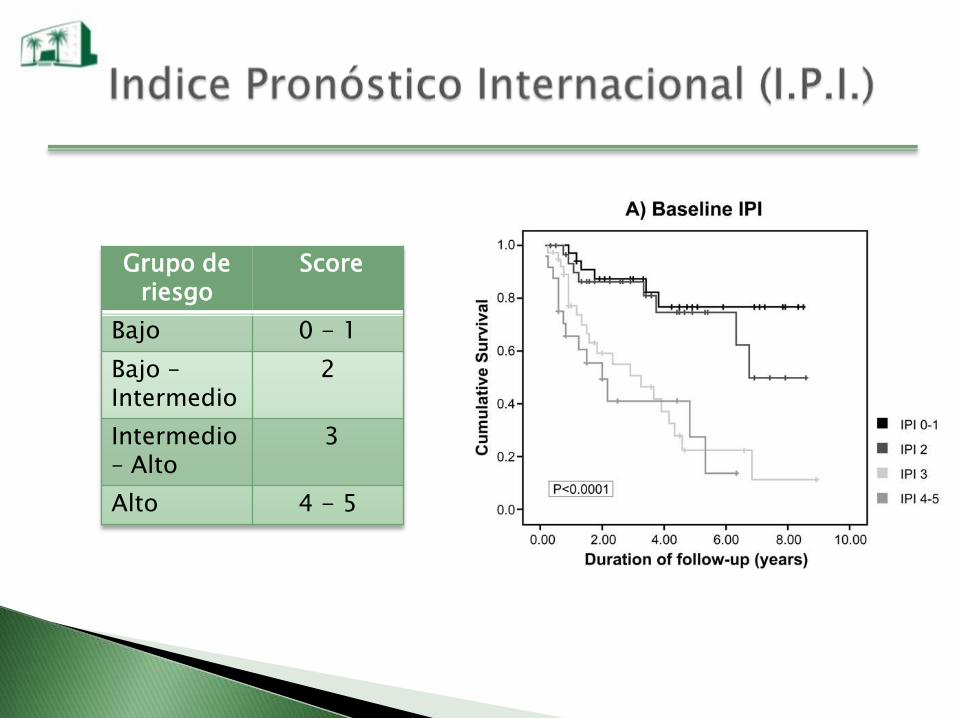
Grupo de riesgo
Score
Bajo 0 - 1
Bajo –Intermedio
2
Intermedio– Alto
3
Alto 4 - 5
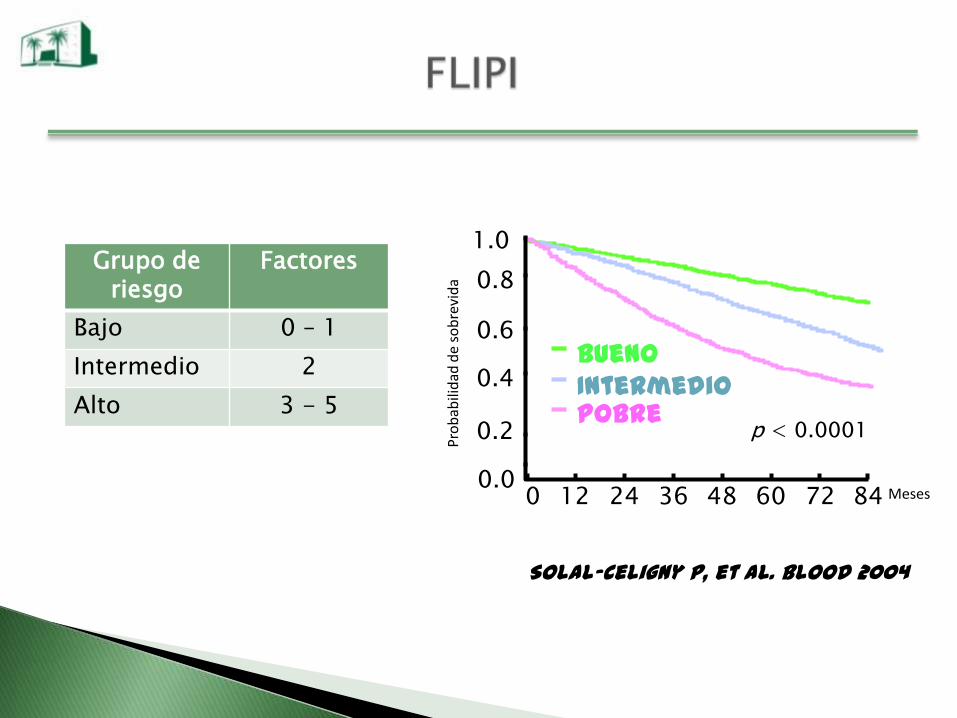
Solal-Celigny P, et al. Blood 2004
1.0
0.00
0.4
0.2
0.8
0.6
12 24 36 8448 60 72
p < 0.0001P
rob
abili
dad
de
sob
revi
da
Bueno
IntermedioPobre
Meses
Grupo de riesgo
Factores
Bajo 0 – 1
Intermedio 2
Alto 3 - 5

Quimioterapia con agentes únicos o combinados
Quimio-inmunoterapia: combinación de QT con Ac Monoclonales
Radioterapia
Transplante de progenitores hematopoyéticos
Cirugía es de utilidad para obtención de biopsias para diagnóstico

Neoplasia linfoide B madura
Leucemia más común en adultos
Edad media 70 años
50% de los pacientes asintomáticos al diagnóstico

Síntomas generales
Adenopatías
Hepatoesplenomegalia
Citopenias inmunes
Predisposición a infecciones

5000/mm3 linfocitos clonales en S.P
Inmunofenotipo: CD 5
CD19 CD20 CD23
Igs sup baja intensidad
Biopsia de Médula Osea >30% de linfocitos (no es requisito para diagnóstico)
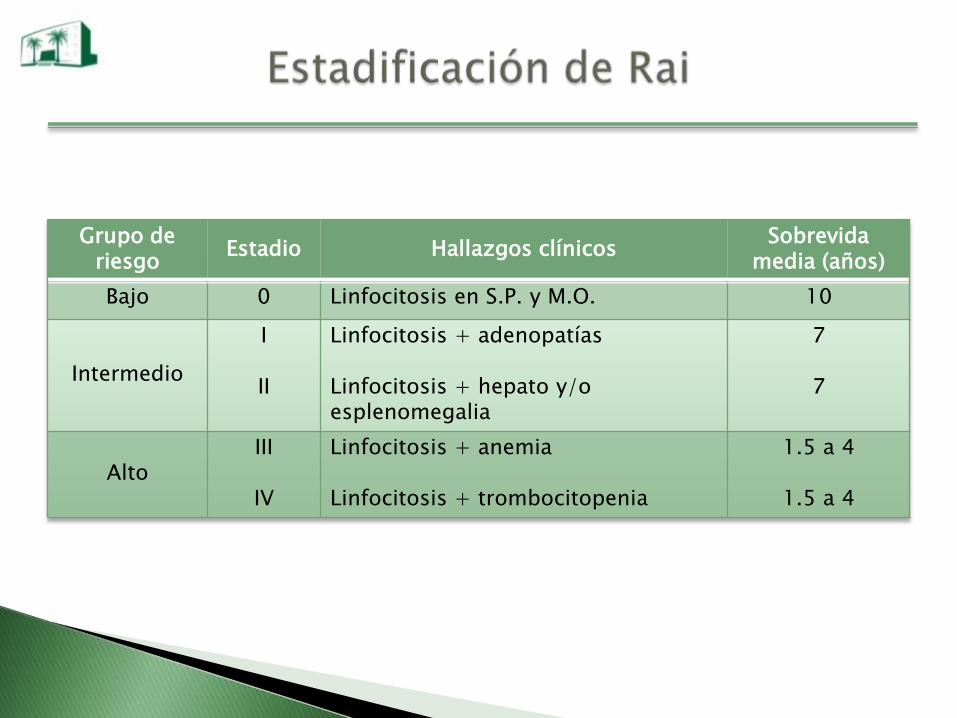
Grupo de riesgo
Estadio Hallazgos clínicosSobrevida
media (años)
Bajo 0 Linfocitosis en S.P. y M.O. 10
Intermedio
I
II
Linfocitosis + adenopatías
Linfocitosis + hepato y/o esplenomegalia
7
7
AltoIII
IV
Linfocitosis + anemia
Linfocitosis + trombocitopenia
1.5 a 4
1.5 a 4
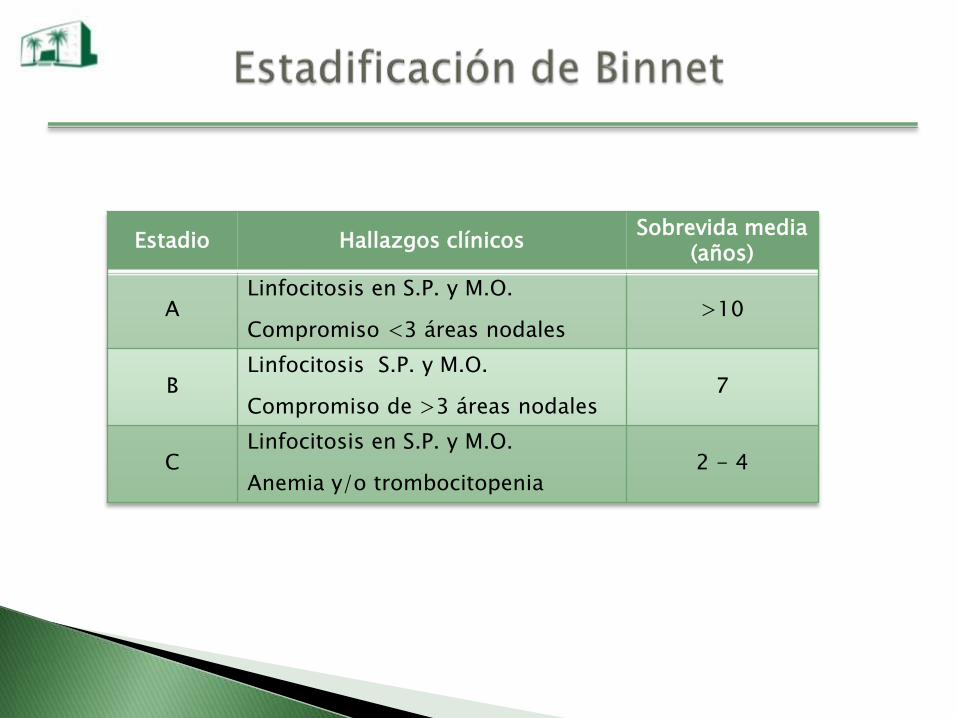
Estadio Hallazgos clínicosSobrevida media
(años)
ALinfocitosis en S.P. y M.O.
Compromiso <3 áreas nodales>10
BLinfocitosis S.P. y M.O.
Compromiso de >3 áreas nodales7
CLinfocitosis en S.P. y M.O.
Anemia y/o trombocitopenia2 - 4

Sexo (masculino peor pronóstico)
Estadio
Tiempo de duplicación de linfocitos
B2microglobulina
LDH
Expresión ZAP70
Estado mutacional de Igs
Alteraciones genéticas (deleción 17p y 11p)
Comorbilidades

La LLC es una enfermedad indolente, crónica e incurable
Hay un grupo de pacientes que no progresa y tiene una expectativa de vida igual a la de la población sana de la misma edad
No se puede predecir qué pacientes tendrán progresión de enfermedad y cuáles no

¿Qué pacientes requieren tratamiento?
Estadios avanzados
Síntomas B
Enfermedad voluminosa sintomática
Citopenias inmunes que no responden a corticoides
Tiempo de duplicación de linfocitos <6 meses

*FCR(fludarabina-ciclofosfamida-rituximab)*R Cl (rituximab-clorambucil)*R Fl (rituximab-fludarabina)
*R B (rituximab-bendamustine)
Paso rápido (go-go) Paso lento (slow go) Reposo (no go)
Independiente
Sin comorbilidades
Expectativa de vida normal
Función orgánica alterada
P.S. >2
Expectativa de vida >6 meses
Comorbilidades importantes
Expectativa de vida <6 meses
Tratamiento intensivoFCR*
Tratamiento moderadoR Cl* – R Fl* – R B*
Cuidados paliativos


Neoplasias que se originan a partir de la
expansión clonal de células plasmáticas
que van a producir una Ig monoclonal
denominada componente M o paraproteína

Gammapatía Monoclonal de Significado Incierto
Mieloma Múltiple: Smoldering
Sintomático
No Secretor
Leucemia de Células Plasmáticas
Plasmocitoma Solitario: Oseo o Extraóseo
Amiloidosis
Mieloma Osteoesclerótico: Síndrome POEMS
Macroglobulinemia de Waldeström
Enfermedad de Cadenas Pesadas
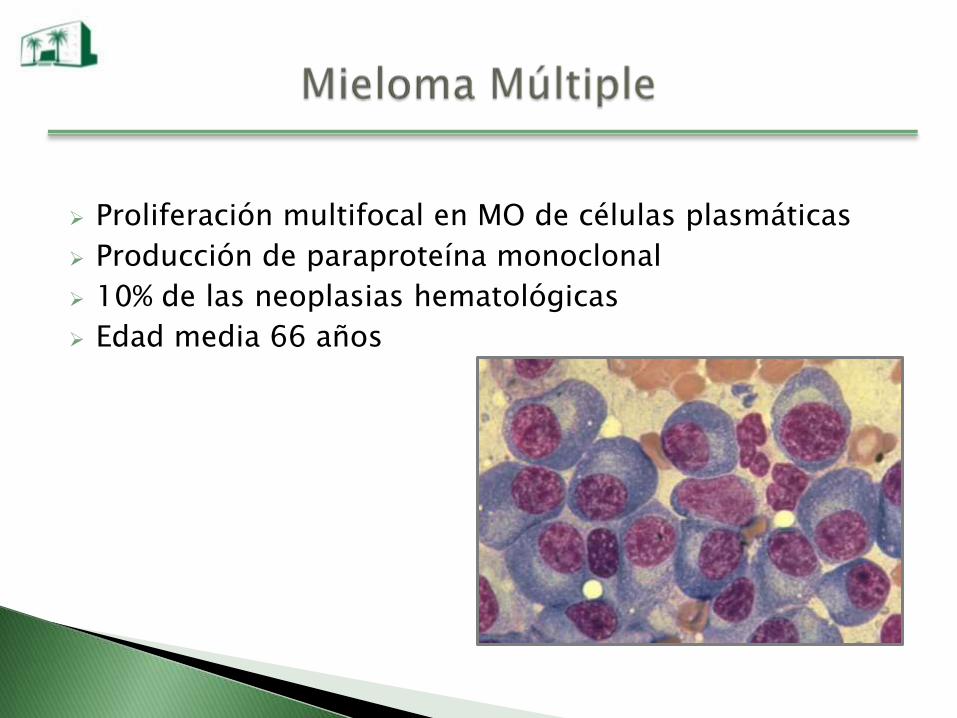
Proliferación multifocal en MO de células plasmáticas
Producción de paraproteína monoclonal
10% de las neoplasias hematológicas
Edad media 66 años

Dolor por alteraciones óseas
Anemia
Hipercalcemia
Alteraciones renales
Predisposición a las infecciones
Aumento de la VES
Gammapatía monoclonal

Proteinograma por Electroforesis
Inmunofijación para caracterizar CM (Ig G, IgA, IgM, cadenas livianas κ y λ)
Cuantificación de Ig G, IgA, IgM
Proteína de Bence Jones e Inmunofijación en orina de 24hs
Cadenas livianas libres
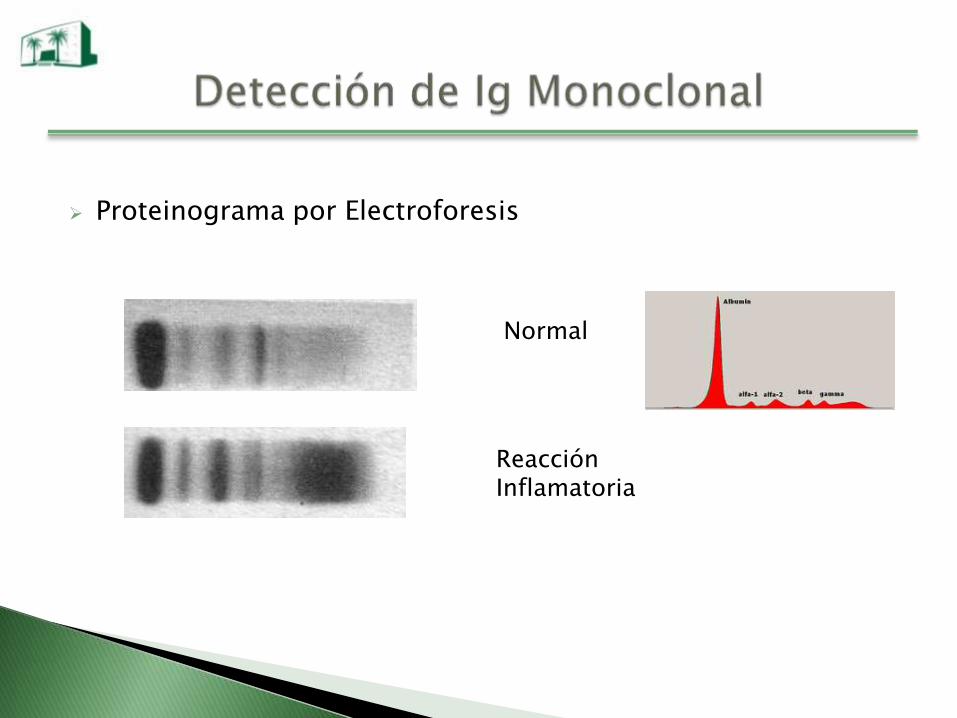
Proteinograma por Electroforesis
Normal
Reacción Inflamatoria
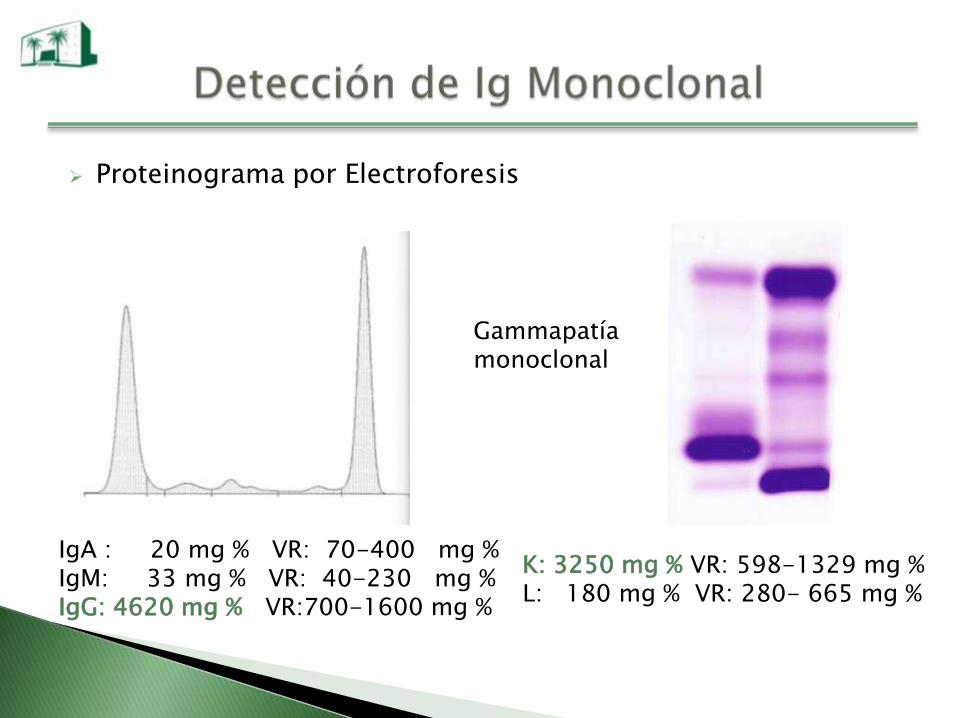
Proteinograma por Electroforesis
Gammapatíamonoclonal
IgA : 20 mg % VR: 70-400 mg %IgM: 33 mg % VR: 40-230 mg %IgG: 4620 mg % VR:700-1600 mg %
K: 3250 mg % VR: 598-1329 mg %L: 180 mg % VR: 280- 665 mg %
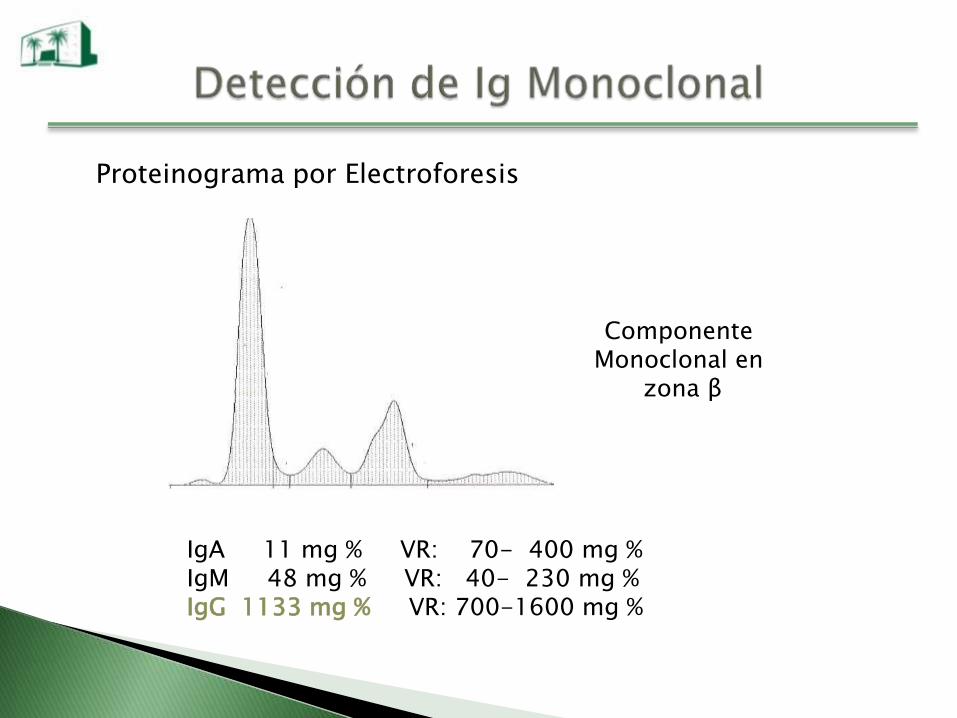
Proteinograma por Electroforesis
Componente Monoclonal en
zona β
IgA 11 mg % VR: 70- 400 mg %IgM 48 mg % VR: 40- 230 mg %IgG 1133 mg % VR: 700-1600 mg %
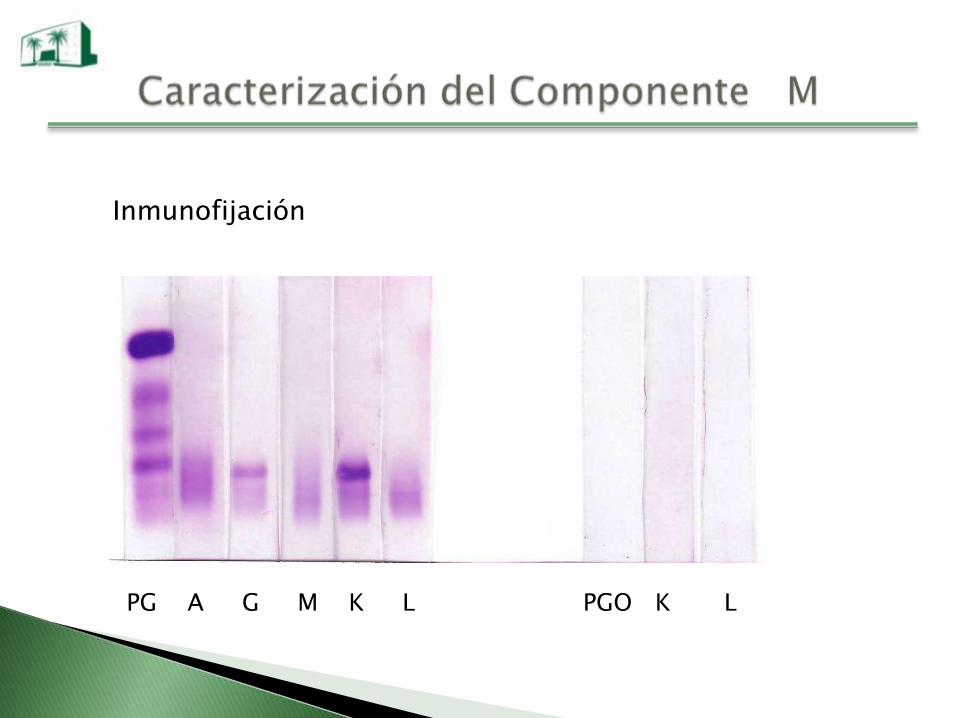
Inmunofijación
PG A G M K L PGO K L
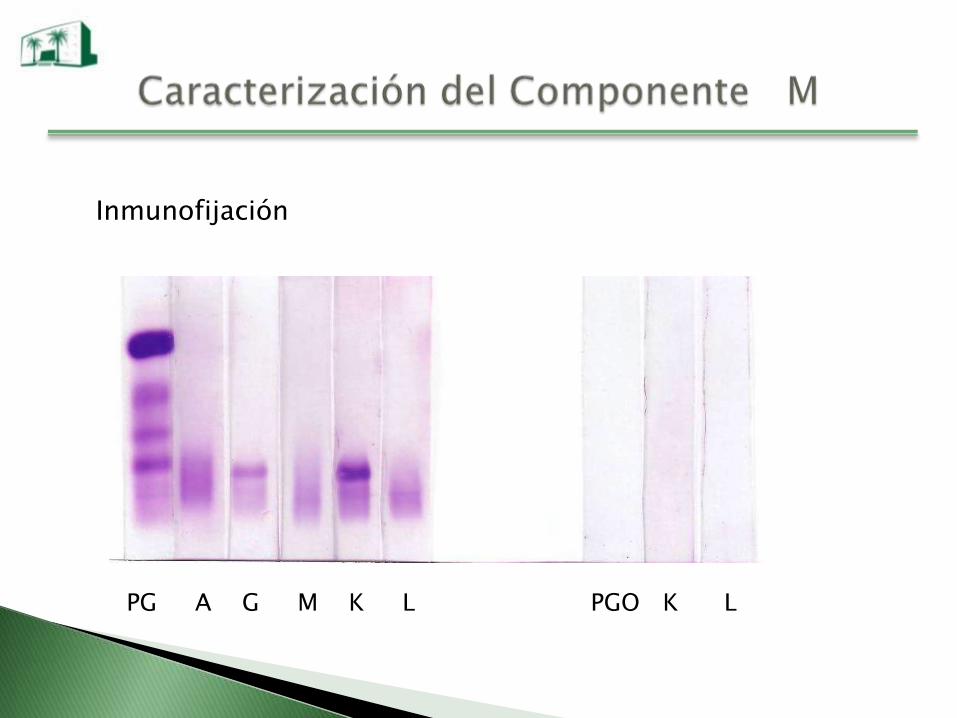
Inmunofijación
PG A G M K L PGO K L

Una vez identificado el CM se lo debe cuantificar y también al
resto de la Igs ya que las Igs normales pueden estar disminuidas
Valores Normales (mg/dl)
Ig G 700 - 1600
Ig A 70 - 400
Ig M 40 - 230
Kappa 598 – 1329
Lambda 280 - 665
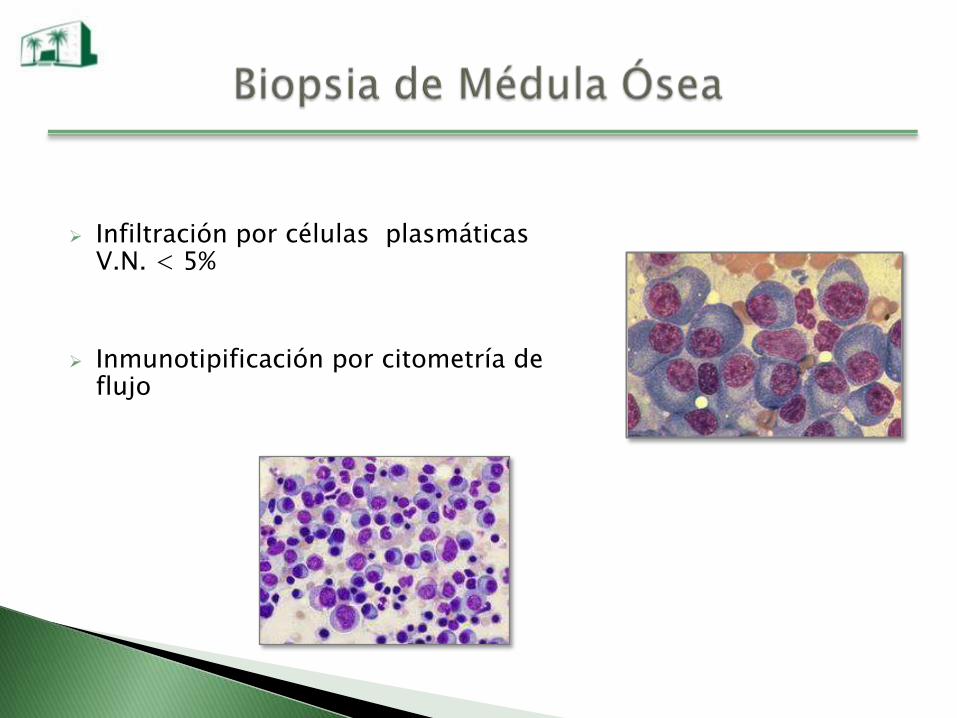
Infiltración por células plasmáticas V.N. < 5%
Inmunotipificación por citometría de flujo
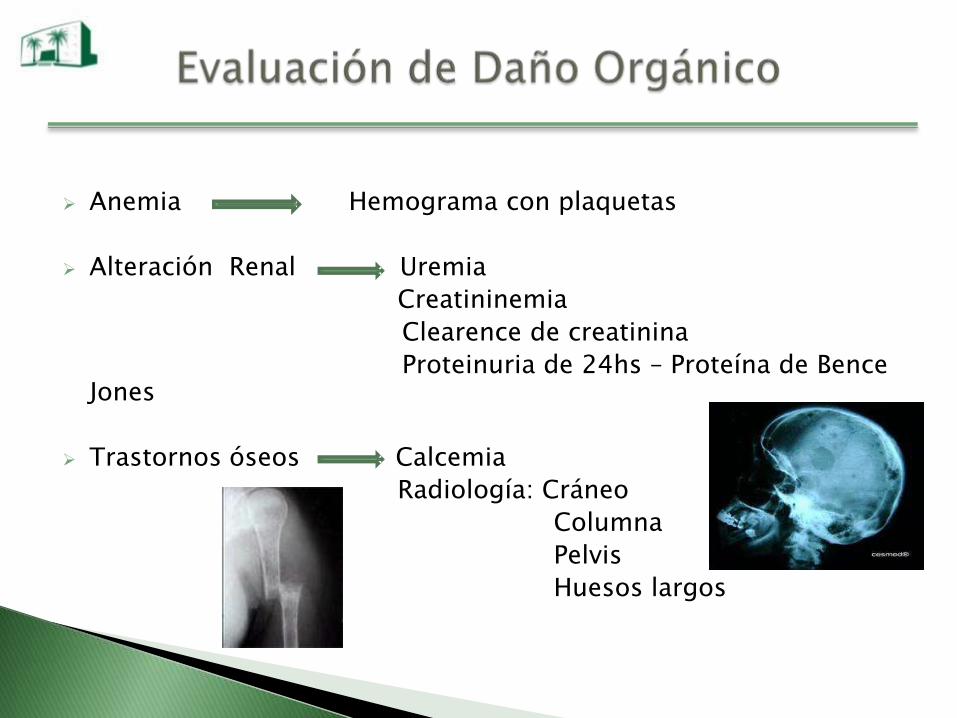
Anemia Hemograma con plaquetas
Alteración Renal Uremia
Creatininemia
Clearence de creatinina
Proteinuria de 24hs – Proteína de Bence Jones
Trastornos óseos Calcemia
Radiología: Cráneo
Columna
Pelvis
Huesos largos

Criterios diagnósticos de la GMSI
• Pico monoclonal < 3 g/dl proteína monoclonal en orina < 50 mg/día
• < 10% células plasmáticas en biopsia de médula ósea• •
• Sin evidencias de compromiso orgánico ni síntomas asociados
Criterios diagnósticos del MM Smoldering
� Pico monoclonal en sangre ≥ 3g/dl y/o
� ≥ 10% de células plasmáticas en médula ósea
• Sin evidencias de compromiso orgánico ni síntomas asociados
Criterios diagnósticos del MM sintomático
• Pico monoclonal en sangre y/u orina
• Infiltración de la médula ósea por células plasmáticas o plasmocitoma
• Afectación de órganos o tejidos: lesiones óseas, anemia, hipercalcemia, lesiones óseas líticas, insuficiencia renal, hiperviscosidad, amiloidosis o infecciones recurrentes
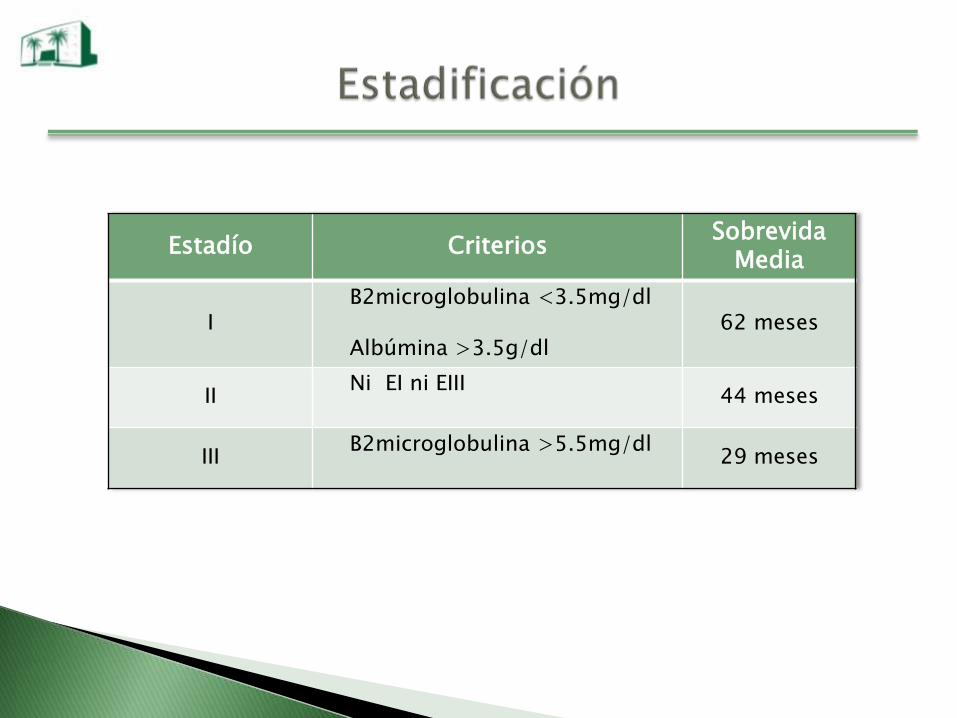
Estadío CriteriosSobrevida
Media
IB2microglobulina <3.5mg/dl
Albúmina >3.5g/dl62 meses
IINi EI ni EIII
44 meses
IIIB2microglobulina >5.5mg/dl
29 meses

Variante clínica
Estadío
Función renal
LDH
Morfología plasmoblástica
Citogenética : deleción 17p
t(4;14)

GMSI control clínico
Plasmocitoma Solitario radioterapia localizada
MM Smoldering control clínico
MM Sintomático tratamiento sistémico
Leucemia de Cél.Plasmáticas tratamiento sistémico
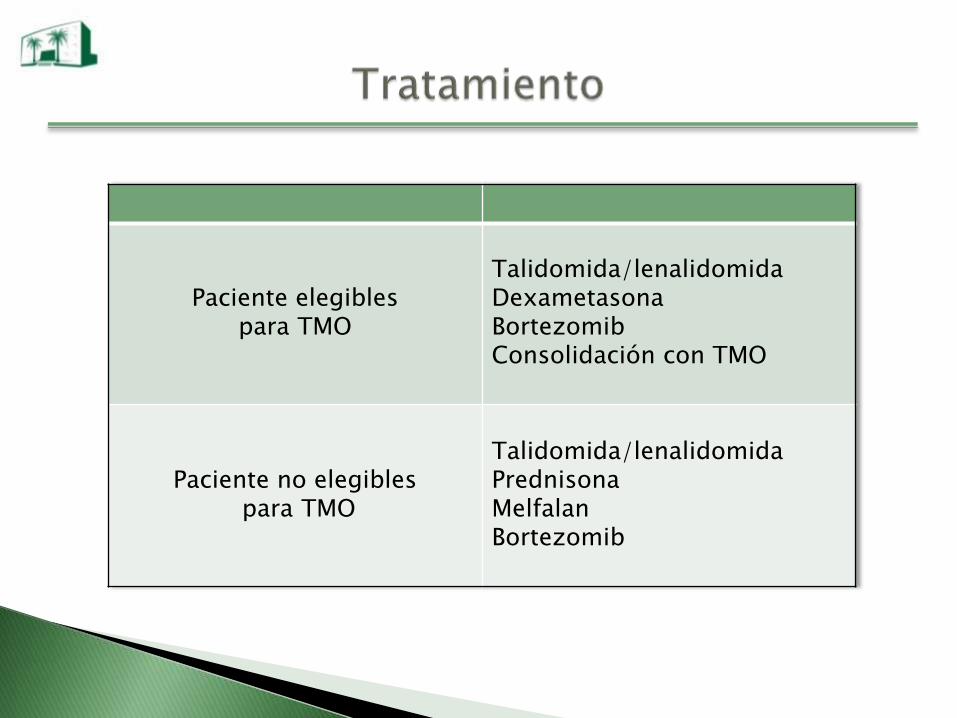
Paciente elegibles para TMO
Talidomida/lenalidomidaDexametasonaBortezomibConsolidación con TMO
Paciente no elegiblespara TMO
Talidomida/lenalidomidaPrednisonaMelfalanBortezomib

Hipercalcemia: hidratación
corticoidesbifosfonatos
Lesiones óseas: bifosfonatos
vertebroplastia
tto. quirúrgico de las fracturas patológicas
radioterapia local
Alt. Renal: hidratación adecuada
evitar AINE, contraste
plasmaféresis
Prevención de infecciones
Prevención de TEV