
Guía para la realización de validación de métodos de ensayo
14
ESTABLECE GUlA TE CNICA PARA LA R EALIZACI ON DE LA VAUDACION DE METODOS DE ENS AYO. RESOLU CION EXENT A w 20. ENF 2 015 SANTI AGO, VISTOS: Providencia lnterna W 2325 de Ia Jefa de Ia Asesoria Juridica; y TENIENDO PRES ENTE: lo dispuesto en Ia l ey Organica Constitucional W 18.575 de "Bases General es de Ia Administraci6n del Estado"; en Ia Ley N o 19.880 de " Base de los Procedimientos Administrativos que rigen los Act os de l os 6rganos de Ia Administraci6n del Estado"; C6digo Sanitario; Decreto Supremo No 3 de 2010, del Ministerio de Salud, que aprueba el "Reglame nto del Sistema Nacional de Control de Productos Farmaceuticos de Uso Humano"; en los articulos 59 letra a) y b), 60 y 611etra b) del Decreto con Fuerza de Ley W 1 de 2005, que fija el texto ref undido, coordinado y sistematizado del Decreto Ley W 2763 de 1979 y de l as Leyes W 18.933 y W 18.469"; Resoluci6n Exenta W 607 del 2014 de l Ministerio de Salud; asi como lo establecido en Ia Resoluci6n W 1.600 de 2008, de Ia Contraloria General de Ia Republica; Decreto Exento N2 27 de 2012 del Ministerio de Salud "Norma Tecnica W 131 que define los criteri os destinados a establecer Ia equivalencia terapeutica en productos farmaceuticos en Chile", Decreto Exento N!:! 28 de 2012 Mini sterio de Sa lud "Nor ma Tecnica W 127 sobre Buenas Prckticas de Manufactura (BPM)" y Decreto Exento N!:! 543 de 2013 Ministerio de Salud, "Norma Tecnica W 139 so bre Buenas Practicas de Laboratorio (BPL)" ; dicto Ia siguiente: CONSIDERANDO: 1.- La importancia de Ia validaci6n de los metodos de ensayo, es que a traves de esta, es posible establecer el procedimiento y las condiciones en la s que deben realizar se, confiando as i, que l os da t os obtenidos cumplen co n Ia ca lidad deseada, brindando seguridad y respaldo. 2.- De acuerdo a las Buen as Practicas de Manufactura (BPM) y de Laboratorio (BPL), Ia validaci6n debe aplicarse en todas las etapas de fabricaci6n y control de un producto farmaceutico, esto es: manufactura, control de procesos control y aseguramiento de Ia calidad. RE S OLU C ION 1.- APRUEBESE como "G U[A TECNICA PARA R EALIZACION DE LA VALIDACION DE METODOS DE ENSAYO" el sig uiente docume nto que se reproduce fntegramente. " GUIA TE CNICA PARA REAUZACION DE LA VALIDACION DE METODOS DE E NSAYO" 1) INTRODUCCION: La validaci6n de los metodos de ensayo se entiende como una acci6n o un procedimiento destinado a establecer pruebas documentales que demuestren cientfficamente que un metoda analltico tiene las caracteristicas de desempefio adecuadas para cumplir los requerimientos de las aplicaciones analfticas prete ndidas, esenc ia lmente en terminos de su exactitud y precision. 2 01
Transcript of Guía para la realización de validación de métodos de ensayo

ESTABLECE GUlA TECNICA PARA LA REALIZACION DE LA VAUDACION DE METODOS DE ENSAYO.
RESOLUCION EXENTA w 2 0. ENF 2 015 ,.~
SANTIAGO,
VISTOS: Providencia lnterna W 2325 de Ia Jefa de Ia Asesoria Juridica; y TENIENDO PRESENTE: lo dispuesto en Ia l ey Organica Constitucional W 18.575 de "Bases Generales de Ia Administraci6n del Estado"; en Ia Ley N o 19.880 de "Base de los Procedimientos Administrativos que rigen los Actos de los 6rganos de Ia Administraci6n del Estado"; C6digo Sanitario; Decreto Supremo No 3 de 2010, del Ministerio de Salud, que aprueba el "Reglamento del Sistema Nacional de Control de Productos Farmaceuticos de Uso Humano"; en los articulos 59 letra a) y b), 60 y 611etra b) del Decreto con Fuerza de Ley W 1 de 2005, que fija el texto refundido, coordinado y sistematizado del Decreto Ley W 2763 de 1979 y de las Leyes W 18.933 y W 18.469"; Resoluci6n Exenta W 607 del 2014 del Ministerio de Salud; asi como lo establecido en Ia Resoluci6n W 1.600 de 2008, de Ia Contraloria General de Ia Republica; Decreto Exento N2 27 de 2012 del Ministerio de Salud "Norma Tecnica W 131 que define los criterios destinados a establecer Ia equivalencia terapeutica en productos farmaceuticos en Chile", Decreto Exento N!:! 28 de 2012 Ministerio de Sa lud "Norma Tecnica W 127 sobre Buenas Prckticas de Manufactura (BPM)" y Decreto Exento N!:! 543 de 2013 Ministerio de Salud, "Norma Tecnica W 139 sobre Buenas Practicas de Laboratorio (BPL)" ; dicto Ia siguiente:
CONSIDERANDO:
1.- La importancia de Ia validaci6n de los metodos de ensayo, es que a traves de esta, es posible establecer el procedimiento y las condiciones en las que deben realizarse, confiando asi, que los datos obtenidos cumplen con Ia calidad deseada, brindando seguridad y respaldo.
2.- De acuerdo a las Buenas Practicas de Manufactura (BPM) y de Laboratorio (BPL), Ia validaci6n debe aplicarse en todas las etapas de fabricaci6n y control de un producto farmaceutico, esto es: manufactura, control de procesos control y aseguramiento de Ia calidad.
RE SOLU CION
1.- APRUEBESE como "GU[A TECNICA PARA REALIZACION DE LA VALIDACION DE METODOS DE ENSAYO" el siguiente documento que se reproduce fntegramente.
"GUIA TECNICA PARA REAUZACION DE LA VALIDACION DE METODOS DE ENSAYO"
1) INTRODUCCION:
La validaci6n de los metodos de ensayo se entiende como una acci6n o un procedimiento destinado a establecer pruebas documentales que demuestren cient fficamente que un metoda analltico tiene las caracteristicas de desempefio adecuadas para cumplir los requerimientos de las aplicaciones analfticas pretendidas, esencia lmente en terminos de su exactitud y precision.
201

2} REQUISITOS Validaci6n de Ia Metodologia Analitica de los ensayos rea lizados a los medicamentos
(materias primas y productos terminados) se encuentra exigido segun lo establecido en:
1. Reglamento del Sistema Nacional de Control de Productos Farmaceuticos, Decreta Supremo MINSAL NQ 03 del af\o 2010:
Titulo II del Registro Sanitaria de las Especialidades Farmaceuticas y otros productos
farmaceuticos: Art. 32° y 40° Parrafo segundo: De los requisites del Registro Sanitaria,
Titulo VI Laboratories Farmaceuticos: Art. 145° Parrafo quinto: los procedimientos y registros; Art. lSSQ Parrafo sexto: del personal y las responsabilidades; Art. 1872 Parrafo cuarto: de Ia exenci6n de control de calidad y del control de serie
2. Norma Tecnica W 131, nominada "Norma que define los criterios destinados a establecer Ia equivalencia terapeutica en productos farmaceuticos en Chile (EQT)"- Decreto Exento MINSAL NQ 27 de 2012.
3. Norma Tecnica W 127 de Buenas Practicas de Manufactura (BPM)- Decreto Exento MINSAL N2
28 de 2012.
4. Norma Tecnica W 139 de Buenas Pn!cticas de Laboratorio (BPL) - Decreta Exento MINSAL NQ
543 de 2013. 3) ALCANCE
Los conceptos y terminos utilizados en esta guia son de aplicaci6n general y estan dirigidos principalmente para ser aplicados sobre los metodos analiticos para el adecuado control de ca lidad en:
Materias primas Materiales de envase
Productos en estadios de producci6n intermedios o terminados
Estudios de estabilidad
Estudios de investigaci6n y desarrollo galenico.
Estudios de Equivalencia Terapeutica In Vitro.
Los Metodos Analiticos (MA) o Metodo de Ensayo (ME) para estudios de metabolismo, farmacocinetica, biodisponibilidad, valoraciones microbiol6gicas y biol6gicas pueden requerir otras exigencias no contempladas en esta Gufa.
4) CONCEPTOS GENERALES: Anal ito Principia activo o componente medido por el M E.
Blanco Matriz en Ia cual se mide el analito sin este.
Buenas Pr<kticas de Conjunto de reglas, procedimientos operatives y practicas que Laboratorio (BPL) garantizan que los datos generados por un sistema de control de
calidad son reproducibles y representativos, asegurando Ia validez y confiabilidad de los resultados.
Buenas Pr<kticas de Norm as tecnicas ' . mm1mas establecidas para todos los Manufactura (BPM) procedimientos destinados a garantizar Ia calidad uniforme y
satisfactoria de los productos farmaceuticos, dentro de los limites aceptados y vigentes para cada uno de ellos.
Ca li ficaci6n del disef\o (DQ, Colecci6n documentada de actividades que definen las por sus siglas en ingles) especificaciones operacionales y funcionales del instrumento y
criterios para Ia selecci6n del vendedor, basandose en el uso previsto del instrumento.
Calificaci6n de instalaci6n {IQ, La ejecuci6n de pruebas para asegurar que los equipos analiticos
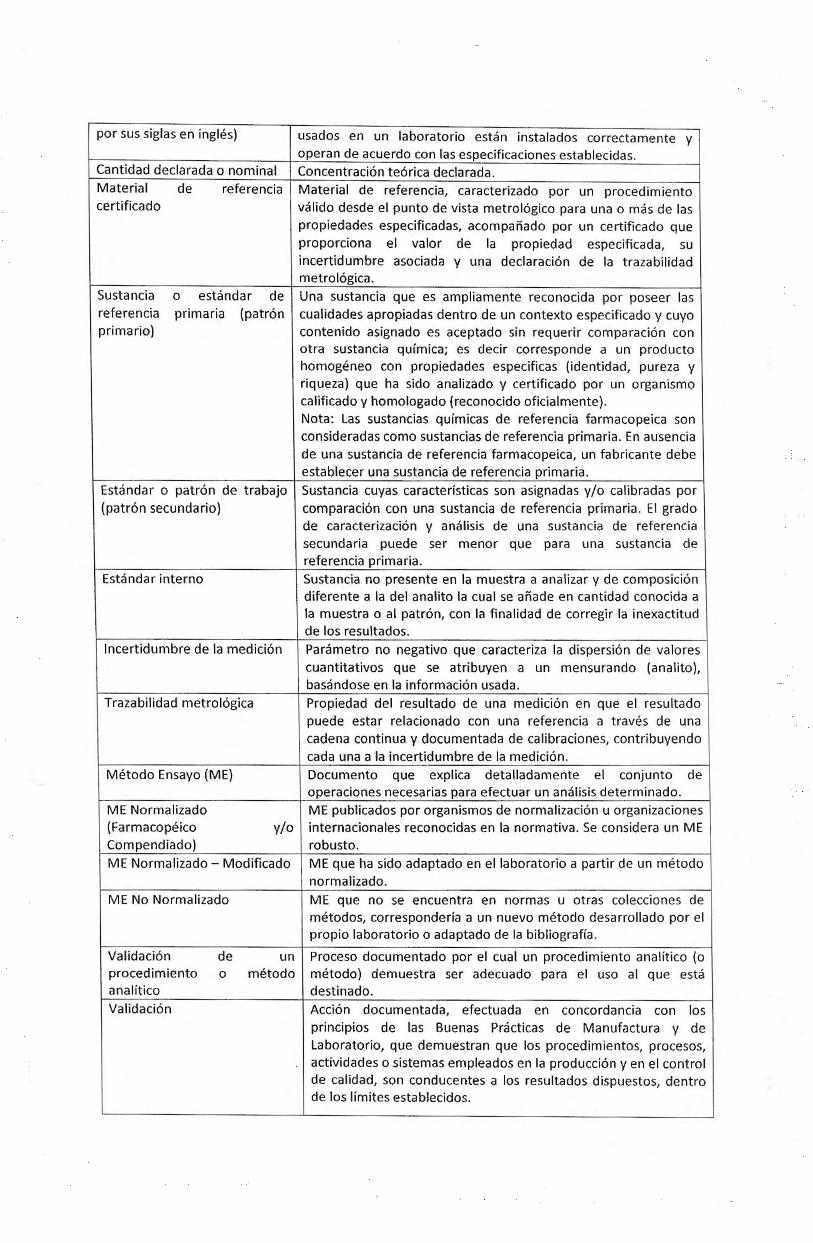
por sus siglas en ingles) usados en un laboratorio est an instalados correctamente y operan de acuerdo con las especificaciones establecidas.
Cantidad declarada o nominal Concentracion teorica declarada. Material de referencia Material de referencia, caracterizado por un procedimiento certificado valido desde el punto de vista metrologico para una 0 mas de las
propiedades especificadas, acompanado por un certificado que propordona el valor de Ia propiedad especificada, su incertidumbre asodada y una declaracion de Ia trazabilidad metrologica.
Sustanda 0 estandar de Una sustancia que es ampliamente reconodda por poseer las referenda prima ria (patron cualidades apropiadas dentro de un contexto especificado y cuyo prima rio) contenido asignado es aceptado sin requerir comparaci6n con
otra sustancia qufmica; es decir corresponde a un producto homogeneo con propiedades especificas (identidad, pureza y riqueza) que ha sido analizado y certificado por un organismo calificado y homologado (reconocido oficialmente). Nota: Las sustancias quimicas de referencia farmacopeica son consideradas como sustandas de referenda primaria. En ausencia de una sustanda de referenda farmacopeica, un fabricante debe establecer una sustanda de referenda primaria.
Estandar o patron de trabajo Sustanda cuyas ca racteristicas son asignadas y/o calibradas por (patron secundario) comparacion con una sustancia de referencia primaria. £1 grado
de caracterizad6n y analisis de una sustancia de referenda secunda ria puede ser me nor que para una sustancia de referenda primaria.
Estandar interno Sustancia no presente en Ia muestra a analizar y de composicion diferente a Ia del analito Ia cual se anade en cantidad conocida a Ia muestra o al patron, con Ia finalidad de corregir Ia inexactitud de los resultados.
lncertidumbre de Ia medicion Parametro no negativo que caracteriza Ia dispersion de valores cuantitativos que se atribuyen a un mensurando (ana lito), basandose en Ia informacion usada.
Trazabilidad metrol6gica Propiedad del resultado de una medid6n en que el resultado puede estar reladonado con una referenda a traves de una cadena continua y documentada de calibraciones, contribuyendo cada una a Ia incertidumbre de Ia medici6n.
Metodo Ensayo (ME) Oocumento que explica detalladamente el con junto de operadones necesarias para efectuar un analisis determinado.
ME Normalizado ME publicados por organismos de normalizaci6n u organizaciones (Fa rmacopeico y/o internadonales reconoddas en Ia normativa. Se considera un ME Compendiado) robusto. ME Normalizado- Modificado ME que ha sido adaptado en ellaboratorio a partir de un metodo
normalizado. ME No Normalizado ME que no se encuentra en normas u otras colecdones de
metodos, corresponderia a un nuevo metodo desarrollado por el propio laboratorio o adaptado de Ia bibliografia.
Validadon de un Proceso documentado por el cual un procedimiento analitico (o procedimiento 0 metodo metodo) demuestra ser adecuado para el uso al que est a analitico destinado. Validacion Acci6n documentada, efectuada en concordanda con los
prindpios de las Buenas Practicas de Manufactura y de Laboratorio, que demuestran que los procedimientos, procesos, actividades o sistemas empleados en Ia produccion yen el control de calidad, son conducentes a los resultados dispuestos, dentro de los limites establecidos.
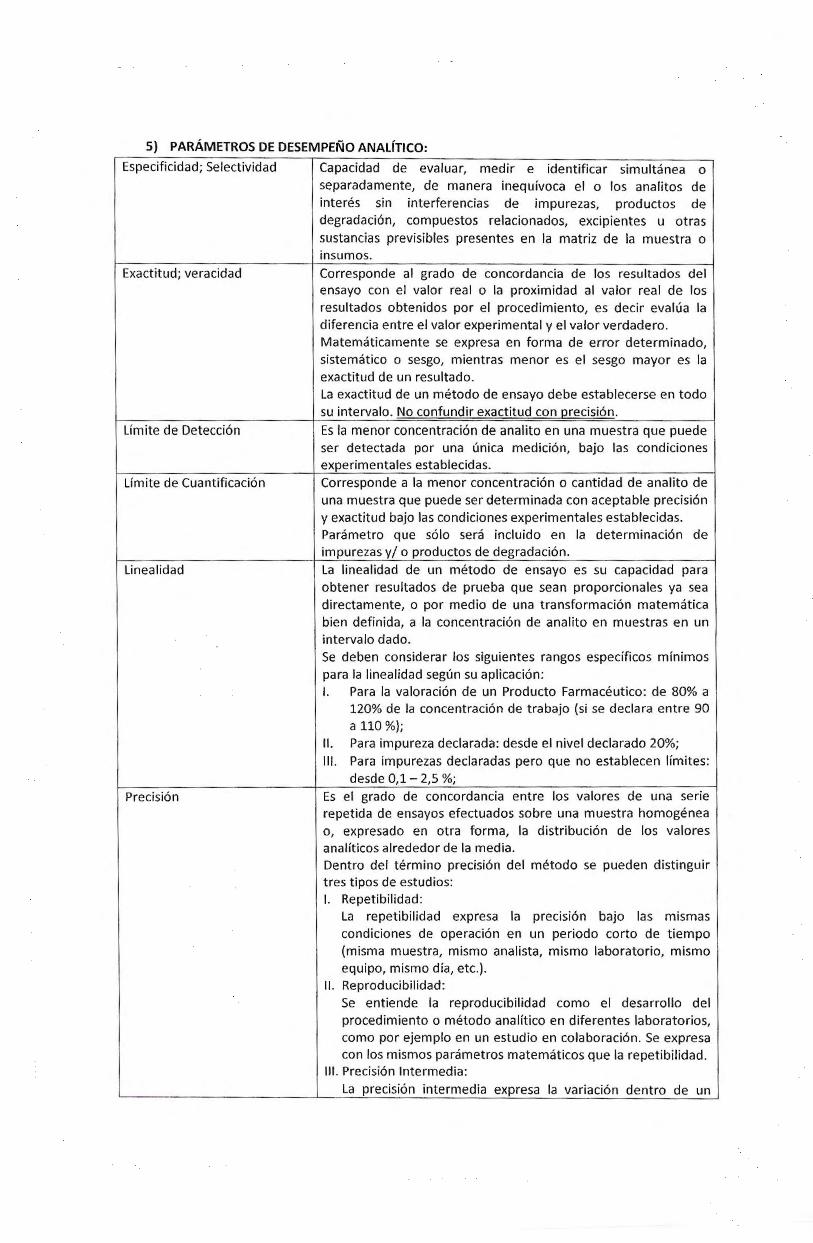
5) PARAMETROS DE DESEMPENO ANALfTICO:
Especificidad; Selectividad
Exactitud; veracidad
Limite de Detecci6n
Limite de Cuantificacion
Linealidad
Precision
Capacidad de evaluar, medir e identificar simultanea o separadamente, de manera inequivoca el o los analitos de interes sin interferencias de impurezas, productos de degradacion, compuestos relacionados, excipientes u otras sustancias previsibles presentes en Ia matriz de Ia muestra o insumos. Corresponde al grado de concordancia de los resultados del ensayo con el valor real o Ia proximidad al valor real de los resultados obtenidos por el procedimiento, es decir evalua Ia diferencia entre el valor experimental y el valor verdadero. Matematicamente se expresa en forma de error determinado, sistematico o sesgo, mientras menor es el sesgo mayor es Ia exactitud de un resultado. La exactitud de un metodo de ensayo debe establecerse en todo su intervalo. No confundir exactitud con precision.
Es Ia menor concentraci6n de analito en una muestra que puede ser detectada por una unica medicion, bajo las condiciones experimentales establecidas. Corresponde a Ia menor concentracion o cantidad de analito de una muestra que puede ser determinada con aceptable precision y exactitud bajo las condiciones experimentales establecidas. Parametro que solo sera incluido en Ia determinacion de impurezas y/ o productos de degradacion. La linealidad de un metodo de ensayo es su capacidad para obtener resultados de prueba que sean proporcionales ya sea directamente, o por medio de una transformacion matematica bien definida, a Ia concentracion de analito en muestras en un intervalo dado. Se deben considerar los siguientes rangos especificos minimos para Ia linealidad segun su aplicaci6n: I. Para Ia valoracion de un Producto Farmaceutico: de 80% a
120% de Ia concentracion de trabajo (si se declara entre 90 a 110 %);
II. Para impureza declarada: desde el nivel declarado 20%; Ill. Para impurezas declaradas pero que no establecen limites:
desde 0,1 - 2,5 %;
Es el grado de concordancia entre los valores de una serie repetida de ensayos efectuados sobre una muestra homogenea o, expresado en otra forma, Ia distribucion de los valores analiticos alrededor de Ia media. Dentro del termino precision del metodo se pueden distinguir tres t ipos de estudios: I. Repetibilidad:
La repetibilidad expresa Ia precision bajo las mismas condiciones de operacion en un periodo corto de tiempo (misma muestra, mismo analista, mismo laboratorio, mismo equipo, mismo dia, etc.).
II. Reproducibilidad: Se entiende Ia reproducibilidad como el desarrollo del procedimiento 0 metodo analitico en diferentes laboratorios, como por ejemplo en un estudio en colaboracion. Se expresa con los mismos parametros matematicos que Ia repetibilidad.
Ill. Precision lntermedia: La precision intermedia expresa Ia variacion dentro de un
laboratorio por ejemplo en diferentes dias, con diferentes analistas 0 con equipo diferente dentro del mismo laboratorio, con Ia misma muestra homogenea.
Robustez La robustez de un metoda de ensayo esta definida como Ia medida de su capacidad para permanecer inalterado ante pequenas pero deliberadas variaciones en ciertos parametros, proporcionando una idea de su fiabilidad durante su empleo en rutin a.
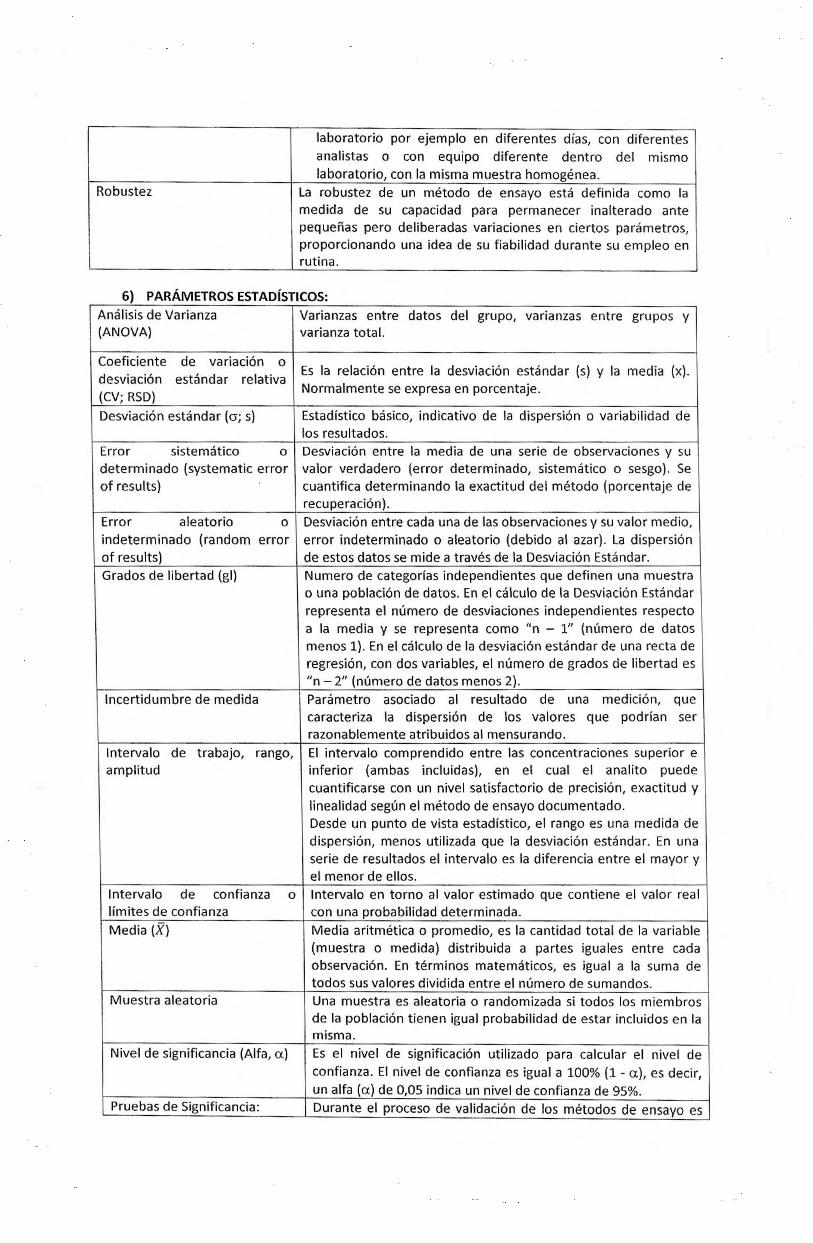
6) PARAMETROS ESTADJSTICOS:
Analisis de Varianza Varianzas entre datos del grupo, varianzas entre grupos y (AN OVA) varianza total.
Coeficiente de variacion 0 Es Ia relacion entre Ia desviacion estandar (s) y Ia media (x).
desviacion estandar relativa (CV; RSD)
Normalmente se expresa en porcentaje.
Desviacion estandar {cr; s) Estadistico basico, indicativa de Ia dispersion o variabilidad de los resultados.
Error sistematico 0 Desviacion entre Ia media de una serie de observaciones y su determinado (systematic error valor verdadero (error determinado, sistematico o sesgo). Se of results) cuantifica determinando Ia exactitud del metoda (porcentaje de
recuperacion). Error aleatorio 0 Desviacion entre cada una de las observaciones y su valor medio, indeterminado {random error error indeterminado o aleatoric {debido al azar). La dispersion of results) de estos datos se mide a traves de Ia Desviacion Estandar. Grados de libertad (gl) Numero de categorlas independientes que definen una muestra
o una poblacion de datos. En el calculo de Ia Desviacion Estandar representa el numero de desviaciones independientes respecto a Ia media y se representa como "n - 1" (numero de datos menos 1). En el calcu lo de Ia desviacion estandar de una recta de regresion, con dos variables, el numero de grados de libertad es "n- 2" (numero de datos menos 2).
lncertidumbre de medida Parametro asociado al resultado de una medicion, que caracteriza Ia dispersion de los valores que podrian ser razonablemente atribuidos al mensurando.
lntervalo de trabajo, rango, El intervalo comprendido entre las concentraciones superior e amplitud inferior (am bas incluidas), en el cual el ana lito puede
cuantificarse con un nivel satisfactorio de precision, exactitud y linealidad segun el metoda de ensayo documentado. Desde un punto de vista estadistico, el rango es una medida de dispersion, menos utilizada que Ia desviacion estandar. En una serie de resultados el intervalo es Ia diferencia entre el mayor y el menor de ellos.
Intervale de confianza 0 lntervalo en torno al valor estimado que contiene el valor real limites de confianza con una probabilidad determinada. Media (X) Media aritmetica o promedio, es Ia cantidad total de Ia variable
(muestra o medida) distribuida a partes iguales entre cada observacion. En terminos matematicos, es igual a Ia suma de todos sus valores dividida entre el numero de sumandos.
Muestra aleatoria Una muestra es aleatoria o randomizada si todos los miembros de Ia poblacion tienen igual probabilidad de estar incluidos en Ia misma.
Nivel de significancia (Aifa, u) Es el nivel de significacion utilizado para calcular el nivel de confianza. El nivel de confianza es igual a 100% (1 - u), es decir, un alfa (u) de 0,05 indica un nivel de confianza de 95%.
Pruebas de Significancia: Durante el proceso de validacion de los metodos de ensayo es

frecuente utilizar pruebas de significancia estadisticas, las mas comunes son: 1. Prueba t-Student para identificar errores sistematicos (sesgo). 2. Prueba F-Fisher para identificar errores aleatorios
(precisiones). AI hacer una prueba de significancia se comprueba Ia veracidad de una hip6tesis experimental, llamada "hip6tesis alternativa" (H1, si hay diferencia,) con respecto a Ia hip6tesis nula (H0, no hay diferencia) . Es Ia H1 Ia que determina el numero de colas. Si Ia H1 contiene Ia frase "mayor que" o "menor que", Ia prueba es de una-cola. Si Ia H1 contiene Ia frase "noes igual que", Ia prueba es de dos-co las.
Resultado Aberrante: Corresponde a cualquier valor extremo que ocurra durante Ia ejecuci6n de una prueba. En situaciones normales estos resultados t ienen una frecuencia aleatoria menor al 1 %.
Varianza (S) Se define como el cuadrado de Ia desviaci6n estandar y su utilidad es que facilita el calculo de Ia propagaci6n de errores.
7) DESARROLLO DE LOS METODOS ENSAYO: Un metodo analitico se desarrolla para probar o ensayar una caracteristica definida de un
principia activo o Ia de un producto farmaceutico frente a los criterios de aceptaci6n establecidos para esa caracte ristica . lnicia lmente en el desarrollo de un nuevo metodo analitico, Ia elecci6n de Ia instrumentaci6n y metodologia analitica debera seleccionarse con base a Ia finalidad prevista y el alcance del metodo analitico. Los parametros que pueden ser evaluados durante el desarrollo del metodo de analisis son: Especificidad, Linealidad, Limite de detecci6n (LD) y Limite de cuantificaci6n (LC), Exactitud y Precision.
Durante las eta pas iniciales del desarrollo de el o los metodos de analisis, se recomienda Ia eva luaci6n de Ia robustez o solidez de estos porque esta caracteristica puede ayudar a decidir que metodo va a se leccionar de manera definitiva.
8) DOCUMENTAR METODOS DE ENSAYO:
Los metodos de ensayos deben estar documentados y deben incluir Ia descripci6n de los metodos analiticos con el su ficiente detalle como para permitir que un ana lista competente pueda reproducir las condiciones necesarias y obtener resultados dentro de los criterios de aceptaci6n establecidos. Tambien debe incluir en Ia descripci6n los aspectos de los metodos analiticos que requieren una atenci6n especial.
Un Metodo de Ensayo o Metodologfa Analitica, puede ser referenciado a Metodos Normalizados o compendiados de fuentes reconocidas por el OS N2 03/10, en su articulo 33° (Farmacopeas oficiales), solo si el metodo de ensayo que se hace referenda no se modifica mas alia de lo permitido en el metodo publicado.
Cuando el metodo de ensayo utilizado sea un metodo "Normalizado-Modificado" o uno "No Normalizado" debe proporcionar en detalle el o los procedimientos para llevarlo a cabo.
La siguiente tabla contiene info rmacion esencial que debe incluir un Metodo de Ensayo Documentado· A. Principia I Alcance
B. Aparatos I Equipos
C. Parametros de funcionamiento
Una descripci6n de los pnncipios basicos del ensayo analitico/ tecnologia (separaci6n, detecci6n, etc.); analito objetivo (s) y tipo de muestra (s) ej., materia prima, producto terminado, impurezas o compuestos relacionados). Todos los equipos y componentes necesarios y ca lificados (ej., tipo de instrumento, detector, tipo de columna, dimensiones, columna alternativa, tipo de fi ltro, etc.). Ajustes 6ptimos ca lificados y rangos (ajustes
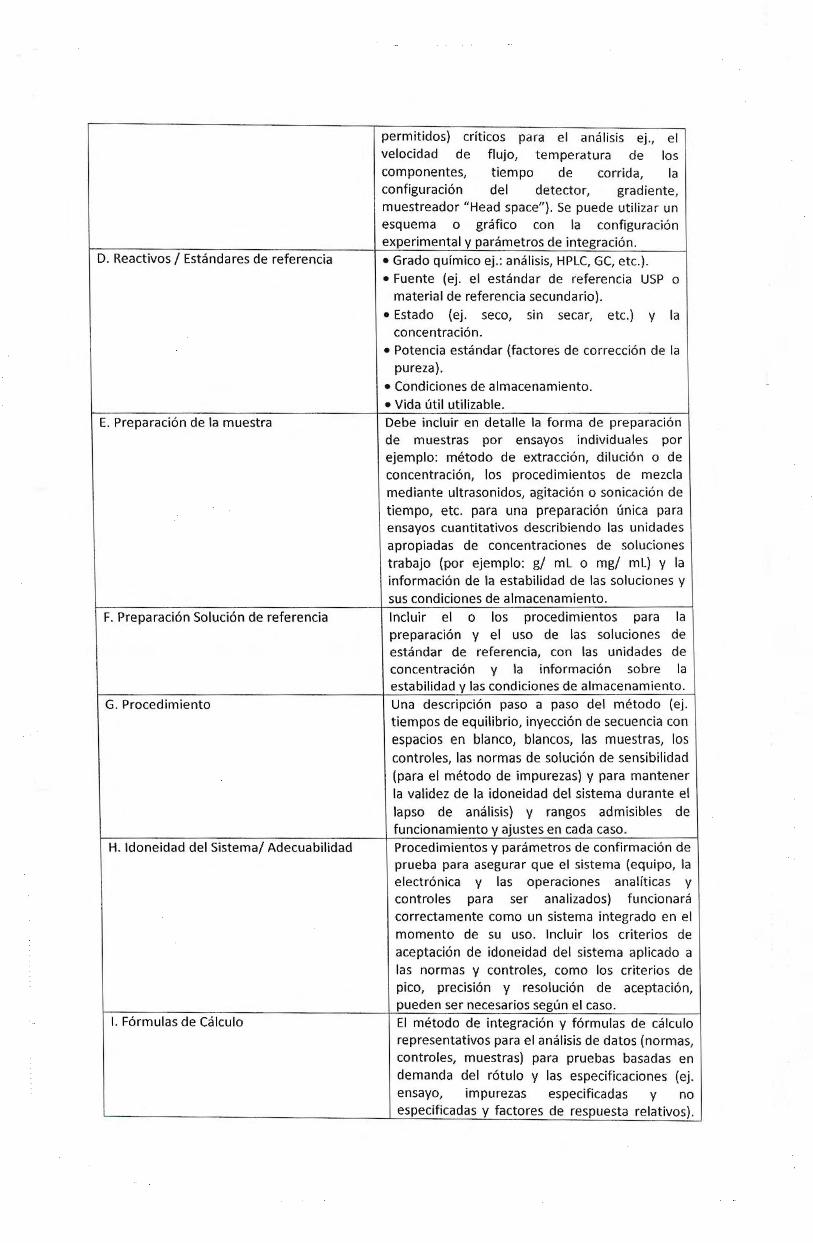
D. Reactivos I Estandares de referenda
E. Preparacion de Ia muestra
F. Preparacion Solucion de referencia
G. Procedimiento
H. ldoneidad del Sistema/ Adecuabilidad
I. Formulas de Calculo
permitidos) crlticos para el analisis ej ., el velocidad de flujo, temperatura de los componentes, tiempo de corrida, Ia configuraci6n del detector, gradiente, muestreador "Head space"). Se puede utilizar un esquema o gratico con Ia configuraci6n experimental y parametros de integracion.
• Grado quimico ej.: analisis, HPLC, GC, etc.). • Fuente (ej. el estandar de referenda USP o
material de referenda secundario).
• Estado (ej. seco, sin secar, etc.) y Ia concentracion.
• Potencia estandar (factores de correccion de Ia pureza).
• Condiciones de almacenamiento. • Vida util utilizable. Debe incluir en detalle Ia forma de preparacion de muestras por ensayos individuales por ejemplo: metodo de extraccion, diluci6n 0 de concentraci6n, los procedimientos de mezcla mediante ultrasonidos, agitadon o sonicaci6n de tiempo, etc. para una preparacion (mica para ensayos cuantitativos describiendo las unidades apropiadas de concentraciones de soluciones trabajo (por ejemplo: g/ mL o mg/ mL) y Ia informacion de Ia estabilidad de las soluciones y sus condiciones de almacenamiento. lncluir el o los procedimientos para Ia preparaci6n y el uso de las soluciones de estandar de referenda, con las unidades de concentracion y Ia informacion sobre Ia estabilidad y las condiciones de almacenamiento. Una descripcion paso a paso del metodo (ej . tiempos de equilibria, inyecci6n de secuencia con espacios en blanco, blancos, las muestras, los controles, las normas de solucion de sensibilidad (para el metodo de impurezas) y para mantener Ia validez de Ia idoneidad del sistema durante el lapso de analisis) y rangos admisibles de funcionamiento y ajustes en cada caso. Procedimientos y parametros de confirmacion de prueba para asegurar que el sistema (equipo, Ia electronica y las operaciones analiticas y controles para ser analizados) funcionara correctamente como un sistema integrado en el momento de su uso. lncluir los criterios de aceptaci6n de idoneidad del sistema aplicado a las normas y controles, como los criterios de pico, precision y resoluci6n de aceptacion, pueden ser necesarios segun el caso. El metodo de integraci6n y formulas de calculo representativos para el analisis de datos (normas, controles, muestras) para pruebas basadas en demanda del rotulo y las especificaciones (ej. ensayo, impurezas especificadas y no especificadas y factores de respuesta relativos).

J. Reporte de datos
Esto incluye una descripcion de las transformaciones o las formulas matematicas utilizadas en el amWsis de datos (transformaciones o analisis estadlstico), junto con una justificacion cientffica para cualquier factor de correccion utilizado. Una presentacion de datos numericos que sean consistentes con las capacidades instrumentales y criterios de aceptaci6n. Debe indicar que formato utilizar para informar de los resultados por ejemplo: % de lo rotulado, peso I peso, y peso/ volumen, etc. con el numero especffico de cifras significativas necesarias. Para los metodos cromatograticos, debe incluir los tiempos de retenci6n (RT) para Ia identificacion con base de comparacion estandar de referencia, tiempos relativos de retenci6n (RRT) (conocidos y desconocidos impurezas) rangos aceptables y resultados de Ia muestra de informes criterios. Debe incluir siempre los valores para los criterios de aceptacion, tolerancia, exclusion y rechazo.
9) MATERIAL Y ESTANDARES DE REFERENCIA:
Para efectos de aplicar las indicaciones contenidas en esta gufa, se entendera que los Estandares de Referenda corresponden a un material altamente caracterizado que se utiliza para garantizar Ia identidad, Ia potencia, Ia calidad y Ia pureza de los analitos que se mediran, tales como: principios activos, impurezas relacionadas, disolventes residuales, excipientes, entre otros.
Para asegurar el mantenimiento de las caracterfsticas y homogeneidad de este material se debe procurar mantener de manera estricta las condiciones de almacenamiento, cond iciones de uso, y las instrucciones de manejo para evitar generacion de impurezas o analisis incorrectos. Se debe mantener un control estricto con los respectivos registros de este material para asf garantizar Ia exactitud y ajustarse a un sistema valido de medidas.
10) ENSAYOS DE APTITUD, VERIFICACION Y VALIDACION DE METODOS ENSAYO:
A. Ensayos de Aptitud de ME: El ensayo de aptitud del sistema es una parte integral de muchos metodos analfticos
instrumentales. Los ensayos estan basados en el hecho que el equipo, Ia parte electronica, las operaciones analiticas y las muestras a ser analizadas contribuyen al sistema. El ensayo de aptitud del sistema que va a ser aplicado depende del tipo de procedimiento a usar.
Los ensayos de aptitud se emplean para Ia verificaci6n de metodos normalizados o procedimientos analfticos validados y deben ser realizados antes de cada uno de los analisis. Siempre que los criterios de aptitud del sistema se cumplan, el metodo o procedimiento se considera adecuado para el proposito previsto.
Para los metodos cromatograficos instrumentales ningun analisis de muest ras es considerado aceptable a menos que se haya demostrado Ia aptitud del sistema.
B. Verificaci6n de ME Normalizados: Se considera que los ME Normalizados estan validados para el uso previsto como se
establece en la(s) monograffa(s). Sin embargo, en el laboratorio donde se utilicen se debe confirmar, por ejemplo, que para un producto farmaceutico terminado que se analiza por primera vez, no surgen interferencias a partir de los excipientes presentes, o que para un principia activo, las impurezas que aparecen a partir de una nueva ruta de sfntesis esten diferenciadas adecuadamente. Si el metodo normalizado se adapta para otro uso, debe validarse para tal uso y asf demostrar que es apto para el mismo.
En Ia verificaci6n se deben evaluar diversos elementos, tales como el efecto de Ia matriz sobre Ia recuperaci6n de impurezas y farmacos desde Ia matriz de producto farmaceutico, Ia

aptitud de las columnas y condiciones cromatograficas, y Ia adecuada respuesta de Ia senal del detector, entre otros.
Para el proceso de verificacion se pueden usar algunas de las caracteristicas de desempeno analitico que se enumeran en el punta C. La Verificacion puede requerir o no Ia ejecucion, en condiciones rea les, del metoda para cada una de los parametros de desempeno, siendo Ia evaluacion de especificidad un parametro clave en Ia verificacion de que un metoda farmacopeico es apto para usar en Ia valoracion de principia activo. El grado y extension del proceso de verificaci6n puede depender del t ipo de metoda (pasos especificos y matrices) y los equipos o instrumentos asociadas.
No se requiere verificaciones ni va lidacion para metodos normalizados basicos tales como (pero no limitados a): apariencia, cenizas sulfatadas, densidad, dureza, friabilidad, indice refraccion, masa, metodos de quimica humeda, perdida por secado, pH, tiempo de desintegracion, viscosidad y volumen.
C. Va lidaci6n de metodos de ensayo "Normalizados-Modificados" y "No Normalizados"
Validacion del metoda analitico es el proceso para demostrar que un procedimiento analitico es adecuado para los fines previstos. La metodologia y el objetivo de los procedimientos analiticos deben estar claramente definidos y entendidos antes de iniciar los estud ios de va lidacion. La validacion de datos deben generarse en virtud de un protocolo aprobado se debe contar con Ia descripci6n del metoda de ensayo de cada prueba caracteristica incluyendo los criterios de aceptaci6n debidamente justificados, todo esto se debe operar siguiendo las BPL y/o BPM.
o. Parametros de desempeno analitico (Validaci6n Metoda de Ensayo) : Los parametros de validacion son·
Especificidad/ Selectividad
Linea lid ad
Precision
cQue·hacer? Analisis simultaneo de: • Un blanco de reactivos; • Una matriz natural o sintetica libre de analito;
• Analito en un solvente adecuado; • La misma matriz con el agregado de una
cantidad conocida de analito o una muestra rea l; y
• Potenciales interferencias en presencia de ana lito.
Cantidad de puntas: ~ 5 concentraciones, lgualmente espaciadas. Cantidad de replicados por concentraci6n: de 3 a 5 replicas independientes Usando analito puro o analito en matriz Medidos aleatoriamente
1) Repetibilidad: Preparar 3 niveles de concentraciones independientes (alto-medio-bajo) analizar en sextuplicado.
2) Precision intermedia Preparar 3 niveles de concentraciones independientes {alto-medio-bajo) analizar en sextuplicado. Modificando: dia o instrumento o analista o
cC6mo se evalua?
Se debe garant izar el buen funcionamiento del metoda, y que este distingue los efectos de las impurezas o interferentes, las sustancias que reaccionan entre si, etc., que podrian estar presentes en Ia matriz
Eva luacion visual para definir t ipo curva (lineal, exponencial, logaritmo etc.) Evaluacion estadistica de Ia curva (ej. cuadrados minimos) Gratico de los residuos Coeficiente de correlacion (R2
)
>0,99 Pendiente y ordenada al origen (dependiente de Ia concentracion de trabajo) Repetibilidad: La precision de un metoda se expresa cuantitativamente como "imprecision" a traves del calculo de Ia desviacion estandar (s) o coeficiente de variacion (%) de una serie de resultados. Precision intermedia: Calcular
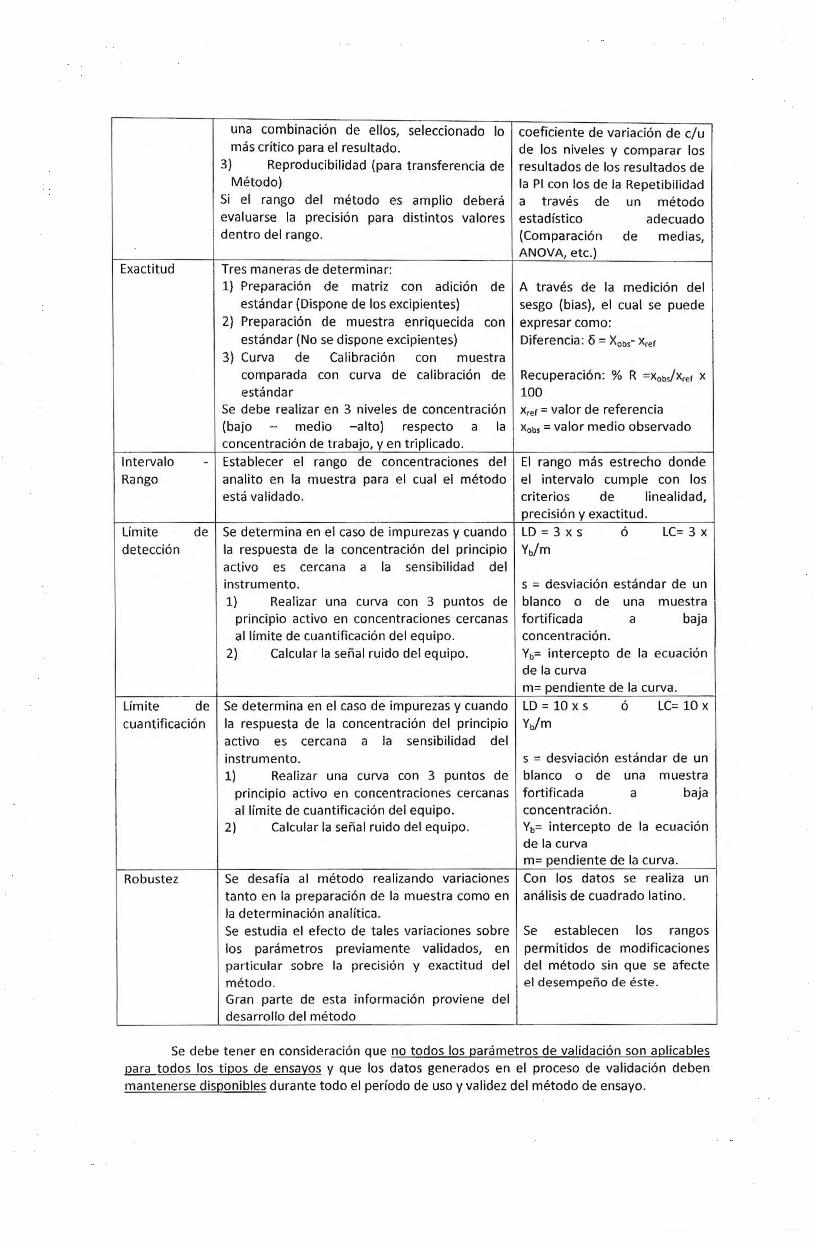
Exactitud
lntervalo Rango
Umite detecci6n
Umite
una combinaci6n de ellos, seleccionado lo mas critico para el resultado.
3) Reproducibilidad (para transferencia de Metoda)
Si el rango del metodo es amplio debera evaluarse Ia precision para distintos valores dentro del rango.
Tres maneras de determinar: 1) Preparacion de matriz con adicion de
estandar (Dispone de los excipientes) 2) Preparaci6n de muestra enriquecida con
estandar (Nose dispone excipientes) 3) Curva de Calibracion con muestra
comparada con curva de calibracion de estandar
Se debe realizar en 3 niveles de concentracion (bajo - media -alto) respecto a Ia concentracion de trabajo, yen triplicado.
- Establecer el rango de concentraciones del analito en Ia muestra para el cual el metodo esta validado.
de Se determina en el caso de impurezas y cuando Ia respuesta de Ia concentracion del principia activo es cercana a Ia sensibilidad del instrumento. 1) Realizar una curva con 3 puntas de
principio activo en concentraciones cercanas al limite de cuantificacion del equipo.
2) Calcular Ia sefial ruido del equipo.
coeficiente de variaci6n de c/u de los niveles y comparar los resultados de los resultados de Ia PI con los de Ia Repetibilidad a traves de un metodo estad lstico adecuado (Comparaci6n de medias, ANOVA, etc.)
A traves de Ia medicion del sesgo (bias), el cual se puede expresar como: Diferencia: 6 = Xobs- Xref
Recuperacion: % R =XobJXref x 100 Xref = valor de referenda Xobs = valor medio observado
El rango mas estrecho donde el intervalo cumple con los criterios de linealidad, precision y exactitud.
LD = 3 x S 6 LC= 3 X
Yb/m
s = desviacion estandar de un blanco o de una muestra fortificada a baja concentraci6n. Yb= intercepto de Ia ecuaci6n de Ia curva m= pendiente de Ia curva.
de Se determina en el caso de impurezas y cuando LD = 10 x s 6 LC= 10 x
cua ntificacion Ia respuesta de Ia concentracion del principia Yb/m
Robustez
activo es cercana a Ia sensibilidad del instrumento. 1) Realizar una curva con 3 puntos de
principia activo en concentraciones cercanas al limite de cuantificacion del equipo.
2) Calcular Ia sefial ruido del equipo.
Se desafla al metodo rea lizando variaciones tanto en Ia preparacion de Ia muestra como en Ia determinacion anaHtica. Se estudia el efecto de tales variaciones sobre los parametros previamente validados, en particular sobre Ia precision y exactitud del metodo. Gran parte de esta informacion proviene del desarrollo del metodo
s = desviaci6n estandar de un blanco o de una muestra fortificada a baja concentraci6n. Yb= intercepto de Ia ecuaci6n de Ia curva m= pendiente de Ia curva. Con los datos se realiza un analisis de cuadrado Iatino.
Se establecen los rangos permitidos de modificaciones del metodo sin que se afecte el desempeno de este.
Se debe tener en consideraci6n que no todos los parametros de validacion son aplicables para todos los tipos de ensayos y que los datos generados en el proceso de validaci6n deben mantenerse disponibles durante todo el perfodo de uso y validez del metodo de ensayo.
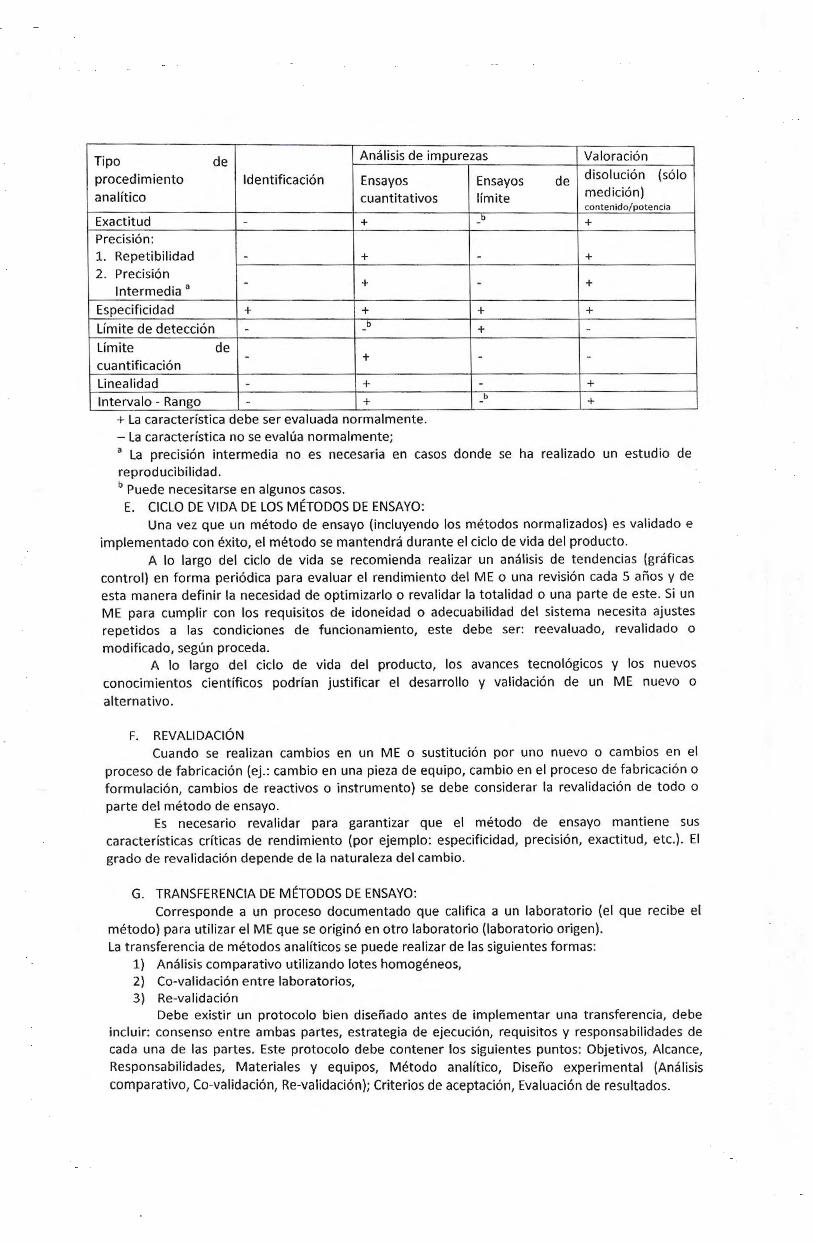
Tipo de Analisis de impurezas Valoracion
procedimiento ldentificacion Ensayos Ensayos de disolucion (solo
analftico cuantitativos If mite medicion) contenido/potencia
Exactitud - + y + Precision: 1. Repetibilidad - + - + 2. Precision
lntermedia a - + - +
Especificidad + + + + Umite de deteccion b - -
Limite de cuantificacion
- +
Linealidad - + Intervale - Rango - +
+ La ca racterist1ca debe ser evaluada normalmente. - La caracteristica no se evalua normalmente;
+ -
- -
- + b + -
a La precision intermedia no es necesaria en casos donde se ha realizado un estudio de reproducibilidad. b Puede necesitarse en algunos casos.
E. CICLO DE VIDA DE LOS METODOS DE ENSAYO: Una vez que un metodo de ensayo (incluyendo los metodos normalizados) es validado e
implementado con exito, el metodo se mantendra durante el ciclo de vida del producto. A lo largo del ciclo de vida se recomienda rea lizar un analisis de tendencias (graficas
control) en forma periodica para evaluar el rendimiento del ME o una revision cada 5 af\os y de esta manera definir Ia necesidad de optimizarlo o revalidar Ia totalidad o una parte de este. Si un ME para cumplir con los requisites de idoneidad o adecuabilidad del sistema necesita ajustes repetidos a las condiciones de funcionamiento, este debe ser: reevaluado, revalidado o
modificado, segun proceda. A lo largo del ciclo de vida del producto, los avances tecnologicos y los nuevos
conocimientos cientificos podrian justificar el desarrollo y validacion de un ME nuevo o
alternative.
F. REVALIDACION Cuando se realizan cambios en un ME o sustitucion por uno nuevo o cambios en el
proceso de fabricacion (ej.: cambio en una pieza de equipo, cambio en el proceso de fabricacion o formulacion, cambios de reactivos o instrumento) se debe considerar Ia revalidacion de todo o
parte del metodo de ensayo. Es necesario revalidar para garantizar que el metodo de ensayo mantiene sus
caracteristicas crfticas de rendimiento (por ejemplo: especificidad, precision, exactitud, etc.) . El grado de revalidaci6n depende de Ia naturaleza del cambio.
G. TRANSFERENCIA DE METODOS DE ENSAYO: Corresponde a un proceso documentado que califica a un laboratorio (el que recibe el
metodo) para utilizar el ME que se origino en otro laboratorio (laboratorio origen). La transferencia de metodos analfticos se puede realizar de las siguientes formas:
1) Analisis comparative utilizando totes homogeneos, 2) Co-validaci6n entre laboratories, 3) Re-validaci6n
Debe existir un protocolo bien disenado antes de implementar una transferencia, debe incluir: consenso entre ambas partes, estrategia de ejecuci6n, requisites y responsabilidades de cada una de las partes. Este protocolo debe contener los siguientes puntos: Objetivos, Alcance, Responsabilidades, Materiales y equipos, Metodo analftico, Diseno experimental (Analisis comparative, Co-validaci6n, Re-validacion); Criterios de aceptaci6n, Evaluacion de resultados.

H. REPORTE DE VERIFICACION o VALIDACION o RE-VALIDACION o TRANSFERENCIA:
Finalmente para documentar todos los resultados obtenidos se debe emitir un Reporte o lnforme de Verificaci6n o Validaci6n o Re-Validaci6n o Transferencia segun sea el caso, este documento se considera Ia finalizaci6n del proceso y debe estar en concordancia con el protocolo respectivo asi como firmado por el personal ttknico responsable. Este documento debe contener claramente Ia conclusion final si el metoda esta calificado para el prop6sito propuesto.
La siguiente tabla contiene informacion esencial que debe incluir un Reporte o lnforme de Verificacion o Validacion o Re-Validaci6n o Transferencia Documentado:
A. Objetivo Declarar el objetivo de Ia validacion B. Alcance Declaraci6n del alcance del metoda de ensayo c. Principia activo I matriz ldentificar analito y matriz D. Metoda ensayo ldentificar M E con nombre y c6digo E. Equipos y Especificaciones Nombre, marca, mode to, numero de
identificacion, tipo de columnas, tipo de detectores, etc.
F. Estandares de Referenda Nombre, marca, procedencia, codigo de identificacion, valores asignadas, unidades, etc.
G. Resultados (de cad a uno de los lncluir una tabla con los resultados, tolerancias parametros) y analisis estadisticos. y calificaci6n. . .
H. Registros derivados de Ia validaci6n Citar los registros o reportes entregados por el (trazabilidad a datos crudos) equipo y copia de las planillas de calculos
generadas en el proceso analftico. I. Conclusiones Declaracion del campo de aplicacion del
metoda: analito, rango de trabajo, unidades, matrices y advertencias ace rca de Ia interferencia conocida en las cuales se ha demostrado Ia validaci6n.
J. Responsables ldentificar personal que realiz6 Ia validaci6n.
K. Revision y V!!B!! Superior ldentificar superior jerarquico que valid a y califica los resultados informados.
L. Fecha lnicio I Fecha de Termino Registrar las fechas de inicio y fin del proceso. validaci6n
11) REFERENCIAS: Farmacopea de Estados Unidos I Formulario Nacional
Capitulo General <621> Cromatografia; Capitulo General <1010> Datos analfticos- Interpretacion y Tratamiento; Capitulo General <1224> Transferencia de los procedimientos analfticos; Capitulo General <1225> Validacion de los procedimientos compendiados; Capitulo General <1226> Verificaci6n de Procedimientos compendiados; Advertencias y requisitos generales; y 7. Resultados de ensayo
Guia para Ia industria: Conferencia lnternacional sobre Armonizaci6n (ICH) Q2 (R1) Validacion de los procedimientos analfticos: Texto y Metodologia (marzo de 1995, mayo de 1997) Q3A (R2) lmpurezas en nuevas sustancias de drogas (junio de 2008) Q3B (R2) Las impurezas en Nuevos Productos Farmaceuticos (agosto de 2006) Q3C (R5) lmpurezas: Solventes Residuales (Febrero de 2011)
Guidance for Industry (FDA) Analytical Procedures and Methods Validation for Drugs and Biologics (Draft February 2014)

Serie de lnformes Tecnicos de Ia OMS, No. 957, 2010 Anexo 1 Buenas practicas de Ia OMS para laboratories de control de calidad de productos farmaceuticos
Serie de lnformes Tecnicos de Ia OMS, No. 823, 1992 Comite de Expertos de Ia OMS en Especificaciones para las Preparaciones Farmaceuticas 32°
In forme
ANOTESE, COMUNIQUESE Y PUBLIQUESE
Resol. Al/N"86 22/ 01/15 Distribuci6n:
Oireccion Asesoria Juridica Departamento ANAMED Subdepto. Registro y Autorizaciones Sanitarias Subdepto Laboratorio Nacional de Cont rol Subdepto. lnspecci6n Subdepto Biofarmacla
Gest i6n de Tramites ... ~ . ) , • >~-
.. ......... .. {
.___ ·-
Avda. Marathon Nl! 1000, Nuiioa - Casilla 48 - Fono 5755100- Fax 56-2-5755684- Santiago, Chile.



















