
Fisiopatologia de la hipoxia disóxica
35
FISIOPATOLOGÍA DE LA HIPOXIA DISÓXICA Cortez Cárdenas, Fabiola Diana UNIVERSIDAD RICARDO PALMA FACULTAD DE MEDICINA HUMANA 1
-
Upload
fabiola-cortez -
Category
Documents
-
view
638 -
download
9
Transcript of Fisiopatologia de la hipoxia disóxica

FISIOPATOLOGÍA DE LA HIPOXIA
DISÓXICA
Cortez Cárdenas, Fabiola Diana
UNIVERSIDAD RICARDO PALMAFACULTAD DE MEDICINA HUMANA
1
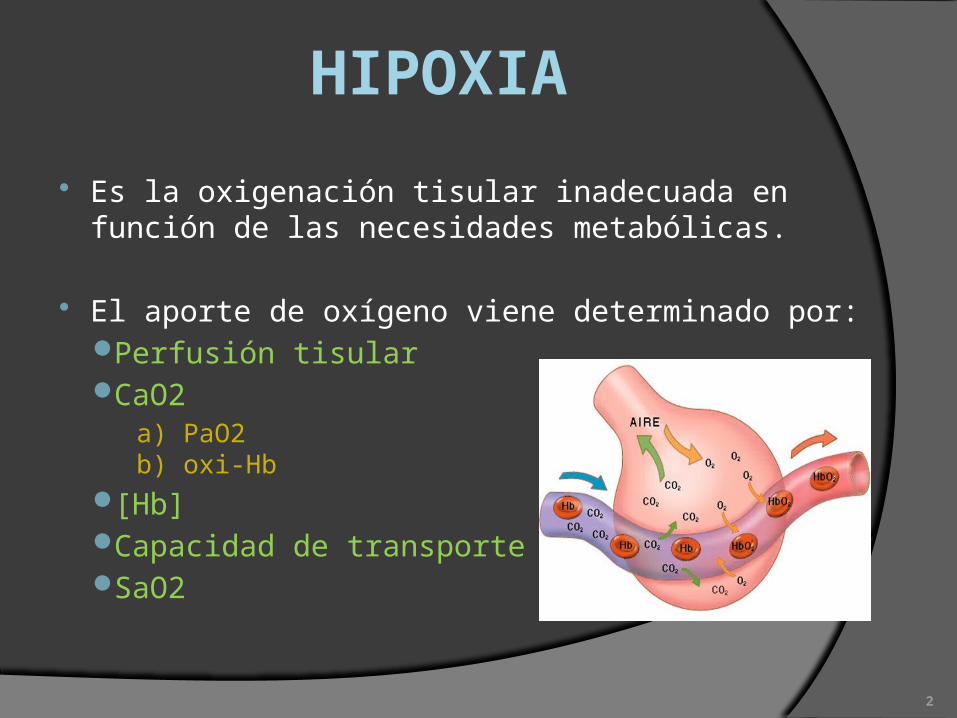
HIPOXIA
Es la oxigenación tisular inadecuada en función de las necesidades metabólicas.
El aporte de oxígeno viene determinado por:Perfusión tisularCaO2
a) PaO2b) oxi-Hb
[Hb]Capacidad de transporteSaO2
2

3
TIPOS DE HIPOXIA
H. hipoxémica H. anémica H. disóxica
H. isquémica
Toda hipoxemia implica hipoxia (hipoxia hipoxémica), no toda hipoxia
se debe a hipoxemia.

4
APORTE DE O2
H. circulatoria: déficit local o generalizado del flujo sanguíneo
H. anémica: déficit cuantitativo o cualitativo del número de hematíes y/o hemoglobina
H. disóxica: utilización celular inadecuada de O2 por las mitocondrias
Producto del gasto cardíaco por el contenido arterial de O2 (CaO2).

5
Exposición aguda a CO

6
CAUSAS DE HIPOXIA DISÓXICA
1. Intoxicación con –CN
2. Exposición aguda a CO
3. Shock séptico

7
HIPOXIA GENERADA POR LA EXPOSICIÓN A CO
1. Hipoxia anémica por alteración de la Hb
2. Hipoxia disóxica en exposición aguda

8
MECANISMOS GENERALES DE COMPENSACIÓN DE LA HIPOXIA
1. Incremento de la ventilación
2. Incremento del GC
3. Mayor liberación tisular de O2
4. Redistribución del flujo
5. Policitemia

9
MECANISMOS DE COMPENSACIÓN DE LA HIPOXIA: H. DISÓXICA
No hay porque es demasiado rápida

10
MECANISMO FISIOPATOLÓGICO

11
INHIBIDORES DE LA CADENA RESPIRATORIA
Tres grupos principales según el sitio de la cadena respiratoria donde actúan:
1. Sobre la NADH-deshidrogenasa, (Inhibidores del sitio I)
a) Barbitúricosb) Piericidina A c) Rotenona

12
3. Actúan sobre el Hemo a3 de la citocromo oxidasa (inhibidores de sitio IV)
a) Cianuro
b) Monóxido de carbono
c) H2S
2. Actúa bloqueando la transferencia de electrones entre el citocromo b y el citocromo c1. (inhibidores de sitio III)
a) Antimicina

13
INTOXICACIÓN CON –CN
Forma complejos estables con iones metálicos, teniendo una gran afinidad por el hierro férrico (fe+++).
El complejo que forma con el metal, inactiva ciertas enzimas, siendo la más importante la citocromo-oxidasa.
La inhibición de la citocromo-oxidasa produce anoxia citotóxica la glucólisis pasa de aerobia a
anaerobia Se inhibe la transformación del piruvato en
el ciclo de Krebbs: hiperproducción de lactato acidosis metabólica severa

14
Otros mecanismos
Alteración del metabolismo de la vit. B12
Afectación miocárdica Liberación de aminas
biógenas Vasocontricción arteriolar y/o
coronaria.

15
CUADRO CLÍNICO DE LAINTOXICACIÓN POR CIANURO
1a FASE Vértigo Debilidad Náusea Vómito Ebriedad
Cefalea Taquipnea Arritmia Ansiedad intensa Rigidez de la
mandíbula (trismus)
SE PRESENTA EN TRES FASES:

16
2a FASE Confusión mental Convulsiones tónico-
clónicas Opistótonos Coma superficial Hipertensión arterial

17
3a FASE Cianosis Coma profundo Midriasis Taquicardia Incontinencia
de esfínteres
Edema agudo del pulmón
Shock Paro cardio-
respiratorio

EXPOSICIÓN A CO
Exógenas: ambiente laboralel hogar contaminación ambiental.
Tabaquismo: Importante fuente de
exposición crónica a CO

19
Endógenascatabolismo de los pigmentos sanguíneos,

20
EXPOSICIÓN AGUDA A CO
El CO ingresa al organismo vía inhalatoriaen la sangreel resto se une a proteínas transportadoras
de O2 La afinidad de esta última por el CO es
200 a 250 veces mayor que por O2.
21
se une a las enzimas del grupo Hem de la hemoglobina, desplazando al oxigeno de la misma.
se forma en la sangre un complejo que se denomina
carboxihemoglobina

22
23
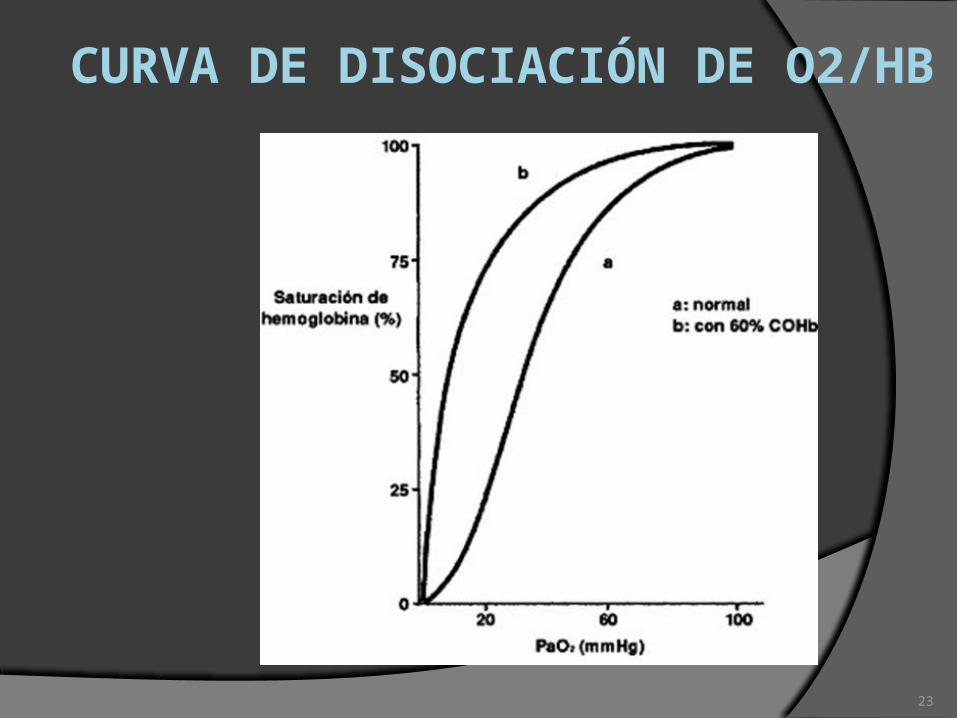
CURVA DE DISOCIACIÓN DE O2/HB

24
En intoxicación aguda grave a este tóxico, se produce unión a la mioglobina tisular a los citocromos a3 y P450.
Este evento crea un reservorio, produciendo una intoxicación persistente, aún con oxigenoterapia.

25
OTROS MECANISMOS:
Daño por reperfusión del sistema nervioso central (SNC)
Inhibe la síntesis aeróbica de adenosina trifosfato
peroxidación lipídica- Enz. Xantina DHA- Hipoxantinas superoxidos- Peroxidonitritos AGI

26
TRES PERIODOS CLINICOS:
ESTADIO INICIAL (12-25%): Síntomas inespecíficos
náuseas, vómitos, trastornos visuales, cefalea a veces diarrea, especialmente en niños.
casos de angina de pecho

27
ESTADIO MEDIO (25-40% ):
Confusión, irritabilidad e impotencia muscular.
Trastornos en la conducta y obnubilación.
Pueden objetivarse alteraciones en el electrocardiograma (ECG).

28
ESTADIO DE COMA (40-45%):
Hiperreflexia, hipertonía, reflejo de la planta del pie en extensión.
En ocasiones aparecen convulsiones e hipertermia.
Pueden aparecer hipotensión e infarto de miocardio
Cifras superiores al 60% de COHb son potencialmente letales.

29
SHOCK SÉPTICO

30

31
MANIFESTACIONES CLINICAS
DISFUNCION RESPIRATORIA Aparición de taquipnea o hiperventilación e hipoxemia. La sepsis provoca demandas extremas a los
pulmones, requiriendo un volumen minuto alto la resistencia en la vía aérea aumentada por
broncoconstricción Casi el 85% de los pacientes necesitan ventilación
mecánica de 7 a 14 días Detectándose en la radiografía de tórax infiltrados
algodonosos alveolointersticiales

32
DISFUNCION METABOLICA
La situación de shock se produce por un inadecuado aporte del sustrato metabólico,
En un primer momento el consumo de oxígeno tisular es normal o está aumentado en dependencia del aporte, para luego estar disminuido.
Otras alteraciones metabólicas encontradas en la sepsis son: hiperglucemia (fase precoz)hipoglucemia (fase tardía)

33
LAS CITOQUINAS
Estos péptidos dan lugar a complejas reacciones inmunológicas que pueden conducir al fallo multiorgánico y, potencialmente, a la muerte.
Se han descrito 18 citoquinas1. TNF-α, la IL-1 y la IL-8;
2. IL-6;
3. IL-4, IL-10 y la IL-13,
4. El interferón (INF-γ) aumenta la actividad del TNF-α e induce la síntesis:
OXIDO NITRICO

34
LA RESPUESTA INFLAMATORIA
El interferón y la IL-1 estimulan la síntesis y liberación endotelial de óxido nítrico.
Todos estos mediadores junto con el complemento activado
inducen la quimiotaxis de neutrófilos en los órganos diana (pulmón, hígado y riñón), dando lugar a su activación.
La activación del complemento da lugar además a la degranulación de los mastocitos, liberándose histamina y serotonina la activación del sistema kalikreína (K-K)con la producción de bradikinina

35
DISFUNCIÓN METABOLICA
La taquipnea y la hiperventilación se deben al TxA2, la PGE2 y la prostaciclina.
La histamina, los leukotrienos LTC4, LTD4 y LTE4, la PGF2 y el TxA2originan un incremento de la resistencia de la vía aérea
El NO inhibe la respiración mitocondrial, originando una alteración de la utilización tisular del oxígeno.
DISFUNCIÓN RESPIRATORIA


















