TEMA 9-1.Mètodes generals d´estudi de les eritropaties. TEMA 9-2. Anèmia microcítica

HOSPITAL UNIVERSITARIARNAU DE VILANOVA
LLEIDA
Mª José Panadés

HISTÒRIA CLÍNICA (2002)p Dona de 33 anys, natural de la Seu d’Urgell.p Sense AF ni AP d’interès.p Febrícula i anèmia des de fa 1 any.p Síndrome nefròtica des de fa 3 mesos, amb:
- Edemes i anasarca- Proteïnúria Bence-Jones (-)- Funció renal normal
BIÒPSIA RENAL
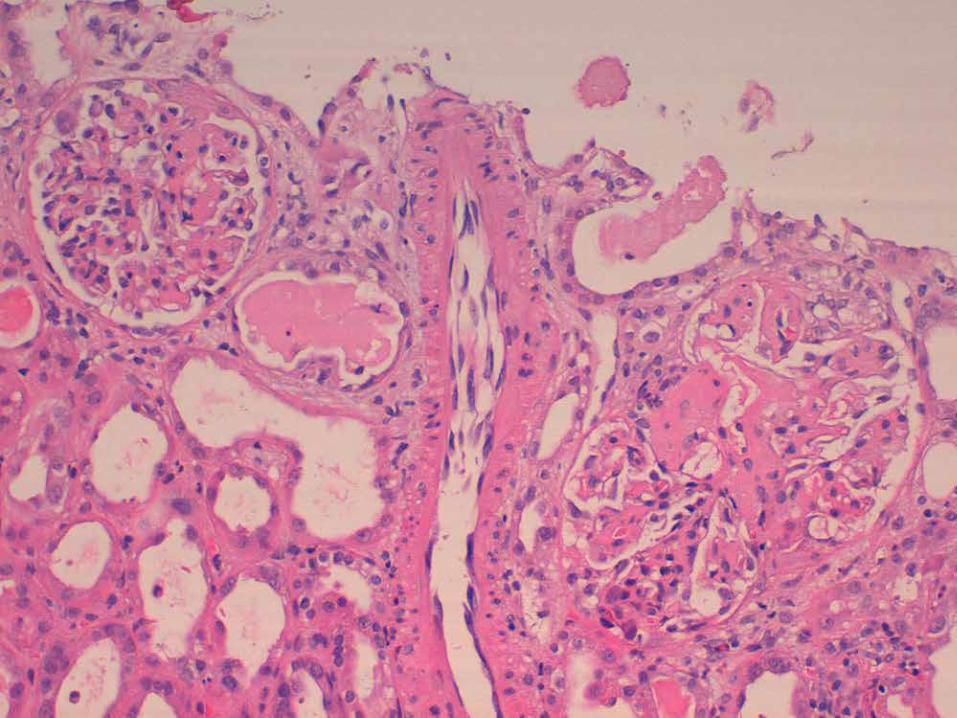


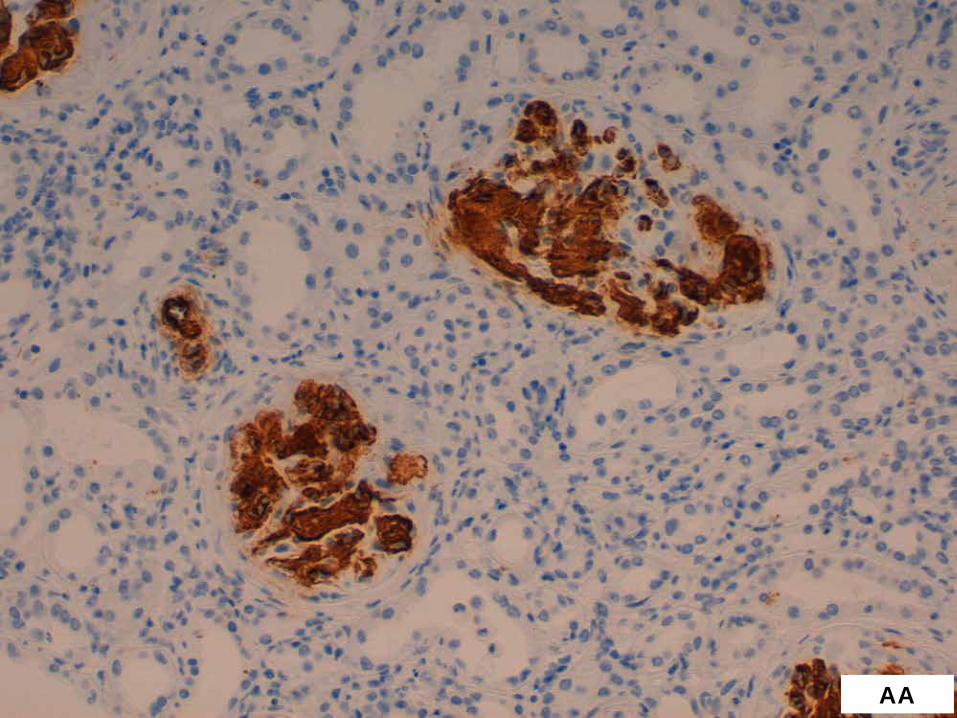
AA

DIAGNÒSTIC
p AMILOÏDOSI RENAL AA

ANALÍTICA
p Anèmia microcítica-normocròmicap Trombocitosip Augment de la VSG i proteïna Cp Hipergammaglobulinèmia policlonalp Hipoalbuminèmiap Serologia Hepatitis B, C i VIH (-)

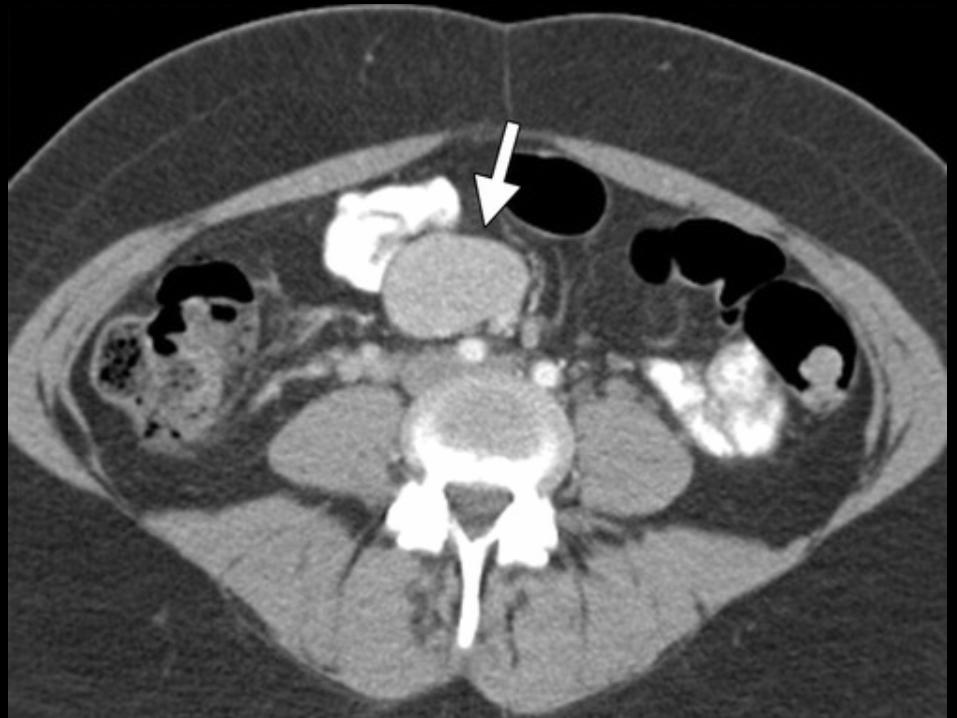
p TAC TORÀCIC: NORMALp TAC ABDOMINAL:
MASSA RETROPERITONEAL: EXÈRESI









AMILOIDE A

BCL2
IgD

CD138

CD138

KAPPA

LAMBDA

IgG

IgG4

CD34

HHV8

ANÀLISI MOLECULAR DE CLONALITAT AMB LIMFÒCITS B PER ALS GENS IgH: NO CLONAL

DIAGNÒSTIC
p MALALTIA DE CASTLEMAN LOCALITZADA(cèl·lules plasmàtiques)
p AMILOÏDOSI AA

FORMES CLÍNIQUES
p UNICÈNTRICA:Asimptomàtica. Bon pronòstic després d’extirpar la lesióTòrax (24%), Abdomen (18%), Retroperitoneu (14%)
p MULTICÈNTRICA:Simptomàtica. Pitjor pronòstic.Limfoadenopatia generalitzadaHepatoesplenomegàliaVessamentsHipergammaglobulinèmia policlonalTrombocitosi, anèmia
FREQÜENT ASSOCIACIÓ AMB IMMUNOSUPRESSIÓ(FORMA MULTICÈNTRICA)

FORMES HISTOLÒGIQUES
p HIALOVASCULAR:- Més freqüent a CD unicèntrica- Fol·licles hiperplàsics/atròfics- Proliferació vascular interfol·licular- Penetració de vasos hialinitzats en els centres germinals
p CÈL·LULES PLASMÀTIQUES:- Més freqüent en CD multicèntrica- Fol·licles hiperplàsics/atròfics- Infiltració per plasmàtiques policlonals, interfol·licular- Immunoblastes (plasmoblastes) a la zona del mantell i
centres germinals, que formen acúmuls (microlimfomes).
p MIXTA:- Més freqüent en CD multicèntrica

ETIOLOGIA I PATOGÈNESI
Estímul antigènic (HHV8)
Excés de producció de IL-6 i VEGF
Canvis reactius exagerats:- Hiperplàsia dels centres germinals- Infiltració per plasmàtiques- Augment de la vascularització- Trombocitosi

MALALTIA CASTLEMAN MULTICÈNTRICAESTATUS HHV8
p HHV8 POSITIU:Freqüent associació amb immunosupressió
VIH(+)----HHV8(+): 100% casosVIH(-)-----HHV8(+): 40-50% (endèmics)
p HHV8 NEGATIU:MCD IDIOPÀTIC (NOS)MCD IDIOPÀTIC amb factors TAFRO

SÍNDROME TAFRO (CASTLEMAN-KOJIMA)
p Trombocitopènia (T)Anasarca (A)Febre (F)Fibrosi reticulínica a moll de l’os (R)Organomegàlia (O)
p Morfologia de Castleman amb fol·licles atròficsp Absència d’hipergammaglobulinèmiap Curs clínic més agressiu

PATOGÈNESI SÍNDROME TAFRO
p Entitat diferent?
p MCD idiopàtica atípica?- Associada a: M. autoimmunes
M. infecciosesM. hematològiquesM. relacionada a IgG4

HISTÒRIA CLÍNICA (2016)p Home de 30 anys, natural de Gàmbiap Ingressa l’any 2013 per:
-SVCS, amb: massa mediastínicaadenopaties petitesvessament pleural/pericàrdic
-Analítica: no hipergammaglobulinèmiano trombocitosiVIH (-)
-Sospita clínica: limfoma-Biòpsia massa mediastínica: negatiu-Tractament: corticoides
p Reingressa al gener de l’any 2016 per:-Mal estat general-Augment de la mida de les adenopaties-Massa mediastínica estable-Vessament pleural/pericàrdic







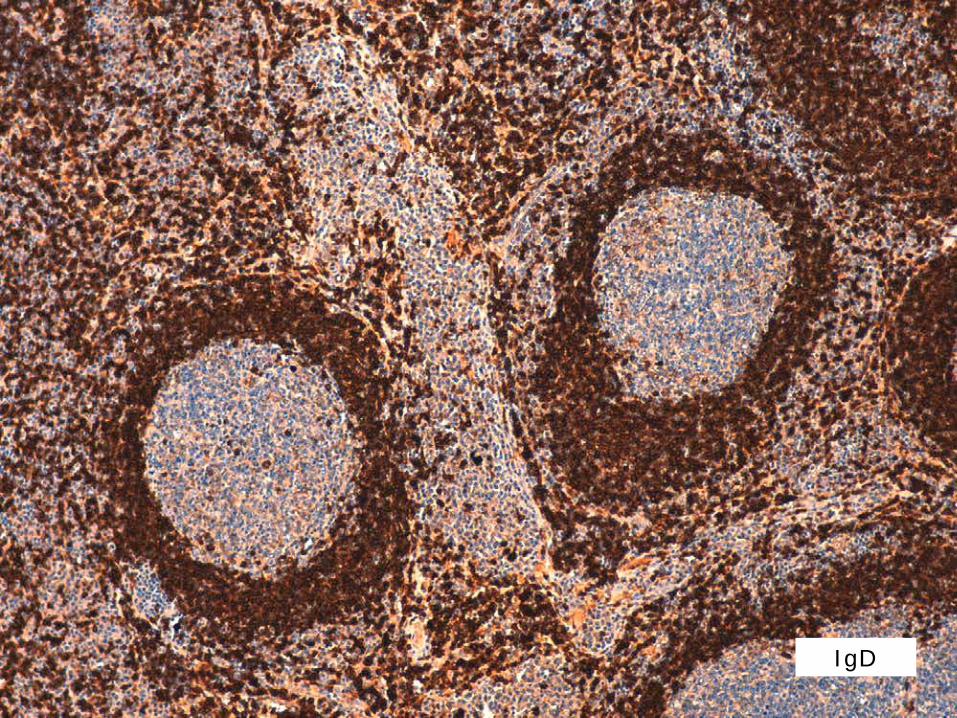
INMUNOHISTOQUÍMIA 1
BCL2
CD21CD31
IgD

INMUNOHISTOQUÍMIA 2
CD138
HHV8IgG4IgG
LAMBDAKAPPA


DIAGNÒSTIC
MALALTIA DE CASTLEMAN MULTICÈNTRICA IDIOPÀTICA (HHV8-)

COMPLICACIONS
p Sarcoma de Kaposi: 70%p Limfoma no Hodgkin: 15-20%
- L.D.C.G.B- L. plasmoblàstic
p S. de POEMSp Amiloïdosi

COMPLICACIONS RENALS DE LA MALALTIA DE CASTLEMAN
p Microangiopatia trombòtica-like: 60%p Amiloïdosi AA : 20%p Nefritis intersticial : 13%p Glomerulopaties

EVOLUCIÓ DE L’AMILOÏDOSI RENAL(després de la exèresi de la lesió)
p Desaparició del S. nefròtic p Disminucióp Persistència
p Dipòsits del material amiloïde: ???

EVOLUCIÓ DEL NOSTRE CAS
p 2010: funció renal normalmicroalbuminúria residual
p 2013: no microalbuminúria
p 2015: ALTA