
FARMACOLOGIA: AINES OPIOIDES
79
FARMACOLOGÍA DEL DOLOR Y LA INFLAMACIÓN AINES Y OPIÁCEOS Luis E. Esteves Lecaros Universidad César Callejo Lima Norte EAP enfermería
-
Upload
charlesarles -
Category
Health & Medicine
-
view
5.138 -
download
6
description
Este trabajo se presenta con la finalidad de educar y enseñar en el área de salud las diversas funciones de los fármacos y sus propiedades.
Transcript of FARMACOLOGIA: AINES OPIOIDES

FARMACOLOGÍA DEL DOLOR Y LA INFLAMACIÓNAINES Y OPIÁCEOS
Luis E. Esteves LecarosUniversidad César Callejo Lima NorteEAP enfermería

Antiinflamatorios no esteroideos

ANTIINFLAMATORIOS NO ESTEROIDEOS
Incluyen diversos compuestos sin relación química alguna
El prototipo es la aspirina
Inhiben a la ciclooxigenasa

ANTIINFLAMATORIOS NO ESTEROIDEOS
MECANISMO DE ACCIÓN Inhiben la síntesis de Prostaglandinas

Prostaglandinas:
TxA2 (1975) Prostaciclina (1976) Leucotrienos (1983) Misoprostol (Cytotec®)
Alprostadil (Caveryect®)
Epoprostenol (Flolan ®)

ANTIINFLAMATORIOS NO ESTEROIDEOS MECANISMO DE ACCIÓN
INFLAMACION FASES:
Transitoria aguda Subaguda tardía Proliferativa crónica

ANTIINFLAMATORIOS NO ESTEROIDEOS INFLAMACION
Eritema Edema Dolor Intervienen sustancias como
IL-1, TNF, IL-2, IL-6, IL-8, Factores de crecimiento
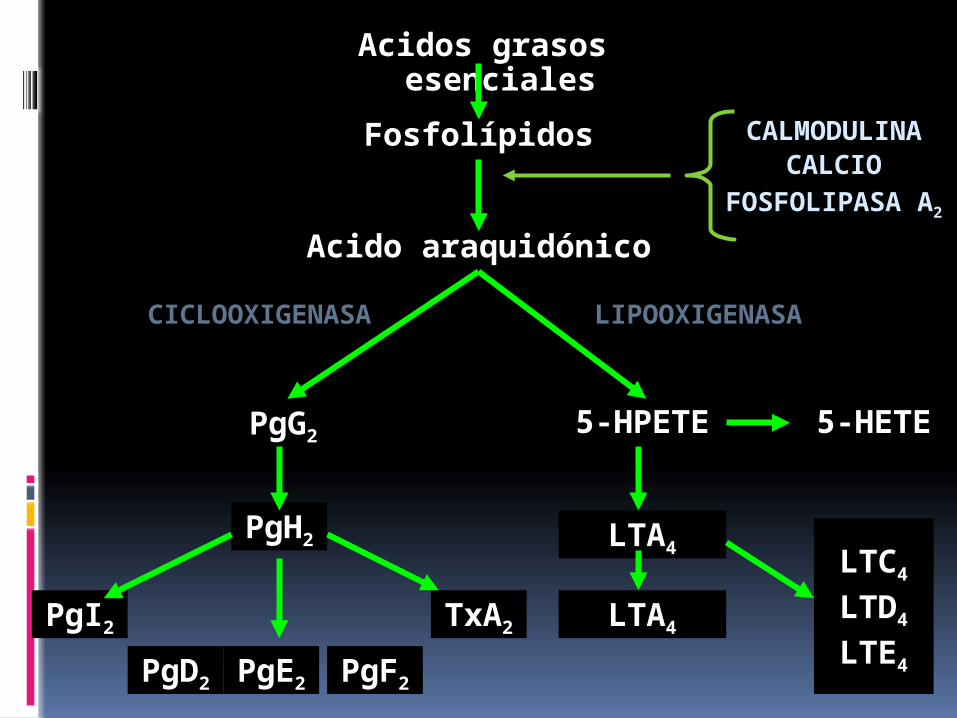
Acido araquidónico
Acidos grasos esenciales
Fosfolípidos
LIPOOXIGENASACICLOOXIGENASA
5-HPETEPgG2
PgH2
PgF2PgE2PgD2
PgI2 TxA2
LTC4
LTD4
LTE4
LTA4
LTA4
5-HETE
CALMODULINACALCIO
FOSFOLIPASA A2
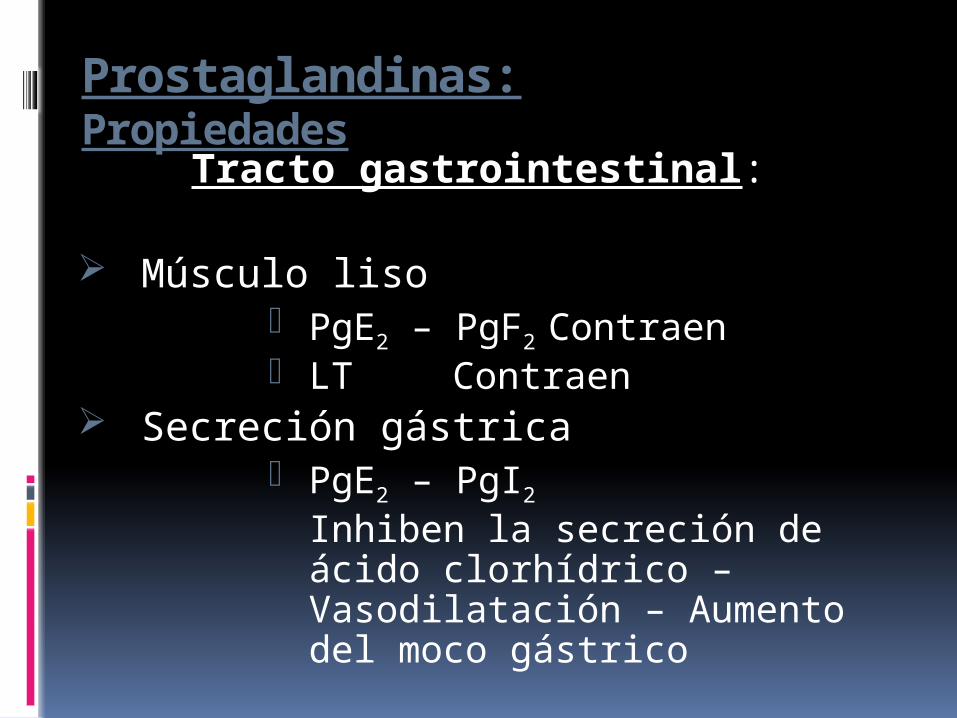
Prostaglandinas:Propiedades
Tracto gastrointestinal:
Músculo liso PgE2 – PgF2 Contraen LT Contraen
Secreción gástrica PgE2 – PgI2
Inhiben la secreción de ácido clorhídrico – Vasodilatación – Aumento del moco gástrico

Pgs: PropiedadesSangre:
LTC4 y LTD4 Exudado de plasma a nivel capilar
PgE2 – PgD2 Inhibe agregación plaquetaria
Prostaciclina Inhibe agregación plaquetaria
TxA2 Inductor de la agregación Pks. LTC4 y LTD4 Quimiotaxis de PMN. PgE2 – PgI2 Inhiben la liberación de
histamina
PgD2 5-HPETE Favorecen la liberación de
histaminaHETE

Pgs: Propiedades
Sistema cardiovascular: PgI2 – PgE2 Vasodilatador y aumento del
GC TxA2 Vasoconstrictor
Riñón: PgI2 – PgE2 Aumenta el flujo renal TxA2 Disminuye el flujo renal
Sistema Nervioso Central (SNC) PgE2 Produce fiebre a nivel del
hipotálamo

Pgs: Propiedades
Terminaciones nerviosas aferentes: PgE2 Produce dolor
Músculo liso bronquial: LTC4 y LTD4 Potentes
broncoconstrictores
Útero PgF2 Contrae PgE2 Contrae --------- relaja PgI2 Relaja

ANTIINFLAMATORIOS NO ESTEROIDEOS DOLOR
La bradicinina, el TNF alfa, IL-1, IL-8 liberan prostaglandinas y otros mediadores que estimulan la hiperalgesia
FIEBRE IL-1ß, IL-6, Interferón alfa y ß, TNF alfa
incrementan síntesis de PGE2 en órganos periventriculares cerebrales, área preóptica

BIOSINTESIS DE PROSTAGLANDINAS

BIOSINTESIS DE PROSTAGLANDINAS
Se intensifica por la acción de hormonas, autacoides y otras sustancias al interactuar con receptores de membrana dependientes de Proteína G
Esta acción activa en forma directa a las fosfolipasas C y A2

BIOSINTESIS DE PROSTAGLANDINAS
La fosfolipasa A2 hidroliza el enlace éster de los fosfolípidos de membrana liberando ácido araquidónico.
La fosfolipasa C desdobla el enlace fosfodiéster, formando 1,2 diacilglicérido y posteriormente se libera el ácido araquidónico.

BIOSINTESIS DE PROSTAGLANDINAS VIAS ENZIMATICAS
Vía de la ciclooxigenasa
Vía de la lipooxigenasa
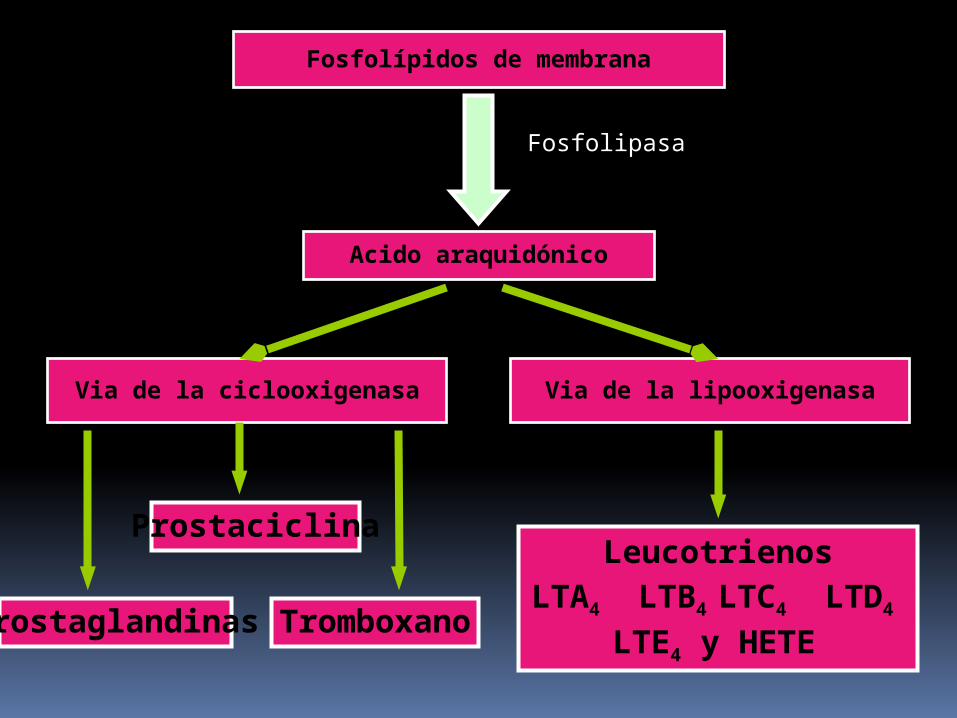
Fosfolípidos de membrana
Acido araquidónico
Fosfolipasa
Via de la ciclooxigenasa Via de la lipooxigenasa
Leucotrienos
LTA4 LTB4 LTC4 LTD4
LTE4 y HETE
Prostaciclina
Prostaglandinas Tromboxano

Prostaglandinas y dolor
Las prostaglandinas causan dolor si se inyectan por vía intradérmica
La bradiquina y la histamina dan efectos mas inmediatos e intensos
Las prostaglandinas sensibilizan las terminaciones nerviosas
Las prostaglandinas amplifican el dolor y tienen resultados mas duraderos
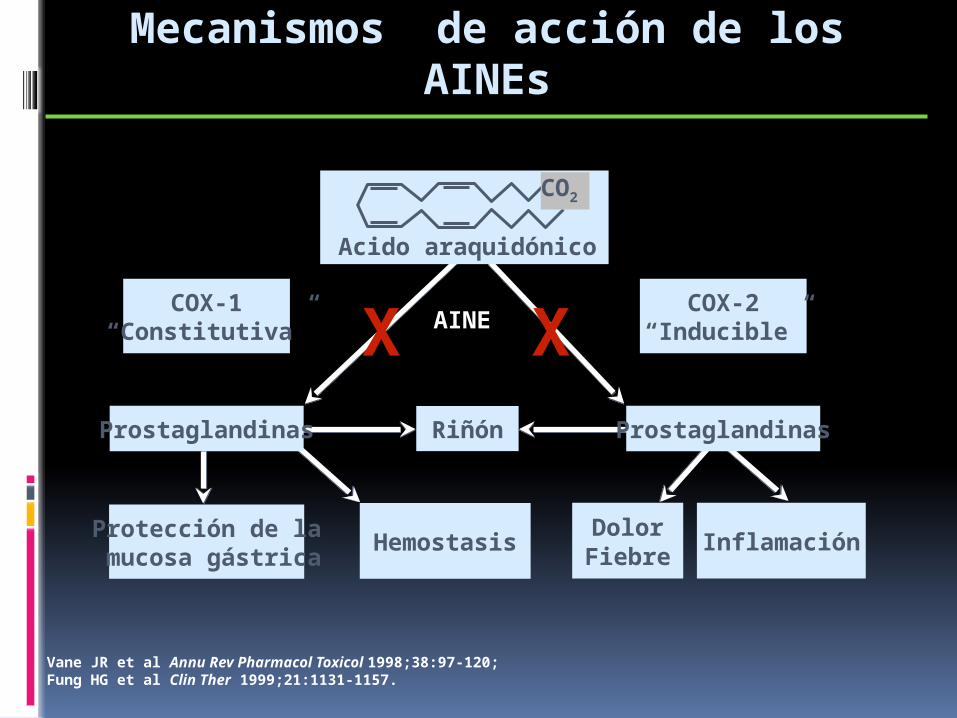
Acido araquidónico
AINE
Protección de la mucosa gástrica
Hemostasis
COX-1“Constitutiva”
COX-2“Inducible”
Vane JR et al Annu Rev Pharmacol Toxicol 1998;38:97-120; Fung HG et al Clin Ther 1999;21:1131-1157.
Riñón
Mecanismos de acción de los AINEs
Prostaglandinas Prostaglandinas
CO2
X X
DolorFiebre
Inflamación

ANTIINFLAMATORIOS NO ESTEROIDEOS CLASIFICACION
DERIVADOS DEL ACIDO SALICILICO: Aspirina
DERIVADOS DEL PARA-AMINO FENOL: Paracetamol
INDOL: Indometacina ACIDO
HETEROARILACETICOS: Ketorolaco, Diclofenaco, Tolmetín

CLASIFICACION ACIDOS ARILPROPIONICOS:
Ibuprufeno, Naproxeno FENAMATOS: Acido
mefenámico ACIDOS ENOLICOS: Oxicam,
pirozalidindionas ALCANONAS: Nabumetona NICOTÍNICO: Clonixilato de
lisina
ANTIINFLAMATORIOS NO ESTEROIDEOS

Nuevos aines:COXIBS
Derivados tricíclicos de sulfonaEtoricoxib (Arcoxia®)Rofecoxib (Vioxx®)
SulfonamidasValdecoxib (Valdure®)Celecoxib (Celebra®)
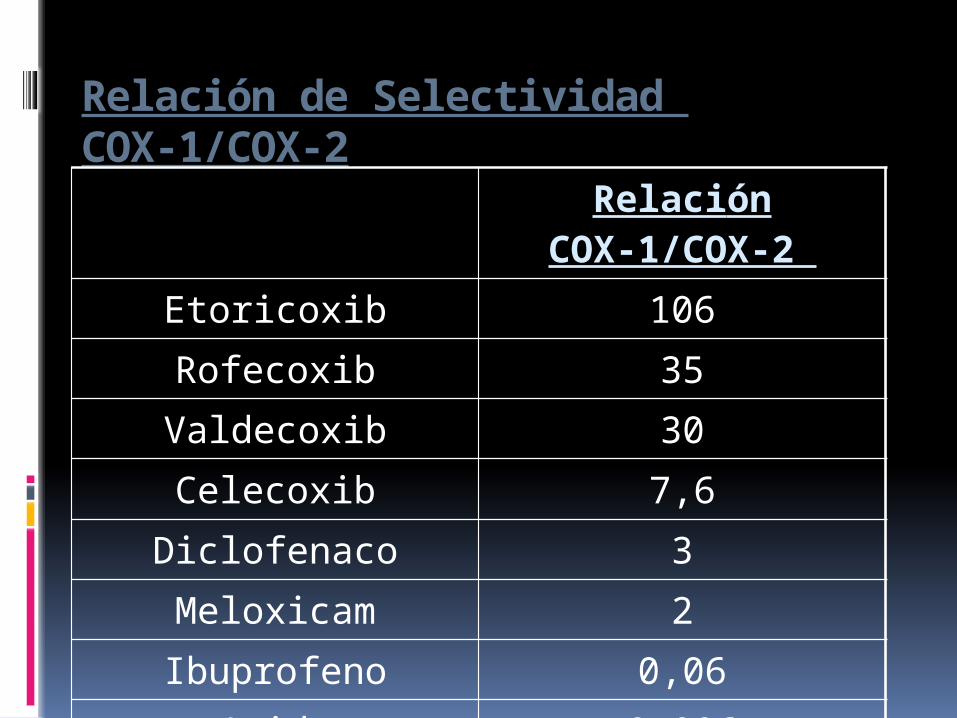
Relación de Selectividad COX-1/COX-2
Relación COX-1/COX-2
Etoricoxib 106Rofecoxib 35Valdecoxib 30
Celecoxib 7,6
Diclofenaco 3
Meloxicam 2
Ibuprofeno 0,06
Acido acetilsalicilico 0,006

Aines de acción breve
Acción breve: 2 – 3 horas
AAS (Aspirina® – Rhonal® – Ecotrin®)
Diclofenaco (Voltaren® - Cataflam®)
Ibuprofeno (Motrín® – Advil® – Dorival®)
Indometacina (Indocid®)

Aines de acción intermedia
Acción intermedia: 10 – 12 horas
Flurbiprofeno (Ansaid®)
Naproxeno (Naprosyn® – Flanax®)
Sulindaco (Clinoril®)

Aines de acción prolongada
Acción prolongada: Más de 24 horas
Piroxicam (Feldene®)Tenoxicam (Tilcotil®)
Meloxicam (Mobicox®)
Coxibs

ANTIINFLAMATORIOS NO ESTEROIDEOS EFECTOS TERAPEUTICOS
Son antiinflamatorios, antipiréticos y analgésicos
Disminuyen temperatura sólo en estados febriles
Se usan en trastornos musculoesqúeléticos
Dismenorrea primaria

ANTIINFLAMATORIOS NO ESTEROIDEOS EFECTOS COLATERALES
Ulcera e intolerancia en vías GI
Bloqueo de la agregación plaquetaria
Inhibición de la motilidad uterina
Inhibición de función renal Reacciones de
hipersensibiliudad

Interacciones
Warfarina (hemorragias) Quinolonas (convulsiones) Diuréticos de asa (antagonismo) Inhibidores de ECA (hiperkalemia) β-bloqueadores Metotrexato Litio

Salicilatos
Absorción en estómago y duodenoSe unen a proteínas (Albúmina)Metabolismo hepático y excreción
renalVida media: 3 – 6 horasDosis bajas: 1 – 2 g / dia AnalgesiaDosis altas: 4 – 5 g / dia
AntiinflamatorioNivel serico: 20 – 30 mg / dl

Ibuprofeno
Absorción gastrointestinal rápida Nivel plasmático máximo en 1-2
horas Vida media de 2 horas Unión a proteínas plasmáticas
(99%) Excreción renal rápida y completa Molestias gastrointestinales (5-15%) Dosis antiinflamatoria 2400 mg/día
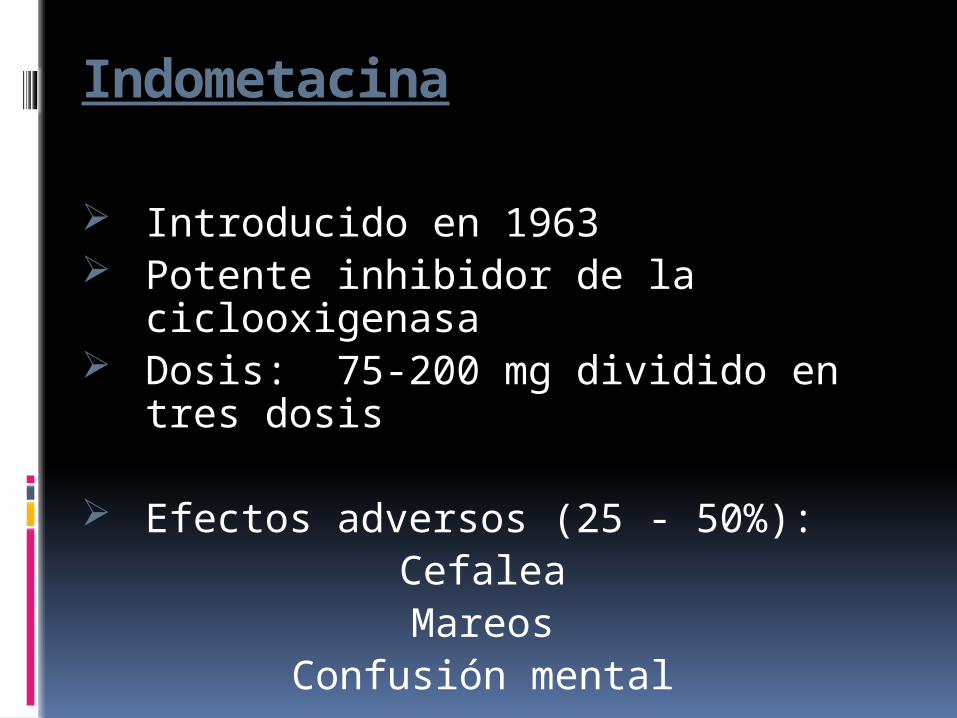
Indometacina
Introducido en 1963 Potente inhibidor de la
ciclooxigenasa Dosis: 75-200 mg dividido en tres
dosis
Efectos adversos (25 - 50%):CefaleaMareos
Confusión mental

Sulindaco
Prodroga Vida media de 18 horas Menor toxicidad renal Dosis:
200 mg bid VO
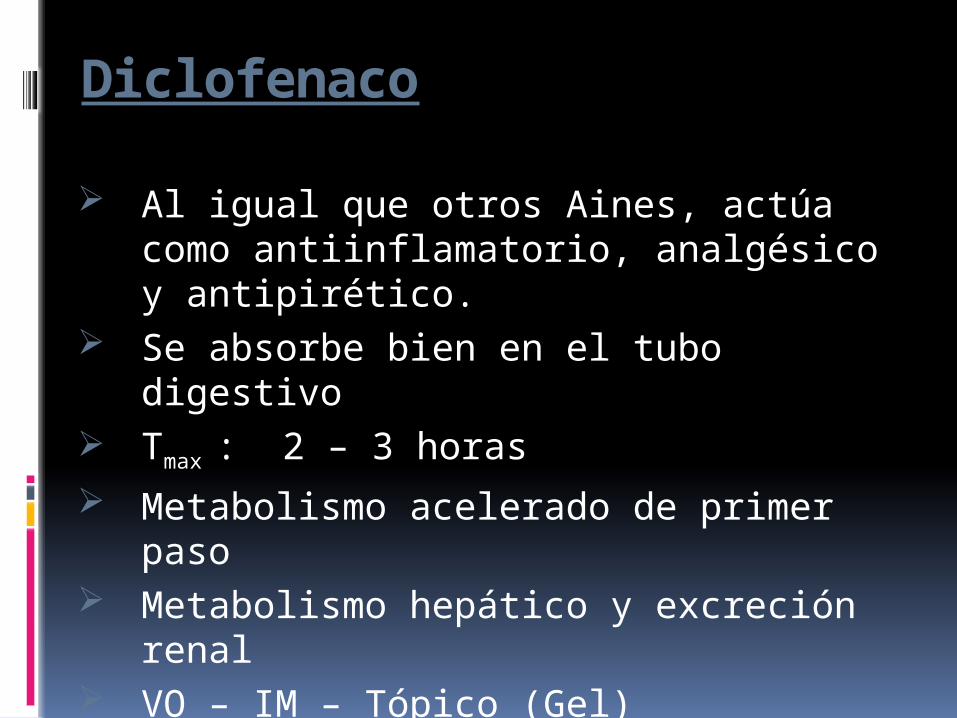
Diclofenaco
Al igual que otros Aines, actúa como antiinflamatorio, analgésico y antipirético.
Se absorbe bien en el tubo digestivo Tmax : 2 – 3 horas Metabolismo acelerado de primer paso Metabolismo hepático y excreción
renal VO – IM – Tópico (Gel) Dosis:
150 mg al día

Oxicams
PiroxicamTenoxicamMeloxicam
Vida media larga: 45 horas Una sola dosis diaria Mejor tolerancia que AAS o
indometacina
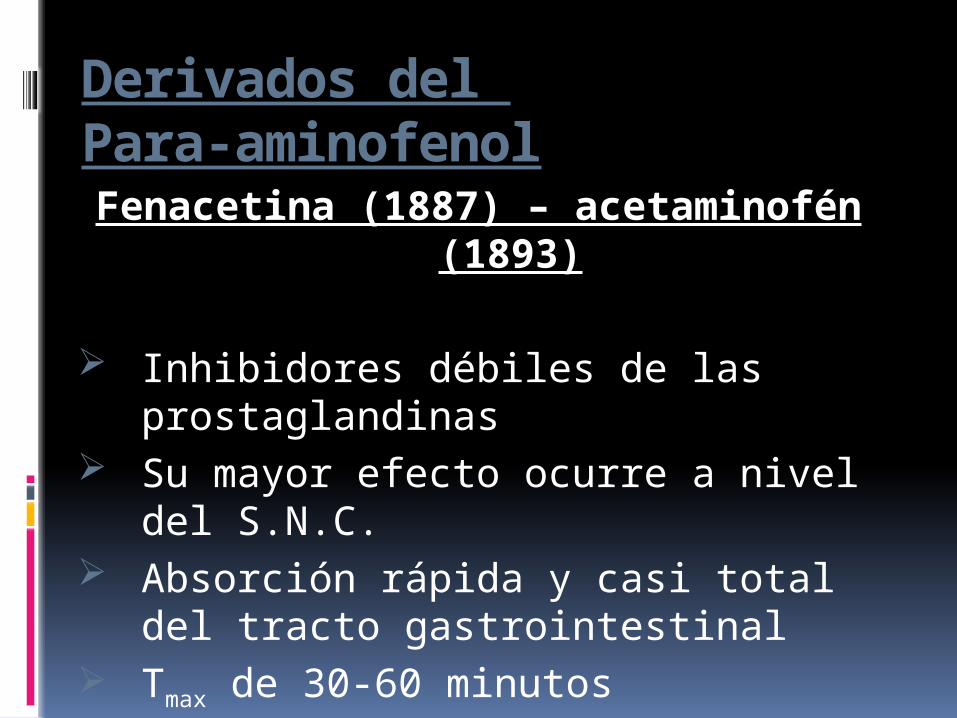
Derivados del Para-aminofenolFenacetina (1887) – acetaminofén
(1893)
Inhibidores débiles de las prostaglandinas
Su mayor efecto ocurre a nivel del S.N.C.
Absorción rápida y casi total del tracto gastrointestinal
Tmax de 30-60 minutos
T1/2 de 2 horas

Efectos adversos
Fenacetina • Metahemoglobinemia• Anemia hemolitica• Nefrotoxicidad
Acetaminofen• Erupciones cutáneas• Necrosis hepática (Mayor de 10 g)
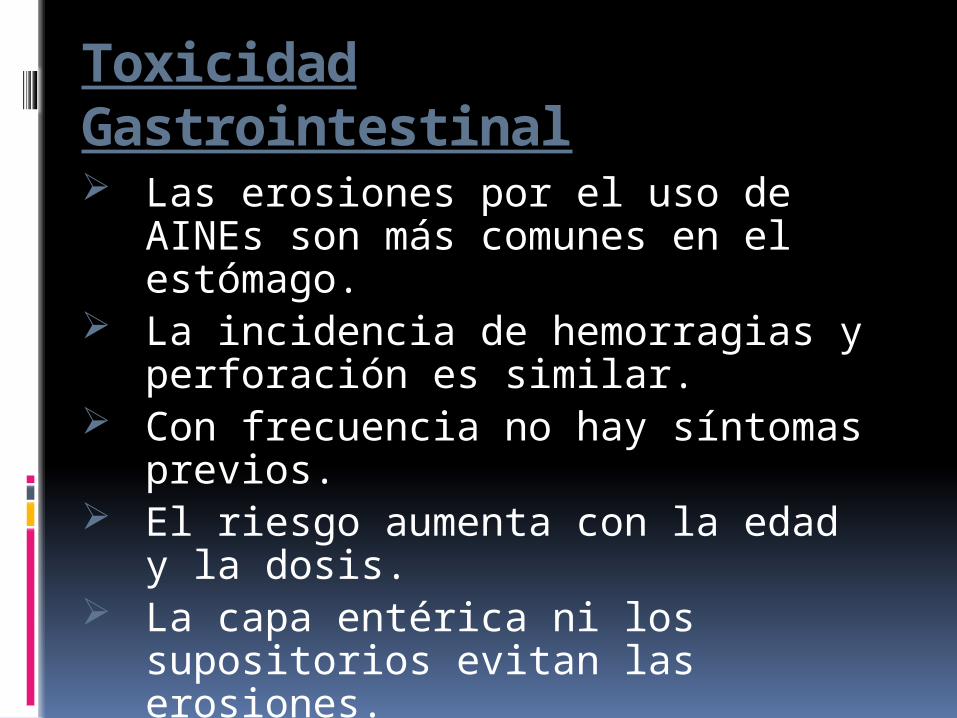
Toxicidad Gastrointestinal
Las erosiones por el uso de AINEs son más comunes en el estómago.
La incidencia de hemorragias y perforación es similar.
Con frecuencia no hay síntomas previos.
El riesgo aumenta con la edad y la dosis.
La capa entérica ni los supositorios evitan las erosiones.
Uso de Aines selectivos COX-2.

Síndromes renales asociados con AINEs
1. Insuficiencia renal aguda2. Daño renal crónico3. Síndrome nefrótico4. Nefritis intersticial5. Alteraciones en el metabolismo del
agua6. Alteraciones en la homeostasis del
sodio y del potasio

Acido Acetilsalicílico (A.A.S.) SALICISMO
Tinnitus – sorderaHipertermia – sudoración
Hiperventilación Alcalosis respiratoria
Acidosis respiratoria y metabólicaConvulsión y coma.

Opioides

Consideraciones Generales
Opioide Productos derivados del opio (morfina,
codeína, tebaína, papaverina) Derivados de papaver somniferum
Opiáceo Sustancias endógenas o exógenas con
afinidad por los receptores opioides

Consideraciones Generales
•μ: activados por morfina ocasionando analgesia supraespinal, depresión respiratoria, bradicardia, miosis, dependencia física, disminución de motilidad intestinal, euforia, sedación e hipotermia (efectos agonistas típicos)•: activado por la ketaciclazocina; media la analgesia espinal, sedación, miosis y depresión de reflejos flexores•σ: activado por N-alilnormetazocina; media la disforia , alucinaciones, midriasis, taquicardia y la activación respiratoria. •Este último tipo de receptor ha dejado de ser considerado un receptor opioide debido a la relación que guarda con alucinógenos como la fenciclidina y la ketamina, los cuales se hallan vinculados con el glutamato.
Inicialmente se reconocieron 3 tipos de recepto
res:

Consideraciones Generales
Identificación por Hughs & cols en 1975 de los dos primeros ligandos endógenos (péptidos opioides endógenos): metionina-encefalina leucina-encefalina
Capaces de interactuar directamente con el receptor para opioide** δ mayor afinidad que la morfina

Consideraciones Generales
Posteriormente se identificaron otros péptidos como la b-endorfina, con afinidad para el receptor μ y la dinorfina-A, con mayor afinidad por el receptor .
Los últimos péptidos descubiertos han sido los tetrapéptidos endorfina 1 y endorfina 2, con gran selectividad sobre el receptor μ.
Cada tipo básico de receptor tiene subtipos propuestos: δ1, δ 2, μ 1, μ 2, 1, 2, y 3 para los receptores δ, y μ, respectivamente.

Sistema opioide endógeno
Inicialmente se identificaron tres familias de POE genéticamente independientes: encefalinas, endorfinas y dinorfinas.
Incluyen unos 20 péptidos con actividad opioide, originados a partir de moléculas precursoras inactivas (propio-melanocortina, pro-encefalina y pro-dinorfina).
Las similitudes en la organización de sus genes sugieren un antecesor común.


Caracterización molecular de los receptores opioides
Los efectos antinociceptivos de los
opioides están mediados por la unión a proteínas
específicas de membrana (RO), localizadas a nivel supraespinal, espinal y
periférico.

Caracterización molecular de los receptores opioidesLos receptores opioides se diferencian entre sí por su configuración, distribución anatómica y afinidad a los opioides.
Todos ellos producen antinocicepción al ser activados por agonistas opioides, aunque parece que pueden modular distintos tipos de dolor.

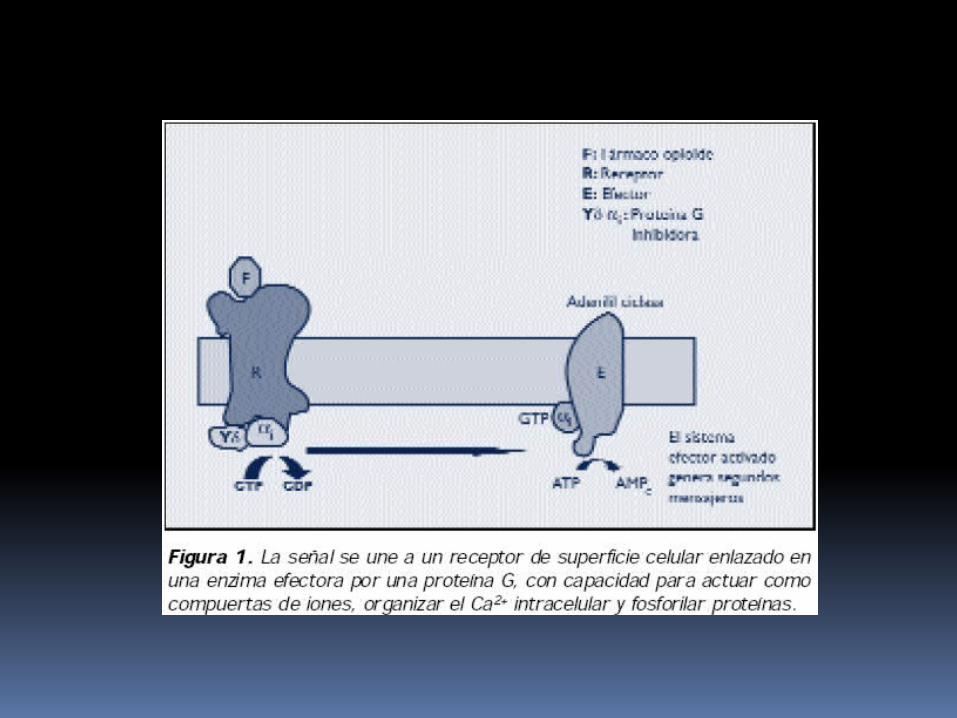
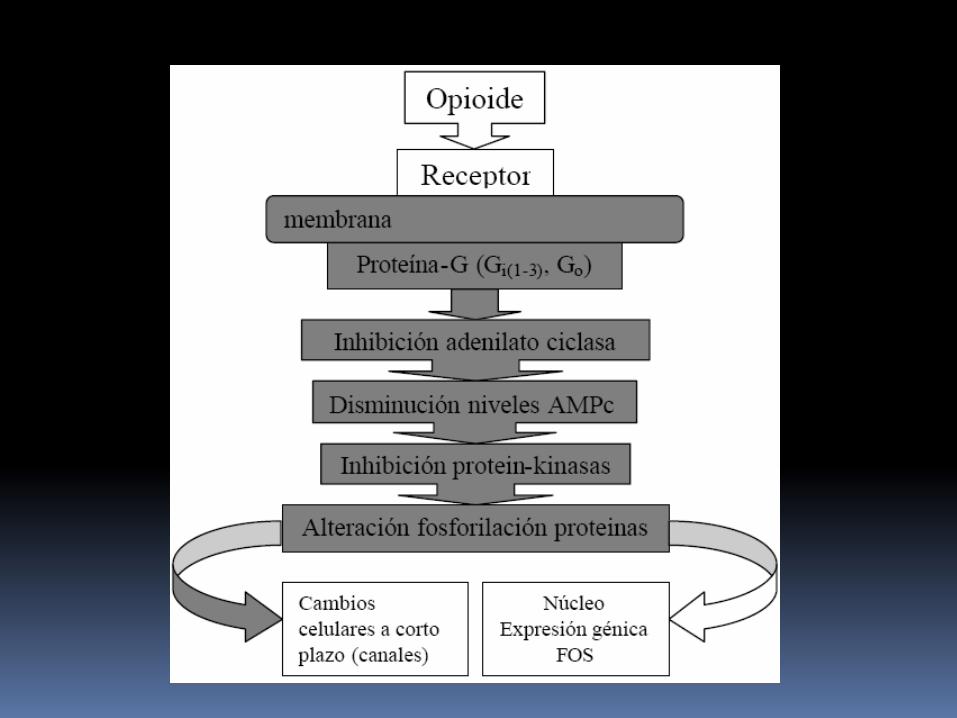
Caracterización molecular de los receptores opioides La mayoría de RO actúan a través de la proteína G de
membrana, inhibiendo la adenilato ciclasa.
Esto produce una disminución en los niveles intracelulares de segundos mensajeros (AMPc) y altera la fosforilación de proteinas intracelulares y como consecuencia se producen respuestas celulares a corto plazo (modificando la permeabilidad de los canales iónicos de membrana, sobre todo para K+ y Ca++, disminuyendo la excitabilidad neuronal); y a nivel del núcleo se producen cambios en la expresión de genes (aumentando o disminuyendo).
Estas modificaciones en la expresión de genes pueden contribuir a explicar los fenómenos de tolerancia y dependencia, que ocurren tanto con opioides como otras sustancias de abuso.

Receptores opioides periféricos La distribución de los RO en el tejido
nervioso muestra que estos se sitúan en las capas más superficiales de asta posterior de la médula espinal y a nivel cerebral, siendo su concentración mayor a nivel de la estructuras límbicas, núcleos del tálamo y áreas de control de funciones viscerales.

Receptores opioides periféricos Existe evidencia de expresión a nivel periférico:
ganglios de la raíz dorsal, células endocrina y en el sistema inmune; por tanto se han demostrado RO a nivel pre, post y extrasináptico.
En el SNP se han encontrado RO en fibras nerviosas sensoriales y simpáticas de piel y articulaciones, en los plexos submucosos del intestino, vejiga urinaria y en los conductos deferentes.
A nivel gastrointestinal los RO se expresan en condiciones normales (RO constitutivos), pero en otras estructuras como en piel y articulaciones, sólo se expresan después de una lesión tisular y en presencia de cambios inflamatorios.

Farmacología clínica de los opioides Los POE y los fármacos opioides producen analgesia al unirse
a los mismos receptores; sin embargo esta unión es diferente en función de si se trata de opioides alcaloides (morfina), no alcaloides (fentanilo) o péptidos (POE y análogos)
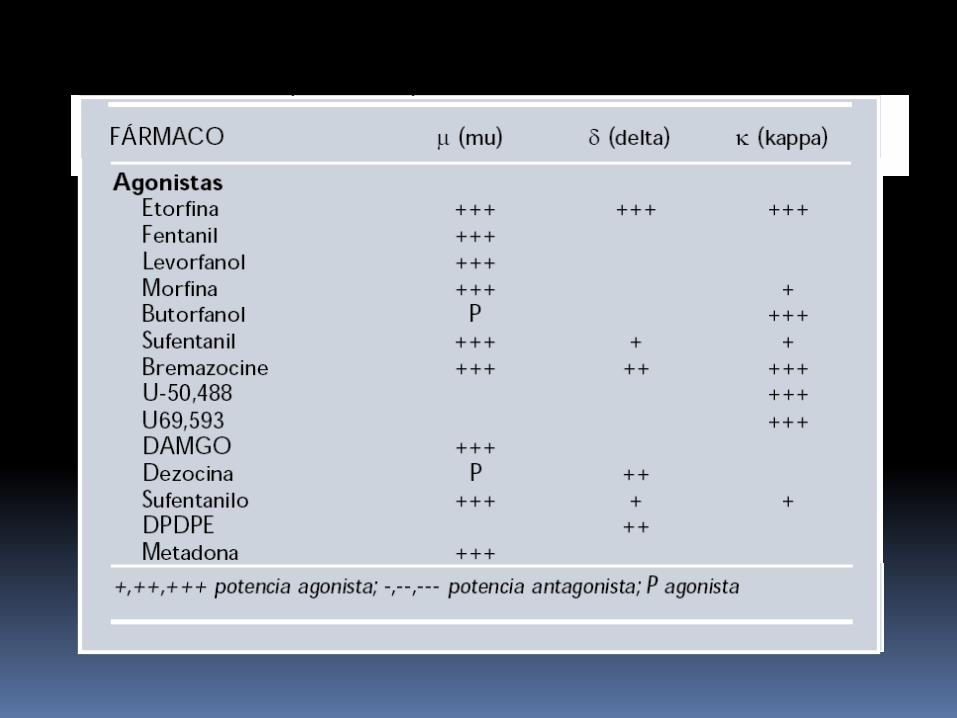
En la clínica habitualmente se utilizan los analgésicos opioides, los cuales se pueden clasificar utilizando diferentes criterios:
origen: naturales, sintéticos, semisintéticos estructura química: fenantrenos, fenilpiperidinas,
benzomorfanos, morfinanos; intensidad de dolor que pueden suprimir: débiles, potentes; tipo de interacción con el receptor afinidad por los receptores (μ, y δ) eficacia (agonistas, antagonistas, agonistas parciales, agonistas-
antagonistas) duración de acción: corta, ultracorta, retardada.

Farmacología clínica de los receptores opioides La afinidad de los opioides por los receptores es
relativa, de tal forma que un opioide puede desplazar a otro del receptor al que se ha unido.
Los agonistas-antagonistas y los agonistas parciales muestran un “efecto techo” para la analgesia que limita su uso; sin embargo producen menos depresión respiratoria y dependencia.
Los agonistas-antagonistas pueden provocar un síndrome de abstinencia cuando se administran a pacientes que han recibido agonistas puros.


Farmacología clínica de los receptores opioides Los opioides poseen múltiples efectos
farmacológicos, además del analgésico, a causa de la amplia distribución de los RO en el organismo: somnolencia, depresión respiratoria, miosis, emesis,
retención urinaria, constipación, prurito, euforia; además tras su administración repetida puede aparecer tolerancia y dependencia.
Características importantes de los opioides: Su respuesta farmacológica se modifica en función
de la presencia de dolor Los efectos analgésicos y no analgésicos ocurren de
forma simultánea.

Farmacología clínica de los receptores opioides
La analgesia de los opioides es dosis-dependiente y la respuesta a los agonistas no tiene efecto techo, la aparición de efectos indeseables es el factor que limita el incremento de la dosis.
Los analgésicos opioides poseen un índice terapéutico relativamente pequeño por lo que para obtener un ligero incremento en la analgesia se puede producir con cierta facilidad la aparición de depresión respiratoria.
La administración simultanea de otros depresores del SNC favorece la aparición de sedación, depresión respiratoria, hipotensión y constipación.
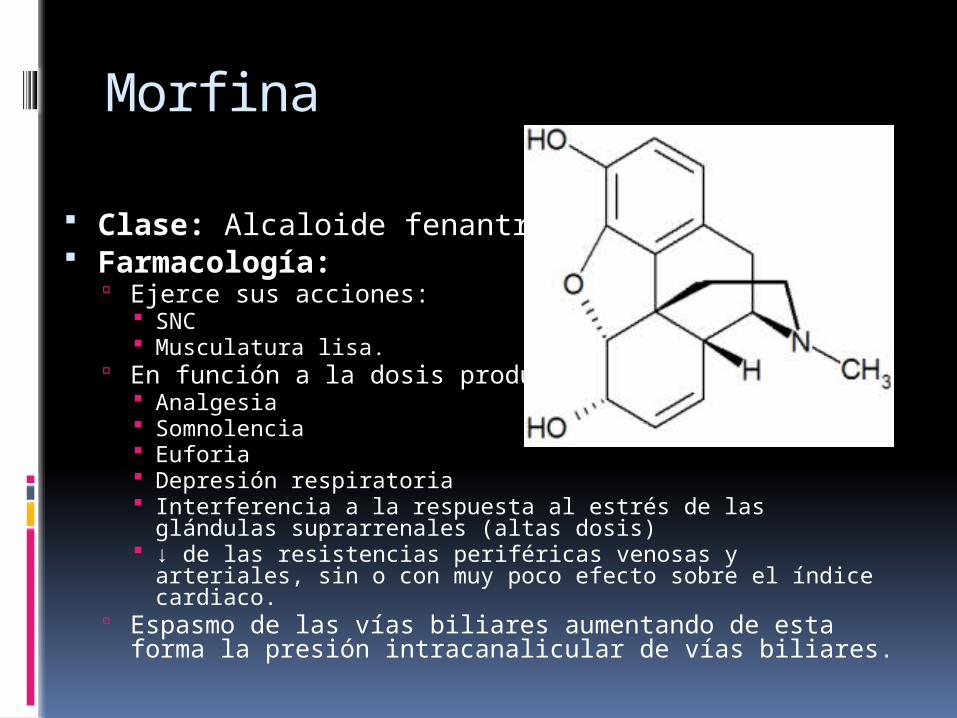
Morfina
Clase: Alcaloide fenantreno Farmacología:
Ejerce sus acciones: SNC Musculatura lisa.
En función a la dosis produce: Analgesia Somnolencia Euforia Depresión respiratoria Interferencia a la respuesta al estrés de las glándulas
suprarrenales (altas dosis) ↓ de las resistencias periféricas venosas y arteriales, sin o
con muy poco efecto sobre el índice cardiaco. Espasmo de las vías biliares aumentando de esta forma
la presión intracanalicular de vías biliares.

Morfina
Se presenta depresión respiratoria máxima en plazo de 5 a 10 min de su administración IV, o de 30 a 90 min después de su administración IM o SC. de la capacidad de reacción de los centros respiratorios del
tallo encefálico al CO2. Los opiodes deprimen también los centros pontinos y bulbares
que participan en la ritmicidad respiratoria.
el flujo sanguíneo cerebral metabolismo cerebral PIC
Provoca náusea y vómito por efecto directo (SNC) Libera histamina y puede causar prurito después de su
administración oral o sistémica.
Morfina
Dosis:
Analgesia: IV, 2.5-15 mg (en niños, 0.05-0.2 mg/kg, 15 mg como dosis
máxima) IM/SC, 2.5-20 mg (en niños, 0.05-0.20 mg/kg, 15 mg como
dosis máxima) VO, 10-30 mg cada 4 horas.
Como inductor anestésico: IV 1 mg/kg Epidural: bolo de 2-5 mg (40-100 mcg/kg) diluído en 10 ml
de solución salina o anestésico local Infusión, 0.1-1 mcg/h (2-20 mcg/kg/h) Espinal, 0.1-1 mg (4-20 mcg/kg)
Morfina
Eliminación: 90% renal; en forma conjugada; como morfina-3-
glucurónido. El 90 % de la dosis aparece en la orina durante las 24 hrs. y
la mayor parte lo hace en las primeras 6 horas. Metabolismo hepático: citocromo P450 2D6 Principales metabolitos
3-glucurónido 6-glucurónido 3,6-glucurónido.
Si se administran dosis muy altas de morfina, el 3-glucurónido antagoniza los efectos de la morfina produciendo hiperalgesia y mioclono.
Se elimina una pequeña cantidad en el estómago y la bilis, la cual aparece en las heces. Se encuentran trazas en el sudor y en la leche.

Morfina
Efectos secundarios: Hipotensión e hipertensión arterial sistémica Bradicardia Arritmias Rigidez torácica (torax leñoso) Broncoespasmo, laringoespasmo Visión borrosa Síncope Euforia, disforia Retención urinaria, efecto antidiurético, espasmo ureteral Espasmo de vías biliares Constipación, anorexia, náusea, vómito, y retraso del
vaciamiento gástrico Miosis, rigidez torácica, prurito y urticaria.

Fentanil
Clase: Fenilpiperidinas Farmacología:
Potente opiode agonista. Primordialmente agonista μ 75-125 veces más potente que la morfina Liposoluble Se une en un 84% a las proteínas
plasmáticas No libera histamina La depresión respiratoria es dosis
dependiente y dura más que el efecto analgésico.

Fentanil
↓ flujo sanguíneo cerebral presión intraocular, miosis
bradicardia vagal, depresión respiratoria ↑
resistencias del árbol traqueobronquial, secreción de la hormona antidiurética, retención urinaria, presión intrabiliar, espasmo del músculo liso biliar
náusea, vómito, disminución de la peristalsis, disminución del vaciamiento gástrico,
rigidez muscular torácica.

Fentanil
Fentanil
Interacción y toxicidad: Depresión respiratoria y cardiovascular
Se potencia con: hipnótico-sedantes, narcóticos, anestésicos volátiles,
N2O
↑ anfetaminas, inhbidores de la MAO, fenotiazinas,
antidepresivos tricíclicos
La analgesia aumenta y prolonga su duración con los alfa-2 agonistas (epinefrina, clonidina).
Fentanil
Farmacocinética: Latencia: IV, en 30 seg; IM, <8 min; epidural/espinal, 4-10 min. Efecto máximo: IV, 5-15 min; IM, <15 min; epidural/espinal, <30 min. Duración: IV, 30-60 min; IM, 1-2 hrs; epidural/espinal, 1-2 hrs.
Metabolismo hepático. Por dealquilación, hidroxilación, e hidrólisis amida a
metabolitos inactivos que se excretan por la bilis y la orina
Eliminación renal

Nalbufina
Clase: opioides semisintéticos Farmacología:
μ antagonista y agonista agonista tan potente como la morfina como antagonista tiene la cuarta parte de la potencia de
nalorfina analgesia profunda miosis sedación limitada depresión respiratoria ↑ PIC reacciones de supresión náusea vómito espasmo del esfínter de Oddi, cólico biliar.

Nalbufina
Dosis: Sedación/analgesia: IV/IM/SC, 5-10 mg (0.1-0.3
mg/kg). Inducción: IV, 0.3-3 mg/kg. Analgesia intravenosa controlada por el paciente:
bolo de 1 a 5 mg (0.02-0.1 mg/kg); Infusión, 1-8 mg/hr (0.02-0.15 mg/kg/h).
Eliminación: alrededor de un 7% de nalbufina se excreta por orina sin modificar junto con dos productos metabólicos. También en materia fecal. Se metaboliza en el hígado.

Nalbufina
Farmacocinética: Latencia: IV, 2-3 min, e IM/SC <15 min.
Efecto máximo: IV, 5-15 min. Duración: IV/IM/SC, 3-6 hrs. La vida plasmática media es alrededor
de 5 horas.

Naloxona
Clase: Sintético (n-alil derivado de oximorfona)
Farmacología: antagonista puro de receptores μ, δ y 10 a 20 veces más activo que la nalorfina revierte: efecto depresor de los morfínicos espasmo de vías biliares por opiodes tratamiento para la sobredosificación de
clonidina, codeína, dextrometorfán, difenoxilato y propoxifeno

Naloxona
Dosis: Como tratamiento y profilaxis de los
efectos secundarios de los opiáceos: IV/IM/SC, 0.1-0.8 mg; en infusión, 50-250
mcg/hr (1-5 mcg/kg/hr); la rapidez de infusión debe ser menor de 125 ml/hr si 1-2 ámpulas de naloxona (ámpula = 0.4 mg/1 ml) se diluyen en 1000 ml de la solución intravenosa actual del paciente.

Naloxona
Eliminación: Metabolismo hepático (95%)
Por conjugación con ácido glucurónico; se producen otros metabolitos en cantidades muy pequeñas.

Naloxona
Farmacocinética: Latencia: IV, 1-2 min; IM/SC, 2-5 min. Efecto máximo: IV/IM/SC, 5-15 min. Duración: IV/IM/SC, 1-4 hr.
Interacción y Toxicidad: reversión de la analgesia producida por opiáceos ↑ la actividad del SNS
taquicardia, hipertensión arterial, edema pulmonar y arritmias cardiacas
produce náusea y vómito relacionados con la rapidez de adminIstración y la dosis.

GRACIAS



![FARMACOLOGIA Antibioticos AINES [Autoguardado]](https://static.fdocuments.ec/doc/165x107/577c77d01a28abe0548d95a5/farmacologia-antibioticos-aines-autoguardado.jpg)














