
Hidrometalurgia
183
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia http://www.ivan.cl 2007 Universidad de Atacama 1 Por hidrometalurgia se entiende los procesos de lixiviación selectiva ( disolución ) de los componentes valiosos de las menas y su posterior recuperación de la solución por diferentes métodos. El nombre de hidrometalurgia se refiere al empleo generalizado de soluciones acuosas como agente de disolución. La hidro-electrometalurgia comprende el conjunto de procesos de lixiviación y precipitación por medio de electrólisis, donde los procesos electroquímicos son precedidos por los procesos hidrometalúrgicos. Hay tres principales etapas de los procesos hidrometalúrgicos : (1) Disolución del componente deseado presente en la fase sólida. (2) Concentración y/o purificación de la solución obtenida. (3) Precipitación del metal deseado o sus compuestos. Los reactivos químicos empleados ( agentes lixiviantes ) deben reunir muchas propiedades para poder usarse, por ejemplo : no deben ser muy caros, deben ser fácilmente recuperables y deben ser bastante selectivos para disolver determinados compuestos. Los procesos de lixiviación y purificación de la solución corresponden a las mismas operaciones que se practican en el análisis químico, solamente que a escala industrial. En estos apuntes sólo se presenta los procesos hidro-electrometalúrgicos de algunos de los metales más comunes en Chile: el cobre y el oro. Se recomienda al estudiante referirse a las varias publicaciones que se publicaron estos últimos años en el campo de la hidrometalurgia para profundizar los temas que le interesan. Introduccióna la hidrometalurgia
-
Upload
pablo-reyes -
Category
Documents
-
view
11.894 -
download
17
Transcript of Hidrometalurgia

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama1
Por hidrometalurgia se entiende los procesos de lixiviación selectiva ( disolución )de los componentes valiosos de las menas y su posterior recuperación de lasolución por diferentes métodos. El nombre de hidrometalurgia se refiere alempleo generalizado de soluciones acuosas como agente de disolución.
La hidro-electrometalurgia comprende el conjunto de procesos de lixiviación yprecipitación por medio de electrólisis, donde los procesos electroquímicos sonprecedidos por los procesos hidrometalúrgicos.
Hay tres principales etapas de los procesos hidrometalúrgicos :
( 1 ) Disolución del componente deseado presente en la fase sólida.( 2 ) Concentración y/o purificación de la solución obtenida.( 3 ) Precipitación del metal deseado o sus compuestos.
Los reactivos químicos empleados ( agentes lixiviantes ) deben reunir muchaspropiedades para poder usarse, por ejemplo : no deben ser muy caros, deben serfácilmente recuperables y deben ser bastante selectivos para disolverdeterminados compuestos. Los procesos de lixiviación y purificación de la solucióncorresponden a las mismas operaciones que se practican en el análisis químico,solamente que a escala industrial.
En estos apuntes sólo se presenta los procesos hidro-electrometalúrgicos dealgunos de los metales más comunes en Chile: el cobre y el oro. Se recomienda alestudiante referirse a las varias publicaciones que se publicaron estos últimosaños en el campo de la hidrometalurgia para profundizar los temas que leinteresan.
Introduccióna lahidrometalurgia

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama2
2.1. Reacción Química
En la práctica, se acostumbra escribir una reacción química, bajo la forma :
dDcCbBaA (1)
En (1), a,b,c,d : Coeficientes estequiométricosA,B : ReactantesC,D : Productos
El valor de la energía libre estándar de la reacción (G°) puede determinarseconociendo los potenciales químicos estándares o las energías libres de formaciónde los reactantes y productos :
Donde i son los coeficientes estequiométricos de la reacción.
FundamentosTermodinámicos

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama3
En este caso :
(2)
En (2), G° : Variación de energía libre estándar de la reacción (1)(cal/mol o joule/mol)a,b,c,d : Coeficientes estequiométricos
: Energías libres estándar de formación (opotenciales químicos ) de los compuestosque participan en la reacción.En las tablas, se escriben también G° o F°.
Se usa la convención de LATIMER.
a 298 K ( 25°C )
Utilizando los estados estándares, la variación de energía libre (G) de estareacción, puede ser expresada como :
(3)
En (3), G° : Energía libre estándar de la reacciónaA,aB,aC,aD : Actividades de los productos y reactantesT : Temperatura (K)R : Constante de los gases ( 1.987 cal/mol.K )

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama4
Al equilibrio, no hay variación de energía libre : G = 0.Por lo tanto :
(4)
A 25°C G° en cal/mol) :
(5)
La ecuación (4) define K, constante de equilibrio de la reacción considerada a latemperatura T :
(6)
En (6), a,b,c,d : Coeficientes estequiométricosaA,aB,aC,aD : Actividades de los productos y reactantes. Ensoluciones diluidas, las actividades de las especies disueltas seaproximan por su concentración molar (mol/litro). La actividad deun solido o líquido puro es 1.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama5
Ejemplo
Se considera la reacción CuO + 2H+ <=> Cu2+ + H2O
(Concentraciones en mol/litro)
2.2. Reacción Electroquímica
Por lo general, una reacción electroquímica (o semi reacción), es decir en queparticipan además electrones, se escribe en el sentido de la reducción (captaciónde electrones) :
(7)
En (7), Ox. : Especie oxidanteRed. : Especie reductoran : Número de electrones que participan en la reacción

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama6
La condición de equilibrio para un sistema electroquímico es :
(8)
Ecuación de NERNST
En (8), E : Potencial de la reacción electroquímica (V)R : Constante de los gases ( 1.987 cal/mol.K )T : Temperatura ( K )n : Número de electrones que participan en la reacciónF : Constante de Faraday ( 23060 cal/volt.equival. o 96500 Coulomb )[Ox] : Actividad de Ox.[Red] : Actividad de Red.E° : Potencial estándar de la reacción electroquímica (V)
Nota : En ciertos libros, la ecuación (8) se escribe bajo la forma :
Reemplazando en (8) los valores numéricos de R y F, expresando esa ecuaciónen términos de logaritmo decimal y a 25°C, se llega a :
(9)
E° es el potencial estándar de la reacción electroquímica, es decir cuando todoslos compuestos que participan en la reacción están en su estado estándar (sólidos y líquidos puros, especies disueltas en una concentración de 1 mol/litro).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama7
(10)
o
En(10), i : Coeficientes estequiométricos de la reaccióni : Energias libres de formaciónn : Número de electrones que participan en la reacciónF : Constante de Faraday ( 23060 cal/volt.equivalente o 96500 Coulomb )
E° puede calcularse por la formula (10), pero en general se prefiere obtenerlodirectamente en tablas (Tabla ..) y escalas de potenciales estándares ( Fig. ..).
Ejemplo
Se considera la semi reacción :
Cu2+ + 2 e- <=> Cu
Resultados para diferentes concentraciones de cobre :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama8
2.3. Diagramas Eh - pH
2.3.1. Introducción
La forma más conveniente de representar la termodinámica de sistemas acuososes en forma gráfica en los diagramas de Pourbaix o diagramas potencial - pH.Estos diagramas son ampliamente utilizados por los hidrometalurgistas, por cuantopermiten visualizar posibilidades de reacciones sin tener que recurrir al cálculotermodinámico para los fenómenos que ocurren en medio acuoso.
Una importante restricción en la aplicación práctica de los diagramastermodinámicos, es que predicen tendencias a que ocurran fenómenos, pero no lavelocidad con que éstos puedan ocurrir. En la práctica las velocidades de reacciónpueden variar desde valores tan altos que son controlados por limitaciones en latransferencia de masa, a valores tan bajos que se requieren períodos geológicospara observar en forma directa el fenómeno. La cinética extremadamente lenta enalgunas reacciones conduce a que algunas fases sólidas existan en condicionesfuera de su rango de estabilidad termodinámica o que fases sólidas no se formenen condiciones termodinámicas favorables y lo hagan otras en su lugar (fasesmetaestables) (ejemplo : precipitación de hidróxido de hierro). En este caso, es aveces útil utilizar diagramas Eh - pH modificados que consideren las fasesmetaestables.
Existen Atlas de diagramas Eh - pH (Pourbaix). En este curso, se pretendemostrar como interpretar y utilizar esos diagramas, por lo cual se van a construir amodo de ejemplos el diagrama Eh - pH del agua y del cobre.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama9
2.3.2. Diagrama Eh - pH del agua
Puesto que se está considerando el equilibrio termodinámico de especies ensolución acuosa, es relevante incluir en los diagramas Eh - pH los límites deestabilidad del agua.
Las semi reacciones a considerar son :
En medio ácido, Oxidación 2 H2O O2 + 4 H+ + 4 e - E° = 1.23 V
Reducción 2 H++ 2 e H2 E° = 0.00 V
En medio básico, Oxidación 4 OH- O2 + 2 H2O + 4 e- E° = 0.401 V
Reducción 2 H2O + 2 e- H2 +2 OH- E° = -0.83 V
Para calcular un diagrama de Pourbaix, se utilizan las ecuaciones de lasreacciones en medio ácido, las cuales están directamente relacionadas con laconcentración en iones H+ y el pH.
* Oxidación 2 H2O <=> O2 + 4 H+ + 4e- E° = 1.23 V
(11)
* Reducción 2 H+ + 2 e- <=> H2 E° = 0 V
(12)
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama10
O2
H2O
H+
Acido yoxidante
Básico yoxidante
Básico yreductor
Acido yreductor
pH
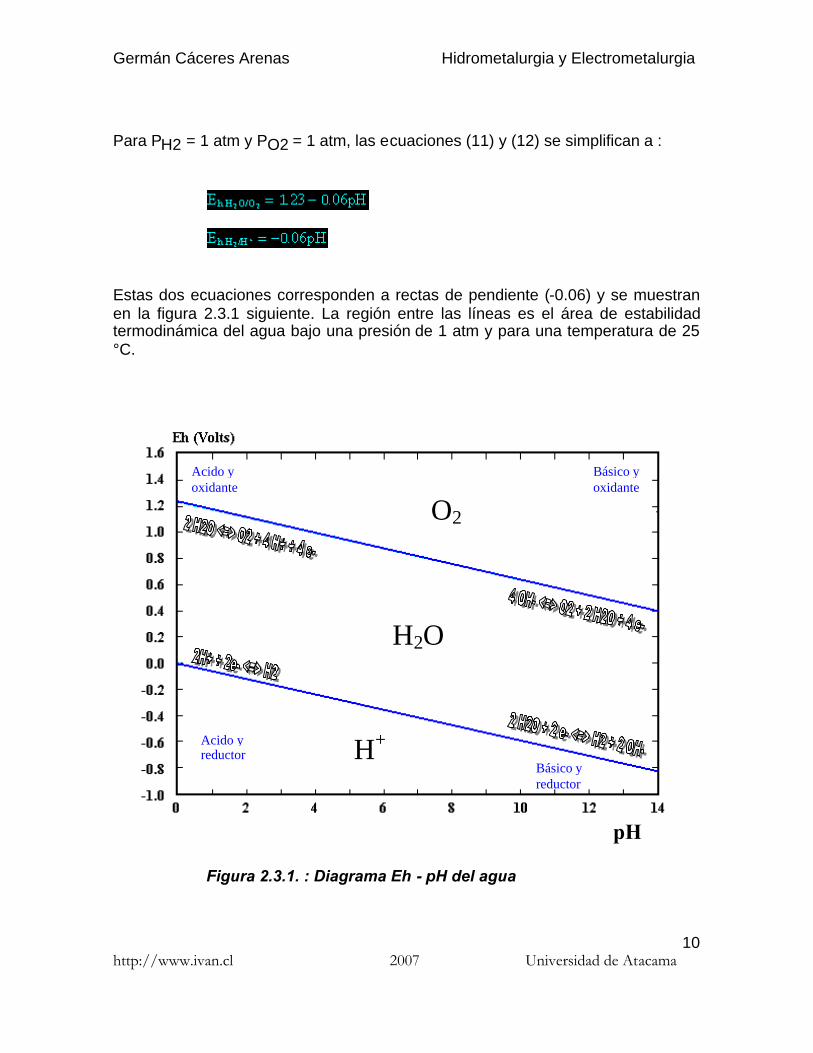
Para PH2 = 1 atm y PO2 = 1 atm, las ecuaciones (11) y (12) se simplifican a :
Estas dos ecuaciones corresponden a rectas de pendiente (-0.06) y se muestranen la figura 2.3.1 siguiente. La región entre las líneas es el área de estabilidadtermodinámica del agua bajo una presión de 1 atm y para una temperatura de 25°C.
Figura 2.3.1. : Diagrama Eh - pH del agua

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama11
2.3.3. Construcción del diagrama Eh - pH del cobre enagua
Se presenta a continuación la construcción - simplificada - y uso del diagrama Eh -pH para el caso del sistema Cu - H2O. Consideramos actividades unitarias paratodas las especies metálicas en solución. Este diagrama es adecuado paraanalizar la lixiviación de óxidos simples como tenorita (CuO) y cuprita (Cu2O) o decobre nativo.
Tabla 2.3.1 :
Especies consideradas en el diagrama Eh - pH de Cu - H2O.
EspecieEnergia libre de
formación °(calorias)
Estado deoxidación del Cu Denominación
Cu 0 0 cobre nativo
Cu2O -34.950 1 cuprita (rojo)
CuO -30.400 22 tenorita (negro)
Cu+ 12.000 1 ion cuproso
Cu2+ 15.530 2 ion cuprico
HCuO2-
-61.420 2ion hidrogenuro
de cuprato
Se consideran sucesivamente las diferentes reacciones entre las especiesconsideradas para el diagrama Eh - pH del cobre. Cada reacción corresponde auna línea de equilibrio en el diagrama de Pourbaix.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama12
2.3.3.1. Reacciones en que participan H+, pero no e-
Reacciones químicas propiamente tal, dependen sólo del pH.
CuO + 2 H+ <=> Cu2+ + H2O
(13)
En este caso, se consideran actividades unitarias para todas las especiesmetálicas en solución.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama13
Figura 2.3.2. Representación del equilibrio químico Cu2+/CuO,cuando [Cu2+]=1mol/l.
La figura 2.3.2 muestra que si la concentración de Cu2+ es de 1 mol/litro, el oxidode cobre CuO precipita a un pH igual o superior a 3.8, independientemente del Eh.
CuO + H2O <=> HCuO2- + H+ G° = 25670 cal
A partir del G°, se calcula la relación de equilibrio siguiente :
Este pH es mayor que 14, el límite superior de un diagrama Eh - pH convencional.=> El CuO se considera como estable hasta pH 14.
CuO + H2O <=> CuO22- + 2H+
A partir del G°, se calcula la relación de equilibrio siguiente :
del CuO
Zona deestabilidad
Eh
pH3.8
Zona deestabilidaddel Cu(2+)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama14
Este pH es mayor que 14, el límite superior de un diagrama Eh - pH convencional.=> El CuO se considera como estable hasta pH 14.
HCuO2- <=> CuO22- + H+
A partir del G°, se calcula la relación de equilibrio siguiente :
Si hay 2 especies en solución, la línea de equilibrio se define donde lasactividades de las dos especies son iguales.
Figura 2.3.3. Representación del equilibrio químico HCuO2- <=>
CuO22- + H+
Ese equilibrio no se dibuja en el diagrama de Pourbaix porque HCuO22- no existea pH < 18.95 ! En la construcción del diagrama, algunas líneas generadasmediante los cálculos termodinámicos deben ser eliminados total o parcialmente,ya que representan equilibrios que no tienen significado en la práctica.
Eh
pH13.15
HCuO2- CuO2
2-
[HCuO2(-)] >> [CuO2(2-)] [HCuO2(-)] << [CuO2(2-)]

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama15
Cu2O + 2 H+ <=> 2 Cu+ + H2O
pH = -0.85 Al equilibrio, si [Cu+] = 1 mol/l.
=> El Cu2O debería existir para todo pH superior a -0.85, pero no es así porque el
Cu+ no existe en solución, como se muestra en el capítulo siguiente.
2.3.3.2. Reacciones en que participan e-, pero no H+
Reacciones electroquímicas propiamente tal, dependen sólo del Eh.
Cu2+ + e- <=> Cu+ E = 0.15 V
Cu+ + e- <=> Cu E = 0.52 V
Cu2+ + 2e- <=> Cu E = 0.34 V
Se puede observar el la figura 3.3.4. que hay un conflicto de equilibrios, no esposible que un especie se mantenga en 2 campos termodinámicos. El ion cuproso(Cu+) no es estable en soluciones acuosas, transformándose en Cu2+ y Cu°según la reacción de DISMUTACION :
2 Cu+ => Cu2+ + Cu° (14)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama16
Figura 3.3.4. Inestabilidad del ion Cu+por dismutación
2.3.3.3. Reacciones en que participan e- y H+
Reacciones electroquímicas, dependen del Eh y del pH.
Cu2+ + H2O + 2 e- <=> Cu2O + 2 H+
E = 0.20 + 0.06 pH
2 CuO + 2 H+ + 2 e- <=> Cu2O + 2 H2O
E = 0.67 - 0.06 pH
Cu2O + 2 H+ + 2 e- <=> 2 Cu + 2 H2O
E = 0.47 - 0.06 pH
Eh
pH
0.52
0.34
0.15
Cu+
Cu °
Cu2+
Cu °
Cu2+
Cu+
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama17
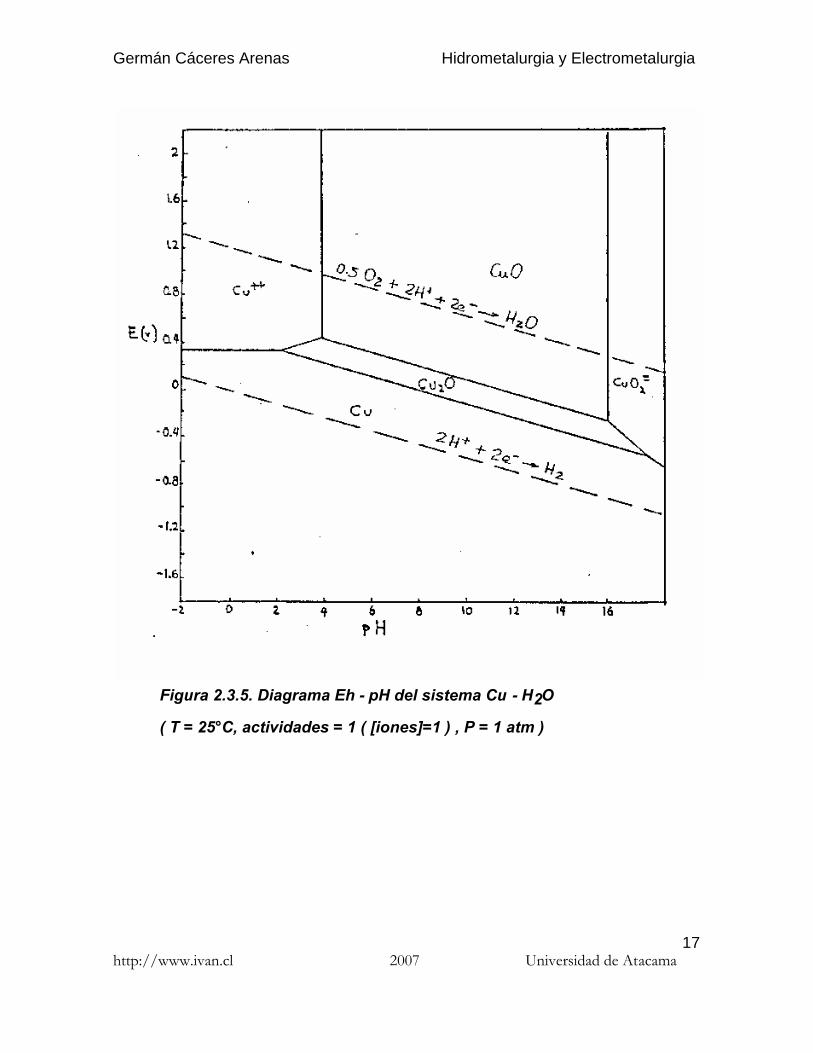
Figura 2.3.5. Diagrama Eh - pH del sistema Cu - H2O
( T = 25°C, actividades = 1 ( [iones]=1 ) , P = 1 atm )

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama18
2.3.4. Interpretación del diagrama Eh-pH del cobre enagua
2.3.4.1. Interpretación
La disolución de los óxidos simples de cobre es termodinámicamente posibleen el dominio ácido y en presencia de oxidantes. La tenorita (CuO) sólonecesita condiciones de pH, mientras que en esas condiciones, la cuprita(Cu2O) necesita además la presencia de un agente oxidante ( iones Fe3+, O2,u otros ).
Las reacciones son :
CuO + 2 H+ <=> Cu2+ + H2O
yCu2O + 2H+ <=> 2 Cu2+ + H2O + 2 e-
Ox. + 2 e- <=> Red.
Cu2O + 2 H+ + Ox. <=> 2 Cu2+ + Red. + H2O
en que Ox. representa un agente oxidante cualquiera.
En forma inversa, al estar el Cu2+ en solución, y para poder permanecer enella, necesita de una cierta acidez libre, evitándose de esta manera suposterior precipitación a pH >4.
A través de todo el rango de pH, el cobre metálico es termodinámicamenteestable estando en contacto con agua, pero en ausencia de oxigeno (u otrooxidante).
La precipitación electrolítica se puede realizar aplicando al cátodo un potencialinferior a 0.34 V. De esta forma el cobre Cu2+ se reduce en el cátodo deacuerdo a :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama19
Cu2+ + 2e- <=> Cu° (cátodo)
2.3.4.2. Influencia de la concentración de los iones
El diagrama ha sido trazado para actividades unitarias. Si se traza para otrasactividades, por ejemplo 10-6 (diagramas de corrosión), aumentan el dominio deestabilidad de los iones, pero el diagrama mantiene su forma produciéndose sólodesplazamientos paralelos de las rectas que limitan a estos iones.
Por ejemplo, reemplazando en (13) el valor numérico de [Cu2+]=10-6,se llega a :
=> pH = 6.95
En este caso, el pH de precipitación de CuO aumenta hasta 6.95; se amplia elrango de estabilidad de los iones en solución.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama20
3.1. Cinética de Reacciones
3.1.1. Introducción
El hecho de que una reacción sea termodinamicamente posible (G<0), no essuficiente para predecir si la reacción va a pasar en una escala de tiemporazonable. Eso depende de la cinética de la reacción. Este factor es muyimportante para la concepción y la evaluación de la rentabilidad económica detodos los procesos hidrometalurgicos. También en las plantas en operación,optimizar la cinética resulta generalmente en un mejoramiento del proceso.
De esta forma, los productos finales de una operación hidrometalúrgica van aestar condicionados generalmente por condiciones de tipo cinético.
Por ejemplo, la lixiviación de la calcopirita y otros sulfuros con sulfato férrico estermodinamicamente factible ( Existen discrepancias respecto a la reacción consulfato férrico, también occurre la reacción CuFeS2 + 4 Fe3+ => Cu2+ + 5 Fe3+ +2 S° ) :
Lixiviación

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama21
CuFeS2 + 8 H2O => Cu2+ + Fe3+ + 2 SO42- + 17 e- + 16 H+
17 Fe+3 +17 e- => 17 Fe2+---------------------------------------------------------------------------------G<0CuFeS2 + 16 Fe+3 + 8 H2O => Cu2+ + 17 Fe2+ + 2 SO42- + 16 H+
Pero en la práctica, después de 100 días de lixiviación, sólo se alcanza a poner ensolución un 5 % de la calcopirita, 50 % de calcosina y 80% de covelina.
3.1.2. Definiciones
3.1.2.1. Reacción homogénea - heterogénea
Reacción Homogénea : Reacción química u electroquímica en la cual todos losproductos y reactantes pertenecen a una sola y misma fase.
Ejemplo : MnO4- + 5 Fe2+ + 8 H+ <=> Mn2+ + 5 Fe3+ + 4 H2O
Reacción Heterogénea : Una reacción es heterogénea si tiene lugar en dos omás fases.
Ejemplo : NaCl (sólido) <=> Na+ + Cl- (solución)
3.1.2.2. Velocidad de una reacción
En general, las reacciones químicas se pueden escribir :
A + B => C + Do A => Bo A => B => Co

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama22

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama23
La velocidad de una reacción (v) es el número de moles transformados por unidadde tiempo del componente i involucrado en la reacción química : .
La velocidad se puede definir de varias formas :
(a) Basada en la unidad de volumen de fluido reaccionante
(b) Basada en la unidad de volumen del reactor
(c) Basada en la unidad de superficie interfacial en un sistema de dosfluidos o basada en la unidad de superficie del sólido en los sistemaslíquido - sólido (reacción heterogénea):
(d) Basada en la unidad de masa del sólido en los sistemas fluido-sólido
dtdNi

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama24
3.1.3. Etapas de una reacción
En general, los sistemas hidrometalúrgicos están caracterizados por sistemasheterogéneos, es decir , sus reacciones tienen lugar en una interface en la cualocurre transferencia de materia de una fase a la otra.
Figura 3.1.1. Esquema de una reacción de lixiviación condisolución completa del mineral.
Las reacciones heterogéneas son controladas por la velocidad de la reacciónquímica propiamente tal o por la transferencia de masa de los diversos reactanteshacia la superficie de contacto de los dos fases. En la figura 3.1.1. se muestra unmodelo simplificado de lixiviación, con disolución completa del mineral. Este casorepresenta la disolución de especies puras que no forman residuos sólidos comoproducto de la reacción, tales como el oro en cianuro, la chalcantita en ácidosulfúrico,...

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama25
Las etapas principales de una reacción son :
(1) Transporte de masa de los reactantes gaseosos a través de la interfasegas - líquido y posterior disolución (cuando es el caso).
(2) Transporte de masa de los reactantes a través de la capa límitesolución - sólido, hacia la superficie del sólido.
(3) Reacción química o electroquímica en la superficie del sólido,incluyendo adsorción y desorpción en la superficie del sólido y/o através de la doble capa electroquímica.
(4) Transporte de masa de los especies producidos a través de la capalímite hacia el seno de la solución.
La etapa controlante de una reacción es la de velocidad más lenta ( R.D.S. : RateDetermining Step). El control de la reacción global puede ser :
- Por transporte de masa ( etapa 1, 2 o 4 )- Por reacción química (etapa 3)- Mixto
Las reacciones homogéneas son generalmente más rápidas que las reaccionesheterogéneas, ya que necesitan transporte de masa en una sola fase y que lasespecies en solución reaccionan rápidamente. Por otra parte, las reaccionesheterogéneas implican el transporte de masa a través del límite entre dos fases, loque a veces es la etapa controlante de las reacciones. Las reacciones másimportantes en hidrometalurgía son heterogéneas, y a veces son controladas porel transporte de masa ( difusión).
En la figura 3.1.2. se muestra un modelo simplificado de lixiviación, con formaciónde una capa de residuo poroso. Este caso es tal vez el más frecuente enlixiviación. La reacción de disolución de las especies sulfuradas de cobre coniones férricos, corresponde a este caso. La lixiviación de minerales de baja ley, enlas que el material estéril o ganga constituye la fracción mayoritaria, puedenconsiderarse también en ese grupo. La "capa" que se forma representa el estérildel mineral, mientras la disolución se propaga hacia el interior de la partícula.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama26
Figura 3.1.2. Esquema de una reacción de lixiviación conformación de una capa porosa.
En este modelo, hay dos etapas adicionales :
(5) Difusión del reactivo a través de la capa sólida producida por la reacción(producto poroso)
(6) Transporte de masa de las especies solubles, productos de la reacción,a través de la capa sólida formada, hacia el seno de la solución.
5
6

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama27
3.1.4. Modelización de la cinética
¿ Que es un modelo ? Algo que se acerca a la realidad, una representaciónnecesariamente imperfecta de ésta.
La ecuación general utilizada para describir la cinética de una reacción
es :
(1)
Ci : Concentración de la especie i (sólo reactantes)k : Constante de velocidadn : orden de la reacción (= n1 + n2 + n3 +...)
En el caso de que exista sólo un reactante en solución, la ecuación (1) sesimplifica a :
(2)
C : concentración de la especie reactante ( si hay una solaespecie reactante en solución )
k : constante de velocidadn : orden de la reacciónt : tiempo
La velocidad de cualquier reacción es proporcional a la concentración delreactante, elevado al orden de la reacción. Esta expresión puede ser integradapara mostrar la evolución de la concentración con el tiempo para una ciertaconstante de velocidad y un cierto orden de reacción. La mayoría de la reacciones
C
C
n
n
kdtCd
dtCd
dtCVd
VdtdN
Vv
kv
11

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama28
en hidrometalurgia son de primer orden. Si la reacción es de primer orden ( n = 1),entonces se llega a :
e ktet CC 0 (3)
o
303.2log
0
ktCC
La constante de velocidad ( k ) de cualquier reacción puede ser expresada por laecuación de ARRHENIUS :
(4)
Ea : Energía de activación de la reacciónR : Constante de los gases (8.314 J/molK)T : Temperatura absoluta (K)A : Constante relacionada con la frecuencia de colisiones de
las especies en solución.
Experimentalmente, el valor de Ea de una reacción puede ser determinado en undiagrama log k versus 1/T, calculando la pendiente de la recta ( Ea/R ) (fig. 3.1.3).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama29
Figura 3.1.3. Determinación experimental de la energía deactivación de una reacción.
Control químico <=> Ea > 40 kJ/mol Control difusional <=> Ea = 5 a 20 kJ/mol
Practicamente, eso implica que la velocidad de una reacción controladaquímicamente se multiplica por 2 cuando la temperatura aumenta de 10 °C.
Ejemplo :
Una reacción química a 20 °C tiene una constante de velocidad k1=v
[mol/s.cm2], y a 40°C la constante de velocidad aumenta a k2 = 4v
(v=const). Determinar su energía de activación.
T (°C) T(K) K ln K 1/T -Ea/R
20 293 v ln v 0.00341
40 313 4v ln 4 + ln v 0.00319
-
6,300
=> Ea = 6300 * 8.314 [J/molK] / 1000 [J/kJ] = 52.3 kJ/mol
=> Control químico (Ea > 40 kJ/mol)
ln k
1/T
Ea/R
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama30
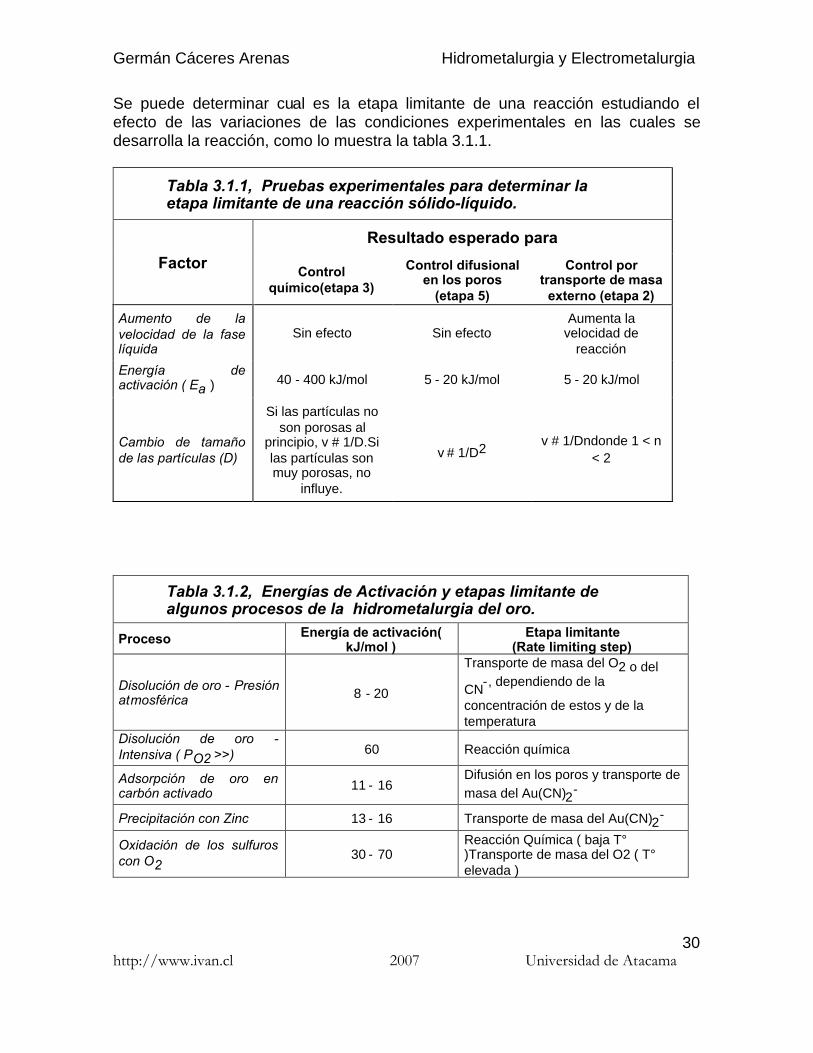
Se puede determinar cual es la etapa limitante de una reacción estudiando elefecto de las variaciones de las condiciones experimentales en las cuales sedesarrolla la reacción, como lo muestra la tabla 3.1.1.
Tabla 3.1.1, Pruebas experimentales para determinar laetapa limitante de una reacción sólido-líquido.
Resultado esperado para
Factor Controlquímico(etapa 3)
Control difusionalen los poros
(etapa 5)
Control portransporte de masa
externo (etapa 2)
Aumento de lavelocidad de la faselíquida
Sin efecto Sin efectoAumenta la
velocidad dereacción
Energía deactivación ( Ea ) 40 - 400 kJ/mol 5 - 20 kJ/mol 5 - 20 kJ/mol
Cambio de tamañode las partículas (D)
Si las partículas noson porosas al
principio, v # 1/D.Silas partículas sonmuy porosas, no
influye.
v # 1/D2 v # 1/Dndonde 1 < n< 2
Tabla 3.1.2, Energías de Activación y etapas limitante dealgunos procesos de la hidrometalurgia del oro.
Proceso Energía de activación(kJ/mol )
Etapa limitante(Rate limiting step)
Disolución de oro - Presiónatmosférica 8 - 20
Transporte de masa del O2 o del
CN-, dependiendo de la
concentración de estos y de latemperatura
Disolución de oro -Intensiva ( PO2 >>) 60 Reacción química
Adsorpción de oro encarbón activado 11 - 16
Difusión en los poros y transporte demasa del Au(CN)2-
Precipitación con Zinc 13 - 16 Transporte de masa del Au(CN)2-
Oxidación de los sulfuroscon O2
30 - 70Reacción Química ( baja T°)Transporte de masa del O2 ( T°elevada )

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama31
3.1.5. Difusión
3.1.5.1. Introducción
La difusión en fase homogénea es la etapa limitante más común en una reacciónhidrometalurgica. Básicamente, la difusión es un proceso que tiende a igualarconcentraciones dentro de una fase. El potencial que provoca la difusión es elgradiente de concentración dentro de la fase, tal como, por ejemplo, el potencialque provoca la transferencia de calor es el gradiente de temperatura.
Cuando empieza una reacción (por ejemplo consumo de un reactante en lasuperficie de un sólido), un perfil de concentración se desarrolla poco a poco cercade la interfase sólido - líquido (ver fig. 3.1.4 ).
C = C( t, x )
En reacciones de disolución o precipitación de un sólido, la difusión a través deuna zona adyacente a la interfase sólido - líquido puede ser controlante de lavelocidad. En condiciones de agitación constante, el espesor de esta zonapermanece constante y pronto se obtiene una condición de estado estacionario enla cual la cantidad de material que entra en la zona es igual a la que deja la zona.
vconsumo del reactivo = vaporte por difusióny
C = C( x )
Figura 3.1.4, Perfilde concentración deun reactante ( quese consume) cercade la interfase sólido- líquido en funcióndel tiempo y de ladistancia.
ConcentraciónReactante
Distancia de la interfase(x)
00
Sólido
C°t=0
t
Cs

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama32
3.1.5.2. Primera ley de FICK
La primera ley de FICK relaciona la cantidad de material que difunde por unidadde tiempo en una dirección perpendicular a un plano de referencia de área unitariacon el gradiente de concentración de este:
(5)
J : Flujo de material a través de un plano de referencia. [moles cm-2 s-1 ]
C : Concentración de la especie disuelta [ moles cm-3 ]x : Coordenada de posición (medida perpendicularmente al
plano de referencia)D : Coeficiente de difusión de la especie considerada en
solución acuosa. En el sistema CGS, D tiene lasdimensiones [cm2/s]. En la mayoría de los sistemashidrometalúrgicos se introduce poco error al considerar elcoeficiente de difusión constante e independiente de laconcentración. ( Valor aprox. = 10-6 cm2/s )
3.1.5.3. Capa de difusión
La capa de difusión es una delgada capa de líquido adyacente a la interfase sólido- líquido y que prácticamente se adhiere al sólido, debido a que es necesario quela velocidad de la solución sea nula en la interfase con el sólido. El transporte demasa de especies disueltas a través de esa capa de líquido adherido al sólido sehace por difusión.
NERNST propone la siguiente aproximación de la ecuación de Fick (5), porlinearización en x=0 :
(6)
C0 : Concentración de la especie en el seno de la soluciónCs : Concentración de la especie en la superficie del sólidox : Espesor de la capa de difusión de Nernst
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama33
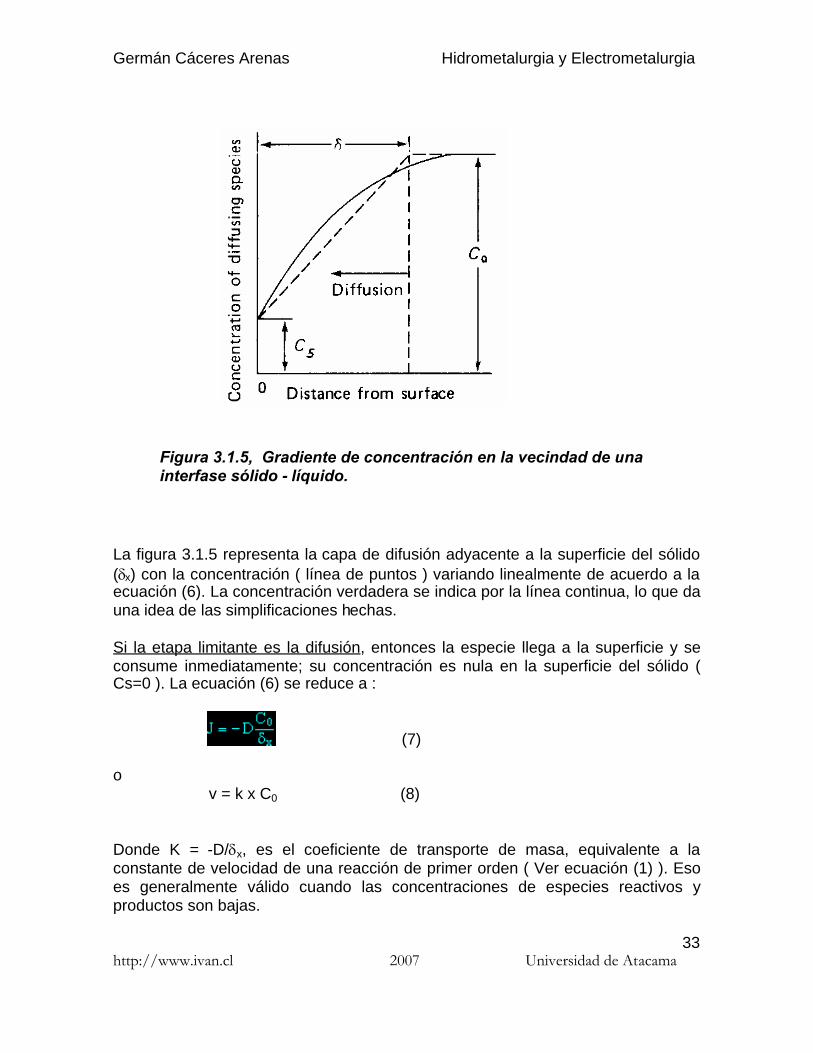
Figura 3.1.5, Gradiente de concentración en la vecindad de unainterfase sólido - líquido.
La figura 3.1.5 representa la capa de difusión adyacente a la superficie del sólido(x) con la concentración ( línea de puntos ) variando linealmente de acuerdo a laecuación (6). La concentración verdadera se indica por la línea continua, lo que dauna idea de las simplificaciones hechas.
Si la etapa limitante es la difusión, entonces la especie llega a la superficie y seconsume inmediatamente; su concentración es nula en la superficie del sólido (Cs=0 ). La ecuación (6) se reduce a :
(7)
ov = k x C0 (8)
Donde K = -D/x, es el coeficiente de transporte de masa, equivalente a laconstante de velocidad de una reacción de primer orden ( Ver ecuación (1) ). Esoes generalmente válido cuando las concentraciones de especies reactivos yproductos son bajas.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama34
El transporte de masa a través de la capa de difusión puede ser aumentado :
(1) Reduciendo el espesor de la capa de difusión.
(2) Aumentando el gradiente de concentración, esto es aumentando laconcentración de la solución (C0) .
(3) Aumentando la temperatura de la solución.
(4) Aumentando la superficie de contacto.
El espesor de la capa de difusión depende de la rugosidad del sólido, de laviscosidad de la solución, de la velocidad de agitación y del grado de turbulencia yfuerzas de cizalle. x disminuye cuando aumenta el grado de turbulencia.
Pobre agitación => x > 0.5 mm ( ej.: lixiviación en pilas )Buena agitación => x = +/- 0.01 mm
Un aumento de temperatura no necesariamente aumenta la velocidad de unproceso global ya que , por ejemplo, la solubilidad de los gases disminuye con latemperatura.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama35
3.1.5.4. Difusión a través de un producto poroso
La difusión a través de la capa de sólido que se va formando como producto de lareacción sobre el núcleo reaccionante y/o a través de la capa de partículas inerteses un factor importante en prácticamente todos los procesos heterogéneos.Cuando los poros son grandes, que es el caso que interesa en sistemashidrometalúrgicos, es válida la ley de Fick expresada en este caso como :
(9)
Donde C es la concentración en la solución y Df es el COEFICIENTE DEDIFUSIÓN EFECTIVO, dado por :
(10)
D : Coeficiente de difusión de la especie considerada ensolución acuosa
E : Porosidad de la capa de producto (varia de 0 a 1) : Tortuosidad de la capa de producto ( En general, fluctúa
entre 2 y 10 )
3.1.6. Transporte de masa en el seno de la solución
Si el sistema es bien agitado (agitador, convección,...), el transporte de masa esmuy eficiente y no limita la velocidad de la reacción. La solución se considerahomogénea ( C, pH, ... ).Sin embargo, algunos procesos, como la lixiviación en pila, no pueden siempre seroperados para mantener una solución homogénea.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama36
3.1.7. Absorpción de gases en líquidos
La termodinámica determina la concentración de un gas disuelto en solucióndespués de un tiempo de contacto gas - líquido suficiente para alcanzar elequilibrio. Por ejemplo, si PO2 = 0.21 atm, [O2]disuelto = 8 mg/l (25 °C).
La absorción de los gases en líquidos puede ser la etapa limitante si hay un granconsumo de oxigeno y cuando la cinética de reacción sólido - solución es rápida (ej. : Oxidación de los sulfuros a presión o biológicamente ).
La velocidad de absorción de gases en un líquido depende de :
(1) Superficie de contacto gas - líquido efecto positivo(Mayor cuando hay muchas burbujas pequeñas )(2) Velocidad relativa de las burbujas efecto positivo(3) Presión parcial del gas efecto positivo(4) Grado de agitación efecto positivo(5) Temperatura efecto negativo
3.1.8. Influencia de las características de laspartículas
3.1.8.1. Tamaño de las partículas
La superficie de reacción aumenta cuando disminuye el tamaño de las partículas.
3.1.8.2. Forma y textura de las partículas
El efecto de la forma de las partículas es poco importante en hidrometalurgía, ymuy difícil de modelizar.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama37
3.1.8.3. Factores mineralógicos
La reactividad de los granos de minerales es fuertemente afectada por factorestales que orientación cristalina, inclusiones, dislocaciones e impurezas.
3.1.8.4. Porosidad
La velocidad de cualquier reacción aumenta con la porosidad, ya que la difusiónes más fácil y/o hay una mayor superficie de reacción.
3.1.8.5. Efectos galvánicos
Cuando dos minerales conductores están en contacto eléctrico en una solución, elde potencial más electronegativo se va a disolver preferentemente.
Figura 3.1.6. Efecto galvánico en la disolución de arsenopirita encontacto con pirita.
3.1.8.6. Efecto de las especies que compiten con la especie valiosa

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama38
Minerales otros que el mineral valioso, si están presentes en la mena, puedeneventualmente consumir los reactivos de lixiviación, disminuyendo susconcentraciones y la velocidad de reacción. Por ejemplo, los carbonatosconsumen el ácido sulfúrico en la lixiviación de cobre y los sulfuros consumen elcianuro en la disolución de oro.
3.2. Práctica de la lixiviación
3.2.1. Sistemas de lixiviación
En general, la práctica industrial de la lixiviación presenta diferentes sistemas deoperación que se seleccionan de acuerdo a factores técnicos y económicos en elanálisis de un proyecto, algunos de los cuales son :
- ley de la especie de interés a recuperar
- reservas de mineral
- caracterización mineralógica y geológica
- comportamiento metalúrgico
- capacidad de procesamiento
- costos de operación y de capital
- rentabilidad económica, ...
Una forma de clasificar los métodos de lixiviación es :
Lixiviación de lechos fijos :
- in situ, in place
- en botaderos
- en pilas
- en bateas
Lixiviación de pulpas :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama39
- por agitación, a presión ambiente
- en autoclaves
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama40
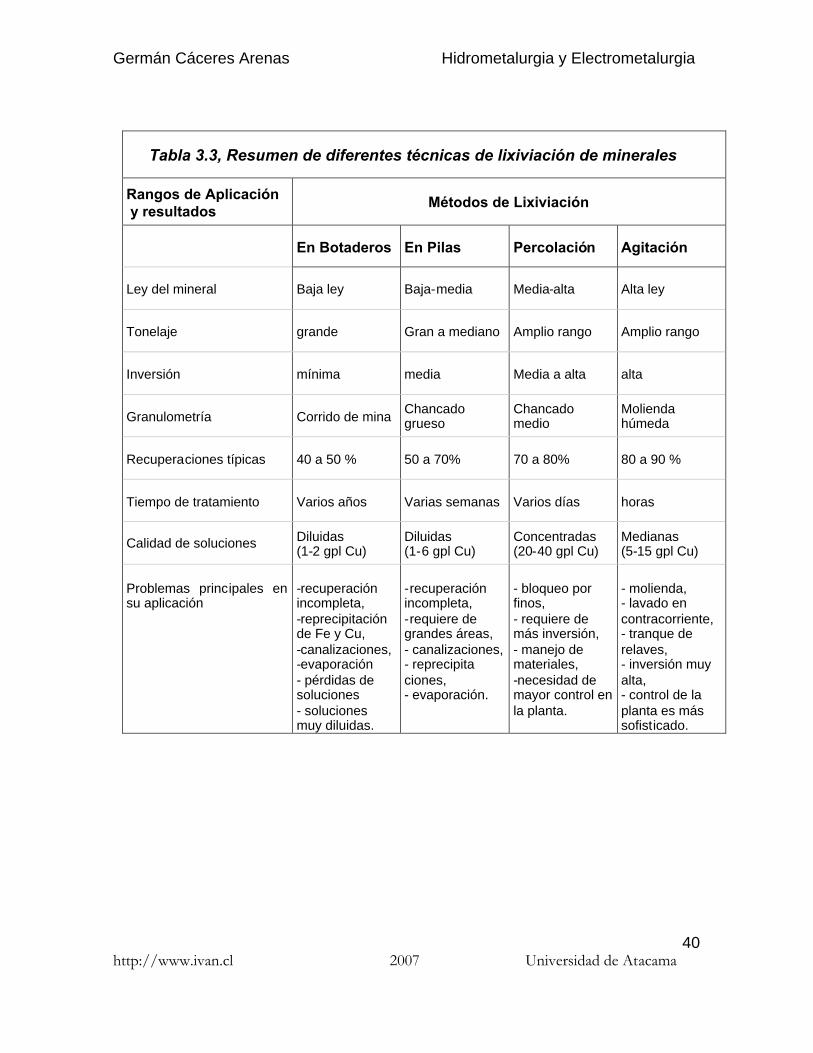
Tabla 3.3, Resumen de diferentes técnicas de lixiviación de minerales
Rangos de Aplicacióny resultados
Métodos de Lixiviación
En Botaderos En Pilas Percolación Agitación
Ley del mineral Baja ley Baja-media Media-alta Alta ley
Tonelaje grande Gran a mediano Amplio rango Amplio rango
Inversión mínima media Media a alta alta
Granulometría Corrido de mina Chancadogrueso
Chancadomedio
Moliendahúmeda
Recuperaciones típicas 40 a 50 % 50 a 70% 70 a 80% 80 a 90 %
Tiempo de tratamiento Varios años Varias semanas Varios días horas
Calidad de soluciones Diluidas(1-2 gpl Cu)
Diluidas(1-6 gpl Cu)
Concentradas(20-40 gpl Cu)
Medianas(5-15 gpl Cu)
Problemas principales ensu aplicación
-recuperaciónincompleta,-reprecipitaciónde Fe y Cu,-canalizaciones,-evaporación- pérdidas desoluciones- solucionesmuy diluidas.
-recuperaciónincompleta,-requiere degrandes áreas,- canalizaciones,- reprecipitaciones,- evaporación.
- bloqueo porfinos,- requiere demás inversión,- manejo demateriales,-necesidad demayor control enla planta.
- molienda,- lavado encontracorriente,- tranque derelaves,- inversión muyalta,- control de laplanta es mássofisticado.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama41
3.2.2. Lixiviación in situ - in place
La lixiviación IN PLACE se refiere a la lixiviación de residuos fragmentadosdejados en minas abandonadas, mientras la lixiviación IN SITU se refiere a laaplicación de soluciones directamente a un cuerpo mineralizado.
Por lo general, estas operaciones presentan actualmente un gran interés por losbajos costos de inversión y operación que se requieren, y que posibilitan recuperarvalores metálicos que de otra manera no podrían ser extraídos. Los bajos costosson consecuencia de evitar o al menos disminuir los costos de extracción minera,el transporte del mineral a la planta y de los desechos finales del proceso, y laconstrucción de una planta de lixiviación. Generalmente, la recuperación es baja (< 50% ).
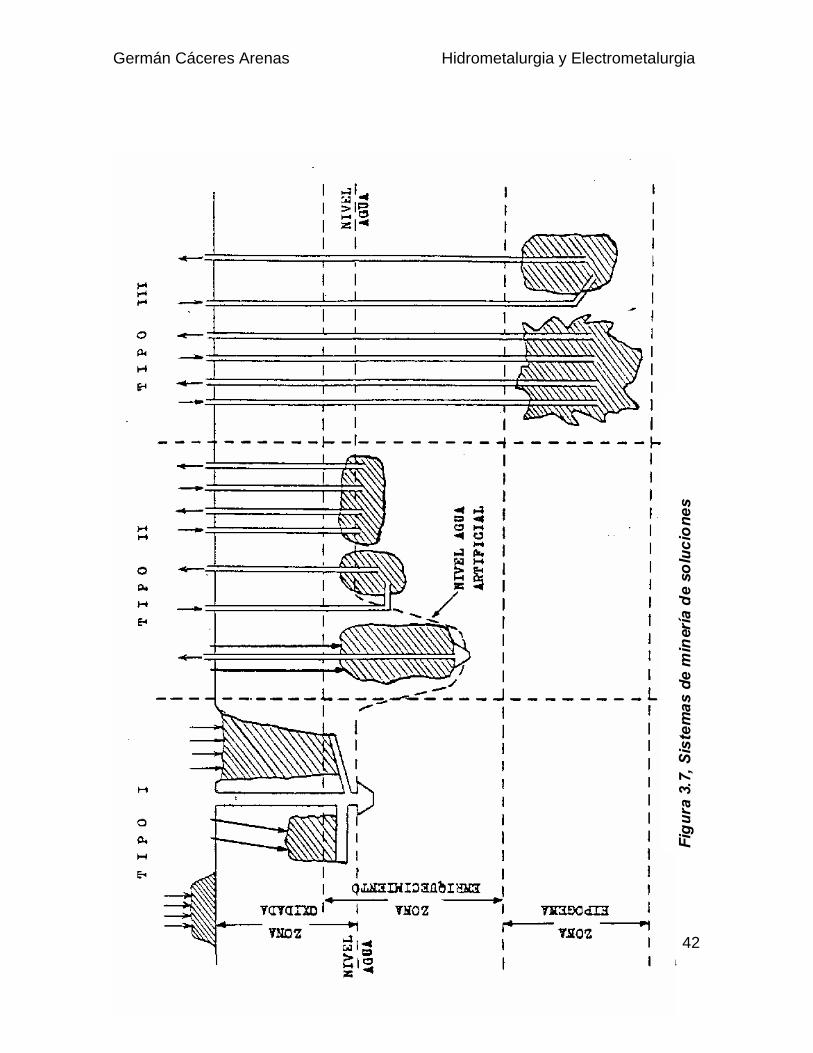
Dependiendo de la zona a lixiviar, que puede ser subterránea o superficial, sedistinguen tres tipos de lixiviación in situ, como se puede visualizar desde Fig 3.7.
Tipo I : Se trata de la lixiviación de cuerpos mineralizados fracturados situadoscerca de la superficie, sobre el nivel de las aguas subterráneas. Puedeaplicarse a minas en desuso, en que se haya utilizado el "block caving",o que se hayan fracturado hidráulicamente o con explosivos (IN PLACELEACHING).
Tipo II : Son lixiviaciones IN SITU aplicadas a yacimientos situados a ciertaprofundidad bajo el nivel de aguas subterránea, pero a menos de 300 -500 m de profundidad. Estos depósitos se fracturan en el lugar y lassoluciones se inyectan y se extraen por bombeo.
Tipo III : Se aplica a depósitos profundos, situados a más de 500 m bajo elnivel de aguas subterráneas
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama42
Fig
ura
3.7,
Sis
tem
asde
min
ería
de
solu
cio
nes

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama43
3.2.3. Lixiviación en botaderos (Dump leaching)
Esta técnica consiste en lixiviar lastres, desmontes o sobrecarga de minas de tajoabierto, los que debido a sus bajas leyes (por ej. < 0.4% Cu) no pueden sertratados por métodos convencionales. Este material, generalmente al tamaño "runof mine" es depositado sobre superficies poco permeables y las solucionespercolan a través del lecho por gravedad. Normalmente, son de grandesdimensiones, se requiere de poca inversión y es económico de operar, pero larecuperación es baja (por ej. 40-60 % Cu) y necesita tiempos excesivos paraextraer todo el metal. Las soluciones se alimentan generalmente por aspersión.
Normalmente la lixiviación en botaderos es una operación de bajo rendimiento(pero también de bajo costo). Entre las diferentes razones para ello se puedemencionar :
- Gran tamaño de algunas rocas (> 1 m).
- Baja penetración de aire al interior del botadero.
- Compactación de la superficie por empleo de maquinaria pesada.
- Baja permeabilidad del lecho y formación de precipitados (yeso, ...)
- Excesiva canalización de la solución favorecida por la heterogeneidadde tamaños del material en el botadero.
Figura 3.8,Botaderos.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama44
3.2.4. Lixiviación en batea (por percolación)
Esta técnica consiste en contactar un lecho de mineral con una solución acuosaque percola e inunda la batea o estanque. Un esquema de equipo empleado enlixiviación en batea se presenta en Fig. 3.9.
Los minerales a tratar por este método deben presentar contenidos metálicos altoso muy altos, debiendo ser posible lixiviar el mineral en un período razonable (3 a14 días) y en trozos de tamaño medio con tonelajes suficientes de mineralpercolable en el yacimiento que permitan amortizar la mayor inversión inicial querequiere este tipo de proceso.
Ya que esos minerales no existen más, es una tecnología antigua actualmente endesuso.
Figura 3.9, Equipos de lixiviación en batea.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama45
3.3. Lixiviación en pilas (heap leaching)
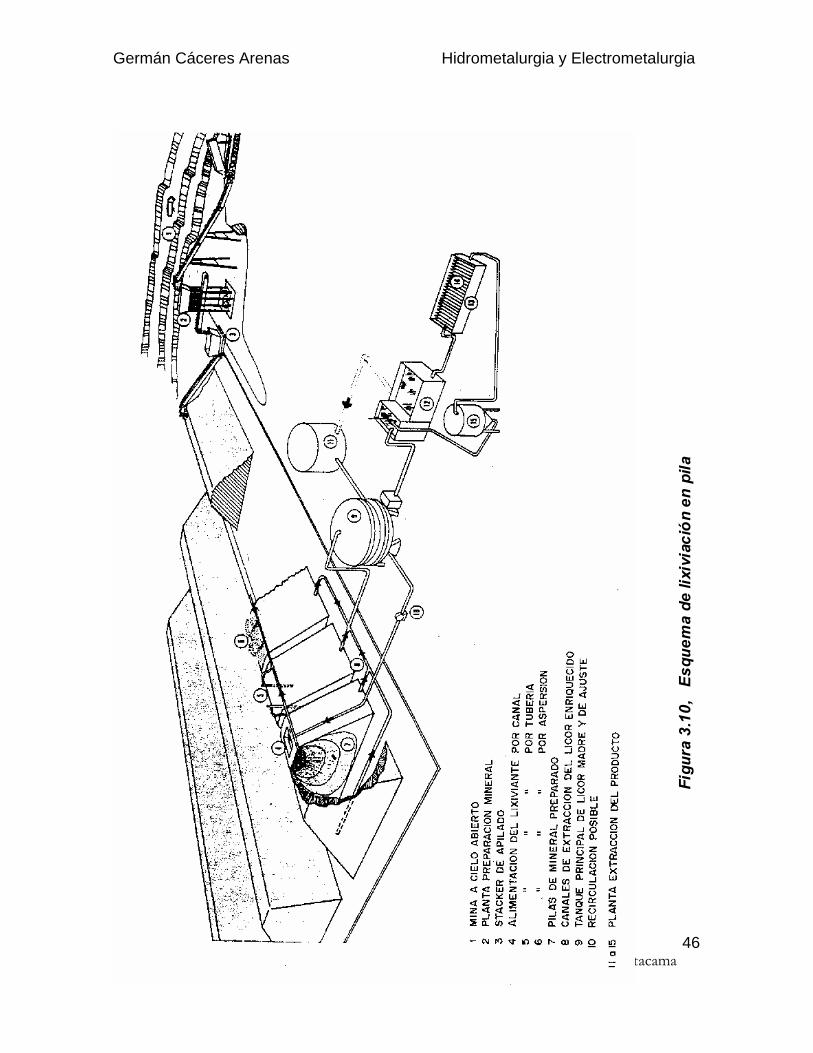
3.3.1. Descripción
El esquema general del proceso se puede observar en la Fig. 3.10. El mineralprocedente de la explotación, a cielo abierto o subterránea, debe ser ligeramentepreparado en una planta de chancado y/o aglomeración, para conseguir unagranulometría controlada que permita un buen coeficiente de permeabilidad.
Una vez preparado el mineral, se coloca en montones de sección trapezoidal yaltura calculada para proceder a su riego con la solución lixiviante. Tras percolar através de toda la pila, se recolectan los líquidos enriquecidos (solución rica) que sellevan a la planta de proceso de recuperación de la sustancia mineral (sal o metal).Las aguas sobrantes del proceso vuelven a ser acondicionadas para serrecicladas hacia las pilas. También en algunos casos es preciso añadir aguanueva, para reponer las fuertes pérdidas de evaporación del circuito.
Se denomina cancha de lixiviación a la superficie de apoyo de la pila donde secoloca la impermeabilización. Cuando la cancha es recuperada para reutilizarlacon un nuevo mineral se trata de lixiviación en PILAS DINAMICAS, mientras que siel terreno no es recuperado y, por lo tanto, el mineral agotado queda en eldepósito como nueva base para otra pila, se está en la lixiviación en PILASESTATICAS o PERMANENTES.
La solución rica (S.R. o P.L.S. : pregnant leach solution) es generalmente impura ydiluida y debera ser purificada y concentrada antes de recuperar el metal. En lahidrometalurgia del cobre, eso se realiza mediante la extracción por solventeseguida por la electrodepositación del cobre. La solución rica sólo contiene 4 - 6g/l Cu y 1 - 2 g/l H2SO4 y es impura ( 5 g/l Fe, SiO2, Al2O3 coloides, sólidos ensuspensión, ...)
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama46
Figu
ra3.
10,
Esq
uem
ade
lixiv
iaci
ón
enpi
la

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama47
3.3.2. Construcción de las pilas
El diseño de las pilas debe tener en cuenta los siguientes factores :
- La calidad del patio o base de apoyo (impermeable)
- Las facilidades de riego y recolección o drenaje del efluente.
- La estabilidad de la pila seca y saturada en agua
- Los tanques (piscinas) de soluciones ricas y pobres
- La forma de apilamiento o deposición del material lixiviable(Compactación, homogeneidad, ...)
3.3.2.1. Preparación de la base de las pilas
Se necesita disponibilidad de amplias superficies de terreno relativamente llanas (menos de 10% de pendiente ). La cancha debe ser considerada con su sistema deimpermeabilización, para controlar las pérdidas de soluciones y evitarcontaminaciones del medio ambiente. El sistema consiste en :
- Una base firme y consolidada, debidamente preparada- Una capa de lecho granular sobre el que apoyar suavemente la lámina- La lámina o capa de impermeabilización- Un conjunto de drenaje o capa de recolección de líquidos- Una capa protectora del sistema
Generalmente, las membranas o láminas de impermeabilización del patio songeomembranas de origen sintético (láminas de plástico : polietileno de altadensidad o PVC de 1 a 1.5 mm o polietileno de baja densidad de 0.2 a 0.3 mm deespesor) pero también pueden ser materiales arcillosos compactados sobre elpropio terreno, hormigón, asfalto, etc.. Se pueden disponer de membranas osellados simples, dobles o triples, de acuerdo con el número de capasimpermeables o membranas que se hayan utilizado.
Una parte importante de la construcción de la pila es el sistema de recolección dela solución rica, que, en general consta de grava o material filtrante sobre la láminay tuberías perforadas drenantes de plástico.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama48
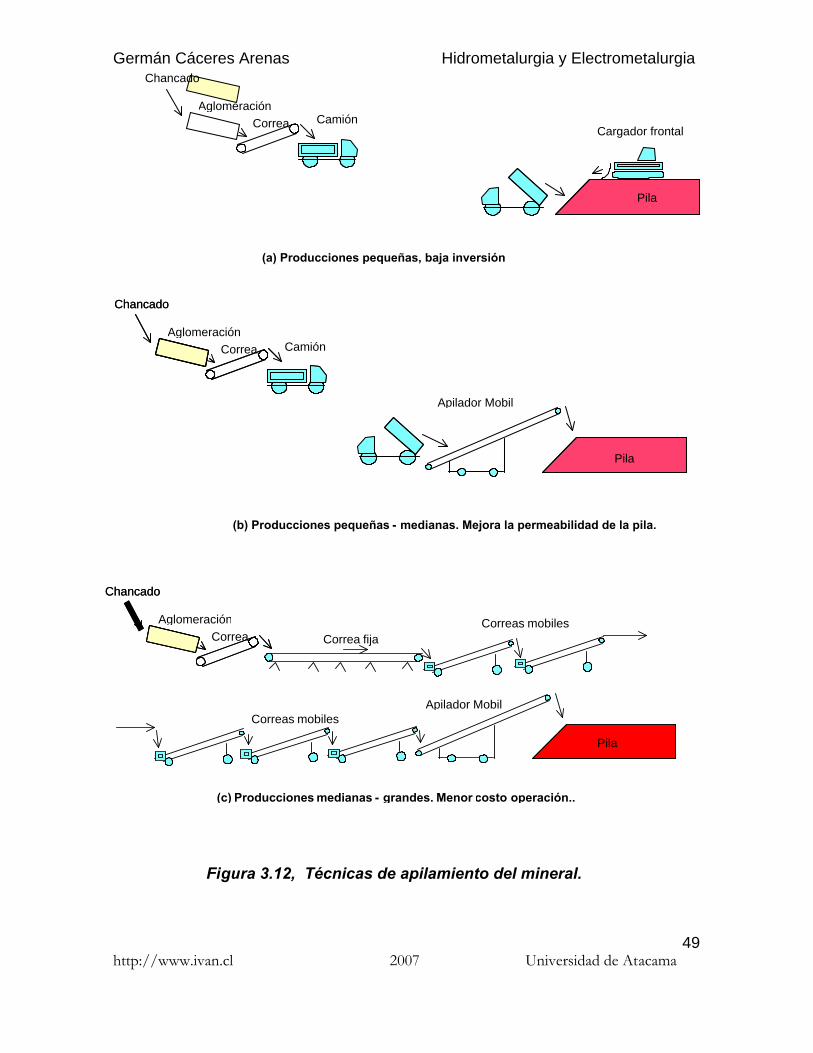
3.3.2.2. Técnicas de apilamiento del mineral
El uso de cargadores frontales y camiones (Fig. 3.12a) a sido desplazado, en losúltimos proyectos, por apiladores autopropulsados de bajo perfil de carga, comoen el caso de Lince, para tonelajes de 5000 a 10000 tpd (Fig. 3.12b). Paratonelajes mayores, 17500 tpd en Quebrada Blanca por ejemplo, se ha adoptado elsistema de correas cortas y móviles (grass hoppers) que se articulan flexiblementeen secuencia para transportar el mineral desde el aglomerador hasta el apiladormóvil que construye la pila (Fig. 3.12c). Para tonelajes aún mayores, como en ElAbra 125000 tpd, se implementaron otros tipos de equipos (apiladores sobreorugas, ...).
La altura de la pila fluctúa entre 2.5 m para sistemas de camión y cargador frontal,hasta 10 m para apiladores.
3.3.2.3. Riego de la pila
El riego de las pilas se puede realizar fundamentalmente por dos procedimientos :por aspersión o por distribución de goteo, este último siendo recomendable encaso de escasez de líquidos y bajas temperaturas (Fig 3.13 a y b). En la industria,se utiliza generalmente una tasa de riego del orden de 10 - 20 litros/h.m2.
El riego tiene que ser homogeneo.
(a) Por aspersión (b) Por goteo
Figura 3.13, Técnicas de irrigación de las pilas
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama49
Figura 3.12, Técnicas de apilamiento del mineral.
Chancado
AglomeraciónCorrea Camión
Cargador frontal
Pila
(a) Producciones pequeñas, baja inversión
Apilador Mobil
Chancado
AglomeraciónCorrea Camión
Chancado
Pila
(b) Producciones pequeñas - medianas. Mejora la permeabilidad de la pila.
Apilador Mobil
Chancado
AglomeraciónCorrea
Chancado
Correa fijaCorreas mobiles
Correas mobiles
Pila
(c) Producciones medianas - grandes. Menor costo operación..

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama50

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama51
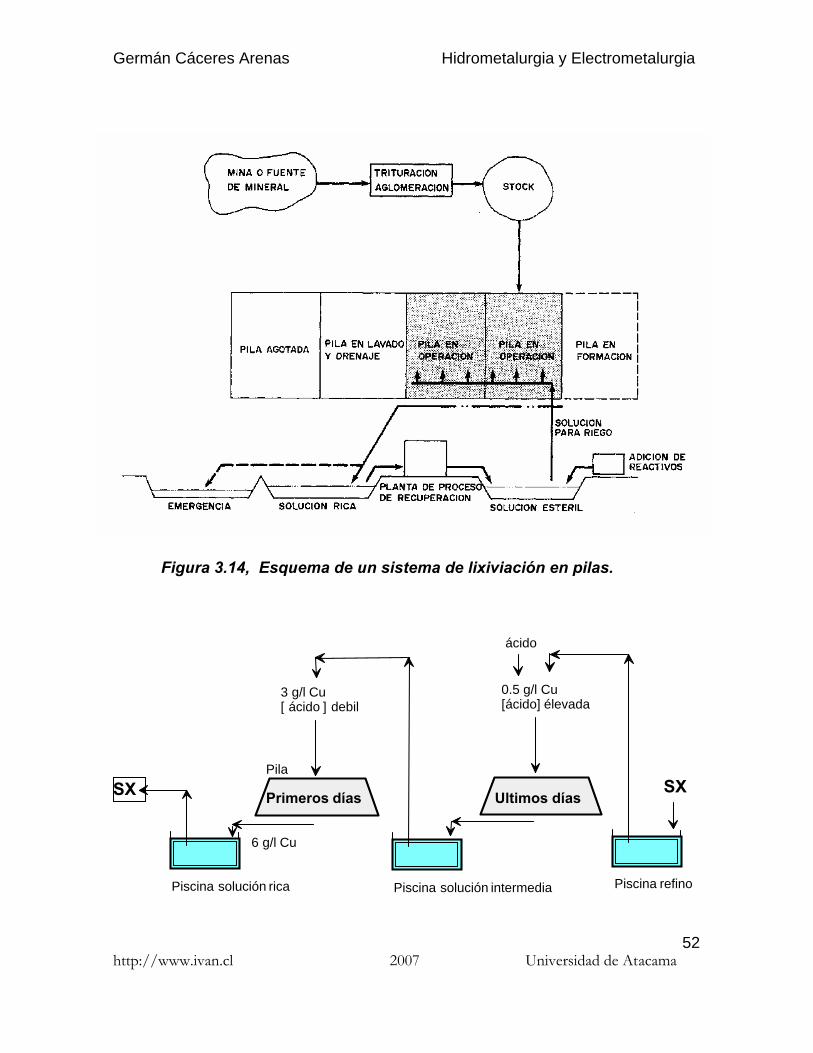
3.3.3. Operación de las pilas
Al inicio de la operación, se deben disponer como mínimo de dos pilas,comenzándose con el riego de la primera de ellas. En un principio se obtendrá unaalta concentración (Cmx) en la solución, que irá descendiendo hasta un valor pordebajo de la concentración media (Cmd) de diseño. En este momento se ponesimultáneamente en operación la segunda pila, con dos sistemas posibles :
a) Lixiviación de las dos pilas con obtención de una única solución ricafinal.
b) Lixiviación de la primera pila con producción de solución intermedia(pobre), que se recicla a la segunda pila nueva en donde se obtiene lasolución rica (Fig. 3.15). Este segundo sistema se generalizo, ya quepermite alargar el tiempo de lixiviación de las pilas y/o disminuir elcaudal de solución rica y entonces el tamaño de la planta de SX.
Cuando la primera pila alcanza el valor mínimo económico, se procede al lavadocon agua fresca y drenaje hasta el agotamiento, yendo esta solución al depósito opiscina de solución estéril para recirculación al sistema. Al mismo tiempo se poneen operación una nueva pila.
Según las disponibilidades de área, la pila agotada se puede cargar y transportar aun vertedero cercano (PILA DINAMICA o REMOVIBLE) o puede servir de basepara la formación de una nueva pila (PILA PERMANENTE).
La tendencia se desplaza al uso de pilas permanentes, para evitar los costosasociados a los movimientos de materiales residuales y aminorar las perdidas desolución por filtración a través de la lámina de plástico. Por ejemplo, en MantosVerde, se planea subir hasta 6 pisos de 3 m cada uno.
Si el tiempo de lixiviación no es suficiente, la recuperación baja. Es un problema,porque no es posible aumentar el tiempo sin aumentar el area de la cancha delixiviación.
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama52
Figura 3.14, Esquema de un sistema de lixiviación en pilas.
SX
0.5 g/l Cu[ácido] élevada
Ultimos días
Piscina refino
ácido
Piscina solución intermedia
3 g/l Cu[ ácido ] debil
Primeros días
6 g/l Cu
Piscina solución rica
SXPila

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama53
Figura 3.15, Reciclage de la solución lixiviante en contra -corriente.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama54
3.3.4. Chancado del mineral
El chancado del mineral debe cumplir con tres objetivos :
(1) Ser lo suficiente fino para que la mayoría de la especie metálica valiosaeste expuesta a la acción de la solución lixiviante.
Por ej. : 100 % bajo 3/4"
(2) No puede producir demasiado partículas finas para no alterar lapermeabilidad de la pila. ( Por convención, se llama fina toda partículabajo 100 mallas) “Material arcilloso”
Por ej. : partículas finas < 10%
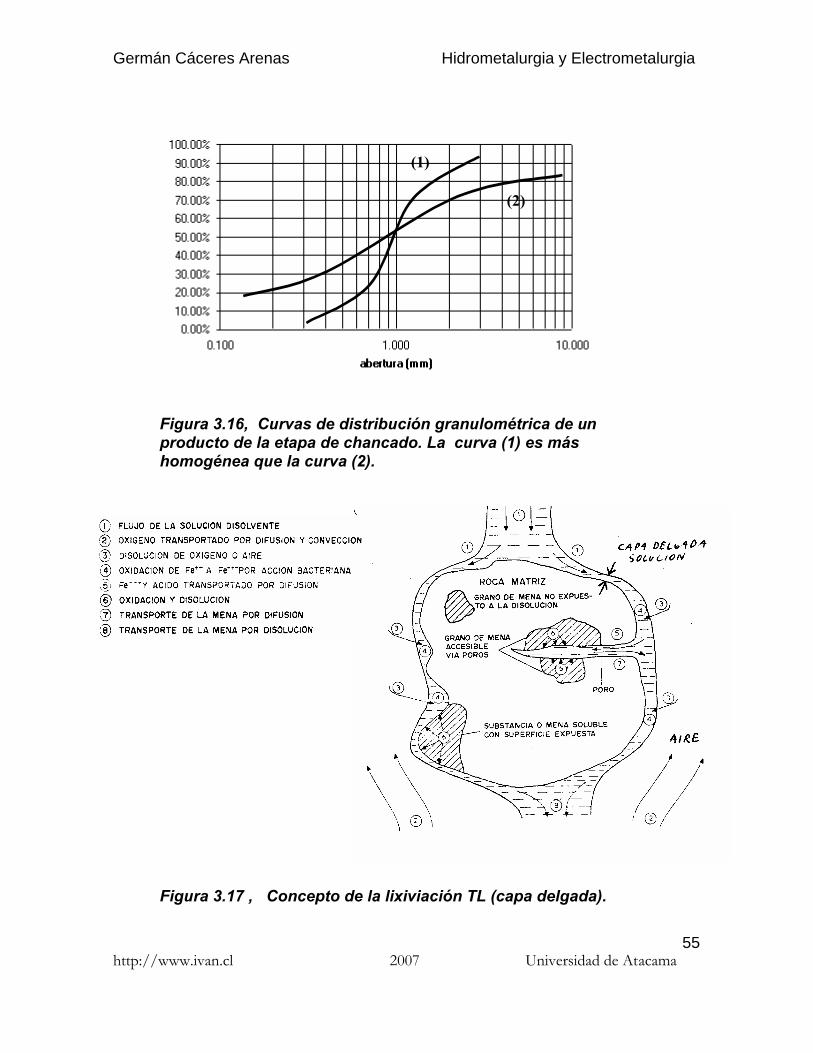
(3) El mineral chancado debe ser el más homogéneo posible, todas laspartículas siendo comprendidas en un estrecho rango de tamaño (Fig.3.16).
Tabla 3.4 , Escala de tamaños de partículas, en pulgadas, mallasTyler y mm.
16/16" 8/8" 4/4" 2/2" 1" 25.4 mm3/4" 19.0 mm FRACCION
8/16" 4/8" 2/4" 1/2" 12.7 mm3/8" 9.5 mm GRUESA
4/16" 2/8" 1/4" 6.4 mm3/16" 4.8 mm2/16" 1/8" 3.2 mm FRACCION...
MEDIA
4 mallas 4.75 mm...
FRACCIONFINA
100mallas
150 mm
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama55
Figura 3.16, Curvas de distribución granulométrica de unproducto de la etapa de chancado. La curva (1) es máshomogénea que la curva (2).
Figura 3.17 , Concepto de la lixiviación TL (capa delgada).
(1)
(2)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama56
3.3.5. Aglomeración
3.3.5.1. Lixiviación TL (Thin Layer)
Este concepto revolucionó la industria de lixiviación del cobre (Pudahuel 1980).Consiste en impedir la acumulación de solución en la pila. Al contrario de lalixiviación en bateas, la pila no se inunda. La solución escurre sobre las partículasde minerales, formando una capa delgada de líquido (Fig. 3.17)
3.3.5.2. Permeabilidad del lecho
Se necesita que el lecho de partículas que conforman la pila sea bien permeable,para asegurar una buena percolación y dispersión de la solución lixiviante en lapila, sin escurimiento preferencial. También, las pilas podrían derrumbarse sihabía acumulación de agua en la pila.
La permeabilidad del lecho de mineral es mayor si :
- Las partículas son de tamaño suficientemente grande
- No hay acumulación de partículas finas
- El tamaño de las partículas es homogéneo en la pila
- No hay compactación de la pila por maquinaria pesada
3.3.5.3. Proceso de aglomeración
De lo anterior, se deduce que se tiene que reducir la cantidad de partículas finasen la pila para aumentar su permeabilidad. Hoy en día, el proceso más empleadopara solucionar el problema de los finos es la aglomeración.
El procesos de aglomeración consiste en esencia en la adhesión de partículasfinas a las gruesas, que actúan como núcleos o la aglomeración de los finos conlos finos, a partir de la distribución de tamaños en la alimentación (Fig. 3.18).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama57
Figura 3.18, Concepto de aglomeración.
Aglomeración por humedad
El proceso más simple de aglomeración es humedecer el mineral con líquido,hasta alcanzar un contenido de agua que origine una tensión superficial suficiente,para que al colisionar las partículas entre sí, los finos se adhieran a los tamañosgruesos. Se forma un puente líquido entre las partículas.
El cálculo teórico de la humedad óptima es casi imposible y depende de muchosfactores como la mineralogía del mineral, contenido de finos, arcillas, ... Puede serde 6 - 8 % para minerales muy limpios, hasta un 10-15 % H2O para materialesnormales.
Aglomeración por adherentes
Existen ciertos materiales que pueden mejorar la adherencia de las partículasfinas a las gruesas, prolongando esta unión tanto en la manipulación como en laoperación de lixiviación.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama58
En el caso de la lixiviación del cobre, la aglomeración (o curado) se realiza con elmismo lixiviante ácido en un tambor rotatorio (Fig. 3.19). Primero, se humecta elmineral (+/- 4%) con agua o solución pobre (refino). Después, se agrega ácidosulfúrico concentrado (+/- 30 kg/t o 3%), este ácido ataca el mineral y generacompuestos cementantes entre las partículas.
Además de la aglomeración, ocurren reacciones químicas conduciendo a laformación de sulfatos de cobre y hierro (curado propiamente tal). Estas reaccionesson exotérmicas y generan mucha calor. Por ejemplo :
CuO + H2SO4 => CuSO4 + H2OCuSiO3 + H2SO4 => CuSO4 + SiO2 + H2O ........
Después de la aglomeración en el tambor rotatorio, se deja reposar el mineraldurante 24 h en la pila, para que se completen las reacciones químicas y que seadhieren entre sí las partículas en la misma pila.
En el caso de la aglomeración de minerales de oro y plata, los aglomerantes sonnormalmente el cemento y la cal. Estos reactivos mejoran la adhesión de laspartículas entre sí, y también aumentan el pH del mineral para su posteriorcianuración.
Equipos
El equipo más común es el tambor aglomerador. Consiste en un cilindroinclinado girando a baja velocidad, ocasionando el deslice (cascada) y laaglomeración del mineral previamente mojado con agua y/o adherentes (Fig 3.19).Se practica también la aglomeración en depósitos (stock), en cintastransportadoras y en platos.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama59
Figura 3.19, Aglomeración en tambor rotatorio.
3.3.6. Variables del proceso
Se pueden estudiar el efecto de varias variables operacionales sobre larecuperación del metal valioso y la cinética realizando pruebas de laboratorio encolumnas.
Las principales variables son :
La granulometría
La altura de la pila La tasa de riego [l/h.m2] o [l/h.T] La concentración en ácido de la solución de riego El tiempo de lixiviación Depende de la cinética (lix. química : 1 a 2 meses; lix. bacterial : 3 a 12meses)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama60
Todos estos factores están relacionados entre si.
Por ejemplo, si se aumenta la altura de la pila, la concentración en ácido delprimer metro es buena, pero baja a medida que la solución percola en la pila y elcobre de los estratos inferiores no se lixivia bien. Se puede aumentar laconcentración en ácido de la solución de lixiviación, para salir de la pila con +/- 3g/l [H+], pero más ácido se da a la ganga y más se come, así que se va aincrementar el consumo de ácido. Otra posibilidad es aumentar la tasa deriego,pero existe un riesgo de inundar la pila.
3.3.7. Diseño de las pilas
En este párrafo, se considera el diseño de una operación mediana de lixiviación deóxidos de cobre, por ejemplo Mantos Verde (III región).
3.3.7.1. Datos
Capacidad de la planta : 48000 tCu/año = 4000 tCu/mes = 133 tCu/día
Ley del mineral : 0.95 % CuT
(0.80% Cu soluble + 0.15% Cu insoluble)
Fierro : 5%
Consumo de ácido : 3.5 kg ácido/kg Cu producido
Recuperación en la pila : 80% CuT en 2 meses (le da las pruebas piloto)
Granulometría : 100% < 3/8"
Altura de la pila : 5 m (parámetro de diseño)
Densidad aparente del mineral en la pila : 1.45 t/m3 (material chancado)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama61
3.3.7.2. Capacidad de la planta de chancado
Se recupera 80% x 9.5 kg Cu/TMS = 7.6 kg Cu/TMS (TMS = Tonelada MétricaSeca).
Entonces, se tiene que procesar
Figura 3.20 , Diagrama de flujo simplificado de una planta delixiviación de cobre.
Plantarecup=80%
17500TMS /día0.95%C u
M ineral Abotadero17367T M S/día
C átodos133T M C u/día

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama62
3.3.6.2. Superficie de terreno
El ciclo de lixiviación de una pila de mineral es de 2 meses. Entonces, el stock demineral en la planta es de 17500 TMS/día x 60 días = 1.050.000 TMS.
Si se consideran pilas rectangulares (aproximación) de 5 metros de altura, sepuede almacenar 1.45 TMS/m3 x 5 m = 7.25 TMS/m2.
La superficie de las pilas en funcionamiento es de
Pero todos los días, hay por lo menos una pila en carga, otra en descarga y senecesita espacio para el movimiento de las máquinas. La práctica indica que esosespacios ocupan un 10% de la superficie de las pilas en funcionamiento. Senecesita entonces una superficie total de terreno de 144827 m2 x 110% = 159310m2.
Eso corresponde a una área de 400 m x 400 m, o 200 m x 800 m, o 100 m x 1600m.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama63
3.4. Lixiviación por agitación
3.4.1. Descripción
La lixiviación por agitación se utiliza en los minerales de leyes más altas, cuandolos minerales generan un alto contenido de finos en la etapa de chancado, ocuando el mineral deseado está tan bien diseminado que es necesario molerlopara liberar sus valores y exponerlos a la solución lixiviante. Es también el tipo detécnica que se emplea para lixiviar calcinas de tostación y concentrados.
Se recurre a la agitación mediante burbujeo o bien a la agitación mecánica paramantener la pulpa en suspensión hasta que se logra la disolución completa,siendo el tiempo de contacto de los sólidos con la solución del orden de horascomparado con el proceso de lixiviación en pilas que requiere meses. Losagitadores mecánicos son simplemente impulsores colocados en el interior deltanque (Fig. 3.21a), mientras que los tanques agitados con aire son a menudotanques de tipo "Pachuca" (Fig. 3.21b).
Sus ventajas comparativas con otros métodos de lixiviación son :
- Alta extracción del elemento a recuperar
- Tiempos cortos de procesamiento (horas)
- Proceso contínuo que permite una gran automatización
- Facilidad para tratar menas alteradas o generadoras de finos
Sus desventajas son :
- Un mayor costo de inversión y operación
- Necesita una etapa de molienda y una etapa de separación sólido-líquido (espesamiento y filtración)
En la región de Atacama, Chile, se puede mencionar la planta "La Coipa",propiedad de Minera Mantos de Oro y mayor productora de plata del mundo.Utiliza el proceso de cianuración de oro y plata por agitación en ocho tanques en

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama64
serie. Procesa 16000 t/día de mineral para producir 8214 kg/año de oro y 3315t/año de plata (1993).
Figura 3.21, Equipos de lixiviación por agitación
3.4.2. Variables del proceso
El análisis de las variables de la lixiviación por agitación en sistemas industriales,para la definición y optimización del proceso, debe necesariamente hacer confluiraspectos técnico, operacionales y económicos.
3.4.2.1. Granulometría
El grado de molienda debe ser lo suficiente para exponer, por lo menospartialmente, la superficie del mineral valioso a la acción de la solución lixiviante.Depende del tipo de mineral y de sus caracteristicas mineralogicas. Deberaconsiderarse un tamaño tal que no contenga un exceso de gruesos (> 2 mm) que

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama65
produzca problemas en la agitación (embancamiento, aumento de la potencia delagitador) y que por otra parte, no contenga un exceso de finos (menos de 40% <75 micrones) , que dificulten la separación sólido-líquido posterior de la pulpalixiviada. Debido a lo anterior, y además, para disminuir los consumos de energíapor concepto de molienda y los costos de filtración y decantación, la agitación sedeberá tratar de realizarla al mayor tamaño que la operación lo permita.
Tabla 3.5, Tamaño de algunos minerales para la lixiviación poragitación
Tamaño de lixiviaciónMineral(mm) (mallas ASTM)
Cobre oxidado 0.83 20Oro 0.25 60Conc. de oro(sulfuros) 0.044 325
Calcinados dezinc 0.074 200
3.4.2.2. Tiempo de lixiviación
La economía del proceso de lixiviación es función del grado de disolución oporcentaje de extracción del mineral valioso. Sin embargo, esto no es tanimportante como el tiempo necesario para una extracción aceptable, es decir lavelocidad de disolución.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama66
Figura 3.21, Porcentaje de extracción en función del tiempo.
La figura 3.21 muestra una curva típica entre estos dos parámetros. Existe alprincipio una extracción rápida, que decrece posteriormente al máximo obteniblepara un tamaño dado de partícula. Esta curva se puede obtener de pruebas delixiviación en botellas en el laboratorio.
3.4.2.3. Mineralogía del mineral
El tamaño y la disposición de la especie valiosa influye el grado de moliendanecesario para exponer esta especie a la solución lixiviante (Tabla 3.5).
La arcillas son una familia de minerales, alumino-silicatos, existen en todos lasmenas y producen partículas muy finas (algunos micrones). La presencia demuchas arcillas puede impedir una buena filtración del relave.
3.4.2.4. Otras variables
La lixiviación se realiza a temperatura ambiente (o en autoclaves).
La concentración de reactivos debe ser optimizada según el tipo de operación.
El porcentaje de sólidos debe ser en la mayoría de los casos lo más alto posiblepara alcanzar una alta concentración del ion metálico en la solución de lixiviación,minimizar los costos de inversión en el circuito de lixiviación por menor capacidadvolumétrica y reducir el tamaño y costo subsecuente de espesamiento y filtración.El porcentaje de sólidos en la pulpa varia entre 20 y 50%.
El porcentaje de sólidos se calcula por el peso del mineral en la
pulpa. Por ejemplo, si una pulpa es constituida por 1 kg de mineral en
2 litros de
agua, su

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama67
La velocidad de agitación debe ser lo suficiente alta para mantener los sólidos ensuspención, para que no decanten. Una velocidad de agitación alta tiende afavorecer la cinética de la reacción, pero tiene un costo energetico apreciable (Fig.3.22). Favorece también la disolución de gases en la solución. Existen variosdiseños de agitadores (Fig. 3.23).
Figura 3.22, Efecto de la agitación en la velocidad de lixiviación
Figura 3.23, Varios diseños de turbinas.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama68
3.4.3. Diseño
Se considera como ejemplo un sistema de lixiviación contínua industrialconstituido de varios estanques en serie (Fig. 3.24).
Datos
Número de estanques : 8
Capacidad : 15000 t/d = 625 t/h
% sólidos : 33.33%
grado de molienda : 100% < 60 mallas ASTM
densidad real del mineral : 2.8 g/cm3 = 2.8 t/m3
Tiempo de lixiviación : 24h
(determinado por la curva grado de lixiviación/tiempo)
sólido - líquidode separación
Mineralmolido
SoluciónPulpa
Pulpa a etapa
Figura 3.24, Esquema de un sistema contínuo industrial

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama69
Cálculo de la cantidad de agua en la pulpa :
Cálculo del flujo de pulpa (sólidos + agua) :
Cálculo del volumén de cada uno de los 8 estanques (Fig. 3.24) :
Volumén de un estanque cilíndrico :
Entonces, si consideramos 8 estanques cilindricos tales que h = 1.5d,

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama70
3.5. Separación sólido-líquido
3.5.1. Introducción
En una planta de lixiviación en pilas, la solución que escurre de las pilas esbastante clara y contiene muy pocos sólidos en suspensión. En la mayoría de loscasos, no hay etapa específica de separación sólido - líquido. La solución rica sealmacena en piscinas donde pueden decantar los pocos sólidos que contiene.
Al contrario, la lixiviación por agitación produce una pulpa consistente enpequeñas partículas sólidas en suspensión en la solución. Una etapa deseparación sólido - líquido es absolutamente necesaria para sacar los sólidos de lasolución y enviarlos al tranque de relaves, mientras la solución clarificada puedepasar una posterior etapa de recuperación de valores.
Hay dos métodos de separación sólido - líquido :
- El espesamiento (hasta 55% sólidos)
- La filtración (hasta 92% sólidos)
Alimentación (Pulpa)Solución claraSolución clara
Pulpa más concentrada(40 - 60 % sólidos)
Solución
Decantación sólidos
Figura 3.25, Esquema de un espesador

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama71
3.5.2. Espesadores
Los espesadores son equipos que efectúan la separación líquido - sólido pordecantación de los sólidos en un estanque grande. Un esquema se presenta en lafigura 3.25.
3.5.3. Lavado en circuitos de decantación encontracorriente (DCC)
El sólido (barro decantado) siempre viene acompañado de solución deimpregnación (+/- 40 %), y está contiene valores, por lo cual es imperativorecuperarla.
Después de la lixiviación dinámica, la pulpa que se obtiene del último agitadordebe pasar por etapas de separación sólido - líquido y lavado, antes de desecharlos sólidos lixiviados. El lavado se puede realizar en varios espesadores, en loscuales el agua de lavado y la pulpa fluyen en contracorriente (Fig. 3.26). Delprimer espesador sale la solución exenta de sólidos que continua el proceso y delúltimo, el mineral agotado o relave.
A las operaciones de lavado de los sólidos en contracorriente, se les denominacomo etapas DCC, que significa decantación en contracorriente.
Figura 3.26 Circuito de decantación en contra corriente DCC.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama72
3.5.3.1. Determinación de la eficiencia de lavado por balance másicoen circuitos DCC.
Operacionalmente, el balance másico del sistema permite determinar la eficienciade lavado en sistemas DCC. Su aplicación se visualizará a través del siguienteejemplo (Fig. 3.27).
Bases :
1000 t/d mineral
55% sólidos en la descarga delos espesadores
Razón de lavado RL = 2.5 (aguade lavado / flujo de solución delix. acompañando a los sólidos)
55% sólidos en la alimentación
Figura 3.27, Balance de masa en un circuito DCC.
Volumen de líquidos, m3/d:
Alimentación A = 1000 x (100 - 55)% = 818Descarte D = 818Agua de lavado W = 818 x 2.5 = 2000Solución rica SF = 2000

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama73
Cálculo de las concentraciones :o
800 C0 + 2000 C2 = 2800 C1 2 C0 + 5 C2 = 7 C1800 C1 + 2000 C3 = 2800 C2 2 C1 + 5 C3 = 7 C2800 C2 + 2000 C4 = 2800 C3 2 C2 + 5 C4 = 7 C3800 C3 + 2000 x 0 = 2800 C4 2 C3 + 0 = 7 C4
Por lo tanto,
La eficiencia de lavado queda dada por :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama74
3.5.3.2. Ecuación de PAGE para cálculos DCC
PAGE ha desarrollado y comprobado una ecuación generalizada, definida entérminos de razón de lavado y número de etapas, para el caso más simple, peromás común en DCC (Fig. 3.26), asumiendo que no hay disolución en losespesadores y que la mezcla de la pulpa y solución de lavado es completa encada etapa.
Cn : concentración metal en la etapa nCL : concentración metal en la alimentación al primer
espesadorCW : concentración metal en el agua o solución de lavadoRL : razón de lavado = Qw / QLn : número de etapas de lavadoQW : flujo de la solución de lavadoQL : flujo de la solución de lixiviación (alimentación)
3.5.4. Filtros
El empleo de filtros en hidrometalurgia permite recuperar una cantidad adicionalde líquido retenido a partir de precipitados o de pulpas de 55-60% en sólidosprocedentes de espesamiento como también tratar directamente soluciones delixiviación, a objeto de disminuir la cantidad de sólidos en suspensión que puedenafectar procesos posteriores de tratamiento.
Filtros de vacío : - de tambor (Fig. 3.28)- de discos (o cerámica)- horizontal, de cinta
Filtros de presión : - prensa- stellar- de arena
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama75
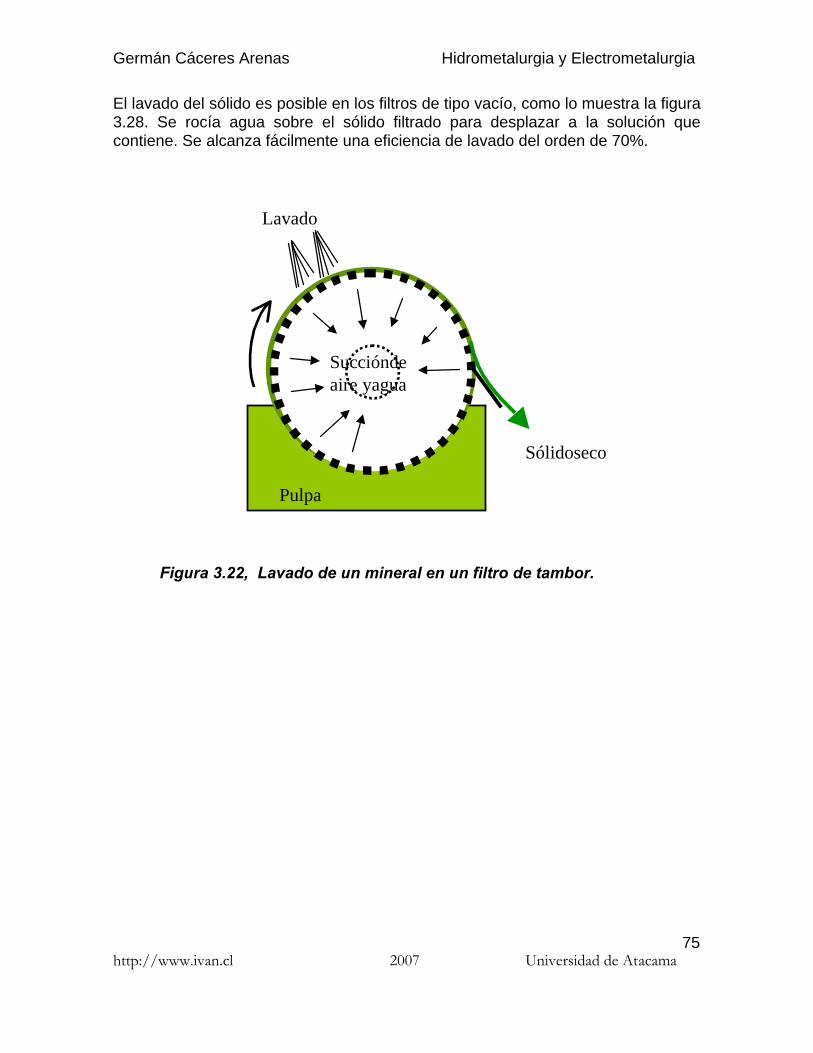
El lavado del sólido es posible en los filtros de tipo vacío, como lo muestra la figura3.28. Se rocía agua sobre el sólido filtrado para desplazar a la solución quecontiene. Se alcanza fácilmente una eficiencia de lavado del orden de 70%.
Figura 3.22, Lavado de un mineral en un filtro de tambor.
Pulpa
Succióndeaire yagua
Sólidoseco
Lavado

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama76
4.1. Introduccion
En un proceso de purificación o de concentración, siempre hay dos fases encontacto ( líquido - sólido o líquido - líquido). Se trata de un proceso de :
Purificación si la impureza va a la otra faseConcentración si el elemento deseado va a la otra fase
Los procesos de purificación y/o concentración son muy variadosy dependen de :
- la naturaleza del elemento deseado- las impurezas presentes en la solución- el tipo de proceso de recuperación- el grado de pureza deseado para el producto final
Los procesos de purificación y/o concentración se pueden dividir en variascategorías :
- Hidrólisis- Cementación- Precipitación de un compuesto específico- Extracción por solventes
Purificacióny concentración
dela solución
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama77
- Resinas de intercambio iónico
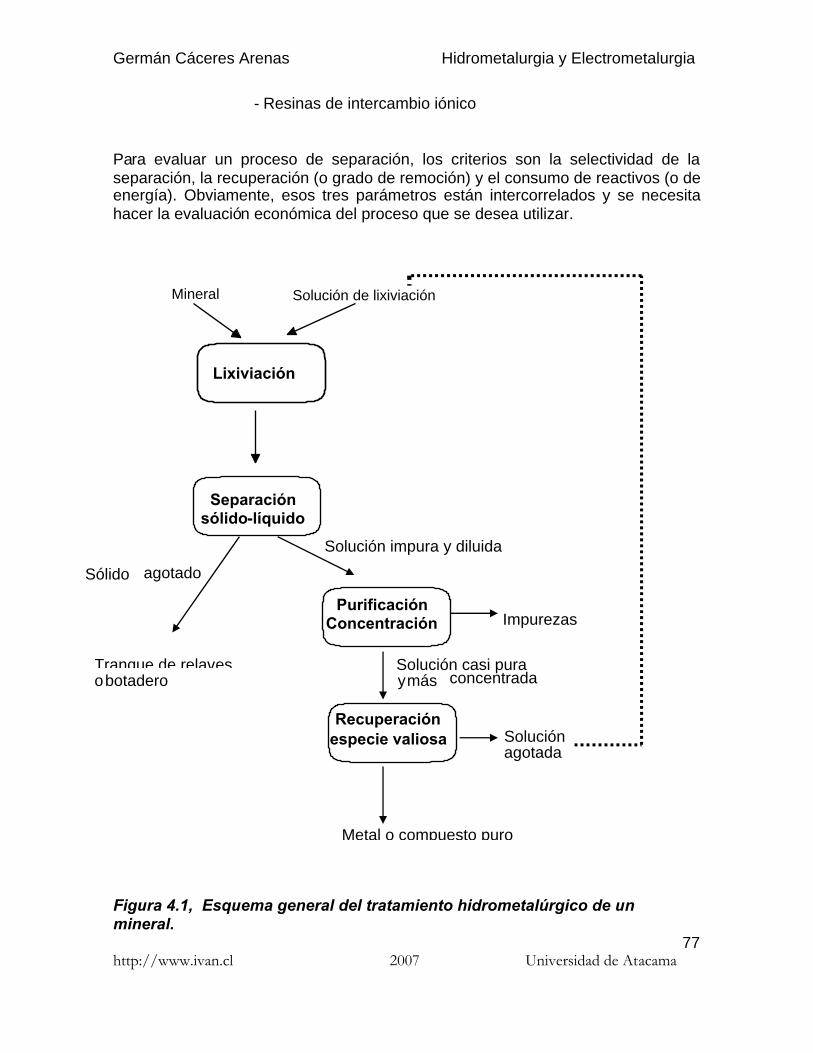
Para evaluar un proceso de separación, los criterios son la selectividad de laseparación, la recuperación (o grado de remoción) y el consumo de reactivos (o deenergía). Obviamente, esos tres parámetros están intercorrelados y se necesitahacer la evaluación económica del proceso que se desea utilizar.
Figura 4.1, Esquema general del tratamiento hidrometalúrgico de unmineral.
Mineral Solución de lixiviación
Lixiviación
Separaciónsólido-líquido
PurificaciónConcentración
Recuperaciónespecie valiosa
Solución impura y diluida
Sólido agotado
Tranque de relavesobotadero
Solución casi puraymás concentrada
Metal o compuesto puro
Impurezas
Soluciónagotada

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama78

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama79
En este capítulo, se mostrarán como ejemplos las etapas de purificación de unasolución de zinc en ácido sulfúrico antes de su electroobtención. El proceso semuestra en la figura 4.2.
Figura 4.2, Esquema de purificación de una solución paraelectroobtención de zinc.
Tabla 4.2, Impurezas en la solución de lixiviación del zinc.
Antes depurificación
(mg/l)
Después depurificación
(mg/l)Fe mucho -Cu 1000 <0.1
Cd 1000 0.1 - 0.2
Co 30 - 50 0.1 - 0.2
Ni 30 - 50 trazas
Sb, As 0.1 < 0.01
Ge < 0.1 <0.01
Solución impura
Hidrolisis
Cementación
PrecipitaciónCompuestos
Solución puraa electroobtención
FeCoprecipitaciónde Ge y As
Cu y Cd
Ni y Co

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama80
4.2. Hidrolisis
4.2.1. Precipitación de hidroxidos
Los diagramas Eh - pH muestran que los iones metálicos existen en solucionesacuosas sólo dentro de ciertos límites de pH. La manera más sencilla de separarun metal de su solución es mediante la alcalinización de la solución, dando lugar asu precipitación como hidóxido.
Por ejemplo : Fe3+ + 3 OH- => Fe(OH)3 (floculos café / rojos)
La tabla 4.1 muestra el pH de precipitación de los hidróxidos de algunos metalesen agua. Los fosfatos y arseniatos básicos, si se encuentran en la solución,precipitan, a veces, más rápidamente que los hidróxidos y con pH más bajo.
Tabla 4.1, pH de precipitación de algunoshidróxidos (C. EK).
Concentracióndel ion metálico
10-2 mol/l 10-5 mol/lFe3+ 2.2 3.2Al3+ 3.6 4.6Fe2+ 5.5 7.2
Cu2+ 5.6 7.3
Zn2+ 6.6 8.3
Ni2+ 6.7 8.4
Co2+ 6.8 8.5
Cd2+ 7.0 8.7
Pb2+ 7.4 8.9
Mn2+ 8.4 10.1
Mg2+ 9.7-10.8 11.2 - 12.3

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama81
4.2.2 Hidrolisis
Los procesos de hidrólisis se basan fundamentalmente en el mismo tipo dereacción de precipitación de hidróxido, con la diferencia de que en la mayor partede los casos no se termina la reacción en un hidróxido sino en una sal básica, enuna sal doble o hasta en un óxido.
Dentro de este tipo de reacciones, la que tiene mayor interés industrial es lahidrólisis del hierro, utilizado comúnmente como un método de eliminación de estaimpureza en circuitos hidrometalúrgicos.
4.2.3 Hidrolisis del fierro
Existen básicamente tres tipos de procesos para la eliminación del hierro acuoso :precipitación como jarosita, goetita y hematita. Estos dos últimos procesosproducen precipitados más fácil de filtrar y decantar.
Jarositas
Las jarositas son compuestos que tienen la siguiente formula : MFe3(SO4)2(OH)6,
en la que M puede ser Na+, K+, NH4+, Ag+, Rb+, Pb2+ y H+. La precipitación delFe como tal se consigue entre 80 y 100 °C, con adición de un álcali y con controlde la acidez. Para la formación de jarosita de amonio, la siguiente reacción tienelugar :
3 Fe3+ + 2 SO4 2- + NH4+ + 6 H2O => NH4Fe3(SO4)2(OH)6 + 6 H+
Con la exposición a la intemperie en los tranques de relaves, la jarosita setransforma gradualmente en hematita y debido a sus propiedades de adsorción yretención de agua, se produce una eliminación (disolución) de metales solublesque ocasionan un problema ecológico.
Goetita
La precipitación del hierro como goetita se realiza mediante la siguiente reaccióngeneral :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama82
Fe3+ + 2 H2O => FeOOH + 3 H+
La temperatura de la precipitación oscila entre 20 y 100 °C, el pH entre 2 y 4 y elpotencial debe ser oxidante con el fin de mantener el hierro en forma férrica. Lacinética de precipitación es lenta, dependiendo de la concentración de Fe inicial ydel nivel de la impureza a eliminar. El tipo de cristal que se obtiene es de mejorescaracterísticas para la separación sólido - líquido.
Hematita
La precipitación del hierro como hematita se fundamenta en la siguiente reacción :
2 Fe3+ + 3 H2O => Fe2O3 + 6H+
Los factores de mayor importancia en este método son la temperatura,concentración de hierro y la concentración de ácido. Dependiendo de estosfactores se pueden formar una serie de compuestos, pudiéndose señalar engeneral, que las hematitas tienden a formarse sobre 140 °C.
4.3. Cementacion
La cementación de un metal a partir de una solución, depende de una reacción dedesplazamiento en la cual un metal menos noble reduce a los iones del metal porprecipitar al estado metálico. Entonces, éste sale de la solución, y los iones delmetal menos noble entran en la solución para substituirlos.
Por ejemplo :
Cu2+ + Zn => Cu + Zn2+
El metal utilizado debe tener un potencial de electrodo inferior al potencial delmetal que se desea precipitar.
La reacción se produce a la superficie del metal, que se agrega habitualmente enforma de polvo a la solución impura (Fig. 4.3). Para alcanzar velocidades dereacción aceptables con impurezas que se encuentran en bajas concentraciones,

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama83
se tiene que agregar un exceso de metal, lo que conduce a la obtención decementos impuros. Ademas, estos cementos contienen todos los metales depotencial superior al metal agregado.
Cu
Zn
Zn2+
Cu2+
e-
Figura 4.3, Cementación de cobre en la superficie de unapartícula de zinc.
El mejor metal utilizado para cementar las impurezas de una solución esgeneralmente el mismo metal que se desea recuperar desde la solución pura. Así,no se agrega otra impureza a la solución. Por ejemplo, el cadmio y el cobrecontenidos en las soluciones de lixiviación de concentrados de zinc se cementancon polvo de zinc.
Ademas de utilizarse como método de purificación de soluciones, la cementaciónpuede utilizarse para recuperar el metal valioso a partir de soluciones diluidas ( ej :cementación de cobre por chatarra de hierro).
4.4. Precipitacion de un compuestoespecifico
Cuando otros métodos no son aplicables, se pueden usar reactivos específicos deprecipitación. Estas reacciones son generalmente complejas, necesitancondiciones de trabajo estrictas y consumen reactivos costosos. Por ejemplo, seusa el nitrosobetanaphtol para precipitar el cobalto contenido en las soluciones deelectroobtención de zinc.
Se puede aplicar todas las reacciones que estudiaron en el ramo de química en laenseñanza media tales como precipitación de sulfuros, formación de sales, ...
Por ejemplo : AgNO3 + NaCl => AgCl (precipitado) + NaNO3

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama84
Es importante el fenómeno de envejecimiento de un precipitado, favorecido poruna temperatura alta y un tiempo de contacto solución-precipitados largo. Elenvejecimiento consiste en la disolución de las partículas más finas y formación deprecipitados más gruesos, generalmente acompañado por un cambio en laestructura cristalina de estos. Mejora la insolubilidad del precipitado y su posteriorfiltración.
4.5. Fenomeno de coprecipitación
La coprecipitación es cuando hay precipitación de dos o más elementos al mismotiempo. El elemento minoritario puede ser disuelto en la matriz cristalina delelemento mayoritario, o absorbido en su superficie.
Esto puede ser ventajoso, ya que la la concentración del elemento minoritario enla solución puede bajar a niveles mucho más bajos que por precipitación de esteelemento en forma pura.
Por ejemplo, en la hidrometalurgia del zinc, impurezas como el Ge y el Ascoprecipitan con un "hidróxido" (*) de hierro , probablemente por absorción en lasuperficie de los floculos de "hidróxido" de hierro. ( (*): Existen varias formas deprecipitar el hierro : como jarosita, goetita,...)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama85
4.6. Extracción por solventes
4.6.1. Introducción
Uno de los métodos de purificación que ha tenido un espectacular desarrollo eneste último tiempo, ha sido el proceso de extracción por solventes o proceso SX.Ello se ha traducido en la instalación de numerosas plantas que operanactualmente en el mundo en la separación, purificación y concentración de más deuna treintena de elementos químicos, como cobre, niquel, cobalto, zinc, uranio,molibdeno, tungsteno, vanadio, tierras raras, zirconio, hafnio, niobio, tantalio, boro,germanio, arsénico, renio, torio, el grupo de los metales del platino, berilio y otros.
En estos apuntes, para más claridad, se tomará como ejemplo la extracción porsolventes del cobre, de uso masivo en Chile.
4.6.2. Proceso SX - EW de Cobre
El proceso de extracción por solventes, conocido en la hidrometalurgia del cobretambién como SX (del inglés Solvent Extracction), consiste en la extracciónselectiva del cobre contenido en las soluciones de lixiviación mediante un solventeorgánico, para luego transferirlo a una solución de sulfato de cobre pura yconcentrada, denominada electrolito rico.
4.6.2.1. Descripción general
La planta SX recibe la solución rica generada en la etapa de lixiviación en pilas deminerales de cobre. Esta solución se caracteriza por tener una baja concentraciónde cobre disuelto, junto con impurezas como el Fe, Cl, Al, Mn, Mg, Na y otrosdisueltos durante el proceso (Figura 4.4).
El objetivo del proceso SX es extraer selectivamente el cobre contenido en estasolución rica impura, mediante intercambio ionico entre la fase acuosa (solución
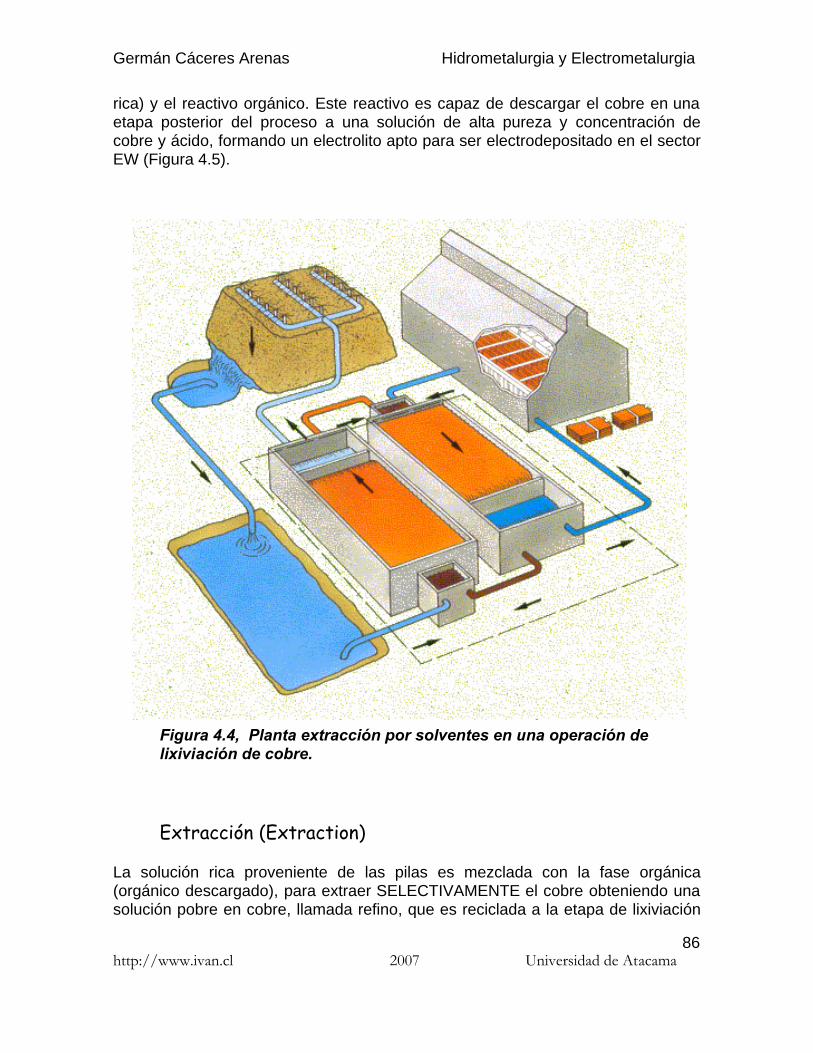
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama86
rica) y el reactivo orgánico. Este reactivo es capaz de descargar el cobre en unaetapa posterior del proceso a una solución de alta pureza y concentración decobre y ácido, formando un electrolito apto para ser electrodepositado en el sectorEW (Figura 4.5).
Figura 4.4, Planta extracción por solventes en una operación delixiviación de cobre.
Extracción (Extraction)
La solución rica proveniente de las pilas es mezclada con la fase orgánica(orgánico descargado), para extraer SELECTIVAMENTE el cobre obteniendo unasolución pobre en cobre, llamada refino, que es reciclada a la etapa de lixiviación
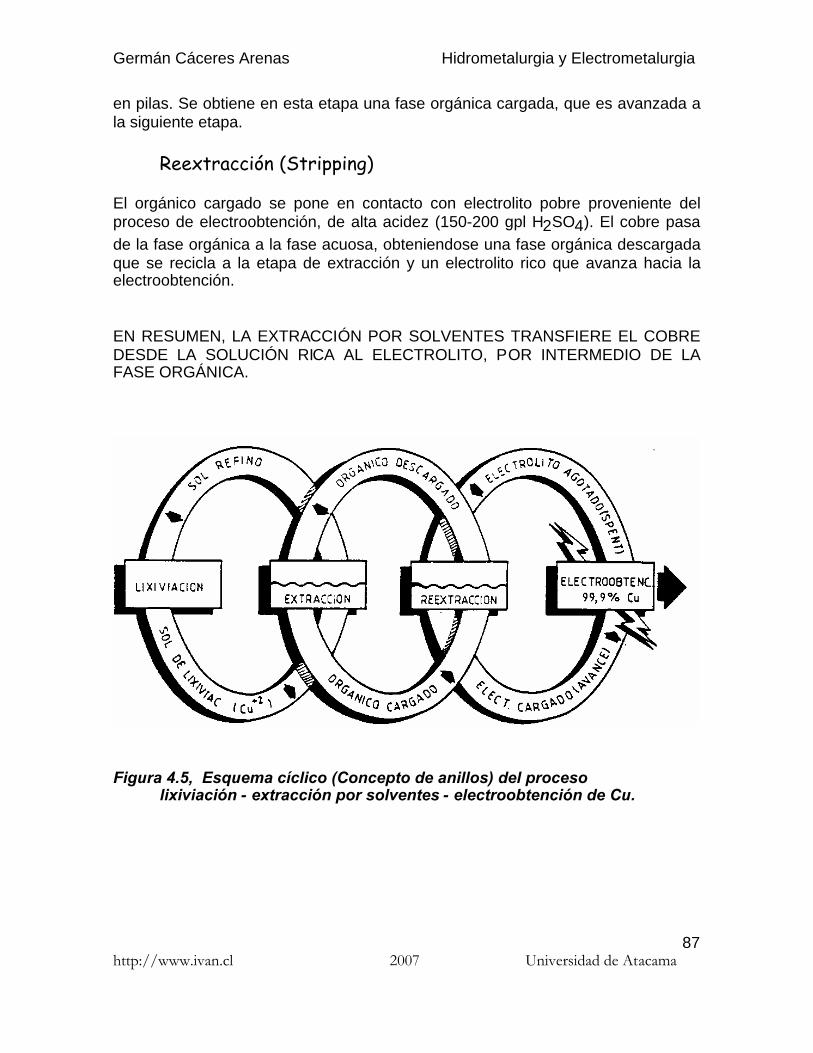
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama87
en pilas. Se obtiene en esta etapa una fase orgánica cargada, que es avanzada ala siguiente etapa.
Reextracción (Stripping)
El orgánico cargado se pone en contacto con electrolito pobre proveniente delproceso de electroobtención, de alta acidez (150-200 gpl H2SO4). El cobre pasade la fase orgánica a la fase acuosa, obteniendose una fase orgánica descargadaque se recicla a la etapa de extracción y un electrolito rico que avanza hacia laelectroobtención.
EN RESUMEN, LA EXTRACCIÓN POR SOLVENTES TRANSFIERE EL COBREDESDE LA SOLUCIÓN RICA AL ELECTROLITO, POR INTERMEDIO DE LAFASE ORGÁNICA.
Figura 4.5, Esquema cíclico (Concepto de anillos) del procesolixiviación - extracción por solventes - electroobtención de Cu.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama88
4.6.2.2. Mecanismo de la transferencia de cobre
El proceso SX se basa en la siguiente reacción REVERSIBLE de intercambioionico :
Cu2+(A) + 2 RH(O) <=> R2Cu(O) + 2 H+(A)
en la cual el sentido de reacción está controlado por la acidez o pH de la soluciónacuosa.
En la etapa de extracción, el reactivo orgánico se contacta con la soluciónacuosa impura de lixiviación y extrae selectivamente desde la fase acuosa losiones de cobre, incorporandolos en la fase orgánica. El ion cúprico reacciona conel extractante formando un compuesto organometálico insoluble en agua,totalmente soluble en el solvente orgánico (Kerosene, ...), con la cual se producela extracción del cobre desde la fase acuosa a la orgánica. Mediante estemecanismo, cada ion de cobre se intercambio con dos iones de hidrogeno quepasan a la fase acuosa donde se regenera ácido sulfúrico en una proporción de1.54 (kg de ácido / kg de cobre).
Figura 4.6, Esquema de un mezclador – sedimentador paraoperaciones de extracción y reextracción.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama89
La etapa de reextracción ocurre por efecto del cambio de acidez en la faseacuosa, revertiendo la reacción y generando un electrolito de alta pureza y altaconcentración en cobre.
R2Cu(O) + 2 H+(A) <=> Cu2+(A) + 2 RH(O)
4.6.2.3. Configuración de plantas SX
Las plantas de extracción por solventes, tanto en la extracción como en lareextracción, están configuradas en UNA, DOS O TRES ETAPAS DE CONTACTOde las fases acuosa y orgánica, con el objeto de aumentar el tiempo de contactoentre ambas fases y mejorar la transferencia de cobre. El contacto de las fasesorgánica y acuosa se realiza EN CONTRACORRIENTE.
Además, en algunos casos, se debe incorporar una etapa de lavado del orgánicocargado, con el objeto de eliminar impurezas dañinas en la electroobtención, porejemplo el cloro.
Se presenta en la figura 4.7 una configuración típica de una planta de extracciónpor solventes, compuesta por 2 etapas de extracción y 1 etapa de reextracción (2E-1S). La figura 4.8 representa una planta 3 E-2 S.
Lixiviación Electro-obt.
E1
E2S1
Extracción Reextracción
SR
RFOSC
SRFOC OD
ED EC
OC

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama90
Figura 4.7, Configuración 2 E - 1 S (o 2 E - 1 R E).
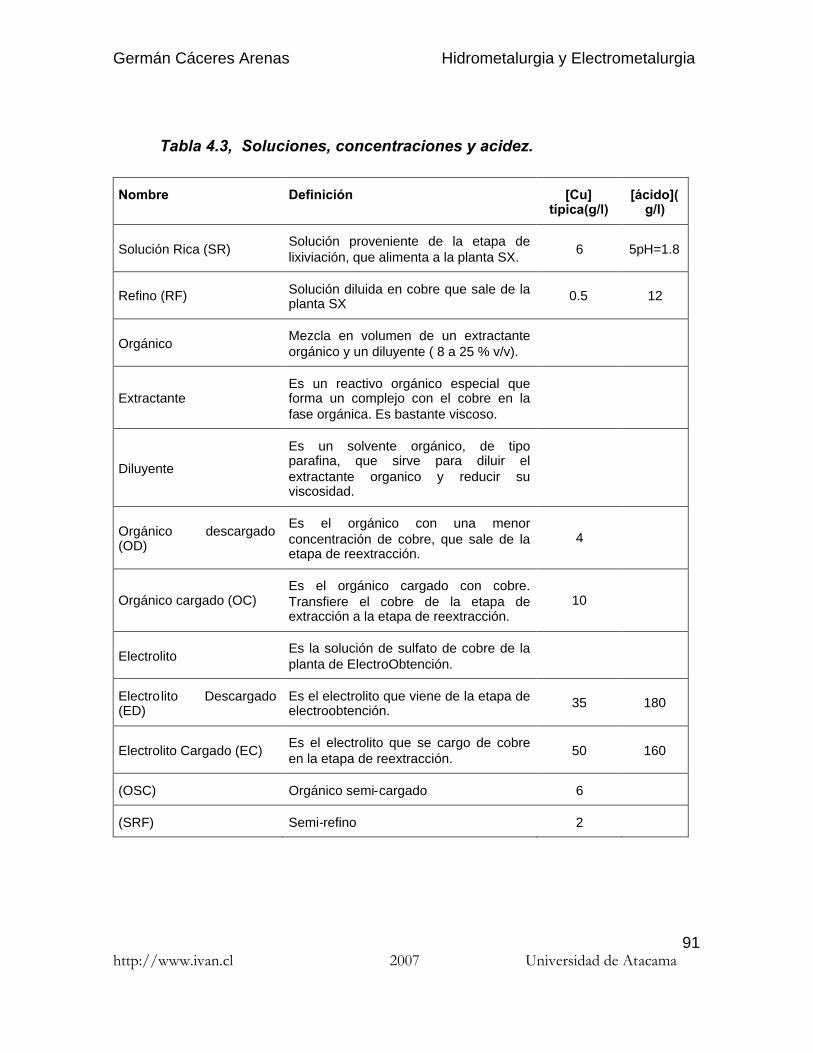
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama91
Tabla 4.3, Soluciones, concentraciones y acidez.
Nombre Definición [Cu]típica(g/l)
[ácido](g/l)
Solución Rica (SR) Solución proveniente de la etapa delixiviación, que alimenta a la planta SX. 6 5pH=1.8
Refino (RF) Solución diluida en cobre que sale de laplanta SX 0.5 12
Orgánico Mezcla en volumen de un extractanteorgánico y un diluyente ( 8 a 25 % v/v).
ExtractanteEs un reactivo orgánico especial queforma un complejo con el cobre en lafase orgánica. Es bastante viscoso.
Diluyente
Es un solvente orgánico, de tipoparafina, que sirve para diluir elextractante organico y reducir suviscosidad.
Orgánico descargado(OD)
Es el orgánico con una menorconcentración de cobre, que sale de laetapa de reextracción.
4
Orgánico cargado (OC)Es el orgánico cargado con cobre.Transfiere el cobre de la etapa deextracción a la etapa de reextracción.
10
Electrolito Es la solución de sulfato de cobre de laplanta de ElectroObtención.
Electrolito Descargado(ED)
Es el electrolito que viene de la etapa deelectroobtención. 35 180
Electrolito Cargado (EC) Es el electrolito que se cargo de cobreen la etapa de reextracción. 50 160
(OSC) Orgánico semi-cargado 6
(SRF) Semi-refino 2
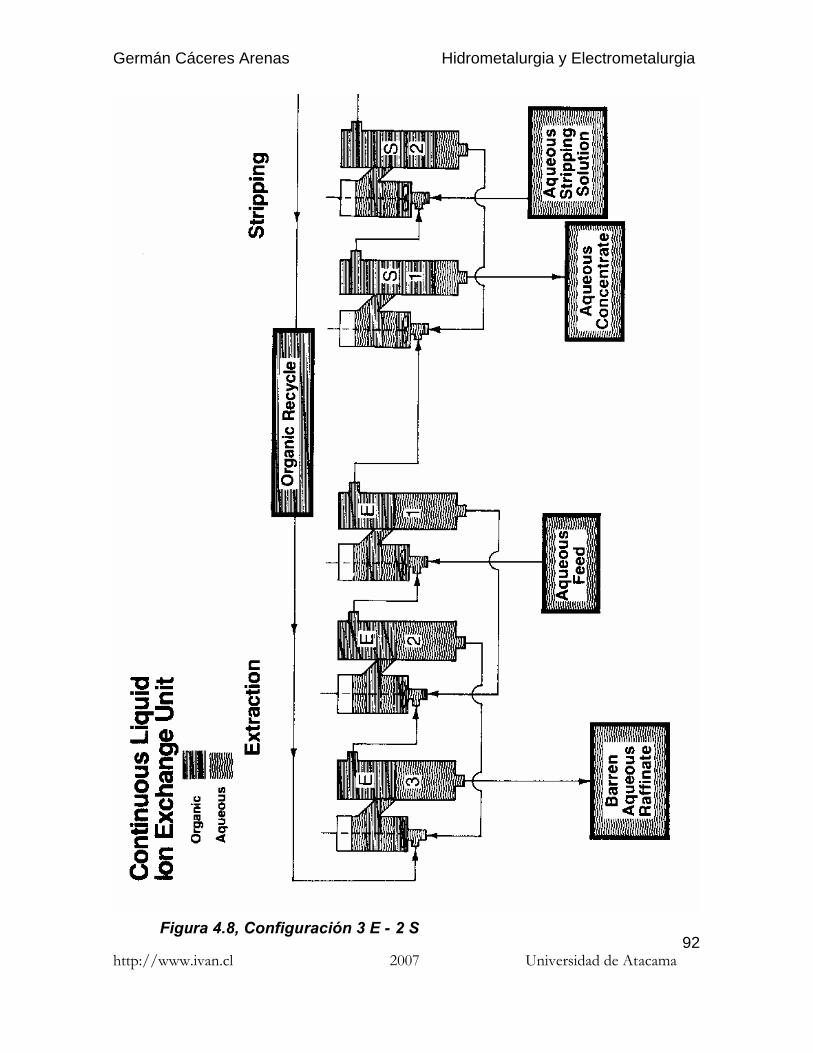
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama92
Figura 4.8, Configuración 3 E - 2 S

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama93

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama94
4.6.3. Conceptos Fundamentales
4.6.3.1. Coeficiente de distribución
Un sistema de extracción por solventes implica una transferencia de masa delelemento acuoso desde una fase a otra. La extracción o la reextracción seproduce por la dispersión de una fase en la otra en forma de pequeñas gotitas,que favorece la transferencia de materia y se realiza por medio de agitaciónmecánica.
Al contactar una solución acuosa que contiene al elemento M que interesa extraercon una fase orgánica inmiscible, el elemento se distribuirá entre las dos faseshasta alcanzar el equilibrio. La concentración del elemento en el equilibriodependerá de su afinidad relativa (o solubilidad) por ambas fases.
La razón entre la concentración del metal en la fase orgánica y acuosa, en elequilibrio, define al coeficiente de distribución D (o coeficiente de extracción EaO) :
(4.1)
El coeficiente de re-extracción (stripping) se define del mismo modo :
(4.2)
4.6.3.2. Selectividad
La existencia de más de una especie química en solución dará lugar a que dichasespecies se distribuyan entre las fases acuosa y orgánica de acuerdo a susrespectivas solubilidades. Las diferencias en las solubilidades entre ambas fasesse pueden aprovechar para extraer las especies más solubles y separarlas de lasmenos solubles. de este modo, se puede establecer el concepto de factor deseparación como la relación de los coeficientes de distribución de dos especiesdistintas (DM y DN), que realmente mide la posibilidad de separación de lasespecies M y N y que se conoce con el nombre de selectividad.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama95
(4.3)
4.6.3.3. Influencia del pH
En el caso de agentes de extracción que formen especies químicas con iones decobre en solución, la reacción de extracción por solventes se puede escribir como:
Cu2+(A) + 2 RH(O) <=> R2Cu(O) + 2 H+(A) (4.4)
Con una constante de reacción definida por :
(4.5)
Reemplazando (4.1) en (4.5) se tiene la siguiente expresión para la constante K :
(4.6)
D : Coeficiente de distribuciónF : Producto de los coeficientes de
actividad
K es una característica del sistema y sólo depende de la temperatura. Aplicandologaritmos a esta última expresión, resulta :
0)log(22log HRpHFK
D (4.7)
que relaciona el coeficiente de distribución con el pH e indica que la extracciónesta afectada por el pH de la solución. En la figura 4.9 se puede observar este
N
M
DD
S
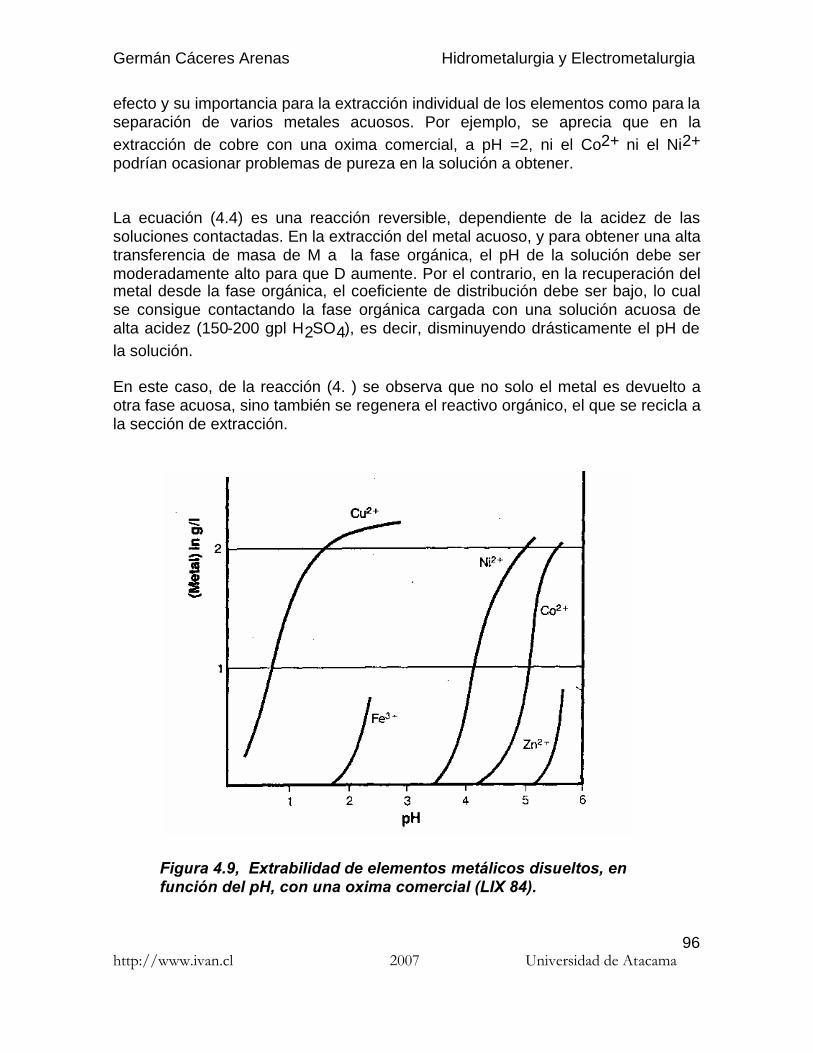
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama96
efecto y su importancia para la extracción individual de los elementos como para laseparación de varios metales acuosos. Por ejemplo, se aprecia que en laextracción de cobre con una oxima comercial, a pH =2, ni el Co2+ ni el Ni2+podrían ocasionar problemas de pureza en la solución a obtener.
La ecuación (4.4) es una reacción reversible, dependiente de la acidez de lassoluciones contactadas. En la extracción del metal acuoso, y para obtener una altatransferencia de masa de M a la fase orgánica, el pH de la solución debe sermoderadamente alto para que D aumente. Por el contrario, en la recuperación delmetal desde la fase orgánica, el coeficiente de distribución debe ser bajo, lo cualse consigue contactando la fase orgánica cargada con una solución acuosa dealta acidez (150-200 gpl H2SO4), es decir, disminuyendo drásticamente el pH dela solución.
En este caso, de la reacción (4. ) se observa que no solo el metal es devuelto aotra fase acuosa, sino también se regenera el reactivo orgánico, el que se recicla ala sección de extracción.
Figura 4.9, Extrabilidad de elementos metálicos disueltos, enfunción del pH, con una oxima comercial (LIX 84).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama97
4.6.4. Determinación del número de etapas deextracción y re-extracción
4.6.4.1 Isoterma de Distribución
La ISOTERMA DE DISTRIBUCIÓN es un gráfico de la concentración de laespecie extraída en la fase orgánica versus la concentración de la misma en faseacuosa, en el equilibrio y a una temperatura dada, y puede ser preparada paraextracción como para reextracción.
Normalmente la forma más utilizada para construir la isoterma de extracción escontactar en diferentes proporciones el acuoso y el orgánico, y analizar por elelemento metálico una vez logrado el equilibrio. La fases orgánicas y acuosas seponen en contacto en un embudo decantador (250 cc) y se agita durante 3minutos. Luego se deja decantar las fases. Generalmente se emplean razonesO/A de 10/1, 5/1, 2/1, 3/2, 1/1, 1/2 y 1/5.
La figura 4.11 muestra la construcción de una isoterma de distribución cuando seponen en contacto A ml de solución rica conteniendo X0 g/l de metal disuelto conO ml de orgánico fresco. Despues de la puesta en equilibrio, el refino contiene X1g/l y el orgánico Y1 g/l de metal. El balance de masa se puede expresar como :
y para cada razón existe un coeficiente de distribución diferente :
PUNTO DE SATURACIÓN : Composición del extractante dónde el a alcanzado sucapacidad máxima de carga (a la derecha del gráfico).
Isotermas de extracción y de re-extracción pueden ser generados de manerasimilar (ver laboratorio). Los isotermas son válidos solamente para condiciones
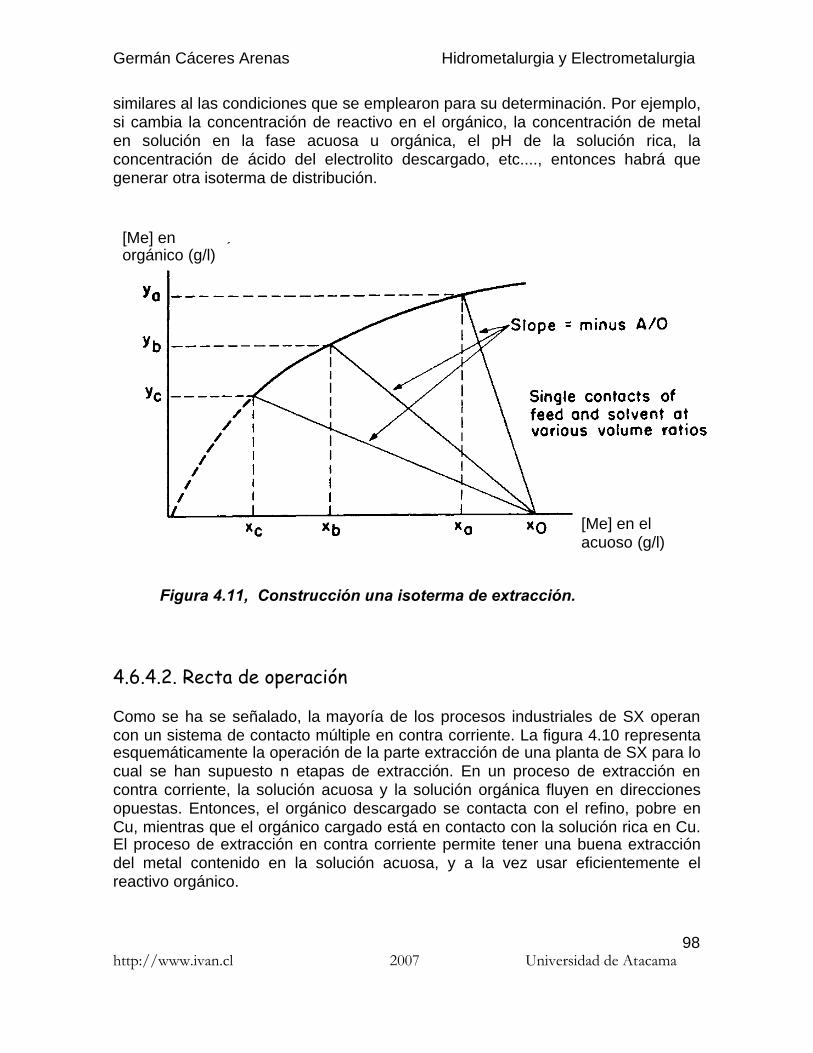
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama98
similares al las condiciones que se emplearon para su determinación. Por ejemplo,si cambia la concentración de reactivo en el orgánico, la concentración de metalen solución en la fase acuosa u orgánica, el pH de la solución rica, laconcentración de ácido del electrolito descargado, etc...., entonces habrá quegenerar otra isoterma de distribución.
Figura 4.11, Construcción una isoterma de extracción.
4.6.4.2. Recta de operación
Como se ha se señalado, la mayoría de los procesos industriales de SX operancon un sistema de contacto múltiple en contra corriente. La figura 4.10 representaesquemáticamente la operación de la parte extracción de una planta de SX para locual se han supuesto n etapas de extracción. En un proceso de extracción encontra corriente, la solución acuosa y la solución orgánica fluyen en direccionesopuestas. Entonces, el orgánico descargado se contacta con el refino, pobre enCu, mientras que el orgánico cargado está en contacto con la solución rica en Cu.El proceso de extracción en contra corriente permite tener una buena extraccióndel metal contenido en la solución acuosa, y a la vez usar eficientemente elreactivo orgánico.
[Me] enorgánico (g/l)
[Me] en elacuoso (g/l)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama99
EtapaN°1
EtapaN°2
EtapaN°3
EtapaN°n
A, X0
O, Y1
A, X1
O, Y2
A, X2
O, Y3
A, X3
O, Y4
A, X(n-1)
O, Y(n)
A, X(n)
O, Y(n+1)
A : Flujo de acuoso (m3/h)O : Flujo de orgánico (m3/h)X(n) : Concentración de metal en el acuoso que sale de la etapa n (g/l)Y(n) : Concentración de metal en el orgánico que sale de la etapa n (g/l)
Figura 4.10, Esquema de una extracción en contra corriente
El balance de masa para la primera etapa es :
y para la etapa N° n :
(4.8)
El balance de masa global para n etapas es :
(4.9)
La composición de la fase orgánica que entra en la etapa n es una función linealde la composición de la fase acuosa que abandona la nsima etapa, y vise versa.La ecuación (4.9) es la ecuación de una linea recta llamada RECTA DE

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama100
OPERACIÓN en el diagrama de McCabe-Thiele, el cual se describe en el parafoposterior.
4.6.4.3. Diagrama de McCabe-Thiele.
La combinación de la isoterma de distribución y la linea de operación constituye elDIAGRAMA DE OPERACIÓN o DIAGRAMA DE MCCABE-THIELE, el que esutilizado para ESTIMAR EL NÚMERO TEÓRICO DE ETAPAS o pisos en unsistema SX. La isoterma de distribución es fija, y depende sólo de la química desistema (concentración de reactivos, pH, ...). La linea de operación se basa en elbalance de masa, varia con la razón A/O y las concentraciones de metal en laentrada y salida del sistema.
Un diagrama típico de operación se presenta en la figura 4.12. Se supone que elorgánico descargado que entra en la última etapa de extracción contiene 1.80 g/lCu y que los flujos del orgánico y del acuoso son iguales (razón A/O =1).Conociendo eso, una recta de operación puede ser determinada, partiendo delpunto dónde el orgánico descargado intercepta a la isoterma de extracción ydibujando la recta hacia arriba y la derecha con una pendiente igual a la razón A/O(=1 en este caso).
Para determinar el número de etapas de extracción, se dibuja una linea verticalrepresentando la concentración de la solución rica (feed = 2.50 g/l Cu) hasta queintercepte la recta de operación. Después, se traza una recta horizontal hasta laisoterma de extracción y de ahí una recta vertical hasta la recta de operación,creando de esa forma un escalón correspondiente a la primera etapa deextracción. Se repite este procedimiento para crear un segundo escalóncorrespondiente a la segunda etapa de extracción. En este sistema, un refino de0.22 g/l Cu y un orgánico cargado de 4.24 g/l Cu se predicen para una extracciónen dos etapas.
Aunque el diagrama de McCabe-Thiele que se muestra en la figura 4.12representa solamente una aproximación del sistema, resulta muy útil paradeterminar el número de etapas necesarias para alcanzar una cierta extracción.Un diagrama de McCabe-Thiele más preciso podría dibujarse a partir de la figura4.12, dibujando la recta de operación a partir de un punto situado a igual distanciade la isoterma de extracción y de la recta vertical representando la composicióndel refino. Entonces, se hace de nuevo el diagrama de McCabe-Thiele, y seobtienen valores de 0.15 g/l Cu en el refino y 4.17 g/l Cu en el electrolito cargado.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama101
La construcción de un verdadero diagrama de McCabe-Thiele es un procesoiterativo.
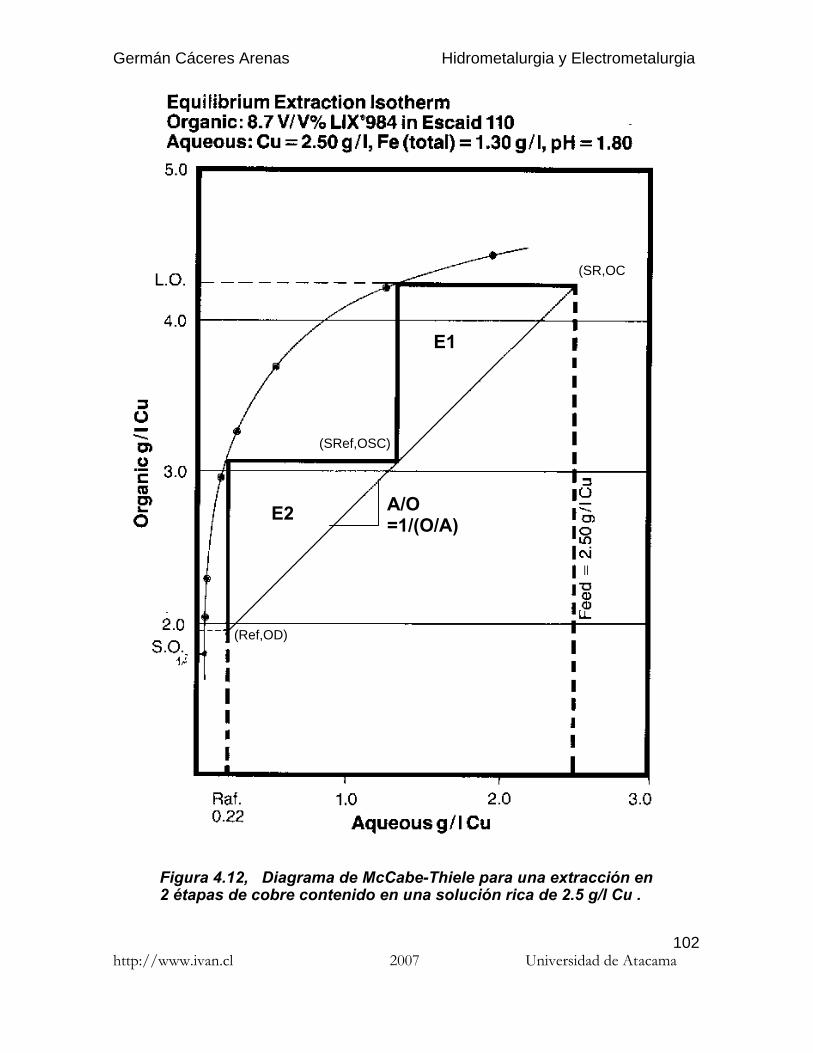
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama102
Figura 4.12, Diagrama de McCabe-Thiele para una extracción en2 étapas de cobre contenido en una solución rica de 2.5 g/l Cu .
E1
E2 A/O=1/(O/A)
(SR,OC)
(Ref,OD)
(SRef,OSC)
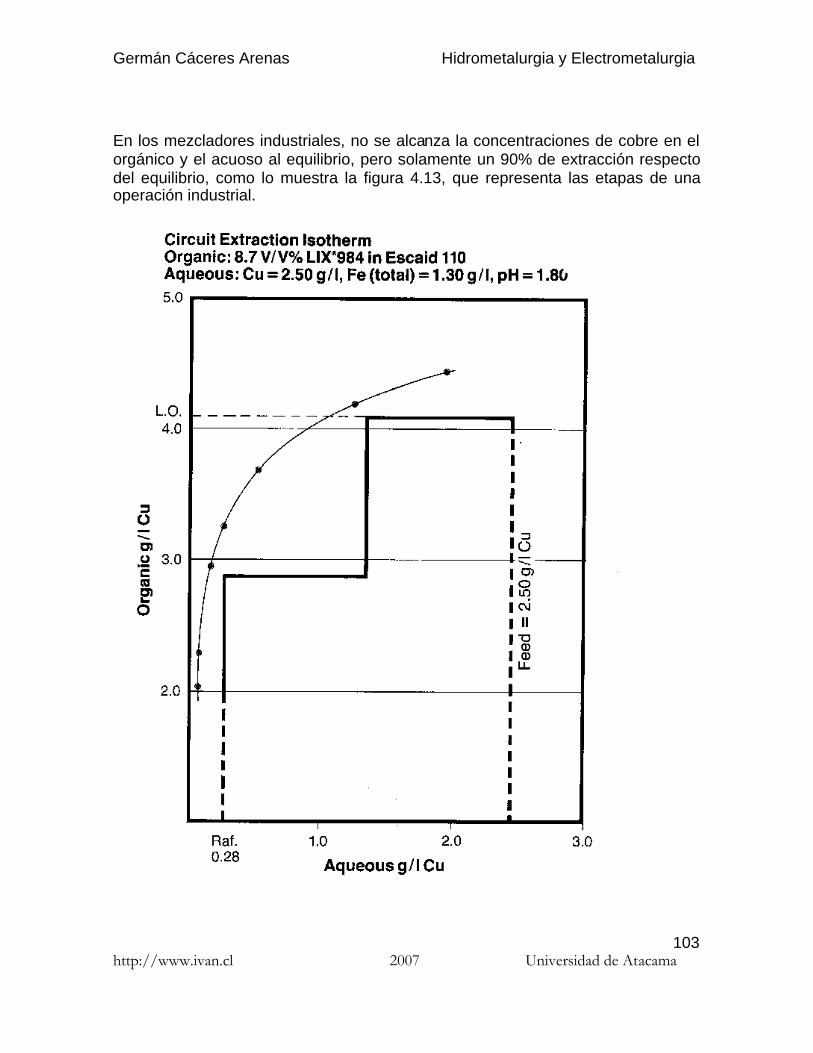
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama103
En los mezcladores industriales, no se alcanza la concentraciones de cobre en elorgánico y el acuoso al equilibrio, pero solamente un 90% de extracción respectodel equilibrio, como lo muestra la figura 4.13, que representa las etapas de unaoperación industrial.
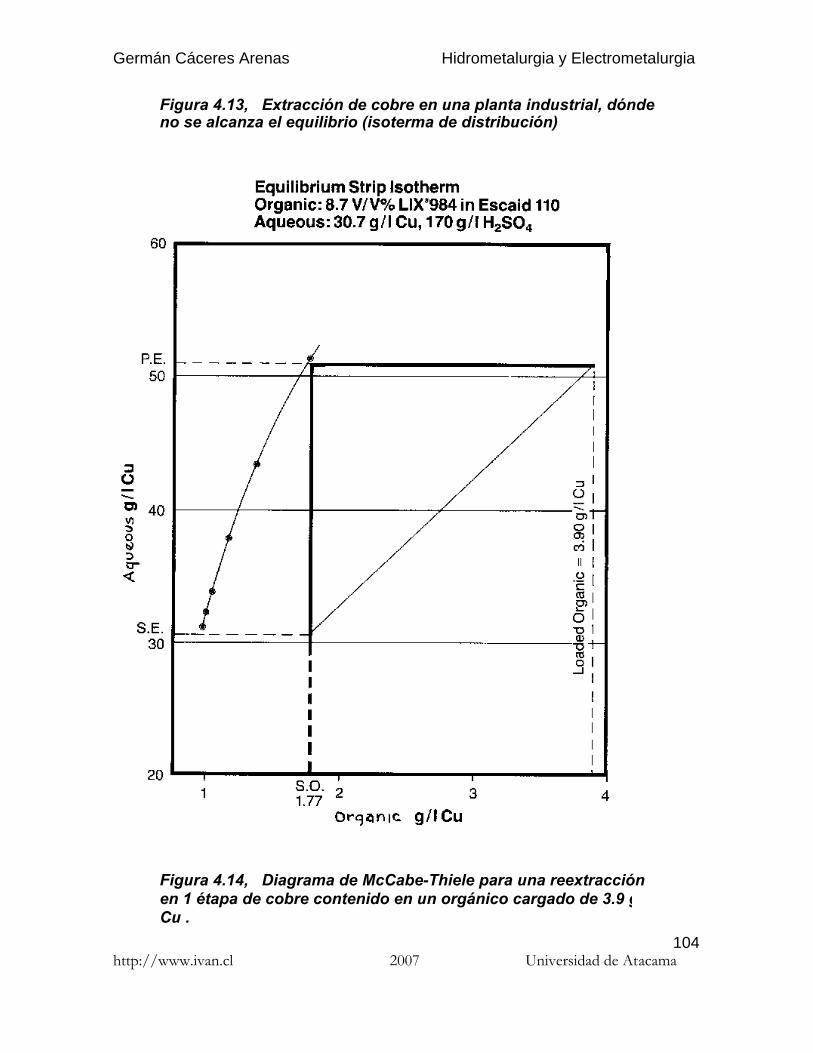
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama104
Figura 4.13, Extracción de cobre en una planta industrial, dóndeno se alcanza el equilibrio (isoterma de distribución)
Figura 4.14, Diagrama de McCabe-Thiele para una reextracciónen 1 étapa de cobre contenido en un orgánico cargado de 3.9 g/lCu .

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama105

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama106
4.6.5. Reactivos utilizados en SX de cobre
4.6.5.1. Química de los extractantes
Los extractantes se disuelven en un solvente orgánico tipo parafina, generalmenteen una proporción de 8 a 25 % v/v (% en volumen).
Los extractantes industriales forman complejos de Cu solubles en la fase orgánica.Actuan según un mecanismo de chelación : toman el ion de cobre entre sus "pinzas " (Fig. 4.16).
Cu2+(A) + 2 RH(O) <=> R2Cu(O) + 2 H+(A)
Figura 4.15, Estructura química general de las Oximas utilizadascomercialmente para la recuperación de cobre.
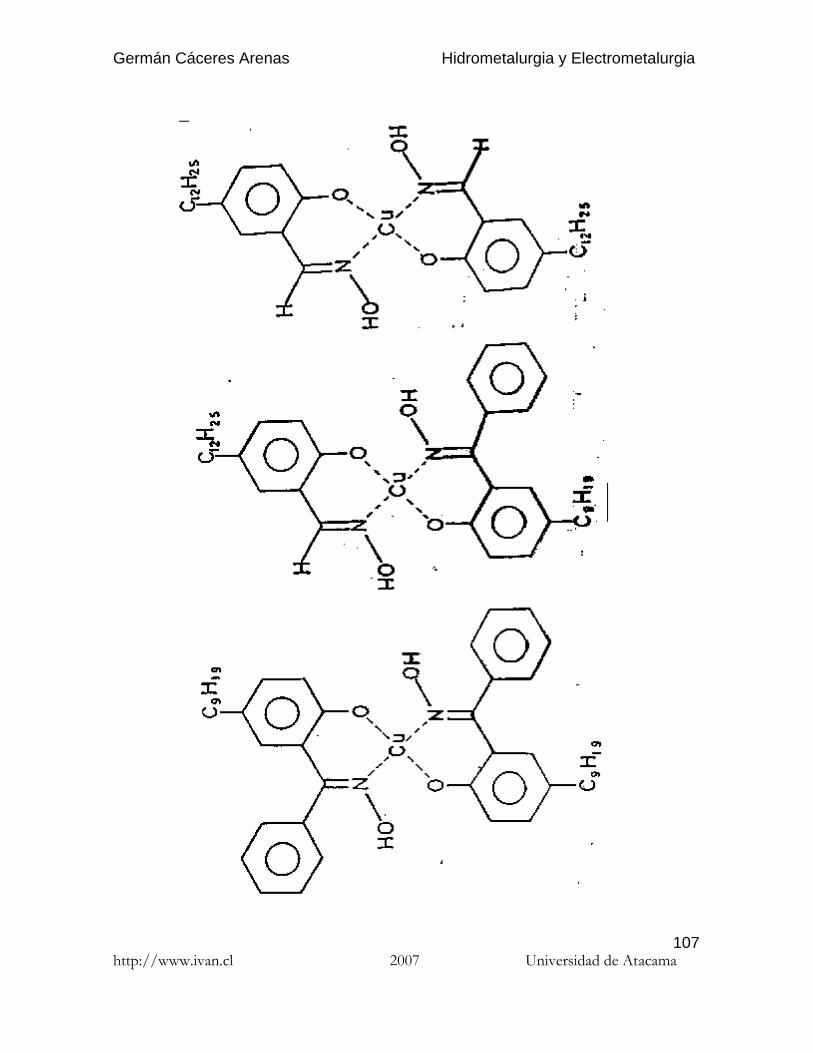
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama107

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama108
Figura 4.16, Estructura de los complejos formados.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama109
4.6.5.2. Tipos y características de reactivos
Reactivos extractantes
Los tipos más usados son : LIX 984N (Henkel), PT 5050 (Acorga) y MOC 45(Allied Signal).
Sus características principales son (en promedio) :
Densidad : 0.91 g/cc
Color : ámbar
Punto de inflamación : 70 °C
Selectividad Cu/Fe : > 2000
Carga máxima (a 10% v/v) : 5.2 g/l de Cu a pH =2
Transferencia neta (10% v/v) : 3.0 g/l de Cu
Separación de fases : < 90 segundos
Cinética de extracción : > 95% en 60 segundos
Diluyentes
Los tipos de diluyentes más usados son : ESCAID 103 (Exxon) y OXIMIN(Oxiquim).
Sus características principales son :
Densidad : 0.81 g/cc
Punto de inflamación : 79 °C
Modificadores
Los modificadores son compuestos que aumentan la solubilidad del extractanteorgánico y del metal en la fase orgánica. Mejoran la velocidad de separación defases y extracción de cobre, favoreciendo la coalescencia.
Los modificadores más usados son el TRIDECANOL y el ESTER.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama110
4.6.5.3. Tipos de extractantes
Ketoximas
Las KETOXIMAS fueron los primeros reactivos usados comercialmente para laextracción de cobre desde soluciones diluidas y fueron usados exclusivamentedurante 12 años. Sus principales propiedades eran : excelente separación defases, bajas pérdidas de orgánico por arrastre y baja formación de crud. Su uso,sin embargo fue limitado, debido a dos desventajas principales : Extractantemoderados de cobre y cinética lenta a bajas temperaturas. El reactivo típico fue elLIX 64N.
Salicilaldoximas
Para superar estas desventajas se desarrollaron las SALICILALDOXIMAS, lo quepermitió reducir circuitos 4E+3S o 3E +2S a 2E+2S o 2E + 1S, incluso para altasconcentraciones de cobre y bajo pH (40 gpl Cu y pH 1.5). Sin embargo, estosreactivos son extractantes tan fuertes que requieren un modificador para realizareficientemente la reextracción.El uso de modificadores (tridecanol o nonifenol) presentan las siguientesdesventajas : hidrólisis y degradación del reactivo, mayor transferencia de Fe alelectrolito, mayor pérdida de orgánico por arrastre físico y mayor formación decrud. Reactivos típicos son el LIX 860 y el Acorga PT-5050.
Mezclas Salicilaldoximas - Ketoximas.
Estas mezclas no contienen modificadores. El LIX 984, por ejemplo, es unamezcla de LIX 860 (salicilaldoximas) y LIX 84 (Ketoxima). Combina la capacidadextractiva y cinética rápida de la salicilaldoxima con la estabilidad y propiedadesfísicas de las Ketoximas.
Una lista de los reactivos actualmente en uso se indica en la tabla 4.4.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama111
Tabla 4.4, Reactivos en uso actualmente
(Revista Mineria Chilena, sept. 1994)
Reactivo Extractante Modificador
Acorga PT5100 Aldoxima Nonil Fenol
PT5050 Aldoxima Tridecanol
M5640 Aldoxima Ester
LIX 84 Ketoxima No
984 Mezcla No
622 Aldoxima Tridecanol
860 Aldoxima No
MOC 45 Ketoxima No
55 Aldoxima No
4.6.6. Arrastres
4.6.6.1. Continuidad
Al mezclarse en un mixer 2 fases inmiscible, una de ellas debe encontrarsedispersa en la otra. Se presentan dos casos :
(1) Cuando la fase acuosa está dispersa en la fase orgánica, se habla decontinuidad orgánica, y
(2) cuando la fase orgánica está dispersa en la fase acuosa, se habla decontinuidad acuosa (Fig. 4.17)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama112
O
A
A
O
Continuidad orgßnica Continuidad acuosa
Figure 4.17, Tipos de continuidad.
4.6.6.2. Recuperación del orgánico
El principal costo de operación en una planta de SX es la reposición deextractante, el cual se pierde por arrastre físico en el refino y electrolito. Diferentessistemas han sido implementados para reducir estas pérdidas.
Electrolito
El mayor énfasis se ha puesto en el electrolito, puesto que el orgánico afecta lacalidad de los cátodos. La práctica normal es pasar el electrolito por filtros rellenoscon sílice, granate y antracita, el cual ha demostrado ser insuficiente por sí solo.Por lo tanto, se han desarrollado e implementado diferentes combinaciones parareducir la transferencia del orgánico al electrolito, por ejemplo :
Uso de post-decantadores o columnas de flotación convencionales o Jamesonantes de los filtros.
Uso de celdas de sacrificio en electroobtención.
Refino
El método normal es recuperar el orgánico arrastrado desde la piscina de refino.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama113
4.6.6.3. Remoción de acuoso
De manera similar a los arrastres de orgánico, existen arrastres de acuoso en lafase orgánica, los que constituyen la principal fuente de contaminación delelectrolito. El Cl y Mn, además de Fe, son actualmente las mayores impurezas. Elmétodo normal de remoción de acuoso es el diseño del estanque de orgánico contiempo suficiente y facilidades para eliminar periódicamente el acuoso acumulado.Este método puede ser suficiente para el Fe, pero no así para el Cl y Mn, cuyasconcentraciones permisibles en el electrolito son muy inferior a las de Fe (30 ppmCl y 100 ppm Mn vs 1.5-2.0 gpl Fe). En este caso se pueden implementar lossiguientes métodos :
Uso de una o más etapas de LAVADO. Diseño del estanque de orgánico como post-decantador. Uso de coalescedor del tipo desarrollado por Chuquicamata. Combinación de cualquiera de las alternativas anteriores, dependiendo de la
concentración de las impurezas presentes.
4.6.6.4. Diseño alternativo de Mezcladores Decantadores
Además de los diseños de Krebs y CMS de Davy, recientemente Outokumpu hapropuesto el VSF (agitación muy suave). Las principales ventajas atribuidas a estediseño son :
Menores arrastres de orgánico y acuoso. Menor formación de crud.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama114
5.1. Introducción
5.1.1. Definición
La ELECTROMETALURGIA consiste en la producción de depósitos metálicosmediante la aplicación de la ENERGIA ELECTRICA.
Se distingue :
La electrometalurgia en solución acuosa :Aplicada fundamentalmente a la producción de Cu, Zn, Ni, Co, Pb, Ag, Au yotros metales menores ( Cd, Cr, Mn, Ga, Ti, Te ).
La electrometalurgia en sales fundidas :Aplicada principalmente a la producción de Al, Li, Mg, Na, K y otros metalesmenores (Tierras raras, Ti, V, W, Zr, Th).
Electrometalurgia

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama115
Figura 5.1 : Producción Mundial aprox. de metales por electrolisis
(Fuente : Journal of Metals - Enero 1985)
5.1.2. Procesos electrometalúrgicos
Según el tipo de depósito obtenido, se distinguen los siguientes PROCESOSELECTROMETALURGICOS :
ELECTROOBTENCION (Electrowinning) de metales :
Consiste en la extracción de metales a partir de soluciones, en forma dedepósitos metálicos puros, densos y compactos o depósitos metálicos en polvo(pulvi-electrometalurgia) o bien, depósitos de compuestos metálicos (óxidos,hidróxidos o sales).
ELECTROREFINACION (Electrorefining) de metales :
Consiste en la obtención de depósitos metálicos de alta pureza a partir de unmetal impuro.
GALVANOPLASTIA (Electroplating) :
Consiste en recubrimientos metálicos delgados con fines anticorrosivos oestéticos (cromados).
ELECTROCONFORMADO (Electroforming) :
Consiste en la elaboración de piezas metálicas especiales por vía electrolítica.
0 1 2 3 4 5 6 7Log. Producción anual (tm/año)
GaIn
TlTe
SbBi
CrAu Cd Co Mn Sn Pb
Ag Ni ZnCu

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama116
5.2 Conceptos Fundamentales
5.2.1. Celdas de electrólisis
Los procesos electrometalúrgicos tienen lugar en unidades llamadas CELDAS DEELECTROLISIS, las cuales se agrupan para constituir la nave o plantaelectrolítica. Una celda de electrólisis está constituida por :
La celda misma : Es un recipiente que contiene el electrolito y los electrodos.En algunos casos, la celda puede ser constituida por dos mitades, conectadasentre sí por un puente salino.
El electrolito : Un medio acuoso, que contiene los iones del metal a depositar yotros iones que migran permitiendo el paso de la corriente entre los electrodos.
El ánodo : Material sólido conductor en cuya superficie se realiza un procesode oxidación con liberación de electrones.
Ejemplo : Zn => Zn2+ + 2 e-
El cátodo : Electrodo sólido conductor en cuya superficie se realiza un procesode reducción con los electrones provenientes del ánodo.
Ejemplo : Cu2+ + 2 e- => Cu
ENERGIA
ELECTRICA
=> PROCESO
ELECTROMETALURGICO
DEPOSITO
METALICO
=>
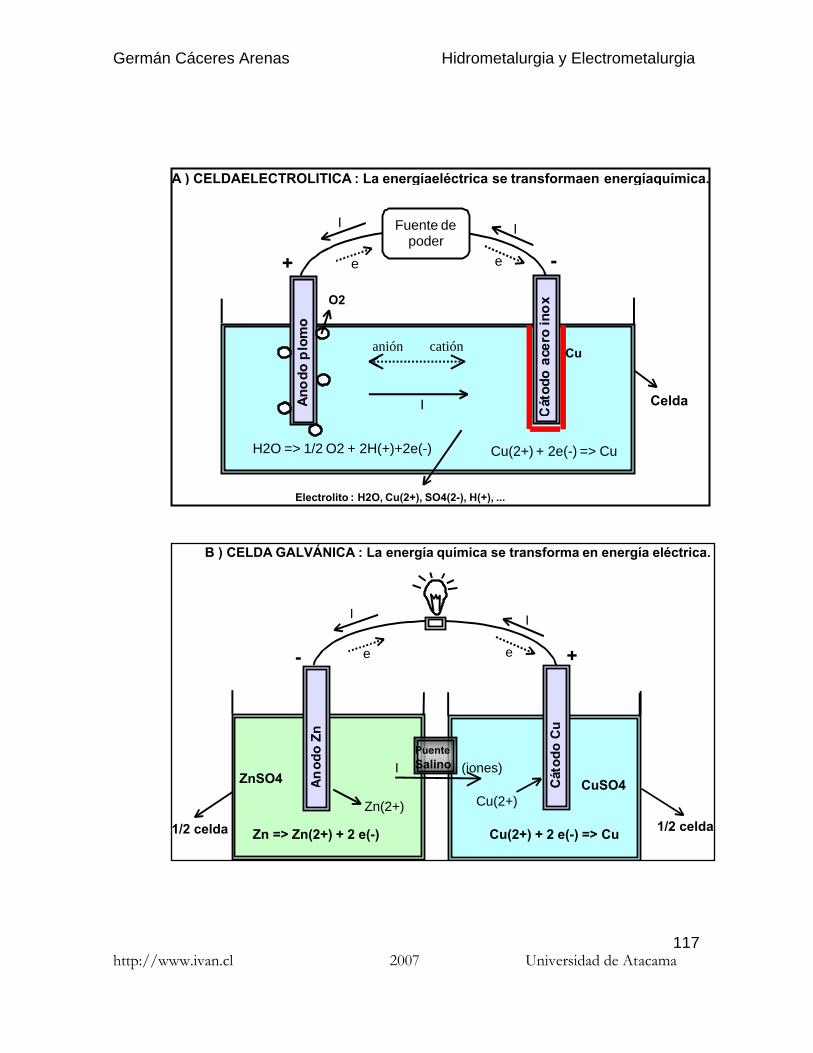
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama117
A ) CELDAELECTROLITICA : La energíaeléctrica se transformaen energíaquímica.
Electrolito : H2O, Cu(2+), SO4(2-), H(+), ...
Ano
dop
lom
o
Cát
odo
acer
oin
ox
I
anión
+
I I
ee
H2O => 1/2 O2 + 2H(+)+2e(-) Cu(2+) + 2e(-) => Cu
Celda
-
Cu
O2
Fuente depoder
catión
B ) CELDA GALVÁNICA : La energía química se transforma en energía eléctrica.
I I
ee
An
odo
Zn
Cát
odo
Cu
PuenteSalino
1/2 celda 1/2 celdaZn => Zn(2+) + 2 e(-) Cu(2+) + 2 e(-) => Cu
ZnSO4 CuSO4
Zn(2+) Cu(2+)
I
- +
(iones)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama118
Figura 5.2 : Celdas electrolitica y galvánica
( ej. : electrodepositación de cobre y pila de Daniel (1830) ).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama119
Proceso Celda EW Pila
Anodo Oxidación + -Cátodo Reducción - +
5.2.2. Proceso electroquímico
Un proceso de naturaleza electro-química se caracteriza por presentar larealización simultánea de dos reacciones denominadas anódicas y catódicas. Enla primera sucede una transformación química de oxidación y se liberanelectrones. La reacción catódica involucra un proceso químico de reducción conparticipación de los electrones liberados en el ánodo y que viajan porCONDUCTORES ELECTRÓNICOS (cables) que unen el cátodo con el ánodo.
En la solución, no hay desplazamiento de electrones, sino que los iones sedesplazan en la solución. Los aniones (-) van hacia el electrodo de carga positiva ylos cationes (+) hacia el electrodo de carga negativa. El electrolito es unCONDUCTOR IONICO.
Los procesos electroquímicos pueden ser clasificados en dos tipos según sean ono espontáneos. Los primeros suceden en forma natural y la celda se denominaGALVÁNICA o PILA. Los no espontáneos se realizan por medio de la aplicaciónde corriente externa y se realizan en una celda llamada ELECTROLITICA. En lafigura 5.2, se ilustran ejemplos de procesos galvánicos y electrolíticos.
Los procesos de electrodepositación de metales no son espontáneos y necesitanun aporte de energía eléctrica para ser forzados a ocurrir, por lo cual se estudianlas celdas electrolíticas en estos apuntes. La FUENTE DE ENERGIA ELECTRICAdebe proporcionar corriente continua o directa (DC) a la celda, permitiendo el flujoforzado de electrones entre el ánodo y el cátodo dónde son consumidos. En formasimple, la fuente de energía actúa como bomba impulsora de electrones quefluyen por los conductores y los electrodos.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama120
5.3. Cantidad de metal depositado o disuelto
5.3.1. Ley de Faraday
La ley de Faraday establece que la masa de metal depositado es proporcional a lacantidad de corriente que circula a través de la celda y al tiempo de operación dela electrólisis.
Se expresa así :
mF : masa depositada (g)M : Peso molecular del metal depositadon : Valencia del ion metálico en la soluciónF : Constante de Faraday (96487 Coulomb/equivalente)
(1 coulomb = 1 A x 1 s)I : Corriente que circula (A)t : Tiempo de operación de la electrólisis (s)
El equivalente electroquímico (EEQ) de la substancia transformada, se definecomo la cantidad de sustancia que es afectada por el paso de una corriente de 1 Aen 1 hora. El equivalente electroquímico de un metal queda determinado por lassiguientes constantes :

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama121
Para el caso del cobre (EEQ = 1.18 kg/kA.h), la expresión anterior queda :
mF : masa de cobre depositada (kg)I : Corriente que circula (kA)t : Tiempo de operación de la electrólisis (h)
5.3.2. Eficiencia de corriente
La masa que se obtiene con la ecuación de Faraday (mF) es teórica oestequiométrica, ya que considera que toda la corriente que circula se aprovechasolo para depositar el metal, pero en los procesos reales de EW de cobre, hayreacciones parásitas y la masa depositada realmente (mR) es menor a lo que seesperaba.
La EFICIENCIA DE CORRIENTE se define como la razón entre la cantidad decobre depositada y la que se debería haber depositado teóricamente según la leyde Faraday :
Como concepto, indica la fracción de corriente que es efectivamente utilizada en lareacción de depositación de cobre. Así, por ejemplo, si la EC es de 0.8 (80%),significa que solamente el 80% de la corriente está siendo útilmente utilizado y el20% restante está siendo empleado en reacciones paralelas o parasitarias, fugas,etc.
Por ejemplo, en EW de Cu,
Reacción principal : Cu2+ + 2 e- => Cu 97 % IReacción parásita : Fe3+ + 1 e- => Fe2+ 3 % I

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama122
5.4. Aspectos termodinámicos
Los aspectos termodinámicos de los PROCESOS ELECTROMETALURGICOSestán basados en la TERMODINAMICA ELECTROQUIMICA que estudia losprocesos de electrodo en equilibrio y cuyas conclusiones más importantes,relacionadas con los procesos de electroobtención y electrorefinación de metales,se resumen en la escala normal de potenciales, la ley de Nernst (capitulo 2.2) y enlos diagramas de equilibrios potencial - pH (diagramas de Pourbay, capitulo 2.3).
5.4.1. Potencial de electrodo
Se llama electrodo, el sistema bi-fásico constituido por un conductor electrónico(metal o semiconductor) sumergido en un conductor ionico (electrolito), como semuestra en la figura 5.3.
Cuando no hay corriente, se crea una doble capa eléctrica en la interfaceelectrodo/solución. Entre el conductor electrónico y el conductor ionico seestablece una diferencia de potencial , que es una manifestación del equilibrioentre el metal en el electrodo y sus iones en solución :
= metal -solución
Llamado potencial absoluto del electrodo, que no se puede medir y que sólo sepuede comparar su valor con otro potencial absoluto de electrodo que sea fijo yreproducible, llamado potencial absoluto de referencia, ref.
Se llama entonces potencial relativo o simplemente potencial del electrodo, ladiferencia de potenciales absolutos de este electrodo y del electrodo de referencia:
E = metal -ref = -ref
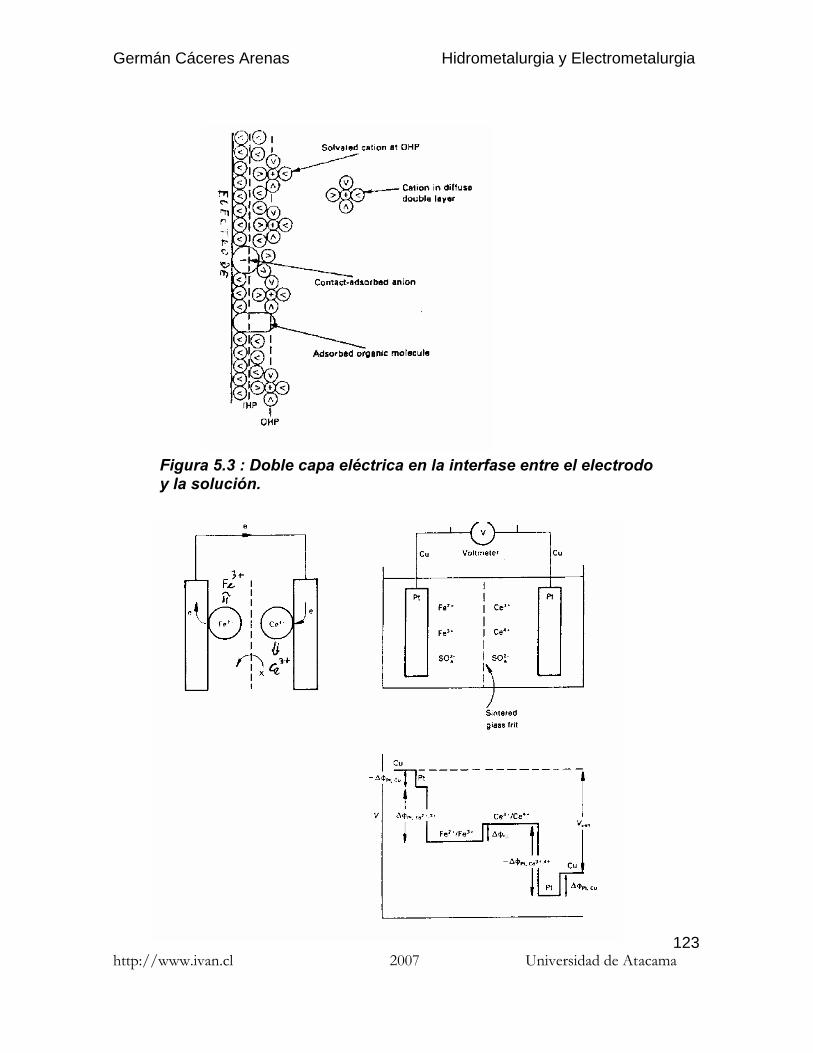
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama123
Figura 5.3 : Doble capa eléctrica en la interfase entre el electrodoy la solución.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama124
Figura 5.4 : Reacciónentre el fierro (II) y elcerio (iV), y voltajeabsoluto en cadapunto de la celda.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama125
5.4.2. Electrodos de referencia
El electrodo de referencia utilizado para todos los cálculos termodinámicos es elelectrodo normal a hidrogeno, constituido por un electrodo de Pt sobre la cual seproduce un despredimiento de hidrógeno, en medio ácido ....
Por convención, el ENH tiene un potencial E = 0.
ENH H+/H2 E = 0 mV
Sin embargo, en los laboratorios, se usan otros electrodos de referencia deutilización más fácil y que tiene un potencial fijo respecto al ENH :
ECS Hg... E = ... Plata Ag/AgCl/Cl- E = 208 mV
En la práctica, no se habla de E, pero delpotencial que tiene un electrodo con respecto auna cierta referencia.
Por ejemplo, E°Cu/Cu2+
= 340 mV con respecto a ENH= 132 mV con respecto a Ag/AgCl/Cl-
5.4.3. Escala normal de potenciales
Para construir la escala normal de potenciales de electrodos, se tomó porconvención el electrodo normal de hidrógeno como referencia. Se midió entonces ,en condiciones experimentales estándar ([iones] =1M, T = 298 K, P= 1 atm) elpotencial de electrodo E°, de todos los electrodos metálicos y no metálicos (Figura5.5).
E (mV)
E° Cu/Cu2+
Eref Ag/AgCl/Cl-
ENH
340
208
0
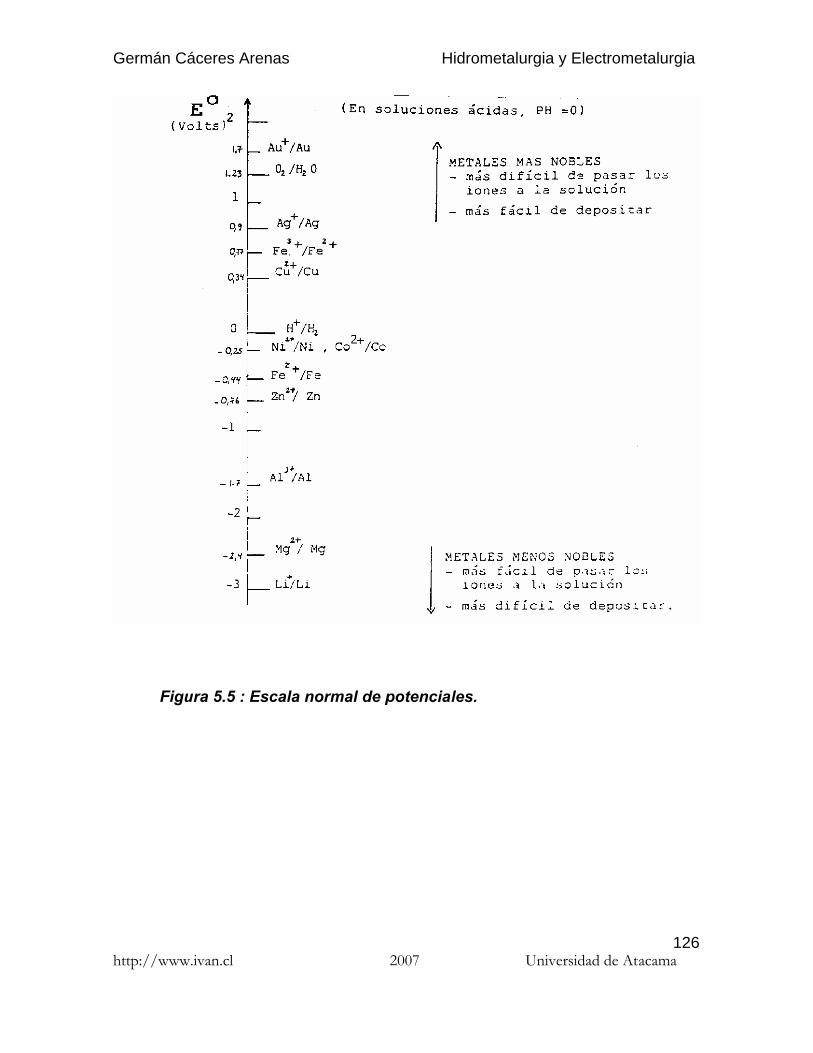
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama126
Figura 5.5 : Escala normal de potenciales.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama127
5.5. Aspectos cinéticos
Los aspectos cinéticos de los procesos electrometalurgicos están basados en laCINETICA ELECTROQUIMICA que estudia los procesos de electrodos cuandoéstos se encuentran fuera del equilibrio; es decir cuando a través del electrodocircula una corriente I.
5.5.1. Densidad de corriente
Intensidad de corriente que fluye o pasa por unidad de superficie de electrodo.
SI
i
i : densidad de corriente (A/m2)I : corriente que pasa por el electrodo (A)S : superficie del electrodo (m2)
La densidad de corriente es proporcional a la velocidad de la reacción que seproduce sobre el electrodo (ver ley de Faraday).
=> En electrometalurgia, la densidad de corriente i es equivalente a la velocidadde la reacción.
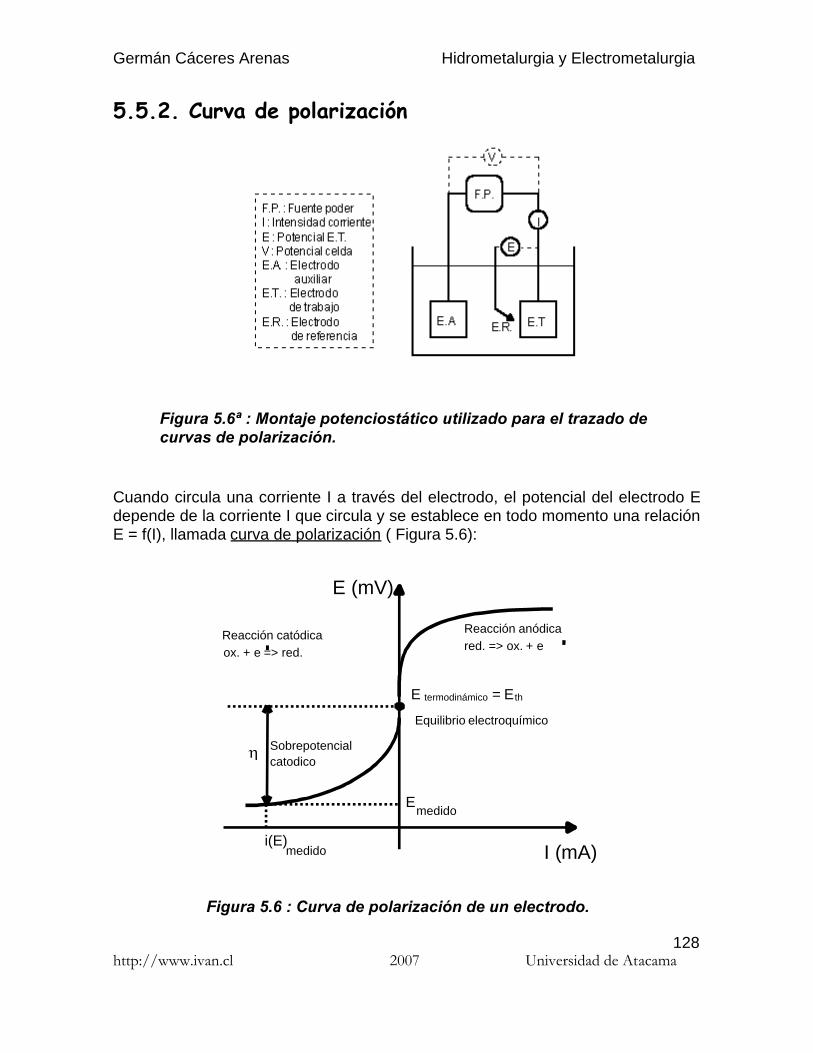
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama128
5.5.2. Curva de polarización
Figura 5.6ª : Montaje potenciostático utilizado para el trazado decurvas de polarización.
Cuando circula una corriente I a través del electrodo, el potencial del electrodo Edepende de la corriente I que circula y se establece en todo momento una relaciónE = f(I), llamada curva de polarización ( Figura 5.6):
Reacción anódicared. => ox. + e
Reacción catódicaox. + e => red.
E (mV)
I (mA)
E termodinámico = E th
i(E)
E
Sobrepotencialcatodico
medido
medido
Equilibrio electroquímico
Figura 5.6 : Curva de polarización de un electrodo.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama129
En consecuencia, el paso de corriente I, o mejor el paso de la densidad decorriente i, modifica el potencial de equilibrio del electrodo o potencialtermodinámico, Eth (dado por la formula de Nernst) en un valor denominadosobrepotencial, dado por :
(i) = E(i) - Eth
depende de i (figura 5.6).
Se llama potencial de abandono Ei=0 al potencial que tiene un electrodo cuando lacorriente es nula. Ei=0 es igual a Eth solamente si el electrodo participa en unareacción bien definida ( ej. : depositación y disolución del cobre); sino Ei=0 y Ethson diferentes ( ej. : desprendimiento de oxigeno sobre ánodo de plomo omedición del Eh sobre electrodo de platino).
Se llama polarización , la diferencia entre el potencial del electrodo cuando pasauna cierta corriente y el potencial al abandono :
(i) = E(i) - Ei=0
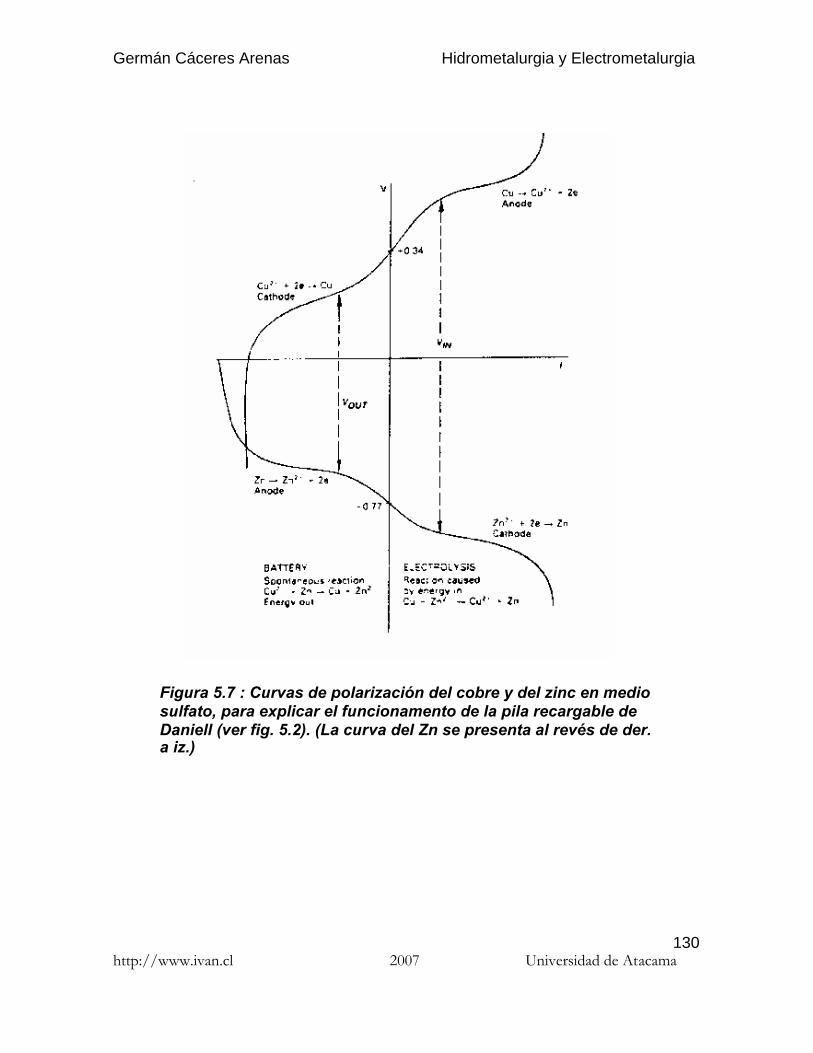
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama130
Figura 5.7 : Curvas de polarización del cobre y del zinc en mediosulfato, para explicar el funcionamento de la pila recargable deDaniell (ver fig. 5.2). (La curva del Zn se presenta al revés de der.a iz.)
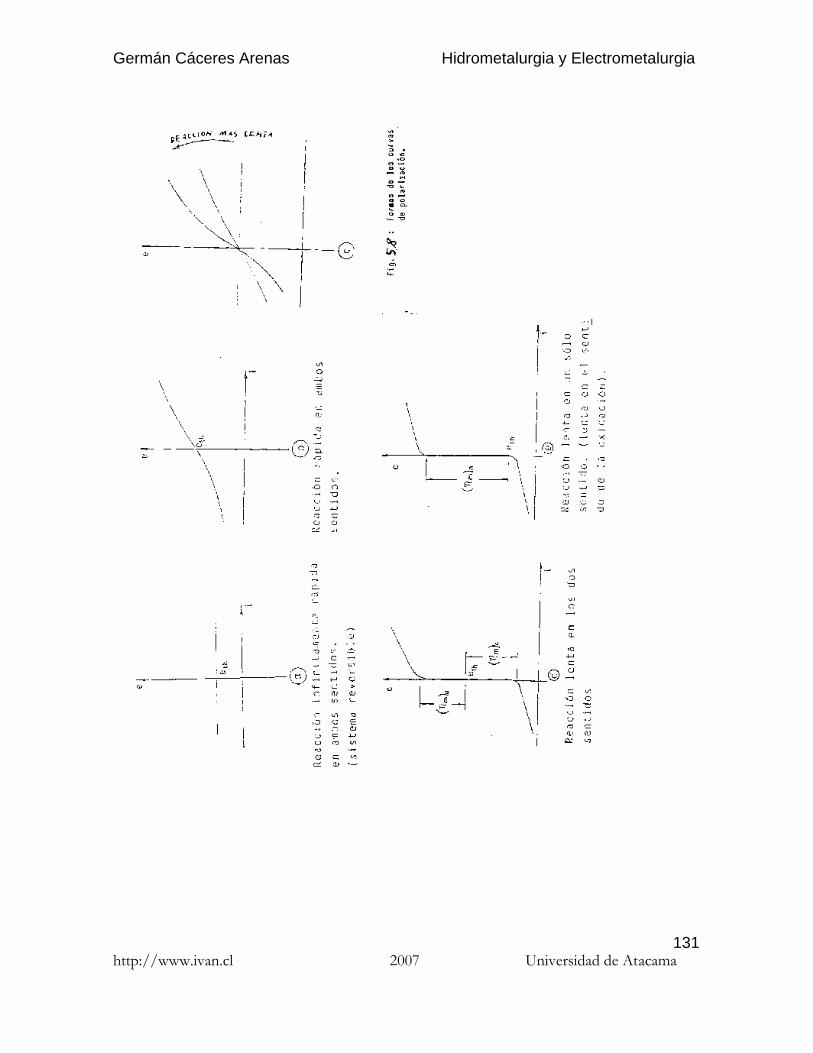
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama131

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama132
5.5.3. Ley de Tafel
La ley de Tafel (1905) es una ecuación empírica que describe las curvas depolarización.
Figura 5.10 : Curvas de Tafel (Coordenadas lineales).

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama133
5.5.4. Mecanismos de la reacción de electrodo
Las reacciones de electrodos se caracterizan por realizarse en una interfase sólido/ acuoso donde se intercambia materia y cargas eléctricas. La velocidad de unareacción de electrodo depende esencialmente de dos procesos básicos o etapasdel mecanismo de la reacción :
Transporte de especies reactantes desde el seno del electrolito hacia lasuperficie del electrodo.
Transferencia o entrega de electrones en la superficie del electrodo.
La etapa más lenta determina la cinética de la reacción y se denomina etapacontrolante.
5.5.5. Régimen de difusión pura
En esta situación, la etapa lenta es el transporte de la especie reactante por lacapa limite adyacente al electrodo (control por difusión). La concentración dereactante en el sitio mismo de reacción es cercana a cero, y la velocidad de lareacción va a depender de la rapidez con que se acerque el reactante al sitio dereacción, lo que esta determinado por el transporte por difusión según la ley deFick :
i : densidad de corriente límite ( cuando se impone unpotencial muy grande a la celda de electrólisis)
D : coeficiente de difusiónC : concentración del ion en solución : espesor de la capa limite
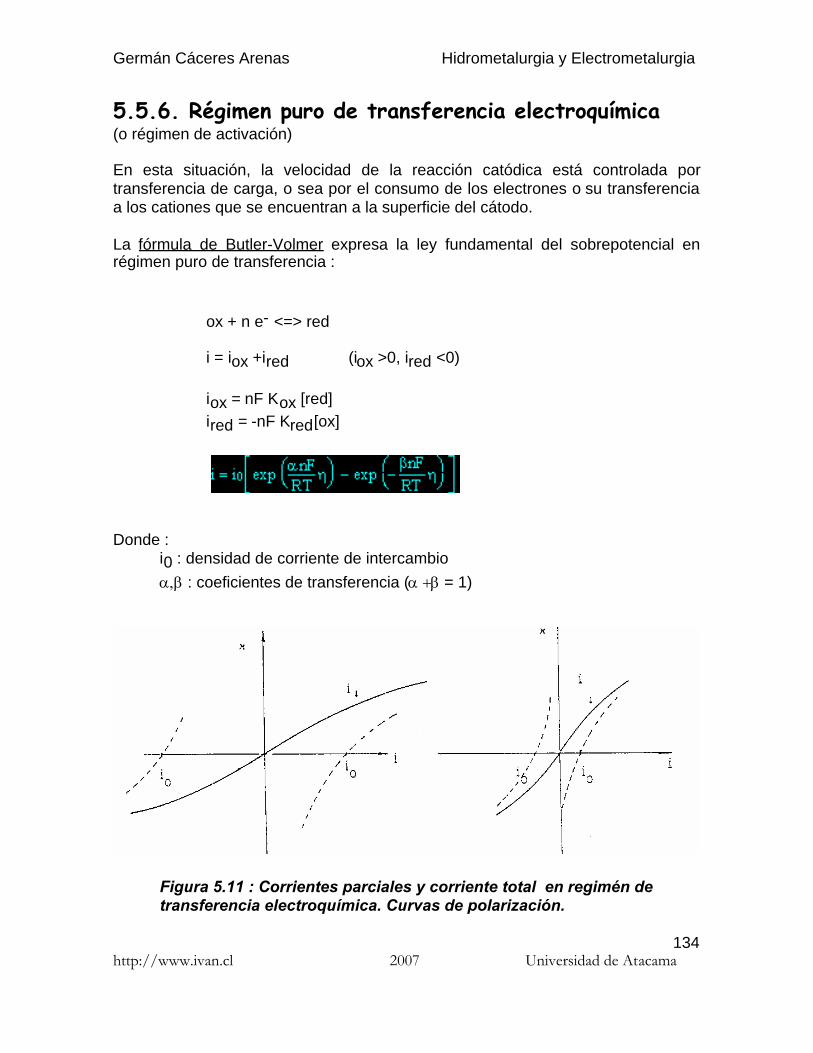
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama134
5.5.6. Régimen puro de transferencia electroquímica(o régimen de activación)
En esta situación, la velocidad de la reacción catódica está controlada portransferencia de carga, o sea por el consumo de los electrones o su transferenciaa los cationes que se encuentran a la superficie del cátodo.
La fórmula de Butler-Volmer expresa la ley fundamental del sobrepotencial enrégimen puro de transferencia :
ox + n e- <=> red
i = iox +ired (iox >0, ired <0)
iox = nF Kox [red]ired = -nF Kred[ox]
Donde :i0 : densidad de corriente de intercambio: coeficientes de transferencia (= 1)
Figura 5.11 : Corrientes parciales y corriente total en regimén detransferencia electroquímica. Curvas de polarización.
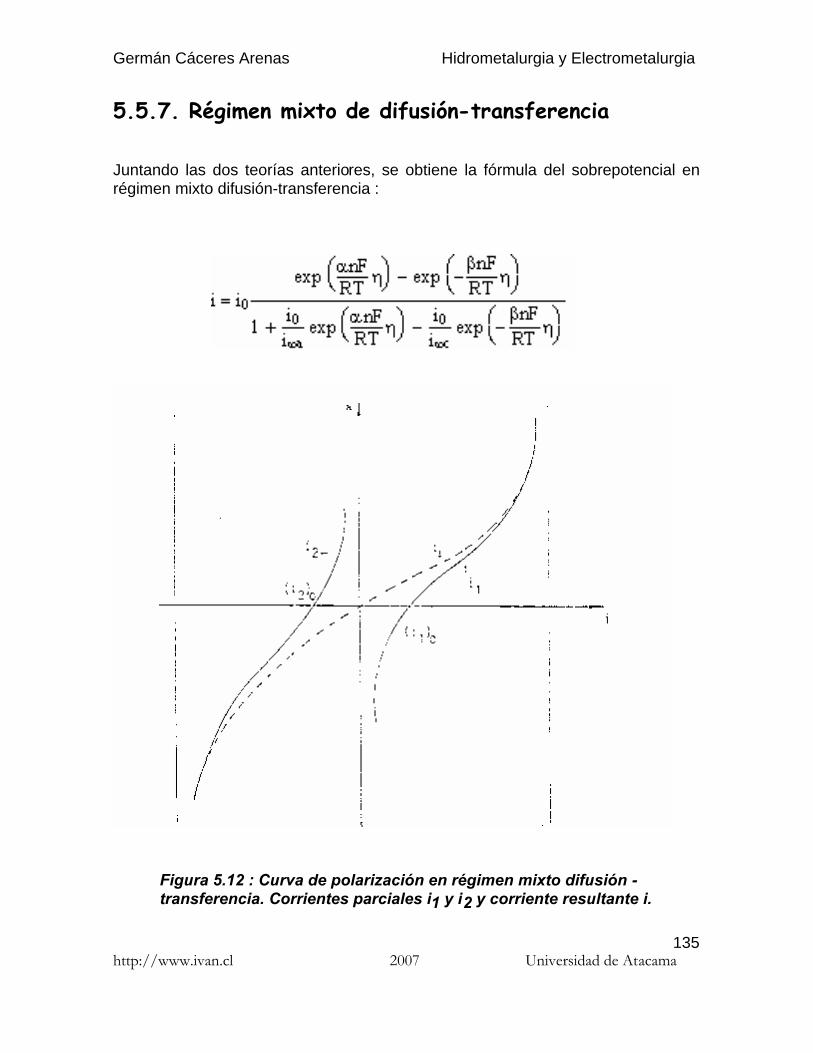
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama135
5.5.7. Régimen mixto de difusión-transferencia
Juntando las dos teorías anteriores, se obtiene la fórmula del sobrepotencial enrégimen mixto difusión-transferencia :
Figura 5.12 : Curva de polarización en régimen mixto difusión -transferencia. Corrientes parciales i1 y i2 y corriente resultante i.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama136
5.6. Electroobtención de cobre(Electrowinning)
5.6.1. Descripción del proceso
El proceso de electroobtención de cobre consiste básicamente en latransformación electroquímica del cobre disuelto en un electrolito en cobremetálico depositado en un cátodo, mediante la utilización de energía eléctricaproveniente de una fuente externa. El cobre iónico (Cu2+) del electrolito esdepositado selectivamente sobre la superficie del cátodo y a la vez sedescompone agua en oxigeno y ácido sulfúrico en la superficie de ánodosinsolubles de plomo. Este proceso electrolítico y las reacciones involucradas sepresentan esquemáticamente en la figura 5.13 . Dado que el cobre es más bien unmetal noble ( E° = 0.34 V), el proceso de electroobtención es relativamente simpley puede ser realizado sin peligro de desprendimiento de hidrogeno ( E° = 0V).
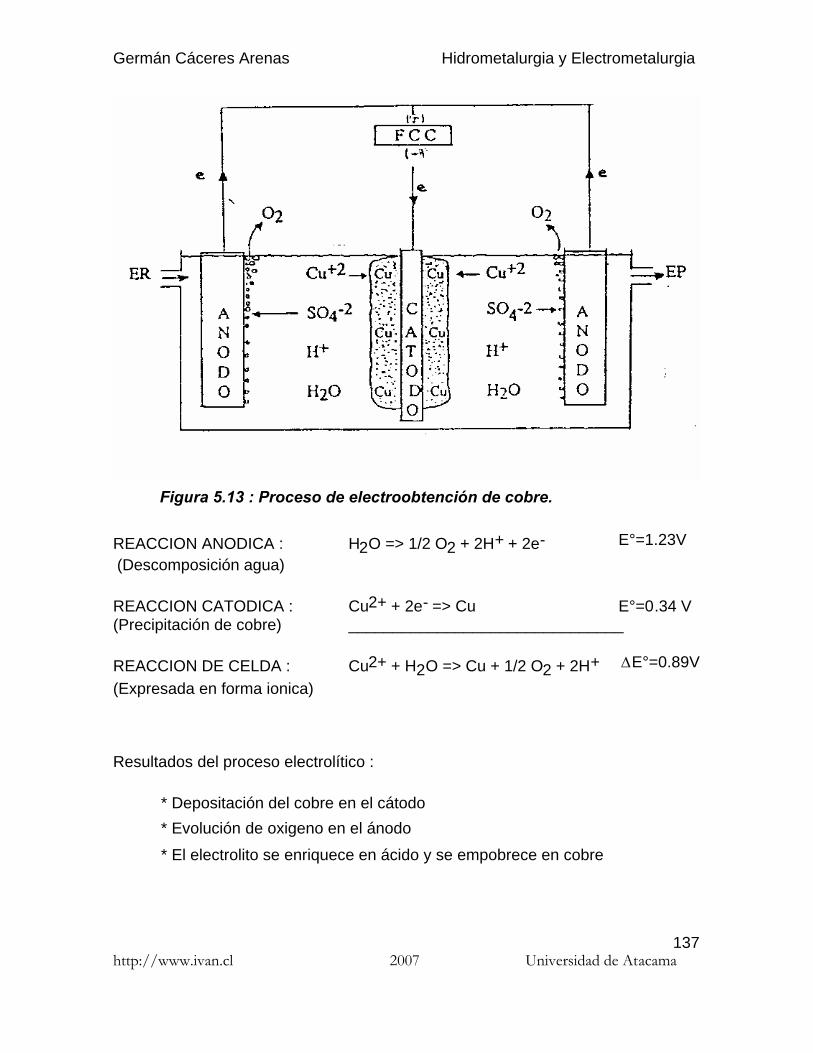
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama137
Figura 5.13 : Proceso de electroobtención de cobre.
REACCION ANODICA : H2O => 1/2 O2 + 2H+ + 2e- E°=1.23V
(Descomposición agua)
REACCION CATODICA : Cu2+ + 2e- => Cu E°=0.34 V(Precipitación de cobre) _______________________________
REACCION DE CELDA : Cu2+ + H2O => Cu + 1/2 O2 + 2H+ E°=0.89V
(Expresada en forma ionica)
Resultados del proceso electrolítico :
* Depositación del cobre en el cátodo
* Evolución de oxigeno en el ánodo
* El electrolito se enriquece en ácido y se empobrece en cobre

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama138
El proceso de EW, se lleva a cabo en una celda electrolítica, donde circulacontinuamente el electrolito acuoso que contiene disuelto CuSO4 y H2SO4 y queproviene de la planta SX para el caso de nuestro interés en este curso. En la celdaocurren reacciones electroquímicas de oxidación - reducción provocadas por laenergía eléctrica. En el cátodo el ion cúprico es reducido a cobre por loselectrones suplidos por la corriente y que vuelven dicho electrodo de polaridadnegativa. En el electrodo positivo hay un déficit de electrones, y se descomponeagua generándose oxigeno gaseoso que burbujea en la superficie del ánodo yademás ácido sulfúrico, de acuerdo a la reacción neta global :
REACCION DE CELDA O GLOBAL : CuSO4 + H2O => Cu + H2SO4 + 1/2 O2(Expresada en forma molecular)
Como resultado de la estequiometria de la reacción anterior, por cada tonelada decobre depositada en los cátodos,
Se generan : 1 T Cobre metálico1.54 T ácido sulfúrico0.25 T oxigeno (5.6 Nm3)
Se descomponen : 0.28 T aguaSe consumen : 1900 - 2300 kw/h (costo : +/- 20$pesos / kw/h)
El electrolito es una solución ácida de sulfato de cobre que contiene entre 30 y 50g/l de Cu2+ y 130 a 160 g/l de H2SO4; la temperatura de trabajo es del orden de40 °C. En el proceso de EW, otras sustancias denominadas aditivos (sulfato decobalto y guarfloc) pueden ser agregadas al electrolito para mejorar los resultadosdel proceso.
La densidad de corriente se mantiene entre 250 y 300 A/hm2, para tener laproducción más alta posible compatible con una buena calidad del depósito.
En cada celda, los cátodos de acero inoxidable (1 m2) se posicionan entre dosánodos de plomo aleado y permanecen alrededor de 6-7 días recibiendo cobre ensus dos caras, logrando cosechar un peso de 45 a 55 kg de cobre catódico porcara. Los cátodos cosechados cosechados posteriormente se someten a lasoperaciones de lavado, despegue de las láminas de cobre, encerado del bordeinferior y retorno a la celda.
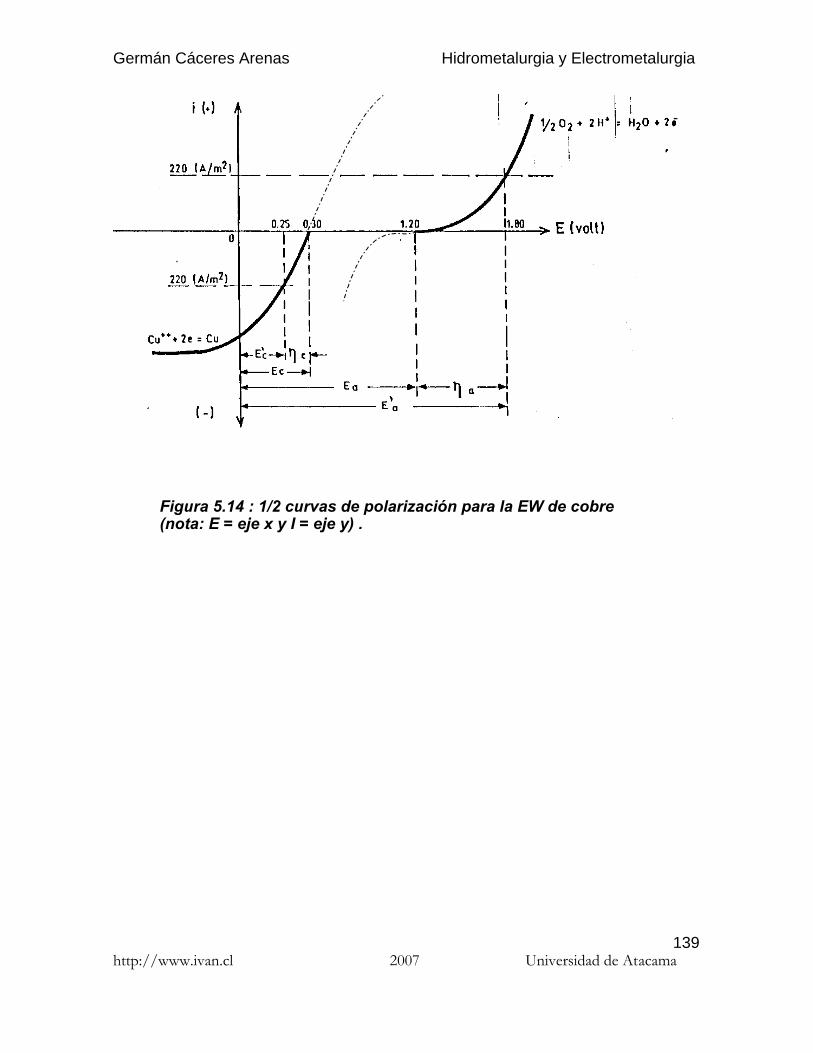
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama139
Figura 5.14 : 1/2 curvas de polarización para la EW de cobre(nota: E = eje x y I = eje y) .

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama140
5.6.2. Composición del voltaje de celda.
Eth = Ethanódico - Ethcatódico
Vcelda = Eth + A + |c| + RI + pérdidas
= +/- ( 0.89 + 0.6 + 0.1 + 0.4 + 0.1) V= +/- 2.1 V
Ethanódico : Potencial termodinámico ánodo (Nernst)Ethcatódico : Potencial termodinámico cátodoR : Resistencia electrolito (m2)I : Densidad de corriente (A/m2)Pérdidas : perdidas en los conductores externos (contacto).
5.6.3. Calidad de los cátodos producidos
La EW, como etapa final del proceso Hidrometalurgico, tiene entre sus objetivosproducir cátodos de cobre de alta pureza para maximizar los resultadoseconómicos de venta del producto. Los procesos LX/SX/EW han logrado undesarrollo y potencialidad para producir cobre de alta pureza con una calidadsuperior o similar al cobre electrorefinado. El cátodo “grado A” contiene más de99.96 % Cu.
En general, los cátodos electroobtenidos producidos por medio de SX/EWpresentan bajos niveles de impurezas de baja tolerancia, como son los elementos:arsénico (As), selenio (Se), bismuto (Bi) y antimonio (Sb). Las impurezas que másproblemas presentan son el plomo (Pb), azufre (S) y fierro (Fe). Los niveles deestas últimas impurezas en los cátodos, están influenciadas significativamente porla práctica operacional empleada en las plantas de electroobtención.
Debemos tener presente que tanto el plomo como el azufre, son elementos queobligatoriamente deben estar en la celda para el funcionamiento del proceso. Elprimero es el principal constituyente del ánodo que se ubica cercano al cátodo, yel azufre es uno de los elementos del ácido sulfúrico y de los iones sulfatos que seencuentran disueltos en el electrolito..

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama141
La calidad química de los cátodos está ligada fuertemente a la calidad física oapariencia presentada por el deposito; estableciéndose en la práctica operacional,que un deposito liso, denso y coherente, presenta mejor calidad química que otrorugoso, poroso e incoherente. Eso se debe a que, en el primer caso, la soluciónque contiene iones sulfatos y partículas de plomo, no queda trampeada enposibles huecos del depósito.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama142
5.7. Electrorefinación de cobre
5.7.1. Introducción
La ELECTROREFINACION de un metal consiste en la disolución anódica delmetal impuro y el posterior depósito catódico de este metal puro.
El proceso de refinación acuosa del cobre es el más importante, puesprácticamente toda la producción de cobre por pirometalurgia es electrorefinada. Amenor escala, se practica también la electrorefinación de Pb, Ni, Ag, Au y otrosmetales menores.
La electrorefinación de cobre tiene dos objetivos :
Eliminar las impurezas que perjudican las propiedades electricas y mecánicasdel cobre, tales como : As, Sb, Bi, O, S, Se, Te, Fe, Co, Ni y Pb.
Recuperar las impurezas valorizables, como : Au, Ag, metales del grupo del Pty Se.
5.7.2. Descripción del proceso
Los ánodos se moldean en la fundición y son de cobre impuro (99.4 a 99.8 % Cu)y los cátodos son láminas de cobre puro (99.98 % Cu) u de acero inoxidable sobreel cual se va a depositar el cobre puro.
REACCION ANODICA : Cu => Cu2+ + 2e- E° = 0.34 V(Disolución de cobre)
REACCION CATODICA : Cu2+ + 2e- => Cu E° = 0.34V(Precipitación de cobre) _______________________________
REACCION DE CELDA O GLOBAL : Cu => Cu E = 0 V(Expresada en forma ionica)
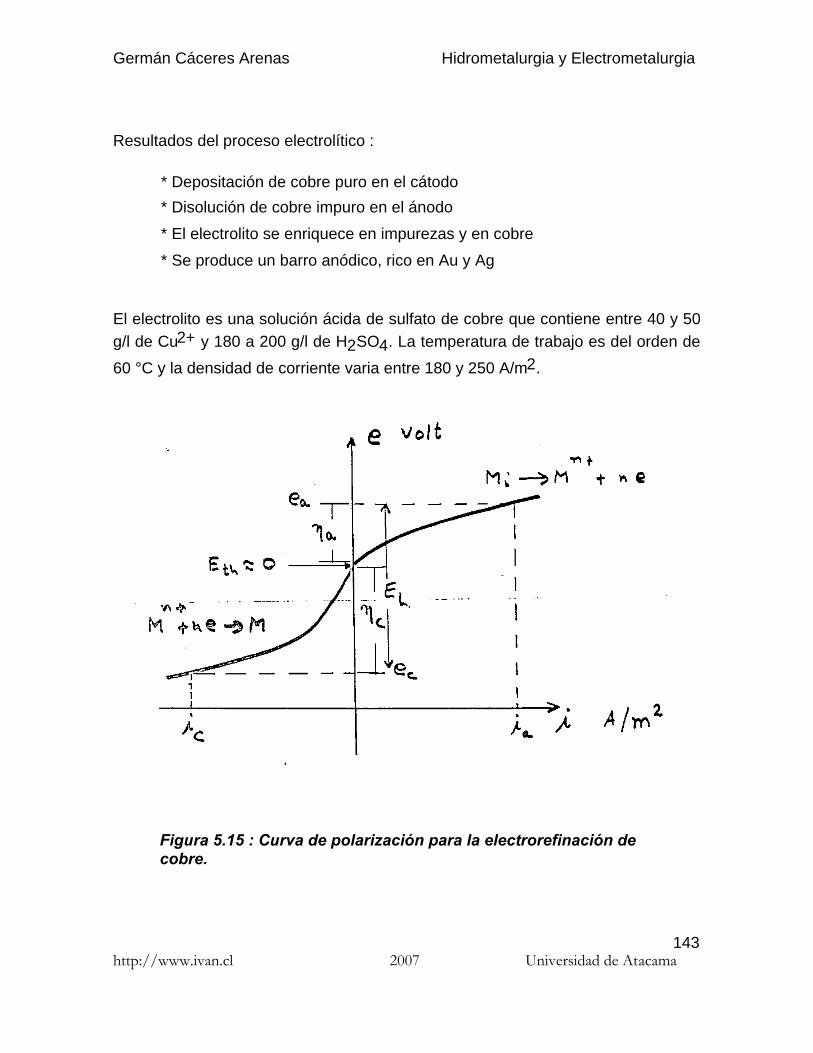
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama143
Resultados del proceso electrolítico :
* Depositación de cobre puro en el cátodo
* Disolución de cobre impuro en el ánodo
* El electrolito se enriquece en impurezas y en cobre
* Se produce un barro anódico, rico en Au y Ag
El electrolito es una solución ácida de sulfato de cobre que contiene entre 40 y 50g/l de Cu2+ y 180 a 200 g/l de H2SO4. La temperatura de trabajo es del orden de
60 °C y la densidad de corriente varia entre 180 y 250 A/m2.
Figura 5.15 : Curva de polarización para la electrorefinación decobre.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama144
5.7.3 Comportamiento de las impurezas
Tabla 5.1 :
Rangos de composición industrial de ánodos y cátodos de cobre.
Elemento Ánodos( rango de %)
Cátodos(rango de %)
Cu 99.4 - 99.8 > 99.99O 0.1 - 0.3 no analisadoNi < 0.5 <0.0010Pb < 0.1 < 0.0005As < 0.3 < 0.0002Sb < 0.3 < 0.0002Se < 0.02 < 0.0002Fe 0.002 - 0.03 0.0002 – 0.0020Te < 0.001 < 0.0001S 0.001 - 0.003 0.0004 - 0.0010Bi < 0.01 < 0.0001Ag < 0.1 0.0005 - 0.001Au <0.005 < 0.00001
Durante el electrólisis, el cobre y los metales menos nobles que él : As, Sb, Bi, Fe,Co, Ni, Pb, pasan desde el ánodo a la solución; mientras que los metales másnobles como oro y plata, y los sulfuros, selenurios y teluros de cobre y plata, muyrefractarios a la disolución electroquímica, no se disuelven, decantan y pasan albarro anódico.
El cobre es depositado sobre el cátodo; mientras que los metales menos noblescomo Fe, Ni y As permanecen en la solución. Un circuito de purificación delelectrolito permite eliminar estos elementos y evitar así que su concentraciónaumente en solución con el tiempo. El electrolito circula en circuito cerrado en laplanta de electrorefinación.
Ánodo :
cobre : Cu => Cu2+ + 2e-
- nobles : Fe => Fe2+ + 2e-+nobles : Au <=> Au sólido que decanta al fondo de la celda.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama145
Cátodo :
cobre : Cu2+ +2e- => Cu-nobles : Fe2+ <=> Fe2+ : no se depositan+nobles : No están en solución
Además, para mejorar la calidad del depósito catódico, se agrega al electrolitosustancias orgánicas en pequeñas cantidades, llamadas inhibidores decristalización, tales como : tiourea, cola y avitone.
Finalmente, como la concentración de cobre aumenta en el electrolito, debido a ladisolución química del óxido cuproso contenido en el ánodo, es necesario que unaparte del electrolito sea sometida al proceso de electroobtención para mantener laconcentración de cobre constante en solución.
Disolución química (ánodo) :
Cu2O + 2 H+ => 2 Cu+ + H2O
2 Cu+ => Cu2+ + Cu(precipitado) (dismutación)
5.8. Práctica industrial
5.8.1. Circuito hidráulico
La figura 5.16 muestra el circuito hidráulico de una planta deELECTROOBTENCIÓN DE COBRE. El electrolito rico, proveniente del proceso deSX, se bombea a las celdas en paralelo, dónde se deposita parte del cobredisuelto, y vuelve al proceso de SX.
El estanque de recirculación permite el mezclado del electrolito rico, provenientede la SX, con parte del electrolito pobre que se recicla a las celdas. Esta

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama146
recirculación permite ajustar el balance de cobre en el electrolito que sufre unafuerte variación en la etapa de SX y muy leve en la electroobtención.
En el caso de la ELECTROREFINACIÓN DE COBRE, el electrolito circula encircuito cerrado en la planta. Solamente una pequeña parte del caudal delelectrolito pasa en un circuito de purificación para bajar el nivel en impurezas delelectrolito.
5.8.2. Circuito eléctrico
Los circuitos eléctricos de las plantas de electroobtención y electrorefinación decobre son similares; a pesar de que el voltaje de celda es mayor en el primer caso.
La corriente eléctrica continua proviene de un rectificador y alimenta a las celdasen serie. A dentro de cada celda, la corriente atraviesa los electrodos en paralelo(figura 5.16 ).
Entonces, la corriente y el potencial que debe entregar el rectificador están dadospor :
Vrectificador = Vcelda x Número de celdas
Irectificador = Nc x Sc x Icorriente x 2
Vcelda : Potencial entre los cátodos y los ánodos de la celda(V)
Nc : Número de cátodos por celda
Sc : Superficie de cada cara de un cátodo (m2)
Icorriente : Intensidad de corriente (A/m2)
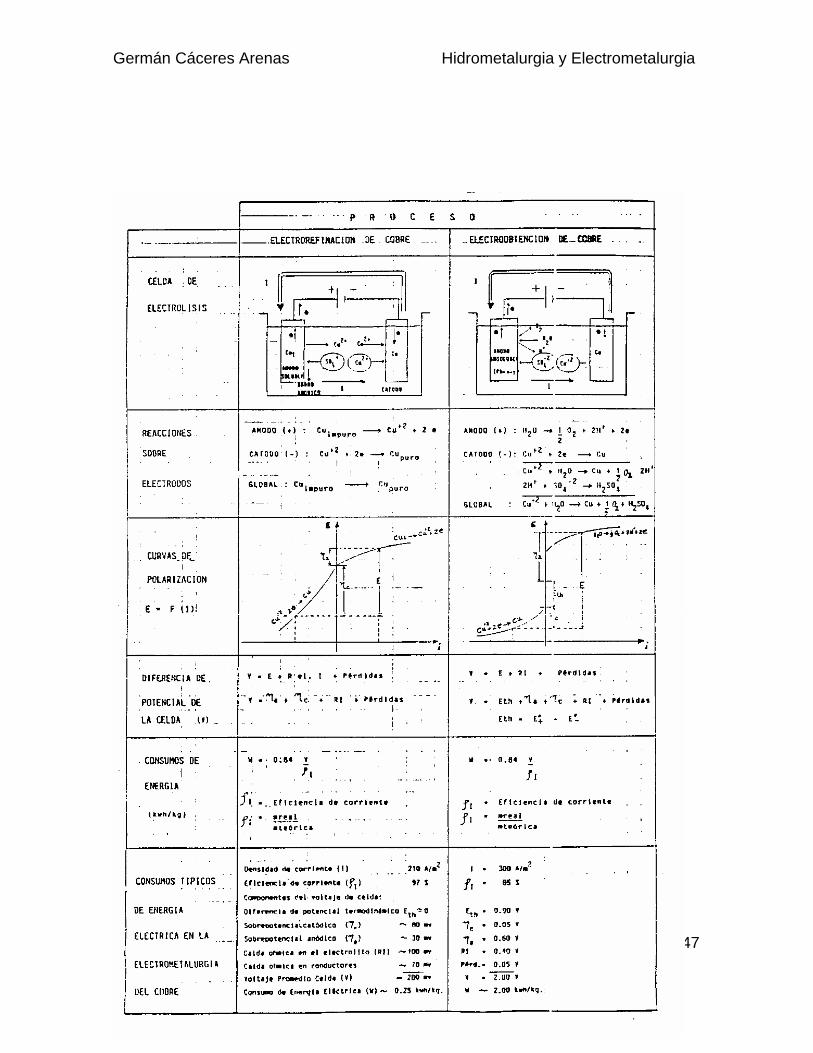
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama147

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama148
Fig
ura
5.16
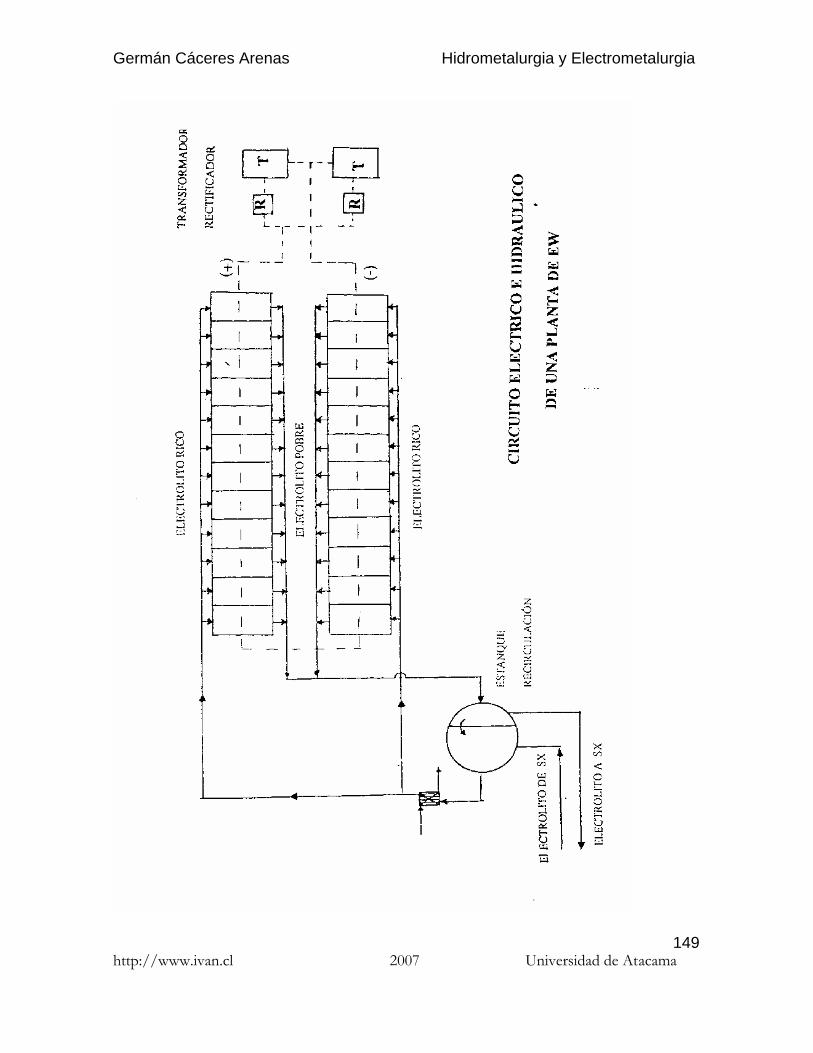
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama149

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama150
6.1. Introducción
Como lo muestra el diagrama de equilibrio Potencial – pH del sistema Au – H2O a25 °C, representado en la figura 6.1, el oro es un metal muy noble, es decir,difícilmente oxidable porque su dominio de estabilidad recubre todo el dominio deestabilidad del agua.
En la naturaleza, el oro se encuentra sobretodo al estado nativo, diseminado enrocas cuarzíferas, en vetas auríferas y en los depósitos aluvionales o placeres,provenientes de la desagregación de estas rocas.
El oro se encuentra asociado principalmente a los minerales de plata y cobre. Else encuentra a menudo también asociado a minerales de fierro, plomo-zinc ysulfoarseniuros más complejos.
El contenido de oro en los minerales varía enormemente; pero, normalmente seexplota minerales que tienen una ley de entre 1 y 10 g/t.
El tratamiento de los minerales de oro se hace, en términos generales, mediantelos procesos clásicos de flotación, concentración gravitacional y cianuración (figura6.2)
Cianuraciónde minerales
deoro.
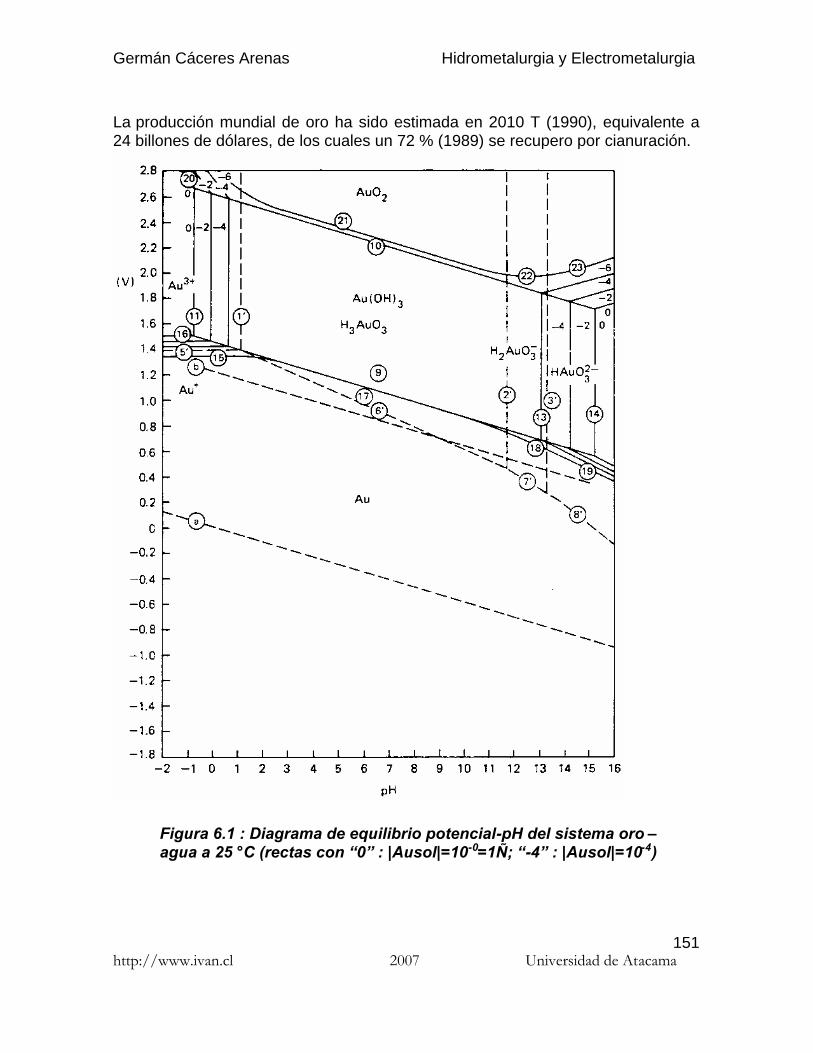
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama151
La producción mundial de oro ha sido estimada en 2010 T (1990), equivalente a24 billones de dólares, de los cuales un 72 % (1989) se recupero por cianuración.
Figura 6.1 : Diagrama de equilibrio potencial-pH del sistema oro –agua a 25 °C (rectas con “0” : |Ausol|=10-0=1Ñ; “-4” : |Ausol|=10-4)
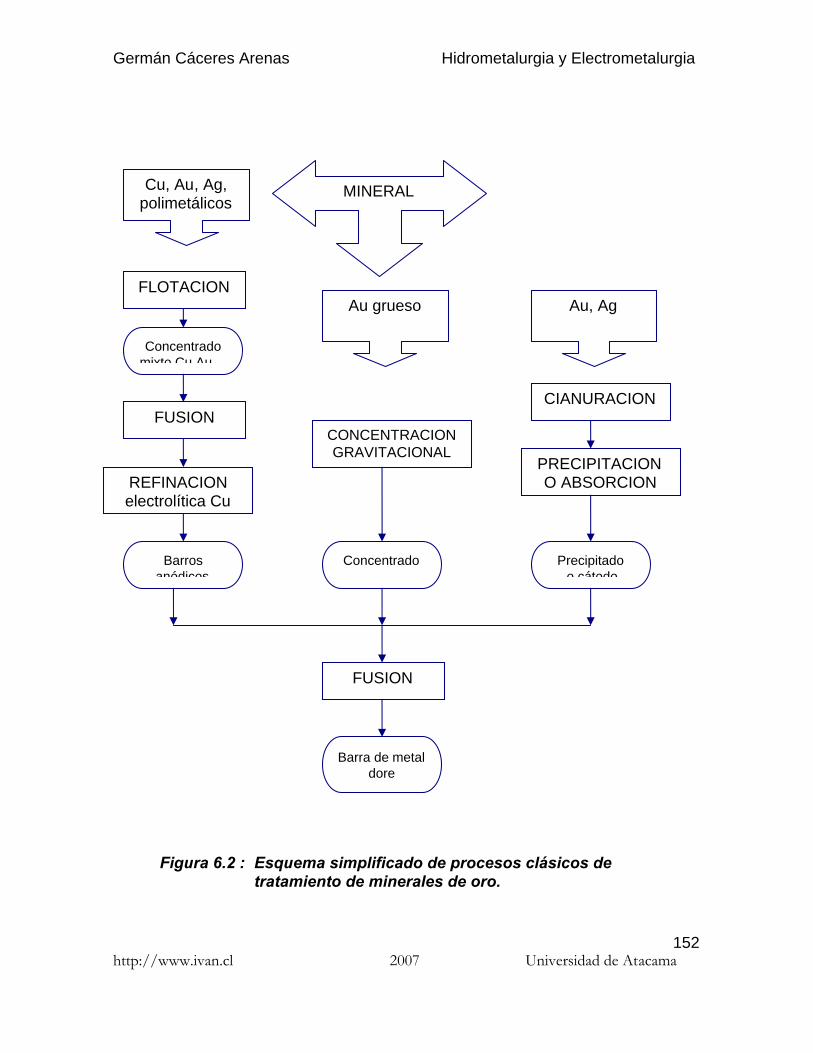
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama152
Figura 6.2 : Esquema simplificado de procesos clásicos detratamiento de minerales de oro.
MINERALCu, Au, Ag,polimetálicos
Au, AgAu gruesoFLOTACION
FUSIONCONCENTRACIONGRAVITACIONAL
REFINACIONelectrolítica Cu
CIANURACION
PRECIPITACIONO ABSORCION
Concentradomixto Cu,Au, ..
Barrosanódicos
Concentrado Precipitadoo cátodo
FUSION
Barra de metaldore
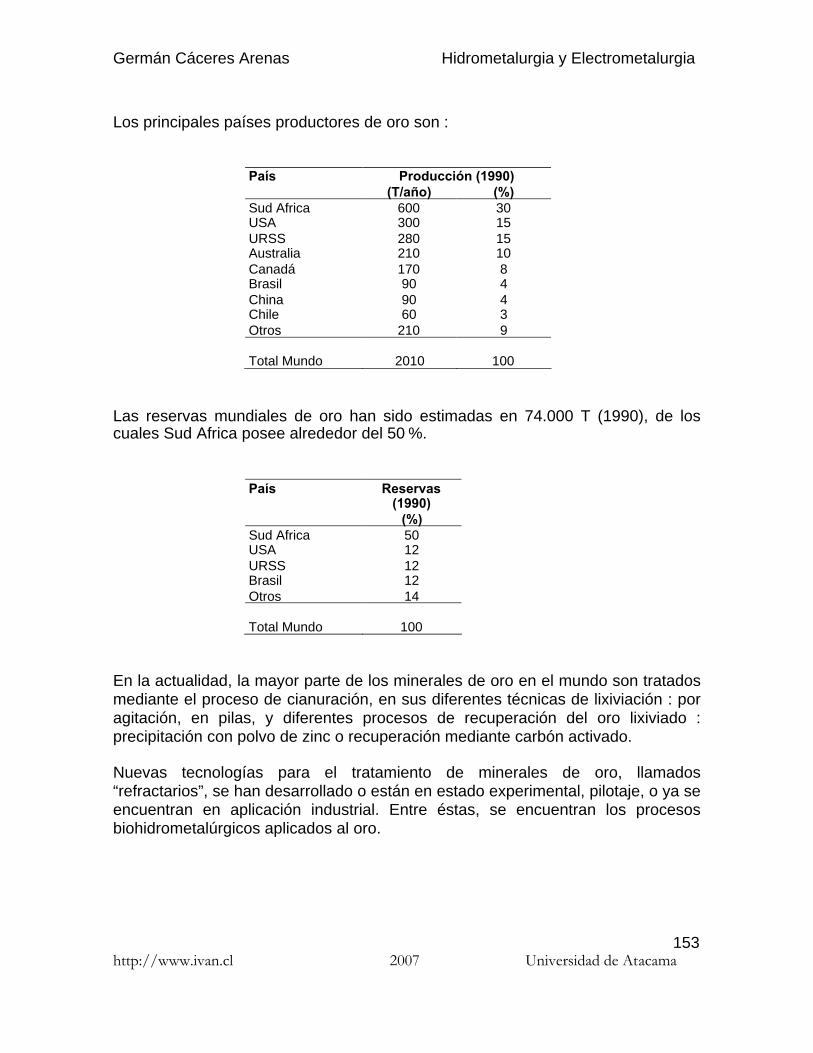
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama153
Los principales países productores de oro son :
País Producción (1990)(T/año) (%)
Sud Africa 600 30USA 300 15URSS 280 15Australia 210 10Canadá 170 8Brasil 90 4China 90 4Chile 60 3Otros 210 9
Total Mundo 2010 100
Las reservas mundiales de oro han sido estimadas en 74.000 T (1990), de loscuales Sud Africa posee alrededor del 50 %.
País Reservas(1990)
(%)Sud Africa 50USA 12URSS 12Brasil 12Otros 14
Total Mundo 100
En la actualidad, la mayor parte de los minerales de oro en el mundo son tratadosmediante el proceso de cianuración, en sus diferentes técnicas de lixiviación : poragitación, en pilas, y diferentes procesos de recuperación del oro lixiviado :precipitación con polvo de zinc o recuperación mediante carbón activado.
Nuevas tecnologías para el tratamiento de minerales de oro, llamados“refractarios”, se han desarrollado o están en estado experimental, pilotaje, o ya seencuentran en aplicación industrial. Entre éstas, se encuentran los procesosbiohidrometalúrgicos aplicados al oro.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama154
6.2 Reacciones de la cianuración
6.2.1 Aspectos termodinámicos
La reacción de disolución del oro por el cianuro, conocida como la ecuación deElsner (1850), es :
4 Au + 8 CN- + O2 + 2 H2O 4 Au(CN)2- + 4 OH-
Es la resultante de 2 reacciones electroquímicas que se producensimultáneamente.
a) La reacción de oxidación del oro en medio cianuro, que se produce en un sitioanódico, de polaridad negativa :
Au + 2 CN- => Au(CN)2- + e- E° = -0.6 V
b) La reacción de reducción del oxígeno disuelto en la solución, que se produce enun sitio catódico, cuya polaridad es positiva :
O2 + 2 H2O + 4 e- => 4 OH- E° = 0.4 V

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama155
Figura 6.3 : Esquema de disolución del oro en medio cianuro
Au SOLUCION
O2O2 + 2 H2O + 4 e- => 4 OH -
OH-
Zona catódicae-
Zona anódica
Au(CN)2-
Au + 2 CN - => Au(CN)2- + e-
CN -

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
http://www.ivan.cl 2007 Universidad de Atacama156
Desde el punto de vista termodinámico, y tal como se representa en la figura 6.4,la reacción de disolución del oro se realizará si el potencial de reducción deloxígeno disuelto, Ered, es superior al potencial de oxidación del oro, Eox.
Figura 6.4 : Potencial delas reacciones parciales dedisolución del oro.
Cátodo (+) :
O2 + 2 H2O + 4 e- => 4 OH-
E° = 0.4 V
42log
406.0
4.0
OH
PE O
red
Anodo(-) :
Au + 2 CN- => Au(CN)2- + e-
E° = -0.6 V
2
2)(log06.06.0
CN
CNAuEox
Ered
Eox
O2
OH-
Au(CN)2-
Au

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 157
Como se necesita que Ered > Eox, se obtiene :
42log
406.0
4.0
OH
PO > 2
2)(log06.06.0
CN
CNAu
17)(
log2
4
2
2
CNAuOH
CNPO
De lo anterior, se deduce que las mejores condiciones termodinámicas paradisolver el oro son :
a) Presión parcial de oxígeno elevadab) Concentración de cianuro elevadac) Concentración de iones OH- baja (pH moderamente alcalino)
Sin embargo, las condiciones industriales de cianuración son :
a) Presión parcial de oxígeno constante (0.21 atm)b) Concentración de cianuro bajac) pH elevado
Estas dos últimas condiciones son para evitar la hidrólisis del cianuro y suposterior volatilización, según la reacción :
CN- + H2O => HCN + OH-
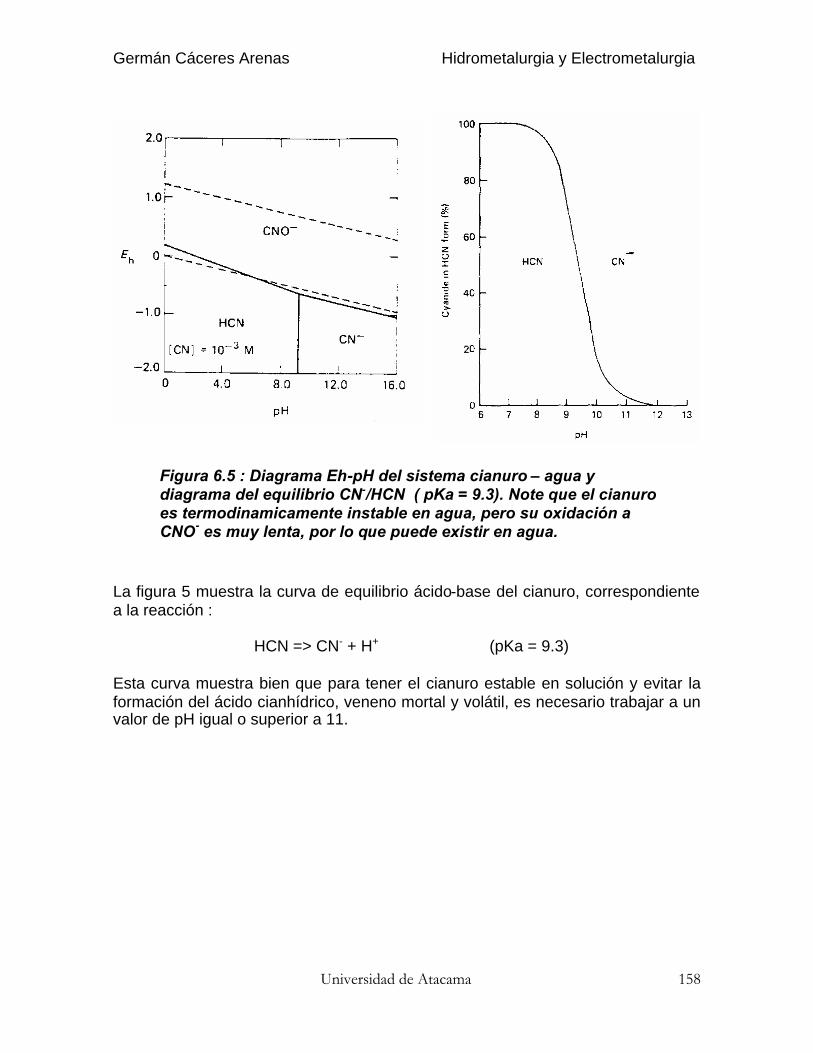
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 158
Figura 6.5 : Diagrama Eh-pH del sistema cianuro – agua ydiagrama del equilibrio CN-/HCN ( pKa = 9.3). Note que el cianuroes termodinamicamente instable en agua, pero su oxidación aCNO- es muy lenta, por lo que puede existir en agua.
La figura 5 muestra la curva de equilibrio ácido-base del cianuro, correspondientea la reacción :
HCN => CN- + H+ (pKa = 9.3)
Esta curva muestra bien que para tener el cianuro estable en solución y evitar laformación del ácido cianhídrico, veneno mortal y volátil, es necesario trabajar a unvalor de pH igual o superior a 11.
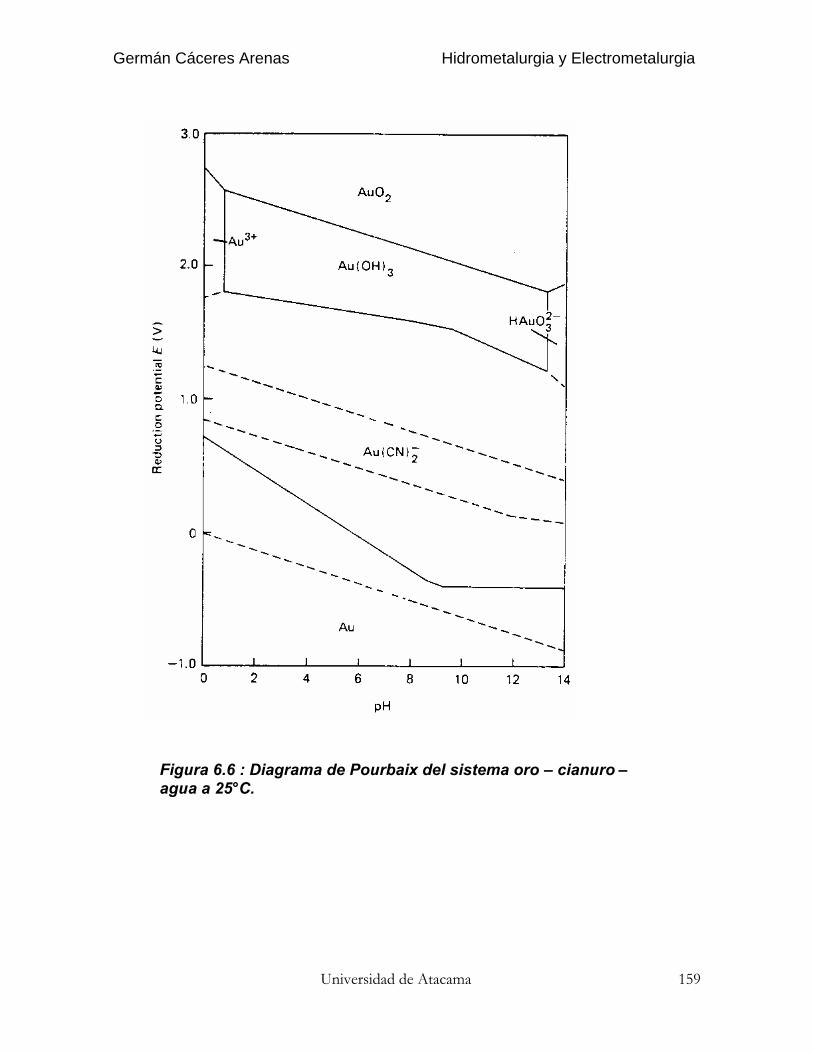
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 159
Figura 6.6 : Diagrama de Pourbaix del sistema oro – cianuro –agua a 25°C.
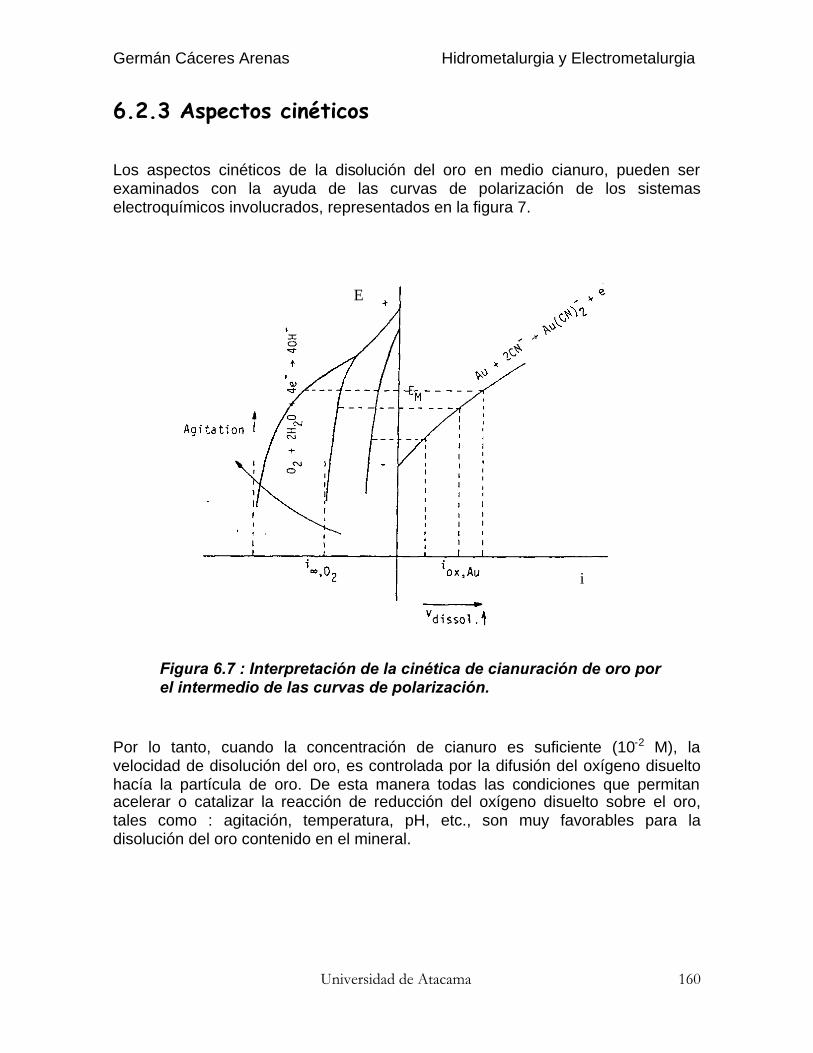
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 160
6.2.3 Aspectos cinéticos
Los aspectos cinéticos de la disolución del oro en medio cianuro, pueden serexaminados con la ayuda de las curvas de polarización de los sistemaselectroquímicos involucrados, representados en la figura 7.
Figura 6.7 : Interpretación de la cinética de cianuración de oro porel intermedio de las curvas de polarización.
Por lo tanto, cuando la concentración de cianuro es suficiente (10-2 M), lavelocidad de disolución del oro, es controlada por la difusión del oxígeno disueltohacía la partícula de oro. De esta manera todas las condiciones que permitanacelerar o catalizar la reacción de reducción del oxígeno disuelto sobre el oro,tales como : agitación, temperatura, pH, etc., son muy favorables para ladisolución del oro contenido en el mineral.
E
i

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 161
6.2.4 Práctica de la cianuración
Debido a la gran variedad de minerales de oro, existe una multitud de formas deponer en práctica la cianuración de minerales de oro, en combinaciones con otrosprocesos tales como concentración gravimétrica, flotación, amalgamación,preoxidación, etc.
Hay tres tipos de minerales :
Free millling : Muy aptos a la cianuraciónComplejos : Necesitan pretratamientos y/o operación especialesRefractarios : Imposibles o muy difíciles de cianurar
Tabla 6.1 : Comparación entre la cianuración por agitación ycianuración en pilas (valores típicos)
CIANURACIONPOR AGITACION
CIANURACIONEN PILAS
Tipos de minerales Varios Oro asequible a las solucionessin moler
Granulometría mineral 80 % < 150 a 45 m 1”pH 9.5 a 11.5 9.5 a 11.5 (CaO)concentración NaCN <1 g/l < 0.3 g/lOxígeno burbujas aireación naturaltiempo de cianuración horas – días mesesEtapas complementarias Lavado por decantación en
contracorrienteDecantación de los sólidos
Aglomeración
Costo alto bajoRecuperación de oro 70 –90 % 50 – 80 %Procesos de recuperación deoro asociados
Merryll-CroweCarbón activadoCarbón en pulpa (CIP)Resina en pulpa (RIP)Carbón en lixiviación (CIL)Resina en lix. (RIL)
Merryll-CroweCarbón activado

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 162
6.3 Purificación y concentración de lasolución
Desde los años 80, la adsorción sobre carbono activado es un procesouniversalmente empleado para obtener una solución más pura y concentrada deoro, de forma similar a la SX en lixiviación de cobre. El oro se absorbe sobre elcarbono a partir de una solución diluida, y se hace su elución en una soluciónmucho más concentrada y pura antes de precipitarlo.
En algunas plantas, no se hace esta etapa de purificación - concentración, y serealiza la cementación de oro con zinc directamente a partir de la solución diluida.
6.3.1 Propiedades del carbón activado
El carbón activado, se fabrica calentando materiales orgánicos (carbón, madera,cáscaras de coco, etc.) hasta temperaturas de 700-1000 °C en una atmósferacontrolada, de vapor de agua, dioxido de carbono y/o oxígeno.
De estructura grafítica, tiene una superficie específica extremadamente grande (>1000 m2/g) debido a una estructura íntimamente porosa.
Los poros pueden ser clasificados según su tamaño característico :
Macroporos x > 100-200 nm Mesoporos 1.6 < x < 100-200 nm Microporos x < 1.6 nm
Las propiedades más importantes del carbón activado son : la capacidad deadsorción, la velocidad de adsorción, la dureza y resistencia a la abrasión, lacaracterísticas de reactivación y el tamaño.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 163
Por su gran superficie específica y sus características químicas, el carbón activadoes capaz de adsorber al oro presente en solución, según mecanismos diversos,pero en los cuales predominaría la adsorción en asociación con un catión metálicotal como el Ca2+:
Mn+ + n Au(CN)2- Mn+[Au(CN)2
-]n (adsorbido)
FENOMENO DE ADSORCION : Se define como la adhesión de sustanciasdisueltas en la superficie de sólidos, con los cuales se hallan en contacto. Laadsorción puede deberse a causas físicas (fuerzas coulómbicas, hidrofobicidad,etc.). También puede ocurrir adsorción química, la que se manifiesta a través deuna interacción química con las moléculas de la superficie del sólido.
Otras aplicaciones del carbón activado en metalurgia extractiva :
Adsorción de reactivos de flotación Modificador del potencial oxidante de una pulpa o solución (reductor).
Por ejemplo para no degradar el HS- y S2- en flotación Catalizador, por ejemplo de la reducción del Fe+3 con SO2. Eliminación de contaminantes orgánicos en soluciones de SX, aguas,
etc. Adsorción de iones metálicos : complejos Au(CN)2- y Ag(CN)2
2-, U,metales pesados en aguas, ...
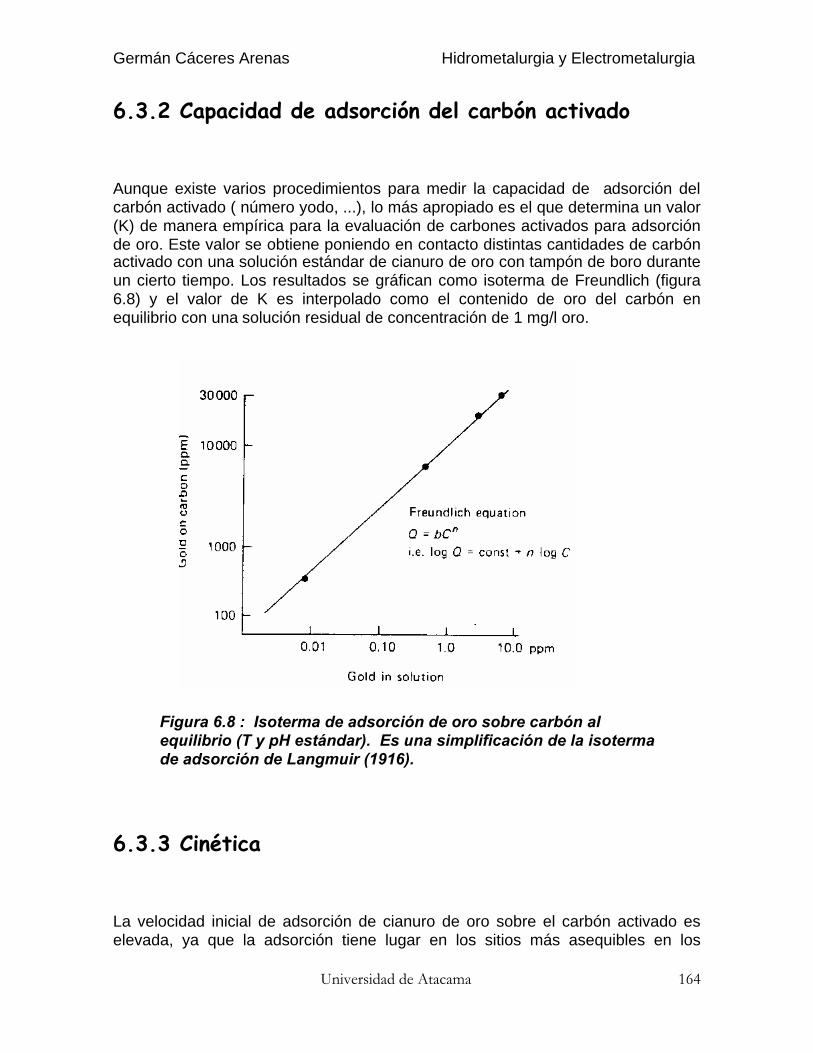
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 164
6.3.2 Capacidad de adsorción del carbón activado
Aunque existe varios procedimientos para medir la capacidad de adsorción delcarbón activado ( número yodo, ...), lo más apropiado es el que determina un valor(K) de manera empírica para la evaluación de carbones activados para adsorciónde oro. Este valor se obtiene poniendo en contacto distintas cantidades de carbónactivado con una solución estándar de cianuro de oro con tampón de boro duranteun cierto tiempo. Los resultados se gráfican como isoterma de Freundlich (figura6.8) y el valor de K es interpolado como el contenido de oro del carbón enequilibrio con una solución residual de concentración de 1 mg/l oro.
Figura 6.8 : Isoterma de adsorción de oro sobre carbón alequilibrio (T y pH estándar). Es una simplificación de la isotermade adsorción de Langmuir (1916).
6.3.3 Cinética
La velocidad inicial de adsorción de cianuro de oro sobre el carbón activado eselevada, ya que la adsorción tiene lugar en los sitios más asequibles en los
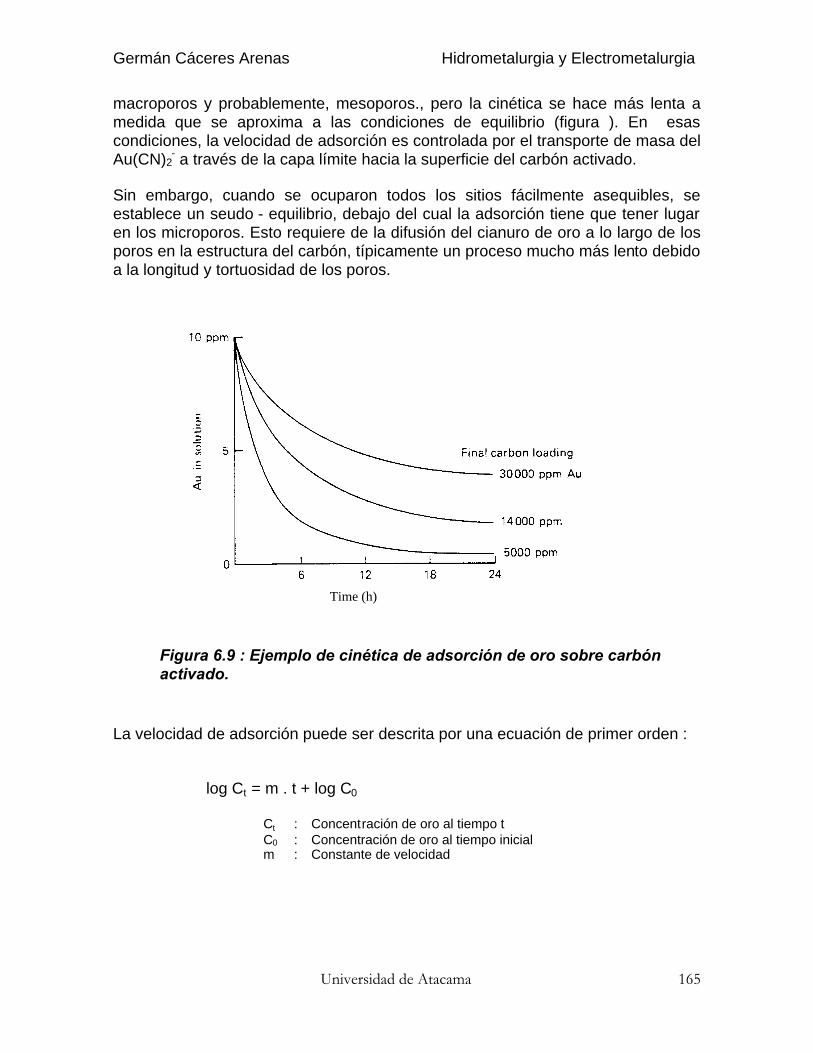
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 165
macroporos y probablemente, mesoporos., pero la cinética se hace más lenta amedida que se aproxima a las condiciones de equilibrio (figura ). En esascondiciones, la velocidad de adsorción es controlada por el transporte de masa delAu(CN)2
- a través de la capa límite hacia la superficie del carbón activado.
Sin embargo, cuando se ocuparon todos los sitios fácilmente asequibles, seestablece un seudo - equilibrio, debajo del cual la adsorción tiene que tener lugaren los microporos. Esto requiere de la difusión del cianuro de oro a lo largo de losporos en la estructura del carbón, típicamente un proceso mucho más lento debidoa la longitud y tortuosidad de los poros.
Figura 6.9 : Ejemplo de cinética de adsorción de oro sobre carbónactivado.
La velocidad de adsorción puede ser descrita por una ecuación de primer orden :
log Ct = m . t + log C0
Ct : Concentración de oro al tiempo tC0 : Concentración de oro al tiempo inicialm : Constante de velocidad
Time (h)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 166
6.3.4 Selectividad
El carbón activado es altamente selectivo para la adsorción de Au y Ag en vez deotros metales, con la notable excepción del mercurio. El orden de preferencia paravarios complejos de metales se indica a continuación :
Au(CN)2- > Hg(CN)2 > Ag(CN)2
- > Cu(CN)32- > ZN(CN)4
2- > Ni(CN)42- > Fe(CN)6
4-
6.3.5 Factores afectando la adsorción
Factores físicos
Variable Efecto
Tipo de carbónactivado
Varios
Tamaño de laspartículas de carbón
Un tamaño mayor disminuye la velocidad de adsorción, por lo menosdurante la primera etapa.
Agitación Una agitación más fuerte aumenta la velocidad de adsorción, porque lamayoría de los sistemas industriales son operados solamente hasta elseudo - equilibrio.
Densidad de pulpa Disminuye la cinética

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 167
Factores químicos
Variable Efecto
Temperatura La adsorción de oro sobre carbón activado es exotérmica, un aumento entemperatura disminuye la capacidad de carga, lo que explica el fenómenode desorción a temperatura más alta.
Al contrario, favorece la cinética.
Concentración de oro Aumenta la cinética y la capacidad de carga
Concentración decianuro
Disminuye la cinética y la capacidad de carga
pH solución En el rango 9 – 11, tiene poco efecto
Fuerza iónica Aumenta la cinética y la capacidad de carga
Concentración deotros metales
Si hay otros metales que compiten por los sitios activos, eso disminuye lacinética y la capacidad de carga para el oro
Deterioro del carbón El deterioro, o envenenamiento, del carbón por adsorción, precipitación obloqueo físico de otra especie en solución, pueden tener un efecto muygrave sobre la eficiencia de adsorción
Figura 6.10 : Isotermas de adsorción a distintas temperaturas(Condiciones experimentales : 50 ml solución, 0.25 g carbón, 2.8g/l CaCl2, 0.5 g/l KCN)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 168
6.3.6 Elución
El carbón activado previamente cargado con oro y otros metales en el proceso deadsorción debe ser sometido a una etapa de elución para desorber los metales delcarbón. Eso produce un volumen pequeño de solución con alta concentración enoro, adecuada para su posterior recuperación, y permite al carbón ser recicladohacia el circuito de adsorción.
El proceso de desorción ocupa al revés el mismo fenómeno que el proceso deadsorción, y factores que inhiben la adsorción van a favorecer la desorción
Mn+[Au(CN)2-]n (adsorbido) Mn+ + n Au(CN)2-
En la practica, generalmente la elución se realiza poniendo el carbón en unacolumna en contacto con un flujo acendiente de una solución caliente y con altaconcentración en cianuro.
Variable Efecto
Temperatura Es el factor más importante; la velocidad de elución a 180 °C es 8 vecesmayor a la velocidad de elución a 90 °C.
Concentración decianuro
Favorece la desorción, porque los iones CN– compiten con el oro para lossitios de adsorción
Fuerza iónica Debe ser la más baja posible. Por ejemplo, es posible desorber el oro conagua desionizada pura.
pH Un pH alto favorece la desorción, porque los iones OH– compiten con eloro para los sitios de adsorción
Solventes orgánicos Efecto positivo porque aumentan la actividad de otras especies iónicas ensolución, como el CN–
Concentración de oroen la solución
Disminuye la velocidad y eficiencia de la elución
Otros metales La elución de Cu, Ag y Hg se hace preferencialmente al oro, lo queposibilita eventualmente realizar una elución en dos etapas parasepararlos del oro.
Posteriormente, el carbón debe ser sometido a una etapa de limpieza química(HCl,..) y una etapa de reactivación en un horno a 650-750 °C con vapor de agua.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 169
6.3.7 Aspectos de ingeniería
Las etapas comunes del ciclo del carbón en la mayoría de las plantas son :
Adsorción del complejo de cianuro y oro sobre el carbón activado Lavado con agua para sacar la solución de impregnación Desorpción (elución), generalmente con soluciones 0.2 % NaCN y 1
% NaOH a 90 °C Lavado con ácido para sacar precipitados de CaCO3 Lavado con agua Secado, calentamiento a 700 °C durante 30 min en ausencia de aire
y templado Reciclaje a la etapa de adsorción
Dos factores contribuyen a la elección del proceso de adsorción :
Las propiedades de filtración de la pulpa La presencia de materia orgánica en el mineral
Como resultado, los siguientes procesos son utilizados :
Adsorción en columnas : Cuando la pulpa es fácil de filtrar o cuando se utiliza lacianuración en pilas.
Carbón en pulpa (CIP) : Cuando el mineral contiene mucha arcilla. La pulpa y lospelets de carbón son agitados en uno o varios estanques ; cuando la adsorción secompletó, el carbón, más grueso, es separado de la pulpa por tamizaje.
Carbón en lixiviación (CIL) : Cuando el mineral contiene materia orgánica sobrela cual podría adsorberse el oro, con las consiguientes perdidas que esorepresenta. El carbón se agrega directamente a la etapa de cianuración, de talmanera que pueda adsorber el Au(CN)2- en cuando se forme.
La adsorción y la elución son procesos lentos. Típicamente, es necesario 24 hpara adsorber el oro de una solución con una concentración inicial de 10 ppm Au u50 h para la elución. Una tonelada de carbón adsorbe alrededor de 10 kg de oro.Aproximadamente 4 litro de solución son suficientes para la elución de 1 kg decarbón.
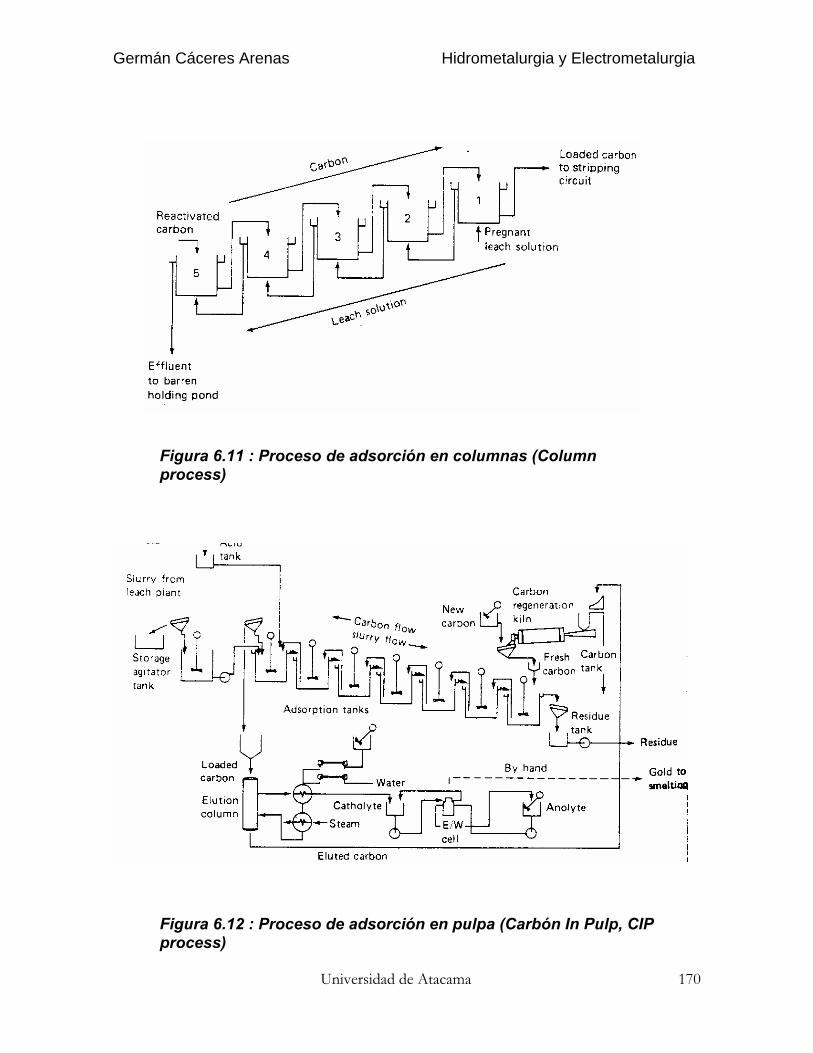
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 170
Figura 6.11 : Proceso de adsorción en columnas (Columnprocess)
Figura 6.12 : Proceso de adsorción en pulpa (Carbón In Pulp, CIPprocess)

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 171
6.4 Recuperación de oro
La electroobtención y la cementación con zinc son los dos procesos utilizadospara la recuperación de oro metálico a partir de las soluciones concentradasprovenientes de la elución del carbón activado.
También, la cementación con zinc todavía se aplica directamente a la solucióndiluida proveniente directamente de la etapa de lixiviación cuando :
a) Los minerales contienen mucha plata (Ag), la que saturaríarápidamente el carbón activado.
b) Minerales con altos contenidos de arcillas u otras especies queinterfieren con la adsorción sobre carbón activado.
c) Operaciones a pequeña escala
6.4.1 Precipitación con zinc (proceso Merryll-Crowe)
Comportamiento anódico del zinc
El zinc se disuelve en solución según la siguiente semi-reacción para formar uncomplejo estable con el cianuro :
Zn + 4 CN- Zn(CN)42- + 2 e-
E = -1.25 +0.0295 log [Zn(CN)42- ] + 0.118 pCN (V)
En condiciones de baja concentración en cianuro, alta concentración en zinc yalcalinidad, el hidróxido de zinc Zn(OH)2 puede precipitar (figura 6.13). Estefenómeno es muy poco deseable, porque una capa de este hidróxido puede cubrirla superficie de zinc, causando su pasivación y impidiendo la precipitación de oro.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 172
Figura 6.13 : Diagrama de Pourbaix para el sistema Zn – H2O – CN-
a 25 °C, además de algunos equilibrios entre Au - H2O – CN- . Sevisualiza que un aumento en la concentración en Zn (arriba haciaabajo) provoca la formación de una zona de pasivación deZn(OH)2

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 173
Semi - reacción catódica
La semi - reacción catódica que acompaña a la oxidación del zinc puede ser lareducción y precipitación de oro y de los otros metales preciosos, pero tambiénuna de todas las poco deseables reacciones parásitas, incluyendo la reduccióndel agua, oxígeno y otras especies en solución.
Reducción del oro
Au(CN)2- + e- Au + 2 CN-
E = -0.60 + 0.118 log [CN-] + 0.059 log [Au(CN)2-] (V)
La figura 6.13 muestra que en todo rango de pH, este potencial es mucho mayor alpotencial de oxidación del zinc, indicando una fuerza termodinámica importantepara la precipitación de oro.
Figura 6.14 : Representación esquemática del mecanismo deprecipitación de oro sobre partículas de zinc. Note que las semi –reacciones anódica y catódica tienen lugar en distintos sitios.
Reducción de otros metales : Hg , Ag, Cu, Ni, Co, ....
Reducción de agua y oxígeno
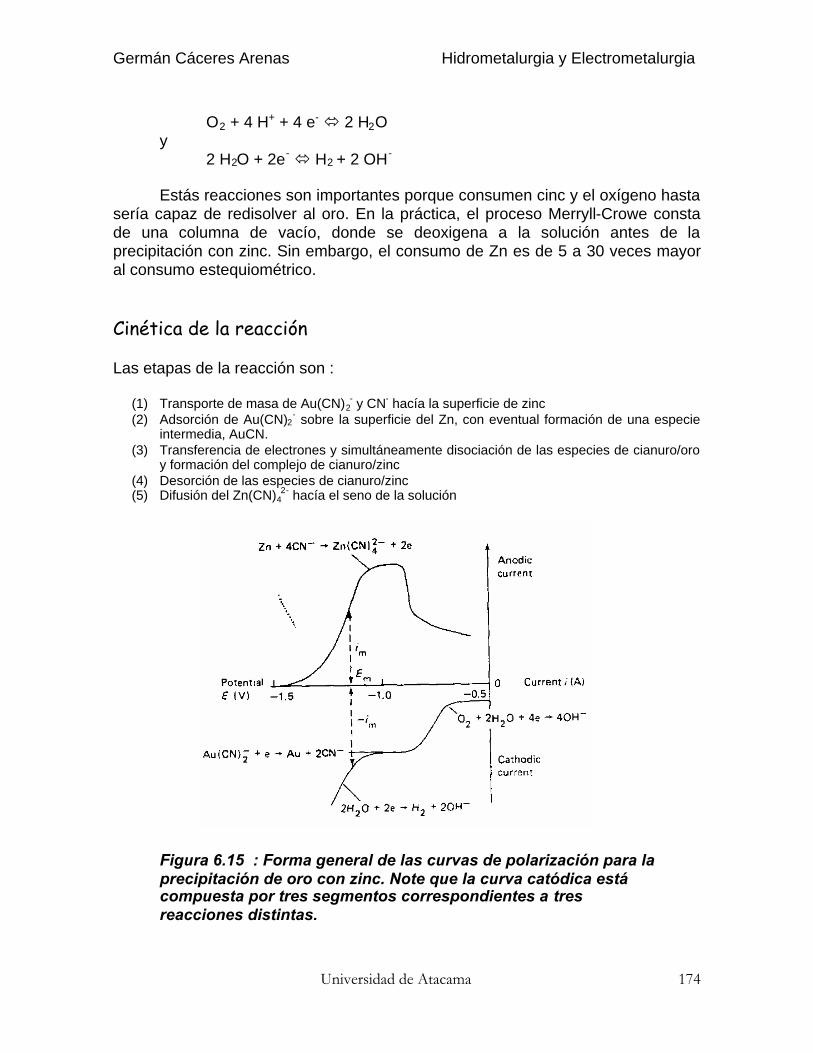
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 174
O2 + 4 H+ + 4 e- 2 H2Oy
2 H2O + 2e- H2 + 2 OH-
Estás reacciones son importantes porque consumen cinc y el oxígeno hastasería capaz de redisolver al oro. En la práctica, el proceso Merryll-Crowe constade una columna de vacío, donde se deoxigena a la solución antes de laprecipitación con zinc. Sin embargo, el consumo de Zn es de 5 a 30 veces mayoral consumo estequiométrico.
Cinética de la reacción
Las etapas de la reacción son :
(1) Transporte de masa de Au(CN)2- y CN- hacía la superficie de zinc
(2) Adsorción de Au(CN)2- sobre la superficie del Zn, con eventual formación de una especie
intermedia, AuCN.(3) Transferencia de electrones y simultáneamente disociación de las especies de cianuro/oro
y formación del complejo de cianuro/zinc(4) Desorción de las especies de cianuro/zinc(5) Difusión del Zn(CN)4
2- hacía el seno de la solución
Figura 6.15 : Forma general de las curvas de polarización para laprecipitación de oro con zinc. Note que la curva catódica estácompuesta por tres segmentos correspondientes a tresreacciones distintas.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 175
En condiciones industriales, la etapa limitante que determina la cinética es ladifusión de Au(CN)2-, porque su concentración es muy baja. Sin embargo, como seutiliza polvo de zinc, de muy grande superficie específica, la reacción global esrápida.
6.4.2 Electrodepositación de oro
Semi – reacción catódica
Reducción catódica del oro
Au(CN)2- + e- Au + 2 CN-
Reducción de otros metales : Hg , Pb, Ag, Cu, Ni, Co, ....
Reducción de agua y oxígeno
O2 + 4 H+ + 4 e- 2 H2Oy
2 H2O + 2e- H2 + 2 OH-
=> La eficiencia de corriente es baja, pero eso no tiene importancia para laproducción de oro, porque el volumen de producción es bajo.
Semi-reacción anódica
La reacción principal es :
H2O O2 + 4 H+ + 4 e-
También una pequeña parte del cianuro puede ser oxidado a cianato, pero lareacción es lenta :
CN- + 2OH- CNO- + H2O + 2 e- E° = -0.97 V

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 176
Cinética de la reacción
La electrodepositación de oro es controlada electroquímicamente hasta unpotencial del orden de –0.85 a –1.0 V, dependiendo de las propiedades de lasolución (Butler Volmer). A potencial más negativo, la velocidad de depositaciónes controlada por la difusión de Au(CN)2
- hacia el cátodo (figura ).
Generalmente, la celdas operan con concentraciones bajas de oro, a un voltajedónde la etapa limitante es la difusión de Au(CN)2-, pero no demasiado bajo paraminimizar las reacciones parásitas como la producción de H2.
Configuración de las celdas de EW
Generalmente, los ánodos son placas de acero inoxidable perforadas, mientrasque los cátodos son de birutilla de fierro, para maximizar la superficie catódica, yaque la etapa controlante es la difusión de cianuro de oro.
La recuperación de oro varia entre 60 y 99 % según el tiempo del ciclo deelectrodepositación.
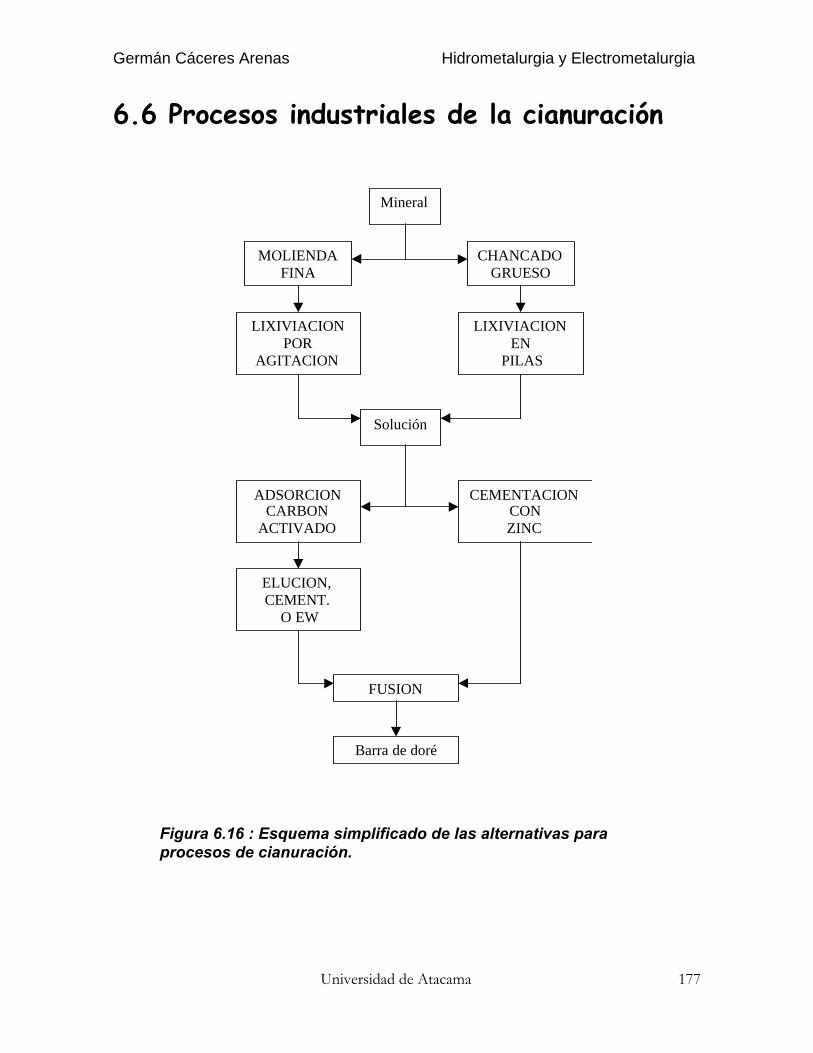
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 177
6.6 Procesos industriales de la cianuración
Figura 6.16 : Esquema simplificado de las alternativas paraprocesos de cianuración.
Mineral
MOLIENDAFINA
CHANCADOGRUESO
LIXIVIACIONPOR
AGITACION
LIXIVIACIONEN
PILAS
Solución
ADSORCIONCARBON
ACTIVADO
CEMENTACIONCONZINC
ELUCION,CEMENT.
O EW
FUSION
Barra de doré

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 178
6.6.1 Ejemplo : Planta East Driefontein (1973)
Figura 6.17 : Diagrama de flujo de la planta East Driefontein (1973)
Mineralogía : Conglomerado del Witwatersrand – cuarzo. Partículas finas de oro nativo y electrumen ganga silicea, pero con presencia de un poco de pirotita.
Producción : 8000 tpd Ley mineral : 8 g/t Recuperación oro : 98.5 %
Molienda : 78 % < 75 m Pre-oxidación de la pirotita : 10 h a pH 11
[NaCN] : 0.25 g/l Tiempo residencia : 40 h

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 179
6.7 Caso de aplicación industrial :cianuración en pilas de relaves de oro
Se muestran a modo de ejemplo los resultados de operación industrial de unapequeña planta de cianuración de relaves de oro, provenientes de antiguas faenasde flotación.
El relave contiene 5 g/T de oro y 0.4 % de cobre; tiene una granulometría de 80 %-200# y está compuesto de una mezcla de minerales de fierro y cobre, con sílice yarcillas como ganga. El oro se encuentra al estado libre y en partículas muy finas,inferiores a 50 micrones.
La planta tiene una capacidad de tratamiento de 200 T/día, y las reservas derelaves son del orden de 100.000 toneladas; por lo tanto el funcionamientoprevisto para esta planta es de 11½ años.
El proceso de tratamiento, estudiado y puesto a punto en la Universidad deAtacama, se muestra en la figura 9.
Las condiciones de operación y los resultados obtenidos fueron los siguientes(1980) :
Capacidad de tratamiento : 200 T/díaConsumos :
cianuro de sodio : 3 kg/Tcal : 15 kg/Tcemento : 15 kg/T
Tasa de riego : 10 l/h/m2
Concentración de NaCN : 1 g/lpH : 11Consumo de zinc : 80 g/g de oroRecuperación de oro : 60 %, o sea 3 g/T relavesPureza barra de oro : 97 % Au
El balance económico fue el siguiente :
Total gasto : 25 US$/T relaveTotal ingreso : 36 US$/T relaveUtilidad : 11 US$/T relave
Utilidad total del proyecto : US$ 1.100.000
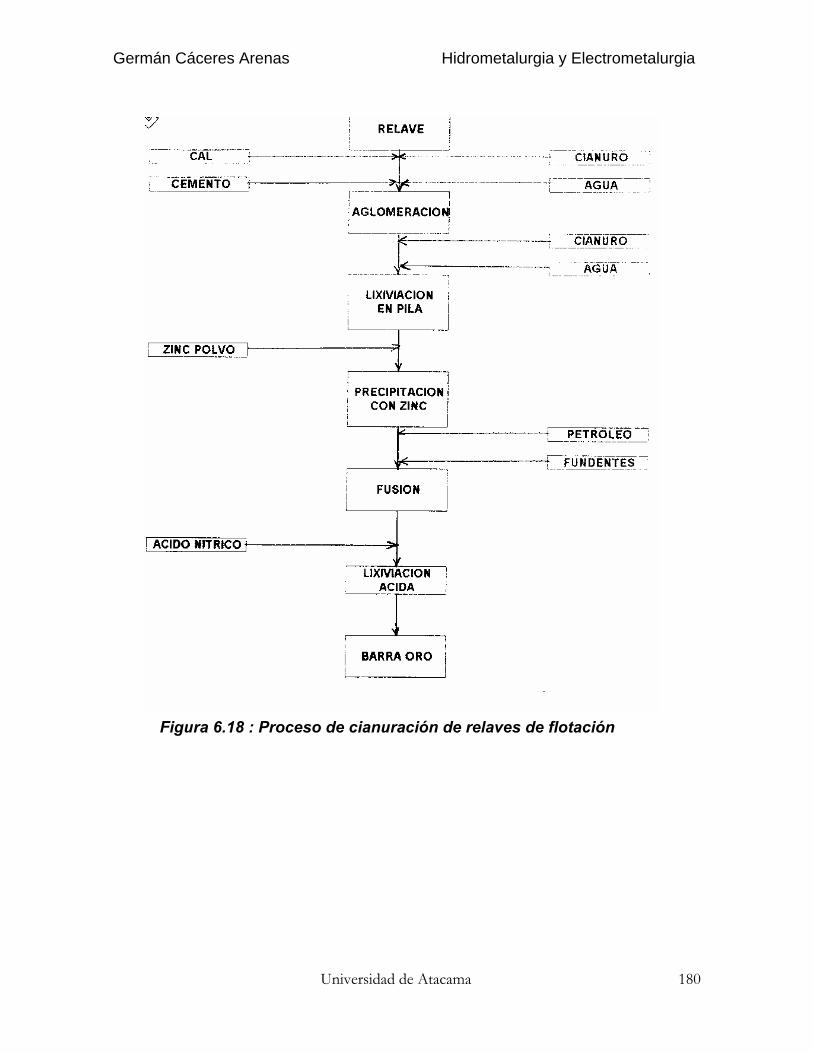
Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 180
Figura 6.18 : Proceso de cianuración de relaves de flotación

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 181
7.1. Introducción
(1) Esteban M. Domic M., " Revisión de los mas Recientes Proyectos Chilenos deLixiviación de Cobre ", V° Encuentro Minero de Tarapaca, pp 293-307, UniversidadArturo Prat, Iquique, 1995.
(2) " Plantas y Proyectos LIX-SX-EW en Chile ", Minería Chilena N°171, pp 13-23,Septiembre 1995.
7.2. Fundamentos termodinamicos
(1) G. Cáceres, " Hidrometalurgia y Electrometalurgia ", Curso de capacitación,Universidad de Atacama, Copiapó, 1992.
(2) F. Habashi, " A Textbook of Hydrometallurgy ", Metallurgie Extractive Quebec, 1993.
(3) A. Reghezza I., " Fisico-Química de la Hidrometalurgia ", Universidad de Concepción,Concepción, 1987.
(5) S. Zumdahl, " Chemistry ", D.C. Heath and Company, Lexington, Massachusetts,1993.
Bibliografía

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 182
7.3. Lixiviación
(1) Martin Col & al (RAHCO), " Manejo de Materiales en Proyectos de Lixiviación ",Minería Chilena N°191, pp 109-123, Mayo 1997.
(2) Marcos Gonzalez, " Curado-Aglomeración-Lixiviación ", Curso de capacitación,Universidad de Atacama, Copiapó, 1994.
(3) John Marsden & Lain House, " The Chemistry of Gold Extraction ", Ellis HorwoodLimited, 1992.
(4) Paul E. Queneau & al, " Hydrometallurgy - a short course ", TMS-AIME ContinuingEducation Committee.
(5) Andrés Reghezza, " Aspectos Tecnologicos de la Lixiviación ", Universidad deConcepción, Concepción, 1987.
(6) Gabriel Zárate, " Beneficio de Minerales de Cobre ", in Minería Chilena N°159, pp 37-57, Septiembre 1994.
(7) " Minería Química ", Instituto Tecnológico Geominero de España ITGE, 1994.
7.4. Purificacion de soluciones
(1) Juan Astudillo, " Extracción por Solventes ", Curso de Capacitación, Universidad deAtacama, Copiapó, 1994.
(2) Germán Cáceres A., " Extracción por Solventes y Electro-obtención ", curso decapacitación para operadores, Universidad de Atacama, Copiapó, 1996.
(3) C. Ek, " Metallurgie des Metaux autres que le Fer ", Université de Liege, 1975.
(4) Denis Goffaux, " Procesamiento de Minerales - secunda parte : Metalurgia ",Universidad de Atacama, Copiapó, 1993.
(5) Gary A. Kordosky, " The Chemistry of Metals Recovery Using LIX Reagents ", MineralIndustry Division Red Book, Henkel Corporation, Tucson, 1990.
(6) Paul E. Queneau & al, " Hydrometallurgy - a short course ", TMS-AIME ContinuingEducation Committee.
(7) Andrés Reghezza I., " Fundamentos y Aplicaciones de Extracción por Solventes ",Universidad de Concepción, Concepción, 1987.
(8) Andrés Reghezza, " Precipitación de Soluciones ", Universidad de Concepción,Concepción, 1987.
(9) Gabriel Zárate, " Beneficio de Minerales de Cobre ", in Minería Chilena N°159, pp 37-57, Septiembre 1994.
(10) " Seminario y Capacitación a Nivel de Laboratorio Hidrometalurgico en Extracción porSolventes y Reactivos LIX ", Harting S.A., Santiago, 1996.

Germán Cáceres Arenas Hidrometalurgia y Electrometalurgia
Universidad de Atacama 183
7.5. Electroobtención - Electrorefinación
(1) Alonso Arenas, " Electroobtención de Cobre ", Curso, Universidad Católica del Norte,Antofagasta, 1995.
(2) Germán Cáceres & Hugo Carcamo, " Electrometalurgia ", Curso de capacitación,Universidad de Atacama, Copiapó, 1988.
(3) G. Cáceres, " Hidrometalurgia y Electrometalurgia ", Curso de capacitación,Universidad de Atacama, Copiapó, 1992.
(4) Germán Cáceres, " Electroobtención ", Curso de capacitación, Universidad deAtacama, Copiapó, 1994.
(5) Jaime Izquierdo L., " Electroobtención de Cobre (EW) ", Mining edición Español, pp27-29, abril 1997.
(6) Terkel Rosenquist, " Fundamentos de Metalurgia Extractiva ", ed. Limusa, México,1987.
(7) Zbigniew Szczygiel Jordens, " Metalurgia No Ferrosa ", ed. Limusa, México, 1984.
7.6. Cianuración de Minerales de Oro
(1) Germán Cáceres, " Cianuración de Minerales de Oro ", Seminario presentado en laUniversidad de Chile, Santiago, 1993.
(2) G. Cáceres, W. Silva & D. Guzman, " Fundamentos de cianuración y Precipitación deoro y plata ", Curso de capacitación, Universidad de Atacama, Copiapó, 1989.
(3) John Marsden & Lain House, " The Chemistry of Gold Extraction ", Ellis HorwoodLimited, 1992.
(4) D van Zyl, I. Hutchinson & J. Kiel Editors, " Introduction to Evaluation, Design andOperation of Precious Metal Heap Leaching Projects, Society of Mining Engineers,Littleton, Colorado, 1988.


















