
g INSTITUTO DE QUÍMICA€¦ · Cuando el diablo pase la factura O si alguna vez me faltas tu....
192
g INSTITUTO DE QUÍMICA ' _· -_ ·Y ·- "'\ " . . j . JBI BlrOTECA INSTITUTO DE QUíMICA Unlversidad8 (]e Sao Paulo Z!4CJ? ESTUDOS VISANDO À SÍNTESE DE SESQUITERPENOS BACANOS TIAGO DE OLIVEIRA VIEIRA Tese de Doutorado Orientadora: Helena Maria Carvalho Ferraz São Paulo - SP 2005
Transcript of g INSTITUTO DE QUÍMICA€¦ · Cuando el diablo pase la factura O si alguna vez me faltas tu....

g INSTITUTO DE QUÍMICA r ~ :-- ' _· -_ ·Y ·- "'\ " . . ,._-_.~~) j .
JBI BlrOTECA INSTITUTO DE QUíMICA Unlvers idad8 (]e Sao Paulo
Z!4CJ?
ESTUDOS VISANDO À SÍNTESE DE
SESQUITERPENOS BACANOS
TIAGO DE OLIVEIRA VIEIRA
Tese de Doutorado
Orientadora: Helena Maria Carvalho Ferraz
São Paulo - SP
2005

DEDALUS - Acervo - CQ
lllllllllllllllllllllllllllllllllllllllllllllllll~ III~ 11111111
30100011247
Ficha Catalográfica Elaborada pela Divisào de Biblioteca e
Documentaçào do Conjunto das Químicas da USP.
Vieira , Ti ago de Oliveira V658e Estudos visando à síntese de sesquiterpenos bacanos / Tiago
de Oliveira Vieira. São Paulo, 2005. 179p .
Tese (doutorado) - Instituto de Química da Universidade de São Paulo . Departamento de Química Fundamental.
Orientador : Ferraz , Helena Maria Carvalho
1. Síntese : Química orgânica 2 . Sesquiterpeno I. T. 11. Ferraz, Helena Maria Carvalho, orientador.
547.2 CDD


Às vezes duvido se
uma vida calma e tranqüila
teria sido conveniente para mim - e no entanto
às vezes anseio por isso.
Lord Byron
"Há muito tempo, abandonei a noção de uma vida sem tempestades, ou de um mundo sem
estações secas e assassinas. A vida é por demais complicada, é constante demais nas suas
mudanças para ser diferente do que realmente é. Eu sou, por natureza, instável demais para
ter outra atitude a não ser a de uma profunda desconfiança diante da grave artificialidade
inerente a qualquer tentativa de exercer um controle excessivo sobre forças essencialmente
incontroláveis. Sempre haverá elementos perturbadores, propulsores; e eles estarão sempre
presentes até o momento em que, nas palavras de Lowell, o relógio for retirado do pulso. No
final das contas, são os momentos isolados de inquietude, de desolação, de fortes
convlcçoes e entusiasmos enlouquecidos, que caracterizam nossa vida, que mudam a
natureza e a direção do trabalho e que dão colorido e significado final ao amor e às
amizades. "
Kay R. Jamison em Uma Mente Inquieta
"Sentia-se muito jovem; e, ao mesmo tempo, indizivelmente velha. Passava como uma
navalha através de tudo; e ao mesmo tempo ficava de fora, olhando. Tinha a perpétua
sensação, enquanto olhava os carros, de estar fora, longe e sozinha no meio do mar;
sempre sentira que era muito, muito perigoso viver, por um só dia que fO$se. "
Quem passou pela vida em branca nuvem,
E em plácido repouso adormeceu;
Quem não sentiu o frio da desgraça,
Quem passou pela vida e não sofreu;
Foi espectro de homem, não foi homem,
Só passou pela vida, não viveu.
Ilusões da Vida - Francisco Otaviano
Virginia Woolf em Mrs.Dalloway

Sendo um pouco mais positivo e otimista ...
Cuando pierda todas las partidas Cuando duerma con la soledad
Cuando se me cierren las salidas Y la noche no me deje en paz.
Cuando sieflta miedo deI silencio Cuando cueste mantenerse en pié Cuando se rebelen los recuerdos
Y me pongan contra la pared.
Resistiré, erguido frente a todo Me volveré de hierro para endurecer la pieI
Yaunque los vientos de la vida soplen fuerte Soy como el junco que se dobla
pero siempre sigue en pié.
Resistiré para seguir viviendo Soportaré los golpes y jamas me rendiré
Y aunque los suenos se me rompan en pedazos Resistiré, Resistiré ...
Cuando el mundo pierda toda magia Cuando mi enemigo sea yo
Cuando me apufíale la nostalgia Y no reconozca ni mi voz
Cuando me amenace la locura Cuando en mi moneda salga cruz Cuando el diablo pase la factura
O si alguna vez me faltas tu.
Resistiré, erguido frente a todo Me volveré de hierro para endurecer la pieI
Y aunque los vientos de la vida soplen fuerte soy como el junco que se dobla
pero siempre sigue en pié.
Resistiré, para seguir viviendo Soportaré los golpes y jamas me rendiré
Y aunque los suenos se me rompan en pedazos Resistiré, Resistiré ...
Resistiré - Carlos Toro e Manuel de la Calva

Dedico esta tese à minha mãe, Sandra,
que não só me trouxe ao mundo,
mas também me educou,
sempre apoiou minhas decisões e fez
com que fosse possível dar mais esse passo.

À minha irmã, )úlia, e ao meu padrasto, lô,
pelos agradáveis anos que convivemos,
pelo suporte financeiro e emocional,
por sempre torcerem por mim
e por terem tornado possível,
em diversos sentidos, a execução desta tese.

Ag radeci mentos
" ... As coisas estão tão esquisitas hoje em dia, que a gente, incrível, a gente anda
ressabiado de dizer que gosta das pessoas. Então a gente inventa coisas, entende? A
gente inventa que é tímido e que não encontra jeito de dizer. A gente inventa que está
ocupado e que um dia, sei lá, vai ter tempo de sentar e conversar. Ai, de repente, você
se toca que não tem mais nada para ser feito, é tarde paca. "
Elis Regina, Programa Ensaio 1973.
Gosto muito desta fala da Elis e decidi que iria agradecer - ae coração aberto e sem
vergonha de incorrer na pieguice - a todas as pessoas que foram importantes em minha
vida ao longo destes anos de USP. Aí vai a longa lista ...
À Prof. Helena Ferraz (Marilena) - a quem devo toda a minha formação em síntese
orgânica e acadêmica -, pelos quase dez anos de convívio desde a iniciação científica,
pela orientação cuidadosa, pela liberdade dada para eu expressar minhas idéias
químicas, pelo incentivo sempre constante, pelas muitas cervejas bebidas juntos, pelos
dias acolhedores que passei em sua casa, pelos papos filosóficos - quando eu estava
deveras reticente em relação ao sentido de tudo e da vida -, pelas dicas de gramática -
principalmente quanto às crases - e literatura, por sempre me incitar a viver outras
coisas além da química, pela amizade, enfim.
Ao Prof. Luiz Fernando - meu co-orientador, em caráter informal, com quem também
tive a oportunidade de ser supervisionado no laboratório quando eu era IC e ele aluno de
doutorado -, por todas as sugestões referentes ao trabalho experimental e ao
andamento do projeto.
Aos colegas de laboratório pelo convívio agradável. Os dividi em dois blocos, os antigos e
os atuais, dispostos em ordem não alfabética - espero não ter esquecido de ninguém.
Antigos: Mônica, Elena, Myrian, Fernando Beleza, Ricardo, Luiz Sidney, Luiz Rogério,
Alberto, Andréa Mané, Andréa Fofi, Eliseu, Elias, Rosana, Caio, Fátima, Patrícia, Walter,
Cláudio, Karina e Raquel. Atuais: Graziela, Eliane, Fernanda, Marcus, Samir, Carla ,
Alexsandra, Ramón, Jaqueline, Mariza, Olga e Fabiana.
Aos meus estagiários Cláudia, Juliano e Pedro, por terem me dado a oportunidade de
aprender a transmitir conhecimento teórico e experimental, por terem me ajudado, cada
um a sua maneira, no desenvolvimento da parte experimental, e, por fim, por terem tido
muita paciência.

À Dona Rosa, pelos inúmeros momentos de descontração - que às vezes provocavam
risadas descontroladas logo no período da manhã -, pelo bom humor sempre constante
e, sobretudo, pela lição de vida.
Aos "primos" do laboratório ao lado - em especial Priscila, Takeo e Juliano-, pelos
empréstimos de reagentes, pelos desabafos e, óbvio, pelas festas.
Ao Peta, amigo desde o primeiro ano da graduação, pela confiança em mim como seu
confidente, por todas as discussões teóricas de química - ele é realmente um excelente
químico orgânico -, pela sua visão otimista perante a vida e a ciência.
Ao meu "afilhado" George, pela amizade, pelos bons tempos da faculdade, em que
fizemos grupo de laboratório, e pela aquela prova de cálculo llI.
Aos amigos do tempo de graduação - em especial, Sérgio, Bianca, Ladrão, Gustavo,
Chandelle, Fabiana, Denise e Marcelo (in memoriam) - por terem tornado o curso mais
ameno através da várias festas, jogos de truco e de sinuca no centro acadêmico.
Ao Prof. Teng, por todas as dicas cinematográficas, por todos os DVDs emprestados e por
todas as discussões filosóficas acerca da ciência.
Ao Prof. Borin, pelos tempos de "naquela mesa", em comprávamos livro na física e
éramos mais felizes.
Ao Prof. Hélio Stefani e aos seus alunos (Diogo, Iguatemi, Cláudio e Rodrigo) pelos
reagentes cedidos - em especial o cloreto de alumínio que salvou minha síntese.
Ao Prof. Leandro e ao Dr. André, do bloco zero, pelo trabalho em colaboração sobre as
resoluções enzimáticas.
Aos funcionários da SPG, Central Analítica, Biblioteca e Xerox, em especial Cibele, Milton,
Emiliano, Adriana, Fernando, Márcio, Luzia, Chico e Jailton, por todo o suporte - tão
essencial - ao longo destes anos.
Ao pessoal da manutenção - em especial Dorian, Walter, Zé Tavolaro e Valério (in
memoriam) -, por todos os favores e galhos quebrados e, também, por me aceitarem
como convidado em suas festas.

Ao Joaquim, por todos os churrascos em que fui seu auxiliar, pelas discussões filosóficas
(de botequim), pela enologia - ou será enolatria? -, por toda ajuda no laboratório, e até
mesmo por aquele THF que ele disse que beberia se não ficasse azul, e não ficou.
À Bombom, por ter aprendido a ser mais paciente comigo e pelo tempo que passamos
juntos no laboratório - inclusive nos sábados mais preguiçosos, quando o sol era um
convite à piscina, e continuávamos acorrentados à bancada.
À Aguilar Fraude - minha amiga também neurótica quanto à organização do laboratório e
tratamento de solventes e reagentes -, pelo convívio agradável, apesar das eventuais
discussões, pelas lamentações de ambas as partes quando uma reação não funcionava -
e foram muitas - por conseguir me entender e por ser tão parecida comigo.
À Pepi (Érika), minha confidente e cúmplice mais do que paciente, pelas crises que me
ajudou a superar, pelas cervejas - muitas, por sinal -, por todos os presentes, pelo
carinho constante, por tentar me entender e, por fim, por me mostrar como é importante
ter amigos.
À Sônia Ula-Ula, pela amizade, pelos anos de Vai-Vai, pelo saudosismo, pelos incontáveis
almoços de domingo e pela Aquela feijoada que finalmente saiu.
À "irmã" Bete, por todos os bons momentos vividos tanto no Brasil quanto na Alemanha,
pelos dias de "penúria" no 3114 - que se tornaram mais amenos com aquelas
cervejinhas -, pela confiança e amizade incondicional e pelos "contrabandos" de
guloseimas, apetrechos de cozinha e especiarias trazidos diretamente do velho mundo.
À Martinha, pela amizade sempre fiel - que sempre soube tolerar meus momentos mais
implicantes e chatos -, por estar sempre presente mesmo à distância, nos últimos
tempos, para compartilharmos nossas vitórias e agruras. Nunca esquecerei daquele
pedaço de picanha e das incontáveis garrafas de vinho que bebemos - imagino que ela
também não.
Ao Alexandre - fanático por Almodóvar como eu -, pelos bons filmes que assistimos
juntos, por sua cultura cinematográfica - mesmo com as falhas quanto aos clássicos -,
por ter me disponibilizado um dos melhores filmes do Almodóvar (Qué he hecha Ya para
merecer esta?), que eu ainda não havia visto.

À Morcega (Alessandra), pelos armários que montamos e desmontamos - um deles não
conseguimos remontar -, pela paciência de aturar morar comigo por quase três anos,
pelos empréstimos de carro, pelo incentivo nos meus momentos mais tristes e, por fim,
pela minha nova vida.
À Carranca (Adriana), por sua contagiante alegria de viver, pelo companheirismo, pelos
bolos que fizemos e comemos, por me ensinar que vale a pena viver, pelo um ano e
meio que moramos juntos e, sobretudo, por aturar minhas manias e implicâncias.
À Valquíria , por compartilhar comigo a vida já há dez anos, por ser minha alma gêmea,
ou melhor, minha versão feminina, pelas tortinhas de morango - acompanhadas sempre
de capuccino LIGHT na Saint Etienne -, pelos sanduíches de tomate seco, pelas
incontáveis calorias que ingerimos juntos, pela amizade incondicional - sempre nos
apoiamos até mesmo nos momentos mais Beck Bloom - e por me chamar para a
realidade nos momentos mais "surtados".
Ao Babá Akitunde - meu babalorixá e amigo -, por ter cuidado de mim desde 1999, pelo
carinho sempre presente, pelos conselhos mais que valiosos e, por fim, por ter me
iniciado para Ogun.
À minha família de santo - Tatá, Ominlesi, Toloji, Oyatolu, Ekedi, Alamefá, Odetolá,
Fagbenlé, Ominade, Oyadeji, Babátogun, Tomori, Bandeie (e família) e Akinielé -, pelo
suporte religioso e emocional, pela amizade incondicional e por me ajudarem a superar
os momentos mais difíceis que vivi nos últimos tempos.
À minha família da Bahia nas figuras de vó Ziza (in memoriam), tia Nílbia, vó Núrbia, tia
Dedé, tia Gaua, tia Sônia, tia Vera, tio Fernando, César, Isa, Sheila e Adel, por todo
carinho, mesmo à distância, e pela eterna torcida quanto ao meu futuro.
À FAPESP pela bolsa concedida.

Índice
Resumo
Abstract
1.Introdução
1.1.Sobre a (+ )-Baquenolida-A
1.2.Sínteses da Baquenolida-A
1.2.1.Síntese de Evans
1.2.2.Síntese Biomimética de Hayashi
1.2.3.Síntese Formal de Silva
1.2.4.Síntese de Greene
1.2.S.Síntese de Back
1.2.6.Síntese Formal de Kozmin
1.2.7.Síntese de Reddy
1.2.8.Síntese da (- )-7-Epi-baquenolida-A
1.3.Reações Mediadas por Sais de Tálio(III)
1.3.1.lntrodução
1.3.2.Reações de Ciclização
1. 3.3. Reações de Contração de Anel
2.0bjetivos
3.Planejamento Sintético
4. Resultados e Discussão
4.1.Estudos Visando à Síntese da nor-Baquenolida-A
4.1.1. Preparação do Material de Partida
4.1.2.Construção do Sistema cis-Hidrindânico
4.1.3.Construção do Centro Quaternário
4.1.4.Construção do Anel Espirolactônico
4.1.4.1. Primeira Abordagem
4.1.4.2.Segunda Abordagem
4.2.0utros Estudos Visando à Construção de cis-Hidrindanos
4.2.1.lntrodução
4.2.2.Preparação do Aduto 67
4.2.3.Reação da Olefina 68 com TTN
4.2.4.Reação do 4-Alquenol 71 com TTN
Índice
ii
1
1
3
3
5
6
7
9
10
12
13
16
16
17
18
21
22
24
24
24
27
29
31
31
33
38
38
39
39
40

Índice
4.2.5.Reação de Contração de Anel da Olefina 74 com TTN 44
4.3.Estudos Visando à Síntese da Baquenolida-A 46
4.3.1.Introdução 46
4.3 .2.Definição da Relação eis entre as Metilas da Baquenolida-A 48
4.3.2.1.Primeira Abordagem 48
4.3.2.2.Segunda Abordagem 50
4 .3.3.0utra Possível Rota Sintética para a Baquenolida-A 52
4.3.3.1.Introdução 52
4.3.3 .2.Preparação da octalona 91 58
4.3.3.3.Reação de Contração de Anel da Octalona 91 64
4.3.3.4.Tentativas de Contração de Outras Cetonas 66
4.3.3.5.Tentativas de Contração do Dieno 105 67
4.4.Resolução Enzimática de eis-Octalinas 69
4.4.1.Introdução 69
4.4.2.Preparação dos Materiais de Partida 70
4.4.3.Resolução Cinética: O "Screening" 71
4.4.4.Resolução Cinética dos cis-Octalóis com Novozyme 435 72
5.Conclusão 77
6.Parte Experimental 79
Considerações Gerais - Avis aux Lecteurs 79
6.1.nor-Baquenolida-A 81
6.2.0utros eis-Hidrindanos 88
6.3 .Baquenolida-A 93
6.4.Resolução Enzimática 104
7.Espectros de RMN 110
8.Referências 173
Curriculum Vitae 178

Resumo
Nesta tese, efetuamos estudos visando à síntese de sesquiterpenos bacanos, cuja
etapa chave consistiu na construção do sistema cis-hidrindânico, através de reação de
contração de anel de cis-octalinas e 2-octalonas mediada por trinitrato de tálio (ITN).
Apenas as cis-octalinas como, por exemplo, o cis-4a-metil-l,2,3,4,4a,5,8,8a-octa
hidronaftaleno e o cis-4a, 7-dimetil-l,2,3,4,4a,5,8,8a-octa-hidronaftaleno, foram
passíveis de reação de contração de anel em rendimentos satisfatórios; já a cis-5,10-
dimetil-l(9)-octal-2-ona levou ao produto de contração em baixo rendimento.
Tentamos utilizar a reação de cis-4a-metil-l,2,3,4,4a,5,8,8a-octa-hidronaftaleno
com ITN na síntese da nor-baquenolida-A, porém não conseguimos completar a síntese
desta, pois não foi possível efetuar a última etapa sintética, nas várias abordagens
testadas.
Grandes esforços também foram empregados na preparação diastereosseletiva da
cis-5,10-dimetil-l(9)-octal-2-ona através de três abordagens diferentes que foram
investigadas, sendo duas delas com êxito. Contudo, o baixo rendimento (38%) da etapa
de contração de anel da cis-5,10-dimetil-l(9)-octal-2-ona não permitiu a continuação da
rota sintética proposta para a baquenolida-A.
Também realizamos a resolução cinética de três diferentes cis-octalóis - que
foram preparados através da reação de Diels-Alder seguida de redução diastereosseletiva
- com a lipase Novozym 435, e os produtos resolvidos foram obtidos em excelentes
rendimentos isolados (~ 40% para cada enantiômero) e excelentes excessos
enantioméricos (~ 98%).

Abstract
Abstract
In this thesis, we have developed studies towards the synthesis of sesquiterpenes
bakkanes, which key step consisted on the construction of the cis-hydrindanic system
through a thallium(III) mediated ring contraction reaction of cis-decalins and 2-
octalones. Only the cis-octalins, such as the 1,2,3,4,4a,5,8,8a-octahydro-4a
methylnaphthalene and the 1,2,3,4,4a,5,8,8a-octahydro-4a,7-dimethylnaphthalene,
were able to be ring contracted in satisfactory yields; the 4,4a,5,6,7,8-hexahydro-4a,5-
dimethylnaphthalen-2(3H)-one, however, furnished the ring contraction product in low
yield.
We tried to use the reaction of 1,2,3,4,4a,5,8,8a-octahydro-4a-
methylnaphthalene with TIN in the synthesis of nor-bakkenolide-A, but we could not
accomplish the synthesis because it was not possible to make the last step of the
sequence, in ali tested approaches.
Great efforts were made in the diastereoselective preparation of the 4,4a,5,6, 7 ,8-
hexahydro-4a,5-dimethylnaphthalen-2(3H)-one, through three different approaches that
were investigated, being two of them with profit. However, the low yield (38%) of the
ring contraction reaction of 4,4a,5,6, 7,8-hexahydro-4a,5-dimethylnaphthalen-2(3H)-one,
precluded the continuation of the synthetic rout proposed to the bakkenolide-A.
We have also performed the kinetic resolution of three different cis-octalols - that
were prepared through Diels-Alder reaction followed by diastereoselective reduction -
with the Novozym 435 lipase, and the resolved products were isolated in excellent yields
(~40% for each enantiomer) and excellent ee's (~ 98%).
li

Introdução
1.Introdução
1.i.Sobre a (+ )-Baquenolida-A*
A (+ )-baquenolida-A (1) (Figura 1) é uma p-metileno-y-espirolactona
sesquiterpênica - primeiro membro da família dos bacanos - que foi isolada em 1968,
por dois grupos independentes,1,2 da planta japonesa Petasites japonicus. Foi batizada
pelos autores como baquenolida ou fuquinanolida, em virtude do nome local da planta
(Bakke ou Fuki) da qual foi isolada. Esta substância apresenta atividade citotóxica e
antitumoral,3,4 além de impedir o crescimento larval e de ser um pesticida naturaI. 5-11
# Cpp (+)-1 1
Figura 1
Embora não seja determinada a biossíntese dos bacanos - um esqueleto
rearranjado em relação às unidades isoprênicas - acredita-se que eles derivem dos
eremofilanos,2,12 através de um rearranjo envolvendo uma contração de anel, que, por
sua vez, derivam dos eudesmanos, através de uma migração 1,2 de metila, como
sugerido no Esquema 1,
eudesmanos
Esquema 1
1 H 9
eremofilanos
13
11 "'-
12 • 8
9 11
H 12 bacanos
13
• Para efeito de melhor visualização e didatismo, optamos por adotar a representação da direita na Figura 1
para a baquenolida-A, para todas as sínteses.racêmicas, bem como para os nossos resultados apresentados ao
longo dessa tese . Para as sínteses assimétricas, mostramos a representação que condiz com a configuração
absoluta da baquenolida-A, ou seja, a da esquerda na Figura 1.

Introdução
Foram isolados, até o momento, aproximadamente cinquenta bacanos, tendo sido
a maioria em plantas do Japão e Taiwan. Também foram encontrados bacanos em
plantas da China, República Tcheca, Suíça, França, Irã, Noruega, Escócia e Espanha, e
em ortocorais do Oceano Índico. 13
A relação eis entre as meti las, bem como a junção eis entre os anéis de cinco e
seis membros, é uma característica estereoquímica marcante desta classe de compostos,
além do fato de que a configuração relativa do centro quaternário do anel de cinco
membros é aquela em que o grupo carboxílico está em eis com as meti las - só existe um
bacano com a estereoquímica oposta: o palmosolídeo-C, Outros exemplos de bacanos,
com diferentes oxidações, estão mostrados na Figura 2.
~~~ 6-{y
O
6ct)"olA "H O
OAc OH 0f(~
O
Palmosolideo-C Homoginolídeo-B Acetóxi-fuquinanolida Baquenolida-C
Figura 2
Até o momento foram publicadas quatro sínteses totais racêmicas,14-19 uma
biomimética,2° duas formais21,22 e uma total assimétrica 23 da baquenolida-A (1), sendo a
primeira em 1973 e a última em 2004. Recentemente, foram publicados dois artigos
versando sobre a síntese dos bacanos: o primeiro, um artigo de revisão escrito por Silva
Jr. 13 e o outro um artigo completo sobre as sínteses dos bacanos efetuadas no grupo de
Greene. 24
Descreveremos, detalhada mente, nos itens que se seguem, as referidas sínteses
da baquenolida-A em ordem cronológica. Mostraremos, também, um pouco da química
de tálio(IlI) que vem sendo desenvolvida em nosso grupo de pesquisa, uma vez que
utilizamos uma reação de contração de anel, mediada por estes sais, nos estudos
visando à síntese de bacanos.
2

Introdução
1.2.Sínteses da Baquenolida-A
1.2.1.síntese de Evans
A primeira síntese de 1 (Esquema 2), concluída cinco anos após o seu isolamento,
foi efetuada por Evans e coI./4,15 numa seqüência de treze etapas. Os autores partiram
da 2,3-dimetilciclo-hexanona e, após quatro etapas - rota esta anteriormente descrita
por Piers25 -, obtiveram acetona 3, na forma de uma mistura diastereoisomérica 4: 1.
Após degradação oxidativa, separação das dicetonas isoméricas 4 e 5, condensação
aldólica e hidrogenação, a hidrindanona 6, material de partida chave para a síntese de 1,
foi obtida.
A etapa chave da síntese de Evans consiste de um rearranjo [2,3]-sigmatrópico
mediado por hidreto de sódio do intermediário 8 (sintetizado em três etapas), gerando o
centro quaternário - que dará origem ao anel espirolactônico - em rendimento razoável e
total estereosseletividade, o que indica que o rearranjo deve ocorrer através da face
convexa da molécula. O mecanismo de tal transformação está mostrado no Esquema 3.
10 foi convertido em 1 via oxidação alílica, mediada por ácido selenioso, seguida de
lactonização in situ do álcool formado, como mostrado no Esquema 2 .
Os pontos fracos da síntese de Evans são: a baixa estereosseletividade (4:1) no
estabelecimento da relação eis entre as metilas, na etapa de alquilação da cetona 2 -
embora tenha sido possível a separação posterior das dicetonas isoméricas 4 e 5; o
baixo rendimento na última etapa sintética; e o grande número de etapas envolvidas. O
que há de mais elegante nesta síntese é, de fato, o rearranjo [2,3]-sigmatrópico que
ocorre com total estereosseletividade, embora em rendimento moderado.
3

~ NaOMe.<j>H .. Ur HC02Me O O
Esquema 2
nC4HgSH .. Ç( CHSnC4Hg
2
Introdução
/C .. W KOtSu
CHSnC4Hg
(84%)
KOH. Ctr OS04. Na104.. ~ + ~ tSuOH. H20 U o ~ l~o ~
tSuOK
3 (76%)
4:1 (cis :trans)
Cb=o H2/Pd-C.
(63%)
Cb=O H
6 (93%)
4
~Li
5
(93%. 1 :4)
~ 'L PBr,. ~ /
l+XOH ~ H
NaH
7 Ct0 NaH
~~SMe lVr
(60%) CP<; t:i9Q.... -"/. SMe
H S 8 (75%) A
(2 etapas) MeS ~N-NHTs
H,SeO,. ~ H 11
1 (39%)
9 (62%)
Esquema 3
HgCI2 li H
10 (70%)
O ~S-NHNHCSMe
7
Jb MeSyN-NHTS Jb ~IN=NTS -rb t1SMe
LJ ~S NaH. LJ ~S) .> lJ ;L VJ=NTs -iX'SMe 8 9
4

Introdução
1.2.2.Síntese Biomimética de Hayashi
Ainda em 1973, Hayashi e col. 20 publicaram uma síntese biomimética parcial de 1
(Esquema 4), numa seqüência de quatro etapas partindo da enona natural fuquinona,
isolada da Caca/ia hastata. Deve-se salientar que a biossíntese da baquenolida-A, bem
como dos bacanos em geral, não foi determindada; entretanto, os autores acreditam que
a fuquinona é o precursor biossintético de 1.
Assim, "imitando" a natureza, a fuquinona foi epoxidada em rendimento razoável;
o epóxido 11 foi submetido à reação de contração de anel, que ocorre através de
rearranjo do tipo Favorskii, fornecendo uma mistura de três produtos, em que 13 é
proveniente de 12; o álcool terciário 14 foi desidratado na presença de cloreto de tionila
levando à olefina 15, que foi oxidada com dióxido de selênio formando uma mistura do
produto natural desejado, a baquenolida-A, e do aldeído 16. Este pôde ser convertido em
1 através de redução com boro-hidreto de sódio, em 47% de rendimento - melhorando
assim o rendimento global da transformação -, como mostrado no Esquema 4.
Vale a pena ressaltar que Hayashi e col. 20 separaram os dois epóxidos
diastereoisoméricos 11 e efetuaram, em separado, o rearranjo de Favorskii; ambos os
isômeros levaram à mesma proporção de produtos (com a mesma configuração
absoluta). Os autores não explicaram a distribuição dos produtos formados e, tampouco,
a estereosseletividade da referida transformação.
O maior problema da síntese de Hayashi é a baixa regiosseletividade na etapa de
contração de anel via rearranjo de Favorskii, em que o produto 14 desejado é obtido
como componente minoritário da mistura. Outro ponto fraco é o baixo rendimento da
etapa de lactonização envolvendo dióxido de selênio como oxidante.
5

Introdução
Esquema 4
NaOH ~ H
?=ó O H
30% H20 2 •
Fuquinona 11 (69%)
jNaoH MeOH
+ \ ~ Me02C>CÓ M~ + --.,.r'
2 HÓ \
H01"'~ Me02W
12 (42%)
Me02C, ~ OHC~~ + #
16 (57%) 1 (33%)
NaBH4 j (47%)
1.2.3.Síntese Formal de Silva
13 (16%)
• Se02 AcOH
H
14 (42%)
j SOCI2
Me02C., ~ ~~ H
15 (89%)
Em 1984, Silva descreveu, em sua tese de doutorad021 orientada por Petragnani,
a síntese formal de 1, como mostrado no Esquema 5, em que a etapa chave consistiu de
uma reação de contração de anel via rearranjo fotoquímico de Wolff. Assim como
Evans,14,15 os autores partiram da 2,3-dimetilciclo-hexanona e, após 4 etapas - anelação
de Robinson, bloqueamento da posição a-carbonila e hidrogenação catalítica - a decalona
17 foi obtida e transformada na a-diazocetona 18 correspondente através de tratamento
com nitreto de p-toluenossulfonila. Sem purificação prévia, 18 foi submetida à irradiação
e forneceu o produto de contração de anel 19, em 75% de rendimento, cujo enolato
formado na presença de LDA foi adicionado ao formaldeído. Após adição de metil lítio, o
6

Introdução
dial 20 foi obtido e o álcool primário foi oxidado e esterificado, levando ao hidróxi-ester
14. Desidratação da hidroxila terciária com cloreto de tionila deu origem à olefina 15
que, segundo o procedimento de Hayashi20 poderia ser convertida em duas etapas na
baquenolida-A.
Esquema 5
eX O (ti H
1)~ (ti H H2S04 cu NaOMe h-
H2' py
2) NaOMe ~ HC02Et ~ 10% Pd/C O O H O
(30%) (79%) 17 (89%)
p-TsN:l .. fuN2 hv CP- 1) LDA, HCHO (pçoH
Et3N ~ C02Me 2) MeLi .. "'lÇ O H OH H H
18 19 (75%, 2 etapas) 20 (64%)
1)Janes d=xC02Me SOCI,. (PC02Me Hayashl . Cpp .. 2) CH2N2
H il H roH
14 (44%) 15 (92%) (±)-1
O maior problema da síntese de Silva talvez seja a seqüência que leva de 19 a
15, durante a definição do centro quaternário em C-7, em que cinco etapas sintéticas
foram gastas. Não podemos deixar de mencionar também o baixo rendimento na
primeira etapa da seqüência sintética, a anelação de Robinson.
1.2.4.Síntese de Greene
Em 1985, Greene e col. 16 publicaram a síntese racêmica de 1 - primeiro bacano
de uma série que foi sintetizado por este grupo - num trabalho em colaboração com
Timathy J. Brocksom, da Universidade Federal de São Carlos-SP, em seis etapas. Já em
1988,23 este mesmo grupo publicou a síntese assimétrica de 1, numa rota idêntica porém
7

Introdução
partindo-se do (S)-l,6-dimetil-l-ciclo-hexeno, cuja preparação custou cinco etapas
sintéticas. Como as duas rotas só diferem na preparação do material de partida,
mostraremos exclusivamente a rota assimétrica.
A 2-metil-2-ciclo-hexenona foi assimetricamente reduzida ao álcool
correspondente que foi transformado na respectiva carbamida. Esta, através de
acoplamento com o dimetilcuprato de Guillman, com total inversão de configuração,
levou ao (S)-l,6-dimetil-l-ciclo-hexeno (21) em ótimo rendimento. A reação de
cicloadição entre 21 e dicloroceteno, gerado in situ na presença de zinco, levou ao biciclo
22 em ótimo rendimento. Em três etapas, efetuadas "one-pot", 22 foi clivado
oxidativamente e posteriormente transformado no respectivo diiodeto 23 que, então, foi
submetido à reação de dupla adição com o intermediário 24, levando a 2S como uma
mistura diastereosomérica na proporção de 3: 1. A mistura de 2S foi tratada com HF e
levou a (+)-1, juntamente com o seu epímero em C-7, numa razão de 3:1, conforme
mostrado no Esquema 6.
Esquema 6
~ o
1)SOCI2 ~ 2) 2,4,6-colidina' U
1) LiAIH4 (-)-N-metilefedrina
• 2-(etilamino )piridina
OCONHPh ~ U Me2CuLi. U 2) O=C=N-Ph
(49%,2 etapas) (66%, 2 etapas) 21 (90%)
m 1) Buli
CCI3COCI .. CI 2) AC20 .. Zn, POCI3 3) RU02' NalO4
O H "one-pot" 22 (89%)
Me02cq) TBSO\( H
LiHMDS .. + ----.!:iL ~C02Me
OTBS TBSO 24
I I
H02C"r) 1) LiAIH4
HO C" 2) TMSI
2 H
# (+)-1
~ H +
3:1 (59%)
.. ./I'r) 1'''- '.
k, ...... ",. H 23
rendimento não mencionado
8

Introdução
o maior problema da síntese de Greene é a baixa estereosseletividade (3: 1) na
etapa de formação do centro quaternário do anel espirolactônico; entretanto, a
abordagem é bastante convergente e insere todos os carbonos que faltavam de uma só
vez.
1.2.S.Síntese de Back
Outra abordagem muito elegante e convergente visando à síntese de 1, que
também lançou mão de uma reação de cicloadição, foi recentemente publicada por Back
e Payne. 17,18 A etapa chave da síntese consistiu de uma reação de Diels-Alder
intramolecular de um trieno altamente funcionalizado, levando à construção do esqueleto
cis-hidrindânico numa única etapa.
Desta maneira, três fragmentos (26, 27 e 28) foram preparados, através de
transformações triviais, e foram acoplados, levando ao trieno 29, que sofreu reação
térmica de Diels-Alder, levando ao aduto 30 como uma mistura de isômeros em
rendimento moderado. A hidrogenação catalítica de 30 não só reduziu a dupla ligação
remanescente, mas também desprotegeu o grupamento benzi la , levando a um álcool
intermediário que, após tratamento ácido, prontamente sofreu reação de lactonização,
em excelente rendimento. Após olefinação de Wittig, a baquenolida-A foi obtida como
componente majoritário de uma mistura de quatro isômeros, como mostrado no
Esquema 7.
Esta síntese, de modo geral, apresenta bons rendimentos para a maioria das
etapas sintéticas. O único ponto fraco que deveria ser salientado é a baixa
diastereosseletividade da etapa chave - a reação de Diels-Alder - em que há formação
dos quatro possíveis diastereoisômeros, além de um rendimento apenas moderado.
9

Introdução
I 1) CH2H2 •
~ 2) LiAIH4 H02C 3)PBr3 Br~
26 (50%)
o O
Esquema 7
o 2) PBr3 1) ipr2CONLi
O
.. ~ ~
o
~ NaH E~ NaH,26. ~ NaH,27
CI BnOH
BnO BnO f BnO
O 28 (83%)0 O ( 85%) 29 (92%)
190°C, PhMe • BHT ~ OEt
:::::,... H O OBn
1) H2' Pd/C .. 2) HCI6N ~ Ph3P=CH2 ..
30 (54%) (93%)
~ + + ~+ H J 1
(62%,54 19 16 11 )
1.2.6.Síntese Formal de Kozmin
Recentemente, Reddye Kozmin22 publicaram uma abordagem muito interessante,
baseada numa reação de metátese de um enino, para a síntese de eremofilanos. Nesta
publicação, os autores efetuaram a síntese da fuquinona, precursor biossintético da
baquenolida-A, como mostrado por Hayashi/o podendo então ser considerada como uma
síntese formal de 1.
Na primeira etapa - que consiste da reação de Diels-Alder, catalisada por dicloreto
de metilalumínio, entre o aldeído tíglico e o dieno 31 - três dos quatro centros
estereogênicos de 1 foram estabelecidos e o aduto 32 foi obtido em ótimo excesso
diastereoisomérico e excelente rendimento. Após homologação do aldeído através de
olefinação de Wittig, seguida por oxidação e esterificação, o éster 33 foi obtido em 74%
10

Introduçao
de rendimento. Este foi submetido à reação de hidrogenação - em que ocorreu redução
da dupla ligação e desproteção do álcool benzílico -, oxidação de Swern e olefinação de
Wittig levando, por fim, à olefina 34 em ótimo rendimento. Esta foi convertida no enino
35 que, por sua vez, foi tratado com catalisador de Grubbs de segunda geração - para
promover a referida reação de metátese - e ácido fluorídrico para efetuar a desproteção
do éter enólico de silício intermediário 36, levando então à enona 37 em 85% de
rendimento. A enona 37, após quatro etapas - adição de metil lítio, hidroboração,
oxidação com reagente de Jones e desidratação do álcool terciário com cloreto de tionila
- pôde ser convertida na fuquinona em rendimento bastante satisfatório, como mostrado
no Esquema 8.
Esquema 8
~HO OBn
~ d \CHO MeAICI2. '
31
1) H2' Pd/C. 2) Swern
~ "'I/'-OBn
32 (86%) (92:8 cis:trans)
(j"\\'-.....C02Me Ph3P=CH2.
" ..... CHO 'I .....
1\
Mes,NyN'Mes CI/'Ru=..
CI ....... I Ph PCY3
1) Ph3P=CHOMe .. 2) Cr03' H2S04 3) CH2N2
~"\\'-.....C02Me U"'I~
34 (75%, 3 etapas)
~"\\'-.....C02Me U"'I/'-OBn 33 (74%, 3 etapas)
1) CH2Br2, LiTMP. 2) LiHMDS; nBuLi
TIPSOTf
HF ~,\\~ U:"I~OTIPS .. Ctr
PS
MeCN Ct1 35 (73%)
MeLi (tr0H 1)BH3 I --':'2,L) J=.;o----'nL-es--
3) SOCI2, PY H (85%)
H 36
etC H O
Fuquinona
(61%,3 etapas)
H 37 (85%)
A abordagem de Kozmin, embora elegante e atual - haja vista a utilização de uma
reação de "ring-closing metathesis" (ReM) entre um alcino e um alceno, em que não há
11

Introdução
perda de carbonos - conta com um número muito grande de etapas (leia-se quinze)
para chegar à fuquinona que, para ser convertida em 1, consumiria mais quatro etapas,
segundo a abordagem de Hayashi. 2o Outro fato que dever ser mencionado é que o dieno
31, utilizado na primeira etapa da síntese, não é comercial e são necessárias três etapas
para prepará-lo, apesar dos autores não mencionarem tal fato.
Embora a reação de metátese olefínica seja de vasta aplicação em química
sintética, assim como seus mecanismos sejam relativamente bem estabelecidos,
julgamos por bem ilustrar simplificadamente, no Esquema 9, o processo pelo qual ocorre
a reação de metátese do alcenino 35, levando ao éter enólico de silício 36.
Esquema 9
d , ,~,- d ,'",-, Ct=n0TIPS .' ~ "Ru". .' ~ Ea - ~ "" OTIPS "" OTIPS "Ru" [2,2)
35 l l,"Ru" H
1.2.7.Síntese de Reddy
CtfPS H 36
A síntese mais recente da baquenolida-A foi publicada em 2004 por Reddy,19
colaborador de Kozmin na síntese da fuquinona 22 (cf Esquema 8). A etapa chave
consistiu, também, de uma reação de Diels-Alder - aliás, muito parecida com a descrita
anteriormente por Kozmin 22 - entre o aldeído tíglico e a dienona 39, preparada em uma
única etapa, a partir do divinil carbinol 38, através de um rearranjo de Claisen. O aduto
40 sofreu condensação aldólica, levando à enona 41 que, por sua vez, foi hidrogenada
sobre óxido de platina dando origem à hidrindenona 42. Esta foi seletivamente
carboxilada, fornecendo 43, que sofreu olefinação de Wittig para instaurar o grupamento
metileno. Por fim, a olefina 15 foi convertida em 1 através de oxidação alílica mediada
por dióxido de selênio, seguida de redução com boroidreto de sódio, num procedimento
muito similar ao utilizado por Hayashif2° como mostrado no Esquema 10,
12

Introdução
Esquema 1.0
OMe O
OH
~ A
Hg(OAch 150°C
[~l ~ 85-90% 39 38
39
d,'CHO ~o r CHO MeAICI2., , lSQt:L '\ I ~ ",,~ ~
H II H 40 (48%) O 41
!:b.... Pt02
(p"l H
42 (95%)
)( C ~
1) HMDS, d:>tOMe Ph,P=CH,. d:>t d:>t TMSI lo
OMe 1 ) Se02 • O 2) MeU; o'r 2) NaBH4 o-r
CNC02Me H 7i H J H J O 43 (64%) 15 (64%) 1 (58%)
A síntese de Reddy conta com uma abordagem extremamente inteligente,
envolvendo um número curto de etapas. Entretanto, os rendimentos, de modo geral, são
moderados.
1.2.S.Síntese da (-)-7-Epi-baquenolida-A
Em 1998, numa tentativa de síntese assimétrica de 1, através de uma abordagem
de espiro-anelação radicalar, Srikrishna e co I. 26 efetuaram a síntese da (- )-7-epi-
baquenolida-A em doze etapas (Esquema 11 ). Para tal, os autores investiram na
preparação da hidrindanona (+)-6, já preparada anteriormente por Evans, porém numa
versão racêmica. Partindo-se da R-carvona, através de uma seqüência de adição de
Michaeljinterceptação do enolato intermediário com brometo de alila, oxidação de
Wacker - em que foi possível isolar o isômero 44 desejado em 70% de rendimento - e
rearranjo de Criegee seguido de tratamento com DSU, foi obtida a dicetona 45. Após três
etapas - hidrogenação catalítica, reação de condensação aldólica e, mais uma vez,
hidrogenação catalítica - a hidrindanona enantiomericamente pura (+)-6 foi obtida em
13

Introdução
rendimento global de 26%. Esta foi convertida em um éter enólico através de olefinação
de Wittig e, após tratamento com NBS na presença de álcool propargílico, o bromo acetal
46 foi obtido em ótimo rendimento. Através de uma ciclização radicalar do tipo 5-exo-
dig, mediada por hidreto de tributil estanho - gerando in situ pelo par cloreto de tributil
estanho e ciano boro-hidreto de sódio - o espirociclo foi construído e, após hidrólise do
acetal 47 com posterior oxidação do lactol 48, mediada por clorocromato de piridínio
(PCC) suportado em sílica, a (- )-7-epi-baquenolida-A foi obtida.
Esquema 11
&0 MeLi,Cul; ~ HMPT, • PdCI2
~" .. ~Br I" O CuCI, O2
(89%)
~(W~rCtY (13%) 44 (70%)
~ (R)-carvona
-=-O.>L-iO..:...2~' --:M::-:eO::-:H:-:---= __ • ~ O' ~ p-N02-C6H4COCI, Py ,·UI"'\ ~ ~",·UI"'\O ~ O
AcO' O I ~CtY
45 (55%, 2 etapas)
d:>= H d=>= 1) Ph,P=CHOMe. 1) H2' Pd/C • O ~ O 2) NBS, =---\, 2) KOH, MeOH ;:,-.... Pd/C OH ~) H
(80%) (+)-6 (95%)
nBu3SnCI, NaCNBH3 AIBN, IBuOH •
HCI3N ..
47 (90%) 48
46 (86%)
PCC .. Si02
(59%,2 etapas) (-)-7 -epi-baquenolida-A
Embora este protocolo de espirociclização radicalar seja bastante eficiente, os
autores desconsideraram os efeitos estéricos envolvidos em sistemas bicíclicos de junção
eis, em que, assim como na síntese de Evans,14,15 a formação da ligação quaternária do
espirociclo ocorre pela face convexa (menos impedida) da molécula, como mostrado no
Esquema 12.
14

Introdução
Esquema 12
ft~ M~ O - trJ· tBUOH·W 47
Afora os aspectos estereoquímicos negativos da síntese de Srikrishna, devemos
mencionar a perda de três átomos de carbono, que ocorre através do rearranjo de
Criegee, durante a preparação da hidrindanona (+)-6; porém, esta isopropenila que é
perdida é a responsável pela diastereosseletividade na etapa de adição de Michael. À
parte isso, esta síntese, embora longa, conta com bons rendimentos ao longo da maioria
das etapas.
Os mesmos autores27 empregaram esta metodologia na síntese da nor-
baquenolida-A (49), que foi realizada em doze etapas, como mostrado no Esquema 13.
49 foi obtida, juntamente com seu isômero em C-7 (49'), numa proporção de 5: 1.
Esquema 13
1) MeC(OEth cjb:0 EtC02H cat.
2) NaOH/MeOH· '~ OH v LiAlH4 • Y 1) (COCI)~ 2) CH2N2
O (95%) OH
crt:2
CUS04 ~ lâmpada-W· ~O
(48%, 3 etapas)
Ph3P=CHOMe. CÍ>=1 NBS
H OMe ~ (96%) OH
nSu3SnCI, NaCSNHJ. AISN,lSuOH
MeO @ Jones.
(90%)
(60%)
Li, NH,. dJo H
(80%)
@.o H Bc)
( 93%) !J;
cpp + cpp 49 49'
(95%, 5 : 1)
15

Introdução
1.3.Reações Mediadas por Sais de Tálio(III)
1.3.1.Introdução
"( ... ) one thing a/ways happens sooner or /ater:
the hair fal/s out. Thallium used to be used for
depi/ation at one timeparticu/ar/y for chi/dren with
ringworm. Then it was found to be dangerous. But it's
occasional/y given internal/y, but with very carefu/
dosage going by the weight of the patient. It's mainly
used nowadays for rats, I believe. It's tasteless,
so/uble, and easy to buy. There's only one thing:
poisoning mustn 't be suspected. "
Pale Horse - Agatha Christie
Nosso grupo de pesquisa vem se dedicando já há quase duas décadas à aplicação,
em síntese orgânica, de sais de tálio(III) - especificamente trinitrato e triacetato (TIN e
TIA, respectivamente) -, versáteis reagentes capazes de promover uma vasta gama de
rearranjos oXidativos, conforme mostrado em diversos artigos de revisão. 28-33
Limitar-nas-emas a uma curta discussão e revisão a respeito da química do tálio,
uma vez que recentemente já publicamos dois artigos de revisã028,29 sobre o assunto, e
também pelo fato deste não ser o foco de estudo dessa tese. Entretanto, julgamos ser
conveniente e oportuno mostrar algumas das reações de contração de anel - reação da
qual fizemos uso em nossos estudos visando à síntese de bacanas - e de ciclização,
mediadas por sais de tálio(III), que foram desenvolvidas no nosso grupo de pesquisa.
Vale a pena ressaltar também que o desenvolvimento da química do tálio teve início na
década de 60, mas foi nos anos 70 que McKillop e Taylor - os quais informalmente
apelidamos de "os pais do tálio" - a exploraram amplamente.
Ao longo destes anos, foi efetuada uma série de estudos sistemáticos que
culminaram na síntese total de alguns produtos naturais . A seguir mostraremos alguns
exemplos ilustrativos.
16

Introdução
1.3.2.Reações de Ciclização
Tudo começou em meados da década de oitenta, com o estudo de ciclização de
álcoois monoterpênicos mediada por TIA, reações que foram feitas pelas mãos da
própria Helena Ferraz. 34 Outros substratos foram investigados35 e este protocolo foi
aplicado, com êxito, na síntese da lactona monoterpênica (+ )-mintlactona36 e do
feromônio trans-pitiol,37 como mostrado resumidamente no Esquema 14.
Esquema 14
Á -Iffi.. U OH
~ Isopulegol
YlOH
Sulcatol
(92%)
TTA • ouTTN
1) SOCI2•
2) PCC
O Mintlactona
,,"Ç)yOH trans-Pitiol
(71%)
Deve-se mencionar que se trata, formalmente, de uma ciclização eletrofílica,
porém, diferentemente de outros eletrófilos como, por exemplo, iodo, bromo, selênio,
mercúrio e telúrio, o tálio não se incorpora ao produto final.
Recentemente, foi estudada a reação de 3-alquenóis derivados de ciclo-
hexanonas. 38 Os produtos de ciclização correspondentes se mostraram excelentes
precursores de lactonas de anel médio, através de clivagem oxidativa mediada por
rutênio, como mostrado no Esquema 15. 39
Esquema 15
:yCnH TTN COC!H • ~"" O
(69%)
O
RuCI3.nH20. A Nal04 O
O (69%)
17

Introdução
Ácidos carboxílicos insaturados também são passíveis de sofrer reação de
ciclização, levando a 8-hidroxi-y-lactonas, na presença de sais de tálio(III), porém em
rendimentos moderados e baixa diastereosseletividade.4o No Esquema 16 está mostrado
um exemplo ilustrativo.
Esquema 16
~C02H TTN. ~OH + ~OH O O O O
(57%.1:1)
1.3.3.Reações de Contração de Anel
Reações de contração de anel também foram alvo de estudo em nosso grupo de
pesquisa, uma vez que anéis de cinco membros fazem parte de uma infinidade de
produtos naturais e sintéticos de pronunciada atividade biológica. Diferentes protocolos
podem ser empregados para obter tal unidade estrutural, e as reações de contração de
anel são, sem sombra de dúvidas, uma excelente abordagem, como revisado
recentemente por Silva Jr. 41
O primeiro estudo consistiu na preparação de ácidos ciclo-pentanocarboxílicos a
partir de ciclo-hexanonas, em que os aspectos mecanísticos foram amplamente
discutidos com base na estereoquímica dos produtos formados. De modo geral, a referida
transformação foi alcançada em excelentes rendimentos, contanto que não houvesse
grupos alquila na posição a-carbonila. 42
Em relação a sistemas bicíclicos, foi estudada a reação de contração de anel de
trans e de cis-decalonas. 43 No primeiro caso, trans-hidrindanos44 foram obtidos em
excelentes rendimentos e total diastereosseletividade, como exemplificado no Esquema
17.
18

Esquema 17
fi TIN, CH2CI2~ t.a., 24h : O
H cb"C0
2H
H (93%)
Introdução
Porém, para as cis-decalonas os resultados não foram tão satisfatórios: os
produtos de contração de anel foram obtidos com baixa diastereo- e regiosseletividade,
apesar do bom rendimento da transformação. 43 Não abordaremos, por enquanto,
aspectos mecanísticos das reações de contração de anel mediadas por tálio(III), mas
devemos lembrar que estas reações ocorrem, para as cetonas, via forma enólica. É,
portanto, em virtude da mobilidade do sistema cis-decalínico - que possibilita então duas
formas enólicas - que podemos explicar a baixa regiosseletividade da reação, como
mostrado no Esquema 18.
Esquema 18
fu . . fu + fu TTN, CH2Clz • // 24h, t.a.
H O H OH H OH
Cp + CtrC02H
H C02H H
(2: 1,95%)
Visando à obtenção de inda nos funcionalizados, três protocolos foram por nós
amplamente investigados: contração de anel de a-tetralonas, de l,2-di-hidronaftalenos e
de 3-alquenóis. As a-tetralonas não se mostraram bons candidatos a precursores de
indanos, uma vez que os produtos de contração de anel foram obtidos em rendimentos
apenas moderados, juntamente com produtos de a-oxidação (Esquema 19),45 num
protocolo em que se fez uso de TTN suportado em Montmorilonite K-10.
19
Introdução
Esquema 19
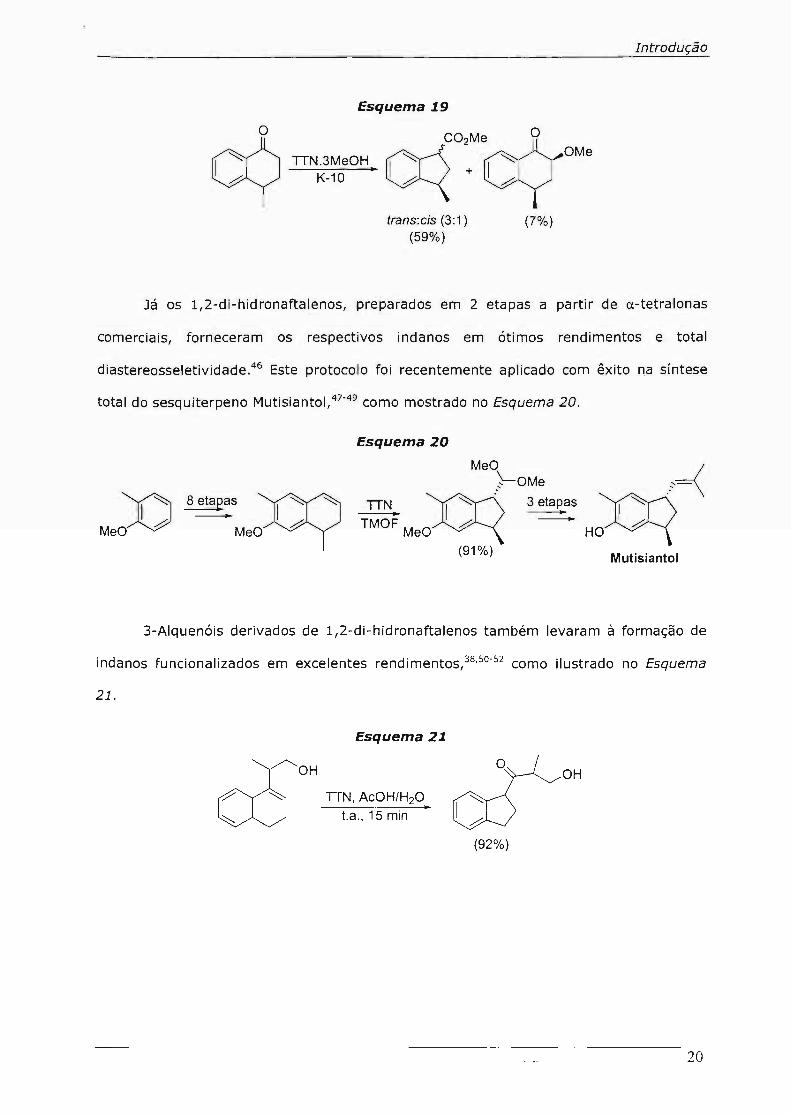
o cy C02Me O
TIN.3MeOH. 0=<-....::::: ~OMe K-10 // + I //
trans:cis (3:1) (7%) (59%)
Já os l,2-di-hidronaftalenos, preparados em 2 etapas a partir de a-tetralonas
comerciais, forneceram os respectivos indanos em ótimos rendimentos e total
diastereosseletividade.46 Este protocolo foi recentemente aplicado com êxito na síntese
total do sesquiterpeno Mutisiantol,47-49 como mostrado no Esquema 20.
~ Meo~
8~S lí:íI Meo~
Esquema 20
MeO )-OMe
~ YY\ ~as TMOFMeo~ -
(91%)
JíYÇ=< HO~
Mutisiantol
3-Alquenóis derivados de l,2-di-hidronaftalenos também levaram à formação de
indanos funcionalizados em excelentes rendimentos,38,so-s2 como ilustrado no Esquema
21.
Esquema 21
cO°H
-;;/ I -....::::: TIN, AcOH/H20. ~ t.a., 15 min
OH
(92%)
20

Objetivos
2.0bjetivos
o objetivo geral desta tese foi realizar estudos que propiciassem o
estabelecimento de uma abordagem eficaz para a síntese de bacanos. A metodologia
proposta inclui como etapa-chave uma reação de contração de anel mediada por
tálio(III), que forneceria a unidade cis-hidrindânica presente no esqueleto dos bacanos.
O alvo sintético escolhido foi a baquenolida-A - membro mais simples da família
dos bacanos; contudo, nos pareceu importante investigar antes a síntese de um
composto modelo, a nor-baquenolida-A. Portanto, o objetivo específico desta tese foi o
de efetuar estudos visando à síntese destas duas moléculas.
21

Planejamento Sintético
3.Planejamento Sintético
Quando propusemos a síntese da baquenolida-A nos focamos em três aspectos
estereoquímicos da molécula: a fusão eis entre os anéis de cinco e de seis membros, a
relação eis entre as meti las, e a estereoquímica relativa do centro quaternário em C-7,
em que o grupamento carboxílico está também em eis com as meti Ias.
Para alcançar o esqueleto cis-hidrindânico da baquenolida-A, imaginamos ser
possível lançar mão de uma reação de contração de anel como etapa chave. Como já
sabido, olefinas cíclicas são substratos passíveis de sofrer rearranjo oxidativo com sais de
tálio(III),28,29,33 levando a produtos de contração de anel, sendo possível assim obter o
esqueleto central da molécula.
Desta maneira, propusemos a seguinte retrossíntese (Esquema 22): o anel
espirolactônico de 1 poderia ser alcançado através de reação de lactonização, seguida de
olefinação, do ceto éster 43 - mas a seqüência poderia ser invertida; 43 seria fruto de
uma reação de carboxilação de uma metil cetona preparada a partir de 49 que, por sua
vez, seria proveniente de uma reação de contração de anel, mediada por TI3+, da olefina
50, oriunda de uma reação de Diels-Alder, seguida de algumas transformações de
grupos funcionais.
Esquema 22
Olefinação Cpp Lactonização >
1
~1-oMe l+J);
H cf 43
~ Contração Cb Diels-Alder CHO i > >
de Anel IGF
H H 50 51
IGF :>
Carboxilação
o
~
22

Planejamento Sintético
Antes de iniciar a síntese de 1, julgamos por bem testar a rota sintética proposta
para 1 em um composto modelo, a nor-baquenolida-A (49), que difere de 1 apenas pela
ausência da metila em C-15. Desta maneira, propusemos a rota descrita no Esquema 23
para a síntese de 49.
O
lf'+) rj::rCHO
H 54
cbtoMe
H li 56 O
Esquema 23
O
Ácido de Cb Wolff- cj) T1 3+ • • -Lewis Kishner
H H 52 53
1) MeU • ~
H
1) HMDS, TMSI • 2) MeU
Me02CCN 2) Jones
55
cbto
H 1~ 57 O
1) HMDS, TMSI •
2) OS04' NMO 3) W
Wittig .. cbb H J 49
Limitar-nos-emos, por enquanto, a esta proposta sintética para 49 e não
mostraremos, portanto, a rota que inicialmente imaginamos para a baquenolida-A. Ao
longo do capítulo intitulado "Resultados e Discussão", discutiremos mais detalhadamente
as alterações efetuadas nas referidas rotas sintéticas, bem como as rotas alternativas
que também foram investigadas.
Alea jacta est.
23

Resultados e Discussão
4. Resultados e Discussão
4.1.Estudos Visando à Síntese da nor-Baquenolida-A
4.1.1.Preparação do Material de Partida 53
A abordagem óbvia para obtenção de anéis de seis membros fundidos com junção
cis é a reação de Diels-Alder. Teoricamente, seria possível preparar a olefina S3 (cf
Esquema 23) através da reação entre 1-metil-1-ciclo-hexeno e butadieno; todavia, tanto
o dieno como o dienófilo são desativados e, portanto, tal protocolo deveria envolver
condições drásticas como, por exemplo, alta pressão e temperatura. Desta maneira,
decidimos por um dienófilo ativado (2-metilciclo-hexen-2-ona) e assim poderíamos
efetuar uma reação de Diels-Alder catalisada por áCido de Lewis. Porém, tivemos que
incluir mais uma etapa sintética para realizar a redução total da carbonila presente no
aduto obtido.
Muito embora a 2-metilciclo-hexen-2-ona seja comercial, ela foi preparada a partir
da 2-metilciclo-hexanona, através de uma seqüência de a-bromação termodinâmica
seguida de desidro-halogenação, em 68% de rendimento global (Esquema 24).53
o
cY NBS, CCI4
1'1, 13h
Esquema 24
o
Cf Li2C03, LiBr Br --~~~------
DMF, 130°C, 3h & (68%,2 etapas)
Apesar da reação entre a 2-metilciclo-hexen-2-ona e o butadieno - catalisada por
tricloreto de alumínio em 10% - ser descrita na literatura,54,55 encontramos dificuldades
pouco usuais na repetição deste protocolo. Porém, aumentando a quantidade do ácido de
Lewis para 50 mol% conseguimos obter o aduto S2 desejado, em escala de multigramas,
em 76% de rendimento, como mostrado no Esquema 25.
24

Resultados e Discussão
&+) Esquema 25
0,5 eq. AICI3 CH2CI2, t.a. 24h
hidroquinona
o
OlBLIOTECA ~NSTITUTO DE QUíMiCA Universidade d'2 S~;o PilU!O
Cb H
52 (76%)
Devido à presença de uma ligação dupla no aduto 52, descartamos a possibilidade
de efetuar a redução da carbonila através de um protocolo que envolvesse condições
ácidas como, por exemplo, a redução de Clemmensen e optamos então pela redução de
Wolff-Kishner.
A redução de Wolff-Kishner - que parece ser muito simples e que consiste do
tratamento do composto carbonílico com hidrazina, para formar a hidrazona, que é in
situ decomposta pela ação de base, levando ao hidrocarboneto correspondente -
ofereceu algumas dificuldades pouco usuais logo de início.
Segundo procedimento clássico, uma mistura do composto carbonílico, hidrazina
(10 equivalentes) e KOH (12 equivalente) é refluxada em etilenoglicol ou trietilenoglicol
até redução completa. Quando esta condição foi utilizada, o produto foi obtido em
baixíssimo rendimento (13%). Outra possibilidade é formar primeiro a hidrazona,
refluxar, em balão aberto, acima de 200°C para eliminar o excesso de hidrazina, para
então adicionar a base, que pode ser KOH ou Na2C03.56 Mais uma vez o rendimento foi
baixo e houve formação de subprodutos cuja caracterização estrutural não foi possível.
Tentativas de mudança da hidrazina para a tosil-hidrazina,57 o que implicou em
uma etapa a mais de redução com boro-hidreto de sódio, também não levaram a
resultados satisfatórios (Esquema 26).
O
Cb H 52
TsNHNH 2
EtOH, 6, 4h
Esquema 26
TsHN'N
.. Cb NaBH4 , AcOH. ~ 70°C,6h ~
H H 53 (15%, 2 etapas)
25

Resultados e Discussão
Paralelamente, tentamos efetuar um outro protocolo que não passasse pela
formação de hidrazonas. Nesta "abordagem, acetona 52 foi seletivamente reduzida ao
álcool 58 (comentaremos posteriormente - nos Esquemas 52 e 53 - os aspectos
estereoquímicos destas reduções para um substrato análogo a 52), e este foi tratado
com cianoboro-hidreto de sódio na presença brometo de zinco;s8 contudo, houve apenas
recuperação do material de partida (Esquema 27).
ÓJ H 52
NaBH4 , MeOH -78 °c, 1h ..
Esquema 27
OH
Cb ZnBr2' NaBH3CN Et20, t.a., 24h • recuperação de 58
H 58 (95%)
Por fim, decidimos retomar as condições clássicas e diminuir a quantidade dos
reagentes pela metade, além da seguinte modificação: primeiro formou-se a hidrazona,
depois adicionou-se KOH. Após otimização, foi possível obter o produto desejado em
80% de rendimento, como mostrado no Esquema 28.
O
Cb H 52
Esquema 28
1) 5 eq. NH2NH2.H20 trietilenoglicol 130°C, 1,5 h
2) 6 eq . KOH 130°C,4,5h
cb H
53 (80%)
Desta maneira, foi possível obter a olefina 53 numa seqüência de quatro etapas
em bons rendimentos.
26

Resultados e Discussão
4.1.2.Construção do Sistema cis-Hidrindânico
Passamos então ao estudo da reação de contração de anel 53, em condições já
bem estabelecidas por nosso grupo de pesquisa.46 Assim, quando a olefina 53 foi tratada
com TIN em trimetilortoformiato (TMOF), o produto de contração de anel 59 foi obtido.
Contudo, enfrentamos dificuldades na purificação do acetal formado, que se mostrou
muito lábil e espontaneamente hidrolisou para o aldeído 54 durante a coluna
cromatog ráfica.
Optamos então por efetuar a hidrólise do produto bruto da reação com ácido
trifluoroacético,59 para obter diretamente o aldeído 54. Mais uma vez, o produto formado
se mostrou pouco estável, impossibilitando uma caracterização espectroscópica
completa. Decidimos dar continuidade à seqüência sintética, com a reação de adição de
metil lítio à carbonila do aldeído, chegando, por fim, ao álcool 60, que é estável. Esta
seqüência de três etapas reacionais foi efetuada em 68% de rendimento global e está
descrita no Esquema 29.
cb H 53
Esquema 29
t.a. 3,5h
~re ~ ·OMe
TTN, TMOF •
H 59
d=>CHO MeLi, EbO .. O °C, 1,5 h ~
H
H H 54 60 (68%, 3 etapas)
TFA, CHCI~ t.a., 2 h
O álcool 60 foi obtido como uma mistura de quatro diastereoisômeros, o que já
era de se esperar, uma vez que nesta seqüência sintética 2 centros estereogênicos foram
gerados.
A etapa seguinte - a oxidação do álcool 60 à cetona 55 - foi efetuada utilizando-
se reagente de Jones,60 em 94% de rendimento (Esquema 30).
27

Esquema 30
~~r ~Jl ~ " Jones. ~ "
H 60
H 55 (94%)
Resultados e Discussão
Acetona 55 foi obtida como uma mistura de diastereoisômeros na proporção de
5: 1, porém não nos preocupamos em determinar a estereoquímica relativa. Na realidade,
esta estereoquímica é pouco relevante uma vez que na próxima etapa é que a
estereoquímica correta deste centro será definida.
Em escala de 10 mmol, estas quatro etapas foram efetuadas consecutivamente,
sem purificação do álcool 60, e foi possível isolar a metil cetona 55 em 65% de
rendimento global, de onde podemos inferir um rendimento médio por etapa de cerca de
80%.
o mecanismo proposto, e aceito, para a contração de olefinas, como mostrado em
três dimensões no Esquema 31 para 53, é o seguinte: i) o primeiro passo é o ataque
eletrofílico do tálio à ligação dupla, com perda de um dos ligantes, formando um
complexo do tipo 1t, o íon talônio; ii) depois ocorre a abertura trans-diaxial do íon talônio
por uma molécula de metanol, proveniente da hidrólise do TMOF; para ocorrer o
rearranjo oxidativo tipo Wagner-Meerwein, há que haver inversão da conformação, de
modo a alcançar a anti-periplanaridade necessária entre a ligação que vai migrar e a
ligação carbono-tálio; iii) ocorre a migração formal com expulsão e concomitante redução
do tálio(III) a tálio(I); iv) por fim, o íon oxônio é atacado por uma molécula de metanol,
levando ao produto final de contração na forma de um acetal.
Com base no mecanismo da reação, podemos sugerir que o isômero formado em
maior proporção seja o de configuração relativa eis entre a meti la em C-14 e o grupo
acetal. Para explicar a formação do isômero trans, temos que lançar mão de um
mecanismo iônico, em que haja a formação de um carbocátion formal, através da quebra
da ligação carbono-tálio, antes da migração ocorrer.
28

Resultados e Discussão
OMe
H+OMe OMe
TMOF
Esquema 31
H20
~
o )l + 2 MeOH
H OMe
ff: TIX3 H /'------=x- )#
X'TI'X
H 1\ -H+ - ff:iIX2
H = ~ I ÇX
53 X=ON02
\ HOMe
OMe TI /' OMe ~.
H
-TI X -X" ~~Me
H \
-H+ -- ~OMe H OMe
~OMe H OMe
59 HOMe
4.1.3.Construção do Centro Quaternário
A próxima etapa consiste da formação do centro quaternário, que pode ser feita
através da carboxilação de um enolato de lítio. Entretanto, há que se preparar o enolato
termodinâmico, o que não pode ser feito, portanto, com LDA. Tendo isso em mente, o
que parece óbvio, e foi utilizado na síntese da homoginolídeo-A por Mori,61 é preparar o
éter enólico de silício termodinânico, efetuar a troca silício-lítio com MeU, para depois
efetuar a etapa de carboxilação.
Na preparação de 61, utilizamos um procedimento caro porém deveras eficiente:
iodotrimetilsilano (TMSI) e hexametildisilazano (HMDS) como base. Não tivemos
problema algum, e 61 foi obtido em 95% de rendimento como uma mistura 1: 1 dos
isômeros geométricos E:Z, como mostrado no Esquema 32.
~ H
55
Esquema 32
TMSI. HMDS • CH 2CI 2 • O oCo 1 h ct>=tMS
H 61 (95%)
29

Resultados e Discussão
A etapa de carboxilação, também já descrita61 para um substrato análogo e que
supostamente não apresentaria complicações experimentais, ofereceu dificuldades pouco
usuais. Não foi possível efetuar a troca silício-lítio em 61 com MeU nas condições
descritas, isso é, utilizando-se 2 equivalentes do organolítio a -40°C. Contudo,
conseguimos efetuar a transformação desejada utilizando dois equivalentes de MeU a O
OCo Vale a pena ressaltar que o acompanhamento da reação foi efetuado através da
verificação da formação da cetona 55, que é o produto de hidrólise do enolato de lítio,
que é mais sensível à água do que o éter enólico de silício.
Superada esta barreira, a carboxilação foi feita pela adição in situ de
cianoformiato de meti la ao enolato de lítio intermediário, e o ceto éster 56 foi obtido em
apenas 20% rendimento. Porém, aumentando a quantidade de MeU para três
equivalentes e mantendo a temperatura a -40°C, foi possível obter o ceto éster em 47%
de rendimento, como mostrado no Esquema 33.
cj)=(TMS H 61
Esquema 33
1) MeU, THF -40 °C, 30 min
• 2) CNC02Me
THF, -40 °C, 1h cOt°Me
H 1/ O
56 (47%)
Após otimização, foi possível obter 56 em 54% de rendimento, partindo-se da
metil cetona 55. Vale a pena ressaltar que Mori,61 na síntese do homoginolídeo-A, obteve
rendimentos na faixa de 60% para a mesma etapa de carboxilação.
Acredita-se61 que a estereoquímica proposta para o centro quaternário é a
descrita acima, com base na conformação do enolato formado, em que o ataque deve
ocorrer pela face convexa e, portanto, menos impedida da molécula, como mostrado na
Figura 3.
30

H~r LJ ~)-OLi
Figura 3
4.1.4.Construção do Anel Espirolactônico
4.1.4.1.Primeira Abordagem
Resultados e Discussão
Para a formação do espirociclo da nor-baquenolida-A planejamos efetuar a 0.-
hidroxilação da metil cetona 56, seguida de lactonização, através de catálise ácida. 52
Para efetuar a a.-hidroxilação preparamos o éter enólico de silício 62 - de maneira
análoga à preparação de 61 (Esquema 32), uma vez que já estávamos habituados a este
procedimento e o tratamos com tetróxido de ósmio, em condições catalíticas,
utilizando N-óxido de morfolina (NMO) como co-oxidante. Após "work-up" e tratamento
com Hei, aceto lactona 57 foi obtida em 40% de rendimento, para as três etapas, após
purificação. Foram recuperados também cerca de 20% do ceto éster 56, proveniente da
hidrólise do éter enólico de silíco 62. No Esquema 34 está resumido este parágrafo.
cbtoMe
H 7i 56 O
O
ctX= OMe
"í/"OH H O
63
Esquema 34
TMSI, HMDS .. CH 2CI2, t.a., 3h
HCI cat. Et20, t.a., 1,5 h
ct>toMe
H r 62 OTMS
cbb H cf
57 (40%, 3 etapas)
OS04, H20, THF. NMO, t.a., 40 h
+ 56 (20%)
31

Resultados e Discussão
Em termos mecanísticos, a a-hidroxilação pode ser entendida da seguinte forma:
o éter enólico de silício 62 édi-hidroxilado, formando uma espécie de hemicetal sililado
64 que, após hidrólise, é convertido na a-hidróxi cetona 63 (Esquema 35).
r"h~oMe ~}~~
62 OTMS
0504 •
H20
Esquema 35
r"h~oMe 4J~OH
H HO OTMS 64
H+ --- cbtoMe
H l/"o H
63 O
Tentativas de outras condições reacionais para a oxidação com tetróxido de ósmio
foram pouco frutíferas, levando a resultados similares quando THF foi substituído por
uma mistura acetona/t-butanoI. 63
Embora o procedimento utilizado seja satisfatório, não é o ideal, pois a quantidade
de tetróxido de ósmio é grande (13 mol%), e ele é manuseado na forma de um sólido
que apresenta grande volatilidade e toxicidade. Em virtude disso decidimos tentar utilizar
uma fonte de ósmio "estabilizada", AD-mix-a, embora seja um reagente utilizado para di-
hid roxilação assi métrica. 64
Quando efetuamos a referida oxidação do éter enólico de silício 62 com AD-mix-a,
obtivemos, para a nossa surpresa, exclusivamente o produto de hidrólise de 62, isto é, o
ceto éster 56, como mostrado do Esquema 36.
r"h~oMe ~r
H OTMS 62
Esquema 36
AD-mix-a
o
~OMe H O
56 (90%)
Uma vez que já tínhamos em mãos aceto lactona 57 e só faltava uma etapa
sintética para chegar à nor-baquenolida-A, demos continuidade à síntese proposta, ou
seja, tentamos a olefinação de Wittig.
32

Resultados e Discussão
Utilizamos o procedimento descrito por Mori para a síntese do homoginolídeo-A,61
em que uma solução da ceto lactona foi tratada com quatro equivalentes de uma solução
da fosforana apropriada (Ph 3P=CH 2), gerada previamente em outro balão reacional.
Neste protocolo, não foi possível obter a nor-baquenolida-A: houve apenas recuperação
do material de partida e formação de produtos de decomposição (Esquema 37), cuja
determinação estrutural não foi possível.
Esquema 37
cPÇ0
. ° H;;-J 57 °
<P3P=CH2 (4 eq.) •
Et20, O °C, 6h
4.1.4.2.Segunda Abordagem
Produtos de decomposição
+ 57
Tendo em vista as dificuldades enfrentadas não só na etapa de a-hidroxilação do
ceto éster, bem como na olefinação da ceto lactona 57, imaginamos ser possível inverter
a ordem da seqüência sintética e efetuar a reação de Wittig na porção cetônica de 56,
para então efetuar a lactonização - que poderia ser feita através de oxidação alílica -
chegando, por fim, à nor-baquenolida-A.
A olefinação de Wittig de 56 também apresentou dificuldades pouco usuais, tendo
sido possível obter o produto desejado 65 em baixo rendimento (33%) mesmo utilizando
quatro equivalentes da fosforana. 19 Através de modificação do procedimento clássico,61
ou seja, realizando-se a adição da fosforana, gerada em um balão à parte, ao ceto éster
56, foi possível obter 65, após laboriosa otimização, em rendimento superior (60%) e
bastante satisfatório para reações de Wittig com fosforanas não estabilizadas. As
condições otimizadas para tal transformação estão mostradas no Esquema 38. Vale a
pena ressaltar que foram recuperados cerca de 24% do material de partida e, portanto,
se levarmos em consideração tal fato, podemos chegar a um rendimento de 79%.
33

Esquema 38
ePC°. " OMe ~3P=CH2 (4 eq.l
íi Et20, O °c, 8h H 56 °
Resultados e Discussão
cPtoMe
Hr 65
(60%, 79% brmp)
o aumento do tempo reacional, da temperatura, ou mesmo da quantidade de
fosforana, não levou a resultados melhores; muito pelo contrário, houve diminuição no
rendimento global e em alguns casos foi obtido o produto de descarboxilação, isto é, a
metil cetona 55.
Devemos lembrar que esta mesma abordagem foi utilizada em algumas sínteses
da baquenolida-A e tais complicações não foram evidenciadas. Para explicar a baixa
reatividade de 56, recorremos a um modelo molecular tridimensional comparativo. O
ceto éster 43, precursor da baquenolida-A, pode ser representado em duas
conformações (A e B, Esquema 39). Em ambas as conformações os substituintes do anel
de seis membros ocupam duas posições axiais e duas equatoriais e, portanto, A e B
seriam de estabilidade comparável. Entretanto, acreditamos que o equilíbrio está
deslocado em direção a B, uma vez que há alívio da tensão estérica entre a metil cetona
e o anel de seis membros. Portanto, a carbonila estaria mais disponível a qualquer tipo
de ataque nucleofílico.
Ao extrapolar este raciocínio para o ceto éster 56, vemos que a conformação mais
estável poderia muito bem ser aquela em que a metil cetona está voltada para a face
côncava da molécula, já que dois dos três grupos substituintes ocupam posição
equatorial (veja as conformações A' e B' no Esquema 39). Sendo assim, neste caso o
ataque da fosforana à carbonila ficaria mais dificultado, conferindo uma menor
reatividade ao sistema, como evidenciado experimentalmente. Se non ti: vero, ti: ben
trovato.
34

Resultados e Discussão
Esquema 39
ax
ax~ ~ 43 ~OMe
COMe
=----e~q ~ ~OMe
COMe
A
~~ 56 ~OMe
COMe A'
Favorecida
8 Favorecida
ax
~q~ ~OMe
COMe
8'
Tendo em mente os argumentos acima discutidos, julgamos desnecessário
investigar outras reações de olefinação como, por exemplo, Petasis ou Paterson - ou
mesmo protocolos mais recentes, utilizando catalisador de Wilkinson e TMSCHN2.65
Para efetuar a etapa de lactonização, pareceu-nos interessante empregar as
condições utilizadas por Hayashi,2o e recentemente otimizadas por Reddy/9 na síntese da
baquenolida-A. Neste protocolo, lançou-se mão de uma oxidação alílica mediada por
dióxido de selênio em ácido acético aquoso, seguida de tratamento com boro-hidreto de
sódio para reduzir o aldeído - proveniente de um processo de oxidação exaustiva -
formando, assim, um hidróxi éster que lactonizou espontaneamente no meio reacional
(Esquema 4).
Infelizmente não conseguimos adaptar as referidas condições experimentais para
a lactonização de 65. Recuperamos apenas o material de partida, parcialmente
decomposto, após 24 horas de aquecimento a 150 °c, como mostrado no Esquema 40.
O
~OMe 65
Esquema 40
8e02' AcOH. 150 oC,24h Material de Partida
35

Resultados e Discussão
Alternativamente, tentamos efetuar a lactonização através de um protocolo em
que não estivesse envolvida uma oxidação alílica. Para tal, preparamos o epóxido 66, em
excelente rendimento, pelo tratamento da olefina 6S com Oxone®,66 como mostrado no
Esquema 41.
cPtoMe
Hr 65
Esquema 41
Oxone®, acetona NaHC03, H20
t.a.,3h cPtoMe
H~ 66 (95%)
Tendo em mãos o epóxido 66, imaginamos ser possível realizar sua abertura,
seguida de lactonização in situ, através de um mecanismo iônico67 - que pode ser visto
no Esquema 42 - ou radicalar. 68
Esquema 42
o
ct>ÇOMe
6~ &:)
o
-cttoMe
0-
o
Lactonização. ~.b -OMe ~r
51
Na primeira tentativa, utilizamos como base não nucleofílica o amideto de lítio da
isopropilciclo-hexilamina (LICA) e apenas ocorreu a recuperação do material de partida,
como mostrado no Esquema 43.
cPtoMe
H~ 66
Esquema 43
LlCA, THF -78°C a O cC, 3h"
Recuperação do MP
36

Resultados e Discussão
Na segunda tentativa, ou seja, a abordagem radicalar, lançamos mão de um
protocolo bastante elegante que faz uso de um titanoceno. 68 Contudo, também não foi
possível aplicar estas condições para o epóxido 66 (Esquema 44).
Esquema 44
dXOMe
Hto CP2 TiCI2, Zn , THF . Argónio, t.a., 24h
Recuperação do MP
66
Em virtude desta falta de reatividade, resolvemos interromper os estudos visando
à síntese da nor-baquenolida-A, deixando esta última etapa em "standby"; todavia, não
podemos deixar de salientar que estes estudos serão continuados em nosso grupo de
pesquisa.
-..~
.) I

Resultados e Discussão
4.2.0utros Estudos Visando à Construção de cis-Hidrindanos
4.2.1.Introdução
Antes de discutir os esforços despendidos em relação à síntese da baquenolida-A,
mostraremos alguns estudos que fizemos visando à construção de cis-hidrindanos.
Imaginando ser possível encurtar a síntese da nor-baquenolida-A, pensamos em
estudar a reação de contração de anel de substratos que contivessem ligação dupla
trissubstituída. O único exemplo estudado em nosso grupo de pesquisa foi a reação de 4-
alquil-l,2-di-hidronaftalenos, sendo que estes substratos não se mostraram bons
candidatos ao rearranjo oxidativo com sais de tálio; simplesmente foram obtidos
produtos de adição, como exemplificado no Esquema 45. 46
có TIN, MeOH
t.a. , 30 min
Esquema 45
cODMe
I -.....::::::: ." OMe "..;::; +
(38%)
~Me ~. ,\OMe
(31 %)
Entretanto, existem precedentes na literatura32,69 de reação de contração de
olefinas cíclicas que contém duplas trissubstituídas com sais de tálio(III) (Esquema 46),
por isso decidimos investigar outros substratos que não contivessem um anel aromático
em sua estrutura.
Esquema 46
O
0-0 TTN cYu MeOH
(50%)
qy TIA ~ AcOH
(76%)
38

Resultados e Discussão
4.2.2.Preparação do Aduto 67
Preparamos então o aduto de Diels-Alder 67, através da cicloadição entre a 2-
metil-2-ciclo-hexenona e isopreno, catalisada por tricloreto de alumínio - de maneira
análoga à preparação de 52, material de partida da síntese da nor-baquenolida-A -
como descrito no Esquema 47. Não conseguimos reproduzir o rendimento de 88%
descrito pelos autores que, inclusive, não relataram o método de purificação utilizado; e
é de antemão sabido que há formação de material polimérico.
Esquema 47
(~J/ + Á t.a.24h
o m Alel3, benzeno •
H 67 (54%)
4.2.3.Reação da Olefina 68 com TTN
Cetonas cíclicas reagem com tálio(III) fornecendo produtos de contração de anel
ou de a_oxidação. 42,44,4s .7o Levando tal fato em consideração, a carbonila de 67 foi
protegida na forma clássica de seu derivado dioxolano 68, em 91% de rendimento
(Esquema 48), para então iniciar os primeiros testes com TTN.
Esquema 47
o
,~ ~
HOCH2CH20H. ~H 9h. o~ APTS, Dean-Slark, 2h
H H 67 68 (91 %)
o cetal 68 foi submetido então às condições clássicas de contração de olefinas
com tálio(III), isto é, TTN em MeOH. Entretanto, ocorreu desproteção da carbonila, com
39

Resultados e Discussão
formação de dois produtos de adição à dupla ligação (69 e 70, Esquema 49). Mais uma
vez foi possível comprovar a· resistência à contração de anel de ligações duplas
trissubstituídas em sistemas cíclicos.
Esquema 49
("'o
°m I TTN, MeOH~ t.a., 15 min
H
o ~OMe
~ON02+ 69 (25%) 68
~OMe ~OMe
H 70 (30%)
A presença do grupo nitrato em 69 pôde ser confirmada através das bandas
características de absorção forte no infravermelho em 1607 e 1300 cm- 1 mas a
estereoquímica relativa dos compostos 69 e 70 não foi determinada.
Como a desproteção da carbonila ocorreu espontaneamente - devemos lembrar
que há liberação de ácido nítrico no meio reacional durante a reação com TTN -
decidimos preparar outros substratos que não contêm a função carbonila, a partir de 67.
4.2.4.Reação do 4-Alquenol 71 com TTN
3-Alquenóis contendo ligação dupla t rissubstituída são passíveis de contração de
anelou reação de ciclização, dependendo da natureza do substrato, como já discutido na
introdução desta tese.
Como se pode observar no exemplo mostrado no Esquema 49, o material de
partida contém uma ligação dupla trissubstituída, e mesmo assim, só é observada a
formação do produto de contração de anel. Acreditamos, portanto, que o grupo
hidroxílico deve desempenhar um papel fundamental durante o rearranjo oxidativo, que
certamente deve ser o de coordenação intramolecular com o tálio. No Esquema 50 está
descrito, simplificada mente e levando em conta a estereoquímica da reação, o
mecanismo de contração com TTN. 5 1
40

Esquema 50
~~OH
TTN - X
~ TI', X
X= ON02 O'H HO
OH
Resultados e Discussão
(j ~Lr
HO~ O
Sendo assim, pensamos ser possível lançar mão desta complexação para alcançar
a contração do sistema cis-octalínico. Desta maneira, precisávamos primeiro efetuar a
redução estereosseletiva da carbonila de 67. A redução de 67 com boro-hidreto de sódio
em metanol, à temperatura ambiente, levou a uma mistura 5: 1 dos álcoois trans e eis
em 88% de rendimento. Quando a reação foi efetuada a -78°C, obteve-se uma mistura
12: 1 dos referidos álcoois 71 e 71' em 92% de rendimento, como mostrado no
Esquema 51.
fu H 67
Esquema 51
NaBH4 , Me0l:l -78°C, 1 h
OH OH
m+fu H 71
H 71 '
12 1 (92%)
Uma possível explicação para a estereosseletividade destas reações de redução é
proposta a seguir. Em primeiro lugar, devemos lembrar que estamos lidando com
sistemas cis-decalínicos, em que a mobilidade dos dois anéis é grande, pelo menos
quando comparado com o sistema trans-decalínico. Fazendo o modelo molecular dos dois
álcoois (Esquema 52), e levando em consideração as interações 1,3-diaxiais entre os
grupos substituintes, intuitivamente atribuímos ao álcool trans a maior estabilidade. Com
base nos resultados das reações efetuadas em diferentes temperaturas (t.a. e -78°C)
somos levados a acreditar que o álcool trans é não só o produto termodinâmico como
também o cinético.
41

Resultados e Discussão
Esquema 52
H OH
tmns.;iê r :/f H 1t eis: 71 'I OH 71'
'I H
1 eq, 3 ax 3 eq, 1 ax 2 eq, 2 ax 2 eq, 2 ax
Para a redução de ciclo-hexanonas, tanto o modelo de Felkin-Ahn, quanto o de
eram, falham na predição do produto favorecido; ambos os modelos indicam o álcool de
configuração relativa eis, enquanto experimentalmente se observa o trans como
majoritário, dependendo, é claro, da natureza do substrato. Posto isso, há que se levar
em consideração fatores conformacionais e não esquecer da angulação de
aproximadamente 110° em que ocorre o ataque do hidreto (trajetória de Bürgi-Dunitz).71
Vendo a molécula pelo ângulo da carbonila, temos que a conformação mais
estável é aquela em que a metila ocupa uma posição equatorial (Esquema 53), em que o
impedimento estérico deste grupo é pouco pronunciado. Desta maneira, o hidreto
atacará preferencialmente em axial, levando ao álcool equatorial - e mais estável
portanto trans.
~o Esquema 53
~o jieq
mais estável
H
NaBH4 • ~OH
71
Preparado o álcool 71 em ótima diastereosseletividade, efetuamos a sua reação
com TrN em TMOF e, para a nossa surpresa, obtivemos o éter cíclico 72 em 63% de
rendimento como único produto formado, conforme mostrado no Esquema 54,
42

Resultados e Discussão
Esquema 54
OH
m H
t.a., 30 min
~.,\OMe
~ H
TIN, TMOF ..
71 72 (63%)
Vista em 3 dimensões talvez seja mais fácil de entender, em termos mecanísticos,
esta reação de ciclização. Assim, em primeiro lugar ocorre o ataque nucleofílico da
ligação dupla ao TIN, formando o íon talônio I (uma espécie de complexo n).
Posteriormente, ocorre a abertura trans-diaxial de talônio - o que só pode ocorrer
através da conformação menos estável: aquela em que a hidroxila está em axial -
através do ataque intramolecular da hidroxila, levando ao aduto oxitaliado li, que sofre
solvólise, originando finalmente o produto de ciclização 72 (Esquema 55). Cabe ressaltar
que a estereoquímica do álcool é fundamental para que a ciclização ocorra, uma vez que
a hidroxila se encontra bem perto da ligação dupla. O isômero 71' (hidroxila em cis com
a metila angular) não atingiria a conformação adequada para a reação ocorrer.
Esquema 55
~O H TIX HO~ - H+ J?H 3. ( _ -X
-- X= ON02 /\",'7 (\+/ 71 /TI, /TI-...
X X X X
MeOH J?H -TI X • -HX
MeO
Meo~ 72
11
Num primeiro momento, este resultado pode até parecer surpreendente, mas há
precedentes na literatura de ciclização de 4-alquenóis com sais de mercúrio72 e de
tálio. 35,37,73 No Esquema 56 está mostrada uma reação de ciclização de um substrato
similar a 71 com acetato de mercúrio(II).72 Vale a pena ressaltar que estas reações de
oxidação com sais de mercúrio se procedem por um mecanismo análogo ao dos sais de
tálio(III).
43

Resultados e Discussão
Esquema 56
tb 1) HgO(Ac),. THFIH,O ~H9CI NaBH4 • ~ 2) NaCI • 0-· ----
H H
H
(69%,2 etapas)
1) HgO(Ac),. MeOHIH O ~ NaBH4 • ctb 2 I
2) NaCI • 0-· ----H HgCI H
(86%,2 etapas)
Apesar da natureza interessante do triciclo 72, não encontramos até o momento
nenhuma classe de compostos com o mesmo esqueleto, para que pudéssemos propor
uma possível aplicação sintética para tal reação.
4.2.5.Reação de Contração de Anel da Olefina 74 com TTN
Por fim, efetuamos a redução total da carbonila de 67 (Esquema 57) nas mesmas
condições de Wolff-Kishner utilizadas para o aduto 52, nos estudos visando à na síntese
da nor-baquenolida-A (Esquema 28).
Esquema 57
O m 1)5eq.NH,NH,H,O I trietilenoglicol •
130 °C, 1,5 h m H 2) 6 eq. KOH H
67 130°C,4,5h 73 (85%)
Realizamos a reação da olefina 73 com TTN e TTA em diversos solventes, a saber:
metanol, TMOF, diclorometano, ácido acético e ácido acético-água (1: 1). Para os três
primeiros solventes, houve recuperação do material de partida, mesmo após longos
tempos de reação (até 48 horas), parcialmente decomposto. Para os dois últimos, houve
formação de uma mistura complexa de produtos. Resolvemos, então, efetuar a reação
44

Resultados e Discussão
em metanol e adicionar apenas algumas gotas de ácido acético e, para nossa surpresa,
foi possível obter o produto de contração, embora em rendimento baixo, uma vez que
houve a formação de uma gama bastante grande de produtos minoritários. A metil
cetona 55 foi obtida na forma de uma mistura 1: 1 dos isômeros eis e trans, como
mostrado no Esquema 58.
Esquema 58
~ TTN,MeOH. ~ AcOHcat.
H t.a., 2h
73
(j:)Á H
55 (40%) cis:trans (1: 1)
Para efeitos sintéticos, esta ausência de diastereosseletividade não é relevante,
uma vez que na nossa proposta para a síntese de bacanos, a estereoquímica do centro
quaternário em C-7 é determinada a posteriori. Por outro lado, o rendimento da reação
não é satisfatório, embora tenha sido possível obter numa só etapa a metil cetona S5 -
deve-se lembrar que na síntese da nor-baquenolida-A são necessárias quatro etapas
para se chegar a S5 (Esquemas 29 e 30). De qualquer maneira, estes estudos devem ser
continuados em nosso grupo de pesquisa, esperando obter resultados mais conclusivos.
45

Resultados e Discussão
4.3.Estudos Visando à Síntese da Baquenolida-A
4.3.1.Introdução
Tendo em vista os estudos descritos no item 4.1 - isto é, as tentativas de síntese
da nor-baquenolida-A - imaginamos ser possível aplicar a mesma seqüência para a
síntese de 1, em que se empregaria também uma reação de contração de anel, mediada
por tálio(III), da olefina 50, como mostrado na rota sintética descrita no Esquema 59 (cf
Esquema 23 para melhor entendimento da rota sintética aqui proposta).
Esquema 59
Cb TIN, ~CHO 1) MeU. (tyl Selapas ~O - 'r 2) Jones -- H h H H H
51 50 42 1
o ponto crítico da preparação da olefina 51 é o estabelecimento da relação eis
entre as metilas em C-14 e C-15. Seria natural pensar que poderíamos preparar, numa
única etapa, um aduto de Diels-Alder que contivesse o sistema bibíclico e as duas meti las
em relação eis. Assim, teríamos de partir do 1,6-dimetilciclo-hexeno e butadieno, ou
seja, uma reação de Diels-Alder tipo ciclo-hexeno/butadieno, em que o dieno e o
dienófilo são desativados, existindo um grande "gap" de energia entre HOMO e LUMO.
Reações como estas só acontecem em fase gasosa e a altas pressões e temperaturas, e
não encontramos nenhuma descrição na literatura para a referida reação. Além do mais,
não teríamos controle algum sobre a relação entre as meti Ias.
Apenas a título de curiosidade, mostramos a seguir duas possíveis rotas sintéticas
que levariam inequivocamente a esta relação estereoquímica. Na primeira (Esquema 60),
valer-nos-íamos de uma adição conjugada de dimetilzinco74 ou dimetilcuprato de lítio75,76
à enona 75, prepara através da reação de cicloadição regiosseletiva de 74 com o dieno
de Danishefsky. Adição conjugada a 75 levaria à cetona 76, com relação correta entre as
46

Resultados e Discussão
meti las, como previ,sto por nós com base em modelo molecular, levando em consideração
o impedimento relativo entre as faces. Redução dos grupos éster e cetona aos álcoois
correspondentes, seguida de redução com zinco em ácido acético, levaria, por fim, à
olefina 51.
Esquema 60
OMe MeO C02Me C02Me
UC02Me '"'OTMS · m1-m I I +
Me2M~ C02Me TMSO H O H
75 R) 74 75
me m Cb O
Me2Zn M= Zn ou CuLi Ni(acach • I LiAIH4 • I Zn
ou Me2CuLi AcOH O H HO H H
76 51
Na segunda abordagem (Esquema 61), prevalecer-nos-íamos da reação de Diels
Alder, descrita por Kozmin,22 para a preparação de 32. Homologação de 32, através de
reação de Wittig seguida de hidrólise, levaria ao aldeído 77. Após hidrogenação catalítica
e oxidação seletiva, o di-aldeído 78 seria obtido e poderia ser facilmente transformado
no dieno 79, substrato adequado para reação de metátese, que daria origem então à
olefina 51.
Esquema 61
"=(HO OSn v-r d\CHO MeAICIz. .' 1) Ph3P=CHOMe
2) W • ~·"\"-..CHO ~""~OBn
H
1) H2' Pd/C. 2) Swern d'\\"-.. . CHO
",, / CHO
H 78
~ ""~OBn H 32
Ph3P=CH 2• ~ .. ,\~ ~"'/~
H 79
77
Me'.'ese. Cb H 51
47

Resultados e Discussão
Embora estas rotas sintéticas se mostrem interessantes e promissoras, elas são
um pouco longas e muito elabOradas, uma vez que se trata da preparação do material de
partida numa versão racêmica. Assim, decidimos investir em uma rota mais simples, que
será discutida e mostrada no próximo item.
De mais a mais, devemos comentar que durante nossos estudos visando à síntese
de bacanos, Reddy 19 publicou a síntese da baquenolida-A (Esquema 10) na qual a meti I
cetona 42 foi preparada; portanto, a síntese desta cetona, conforme a rota descrita no
Esquema 59, poderia ser caracterizada como uma síntese formal de 1.
4.3.2.Definição da Relação eis entre as Metilas da Baquenolida-A
4.3.2.1.Primeira Abordagem
Tendo sido possível a obtenção de álcool 71 - cuja hidroxila é trans em relação à
metila - em excelentes rendimento e diastereosseletividade, imaginamos efetuar
inversão do centro carbinólico, através de uma reação de acoplamento com um cuprato,
o que levaria ao estabelecimento da relação eis entre as meti las da baquenolida-A.
Assim, preparamos o tosilato 80, em rendimento bastante satisfatório, segundo
condições clássicas: cloreto de tosila e piridina como base77 (Esquema 62).
OH
fu H 71
Esquema 62
TsCl, py .. O °C, 24h
OTs
fu H
81 {62%}
Todas as tentativas de acoplamento do tosilato 80 com dimetilcuprato de lítio
foram infrutíferas (Esquema 63) e atribuímos esta falta de reatividade ao fato do centro
a ser invertido ser um carbono tipo neopentílico, portanto muito impedido. Poderíamos
48

Resultados e Discussão
tentar fazer o acoplamento com um reagente de Grignard, mas teríamos o mesmo efeito
estérico. O mais surpreendente foi o fato de não ter sido obtido nem mesmo o produto
de eliminação do grupo tosila.
Esquema 63
OTs m Me2CuLi Et
20, 0C, 3h- Material de Partida
H 80
Mesmo assim, imaginamos que em condições mais drásticas talvez fosse possível
efetuar a inversão do centro carbinólico. Deste modo, planejamos a reação do tosilato
com cianeto, que ocorre exclusivamente via mecanismo SN2. Através de transformações
de grupo funcional, chegaríamos à olefina de interesse 81 (Esquema 64).
OTs m KCN•
H 80
CN
fu H
Esquema 64
HO
1) NaOH -~'/',.I 2)LiAIH4 -~
H
1) MsCI • 2) LiB(EthH fu
H 81
Testamos a reação do tosilato 80 com cianeto de potássio e de sódio e, mesmo
após aquecimento em DMSO por 48 horas, este se mostrou resistente à reação de
substituição (Esquema 65).
Esquema 6S
OTs m KCNouNaCN Material de Partida
H 80
Em vista destes resultados, abandonamos esta abordagem e iniciamos uma outra,
que será discutida no item subseqüente.
49

Resultados e Discussão
4.3.2.2.Segunda Abordagem
Alternativamente, imaginamos ser possível adaptar um procedimento descrito por
Paquette78 para a definição da relação eis entre as meti Ias. Assim, a meti la seria colocada
de maneira indireta, envolvendo a homologação da carbonila através da seguinte
seqüência: reação de Wittig e hidrólise do enol éter 82 (ou 83), levando ao aldeído 84
(ou 85). 84 (ou 85) poderia ser submetido à reação de Wolff-Kishner, levando
diretamente à olefina 81 (ou 51). No Esquema 66 está descrita a seqüência sintética
proposta.
Esquema 66
o MeOJl,~ fu Ph3P=CHOCH3 , Lu H R H R
HCI -MeOH ~R Wolff-Kishner, fuR
R= Me (67) R= H (52)
82 83
H H 84 85
81 51
Efetuamos então a reação de Wittig da cetona 67 com cloreto de (metóximetil)-
trifenilfosfônio, utilizando hexametildissilazida de potássio (KHMDS) e tratamento com
Hei no próprio meio reacional, como descrito na literatura. 78 Para nossa surpresa, não
obtivemos o aldeído 84 e, sim, o derivado tricíclico 86 em 37% de rendimento, como
mostrado no Esquema 67.
Esquema 67
° + CI-m 1) Ph3PCH20CH3
I KH~DS, THF • -30 C a La., 17h
2) HCI 4N, MeOH t.a ., 24 h
H 67
OH di = ~/H H HO;L-~
86 (37%)
50

Resultados e Discussão
A formação de 87 pode ter se dado através do mecanismo mostrado no Esquema
68, em que a etapa de ciclização supostamente ocorre durante a hidrólise do éter enólico
intermediário 83.
Esquema 68
H I
Me«.~OH
fu fu-MeOH H H 87 87'
H~-86 ~
H ~ +
HO ..J
H 89
+ ~OH
fu H 88
III
6/H
~~ +
88
o primeiro passo do mecanismo descrito trata-se da hidratação da ligação dupla
do enol éter, dando origem ao hemi-acetal protonado 87, que pode eliminar tanto água
como metanol. No atual caso ocorreu a segunda possibilidade, valendo a pena ressaltar
que estas reações são todas de equilíbrio. Assim, há formação do aldeído protonado 88,
que sofre ataque nucleofílico da ligação dupla, gerando o carbocátion terciário 89, que
leva ao triciclo 86, através da perda de um próton.
Entretanto, quando a octalona 52, que não contém dupla ligação trissubstituída,
foi submetida às reações condicionais descritas acima/8 o aldeído 85 foi obtido como
uma mistura 5: 1 dos dois possíveis isômeros (Esquema 69), cujas configurações
relativas não foi possível determinar, embora acreditemos que o isômero majoritário seja
trans. Tal suspeita é fruto da formação do triciclo 86 - obtido através da hidrólise do éter
enólico 82, que contém a dupla ligação substituída -, uma vez que o rearranjo do oxônio
para um carbocátion terciário só seria possível se o grupo estiver em trans com a metila
e em axial.
51

tb H 52
+ cr Ph3PCH20CH3
KHMDS, THF -30°C a t.a., 19h
..
Esquema 69
Me~
H 83
Resultados e Discussão
O~H
HCI4N. MeOH. ~ t.a., 6h ~
H 85
(91%,5:1)
Como a reação apresentou baixa diastereosseletividade, e não foi possível a
determinação da estereoquímica relativa dos produtos formados, resolvemos abandonar
esta rota sintética.
4.3.3.0utra Possível Rota Sintética para a Baquenolida-A
4.3.3.1.Introdução
Tendo em vista as dificuldades de estabelecer a relação eis entre as meti las do
anel cis-octalínico que daria origem ao esqueleto central da baquenolida-A, propusemos
uma rota alternativa em que partiríamos da 2-octalona 90 e, em quatro etapas, teríamos
efetuado a síntese formal de 1, já que 42 é intermediário da síntese de Reddy (Esquema
10),19 como mostrado no Esquema 70.
Esquema 70
fu nt>.. (b"C02Me
° 90 91
1) NaOH 2) LiH;
MeLi 3) [H]
(p.l H 42
setapas(pt0 - y · H) Baquenolida-A
Antes de descrevermos as três abordagens que utilizamos para a preparação da
octalona 90, faremos uma breve descrição das rotas descritas na literatura até o
52

Resultados e Discussão
momento. Mostraremos, nos itens que se seguem, tanto as sínteses racêmicas como as
assimétricas de 90, uma vez que também tencionávamos uma abordagem assimétrica
para a baquenolida-A.
As sínteses racêmicas de 90
o protocolo óbvio para a preparação de 901 é a anelação Robinson direta entre a
2,3-dimetilciclo-hexanona e a metil vinil cetona (MVK), catalisada por base. Neste
procedimento/9-82 a base utilizada foi metóxido de sódio e os resultados não foram muito
satisfatórios, uma vez que 90 foi obtida em apenas 15% de rendimento e numa
proporção 3:2 para com o seu diastereoisômero trans 90', como ilustrado no Esquema
71.
Esquema 71
eX NaOMe .. fuo+fuo MVK
90 90' (15%.3:2)
Também num protocolo de anelação de Robinson, Zoretic e Golen83 efetuaram a
preparação de 90, em duas etapas, através de uma adição de Michael catalisada por
ácido, seguida de condensação aldólica, mediada por base, das dicetonas intermediárias.
Segundo os autores, foi obtida uma mistura 9: 1, em 33% de rendimento das octalonas
diastereoisoméricas 90 e 90', como mostrado no Esquema 72
Esquema 72
(( H,SO'. ~ MVK lÁo · .
O
NaOMe .
-
fuo+fuo 90 90'
(33%.9:1)
53
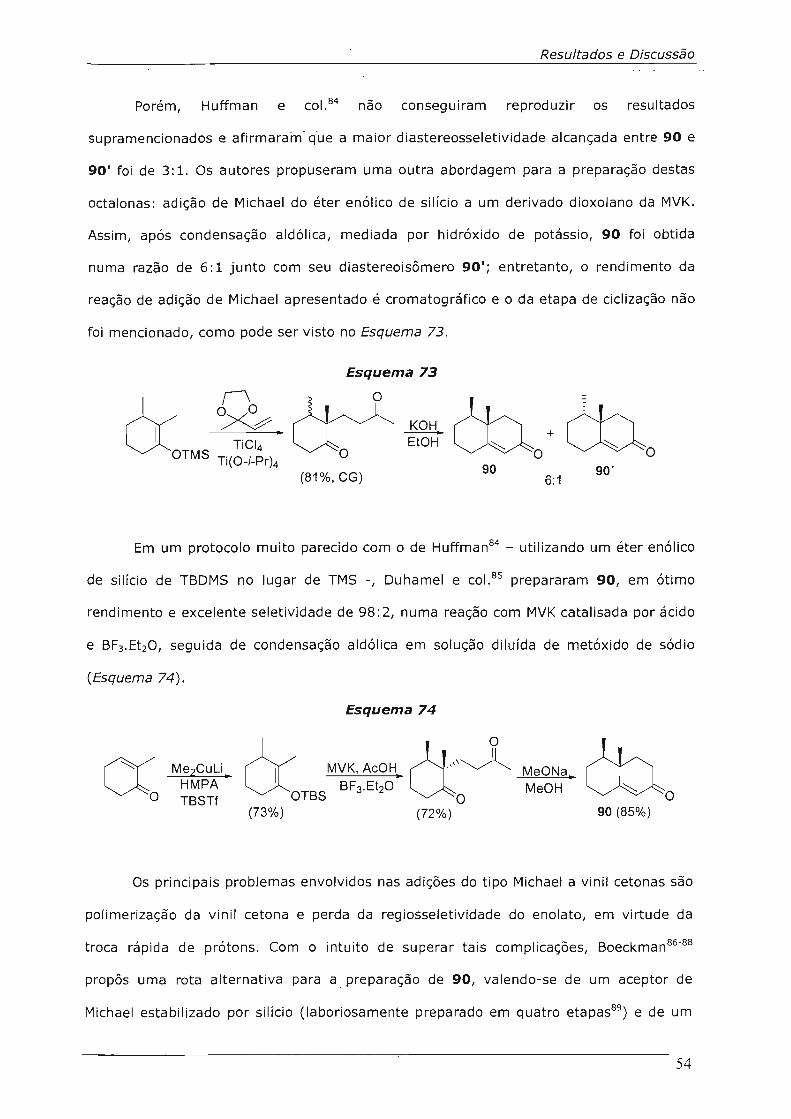
Resultados e Discussão
Porém, Huffman e col. 84 não conseguiram reproduzir os resultados
supramencionados e afirmaram- qúe a maior diastereosseletividade alcançada entre 90 e
90' foi de 3: 1. Os autores propuseram uma outra abordagem para a preparação destas
octalonas: adição de Michael do éter enólico de silício a um derivado dioxolano da MVK.
Assim, após condensação aldólica, mediada por hidróxido de potássio, 90 foi obtida
numa razão de 6: 1 junto com seu diastereoisômero 90'; entretanto, o rendimento da
reação de adição de Michael apresentado é cromatográfico e o da etapa de ciclização não
foi mencionado, como pode ser visto no Esquema 73.
Esquema 73
n
eX ~. I TiCI4 OTMS Ti(O-i-Pr)4 ~ O
(81%, CG)
KOH -EtOH Cho+mo 90 90'
6:1
Em um protocolo muito parecido com o de Huffman84 - utilizando um éter enólico
de silício de TBDMS no lugar de TMS - Duhamel e col. 85 prepararam 90, em ótimo
rendimento e excelente seletividade de 98:2, numa reação com MVK catalisada por ácido
e BF3 .Et20, seguida de condensação aldólica em solução diluída de metóxido de sódio
(Esquema 74).
ex Me2CuLi. HMPA TBSTf
Esquema 74
eX MVK, AcOH. I BF3.Et20
OTBS ct~
O (73%) (72%)
MeONa. MeOH Cho
90 (85%)
Os principais problemas envolvidos nas adições do tipo Michael a vinil cetonas são
polimerização da vinil cetona e perda da regiosseletividade do enolato, em virtude da
troca rápida de prótons. Com o intuito de superar tais complicações, Boeckman 86-88
propôs uma rota alternativa para a _ preparação de 90, valendo-se de um aceptor de
Michael estabilizado por silício (laboriosamente preparado em quatro etapas89) e de um
54

Resultados e Discussão
enolato de cobre preparado regiosseletivamente através da alquilação da 2-metil-2-ciclo-
hexenona (preparada em 2 etapas), como mostrado no Esquema 75.
Esquema 75
SiMe3
ex Me2CuLi. ÓC O OLiCuMe
1) ~O Me3Siu) --..!.---..... I 2) NH4CI, NH40H O
KOHaq,_ ~ MeOH ~O
OH 90 (43-57%)
Piers e col. 80 prepararam 90, diastereoisomericamente pura, através de uma rota
longa, entretanto com alta seletividade na etapa de definição da relação eis entre as
metilas, e envolvendo transformações com rendimentos médios muito superiores às
rotas anteriores, conforme descrito no Esquema 76.
Esquema 76
~ MeONa U o HC02Et ~ SH(CH2),CH 3•
~O p-TSA
~ t-BuOK, t-BuOH ~O CH 3CHBrC02Et"
/l"\ ~o C02EI
CHOH (90%)
KOH aq J-J.",\ HOCH2CH20H- U C02H
O (90%)
AC20 _ NaOAc
Recristalizacão. fuo 1) MeLi
2) KOH
(80%)
CHS(CH2hCH3
(87%) CHS(CH2hCH3
(86%)
Coo+ fuo (85%,9:1)
mo 90 (70%)
Por fim, Bonjoch e col. 90 prepararam 90 numa sequência de 8 etapas e análoga à
de Piers,80 porém partindo-se da 3-metilciclo-hexanona e utilizando uma enamina como
grupo bloqueador da posição a da carbonila, em rendimento total de 24%.
55

Resultados e Discussão
As sínteses assimétricas de 90
(+ )-90 é o enantiômero que poderia ser usado na síntese da (+ )-baquenolida-A e
há apenas duas sínteses laboriosas publicadas até o momento. 78,91 A primeira delas, de
seis etapas, foi descrita por Paquette78 (Esquema 77), partindo-se da (+ )-cetona de
Wieland-Miescher (92), cuja preparação assimétrica catalisada por 5-(-)-prolina é bem
estabelecida. 92 Vale a pena ressaltar que Bui e Barbas 11193 recentemente mostraram a
preparação "one-pot" de 92, entretanto em rendimento moderado (49%) e baixo
excesso enantiomérico (76%).
Esquema 77
O O O
~ S-(-)-prolina.. ~ HSCH2CH2SH.. ~ 1)Ph3P=CHOCH3 oN MVK O~ p-TSA (S~ 2) HCI4N
~0:0
(fLJ 7:1 (cis:trans)
(93%)
92 (57%) '\.-S (99%)
3) LiBHEt3 (P:) TTN oJJ) 1) NaBH4 2) MeS02C1 ..
(72%) (+)-90 (93%)
Na rota descrita por Jenniskens e De Groot91 para a preparação de (+)-90, que
envolve nove etapas (Esquema 78), parte-se de S-carvona e, é portanto, a isopropenila a
responsável pela diastereosseletividade das transformações químicas. O material de
partida é caro, pois não se trata do enantiômero abundante da carvona, e há perda de
três carbonos durante o rearranjo de Criegee sendo, portanto, uma síntese de baixa
economia de átomos.
56
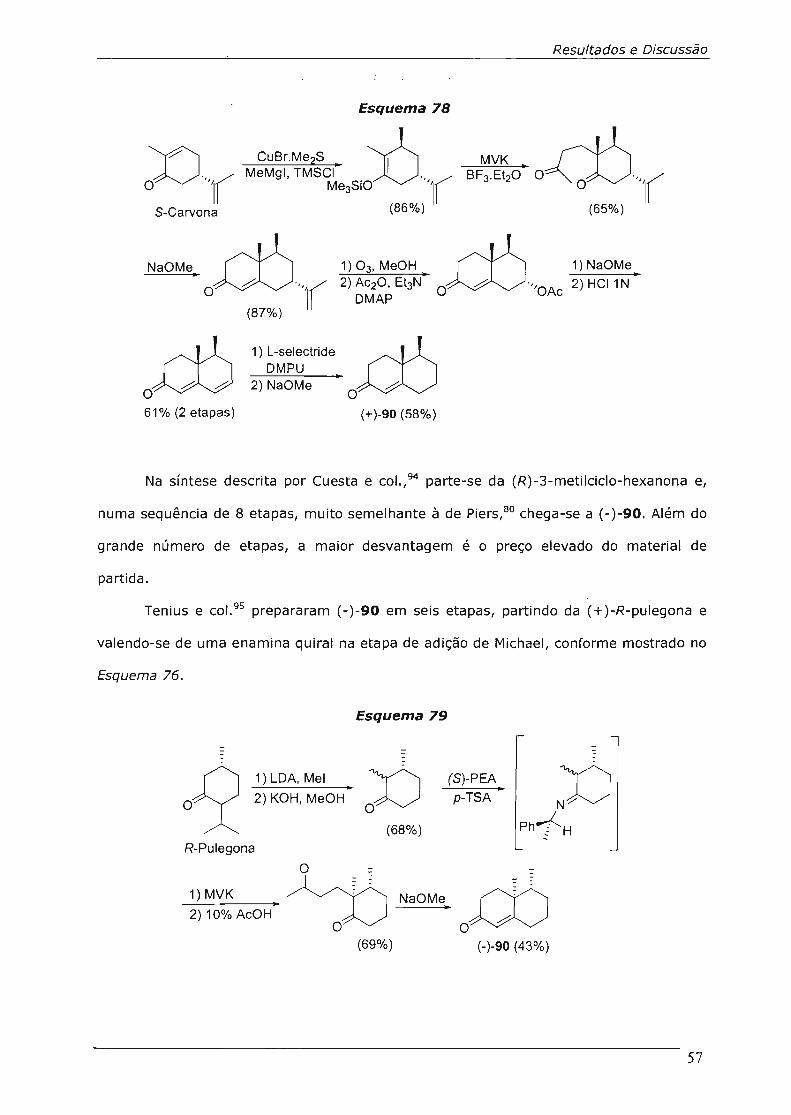
Resultados e Discussão
):)y S-Carvona
Esquema 78
CuBr.Me,S. + MeMgl, TMSCI . "A).",(
Me3SIO I (86%)
MVK • Jj:).,,( BF3·Et20 O O I
(65%)
NaOMe ~ 1) O,. MeOH ~ 1) NaOMe
.. O~""( 2) AC20, Et3N" O~" 'OA 2) HCI 1 N ..
oill 61 % (2 etapas)
(87%) I DMAP c
1) L-selectride có DMPU ..
- ~ 2) NaOMe O~
(+)-90 (58%)
Na síntese descrita por Cu esta e col.,94 parte-se da (R)-3-metilciclo-hexanona e,
numa sequência de 8 etapas, muito semelhante à de Piers,8o chega-se a (-)-90. Além do
grande número de etapas, a maior desvantagem é o preço elevado do material de
partida.
Tenius e co I. 95 prepararam (-)-90 em seis etapas, partindo da (+ )-R-pulegona e
valendo-se de uma enamina quiral na etapa de adição de Michael, conforme mostrado no
Esquema 76.
O~ 1) LDA, Mel
2)KOH,MeOH
R-Pulegona
Esquema 79
;o (68%)
O -
1)MVK ~ 2) 10% AcOH· oN NaOMe.
(69%)
)J (S)-PEA .. p-TSA
Ph-:Z ::: H
-
00) (-)-90 (43%)
57

Resultados e Discussão
Apesar de não estar mostrado no Esquema 79, a etapa de adição de Michael leva
à formação de subprodutos, que são carregados até a última etapa, daí o motivo do
baixo rendimento desta.
4.3.3.2.Preparação da Octalona 90
Procedimento de Zoretic
Dentre todos os métodos de preparação de 90 descritos no item anterior,
julgamos melhor tentar repetir as condições descritas por Zoretic,83 uma vez que era a
rota mais simples e curta e com suposta alta seletividade.
o material de partida, a 2,3-dimetilciclo-hexanona, por não ser comercial, foi
preparado em grande escala através da oxidação direta do 2,3-dimetilciclo-hexanol com
reagente de Jones,60 em ótimo rendimento (Esquema 80).
eXH Esquema 80
Cr203/H2S04
acetona, O°C eX (80%)
A 2,3-dimetilciclo-hexanona foi submetida à reação de anelação de Robinson com
MVK em meio de benzeno e ácido sulfúrico. Foi necessária etapa posterior de ciclização
da dicetona 93 em metóxido de sódio. Em todas as tentativas, não foi possível conseguir
rendimento superior a 23%, e mais, a diastereosseletividade, determinada por lH-RMN,
foi de 4: 1 e não de 9: 1, como publicado pelos autores. Os resultados estão resumidos no
Esquema 81.
58

Resultados e Discussão
~ MVK,H2S04 • Ur PhH, GOC, 16 h O
Esquema 81
n ! .~} ;~, ., j ::,' ,t -f L~~ C~~ ... L~
f,}lr:: " ~ - l '~r~)': ;' ,:~ ;;:-;E C~U~t.~tC.: , t _hl;\lf;;:-~;d~ (\ ~:~ (h~ ~3 J.o \~';' . ,,!~~
~ MeONa/MeOH ~ ~ 45°C, 1,5h ~O
O 93 90
cis:trans (4:1) (23%,2 etapas)
A preparação mencionada acima apresenta alguns fatores complicadores, como,
por exemplo, a extensa polimerização da MVK, que é utilizada em grande excesso,
tornando-se necessária a utilização de agitação mecânica. Após extração, o material
pOlimérico formado é destilado na trompa de vácuo, para retirar a 2,3-dimetilciclo-
hexanona não consumida (ca. de 30 %) e depois à pressão reduzida, para fornecer a
mistura de 90 e das dicetonas 93. Outros subprodutos da reação são as cetonas
bicíc/icas (ca. 5% conforme descrito pelos autores) mostradas na Figura 4, Tendo em
vista o baixo rendimento da transformação e, sobretudo, a baixa seletividade, decidimos
testar outra rota de preparação da 2-octalona 90.
~~ Figura 4
Procedimento de Duhamel Adaptado
Em vista da baixa estereosseletividade do procedimento de Zoretic, decidimos
preparar 90, utilizando a segunda parte do protocolo descrito por Duhamel et al. 8S
(Esquema 74). Como na época não tínhamos TBSTf em nosso laboratório, optamos por
preparar o éter enólico de silício na forma de TMS, a partir da 2,3-dimetilciclo-hexanona,
já preparada em grande escala.
59

Resultados e Discussão
Várias são as possibilidades de preparação de éteres enólicos de silício
termodinâmicos, como descrito por Krafft e Holton96. Seguindo um procedimento
clássico/7,98 os éteres enólicos de silício termodinâmico 94 e cinético 95foram obtidos na
forma de uma mistura 1: 1 (Esquema 82).
eX Esquema 82
TMSCI, Nal, EtN:}.
CH3CN, t.a., 3,5h cXTM; CxTMS 94 95
(80%, 1 : 1)
Tendo em vista a baixa regiosseletividade supramencionada, empregamos uma
outra condição99,lOo para a preparação de 94; este foi obtido junto com ca. de 20%,
determinado por CG, dos outros isômeros cinéticos 95 em 92% de rendimento (Esquema
83).
Esquema 83
eX HMDS, TMSI cXTM; CxTMS .. CH2CI2, t.a., 1,5h
94 95
(92%,4: 1)
Apesar da seletividade não ser das melhores, decidimos prosseguir usando esta
mistura de éteres enólicos de silício. Vale a pena lembrar que Duhamel et al. 8S obtiveram
os enóis éteres de silício de análogos a 94 e 95 (TBDS no lugar de TMS) em uma
proporção de 97:3.
Sendo assim, a mistura de 94 e 95 em nitrometano foi tratada com MVK, ácido
dibenzoiltartárico (DBAT) e BF3 .Et20, a -20°C. Em todas as tentativas, houve
principalmente desproteção do éter enólico de silício e apenas 20-30% de formação do
produto de adição. Trata-se de uma reação extremamente sensível, de modo que vários
fatores podem ter influenciado: controle da temperatura do banho criogênico, traços de
60

Resultados e Discussão
água no solvente e problemas de secagem do DBAT, que é relativamente higroscópico.
Há uma grande desvantagem neste método, que, de certa forma, inviabiliza sua
extensão a maiores escalas: a quantidade elevada de nitrometano utilizado, solvente
caro, pelo menos no Brasil.
Em vista destes resultados desanimadores, adaptamos o procedimento descrito
por Jenniskens,91 uma vez que o enol éter de silício empregado também era de TMS. As
condições experimentais utilizadas estão resumidas no Esquema 84.
Esquema 84
ex ~ MVK, CH3N02 •
I + Ur í-PrOH, BF3·Et20 OTMS OTMS -65°C, 2 h
94 95 4:1
~ O
93 (48%, 2 etapas)
MeONa/MeOH 45°C, 1h
• fuo 90
cís:trans (9: 1) (72%)
Deve-se ressaltar que o rendimento das primeiras duas etapas (48%) pode ser
perfeitamente aceitável, tendo em vista a pureza do silil enol éter intermediário (80%), e
o fato de que sempre é recuperada 2,3-dimetilcicloexanona, proveniente da desproteção
dos éteres enólicos de silício 94 e 95. Então, 90 foi obtida junto com seu isômero 90'
numa proporção de 9: 1. Portanto, acreditamos que este seja um bom método para a
preparação de 90, que foi feita em escala de multigramas.
A diastereosseletividade observada pode ser facilmente racionalizada com base na
análise conformacional dos enolatos intermediários formados (Esquema 85). A reação via
o confôrmero B permite alívio da interação do tipo A(1,2), como discutido por Johnson,lOl
entre os grupos meti la e favorece uma aproximação axial do agente alquilante, em
virtude de fatores estereoeletrônicos, levando assim, à relação eis observada entre as
meti las, ou seja, adição trans, quase que total, do eletrófilo.
61

Resultados e Discussão
Esquema 85 -
TM0'v~t A .. frF dE
E+ Me E O
ataque axial por baixo
11 A(1.2)
I ~
El~,H --
B
E Ce O~ TMSOYv/ ~e .. O
ataque axial por cima
o procedimento descrito nos Esquemas 83 e 84 para a preparação de 90 é
bastante sucinto, entretanto envolve reagentes caros como, por exemplo,
iodotrimetilsilano e nitrometano. Além disso, não só a seletividade na preparação do éter
enólico de silício termodinâmico 94 é baixa (4: 1), como também o próprio rendimento da
etapa de adição de Michael não é tão satisfatório. Além do mais, a escala tem de ser
limitada (máximo de 80 mmol) em virtude da manipulação de grandes volumes de
solvente (Vide Parte Experimental). Estes já seriam motivos suficientes para a alteração
da rota sintética.
Procedimento de Cuesta e Piers adaptado
Com base nos motivos supramencionados, decidimos efetuar a preparação de 90
adaptando o procedimento descrito por Cuesta et al. 94 para a síntese assimétrica de (-)-
90, numa seqüência muito parecida com a descrita por Piers et aI. 80 (Esquema 76).
Partimos, mais uma vez, da 2,3-dimetilciclo-hexanona, e efetuamos o bloqueio da
posição alfa através de reação de formilação, seguida de transformação na respectiva
62

Resultados e Discus~ão
enamina 96 em 73% de rendimento. Esta foi então alquilada com 3-bromoacetato de
etila na única posição alfa disponível. O aduto foi então tratado com Hei, com intuito de
hidrolisar a função enamina, e em seguida tratado com NaOH para hidrolisar o éster,
fornecendo, assim, o ácido 97 em 78% de rendimento. Este foi transformado numa
mistura epimérica (6: 1) das enóis lactonas 98 e 98' em 75%. As lactonas puderam ser
separadas por recristalização em hexano, sendo possível obter 98 pura em 62% de
rendimento. Reação de 98 com MeU, seguida de tratamento com KOH metanólico, levou
à octalona 91 em 60%. O resumo da rota sintética, em rendimento total de 16%, está
mostrado no Esquema 86.
Esquema 86
Cc • HC02Et, t.a., 14h
MeONa, PhH PhNHCH3, PhH • Dean-Stark, 3,5 h
N/ I
73% (2 etapas) Ph 96
1 ) t-BuOK, t-BuOH BrCH2CH2C02Et t.a., 2 h ~.'\\,\
U o C02H
97 (78%)
AC20,AcONa. ~ ó,2h Ur"\~ 2) HCI 10%, !l, 30 min
3) NaOH 10%, ó, 5h
Recristalização.. ~o 98 (62%)
1) MeU, Et20 t.a., 1,5 h •
2) KOH, MeOH !l,2 h
O O 98 + 98' (75%) cis:trans (6:1)
fuo 90 (60%)
A transformação da enol lactona 98 em 90 pode ser facilmente racionalizada
como sendo a abertura de um éster cíclico para formar a dicetona 93 (cf Esquema 84),
seguida de condensação aldólica intramolecular, como nas preparações descritas
anteriormente, porém utilizando KOH como base. A vantagem do uso de KOH metanólico
é a eliminação das condições anidras.
63

Resultados e Discussão
4.3.3.3.Reação de. Contração de Anel da Octalona 90
Mincione et al. 10 2 mostraram a contração de anel de l-octalonas, 2-octalonas e
enonas esteroidais em rendimentos bastante variáveis. Como mostrado no Esquema 87,
a 2-octalona 99, quando tratada com TIN em uma mistura de trimetilortoformiato
(TMOF) e MeOH, forneceu o produto de contração 100 em 61 % de rendimento.
Esquema 87
mo TTN, TMOF (j:) .. 'C0 2Me MeOH, O°C, 30 min
99 100 (61%)
Aplicando as mesmas condições experimentais de Mincione para a octalona 90, foi
possível isolar, de uma mistura de produtos e em apenas 36% de rendimento, o produto
de contração 91, como mostrado no Esquema 88.
Esquema 88
fuo TTN , TMOF ..
MeOH, O°C, 5 min
90 (b"'C0
2Me
91 (36%)
Utilizando apenas TMOF como solvente, 91 foi obtido em 38% de rendimento, ou
seja, não houve melhora no método. Outros solventes, como, por exemplo, CH2C1 2 e
HCI04 foram testados e houve recuperação do material de partida, no primeiro caso, e
formação de mistura complexa de produtos, no segundo caso.
É curioso notar que a reação de contração de anel da octalona 101, que também
não contém uma metila em C-8, levou à formação do produto de contração 102 em
apenas 36% de rendimento, como mostrado no Esquema 89. 102
64

Esquema 89
mo .. TTN, TMOF MeOH, O°C, 30 min
101
Resultados e Discussão
(:b'''C02Me 102 (36%)
Embora não saibamos exatamente o porquê desta diferença de comportamento,
nos parece claro que a metila em C-8 desempenha algum papel que favorece a contração
de anel de 99. Aventamos a proposta de que a ausência da metila em C-8
eventualmente possibilitasse a formação de uma outra forma enólica (Figura 5) que
originaria outros produtos de adição à dupla ligação. De qualquer forma, não podemos
ser categóricos em afirmar tal fato, uma vez que não foi possível o isolamento, e
tampouco a caracterização, dos subprodutos; ademais, não houve formação de nenhum
subproduto em proporções relevantes, como comprovado por análise por cromatografia
gasosa.
R mOH R= H ou Me
Figura 5
Tendo em vista este resultado pouco satisfatório, decidimos procurar outras
condições para a contração de anel de 90. Como é bem conhecido na literatura,l03
compostos de iodo hipervalente promovem rearranjos oxidativos, como, por exemplo,
contrações de anéis de cetonas cíclicas. Exemplos de contração de cetonas a,p-
insaturadas com iodobenzeno diacetato (IBD) estão mostrados no Esquema 90. 104
Esquema 90
HCI04 , t. 8. ~ LI UR
o
~ U llAR
Phl(AcOh, TEOF •
R= H, Me, OMe, CI 58-74%
65

Resultados e Discussão
Nas mesmi3s condições referidas acima e também trocando trietilortoformiato
(TEOF) por TMOF, 90 sofreu decomposição. Na ausência de ácido como catalisador, 90
não reagiu com IBO, mesmo após 24 horas de reação, em metanol, etanol, TMOF ou
TEOF. Fizemos o "branco" da reação e, na ausência de IBO, o substrato também sofreu
decomposição, em menos de cinco minutos, em meio de ácido perclórico ou sulfúrico.
Assim, não foi possível contração de 90 em nenhuma das condições testadas.
Em virtude do baixo rendimento (38%) da etapa chave da síntese proposta para a
baquenolida-A, isto é, a reação de contração de anel da octalona 90, decidimos
abandonar esta seqüência sintética.
4.3.3.4.Tentativas de Contração de Outras Cetonas
Pensamos também na possibilidade de utilizar a cetona de Wieland-Miescher (92),
bem como seu respectivo álcool 103, como possíveis materiais de partida para a síntese
da baquenolida-A. Embora comercial, preparamos 92 em duas etapas, através de
procedimentos clássicos, a partir da l,3-ciclo-hexanodiona (Esquema 91).105,106
60
KOH, Mel .. MeOH, H20 ~, overnight
Esquema 91.
o
Cc 1)KOH,MeOH MVK,~, 3h
O 2) pirrolidina, <l>H Dean-Stark, 1 h
50%
fuo 92 (50%)
Por sua vez, 103 foi obtido através do protocolo clássico de redução seletiva de
92 com boro-hidreto de sódio em etanol 107 (Esquema 92).
fuo 92
Esquema 92
NaBH4 , EtOH O °C, 1 h .. fuo
103 (67%)
66

Resultados e Discussão
Muito embora a preparação dos materiais de partida 92 e 103 tenha sido trivial,
as tentativas de contração deles com TTN levaram à formação de uma mistura complexa
de produtos em todas os solventes testados (MeOH, TMOF e CH 2CI2 ).
4.3.3.s.Tentativas de Contração do Dieno 105
A migração vinílica de dienos no rearranjo oxidativo com sais de tálio(III),
geralmente ocorre em conjunto com a formação de produtos de adição 1,2 e 1,4.108-110
Um exemplo de reação do l,3-ciclo-hexadieno com TTN está mostrado no Esquema
93. 110
O°C, 15 min o TTN, MeOH/CH2CI2 •
Esquema 93
0---\oMe +
OMe (31%)
ÇtOM: OMe (10%)
~e OMe
(12%)
Rigby e Pigge ll1 aplicaram, entretanto, a contração de um dieno na síntese da
(+ )-ferruginina, como mostrado no Esquema 94.
Esquema 94
E ,C02R* I .' H+N+H TTN, MeOH.
V MeO
E=C02Me R*=( -)-8-fenilmentil
E
1)1 ."C02R*
H N H
h-
OMe (85%)
Com base nos resultados de Rigby 111 e no baixo rendimento obtido para a
contração de 90, decidimos investigar a reação de TTN com o dieno conjugado 104, que
seria facilmente preparado em duas etapas a partir de 90, como mostrado no Esquema
95.
67

Resultados e Discussão
Esquema 95
fuo
NaBH,. ~ ~ ~ UJOH
lA) 90 104
Como a preparação da octalona 90 é bastante laboriosa, decidimos preparar um
substrato análogo, que difere apenas de uma metila em C-S isto é, a o 2-octalona 101,
para então preparar o dieno lOS, análogo a 104. 101 foi preparada segundo
procedimento clássico de anelação de Robinson entre a 2-metilciclo-hexanona e metilvinil
cetona,112 em rendimento apenas moderado, como mostrado no Esquema 96 .
Esquema 96
&+f H2S04 • ~ PhH,~,16h ~O
101 (40%)
A seguir, o dieno lOS foi preparado, em ótimo rendimento, através da redução de
101 com boro-hidreto de sódio - sem necessidade de adição de tricloreto de cério -
seguida de desidratação, em meio de ácido fosfórico, do álcool alílico intermediário
formado, como mostrado no Esquema 97.
Esquema 97
~ NaBH4 , MeOH.
~O 1h,t.a.
~ H3P04 (85%) ..
~OH PhH,~,1h 101
d:) 105
(84%, 2 etapas)
Na tentativa de contração de lOS mais uma vez nos deparamos com resultados
nada satisfatórios; isto é, quando lOS foi tratado com TTN em MeOH, TMOF,
MeOH/CH2C1 2, EtOH ou TEOF, em todos os casos houve formação de uma mistura
complexa de produtos.
68

Resultados e Discussão
Em vista destes resultados pouco animadores, decidimos também abandonar esta
rota para a síntese da baquenolida-A.
4.4.Resolução Enzimática de cis-Octalinas'
4.4.1.Introd ução
o sistema eis-decalínico está presente numa vasta gama de classes de produtos
naturais, como, por exemplo, cis-clerodanos, calirienos, telepoganos, cadinanos,
eremofilanos e valerononanos.1l3 A síntese assimétrica de anéis decalínicos de junção eis
ainda é restrita e poucos trabalhos nesta área, até o momento, foram realizados. 11 3
Como exemplo representativo, podemos citar a abordagem que envolve a aplicação de
ácidos de Lewis quirais como catalisadores de reações de Diels-Alder iônicas.1l4
É bem verdade que a biocatálise vem se mostrando uma boa ferramenta
alternativa para a obtenção de moléculas quirais, através de reações enzimáticas
mediadas por lipases, como, por exemplo, a dessimetrização de cis-octalinas. llS
Durante os nossos estudos visando à síntese de bacanos, os álcoois 58 e 71
foram preparados em ótimos rendimentos e excelentes diastereosseletividades. Tendo
em vista a importância do sistema cis-octalínico, imaginamos ser possível realizar a
resolução enzimática dos referidos octaló is, com intuito de obter representantes desta
classe de moléculas enantiomericamente enriquecidos.
Como a resolução enzimática não é o alvo central dos nossos estudos, limitar-nos-
emos à discussão sucinta dos resu ltados obtidos, como descrito a seguir .
. Deve-se salientar que este traba lho foi efetuado em colaboração com o Dr. André Porto e o agora Prof. Dr.
Leandro Andrade (outrora colaboradores do Prof . Dr. Comasseto no progra ma de Jovem Pesquisador da
FAPESP) .
69

Resultados e Discussão
4.4.2.Preparação dos Materiais de Partida
A preparação dos álcoois 58 e 71 já foi descrita em itens anteriores (Esquema 27
e 50). Além destes dois substratos, preparamos o álcool 107, através da redução
seletiva do aduto de Diels-Alder 106, cuja preparação está mostrada no Esquema 98.
Esquema 98
&+X AICI3 , benzeno •
t.a.24h 0x H
106 (36%)
A redução das cetonas 52, 67 e 106 está sumarizada no Esquema 99.
C~b(1 H R2
racêmico 52: R1=R2=H
67: R1=H, R2=Me
106: R1=R2=Me
Esquema 99
NaBH4 , MeOH -78 DC,1h
•
OH
~R1 ~R2
H racêmico 58 (95%)
71 (92%)
107 (93%)
Os acetatos 108, 109 e 110, utilizados como padrões racêmicos, foram
preparados através da clássica 116 acetilação dos álcoois 58, 71 e 107, respectivamente,
como mostrado no Esquema 100.
OH
(trR' H R2
racêmico 58: R1=R2=H
71: R1=H, R2=Me
107: R1=R2=Me
Esquema 100
AC20,DMAP Et3N, 30 min, U;
OAc
~R1 ~R2
H racêmico 108 (85%)
109 (83%)
110 (91%)
70

Resultados e Discussão
4.4.3.Resolução Cinética: O "Screening"
Com o intuito de identificar a melhor lipase, dentre as disponíveis em nossos
laboratórios, para efetuar a resolução cinética dos referidos octalóis, escolhemos
aleatoriamente 71 para fazer o estudo de "screening" com nove diferentes enzimas.
As lipases utilizadas foram: CAl-B (Novozyme 435 - Candida antarctica tipo B
imobilizada - 10,000 PlUjg); CAl-A (Novozyme 735, liquida); CAl-B l (Novozyme,
líquida); Lipozyme Tl 100l (Novozyme, líquida); Lipozyme 1M (Novo Nordisk - Mucor
miehei imobilizada); Lipolase 100T (imobilizada); Lipase E.C. 3.1.1.3 (Sigma - Candida
rugosa liofilizada); Lipase E.C. 3.1.1.3 (Sigma- Mucor javanicus liofilizada) e Lipase E.C.
3.1.1.3 (Sigma-Type II - pâncreas de porco).
Conforme pode ser visto na Tabela 1, apenas quatro das nove enzimas testadas
se mostraram ativas para a resolução cinética do álcool 71 (entradas 1, 2, 5 e 7).
Contudo, apenas CAl-B e CAl-A (entradas 1 e 2) mostraram resultados satisfatórios, em
que foi possível obter o acetato 109b em excelente excesso enantiomérico (98%).
Porém, só com a CAl-B (entrada 1) foi possível obter o álcool 71a em excesso
enantiomérico razoável (82%).
71

Resultados e Discussão
Tabela 1. Resolução Cinética do octalol 71 com diferentes lipases. a
OH OH OH AcO m+ fu Lipase m fu hexano, 32°C +
H H -::7'OAc H H 71a 71b 71a 109b
Entrada Enzimas c e.e. e.e. E
(Lipases) 71b 71a 10gb
1 Novozyme 435 CAl-B 45 82 >98 510
2 Novozyme 735 CAl-A 14 16 >98 232
3 CAl-B l
4 Lipozyme Tl 100l
5 Lipozyme IM 2 4 >98 Nc
6 Lipolase
7 Lipase E.C. 3.1.1.3 9 10 >98 Nc
(Candida rugosa)
8 Lipase E.C. 3.1.1.3
(Mucor javanicus)
9 Lipase E.C. 3.1.1.3
(Pâncreas de Porco)
aA reação foi efetuada a 32°(, usando (±)-cis-octalol 71 (3 IlL), acetato de vinila (60 IlL), hexano
(600 IlL) e uma lipase (6 mg). c (%) - conversão calculada a partir da seguinte equação: c =
(ees)/(eep + ees); ee (%) - excesso enantiomérico; E - razão enantiomérica.
4.4.4.Resolução Cinética dos cis-Octalóis com Novozyme 435
Tendo sido encontrada a melhor enzima para a resolução cinética, efetuamos um
acompanhamento periódico da resolução cinética dos álcoois 58, 71 e 107 com
Novozyme 435 em duas temperaturas diferentes (32 e 40°C). Os resultados obtidos
estão mostrados nas Tabelas 2, 3 e 4.
72
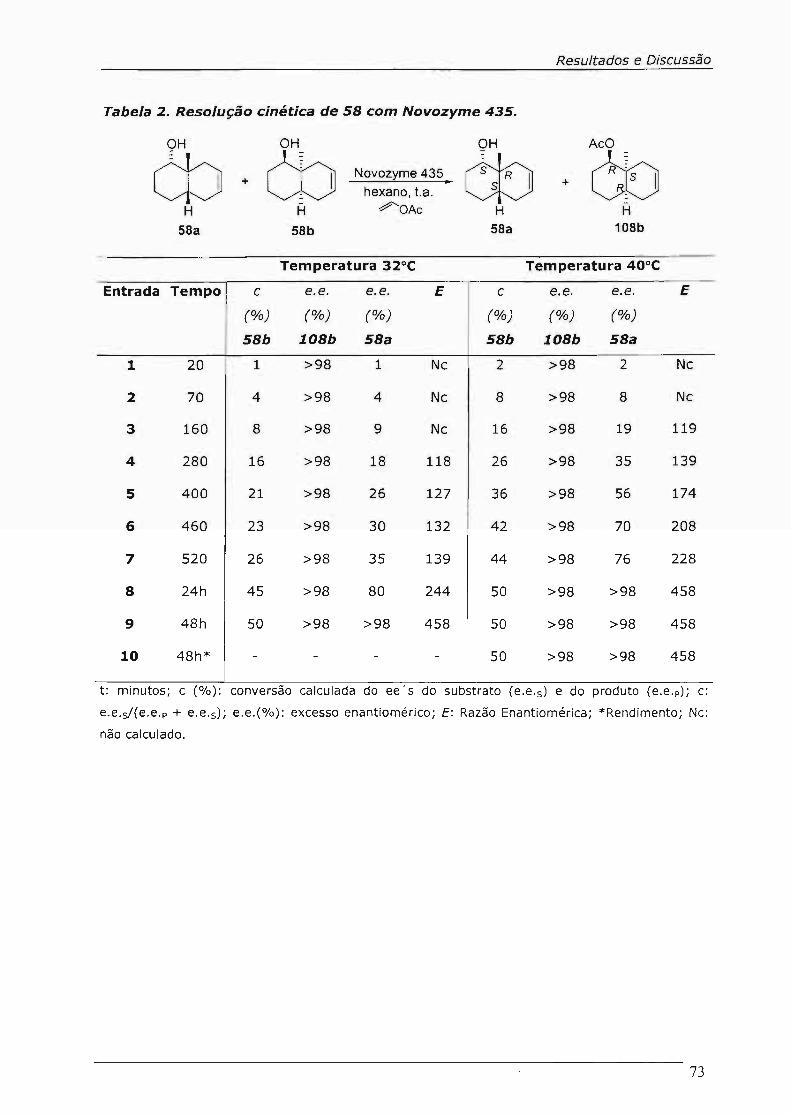
Resultados e Discussão
Tabela 2. Resolução cinética de 58 com Novozyme 435.
OH OH OH AcO
fi + CO Novozyme 435 ób + ób .. hexano, t.a.
H H -::7'OAc H H 58a 58b 58a 108b
Temperatura 32°C Temperatura 40°C
Entrada Tempo I c e.e. e.e. E c e.e. e.e. E
(%) (%) (%) (%) (%) (%)
58b 108b 58a 58b 108b 58a
1 20 1 >98 1 Nc 2 >98 2 Nc
2 70 4 >98 4 Nc 8 >98 8 Nc
3 160 8 >98 9 Nc 16 >98 19 119
4 280 16 >98 18 118 26 >98 35 139
5 400 21 >98 26 127 36 >98 56 174
6 460 23 >98 30 132 42 >98 70 208
7 520 26 >98 35 139 44 >98 76 228
8 24h 45 >98 80 244 50 >98 >98 458
9 48h 50 >98 >98 458 50 >98 >98 458
10 48h* 50 >98 >98 458
t: minutos; c (%): conversão calculada do ee ' 5 do substrato (e.e.s) e do produto (e.e.p); c:
e.e.s/(e.e.p + e.e.s); e.e.(%): excesso enantiomérico; E: Razão Enantiomérica; *Rendimento; Nc:
não calculado.
73

Resultados e Discussão
Tabela 3. Resolução cinética de 71 com Novozyme 435.
OH OH OH AcO m+ fu Lipase m fu hexano, 32°C +
H H rOAc H H 71a 71b 71a 73b
Temperatura 32°C Temperatura 40°C
Entrada Tempo c e.e. e.e. E c e.e. e.e. E
(%) (%) (%) (%) (%) (%) 71b 109b 71a 71b 109b 71a
1 20 4 >98 4 Nc 4 >98 4 Nc
2 70 8 >98 5 Nc 8 >98 8 Nc
3 160 8 >98 8 Nc 18 >98 21 121
4 280 14 >98 16 16 27 >98 36 140
5 400 18 >98 22 22 34 >98 51 164
6 460 22 >98 28 28 37 >98 58 178
7 520 23 >98 30 30 39 >98 63 189
8 24h 44 >98 78 78 50 >98 97 419
9 48h 50 >98 97 97 50 96 96 193
10 48h* 48 >98 92 327
t: minutos; c (%): conversão calculada do ee ' s do substrato (e.e.s) e do produto (e.e.p): c:
e.e.s/(e.e.p + e.e.s); e.e .(%): excesso enantiomérico; E: Razão Enantiomérica ; *rendimento; Nc:
não calculado.
74

Resultados e Discussão
Tabela 4. Resolução cinética de 107 com Novozyme 435.
OH OH OH AcO
cb> ltx Novozyme 435 ctx óbc • + hexano, t.a.
H H -:?'OAc H H 107a 107b 107a 110b
Temperatura 32°C Temperatura 40°C
Entrada Tempo c e.e. e.e. E c e.e. e.e. E (%) (%) (%) (%) (%) (%)
107b 110b 107a 107b 110b 107a 1 20
2 70 2 >98 1 Nc
3 160 2 >98 1 Nc
4 280 2 >98 2 Nc 4 >98 3 Nc
5 400 4 >98 3 Nc 4 >98 3 Nc
6 460 4 >98 3 Nc 10 >98 7 Nc
7 520 4 >98 3 Nc 10 >98 8 Nc
8 24h 10 >98 11 110 32 >98 46 156
9 48h 19 >98 23 123 32 >98 46 156
10 48h* 50 >98 >98 458
t: minutos; c (%): conversão calculada do ee's do substrato (e.e,s) e do produto (e,e.p): c:
e.e.s/(e.e.p + e.e.s); e.e.(%): excesso enantiomérico; E: Razão Enantiomérica; *rendimento; Nc:
não calculado.
Uma vez otimizadas as condições experimentais - 48 horas de reação a 40 DC -, as
reações foram repetidas sem a retirada de alíquotas, e os produtos foram separados
cromatograficamente. Como pode ser visto no Esquema 101, os rendimentos dos
produtos resolvidos isolados, bem como os respectivos excessos enantioméricos foram
excelentes.
75

OH
~R1 ~R2
H racêmico
58: R1=R2=H
71: R1=H, R2=Me
107: R1=R2=Me
Esquema 101
QH
Novozyme435 ~R1 hexano ~ + ~OAc H R2
58a (40%, 98% ee)
71a (41%, 92% ee)
107a (41%, 98% ee)
Resultados e Discussão
~R1 ~R2
H
108b (40%, 98% ee)
109b (41%, 98% ee)
110b (47%, 98% ee)
Os valores de rotação específica encontrados foram: 58a [a]o25= +6.6° Cc 8.6,
CHCI3); 108b [a]o25= -24,0° Cc 3.25, CHCI3); 71a [a]o25= -1.6° Cc 3.02, CHCI3); 10gb
[a]o25= -8.5° (c 2.95, CHCI3); 107a [a]o25= -8.0° Cc 1.35, CHCI3); 110b [a]o25= +13.5°
Cc 2.73, CHCI3).
A determinação da configuração absoluta de 107a como sendo S/S/R foi efetuada
através da comparação do valor da rotação específica obtido com o descrito na literatura
C -9. 7°, c 1.49, CHCI3).114 A configuração absoluta dos demais compostos foi atribuída
por extrapolação. Uma vez que usamos a mesma enzima para a resolução cinética dos
três álcoois, que diferem entre si pela presença de grupos metilas na dupla ligação,
acreditamos que tal extrapolação possa ser feita; e mais, o centro carbinólico a ser
resolvido se encontra distante da ligação dupla.
76

Conclusão
5.Conclusão
Apesar dos longos esforços investidos na síntese tanto da nor-baquenolida-A (49)
como da baquenolida-A (I), não conseguimos completar as referidas seqüências
sintéticas propostas.
Durante a síntese de 49 enfrentamos algumas dificuldades pouco usuais que
foram sanadas após muito empenho de bancada, com exceção da última etapa, que
deverá ser re-estudada e completada, assim que possível, em nosso grupo de pesquisa.
Dentre as várias abordagens investigadas para a síntese da baquenolida-A,
nenhuma delas mostrou resultados satisfatórios, uma vez que foi difícil efetuar a etapa
chave de contração de anel dos vários substratos testados. A única reação de contração
com TTN que ocorreu foi a da octalona 90 (Esquema 88), porém em baixo rendimento; o
que de fato foi surpresa, pois, afinal, tal metodologia havia sido empregada para uma 2-
octalona regio-isomérica de 90 (Esquema 87). Tal era nossa confiança no funcionamento
desta reação, que despendemos tempo relativamente longo de bancada na otimização de
uma rota que levasse à obtenção da octalona 90 em bom rendimento e boa
diastereosseletividade; e tal propósito foi alcançado (cf item 4.3.3.2).
Quanto à resolução cinética dos cis-octalóis, ótimos resultados foram alcançados,
já que os produtos resolvidos com a lipase Novozyme 435 foram obtidos em excelentes
rendimentos isolados e também excelentes excessos enantioméricos, através de um
protocolo de grande praticidade laboratorial.
De certa maneira, os objetivos específicos aos quais nos propusemos não foram
completamente alcançados; todavia, através dos estudos apresentados nesta tese,
demonstramos, pela primeira vez, a possibilidade de preparar o sistema cis-hidrindânico
- unidade estrutural central dos bacanos - através de uma reação de contração de anel
mediada por tálio(III) de cis-octalinas; portanto, em parte alcançamos o objetivo geral a
que esta tese se propunha .
77

Conclusão
Esta reação poderia muito bem ser aplicada na contração de anel da cis-octalina
51 (Esquema 59) - que já contém as meti las com a configuração relativa cis entre si - e
assim seria possível completar pelo menos a síntese formal da baquenolida-A. Porém,
por uma questão de tempo, as propostas sintéticas para 51, descritas nos Esquemas 60
e 61, não foram efetuadas. Ademais, a publicação de Reddy 19 versando sobre a síntese
de 1 nesse ínterim, também nos tirou o incentivo de continuar com tal proposta.
Two roads diverged in a yel/ow wood,
And sorry I could not traveI both
And be one traveler, long I stood
And looked down one as far as I could
To where it bent in the undergrowth;
Then took the other, as just as fair,
And having perhaps the better claim,
Because it was grassy and wanted wear;
Though as for that the passing there
Had worn them real/y about the same.
And both that morning equal/y lay
In leaves no step had trodden black.
Oh, I kept the first for another day!
Yet knowing how way leads on to way,
I doubted if I should ever come back.
I shall be telling this with a sigh
Somewhere ages and ages hence:
Two roads diverged in a wood, and l
I took the one less traveled by,
And that has made ali the difference.
The Road not Taken - Robert Frost
E agora José?
A festa acabou,
a luz apagou,
o povo sumiu,
a noite esfriou,
e agora José?
e agora você,
você que é sem nome,
que zomba dos outros,
você que faz versos,
que ama, protesta,
e agora, José?
Carlos Drummond de Andrade
78

Parte Experimental
6.Parte Experimental
Considerações gerais - Avis aux lecteurs
As reações foram acompanhadas utilizando-se um cromatógrafo a gás modelo HP
6890 (coluna capilar HP-5 de natureza apoIar, contendo 5% de difenil- e 95% de dimetil
polisiloxano). Também foi utilizada cromatografia em camada delgada, empregando-se
placas de sílica gel do tipo 60-F254 sobre alumínio, comercializadas pela Merck, as quais
foram reveladas com iodo, com luz ultravioleta, com solução de vanilina ou com solução
de para-anisaldeído.
As purificações em coluna cromatográfica e coluna cromatográfica flash foram
realizadas utilizando-se sílica gel Acros 80-230 Mesh e 200-400 Mesh, respectivamente.
Os reagentes e solventes foram tratados e/ou secos antes do uso, quando
necessário, conforme os métodos usuais.
Os pontos de fusão - determinados em um aparelho Buchi, modelo B-545 - não
foram corrigidos.
Os espectros no infravermelho, os espectros de massas e as análises elementares
foram realizados pelos técnicos da Central Analítica do Instituto de Química da USP,
utilizando-se os seguintes aparelhos, respectivamente: Perkin-Elmer 1750-FT, Finingan
MAT INCOS 50B acoplado a um cromatógrafo a gás Varian 3400 e Perkin-Elmer
2400/CHN.
As análises de Espectroscopia de Massas de Alta Resolução (EMA R) foram
realizadas na Central Analítica do Instituto de Química da Universidade Estadual de
Campinas.
Os espectros de Ressonância Magnética Nuclear foram realizados utilizando os
aparelhos Varian INOVA 300, Bruker AC-200, Bruker OPX-300 e Bruker OPX-500. As
amostras foram preparadas utilizando COCI3 como solvente e tetrametilsilano como
padrão interno. Para fins de atribuição dos sinais dos espectros de RMN de lH e de 13C, a
numeração dos compostos foi feita arbitrariamente. Apenas para os compostos inéditos
79

Parte Experimental
foi efetuada a atribuição dos sinais e caracterização completa - leia-se lH-RMN, 13C-RMN,
massa, IVe CHN. Os espectros de lH-RMN e de 13C-RMN encontram-se em anexo, junto
com os de intermediários sintéticos de relevância, cujos dados estão listados na parte
experimental, mesmo que já previamente descritos na literatura.
Os compostos foram nomeados utilizando o programa CS ChemDraw Ultra®
(versão 6.0), o qual utiliza o programa para nomenclatura de compostos orgânicos
AutoNom (versão 2.1) sob licença da Beilstein In forma tionssystem e (Copyright® 1988-
1998, Bei/stein Institut fuer Literatur der Organischen Chemie).
80

Parte Experímental
6.1.nor-Baquenolida-A
6.l.l.Preparação da 2-metilciclo-hexen-2-ona53
o Uma solução da 2-metilciclo-hexanona (15,0 g; 0,134 moi) e NBS recém
~ recristalizada (23,8 g; 0,134 moi) em tetracloreto de carbono (150 mL) foi
agitada e refluxada sob atmosfera de nitrogênio por 13 h. A suspensão
resultante foi filtrada sob Celite®, para a remoção da succinimida formada. O sólido foi
lavado com éter (150 mL) e o filtrado foi evaporado à pressão reduzida. O resíduo foi
dissolvido em DMF seco (300 mL) e foram adicionados Li2C03 (29,7 g; 0,400 moi) e LiBr
(34,9 g; 0,400 moi). A suspensão resultante foi refluxada por 3 horas sob atmosfera de
nitrogênio. A mistura reacional foi resfriada, o sólido decantado, a fase líquida foi diluída
com água (400 mL) e extraída com éter (300 + 2 x 200 mL). A fase etérea foi lavada
com água, solução saturada de NaCI, secada com MgS04 , e o solvente foi evaporado à
pressão reduzida. O produto bruto foi destilado à pressão atmosférica (PE: 172-176 0c),
rendendo 9,92 g (90,0 mmol, 67%) da 2-metilciclo-hexen-2-ona, na forma de um líquido
levemente amarelado.
6.l.2.Preparação da cis-8a-metil-3,4,4a,S,8,8a-hexa-hidro-2H-naftalen-l-ona
(52)54
(b H 52
A uma suspensão de AICI 3 (10,0 g; 75,0 mmol) em diclorometano seco (150
mL), em banho de gelo, foi adicionada a 2-metilciclo-hexen-2-ona (16,5 g;
0,150 moi) . O AICI3 foi gradualmente dissolvido para formar uma solução de
coloração amarela. Adicionou-se uma ponta de espátula de hidroquinona e
1,3-butadieno (30 g; 0,60 moi) recém condensado. O balão foi tampado, o banho de gelo
foi retirado, e a solução foi agitada a temperatura ambiente por 24 horas. O solvente foi
removido à pressão reduzida e o resíduo foi particionado entre água e éter. A fase etérea
foi lavada com água, solução saturada de NaCI, secada com MgS04 , e o solvente foi
81

Parte Experimental
evaporado à pressão reduzida. O produto bruto foi destilado à pressão reduzida (PE: 66-
69°C a 0,5 mmHg) rendendo 18,9 9 de 52 (0,114 mmol, 76%) na forma de um líquido
levemente amarelado.
lH-RMN (200 MHz, 8):1,05 (s, 3H); 1,59-2,70 (m, 11H); 5,55-5,69 (m, 2H).
13C-RMN (50 MHz,8): 20,4; 25,1; 27,7; 28,0; 31,2; 37,1; 40,9; 47,6; 122,9; 127,1;
214,9.
6.l.3.Preparação do cis-4a-metil-l,2,3,4,4a,5,8,8a-octa-hidronaftaleno (53)
cb A uma solução da cetona 52 (3,27 g; 20,0 mmol) em trietilenoglicol (20 mL)
foi adicionado NH2NH2 .H20 (5,0 mL; 5,0 g; 0,10 moi) e a solução resultante H 53 foi refluxada a 130 °C por 1,5 horas. A mistura reacional foi resfriada para
então se adicionar KOH em pastilhas (6,7 g; 0,12 mmol) e depois foi refluxada a 130 °C
por mais 4,5 horas. A mistura reacional foi vertida em gelo e acidificada com HCI 6M. A
fase aquosa foi extraída com pentano (3 X 150 mL) e a fase orgânica foi lavada com
solução saturada de cloreto de sódio, secada com MgS04 e o solvente foi evaporado à
pressão reduzida. O produto bruto foi purificado em coluna cromatográfica de sílica (80-
230 mesh), eluindo-se com pentano, rendendo 2,41 9 de 53 (16,0 mmol; 80%) como
um líquido incolor.
lH-RMN (200 MHz, ppm): 0,89 (s, 3H); 1,09-1,70 (m, 11H); 2,23-2,39 (m, 2H); 5,47-
5,59 (m, 2H).
13C-RMN (50 MHz, ppm): 22,6; 26,7; 28,7; 29,6; 30,1; 31,6; 31,9; 39,6; 40,3; 124,2;
124,7.
CAS: 21789-58-2
82

Parte Experimental
6.1.4.Preparação da 1-(3a-metil-octa-hidro-inden-2-il)-etanona (55)
Primeira Parte:
cÍ>-CHO H 54
A uma solução da olefina 53 (1,50 g; 10,0 mmol) em
trimetilortoformiato (50 mL) foi adicionado TTN.3H 20 (5,11 g; 11,5
mmol) e agitou-se à temperatura ambiente por 3,5 horas. Observou-
se uma precipitação abundante durante este período. O precipitado de TIN03 formado foi
filtrado, sob pressão, numa coluna curta de sílica, eluindo-se com diclorometano. O
filtrado foi sucessivamente lavado com água e solução saturada de cloreto de sódio e
secado com MgS04 • O solvente foi evaporado à pressão reduzia e o produto bruto foi
dissolvido em clorofórmio (30 mL). A esta solução adicionou-se uma mistura 1: 1 de ácido
trifluoroacéticojágua (10 mL) a O °C e agitou-se por 2 horas à temperatura ambiente.
Adicionou-se água e a fase aquosa foi extraída com clorofórmio. A fase orgânica foi
sucessivamente lavada com solução saturada de NaHC03 , solução saturada de NaCl,
seca da com MgS04 , e o solvente foi evaporado a pressão reduzida.
Segunda Parte:
cjyl H 55
O produto bruto foi dissolvido em éter etílico anidro (40 mL) e então
adicionou-se, a -78°C, uma solução 1,6 M de MeLi em éter etílico (10,0
mL; 16,0 mmol). A solução foi agitada por 1,5 horas à temperatura
ambiente e a mistura reacional foi vertida em gelojNH4 CI. A fase aquosa foi extraída com
éter. A fase etérea foi lavada com água, solução saturada de NaCl, secada com MgS04 , e
o solvente foi evaporado à pressão reduzida. Dissolveu-se o álcool em acetona (40 mL),
resfriou-se a solução em banho de gelo e titulou-se lentamente, sob agitação, com a
solução do reagente de Jones 8M (20 mL). Após a adição, a mistura foi agitada por 15
minutos e o excesso de oxidante foi destruído com isopropanol. A mistura foi
particionada em H20 e AcOEt e a fase orgânica lavada com água, solução saturada de
NaHC03 , solução saturada de NaCI e seca com MgS04 . O solvente foi evaporado à
pressão reduzida e o produto bruto foi purificado em coluna cromatográfica de sílica flash
83

Parte Experimental
(200-400 mesh), com eluição gradiente de uma mistura acetato/hexano (5-25%),
rendendo 1,18 g de 55 (6,55 moi; 65%) como um óleo incolor. 55 foi obtida como uma
mistura 5: 1 de diastereoisômeros. Os dados espectroscópicos para o isômero majoritário
estão listados a seguir.
lH-RMN (300 MHz, 8): 0,98 (s, 3H); 1,16-2,00 (m, 13H); 2,99-3,12 (m, 1H); 2,15
(s, 3H).
13C- RMN (75 MHz, 8): 21,S; 22,1; 25,2; 25,7; 28,6; 30,9; 33,3; 41,0; 42,2; 44,2; 48,S;
211,0.
IVfilme (v, cm- 1): 1711.
M/Z: 180 (M+, 13%); 81 (100%).
CAS: 33020-76-7
6.l.s.Preparação do 2-acetil-3a-metil-octa-hidro-indeno-2-carboxilato de metila
(56)61
~10 8 O 11 A uma solução da cetona 55 (0,721 g; 4,00 mmol) em
2 59 ",12 oMe diclorometano anidro (40 mL) foram adicionados sob agitação,
3 7/ 13 4 H 6 O atmosfera de Nz e a -40°C, hexametildissilasano (4,2 mL; 20
56 mmol) e iodotrimetilsilano (1,42 mL; 10,0 mmol). A solução
levemente amarelada foi agitada por 10 minutos a -40°C e 1 hora a O cc. A mistura
reacional foi vertida em uma mistura pentano/solução saturada de NaHC03 . A fase
aquosa foi extraída com pentano (3 x 50 mL) e a fase orgânica foi lavada com solução
saturada de cloreto de sódio e secada com MgS04 • O solvente foi evaporado à pressão
reduzida e o produto bruto foi dissolvido em THF anidro (40 mL). A esta solução foi
adicionada, a -40°C e sob atmosfera de Nz, uma solução 1,6 M de MeU em éter etílico
(7,5 mL; 12 mmol) e agitou-se por 30 minutos. Adicionou-se uma solução de
cianoformiato de meti la (l,39g; 12,0 mmol) em THF (5 mL) e a solução resultante foi
agitada por 1 hora. A mistura reacional foi vertida em gelo e a fase aquosa foi extraída
84

Parte Experimental
com érter etílico. A fase etérea foi lavada com solução saturada de NaHC03, solução
saturada de NaCI e secada com MgS04. O solvente foi evaporado à pressão reduzida e o
produto bruto foi purificado em coluna cromatográfica de sílica flash (200-400 mesh),
com eluição gradiente de uma mistura acetatojhexano (10-30%), fornecendo 0,515 9 de
56 (2,16 mmol; 54%) como um sólido branco amorfo. Todo o material utilizado após a
etapa de carboxilação foi lavado com solução saturada de sulfato ferroso, com o intuito
de promover a complexação dos íons cianeto gerados no meio reacional!
lH-RMN (300 MHz, 8):1,03 (s, 3H, - HClO ); 1,1-1,6 (m, 8H - HC1 + HC2 + HC3 + HC4 );
1,74-1,82 (m, 1H - HCs ); 1,92 (d, J= 13,8 Hz, 1H - HCs ); 2,13 (s, 3H - HC13); 2,22-
2,27 (m, 2H - HC6 ); 2,44 (d, J=13,8 Hz, 1H - H 'Cs); 3,72 (s, 3H - OCH3 ).
13C-RMN (75 MHz,8): 33,6 (C1); 21,2 (C2); 22,3 (C3)*; 24,6 (C4); 44,4 (Cs); 36,1 (C6);
64,6 (C7); 45,8 (Cs); 40,6 (C9); 26,6 (ClO)*; 174,7 (Cll); 203,6 (C12); 25,6 (C13)*; 52,7
(OCH3). *os sinais podem estar trocados entre si.
IV (v, cm- 1): 1711; 1746.
CHN: calculado (C 70.46; H 9.30), obtido (C 70,03; H 9,12).
6.1.6.Preparação da ceto lactona 57
2~1 3~í;:J
4 H 6 1111 12 57 O
A uma solução do ceto éster 56 (0,184 g; 0,773 mmol) em
diclorometano anidro (8 mL) foram adicionados sob agitação,
atmosfera de N2 e a ° cC, hexametildissilasano (0,82 mL; 0,62 g; 3,9
mmol) e iodotrimetilsilano (0,28 mL; 0,39 g, 1,9 mmol). A solução
levemente amarelada foi agitada por 10 minutos a ° cC e 3 horas a temperatura
ambiente. A mistura reacional foi vertida em uma mistura pentanojNaHC03 . A fase
aquosa foi extraída com pentano e a fase orgânica foi lavada com solução saturada de
cloreto de sódio e secada com MgS04. O solvente foi evaporado à pressão reduzida e o
produto bruto dissolvido em THF (9 mL). A esta solução foram sucessivamente
adicionados água (3 mL), NMO (0,21 g; 1,5 mmol) e OS04 (19,6 mg; 7,73 x 10-2 mmol).
85

Parte Experimental
A suspensão resultante foi agitada por 40 horas à temperatura ambiente e então se
adicionou metabissulfito de sódio sólido. A suspensão preta obtida foi filtrada sobre celite
e o sólido lavado com diclorometano. O filtrado foi concentrado à pressão reduzida e
particionado entre água e éter etílico. A fase etérea foi lavada com água e solução
saturada de cloreto de sódio. O solvente foi evaporado e o resíduo foi dissolvido em éter
etílico (10 mL) e adicionou-se Hei 10% (5 gotas). A solução resultante foi agitada por 2
horas a temperatura ambiente, lavada com solução saturada de NaHC03 e secada com
MgS04 • O solvente foi evaporado à pressão reduzida e o produto bruto foi purificado em
coluna cromatográfica de sílica flash (200-400 mesh), com eluição gradiente de uma
mistura acetato de etila/hexano (5-30%), fornecendo 0,0668 g de 57 (0,300 mmol;
39%) como um sólido branco amorfo.
lH-RMN (500 MHz, 8): 1,15 (s, 3H - HClO); 1,20-1,60 (m, 8H - HC1 + HC2 + HC3 + HC4 );
1,7-1,8 (m, lH - HCs); 1,87 (d, J=13,4 Hz, lH - HCs); 1,92 (d, J=13,4 Hz, lH - H'Cs);
2,1-2,2 (m, 2H - HC6 ); 4,62 (5, 2H - HC13).
13C-RMN (50 MHz, 8): 32,8 (C1); 22,3 (C2)*; 20,7 (C3)*; 24,0 (C4)*; 45,3 (Cs);40,l (C6);
51,7 (C7); 49,2 (Cs); 42,2 (C9); 25,0 (ClO); 210,5 (C11); 72,3 (C12); 179,0 (C13);. *os
sinais podem estar trocados entre si.
IV (v, em-I): 1747; 1794.
eHN: calculado (e 70,24; H 8,16), obtido (e 69,90; H 8,13).
6.1.7.Preparação do octaidro-3a-metil-2-(prop-l-en-2-il)-lH-indeno-2-
carboxilato de metila (65)
Preparação da solução 0,1 mM da fosforana: A uma suspensão de cloreto de metil
trifenilfosfônio (0,79 g; 2,1 mmol) em éter (20mL) foi adicionado BuLi 2,2SM (0,90 mL;
2,0 mmol) sob atmosfera de N2 e a -20 0e. A suspensão amarela foi agitada por 30 min e
deixou-se decantar o sólido.
86

Parte Experimental
A uma solução do ceto éster 56 (0,153 g; 0,645 mmol) em éter
~10 8011
2 gOMe
3 4 5H
6'f 13 anidro anidro (7 mL) foi adicionada sob agitação, atmosfera de N2 e
a O °C, uma solução de fosforana recém preparada (13 mL) e 65 14
mistura reacional foi agitada a esta temperatura por 8 horas, para
então se adicionar água. A fase aquosa foi extraída com éter e a fase orgânica foi lavada
com água, solução saturada de cloreto de sódio e seca com MgS04 • O solvente foi
evaporado à pressão reduzida e o produto bruto foi purificado em coluna cromatográfica
de sílica flash (200-400 mesh), com eluição gradiente de uma mistura acetatojhexano
(5-10%), fornecendo 91,5 mg de 65 (0,387 mmol; 60%) como um líquido incolor. Se
levarmos em consideração recuperação de 24% (36,8 mg, 0,154 mmol) do material de
partida (56), o rendimento pode ser entendido como 79%.
lH-RMN (300 MHz, o): 1,03 (s, 3H - HClO ); 1,2-1,7 (m, 9H - HC1 + HC2 + HC3 + HC4 +
HCs); 1,72 (si, 3H - HC13 ); 1,83 (d, )=13,9 Hz, 1H - HC8 ); 2,04 (tap , )=12,8 Hz, 1H -
HC6); 2,17 (d, )=13,9 Hz, 1H - H 'C8 ); 2,25 (dd, )=12,8 e 6,3 Hz, 1H - H 'C6); 3,67 (s,
3H - OCH3 ); 4,85 (quintetoap, )=1,3 Hz, 1H - HC6); 4,96 (si, 1H - HC14 ).
13C-RMN (75 MHz, o): 35,2 (C1); 21,2 (C2)*; 22,6 (C3)*; 24,5 (C4)*; 44,0 (Cs); 38,2
(C6); 57,6 (C7); 48,7 (C8); 39,6 (Cg); 25,7 (ClO); 177,5 (Cll ); 147,6 (C12); 21,1 (C13);
110,0 (C14); 52,3 (OCH3). *os sinais podem estar trocados entre si.
MjZ: 236 (M+, 28%); 95 (100%).
87

Parte Experimental
6.2.0utros cis-Hidrindanos
6.2.1.Preparação da 6,8a-dimetil-3,4,4a,S,8,8a-hexa-hidro-2H-naftalen-l-ona
(67)54
fu H 67
A uma suspensão de AICI3 (3,30 g; 25,0 mmol) em benzeno seco (120
mL) foi adicionada a 2-metilciclo-hexen-2-ona (11,0 g; 0,100 moi). O
AICI 3 formou uma pasta no fundo do balão e então se adicionou, em
banho de gelo, isopreno (20 mL; 0,40 moi). O balão foi tampado e o
banho de gelo foi retirado, e a solução agitada a temperatura ambiente por 24 horas. O
solvente foi evaporado à pressão reduzida e o resíduo foi particionado entre água e éter.
A fase etérea foi lavada com água, solução saturada de NaCl, secada com MgS04 , e o
solvente foi evaporado à pressão reduzida. O produto bruto foi destilado em trompa de
vácuo (PE: 135-140 0c) rendendo 9,54 g de 67 (53,S mmol, 54%) na forma de um
líquido levemente amarelado.
lH-RMN (300 MHz, 8): 1,09 (s, 3H); 1,55-1,77 (m, 9H); 1,98-2,61 (m, 5H); 5,29-5,34
(m, 1H).
13C-RMN (75 MHz,8): 20,S; 23,7; 25,2; 28,0; 31,7; 33,1; 37,2; 41,7; 47,S; 116,9;
130,9; 215,5.
6.2.2.Preparação do 6,8a-dimetil-3,4,4a,S,8,8a-hexa-hidro-2H-naftalen-l-ona-
dixolano (68)
13
13 '("011 9
°W 2 1 10 I 7
3 6 12 4 H
Uma solução da cetona 67 (1,78 g; 10,0 mmol), etilenoglicol
(l,86g; 30,0 mmol) e ácido p-toluenosulfônico (0,019 g; 0,10
mmol) em benzeno seco (30 mL) foi refluxada por 2 horas em um
68 aparelho de Dean-Stark. Após resfriamento, foi adicionada solução
saturada de NaHC03 e a fase aquosa foi extraída com AcOEt, lavada com água, solução
88

Parte Experimental
saturada de NaCI, secada com MgS04 , e o solvente foi evaporado à pressão reduzida. O
produto bruto foi purificado em coluna de sílica flash (200-400 mesh), eluindo-se com
uma mistura hexanojAcOEt (75:25), rendendo 2,02g de 68 (9,09 mmol; 91%) na forma
de um óleo incolor.
lH-RMN (300 MHz, 8): 0,87 (s, 3H - HCII ); 1,2-1,8 (m, 9H - HC2 + HC3 + HC4 + HCs +
HC9 ); 1,63 (si, HCI2 ); ,15-2,27 (m, 2H - HC6 ); 3,9-4,0 (m, 4H - HCI3 + HC13 ' ); 5,24-
5,26 (m, 1H - HC8 ).
13C-RMN (75 MHz, 8): 113,0 (CI ); 34,0 (C2); 22,9 (C3); 28,6 (C4)*; 38,2 (Cs); 30,1
(C6)*; 130,5 (C7); 117,9 (C8); 30,4 (C9)*; 40,4 (ClO); 18,1 (Cll ); 23,7 (C12); 64,9 e 65,1
(C13 e C13 ) . *os sinais podem estar trocados entre si.
6.2.3.Preparação da 6,7-dimetóxi-6,8a-dimetil-octa-hidronaftalen-l-ona (70) e
da 7-metóxi-6,8a-dimetil-6-nitróxi-octa-hidronaftalen-l-ona (69)
O 11
~.,\OMe
2 1 10
3 5 .: OMe 4 H 6 =
70 12
A uma solução do cetal 68 (0,334 g; 1,50 mmol) em MeOH (7,5 mL)
adicionou-se TTN.3H 20 (0,73 g; 1,65 mmol) e este foi prontamente
dissolvido. Alguns minutos após a adição de TTN observou-se uma
O 11
~8\OMe .'
2 1 10
3 5 : ON02 4 H 6 =
69 12
precipitação abundante e deixou-se reagir por 15 minutos. A
suspensão foi filtrada, sob pressão, em uma pequena coluna de
sílica (10 cm), eluindo-se com CH 2C1 2 (100 mL). A solução foi lavada
com água, solução saturada de NaCI e secada com MgS04 • O
solvente foi evaporado à pressão reduzida e o produto bruto purificado em coluna
gradiente de sílica flash (200-400 mesh), eluindo-se com AcOEtjhexano (30-40%).
Foram obtidas 0,0833 g de 70 (0,370 mmol; 25%) como um sólido amorfo e 0,108 g de
69 (0,450 mmol; 30%) na forma de um sólido branco.
70 - lH-RMN (200 MHz, 8): 0,87 (dd, J= 13,2 e 11,8 Hz, 1H - HaxC9 ); 1,16 (s, 3H -
HCll ); 1,26 (s, 3H - HCI2 ); 1,41-2,32 (m, 8H - HC3 + HC4 + HCs + HC6 ); 2,48-2,66 (m,
89

Parte Experimental
2H) - HC2); 3,24 (5, 3H - OCH3 ); 3,33 (dd, J= 10,7 e 4,8 Hz, 1H - HCs ); 3,46 (5, 3H -
OCH3 ).
RMN- 1H (50 MHz, 5): 214,0 (Cl ); 37,8 (C2)*; 22,0 (C3); 25,5 (C4); 41,8 (Cs); 37,4 (C6)*;
77,8 (C7); 80,3 (Cs); 36,1 (C9)*; 49,4 (C10); 15,2 (Cll ); 26,6 (C12); 49,1 e 57,3 (OCH3).
*os sinais podem estar trocados entre si.
69 - lH-RMN (200 MHz, 8): 0,98 (dd, J= 13,6 e 11,9 Hz, 1H - HaxC9 ); 1,14-2,32 (m, 8H -
HC3 + HC4 + HCs + HC6 + HeqC9); 1,28 (5, 3H - HCll ); 1,59 (5, 3H - HC12 ); 2,52-2,62
(m, 2H - HC2 ); 3,42-3,54 (m, 1H - HCs); 3,46 (5, 3H - OCH3 ).
lH-RMN (50 MHz, 8): 213,4 (C l ); 37,3 (C2)*; 21,9 (C3); 25,3 (C4); 41,8 (Cs); 36,3 (C6)*;
95,8 (C7); 78,3 (Cs); 36,4 (C9)*; 49,0 (C10); 15,4 (Cll ); 26,3 (C12); 58,4 (OCH3). *os
sinais podem estar trocados entre si.
IV (v, cm- 1): 1696; 1607, 1300.
6.2.4.Preparação do trans-6,8a-Dimetil-1,2,3,4,4a,S,8,8a-octa-hidronaftalen-1-
01 (71)
~ºH 1 A uma solução da octalona 67 (1,79 g; 10,1 mmol) em MeOH (50 mL) . 8
3 1 10 I 7 mantida a -78°C foi adicionado NaBH4 (0,38 g; 10,0 mmol) sólido,
4 H 6 12 numa única porção. A suspensão foi agitada por uma hora para então 71
se adicionar água (30 mL). A fase aquosa foi extraída com AcOEt (3 x
30 mL), lavada com solução saturada de NaCI e secada com MgS04 • O solvente foi
evaporado à pressão reduzida, para render 1,67 9 de 71 (9,24 mmol; 92%) na forma de
um óleo incolor.
lH-RMN (300 MHz, 8): 1,01 (5, 3H - HCll ); 1,2-1,7 (m, 10H - HC2 + HC3 + HC4 + HCs +
HC6 + OH); 1,64 (si, 3H - HC12 ); 2,13-2,33 (m, 2H - HC9 ); 3,32 (dd, J= 11,1 e 4,4 Hz,
1H - HC1 ); 5,27 (51, 1H - HCs).
90

Parte Experimental
13C-RMN (75 MHz, õ): 78,2 (C1); 34,0 (C2); 23,7 (C3)*; 24,0 (C4)*; 39,9 (Cs); 28,9
(C6J**; 130,7 (C7); 117,7 (Cs); 30,6 (C9)**; 36,5 (ClO); 26,1 (Cll )*; 23,4 (C12)*. *os
sinais podem estar trocados entre si. **os sinais podem estar trocados entre si.
IVfi1me (v, cm' l): 3388.
6.2.S.Preparação do 10-Metóxi-1,8-dimetil-2-oxa-triciclo[S.3.1.03,8] undecano
(72)
11
~g~,\OMe
2 I 10
A uma solução do álcool 71 (0,275 g; 1,53 mmol) em TMOF (7,5 mL)
0- · -___ 7 foi adicionado TIN.3H 20 (0,75 g; 1,68 mmol) e este foi prontamente 3
4 H 6 12 dissolvido. Alguns minutos após a adição de TIN, uma precipitação 72
abundante foi observada e deixou-se reagir por 30 minutos. A
suspensão foi filtrada em uma pequena coluna de sílica (10 cm), eluindo-se com CH 2Cb
(100 mL). A solução foi lavada com água, solução saturada de NaCl e seca sob MgS04 • O
solvente foi evaporado à pressão reduzida e o produto bruto purificado em coluna
gradiente de sílica flash (200-400 mesh), eluindo-se com hexanojAcOEt (7:3). Foram
obtidas 0,202 g de 72 (0,961 mmol; 63%) como um líquido fluido incolor.
lH-RMN (300 MHz, 8): 0,86 (s, 3H - HCll ); 1,15 (s, 3H - HC13 ); 1,24-1,33 (m, 4H - HC2
+ HC3 + HeqC6 +HeqC9 ); 1,45-1,77 (m, 4H - H'C2 + HC4 + HCs); 1,79-1,95 (m, lH -
H 'C3 ); 1,87 (dd, J= 13,6 e 9,2Hz, lH - HaxC9 ); 2,07 (dd, J= 14,0 e 11,5 Hz, lH -
HaxC6 ); 3,23 (ddd, J= 9,3; 4,8 e 1,9 Hz, lH - HCs); 3,33 (s, 3H - OCH3 ); 3,55-3,57 (m,
lH - HC1 ).
13C-RMN (75 MHz, 8): 76,0 (C1); 26,7 (C2); 14,2 (C3); 27,2 (C4); 33,8 (Cs); 32,5 (C6);
70,2 (C7); 80,7 (Ca) ; 42,4 (C9); 33,8 (ClO); 22,0 (C11)*; 23,5 (C12)*. *os sinais podem
estar trocados entre si.
IVfi1me (v, cm'l): 1443, 1463.
M/Z: 210 (M+, 45%); 94 (100%).
CHN: calculado (C 74.24; H 10.54), obtido (C 74,03; H 10,72).
91

Parte Experimental
6.2.6. Preparação do cis-4a-metil-l,2,3,4,4a,5,8,8a-octa-hidronafta leno (73)
11 9 A uma solução da cetona 67 (0,891 g; 5,00 mmol) em trietilenoglicol
$ (5 mL) foi adicionado NH,NH,.H,O (1,25 mL; 25,0 mmol). A solução
4 H 6 12 resultante foi refluxada a 130°C por 1,5 horas. A mistura reacional foi 73
resfriada para então se adicionar KOH em pastilhas (1,67 g; 30,0
mmol) e depois refluxada a 130°C por 4,5 horas. A mistura reacional foi vertida em gelo
e acidificada com HCI 6M. A fase aquosa foi extraída com pentano (3 X 50 mL) e a fase
orgânica foi lavada com solução saturada de cloreto de sódio, seca com MgS04 e o
solvente foi evaporado à pressão reduzida. O produto bruto foi purificado em coluna
cromatográfica de sílica (80-230 mesh), eluindo-se com pentano, rendendo 0,696 g de
73 (4,24 mmol; 85%) como um líquido incolor.
lH-RMN (200 MHz, 8): 0,87 (s, 3H); 1,13-1,70 (m, 8H); 1,67 (s, 3H); 2,18-2,39 (m, 2H);
5,24 (si, 1H).
13C-RMN (50 MHz, 8): 22,4; 23,7; 26,4; 28,S; 29,5; 31,3; 31,5; 34,8; 39,0; 40,5; 118,4;
130,5.
6.2.7.Preparação da 1-{3a-metil-octa-hidro-inden-2-i1)-etanona (55)
~ A uma solução da olefina 74 em MeOH (3 mL) AcOH (10 gotas)
adicionou-se TTN.3H 20 (0,25 g; 0,56 mmol) e este foi prontamente
H 55
dissolvido. Deixou-se reagir por 2 horas e a suspensão foi filtrada, sobre
pressão, em uma pequena coluna de sílica (10 cm), eluindo-se com CH 2C1 2 (100 mL). A
solução foi lavada com água, solução saturada de NaCl e secada com MgS04 • O solvente
foi evaporado à pressão reduzida e o produto bruto purificado em coluna gradiente de
sílica flash (200-400 mesh), eluindo-se com AcOEtjhexano (10-30%). Foram obtidas
40,4 mg de 55 (0,224 mmol; 40%), como uma mistura 1: 1 (cis:trans), na forma de um
líquido incolor.
92

Parte Experimental
6.3.Baquenolida-A
G.3.1.preparação do éster G,8a-dimetil-l,2,3,4,4a,5,8,8a-octa-hidronaftalen-l-iI
tolueno sulfônico (80)
o I I
A uma solução do álcool 71 (0,754 g; 4,18 mmol) em piridina
)JS"-O '-':: 11 _ 11 I O : 9 8
~m 4 H 6 12
seca (13 mL) foi adicionado, sob agitação e atmosfera de
nitrogênio, cloreto de tosila (1,19 g; 6,27 mmol). A solução foi
agitada por 24 horas e a mistura diluída com água gelada. A 80
fase aquosa foi extraída com éter e os extratos orgânicos
combinados foram lavados com solução saturada de CUS04, água, solução saturada de
cloreto de sódio, e secada com MgS04. O solvente foi evaporado à pressão reduzida e o
produto bruto purificado em coluna de sílica flash (200-400 mesh), eluindo-se com uma
mistura hexano/acetato (4:1), rendendo 0,866 g de 80 (2,59 mmol, 62%) na forma de
um sólido branco.
lH-RMN (200 MHz, 8): 0,82 (s, 3H - HC11 ); 1,2-1,89 (m, 9H - HC2 + HC3 + HC4 + HCs +
HC6 ); 1,60 (s, 3H - HC12 ); 2,1-2,3 (m, 2H - HCg ); 2,43 (s, 3H - HCPh); 4,33 (dd, J= 10,8
e 5,5 Hz, 1H - HC1 ); 5,21 (si, 1H - HCs); 7,3 e 7,8 (4H - grupo tosila).
13C-RMN (50 MHz, 8): 90,1 (C1); 33,8 (C2); 23,2 (C3)*; 26,4 (C4)**; 40,1 (Cs); 28,1
(C6)**; 130,2 (C7); 117,1 (Cs); 28,3 (Cg)**; 36,6 (ClO); 23,4 (Cll )*; 23,7 (C12)*;
21,5/127,6/ 129,6/135,0/144,2 (grupo tosila). *os sinais podem estar trocados entre si. **05
sinais podem estar trocados entre si.
93

Parte Experimental
6.3.2. Preparação do 6,8a-dimetil-l,2,3,4,4a, 7 ,8,8a-ocata-hidro-l, 7-
metanonaftalen-9-ol (86)
3
8 7
12
A uma solução do cloreto de (metóximetil)-trifenilfosfônio (1,03 g;
3,00 mmol) em THF (10 mL) foi adicionada, a -30°C e sob nitrogênio,
uma solução 0,5 M de KHMDS em tolueno (5,0 mL; 2,50 mmol). A
solução de cor vermelho intenso foi agitada a O °C por 15 minutos,
para então se adicionar uma solução da cetona 67 (0,178 g; 1,00 mmol) em THF (2,5
mL). A mistura reacional foi agitada por 19 horas à temperatura ambiente e para então
se adicionar, a O °C, uma solução 1: 1 de MeOHjTHF (2,5 mL) e HCI 4N (2,5 mL). A
solução foi agitada à temperatura ambiente por mais 24 horas e depois vertida em água
(10 mL) e a fase aquosa foi extraída com éter (4 x 30 mL). A fase etérea foi lavada com
água, solução saturada de cloreto de sódio, e secada com MgS04 . O solvente foi
evaporado à pressão reduzida e o produto bruto purificado em coluna de sílica flash
(200-400 mesh), eluindo-se com uma mistura hexanojacetato (4: 1), rendendo 0,0708 9
de 86 (0,368 mmol, 37%) na forma de um óleo incolor.
lH-RMN (200 MHz, 8): 1,21 (s, 3H - HCll ); 1,2-2,1 (m, 12 H - HC1 + HC2 + HC3 + HC4 +
HCs + HCs+ HC9 + OH); 1,73 (t, J=1,3 Hz, 3H - HC12 ); 3,70 (51, lH - HC13 ); 4,90 (51, lH
- HC6 ).
13C-RMN (50 MHz,8): 49,9 (C1); 22,8 (C2)*; 16,7 (C3); 26,2 (C4)*; 42,0 (Cs); 124,7
(C6); 139,1 (C7); 55,9 (Cs); 43,7 (C9); 40,3 (ClO); 22,3 (Cll )**; 24,5 (C12)**. *os sinais
podem estar trocados entre si. **os sinais podem estar trocados entre si.
94

Parte Experimental
6.3.3.Preparação do 8a-metil-l,2,3,4,4a,5,8,8a-octa-hidronaftaleno-l-
carbaldeído (85)
2b H 85
A uma solução do cloreto de (metóximetil)-trifenilfosfônio (3,09 g; 9,00
mmol) em THF (20 mL) foi adicionado, a -30°C e sob nitrigênio, solução
0,5 M de KHMDS em tolueno (15,0 mL; 7,5 mmol). A solução de coloração
vermelha intensa foi agitada a O °C por 15 minutos, para então se adicionar
uma solução da cetona 52 (0,493 g; 3,00 mmol) em THF (2,5 mL). A mistura reacional
foi agitada por 19 horas à temperatura ambiente e foi então adicionada, a O °C, uma
solução 1: 1 de MeOHjTHF (7,5 mL) e HCI 4N (7,5 mL). A solução foi agitada à
temperatura ambiente por 24 horas e depois vertida em água (10 mL) e a fase aquosa
foi extraída com éter (4 x 50 mL). A fase etérea foi lavada com água, solução saturada
de cloreto de sódio, e secada com MgS04 • O solvente foi evaporado e o produto bruto
purificado por destilação horizontal (0,40 mmHg; /V150 0c), rendendo 0,485 g de 85
(2,72 mmol, 91%) na forma de um óleo incolor.
lH-RMN (200 MHz, 8): 1,14 (s, 3H); 1,33-3,48 (m, 13H); 5,47-5,61 (m, 2H); 9,81 (d, J=
3,8 Hz).
6.3.4.Preparação da 2,3-dimetilciclo-hexanona60
Preparação do Reagente de Jones:a 25,0 g de er03, mantidos em banho de gelo, foram
adicionados vagarosamente e sob agitação 22 mL de H2S04 concentrado. A solução
obtida foi diluída a 200 mL com água destilada. O reagente é estável e pode ser estocado
em vidro âmbar por tempo indeterminado.
~ Dissolveu-se 2,3-dimetilciclo-hexanol (40,1 g; 0,31 moi) em acetona (150
Ur mL). Resfriou-se a solução em banho de gelo e titulou-se lentamente, sob
O agitação, com a solução do reagente de Jones (/V80 mL). Após a adição, a
mistura foi agitada por 15 minutos e o excesso de oxidante foi destruído com
95

Parte Experimental
isopropanol. A mistura foi particionada em H20 e AcOEt e a fase orgânica lavada com
água, solução saturada de NaHC03 , solução saturada de NaCI e secada com MgS04 • O
solvente foi evaporado à pressão reduzida e o produto bruto destilado em trompa de
vácuo (PE: 79-80 0c), obtendo-se 31,15 g da 2,3-dimetilcicloexanona (0,25 moi; 80%)
como um óleo incolor.
6.3.S.Preparação da (±)-cis-S,10-0imetil-l(9)-octal-2-ona (90): Método de
Zoretic83
fuo
A uma solução de 2.3-dimetilciclo-hexanona (19,0 g; 0,15 moi) e
metilvinilcetona (0,15 moi; 12,1 mL) em benzeno (100 mL), resfriada a O
90 DC, foram adicionados, sob agitação mecânica, H2S04 concentrado (3 mL)
Deixou-se sob agitação por 2 horas à mesma temperatura e adicionou-se então uma
segunda porção de MVK (0,075 moi; 6,1 mL) e H2S04 concentrado (1 mL). Agitou-se por
mais 2 horas e adicionou-se uma porção final de MVK (0,075 moi; 6,1 mL). Agitou-se por
mais 12 horas a O°e. A mistura reacional foi diluída em éter (400 mL) e o material
polimérico foi lavado com 2 porções de 200 mL de éter. A fase etérea foi lavada com
NaOH 1N (200 mL) e com solução saturada de NaCI (2 x 200 mL). A fase aquosa foi re-
extraída com éter (2 x 200 mL) e as fases etéreas foram secadas com MgS04 • O solvente
foi evaporado à pressão reduzida e a 2,3-dimetilcicloexanona que não reagiu foi
recuperada (5,2 g) através de destilação em trompa de vácuo. O restante foi destilado à
pressão reduzida (75-100 °C, 0,4 mmHg), fornecendo 6,75 g. Foi preparada uma solução
de metóxido de sódio (1,1 g em 100 mL de MeOH anidro) e adicionou-se ao destilado. A
solução foi refluxada por 1 hora entre 45-50 0e. Resfriou-se a solução, adicionou-se água
e a fase orgânica foi extraída com éter, lavada com solução saturada de NaCI e secada
com MgS04 • O solvente foi evaporado à pressão reduzida, e o produto bruto destilado à
pressão reduzida (100-108 D C, 1 mmHg), fornecendo 6,15 g de 90 (0,035 moi, 23%)
como um óleo incolor.
96

Parte Experimental
6.3.6.Preparação da (±)-cis-S,10-Dimetil-l(9)-octal-2-ona (90): Método de
Duhamel adaptado85
ct~ O
93
Primeira parte:
A uma solução contendo 2,3-dimetilciclo-hexanona (10,1 g; 0,08 mmol)
e l,l,l,3,3,3-hexametildissilazano (19,8 g; 0,12 moi) em CH 2Cb anidro
(200 mL), resfriada à O°C e sob nitrogênio, foi vagarosamente
adicionado iodotrimetilsilano (24,2 g; 0,12 moi). A mistura foi agitada por 10 minutos a
OOC e 1,5 horas à temperatura ambiente. A mistura reacional foi diluída em hexano e
filtrada sob Florisil (60-100 mesh). O solvente foi evaporado, rendendo 15,7 9 de uma
mistura de sililenoléteres. Esta massa bruta foi dissolvida em CH 2C1 2 anidro (80 mL) e
adicionou-se CH)N02 (9,66 g; 0,16 moi), para então ser resfriada a -78°e. Adicionou-se
então uma mistura de MVK (11,06 g; 0,16 moi) e isopropanol (9,50 g; 0,16 moi) e
agitou-se por 30 minutos. A esta solução foi adicionado, vagarosamente, BF).Et20 (11,23
g; 0,079 moi). Elevou-se a temperatura para -65°C e agitou-se por mais 2 horas.
Retirou-se o banho refrigerante e adicionou-se solução saturada de NaHCO) (80 mL). A
fase orgânica foi extraída com CH 2C1 2 (3 x 100 mL), lavada com água, solução saturada
de NaCI e saca sob MgS04. O solvente foi evaporado à pressão reduzida e o produto
bruto destilado a pressão reduzida (103-110°C, 0,8 mmg), rendendo 7,51g de 93 (38,26
mmol; 48%) como um óleo incolor.
lH-RMN (200 MHz,8): 0,92 (d J=6,6Hz, 3H); 1,02 (s, 3H); 1,51-2,60 (m, 11H); 2,16 (s,
3H).
13C-RMN (50 MHz,8): 15,4; 18,6; 24,5; 29,1; 29,4; 29,9; 38,3; 38,8; 51,4; 208,8;
215,5.
97

Parte Experimental
Segunda Parte:
fuo
Sódio metálico (0,98 g; 42,S mmol) foi dissolvido em metanol anidro
(140 mL). A esta solução foi adicionada a dicetona 93 (7,51 g; 38,3). A
90 mistura foi refluxada por 1 hora a 45°C, para então se adicionar solução
saturada de NaCI (140 mL). A fase orgânica foi extraída com éter etílico e secada com
MgS04 • O solvente foi evaporado à pressão reduzida e o produto bruto destilado à
pressão reduzida (108-112°C, 1,1 mmHg), fornecendo 4,35 g de 90 (24,4 mmol; 64%)
como um óleo incolor.
lH-RMN (200 MHz,8): 0,92 (d J=5,7 Hz, 3H); 1,11 (s, 3H); 1,31-2,47 (m, 11 H); 5,73
(s, 1H).
13C-RMN (50 MHz,8): 15,2; 16,0; 26,6; 30,S; 33,4; 34,0; 35,6; 39,0; 43,2; 124,0;
171,2; 199,5.
6.3.7. Preparação da (±)-cis-S,10-Dimetil-l(9)-octal-2-ona (90): Método de
Piers adaptado8o,94
Primeira Parte:
"N 96 I
~
A uma suspensão de NaOMe - preparado a partir da evaporação de uma
solução de sódio metálico (6,6 g; 0,28 moi) e MeOH (85 mL) - em benzeno
o seco (160 mL), foi adicionada, em banho de gelo, gota a gota e sob
agitação mecânica, 2,3-dimetilmetilciclo-hexanona (14,1 g; 0,11 moi). Após
10 minutos foi adicionado, gota a gota, formiato de etila (14,1 g; 15,3 mL; 0,19 moi). A
mistura reacional foi agita por 14 h a temperatura ambiente para então se adicionar água
gelada (150 mL). A fase orgânica foi separada e foi lavada com NaOH a 10%. Os
extratos aquosos foram neutralizados com HCI 6N e extraídos com éter etílico (3 X 150
mL). Os extratos orgânicos combinados foram lavados com água, solução saturada de
NaCI e secados com MgS04 • O solvente foi evaporado à pressão reduzida. O resíduo foi
dissolvido em benzeno seco (190 mL) e a esta solução foi adicionada N-metilanilina (11,8
98

Parte Experimental
g; 12,0 mL, 0,11 moi), para então se refluxar por 3,5 h em um aparelho de Dean-Stark.
o solvente foi evaporado e o' produto bruto destilado à pressão reduzida (PE: 158-160
CC; 0,6 mmHg), rendendo 19,6 g de 96 (0,081 moi; 73%), na forma de um óleo
alaranjado viscoso.
Segunda Parte:
(tC02H
A uma suspensão de t-ButOK (43,8 g; 0,39 moi) em t-ButOH (330 mL)
foi adicionada a enamina 96 (32,2 g; 0,13 moi). A solução resultante
97 foi agitada por 20 minutos para então se adicionar, gota a gota e sob
atmosfera de N2 , 3-bromopropionato de etila (51,2 g; 36,0 mL; 0,28 moi). A reação é
bastante exotérmica! A solução resultante foi agitada por 2 h a temperatura ambiente e
depois concentrada à pressão reduzida. O resíduo foi vertido em água/gelO e a fase
aquosa foi extraída com éter, lavada com água, solução saturada de NaCI e secada com
MgS04 . O solvente foi evaporado à pressão reduzida e o resíduo refluxado por 30
minutos com HCI a 10% (150 mL). A fase aquosa foi extraída com éter. O solvente foi
evaporado e o resíduo tratado com NaOH a 10% (150 mL) e a solução resultante
refluxada por 5 h. A solução foi resfriada e acidificada com HCI 6N e extraída com éter. A
fase orgânica foi lavada com água, solução saturada de NaCl e secada com MgS04 • O
solvente foi evaporado à pressão reduzida e o produto bruto destilado à pressão reduzida
(PE: 160-165 cC; 1,0 mmHg) rendendo 20,1 g de 97 (0,10 moi; 78%), na forma de um
óleo incolor viscoso.
Terceira Parte:
Coo A uma solução do ácido 97 (20,1 g; 0,101 moi) em anidrido acético,
recém destilado (100 mL), foi adicionado acetato de sódio (4,1 g; 0,030
98 moi). A solução resultante foi refluxada por 2 h e o anidrido acético
destilado em trompa de vácuo. O resíduo foi particionado entre água e éter. A fase
orgânica foi lavada com solução saturada de NaHC03 , solução saturada de NaCl, seca da
com MgS04 , e o solvente foi rotoevaporado. O produto bruto foi destilado à pressão
99

G.\ ~.
BIBLIOT ECA INSTITIITO DE QUíMICA
Umversi,dade de São Paulo
Parte Experimental
reduzida (PE: 110-114 °C; 1,0 mmHg) rendendo 13,5 9 de uma mistura 6:1 das enol
lactonas epiméricas 98 e 98'- (75,0 mmol; 75%). A mistura foi recristalizada de hexano
gelado, fornecendo 8,40 9 de 98 (46,6 mmol, 62%), na forma de um sólido branco (PF:
47,0 - 47,1) .
l H-RMN (300 MHz, õ): 0,95 (d, J= 6,6 Hz, 3H); 1,04 (s, 3H); 1,47-1,68 (m, 4H); 1,84-
1,92 (m, 1H); 2,01-2,14 (m, 2H); 2,61-2,66 (m, 2H); 5,28 (t, J= 4,2 Hz) .
13C-RMN (75 MHz, õ): 15,0; 17,2; 22,8; 26,2; 27,6; 30,7; 34,8; 39,1; 106,3; 154,5;
168,5.
Quarta Parte:
A uma solução da enol lactona 98 (1,04 g; 5,77 mmol) em éter seco (10
fuo mL) à -20°C foi rapidamente adicionado MeU l,6M (5,8 mL; 9,3 mmol).
90 A solução resultante foi agitada à temperatura ambiente por 1,5 h e
~ então vertida em HCI 1N (5 mL). A fase aquosa foi extraída com éter e concentrada, para
~ '\l . então se adicionar KOH aquoso 2,5N (6 mL) e MeOH (54 mL). A mistura foi refluxada por
2 h, o solvente foi evaporado à pressão reduzida e o resíduo particionado entre água e
éter. A fase orgânica foi lavada com água, solução saturada de NaCl, secada com MgS04 ,
e o solvente foi evaporado à presão reduzida. O produto bruto foi purificado em coluna
de sílica flash (200-400 mesh) eluindo-se com uma mistura hexanojacetato 7:3,
rendendo 0,613 9 de 90 (3,44 mmol; 60%) na forma de um óleo incolor.
6.3.S.Preparação do 7,7a-Dimetil-2,4,S,6,7,7a-1H-indeno-2-carboxilato de
metila (91)102
C±:> ''' CO,Me
91
A uma solução de 90 (208,8 mg; 1,17 mmol) em TMOFjMeOH (4:3)
(7 mL) adicionou-se TTN (0,62 g; 1,41 mmol) e este foi
prontamente dissolvido. 2 minutos após a adição de TTN observou-
se uma precipitação abundante e deixou-se reagir por 30 minutos. A suspensão foi
100

Parte Experimental
filtrada em uma pequena coluna de sílica (10 cm), eluindo-se com CH 2CI 2 (100 ml). A
solução foi lavada com água, solução saturada de NaCl e secada com MgS04 • O solvente
foi evaporado à pressão reduzida e o produto bruto purificado em coluna gradiente de
sílica flash (230-400 mesh), eluindo-se com AcOEt/hexano (0-20%). Foram obtidas 88,8
mg de 91 (0,43 mmol; 36%) como um óleo incolor. Em um procedimento análogo,
empregando apenas TMOF como solvente, 91 foi obtido em 38% de rendimento.
lH-RMN (200 MHz,8): 0,84 (d J=6,lHz, 3H); 0,91 (s, 3H); 1,12-1,46 (m, 4H); 1,71-2,10
(m, 4H); 2,27-2,33 (m, 1H); 3,51-3,55 (m, 1H); 3,67 (s, 3H).
13C-RMN (50 MHz,8):16,6; 17,0; 25,8; 26,7; 30,4; 42,9; 44,1; 47,6; 49,6; 51,7; 117,1;
153,7; 176,1.
6.3.9.Preparação da 2-metil-1,3-ciclo-hexanodiona1os
& Uma mistura de KOH (30,0 g; 0,53 mal) e 1,3-cicloexanodiona (60,0 g; 0,535
mal) em solução aquosa de metanol a 50% (180 mL) foi refluxada até
dissolução total da cetona. Após resfriamento foi adicionado iodeto de meti la
(88,8 g; 0,62 mal) e a solução foi refluxada "overnight". A mistura reacional foi resfriada
e o precipitado formado foi filtrado. O filtrado foi evaporado à pressão reduzida e os dois
resíduos foram dissolvidos em solução aquosa de NaOH a 3% (540 mL). A solução
resultante foi lavada com éter (2 x 15 mL) e acidulada com HCI 4N até pH 4. O
precipitado foi filtrado e recristalizado de metanol a quente, e secado a vácuo, para
render 33,7 9 da 2-metil-1,3-cicoexanodiona (0,267 mal; 50%).
101

Parte Experimental
6.3.10.Preparação da cetona de Wieland-Miescher (92)106
fuo
Uma mistura da 2-metil-1,3-ciclo-hexanodiona (9,00 g; 71,4 mmol), MVK
(7,50 g; 0,107 moi) e KOH (uma pastilha) foi dissolvida em MeOH seco
92 (30 mL) e refluxada por 3 horas. A solução foi resfriada e o metanol e o
excesso de MVK foram evaporados à pressão reduzida e o resíduo dissolvido em benzeno
seco (30 mL). A solução foi refluxada por 30 minutos em aparelho de Dean-Stark e
depois resfriada à temperatura ambiente, para então se adicionar pirrolidina (1 mL)
refluxar-se por mais 1 hora. Após resfriamento, a solução foi diluída em éter e foi lavada
com HCI 0,15 M. A fase aquosa foi extraída com éter e os extratos orgânicos combinados
foram lavados sucessivamente com água, solução saturada de cloreto de sódio e secados
com MgS04 • O solvente foi evaporado à pressão reduzida e o resíduo filtrado sob sílica
(60-230 mesh), eluindo-se com uma mistura 9: 1 de hexanojacetato. O filtrado foi
evaporado e o produto destilado a pressão reduzida (PE: 120-130 °C; 0,2 mmHg) e
cristalizado de éter, para render 6,34 g de 92 (35,7 mmol; 50%), como um sólido
branco.
6.3.11. Prepa ração da S-j3-h idróxi-4a-meti 1-4,4a,S,6, 7 ,8-hexa-h id ro-3H-naftalen-
2-ona (103)107
fuo 103
A uma solução de 92 (1,78 g; 10,0 mmol) em etanol seco (13 mL), foi
adicionada gota a gota e a O °C, por um período de 45 minutos, uma
solução de boro-hidreto de sódio em etanol (25 mL). A solução foi
agitada por mais 15 minutos e a reação foi interrompida pela adição de algumas gotas de
ácido acético. O solvente foi evaporado à pressão reduzida e o resíduo foi dissolvido em
água. A fase aquosa foi extraída com diclorometano e os extratos orgânicos combinados
foram lavados sucessivamente com água, solução saturada de cloreto de sódio e secado
com MgS04 . O solvente foi evaporado e o produto bruto foi purificado por cromatografia
102

Parte Experimental
em coluna de sílica .flash (200-400 mesh), com eluição gradiente de acetato/hexano (10-
50%), para render 1,20 9 de 103 (6,70 mmol; 67%).
6.3.12 .. Preparação da 10-Metil-l(9)-octal-2-ona (101)112
mo Refluxou-se suavemente uma mistura de 2-metilcicloexanona (29,0 g;
0,26 moi), metilvinilcetona recém destilada (23,3 g; 0,32 moi), H2S04
101 concentrado (0,2 mL) e benzeno seco (70 mL) por 16 horas. A solução
resultante foi resfriada, diluída com 100 mL de hexano, lavada com solução aquosa de
KOH a 5%, solução saturada de NaCl, secada com MgS04 e o solvente foi evaporado à
pressão reduzida. O produto bruto foi destilado à pressão reduzida (80-95°C, 0,6 mmHg)
fornecendo uma mistura de 101 e sua dicetona intermediária. Esta mistura de produtos
foi refluxada por 6 horas em benzeno seco contendo 5 gotas de H2S04 concentrado.
Seguiu-se o mesmo work-up descrito acima e o produto foi destilado à pressão reduzida
(77-80 0 C, 0,5 mmHg), fornecendo 17,0 9 (0,10 moi, 40%) de 101 como um óleo
incolor.
lH-RMN (300 MHz,8): 1,25 (s, 3H); 1,30-1,47 (m, 2H); 1,64-1,95 (m, 6H); 2,21-2,57
(m, 4H); 5,72 (m, 1H).
13C-RMN (75 MHz, 8) : 21,7; 22,0; 27,1; 32,7; 33,9; 35,9; 38,0; 41,S; 124,0; 170,4;
199,5.
6.3.13.Preparação do 4a-Metil-l,2,3,4,4a,S-hexa-hidronaftaleno (105)
cb A uma solução de 10-metil-1(9)-octal-2-ona (106) (3,29 g; 20,0 mmol) em
metanol (100 mL) foi adicionado boro-hidreto de sódio (0,76 g; 20,0 mmol) .
105 A solução foi agitada por 1 hora e adicionaram-se 100 mL de água. A fase
orgânica foi extraída com acetato de etila, lavada com solução saturada de NaCI e secada
103

Parte Experimental
com MgS04 • O solvente foi evaporado à pressão reduzida e o resíduo dissolvido em THF
(30 mL). Adicionou-se H3P04 85% (5 mL) e refluxou-se por 1 hora . Após resfriamento a
mistura foi neutralizada com NaHC03 sólido e a fase orgânica foi extraída com acetato de
etila, lavada com água, solução saturada de NaCI e secada com MgS04 • O solvente foi
evaporado à pressão reduzida , fornecendo um óleo levemente amarelado que foi filtrado
numa coluna curta de sílica (60-200 mesh), eluindo-se com hexano, fornecendo então
2,49 9 de 105 (16,8 mmol; 84%) como um óleo incolor.
lH-RMN (200 MHz,o) : 1,01 (s, 3H); 1,26-2,15 (m, 10H); 5,37-5,41 (m, 1H); 5,57-5,61
(m, 1H); 5,91-5,96 (M, 1H).
13C- RMN (50 MHz,o): 18,5; 23,1; 23,2; 25,7; 32,3; 37,0; 37,6; 123,3; 125,7; 128,8;
140,9.
6.4.Resolução Enzimática
6.4.1.Preparação do 8a-metil-l,2,3,4,4a,5,8,8a-octa-hidronaftalen-l-ol (58)
QH11 A uma solução da octalona 52 (1,00 g; 6,11 mmol) em MeOH (30 mL)
2~ 8 mantida a -78°C foi adicionado NaBH4 (0,23 g; 6,11 mmol) sólido. A 3~7
4 H 6 suspensão fo i agitada por uma hora para então se adicionar água (30 58
mL). A fase aquosa foi extraída com AcOEt (3 x 30 mL), lavada com
solução saturada de NaCl e seca sob MgS04 . O solvente foi evaporado à pressão
reduzida, para render 0,962 9 de 58 (5,79 mmol; 95%) na forma de um óleo incolor.
lH-RMN (200 MHz, ppm): 1,05 (s, 3H); 1,22-1,77 (m, 10H); 2,13-2,39 (m, 2H); 3,31
(dd, J= 10,7 e 4,6 Hz, 1H); 5,50-5,64 (m, 2H) .
104

Parte Experimental
6.4.2.Preparação do éster 8a-metil-l,2,3,4,4a,5,8,8a-octa-hidronaftalen-l-il
acético (108)
o A uma solução do álcool 58 (0,332 g; 2,02 mmol) e DMAP (0,049 g;
13~? 11 9 0,404 mmol) em trietilamina (2 mL), sob atmosfera de nitrogênio, foi
2~ 8 adicionado o anidrido acético (0,57 mL; O,62g; 6,02 mmol). A reação 3lJK.)7 4 H 6 foi agitada por 30 minutos a temperatura ambiente para então se
108 adicionar metanol (2 mL) . O solvente foi evaporado e o resíduo particionado entre água e
acetato de etila. A fase orgânica foi lavada com solução saturada de cloreto de sódio,
seca sob MgS04 e o solvente foi evaporado . O produto bruto foi purificado em coluna de
sílica flash (200-400 mesh), eluindo-se com uma mistura hexanojacetato (85%),
rendendo 0,352 9 de 108 (1,69 mmol; 84%) como um óleo incolor.
lH-RMN (200 MHz, ppm): 0,93 (s, 3H); 1,24-1,82 (m, 9H); 2,27-2,39 (m, 2H); 4,56 (dd,
J= 10,5 e 4,8 Hz); 5,50-5,62 (m, 2H)
6.4.3. Preparação do éster 6,8a-dimetil-l, 2,3,4,4a,5,8,8a-octa-hidronaftalen-l-iI
acético (109)
o
14~º 11 m9
1 10 I 8
3 57
4 H 6 12
A uma solução do álcool 71 (0,578 g; 3,21 mmol) e DMAP (0,078
g; 0,642 mmol) em trietilamina (3,5 mL), sob atmosfera de
nitrogênio, foi adicionado o anidrido acético (0,90 mL; 0,98 g; 9,63
mmol). A reação foi agitada por 30 minutos a temperatura 109
ambiente para então se adicionar metanol (2 mL) . O solvente foi
evaporado e o resíduo particionado entre água e acetato de etila. A fase orgânica foi
lavada com solução saturada de cloreto de sódio, secada com MgS04 e o solvente foi
evaporado. O produto bruto foi purificado em coluna de sílica flash (200-400 mesh),
eluindo-se com uma mistura hexanojacetato (85%), rendendo 0,595 g de 109 (2,68
mmol; 83%) como um óleo incolor.
105

Parte Experimental
lH-RMN (300 MHz, 8):0,89 (s, 3H - HCll ); 1,2-1,8 (m,9H - HC2 + HC3 + HC4 + HCs +
HC6 ); 2,04 (s, 3H - HC12 ); 2,25-2,32 (m, 2H - HCg ); 4,58 (dd, J= 11,2 e 4,6 Hz, 1H -
HC1); 5,22-5,27 (m, 1H - HCa).
13C-RMN (75 MHz, 8): 80,1 (Cl ); 33,9 (C2); 23,7 (C3)*; 26,9 (C4)**; 39,9 (Cs); 28,6
(C6)**; 130,5 (C7); 117,5 (Ca); 27,0 (Cg)**; 35,8 (C10); 23,3 (Cll )*; 23,6 (C12)*; 170,9
(C13); 21,2 (C14). *os sinais podem estar trocados entre si. **os sinais podem estar trocados
entre si.
IVfi1me (v, cm- 1): 1736
6.4.4. Preparação da cis-3,4,4a,S,S,Sa-hexa-hidro-6, 7,Sa-trimetilnaftalen-1-
(2H)-ona (106)
cjx A uma suspensão de AICb (0,40 g; 7,50 mmol) em benzeno seco (20 mL),
H 106
em banho de gelo, foi adicionada a 2-metilciclo-hexen-2-ona (1,10 g;
10,0 moi) e houve a formação de uma pasta no fundo do balão.
Adicionou-se uma ponta de espátula de hidroquinona e 2,3-dimetil-1,3-
butadieno (3,28g; 40,0 mmol) . O balão foi tampado, o banho de gelo foi retirado, e a
solução foi agitada a temperatura ambiente por 24 horas. O solvente foi removido à
pressão reduzida e o resíduo foi particionado entre água e éter. A fase etérea foi lavada
com água, solução saturada de NaCl, secada com MgS04 , e o solvente foi evaporado à
pressão reduzida. O produto bruto foi purificado em coluna de sílica flash (200-400
mesh), eluindo-se com uma mistura hexano/acetato (85%), rendendo 0,649 g de 106
(3,64 mmol; 36%) como um óleo incolor.
lH-RMN (200 MHz,8): 1,09 (s, 3H); 1,47-2,63 (m, 11H); 1,62 (s, 6H).
13C-RMN (50 MHz,8): 18,9; 19,0; 20,6; 25,1; 27,7; 34,7; 37,2; 37,6; 41,6; 48,S; 121,3;
122,5; 215,6.
106

Parte Experimental
6.4.S.Preparação do 1,2,3,4,4a,S,8,8a-octa-hidro-6,7,8a-trimetilnaftalen-l-01
(107)
OH
dx A uma solução da octalona 106 (0,310 g; 1,74 mmol) em MeOH (9 mL)
mantida a -78°C foi adicionado NaBH4 (46,0 mg; 1J4 mmol) sólido . A
H 107
suspensão foi agitada por uma hora para então se adicionar água (30
mL). A fase aquosa foi extraída com AcOEt (3 x 30 mL), lavada com
solução saturada de NaCI e secada com MgS04 • O solvente foi evaporado à pressão
reduzida, para render 0,292 9 de 107 (1,62 mmol; 95%) na forma de um óleo incolor.
6.4.6.Preparação do éster 1,2,3,4,4a,S,8,8a-octa-hidro-6,7,8a-trimetilnaftalen-
l-iI acético (110)
O
15~ct0t? 11 9 8 13 2 1 10 I 7
3 5 12 4 H 6
110
A uma solução do álcool 107 (0,121 g; 0,67 mmol) e DMAP (0,016
g; 0,135 mmol) em trietilamina (1 mL), sob atmosfera de
nitrogênio, foi adicionado o anidrido acético (0,19 mL; 0,21 g;
2,01 mmol). A reação foi agitada por 30 minutos a temperatura
ambiente para então se adicionar metanol (1 mL) . O solvente foi evaporado e o resíduo
particionado entre água e acetato de etila. A fase orgânica foi lavada com solução
saturada de cloreto de sódio, secada com MgS04 e o solvente foi evaporado. O produto
bruto foi purificado em coluna de sílica flash (200-400 mesh), eluindo-se com uma
mistura hexanojacetato (85%), rendendo 0,144 9 de 110 (0,61 mmol; 91%) como um
óleo incolor.
107

Parte Experimental
6.4.7.Procedimento Geral para a Resolução Enzimática
A um Erlenmeyer contendo hexano (20 mL, pureza de HPLC), acetato de vinila (1
mL) e Novozyme 435 (200 mg) foi adicionado o álcool apropriado (58 - 0,195 9 ou 71 -
0,280 9 ou 107 - 0,280 g). A mistura reacional foi agitada por 48 h a 40°C (160 rpm)
para então ser filtrada e o solvente ser evaporado à pressão reduzida. O resíduo foi
purificado em coluna de sílica flash (200-400 mesh), eluindo-se com uma mistura
hexanojacetato (85%). Os compostos foram analisados em uma coluna capilar quiral de
(3-ciclodextrina. Condições do CG: injetor - 200°C; detector - 220 cC; pressão - 100 kPa
(lS,4aS,BaR)-Ba-Metil-l,2,3,4,4a,5,B,Ba-octa-hidronaftalen-l-ol (5Ba)
OH
ób H
58a
Forno: 100-180°C, razão l°Cjmin;
Tempo de retenção: $= 25.5 min, R= 26.3 min - e.e.= 98%;
Coluna: CHIRASIL DEX CB;
[0.]D25= +6,6° (c 8.6, CHCI 3).
(1 R,4aR,BaS )-Éster Ba-metil-l,2,3,4,4a,5,B,Ba-octa-hidronaftalen-l-iI acético
(lOBb)
Ób H
108b
Forno: 100-l80°C, razão l°Cjmin;
Tempo de retenção: $= 20.9 min, R = 22.8 min - e.e. = 98%;
Coluna: CHIRASIL DEX CB;
[0.[D25= -24,0° (c 3.25 CHCI3).
108

Parte Experimental
(lS,4aS, BaR)-6, Ba-:Dimeti/-1, 2, 3,4, 4a, 5, B, Ba-octa-hidrona ftalen-1-ol (71a)
~ H
71a
Forno: 100-180°C, razão l°Cjmin;
Tempo de retenção: S= 27.0 min, R= 28 .8 min - e.e. = 98%;
Coluna: CHIRASIL DEX CB;
[a]D2s = -1,6° (c 3.02, CHCI3).
(lR,4aR,BaS)-Éster 6,Ba-dimetil-1,2,3,4,4a,5,B,Ba-octa-hidronaftalen-1-iI
acético (10gb)
AcO
ótl H
109b
Forno: 100-180°C, razão 10°Cjmin;
Tempo de retençao: S= 64.4 min, R= 64.8 min - e.e= 92%;
Coluna: GAMA DEX™ (Supelco);
[a]D2S = - 8,5° (c 2.95, CHCI3).
(lS,4aS,BaR)-1,2,3,4,4a,5,B,Ba-Octa-hidro-6, 7,Ba-trimeti/naftalen-1-ol (10 7a)
QH Forno: 100-150°C, razão 2°Cjmin; crx Tempo de retençao: S~ 23.8 min, R~ 25.1 min - e.e= 98%;
H Coluna: CHIRASIL DEX CB;
107a [a[D2S= -8,0° (c 1.35, CHCI3).
(1 R,4aR,BaS )-Éster 1,2,3,4,4a,5,B,Ba-octa-hidro-6, 7,Ba-trimetilnaftalen-1-il
acético (11 Ob)
AcO
ób: H
110b
Forno: 100-180°C, razão 10°Cjmin;
Tempo de retençao: S= 66.8 min, R= 67.0 min - e.e= 98%;
Coluna: GAMA DEX™ (Supelco);
[a ] D2S = +13,5° (c 2.73, CHCI3) .
109

o I I --------------------------~----------------------------
NWlI ap SOJ:a:>adS3" L

integrol
~ 2.0000 .... 1---__ [
:' ....
... ::t I ~ ~ :' ~ c:.
N Q Q
~ !-> .... ::t
~
~ !-> .? c:.
~ VI !v ....
2.0288
!v c:.
..... J-----
t., 12.132
°lned o!:'s ap apcplSlaMUn
\f:J!~Jlno ::~a OJ.f11I.lSNf
'1:):11011818
-7.2535
j5.6431 5.6277 5.5926 5.5817 5.5400 5.5334 5.5246
-5.4742
~2.39J5 1.3213 1.3016 2.1467 1.2292 1.7070 1.6982 1.6719 1.6346 1.6148 1.5534 1.5227 1.5029 1.4854 1.4678 1.4547 1.4349 1.4174 1.3976 1.3384 1.3252 1.2989 1.2660 1.2353 1.2177 1.1980 1.1585 1.1321 1./146 1.0904
'\:0.8974 ..(}.OOOO
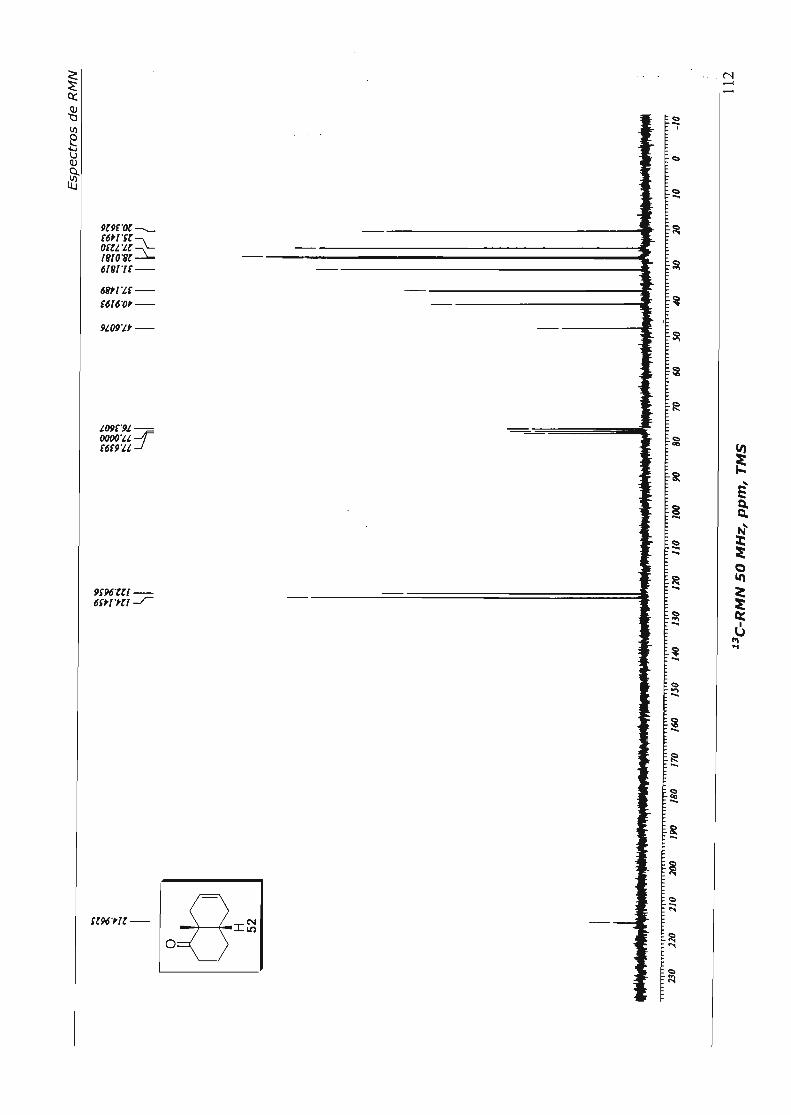
...... ...... N·
-~
~
~
~
~
~
~
~
-c::>
c::>
, -c::>
1
~ ,
~
1
l
~
~
!
,
. ..,
~ ~
t
!'
F--
~
l
~
t-
--
-
~
-
-
-214.9625
-../124.1459 -122.9656
r 77.6393 r77.()()()() -76.36(J7
-47.6076
-40.9/93 -37.1489
-31.18/9 ___ 28.0181 -.. "'-27.7230
'-25.1493 ---20.3626

.. ::t I ~ ~ ~ N o o ~ ::t ,'"
" " 3 ,
~ til
/nt~graJ
~
'"
... Q
!-> ....
... Q
Iv i...
l5.6431 5.6277 5.5926 5.5817 5.5400 5.5334 5.5246
-5.4742
~2.39J5 2.3213 2.3016 2.2467 2.2292 1.7070 1.6982 1.6719 1.6346 1.6148 1.5534 1.5227 1.5029 1.4854 1.4678 1.4547 1.4349 1.4174 1.3976 /.3384 1.3252 1.2989 1.2660 1.2353 1.2177 1.1980 1.1585 1.1321 /./146 10904
C 0:8974 ..().OOOO

..,
~
~
i ~
r-
•
~==-
...
~
~
..;
~
~ to
s= -;
p..
'" , i"
"l
-
-
~I
- 8-
~ L 124.7438 114.1510
í 77.9()93 =:Ir 77.1700 -76.6307
.-r 40.2712 -39.5991
~31.9437
t31.5830 30.()9}3 29.6159
\\1-28.7470 t 26.7471
22.6325

.. "
cjyl H 55
Ol N co N
rumruo_mro ~~V-N~m~mom~~~wmrororn ~mM~N- m~Mru_~~N~oNm~mom_WN~ ~vm~~N~~_m~V~~~OOMNomm~~~mro MMNOOO m~~~~~~~~~ •• MMMMMOlOl
MMMMMMNN 00
~~~~~~
( r
.,./1> j .
f I
I I
I , , I
, I · r !! t I ( I i ! I
! I r I' I 1/.1111 I f i! I' l i r r ! I· I I { ! l f
@~ ~o ~ {!m!o {lm lD~lD~~~~~ -~-o ro~mw~m~v -~-o W~ONruN~M -ru-o rnruruo~mm-. . . . . . . . . . . . 000- N-ru-N~-M
' -- l--r -.----,----,--,-~-r--T- '__r- I I I I I I 1-''' ' " 9 8 765 432 O
lH-RMN 300 MHz, ppm, TMS
Espectros de RMN
Current NAHE EXPNO PROCNO
Data Parameters Tlago
I
F2 - AeQulsit ion Para..eters Date 20040916 Ti~e 9 . 41 INSTRUM .peel
PROBHO 5 ... Dual I3CI PLLPRQG zg30
TO 65536 S<l. vENT COCl3
tIS 16
OS SWH 3378 .378 Hz
FIO~S 0.051550 Hz
AG 9 .6993780 see
RG 80 .6
OW 148 . 000 usee
DE 6 . 00 usee
TE 300 . 0 K
OI I. 00000000 see
........ CHANNEl fi ••••••• -
IUCI IH PI 9 .50 usee PU 3 . 00 dB SFOI 300 . \314218 !tiz
F2 - Proeessinq para..elers SI 32768 SF 300 . 1299991 !tiz WOW EH SSB LB G8 PC
0.30 Hz O
1.00
LO NI4R plot parar.eters CX 23 . 00 ell CY 12 .50 ell FIP 10 . 000 pplII FI 3001.30 Hz F2P -O . 500 pp~ F2 -150 .07 Hz Pl'HCH O. 45652 pp~/e~ HZCH \37 . 0 1587 Hz/ell
115

.-.-
--.
I 22
5
01
<0
C
D
01
0
1 "-
O
O
C\J
C
\J V 20
0
~Mm~M~~MwwmMM~wo_mru~mm
ruo~mO~M~ru~mM~~~Mrn~~~~M
~o~m~ruNO~NMOrn~mm~Mrnru_~
. ..
..
..
...
..
~~~OO~~N-rnçMruOOOWW~~Nruru~
~~~~~~~qMMMMMruruNrurururururu
~~~\~
50
13
C-R
MN
75
MH
z,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cu
rren
t N
AHE
fKPN
O
PROC
NO
Da
ta
Par
alle
ters
T
iaoo
2
AC
Qu\
sHio
n ·P
aral
(!te
rs
200~0916
9.5
0
F2
-o.
t. T
ille
IH
ST
FU
I PR
OOttl
I'U
LPOO
G TO
SO
LVEN
T NS
os
SN
H F

rh~o
Me
~ír
56
O
(Tl .....
tO
(\J
~ruooM~m~mW~OONommoruIDom~w~o
~m~m~w ~M~ _
_ ruMmMwwmo~oo
o ~_OW~VNM~mONN_mmmWVqM~~MO
~~~NruruN_mmw~VV~MMMMMMru_oo
....
.........
~~~
I ~ I! i i
r ' 1/ I ' , I
i I I I } I j
I i I
}:
!
!lI I
I') I
i 1,
r
1I
I,
""
.-.---____
~_J
1 llLtW~ __ ~
I! ..... ~ tO
(\J~
~~~~~~ ..... ~
~~ o
m~~~O~-OMmmo
~
~wMMoruv-Nm-~
o M~ooomNm~ID--
. .
.....
..
..
. .
M
__
M
_
_ O
_v
NN
OO
,-·,
,-r--
-.-,-
,-,-
-,
I UI
I
I I
I T
-r-
'----'
-9
8 7
65
4
32
O
lH-R
MN
30
0 M
Hz,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cu
rren
t D
at.
Par
•••
ter.
NA
ME
r lag
o EX
PNO
4
PROC
HO
I
F2
-A
cqu
lsit
ion
P.r
a..
,ters
O
at.
2004
0917
11
••
7.5
3 IN
STfU
4 sp
eet
PRO
BH
l 5
.. D
ual
13C
/ PU
LPR
QG
Z
930
TO
6553
6 SO
LVEN
T co
el3
NS
32
OS
O
SI
/H
3272
.251
H
z Fl
OR
ES
0.0
4993
\ H
z ~Q
10.0
1395
03 s
ee
RG
32
2.5
OW
15
2. B
OO
use
e DE
6
.00
use
e TE
30
0.0
K
OI
I. 0
0000
000
see
CH
AN
Ifl.
f \
••••
••••
lfJC
I
IH
PI
9.5
0
us.
e
PU
3
.00
dB
SFO
I 30
0.1
3\36
55 !
tiz
F2
-P
roe.
ssin
g p
.r ..
.. t.r
s
SI
3276
8 SF
30
0.13
0003
7 !t
iz
1101
1 EM
SS
B LB
0
.30
Hz
GB
O
PC
1.0
0
\O lH
l p
lot
par
a ...
ter.
CX
2
3.0
0 e~
CY
12.5
0 C
. F
IP
10.0
00 pp~
FI
3001
.30
Hz
F2P
-o. 5
00 pp~
F2
-150
.07
Hz
PPH
CH
O 4
5652
PP~/c~
HZC
H
137
.015
87
Hz/
clI
117
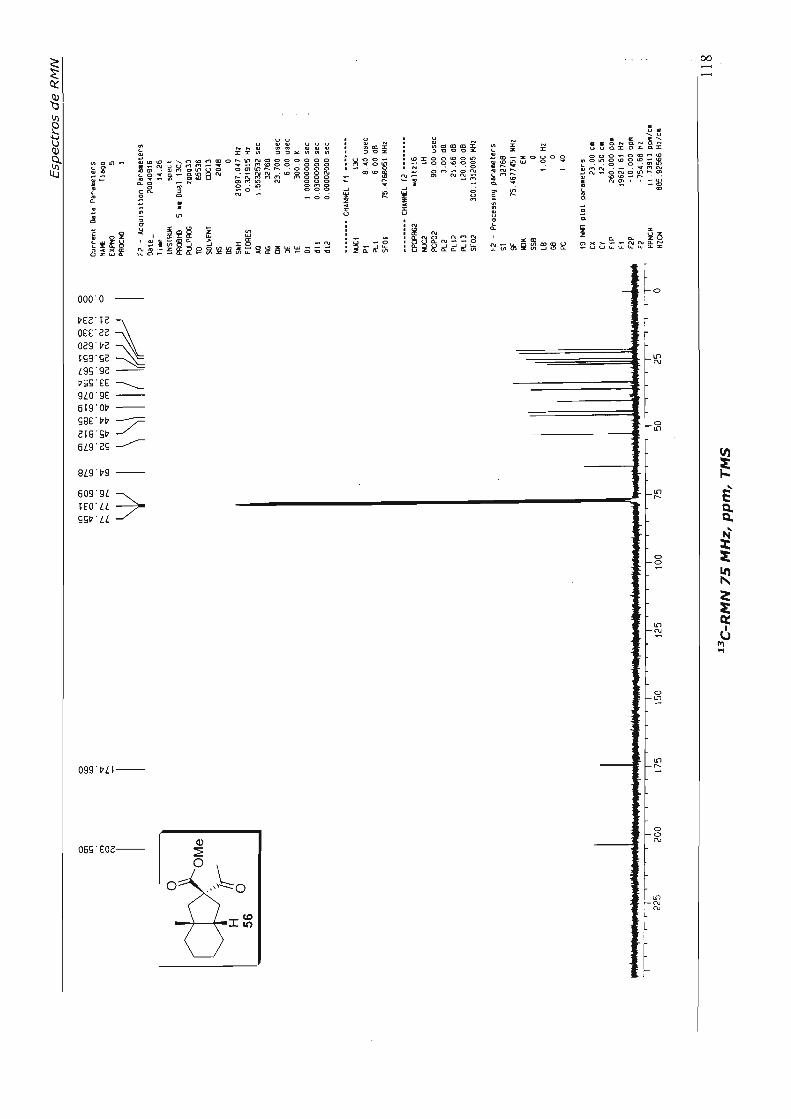
o cn In
f") o
I ~OMe
56 O
o ID ID ... ......
In cn In f") o ... o ID
r-- r-- ID r-- r-- r--
<Xl ...... ID
.... ID
0'1 C\J U'1 m c.D 'Q" ,..... o o .... o ...... CC_"-ll1WlClC\J('T1(T) o c.DCD(T'J(,!)otOlnUlUl(T1(\J o ruUloq-OtDCT1WL!"1'V(\J o t.n'Q'~'q"(T'l(T')ruNC\JNC\J
"V I \ \\ 11 / \~ I
L. .... I I I I I I I I I · ·~ '-'- .--.---."
225 200 175 150 125 100 75 50 25 O
J3C-RMN 75 MHz, ppm, TMS
Espectros de RMN
Current Data Paralleters HAHE Tlago EXPNO 5 PROCNO
F2 - AcQuisit ion Para.eters Date 200_0916
Ti.e 14 . 26 INS lRUH spect PROfltfJ 5 00 Dual IX/ PULPOO:; IOpo30 TO 65536 SOLVENT mCl3
IIS 2048
OS O
SHH 21097 .0H HI FlORES 0 . 321915 HI
AO I .5532532 see RG 32768 OH 23.700 usee
DE 6 . 00 u.ee
TE 300 . O K
OI . 00000000 see
dll O . 03000000 see
dl2 0 . 00002000 see
•••••••• CHAt«'L f I ••••••••
IfJCI \3C PI 8 . 40 usee PU 6 .00 d8 SFOI 75 . 4768051 HHz
•••••••• CHANNEL f 2 •••• • •••
CPIPRG2 •• 11216 IfJC2 IH peP02 90 . 00 usee
PL2 3 .00 d8 Pll2 21 .66 d8
PLI3 120 .00 dB Sf02 300 1312005 HHz
F2 Processing para.eters 51 32768 51' 75 . 4677451 HHz
HeH SSB LB GB PC
EM O
.00 HI
. 40
tO NJr41 plot para.elers CX 23 .00 ca CY 12 .50 eM FIP 260 . 000 ppo
FI 19621 .61 Hz F2P -10 . 000 PP~ F2 -754 .68 Hz PPHCH 11 . 73913 ppN/ eo
HZCH 885 . 92566 Hz /e o
118

rhJ-
oM
e
~ír
56
O
..• ~
'--r-
,---'---"'--~r--
'--r-----r-·_
-"
180
160
C>
In
r- tD
C\J
In
1
.ü
L~
.,.. ."
r-r
1 I
, ,
140
120
100
80
60
DE
PT
-13
5 7
5 M
Hz,
pp
m,
TM
S
....
(T
l _
alL
C1
CO
cn
(T
l
.. r
-cn
,...
...(
T')
C'\
J(\
JN
LC
'J
~
CD
,.
....
.Il1
CD
IllC
\J(T
')(T
) C
D
(Tl
OL
Ol.
OU
ltO
MN
.
. .
. .
. In
...
. c.o
(T
1
tO
lCl
""Q'
" C
\J
......
(TI
(T1
C
\I ('
\J
C\J
C
\J
C\I
\/ \1
\V
~
. ....
, ,
,-~-.-
40
20
o
Esp
ect
ros
de
RM
N
Cu
rren
t C
itA
P
.arM
eter
s H
AtE
. n
AQ
O
EXPI
«J
6 p
roo
oo
f2
-A
CQ
u15k
tlon
P
ar'
lIf:
teN
o
.te
2004
0917
li ..
I'
50
IltS
fRlI
oI
spec
t P
RJ!
t()
5 .. l
lJal
13
C/
P\.L
PRO
G
oopt
l15
TO
6653
6 S<
X. 'r
UIT
C
OC
13
HS
2070
DS
8 S
lti
1798
5.6
11
HI
FlO
lES
0
.27"
39
Hl
AO
l.8
21
95
08
see
AG
32
768
OM
27.8
00 u
sec
DE
6.0
0 us
ec
TE
300
.0
K
CltS
T2
1'5
.000
0000
O
I o .
5000
0000
I te
02
0
.00
3'4
82
8 .
oe
012
0.0
0002
000
see
DEL T
A 0
.000
0107
0 lt
e
••••
•• ••
CIW
f<E
L fi
••
••••
••
tu:
I 13
C P
l 8
.40
use
c p2
16
.80
usec
P
U
6.0
0 d
B
SFO
I 75
. '75
2953
lti
z
••••
••••
CH
AIfE
L f 2
•••
••••
•
CPI
lI'R
G2
..alt
1l6
tu
:2
IH
P3
10.5
0 u
sec
p,
21.0
0 us
ec
PO
'02
90 .
00
UH
C
PL2
3.0
0 0
8
PL
I2
21.6
6
08
SF
02
300
.131
2005
1H
Z
F'2
-P
roce
ss 1
n9 p
ara
. ter.
SI
32
768
Sf
75.4
67
H6
3IH
Z
MIlH
E
" ss
e O
LO
1
.00
Hl
G8
O
PC
1..
0
10 "
"'
plo
t p
.r •• te
rs
ex
23
.00
c.
Cy
6.0
0 e.
FlP
21
0.0
00 p
p.
FI
15
8.a
.23
HI
f2P
-1
0.0
00
pp
. f2
-7
5'.
68 H
z PI
't4CH
9
.585
22
ppo
/e.
HZC
l<
721.
865
36 H
l/e
.
119

U') <D U') <D
'"
I
cp?J 57 O
1 1 8 7
NOlOlC\.lCTI 'q' (Jl ,.... Q)
LClIl1- m c.o U1 C\J N m WtDWlOlLl
~;;
~I
~O~NO_M~MmOOMmwmmONN~WO ~Mm~mo~~mommmONOM-~mo-o N~M~~~mm-NM~MO~Nm~N~~ruo ~ww~~~omwm~ __ oommm~~mo
oommmm~~~~~~~MMM--mo
~~~
nl
lH-RMN 500 MHz, ppm, TMS
Espectros de RMN
Current HAME EXPIlO PROCHO
Data Par.~eter. Hago
2
f2 - AcQulsltlon Par ... ters Date 200.0.15 Ti.. 8 .52 INSTlU4 .peet PROBHO 5 .. UI IX Z Pll.PAOG TO 5a.VENT NS OS SNH fIORES ~Q
RG OH (J::
TE OI
'930 65536 COC13
32
5307 .855 H' 0 .080991 H'
6 . 1735411.ee 161 .3
94.200 u.ee 6 .00 u.ee
300 .0 K I .00000000 .ee
••• ••••• OiAlfEl f I ••••••••
tfJCl IH PI 1\.00 usee PU 0 .00 dB SfOI 500 . \320442 1t1,
f2 - Proeesslng paralleters SI 32768 Sf 500 . \30010. Ittl NDII EM SSB O
LB 0 .00 H'
GB o PC \.00
lO tH\ plot para~ters CX 23 .00 e. CY 12 .50 e. flP 9 .000 pp. fi .50\.17 Hz
f2P - o .500 pp~ f2 -250 .06 H' PPMCH O. 41304 pp~/e. HZCH 206.57542 Hlle ..
120

-210.4704
-179.0454
.... .... ~
... IN
t;' ::o .... ..... ~
~
~
'" Q
~ ::t .... , .... ~ ~ ~
~
~ f-77.6393 00
77.0000 ~ ti) --76.3771
-72.3JJ6
Q\ ~
-----51.6567 -49.1814 -45.2963 -42.1980 .... """-40.0997 ~
-32.8049
~24.9691 1:23.9364
22.3463 20.7070
[AI \:l (ti () ,..,. d ti)
Q. (ti
:::o IV ~ -<

_o,
cPto
Me
~5 r
C\J
C
\J
lCl
tO
C\J
I
"'-'-
,1
r-r
-'--
'-'--
-" 9
8 T
1
7 6
oo~m~~_~voommm_~WW~NONm~mM~~v~~OO
o~ro~~NMm~~wm~O~N~~ O~~OM
-m mruo~-o
~o~-~~mNww~~womOO~No~~~o~~~mmM~OO
ww~~~~-om~~-m~~~~ONN---oom~w~~~~o
mmwmmm~~WWWN-_oomm~~~~~~~MMMMMOOO
. ...
........
. ..
~~LL1:~~~~?
f (
I1
! )
JI i
! I (
i,
I ! I (
i
,
I I , I I , l I i I i (
I ; I' I
; I
,'J
i il:
I
," f
' 1
; J
oi I
,
~~~ ~~
~~(~~~
~~ I
• --,
---,
-',
1 1
5 4
3 2
lH-R
MN
30
0 M
Hz,
pp
m,
TM
S
"'-
T I
"T---
,.-.
O
Esp
ect
ros
de
RM
N
Curr~nt
Dat
a P
aral
lete
rs
NA~E
fiag
o
EXPN
O I
PAOC
NO
I
F2
-I<
Cqu
lslt
lon
P
ara .
.. te
rs
Dat
e 200~1202
fill
' 13
.26
INST
fU4
speet
PRO
BHl
5 l1
li
Oua
l 13
C/
PULP
ROG
Z93
0 TO
65
536
SOlV
EIH
CO
C13
NS
32
OS
SWH
3378
,378
Hz
F
IDR
õS
0.0
5155
0 H
z AQ
9.
6993
780
see
RG
574
.7
OH
I ~8
. 000
u
see
[lO
6.0
0 u
sec
TE
300
.0
K
OI
I. 0
0000
000
see
••••
•••
• CH
ANNE
L f
i'"
••
•••
NU
CI
IH
PI
9.5
0 u
see
PU
3
.00
dB
SFO
I 30
0.1
3137
53 l
tiz
F2
-P
roee
ssin
g p
ara
..ete
r.
SI
3276
8 SF
300
.13000~6
MUZ
HOH
E~
SSB
o LB
0
.30
Ht
GB
o PC
1
.00
10
NIf1
P
lot
par
a ...
. ters
ex
23
.00
ell
CY
12.5
0 el
l F
IP
10.0
00
ppll
F
I 30
01. 3
0 HZ
F2
P -0
.500
pp
ll
F2
PPH
CH
HZCH
-150
.07
Hz
O. ~5652
ppll
/ell
13
7.0
15
87
H
z/e
",
5= z
CD
-cn
~
-f
"'-
OJ
~, -f
a.
C
r ru
-f
ito
a.C
O
(l)
m
(J) o
-I
Cl)
. c:
o
_.m
-0
3:
(")
ru
()
c Õ'
:> >
122

Ir>
Ir
> '" r r-
cPto
Me
~5 r
l'-
r,-~
--~
'
200
17
5
<O
o <O
..... ..
lO
<O '" Dl o
-ro
..
C\I
'"
...
.. .. "'I
r>
r-<
O
<O
........
.......
\V
(1
"l"
Cf"C
\J"Q
"a
ltn
m
.......
CT
llD
'Q'
oq
-mlD
Lt'
lOlC
O(T
')N
CT
lL!l
C1
1C
D
tOC
\JtD
CT
ll!1
-ru
""
"'l
l1t.
nC
\JO
C-N
CD
MO
l«l\
CII.C
'l"
'Q'C
\I
....
-
U1
U"
')"
Q'<
q'"
(T
)(T
')f"
'l(\J
(\J
ru
ru
ru
\ \ \ \
\! ( \1 (~
,--,
-I
, T
----
-,-1
" ---
-,--
-, 1
'---'
---"
1--'
1 ,
-,---.
,' .--~
150
125
100
75
50
25
o
13
C-R
MN
75
MH
z, p
pm
, T
MS
Esp
ect
ros
de
RM
N
Cu
rren
t ~~ME
EXP
NO
PRO
ChO
DaU
P
araa
eler
s n~Qo 2
f2
-A.cQu~sition
Par
elle
ters
o.
to_
200~1201
11
..
13.4
6 IN
Slr
uot
spee
t PR~t{)
5 ••
Ouo
J IX
/ PU
LPAO
G
zgpg
30
10
6553
6 SO
LYEI
II aJ
CIJ
NS
20
48
OS
o SM
H 18
939.
395
Hz
FlO
RE
S
0.2
8699
2 H
z ~o
I. 7302oo~
soe
RG
3276
8 OM
2
6.4
00
use
e
OC
6.0
0 us
ec
IE
300
. O K
O
I 00
0000
00 .
ec
~II
0.0
3000
000
.ec
~12
0.0
0002
000
.ec
•••• •••• C
HA
tf<tL
11
••
••••
••
MUC
I 13
C
PI
8.~Ousee
PU
6
.00
~8
SfO
I 75
.~7610~2
MHz
•••••••• C
HA
IfE
L
12 ••
••••
••
CP!
I'RG
2 •• lt
,16
N
JC2
IH
PCPO
Z 90
.00
U5
ee
PL2
3.0
0 d
8 P
Ll2
21
.66
d8
PU
) 12
0.0
0 dB
S
f02
300
.131
2005
HH
'
f2
Pro
cess
ing
oara
lllf
ters
SI
32
76B
$
f 75
~677 ~g~
HH
' NO
W
EM
SSB
O
lB
.00
H'
GB
O
PC
1.4
0
10 k
HR
P lo
t p
&f'
a.e
ters
CX
2
3.0
0 c
o
cv
F IP
F
I F2
P F
l PP
HC
H
HlC
M
t2.5
0 C
RI
23
0.0
00
pp
. 17
357
58 H
, -
10.0
00 p
p.
-75~
.68
H,
!O. ,
0.1
8 p
pl'l
/cJI
78
7 . ~89S6
H,l
eo
123

o ...
.. ~r-...
q-m
_ .
... C"1,....,~
.... O
lm
Ln
O
'l(T
1(T
1'q
"lC
lf'1
CD
~
NU
lO
'}
.....
ru
r-..
.lC
llllC
\JO
...
....
..
. C1
l {
\JC
D(T
l Q
Jl!
1U
1'q
"N
..
....
..
o \f
1
'q"
"'q""
M
(T
l C
\I
("\J
(\
J
C'\J
C
\J
\ 1I \
I \\I
f
~~OM
e ~íT -I
.. .....
... ..
. ~
,l
.J.
~l
.....
.t.
,'" ~
IILJ
....
• u
• o ...
.......
.... ~ , •.
'li
....
. ..,.
. ".
....
... '
1'
""., '
' .
., ,
T
1"""
o
r
-,~
I .--
,.--
, I
I I
I I
I I
I I
200
180
160
140
120
100
80
60
40
20
o
DE
PT
-13
5 7
5 M
Hz,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cur
rent
Oat
. P
.r • .
elt
rs
HAHE
1
10
;0
E~
3 P
MO
«l
f2
-A
c.q
uls
ltlo
n P
ara
.ete
rs
Oo
t<-
2004
1130
li ..
15
,23
II
CST
IU4
spec
t P
mIH
l 5
_ D
Jol
llel
PU.P
RO
G
cltp
t 135
10
85
53
6
SQ..V
ENT
CIlC
\3
ICS
1994
OS
4
SWH
18
1l5
,941
H
2
rI~S
0,2
76
42
7 H
Z
Aa
8088
436
.ee
RG
1638
4 0\
1 2
7,6
00 us
ee
DE
6,0
0 us
ee
TE
300
, o K
CI
CST2
\45
~ 000
0000
OI
I. 0
00
00
00
0 s
ee
d2
0,0
03
«8
26
5ee
d
l2
0,0
0002
000
see
DE
lTA
o
~ 000
0107
0 se
e
._ ••
•••
0W
fEI.
f I
....... .
HOC
I Il
C
Pl
8,4
0 us
ee
p2
16
~80
usee
P
U
6.0
0 d
8
51'0
I 75
.475
2958
MH2
• ....... C
HA
1f6
. f 2
..
...
.. .
CPIlP
RG2
•• 1
t 216
H
OC
2 IH
P3
10.5
0 us
ec
p4
21
.00
u
sec
PCP0
2 90
.00
us
ee
Pl2
3
.00
dB
PL
l2
21,6
6 d
B
SF
02
300
\31
20
05
O4I
i2
F2
-Pr
ocf:
S51n
o p
araa
ettr
s S
I 3
27
68
51'
75.4
6774
93
MH
2
NOW
EM
SSB
LB
1
,00
H2
G8
o PC
I.
40
10 t
+4R
plo
t pa
ra.e
ters
CX
23
~00
ea
CY
FIP
F
I f2
P f2
PP
MCH
HZ
CH
7.0
0 c.
22
0,0
00 p
p.
1660
2 9
0 H
2 -1
0,0
00 p
p.
-754
~68
H2
lO.O
OOOO
pp
.. /c
lI
75"
67
7 .. 9
HZ
/CfI
124

fu
H
67
'<q
OI
OI
N .....
,...
. ('
\J M
'"
~
I.ll
N
oq
'
-o
o O
I (T
l (T
1
(T)
N
lOlt
'l
lO
lO
\}Y
( I )
-~~-owm~oru~o-mw~~M-om~o
vwm~OO~~~N~~-mw~~~o~-w~o
~M _~O~~~~~NmmW(TlNN_Ol.llmo
~~(T)MOO~~~~~~~WWWWWWWNOO
..
...
.
..
NN
NN
NN
-o
! I
(
I ( !
r I
I I
. I I
I
,/li
!'l!
/
( liJ
IJ J
1l
l J
~~J!JJI J~
!~\ ~~!3
~)~\ ~I~~~
~) T
---'
-T
T
~ r'
-íT
I i
I i
, I
i i
I i
i i
, I
i f
I i
, I
i i
I i
i i
, i
• I
i i
I i
9 B
7
65
4
32
1
o
lH-R
MN
30
0 M
Hz,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cu
rren
t D
ata
Para~eters
NAM
E T
lag
o
EXPN
O
1 PR
OCN
Q
I
F2
-
AC
Qu
i 5 j
l i c
n
Par
ame
lers
Dat
e 50
0000
T
I..,
2
2.5
6
IHS
T_
5
pee
l PR
OO
IfJ
5 .. O
U81
13
PU
.PR
OG
19
30
TO
3276
8 Sl
l.VE
NT
C
OC
l3
HS
32
OS
O
SNH
3960
.892
ti
l FI
IJIE
S 0
.121
487
HI
AO
4.1
1571
07 s
ee
RG
&I
OM
125
.600
use
c DE
6
.00
U5B
C
T€
300
.0 K
01
I.
0000
0000
ale
P
l 8
.00
use
e DE
6
.00
usec
SF
DI
300
.131
3270
!fi
z
IM: I
IH
P
U
1.00
118
F2
-!'r
oces
, ill
Q
par
alll
eler
s S
I 32
768
Sf
300
. 129
9937
lfi
2 IID
W
EM
SS8
O
L8
0
.30
Hl
se
o PC
1.
00
10 N
I4P
plo
l p
aral
lele
rs
ex
22
.50
CII
FlP
10
.000
PPI
I
FI
3001
. 30
HZ
F2P
'0
.500
PO
lI F
ê -1
50.0
7 H
I P
PI0
4 O
. ~6667
pDJ\
/ell
HZ
CH
140
.060
67 tI
l/e~
125
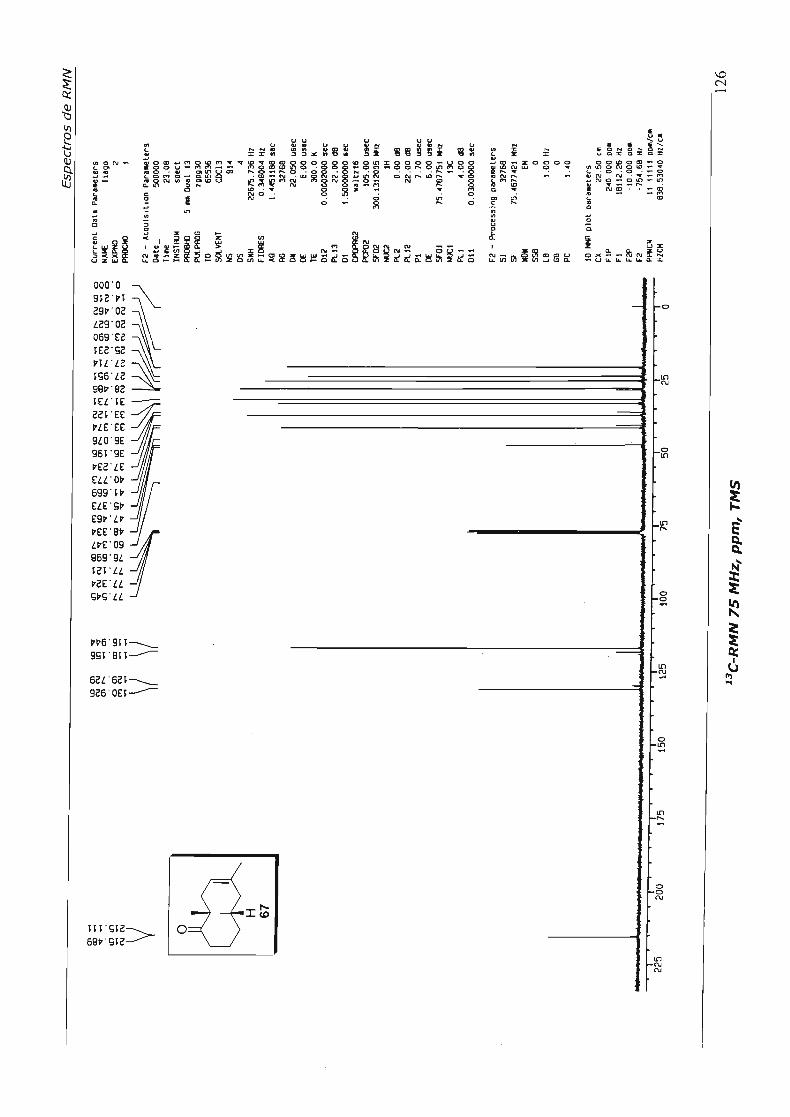
I m
-co
-
.... LO
LO v o m
I
lDal
UJ~
NC
\I
ll'J
"Q'
01
,...
..
-O
)
..
" 0
01
E
DU
)
~~ \/
\I
11 J
~~~m~~~MmM~WW~N-W-~_O~NWO
~NNm~mW"""Ul~~m~~NMm~~~mN~_O
~~ WM~~~W~N~OM_""""Q'm"""NW~~NO
,.....,.....,.....wom,.....lCJ-O,.....ww~M-m,.....,.....~~oo~o
~,.....,.....,.....w~~~~vmmm~~mNruNNNNN_
~~~\~/
J I
I I
I I
I I
I I
I 22
5 20
0 17
5 15
0 12
5 10
0 75
50
25
O
13
C-R
MN
75
MH
z,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
eUrr
enL
Oal
a P
aro
,..,
lers
NA
ME
TlI
go
EXPN
O
2 PR
OCH
O
F2
-A
tQui
Sll
ion
Par
o""
ters
D
ate_
50
0000
T
lIIe
23.0
8 IN
STfU
N
soo
ct
PRO
BIfJ
5
...
Dua
l 13
PU
LPR
OG
19
p930
TO
65
536
SOLV
ENT
CO
el3
NS
91~
OS SIIH
22
675
. 736
HZ
FlO
RE
S 0
.~6004
HZ
AO
I. 4
4511
88 s
ee
RG
3276
8 0_
22
.050
u
sec
Df
6.0
0 us
e c
TE
30
0.0
K
012
O • 0
0002
000
sec
PU
3
22.0
0 di
! 01
1
.500
0000
0 n
e
CPI
J'IIG
2 .a
ltzl6
PC
P02
105
.00
use
c S
F02
30
0.1
3120
05 l
IiZ
N
u:2
IH
Pl2
0
.00
dB
P1
l2
22
.00
di!
PI
7.7
0 us
ec
DE
6.0
0 u
ne
SFO
I 75
.476
7751
!ti
z N
UC
t 13
C
PU
4
.00
de
011
0.0
3000
000
sec
F2
-P
rQce
ssin
g pa
raoo
eler
s S
I 32
768
SF
75.4
6774
21
14Hz
NOW
EH
55
8 O
lB
1
.00
Hz
GB
O
pe
1.4
0
10 N
Ifl
plo
l 08
raoo
eter
s ex
22
.50
c"'
FIP
FI
F2
P F
2 PP
HCM
H
leH
240
.000
D
O"
1811
2.2
6 Hz
-
10.0
00
Do
a
-754
.68
Hz
11
1111
1 O
Oll
/C
.
838.
5304
0 Hz
/ca
126

('o
°m H 68
.... O! ..... ru
.....
Nm~_N~mw~~_~m~N~~mmm~om~w~-ru~~mW~~NO ~~m~o~~~~m~m~~o~~~~m~N~Nmo~--m-~-mMO ~~~~~~~~~~~ruN_~_o_om~~~~mmmm~~Nooro~o NNNNNmmmmmmmmmNruN~~mw~ww~~~~~~~~MNmo ~~~~~~~M~~~~~MNNN _________________ OO
/ i I
I I II
1I
1
I I ! (
I I
i I 1I1
I I I I I / .! I I I I :
J
I r r J I J r
_ _ _ --'--____ J! J I ~u Ju [5\ I~\!~I ~ 15 ~~~ !5\
'I' 'I' 'I'" 'I' 'I' 'I'" 'I' ." 'I"" 'I' 'I' 9 8 7 6 5 4 3 2 t o
lH-RMN 300 MHz, ppm, TMS
Espectros de RMN
Curr@nt Data Paralleters NANE T ; .go EXPHO 3 PAOCHO I
F2 - ACQuis i tion Pa ra~el.rs Dat e_ 500000 TlIOIe 2 . 11 INST~ SDec t PR06H1 5 _ Oual 13
Pl1.PROG 2930 TO 32768 S!X. VENT COel3 NS 32 OS o SMH flORES AO RG OM DE TE DI PI DE SFOI NUCI PU
3980 . 892 Hl 0 . 121487 H2
~ . 1157107 see &4
125 .600 usec. li .00 usec
300 .0 K 1 . 00000000 see
8 .00 U6ec 6 . 00 usee
300 . 1313~ ltiZ IH
1.00 CIlI
F2 - Process i ng par arelers SI 32768 SI' 300 . 1299997 HItl 11011 EM SS8 O LB 0 .30 Hz
GIl o PC 1.00
lO NNR plot paraMeter s ex 22.50 CI
F1P 10 .000 PPM F! 300 I. 30 H2 F2P -o .500 pp. F2 -150 . 07 Hl PPMCM o . <46667 POli / C. HZCM 1~0 . 06067 H2/CIi
127

("o °m
I
I
~NNo~ru~orn~~-moN~-rum~-_ruo~MmM~~m~wo
m-~~~~o~mmw~mwo~omomooo_~w~~~ro_~ru~o
~~~mwmOOO~NO~O~~WMMruN~O-Moru~Mm~mN_O
. .
. .
. . ..
........
......
. ..
. g~~~~~~~~~~~~~~~;~~~~M~~~rore~~~~~~o
~~íV~~~~~;
.,
.1 1
I I
I I
I I
I I
I I
225
200
175
150
125
100
75
50
25
o
13
C-R
MN
75
MH
z,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cur
r2nt
Dat
a Pa
ral!l
€ter
s N
AH
E EX
PNO
PRO
CNO
110g
0
F2
-A
CQ
uisi
t10n
P
aram
eter
s D
ote
5000
00
Till
e 4
. OI
IhST
RU
M
spec
t PR
OO
HO
5 ..
. O
ua
l 13
P
QP
RO
G
19p9
30
TO
6553
6 S<
l. VE
NT
CD
Cl3
tIS
2048
OS
A
SNH
22
67
5.7
36
Hl
FlO
RES
0.
3AG
004
Hz
AO
1.4
45
11
88
,ec
RG
32
76B
OM
2
2.0
50
use
c DE
6
.00
use
c TE
3
00
.0 K
01
2
0.0
00
02
00
0
sec
PU
3
22
.00
dB
O
I . 5
0000
000
5 ec
CP
OPA
G2
"alt
zl6
PC
P02
105.
00 u
se c
Sf0
2
300
1312
005 ~z
NUC2
IH
P
l2
0.0
0 dB
P
l12
22
.00
dB
1'
1 7
.70
us
ec
DE
6.0
0 u
sec
SfO
I 75
47
6775
1 11
Hz
NlIC
I 13
C
PU
4
.00
d8
011
0300
0000
se
c
F2 -
PrD
cess
Ing
par
."",
lers
51
SF
NOW
SSB
LB
GIl
PC
10 N
NR p
lot
ex
FIP
fI
F2
P f2
f'f
'HCM
H
ICH
32
76
8
75.4
67
74
38
11
Hz
EM O
.00
Hl
O
l. 4
0
para,,~ter5
. 2
2.5
0 c-
24
0.0
00
pp_
1
81
12
.26
H
l -
10.0
00 OO~
75
4.6
8
Hl
11
1111
1 po
"/cl
I B
3B
.53
04
0
Hz
feIO
128

';---
.. :fo.. ....
~ I
::a ~ ~ :fo..
<:> N Q Q
~ !o'>
~ ....
... 2.8601 \.
~ 1.0944
'tJ 'tJ
2.8503
~ !o'> <:>
~ til
'" .... .r-'c:-
2.0388
.~ 2.3671
3.0908
~C 3.1437
2.9813 ,-C- ... 2.9254 ::=k.--- 1.0000 ~_
;--
~I
11'"
O s: Cll
---
-7.3369
3.4600 3.4140 3.3723 3.3481 3.3152 3.2911 3.2472 2.6636 1.6022 1.5868 1.5780 1.5605 2.5341 2.5100 2.4837
~1.3191 2.2950 1.1511 2.2191 2.1875
2/2.1590 ~2.1370
L1.9U7
~~:;~~~ 1.9066 1.8891 1.8759 1.8671 1.8474 1.8255 1.5205 1.5073 1.4722 1.4415 1.4152 1.3493 1.1594 1.2111 1.1629 0.9391 0.8798 0.8732 0.8140
-0.0000

, .....
'-1
~ ..; ... .... I O m
OM
e
~ O
Me
H
-
70
I
..l
'1'-"
,
•
~~~g~
.... ..,
..,g .
.. gl:::l:::~~
~I I II
.J.t.
O"
. I
-.,
1 •
-..
C>
..,~
... ..
...
..,
\O
:::!
~;;;
....
~~
I ~
I I .JI
. o,
' .'
~ ~~1i
00
0
. ...
......
.. ....
.; "...
..: \(
) "" "lfY)~
~V')h
C
~~~ ~
"'1
11
'\0
..
""
"Ô"'~
....)
........
"'" --..
.
Esp
ect
ros
de
RM
N
I Irr
I(
(
I ' I
I
I I
I
I
i.i!
!Ji.~
, "
T-"
'[
""-'
-.. '
1fT
.' T
il 1
11
" III i i'
l i' i i
i i
i i
i i i
i I.
i III i
I' I
i I
i I
i i
i i
i j
I i.
i i
j i i I
I i
i I
i i
i 'i i li'
i i i,
i I i
i j'
I i i
i i li i
i i li f
11
1' i
i i
i I
i i
i I l
i li i i, i
i I i
i i
'.11
11
r I
i li i
li
I i
11 i
i i
i'
i ••
1 I
r i I
i I
,11
1 I
i i i'
III
I i I i
i i
i I
i I
i i
r i II
I i
i i
i i i 1
i i i,
i li i I'
i li I
i i
i i li i
i i
i I
I i
i I
i i
i i
i i
i i
i i
I i
i i
i
120
110
100
/90
/80
/70
160
150
140
130
120
/lO
10
0 90
80
70
60
JO
4(
) 30
10
10
O
130
13
C-R
MN
50
MH
z,
pp
m,
TM
S
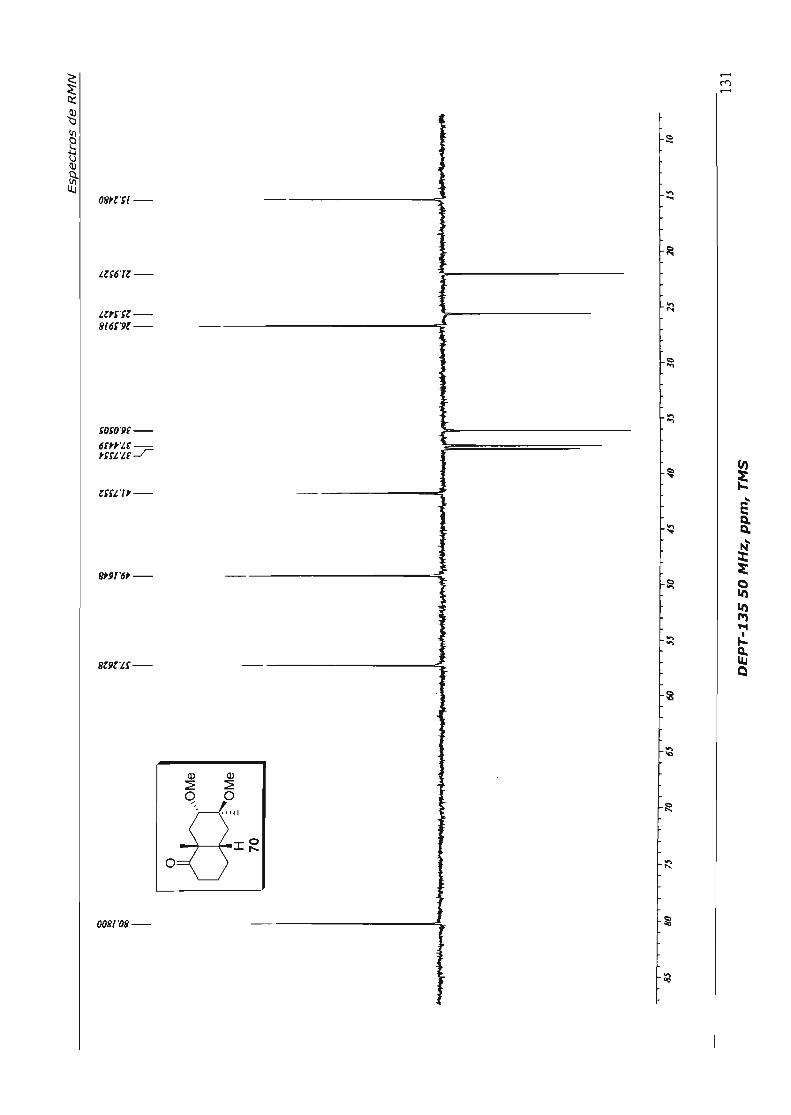

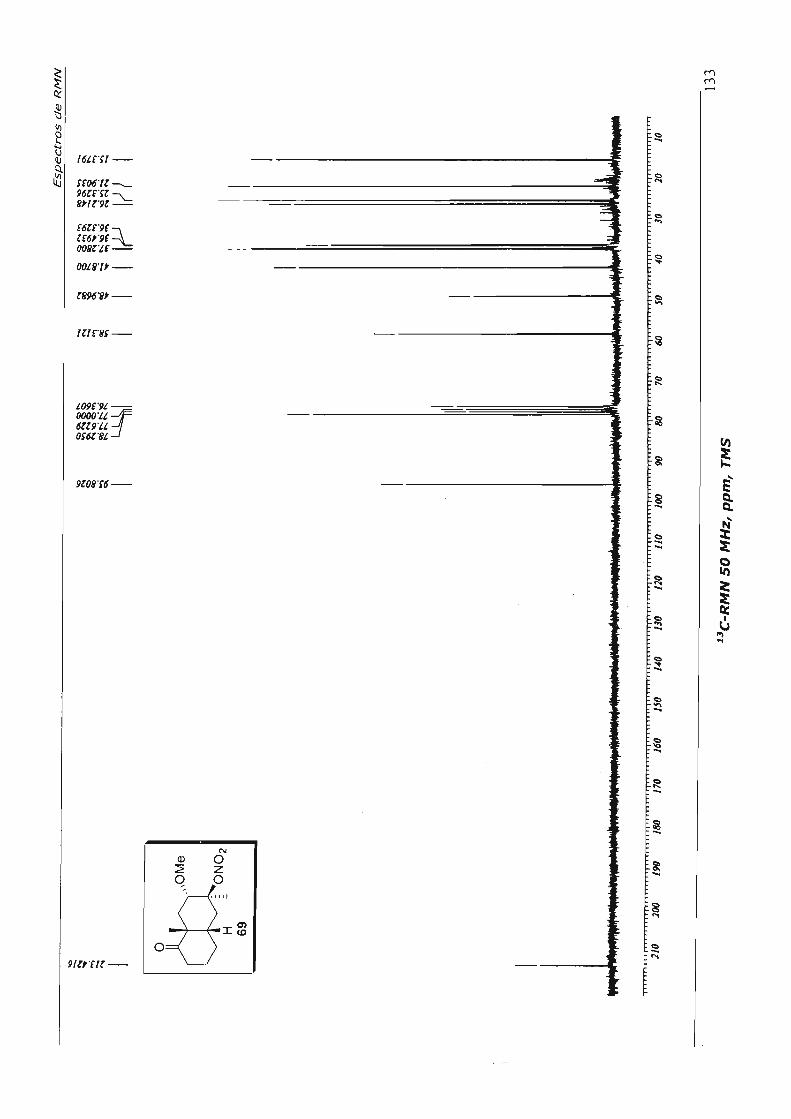
Espectros de RMN '<>
~ ~ "" /
o Ah·"OMe
~ON02 H = 69
~ ~
/
QO\:St>-...
~6~~ ~~~~
w..,)1
..... '" ..... .... ~
/
i ~
I
o 8"'''''' ~ ~~:;: ..... '" 'Ó 'Ci .,.. .., ... .., //~
~~~ ;;;~~ \IÓ"'''''; "' ........ /( (
I I
..... ~ ....,
'" .....
li ' I li • i i I li li 1# li i i li f li li i i ij i i " " i i li i i li i i i i li i li " li li " i i 11 li " fi " i i I 11 i I i i ti i i li 11 li i i li li li li i i ; li li li " li " i i li 11 i i li i i 11 .. li i " 11 li li li • li I" • i li , i li li I " li i i 11 li " li i i li li li i i " i 11 li li i i 11 i i I i i I " li li " i
210 ]()() 199 180 170 160 150 140 130 120 110 /00 90 80 70 60 50 40 30 ]0 /0
13C-RMN 50 MHz, ppm, TMS 133
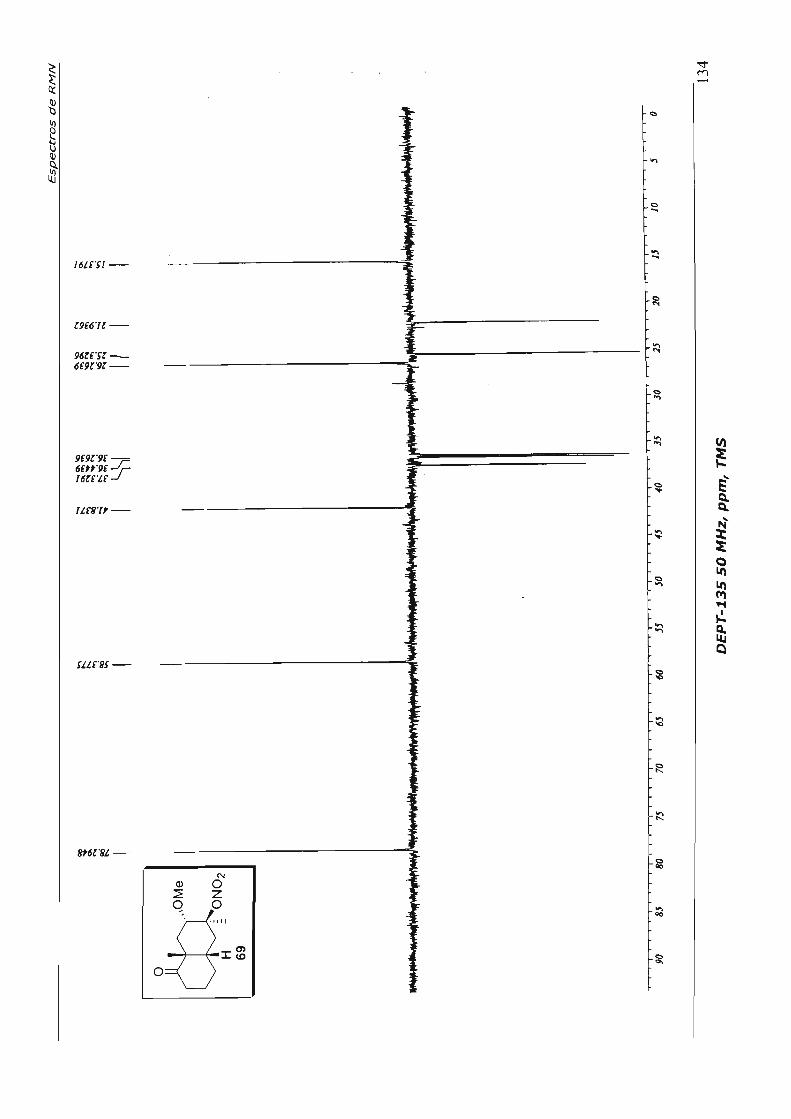
Espectros d e RMN
~ ~ .... .... 0.10 ~~ ~ -t-. 0. ...... o-
o. t-. ~ .... ~ ... lO"" ... .....
'" ... ... ~ .... ....... o. ... ~ ~ ... ....:,.;,.; ,.;.,; ... .,;
~ ............ "' .... .... ....
I I ~~I 1/ I o
mOMe
12
I
, I I ' I I i j I I I i i i i I i I i t i I ( i i i i i i i i I i I i i I i i i I i i i i I ' I I i 1 .9
9(J 85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 5 O
134 DEPT-135 50 MHz, ppm, TMS

OH
m H 71
4_ ,, ·_-Õ·~_·"- - ,
9
o (I) Ln rru r-
I
o ..... UJ ru ru ... CD o ..... ..... r- <D ru ru ru
\V
I~I
~~mm~ru~mrn~m~illoru-~m ~ruruvo ooomru~ru~~m~oo~~-ooo~mo~~~~o m~m~~mm~rumruW~~~wwrummç~~o ~~ mm oooo~~mroNm~~~~Mru -o MM~ru ~~~ww~w~~rururururururuooo
~~\/~
j
I!I ~IM~~~~)) 1 I I I ,--,------r- 1 I .,- .......,-- -- ,-- r --, --l 865 4 3 2 O
lH-RMN 300 MHz, ppm, TMS
Espectros de RMN
Current Data Parameters NAWE Tiaga EXPNO I PROCNO
F2 - AcQu is i tion Parafte ters Oat. 20050401 TI~e 15 .55 INSTRUM sp.et PROBHl 5 "" Dual lJe / P\JlPRDG Z9 30 TO 32768 SOLVENT CDel3 NS 16 OS O
SWH 3453 .039 Hz flORES 0 . 105378 Hz ~O 4 . 7448564 s.e RG 128 OH 144 .800 "see DE 6 .00 "s.e TE 300 .0 K
DI 00000000 see
. .. ••••• CHmi'EL f I • _. - - ---NUeI IH PI 9 .50 "s.e PU 3 .00 dB SfOI 300 . 1313823 ltiz
f2 - Proe",sing para""ters SI 16384 Sf 300 . 1300012 ltiz IoUH EM SSB LB GB PC
0 . 10 Hz O
1.00
10 tHl plot para""ters ex 23 .00 eOl CY 12 .50 e~ FIP 10 .000 pp. f i 3001 . 30 Hz F2P -o. 500 pp~ F2 - 150 .07 Hz PPMCM o . 45652 pp./e~ HZCM 137 . 0158 7 Hz /e~
135

OH
m H 71
r--r--r----.-.- - í - r-.,--- I I 200 175
o co ("I"') Ol <.O ('\Jll1t"--ru'VfTlO""'lt.n co (CCTlUl"I;J (\JlO-(\JlC)Cf)M(D(T') "- lO '<10<ll OlCDOfJJ(DOOlO"Q'" o "- m,...,,....,LO mtooq-OaJlOV(T'l(T'l fT1 ,.....,...,,.... ,......
(TlfTl("")fTlrur\Jru(\JC\J
\~ \\\\I/P
l -- -"- '- -'---r-r-' -,--r-r--r--'
150 125 100 I I I -'·-- T- - , - \
75 50 25 o
13C-RMN 75 MHz, ppm, TMS
Espectros de RMN
Correot Oata Para. eters N~1t' T 1000 EKPNO 2 PROCNO
f2 - AcQuisition Para.etll!r!i [)ate 20050401 filie 15 .57 INSTRlH soect PROOtfl •• Dual 13<:1 PULPAO;; 2gp030 TO 65536 SOLVENT COCI3 HS 2054 OS 5NH 19607 . 84 4 H2 F[[JlES 0 . 299192 H2 ~o .6712180 See ~ 32768 DI! 25 . 500 usee ~ 6 .00 usee TE 300.0 K DI . 00000000 see dll 0 . 03000000 see dl2 0 . 00002000 see
•••••• • • CHAffl:'L f I ••••••• •
ItJCI 13C PI 10 .50 usee PlI 6 . 00 dB SFOI 75 4760505 HH2
•••• ••• • CHANHEL f 2 •• • •••• •
CPOPAG2 •• It'16 1tJC2 IH PCP02 100 . 00 usee PL2 2 .00 de PLI2 20 . 79 dB
PL 13 120 .00 dB
Sf02 300 . 1312005 HHz
F2 - Prccess \no paralltlers 51 3276B SF 75 4671452 HHz IIlH EM SSB LB GB PC
3 .00 H2 o
t. 40
10 ht4\ plot parallleters Cx 23 .00 em Cy FIP FI F2P F2 PPHeM 'iZCN
U .50 co 230 . 000 PPII
17357 .58 H' - 10 . 000 ppm -754 .68 Hl
10 . .0-4 78 [lpll / CII
787 . '89'.)0 H2 /c o
136

Gt(.,'OMe 0- · - ---
H 72
C\I \O co C\I ,...
~~~~m~~~m~~~~~~~~~m;~~~m~~~g ~~~~~N_~~w~o~~Nm~~~~~omm~~IDw ~~~~NNN ooommmm~~~~~~~NNNN~m
MMMMMMM .C\INNN---------------- O
~L~~~~
í I I
J I J
I I
I I, ( 1 11' I .~ I i fI I . I J I I f J i' i
! I
'------_------J. IJ~~~ )~ ~ ll~/' I ~ ~J I
Isl~ ~I 1~1~151~1515 !I!!\ '~' 'ê' '~'" 'à' "'~' '1"'" "~' "" '~' ""t' 'b"
lH-RMN 300 MHz, ppm, TMS
Espectros de RMN
(urrent DaU Parameters NAIE Tllgo EXPNO 3 PROCNO
F2 - ACQulsition Paratoelers Date_ 500000 Tille 23 . 12 IHSTIU4 511ee t PRLIIHO 5 .. Dual 13 P\.l.PROG zg30 TO 65536 SOl YENT aJC13 HS 32 OS O Sltl 3B81 .988 H1 fl1JlE5 O . 05923~ H2 AO RG OM DE TE DI
B . ~~I096e sec 57
128 .BOO u6ec 6 . 00 ~gec
300 . 0 K I. 00000000 lec
PI 8 .00 usec DE 6 .00 usec SFDI 300 . 1313532 14HZ NUCI IH PU 1.00 dB
F2 - Processing per_ters 51 32768 SF 300 . 1299977 !fiZ IIDII EM SSB O LB GB PC
0 .30 Hz o
1.00
10 NIII plot oaralleters ex 22.50 C.
flP FI F2P F2 PPIOI HZCN
10 .000 pp. 3001.30 H2
- 0 .500 pPII
- 150 . 07 H2 O. ~6667 pp./c.
140 .06067 HI/C.
137

~. ,\OMe
~ H 72
~~ON~om~~~~~~rn~~_~~~o~~m~ ~~m~~omm~o~m~~~~_~NN_~~W~ ruoruoomruwmMm~M~Mru~ru~wmru~m~ O~~NO~o-~~w~m~~N~N~~mO~NN ~O~~~~~OW~NN~N_~WW~~ __ O~~
~7~~~~
I I I I I I I I I ........ ~-___ ~........-,"Tl"W'- ~ ..... ---.-.. .; c •• ,
225 200 175 150 125 100 75 50 25 O
13C-RMN 75 MHz, ppm, TMS
Espectros de RMN
Curr~nt Data Parameters NAIE EXPHO PROCHO
Tlago
F2 - AeQuisillon Para""'lers Oate_ 500000 Ti IM! 23. ~O INSTIU4 speet PROIII(J 5 "" Dual 13 PU.PAOG zgpg30 TO 65536 Sa..VENT COCI3 NS 823 OS ~
SI/H 22675 .736 Hz FlIJlE5 O. ).4600~ Hz AO I. ~~SI18B see RG 32768 01/ 22 .050 usec DE 6.00 usec TE 300 .0 K 012 O • 00002000 see PU3 22 .00 dB OI .50000000 see CPIlPR62 .altzl6 PCP02 105.00 usec Sf02 300 . 1312005 lt1z t«JC2 IH Pl2 0 .00 dB PU2 22 .00 dB PI 7 .70 usec llE 6 .00 use c SfOI 75 . ~ 767751 MHz NUCI 13C PlI ~ . 00 dB 011 O . 03000000 see
F2 - Proee561 n9 par81!1e ters SI 32768 Sf 75 . ~6T7.97 14Hz ICIlM EM sse O lB GB PC
t . 00 Hz
O I. 40
10 NNR plot paralleters CX 22.50 e. FI P 2~0 . 000 pp_ FI F2P F2 PPIOI HICM
18112 .26 Hz ' 10 .000 po_ - 75~ . 68 HZ
11 . 11 111 PPII/C. 838 .53052 Hz/e ..
138
~.,\OMe
~ H 72
<XI .... NM OlM 10 o 010 <XI .....
I I
M NC1l-0C""1o CDf.D ..... OlfT1-,......-NCDtD rn t"'1C'\J"'VC\Joq,....m-~ ...,-COlllN,....,;OlC\J
lO C\I(T'JC'U,....,t.O(T)~...,.
'" 'q'(T")(TlNNNN-
I \ \\ ~ lI!
I I I I I I I I I I 225 200 175 150 125 100 75 50 25 O
DEPT-135 75 MHz, ppm, TMS
Espectros de RMN
CUl'rent Data Parallleters ""ME TUgo EXPNO 5 PROCNO 1
F2 - AcqUlsitlon Plr • ..,ters o.te 500000
T \"" 0 . 15 lNSTR\J4 speet PAOBHl 5 _ Dual !3
PlILPROG Oeoll35 TO 65536 so..VEHT a:JC13 MS 107~
OS B
SIIH 21097.0.7 Hz F1O!1:S 0.321915 Hz
AIl 1 . 5532532 see
I\G 32768 lJIj 23.700 use e DE 6 .00 usee TE 300.0 K
Pl 7.70 usee DELTA O • 0000096 see DI \. 50000000 see DI2 0.00002000 sec Pl2 0.00 OS
P3 8.60 usee 51'02 300. 1312005 MHz HUC2 IH
D2 O.003571~3 ,ee P. 17 . 20 usee 51'01 75 . ~76020. Iftz tU:1 13C
PU •. 00 áB
P2 15.40 usee PL12 22.00 OB
DE 6.00 usee cro>RG2 waltzl6
PCP02 105 .00 usee
F2 - Proeesslng pare..eters SI 3276B 51' 75 . 4677 .95 ~Hz
MO" EM SS6 O LB .00 Hz G8 O PC .• 0
10 HMA plot par_ters CX 22.50 e. FIP FI F2P f2 PPMCM HZC~
240.000 pp" 16112.26 Hz -10.000 po" -75-4 .66 Hz
t\.Il 111 pp"lc~ 836.53052 Hz/ca
139

~.,\OMe
~ H 72
-1
r-- '
. • -", . ~. , .
e • . =: ..w. , .1.
~
I~, • I " iP. " I
•• u' •• 00 '.
CZI li
. _-.- ----.- . I
I • I
I I I I I I I 4 . 0 3 . S 3 .0 2 .5 2 . 0 I . S ' 1 .0
COSY 300 MHz, ppm, TMS
1.0
1.5
2 . 0
2 .5
3 .0
3 .5
4 .0
ppm
EspecCros de RMN
CurTe1Il o.u P.r ... t .... ...... li 'OI)
E"'" ,. ....,., Fé? - AcQulslllon Pr_trrs
OI .. 500000 TI .. 7, " 'NSlFUC 'Me l
""""" 5 _ au.1 13 .......,. tos.,.s 10 .... 5a.'/Ellr CIlC1J MS J2 OS ,. SIt! 2Il5J . 1I31 Hl riMES 1. )U497 Hl
'" O !J588S96 M e 06 90 .5 .. 175 .200 UNe
tE 6 .00 UHt rE lOO .O ti: D. t .~"c ., 8 . 00 une DO O. OOOOO]()(i Ne. tE 6.00 Uln: "OI 300 . ntn02 ... , IUI 'H .\.1 L«! a8 11 .. O .~C! .. c
fi - Ac""isll!on O ... ..elerl ICJCI I 10 '28 "OI )00 . t12t tttl fllRS 22.25l!W.16J K:I
S1I 9.~ c.-
f2 - Pr"1Itn,'1"I9 par"_et.,., SI .... 6f lDO . t.2SN963 tttz .. SINO SS! • LI OC-(iHJ:
'" • PC .. <O
FI - Pro" .. lng DW'.net's SI U'2' lC2 CE .. )00 . 1)08]62 liII1l .. 5'''' SS8 •
LI iL C-O 1041
til <
2D ~ plot D ....... l trs CW, I~ . c.o c. CXt 15. 00 c.
' ...... • . 278 _ , .... IZ'I-4 .MHI
f2OH1 a, 7U_
''''' 211 &8 ttl flPl9 • . 2'81.-FJLO 12'9 • . 85 ", nPHI O. M] ~ flMI m_ ~1 Hl
''''''''''' C.2.J772 "'"'~ 'lt1lDC 11 . ).0170] ""Iu
"~ O. ll896 rroa/t.a ""IOf 71 71899 Hl/UO
140

r7h."OMe
~ H 72
I ~I I 1 lld li .I
t I t' 4'
t I ,
a ,
~ I A , I 1 I 1
80 60 40 20
HETCOR 300 X 75 MHz, ppm, TMS
1.0
~ 1.5
I 2.0
2.5
.. 3 .0
3 .5
Dpm
Espectros de RMN
CI./ff."t PIta ' ..... lrs MME TI., .- . -...
n · ...... HI.,. ...... ~ ... ..... -n. 101 1XSl..... ...-::t -..cI ,_ DAI fi OI&.- ..." lO .... a,w.t CD:tI .. .. .. lO .......... JIZOM.I: nOllO I ....... '" .. 0 ,1fI1062 M e
'li It!ll!!l . ' .. 14 ."nec EE 1.10 IIIttt TE •. 011 011 O. QlIiIOOOOOtK DI I .MCIOOOOCI Mo(
DII O.OIIIIIOOO.c IIU 0 .00 • ,., ' .10 ....: FQI "". lJI11Zl...,
""" 'H I» O • ..ooJOO IIK "l ., ..... ar O.oaseeao ..c lIt 7.10~
D3 O.MInOOOO Me 1Il12 22 .oa • ar •. 00 lI6K RI 7'5 . .. ~1tQo MI IX 'lI ...... ~ _I"'. JICJIDI Ift."..-: .... O.OIJ:tI7«11 ftC
..,. FI - ~I.ltl~ ..,-..t ....
• lO '"' .. F\IIIU
'" ." lOCI . Ul1311ttz
22 . ....at40Ml I .BU_
fi! - PnK'n6 l ftQ ,.-ur-. " .... • 75 .. n...o 11ft,. .. os ... la • U 0 ."1'Q
• f'C 1 . .0
It-~'"' ..... ~ ..... SI 1124 ra IF ,. JDO , ll'9IW! .." .. asnE ... O lt ' . 00 lU
• 1D_"lol..,. ... t ....
CU " .00 CII 011 \'.00 ~ nPlO 10 .0«1_ ,"&,O l57J') . iO 141
"~I t~1OII
'2MI 1'601141 rl"lO ]." 00II fiLO 1111 10 141 '1 .... ( 1500_ .... , '''''''''' .... '00 " ..... '1"'CJ1
150 .01 .... ,lUa. _Ir:.a) J05t. ",/ç •
O llJI2 tllllA/te. t6'51f "l/c-
14l


m
...... .......l ' r' , .. , ,
~ ~
~
I
~ ~
I
I
~-.
~I~ ~~I!!
~I
1\\
, ..... ~ I. U ' I' .,...... .....
13C-RMN 50 MHz, ppm, TMS
Espectros de RMN
II ~ifi~iii~ ~:II ~;;;;lIi:l!~~~
~I \y /((((
I
11
\1
I I I
I
.Jl LI! .....L .... . r ."
143

integral
?o ....
?o Q c::.
2.0000 -.r 7.8108
0=(/)=0 -7.7713 /
:'" " 10
.... -.J; 7.3391
2.10901 gI ~7.29U
-7.1777
:'" c::.
~ ....
~ c::.
~ ... .... ::t I
~ 0.9853 -5.2109
~ ~ ~ c::.
N Q Q
~ ,.. j.4.3706 .... 4.3442
::t 1.0045 -4.3179
.... 1'11 - . 4.2894
'ti ,.. 'ti c::.
.... 3
~ !-'> (I) ....
!-'> c::.
'" .... 5.0622 -1.4332 ~1.2643
1.0535 -1.1853
'" -2.0417
c::. ~ 1.8715
~l~~l "-t., 13.478
1.7070
ll~: 1.6017
"-
1.4910
e:, 1.3186
3.0388
1.2923 't 1.2704 1.1143
!=>
0.8272 [hl
.... "\:l Cb C)
~ O
!=> ti)
c::. ~ ~ -~
~ ~

... ~
... .... <:>
-... ~
~-
... ... <:>
... ~ --... ... <:>
... 2
... IA!
? :!: ::o ~ ~ U1
~ Q
~ ~ ,
~ ~ ~
~
~ (I)
~
~
~
-
~~ i~
F
1
F-
-
-
-
-
-
-
Q ~
=cn=O / _O'~ o
~I>=> -144,1617
-/34,9980 í/30,1949
::!C. 119,5720 I 117,6540
127,5721
-IJ7,IJ34
-90,0979
,77,6393 ~77,OOOO -76,3771
-40,1314
-96,6407
-33,7720
:::c: 28,2640 'e 28,1000
26,4443 :l-- 23.6576 lt23.4281
\::::13.1477 21.4937

-7.17/1
~ o
1.(}()()() -4.9015
... :z: ~ I .... ::o 3: ~ N ~ () o ()
3: 1.0758 ..L. 3.7080
3.6970
~ !-oi ." ... :g ~
~ !-oi
1.0811 o 1.0492
1.9878 cn 1.9659
1.9154 1.8198
!-oi ... 1.8057 1.7750 1.7509 1.7377 1.7311
!-oi 1.7101 o
~1.6433 1.6017
16.896 1.5644
!'-
3'1.5171 -1.5073 ... I.4854
1.4700 I.4305
3.2933 1.4064
!'-1.3152
o 1.3164 1.3055 1.1879 1.2571 1.2396
!=> ... 1.1243 1.1001 1.1826
~ 1.1650 1.1409 '\j
!=> Cb
<:) \: 1.1212 C')
O.(}()()() ~ C 11)
~ ~
.+:o-~
0\ <

-/39.0635
-/24.6705
-./82.6555 ,77.6393
dr 77.0000 -76.377/
-55.9843
-49.0829
~=======================:-_______ -.r 43.6569 ~ ___ 42.0012
-4Q.26J5
~;:;:====~~~;;;;;;;;;;;;~~~~=~~~;;;;~~;;;;;;;;::;;;;;;;;;;;;:===-__ - 26. /82/ ~ ~1M1I6 '- 22.7395
12./822 ~J6.7070
~
o ~ ~ ";i .... w
"" "" Co) ;:!
:t ~ ... ~ ~
~ t
~ ln
~
-~ 00
-:i F
~
~ ....
,;
~
~ ~
~
~
~ ~
~
..;
.-I
~=-
oi j
..., ..... F
... ~
~ ~
~ ~ i ... ...,~
J. '9 F-
-.i
.. ~ ~
"
~I __ -4-
-
-124.7031
-82.6555
-55.9188
-1/9.0338
-1/3.6731/ -1/1.981/9
~26.1986 ~2M773 -,22.7725 -22.3135
-16.7399
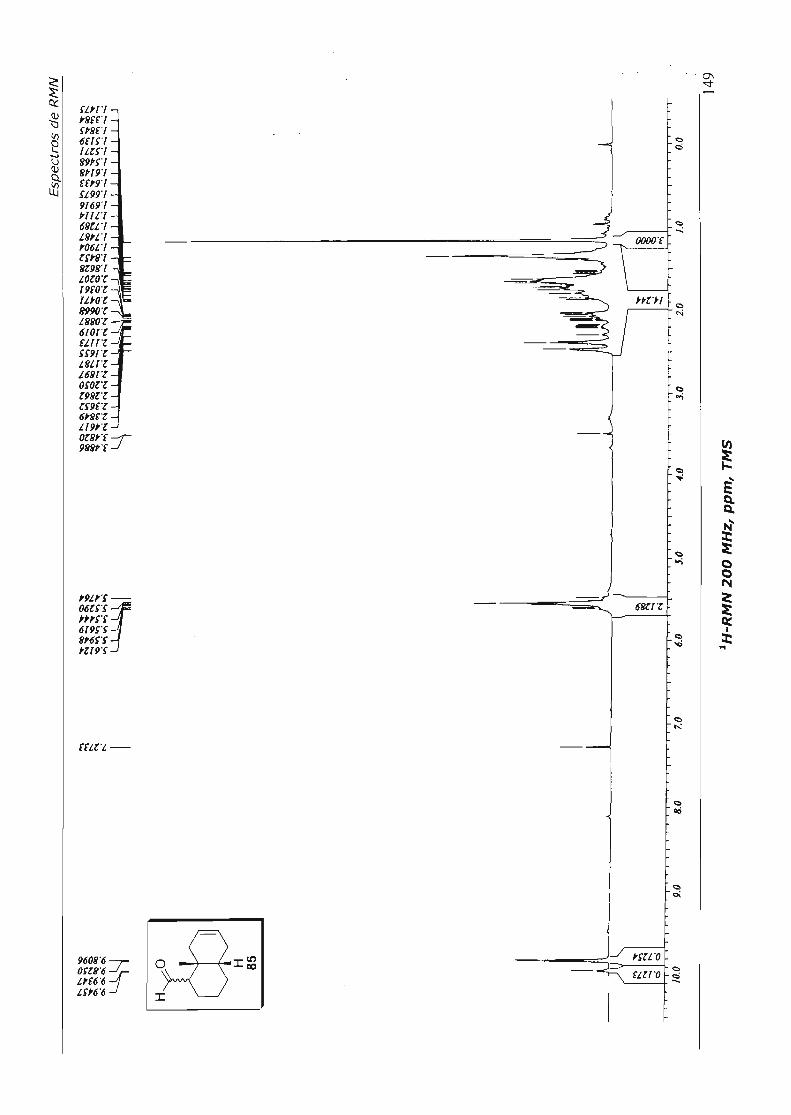
.... +-----,. ~ 0.1273
0.725-1
.. l'" ::r.: Q
I ::a l 2.1289 ~ N Q Q !-"
l C)
::r.: ,N
'ti 'tJ
~ ,...
~ Q
til
!-'> C)
!'> <:>
14.244
3.0000 .... c
!::> Q
+:>. \O .
r 9.9457 L 9.9347 :c: 9.8]50
9.8096
-7.1733
15.6/2< 5.5948 5.5619 5.5-144 5.5290
-5.476-1
~H886 H820 1.4617 2.3849 1.365] ].1861 1.1050 1.1897 1.1787 1.1655 1.1173 1.1019
~i= 2.0471 1.0361 1.0107 1.8628 1.8452 1.7904 1.7487 1.7189 1.7114 1.6916 1.6675 1.6433 1.6148 1.5-168 1.5271 1.5139 1.3845 1.3384 1.1475
D;1 \:) C'll ()
sr Ci V)
~ ):)
::3: <:

~ ~Q:-
":-"7.3172
O ~O ;-I o
~
IA ~
IA o ... :z:
I ~ t l 2.6021 ~ 2.5561
'" 2.'2'4 C)
~ 2.4105 C) 2.4441
l 2.4179
~ 2.3915 2.3674 ... Iof 2.3411 :g ~ 2.3147
~ 2.2972 2.2'33
~ \oi 2.2l92 • 2.1561
til 2.1304 2.0756 2.051' 1.9922 1.9639
......,;.- 1.9374 1.9191
"" 14.770 1.193' o 1.8693 1.1408 1.1111 - 1.7916 \.to 1.7614 1.7421 1.7ll3 -- 1.6960 -b 6.0000
_ . 1.6697 1.~11 1.6148 1.d039 e 1.'644 1.'117 1.0107 1.0378 [h1 o 1.0110 • 0.9413 \:)
,,- Cll n q-o til
Q. Cll
:::tI ~
VI :< o
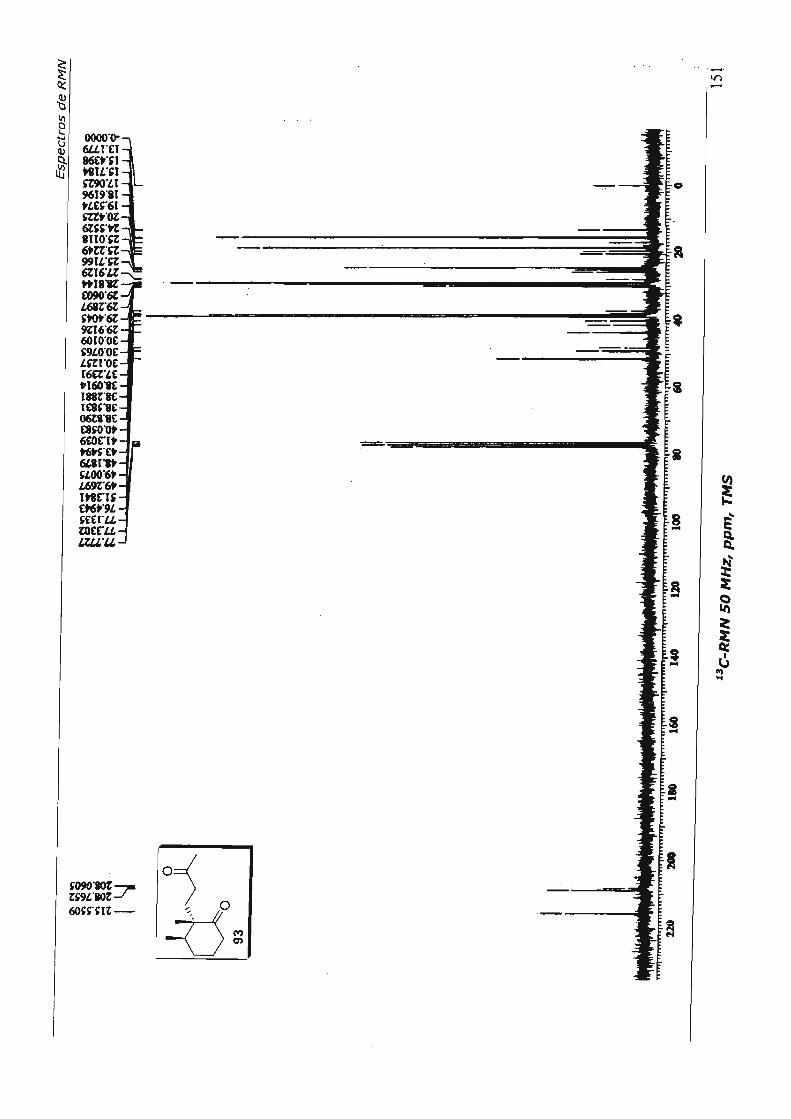
~~----------------~------
-VI .-
-21'.5509 r 208.1652
--208.0605
17.1727 17.3302 77.1335 76.4943 51.3841 49.2697 49.0073 41.1819 43.5494 41.3039 40.0383 38.1290 38.'831 38.2881 38.0914 37.2391 30.12$7 30.0765 30.0109 29.9126 29.4045 29.2197 29.0603 21.8144
"'-21.9129 2$.7166 2$.2249 2$.0118 24.'529 20.4225 19.5374 18.6196 17.0625 1'.7184 C\l 15.4398 'ti 13.1779 ~ -0.0000 S'
~ ~ ~
~
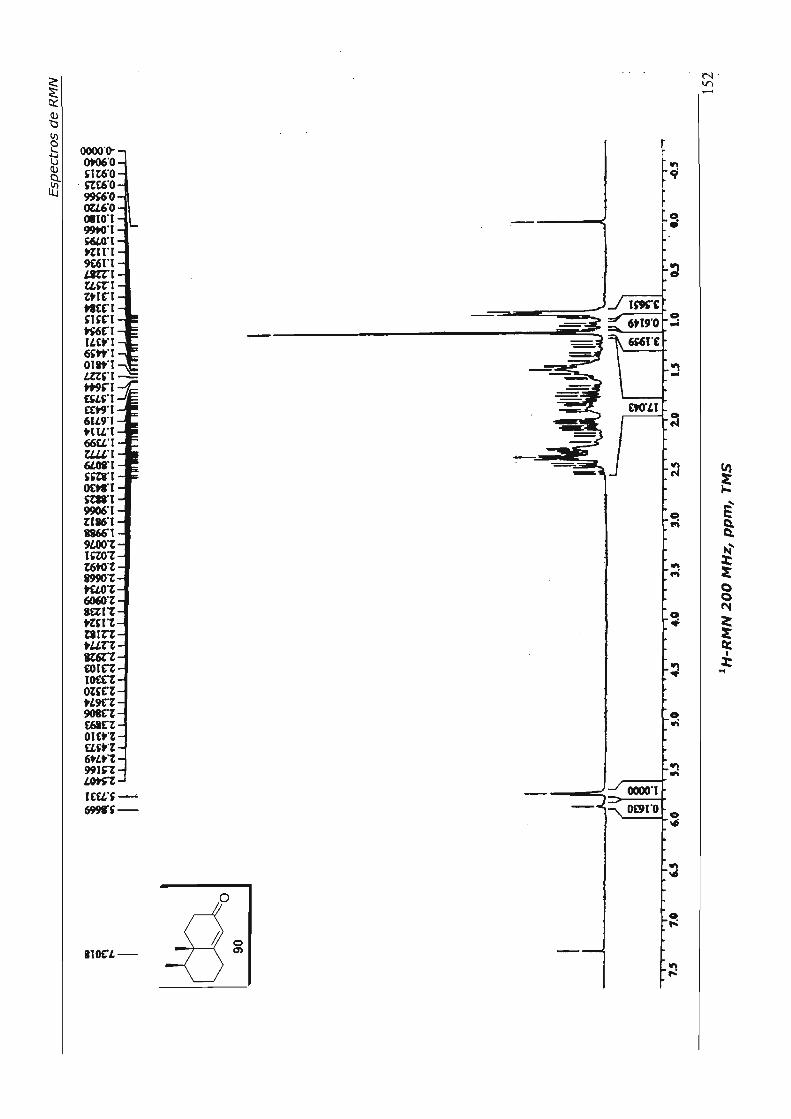
-7.3018
fi' o 0.1630 ._,.8669 1.0000 -'.7331
~ 1.S407
I.. 2.5166 2.4749 2.4'73 2.4310
~ 1.3193 o 2.3806 2.3674 2.3'20
... ~ 2..3301
::t 2.3103 I 2.2928 ~ 1.2174 ~ 2.2182 ~ ~ 2.1524 o ..., 2.1238 Q 2.0909 Q 2.0734
~ w 2.0668
~ '" 2.0492 2.0231 ... 2.0076
'ti 1.9981 'ti
~ 1.9112 1.9066 1.1125
~ 1.1430
(n 1.12!' 1.8079 1.1712 1.7399 1.7114
t" o 1.6719
17.043 1.6433 ~1.S7g
1.-'644 - -1.5227 I.. 1.4110
1.44'9 3.1959 1.4371
- 0.614' 1.3954
o - 1.3'15 3-"'1 1.3384
1.3142 1.l$71
f! 1.2217 1.1936 1.1124 1.0795
" ~ 1.0466
o 1.0110 G.9720 0.9566 [;\1
e 0.9325 . 0.9215 "tl
('l) 0.9040 (')
~.OOOO Si o ti)
~ ~
Vl ~ · N

1 f ~ r ~ IE t ~
.-~ t:
t ~ i
-ir
i ~
~ • -... a '" ? .
:o ~
~ til li Q
~ ~ p..--.
~ ~
~ ...
~ -~
8
F
~
~ t :i
~ cn I ~
..
~ t .,
~ .. ~
~ ~-.:; r-
~ ~
~
o ~---~ ~ ..
~.
Olned O?S ap apep!sJol\lUn
V:lIWlnO 30 OlOlllSNI \f~310118IB
.. ~.
-
8& O
-
-199-'296
---124.0505 ·~1l1.2647
117.7241
. 17.2981 17.1014 76.4623
44.933' 43.1101 40.9023 39.0341 31.7556 3'.5764 35.1661 34.9046
I: 34.0197 33.5444 33.3642 30.3128 29.3657 21.5791 21-'136 26.5199 20.S608 16.0430 15.2236 ~
\:14.3223 "1::í -4.0000 !b
~ g-~ ~ );)
~

CçC02H
97
/
~~(
o o OI N
....
~ere~m;~m~~~~~~~m~~~g ~mw.NoOl""W.NO~eD""""~~NO ~~~~~~~~~~eD~~~""~~~~~ NNNNNNNN~--~ 000
lu
~~ ~~ .... ~ ~~~O ..... W(T) .... Oal eDcDN .... ,....t,D-ON ..... anUlCD ","".-tOU') NC\JNN •• • o ' • • ••
O ...... O-N 0 ..... 0 .....
"'''''''''''' '" I'"'''''''''''''''' I'"'''''''''''''''' I""""""""'" I'"'''''''''''''''' I'"'''''''''''''''' I""""" i""'''' I""'" ,Ti 12 10 B 6 " 2 O
lH-RMN 300 MHz, ppm, TMS
Espectros de RMN
CUrrent DIta Paralleter~ NAME Tiaga EXPI«l 3 PROCNO I
F2 - Aequl<ian Pera .. tera Olte_ 500000 Ti.. 1.~3
IHSTRUM speet PR(IIHJ 5 _ Duel 13
PlA.PROG zg3D TO 65536 SIl.VENT coel3 NS 2~
os o Sltl ~562 . O~~ Hz FlIJlES o .069611 Hz AQ 7 . 1827955 ,ee RG ~ OM 109 . 600 usee DE 6 .00 use e TE 300 .0 K OI 1.50000000 sec Pl 8 .00 usee DE 6 .00 u,ec SFDI 300 . 1319591 NH2 IAJCI IH PU 1.00 dO
F2 - Praeass ing pere_ters SI ~7~ SF 300 . 1299967 If-IZ _011 EM SSB o LO G8 PC
0 .30 Hz o
1.00
10 IN'! plot poroMlers o ~ .5O ~
F1P 1~ . 139 pp. fi ~2~3.48 Hz F2P -1.000 pp. F2 -300.13 Hz PPMCI4 0 . 6728-4 pp_/e. HZCII 20 I. 9381~ Hz/e_
154

lD m ... LO
r CçC02H
97
..... lD OI .... lD N
OI OI ........ .......... v ~~~~~N~~~~~~~~g~~~ffi~gg~ VNO~~~~~~_~~~m~~~~~~~~o . . . . . . . . . . . . . . . . . ~~~~~~~~~ffl~rerorere~~~~~~~?
~~~p~;
,bc; ~Ân .. l~ .1_ -., --- ......... ..,.. : • ~.........,...r---..-... C ;' 4 '; *4
J3C-RMN 75 MHz, ppm, TMS
Espectros de RMN
Currllnt Data Paralletsrs NAME Tiago EXPI«l 4 I'AIJCHO I
F2 - AcQulsltion Parallelers Date 500000 TI .... 1.49 INSTAU14 speel PIUIHO 5 "'" Dual 13 PlA.PROG zgpg30 TO 65536 Sll.VENT CDC13 NS I~B9
OS 4 SNH 21097 .047 H2 flORES 0 . 321915 Hz AO I .5532532 see AS 32768 DN 23 .700 usee DE 6 .00 usee TE 300 .0 K 012 O • 00002000 see PU3 22 .00 dB DI I . 00000000 see CP!FRG2 lIalLzl6 PCP02 105 .00 usee SF02 300.1312005 HHz NOC2 IH Pl2 0 .00 dB Pl12 22.00 dB 1'1 7 .70 usee DE 6 . 00 use e SFOt 75 .• H6397B lfiz MJCI BC PU 4 .00 dB 011 0.03000000 see
F2 - Flroees6ing paralleters SI 32768 SF 75 . 4677513 MHz MDN EM SSB O L8 1.00 !lz GB O PC 1 . 40
lO NNR plot p8I'alleters ex 22.50 e_ FIP 2~0 . 000 ppll fi 18112 . 26 Hz F2P -10 .000 PPII F2 -754.68 Hz PPMCN 11. 11111 PPII/ell
IItU'! 838 .53052 Hz/c.
155

Ctto 98
9
-... ~ ...,
I ~!!!~ lt1 - OI OI aJ 10 NN N
W ~~~~~~~m~~~~M~~Nmruw~m~-N O~~N~~mNW~W~O~~~.-~m~o~N.N_~_~.m.mo~_~~m~Om~NmO ~.~~ __ N_OOmm~w.~ __ ~m~m.~ ~1O~~~~~-_~~~omlOlt1lt1lt1lt1 ... ocncncn
ttL~~~dY3
o
lH-RMN 300 MHz, ppm, TMS
___ E_sLpectros de RMN
Current Data Para_tars NAME Bago EXPNO I PROCIIO
Fi! - ACQuisition Paraaelers Da te _ 500000 Ti.. 1.30 INSTAUN specl PAOIIt() 5 .... OU.I 13 PlJ..PROG 1930 TO 32768 SCl.VENT coeI 3 N5 32 05 O Slti 3531.073 Hz FlORES 0.107760 Hl AQ ~ . 6399989 see RG 6~
OW 1~1 . 600 usee DE 6 .00 usec TE 300 .0 K OI I. 00000000 sec Pl B.OO usec DE 6 .00 use e SFDl 300 . IJHBI6 ltiz NUCl IH PU 1.00 d8
F2 ·- Processing p.r .... ters SI ~7~
SF 300 . 1299B81 Ittt WOW EN SSB O LB 0 .30 Hz G8 O PC 1.00
10 NNR plot par ... ters ex 22 .50 C. FiP 10 .000 ppll FI 3001 .30 Hz F2P -0 .500 pp" Fi! -150 .07 Hz PPMCH O. ~6667 pp"/CII HZCM 1.0 . 060~ Hz I c.
156

-
'" In In
a:J 10 ....
I"l .... In ... ~
I
.... 10 C\I
10 o ....
1"l1na:J1"l C\l0l0l .... "II:f-01U1 .... ........ 1010 ",...,r--.,....
~
,.....lC01 ......... COOO1 ...... lOaJ ...... 01lCl"" ..... ooq-,..... .. coooq-I.D_ ...... ,..... O-CO"""'OlllCDN-O(QOlNO,...... . . .. . .. ... .. . . . O1CD..,om ...... tOtDw~C\Jo ...... l.n~ C'l(Tlt"1~NC\I('\I(\JNruC'\lC\J __ _
~~~
~o 98
I
I
_l Wl1lUJ I i I I I I I I
?nn 175 150 125 100 75 50 225 25 O
13C-RMN 75 MHz, ppm, TMS
Espectros de RMN
Current Data Para.eters NAIE 1Ia;0 EXPHO Z PROCNO
FZ - ACQuisition Paraooelers Date_ 500000 Tille I. 35 rN5TflJM spect PROOHO 5 ... Oual 13 PlA.PROG zgpg30 TO 65536 so..VEMT NS os SIIH flORES AQ RG ON DE TE DIZ PU3 OI CP!J>RG2 PCP02 SF02 NUC2 Pl2 PLI2 PI DE SfOI NUCI PLI ali
COCI3 IZZ~
~
22675.736 H2 0 . 3-4600~ Hz . -4-451188 sec
32768 2Z ,OSO usec
6.00 usec 300 . 0 K
0.00002000 sec 22.00 dB
I. 50000000 sec nltzl6
105.00 usec 300.1312005 NH2
IH 0 .00 dB
22.00 dB 7 . 70 usec 6 .00 usec
75 . -4767751 ltiz 13C
-4 .00 dB 0 . 03000000 sec
F2 - Processlng parsllleters 51 32768 SF 75. -4677573 ltiz
~ EM S~ o LB 1.00 Hz G6
PC o
1.-40
la ~ plot para..,ters CX 22.50 CII
FIP FI f2P FZ PPIICN HICH
2~0 . OOO ppm IBI12.26 m -10.000 PPII
-75~.68 Hz 11 . 11111 ppII/CII
836 .53058 m/clI
157

!lO
~g:-....
!lO ~
--()
:-I o .... I\)
~ -7.2909 (1)
:-I ~
P. ....
0\ o
.... 0.0389 V. ~'.3$3' 0.0555 --5.2658
... -5.1692 ::t .... 1.0000 I o ~ :t 3.7870 ~ ...
V. 3.7760 N 3.7584 Q 3.6882 Q
:t ... 3.6027 C
0.6495 3.5~
~ 3.5325 3.5149 ... 3.3549
:g w 13.2231
V. 1 . .2442 3.1946
~ 2.3279 . 0.3661 2.2665
~ w 2.0975 o 2.0100
til 2.0558 2.0339
!'" 1.9944 .... . 1.9'17
1.3840 1.9110 1.8672
~ 1.8474 5.9683 ~1.1013
\:1.7289 1.7092 -V. ~1.4656
6.1287 1.4283 1.3954
~ 1.3109 1.3823 1.3450
·7.6543 1.3174 1.3099
~ 1.2923
.... 1.2769 1.2594 1.2089
g 1.1497 1.1212 1.0619 1.0312
W ~ 1.0071 .... 0.9786 "tJ 0.9083
(b ()
0.8601 ,..,.
.!. 0.8294 a o ~.OOOO
11'1
~ ::tl - ~
VI :< ·00 .
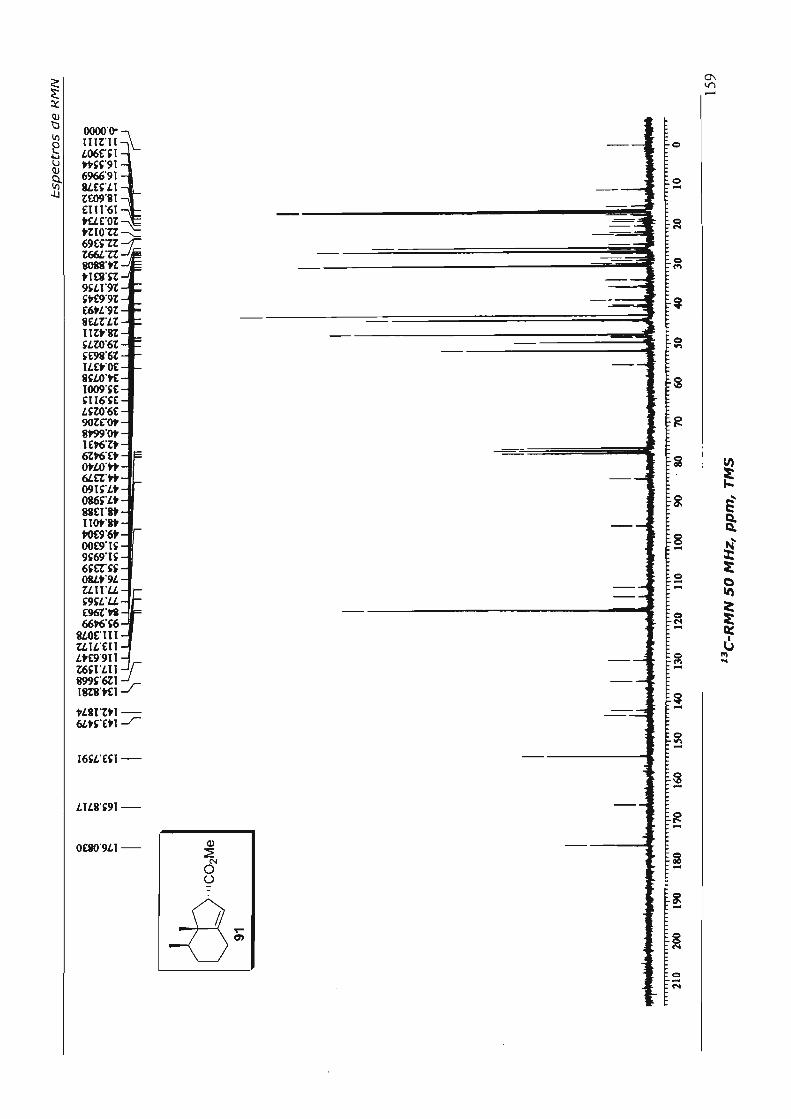
-' N -;~ Q
~ -! -Cf
~
-t ... w
~ ;:Q ~ ::t
'" C)
~
~ , :g ~ ~ tn !
Cf
S
.... o
t
w o
~ ~
-o
o
---
~....:
~-
~.-
~-~-
f--
r--
P--
~-
~
~
:=-~.-
---0----
F--
-
--
-
-176.0830
-165.8717
-153.7591
-.-r 143.5479 -142.1874
.....r 134.8281 í 129.5668
Jr117.1592 116.6347 113.7172 111.3078 95.9499 84.2963 77.7565 77.1172 76.4780 55.2359 51.6956 51.6300 49.6304 48.4011 48.1388 47.5980 47.5160 44.2379 44.0740 43.9429 42.9431 40.6648 40.3206 39.0257 35.911.5 3.5.6001 34.07.58 30.4371 29.863.5 29.0275 28.4211 27.2738 26.7493 26.634.5 26.17.56 2.5.8314 24.8808
!!!Jr 22.7992 J-22.5369 ~22.0124
20.3734 19.1113 18.6032 17.5378 16.9969 16 . .5.544 15.3907 t 11.2111 .0.0000

00 O
~ ~s=~ IA Co) Ij -7.3369
~ e O
0\ V.
0\ o )[5.7879
1.0000 4.1380 4.1007 3.4710
IA V.
3.4491 3.4161 3.3942 3.2955
u. 2.4661 Q 2.4420
2.4332 2.4157 ...
:z:: ... I V.
2.4047
::a 2.3805 2.3542
:t 2.3147 ~ ... ~ Q
2.3060
Q 2.2884 2.2796
Q 2.2555 :t w
~ V. 2.2379
2.1288 2.2138 . 2.1897
O O
2.1699
3 w 2.1655
. Q 2.1458 2.M80
~ 2.0492
rn ~
1.9220 1.8891
IA 1.8584 5.3734 1.8430
1.8276 ~ 0.4267 e
1.7618 1.7443
~1.7048
... ;k 1.6872
u. 1.6412 1.6236 l.5271
3.0261 1.5029 ... Q
1.4810 1.4656 1.4437 1.4217 1.3998 p
u. 1.3779 1.3"9 1.3362 1.3120
e o 1.2967 [hl 1.2616 'tl 1.2462 rtl 1.2089 C')
0.0000 Si C ti}
~ ~
..-0\ ~ . 0 .

-::
-~
-~
-~
-N o
-g
00 o
~
~
N o
o
-0\
1
--
-
!:'""
r---
--
t:-
L
r-
-
-
-
-199.8S9S
-169.1446
-12H047
j77.901S 77.6392 77.2131 77.0000
-76.3772
-60.2822
-41.5976
-34.0746 ~33.SS01 "\\....31.9603
'-30.0S9O '-23.0933
-IS.2097 ~ 14.0132
-~.lg0S
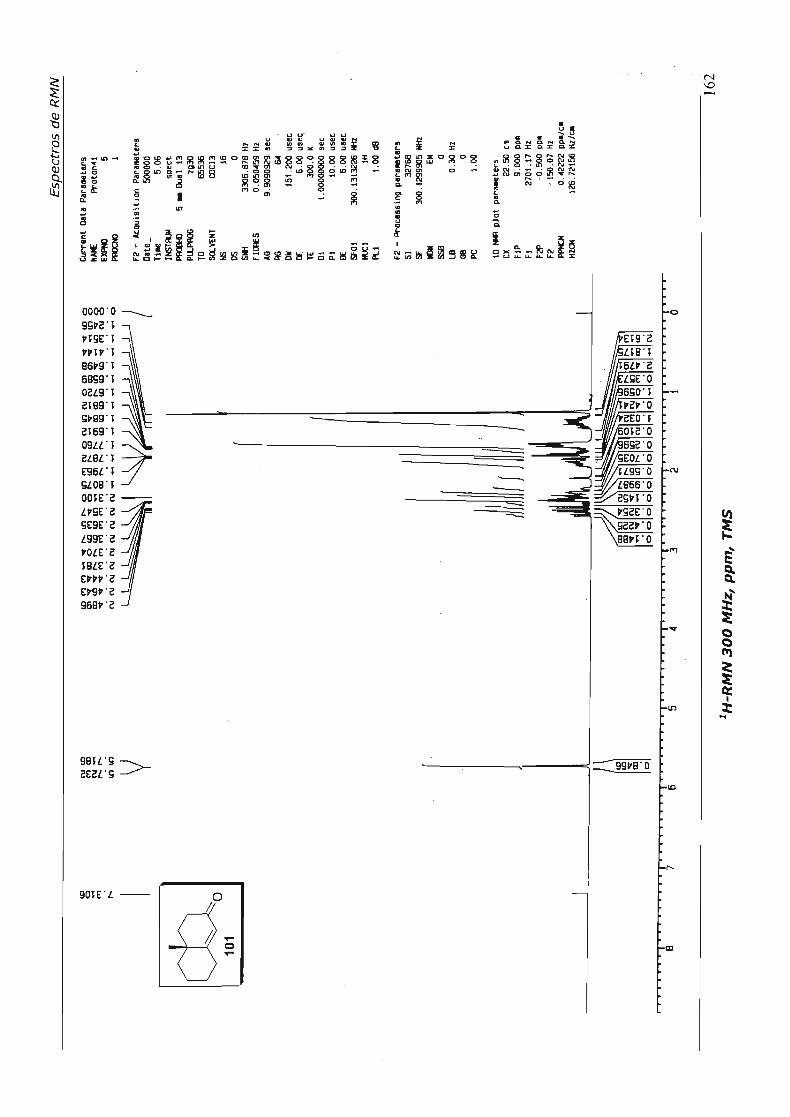
10
o .... P
l ......
mo
101
C\I
CD
Pl
CD
C\I
....
,....
,....
U1
U1 v !~~
~~~~~~~~g~~~~~~~~mm;~mg
CDCD~""''''''CDCDU1
.... omm,....mmCD''''''U1~_U1~O
~ •• P
lPlP
lPlP
lPlC
D""''''''''''''IO
IO
CD
IO
CD
IO
.PlN
O
. .
. .
. .
. N
NC
\lN
C\l
NN
C\l
N..
..
o
L~~I~~~/
lH-R
MN
30
0 M
Hz,
pp
m,
TM
S
Esp
ect
ros
de
RM
N
Cu
rren
l D
ata
Par
alll
lllr
s N
AIE
Proton~ 1
EX
PI«l
5
PRO
CI«l
1
F2 -
AC
Qui
s i t
ion
O
ale
Par
alll
lter
s 50
0000
5
.06
TI.
e
INST
FtJM
PR
IIIH
l PI
1.PR
OG
TO
sa
..VEN
T NS
OS
S
Iti
FH
IlE5
AO
AG
DII
11:
TE
spec
t 5
_ D
ual
13
Ig30
65
536
CD
Cl3
16
O
3306
.878
Itl
0.050~59
Hz
9.9
09O
929
see
~
151
.200
u
se,
6.0
0 u
se,.
300
.0 K
01
1.
0000
0000
set
P
I 10
.00
usec
11
: 6
.00
usec
SF
OI
300
.131
3226
NHl
N
lX:1
IH
P
U
1.0
0 d
B
F2 -
I'ro
cess
ing
para
.lers
S
I 327~
SF
300
. 129
9905
NHz
I(J
W
EM
SSB
O
La
0.3
0 H
z G8
O
PC
1.
00
10 N
4R p
lot
par_
lers
ex
22
.50
C~
FIP
9
.00
0
ppa
FI
2701
.17
Hz
F2P
-0.5
00 p
p~
F2
-15
0.0
7 H
z P
PK
J4
O . ~2222
pp
a/ca
HZ
CM
126
.721
56 H
z/,a
162

ru
lI)
ID
lD"'f
O
~gst:~m~~~
-"'
f lI
)
-0'
1 ...
...
In
"'
f o
"'l
0l1
Cl
""if
01
alO
1c
.c
......
O
UJ
ai
..
.....
. o
"'f
..... "
''''
......
,...
.U'l
t"lN
,...
....
N_
0'
1 !:::
ru
....
........
.....
""""
(T)(
T)(
T)r
uru
ru
-- I
\f
\\\11
/ f
mo
101
I"""'~
2~
~Àn
.~o;
,L
.L
-: ";'
:::7'-
;=:
-;q:.
; ~
13
C-R
MN
75
MH
z, p
pm
, T
MS
Esp
ect
ros
de
RM
N
Cu
rren
t D
ata
Par
arae
ters
NA
ME
13C
19
EXPN
O PR
OCN
O
F2
-A
CQ
uis
ltia
n
Par
alH
lter
s O
ate_
50
0000
Tl
IIe
~.~I
INST
RUM
sp
eet
PRO
OIfl
5
_ D
ual
13
PULP
ROG
zg
PQ30
TO
32
768
SOLV
ENT
CO
CI)
N
S 20
48
05
O
sNH
2109
7.0
H H
z Fl
IlRE
S 0.6~3B31
Hz
AO
0.7
7665
16
aee
RG
2317
0.5
OM
2
3.7
00
use
e DE
6
.00
use
e TE
30
0.0
K
01
2 O
.000
0200
0 se
e P
U3
2
2.0
0 d
B 01
.0
0000
000
8ee
C
P!PA
G2
wal
tz 1
6 PC
PIl2
10
5. 0
0 u
see
SF
02
30
0.1
31
20
05
'ti
l N
UC2
IH
PL
2 0
.00
dB
P
U2
2
2.0
0
da
PI
8.0
0
use
e DE
6
.00
use
e SF
OI
75.4
7677
51
NH
z N
UCI
13
C
PU
4
.00
dS
01
1 0
.030
0000
0 le
e
F2
-P
raee
ssin
g
par
arn
eter
s S
I 16
384
SF
75
.46
77
55
3 '
tiz
~Oll
EM
ss
a O
LB
I.
00
Hz
GB
O
PC
0.1
0
10 N
MR
plo
t p
arar
aete
rs
ex
22
.50
c.
F
I P
259
. 000
PO
li F
I 1~6. 1
5 Hz
F2
P -1
0.0
00
Pal
l F2
-75~.68
Hz
PPM
CN
11.9
5556
po
li/e
_
Hl!;
M
902
.258
97 H
z le.
163
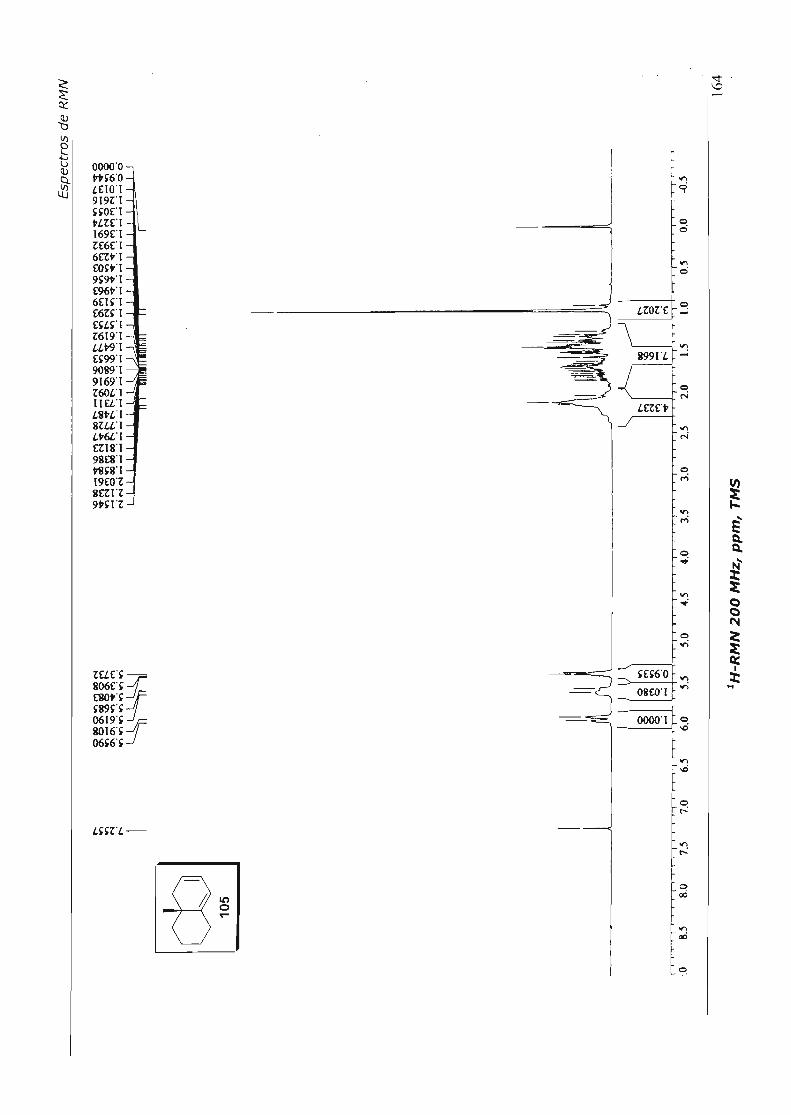
:1 8-
u,-..... o
oc UI
o
-.I u,
-7.2557
-..l o
f3\ u,
);5.9590 f3\ di: 5.9108 o 1.0000
J5
.619O
5.568S 1.0380 ~5.4083 .. ..... 5.3908
:z: u, 0.9535 5.3732 I
:ti l ~ ~ o
N <:) <:) ~
l .....
:z: ,N • 'ti o 'ti
~ ..... u,
~ 2.1546 2.1238
U) ..... 2.0361 o 1.8584
1.8386 1.8123
IV 1.7947 u, 1.7728
4.3237 1.7487 1.7311 IV
o 1.7092 1.6916 1.6806
7.1668 1.6653 u, 1.6477
1.6192 l.S753
o 3.2027 1.5293 1.5139 1.4963 1.4656
o u, 1.4503
1.4239 1.3932
o 1.3691 o 1.3274
1.3055 1.2616
[Xl 6 1.0137 u, 0.9544 "b
0.0000 (b r) ...,. ti Vl
Q. (b
::tl
0\ ~ .+:-<
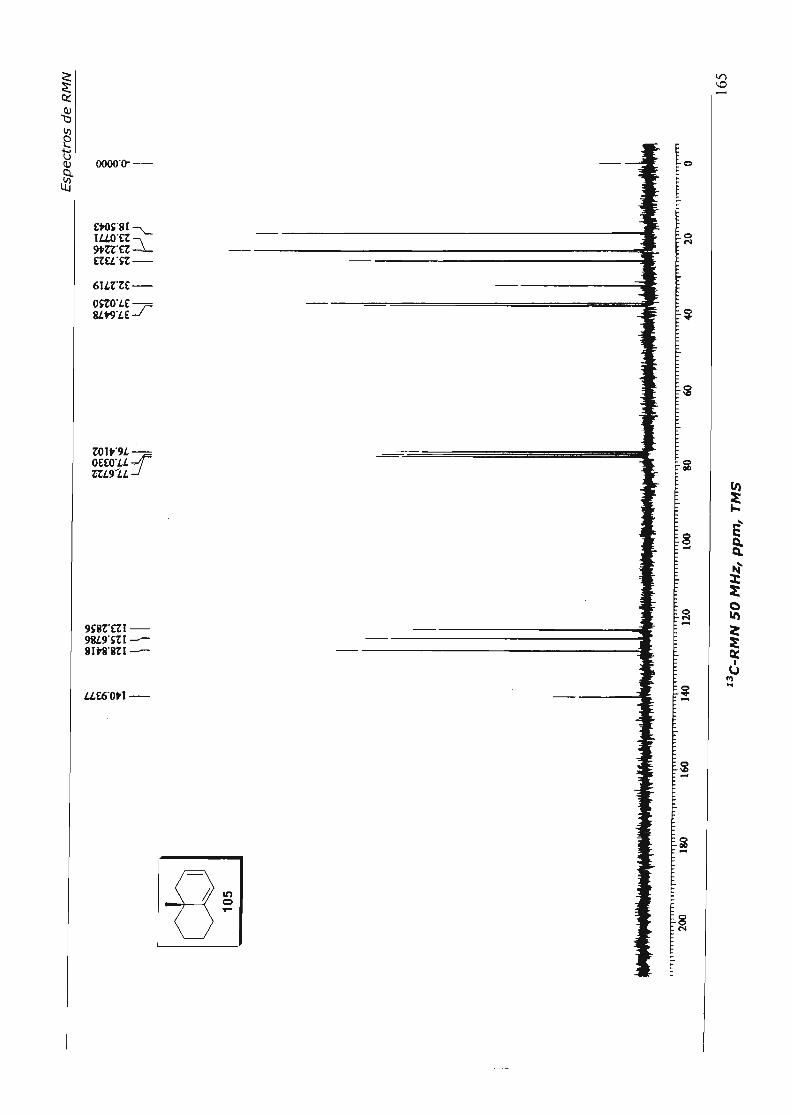
.. lu t"') I
::o ~ ~ \11 C)
~ ::t
.......
:g ~
~ cn
0\ Vt
~
N e
8
~
o
~
~ r
~ f ~
"'!
~
~
~
~
l~ ~ ~
o-
~
~
~
~
..:i ~
...
~ ~
~
~ ~
1 ~ Í"-
~ ~
~ ~ t-
1 r ~ t 'i
1 to f--
-'!
-
--
-
--
-
-
-140.9377
-128.8418 --125.6786 -123.2856
. r .71.6722 ",r-77.0330 -76.4102
J37.6478 -37.0250
-32.2719
-~.OOOO
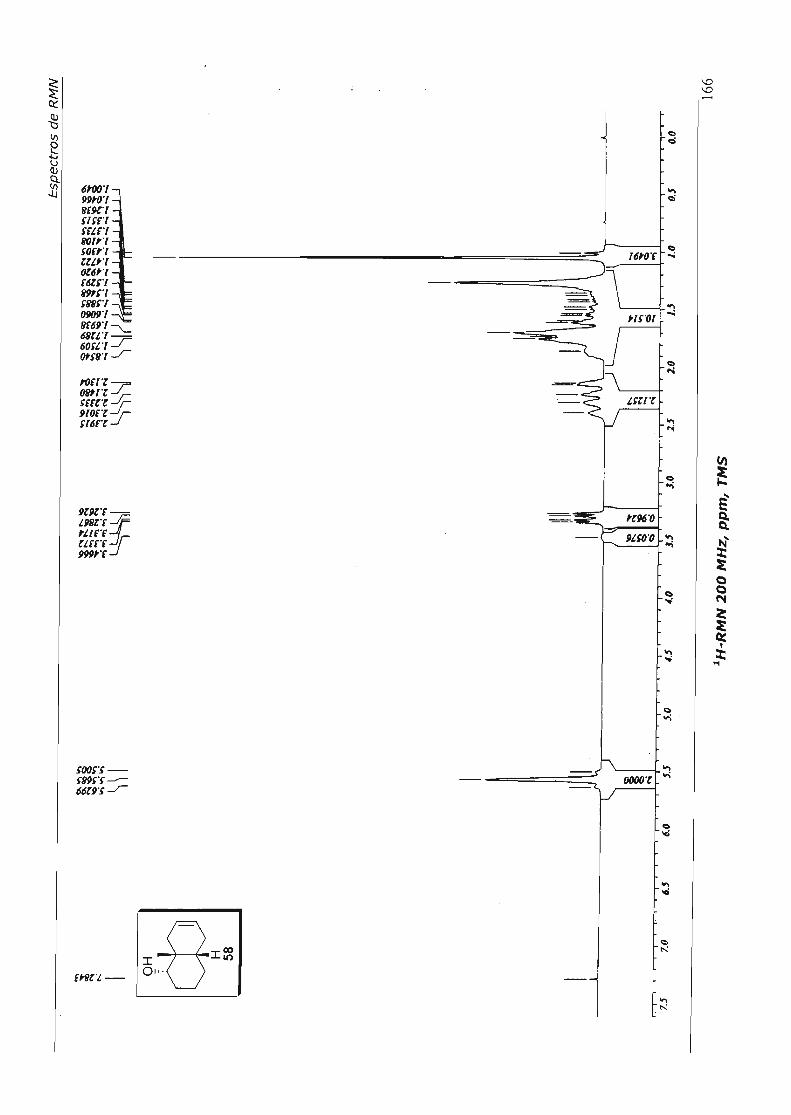
- 7.2843
5.6299 - 5.5685 - 5.5005
N14121 ap
soip
ads2
O
,„ 2.0000
o o
(/)
^Oh _
o
ú10.0576
0.9624
aJ
2.1257
10.514
3.0491 et,
L
Crt 03
%3.4666 J 3.3372
3.3174 3.2867
- 3.2626
r- 2.3915 -1/- 2.3016 f2.2335 7- 2.1480
' 2.1304
_/- 1.8540 _r- 1.7509 - 1.7289 -"\-- 1.6938
‘
11.:88 55 46850 1 1.5293
11 ..449722 2° 1.4305
1.4108 1.3735 1.3515 1.2638 1.0466 1.0049

uI co
r- 124.0148 123.8181
j78.0983 77.6393
- 77.0000 76.3771
39.2636 - 37.1325
- 30.5753 -\"-- 28.9688 ""\`-- 28.6246
`-• 25.8870 23.9526 23.2969
N14 d
ap
soip
adsg

2.0433
0.1014
0.8932
- 7.2821
5.6190 5.5685 5.5597 5.5531
- 5.5005
__,- 5.2240 4.7084 4.6931 4.6755 4.6053 4.5812
J- 4.5527 -- 4.5285
-/ O
.. 2.0493
N," 3.1612
9.6482
o _ 3.0000
_
C\ 00
2.3981 2.3674 2.3498 2.3235 2.2972 2.2708 2.0822 2.0712 2.0624 2.0449 1.8255 1.8035 1.7552 1.7333 1.6938 1.6785 1.6521 1.6368
1.6192 1.6039 1.5973 1.5687 1.5468 1.5161 1.4985 1.4876 1.4656 1.4393 1.4261 1.4108 1.4042 1.3910 1.3844 1.3537 1.3098 1.2901 1.2396 0.9281
Nkld
ap scup
ads2

CD
ID
02 CD
7.26969
CTI
_<5:15.26816 5.25596
4.61426 4.59880
\:4.57741 4.56089
2.31330 2.26138 2.05658 2.05547 2.03846 1.73294 1.72267 1.69262 1.68372 1.66813
1.59774 1.58674 1.53314 1.38173 1.37081 1.28561 1.27579 1.25759 1.25102 0.89360 0.00000
1.0781 -
1.0000,,- a. -
CJ
2.1383'\._ __------
ru
2.5400,„ - n.
2.2103,G5-',= 0.1639/-. 3.3358///-
F.), 3 z,, -, .-2 ..-- r, ..,- "P, @ G Li 2p V. 4 Zl 22 g lâ R.- R W1 g C; :. tTI eS;., -e rn z r-, x n
,... ..... 'n P E'2.2 ,,', m
: 1 N' - : ,'
1 6 I P 5 2 c c.
% ,... a., ,2 ta, r O 5 .... -o , O O co g
.§ ... ..O•• o ... ., • 00, O ...,. i ... o
17, O o n.3 ', O O o O 1:1 -o E
§ A R O 2? F' R; m 5' §w tl ia 'k', a, o 2 4f m .» 2 E2 m .
,• • e m• m ... , ... a O 01 cu o M1, o et.
Lna .....o ruo, r; il"; • .4.21 • O • n
01 01 O O lu o fl, O 8 O eziN2,.. O O • O O .1à tu to o win ""w ..... g UI . Oo ea
aaa000 .0 0S-00000002,14100ruwenaial,.-00
SoS0212n a o a ra a I= a
''S 0
il 2 E ' 2 2 R ''' . , .
-.. ' f - . . , ,. e a . .... ,,, o .
n , n ff n n II "
NN
d ap so
ipadsg

0
170.881
131.310 130.465
'--130.132
mç117.787 117.512 117.379 116.895
80.099 80.019 77.426 77.206 77.002 76.579 41.623 39.948 39.036 35.813 33.887 31.690 29.079 28.575 28.336 27.047 26.926 26.676 23.864 23.704 23.595 23.377 23.310 21.169 20.670
,s+ N •-• -• a•C O
, x -,, -, -, -.., r, ,... 2 2.2g5,,,2 z, 0 12 gl X A n1 0.... i_12 ,-. ,r0 0. R .3. ..., -,1 51./1 51 ,,, Z. p. a% 6. ,,, -n ,... r- rr, ..... _ 2 . , Ed . -.. - - . ,.._ N N N
Ir n) no.ã Ia N.) 1.4,
M tn
53 .,, -. -I te ri, I u5 a .... li x s
O
,. 2 N -42 g g 1
Dr•-•
6"
.... R . - . co . . o -.., 5' O ...J
1, r.
CD W + .... g)
fla W LO W
',.. ff g 8 O . O
• O fs., §
-C eu n
a -.. • ,cowg ... -o w ... ...• .., ,.., R r:11 ;: O . . , ,.., -.-...,, LJ (X a L11
LJ - a 0 O a La ...J
0 O a 8
...... --I8;•088.1. 6 IN •-• 01 •
10. 6 m át ,-;, R ., • O IL 8 6 N 6 N I •
u, -. ign, O N 0 GT, O Eà'Nii,':•-siSSISSa'SSgb8g-g2E- 2' g . g"' E si 5 rz 2- rZigã
•.. O.
•• .~,
NI •-... 00 Cl cn Ca O 00,00Z(Ja3 - - LLe O Lr, •••• 40 "5
•••• f 2 2 a f 5 5 c% c9i. g g 2 8;2 " 5 g 2 '1 n N O O R n n II mn 0
n es n n n
O NN
d a
p s
odp
ads2
zo na rw ó 1,1 ig 3
51

- 7.2800
26950 2.6818 2.6348 2.6182 2.6016 2.5809 2.5622 25290 2.4668 2.3796 2.3569
2.3.181 2.3278
_7- 2.2552 _/- 2.1722 _/- 2.0601 2.0435 12.0020
1.9854
1
1.9847 1.9460 1.9191 1.7552 1.7240 1.7178 1.6742 1.8556 1.6182 1.5581 1.4709 1.0850
- 0.0000
NW
H a
p s
wp
adsg

NN
d a
p s
wp
ad
sg
- 215.5812
122.4823 121.2858
77.8392 77.0000 763808
- 48.4614
--- 41.8467 /- 37.5820
37.1722 34.8974
27.7316 25.1092
20.5655 18.9957 18.9302

£L1
'1999 '6Z
'8861 'a7 ualpayeLiai 'c '± 'wos)pcmg f'd 'c 1seiclaa loglao3 f*3 *v 'auaaig (EZ)
•09817 '69 'VOU *WaL0 '5-10 •C 'V 's 'ulwzoN 1.*g *C1 `ÁPIDal (ZZ)
'17861 'olned oeS ap apepsJamun 'opeJmnoa ap asal T 'A 'D leAl!S ea (TZ)
'908Z 'TZ 'EL61 '09 'wJecíd 'ulPLID 'H 1!Liseqns4HAJ H 1e.mweleN f'N 1NseÁeH (OZ)
'517EE IVOOZ 'S 'a IÁJDIDa2i (6T)
T9E17 '99 'IOOZ 'iliPL/3 '510 'C '3 'c 'auÁed 1.'0 'A 'opebles-eÁeN f*D '1De9 (8T)
'E99 '6661 '4-7.97 •3 .1 'auÁed f*D '>foe9 (LI)
'TZS 'S861
'3Upp wseig 'peDV 'uv •E J. 111JOS)1D0.19 1s9Jclaa 1.'j 101.11a00 f*3 *v 1auaaJD (9T)
'ES179 '66 'LL61 'DOS 'LlIPLID "WV V. '3 *D ismaipuv 1.'i .D 'swis f•V 'a 1suen3 (9T)
'16917'£L61 *-7.737 uwpayegai 'i'D Sw!S f'y 'a 'sueÁ3 (17T)
'TL9 'IOOZ sIsati-luAS '3 '1 eAlIS (ET)
'96LZ 'ZL,61Ái-lsiwaypo4Átid 'A 'incuaH 1ueLuoi lemoN 'Áu4onoN (ZT)
'56 'Zr '17861 7owo_ite 7c1c1V T.1 '40.imeN 1.•c'eulewJeH (Ti)
176I
'ZTT '1661 'iowo4u2 7c1c/v C C 'eulew.ieH :•9 •vi 1uewsi 1.•0 Ilno>1 14cumeN (Ou
'T6 £' '8861 '1142/30-/M 'tipaillod
'5./0 'waLID sul ">IneN ad 'V ilIdeum ç 1eyiewJeH 1.3 11Ázsoig 'ilsu!scn:i (6)
'SOT 'E£' '8861 'mePo-M1
.cpamod .z.12 .5.10 •wato 'sul ',IneAl ad 'c 12q4etweH C 'IODaJN 1,•/ leno)paiN (8)
'LZE 1£8 '8861 'no/sowaL/09
70w0-7u2 e40V 'g 'zpzcua 1,,kulonoN f•I 1eglet.weH 11Ázsoig f'r '4cumeN
'66 'Zr '17861 70.33 .4sÁs •wat.po!9, l 1Áu4onoN 1.•c leglewJeH :'r 14climeN (9)
176E
'86 '17861 yddv 'C l ii,kuionoN 1.•C 'auletweH :'3 11Ázsolg Ç 14cumeN (9)
'86TI 'OE 'Z861 'ling •wJeLid 'watlp 'H luseqns4NAJ '•'>1 ligseÁaH 1.•>i 'oue>I (17)
'ETLI
'9L61 Msfi.vatpo_Mtid *1 'v Juosa!wer f'd 'g 1JauJr-u. '.'H •3 IDR2:1 f'2J 'D Juosa!wer (E)
'8TE '8961 'Pu/ 'waL/D
'S leanwnsleà f•ki 'iuseÁeqo>1 :•S 'e.mweleN 1.•vi It.iseÁeH !'i 'w!52)121 1.'>1 'eÁeN (Z)
'692 '8961 '-1-7a7 uaipayeilai
•A 12.1eLie4!>1 f'D •IA1 Spoom 1..1 '012>i '..>1 ialeueAt.is 1.'23 'ePou0 N jacIV (i)
sepugJeJaW8
sepuaJajad

Referências
(24) Brocksom, T. J.; Coelho, F.; Deprés, J.-P.; E., G. A.; De Lima, M. E. F.; Hamelin,
O.; Hartmann, B.; Kanazawa, A. M.; Yanyun, W. J. Am. Chem. Soe. 2002, 124,
15313.
(25) Piers, E. ; Britton, R. W. ; de Waal, W. Cano J. Chem. 1969, 47, 831.
(26) Srikrishna, A. ; Reddy, T. J.; Nagaraju, S.; Sattigeri, J. A. Tetrahedron Lett. 1994,
35,7841.
(27) Srikrishna, A.; Nagaraju, S.; Sharma, V. R. J. Chem. Soe., Cehm. Commu. 1993,
285.
(28) Ferraz, H. M. c.; Silva, L. F., Jr.; Vieira, T. d. O. Synthesis 1999, 2001.
(29) Ferraz, H. M. c.; Silva, L. F., Jr. Quím. Nova 2000, 23, 216.
(30) McKillop, A.; Taylor, E. C. In Organíe Synthesís by Oxidation with Metal
Compounds; Miss, W. J., De Jonge, Cornelis, R. H. 1., Eds.; Plenum: New York,
1986, p 695.
(31) McKillop, A.; Taylor, E. c.; Hunt , J. D.; Kienzle, F. Tetrahedron Lett. 1970, 5275.
(32) McKillop, A.; Hunt, J. D.; Kienzle, F.; Bigham, E.; Taylor, E. C. J. Am. Chem. Soe.
1973, 95, 3635.
(33) Vieira, T. O. Synlett 2002, 1017.
(34) Ferraz, H. M. C.; Brocksom, T. J.; Pinto, A. c.; Abla, M. A.; Zocher, D. T. H.
Tetrahedron Lett. 1986, 27, 811.
(35) Ferraz, H. M. c.; Ribeiro, C. M. R.; Grazini, M. V. A.; Brocksom, T. J.; Brocksom,
U. Tetrahedron Lett. 1994, 35, 1497.
(36) Ferraz, H. M. c.; Grazini, M. V. A.; Ribeiro, C. M. R.; Brocksom, U.; Brocksom, T.
J. J. Org. Chem. 2000, 65, 2606.
(37) Ferraz, H. M. C.; Sano, M. K.; Ribeiro, C. M. R. Quim. Nova 1993, 16, 548.
(38) Ferraz, H. M. c.; Longo, L. S., Jr.; Zukerman-Schpector, J. J. Org. Chem. 2002,
67, 3518.
(39) Ferraz, H. M. C.; Longo Jr., L. S. Org. Lett. 2003, 5, 1337.
(40) Ferraz, H. M. c.; Ribeiro, C. M. R. Synth. Commun. 1992,22, 399.
(41) Silva Jr, L. F. Tetrahedron 2002, 58, 9137.
(42) Ferraz, H. M. c.; Silva, L. F., Jr. Tetrahedron Lett. 1997, 38, 1899.
(43) Ferraz, H. M. c.; Silva, L. F., Jr. J. Braz. Chem. Soe. 2001,12,548.
(44) Ferraz, H. M. c.; Silva, L. F., Jr. J . Org. Chem. 1998,63, 1716.
(45) Ferraz, H. M. c.; Silva, L. F., Jr.; Aguilar, A. M. ; Vieira, T. O. J. Braz. Chem. Soe.
2001, 12, 680.
(46) Ferraz, H. M. c.; Silva, L. F., Jr.; Vieira, T . O. Tetrahedron 2001,57,1709.
(47) Ferraz, H. M. c.; Aguilar, A. M.; Silva Jr, L. F. Synthesis 2003,1031.
(48) Ferraz, H. M. c.; Aguilar, A. M.; Silva Jr, L. F. Tetrahedron 2003, 59,5817.
(49) Silva Jr, L. F.; Sousa, R. M. F.; Ferraz, H. M. c.; Aguilar, A. M. J . Braz. Chem.
Soe., no prelo.
174

SL1
• LL9 '9£'1161 'Luati.3 '510 'C 'NQI 'C 'Unis-lel/4 (9L)
'T9Z 't' '0861 9 'pueqs• .wat.0 e_Pv 'S 'uaueuuad (5L)
'0017L 1£00Z .1-.11a11.3 '510 TC ilipoNog :•v izaiezuoD '.•c lelsanj 1.•S 'zela (171)
'0855 '55 '0661 'LL/Pqp '5i0 'C 'kl 'Jnod ',()IsAcoo>1 (EL)
•EVIT
'SL61 "4-797 •waL0 JeJeLielN W 'nz!weun3 1.•ki louan 'epaew 'eunleõeN (ZL)
't766I
'suo!4!ID3 leuopewalui TIIH-Wzn:IDDIA1 :SISPLAUÁS D!ue510 '9 'W '141!wS (IL)
'6LLI '9961 '-7-7P7 uoipayeAai 'M igDON '),1 16-lacl!M (OL)
'6E17Z 11.5 '8161 'udt- 'Dos •wato *ling *s 'anoui '.•W '041 Ilez!>10S (69)
'SLZ5 '69 '1700Z 'Watij '510 T lienopues 1..3 'ofawJa9 (99)
'EL6T I0Ined 02S ap aPepsJanwn lopeAsaw ap oe5e4Jass!CI 'D 'W 'H izenaj (L9)
'TZOS '1.17
'OOOZ '-7-7P7 uctipayeAai 'H 1Jall-la!A !'0 le-l!a!A '.'D 'w 'H izelia3 (99)
'1061 ISOOz sysacpuÁs *H 'legai 1.•A sa4anbed (59)
'99LZ 'LS 'Z661 'wP11.3
*L- 15ueqz f•a 'nx '.1/4-•Z 'Buem .̀.N 'emelpow '5uomm
'6uoar 1.•[. '6unieH 'D 'ou!dspj 1.'3 'A 'weuuag 1../NA 'Euaqwv 1.'9 issaicliet4S (179)
'L09 'ZZ '1861 '4.7a7 ualpayeAai M W 'uosuuor 1.'m 1>llsewoi 11D!w-I0DDIA1 (E9)
'61755 '80T
'9861 'DOS 'Wal-ID 'WV T '3-'d 1wrIS illalD!N T '14aue4S 'Liewpaog (Z9)
'9178 '9661 sisatpuÁs •À 'ew!usns4evi '-'>1 1P0W (T9)
'6L 'Z171Z961 '114uÁs *510 •ci lJei(aNDS 1.'j 'G IJa11aJu!al>1 (09)
'6617 ISL6T '4-137 uaipayeilai 'M-'J .3 Jupequalmi f.y 'uos!II3 (65)
'LL17
'L6 'SL61 'DOS 'wat.ID •wv 'S 'm Juosuuor f'D •D 'smaipuv f•v b luew>110A (85)
'Z99E 'S6 '£161 'watID '510 T '3 '9 Iljoue/uew 'D IPIsmal!W Isu!LID4nH (L5)
'170171 '8t7 '£861 'w9L/D '510 1" '1A1 '9 '450-11'.' 'N 'jjnwnl-IDS (95)
'17ZZI 'ES '8861
•wato '510 T '3 '4-19)1uaM f'v lipPnej_ '..3 Jozzld Iiiaribupd '3 Illa6uV (55)
'566Z
'8t' '5161 'udC 'Dos •waLID •À JeJeue4!>i '0uafl •I-1 '24250 f•I 'eumie6eN (175)
'9889 'ZS, '1661 '11-/PLID '510
'>1 •d '5ueND •N;( •r luewaaJ3 'c 114D!saPe1 f•W '3 14allat-P-lad
M 'S IllamaN 'Cl •d luosum •LAJ iolcueN ,.S-•H Jus :•A luat-o H 'G lenH (ES)
•olaid ou isainDalokv •A 'w lanaÁeJj f.j eAl!S (Z5)
'6E66 'LS 'IOOZ uoipayeAaj ''3 '1 'eAl!S J 'W 'H lze_uad (I5)
'T 5L 'OE 'OOOZ
•unwwo3 •LnuÁs •0 12-1!a!A 1'3 '1 leAl!S 'v 'smues 1.•D 'yd 'H Ize.ua3 (Os)
sepup,Jajad

9LI
'0E9T '0161
•unwwo3 .wato i•Dos 'Waqj "C '>j 1.'v 'elegei '.•s JeJnwan (90T)
'TZ9 '0961 .3 'Dos •waqj '5 'C '45Jrla411-1M f'D '9 'D 'api(og (LOT)
'9817 'EL6I S 7103 'LPUÁS "5-10 's 'vsj 'uewmaN 1.•s 'ue.ipuet.peweu (90T)
'69L 'L9 S961 'illaqj •ma5uv •I-1 '-ial4a4S (50T)
'99Z116661 sisay_luÁs iew.seA (POT)
*E9 '8Z 'S661 e-Pv epiwit.p.uplV 'O Ilisele.td (E0T)
'LE
'511. 'S861 72-11. "zzeD '1 'tAi 'asailaDJOJ '9 'I 'In Iliapp\og i.•3 lauopuM (ZOT)
'SLE '89 '9961 'na¿/ •watip 'uosugor (TOT)
'LI8Z 'L '8661 ti-lawwÁsv :uwpayeAai 'd 'auouej 1.'d 'neau6ea (00T)
'OEL '6161 sisaipuÁs •43 'ueampinj :*C1 IJallnAl (66)
'L17E17 '6E '8661 'a7 ucupayeaaj 's ipoki :'s ILIDa)121 11115121 (86)
'6D 'IOZ
'0861 *waLo yejawoue5J0 'ffnocina :•(:) la4.Jode1 1..3 isau!inovi '.'d ineezej (L6)
'517E1 'PE '£961 '-7-7P7 uwpayeijai •xt '2:1 '110110H '3 '14 11-11e-IN (96)
'6L5 'Z'T ITOOz Á.LiawwÁsv 'I 1Jopaclip5-uewJa>InZ
'fliaDeJej f'2:f 'd lelsop :'1A1 'S 'smual :'W .3 isolues '.'y JoieualpS (56)
'S9EE 'OT '6661 MawwÁsv :uarpayei4ai .1 'poCuog izaRzuoD 1.'x 1e4sanD (176)
'T569 'Ti 1000Z '-7.7a7 ucupaye-Lial. 'A 'D secpeg :•11r19 (E6)
'LE 'E9 'S861 'y-IuÁs '5.i 14s.u33 1.•ci 1.101pelpSq3n9 (Z6)
*L199 'PS '8661 ucupayeAai 'V1400-19 aa :',C1 'H isualsiuual (T6)
'6995 'TP '000Z '-7.7P7 uwpayeAai 'y izai9zuop izeja 1.•c 'eqsanj f'c 'tpcguog (06)
'ZST
'85 '8161 'LIAS '5.10 *N 1.•£i iwaueD W 'a iwnls f'm ueuppaos (68)
'5Z6 '6E '£961 uwpayeilai "Jr uew)paog (88)
'85T '85 '8161 •uÁs• '5 *g 1waueD 'wn¡s uewlDa09 (L9)
'L989 '56 'E161 'Dos •wat40 'LLIV ')1'211•JI ueuDpao9 (98)
'L9E 'Z661 T •sue-1.1
uppad •Dos wayj '1A1-*C 'uinbauuaH 1.•5 Juipuerna laweynC1 (Se)
'99Z17 'OS 'S861 u/Pq.3 *5-/0 'C 'A 'V '115142S :1/5J 'S 'splod 1.*AA luew4r1H (178)
'1755E '917 '1861 "LuaL0 *5-/0 *C1 luawz4leS 'Nzi 'C lual0D 'd IP!la-10Z (E9)
'5517E '05 IZZ61 'a/PL./3 *ueD 'V '9 '44a>1Dag 5HuDawez '.'9 *?J lÁllaN (Z9)
'0T17E 'T161 (D) •Dos •wat40 •\7, JaDuaucu f•u 1JaPuld (T9)
*L0E17 217'6961 wa4.3 veD 'M ileeM aP '?21 iu041!-19 is-la!d (08)
'1517E '8961 '4.7a7 uo-ipayeiJai *D 'uosspno 'uuewnaN-1Due.13 f•D `Ja6Ja9 (6L)
'L9EE 191-T 't'661
•Dos •way.3 'S '5uem •D 'frj .D iodd!IINd• '6u2M Iallanbad (8L)
'Z'fr '9861 uoipayeAai 1.•>1 'PON (££)
SP,1.9U9JPiPd

LLI
*6Z8T '017'SL6T
•waqj '5.10 •1- 'D 'lenzseig 'Pew4V 1.•H f 'Jassnvi w •9 *1 'kunkl (9IT)
•L8S£
'SI 't700z MaWUJÁSV :U0.113PLIEWPi isanbDer f•D 'ausapJnoj :•?:j '1.1anauelp (ST1)
*zzaz 'r
'cooz 70W01g .5-10 3 'uosl!M !*d T 'weyeJD 1.•1 f 'alem •I luosiapuV (i7IT)
•L6T6 'T9 '500Z uwpayenai 's
f*u •S'JaÁI 1.*A '46u!S (ETT)
'566-17'1L61
.4.1a7 uoipayeAai *v lounoddop :*3 *1 luinwpki :*3 T S11I3 H 3 l>130oLgeaH (ZIT)
•Z6EL '0915661 "waL0 '5J0 T*3 *A '66!d f*H 'ÁcI5121 (ITT)
'L66 '186T *-71a7 way0 'S 1213!4s!N fiAj '!we)ieJniAj (OTT)
'T6Z 1LE 'T861 uai/33142/4a/ w loue)10 :*v le4eqei f•H 'ÁusoÁRAJ :'s 'eunwan (601)
SEIDU91Pjad


















