
Equilibrios Iónicos y Sus Aplicaciones Analíticas
417
-
Upload
pablo-cardella -
Category
Documents
-
view
1.577 -
download
348
description
El objetivo de este libro es introducir al alumno en los aspectos más relevantes de los equilibrios iónicos en disolución, base de numerosos procesos analíticos, y en sus aplicaciones analíticas cuantitativas más características. Por su carácter fundamental, el contenido del mismo forma parte de losprogramas de las asignaturas básicas de Química Analítica que se imparten en numerosas titulaciones, tales como Química, Ingeniería Química, Farmacia, Bioquímica, Ciencias Ambientales, Ciencia y Tecnología de los Alimentos, Geología, Biología, etc. En el texto, a la vez que se consideran los diversos tipos de equilibrios iónicos, se resalta la relación entre los mismos y sus aplicaciones analíticas más relevantes. Se pretende además establecer las bases del conocimiento de estos equilibrios con objeto de que el alumno los utilice posteriormente al desarrollar metodologías analíticas de muy distinta índole en otras disciplinas
Transcript of Equilibrios Iónicos y Sus Aplicaciones Analíticas


EQUILIBRIOS IÓNICOS Y SUS APLICACIONES
ANALÍTICAS

PROYECTO EDITORIAL
BIBLIOTECA DE QUÍMICAS
Director:
Carlos Seoane Prado

EQUILIBRIOS IÓNICOS Y SUS APLICACIONES
ANALÍTICAS
Manuel SilvaCatedrático de Química Analítica
Universidad de Córdoba
José BarbosaCatedrático de Química Analítica
Universidad de Barcelona
Departamento de Ingeniería Químicade la Universitad Autónoma de Barcelona
EDITORIALSINTESIS

Reservados todos los derechos. Está prohibido, bajo las sanciones penales y el resarcimiento civil previstos en las leyes, reproducir, registrar o transmitir esta publicación, íntegra o parcialmente
por cualquier sistema de recuperación y por cualquier medio, sea mecánico, electrónico, magnético,electroóptico, por fotocopia o por cualquier otro, sin la autorización previa por escrito de
Editorial Síntesis, S. A.
© Manuel SilvaJosé Barbosa
© EDITORIAL SÍNTESIS, S. A.Vallehermoso, 34 - 28015 Madrid
Teléf.: 91 593 20 98http://www.sintesis.com
ISBN: 978-84-975602-5-2
Impreso en España - Printed in Spain
Consulte nuestra página web: www.sintesis.comEn ella encontrará el catálogo completo y comentado
Depósito Legal: M. 35.627-2008

A nuestras Mercedes

6 Cinética química aplicada

PREFACIO ....................................................................................................................................... 13
PRÓLOGO ....................................................................................................................................... 15
1. INTRODUCCIÓN AL EQUILIBRIO QUÍMICO .............................................................. 17
1.1. Cinética y equilibrio químico ........................................................................................ 181.2. Interacciones entre solutos iónicos y el agua: actividad y coeficiente de actividad 19
1.2.1. Cálculo del coeficiente de actividad. Leyes de Debye-Hückel ...................... 201.2.2. Variación de los coeficientes de actividad con la fuerza iónica del medio ... 221.2.3. Cálculo de constantes de equilibrio estequiométricas a partir de las termo-
dinámicas ............................................................................................................... 221.3. Tipos de equilibrio en Química Analítica ................................................................... 24
1.3.1. Equilibrios homogéneos ...................................................................................... 241.3.2. Equilibrios heterogéneos ..................................................................................... 261.3.3. Uso analítico del equilibrio químico .................................................................. 27
1.4. Estudio sistemático del equilibrio químico ................................................................. 271.4.1. Balance de masas y de cargas ............................................................................. 281.4.2. Balance protónico ................................................................................................. 291.4.3. Simplificaciones y errores .................................................................................... 30
Cuestiones .................................................................................................................................... 32Seminarios: problemas numéricos ............................................................................................ 33
2. EQUILIBRIOS ÁCIDO-BASE .............................................................................................. 37
2.1. Introducción .................................................................................................................... 382.2. Teoría protónica de Bronsted y Lowry ........................................................................ 39
2.2.1. Influencia del disolvente ...................................................................................... 392.2.2. Autoprotolisis y escala de pH de un disolvente ............................................... 40
2.3. Fuerza de ácidos y bases ................................................................................................ 422.3.1. Efecto nivelador del disolvente .......................................................................... 452.3.2. Interacción con el disolvente .............................................................................. 45
2.4. Equilibrios ácido-base de protolitos fuertes. Cálculo del pH ................................... 462.4.1. Ácido fuerte .......................................................................................................... 462.4.2. Base fuerte ............................................................................................................. 482.4.3. Mezclas de protolitos fuertes .............................................................................. 48
Índice 7
ÍNDICE

2.5. Equilibrios ácido-base de protolitos débiles monopróticos ...................................... 492.5.1. Ácidos débiles monopróticos .............................................................................. 492.5.2. Bases débiles monopróticas ................................................................................ 522.5.3. Cálculo del pH de disoluciones de sales ............................................................ 532.5.4. Mezclas de protolitos monopróticos .................................................................. 58
2.6. Equilibrios ácido-base de protolitos débiles polipróticos ......................................... 632.6.1. Ácidos débiles dipróticos y polipróticos ............................................................ 632.6.2. Bases débiles dipróticas ....................................................................................... 662.6.3. Anfolitos ................................................................................................................ 682.6.4. Sales de ácidos polipróticos ................................................................................. 69
2.7. Disoluciones reguladoras ............................................................................................... 712.7.1. Cálculo del pH de una disolución reguladora .................................................. 742.7.2. Preparación de disoluciones reguladoras .......................................................... 752.7.3. Capacidad reguladora .......................................................................................... 792.7.4. Disoluciones amortiguadoras con compuestos polipróticos ........................... 822.7.5. Disoluciones amortiguadoras que contienen más de un protolito ................. 82
Anexo. Expresiones simplificadas para el cálculo del pH de disoluciones de diferentes protolitos .......................................................................................................................... 84
Cuestiones .................................................................................................................................... 85Seminarios: problemas numéricos ............................................................................................ 87
3. REPRESENTACIÓN GRÁFICA DEL EQUILIBRIO ÁCIDO-BASE ......................... 103
3.1. Introducción .................................................................................................................... 1043.2. Diagrama logarítmico de concentración ...................................................................... 1043.3. Diagrama logarítmico de un protolito fuerte. Cálculo del pH ................................. 1053.4. Diagrama logarítmico de un protolito débil monoprótico ........................................ 106
3.4.1. Determinación del pH de una disolución de un ácido débil monoprótico ... 1093.4.2. Determinación del pH de una disolución de una base débil monoprótica ... 109
3.5. Diagrama logarítmico de una mezcla de dos protolitos débiles monopróticos. Cálculo del pH ................................................................................................................ 110
3.6. Diagrama logarítmico de la disolución de una sal. Cálculo del pH ......................... 1113.7. Diagrama logarítmico de protolitos dipróticos ........................................................... 112
3.7.1. Determinación del pH de disoluciones de diferentes protolitos dipróticos . 1153.8. Diagrama logarítmico de protolitos polipróticos ....................................................... 1163.9. Diagramas de distribución de especies en función del pH ...................................... 117
Cuestiones .................................................................................................................................... 122Seminarios: problemas gráficos ................................................................................................ 124
4. VALORACIONES ÁCIDO-BASE ........................................................................................ 127
4.1. Aspectos generales de las valoraciones ....................................................................... 1284.2. Introducción a las valoraciones ácido-base ................................................................. 131
8 Equilibrios iónicos y sus aplicaciones analíticas

4.3. Indicadores ácido-base ................................................................................................... 1324.4. Curvas de valoración ...................................................................................................... 135
4.4.1. Valoración de protolitos monopróticos ............................................................. 1364.4.2. Valoración de protolitos polipróticos ................................................................. 151
4.5. Preparación de disoluciones valorantes ....................................................................... 1584.5.1. Disolución estándar de HCl 0,1 mol L–1 ............................................................ 1604.5.2. Disolución estándar de NaOH 0,1 mol L–1 ....................................................... 161
4.6. Aplicaciones .................................................................................................................... 1624.6.1. Determinación de la acidez de un vinagre comercial ...................................... 1624.6.2. Determinación de acidez en alimentos .............................................................. 1634.6.3. Determinación de compuestos nitrogenados .................................................... 163
Cuestiones .................................................................................................................................... 166Seminarios: problemas numéricos ............................................................................................ 169
5. EQUILIBRIOS DE FORMACIÓN DE COMPLEJOS ..................................................... 191
5.1. Introducción ..................................................................................................................... 1925.2. Constantes de equilibrio ................................................................................................. 194
5.2.1. Factores que determinan la estabilidad de los complejos ............................... 1975.2.2. Reacciones de desplazamiento ........................................................................... 198
5.3. Cálculo de las concentraciones de especies en el equilibrio de formación de com-plejos ................................................................................................................................. 1995.3.1. Diagramas de distribución de especies .............................................................. 201
5.4. Introducción a los equilibrios concurrentes ................................................................ 2015.5. Equilibrios concurrentes de acidez y complejación ................................................... 202
5.5.1. Tratamiento general de la influencia del pH en la formación de complejos 2035.5.2. Coeficientes de reacción secundaria .................................................................. 2045.5.3. Constantes condicionales de formación de complejos .................................... 2065.5.4. Influencia de otros equilibrios concurrentes ..................................................... 207
Cuestiones .................................................................................................................................... 219Seminarios: problemas numéricos ............................................................................................ 210
6. VALORACIONES DE FORMACIÓN DE COMPLEJOS ............................................... 219
6.1. Introducción .................................................................................................................... 2206.2. Curvas de valoración con EDTA .................................................................................. 222
6.2.1. Influencia de reacciones secundarias del ion metálico .................................... 2286.2.2. Influencia de reacciones secundarias del ligando ............................................. 230
6.3. Indicadores metalocrómicos .......................................................................................... 2336.4. Tipos de valoraciones complexométricas .................................................................... 2376.5. Aplicaciones .................................................................................................................... 239
Cuestiones .................................................................................................................................... 241Seminarios: problemas numéricos ............................................................................................ 243
Índice 9

7. EQUILIBRIOS DE PRECIPITACIÓN ................................................................................. 249
7.1. Introducción .................................................................................................................... 2507.2. Solubilidad y producto de solubilidad ......................................................................... 2517.3. Factores que afectan a la solubilidad ........................................................................... 252
7.3.1. Influencia de la fuerza iónica .............................................................................. 2547.3.2. Efecto de ion común ............................................................................................ 255
7.4. Representación gráfica de los equilibrios de precipitación ....................................... 2557.5. Precipitación fraccionada ............................................................................................... 2587.6. Equilibrios concurrentes de acidez, complejación y precipitación. Producto de solu-
bilidad condicional .......................................................................................................... 2597.6.1. Reacciones secundarias de protonación del anión ........................................... 2607.6.2. Reacciones secundarias de hidrólisis del metal ................................................ 2627.6.3. Reacciones secundarias de complejación .......................................................... 2647.6.4. Influencia conjunta de reacciones secundarias de acidez y complejación .... 267
Cuestiones .................................................................................................................................... 269Seminarios: problemas numéricos ............................................................................................ 270
8. VALORACIONES DE PRECIPITACIÓN .......................................................................... 281
8.1. Introducción .................................................................................................................... 2828.2. Curvas de valoración ...................................................................................................... 282
8.2.1. Valoración de mezclas .......................................................................................... 2858.3. Indicadores de precipitación ......................................................................................... 287
8.3.1. Método de Mohr ................................................................................................... 2888.3.2. Método de Volhard .............................................................................................. 2888.3.3. Indicadores de adsorción ..................................................................................... 289
8.4. Aplicaciones .................................................................................................................... 291Cuestiones .................................................................................................................................... 292Seminarios: problemas numéricos ............................................................................................ 293
9. GRAVIMETRÍAS ..................................................................................................................... 297
9.1. Introducción .................................................................................................................... 2989.2. Factor gravimétrico y sensibilidad ................................................................................ 2999.3. Reactivos orgánicos y gravimetrías .............................................................................. 3019.4. Aspectos experimentales de interés en gravimetrías ................................................. 3029.5. Métodos gravimétricos de calcinación ......................................................................... 305
9.5.1. Determinaciones por formación de óxidos hidratados ................................... 3059.5.2. Determinaciones por formación de compuestos salinos ................................. 307
9.6. Métodos gravimétricos de desecación ......................................................................... 3099.6.1. Determinación de plomo como cromato de plomo ......................................... 3099.6.2. Determinación de níquel con dimetilglioxima ................................................. 310
10 Equilibrios iónicos y sus aplicaciones analíticas

Cuestiones .................................................................................................................................... 311Seminarios: problemas numéricos ............................................................................................ 312
10. EQUILIBRIOS DE OXIDACIÓN-REDUCCIÓN ............................................................. 315
10.1. Introducción .................................................................................................................... 31610.2. Estudio de reacciones redox: celdas electroquímicas ................................................ 317
10.2.1. Celdas galvánicas .............................................................................................. 31810.2.2. Celdas electrolíticas ......................................................................................... 320
10.3. Potencial y carácter químico redox: ecuación de Nernst ........................................... 32110.4. Medida del potencial redox: electrodos de referencia ............................................... 322
10.4.1. Electrodo estándar de hidrógeno ................................................................... 32310.4.2. Electrodo de Ag/AgCl ..................................................................................... 323
10.5. Potencial estándar ........................................................................................................... 32410.6. Constante de equilibrio de una reacción redox .......................................................... 32610.7. Potencial de equilibrio ................................................................................................... 32810.8. Sistemas poliredox .......................................................................................................... 33010.9. Disoluciones reguladoras redox .................................................................................... 332
10.10. Sistemas redox del agua ................................................................................................. 33310.11. Representación gráfica del equilibrio redox ............................................................... 33510.12. Equilibrios concurrentes: potencial condicional ......................................................... 337
10.12.1. Equilibrios concurrentes redox/ácido-base ................................................... 33710.12.2. Equilibrios concurrentes redox/formación de complejos ........................... 34310.12.3. Equilibrios concurrentes redox/precipitación .............................................. 34710.12.4. Equilibrios concurrentes con varias interacciones ....................................... 348
Cuestiones ................................................................................................................................. 354Seminarios: problemas numéricos .......................................................................................... 355
11. VALORACIONES DE OXIDACIÓN-REDUCCIÓN ....................................................... 369
11.1. Introducción .................................................................................................................... 37011.2. Curvas de valoración ...................................................................................................... 371
11.2.1. Influencia de reacciones secundarias ............................................................. 37611.3. Indicadores redox ........................................................................................................... 37811.4. Normalidad versus molaridad en valoraciones redox ................................................ 38211.5. Aplicaciones .................................................................................................................... 383
11.5.1. Tratamiento previo en volumetrías redox ..................................................... 38411.5.2. Valoraciones con permanganato (permanganimetrías) .............................. 38611.5.3. Valoraciones con dicromato (dicromatometrías) ......................................... 39111.5.4. Valoraciones en las que interviene el yodo .................................................. 393
Cuestiones .................................................................................................................................... 399Seminarios: problemas numéricos ............................................................................................ 402
Índice 11


La enseñanza de los principios básicos de la Química Analítica, en los diferentes planes de estu-dio que se imparten en las distintas universidades españolas, se desarrolla esencialmente dentro deuna materia troncal denominada “Química Analítica”. En ella se establece el primer contacto delalumno con nuestra disciplina, contacto que no siempre es idóneo dado que, por una parte, el alum-no no siempre accede a la universidad con una formación química adecuada y, por otra, a que no dis-pone de libros de texto que se adapten a los programas que generalmente impartimos, aquellos cuyalabor docente se concentra en estos cursos iniciales de la licenciatura. Existen diversos textos, gene-ralmente traducciones, que por su propia naturaleza enfocan el estudio de estos principios básicos,tales como los equilibrios iónicos en disolución, con una concepción diferente a la sistemática segui-da en la mayoría de las universidades españolas, con lo que el alumno ha de extraer y ordenar de losmismos la información necesaria. En muchos casos, además, no se incluyen problemas numéricos, degran importancia para este tipo de materia, totalmente resueltos, o incluso no se indica el resultadofinal de los mismos.
Ante esta situación, nuestro propósito a la hora de elaborar este libro es enseñar al alumno losfundamentos de la Química Analítica, considerando la experiencia adquirida en los numerosos añosde impartición de la asignatura Química Analítica en las licenciaturas de Química y Farmacia de lasUniversidades de Córdoba y Barcelona, y las opiniones recogidas de otros compañeros de diferen-tes universidades. En este sentido, el texto se ha estructurado en cuatro bloques fundamentales querecogen el estudio de los diferentes equilibrios iónicos y sus correspondientes aplicaciones analíticascuantitativas en valoraciones, a pesar de que toda esta materia se ha dividido en dos asignaturas enciertas universidades. Este enfoque integral conlleva un sencillo aprendizaje y un mayor aprovecha-miento de estos conceptos, no sólo para el alumnado de la licenciatura en Química, sino para aque-llos de ETS de Ingenieros, Farmacia, Ciencias Ambientales, Ciencia y Tecnología de los Alimentos,Bioquímica y otras titulaciones que requieran de estos principios básicos de Química Analítica.
Cada capítulo está organizado de manera que comienza con una indicación de los objetivos quese pretenden alcanzar en el mismo. Tras una introducción se aborda su desarrollo sistemático, intro-duciendo los diferentes conceptos y resaltando aquellas expresiones y aspectos de mayor interés conla ayuda de figuras, cuadros y recuadros. Los términos y símbolos utilizados en la exposición se corres-ponden con las indicaciones recogidas en el Compendium of Analytical Nomenclatura. DefinitiveRules editado por la IUPAC en 1997. El capítulo concluye con la propuesta de una serie de cuestio-nes a modo de resumen y con la propuesta de un número apreciable de problemas numéricos distri-
PREFACIO

buidos de acuerdo con la materia tratada. Se resuelven de forma pormenorizada en cada apartadolos problemas más significativos y se propone al alumno la resolución de otros similares, de los quese indica su resultado, con objeto de que pueda constatar su nivel de conocimiento sobre la materia.
Sacar a la luz un libro siempre es trabajo de equipo. Así, queremos mostrar nuestro agradecimientoa los profesores José Luis Beltrán, Gemma Fonrodona y Jacinto Guiteras de la Universidad de Bar-celona por la revisión sistemática del texto, y al profesor Miguel Valcárcel por su inestimable ayudadada su experiencia en la enseñanza de la Química Analítica y por sus grandes esfuerzos para queeste texto viese la luz, todos ellos compañeros en nuestras tareas docentes desarrolladas en diversaslicenciaturas de las Universidades de Córdoba y Barcelona, y también a Carlos Seoane, coordinadorde la colección Biblioteca de Químicas de la editorial Síntesis, así como a la citada editorial por ladiligencia y esmero que ha puesto en la presente publicación.
Este libro se lo dedicamos a todos aquellos alumnos que lo hagan servir como ayuda en sus estu-dios y a aquellos compañeros que decidan hacer uso de él como texto de referencia en su docen-cia sobre nuestra disciplina. Evidentemente, surgirán comentarios, sugerencias, críticas, etc. queesperamos y agradecemos de antemano que se nos hagan llegar con objeto de mejorar la presenteedición.
14 Equilibrios iónicos y sus aplicaciones analíticas

La propuesta para prologar este texto me lle-gó mientras revisaba los datos del estudio reali-zado por el UK Analytical Partnership (UKAP)que, acerca de la innovación en Química Analíti-ca, han aparecido en el número de febrero de 2002 de la revista Chemistry in Britain (y quepueden ampliarse en la dirección de la redwww.ukap.org). Dichos datos muestran cómo sor-prendentemente la Química Analítica españolase sitúa a la cabeza de los países considerados(todos los importantes desde un punto de vistaindustrial y tecnológico) en lo que se refiere apublicaciones científicas de calidad en relación alProducto Interior Bruto. En otras palabras, losquímicos analíticos actuales parecen ser los quemejor rentabilizan en producción científica losrecursos que España pone a su disposición. Obvia-mente, estos datos no deberían engañarnos. Escierto que, de acuerdo con ellos, nuestra rentabi-lidad científica es superior a la de Estados Uni-dos, Japón y el resto de los países europeos, peroeso no quiere decir que en España se inviertamucho dinero en investigación científica. Por elcontrario, se trata de un cociente y, en conse-cuencia, el valor del mismo crece al disminuir eldenominador (el PIB). En todo caso, el creci-miento del porcentaje de publicaciones corres-pondiente a nuestro país a lo largo de los últimosveinte años ha sido prácticamente de un 4% mien-tras que para otros países parece estancado oincluso disminuye (caso del Reino Unido).
Lamentablemente, los datos de los cocientes entreel número de patentes y el número de publica-ciones nos devuelven a la cruda realidad al situar-nos al final de la lista de los países considerados.
Independientemente de la interpretación quepudiera darse a este estudio, lo cierto es que laQuímica Analítica española ha cambiado radi-calmente en los últimos 25 años. Su proyeccióninternacional ha crecido de manera notoria y sereconoce que lo ha hecho con mayor pendienteque para el resto de las disciplinas químicas ennuestro país. Varias generaciones de químicosanalíticos han protagonizado un salto cualitati-vo que pocos esperaban y que, probablemente,ha sorprendido a la mayoría de nuestros colegas.Lógicamente, este salto cualitativo (y cuantitati-vo) debería hacerse notar también en la docen-cia, y efectivamente así ha sido. Se acepta uni-versalmente que en la universidad solamentepuede haber buena docencia si hay buena inves-tigación, por tanto, la mejora de condiciones,planteamientos y resultados del conjunto de lainvestigación químico analítica española se hatraducido en una mejor docencia, de más cali-dad, más innovadora y al nivel de cualquiera delos países con los que nos relacionamos o com-petimos. Sin embargo, existe una laguna todavíaimportante en lo que se refiere a los textos docen-tes universitarios.
Durante años, hemos venido utilizando ennuestros cursos textos traducidos o directamen-
PRÓLOGO

te los textos en inglés. Se han producido loablesesfuerzos por mantener y actualizar algunos delos textos clásicos de la Química Analítica espa-ñola, pero su número es relativamente escaso yla cobertura claramente insuficiente para la actualcarga docente de la disciplina y el desarrolloalcanzado en todos los órdenes. Por tanto, debeanimarse a los profesores de Química Analíticade este país a que asuman el compromiso histó-rico de elaborar nuevos textos docentes en nues-tro idioma y dirigidos a nuestros alumnos, con-siderando las peculiaridades de nuestros planesde estudio actuales y nuestra propia forma de sery manifestarnos.
Los profesores Silva y Barbosa pertenecen aesas generaciones de químicos analíticos espa-ñoles que han contribuido al aparente milagrode nuestra disciplina. Su categoría como cientí-ficos y el nivel de calidad de su investigación essobradamente conocido y reconocido a nivelinternacional. Además, acumulan una ampliaexperiencia docente y una notable inquietud pormejorar cada día su mensaje al alumno y repre-sentan grupos de trabajo en dos de las universi-dades españolas con mejor nivel en el campo dela Química Analítica. Por tanto, era razonableesperar un texto de gran calidad como fruto desu colaboración. Efectivamente, este texto sobreEquilibrios iónicos y sus aplicaciones analíticasno es un libro más sobre “equilibrios”. En pri-mer lugar, porque nace con una clara vocaciónde texto universitario (basta considerar los másde 350 problemas, de los que aproximadamen-te una tercera parte han sido resueltos con grandetalle, y las más de cien cuestiones que inclu-ye) que aspira a ser utilizado de forma intensi-va y continuada por el alumno. En segundolugar, porque representa la tendencia actual dela química analítica a explicar los equilibrios nocomo algo aislado o un elemento formativo inde-pendiente para el alumno, sino conectados direc-tamente con las aplicaciones analíticas conven-cionales de tipo cuantitativo. De este modo, elalumno aprecia de forma inmediata la utilidadde lo que va aprendiendo y lleva al terreno de
la realidad material los conocimientos abstrac-tos que adquiere. En tercer lugar, porque estáescrito con un lenguaje directo y simple, resal-tando los conceptos y apoyándose en las ecua-ciones cuando es necesario, pero sin abrumar alalumno con interminables demostraciones.Todos los aspectos formales y pedagógicos sehan cuidado; la tipografía, la equilibrada distri-bución del espacio, atención y detalles entre lasdiversas aproximaciones a utilizar, etc., contri-buyen a facilitar la lectura y el estudio de estetexto.
Especialmente destacables son las coleccionesde cuestiones y problemas. Las primeras tienen enmuchos casos una estructura de tipo test que faci-lita al alumno un chequeo rápido del nivel de apro-vechamiento de su estudio para cada capítulo. Enel caso de los problemas, un elemento imprescin-dible en el estudio de esta materia, los autores hanefectuado un notable esfuerzo de reunión y siste-matización de este tipo de elementos pedagógicos.El hecho de que, prácticamente en todos los casos,el alumno disponga de varios problemas repre-sentativos de cada tipo resueltos paso a paso y deotros muchos de tipo similar, mediante los cualescomprobar hasta qué punto ha entendido los con-ceptos y puede lograr una destreza suficiente en elmanejo de las herramientas analíticas que se le pro-porcionan, constituye, sin duda, uno de los ele-mentos más destacables del texto. Además, suextensión lo hace viable como texto de un cursobásico de equilibrios en Química Analítica pero,también, como un libro de consulta para el alum-no en cursos posteriores.
En definitiva, considero este texto no sola-mente recomendable para los alumnos de la dis-ciplina sino también, lógicamente, para quienestengamos que impartirla. Igualmente, aplaudo lainiciativa de la editorial Síntesis al abordar unaempresa ambiciosa destinada a dotar al merca-do editorial español de textos universitarios degran calidad, actuales y adaptados a la realidadde nuestra enseñanza.
Rafael Cela Torrijos
16 Equilibrios iónicos y sus aplicaciones analíticas

1
1.1. Cinética y equilibrio químico1.2. Interacciones entre solutos iónicos y el
agua: actividad y coeficiente de actividad1.3. Tipos de equilibrio en Química
Analítica1.4. Estudio sistemático del equilibrio
químicoCuestionesSeminarios: problemas numéricosINTRODUCCIÓN
AL EQUILIBRIOQUÍMICO

1.1. Cinética y equilibrio químico
Toda reacción química, sea cual sea su natura-leza, transcurre a una velocidad finita tendente aalcanzar una posición final de equilibrio, por lo quese pueden distinguir en la misma dos zonas o regio-nes: una región cinética (o dinámica) y como taldependiente del tiempo en el que el sistema seaproxima al equilibrio, y otra región de equilibrio(o estática) que se origina después de que todos losprocesos del sistema hayan alcanzado el equilibrio.Ambas regiones están íntimamente relacionadasy suministran información para la resolución devariados problemas en Química Analítica.
La región cinética puede caracterizarse por lavelocidad de reacción, que se define como elnúmero de moles consumidos o formados porunidad de volumen y unidad de tiempo. Estavelocidad de reacción depende de la concentra-ción de las especies reaccionantes, de las afini-dades químicas de las mismas y de la tempera-tura, dado que ésta incide positivamente sobreel número de colisiones entre las especies queintervienen en la reacción química. Así, por ejem-plo, para la reacción irreversible
A + B → C + D [1.1]
la velocidad de reacción en un tiempo t vienedada por la derivada de la concentración de cual-quiera de las especies implicadas en la reaccióncon respecto al tiempo:
[1.2]
El signo negativo en las derivadas corres-pondientes a las especies A y B se debe a su desa-parición a medida que progresa la reacción.
Como se ha indicado, la velocidad de reac-ción es proporcional a la concentración de cual-quiera de las especies implicadas en la misma,reflejándose esta dependencia en la correspon-diente ecuación de velocidad:
[1.3]
donde k1 es la constante de velocidad. De mane-ra similar, para la reacción polimolecular
a A + b B → c C + d D [1.4]
se puede establecer la siguiente ecuación develocidad:
[1.5]
en la que, como en la ecuación [1.3], la suma delos exponentes de las concentraciones implica-das se conoce como orden de reacción global,mientras que el exponente de cada una de estasconcentraciones recibe el nombre de orden par-cial de reacción. Así, la ecuación [1.3] será glo-balmente de segundo orden, y de primer ordenen cada uno de sus componentes.
Hasta ahora se han considerado reaccionesirreversibles, sin embargo, ésta no es la situación
Velocidad[A]
[B][A] [B]
= − =
= − =
1
11
ad
dt
bd
dtk a b
Velocidad[A]
[B][A][B]
= − =
= − =
ddt
ddt
k1
Velocidad[A]
[B] [C] [D]
= − =
= − = =
ddt
ddt
ddt
ddt
18 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Introducir al alumno en los aspectos básicos del equi-librio químico.
• Describir el comportamiento de las sustancias en situa-ciones reales (no ideales).
• Diferenciar los distintos tipos de equilibrio en Quí-mica Analítica.
• Establecer las pautas para abordar el estudio siste-mático del equilibrio.

más real ya que la mayoría de las reacciones deinterés en Química Analítica son reversibles. Eneste caso, para la reacción
a A + b B →← c C + d D [1.6]
se pueden definir dos tipos de velocidades dereacción: velocidad de reacción directa, que res-ponde a la formación de los productos C y D yque por tanto es proporcional a las concentra-ciones de las sustancias reaccionantes A y B:
[1.7]
y velocidad de reacción inversa, que origina la for-mación de los reactivos A y B a partir de loscorrespondientes productos de la reacción C y D:
[1.8]
FIGURA 1.1. Evolución de las velocidades de una reacciónquímica hasta alcanzar el equilibrio.
Al comienzo de la reacción, VDirecta será muyelevada, mientras que VInversa será prácticamentecero, pero a medida que transcurre la misma
ambas velocidades tenderán a coincidir llegandoun momento en que se igualen, instante en el quese alcanza el equilibrio químico. Hay que indicarque este equilibrio no es un equilibrio estáticosino dinámico, ya que simultáneamente se estáoriginando la formación de los productos y la con-versión de éstos en las correspondientes sustan-cias reaccionantes. En la figura 1.1 se muestra grá-ficamente la evolución temporal de ambasvelocidades hasta alcanzar el estado de equilibrio.
Como se ha indicado, en el momento en quese alcanza el equilibrio químico VDirecta = VInversa,por tanto:
[1.9]
y tras operar se tiene:
[1.10]
expresión que proporciona la constante de equili-brio(K) de una reacción que se corresponde conla ley de acción de masas de Guldberg y Waage.Esta constante de equilibrio sólo depende de latemperatura. Finalmente, hay que indicar que aun-que la reacción en estudio transcurra en varias eta-pas más o menos complejas y las expresiones delas velocidades puedan ser diferentes a las indi-cadas, la constante de equilibrio responderá entodos los casos a la expresión de la ecuación [1.10].
1.2. Interacciones entre solutos iónicosy el agua: actividady coeficiente de actividad
La expresión de la constante de equilibriodefinida por la ecuación [1.10] predice que elcociente de concentraciones de productos y reac-tivos es un valor constante, en un sistema en equi-librio, para una determinada reacción química.
K
kk
c d
a b= =1
2
[C] [D][A] [B]
k ka b c d1 2[A] [B] [C] [D]=
V k c dInversa [C] [D]= 2
V k a bDirecta [A] [B]= 1
Capítulo 1: Introducción al equilibrio químico 19

Esta definición es adecuada para un sistema hipo-tético que se comporta idealmente, por lo que enlos sistemas químicos reales no siempre resultaválida dicha ecuación.
De hecho, la expresión de la constante de equi-librio, tal como está planteada, considera las molé-culas de las sustancias reaccionantes como partí-culas independientes sin interacción entre ellas.Esto puede ser aceptable cuando se trate de molé-culas sin carga y a bajas concentraciones. Sin embar-go, cuando se tienen especies iónicas en disolución,hay que tener en cuenta como mínimo las interac-ciones electrostáticas entre las mismas: atracciónentre partículas de carga opuesta y repulsión entrepartículas de la misma carga. Por otra parte, pue-den existir además otras interacciones, como ion-dipolo, formación de pares iónicos, etc. Estas inte-racciones son la causa principal del comportamientono ideal en las disoluciones de electrolitos. Comoconsecuencia, la constante de equilibrio definidapor la ecuación [1.10] solamente será cierta en unascondiciones en las que las partículas estén lo sufi-cientemente alejadas unas de otras como para quesu interacción sea mínima y despreciable, es decir,a concentraciones muy diluidas.
Teniendo en cuenta estas consideraciones sedefine un nuevo parámetro, llamado actividad(ai), que es una medida de la “concentración efec-tiva” de las especies en equilibrio, de manera queel cociente de actividades de las especies de unsistema en equilibrio sí es constante para cual-quier condición experimental del mismo, siem-pre que la temperatura permanezca constante.
Considerando las actividades de las diferen-tes especies en lugar de sus concentraciones, laconstante de equilibrio del sistema descrito en laecuación [1.10] toma la forma:
[1.11]
A esta constante, que relaciona actividades deproductos y sustancias reaccionantes, se le deno-mina constante de equilibrio termodinámica (KT),mientras que las constantes de equilibrio expre-
sadas en términos de concentración (ecuación[1.10]), se denominan constantes de equilibrio este-quiométricas (K). Ambas constantes pueden rela-cionarse mediante el coeficiente de actividad (γi),que se define como el factor que relaciona la con-centración de una especie con su actividad:
[1.12]
Así, teniendo en cuenta los coeficientes deactividad, se puede relacionar el valor de unaconstante de equilibrio termodinámica con sucorrespondiente estequiométrica:
[1.13]
1.2.1. Cálculo del coeficiente de actividad. Leyes de Debye-Hückel
Si bien Bronsted dedujo una expresión empí-rica que permitía obtener el coeficiente de acti-vidad medio de electrolitos uni-univalentes (porejemplo, NaCl) en disoluciones diluidas, fueronP. Debye y E. Hückel quienes desarrollaron entre1923 y 1924 las teorías que permiten explicar elcomportamiento de las disoluciones diluidas deelectrolitos y calcular el coeficiente de actividadde las especies en unas condiciones experimen-tales determinadas.
La primera teoría de Debye-Hückel, en la quese considera solamente la energía de interacciónelectrostática entre iones, supone que el coefi-ciente de actividad para un determinado iondepende de su carga y de la concentración y car-ga del resto de los iones en disolución a través dela fuerza iónica (I). Esta fuerza iónica se calculacomo la semisuma de los productos de las con-centraciones de cada uno de los iones presentes(Ci) por sus respectivas cargas al cuadrado (zi
2):
K
K
T Cc
Dd
Aa
Bb
cCc d
Dd
aAa b
Bb
Cc
Dd
Aa
Bb
= =
= =
a aa a
[C] [D][A] [B]
γ γγ γ
γ γγ γ
a Ci i i= γ
KT Cc
Dd
Aa
Bb
= a aa a
20 Equilibrios iónicos y sus aplicaciones analíticas

[1.14]
La expresión del coeficiente de actividadpara un determinado ion de carga zi viene dadapor:
[1.15]
donde A es igual a:
[1.16]
siendo ε la constante dieléctrica, e la carga delelectrón, NA el número de Avogadro, k la cons-tante de Boltzmann y T la temperatura absolu-ta. Para disoluciones acuosas a 25 °C, el pará-metro A tiene un valor de 0,512.
La ecuación [1.15] es la expresión de la leylímite de Debye-Hückel, que permite calcularvalores en buena concordancia con los resulta-dos experimentales, sólo para disoluciones conconcentraciones bajas de electrolito (con valo-res de I < 0,01 mol L–1). Esto es debido a que nose han tenido en cuenta una serie de factores,tales como la variación de la constante dieléc-trica del disolvente en disoluciones de electro-litos, o que se consideran los iones como cargaspuntuales (es decir, que no ocupan un volumen),etc. En una segunda aproximación de la teoríade Debye-Hückel, se corrige la ecuación [1.15]para dar lugar a una expresión (ley ampliada)donde se tienen en cuenta las dimensiones delos iones:
[1.17]
El término a es la distancia mínima de apro-ximación de los centros de los iones (equivalen-te al radio del ion solvatado), aunque la diferen-
te solvatación de los iones da lugar a que su valorreal sea variable (por ejemplo, se toman valoresentre 3 y 5 Å, siendo 1 Ångstrom = 10–10 m). Elparámetro B depende de la constante dieléctricadel medio y de la temperatura:
[1.18]
En disoluciones acuosas a 25 °C, el valor deB es de 0,328 × 1010. Esta segunda aproximaciónde Debye-Hückel puede aplicarse, con una bue-na concordancia con los datos experimentales,para disoluciones de concentración igual o infe-rior a 0,05 – 0,1 mol L–1.
Como el valor del radio iónico es difícil deconocer exactamente para cada ion, se utilizatambién una aproximación (debida a Güntelberg),en la que se considera que el producto a × B enla ecuación [1.17] es igual a la unidad (supone quepara todos los electrolitos a = 3,04 Å a 25 ºC).Esta aproximación se conoce como ley ampliadasimplificada de Debye-Hückel:
[1.19]
Las leyes límite y ampliada de Debye-Hückelpredicen una disminución del coeficiente de acti-vidad al aumentar la concentración de electrolito.Sin embargo, para disoluciones relativamenteconcentradas se observa experimentalmente unaumento en el coeficiente de actividad, debido aun incremento de las fuerzas de repulsión entrelos iones solvatados. Para compensar este efec-to, se introduce un término empírico (C) en laley ampliada de Debye-Hückel, ecuación [1.17],que corrige este efecto:
[1.20] log
A B
C γ iiz I
a II= −
++
2
1
log
,
γ iiz I
I= −
+0 512
1
2
Be N
k T
T K
A= =
×
2 1.000
=5,03 10
(L mol ) m
11–1 1/2 –1
ε
ε
log
A B
γ iiz I
a I= −
+
2
1
A ek T
e Nk T
T K
A= =
= ×
12,303
2
8
1.000
( ) (L mol )
2 2
–3/2 –1 1/2 3/2
επ
ε
ε1 825 106,
log γ i iA z I= − 2
I C zi i= ∑1
22
Capítulo 1: Introducción al equilibrio químico 21
Esta última ecuación se conoce como latercera aproximación de Debye-Hückel, quepermite predecir el comportamiento de los elec-trolitos a concentraciones superiores a 1,0 molL–1; no obstante, no es de aplicación general yaque los valores de a y C son empíricos y por tan-to se deben determinar para cada sistema enparticular.
Para el caso de que el medio no sea acuoso,se debe tener en cuenta la densidad del disol-vente, de manera que para obtener el valor delcoeficiente de actividad se calculan los coefi-cientes de Debye-Hückel de la ecuación [1.17] apartir de las expresiones:
[1.21]
[1.22]
siendo εs y ρs la constante dieléctrica y la densi-dad del disolvente en el que tiene lugar el equi-librio considerado, respectivamente, y εw y ρw losvalores correspondientes para el agua. La ecua-ción [1.22] sólo es aplicable a 25 ºC.
Las ecuaciones [1.21] y [1.22] permiten, deacuerdo con la IUPAC, el cálculo de los coefi-cientes de actividad de las especies iónicas enmedios hidroorgánicos, como las mezclas meta-nol-agua, acetonitrilo-agua, etc., y en medios noacuosos, como metanol, tetrahidrofurano, etc.
1.2.2. Variación de los coeficientes de actividadcon la fuerza iónica del medio
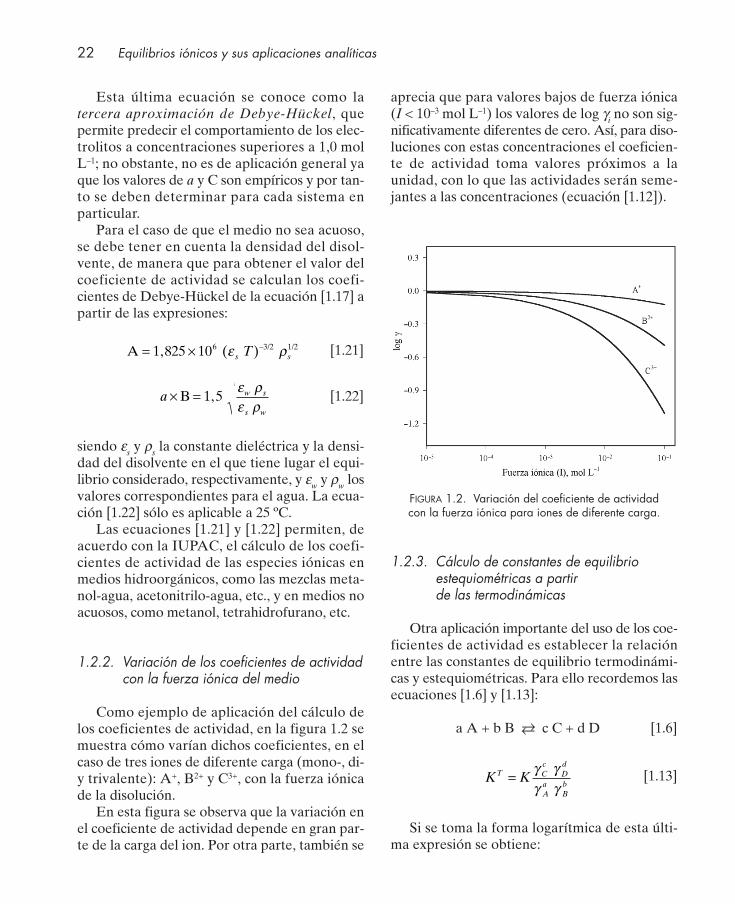
Como ejemplo de aplicación del cálculo delos coeficientes de actividad, en la figura 1.2 semuestra cómo varían dichos coeficientes, en elcaso de tres iones de diferente carga (mono-, di-y trivalente): A+, B2+ y C3+, con la fuerza iónicade la disolución.
En esta figura se observa que la variación enel coeficiente de actividad depende en gran par-te de la carga del ion. Por otra parte, también se
aprecia que para valores bajos de fuerza iónica(I < 10–3 mol L–1) los valores de log γi no son sig-nificativamente diferentes de cero. Así, para diso-luciones con estas concentraciones el coeficien-te de actividad toma valores próximos a launidad, con lo que las actividades serán seme-jantes a las concentraciones (ecuación [1.12]).
FIGURA 1.2. Variación del coeficiente de actividad con la fuerza iónica para iones de diferente carga.
1.2.3. Cálculo de constantes de equilibrio estequiométricas a partirde las termodinámicas
Otra aplicación importante del uso de los coe-ficientes de actividad es establecer la relaciónentre las constantes de equilibrio termodinámi-cas y estequiométricas. Para ello recordemos lasecuaciones [1.6] y [1.13]:
a A + b B →← c C + d D [1.6]
[1.13]
Si se toma la forma logarítmica de esta últi-ma expresión se obtiene:
K KT C
cDd
Aa
Bb
= γ γγ γ
a w s
s w
× =B 1,5
ε ρε ρ
A 1,825 10 ( ) 6 –3/2 1/2= × ε ρs s T
22 Equilibrios iónicos y sus aplicaciones analíticas

[1.23]
y si además se tiene en cuenta la ley de Debye-Hückel ampliada y simplificada, dada por laecuación [1.19], que es una aproximación gene-ral válida para calcular el coeficiente de activi-dad de una disolución con valores de fuerza ióni-ca inferiores o iguales a 0,05 – 0,1 mol L–1, seobtiene la siguiente expresión:
[1.24]
que, reordenando términos, queda simplificada a:
[1.25]
Esta ecuación proporciona, finalmente, larelación entre la constante de equilibrio termo-dinámica y estequiométrica para una reacciónquímica determinada, una vez se aplica la leyampliada simplificada de Debye-Hückel. En estascondiciones, hay que destacar que la variaciónde K es solamente función de la fuerza iónica delmedio, de los coeficientes estequiométricos delas distintas especies en equilibrio y de sus car-gas elevadas al cuadrado.
La expresión [1.25] puede escribirse de for-ma más general como:
[1.26]
donde el término ∆(vi zi2 ) indica el incremento
entre los valores obtenidos al sumar los pro-ductos de los coeficientes estequiométricos (vi)
de cada especie multiplicados por el cuadradode su carga para los productos de la reacciónrespecto al mismo valor obtenido para los reac-tivos:
[1.27]
Como ejemplo práctico de este tratamien-to, se estudia la variación de la constante dedisociación del ácido acético en función de lafuerza iónica. La reacción de disociación de esteácido en medio acuoso viene dada por la expre-sión:
CH3COOH →←→← H+ + CH3COO–(KT
a = 10 –4,75)[1.28]
Conocida la constante termodinámica de equi-librio (a 25 ºC), se puede calcular la constanteestequiométrica para diferentes valores de fuer-za iónica haciendo uso de la ecuación [1.26], conlo que se tiene:
[1.29]
dado que en este caso ∆(vi zi2 ) = (1 × 12 + 1 × 12
– 1 × 02) = 2. Al aplicar la ecuación [1.29] a estesistema para diferentes valores de fuerza iónicase calculan los valores de Ka que se representanen la figura 1.3, en la que se observa que paravalores bajos de fuerza iónica las diferencias entrela constante estequiométrica y la termodinámi-ca son poco significativas.
Este hecho permite suponer que el compor-tamiento de los electrolitos en disolución muydiluida (y en condiciones de baja fuerza iónica)es similar al comportamiento ideal. Esto implicaque el valor de los coeficientes de actividad esprácticamente la unidad para las diferentes espe-cies en el sistema (es decir, la actividad de unaespecie se asimila al valor de su concentración
log log
A
K KI
Ia aT= +
+×
12
∆(
) )
ν
ν ν
i i
i i i i
z
z z
2
2 2
)
( (productos reactivos
=
= −∑ ∑
log log –
A ) K K
II
zTi i=
+12∆(ν
log log –
–A
( –
K K
II
c z d z a z b z
T
C D A B
=
++ −
12 2 2 2 )
log log –A
–A
A A
K K cz I
Id
z II
az I
Ib
z II
T C D
A B
=+ +
+
++
++
2 2
2 2
1 1
1 1
log log log
+ log – log – log A
K K c
d a b
TC
D B
= + +γ
γ γ γ
Capítulo 1: Introducción al equilibrio químico 23

molar) y que se puede tomar el valor de la cons-tante de equilibrio termodinámica como equi-valente al de la constante de equilibrio este-quiométrica.
Esta aproximación se realizará de aquí enadelante en este libro (salvo en casos en que seindique lo contrario), lo que supone considerarque las condiciones experimentales permitentrabajar directamente con las constantes deequilibrio termodinámicas. Por ello, no se harádistinción entre KT y K.
FIGURA 1.3. Variación de la constante de disociacióndel ácido acético con la fuerza iónica.
1.3. Tipos de equilibrioen Química Analítica
En Química Analítica se pueden distinguirbásicamente dos tipos de equilibrio, homogé-neos y heterogéneos, regidos por su correspon-diente constante de equilibrio. Los primeros tie-nen lugar en una única fase, generalmente líqui-da, mientras que en los segundos intervienen doso más fases, bien porque en el curso del procesose genere una segunda fase, como ocurre en lasreacciones de precipitación, bien porque ambasfases sean diferentes, como es el caso de la extrac-ción líquido-líquido.
1.3.1. Equilibrios homogéneos
Entre los equilibrios homogéneos pueden dis-tinguirse los siguientes tipos:
A) Equilibrio ácido-base
Son aquellos en los que tiene lugar un inter-cambio de protones (teoría de Bronsted) o unintercambio de pares de electrones (teoría deLewis). El equilibrio ácido-base está reguladopor la constante de acidez (Ka) o constante dedisociación. Así, para la reacción de disociaciónde un ácido:
HA + H2O →← A– + H3O+ [1.30]
la constante de acidez se expresa como:
[1.31]
También se puede considerar la constante debasicidad (Kb), que es la que corresponde a laprotonación de una base:
B + H2O →← HB+ + OH– [1.32]
[1.33]
En este contexto se puede también considerarel equilibrio ácido-base correspondiente a la auto-protolisis del disolvente, ya que ciertos disolven-tes pueden actuar como ácido o como base, sien-do el caso más característico el agua. La constantede este equilibrio se denomina constante de auto-protolisis y, en el caso particular del agua, recibeel nombre de producto iónico del agua (Kw):
H2O + H2O →← H3O+ + OH– [1.34]
[1.35] Kw = + −[H O ][OH ]3
Kb =
+ −[HB ][OH ][B]
Ka =
− +[A ][H O ][HA]
3
24 Equilibrios iónicos y sus aplicaciones analíticas

B) Equilibrio de complejación
En sentido amplio, la complejación consisteen la asociación de dos especies que pueden exis-tir aisladamente. Esta definición es tan ampliaque incluye a la mayor parte de las reaccionesanalíticas. Por ejemplo, en este sentido, las reac-ciones:
CH3COO– + H+ →← CH3COOH [1.36]
Ag+ + Cl– →← AgCl(S) [1.37]
pueden ser consideradas reacciones de com-plejación, aunque de hecho se trate de reaccio-nes ácido-base y de precipitación, respectiva-mente.
En un sentido más estricto, se define comocomplejación el proceso en el que se produce latransferencia de uno o más pares de electronesdesde una especie dadora (átomo cargado nega-tivamente o molécula), llamada ligando, hacia unaespecie aceptora. Aun así, el equilibrio ácido-basepodría ser considerado como un caso particulardel equilibrio de complejación, ya que, según lateoría de Lewis, los ligandos serían bases y lasespecies aceptoras serían ácidos. Para evitar estacircunstancia, por lo general se acepta que la com-plejación se limita al caso de la formación de losllamados complejos de coordinación, en los quela especie que acepta pares de electrones es union metálico (complejos metálicos).
La reacción de complejación entre un ionmetálico (M) y n moléculas de un ligando (L) sepuede enfocar desde dos perspectivas comple-mentarias:
1. Considerando el equilibrio global de for-mación del complejo, esto es, la formaciónde un único complejo de máxima este-quiometría:
Mn+ + n L– →← MLn [1.38]
este equilibrio viene regido por su cons-tante de formación global, (βn):
[1.39]
2. A partir de la formación escalonada de losdiferentes complejos, que para n = 2 daríalugar a los siguientes equilibrios:
M2+ + L– →← ML+ [1.40]
ML+ + L– →← ML2 [1.41]
siendo sus constantes respectivas:
[1.42]
[1.43]
las constantes K1 y K2 se denominan cons-tantes de formación sucesivas, y se intuyeclaramente que el producto de las mismascorresponde a la constante de formaciónglobal.
C) Equilibrio de oxidación-reducción
En un equilibrio de oxidación-reducción (deno-minado también equilibrio redox) se produce unintercambio de electrones. Se conoce como oxi-dación el proceso en que una especie pierde elec-trones y como reducción el proceso en que losgana. Una reacción de oxidación-reducción pue-de, por lo tanto, ser considerada la combinaciónde dos semirreacciones, o pares redox, una en laque un compuesto, denominado el oxidante (Ox1),se reduce, y otra en la que un segundo compues-to, denominado el reductor (Red2), se oxida:
Ox1 + n1 e →← Red1
Red2→← Ox2 + n2 e [1.44]
n2 Ox1 + n1 Red2→← n2 Red1 + n1 Ox2
K2
MLML L
= + −
[ ][ ][ ]
2
K1
MLM L
=+
+ −
[ ][ ][ ]2
βn
nn n
= + −
[ ][ ][ ]
MLM L
Capítulo 1: Introducción al equilibrio químico 25

La constante de equilibrio de la reaccióndepende de las respectivas capacidades oxi-dante y reductora de los compuestos implica-dos en la misma, caracterizadas por el llamadopotencial estándar o normal (E0). Como semuestra en el capítulo que trata sobre equili-brios de oxidación-reducción, dicha constanteresponde a la expresión:
[1.45]
en la que E10 y E2
0 son los potenciales normales delos sistemas redox implicados en el equilibrio.
1.3.2. Equilibrios heterogéneos
Los equilibrios heterogéneos, por su parte,son aquellos en que se ven implicadas dos o másfases. Los casos más habituales en QuímicaAnalítica son los de precipitación, consistentesen la aparición de una fase sólida, y los de dis-tribución, en los que se produce transferenciade una especie en una fase líquida a otra faseque puede ser líquida (extracción líquido-líqui-do) o sólida (extracción líquido-sólido e inter-cambio iónico).
A) Equilibrio de precipitación
La precipitación, o bien el proceso inverso dedisolución de un sólido iónico, puede represen-tarse mediante la reacción:
n Mm+ + m Nn– →← MnNm (S) [1.46]
La constante que regula este equilibrio sedenomina constante del producto de solublidado, más sencillamente, producto de solubilidad yse representa por Ks:
[1.47]
B) Extracción líquido-líquido
La distribución líquido-líquido, también cono-cida como extracción líquido-líquido, es un pro-ceso en el que un soluto se transfiere desde unafase líquida (generalmente acuosa) hacia otra faselíquida inmiscible (orgánica). Este proceso de trans-ferencia se evalúa mediante la constante de distri-bución (KD) que se define como la relación entrela concentración de una especie en una de las fasesdividida por su concentración en la otra fase. Sicomo se ha indicado, una de las fases es un disol-vente orgánico (org) y la otra agua (aq), por con-venio la concentración en la fase orgánica figuraen el numerador. Así, para un proceso del tipo:
Aaq→← Aorg [1.48]
puede definirse la constante de distribución como:
[1.49]
C) Extracción líquido-sólido
La extracción líquido-sólido conocida comoextracción en fase sólida, se basa en la distribuciónde un soluto desde una fase líquida a una fase sóli-da (sólido sorbente), la cual presenta una elevadaárea superficial para favorecer el proceso de trans-ferencia de materia. Este equilibrio de distribución:
Aaq→← As [1.50]
donde s indica fase sólida, se suele regir por laecuación de Freundlich, similar a la isoterma deadsorción de Freundlich deducida para la adsor-ción de gases sobre sólidos:
[1.51]
donde K es la constante de equilibrio, n un pará-metro de valor constante, q la cantidad de masade soluto adsorbida por unidad de masa de adsor-bente y C la concentración de soluto en disolu-ción acuosa.
q KC n=
K
A
AD =[ ]
[ ]org
aq
Ksm n n m= + −[ ] [ ]M N
K K
E En nlog
,= −1
020
1 20 059
26 Equilibrios iónicos y sus aplicaciones analíticas

D) Intercambio iónico
El proceso de intercambio iónico consiste en ladistribución de un soluto desde una fase líquida auna fase sólida constituida por una resina cambia-dora de iones, que son sustancias de naturalezaorgánica de alto peso molecular (R) que contie-nen un gran número de iones positivos o negati-vos de pequeño volumen fácilmente intercambia-bles. Así, para el equilibrio de intercambio:
RM + N+ →← RN + M+ [1.52]
la constante de equilibrio viene expresada por:
[1.53]
donde las concentraciones en resina se expresanen miliequivalentes de ion intercambiado por gra-mo de resina. Esta constante de equilibrio se sue-le denominar coeficiente de selectividad y se repre-senta como se indica en la ecuación [1.53] por KM
N.
1.3.3. Uso analítico del equilibrio químico
Los equilibrios homogéneos y heterogéneosdescritos en las secciones precedentes constituyenel fundamento de diferentes métodos analíticosque permiten la determinación tanto cuantitativacomo cualitativa de una gran variedad de especies.El estudio en profundidad de los equilibrios homo-géneos ácido-base, complejación y redox, así comodel equilibrio heterogéneo de precipitación, cons-tituye un primer paso fundamental para poderabordar con las suficientes garantías el estudio desus aplicaciones analíticas cuantitativas, esto es, lascorrespondientes técnicas volumétricas y gravi-métricas, estas últimas en el caso concreto de losequilibrios heterogéneos de precipitación.
En esta obra se presenta el estudio de los dife-rentes equilibrios y sus correspondientes apli-caciones en el siguiente orden: ácido-base, for-mación de complejos, precipitación y oxidación-
reducción. Esta ordenación conceptual, aunquerompe el esquema de división de los equilibriosentre homogéneos y heterogéneos, se ha escogi-do porque en los tres primeros tipos de equilibriolas diferentes especies involucradas no presentancambio en su estado de oxidación. Además, enestos tres equilibrios se origina la asociación dedos especies, que pueden existir aisladamente, paraformar una nueva o bien la disociación de ésta enaquéllas; este comportamiento responde al con-cepto de complejación en su sentido más ampliodado en el apartado 1.3.1.
Los equilibrios heterogéneos de distribuciónindicados en el apartado 1.3.2, tales como extrac-ción líquido-líquido, extracción líquido-sólido eintercambio iónico, constituyen el fundamentode diferentes técnicas de separación y, por tan-to, son de gran interés en procesos de precon-centración de diferentes especies o bien para laeliminación de interferencias (clean-up) con elobjetivo de separar al analito(s) de los compo-nentes de una muestra. Un tratamiento en pro-fundidad de estos equilibrios y, en consecuencia,de las técnicas analíticas derivadas está fuera delobjetivo de esta obra, ya que el alumno aborda-rá el estudio de estos equilibrios y sus aplicacio-nes analíticas en curso posterior.
1.4. Estudio sistemáticodel equilibrio químico
Para abordar el estudio de los diferentes tiposde equilibrio químico es conveniente desarrollaruna sistemática que permita el cálculo de las con-centraciones de las diferentes especies presentesen él. Este proceso de cálculo se basa en conse-guir un número de ecuaciones igual al númerode especies que se desea cuantificar, o sea alnúmero de incógnitas que se plantean. Estasincógnitas son las concentraciones en equilibriode todas las especies presentes en la disolucióny su número variará en función del compuesto ocompuestos presentes. La información que sedebe considerar, para obtener el número de ecua-ciones que relacionen satisfactoriamente las espe-
KMN =
+ +
+ +
[ ][ ]
[ ][ ]
N M
M Nres aq
res aq
Capítulo 1: Introducción al equilibrio químico 27

cies a cuantificar y calcular su concentración, seconcreta en los siguientes puntos:
1. Descripción de todas las reacciones que tie-nen lugar en la disolución considerada acom-pañadas de sus respectivas constantes deequilibrio. Así, por ejemplo, en el caso deequilibrios ácido-base, es necesario consi-derar los equilibrios de disociación de áci-dos y bases, autoprotolisis del disolvente, etc.
2. Considerar los balances que puedan estable-cerse entre las especies a cuantificar, talescomo el balance de masas y el balance de car-gas. En el caso concreto de los equilibrios áci-do-base es interesante considerar, además delos balances indicados, el balance protónico.
3. Una vez obtenida esta información se pue-de resolver directamente el sistema de ecua-ciones planteado para conseguir las con-centraciones de todas las especies implicadasen el(los) equilibrio(s) objeto de estudio, obien llevar a cabo una serie de aproxima-ciones previas con vistas a abordar su reso-lución de manera más simple e inmediata.
1.4.1. Balance de masas y de cargas
El balance de masas surge del hecho de quela materia que interviene en una reacción quí-mica permanece constante al tener lugar dichareacción. Por tanto, la concentración analítica deuna sustancia determinada será igual a la sumade las concentraciones de las especies en las quepuede encontrarse en disolución, en virtud de losequilibrios que tengan lugar.
Se distingue, pues, entre concentración analíti-ca y concentración de equilibrio, de manera que laconcentración analítica (C), expresada en moles desustancia por litro de disolución, es independientede las condiciones experimentales en las que sedesarrolla el equilibrio químico. La concentraciónde equilibrio, expresada también en mol L–1, serefiere a las diferentes especies en que una sus-tancia puede existir en disolución merced a undeterminado equilibrio químico. Esta concentra-
ción se indica entre corchetes y depende de las con-diciones que regulan dicho equilibrio. Así, en elcaso de los equilibrios ácido-base la concentraciónanalítica es independiente del pH mientras que lasconcentraciones de equilibrio dependen de él,excepto en el caso de ácidos y bases fuertes.
Como ejemplo de la aplicación de este balan-ce de masas, vamos a considerar una disoluciónde ácido acético preparada a partir de la adiciónde 0,1 moles de este ácido en un litro de agua. Eneste caso, la concentración analítica es 0,1 mol L–1
y está relacionada con las concentraciones de equi-librio de las diferentes formas que pueden existiren disolución para este ácido: ácido acético e ionacetato, mediante el balance de masas. Así:
Concentración analítica:
CCH3COOH= 0,1 mol L–1 [1.54]
Equilibrio ácido-base:
CH3COOH + H2O →←→← CH3COO– + H3O
+ [1.55]
Balance de masas:
CCH3COOH = 0,1 mol L–1 == [CH3COOH] + [CH3COO–] [1.56]
Es evidente que al cambiar el pH del medio(condición experimental) se modificarán las con-centraciones de ácido acético y de ion acetatoconforme al equilibrio de la expresión [1.55],aunque su suma, balance de masas, seguirá sien-do en todos los casos constante e igual a la con-centración analítica de ácido acético: 0,1 mol L–1.En el cuadro 1.1 se muestran otros ejemplos deaplicación de este balance de masas a equilibriosácido-base y de complejación.
El balance de cargas se fundamenta en que unadisolución debe cumplir la condición de electro-neutralidad, esto es, la carga total de iones posi-tivos tiene que ser igual a la carga total de losiones negativos. Así pues, la suma de las concen-
28 Equilibrios iónicos y sus aplicaciones analíticas

traciones de todas las especies cargadas positiva-mente multiplicadas por su carga, ha de ser iguala la suma de las concentraciones de las especiescargadas negativamente multiplicadas por su car-ga. Hay que indicar que para establecer estebalance siempre se ha de considerar el equilibriode autoprotolisis del disolvente además del pro-pio equilibrio o equilibrios objeto de estudio.
Al aplicar este balance de cargas al ejemploanterior se deben considerar los siguientes equi-librios:
Equilibrio ácido-base:
CH3COOH + H2O →←→← CH3COO– + H3O
+ [1.55]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [1.57]
y por tanto el balance de cargas viene dado por:Balance de cargas:
[H3O+] = [CH3COO–] + [OH–] [1.58]
Otros ejemplos de la aplicación de este balan-ce de cargas a equilibrios ácido-base y de com-plejación se muestran en el cuadro 1.2.
1.4.2. Balance protónico
Este balance, sólo aplicable a equilibrios áci-do-base, es consecuencia directa de la teoría pro-tónica en la que se considera que los protonescedidos deben ser exactamente los mismos quelos protones aceptados. De acuerdo con ello,debe cumplirse que la suma de las concentra-ciones de las especies que son resultado dehaber ganado protones multiplicada por elnúmero de protones ganados, ha de ser igual ala suma de las concentraciones de las especiesresultantes de haber perdido protones multipli-cada por el número de protones perdidos. Debi-do a su propia definición, hay que destacar queen el balance protónico no aparecerán nunca lasespecies de partida de los equilibrios implica-dos, pero sí aquellas especies que resulten a par-tir de ellas por haber ganado o perdido proto-nes. Así pues, para expresar el balance protónico
Capítulo 1: Introducción al equilibrio químico 29
CUADRO 1.1Ejemplos de aplicación del balance de masas en equilibrios iónicos
Sustancia Equilibrios Balance de masas
Ácido fuerte: HCl HCl + H2O → Cl– + H3O+ CHCl = [Cl–]
Ácido débil: CH3COOH CH3COOH + H2O →← CH3COO– + H3O+ CCH3COOH = [CH3COO–] + [CH3COOH]
Ácido diprótico: H2CO3 H2CO3 + H2O →← HCO3̄ + H3O+ CH2CO3
= [CO32¯] + [HCO3̄ ] +[ H2CO3]
HCO3̄ + H2O →← CO32¯ + H3O
+
Base débil: NH3 NH3 + H2O →← NH4+ +OH¯ CNH3
= [NH3] + [NH4+]
Sal: NH4CH3COO NH4CH3COO → NH4+ + CH3COO¯ CNH4
+ = [NH3] + [NH4+]
NH4+ + H2O →← NH3 + H3O
+ CCH3COOH¯= [CH3COO¯] + [CH3COOH]
CH3COO¯ + H2O →← CH3COOH + OH¯ CSal = [NH3] + [NH4+] = [CH3COO¯] + [CH3COOH]
Complejo ML2: Ag(NH3)3+ Ag+ + NH3
→← AgNH3+ CAg = [Ag+] + [AgNH3
+ ] + [Ag(NH3)2+]
AgNH3+ + NH3
→← Ag(NH3)2+ CNH3
= [NH3] + [AgNH3+ ] + 2 [Ag(NH3)2
+] + [NH4+]
NH4+ + H2O →← NH3 + H3O
+

se deben considerar únicamente las especies pro-ducto de los equilibrios implicados y no las espe-cies iniciales.
El balance protónico puede sustituir con éxi-to al balance de cargas para el cálculo de las con-centraciones de las especies implicadas, en lamayoría de los casos que se pueden plantear y esel resultado de combinar adecuadamente elbalance de masas y el balance de cargas. En elejemplo que nos ocupa, las especies producto delos equilibrios implicados conforme a las ecua-ciones [1.55] y [1.57] son los iones: hidronio(H3O
+), hidroxilo (OH– ) y acetato (CH3COO–).De acuerdo con la definición del balance protó-nico, éste viene dado por:
Balance protónico:
[H3O+] = [CH3COO–] + [OH–] [1.59]
y como se aprecia es idéntico al balance de cargasindicado en la ecuación [1.58]. En el cuadro 1.3 semuestran otros ejemplos de la aplicación de estebalance a diferentes equilibrios ácido-base.
Hay que indicar que el balance protónico y elbalance de cargas pueden coincidir (como en elejemplo considerado) o no en algunos casos dedisoluciones simples (comparar los balances mos-trados en los cuadros 1.2 y 1.3). Para ácidos pro-tonados tipo HB+ o bases neutras B no coinciden.
1.4.3. Simplificaciones y errores
La utilización de la sistemática propuesta conla aplicación de los balances indicados permiteel cálculo de todas las concentraciones de lasespecies implicadas en los equilibrios, aunque enciertos casos se obtienen expresiones matemáti-
30 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 1.2Ejemplos de aplicación del balance de cargas en equilibrios iónicos
Sustancia Equilibrios Balance de cargas
Ácido fuerte: HCl HCl + H2O → Cl– + H3O+ [H3O
+] = [OH¯] + [Cl¯]
H2O + H2O →← H3O+ + OH¯
Ácido débil: CH3COOH CH3COOH + H2O →← CH3COO– + H3O+ [H3O
+] = [OH¯] + [CH3COO–]
H2O + H2O →← H3O+ + OH¯
Ácido diprótico: H2CO3 H2CO3 + H2O →← HCO3̄ + H3O+ [H3O
+] = [OH¯] + 2[CO32¯] + [HCO3̄ ]
HCO3̄ + H2O →← CO32¯ + H3O
+
H2O + H2O →← H3O+ + OH¯
Base débil: NH3 NH3 + H2O →← NH4+ +OH¯ [H3O
+] + [NH4+] = [OH¯]
H2O + H2O →← H3O+ + OH¯
Sal: NH4CH3COO NH4CH3COO → NH4+ + CH3COO¯ [H3O
+] + [NH4+] = [OH¯] + [CH3COO¯]
NH4+ + H2O →← NH3 + H3O
+
CH3COO¯ + H2O →← CH3COOH + OH¯
H2O + H2O →← H3O+ + OH¯
Complejo ML2: Ag+ + NH3→← AgNH3
+ [H3O+] + [Ag+] + [AgNH3
+] + [Ag(NH3)2+] =
[Ag(NH3)2] NO3 AgNH3+ + NH3
→← Ag(NH3)2+ = [OH¯] + [NO3̄ ]
NH4+ + H2O →← NH3 + H3O
+
H2O + H2O →← H3O+ + OH¯

cas cuya resolución presenta un cierto grado dedificultad. En estas circunstancias, la aplicaciónde simplificaciones adecuadas y asumiendo siem-pre un error controlado asociado a la misma, pue-de facilitar considerablemente la resoluciónnumérica del problema.
Así, por ejemplo, en el caso concreto de losequilibrios ácido-base, para resolver el problemaque se plantea con más frecuencia, como es elcálculo del pH de una disolución, la aplicación dela sistemática propuesta conduce a expresionesmatemáticas que pueden ser simplificadas, en basea criterios químicos, despreciando unas concen-traciones frente a otras o frente a valores de cons-tantes, de manera que el grado de complicaciónmatemática a resolver sea como máximo unaecuación de segundo grado.
Al llevar a cabo las simplificaciones comenta-das, se suele tomar el criterio de que el error per-mitido sea inferior al 5%. Ello implica que una con-centración (B) de especie o valor de constante deequilibrio, será despreciable frente a otra concen-
tración (A) o constante de equilibrio, si el valor deesta última es como mínimo 20 veces mayor quela concentración o constante a despreciar. Esto es:
A + B ≈ A Si B ≤ 5100
A ⇒⇒ A ≥ 20 B
[1.60]
En términos logarítmicos una diferencia de20 veces implica que existe una diferencia de 1,3unidades logarítmicas entre las cantidades quese comparan. Así, B será despreciable frente aA, admitiendo un error del 5%, si se cumple
log A ≥ log 20 + log B ⇒⇒ log A ≥ 1,3 + log B [1.61]
y de igual forma, log B será despreciable frentea log A, si éste es al menos 1,3 unidades mayor alog B. En estas ecuaciones, A y B son dos mag-nitudes numéricas que pueden corresponder aconcentraciones de especies, a constantes o a susproductos.
Capítulo 1: Introducción al equilibrio químico 31
CUADRO 1.3Ejemplos de aplicación del balance protónico en equilibrios ácido-base
Sustancia Equilibrios Balance protónico
Ácido fuerte: HCl HCl + H2O → Cl¯ + H3O+ [H3O
+] = [OH¯] + [Cl¯]
H2O + H2O →← H3O+ + OH¯
Ácido débil: CH3COOH CH3COOH + H2O →← CH3COO– + H3O+ [H3O
+] = [OH¯] + [CH3COO¯]
H2O + H2O →← H3O+ + OH¯
Ácido diprótico: H2CO3 H2CO3 + H2O →← HCO3̄ + H3O+ [H3O
+] = [OH¯] + 2[CO32¯] + [HCO3̄ ]
HCO3̄ + H2O →← CO32¯ + H3O+
H2O + H2O →← H3O+ + OH¯
Base débil: NH3 NH3 + H2O →← NH4+ +OH¯ [H3O
+] + [NH4+] = [OH¯]
H2O + H2O →← H3O+ + OH¯
Sal: NH4CH3COO NH4CH3COO → NH4+ + CH3COO¯ [H3O
+] + [CH3COOH] = [NH4+] + [OH¯]
NH4+ + H2O →← NH3 + H3O
+
CH3COO¯ + H2O →← CH3COOH + OH¯
H2O + H2O →← H3O+ + OH¯

32 Equilibrios iónicos y sus aplicaciones analíticas
1.1. ¿Cuántas regiones se pueden diferenciar en una reacción química y cómo se denominan?
1.2. Definir velocidad de reacción.
1.3. ¿Qué parámetros conforman una ecuación de velocidad?
1.4. ¿Cuándo se alcanza el equilibrio químico desde un punto de vista cinético?
1.5. Diferenciar entre concentración efectiva y concentración analítica.
1.6. ¿En qué condiciones se puede trabajar con constantes de equilibrio estequiométricas?
1.7. ¿Qué es la fuerza iónica?
1.8. ¿En qué condiciones experimentales utilizaría la ley límite de Debye-Hückel para el cálculo de los coeficien-tes de actividad?
1.9. Explicar si son verdaderas o falsas las siguientes afirmaciones. En disoluciones diluidas:
a) los coeficientes de actividad son siempre inferiores a la unidad,b) los coeficientes de actividad son siempre distintos de la unidad.
1.10. ¿Qué es un complejo de coordinación?
1.11. Clasificar los siguientes equilibrios marcando con una X:
Equilibrio Homogéneo Heterogéneo
Extracción líquido-líquido
Formación de complejos
Intercambio iónico
Autoprotolisis
Precipitación
1.12. ¿Qué característica técnica es fundamental para favorecer el proceso de transferencia de materia en unequilibrio de distribución líquido-sólido?
1.13. ¿Qué diferencias y analogías se pueden establecer entre el balance de cargas y el balance protónico?
1.14. Diferenciar los términos de concentración analítica y concentración de equilibrio.
1.15. Establecer los balances de masas y cargas de una disolución 0,01 mol L–1 de carbonato de potasio.
1.16. Establecer los balances de cargas y de masas al disolver 0,1 moles de hidróxido de bario en un litro de aguadestilada.
Cuestiones

1.17. Establecer los balances de cargas y protónico al disolver 0,5 moles de carbonato de amonio en 500 mL deagua destilada.
1.18. Establecer los balances de cargas y protónico de una disolución 0,1 mol L–1 de sulfato de sodio a pH 5,0(Ka2 H2SO4 = 10–1,9).
Capítulo 1: Introducción al equilibrio químico 33
Seminarios: problemas numéricos
• Objetivo pedagógico:Introducción al empleo de los conceptos de fuerzaiónica, coeficientes de actividad y su incidencia en elcálculo de constantes estequiométricas.
Fuerza iónica
1. Calcular la fuerza iónica que proporciona una diso-lución 0,1 mol L–1 de cloruro de sodio.
De acuerdo con la ecuación [1.14] y considerandola concentración y carga de las especies en las que sedisocia este electrolito fuerte (disociación total) setiene:
NaCl → Na+ + Cl– ⇒ [Na+] = 0,1 mol L–1
[Cl–] = 0,1 mol L–1
2. Calcular la fuerza iónica de una disolución 0,1 molL–1 de cloruro de bario.
De forma similar al problema anterior se puedeestablecer:
BaCl2 → Ba2+ + 2Cl– ⇒ [Ba2+] = 0,1 mol L–1
[Cl–] = 0,2 mol L–1
3. Calcular la fuerza iónica de una disolución 0,3 molL–1 de cloruro de sodio.
Siguiendo el procedimiento indicado en problemasanteriores se tiene:
NaCl → Na+ + Cl– ⇒ [Na+] = 0,3 mol L–1
[Cl–] = 0,3 mol L–1
A la vista de este resultado y comparando los encon-trados en los problemas anteriores, se observa, con-forme a la ecuación [1.14], cómo un incremento en lacarga de los iones que constituyen el electrolito inci-de más acusadamente sobre la fuerza iónica que ésteproporciona, que un aumento de su concentración.
4. Calcular la fuerza iónica que proporcionan las si-guientes disoluciones: a) sulfato de cobre 0,01 mol L–1;b) fosfato trisódico 0,05 mol L–1; c) carbonato de pota-sio 0,1 mol L–1; y d) una mezcla que es 0,05 mol L–1 encloruro de potasio y 0,1 mol L–1 en sulfato de sodio.Sol.: a) 0,04; b) 0,3; c) 0,3; d) 0,35.
Coeficientes de actividad
5. Calcular el coeficiente de actividad del ion Ba2+ enuna disolución 0,05 mol L–1 de nitrato de bario.
Dado el valor de la concentración de electrolito(0,05 mol L–1) y conforme a lo expuesto en el aparta-do 1.2.1 se ha de utilizar la ley ampliada simplificadade Debye-Hückel para el cálculo del coeficiente deactividad del ion Ba2+, esto es la ecuación [1.19], cono-cida previamente la fuerza iónica de la disolución. Así:
Ba(NO3)2 → Ba2+ + 2 NO3̄ ⇒ [Ba2+] = 0,05 mol L–1
[NO3̄] = 0,1 mol L–1
I C zi i= = × + × =∑1
212
0 3 1 0 3 1 0 32 2 2 1( , , ) , mol L–
I C zi i= = × + × =∑12
12
0 1 2 0 2 1 0 32 2 2 1( , , ) , mol L–
I C z z zi i= = × + ×( ) =
= × + × =
− +− +∑1
212
12
0 1 1 0 1 1 0 1
2 2 2
2 2 1
[ ] [ ]
( , , ) ,
Cl Na
mol L
Cl Na
–

de donde el coeficiente de actividad γBa2+ = 0,269,
lo que pone de manifiesto que el ion bario en esta diso-lución presenta un comportamiento alejado del ideal.
6. Calcular el coeficiente de actividad del ion Fe3+ enuna disolución 0,01 mol L–1 de cloruro de sodio.
Para la resolución de este problema, se puede seguirun razonamiento similar al expuesto en el anterioraunque en este caso haciendo uso de la ley límite deDebye-Hückel, ecuación [1.15], dada la concentra-ción de electrolito. Así:
NaCl → Na+ + Cl– ⇒ [Na+] = 0,01 mol L–1
[Cl–] = 0,01 mol L–1
7. Calcular el coeficiente de actividad de los siguientesiones: a) Ca2+ en una disolución 0,03 mol L–1 en nitra-to de sodio; b) Cl– en una disolución 0,1 mol L–1 desulfato de sodio; y c) Ni2+ en una disolución 0,05 molL–1 de cloruro de bario.Sol.: a) 0,50; b) 0,66; c) 0,27.
Constantes estequiométricas
8. Calcular la constante de disociación del ácido acéticoen una disolución de cloruro de sodio 0,1 mol L–1.CH3COOH (Ka
T = 10–4,75).
En este problema se aborda la variación que experi-menta la constante de disociación estequiométrica deun ácido débil, como el acético, al cambiar la fuerzaiónica del medio. Para este ácido, la relación entre las
constantes termodinámica y estequiométrica en fun-ción de la fuerza iónica viene dada por la ecuación [1.29]:
Al sustituir el valor de
en la expresión anterior se tiene:
Del valor encontrado para log Ka se deduce unincremento de la constante estequiométrica con rela-ción a la termodinámica (10–4,5 > 10–4,75), lo que impli-ca un aumento del carácter ácido del acético. Asípues, al adicionar un electrolito a una disolución deun ácido (o base) débil se incrementa su carácter áci-do (o básico), siendo esta dependencia directamen-te proporcional a la fuerza iónica que dicho electro-lito suministra a la disolución.
9. Se mezclan 25 mL de una disolución de amoniaco0,12 mol L–1 con 50 mL de nitrato de potasio 0,3 molL–1. Calcular la constante de basicidad del amoniacoen estas condiciones. NH3 (Kb
T= 10–4,75).Sol.: Kb = 10–4,43
10. Calcular el aumento del grado de disociación de un áci-do débil HA (Ka
T = 10–4) cuando una disolución 0,1 molL–1 del mismo se hace 0,2 mol L–1 en cloruro de potasio.
De acuerdo con el equilibrio de disociación de esteácido débil, HA + H2O →← A– + H3O
+ , cuya concen-tración analítica es CHA y suponiendo que su grado dedisociación viene dado por α ([A–] = [H3O
+] = CHA
α) su constante de equilibrio en función de este gra-do de disociación se puede expresar por:
Haciendo uso de esta ecuación y sustituyendo enella los valores de Ka
T y Ka, se puede determinar fácil-mente la variación del grado de disociación debido
K
C CC
C= = × ×−
= ×−
− +[A ][H O ][HA]
3 HA HA
HA
HA( )( )( )
α αα
αα1 1
2
log log
, ,,
,,Ka = ++
× = −−100 512 0 11 0 1
2 4 504 75
I C zi i= = × + × =∑1
212
0 1 1 0 1 1 0 12 2 2 1( , , ) , mol L–
log log
,K K
IIa a
T= ++
×0 5121
2
log , , , ,γ γFe Fe3 30 512 3 0 01 0 46 0 352
+ += − × = − ⇒ =
I C zi i= = × + × =∑12
12
0 01 1 0 01 1 0 012 2 2 1( , , ) , mol L–
log,
, ,,
,
γBa2
0 512 21
0 512 4 0 151 0 15
0 57
2
+ = − ×+
=
= − × ×+
= −
II
I C zi i= = × + × =∑12
12
0 05 2 0 1 1 0 152 2 2 1( , , ) , mol L–
34 Equilibrios iónicos y sus aplicaciones analíticas

a la fuerza iónica que proporciona el electrolito.Haciendo uso de la expresión [1.29] se puede cal-cular el valor de Ka que resulta ser de 10–3,68, y portanto:
de donde α1 = 0,001 o bien 0,1%
de donde α2 = 0,002 o bien 0,2%
Se origina por tanto un incremento del 100% delgrado de disociación de este ácido débil debido a lapresencia del electrolito.
11. ¿Cuántos gramos de cloruro de sodio hay que añadira 500 mL de una disolución 0,01 mol L–1 de ácido acé-tico para disminuir su pKa en 0,1 unidades?CH3COOH (Ka
T = 10–4,75).Peso molecular: NaCl = 58,5.
La disminución del pKa responde a la presenciadel electrolito. Si éste es 4,75 conforme al valor deKa
T, el estequiométrico será: 4,75 – 0,1 = 4,65 y portantoKa= 10–4,65. Mediante la ecuación [1.29] se pue-de calcular el valor de la fuerza iónica en estas con-diciones:
de donde I = 0,012 mol L–1. Considerando la expre-sión de la fuerza iónica para este electrolito se obser-va que ésta se corresponde con su concentración,por lo que para conseguir una concentración deNaCl de 0,012 mol L–1 en 500 mL, se han de añadir0,351 g de cloruro de sodio a la disolución de ácidoacético.
12. Calcular el producto de solubilidad del hidróxido dealuminio en un medio que contiene como electrolitonitrato de sodio 0,15 mol L–1, cuyo producto de solu-bilidad termodinámico es 2 × 10–32.
El equilibrio heterogéneo de precipitación delhidróxido de aluminio viene regido por una constanteque se denomina producto de solubilidad conformea la ecuación [1.47]:
Al3+ + 3OH– →← Al(OH)3 (S)
KST = [Al3+] [OH–]3
Para calcular el valor de esta constante a una deter-minada fuerza iónica se puede hacer uso de la ecua-ción general [1.26] considerando que Ks correspon-de al equilibrio de disociación del precipitado y que,por tanto, los iones constituyen el producto de la reac-ción. En estas condiciones, la ecuación [1.26] se pue-de escribir como:
dado que al tratarse de un preci-pitado.
Al sustituir los valores de
y de
en la ecuación anterior se tiene:
de donde Ks = 10–30. Se origina un aumento del pro-ducto de solubilidad y por tanto el hidróxido de alu-minio es más soluble en estas condiciones.
13. Calcular el producto de solubilidad de los siguientesprecipitados en las condiciones experimentales quese indican: a) sulfato de bario (KS
T = 1,3 × 10–10) encloruro de potasio 0,1 mol L–1; b) cloruro de plata(KS
T = 1,8 × 10–10) en nitrato de bario 0,02 mol L–1; yc) yoduro de plomo (KS
T = 7,1 × 10–9) en percloratode sodio 0,05 mol L–1.Sol.: a) 1,2 × 10–9; b) 2,9 × 10–10; c) 2,6 × 10–8
log log( )
, ,,
Ks = × ++
× = −−2 100 512 0 151 0 15
12 3032
( )ν i iz2 2 21 3 3 1 12productos = × + × =∑
I C zi i= = × + × =∑1
212
0 15 1 0 15 1 0 152 2 2( , , ) , mol L–1
( )ν i iz2 0reactivos =∑
log log ( )K K
A II
zs sT
i i= ++ ∑1
2ν productos
log log
,, ,10 100 5121
24 65 4 75− −= ++
×II
100 11
3 68 2
2
− = ×−
. , αα
100 11
4 1
1
− = ×−
, αα
Capítulo 1: Introducción al equilibrio químico 35


2
2.1. Introducción2.2. Teoría protónica de Bronsted y Lowry2.3. Fuerza de ácidos y bases2.4. Equilibrios ácido-base de protolitos
fuertes. Cálculo del pH2.5. Equilibrios ácido-base
de protolitos débiles monopróticos2.6. Equilibrios ácido-base
de protolitos débiles polipróticos2.7. Disoluciones reguladorasAnexo. Expresiones simplificadas para
el cálculo del pH de disoluciones de diferentes protolitos
CuestionesSeminarios: problemas numéricos
EQUILIBRIOSÁCIDO-BASE

2.1. Introducción
El concepto de ácido y de base, uno de los másantiguos dentro de la Química, ha experimenta-do una importante evolución a lo largo del tiem-po auspiciada por el propio acontecer del de-sarrollo científico. Inicialmente, la palabra ácidose utilizó para caracterizar el sabor agrio de cier-tas sustancias como por ejemplo el vinagre y fueen el siglo XVII cuando el científico británicoRobert Boyle estableció su definición conside-rando el poder disolvente de estas sustancias y sucapacidad para colorear de rojo el tornasol (tinterosa que se obtiene de determinados líquenes).Por el contrario, las bases presentan unas carac-terísticas opuestas, tales como sabor amargo, colo-rean el tornasol de azul y tienen tacto jabonoso.En el siglo XVIII, Antoine Laurent de Lavoisier,químico francés considerado el fundador de la quí-mica moderna, fue el primero que estableció unarelación entre la composición química de una sus-tancia y su carácter ácido. En su obra Considera-ciones sobre la naturaleza de los ácidos en 1778asigna el carácter ácido a la presencia de oxígenoen una sustancia. Posteriormente, otro químicofrancés, Claude Louis Berthollet descubrió quealgunos ácidos, como por ejemplo HCN y H2S, nocontenían oxígeno en contra de la opinión deLavoisier. Pronto se descubrió que el elementocomún a todos los ácidos era el hidrógeno y fue elquímico alemán Justus Liebig en 1838 el que defi-nió a los ácidos como compuestos hidrogenadosen los que el hidrógeno podía reemplazarse pormetales.
Estas teorías no explicaban aún de manerasatisfactoria la diferencia de fuerza existente entre
los diversos ácidos. Los conocimientos moder-nos de los ácidos y las bases parten de 1834, cuan-do el físico inglés Michael Faraday descubrió queácidos, bases y sales eran electrólitos por lo que,disueltos en agua se disocian en partículas concarga o iones que pueden conducir la corrienteeléctrica. En 1884, el químico sueco Svante Arrhe-nius, y más tarde el químico alemán Wilhelm Ost-wald, definieron a los ácidos como sustancias queen agua proporcionan iones hidrógeno y comobases aquellas sustancias que en agua dan ioneshidroxilo. Esta teoría, permite explicar de mane-ra satisfactoria la mayor parte de los fenómenosderivados de los equilibrios ácido-base en diso-luciones acuosas, sin embargo, persisten ciertoshechos que contradicen la teoría de Arrhenius oteoría clásica.
Una teoría más satisfactoria es la que formu-laron en 1923 el químico danés Johannes Brons-ted y, paralelamente, el químico británico Tho-mas Lowry que permite el tratamiento de losequilibrios ácido-base no sólo en agua sino tam-bién en otros medios, denominados medios noacuosos. Según estos autores, en toda reacciónácido-base se produce un intercambio de proto-nes, de ahí que a esta teoría se le llame, también,teoría protónica. Las especies que participan enestos procesos, dadores y aceptores de protones,se conocen con el nombre genérico de ácidos ybases.
Una teoría más general, es la teoría ácido-baseelectrónica, desarrollada por el químico esta-dounidense Gilbert N. Lewis, según la cual en lasinteracciones ácido-base, ácidos son las sustan-cias capaces de aceptar electrones de las bases,las que se caracterizan por poseer pares de elec-
38 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Introducir al alumno en las diferentes teorías ácido-base mostrando el papel decisivo del disolvente enel desarrollo y uso de las mismas.
• Descripción de una metodología sistemática para elcálculo del pH de diferentes disoluciones de protolitos
y de las concentraciones de las especies existentes enestos equilibrios.
• Estudio de las características de las disoluciones regu-ladoras de diferentes protolitos con especial énfasisen las formas de preparación de las mismas.

trones libres. Aunque esta teoría electrónica tie-ne posibles aplicaciones más amplias, la teoríade Bronsted y Lowry resulta más adecuada parael tratamiento de los equilibrios ácido-base enQuímica Analítica.
2.2. Teoría protónica de Bronstedy Lowry
Según la teoría protónica enunciada por Brons-ted y Lowry, ácido es toda sustancia capaz de cederprotones y base toda sustancia capaz de aceptar-los. En toda interacción ácido-base al haber inter-cambio de protones debe existir una especie quelos ceda y, simultáneamente, otra que los acepte.Este proceso se puede representar por la ecuación:
ácido1 + base2→← base1 + ácido2 [2.1]
El resultado es una reacción ácido-base, en laque el ácido1 cede un protón a la base2 y estassustancias a su vez se transforman en la base1 yen el ácido2 respectivamente. El ácido1 forma conla base1 un par ácido-base o sistema conjugado.Todas las especies químicas que participan en elequilibrio anterior, moléculas o iones, se deno-minan protolitos, o sea los ácidos y las bases sonprotolitos. De las consideraciones anteriores sededuce que un ácido poseerá un protón más quesu base conjugada, independientemente de sucarga, por lo que un ácido podrá ser una molé-cula neutra, un catión o un anión. Así pues, enlos siguientes pares ácido-base:
ácido →← base + H+
CH3COOH →← CH3COO– + H+
NH4+ →← NH3 + H+
H3PO4→← H2PO4̄ + H+ [2.2]
H2CO3→← HCO3̄ + H+
HCO3̄ →← CO32¯ + H+
las especies CH3COOH, NH4+ y HCO3̄ se com-
portan como ácidos y concretamente como áci-dos monopróticos ya que sólo pueden ceder unprotón, mientras que las especies CH3COO–,
NH3, H2PO4̄ y HCO3̄ son bases monopróticas alpoder aceptar sólo un protón. Igualmente, se pue-de considerar a la especie H2CO3 como un ácidodiprótico al poder ceder dos protones, a la espe-cie CO3
2¯ como una base diprótica y a la especieH3PO4 como un ácido triprótico.
Como puede observarse en los dos últimosejemplos de pares ácido-base considerados en[2.2], la especie HCO3̄ puede actuar como baseo como ácido. Este tipo de moléculas o iones reci-be el nombre de anfolitos y decimos que tienenun comportamiento anfiprótico.
2.2.1. Influencia del disolvente
El concepto ácido-base es relativo, ya que elgrado en que transcurre la reacción ácido-base[2.1] depende de las tendencias relativas de losdos ácidos para ceder protones o de las dos basespara aceptarlos. Así, por ejemplo, el ácido acé-tico se comporta como ácido frente al agua:
CH3COOH + H2O →← CH3COO– + H3O+
[2.3]
y sin embargo, presenta carácter básico frente alácido perclórico:
HClO4 + CH3COOH →← ClO4̄ + CH3COOH2+
[2.4]
En consecuencia, la pregunta inmediata quese genera es: ¿a qué sustancias le damos el nom-bre de ácido y cuáles serán bases? Para resolvereste problema, y dado el carácter relativo del con-cepto ácido-base, se ha de tomar una referencia:el disolvente de trabajo, generalmente el agua,que es el más importante y más frecuentementeutilizado. De manera que, al trabajar en medioacuoso, se denomina ácido aquella sustancia quelo es frente al agua:
CH3COOH + H2O →← CH3COO– + H3O+
H2PO4̄ + H2O →← HPO42¯ + H3O
+ [2.5]NH4
+ + H2O →← NH3 + H3O+
Capítulo 2: Equilibrios ácido-base 39

y base a la que se comporte como tal frente a estedisolvente:
NH3 + H2O →← NH4+ + OH–
[2.6]HCO3̄ + H2O →← CO3
2¯ + OH–
Así pues, el comportamiento ácido-base deun determinado compuesto viene dado por laespecie con la que interacciona. Si, como general-mente ocurre, se trabaja con disoluciones, el com-portamiento de una sustancia dependerá en granparte del disolvente en que está disuelta.
Como se observa en las reacciones [2.5] y [2.6],el agua es un disolvente anfiprótico, es decir, capazde aceptar o ceder protones, dando a su vez ioneshidronio (H3O
+) en [2.5] e iones hidroxilo (OH–)en [2.6]. Otros disolventes anfipróticos muy co-munes son el metanol y etanol, de característicasácido-base semejantes al agua y de ahí que se lesdenomine disolventes anfipróticos neutros. Enestos disolventes se tienen equilibrios ácido-basesemejantes a los que se presentan en agua:
HNO2 + CH3CH2OH →← CH3CH2OH2+ + NO2̄
NH3 + CH3OH →← NH4+ + CH3O
– [2.7]
Los disolventes anfipróticos pueden presentartendencia a ceder protones y en tal caso reciben elnombre de disolventes protógenos, como el ácidoacético glacial, o tener propiedades básicas y deno-minarse disolventes protófilos como el amoniacoo las aminas. Los primeros exaltan las propieda-des básicas de los compuestos y los segundos laspropiedades ácidas. En el cuadro 2.1 se muestra,a modo de ejemplo, el distinto carácter ácido-baseque presenta la urea en diversos disolventes.
Este diferente comportamiento ácido-basepuede explicarse de forma cualitativa conside-rando que al pasar de un disolvente a otro cam-bian las condiciones del medio que determinanla existencia de las diferentes especies en diso-lución, como la polaridad, constante dieléctrica,capacidad dadora o aceptora de formación depuentes de hidrógeno, etc., lo que favorece o difi-culta los procesos de transferencia de protones.
2.2.2. Autoprotolisis y escala de pHde un disolvente
Los disolventes anfipróticos, al poder actuarcomo ácido y como base, experimentan autoio-nización o autoprotolisis, que no es más que otroejemplo de comportamiento ácido-base, comoilustran las reacciones siguientes, todas ellas reac-ciones de autoprotolisis:
H2O + H2O →← H3O+ +OH– [2.8]
CH3OH + CH3OH →←→← CH3OH2
+ + CH3O– [2.9]
CH3CH2OH + CH3CH2OH →←→← CH3CH2OH2
+ + CH3CH2O– [2.10]
CH3COOH + CH3COOH →←→← CH3COOH2
+ + CH3COO– [2.11]
NH3 + NH3→← NH4
+ + NH2̄ [2.12]
El ion hidronio, H3O+, formado en la auto-
protolisis del agua, consta de un protón unido a
40 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 2.1Diferente carácter ácido-base de la urea según el disolvente utilizado. Urea: H2N-CO-NH2 (BH)
Carácter ácido-base Disolvente Reacción ácido-base Par ácido-base
Base muy débil Agua BH + H2O →← BH2+ + OH– BH2
+ / BHBase débil Acético glacial BH + CH3COOH →← BH2
+ + CH3COO– BH2+ / BH
Acido débil Amoniaco líquido BH + NH3 →← B– + NH4
+ BH / B–

una molécula de agua. También existen hidratossuperiores, como H5O2
+ y H9O4+, pero son menos
estables que H3O+ en varios órdenes de magni-
tud. En disoluciones acuosas sólo existen prácti-camente protones hidratados debido a la grandensidad de carga del H+. Por ello, muchas vecesse utiliza la notación H3O
+ para representar alprotón en medio acuoso, aunque también se uti-liza la notación simplificada H+. En los capítulosde esta obra correspondientes a equilibrios yvolumetrías ácido-base se utilizará la notaciónH3O
+, mientras que en los restantes por simpli-cidad se hará uso de la notación H+.
La constante de equilibrio de la reacción deautoprotolisis del agua, ecuación número [2.8],se expresa según la relación:
[2.13]
y al considerar que la actividad de una sustanciapura es la unidad, se obtiene:
[2.14]
El valor de esta constante de autoprotolisisdel agua a 25 ºC es de Kw = 10–14, valor muy bajoque permite sustituir las actividades por concen-traciones:
[2.15]
De acuerdo con el equilibrio [2.8], en aguapura las concentraciones de los iones hidronio ehidroxilo son iguales: [H3O
+ ] = [OH–] y al susti-tuir en la ecuación [2.15] se tiene:
[2.16]
es decir en agua pura, medio que se consideraneutro, la concentración de ambos iones es 10–7
mol L–1.
Como se deriva de la propia definición de áci-do, la concentración de iones hidronio determi-na la acidez de la disolución. De la expresión[2.15] se deduce que si aumenta la concentraciónde ion hidronio en agua, la concentración de ionhidroxilo disminuirá. Se dice que la disolución esácida, si la concentración de iones hidronio essuperior a 10–7 mol L–1 y por tanto la concentra-ción de iones hidroxilo inferior a 10–7 mol L–1. Alcontrario, para una disolución básica [OH–] > 10–7
mol L–1 y [H3O+] < 10–7 mol L–1. A fin de simpli-
ficar los números a manejar para expresar la aci-dez o basicidad, Sorensen propuso indicar la aci-dez de una disolución por medio de la variablepH, definida como el logaritmo cambiado de sig-no de la actividad de los iones hidronio:
[2.17]
En disoluciones diluidas, con las que nor-malmente se trabaja, se puede sustituir la acti-vidad de iones hidronio por su concentración,de manera que:
[2.18]
El símbolo p es un operador matemático apli-cado en química a diferentes magnitudes, que indi-ca aplicar el logaritmo y multiplicar por –1. El pHserá pues una medida de la acidez o basicidad deuna disolución, de manera que a pH inferior a 7las disoluciones serán ácidas, con pH = 7 son neu-tras y a pH superior a 7 serán básicas. Con vistasa comparar la acidez o basicidad de diferentesdisoluciones en función del pH hay que tener encuenta que no existe proporcionalidad directaentre los valores de pH y la concentración de ioneshidronio, como se muestra en el recuadro 2.1.
Teniendo en cuenta la expresión [2.15],correspondiente a la constante de autoprotólisisdel agua, al aplicar el operador p, se tiene quepKw = 14. El logaritmo cambiado de signo de la constante de autoprotolisis de un disolvente(pKap), indica el intervalo de pH de trabajo del
pH [H O ]3= − +log
pHH O3
= − +loga
[H O ] [OH ]3+ − −= = =Kw 10 7
Kw = =+ − −[H O ][OH ] 10314
K a aw = ×+ −H O OH3
K
a a
aw =
×+ −H O OH
H O2
3
2
Capítulo 2: Equilibrios ácido-base 41

mismo. Así, en agua el intervalo de pH es de 0 a14. De igual forma, para el metanol con unaconstante de autoprotolisis, Kap = 10–16,9 o sea conun pKap = 16,9, el intervalo de pH es de 0 a 16,9.En el cuadro 2.2 se muestran los valores de pKappara los disolventes más significativos. Se obser-va una gran diversidad de valores conforme alcarácter de estos disolventes, desde el tetrahi-drofurano cuyo pKap es de 34,7 con un ampliointervalo de pH (0 – 34,7), al ácido fórmico conun pKap = 6,2, cuyo intervalo de pH de 0 a 6,2, esmuy reducido.
2.3. Fuerza de ácidos y bases
La tendencia de un ácido a ceder protones yde una base a aceptarlos es una medida de sufuerza. Ahora bien, tal como ya se ha comenta-do, el concepto ácido-base es relativo y la fuer-za de un ácido o una base no puede ser determi-nada por sí sola, sino que se ha de comparar conotra sustancia. El compuesto de referencia, porantonomasia, es el agua, ya que es en este medio
donde se trabaja en la inmensa mayoría de lasocasiones. Así, se consideran ácidos fuertes enagua, aquellos protolitos que, a concentracionesmoderadas, se encuentran totalmente disociadosen medio acuoso. Igualmente, se define comobase fuerte aquel protolito que, a concentracio-nes no elevadas, experimenta una protolisis total.
Los demás ácidos o bases son débiles y reac-cionan de forma incompleta con el agua, tal comose representa por el equilibrio siguiente:
ácido + H2O →← base + H3O+ [2.19]
El valor de la constante de equilibrio de estareacción, llamada constante de disociación o deacidez, es una medida cuantitativa de la fuerzadel ácido en disolución acuosa:
[2.20]
que para disoluciones diluidas se puede escribircomo:
K
a a
aa =× +base H O
ácido
3
42 Equilibrios iónicos y sus aplicaciones analíticas
Se exponen diversos ejemplos de cómo varía el pH de una disolución en función de diversos incrementos en laconcentración de iones hidronio y viceversa.
1. Al duplicar la concentración de iones hidronio de una disolución los valores de pH disminuyen conforme a:
[H3O+], mol L–1 1,0 × 10–5 2,0 × 10–5 4,0 × 10–5 8,0 × 10–5 1,6 × 10–4
pH 5,0 4,7 4,4 4,1 3,8
En resumen, cada vez que se duplica la concentración de iones hidronio el pH disminuye 0,3 unidades.
2. Al incrementar el pH de una disolución en una unidad se originan diferentes incrementos de [H3O+] cuyo
valor depende del pH inicial de dicha disolución:
pHinicial pHfinal ∆pH [H3O+]inicial [H3O
+]final ∆ [H3O+]
1,0 2,0 1,0 10–1 mol L–1 10–2 mol L–1 9 × 10–2 mol L–1
4,0 5,0 1,0 10–4 mol L–1 10–5 mol L–1 9 × 10–5 mol L–1
7,0 8,0 1,0 10–7 mol L–1 10–8 mol L–1 9 × 10–8 mol L–1
Se observa cómo incrementos iguales de pH comportan diferentes incrementos de [H3O+].
RECUADRO 2.1

[2.21]
En el caso de que Ka sea superior a la unidad,el equilibrio está desplazado totalmente hacia laderecha y se habla de ácidos fuertes, como es elcaso de los ácidos HClO4, HNO3 y HCl. Las basesconjugadas de estos ácidos serán muy débiles yde hecho los iones ClO4̄, NO3̄ y Cl– son bases tandébiles que no reaccionan con el agua.
Otros ácidos como por ejemplo CH3COOH,H2PO4̄ y NH4
+, presentan valores pequeños deKa, lo que implica que el equilibrio [2.19] estádesplazado hacia la izquierda.
H2PO4̄ + H2O →← HPO42¯ + H3O
+ [2.5]
[2.22]
La sustancia será tanto más ácida cuantomayor sea su constante de acidez, Ka, o lo que eslo mismo, menor sea su pKa. Así, si los pKa delos ácidos CH3COOH, H2PO4̄ y NH4
+ son res-pectivamente 4,75, 7,21 y 9,25, éste será tambiénel orden de su fuerza ácida. En el cuadro 2.3 semuestran los valores de pKa de diferentes ácidos.
De manera similar para una base y en diso-lución diluida, se puede escribir:
base + H2O →← ácido + OH– [2.23]
cuya constante de equilibrio o constante de basi-cidad viene dada por:
[2.24]
Es importante señalar que existe una serieinterminable de ácidos y bases para los que 0 <pK < 14, denominados ácidos y bases débiles.
Kb =−[ ]
[ ]ácido [OH ]
base
Ka = =
− +
−−[HPO ][H O ]
[H PO ]42
3
2 4
,10 7 21
Ka =
+[ ][[ ]
base H O ]ácido
3
Capítulo 2: Equilibrios ácido-base 43
CUADRO 2.2Valores de pKap de los disolventes más significativos
ANFIPRÓTICOS
Neutros Protógenos Protófilos
Disolvente pKap Disolvente pKap Disolvente pKap
Agua 14,2 Ácido fórmico 6,2 Formamida 16,8Etilenglicol 15,8 Ácido acético 13,9Metanol 16,5Etanol 18,7n-Propanol 19,2Isopropanol 22,0n-Butanol 21,6t-Butanol 28,5
APRÓTIDOS DIPOLARES
Protófilos Protófobos
Disolvente pKap Disolvente pKap
N,N-Dimetilformamida 27,0 Acetonitrilo 33,6Tetrahidrofurano 34,7 Nitrobenceno 26,5
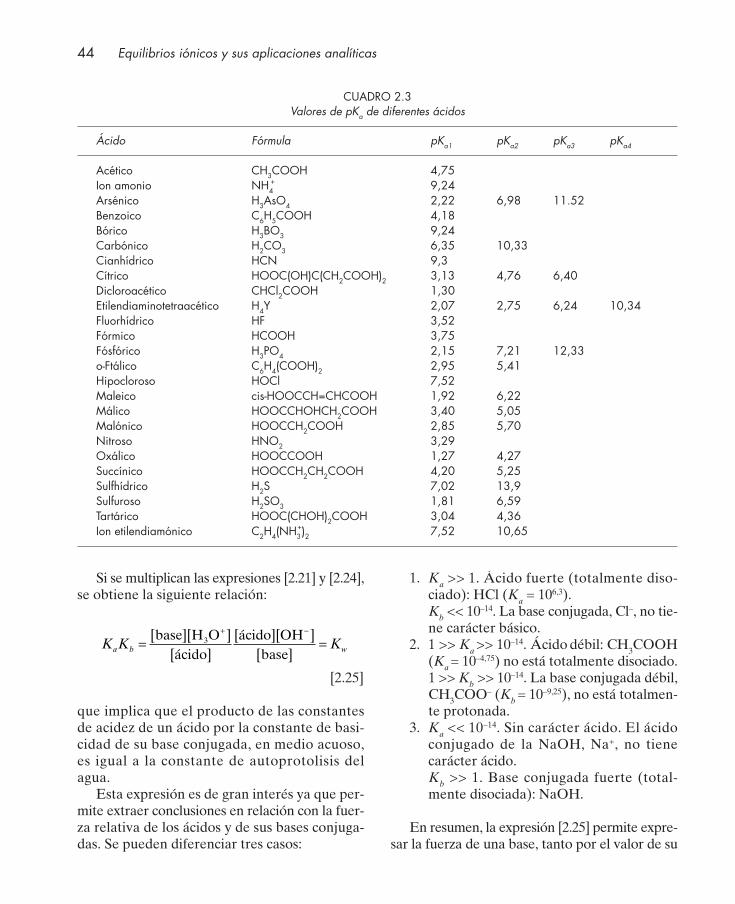
Si se multiplican las expresiones [2.21] y [2.24],se obtiene la siguiente relación:
[2.25]
que implica que el producto de las constantesde acidez de un ácido por la constante de basi-cidad de su base conjugada, en medio acuoso,es igual a la constante de autoprotolisis delagua.
Esta expresión es de gran interés ya que per-mite extraer conclusiones en relación con la fuer-za relativa de los ácidos y de sus bases conjuga-das. Se pueden diferenciar tres casos:
1. Ka >> 1. Ácido fuerte (totalmente diso-ciado): HCl (Ka = 106,3).Kb << 10–14. La base conjugada, Cl–, no tie-ne carácter básico.
2. 1 >> Ka >> 10–14. Ácido débil: CH3COOH(Ka = 10–4,75) no está totalmente disociado.1 >> Kb >> 10–14. La base conjugada débil,CH3COO– (Kb = 10–9,25), no está totalmen-te protonada.
3. Ka << 10–14. Sin carácter ácido. El ácidoconjugado de la NaOH, Na+, no tienecarácter ácido.Kb >> 1. Base conjugada fuerte (total-mente disociada): NaOH.
En resumen, la expresión [2.25] permite expre-sar la fuerza de una base, tanto por el valor de su
K K Ka b w= =
+ −[ ][[ ]
[ ][ ][ ]
base H O ]ácido
ácido OHbase
3
44 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 2.3Valores de pKa de diferentes ácidos
Ácido Fórmula pKa1 pKa2 pKa3 pKa4
Acético CH3COOH 4,75Ion amonio NH4
+ 9,24Arsénico H3AsO4 2,22 6,98 11.52Benzoico C6H5COOH 4,18Bórico H3BO3 9,24Carbónico H2CO3 6,35 10,33Cianhídrico HCN 9,3Cítrico HOOC(OH)C(CH2COOH)2 3,13 4,76 6,40Dicloroacético CHCl2COOH 1,30Etilendiaminotetraacético H4Y 2,07 2,75 6,24 10,34Fluorhídrico HF 3,52Fórmico HCOOH 3,75Fósfórico H3PO4 2,15 7,21 12,33o-Ftálico C6H4(COOH)2 2,95 5,41Hipocloroso HOCl 7,52Maleico cis-HOOCCH=CHCOOH 1,92 6,22Málico HOOCCHOHCH2COOH 3,40 5,05Malónico HOOCCH2COOH 2,85 5,70Nitroso HNO2 3,29Oxálico HOOCCOOH 1,27 4,27Succínico HOOCCH2CH2COOH 4,20 5,25Sulfhídrico H2S 7,02 13,9Sulfuroso H2SO3 1,81 6,59Tartárico HOOC(CHOH)2COOH 3,04 4,36Ion etilendiamónico C2H4(NH3
+)2 7,52 10,65

constante de basicidad, Kb, como por el valor dela Ka de su ácido conjugado, ya que Ka × Kb = 10–14.
2.3.1. Efecto nivelador del disolvente
La tendencia de un disolvente a aceptar o aceder protones, determina la fuerza del ácido ode la base disueltos en él. Por ejemplo, los áci-dos HClO4, HNO3 y HCl son ácidos fuertes enagua, lo que implica que al disolver una mismacantidad (expresada en moles) de estos ácidosen este disolvente se obtendrán disoluciones quecontengan la misma cantidad de H3O
+, ademásde los aniones correspondientes. Esto nos indi-ca que estos ácidos se comportan como si tuvie-sen la misma fuerza y se dice entonces que eldisolvente, agua, ejerce un efecto nivelador sobrela fuerza de los mismos.
Sin embargo, si en lugar de agua, se utiliza áci-do acético anhidro como disolvente, medio quees aceptor de protones más débil que el agua,ninguno de los ácidos HClO4, HNO3 o HCl sedisocia por completo, teniendo lugar en cambioequilibrios como:
HClO4 + CH3COOH →← ClO4̄ + CH3COOH2+
[2.4]
HCl + CH3COOH →← Cl– + CH3COOH2+
[2.26]
De estos dos equilibrios, el primero está másdesplazado hacia la derecha, con una constantede 10–4,7, que se corresponde con el pKa delHClO4 en ácido acético anhidro. El HClO4 esunas 5.000 veces más fuerte que el HCl en áci-do acético anhidro. Se dice, así, que el ácido acé-tico se comporta como un disolvente diferencia-dor frente a estos dos ácidos, por cuanto en estemedio se ponen de manifiesto sus diferencias deacidez. El agua en cambio es un disolvente nive-lador de los ácidos HClO4, HNO3 y HCl ya quetodos ellos se disocian completamente en ella y por tanto no presentan diferencias de fuerzaácida.
De manera similar, compuestos sólidos comoNaOH, KOH y LiOH, se disuelven y disociantotalmente en agua, dando OH–, que es la basefuerte más importante en agua. Sin embargo, lamayoría de bases son débiles en medio acuoso,entre ellas las aminas. Por ejemplo, la n-butila-mina (pKa = 10,61) y la dietilamina (pKa = 6,56)son bases débiles en agua, sin embargo en medioácido acético anhidro se comportan como basesfuertes. Se dice, entonces, que estas dos basesestán niveladas en medio acético, disolvente nive-lador de bases y que se pueden diferenciar porsu distinta fuerza en medio acuoso.
2.3.2. Interacción con el disolvente
Según el tratamiento clásico de Arrhenius, lainteracción entre las moléculas de agua y losiones de una sal se denomina hidrólisis. Así, enel caso del acetato de sodio, los iones Na+ noreaccionan con el agua, es decir no sufren hidró-lisis, por provenir de una base fuerte, pero sí inte-raccionan con el agua los iones acetato, que pro-ceden de un ácido débil:
CH3COO– + H2O →← CH3COOH + OH– [2.5]
En esta reacción se originan iones OH– y ladisolución será por tanto básica. El equilibrio dehidrólisis se describe cuantitativamente median-te la correspondiente constante de equilibrio,que recibe el nombre de constante de hidrólisis(Kh) y que para disoluciones diluidas toma la forma:
[2.27]
Según la teoría protónica, la hidrólisis es tansolo un caso particular de intercambio de proto-nes, en el que intervienen el agua y un protolitocargado. Así, el equilibrio [2.5] viene tambiéncuantificado por la constante de basicidad del
Kh =
−
−
[CH COOH][OH ][CH COO ]
3
3
Capítulo 2: Equilibrios ácido-base 45

acetato, Kb (ver ecuación [2.24]), que será idén-tica a Kh (ecuación [2.27]).
Si en la expresión [2.27] se sustituye [OH–] =Kw /[H3O
+] (ecuación [2.15]) se obtiene:
[2.28]
y por lo tanto:
Kw = Ka Kh [2.29]
o bien
Kw = Ka Kb [2.25]
De este tratamiento se deduce que tanto laKh como Kb se pueden calcular a partir de Ka, obien Ka puede ser obtenida si se conoce Kb o loque es lo mismo, Kh.
De esta breve comparación entre la teoría clá-sica y la protónica, puede deducirse que estaúltima da una explicación más sencilla y genera-lizada de los diferentes tipos de reacciones áci-do-base, uniéndolas bajo el nombre común deprotolisis.
2.4. Equilibrios ácido-basede protolitos fuertes.Cálculo del pH
Los equilibrios ácido-base de protolitos fuer-tes (ácidos y bases fuertes) están totalmente des-plazados hacia las especies disociadas, tal comose ha comentado anteriormente. Así, para repre-sentar estos equilibrios se utilizará una sola fle-cha indicando que éste se encuentra totalmen-te desplazado hacia la derecha. En este estudiose desarrollará la sistemática a seguir para deter-minar la concentración de todas las especiesimplicadas en cada uno de estos equilibrios, fun-damentalmente el cálculo del pH de sus disolu-ciones, así como de posibles mezclas de estosprotolitos.
2.4.1. Ácido fuerte
Para estudiar cuantitativamente el caso de ladisociación de un ácido fuerte y determinar laconcentración de las especies presentes en estesistema, se procede de acuerdo con la sistemáti-ca general descrita en el capítulo 1:
1. Se describen las reacciones implicadas enel sistema.
2. Se consideran las expresiones de las cons-tantes de las mismas.
3. Se aplican los balances ya comentados:balance de masas y de cargas o protónico.
Si se toma como ejemplo de ácido fuerte elHCl se tiene:
Estudio sistemático del equilibrio ácido-basede un ácido fuerte
Reacciones y constantes implicadas:
Reacción ácido-base:
HCl + H2O → Cl– + H3O+ [2.30]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Kw = [H3O+ ] [OH–] =10–14 [2.15]
Balance de masas:
CHCl = [HCl] + [Cl–] ≈ [Cl–] [2.31]
La concentración de la forma molecular [HCl] podrá des-preciarse por tratarse de un ácido fuerte y por tanto estartotalmente disociado.
Balance de cargas y balance protónico:
[H3O+] = [Cl–] + [OH–] [2.32]
(El balance de cargas coincide exactamente con elbalance protónico.)
K K
K KKh b
w w
a
= = =− +
[CH COOH][CH COO ][H O ]
3
3 3
46 Equilibrios iónicos y sus aplicaciones analíticas

Dado que se trata de un ácido fuerte, fijada suconcentración analítica (CHCl), las incógnitas seránlas concentraciones de las especies presentes enel sistema, es decir [H3O
+], [OH–] y [Cl–]. Se tie-nen pues, tres incógnitas y tres expresiones quelas relacionan: Kw [2.15], el balance de masas [2.31]y el balance de cargas [2.32], por tanto se puederesolver el problema de forma cuantitativa.
Así, considerando el balance de cargas yteniendo en cuenta que [Cl–] = CHCl, al sustituiresta condición en la expresión del balance demasas se tiene:
[H3O+] = CHCl + [OH–] [2.33]
Se deben considerar ahora las posibles sim-plificaciones de la ecuación obtenida. En estaecuación, se puede despreciar la concentraciónde iones hidroxilo frente a la concentración ana-lítica del ácido, simplemente considerando quetenemos una disolución de ácido y por tanto la[OH–] será muy baja. De esta forma, si [OH–] <<CHCl la expresión [2.33] se transforma en:
[2.34]
Conocidas [H3O+] y [Cl–], la incógnita que
queda por resolver, [OH–], se obtiene utili-zando la tercera expresión de que disponemos:constante de autoprotolisis (ecuación [2.15]), demanera que:
[2.35]
Siempre que se introduzca una simplificaciónse tendrá que comprobar que ésta es apropiada,antes de aceptar el valor numérico obtenidocomo correcto. En este caso, la simplificaciónintroducida [OH–] << CHCl será correcta siempreque la concentración analítica del ácido no seaextremadamente baja. Baste considerar la clási-ca pregunta: ¿qué pH tiene una disolución deácido fuerte de CHCl = 10–8 mol L–1? Si se haceuso de la expresión [2.34] se obtendría un valor
de pH de 8,0, lo cual es una incongruencia dadoque la disolución de un ácido no puede presen-tar bajo ningún concepto un pH básico. En estassituaciones, la disolución de ácido fuerte es muydiluida, la simplificación considerada en la ecua-ción [2.33] no es válida y hay que hacer uso deotra expresión para calcular el pH de la disolu-ción del ácido fuerte como se muestra a conti-nuación.
Si la disolución de ácido fuerte es muy dilui-da, con una concentración analítica igual o infe-rior a 10–6,5 mol L–1, la concentración de ioneshidroxilo, procedentes de la autoprotolisis delagua, ya no será despreciable frente a esta con-centración en la expresión [2.33], dado que alaplicar la ecuación [2.35] se tiene que [OH–] =10–7,5 mol L–1. En estas condiciones se ha de con-siderar la expresión de la constante de autopro-tolisis del agua, ecuación [2.15], y sustituir [OH–]procedente de la misma en la expresión [2.33],con lo que se obtiene:
[2.36]
de donde:
[H3O+]2 – CHA [H3O
+] – Kw = 0 [2.37]
y despejando [H3O+]:
[2.38]
Esta expresión es la adecuada para el cálcu-lo de [H3O
+] sea cual sea la concentración analí-tica del ácido fuerte.
A pesar del carácter general de la ecuación[2.38], hay que tener en cuenta que los ácidos quegeneralmente se emplean en el laboratorio ana-lítico no se encuentran normalmente a concen-traciones inferiores a 10–3 mol L–1, y por tanto laexpresión de mayor importancia práctica para elcálculo del pH de disoluciones de ácidos fuerteses la ecuación [2.34]:
[H O ]3
HA HA+ =+ + ×C C Kw
2 4
2
[H O ]
[H O ]3 HA3
++
= +CKw
[OH ][H O ]3 HCl
−+= =K K
Cw w
[H O ]3 HCl+ = C
Capítulo 2: Equilibrios ácido-base 47

[2.34]
expresión muy sencilla y conocida que permiteel cálculo inmediato de [H3O
+] en estos medios.
2.4.2. Base fuerte
En el caso de disoluciones de bases fuertes seprocede de manera similar. Si se toma comoejemplo el NaOH se tiene:
Estudio sistemático del equilibrio ácido basede una base fuerte
Reacciones y constantes implicadas:
Reacción ácido-base:
NaOH → OH– + Na+ [2.39]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH–
Kw = [H3O+ ] [OH–] =10–14 [2.15]
Balance de masas:
CNaOH = [NaOH] + [Na+] ≈ [Na+] [2.40]
La concentración de la forma molecular [NaOH] podrá des-preciarse por tratarse de una base fuerte y por tanto estartotalmente disociada.
Balance de cargas y protónico:
[H3O+] + [Na+] = [OH–] [2.41]
(El balance de carga coincide exactamentecon el balance protónico.)
Mediante un tratamiento similar al realizadopara ácidos fuertes se tiene:
1. Por combinación de las ecuaciones corres-pondientes al balance de cargas y balancede masas se llega a la expresión:
[H3O+] + CNaOH = [OH–] [2.42]
2. Simplificando esta ecuación, en base a[H3O
+] << CNaOH se transforma en:
[2.43]
3. Si la disolución de base fuerte es muy dilui-da, la concentración de iones hidronio serácomparable con la concentración analíti-ca de NaOH, esto es, CNaOH y por tanto noserá despreciable. Como en el caso ante-rior, se tendrá que resolver la ecuación desegundo grado
[2.44]
similar, a la encontrada para ácidos fuertes,que resulta de sustituir en la ecuación [2.42],la expresión correspondiente a la [OH–]obtenida al considerar la autoprotolisis delagua, ecuación [2.15].
2.4.3. Mezclas de protolitos fuertes
Si en una disolución están presentes más de unácido fuerte (o más de una base fuerte) la expre-sión [2.33] (o la expresión [2.42] en el caso de basesfuertes) se transforma en las expresiones:
1. Mezcla de ácidos fuertes:HCl + HNO3 + ...
[H3O+] = [OH–] + CHCl + CHNO3
+ ... [2.45]
2. Mezcla de bases fuertes:NaOH + KOH + ...
[H3O+] + CNaOH + CKOH + ... = [OH–] [2.46]
En el caso de que sus concentraciones analí-ticas no sean excesivamente bajas, se podrá depre-ciar la [OH–] en la expresión [2.45] o la [H3O
+] enla expresión [2.46], de manera que se obtendránlas ecuaciones:
[H O ]–
3NaOH NaOH+ =
+ + ×C C Kw2 4
2
CNaOH [OH ]= −
[ ]H O3 HA+ = C
48 Equilibrios iónicos y sus aplicaciones analíticas

[2.47]
[2.48]
mientras que si se trata de mezclas de ácidos obases fuertes de concentraciones extremada-mente diluidas se tendrá que tener en cuenta laautoprotolisis del agua para calcular el pH de lasmismas, caso este último de escasa importanciapráctica.
2.5. Equilibrios ácido-base de protolitosdébiles monopróticos
Los protolitos débiles monopróticos, ácidosy bases débiles capaces de ceder o aceptar unsolo protón, se caracterizan por el hecho de noestar totalmente disociados en disolución acuo-sa. El estudio de estos equilibrios y en definiti-va del cálculo del pH de sus disoluciones se vaa dividir en cuatro apartados atendiendo alcarácter de los mismos: 1) ácidos débiles, 2) basesdébiles, 3) sales, al considerar que éstas proce-den de la mezcla estequiométrica de ácidos ybases, y 4) mezclas de ácidos y bases de diferentenaturaleza.
La sistemática a seguir con objeto de lograruna expresión útil para el cálculo del pH de lasdisoluciones de estos protolitos, se fundamentaen las siguientes etapas:
1. Descripción de las reacciones implicadasen el sistema en estudio con sus corres-pondientes constantes de equilibrios.
2. Realización de los correspondientes balan-ces: masas, cargas y protónico.
3. Desarrollo de las expresiones que per-mitan calcular las concentraciones deequilibrio de las especies implicadas enel par ácido-base en función del pH y dela concentración analítica mediante lacombinación del balance de masas y dela constante del equilibrio ácido-base.
4. Sustitución de estas expresiones en elbalance de cargas o bien en el balance pro-tónico.
5. Realización de las simplificaciones opor-tunas con objeto de conseguir la expresiónfinal para el cálculo del pH de la disolu-ción del correspondiente protolito, com-probando la validez de las mismas.
2.5.1. Ácidos débiles monopróticos
En este tipo de protolitos (por ejemplo, unácido débil HA), existe un equilibrio de disocia-ción que puede expresarse mediante la reacción:
HA + H2O →← A– + H3O+ [2.49]
y está regido por la correspondiente constantede equilibrio, Ka, denominada constante de diso-ciación o constante de acidez, la cual queda defi-nida en disoluciones diluidas por la expresión:
[2.50]
Para el estudio sistemático del equilibrio ácido-base de estos protolitos se sigue el proce-dimiento indicado anteriormente, consideran-do, por ejemplo, que se parte de una disoluciónacuosa de un ácido débil monoprótico, HA, deconstante de disociación, Ka, y de concentraciónanalítica CHA. Se tiene por tanto:
Estudio sistemático del equilibrio ácido-basede un ácido débil monoprótico
Reacciones y constantes implicadas:
Reacción ácido-base:
HA + H2O →← A– + H3O+ [2.49]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Ka =
− +[A ][H O ][HA]
3
[OH NaOH KOH− = + +] ...C C
[H O ]3 HCl HNO
+ = + +C C3
...
Capítulo 2: Equilibrios ácido-base 49

Existen cuatro especies distintas en disolución: HA, A–, H3O+
y OH– lo que obliga a plantear cuatro ecuaciones inde-pendientes.
Constante de disociación del ácido:
[2.50]
Autoprotolisis del agua:
Kw = [H3O+] [OH–] [2.14]
Balance de masas:
CHA = [HA] + [A–] [2.51]
Balance de cargas y balance protónico:
[H3O+] = [A–] + [OH–] [2.52]
A) Cálculo de las concentracionesen el equilibrio
Como se ha indicado anteriormente, es útildesarrollar ecuaciones que permitan expresar laconcentración de equilibrio de las distintas espe-cies en disolución, [HA] y [A–], en función de losdatos que normalmente se conocen (la concen-tración analítica y las constantes de equilibrio) ydel pH o [H3O
+]. Así pues, para calcular [HA],la ecuación de la constante de disociación [2.50]se puede escribir como:
[2.53]
y al sustituir esta expresión en el balance demasas, ecuación [2.51], se obtiene:
[2.54]
que al despejar [HA] queda:
[2.55]
expresión que permite calcular [HA] si se cono-ce Ka, para una concentración CHA y a un pHdeterminado.
La ecuación correspondiente a [A–] puedededucirse de forma equivalente. Dado que a par-tir de la ecuación [2.50]:
[2.56]
la sustitución de [HA] en el balance de masas ori-gina la ecuación:
[2.57]
y al despejar [A–] se obtiene:
[2.58]
Así pues, las expresiones [2.55] y [2.58] per-miten calcular las concentraciones de las espe-cies de protolitos débiles en disolución acuosa,[HA] y [A–], a partir de los datos que general-mente se conocen, Ka, CHA y el pH de la disolu-ción, dato este último de enorme importancia.
B) Cálculo del pH de una disoluciónde un ácido débil monoprótico
En el caso de un ácido débil monoprótico, tipoHA, el balance protónico, idéntico al de cargas(ecuación [2.52]), indica que [H3O
+] presente en
[A ]
H O
HA
3
−+
=+
C KK
a
a[ ]
CK
K
a
a
HA3
3
H O ][A ]A
AH O
= + =
= +
+ −−
−+
[[ ]
[ ][ ]
1
[HA
H O A ]3][ ][=
+ −
Ka
[HA]
[H OH O
HA 3
3
=+
+
+
CKa
][ ]
CK
K
a
a
HA3
3
HAHA]
[H O ]
HA]H O
= + =
= +
+
+
[ ][
[[ ]
1
[A ]
HA][H O ]3
−+
= Ka [
Ka =− +[A ][H O ][HA]
3
50 Equilibrios iónicos y sus aplicaciones analíticas

la disolución es la suma de los hidronios que pro-ceden de la disociación del ácido, o sea [A–], máslos hidronios que proceden de la autoprotolisisdel agua, es decir, [OH–], ya que el agua, al diso-ciarse, por cada H3O
+ dará un OH–. El que laaportación de hidronios procedentes del agua seadespreciable dependerá de las condiciones de ladisolución.
Teniendo en cuenta el balance de cargas y sus-tituyendo sus términos [A–] y [OH–] en funciónde [H3O
+], considerando para ello las ecuaciones[2.58] y [2.14], respectivamente, se obtiene laecuación general:
[2.59]
Esta ecuación de tercer grado, puede simpli-ficarse en la mayoría de las ocasiones, con lo quese facilita el cálculo del pH.
Así, al tratarse de la disolución de un ácidodébil monoprótico, por definición el medio seráácido, con lo que [OH–] será despreciable en elbalance de cargas, ecuación [2.52], por lo que éstese transforma en:
[H3O+] = [A–] [2.60]
Esta ecuación simplificada del balance decargas también responde al siguiente razona-miento que considera las reacciones que se tie-nen en el sistema; es decir, la correspondiente ala disociación del ácido y la autoprotolisis delagua, ecuaciones [2.49] y [2.8]. En efecto, lainclusión de [OH–] en el balance de cargas (ecua-ción [2.52]) proviene de la autoprotolisis delagua, ya de por sí muy poco disociada, Kw = 10–14,pero además en este caso se añade un ácido, esdecir, se adiciona H3O
+, con lo que la reacciónde autoprotolisis, ecuación [2.8], se desplazarámás hacia la izquierda, por lo que [OH–] tende-rá a ser despreciable.
El despreciar, por tanto, [OH–] en el balancede cargas supone eliminar el último término en laecuación [2.59], con lo que ésta se transforma en:
[2.61]
Esta ecuación de segundo grado puede resol-verse o bien considerar una nueva simplificación,posible en muchos casos, al tener en cuenta que,para un ácido débil, generalmente se cumple que[H3O
+] >> Ka (ver problema 1), con lo que pue-de aceptarse que [H3O
+] + Ka ≈ [H3O+] y la ecua-
ción [2.61] se reducirá a:
[2.62]
de donde:
[2.63]
expresión muy sencilla, que permite calcular elpH de la disolución de un ácido débil y que indi-ca que la acidez de esta disolución aumenta conla concentración y la fuerza del ácido.
Si el cálculo del pH se hace utilizando la ecua-ción simplificada [2.63], se ha de comprobar quelas aproximaciones realizadas realmente se cum-plen. Así:
1. Si no se cumple que [H3O+] >> Ka, deberá
utilizarse la ecuación [2.61].2. Si la influencia de la autoprotolisis del
agua no es despreciable, debe conside-rarse la ecuación general [2.59]. Esto últi-mo sólo ocurre en los casos en que no secumple que CHA Ka >> Kw , lo cual puedetener su origen en:
a) CHA muy pequeña. Situación de escasointerés práctico, ya que raramente seutilizan ácidos a concentraciones infe-riores a 10–3 M.
b) Ka muy pequeña (ácidos muy débiles).En este caso, en la ecuación [2.59] sepuede considerar que [H3O
+] >> Ka enel denominador del segundo término,quedando ésta simplificada a:
[H O ] 3 HA+ = C Ka
[H O ] 3 HA+ =2 C Ka
[H O ]
H O ]3HA
3
++
=+
C KK
a
a[
[H O ]
[H O ] [H O ]3HA
3 3
++ +
=+
+C KK
Ka
a
w
Capítulo 2: Equilibrios ácido-base 51

[2.64]
ecuación de segundo grado que permite calcularfácilmente el pH de las disoluciones de estos pro-tolitos:
[2.65]
En resumen, la eliminación de algunos de lostérminos del balance cargas y/o protónico redu-ce el tiempo necesario para resolver las ecuacio-nes y en la mayoría de los casos proporcionaresultados correctos. Sin embargo, en algunasocasiones existe el riesgo de eliminar términosque ejercen una pequeña influencia, pero no des-preciable, en el pH final de la disolución y nosiempre es fácil apreciar que la simplificación noera válida. El problema radica no tanto en elvalor numérico del pH, que por lo general no serámuy diferente del valor correcto, sino en que unasimplificación excesiva puede llevar a generali-zaciones erróneas, como se verá más adelantepara algunos casos.
2.5.2. Bases débiles monopróticas
En una disolución de una base débil mono-prótica, B, como por ejemplo el NH3, de con-centración analítica CB mol L–1 y constante deacidez Ka, tienen lugar las siguientes reacciones:
Estudio sistemático del equilibrio ácido-basede una base débil monoprótica
Reacciones y constantes implicadas:
Reacción ácido-base. Protonación de la base:
B + H2O →← HB+ + OH– [2.66]
Autoprotolisis del agua:
H2O + H2O →← H3O+ + OH– [2.8]
Existen cuatro especies distintas en disolución: BH+, B, H3O+
y OH– y es necesario plantear cuatro ecuaciones indepen-dientes.
Constante de acidez del ácido conjugado:
[2.67]
Constante de autoprotolisis:
Kw = [H3O+] [OH–] [2.14]
Balance de masas:
CB = [HB+] + [B] [2.68]
Balance de cargas y balance protónico:
[HB+] + [H3O+] = [OH–] [2.69]
Es importante resaltar que, pese a tratarse deuna disolución de una base, se emplea la cons-tante de disociación del ácido conjugado, HB+,a fin de unificar las ecuaciones y el tratamientoa seguir. Por otra parte, al tratarse de una basedébil monoprótica del tipo B, cuya forma proto-nada tiene carga positiva y cuya forma disociadaes neutra, el balance de cargas y el balance pro-tónico coinciden.
De manera similar al caso de un ácido débilmonoprótico, por combinación de las expresionesdel balance de masas, ecuación [2.68] y constantede acidez, ecuación [2.67], se pueden deducir fácil-mente las siguientes expresiones para el cálculo delas concentraciones de HB+ y B en el equilibrio:
[2.70]
[2.71]
similares a las expresiones [2.55] y [2.58].
[B
H OB
3
][ ]
=++
C K
Ka
a
[HB ] [H O ]
[H O ]B 3
3
++
+=+
C
Ka
Ka =+
+
[B][H O ][HB ]
3
[H O ]3 HA+ = +C K Ka w
[H O ]
[H O H O3HA
3 3
++ +
= +C K Ka w
] [ ]
52 Equilibrios iónicos y sus aplicaciones analíticas

Al reemplazar los términos de la expresióndel balance protónico, ecuación [2.69] por lasexpresiones que los relacionan con las constan-tes de equilibrio, la concentración analítica y la[H3O
+], se obtiene:
[2.72]
Para calcular el pH de la disolución de base con-siderada y al igual que ocurría para el caso de diso-luciones de ácidos débiles, casi siempre es posiblesimplificar directamente el balance protónicomediante la supresión de alguno de sus términos.
Así, es evidente que en una disolución de unabase no excesivamente diluida y que no sea extre-madamente débil (es decir, en una disolución bási-ca de interés práctico), se cumplirá que [H3O
+] <<10–7, por ser una disolución básica y porque el equi-librio de disociación del agua, único equilibriogenerador de H3O
+ en este caso, estará aún másdesplazado hacia la izquierda que en agua pura.Por lo tanto, la ecuación [2.69] se reducirá a:
[HB+] = [OH–] [2.73]
Al considerar la ecuación [2.70], que propor-ciona la concentración de equilibrio de la espe-cie ácida de la base, y la [2.14], expresión de Kw,y sustituyendo ambas en la [2.73], se tiene:
[2.74]
Esta expresión de segundo grado se puederesolver fácilmente o bien simplificar teniendoen cuenta que en una disolución de base débilgeneralmente se cumple que Ka >> [H3O
+], loque implica que [H3O
+] + Ka ≈ Ka y por tanto laecuación [2.74] se simplifica a:
[2.75]
de donde:
[2.76]
Esta ecuación permite el cálculo del pH deuna disolución de una base débil e indica que[H3O
+] disminuirá (aumentará la basicidad) cuan-do la Ka sea menor (aumente la fuerza básica delprotolito) o la concentración analítica sea mayor.
Una vez obtenido el pH de la disolución, paradarlo por válido hay que comprobar que las apro-ximaciones realizadas son correctas. Así, se hade comprobar que realmente Ka >> [H3O
+]. Deno ser así, se debe aplicar la ecuación [2.74] paraobtener un valor correcto. En el caso, sin interéspráctico, de que la autoprotólisis del agua no seadespreciable y se hayan de tener en cuenta losH3O
+ procedentes del agua, con lo que [H3O+]
no será despreciable frente a [HB+] en la ecua-ción [2.69], se tendrá que aplicar la ecuacióngeneral [2.72], o bien una semejante a la obteni-da para el caso de ácidos muy débiles, tal como:
[2.77]
en base a razonamientos similares y de fácildeducción.
2.5.3. Cálculo del pH de disoluciones de sales
Al mezclar cantidades estequiométricas de unácido y una base monopróticos, tiene lugar unareacción de neutralización que da lugar a la for-mación de una sal. En función del tipo de ácido yde base, pueden darse cuatro casos claramentediferenciados: 1) sales de ácido fuerte y base fuer-te, denominadas sales neutras, cuya disolución enagua origina un pH neutro; 2) sales de ácido fuer-te y base débil, sales ácidas, cuyas disoluciones tie-nen carácter ácido; 3) sales de ácido débil y basefuerte, conocidas como sales básicas, por el pH
[H O ]3
B
+ =+
K KC K
w a
a
[H O ]3B
+ = K KCw a
CK
K
a
wB 3
3
H OH O
[ ][ ]
+
+=
C
K
K
a
wB 3
3 3
H OH O H O
[ ][ ] [ ]
+
+ ++=
C
K
K
a
wB 3
33
3
H OH O
H O ]H O
[ ][ ]
[[ ]
+
++
+++ =
Capítulo 2: Equilibrios ácido-base 53

que poseen sus disoluciones; y 4) sales de ácidodébil y base débil, cuyo pH puede ser moderada-mente ácido, o moderadamente básico, según lasconstantes de acidez implicadas.
Como norma general, puede decirse que loque determina que la disolución de una sal seaácida o básica, es el carácter ácido-base del másfuerte de los compuestos que la originan. A con-tinuación se describe el tratamiento a seguirpara calcular el pH en cada uno de los casosindicados.
A) Sal de ácido fuerte y base fuerte
Un ejemplo de este tipo de sales es el NaCl, queproviene de la reacción entre el HCl y el NaOH.Esta sal está totalmente disociada en disoluciónacuosa, con lo que se tendrán las reacciones:
NaCl → Na+ + Cl– [2.78]
H2O + H2O →← H3O+ + OH– [2.8]
Dado que ni el Cl– (“base” conjugada de unácido fuerte y, por lo tanto, sin carácter básico),ni el Na+ (“ácido” conjugado de una base fuertey, en consecuencia, sin carácter ácido) reaccio-nan con el agua, es evidente que:
[Na+] = [Cl–] [2.79]
y dado que, según el balance de cargas:
[Na+] + [H3O+] = [Cl–] + [OH–] [2.80]
puede comprobarse que:
[2.81]
es decir, la disolución será neutra.Una manera más rápida de deducir que el
pH de esta disolución será neutro, consiste enconsiderar que tanto el Cl– como el Na+ no reac-cionan con el agua, con lo que los H3O
+ de la
disolución procederán de la autoprotolisis deeste disolvente, que no estará influenciada, loque supone pH = 7.
B) Sal de ácido fuerte y base débil
Ejemplo de este tipo de sales se puede consi-derar el NH4Cl, formado por la reacción de HCly NH3. En agua la sal está totalmente disociada:
NH4Cl → NH4+ + Cl– [2.82]
y, si bien el Cl– no tiene carácter básico, el NH4+
es un ácido débil (pKa = 9,24) por lo que se tienela reacción:
NH4+ + H2O →← NH3 + H3O
+ [2.83]
que confiere un pH ácido a la disolución.El cálculo del pH de la disolución de esta sal,
por tanto, se circunscribe a determinar el quepresenta una disolución de un ácido débil, queya ha sido tratado con detalle, aunque en estecaso se da la circunstancia de que la forma pro-tonada tiene carga positiva (NH4
+), mientras quela forma disociada (NH3) es neutra, por lo quelos balances de carga y protónico no coinciden,como se muestra a continuación.
Estudio sistemático del equilibrio ácido-base de una salde ácido fuerte y base débil
Reacciones y constantes implicadas:
Disociación de la sal:
NH4Cl → NH4+ + Cl– [2.82]
Reacción ácido-base:
NH4+ + H2O →← NH3 + H3O
+ [2.83]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
[H O ] [OH ] pH3+ − −= = = ⇒ =Kw 10 77
54 Equilibrios iónicos y sus aplicaciones analíticas

En la disolución existirán cinco especies diferentes: NH4+,
NH3, Cl–, H3O+ y OH– por lo que serán necesarias cinco
ecuaciones independientes.
Constante de disociación del ácido:
[2.84]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balances de masas:
Csal = [NH4+]+ [NH3]; Csal = [Cl–] [2.85]
Balance de cargas:
[NH4+] + [H3O
+] = [Cl–] + [OH–] [2.86]
Balance protónico:
[H3O+] = [NH3] + [OH–] [2.87]
A partir del balance protónico, la resoluciónes idéntica a la descrita anteriormente para elcaso general de una disolución de ácido débilmonoprótico. Así, al sustituir en este balance laexpresión para [NH3] en función del pH y Ka:
[2.88]
ecuación similar a la [2.58] correspondiente a [A–]para un ácido débil monoprótico, y para [OH–]conforme a la autoprotolisis del agua, se tiene:
[2.89]
Esta ecuación general, similar a la [2.59] si CHA = Csal, permite calcular el pH de la disoluciónde una sal de ácido fuerte y base débil. Si se lle-
van a cabo las mismas simplificaciones indica-das en el apartado correspondiente al cálculodel pH de un ácido débil monoprótico, se obtie-ne la expresión final:
[2.90]
C) Sal de ácido débil y base fuerte
Un ejemplo de este tipo de sales es elCH3COONa, que puede obtenerse por reacciónde CH3COOH (pKa = 4,75) con NaOH. El estu-dio sistemático del equilibrio ácido-base de estasal se muestra a continuación:
Estudio sistemático del equilibrio ácido-base de una salde ácido débil y base fuerte
Reacciones y constantes implicadas:
Disociación de la sal:
CH3COONa → Na+ + CH3COO– [2.91]
Reacción ácido-base:
CH3COO– + H2O →← CH3COOH + OH– [2.92]
Autoprotolisis del disolvente:
H2O + H2O →← H3O++ OH– [2.8]
Dado que en esta disolución existen cinco especies distin-tas: CH3COOH, CH3COO–, Na+, H3O
+ y OH– serán nece-sarias cinco ecuaciones independientes.
Constantes de disociación del ácido:
[2.93]
Autoprotolisis del disolvente:
Kw = [H3O+][OH–] [2.14]
Balances de masas:
Csal = [CH3COOH] + [CH3COO–]; Csal = [Na+] [2.94]
Ka =
− +[CH COO ][H O ][CH COOH]3 3
3
[H O ]3 sal+ = C Ka
[H O ][H O ] [H O ]3
sal
3 3
++ +=
++C K
K
Ka
a
w
[NH ][H O ]3
sal
3
=++
C K
Ka
a
Ka =
+
+
[NH ][H O ][NH ]
3 3
4
Capítulo 2: Equilibrios ácido-base 55

Balance de carga:
[Na+] + [H3O+] = [CH3COO–] + [OH–] [2.95]
Balance protónico:
[CH3COOH] + [H3O+] = [OH–] [2.96]
Como en el caso anterior resulta evidente queel balance protónico no puede coincidir con el decargas, ya que en éste han de aparecer las concen-traciones de Na+ y de CH3COO–, que son las espe-cies inicialmente disueltas y que, en consecuencia,no se forman por pérdida o ganancia de protones,como ocurre en el caso de las especies que consti-tuirán el balance protónico, ecuación [2.96].
Para calcular el pH de estas disoluciones seconsidera el balance protónico y se procede aun tratamiento idéntico al descrito en el apar-tado 2.5.2 para el caso de una base débil mono-prótica del tipo B. La ecuación final que resul-ta para el cálculo del pH es:
[2.97]
similar a la ecuación [2.76] tras realizar las corres-pondientes simplificaciones en la ecuación general.
Como en el apartado anterior, se ha utilizadoel balance protónico para conseguir una expre-sión útil para el cálculo del pH que presentan lasdisoluciones de las sales estudiadas, aunque tam-bién se podría emplear el balance de cargas. Sinembargo, para el caso de disoluciones de sales,es más recomendable el uso del balance protó-nico, ya que como se muestra en el recuadro 2.2,las simplificaciones del balance de cargas en estoscasos pueden conducir a resultados absurdos.
D) Sal de ácido débil y base débil
Consideraremos en este caso como ejemploel HCOONH4, obtenido por reacción de ácidofórmico (pKHCOOH = 3,75) con amoniaco, NH3
(pKNH4+ = 9,24). En disolución acuosa esta sal está
totalmente disociada:
HCOONH4 → HCOO– + NH4+ [2.98]
y, dado que el ion formiato tiene carácter básicopor tratarse de la base conjugada de un ácido débily el amonio es un ácido débil, el estudio sistemá-tico de este equilibrio ácido-base viene dado por:
Estudio sistemático del equilibrio ácido-base de una salde ácido débil y base débil
Reacciones y constantes implicadas:
Disociación de la sal:
HCOONH4 → HCOO– + NH4+ [2.98]
Reacciones ácido-base:
NH4+ + H2O →← NH3 + H3O
+ [2.83]
HCOO– + H2O →← HCOOH + OH– [2.99]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen seis especies distintas en disolución: NH4+, NH3, HCO-
OH, HCOO–, H3O+ y OH– y son necesarias seis ecuacio-
nes independientes.
Constantes ácido-base:
[2.84]
[2.100]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balances de masas:
Csal = [NH4+] + [NH3̄ ]; Csal= [HCOOH] + [HCOO–] [2.101]
KHCOOH3[HCOO ][H O ]
[HCOOH]=
− +
KNH
3 3
44
[NH ][H O ][NH ]+ =
+
+
[H O ]3sal
+ = K KC
w a
56 Equilibrios iónicos y sus aplicaciones analíticas

Balance de cargas:
[NH4+]+ [H3O
+] = [HCOO–] + [OH–] [2.102]
Balance protónico:
[HCOOH] + [H3O+] = [NH3] + [OH–] [2.103]
Aplicando la misma sistemática que en los casosanteriores y sustituyendo las concentraciones de
las especies ácida, HCOOH, y básica, NH3, por susexpresiones en función de la concentración analí-tica, constantes de acidez y pH, se tiene la siguien-te ecuación de cuarto grado:
[2.104]
[ ]
[ ][ ]
[ ] [ ]
C
KC K
K
Kw
sal 3
3 HCOOH3
sal NH
3 NH 3
H OH O
H O
H O H O
+
++
+ +
++ =
=+
++
+
4
4
Capítulo 2: Equilibrios ácido-base 57
Estudio comparativo del uso del balance de cargas y del balance protónico para el cálculo del pH de la disolución de una sal
Sea una sal sódica, tipo NaA, que en disolución se corresponde con una disolución de base débil, A–, de con-centración Csal, a la que le corresponde una constante de disociación Ka del ácido conjugado HA. Realizandoun tratamiento paralelo para ambos balances se tiene:
Balance de cargas: Balance protónico:
[Na+] + [H3O+] = [A–] + [OH–] [HA] + [H3O
+] = [OH–]
Expresión general:
Simplificaciones:
Por ser una base relativamente concentrada: Csal >> [H3O+] y [A–] >> [OH–]
En una base se tiene que Ka >> [H3O+]
Csal = Csal
Se observa cómo a partir del balance de cargas se obtiene una expresión que carece de utilidad, mientras que apartir del balance protónico se llega a una ecuación válida cuya expresión final corresponde a la ecuación [2.97]Para obtener una expresión útil a partir del balance de cargas, sería necesario asumir que [A–] ≈ [OH–] y queKa ≈ [H3O
+], aproximaciones no siempre lógicas en el caso de sales relativamente concentradas. En este casose obtiene una ecuación de segundo grado que conduce a un resultado correcto.
CK
K
a
wsal H OH O
[ ][ ]
3
3
+
+=
CK
K
a
wsal H OH O H O
[ ][ ] [ ]
3
3 3
+
+ ++=C
C KK
a
asal
sal
H O=
++[ ]3
CK
K
a
wsal H OH O
H OH O
[ ][ ]
[ ][ ]
3
33
3
+
++
+++ =
C
C KK
Ka
a
wsal
salH OH O H O
+ =+
+++ +[ ]
[ ] [ ]33 3
RECUADRO 2.2

Sin embargo, y también como en apartadosanteriores se pueden llevar a cabo una serie desimplificaciones que reducen la complejidad dela misma.
Así, es fácil aceptar que, dado que la disolu-ción contiene un ácido y una base, el pH no seráexcesivamente ácido ni básico y que, en conse-cuencia:
[HCOOH] >> [H3O+] y [NH3] >> [OH–]
[2.105]
y la expresión del balance protónico se reduce a:
[HCOOH] = [NH3] [2.106]
con lo que obtenemos:
[2.107]
Expresión de segundo grado que permite calcu-lar con facilidad el pH de la disolución de sal con-siderada. Esta aproximación se cumple general-mente. Si además se considera que:
[2.108]
la expresión [2.107] puede simplificarse a:
[2.109]
de donde:
[2.110]
Esta ecuación indica que si todas las simplifi-caciones son válidas, el pH sólo depende de lasconstantes de acidez del ácido y de la base en cues-tión. Sin embargo, es importante resaltar que paraque la ecuación simplificada [2.110] sea válida,
es condición necesaria que el pH sea moderado,aunque no suficiente ya que debe cumplirse ade-más que CsalKNH4
+ >> Kw (cosa que ocurre habi-tualmente) y que Csal >> KHCOOH, lo que en oca-siones no se cumple.
2.5.4. Mezclas de protolitos monopróticos
En este apartado se aborda el estudio de losequilibrios ácido-base correspondientes a mezclasno estequiométricas de protolitos (ácidos o bases)monopróticos, así como la deducción de las expre-siones correspondientes para el cálculo del pH desus disoluciones. Considerando el carácter de estosprotolitos se pueden diferenciar cuatro tipos demezclas: 1) ácido fuerte + ácido débil; 2) mezclade dos ácidos débiles; 3) base fuerte + bases débil;y 4) mezclas de dos bases débiles.
A) Mezcla de ácido fuerte y ácido débil
Si una disolución contiene un ácido fuerte,HA1, a concentración C1 y un ácido débil HA2,de constante de acidez K2, a concentración C2,tendrán lugar en su seno las siguientes reaccionesque conforman el siguiente sistema ácido-base:
Estudio sistemático del equilibrio ácido-base de una mezclade un ácido fuerte y un ácido débil
Reacciones y constantes implicadas:
Reacciones ácido-base:
HA1 + H2O → A1̄ + H3O+ [2.111]
HA2 + H2O →← A2̄ + H3O+ [2.112]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen cinco especies distintas en disolución: A1̄ , HA2 , A2̄ ,H3O
+ y OH–, lo que obliga a plantear cinco ecuaciones inde-pendientes.
[H O ]3 HCOOH NH
+ = +K K4
[ ]H O[H O ]
3
HCOOH
NH
3
+
+=
+
K
K4
K KHCOOH 3 NHH O> >+
+[ ]4
CK
C K
Ksal 3
3 HCOOH
sal NH
3 NH
[H OH O ] H O
+
+ ++=
++
+
][ [ ]
4
4
58 Equilibrios iónicos y sus aplicaciones analíticas

Constante ácido-base:
[2.113]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balance de masas:
C1 = [A1̄ ] [2.114]
C2 = [HA2] + [A2̄ ] [2.115]
Balance de cargas y balance protónico:
[H3O+] = [A1̄ ] + [A2̄ ] +[OH¯] [2.116]
En este caso el balance de cargas y el balan-ce protónico coinciden. Al sustituir los términosdel balance protónico considerando las ecuacio-nes [2.114], [2.58] y [2.14] se obtiene la ecuacióngeneral:
[2.117]
Como en otros casos, se pueden llevar a cabosimplificaciones de gran interés práctico. Así, enla ecuación del balance protónico [OH–] será des-preciable al ser una disolución de ácidos y en elcaso general en la práctica en que la contribu-ción del ácido débil sea despreciable, este balan-ce queda reducido a:
[2.118]
que al tratarse de un ácido fuerte toma la forma:
[2.119]
Esta expresión viene a decir que el pH de unadisolución que contiene un ácido fuerte y un áci-do débil está determinado por el ácido fuerte, siem-
pre que su concentración sea lo suficientementeelevada para permitir despreciar la contribucióndel ácido débil y de la autoprotolisis del agua.
B) Mezcla de dos ácidos débiles monopróticos
El equilibrio ácido-base de una disolución quecontiene dos ácidos débiles, HA1 de constantede disociación K1 y a concentración C1 y HA2 deconstante de disociación K2 y a concentración C2,se puede esquematizar conforme a las siguientesreacciones:
Estudio sistemático del equilibrio ácido-base de una mezclade dos ácidos débiles monopróticos
Reacciones y constantes implicadas:
Reacciones ácido-base:
HA1 + H2O →← A1̄ + H3O+ [2.120]
HA2 + H2O →← A2̄ + H3O+ [2.121]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.14]
Existen seis especies distintas en disolución: HA1 , A1̄ , HA2 ,A2̄ , H3O
+ y OH– lo que obliga a plantear seis ecuacionesindependientes.
Constantes ácido-base:
[2.122]
[2.123]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balances de masas:
C1 = [HA1] + [A1̄ ] [2.124]C2 = [HA2] + [A2̄ ] [2.125]
K2 =− +[A ][H O ][HA ]2 3
2
K1 =
− +[A ][H O ][HA ]
1 3
1
[H O ]3+ = C1
[H O ] [A ]3 1–+ =
[H O ][H O ] [H O ]3
3 3
++ += +
++C
C K
K
Kw1
2 2
2
K 2 =− +[A ][H O ][HA ]2 3
2
Capítulo 2: Equilibrios ácido-base 59

Balance de cargas y balance protónico:
[H3O+] = [A1̄ ] + [A2̄ ] +[OH¯] [2.126]
Al tratarse de especies ácidas neutras elbalance de cargas y el balance protónico coinci-den. Al sustituir en esta expresión los términos[A1̄], [A2̄] y [OH¯] en función de las ecuaciones[2.58] y [2.14] se obtiene la siguiente ecuacióngeneral:
[2.127]
El cálculo del pH de una disolución de dosácidos débiles, puede simplificarse consideran-do que la disolución será ácida, con lo que [OH–]será despreciable, es decir la contribución de laautoprotolisis del agua es despreciable, por loque la ecuación [2.127] se transforma en:
[2.128]
Por otra parte, si por ejemplo, C1K1 >> C2K2 ,la contribución del ácido HA2 será despreciable,con lo que se tiene:
[2.129]
ecuación de segundo grado que puede resolver-se fácilmente, pero si, como ya se ha comenta-do para ácidos débiles, puede aceptarse, comoocurre generalmente, que [H3O
+] >> Ka, se lle-ga a la expresión más simple:
[2.130]
lo que significa que en una disolución que contie-ne dos ácidos débiles el pH viene determinado por
el que tiene el término CKa más elevado. Dichode otra manera, el que la contribución de un áci-do al pH final de una disolución sea despreciableo no depende de su fuerza y también de su con-centración relativa.
C) Mezcla de una base fuerte y débil
De forma similar al estudio realizado para unadisolución que contiene un ácido fuerte en mez-cla con un ácido débil, se pueden establecer lassiguientes expresiones considerando una disolu-ción que contiene una base fuerte, B1OH a con-centración C1 y una base débil, B2, a concentra-ción C2 y constante de disociación K2:
Estudio sistemático del equilibrio ácido-base de una mezclade una base fuerte con una base débil
Reacciones y constantes implicadas:
Reacciones ácido-base:
B1OH → B1+ + OH– [2.131]
B2 + H2O →← HB2+ + OH– [2.132]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen cinco especies distintas en disolución: B1+, B2 , HB2
+,H3O
+ y OH–, lo que obliga a plantear cinco ecuaciones inde-pendientes.
Constante ácido-base:
[2.133]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balances de masas:
C1 = [B1+] [2.134]
K2 =
+
+
[B ][H O ][HB ]
2 3
2
[H O ]3+ = C K1 1
[H O ]
H O ]33
++
=+
C KK
1 1
1[
[H O ]
[H O ] [H O ]33 3
++ +
=+
++
C KK
C KK
1 1
1
2 2
2
[H O ]
[H O ] H O ] H O33 3 3
++ + +
=+
++
+C KK
C KK
Kw1 1
1
2 2
2[ [ ]
60 Equilibrios iónicos y sus aplicaciones analíticas

C2 = [HB2+] +[B2] [2.135]
Balance de cargas y protónico:
[B1+] + [HB2
+] + [H3O+] = [OH¯] [2.136]
Al sustituir los términos del balance protóni-co en función de las ecuaciones [2.134], [2.70] y[2.14], se obtiene la siguiente ecuación general:
[2.137]
que puede simplificarse fácilmente, al igual queen los casos anteriormente considerados.
Así, teniendo en cuenta, en este balance pro-tónico, que [H3O
+] en una disolución de bases serádespreciable y que además si la concentración debase fuerte no es sumamente baja, se cumplirá que[HB2
+] << [B1+], éste se transforma en:
[B1+] = [OH¯] [2.138]
por lo que la ecuación [2.137] queda como:
[2.139]
o bien
[2.140]
De esta expresión se deduce que el pH de unamezcla de una base fuerte y una base débil vie-ne determinado únicamente por la base fuerte,siempre que ésta sea lo suficientemente concen-trada.
En este punto es interesante resaltar que enmuchas ocasiones la mezcla puede estar consti-tuida por una base fuerte, por ejemplo NaOH deconcentración C1 y una sal de un ácido débil,NaA, de concentración C2. En este caso el balan-ce de cargas, dado por:
[Na+] + [H3O+] = [A–] + [OH–] [2.141]
en el que aparecen especies disueltas inicial-mente, como Na+ y A–, no coincide con el pro-tónico. Sin embargo, la deducción de este balan-ce protónico es fácil si se combina el balance decargas, ecuación [2.414], con los de masas:
[Na+] = C1 + C2 [2.142]
[HA] + [A–] = C2 [2.143]
con lo que se obtiene:
(C1 + C2) + [H3O+] = (C2 – [HA]) + [OH–]
[2.144]
es decir,
C1 + [HA] + [H3O+] = [OH–] [2.145]
En este balance protónico, los términos[H3O
+] y también en la mayoría de ocasiones, alser una base débil, [HA] serán despreciables, conlo que resulta una expresión idéntica a la ante-riormente obtenida, tal como la ecuación [2.138].
D) Mezcla de dos bases débiles monopróticas
En una disolución que contiene dos basesdébiles, B1 con una constante de disociación K1y una concentración C1 y B2 con una constantede disociación K2 y una concentración C2, tienenlugar las siguientes reacciones:
Estudio sistemático del equilibrio ácido-base de una mezclade dos bases débiles
Reacciones y constantes implicadas:
Reacciones ácido-base:
B1 + H2O →← HB1+ + OH– [2.146]
B2 + H2O →← HB2+ + OH– [2.147]
[H O3
+ =]KC
w
1
C
Kw1 =
[H O ]3+
C
CK
Kw1
2
2
++
+ =+
++
+
[[ ] [
H O ]H O
[H O ]H O ]
3
33
3
Capítulo 2: Equilibrios ácido-base 61

Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen seis especies distintas en disolución: B1, HB1+, B2,
HB2+, H3O
+ y OH–, lo que obliga a plantear seis ecuacionesindependientes.
Constantes ácido-base:
[2.148]
[2.149]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH–] [2.14]
Balances de masas:
C1 = [HB1+] + [B1] [2.150]
C2 = [HB2+] + [B2] [2.151]
Balance de cargas y protónico:
[HB1+] + [HB2
+] + [H3O+] = [OH–] [2.152]
Al sustituir los términos del balance protóni-co en función de las concentraciones analíticas,constantes de equilibrio y [H3O
+], se obtiene laecuación general:
[2.153]
Se tiene una ecuación mucho más sencilla, sise considera que, en el balance protónico, [H3O
+]será despreciable al ser una disolución de bases:
[2.154]
y además si se considera, como ocurre general-mente, que C1 /K1 >> C2 /K2 (siendo la base B1 másfuerte, K1 más pequeña, y a concentraciones nomuy diluidas), se puede eliminar el término HB2
+
correspondiente a B2 en este balance, quedando:
[2.155]
Por otra parte, se eliminaría el término B1 sise cumpliese que C1/K1 << C2/K2. La ecuación[2.155] de segundo grado, como en otros casos,puede reducirse aún más, al cumplirse en estasdisoluciones que [H3O
+] << K1, con ello:
[2.156]
de donde:
[2.157]
Expresión idéntica a la obtenida para el casode una base débil y que indica que en una mez-cla de dos bases débiles, a concentraciones noexcesivamente diluidas, el pH de la disoluciónestará determinado por una de ellas, siempre quese cumplan las condiciones de que C1/K1 >> C2/K2. En estos casos, las contribuciones de lasegunda base y del agua serán despreciables.
Finalmente reseñar que en muchos casos lamezcla de dos bases débiles puede contener lassales de dos ácidos débiles, por ejemplo NaA1 yNaA2. El balance de cargas correspondiente aesta disolución viene dado por:
[Na+] + [H3O+] = [A1̄] + [A2̄] + [OH¯] [2.158]
el cual, claramente, no coincide con el balanceprotónico expresado por:
[HA1] + [HA2] + [H3O+] = [OH¯] [2.159]
[ ]H O3
+ = K KCw a1
1
CK
Kw1
1
[ ][ ]
H OH O
3
3
+
+=
CK
Kw1
1
[ ][ ] [
H OH O H O ]
3
3 3
+
+ ++=
CK
CK
Kw1
1
2
2
[ ][ ]
[ ][ ]
H OH O
H OH O [H O ]
3
3
3
3 3
+
+
+
+ +++
+=
[ ]
[ ][C
KC
KKw
1
1
2
2
H OH O
H O ][H O ]
[H O ][H O ]
3
3
3
3
33
+
+
+
+
++
++
++
+ =
K2 =
+
+
[B ][H O ][HB ]
2 3
2
K1 =
+
+
[B ][H O ][HB ]
1 3
1
62 Equilibrios iónicos y sus aplicaciones analíticas

Como en otros casos, este balance protónicopuede deducirse fácilmente a partir de las reac-ciones que tienen lugar, considerando las especiesque ganan o pierden protones, o mediante la com-binación de los balances de materia y carga. Si serealizan simplificaciones en este balance protó-nico, se tiene:
1. Si se considera que [H3O+] será desprecia-
ble al ser una disolución de bases:
[2.160]
2. Si se considera como ocurre generalmen-te que C1 /K1 >> C2 /K2:
[2.161]
Al sustituir, en este balance protónico sim-plificado, [HA1] y [OH–] como en casos anterio-res, se tiene una ecuación idéntica a la [2.156], dela que se obtiene finalmente la expresión que per-mite calcular el pH de esta disolución.
2.6. Equilibrios ácido-base de protolitosdébiles polipróticos
El estudio de protolitos débiles polipróticoses equivalente al descrito para los monopróticos,aunque, evidentemente, las ecuaciones son máscomplicadas. Para abordar su estudio sistemáti-co se van a considerar cuatro apartados confor-me el carácter de estos protolitos: ácidos dipró-ticos y polipróticos, bases dipróticas, anfolitos ysales de ácidos polipróticos.
2.6.1. Ácidos débiles dipróticos y polipróticos
De forma similar al estudio realizado sobre áci-dos débiles monopróticos, se va a considerar ladeducción en primer lugar de las expresiones corres-pondientes para el cálculo de las concentracionesen el equilibrio de las diferentes especies presentesen el mismo, y en segundo lugar de las ecuacionespara el cálculo del pH de sus disoluciones.
A) Cálculo de las concentraciones de las especiesimplicadas en el equilibrio
Como se ha indicado, el objetivo de esteapartado es conseguir las ecuaciones que per-miten calcular la concentración de cualquierade las formas en que puede estar un ácido poli-prótico en disolución, conociendo la concen-tración analítica, las constantes de los equili-brios implicados y la [H3O
+].El tratamiento a desarrollar es similar al
seguido para ácidos monopróticos, esto es, por combinación del balance de masas y de lasexpresiones de las constantes de disociación ácido-base.
Por ejemplo, para un ácido diprótico H2Acuya concentración analítica es CH2A
, el balancede masas viene dado por:
[2.162]
siendo sus constantes ácido-base:
[2.163]
[2.164]
A partir de estas ecuaciones se puede dedu-cir que:
[2.165]
que, después de simplificar y reordenar, queda:
[2.166] [ ]
[ ]
[ ] [ ]H A
H O
H O H O2H A 3
3 3
2=+ +
+
+ +
C
K K Ka a a
2
21 1 2
CK
K K
a
a a
H A 22
3
2
3
2H A
[H AH O
H AH O
= + +
+
+
+
[ ]]
[ ]
[ ][ ]
1
1 22
Ka 2 =
− +
−
[A ][H O ][HA ]
23
Ka1 =
− +[HA ][H O ][H A]
3
2
CH A 2
22
H A] [HA ] [A ]= + +− −[
[HA ] [OH ]1 = −
[HA ] [HA ] [OH ]1 2+ = −
Capítulo 2: Equilibrios ácido-base 63

Las ecuaciones para las formas monodisocia-da y totalmente disociada pueden deducirsesiguiendo un razonamiento similar. Así:
[2.167]
que, una vez reordenada y simplificada, queda:
[2.168]
y en el caso de la especie totalmente disociadapuede deducirse que:
[2.169]
que lleva a la expresión:
[2.170]
Comparando las expresiones obtenidas paracalcular las concentraciones en el equilibrio delas especies implicadas en función de la concen-tración analítica del ácido, constantes de acido-base y [H3O
+], con las obtenidas para un ácidomonoprótico, ecuaciones [2.55] y [2.58], se obser-va una cierta tendencia o secuencia entre las mis-mas, como se muestra a continuación:
1. Equilibrio ácido-base de un ácido débilmonoprótico
[2.55]
[2.58]
2. Equilibrio ácido-base de un ácido débildiprótico:
[2.166]
[2.168]
[2.170]
3. Equilibrio ácido-base de un ácido débiltriprótico. Se pueden deducir fácilmentelas siguientes expresiones:
[2.171]
[2.172]
[2.173]
[2.174]
Puede comprobarse que estas expresiones tie-nen un denominador común, que para un ácidoque puede perder n protones, HnA, consiste enun polinomio de n + 1 términos que presenta laforma general:
[A ]
[H O ] [H O ] [H O ]3 H A
3 3 3
3−+ + +
=+ + +
C K K K
K K K K K Ka a a
a a a a a a
1 2 3
31
21 2 1 2 3
[ ]HA
[H O ]
[H O ] [H O ] [H O ]2 H A 3
3 3 3
3−+
+ + +=
+ + +C K K
K K K K K Ka a
a a a a a a
1 2
31
21 2 1 2 3
[H A
[H O ]
[H O ] [H O ] [H O ]2H A 3
3 3 3
3−+
+ + +=
+ + +]
C K
K K K K K Ka
a a a a a a
12
31
21 2 1 2 3
[H A
[H O ]
[H O ] [H O ] [H O ]33
3 3 3
]=+ + +
+
+ + +
C
K K K K K KH A
a a a a a a
3
3
31
21 2 1 2 3
[ ]
[ ] [ ]A
H O H O2 H A
3 3
2−+ +
=+ +
C K K
K K Ka a a
1 2
21 1 2
[ ]
[ ]
[ ] [ ]HA
H O
H O H OH A 3
3 3
2−+
+ +=
+ +C K
K K Ka
a a a
1
21 1 2
[H A]
H O
H O H O2H A 3
3 3
2=+ +
+
+ +
C
K K Ka a a
[ ]
[ ] [ ]
2
21 1 2
[ ]
[ ]A
H O
HA
3
−+
=+
C KK
a
a
[HA]
[H O ]H O
HA 3
3
=+
+
+
CKa[ ]
[ ]
[ ] [ ]A
H O H OH A
3 3
22 1 2
21 1 2
−+ +
=+ +C K K
K K Ka a
a a a
CK K
K
a a
a
H A
23
2
1 2
23
2
2
2
[A ][H O ]
A ][H OA
= +
+ +
− +
− +−[ ]
[ ]
[ ]
[ ]
[ ] [ ]HA
H O
H O H OH A 3
3 3
2−+
+ +=
+ +C K
K K Ka
a a a
1
21 1 2
CKa
a
H A3
1
2
3
2
HA ][H O
HAK HA
H O ]
= +
+ +
− +
−−
+
[ ]
[ ][ ]
[
64 Equilibrios iónicos y sus aplicaciones analíticas

[2.175]
y que los términos del numerador se correspon-den con cada uno de los de este polinomio de for-ma ordenada, multiplicado por la concentraciónanalítica, en este caso CHnA
. Así, la expresión quepermite calcular la concentración de la especieHnA, presenta en su numerador el primer tér-mino de este polinomio multiplicado por CHnA
,mientras que sería CHnA
Ka1[H3O+]n–1 para [Hn–1A]
y así sucesivamente. De esta manera resulta fácilescribir la expresión correspondiente a cualquierespecie de un ácido poliprótico sin necesidad deuna deducción completa. Así, por ejemplo, laexpresión para calcular la concentración en elequilibrio de la forma más disociada de un áci-do tetraprótico, como es el ácido etilendiami-notetraacético (H4Y, EDTA) (ver capítulo deequilibrios de formación de complejos) respon-de a la ecuación:
[2.176]
B) Cálculo del pH de una disoluciónde un ácido diprótico
En una disolución de un ácido diprótico H2Acuya concentración analítica es CH2A
, pueden dar-se los siguientes equilibrios con sus correspon-dientes constantes ácido-base y balances demasas, cargas y protónico:
Estudio sistemático del equilibrio ácido-basede un ácido débil diprótico
Reacciones y constantes implicadas:
Reacciones ácido-base:
H2A + H2O →← HA– + H3O+ [2.177]
HA– + H2O →← A2– + H3O+ [2.178]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen cinco especies distintas en disolución: H2A, HA–, A2–,H3O
+ y OH– y por lo tanto serán necesarias cinco ecuacio-nes independientes para resolver el problema.
Constantes ácido-base:
[2.163]
[2.164]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH¯] [2.14]
Balance de masas:
CH2A= [H2A] + [HA¯] + [A2¯] [2.179]
Balance de cargas y protónico:
[H3O+] = [HA¯] + 2[A2¯] + [OH¯] [2.180]
Como en casos anteriores, para ácidos cuyaforma molecular es neutra y cuyas formas diso-ciadas están cargadas negativamente, coincidenel balance de cargas y el balance protónico. Eltérmino [A2–] va multiplicado por 2 en ambosbalances, porque cada mol de esta especie endisolución da origen a dos moles de cargas posi-tivas, o bien porque por cada mol de esta espe-cie, que aparece por disociación de H2A, debenaparecer dos moles de iones H3O
+, según se con-sidere el balance de cargas o el balance protóni-co, respectivamente.
Al sustituir cada término de la ecuación del balance protónico por las correspondientesexpresiones que los relacionan con la concen-tración analítica, las constantes de equilibrio y[H3O
+], se obtiene una ecuación general de cuar-to grado, de difícil resolución. Siguiendo la mis-ma sistemática que en el caso de los protolitos
Ka2 =− +
−
[A ][H O ][HA ]
23
Ka1 =− +[HA ][H O ][H A]
3
2
[Y
H O H O H O H O4 H Y
3 3 3 3
4−+ + + +
=+ + + +
][ ] [ ] [ ] [ ]
C K K K K
K K K K K K K K K Ka a a a
a a a a a a a a a a
1 2 3 4
41
31 2
21 2 3 1 2 3 4
[ ] [ ] [ ]
H O H OH O ... ...
3 3
3
+ + −
+ −
+ ++ + +
na
n
a an
a a an
K
K K K K K1
1
1 22
1 2
Capítulo 2: Equilibrios ácido-base 65

monopróticos, podemos obtener ecuaciones mássencillas considerando diversas simplificacionesen la expresión [2.180] del balance protónico.Así:
1. El término [OH–] será despreciable al seruna disolución de un ácido y por tanto ladisolución será ácida por definición.
2. Si se consideran los equilibrios de diso-ciación de H2A, el ácido cede el primerprotón con la extensión que indica Ka1,pero siempre le costará más ceder unsegundo protón, con lo que Ka1 >> Ka2 y[HA–]>>[A2–]. Ello implica que el térmi-no 2 [A2–] en el balance protónico será enla mayoría de los casos despreciable.
De esta manera, la ecuación [2.180] se sim-plifica a:
[2.181]
y teniendo en cuenta la expresión [2.168], se tiene:
[2.182]
en la que el término con Ka2 en el denominador,puede también simplificarse teniendo en cuentaque [H3O
+] >> Ka2, de manera que:
[2.183]
Expresión idéntica a la obtenida para el casode ácidos débiles monopróticos y aplicando lamisma simplificación, es decir:
[2.184]
se obtiene finalmente:
[2.185]
ecuación que es idéntica a la de un ácido mono-prótico. Esto significa que, si las dos constantesde disociación son suficientemente diferentes, unácido diprótico se comporta como un ácidomonoprótico de constante de acidez Ka1.
En algunas ocasiones se observa que [H3O+],
calculada mediante la ecuación simplificada ante-rior, no es mucho mayor que Ka1, pero por logeneral es mucho mayor que Ka2 y el pH es losuficientemente ácido para que [OH–] siga sien-do despreciable frente a la concentración de HA–.En tal caso basta con resolver la ecuación desegundo grado obtenida, ecuación [2.183], quepuede escribirse como:
[2.186]
2.6.2. Bases débiles dipróticas
Un ejemplo de base diprótica sería la sal sódi-ca de un ácido diprótico, Na2A. En agua, estecompuesto, de concentración analítica Csal, esta-ría totalmente disociado en iones Na+, que no tie-nen carácter ácido y A2–, que es la base conjuga-da de un ácido débil. Por lo tanto, se puedeplantear el siguiente estudio sistemático de esteequilibrio ácido-base:
Estudio sistemático del equilibrio ácido-basede una base débil diprótica
Reacciones y constantes implicadas:
Reacciones ácido-base:
A2– + H2O →← HA– + OH– [2.187]
y posteriormente puede llegar a producirse:
HA– + H2O →← H2A + OH– [2.188]
aunque generalmente en una extensión muy inferior.
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
[H O H O3 3 H A2
+ ++ − =] [ ]21 1 0K C Ka a
[H O ]3 H A2
+ = C Ka1
[H O ]3 1+ > Ka
[ ]H O
[H O ]3H A
3
2++
=+
C K
Ka
a
1
1
[ ]
[ ]
[ ]H O
H O
H O [H O ]3H A 3
3 3
2++
+ +=
+ +C K
K K Ka
a a a
1
21 1 2
[H O ] [HA ]3+ −=
66 Equilibrios iónicos y sus aplicaciones analíticas

Existen seis especies distintas en disolución: H2A, HA–, A2–,Na+, H3O
+ y OH– y por lo tanto serán necesarias seis ecua-ciones independientes para resolver el problema.
Constantes ácido-base:
[2.163]
[2.164]
Autoprotólisis del disolvente:
Kw = [H3O+] [OH¯] [2.14]
Balance de masas:
Csal = [H2A] + [HA–] + [A2–]; 2 Csal = [Na+][2.189]
Balance de cargas:
[Na+] + [H3O+] = [HA–] + 2 [A2–] + [OH–]
[2.190]
Balance protónico:
2 [H2A] + [HA–] + [H3O+] = [OH–] [2.191]
A diferencia del caso anterior, el balance decargas difiere del balance protónico, y como vie-ne siendo habitual, en estas condiciones, se uti-lizará el balance protónico como punto de parti-da para obtener una expresión que permita elcálculo del pH de las disoluciones de estas basesdébiles dipróticas. En este balance, el término[H2A] está multiplicado por 2 porque por cadamolécula de esta especie que se forme por pro-tonación de A2– se ganan dos protones.
Al expresar la concentración de todas lasespecies que aparecen en el balance protónicoen función de la concentración analítica, las cons-tantes de equilibrio ácido-base y [H3O
+], se obtie-ne una ecuación general de cuarto grado, de difí-cil resolución. Sin embargo, es fácil simplificar elbalance protónico antes de llevar a cabo estassustituciones. Así:
1. La concentración de ion H3O+ será peque-
ña al tratarse de una disolución de unabase, si ésta no es extremadamente débilo se encuentra muy diluida.
2. La concentración de H2A deberá ser des-preciable frente a la de HA– siempre queKa1 y Ka2 sean suficientemente diferentes.
En estas condiciones el balance protónico setransforma en:
[HA–] = [OH–] [2.192]
y considerando la expresión de [HA–] y Kw, ecua-ciones [2.168] y [2.14], respectivamente, estebalance se transforma en:
[2.193]
Expresión que, al tratarse de una base, pue-de considerarse que [H3O
+] es muy baja y [H3O+]
<< Ka1 con lo que:
[2.194]
o bien:
[2.195]
Si además se cumple que [H3O+] << Ka2, esta
ecuación se simplifica a:
[2.196]
de donde:
[2.197][ ]H O3sal
+ = K KCa w2
CK
K
a
wsal 3
3
H OH O
[ ][ ]
+
+=2
C
K
K
a
wsal 3
3 3
H O ]H O [H O
[[ ] ]
+
+ ++=
2
C K
K K K
Ka
a a a
wsal 3
3 3
H OH O H O
1
1 1 2
[ ][ ] [ ]
+
+ ++=
C K
K K K
Ka
a a a
wsal 3
3 3 3
H OH O [H O ] [H O ]
12
1 1 2
[ ][ ]
+
+ + ++ +=
Ka2 =
− +
−
[A ][H O ][HA ]
23
Ka1 =− +[HA ][H O ][H A]
3
2
Capítulo 2: Equilibrios ácido-base 67

En algunas ocasiones el resultado obtenidoutilizando esta ecuación simplificada indica que[H3O
+] ≈ Ka2. Si el resto de aproximaciones siguensiendo válidas, basta recalcular [H3O
+] median-te la ecuación [2.195], que puede reescribirsecomo:
[2.198]
2.6.3. Anfolitos
Un anfolito es un compuesto que puedeactuar simultáneamente como ácido y como base.Un ejemplo típico de anfolito es la sal ácida deun ácido diprótico, por ejemplo NaHA. Para unadisolución de un anfolito de concentración ana-lítica Canfolito, se puede establecer como en otroscasos el siguiente estudio sistemático:
Estudio sistemático del equilibrio ácido-basede un anfolito
Reacciones y constantes implicadas:
Reacciones ácido-base:
En agua, esta sal está totalmente disociada:
NaHA → Na+ + HA– [2.199]
y el anión HA– puede experimentar las reacciones:
HA– + H2O →← A2– + H3O+ [2.178]
en la que actúa como ácido con constante de equilibrio Ka2 y
HA– + H2O →← H2A + OH– [2.188]
en la que se comporta como una base con un equilibrio regi-do por la constante Ka1.
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Hay un total de seis especies en disolución: H2A, HA–, A2–,Na+, H3O
+ y OH–, lo que obliga a plantear un total de seisecuaciones independientes para resolver el problema.
Constantes ácido-base:
[2.163]
[2.164]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH¯] [2.14]
Balance de masas:
Canfolito = [H2A] + [HA–] + [A2–]; Canfolito = [Na+][2.200]
Balance de cargas:
[Na+] + [H3O+] = [HA–] + 2 [A2–] + [OH–] [2.190]
Balance protónico:
[H2A] + [H3O+] = [A2–] + [OH–] [2.201]
Como en otras ocasiones es interesante rea-lizar simplificaciones en el balance protónico con objeto de obtener la expresión válida para elcálculo del pH de esta disolución. Así, en muchoscasos es posible aceptar la hipótesis de que lasconcentraciones de H3O
+ y de OH– serán des-preciables frente a [H2A] y [A2–] respectivamen-te, ya que al tener una disolución de un anfolito,el medio no será ni fuertemente ácido ni fuerte-mente básico. En estas condiciones el balanceprotónico se simplifica a:
[H2A] = [A2–] [2.202]
y considerando las ecuaciones [2.166] y [2.170]se transforma en:
[2.203]
]C
K K KC K K
K K K
a a a
a a
a a a
anfolito 3
3 3
anfolito
3 3
[H O ][H O [H O ]
[H O ] [H O ]
+
+ +
+ +
+ +=
=+ +
2
21 1 2
1 22
1 1 2
Ka2 =− +
−
[A ][H O ][HA ]
23
Ka1 =
− +[HA ][H O ][H A]
3
2
C K K Kw w asal 3 3H O H O[ ] [ ]+ +− − =22 0
68 Equilibrios iónicos y sus aplicaciones analíticas

Simplificando Canfolito y los denominadores,se tiene:
[2.204]
lo que significa que, para la mayor parte de losanfolitos, el pH de su disolución depende única-mente del valor de las constantes de disociacióny es, por tanto, independiente de la concentra-
ción. Esta expresión simplificada deja de cum-plirse si la concentración analítica del anfolito,Canfolito, o Ka2 son muy pequeñas y si Ka1 es relati-vamente grande.
Si estas premisas no son correctas, la ecua-ción general no puede simplificarse. Un ejem-plo de una disolución donde puede haber pro-blemas de simplificaciones se expone en elrecuadro 2.3.
[H O ]3+ = K Ka a1 2
Capítulo 2: Equilibrios ácido-base 69
Se trata de evaluar las simplificaciones que se puedenllevar a cabo en el cálculo del pH de una disolución de unanfolito. Sea una disolución de NaH2PO4 de concentración0,1 mol L–1 (pKa1 = 2,15; pKa2 = 7,21; pKa3 = 12,32). Elbalance protónico será:
[H3PO4] + [H3O+] = [HPO4
2¯] + 2[PO43¯] + [OH¯] [2.205]
En una simplificación sobre este balance protónico, seráfácil aceptar que probablemente:
[H3PO4] >> [H3O+]
y también que:
[HPO42¯] >> 2[PO4
3¯] + [OH¯]
con lo cual la ecuación se reduce a:
[H3PO4] = [HPO42¯] [2.206]
ecuación similar a la [2.205] que, una vez resuelta da:
[H3O+]2 = Ka1 Ka2 [2.207]
es decir, pH = 4,68 y permite concluir que el pH de la diso-lución de este anfolito no depende de su concentración.
Con este valor de pH no resulta fácil apreciar que se hacometido un error. Sin embargo, al considerar las simplifica-ciones sistemáticas sobre el balance protónico completo se com-prueba que, de hecho, hay que resolver la ecuación
[H3PO4] + [H3O+] = [HPO4
2¯] [2.208]
ya que [H3O+] puede no ser despreciable frente a [H3PO4],
lo que conduce a la expresión final
[2.209]
y a un pH = 4,69.
La influencia del error en [H3O+] puede parecer des-
preciable y en este caso lo es, ya que la diferencia entre elvalor erróneo y el correcto es del orden del 3%. Lo real-mente importante es que el pH de la disolución de este anfo-lito depende de su concentración y el no ser conscientes deello puede provocar errores más graves. Por ejemplo, parauna disolución de NaH2PO4 de concentración 0,01 mol L–1,el pH obtenido mediante la ecuación aproximada [2.207]es de 4,68, mientras que la ecuación exacta [2.209] pro-porciona un pH de 4,80, lo que supone una diferencia supe-rior al 30% en el valor de [H3O
+].
[H O ]
3anfolito
anfolito
+ =+
2 1 2
1
C K KC K
a a
a
RECUADRO 2.3
2.6.4. Sales de ácidos polipróticos
Cuando un ácido diprótico, H2A, reaccionacon una base fuerte, pueden producirse dos salesdistintas, NaHA y Na2A. Estos casos ya han sidocomentados, ya que la primera es un anfolito yla segunda es una base dibásica. Lo mismo ocu-rre con un triprótico. En función de que se tratede la especie NaH2A, Na2HA o Na3A el cálculo
corresponderá al de un anfolito para las dos pri-meras especies o al de una base triprótica.
Es más complicado el caso de las sales de unácido poliprótico con una base débil. Comoejemplo de dificultad relativamente elevada, seconsiderará el cálculo del pH de una disolución0,1 mol L–1 (CP) de (NH4)2HPO4. Como en otrasocasiones se puede establecer el siguiente estu-dio sistemático:
Estudio sistemático del equilibrio ácido-base de una salde ácido triprótico: (NH4)2HPO4
Reacciones y constantes implicadas:
Este compuesto está totalmente disociado en amonio e hidro-genofosfato:
(NH4)2HPO4 → 2NH4+ + HPO4
2¯ [2.210]
Reacciones ácido-base:
NH4+ + H2O →← NH3 + H3O
+ [2.5]
HPO42¯ + H2O →← PO4
3¯ + H3O+ [2.211]
HPO42¯ + H2O →← H2PO4̄ + OH¯ [2.212]
H2PO4̄ + H2O →← H3PO4 + OH¯ [2.213]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH¯ [2.8]
Se tiene ocho especies distintas en disolución: NH4+, NH3,
H3PO4, H2PO4̄ , HPO42¯, PO4
3¯, H3O+ y OH– y la necesidad
de plantear ocho ecuaciones independientes.
Constantes ácido-base:
[2.214]
[2.215]
[2.216]
[2.217]
Autoprotolisis del disolvente:
Kw = [H3O+] [OH¯] = 10¯14 [2.14]
Balances de masas:
Cp = [H3PO4] + [H2PO4̄ ] + [HPO42¯] + [PO4
3¯];
2 CP = [NH4+] + [NH3]
[2.218]
Balance de cargas:
[NH4+] + [H3O
+] = [H2PO4̄ ] + 2[HPO42¯] + 3[PO4
3¯] + [OH¯][2.219]
Balance protónico:
2[H3PO4] + [H2PO4̄ ]+ [H3O+] = [NH3] + [PO4
3¯] + [OH–][2.220]
Sustituyendo las concentraciones de las espe-cies que aparecen en el balance protónico por lasexpresiones que las relacionan con la concen-tración analítica de sal, las constantes de equili-brio y [H3O
+], se obtiene una expresión enor-memente complicada, la cual afortunadamenteno es necesario aplicar en la inmensa mayoría delas ocasiones.
En efecto, se pueden llevar a cabo las siguien-tes simplificaciones en el balance protónico:
1. Los términos [H3O+] y [OH–] serán des-
preciables, ya que es aceptable que la diso-lución de una sal no sea ni fuertemente áci-da ni fuertemente básica.
2. Considerando los equilibrios que tienenlugar es este sistema, debe formarse pri-mero H2PO4̄ a partir de la especie inicialHPO4
2¯. Con una constante muy baja y porprotonación del H2PO4̄ formado, se origi-na H3PO4, con lo que puede considerarseque [H2PO4̄] >> [H3PO4].
3. Si se tiene en cuenta las reacciones [2.5] y[2.211] con Ka >> Ka3, se deduce que [NH3]>> [PO4
3¯].
En estas condiciones, el balance protónicoqueda reducido a:
[2.221]
Sustituyendo estas especies por las expresio-nes que las relacionan con las constantes de equi-librio, las concentraciones de sal CP y [H3O
+], sellega a:
[H PO ] [NH ]2 4 3− =
Ka3
12 3210= =− +
−−[PO ][H O ]
[HPO ]43
3
42
,
Ka2
7 2110= =− +
−−[HPO ][H O ]
[H PO ]42
3
2 4
,
Ka12 1510= =
− +−[H PO ][H O ]
[H PO ]2 4 3
3 4
,
Ka = =+
+−[NH ][H O ]
[NH ]3 3
4
,10 9 24
70 Equilibrios iónicos y sus aplicaciones analíticas

[2.222]
Si se tiene en cuenta que el medio no seráfrancamente ácido ni básico, los términos [H3O
+]3
y Ka1Ka2Ka3 (Ka3 es muy baja) pueden despre-ciarse, con lo que se obtiene la expresión:
[2.223]
En esta expresión, de segundo grado y portanto fácilmente aplicable, puede considerarseque Ka2 > [H3O
+] > Ka condición ya más restrin-gida, que permite su simplificación a:
[2.224]
Esta ecuación demuestra que si CP es lo sufi-cientemente elevada (en este caso CP = 10–1 molL–1), el pH depende únicamente de las reaccionesde disociación del NH4
+ y de formación del H2PO4̄,lo que resulta lógico ya que el NH4
+es un ácido másfuerte que el HPO4
2¯ o el agua y el HPO42¯ es una
base más fuerte que el H2PO4̄ o el agua.En el ejemplo considerado se obtendría
[H3O+] = 8,42 × 10–9, es decir, pH = 8,07. Este
resultado indica que no todas las simplificacio-nes eran correctas, ya que la diferencia entrepKa2 y pH y entre pH y pKa no supera las 1,3unidades logarítmicas que se requieren para queuna simplificación sea considerada válida. Aunasí, es importante hacer notar que Ka1 >> Ka2 ≈[H3O
+] ≈ Ka >> Ka3 y por lo tanto las aproxima-ciones que consideran que las concentracionesde H3PO4, H3O
+, PO43¯y OH– son despreciables
frente a las de H2PO4̄ y NH3, siguen siendo total-mente válidas. Así pues, si interesa obtener unvalor más exacto, basta con resolver la ecuación[2.223] de segundo grado, que da como resulta-do [H3O
+] = 8,71 × 10–9, es decir, pH = 8,06. Este
valor, que difiere en poco más de un 3% delobtenido mediante la ecuación simplificada, con-firma que el resto de las hipótesis eran total-mente válidas.
2.7. Disoluciones reguladoras
Se denominan disoluciones reguladoras aque-llas disoluciones que por su composición admitenla adición de cantidades moderadas de ácido o debase sin modificar de forma significativa su valorde pH. Estas disoluciones, que también reciben elnombre de disoluciones tampón o disolucionesamortiguadoras, presentan una gran importanciaen procesos químicos, biológicos y fisiológicos don-de es necesario mantener los valores de pH de lasdisoluciones dentro de márgenes muy reducidos.
En general, se pueden considerar dos gran-des tipos de disoluciones reguladoras:
1. Disoluciones de ácidos fuertes o de basesfuertes a concentraciones relativamenteelevadas.
2. Disoluciones que contienen un ácido débily su base conjugada a concentracionessemejantes.
En el primer caso, el ácido fuerte o la basefuerte de la disolución actúan como reguladordel pH por su concentración, o sea por la eleva-da cantidad de H3O
+ o de OH– que se tiene en elmedio. La adición de una pequeña cantidad deácido o de base modificará muy poco la concen-tración total de H3O
+ o de OH– presente. En estecaso, la capacidad de amortiguación del pH,depende de la concentración inicial de ácido fuer-te o base fuerte. A mayor concentración de áci-do o de base, se originará una menor variacióndel pH y por tanto mayor amortiguación. En elrecuadro 2.4 se muestra un ejemplo de este tipode disoluciones reguladoras.
Las disoluciones reguladoras más utilizadas,de enorme importancia en los sistemas básicospara la vida que requieren regular el pH, estánconstituidas por un ácido y su base conjugada.
[ ]H O3+ = 2 2K Ka a
C
K
C K
Ka
a
a
P 3
3
P
3
H OH O H O
[ ][ ] [ ]
+
+ ++=
+2
2
C KK K K K K K
C KK
P a
a a a a a a
P a
a
12
31
21 2 1 2 3
2
[H O ][H O ] [H O ] H O
H O
3
3 3 3
3
+
+ + +
+
+ + +=
=+
[ ]
[ ]
Capítulo 2: Equilibrios ácido-base 71

La adición moderada de ácido o base a estasdisoluciones modifica ligeramente su pH. Laacción reguladora de pH de estas disolucioneses especialmente importante si el ácido y baseconjugada están presentes a concentracionesequimoleculares.
Para el cálculo del pH de este tipo de diso-luciones se utiliza frecuentemente la ecuaciónde Henderson-Hasselbalch, que de hecho no esmás que aquella que resulta de la aplicacióndirecta de la correspondiente constante de aci-dez. En efecto para un ácido débil monopróti-co, su constante de acidez viene dada por laexpresión [2.50]:
[2.50]
A partir de ella resulta evidente que:
[2.225]
ecuación que escrita en forma logarítmica tomala forma:
[2.226] pH
[A ][HA]
= +−
pKa log
[H O ]
[HA][A ]3
+−
= Ka
Ka =
− +[A ][H O ][HA]
3
72 Equilibrios iónicos y sus aplicaciones analíticas
Se expone, en este ejemplo, la variación de pH que seproduce cuando se adiciona una pequeña cantidad de NaOHa una disolución de HCl y a otra de agua pura, con objetode constatar la capacidad de amortiguación del pH de ladisolución de HCl. En concreto, se considera la adición de0,1 mL de disolución de NaOH 0,1 mol L–1 a: 1) 100 mL deagua y 2) 100 mL de disolución de HCl 0,1 mol L–1.
1. En este caso, el pH inicial corresponderá al del aguapura, esto es pH = 7,00. Después de la adición de 0,1mL de disolución de NaOH 0,1 mol L–1 a 100 mL de aguase puede establecer el siguiente balance de cargas:
[H3O+] + [Na+] = [OH–]
en el cual se puede despreciar [H3O+] ya que se trata de
la disolución una base. En estas condiciones se tiene:
de donde pH = 10,00. Se ha producido, por tanto,un incremento de pH de 3 unidades.
2. Cuando se adiciona una pequeña cantidad deNaOH a 100 mL de una disolución de HCl 0,1 mol L–1,el pH inicial vendrá dado por:
[Cl–] = [H3O+] = 10–1 mol L–1
y por tanto pH = 1,00. Después de la adición de 0,1mL de disolución de NaOH 0,1 mol L–1 a esta diso-lución de HCl se puede establecer el siguiente balan-ce de cargas:
[H3O+] + [Na+] = [OH–] + [Cl–]
en el cual se puede despreciar [OH–] ya que se tra-ta de la disolución de un ácido fuerte. Las concen-traciones de los iones Cl– y Na+ en este balance vie-nen dadas por:
de donde:
y por tanto pH = 1,001. De lo que se deduce que lavariación de pH es de 0,001 unidades quedandoclaramente constatada la capacidad de amortigua-ción del pH de esta disolución de HCl.
[ ] [ ] – [ ] ,H O Cl Na – 9,99
= 9,98 mol L3
–
1
= = × ×
×
+ − −
− −
9 99 10 10
10
2 5
2
[ ] , ,,
,Na mol L+ 1= ×+
= × − −0 1 0 1100 0 1
9 99 10 5
[ ] ,,
,–Cl mol L 1= ×+
= × − −100 0 1100 0 1
9 99 10 2
[ ]
[ ]–
–
–N OOH
mol L31+ − −= = =Kw 10
1010
14
410
[ ] [ ] , ,,
,–Na OH mol L 1+ − − −= = ×+
= × ≈0 1 0 1100 0 1
9 99 10 105 4
RECUADRO 2.4

Esta expresión permite establecer las siguien-tes conclusiones:
1. Una disolución acuosa que, en el equilibrio,contenga cantidades equimolares de ácidoy base conjugada tiene un pH = pKa. Estaconclusión se deduce fácilmente de estaecuación, ya que la condición [HA] = [A–]implica que el último término valga cero.
2. El pH de la disolución reguladora no cam-bia al diluir la disolución. En efecto, el últi-mo término de dicha ecuación es un cocien-te que no varía al variar la concentración dela disolución, por tanto el pH no variará trasun proceso de disolución. Esto se cumplirásiempre que las disoluciones sean de proto-
litos adecuadamente débiles y lo suficiente-mente diluidas para poder despreciar loscoeficientes de actividad, si no es así debe-rá esperarse un cierto cambio de pH.
3. El pH de la disolución reguladora nocambia apreciablemente al añadir unapequeña cantidad de ácido o de base fuer-te. Para ilustrar esta propiedad se puedeconsiderar el ejemplo mostrado en elrecuadro 2.5.
La ecuación de Henderson-Hasselbalch per-mite una estimación del pH de disoluciones regu-ladoras relativamente sencillas. Sin embargo, paraprofundizar más en este tema y facilitar el cálcu-lo del pH para el caso de ácidos polipróticos o de
Capítulo 2: Equilibrios ácido-base 73
En este ejemplo se trata de evaluar la capacidad de amor-tiguación del pH de una disolución formada por ácido acéti-co y acetato de sodio al añadirle una pequeña cantidad deNaOH y, de manera similar a lo expuesto en el recuadro 2.4,comparar estos resultados con los obtenidos cuando se adi-ciona esa misma cantidad de NaOH a una disolución de aguapura. En concreto, se considera la adición de 0,05 moles deNaOH a: 1) 1 litro de agua y 2) 1 litro de disolución regula-dora preparada con ácido acético y acetato de sodio.
1. En este caso, el pH inicial corresponderá al del aguapura, esto es pH = 7,00. Después de la adición de0,05 moles de NaOH a 1 litro de agua se puedeestablecer el siguiente balance de cargas:
[H3O+] + [Na+] = [OH–]
en el cual se puede despreciar [H3O+], como en el
caso anterior, ya que se trata de la disolución unabase. Se tiene por tanto:
de donde pH = 12,69. Se ha producido, por tanto,una variación de pH de +5,69 unidades.
2. Cuando se adicionan 0,05 moles de NaOH a 1 litrode disolución reguladora preparada con 0,1 mol L–1
de ácido acético (pKa = 4,75) y 0,1 mol L–1 de ace-tato de sodio, el pH inicial se calcula mediante laexpresión [2.229], y dado que las concentracionesde ácido y base conjugada son iguales se obtiene:pH = pKa = 4.75.
Después de añadir 0,05 moles de NaOH y deacuerdo con la reacción ácido-base que origina trasla adición de esta base fuerte:
CH3COOH + NaOH → CH3COO¯ + H2O
se tiene:
[CH3COOH] = 0,1 – 0,05 = 0,05 mol L–1
[CH3COO–] = 0,1 + 0,05 = 0,15 mol L–1
Al sustituir estas concentraciones en la ecuación[2.226] se obtiene:
con lo que la diferencia de pH es de +0,48 unida-des, mostrándose así la capacidad de amortiguacióndel pH de esta disolución reguladora.
pH pKa= + = + =
−
log , log ,,
[CH COO ][CH COOH]
5,233
3
4 75 0 150 05
[H O ]
[OHmol L3
1+−
−− −= = =Kw
] ,,10
0 0510
1412 69
[Na ] [OH ] mol L 1+ − −= = 0 05,
RECUADRO 2.5

mezclas complejas, es mejor realizar un trata-miento sistemático de los equilibrios implicados,siguiendo un esquema semejante al realizadoanteriormente para el cálculo del pH de dife-rentes disoluciones de protolitos. Se ha de teneren cuenta, de todas formas, que en los casos dedisoluciones reguladoras de pH las incógnitassuelen ser las concentraciones de ácido o de baseimplicadas o bien los volúmenes de disoluciónde las mismas que hay que mezclar para conse-guir un determinado valor de pH.
2.7.1. Cálculo del pH de una disoluciónreguladora
Sea una disolución reguladora típica, prepara-da por mezcla de un volumen, VHA, de un ácidodébil monoprótico HA de concentración analíti-ca CHA con un volumen, VNaA, de su base conjuga-da NaA cuya concentración analítica es CNaA. Unavez preparada la disolución reguladora su volu-men final, VT, vendrá dado por: VT = VHA + VNaA.El estudio sistemático de los equilibrios ácido-baseimplicados en la preparación de esta disoluciónreguladora se muestra a continuación.
Estudio sistemático de los equilibrios ácido-base implica-dos en la preparación de una disolución reguladora a partir
de un ácido débil y su base conjugada
Reacciones y constantes implicadas:
Reacciones ácido-base:
NaA → A– + Na+ [2.227]
HA + H2O →← A– + H3O+ [2.49]
A– + H2O →← HA + OH– [2.228]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen cinco especies diferentes en disolución: Na+, HA, A–,H3O
+ y OH–, y por tanto para resolver de forma completa elsistema se necesitan cinco ecuaciones independientes.
Constante de disociación del ácido:
[2.50]
Autoprotolisis del agua:
Kw = [H3O+] [OH–] [2.14]
Balance de masas: se ha de tener en cuenta el ácido CHA yla base añadidos, CNaA
[2.229]
[2.230]
Balance de cargas:
[Na+] + [H3O+] = [OH–] + [A–] [2.231]
Estas expresiones permiten el cálculo del pHque proporciona la disolución reguladora. Eneste caso es mejor utilizar el balance de cargas,en lugar del balance protónico, para que las sim-plificaciones pertinentes no conduzcan a resul-tados absurdos. Así, sustituyendo en la expresióndel balance de cargas, las concentraciones de Na+
(balance de masas), A– y OH– en función de[H3O
+] según las ecuaciones [2.58] y [2.14], res-pectivamente, se tiene:
[2.232]
Esta ecuación se puede simplificar en la mayo-ría de los casos, ya que para que una disolucióntampón sea útil debe ser relativamente concen-trada y raramente se preparan tampones muyácidos o muy básicos. Así pues, en general esposible asumir que las concentraciones de [H3O
+]y [OH–] podrán despreciarse en el balance de car-gas, con lo que éste quedaría como:
CK C K
Kw a
ab 3
3 3
[H O ]H O [H O ]
+ = ++
++ +[ ]
C C CC V C V
V
a b
T
= + = + =
=× + ×
−[HA] [A ]
HA HA NaA NaA
C
C VVb
T
= =×+[Na ] NaA NaA
Ka =
− +[A ][H O ][HA]
3
74 Equilibrios iónicos y sus aplicaciones analíticas

[Na+] = [A–] [2.233]
y al sustituir:
[2.234]
Ecuación que puede reordenarse y teniendoen cuenta que C – Cb = Ca, toma la forma:
[2.235]
expresión idéntica a la que puede obtenerse apartir de la constante de equilibrio, ecuación[2.50]. En el recuadro 2.6 se muestra un ejemplopráctico de disolución reguladora preparada con-forme al procedimiento descrito.
2.7.2. Preparación de disoluciones reguladoras
Hay tres opciones básicas que permiten obte-ner una disolución que contenga cantidades
apreciables de un ácido y de su base conjugada,como se muestra en la figura 2.1:
1. La considerada en el apartado anterior,mezclando un ácido débil y su base conju-gada (HA + NaA).
2. Mezcla de un ácido débil y de una canti-dad inferior de base fuerte (HA + NaOH).
3. Mezcla de una base débil y de una canti-dad inferior de ácido fuerte (NH3 + HCl,NaA + HCl).
En el apartado anterior se ha indicado cómo secalcula el pH para una disolución del tipo 1 y a con-tinuación se muestra el estudio sistemático a rea-lizar para el cálculo del pH de las disoluciones delos dos últimos tipos, y se muestran ejemplos prác-ticos de cada uno de estos casos en el recuadro 2.6.
Sea una disolución reguladora preparada pormezcla de un volumen, VHA, de un ácido débilmonoprótico HA de concentración analítica CHA,con un volumen, VB, de una disolución de NaOHcuya concentración analítica es CB. Una vez pre-parada esta disolución tampón su volumen final,VT, vendrá dado por: VT = VHA + VB. El estudio
[H O ]3a
b
HA HA
NaA NaA
+ = = ××
KCC
KC V
C Va a
CC K
Ka
ab
3H O ]=
++[
Capítulo 2: Equilibrios ácido-base 75
FIGURA 2.1. Formas de preparaciónde 1 litro de una disolución regula-dora 0,1 mol L–1 en ácido acéticoy 0,1 mol L–1 en acetato de sodio.

sistemático de los equilibrios implicados en lapreparación de esta disolución reguladora semuestra a continuación.
Estudio sistemático de los equilibrios ácido-base implicadosen la preparación de una disolución reguladora a partirde un ácido débil y de una cantidad inferior de base fuerte
Reacciones y constantes implicadas:
Reacciones ácido-base:
HA + NaOH → NaA + HA [2.236]
NaA → A– + Na+ [2.227]
HA + H2O →← A– + H3O+ [2.49]
A– + H2O →← HA + OH– [2.228]
Autoprotolisis del disolvente:
H2O + H2O →← H3O++ OH– [2.8]
Existen cinco especies diferentes en disolución: Na+, HA,A–, H3O
+ y OH– y como en el caso anterior, para resolver
de forma completa el sistema se necesitan cinco ecuacio-nes independientes.
Constante de disociación del ácido:
[2.50]
Autoprotolisis del agua:
Kw = [H3O+] [OH–] [2.14]
Balance de masas: se ha de tener en cuenta el ácido CHA yla NaOH añadidos, CB
[2.237]
[2.238]
Balance de cargas:
[Na+] + [H3O+] = [OH–] + [A–] [2.231]
C C VVT
aHA HAHA] [A= + = ×−[ ]
C C VVT
bB B[Na ]= = ×+
Ka =− +[A ][H O ][HA]
3
76 Equilibrios iónicos y sus aplicaciones analíticas
Se muestra en este ejemplo el cálculo del pH que pre-senta diferentes disoluciones reguladoras preparadas con-forme a los casos indicados en la figura 2.1.
Caso 1. La disolución reguladora se prepara mezclan-do la disolución de un ácido HA (VHA = 100 mL, pKa = 5,0 yCHA = 0,09 mol L–1) con la de su base conjugada NaA (VNaA = 110 mL y CNaA = 0,1 mol L–1). Para el cálculo del pHde la disolución resultante se considera la ecuación [2.235]:
y por tanto, pH = 5,08.
Caso 2. Se mezcla un volumen, VHA = 100 mL, de unadisolución de ácido débil pKa= 5,0 y CHA = 0,19 mol L–1,con un volumen, VB = 90 mL, de una disolución de NaOHCB = 0,1 mol L–1. Para calcular el pH de la disolución regu-ladora resultante se hace uso de la ecuación [2.240]:
resultando un pH = 4,95.
Caso 3. Se mezcla un volumen, VNaA = 100 mL, de unadisolución de la base débil NaA, pKa= 5,0 y CNaA = 0,19mol L–1, con un volumen, VHCl = 90 mL, de una disoluciónde HCl, CHCl = 0,1 mol L–1. Para calcular el pH de la diso-lución reguladora resultante se emplea la ecuación [2.247]:
de donde pH = 5,04.
[H O ]3HCl HCl
NaA NaA HCl HCl
+×
− −
=× − ×
=
= ×× − ×
=
KV C
V C V Ca
10 90 0 1100 0 19 90 0 1
105 5 04,, ,
,
[H O ]3HA HA B B
B B
+
− −
= × − ××
=
= × − ××
=
K C V C VC Va
10 100 0 19 90 0 190 0 1
105 4 95, ,,
,
[H O ]3
HA HA
NaA NaA
+ − −= ××
= ××
=K C VC Va 10 100 0 09
110 0 1105 5 08,
,,
RECUADRO 2.6

Como en el caso anterior, el balance de car-gas se puede simplificar a:
[Na+] = [A–] [2.233]
y al sustituir:
[2.239]
y despejar [H3O+] se tiene finalmente:
[2.240]
expresión que permite calcular directamente elpH de estas disoluciones reguladoras.
Como tercer caso, se va a contemplar el cálcu-lo del pH de una disolución reguladora prepara-da por mezcla de una base débil y de una canti-dad inferior de ácido fuerte. Sea, por ejemplo,una disolución reguladora preparada por mezclade un volumen, VNaA, de una base débil mono-prótica NaA de concentración analítica CNaA conun volumen, VHCl, de una disolución de HCl cuyaconcentración analítica es CHCl. Una vez prepa-rada esta disolución reguladora su volumen final,VT, vendrá dado por: VT = VNaA + VHCl. El estu-dio sistemático de los equilibrios implicados enla preparación de esta disolución reguladora semuestra a continuación.
Estudio sistemático de los equilibrios ácido-base implicadosen la preparación de una disolución reguladora a partirde una base débil y de una cantidad inferior de ácido fuerte
Reacciones y constantes implicadas:
Reacciones ácido-base:
NaA + HCl → NaCl + HA [2.241]
NaCl → Cl– + Na+ [2.78]
HA + H2O →← A– + H3O+ [2.49]
A– + H2O →← HA + OH– [2.228]
Autoprotolisis del disolvente:
H2O + H2O →← H3O+ + OH– [2.8]
Existen seis especies diferentes en disolución: Cl–, Na+, HA,A–, H3O
+ y OH– y como en el caso anterior, para resolverde forma completa el sistema se necesitan seis ecuacionesindependientes, aunque para el cálculo del pH sólo sonnecesarias las siguientes.
Constante de disociación del ácido:
[2.50]
Autoprotolisis del agua:
Kw = [H3O+] [OH–] [2.14]
Balance de masas: se ha de tener en cuenta la base CNaA yel HCl añadidos, CHCl
[2.242]
[2.243]
Balance de cargas:
[Na+] + [H3O+] = [OH–] + [A–] + [Cl–] [2.244]
Al igual que en casos anteriores, el balancede cargas se puede simplificar a:
[Na+] = [A–] + [Cl–] [2.245]
y sustituyendo en esta expresión las concentra-ciones implicadas a partir del balance de cargasy de la ecuación [2.58] para la concentración deA– se tiene:
[2.246]CC K
KCa
ab
b
3aH O
=+
++[ ]
C
C VVT
bNaA NaA[Na ] [HA] [A ]= = + =
×+ −
C
C VVT
aHCl HCl[Cl ]= =
×−
Ka =
− +[A ][H O ][HA]
3
[H O ]
3a b
b
HA HA B B
B B
+ = − =
= × − ××
KC C
C
KC V C V
C V
a
a
CC K
Ka
ab
a
3H O=
++[ ]
Capítulo 2: Equilibrios ácido-base 77

y despejando la concentración de H3O+:
[2.247]
Hay que indicar que las expresiones deduci-das para el cálculo del pH de disoluciones regu-ladoras sólo son válidas cuando lo sean las sim-plificaciones efectuadas, es decir para el casomuy general de disoluciones tampón relativa-mente concentradas y a la vez no muy ácidas omuy básicas. Se debe considerar, de todas for-mas, que raramente se resolverá la ecuación[2.232] completa, ya que si la disolución es muyácida el término [OH–] será despreciable o biensi es muy básica, el término [H3O
+] podrá notenerse en cuenta.
• Disoluciones reguladoras constituidaspor un ácido con pKa relativamente bajo
Una excepción al caso más general, ya comen-tado, de disoluciones tampón, se tiene cuando seconsideran disoluciones reguladoras constituidaspor un ácido de pKa relativamente bajo, comopor ejemplo pKa = 2,0, y su base conjugada.
En este caso, en el balance de cargas corres-pondiente no se puede despreciar [H3O
+] portener el ácido el citado valor de pKa, y por tantopara el caso general de una disolución tampónpreparada a partir de un ácido débil monopróti-co (HA) y su base conjugada (NaA) se tendríael siguiente balance de cargas:
[H3O+] + [Na+] = [A–] [2.248]
una vez despreciada la concentración de OH– enel mismo, dado el carácter ácido de la disoluciónreguladora. Sustituyendo en este balance las con-centraciones de Na+ (ecuación [2.229] y A– (ecua-ción [2.58]) y considerando que C = Ca + Cb, setiene:
[2.249]
y al operar se obtiene la siguiente ecuación desegundo grado:
[2.250]
la que expresada en función de las concentra-ciones analíticas y volúmenes de disolución deácido y base conjugada, mezclados para tener ladisolución reguladora, toma la forma:
[2.251]
En el caso práctico de una disolución regula-dora formada por 100 mL de un ácido HA, de pKa= 2,0 y concentración CHA = 0,1 mol L–1 y 100 mLde su base conjugada, NaA, también a concentra-ción 0,1 mol L–1, al aplicar la ecuación simplifica-da [2.235] se obtendría un valor de pH de 2,0 mien-tras que haciendo uso de la ecuación [2.251] el valorde pH es 2,13. Si, por otra parte, la disolución regu-ladora anterior se diluye a un litro con agua desti-lada, el valor de pH que proporciona la mismahaciendo uso de la ecuación [2.251] es de 2,38.
A la vista de los ejemplos anteriores, se obser-va que para casos en que la disolución tampónestá constituida por un ácido con un pKa relati-vamente muy bajo o muy alto, el pH de la diso-lución varía con la concentración y/o dilución.
También se ha de indicar que en estos casostampoco se cumple la condición de pH = pKacuando se mezclan cantidades equimoleculares deácido y base conjugada, tal como se deducía parael caso general, cuando el pKa no es ni muy bajoni muy alto relativamente. Que se obtengan resul-tados de pH = 2,13 o en el caso de la disolucióndiluida de 2,38, distintos de pH = 2,0, se explica sise tiene en cuenta que las concentraciones de HA
[ [H O ] H O ]3NaA NaA
NaA HA3
HA HA
NaA HA
+ ++ + ×+
−
− ×+
=
2
0
KC VV V
C VV V
K
a
a
[ ] ( )H O [H O ]3 b 3 a+ ++ + − =2 0K C C Ka a
[( )[ ]
H O ]H O3 b
a b
3
+++ = +
+C
C C K
Ka
a
[H O ]3a
b a
HCl HCl
NaA NaA HCl HCl
+
×
=−
=
=× − ×
KC
C C
KV C
V C V C
a
a
78 Equilibrios iónicos y sus aplicaciones analíticas

y A– en el equilibrio son, en estos casos, distintasde las iniciales. Así, si se sustituye en las expre-siones [2.58] y [2.55], correspondientes a las con-centraciones de A– y HA, respectivamente en fun-ción del [H3O
+] y Ka, el valor de C se tiene:
[2.252]
[2.253]
y al sustituir en las mismas las condiciones dadasen el caso en que el pH = 2,38, dichas concentra-ciones en el equilibrio son: [A–] = 1,4 × 10–2 molL–1 y [HA] = 5,8 × 10–3 mol L–1.
En cambio para ácidos con pKa ni muy bajo nimuy alto, se cumple que al mezclar el ácido con labase conjugada a la misma concentración, en elequilibrio [HA] = [A–] y pH = pKa, como se esta-blece en la ecuación de Henderson-Hasselbalch.
2.7.3. Capacidad reguladora
Desde un punto de vista semicuantitativo,puede saberse que una disolución reguladora ten-drá una mayor capacidad tampón, cuanto mayorsean las concentraciones de sus componentes,como queda reflejado en el recuadro 2.7.
Desde un punto de vista cuantitativo la capa-cidad reguladora, representada mediante la le-tra β, se calcula a partir de la ecuación:
[2.254]
para la adición de una base, y por
[2.255]
para la adición de un ácido. El signo menos de laexpresión, en este caso, se introduce por razonesformales, ya que un aumento de la concentraciónde ácido fuerte provocará una disminución depH, y una capacidad amortiguadora negativacarece de sentido.
β = − dCd
HA
pH
β = dCd
B
pH
[HA][H O
H O
HA HA NaA NaA
T3
3
=
× + ×
+
+
+
V C V CV
Ka
]
[ ]
[A ]
[H O ]
HA HA NaA NaA
T
3
−+=
× + ×
+
V C V CV
K
K
a
a
Capítulo 2: Equilibrios ácido-base 79
Se trata de evaluar la capacidad reguladora de unadisolución en función de la concentración de los compo-nentes (HA/NaA, pKa = 4,0) de la misma, mediante lasvariaciones de pH que se observan al adicionar a esta diso-lución, por ejemplo, cantidades moderadas de NaOH.
Se va a considerar en todos los casos que esta diso-lución tampón se prepara por mezcla de cantidades equi-moleculares de HA y NaA, por tanto conforme a la ecua-
ción [2.226], pHinicial = pKa = 4,0. Por otra parte, en todoslos casos se supone la adición de 0,01 moles de NaOHcon objeto de observar la variación que se origina en elpH de la disolución tampón. Se obtienen los resultadosque se muestran a continuación (mediante el empleo dela ecuación [2.236]), cuando se llevan a cabo las siguien-tes experiencias haciendo uso de distintas disolucionesreguladoras:
RECUADRO 2.7
Disolución CHA, mol L–1 CNaA, mol L–1 pHinicial pHfinal ∆ pH
1 0,050 0,050 4,0 4,18 + 0,182 0,025 0,025 4,0 4,38 + 0,383 0,015 0,015 4,0 4,70 + 0,70
Se observa que la máxima capacidad reguladora la presenta aquella disolución tampóncon mayor concentración en sus componentes.

Teniendo en cuenta la expresión [2.254], lacapacidad tampón o amortiguadora de pH de unadisolución es el cociente entre la cantidad de baseañadida y la variación de pH provocada en ladisolución al añadir una cantidad infinitesimalde base. Esta capacidad tampón indica, por tan-to, la resistencia de una disolución a los cambiosde pH y que cuanto mayor sea su valor, mayorserá la cantidad de ácido fuerte o de base fuerteque habrá que añadir para provocar una deter-minada variación de pH. Es conveniente estu-diar de qué parámetros depende dicha función ycómo varía al hacerlo el pH.
Supongamos que se dispone de una disoluciónamortiguadora que contiene un ácido débil HA ysu base conjugada, NaA; el balance de cargas será:
[Na+] + [H3O+] = [OH–] + [A–] [2.231]
Al añadir una base fuerte, por ejemploNaOH, aumentarán las concentraciones de Na+,A– y OH– y disminuirá la de H3O
+. Si la adiciónes infinitesimal, podrá aceptarse que:
[2.256]
y al dividir esta ecuación por d pH se obtiene:
[2.257]
que puede interpretarse en el sentido de que, β total de una disolución es una cantidad queresulta de la suma de la capacidad amortigua-dora de la pareja HA–A–, más las de las pare-jas H3O
+–H2O y H2O–OH–. Es decir:
[2.258]
Es conveniente desarrollar ecuaciones quepermitan evaluar cómo varían las distintas capa-cidades amortiguadoras en función del pH.
En el caso de βbase, es decir, de la correspon-diente a la pareja HA–A–, puede deducirse losiguiente:
[2.259]
Derivando la expresión para [A–], conformea la ecuación [2.58]:
[2.58]
con respecto a [H3O+] se tiene:
[2.260]
y al sustituir en la expresión [2.259] resulta final-mente:
[2.261]
Esta expresión indica que la capacidad amor-tiguadora de la pareja HA–A– es directamente pro-porcional a su concentración analítica y depende,aunque no de una manera lineal, de Ka y del pH.Si esta expresión se multiplica y divide por la con-centración C y se separan términos, se obtiene:
[2.262]
β βbase base
3
3 3
H OH O H O
= =
=+ +
+
+ +
CCC
K
C K
K Ca
a
a
2 31
,[ ]
[ ] [ ]
βbase3
3
H O
H O=
+( )+
+2 3
2,
[ ]
[ ]
C K
K
a
a
dd
C K
Ka
a
[ ][ ] [ ]
AH O H O3 3
−
+ += −
+( )2
[ ]
[A
H O ]3
−+
=+
C KK
a
a
βbase 33
, [H O[A
[H O= − +
−
+2 3 ]
]]
d
d
βbase3
3
ApH
[A[H O
,[A[H O
= =−
=
= −
− −
+
−
+
dd
d
d
d
d
[ ] ]log ]]
ln ]2 3
β β β β= + ++ −H O OH base
3
β = = − +
+ +
+ +
− −
dd
dd
dd
dd
[ ] [
[ ] [
NapH
H O ]pH
OHpH
A ]pH
3
d d d d[ ] [ ] [ ]Na H O OH [A ]3+ + − −= − + +
80 Equilibrios iónicos y sus aplicaciones analíticas
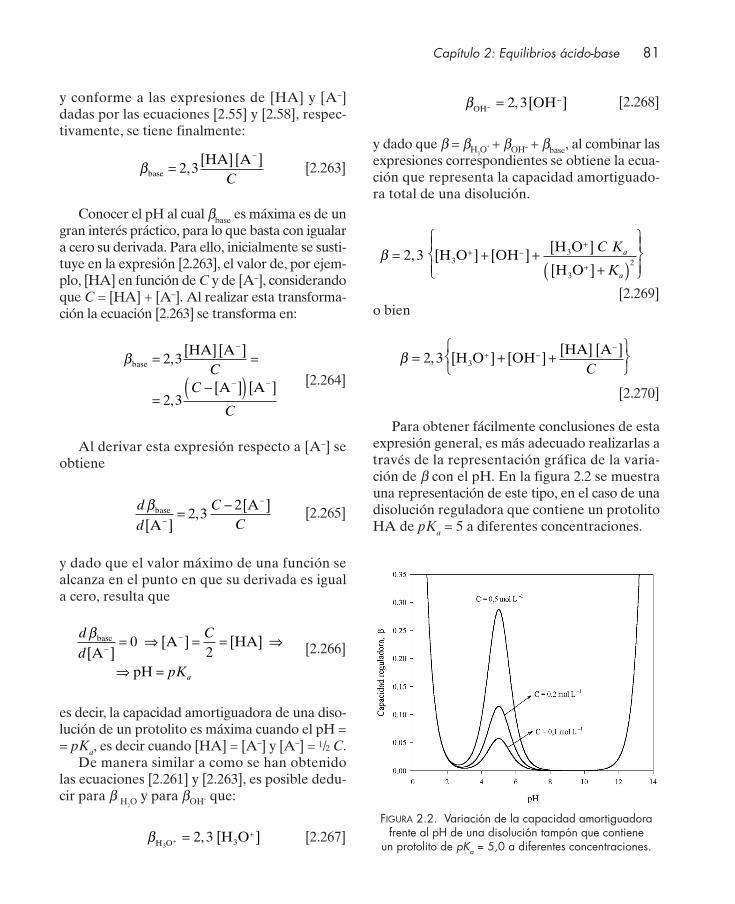
y conforme a las expresiones de [HA] y [A–]dadas por las ecuaciones [2.55] y [2.58], respec-tivamente, se tiene finalmente:
[2.263]
Conocer el pH al cual βbase es máxima es de ungran interés práctico, para lo que basta con igualara cero su derivada. Para ello, inicialmente se susti-tuye en la expresión [2.263], el valor de, por ejem-plo, [HA] en función de C y de [A–], considerandoque C = [HA] + [A–]. Al realizar esta transforma-ción la ecuación [2.263] se transforma en:
[2.264]
Al derivar esta expresión respecto a [A–] seobtiene
[2.265]
y dado que el valor máximo de una función sealcanza en el punto en que su derivada es iguala cero, resulta que
[2.266]
es decir, la capacidad amortiguadora de una diso-lución de un protolito es máxima cuando el pH == pKa, es decir cuando [HA] = [A–] y [A–] = 1/2 C.
De manera similar a como se han obtenidolas ecuaciones [2.261] y [2.263], es posible dedu-cir para β H2O
y para βOH¯ que:
[2.267]
[2.268]
y dado que β = βH3O+ + βOH¯ + βbase, al combinar las
expresiones correspondientes se obtiene la ecua-ción que representa la capacidad amortiguado-ra total de una disolución.
[2.269]o bien
[2.270]
Para obtener fácilmente conclusiones de estaexpresión general, es más adecuado realizarlas através de la representación gráfica de la varia-ción de β con el pH. En la figura 2.2 se muestrauna representación de este tipo, en el caso de unadisolución reguladora que contiene un protolitoHA de pKa = 5 a diferentes concentraciones.
FIGURA 2.2. Variación de la capacidad amortiguadorafrente al pH de una disolución tampón que contiene
un protolito de pKa = 5,0 a diferentes concentraciones.
β = + +
+ −−
2 3,[ ]
[H O ] [OH ]HA] [A
3 C
β = + ++( )
+ −
+
+2, 3 [H O ] [OH ]
H O
H O3
3
3
2
[ ]
[ ]
C K
Ka
a
βOH OH− = −2 3, [ ]
βH O 33
, [H O ]+ = +2 3
d
d
C
pKa
βbase
AA HA]
pH[ ]
[ ] [−−= ⇒ = = ⇒
⇒ =
02
d
d
CC
βbase
AA
[ ],
[ ]−
−
= −2 3
2
βbase
[HA] [A
A A
= =
=−( )
−
− −
2 3
2 3
,]
,[ ] [ ]
CC
C
βbase 2,3HA] [A ]=
−[C
Capítulo 2: Equilibrios ácido-base 81

A partir de las curvas obtenidas es posibleextraer las siguientes conclusiones:
1. La capacidad amortiguadora es máximacuando pH = pKa.
2. El valor de β es adecuado en un intervaloaproximadamente igual a pKa ± 1.
3. β alcanza valores elevados a pH muy áci-do o muy básico, debido a la acción amor-tiguadora de los iones H3O
+ y OH–, res-pectivamente, aunque este valor estotalmente independiente de la concen-tración del protolito.
4. La capacidad tampón aumenta al aumen-tar la concentración de los componentesde la disolución reguladora.
Estas conclusiones son de gran importanciapráctica, ya que indican claramente que, porejemplo, con un protolito de pKa = 5 será posiblepreparar tampones válidos entre pH = pKa ± 1,es decir entre pH = 4 y pH = 6 aproximadamen-te, aunque la β máxima se obtendrá a pH = pKa.Por otra parte, para preparar una disoluciónamortiguadora de, por ejemplo, pH = 6, lo idealseria utilizar un protolito de pKa = 6, si ello esposible, pero cualquier ácido cuyo pKa cumpla lacondición 5 < pKa < 7 permitirá obtener un tam-pón válido, aunque, evidentemente, con una βinferior a la del caso ideal.
2.7.4. Disoluciones amortiguadorascon compuestos polipróticos
Un ácido poliprótico presentará tantas zonastamponadas como pareja ácido débil/base con-jugada sea capaz de formar, es decir, tantas comoprotones disociables. Si las constantes de diso-ciación son muy diferentes, hecho frecuente enestos protolitos, será posible, en la práctica, des-preciar la contribución de los restantes equili-brios y, en consecuencia, considerar el problemacomo si la disolución amortiguadora se hubierapreparado a partir de un ácido débil monopróti-co y su base conjugada.
Así, por ejemplo, el ácido fosfórico, H3PO4,con pKa1 = 2,15, pKa2 = 7,21 y pKa3 = 12,33 per-mite preparar disoluciones reguladoras en lossiguientes intervalos de pH:
pH = pKa1 ± 1 = 2,15 ±1Intervalo de pH ≈ 1,2 – 3,2
pH = pKa2 ± 1 = 7,21 ± 1Intervalo de pH ≈ 6,2 – 8,2
pH = pKa3 ± 1 = 12,33 ±1Intervalo de pH ≈ 11,3 – 13,3
y, por lo tanto, su capacidad amortiguadora ten-drá tres máximos, a los valores de pH que coin-cidan con los distintos pKa. Si, por el contrario,las constantes de disociación no son muy dife-rentes, será necesario tener en cuenta la contri-bución de más de un equilibrio.
En cualquier caso el tratamiento sistemáticoes similar al que se ha comentado en el apartado2.7.2, si bien las ecuaciones podrían ser más com-plicadas. En el recuadro 2.8 se muestra un ejem-plo de preparación de este tipo de disolucionesreguladoras.
2.7.5. Disoluciones amortiguadorasque contienen más de un protolito
Si una disolución tampón contiene más deun protolito cuyas constantes de acidez son muydiferentes, tendrá dos zonas tamponadas clara-mente diferenciadas, teniendo en cuenta en cadauna de ellas el equilibrio ácido-base correspon-diente a cada uno de los protolitos. En cambio,si las constantes de disociación son similares,habrá que considerar la influencia de ambosprotolitos en el cálculo del pH de dicha disolu-ción reguladora.
Por ejemplo, si una disolución contiene dosácidos, HA1 a concentración CHA1
y de constan-te de disociación K1 y HA2, a concentración CHA2
y de constante de disociación K2 y un volu-men Va de esta disolución se mezcla con un
82 Equilibrios iónicos y sus aplicaciones analíticas

volumen VB de una base fuerte, como NaOH,de concentración CB, el balance de cargascorrespondiente será:
[2.271]
Si, como ocurre generalmente, [H3O+] es des-
preciable frente a [Na+] e igualmente [OH–] lo esfrente a [A1̄] y/o [A2̄], la ecuación se reduce a:
[2.272]
tras realizar las sustituciones oportunas para lasconcentraciones de Na+, A1̄ y A2̄. Esta ecuaciónpermite realizar los cálculos en la zona en que lacapacidad amortiguadora de la disolución depen-de de ambos protolitos.
V C
V C K
K
V C K
Ka a
B BHA
3
HA
3[H O ] H O2× =
× ×+
+× ×
++ +1 1
1
2
2[ ]
[Na ] [H O ] [A ] [A ] [OH ]3 1 2+ + − − −+ = + +
Capítulo 2: Equilibrios ácido-base 83
RECUADRO 2.8
Para ilustrar la preparación de disoluciones regula-doras con compuestos polipróticos, se ha tomado comoejemplo una disolución tampón preparada por mezcla deNaH2PO4 y Na2HPO4 y de pH ≈ pKa2. Un planteamientoriguroso para resolver el problema, como en los casosindicados en el apartado 2.7.2, requiere de siete ecua-ciones independientes, ya que pueden existir siete espe-cies distintas en disolución: H3PO4, H2PO4̄ , HPO4
2¯, PO43¯,
Na+, H3O+ y OH–. Sin embargo, como se ha indicado en
otras ocasiones, en la mayoría de las disoluciones amor-tiguadoras puede aceptarse que las concentraciones deH3O
+ y OH– son despreciables frente a las de los demásiones presentes en la disolución, especialmente si la con-centración del protolito es relativamente elevada y el pHno es muy ácido o muy básico, con lo que el balance decargas:
[Na+] + [H3O+] = [H2PO4̄ ] + 2[HPO4
2¯] + 3[PO43¯] + [OH¯]
puede simplificarse si se tiene en cuenta que el equilibrio aconsiderar más relevante será:
H2PO4̄ + H2O →← HPO42¯ + H3O
+
y el pH = pKa2 = 7,21. Con lo que resulta:
[Na+] = [H2PO4̄ ] + 2[HPO42¯]
de donde se deduce fácilmente que:
expresión equivalente a la ecuación [2.235] para el cálcu-lo de pH de disoluciones tampón preparadas a partir de unácido débil monoprótico y su base conjugada.
[H O ]3H PO H PO
HPO HPO
a
b
2 4 2 4
42
42
+ =×
×=
− −
− −
KC V
C VK C
Ca a2 2

84 Equilibrios iónicos y sus aplicaciones analíticas
ANEXO
Expresiones simplificadas para el cálculo del pH de disoluciones de diferentes protolitos
Protolito Ejemplo Expresión
Ácido fuerte HCl
Base fuerte NaOH
Mezcla de ácidos fuertes HCl + HNO3
Mezcla de bases fuertes NaOH + KOH
Ácido débil monoprótico CH3COOH
Base débil monoprótica NH3
Sal de ácido fuerte y base fuerte NaCl
Sal de ácido fuerte y base débil NH4Cl
Sal de ácido débil y base fuerte CH3COONa
Sal de ácido débil y base débil HCOONH4
Mezcla de ácidos fuerte y débil HCl + CH3COOH
Mezcla de dos ácidos débiles CH3COOH + H3BO3
Mezcla de bases fuerte y débil NaOH + NH3
Mezcla de dos bases débiles NH3 + C6H5NH2
Ácido débil diprótico H2CO3
Base débil diprótica Na2CO3
Anfolito NaHCO3 [H O3+ =] K Ka a1 2
[ ]H O3sal
+ = K KCw a 2
[ ]H O3 H CO2 3
+ = C Ka1
K K
NH C H NH4 6 5 3+ +<<
[H O ]3
NH
NH3
+ =+K K
C
w4
[ ]H O3
NaOH
+ = KC
w
K KCH COOH H BO3 3 3
>>
[H O ] 3 CH COOH CH COOH3 3
+ = C K
[H O ]3 HCl+ = C
[H O3 HCOOH NH4
+ = +] K K
[H O ]3CH COOH
sal
3+ =K K
Cw
[H O3 sal NH+ = +] C K
4
[H O ] 10 pH 737+ −= = ⇒ =Kw
[H O ]3
NH
NH
4
3
+ =+K K
C
w
[H O ] 3 CH COOH CH COOH3 3
+ = C K
KC Cw
[ ]H O3NaOH KOH+
= +
[H O3 HCl HNO3
+ = +] C C
[H O ]3
NaOH
+ = KC
w
[H O3 HCl+ =] C
2.1. En disolución acuosa la base B es una base débil. ¿En cuál de los disolventes indicados se incrementaría lafuerza de dicho ácido? (Marcar con una X.)
[ ] Ácido acético[ ] Acetonitrilo[ ] Tetrahidrofurano
2.2. ¿Qué tipo de simplificación se utiliza generalmente para conseguir la expresión final del cálculo del pH dela disolución de un ácido débil monoprótico? ¿Y para una base débil monoprótica? ¿Por qué?
2.3. ¿En qué tipos de protolitos es necesario utilizar el balance protónico en lugar del balance de cargas paracalcular el pH de sus disoluciones?
2.4. Indicar qué sales, de las relacionadas a continuación, no originan variación de pH cuando se disuelven enagua (marcar con una X):
[ ] KCl [ ] NaCH3COO
[ ] NH4Cl [ ] NH4CH3COO
2.5. Identificar las siguientes especies, en relación con sus propiedades ácido-base:
Tipo Reacción en aguaEspecie pKa (Ácido/base/sal) Ácida Neutra Básica
KNO3 HNO3 , ∞KOH, ∞
CaCl2 HCl, ∞Ca(OH)2, ∞
NH4NO2 HNO2, 3,29
NH4+, 9,24
NaH2BO3 H3BO3, 9,24
NaOH, ∞KOH KOH, ∞
2.6. Indicar la variación de pH (con respecto a agua pura) que se origina al disolver las siguientes sales en agua:
Variación de pH
Se incrementa No varía DisminuyeCloruro de sodioAcetato de potasioCloruro de amonioAcetato de amonio
Capítulo 2: Equilibrios ácido-base 85
Cuestiones

2.7. Clasificar los siguientes compuestos químicos (marcar con una cruz en el correspondiente casillero):
Ácido Base Sal AnfolitoNaHCO3
Ba(OH)2
NH4CH3COOH3PO4
KHSO4
2.8. Al diluir al doble una disolución 0,1 M de bicarbonato sódico, el pH de la misma (marcar con una X)
[ ] Disminuye al doble [ ] No varía [ ] Aumenta al doble
¿Por qué?
2.9. ¿Qué datos son imprescindibles para calcular la concentración de todas las especies presentes en unadisolución de ácido fosfórico?
[ ] Concentración inicial del ácido[ ] Ka1 , Ka2 y Ka3
[ ] pH de la disolución[ ] Fuerza iónica[ ] Temperatura
2.10. Se desea preparar una disolución reguladora de pH = 5,0 y se dispone de los siguientes pares ácido-base:
1) Par 1: pKa = 4,0. 2) Par 2: pKa = 4,5. 3) Par 3: pKa = 7,5. ¿Qué par ácido-base utilizaría y por qué?
2.11. Al diluir al doble una disolución reguladora de ácido acético-acetato de sodio (marcar con una X):
[ ] Cambia el pH [ ] Aumenta la capacidad reguladora[ ] Disminuye la capacidad reguladora [ ] Varía la proporción [CH3COOH]/[CH3COO–]
2.12. ¿Cómo se prepararía una disolución reguladora ácido-base de pH 4,0? (Marcar con una X.)
[ ] Mezclando volúmenes iguales de disoluciones equimoleculares de un ácido HAy una base NaA (pKa = 4,0)
[ ] Preparando una disolución de un ácido HA (pKa = 4,0)[ ] Mezclando volúmenes iguales de disoluciones equimoleculares de un ácido HB
y una base NaB (pKa = 7,0)[ ] Mezclando volúmenes iguales de disoluciones equimoleculares de un ácido HA y NaOH (Ka = 10–4)
2.13. Indicar (marcar con una X) las características atribuibles a una disolución reguladora:
[ ] Su pH no varía significativamente al añadir una pequeña cantidad de base[ ] Su pH no varía significativamente al añadir una pequeña cantidad de ácido[ ] Se emplea como estándar químico-analítico primario[ ] Su pH no varía con la dilución
2.14. ¿De qué depende la capacidad reguladora de pH de una disolución? ¿Cómo se cuantifica?
86 Equilibrios iónicos y sus aplicaciones analíticas

• Objetivo pedagógico:Introducción al cálculo del pH de disoluciones dediversos tipos de protolitos (fuertes, débiles y sus mez-clas) así como de las concentraciones de las diferen-tes especies presentes en estos equilibrios. Finalmentese resuelven y proponen ejercicios encaminados a lapreparación de disoluciones reguladoras.
Ácidos y bases (fuertes y débiles) monopróticos
1. Calcular el pH de las siguientes disoluciones: a) HCl0,1 mol L–1 ; b) KOH 0,03 mol L–1 ; c) CH3COOH 0,01 mol L–1 y d) NH3 0,05 mol L–1. CH3COOH (pKa = 4,75), NH+
4(pKa = 9,24).
a) Para el HCl, protolito fuerte, se pueden establecerlos siguientes reacciones ácido-base y balances:
HCl + H2O → H3O+ + Cl–
H2O + H2O←→ H3O
+ + OH–
Balance de masas (BM): CHCl = 0,1 = [Cl–]Balances de carga y protónico (BC): [H3O
+] = [Cl–] + [OH–]
Si se considera que la concentración de ioneshidroxilo es despreciable en el balance protónicoal tratarse de una disolución de un ácido fuerte,al combinar ambos balances se tiene:
[H3O+] = [Cl–] = CHCl = 0,1 mol L–1
de donde pH = 1,0.
b) Para una disolución de una base fuerte como esel caso del hidróxido de potasio de concentra-ción analítica 0,03 mol L–1, de manera similar alcaso anterior se puede establecer:
KOH → K+ + OH–
H2O + H2O←→ H3O
+ + OH–
BM : CKOH = 0,03 = [K+]
BC: [H3O+] + [K+] = [OH–]
En este caso se puede considerar despreciable laconcentración de iones hidronio en balance protó-nico, y al combinar ambos balances se tiene:
[OH–] = [K+] = 0,03 mol L–1
de donde pH = 12,48.
c) Para calcular el pH de esta disolución de ácidoacético se sigue el procedimiento sistemático habi-tual:
CH3COOH + H2O←→ H3O
+ + CH3COO–
H2O + H2O ←→ H3O+ + OH–
BM: = 0,01 =
= [CH3COOH] + [CH3COO–]
BP: [H3O+] = [CH3COO–] + [OH–]
Al tratarse de un ácido se puede despreciar la[OH–] en el balance protónico y sustituyendo enéste la [CH3COO–] obtenida al combinar lasexpresiones del balance de masas y constante deacidez se tiene:
[H3O+] =
Si se desprecia el valor de Ka frente a la [H3O+]
en el denominador de la expresión anterior setiene:
[H3O+] = 10–3,37 mol L–1
de donde pH = 3,37. Esta simplificación es comúnpara la gran mayoría de ácidos débiles monopró-ticos, HA, ya que al estar el equilibrio de diso-ciación de dicho ácido desplazado hacia la izquier-da se cumple que [HA] >> [A–]. Si la expresiónde la constante de disociación de dicho ácido setransforma en:
Ka
[H O ][A ][HA]3
+
−=
0 01 10 4 75, ,× =−
0 01 1010
4 75
4 75
,[
,
,
×+
−
+ −H O ]3
CCH COOH3
Ka = =− +
−[CH COO ][H O ][CH COOH]
3 3
3
10 4 75,
Capítulo 2: Equilibrios ácido-base 87
Seminarios: problemas numéricos

al sustituir en ella la condición anterior, resultaevidente que [H3O
+] >> Ka.Para dar como válido este valor de pH se ha de
comprobar si las simplificaciones realizadas soncorrectas: 1) simplificar la [OH–] en el balanceprotónico es correcto a tenor del valor encontra-do para la misma: 10–10,63 mol L–1 , y 2) el valor deKa (10–4,75) es inferior al 5% del valor encontradopara la [H3O
+], esto es: 10–3,37 > 20 × 10–4,75 = 10–3,45,por lo que esta simplificación también es acepta-ble.
d) Para una base débil como el amoniaco se puedeestablecer:
NH3 + H2O ←→ + OH–
H2O + H2O ←→ H3O+ + OH–
BM: = 0,05 = + [NH3]
BP: [H3O+] + = [OH–]
Al tratarse de una base se puede despreciar la[H3O
+] en el balance protónico y sustituyendo enéste la [CH3COO–] obtenida al combinar lasexpresiones del balance de masas y constante deacidez se tiene:
De manera similar al caso anterior, en éste sepuede despreciar el valor de la [H3O
+] frente a Ka
en el denominador de la expresión anterior, porlo que ésta se transforma en:
[H3O+] = 10–10,97 mol L–1
de donde pH = 10,97. Para dar como válido este valor de pH se ha de
comprobar si las aproximaciones realizadas soncorrectas, especialmente si el valor encontradopara la [H3O
+] es inferior al 5% de Ka. Dado que10–9,24 > 20 × 10–10,97 = 10–9,67 la aproximación esaceptable y el valor de pH correcto.
2. Calcular el pH de las siguientes disoluciones: a) HF0,01 mol L–1; b) HCN 0,1 mol L–1 y c) HCOOH 0,001mol L–1. HF (pKa = 3,52), HCN (pKa = 9,3) y HCOOH (pKa = 3,75).Sol.: a) 2,80; b) 5,15; c) 3,47.
3. Una disolución 0,1 mol L–1 de un ácido débil, HA,tiene un pH de 1,7. Calcular su constante de acidez.Sol.: 2,30.
4. Calcular el pH de una disolución 10–5 mol L–1 de áci-do bórico. H3BO3 (pKa = 9,24).Sol.: 6,90.
5. ¿A qué volumen debe diluirse 1 litro de una disolu-ción de un ácido débil monoprótico HA en agua, paraconseguir que la concentración de iones hidronio seala mitad de la concentración original?
La expresión que permite calcular el pH de una diso-lución de un ácido débil monoprótico en función de suconcentración inicial viene dada por la ecuación [2.23]:
De manera general, para dos condiciones experi-mentales en las que se encuentre el ácido HA se pue-de establecer:
y al aplicar el requisito indicado para la relación deiones hidronio entre ambas
[H3O+]final = 1/2 [H3O
+]inicial
se tiene:
= 1/2
Al operar se llega a la relación:
(CHA) final = 1/4 (CHA) inicial
por lo que al partir de un litro de disolución de HA,ésta se ha de diluir cuatro veces, esto es añadir 3 litrosde agua, siempre que se puedan aplicar las ecuacio-nes simplificadas.
( )C KaHA inicial[ ] ( )H O3 final HA final+ = C Ka
[H O3 final HA final+ =] ( )C Ka
[H O3 inicial HA inicial+ =] ( )C Ka
[H O ]3 HA+ = C Ka
10 100 05
14 9 24− −×=
,
,
0, 05 [H O ][H O ] 10
10[H O ]
3
39,24
14
3
+
+ −
−
++=
[NH ]4+
[NH ]4+CNH3
Ka = = −[NH ][H O ][NH
3 3+
4+]
,10 9 24
NH4+
88 Equilibrios iónicos y sus aplicaciones analíticas

6. Calcular el cambio que experimenta el pH de unadisolución de ácido fórmico de pH 3,0 cuando sediluye con un volumen de agua destilada doble elsuyo. HCOOH (pKa = 3,75).
Antes de efectuar el proceso de dilución se tieneuna disolución de ácido fórmico:
HCOOH + H2O ←→ HCOO– + H3O+ Ka = 10–3,75
de pH = 3,0 y cuya concentración se puede calcularhaciendo uso de la ecuación [2.61]:
dado que Ka no se puede despreciar frente a [H3O+]
en el denominador de esta expresión. En efecto, Ka > 5% [H3O
+]: 10–3,75 > 5 × 10–5. En estas condiciones la concentración inicial de áci-
do fórmico, haciendo uso de esta expresión, vienedada por:
6,62 × 10–3 mol L–1
Al diluir con un volumen de agua destilada dobledel inicial la concentración de ácido fórmico dismi-nuye por un factor de tres, por lo tanto su concen-tración final será:
CHCOOH = (6,62 × 10–3) / 3 = 2,21 × 10–3 mol L–1
El pH de esta disolución se puede calcular hacien-do uso de la expresión ya indicada:
de donde pH = 3,26.
7. Calcular el cambio de pH que se produce en cada delas siguientes disoluciones al diluirlas cinco veces conagua: a) HCl 0,02 mol L–1; b) NaOH 0,3 mol L–1; c) NH3 0,05 mol L–1 y d) CH3COOH 0,1 mol L–1. CH3COOH (pKa = 4,75), (pKa = 9,24).Sol.: a) Se incrementa 0,7 unidades, de 1,7 a 2,4;
b) disminuye 0,7 unidades, de 13,5 a 12,8; c) dis-minuye 0,35 unidades, de 10,97 a 10,62; d) seincrementa 0,35 unidades, de 2,87 a 3,22.
8. Calcular la variación que experimenta el pH de la diso-lución de una base débil B cuando su concentraciónpasa de 10–2 a 10–6 mol L–1. BH+ (pKa = 7,20).Sol.: disminuye 2,09 unidades de pH, esto es de 9,60
a 7,51.
9. ¿Cuál es la relación de ácido y base conjugada en unadisolución de amoniaco de pH 8,5.
(pKa = 9,24).Sol.: .
10. Calcular las concentraciones de las especies presen-tes en el equilibrio de un ácido débil monoprótico,HA, cuya concentración analítica es 0,02 mol L–1 y elpH de 3,5. HA (pKa = 4,5).
De acuerdo con lo expuesto en el apartado 2.5.1 y atenor de los equilibrios correspondientes y del balancede masas y la expresión de la constante, se puede haceruso de las expresiones [2.55] y [2.58] para el cálculo delas concentraciones de HA y A– en el equilibrio:
Dados los valores de [H3O+] y Ka, esta última no se
puede despreciar frente a [H3O+] en el denominador
de estas expresiones (Ka > 5% [H3O+]), por lo que se
han de emplear tal cual. Así:
Sales de protolitos monopróticos
11. Calcular el pH de una disolución 1,0 mol L–1 de clo-ruro de amonio.
(pKa = 9,24).
En disolución el cloruro de amonio, sal de ácido fuer-te y base débil, se encuentra totalmente disociado:
NH4Cl → Cl– + NH4+
NH4+
[A ]0,02 1010 10
1,8 10 molL4,5
3,5 4,53 1−
−
− −− −= ×
+= ×
[HA]0,02 1010 10
1,8 10 molL3,5
3,5 4,52 1= ×
+= ×
−
− −− −
[A ]H O
HA
3
−+
=+
C KK
a
a[ ]
[HA][H O
H OHA 3
3
=+
+
+
CKa
][ ]
[NH ]/[NH ] 5, 54 3+ =
NH4+
NH4+
[H OH O H O3
HCOOH
3 3
++
− −
+ −=
+=
× ×+
][ ]
,[ ]
,
,
C KK
a
a
2 21 10 1010
3 3 75
3 75
⇒ = HCOOHC
1010
10 10 3 HCOOH
3,75
3 3,75−
−
− −=
+⇒C
[H O
H O3HCOOH
3
++
=+
][ ]C K
Ka
a
Capítulo 2: Equilibrios ácido-base 89

En esta disolución se pueden establecer los siguien-tes equilibrios:
Cl– + H2O → No reaccionan: ion sin carácter bási-co (procede de ácido fuerte)
+ H2O ←→ NH3 + H3O+
H2O + H2O ←→ H3O+ + OH– Kw = [H3O
+] [OH–] = 10–14
y los correspondientes balances de masa y protónico:
BM : Csal = 1,0 = [Cl–] = + [NH3]
BP: [H3O+] = [OH–] + [NH3]
Dado que esta sal procede de ácido fuerte y basedébil tendrá carácter ácido en disolución, por lo que[OH–] se podrá despreciar en el balance protónico,quedando éste como: [H3O
+] = [NH3]. Al combinarlas expresiones del balance de masas y constante deacidez, se puede deducir una expresión para la [NH3]que sustituida posteriormente en el balance protóni-co origina:
En esta expresión se puede considerar que [H3O+]
>> para simplificar cálculos, quedando final-mente:
mol L–1
de donde pH = 4,62.Este valor de pH obtenido confirma que la simplifi-
cación realizada es correcta ya que [H3O+] = 10–4,62 >>
= 10–9,24.
12. Calcular el pH de las siguientes disoluciones: a) HCO-ONa 0,1 mol L–1; b) NH4NO3 0,1 mol L–1; c) NH4NO2
0,1 mol L–1; d) CH3COONH4 0,1 mol L–1; e) HCO-ONH4 0,1 mol L–1. HCOOH (pKa =3,75); CH3COOH (pKa = 4,75);
(pKa = 9,24); HNO2 (pKa = 3,29).Sol.: a) 8,37; b) 5,12; c) 6,26; d) 7,0; e) 6,50.
13. Una disolución de cloruro de amonio tiene un pH de4.82. Calcular su concentración analítica.
(pKa = 9,24).Sol.: 0,398 mol L–1.
14. ¿Cuál es la concentracion analítica de una disoluciónde acetato de sodio que tiene un pH de 8,42.CH3COOH (pKa = 4,75).Sol.: 1,23 × 10–2 mol L–1.
15. Una disolución 0,1 mol L–1 de sal sódica de un ácidodébil monoprótico tiene un pH de 10.¿Cuál será elpH de una disolución 0,02 mol L–1 de dicha sal?
Para determinar el pH de una disolución de la salsódica de un ácido débil monoprótico (base débil) sesigue un procedimiento similar al utilizado en proble-mas anteriores, llegando finalmente a la expresión [2.99]
la cual en las condiciones iniciales dadas en el pro-blema permite en este caso calcular la constante deacidez del ácido débil monoprótico del cual se deri-va la sal. De esta forma:
de donde Ka = 10–7. Al comparar el valor obtenido deKa con la [H3O
+] se observa la correcta aplicación dela ecuación [2.99] ya que al ser Ka >> [H3O
+] se cum-ple la condición de simplificación que conduce a lamisma.
Para el cálculo del pH que presenta una disolución0,02 mol L–1 de esta sal, se hace de nuevo uso de laexpresión [2.99], aunque en este caso la [H3O
+] es laincógnita. Así se tiene:
de donde pH = 9,65.
16. Se preparan tres disoluciones de KOH, NH3 y KCN.¿Qué concentración analítica debe tener cada una deellas para que el pH sea en todos los casos 10,5. HCN (pKa = 9,3); (pKa = 9,24).Sol.: [KOH] = 3,16 × 10–4 mol L–1; [NH3] = 6,07 × 10–3
mol L–1; [KCN] = 5,33 × 10–3 mol L–1.
17. Calcular la concentración analítica de las disolu-ciones de los siguientes protolitos: CH3COOH, HCly NH4Cl si todas ellas se encuentran a pH 5,0.
NH4+
[H O10 10
0,022,23 10 molL3
14 710 1+
− −− −= = ×]
1010
0 110
14−
−=
Ka
,
[H O3+ =]
K KC
w a
Sal
NH4+
NH4+
KNH4+
[ ]H O 1,0 10 103 Sal NH
9,24 4,62
4
+ − −= = × =+C K
KNH4+
[H O ]H O ]3
Sal NH
3 NH4
++=
++
+
C K
K4
[
[NH ]4+
KNH3 3
44
[NH ][H O ][NH ]
+ = =+
+−10 9 24,
NH4+
90 Equilibrios iónicos y sus aplicaciones analíticas

CH3COOH (pKa = 4,75); (pKa = 9,24).
Sol.: [CH3COOH] = 1,55 × 10–5 mol L–1; [HCl] = 1,0 × 10–5 mol L–1; [NH4Cl] = 0,17 mol L–1.
18. Se tienen dos disoluciones: una de cianuro de pota-sio cuya concentración es 10–2 mol L–1 y otra de for-miato de sodio. ¿Qué concentración analítica debetener esta última para que la concentración de ioneshidronio de la primera sea 105 veces mayor que la deiones hidroxilos de la segunda?HCN (pKa = 9,3); HCOOH (pKa = 3,75).
En ambos casos se trata de disoluciones de ácidodébil y base fuerte con lo que inicialmente se puedehacer uso de la expresión [2.99] para el cálculo delpH de las mismas:
Para cada una de las disoluciones se tiene:
= 10–10,65 mol L–1
Al sustituir la condición dada por el problema:
y por tanto
[H3O+] = 10–8,35 mol L–1
en esta última ecuación se tiene:
de donde CFormiato sódico = 0,089 mol L–1.
Mezclas de protolitos monopróticos
19. Calcular el pH de las siguientes disoluciones: a) 0,1mol L–1 en HCl y 0,1 mol L–1 en CH3COOH y b) 0,1mol L–1 en HCOOH y 0,1 mol L–1 en CH3COOH. HCOOH (pKa = 3,75); CH3COOH (pKa = 4,75).
a) Los equilibrios ácido-base y balances implicadosen este sistema son los siguientes:
HCl + H2O → Cl– + H3O+
CH3COOH + H2O←→ CH3COO– + H3O
+
H2O + H2O←→ H3O
+ + OH–
BM: CHCl = 0,1 = [Cl–]
BP: [H3O+] = [OH–] + [Cl–] + [CH3COO–]
En el balance protónico se pueden despreciarlas concentraciones de OH– (se trata de una mez-cla de ácidos y por tanto su pH será ácido) y deCH3COO– frente a la de Cl– ya que este últimoprocede de un ácido fuerte y los ácidos están a lamisma concentración inicial.
En tal caso se tiene:
BP: [H3O+] = [Cl–] = CHCl = 0,1 mol L–1
de donde pH = 1,0.
b) En esta mezcla de ácidos débiles se pueden esta-blecer los siguientes equilibrios ácido-base ybalances:
HCOOH + H2O←→ HCOO– + H3O
+
CH3COOH + H2O←→ CH3COO– + H3O
+
H2O + H2O←→ H3O
+ + OH–
BM:
BP: [H3O+] = [OH–] + [HCOO–] + [CH3COO–]
Realizando las sustituciones oportunas en elbalance protónico de las concentraciones deHCOO– y CH3COO– (por combinación del balan-ce de masas y su constante de acidez) y despre-ciando la de OH– ,como en el caso anterior, setiene:
Al despreciar Ka en el denominador de ambostérminos frente a [H3O
+], como es común paraácidos débiles monopróticos, y reagrupando tér-minos se llega a:
[H O ]
[H O ] H O ]3CH COOH CH COOH
3 CH COOH
HCOOH HCOOH
3 HCOOH
3 3
3
++ +
=+
++
C K
KC K
K[
CCH COOH 3 330,1 [CH COOH] [CH COO ]= = + −
CHCOOH [HCOOH] [HCOO ]= = + −0 1,
CCH COOH 3 33[CH COOH] [CH COO ]= = + −0 1,
1010 108 35
14 3 75−
− −
= ×,,
CFormiato sódico
[OH Formiato sódico− −=] ,10 5 65
[ ] [ ],H O OH3 Cianuro sódico Formiato sódico+ − −= =10 1010 65 5
[H O ]3 Formiato sódico
Formiato sódico
+− −
= ×10 1014 3 75,
C
[H O ]3 Cianuro sódico+
− −
−=
,10 1010
14 9 3
2
[H O ]3Sal
+ = K KC
w a
NH4+
Capítulo 2: Equilibrios ácido-base 91

y sustituyendo los valores dados se transforma en:
de donde pH = 2,35.
20. Calcular el pH de las siguientes disoluciones: a) 0,05mol L–1 en HCl y 0,1 mol L–1 en HF; b) 0,1 mol L–1 enCH3COOH y 0,075 mol L–1 en NH4Cl; c) 0,15 mol L–1
en HF y 0,1 mol L–1 en CH3COOH. HF (pKa = 3,52); CH3COOH (pKa = 4,75); (pKa = 9,24).Sol.: a) pH = 1,30; b) 2,87; c) 2,16.
21. Calcular el pH de la disolución que se obtiene al mez-clar 10 milimoles de un ácido débil monoprótico (HA)con 5 milimoles de otro ácido (HB) en un litro de agua. HA (pKa = 3,75); HB (pKa = 7,5).Sol.: 2,90.
22. Calcular el pH de las siguientes disoluciones: a) 0,1mol L–1 en NaOH y 0,05 mol L–1 en NH3; b) 0,1 molL–1 NaH2BO3 y 0,05 mol L–1 NaNO2.
(pKa = 9,24) ; H3BO3 (pKa = 9,24); HNO2 (pKa
= 3,29).Sol.: a) 13,0; b) 11,12.
23. Calcular el pH de una mezcla de 40 mL de amonia-co 0,05 mol L–1 y 10 mL de metilamina 0,1 mol L–1.
(pKa = 9,24); (pKa = 10,66).
Al mezclar ambas disoluciones sus concentracionesanalíticas tras la disolución que se origina viene dada por:
El estudio de este sistema ácido-base implica lossiguientes equilibrios y balances:
NH3 + H2O←→ + OH–
CH3NH2 + H2O←→ + OH–
H2O + H2O←→ H3O
+ + OH–
BM:
BP:
Al sustituir los términos del balance protónico enfunción de las concentraciones analíticas y constan-tes de equilibrio y al despreciar [H3O
+] frente a [OH–]por tratarse de una mezcla de bases, se tiene:
Al ser la metilamina una base más fuerte que elamoniaco y conforme a las concentraciones analíti-cas de ambas se tiene que:
y por tanto la expresión anterior se transforma en:
cuya resolución proporciona un valor de [H3O+] de
3,63 × 10–12 mol L–1 y por tanto un pH = 11,44.
24. Calcular el pH de una disolución que contiene 0,03mol L–1 de metilamina y 0,01 mol L–1 de amoniaco.
(pKa = 10,66), (pKa = 9,24).Sol.: 11,55.
25. Calcular el pH de la disolución resultante al: a) mez-clar 60 mL de HCl 6,0 mol L–1 con 75 mL de Ba(OH)2
3,0 mol L–1 ; b) diluir a 100 mL una mezcla de 50 mLde ácido acético 0,1 mol L–1 y 20 mL de NaOH 0,1mol L–1. CH3COOH (pKa = 4,75).Sol.: a) 13,82; b) 4,57.
26. Calcular el pH de la disolución obtenida al mezclar1 mol de HCl, 1 mol de Ba(OH)2 y 1,5 moles de clo-ruro de amonio, diluyendo esta mezcla en agua has-ta obtener un litro de disolución.
(pKa = 9,24).Sol.: 9,54.
27. Calcular el pH de las siguientes disoluciones: a) 0,1mol L–1 en NH4Cl y 0,05 mol L–1 en NaNO2 y b) ladisolución que se obtiene al mezclar volúmenes igua-
NH4+
NH4+CH NH3 3
+
C
KKwCH NH 3
3 CH NH 3
3 2
3 2
[H O ]
[H O ] [H O ]
+
+ ++=
C KNH NH3 3/ , / ,,= = ×−0 02 10 3 47 109 24 7
C KCH NH CH NH3 2 3 2/ , / ,,= = ×−0 04 10 1 82 1010 66 9
C
K
C
KKwNH 3
3 NH
CH NH 3
3 CH NH 3
3
3
3 2
3 2
[H O ]
[H O ]
[H O ]
[H O ] [H O ]
+
+
+
+ +++
+=
[NH ] [CH NH ] [H O ] [OH ]4 3 3 3+ + + −+ + =
CCH NH 3 3 3 23 20, 02 [CH NH ] [CH NH ]= = ++
CNH 4 330, 04 [NH ] [NH ]= = ++
CH NH3 3+
NH4+
[CH NH ]
10 0,150
0, 02 mol L3 31+ −=
×=
[NH ]
40 0, 0550
0, 04 mol L31=
×= −
CH NH3 3+NH4
+
NH4+
NH4+
[H O ] mol L31+ − − − − − −= × + × =10 10 10 10 101 4 75 1 3 75 4 7, , ,
[H O ]3 CH COOH CH COOH HCOOH HCOOH3 3+ = +C K C K
92 Equilibrios iónicos y sus aplicaciones analíticas

les de ácido trimetilacético 0,2 mol L–1 y amoniaco0,2 mol L–1.
(pKa = 9,24); HNO2 (pKa = 3,29); (CH3)3CCO-OH (pKa = 5,03).Sol.: a) 6,11; b) 7,14.
Protolitos polipróticos
28. Calcular el pH de las siguientes disoluciones: a) áci-do tartárico 0,1 mol L–1; b) NaHCO3 0,1 mol L–1; c)H3PO4 0,1 mol L–1. Ácido tartárico, H2T (pKa1 = 3,04, pKa2 = 4,36), H2CO3
(pKa1 = 6,35, pKa2 = 10,33), H3PO4 (pKa1 = 2,15, pKa2
= 7,21, pKa3 = 12,33).
a) Para este ácido diprótico, H2T, se pueden esta-blecer los siguientes equilibrios ácido-base:
H2T + H2O ←→ HT– + H3O+
HT– + H2O ←→ T2– + H3O+
H2O + H2O ←→ H3O+ + OH–
Los balances de masa y protónico son:
BM:
BP:
En este balance protónico se puede despreciarla concentración de OH– al tratarse de la disoluciónde un ácido, así como la de T2– frente a la de HT–
ya que Ka1/Ka2 > 20. Al sustituir en el mismo [HT–]en función de la concentración analítica, de H3O
+
y de las constantes de acidez se tiene la expresión:
en la que se puede despreciar el término con Ka2
en el denominador quedando:
La resolución de esta ecuación de segundo gra-do proporciona un valor de pH = 2,04.
b) En este caso se trata de la disolución de un anfo-lito. Mediante el uso de la sistemática habitual se
puede llegar al siguiente balance protónico sim-plificado:
el cual se transforma, conforme a lo indicado enel apartado 2.6.3, en:
quedando finalmente:
y por tanto pH = 8,34.
c) Para calcular el pH de esta disolución de ácido fos-fórico y considerando los valores relativos de susconstantes de acidez, el problema se puede enfo-car como si se tratase de un ácido monoprótico ypor tanto la expresión para el cálculo del pH sería:
Al sustituir en la misma los valores de concen-tración analítica y Ka1 y resolver la ecuación desegundo grado resultante, se tiene un pH = 1,63.
29. Calcular el pH de las siguientes disoluciones: a) ftala-to ácido de potasio 0,1 mol L–1; b) Na3PO4 0,1 mol L–1;c) NH4HCO3 0,1 mol L–1; d) (NH4)2HPO4 0,1 mol L–1. Ácido ftálico, H2Ft (pKa1 = 2,95, pKa2 = 5,41), H2CO3
(pKa1 = 6,35, pKa2 = 10,33), H3PO4 (pKa1 = 2,15, pKa2
= 7,21, pKa3 = 12,33). Sol.: a) 4,18; b) 12,56; c) 7,78; d) 8,06.
30. Indicar cuáles son las especies mayoritarias en lasdisoluciones que se obtienen cuando se mezclan: a)20 mL de H2CO3 10–2 mol L–1 + 10 mL de NaOH 10–2
mol L–1; b) 20 mL de H2CO3 10–2 mol L–1 + 20 mL deNaOH 10–2 mol L–1; c) 20 mL de H2CO3 10–2 mol L–1
+ 40 mL de NaOH 10–2 mol L–1; d) 20 mL de H2CO3
10–2 mol L–1 + 20 mL de Na2CO3 10–2 mol L–1; e) 20mL de H2CO3 10–2 mol L–1 + 20 mL de NaHCO3 10–2
mol L–1.
[H O ]
[H O ]3H PO 1
3 1
3 4++
=+
C K
Ka
a
[H O ]
molL3 1 2
1
+ − −
− −
= = × ==
K Ka a 10 10
10
6 35 10 33
8 34
. .
,
=+ ++ +
C K K
K K Ka a
a a a
anfolito 1 2
32
1 3 1 2[H O ] [H O ]
C
K K Ka a a
anfolito 32
32
1 3 1 2
[H O ][H O ] [H O ]
+
+ ++ +=
[H CO ] [CO ]2 3 32= −
[H O ]
[H O ]0,1 10
[H O ] 103H T 1
3 1
3,04
33,04
2++
−
+ −=
+=
×+
C K
Ka
a
[H O ][H O ]
[H O ] [H O ]3H T 1 3
32
1 3 1 2
2++
+ +=
+ +C K
K K Ka
a a a
[H O ] [HT ] 2 [T ] [OH ]32+ − − −= + +
CH T 22
20,1 [H T] [HT ] [T ]= = + +− −
NH4+
Capítulo 2: Equilibrios ácido-base 93

H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: a) H2CO3/NaHCO3; b) NaHCO3; c) Na2CO3;
d) NaHCO3; e) H2CO3/NaHCO3.
31. En una disolución de etilendiamina (NH2CH2CH2
NH2 = B), ¿cuáles son las especies predominantes apH: a) 2,00; b) 7,52; c) 9,09; d) 10,65; e) 13,0?
(pKa1 = 7,52, pKa2 = 10,65).Sol.: a) H2B
2+; b) H2B2+, HB+; c) HB+; d) HB+, B; e) B.
32. Calcular el pH de una disolución de ácido carbónico0,05 mol L–1 a partir de los siguientes datos: a) el pHde una disolución 0,1 mol L–1 de bicarbonato de sodioes 8,34 y b) el pH de una disolución de carbonato desodio 0,1 mol L–1 es 11,67.
Para calcular el pH de la disolución dada de ácidocarbónico es necesario conocer las constantes de aci-dez de este protolito. Aunque el problema no pro-porciona como dato el valor de las mismas, sí ofrecela posibilidad de su cálculo a partir de dos experien-cias. En efecto, en el apartado a) al tratarse de unadisolución de un anfolito y considerando que el pHde la misma responde a la expresión:
Por otra parte, en el apartado b) se conoce el pH deuna disolución de carbonato de sodio, base débil dipro-tónica, el cual se calcula a partir de la expresión [2.200]:
Al sustituir en ambas expresiones los valores depH y de concentración analítica de carbonato de sodiose llega al siguiente sistema de ecuaciones:
cuya resolución proporciona los siguientes valores depKa: pKa1 = 6,35 y pKa2 = 10,34.
Para calcular el pH de la disolución de ácido car-bónico, teniendo en cuenta los valores de sus cons-tantes de acidez, se puede tratar como un ácido mono-prótico débil y hacer uso de la expresión:
de donde pH = 3,82.
33. Calcular el pH de una disolución 0,1 mol L–1 del ácidoH2A a partir de los siguientes datos experimentales:a) el pH de una disolución 0,05 mol L–1 de A2– es 10,0y b) el pH de una disolución 0,1 mol L–1 de HA– es 6,0.Sol.: 2,85.
34. Calcular la concentración de cada una de las especies:H2CO3, y en una disolución de pH 7,0 en la que [H2CO3] + [ ] + = 10–2 mol L–1.H2CO3 (pKa1 = 6,35, pKa2 = 10,33).
Dado que se dispone de la concentración analíticade esta disolución y del pH de la misma, para calcularlas concentraciones de las especies presentes en la mis-ma derivadas del ácido carbónico se puede hacer usode las expresiones generales deducidas en el apartado2.6.1.A correspondiente al cálculo de las concentra-ciones en el equilibrio para un ácido débil diprotónico.Estas expresiones son:
Al sustituir en estas expresiones los valores cono-cidos de [H3O
+], Ka1 y Ka2 se tienen los siguientes valo-res de concentración para cada una de las especies:
1,83 × 10–3 mol L–1
8,16 × 10–3 mol L–1
3,82 × 10–6 mol L–1
35. El pH de una disolución 0,1 mol L–1 de ftalato ácidode potasio (KHFt) es 4,18 y el pH de una disolución0,05 mol L–1 de ftalato de potasio (K2Ft) es 9,05. Cal-cular: a) El pH de una disolución 0,1 mol L–1 de áci-do ftálico y b) las concentraciones de los iones Ft2– yHFt– así como de H2Ft cuando el pH sea 4,0 y la con-centración analítica de ácido ftálico es de 0,1 mol L–1.Sol.: a) 1,98; b) [H2Ft] = 7,90 × 10–3 mol L–1; [HFt–] =
8,86 × 10–2 mol L–1; [Ft2–] = 3,45 × 10–3 mol L–1.
36. En una disolución de carbonato de sodio en la que[H2CO3] +[HCO–
3] + [CO2–3 ] = 0,1 mol L–1, ¿a qué valor
de pH será [CO2–3 ] = 10–2 mol L–1?
H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: 9,38.
[CO ]32− =
[HCO ]3− =
[H CO ]2 3 =
[ ]
[ ] [ ]CO
H O H O32–
3 3
=+ ++ +
C K KK K K
a a
a a a
1 2
1 1 2
[HCO ]
[H O ][H O ] [H O ]3
1 3
32
3
−+
+ +=
+ +C K
K K Ka
a a a1 1 2
[H CO ]
[H O ][H O ] [H O ]2 3
32
32
1 3 1 2
=+ +
+
+ +
CK K Ka a a
[CO ]32−HCO3
−
CO32−
HCO3−
[H O ] 0,05 10 1,51 10 molL3 H CO 16,35 4 1
2 3
+ − − −= = × = ×C Ka
10
1011 6714
−−
=, Ka2
0,1
10–8,341 2= K Ka a
[H O ]32
CO32–
+ = K KC
a w
[H O ]3 1 2+ = K Ka a
BH22+
94 Equilibrios iónicos y sus aplicaciones analíticas

37. Se mezclan 50 mL de carbonato de sodio 0,02 mol L–1
y 50 mL de ácido sulfúrico 0,01 mol L–1. Calcular: a)el pH de la disolución resultante; y b) determinar laconcentración de cada una de las especies presentesen la disolución. H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: a) 8,34; b) [SO2
–4] = 5,0 × 10–3 mol L–1, [CO2–3 ] =
1,02 × 10–4 mol L–1, [HCO–3] = 9,78 × 10–3 mol L–1,
[H2CO3] = 1,02 × 10–4 mol L–1, [Na+] = 0,02 mol L–1.
38. A 25 mL de una disolución de ácido maléico 0,1 molL–1 se añaden 25 mL de una disolución 0,2 mol L–1
de NH3. Calcular el pH de la disolución resultante. Ácido maleico, H2M (pKa1 = 1,92, pKa2 = 6,22).Sol.: 7,58.
39. Calcular el pH de las disoluciones que se obtienen almezclar 50 mL de una disolución 0,1 mol L–1 de un áci-do triprótico H3A con los siguientes volúmenes deNaOH 0,1 mol L–1: a) 25 mL; b) 50 mL; c) 75 mL; d)100 mL; e) 125 mL; f) 150 mL; g) 200 mL. H3A (pKa1 = 4,0; pKa2 = 7,0; pKa3 = 10,0).Sol.: a) 4,0; b) 5,5; c) 7,0; d) 8,5; e) 10,0; f) 11,2; g) 12,3.
40. Calcular el pH de una disolución que se obtiene almezclar 50 mL de una disolución de ftalato ácido depotasio 0,1 mol L–1 con: a) 25 mL de HCl 0,1 mol L–1
y b) 25 mL de HCl 0,2 mol L–1. Ácido ftálico, H2Ft (pKa1 = 2,95, pKa2 = 5,41).Sol.: a) 2,95; b) 1,82.
Disoluciones reguladoras
41. Se desea preparar una disolución reguladora de pH= 5,0. a) ¿Cuántos mL de acetato de sodio 0,1 mol L–1
se han de añadir a 10 mL de ácido acético 0,1 mol L–1
para obtener dicha disolución?, b) ¿cuántos mL dehidróxido de sodio 0,1 mol L–1 habría que añadir alos 10 mL de ácido acético para obtener el mismopH?, c) ¿cuál debería ser el volumen de NH3 0,1 molL–1? Justificar los resultados obtenidos. CH3COOH (pKa = 4,75), NH+
4 (pKa = 9,24).
a) Los equilibrios ácido-base implicados en la pre-paración de esta disolución reguladora son lossiguientes:
CH3COOH + H2O ←→ CH3COO– + H3O+
CH3COO– + H2O ←→ CH3COOH + OH–
H2O + H2O ←→ H3O+ + OH–
Se pueden establecer además los siguientesbalances de masa y de carga:
BM:
BC: [H3O+] + [Na+] = [CH3COO–] + [OH–]
En general, en estos balances de carga se pue-de despreciar [H3O
+] y [OH–] frente a las con-centraciones de los otros componentes, con lo cualse simplifica a:
BC: [Na+] = [CH3COO–]
Sustituyendo las concentraciones de Na+ y deCH3COO– a partir de los balances de masa y con-siderando en el caso de ion acetato la expresiónde la constante de acidez, se tiene:
o bien
En estas expresiones se puede simplificar eldenominador (VAcetato + 10), y por tanto:
de donde VAcetato = 17,76 mL.
b) En este caso se tienen los siguientes equilibriosácido-base con sus correspondientes balances:
CH3COOH + H2O ←→ CH3COO– + H3O+
NaOH → Na+ + OH–
H2O + H2O ←→ H3O+ + OH–
BM:
BC: [H3O+] + [Na+] = [CH3COO–] + [OH–]
C
VCH COOH 3 3NaOH
3[CH COOH] [CH COO ]= + =
×+
− 10 0 110
,
CVVNaOH
NaOH
NaOH
[Na ]= =×+
+ 0 110,
V
VAcetato
Acetato+ =× +[ ]
+
−
− −10
10 1 10
10 10
4 75
5 4 75
( ) ,
,
VV
VV V
Acetato
Acetato
Acetato
Acetato Acetato×+
=
×+
+ ×+
×
+
−
− −
0 110
0 110
10 0 110
10
10 10
4 75
5 4 75
,, , ,
,
[Na ]
H O3
++
=+
C KK
a
a[ ]
C C C= + = + −NaCH COO CH COOH 3 33 3
[CH COOH] [CH COO ]
C
VCH COOH 3 Inicial 3 InicialAcetato
3[CH COOH] CH COO= + = ×
+−[ ]
,10 0 110
CVVNaCH COO
Acetato
Acetato3
[Na ]= = ×+
+ 0 110,
Capítulo 2: Equilibrios ácido-base 95

De forma similar al caso anterior, este balancede cargas se simplifica a:
BC: [Na+] = [CH3COO–]
y sustituyendo las concentraciones de Na+ y deCH3COO– se tiene igualmente:
de donde VNaOH = 6,40 mL.
c) Cuando se adiciona amoniaco a la disolución deácido acético para conseguir la disolución regu-ladora, se pueden establecer los siguientes equi-librios y balances:
CH3COOH + H2O ←→ CH3COO– + H3O+
NH3 + H2O ←→ + OH–
H2O + H2O ←→ H3O+ + OH–
BM:
BC: [H3O+] + = [CH3COO–] + [OH–]
Al simplificar este balance y sustituir las con-centraciones implicadas en el mismo se tiene: BC: = [CH3COO–]
A tenor del valor de pH de la disolución regu-ladora se puede despreciar frente a [H3O
+]
y por tanto la expresión anterior se transformaen:
con lo que
Dado que a este valor de pH el amoniaco aña-dido se encuentra prácticamente como ion amo-nio, esto es, totalmente disociado, su comporta-miento es similar al de una base fuerte y de aquíque el volumen necesario del mismo para formarla disolución reguladora sea idéntico al encon-trado cuando se utiliza una disolución de NaOH.
42. ¿Qué volumen de disolución de NaOH 6,0 mol L–1
debe añadirse a 500 mL de ácido acético 0,25 mol L–1
para que resulte una mezcla de pH 5,0?CH3COOH (pKa = 4,75).Sol.: 13,34 mL.
43. Se necesita preparar una disolución reguladora depH 3,20 y se dispone de dos ácidos débiles HA1 y HA2
y de sus sales alcalinas. ¿Qué ácido y que sal y en quéproporción se han de mezclar para preparar dichadisolución tampón. HA1 (pKa = 3,0), HA2 (pKa = 5,0).Sol.:
44. a) ¿Qué volumen de HCl 5,0 mol L–1 debe añadirsea 1.500 mL de una disolución de acetato de sodio 0,2mol L–1 para preparar una disolución tampón de 4,0?b) ¿Qué cantidad de H+, expresada en milimoles, exis-tirá en la disolución final tras añadir el HCl? c) ¿Cuálserá el pH si esta disolución se diluye con agua des-tilada hasta 2.000 mL?CH3COOH (pKa = 4,75).Sol.: a) 50,94 mL; b) 0,155 milimoles; c) 4,0.
45. Una disolución reguladora de pH 5,35 contiene ácidoacético y acetato de potasio, siendo el número total demoles de ácido y de sal 0,15. Calcular el número demoles de ácido acético y de acetato de sodio separa-damente. CH3COOH (pKa = 4,75).Sol.: 0,12 moles de KCH3COOH y 0,03 moles de
CH3COOH.
46. Determinar el volumen de HCl 0,2 mol L–1 que sedebe añadir a 50 mL de dietanolamina (R2NH) 0,2mol L–1 para obtener una disolución tampón de pH= 8,6.
(pKa = 8,90).Sol.: 33,3 mL.
47. Indicar, razonando la respuesta, cuáles de las siguien-tes mezclas corresponden a disoluciones tampón ycuales son las especies predominantes: a) 0,1 mol L–1
de NH4Cl y 0,1 mol L–1 de NH3; b) 0,1 mol L–1 de HCl
R NH2 2+
[A ]/[HA ] 1, 58.1 1− =
VNH36, 40 mL.=
VNH3
0 110 0 1 10
10 10
4 75
5 4 75× =
× ×+
−
− −,
, ,
,
KNH4+
V
V
K
VK
K
NH
NH3
3 NH
NHCH COOH
3 CH COOH
3
3
4
3
3
H O
H O H O
×+
+=
×+
+
+
+ ++
0 1
1010 0 1
103
,[ ]
[ ]
,
[ ]
[NH ]4+
[NH ]4+
C
VCH COOH 3 3NH
3
3
[CH COOH] [CH COO ]= + =×
+− 10 0 1
10,
C
V
VNH 3 4NH
NH3
3
[NH ] [NH ]= + =×+
+ 30 1
10
,
NH4+
VV
VNaOH
NaOH
NaOH×+
=
×+
×
+
−
− −
0 110
10 0 110
10
10 10
4 75
5 4 75
,, ,
,
[Na ]H O
CH COOH
3
3++=
+C K
Ka
a[ ]
96 Equilibrios iónicos y sus aplicaciones analíticas

y 0,1 mol L–1 de NaOH; c) 0,1 mol L–1 de CH3COOHy 0,1 mol L–1 de NH3; d) 0,2 mol L–1 de CH3COOH y0,1 mol L–1 de NH3; e) 0,1 mol L–1 de CH3COOH y0,2 mol L–1 de NH3. f) ¿Cuántos mL de HCl 0,1 molL–1 habría que añadir a 100 mL de NH3 0,1 mol L–1
para obtener una disolución reguladora de pH = 10?g) ¿Qué pH se obtendrá si se añade 1 mL de NaOH1 mol L–1 a 100 mL de la disolución del apartado ante-rior? (Puede despreciarse el efecto de la dilución). NH+
3 (pKa = 9,24), CH3COOH (pKa = 4,75).
a) Al mezclar disoluciones de NH4Cl y NH3, esto esde un ácido débil y su base conjugada, se obtieneuna disolución reguladora. Al ser ambas disolu-ciones equimoleculares el pH de la misma coin-cide con su pKa = 9,24, presentando por esta cir-cunstancia la máxima capacidad reguladora.
b) La mezcla de HCl y de NaOH, ambos a una con-centración de 0,1 mol L–1, da lugar a la formaciónde una sal, cloruro de sodio.
c) Esta mezcla también da lugar a una sal, acetatode amonio, en este caso de ácido y base débil.
d) A pesar de partir de los mismos componentes queen el apartado anterior, en este caso se origina unadisolución reguladora al estar en exceso la con-centración de ácido acético frente a la de amonia-co. En efecto, tras la mezcla se tiene CH3COOH0,1 mol L–1 y NH4CH3COO 0,1 mol L–1.
e) Este caso, opuesto al anterior, la mezcla da lugara la disolución tampón NH+
4/NH3 en presencia deiones acetato.
f) Para calcular los mL de HCl 0,1 mol L–1 que sehan de añadir a 100 mL de NH3 0,1 mol L–1 paraformar una disolución reguladora NH+
4/NH3 depH 10,0 se sigue el procedimiento habitual indi-cado en el problema 41, esto es, establecer laexpresión adecuada a partir de los equilibrios áci-do-base implicados en dicha disolución tampóny de los balances de masa y de carga. Todo elloconduce, como se ha indicado en el apartado 2.7.2,a la expresión [2.247]:
en la que al sustituir los valores dados en el pro-blema se tiene:
de donde VHCl = 14,82 mL.
g) Al añadir NaOH a la disolución reguladora ante-rior, esta base fuerte reacciona con el ion amoniode acuerdo con:
NH+4 + OH– ←→ NH3 + H2O
con lo cual al ser superior la concentración de sucomponente básico su pH deberá de incrementar-se. Así, antes de añadir 1 mL de NaOH 1 mol L–1
y considerando los datos y resultados del apartadoanterior, la disolución tampón estaba formada por:
mmol NH+4 = mmol HCl añadidos =
= 14,82 × 0,1 = 1,482 mmolmmol NH3 = mmol NH3 (iniciales) – mmol HCl
= 100 × 0,1 – 14,82 × 0,1 = 8,518 mmol
en un volumen final de 100 + 14,82 = 114,82 mL.Dado que de acuerdo con el enunciado del pro-
blema, 1 mL de NaOH 1 mol L–1 (1 × 1 = 1 mmolde NaOH) se añade a 100 mL de la disoluciónreguladora y no al total de la misma, se ha de efec-tuar una corrección de volumen tal como:
mmol mmol
mmol NH3 = mmol
Finalmente, tras la adición de NaOH y deacuerdo con el equilibrio ácido-base:
NH+4+ OH– ←→ NH3 + H2O
se tiene:
mmol NH+4 = 1,291 – 1 = 0,291 mmol
mmol NH3 = 7,418 + 1 = 8,418 mmol
Al sustituir estos valores en la expresión delcálculo del pH se llega a:
de donde pH = 10,70.
48. Justificar la diferente capacidad reguladora de las diso-luciones que se obtienen al mezclar: a) 25 mL de ácidoacético 0,2 mol L–1 + 25 mL de acetato de sodio 0,2 molL–1; b) 25 mL de ácido clorhídrico 0,1 mol L–1 + 25 mL
[H O ]3
+ −= 100 2918 418
9 24, ,,
8 518
100114 82
7 418,,
,=
NH4
+ = =1 482100
114 821 291,
,,
10 10
0 1100 0 1 0 1
10 9 24− −=×
× − ×, ,
, ,V
VHCl
HCl
[H O ]3
HCl HCl
Base débil Base débil HCl HCl
+ =×
× − ×K
V C
V C V Ca
Capítulo 2: Equilibrios ácido-base 97

de ácido acético 0,1 mol L–1; c) 25 mL de hidróxido desodio 0,1 mol L–1 + 25 mL de ácido acético 0,2 mol L–1. CH3COOH (pKa = 4,75).Sol.: a) CH3COOH/NaCH3COO, tampón, concen-
tración 0,1 mol L–1; b) CH3COOH/HCl; c)CH3COOH/ NaCH3COO, tampón, concentra-ción 0,05 mol L–1.
49. A cuatro fracciones de 50 mL de una disolución 0,05mol L–1 de acetato de amonio se añaden: a) 50 mL deHCl 0,05 mol L–1; b) 25 mL de HCl 0,05 mol L–1; c)50 mL de NaOH 0,05 mol L–1; d) 25 mL de NaOH0,05 mol L–1. Indicar en qué casos se obtiene una diso-lución tampón, razonando la respuesta. e) Calcularel volumen de HCl 0,05 mol L–1 que hay que añadira 50 mL de acetato de amonio 0,05 mol L–1 para pre-parar una disolución reguladora de pH 4,3. CH3COOH (pKa = 4,75), NH+
4 (pKa = 9,24).Sol.: Son tampón: b) (CH3COOH/CH3COO–) y d)
(NH+4/NH3); e) 36,9 mL.
50. Una disolución reguladora es 0,1 mol L–1 en ácidoacético y 0,1 mol L–1 en acetato de sodio. Calcular elpH de dicha disolución después de añadirle: a) 1 mLde disolución de HCl 10 mol L–1 por litro, y b) 1 mLde disolución de NaOH 5,0 mol L–1 por litro. Des-preciar el cambio de volumen que se produce por laadición de HCl o NaOH. CH3COOH (pKa = 4,75).Sol.: a) 4,66; b) 4,79.
51. Se dispone en el laboratorio de las siguientes disolu-ciones (sin limitación de volumen): NaOH 0,05 molL–1, HCl 0,1 mol L–1, CH3COOH 0,02 mol L–1 yCH3COONa 0,05 mol L–1. a) Indicar las diferentesformas de preparar una disolución reguladora de pH= 5,0. b) En cada uno de los casos, calcular los volú-menes de las disoluciones seleccionadas para prepa-rar 100 mL de la citada disolución reguladora. CH3COOH (pKa = 4,75).Sol.: a) Mezclando CH3COOH + NaCH3COO,
NaCH3COO + HCl y CH3COOH + NaOH. b)58,14 mL de CH3COOH 0,02 mol L–1 + 41,86 mLde NaCH3COO 0,05 mol L–1; 79,55 mLCH3COOH 0,02 mol L–1 + 20,45 mL de NaOH0,05 mol L–1; 84,85 mL NaCH3COO 0,05 mol L–1
+ 15,15 mL HCl 0,1 mol L–1.
52. Se dispone de una disolución de cloruro de amonio0,05 mol L–1. Calcular los gramos de NaOH que hayque añadir a 500 mL de la disolución de cloruro deamonio para conseguir una disolución reguladora
cuyo pH sea el doble del que presenta inicialmentela disolución de cloruro de amonio. NH+
4 (pKa = 9,24). Pesos atómicos: H = 1, O = 16, Na = 23.Sol.: 0,952 g.
53. ¿Qué disoluciones tampón será posible preparar sise dispone de fluoruro de amonio 0,1 mol L–1, ácidoclorhídrico 0,1 mol L–1 e hidróxido de sodio 0,1 molL–1? ¿Entre qué valores aproximados de pH seránefectivas estas disoluciones? b) ¿A qué valores de pHtendrán la máxima capacidad reguladora?HF (pKa = 3,52), NH+
4(pKa = 9,24).Sol.: a) HF/F– 2,52 – 4,52 y NH+
4/NH3 8,24 – 10,24; b) 3,52 y 9,24, respectivamente.
54. ¿Qué volumen de amoniaco 0,2 mol L–1 sería necesa-rio añadir a 25 mL de ácido fórmico 0,1 mol L–1 paraobtener una disolución tampón de pH = 4,0? NH+
4 (pKa = 9,24); HCOOH (pKa = 3,75). Sol.: 8,03 mL.
55. Calcular el volumen de NaOH 0,1 mol L–1 que habríaque añadir a 25 mL de una disolución 0,1 mol L–1 deácido acético y 0,1 mol L–1 en ácido fórmico para obte-ner una disolución reguladora de pH 4,25. CH3COOH (pKa = 4,75), HCOOH (pKa = 3,75).Sol.: 25 mL.
56. Se prepara una disolución reguladora disolviendo26,5 g de carbonato de sodio en 500 mL de agua, aña-diendo 125 mL de HCl 1,0 mol L–1 y diluyendo lamezcla a un litro. Calcular el pH de la disolución. H2CO3 (pKa1 = 6,35, pKa2 = 10,33). Pesos atómicos: C = 12, O = 16 y Na = 23.Sol.: 10,33.
57. El sistema HCO–
3/H2CO3 es de gran importancia parala regulación del pH de la sangre humana entre 7,35y 7,45. ¿Cuál debe ser la relación [HCO–
3]/[H2CO3]en la sangre para que el valor de pH sea de 7,4?H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: [HCO–
3]/[H2CO3] = 11,2.
58. Indicar cuáles son las especies mayoritarias y el pH delas disoluciones que se obtienen cuando se mezclan:a) 50 mL de ftalato de sodio (Na2Ft) 10–1 mol L–1 + 25mL de HCl 10–1 mol L–1; b) 50 mL de Na2Ft 10–1 molL–1 + 50 mL de HCl 10–1 mol L–1; c) 50 mL de Na2Ft10–1 mol L–1 + 75 mL de HCl 10–1 mol L–1; d) 50 mL de Na2Ft 10–1 mol L–1 + 100 mL de HCl 10–1 mol L–1;e) 50 mL de Na2Ft 10–1 mol L–1 + 50 mL de H2Ft 10–1
98 Equilibrios iónicos y sus aplicaciones analíticas

mol L–1. f) Calcular el volumen de ácido clorhídrico0,2 mol L–1 que es necesario añadir a 50 mL de ftala-to de sodio 0,1 mol L–1 para obtener una disoluciónreguladora de pH = 5,5. (H2Ft: pKa1 = 2,95, pKa2 = 5,41).
a) La mezcla de 50 mL Na2Ft 10–1 mol L–1 (50 × 0,1 =5 mmol) con 25 mL de HCl 10–1 mol L–1 (25 × 0,1= 2,5 mmol) origina tras su reacción, Ft2– + H+ ←→HFt–, una disolución que contiene cantidades equi-moleculares (2,5 mmol) de Ft2– y HFt– dando lugarpor tanto a una disolución reguladora en la que pH = pKa2 = 5,41.
b) Cuando, como es el caso, se mezclan cantidadesequimoleculares de Ft2– y HCl se obtiene la diso-lución del anfolito HFt– cuyo pH viene dado por:pH = 1/2 (pKa1 + pKa2) = 4,18.
c) En este caso se adiciona una cantidad de HCl (75mL de HCl 0,1 mol L–1 = 7,5 mmol) en exceso conrespecto a Ft2– (5 mmol), con lo cual se tendrá unadisolución tampón formada por las especies HFt– yH2Ft (2,5 mmol de cada) con lo cual pH = pKa1 =2,95.
d) Cuando se adicionan 100 mL de HCl 0,1 mol L–1
todo el ftalato de sodio se transforma en ácido ftá-lico, esto es se tienen 5 mmol de H2Ft en 150 mL,con lo cual [H2Ft] = 0,033 mol L–1 y el pH de estadisolución se calcula haciendo uso de la expresión:
de donde [H3O+] = 5,55 × 10–3 mol L–1 y pH = 2,26.
e) Finalmente cuando se mezclan cantidades equi-moleculares de Na2Ft y H2Ft (50 mL 0,01 mol L–1
= 5 mmol) se origina una disolución que contie-ne al anfolito HFt– cuyo pH es idéntico al indica-do en el apartado b), esto es, pH = 4,18.
f) Para abordar la resolución de este apartado sesigue el procedimiento utilizado en otras ocasio-nes. Así, se tiene:
Equilibrios ácido-base implicados en la disolu-ción reguladora:
Na2Ft → Na+ + Ft2–
Ft2– + H2O ←→ HFt– + OH–
H2O + H2O ←→ H3O+ + OH–
Balances de masas y de carga:
BM:
BC: [Na+] + [H3O+] = [HFt–] + 2 [Ft2–] + [OH–] + [Cl–]
En este balance de cargas se pueden despreciar lasconcentraciones de H3O
+ y OH–, como es común enel caso de disoluciones reguladoras. En estas condi-ciones, al sustituir en el balance de cargas las con-centraciones de las especies implicadas en el mismose tiene:
Al sustituir los valores de [H3O+], Ka1 y Ka2 en la
expresión anterior, se observa que [H3O+]2 en el deno-
minador es despreciable frente a los otros términos,quedando la expresión al simplificar el término (50+ VHCl), como:
de donde, al resolver, VHCl = 11,21 mL.
59. Se desea preparar 100 mL de una disolución tampónde pH = 3,0. Explicar cómo se haría si se dispone delas siguientes disoluciones: a) Ácido malónico 0,1 molL–1 , hidrogeno malonato de sodio 0,1 mol L–1, ácidoclorhídrico 0,1 mol L–1 y ácido fórmico 0,1 mol L–1 ;b) Calcular el pH de la disolución que se obtiene almezclar 50 mL de ácido malónico 0,2 mol L–1 con elmismo volumen de amoniaco 0,4 mol L–1. Ácido malónico (pKa1 = 2,85, pKa2 = 5,70), HCOOH(pKa = 3,75), NH+
4 (pKa = 9,24).Sol.: a) ácido malónico/ hidrógeno malonato de sodio,
hidrógeno malonato de sodio/ ácido clorhídri-
2 5 10 1010 10 10 10
2 95 5 41
2 95 5 5 2 95 5 41+
× × ×× + ×
− −
− − − −
, ,
, , , ,
10 0 25 10 10
10 10 10 10
2 95 5 5
2 95 5 5 2 95 5 41= × +
× ×× + ×
+− −
− − − −VHCl ,
, ,
, , , ,
550
2 1 2
21 1 2
+ +( )
+ ++ +
VK K
K K K
a a
a a a
HCl
3 3H O H O[ ] [ ]
550 1
21 1 2
+ +( )
+ ++
+
+ +
VK
K K K
a
a a a
HCl3
3 3
H O
H O H O
[ ]
[ ] [ ]
1050
0 250+
=×
++
VV
VHCl
HCl
HCl
,
CVSal 2
2
HCl
H Ft HFt ] [F= + + = ×+
− −[ ] [ ],50 0 1
50
C
VVHCl
HCl
HCl
Cl= =×
+−[ ]
,0 250
[Na ]HCl
+ =× ×
+2 50 0 150
,V
[H O ]H Ft
H O H O32
3 3
++
−
+ −=
+=
×+
[ ][ ]
,[ ]
,
,
KK
a
a
1
1
2 95
2 95
0 033 1010
Capítulo 2: Equilibrios ácido-base 99

co, hidrógeno malonato de sodio/ ácido fórmi-co; b) 7,32.
60. Se desea preparar un litro de disolución reguladoraNa2CO3/NaHCO3 con un pH inicial de 9,70 y de talcomposición que al añadir 60 milimoles de un ácidofuerte monoprótico, el pH se mantenga en 9,30. ¿Cuá-les son las concentraciones mínimas necesarias decarbonato de sodio y bicarbonato de sodio para pre-parar la citada disolución tampón?H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: Na2CO3 0,466 mol L–1 y NaHCO3 0,109 mol L–1.
61. A dos alícuotas de 50 mL de una disolución de bicar-bonato de sodio 0,1 mol L–1 se le adiciona por sepa-rado: a) 20,0 mL de disolución de ácido clorhídrico0,01 mol L–1 y b) 25,0 mL de disolución de hidróxidode potasio 0,03 mol L–1. Calcular: a) los incrementosde pH que se producirán en ambos casos, y b) indi-car en qué caso se obtiene una disolución tampón conmayor capacidad reguladora. H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: a) –0,61 y +1,24, respectivamente; b) cuando se
añade NaOH.
62. ¿Cuántos mL de HCl 0,2 mol L–1 se han de añadir a 50mL de etilendiamina (NH2CH2CH2NH2 = B) 5 × 10–2
mol L–1 para obtener una disolución reguladora de pH= 7,20? Indicar si es posible obtener una disoluciónreguladora de pH = 5,0 a partir de disoluciones de eti-lendiamina y HCl. Justificar la respuesta. NH2+
2 (pKa1 = 7,52, pKa2 = 10,65).
En la preparación de esta disolución tampón se tie-nen los siguientes equilibrios ácido-base:
HCl + H2O → Cl– + H3O+
+ H2O ←→ BH+ + H3O+
BH+ + H2O ←→ B + H3O+
H2O + H2O ←→ H3O+ + OH–
y se pueden establecer los correspondientes balances:
BM:
BC: [H3O+] + 2 + [BH+] = [OH–] + [Cl–]
Este balance de cargas puede simplificarse a:
2 + [BH+] = [Cl–]
Sustituyendo en este balance las respectivas con-centraciones se tiene:
A pH 7,2 el valor de Ka1 Ka2 es despreciable en eldenominador de la expresión anterior, por lo que éstase transforma, tras sustituir los valores de CB, CHCl,[H3O
+] y Ka1, en:
de donde VHCl = 20,95 mL.Por otra parte, no se puede obtener una disolu-
ción reguladora de pH = 5,0 a partir de disolucio-nes de etilendiamina y HCl ya que a este valor depH tendríamos sólo en disolución la especie y una cantidad inapreciable de su base conjugadaBH+.
63. Se dispone en el laboratorio de las siguientes disolu-ciones sin limitación de volumen: H2A 0,1 mol L–1,NaHA 0,1 mol L–1 y Na2A 0,05 mol L–1. Calcular: a)¿Cuál sería el pH de la disolución resultante al mez-clar 20 mL de Na2A 0,05 mol L–1 con 10 mL de H2A0,1 mol L–1?, y b) ¿Cómo se prepararían 100 mL deuna disolución reguladora de pH 9,0?H2A (pKa1 = 4,0, pKa2 = 10,0).Sol.: a) 7,00; b) 16,67 mL Na2A 0,05 mol L–1 + 83,33
mL NaHA 0,1 mol L–1.
64. Se desea preparar una disolución tampón de pH = 6,5y se dispone de las siguientes disoluciones: HCl, HCO-OH, H3PO4, CH3COOH, NaH2PO4, NH4Cl, Na2HPO4,NH3, Na3PO4 y NaOH. a) Enumerar todas las posiblescombinaciones binarias que permitirían obtener untampón con una capacidad amortiguadora razonable-mente alta. b) Calcular el volumen de NH3 0,2 mol L–1
que se debe añadir a 50 mL de ácido succínico 0,1 molL–1 para preparar una disolución tampón de pH 6,05.HCOOH (pKa = 3,75), H3PO4 (pKa1 = 2,15, pKa2 = 7,21,pKa3 = 12,33), CH3COOH (pKa = 4,75), NH+
4 (pKa =9,24), HOOCCH2CH2CH2COOH (pKa1 = 4,20, pKa2 =5,25).
BH22+
2 550
2 10 10 10
10 10 100 2
50
7 2 2 7 2 7 52
7 2 2 7 2 7 52
,( )
( ),
, , ,
, , ,
+× + ×( )
+ ×=
×+
− − −
− − −
V VV
HCl HCl
HCl
C K
K K KC
a
a a a
B 3 3
3 3HCl
H O ] H O ]
[H O ] H O ]
2 21
21 1 2
[ [
[
+ +
+ +
+( )+ +
=
[BH ]22+
[BH ]22+
C
VB 22
HCl
[BH ] [BH ] [B]= + + =× ×
++ +
−50 5 1050
2
CV
VHClHCl
HCl
[Cl= = ×+
− ],0 2
50
BH22+
100 Equilibrios iónicos y sus aplicaciones analíticas

Sol.: a) H3PO4/NaOH, H3PO4/ NH3, H3PO4/ Na3PO4,H3PO4/ Na2HPO4, NaH2PO4/NaOH, NaH2PO4/NH3, NaH2PO4/ Na2HPO4, NaH2PO4/ Na3PO4,Na2HPO4/ HCl, Na2HPO4/HCOOH, Na2HPO4/CH3 COOH, Na3PO4/HCl, Na3PO4/HCOOH,Na3PO4/C H3COOH; b) 46,49 mL.
65. Indicar cuáles serán las especies mayoritarias en lasdisoluciones que se obtienen al mezclar: a) 25 mL deNaHCO3 10–1 mol L–1 + 25 mL de Na2CO3 10–1 molL–1; b) 25 mL de NaHCO3 10–1 mol L–1 + 25 mL deNaOH 10–1 mol L–1; c) 25 mL de NH4Cl 10–1 mol L–1
+ 25 mL de NaCH3COO 10–1 mol L–1; d) 50 mL deH3PO4 10–1 mol L–1 + 75 mL de NaOH 10–1 mol L–1.Razonar cuáles de estas mezclas corresponden a diso-luciones tampón y calcular su pH. H2CO3 (pKa1 = 6,35, pKa2 = 10,33), NH+
4 (pKa = 9,24),CH3COOH (pKa = 4,75), H3PO4 (pKa1 = 2,15, pKa2 =7,21, pKa3 = 12,33).Sol.: a) NaHCO3/Na2CO3 pH = 10,33; b) Na2CO3; c)
NH4CH3COO; d) NaH2PO4/Na2HPO4 pH = 7,21.
66. Se dispone de disoluciones 0,1 mol L–1 de los siguien-tes compuestos: HCl, NaOH, NaH2PO4, H3BO3,Na2HPO4 y NH3. Explicar cualitativamente cómo sepueden preparar disoluciones tampón de a) pH 6,90 yb) pH 8,4. Justificar la respuesta. c) ¿Se puede esperarque una de las disoluciones tampón del apartado ante-rior sea más efectiva que otra? Razonar la respuesta. H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33), H3BO3
(pKa = 9,24), NH+4 (pKa = 9,24).
Sol.: a) pH = 6,9: NaH2PO4/Na2HPO4; NaH2PO4/NaOH; NaH2PO4/NH3, Na2HPO4/ HCl; b) pH= 8,4: H3BO3/ NaOH; NH3/HCl; c) Serán másefectivas las de pH 6,9.
67. ¿Cuántos mL de NaOH 0,1 mol L–1 habría que aña-dir a 25 mL de H3PO4 0,1 mol L–1 para obtener unadisolución reguladora de pH 7,0? H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33).
Cuando se mezclan disoluciones de NaOH y H3PO4
se origina la siguiente reacción ácido-base:
2 H3PO4 + 3 NaOH ←→ NaH2PO4 + + Na2HPO4 + 3 H2O
y se pueden establecer los balances de masas y de car-gas que se indican a continuación:
BM:
BC:
En este balance de cargas se puede despreciar: 1)las concentraciones de H3O
+ y de OH– dado queambas son iguales (10–7 mol L–1), y 2) la concentra-ción de PO3–
4 a tenor del valor del pH que presenta ladisolución reguladora. Al sustituir en este balance lasconcentraciones respectivas se transforma en:
+
+ 2
En el denominador de los términos del segundomiembro de esta igualdad se puede despreciar [H3O
+]3
y Ka1 Ka2 Ka3 dado el valor de pH de la disolucióntampón. En estas condiciones la expresión anteriorse transforma en:
+
+ 2
y al sustituir los valores de concentraciones, y cons-tantes de acidez se tiene:
de donde VNaOH = 34,5 mL.
68. Se dispone de una disolución de H3PO4 0,5 mol L–1 yde otra de NaOH 3,0 mol L–1. ¿Cómo se prepararían1.250 mL de una disolución reguladora de pH 7,3?H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33).
+× × ×
× + × ×
− − −
− − − − −
5 10 10 1010 10 10 10 10
7 2 15 7 21
7 2 2 15 7 2 15 7 21( )
, ,
, , ,
VNaOH × =
× ×× + × ×
+− −
− − − − −0 1
2 5 10 1010 10 10 10 10
7 2 15
7 2 2 15 7 2 15 7 21,
,( )
,
, , ,
C K K
K K Ka a
a a a
H PO 3
3 3
3 4[H O
H O H O
+
+ ++]
[ ] [ ]1 2
21 1 2
C
C K
K K Ka
a a aNaOH
H PO 3
3 3
3 4[H O
H O H O=
+
+
+ +
]
[ ] [ ]
21
21 1 2
C K K
K K K K K Ka a
a a a a a a
H PO 3
3 3 3
3 4[H O ]
H O H O H O
+
+ + ++ + +1 2
3 21 1 2 1 2 3[ ] [ ] [ ]
C
C K
K K K K K Ka
a a a a a aNaOH
H PO 32
33
3 3
3 4[H O ]
H O ] [H O ] H O=
+ + +
+
+ + +1
21 1 2 1 2 3[ [ ]
3[PO ] [OH ]43− −+ +
[Na ] [H O ] [H PO ] 2[HPO ]3 2 4 42+ + − −+ = + +
V42
43
NaOH
[HPO ] [PO ]+ + =×
+− − 25 0 1
25,
CH PO 3 4 2 43 4
[H PO ] [H PO ]= + +−
C
VVNaOH
NaOH
NaOH
[Na ]= =×
++ 0 1
25,
Capítulo 2: Equilibrios ácido-base 101

Sol.: 993,1 mL de H3PO4 0,5 mol L–1 y 256,9 mL deNaOH 3,0 mol L–1.
69. Calcular el pH de la disolución reguladora que resul-ta al mezclar 25 mL de NaH2PO4 0,1 mol L–1 y 10 mLde NaOH 0,1 mol L–1. H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33).Sol.: 7,03.
70. Calcular la relación molar entre NaOH y ácido cítri-co necesaria para obtener un tampón fisiológico depH 6,40.
Ácido cítrico (pKa1 = 3,13, pKa2 = 4,76, pKa3 = 6,40).Sol.: [NaOH]/[Ácido cítrico] = 2,5.
71. Calcular el volumen de NH3 1 mol L–1 que se ha de añadir a 100 mL de disolución de ácido cítrico0,1 mol L–1 para obtener una disolución reguladorade pH 5,0. Ácido cítrico (pKa1 = 3,13, pKa2 = 4,76, pKa3 = 6,40),NH+
4 (pKa = 9,24).Sol.: 15,87 mL.
102 Equilibrios iónicos y sus aplicaciones analíticas

3
3.1. Introducción3.2. Diagrama logarítmico de concentración3.3. Diagrama logarítmico de un protolito
fuerte. Cálculo del pH3.4. Diagrama logarítmico de un protolito débil
monoprótico3.5. Diagrama logarítmico de una mezcla
de dos protolitos débiles monopróticos.Cálculo del pH
3.6. Diagrama logarítmico de la disoluciónde una sal. Cálculo del pH
3.7. Diagrama logarítmico de protolitosdipróticos
3.8. Diagrama logarítmico de protolitospolipróticos
3.9. Diagramas de distribución de especiesen función del pH
CuestionesSeminarios: problemas gráficos
REPRESENTACIÓNGRÁFICA
DEL EQUILIBRIOÁCIDO-BASE

3.1. Introducción
El uso de representaciones gráficas para elestudio de equilibrios iónicos en disoluciónconstituye una herramienta muy útil, ya quepermiten visualizar de manera global el com-portamiento de las diferentes especies impli-cadas en los distintos tipos de equilibrio, fren-te a una determinada propiedad característicade los mismos (pH en equilibrios ácido-base,potencial en un sistema redox, etc). Estos méto-dos gráficos constituyen una valiosa ayuda a losmétodos de cálculo numérico, utilizados en elestudio de los diferentes equilibrios iónicos,sobre todo para el caso de sistemas complejos,ya que la resolución numérica de las ecuacio-nes requiere, en el mejor de los casos, simplifi-caciones que no siempre son inmediatas, mien-tras que en otras ocasiones es necesario resolverecuaciones más o menos complejas. Como talayuda, ambas metodologías (numérica y gráfi-ca) son complementarias y por tanto se han deutilizar de forma ponderada.
Una propiedad peculiar de muchos sistemasquímicos es que dependen de una misma varia-ble. El desplazamiento de un equilibrio ácido-base, por ejemplo, depende exclusivamente dela concentración de iones hidronio en disolu-ción, es decir, de su pH, mientras que en siste-mas redox la variable es la concentración deelectrones o bien el potencial redox del mismo.Estas variables se conocen con el nombre devariable principal. En los métodos gráficos, serepresentan de forma adecuada las variables delsistema frente a la variable principal. Así, sepuede hacer uso de diagramas logarítmicos deconcentración, en los que se representan los
logaritmos de las concentraciones de las espe-cies que existen en disolución en función de lavariable principal, o bien de diagramas de dis-tribución que permiten visualizar la variaciónde la concentración de las diferentes especiespresentes en el equilibrio, expresadas como frac-ciones molares, en función asimismo de dichavariable principal.
3.2. Diagrama logarítmicode concentración
En el caso de los equilibrios ácido-base,teniendo en cuenta que en las reacciones de neu-tralización la [H3O
+] puede cambiar en un factorde un millón, se ha de utilizar la escala logarít-mica en las representaciones gráficas, con lo cualse tienen los diagramas logarítmicos de concen-tración, que en definitiva son gráficas donde serepresentan los logaritmos de las concentracio-nes de todas las especies protolíticas que existenen la disolución considerada, en función del pH.Para trazar un diagrama logarítmico se repre-senta en el eje de ordenadas log [X], donde Xrepresenta a cada una de las especies protolíti-cas presentes en el equilibrio frente al pH, queactúa como variable principal y que se representaen el eje de abscisas. La utilidad de este tipo dediagrama es doble, ya que:
1. Permite la visualización de la concentra-ción de las diferentes especies en equili-brio y efectuar de forma rápida y seguralas simplificaciones adecuadas, esto es deci-dir cuáles son las especies relevantes y cuá-les se pueden despreciar sin problemas.
104 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Mostrar al alumno el potencial de las representacio-nes gráficas en equilibrios ácido-base.
• Intensificar sus conocimientos sobre estos equili-brios, mostrándole cómo estos diagramas permiten
visualizar globalmente el comportamiento de lasespecies implicadas en los mismos.
• Inculcar el carácter complementario de estos diagra-mas frente a los procedimientos de cálculo numérico.
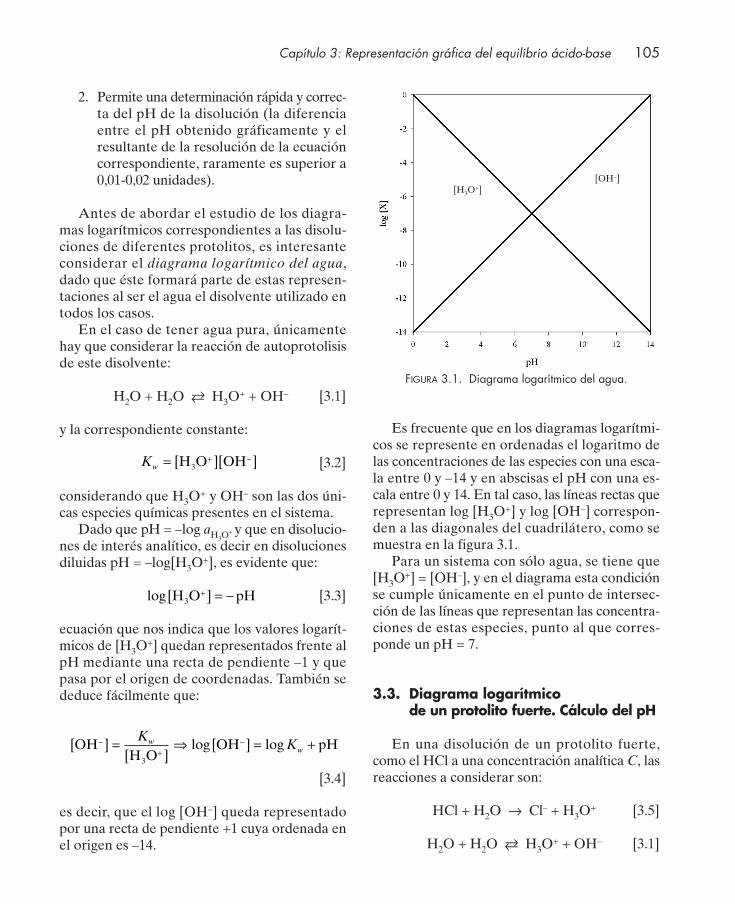
2. Permite una determinación rápida y correc-ta del pH de la disolución (la diferenciaentre el pH obtenido gráficamente y elresultante de la resolución de la ecuacióncorrespondiente, raramente es superior a0,01-0,02 unidades).
Antes de abordar el estudio de los diagra-mas logarítmicos correspondientes a las disolu-ciones de diferentes protolitos, es interesanteconsiderar el diagrama logarítmico del agua,dado que éste formará parte de estas represen-taciones al ser el agua el disolvente utilizado entodos los casos.
En el caso de tener agua pura, únicamentehay que considerar la reacción de autoprotolisisde este disolvente:
H2O + H2O →← H3O+ + OH– [3.1]
y la correspondiente constante:
[3.2]
considerando que H3O+ y OH– son las dos úni-
cas especies químicas presentes en el sistema.Dado que pH = –log aH3O
+ y que en disolucio-nes de interés analítico, es decir en disolucionesdiluidas pH = –log[H3O
+], es evidente que:
[3.3]
ecuación que nos indica que los valores logarít-micos de [H3O
+] quedan representados frente alpH mediante una recta de pendiente –1 y quepasa por el origen de coordenadas. También sededuce fácilmente que:
[3.4]
es decir, que el log [OH–] queda representadopor una recta de pendiente +1 cuya ordenada enel origen es –14.
FIGURA 3.1. Diagrama logarítmico del agua.
Es frecuente que en los diagramas logarítmi-cos se represente en ordenadas el logaritmo delas concentraciones de las especies con una esca-la entre 0 y –14 y en abscisas el pH con una es-cala entre 0 y 14. En tal caso, las líneas rectas querepresentan log [H3O
+] y log [OH–] correspon-den a las diagonales del cuadrilátero, como semuestra en la figura 3.1.
Para un sistema con sólo agua, se tiene que[H3O
+] = [OH–], y en el diagrama esta condiciónse cumple únicamente en el punto de intersec-ción de las líneas que representan las concentra-ciones de estas especies, punto al que corres-ponde un pH = 7.
3.3. Diagrama logarítmicode un protolito fuerte. Cálculo del pH
En una disolución de un protolito fuerte,como el HCl a una concentración analítica C, lasreacciones a considerar son:
HCl + H2O → Cl– + H3O+ [3.5]
H2O + H2O →← H3O+ + OH– [3.1]
[ ]
[ ]log[ ] logOH
H OOH pH
3
−+
−= ⇒ = +KKw
w
log[H O ] pH3+ = −
Kw = + −[H O ][OH ]3
Capítulo 3: Representación gráfica del equilibrio ácido-base 105
[H3O+]
[OH–]
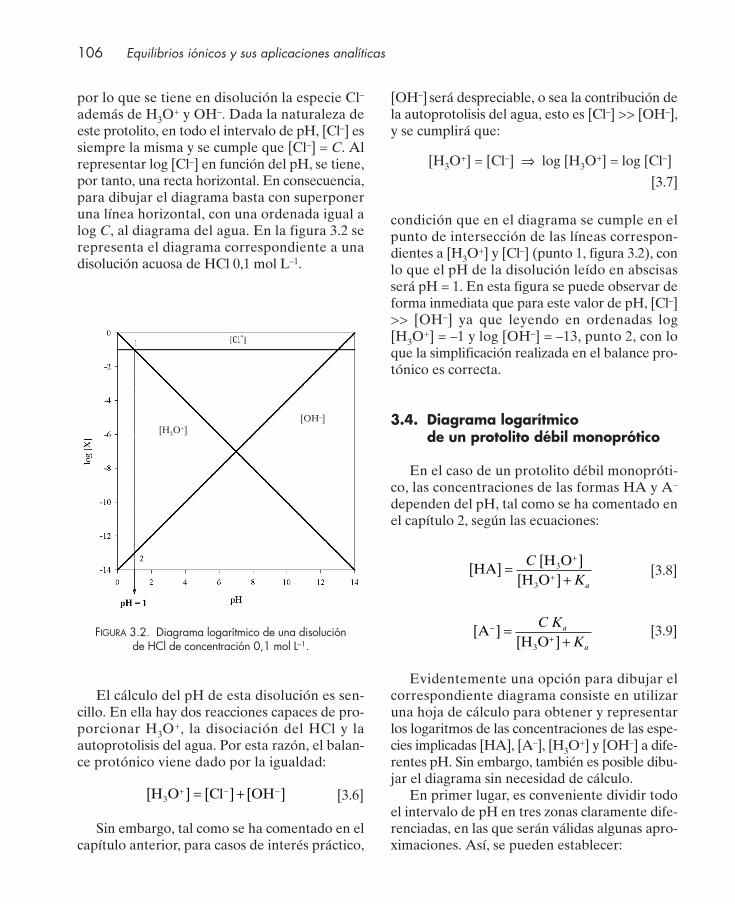
por lo que se tiene en disolución la especie Cl–
además de H3O+ y OH–. Dada la naturaleza de
este protolito, en todo el intervalo de pH, [Cl–] essiempre la misma y se cumple que [Cl–] = C. Alrepresentar log [Cl–] en función del pH, se tiene,por tanto, una recta horizontal. En consecuencia,para dibujar el diagrama basta con superponeruna línea horizontal, con una ordenada igual alog C, al diagrama del agua. En la figura 3.2 serepresenta el diagrama correspondiente a unadisolución acuosa de HCl 0,1 mol L–1.
FIGURA 3.2. Diagrama logarítmico de una disolución de HCl de concentración 0,1 mol L–1.
El cálculo del pH de esta disolución es sen-cillo. En ella hay dos reacciones capaces de pro-porcionar H3O
+, la disociación del HCl y laautoprotolisis del agua. Por esta razón, el balan-ce protónico viene dado por la igualdad:
[3.6]
Sin embargo, tal como se ha comentado en elcapítulo anterior, para casos de interés práctico,
[OH–] será despreciable, o sea la contribución dela autoprotolisis del agua, esto es [Cl–] >> [OH–],y se cumplirá que:
[H3O+] = [Cl–] ⇒ log [H3O
+] = log [Cl–]
[3.7]
condición que en el diagrama se cumple en elpunto de intersección de las líneas correspon-dientes a [H3O
+] y [Cl–] (punto 1, figura 3.2), conlo que el pH de la disolución leído en abscisasserá pH = 1. En esta figura se puede observar deforma inmediata que para este valor de pH, [Cl–]>> [OH–] ya que leyendo en ordenadas log[H3O
+] = –1 y log [OH–] = –13, punto 2, con loque la simplificación realizada en el balance pro-tónico es correcta.
3.4. Diagrama logarítmicode un protolito débil monoprótico
En el caso de un protolito débil monopróti-co, las concentraciones de las formas HA y A–
dependen del pH, tal como se ha comentado enel capítulo 2, según las ecuaciones:
[3.8]
[3.9]
Evidentemente una opción para dibujar elcorrespondiente diagrama consiste en utilizaruna hoja de cálculo para obtener y representarlos logaritmos de las concentraciones de las espe-cies implicadas [HA], [A–], [H3O
+] y [OH–] a dife-rentes pH. Sin embargo, también es posible dibu-jar el diagrama sin necesidad de cálculo.
En primer lugar, es conveniente dividir todoel intervalo de pH en tres zonas claramente dife-renciadas, en las que serán válidas algunas apro-ximaciones. Así, se pueden establecer:
[ ]A
[H O ]3
−+
=+
C KK
a
a
[HA]
[H O ][H O ]
3
3
=+
+
+
CKa
[H O ] [Cl ] [OH ]3+ − −= +
106 Equilibrios iónicos y sus aplicaciones analíticas
[H3O+]
[OH–]

Zona 1: pH << pKa ⇒ [H3O+] >> Ka
Cuando esta aproximación sea aplicable, pue-de comprobarse que:
[3.10]
de donde:
[3.11]
La [HA] no depende del pH en esta zona, conlo que vendrá representada por una recta hori-zontal en el diagrama como se observa en la figu-ra 3.3. Por otra parte, en esta zona:
[3.12]
y por tanto:
[3.13]
La variación de log [A–] respecto al pH, secorresponde con una recta de pendiente +1 y deordenada en el origen log CKa, como se apreciaen la figura 3.3.
Estas ecuaciones son válidas mientras se cum-pla que [H3O
+] >> Ka. Si se considera que es admi-sible un error de hasta un 5%, Ka será desprecia-ble frente a [H3O
+], si [H3O+] ≥ 20 Ka y a escala
logarítmica: log [H3O+] ≥ 1,3 log Ka o bien pH <
pKa –1,3. Sin embargo, a efectos prácticos y conel fin de facilitar el trazado del diagrama logarít-mico, se suele considerar que las ecuaciones ante-riores son válidas hasta un valor de pH = pKa –1aproximadamente.
Zona 2: pH >> pKa ⇒ [H3O+] << Ka
En esta zona es posible comprobar que:
[3.14]
de donde:
log [HA] = log (C/Ka) – pH [3.15]
La [HA] varía con el pH según una recta dependiente –1. Mientras que:
[3.16]
y por lo tanto:
[3.17]
expresión que corresponde a una línea horizon-tal en el diagrama logarítmico.
Teniendo en cuenta las consideraciones ante-riores estas ecuaciones serán válidas de formaaproximada para pH ≥ pKa +1, como se indicaen la figura 3.3.
Zona 3: pH ≈ pKa ⇒⇒[H3O
+] ≈ Ka (pH = pKa ± 1)
En el intervalo de pH comprendido entrepKa ± 1 no es posible aceptar ninguna de lashipótesis anteriores y las líneas que represen-tan los logaritmos de las concentraciones deHA y A– dejan de ser rectas. Sin embargo, esposible dibujarlas de manera aproximada si setiene en cuenta que para pH = pKa o bien[H3O
+] = Ka se tiene que:
[3.18]
y al sustituir esta condición en las expresiones[3.8] y [3.9] se tiene respectivamente:
[3.19]
[3.20] [A A− −= ⇒ = −] log[ ] log ,
CC
20 3
[HA] [HA] ,= ⇒ = −C
C2
0 3log log
[ ] [ ]H O H O3 3+ ++ = =K Ka a2 2
log[ ] logA− = C
[A
[H O ]3
−+
=+
≈ =]C K
KC KK
Ca
a
a
a
[HA]
[H O ][H O ]
[H O ]3
3
3=+
≈+
+
+CK
CKa a
log log[A ] pH− = +C Ka
[A ]
[H O ] [H O ]3 3
−+ +
=+
≈C KK
C Ka
a
a
log log[HA] = C
[HA]
[H O ][H O ]
[H O ][H O ]
3
3
3
3
=+
≈ =+
+
+
+
CK
CC
a
Capítulo 3: Representación gráfica del equilibrio ácido-base 107
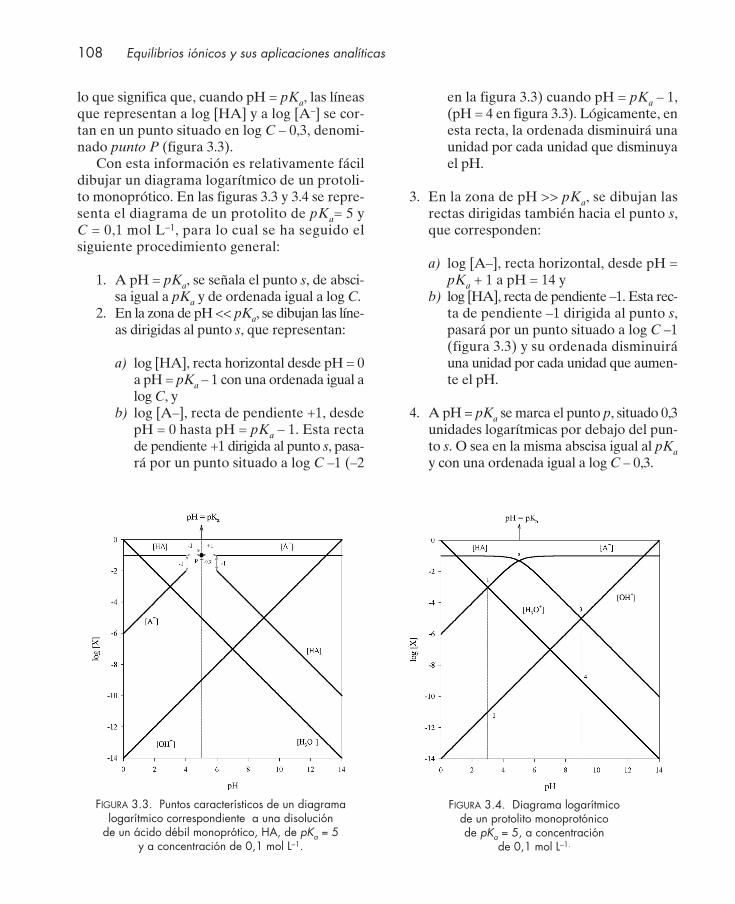
lo que significa que, cuando pH = pKa, las líneasque representan a log [HA] y a log [A–] se cor-tan en un punto situado en log C – 0,3, denomi-nado punto P (figura 3.3).
Con esta información es relativamente fácildibujar un diagrama logarítmico de un protoli-to monoprótico. En las figuras 3.3 y 3.4 se repre-senta el diagrama de un protolito de pKa= 5 yC = 0,1 mol L–1, para lo cual se ha seguido elsiguiente procedimiento general:
1. A pH = pKa, se señala el punto s, de absci-sa igual a pKa y de ordenada igual a log C.
2. En la zona de pH << pKa, se dibujan las líne-as dirigidas al punto s, que representan:
a) log [HA], recta horizontal desde pH = 0a pH = pKa – 1 con una ordenada igual alog C, y
b) log [A–], recta de pendiente +1, desdepH = 0 hasta pH = pKa – 1. Esta rectade pendiente +1 dirigida al punto s, pasa-rá por un punto situado a log C –1 (–2
en la figura 3.3) cuando pH = pKa – 1,(pH = 4 en figura 3.3). Lógicamente, enesta recta, la ordenada disminuirá unaunidad por cada unidad que disminuyael pH.
3. En la zona de pH >> pKa, se dibujan lasrectas dirigidas también hacia el punto s,que corresponden:
a) log [A–], recta horizontal, desde pH =pKa + 1 a pH = 14 y
b) log [HA], recta de pendiente –1. Esta rec-ta de pendiente –1 dirigida al punto s,pasará por un punto situado a log C –1(figura 3.3) y su ordenada disminuiráuna unidad por cada unidad que aumen-te el pH.
4. A pH = pKa se marca el punto p, situado 0,3unidades logarítmicas por debajo del pun-to s. O sea en la misma abscisa igual al pKay con una ordenada igual a log C – 0,3.
108 Equilibrios iónicos y sus aplicaciones analíticas
FIGURA 3.3. Puntos característicos de un diagramalogarítmico correspondiente a una disolución
de un ácido débil monoprótico, HA, de pKa = 5y a concentración de 0,1 mol L–1.
FIGURA 3.4. Diagrama logarítmicode un protolito monoprotónicode pKa = 5, a concentración
de 0,1 mol L–1.

5. Se unen los dos segmentos rectos querepresentan log [HA] en las zonas de pH<< pKa y de pH >> pKa mediante una cur-va que pase por el punto p.
6. Análogamente, se unen los segmentos rec-tos que representa log [A–] en las zonas depH << pKa y de pH >> pKa mediante unacurva que pase por el punto p.
Es evidente que, cuando un diagrama se dibu-ja por el procedimiento descrito, la zona com-prendida entre pH = pKa ± 1, en la que las líne-as que representan log [HA] y log [A–] se han detrazar a mano alzada, es poco adecuada paraobtener información cuantitativa, aunque lainformación cualitativa que suministra es total-mente válida. En cambio, las zonas más alejadasdel pKa, en las que las concentraciones de las dis-tintas especies están representadas por líneas rec-tas, proporcionan información cuantitativacorrecta; de hecho, la diferencia entre el valorde pH calculado numéricamente y el determi-nado mediante un diagrama logarítmico biendibujado raramente es superior a 0,01-0,02 uni-dades.
3.4.1. Determinación del pH de una disoluciónde un ácido débil monoprótico
Tal como se ha indicado en el capítulo 2, enuna disolución de un ácido débil monoprótico,HA, se han de considerar los siguientes equili-brios ácido-base:
HA + H2O →← A– + H3O+ [3.21]
H2O + H2O →← H3O++ OH– [3.1]
y el balance protónico vendrá dado por laexpresión
[H3O+] = [A–] + [OH–] [3.22]
Como ya se explicó en el capítulo 2 al conside-rar el cálculo numérico del pH, si en la expresión
anterior se desprecia la autoprotolisis del agua,generalmente despreciable, se cumplirá que:
[3.23]
Si se considera la figura 3.4, donde están repre-sentadas las especies químicas que se tienen enel sistema constituido por un ácido de pKa = 5 yC = 0,1 mol L–1, la igualdad de la ecuación ante-rior se cumple en el punto 1, que corresponde aun pH = 3, leído en abscisas, que será el pH de la disolución.
Sin embargo, para asegurar que el pH finales el que determina el punto 1 se ha de com-probar que la aproximación realizada, [A–] >>[OH–], sea cierta. En el ejemplo considerado, esevidente que la [OH–] = 10–11, que viene dadapor la ordenada del punto 2, es muy inferior a[A–] = 10–3, dada por la ordenada del punto 1.Puede ser de interés resaltar que los OH– de la disolución pueden provenir únicamente de laautoprotolisis del agua (cada OH– dará unH3O
+), ello implica que [H3O+] proveniente del
agua será igual a 10–11 y por tanto despreciablefrente a [H3O
+] que provienen, de la disociacióndel ácido, igual a 10–3.
Para que [A–] >> [OH–], se ha de cumplir queel segmento 1-2 sea mayor o igual a 1,3 (en elejemplo es mucho mayor), o lo que es lo mismoque [A–] >> 20 [OH–].
3.4.2. Determinación del pH de una disoluciónde una base débil monoprótica
Si se considera una disolución de base débilmonoprótica del tipo NaA, a concentración 0,1mol L–1, y que proviene de un ácido, HA, depKa = 5, esta sustancia estará totalmente diso-ciada en agua y los equilibrios a tener en cuen-ta, de acuerdo con lo indicado en el capítulo 2,serán:
A– + H2O →← HA + OH– [3.24]
H2O + H2O →← H3O+ + OH– [3.1]
[H O ] [A ]3+ −=
Capítulo 3: Representación gráfica del equilibrio ácido-base 109

En estos equilibrios, no aparece la especieNa+, ya que este catión no tiene carácter ácido yno reacciona con el agua. El balance protónicovendrá dado por la expresión:
[H3O+] + [HA] = [OH–] [3.25]
Si se desprecian los iones H3O+, únicamente
aportados en este medio por el agua, frente a[HA], se cumplirá que:
[3.26]
Si se observa la figura 3.4, en la que tambiénse representan todas las especies de la disoluciónde la base en estudio, la igualdad [HA] = [OH–]se cumple en el punto 3 al que le corresponde unpH = 9 (leído en abscisas), que será el pH de ladisolución.
Para comprobar que el pH obtenido es elcorrecto, se ha de cumplir a este valor de pH que[HA] >> [H3O
+]. Se observa en la figura 3.4 quela [HA] = 10–5 mol L–1 (ordenada del punto 3),mientras que [H3O
+] = 10–9 mol L–1 (ordenadadel punto 4), por lo que [HA] >> [ [H3O
+] y lasimplificación es correcta. En general, [H3O
+]será despreciable frente a [HA], siempre que el segmento 3-4 sea mayor o igual a 1,3 o bien[HA] >> 20 [H3O
+].
3.5. Diagrama logarítmico de una mezclade dos protolitos débiles monopróticos. Cálculo del pH
Para dibujar el diagrama de una mezcla dedos protolitos débiles monopróticos, basta consuperponer, en la misma gráfica, los diagramasde cada uno de los protolitos individuales a laconcentración correspondiente. En la figura 3.5se representa el correspondiente a una mezclade HCOOH 0,1 mol L–1 (pKa = 3,75) y deCH3COOH 0,05 mol L–1 (pKa = 4,75).
El pH de esta mezcla se determina medianteel procedimiento descrito anteriormente. En estadisolución, hay que considerar tres equilibrios:
HCOOH + H2O →←→← HCOO– + H3O
+ [3.27]
CH3COOH + H2O →←→← CH3COO– + H3O
+ [3.28]
H2O + H2O →← H3O+ + OH– [3.1]
y el balance protónico vendrá dado por laexpresión:
[H3O+] = [HCOO–] +
+[CH3COO–] + [OH–] [3.29]
FIGURA 3.5. Diagrama logarítmico de una mezcla de dosprotolitos monoprotónicos. HCOOH 0,1 mol L–1 (pKa = 3,75)
y CH3COOH 0,05 mol L–1 (pKa = 4,75).
Para obtener el pH de esta disolución se hade considerar qué términos del segundo miem-bro de la expresión del balance protónico, sondespreciables, y comprobar más tarde que real-mente lo son. En principio, [OH–] en una diso-lución de dos ácidos será despreciable y la expre-sión [3.29] se transforma en:
[H3O+] = [HCOO–] + [CH3COO–] [3.30]
[HA] [OH ]= −
110 Equilibrios iónicos y sus aplicaciones analíticas

De los ácidos disueltos se disociará más aquelque sea más fuerte, así pues cabe pensar que[HCOO–] será mayor que [CH3COO–] y se cum-plirá por tanto:
[3.31]
Esta igualdad se cumple en el punto 1 de lafigura 3.5, al que corresponde un pH = 2,4 queserá el pH de la disolución. Para comprobar quelas aproximaciones realizadas son correctas, seobserva que [OH–] es muy inferior a [HCOO–],[HCOO–] = [H3O
+] = 10–2,4, mientras que [OH–]= 10–11,6 (ordenada del punto 2). También se cum-ple que [CH3COO–] << [HCOO–], ya que el seg-mento 1–3, es mayor que 1,3. Así pues, el pHobtenido es el correcto, aunque debe tenerse encuenta que así como los iones H3O
+ procedentesdel agua, son francamente despreciables, los pro-cedentes del ácido acético, iguales a [CH3COO–]pueden no ser serlo según las concentraciones delos ácidos y en estos casos se deberá proceder alcálculo numérico.
3.6. Diagrama logarítmicode la disolución de una sal.Cálculo del pH
De los cuatro tipos de sales posibles, ya sehan comentado tres, aunque no de forma explí-cita. En efecto, se han considerado los siguien-tes casos: 1) Sal de ácido fuerte + base fuerte(tipo NaCl), que origina una disolución neutra(diagrama del agua). 2) Sal de ácido débil y basefuerte (tipo NaA) que corresponde al diagramade la disolución de una base débil monoprótica.3) Sal de ácido fuerte y base débil (tipo NH4Cl)cuyo diagrama logarítmico corresponde al de unácido débil monoprótico.
Conforme a los diferentes tipos de salesexpuestos en el capítulo 2, sólo falta por comen-tar el caso de una sal de un ácido débil y unabase débil, como es, por ejemplo, HCOONH4,sal de ácido fórmico (pKa = 3,75) y amoníaco(pKa = 9,24).
En la figura 3.6 se representa el diagramalogarítmico de una disolución 0,1 mol L–1 de for-miato de amonio, que no es más que la super-posición de los diagramas correspondientes aHCOO– 0,1 mol L–1 y NH4
+ 0,1 mol L–1, especiesen las que la sal se disocia en agua.
FIGURA 3.6. Diagrama logarítmico de una disolución0,1 mol L–1 de HCOONH4. HCOOH (pKa = 3,75)
y NH4– (pKa = 9,24).
Las reacciones que se dan son las siguientes:
HCOO– + H2O →← HCOOH + OH– [3.32]
NH4+ + H2O →← NH3 + H3O
+ [3.33]
H2O + H2O →← H3O+ + OH– [3.1]
lo que conduce al balance protónico:
[HCOOH] + [H3O+] = [NH3] + [OH–] [3.34]
Esta expresión del balance protónico puedesimplificarse, teniendo en cuenta que, en las reac-ciones de hidrólisis que se producen, se obtienen[H3O
+] y [OH–], que lógicamente reaccionan entresí (el pH no será tan ácido como determinaría el
[H O ] [HCOO ]3+ −=
Capítulo 3: Representación gráfica del equilibrio ácido-base 111

ion NH4+, ni tan básico como en una disolución de
HCOO–) y se puede considerar que [H3O+] <<
[HCOOH] y que [OH–] <<[NH3] , con lo que laexpresión del balance protónico quedaría simpli-ficada a:
[3.35]
Esta igualdad se cumple en el punto 1 de lafigura 3.6, al que corresponde un pH = 6,5, queserá el pH de la disolución siempre que se cum-plan las aproximaciones comentadas. En efecto,el segmento 1-2 es mayor que 1,3 con lo que[H3O
+] << [HCOOH] y todavía mayor es el seg-mento 1-3, con lo que [OH–] << [NH3]. Hay quetener en cuenta que las concentraciones de lasespecies se leen siempre en ordenadas. Así pues,el pH = 6,5 obtenido es perfectamente correcto.
Como resumen, en el recuadro 3.1 se exponeel procedimiento general a seguir para determi-nar el pH a partir de un diagrama logarítmico.
3.7. Diagrama logarítmicode protolitos dipróticos
En el caso de protolitos dipróticos, para laconstrucción del diagrama logarítmico de con-centración se divide la escala pH en cinco zonasbien diferenciadas:
Zona 1: pH << pKa1 << pKa2 ⇒⇒ [H3O
+] >> Ka1 >> Ka2 ⇒⇒ [H3O
+]2 + Ka1[H3O+] + Ka1Ka2 ≈ [H3O
+]2
En esta zona se cumple:
[3.36]
de donde:
[]
] ]
]
H A][H O
[H O [H O
[H O ][H O
23
3 3
3
3
=+ +
≈ =
+
+ +
+
+
CK K K
CC
a a a
2
21 1 2
2
2
[HCOOH] [NH ]3=
112 Equilibrios iónicos y sus aplicaciones analíticas
El procedimiento para determinar el pH de una disolucióna partir del diagrama logarítmico del sistema consta de lospasos siguientes:
1. Se dibuja el diagrama logarítmico de concentracióncorrespondiente en el que se representan todas lasespecies presentes en la disolución.
2. Se obtiene el balance protónico del sistema ácido-base.
3. Se simplifica esta expresión del balance protónico demanera que nos quede un término en cada miembrode la ecuación. En este proceso es de interés resal-tar que los términos [H3O+] y [OH–], cuando no estánsolos en un miembro, provienen de la autoprotolisisdel agua en disoluciones de bases y ácidos respec-tivamente y en consecuencia serán despreciables fren-te a otras concentraciones. También en el caso de lassales de ácidos y bases débiles, serán menores losvalores de [H3O+] y [OH–] que las de otras concen-traciones al reaccionar estos iones con los genera-dos por la hidrólisis.
4. Se señala el punto del diagrama en el que se cum-ple la igualdad obtenida con el balance protónicosimplificado. La abscisa de este punto nos dará elpH de la disolución.
5. Finalmente, se confirma que el pH determinado esel correcto, comprobando que las concentracionesde las especies, que han desaparecido al simplifi-car el balance protónico eran realmente despre-ciables. Si es así, el pH real de la disolución es eldeterminado por el punto de intersección que cum-ple el balance protónico. De no ser así, el pH exac-to debe ser calculado mediante un procedimientonumérico.
(Existen métodos de corrección que permiten obtenervalores exactos de pH a partir del diagrama logarítmi-co, incluso en los casos en que más de una reacciónejerce una influencia importante en la [H3O+]. Sin embar-go, dichos métodos son engorrosos y no se comentarán,ya que en estas circunstancias es mejor recurrir al cálcu-lo numérico.)
Procedimiento general para el cálculo del pH a partir de diagramas logarítmicos de concentración
RECUADRO 3.1

[3.37]
ecuación que corresponde a una recta horizon-tal en el diagrama logarítmico de concentración.Por otra parte, para la especie HA– se tiene:
[3.38]
y tomando logarítmos:
[3.39]
expresión que corresponde a una recta de pendiente+1. Finalmente, para la especie A2– se llega a:
[3.40]
[3.41]
ecuación de una recta de pendiente +2 en el dia-grama logarítmico de la concentración.
Zona 2: pH = pKa1 << pKa2 ⇒⇒ [H3O
+] = Ka1 >> Ka2 ⇒ [H3O+]2 +
+ Ka1[H3O+] + Ka1Ka2 ≈ 2 [H3O
+]2 = 2 Ka1[H3O+]
Siguiendo un procedimiento similar al esta-blecido anteriormente y aplicando en las expre-siones de las concentraciones de H2A, HA– y A2–
las simplificaciones correspondientes se tiene:
[3.42]
[3.43]
[3.44]
[3.45]
[3.46]
[3.47]
A tenor de las expresiones [3.43], [3.45] y[3.47] se puede establecer que cuando pH = pKa1,las líneas de log [H2A] y log [HA–] se cortan enun punto situado 0,3 unidades logarítmicas pordebajo del punto del sistema, mientras que lalínea correspondiente al log [A2–] corresponde auna recta de pendiente +1.
Zona 3: pKa1 << pH << pKa2 ⇒⇒ Ka1 >> [H3O
+] >> Ka2 ⇒⇒ [H3O
+]2 + Ka1[H3O+] + Ka1Ka2 ≈ Ka1 [H3O
+]
Operando de forma similar se tiene:
[3.48]
[3.49]
[3.50]
[HA ]H O
H O H O ]
H OH O
3
3 3
3
3
−+
+ +
+
+
=+ +
≈
≈ =
C KK K K
C KK
C
a
a a a
a
a
12
1 1 2
1
1
[ ][ ] [
[ ][ ]
log[H A log log pH2 ] = − −C Ka1
[H A][H O
[H O [H O
[H O[H O
[H O
23
3 3
3
3
3
=+ +
≈
≈ =
+
+ +
+
+
+
CK K K
CK
CK
a a a
a a
]] ]
]]
]
2
21 1 2
2
1 1
log[ ] log log ,A pH2− = + − +C Ka 2 0 3
[A ][H O ] [H O ]
2 [H O ] 2[H O ]
2
32
3
3 3
−+ +
+ +
=+ +
≈
≈ =
C K KK K K
C K KK
C K
a a
a a a
a a
a
a
1 2
1 1 2
1 2
1
2
log[ ] log ,HA− = −C 0 3
[HA ][H O ]
[H O ] [H O ]
[H O ]2 [H O ] 2
3
32
3
3
3
−+
+ +
+
+
=+ +
≈
≈ =
C KK K K
C KK
C
a
a a a
a
a
1
1 1 2
1
1
log[H A log 0,32 ] = −C
[H A][H O
[H O [H O
[H O[H O
23
3 3
3
3
=+ +
≈
≈ =
+
+ +
+
+
CK K K
C Ca a a
]] ]
]]
2
21 1 2
2
22 2
log[ ] log log
log
A
pH
2− = + +
+ +
C K
Ka
a
1
2 2
[A ]H O H O
H O
2
3 3
3
−+ +
+
=+ +
≈
≈
C K KK K K
C K K
a a
a a a
a a
1 22
1 1 2
1 22
[ ] [ ]
[ ]
log ] log log[HA pH− = + +C Ka1
[ ][ ]
[ ] [ ][ ]
[ ] [
HAH O
H O H OH O
H O H O ]
3
32
3
3
3 3
−+
+ +
+
+ +
=+ +
≈
≈ =
C K
K K K
C K C K
a
a a a
a a
1
1 1 2
12
1
log log[H A]2 = C
Capítulo 3: Representación gráfica del equilibrio ácido-base 113

[3.51]
[3.52]
[3.53]
Las expresiones mostradas en [3.49], [3.51]y [3.53] corresponden a rectas de pendiente –1,0 (línea horizontal) y +1 en el diagrama loga-rítmico.
Zona 4: pKa1 << pH = pKa2 ⇒⇒ Ka1 >> [H3O
+] = Ka2 ⇒⇒ [H3O
+]2 + Ka1[H3O+] + Ka1Ka2 ≈
≈ 2 Ka1[H3O+] = 2 Ka1Ka2
En esta zona se tiene:
[3.54]
[3.55]
[3.56]
[3.57]
[3.58]
[3.59]
Lo que significa que si pH = pKa2, las líneas querepresentan log [HA–] y log [A2–] se cortan en unpunto situado 0,3 unidades logarítmicas por deba-jo del punto del sistema, mientras que el log [H2A]viene representado por una línea de pendiente –1.
Zona 5: pKa1 << pKa2 << pH⇒ Ka1 >> Ka2 >> [H3O
+] ⇒⇒ [H3O
+]2 + Ka1[H3O+] + Ka1Ka2 ≈ Ka1Ka2
En esta zona se cumple:
[3.60]
[3.61]
[3.62]
[3.63]
[3.64]
[3.65]
Las expresiones [3.61], [3.63] y [3.65] corres-ponden a rectas de pendiente –2, –1 y 0 (líneahorizontal) en el diagrama logarítmico.
Conforme a las expresiones obtenidas, el dia-grama logarítmico de concentración para un pro-tolito diprótico (por ejemplo, H2A de pKa1 = 4 ypKa2 = 8, a C = 0,1 mol L–1, en la figura 3.7) seconfecciona en base a las siguientes etapas:
log[ ] logA2− = C
[ ][ ] [ ]
AH O H O
2
3 3
−+ +
=+ +
≈
≈ =
C K KK K K
C K KK K
C
a a
a a a
a a
a a
1 22
1 1 2
1 2
1 2
log[ log logHA ] pH− = − −C Ka 2
[HA ]H O ]
[H O ] [H O ]
H O H O
3
3 3
3 3
−+
+ +
+ +
=+ +
≈
≈ =
C KK K K
C KK K
CK
a
a a a
a
a a a
12
1 1 2
1
1 2 22
[
[ ] [ ]
log log log
log
[H A]
pH2 = − −
− −
C K
Ka
a
1
2 2
[H A][H O
[H O [H O
[H O ]
23
3 3
3
=+ +
≈
≈
+
+ +
+
CK K K
CK K
a a a
a a
]] ]
2
21 1 2
2
1 2
log[ ] log ,A2− = −C 0 3
[ ][ ] [ ]
AH O H O
2
3 3
−+ +
=+ +
≈
≈ =
C K KK K K
C K KK K
C
a a
a a a
a a
a a
1 22
1 1 2
1 2
1 22 2
log[ log ,HA ]− = −C 0 3
[HA ][H O ]
H O ] H O ]
H O ]H O
3
3 3
3
3
−+
+ +
+
+
=+ +
≈
≈ =
C KK K K
C KK
C
a
a a a
a
a
12
1 1 2
1
12 2
[ [
[[ ]
log log log ,[H A] pH2 = − − −C Ka1 0 3
[] ]
]]
]
H A][H O ]
[H O [H O
[H O[H O
[H O
23
3 3
3
3
3
=+ +
≈
≈ =
+
+ +
+
+
+
CK K K
CK
CK
a a a
a a
2
21 1 2
2
1 12 2
log[ log logA ] pH2− = + +C Ka 2
[A ][H O ] [H O ]
[H O ] H O ]
2
3 3
3 3
−+ +
+ +
=+ +
≈
≈ =
C K KK K K
C K KK
C K
a a
a a a
a a
a
a
1 22
1 1 2
1 2
1
2
[
log[ ] logHA− = C
114 Equilibrios iónicos y sus aplicaciones analíticas
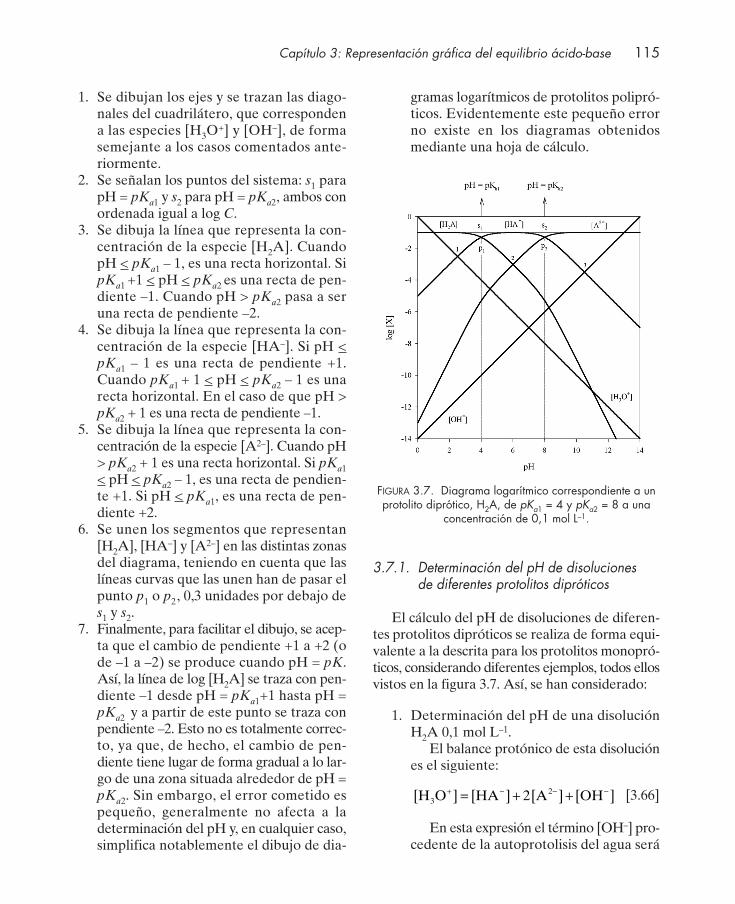
1. Se dibujan los ejes y se trazan las diago-nales del cuadrilátero, que correspondena las especies [H3O
+] y [OH–], de formasemejante a los casos comentados ante-riormente.
2. Se señalan los puntos del sistema: s1 parapH = pKa1 y s2 para pH = pKa2, ambos conordenada igual a log C.
3. Se dibuja la línea que representa la con-centración de la especie [H2A]. CuandopH < pKa1 – 1, es una recta horizontal. SipKa1 +1 < pH < pKa2 es una recta de pen-diente –1. Cuando pH > pKa2 pasa a seruna recta de pendiente –2.
4. Se dibuja la línea que representa la con-centración de la especie [HA–]. Si pH <pKa1 – 1 es una recta de pendiente +1.Cuando pKa1 + 1 < pH < pKa2 – 1 es unarecta horizontal. En el caso de que pH >pKa2 + 1 es una recta de pendiente –1.
5. Se dibuja la línea que representa la con-centración de la especie [A2–]. Cuando pH> pKa2 + 1 es una recta horizontal. Si pKa1< pH < pKa2 – 1, es una recta de pendien-te +1. Si pH < pKa1, es una recta de pen-diente +2.
6. Se unen los segmentos que representan[H2A], [HA–] y [A2–] en las distintas zonasdel diagrama, teniendo en cuenta que laslíneas curvas que las unen han de pasar elpunto p1 o p2, 0,3 unidades por debajo des1 y s2.
7. Finalmente, para facilitar el dibujo, se acep-ta que el cambio de pendiente +1 a +2 (ode –1 a –2) se produce cuando pH = pK.Así, la línea de log [H2A] se traza con pen-diente –1 desde pH = pKa1+1 hasta pH =pKa2 y a partir de este punto se traza conpendiente –2. Esto no es totalmente correc-to, ya que, de hecho, el cambio de pen-diente tiene lugar de forma gradual a lo lar-go de una zona situada alrededor de pH =pKa2. Sin embargo, el error cometido espequeño, generalmente no afecta a ladeterminación del pH y, en cualquier caso,simplifica notablemente el dibujo de dia-
gramas logarítmicos de protolitos polipró-ticos. Evidentemente este pequeño errorno existe en los diagramas obtenidosmediante una hoja de cálculo.
FIGURA 3.7. Diagrama logarítmico correspondiente a unprotolito diprótico, H2A, de pKa1 = 4 y pKa2 = 8 a una
concentración de 0,1 mol L–1.
3.7.1. Determinación del pH de disolucionesde diferentes protolitos dipróticos
El cálculo del pH de disoluciones de diferen-tes protolitos dipróticos se realiza de forma equi-valente a la descrita para los protolitos monopró-ticos, considerando diferentes ejemplos, todos ellosvistos en la figura 3.7. Así, se han considerado:
1. Determinación del pH de una disoluciónH2A 0,1 mol L–1.
El balance protónico de esta disoluciónes el siguiente:
[3.66]
En esta expresión el término [OH–] pro-cedente de la autoprotolisis del agua será
[H O ] [HA ] 2[A ] [OH ]32+ − − −= + +
Capítulo 3: Representación gráfica del equilibrio ácido-base 115

despreciable y en la mayoría de los casos 2[A2–] << [HA–] al formarse A2– tras la segun-da disociación, a partir de HA–. Así puescabe esperar que se cumpla que:
[3.67]
Esta igualdad se cumple en el punto 1del diagrama, figura 3.7, con lo que el pHde esta disolución será 2,5. Se observa enesta figura que en efecto a este pH, [OH–]es muy inferior a [HA–], [OH–] = 10–11,5 , y2 [A2–] << [HA–] ya que [A2–] ≈ 10–8 molL–1 leída en ordenadas, con lo que se con-firma la simplificación realizada.
2. Determinación del pH de una disoluciónde NaHA 0,1 mol L–1.
El balance protónico de la disoluciónde este anfolito, como se mostró en el capí-tulo 2 al considerar el cálculo del pH deforma numérica, viene dado por:
[3.68]
En esta expresión cabe pensar que[H3O
+] << [HA–] y [OH–] << [A2–], ya queel medio no será muy ácido ni muy básico,con lo que se cumplirá que:
[3.69]
expresión que se cumple en el punto 2 deldiagrama de la figura 3.7, al que le corres-ponde un pH = 6. A este pH, como indicael punto 2, [H2A] = [A2–] ≈ 10–3 mol L–1 leí-do en ordenadas y por tanto [HA–] >>[H3O
+] = 10–6 mol L–1 y [A2–] >> [OH–] =10–8 mol L–1.
3. Determinación del pH de una disoluciónde Na2A 0,1 mol L–1.
El balance protónico para una disolu-ción de esta sal, como se ha indicado en elcapítulo 2, será:
[3.70]
En esta expresión el término [H3O+] será
despreciable al ser la disolución de una base
y por otra parte 2 [H2A] << [HA–] en lamayoría de los casos, ya que la especie H2Ase origina a partir de HA– considerando elequilibrio que rige K2 generalmente peque-ño. Así pues se ha de cumplir que:
[3.71]
Esto se cumple en el punto 3 de la figu-ra 3.7 al que le corresponde un pH = 10,5.A este pH, en efecto, como muestra suordenada [HA–] = 10–3,5 mol L–1 = [OH–] yclaramente las concentraciones [H3O
+] y[H2A] están muy por debajo de este valor,muy inferior a 1,3 unidades en el diagrama:[H3O
+] = 10–10,5 mol L–1 y [H2A] algo mayor,en ambos casos leídas en ordenadas.
3.8. Diagrama logarítmicode protolitos polipróticos
En el caso de un protolito triprótico, siguien-do un tratamiento similar al realizado para losotros protolitos estudiados se pueden establecerdiferentes zonas de predominio para la construc-ción del correspondiente diagrama logarítmicode la concentración. En el cuadro 3.1 se muestranlas diferentes zonas estudiadas así como las expre-siones logarítmicas correspondientes para cadauna de las especies presentes en la disolución.
Es fácil comprobar a partir de las expresio-nes mostradas en el cuadro 3.1 que los diagra-mas logarítmicos ofrecen un comportamientosistemático que permite dibujarlos sin que seanecesaria una deducción completa. Así porejemplo, la línea de log [H3A], con pendiente 0 cuando pH < pK1, pasa a tener pendiente –1cuando pK1 + 1 < pH < pKa2 – 1, posteriormen-te a pendiente –2 y finalmente pendiente –3; lalínea de log [HA–], de pendiente +1 a pH <pKa1, después tiene valores de 0, –1 y –2 y asísucesivamente. En la figura 3.8 se representa eldiagrama logarítmico de un ácido triprótico depKa1 = 4, pKa2 = 7 y pKa3 = 10 y de concentra-ción analítica 0,1 mol L–1.
[HA ] [OH ]− −=
[H O ] [HA ] 2[H A] [OH ]3 2+ − −+ + =
[H A] [A ]22= −
[H O ] [H A] [A ] [OH ]3 22+ − −+ = +
[H O ] [HA ]3+ −=
116 Equilibrios iónicos y sus aplicaciones analíticas

FIGURA 3.8. Diagrama logarítmico correspondiente a unprotolito triprótico, H3A, de pKa1 = 4, pKa2 = 7 y pKa3 = 10
a una concentración analítica de 0,1 mol L–1.
El estudio de la determinación del pH de diso-luciones de diferentes protolitos polipróticos se lle-va a cabo de manera similar a la expuesta en casosanteriores, esto es: 1) establecimiento del balanceprotónico, 2) simplificación del mismo (punto delsistema), 3) cálculo gráfico del pH, y 4) confirma-ción de que la simplificación es correcta. En el recua-dro 3.2 se muestra de forma esquemática el cálculodel pH de diferentes disoluciones de protolitos poli-próticos haciendo uso del diagrama logarítmico dela concentración mostrado en la figura 3.8.
3.9. Diagramas de distribuciónde especies en función del pH
Otra manera de representar gráficamente lasconcentraciones de las especies que intervienenen los equilibrios que tienen lugar en un siste-ma determinado, es mediante los diagramas dedistribución. Un diagrama de distribución con-siste en una representación gráfica que permitevisualizar la variación de concentración de las
Capítulo 3: Representación gráfica del equilibrio ácido-base 117
CUADRO 3.1Expresiones logarítmicas empleadas en la construcción
de un diagrama logarítmico de concentraciónde un protolito poliprótico
1. pH << pKa1 << pKa2 << pKa3
log [H3A] ⇒ Recta horizontallog [H2A–] ⇒ Recta pendiente +1log [HA2–] ⇒ Recta pendiente +2log [A3–] ⇒ Recta pendiente +3
2. pH << pKa1 << pKa2 << pKa3
log [H3A] = log C – 0,3log [H2A–] = log C – 0,3log [HA2–] ⇒ Despreciablelog [A3–] ⇒ Despreciable
3. pKa1 << pH << pKa2 << pKa3
log [H3A] ⇒ Recta pendiente –1log [H2A–] ⇒ Recta horizontallog [HA2–] ⇒ Recta pendiente +1log [A3–] ⇒ Recta pendiente +2
4. pKa1 << pH = pKa2 << pKa3
log [H3A] ⇒ Despreciablelog [H2A–] = log C – 0,3log [HA2–] = log C – 0,3log [A3–] ⇒ Despreciable
5. pKa1 << pKa2 << pH << pKa3
log [H3A] ⇒ Recta pendiente –2log [H2A–] ⇒ Recta pendiente –1log [HA2–] ⇒ Recta horizontallog [A3–] ⇒ Recta pendiente +1
6. pKa1 << pKa2 << pH << pKa3
log [H3A] ⇒ Despreciablelog [H2A–] ⇒ Despreciablelog [HA2–] = log C – 0,3log [A3–] = log C – 0,3
7. pKa1 << pKa2 << pKa3 << pH
log [H3A] ⇒ Recta pendiente –3log [H2A–] ⇒ Recta pendiente –2log [HA2–] ⇒ Recta pendiente –1log [A3–] ⇒ Recta horizontal

diferentes especies, expresada según una ciertafunción αi, respecto al pH.
La función αi representa la fracción molar dela especie i y se define como la relación entre laconcentración en el equilibrio de dicha especiey la concentración analítica.
[3.72]
La construcción de este tipo de diagramasrequiere del conocimiento de las constantes ácido-base del protolito.
A continuación, como ejemplo, se deduce eldiagrama de distribución para un ácido débilmonoprótico, HA. El equilibrio de disociaciónde este ácido:
HA + H2O →← A– + H3O+ [3.21]
viene regido por la constante:
[3.73]
siendo su balance de masas el siguiente:
CHA= [HA] + [A–] [3.74]
Según la definición dada de fracción molar,para cada de las especies del ácido HA presen-tes en la disolución se tiene:
[3.75]
[3.76]αA
HA
–
A=−[ ]
C
αHA
HA
HA= [ ]C
Ka =
+ −[H O ][A ][HA]
3
α i C
= [i
i
]
118 Equilibrios iónicos y sus aplicaciones analíticas
1. Disolución de H3A 0,1 mol L–1
Balance protónico:
Punto 1 del diagrama (figura 3.8)
pH = 2,5
2. Disolución de NaH2A 0,1 mol L–1
Balance protónico:
Punto 2 del diagrama (figura 3.8):
pH = 5,4
3. Disolución de Na2HA 0,1 mol L–1
Balance protónico:
Punto 3 del diagrama (figura 3.8):
pH = 8,4
4. Disolución de Na3A 0,1 mol L–1
Balance protónico:
Punto 4 del diagrama (figura 3.8):
pH = 11,5
[HA ] [OH ]2− −=
[H O ] 3[H A] 2[H A ] [HA ] [OH ]3 3 22+ − − −+ + + =
[H A ] [A ]23-− =
[H O ] 2[H A] [H A ] [A ] [OH ]3 3 23-+ − −+ + = +
[H A] [HA ]32-=
[H O ] [H A] [HA ] 2[A ]3 32- 3-+ + = + +
[H O ] [H A ]3 2+ −=
[H O ] [H A ] 2[HA ] 3[A ] [OH ]3 22- 3-+ − −= + + +
Cálculo del pH de disoluciones de diferentes protolitos polipróticos
Se muestran a continuación de forma esquemática las expresiones a utilizar para el cálculo del pHde disoluciones de diferentes protolitos polipróticos, tomando como base la figura 3.8
RECUADRO 3.2

y además, es fácilmente demostrable que la sumade fracciones molares es igual a la unidad, esdecir:
[3.77]
Con objeto de relacionar las fracciones mola-res de cada especie con el pH es necesario susti-tuir en las expresiones [3.75] y [3.76] las concen-traciones en el equilibrio de las especies HA yA– por sus expresiones en función de [H3O
+], CHAy Ka, [3.8] y [3.9], respectivamente:
[3.8]
[3.9]
Sustituyendo estas expresiones en las ecua-ciones [3.75] y [3.76] se obtiene:
[3.78]
[3.79]
De forma similar a los diagramas logarítmicosde la concentración, para la construcción del diagrama de distribución se pueden considerardiversos puntos característicos correspondientes avalores significativos de pH. A continuación semuestran estos puntos del sistema y el valor quetoman las fracciones molares para las especies HAy A– haciendo uso de las expresiones [3.78] y [3.79].
1) pH = pKa ⇒ [H3O+] = Ka
[3.80]
[3.81]
2) pH = pKa – 1 ⇒ [H3O+] = 10 Ka
[3.82]
[3.83]
3) pH = pKa – 2 ⇒ [H3O+] = 100 Ka
[3.84]
[3.85]
4) pH = pKa + 1 ⇒ 10 [H3O+] = Ka
[3.86]
[3.87]
αA3
3
3
3 3
–
H O[H O
10 [H O ]10 [H O ] [H O ]
=+
=
= ×× +
= =
+
+
+
+ +
[ ]]
,
Ka
1011
0 9
αHA3
3
3
3 3
H OH O ]
[H O ]10 [H O ] [H O ]
=+
=
=× +
= = ≈
+
+
+
+ +
[ ][
, ,
Ka
111
0 09 0 1
αA3H O ]
− =+
=
=× +
= = ≈
+
KK
KK K
a
a
a
a a
[
, ,100
1101
0 009 0 01
αHA
3
3
H OH O
=+
= ×× +
= =+
+
[ ][ ]
,K
KK Ka
a
a a
100100
100101
0 99
αA
3H O− =
+=
× += = ≈
+
KK
KK K
a
a
a
a a[ ], ,
10111
0 09 0 1
αHA
3
3
H OH O
=+
= ×× +
= =+
+
[ ][ ]
,K
KK Ka
a
a a
1010
1011
0 9
αA
3[H O ]− =
+=
+= =
+
KK
KK K
a
a
a
a a
12
0 5,
αHA
3
3
3
3 3
[H O ][H O ]
[H O ][H O ] [H O ]
=+
=+
= =+
+
+
+ +Ka
12
0 5,
αA
3H O− =
++
KK
a
a[ ]
αHA
3
3
H OH O
=+
+
+
[ ][ ] Ka
[ ]A
[H O ]3
−+
=+
C KK
a
a
[HA]
[H O ][H O ]
3
3
=+
+
+
CKa
α αHA A+ =− 1
Capítulo 3: Representación gráfica del equilibrio ácido-base 119

5) pH = pKa + 2 ⇒ 100 [H3O+] = Ka
[3.88]
[3.89]
La representación del valor de las expresiones[3.80] – [3.89] en función del pH se obtiene el dia-grama de distribución, el cual para un ácido mono-prótico de pKa = 4,0 se muestra en la figura 3.9. Estediagrama permite conocer en todo momento la frac-ción molar de cada una de las especies a un deter-minado valor de pH y por ende su concentraciónen disolución a partir de la concentración analítica.
FIGURA 3.9. Diagrama de distribución de un ácido débil monoprótico de pKa = 4.
De forma semejante se puede actuar para obte-ner el diagrama de distribución de un ácido dipró-tico, teniendo en cuenta que para las especies aconsiderar las fracciones molares vienen dadas por:
[3.90]
[3.91]
[3.92]
Al sustituir las concentraciones de las espe-cies H2A, HA– y A2– por las expresiones que lasrelacionan con [H3O
+], CH2A, Ka1 y Ka2, deducidas
en el capítulo 2, las ecuaciones para α H2A, αHA¯ y
αA2– en función del pH vienen dadas por:
[3.93]
[3.94]
[3.95]
Introduciendo las simplificaciones adecuadasen función de la zona de pH sobre el denomina-dor, D = [H3O
+]2 + [H3O+] Ka1 + Ka1 Ka2, de estas
expresiones se pueden obtener fácilmente losvalores de las fracciones molares para cada espe-cie. Estas simplificaciones son las siguientes:
1. [H3O+] > Ka1 > Ka2
D = [H3O+]2
2. 100 Ka1 > [H3O+] > 0,01Ka1
D = [H3O+]2 + [H3O
+] Ka1
3. 100 Ka2 > [H3O+] > 0,01Ka2
D = [H3O+] Ka1 + Ka1 Ka2
4. [H3O+] < Ka2
D = Ka1 Ka2
αA
32
3
2
H O ] [H O− =
+ ++ +
K KK K K
a a
a a a
1 2
1 1 2[ ]
αHA
3
32
3
[H OH O ] [H O ]
− =+ +
+
+ +
][
KK K K
a
a a a
1
1 1 2
αH A
3
32
32
H OH O ] [H O ]
=+ +
+
+ +
[ ][
2
1 1 2K K Ka a a
αA
HA
2–
A=−[ ]2
C
αHA
HA
HA=−[ ]
C
αH A
2
HA2
H A= [ ]C
αA3
3
3
3 3
–
[H OH O
100 [H O ]100 [H O ] [H O ]
=+
=
= ×× +
= =
+
+
+
+ +
][ ]
,
Ka
100101
0 99
αHA3
3
3
3 3
H OH O
[H O ]100 [H O ] [H O ]
=+
=
=× +
= = ≈
+
+
+
+ +
[ ][ ]
, ,
Ka
1101
0 009 0 01
120 Equilibrios iónicos y sus aplicaciones analíticas

En el cuadro 3.2 se muestran los valores delas fracciones molares de cada una de las espe-cies en disolución en función del pH para valo-res característicos del mismo que permiten dibu-jar el diagrama de distribución mostrado en lafigura 3.10 para el caso concreto de un ácidodiprótico de pKa1 = 4 y pKa2 = 8.
Hay que reseñar finalmente, que la proximi-dad entre los valores de pKa modifica en mayor
o menor medida la forma del diagrama de mane-ra que la fracción molar de la especie intermediaHA– disminuye y su máximo por tanto no alcan-za el valor cercano a la unidad. Este comporta-miento se observa claramente en la figura 3.11,que corresponde al diagrama de distribución deun ácido diprótico cuyos valores de pKa son:
pKa1 = 5,0 y pKa2 = 7,0
Capítulo 3: Representación gráfica del equilibrio ácido-base 121
CUADRO 3.2
Valores de fracciones molares de las especies presentes en la disolución de un protolito diprótico
Puntos característicos del diagrama αH2A αHA¯ αA 2¯
pH = pKa1 –2 [H3O+] = 100 Ka1 0,99 0,01 DespreciablepH = pKa1 –1 [H3O+] = 10 Ka1 0,9 0,1 DespreciablepH = pKa1 [H3O+] = Ka1 0,5 0,5 DespreciablepH = pKa1 +1 10 [H3O+] = Ka1 0,1 0,9 DespreciablepH = pKa1 +2 100 [H3O+] = Ka1 0,01 0,99 DespreciablepH = pKa2 –2 [H3O+] = 100 Ka2 Despreciable 0,99 0,01pH = pKa2 –1 [H3O+] = 10 Ka2 Despreciable 0,9 0,1pH = pKa2 [H3O+] = Ka2 Despreciable 0,5 0,5pH = pKa2 +1 10 [H3O+] = Ka2 Despreciable 0,1 0,9pH = pKa2 +2 100 [H3O+] = Ka2 Despreciable 0,01 0,99
FIGURA 3.10. Diagrama de distribución de un ácido diprótico de pKa1 = 4 y pKa2 = 8.
FIGURA 3.11. Diagrama de distribución de un ácido diprótico de pKa1 = 5 y pKa2 = 7.
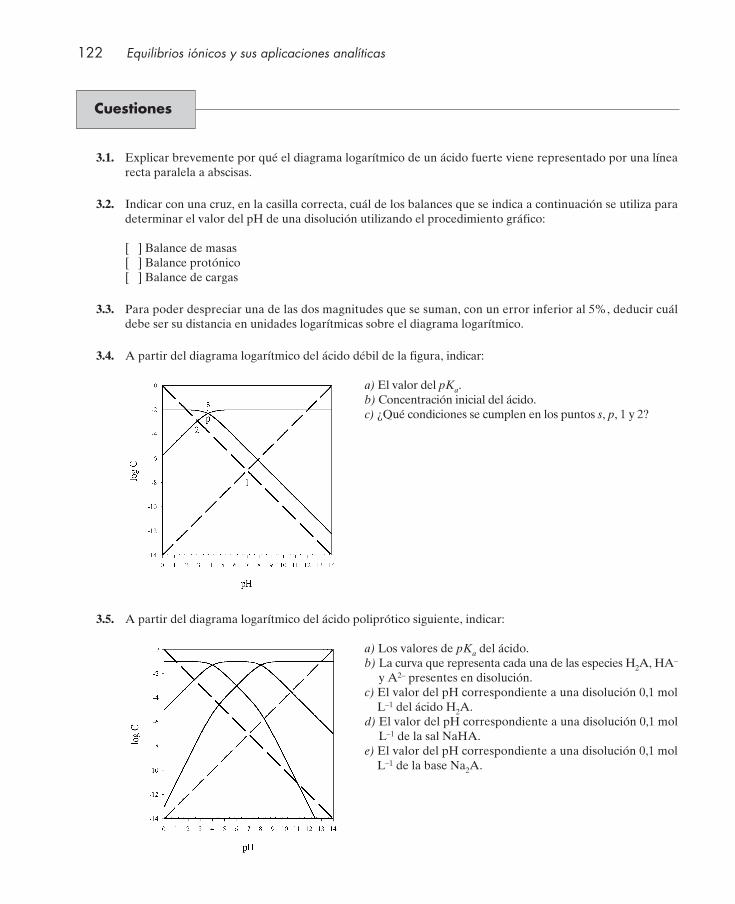
122 Equilibrios iónicos y sus aplicaciones analíticas
3.1. Explicar brevemente por qué el diagrama logarítmico de un ácido fuerte viene representado por una línearecta paralela a abscisas.
3.2. Indicar con una cruz, en la casilla correcta, cuál de los balances que se indica a continuación se utiliza paradeterminar el valor del pH de una disolución utilizando el procedimiento gráfico:
[ ] Balance de masas[ ] Balance protónico[ ] Balance de cargas
3.3. Para poder despreciar una de las dos magnitudes que se suman, con un error inferior al 5%, deducir cuáldebe ser su distancia en unidades logarítmicas sobre el diagrama logarítmico.
3.4. A partir del diagrama logarítmico del ácido débil de la figura, indicar:
a) El valor del pKa.b) Concentración inicial del ácido.c) ¿Qué condiciones se cumplen en los puntos s, p, 1 y 2?
3.5. A partir del diagrama logarítmico del ácido poliprótico siguiente, indicar:
a) Los valores de pKa del ácido.b) La curva que representa cada una de las especies H2A, HA–
y A2– presentes en disolución.c) El valor del pH correspondiente a una disolución 0,1 mol
L–1 del ácido H2A.d) El valor del pH correspondiente a una disolución 0,1 mol
L–1 de la sal NaHA.e) El valor del pH correspondiente a una disolución 0,1 mol
L–1 de la base Na2A.
Cuestiones
3.6. A partir del diagrama logarítmico anterior indicar qué especie o especies son las predominantes a lossiguientes valores de pH
pH Especies
2,0
4,0
6,0
8,0
10,0
12,0
3.7. A partir del diagrama logarítmico anterior, indica cuál es la concentración de cada una de las especiespresentes a pH = 7.
3.8. ¿Qué información se puede obtener a partir del diagrama de distribución de especies?
3.9. A partir del diagrama de distribución de la figura, indicar:
a) ¿Que pKa tiene este ácido?b) ¿Cuál es la especie que predomina a pH = 6,5?
3.10. Dibujar aproximadamente cuál sería la forma de un diagrama de distribución de un ácido diprótico de pKa1 = 2,5 y pKa2 = 8,3.
3.11. ¿Cómo se modifica la forma del diagrama de distribución de un ácido diprótico, cuando los valores de pKaestán relativamente próximos, como por ejemplo pKa1 = 4,8 y pKa2 = 7,3?
3.12. A partir del diagrama de distribución de la cuestión 9, indicar en qué intervalo de pH se obtendría una diso-lución tampón con capacidad reguladora elevada.
Capítulo 3: Representación gráfica del equilibrio ácido-base 123
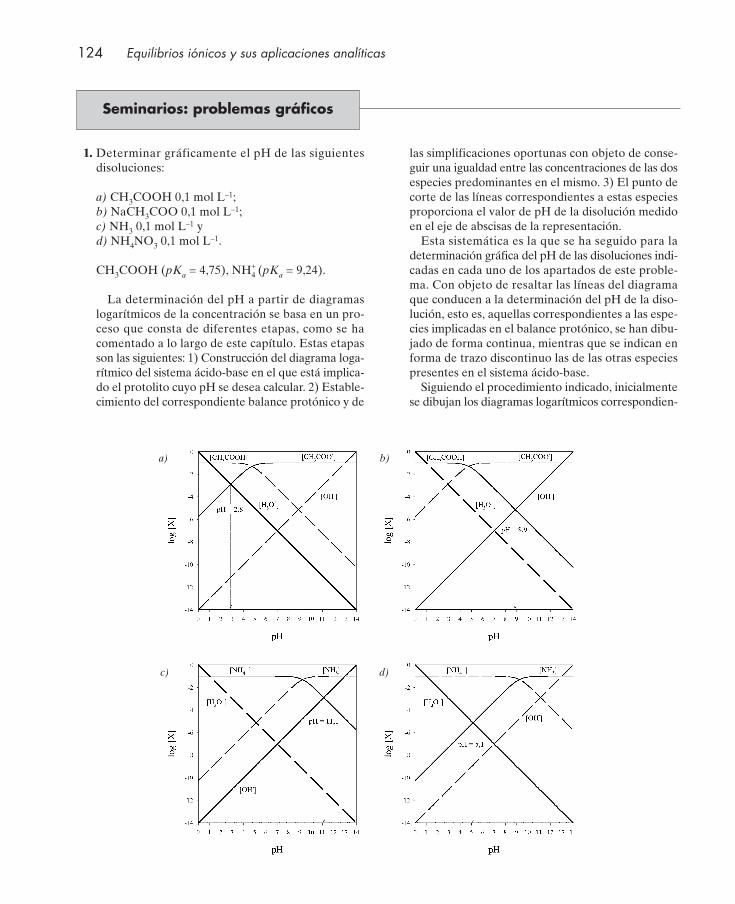
124 Equilibrios iónicos y sus aplicaciones analíticas
1. Determinar gráficamente el pH de las siguientesdisoluciones:
a) CH3COOH 0,1 mol L–1;b) NaCH3COO 0,1 mol L–1; c) NH3 0,1 mol L–1 yd) NH4NO3 0,1 mol L–1.
CH3COOH (pKa = 4,75), NH4+ (pKa = 9,24).
La determinación del pH a partir de diagramaslogarítmicos de la concentración se basa en un pro-ceso que consta de diferentes etapas, como se hacomentado a lo largo de este capítulo. Estas etapasson las siguientes: 1) Construcción del diagrama loga-rítmico del sistema ácido-base en el que está implica-do el protolito cuyo pH se desea calcular. 2) Estable-cimiento del correspondiente balance protónico y de
las simplificaciones oportunas con objeto de conse-guir una igualdad entre las concentraciones de las dosespecies predominantes en el mismo. 3) El punto decorte de las líneas correspondientes a estas especiesproporciona el valor de pH de la disolución medidoen el eje de abscisas de la representación.
Esta sistemática es la que se ha seguido para ladeterminación gráfica del pH de las disoluciones indi-cadas en cada uno de los apartados de este proble-ma. Con objeto de resaltar las líneas del diagramaque conducen a la determinación del pH de la diso-lución, esto es, aquellas correspondientes a las espe-cies implicadas en el balance protónico, se han dibu-jado de forma continua, mientras que se indican enforma de trazo discontinuo las de las otras especiespresentes en el sistema ácido-base.
Siguiendo el procedimiento indicado, inicialmentese dibujan los diagramas logarítmicos correspondien-
Seminarios: problemas gráficos
a) b)
d)c)
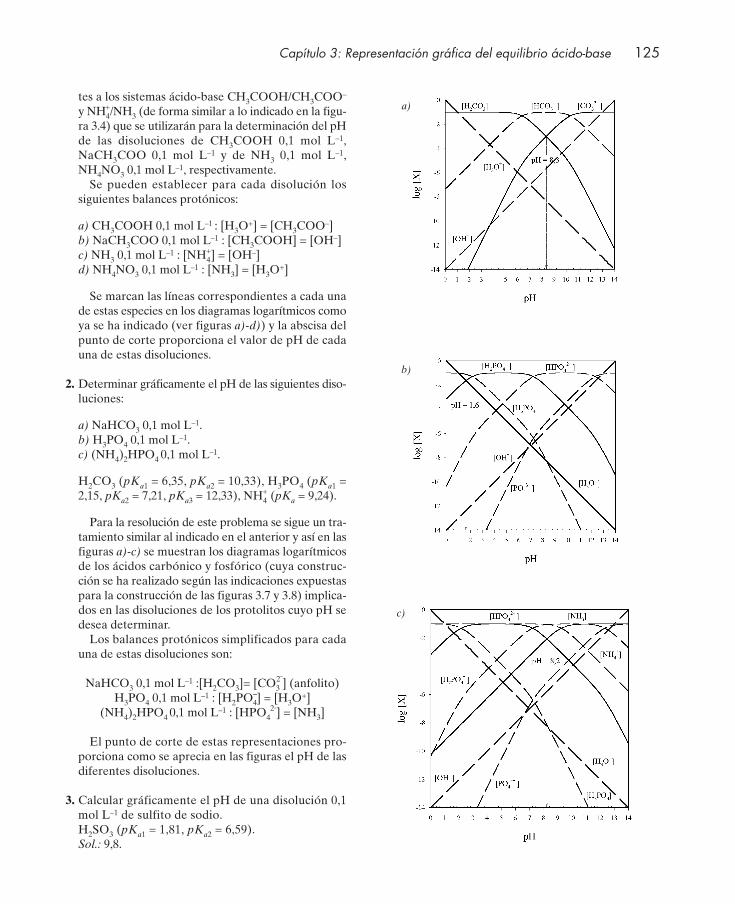
tes a los sistemas ácido-base CH3COOH/CH3COO–
y NH4+/NH3 (de forma similar a lo indicado en la figu-
ra 3.4) que se utilizarán para la determinación del pHde las disoluciones de CH3COOH 0,1 mol L–1,NaCH3COO 0,1 mol L–1 y de NH3 0,1 mol L–1,NH4NO3 0,1 mol L–1, respectivamente.
Se pueden establecer para cada disolución lossiguientes balances protónicos:
a) CH3COOH 0,1 mol L–1 : [H3O+] = [CH3COO–]
b) NaCH3COO 0,1 mol L–1 : [CH3COOH] = [OH–]c) NH3 0,1 mol L–1 : [NH4
+] = [OH–]d) NH4NO3 0,1 mol L–1 : [NH3] = [H3O
+]
Se marcan las líneas correspondientes a cada unade estas especies en los diagramas logarítmicos comoya se ha indicado (ver figuras a)-d)) y la abscisa delpunto de corte proporciona el valor de pH de cadauna de estas disoluciones.
2. Determinar gráficamente el pH de las siguientes diso-luciones:
a) NaHCO3 0,1 mol L–1.b) H3PO4 0,1 mol L–1.c) (NH4)2HPO4 0,1 mol L–1.
H2CO3 (pKa1 = 6,35, pKa2 = 10,33), H3PO4 (pKa1 =2,15, pKa2 = 7,21, pKa3 = 12,33), NH4
+ (pKa = 9,24).
Para la resolución de este problema se sigue un tra-tamiento similar al indicado en el anterior y así en lasfiguras a)-c) se muestran los diagramas logarítmicosde los ácidos carbónico y fosfórico (cuya construc-ción se ha realizado según las indicaciones expuestaspara la construcción de las figuras 3.7 y 3.8) implica-dos en las disoluciones de los protolitos cuyo pH sedesea determinar.
Los balances protónicos simplificados para cadauna de estas disoluciones son:
NaHCO3 0,1 mol L–1 :[H2CO3]= [CO32¯] (anfolito)
H3PO4 0,1 mol L–1 : [H2PO4̄] = [H3O+]
(NH4)2HPO4 0,1 mol L–1 : [HPO42¯] = [NH3]
El punto de corte de estas representaciones pro-porciona como se aprecia en las figuras el pH de lasdiferentes disoluciones.
3. Calcular gráficamente el pH de una disolución 0,1mol L–1 de sulfito de sodio. H2SO3 (pKa1 = 1,81, pKa2 = 6,59).Sol.: 9,8.
Capítulo 3: Representación gráfica del equilibrio ácido-base 125
c)
b)
a)
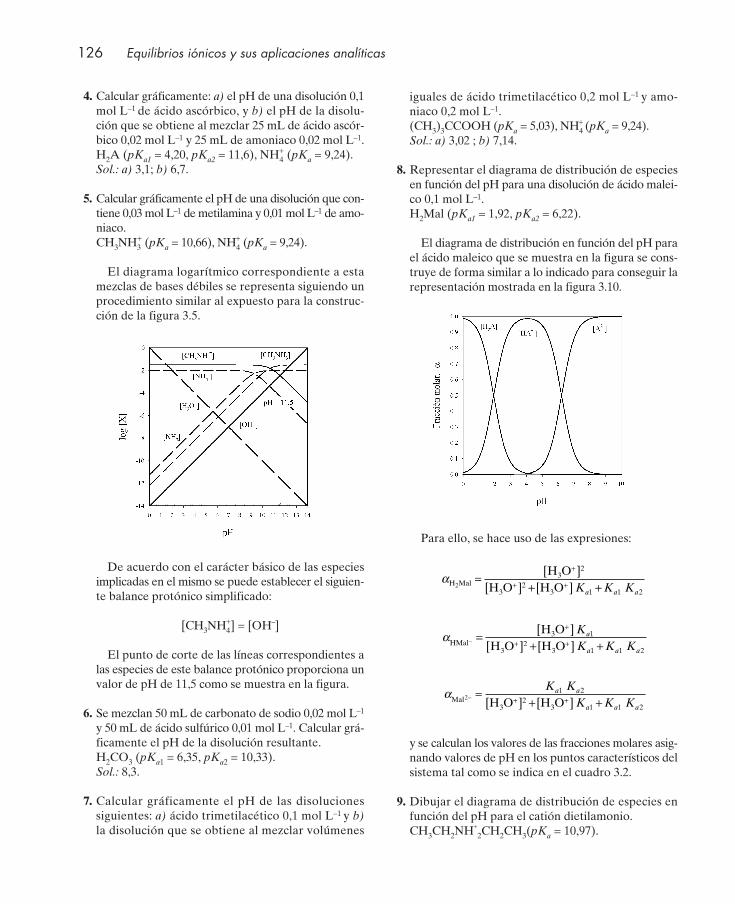
4. Calcular gráficamente: a) el pH de una disolución 0,1mol L–1 de ácido ascórbico, y b) el pH de la disolu-ción que se obtiene al mezclar 25 mL de ácido ascór-bico 0,02 mol L–1 y 25 mL de amoniaco 0,02 mol L–1. H2A (pKa1 = 4,20, pKa2 = 11,6), NH4
+ (pKa = 9,24).Sol.: a) 3,1; b) 6,7.
5. Calcular gráficamente el pH de una disolución que con-tiene 0,03 mol L–1 de metilamina y 0,01 mol L–1 de amo-niaco. CH3NH3
+ (pKa = 10,66), NH4+ (pKa = 9,24).
El diagrama logarítmico correspondiente a estamezclas de bases débiles se representa siguiendo unprocedimiento similar al expuesto para la construc-ción de la figura 3.5.
De acuerdo con el carácter básico de las especiesimplicadas en el mismo se puede establecer el siguien-te balance protónico simplificado:
[CH3NH4+] = [OH¯]
El punto de corte de las líneas correspondientes alas especies de este balance protónico proporciona unvalor de pH de 11,5 como se muestra en la figura.
6. Se mezclan 50 mL de carbonato de sodio 0,02 mol L–1
y 50 mL de ácido sulfúrico 0,01 mol L–1. Calcular grá-ficamente el pH de la disolución resultante. H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: 8,3.
7. Calcular gráficamente el pH de las disolucionessiguientes: a) ácido trimetilacético 0,1 mol L–1 y b)la disolución que se obtiene al mezclar volúmenes
iguales de ácido trimetilacético 0,2 mol L–1 y amo-niaco 0,2 mol L–1. (CH3)3CCOOH (pKa = 5,03), NH4
+ (pKa = 9,24).Sol.: a) 3,02 ; b) 7,14.
8. Representar el diagrama de distribución de especiesen función del pH para una disolución de ácido malei-co 0,1 mol L–1. H2Mal (pKa1 = 1,92, pKa2 = 6,22).
El diagrama de distribución en función del pH parael ácido maleico que se muestra en la figura se cons-truye de forma similar a lo indicado para conseguir larepresentación mostrada en la figura 3.10.
Para ello, se hace uso de las expresiones:
y se calculan los valores de las fracciones molares asig-nando valores de pH en los puntos característicos delsistema tal como se indica en el cuadro 3.2.
9. Dibujar el diagrama de distribución de especies enfunción del pH para el catión dietilamonio.CH3CH2NH+
2CH2CH3(pKa = 10,97).
αMal
32
32
[H O ] [H O− =
+ ++ +
K KK K K
a a
a a a
1 2
1 1 2]
αHMal
3
32
3
H OH O ] [H O ]
− =+ +
+
+ +
[ ][
KK K K
a
a a a
1
1 1 2
αH Mal
3
32
32
H O[H O ] [H O ]
=+ +
+
+ +
[ ]2
1 1 2K K Ka a a
126 Equilibrios iónicos y sus aplicaciones analíticas

4
4.1. Aspectos generales de las valoraciones4.2. Introducción a las valoraciones ácido-base4.3. Indicadores ácido-base4.4. Curvas de valoración4.5. Preparación de disoluciones valorantes4.6. AplicacionesCuestionesSeminarios: problemas numéricos
VALORACIONESÁCIDO-BASE

4.1. Aspectos generalesde las valoraciones
Las valoraciones constituyen un amplio ydiverso grupo de métodos analíticos con unagran tradición en el análisis químico cuantitati-vo. Una valoración es una técnica analítica basa-da en una reacción química (reacción de valora-ción), mediante la cual se determina la cantidadde una sustancia (analito) presente en una mues-tra, por adición de una cantidad conocida de otrasustancia (valorante), en presencia de un siste-ma que permita identificar (indicador) en quémomento se ha consumido la totalidad del ana-lito por adición de la cantidad estequiométricade valorante (punto final). Una vez conocida lacantidad de valorante que ha reaccionado cuan-titativamente con el analito, la cantidad de ésteen la muestra se determina fácilmente en basea la relación estequiométrica implicada en lareacción de valoración. En la mayoría de loscasos, la cantidad de analito se determina a par-tir de la medida correcta del volumen de diso-lución valorante gastado (disolución valorante,de concentración conocida) y en tal caso la valo-ración recibe el nombre de volumetría. Por lotanto, los términos valoración y volumetría noson sinónimos ya que el primero es un concep-to general que engloba al segundo, puesto quela cantidad de valorante consumido en una valo-ración se puede también determinar por otrosmétodos haciendo uso de dispositivos instru-mentales adecuados.
La reacción de valoración puede ser de muydiversa índole, como por ejemplo: ácido-base,formación de complejos, precipitación y redox(que se corresponden con los diferentes equili-
brios iónicos contemplados en este libro); ade-más se pueden considerar reacciones de adición,sustitución, condensación, etc., en las que estánimplicados compuestos orgánicos no-electrolitos.La naturaleza de la reacción de valoración es elcriterio más común para establecer los distintostipos de valoraciones; así se tienen valoracionesácido-base, valoraciones redox, valoraciones desustitución, etc. La reacción de valoración ha de cumplir una serie de requisitos con objeto deconseguir resultados analíticos fiables con senci-llez y rapidez:
1. Su estequiometría ha de estar definida enlas condiciones experimentales en las quese lleve a cabo la valoración, ya que comose ha indicado, en ella se sustentan bási-camente los cálculos encaminados a deter-minar la concentración de analito en lamuestra. La relación estequiométrica seha de constatar en las valoraciones redox,ya que el analito puede poseer diferentesestados de oxidación; en este caso, antesde llevar a cabo su valoración es comúnrealizar un tratamiento previo con un reac-tivo oxidante o reductor, según el caso, conobjeto de asegurar el estado de oxidacióndel mismo. Tal es el caso del uso del reduc-tor de Jones (amalgama de cinc) para ase-gurar el estado de oxidación +2 del hierroantes de su valoración con permanganatoo dicromato.
2. Ha de poseer una constante de equilibrioelevada para garantizar que la reacción seacompleta. En general, las reacciones devaloración presentan una constante deequilibrio alta, esto es, se encuentran des-
128 Equilibrios iónicos y sus aplicaciones analíticas
• Describir las características esenciales de las valo-raciones y en concreto de las valoraciones ácido-base: indicadores, curvas de valoración y aplica-ciones.
• Exponer las diferentes alternativas de curvas de valo-ración, así como de los tipos de indicadores emplea-dos en cada caso.
• Mostrar las aplicaciones más importantes de este téc-nica analítica cuantitativa.
OBJETIVOS

plazadas hacia la derecha; sin embargo, enciertas valoraciones de precipitación se pue-den originar errores por exceso si el preci-pitado que se forma en el transcurso de lareacción no es lo suficientemente insolu-ble en las condiciones de la valoración.
3. La reacción de valoración debe ser rápi-da, esto es con una cinética adecuada enlas condiciones experimentales de la valo-ración. Las reacciones ácido-base y de for-mación de complejos presentan en gene-ral la rapidez deseable para su uso envaloraciones, sin embargo las valoracionesde precipitación y redox presentan en cier-tos casos una cinética lenta, especialmen-te en las cercanías del punto final de lasmismas. No es de extrañar, por tanto, quealgunos procedimientos experimentales envaloraciones redox requieran calentar lamuestra e incluso de la presencia de cata-lizadores, como por ejemplo la valoraciónde oxalato con permanganato que se llevaa cabo en caliente y en presencia de Mn2+
que actúa como catalizador.4. La disponibilidad de sistemas indicadores
del punto final es uno de los aspectos queha condicionado el uso analítico de losdiferentes tipos de valoraciones. Así, lasvaloraciones ácido-base son las que pre-sentan un mayor campo de aplicación yaque existen una gran variedad de indica-dores, hecho que puede atribuirse a quees común que una sustancia presente pro-piedades ácido-base. Son menos numero-sas las sustancias con características redoxy mucho menos aquellas con capacidad deformación de complejos o de formar espe-cies insolubles.
5. La selectividad es una propiedad analíticabásica, deseable para cualquier métodoanalítico. Las valoraciones en general noson muy selectivas; si bien, en ciertos casosmediante una apropiada selección de lascondiciones experimentales se puede con-seguir el grado de selectividad adecuadoal problema analítico a resolver. Así, por
ejemplo, es posible la determinación decalcio en aguas en presencia de magnesiomediante valoración complexométrica conácido etilendiamino tetracético (EDTA)si ésta se lleva a cabo a pH = 12, dado quea este pH el magnesio precipita comohidróxido y no reacciona con el EDTA.
El analito es la sustancia objeto de determi-nación en la valoración. En general, como semuestra en la figura 4.1, éste se coloca en el enler-meyer junto con el sistema indicador del puntofinal y aquellos otros ingredientes necesarios parael desarrollo de la valoración, como por ejemploel tampón de pH adecuado para que transcurrala reacción correctamente. Este dispositivo en elque el valorante reacciona con el analito, recibeel nombre de valoración directa y puede utilizar-se siempre que se cumplan perfectamente losrequisitos descritos anteriormente, referentes aque la reacción ha de ser estequimétrica, cuanti-tativa, rápida, selectiva y se disponga de sistemasindicadores de punto final adecuados. Cuandofalla alguno de estos requisitos, generalmente serecurre a un procedimiento denominado valora-ción por retroceso, para la determinación del ana-lito, en el que se añade un exceso medido de valo-rante y se determina este exceso. Así, por ejemplo,si la reacción de valoración es muy lenta, previa-mente se desarrolla la misma en el propio enler-meyer por adición de un exceso conocido de ladisolución de valorante, R, al analito, A:
A + R exceso controlado → B + R sin reaccionar [4.1]
En estas condiciones al existir un exceso deR la reacción transcurre en un menor tiempo.Posteriormente, se determina R sin reaccionar (noconsumido por A) mediante un nuevo valoran-te, R′, añadido desde la bureta:
R′ + R sin reaccionar → P [4.2]
La cantidad de analito se deduce por diferen-cia entre las cantidades puestas en juego de los dosestándares químico-analíticos utilizados: R y R′.
Capítulo 4: Valoraciones ácido-base 129

Pueden también utilizarse las llamadas valo-raciones indirectas, en las que el analito no reac-ciona directamente con el valorante. Este pro-cedimiento se lleva a cabo a través de unareacción auxiliar del analito por la que se pro-duce una cantidad estequiométrica de un tercerreactivo, que es el que se valora con la disoluciónpatrón. Tanto las valoraciones por retrocesocomo las indirectas se utilizan cuando no puederealizarse la valoración directa.
FIGURA 4.1. Dispositivo instrumental utilizado en el desarro-llo de valoraciones directas.
La naturaleza del valorante depende del ana-lito a determinar, y en ocasiones, esta naturale-za, se suele utilizar como criterio para denomi-nar a las valoraciones; así se tienen valoracionesacidimétricas, alcalimétricas, iodométricas, etc.Una valoración acidimétrica consiste, por tanto,en la valoración de un analito con carácter bási-co, ya que se utiliza como valorante una disolu-ción de un ácido de concentración conocida. Ladisolución de valorante se ha de preparar a par-tir de un estándar químico-analítico primario opatrón primario (sustancia química de elevadapureza). La pesada exacta de una cantidad de esta
sustancia y su posterior disolución en un matrazaforado, proporciona la disolución valorante deconcentración exactamente conocida. Cuando nose puede utilizar un patrón primario (no está dis-ponible, es caro, etc.) se ha de recurrir al uso deun patrón secundario, el cual se valora con unestándar primario. Tras su estandarización, escomún expresar la concentración de la disoluciónde valorante de acuerdo con:
[Valorante]real = [Valorante]aprox × f [4.3]
siendo [Valorante]aprox la concentración de ladisolución valorante aproximada, que se tieneantes de su estandarización y f el factor volumé-trico. De acuerdo con la expresión anterior, f esel factor por el que hay que multiplicar la con-centración aproximada de la disolución de valo-rante para obtener su concentración real.
Los sistemas indicadores del punto final enlas valoraciones pueden ser visuales e instru-mentales. En los primeros, el indicador es unasustancia que participa en la reacción de valo-ración originando un cambio brusco percepti-ble (color, turbidez, precipitado, etc.) en el pun-to final de la misma, lo que permite interrumpirla adición de la disolución valorante. Cuando elcurso de la reacción de valoración se monitori-za mediante una técnica instrumental, en basea una magnitud físico-química relacionada conel analito, valorante o producto de la reacción,se tienen los denominados indicadores instru-mentales. En esta obra se abordará sólo el estu-dio de aquellas valoraciones con detección visualdel punto final.
Como se ha indicado, el objetivo de una valo-ración es determinar la cantidad de analito pre-sente en la muestra a partir de la cantidad de valo-rante estequiométricamente equivalente. El puntoen el que se alcanza esta condición se denominapunto de equivalencia o, frecuentemente, puntoestequiométrico. Sin embargo, en la práctica, laconclusión de la valoración depende del sistemaindicador utilizado; el punto en que un indicadorproporciona el cambio visual en el que se da porfinalizada la valoración, se conoce como punto
130 Equilibrios iónicos y sus aplicaciones analíticas

final. Es importante resaltar que un indicador viracuando una determinada magnitud (pH, poten-cial redox, etc.) llega al valor necesario para queello se produzca, independientemente de que sehaya alcanzado o no el punto de equivalencia. Porlo tanto, la exactitud de los resultados y, en con-secuencia, la calidad del análisis dependerán dela selección de un indicador adecuado, que garan-tice una buena correspondencia entre el puntofinal y el punto de equivalencia. De hecho, todavaloración realizada con un indicador visual impli-ca un error total que es la combinación de treserrores independientes:
1. Error químico provocado por el hecho deque el punto final puede no coincidir conel punto de equivalencia.
2. Error de discriminación visual, que apare-ce por la limitada capacidad del ojo huma-no para distinguir cambios de colores.
3. Error del indicador, provocado por la cir-cunstancia de que el indicador es una sus-tancia que también consume una pequeñacantidad de valorante para poner de mani-fiesto el punto final. Por lo general loserrores de discriminación visual y de indi-cador son despreciables frente al error quí-mico.
Una curva de valoración es la representacióngráfica de una variable principal (pH, potencial,absorbancia, etc.) relacionada con la concentra-ción de los componentes de la reacción de valo-ración, en función del volumen de valorante aña-dido. Según la relación entre la variable principaly la concentración de estos reaccionantes, se ori-ginan dos tipos de curvas de valoración: linealesy logarítmicas, en estas últimas, la variable prin-cipal está relacionada con la concentración de loscomponentes de la reacción de valoraciónmediante una función logarítmica. Así, se siguela variación de pH (–log [H3O
+]) en valoracio-nes ácido-base, de pM (–log [Mn+]) en valora-ciones de formación de complejos y de potencial(E = E 0 + 0,059/n log [Ox]/[Red]) en valoracio-nes redox. Estas curvas tienen forma sigmoidal
(creciente o decreciente), según varíe la variableseguida en el transcurso de la valoración.
En el caso de las valoraciones visuales es tam-bién interesante construir curvas de valoraciónteóricas, ya que a partir de ellas se pueden ex-traer diversas conclusiones, como por ejemplo:evaluar la exactitud de la determinación, selec-cionar el indicador visual más apropiado para lamisma, o bien ajustar las condiciones experi-mentales de manera que se mejore el curso de lavaloración.
Finalmente, se ha de indicar que las valora-ciones con indicación visual del punto final, apesar de su consideración de técnicas clásicas enel contexto actual de la Química Analítica, sonuna importante alternativa frente a las técnicasinstrumentales dada su sencillez, rapidez y bajocoste, para abordar la resolución de diferentesproblemas analíticos. En muchos manuales per-sisten aún hoy en día métodos estándar volumé-tricos para la determinación de diferentes anali-tos en diferentes tipos de muestras.
4.2. Introducción a las valoracionesácido-base
Las valoraciones ácido base, también conoci-das como volumetrías de neutralización, consti-tuyen un procedimiento rápido y útil para el aná-lisis cuantitativo de sustancias con propiedadesácidas o básicas. En las valoraciones ácido-basese utilizan las propiedades de diferentes protoli-tos para determinar el contenido en ácido o enbase de una disolución. La técnica experimentales muy sencilla ya que consiste básicamente enañadir cantidades medidas de una disoluciónpatrón de ácido fuerte (por ejemplo HCl) parala determinación de bases, mientras que para ladeterminación de ácidos se utiliza una disoluciónpatrón de una base fuerte (por ejemplo NaOH).Es de resaltar que, mientras que el analito adeterminar en una muestra de interés vieneimpuesto, el valorante a utilizar se escoge. Asípues, los valorantes siempre serán ácidos fuertes(HCl, H2SO4 en disolución acuosa o HClO4 en
Capítulo 4: Valoraciones ácido-base 131

medios no acuosos) o bien bases fuertes (NaOHo KOH en medios acuosos).
Una valoración ácido-base se puede considerarcomo la interacción entre dos parejas formadas porun ácido y una base y sus especies conjugadas:
ácido1 + base2→← base1 + ácido2 [4.4]
Cuando la reacción se lleva a cabo en medioacuoso, se puede escribir como:
H3O+ + OH– →← 2 H2O [4.5]
Si se considera la valoración de un ácido, o deuna base, con una disolución patrón o valoran-te, el objetivo de la misma es determinar la can-tidad de valorante químicamente equivalente ala cantidad de ácido, o bien de base, presente.Durante una gran parte de la valoración el pHde la disolución cambia lentamente, pero en laregión del punto de equivalencia el cambio depH es muy acusado. Por esta razón, la represen-tación gráfica del pH en función del % valoradoo del volumen de valorante adicionado (curva devaloración) es de gran interés, ya que permiteevaluar la exactitud con la que, teóricamente,puede llevarse a cabo la valoración y tambiénseleccionar el indicador más adecuado.
Antes de abordar el estudio de la valoraciónde diferentes tipos de protolitos, esto es de laconstrucción de las correspondientes curvas devaloración teóricas, es interesante considerar losaspectos, más generales, de interés de los indi-cadores ácido-base más usuales con objeto dedisponer de los argumentos necesarios para lle-var a cabo la selección del mismo para cada unade las valoraciones de interés.
4.3. Indicadores ácido-base
En las valoraciones ácido-base, un indicadorvisual es, generalmente, un ácido débil que pre-senta el equilibrio:
HIn + H2O →← In– + H3O+ [4.6]
y que se caracteriza por el hecho de que la espe-cie HIn tiene un color diferente al de la especieIn–, lo que origina un cambio de color en funcióndel pH.
Los indicadores visuales utilizados en valora-ciones ácido-base son compuestos orgánicos condobles enlaces conjugados, en los que la pérdidade un protón provoca cambios estructurales quese traducen en una mayor o menor movilidad deelectrones, lo que trae consigo posibles cambiosde color. Al tratarse de ácidos débiles, su equili-brio, representado por la reacción [4.6], vieneregido por una constante, denominada constan-te de disociación del indicador (KHIn):
[4.7]
que también puede escribirse como:
[4.8]
o bien en forma logarítmica:
[4.9]
Para un indicador visual, se define el interva-lo de transición (zona de viraje), como aquellaregión, de la escala de pH, en la que el indicadormanifiesta el cambio de color. Este intervalo detransición depende de la capacidad de percep-ción del ojo humano para distinguir claramenteuna forma coloreada en presencia de la otra, loque está íntimamente relacionado con la relaciónde concentraciones de ambas formas de indica-dor. Para establecer este intervalo de transición,se acepta, de forma aproximada, que en el casode una mezcla de colores, el ojo humano sólodetecta un cambio de tonalidad cuando la rela-ción entre las concentraciones de las dos espe-cies coloreadas varía entre 10 y 0,1.
pH
[In ][HIn]HIn= +
−
pK log
[H O ]
[HIn][In ]3 HIn
+−
= K
KHIn
3[In ][H O ][HIn]
=− +
132 Equilibrios iónicos y sus aplicaciones analíticas

Es decir, si
[4.10]
se observará el color de la forma disociada (In–)del indicador, y si
[4.11]
se observará el color de la forma molecular (HIn)del indicador. En el intervalo entre ambas con-diciones, esto es, si
[4.12]
se observará un color que será mezcla de los colo-res correspondientes a ambas formas del indica-dor. Resulta, por tanto, que el intervalo de vi-raje (o de transición) de un indicador visualcorresponde al intervalo de pH, resultante decombinar las expresiones [4.9] y [4.12]:
[4.13]
es decir, tiene lugar dos unidades de pH alrede-dor del valor del pK del indicador. Esto signifi-ca que, por ejemplo, como se muestra en la figu-ra 4.2, con un indicador de pKHIn = 5 y con laespecie molecular (HIn) de color rojo y la formadisociada (In–) de color amarillo, puede acep-tarse aproximadamente que para:
— pH ≤ 4, la disolución será de color rojo.— pH ≥ 6, la disolución será de color amarillo.— 4 < pH < 6 la disolución será de color
naranja, que corresponde a una mezcla derojo y amarillo, aunque el tono depende-rá del pH (más rojizo a pH < 5 y más ama-rillento a pH > 5).
El tratamiento realizado para establecer el inter-valo de transición de un indicador no es más queuna generalización, dado que como ya se ha indi-cado la sensibilidad del ojo humano no es la mismapara distinguir todos los colores. En consecuencia,el intervalo de viraje de un indicador determinadopuede ser ligeramente superior o inferior a dos uni-dades de pH o puede no estar perfectamente cen-trado alrededor del valor del pKHIn, en función delcolor concreto de sus formas molecular y disocia-da. Este hecho es especialmente importante en losindicadores para los que una de la especies, impli-cadas en el cambio de color, es incolora. ∆ pH HIn= ±pK 1
0 1 10 1 1,
[In ][HIn]
– log[In ][HIn]
< < ⇒ < <
− −
[In ][HIn]
0, 1 log[In ][HIn]
1− −
≤ ⇒ ≥ −
[In ][HIn]
10 log[In ][HIn]
1− −
≥ ⇒ ≥
Capítulo 4: Valoraciones ácido-base 133
FIGURA 4.2. Fundamentode los indicadores
ácido-base.

En el cuadro 4.1 se muestran algunos de losindicadores más usuales en valoraciones ácido-base, junto con su valor de pKHIn, intervalo detransición, colores implicados en el cambio y con-centración óptima de la disolución de indicadora utilizar.
Los indicadores mostrados en el cuadro 4.1cubren prácticamente todo el intervalo de pH, yen consecuencia permitirán los diversos tipos devaloraciones ácido-base que se discutirán en lossiguientes apartados de este capítulo. Desde unpunto de vista estructural, estos indicadores sepueden clasificar en tres grupos o familias impor-tantes: 1) indicadores del tipo de las ftaleínas, 2)indicadores del tipo de las sulfoftaleínas, y 3) indi-cadores azoicos.
Los indicadores del tipo de las ftaleínas, cuyoejemplo más significativo es la fenolftaleína, pre-sentan una estructura de lactona (figura 4.3a).Estos indicadores para los que la especie ácidaes incolora, tienen su intervalo de transición enla zona básica y por lo tanto son útiles para la
valoración de protolitos con carácter ácido(fenolftaleína y timolftaleína en cuadro 4.1). Seemplean en disolución etanólica ya que son prác-ticamente insolubles en agua.
Los indicadores del tipo de las sulfoftaleínasson de los más utilizados en valoraciones ácido-base, dado que presentan un cambio brusco decolor entre sus formas en el punto final, lo quereduce el posible error de discriminación visual.La introducción en la estructura de estos indica-dores de un grupo sulfónico, además de la pérdi-da del anillo de lactona, frente a los indicadoresde tipo ftaleínas, les confiere una mayor solubili-dad en agua. El rojo fenol presenta la estructuratípica de este tipo de indicadores (figura 4.3b).Como se muestra en el cuadro 4.1, rojo cresol,azul de timol, azul de bromofenol y verde de bro-mocresol presentan su intervalo de transición enla zona ácida de la escala de pH (son adecuadospara valorar compuestos cuyo punto de equiva-lencia se encuentre en zona ácida, como las basesdébiles); rojo cresol, púrpura de m-cresol y azul
134 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 4.1Valores de pKHIn, intervalo de transición, cambio de color y concentración óptima
de uso de los indicadores más utilizados en valoraciones ácido-base
Indicador pKHIn Intervalo Cambio de color Preparación de transición (ácido-base) de disolución HIn
Rojo cresol 0,2 – 1,8 Rojo – Amarillo 0,04% en aguaAzul de timol 1,65 1,2 – 2,8 Rojo – Amarillo 0,04% en aguaNaranja de Metilo 3,46 3,1 – 4,4 Rojo – Amarillo 0,04% en aguaAzul de Bromofenol 4,10 3,0 – 4,6 Amarillo – Púrpura 0,04% en aguaVerde de Bromocresol 4,90 4,0 – 5,6 Amarillo – Azul 0,1% (etanol 20%)Rojo de metilo 5,00 4,4 – 6,2 Rojo – Amarillo 0,1% en etanolPúrpura de Bromocresol 6,40 5,2 – 6,8 Amarillo – Púrpura 0,04% en aguaAzul de bromotimol 7,30 6,0 – 7,6 Amarillo – Azul 0,04% en aguaRojo Neutro 7,40 6,8 – 8,0 Rojo – Naranja 0,1% (etanol 70%)Rojo Fenol 8,00 6,4 – 8,2 Amarillo – Rojo 0,04% en aguaRojo Cresol 8,46 7,0 – 8,8 Amarillo – Púrpura 0,04% en aguaPurpura de m-cresol 8,32 7,4 – 9,0 Amarillo – Púrpura 0,04% en aguaAzul de timol 9,2 8,0 – 9,6 Amarillo – Azul 0,04% en aguaFenolftaleína 9,6 8,2 – 9,8 Incoloro – Púrpura 0,1% en etanolTimolftaleína 9,7 9,3 – 10,5 Incoloro – Azul 0,1% (etanol 50%)Violeta Azo 12,00 11,0 – 13,0 Amarillo – Violeta 0,5% en agua

de timol son útiles para la valoración de ácidos(intervalo de transición en la zona básica); mien-tras que púrpura de bromocresol, azul de bro-motimol y rojo fenol son igualmente sensibles aácidos y bases.
Los indicadores azoicos presentan la estruc-tura básica del p-amino-azobenceno o p-dimeti-lamino-azobenceno, ambas muy insolubles enagua, por lo que se han introducido sustituyen-tes polares en las mismas, como por ejemplo gru-pos sulfónicos (naranja de metilo, figura 4.3c) ogrupos carboxílicos (rojo de metilo), con objetode favorecer dicha solubilidad en agua. En el cua-dro 4.1 se incluyen tres indicadores azoicos cuyosintervalos de transición se sitúan en diferenteszonas de pH: ácida (naranja y rojo de metilo) ybásica (violeta azo).
Para facilitar la selección de un indicador es de interés definir el concepto de región de equivalencia. En sentido estricto, esta regióncorresponde a la parte de la curva de valoracióncomprendida entre el 99,9% y el 100,1% de valo-ración, es decir, entre la adición del 99,9% delvalorante necesario para alcanzar el punto deequivalencia y la adición de un exceso del 0,1%.
Cualquier indicador que vire dentro de esta zonapermitirá realizar la valoración con un error quí-mico inferior al 0,1%. Sin embargo, este criterioes demasiado restrictivo para los protolitos débi-les, que se caracterizan por el hecho de que elcambio de pH alrededor del punto de equiva-lencia es relativamente pequeño, por lo que enestos casos es necesario aceptar un error del 1%y definir la zona de equivalencia entre el 99% yel 101% de valoración.
4.4. Curvas de valoración
Una curva de valoración ácido-base, o curvade neutralización, consiste en la representaciónde la variación de un parámetro de la disolu-ción, en este caso el pH, en función del volumende valorante añadido, o bien de la fracción valo-rada, F, que se define por:
F = Cantidad de valorante añadidoCantidad de valorante necesaria
para alcanzar el punto de equivalencia
Capítulo 4: Valoraciones ácido-base 135
FIGURA 4.3. Estructura química de algunos de los indicadores ácido-base más utilizados: a) fenolftaleína, b) rojo fenol, y c) naranja de metilo.
a) b)
c)

La fracción valorada se expresa en tanto poruno o como porcentaje en tanto por ciento, si suvalor se multiplica por 100.
Existen diversos procedimientos para la cons-trucción de las curvas de neutralización; sinembargo, el más sencillo consiste en descompo-ner la curva en varias zonas y desarrollar ecua-ciones simplificadas para cada una de ellas. Elesquema general que se va a utilizar en este libropara el estudio de las diferentes valoraciones ácido-base consta de los siguientes puntos:
1. Se usa un ejemplo para estudiar la valora-ción de un determinado tipo de protolito.
2. Se consideran las zonas de interés de lacurva de valoración. En el caso de un pro-tolito monoprótico estas zonas son:
— Punto inicial (antes de añadir la diso-lución de valorante).
— Antes del punto de equivalencia (zonacon defecto de valorante).
— Punto de equivalencia.— Después del punto de equivalencia
(zona con exceso de valorante).
En cada una de estas zonas se conside-ran las especies predominantes, y se reali-za el cálculo del pH de las disoluciones quese tienen.
3. Tras el trazado de la curva de valoración[pH = f(% valorado) o bien pH = f(mLvalorante)], se discute la selección del indi-cador más adecuado para llevar a cabo lavaloración en el laboratorio.
4.4.1. Valoración de protolitos monopróticos
En este estudio se abordará la valoración delos siguientes tipos de protolitos monopróticos:
1. Ácido fuerte y base fuerte.2. Ácido débil y base débil.3. Mezcla de protolitos.
En todos los casos, el valorante seleccionadoes un protolito fuerte con objeto de que la varia-ción del pH en las inmediaciones del punto deequivalencia sea lo más grande posible, lo queproporciona una mayor exactitud al proceso devaloración.
Dado que las valoraciones constituyen porsí mismas las aplicaciones cuantitativas de losequilibrios ácido-base, la deducción de las expre-siones a utilizar para el cálculo del pH en lasdiferentes zonas de la curva de valoración no sellevará a cabo detalladamente, ya que ésta seencuentra pormenorizada en el capítulo 2 deeste texto.
A) Valoración de un ácido fuerte
Sea la valoración de un ácido fuerte (porejemplo HCl de concentración CHCl) con unabase fuerte (por ejemplo NaOH, de concentra-ción Cvalorante). La reacción de valoración vendrádada por:
HCl + NaOH → NaCl + H2O [4.14]
siendo VHCl y Vvalorante los volúmenes de analito avalorar y de valorante añadido, respectivamente.
Para llevar a cabo los cálculos necesariospara el desarrollo teórico de la curva de valo-ración se aplican los balances de masas y de car-gas o protónico que se muestran a continuacióny que deberán simplificarse de forma adecuadasegún la zona o punto de la curva de valoraciónconsiderada.
Zona 1: punto inicial: (F = 0; Vvalorante = 0)
Balance de masas:
Ca = CHCl = [Cl–] [4.15]
Balance de cargas:
[H3O+] = [OH–] + [Cl–] [4.16]
136 Equilibrios iónicos y sus aplicaciones analíticas

Antes de cualquier adición de valorante, sóloestá presente la cantidad inicial de ácido, por lo queel pH depende únicamente de su concentración.Aplicando el balance de cargas y simplificandode la forma adecuada, resulta:
[H3O+] = [OH� –] + [Cl–] [4.17]
de donde:
[4.18]
Zona 2: antes del punto de equivalencia(0 < F < 1; 0 < Vvalorante < Vequivalencia)
Balance de masas:
[4.19]
Balance de cargas:
[H3O+] + [Na+] = [OH–] + [Cl–] [4.20]
En esta zona se adicionan cantidades de valo-rante, siempre inferiores a la cantidad corres-pondiente a la neutralización del ácido inicial. ElpH en esta zona depende de la concentración dela especie no neutralizada, esto es del HCl sinvalorar y por tanto:
[H3O+] = [Cl–] – [Na+] [4.21]
o bien:
[4.22]
Si se expresa en función de la fracción valo-rada, F esta expresión se transforma en:
[4.23]
y al operar se tiene:
[4.24]
Zona 3: punto de equivalencia(F = 1; Vvalorante = Vequivalencia)
En este punto la cantidad de valorante aña-dido es estequiométricamente equivalente a lacantidad de analito. El pH depende de la espe-cie resultante de la total neutralización del áci-do, en este caso NaCl, se cumple por tanto que[Na+] = [Cl–] y aplicando esta condición al balan-ce de cargas, se obtiene que:
[4.25]
De esta expresión se deduce que el pH en elpunto de equivalencia de la valoración de un áci-do fuerte con una base fuerte es neutro y por tan-to independiente de las concentraciones de áci-do a valorar (analito) y de base utilizada comovalorante.
Zona 4: exceso de valorante(F > 1; Vvalorante > Vequivalencia)
Después del punto de equivalencia, lassiguientes adiciones de valorante no podrán neu-tralizar ya al analito, por lo que el pH depende-rá sólo del exceso de valorante añadido. Apli-cando el balance de cargas se obtiene:
[H�3O+] + [Na+] = [OH–] + [Cl–] [4.26]
sustituyendo:
[4.27]
V CV V
K V CV V
w
valorante valorante
valorante HCl
3
HCl HCl
valorante HCl[H O ]
×+
=
= + ×++
[H O ] [OH ]
pH3
+ −
−
= = == ⇒ =
Kw
,10 7 07
[H O ]3HCl HCl
HCl valorante
a
+ = − ×+
=
= −
( )
( )
1
1
FV C
V VF C
[H O ]
( )3
HCl HCl HCl HCl
HCl valorante
+ = × − ×+
V C F V CV V
[H O ]3
HCl HCl valorante valorante
HCl valorante
+ = × − ×+
V C V CV V
CV C
V VaHCl HCl
HCl valorante
– [Cl ]= ×+
=
[H O ]3 a+ = C
Capítulo 4: Valoraciones ácido-base 137

de donde:
[4.28]
o bien en función de F:
[OH–] = [Na+] – [Cl–]== (F – 1) Ca [4.29]
[4.30]
En el cuadro 4.2 se indican los datos corres-pondientes a la valoración de una disolución deácido clorhídrico 0,1 mol L–1 con hidróxido de sodio, sin tener en cuenta la dilución, es decirVfinal = VHCl y teniendo en cuenta la dilución , esdecir Vfinal = VHCl + Vvalorante. Puede observarse queen este caso el efecto de la dilución es significati-vo desde el punto de equivalencia en adelante.
[H O ][OH ]3
a
+−= =
−( )K K
F Cw w
1
[H O ]3
valorante HCl
valorante valorante HCl HCl
+ =
= +× − ×
K V VV C V C
w( )
138 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 4.2Valoración de 10 mL de HCl 0,1 mol L–1 con NaOH 0,1 mol L–1
% Valoración Vvalorante (mL) pH pH*
0 0,0 1,00 1,0010 1,0 1,09 1,0450 5,0 1,48 1,3090 9,0 2,28 2,0099 9,9 3,29 3,0099,9 9,99 4,30 4,00
100 10,0 7,00 7,00100,1 10,01 9,70 10,00101 10,1 10,70 11,00110 11,0 11,68 12,00150 15,0 12,30 12,70200 20,0 12,52 13,00
* Valores calculados sin tener en cuenta la dilución provocada por la adición de valorante.
En la figura 4.4 se representan las curvas devaloración correspondientes a la valoración dedisoluciones de ácido clorhídrico de concentra-ción 0,1 (curva de valoración en línea continua)y 0,01 mol L–1 (curva de valoración en línea dis-continua) con hidróxido de sodio. Ambas sonsimétricas con respecto al eje horizontal quepasa por el valor de pH correspondiente al pun-to de equivalencia (pH = 7,0). Se observa que aldisminuir la concentración del analito, el cam-bio de pH alrededor del punto de equivalenciaes menor.
Puede comprobarse que en el caso de la valo-ración de HCl 0,1 mol L–1 con hidróxido de sodio0,1 mol L–1 (curva continua en figura 4.4), la regiónde equivalencia considerando un error inferior al± 0,1% abarca desde pH 4,3 a pH 9,7, por lo quees fácil seleccionar un indicador que vire dentrode este intervalo. Por ejemplo, el azul de bromoti-mol sería perfecto y compuestos como rojo de me-tilo, púrpura de bromocresol, rojo neutro, rojo fenol,rojo cresol, púrpura de m-cresol y azul de timol,tendrían también todo su intervalo de transiciónincluido en la zona de equivalencia del ± 0,1%.
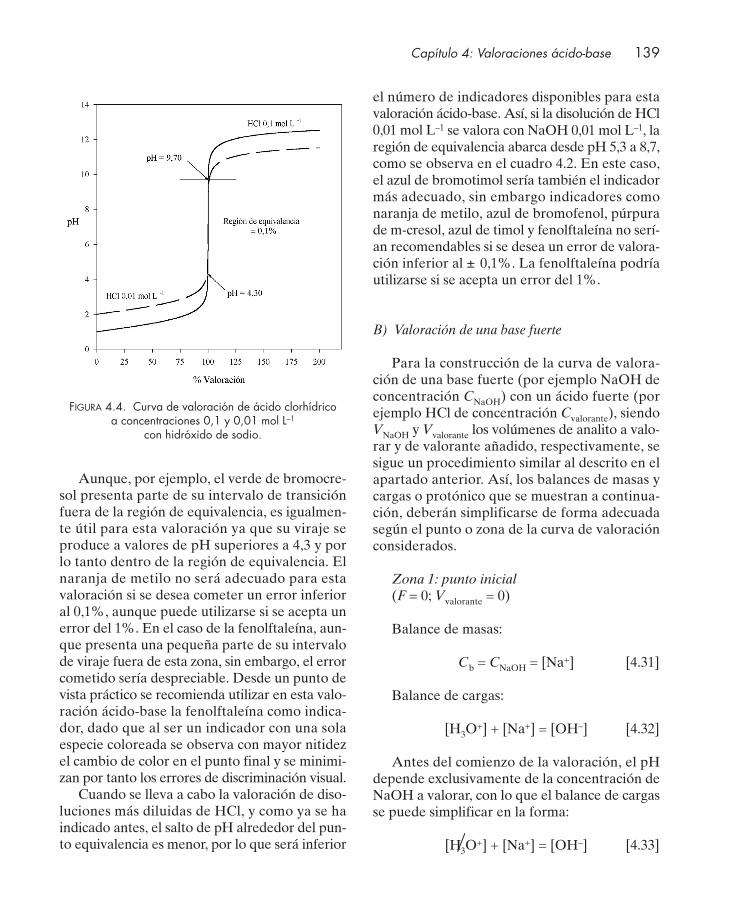
FIGURA 4.4. Curva de valoración de ácido clorhídricoa concentraciones 0,1 y 0,01 mol L–1
con hidróxido de sodio.
Aunque, por ejemplo, el verde de bromocre-sol presenta parte de su intervalo de transiciónfuera de la región de equivalencia, es igualmen-te útil para esta valoración ya que su viraje seproduce a valores de pH superiores a 4,3 y porlo tanto dentro de la región de equivalencia. Elnaranja de metilo no será adecuado para estavaloración si se desea cometer un error inferioral 0,1%, aunque puede utilizarse si se acepta unerror del 1%. En el caso de la fenolftaleína, aun-que presenta una pequeña parte de su intervalode viraje fuera de esta zona, sin embargo, el errorcometido sería despreciable. Desde un punto devista práctico se recomienda utilizar en esta valo-ración ácido-base la fenolftaleína como indica-dor, dado que al ser un indicador con una solaespecie coloreada se observa con mayor nitidezel cambio de color en el punto final y se minimi-zan por tanto los errores de discriminación visual.
Cuando se lleva a cabo la valoración de diso-luciones más diluidas de HCl, y como ya se haindicado antes, el salto de pH alrededor del pun-to equivalencia es menor, por lo que será inferior
el número de indicadores disponibles para estavaloración ácido-base. Así, si la disolución de HCl0,01 mol L–1 se valora con NaOH 0,01 mol L–1, laregión de equivalencia abarca desde pH 5,3 a 8,7,como se observa en el cuadro 4.2. En este caso,el azul de bromotimol sería también el indicadormás adecuado, sin embargo indicadores comonaranja de metilo, azul de bromofenol, púrpurade m-cresol, azul de timol y fenolftaleína no serí-an recomendables si se desea un error de valora-ción inferior al ± 0,1%. La fenolftaleína podríautilizarse si se acepta un error del 1%.
B) Valoración de una base fuerte
Para la construcción de la curva de valora-ción de una base fuerte (por ejemplo NaOH deconcentración CNaOH) con un ácido fuerte (porejemplo HCl de concentración Cvalorante), siendoVNaOH y Vvalorante los volúmenes de analito a valo-rar y de valorante añadido, respectivamente, sesigue un procedimiento similar al descrito en elapartado anterior. Así, los balances de masas ycargas o protónico que se muestran a continua-ción, deberán simplificarse de forma adecuadasegún el punto o zona de la curva de valoraciónconsiderados.
Zona 1: punto inicial(F = 0; Vvalorante = 0)
Balance de masas:
Cb = CNaOH = [Na+] [4.31]
Balance de cargas:
[H3O+] + [Na+] = [OH–] [4.32]
Antes del comienzo de la valoración, el pHdepende exclusivamente de la concentración deNaOH a valorar, con lo que el balance de cargasse puede simplificar en la forma:
[H�3O+] + [Na+] = [OH–] [4.33]
Capítulo 4: Valoraciones ácido-base 139

de donde:
[4.34]
Zona 2: antes del punto de equivalencia(0 < F < 1; 0 < Vvalorante < Vequivalencia)
Balance de masas:
[4.35]
Balance de cargas:
[H�3O+] + [Na+] = [OH–] + [Cl–] [4.36]
En esta zona, la cantidad de HCl añadido serásiempre inferior a la de NaOH a valorar, por loque el pH en la misma dependerá de la concen-tración de la especie no neutralizada, esto es delNaOH sin valorar, y por tanto:
[OH–] = [Na+] – [Cl–] [4.37]
o bien:
[4.38]
En el caso de hacer uso de la fracción valora-da, F, se tiene:
[4.39]
Zona 3: punto de equivalencia(F = 1; Vvalorante = Vequivalencia)
En este punto la cantidad de valorante aña-dido es estequiométricamente equivalente a la
cantidad de analito. El pH como en el caso de lavaloración de HCl con NaOH viene dado por:
[4.25]
Zona 4: exceso de valorante(F > 1; Vvalorante > Vequivalencia)
Después del punto de equivalencia, el pHdependerá sólo del exceso de valorante añadido.Su concentración afectada por la propia dilucióndeterminará el pH de la valoración en esta zona.Aplicando el balance de cargas se obtiene:
[H3O+] + [Na+] = [OH� –] + [Cl–] [4.40]
de donde:
[4.41]
CUADRO 4.3Valoración de NaOH con HCl
Valores de pH en la valoración de
NaOH 0,1 NaOH 0,01 % Valoración mol L–1 mol L–1
0 13,00 12,0010 12,91 11,9150 12,52 11,5290 11,72 10,7299 10,70 9,7099,9 9,70 8,70
100 7,00 7,00100,1 4,30 5,30101 3,30 4,30110 2,32 3,32150 1,70 2,70200 1,48 2,48
[H O ]3
valorante valorante NaOH NaOH
valorante NaOH
+ =
= × − ×+
V C V CV V
[H O ] [OH ]pH
3+ −
−= = =
= ⇒ =Kw
10 7 07 ,
[H O ]3NaOH valorante
NaOH NaOH
b
+ =−
× +×
=
=−
KF
V VV C
KF C
w
w
( )
( )
1
1
[( )
H O ]3
NaOH valorante
NaOH NaOH valorante valorante
+ =
= +× − ×K V V
V C V Cw
C
V CV Vb
NaOH NaOH
NaOH valorante
[Na ]= ×+
= +
[H O ]3b
+ = KC
w
140 Equilibrios iónicos y sus aplicaciones analíticas
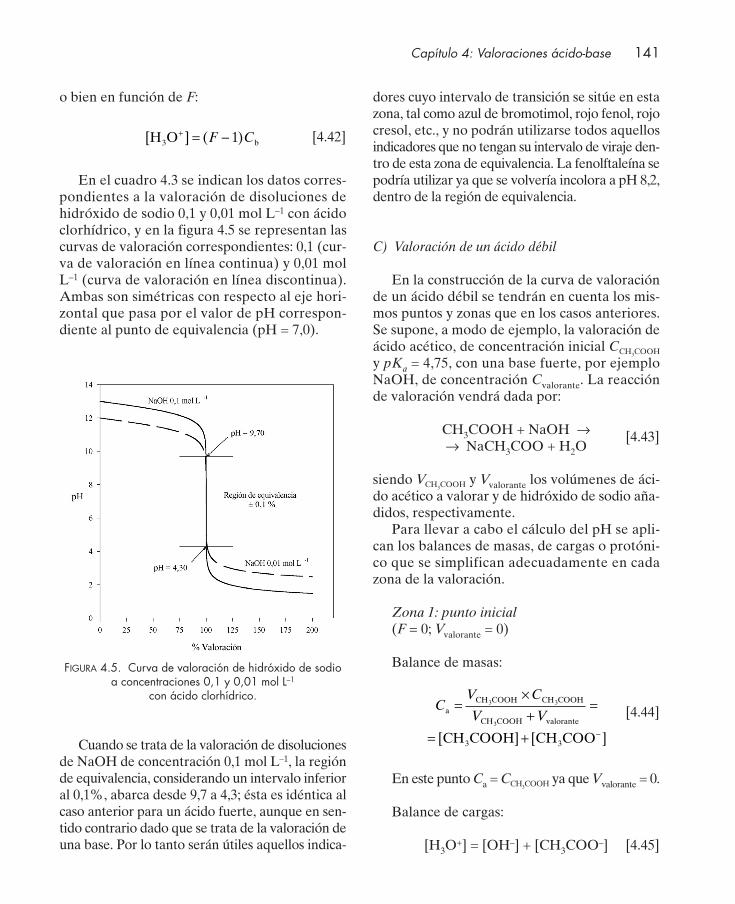
o bien en función de F:
[4.42]
En el cuadro 4.3 se indican los datos corres-pondientes a la valoración de disoluciones dehidróxido de sodio 0,1 y 0,01 mol L–1 con ácidoclorhídrico, y en la figura 4.5 se representan lascurvas de valoración correspondientes: 0,1 (cur-va de valoración en línea continua) y 0,01 molL–1 (curva de valoración en línea discontinua).Ambas son simétricas con respecto al eje hori-zontal que pasa por el valor de pH correspon-diente al punto de equivalencia (pH = 7,0).
FIGURA 4.5. Curva de valoración de hidróxido de sodioa concentraciones 0,1 y 0,01 mol L–1
con ácido clorhídrico.
Cuando se trata de la valoración de disolucionesde NaOH de concentración 0,1 mol L–1, la regiónde equivalencia, considerando un intervalo inferioral 0,1%, abarca desde 9,7 a 4,3; ésta es idéntica alcaso anterior para un ácido fuerte, aunque en sen-tido contrario dado que se trata de la valoración deuna base. Por lo tanto serán útiles aquellos indica-
dores cuyo intervalo de transición se sitúe en estazona, tal como azul de bromotimol, rojo fenol, rojocresol, etc., y no podrán utilizarse todos aquellosindicadores que no tengan su intervalo de viraje den-tro de esta zona de equivalencia. La fenolftaleína sepodría utilizar ya que se volvería incolora a pH 8,2,dentro de la región de equivalencia.
C) Valoración de un ácido débil
En la construcción de la curva de valoraciónde un ácido débil se tendrán en cuenta los mis-mos puntos y zonas que en los casos anteriores.Se supone, a modo de ejemplo, la valoración deácido acético, de concentración inicial CCH3COOH
y pKa = 4,75, con una base fuerte, por ejemploNaOH, de concentración Cvalorante. La reacciónde valoración vendrá dada por:
CH3COOH + NaOH →→ NaCH3COO + H2O
[4.43]
siendo VCH3COOH y Vvalorante los volúmenes de áci-do acético a valorar y de hidróxido de sodio aña-didos, respectivamente.
Para llevar a cabo el cálculo del pH se apli-can los balances de masas, de cargas o protóni-co que se simplifican adecuadamente en cadazona de la valoración.
Zona 1: punto inicial(F = 0; Vvalorante = 0)
Balance de masas:
[4.44]
En este punto Ca = CCH3COOH ya que Vvalorante = 0.
Balance de cargas:
[H3O+] = [OH–] + [CH3COO–] [4.45]
CV C
V VaCH COOH CH COOH
CH COOH valorante
3 3
3 3
3
[CH COOH] [CH COO ]
=×+
=
= + −
[H O ]3 b+ = −( )F C1
Capítulo 4: Valoraciones ácido-base 141

Antes de cualquier adición de valorante, elpH depende únicamente de la concentración deácido acético y de la constante de disociación dedicho ácido. Aplicando el balance de cargas, eneste caso igual que el balance protónico y sim-plificado de forma adecuada resulta:
[H3O+] = [OH� –] + [CH3COO–] [4.46]
y por tanto:
[4.47]
Si como es común [H3O+] >> Ka, la expresión
anterior se reduce a:
[4.48]
Zona 2: antes del punto de equivalencia(0 < F < 1; 0 < Vvalorante < Vequivalencia)
Se van adicionando cantidades de valorante,siempre inferiores a la correspondiente a la neu-tralización del ácido inicial, por lo que en esta zonacoexisten las formas protonada y disociada del pro-tolito, lo que constituye una zona tamponada. ElpH en esta zona se calcula o bien a partir del balan-ce de cargas, o bien utilizando la expresión de Hen-derson-Hasselbalch cuando todas las simplifica-ciones sean válidas, caso más usual:
[4.49]
de donde se deduce que la [H3O+] en función de
los volúmenes y concentraciones resulta ser:
[4.50]
Si el cálculo del pH se realiza en función dela fracción valorada se obtiene la expresión:
[4.51]
Zona 3: punto de equivalencia(F = 1; Vvalorante = Vequivalencia)
En el punto de equivalencia la cantidad deNaOH añadida es estequiométricamente equi-valente a la cantidad de ácido acético. El pHdepende de la especie resultante, en este casoNaCH3COO. Este valor de pH se puede calcu-lar mediante la expresión simplificada siguiente,siempre que se cumpla que [H3O
+] << Ka.
[4.52]
Zona 4: exceso de valorante(F > 1; Vvalorante > Vequivalencia)
Después del punto de equivalencia, la varia-ción de pH dependerá sólo del exceso de valo-rante añadido, esto es NaOH, y aplicando elbalance de cargas:
[H�3O+] + [Na+] = [OH–] + [CH3COO–] [4.53]
de donde:
[OH–] = [Na+] – [CH3COO–] [4.54]
y se puede deducir:[4.55]
o bien en función de F:
[4.30][H O ]OH 1
3
a
+−= =
−( )K K
F Cw w
[ ]
[H O ])
3valorante CH COOH
valorante valorante CH COOH CH COOH
3
3 3
+ =+
× − ×K V V
V C V Cw (
[H O ]3a
+ = K KCw a
[H O ]
(1 )3
+ = −K
FFa
[H O ]3CH COOH CH COOH valorante valorante
valorante valorante
3 3+ =× − ×
×K
V C V C
V Ca
[H O ]
[CH COOH][CH COO ]3
3
3
+−= Ka
[H O ]3+ = C Ka a
[H O ]
H O ]33
++=
+C K
Ka a
a[
142 Equilibrios iónicos y sus aplicaciones analíticas
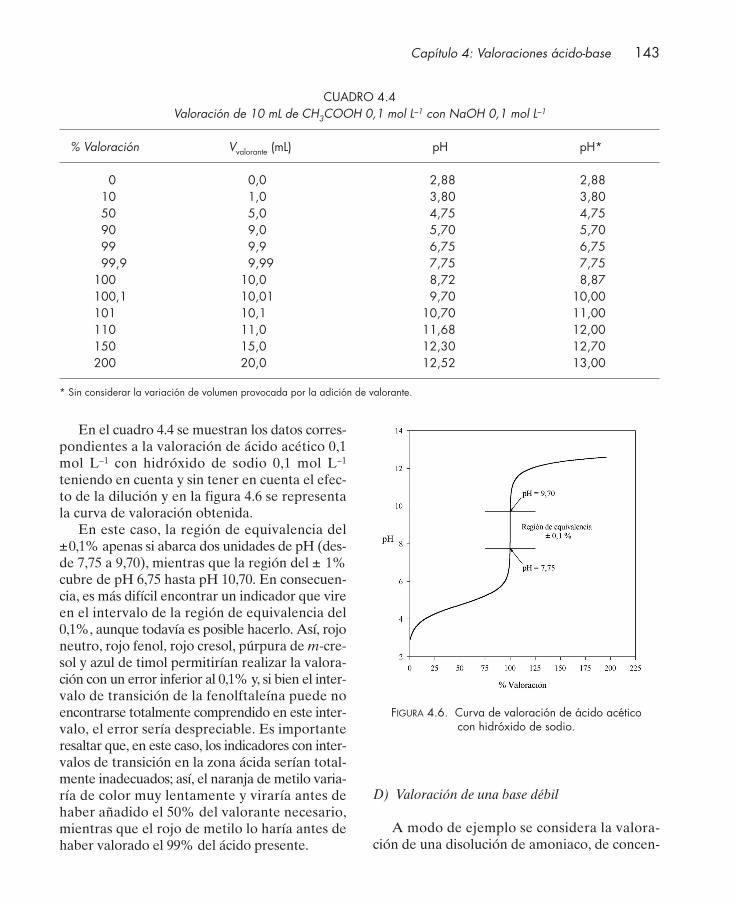
Capítulo 4: Valoraciones ácido-base 143
CUADRO 4.4Valoración de 10 mL de CH3COOH 0,1 mol L–1 con NaOH 0,1 mol L–1
% Valoración Vvalorante (mL) pH pH*
0 0,0 2,88 2,8810 1,0 3,80 3,8050 5,0 4,75 4,7590 9,0 5,70 5,7099 9,9 6,75 6,7599,9 9,99 7,75 7,75
100 10,0 8,72 8,87100,1 10,01 9,70 10,00101 10,1 10,70 11,00110 11,0 11,68 12,00150 15,0 12,30 12,70200 20,0 12,52 13,00
* Sin considerar la variación de volumen provocada por la adición de valorante.
En el cuadro 4.4 se muestran los datos corres-pondientes a la valoración de ácido acético 0,1mol L–1 con hidróxido de sodio 0,1 mol L–1
teniendo en cuenta y sin tener en cuenta el efec-to de la dilución y en la figura 4.6 se representala curva de valoración obtenida.
En este caso, la región de equivalencia del±0,1% apenas si abarca dos unidades de pH (des-de 7,75 a 9,70), mientras que la región del ± 1%cubre de pH 6,75 hasta pH 10,70. En consecuen-cia, es más difícil encontrar un indicador que vireen el intervalo de la región de equivalencia del0,1%, aunque todavía es posible hacerlo. Así, rojoneutro, rojo fenol, rojo cresol, púrpura de m-cre-sol y azul de timol permitirían realizar la valora-ción con un error inferior al 0,1% y, si bien el inter-valo de transición de la fenolftaleína puede noencontrarse totalmente comprendido en este inter-valo, el error sería despreciable. Es importanteresaltar que, en este caso, los indicadores con inter-valos de transición en la zona ácida serían total-mente inadecuados; así, el naranja de metilo varia-ría de color muy lentamente y viraría antes dehaber añadido el 50% del valorante necesario,mientras que el rojo de metilo lo haría antes dehaber valorado el 99% del ácido presente.
FIGURA 4.6. Curva de valoración de ácido acéticocon hidróxido de sodio.
D) Valoración de una base débil
A modo de ejemplo se considera la valora-ción de una disolución de amoniaco, de concen-

tración inicial CNH3y pKa = 9,24, con un ácido fuer-
te, HCl, de concentración Cvalorante. La reacciónde valoración vendrá dada por:
NH3 + HCl → NH4Cl [4.56]
siendo VNH3y Vvalorante los volúmenes de la diso-
lución de amoniaco a valorar y de ácido clorhí-drico añadido, respectivamente. Como en otroscasos, la deducción de las expresiones para elcálculo del pH en cada una de las zonas de lacurva de valoración se basa en los balances demasas, de carga y protónico que deben ser sim-plificados de manera adecuada.
Zona 1: punto inicial(F = 0; Vvalorante = 0)
Balance de masas:
Cb = [NH3] + [NH4+] [4.57]
Balance de carga:
[H�3O+] + [NH4
+] = [OH–] [4.58]
En este punto, en la disolución sólo está pre-sente la cantidad inicial de base débil, por lo que elpH depende únicamente de su concentración y dela constante de disociación de la especie presente.Aplicando el balance de cargas o el balance pro-tónico simplificado de la forma adecuada resulta:
[H�3O+] + [NH4
+] = [OH–] [4.59]
[4.60]
y si como es común para el caso de una basedébil, [H3O
+] << Ka, la expresión anterior sereduce a:
[4.61]
Zona 2: antes del punto de equivalencia(0 < F < 1; 0 < Vvalorante < Vequivalencia)
En esta zona comienza la adición de HCl y seva neutralizando progresivamente el amoniaco,por lo que coexisten las formas protonada y diso-ciada del protolito, lo que constituye una zonatamponada. El pH en esta zona se calcula a par-tir del balance de cargas:
[H3O+] + [NH4
+] = [OH–] + [Cl–] [4.62]
o bien utilizando la expresión de Henderson-Has-selbalch. En este último caso, se tiene:
[4.63]
o bien:
[4.64]
Si el cálculo del pH se realiza en función dela fracción valorada se hace uso de la expresión:
[4.65]
Zona 3: punto de equivalencia(F = 1; Vvalorante = Vequivalencia)
En el punto de equivalencia se añade la can-tidad estequiométrica de HCl, por lo que la espe-cie resultante es NH4Cl. El pH en este punto sepuede calcular aplicando el balance protónicocorrespondiente y si se cumple también la simpli-ficación para un ácido débil de que [H3O
+] >> Ka,se obtiene:
[4.66][H O ]3 b+ = C Ka
[ ]H O(1 )3
+ =−
KF
Fa
[H O ]3NH NH valorante valorante
valorante valorante
3 3+ =× − ×
×K
V C V C
V Ca
[ ]
[ ][ ]
H ONHNH3
4
3
++
= Ka
[ ]H O3b
+ = K KCw a
C
K
K
a
wb 3
3 3
H OH O H O ]
[ ][ ] [
+
+ ++=
144 Equilibrios iónicos y sus aplicaciones analíticas
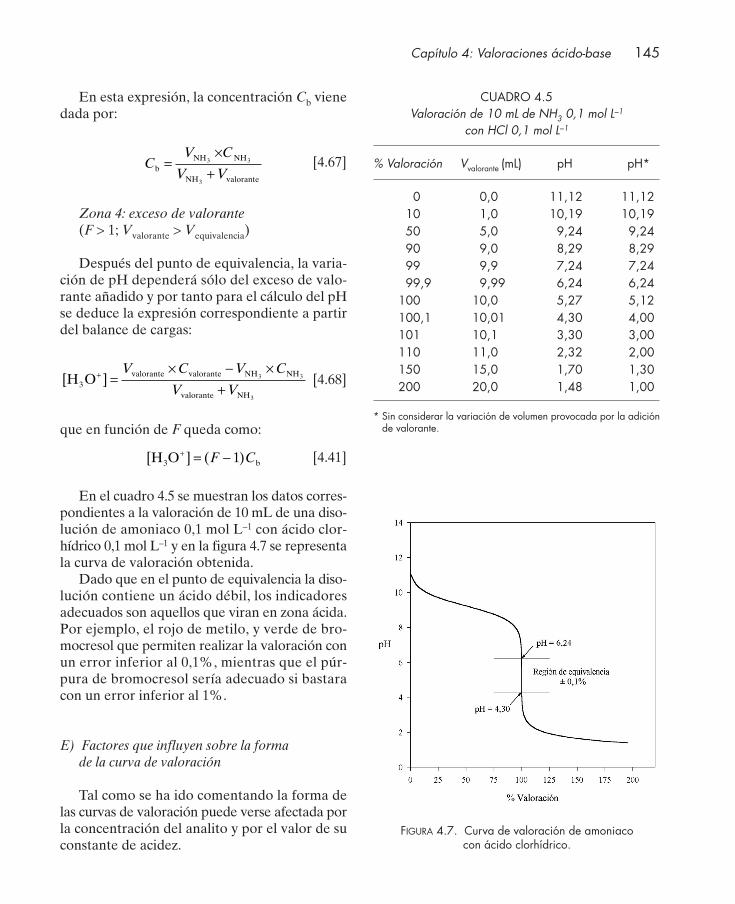
En esta expresión, la concentración Cb vienedada por:
[4.67]
Zona 4: exceso de valorante(F > 1; Vvalorante > Vequivalencia)
Después del punto de equivalencia, la varia-ción de pH dependerá sólo del exceso de valo-rante añadido y por tanto para el cálculo del pHse deduce la expresión correspondiente a partirdel balance de cargas:
[4.68]
que en función de F queda como:
[4.41]
En el cuadro 4.5 se muestran los datos corres-pondientes a la valoración de 10 mL de una diso-lución de amoniaco 0,1 mol L–1 con ácido clor-hídrico 0,1 mol L–1 y en la figura 4.7 se representala curva de valoración obtenida.
Dado que en el punto de equivalencia la diso-lución contiene un ácido débil, los indicadoresadecuados son aquellos que viran en zona ácida.Por ejemplo, el rojo de metilo, y verde de bro-mocresol que permiten realizar la valoración conun error inferior al 0,1%, mientras que el púr-pura de bromocresol sería adecuado si bastaracon un error inferior al 1%.
E) Factores que influyen sobre la formade la curva de valoración
Tal como se ha ido comentando la forma delas curvas de valoración puede verse afectada porla concentración del analito y por el valor de suconstante de acidez.
FIGURA 4.7. Curva de valoración de amoniaco con ácido clorhídrico.
[ ] ( )H O3 b+ = −F C1
[ ]H O3valorante valorante NH NH
valorante NH
3 3
3
+ =× − ×
+V C V C
V V
CV C
V VbNH NH
NH valorante
3 3
3
=×
+
Capítulo 4: Valoraciones ácido-base 145
CUADRO 4.5Valoración de 10 mL de NH3 0,1 mol L–1
con HCl 0,1 mol L–1
% Valoración Vvalorante (mL) pH pH*
0 0,0 11,12 11,1210 1,0 10,19 10,1950 5,0 9,24 9,2490 9,0 8,29 8,2999 9,9 7,24 7,2499,9 9,99 6,24 6,24
100 10,0 5,27 5,12100,1 10,01 4,30 4,00101 10,1 3,30 3,00110 11,0 2,32 2,00150 15,0 1,70 1,30200 20,0 1,48 1,00
* Sin considerar la variación de volumen provocada por la adiciónde valorante.

Las ecuaciones simplificadas deducidas anteriormentese basan en la hipótesis de que tanto el protolito que sevalora como su base (o ácido) conjugado, son lo sufi-cientemente débiles como para no reaccionar significa-tivamente con el agua, por lo que puede aceptarse que,por ejemplo, cuando se ha añadido la cantidad de valo-rante necesaria para neutralizar un X% de un ácido HA,se cumple que:
Sin embargo, hay casos en que esto no es cierto. Porejemplo, una disolución 0,1 mol L–1 de un ácido HA depKa = 1,9 tiene un pH = 1,63 y a este valor de pH secumple que:
lo que significa que el ácido se encuentra disociado enun 35%. Si, al calcular la curva de valoración, se utili-za la ecuación simplificada:
se obtienen los siguientes valores de pH:
VHA = 10 mL CHA = 0,1 mol L–1 CNaOH = 0,1 mol L–1
% Valoración Vvalorante (mL) pH
10 1 0,95
50 5 1,90
90 9 2,85
99 9,9 3,90
Es evidente que, como mínimo, el valor de pH corres-pondiente al 10% de valoración es incorrecto. La razónes la siguiente: para calcularlo se ha supuesto que, eneste punto, el 10% del protolito está en forma disociaday el 90% en forma molecular; sin embargo, si antes deiniciar la valoración la concentración de la forma diso-ciada ya es el 35% de la concentración analítica, paraque esta proporción se redujera al 10% sería necesarioañadir un ácido, no una base y esto es lo que se obtie-ne mediante la ecuación simplificada.
Para obtener resultados correctos el procedimientosería el siguiente:
Balance de cargas:
En medio ácido puede simplificarse a:
y al sustituir se tiene la expresión:
que se corresponde con la ecuación de segundo grado
en la que VT = VHA + Vvalorante, la cual proporciona losresultados correctos que se indican a continuación:
VHA = 10 mL CHA = 0,1 mol L–1 CNaOH = 0,1 mol L–1
% Valoración Vvalorante (mL) pH % disociación
10 1 1,64 35,3
50 5 2,11 61,7
90 9 2,96 92,1
99 9,9 3,99 99,2
V V C V KV C V C K
e a
a
T 3 valorant valorante T 3
valorante valorante HA HA
H O H O[ ] ( )[ ]( )
+ ++ + ++ − =
2
0
V CV V
V CV V
K
K
a
a
valorante valorante
valorante HA3
HA HA
valorante HA
3
H OH O
×+
+ =
×+
++
+[ ][ ]
[Na ] [H O ] [A ]3+ + −+ =
[Na ] [H O ] [A ] [OH ]3+ + − −+ = +
[ ]H O3
HA HA valorante valorante
valorante valorante
+ =× − ×
×K
V C V CV Ca
[ ] [ ]
[ ][ ]HA H O
H O0,065 HA 65
100HA 3
3HA=
+= ⇒ =
+
+
CK
Ca
[ ]
[ ]]A
H O0,035 [A 35
100HA a
3HA
−+
−=+
= ⇒ =C KK
Ca
[A ] X
100[HA] 100 X
100HA HA− = ⇒ = −C C
146 Equilibrios iónicos y sus aplicaciones analíticas
Curvas de valoración de protolitos monopróticos relativamente fuertes
RECUADRO 4.1
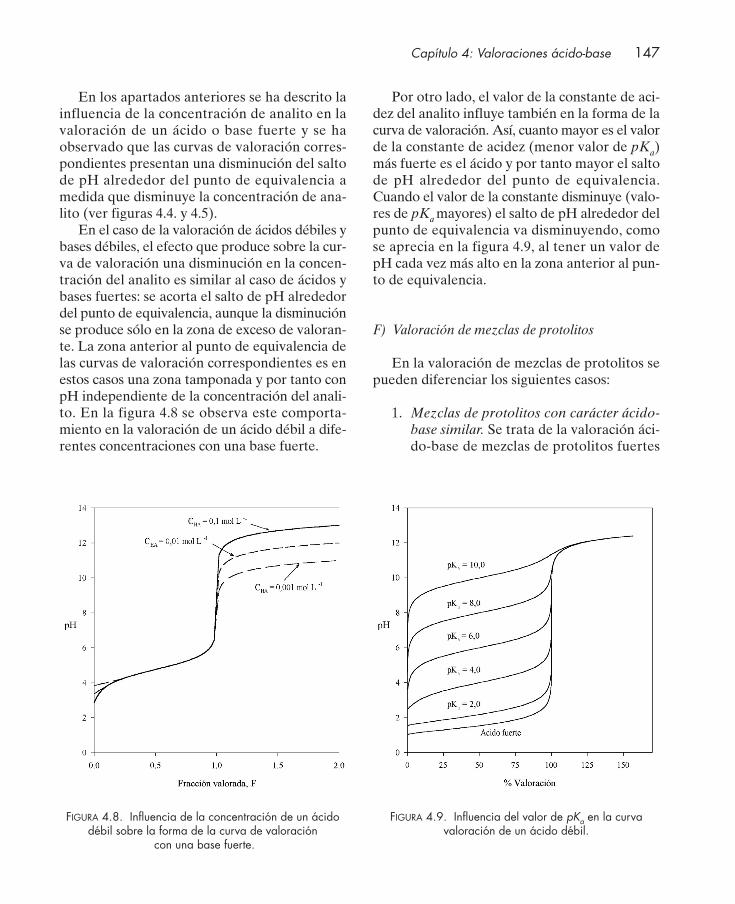
En los apartados anteriores se ha descrito lainfluencia de la concentración de analito en lavaloración de un ácido o base fuerte y se haobservado que las curvas de valoración corres-pondientes presentan una disminución del saltode pH alrededor del punto de equivalencia amedida que disminuye la concentración de ana-lito (ver figuras 4.4. y 4.5).
En el caso de la valoración de ácidos débiles ybases débiles, el efecto que produce sobre la cur-va de valoración una disminución en la concen-tración del analito es similar al caso de ácidos ybases fuertes: se acorta el salto de pH alrededordel punto de equivalencia, aunque la disminuciónse produce sólo en la zona de exceso de valoran-te. La zona anterior al punto de equivalencia delas curvas de valoración correspondientes es enestos casos una zona tamponada y por tanto conpH independiente de la concentración del anali-to. En la figura 4.8 se observa este comporta-miento en la valoración de un ácido débil a dife-rentes concentraciones con una base fuerte.
Por otro lado, el valor de la constante de aci-dez del analito influye también en la forma de lacurva de valoración. Así, cuanto mayor es el valorde la constante de acidez (menor valor de pKa)más fuerte es el ácido y por tanto mayor el saltode pH alrededor del punto de equivalencia.Cuando el valor de la constante disminuye (valo-res de pKa mayores) el salto de pH alrededor delpunto de equivalencia va disminuyendo, comose aprecia en la figura 4.9, al tener un valor depH cada vez más alto en la zona anterior al pun-to de equivalencia.
F) Valoración de mezclas de protolitos
En la valoración de mezclas de protolitos sepueden diferenciar los siguientes casos:
1. Mezclas de protolitos con carácter ácido-base similar. Se trata de la valoración áci-do-base de mezclas de protolitos fuertes
Capítulo 4: Valoraciones ácido-base 147
FIGURA 4.8. Influencia de la concentración de un ácidodébil sobre la forma de la curva de valoración
con una base fuerte.
FIGURA 4.9. Influencia del valor de pKa en la curva valoración de un ácido débil.

o bien de protolitos débiles con valoresde pKa semejantes, como por ejemplo lamezcla de dos ácidos o bases fuertes obien de ácidos o bases débiles. En amboscasos es imposible conseguir la resoluciónde estas mezclas por valoración ácido-base, puesto que la fuerza similar que pre-sentan estos protolitos impide su dis-criminación. La curva de valoración obte-nida presenta sólo un salto en el puntofinal que es el correspondiente a la neu-tralización de los dos protolitos. La for-ma de la curva de valoración obtenidaserá similar a las obtenidas para un soloprotolito, en cada caso.
2. Mezclas de protolitos de distinta fuerza.En este caso se podrá conseguir la reso-lución de la mezcla de protolitos median-te valoración ácido-base siempre que setengan mezclas de protolitos fuertes ydébiles o bien mezclas de protolitos débi-les con valores de pKa muy diferentes. Sevalora siempre en primer lugar el proto-lito de mayor fuerza (ácido/base fuerte obien el protolito débil con menor valor depKa) y posteriormente el protolito másdébil. La curva de valoración presenta dossaltos, y en estas zonas se detectan los dospuntos de equivalencia, correspondientesa la valoración de cada componente en lamezcla. La valoración de mezclas de pro-tolitos débiles con valores de pKa muydiferentes es similar a la valoración deprotolitos polipróticos (si las concentra-ciones de los protolitos son iguales) aspec-to que se tratará en el próximo apartadode este capítulo.
Sea la valoración de una mezcla formada porun volumen VHCl mL de HCl de concentraciónCHCl y un volumen VCH3COOH mL de CH3COOHde concentración CCH3COOH con una base fuerte(NaOH de concentración Cvalorante). Las reaccio-nes de valoración son las siguientes:
HCl + NaOH → NaCl + H2O [4.14]
CH3COOH + NaOH →→ NaCH3COO + H2O
[4.43]
siendo Vvalorante el volumen de hidróxido de sodioañadido.
Como en otros casos, los cálculos necesariospara la construcción de la curva de valoración sebasan en los correspondientes balances de masas,de cargas y protónico que deberán simplificarsede forma adecuada según la zona o punto de lamisma. Sin embargo, para el caso de valoracio-nes de mezclas de protolitos es mejor utilizarvolúmenes y no fracción valorada.
Balance de masas (Caf es la concentración deácido fuerte y Cad es la concentración de ácidodébil):
[4.69]
[4.70]
Balance de cargas o balance protónico en el pun-to inicial:
[H3O+] = [OH–] + [Cl–] + [CH3COO–] [4.71]
Balance de cargas cuando ya se ha iniciado lavaloración:
[H3O+] + [Na+] = [OH–] +
+ [Cl–] + [CH3COO–] [4.72]
En esta valoración se pueden diferenciar lassiguientes zonas o puntos característicos de lamisma:
Zona 1: punto inicial
Antes de la adición de valorante se tiene lamezcla de ácido fuerte y ácido débil. De acuer-do con lo indicado en el capítulo 2, el pH de la
CV C
V V VadCH COOH CH COOH
HCl CH COOH valorante
3 3
3 3
3
[CH COOH] [CH COO ]
=×
+ +=
= + −
CV C
V V VafHCl HCl
HCl CH COOH valorante
–
3
= [Cl ]= ×+ +
148 Equilibrios iónicos y sus aplicaciones analíticas

disolución depende exclusivamente de la con-centración de ácido fuerte presente en la mismasi como generalmente ocurre CHCl × [H3O
+] >>CCH3COOH × Ka en este caso, el balance de cargascoincide con el protónico y se expesa como:
[H3O+] = [OH–] + [Cl–] + [CH3COO–] [4.73]
de donde:
[4.74]
expresión muy similar a la utilizada en esta zonapara la valoración de HCl con NaOH.
Zona 2: antes del punto de equivalenciadel ácido fuerte
En esta zona se adicionan cantidades deNaOH siempre inferiores a la cantidad corres-pondiente a la neutralización del ácido fuerte. ElpH en esta zona depende de la concentración dela especie no neutralizada, esto es del HCl sinvalorar y de la presencia de ácido acético. Des-de un punto de vista general, el balance de car-gas en esta zona se puede expresar por:
[H3O+] + [Na+] =
= [Cl–] + [CH3COO–] + [OH� –] [4.75]
y por tanto:
[H3O+] = [Cl–] – [Na+] + [CH3COO–] [4.76]
o lo que es lo mismo:
[4.77]
Al comienzo de la valoración sólo es signifi-cativo el primer término de esta expresión, mien-
tras que en las inmediaciones del punto de equi-valencia, hay que aplicar la expresión completa.
Zona 3: punto de equivalencia del ácido fuerte
En este punto la cantidad de NaOH añadi-da es estequiométricamente equivalente a lacantidad de ácido fuerte. Sin embargo, el pH noviene dado por el que proporciona la sal for-mada en este punto (NaCl), sino por el ácidodébil presente en el mismo sin valorar. Así, elpH depende de la concentración de ácido acé-tico (teniendo en cuenta la dilución que ha sufri-do debido a la valoración del HCl) y de su cons-tante de acidez. Aplicando el balance de cargas,y considerando que en el punto de equivalen-cia del HCl, [Cl–] = [Na+] y que [OH–] se puededespreciar, pues se trata de la disolución de unácido, se tiene:
[H3O+] + [Na�+] =
= [OH� –] + [Cl�–] + [CH3COO–] [4.78]
de donde:
[4.79]
Esta expresión corresponde a la ya deducidapara el cálculo del pH en el punto incial de la valo-ración de un ácido débil con una base fuerte.
Zona 4: antes del punto de equivalenciadel ácido débil
Se añade NaOH suficiente para neutralizartodo el ácido fuerte, pero en menor concentracióna la necesaria para neutralizar el ácido acético.En esta zona coexisten las formas protonada ydisociada del ácido acético, lo que constituye unazona tamponada, y el pH se calcula utilizando laexpresión del balance de cargas que viene dadapor:
[H3O+] + [Na+] =
= [CH3COO–] + [Cl–] + [OH–][4.80]
[ ]H O3 ad+ = C Ka
[ ]
[ ]H O
H O3HCl HCl valorante valorante
HCl CH COOH valorante
ad
33
++= × − ×
+ ++
+V C V C
V V VC K
Ka
a
[ ]H O3HCl HCl
HCl CH COOH3
+ = ×+
V CV V
Capítulo 4: Valoraciones ácido-base 149

de donde se deduce que:
[4.81]
Esta expresión es adecuada para la mayor par-te de esta zona, sin embargo se obtienen valoresanómalos de pH cuando se aplica muy al comien-zo de la misma, esto es, tras el punto de equiva-lencia del ácido fuerte. En este caso se reco-mienda hacer uso de un balance de cargas menossimplificado, como en la zona 2, para obtener unaexpresión útil para el cálculo del pH.
Zona 5: punto de equivalencia del ácido débil
En este segundo punto de equivalencia se aña-de la cantidad estequiométrica de NaOH corres-pondiente al ácido clorhídrico y al ácido acéticopresentes. El pH depende de la especie resul-tante de la total neutralización del ácido acético:NaCH3COO, ya que el NaCl formado no influ-ye en el valor del pH. Este valor de pH se pue-de calcular a partir del balance protónico y sim-plificando adecuadamente se obtiene:
[4.82]
Zona 6: exceso de valorante
Después del punto de equivalencia, el pHdependerá sólo del exceso de valorante añadido.Su concentración afectada por la dilución deter-minará el pH de la valoración en esta zona. Apli-cando el balance de cargas:
[H3O+] + [Na+] =
= [OH–] + [Cl–] + [CH3COO–] [4.83]
convenientemente simplificado, se obtiene:
[4.84]
En el cuadro 4.6 se muestran los datos co-rrespondientes a la valoración de una mezcla de10 mL de una disolución de ácido clorhídrico 0,1 mol L–1 y 10 mL de ácido acético 0,1 mol L–1
con hidróxido de sodio 0,1 mol L–1 y en la fi-gura 4.10 se representa la curva de valoraciónobtenida.
CUADRO 4.6Valoración de una mezcla constituida por 10 mL
de HCl 0,1 mol L–1 y10 mL de CH3COOH 0,1 mol L–1
con NaOH 0,1 mol L–1
VNaOH pH
0,0 1,301,0 1,375,0 1,709,0 2,469,9 3,029,99 3,10
10,0 3,1110,01 3,1210,1 3,2111,0 3,8115,0 4,7519,0 5,7019,9 6,7419,99 7,7520,0 8,5720,01 9,4020,1 10,4021,0 11,3925,0 12,0530,0 12,30
El primer punto de equivalencia de la curvaes prácticamente imposible de detectar con erro-res aceptables desde un punto de vista cuanti-tativo, ya que la región de equivalencia de ±1%abarca únicamente 0,2 unidades de pH. En cier-tos casos, asumiendo un mayor error en la valo-ración se podría utilizar el azul de timol como
[ ](
( )H O
)3
valorante HCl CH COOH
valorante valorante HCl HCl CH COOH CH COOH
3
3 3
+ =+ +
× − × + ×K V V V
V C V C V Cw
[ ]H O3
ad
+ = K KCw a
[H O ]3
CH COOH CH COOH HCl HCl valorante valorante
valorante valorante
3 3+ =× + × − ×
×K
V C V C V C
V Ca
150 Equilibrios iónicos y sus aplicaciones analíticas
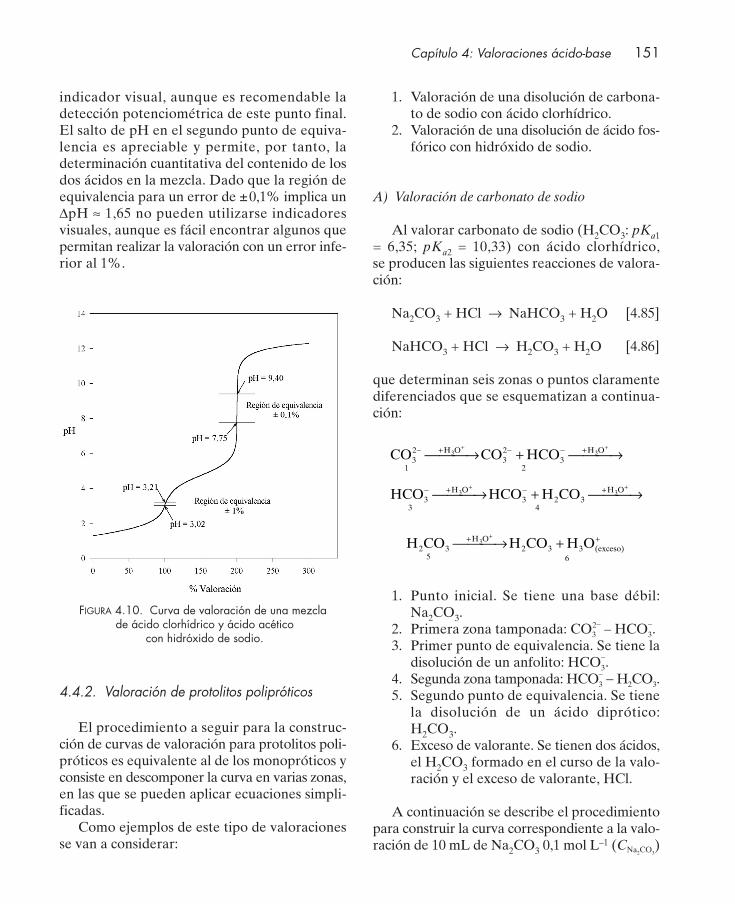
indicador visual, aunque es recomendable ladetección potenciométrica de este punto final.El salto de pH en el segundo punto de equiva-lencia es apreciable y permite, por tanto, ladeterminación cuantitativa del contenido de losdos ácidos en la mezcla. Dado que la región deequivalencia para un error de ±0,1% implica un∆pH ≈ 1,65 no pueden utilizarse indicadoresvisuales, aunque es fácil encontrar algunos quepermitan realizar la valoración con un error infe-rior al 1%.
FIGURA 4.10. Curva de valoración de una mezclade ácido clorhídrico y ácido acético
con hidróxido de sodio.
4.4.2. Valoración de protolitos polipróticos
El procedimiento a seguir para la construc-ción de curvas de valoración para protolitos poli-próticos es equivalente al de los monopróticos yconsiste en descomponer la curva en varias zonas,en las que se pueden aplicar ecuaciones simpli-ficadas.
Como ejemplos de este tipo de valoracionesse van a considerar:
1. Valoración de una disolución de carbona-to de sodio con ácido clorhídrico.
2. Valoración de una disolución de ácido fos-fórico con hidróxido de sodio.
A) Valoración de carbonato de sodio
Al valorar carbonato de sodio (H2CO3: pKa1= 6,35; pKa2 = 10,33) con ácido clorhídrico, se producen las siguientes reacciones de valora-ción:
Na2CO3 + HCl → NaHCO3 + H2O [4.85]
NaHCO3 + HCl → H2CO3 + H2O [4.86]
que determinan seis zonas o puntos claramentediferenciados que se esquematizan a continua-ción:
1. Punto inicial. Se tiene una base débil:Na2CO3.
2. Primera zona tamponada: CO32− – HCO3
−.3. Primer punto de equivalencia. Se tiene la
disolución de un anfolito: HCO3−.
4. Segunda zona tamponada: HCO3− − H2CO3.
5. Segundo punto de equivalencia. Se tienela disolución de un ácido diprótico:H2CO3.
6. Exceso de valorante. Se tienen dos ácidos,el H2CO3 formado en el curso de la valo-ración y el exceso de valorante, HCl.
A continuación se describe el procedimientopara construir la curva correspondiente a la valo-ración de 10 mL de Na2CO3 0,1 mol L–1 (CNa2CO3
)
H CO H CO H O2 3
5
H O2 3 3 (exceso)
6
3+ ++
→ +
CO CO HCO
HCO HCO H CO
32
1
H O32
32
H O
33
H O3 2 3
4
H O
3 3
3 3
− + − − +
− + − +
+ +
+ +
→ + →
→ + →
Capítulo 4: Valoraciones ácido-base 151

con HCl 0,1 mol L–1 (Cvalorante). Los balances autilizar son los balances de masas, de cargas y elbalance protónico:
Balance de masas:
[4.87]
[Na+] = 2 × Cb [4.88]
[4.89]
Balance de cargas:
[4.90]
siendo VT el volumen total de la disolución encada momento de la valoración.
Zona 1: punto inicial(Vvalorante = 0; F = 0)
Inicialmente se tiene la disolución de una saly por tratarse de una base débil, para el cálculodel pH es conveniente utilizar el balance protó-nico, que en este caso es:
[4.91]
Dado que el pH esperado para esta disoluciónes básico, cabe suponer que [H2CO3] y [H3O
+]serán despreciables frente a [HCO3
−]. La ecuaciónse puede simplificar fácilmente, ya que el Na2CO3es la base conjugada del NaHCO3, que como áci-do actúa con Ka2, por lo que puede aceptarse que[H3O
+] < Ka2 << Ka1. Si esta hipótesis es correcta,el pH se calcula haciendo uso de la expresión:
[4.92]
Mediante esta expresión se obtiene un valorde pH = 11,66 lo que indica que en este casotodas las aproximaciones realizadas son válidas.
Zona 2: primera zona tamponada(0 < Vvalorante < Vequivalencia(1); 0 < F < 1)
Al tratarse de un tampón, es aconsejable uti-lizar el balance de cargas:
[4.90]
Esta ecuación se puede simplificar teniendoen cuenta las consideraciones ya desarrolladas alestudiar las disoluciones amortiguadoras de pH.Así, al ser una disolución tampón, no será ni muyácida ni muy básica, en el caso más general, conlo que las [H3O
+] y [OH–] serán despreciables yse podrá escribir:
[4.93]
y aplicando a esta expresión los balances demasas se obtiene la expresión:
[4.94]
de donde:
[4.95]
Esta ecuación general es útil para toda estazona de la curva de valoración y especialmente
[ ]
[ ]
H O 2
H O 03
2b a
3 b a a 1 2
+
+
−( ) +
+ −( ) − =
C C
K C C C K Ka a a1
2
H O 2
H O H Ob
b 3 1 2
3 3 1a× =
+( )+ +
++
+ +CC K K K
K K KC
a a a
a a a
1
21 2
[ ]
[ ] [ ]
[Na ] [HCO ] 2[CO ] [Cl ]3 32–+ − −= + +
[Na ] [H O ][HCO ] 2[CO ] [Cl ] [OH ]
3
3 32–
+ +
− − −
+ == + + +
[ ]H O32
b
+ = K KCa w
2[H CO ] [HCO ] [H O ] [OH ]2 3 3 3+ + =− + −
[H O ] [Na ][OH ] [HCO ] 2[CO ] [Cl ]
3
3 32-
+ +
− − −+ =
= + + +
[ ]Cl valorante valorante
Ta
− = × =V CV
C
[H CO ] [HCO ] [CO2 3 3 32
Na CO Na CO
Tb
2 3 2 3
+ + =
=×
=
− − ]V C
VC
152 Equilibrios iónicos y sus aplicaciones analíticas

en las cercanías del punto de equivalencia. Sinembargo, su empleo desde un punto de vista prác-tico es, por lo general, escaso, y por ello el uso deuna ecuación simplificada proporciona resultadosaceptables para poder deducir conclusiones de inte-rés sobre la curva de valoración. Así, el término[H3O
+]2 es despreciable en la ecuación [4.94], altener una mezcla de las especies HCO3
− y CO32− y
pH ≈ pKa2, con lo que ésta puede simplificarse a:
[4.96]
que puede reordenarse fácilmente en la forma:
[4.97]
obteniéndose una expresión sencilla que permi-te calcular los valores de pH en la primera zonatamponada.
Zona 3: primer punto de equivalencia(100% de valoración)(Vvalorante = Vequivalencia (1) = 10 mL; F = 1)
En este punto la disolución contiene hidro-genocarbonato de sodio a una concentración 0,05 mol L–1, ya que VT = VNa2CO3
+ VHCl = 20 mL.Dado que se trata de un anfolito, el pH se calcu-la a partir del correspondiente balance protónico:
[4.98]
La disolución contiene simultáneamente unácido de pKa2 = 10,33 y una base de pKa1 = 6,35,por lo tanto, Ka1 >> [H3O
+] >> Ka2 y el pH espe-rado no será ni muy ácido ni muy básico, por loque podrá aceptarse que [H3O
+] y [OH–] serándespreciables frente a [H2CO3] y [CO3
−], respec-tivamente. Si estas simplificaciones son válidas,como generalmente ocurre, se obtiene la ecua-ción simplificada:
[4.99]
y un valor de pH = 8,34.
Zona 4: segunda zona tamponada(Vequivalencia (1) = 10 < Vvalorante < Vequivalencia(2) == 20 mL; 1 < F < 2)
Para el cálculo del pH en esta zona, se parte,como es común, del balance de cargas dado enla ecuación [4.90]:
[4.90]
Al sustituir en este balance los correspon-dientes balances de masas y despreciar los tér-minos [H3O
+] y [OH–], como en otros casos, seobtiene una expresión idéntica a la obtenida enla anterior zona tamponada:
[4.94]
Las simplificaciones a aplicar en esta zona,son diferentes a las de la anterior zona tampo-nada, ya que el tampón está constituido por unamezcla de H2CO3 − HCO3
−, por lo que pH ≈ pKa1y, en consecuencia, el término Ka1 Ka2 del deno-minador de esta ecuación es ahora el que se pue-de despreciar. Con estas aproximaciones, la ecua-ción general se reduce a:
[4.100]
y puede reordenarse en la forma:
[4.101][ ]H O23
a b
b a1
+ = −−
C CC C
Ka
21
× =+
++
+CC
KC
ab
b 3
3a
H OH O
[ ][ ]
2H O 2
H O H Ob
b 3 1 2
3 3 1a× =
+( )+ +
++
+ +CC K K K
K K KC
a a a
a a a
1
21 2
[ ]
[ ] [ ]
[Na ] [H O ] [HCO ]2[CO ] [Cl ] [OH ]
3 3
32–
+ + −
− −
+ = ++ + +
[ ]H O3 1 2+ = K Ka a
[H CO ] [H O ] [CO ] [OH ]2 3 3 32+ = ++ − −
[ ]H O3a
b a2
+ =−
CC C
Ka
2
2 2
2
× = ++
++
+CC K
KCa
ab
b 3
3a
H OH O
([ ] )[ ]
Capítulo 4: Valoraciones ácido-base 153

Zona 5: segundo punto de equivalencia(200% de valoración)(Vvalorante = Vequivalencia (2) = 20 mL; F = 2)
En este punto la disolución contiene ácido car-bónico a una concentración 0,033 mol L–1 (ya queVT = VNa2CO3
+ VHCl = 30 mL). El pH se calcula apartir del correspondiente balance protónico
[4.102]
que puede simplificarse fácilmente, ya que al tra-tarse de una disolución de un ácido débil de pKa1= 6,35 puede aceptarse que [H3O
+] > Ka1 >> ka2lo que equivale a decir que tanto [CO3
2–] como[OH–] serán despreciables frente a [HCO3
–], loque permite obtener la siguiente ecuación sim-plificada:
[4.103]
que proporciona un valor de pH = 3,91 en estepunto de equivalencia.
Zona 6: exceso de valorante(Vvalorante > Vequivalencia (2) = 20 mL; F > 2)
En esta zona se da una mezcla de dos ácidos,el carbónico procedente del carbonato de sodioinicial y el exceso de ácido clorhídrico. El balan-ce de cargas, representado por la ecuación [4.90],de esta disolución es idéntico al de las zonasanteriores:
[4.90]
pero la diferencia radica en que el pH de estadisolución será francamente ácido, por lo quepodrá aceptarse que [HCO3
−], [CO32−] y [OH–]
serán despreciables frente a [Cl− ], con lo que elbalance se reduce a:
[4.104]
ecuación que permite obtener los valores de pHen la zona de exceso de valorante.
En el cuadro 4.7 se muestran los datos corres-pondientes a la valoración de una disolución deeste protolito diprótico a concentración 0,1 mol L–1
con ácido clorhídrico 0,1 mol L–1 y en la figura 4.11se representa la curva de valoración obtenida.
Los valores obtenidos que se dan en el cuadro4.7 permiten llegar a las siguientes conclusiones:
1. La ecuación simplificada proporcionaresultados correctos, salvo en las inme-diaciones de los dos puntos de equivalen-cia, ya que al usar la ecuación simplifica-da se supone como cierto que, porejemplo, para un 99,9% de valoración,
[4.105]
y que para un 100,1% de valoración
[4.106]
Sin embargo, es fácil comprobar que a pH= 8,34 (valor que corresponde al primerpunto de equivalencia),
[4.107]
por lo que el resultado de la ecuación sim-plificada sencillamente indica que para teneruna concentración de hidrogenocarbonatosuperior a la que existe en el punto de equi-
[ ]
[ ]
[ ]
HCO98
100
H CO1
100
CO1
100
3 b
2 3 b
32
b
−
−
=
=
=
C
C
C
[HCO ],
[H CO ],
3 b
2 3 b
− =
=
99 91000 1100
C
C
[HCO,
CO,
3 b
32
b
−
−
=
=
]
[ ]
99 91000 1100
C
C
[ ] [ ] [ ]H O Cl Na3 a b+ − += − = −C C2
[Na ] [H O ] [HCO ]2[CO ] [Cl ] [OH ]
3 3
32–
+ +
− −
+ = ++ + +
–
[ ]H O3 b+ = C Ka1
[H O ] [HCO ] 2[CO ] [OH ]3 3 32-+ − −= + +
154 Equilibrios iónicos y sus aplicaciones analíticas
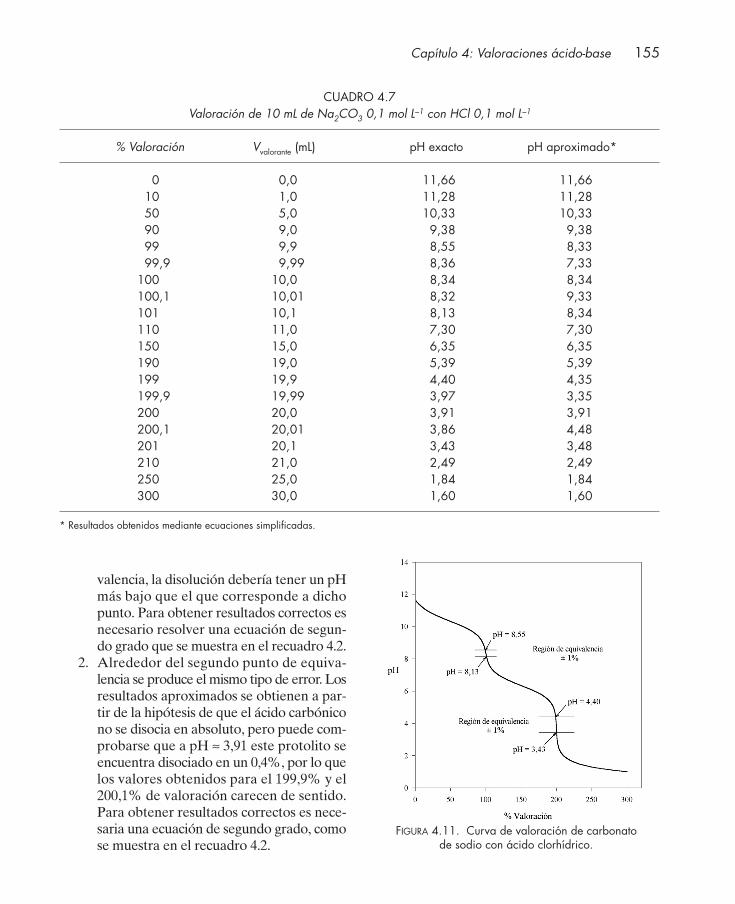
valencia, la disolución debería tener un pHmás bajo que el que corresponde a dichopunto. Para obtener resultados correctos esnecesario resolver una ecuación de segun-do grado que se muestra en el recuadro 4.2.
2. Alrededor del segundo punto de equiva-lencia se produce el mismo tipo de error. Losresultados aproximados se obtienen a par-tir de la hipótesis de que el ácido carbónicono se disocia en absoluto, pero puede com-probarse que a pH ≈ 3,91 este protolito seencuentra disociado en un 0,4%, por lo quelos valores obtenidos para el 199,9% y el200,1% de valoración carecen de sentido.Para obtener resultados correctos es nece-saria una ecuación de segundo grado, comose muestra en el recuadro 4.2.
Capítulo 4: Valoraciones ácido-base 155
CUADRO 4.7Valoración de 10 mL de Na2CO3 0,1 mol L–1 con HCl 0,1 mol L–1
% Valoración Vvalorante (mL) pH exacto pH aproximado*
0 0,0 11,66 11,6610 1,0 11,28 11,2850 5,0 10,33 10,3390 9,0 9,38 9,3899 9,9 8,55 8,3399,9 9,99 8,36 7,33
100 10,0 8,34 8,34100,1 10,01 8,32 9,33101 10,1 8,13 8,34110 11,0 7,30 7,30150 15,0 6,35 6,35190 19,0 5,39 5,39199 19,9 4,40 4,35199,9 19,99 3,97 3,35200 20,0 3,91 3,91200,1 20,01 3,86 4,48201 20,1 3,43 3,48210 21,0 2,49 2,49250 25,0 1,84 1,84300 30,0 1,60 1,60
* Resultados obtenidos mediante ecuaciones simplificadas.
FIGURA 4.11. Curva de valoración de carbonato de sodio con ácido clorhídrico.

También es importante resaltar que paraun 199% y un 201% de valoración, la ecua-ción de segundo grado proporciona un pHligeramente distinto al obtenido mediantela ecuación simplificada, lo que significaque en estos puntos la disociación del áci-do carbónico influye en el pH final de ladisolución. Cuando el exceso de valorantees superior al 1%, ambas ecuaciones pro-porcionan los mismos valores, lo que indi-ca que, al aumentar el exceso de ácido fuer-te, la contribución del ácido carbónico alpH final es totalmente despreciable.
3. El primer punto de equivalencia de la cur-va no puede utilizarse para un análisiscuantitativo con indicadores visuales, yaque la zona de equivalencia de ±1% com-prende únicamente 0,4 unidades de pH,aunque sí es posible determinar este pun-to final potenciométricamente.
4. Para el segundo punto de equivalencia, elúnico indicador utilizable sería el naranjade metilo, aunque se cometería un errorsuperior al 1%. Sin embargo, este proble-ma puede solucionarse fácilmente si se hier-ve la disolución durante unos minutos cuan-do el indicador adquiere el color de la forma
ácida; ello provoca un aumento de pH deri-vado del desplazamiento de la reacción dedescomposición del ácido carbónico pre-sente (H2CO3 → CO2↑ + H2O) al disminuirla solubilidad del CO2 (gas) y evaporarse elCO2 de la disolución. Con ello, la disoluciónde HCO3
− + H2CO3 que se tiene en las cer-canías del punto final, pasa a contener sóloHCO3
−, y en consecuencia, el pH aumentaconsiderablemente hasta ser el correspon-diente a una disolución de HCO3
−. Despuésde enfriar, se prosigue la adición de valo-rante y se repite el proceso hasta que la ebu-llición no provoque la desaparición del colorde la forma ácida del indicador, lo que sig-nifica que la disolución ya contiene un lige-ro exceso de ácido fuerte. Mediante esta téc-nica es posible determinar el punto final conuna buena exactitud y utilizar indicadorescomo el rojo de metilo, que sin esta técnicaserían inadecuados. Evidentemente, tam-bién cabe utilizar un método potenciomé-trico para la detección del punto final.
5. Un aspecto relevante es la importancia realde los resultados exactos obtenidos median-te la ecuación de segundo grado. Aunquepueden tener interés teórico, su interés
156 Equilibrios iónicos y sus aplicaciones analíticas
Primer punto de equivalencia
A partir del balance de cargas se obtiene:
de donde:
Segundo punto de equivalencia
A partir del balance de cargas se obtiene la expresión:
que, una vez reordenada, queda:
V C V K C C Ka aT 3 b T a 3 aH O H O[ ] ( )[ ]+ ++ + − − =21 12 0
2 1
1
CC K
KCa
ab 3
b
3aH O
H O+ =
+++
+[ ][ ]
( )[ ]
( )[ ]
2
0
2
1 1 2
C C
K C C C K Ka a a
b a 3
b a 3 a
H O
H O
− +
+ − − =
+
+
2
21 1 22
1 1 2
CC K K K
K K Ka a a
a a ab
b 3
3 3
H OH O H O
=+
+ +
+
+ +
( [ ][ ] [ ]
Ecuaciones a aplicar en las inmediaciones de los puntos de equivalencia de la valoraciónde carbonato de sodio con ácido clorhídrico
RECUADRO 4.2

práctico es, frecuentemente, escaso. Porejemplo, en el caso de la curva de valora-ción de carbonato de sodio, si el pH parael 90% de valoración es de 9,38, para el100% es de 8,34 y para el 110% de 7,30, esevidente que la zona de equivalencia de±1% será muy inferior a 2 unidades de pHy, por lo tanto, no será posible la utilizaciónde un indicador visual; dado que cualquierpunto comprendido entre el 90 y el 110%de valoración deberá tener un pH 9,38 y7,30, un cálculo exacto es innecesario, sal-vo que se desee evaluar la exactitud posi-ble si se recurre a una detección potencio-métrica del punto final.
B) Valoración de ácido fosfórico
Para la construcción de la curva de valoraciónde este protolito poliprótico se hace uso de unplanteamiento equivalente al de los casos más sen-cillos, aunque a medida que aumenta el númerode equilibrios de disociación es posible que algu-nas constantes tengan valores muy próximos, loque puede impedir las simplificaciones y obligara resolver ecuaciones relativamente complejas.
Se toma como ejemplo la valoración de 10 mLde una disolución de ácido fosfórico 0,1 mol L–1
(H3PO4: pKa1 = 2,15; pKa2 = 7,21; pKa3 = 12,32)con hidróxido de sodio 0,1 mol L–1. Al añadir elvalorante se producirán las siguientes reaccionesde valoración:
H3PO4 + NaOH →→ NaH2PO4 + H2O
[4.108]
NaH2PO4 + NaOH →→ Na2HPO4 + H2O
[4.109]
Na2HPO4 + NaOH →→ Na3PO4 + H2O
[4.110]
De acuerdo con estas reacciones la curva devaloración puede descomponerse en ocho zonas,que se esquematizan a continuación:
1. Punto inicial: corresponde a la disoluciónde un ácido triprótico, y se cumplirá que:
[H3O+] > Ka1 >> Ka2 >> Ka3
2. Zona tamponada: la disolución contienemezclas en diversas proporciones deH3PO4 y H2PO4
– , con lo que:
[H3O+] ≈ Ka1 >> Ka2 >> Ka3
3. Primer punto de equivalencia: la disolucióncontiene únicamente H2PO4
–, un anfolito,con lo que:
Ka1 > [H3O+] > Ka2 >> Ka3
4. Zona tamponada: la disolución contienemezclas en diversas proporciones deH2PO4
– y HPO42– , con lo que:
Ka1 >> [H3O+] ≈ Ka2 >> Ka3
5. Segundo punto de equivalencia: la disolu-ción contiene únicamente HPO4
2–, un anfo-lito, por lo que:
Ka1 >> Ka2 > [H3O+] > Ka3
6. Zona tamponada: la disolución contienemezclas en diversas proporciones deHPO4
2– y PO43–, por lo que:
Ka1 >> Ka2 >> [H3O+] ≈ Ka3
7. Tercer punto de equivalencia: la disolucióncontiene una base, PO4
3–, lo que permitesuponer que:
Ka1 >> Ka2 >> Ka3 > [H3O+]
H PO H PO H PO
H PO H PO HPO
HPO HPO PO
PO PO NaOH
3 41
NaOH3 4 2 4
2
NaOH
2 43
NaOH2 4 4
2
4
NaOH
42
5
NaOH42
43
6
NaOH
43
7
NaOH43
exceso
8
+ − +
− + − − +
− + − − +
− + −( )
→ + →
→ + →
→ + →
→ +
Capítulo 4: Valoraciones ácido-base 157

8. Exceso de valorante: se trata de una mez-cla de dos bases, Na3PO4 e hidróxido desodio, con lo que:
Ka1 >> Ka2 >> Ka3 > [H3O+]
De manera análoga a los expuesto en otroscasos, a partir de los balances de masas, cargas yprotónico y haciendo uso de las simplificacionesindicadas se pueden conseguir fácilmente expre-siones útiles para el cálculo del pH en cada unade estas zonas de la curva de valoración.
En el cuadro 4.8 se muestran los resultadoscorrespondientes a una curva de valoración deH3PO4 con hidróxido de sodio, obtenidos median-te ecuaciones simplificadas ya discutidas en loscapítulos de equilibrio ácido-base, junto a los valo-res correctos proporcionados por ecuaciones nosimplificadas.
Puede observarse que en algunos casos losresultados de las ecuaciones simplificadas pro-porcionan resultados absurdos (fácilmentedetectables) o incorrectos; pero, por lo general,ello no afecta a la validez de las conclusiones.En la figura 4.12 se representa la curva de valo-ración obtenida.
A la vista de los resultados, se obtienen lassiguientes conclusiones:
1. El primer protón del ácido fosfórico, aun-que corresponde a un ácido relativamen-te fuerte (pKa1 = 2,15), produce un cambiode pH pequeño (la zona de equivalenciade ± 1% abarca desde pH 4,17 hasta pH5,25), ya que en el primer punto de equi-valencia existe un anfolito con pKa1 = 2,15y pKa2 = 7,21, lo que provoca que el pH deeste punto sea francamente ácido. Comoconsecuencia, no será posible detectar elprimer punto final de esta curva median-te indicadores visuales con un error infe-rior al 1%, si bien el verde de bromocre-sol permitiría detectarlo con un errorligeramente superior al 1%, lo que puedeser tolerable en muchos casos.
FIGURA 4.12. Curva de valoración de ácido fosfóricocon hidróxido de sodio.
2. En el segundo punto de equivalencia, exis-te otro anfolito (HPO4
2−) de pKa2 = 7,21 y pKa3 = 12,32, por lo que el pH en este pun-to es sólo moderadamente alcalino y la altu-ra del salto es escasa (la zona de equivalen-cia de ±1% abarca desde pH 9,16 a pH10,16) por lo que no es posible detectar elpunto final mediante un indicador visual,aunque la fenolftaleína o la timolftaleína po-drían ser válidas para algunas aplicaciones.
3. La neutralización del tercer protón del áci-do fosfórico no provoca, prácticamente,cambio de pH, como era de esperar parael caso de un ácido extremadamente débil(pKa3 = 12,32), por lo que no será posiblela determinación de este protón medianteuna volumetría de neutralización.
4.5. Preparación de disolucionesvalorantes
En el estudio desarrollado en el apartado ante-rior sobre la valoración de diferentes tipos de pro-
158 Equilibrios iónicos y sus aplicaciones analíticas
tolitos, se ha indicado que para conseguir unamayor exactitud en el proceso de valoración, lavariación del pH en las cercanías del punto deequivalencia debe ser lo más grande posible. Estacondición, muy importante desde un punto devista práctico, se consigue mediante el empleo dedisoluciones valorantes constituidas por protoli-tos fuertes. Así, es común utilizar un ácido fuer-
te (ácido clorhídrico o ácido sulfúrico) y una basefuerte (hidróxido de sodio) para la valoración debases y ácidos, respectivamente. Dado que estassustancias no se pueden obtener comercialmen-te con la pureza adecuada para su empleo comoestándar o patrón primario, sus disoluciones sehan de estandarizar, previamente a su uso, reali-zando valoraciones con un patrón primario.
Capítulo 4: Valoraciones ácido-base 159
CUADRO 4.8Valoración de10 mL de H3PO4 0,1 mol L–1 con NaOH 0,1 mol L–1
% Valoración Vvalorante (mL) pH exacto pH aproximado*
0 0,0 1,63 1,5810 1,0 1,76 1,2050 5,0 2,29 2,1590 9,0 3,17 3,1099 9,9 4,17 4,1599,9 9,99 4,64 5,15
100 10,0 4,71 4,68100,1 10,01 4,78 4,21101 10,1 5,25 5,21110 11,0 6,26 6,26150 15,0 7,21 7,21190 19,0 8,16 8,16199 19,9 9,16 9,21199,9 19,99 9,60 10,21200 20,0 9,66 9,77200,1 20,01 9,72 9,32201 20,1 10,16 10,32210 21,0 11,12 11,37250 25,0 11,85 12,32290 29,0 12,12 13,27299 29,9 12,16 14,32299,9 29,99 12,17 15,32300 30,0 12,17 12,36300,1 30,01 12,17 9,40301 30,1 12,17 10,40310 31,0 12,21 11,39350 35,0 12,34 12,05400 40,0 12,45 12,30
* Resultados obtenidos mediante ecuaciones simplificadas.

4.5.1. Disolución estándar de HCl 0,1 mol L–1
El ácido clorhídrico es el ácido más utilizadoen valoraciones ácido-base. Aunque no es están-dar primario o patrón primario. Sus disolucionesforman mezclas azeotrópicas que pueden utili-zarse en ciertas condiciones para preparar diso-luciones valoradas por pesada. Sin embargo, lomás común es llevar a cabo la estandarización delas mismas mediante valoración con un patrónprimario, de características básicas, tal como elcarbonato de sodio.
El Na2CO3 se encuentra en el comercio conuna alta pureza (99,999%) y sólo requiere unsecado posterior a 150 °C durante unas dos horaspara su empleo directo como patrón primario enla valoración de HCl. Para llevar a cabo la estan-darización de una disolución de HCl 0,1 mol L–1,una cantidad exactamente pesada (con precisiónde hasta la décima de mg) de carbonato de sodio(entre 0,20 y 0,25 g) se disuelve en 50 mL de aguadestilada en un enlermeyer de 300 mL. Se aña-den una o dos gotas de disolución de fenolftaleí-na al 1% en etanol y se comienza la valoraciónpor adición de volúmenes de la disolución de HCldesde una bureta de 50 mL. Cuando se decolo-ra la fenolftaleína (púrpura → incoloro), justo amitad de la valoración:
Na2CO3 + HCl → NaHCO3 + H2O [4.111]
se adicionan varias gotas de disolución acuosa derojo de metilo y se continua la valoración hastaviraje del indicador (amarillo → rojo)
NaHCO3 + HCl → H2CO3 + H2O [4.112]
Como se ha indicado en el apartado 4.4.2.A,se ha de hervir la disolución durante unos minu-tos cuando el indicador adquiere el color de laforma ácida (rojo) para obtener resultados exac-tos en esta valoración. Por otra parte, dado queel primer punto de equivalencia de esta valora-ción es difícil de detectar con indicadores visua-les (ver apartado 4.4.2.A), no se aconseja haceruso del volumen correspondiente al viraje de la
fenolftaleína ya que se obtienen errores apre-ciables desde un punto de vista cuantitativo. Apartir del volumen de ácido consumido en estavaloración y de la cantidad de carbonato de sodiopesada se determina la concentración real de ladisolución de HCl.
De acuerdo con lo indicado en el apartado4.1, es común expresar la concentración de ladisolución de valorante, en este caso HCl, deacuerdo con:
Creal = Caprox × f [4.113]
siendo Caprox la concentración aproximada de ladisolución HCl, Creal su concentración real y f elfactor volumétrico. De acuerdo con esta expre-sión, f es el factor por el que hay que multiplicarla concentración aproximada de la disolución devalorante para obtener su concentración real.
De acuerdo con la reacción de valoraciónglobal:
Na2CO3 + 2 HCl → H2CO3 + 2 NaCl [4.114]
se puede establecer que si se parte de Na2CO3sólido:
[4.115]
y para calcular el factor:
[4.116]
En general, f oscila alrededor de 1, si bien hayque reseñar que lo importante es su correcta deter-minación, y por tanto, su mayor o menor cercaníaa 1 no es una indicación de que la disolución deHCl esté mejor o peor estandarizada. En el casode una disolución de ácido sulfúrico el proceso depreparación y estandarización sería análogo.
f
CC
= real
aproximada
gNa CO1 mol Na CO
Peso molecular Na CO2 moles HCl
1 mol Na CO1.000 mL/L
(mL)
2 32 3
2 3
2 3 HCl consumidoreal
× ×
× × =V
C
160 Equilibrios iónicos y sus aplicaciones analíticas

4.5.2. Disolución estándar de NaOH 0,1 mol L–1
Las disoluciones de NaOH se utilizan gene-ralmente para la valoración de diversos tipos deácidos. El hidróxido de sodio no es estándar opatrón primario y por tanto no es posible pre-parar sus disoluciones por pesada y se ha de re-currir a la estandarización de sus disoluciones.Los patrones primarios más utilizados para lavaloración de disoluciones de NaOH son el fta-lato ácido de potasio y el ácido sulfámico, el cualorigina en agua sulfato ácido de amonio que esrealmente el patrón primario. De ambos están-dares se recomienda utilizar ftalato ácido de pota-sio para valorar disoluciones de NaOH.
El ftalato ácido de potasio (KHFt) se encuen-tra en el comercio con una pureza de 99,98%.Para poder utilizarlo como patrón primario seha de desecar en estufa. Se ha de evitar sobre-pasar los 125 °C en este tratamiento ya que seoriginan transformaciones indeseables: el ftala-to ácido de potasio se transforma en ftalato neu-tro y ácido ftálico y sublima algo de anhídricoftálico para llevar a cabo la estandarización deuna disolución de NaOH 0,1 mol L–1 con estepatrón primario, conforme con la reacción devaloración:
Una cantidad exactamente pesada (con unaprecisión de hasta la décima de mg) de estepatrón primario (entre 0,80 y 0,90 g) se disuel-ve en 50 mL de agua destilada en un enlermeyerde 300 mL. Se añaden una o dos gotas de diso-lución de fenolftaleína y se comienza la valora-ción mediante la adición de la disolución deNaOH desde la bureta de 50 mL. El punto finalviene dado por la aparición de un color rosa opúrpura débil.
La concentración real de la disolución deNaOH se determina haciendo uso de una expre-sión similar a la [4.115], que en este caso vienedada por:
[4.117]
Análogamente puede definirse el factor de lavaloración con la expresión [4.116].
Un aspecto de gran interés en la preparacióny uso de las disoluciones de NaOH es evitar lacarbonatación de las mismas, dado que el hidró-xido de sodio es delicuescente y tiene gran ten-dencia a absorber CO2. Así, para preparar 1l deuna disolución de NaOH 0,1 mol L–1 se pesa enuna vaso de precipitados una cantidad de NaOHque sea aproximadamente un 50% superior a lacantidad teórica (unos 6 g de NaOH). Para disol-ver la corteza de Na2CO3 que puede envolver laslentejas, se añaden 2-3 porciones de aproxima-damente 2 mL cada una de agua destilada, se agi-ta suavemente y se tira el líquido sobrenadante.Las lentejas que quedan en el vaso de precipita-dos se disuelven en agua destilada y se llevan aun volumen de un litro que se transvasa y con-serva en un frasco de plástico. Cuando se deseaconservar la disolución de NaOH 0,1 mol L–1 porun espacio de tiempo prolongado, se han de tomarlas precauciones necesarias para evitar su carbo-natación haciendo uso de recipientes especialesque evitan su contacto con el CO2 atmosférico.
Cuando no se toman las precauciones ade-cuadas en la conservación de la disolución deNaOH y por tanto ésta se encuentra ligeramen-te carbonatada, se pueden originar errores en lavaloración que están directamente relacionadoscon el indicador utilizado en la misma. La reac-ción de carbonatación responde a:
[4.118]
2 NaOH CO
Na CO H O2
2 3 2
+ →→ +
gKHFt1 mol KHFt
Peso molecular KHFt1 mol NaOH1 mol KHFt
1.000 mL/L(mL)NaOH consumido
real
× ×
× × =V
C
Capítulo 4: Valoraciones ácido-base 161

Si esta disolución de NaOH parcialmente car-bonatada se utiliza en la valoración de un ácido(por ejemplo HCl) utilizando rojo de metilo(intervalo de transición: 4,4 – 3,1) como indica-dor se originan las siguientes reacciones de valo-ración:
NaOH + HCl →→ H2O + NaCl [4.119]
Na2CO3 + 2 HCl →→ H2CO3 + 2 NaCl [4.120]
Dado que por cada 2 moles de NaOH que secarbonata se origina un mol de Na2CO3 y se con-sumen 2 moles de HCl en la valoración, el efec-to de la carbonatación no se manifiesta en el pro-ceso de valoración, y por tanto no se comete erroral utilizar estas disoluciones de NaOH.
Si se utiliza fenolftaleína como indicador setienen las siguientes reacciones de valoración:
NaOH + HCl →→ H2O + NaCl [4.119]
Na2CO3 + HCl →→ NaHCO3 + NaCl [4.121]
En este caso, 2 moles de NaOH originan unmol de Na2CO3 y sólo se consume para su valo-ración un mol de HCl. Se origina por tanto unerror debido a la carbonatación de la disoluciónde NaOH. Este error es por exceso, dado que serequiere un mayor volumen de la disolución deNaOH carbonatada para valorar todo el ácidopresente.
Aunque utilizando como indicador rojo demetilo se evitan los errores debido a la carbonata-ción de las disoluciones de NaOH, este indicadorno es adecuado para las valoraciones de ácido débi-les, y por tanto se ha de utilizar fenolftaleína u otrocon un intervalo de transición similar. En definiti-va, se han de extremar las precuaciones en la con-servación de las disoluciones de hidróxido de sodiopara evitar los errores asociados a la carbonata-ción de las mismas.
4.6. Aplicaciones
Las aplicaciones de las valoraciones ácido-base son muy numerosas, siendo de especial inte-rés en el ámbito agroalimentario, en el cual cons-tituyen un apartado significativo en el contextode las técnicas analíticas recomendadas por orga-nismos internacionales para el control de calidadde diversos productos. En este sentido, las apli-caciones que se comentan a continuación cons-tituyen ejemplos significativos de determinacio-nes en esta área de aplicación, tales comodeterminación de: 1) acidez de un vinagre comer-cial; 2) acidez en otros tipos de alimentos y 3)compuestos nitrogenados tanto inorgánicos comoorgánicos.
4.6.1. Determinación de la acidezde un vinagre comercial
El vinagre es un líquido ácido obtenido a par-tir de sustancias que contienen almidón o azú-cares mediante fermentación. El vinagre ya eraconocido en las antiguas civilizaciones orienta-les, donde fue utilizado en ciertas ocasionescomo bebida refrescante; posteriormente en laantigua Grecia y Roma fue considerado comoun remedio para ciertas afecciones. Existendiversos tipos de vinagre según su procedencia.Así se tiene vinagre de sidra a partir de la man-zana, de vino (uva), de malta (cebada) y de azú-car (azúcar, melazas o jarabes). En todos loscasos, se origina a partir de procesos microbio-lógicos. Así, las bacterias pertenecientes a lasespecies acetobacter presentes en vinos fermen-tados, zumos de manzana, etc, actúan sobre eletanol presente en estos alimentos originandoen diversas etapas su oxidación a ácido acético,siendo de gran importancia en este proceso lapresencia de oxígeno.
La determinación de la acidez en un vinagrecomercial constituye una aplicación de la valo-ración de un ácido débil con una base fuerte. Parallevar a cabo esta valoración, se toma un volu-men de vinagre y se diluye a unos 50 mL con
162 Equilibrios iónicos y sus aplicaciones analíticas

agua destilada. Posteriormente se valora con ladisolución de NaOH utilizando fenolftaleínacomo indicador. Los resultados se expresan enmg de ácido acético por 100 mL de muestra.
4.6.2. Determinación de acidez en alimentos
El contenido total de ácidos en un alimento esuno de los ensayos más simples que se puedenrealizar para controlar su pureza. La mayoría delos ácidos utilizados en la industria alimentaria sepueden valorar por métodos simples, ampliamen-te descritos en la bibliografía. La acidez en un ali-mento se puede expresar de diversas formas,como se muestra a continuación.
La acidez total valorable (TTA) se determinafrecuentemente por valoración con NaOH 0,1mol L–1 usando fenolftaleína o azul de bromoti-mol como indicador. La detección exacta del pun-to final a veces es difícil debido a la presenciafundamentalmente de colores oscuros en el ali-mento. En este caso, se puede añadir una canti-dad extra de indicador o bien llevar a cabo unadilución previa con agua, si la sensibilidad de lavaloración lo permite, aunque este problema seresuelve fácilmente haciendo uso de la detecciónpotenciométrica del punto final. El valor de TTApara cada alimento se presenta en términos delácido predominante en el mismo, por ejemplo,ácido láctico en leche, cítrico en la mayoría delos frutos, málico en manzanas, acético en vina-gre, etc. El principal ácido volátil presente en losalimentos es el ácido acético, presente en sal-mueras, adobos, escabeches, etc. La acidez volá-til (VA) se puede determinar por diferencia alvalor de TTA después de evaporar la muestravarias veces, de forma que sólo permanezca enla misma el ácido fijo (FA), de tal modo que:
%VA = %TTA – %FA [4.122]
El contenido de ácido volátil también se pue-de determinar de manera directa en el propiodestilado de la muestra. Los resultados se expre-san como contenido de ácido acético.
La acidez volátil es, por ejemplo, una medidade la corrección del proceso de fermentación delos vinos nuevos y del mantenimiento de la cali-dad de los vinos viejos. Un aumento apreciablede la acidez volátil durante el almacenamientoindica contaminación bacteriana y obliga a la pas-teurización inmediata y/o a la adición de 100-150µg mL–1 de SO2. Los vinos nuevos no contami-nados deben tener una acidez volátil inferior a0,05 g 100 mL–1, mientras que el valor corres-pondiente a vinos envejecidos no contaminadoses del orden de 0,08 g 100 mL–1.
En productos tales como escabeches, sal-mueras, cremas de ensaladas, etc., es usual cal-cular el porcentaje de ácido volátil en la fase acuo-sa (VAqa), parámetro que viene determinado porla expresión:
[4.123]
donde VA es el porcentaje de acidez volátil en elproducto, W es el porcentaje de agua en la mues-tra y VA + W es virtualmente equivalente a lapérdida total de materia volátil durante la eva-poración. Para un producto cuya conservacióndepende del contenido de ácido acético, el valorde VAaq debe ser al menos inferior al 3,5%.
4.6.3. Determinación de compuestos nitrogenados
La determinación de sustancias nitrogenadaspor valoraciones ácido-base es muy importantedesde un punto de vista práctico y abarca tantoa compuestos inorgánicos como orgánicos. En elámbito de la determinación de compuestos nitro-genados inorgánicos se aborda la determinaciónde sales amónicas y de nitratos y nitritos de granimportancia en abonos.
Por otra parte, la determinación de compues-tos orgánicos nitrogenados mediante el conocidométodo de Kjeldahl, es hoy por hoy un referen-te para el análisis de proteínas y piensos, funda-mentalmente.
VA
VAVA Waq 100=
+×
Capítulo 4: Valoraciones ácido-base 163

Estas determinaciones se fundamentan en tresetapas básicas:
1. Tratamiento de la muestra para transfor-mar el compuesto nitrogenado en salamónica.
2. Destilación del amoniaco previamente for-mado y recogida del mismo en una diso-lución ácida. Habitualmente se utiliza unadisolución de ácido bórico al 4%.
3. Valoración del borato de amonio forma-do con una disolución patrón de ácidofuerte, generalmente HCl 0,1 mol L–1.
La reacción química que se utiliza para trans-formar el compuesto nitrogenado en amoniacodepende de la propia naturaleza de la muestra.Así, en el caso de sales amónicas se tiene:
NH4R + NaOH →→ NH3 + NaR + H2O [4.124]
mientras que para reducir los iones nitrato y nitri-to a amoniaco se utiliza un reductor tal como laaleación Devarda (50% Cu, 45% Al y 5% Zn) enmedio básico y posterior tratamiento con NaOH.
En el caso de compuestos orgánicos nitroge-nados, se requiere un proceso de mineralización yuno de destilación. En la mineralización, se calien-ta durante unos 20-45 minutos el compuesto orgá-nico con ácido sulfúrico concentrado en presenciade un catalizador y de K2SO4 anhidro. Aunque seha comprobado que el mercurio es el mejor cata-lizador para esta digestión, en la actualidad no seutiliza debido a su elevada toxicidad y en su lugarse emplea sulfato de cobre. En estas condiciones,el carbono presente en el compuesto orgánico seoxida a CO2 y el nitrógeno queda en disolucióncomo sal amónica dada la acidez del medio. Enuna segunda etapa, estas sales amónicas por adi-ción de un exceso de NaOH de acuerdo con lareacción [4.124] se transforman en amoniaco quese destila. Para llevar a cabo este proceso se utili-za un matraz especial con un cuello largo con obje-to de evitar proyecciones que se denomina matrazde Kjeldahl (figura 4.13). Después de la minerali-
zación el matraz, como se muestra en la figura, seacopla a un sistema de destilación para separar elamoniaco formado y que finalmente se recogesobre la disolución de ácido bórico.
FIGURA 4.13. Sistema de Kjeldahl para la determinaciónde compuestos nitrogenados. a) Preparación de la muestra;
b) Destilación y recogida del amoniaco.
Cuando la disolución ácida que se utiliza pararecoger el amoniaco es de ácido bórico, no esnecesario que esté estandarizada, ya que al seréste un ácido muy débil, se puede valorar per-fectamente el borato amónico formado con áci-do clorhídrico, de acuerdo con la reacción:
NH4H2BO3 + HCl → H3BO3 + NH4Cl [4.125]
Dado que el pH de la disolución de ácidobórico es aproximadamente 4,7, y que éste seencuentra en exceso frente a la sal amónica for-mada, al añadir rojo de metilo a la disolución éstatomará inicialmente un color amarillo. Cuandose adiciona HCl, el color no varía hasta que seha valorado todo el borato amónico. Una gotaen exceso de HCl hará virar al indicador toman-do la disolución un color rojo.
El uso combinado de los métodos propuestospara sales amónicas y nitratos, permite la deter-minación de este último en abonos.
164 Equilibrios iónicos y sus aplicaciones analíticas
a) b) NaOH

Generalmente, estos productos contienen salesamónicas y nitratos, y por tanto, el amoniaco des-prendido procede de ambas especies. Sin embar-go, es posible su diferenciación si se llevan a cabodos determinaciones en la misma muestra con-forme a:
1. Una porción de la muestra se trata condisolución de NaOH y de acuerdo con laecuación [4.121] el amoniaco desprendidoprocede sólo de las sales amónicas pre-sentes en la misma.
2. Otra porción similar se trata con aleaciónDevarda en medio básico y el amoniacodesprendido será proporcional al conteni-do de sales amónicas y nitrato en el abono.
3. Finalmente, por diferencia entre ambasdeterminaciones se calcula el contenido denitrato en el abono.
En el caso de la determinación de compues-tos orgánicos nitrogenados, una vez determina-do el contenido de nitrógeno en la muestra, elporcentaje de proteínas en la misma se calcula enbase al empleo de un factor empírico que depen-de del aminoácido mayoritario presente en dichaproteína.
En general, este factor es de 6,25, lo que equi-vale a considerar que todas las proteínas estáncompuestas por un 16% de nitrógeno:
% Proteínas = % Nitrógeno × 6,25 [4.126]
Sin embargo, el rango de composición de nitró-geno en las proteínas oscila entre el 15 y el 18%,por lo que es común que en productos específi-cos este factor sea diferente. Así, toma valores de5,18 para almendras; 5,30 para cocos y demás fru-tos secos; 5,46 para cacahuetes; 5,70 para harinade trigo y 6,38 para leche y productos lácteos.
En la actualidad están comercializados equi-pos automatizados para la determinación denitrógeno orgánico en diversas muestras median-te el método de Kjeldahl, tal como el modeloUDK 140 de la firma Gomensoro, S.A., que semuestra en la figura 4.14.
FIGURA 4.14. Dispositivo comercial para la determinaciónde nitrógeno mediante el método de Kjeldahl
(modelo UDK 140 de la firma Gomensoro, S. A.). Reproducido con permiso de esta firma comercial.
Este dispositivo permite llevar a cabo todaslas operaciones excepto la digestión con ácidosulfúrico, que se realiza en un digestor que per-mite el tratamiento simultáneo de diversas mues-tras (6-20 muestras). Posteriormente, el recipientede vidrio en el cual se ha llevado a cabo la diges-tión se coloca en la unidad Kjeldahl que realizalas siguientes funciones:
1. Adiciona la disolución de NaOH con obje-to de transformar las sales amónicas enamoniaco.
2. Una corriente de vapor arrastra al amo-niaco formado que condensa en el reci-piente de valoración.
3. El borato amónico formado se valorapotenciométricamente con disolución deHCl utilizando un valorador automáticoen la modalidad de punto final prefijado,en este caso al valor de pH del ácido bóri-co (pH = 4,7).
Capítulo 4: Valoraciones ácido-base 165

4.1. Definir la zona de viraje de un indicador de neutralización (ácido-base).
4.2. Explicar brevemente por qué el pH varía lentamente en algunas zonas de una curva de valoración.
4.3. La fenolftaleína (8,2-9,8) es un indicador adecuado para la valoración de (marcar con una X lo correcto):
[ ] Ácidos fuertes con bases fuertes[ ] Ácidos débiles con bases fuertes[ ] Bases fuertes con ácidos fuertes[ ] Bases débiles con ácidos fuertes
4.4. ¿Qué condición ha de cumplir un indicador para poderse utilizar en la valoración de un ácido débil conNaOH?
4.5. ¿De qué depende el salto de pH en el punto de equivalencia en una valoración de un ácido con una base?
[ ] Fuerza del ácido[ ] Indicador utilizado[ ] Concentración del ácido[ ] Temperatura
4.6. En la tabla siguiente se dan los intervalos de viraje de tres indicadores:
Indicador Intervalo de viraje
Fenolftaleína (F) 8,0-9,6
Rojo de metilo (RM) 4,8-6,0
Naranja de metilo (NM) 3,1-4,4
Cuál o cuáles se podrían utilizar para:
1. Valoración de HCl con NaOH 0,1 mol L–1.2. Valoración de Na2CO3 con HCl 0,1 mol L–1.3. Valoración de Na2HPO4 con HCl 0,1 mol L–1.4. Determinación de bórax por valoración con HCl 0,1 mol L–1.5. Determinación de sales amónicas mediante la destilación del amoniaco formado por adición de un ex-
ceso de NaOH, recogida del amoniaco sobre un exceso de H2SO4 estandarizado y valoración con NaOH0,1 mol L–1 del exceso de H2SO4.
H2CO3 (pKa1 = 6,35, pKa2 = 10,33); H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33); NH4+ (pKa = 9,24);
H3BO3 (pKa = 9,24).
4.7. La gráfica siguiente muestra la curva de valoración de un ácido con hidróxido de sodio. ¿De qué tipo de áci-do se trata? ¿Podría utilizarse un indicador de pKa = 8,3 para la determinación del punto final?
166 Equilibrios iónicos y sus aplicaciones analíticas
Cuestiones
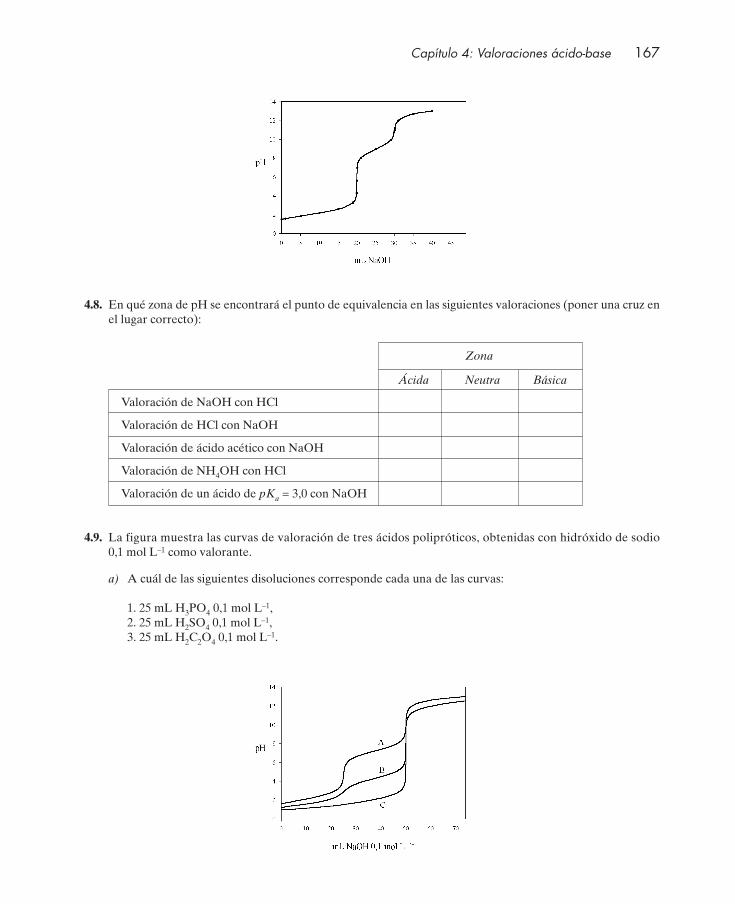
4.8. En qué zona de pH se encontrará el punto de equivalencia en las siguientes valoraciones (poner una cruz enel lugar correcto):
Zona
Ácida Neutra Básica
Valoración de NaOH con HCl
Valoración de HCl con NaOH
Valoración de ácido acético con NaOH
Valoración de NH4OH con HCl
Valoración de un ácido de pKa = 3,0 con NaOH
4.9. La figura muestra las curvas de valoración de tres ácidos polipróticos, obtenidas con hidróxido de sodio 0,1 mol L–1 como valorante.
a) A cuál de las siguientes disoluciones corresponde cada una de las curvas:
1. 25 mL H3PO4 0,1 mol L–1, 2. 25 mL H2SO4 0,1 mol L–1, 3. 25 mL H2C2O4 0,1 mol L–1.
Capítulo 4: Valoraciones ácido-base 167

b) Proponer métodos volumétricos ácido-base para la determinación de los ácidos anteriores, comentandoqué indicador sería el más adecuado y el volumen de valorante que se necesitaría en cada caso.
Indicador pK
Rojo de metilo 5,0
Verde de bromocresol 4,7
Azul de bromotimol 7,1
Fenolftaleína 9,2
H3PO4 (pKa1 = 2,1, pKa2 = 7,2, pKa3 = 12,3); H2SO4 (pKa2 = 1,9); H2C2O4 (pKa1 = 1,3 pKa2 = 4,3).
4.10. Comentar las diferencias más significativas entre las curvas de valoración que se obtendrían al valorar unabase monoprótica fuerte y una débil utilizando un ácido fuerte como valorante. Razonar la forma que ten-drían las curvas que se obtendrían al valorar: 1) una disolución de maleato de sodio (Na2M) con HCl; 2) unamezcla equimolar de ácido sulfúrico y ácido maleico con NaOH; y 3) una mezcla equimolar de carbonatode sodio e hidrogenocarbonato de sodio con HCl.
H2SO4 (pKa2 = 1,9); HOOC-CH=CH-COOH (pKa1 = 2,30, pKa2 = 6,26); H2CO3 (pKa1 = 6,35, pKa2 = 10,33).
4.11. Dibujar, de forma intuitiva, las curvas de valoración de las siguientes disoluciones utilizando los valorantesque se indican:
Valorante Disolución
NaOH HCl + HA
HCl Na2B + NaHBNaOH + Na2B HA (pKa = 7,0); H2B (pKa1= 6, pKa2 = 10).
4.12. Suponiendo que una especie sólida HA tiene las características de un patrón primario, ¿para qué se utiliza-rá? (Marcar con una X.)
[ ] Para estandarizar disoluciones de bases[ ] Para estandarizar disoluciones de ácidos[ ] Para ambos fines
4.13. ¿Cuándo influye la carbonatación de las disoluciones de NaOH cuando éstas se emplean para valorar áci-dos fuertes?
[ ] Cuando se emplea fenolftaleína[ ] Cuando se emplea naranja de metilo[ ] Cuando se realiza la valoración en ausencia de luz solar
Indicar el sentido del error debido a la carbonatación:
[ ] Por exceso [ ] Por defecto
168 Equilibrios iónicos y sus aplicaciones analíticas

4.14. Indicar, de las siguientes sustancias, cuáles se pueden utilizar como patrón primario para estandarizar unadisolución de NaOH:
[ ] Ácido nítrico [ ] Carbonato de sodio[ ] Ácido sulfámico [ ] Ftalato ácido de potasio[ ] Ácido sulfúrico [ ] Ácido perclórico
4.15. Para determinar la acidez de un vinagre se realiza una valoración ácido-base con NaOH como valorante.Previamente la disolución de NaOH se estandariza con ftalato ácido de potasio. ¿Cuál es el estándar secun-dario? ¿Cuál es el estándar primario o patrón primario? ¿Cuáles son los estándares químicos?
Capítulo 4: Valoraciones ácido-base 169
Seminarios: problemas numéricos
• Objetivo pedagógico.Desarrollo de diferentes tipos de problemas sobre laconstrucción de curvas de valoración de diferentes pro-tolitos y sus mezclas así como acerca del uso y selec-ción de indicadores ácido-base. Finalmente se resuel-ven y proponen ejercicios relacionados con lasaplicaciones de las valoraciones ácido-base.
Indicadores
1. En la valoración de un ácido orgánico débil (HA) condisolución de NaOH se utiliza un indicador cuyopKHIn es 5,3. Suponiendo que se observa el cambiode color del indicador cuando se ha transformado el20% en la forma básica, calcular: 1) el pH en el pun-to final de la valoración; 2) la relación [HA]/[A–] endicho punto final; y 3) el error que se comete en lavaloración.HA (pKa = 4,7).
El punto final de la valoración vendrá dado por elcambio de color del indicador, y como éste se pro-duce cuando se ha transformado el 20% del mismoen la forma básica, quedará el 80% sin transformar,esto es, como forma ácida. De acuerdo con el equili-brio ácido-base del indicador:
HIn + H2O →← In– + H3O+
al sustituir en la expresión de la constante de acidez lascondiciones del indicador en el punto final se tiene:
de donde [H3O+] = 2,0 × 10–6 mol L–1 y pH = 4,70.
Dado que el valor de pH en el punto final coinci-de con el pKa del ácido débil a valorar, se tendrá eneste punto que [HA] = [A–].
Si en el punto final la concentración de ácido débilsin valorar es igual a la de base formada, esto es, la de ácido valorado, el error de valoración será del 50% con signo negativo ya que queda ácido sinvalorar.
2. Un indicador ácido débil (HIn) cambia de color parapH = 9,4 cuando las 2/5 partes del mismo se encuen-tran en la forma básica. Determinar: 1) la constantede disociación del indicador; 2) su zona de viraje; y3) en qué tipo de valoraciones puede utilizarse.Sol.: 1) 2,65 × 10–10; 2) 8,6 – 9,6; 3) valoración de áci-
do débiles con base fuerte.
3. Un indicador visual ácido-base que tiene un interva-lo de transición de 1,2 a 2,9 cambiando de incoloro avioleta. a) ¿Servirá para valorar una base débil conun ácido fuerte?, ¿por qué?; b) ¿cuál será el color queadquirirán las siguientes disoluciones al añadirles unasgotas de indicador: 1) disolución 0,01 mol L–1 de unácido HA; 2) disolución 0,1 mol L–1 de NaA?HA (pKa = 4,5).Sol.: a) No; b) 1: violeta, 2: violeta.
10
20[H O ]80
5,3 3−+
=
KHIn3[In ][H O ]
[HIn]=
− +

4. Un sistema HIn actúa como indicador ácido-base.Una disolución de este indicador tiene un pH de 4,0cuando la relación entre las formas ácida (HIn) y bási-ca (In–) es de 8,5. Este indicador se utiliza para valo-rar 40 mL de una disolución de carbonato de sodio0,01 mol L–1 con una disolución de HCl 0,025 mol L–1.¿Qué volumen de la misma se necesitará para el vira-je del indicador añadido a la disolución de carbona-to de sodio?H2CO3 (pKa1 = 6,35, pKa2 = 10,33).
Inicialmente se ha de caracterizar al indicador queposteriormente se utiliza en la valoración de carbo-nato de sodio con ácido clorhídrico. A partir del equi-librio ácido-base del indicador:
HIn + H2O →← In– + H3O+
y al sustituir en la expresión de la constante de aci-dez las condiciones dadas para conseguir con el mis-mo un pH = 4, se tiene:
de donde pKa = 4,93. Su intervalo de transición será3,93-5,93, por lo tanto adecuado para la valoraciónde carbonato de sodio con ácido clorhídrico.
Dado este intervalo de transición se conseguirá lavaloración de carbonato de sodio hasta ácido carbó-nico, esto es:
Na2CO3 + 2 HCl → H2CO3 + 2 NaCl
De acuerdo con esta reacción de valoración:
mmol de HCl para valorar Na2CO3== 2 × mmol iniciales de Na2CO3
VHCl × 0,025 = 2 × 40 × 0,01
de donde VHCl = 32 mL. Al añadir este volumen dela disolución de HCl se produce el viraje del indi-cador.
5. A 100 mL de una disolución ácida se añaden 0,5 mLde disolución al 0,1% (m/v) de un indicador cuyo pesomolecular es 250. Suponiendo que el indicador es unácido monoprótico y que el cambio de color se per-cibe cuando el 50% del mismo ha pasado a la formabásica, calcular los mL de disolución NaOH 0,1 molL–1 necesarios para hacer virar el indicador.Sol.: 0,01 mL.
6. Se valoran 50 mL de una disolución 0,02 mol L–1 de unácido monoprótico de pKa = 5,0 con disolución 0,02mol L–1 de NaOH. Se utiliza un indicador que vira apH = 8,40. Calcular el % de error en la valoración.Sol.: –0,04%
7. El intervalo de transición del rojo de metilo (rojo →amarillo) está comprendido entre 4,4 y 6,2. Calcularlos gramos de acetato de sodio que hay que añadir a25 mL de una disolución de ácido acético 0,01 molL–1 para que esta tome el color rojo. Despreciar cual-quier posible efecto de dilución.CH3COOH (pKa = 4,75).Peso molecular: NaCH3COO = 82.
Antes de adicionar acetato de sodio la disoluciónde ácido acético 0,01 mol L–1 presenta un pH que sedetermina en base a la expresión:
de donde pH = 3,34, y por tanto la disolución de áci-do acético tras adicionar rojo de metilo tomará uncolor rojo.
Para conseguir que el indicador vire a color ama-rillo será necesario alcanzar un pH de 6,2 que se con-sigue con la adición de acetato de sodio. En definiti-va se trata de preparar una disolución reguladora depH 6,2. Si se hace uso de la expresión de Henderson-Hasselbalch:
se tiene al sustituir las condiciones dadas en el pro-blema:
de donde [CH3COO–] = 0,282 mol L–1.
10 10
0, 01[CH COO ]
6,2 4,75
3
− −−
=
[ ]H O
[CH COOH][CH COO ]3
3
3
+−
= Ka
[ ]
, ,,
H O
molL
3 CH COOH
1
3
+
− − −
= =
= × = ×
C Ka
0 01 10 4 22 104 75 4
KHIn
InHIn
= =
= = ×
− −
−−
[ ][ ]
,,
10
108 5
1 18 10
4
45
KHIn
3In H OHIn
=− +[ ][ ][ ]
170 Equilibrios iónicos y sus aplicaciones analíticas

A partir de esta concentración de acetato de sodiose puede calcular fácilmente la cantidad a añadir deesta sal a la disolución de ácido acético:
de donde se deduce que hay que adicionar 578,1 mgde NaCH3COO para conseguir hacer virar al indica-dor rojo de metilo.
8. Se valoran 100 mL de ácido acético con disolución0,1 mol L–1 de NaOH con un indicador HIn. Despuésde la adición de 50 mL de NaOH se encontró que larelación entre las concentraciones de las formas bási-ca y ácida del indicador era de 0,6. Calcular la mola-ridad inicial del ácido acético.InH (pKHIn = 5,2), CH3COOH (pKa = 4,75).Sol.: 0,079 mol L–1.
Curvas de valoración
9. Se disuelven 0,610 g de ácido benzoico en 500 mL deagua y se valoran con NaOH 0,5 mol L–1. Calcular elpH de la disolución: a) al comienzo de la valoración,b) cuando se ha neutralizado el 50%, y c) en el pun-to de equivalencia.C6H5COOH (pKa = 4,18).Peso molecular: ácido benzoico = 122.
Antes de llevar a cabo la valoración del ácido ben-zoico (HBz) es necesario determinar la concentraciónde la disolución del mismo que se desea valorar:
La reacción de valoración con una base fuerte será:
HBz + NaOH → NaBz + H2O
a) En el punto inicial de la valoración se tendrá ladisolución de ácido benzoico 0,01 mol L–1, ácidodébil, cuyo pH vendrá dado por:
Balance protónico: [H3O+] = [OH–] + [Bz–]
si [H3O+] >> Ka, se tiene:
A tenor del valor calculado para [H3O+], se
observa que Ka no es despreciable frente al mis-mo y por tanto en la expresión para el cálculo delpH se transforma en la siguiente ecuación desegundo grado:
en la que al sustituir los valores de CHBz y Ka seobtiene pH = 3,11.
b) Cuando se ha neutralizado el 50% del ácido se tie-ne una zona tamponada en la curva de valoraciónen la que [HBz] = [Bz–], y de acuerdo con la ecua-ción de Henderson-Hasselbalch, pH = pKa = 4,18.
c) En el punto de equivalencia se tiene una disolu-ción de benzoato de sodio (NaBz) de acuerdo conla reacción de valoración indicada. Esta sal sedisocia totalmente en agua, y el pH de esta diso-lución se calcula en base a:
Balance protónico: [H�3O+] + [HBz] = [OH–]
si [H3O+] << Ka, se tiene:
expresión en la que hay que sustituir adecuada-mente CHBz dado que en el punto de equivalen-cia ésta es inferior a la inicial debido al efecto dedilución. Dado que se valoran 500 mL de HBz0,01 mol L–1 con disolución de NaOH 0,5 mol L–1,el volumen de disolución de hidróxido de sodioañadido en el punto de equivalencia será:
500 0 01 0 510
× = × ⇒⇒ =
, , V
VNaOH
NaOH mL
[ ]H O3
HB
+ = K KC
w a
z
C
KKz
a
wHB 3
3 3
H OH O H O
[ ][ ] [ ]
+
+ ++=
[ ] [ ]H O H O3 3 HB+ ++ − =2 0K C Ka z a
[H O3 HBz+
− −
= × == × =
]
, , ,
C Ka
0 01 10 104 18 3 09
[ ][ ]
H OH O3
HBz
3
++= ×
+C K
Ka
a
[ ]
, /,
,
HBzmoles
L
mol L 1
= =
= = −0 610 1220 500
0 01
0, 282
cantidad(mg)/8225
=
Capítulo 4: Valoraciones ácido-base 171

En estas condiciones,
y al aplicar en la expresión para el cálculo del pHse tiene pH = 8,09.
10. 20 mL de ácido acético 0,5 mol L–1 se diluyen con aguahasta un volumen final de 100 mL y la disolución resul-tante se valora con NaOH 0,5 mol L–1. Construir la cur-va de valoración. Despreciar el efecto de la dilución.CH3COOH (pKa = 4,75).Sol.:
F 0 0,1 0,5 0,9 0,99
pH 2,88 3,80 4,75 5,70 6,75
F 1,0 1,01 1,1 1,5 2,0
pH 8,88 11,00 12,0 12,70 13,0
11. Una disolución 0,01 mol L–1 de NaH2BO3 se valoracon HCl. Representar la curva de valoración.H3BO3 (pKa = 9,24).Sol.:
F 0 0,1 0,5 0,9 0,99
pH 10,62 10,19 9,24 8,29 7,24
F 1,0 1,01 1,1 1,5 2,0
pH 5,62 4,00 3,00 2,30 2,00
12. El fenoprofeno, de fórmula C15H14O3, se comporta,en disolución acuosa, como un ácido monoprótico(HA) de pKa = 7,3. Se valoran 50 mL de una disolu-ción 0,1 mol L–1 de la sal sódica del fenoprofeno(NaA) con HCl 0,5 mol L–1. Dibujar la curva de valo-ración despreciando el efecto de la dilución e indicarde los indicadores mostrados en la tabla, cuál sería eladecuado para determinar el punto final de la valo-ración con un error inferior al 1%.
Indicador pKa
Azul de timol 1,6
Naranja de metilo 3,4
Azul de bromofenol 4,1
Rojo de clorofenol 6,2
Timolftaleína 10,0
Sol.: Zona de equivalencia ±1%: pH = 5,3 – 3,0. Indi-cador adecuado: Azul de bromofenol.
13. Representar la curva de valoración que se obtienecuando se valoran 50 mL de ácido dicloroacético 0,05mol L–1 con una disolución 0,5 mol L–1 de NaOH.Despreciar el efecto de la dilución.CHCl2COOH (pKa = 1,30).
Para construir la curva de valoración de este ácidodébil (HA) se considerarán las cuatro zonas caracte-rísticas de la misma: zona 1. Punto inicial; zona 2.Antes del punto de equivalencia; zona 3. Punto deequivalencia; y zona 4. Después del punto de equi-valencia. Para llevar a cabo estos cálculos se ha derecurrir a los balance de masas y cargas. La reacciónde valoración será:
CHCl2COOH + NaOH → NaCHCl2COO + H2O
Balance de masas: CHA = 0,05 = [HA] + [A–]
Balance de cargas: [H3O+] = [OH–] + [A–]
Zona 1: punto inicial. En este caso, el balance decargas se simplifica a:
[H3O+] = [OH–] + [A–]
y al sustituir se tiene:
Dado que el valor de Ka es elevado, no se puedesimplificar frente a [H3O
+] en el denominador de laexpresión anterior, y ésta se transforma en:
y al sustituir los valores de CHA = 0,05 mol L–1 y Ka =10–1,30 se tiene un valor de pH para la disolución deácido dicloroacético de 1,51.
Zona 2: antes del punto de equivalencia. En estecaso, no se puede hacer uso en esta zona tamponadade la expresión de Henderson-Hasselbalch dado elvalor relativamente elevado de Ka, por lo tanto se hade recurrir el balance de cargas, que en esta zona vie-ne dado por:
[ ] [ ]H O H O3 3 HA+ ++ − =2 0K C Ka a
[ ]
[ ]H O
H O3HA
3
++
= ×+
C KKa
a
C zHB1molL= ×
+= −500 0 01
500 100 0098
,,
172 Equilibrios iónicos y sus aplicaciones analíticas

[H3O+] + [Na+] = [OH–] + [A–]
en el cual se puede despreciar [OH–]. Este balance setransforma en:
y al operar queda finalmente:
Al sustituir en esta expresión los valores conocidosde CHA, CNaOH, VHA, Ka y variables de 0 < VNaOH < 4,95(se necesitan 5,0 mL de NaOH 0,5 mol L–1 para alcan-zar el punto de equivalencia) se obtienen los siguien-tes valores significativos de F en función del pH:
F 0,1 0,5 0,9 0,99
pH 1,56 1,85 2,59 3,60
Zona 3: punto de equivalencia. En este punto setiene la sal sódica correspondiente al ácido valorado,y por tanto se ha de recurrir al balance protónico:
[H3O+] + [HA] = [OH–]
el cual se transforma en:
En este balance protónico se puede despreciar [H3O+]
frente a Ka dado que se trata de una disolución de unasal con carácter básico y además el valor de Ka es rela-tivamente elevado. En estas condiciones se tiene:
de donde:
y por tanto pH = 7,15.
Zona 4: después del punto de equivalencia. En estazona de exceso de valorante, el balance de cargas será:
[H3O+] + [Na+] = [OH–] + [A–]
y se podrá despreciar [H3O+] dado que se tiene un
exceso de la disolución de NaOH que se utiliza comovalorante. Este balance se transforma en:
[OH–] = [Na+] – [A–]
Sustituyendo los valores de Kw, [Na+] y [A–] en estaexpresión se tienen los siguientes valores significati-vos de F en función del pH:
F 1,01 1,1 1,5 2,0
pH 10,70 11,70 12,40 12,70
14. Calcular los pH que corresponden a los puntos deequivalencia de la valoración de una disolución 0,1mol L–1 de ácido sulfuroso con hidróxido de sodio.Despreciar la variación de volumen. H2SO3 (pKa1 = 1,81, pKa2 = 6,59).Sol.: 4,23 y 9,79.
15. Representar la curva de valoración que se obtienecuando se valoran 25 mL de una disolución 0,05 molL–1 de ácido malónico (H2M) con NaOH 0,1 mol L–1.Despreciar el efecto de la dilución.HOOCCH2COOH (pKa1 = 2,85, pKa2 = 5,70).
En esta valoración de un ácido diprótico con unabase fuerte se tienen las siguientes reacciones de va-loración:
HOOCCH2COOH + NaOH →→ NaOOCCH2COOH + H2O
NaOOCCH2COOH + NaOH →→ NaOOCCH2COONa + H2O
[ ]H O
[Na ] [A ]3+
+ −=−
Kw
[ ],
,
,
H O
mol L
3HA
1
+−
−
− −
=+
=+
=
= ×
KCK
w
a
1
10
10 0510
7 07 10
14
1 3
8
[ ]
[ ]H O
H O3HA
3
++
+
=1CK
K
a
w
[ ]
[ ][ ]
[ ]H OH O
H OOH3
HA 3
3
++
+−+
+=C
Ka
[ ] [ ]H O H O3 3NaOH NaOH
HA
NaOH NaOH
HAHA
+ ++ + ×
+
+ × −
=
2
0
KV C
V
KV C
VC
a
a
[ ]
[ ]
H O
H O
3NaOH NaOH
HA
HA
3
+
+
+ × =
= ×+
V CV
C K
Ka
a
Capítulo 4: Valoraciones ácido-base 173

174 Equilibrios iónicos y sus aplicaciones analíticas
Como en otras ocasiones, a partir de los balancesde masas y de cargas o protónico, según el caso, sepueden conseguir las expresiones útiles para el cálcu-lo del pH en las diversas zonas de interés de esta cur-va de valoración.
Balance de masas:
Balance protónico:
Zona 1: punto inicial de la valoración. Se tiene unadisolución de ácido malónico y por tanto [OH–] y[M2–] se pueden despreciar en el balance protónico,quedando éste como:
Dado que Ka2 << Ka1, el producto Ka1 Ka2 se pue-de despreciar en el polinomio del denominador, que-dando esta expresión reducida a:
que se transforma en la expresión de segundo grado:
cuya resolución proporciona un valor de pH de 2,13.
Zona 2: antes del primer punto de equivalencia. Enesta primera zona tamponada de la valoración se pue-de establecer el siguiente balance de cargas:
En este balance se puede despreciar [OH–] y con-siderar que:
por lo que se transforma en:
Como en la zona anterior se puede despreciar eltérmino Ka1 Ka2 en el polinomio del denominador deesta expresión, con lo que ésta se transforma, tras rea-grupar términos, en:
Al variar el valor de F en el intervalo 0 < F < 1 seobtienen los correspondientes valores de pH paraesta zona de la curva de valoración. A continuaciónse muestran algunos valores significativos:
F 0,1 0,5 0,9 0,99
pH 2,30 2,88 3,74 4,09
Zona 3: primer punto de equivalencia. En este pun-to se tiene la disolución de un anfolito NaHM, cuyopH se calcula en base a la siguiente expresión:
de donde pH = 4,68.
Zona 4: antes del segundo punto de equivalencia.Se tiene de nuevo una zona tamponada, en este casoconstituida por el par ácido-base HM–/M2–. El balan-ce de cargas:
se simplifica a:
que se transforma en:
En esta expresión se puede despreciar [H3O+]2 en
el polinomio del denominador, y tras reagrupar tér-minos queda en la forma:
F C
C K K K
K K Ka a a
a a aH M
H A 3
3 32
2H O
H O H O=
+( )+ +
+
+ +
[ ]
[ ] [ ]1 1 2
21 1 2
2
[Na ] [HM ] 2 [M ]2+ − −= +
[H O ] [Na ] [OH ] [HM ] 2 [M ]32+ + − − −+ = + +
[ ], , ,
H O
molL3
1
+
− − − −
= == × =
K Ka a1 2
2 88 5 68 4 2810 10 10
F F K K Ka a a[ ] ( ) [ ]H O H O3 3+ ++ − − =2
1 1 21 2 0
F C
C K K K
K K Ka a a
a a aH M
H A 3
3 32
2H O
H O H O=
+( )+ +
+
+ +
[ ]
[ ] [ ]1 1 2
21 1 2
2
[H O ] [Na ]3 H M2
+ +<< = F C
[H O ] [Na ] [OH ] [HM ] 2 [M ]32+ + − − −+ = + +
[ ] [ ]H O H O3 3 H M2
+ ++ − =21 1 0K C Ka a
[ ]
[ ]H O
H O3H A
3
++
=+
C K
Ka
a
2 1
1
[ ]
[
[ ] [ ]H O
H O ]
H O H O3H A 3
3 3
++
+ +=
+ +C K
K K Ka
a a a
2 1
21 1 2
[H O ] [OH ] [HM ] 2 [M ]32+ − − −= + +
CH M 2
22
0, 05 [H M] [HM ] [M ]= = + +− −

de donde:
Al sustituir en esta expresión valores de F com-prendidos en el intervalo 1 < F < 2, se obtienen los valo-res de pH necesarios para construir esta zona de la cur-va de valoración. Algunos valores significativos son:
F 1,1 1,5 1,9 1,99
pH 4,73 5,68 6,63 7,67
Zona 5: segundo punto de equivalencia. En estesegundo punto de equivalencia se tiene la disoluciónde la sal Na2M y para calcular el pH se puede haceruso de la expresión típica para disoluciones de basesdipróticas:
de donde pH = 9,19.
Zona 6: exceso de valorante. En esta zona final dela curva de valoración, el balance de cargas se sim-plifica a:
que se transforma en:
En este caso se pueden despreciar los términos[H3O
+]2 + Ka1 [H3O+] en el polinomio, y por tanto
tras operar, la expresión para el cálculo del pH vienedada por:
Mediante esta expresión de pueden calcular lasparejas de valores de F y pH que permiten construiresta zona de la curva de valoración. Algunos valoressignificativos son:
F 2,01 2,1 2,5 3,0
pH 10,70 11,70 12,40 12,70
16. Dibujar la curva de valoración de una disolución 0,1mol L–1 de ácido oxálico con hidróxido de sodio.¿Sería posible utilizar los dos puntos de equivalen-cia como puntos finales de una valoración con indi-cadores visuales? Puede despreciarse la variaciónde volumen.HOOC–CH2–CH2–COOH (pKa1 = 1,27, pKa2 = 4,27).Sol.:
F 0 0,1 0,5 0,9 0,99 1,0 1,01 1,1
pH 1,29 1,35 1,67 2,38 2,71 2,77 2,83 3,37
F 1,5 1,9 1,99 2,0 2,01 2,10 2,50 3,0
pH 4,27 5,22 6,27 8,64 11,07 12,0 12,70 13,0
Sólo se podría utilizar el segundo punto de equi-valencia con indicadores visuales.
17. Una disolución 0,01 mol L–1 de un cloruro de ami-noácido, H2Am+Cl–, se valora con NaOH 0,03 molL–1. La valoración se sigue potenciométricamente yse obtiene la curva que se muestra en la figura.
[ ]( )
H O3H M2
+ =−
KF C
w
2
KF C
C K K
K K K
w
a a
a a a
[ ]
[ ] [ ]
H O
H O H O
3H M
H A
3 3
2
2
+
+ +
= −
−+ +2 1 2
21 1 2
[Na ] [OH ] 2 [M ]2+ − −= +
[H O
molL
3H M
1
2
+
− −− −
= =
= × = ×
]
,,
,
K KC
w a 2
14 5 681010 10
0 056 46 10
[ ]H O3
+ = −−
KF
Fa22
1
F F K Ka a[ ] [ ]H O H O3 3+ ++ = +2 22
Capítulo 4: Valoraciones ácido-base 175

a) Hacer una estimación de los valores de pKa.b) Comprobar, mediante un cálculo numérico, que
el valor del pH en el punto inicial y en cada unode los puntos de equivalencia concuerda con elobtenido a partir de los valores estimados de lasconstantes de acidez.
c) ¿Sería posible utilizar indicadores visuales en estadeterminación? En caso afirmativo, enumerar losrequisitos que deben cumplir.
Sol.: a) pKa1 = 6,0; pKa2 = 9,0; b) 4,0, 7,5 y 10,58; c) No.
18. En la valoración de 50 mL de una disolución fosfatode sodio 0,1 mol L–1 con HCl 1,0 mol L–1:
a) ¿Cuál sería el salto más adecuado para la deter-minación de la concentración de fosfato de sodio,si el punto final se determina mediante un indi-cador visual?
b) ¿Sería posible realizar determinación con un errorinferior al 0,1%?
Razonar las respuestas.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,33).Sol.: a) El segundo salto perceptible; b) No es posible.
19. Representar la curva de valoración que se obtiene cuan-do se valoran con NaOH 0,1 mol L–1 25 mL de una diso-lución que contiene HCl 0,01 mol L–1 y ácido maléico0,02 mol L–1. Despreciar el efecto de la dilución.cis-HOOCCH=CHCOOH (pKa1 = 1,92, pKa2 = 6,22).
Se tiene la valoración de una mezcla formada porun ácido fuerte (HCl) y un ácido débil diprótico (áci-do maleico, H2M). Las reacciones de valoración sonlas siguientes:
HCl + NaOH → NaCl + H2O
HOOCCH=CHCOOH + NaOH →→ NaOOCCH=CHCOOH + H2O
NaOOCCH=CHCOOH + NaOH →→ NaOOCCH=CHCOONa + H2O
Los balances de masas a emplear en cada una delas zonas de la curva de valoración serán:
Balances de masas:
CHCl = 0,01 = [Cl–]
CMaleico= 0,02 = [H2M] + [HM–] + [M2–]
Zona 1: punto inicial. En este caso, el balance pro-tónico viene dado por:
[H3O+] = [OH–] + [Cl–] + [HM–] + 2 [M2–]
donde se pueden despreciar las concentraciones deOH– y de M2–, quedando al sustituir del balance de masas:
En esta zona, generalmente [H3O+] > Ka1 > Ka2, y por
tanto el denominador de la expresión anterior se trans-forma en [H3O
+]2 + Ka1 [H3O+], y al operar se tiene:
o bien:
de donde al sustituir los valores conocidos y resolverse obtiene un valor de pH de 1,74.
Zona 2: antes del primer punto de equivalencia. Esen esta zona donde empieza a valorarse el ácido fuer-te y el primer protón del ácido maleico dado el valorde Ka1 con lo que se puede establecer el siguientebalance de cargas:
[H3O+] + [Na+] = [OH–] + [Cl–] + [HM–] + 2 [M2–]
en el cual se pueden despreciar las concentracionesde OH– y de M2–, como en el caso anterior, quedan-do al sustituir del balance de masas:
[ ],
[ ]
[ ] [ ]
H O
H O
H O H O
3NaOH
HClH M 3
3 3
2
+
+
+ +
+ × =
= ++ +
V
CC K
K K Ka
a a a
0 125
1
21 1 2
[ ] ( )[ ]( )
H O H O3 HCl 3
HCl H M2
+ ++ − −− + =
21
1 0K C
K C Ca
a
[ ]
[ ]H O
H O3 HClH M
3
2++= +
+C
C K
Ka
a
1
1
[ ][ ]
[ ] [ ]
H OH O
H O H O
3 HCl
H M 3
3 3
2
+
+
+ +
= +
++ +
C
C K
K K Ka
a a a
1
21 1 2
[ ]
,Na NaOH NaOH
Inicial
NaOH+ = × = ×V CV
V 0 125
176 Equilibrios iónicos y sus aplicaciones analíticas

En este caso se puede llevar a cabo la siguienteaproximación: [H3O
+] ≈ Ka1 > Ka2, por lo que la expre-sión anterior se transforma en:
de donde al operar y reunir términos se tiene unaexpresión de segundo grado tal como:
Al aplicar en esta expresión valores de VNaOH com-prendidos en el intervalo 0 < VNaOH < 7,5 se obtienenlos valores de pH correspondientes a esta zona de lavaloración. Algunas parejas significativas de estosvalores se muestran a continuación:
mL NaOH 0,25 1,25 2,25 2,50
pH 1,76 1,86 1,95 1,97
mL NaOH 3,00 5,00 7,00 7,40
pH 2,03 2,34 3,11 3,82
Zona 3: primer punto de equivalencia. En este pun-to de equivalencia se tienen las sales NaCl e hidro-genomaleato de sodio. En este caso el balance pro-tónico será:
[H3O+] + [H2M] = [OH–] + [M2–]
en el cual se puede despreciar la concentración de OH–
y si Ka1 > [H3O+] > Ka2, el polinomio de segundo gra-
do se puede aproximar a [H3O+] Ka1 con lo cual:
y tras operar se llega:
de donde pH = 4,07.
Zona 4: antes del segundo punto de equivalencia.En esta zona se origina la valoración del segundo pro-tón del ácido maléico. El balance de cargas será:
[H3O+] + [Na+] = [OH–] + [Cl–] + [HM–] + 2 [M2–]
en el cual se puede despreciar [OH–]. Sustituyendoen este balance se tiene:
Si se considera en esta zona que Ka1 > [H3O+] ≈
Ka2, el polinomio de segundo grado se puede aproxi-mar a: [H3O
+] Ka1 + Ka1 Ka2 y por tanto la expresiónanterior se transforma en:
y tras operar resulta:
Valores significativos de pH para esta zona de lacurva de valoración se muestran a continuación.
mL NaOH 7,60 8,00 10,00 12,00 12,45
pH 4,56 5,27 6,22 7,17 8,22
Zona 5: segundo punto de equivalencia. En estepunto se tiene la mezcla de sales formadas tras la valo-ración de ambos ácidos: NaCl y Na2M. El balanceprotónico en este punto viene dado por:
[H3O+] + [HM–] + 2 [H2M] = [OH–]
Dado que nos encontramos en una zona de la cur-va de valoración francamente básica se cumple que
[ ],
[ ]
,
H O H O3NaOH
HCl H M 3
NaOHHCl H M
2
2
+ ++ + × − −
+
+ × − −
=
22
2
0 125
0 125
2 0
KV
C C
KV
C C
a
a
[ ],
[ ][ ]
H O
H OH O
3NaOH
HCl H M3
32
+
+
+
+ × =
= + ++
V
C CK
Ka
a
0 125
2 2
2
[ ],
[ ][ ] [ ]
H O
H OH O H O
3NaOH
HCl H M3
3 32
+
+
+ +
+ × =
= + ++ +
V
C CK K K
K K Ka a a
a a a
0 125
21 1 22
1 1 2
[ ]
,,
,, ,
,
H O
molL
3H M
H M
1
2
2
+
− −
−− −
=+
=
= × ×+
= ×
C K K
K Ca a
a
1 2
1
1 92 6 22
1 9250 02 10 10
10 0 028 51 10
[ ][ ]
[ ] [ ]H O
H O
H O H O3H M 3
3
H M
3
2 2++
+ ++ =
C
K
C K K
Ka
a a
a
2
1
1 2
1
[ ],
[ ]
,
H O H O3NaOH
HCl 3
NaOHHCl H M2
+ ++ + × −
−
− × − −
=
21
1
0 125
0 125
0
KV
C
KV
C C
a
a
[ ]
,[ ]
H OH O3
NaOHHCl
H M
3
2+++ × = +
+V
CC K
Ka
a
0 125
1
1
Capítulo 4: Valoraciones ácido-base 177

Ka1 > Ka2 > [H3O+], y por tanto, el polinomio de
segundo grado se puede aproximar a: [H3O+] Ka1 +
Ka1 Ka2 . En estas condiciones, el balance protónicose transforma en:
de donde:
Sustituyendo en esta expresión los valores de CH2M,
KW y Ka2 se obtiene un valor de pH en este punto deequivalencia de 9,17.
Zona 6: exceso de valorante. Cuando se han valo-rado ambos ácidos, al añadir un exceso de NaOH, sepuede establecer el siguiente balance de cargas:
[H3O+] + [Na+] = [OH–] + [Cl–] + [HM–] + 2 [M2–]
En este balance de cargas se pueden despreciar[H3O
+] y [HM–], por lo que se transformaría en:
y al reordenar términos:
Esta expresión permite calcular el pH en esta zonade la curva de valoración. Algunos valores significa-tivos se muestran a continuación:
mL NaOH 12,55 12,70 13,00 15,00 20,00
pH 10,58 11,18 11,58 12,28 12,76
20. Representar la curva de valoración que se obtiene alvalorar con HCl 0,5 mol L–1 10 mL de una disolución
0,2 mol L–1 en NaOH y 0,1 mol L–1 en Na2CO3. Des-preciar el efecto de la dilución.H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.:
mL HCl 0 0,20 0,40 2,00 3,00
pH 13,30 13,28 13,26 13,00 12,32
mL HCl 4,00 4,20 5,00 5,80 6,00
pH 11,65 11,21 10,33 9,37 8,34
mL HCl 6,20 7,00 7,80 8,00 8,20
pH 7,30 6,35 5,40 3,68 2,00
mL HCl 9,00 10,00
pH 1,30 1,00
21. Una disolución contiene 0,01 mol L–1 de ácido clor-hídrico y 0,01 mol L–1 de ácido bórico. En base a lacurva correspondiente a su valoración con hidróxidode sodio, explicar si sería posible la determinación deambos ácidos mediante una valoración directa.H3BO3 (pKa = 9,24).Sol.: No sería posible.
22. Calcular: a) el pH de una disolución que contiene 0,2mol L–1 de HCl y 0,1 mol L–1 de ácido maleico y b) elpH en los diferentes puntos de equivalencia de la valo-ración de esta mezcla con hidróxido de sodio. (Puededespreciarse el efecto de la dilución.) Proponer unmétodo que permita determinar la concentración deácido clorhídrico y de ácido maleico en una mezcla.HOOC–CH= CH–COOH (pKa1 = 2,30, pKa2 = 6,26).Sol.: a) 0,70; b) 1,70, 4,29 y 9,63.
23. 25 mL de una mezcla de ácido acético 0,05 mol L–1 ycloruro de amonio 0,01 mol L–1 se valoran con NaOH0,1 mol L–1. Construir la curva de valoración. Des-preciar el efecto de la dilución.CH3COOH (pKa = 4,75), NH4
+ (pKa = 9,24).Sol.:
mL NaOH 0 1,25 6,25 11,25 12,50 12,75
pH 3,03 3,80 4,75 5,70 7,34 8,29
mL NaOH 13,75 14,75 15,00 15,25 18,00 20,00
pH 9,24 10,19 10,62 11,0 12,81 12,90
24. En la curva de valoración de una disolución que con-tiene 0,1 mol L–1 de un ácido HA y 0,1 mol L–1 de un
VC
VC C K K K Ka w w a
NaOHHCl 3
NaOHHCl H M 3
H O
H O2
× −
+
+ × − −
−
− =
+
+
0 125
0 125
2 0
2
2 2
,[ ]
,[ ]
K V
CC K
K
w
a
a
[ ],
[ ]
H O
H O
3
NaOH
HClH M
3
2
+
+
= × −
− −+
0 125
2 2
2
C K K Kw w aH M 3 32
[H O ] H O+ +− − =22 0[ ]
C
KK
a
wH M 3
3 3
2[H O ]
H O H O
+
+ ++=
[ ] [ ]2
178 Equilibrios iónicos y sus aplicaciones analíticas

ácido H2B con hidróxido de sodio, indicar razonada-mente qué saltos de pH de esta curva se podrían uti-lizar para la determinación de la concentración de estosácidos. Si se dispone de disoluciones patrón de NaOH0,1 mol L–1 y HCl 0,1 mol L–1, proponer un procedi-miento para determinar la concentración de los com-ponentes de las mezclas siguientes: 1) HA + H2B y 2)H2B + NaHB.HA (pKa = 2,0), H2B (pKa1 = 6,40, pKa2 = 10,8).
25. Una muestra de ácido débil monoprotónico que pesa1,25 g se disuelve en 50 mL de agua. Se gastan 41,2mL de NaOH 0,09 mol L–1 para alcanzar el puntofinal de la valoración. En el curso de la misma seobserva que en el momento de añadir 8,24 mL deNaOH el pH vale 4,30. Calcular: 1) el peso molecu-lar del ácido; 2) su constante de acidez; y 3) el pH enel punto de equivalencia de la valoración.
Cuando se alcanza el punto final de la valoracióndel ácido monoprótico se cumple que:
mmol NaOH añadidos = mmol HA valorados41,2 × 0,09 = 50 × CHA
de donde CHA = 0,074 mol L–1.Dado que esta disolución se ha preparado disol-
viendo 1,25 g del ácido en 50 mL de agua destilada:
y por tanto el peso molecular del ácido HA será: 337,8.Cuando se añaden 8,24 mL de la disolución de
NaOH 0,09 mol L–1 nos encontramos en la zona de lacurva de valoración anterior al punto de equivalen-cia. Se tiene por tanto una zona tamponada consti-tuida por las especies HA sin valorar y NaA formadadurante la valoración por adición de NaOH. Así,
[HA] sin valorar =
[A–] formada =
Si se sustituyen estas concentraciones en la expre-sión de la constante de acidez se tiene:
En el punto de equivalencia se tiene una disoluciónde la sal NaA, cuya concentración será:
El balance protónico para esta disolución vienedado por:
[H3O+] + [HA] = [OH–]
en el cual se puede despreciar [H3O+], quedando al
sustituir a partir del balance de masas y de la expre-sión de Ka
Dado que Ka >> [H3O+], la expresión anterior se
transforma en:
de donde pH = 8,76.
26. Un ácido débil monoprótico tiene una constante deacidez de 2,0 × 10–4. 200 mL de una disolución 0,05mol L–1 de este ácido se valoran con NaOH 0,25 molL–1. Calcular el pH: 1) antes de comenzar la valora-ción; 2) en el punto de equivalencia; y 3) cuando sehan neutralizado las 4/5 partes del ácido. Sol.: 1) 2,5; 2) 8,16; 3) 4,3.
27. Se valoran 100 mL de NH3 0,1 mol L–1 con HCl 0,5 mol L–1. 1) ¿Qué pH tiene la disolución cuandose han añadido 18 mL de ácido? y 2) ¿Cuántos mLde ácido deben añadirse para que [H3O
+] = 3 KNH4+?
NH4+ (pKa = 9,24).
Sol.: 1) 8,29; 2) 10 mL.
[ ],
,
,
H O
mol L
3W
A
1
+− −
−
− −
= = × ××
=
= ×−
K KCa 1 25 10 10
4 06 10
1 75 10
5 14
2
9
C
KK
a
wA 3
3 3
H O
H O H O−
+
+ ++=
[ ]
[ ] [ ]
C
A1mol L− = ×
+= × − −50 0 74
50 41 24 06 10 2,
,,
Ka = =
= × = ×
− +
−−
[A ][H O ][HA]
3
0 74 102 96
1 25 104 3
5,,
,,
8 24 0 0950 8 24
0 7458 24
, ,,
,,
×+
= −mol L 1
50 0 074 8 24 0 0950 8 24
2 9658 24
× − ×+
= −, , ,,
,,
mol L 1
CPm
VPm
HA
mg HA HA(mL)
= = =
=
0 074
1 25050
,/
. /
Capítulo 4: Valoraciones ácido-base 179

28. Se valoran 50 mL de un ácido débil HA con NaOH0,1 mol L–1. Se gastan 40 mL de esta disolución valo-rante para alcanzar el punto final. Si durante la valo-ración el pH es 4,0 cuando se han añadido 20 mL deNaOH 0,1 mol L–1, calcular el pH en el punto finalde esta valoración.Sol.: 7,82 mL.
29. Una muestra líquida que pesa 100 g contiene un 10%de HCl y un 5% de ácido acético. Un gramo de estadisolución se diluye a 100 mL y se valora con unadisolución de NaOH, gastándose 10 mL de la mismacon un indicador que vira a pH = 3,5. Calcular: 1) lamolaridad de la disolución de NaOH; 2) el volumende la misma disolución de NaOH que se gastaría alvalorar una disolución de 100 mL que contiene 1,5gramos de la muestra ácida utilizando un indicadorcuyo viraje se produce a pH = 9,0.CH3COOH (pKa = 4,75).Pesos atómicos: Cl = 35,5; C = 12; H = 1,0; O = 16,0.
En la primera valoración, a tenor del indicador uti-lizado, se valora sólo el ácido fuerte, esto es el HCl.Al partir de 1 g de muestra ácida y considerando queel contenido de HCl es del 10%, se valorarán 0,1 g obien 100 mg. Esta cantidad de ácido fuerte implicaconsiderando el peso molecular del HCl (36,5):
Para valorar estos milimoles de HCl se gastarán:
2,74 = 10 × CNaOH
de donde la concentración de la disolución de NaOHes 0,274 mol L–1.
Con esta disolución de NaOH se valora otra alí-cuota de la muestra ácida, en este caso de 1,5 g, conun indicador que vira a pH 9,0, por lo que se valora-rán ambos ácidos conjuntamente. La cantidad de cadauno de ellos presente en la muestra será:
La valoración conjunta de ambos ácidos requeri-rá: 4,11 + 1,25 = 5,36 mmol de NaOH 0,274 mol L–1,por lo que se consumirán:
30. Se valora con HCl 0,1 mol L–1 25 mL de una disolu-ción que contiene una mezcla de las sales NaHA yNa2A. De la curva de valoración se conocen dos pun-tos: 1) al añadir 1,25 mL de HCl el pH es 6,0 y 2) alañadir 12,5 mL de HCl el pH es 4,5. Calcular el volu-men de la disolución de HCl 0,1 mol L–1 necesariopara la valoración completa de ambas sales.H2A (pKa1 = 3,0, pKa2 = 6,0).Sol.: 35 mL.
31. Se valoran 40 mL de una disolución que contieneNa2CO3 y NaHCO3 con NaOH 0,01 mol L–1 gastán-dose 20 mL de esta disolución para alcanzar el pun-to final de la valoración. Si el pH en dicho punto finales de 10,78, calcular las concentraciones de Na2CO3y NaHCO3 en la disolución expresada en mol L–1.H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Pesos atómicos: H = 1,01; Na = 23,00; C = 12,00; O = 16,00.Sol.: [NaHCO3] = 5,0 × 10–3 mol L–1,
[Na2CO3] = 3,0 × 10–3 mol L–1.
32. Una muestra impura de NaH2PO4 se disuelve en 100mL de agua y se valora con una disolución de NaOH0,1 mol L–1 gastándose 45,0 mL de la misma utilizan-do fenolftaleína indicador. ¿Cuál será el pH cuando sehan añadido sólo 20,0 mL de la disolución de NaOH?H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32).Sol.: 7,11.
Aplicaciones
33. La utilización del ácido oxálico, H2C2O4. 2H2O, comopatrón para valorar bases tiene el inconveniente delagua de cristalización. Es difícil tener el ácido con dosmoléculas de agua exactamente. Una muestra de áci-do oxálico que pesa 1,225 g consume, al valorar confenolftaleína, 80 mL de NaOH 0,25 mol L–1. Calcularel número de moléculas de agua de cristalización.HOOCCOOH (pKa1 = 4,20, pKa2 = 5,25),Pesos atómicos: C = 12,00; H = 1,01; O = 16,00.
La reacción de valoración del ácido oxálico condisolución de NaOH utilizando fenolftaleína comoindicador será:
mL de disolución de NaOH
5, 360, 274
19, 56 mL= =
mmol CH COOH 1.500
5100
601,25 mmol CH COOH
3
3
=×
=
=
mmol HCl 1.500
10100
36,54,11 mmol HCl
=×
=
=
mmol HCl =
10036,5
2,74 mmol de HCl=
180 Equilibrios iónicos y sus aplicaciones analíticas

H2C2O4 + 2 NaOH → Na2C2O4 + 2 H2O
De acuerdo con esta reacción:
mmol NaOH para valorar H2C2O4 == 2 × mol H2C2O4 presentes
80 × 0,25 = 2 × mol H2C2O4
de donde:
mmol de ácido oxálico presentes en la muestra = 10
Al partir de 1,225 g de ácido oxálico, esto es,1.225 mg y tener 10 mmol de ácido en la muestra,el peso molecular será:
Este peso molecular viene dado por 90 + X × 18, sien-do X el número de moléculas de agua de cristalizaciónque contiene el ácido. Por ello, el valor de X será:
El ácido oxálico presenta por tanto 1,8 moléculasde agua de cristalización en su estructura.
34. Para determinar el contenido de fenoprofeno(C15H14O3, ácido monoprótico, HA) en un productofarmacéutico se toman 5 comprimidos (peso total3,1097 g) y se trituran para homogeneizar la mues-tra. Se pesan 0,5348 g de muestra se disuelve en 25mL de NaOH 0,1 mol L–1. El exceso de NaOH sevalora con 16,1 mL de H2SO4 0,02 mol L–1, utilizan-do timolftaleína como indicador. Calcular: 1) el por-centaje de fenoprofeno en la muestra y 2) la cantidadde fenoprofeno en un comprimido.HA (pKa = 7,3). Pesos atómicos: C = 12,00; H = 1,01; O = 16,00.Sol.: 1) 84,04%; 2) 0,5229 g fenoprofeno/comprimido.
35. Una muestra de 0,3 g de MgO impuro se valora conHCl (3,0 mL de la disolución de HCl equivalen a0,0478 g de Na2CO3). Se añaden 48,0 mL del ácido ala muestra con lo que se sobrepasa el punto final. Elexceso de ácido requiere para su neutralización laadición posterior de 2,40 mL de NaOH 0,4 mol L–1.Calcular el tanto por ciento de MgO de la muestra.Pesos atómicos: C = 12,00; O = 16,00; Na = 23,00;Mg = 24,3.
Al adicionar a una muestra de MgO un ácido fuer-te como es el HCl se tiene:
MgO + 2 HCl → MgCl2 + H2O
La concentración de la disolución de ácido clor-hídrico se determina a partir de la equivalenciaexpresada frente al Na2CO3 conforme a la reacciónde valoración:
Na2CO3 + 2 HCl → Na2CO3 + H2O
De esta forma se puede escribir:
La cantidad de MgO en la muestra expresada entanto por ciento viene dada por:
mmol de HCl añadidos: 48 × 0,3 = 14,4
mmol de NaOH para valorar exceso de ácido == 2,4 × 0,4 = 0,96
mmol de HCl consumidos por MgO == 14,4 – 0,96 = 13,44
mmol de MgO presentes en la muestra = =13,44 / 2 = 6,72
36. Una muestra que pesa 0,542 g y que contiene ácidofosfórico se valora con una disolución de NaOH 0,15mol L–1, consumiéndose 30 mL para el viraje del rojode metilo. Calcular el porcentaje de ácido fosfóricoen la muestra.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: P = 30,97; H = 1,01; O = 16,00.Sol.: 81,3%.
37. Un gramo de acero se calienta en atmósfera de oxí-geno. El CO2 obtenido en la oxidación del carbonose recoge en 80,0 mL de Ba(OH)2 y el exceso de álca-li consume 86,8 mL de HCl 0,1 mol L–1. Si el % decarbono en la muestra de acero es del 0,6%, calcularla molaridad de la disolución de barita.Peso atómico: C = 12,00.Sol.: 6,05 × 10–2 mol L–1.
%MgO = × =6 72 40 3300
100 90 3, ,
, %
47 81
1062
11
30 30
2 33
3
,
,
mgNa COmolNa CO
gmoles HCl
molNa CO mLmolL
2
2
1
×
× × = −
X = − =122 5 90
181 8
,,
Pm = =1 225
10122 5
.,
Capítulo 4: Valoraciones ácido-base 181

38. Una disolución de NaOH se valora utilizando ácidosulfámico (NH2SO3H) como estándar químico-ana-lítico primario. Se gasta en la valoración un númerode mL igual a la décima parte del ácido sulfámicotomado, expresado en mg. Calcular la molaridad dela disolución de NaOH.Pesos atómicos: S = 32,06; H = 1,01; O = 16,00; N = 14,00.
Al ser el ácido sulfámico un ácido monoprotónico,se puede establecer la siguiente equivalencia en suvaloración con NaOH:
mmol NaOH = mmol ácido sulfámico
o bien:
Si se considera la condición dada por el problemay se sustituye en la expresión anterior se tiene:
de donde:
39. Se determina el contenido de fósforo en un acero quepesa 2,5 g. Disuelta la muestra en HNO3 se precipi-ta el fósforo como (NH4)3PO4.12MoO3.2H2O. El pre-cipitado amarillo se disuelve en 20,0 mL de KOH 0,5mol L–1, según la reacción:
→
→
El exceso de KOH consume por retroceso 20,0 mLde HNO3 0,333 mol L–1. ¿Cuál es % de fósforo en elacero?Peso atómico: P = 30,97.Sol.: 0,188%.
40. Una muestra de KHC2O4.H2C2O4.2H2O que pesa0,4212 g se disuelve en agua destilada y se valora conuna disolución de NaOH consumiéndose 48,9 mL dela misma cuando se utiliza fenolftaleína como indica-dor. Calcular la molaridad de la disolución de NaOH.
HOOCCOOH (pKa1 = 4,20, pKa2 = 5,25).Pesos atómicos: C = 12,00; H = 1,01; O = 16,00;K = 39,1.Sol.: 0,102 mol L–1.
41. Una muestra que pesa 1 g contiene un 20% de un áci-do fuerte HA y un 70% de un ácido débil H2B sedisuelve en 1 litro de agua. Se toma una alícuota de50 mL y se valora con una disolución de NaOH. Segasta un volumen de 15 mL de esta disolución de NaOH cuando se utiliza un indicador que vira apH = 5,0. Calcular: 1) la molaridad de la disoluciónde NaOH utilizada como valorante; 2) el volumen deNaOH que se gastaría al valorar una alícuota de 100mL de la mezcla de ácidos utilizando un indicadorcuyo viraje se produzca a pH = 9,0.H2B (pKa1 = 3, pKa2 = 8).Pesos moleculares: HA = 40; H2B = 100.Sol.: 1) 0,04 mol L–1; 2) 47,5 mL.
42. Se toman 0,6 g de una muestra que contiene bórax,ácido bórico y materia inerte (NaCl). Se disuelve enagua, se valora con HCl 0,1 mol L–1 y se consumen20 mL. Se añade manitol a la misma disolución, y sevalora con NaOH 0,1 mol L–1 y se consumen 50 mL.Determinar el tanto por ciento de bórax y ácido bóri-co de la muestra.H3BO3 (pKa = 9,24).Pesos moleculares: Bórax = 381,4; Ácido bórico =61,8.
Al disolver la muestra en agua que contieneNa2B4O7 y H3BO3 como protolitos activos frente alpH se tiene:
Na2B4O7 + 5 H2O → 2 H2BO3
_+ 2 H3BO3 + Na+
H3BO3 + H2O → H2BO3
_+ H3O
+
Al añadir a esta disolución ácido clorhídrico se valo-ra el ion borato formado a partir del bórax conformea la reacción:
H2BO3
_+ HCl → H3BO3 + Cl–
La adición posterior de manitol a esta disoluciónorigina la formación de un complejo con el ácido bóri-co presente en la misma (el inicial de la muestra másel formado a partir del bórax) lo cual exalta el carác-ter ácido de este protolito débil y permite su valora-ción con disolución de NaOH:
H3BO3 + NaOH → H2BO3
_+ Na+ + H2O
12MoO 3NH H PO 10H O42
4 2 4 2− + −+ + +
(NH ) PO .12MoO .2H O 22OH4 3 4 3 2 + −
C
PmNaOH1molL= = = −10 10
97 060 103
,,
mg ácido sulfámico10
mg ácido sulfámico
NaOH× =
=
C
Pm
mL NaOH
mg ácido sulfámicoNaOH× =C
Pm
182 Equilibrios iónicos y sus aplicaciones analíticas

De acuerdo con estas reacciones, cuando se con-sumen 20 mL de HCl 0,1 mol L–1 se valora el bóraxpresente en la muestra.
En este caso se tiene:
de donde se obtiene que el contenido de bórax en lamuestra es del 63,56%.
Tras la adición de manitol y valoración con disolu-ción de NaOH 0,1 mol L–1 se tiene:
mmol H3BO3 totales:
mmol H3BO3 procedentes del bórax:
de donde:
y por tanto el contenido de ácido bórico en la mues-tra es del 10,3%.
43. Una muestra formada por Li2CO3 y BaCO3 que pesa1 gramo, consume 15,0 mL de HCl 1,0 mol L–1 cuandose valora utilizando naranja de metilo como indicador.¿Cuál es el porcentaje de BaCO3 en la muestra?H2CO3 (pKa1 = 6,35, pKa2 = 10,33). Pesos moleculares: Li2CO3 = 74, BaCO3 = 197,3.Sol.: 28,8% Li2CO3 y 71,2% BaCO3.
44. Una mezcla formada por 0,1 g de NaHCO3 y 0,1 g deNaOH se disuelve en agua destilada y se valora conácido sulfúrico 0,1 mol L–1 y fenolftaleína como indi-cador. Calcular los mL de ácido consumidos en lavaloración.H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Pesos atómicos: C = 12,00; H = 1,01; O = 16,00; Na = 23,00.
Cuando la mezcla indicada se disuelve en agua seproduce la siguiente reacción:
NaHCO3 + NaOH → Na2CO3 + H2O
y considerando las cantidades de ambas sustanciaspuestas en juego y sus respectivos pesos moleculares:
se tendrá una disolución final que contiene carbona-to de sodio y un exceso de NaOH. Al valorar estadisolución con ácido sulfúrico 0,1 mol L–1 utilizandofenolftaleína como indicador, se valora la base fuer-te y la base débil queda como HCO3
_. En definitiva
se valora sólo el contenido de NaOH presente en lamuestra, permaneciendo el NaHCO3 como una espe-cie “inerte” en el proceso global. Así:
de donde se consumen 12,5 mL en la valoración conácido sulfúrico.
45. Una muestra que pesa 1,5 gr está formada por NaH-CO3, Na2CO3 y materia inerte consume 20 mL deNaOH 0,05 mol L–1 para su valoración utilizando fenolf-taleína como indicador. Otra muestra de 1,5 gr consu-me 20 mL de HCl 0,1 mol L–1 cuando se valora emple-ando naranja de metilo como indicador. ¿Cuál es elporcentaje de NaHCO3 y Na2CO3 en la muestra?H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Pesos atómicos: H = 1,01; Na = 23,00; C = 12,00; O = 16,00.Sol.: 5,6% NaHCO3 y 3,53% Na2CO3.
46. Una muestra puede contener NaOH, NaHCO3,Na2CO3 o mezclas de estas sustancias. Dos porcionesde 0,1 g de esta muestra se disuelven en agua. Unade ellas, consume 17,96 mL de disolución de HCl0,1038 mol L–1 utilizando fenolftaleína como indica-dor. La otra porción se valora usando naranja de meti-lo como indicador y consume en dicha valoración21,17 mL de disolución del mismo ácido. ¿Qué espe-cies se encuentran presentes en la muestra? ¿Cuál essu tanto por ciento en la muestra original?
100 mg NaOH1 mmol NaOH40 mg NaOH
1 mmol H SO2 mmol NaOH
(mL) 0,1 molL
2 4
H SO1
2 4
× × =
= × −V
mmol NaOH
10040
2,5= =
mmol NaHCO
100106
0,9433 = =
( , , )
,5 0 4 0
61 81
100600
− × ×mmol H BOmg
mmol H BO3 33 3
20 0,1 mmol HCl2 mmol H BO
2 mmol HCl
1 mmol bórax2 mmol H BO
4 mmol H BO1 mmol bórax
4, 0
2 3
2 3
3 3
× × ×
× × =
−
−
50 0,1 mmol NaOH
1 mmol H BO1 mmol NaOH
5, 03 3× × =
20 0,1 mmolHCl2 mmol H BO
2 mmol HCl1 mmol bórax
2 mmol H BO381,4 mg
1 mmol bórax100600
2 3
2 3
× × ×
× × ×
−
−
Capítulo 4: Valoraciones ácido-base 183

H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Pesos atómicos: H = 1,01; Na = 23,00; C = 12,00; O = 16,00.
Las posibles combinaciones entre los componen-tes que podrían estar presentes en la muestra son lassiguientes:
1. Sólo NaOH. No es posible ya que se consumiríael mismo volumen de disolución de HCl al valo-rar la muestra utilizando naranja de metilo yfenolftaleína como indicadores.
2. Sólo Na2CO3. No es posible ya que se gastaría unvolumen doble de HCl 0,1038 mol L–1 al usarnaranja de metilo como indicador en lugar defenolftaleína.
3. Sólo NaHCO3. No es posible ya que el volumende disolución de HCl consumido al utilizar fenolf-taleína como indicador sería cero.
4. Mezcla NaHCO3 + NaOH. No es posible debi-do a que ambas especies son incompatibles endisolución.
5. Mezcla Na2CO3 + NaOH. Mezcla compatible yconforme con los volúmenes de ácido clorhídri-co consumidos utilizando naranja de metilo yfenolftaleína como indicadores.
6. Mezcla NaHCO3 + Na2CO3. Mezcla compatibleaunque no es posible ya que se consumiría unvolumen de disolución de HCl utilizando naran-ja de metilo como indicador superior al doble gas-tado cuando se emplea fenolftaleína.
7. Mezcla NaHCO3 + Na2CO3 + NaOH. Mezclaincompatible debido a la presencia en la mismade NaHCO3 y NaOH.
Una vez deducida de forma cualitativa la compo-sición de la mezcla, conforme a los datos aportadospor el problema se tiene que cuando se valora em-pleando naranja de metilo como indicador, se tienenlas siguientes reacciones de valoración:
Na2CO3 + 2 HCl → H2CO3 + 2 NaCl
NaOH + HCl → NaCl + H2O
mientras que al utilizar fenolftaleína se tiene:
Na2CO3 + HCl → NaHCO3 + NaCl
NaOH + HCl → NaCl + H2O
Se observa que la diferencia de volumen de diso-lución de HCl entre ambas valoraciones correspon-de a la siguiente reacción valoración:
Na2CO3 + HCl → NaHCO3 + NaCl
De esta forma:
de donde el contenido de carbonato de sodio en lamuestra es del 35,32%.
Para determinar el contenido de NaOH en la mues-tra se puede hacer uso, por ejemplo, de la valoraciónutilizando fenolftaleína como indicador:
mmol HCl consumidos: 17,96 × 1,038 = 18,64
mmol Na2CO3 presentes:
mmol NaOH presentes = 18,64 – 3,33 = 15,31
El contenido de NaOH en la muestra vendrá dadopor:
47. Una mezcla que contiene NaOH, Na2CO3 y materiainerte se valora con HCl 0,123 mol L–1. 0,5 g de la mez-cla requieren 28,32 mL para valorarlos en presencia defenolftaleína, y un volumen adicional de 22,04 mL paravalorarlos en presencia de naranja de metilo. Calcularel tanto por ciento en peso de NaOH y Na2CO3.H2CO3 (pKa1 = 6,35, pKa2 = 10,33). Pesos atómicos: H = 1,01; Na = 23,00; C = 12,00; O = 16,00.Sol.: 6,18% NaOH y 57,47% Na2CO3.
48. Una mezcla de Na2CO3 y NaHCO3 que pesa 0,4503 grequiere 30,64 mL de disolución de HCl 0,2 mol L–1
para su valoración con naranja de metilo. Otra mues-tra que pesa 0,9006 g se trata con 50 mL de disoluciónde NaOH 0,2 mol L–1 y 10 mL de BaCl2 1,0 mol L–1.El exceso de disolución de NaOH consume 27,63 mL
15,31 mmolNaOH40 mg
1 mmolNaOH100
1.00061,24%
× ×
× =
(21,17 17, 96) 1, 038 mmol HCl
1 mol Na CO1 mmol HCl
3, 332 3
− × ×
× =
(21,17 17,96) 1,038 mmolHCl1 molNa CO1 mmolHCl
106 mg1 mmol Na CO
1001.000
2 3
2 3
− × ×
× × ×
184 Equilibrios iónicos y sus aplicaciones analíticas

de HCl 0,2 mol L–1 para su valoración. Calcular el tan-to por ciento de Na2CO3 y NaHCO3 en la muestra.H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Pesos atómicos: H = 1,01; Na = 23,00; C = 12,00; O = 16,00.Sol.: 41,7% NaHCO3 y 45,8% Na2CO3.
49. Una disolución contiene HCl y H3PO4. Se toman 50,0mL y se valoran con NaOH 0,1 mol L–1 y naranja demetilo como indicador. Se consumen 27,95 mL. Otrafracción de 50,0 mL se valora con la misma disolu-ción de NaOH, pero con fenolftaleína como indica-dor, y se gastan 49,63 mL. Calcular la concentraciónde los ácidos HCl y H3PO4 en la disolución original.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32).Sol.: CHCl = 0,0125 mol L–1, CH3PO4
= 0,0434 mol L–1.
50. Calcular la concentración de ácido clorhídrico y deácido fosfórico en una disolución en base a las siguien-tes experiencias: 1) 50 mL de la disolución consumen38,2 mL de NaOH 0,1 mol L–1 utilizando naranja demetilo como indicador. 2) 50 mL de la disolución con-sumen 49,5 mL de NaOH 0,1 mol L–1 utilizandofenolftaleína como indicador.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32).Sol.: CHCl = 0,0538 mol L–1, CH3PO4
= 0,0226 mol L–1.
51. 25 mL de una mezcla de ácidos sulfúrico y fosfóricorequieren 38,32 mL de NaOH 0,10 mol L–1 para neu-tralizarse utilizando fenolftaleína como indicador. Otraporción de 10 mL necesita 12,21 mL de la misma diso-lución de NaOH si se utiliza naranja de metilo. Calcu-lar la concentración en g L–1 de cada ácido en la mezcla.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32), H2SO4(pKa2 = 1,92). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00; S = 32,06.
Cuando se lleva a cabo la valoración de una alí-cuota de 25 mL de la mezcla de ácidos con fenolfta-leína como indicador se tienen las siguientes reac-ciones de valoración:
H2SO4 + 2 NaOH → Na2SO4 + 2 H2O
H3PO4 + 2 NaOH → Na2HPO4 + 2 H2O
mientras que para la alícuota de 10 mL utilizandonaranja de metilo como indicador, éstas serán:
H2SO4 + 2 NaOH → Na2SO4 + 2 H2O
H3PO4 + NaOH → NaH2PO4 + H2O
Antes de abordar la resolución de esta mezcla sehan de referir los volúmenes de disolución de NaOHconsumidos en cada valoración a una misma alícuo-ta, ya sean 25 o 10 mL. Así, en el segundo caso, si setomase una alícuota de 25 mL el volumen de NaOHconsumido en la valoración sería:
Comparando ambas valoraciones se observa quela diferencia de volúmenes entre ambas correspon-de a la valoración de un protón del ácido fosfórico,por lo tanto:
de donde la concentración de ácido fosfórico en lamuestra es de 3,06 g L–1.
Para determinar la concentración de ácido sulfúri-co en la muestra se puede partir, por ejemplo, de lavaloración en presencia de naranja de metilo comoindicador. Así,
moles NaOH consumidos:
0,03052 × 0,1= 3,052 × 10–3 moles
moles de H3PO4 valorados:
moles de NaOH para valorar:
H2SO4 = 3,052 × 10–3 – 7,8 × 10–4 = 2,272 × 10–3 moles
De aquí se tiene:
y por lo tanto la concentración de ácido sulfúrico enla muestra es de 4,45 g L–1.
2,272 10 molNaOH1 mol H SO2 mol NaOH
98 g H SO1 mol H SO
1.000 mL1 L
125 mL
3 2 4
2 4
2 4
× × ×
× × ×
−
(0, 03832 0, 03052) 0,1 mol NaOH
1 mol H PO1 mol NaOH
7, 8 10 moles3 4 –4
− × ×
× = ×
( , , ) ,.
0 03832 0 03052 0 111
981
1 0001
125
4 4
4
− × ×
× × × ×
molNaOHmol H POmol NaOH
H POmol H PO
mLL mL
3 3
3
g
VNaOH mL= × =12 21
2510
30 52, ,
Capítulo 4: Valoraciones ácido-base 185
52. Unas disoluciones pueden contener HCl, H3PO4,NaH2PO4 o cualquier combinación compatible deestos compuestos. A partir de los datos de la tablasiguiente, deducir la composición de cada disolucióny los mg de soluto, o solutos, que contiene.
Volumen (mL) de NaOH 0,1158 mol L–1
Disolución Punto final Punto finalVerde de bromocresol Fenolftaleína
1 0,00 18,34
2 8,48 16,96
3 8,48 31,84
4 10,77 10,77
5 24,56 30,00
H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00. Intervalos de viraje: Verde de bromocre-sol 3,8 – 5,4; Fenolftaleína 8,2 – 9,8.Sol.: 1) NaH2PO4, 254 mg; 2) H3PO4, 96 mg; 3)
NaH2PO4/ H3PO4, 207 mg/96 mg; 4) HCl, 45 mg;5) HCl/H3PO4 79,7 mg/62 mg.
53. Tres muestras de 1 g cada una que pueden contenerHCl, Na2PO4, NaH2PO4 y H3PO4 se disuelven en aguay se valoran con una disolución de NaOH 0,2 mol L–1
con los indicadores naranja de metilo y fenolftaleína.Se obtienen los siguientes resultados:
Volumen (mL) NaOH 0,2 mol L–1
Muestra Punto final Punto finalNaranja de metilo Fenolftaleína
1 26,88 39,12
2 8,62 30,06
3 22,62 45,24
Calcular la composición de cada una de las mues-tras expresada en tanto por ciento.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00.
Muestra 1. Dado que se consume en la valoraciónun volumen de disolución de NaOH cuando se utili-za naranja de metilo como indicador superior a la
mitad del gastado con fenolftaleína, esta muestra con-tiene una mezcla de los ácidos HCl y H3PO4. Las reac-ciones de valoración utilizando naranja de metilocomo indicador son:
HCl + NaOH → NaCl + H2O
H3PO4 + NaOH → NaH2PO4 + H2O
mientras que haciendo uso de la fenolftaleína se tiene:
HCl + NaOH → NaCl + H2O
H3PO4 + 2 NaOH → Na2HPO4 + 2 H2O
La diferencia entre ambos volúmenes proporcio-na de acuerdo con estas reacciones de valoración, losmL de NaOH 0,2 mol L–1 necesarios para valorar unprotón del ácido fosfórico. En estas condiciones sepuede establecer:
de donde el contenido de ácido fosfórico en la mues-tra es del 23,99%.
El volumen de NaOH consumido para valorar elácido clorhídrico se puede deducir, por ejemplo, dela valoración efectuada en presencia de naranja demetilo. Así:
mmol NaOH consumidos = 26,88 × 0,2 = 5,376
mmol H3PO4 presentes = (39,12 – 26,88) × 0,2 = 2,448
mmol NaOH para valorar HCl = 5,376 – 2,448 = 2,928
de donde se deduce que el contenido de HCl en lamuestra es del 10,68%.
Muestra 2. En la valoración de esta muestra se con-sume con fenolftaleína un volumen de disolución deNaOH superior al doble del gastado con naranja demetilo, por lo que esta muestra contiene H3PO4 yNaH2PO4. Las reacciones de valoración con ambosindicadores son:
2,928 mmolNaOH1 mmol HCl
1 mmol NaOH36,5 mg HCl1 mmol HCl
1001.000mg
×
× × ×
(39,12 26,88) 0,2 mmolNaOH1 mmol H PO1 mmol NaOH
98 mg H PO1 mmol H PO
1001.000mg
3 4 3 4
3 4
− × ×
× × ×
186 Equilibrios iónicos y sus aplicaciones analíticas

naranja de metilo
H3PO4 + NaOH → NaH2PO4 + H2O
fenolftaleína
H3PO4 + 2 NaOH → Na2HPO4 + 2 H2O
NaH2PO4 + NaOH → Na2HPO4 + H2O
En este caso, la valoración con naranja de me-tilo como indicador proporciona los datos necesariospara poder calcular el contenido de uno de los com-ponentes de la muestra, esto es el ácido fosfórico:
Para determinar el contenido de NaH2PO4 en lamuestra se ha de calcular previamente el volumen dedisolución de NaOH 0,2 mol L–1 consumido para suvaloración. De acuerdo con las reacciones de valo-ración en presencia de cada uno de los indicadoresutilizados, este volumen será:
y por tanto:
de donde el contenido de NaH2PO4 en la muestra esdel 30,77%.
Muestra 3. En la valoración de esta muestra se con-sume con fenolftaleína un volumen doble del gasta-do con naranja de metilo, lo que indica que esta mues-tra sólo contiene ácido fosfórico. A partir del volumende disolución de hidróxido de sodio consumido uti-lizando naranja de metilo como indicador se tiene uncontenido de ácido en la muestra de:
54. Cinco muestras de 1 g cada una que pueden conte-ner HCl, Na2HPO4, NaH2PO4 y H3PO4 se disuelvenen agua y se valoran con una disolución de NaOH 0,4 mol L–1 con los indicadores verde de bromocre-sol (3,8 – 5,4) y fenolftaleína. Se obtienen los siguien-tes resultados:
Volumen (mL) de NaOH 0,4 mol L–1
Muestra Punto final Punto finalVerde de bromocresol Fenolftaleína
1 32,40 32,40
2 22,62 45,24
3 26,88 39,12
4 0,00 20,22
5 8,62 30,06
Calcular la composición de cada una de las mues-tras expresada en tanto por ciento.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00.Sol.: 1) 47,3% HCl; 2) 88,67% H3PO4; 3) 21,37% HCl
y 47,98% H3PO4; 4) 97,03% NaH2PO4; 5) 32,80H3PO4 y 61,52% NaH2PO4.
55. Se toman 10,0 mL de una disolución que contieneH3PO4 y NaH2PO4 y se valoran con una disolución deNaOH 0,05 mol L–1. Se consumen 12,2 mL para alcan-zar el punto final con un indicador de que vira a pH =4,0. Otra muestra de 10,0 mL consume 39,1 mL de lamisma disolución alcalina con un indicador que vira apH = 9,5. Calcular la concentración de ambos compo-nentes en la disolución original.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32).Sol.: CH3PO4
= 0,061 mol L–1; CNaH2PO4= 0,073 mol L–1.
56. Una muestra de 5,0 grs de abono fosfatado que con-tiene una mezcla de NaH2PO4 y Na2HPO4 se disuelveen 1 litro de agua. Para su análisis se toman alícuotasque se valoran de la siguiente forma: 1) una alícuota de25 mL consume 8,5 mL de NaOH 0,05 mol L–1 para suvaloración con un indicador que vira a pH = 9,5. 2) unaalícuota de 50 mL consume 12,5 mL de HCl 0,1 molL–1 para su valoración con un indicador cuyo cambiode color se produce a pH = 4,0. Calcular el tanto porciento de fósforo en la muestra de abono.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: Na = 23; P = 30,97; O = 16,00; H = 1,01.Sol.: 26,01%.
22,62 0,2 mmolNaOH1 mmol H PO1 mmol NaOH
98 mg H PO1 mmol H PO
1001.000mg
44,33%
3 4
3 4
3 4
× × ×
× × =
12,82 0,2 mmolNaOH1 mmol NaH PO
1 mmol NaOH120 mg NaH PO1 mmol NaH PO
1001.000mg
2 4 2 4
2 4
× ×
× × ×
V V VNaH PO fenolfatleína naranjademetilo2 42
30,06 2 8,62 12,82 mL
= − × =
= − × =
8,62 0,2 mmolNaOH1 mmol H PO1 mmol NaOH
98 mg H PO1 mmol H PO
1001.000mg
16,89%
3 4
3 4
3 4
× × ×
× × =
Capítulo 4: Valoraciones ácido-base 187

57. Una muestra puede contener Na3PO4.12H2O oNa2HPO4.12H2O o NaH2PO4.H2O o alguna de susmezclas. Se hacen dos pesadas de 3,0 g de ésta y sevaloran una con HCl 0,5 mol L–1 y la otra con NaOH0,6 mol L–1. En la primera valoración se gastan 14,0mL utilizando naranja de metilo como indicador y no se consume ácido con fenolftaleína. En la se-gunda se gastan 5,0 mL y el indicador es la fenolf-taleína. Calcular la composición de la muestra.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00.
En la primera valoración con HCl 0,5 mol L–1 seconsume sólo volumen de este valorante cuando seutiliza naranja de metilo como indicador, lo que des-carta la presencia de la sal Na3PO4.12H2O en la mues-tra. En este caso la reacción de valoración será:
Na2HPO4 + HCl → NaH2PO4 + NaCl
Cuando se lleva a cabo la valoración con NaOH0,6 mol L–1 empleando fenolftaleína como indicadorse produce la siguiente reacción de valoración:
NaH2PO4 + NaOH → Na2HPO4 + H2O
En resumen, en cada valoración se produce ladeterminación de uno de los componentes de la mues-tra. Así, de la valoración de la muestra con HCl enpresencia se naranja de metilo se tiene:
de donde el contenido de Na2HPO4.12H2O en lamuestra es del 83,52%.
A partir de la valoración con NaOH utilizandofenolftaleína se puede establecer:
y por tanto el contenido de NaH2PO4.H2O en lamuestra es del 13,78%.
58. Dos disoluciones, A y B, pueden contener NaOH,Na3PO4, Na2HPO4 o cualquier mezcla de estas sus-tancias. Al valorar alícuotas de 25 mL de estas diso-luciones con HCl 0,1 mol L–1 y naranja de metilo ofenolftaleína como indicador, se obtienen los resul-tados siguientes:
Disolución Naranja de metilo Fenolftaleína
A 41,30 mL 17,09 mL
B 40,92 mL 24,33 mL
Deducir qué compuestos contiene cada diso-lución y calcular su concentración, expresada en mg L–1.H3PO4 (pKa1 = 2,15, pKa2 = 7,21, pKa3 = 12,32). Pesos atómicos: H = 1,01; Na = 23,00; P = 30,97; O = 16,00.Sol.: A) Na3PO4: 11.206 mg L–1, Na2HPO4: 4.043 mg L–1;
B) NaOH: 1.238 mg L–1, Na3PO4: 10.879 mg L–1.
59. Una muestra que pesa 10 g de una disolución con-tiene H2SO4 y HNO3, se valora con NaOH 0,1 molL–1 y se consumen 40,0 mL utilizando fenolftaleínacomo indicador. Otra muestra de 10 g se analiza,previa reducción de HNO3 , por el método Kjeldahl.El NH3 producido se recoge en 50,0 mL de HCl 0,1 mol L–1 y el exceso de dicho ácido consume 45,0 mL de NaOH 0,1 mol L–1 para su valoración.Calcular los porcentajes de H2SO4 y HNO3 en lamuestra.H2SO4 (pKa2 = 1,92). Pesos moleculares: H2SO4 = 98, HNO3 = 63.Sol.: 1,71% H2SO4 y 0,31% HNO3.
60. Para determinar la capacidad nutriente de una par-tida de abono importado debe conocerse su conteni-do en nitrato y amonio. Para ello se realizan dos expe-riencias: 1) A 2 g de muestra se le añade hidróxidode sodio 6,0 mol L–1 en exceso en un sistema Kjeld-hal. La disolución de ácido bórico al 5% en peso querecoge el destilado consume 42,4 mL de ácido clor-hídrico 0,25 mol L–1 utilizando naranja de metilocomo indicador. 2) A 1 g de muestra se le añade alu-minio metálico en polvo e hidróxido de sodio 6,0 molL–1 en exceso en un sistema Kjeldhal. El destilado serecoge en una disolución de 40 mL de ácido clorhí-drico 0,5 mol L–1. Se consumen 36,5 mL de hidróxi-do de sodio 0,2 mol L–1 cuando se valora su excesocon fenolftaleína como indicador. Expresar en por-
5,0 0,6 mmolNaOH1 mmol NaH PO .H O
1 mmol NaOH137,97 mg NaH PO .H O
1 mmol NaH PO .H O100
3.000mg
2 4 2
2 4 2
2 4 2
× × ×
× ×
14,0 0,5 mmolHCl1 mmol Na HPO .12H O
1 mmol HCl357,97 mg Na HPO .12H O
1 mmol Na HPO .12H O100
3.000mg
2 4 2
2 4 2
2 4 2
× × ×
× ×
188 Equilibrios iónicos y sus aplicaciones analíticas

centaje el contenido de nitrógeno en el abono en tresformas: 1) nitrógeno total, 2) nitrato y 3) amonio.Pesos atómicos: N = 14,00; O = 16,00; H = 1,01.
En la primera experiencia, al añadir a la muestraexclusivamente hidróxido de sodio se origina amo-niaco sólo a partir del amonio presente en la misma:
NH4+ + OH– → NH3 ↑ + H2O
Dado que la amoniaco destilado se recoge sobreácido bórico, se forma borato de sodio, el ácido clor-hídrico consumido equivale al amoniaco producidoy por ende al amonio presente en la muestra. Así,
En la segunda experiencia, al tratar la muestra conaluminio metal en medio básico, tanto el nitratocomo el amonio presentes en la muestra originanamoniaco:
NH4+ + OH– → NH3 ↑ + H2O
3 NO3
_+ 8 Al + 5 OH
_+ 2 H2O → 3 NH3 ↑ + 8 AlO2
_
En este caso, el amoniaco destilado se recogesobre ácido clorhídrico, y el exceso del mismo sinreaccionar se valora con una disolución de hidró-xido de sodio. Dándose estas condiciones se puedeestablecer:
mmol HCl añadidos = 40 × 0,5 = 20 mmol
mmol NaOH (valoración exceso HCl) == 36,5 × 0,2 = 7,3 mmol
mmol HCl (valoración NH3) = 20 – 7,3 = 12,7 mmol
mmol NO3
_+ NH4
+ = mmol HCl (valoración NH3) = = 12,7 mmol
Dado que en ambas experiencias se parte de dife-rentes cantidades de muestra, si se considera que eneste último caso se hubiese partido de 2 g (tal comoen el caso anterior) se tiene:
mmol NO3
_+ NH4
+ = 12,7 × 2 = 25,4 mmol
de donde:
mmol NO3
_= 25,4 – 10,6 = 14,8 mmol
La expresión de estos resultados conforme a lo esta-blecido en el enunciado del problema será:
% Nitrógeno total:
% Nitrato:
% Amonio:
61. Se determina el contenido de nitrógeno en un com-puesto orgánico por valoración ácido–base haciendouso del método de Kjeldahl partiendo de 1,7 g demuestra. ¿Qué concentración ha de tener el ácidoempleado en la neutralización del amoniaco produ-cido para que el número de mL gastados en esta valo-ración multiplicado por 0,1 proporcione el tanto porciento de nitrógeno en la muestra?Pesos atómicos: N = 14,00; H = 1,01.Sol.: 0,121 mol L–1.
62. El nitrógeno contenido en 0,5 g de una sustancia orgá-nica se determina por el método de Kjeldahl. A ladisolución resultante se le añade un exceso de NaOHy se recoge el amoniaco formado: 1) Sobre 150 mLde H2SO4 0,01 mol L–1. Se consumen 10,8 mL de diso-lución de NaOH en la valoración del exceso de H2SO4(1,0 mL de NaOH equivalen a 0,0070 g de ácido oxá-lico hidratado H2C2O4.2H2O). 2) Sobre una disolu-ción de ácido bórico al 4%. Se valora con HCl 0,0992mol L–1 y se consumen 18,15 mL. Calcular en amboscasos el % de nitrógeno en la muestra.
10,6 mmolNH18,04 mg NH1 mmol NH
1002.000mg
9,56%
44
4
++
+× ×
× =
14,8 mmolNO62 mg NO
1 mmol NO100
2.000mg45,88%
33
3
−−
−× ×
× =
25,4 mmol(NO NH )1 mmol N
1 mmol (NO NH )14 mg N
1 mmol N100
2.000mg17,78%
3 43 4
− +− ++ ×
+×
× × =
42, 4 0, 25 mmol HCl1 mmol NH1 mmol HCl
1 mmol NH1 mmol NH
10, 6 mmol NH
3
4
34
× × ×
× =+
+
Capítulo 4: Valoraciones ácido-base 189

HOOCCOOH (pKa1 = 4,20, pKa2 = 5,25).Pesos atómicos: C = 12,00; H = 1,01; O = 16,00.Pesos atómicos: N = 14,00; H = 1,01.
En la primera experiencia, el amoniaco se recogesobre ácido sulfúrico y el exceso de éste se valoracon disolución de hidróxido de sodio. La diferenciaentre las cantidades de ambos protolitos puestas enjuego proporciona el amoniaco desprendido por elcompuesto orgánico. Inicialmente, se ha de calcularla concentración de la disolución de hidróxido desodio utilizada en esta experiencia:
y de aquí, CNaOH = 0,111 mol L–1.
Una vez determinada la concentración de esta diso-lución de NaOH se puede establecer conforme al pro-cedimiento seguido:
mmol H2SO4 = 150 × 0,01 = 1,5 mol
mmol NaOH (valoración exceso H2SO4) == 10,8 × 0,111 = 1,2 mmol
mmol H2SO4 (valoración NH3) = mmol
A partir de esta cantidad de ácido sulfúrico se pue-de estimar un contenido de nitrógeno en el compuestoorgánico de:
Cuando el amoniaco desprendido se recoge sobreuna disolución de ácido bórico, el contenido de nitró-geno, expresado en porcentaje, se puede determi-nar tal como:
63. El amoniaco obtenido en la digestión de 2,0 g deproteína se recoge sobre una disolución saturada deácido bórico. En la valoración posterior de la diso-lución resultante se consumen 42,7 mL de disolu-ción de HCl 0,2596 mol L–1. ¿Cuál es el tanto porciento de nitrógeno en la proteína expresado comosulfato de amonio?Pesos atómicos: N = 14,00; H = 1,01; S = 32,06; O = 16,00.Sol.: 36,62%.
64. Se desea conocer el contenido de nitrato en un abono.Para ello 1 gr de abono se trata en un matraz Kjeldhalcon aluminio metal en medio básico. La disolucióncolectora de amoniaco es 100 mL de HCl 0,2 mol L–1,la cual se valora posteriormente con NaOH 0,3 mol L–1
y fenolftaleína como indicador gastándose 29,63 mL.Calcular el porcentaje de nitrato en el abono.Pesos atómicos: N = 14,00, O = 16,00. Sol.: 68,89%.
18,15 0, 0992 mmol HCl1 mmol NH1 mmol HCl
1 mmol N1 mmol NH
14 mg N1 mmol N
100500 mg
5, 04 %
3
3
× × ×
× × × =
0,9 mmolH SO2 mmol NH
1 mmol H SO1 mmolN
1 mmolNH14 mgN
1 mmolN100
500mg5,04%
2 43
2 4 3
× × ×
× × =
1 51 22
0 9,,
,− =
1,0 mmolNaOH1 mmol H C O .2H O
2 mmol NaOH7,0 mg H C O .2H O
126
NaOH2 2 4 2
2 2 4 2
× × =
=
C
190 Equilibrios iónicos y sus aplicaciones analíticas

5
5.1. Introducción5.2. Constantes de equilibrio5.3. Cálculo de las concentraciones
de especies en el equilibrio de formaciónde complejos
5.4. Introducción a los equilibriosconcurrentes
5.5. Equilibrios concurrentes de acidezy complejación
CuestionesSeminarios: problemas numéricos
EQUILIBRIOSDE FORMACIÓN DE COMPLEJOS

5.1. Introducción
El interés del estudio de los equilibrios de for-mación de complejos reside en sus amplias yvariadas aplicaciones dentro de la Química, y enparticular de la Química Analítica. De hecho,una gran cantidad de metodologías utilizan lasreacciones de formación de complejos con obje-to de mejorar sus características analíticas. Así,por ejemplo, en análisis instrumental se empleala formación de complejos para mejorar las pro-piedades analíticas básicas como la selectividad,enmascarando las especies interferentes, o la sen-sibilidad, empleando sustancias formadoras decomplejos con propiedades absorciométricas ofluorescentes acusadas.
La utilización de complejos se extiende al aná-lisis cualitativo clásico con diversas finalidades,tales como:
1. Separación de iones por disolución selectivade precipitados. Mediante la adición de amo-niaco a una disolución que contiene los pre-cipitados de cloruro y yoduro de plata, esposible la disolución selectiva del AgCl, ypor tanto la separación de los iones cloruroy yoduro, por formación del complejo:
AgCl(S) + 2 NH3→←
→← Ag (NH3)2+ + Cl– [5.1]
dado que el yoduro de plata es insolubleen amoniaco.
2. Reconocimiento de iones. La identificacióndel ion Co2+ se basa en la formación del
complejo con tiocianato de color azul, elcual se intensifica mediante extracción enalcohol amílico, dado que en este mediose favorece su estabilidad:
Co2+ + 4 KSCN + (2 + x)H2O →←→← [Co(SCN)4(H2O)2]
2– [K2(H2O)x]2+ + 2 K+
[5.2]
3. Enmascaramiento de interferencias. El ioncianuro se utiliza para enmascarar al Cu2+
con objeto de evitar su interferencia en laidentificación de cadmio por formacióndel sulfuro de cadmio amarillo, dado queel Cu2+ origina un precipitado de sulfurode cobre de color negro que impide iden-tificar el color amarillo del sulfuro de cad-mio:
2 Cu2+ + Cd2+ + 7 CN– + S2– + 2 OH– →←→← CdS(S) + 2 Cu (CN)6
2 + CNO– + H2O[5.3]
También se pueden considerar numerososejemplos del uso de las reacciones de formaciónde complejos en el análisis cuantitativo clásico,como en las determinaciones volumétricas deiones metálicos utilizando EDTA (ácido etilen-diaminotetraacético) como complejante:
Ca2+ + EDTA →← Ca-EDTA2– [5.4]
o en la determinación gravimétrica de Ni2+ apro-vechando la formación de su complejo insolu-
192 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Describir las aspectos básicos de los equilibrios de for-mación de complejos y las constantes que los rigen:globales y sucesivas.
• Desarrollar las alternativas adecuadas (expresiones ydiagramas gráficos) para el cálculo de la concentra-ción de las diferentes especies presentes en los mismos.
• Introducción al estudio de los equilibrios concurren-tes y en concreto a los de acidez y complejaciónmediante el empleo de los coeficientes de reacciónsecundaria y de constantes condicionales de forma-ción de complejos.

ble con dimetilglioxima (DMG) de color rojointenso:
Ni2+ + DMG →← Ni(DMG)2 [5.5]
Un complejo se define como la asociación dedos o más especies que pueden existir separada-mente. La nueva especie formada –complejo–puede permanecer en disolución o bien partici-par en un proceso de separación de fases (ya seamediante la formación de una fase sólida –preci-pitación– o bien mediante su distribución en otrafase líquida inmiscible en la acuosa –extracciónlíquido-líquido–). En la formación de un complejoparticipan dos tipos de especies; una puede acep-tar uno o más pares de electrones para completarsu estructura electrónica (normalmente orbitalesd vacíos), y que es generalmente un ion metálico.Esta especie se llama especie central o ion central,y se trata de un ácido de Lewis. El segundo tipode especies involucradas son los ligandos, espe-cies que ceden uno o más pares de electrones alion central, y por tanto se tratan de bases deLewis. Este hecho ya indica que cualquier ligan-do posible tendrá al menos un átomo que serácapaz de ceder sus pares de electrones, tal comooxígeno, fósforo, azufre, nitrógeno o halógeno.
La estructura general de un complejo serepresenta conforme a:
donde L indica el ligando y M el ion central.Algunos de los ligandos más comunes puedenser el agua, amoniaco, haluros, etc. La carga delcomplejo (representada como m+) puede serpositiva, negativa, o bien ser eléctricamente neu-tra. En general, los complejos neutros son inso-lubles en agua. El número de enlaces entre un ioncentral y el ligando o ligandos unidos a él se deno-
mina número de coordinación, que depende deltamaño del ion y la naturaleza de los ligandos.Usualmente, el número de coordinación demuchos complejos es de 2, 4 o 6, que correspon-den, respectivamente, a un complejo de geome-tría lineal, tetraédrica u octaédrica.
Los ligandos se pueden clasificar de diferen-tes formas. Así, una primera clasificación se pue-de realizar en función de su carga, con lo que setienen ligandos neutros (NH3, H2O, piridina, etc.),o ligandos cargados (generalmente aniones, comoCN–, Cl–, OH–, ion acetato, etc.). También se pue-de distinguir entre ligandos inorgánicos y orgá-nicos, aunque en general es más práctico clasifi-carlos en función del número de posiciones decoordinación; así, los ligandos monodentados(NH3, H2O, CN–, etc.) presentan una sola posi-ción de coordinación, mientras que los que tie-nen dos o más posiciones de coordinación sedenominan bidentados o polidentados, respecti-vamente. Por ejemplo, la etilendiamina (en), laoxina y la dimetilglioxima (DMG) son ligandosbidentados. El ácido etilendiamino tetracético(EDTA) tiene seis posiciones de coordinación,por tanto se trata de un ligando hexadentado:
La diferencia más importante entre los ligan-dos monodentados y polidentados reside en eltipo de complejos que pueden originar. En el pri-mer caso sólo pueden formar complejos de adi-ción, como por ejemplo: Ag(NH3)2
+, Fe(SCN)2+,mientras que los ligandos polidentados puedendar lugar a los complejos quelatos, en los que elion central se une a una molécula de ligando pormás de un punto de coordinación, como puedeser el caso de los complejos Zn(en)2
2+, Ni(DMG)2y Co(EDTA)2–:
Capítulo 5: Equilibrios de formación de complejos 193

5.2. Constantes de equilibrio
La formación de un complejo determinadoML, a partir de un ligando L y un ion central Mes una reacción reversible, definida como:
M + L →← ML [5.6]
cuya constante de equilibrio:
[5.7]
se denomina constante de formación o constantede estabilidad del complejo. Es decir, cuanto mayores el valor de la constante de formación de uncomplejo, más favorecida está su formación a par-tir de sus componentes (metal y ligando), y portanto el complejo es más estable.
Puede ocurrir, sin embargo, que a partir deun metal y un ligando pueda formarse más de uncomplejo, de diferentes estequiometrías, sobretodo en el caso de los complejos formados conligandos mono- y bidentados. Por ejemplo, el ionCu2+ puede formar con el amoniaco una serie
de complejos de estequiometría 1:1 hasta 1:5(metal:ligando). En los casos donde se puedenformar diferentes complejos no habrá una solaconstante de equilibrio, sino que tendremosvarias. Es necesario, por ello, distinguir entre lasdiferentes reacciones posibles. Por ejemplo, si lainteracción entre un ion central M y un ligandoL da lugar a los complejos 1:1, 1:2 y 1:3; la for-mación de estas especies se produce de formaescalonada de manera que los complejos se for-man sucesivamente, indicando que cada posiblecomplejo se forma a partir del complejo de este-quiometría inmediatamente inferior y el ligan-do libre:
M + L →← ML ML + L →← ML2
ML2 + L →← ML3 [5.8]
En este caso, las constantes correspondientesa estos equilibrios se denominan constantes deformación sucesivas, y vienen definidas por:
[5.9]
Para un caso general, las constantes de for-mación sucesivas correspondientes a la forma-ción de una serie de complejos de estequiome-tria 1:1 hasta 1:n vendrán dadas por:
M + L →← ML [5.10]
ML + L →← ML2 [5.11]
MLn – 1 + L →← Mn [5.12]
En general, el orden de magnitud de las cons-tantes de formación sucesivas es decreciente a
Knn
n
=−
[ML ][ML ][L]1
K2 = [ML ]
[ML][L] 2
K1 = [ML]
[M][L]
K K K1 2 3= = =[ML]
[M][L][ML ]
[ML][L][ML ]
[ML ][L]2 3
2
K = [ML]
[M][L]
194 Equilibrios iónicos y sus aplicaciones analíticas

medida que aumenta el número de coordinación:K1 > K2 > K3 > … > Kn.
Otra posibilidad de definición de las cons-tantes de equilibrio de formación de complejosson las llamadas constantes globales de forma-ción (β), en las que intervienen los componenteslibres M y L de la forma:
M + L →← ML [5.13]
M + 2 L →← ML2 [5.14]
M + n L →← MLn [5.15]
Es fácil deducir que para un cierto metal M,que forma una serie de complejos sucesivos conel ligando L, la relación entre su constante de for-mación global y sucesivas es:
[5.16]
Por tanto, para un sistema metal-ligandodeterminado, es indiferente conocer sus cons-tantes de formación sucesivas o global, ya queconocidas unas podemos obtener la otra y vice-versa. En los cuadros 5.1 y 5.2 se dan los valoresde constantes globales de formación de com-
plejos metálicos con EDTA y con diferentesligandos monodentados.
Como se ha indicado anteriormente, la cons-tante de formación de un complejo puede em-plearse para evaluar la estabilidad del mismo yen definitiva su capacidad para su uso analítico.Sin embargo, los valores de βn no son directa-mente comparables entre sí para dictaminar siun complejo es o no más estable que otro, salvoque tengan igual número de coordinación (este-quiometría). En caso contrario, se ha de utilizarla concentración de ion metálico libre en cadaequilibrio de complejación, como magnitud paraevaluar la estabilidad relativa entre los diferen-tes complejos.
La definición de formación de complejos pro-puesta como la interacción entre un ácido y unabase de Lewis, también se puede aplicar a losequilibrios ácido-base contemplados en el capí-tulo 2 de esta obra. Así, tal como se han defini-do las constantes de acidez o disociación ácidapara un ácido, en el caso de un ácido dipróticoH2A se tiene:
H2A →← HA– + H+ [5.17]
HA– →← A2– + H+ [5.18] Ka 2 =
− +
−
[A ][H ][HA ]
2
Ka1 =
− +[HA ][H ][H A]2
βn nK K K= × × ×1 2 …
βnn
n= [ML ]
[M][L]
β2
22
[ML ][M][L]
=
β1
[ML][M][L]
=
Capítulo 5: Equilibrios de formación de complejos 195
CUADRO 5.1Constantes de formación de complejos metálicos con EDTA
Complejo log K Complejo log K
Al3+– EDTA 16,1 Hg2+– EDTA 21,8Ag+ – EDTA 7,3 Mg2+– EDTA 9,10Ba2+– EDTA 7,8 Mn2+– EDTA 13,8
Ca 2+– EDTA 10,7 Ni2+– EDTA 18,6Cd2+– EDTA 16,5 Pb2+– EDTA 18,0Cu2+ – EDTA 16,3 Sr2+– EDTA 8,6Fe3+– EDTA 25,1 Zn2+– EDTA 16,5
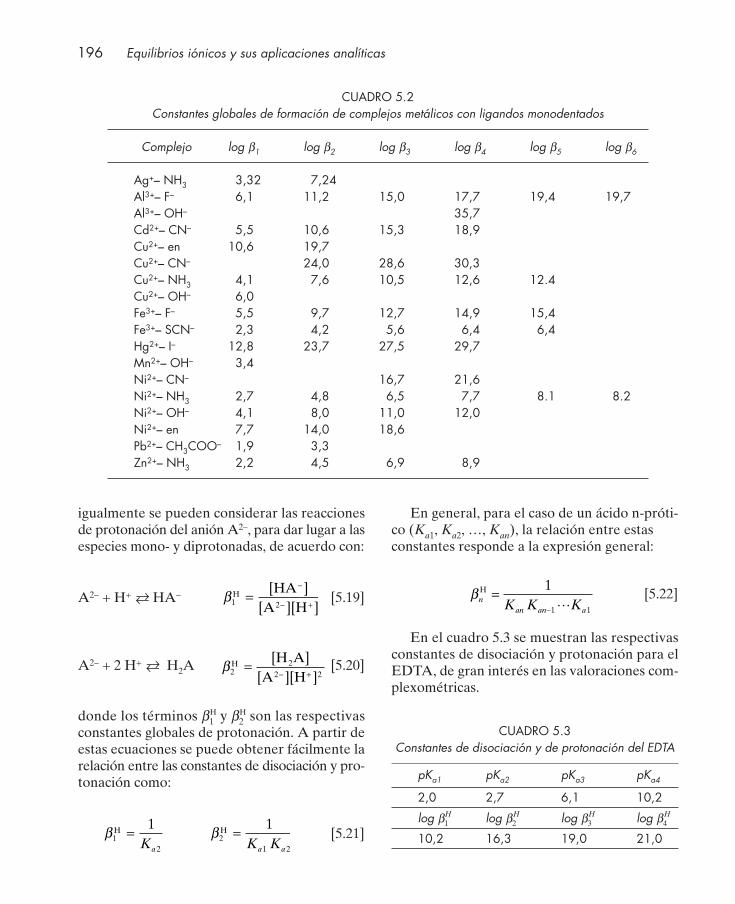
igualmente se pueden considerar las reaccionesde protonación del anión A2–, para dar lugar a lasespecies mono- y diprotonadas, de acuerdo con:
A2– + H+ →← HA– [5.19]
A2– + 2 H+ →← H2A [5.20]
donde los términos β1H y β2
H son las respectivasconstantes globales de protonación. A partir deestas ecuaciones se puede obtener fácilmente larelación entre las constantes de disociación y pro-tonación como:
[5.21]
En general, para el caso de un ácido n-próti-co (Ka1, Ka2, …, Kan), la relación entre estasconstantes responde a la expresión general:
[5.22]
En el cuadro 5.3 se muestran las respectivasconstantes de disociación y protonación para elEDTA, de gran interés en las valoraciones com-plexométricas.
CUADRO 5.3Constantes de disociación y de protonación del EDTA
pKa1 pKa2 pKa3 pKa4
2,0 2,7 6,1 10,2
log β1H log β2
H log β3H log β4
H
10,2 16,3 19,0 21,0
H
–
βnan an aK K K
= 1
1 1�
β2
1 2
1H =K Ka a
β12
1H =Ka
β2
H 22 2
[H A][A ][H ]
=− +
β1
H2
[HA ][A ][H ]
=−
− +
196 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 5.2Constantes globales de formación de complejos metálicos con ligandos monodentados
Complejo log β1 log β2 log β3 log β4 log β5 log β6
Ag+– NH3 3,32 7,24Al3+– F– 6,1 11,2 15,0 17,7 19,4 19,7Al3+– OH– 35,7Cd2+– CN– 5,5 10,6 15,3 18,9Cu2+– en 10,6 19,7Cu2+– CN– 24,0 28,6 30,3Cu2+– NH3 4,1 7,6 10,5 12,6 12.4Cu2+– OH– 6,0Fe3+– F– 5,5 9,7 12,7 14,9 15,4Fe3+– SCN– 2,3 4,2 5,6 6,4 6,4Hg2+– I– 12,8 23,7 27,5 29,7Mn2+– OH– 3,4Ni2+– CN– 16,7 21,6Ni2+– NH3 2,7 4,8 6,5 7,7 8.1 8.2Ni2+– OH– 4,1 8,0 11,0 12,0Ni2+– en 7,7 14,0 18,6Pb2+– CH3COO– 1,9 3,3Zn2+– NH3 2,2 4,5 6,9 8,9

5.2.1. Factores que determinanla estabilidad de los complejos
Existen diferentes factores que influyen en laestabilidad de un complejo, esto es, en el valorde la constante de formación del mismo. Estosfactores dependen, principalmente, del tipo deligando, del ion metálico central, o de ambos ala vez, cuya descripción pormenorizada se des-cribe a continuación.
A) Tamaño y carga del ion metálico
En general, cuanto menor sea el radio del ioncentral y mayor sea su carga, más estables seránsus complejos. Es decir, al aumentar la relaciónentre la carga y el radio iónico del ion metálico cen-tral, aumentará la estabilidad de sus complejos.
B) Fuerza como base del ligando
Dado que un ligando es una base de Lewis, sepuede considerar que cuanto más fuerte sea comobase (posibilidad de ceder sus pares de electro-nes), más estables serán los complejos que forme.
C) Relación entre el metal y el ligando
De acuerdo con Pearson se puede distinguirentre dos tipos de metal y dos tipos de ligando,principalmente. Los iones metálicos pueden serácidos duros o blandos (clase A y clase B, res-pectivamente), y a su vez los ligandos pueden serbases duras y blandas (también clase A y clase B).Así, se tienen:
Ácidos duros: corresponden a metales elec-tropositivos, como los alcalinos, alcalino-térreos,algunos metales de transición del período IV, ade-más de lantánidos y actínidos.
Ácidos blandos: son los más electronegativos,tales como Pt, Au, Hg, Pb y algunos de los meta-les de transición ligeros, en bajo estado de oxi-dación.
Bases duras: son aquellas en las que el átomodador de electrones es N, O o F.
Bases blandas: el grupo dador de electroneses un átomo de P, S, Cl. Es decir, de los mismosgrupos que las bases duras, pero más pesados.
En base a esta clasificación, los complejos másestables se forman entre los pares de ion centraly ligando: ácido duro + base dura y ácido blando+ base blanda.
D) Formación de quelatos
Experimentalmente se observa que los com-plejos quelatos son mucho más estables que susequivalentes complejos de adición. Por ejemplo,si se compara la estabilidad de los complejos deNi2+ con amoniaco y etilendiamina (en) que pre-sentan el mismo número de coordinación (tenien-do en cuenta que el amoniaco es monodentadoy la en es un ligando bidentado):
Ni2+ + 2 NH3→← Ni(NH3)2
2+
βNH3,2 = 6 × 104 [5.23]
Ni2+ + en →← Ni(en)2+
βen,1 = 5 × 107 [5.24]
Ni2+ + 4 NH3→← Ni(NH3)4
2+
βNH3,4 = 3 × 107 [5.25]
Ni2+ + 2 en →← Ni(en)22+
βen,2 = 1,1 × 1014 [5.26]
Ni2+ + 6 NH3→← Ni(NH3)6
2+
βNH3,6= 1,6 × 108 [5.27]
Ni2+ + 3 en →← Ni(en)32+
βen,3 = 4 × 1018 [5.28]
se observa que los complejos quelatos son real-mente mucho más estables que los correspon-dientes complejos de adición, a pesar de que elnúmero de enlaces N–Ni formados entre cadapar de reacciones (5.23 y 5.24, 5.25 y 5.26, 5.27 y5.28) es el mismo, y que la energía de este enla-
Capítulo 5: Equilibrios de formación de complejos 197

ce (en términos de entalpía, ∆H), es práctica-mente la misma ya sea un grupo NH3 o R–NH2el que forme el complejo.
La explicación a este hecho reside en que enrealidad, cuando se tiene un ion metálico en diso-lución acuosa, éste se encuentra solvatado pormoléculas de agua; de hecho, está en forma deaquocomplejo. Por tanto, no se trata estricta-mente de una reacción de formación de un com-plejo, sino de sustitución de ligandos de un com-plejo. Así, las reacciones [5.27] y [5.28] deberíanescribirse:
Ni(H2O)62+ + 6 NH3
→←→← Ni(NH3)6
2+ + 6 H2O[5.29]
Ni(H2O)62+ + 3 en →←
→← Ni(en)32+ + 6 H2O
[5.30]
Si además se tiene en cuenta que la constan-te de equilibrio de una reacción es una medidade la variación de su energía libre, y que el valorde ésta depende también de la variación de entro-pía del proceso:
∆G = ∆H – T ∆S = –RT ln K [5.31]
se observa que aunque el cambio de entalpía dereacción sea prácticamente el mismo en cada caso,no ocurre lo mismo con la variación de entropía,mucho mayor en la formación de complejos que-latos. Por otra parte, también se debería conside-rar el cambio en la solvatación del ligando por elagua, que también daría lugar a una mayor varia-ción en la entropía de la reacción.
A partir de estas consideraciones, es fácildeducir que los ligandos polidentados, capacesde formar complejos quelatos, darán lugar a com-plejos más estables cuanto mayor sea el númerode puntos de coordinación que tengan. Esto esparticularmente cierto en el caso de EDTA, unligando que, gracias a sus 6 puntos de coordina-ción, da lugar a complejos muy estables con unagran cantidad de iones metálicos, de estequio-metría 1:1, y por tanto, de gran aplicación enmétodos cuantitativos.
E) Geometría de los quelatos
La formación de un complejo quelato da lugara una serie de anillos, obtenidos a partir de lapropia molécula de ligando y los enlaces con elion metálico. Teniendo en cuenta las tensionesdentro de estos anillos, podemos deducir que losquelatos más estables serán aquellos en los queestos anillos sean de 5 o 6 eslabones. En el pri-mer caso, cuando en el anillo existen dobles enla-ces, y en el segundo cuando se trata de enlacessencillos. Por otra parte, cuanto mayor sea elnúmero de anillos formados mayor será la esta-bilidad del complejo.
5.2.2. Reacciones de desplazamiento
Las reacciones de desplazamiento se originana partir de un complejo formado por adición deun ion metálico o ligando de diferente naturale-za y constituyen, en definitiva, un reflejo de laestabilidad relativa de diferentes quelatos o com-plejos. Conforme al planteamiento indicado, unareacción de desplazamiento consiste en una com-petencia:
a) Entre dos iones metálicos, M y N, por unligando L, denominada reacción de inter-cambio de metal, o bien:
b) Entre dos ligandos, L y X, por un ionmetálico M, denominada reacción de inter-cambio de ligando.
En el caso de una reacción de intercambio demetal, la reacción de desplazamiento se puedeexpresar por:
ML + N →← NL + M [5.32]
La constante que rige este equilibrio de inter-cambio de metal, KIM , viene dada por:
[5.33] KIM
[NL][M][ML][N]
=
198 Equilibrios iónicos y sus aplicaciones analíticas

la cual se puede expresar en función de las cons-tantes globales de formación de ambos comple-jos, de acuerdo con:
[5.34]⇒
Esta expresión muestra que la extensión dela reacción de desplazamiento depende delvalor de KIM, y en definitiva de los valores rela-tivos de las constantes globales de formaciónimplicadas: βNL y βML. Así, cuanto mayor seaβNL más desplazada estará hacia la derecha lareacción [5.32].
Cuando se trata de una reacción de intercam-bio de ligando, la reacción de desplazamiento seexpresa por:
ML + X →← MX + L [5.35]
cuya constante de equilibrio, KIL, viene dada por:
[5.36]
Esta constante se puede expresar en funciónde las constantes globales de formación de amboscomplejos, de acuerdo con:
[5.37]⇒
Como en el caso anterior, la extensión de lareacción de desplazamiento depende del valorde su constante de equilibrio y en definitiva delos valores relativos de las constantes globales deformación implicadas: βMX y βML. Así, cuantomayor sea βMX más desplazada estará hacia laderecha la reacción [5.35].
Estas reacciones de intercambio presentan ungran interés en el ámbito de la valoraciones com-plexométricas con EDTA, ya que como se mostra-rá en el capítulo siguiente, permiten abordar la valo-ración de ciertos iones metálicos cuya valoracióndirecta con EDTA es imposible de llevar a cabo.
5.3. Cálculo de las concentracionesde especies en el equilibriode formación de complejos
Cuando en un determinado sistema metal-ligando se pueden obtener complejos de dife-rente estequiometría, la formación de uno u otrodepende de los respectivos valores de las cons-tantes de equilibrio y de las concentraciones demetal y ligando en el sistema. Para calcular lasconcentraciones de las diferentes especies pre-sentes en estos equilibrios de formación de com-plejos, se ha seguido un tratamiento similar alexpuesto en el capítulo 2 para los equilibriosácido-base, esto es, combinar el balance de ma-sas, en este caso del ion metálico, con las expre-siones de las constantes de formación de los com-plejos ya sean globales o sucesivas.
Si se toma como ejemplo el sistema formadopor un metal M y un ligando L, que pueden darlugar a la formación de dos especies complejas(ML y ML2), las ecuaciones y constantes globa-les de formación para estos equilibrios son:
M + L →← ML [5.38]
M + 2 L →← ML2 [5.39]
El balance de masas del metal, si se suponeuna concentración total de ion metálico en diso-lución igual a CM, viene dado por:
CM = [M] + [ML] + [ML2] [5.40]
Al sustituir las concentraciones de los com-plejos ML y ML2 en función de las constantes deequilibrio, concentraciones de ligando y metallibre a partir de las ecuaciones [5.38] y [5.39] enel balance de masas, ecuación [5.34], se obtiene:
CM = [M] + β1[M][L] + β2[M][L]2 [5.41]
o bien:
β2 = [ML ]
[M][L]2
2
β1 = [ML]
[M][L]
KIL
MX
ML
= ββ
KIL
[MX][L][ML][X]
[M][M]
= ×
KIL
[MX][L][ML][X]
=
KIM
NL
ML
= ββ
KIM
[NL][M][ML][N]
[L][L]
= ×
Capítulo 5: Equilibrios de formación de complejos 199

CM = [M] (1 + β1 [L] + β2 [L]2 ) [5.42]
La fracción molar (α), de cada una de las espe-cies en las que pueda estar presente el ion metáli-co en disolución (M, ML y ML2), vendrá dada por:
[5.43]
Si se sustituye en las expresiones anteriores elvalor de CM dado por la ecuación [5.42], se obtie-nen las correspondientes fracciones molares enfunción de las constantes globales de formación:
[5.44]
[5.45]
[5.46]
las cuales también pueden escribirse en funciónde las constantes de formación sucesivas de acuer-do con:
[5.47]
[5.48]
[5.49]
Estas ecuaciones indican que el grado de for-mación de cada complejo (representado por sufracción molar), depende únicamente de las cons-tantes de formación y de la concentración deligando libre (es decir, el que no está formandocomplejo).
En general, para un sistema metal-ligandoen el que se puedan formar n complejos suce-sivos, de estequiometrías ML, ML2, …, MLi,…MLn, y con unas constantes globales de for-mación correspondientes β1, β2, …,βi , …βn, lafracción molar de un complejo de estequiome-tria MLi es igual a:
[5.50]
quedando la fracción molar del ion metálico librecomo:
[5.51]
Una vez conocidas las diferentes fraccionesmolares, la concentración de cualquiera de lasespecies en disolución se determina directamen-te multiplicando su fracción molar por la con-centración total en disolución de ion metálico,esto es, su concentración analítica CM:
[5.52]
Esto es cierto siempre y cuando los comple-jos formados no sean polinucleares (casos en losque en una molécula de complejo existe más deun átomo del ion metálico), ni mixtos (en los cua-les coexisten diferentes tipos de ligando en unmismo complejo). En estos casos en particular,la fracción molar dependerá, además, de la con-centración total de metal y de las de los diferen-tes tipos de ligando, respectivamente.
[ ]MLi ML Mi
= ×α C
αβ
M
ii
1
1 [L]=
+=∑i
n
1
α β
βML
ii
ii
i
L
1 L=
+=∑
[ ]
[ ]i
n
1
αML2
LL L
=+ +
K KK K K
1 22
1 1 221
[ ][ ] [ ]
αML
1
1 1 22
[L]1 [L] [L]
=+ +
K
K K K
αM
1 1 221 [L] [L]
=+ +
1K K K
α ββ ββ
β β
ML
M][L]M M][L] M][L
LL L
2
22
1 22
22
1 221
=+ +
=
=+ +
[[ ] [ [ ]
[ ][ ] [ ]
α ββ ββ
β β
ML
M][LM M][L M][L
LL L
=+ +
=
=+ +
1
1 22
1
1 221
[ ][ ] [ ] [ ]
[ ][ ] [ ]
αβ β
β β
M
MM M][L M][L
L [L]
=+ +
=
=+ +
[ ][ ] [ ] [ ]
[ ]
1 22
1 22
11
α α αMM
MLM
ML2
M
[M] [ML] [ML ]2
= = =C C C
200 Equilibrios iónicos y sus aplicaciones analíticas

5.3.1. Diagramas de distribución de especies
En los diagramas de distribución de especies serepresentan los valores de las fracciones molaresde las diferentes especies presentes en el equilibrioen función del logaritmo de la concentración deligando libre en el sistema. Estos diagramas per-miten determinar rápidamente cuales serán lasespecies mayoritarias presentes en la disolución,así como su concentración relativa.
Para su construcción es recomendable utilizarun intervalo aproximado de log[L] comprendidoentre los siguientes valores mínimos y máximos:
log [L]min = – log K1 – 2 [5.53]
log [L]max = – log Kn + 2 [5.54]
En la figura 5.1 se muestra el diagrama de dis-tribución de especies correspondiente al sistemaCu2+–NH3, cuyo desarrollo se ha realizado hacien-do uso de una hoja de cálculo. En el caso de siste-mas más sencillos se puede hacer uso de un pro-cedimiento aproximado similar al utilizado en elcapítulo 3 para el tratamiento gráfico de los equi-librios ácido-base. Así, por ejemplo, una vez esta-blecido el intervalo apropiado de log [L] de acuer-do con las ecuaciones [5.53] y [5.54] se construyeuna tabla de valores de log [L], generados a inter-valos de una unidad, y se calculan haciendo uso delas expresiones adecuadas los valores de las corres-pondientes fracciones molares. Una vez calculadosestos valores, se realiza su representación gráfica.
En esta figura se aprecia una de las principa-les utilidades del diagrama de distribución deespecies, que permite, a partir del conocimientode las constantes de equilibrio, prever cuáles seránlas principales especies, y su concentración rela-tiva, presentes en disolución para una concen-tración de ligando libre determinada. Por ejem-plo, si la concentración de amoniaco libre es muybaja (menor que 10–6 mol L–1), prácticamente nose forman complejos, ya que casi todo el cobreestá como ion Cu2+ libre (su fracción molar es cer-cana a la unidad). A concentraciones de amoniacolibre superiores (aproximadamente 10–3,8 mol L–1),
se observa que la especie más importante es elcomplejo Cu(NH3)
2+ , aunque el ion Cu2+ libre yel complejo Cu(NH3)2
2+ existen en cantidad apre-ciable (aproximadamente la mitad que la especie1:1); igualmente, a esta concentración existe unapequeña proporción del complejo 1:3 (aproxi-madamente una fracción molar de 0,01). Tambiénse observa que a concentraciones moderadas deamoniaco libre (entre 0,01 y 1 mol L–1, aproxi-madamente), la especie predominante es el com-plejo Cu(NH3)4
2+, y además, que será muy difícilobtener el complejo Cu(NH3)5
2+ en disoluciónacuosa, ya que para que éste sea el predominan-te sería necesaria una concentración de amonia-co libre superior a 1 mol L–1 (nótese que estaríaformado casi completamente a concentracionesde ligando libre del orden de 100 mol L–1).
FIGURA 5.1 Diagrama de distribución de especiespara el sistema Cu2+–NH3. Las constantes
de formación sucesivas para los diferentes complejos formados se muestran en el cuadro 5.2.
5.4. Introducción a los equilibriosconcurrentes
Los denominados equilibrios simultáneos oequilibrios concurrentes se originan cuando coe-xisten en una misma disolución dos o más de los
Capítulo 5: Equilibrios de formación de complejos 201

equilibrios, ya sean homogéneos (ácido-base, for-mación de complejos, redox) o heterogéneos (pre-cipitación, extracción líquido-líquido, intercam-bio iónico). Esta concurrencia genera influenciasmutuas de mayor o menor consideración entre lasespecies comunes activas implicadas en los dife-rentes equilibrios. Así, pueden establecerse tantorelaciones binarias del tipo: ácido-base/formaciónde complejos, ácido-base/precipitación, forma-ción del complejos/redox, etc., como ternarias: ácido-base/formación de complejos/redox, ácido-base/precipitación/redox, etc., que son de granimportancia en el desarrollo de numerosos pro-cesos analíticos. Por lo general, estas influenciasmutuas se materializan en desplazamientos de lasdiferentes reacciones químicas según sea la natu-raleza de la especie que participa en las mismas.
Para abordar el tratamiento de estos equilibriosconcurrentes de manera sistemática es necesarioestablecer previamente una jerarquización de suinfluencia mutua, para lo cual se ha consideradola ordenación conceptual de los mismos mostradaen el capítulo 1, esto es: ácido-base, formación decomplejos, precipitación y redox. Conforme a esteenfoque, y como se muestra en la figura 5.2, se con-sidera que el equilibrio ácido-base influye sobretodos los demás; el de formación de complejos lohace sobre el de precipitación y redox; y el de pre-cipitación influye sobre el equilibrio redox.
FIGURA 5.2. Jerarquización de los equilibrios iónicos en disolución.
Este planteamiento es coherente con el estu-dio sistemático de los diferentes equilibrios ióni-cos en esta obra, aunque hay que reconocer queen determinadas circunstancias las influenciasmutuas pueden plantearse en sentido opuesto,tal como se indica por las líneas puntuadas en lafigura. Conforme a lo indicado, en este capítulose aborda a continuación la influencia del pH(equilibrios ácido-base) en los equilibrios de for-mación de complejos.
5.5. Equilibrios concurrentes de acidezy complejación
El pH del medio de reacción influye de mane-ra directa sobre el equilibrio de formación decomplejos por la propia naturaleza de las espe-cies implicadas en el mismo, ya que como se haindicado los iones metálicos y los ligandos sonácidos y bases de Lewis, respectivamente.
En consecuencia, se pueden originar en diso-lución acuosa dos tipos de reacciones competi-tivas con la de formación de complejos:
1. Interacción del ion metálico (ácido) conlos iones hidroxilos (base).
2. Interacción entre el ligando (base) y el ionhidrógeno (ácido).
Así, por ejemplo, en el caso de la formacióndel complejo Ag(NH3)2
+ entre el ion Ag+ y elamoniaco:
Ag+ + 2 NH3→← Ag(NH3)2
+ [5.55]
al ser el amoniaco una base débil, puede reaccio-nar con los iones H+ del medio para dar lugar alion amonio:
NH3 + H+ →← NH4+ [5.56]
Este equilibrio simultáneo indica que enmedio ácido no se tendrá amoniaco en disolu-ción, sino ion amonio. Como éste ya tiene todoslos pares de electrones ocupados no puede actuarcomo ligando. El resultado es que en medio áci-
202 Equilibrios iónicos y sus aplicaciones analíticas

do se destruye el complejo aminado, para darlugar a la base protonada y al ion metálico libre.Por otra parte, en medio básico aumenta la con-centración de iones hidroxilo, por lo que podráreaccionar con el ion metálico originando reac-ciones competitivas de hidrólisis, que disminui-rán la concentración de ion metálico libre endisolución, y por tanto, desplazarán el equilibriode complejación hacia la izquierda. En definiti-va, en ambos casos el pH origina una influencianegativa sobre el equilibrio de formación de com-plejos.
Además de estos equilibrios que dependendel pH, también pueden coexistir otros, en basea la presencia de más de un ligando, o más deun ion metálico, dando lugar a las ya estudiadasreacciones de desplazamiento. Todo ello ponede manifiesto que el cálculo de las concentra-ciones de las diferentes especies en disolución,será más complicado que el realizado a partirde las fracciones molares de cada especiehaciendo uso de las ecuaciones desarrolladas enel apartado 5.3.
5.5.1. Tratamiento general de la influenciadel pH en la formación de complejos
Sea el sistema constituido por un metal y unligando (M2+ y L2–), que pueden dar lugar a laformación de los complejos ML y ML2
2–, y queademás puedan existir: a) las formas protona-das HL– y H2L del ligando, y b) las especieshidroxiladas del metal M(OH)+ y M(OH)2, porejemplo.
Para calcular la concentración de cada una delas especies presentes en disolución en este sis-tema es necesario considerar los siguientes equi-librios con sus respectivas constantes:
H2L →← H+ + HL– [5.57]
HL– →← H+ + L2– [5.58]
M2+ + L2– →← ML [5.59]
M2+ + 2 L2– →← ML22– [5.60]
M2+ + OH– →← M(OH)+
[5.61]
M2+ + 2 OH– →← M(OH)2
[5.62]
Por otra parte, el sistema debe cumplir con lascondiciones de balances de masas para el metaly para el ligando. Estos se plantean a partir delconocimiento de las concentraciones analíticasde metal y ligando (CM y CL, respectivamente) ylas concentraciones de todas las posibles especiesen equilibrio:
CM = [M2+]+ [ML] + [ML22-] +
+ [M(OH)+] + [M(OH)2][5.63]
CL = [L2–] + [HL–] + [H2L] ++ [ML] + 2[ML2
2-] [5.64]
A la vista de estas ecuaciones se puede dedu-cir en una primera aproximación cualitativa, queun medio ácido favorece la protonación del ligan-do, y por tanto disminuye la concentración de laespecie L2– (ligando libre). El resultado final esque las reacciones de complejación [5.59] y [5.60]
β2,OH
22 2
[M(OH) ][M ][OH ]
=+ −
β1,OH 2
[M(OH) ][M ][OH ]
=+
+ −
β2 =
−
+ −
[ML ][M ][L ]
22
2 2 2
β1 2 2
[ML][M ][L ]
=+ −
Ka 2 =
+ −
−
[H ][L ][HL ]
2
Ka1 =
+ −[H ][HL ][H L]2
Capítulo 5: Equilibrios de formación de complejos 203

estarán poco favorecidas, mientras que un mediobásico favorece la existencia de la forma des-protonada del ligando, dando lugar en principioa una mayor tendencia a la formación de com-plejos, pero simultáneamente, también aumen-ta la concentración de los iones hidroxilo, quedan lugar a la formación de especies hidroxila-das del metal, lo que implica una disminución dela concentración de ion metálico libre (M2+), ypor tanto, una disminución de la formación delos complejos ML y ML2
2–. En la práctica, lainfluencia de la formación de especies hidroxi-ladas del metal en medio básico es menos rele-vante en el equilibrio de formación de comple-jos que la protonación del ligando en medioácido, con la excepción de que dicha especiehidroxilada constituya el hidróxido poco solubledel ion metálico. Este último caso, de gran inte-rés analítico, responde realmente al equilibriomixto triple ácido-base/formación de comple-jos/precipitación que será tratado en profundi-dad en el capítulo 7. A pesar de lo expuesto ycon objeto de realizar un enfoque general de lainfluencia del pH en el equilibrio de formaciónde complejos, el tratamiento que se desarrolla acontinuación contempla tanto la influencia deformación de especies hidroxiladas del metalcomo de la protonación del ligando.
La resolución del sistema de ecuaciones plan-teado (ecuaciones [5.57]-[5.64]), permite calcu-lar todas las concentraciones de las especies endisolución, a partir de las constantes de equili-brio, las concentraciones analíticas de metal y deligando y del pH de la disolución. Ello es perfec-tamente posible, ya que se dispone de un siste-ma de 8 ecuaciones con 8 incógnitas. No obstan-te, hay que tener en cuenta que estas ecuacionesno son lineales (es decir, existen términos de con-centración elevados a la primera y segunda poten-cias), y por tanto la resolución exacta obliga alempleo de métodos iterativos de aproximacionessucesivas. Si además se considera que en el mediopueden existir otros posibles ligandos en diso-lución (tales como amoniaco e iones cloruro, loque ocurre por ejemplo al utilizar un tampónNH3/NH4Cl para regular el pH de la disolución),
la resolución matemática de este equilibrio mix-to puede ser larga y complicada.
Si bien existen programas de cálculo especia-lizados que permiten la resolución de sistemascon varios metales y varios ligandos simultánea-mente, es habitual hacer uso de otra estrategiamás sencilla propuesta por Schwarzenbach, queconsiste en el empleo de constantes condiciona-les de equilibrio y de coeficientes de reacciónsecundaria que mantienen la rigurosidad necesa-ria en el tratamiento.
5.5.2. Coeficientes de reacción secundaria
Si se consideran los diferentes equilibriossimultáneos que pueden coexistir en disoluciónen el sistema M-L en estudio, se pueden definircomo reacciones principales las de formación delos complejos principales ML y ML2
2–, mientrasque las otras reacciones presentes en este equi-librio mixto se denominan reacciones secunda-rias, reacciones laterales o reacciones parásitas.
En este sentido, en las ecuaciones del balan-ce de masas ([5.63] y [5.64]), se puede distin-guir entre la concentración de metal que formaparte de los complejos principales, y del restode especies de ion metálico, así como entre laconcentración de ligando formando parte delos complejos principales y del que no formaparte de ellos. Si se denomina [M2+]′ y [L2–]′a las concentraciones de metal y de ligando queno forman parte de los complejos principales,los balances de masas indicados se transformanen:
[M2+]′ = CM – ([ML] + [ML22–]) =
= [M2+] + [M(OH)+] + [M(OH)2][5.65]
[L2–]′ = CL – ([ML] + 2[ML22–]) =
= [L2–] + [HL–] + [H2L] [5.66]
Al sustituir las concentraciones de las espe-cies indicadas en estas ecuaciones, en funciónde las concentraciones de los componentes libresy de las constantes de equilibrio correspon-
204 Equilibrios iónicos y sus aplicaciones analíticas

dientes (ecuaciones [5.57], [5.58], [5.61] y [5.62]),se obtiene:
[5.67]
[5.68]
La ecuación [5.68] se puede expresar tambiénen función de las constantes de protonación delligando. Conforme a la ecuación [5.22] se tiene:
[5.69]
Se define el coeficiente de reacción secunda-ria entre el metal y el ion hidroxilo, αM(OH), comola relación entre la concentración de metal queno forma parte de los complejos principales, lla-mada a veces concentración condicional delmetal, y la de ion metálico libre:
[5.70]
de donde:
[5.71]
Como se puede observar, este coeficientedepende de las constantes de hidrólisis del metaly del pH de la disolución. En general, a valoresde pH bajos, la concentración de ion hidroxiloserá muy pequeña y el coeficiente de reacciónsecundaria entre el metal y los iones OH– seráprácticamente igual a la unidad, lo que se corres-ponde con la no existencia de reacciones secun-darias.
De manera similar se puede definir un coefi-ciente de reacción secundaria entre el ligando y elion hidrógeno, αL(H), como la relación entre laconcentración de ligando que no forma parte delos complejos principales, llamada a veces con-centración condicional de ligando, y la concen-tración de ligando libre:
[5.72]
de donde:
[5.73]
Esta última ecuación también se puede escri-bir en función de las constantes de protonacióndel ligando, resultando la expresión:
[5.74]
muy similar a la ecuación [5.71] correspondien-te al coeficiente de reacción secundaria entre elmetal y el ion hidroxilo.
Como en el caso anterior, este coeficiente dereacción secundaria, entre el ligando y el ionhidrógeno, únicamente depende del pH de ladisolución y de las constantes de acidez del ligan-do. A partir de la ecuación [5.73], se puede apre-ciar que cuando la concentración de H+ de ladisolución sea muy inferior a la segunda cons-tante de disociación (es decir, cuando pH ≥ pKa2+ 2), el valor de este coeficiente es prácticamen-te igual a la unidad, indicando que las reaccionessecundarias con el ion hidrógeno serán despre-ciables y no será necesario tenerlas en cuenta.
α β βL(HH HH H) [ ] [ ]= + ++ +1 1 2
2
[ ] [ ]
] [ ]
αL(H)
H H
[H H
= + + =
= + +
+ +
+ +
12
2
1 2
21 1 2
1 2
K K K
K K KK K
a a a
a a a
a a
[[ ] [ ]
[ ]
αL(H)
2
2
22 2
2
[L ][L ]
L ]L ][H L ][H
L
= ′ =
=+ +
−
−
−− + − +
−
K K Ka a a2
2
1 2
α β βM(OH) 1,OH OH[OH OH= + +− −1 22] [ ],
[ ] ] ]
α
β β
M(OH)
2
2
21,OH
22,OH
2
2
[M ][M ]
M [M ][OH [M ][OH
[M ]
= ′ =
=+ +
+
+
+ + − + −
+
2
[L ] [L H ][L H ] [L2 2 H 2 H 2 2− − + − + −′ = + +] [ ] [ ]β β1 2
[ ][ ] [ ]
L ] [L
H ][L H ][L2 22 2
− −+ − + −
′ = + +K K Ka a a2 1 2
[ ] [ ] [ ][ ]
[ ],
,
M M M OH
M ][OH
2 2OH
2
OH2
+ + + −
+ −
′ = + ++
ββ
1
22
Capítulo 5: Equilibrios de formación de complejos 205

Para un caso general, en el que el ligando pue-de tener una serie de constantes de disociación(Ka1 , Ka2, ..., Kap) y el metal una serie de espe-cies hidroxiladas con unas constantes (β1,OH, β2,OH,..., βq,OH), sus correspondientes coeficientes dereacción secundaria vendrán dados por:
[5.75]
[5.76]
Hay que indicar que los valores de αM(OH) yαL(H) siempre serán iguales o superiores a la uni-dad, y así un incremento de los mismos se corres-ponde con una mayor influencia de la reacciónsecundaria correspondiente, mientras que si suvalor es cercano a la unidad, dicha influencia sepuede despreciar.
5.5.3. Constantes condicionales de formaciónde complejos
El término condicional se aplica a las cons-tantes de equilibrio para indicar que se trata deconstantes aparentes, esto es, que son realmen-te constante sólo en unas determinadas condi-ciones experimentales. Así, en los equilibriosconcurrentes de acidez y complejación se pue-de trabajar en unas condiciones en las que el pHde la disolución es constante, y por tanto, losvalores de αM(OH) y αL(H) también lo serán. Con-forme a este modo de operación, se puede plan-tear un equilibrio de complejación aparente enel cual la formación de los complejos ML y ML2,se origina a partir del ligando e ion metálico queno forman parte de los complejos principales,es decir:
M2+′ + L2–′ →← ML [5.77]
M2+′ + 2 L2–′ →← ML22– [5.78]
Las constantes que rigen estos equilibrios apa-rentes se denominan constantes condicionales deequilibrio, y se representan por β′1 y β′2, respec-tivamente:
[5.79]
[5.80]
Aunque en este caso se han indicado lasconstantes condicionales globales de formaciónde complejos, el tratamiento también se puederealizar haciendo uso de las constantes con-diciones de formación sucesivas indicadas porK′1 y K′2:
[5.81]
[5.82]
Una vez definidas estas constantes condicio-nales de complejación, es interesante establecersu relación con las constantes de equilibrio, nocondicionales. Así, por ejemplo, si en la ecua-ción [5.79], se sustituyen los valores de [M2+]′y [L2–]′ en función de los coeficientes de reac-ción secundaria y de las concentraciones demetal y ligando libre, ecuaciones [5.70] y [5.72],se obtiene la siguiente expresión de la constan-te condicional:
[5.83]
ecuación que, una vez reordenada, toma la for-ma:
[ ]] ]
′ =′ ′
=
=
+ −
+ −
β
α α
1
2
[ML][M ] [L ]
ML[M [L
2 2
M(OH) L(H)2
′ =′
−
−K2
[ML ][ML][L ]
22
2
′ = ′ =′ ′+ −K1 1β [ML]
[M ] [L ]2 2
′ =′ ′
−
+ −β2
[ML ][M ] [L ]
22
2 2 2
′ =
′ ′+ −β1
[ML][M ] [L ]2 2
[ ]
] [ ]
α ββ β
L(H)H
2H H
H
[H H
= + ++ + +
+
+ +
1 1
2 … pp
]
] [ ],
α ββ β
M(OH) 1,OH
2,OH OH
[OH
[OH OH
= + ++ + +
−
− −
12 � q
q
206 Equilibrios iónicos y sus aplicaciones analíticas

[5.84]
[5.85]
Esta expresión muestra la relación existenteentre la constante condicional de complejacióny la constante de equilibrio. Como se puede apre-ciar, la constante condicional depende del valorde β1 y de los valores de los coeficientes de reac-ción secundaria.
Al realizar el mismo tratamiento para la ecua-ción [5.80] se obtiene la expresión:
[5.86]
y si se considera que el ligando y el metal pue-den dar lugar a una serie de complejos, de este-quiometría ML, ML2, …, MLi, ..., MLn, la expre-sión general para la constante condicional globalde formación será:
[5.87]
siendo i la estequiometría de un determinadocomplejo.
Como los valores de αL(H) y αM(OH) seráncomo mínimo la unidad, es evidente que el valorde una constante condicional siempre será igualo menor que su constante de equilibrio corres-pondiente.
Por otra parte, hay que recordar que los valores obtenidos para estas constantes condi-cionales son válidos únicamente para unas condiciones determinadas, que en este caso co-rresponderían a un pH determinado de la diso-lución.
Si como se ha indicado anteriormente la for-mación de especies hidroxiladas con el ion metá-
lico no es significativa frente a la reacción prin-cipal de formación de complejos, αM(OH) → 1,la expresión general para la constante condicio-nal global de formación se transforma en:
[5.88]
5.5.4. Influencia de otros equilibriosconcurrentes
En el apartado anterior se han establecido lasconstantes condicionales en función de posiblesequilibrios simultáneos de protonación del ligan-do e hidrólisis del ion metálico. Sin embargo, eslógico suponer que en muchos casos será nece-sario considerar otros equilibrios adicionales. Porejemplo, si se trabaja a un valor de pH determi-nado será necesario adicionar al medio de reac-ción una disolución reguladora que permita sucontrol, lo que implica la adición de un ácido-base a la disolución, que puede dar lugar a la for-mación de complejos con el ion metálico M2+.Esta situación se origina en las valoracionescomplexométricas de diferentes iones metáli-cos con EDTA, dado que para regular el pH delas mismas se utiliza generalmente una disolu-ción tampón NH4Cl/NH3. La presencia de amo-niaco en el medio establece un equilibrio com-petitivo con el ion metálico dada la capacidaddel NH3 para formar complejos con un grannúmero de iones metálicos. Por otra parte, aun-que con menor relevancia, uno o más de los com-plejos principales del sistema (ML, por ejem-plo) podrían dar lugar a especies de hidrólisisdel tipo MLx(OH)y
–.A tenor de la importancia práctica de cada
uno de estos equilibrios secundarios, sólo seabordará en este estudio la posibilidad de que elmetal M2+, además de experimentar alguna reac-ción de hidrólisis, pueda formar complejos conotros ligandos (X e Y) presentes en la disolución,que dan lugar a las especies: MX, MX2, …, MXjy MY, MY2, …, MYk. En este tratamiento se con-
′ =β βαi i i
1( )L(H)
′ =β βα αi i i
1
M(OH L(H)) ( )
′ =β β
α α2 2 2
1
M(OH L(H)) ( )
′ =β βα α1 1
1
M(OH) L(H)
′ = + −βα α1
[ ]])
ML[M ] [LM(OH L(H)
2 2
Capítulo 5: Equilibrios de formación de complejos 207

sidera que las concentraciones libres de estosligandos son fijas y conocidas e iguales, respec-tivamente, a [X] e [Y]. Por otra parte, se omiti-rán las cargas en las ecuaciones correspondien-tes para mayor sencillez.
En estas condiciones, si los complejos con el ligando principal son ML y ML2, y las especiesde hidrólisis son M(OH) y M(OH)2, el balancede masas para el ion metálico se establece como:
CM = [M] + [ML] + [ML2]+ [M(OH)] ++ [M(OH)2] + [MX] + [MX2] + … [5.89]
... + [MXj] + [MY] + [MY2] + …+ [MYk]
Conforme a la definición de coeficiente dereacción secundaria: relación entre la concentra-ción de metal que no forma parte de los com-plejos principales y la de ion metálico libre, queen este caso ampliado llamaremos αM(TOT), éstevendrá dado por:
[5.90]
y al sustituir del balance de masas se tiene:
[5.91]
Al sustituir en esta ecuación los valores de lasconcentraciones de cada especie en función delas concentraciones libres de sus componentes yde las constantes de formación correspondien-tes, se obtiene:
[5.92]
que, simplificada, queda como:
[5.93]
Si se suman los coeficientes de reacción secun-daria correspondientes a las distintas reaccionessecundarias consideradas individualmente seobtiene:
[5.94]
Por tanto, se puede comprobar que:
[5.95]
Esta expresión indica que, para determinar elcoeficiente de reacción secundaria global del ionmetálico principal, solamente es necesario cono-cer los coeficientes de reacción secundaria corres-pondientes a las reacciones simultáneas con el res-to de ligandos. Para un caso general, en que elmetal M pueda tener reacciones parásitas con zposibles ligandos presentes en la disolución (X1,X2, …, XZ, el coeficiente de reacción secundariaglobal αM(TOT), se obtiene a partir de la expresión:
[5.96]
en la que los coeficientes de reacción secundariaindividuales, se obtienen a partir de las constan-tes de formación entre el metal y esos ligandos, yde sus respectivas concentraciones libres. En estascondiciones, la constante condicional global deformación para un determinado complejo de este-quiometría i viene dada por:
[5.97]′ =β βα αi i i
1
M(TOT) L(H)( )
α α α αM(TOT) M(X1) M(X2) M(XZ) ( 1)= + + + − −... z
α α α αM(TOT) M(OH) M(X) M(Y)= + + − 2
] ]
]
],
α α αβ β
β ββ β
M(OH) M(X) M(Y)
1,OH 2,OH
1,X J,XJ
Y K,YK
[OH [OH
[X [X]
[Y [Y]
+ + == + + +
+ + + ++ + +
3 2
1
��
α ββ β
β β β
M(TOT 1,OH
2,OH X
J,XJ
1,Y K,YK
[OH]
[OH [X
[X] [Y [Y]
)
,] ]
... ]
= + ++ +
+ + + + +
12
1 �
�
αβ β
β β
M(TOT)1,OH 2,OH
X Y
M [M][OH [M][OH
M[M][X [M][Y]
M
=+ + +
+ + + +
[ ] ] ]
[ ]
]
[ ], ,
2
1 1� �
αM(TOT)
M] [M(OH)] [M(OH MXM
MX MY MY
M
= + + + +
+ + + + +
[ ) [ ][ ]
[ ] [ ] [ ]
[ ]
2
� �j k
αM(TOT)
M 2([ML] [ML ])[M]
= − +C
208 Equilibrios iónicos y sus aplicaciones analíticas

Capítulo 5: Equilibrios de formación de complejos 209
Cuestiones
5.1. ¿A qué es igual la constante de formación global del complejo MLn?
[ ] Al producto del número de ligandos y de la concentración libre de ligando
[ ] Al producto de todas las constantes sucesivas de formación
[ ] Al inverso de la constante de disociación global
5.2. Los complejos quelatos son mucho más estables que sus equivalentes complejos de adición por:
[ ] Su diferente estequiometría
[ ] El mayor cambio de entropía que se produce en su formación
[ ] La diferente carga que pueden presentar en disolución
5.3. Si se pone en contacto una disolución del complejo FeF2+ con otra de Al3+ se origina una reacción de des-plazamiento denominada:
5.4. En la formación del complejo ML2, siendo L un anión procedente de un ácido débil, la influencia del pH semanifiesta de la siguiente forma:
[ ] A pH básico se favorece la formación del complejo
[ ] En medio ácido se desfavorece la formación del complejo
[ ] El pH no ejerce influencia en la formación de dicho complejo
5.5. En la reacción de formación del complejo CaY2– a pH = 10 ajustado con una disolución reguladora NH4Cl/NH3,¿qué coeficientes de reacción secundaria se han de considerar?
5.6. En el equilibrio de formación del complejo ML3, αL(H), representa:
[ ] Una forma de considerar la influencia del pH sobre este equilibrio
[ ] La fracción molar del complejo
[ ] El número de coordinación
[ ] La fracción molar de ion metálico
[ ] Un parámetro que permite calcular la constante de formación condicional
5.7. ¿Qué significado tiene [M]′ en la reacción de formación de complejo M + L →← ML
5.8. Se hace uso de la constante condicional de formación de complejos en función del pH cuando:
[ ] El ligando es un anión procedente de un ácido fuerte
[ ] La temperatura ejerce una determinada influencia en el equilibrio
[ ] El ion metálico forma complejos hidroxilados

5.9. El pH es una variable muy importante a la hora de estudiar un equilibrio de formación de complejos. Indi-car si son verdaderas (V) o falsas (F) cada una de estas frases:
[ ] Con el pH se puede incrementar la constante de formación de un complejo
[ ] A pH ácido se puede impedir la formación de un complejo
[ ] Un pH básico no influye en la formación de un complejo
[ ] Si disminuye el pH disminuye la constante de formación condicional
[ ] Si aumenta el pH disminuye la constante de formación condicional
5.10. Indicar la expresión que permite calcular la constante global de protonación para el ligando H2L (Ka1, Ka2).
210 Equilibrios iónicos y sus aplicaciones analíticas
• Objetivo pedagógicoDesarrollo de diferentes tipos de problemas sobre:formación de complejos con ligandos monodentadosy polidentados, cálculo de la concentración de lasdiferentes especies presentes en estos equilibrios yequilibrios concurrentes de acidez y de formación decomplejos.
1. Determinar la concentración de ion Ag+ en una diso-lución de AgNO3 0,01 mol L–1 que también contieneNH3, de concentración 0,1 mol L–1. Despreciar la formación de la especie Ag(NH3)
+.Ag(NH3)2
+ (log β2 = 7,24).
De acuerdo con las condiciones dadas, gran exce-so de amoniaco frente al ion Ag+, se puede despre-ciar la formación del complejo Ag(NH3)
+ y, por tan-to, se tiene la siguiente reacción de formación decomplejos:
Ag+ + 2 NH3→← Ag(NH3)2
+
cuya constante de formación global viene dada por:
Se pueden establecer los siguientes balances demateria:
y llevar a cabo la siguiente aproximación en el balan-ce de materia de la plata a tenor de las concentra-ciones respectivas de Ag+ y NH3:
de donde:
Al sustituir la concentración de complejo en elbalance de materia del amoniaco, se puede calcularla concentración de NH3 libre en disolución:
Por fin, la concentración de plata libre se determinasustituyendo las concentraciones calculadas de com-plejo y amoniaco en la constante de formación global:
de donde [Ag+] = 8,91 × 10–8 mol L–1.
2. El ion Ag+ forma con el amoniaco dos complejos,AgNH3
+ y Ag(NH3)2+, cuyas constantes sucesivas de
β2
7 24 3 2
32 2
100 01
0 08= = =
+
+ +, [ ( ) ]
[ ][ ],
[ ]( , )Ag NHAg NH Ag
[ ] [ ] [ ( ) ] , , ,NH NH Ag NH
molLlibre total3 3 3 2
1
20 1 2 0 01 0 08
= − == − × =
+
−
[ ( ) ] ,Ag NH molL3 210 01+ −≅
[ ] [ ( ) ]Ag Ag NHlibre+ +<< 3 2
[ ] , [ ] [ ( ) ]NH molL NH Ag NHtotal libre31
3 3 20 1 2= = +− +
[ ] , [ ] [ ( ) ]Ag molL Ag Ag NHtotal libre+ − + += = +0 01 1
3 2
β27 24 3 2
32
10= =+
+, [ ( ) ]
[ ][ ]Ag NHAg NH
Seminarios: problemas numéricos

formación son: K1 = 103,32 y K2 = 103,92. Calcular lasconcentraciones de las diferentes especies químicasen disolución cuando se mezclan volúmenes igualesde disoluciones de Ag+ 0,2 mol L–1 y NH3 0,2 mol L–1.Sol.: [Ag+] = [Ag(NH3)2
+] = 0,04 mol L–1; [Ag(NH3)+]
= 0,02 mol L–1; [NH3] = 2,0 × 10–4 mol L–1.
3. A una disolución del ion Cd2+ de concentración 0,01mol L–1 se le añade amoniaco y cianuro de potasio detal forma que la concentración de ambos ligandos traslas correspondientes reacciones de complejación conel ion metálico es 1,0 mol L–1. Calcular la relaciónentre las concentraciones de ambos complejos:[Cd(CN)4
2–] / [Cd(NH3)42+].
Cd(CN)42– (log β4 = 18,9); Cd(NH3)4
2+ (log β4 = 6,85).Sol.: 1,12 × 1012.
4. El ion In3+ y el ion Cl– forman los complejos siguien-tes: InCl2+, InCl2
+ y InCl3. A partir de sus constantesglobales de formación (log β1 = 2,20, log β2 = 3,56 ylog β3 = 3,92), calcular las concentraciones de todaslas especies presentes cuando se prepara una disolu-ción que es 1,00 mol L–1 en HCl y 0,01 mol L–1 enIn(NO3)3. En estas condiciones no es necesario con-siderar las especies de hidrólisis del In3+.
Para la resolución de este ejercicio se parte de losbalances de masa del In3+ y del Cl–. Dado que se cono-cen las concentraciones totales de ambos y las espe-cies que se pueden formar, se tiene un sistema de dosecuaciones en el que existen dos incógnitas (las con-centraciones libres de los iones cloruro e indio), queserá necesario calcular iterativamente:
CCl = [Cl–] + [InCl2+] + 2 [InCl2+] + 3 [InCl3] = 1,00
CIn = [In3+] + [InCl2+] + [InCl2+] + [InCl3] = 0,01
Paso 1: en primer lugar, se puede suponer que todoel metal está complejado en forma del complejo 3:1;a partir de aquí podemos calcular cuál es la concen-tración de ligando libre a partir de su balance de masa:
CCl = [Cl–] + 3 [ InCl3] = [Cl–] + 3 CIn
[Cl–] = CCl – 3 × CIn = 1,00 – 0,03 = 0,97 mol L–1
Paso 2: ahora es posible calcular las concentracio-nes de cada una de las especies de metal, a partir dela concentración total de éste, de las correspondien-tes constantes de formación y de la concentraciónlibre de ion cloruro estimada:
Paso 3: sustituyendo ahora estos valores en elbalance de masa del ion cloruro se obtiene un nuevovalor para la concentración de ligando libre:
Paso 4: este nuevo valor obtenido se utiliza nue-vamente como valor inicial en el Paso 2, repitiendolos cálculos correspondientes. En este caso se obtie-ne el mismo valor de partida, es decir, una concen-tración estimada de ion cloruro de 0,9733 mol L–1,por lo que se considera que el sistema ya está resuel-to. Los valores finales de concentración para cadaespecie serán, por tanto:
[Cl–] = 0,9733 mol L–1
[InCl2+] = 1,37 × 10–4 mol L–1
[InCl3] = 6,81 × 10–3 mol L–1
[H+] = 1,00 mol L–1
[In3+] = 8,88 × 10–7 mol L–1
[InCl2+] = 3,05 × 10–3 mol L–1
[NO3
_] = 0,03 mol L–1
5. Se tiene una disolución 0,85 mol L–1 del complejoHgBr4
2_. Calcular la concentración de los iones Hg2+
y Br– en la disolución, y la cantidad de bromuro desodio que debe añadirse a 500 mL de la disolución
[ClmolL
− − −
− −
= − × − × × −− × × =
] , , , , ,
1 00 1 38 10 2 3 06 103 6 80 10 0 9733
4 3
3 1
[ ] [ ] [ [Cl InCl InCl ] InCl ]Cl2
2 3− + += − − −C 2 3
[ ][ ]
[ ] [ ] [ ] ,
InClCl
Cl Cl ClmolL
In33
3
1 22
33
3 1
16 80 10
=+ + +
=
= ×
−
− − −
− −
Cβ
β β β
[ ][ ]
[ ] [ ] [ ] ,
InClCl
Cl Cl ClmolL
2 In+
−
− − −
− −
=+ + +
=
= ×
Cβ
β β β2
2
1 22
33
3 1
13 06 10
[ ][ ]
[ ] [ [ ] ,
InClCl
Cl Cl ] ClmolL
2In 2
+−
− − −
− −
=+ + +
=
= ×
Cβ
β β β1
1 2 33
4 1
11 38 10
=+ × + +
=
= × − −
0 011 10 0 97 10 0 97 10 0 978 96 10
2 2 3 56 2 3 92 3
7 1
,, ( , ) ( , )
,
, , ,
molL
[ ]
[ ] [ ] [ ]In
Cl Cl ClIn3
1 22
33
11
+− − −=
+ + +=C
β β β
Capítulo 5: Equilibrios de formación de complejos 211

anterior para reducir la concentración de ion Hg2+ ala mil millónesima parte de su valor original. Nota: suponer que solamente se forma el complejoHgBr4
2_.
HgBr42_
(log β4 = 21,6).Peso molecular: NaBr = 102,9.
Las expresiones de los balances de masa de los ionesmercurio y bromuro, en este caso, son las siguientes:
Dado que la concentración total de bromuro es cua-tro veces superior a la de mercurio, a tenor de la rela-ción estequiométrica en el complejo, se puede esta-blecer la igualdad:
CBr = 4 CHg
Sustituyendo en las expresiones de los balances demasa, se tiene:
Si se considera ahora la constante de equilibrio β4, y se sustituye la concentración de ion mercurio librepor la de bromuro, se llega a la siguiente expresión:
Dado que el valor de la constante de formación esmuy elevado, se puede suponer que prácticamentetodo el mercurio está en forma de complejo, y portanto la concentración de ion libre comparada con eltotal es despreciable, quedando la expresión de laconstante de formación como:
En estas condiciones, se puede calcular la concen-tración de mercurio libre directamente, y, a partir deella, la de iones bromuro:
La adición de ion Br_
a la disolución anterior favo-rece aún más la formación del complejo, disminuyen-do la concentración de ion Hg2+, por lo que ésta será:
En estas condiciones la concentración de complejosigue siendo la inicial, esto es, 0,85 mol L–1, con lo quela [Br
_]libre tras la adición del NaBr viene dada por:
de donde [Br_] = 1,07 × 10–2 mol L–1.
La concentración de bromuro añadida correspon-derá a la suma de la libre en el equilibrio tras añadirNaBr y de la necesaria para que el mercurio inicial pasea formar complejo por efecto de ion común, esto es:
Finalmente, la cantidad de NaBr que debe disol-verse en 500 mL de la disolución del complejo (nohay variación de volumen) se calcula de acuerdo con:
y por tanto, hay que adicionar 0,551 g de bromurode sodio.
6. Un cierto ion metálico M2+ forma con el ligando L–
dos complejos, ML+ y ML2, cuyas constantes sucesi-vas de formación son K1 y K2. Demostrar que la máxi-ma concentración de ML+ se obtiene para una con-centración de ligando, tal que .
7. Una disolución del complejo K2HgI4 se prepara disol-viendo 0,03 moles del complejo y diluyendo a 250 mL
[ ]L− = K K1 2
1,07 10NaBr–2× = g /
,103
0 500
[ ] , ( , ) ,Br
mol L–
− − −
−
= × + × × ≅≅ ×
añadida 1 07 10 4 1 5 101 07 10
2 5
2 1
10
0 851 5 10
21 6 42
4 14 4, [ ]
[ ],
, [ ]= =
×
−
+ − − −
HgBrHg ][Br Br2
[[ ] ,
,
Hg ]Hg
mol L
2final
2inicial
–
++ −
−
= = × =
= ×10
1 5 1010
1 5 10
9
5
9
14 1
[ ] [ ] ,Br Hg mol L–1− + −= × = ×4 6 0 102 5
[ ],
,
,Hg
mol L
Hg
–1
24
4
54 21 6
5
5
40 85
4 10
1 5 10
+
−
=×
=×
=
= ×
C
β
β4 4 2 54
=× +
CHg
Hg[ ]
β4 4
2
4 2 54
= = =
=×
=−
×
−
+ −
−
+ +
−
+
+
+
[ ][ ]
[ ]
[ ]
HgBrHg ][Br
[HgBr ][Hg ](4[Hg ])
[HgBr ]4 [Hg ]
Hg
Hg
42
242
2 2 4
42
4 2 5
HgC
[ [Br ] 4 Hg ]2− +=
[ [Br ] [HgBr ] 4 Hg ] [HgBr ] − − + −+ = +4 442 2
42
CBr Br ] [HgBr ] = +− −[ 4 42
CHg2
42Hg ] [HgBr ]= ++ −[
212 Equilibrios iónicos y sus aplicaciones analíticas

con agua destilada. Calcular la concentración de losdiferentes iones en disolución (suponer que solo seforma el complejo K2HgI4).K2HgI4 (log β4 = 29,7).Sol.: [HgI4
2_] = 0,12 mol L–1; [Hg2+] = 2,48 × 10–7 mol L–1;
[I–] = 9,92 × 10–7 mol L–1; [K+] = 0,24 mol L–1.
8. El ion Cd2+ y el Cl– forman los complejos siguientes:CdCl+, CdCl2, CdCl3
_y CdCl4
2_. Sus constantes globa-
les de formación son, respectivamente: log β1 = 1,32,log β2 = 2,22, log β3 = 2,31 y log β4 = 1,85. Calcular lasconcentraciones de todas las especies presentes enuna disolución que es 1,00 mol L–1 en HCl y 0,01 molL–1 en Cd(NO3)2. En estas condiciones no es necesa-rio considerar las especies de hidrólisis del Cd2+.Sol.: [Cl–] = 0,9732 mol L–1;
[Cd2+] = 2,32 × 10−5 mol L–1; [CdCl+] = 4,73 × 10−4 mol L–1;[CdCl2] = 3,65 × 10−3 mol L–1;[CdCl3
_] = 4,37 × 10−3 mol L–1;
[CdCl42_
] = 1,48 × 10−3 mol L–1;[NO3
_] = 0,02 mol L–1; [H+] = 1,00 mol L–1.
9. Calcular la concentración de NH3 en una disolución0,1 mol L–1 del complejo Co(NH3)6
3+, suponiendo queéste es el único complejo formado.Co(NH3)6
3+ (log β6 = 33,6).Sol.: 1,47 × 10–5 mol L–1.
10. La concentración de ion Ag+ libre en una disolución0,1 mol L–1 del complejo Ag(CN)2
_es de 3 × 10–8 mol
L–1. Calcular cuál sería la concentración de ion Ag+ sila concentración de CN– aumenta hasta 10–2 mol L–1.
El equilibrio de formación del complejo de dicia-no argentato (I) viene dado por:
Ag+ + 2 CN– →← Ag(CN)2_
cuya constante de formación global será:
Dado que se conoce la concentración de Ag+, en elequilibrio, 3 × 10–8 mol L–1, y que se parte de una diso-lución 0,1 mol L–1, se tiene:
[Ag+] = 3 × 10–8 mol L–1
CAg = [Ag+] + [Ag(CN)2_]
[Ag(CN)2_] = 0,1 – 3 × 10–8 = 0,0999 mol L–1
CCN_ = [CN_] + 2[Ag(CN)2
_]
[CN–] = 0,2 – 2(0,1– 3 × 10–8) = 6 × 10–8 mol L–1
Al sustituir estas concentraciones en la expresiónde la constante de equilibrio se tiene:
de donde β2 = 9,26 × 1020.Si la concentración de ion cianuro se incrementa
hasta 10–2 mol L–1, la concentración de Ag+ en estascondiciones se determina a partir de la constante deequilibrio, cuyo valor ya es conocido:
con lo que resulta una concentración de ion plata de1,09 × 10–18 mol L–1.
11. Calcular el porcentaje de ion Al3+ en una disolución0,02 mol L–1 del complejo AlF6
3_. ¿Cuál será su valor
si la disolución contiene además NaF, a una concen-tración de 0,05 mol L–1? (Suponer que sólo se formael complejo AlF6
3_).
AlF63_
(log β6 = 19,7).Sol.: 0,94% y 1,25 × 10–10%.
12. En medio ácido fuerte 0,128 mol L–1 la constante dela reacción Fe3+ + SCN– →← FeSCN2+ vale 234. A unadisolución de Fe3+ 0,01 mol L–1 en medio ácido 0,128mol L–1 se le añade tiocianato de sodio hasta que laconcentración [SCN–] + [FeSCN2+] = 0,01 mol L–1.Calcular la concentración de cada una de las especies,Fe3+, SCN– y FeSCN2+, en la disolución resultante.Sol.: [Fe3+] = [SCN–] = 4,7 × 10–3 mol L–1; [FeSCN2+]
= 5,3 × 10–2 mol L–1.
13. Si se supone que las especies Fe3+ y SCN– formanun solo complejo, FeSCN2+: 1) Calcular las concen-traciones de las diversas especies químicas presen-tes en una disolución de Fe3+ y SCN– cuyasconcentraciones analíticas son 10–3 y 0,1 mol L–1,respectivamente. 2) ¿Cuáles serán las concentra-ciones de estas especies si la disolución inicial sediluye 10 veces?FeSCN+ (log β = 2,3).
9 26 10
0 10 01
202 2
,[
[,
[ ] ( , )× = =
−
+ − +
Ag(CN) ]Ag ][CN ] Ag
2
β2 2 8 8 2
0 09993 10 6 10
= =× ×
−
+ − − −
[ ][ ]
,( )
Ag(CN)Ag ][CN
2
β22
2=
−
+ −
[ ) ][ ]
Ag(CNAg ][CN
Capítulo 5: Equilibrios de formación de complejos 213

Sol.:1) [Fe3+] = 5,0 × 10–5 mol L–1; [SCN–] =
= 9,9 × 10–2 mol L–1; [FeSCN2+] = 9,5 × 10–4 mol L–1.2) [Fe3+] = 3,3 × 10–5 mol L–1; [SCN–] =
= 9,9 × 10–2 mol L–1; [FeSCN2+] = 6,7 × 10–4 mol L–1.
14. Si se mezclan disoluciones de Fe3+ 10–3 mol L–1 y deSCN– 0,1 mol L–1 se observa el color rojo del comple-jo FeSCN2+ a partir de una concentración del mismo≥ 10–5,5 mol L–1. Calcular la cantidad de fluoruro desodio que habría que añadir a esta disolución parahacer desaparecer dicha coloración roja por formacióndel complejo FeF2+. Despreciar la influencia del pH.FeSCN2+ (log β = 2,3); FeF2+ (log β = 5,5).
La reacción de desplazamiento que se produceentre ambos complejos de hierro viene dada por:
FeSCN2+ + F– →← FeF2+ + SCN–
De acuerdo con lo expuesto en el apartado 5.3.2,la constante de equilibrio de esta reacción de inter-cambio de ligando se expresa como:
lo que indica que el equilibrio se encuentra despla-zado hacia la formación del complejo FeF2+ debidoa su mayor constante de formación. Se pueden esta-blecer los siguientes balances de materia:
[Fe3+]total = [Fe3+]libre + [FeSCN2+] + [FeF2+][SCN–]total = [SCN–]libre + [FeSCN2+]
[F–]añadido = [F–]libre + [FeF2+]
Dado que se ha de añadir ion fluoruro en la canti-dad necesaria para hacer desaparecer la coloraciónroja de complejo FeSCN2+, y que ésta es perceptiblea concentraciones ≥ 10–5,5 mol L–1, al sustituir estevalor de concentración en los balances de materia deSCN– e Fe3+ se tiene:
0,1 = [SCN–]libre + 10–5,5 ⇒ [SCN–]libre ≅ 0,1 mol L–1
10–3 = [Fe3+]libre + 10–5,5 + [FeF2+] ⇒⇒ [FeF2+] ≅ 10–3 mol L–1
si se considera que la concentración de Fe3+ libre esdespreciable debido a la formación de los complejoscon los iones SCN– y F–. Sustituyendo estas concen-traciones en la expresión de la constante de equilibrio:
de donde [F–]libre = 2,0 × 10–2 mol L–1.La concentración de ion fluoruro añadido se cal-
cula finalmente a partir del balance de materia yaestablecido:
[F–]añadido = [F–]libre + [FeF2+] == 2,0 × 10–2 + 10–3 = 2,1 × 10–2 mol L–1
La concentración de ion fluoruro que hay que aña-dir deberá ser superior a la indicada con objeto deevitar la coloración roja del complejo FeSCN2+.
15. Indicar, razonando las respuestas, qué reacciones ocu-rren cuando se mezclan: 1) LiY3– + Ca2+, 2) BaY2– +Ca2+, 3) FeF2+ + Al3+ y 4) FeF2+ + Y4–.LiY3– (log β = 2,8); CaY2– (log β = 10,7);BaY2– (log β = 7,8); FeY– (log β = 25,1);FeF2+ (log β1 = 5,5); AlF2+ (log β1 = 6,1).Sol.: Se forma YCa2– en 1) y 2); 3) se produce la
reacción de desplazamiento pero no de mane-ra cuantitativa, y 4) se forma FeY–.
16. Se mezclan 10–2 moles de Ba2+, 10–2 moles de Fe3+ y5 × 10–3 moles de EDTA (Y4–), y se diluyen a 1 litro.Calcular las concentraciones de las diferentes espe-cies en disolución. Despreciar la influencia del pH enla formación de los complejos con EDTA.BaY2– (log β = 7,8); FeY– (log β = 25,1).
En la disolución se pueden establecer los siguien-tes equilibrios de formación de complejos con suscorrespondientes constantes:
Ba2+ + Y4– →← BaY2–
Fe3+ + Y4– →← FeY–
Dado que el complejo de hierro es mucho más esta-ble que el de bario y existe defecto de EDTA en elmedio de reacción se puede asumir que únicamentede forma el complejo FeY–. En estas condiciones, losbalances de materia para las especies Fe3+ y el EDTAvienen dados por:
[Fe3+]total = [Fe3+] + [FeY–] = 0,01 mol L–1
[Y4–]total = [Y4–] + [FeY–] = 0,005 mol L–1
βFe Y 3 4
[FeY ][Fe ][Y ]−
−
+ −= = 1025 1,
βBa Y
2
2 4
[BaY ][Ba ][Y ]−
−
+ −= = 107 8,
10
10 0 110
3 23
5 5,
,
,[ ]
= = ×+ −
+ −
−
− −
[FeF ][SCN ][FeSCN ][F ] F
2
2
KIL
2
2fluoruro
tiocianato
[FeF ][SCN ][FeSCN ][F ]
= = = =+ −
+ −
ββ
1010
105 5
2 33 2
,
,,
214 Equilibrios iónicos y sus aplicaciones analíticas

Si se desprecia [Y4–] en el balance de materia delEDTA se tiene que:
[Fe3+] = [FeY–] = 0,005 mol L–1
Al sustituir estas concentraciones en la constantede formación global del complejo FeY– se puededeterminar concentración de EDTA libre:
de donde [Y4–] = 7,94 × 10–26 mol L–1.En el caso del complejo BaY2–, dado que éste prác-
ticamente no se forma, se puede considerar que la[Ba2+] es prácticamente su concentración inicial, esto,es 0,01 mol L–1. La concentración de bario en formacompleja se determina a partir de su constante de for-mación global:
y resulta [BaY2–] = 5,0 × 10–20 mol L–1.En resumen, las concentraciones de las diferen-
tes especies en el equilibrio son:
[Ba2+] = 10–2 mol L–1 ; [Fe3+] = [FeY–] = 5 × 10–3 mol L–1;[Y4–] = 7,9 × 10–26 mol L–1; [BaY2–] = 5 × 10–20 mol L–1 y[Ba2+] = 10–2 mol L–1.
17. Se mezclan 50 mL de una disolución que contiene 0,1mol L–1 de ion Cu2+ con 50 mL de una disolución quecontiene 0,1 mol L–1 de etilendiamina (en). Calcularla concentración de todas las especies presentes enla disolución.Cu(en)2+ (log β1 = 10,6); Cu(en)2
2+ (log β2 = 19,7).Sol.: [en] = 10–9,85 mol L–1 ; [Cu2+] = [Cu(en)2
2+] = 10–2,18
mol L–1; [Cu(en)2+] = 10–1,43 mol L–1.
18. Se prepara una disolución mezclando 200 mL de Ca2+
10–3 mol L–1, 400 mL de Mg2+ 10–3 mol L–1 , 200 mLde EDTA 10–2 mol L–1 diluyendo a un volumen finalde 1 litro. Calcular cuáles son las concentraciones delas diferentes especies químicas presentes en estamezcla.CaY2– (log β = 10,7); MgY2– (log β = 9,1).Sol.: [Ca2+] = 2,86 × 10–12 mol L–1;
[CaY2–] = 2,0 × 10–4 mol L–1; [Y4–] = 1,4 × 10–3 mol L–1;
[MgY2–] = 4,0 × 10–4 mol L–1;[Mg2+] = 2,26 × 10–10 mol L–1.
19. Calcular el valor de la relación [CaY2–] / [LiY3–] cuan-do se mezclan volúmenes iguales de disolución 0,1mol L–1 de LiY3– y 0,2 mol L–1 del ion Ca2+ y se alcan-za el equilibrio.CaY2– (log β = 10,7); LiY3– (log β = 2,8).Sol.: 7,94 × 107.
Equilibrios concurrentes de acidezy formación de complejos
20. Calcular el porcentaje de metal libre ([M2+]) que exis-te en una disolución 0,1 mol L–1 complejo ML2, conel pH ajustado a 4,0.ML2 (log β2 = 6,0); HL (pKa = 4,0).
En este equilibrio concurrente de complejación yacidez se pueden diferenciar las siguientes reaccio-nes regidas por sus respectivas constantes:
M2+ + 2 L– →← ML2
HL →← L– + H+
De acuerdo con el tratamiento realizado en el apar-tado 5.5.1 sobre la influencia del pH en la formaciónde complejos, el enfoque de este equilibrio concu-rrente implica el uso de la constante condicional deformación del complejo, β2′, cuyo valor se determinaa partir de la constante de formación global del mis-mo, β2, y del coeficiente de reacción secundaria delligando, αL(H), dado que el problema sólo considerala influencia del pH en esta especie. Así:
Al sustituir los valores dados en el problema seobtiene el valor de β2′:
La concentración de ion metálico libre, [M2+], eneste equilibrio concurrente se puede determinar a
′ =
+=
+= ×+
−
− −β β2
22
2
6 4 2
4 4 2510 10
10 102 5 10
( )( [ ])
( )( )
,K
Ka
a H
′ = =+ +β β
αβ
22
22
2
2L(H H)
( )( [ ])
K
Ka
a
Ka =
− +[L ][H ][HL]
β2 =
+ −
[ML ][M ][L ]
22 2
βBa Y
2
2 4
[BaY ][Ba ][Y ]
BaY−
−
+ −
−
−= = =
× ×10
0 01 7 94 107 8
2
26, [ ]
, ,
βFe Y 3 4
[FeY ][Fe ][Y ] Y−
−
+ − −= = =10
0 0050 005
25 14
, ,, [ ]
Capítulo 5: Equilibrios de formación de complejos 215

partir de la expresión de la constante condicional deformación global:
de donde, al despreciar [M2+] frente a 0,1 en estaexpresión, se tiene: [M2+] = 4,64 × 10–3 mol L–1. Elporcentaje de metal libre se determina a partir de laexpresión:
el cual al ser inferior al 5% confirma la validez de lasuposición realizada.
21. Calcular el porcentaje de ion Al3+ de una disolución0,1 mol L–1 del complejo AlF6
3_a pH 1,0 (se conside-
ra solamente la formación de este complejo).AlF6
3_(log β6 = 19,7); HF (pKa = 3,52).
Sol.: 0,83%.
22. El ion Ag+ forma con el amoniaco el complejoAg(NH3)2
+ cuya constante de formación global es β2= 107,24. Calcular el pH que ha de tener una disolu-ción 0,1 mol L–1 en amoniaco y en el ion Ag+ para quesólo el 1% de la plata presente en la misma esté enforma compleja. Despreciar la especie Ag(NH3)
+.NH4
+ (pKa = 9,24).Sol.: 2,02.
23. A una disolución del ion Fe3+ de concentración 10–3
mol L–1 se le añade tiocianato de sodio y fluoruro desodio hasta que la concentración analítica de SCN–
es 0,1 mol L–1 y de F– 1,0 mol L–1. Calcular el valorde pH al cual es perceptible la coloración roja delcomplejo FeSCN2+, esto es cuando se concentraciónes ≥ 10–5,5 mol L–1.FeSCN2+ (log β = 2,3); FeF2+ (log β = 5,5);HF (pKa = 3,52).
En este equilibrio concurrente de acidez y forma-ción de complejos se pueden establecer los siguien-tes balances de materia:
[Fe3+]total = [Fe3+] + [FeSCN2+] + [FeF2+] = 10–3 mol L–1
[SCN–]total = [SCN–] + [FeSCN2+] = 0,1 mol L–1
[F–]añadido = [F–]′ + [FeF2+] = 1,0 mol L–1
Dado que para que sea perceptible la concentra-ción de FeSCN2+ debe ser ≥ 10–5,5 mol L–1, al sustituiresta condición en los balances de materia de los ionesFe3+ y SCN– se tiene:
10–3 = [Fe3+]′ + 10–5,5 + [FeF2+]0,1 = [SCN–] + 10–5,5 ≅ [SCN–]
En estas condiciones se puede determinar la con-centración de Fe3+ en disolución sin más que sustituirlos valores de las [FeSCN2+] y [SCN–] en su corres-pondiente constante de formación global:
y resulta [Fe3+] = 1,58 × 10–7 mol L–1.A tenor del valor de esta concentración y de acuer-
do con el balance de materia del ion Fe3+, la concen-tración de FeF2+ se puede estimar en 10–3 mol L–1. Alsustituir esta concentración en el balance de materiadel ion fluoruro se tiene:
[F–]′ = 1,0 – 10–3 = 0,999 mol L–1
En estas condiciones se puede determinar fácilmen-te el pH de la disolución haciendo uso de la constantecondicional de formación global del complejo FeF2+:
de donde pH = 1,83 en base a la simplificación válidade que [H+] >> Ka.
Dado que a valores de pH superiores a 1,83 seincrementa la concentración de ion fluoruro libre endisolución y por tanto su capacidad para formar com-plejos con Fe3+, la coloración roja del complejoFeSCN2+ será perceptible para pH ≤ 1,83.
24. Si se mezcla Ba2+ 10–2 mol L–1, Fe3+ 10–2 mol L–1 yEDTA 1,5 × 10–2 mol L–1, calcular las concentracionesde las diferentes especies químicas en la disolución final.Despreciar la influencia del pH en estos equilibrios.BaY2– (log β = 7,8); FeY– (log β = 25,1).Sol.: [Ba2+] = [BaY2–] = 5,0 × 10–3 mol L–1; [Y4–] =
1,6 × 10–8 mol L–1; [FeY–] = 10–2 mol L–1; [Fe3+]= 4,96 × 10–20 mol L–1.
′ = =+
= × =
=′
=× ×
+
−
+
+
+ −
−
−
β βα
βF(H)
3
H H
FeFFe F
K
Ka
a[ ] [ ]
[ ]
[ ][ ] , ,
, ,10 10
101 58 10 0 999
5 5 3 52
2 3
7
β = = =
+
+ −
−
+
[FeSCN ][Fe ][SCN ]
10[Fe ] 0,1
2
3
5,5
3102 3,
%[
,
,, %
M ][M ]
[ML ] [M ]2
2
22
++
+
−
=+
× =
= × × =
100
4 64 100 1
100 4 643
′ =
′= × = −
+ −
+
+ +β252 5 10
[ML ][M ][L ]
0,1 [M ][M ](2[M ])
22 2
2
2 2 2,
216 Equilibrios iónicos y sus aplicaciones analíticas

25. Determinar cuál deberá ser el pH mínimo para queen una disolución que contiene 10–2 mol L–1 de ionFe3+ y 0,02 mol L–1 de EDTA, el ion metálico estécuantitativamente complejado (más del 99,9%).FeY– (log β = 25,1); EDTA (pKa1 = 2,0; pKa2 = 2,7;pKa3 = 6,1; pKa4 = 10,2).
Conforme a las condiciones dadas en el proble-ma, es fácil calcular las concentraciones de las dife-rentes especies implicadas en este equilibrio, y portanto, la constante condicional de formación delcomplejo FeY–. Así:
Al sustituir estas concentraciones en la expresiónde la constante se tiene:
de donde αY(H) = 1,26 × 1020.Este valor elevado de αY(H) indica que nos encon-
tramos en una zona de pH francamente ácido, conlo cual el EDTA se encontrará en su forma total-mente protonada YH4. Este valor de αY(H) confor-me a lo indicado en el apartado 5.5.2, responde a laexpresión:
de la cual se puede hacer uso sólo del último términodado el valor encontrado para este coeficiente. Así,
siendo log β4Η = 21,0 de acuerdo con los valores dados
de pKa del EDTA. Resolviendo la ecuación anteriorresulta un valor de pH mínimo de 0,22.
26. Justificar la variación de los valores de las constan-tes de formación condicionales de los complejos deCa2+ y Cu2+ con EDTA en función del pH, represen-tadas en la figura.
27. Se prepara una disolución mezclando 100 mL denitrato de calcio 10–2 mol L–1, 100 mL de nitrato debario 10–2 mol L–1 y 500 mL de YH2Na2 2 × 10–2 molL–1 a pH = 7,0. Esta disolución se diluye a un litro yse reajusta el pH a 7,0. Calcular la concentración deY4– en el equilibrio.YCa2– (log β = 10,7); YBa2– (log β = 7,8);EDTA (α4 pH 7,0 = 10–2).Sol.: 2,2 × 10–12 mol L–1.
α βY(H 4H [H) , ]= × ≅ +1 26 1020 4
α β β β βY(HH H H HH H H H) [ ] [ ] [ ] [ ]= + + + ++ + + +1 1 2
23
34
4
′ =′
= ××
=
= × = =
−
+ −
−
−β
βα α
[FeY ][Fe ][Y ]3 4
Y(H) Y(H)
9 99 1010 0 01
9 99 1010
3
5
425 1
,,
,,
[Y ] [Y ] – [FeY ] , – , , mol L
4– 4–total
–
– –1
′ = == × = 0 02 9 99 10 0 013
[FeY ] Fe
mol L
– 3total
–
= × =
= = ×
+
−
99 999 9100
0 01 9 99 10 3 1
, % [ ]
,
, ,
[Fe ] Fe
mol L
3+ 3total
–
= × =
= =
+
−
0 010 1100
0 01 10 5 1
, % [ ]
,
,
Capítulo 5: Equilibrios de formación de complejos 217


6
6.1. Introducción6.2. Curvas de valoración con EDTA6.3. Indicadores metalocrómicos6.4. Tipos de valoraciones complexométricas6.5. AplicacionesCuestionesSeminarios: problemas numéricos
VALORACIONESDE FORMACIÓNDE COMPLEJOS
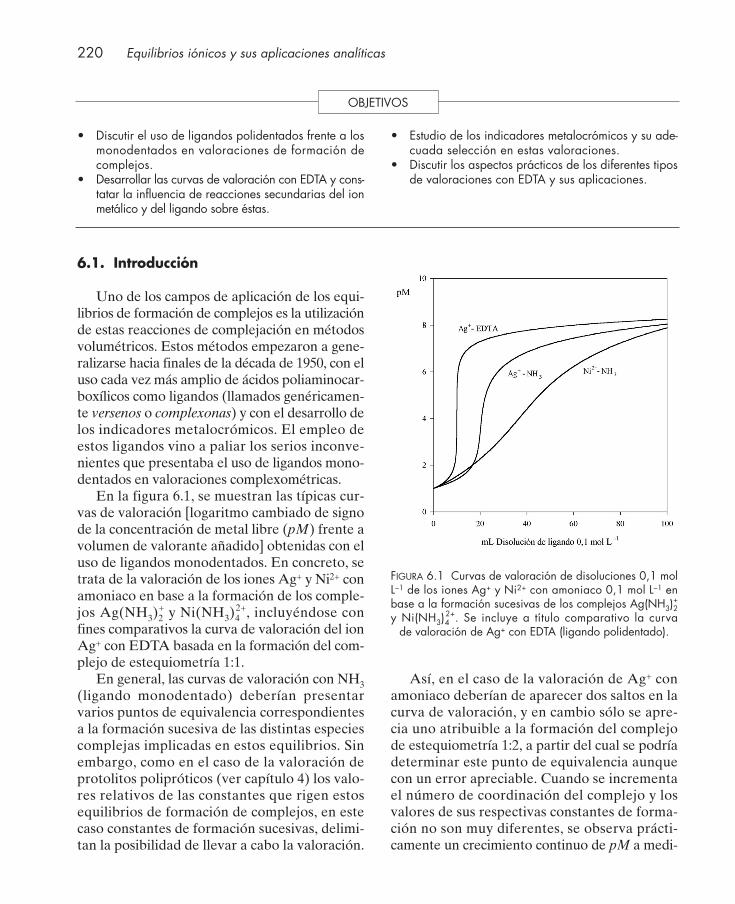
6.1. Introducción
Uno de los campos de aplicación de los equi-librios de formación de complejos es la utilizaciónde estas reacciones de complejación en métodosvolumétricos. Estos métodos empezaron a gene-ralizarse hacia finales de la década de 1950, con eluso cada vez más amplio de ácidos poliaminocar-boxílicos como ligandos (llamados genéricamen-te versenos o complexonas) y con el desarrollo delos indicadores metalocrómicos. El empleo deestos ligandos vino a paliar los serios inconve-nientes que presentaba el uso de ligandos mono-dentados en valoraciones complexométricas.
En la figura 6.1, se muestran las típicas cur-vas de valoración [logaritmo cambiado de signode la concentración de metal libre (pM) frente avolumen de valorante añadido] obtenidas con eluso de ligandos monodentados. En concreto, setrata de la valoración de los iones Ag+ y Ni2+ conamoniaco en base a la formación de los comple-jos Ag(NH3)2
+ y Ni(NH3)42+, incluyéndose con
fines comparativos la curva de valoración del ionAg+ con EDTA basada en la formación del com-plejo de estequiometría 1:1.
En general, las curvas de valoración con NH3(ligando monodentado) deberían presentarvarios puntos de equivalencia correspondientesa la formación sucesiva de las distintas especiescomplejas implicadas en estos equilibrios. Sinembargo, como en el caso de la valoración deprotolitos polipróticos (ver capítulo 4) los valo-res relativos de las constantes que rigen estosequilibrios de formación de complejos, en estecaso constantes de formación sucesivas, delimi-tan la posibilidad de llevar a cabo la valoración.
FIGURA 6.1 Curvas de valoración de disoluciones 0,1 molL–1 de los iones Ag+ y Ni2+ con amoniaco 0,1 mol L–1 enbase a la formación sucesivas de los complejos Ag(NH3)2
+
y Ni(NH3)42+. Se incluye a título comparativo la curva
de valoración de Ag+ con EDTA (ligando polidentado).
Así, en el caso de la valoración de Ag+ conamoniaco deberían de aparecer dos saltos en lacurva de valoración, y en cambio sólo se apre-cia uno atribuible a la formación del complejode estequiometría 1:2, a partir del cual se podríadeterminar este punto de equivalencia aunquecon un error apreciable. Cuando se incrementael número de coordinación del complejo y losvalores de sus respectivas constantes de forma-ción no son muy diferentes, se observa prácti-camente un crecimiento continuo de pM a medi-
220 Equilibrios iónicos y sus aplicaciones analíticas
• Discutir el uso de ligandos polidentados frente a losmonodentados en valoraciones de formación decomplejos.
• Desarrollar las curvas de valoración con EDTA y cons-tatar la influencia de reacciones secundarias del ionmetálico y del ligando sobre éstas.
• Estudio de los indicadores metalocrómicos y su ade-cuada selección en estas valoraciones.
• Discutir los aspectos prácticos de los diferentes tiposde valoraciones con EDTA y sus aplicaciones.
OBJETIVOS

Capítulo 6: Valoraciones de formación de complejos 221
da que progresa la valoración (ver valoraciónde Ni2+ con amoniaco en figura 6.1) sin la apa-rición del salto típico en el punto de equivalen-cia, por lo que es imposible la valoración del ionmetálico. En consecuencia, el uso de los equili-brios de complejación metal-ligando en valora-ciones se limita a aquellos sistemas en los quese forma una sola especie compleja con unaapreciable constante de formación global, talcomo en el caso de ligandos polidentados detipo ácidos poliaminocarboxílicos. Así, en lavaloración de Ag+ con EDTA (ligando poli-dentado) mostrada en la figura 6.1, se observaque la formación del complejo Ag-EDTA deestequiometría 1:1 origina un cambio más defi-nido en pM en el punto de equivalencia que per-mite la determinación cuantitativa de este ionmetálico.
Desde un punto de vista práctico, las valo-raciones requieren de indicadores visuales ade-cuados para conseguir la determinación cuanti-tativa del analito con un error aceptable. Estossistemas de indicación del punto final son muyescasos en valoraciones de formación de com-plejos con el uso de ligandos monodentados.Realmente, la introducción de los indicadoresmetalocrómicos supuso un paso muy importan-te en el desarrollo de este tipo de valoraciones,aunque sólo son adecuados para detectar el pun-to final cuando se emplean ligandos polidenta-dos como valorantes. Aunque con escaso inte-rés en la actualidad, es interesante comentarbrevemente algunos de los indicadores más carac-terísticos que se han utilizado en valoraciones deformación de complejos con el uso de ligandosmonodentados. Uno de los ejemplos más clási-cos es la determinación de cianuros por el méto-do de Liebig basado en la aparición de un com-puesto insoluble (sistema indicador) en el puntofinal. El método consiste en la adición de nitra-to de plata desde la bureta a una disolución quecontiene ion cianuro. Mientras exista algo decianuro sin valorar, tiene lugar la reacción devaloración:
Ag+ + 2 CN– →← Ag(CN)2
_[6.1]
Cuando se alcanza el punto de equivalencia,dado que la concentración de ion Ag+ se incre-menta rápidamente se produce la formación dela especie insoluble de color blanco
Ag(CN)2
_+ Ag+ →← Ag[Ag(CN)2](S) [6.2]
originando una tenue turbidez indicadora delpunto final de la valoración.
Retomando el uso de complexonas (ligandospolidentados) en valoraciones de formación decomplejos, el más sencillo de estos compuestos,como se muestra en la figura 6.2, es el ácido imi-no diacético (IDA). Este ligando da lugar a com-plejos quelatos, ya que presenta tres puntos decoordinación (un grupo amino y dos grupos car-boxilo).
FIGURA 6.2. Estructura química de ligandos polidentados del tipo poliaminocarboxílico.
Tal como se ha comentado en el capítulo ante-rior, uno de los factores de los que depende laconstante de estabilidad de un complejo, es la

posibilidad de formación de quelatos; además,cuanto mayor es el número de anillos de 5 o 6eslabones formados, más estable es el complejo.Por tanto, la utilización de ligandos que permi-tan esta posibilidad, dará lugar a la formación decomplejos muy estables. Es el caso del EDTA(con 6 puntos de coordinación), el que, al formarlos complejos con iones metálicos, da lugar engeneral a 5 anillos (ver capítulo 5), así como delácido etilenglicol-bis-2-amino etiléter tetraacéti-co (EGTA) y del ácido 1,2-diamino ciclohexanotetraacético (DCTA). En este sentido, ademásde EDTA, también se utilizan otros ligandospoliaminocarboxílicos con mayor número de gru-pos de coordinación, como es el caso del ácidodietilen triamino pentaacético (DTPA), que tie-ne 8 puntos de coordinación. Las estructuras deestas complexonas se muestran así mismo en lafigura 6.2.
Otra característica de los ácidos poliamino-carboxílicos como ligandos, es el hecho de tenerun gran número de puntos de coordinación, loque permite que una sola molécula de ligandoocupe todas (o casi todas) las posiciones de coor-dinación de un ion metálico. El resultado es quela estequiometría de los complejos formados es,en general, de 1:1 (complejos ML). Por tanto, lasreacciones de formación de complejos con ácidospoliaminocarboxílicos presentan unas caracterís-ticas que las hacen particularmente apropiadaspara ser aplicadas como reactivos valorantes enmétodos volumétricos, ya que cumplen los reque-rimientos necesarios para toda reacción de valo-ración:
1. Cuantitativa. La constante de formaciónglobal de estos complejos suele ser muyelevada, dando lugar a reacciones cuanti-tativas.
2. Estequiométrica. La estequiometría de lareacción está perfectamente definida (1:1),no existiendo la formación de especies conestequiometría diferente. Debido a los dosfactores indicados, la variación del pM(–log [M]), en las cercanías del punto deequivalencia, es lo suficientemente gran-
de como para determinar el punto final dela valoración con un error aceptable.
3. Rápida. En general, estas reacciones sonademás de cinética rápida (sobre todo conlos iones metálicos divalentes), de formaque se utiliza directamente el ligando comovalorante. En algunos casos (complejoscon Cr3+ o Al3+, por ejemplo) se trata dereacciones lentas, aunque no por ello dejande ser cuantitativas, y se utilizan métodosde valoración por retroceso, previa adiciónde un exceso medido de ligando, como semostrará posteriormente.
4. Disponibilidad de sistema indicador. El usode indicadores metalocrómicos, sensiblesa cambios bruscos de pM, permite la fácildetección del punto final en este tipo devaloraciones.
De los ácidos poliaminocarboxílicos indica-dos en la figura 6.2, es el EDTA el más amplia-mente utilizado en valoraciones complexométri-cas, por lo que en este capítulo se trataránexclusivamente las valoraciones de formación decomplejos en las que se utiliza esta complexonacomo valorante.
6.2. Curvas de valoración con EDTA
El ácido etilendiamino tetracético (EDTA)es un ácido poliprótico que se suele designarcomo H4Y, ya que contiene cuatro hidrógenosdisociables (los correspondientes a los gruposcarboxílicos), cuyos valores de pKa son: pKa1 =2,0; pKa2 = 2,7; pKa3 = 6,1; pKa4 = 10,2. La formatotalmente disociada del EDTA (es decir, el ligan-do libre) se representa como Y4–.
Si a partir de las constantes de disociación delEDTA, se calcula su diagrama de distribución de especies, conforme a lo expuesto en el capí-tulo 2, se obtiene la gráfica representada en lafigura 6.3a, en la que se observa que la formatotalmente desprotonada del ligando se obtienea partir de valores de pH > 12 (como se deducedel valor de su pKa4 = 10,2); por tanto, a valores
222 Equilibrios iónicos y sus aplicaciones analíticas
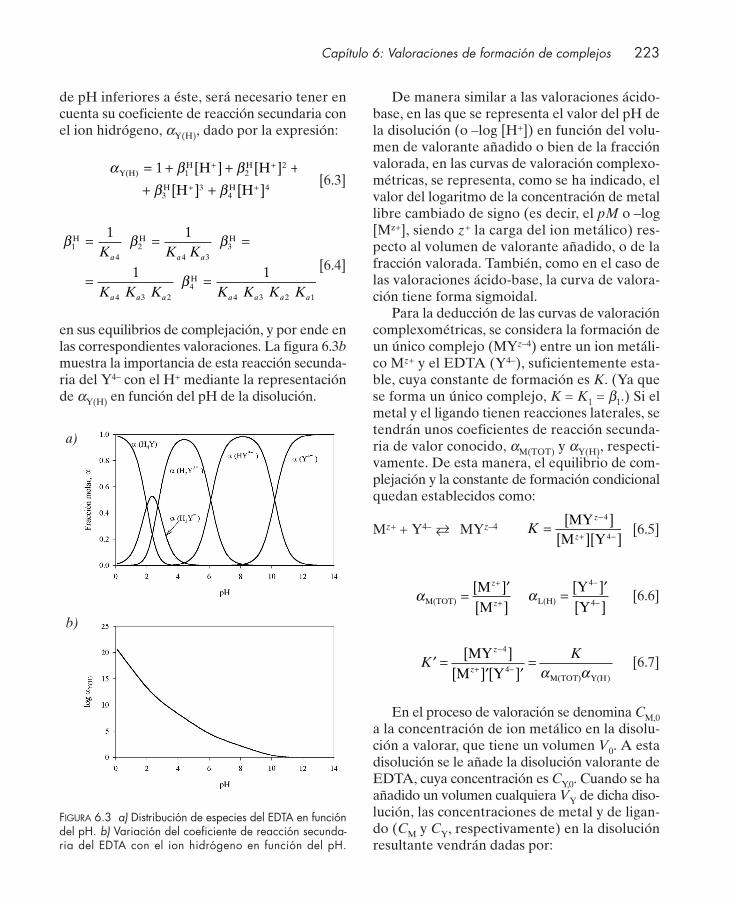
de pH inferiores a éste, será necesario tener encuenta su coeficiente de reacción secundaria conel ion hidrógeno, αY(H), dado por la expresión:
[6.3]
[6.4]
en sus equilibrios de complejación, y por ende enlas correspondientes valoraciones. La figura 6.3bmuestra la importancia de esta reacción secunda-ria del Y4– con el H+ mediante la representaciónde αY(H) en función del pH de la disolución.
FIGURA 6.3 a) Distribución de especies del EDTA en funcióndel pH. b) Variación del coeficiente de reacción secunda-ria del EDTA con el ion hidrógeno en función del pH.
De manera similar a las valoraciones ácido-base, en las que se representa el valor del pH dela disolución (o –log [H+]) en función del volu-men de valorante añadido o bien de la fracciónvalorada, en las curvas de valoración complexo-métricas, se representa, como se ha indicado, elvalor del logaritmo de la concentración de metallibre cambiado de signo (es decir, el pM o –log[Mz+], siendo z+ la carga del ion metálico) res-pecto al volumen de valorante añadido, o de lafracción valorada. También, como en el caso delas valoraciones ácido-base, la curva de valora-ción tiene forma sigmoidal.
Para la deducción de las curvas de valoracióncomplexométricas, se considera la formación deun único complejo (MYz–4) entre un ion metáli-co Mz+ y el EDTA (Y4–), suficientemente esta-ble, cuya constante de formación es K. (Ya quese forma un único complejo, K = K1 = β1.) Si elmetal y el ligando tienen reacciones laterales, setendrán unos coeficientes de reacción secunda-ria de valor conocido, αM(TOT) y αY(H), respecti-vamente. De esta manera, el equilibrio de com-plejación y la constante de formación condicionalquedan establecidos como:
Mz+ + Y4– →← MYz–4 [6.5]
[6.6]
[6.7]
En el proceso de valoración se denomina CM,0a la concentración de ion metálico en la disolu-ción a valorar, que tiene un volumen V0. A estadisolución se le añade la disolución valorante deEDTA, cuya concentración es CY,0. Cuando se haañadido un volumen cualquiera VY de dicha diso-lución, las concentraciones de metal y de ligan-do (CM y CY, respectivamente) en la disoluciónresultante vendrán dadas por:
′ =′ ′
=−
+ −KKz
z
[ ][ ] )
MY[M ] Y M(TOT) Y(H
4
4 α α
αL(H)
[Y ]Y
= ′−
−
4
4[ ] αM(TOT)
[M ][M
= ′+
+
z
z ]
K
z
z=
−
+ −
[MY ][M ][Y ]
4
4
β β β
β
14
24 3
3
4 3 24
4 3 2 1
1 1
1 1
H H H
H
= = =
= =
K K K
K K K K K K K
a a a
a a a a a a a
α β ββ β
Y(H)H
2H
3H
4H
H [H
[H [H
= + + ++ +
+ +
+ +
1 12
3 4
[ ] ]
] ]
Capítulo 6: Valoraciones de formación de complejos 223
a)
b)

[6.8]
El volumen de valorante añadido en el pun-to de equivalencia (Veq), teniendo en cuenta quese forma únicamente el complejo 1:1, es igual a
[6.9]
Para construir la curva de valoración, se repre-senta pM en función de determinados volúme-nes de valorante añadidos, que variarán habi-tualmente entre VY = 0 y VY = 2 × Veq, o bien enfunción de la fracción valorada (F = VY/Veq), encuyo caso ésta varía entre F = 0 y F = 2. Para ello,se tienen en cuenta los balances de materia delmetal y del ligando, que en cualquier punto dela valoración vienen dados por:
CM = [Mz+]′ + [MYz–4] [6.10]
CY = F × CM = [Y4–]′ + [MYz–4] [6.11]
Para llevar a cabo este estudio, se ha seguidoun procedimiento similar al descrito para las cur-vas de neutralización en el capítulo 4, esto es,descomponer la curva de valoración en variaszonas y desarrollar las ecuaciones adecuadas paracada una de ellas. Así, se consideran zonas deinterés de la curva de valoración:
– Punto inicial (antes de añadir la disoluciónde valorante).
– Antes del punto de equivalencia (zona condefecto de valorante).
– Punto de equivalencia.– Después del punto de equivalencia (zona
con exceso de valorante).
En cada una de estas zonas se consideran lasespecies predominantes, y se establecen las expre-siones para el cálculo del pM en las mismas a par-tir de la fracción valorada en cada momento.
Dado que es de gran interés obtener la variaciónde pM en función del volumen de valorante aña-dido, las expresiones deducidas se han transfor-mado adecuadamente haciendo uso de la rela-ción: VY = F × Veq.
Tras el trazado de la curva de valoración, sediscuten ejemplos concretos correspondientes ala influencia de reacciones secundarias en la valo-ración: influencia del pH tanto en el ion metáli-co como en el EDTA.
Zona 1: punto inicial (F = 0)
Antes de cualquier adición de EDTA, sóloestá presente la cantidad inicial de ion metálicoen la disolución, por lo que el pM depende úni-camente de su concentración, esto es:
[6.12]
Zona 2: antes del punto de equivalencia(0 < F < 1)
En esta zona se tiene un exceso de metal, yaque el ligando añadido está por debajo de la rela-ción estequiométrica: CM > CY. Si la constantede formación es lo suficientemente elevada, casogeneral en la práctica, la concentración de ligan-do que no forma parte del complejo principal([Y4–]′) es despreciable frente a la concentraciónde complejo MYz–4, por lo que el balance demasas se reduce a:
CY = F × CM = [Y4–]′ + [MYz–4] ≅ [MYz–4] [6.13]
Combinando las ecuaciones [6.10] y [6.13]se obtiene:
CM = [Mz+]′ + F × CM [6.14]
de donde:
[Mz+]′ = CM – F × CM = CM(1 – F) [6.15]
pMC
C
z= − = −
=
= − +
+log[ ] log
log log
,M M
M(TOT)
M, M(TOT)
0
0
αα
V
C V
CeqM,
Y,
= 0 0
0
C
C V
V VYY, Y
Y
=+0
0 C
C V
V VMM,
Y
=+0 0
0
224 Equilibrios iónicos y sus aplicaciones analíticas

Al sustituir esta ecuación en la [6.6], se tie-ne la expresión para la concentración de metallibre:
[6.16]
expresión de la que se deduce directamente elvalor de pM:
[6.17]
Para obtener la expresión correspondiente alpM en función del volumen de valorante añadi-do basta con sustituir en la ecuación [6.16] elvalor de F por VY/Veq, con lo que resulta:
[6.18]
[6.19]
Se puede considerar que en esta zona, pMsolamente varía con la fracción valorada, o ensu caso con el volumen de EDTA añadido, yaque CM y el coeficiente de reacción secundariason prácticamente constantes. Nótese ademásque en esta zona, antes del punto de equiva-lencia, no tienen influencia las reacciones secun-darias del ligando. En los casos en los que elmetal no tenga reacción secundaria, el términoαM(TOT) es igual a la unidad, y por tanto su loga-ritmo se anula.
Zona 3: punto de equivalencia (F = 1)
En este punto de la valoración se ha añadidola cantidad estequiométrica de ligando, por tanto
CM = CY, y al igualar los balances de masas de lasecuaciones [6.10] y [6.11] se tiene la expresión:
[Mz+]′ + [MYz–4] = [Y4–]′ + [MYz–4] [6.20]
en la que se elimina la concentración de complejoa ambos lados de la ecuación, quedando:
[Mz+]′ = [Y4–]′ [6.21]
Al sustituir esta igualdad en la expresión dela constante condicional (ecuación [6.7]), éstaqueda de la forma:
[6.22]
de donde se puede despejar el valor de [Mz+]′:
[6.23]
Si además se supone que el complejo es muyestable, y que prácticamente todo el metal estáen forma compleja con EDTA, esto es [MYz–4]≅ CM, la concentración de metal libre en el pun-to de equivalencia, tras combinar las ecuaciones[6.6], [6.7] y [6.23] y sustituir en ésta la condicióndada, se calcula como:
[6.24]
y a partir de aquí:
[6.25]pM C
K
= − − +
+ +
12
log12
logM Y(H)
M(TOT)
α
α log log12
12
[ ][ ]
MM
M(TOT) M(TOT)
M
M Y(H)
M(TOT)
zz C
K
C
K
++
= ′ =′
=
=
α α
αα
1
[[ ]
M ]MYz
z
K+
−
′ =′
4
′ =
′
−
+Kz
z
[MY ][M ]
4
2
pM
CV V
V
z= − =
= − + −−
+log ]
log log log
[M
( )M M(TOT)eq Y
eq
α
[ ]
[ ]M
M
M(TOT)
Meq Y
eq
M(TOT)
zz
CV V
V++
= ′ =
−
α α
pM
C F
= − == − + − −
+log[M
log log( ) log(1 )
z
M M(TOT)
]
α
[ ]
[ ]M
M ( F)
M(TOT)
M
M(TOT)
zz C+
+
= ′ = −α α
1
Capítulo 6: Valoraciones de formación de complejos 225

Como se aprecia en esta expresión, el valorde pM en el punto de equivalencia viene deter-minado por la concentración total de metal, laconstante de equilibrio y los coeficientes dereacción secundaria. Si no existen reaccioneslaterales, αM(TOT) = αY(H) = 1 y por tanto suslogaritmos son iguales a cero y se eliminan dela ecuación [6.25].
Zona 4: exceso de valorante (F > 1)
Una vez pasado el punto de equivalencia dela valoración, se está añadiendo un exceso deligando respecto a la cantidad estequiométricade ion metálico. Si el complejo es lo suficiente-mente estable como para poder despreciar sudisociación, se puede considerar que práctica-mente la totalidad de ion metálico está en formacompleja, con lo que los balances de materia demetal y ligando quedan como:
CM = [Mz+]′ + [MYz–4] ≅ [MYz–4] [6.26]
CY = F × CM = [Y4–]′ + [MYz–4] == [Y4–]′ + CM
[6.27]
La combinación de estas ecuaciones permi-te deducir la expresión para calcular la concen-tración de ligando en exceso, que viene dadapor:
[Y4–]′ = CM (F – 1) [6.28]
Al sustituir este valor en la expresión de laconstante condicional (ecuación [6.7]), teniendoen cuenta que prácticamente todo el metal estácomo complejo MYz–4, se tiene la [Mz+]′:
[6.29]
[6.30]
Al combinar esta ecuación con las ecuaciones[6.6] y [6.7] se tiene:
[6.31]
[6.32]
de donde finalmente:
[6.33]
o bien en función del volumen de valorante aña-dido:
[6.34]
Estas expresiones indican que la concentra-ción libre de ion metálico después del punto deequivalencia depende de la constante de equili-brio, el exceso de ligando y de su coeficiente dereacción secundaria, siendo independiente de laconcentración total de metal. Es de interéscomentar que cuando se ha añadido el doble deligando respecto a la relación estequiométrica (F = 2 o VY = 2 Veq), y en el caso de que el ligan-do no tenga reacción secundaria (αY(H) = 1), elvalor de pM coincide con el del logaritmo de laconstante de equilibrio.
En el recuadro 6.1 se resumen las ecuacionesobtenidas para la determinación de pM en cadauna de las zonas de la curva de valoración com-plexométrica con EDTA.
pM
KV V
V
z= − =
= − +−
+log ]
log log log
[M
Y(H)Y eq
eq
α
pM
K F
z= − == − + −
+log ]
log log log
[M
( )Y(H)α 1
[ ]M
( )L(H)z
K F+ =
−α
1
[M ] [M ] 1
( )
1
( )
M(TOT) M(TOT)
M(TOT) L(H)M(TOT)
zz
K F
KF
++
= ′ =′ −
=
=−
α α
α αα
1
1
[M ]
( )z
K F+ ′ =
′ −1
1
′ =
′ ′=
′ −
−
+ − +KCC F
z
z z
[ ][
MY[M ] Y ] [M ] ( )
4M
M4 1
226 Equilibrios iónicos y sus aplicaciones analíticas
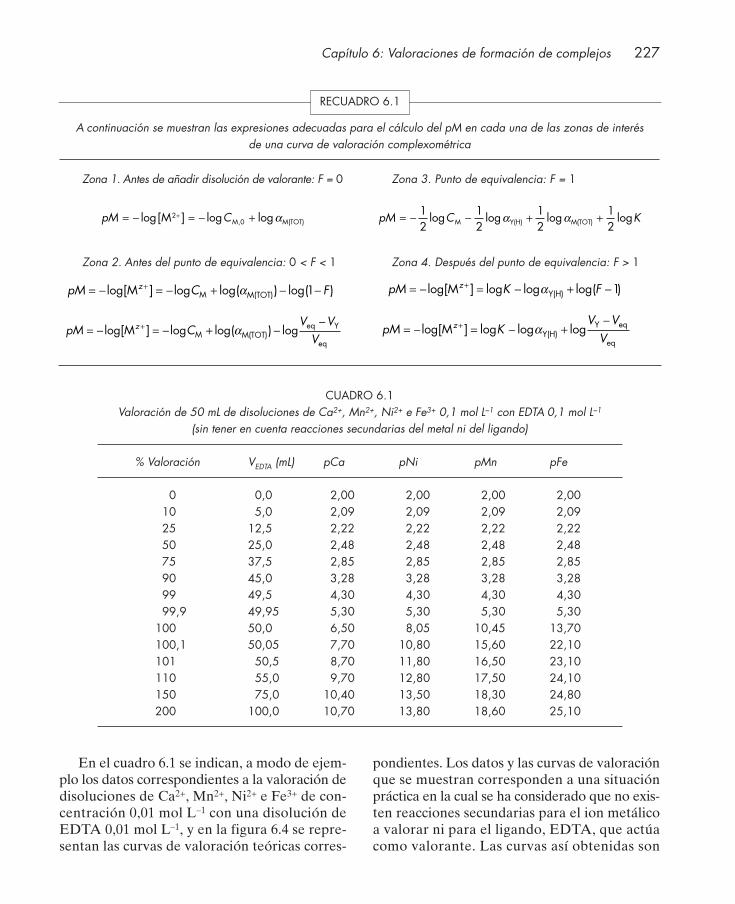
Zona 1. Antes de añadir disolución de valorante: F = 0
Zona 2. Antes del punto de equivalencia: 0 < F < 1
Zona 3. Punto de equivalencia: F = 1
Zona 4. Después del punto de equivalencia: F > 1
pM KV V
Vz= − = − +
−+log ] log log log[M Y(H)Y eq
eqα
pM K Fz= − = − + −+log ] log log log[M ( )Y(H)α 1
pM C K= − − + +1
212
12
12
log log log logM Y(H) M(TOT)α α
pM CV V
Vz= − = − + −
−+log ] log log log[M ( )M M(TOT)eq Y
eqα
pM C Fz= − = − + − −+log ] log log log[M ( ) ( )M M(TOT)α 1
pM C= − = − ++log[ ] log log,M M M(TOT)2
0 α
Capítulo 6: Valoraciones de formación de complejos 227
RECUADRO 6.1
A continuación se muestran las expresiones adecuadas para el cálculo del pM en cada una de las zonas de interésde una curva de valoración complexométrica
En el cuadro 6.1 se indican, a modo de ejem-plo los datos correspondientes a la valoración dedisoluciones de Ca2+, Mn2+, Ni2+ e Fe3+ de con-centración 0,01 mol L–1 con una disolución deEDTA 0,01 mol L–1, y en la figura 6.4 se repre-sentan las curvas de valoración teóricas corres-
pondientes. Los datos y las curvas de valoraciónque se muestran corresponden a una situaciónpráctica en la cual se ha considerado que no exis-ten reacciones secundarias para el ion metálicoa valorar ni para el ligando, EDTA, que actúacomo valorante. Las curvas así obtenidas son
CUADRO 6.1Valoración de 50 mL de disoluciones de Ca2+, Mn2+, Ni2+ e Fe3+ 0,1 mol L–1 con EDTA 0,1 mol L–1
(sin tener en cuenta reacciones secundarias del metal ni del ligando)
% Valoración VEDTA (mL) pCa pNi pMn pFe
0 0,0 2,00 2,00 2,00 2,0010 5,0 2,09 2,09 2,09 2,0925 12,5 2,22 2,22 2,22 2,2250 25,0 2,48 2,48 2,48 2,4875 37,5 2,85 2,85 2,85 2,8590 45,0 3,28 3,28 3,28 3,2899 49,5 4,30 4,30 4,30 4,3099,9 49,95 5,30 5,30 5,30 5,30
100 50,0 6,50 8,05 10,45 13,70100,1 50,05 7,70 10,80 15,60 22,10101 50,5 8,70 11,80 16,50 23,10110 55,0 9,70 12,80 17,50 24,10150 75,0 10,40 13,50 18,30 24,80200 100,0 10,70 13,80 18,60 25,10
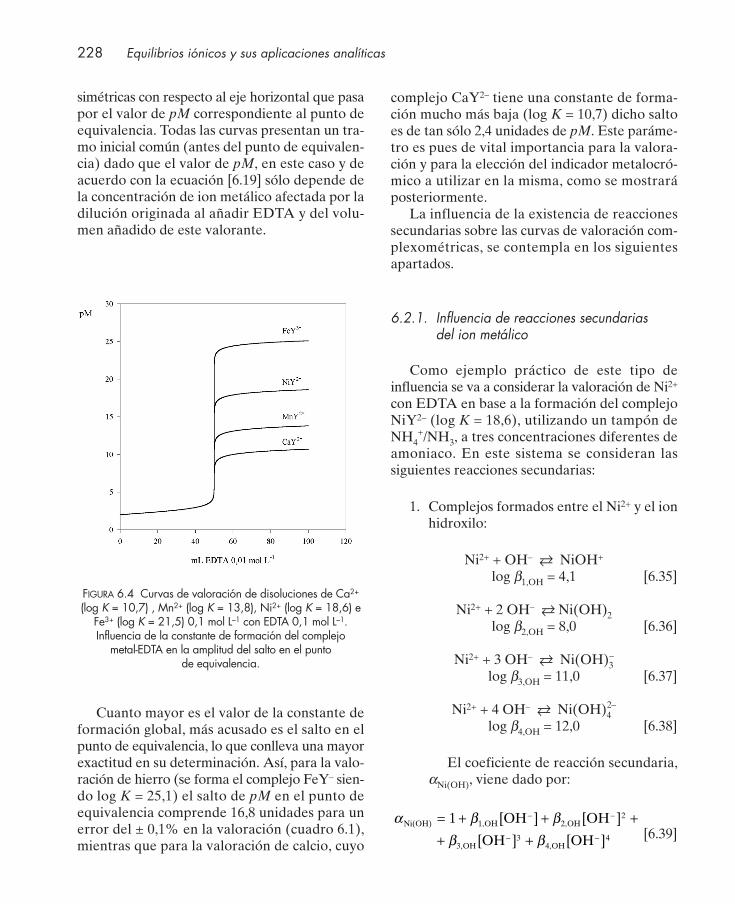
simétricas con respecto al eje horizontal que pasapor el valor de pM correspondiente al punto deequivalencia. Todas las curvas presentan un tra-mo inicial común (antes del punto de equivalen-cia) dado que el valor de pM, en este caso y deacuerdo con la ecuación [6.19] sólo depende dela concentración de ion metálico afectada por ladilución originada al añadir EDTA y del volu-men añadido de este valorante.
FIGURA 6.4 Curvas de valoración de disoluciones de Ca2+
(log K = 10,7) , Mn2+ (log K = 13,8), Ni2+ (log K = 18,6) eFe3+ (log K = 21,5) 0,1 mol L–1 con EDTA 0,1 mol L–1.Influencia de la constante de formación del complejo
metal-EDTA en la amplitud del salto en el puntode equivalencia.
Cuanto mayor es el valor de la constante deformación global, más acusado es el salto en elpunto de equivalencia, lo que conlleva una mayorexactitud en su determinación. Así, para la valo-ración de hierro (se forma el complejo FeY– sien-do log K = 25,1) el salto de pM en el punto deequivalencia comprende 16,8 unidades para unerror del ± 0,1% en la valoración (cuadro 6.1),mientras que para la valoración de calcio, cuyo
complejo CaY2– tiene una constante de forma-ción mucho más baja (log K = 10,7) dicho saltoes de tan sólo 2,4 unidades de pM. Este paráme-tro es pues de vital importancia para la valora-ción y para la elección del indicador metalocró-mico a utilizar en la misma, como se mostraráposteriormente.
La influencia de la existencia de reaccionessecundarias sobre las curvas de valoración com-plexométricas, se contempla en los siguientesapartados.
6.2.1. Influencia de reacciones secundariasdel ion metálico
Como ejemplo práctico de este tipo deinfluencia se va a considerar la valoración de Ni2+
con EDTA en base a la formación del complejoNiY2– (log K = 18,6), utilizando un tampón deNH4
+/NH3, a tres concentraciones diferentes deamoniaco. En este sistema se consideran lassiguientes reacciones secundarias:
1. Complejos formados entre el Ni2+ y el ionhidroxilo:
Ni2+ + OH– →← NiOH+
log β1,OH = 4,1 [6.35]
Ni2+ + 2 OH– →← Ni(OH)2log β2,OH = 8,0 [6.36]
Ni2+ + 3 OH– →← Ni(OH)3_
log β3,OH = 11,0 [6.37]
Ni2+ + 4 OH– →← Ni(OH)42_
log β4,OH = 12,0 [6.38]
El coeficiente de reacción secundaria,αNi(OH), viene dado por:
[6.39]
α β ββ β
Ni(OH) 1,OH 2,OH2
3,OH3
4,OH4
OH OH
OH OH ]
= + + ++ +
− −
− −
1 [ ] [ ]
[ ] [
228 Equilibrios iónicos y sus aplicaciones analíticas

2. Complejos formados entre el Ni2+ y elamoniaco:
Ni2+ + NH3→← Ni(NH3)
2+
log β1, NH3= 2,72 [6.40]
Ni2+ + 2 NH3→← Ni(NH3)2
2+
log β2, NH3= 4,89 [6.41]
Ni2+ + 3 NH3→← Ni(NH3)3
2+
log β3, NH3= 6,55 [6.42]
Ni2+ + 4 NH3→← Ni(NH3)4
2+
log β4, NH3= 7,67 [6.43]
Ni2+ + 5 NH3→← Ni(NH3)5
2+
log β5, NH3= 8,14 [6.44]
Ni2+ + 6 NH3→← Ni(NH3)6
2+
log β6, NH3= 8,17 [6.45]
El coeficiente de reacción secundaria,α Ni(NH3)
, viene dado por:
[6.46]
3. Influencia del pH en la disociación delEDTA, cuyo coeficiente de reacción secun-daria con el ion hidrógeno, αY(H), vienedado de acuerdo con la ecuación [6.3] por:
[6.3]
Una vez considerados todos los equilibriosimplicados en esta valoración, el procedimientoa seguir para conseguir la curva de valoración sebasa en los siguientes puntos:
1. Cálculo de los respectivos coeficientes dereacción secundaria al pH al cual se llevaa cabo la valoración considerando además
la concentración de la disolución regulado-ra NH4
+/NH3 que se utiliza para su ajuste.2. Cálculo de la variación de pM en función
del volumen de EDTA añadido en cadauna de las zonas de la curva de valoraciónhaciendo uso de las ecuaciones corres-pondientes mostradas en el recuadro 6.1.
3. Trazado de la curva de valoración.
El ejemplo considerado consiste en construirla curva de valoración de 50 mL de una disolu-ción de Ni2+ 0,005 mol L–1 con EDTA 0,01 mol L–1
como valorante. En la disolución inicial se regu-la el pH mediante un tampón NH3/NH4Cl depH = 9,25 cuya concentración de amoniaco librees de 0,1, 0,5 y 1,0 mol L–1. Estos valores per-miten considerar que la concentración de NH3no varía apreciablemente al formarse los com-plejos con Ni2+. Suposición que es razonable yaque la concentración total de amoniaco es comomínimo 20 veces superior a la de ion metálicoa valorar.
De acuerdo con el procedimiento indicado,se determinan los distintos coeficientes de reac-ción secundaria implicados en estos equilibriossecundarios. Así, los coeficientes de reacciónsecundaria para el EDTA y para el Ni2+, al sus-tituir en las ecuaciones [6.3] y [6.39] el valor depH al cual se lleva a cabo la valoración (pH =9,25), serán:
αY(H) = 1 + 8,91 + 0,006 + 10–8,75 + 10–16 = 9,91
[6.47]
αNi(OH) = 1 + 0,22 + 0,03 + 10–3,25 + 10–7 = 1,25
[6.48]
Por otra parte, los coeficientes de reacciónsecundaria debido a los complejos aminados deníquel, al sustituir en la ecuación [6.46] serán:
[NH3] = 0,1 mol L–1
α Ni(NH3)= 104,02 [6.49]
[NH3] = 0,5 mol L–1
α Ni(NH3)= 107,00 [6.50]
α β ββ β
Y(H) 1H H
H 3 H 4
1 H H
H H
= + + ++ +
+ +
+ +
[ ] [ ]
[ ] [ ]2
2
3 4
αNi(NH )2,72 4,89 2
6,55 3 7,67 4
8,14 5 8,17 6
3NH NH
NH NH
NH NH
= + + ++ + ++ +
1 10 10
10 10
10 10
3 3
3 3
3 3
[ ] [ ]
[ ] [ ]
[ ] [ ]
Capítulo 6: Valoraciones de formación de complejos 229
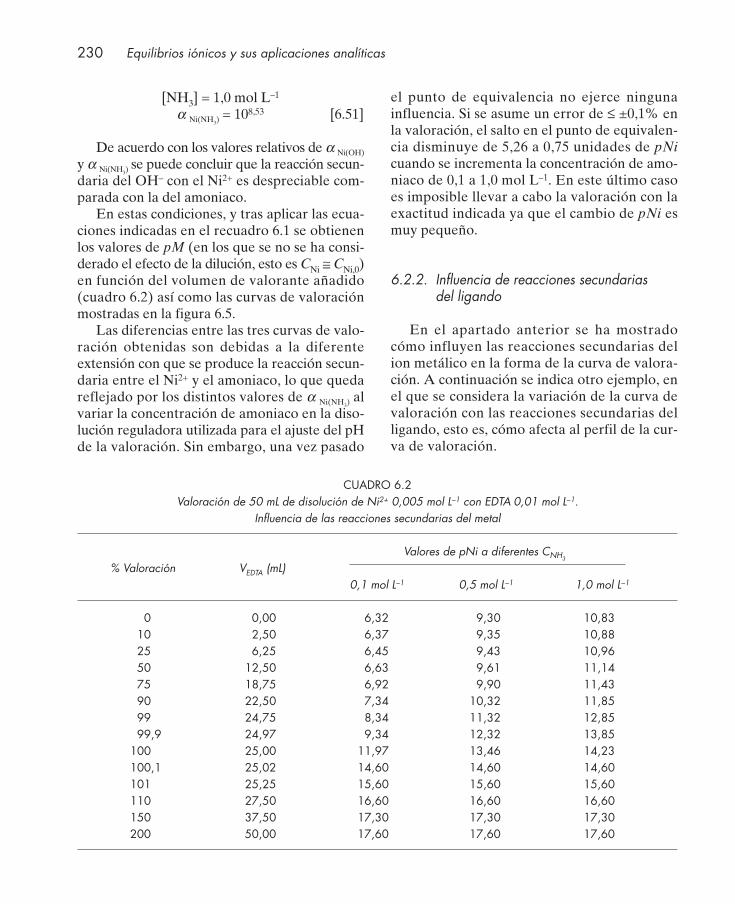
[NH3] = 1,0 mol L–1
α Ni(NH3)= 108,53 [6.51]
De acuerdo con los valores relativos de α Ni(OH)
y α Ni(NH3)se puede concluir que la reacción secun-
daria del OH– con el Ni2+ es despreciable com-parada con la del amoniaco.
En estas condiciones, y tras aplicar las ecua-ciones indicadas en el recuadro 6.1 se obtienenlos valores de pM (en los que se no se ha consi-derado el efecto de la dilución, esto es CNi ≅ CNi,0)en función del volumen de valorante añadido(cuadro 6.2) así como las curvas de valoraciónmostradas en la figura 6.5.
Las diferencias entre las tres curvas de valo-ración obtenidas son debidas a la diferenteextensión con que se produce la reacción secun-daria entre el Ni2+ y el amoniaco, lo que quedareflejado por los distintos valores de α Ni(NH3)
alvariar la concentración de amoniaco en la diso-lución reguladora utilizada para el ajuste del pHde la valoración. Sin embargo, una vez pasado
el punto de equivalencia no ejerce ningunainfluencia. Si se asume un error de ≤ ±0,1% enla valoración, el salto en el punto de equivalen-cia disminuye de 5,26 a 0,75 unidades de pNicuando se incrementa la concentración de amo-niaco de 0,1 a 1,0 mol L–1. En este último casoes imposible llevar a cabo la valoración con laexactitud indicada ya que el cambio de pNi esmuy pequeño.
6.2.2. Influencia de reacciones secundariasdel ligando
En el apartado anterior se ha mostradocómo influyen las reacciones secundarias delion metálico en la forma de la curva de valora-ción. A continuación se indica otro ejemplo, enel que se considera la variación de la curva devaloración con las reacciones secundarias delligando, esto es, cómo afecta al perfil de la cur-va de valoración.
230 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 6.2Valoración de 50 mL de disolución de Ni2+ 0,005 mol L–1 con EDTA 0,01 mol L–1.
Influencia de las reacciones secundarias del metal
Valores de pNi a diferentes CNH3
% Valoración VEDTA (mL)0,1 mol L–1 0,5 mol L–1 1,0 mol L–1
0 0,00 6,32 9,30 10,8310 2,50 6,37 9,35 10,8825 6,25 6,45 9,43 10,9650 12,50 6,63 9,61 11,1475 18,75 6,92 9,90 11,4390 22,50 7,34 10,32 11,8599 24,75 8,34 11,32 12,8599,9 24,97 9,34 12,32 13,85
100 25,00 11,97 13,46 14,23100,1 25,02 14,60 14,60 14,60101 25,25 15,60 15,60 15,60110 27,50 16,60 16,60 16,60150 37,50 17,30 17,30 17,30200 50,00 17,60 17,60 17,60
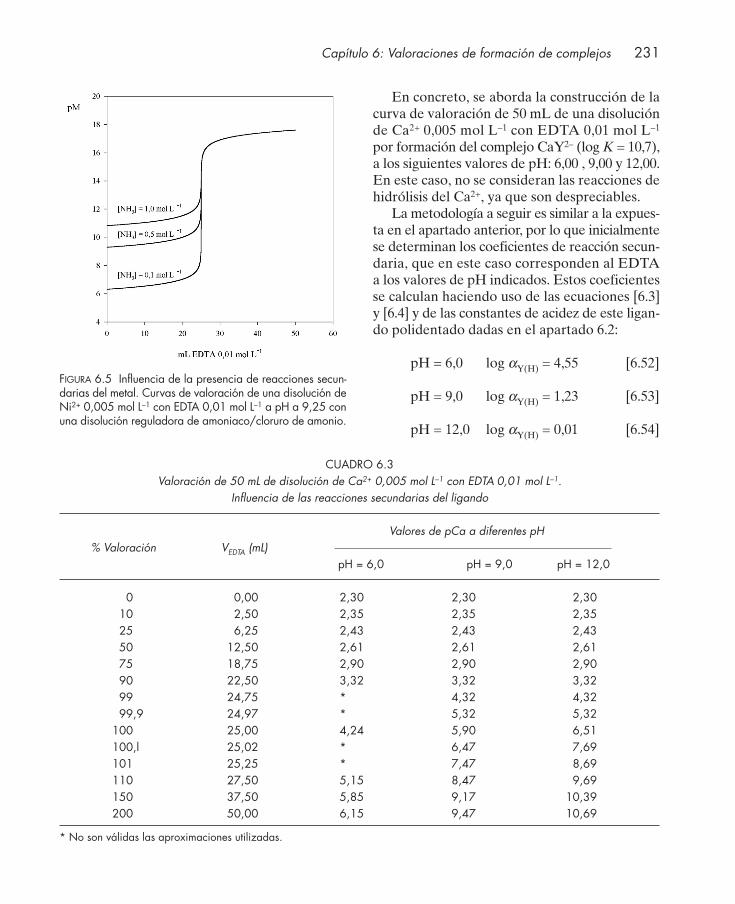
FIGURA 6.5 Influencia de la presencia de reacciones secun-darias del metal. Curvas de valoración de una disolución deNi2+ 0,005 mol L–1 con EDTA 0,01 mol L–1 a pH a 9,25 conuna disolución reguladora de amoniaco/cloruro de amonio.
En concreto, se aborda la construcción de lacurva de valoración de 50 mL de una disoluciónde Ca2+ 0,005 mol L–1 con EDTA 0,01 mol L–1
por formación del complejo CaY2– (log K = 10,7),a los siguientes valores de pH: 6,00 , 9,00 y 12,00.En este caso, no se consideran las reacciones dehidrólisis del Ca2+, ya que son despreciables.
La metodología a seguir es similar a la expues-ta en el apartado anterior, por lo que inicialmentese determinan los coeficientes de reacción secun-daria, que en este caso corresponden al EDTAa los valores de pH indicados. Estos coeficientesse calculan haciendo uso de las ecuaciones [6.3]y [6.4] y de las constantes de acidez de este ligan-do polidentado dadas en el apartado 6.2:
pH = 6,0 log αY(H) = 4,55 [6.52]
pH = 9,0 log αY(H) = 1,23 [6.53]
pH = 12,0 log αY(H) = 0,01 [6.54]
Capítulo 6: Valoraciones de formación de complejos 231
CUADRO 6.3Valoración de 50 mL de disolución de Ca2+ 0,005 mol L–1 con EDTA 0,01 mol L–1.
Influencia de las reacciones secundarias del ligando
Valores de pCa a diferentes pH% Valoración VEDTA (mL)
pH = 6,0 pH = 9,0 pH = 12,0
0 0,00 2,30 2,30 2,3010 2,50 2,35 2,35 2,3525 6,25 2,43 2,43 2,4350 12,50 2,61 2,61 2,6175 18,75 2,90 2,90 2,9090 22,50 3,32 3,32 3,3299 24,75 * 4,32 4,3299,9 24,97 * 5,32 5,32
100 25,00 4,24 5,90 6,51100,l 25,02 * 6,47 7,69101 25,25 * 7,47 8,69110 27,50 5,15 8,47 9,69150 37,50 5,85 9,17 10,39200 50,00 6,15 9,47 10,69
* No son válidas las aproximaciones utilizadas.
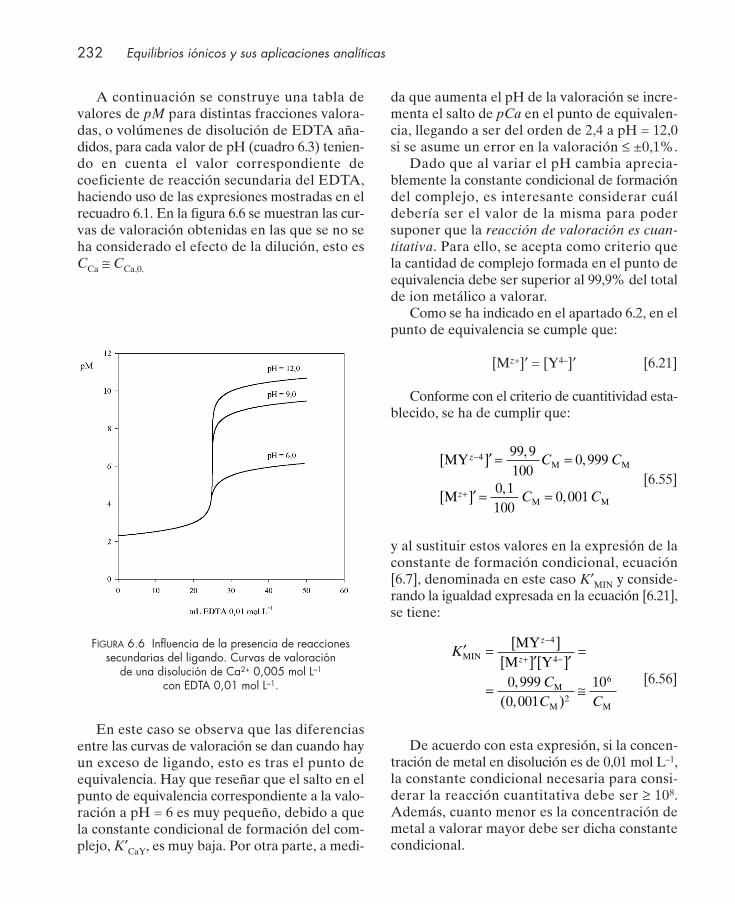
A continuación se construye una tabla devalores de pM para distintas fracciones valora-das, o volúmenes de disolución de EDTA aña-didos, para cada valor de pH (cuadro 6.3) tenien-do en cuenta el valor correspondiente decoeficiente de reacción secundaria del EDTA,haciendo uso de las expresiones mostradas en elrecuadro 6.1. En la figura 6.6 se muestran las cur-vas de valoración obtenidas en las que se no seha considerado el efecto de la dilución, esto esCCa ≅ CCa,0.
FIGURA 6.6 Influencia de la presencia de reaccionessecundarias del ligando. Curvas de valoración
de una disolución de Ca2+ 0,005 mol L–1
con EDTA 0,01 mol L–1.
En este caso se observa que las diferenciasentre las curvas de valoración se dan cuando hayun exceso de ligando, esto es tras el punto deequivalencia. Hay que reseñar que el salto en elpunto de equivalencia correspondiente a la valo-ración a pH = 6 es muy pequeño, debido a quela constante condicional de formación del com-plejo, K′CaY, es muy baja. Por otra parte, a medi-
da que aumenta el pH de la valoración se incre-menta el salto de pCa en el punto de equivalen-cia, llegando a ser del orden de 2,4 a pH = 12,0si se asume un error en la valoración ≤ ±0,1%.
Dado que al variar el pH cambia aprecia-blemente la constante condicional de formacióndel complejo, es interesante considerar cuáldebería ser el valor de la misma para podersuponer que la reacción de valoración es cuan-titativa. Para ello, se acepta como criterio quela cantidad de complejo formada en el punto deequivalencia debe ser superior al 99,9% del totalde ion metálico a valorar.
Como se ha indicado en el apartado 6.2, en elpunto de equivalencia se cumple que:
[Mz+]′ = [Y4–]′ [6.21]
Conforme con el criterio de cuantitividad esta-blecido, se ha de cumplir que:
[6.55]
y al sustituir estos valores en la expresión de laconstante de formación condicional, ecuación[6.7], denominada en este caso K′MIN y conside-rando la igualdad expresada en la ecuación [6.21],se tiene:
[6.56]
De acuerdo con esta expresión, si la concen-tración de metal en disolución es de 0,01 mol L–1,la constante condicional necesaria para consi-derar la reacción cuantitativa debe ser ≥ 108.Además, cuanto menor es la concentración demetal a valorar mayor debe ser dicha constantecondicional.
′ =′ ′
=
= ≅
−
+ −K
CC C
z
zMIN
M
M M
MYM ] [Y ]
,,
[ ][
( )
4
4
2
60 9990 001
10
[ ],
,
[ ],
,
MY
M
M M
M M
z
z
C C
C C
−
+
′ = =
′ = =
4 99 9100
0 999
0 1100
0 001
232 Equilibrios iónicos y sus aplicaciones analíticas

6.3. Indicadores metalocrómicos
Los indicadores visuales utilizados en valora-ciones complexométricas se denominan genéri-camente indicadores metalocrómicos, y, análo-gamente a los indicadores ácido-base, suutilización se basa en cambios de color, que eneste caso no son debidos a cambios del pH de ladisolución, sino a cambios en el valor de pM enla misma. Los indicadores metalocrómicos soncolorantes que poseen varios grupos dadores deelectrones (bases orgánicas) de forma que soncapaces de formar complejos quelatos con el ionmetálico a valorar. Debido a sus característicasácido-base, el pH al cual se lleva a cabo la valo-ración influirá de forma considerable en esteequilibrio de complejación y en definitiva en suempleo en el proceso de valoración.
El mecanismo de acción de estos indicadoresse basa en el siguiente proceso. Sea un indicadormetalocrómico, Inq–, que presenta en disoluciónun determinado color, color 1, y que forma uncomplejo con el ion metálico, Mz+, del tipo MInz–q,cuyo color en disolución denominaremos color 2.Antes de empezar la valoración de Mz+ conEDTA se añade una pequeña cantidad de Inq– ala disolución de ion metálico que toma el color 2debido a la formación de una pequeña cantidadde complejo MInz–q. Al añadir la disolución devalorante se forma el complejo MYz–4 hasta quese consume el ion metálico libre en la disolución.En el punto final, se origina la siguiente reacciónde desplazamiento:
MInz–q + Y4– →← MYz–4 + Inq– [6.57]
acompañada del cambio de color que pone demanifiesto el final de la valoración. Para que estareacción tenga lugar sólo en el punto final, se hade cumplir que el complejo MInz–q sea menosestable que el que forma el ion metálico conEDTA, con objeto de que esté desplazada haciala derecha y sea rápida, al valor de pH al cual selleva a cabo la valoración. Pasado este punto dela valoración, la adición de un exceso de EDTAno origina un cambio de color en la disolución.En la figura 6.7 se muestra de forma esquemá-tica este mecanismo de acción de los indicado-res metalocrómicos.
Como se ha comentado, el pH ejerce unainfluencia considerable sobre la acción de estosindicadores debido a sus propiedades ácido-base,por lo que se deben tener en cuenta sus reac-ciones secundarias con el ion hidrógeno, si sequiere llevar a cabo un estudio cuantitativo sobresu empleo en valoraciones complexométricas.Así, si se considera un indicador del tipo HqInque puede formar un complejo con un ion metá-lico Mz+, se tendrán los siguientes equilibriosconcurrentes:
Capítulo 6: Valoraciones de formación de complejos 233
FIGURA 6.7. Mecanismo deacción de los indicadoresmetalocrómicos.
Mz+ + Inq– MInz–q
H+
HIn(q-1)–
H+
H2In(q-2)–

Para que esta sustancia pueda utilizarse comoindicador metalocrómico, Inq– y MInz–q han detener, como se ha indicado, coloraciones distin-tas, aunque también pueden ser coloreadas lasotras especies del indicador implicadas en suequilibrio ácido-base: HIn(q–1)–, HIn(q–2)–, etc.
Los equilibrios considerados vendrán definidospor la constante de formación del complejo metal-indicador y las constantes de protonación de éste:
[6.58]
[6.59]
A partir de estas últimas, se puede calcular elcoeficiente de reacción secundaria del indicadorrespecto del ion hidrógeno como:
[6.60]
Si se tiene en cuenta que este indicador se uti-lizará para visualizar cambios en el pM de la diso-lución, es decir, en la concentración de ion metá-lico libre, se define por conveniencia su constantecondicional de formación con el metal libre comofunción de su constante de formación KM–In y delcoeficiente de reacción secundaria del indicadorcon el ion hidrógeno, por tanto:
[6.61]
Reordenando esta ecuación se obtiene laexpresión:
[6.62]
en la que tomando logaritmos y cambiando elsigno se tiene finalmente:
[6.63]
Esta ecuación se utiliza como base para obte-ner la zona de viraje de un indicador metalocró-mico. Si como se ha indicado, el color del com-plejo M–In es diferente del color del indicadorno complejado; y si son suficientemente distin-tos, se admite en general que el color que adquie-re la disolución es el de la especie predominan-te. Sin embargo, existirá un intervalo de transicióno zona de viraje (intervalo de pM), en la que elindicador manifiesta el cambio de color a travésde una coloración intermedia. Para establecereste intervalo de transición, al igual que para losindicadores ácido-base, se acepta que el ojohumano sólo detecta un cambio de tonalidadcuando la relación entre las concentraciones delas dos especies coloreadas varía entre 10 y 0,1.Es decir, si
[6.64]
se observará el color de la forma no complejada(Inq–) del indicador, y si
[6.65]
se observará el color del complejo metal-indica-dor (MInz–q). En el intervalo entre ambas con-diciones, esto es, si
[6.66]
[ ]
[ ]
– log[ ]
[ ]
0 1 10
1 1
,In
MInIn
MIn
<′
< ⇒
⇒ <′
<
−
−
−
−
q
z q
q
z q
[ ][ ]
, [ ]
[ ]In
MInlog
InMIn
1q
z q
q
z q
−
−
−
−
′≤ ⇒
′≤
0 1
[ ][ ]
[ ]
[ ]In
MIn10 log
InMIn
1q
z q
q
z q
−
−
−
−
′≥ ⇒
′≥
pM
K
z
q
z q
= =
=′
+ ′
+
− −
– [MIn
MIn
-
M In
log ]
log[ ]
[ ]log
[ ]
[ ]M
MIn [In ]M In
zz q
qK+
−
−−
=′ ′
′ =′
=−
−
+ −−K
Kz q
z qM InM In
In(H)
MIn[M ] [In ]
[ ]α
α ββ β
In(H) In,H
In,2H
In,H
H
H H
= + ++ + +
+
+ +
1 1
2
[ ]
β β
β
In,1H
( )
In,2H 2
( )
In,qH
HInH ][In
H InH In
.........H In]
H In
= =
=
− −
+ −
− −
+ −
+ −
[ ][ ]
[ ][ ] [ ]
[
[ ] [ ]
q
q
q
q
q
q q
1 2
2
K
z q
z qM InMIn
M In−
−
+ −= [ ]
[ ][ ]
234 Equilibrios iónicos y sus aplicaciones analíticas

se observará un color que será mezcla de loscolores correspondientes a ambas formas delindicador.
Al sustituir la condición indicada en la ecua-ción [6.66] en la expresión dada para el pM, ecua-ción [6.63], se tiene:
[6.67]
Como se puede deducir, si se consideran reac-ciones secundarias con el H+, este intervalo depM depende del pH de la disolución. Es necesa-rio tener en cuenta además, que los colorescorrespondientes a las diferentes formas proto-nadas del indicador también variarán con el pH.Este aspecto, de gran importancia a la hora de laelección del indicador, se comenta a continua-ción para algunos de los indicadores más utiliza-dos en estas valoraciones.
La murexida (figura 6.8) presenta un colorrojo-violeta a pH inferior a 9, es violeta entre pH9 y 11, y azul-violeta a pH superior a 11. Da lugara complejos de color rojo (con Ca2+), naranja(con Cu2+) o amarillo (con Ni2+) entre otros. Sise utiliza para la valoración del calcio, sería ade-cuado trabajar a pH superior a 11 dado que el
cambio de color de rojo a azul-violeta seríamucho mas acusado que a otros valores de pH.
El negro de eriocromo T (NET, figura 6.8) esun indicador metalocrómico ampliamente utili-zado en medio básico (disolución amortiguado-ra de NH3/NH4
+ a pH=10) en valoraciones deZn2+, Mg2+o Ca2+ con los que forma complejosde color rojo. Debido a sus equilibrios ácido-base,este indicador presenta color rojo a valores depH inferiores a 6,4, azul entre 6,4 y 11,5 y naran-ja a pH superiores a 11,5. Es evidente, que llevara cabo la valoración a pH = 10 con disoluciónreguladora NH3/NH4
+ conlleva un cambio de coloren el punto final: rojo-azul.
El azul de metiltimol (MTB, figura 6.8) dalugar a complejos de color azul con una seriede iones metálicos a diferentes valores de pH.Por otra parte, debido a sus equilibrios ácido-base, el propio indicador presenta un coloramarillo a pH inferior a 6,5 , es azul entre pH6,5 y 11,5, y a pH superior a 11,5 es de colorgris. A tenor de la distinción de estos colores,este indicador se utiliza a valores de pH infe-riores a 6,5 (cambio de color: azul-amarillo)para la valoración de Pb2+, Zn2+, Mn2+, etc., ysuperiores a 11,5 (cambio de color azul-gris) enel caso de Ca2+, Cd2+ y Cu2+.
∆ pM K= ′ ±−log M In 1
Capítulo 6: Valoraciones de formación de complejos 235
FIGURA 6.8. Estructura química delos indicadores metalocrómicosmás utilizados en valoracionescomplexométricas.

El calcón (figura 6.8) se utiliza como indicadoren las valoraciones con EDTA de los iones Ca2+,Cd2+, Mg2+ y Zn2+ debido a la formación de com-plejos de color rosa. Este indicador presenta bási-camente dos colores en disolución de acuerdo conel pH del medio: rojo a pH < 7,3 y azul a valoressuperiores de pH. En general, las valoraciones delos iones metálicos indicados se lleva a cabo enmedio básico (pH 10-11,5) donde el indicadormanifiesta el cambio de color: rosa-azul.
Hasta ahora se ha expuesto de forma cualitati-va cómo se puede seleccionar el indicador meta-locrómico adecuado para una determinación valo-ración complexométrica, en base a la diferencia decolor entre el complejo metal-indicador y la for-ma no complejada del mismo al valor de pH al cualse lleva a cabo la valoración. Sin embargo, paraque esta elección sea correcta se ha basar en con-sideraciones de tipo cuantitativo, tal como se harealizado en el capítulo 4 correspondiente a lasvaloraciones ácido-base. Por ello, es necesario queel intervalo de viraje del indicador (∆pM) esté den-tro del intervalo de pM en la zona próxima al pun-to de equivalencia (por ejemplo, entre valores deF comprendidos entre 0,999 y 1,001, si se admiteun error del ± 0,1% en la valoración, o entre 0,99y 1,01 si el error asumido es del ± 1%). Ello tam-bién implica que el cambio de pM de la valoraciónen esa zona sea como mínimo de 2 unidades.
Para ilustrar este estudio se ha consideradocomo ejemplo la valoración de una disolución deCu2+ 0,005 mol L–1 con EDTA 0,01 mol L–1 a pH10 obtenido con una disolución reguladoraNH4Cl/NH3 cuya concentración de amoniaco librees 0,1 mol L–1. En primer lugar, se construye lacurva de valoración siguiendo un procedimientosimilar al desarrollado en el apartado 6.2.1.Mediante el uso de las expresiones adecuadas yde los valores de constantes necesarios dados enlos cuadros 5.1-5.3, se determinan los valores delos coeficientes de reacción secundaria implica-dos en esta valoración:
A partir de estos datos y haciendo uso de lasexpresiones [6.12], [6.17], [6.25] y [6.33] se cons-truye la curva de valoración mostrada en la figu-ra 6.9 (no se ha considerado el efecto de diluciónpor adición del valorante) y en el cuadro 6.4 semuestran algunos valores significativos de pCu enfunción del % de valoración o fracción valorada.
CUADRO 6.4Valoración de 50 mL de disolución de Cu2+ 0,005 mol L–1
con EDTA 0,01 mol L–1 a pH 10 ajustado con disoluciónreguladora NH4Cl/NH3 0,03 mol L–1
%Valoración Fracción valorada pCu
0 0,00 8,9010 0,10 8,9525 0,25 9,0350 0,50 9,2075 0,75 9,5090 0,90 9,9099 0,99 10,9099,9 0,999 11,90
100 1,000 13,65100,1 1,001 15,39101 1,01 16,39110 1,10 17,39150 1,50 17,57200 2,00 18,39
Como se observa en el cuadro, el intervalo depCu correspondiente a un error de ±0,1 % en lavaloración está comprendido entre 11,90 y 15,39,por lo que cualquier indicador que posea un valorde log K′Cu–In situado en este intervalo se podríautilizar en principio en esta valoración. En concre-to, el ∆pCu, calculado conforme a la ecuación [6.67]:
[6.68]
se ha de situar en el citado intervalo. En el cua-dro 6.5 se muestran los valores de log K′Cu–In ylos respectivos intervalos de viraje, expresadosen términos de pCu, para algunos indicadoresmetalocrómicos.
∆pCu K= ′ ±−log Cu In 1
log , log ,
log , log ,
KCuY Cu(OH)
Cu(NH ) Y(H)
2
3
− = =
= =
18 8 1 7
6 6 0 41
α
α α
236 Equilibrios iónicos y sus aplicaciones analíticas
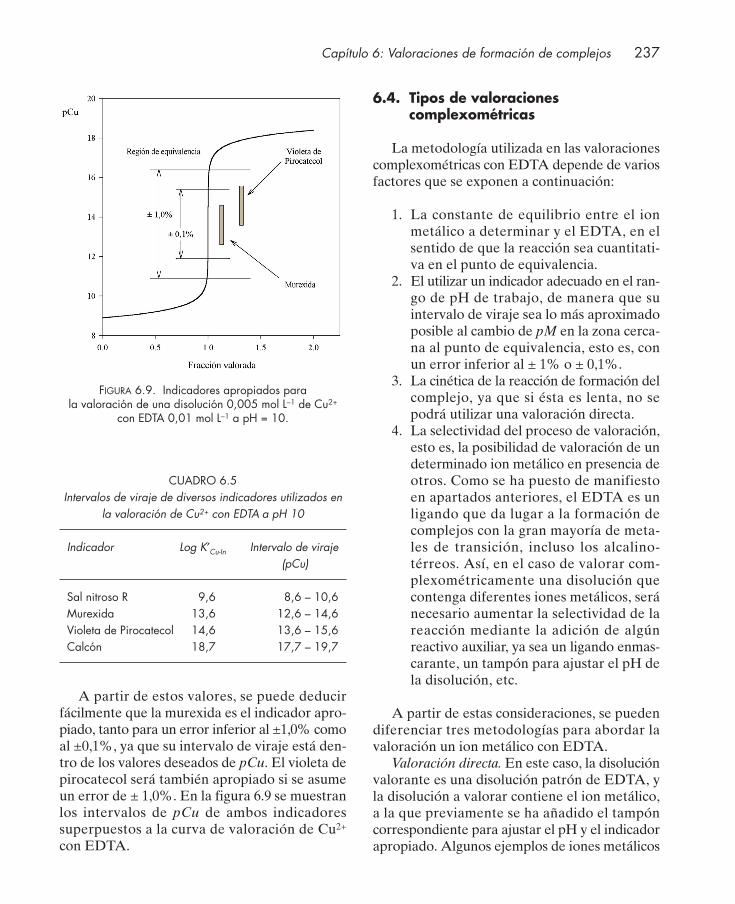
FIGURA 6.9. Indicadores apropiados parala valoración de una disolución 0,005 mol L–1 de Cu2+
con EDTA 0,01 mol L–1 a pH = 10.
CUADRO 6.5Intervalos de viraje de diversos indicadores utilizados en
la valoración de Cu2+ con EDTA a pH 10
Indicador Log K′Cu-In Intervalo de viraje(pCu)
Sal nitroso R 9,6 8,6 – 10,6Murexida 13,6 12,6 – 14,6Violeta de Pirocatecol 14,6 13,6 – 15,6Calcón 18,7 17,7 – 19,7
A partir de estos valores, se puede deducirfácilmente que la murexida es el indicador apro-piado, tanto para un error inferior al ±1,0% comoal ±0,1%, ya que su intervalo de viraje está den-tro de los valores deseados de pCu. El violeta depirocatecol será también apropiado si se asumeun error de ± 1,0%. En la figura 6.9 se muestranlos intervalos de pCu de ambos indicadoressuperpuestos a la curva de valoración de Cu2+
con EDTA.
6.4. Tipos de valoracionescomplexométricas
La metodología utilizada en las valoracionescomplexométricas con EDTA depende de variosfactores que se exponen a continuación:
1. La constante de equilibrio entre el ionmetálico a determinar y el EDTA, en elsentido de que la reacción sea cuantitati-va en el punto de equivalencia.
2. El utilizar un indicador adecuado en el ran-go de pH de trabajo, de manera que suintervalo de viraje sea lo más aproximadoposible al cambio de pM en la zona cerca-na al punto de equivalencia, esto es, conun error inferior al ± 1% o ± 0,1%.
3. La cinética de la reacción de formación delcomplejo, ya que si ésta es lenta, no sepodrá utilizar una valoración directa.
4. La selectividad del proceso de valoración,esto es, la posibilidad de valoración de undeterminado ion metálico en presencia deotros. Como se ha puesto de manifiestoen apartados anteriores, el EDTA es unligando que da lugar a la formación decomplejos con la gran mayoría de meta-les de transición, incluso los alcalino-térreos. Así, en el caso de valorar com-plexométricamente una disolución quecontenga diferentes iones metálicos, seránecesario aumentar la selectividad de lareacción mediante la adición de algúnreactivo auxiliar, ya sea un ligando enmas-carante, un tampón para ajustar el pH dela disolución, etc.
A partir de estas consideraciones, se puedendiferenciar tres metodologías para abordar lavaloración un ion metálico con EDTA.
Valoración directa. En este caso, la disoluciónvalorante es una disolución patrón de EDTA, yla disolución a valorar contiene el ion metálico,a la que previamente se ha añadido el tampóncorrespondiente para ajustar el pH y el indicadorapropiado. Algunos ejemplos de iones metálicos
Capítulo 6: Valoraciones de formación de complejos 237

que se pueden valorar directamente con EDTAse muestran en el cuadro 6.6, en el que ademásse muestra el indicador adecuado para su valo-ración así como la disolución reguladora em-pleada para ajustar el pH al cual se lleva a cabola valoración.
CUADRO 6.6Características de la valoración directa de diversos iones
metálicos con EDTA
Ion metálico Indicador pH Valoración
Ba2+ MTB 9–12 (NH3 diluido)Bi3+ MTB, XO, PAR 2–3 (HNO3 diluido)Ca2+ Murexida 12,0 (NaOH)Co2+ MTB, XO 5–6 (Tampón de hexamina)
Murexida 8,0 (Tampón NH4Cl/NH3)Cu2+ XO 5–6 (Tampón de hexamina)
Murexida 8,0 (NH3 diluido)Mg2+ NET 9–10 (Tampón NH4Cl/NH3)Ni2+ XO 5–6 (Tampón de hexamina)
Murexida 8–9 (Tampón NH4Cl/NH3)Pb2+ XO, MTB 5–6 (Tampón de hexamina)
NET 9–10 (Tampón NH4Cl/NH3)Zn2+ XO, MTB 5–6 (Tampón de hexamina)
NET 9–10 (Tampón NH4Cl/NH3)
XO Naranja de xilenol; PAR: 1-(2-piridilazo)-2-naftol
Valoración por retroceso. Esta metodologíase ha de utilizar cuando no es adecuada la valo-ración directa dado que o bien: 1) no se disponedel indicador adecuado al metal a determinar, 2)no es posible mantener el ion metálico a valoraren disolución al pH de trabajo, dado que porejemplo puede precipitar, o 3) la formación delcomplejo con EDTA es de cinética lenta, comoes el caso de iones tri- o tetravalentes, tales comoCr3+, Al3+, Fe3+, Zr4+, etc.
En este caso el valorante es una disoluciónpatrón de ion metálico (en general, de Zn2+, Mg2+
o Pb2+) en función del pH requerido para valorarla disolución de analito. El procedimiento consis-
te en la adición de un volumen exactamente medi-do de disolución patrón de EDTA a la disolucióna determinar (previo ajuste del pH si es necesario),de manera que se tiene un exceso conocido deligando. En estas condiciones, en el caso de un iondivalente, se origina la siguiente reacción:
M2+ + Y4–(exceso medido)
→←→← MY2– + Y4–
(sin reaccionar) [6.69]
A continuación se ajusta de nuevo el pH (sies necesario), se añade el indicador apropiado yse valora el exceso de EDTA con la disoluciónpatrón de ion metálico. Normalmente se utilizaZn2+ si estamos en medio básico (pH = 9-10), yPb2+ si se trata de una disolución a pH ligera-mente ácido (5-6). La valoración por retroceso,en el caso de utilizar una disolución patrón dePb2+ como valorante consiste en:
Pb2+ + Y4–(sin reaccionar)
→← PbY2– [6.70]
Dado que los complejos con EDTA presentanuna estequiometria 1:1, metal:ligando, el númerode moles de EDTA añadidos (nY) debe ser igual ala suma de los moles de los dos iones metálicos quehan intervenido en las reacciones [6.69] y [6.70]:
nY = nM + nPb [6.71]
A partir de los volúmenes y concentracionesde EDTA y de ion metálico patrón, se tiene final-mente:
[6.72]
Esta modalidad de valoración, es también degran utilidad en el caso de la determinación dealgunos aniones o sales insolubles, como sulfatos.
PbSO4 (S) + Y4–(exceso medido)
→←→← PbY2– + Y4–
(sin reaccionar) + SO42_ [6.73]
En base a una ecuación similar a la [6.72] sedeterminaría el contenido de sulfato en la salinsoluble PbSO4.
n n n C V C VM Y Pb Y Y Pb Pb= − = × − ×
238 Equilibrios iónicos y sus aplicaciones analíticas

Valoración por sustitución. Esta metodologíaes una alternativa a la valoración por retrocesocuando no se dispone de un indicador adecuadopara el metal a determinar por valoración direc-ta. En este caso se utiliza como reactivo auxiliaruna disolución de un complejo Maux-EDTA, cuyaconstante de estabilidad sea baja, pero que sí dis-ponga de un indicador adecuado, en general, pue-den ser los complejos MgY2–, MnY2– o ZnY2–. Ladisolución valorante es EDTA patrón.
El procedimiento en este caso consiste en laadición de una cierta cantidad del complejo auxi-liar (MgY2– por ejemplo) a la disolución pro-blema. Si el complejo MY2– es más estable queel complejo MgY2–, tiene lugar la reacción dedesplazamiento:
M2+ + MgY2– → Mg2+ + MY2– [6.74]
De esta manera se “libera” el ion Mg2+, quepuede ser ahora valorado con EDTA a pH=10utilizando NET como indicador. En esta valora-ción el número de moles de EDTA (volumenconsumido multiplicado por su concentración)proporciona directamente el número de molesde Mg2+ liberados, que será igual al número demoles de metal M2+ a determinar, esto es:
[6.75]
6.5. Aplicaciones
Las valoraciones de formación de complejosse basan casi exclusivamente en la utilización deEDTA como valorante, como ha quedadoexpuesto en el apartado anterior, constituyendola determinación de calcio y magnesio en aguassu principal campo de aplicación. Esta determi-nación es una operación de rutina en el análisisde aguas en el campo de la Química Analíticamedioambiental y en estaciones depuradoras deaguas potables. La presencia y concentración deambos metales como iones metálicos es uno delos factores determinantes de la calidad de lasaguas que se conoce como dureza de las mismas.
Las aguas naturales contienen una cierta con-centración en ambos metales debido principal-mente a su infiltración en las rocas calcáreas,constituidas por carbonatos de calcio y de mag-nesio. De ahí que la dureza de las aguas puedavariar según tipo de agua y procedencia.
La dureza de un agua se define como la sumade las concentraciones de iones metálicos, a excep-ción de los alcalinos y del ion hidrógeno. Sin embar-go, debido a que la concentración de calcio y mag-nesio es superior a la de otros iones metálicos, ladureza se refiere a la suma de las concentracionesde Ca2+ y Mg2+, dureza total, y se expresa general-mente en ppm (mg L–1) de CaCO3. También pue-de expresarse en grados franceses o grado hidroti-métrico (°H o °f), el cual se define como la durezade una disolución que contiene 10 mg CaCO3 L
–1,por lo que 1 ºH equivale a 10,0 ppm de CaCO3.
El método más utilizado para determinar ladureza total de un agua es mediante valoracióncomplexométrica, con EDTA como valorante.Este método permite además diferenciar el con-tenido de calcio y de magnesio en la muestra pro-blema, esto es, la dureza debida al calcio y al mag-nesio, respectivamente.
Para llevar a cabo estas valoraciones es nece-sario disponer de una disolución patrón (valora-da) de EDTA. Para preparar la misma se haceuso de su sal disódica dihidratada, YH2Na2 . 2H2O,sustancia que no se comercializa generalmentecon la pureza adecuada para su utilización comoestándar químico-analítico primario, por lo quees necesario estandarizar las disoluciones deEDTA previamente a su uso como valorante conun estandar químico-analítico primario, tal comoCaCO3, sustancia que se comercializa con unapureza del 99,98%. Para utilizarla como patrónprimario, se ha de desecar en estufa a una tem-peratura de 100-110 °C durante 12 horas y con-servar en desecador. La reacción de valoración
Ca2+ + YH22_ →← CaY2_
+ 2H+ [6.76]
se puede llevar a cabo tanto a pH = 10, ajustadocon disolución reguladora NH4Cl/NH3, aña-diendo una cierta cantidad de complexonato de
n n n C VM Mg Y Y Y= = = ×
Capítulo 6: Valoraciones de formación de complejos 239

magnesio, MgY2– y utilizando NET como indi-cador, o bien a pH = 12 mediante la adición dedisolución de NaOH 1,0 mol L–1 con murexidacomo indicador.
Si se prepara una disolución de EDTA de con-centración aproximada [EDTA]aprox, el factor dela misma, f, que relaciona las concentracionesreal y aproximada de valorante de acuerdo conlo indicado en el capítulo 4, viene dado por:
[6.77]
[6.78]
siendo 100,1 el peso molecular del CaCO3 y mgCaCO3 la cantidad de esta sustancia expresadaen mg que contiene la alícuota de la disoluciónde patrón primario utilizada para estandarizar ladisolución de EDTA. Reordenando la ecuación[6.78] se tiene la expresión que permite calcularel factor de la disolución de EDTA:
[6.79]
Si , como es común, la disolución de EDTA esaproximadamente 0,01 mol L–1, para preparar ladisolución patrón de calcio, se pesan exactamen-te entre 0,35 y 0,45 g de CaCO3 y se disuelven enla menor cantidad de HCl 2 mol L–1 en un vasode precipitados. Se diluye con unos mL de aguay se hierve para expulsar CO2 cuidando de queno se produzcan salpicaduras. Se pasa a un matrazaforado de 100 mL, una vez fría, y se enrasa conagua destilada. Se suele tomar una alícuota de 10mL para la valoración de la disolución de EDTA.
Una vez que se dispone de la disoluciónpatrón de EDTA, la determinación de los distin-tos tipos de dureza de agua se basa en llevar acabo las correspondientes valoraciones directas
a dos valores de pH, 10 y 12, con el uso de losindicadores apropiados en cada caso, NET ymurexida, respectivamente.
Dureza debida al calcio. En este caso, la valo-ración se lleva a cabo a pH = 12 utilizando mure-xida como indicador. El ion magnesio no se valo-ra en estas condiciones, ya que a este valor depH se favorece su precipitación como hidróxido,Mg(OH)2 (S), quedando éste en suspensión. Estosignifica que la reacción:
Mg(OH)2 (S) + Y4– →← MgY2– + 2 OH– [6.80]
está desplazada hacia la izquierda, como se pue-de comprobar fácilmente a tenor de los valoresrelativos del producto de solubilidad del hidró-xido de este ion metálico y de la constante con-dicional de formación del complejo MgY2– a estevalor de pH. Los moles de EDTA consumidosen esta valoración se relacionan directamentecon los de Ca2+ en la muestra de agua.
Dureza total. Este parámetro, como se ha indi-cado, corresponde a la suma de las concentra-ciones de los iones calcio y magnesio en aguas.Su determinación por valoración con EDTA selleva a cabo a pH = 10 utilizando NET como indi-cador, dado que a este valor de pH no se produ-ce la precipitación del hidróxido de magnesio (nose alcanza el producto de solubilidad) y por tan-to la reacción dada en la ecuación [6.80] está des-plazada hacia la derecha.
En esta determinación, los moles de EDTAconsumidos en la valoración, nY, se correspondencon la suma de los moles de calcio, nCa, y de mag-nesio, nMg, presentes en la muestra valorada:
[6.81]
Si se considera que se ha tomado una alícuotade agua, Vmuestra (mL), para su valoración y que éstase ha llevado a cabo con una disolución de EDTAde concentración CY consumiéndose un volumende la misma VY (mL), la dureza total de un aguaexpresada en ppm de CaCO3 se calcula a partir de:
[6.82] mmol EDTA mmolCaCOY Y 3= × =C V
n n nY Ca Mg= +
fV
=× ×
mgCaCOPeso MolecularCaCO [EDTA]
3
3 aprox Y
[ ]
[ ]
EDTAmgCaCO
Peso MolecularCaCO
EDTA
real Y3
3
aprox Y
× = =
= × ×
V
f V
[EDTA] [EDTA]real aprox= × f
240 Equilibrios iónicos y sus aplicaciones analíticas

[6.83]
[6.84]
Dureza debida al magnesio. De acuerdo con loexpuesto en apartados anteriores, este parámetro
correspondiente al contenido de ion magnesio enaguas, se determina fácilmente por diferencia entrela dureza total y la dureza debida al calcio. El pro-ceso experimental, como se muestra en la figu-ra 6.10, implica la valoración de dos alícuotas dela muestra, una a pH = 12 con murexida comoindicador en la que se determina el contenido decalcio en el agua, y otra a pH = 10 utilizando NETcomo indicador en la que se determina la suma deambos iones. Por diferencia entre ambas valora-ciones se determina el contenido de magnesio enla muestra de agua.
Dureza(ppmCaCO )mL
mL3Y Y
muestra
= × ×C VV
( )( )
105
[CaCO ](ppm)mmolCaCO
(mL)
mL)mL
100,1mg1, 0 mmolCaCO
10 g1, 0 mg
33
muestra
Y Y
muestra 3
3
= =
= × × × µV
C VV
(( )
Capítulo 6: Valoraciones de formación de complejos 241
FIGURA 6.10. Fundamento de la determi-nación de los diferentes tipos de durezaen aguas mediante valoración complexo-métrica con EDTA.
Cuestiones
6.1. ¿De qué depende el ∆pM en el punto de equivalencia en una valoración de formación de complejos?
[ ] pH
[ ] Constante de formación
[ ] Concentración analítica de ion metálico
[ ] Temperatura
[ ] Estequiometría del complejo
[ ] Concentración de ligando
6.2. ¿Por qué se prefiere el uso de ligandos polidentados a monodentados en la determinación volumétrica de iones
metálicos por formación de complejos?
6.3. Indicar tres requisitos básicos para que una sustancia pueda actuar como indicador en valoraciones complexométricas.

6.4. La constante de protonación β 2H del Y4– está relacionada con las constantes de acidez del EDTA mediante la
expresión: ...
6.5. Si se desea valorar una disolución de M2+ 0,01 mol L–1 con EDTA, ¿qué valor mínimo ha de tener la constante con-
dicional de formación del complejo metal-EDTA para que la valoración sea cuantitativa?
6.6. La expresión de pM antes del punto de equivalencia en la valoración de Ca2+ con EDTA a pH = 10, ajustado con
una disolución reguladora cloruro de amonio/amoniaco, depende de:
[ ] El indicador utilizado
[ ] Constante de formación del complejo
[ ] Concentración de ion metálico a valorar
[ ] Coeficiente de reacción secundaria del Ca2+ con el amoniaco
[ ] Coeficiente de reacción secundaria del EDTA con el ion hidrógeno
6.7. Para la valoración complexométrica de un ion metálico M2+ se añade un volumen conocido de una disolución están-
dar de EDTA. Posteriormente el exceso de EDTA se valora con disolución estándar de Mg2+. ¿Qué tipo de valo-
ración se ha llevado a cabo?
[ ] Por sustitución
[ ] Por retroceso
[ ] Directa
6.8. Para que una reacción sea adecuada para ser utilizada en volumetrías de formación de complejos es necesario que:
[ ] El ligando forme un complejo coloreado con el ion metálico
[ ] El complejo se forme rápidamente aunque su constante de formación sea pequeña
[ ] El ligando reacciona cuantitativamente con el ion metálico en la relación 1:1
[ ] El complejo formado presente un elevado número de coordinación
[ ] Se emplee un indicador con una gran selectividad frente al ion metálico a valorar
6.9. El complejo AlY3– requiere de un calentamiento previo para su formación. ¿Qué tipo de valoración complexo-
métrica emplearía para la determinación de aluminio en una muestra?
[ ] Valoración por sustitución
[ ] Valoración directa
[ ] Valoración pro retroceso
6.10. Indicar, de los siguientes factores, cuáles influyen positivamente (+) y cuáles de forma negativa (–) en la amplitud
del salto en el punto de equivalencia en valoraciones de formación de complejos:
[ ] Un incremento en el número de coordinación del complejo
[ ] Un incremento en la concentración de ligando valorante
[ ] El uso de un indicador metalocrómico con una amplia zona de viraje
[ ] Un incremento en la constante de formación del complejo formado
[ ] Un incremento del pH
242 Equilibrios iónicos y sus aplicaciones analíticas

6.11. La zona de viraje de un indicador metalocrómico responde a la expresión:
y por tanto depende de:
[ ] Constante de formación del complejo metal-EDTA
[ ] Concentración inicial de ion metálico
[ ] Constante de formación del complejo metal-Indicador
6.12. Indicar (marcando con una X) las características de los tres tipos de valoraciones complexométricas indicadas:
Se usa una disolución patrón de ......................... y se añade
EDTA MY2– M2+
Exceso Bureta Exceso Bureta
Valoración directa
Valoración por retroceso
Valoración por sustitución
6.13. ¿Cómo se selecciona el indicador metalocrómico adecuado para una valoración complexométrica?
6.14. ¿Cómo se suele expresar normalmente la dureza de un agua?
Capítulo 6: Valoraciones de formación de complejos 243
• Objetivo pedagógico:Desarrollo de diferentes tipos de problemas sobre: cur-vas de valoración con EDTA y selección de indica-dores de acuerdo al error asumido en la misma, deter-minación individual de especies en diferentes muestras,y resolución de mezclas de especies por valoración conEDTA con especial énfasis en la determinación de losdiferentes tipos de dureza de aguas.
1. Se valora una mezcla que contiene 0,1 mol L–1 de Cd2+
y 0,1 mol L–1 de Mg2+ con EDTA a pH = 4. Otra alí-cuota se valora en presencia de ion cianuro 1,0 mol L–1
con EDTA a pH = 10,1) Demostrar cuantitativamen-te qué se valora en cada caso. 2) Seleccionar entre losindicadores indicados en la tabla, los más adecuadospara obtener un error inferior al ±1,0% en la valora-ción a pH = 10 en presencia de CN–.
Indicador log K′MgIn (pH = 10)
Negro de eriocromo T 5,4Violeta de pirocatecol 3,5Hidroxifenilazo resorcinol 2,5Negro de eriocromo R 4,1
CdY2– (log K = 16,5); MgY2– (log K = 9,1); EDTA(log αY(H) = 8,6 a pH = 4; log αY(H) = 0,412 a pH = 10);Cd2+ (log αCd(CN) = 18,7 a [CN–] = 1,0 mol L–1).
Para demostrar de forma cuantitativa qué especie sevalora con cada caso se ha de calcular la constante con-dicional de formación del complejo en cada una de lascondiciones experimentales y comparar el valor obte-nido con el mínimo requerido de dicha constante, K′MINpara que la reacción de valoración sea cuantitativa.
Seminarios: problemas numéricos

Valoración a pH = 4,0:
A partir de estos valores se deduce que a este valorde pH sólo se valora la disolución de Cd2+.
Valoración a pH = 10 en presencia de CN– 0,1 molL–1:
En este caso, y a tenor de los valores de K′, se pue-de afirmar que sólo se produce la valoración del ionmagnesio presente en la disolución.
Con objeto de constatar si ambas valoraciones soncuantitativas en las condiciones experimentales dadas,se ha de calcular el valor de K′MIN, que de acuerdocon la ecuación [6.56], viene dado por:
Dado que los valores obtenidos para log K′Cd_Y ylog K′Mg_Y son superiores a K′MIN, se puede afirmarque ambas valoraciones son cuantitativas.
La selección de algunos de los indicadores mostradosen el cuadro como adecuados para la valoración a pH=10 en presencia de ion cianuro, esto es, para la valora-ción de magnesio, para obtener un error inferior al ±1%, se basa inicialmente en el cálculo del intervalo depMg correspondiente a este error asumido. Así:
Error – 1%. F = 0,99. La zona de la valoración esantes del punto de equivalencia, por lo que hacien-do uso de la expresión [6.17], se tiene:
Error + 1%. F = 1,01. En este caso nos encontra-mos después del punto de equivalencia y la expresióna aplicar para el cálculo del pMg, es similar a la ecua-ción [6.33]:
De acuerdo con estos resultados el ∆pMg para unerror de ±1% será:
De los indicadores reseñados en la tabla, sólo elnegro de eriocromo T (∆pM = 4,4 – 6,4) y el negro deeriocromo R (∆pM = 3,1 – 5,1) serán adecuados parala valoración ya que cambian de color en el interva-lo de pMg deseado.
2. Representar la curva de valoración de una disoluciónde Pb2+ 0,01 mol L–1 con EDTA a pH = 5,0, y com-probar si alguno de los siguientes indicadores mos-trados en la tabla es apropiado para esta valoración:
Indicador log K′Pb–In
Azul de Metiltimol 6,0Naranja de Xilenol 7,0
Sol.:
F 0,0 0,5 0,9 0,99 0,999pPb 2,0 2,3 3,0 4,0 5,0F 1,0 1,001 1,01 1,1 1,5 2,0pPb 6,83 8,66 9,66 10,66 11,36 11,66
Se puede hacer uso de ambos indicadores con unerror inferior al ±0,1%.
3. Se valoran 50 mL de una disolución de Mg2+ 0,01 molL–1 con EDTA 0,01 mol L–1 a pH = 10 utilizandonegro de eriocromo T como indicador. El cambio decolor que pone de manifiesto el punto final de la valo-ración se observa cuando la relación MgIn–/HIn2– es0,1. Calcular el error que se comete en esta valora-ción con el signo adecuado.MgY2– (log K = 9,10); MgIn– (log K′Mg–In = 5,4);EDTA (log αY(H) = 0,412 a pH = 10).Sol.: + 0,056%.
4. ¿Qué peso de YH2Na2.2 H2O debe tomarse para pre-parar 1,0 litro de disolución de tal forma que 1,0 mL dela misma sea equivalente a 1,0 mg de sulfato de calcio?Pesos moleculares: YH2Na2.2 H2O = 372,2;CaSO4 = 136,1.
De acuerdo con la reacción de valoración:
→← Ca 2 YH CaY 2 H222 2+ − − ++ +
∆pMg = −3 0 6 7, ,
pMg K F= − + − == − + − =
log log log( )
, , log( , ) ,)αY(H 1
9 1 0 412 1 01 1 6 7
pMg C F= − − − == − − − =
log log( )
log , log( , ) ,M 1
0 1 1 0 99 3 0
′ ≥ = =K
CMINM
10 100 1
106 6
7
,
log log log ( )′ = − ==
− −K KMg Y Mg Y Y H
9,1 – 0, 412 = 8, 69α
loglog log log( ) )
′ == − − ==
−
−
KKCd Y
Cd Y Y H Cd(CN
16, 5 – 0, 412 – 18, 7 = – 2, 61α α
log log log )′ = − ==
− −K KMg Y Mg Y Y(H
9,1 – 8,6 = 0,5α
log log log )′ = − ==
− −K KCd Y Cd Y Y(H
16,5 – 8,6 = 7,9α
244 Equilibrios iónicos y sus aplicaciones analíticas

mmol Ca2+ = mmol EDTA, y considerando que 1,0mL de la disolución de EDTA equivale a 1,0 mg deCaSO4, se tiene:
Igualando ambas expresiones se llega a:
De donde se deduce que se han de pesar 2,735 g dela sal de EDTA para preparar un litro de disolución.
5. Una muestra de un mineral que contiene 0,030 g decarbonato de calcio se disuelve y se diluye a 1,0 litro.50 mL de esta disolución de valoran con una diso-lución de EDTA de tal concentración que 1,0 mLde la misma equivale a 50 µg de calcio. ¿Qué volu-men de la disolución de EDTA se consume en lavaloración?Pesos atómicos: Ca = 40,1; C = 12; O = 16.Sol.: 12,0 mL.
6. Calcular el peso de un mineral de magnesio que debetomarse para que, una vez disuelto, en su valoracióncon EDTA se consuman 10 mL (1,0 mL equivale a5,62 mg de cadmio) suponiendo que el contenido demagnesio en el mineral es del 5%.Pesos atómicos: Cd = 112,4; Mg = 24,3.Sol.: 243,0 mg.
7. A una alícuota de 5,00 mL de una disolución de Ni2+
se le añade una disolución reguladora de amoniaco-cloruro de amonio y 15,00 mL de EDTA 0,050 molL–1. El exceso de EDTA se valora con una disolución0,015 mol L–1 de cloruro de magnesio, de la que seconsumen 21,35 mL. Calcular la concentración de ladisolución de níquel expresada en mg mL–1.Peso atómico: Ni = 58,71.
El problema consiste en la determinación de níquelpor valoración por retroceso, en la que están impli-cadas las siguientes reacciones:
Ni2+ + Y4– (exceso) →← NiY2– + Y4– (sin reaccionar)
Y4– (sin reaccionar) + Mg2+ →← MgY2–
De acuerdo con este procedimiento se puede esta-blecer:
mmol Ni2+ + mmol Mg2+ = mmol Y4–
de donde:
mmol Ni2+ = mmol Y4– – mmol Mg2+ == 15,0 × 0,05 – 21,35 × 0,015 = 0,43 mmol
La concentración de ion níquel en la disoluciónexpresada en mg L–1 se calcula como:
8. Para determinar el contenido de calcio en una caliza setoman 2,500 g de la misma, se tratan convenientemen-te, y se obtiene una disolución de 250 mL. 50 mL deesta disolución consumen 41,2 mL de una disoluciónde EDTA que es aproximadamente 0,05 mol L–1 parasu valoración. 50 mL de esta disolución valorante con-sumen 46,32 mL de una disolución patrón de Zn2+ 0,05mol L–1. Calcular el porcentaje de calcio en la muestra.Peso atómico: Ca = 40,1.Sol.: 15,3%.
9. Una muestra de 1,00 g que sólo contiene MgCO3 sedisuelve en HCl 1,0 mol L–1 y se diluye a 1,0 litro conagua destilada. 50 mL de esta disolución consumen25,0 mL de una disolución de EDTA para su valora-ción. Con esta disolución de EDTA se determina ladureza de un agua. Se toman 200 mL de agua y se con-sumen 15,5 mL de EDTA para su valoración. Expre-sar la dureza del agua en mg L–1 (ppm) de CaCO3.Pesos atómicos: Mg = 24,3; Ca = 40,1; C = 12; H = 1;O = 16.Sol.: 183,8 ppm de CaCO3.
10. Calcular el contenido de cobre en un mineral que, trassu disolución, se valora con EDTA con los siguientesresultados. Muestra: 0,350 g de mineral. Se añaden 20mL de una disolución de EDTA 0,01 mol L–1. El exce-so de EDTA consume 1,5 mL de disolución de nitra-to de plata 0,02 mol L–1 para su valoración.Peso atómico: Cu = 63,54.Sol.: 3,09%.
CNi
1
0, 43 mmol5, 0 mL
58, 71mg1, 0 mmol Ni
5, 05 mg mL
= × =
= −
mmol Ca1, 0
136,1mmol EDTA
1, 0Peso(g)EDTA
372, 2
2+ = = =
= ×
mmol EDTAmol EDTA
LPeso(g)EDTA/372,
Y Y Y= × = × =
= ×
V C V
1 02
1,
mmol Ca
mg CaSOPm CaSO
1, 0136,1
2 4
4
+ = =
Capítulo 6: Valoraciones de formación de complejos 245

11. 50 mL (un exceso) de EDTA 0,022 mol L–1 se aña-den a 25 mL de una disolución de MgCl2. El excesode EDTA gasta en su valoración 20,72 mL de ZnCl20,025 mol L–1. ¿Cuál es la concentración de la diso-lución de MgCl2 expresada en mol L–1?Sol.: 0,0233 mol L–1.
12. Para determinar el contenido de cobre en un mine-ral se toman 2,00 g del mismo y se disuelven 100 mLde una disolución ácida. Se toman 5,0 mL de estadisolución, se añade tampón cloruro de amonio/amo-niaco de pH = 10 y 25 mL de disolución 0,05 mol L–1
del complejo MgY2–. Tras este tratamiento la disolu-ción resultante consume 24,5 mL de una disolución0,01 mol L–1 de EDTA para su valoración utilizandonegro de eriocromo T como indicador. Calcular elporcentaje de cobre en el mineral.CuY2– (log K′ = 15,89); MgY2– (log K′ = 8,69).Peso atómico: Cu = 63,54.
La determinación de cobre se lleva a cabo medianteuna valoración por sustitución a tenor de los valoresrelativos de las constantes condiciones de formaciónde los complejos de Cu2+ y Mg2+ con EDTA. Las reac-ciones implicadas en esta valoración son las siguientes:
Cu2+ + MgY2– (exceso) →←→← CuY2– + Mg2+ + MgY2– (sin reaccionar)
Mg2+ + Y4– →← MgY2–
De acuerdo con estas reacciones se puede estable-cer que:
mmol Cu2+ = mmol Mg2+ = mmol Y4–
por tanto:
mmol Cu2+ = 24,5 × 0,01 = 0,245 mmol
Si se tiene en cuenta que se valoran 5,0 mL de ladisolución de 100 mL en la que se han disuelto 2,000g del mineral de cobre, el porcentaje de éste en lamuestra será:
13. 25 mL de una disolución de EDTA que es aproxi-madamente 0,01 mol L–1 se añaden a 10 mL de clo-
ruro de magnesio 0,01 mol L–1. El exceso de EDTAconsume en su valoración 6,5 mL de disolución decloruro de cinc 0,025 mol L–1. Calcular el factor de ladisolución de EDTA.Sol.: 1,050.
14. Para determinar el contenido de magnesio en un mine-ral se toma 1,000 g del mismo, se trata conveniente-mente, y se obtiene una disolución de 100 mL. 25 mLde esta disolución gastan 20,6 mL de una disolución deEDTA. 50 mL de esta disolución de EDTA consumen46,4 mL de una disolución patrón de Cu2+ 0,05 mol L–1.Calcular el porcentaje de magnesio en el mineral.Peso atómico: Mg = 24,3.Sol.: 9,29%.
15. Una muestra contiene CaCO3, ZnO y materia inerte.Se pesan 2,5000 g, se disuelven en H2SO4 diluido, seneutraliza la disolución con amoniaco, se añade unadisolución amortiguadora de amoniaco-cloruro de amo-nio y se enrasa a 250 mL. Una alícuota de 25 mL de estadisolución consume 21,5 mL de EDTA 0,0500 mol L–1
cuando se utiliza negro de eriocromo T como indica-dor. A otra alícuota de 25 mL se añade KCN, y en estecaso solamente se consumen 16,2 mL de EDTA. Cal-cular el porcentaje de cinc y de calcio en la muestra.Pesos atómicos: Zn = 65,37; Ca = 40,1.
Una vez disuelta la muestra, para determinar sucomposición se llevan a cabo dos valoraciones conEDTA en disolución tampón cloruro de amonio/amo-niaco en ausencia y en presencia de KCN. La adiciónde este ligando enmascara la valoración de cinc porformación de los correspondientes complejos cianu-rados, por lo que se tiene:
a) En ausencia de KCN:
Ca2+ + Y4– →← CaY2–
Zn2+ + Y4– →← ZnY2–
b) En presencia de KCN
Ca2+ + Y4– →← CaY2–
Dado que se utiliza la misma disolución de EDTA,es evidente a partir de estas reacciones que:
VY (Zn2+) = VY (sin KCN) – VY (con KCN) == 21,5 – 16,2 = 5,3 mL
VY (Ca2+) = VY (con KCN) = 16,2 mL
%
,,
,
., %
Cumg Cu2.000
100= × =
=× ×
× =0 245
1005 0
63 54
2 000100 15 57
246 Equilibrios iónicos y sus aplicaciones analíticas

A partir de estos volúmenes se pueden deducirfácilmente los contenidos de cinc y calcio en lamuestra.
16. El sulfato contenido en una muestra de 0,3734 g desulfato de aluminio se precipita en forma de BaSO4.El precipitado se filtra, lava y se disuelve en 50 mLde EDTA 0,02121 mol L–1. El exceso de EDTA con-sume 11,74 mL de una disolución de MgCl2. 25 mLde EDTA consumen 20,65 mL de la disolución deMgCl2. a) Calcular la concentración de la disoluciónde MgCl2. b) Calcular el porcentaje de Al2(SO4)3 enla muestra.Peso molecular Al2(SO4)3: 342.Sol.: a) 0,02568 mol L–1; b) 23,17%.
17. Se pipetean 25 mL de una disolución que contieneAl3+ y Ni2+, y se diluyen a 500 mL. Se toma una alí-cuota de 25 mL, se ajusta el pH a 4,8 y se añaden 25mL de EDTA 0,0188 mol L–1. El exceso de EDTAreacciona con 10,07 mL de Cu2+ 0,00993 mol L–1. Acontinuación se añade un exceso de F– a la mismadisolución (que destruye el complejo Al3+–EDTA),y el EDTA liberado consume 26,30 mL de la disolu-ción de Cu2+. Calcular la concentración de aluminioy níquel en la disolución inicial expresada en g L–1.Pesos atómicos: Al = 26,98; Ni = 58,71. Sol.: 5,64 g L–1 Al3+; 5,13 g L–1 Ni2+.
18. Una disolución de Pb2+ y Bi3+ se valora con EDTA0,01 mol L–1 y 50 mL de la misma consumen 25 mLpara obtener la suma de ambos iones. Otros 50 mLse reducen con Pb según la reacción:
3 Pb + 2 Bi3+ →← 3 Pb2+ + 2 Bi
y se valora de nuevo con EDTA, gastándose ahora30 mL. Calcular las concentraciones de Pb2+ y Bi3+ enla disolución original expresadas en mol L–1.
Como es usual en el caso de la resolución de unamezcla de dos especies por valoración de formaciónde complejos con EDTA, se llevan a cabo dos deter-minaciones en diferentes condiciones experimentales:
1. Inicialmente se valoran ambos iones en base a lasreacciones:
Pb2+ + Y4– →← PbY2–
Bi3+ + Y4– →← BiY–
por lo que se puede establecer:
mmol Pb2+ + mmol Bi3+ = mmol Y4–
o bien:
A + B = mmol Y4– = 25,0 × 0,01 = 0,25 mmol
si se denomina A y B a los mmol de plomo y bis-muto, respectivamente.
2. En estas condiciones experimentales, sólo se valo-ra ion Pb2+: el inicial presente en la muestra y el for-mado a partir de la reacción del plomo metal conel ion bismuto. Así, y teniendo en cuenta la este-quiometría de la reacción de reducción se tiene:
Resolviendo este sistema de ecuaciones y con-siderando que los iones se encuentran en un volu-men de 50 mL se tiene:
19. Para determinar la dureza de un agua se emplea unadisolución de EDTA. 100 mL de agua consumen 15,24mL de valorante. Por otra parte, 100 mL de una diso-lución de Zn2+ de 0,5 g L–1 consumen 32,50 mL de ladisolución de EDTA para su valoración. Expresar ladureza del agua en mg L–1 (ppm) de CaCO3.Pesos atómicos: Ca = 40,1; C = 12; H = 1; O = 16; Zn = 65,37.Sol.: 358,4 ppm de CaCO3.
20. Para estandarizar una disolución de EDTA, se aña-den a un enlermeyer 50 mL de una disolución de Mg2+
de 0,25 g L–1, 10 mL de disolución reguladoraNH4Cl/NH3 y unas gotas del indicador negro de erio-cromo T. Se consumen 27,5 mL de la disolución deEDTA en la valoración. Con esta disolución deEDTA se valora una muestra de 100 mL de agua yse consumen 21,25 mL de valorante. Expresar la dure-
[Bi ]
0,1mmol50 mL
0,002 mol L3 –1+ = =
[Pb ]
0,15mmol50 mL
0,003 mol L2 –1+ = =
A
Bmmol Y = , , = , mmol–+ = ×
32
30 0 0 01 3 04
%
, , ,
., %Ca =
× × ×× =
16 2 0 0525025
40 1
2 500100 12 99
%
, , ,
., %Zn =
× × ×× =
5 3 0 0525025
65 37
2 500100 6 93
Capítulo 6: Valoraciones de formación de complejos 247

za del agua en mg L–1 (ppm) de carbonato de calcio.Pesos atómicos: Ca = 40,1; C = 12; O = 16; Mg = 24,3.Sol.: 387,1 ppm de CaCO3.
21. Calcular la concentración de Ca2+ y de Mg2+ expre-sada en mol L–1 en una disolución a partir de lossiguientes datos: 1) 20,0 mL de esta disolución se valo-ran con EDTA 0,112 mol L–1 a pH = 10 utilizandonegro de eriocromo T como indicador. Se consumen18,15 mL de la disolución de EDTA. 2) A otros 20,0mL de la misma disolución se le añaden 19,0 mL dedisolución de EDTA 0,112 mol L–1 a pH = 12, y elexceso de EDTA se valora con una disolución deCaCl2 0,1021 mol L–1 utilizando murexida como indi-cador. Se gastan 6,00 mL de la disolución de calcio.
En la primera valoración a pH = 10 utilizando negrode eriocromo T como indicador se determina la sumade las concentraciones de ambos iones en la disolución:
Ca2+ + Y4– →← CaY2–
Mg2+ + Y4– →← MgY2–
En estas condiciones los moles consumidos deEDTA serán iguales a la suma de los moles de calcioy magnesio:
mmol EDTA = 18,15 × 0,112 = 2,03 == mmol Ca2+ + mmol Mg2+
En la segunda valoración el exceso de EDTA aña-dido a pH = 12 sólo reacciona con el ion Ca2+:
Ca2+ (muestra) + Y4– (exceso) →←→← CaY2– + Y4– (sin reaccionar)
y posteriormente, se determina este exceso por valo-ración con un patrón de calcio:
Ca2+ (patrón) + Y4– (sin reaccionar) →← CaY2–
Se puede establecer la siguiente relación que permi-te calcular la cantidad de Ca2+ en la muestra problema:
mmol Ca2+ (muestra) = mmol Y4– – mmol Ca2+ (patrón)
mmol Ca2+ (muestra) = 19,0 × 0,112 – 6,0 × 0,1021 == 1,515 mmol
La cantidad de ion Mg2+ en la disolución problemase obtiene por diferencia entre ambas valoraciones:
mmol Mg2+ = 2,033 – 1,515 = 0,518 mmol
Finalmente las concentraciones de ambos iones enla disolución considerando que se valoran 20,0 mLde la misma serán:
22. Una mezcla de CaCO3 y MgCO3 que pesa 1,000 g con-sume 21,86 mL de HCl 1,0 mol L–1 para su valoraciónácido-base utilizando naranja de metilo como indica-dor. Un gramo de esta misma muestra se disuelve enHCl y se diluye a 1,0 litro con agua destilada. Se toman100 mL y se consumen 21,87 mL de una disolución deEDTA para su valoración. Con la disolución anteriorde EDTA se determina la dureza de un agua. Se toman100 mL de agua y se consumen 10 mL de EDTA.Expresar la dureza del agua en ppm (mg L–1) de calcio.Pesos atómicos: Ca = 40,1; Mg = 24,3; C = 12; O = 16.Sol.: 200,5 ppm de Ca.
23. El calcio y magnesio contenidos en un agua se deter-minan mediante valoración con EDTA. Una alí-cuota de 100 mL, ajustada a pH = 10, consume 31,3mL de EDTA 0,0106 mol L–1 cuando se utiliza negrode eriocromo T como indicador. Una segunda alí-cuota de 100 mL se alcaliniza con NaOH y se valoracon el mismo EDTA, consumiendo en este caso 19,2mL utilizando murexida como indicador. a) Deter-minar la dureza total del agua, expresada en mg L–1
de CaCO3. b) Calcular la concentración correspon-diente de calcio y magnesio, expresada en mg L–1.Pesos atómicos: Ca = 40,1; Mg = 24,3; C = 12; O = 16.Sol.: a) 332,1 mg CaCO3 L–1; b) 203,7 mg Ca L–1;
128,4 mg Mg L–1.
24. Para determinar el contenido en calcio y magnesioen un agua de bebida ésta se valora complexométri-camente con EDTA de acuerdo al siguiente proce-dimiento: 1) Se toman 50 mL de agua y se valorancon EDTA 0,005 mol L–1 gastándose 8,25 mL al uti-lizar murexida como indicador. 2) Otra porción de 25mL de agua se valora con EDTA 0,005 mol L–1 gas-tándose 12,5 mL al utilizar negro de eriocromo Tcomo indicador. Expresar el contenido de calcio ymagnesio en el agua analizada en µg mL–1.Pesos atómicos: Ca = 40,1; Mg = 24,3.Sol.: 50,12 µg Ca mL–1 y 9,72 µg Mg mL–1.
[ ]
,, Mg mol L–12 0 518
200 026+ = =
[Ca mol L–12 1 515
200 076+ = =]
,,
248 Equilibrios iónicos y sus aplicaciones analíticas

7
7.1. Introducción7.2. Solubilidad y producto de solubilidad7.3. Factores que afectan a la solubilidad7.4. Representación gráfica de los equilibrios
de precipitación7.5. Precipitación fraccionada7.6. Equilibrios concurrentes de acidez,
complejación y precipitación.Producto de solubilidad condicional
CuestionesSeminarios: problemas numéricos
EQUILIBRIOSDE PRECIPITACIÓN

7.1. Introducción
El equilibrio de precipitación es un tipo deequilibrio heterogéneo, tal como se ha comenta-do en el capítulo 1 y se observa en el cuadro 7.1,que implica la transferencia de materia entre dosfases, una de ellas sólida y la otra líquida, queestán en contacto. La fase líquida contiene unadisolución de iones, mientras que la fase sólida,que se genera in situ, está constituida por un com-puesto químico de composición y fórmula defi-nida. Se define como proceso de precipitación laaparición de una fase sólida en el seno de unadisolución y como precipitado a la fase sólida for-mada en el seno de la misma.
Este equilibrio heterogéneo puede tomar dosdenominaciones diferentes, según el sentido delproceso de transferencia de materia. Así, si seproduce la transferencia en el sentido líquido →sólido, se tendrá un equilibrio heterogéneo de pre-cipitación, mientras que si transcurre en sentidoinverso, sólido → líquido, se trata de un equili-brio heterogéneo de solubilización o disolución.En cualquier caso, el equilibrio es dinámico yaque en la interfase sólido-líquido ocurren simul-táneamente procesos de solubilización y preci-pitación, cuyas velocidades llegan a igualarsecuando se alcanza el equilibrio.
Se puede considerar que se obtiene un preci-pitado, en el seno de una disolución, al aumentarla concentración de una determinada sustancia deforma que se sobrepase el punto de saturación.De esta forma, pueden llegar a precipitar inclusoespecies químicas que se clasifican como solubles.
Los equilibrios heterogéneos de precipitación quese abordan en este capítulo consideran el caso deelectrolitos poco solubles.
CUADRO 7.1Clasificación de los equilibrios heterogéneos
según el tipo de interfase
Interfase Equilibrio
Líquido-líquido Extracción líquido-líquido Diálisis
Líquido-sólido PrecipitaciónExtracción líquido-sólidoIntercambio iónico
Gas-líquido DestilaciónDifusión gasesosa
La cinética juega un papel más preponderan-te que en los equilibrios ácido-base y de forma-ción de complejos. Se puede indicar que, en gene-ral, las reacciones de precipitación son más lentasy el tiempo que tarda en alcanzarse el equilibrioes un factor que debe tenerse presente. Por otrolado, la forma del precipitado es decisiva en elámbito cinético de la solubilización. Si el precipi-tado está recién formado, su solubilización serámás rápida que si se ha dejado envejecer, queimplica una disminución de la superficie específi-ca y, por tanto, del área efectiva de transferenciade materia. Cuando se trata de disolver un sólido,la velocidad del proceso heterogéneo depende desu grado de dispersión (tamaño de partícula); elloda lugar a falsas diferencias de solubilidad que enrealidad no son más que diferencias cinéticas.
250 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Estudio de los aspectos fundamentales de los equi-librios de precipitación: solubilidad y producto desolubilidad. Factores experimentales que afectan alos mismos.
• Explicar las alternativas adecuadas (expresiones ydiagramas gráficos) para el cálculo de la concen-tración de las diferentes especies presentes en este
equilibrio, con especial énfasis en la precipitaciónfraccionada.
• Desarrollo de la metodología a seguir para el estu-dio de los equilibrios concurrentes de acidez y for-mación de complejos mediante el empleo de los coe-ficientes de reacción secundaria y de productos desolubilidad y solubilidad condicionales.

Las reacciones basadas en equilibrios de pre-cipitación suelen utilizarse como fundamento dediferentes procesos de separación, donde se apro-vecha la diferencia de solubilidad entre los com-puestos sólidos para obtener su separación físi-ca. En estos procesos se basan las diferentesmarchas sistemáticas para la separación en gru-pos de mezclas complejas de cationes y aniones.También se utilizan en reacciones de identifica-ción de iones, ya que la presencia de una especiepuede ponerse de manifiesto mediante la apari-ción de un precipitado. Este tipo de reaccioneses, asimismo, la base para procesos cuantitativos,tanto volumétricos como gravimétricos, tal comose describe en otros capítulos.
7.2. Solubilidad y productode solubilidad
Para comprender mejor el comportamientode los equilibrios de precipitación es necesariodefinir el concepto de solubilidad. Ya que cual-quier sólido, por insoluble que sea, está en equi-librio dinámico con sus iones en disolución, sedefine como solubilidad a la concentración desustancia en una disolución saturada a una tem-peratura determinada. Se le asigna la letra S ysuele expresarse en g L–1, mol L–1 o g/100 mL.La solubilidad es un valor característico y cons-tante (para una temperatura dada) de las sus-tancias.
Se entiende por disolución saturada aquellaque no admite más soluto en su seno, a una tem-peratura determinada, mientras que se hablaráde disolución sobresaturada cuando la concen-tración de sustancia disuelta sea superior a lamáxima admitida, con lo que en este caso se tie-ne un equilibrio inestable.
El equilibrio entre un precipitado y sus ionesen disolución, se expresa como:
MmAn(s)→← mM + nA [7.1]
donde se han omitido las cargas de las especiesen disolución para simplificar la expresión.
Este equilibrio viene regido por su constantetermodinámica:
[7.2]
donde el valor de a MmAncorresponde a la activi-
dad de una fase sólida, por lo que se le asigna elvalor unidad. Por otra parte, al tratarse de sus-tancias poco solubles, las disoluciones de sus ionesserán siempre diluidas, con lo que, si no hay otrassustancias, la fuerza iónica será baja y se puedeexpresar la constante KT
S en función de las con-centraciones (mol L–1), tomando así la formausual:
[7.3]
Esta constante recibe el nombre de producto desolubilidad y puede definirse como el productode las concentraciones de los iones producidos enla disolución de un compuesto poco soluble, ele-vados a una potencia igual a su coeficiente este-quiométrico y se designa habitualmente como KS.En el cuadro 7.2 se dan los valores de KS parauna serie de sustancias de interés.
La ecuación del producto de solubilidad per-mite extraer las siguientes conclusiones:
1. Cuanto más aumenta la concentración deuno de los iones, más disminuye la con-centración del otro, y viceversa.
2. A partir de las concentraciones de los ionesen la disolución, se puede predecir si laprecipitación tendrá lugar o no. Si el pro-ducto de las concentraciones elevadas a sucoeficiente estequiométrico es superior alproducto de solubilidad, hay que esperarla aparición de precipitado.
3. La formación de sustancias poco solubleses el fundamento de métodos de separa-ción y de determinación. En este sentido,es útil introducir el concepto de precipita-ción cuantitativa, que significa que una pre-cipitación será completa cuando haya pre-
KSm n= [ ] [ ]M A
Ka aaS
Tm n
m n
= M A
M A
Capítulo 7: Equilibrios de precipitación 251

cipitado el 99,9% del analito, o lo que eslo mismo, que la concentración del ion adeterminar haya disminuido su concentra-ción inicial en al menos un factor de 1.000.
4. Permite deducir la solubilidad de una sus-tancia ya que, para una relación estequio-métrica determinada, cuanto menor sea elproducto de solubilidad más baja será lasolubilidad de la sustancia.
CUADRO 7.2Valores de pKS de algunos compuestos poco solubles
Compuesto pKS Compuesto pKS
Ag3PO4 18,2 CaC2O4 7,9Ag2AsO4 22,0 CaF2 10,3Ag2CrO4 11,9 CaSO4 4,6Ag2C2O4 11,0 Ce(OH)3 19,9Ag2CO3 11,2 Fe(OH)3 39,5Ag2S 48,2 Fe(OH)2 15,1AgCl 9,8 Hg2Cl2 17,2AgBr 12,3 Mg(OH)2 11,1AgI 16,1 Ni(OH)2 15,2AgIO3 7,5 PbCl2 4,1AgOH 7,7 PbI2 8,2AgSCN 11,9 Pb3(PO4)2 43,5Al(OH)3 31,7 PbS 27,1BaCO3 8,3 PbSO4 7,8BaCrO4 9,9 SrSO4 6,5BaSO4 9,9 Zn(OH)2 17,2Bi(OH)3 38,0 ZnS 24,7
Existe una relación entre la solubilidad y el pro-ducto de solubilidad, que es más o menos sencillaen función de la relación estequiométrica que exis-ta entre los iones implicados en el equilibrio deprecipitación. En el caso más general, si se tiene elequilibro de solubilidad dado anteriormente:
[7.1]
cuya constante viene dada por la expresión
[7.3]
la solubilidad viene determinada por las con-centraciones de los iones divididas por sus coe-ficientes estequiométricos:
[7.4]
Si se sustituye [M] y [A] en la expresión [7.3]por sus valores en función de la solubilidad, ecua-ción [7.4], resulta:
[7.5]
de donde se deduce fácilmente que:
[7.6]
De esta expresión se deriva que cuanto menorsea KS, más insoluble es el precipitado, menorserá la solubilidad, S. Sin embargo, al estar inclui-dos en esta relación los coeficientes estequiomé-tricos, para comparar la solubilidad de dos sus-tancias, no pueden usarse únicamente los valoresde KS, salvo que éstas tengan igual estequiome-tría. Se han de utilizar, por tanto, para compararsolubilidades los valores de S. En el recuadro 7.1se muestra un ejemplo en el que se comparan lassolubilidades de dos compuestos poco solublestales como carbonato y cloruro de plata.
7.3. Factores que afectana la solubilidad
Existen diversos factores que modifican lasolubilidad de los precipitados, algunos de ellosafectan al valor del producto de solubilidad mien-tras que otros originan un desplazamiento delequilibrio de precipitación.
Los factores que modifican el valor del pro-ducto de solubilidad son básicamente:
Temperatura. El valor del producto de solu-bilidad aumenta con la temperatura para lamayoría de las sustancias. La disolución de unprecipitado es generalmente un proceso endo-
S
Km n
Sm n
m n= +
K mS nS
m S n S m n SS
m n
m m n n m n m n
=
= = +
( ) ( )
S
m nm nM A
M [A]= =[ ]
KSm n= [ ] [ ]M A
M A M Am n s m n( ) +
252 Equilibrios iónicos y sus aplicaciones analíticas
→←

térmico, dado que la energía reticular consumi-da en el proceso de solubilización es mayor quela energía de hidratación desprendida corres-pondiente a los iones del precipitado.
Disolvente. Las variaciones en el valor de laconstante dieléctrica producen alteraciones en elvalor del producto de solubilidad. En general aldisminuir la constante dieléctrica del medio sefavorece la asociación iónica por lo que dismi-nuye el valor del producto de solubilidad, lo queimplica una disminución en la solubilidad.
Fuerza iónica. Si se aumenta la fuerza iónicade la disolución por la presencia de electrolitosque no contienen iones comunes con el precipi-tado, se origina un aumento de la solubilidad deacuerdo con lo expuesto en el capítulo 1.
Los factores que originan un desplazamientodel equilibrio de precipitación modificando lasolubilidad son fundamentalmente los siguientes:
Efecto del ion común. Se origina al adicionara la disolución sales cuyos iones son comunes alos que intervienen en el equilibrio de precipita-ción. Al aumentar el producto de las concentra-ciones iónicas (por el ion común), el sistema evo-luciona tendiendo a recuperar el equilibrio,produciendo una precipitación o lo que es lo mis-mo una disminución en la solubilidad del com-puesto.
Reacciones secundarias. En el caso de unadisminución de una o ambas concentracionesiónicas, producido por la existencia de equili-brios concurrentes ácido-base, de complejaciónu oxidación-reducción, el sistema tiende a recu-perar el equilibrio produciendo una disolucióndel precipitado o lo que es lo mismo un aumen-to en la solubilidad. La influencia de estas reac-ciones sobre el equilibrio principal se tratarámás adelante.
Capítulo 7: Equilibrios de precipitación 253
De acuerdo con los respectivos valores de KS se podríapensar que el carbonato de plata es ligeramente más inso-luble que el cloruro de plata. Sin embargo, esto no es cier-to a tenor de sus respectivas solubilidades. Para su cálcu-lo se sigue el siguiente procedimiento.
En primer lugar se escriben los correspondientes equi-librios de precipitación:
AgCl(S)→← Ag+ + Cl–
Ag2CO3(S)→← 2 Ag+ + CO3
2_
y sus respectivas constantes del producto de solubilidad:
KS = [Ag+] [Cl–] = 10–9,8
KS = [Ag+]2 [CO32_
] = 10–11,2
Si se tiene en cuenta que la solubilidad S en cada casose puede expresar como:
despejando de las ecuaciones anteriores las concentra-ciones de ion plata e ion cloruro así como de ion plata ycarbonato y aplicando las expresiones obtenidas a la ecua-ción del producto de solubilidad resulta:
de donde se obtienen fácilmente ambas solubilidades
Se observa cómo el cloruro de plata es realmente másinsoluble que el correspondiente carbonato.
S KS
Ag CO–1
2 3mol L= = =
−−
410
4103
1123 3 93
,,
S KSAgCl–1mol L= = =− −10 109 5 4 9, ,
K S S SS ( ) ( ) ( )Ag CO2 Ag CO Ag CO Ag CO3
2 3 2 3 2 3322 4= =
K S S SS ( ) ( ) ( )AgCl AgCl AgCl AgCl2= =
SAgCl Ag Cl= =+ −[ ] [ ]
SAg CO2 3
Ag2
CO= =+
−[ ] [ ]32
Comparar las solubilidades del cloruro de plata (KS = 10–9,8)y carbonato de plata (KS = 10–11,2)
RECUADRO 7.1
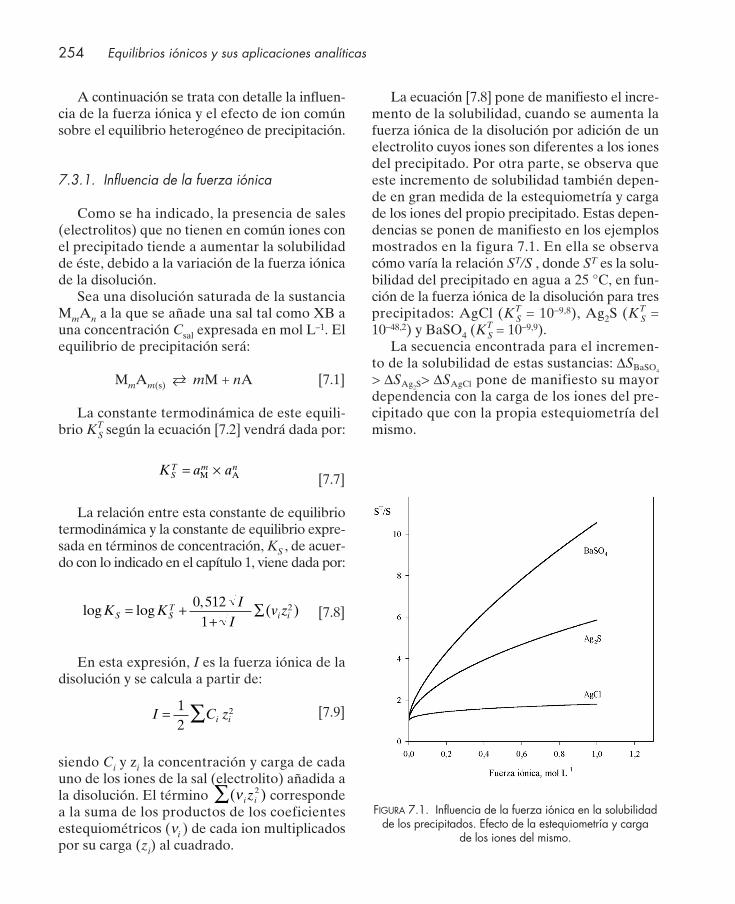
A continuación se trata con detalle la influen-cia de la fuerza iónica y el efecto de ion comúnsobre el equilibrio heterogéneo de precipitación.
7.3.1. Influencia de la fuerza iónica
Como se ha indicado, la presencia de sales(electrolitos) que no tienen en común iones conel precipitado tiende a aumentar la solubilidadde éste, debido a la variación de la fuerza iónicade la disolución.
Sea una disolución saturada de la sustanciaMmAn a la que se añade una sal tal como XB auna concentración Csal expresada en mol L–1. Elequilibrio de precipitación será:
MmAm(s)→← mM + nA [7.1]
La constante termodinámica de este equili-brio KT
S según la ecuación [7.2] vendrá dada por:
[7.7]
La relación entre esta constante de equilibriotermodinámica y la constante de equilibrio expre-sada en términos de concentración, KS , de acuer-do con lo indicado en el capítulo 1, viene dada por:
[7.8]
En esta expresión, I es la fuerza iónica de ladisolución y se calcula a partir de:
[7.9]
siendo Ci y zi la concentración y carga de cadauno de los iones de la sal (electrolito) añadida ala disolución. El término correspondea la suma de los productos de los coeficientesestequiométricos (νi ) de cada ion multiplicadospor su carga (zi) al cuadrado.
La ecuación [7.8] pone de manifiesto el incre-mento de la solubilidad, cuando se aumenta lafuerza iónica de la disolución por adición de unelectrolito cuyos iones son diferentes a los ionesdel precipitado. Por otra parte, se observa queeste incremento de solubilidad también depen-de en gran medida de la estequiometría y cargade los iones del propio precipitado. Estas depen-dencias se ponen de manifiesto en los ejemplosmostrados en la figura 7.1. En ella se observacómo varía la relación ST/S , donde ST es la solu-bilidad del precipitado en agua a 25 °C, en fun-ción de la fuerza iónica de la disolución para tresprecipitados: AgCl (KT
S = 10_9,8), Ag2S (KTS =
10_48,2) y BaSO4 (KTS = 10_9,9).
La secuencia encontrada para el incremen-to de la solubilidad de estas sustancias: ∆SBaSO4
> ∆SAg2S> ∆SAgCl pone de manifiesto su mayor
dependencia con la carga de los iones del pre-cipitado que con la propia estequiometría delmismo.
FIGURA 7.1. Influencia de la fuerza iónica en la solubilidadde los precipitados. Efecto de la estequiometría y carga
de los iones del mismo.
( )ν i iz2∑
I C zi i= ∑1
22
log log
,( )K K
II
v zS ST
i i= ++
∑0 5121
2
K a aST m n= ×M A
254 Equilibrios iónicos y sus aplicaciones analíticas

7.3.2. Efecto de ion común
Al adicionar una sal soluble a la disolución encontacto con un precipitado que tenga un ioncomún con el mismo, se origina una disminuciónde la solubilidad por desplazamiento del equili-brio heterogéneo de precipitación.
Para explicar este efecto de ion común, se haconsiderado un ejemplo concreto como es el cálcu-lo de la solubilidad del cromato de plata en aguay en una disolución a la que se le ha adicionadonitrato de plata 0,1 mol L–1 (Csal). A fin de sim-plificar el cálculo, se desprecia el aumento defuerza iónica que sufre la disolución debido a lasal añadida.
El equilibrio de precipitación será:
Ag2CrO4 (S)→← 2 Ag+ + CrO4
2_ [7.10]
y su constante del producto de solubilidad ven-drá dada por:
[7.11]
Dado que la solubilidad S se puede expresarcomo:
[7.12]
al despejar de esta ecuación las concentracio-nes de ion plata e ion cromato y sustituirlas enla correspondiente al producto de solubilidad,resulta:
[7.13]
de donde se obtiene fácilmente la relación:
[7.14]
Si se realiza un cálculo análogo, pero en pre-sencia de una concentración Csal (0,1 mol L–1) denitrato de plata, se pueden establecer los siguien-tes balances de materia:
[7.15]
[7.16]
Si se considera además que generalmente Csal>> SAg2CrO4
resulta que [Ag+] ≅ Csal, y al sustituiresta condición a la ecuación del producto de solu-bilidad se obtiene:
[7.17]
de donde se deduce que la solubilidad en estascondiciones es:
[7.18]
Comparando los resultados obtenidos en aguay con adición nitrato de plata se observa que lasolubilidad disminuye claramente ya que 10–4,17
>> 10–9,9.
7.4. Representación gráficade los equilibrios de precipitación
La representación gráfica de los equilibriosde precipitación permite obtener una valiosainformación sobre el proceso global de precipi-tación. Se pueden obtener fácilmente las condi-ciones en que tiene lugar el inicio de la precipi-tación o aquellas a las que la precipitación serácuantitativa. El método de representación nodifiere sustancialmente de los diagramas ya estu-diados para otros tipos de equilibrio.
S
KC
SAgCl
sal2
–1
(10(0,1)
10 mol L= = =−
−
)
,,
2
11 99 9
K C SS = ( )sal Ag CrO2 4
2
[ ]CrO Ag CrO24
24
− = S
[ ]Ag 2 Ag CrO sal2
+ = +S C4
SKS
Ag CrO
–1
2 4
10 mol L
= =
= =−
−
410
4
3
11 93 4 17
,,
K S S SS = =( ) ( )2 42
Ag CrO Ag CrO Ag CrO3
2 4 2 4 2 4
SAg CrO2 4
AgCrO= =
+−[ ]
[ ]2 4
2
KS = =+ − −[ ] [ ] ,Ag CrO242 11 910
Capítulo 7: Equilibrios de precipitación 255
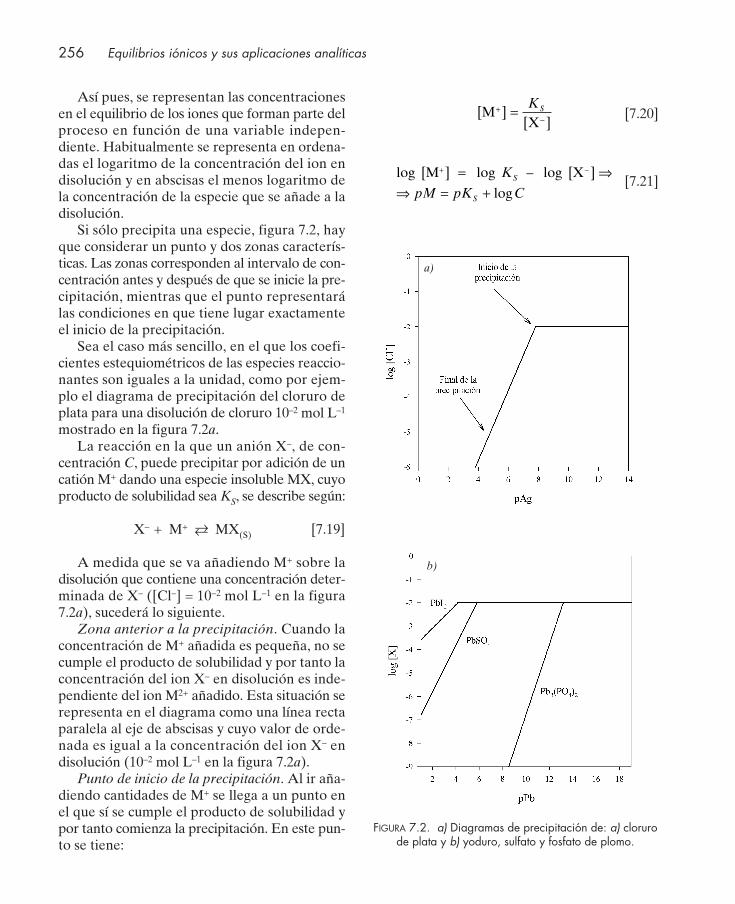
Así pues, se representan las concentracionesen el equilibrio de los iones que forman parte delproceso en función de una variable indepen-diente. Habitualmente se representa en ordena-das el logaritmo de la concentración del ion endisolución y en abscisas el menos logaritmo dela concentración de la especie que se añade a ladisolución.
Si sólo precipita una especie, figura 7.2, hayque considerar un punto y dos zonas caracterís-ticas. Las zonas corresponden al intervalo de con-centración antes y después de que se inicie la pre-cipitación, mientras que el punto representarálas condiciones en que tiene lugar exactamenteel inicio de la precipitación.
Sea el caso más sencillo, en el que los coefi-cientes estequiométricos de las especies reaccio-nantes son iguales a la unidad, como por ejem-plo el diagrama de precipitación del cloruro deplata para una disolución de cloruro 10–2 mol L–1
mostrado en la figura 7.2a. La reacción en la que un anión X–, de con-
centración C, puede precipitar por adición de uncatión M+ dando una especie insoluble MX, cuyoproducto de solubilidad sea KS, se describe según:
X– + M+ →← MX(S) [7.19]
A medida que se va añadiendo M+ sobre ladisolución que contiene una concentración deter-minada de X– ([Cl–] = 10–2 mol L–1 en la figura7.2a), sucederá lo siguiente.
Zona anterior a la precipitación. Cuando laconcentración de M+ añadida es pequeña, no secumple el producto de solubilidad y por tanto laconcentración del ion X– en disolución es inde-pendiente del ion M2+ añadido. Esta situación serepresenta en el diagrama como una línea rectaparalela al eje de abscisas y cuyo valor de orde-nada es igual a la concentración del ion X– endisolución (10–2 mol L–1 en la figura 7.2a).
Punto de inicio de la precipitación. Al ir aña-diendo cantidades de M+ se llega a un punto enel que sí se cumple el producto de solubilidad ypor tanto comienza la precipitación. En este pun-to se tiene:
[7.20]
[7.21]
FIGURA 7.2. a) Diagramas de precipitación de: a) clorurode plata y b) yoduro, sulfato y fosfato de plomo.
log log log
log
[M ] – [X ] + –= ⇒⇒ = +
K
pM pK CS
S
[ ]
[ ]M
X+
−= KS
256 Equilibrios iónicos y sus aplicaciones analíticas
a)
b)

Zona posterior a la precipitación. Una vez ini-ciada la precipitación, la concentración del ionen disolución irá disminuyendo, según una rela-ción lineal entre log [X–] y el valor de pM (espe-cie que estamos utilizando como precipitante)que se obtiene a partir de la expresión del pro-ducto de solubilidad según:
[7.22]
[7.23]
La ecuación [7.23] se corresponde con la ecua-ción de la recta que representa la disminución dela concentración de la especie que está precipi-tando, cuya ordenada en el origen es log KS ycuya pendiente tiene el valor +1.
El final de la precipitación, que no se produ-ce en el sentido estricto, puede considerarse alcan-zado cuando la concentración del ion en disolu-ción, C, se ha reducido a la milésima parte, o sea:
[X–]FINAL = [X–]INICIAL× 10–3 [7.24]
Para el caso más general, en que los coefi-cientes estequiométricos sean diferentes entre sí(ver figura 7.2b), la reacción de equilibrio es:
q Xp– + p Mq+ →← MpXq (S) [7.25]
y su producto de solubilidad vendrá expresadosegún la expresión:
[7.26]
De forma análoga a la descrita para el casoanterior, se deben considerar dos zonas y un pun-to característico.
La zona anterior al inicio de la precipitación,como en el caso anterior, donde el valor de laconcentración del ion a precipitar permanececonstante, se representa por una línea recta hori-zontal con un valor de ordenada que se corres-ponde con la concentración inicial.
El punto característico que marca el inicio dela precipitación se calcula despejando la concen-tración del ion precipitante de la expresión delproducto de solubilidad, ecuación [7.26]:
[7.27]
Finalmente, en la zona de precipitación, laconcentración del ion en disolución va dismi-nuyendo linealmente con el aumento de con-centración del ion precipitante según la expre-sión deducida a partir del producto desolubilidad, ecuación [7.26]:
[7.28]
de donde se deduce fácilmente:
[7.29] pM
ppK
qp
pXS= × − ×1
[ ]
]M
[X
q p S
p q
K+−
=
[ ]
[ ]X
M p S
q pq
K−+
=
KSp q q p= − +[ ] [ ]X M
log [ ] log log
log
X – [M ]+− = == +
K
K pMS
S
[ ]
[ ]X
M−
+= KS
Capítulo 7: Equilibrios de precipitación 257
CUADRO 7.3Ecuaciones deducidas para la zona de precipitación de Ag+ , Hg2
2+ y Pb2+ con cloruros
Ion pKS Equilibrio Ecuación
Ag+ 9,8 Ag+ + Cl_ →← AgCl (S) pAg = pKS— pCl
Hg22+ 17,2 Hg2
2+ + 2Cl_ →← Hg2Cl2 (S) pHg = pKS— 2pCl
Pb2+ 4,1 Pb2+ + 2Cl_ →← PbCl2 (S) pPb = pKS— 2pCl

Algunos autores muestran los diagramas deprecipitación sólo en la zona de precipitación.Así, por ejemplo, en la figura 7.3 se representael diagrama de precipitación de los cloruros deAg+, Hg2
2+ y Pb2+. Las rectas mostradas en estafigura corresponden a las expresiones que semuestran en el cuadro 7.3, que se deducen a par-tir de las ecuaciones del producto de solubilidad.
FIGURA 7.3. Diagrama de precipitación de los cloruros de Ag+, Hg2
2+ y Pb2+.
7.5. Precipitación fraccionada
En ciertas ocasiones, cuando existen en unadisolución varios iones capaces de precipitar fren-te a un reactivo común, es posible conseguir laprecipitación completa de uno de ellos sin quelleguen a precipitar los demás. Esta precipitacióncompleta o cuantitativa corresponde en Quími-ca Analítica a la precipitación de un 99,9% delion que precipita. En la práctica, esto quiere decirque cuando se haya precipitado cuantitativa-mente el primer ion y separado por filtración elcompuesto insoluble formado, en la disoluciónsólo debe quedar como máximo un 0,1% del cita-do ion (su concentración debe ser la milésimaparte de su concentración inicial).
Cuando los productos de solubilidad de los dis-tintos iones frente a un reactivo común sean sufi-cientemente diferentes, se consigue una precipita-ción fraccionada o escalonada, es decir, se producela separación cuantitativa de los mismos de formaescalonada, constituyendo ésta una de las formasmás antiguas de separación química.
Para asegurar una mejor comprensión de esteproceso, se va a considerar como ejemplo la posi-ble precipitación fraccionada de los haluros de pla-ta. Sea, por tanto, una disolución que contiene losiones cloruro, bromuro y yoduro en una concen-tración 0,01 mol L–1. Al ir añadiendo gradualmentenitrato de plata a esta disolución, los tres anionesiniciales reaccionan y dan la correspondiente salde plata según la reacción siguiente:
X– + Ag+ →← AgX(S) [7.30]
donde X– representa a cualquiera de los halurosmencionados. Los productos de solubilidad impli-cados son: AgCl (KS = 10–9,8), AgBr (KS = 10–12,3)y AgI (KS = 10–16,1).
Precipitará en primer lugar el haluro quenecesite menor concentración de plata, la cual sepuede calcular a partir de la expresión del pro-ducto de solubilidad:
[7.31]
De esta expresión se obtienen los siguientesvalores de [Ag+]: 10–7,8, 10–10,3 y 10–14,1 mol L–1 parala precipitación de AgCl, AgBr y AgI, respectiva-mente. A partir de estas concentraciones de plata,es fácil deducir que el primero en precipitar es elyoduro de plata, el segundo el bromuro y por últi-mo el cloruro. En la figura 7.4 se muestra el dia-grama de precipitación de estos haluros de plata.
Para conocer si la precipitación de cada ion escuantitativa, una vez conocido el orden de preci-pitación, se debe calcular cuál es la concentracióndel ion que ha precipitado cuando la concentra-ción de plata sea la necesaria para que precipiteel siguiente, y comparar ambos valores.
[ ]
[ ]Ag
X+
−= KS
258 Equilibrios iónicos y sus aplicaciones analíticas
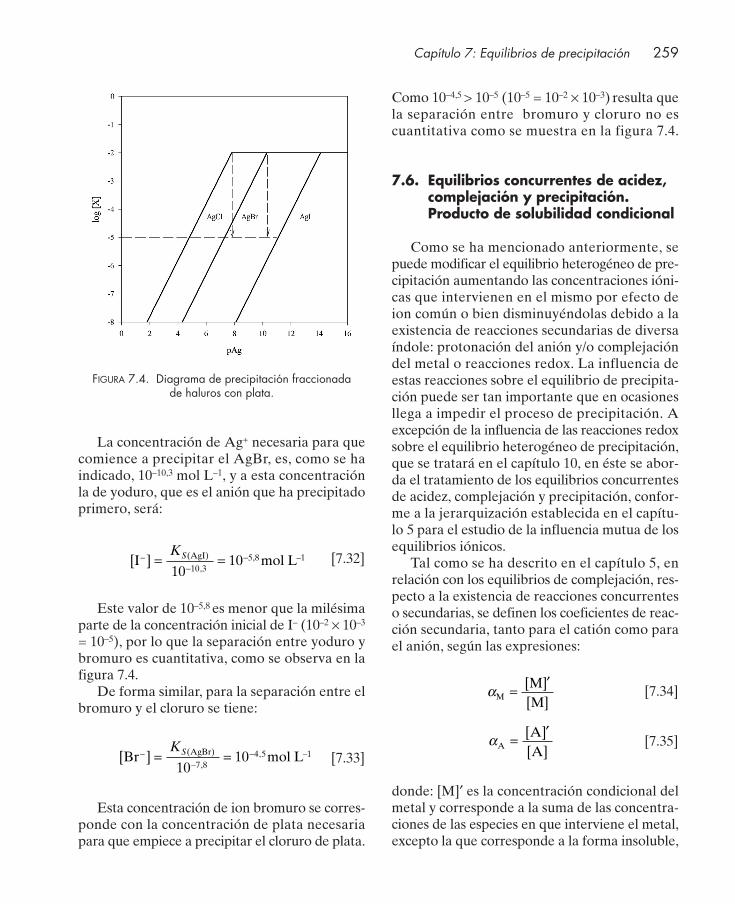
FIGURA 7.4. Diagrama de precipitación fraccionadade haluros con plata.
La concentración de Ag+ necesaria para quecomience a precipitar el AgBr, es, como se haindicado, 10–10,3 mol L–1, y a esta concentraciónla de yoduro, que es el anión que ha precipitadoprimero, será:
[7.32]
Este valor de 10–5,8 es menor que la milésimaparte de la concentración inicial de I– (10–2 × 10–3
= 10–5), por lo que la separación entre yoduro ybromuro es cuantitativa, como se observa en lafigura 7.4.
De forma similar, para la separación entre elbromuro y el cloruro se tiene:
[7.33]
Esta concentración de ion bromuro se corres-ponde con la concentración de plata necesariapara que empiece a precipitar el cloruro de plata.
Como 10–4,5 > 10–5 (10–5 = 10–2 × 10–3) resulta quela separación entre bromuro y cloruro no escuantitativa como se muestra en la figura 7.4.
7.6. Equilibrios concurrentes de acidez,complejación y precipitación.Producto de solubilidad condicional
Como se ha mencionado anteriormente, sepuede modificar el equilibrio heterogéneo de pre-cipitación aumentando las concentraciones ióni-cas que intervienen en el mismo por efecto deion común o bien disminuyéndolas debido a laexistencia de reacciones secundarias de diversaíndole: protonación del anión y/o complejacióndel metal o reacciones redox. La influencia deestas reacciones sobre el equilibrio de precipita-ción puede ser tan importante que en ocasionesllega a impedir el proceso de precipitación. Aexcepción de la influencia de las reacciones redoxsobre el equilibrio heterogéneo de precipitación,que se tratará en el capítulo 10, en éste se abor-da el tratamiento de los equilibrios concurrentesde acidez, complejación y precipitación, confor-me a la jerarquización establecida en el capítu-lo 5 para el estudio de la influencia mutua de losequilibrios iónicos.
Tal como se ha descrito en el capítulo 5, enrelación con los equilibrios de complejación, res-pecto a la existencia de reacciones concurrenteso secundarias, se definen los coeficientes de reac-ción secundaria, tanto para el catión como parael anión, según las expresiones:
[7.34]
[7.35]
donde: [M]′ es la concentración condicional delmetal y corresponde a la suma de las concentra-ciones de las especies en que interviene el metal,excepto la que corresponde a la forma insoluble,
αA
AA
=′[ ]
[ ]
αM
MM
=′[ ]
[ ]
[ ]Br mol L(AgBr)
,, –1−
−−= =
KS
1010
7 84 5
[ ] ,I mol L(AgI)
,–1−
−−= =
KS
1010
10 35 8
Capítulo 7: Equilibrios de precipitación 259

compuesto principal, y [A]′ es la concentracióncondicional del anión y se corresponde a la sumade las concentraciones de las especies en queinterviene el anión, excepto la que correspondea la forma insoluble. [M] y [A] son las concen-traciones de metal y anión procedentes de la diso-lución del precipitado en ausencia de reacciónsecundaria.
En este contexto, para un compuesto insolu-ble MmAn se puede definir su producto de solu-bilidad condicional, KS′, tal como:
[7.36]
de donde:
[7.37]
Dado que αM ≥ 1 y αA ≥ 1 se tiene que KS′ ≥ KS,por lo que la presencia de reacciones secundariassiempre provoca un aumento de la solubilidad delprecipitado. Es decir, la solubilidad condicional essiempre mayor que la solubilidad en condicionestermodinámicas.
Si se toman logaritmos en la expresión [7.37]y se cambia de signo se obtiene:
[7.38]
En resumen, el producto de solubilidad con-dicional permite poner de manifiesto, de unamanera sencilla, el efecto de reacciones secun-darias sobre el equilibrio de precipitación.
Conforme a lo ya indicado, las reaccionessecundarias que se consideran en este tratamientoson las siguientes.
A) Reacciones ácido-base:
Protonación del anión:
An_ + H+ →← HA(n_1)_[7.39]
A(n_1)_+ H+ →← H2A
(n_2)_... [7.40]
Formación de compuestos hidroxilados delmetal:
Mn+ + OH_ →← MOH(n_1)+[7.41]
MOH(n_1)++ OH_ →← M(OH)2
(n_2)+... [7.42]
B) Reacciones de complejación:
Complejación del metal, con el mismo ligan-do con el que precipita o con otro diferente:
Mn+ + L →← ML(n_1)+[7.43]
ML(n_1)++ L →← ML2
(n_2)+...... [7.44]
De acuerdo con estos equilibrios concurren-tes se pueden establecer los siguientes balancesde materia:
[7.45]
[7.46]
A continuación se aborda el estudio detalla-do de cada una de estas influencias sobre el equi-librio heterogéneo de precipitación, haciendo usode un ejemplo concreto en cada caso.
7.6.1. Reacciones secundarias de protonacióndel anión
Todos los precipitados que están constituidospor aniones procedentes de ácidos débiles, talescomo sulfuros, carbonatos, etc., aumentan susolubilidad en medio ácido, ya que al protonar-se el anión disminuye su concentración como ionlibre en la disolución y por tanto el precipitadose disuelve, aumentando su solubilidad.
Sea el equilibrio heterogéneo de precipitación:
MmAn(s)→← mM + nA [7.1]
[ ] [ ] [ ] [ ) ]
[ ] [ ]
( ) ( )
( ) ( )
M M MOH M(OH
ML ML
′ = + + +
+ + +
+ − + − +
− + − +
n n n
n n
12
2
12
2
�
� �
[ ] [ ] [ ] [ ]( ) ( )A A HA H A′ = + + +− − − − −n n n12
2 �
pK pK m nS S′ = − −log logα αM A
′ =K KS Smα αM A
n
′ = ′ ′ =KSm n m m n n[ ] [ ] [ ] [ ]M A M AM Aα α
260 Equilibrios iónicos y sus aplicaciones analíticas

cuya constante de producto de solubilidad vienedada por:
[7.3]
y la solubilidad por:
[7.6]
Si el anión A procede de un ácido débil, alestablecerse los equilibrios de protonacióndados en las expresiones [7.39] y [7.40], su coe-ficiente de reacción secundaria respecto al ionhidrógeno, de acuerdo con lo expuesto en elcapítulo 5, será:
[7.47]
donde β son las respectivas constantes de proto-nación y x el número de protones del ácido poli-protónico formado a partir de la especie An–.
Al sustituir el valor de αA(H) en la expresióndel producto de solubilidad condicional, expre-sión [7.37], se tiene:
[7.48]
y por tanto, la solubilidad, S′, de acuerdo con laecuación [7.6], será:
[7.49]
A continuación se realiza el cálculo numéricocorrespondiente a un caso concreto con objeto deconstatar el aumento de la solubilidad al incre-mentarse la concentración de iones hidrógeno enla disolución, esto es, al disminuir el pH del medio.
Se desea calcular el incremento en la solubi-lidad del carbonato de plata (KS = 10–11,2) en unadisolución reguladora a pH = 4 con relación auna disolución acuosa. El equilibrio heterogéneode precipitación viene dado por:
Ag2CO3(S)→← 2Ag+ + CO3
2_[7.50]
mientras que las reacciones secundarias corres-ponden a la protonación del ion carbonato:
CO32_
+ H+ →← HCO3
_[7.51]
CO32_
+ 2H+ →← H2CO3 [7.52]
En primer lugar se calcula la solubilidad delcarbonato de plata en agua, considerando laexpresión [7.6]:
[7.53]
Con objeto de evaluar la influencia del pH seha de calcular inicialmente el coeficiente de reac-ción secundaria del carbonato a pH = 4 confor-me a la ecuación [7.47]:
[7.54]
siendo:
[7.55]
y Ka1 y Ka2 las constantes de acidez del ácidocarbónico, cuyos valores son: 10–6,3 y 10–10,3, res-pectivamente. Al sustituir estos valores en laexpresión [7.54] y considerar el pH de trabajo,se llega a:
[7.56]
αCO (H
8,6
3
10
),
,
= + × +
+ × ≅
−
−
1 10 10
10 10
10 3 4
16 6 8
β β1
22
1 2
1 1H H= =K K Ka a a
α β βCO (H) 1
H2H
3[H [H= + ++ +1 2] ]
SK
m nS
m nm n= = =+
−−10
410
11 2
3 3 93 1,
, mol L–
′ = ′
+SK
m nS
m nm n
′ = = + + +
+
+ +
+
K K KS Sn
S
xx n
α β β
βA H
H H
H
H H
H
( ) ( [ ] [ ]
[ ] )
1 1 22 �
�
α β β βA(HH H HH H H) [ ] [ ] [ ]= + + + ++ + +1 1 2
2 � xx
S
Km n
Sm n
m n= +
KSm n= [ ] [ ]M A
Capítulo 7: Equilibrios de precipitación 261
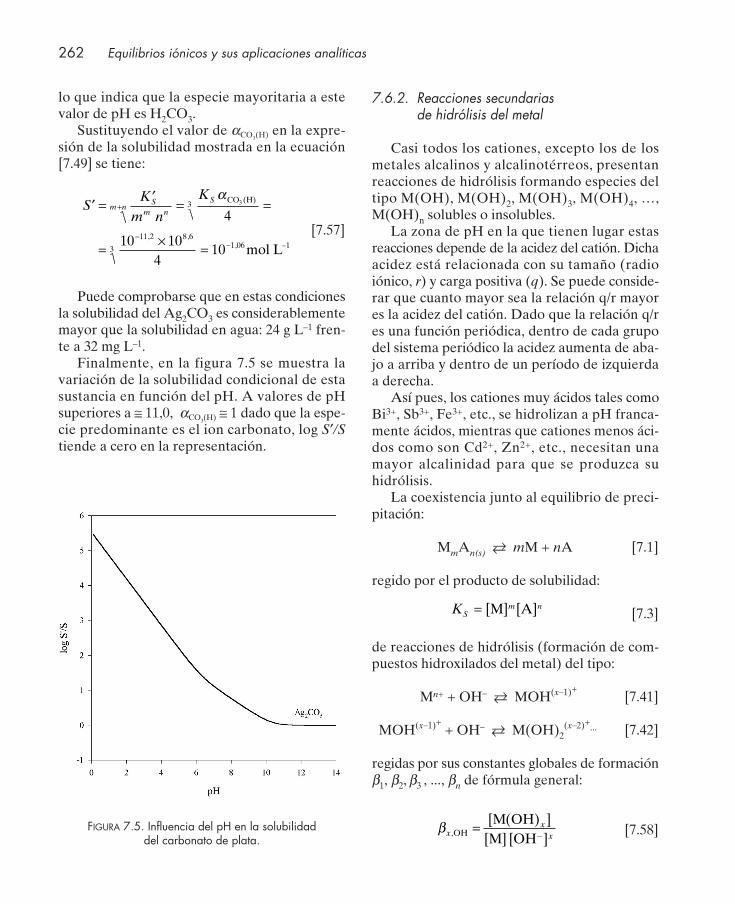
lo que indica que la especie mayoritaria a estevalor de pH es H2CO3.
Sustituyendo el valor de αCO3(H) en la expre-sión de la solubilidad mostrada en la ecuación[7.49] se tiene:
[7.57]
Puede comprobarse que en estas condicionesla solubilidad del Ag2CO3 es considerablementemayor que la solubilidad en agua: 24 g L–1 fren-te a 32 mg L–1.
Finalmente, en la figura 7.5 se muestra lavariación de la solubilidad condicional de estasustancia en función del pH. A valores de pHsuperiores a ≅ 11,0, αCO3(H) ≅ 1 dado que la espe-cie predominante es el ion carbonato, log S′/Stiende a cero en la representación.
FIGURA 7.5. Influencia del pH en la solubilidaddel carbonato de plata.
7.6.2. Reacciones secundariasde hidrólisis del metal
Casi todos los cationes, excepto los de losmetales alcalinos y alcalinotérreos, presentanreacciones de hidrólisis formando especies deltipo M(OH), M(OH)2, M(OH)3, M(OH)4, …,M(OH)n solubles o insolubles.
La zona de pH en la que tienen lugar estasreacciones depende de la acidez del catión. Dichaacidez está relacionada con su tamaño (radioiónico, r) y carga positiva (q). Se puede conside-rar que cuanto mayor sea la relación q/r mayores la acidez del catión. Dado que la relación q/res una función periódica, dentro de cada grupodel sistema periódico la acidez aumenta de aba-jo a arriba y dentro de un período de izquierdaa derecha.
Así pues, los cationes muy ácidos tales comoBi3+, Sb3+, Fe3+, etc., se hidrolizan a pH franca-mente ácidos, mientras que cationes menos áci-dos como son Cd2+, Zn2+, etc., necesitan unamayor alcalinidad para que se produzca suhidrólisis.
La coexistencia junto al equilibrio de preci-pitación:
MmAn(s)→← mM + nA [7.1]
regido por el producto de solubilidad:
[7.3]
de reacciones de hidrólisis (formación de com-puestos hidroxilados del metal) del tipo:
Mn+ + OH_ →← MOH(x_1)+[7.41]
MOH(x_1)++ OH_ →← M(OH)2
(x_2)+... [7.42]
regidas por sus constantes globales de formaciónβ1, β2, β3 , ..., βn de fórmula general:
[7.58] βx
xx,OH
M OHM OH
=−
[ ( ) ][ ] [ ]
KSm n= [ ] [ ]M A
′ = ′ = =
= × =
+
−−
SK
m n
KS
m nm n
S αCO (H)
–1
3
mol L
4
10 104
10
3
11 2 8 6
3 1 06, ,
,
262 Equilibrios iónicos y sus aplicaciones analíticas

nos lleva a definir un coeficiente de reacciónsecundaria o parásita para el metal de la siguien-te forma:
[7.59]
tal como se ha indicado en el capítulo 5 sobre losequilibrios de complejación. En estas condicio-nes el producto de solubilidad condicional vieneexpresado por:
[7.60]
o bien por:
[7.61]
Si αM(OH) >> 1 se tiene que KS′ > KS, lo queimplica que el equilibrio de precipitación descritopor la ecuación [7.1] está desplazado hacia la dere-cha, es decir hacia la disolución del precipitado.
Si como es común en estos casos, el equilibriode precipitación se trata realmente de la forma-ción de un hidróxido insoluble, tal como:
Mn+ + nOH_ →← M(OH)n(S) [7.62]
con el que coexisten los equilibrios concurrentesde formación de especies hidroxiladas del metal,el producto de solubilidad condicional, de acuer-do con al ecuación [7.61] viene dado por:
[7.63]
En esta ecuación, para calcular el valor deαM(OH) no se considera el término correspon-diente a la formación del precipitado, esto es,βn,OH [OH–]n.
Una vez establecida la expresión para el pro-ducto de solubilidad condicional, para el cálculode la solubilidad se ha considerar que en estecaso, el ion hidroxilo es tanto el anión precipi-tante como el ligando implicado en la reacciónsecundaria de hidrólisis del metal. En estas con-diciones, la solubilidad viene dada por la con-centración de metal libre en disolución, esto es:
[7.64]
Al sustituir el valor de KS′ en esta expresiónse tiene:
[7.65]
Como ejemplo concreto de aplicación del tra-tamiento realizado se va a considerar la varia-ción de la solubilidad del hidróxido de cinc enfunción del pH. El equilibrio heterogéneo de pre-cipitación de este hidróxido será:
Zn2+ + 2OH_ →← Zn(OH)2(S)
KS = 10–17,2 [7.66]
Antes de la precipitación del hidróxido, lavariación de la acidez del medio provocará la for-mación de la especie hidroxilada
Zn2+ + OH_ →← Zn(OH)+
β1 = 104,4 [7.67]
en competencia con el ion Zn2+, mientras que apH básico se favorecerá la formación de lasespecies:
Zn2+ + 3OH_ →← Zn(OH)3
_
β3 = 1014,2 [7.68]
Zn2+ + 4OH_ →← Zn(OH)42_
β4 = 1015,5 [7.69]
′ =
+ + + +− − −
−S
KS xx
n
( [ ] [ ] ] )
[ ], ;1 1 2
2β β βOH OH ,OHOH OH [OH
OH
�
[ ]
[ ]M
OHn S
nS
K+−
= ′ =′
′ = = + +
+ + +
−
− −
K K KS S S
xx
α β
β βM(OH) 1,OH
2,OH OH
[OH
[OH OH
( ]
] [ ] ),
12 �
K KS S
xx
= + + +
+
− −
−
( ] ]
] )
1 2β β
β1,OH 2,OH
,OHm
[OH [OH
[OH
�
�
′ =K KS S
mαM(OH)
α β ββ
M(OH) 1,OH 2,OH
OH
[OH [OH
OH
= + + ++
− −
−
1 2] ]
[ ],
�
� xx
Capítulo 7: Equilibrios de precipitación 263

originando la disolución del hidróxido, al com-portarse éste como un anfótero.
En este caso, la expresión de la solubilidad enfunción del pH, de acuerdo con la ecuación [7.65],se puede escribir como:
[7.70]
En la figura 7.6 se muestra la variación de logS′ con el pH. Se observa que a valores de pH áci-do el cinc se encuentra en las formas solubles.La solubilidad va disminuyendo hasta alcanzarla forma insoluble, mientras que a valores de pHfrancamente básico, la presencia de reaccionessecundarias del cinc con el ion hidroxilo produ-ce un aumento de la solubilidad.
FIGURA 7.6. Variación de la solubilidad condicionaldel hidróxido de cinc en función del pH.
7.6.3. Reacciones secundarias de complejación
Dentro de este apartado se pueden considerardos casos significativos: 1) el anión precipitanteforma complejos con otro metal y 2) el catión
forma complejos con un ligando, que puede ser elmismo que constituye el precipitado, como en elcaso de los OH– o bien un ligando diferente. Deambas opciones, las reacciones de complejaciónque afectan al metal son probablemente las quese producen con más frecuencia y por ello seránobjeto de estudio a continuación.
Para abordar el estudio de estos equilibriosconcurrentes, como en los apartados preceden-tes, se han de calcular los valores de los coefi-cientes de reacciones secundaria y posteriormenteel producto de solubilidad condicional. Por com-paración entre los valores de la solubilidad enagua y los calculados a partir de la constante con-dicional, se puede observar el aumento de solu-bilidad que este tipo de reacciones secundariasproduce.
En general, si el metal reacciona con los ioneshidroxilo y con otros ligandos L1, L2, …, Lm pre-sentes en la disolución, se cumplirá que:
[7.71]
y por tanto:
[7.72]
donde m es el número de ligandos con los que elmetal tiene reacciones secundarias. En los casosde importancia práctica el término m – 1 será des-preciable, con lo que el coeficiente de reacciónsecundaria total será la suma de los coeficientesde reacción secundaria parcial.
Es común, en la práctica, que el equilibrioheterogéneo de precipitación entre en compe-tencia con un solo equilibrio de formación de
α α α αM M(OH) M(L ) M(L )1 2
( 1)= + + + − −� m
αM
MMM MOH] [M(OH M(OH
MM(L M(L M(L
MM(L M(L M(L
M
= ′ =
= + + + + +
+ + + + +
+ + + + +
[ ][ ][ ] [ ) ] [ ) ]
[ ][ )] [ ) ] [ ) ]
[ ][ )] [ ) ] [ ) ]
[ ]
2
1 1 2 1
2 2 2 2
�
�
��
n
n
n
′ =
+ + +− − −
−S
KS ( ] ] ] )
[ ]
1 3 4
2
β β β1,OH 3,OH 4,OH[OH [OH [OH
OH
264 Equilibrios iónicos y sus aplicaciones analíticas

complejos, originado por la adición de un ligan-do a la disolución y que, además, la reacciónsecundaria de formación de especies hidroxila-das del metal sea despreciable. Teniendo encuenta estas condiciones, la expresión para αMquedaría reducida a:
[7.73]
o bien a:
[7.74]
de acuerdo con lo indicado en el capítulo 5 enreferencia a los equilibrios de formación decomplejos. En esta expresión los valores de βcorresponden a las respectivas constantes deformación global de cada uno de los complejosformados por el ion metálico con el ligando, y1,2, ... , n es la estequiometría de cada uno deellos.
En estas condiciones, para el equilibrio hete-rogéneo de precipitación:
MmAn(s)→← mM + nA [7.1]
su producto de solubilidad condicional será:
[7.75]
del cual se deduce fácilmente la solubilidad.El siguiente ejemplo hace referencia al caso
de complejación del metal con un ligando dife-rente con el que precipita. Así, se aborda el estu-dio del efecto que produce sobre la solubilidaddel cloruro de plata la presencia de amoniaco 0,1mol L–1 en la disolución.
De acuerdo con la reacción de precipitacióndel cloruro de plata:
AgCl(S)→← Ag+ + Cl_
[7.76]
la solubilidad de la sal en agua será:
[7.77]
Por otra parte, si se adiciona amoniaco a esteequilibrio heterogéneo, el catión plata reaccio-na con este ligando para formar los siguientescomplejos aminados:
Ag+ + NH3→← Ag(NH3)
+ β1= 103,4 [7.78]
Ag+ + 2NH3→← Ag(NH3)2
+ β1= 107,4 [7.79]
Con estos datos se calcula el coeficiente dereacción secundaria para la plata de acuerdo conla ecuación [7.74]:
[7.80]
su producto de solubilidad condicional:
[7.81]
y finalmente su solubilidad en presencia de amo-niaco:
[7.82]
Al sustituir los valores numéricos en la expre-sión anterior resulta un valor de S′ = 10–2,2 mol L–1.Así pues, la solubilidad aumenta de 10–4,9 a 10–2,2
mol L–1 en presencia de amoniaco 0,1 mol L–1. Enla figura 7.7 se observa el incremento exponencialde la solubilidad del cloruro de plata a medida queaumenta la concentración de amoniaco añadidaal medio de reacción.
El ejemplo que se describe a continuaciónhace referencia al caso de complejación del metalcon el mismo ligando con el cual precipita, estoes, una situación similar a la estudiada en el apar-tado anterior referente al fenómeno de anfote-rismo en la precipitación de hidróxidos en medio
′ = ′ = + +S K KS S ( [ ] [ ] )1 1 3 2 32β βNH NH
′ =K KS S αAg(NH )3
α β βAg(NH 3NH NH
31 1 2 3
2) [ ] [ ]= + +
S KSAgCl–1mol L= = =− −10 109 8 4 9, ,
KS = =+ − −[ ] ] ,Ag [Cl 10 9 8
′ =K KS S
mαM(L)
α β β βM(L L L L) [ ] [ ] [ ]= + + + +1 1 22 � n
n
α αM M(L)
M] [ML] [ML MLM
= = + + + +[ ] [ ][ ]
2 � n
Capítulo 7: Equilibrios de precipitación 265
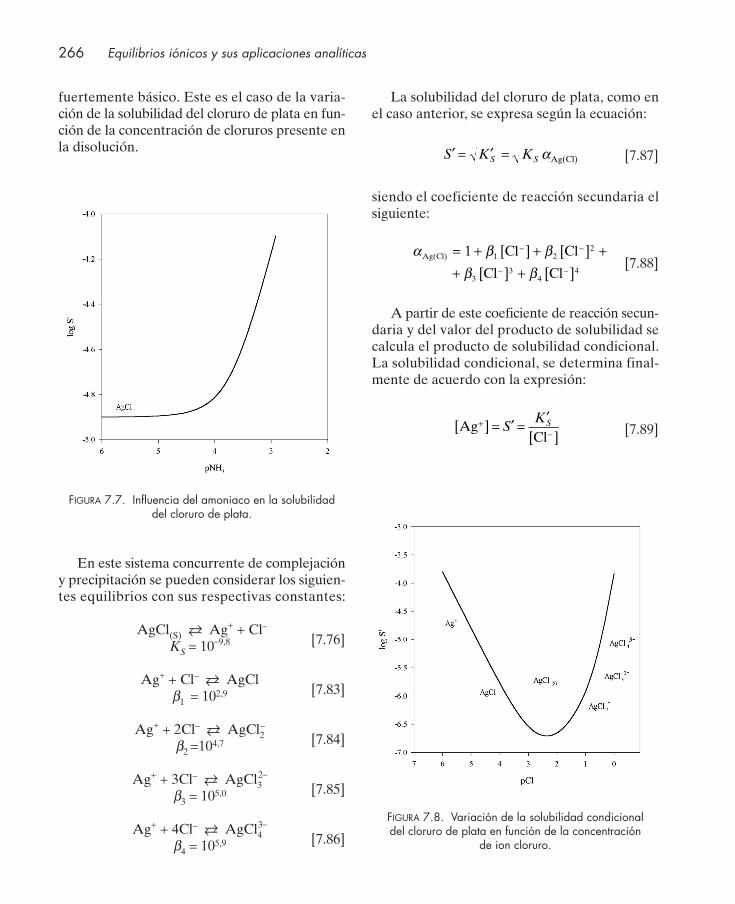
fuertemente básico. Este es el caso de la varia-ción de la solubilidad del cloruro de plata en fun-ción de la concentración de cloruros presente enla disolución.
FIGURA 7.7. Influencia del amoniaco en la solubilidaddel cloruro de plata.
En este sistema concurrente de complejacióny precipitación se pueden considerar los siguien-tes equilibrios con sus respectivas constantes:
AgCl(S)→← Ag+ + Cl_
KS = 10_9,8 [7.76]
Ag+ + Cl_ →← AgClβ1 = 102,9 [7.83]
Ag+ + 2Cl_ →← AgCl2_
β2 =104,7 [7.84]
Ag+ + 3Cl_ →← AgCl32_
β3 = 105,0 [7.85]
Ag+ + 4Cl_ →← AgCl43_
β4 = 105,9 [7.86]
La solubilidad del cloruro de plata, como enel caso anterior, se expresa según la ecuación:
[7.87]
siendo el coeficiente de reacción secundaria elsiguiente:
[7.88]
A partir de este coeficiente de reacción secun-daria y del valor del producto de solubilidad secalcula el producto de solubilidad condicional.La solubilidad condicional, se determina final-mente de acuerdo con la expresión:
[7.89]
FIGURA 7.8. Variación de la solubilidad condicionaldel cloruro de plata en función de la concentración
de ion cloruro.
[ ]
[ ]Ag
Cl+
−= ′ =
′S
KS
α β ββ β
Ag(Cl)21 Cl Cl
Cl Cl
= + + ++ +
− −
− −
1 2
33
44
[ ] [ ]
[ ] [ ]
′ = ′ =S K KS S αAg(Cl)
266 Equilibrios iónicos y sus aplicaciones analíticas

[7.90]
la cual es similar a la desarrollada en el aparta-do anterior para el fenómeno de anfoterismo enla precipitación de hidróxidos.
En la figura 7.8 se representa la variación dela solubilidad condicional del cloruro de platafrente a la concentración de cloruro libre y seobserva una dependencia similar a la mostradaen la figura 7.6 para el fenómeno de anfoteris-mo en la precipitación de hidróxidos de ionesmetálicos. En efecto, inicialmente la solubilidaddisminuye debido a la formación de la especieinsoluble AgCl, y posteriormente, cuando seaumenta la concentración de cloruro el precipi-tado se disuelve por formación de los corres-pondientes complejos, con lo que se incremen-ta la solubilidad.
7.6.4. Influencia conjunta de reaccionessecundarias de acidez y complejación
En este apartado se va a considerar la influen-cia simultánea del pH y de la formación de com-plejos sobre el equilibrio de precipitación. Enconcreto, se contempla el caso más común, queconsiste en la existencia de un ligando que for-ma complejos con una de las especies activas enla precipitación, el ion metálico, y cuyo compor-tamiento depende del pH del medio.
Sea el equilibrio de precipitación:
MmAn(s)→← mM + nA [7.1]
al que se adiciona a un determinado valor de pHun ligando L, anión procedente de un ácido débilHxL.
En general, si se desprecia la reacción secun-daria del ion metálico con los iones hidroxilos yse considera sólo su interacción con el ligando L,el coeficiente de reacción secundaria con esteligando, αM(L), vendrá dado por la expresión [7.74]:
[7.74]
Por otra parte, debido a que la interaccióncon el pH se manifiesta fundamentalmentesobre el ligando L, y por tanto sobre la forma-ción de los complejos metal-ligando, en laexpresión anterior se ha de hacer uso de cons-tantes condicionales, por lo que tal expresiónse transforma en:
[7.91]
De acuerdo con lo expuesto en el capítulo 5,estas constantes condicionales vienen dadas porla expresión:
[7.92]
donde αL(H) es el coeficiente de reacción secun-daria del ligando con el ion hidrógeno dado poruna expresión similar a la [7.47]:
[7.47]
Al combinar las ecuaciones [7.91] y [7.92] setiene:
[7.93]
En estas condiciones, el producto de solubi-lidad condicional será:
[7.75]
del cual es posible calcular fácilmente la solu-bilidad.
′ =K KS S
mαM(L)
α βα
βα
βα
M(LL(H) L(H)
L(H)
L L
L
) [ ]( )
[ ]
( )
[ ]
= + + +
+
1 1 22
2 �
� nn
n
α β β βL(H) 1H
2H H[H [H H= + + + ++ + +1 2] ] [ ]� x
x
′ =β βαi i i
1( )L(H)
α β β βM(L L L L) [ ] [ ] [ ]= + ′ + ′ + + ′1 1 22 � n
n
α β β βM(L L L L) [ ] [ ] [ ]= + + + +1 1 22 � n
n
′ = + + + +− − − −
−S
KS (1 Cl Cl Cl ClCl
2β β β β1 2 33
44[ ] [ ] [ ] [ ] )
[ ]
Capítulo 7: Equilibrios de precipitación 267
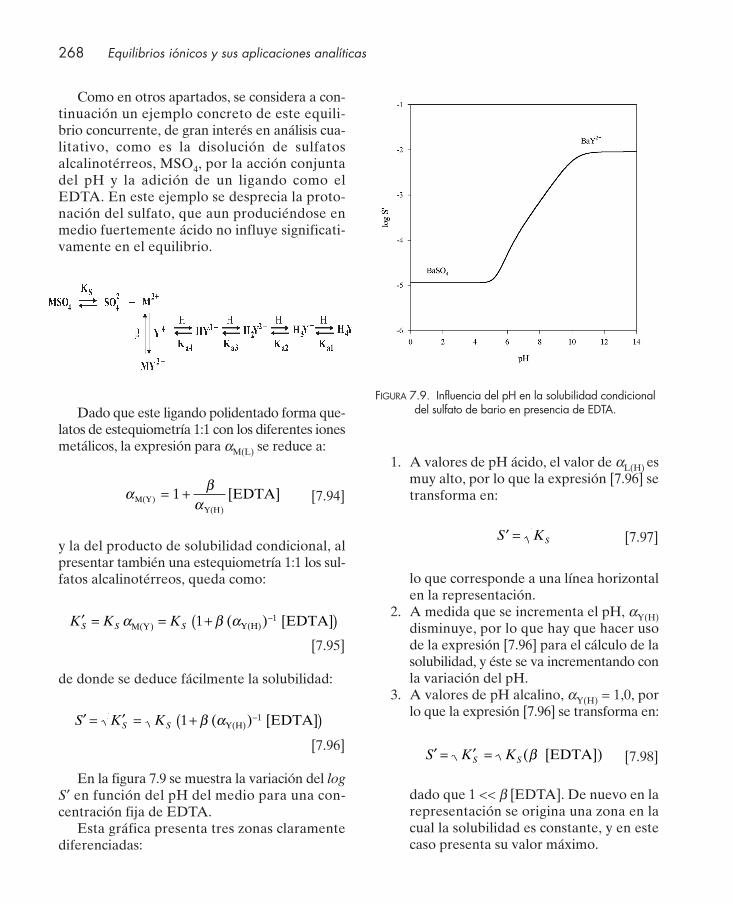
Como en otros apartados, se considera a con-tinuación un ejemplo concreto de este equili-brio concurrente, de gran interés en análisis cua-litativo, como es la disolución de sulfatosalcalinotérreos, MSO4, por la acción conjuntadel pH y la adición de un ligando como elEDTA. En este ejemplo se desprecia la proto-nación del sulfato, que aun produciéndose enmedio fuertemente ácido no influye significati-vamente en el equilibrio.
Dado que este ligando polidentado forma que-latos de estequiometría 1:1 con los diferentes ionesmetálicos, la expresión para αM(L) se reduce a:
[7.94]
y la del producto de solubilidad condicional, alpresentar también una estequiometría 1:1 los sul-fatos alcalinotérreos, queda como:
[7.95]
de donde se deduce fácilmente la solubilidad:
[7.96]
En la figura 7.9 se muestra la variación del logS′ en función del pH del medio para una con-centración fija de EDTA.
Esta gráfica presenta tres zonas claramentediferenciadas:
FIGURA 7.9. Influencia del pH en la solubilidad condicionaldel sulfato de bario en presencia de EDTA.
1. A valores de pH ácido, el valor de αL(H) esmuy alto, por lo que la expresión [7.96] setransforma en:
[7.97]
lo que corresponde a una línea horizontalen la representación.
2. A medida que se incrementa el pH, αY(H)disminuye, por lo que hay que hacer usode la expresión [7.96] para el cálculo de lasolubilidad, y éste se va incrementando conla variación del pH.
3. A valores de pH alcalino, αY(H) = 1,0, porlo que la expresión [7.96] se transforma en:
[7.98]
dado que 1 << β [EDTA]. De nuevo en larepresentación se origina una zona en lacual la solubilidad es constante, y en estecaso presenta su valor máximo.
′ = ′ =S K KS S ( [ ])β EDTA
′ =S KS
′ = ′ = +( )−S K KS S 1 1β α( ) [ ]Y(H) EDTA
′ = = +( )−K K KS S Sα β αM(Y Y(H) EDTA) ( ) [ ]1 1
α β
αM(YY(H
EDTA]))
[= +1
268 Equilibrios iónicos y sus aplicaciones analíticas

7.1. Indicar si son verdaderas (V) o falsas (F) las siguientes afirmaciones:
[ ] A medida que un precipitado envejece es más fácil de disolver[ ] Un compuesto precipita más lentamente al tener una KS menor[ ] En el equilibrio de precipitación ocurren simultáneamente los procesos de precipitación y disolución
7.2. ¿Cuál de los dos precipitados, MA (KS = 10–7) y MA3 (KS = 10–12), será más insoluble?
7.3. Por efecto de ion común se puede:
[ ] Aumentar la solubilidad de un precipitado[ ] Disminuir la solubilidad de un precipitado[ ] No se altera la solubilidad de un precipitado
mientras que por un aumento de temperatura se puede:
[ ] Aumentar la solubilidad de un precipitado[ ] Disminuir la solubilidad de un precipitado[ ] No se altera la solubilidad de un precipitado
7.4. Un electrolito con un ion común con un precipitado, hace que la solubilidad del mismo
[ ] Aumente[ ] Disminuya[ ] No varíe
7.5. ¿Qué factores experimentales permiten modificar el producto de solubilidad y la solubilidad de un com-puesto poco soluble? (Marcar con una X.)
Factor experimental S KS
Fuerza iónicaEfecto de ion comúnTemperaturaPresencia de ligandos complejantes
7.6. Los iones sulfato y Pb2+ forman un precipitado de sulfato de plomo. Cómo afectará a la solubilidad de estaespecie, la adición de:
Aumento de S Disminución de S No se afecta SAcetato de sodioÁcido nítricoCloruro de sodioEtanol
Capítulo 7: Equilibrios de precipitación 269
Cuestiones

7.7. Si se añade NaBr a una disolución en contacto con AgBr sólido:
[ ] Aumentará la solubilidad del bromuro de plata[ ] Se alterará el producto de solubilidad del bromuro de plata[ ] Disminuirá la solubilidad del bromuro de plata
7.8. Las reacciones secundarias de protonación de un anión que forma parte de un precipitado:
[ ] Incrementan su solubilidad[ ] Modifican apreciablemente el valor de KS
[ ] El precipitado se hace más insoluble en medio ácido[ ] El precipitado incrementa su solubilidad en medio básico con respecto a agua destilada
7.9. Indicar con una X aquellos sistemas concurrentes con el equilibrio heterogéneo de precipitación que permitenincrementar la insolubilidad de un precipitado:
[ ] Protonación del anión que forma parte del precipitado[ ] Adición de una sal que contiene uno de los iones del precipitado[ ] Formación de complejos del catión Mn+ que constituye el precipitado[ ] Modificación de la fuerza iónica de la disolución
7.10. ¿Qué constantes son necesarias para evaluar la solubilidad del BaSO4 en una disolución de EDTA a pH = 8?
270 Equilibrios iónicos y sus aplicaciones analíticas
Seminarios: problemas numéricos
• Objetivo pedagógicoDesarrollo de diferentes tipos de problemas sobre losaspectos básicos de los equilibrios heterogéneos deprecipitación y la influencia de diversos factores sobrelos mismos. Estudio de la presencia de diversos equi-librios concurrentes: acidez y formación de comple-jos sobre la solubilidad de los precipitados.
Solubilidad y producto de solubilidad
1. Calcular el producto de solubilidad del Ag3PO4, sabien-do que su solubilidad en agua es de 5,2 × 10–3 g L–1.Peso molecular: Ag3PO4 = 418,61.
Para esta sustancia, se tiene el siguiente equilibrioheterogéneo de solubilidad:
Ag3PO4(S)→← 3 Ag+ + PO4
3_
cuya constante o producto de solubilidad responde ala expresión:
y la solubilidad viene dada por:
Al sustituir las concentraciones de los iones platay fosfato, en función de la solubilidad, en la expre-sión del producto de solubilidad se tiene:
En esta expresión, la solubilidad viene dada enmol L–1.
K S S SS = × = ×( ) ( ) ( )3 273 4
Ag PO Ag PO Ag PO3 4 3 4 3 4
SAg PO3 4
AgPO= =
+−[ ]
[ ]3 4
3
KS = + −[ ] [ ]Ag PO433

Al disponer del valor de solubilidad en g L–1, éstese ha de pasar a mol L–1, antes de sustituir su valoren la expresión del producto de solubilidad. Así,
y finalmente:
2. ¿Qué fracción de ion metálico queda sin precipitarcuando se lleva a pH 10 una disolución 0,1 mol L–1
de Mg2+?Mg(OH)2 (KS = 10–11,1).Sol.: 0,79%.
3. Una mezcla de AgBr y AgSCN se agita con agua des-tilada hasta que se alcanza el equilibrio. Calcular lasconcentraciones de los iones Br–, SCN– y Ag+ en ladisolución en contacto con estos precipitados.AgBr (KS = 10–12,3) , AgSCN (KS = 10–11,9).
Al agitar con agua destilada la mezcla de com-puestos insolubles de plata se establecen los siguien-tes equilibrios de precipitación:
Br– + Ag+ →← AgBr(S) KS = [Br–] [Ag+] = 10–12,3
SCN– + Ag+ →← AgSCN(S) KS = [SCN–] [Ag+] = 10–11,9
La relación existente cuando se alcanza el equili-brio entre los iones Br– y SCN– se puede establecera partir de la relación entre los correspondientes pro-ductos de solubilidad:
Por otra parte, al establecer el balance de cargasen el equilibrio resultante:
[Ag+] = [Br–] + [SCN–]
y sustituir la condición encontrada a partir de la rela-ción de los productos de solubilidad se tiene:
[Ag+] = [Br–] + [SCN–] == 10–0,4 [SCN–] + [SCN–] = 1,398 [SCN–]
Si se multiplica esta expresión por [Ag+]:
[Ag+]2 = 1,398 [SCN–][Ag+] == 1,398 KS (AgSCN) = 1,398 × 10–11,9
De aquí:
De acuerdo con las relaciones establecidas ante-riormente se tiene:
4. La solubilidad del TlSCN a 25 °C es 0,0149 mol L–1 yla del TlCl en las mismas condiciones es de 0,0165mol L–1. ¿Qué composición tiene una disolución satu-rada de ambas sales?Sol.: [Tl+] = 2,22 × 10–2 mol L–1; [Cl–] = 1,22 × 10–2 mol
L–1; [SCN–] = 9,95 × 10–3 mol L–1.
5. Al añadir NaOH a la disolución de un metal triva-lente, empieza a precipitar el hidróxido cuando [M3+]= a y pH = b. ¿Cuál será el valor de pH cuando hayaprecipitado el 99,9% de M3+?
El equilibrio de precipitación para el hidróxido delmetal trivalente viene dado por:
M(OH)3 (S) →← M3+ + 3 OH– KS = [M3+][OH–]3
Si se sustituyen en la expresión del producto desolubilidad los datos dados por el problema cuandocomienza a precipitar el hidróxido se tiene:
Cuando ha precipitado el 99,9% del ion trivalentesu concentración será:
[ ]
,M3 30 1
10010+ −= × =a
a
K a
KaS
wb
= ×
= ×
+
−
−[ ]H
3 14 31010
[ ] ] ,
,
, ,Br [SCN
mol L
− − − − −
− −
= = × × =
= ×
10 10 9 51 10
3 78 10
0 4 0 4 7
7 1
[ ]
[ ],
,,
,SCNAg
mol L−+ −
− −= = × = ×1 398
1 33 101 398
9 51 106
7 1
[ ] , ,,Ag mol L+ − − −= × = ×1 398 10 1 33 1011 9 6 1
K
KS
S
( )
,
,,
AgBr
(AgSCN)
[Ag ][Br ][Ag ][SCN ]
[Br ][SCN ]
= =
= = =
+ −
+ −
−
−
−
−−10
1010
12 3
11 90 4
K SS = × =
= × × = ×− −
27
27 1 24 10 6 38 10
4
5 4 19
( )
( , ) ,
Ag PO3 4
SAg PO
1
3 4
gL
mol418,61 g
mol L
= × × =
= ×
−
− −
5 2 101
1 24 10
3
5
,
,
Capítulo 7: Equilibrios de precipitación 271

y al sustituir en la expresión del producto de solubi-lidad se llega a:
Al igualar las dos expresiones obtenidas para KS,se tiene:
y por tanto 10–b = 10(–pH+1), de donde pH = b + 1.
Influencia de la fuerza iónicay efecto de ion común
6. Comparar la solubilidad del bromuro de plata en aguadestilada y en una disolución de nitrato de sodio 0,3mol L–1.AgBr (KS = 10–12,3).
El problema trata de evaluar el efecto de un elec-trolito inerte (sus iones no forman parte del precipi-tado) sobre la solubilidad del mismo. El equilibrio deprecipitación viene dado por:
AgBr (S)→← Br– + Ag+ KS = [Br–][Ag+] = 10–12,3
En agua destilada, la solubilidad de este precipita-do en función de su producto de solubilidad, se deter-mina como:
La presencia de un electrolito inerte, debido a sufuerza iónica, modifica la solubilidad. La expresión[7.8] pone de manifiesto esta dependencia:
en la que I es la fuerza iónica:
Ci y zi es la concentración y carga de cada uno de losiones del electrolito inerte añadido a la disolución.
El término
corresponde a la suma de los productos de los coefi-cientes estequiométricos (νi ) de cada ion multiplica-dos por su carga (zi) al cuadrado. En este caso su valorserá: 1 × 12 + 1 × 12 = 2.
La fuerza iónica vendrá dada por:
Al sustituir estos valores en la expresión [7.8] setiene:
Se observa cómo se incrementa la solubilidad. Suvalor será:
Finalmente, el incremento de solubilidad expresa-do en porcentaje vendrá dado por:
7. A 1,000 g de un precipitado de AgCl se le añadeseparadamente: 1) 500 mL de agua destilada y 2)500 mL de disolución de NaNO3 0,5 mol L–1. Cal-cular el porcentaje de precipitado que se ha disuel-to en cada caso.AgCl (KS = 10–9,8).Peso molecular: AgCl = 143,34.Sol.: 1) 0,09%; 2) 0,146%.
8. A 2,000 g de un precipitado de AgX se le añaden 500mL de KNO3 1,0 mol L–1. Calcular la cantidad de pre-cipitado que se disuelve (expresada en mg), si en unaexperiencia independiente se disuelven 1,43 mg delmismo precipitado al ponerlo en contacto con 1 litrode agua destilada.Peso molecular: AgX = 120.Sol.: 1,28 mg.
% , %
, ,
,∆S
S SS
=′ −
× =−
× =− −
−100
10 1010
100 51 135 97 6 15
6 15
′ = = =− −S KSAgBr
–1mol L10 1011 94 5 97, ,
log log
, ,,
,,KS = ++
× = −−100 512 0 3
1 0 32 11 9412 3
I = × ×
12
0 3 1 0 3 1 0 32 2 1 ( , + , ) = , mol L–
( )ν i iz2∑
I C zi i= ∑12
2
log log
,( )K K
II
zS ST
i i= ++ ∑0 512
12ν
S KSAgBr–1mol L= = =− −10 1012 3 6 15, ,
K a aS b
= ×
= ×
−
−−
−
−
1010
101010
143
314
3
pH
K a
KaS
w= ×
= ×
−+
−−
−10 101010
3
3
314
3
[ ]H pH
272 Equilibrios iónicos y sus aplicaciones analíticas

9. Un precipitado de BaSO4 se lava con: 1) 100 mL deagua destilada y 2) 100 mL de una disolución de clo-ruro de potasio 0,1 mol L–1. ¿Qué peso de precipita-do se disuelve en cada caso, suponiendo que los líqui-dos de lavado se saturan de BaSO4?BaSO4 (KS = 10–9,9).Peso molecular: BaSO4 = 233,34.Sol.: 1) 0,262 mg; 2) 0,809 mg.
10. Un precipitado de BaSO4 se lava: a) con 100 mL deagua destilada; b) con 100 mL de Na2SO4 0,05 mol L–1.Calcular el peso de precipitado que se disuelve en cadacaso, suponiendo que la disolución de lavado quedasaturada en BaSO4.BaSO4 (KS = 10–9,9).Peso molecular: BaSO4 = 233,34.
La primera parte de este problema es similar a ladel problema 6, y por tanto, la solubilidad en aguadestilada vendrá dada por:
De acuerdo con este valor de solubilidad, se disol-verán 10–4,95/10 = 10–5,95 moles de sulfato de bario en100 mL de agua destilada. Para expresar este valoren g se ha de multiplicar por el peso molecular deesta sal, y así resulta:
g BaSO4 = 10–5,95 × 233,34 = 2,62 × 10–4 g
Cuando el precipitado se pone en suspensión con100 mL de una disolución 0,05 mol L–1 de sulfato desodio, el equilibrio heterogéneo de precipitación:
BaSO4 (S)→← Ba2+ + SO4
2_
se desplaza hacia la izquierda por efecto de ioncomún.
Dado que la concentración de ion sulfato presen-te en la disolución es muy superior a la que procedede la disolución del precipitado debido a su propiasolubilidad, ésta se puede despreciar, y al sustituir enel producto de solubilidad resulta:
KS = 10–9,9 = [Ba2+] [SO42_
] = [Ba2+] × 0,05
La concentración de ion Ba2+, deducida de estaexpresión, es una medida de la solubilidad de este pre-cipitado en presencia de una disolución de sulfato desodio 0,05 mol L–1. El valor que se obtiene para esta
solubilidad es de 2,52 × 10–9 mol L–1, muy inferior sise compara con el obtenido en disolución acuosa.
Para expresar esta solubilidad en peso de precipi-tado disuelto, se realiza un cálculo similar al expues-to anteriormente. Así,
11. Un precipitado de Ni(OH)2 se pone en contacto conun litro de NaOH 0,2 mol L–1 y con 10 mL de aguadestilada. Calcular la relación entre las solubilidadesque se obtienen en cada una de las experiencias.Ni(OH)2 (KS = 10–15,2).Sol.: 3,42 × 106.
12. A un litro de disolución saturada de Ag2CO3 se leañade carbonato de sodio sólido hasta que la con-centración de carbonato es el doble de la de ion pla-ta. ¿Qué cantidad de carbonato de sodio en g L–1 hayque añadir para conseguirlo?Ag2CO3 (KS = 10–11,2).Peso molecular: Na2CO3 = 106.Sol.: 0,0234 g.
Precipitación fraccionada
13. A una disolución 0,5 mol L–1 en Ca2+ y de igual con-centración en Sr2+, se le añade lentamente una diso-lución de sulfato hasta que empieza a precipitar elCaSO4. ¿Cuál es la concentración de Sr2+ en esemomento?CaSO4 (KS = 10–4,6); SrSO4 (KS = 10–6,5).Sol.: 6,3 × 10–3 mol L–1.
14. Se tiene una disolución 0,100 mol L–1 en X– y 0,100mol L–1 en Y2–, dos aniones que forman sales de pla-ta poco solubles. ¿Qué porcentaje de Y2– queda sinprecipitar antes de que empiece a precipitar AgX?AgX (KS = 10–7,1); Ag2Y (KS = 10–19).Sol.: 1,59 × 10–4 %.
15. Indicar el intervalo de pH en el que, aprovechandola insolubilidad de sus hidróxidos, será posible pre-cipitar el 99,99% de Fe3+ contenido en una disolución0,1 mol L–1 de Fe3+ y 0,1 mol L–1 en Mg2+ sin que pre-cipite el Mg2+.Fe(OH)3 (KS = 10–39,5); Mg(OH)2 (KS = 10–11,1).
Los equilibrios heterogéneos de precipitación impli-cados son:
g BaSO g4 =
×× = ×
−−2 52 10
10233 34 5 88 10
98,
, ,
S KSBaSO
–1
4mol L= = =− −10 109 9 4 95, ,
Capítulo 7: Equilibrios de precipitación 273

Fe(OH)3 (S)→← Fe3+ + 3 OH–
Mg(OH)2 (S)→← Mg2+ + 2 OH–
Dado el valor extremadamente bajo de KS Fe(OH)3,
precipitará en primer lugar el hidróxido de Fe3+ y portanto lo hará a un valor de pH inferior al necesariopara que comience la precipitación del hidróxido demagnesio.
Cuando precipita el 99,99% del ion Fe3+ presenteinicialmente en la disolución, su concentración finalserá:
Al sustituir esta concentración en la expresión delproducto de solubilidad:
se puede calcular fácilmente el valor de pH al cualse produce la precipitación completa (99,99%) delhierro:
De esta expresión resulta un valor de pH de 2,5.Para el hidróxido de magnesio, dado que se quie-
re calcular el valor de pH al cual comienza la preci-pitación del mismo, la concentración de este ion serála inicial, esto es, 0,1 mol L–1. De nuevo al sustituiresta concentración en la expresión del producto desolubilidad, se llega a:
De esta expresión se calcula un valor de pH de 8,95.A partir de estos valores se puede indicar que el
intervalo de pH comprendido entre 2,5 y 8,95 per-mite la precipitación cuantitativa del hidróxido deFe3+ sin que precipite el hidróxido de magnesio.
16. A una disolución que es 0,01 mol L–1 en Cl–, Br– y I–
se añade una disolución de AgNO3. a) Indicar el orden
en que precipitarán los diferentes haluros de plata.b) ¿Se puede hacer una precipitación fraccionada deestos haluros de plata? (Considerar una separación del99,9%).AgCl (KS = 10–9,8); AgBr (KS = 10–12,3);AgI (KS = 10–16,1).Sol.: a) Yoduro, bromuro y cloruro; b) De I– y Br–
sí; de Br– y Cl– no.
Influencia del pH en la solubilidad
17. Calcular el pH y la concentración de cloruro de amo-nio necesaria para evitar la precipitación del Mg(OH)2en una disolución que es 0,25 mol L–1 en Mg2+ y 0,5mol L–1 en NH3.Mg(OH)2 (KS = 10–11,1); NH4
+(pKa = 9,24).
El equilibrio de precipitación del hidróxido demagnesio:
Mg(OH)2 (S)→← Mg2+ + 2 OH–
KS = [Mg2+][OH–]2 = 10–11,1
depende claramente del pH del medio, y por tantode la proporción de ion amonio y amoniaco:
NH4+ →← NH3 + H+
si estas especies están presentes en la disolución.El pH al cual se evita la precipitación del hidróxi-
do, es justamente aquel que corresponde al comien-zo de su precipitación, o bien valores inferiores. Enestas condiciones, y a partir del producto de la solu-bilidad, se tiene:
De donde resulta un valor de pH = 8,75.En presencia del par ácido-base NH4
+/NH3 se pue-den establecer los siguientes balances de materia, sise denomina CNH4Cl a la concentración de cloruro deamonio que debe añadirse a la disolución que con-tiene amoniaco para evitar la precipitación del hidró-xido de magnesio.
[ ]
[ ] ,
,,OH
Mgmol L
2–1−
+
−−= = =KS 10
0 2510
11 15 25
Ka = =
+
+−[NH ][H ]
[NH ]3
4
10 9 24,
[ ]
[ ] [ ]
,
OHH H
−−
− +
−
+= = =10
101011 1
1
14Kw
[ ]
[ ] [ ]
,
OHH H
−−
− +
−
+= = =10
101039 5
53
14Kw
KS Fe(OH) OH
310 105 3 39 5= =− − −[ ] ,
[ ]
, ,Fe mol L–13 50 1 99 99
10010+ −=
×=
KS Mg( )
,[ ]OH2Mg ][OH
22 11 110= =+ − −
KS Fe( )
,[ ]OH3Fe ][OH
33 39 510= =+ − −
274 Equilibrios iónicos y sus aplicaciones analíticas

Balances de materia:
[NH4+] + [NH3] = CNH4Cl+ 0,5
[Cl–] = CNH4Cl
Balance de cargas:
[NH4+] + [H+] = [OH–] + [Cl–]
En el balance de cargas se pueden despreciar lasconcentraciones de iones hidrógeno e hidroxilo, dadoque el valor de pH calculado para impedir la preci-pitación del hidróxido de magnesio, pH = 8,75, no esni muy ácido ni muy básico. En estas condicionesdicho balance queda como:
[NH4+] = [Cl–]
Al sustituir en esta expresión las respectivas con-centraciones, se tiene:
o bien:
De esta expresión resulta que hay que añadir clo-ruro de amonio hasta alcanzar una concentración de1,55 mol L–1 o superior, con objeto de que el pH dela disolución sea igual o inferior a 8,75 para impedirla precipitación del hidróxido de magnesio.
18. Se tiene una disolución de Fe2+ de concentración 0,24mol L–1 que contiene cloruro de amonio a una con-centración 3,0 mol L–1. A 100 mL de esta disoluciónse le añade un volumen igual de disolución de amo-niaco. Calcular la concentración mínima que debetener esta disolución de amoniaco para provocar laprecipitación del 20% del Fe3+ presente en la disolu-ción original.Fe(OH)2 (KS = 10–15,1); NH4
+ (pKa = 9,24).Sol.: 1,57 × 10–2 mol L–1.
19. ¿Qué pH debe alcanzar una disolución en contactocon un precipitado de CaC2O4 para que la solubili-dad del mismo se duplique respecto a su solubilidaden agua destilada?H2C2O4 (pKa1 = 1,27, pKa2 = 4,27).Sol.: 3,79.
20. Calcular la solubilidad del oxalato de calcio a pH = 4.CaC2O4 (KS = 10–7,9); H2C2O4(pKa1 = 1,27, pKa2 = 4,27). Sol.: 1,9 × 10–4 mol L–1.
21. Se hace pasar una corriente de H2S a través de unadisolución que es 10–2 mol L–1 en Pb2+ y 10–2 mol L–1
en Zn2+ hasta que queda saturada de H2S (0,1 mol L–1
en H2S). Entre qué límites hay que mantener la con-centración de protones de la disolución para que pre-cipite el 99% del Pb2+ presente sin que llegue a pre-cipitar el Zn2+?PbS (KS = 10–27,1); ZnS (KS = 10–24,7);H2S (pKa1 = 7,02, pKa2 = 13,9).Sol.: [H+] = 3,89 – 2,51 mol L–1.
22. 100 mL de una disolución de Zn2+ es también 0,5 molL–1 en ácido acético, se satura de ácido sulfhídrico(H2S = 0,1 mol L–1). ¿Cuántos mg de Zn2+ quedan sinprecipitar?ZnS (KS = 10–24,7); CH3COOH (pKa = 4,75);H2S (pKa1 = 7,02 , pKa2 = 13,9).Peso atómico: Zn = 65,4.
En esta disolución se tienen los siguientes equili-brios concurrentes:
Equilibrio heterogéneo de precipitación:
ZnS (S)→← Zn2+ + S2– KS = [Zn2+][S2–] = 10–24,7
Equilibrio ácido-base que regula el pH del medio:
CH3COOH →← CH3COO– + H+
Equilibrios ácido–base del anión precipitante:
H2 S →← HS– + H+
HS– →← S2– + H+
La concentración de cinc que se mantiene sin pre-cipitar se corresponde con la solubilidad del precipi-tado en las condiciones dadas, esto es, en equilibrio
Ka2
13 910= =− +
−−[S ][H ]
[HS ]
2,
Ka1
7 0210= =− +
−[HS ][H ][H S]2
,
Ka = =
− +−[CH COO ][H ]
[CH COOH]3
3
10 4 75,
( , ) ,
, ,
CCNH Cl
NH Cl4
40 5 10
10 10
8 75
8 75 9 24
+ ×+
=−
− −
( , )[ ]
[ ]
C
KC
a
NH ClNH Cl
H
H4
4
0 5++
=+
+
Capítulo 7: Equilibrios de precipitación 275

con la concentración de ion sulfuro, la cual dependedel pH del medio que proporciona el ácido acético.
De acuerdo con lo expuesto en el capítulo 2, el pHde la disolución vendrá dado por:
A este valor de pH la concentración de ion sulfu-ro libre se calcula a partir de:
Sustituyendo esta concentración en la expresióndel producto de solubilidad se tiene:
KS = [Zn2+][S2–] = 10–24,7 = [Zn2+] × 10–16,86
de donde
[Zn2+] = 10–7,84 mol L–1
Los mg de cinc que quedan sin precipitar en 100mL de disolución se calculan conforme a:
23. Se desea precipitar completamente como sulfuro unadisolución 0,01 mol L–1 del ion M2+. Se añade H2S has-ta que su concentración analítica es 0,1 mol L–1 y elpH se ajusta saturando la disolución de NaHCO3.¿Qué valor ha de tener el producto de solubilidad dela especie MS para conseguir el fin deseado?H2S (pKa1 = 7,02 , pKa2 = 13,9);H2CO3 (pKa1 = 6,35, pKa2 = 10,33).Sol.: KS = 10–11,6.
24. Se añade AgNO3 a una disolución que contieneC2O4
2_, Br
_y AsO4
3_, con concentraciones analíticas
iguales a 0,01 mol L–1. La disolución tiene pH = 6,0.a) Indicar cuál será el orden de precipitación. b) ¿Seráposible separar cuantitativamente (99,9%) los tresaniones aprovechando la diferente solubilidad de lassales de plata?
Ag2C2O4 (KS = 10–11); AgBr (KS = 10–12,5);Ag3AsO4 (KS = 10–22);H2C2O4 (pKa1 = 1,27, pKa2 = 4,27);H3AsO4 (pKa1 = 2,22, pKa2 = 6,98, pKa3 = 11,52).Sol.: a) bromuro, oxalato y arseniato. b) No.
25. Calcular la solubilidad del fluoruro de calcio en unadisolución 0,1 mol L–1 de fluoruro de sodio a pH = 0.CaF (KS = 10–10,3); HF (pKa = 3,52).Sol.: 0,0549 mol L–1.
Solubilidad y complejación
26. ¿Cuántos gramos de AgI se disolverán en un litro deuna disolución de amoniaco 1,0 mol L–1?AgI (KS = 10–16,1); Ag-NH3 (log β1 = 3,32, log β2 = 7,24).Peso molecular: AgI = 234,8.
Al poner en contacto un precipitado de yoduro deplata con una disolución de amoniaco se origina la diso-lución parcial del mismo debido a la formación de loscorrespondientes complejos aminados de plata. Se tie-nen, por tanto, los siguientes equilibrios concurrentesde precipitación y formación de complejos:
AgI (S)→← I– + Ag+ KS = [I–][Ag+] = 10–16,1
Ag+ + NH3→← Ag(NH3)
+
Ag+ + 2 NH3→← Ag(NH3)2
+
De acuerdo con la expresión [7.82], la solubilidaden este caso viene dada por:
Considerando que β2 >> β1, y a su vez >> 1, laexpresión anterior se transforma en:
En esta expresión, la concentración de amoniaco noes su concentración inicial, ya que parte del mismo seemplea para formar complejos con el ion plata. Así, sepuede establecer el siguiente balance de materia:
′ = × = ×S K KS Sβ β2 32
2 3( ) ( )NH NH
′ = + +S KS [ ( ) ( ) ]1 1 3 2 32β βNH NH
β2
2
32
7 2410= =+
+
[ ) ][ ]
,Ag(NHAg ][NH
3
β1
3
3
3 3210= =+
+
[ ( ) ][ ][ ]
,Ag NHAg NH
10 0 165 41
1 0001
9 45 10
7 84
5
−
−
× × ×
× = ×
, ,,
.,
molL molmg
gmg de cinc
g
[ ][ ] [ ]
, ,
, , ,,
SC K K
K K Ka a
a a a
2 1 2
21 1 2
20 92
5 06 9 55 20 9216 86 1
2
0 1 1010 10 10
10
−+ +
−
− − −−
=× ×
+ +=
= ×+ +
=
H S
–
H H
mol L
[ ]
, , ,
H
mol L
CH COOH
–1
3+
− −
= × =
= × =
C Ka
0 5 10 104 75 2 53
276 Equilibrios iónicos y sus aplicaciones analíticas

En este balance, se puede despreciar la [Ag(NH3)+]
considerando los valores relativos de las constantesde formación β1 y β2, y en estas condiciones la[Ag(NH3)2
+] será la solubilidad, esto es S′, por lo queel balance de materia se transforma en:
y la expresión de la solubilidad en:
A partir de esta ecuación se obtiene un valor desolubilidad de 3,71 × 10–5 mol L–1, o bien de 8,71 mgal multiplicar el valor anterior por el peso moleculardel yoduro de plata y pasar la cantidad obtenida engramos a miligramos.
27. Una disolución es 0,5 mol L–1 en EDTA, 0,01 mol L–1
en Pb2+ y 10–3 mol L–1 en S2–, ¿precipitará el PbS?PbS (KS = 10–27,1); PbY2– (log β = 18,0).Sol.: no precipita.
28. Determinar la solubilidad del AgCl en una disolu-ción 0,1 mol L–1 de NH3.AgCl (KS = 10–9,8); Ag+–NH3 (log β1 = 3,32, log β2 = 7,24).Sol.: 4,75 × 10–3 mol L–1.
29. ¿Precipitará el PbSO4 en una disolución que contie-ne 10–2,30 mol L–1 de Pb2+ al añadir la cantidad este-quiométrica de SO4
2_, si esta disolución contiene, ade-
más, 1 mol L–1 de acetato de amonio?PbSO4 (KS = 10–7,8);Pb–CH3COO (log β1 = 1,9, log β2 = 3,3).
Para poder determinar si precipita o no el sulfatode plomo en estas condiciones se ha de calcular laconcentración de ion Pb2+ libre en disolución tras laformación de los correspondientes complejos con elion acetato, y comprobar posteriormente si se cum-plen las condiciones del producto de solubilidad. Losequilibrios implicados son:
PbSO4 (S)→← Pb2+ + SO4
2_
KS = [Pb2+][SO42_
] = 10–7,8
Pb2+ + CH3COO– →← PbCH3COO+
Pb2+ + 2 CH3COO– →← Pb(CH3COO)2
El coeficiente de reacción secundaria para el ionPb2+, debido a la formación de los correspondientescomplejos con el ion acetato, viene dado por:
y al sustituir los valores dados, resulta:
De acuerdo con el valor de αPb(CH3COO_), el producto
de solubilidad condicional será:
A partir de este valor y considerando la concen-tración dada de ion sulfato se tiene:
Dado que esta concentración de ion Pb2+, necesariapara precipitar el sulfato de plomo, 10–2,2 mol L–1, essuperior a la existente en disolución, 10–2,3 mol L–1,el sulfato de plomo no precipita.
30. Se añade 1,00 g de un precipitado de AgCl a 1,0 litrode una disolución de amoniaco. Calcular la concen-tración mínima de esta disolución de amoniaco paraconseguir la disolución total del precipitado de AgCl.AgCl (KS = 10–9,8); Ag+–NH3 (log β1 = 3,32, log β2 = 7,24). Peso molecular: AgCl = 143,34.Sol.: CNH3
≥ 0,147 mol L–1.
31. Se desea disolver 0,050 g de AgBr en 100 mL de unadisolución de amoniaco. ¿Qué concentración debetener si se supone que sólo se forma el complejoAg(NH3)2
+?AgBr (KS = 10–12,3); Ag(NH3)2
+ (log β2 = 7,24).
[ ]
[ ]
,
,,Pb
SOmol L–12
42
4 5
2 32 210
1010+
−
−
−−=
′= =
KS
′ = × =
= × =
−
− −
K KS S αPb CH COO3( )
, , ,10 10 107 8 3 3 4 5
α
Pb CH COO3( ), , ,
− = + × + × =1 10 1 10 1 101 9 3 3 2 3 3
α β βPb CH COO 3 3CH COO CH COO( ) [ ] [ ]
31 1 2
2− = + +− −
β2
3 310= =+ −
[Pb(CH COO) ][Pb ][CH COO ]
3 22
32
,
β1
1 910= =+
+ −
[PbCH COO ][Pb ][CH COO ]
32
3
,
′ = × − × ′ =
= × − × ′−
S K C S
S
S β2
16 1 7 24
32
10 10 1 0 2
( )
( , ), ,
NH
C SNH3
NH= = + ′1 0 23, [ ]
CNH NH Ag NH Ag NH
31 0 23 3 3 2= = + ++ +, [ ] [ ( ) ] [ ( ) ]
Capítulo 7: Equilibrios de precipitación 277

Peso molecular: AgBr = 187,8.Sol.: 0,907 mol L–1.
32. Una disolución es 0,01 mol L–1 en Ca2+, 0,01 mol L–1
en EDTA y 0,1 mol L–1 en C2O42_
. ¿Precipitará enestas condiciones el ion oxalato?CaC2O4 (KS = 10–7,9).CaY2– log β = 10,7).Sol.: Sí.
33. Demostrar la posibilidad de una separación cuan-titativa (99,9%) entre Fe3+ y Bi3+ (CFe= CBi = 0,01mol L–1), basada en la precipitación de sus hidróxi-dos y en presencia de iones F–, [F–] = 0,1 mol L–1.Calcular el intervalo de pH en que es posible haceresta separación.Fe(OH)3 (KS = 10–39,5); Bi(OH)3 (KS = 10–38,0);Fe3+–F– (log β1 = 6,0; log β2 = 9,1; log β3 = 11,9);BiF2– (log β1 = 1,4).Sol.: 3,18-4,47.
34. Una disolución es 0,01 mol L–1 en Ni2+, 0,01 mol L–1
en Mg2+ y 0,02 mol L–1 en Ce3+. a) ¿Será posible sepa-rar cuantitativamente (99,9%) estos tres iones metá-licos aprovechando la insolubilidad de sus hidróxi-dos? b) Calcular el pH mínimo necesario paraconseguir una precipitación cuantitativa de Ce(OH)3.c) ¿Precipitará el Ni(OH)2 en las condiciones delapartado anterior, si la disolución contiene amonia-co a una concentración de 0,05 mol L–1 ?Ni(OH)2 (KS = 10–15,2); Mg(OH)2 (KS = 10–11,1);Ce(OH)3 (KS = 10–19,9); Ni–NH3 (log β1 = 2,7;log β2 = 4,8; log β3 = 6,5; log β4 = 7,7).Sol.: a) No; b) 8,9; c) No.
Solubilidad, acidez y complejación
35. A 2,00 g de un precipitado de BaSO4 se le añaden 500mL de una disolución de EDTA 0,08 mol L–1 a pH7,0. Calcular el porcentaje de precipitado que se hadisuelto.BaSO4 (KS = 10–9,9); BaY2– (log β = 7,8);EDTA (pKa1 = 2,0; pKa2 = 2,7;pKa3 = 6,1; pKa4 = 10,2).Peso molecular: BaSO4 = 233,34.
En este problema se aborda el cálculo de la solu-bilidad de un precipitado en función de la concen-tración de un ligando que forma complejos con el ionmetálico, y cuya existencia en disolución depende del
pH del medio. Se trata, por tanto, de un sistema con-currente de solubilidad, complejación y acidez, cuyosequilibrios son:
BaSO4 (S)→← Ba2+ + SO4
2_
KS = [Ba2+][SO42_
]=10–9,9
Ba2+ + Y4– →← BaY2–
Y4– + H+ →← YH3–
YH3–+ H+ →← YH22–
YH22–+ H+ →← YH3
–
YH3–+ H+ →← YH4
La solubilidad en estas condiciones viene dada porla ecuación [7.96]:
siendo αY(H):
Al sustituir los valores dados en estas expresionesse tiene:
′ = + × × − ′−S S10 1 10
11 785 4
0 089 9 7 8, ,(. ,
( , )
αY(H)
1.785,4
= + × + × +
+ × + × =
− −
− −
1 10 10 10 10
10 10 10 10
10 2 7 16 3 14
19 21 21 28
, ,
α β β
β βY(H)
H2H
3H
4H
H [H
[H [H
= + + +
+ +
+ +
+ +
1 12
3 4
[ ] ]
] ]
′ = + × ×−
−S KS Ba( ( ) [ ])1 21β αY Y(H) EDTA
β4
4 3 2 1
21110H 4
3
[YH ][YH ][H ]
= = =− + K K K Ka a a a
β3
4 3 2
19110H 3
22
[YH ][YH ][H ]
= = =−
− + K K Ka a a
β2
4 3
16 3110H 2
2
3
[YH ][YH ][H ]
= = =−
− + K Ka a
,
β1
4
10 2110H
3
4
[YH ][Y ][H ]
= = =−
− + Ka
,
β = =
−
+ −
[BaY ][Ba ][Y ]
2
2 4107 8,
278 Equilibrios iónicos y sus aplicaciones analíticas

En esta expresión se ha considerado que la con-centración de EDTA libre es la inicial menos la queforma complejo con el ion Ba2+, esto es, S′.
Resolviendo la ecuación anterior resulta un valorde solubilidad de 5,96 × 10–4 mol L–1. Al poner encontacto el precipitado con sólo 500 mL de disolu-ción de EDTA, se disolverán 5,96 × 10–2/2 = 2,98 ×10–4 mol, o bien 0,0695 g al multiplicar por el pesomolecular el sulfato de bario. Finalmente, el por-centaje de precipitado disuelto, considerando quese parte de 2,000 g, será:
36. Calcular la solubilidad del AgBr en EDTA 10–1 mol L–1
a pH 12,0.AgBr (KS = 10–12,3); AgY3– (log β = 7,3);EDTA (pKa1 = 2,0; pKa2 = 2,7; pKa3 = 6,1; pKa4 = 10,2).Sol.: 9,92 × 10–4 mol L–1.
37. Calcular la solubilidad del sulfato de estroncio enuna disolución que es 0,05 mol L–1 en EDTA y seencuentra a pH = 8.SrSO4 (KS = 10–6,5); SrY2– (log β = 8,6);EDTA (pKa1 = 2,0; pKa2 = 2,7;pKa3 = 6,1; pKa4 = 10,2).Sol.: 0,047 mol L–1.
% precipitado disuelto = × =0 06952 00
100 3 47,,
, %
Capítulo 7: Equilibrios de precipitación 279


8
8.1. Introducción8.2. Curvas de valoración8.3. Indicadores de precipitación8.4. AplicacionesCuestionesSeminarios: problemas numéricos
VALORACIONESDE PRECIPITACIÓN

8.1. Introducción
Las valoraciones de precipitación, junto conlas técnicas gravimétricas, constituyen la base dela aplicación cuantitativa de los equilibrios hete-rogéneos de precipitación. En concreto, en lasvaloraciones de precipitación, se aprovecha unareacción que da lugar a la formación de un pre-cipitado:
M + X →← MX(S) [8.1]
para determinar la concentración de una espe-cie, M, haciéndola reaccionar con una disoluciónvalorante, X, de concentración conocida.
Como en otras valoraciones, la reacción quesirve de base a la misma ha de cumplir lossiguientes requisitos:
1. Cuantitativa. El precipitado que se formaen el transcurso de la reacción debe serlo suficientemente insoluble (pequeñovalor de KS) en las condiciones de la valo-ración para garantizar que la reacción seacompleta.
2. Estequiométrica. La especie insoluble for-mada en el curso de la valoración ha detener una composición definida. Esta con-dición es difícil de conseguir ya que pocosprecipitados obtenidos a partir de una diso-lución compleja (muestra real) tienen unaestequiometría definida.
3. Rápida. Aunque es deseable que el esta-blecimiento del equilibrio heterogéneo seasuficientemente rápido, en la práctica noocurre en muchas reacciones de precipi-
tación y en especial en las cercanías delpunto final de la valoración, o cuando setrabaja con disoluciones diluidas.
4. Disponibilidad de sistema indicador. Esquizás ésta la limitación más importantede este tipo de valoraciones, ya que se dis-pone de pocos indicadores visuales paralas mismas.
A tenor de estos requisitos, se infiere que lasvaloraciones de precipitación tienen una aplica-ción limitada, ya que existen relativamente pocasreacciones de precipitación con indicadores visua-les apropiados que presenten las condiciones ade-cuadas para que puedan ser útiles para la deter-minación de un analito. En general, la valoraciónde haluros con plata o viceversa constituye laaplicación más importante, por lo que estas valo-raciones constituirán la base del desarrollo delpresente capítulo. Finalmente, hay que indicarque, sin embargo, el uso analítico de las valora-ciones de precipitación presenta en la actualidaduna gran importancia en diversos sistemas decontrol de calidad, gracias al empleo de indica-dores físico-químicos, tales como potenciomé-tricos y conductimétricos.
8.2. Curvas de valoración
Una curva de valoración es una gráfica quemuestra la variación de la concentración de unanalito a medida que se va añadiendo la disolu-ción valorante que, en el caso de las valoracio-nes de precipitación, reacciona con el analito paraformar una especie poco soluble.
282 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Describir los aspectos más importantes de este tipo devaloraciones con el empleo de indicadores visuales.
• Desarrollar las curvas de valoración de haluros conplata, y viceversa, así como la resolución de dife-rentes mezclas.
• Estudio de los sistemas indicadores visuales más uti-lizados en este tipo de valoraciones: método de Mohry de Volhard e indicadores de adsorción.

Si se supone que la reacción de precipitación[8.1] cumple todos los requisitos que la hacenapta para su aplicación volumétrica, siendo X elanalito y M el valorante:
M + X →← MX(s) [8.1]
y si se omiten las cargas de las especies reaccio-nantes para simplificar las expresiones utilizadas,la constante de equilibrio, en este caso el pro-ducto de solubilidad, viene dada por:
KS = [M][X] [8.2]
Para construir la curva de valoración de unadisolución de la especie X de concentración, CX,de la que se valora un determinado volumen, VX,con una disolución de valorante M de concen-tración CM, se representa pM (–log [M]) o pX(–log [X]) en función de determinados volúme-nes de valorante añadidos, VM, que variarán entre0 <VM< 2 × Veq, o bien en función de la fracciónvalorada (F = VM/Veq), en cuyo caso ésta variaentre 0 < F < 2. Para ello, se tienen en cuenta losbalances de materia de X y M en cualquier pun-to de la valoración y las constantes de equilibrio,en este caso productos de solubilidad.
En la curva de valoración que se describe acontinuación se considerarán dos puntos rele-vantes y dos zonas características, tal como en lasvaloraciones ácido–base y de formación de com-plejos desarrolladas en capítulos precedentes.
Zona 1: punto inicial (F = 0)
En este punto sólo existe el analito ya que aúnno se ha realizado ninguna adición de valorante.Se tiene que [X] = CX y por tanto:
pX = pCX [8.3]
Zona 2: antes del punto de equivalencia(0 < F < 1)
En esta zona se producen adiciones de valo-rante inferiores a la correspondiente en el punto
de equivalencia, las especies a considerar son elanalito que aún permanece sin valorar y la espe-cie precipitada. En este caso, la concentración deanalito sin valorar en función de F , como en otrasvaloraciones, vendrá dada por:
[X] = CX (1 – F) [8.4]
de donde:
pX = pCX – log (1 – F) [8.5]
Zona 3: punto de equivalencia (F = 1)
Al alcanzar este punto de la valoración se haañadido la cantidad de valorante equivalente ala cantidad de analito, la única especie a consi-derar es el precipitado. La concentración de Xse determina a partir de la solubilidad del preci-pitado, considerando que [X] = [M]. Al aplicaresta condición en la expresión del producto desolubilidad, se llega a:
[8.6]
y por tanto:
[8.7]
Zona 4: exceso de valorante (F > 1)
En esta zona se continúan las adiciones devalorante. Las especies a considerar son la espe-cie precipitada y el exceso de valorante añadido,cuya concentración responde a la expresión:
[M] = CM (F – 1) [8.8]
y al sustituir esta condición en la expresión delproducto de solubilidad, se tiene:
[8.9] [ ]
[ ] ( )X
M M
= =−
K KC F
S S
1
pX pKS= 1
2
[ ]X = KS
Capítulo 8: Valoraciones de precipitación 283

de donde:
pX = pKS – pCM + log (F – 1) [8.10]
En el cuadro 8.1 se recogen las expresiones autilizar en cada zona de la curva de valoración,
cuando ésta se representa en función del volu-men de valorante añadido. En ellas, no se ha con-siderado la disociación de la especie insolubleformada a lo largo de la valoración, ya que habi-tualmente es despreciable salvo en las inmedia-ciones del punto de equivalencia.
284 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 8.1Expresiones a utilizar en cada zona de la curva de valoración cuando se representa pX
frente al volumen de valorante añadido
Punto o Zona Volumen Especies Expresiónañadido presentes
Punto inicial 0 X pX = pCX
Antes del punto de equivalencia 0 < VM < Veq MX, X
Punto de equivalencia Veq MX
Exceso de valorante VM > Veq MX, M pX K V V
V C V CS= +
× − ×( )X M
M M X X
pX pKS= 1
2
[ ]X X X M M
X M
= × − ×+
V C V CV V
En la figura 8.1 se muestra como ejemplo lacurva de valoración de una disolución de cloru-
ros 0,01 mol L–1 con nitrato de plata 0,01 mol L–1
como valorante, con formación de la especie inso-luble AgCl (KS = 10–9.8), y en el cuadro 8.2 losdatos correspondientes a dicha valoración, dedonde se infiere el intervalo de pCl correspon-diente a un error en la valoración de ±1%. Enesta figura también se representa la curva devaloración en función del menos logaritmo de laconcentración de metal, pM en este caso pAg, enlugar de pX. Para ello, las expresiones anterio-res se transforman fácilmente utilizando el pro-ducto de solubilidad.
Hay dos factores que influyen decisivamenteen el perfil de la curva de valoración de precipi-tación; éstos son la concentración de la especiea valorar y el valor del producto de solubilidaddel compuesto insoluble formado. La concen-tración de analito influye de tal manera que unadisminución de su concentración produce un sal-to más pequeño de pX alrededor del punto deequivalencia. En la figura 8.2a se muestran lascurvas de valoración de disoluciones de cloruro
FIGURA 8.1. Curva de valoración de una disolución 0,01 mol L–1 de ion cloruro
con nitrato de plata 0,01 mol L–1.
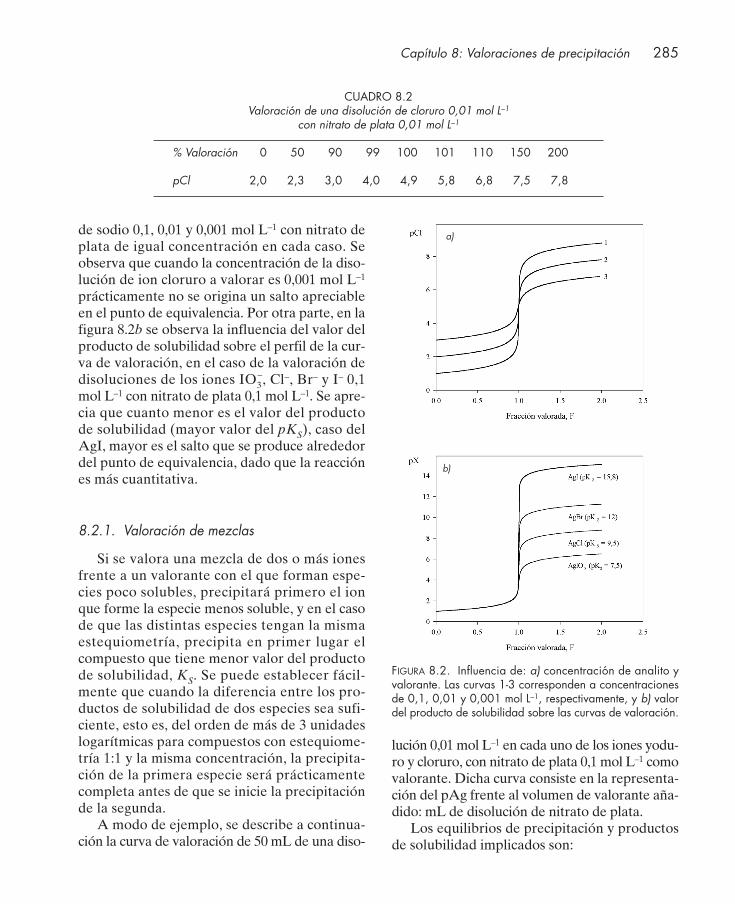
de sodio 0,1, 0,01 y 0,001 mol L–1 con nitrato deplata de igual concentración en cada caso. Seobserva que cuando la concentración de la diso-lución de ion cloruro a valorar es 0,001 mol L–1
prácticamente no se origina un salto apreciableen el punto de equivalencia. Por otra parte, en lafigura 8.2b se observa la influencia del valor delproducto de solubilidad sobre el perfil de la cur-va de valoración, en el caso de la valoración dedisoluciones de los iones IO3
–, Cl–, Br– y I– 0,1mol L–1 con nitrato de plata 0,1 mol L–1. Se apre-cia que cuanto menor es el valor del productode solubilidad (mayor valor del pKS), caso delAgI, mayor es el salto que se produce alrededordel punto de equivalencia, dado que la reacciónes más cuantitativa.
8.2.1. Valoración de mezclas
Si se valora una mezcla de dos o más ionesfrente a un valorante con el que forman espe-cies poco solubles, precipitará primero el ionque forme la especie menos soluble, y en el casode que las distintas especies tengan la mismaestequiometría, precipita en primer lugar elcompuesto que tiene menor valor del productode solubilidad, KS. Se puede establecer fácil-mente que cuando la diferencia entre los pro-ductos de solubilidad de dos especies sea sufi-ciente, esto es, del orden de más de 3 unidadeslogarítmicas para compuestos con estequiome-tría 1:1 y la misma concentración, la precipita-ción de la primera especie será prácticamentecompleta antes de que se inicie la precipitaciónde la segunda.
A modo de ejemplo, se describe a continua-ción la curva de valoración de 50 mL de una diso-
FIGURA 8.2. Influencia de: a) concentración de analito yvalorante. Las curvas 1-3 corresponden a concentracionesde 0,1, 0,01 y 0,001 mol L–1, respectivamente, y b) valordel producto de solubilidad sobre las curvas de valoración.
lución 0,01 mol L–1 en cada uno de los iones yodu-ro y cloruro, con nitrato de plata 0,1 mol L–1 comovalorante. Dicha curva consiste en la representa-ción del pAg frente al volumen de valorante aña-dido: mL de disolución de nitrato de plata.
Los equilibrios de precipitación y productosde solubilidad implicados son:
Capítulo 8: Valoraciones de precipitación 285
CUADRO 8.2Valoración de una disolución de cloruro 0,01 mol L–1
con nitrato de plata 0,01 mol L–1
% Valoración 0 50 90 99 100 101 110 150 200
pCl 2,0 2,3 3,0 4,0 4,9 5,8 6,8 7,5 7,8
a)
b)

I– + Ag+ →← AgI(s)[8.11]
Cl– + Ag+ →← AgCl(s)[8.12]
Con objeto de delimitar las zonas caracterís-ticas de la curva de valoración, se ha de calcularinicialmente el volumen de valorante necesariopara valorar cada haluro:
[8.13]
Debido a que se forman precipitados con lamisma estequiometría, se valora primero el quetenga un menor valor de producto de solubili-dad, en este caso el yoduro y después el cloruro.Al representar la curva respecto a pAg, no sepuede determinar este valor en el punto inicialde la valoración ya que su concentración es evi-dentemente cero. Sí se puede calcular inicial-mente la concentración de plata necesaria paraque se inicie la precipitación de cada haluro, así:
[8.14]
[8.15]
En estas condiciones se pueden establecer lassiguientes zonas en la curva de valoración:
Zona 1: se valora el yoduro(0 < Vplata < 5 mL)
En esta zona de la curva de valoración, laconcentración de yoduro sin valorar viene dadapor:
[8.16]
y por tanto la concentración de ion Ag+ deacuerdo con la expresión del producto de solu-bilidad:
[8.17]
Zona 2: punto de equivalencia del yoduro(Vplata = 5 mL)
En este punto, la concentración de ion platase corresponde con el valor de la solubilidad delyoduro de plata:
[8.18]
lo que implica que pAg = 8,05.
Zona 3: se valora el cloruro(5 < Vplata < 10 mL)
La concentración de ion cloruro sin valorar,tras la valoración del ion yoduro, viene dada porla expresión:
[8.19]
y por tanto:
[8.20]
Zona 4: punto de equivalencia del cloruro(Vplata = 10 mL)
Como en el caso anterior, la concentración deplata en este punto se corresponde con el valor dela solubilidad, en este caso del cloruro de plata:
[Ag( ) (
inicial plata
inicial cloruro yoduro plata plata
+ =+
× + − ×]
( )
)
K V V
V C C V CS
[Cl ]( )inicial cloruro plata plata inicial yoduro
inicial plata
− =× − × − ×
+V C V C V C
V V
[ ] [ ],
Ag I
10 molL
(AgI)
1
+ −
− −
= = =
=
KS
8 05
[ ]( )
Ag inicial plata
inicial yoduro plata plata
+ =+
× − ×K V V
V C V CS
[I ] inicial yoduro plata plata
inicial plata
− =× − ×
+V C V C
V V
[ ]
[ ]
,,Ag
Cl100, 01
10 mol L(AgCl)+−
−− −= = =
KS9 8
7 8 1
[ ]
[ ]
,
AgI
100, 01
10 mol L(AgI) 14,1+−
−− −= = =
KS16 1
1
V CC
inicial haluro
plata
50 0, 010,1
5 mL× = × =
KS = =− + −[ ][ ] ,Cl Ag 10 9 8
KS = =− + −[ ][ ] ,I Ag 10 16 1
286 Equilibrios iónicos y sus aplicaciones analíticas
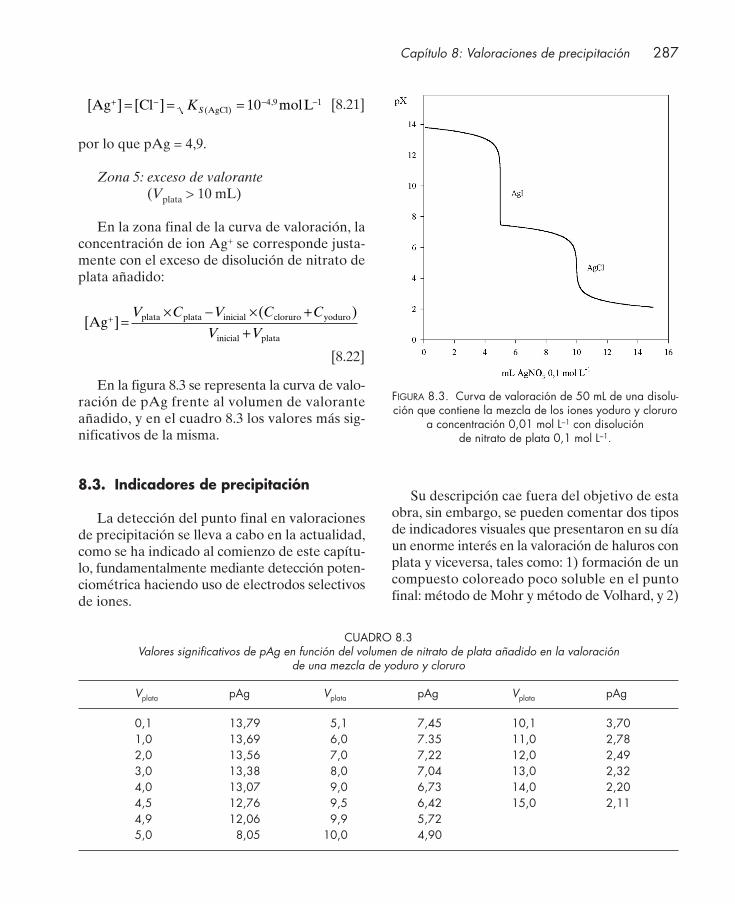
[8.21]
por lo que pAg = 4,9.
Zona 5: exceso de valorante(Vplata > 10 mL)
En la zona final de la curva de valoración, laconcentración de ion Ag+ se corresponde justa-mente con el exceso de disolución de nitrato deplata añadido:
[8.22]
En la figura 8.3 se representa la curva de valo-ración de pAg frente al volumen de valoranteañadido, y en el cuadro 8.3 los valores más sig-nificativos de la misma.
8.3. Indicadores de precipitación
La detección del punto final en valoracionesde precipitación se lleva a cabo en la actualidad,como se ha indicado al comienzo de este capítu-lo, fundamentalmente mediante detección poten-ciométrica haciendo uso de electrodos selectivosde iones.
Su descripción cae fuera del objetivo de estaobra, sin embargo, se pueden comentar dos tiposde indicadores visuales que presentaron en su díaun enorme interés en la valoración de haluros conplata y viceversa, tales como: 1) formación de uncompuesto coloreado poco soluble en el puntofinal: método de Mohr y método de Volhard, y 2)
[ ]Ag( )plata plata inicial cloruro yoduro
inicial plata
+ =× − × +
+V C V C C
V V
[ ] ,Ag ] [Cl 10 molL(AgCl)+ − − −= = =KS
4 9 1
Capítulo 8: Valoraciones de precipitación 287
CUADRO 8.3Valores significativos de pAg en función del volumen de nitrato de plata añadido en la valoración
de una mezcla de yoduro y cloruro
Vplata pAg Vplata pAg Vplata pAg
0,1 13,79 5,1 7,45 10,1 3,701,0 13,69 6,0 7.35 11,0 2,782,0 13,56 7,0 7,22 12,0 2,493,0 13,38 8,0 7,04 13,0 2,324,0 13,07 9,0 6,73 14,0 2,204,5 12,76 9,5 6,42 15,0 2,114,9 12,06 9,9 5,725,0 8,05 10,0 4,90
FIGURA 8.3. Curva de valoración de 50 mL de una disolu-ción que contiene la mezcla de los iones yoduro y cloruro
a concentración 0,01 mol L–1 con disoluciónde nitrato de plata 0,1 mol L–1.

indicadores de adsorción también conocidos comométodo de Fajans.
8.3.1. Método de Mohr
Este método se emplea para la valoracióndirecta de cloruros y bromuros con plata, utili-zando cromato de potasio como indicador. Laaparición de un segundo precipitado, cromato deplata de color rojo, tras el primer exceso de nitra-to de plata, pone de manifiesto el punto final dela valoración. Las reacciones implicadas en esteproceso son:
A) Reacción de valoración:
Cl– + Ag+ →← AgCl(S)
[8.12]
Br– + Ag+ →← AgBr(S)
[8.23]
B) Reacción indicador del punto final:
CrO42– + 2 Ag+ →← Ag2CrO4 (S)
[8.24]
Es evidente que para que el cromato de pota-sio pueda actuar como indicador en estas valo-raciones, las reacciones:
2AgCl(S) + CrO42– →← Ag2CrO4(S) + 2Cl– [8.25]
2AgBr(S) + CrO42– →← Ag2CrO4(S) + 2Br– [8.26]
han de estar lo suficientemente desplazadas haciala izquierda, con objeto de que el ion cromatosólo reaccione con la plata cuando se haya valo-rado todo el cloruro o bromuro presente en lamuestra.
Para determinar el sentido de estas reaccionesbasta con calcular sus respectivas constantes deequilibrio, que de forma general responden a:
[8.27]
siendo X el ion cloruro o bromuro. Si se multi-plica y divide la expresión anterior por [Ag+]2 setiene:
[8.28]
considerando además las expresiones de KS dadasen las ecuaciones [8.12] u [8.23] y [8.24]. Al sus-tituir en esta expresión los valores de KS se obtie-nen los siguientes valores de K: 10–7,7 y 10–12,7,para la valoración de cloruros y bromuros, res-pectivamente. Estos valores son adecuados paraasumir que las reacciones [8.25] y [8.26] estándesplazadas hacia la izquierda, y por tanto, ponende manifiesto la validez de este indicador paraestas valoraciones de precipitación.
La utilización de cromato de potasio en estasvaloraciones está limitada a pH neutro, ya que enmedio ácido el cromato pasa a dicromato, por loque no precipita el cromato de plata y en mediobásico precipita la plata en forma de hidróxidoque evoluciona a óxido, al ser de un metal noble.Finalmente, hay que indicar que los iones yodu-ro no se pueden valorar con nitrato de plata coneste indicador, a pesar de que el AgI es más inso-luble que el AgCl y AgBr, ya que el yoduro deplata adsorbe fuertemente a los iones cromato yla cantidad de cromato necesaria sería ingentedada la gran insolubilidad del AgI.
8.3.2. Método de Volhard
Se utiliza para la determinación directa de pla-ta con tiocianato y utiliza una sal de Fe3+ comoindicador. La determinación se lleva a cabo en
K
KK
S
S
= =−
−
+
+
[ ][ ]
[[ ]
( )( )
XCrO
Ag ]Ag
AgXAg CrO2
2
42
2
2
2
4
K =
−
−
[X ][CrO ]
2
42
KS = =− + −[ ] ,CrO ][Ag42 2 11 910
KS = =− + −[ ] ,Br ][Ag 10 12 3
KS = =− + −[ ] ,Cl ][Ag 10 9 8
288 Equilibrios iónicos y sus aplicaciones analíticas

medio ácido para evitar también la precipitacióndel hidróxido de Fe3+. Las reacciones que tienenlugar son las siguientes:
a) Reacción de valoración:
Ag+ +SCN– →← AgSCN(S) (blanco) [8.29]
b) Reacción indicador del punto final:
Fe3+ + SCN– →← Fe(SCN)2+ (coloración roja)
[8.30]
Sin embargo, la principal aplicación de estemétodo es la determinación por retroceso de halu-ros y su ventaja deriva de que la valoración puedellevarse a cabo en medio ácido y no requiere uncontrol estricto del pH como en otros métodosalternativos. El procedimiento consiste en añadira la muestra un exceso conocido de una disoluciónpatrón de nitrato de plata y determinar su excesomediante una valoración por retroceso con unadisolución patrón de tiocianato.
En el caso de la determinación de cloruros,dado que el cloruro de plata es más soluble queel tiocianato de plata, tiene lugar la siguientereacción alrededor del punto de equivalencia:
AgCl(S) + SCN– → AgSCN(S) + Cl– [8.31]
que conduce a un consumo en exceso de tiocia-nato y consecuentemente a unos valores bajos enla determinación de los cloruros. Este error pue-de evitarse mediante la adición a la disolución delanalito de una pequeña cantidad de nitrobence-no (compuesto orgánico extremadamente tóxicopor inhalación, ingestión y contacto con la piel,con riesgo de efectos acumulativos. Por esta razónno es aconsejable su uso), que cubre la superficiedel precipitado impidiendo su disolución en lavaloración por retroceso con tiocianato. Una alter-nativa a la adición de nitrobenceno, aunqueextremadamente engorrosa, es la filtración delcloruro de plata formado antes de llevar a cabola valoración por retroceso. La adición de nitro-
benceno o la filtración, no es necesaria en la deter-minación de los otros haluros, ya que todos ellosforman con la plata sales más insolubles que lacorrespondiente de tiocianato.
En el caso de la determinación de yoduros,existe el riesgo de oxidación de este anión por elácido nítrico generalmente utilizado para obte-ner el medio ácido. Para evitarlo, es convenien-te añadir el exceso de plata en medio neutro y acontinuación acidificar para llevar a cabo la valo-ración por retroceso.
La aplicación más importante del método Vol-hard es la determinación de los aniones cuya sal deplata es insoluble en medio ácido, ya que en estascondiciones se evita la interferencia de numerososaniones que forman compuestos insolubles con salde plata en medio neutro o ligeramente básico. Porotra parte el procedimiento puede adaptarse a ladeterminación de cualquier anión cuya sal de pla-ta sea insoluble. Para ello basta con añadir el exce-so de plata en medio neutro o ligeramente básico,filtrar el precipitado y, tras acidificar el filtrado conácido nítrico, valorar el exceso de plata.
8.3.3. Indicadores de adsorción
Estos indicadores, introducidos por Fajans yHassel en 1923, cambian de color al ser adsorbidosen la superficie de un precipitado. Su funciona-miento se basa en dos hechos bien conocidos: losprecipitados coloidales, como los que generalmenteforman los haluros con plata, tienen una elevadatendencia a adsorber iones, y un precipitado adsor-be preferentemente los iones que lo forman.
Los indicadores de adsorción son compues-tos orgánicos con un carácter de ácido débil (indi-cadores aniónicos, adecuados para la valoraciónde haluros), o de base débil (indicadores catió-nicos, útiles para la valoración con disolucionesde haluros como valorante). A continuación secomenta el esquema de funcionamiento de unindicador de adsorción aniónico.
Para utilizar un indicador aniónico, de formaHIn, en la valoración de, por ejemplo, cloruros conplata, el pH del medio en que se lleva a cabo la valo-
Capítulo 8: Valoraciones de precipitación 289

ración debe ser lo suficientemente alto para garan-tizar que en la disolución predomine la forma diso-ciada del indicador, In–. Al iniciar la adición del valo-rante, Ag+, se origina un precipitado de AgCl que,debido a su carácter coloidal, a su tendencia aadsorber preferentemente los iones que lo formany a que en la zona de defecto de valorante la con-centración de cloruro es mucho mayor que la deplata, adsorberá iones cloruro; la consecuencia esque las partículas del precipitado adquieren una car-ga negativa, lo que provoca la repulsión de las molé-culas del indicador y la atracción de los iones posi-tivos presentes en la disolución, como por ejemploNa+, tal como se muestra en la figura 8.4a. Supera-do el punto de equivalencia, la concentración decloruro es muy inferior a la de plata y por lo tantoes éste el ion que se adsorbe preferentemente, loque confiere una carga positiva a las partículas delprecipitado; la consecuencia es que, en estas con-diciones, las especies iónicas que existan en disolu-ción constituirán la capa contraiónica negativa, porejemplo iones nitrato, y si en el medio está presen-te el indicador, las moléculas aniónicas del mismoserán atraídas por las partículas de cloruro de pla-ta, envueltas por iones Ag+ y adsorbidas en su super-ficie (figura 8.4b). Este hecho provoca que el pre-cipitado adquiera un color característico y que lacoloración correspondiente al indicador desapa-rezca de la disolución, de acuerdo con la siguientereacción indicador:
Indicador(+ o –) + Precipitado →←→← Indicador(Precipitado) [8.32]
Así, por ejemplo, en el punto final de la valo-ración de cloruros con plata utilizando dicloro-fluoresceína como indicador, el precipitadoadquiere un color rosado (Color 2) y desapare-ce el color amarillo verdoso (Color 1), fluores-cente, de la diclorofluoresceína en disolución.
El mecanismo de funcionamiento de los indi-cadores de adsorción no está claro. Por lo gene-ral se acepta que el cambio de color se debeexclusivamente al hecho de ser adsorbidos porel precipitado. También parece haber una coin-cidencia generalizada en que nunca llega a pre-cipitar la sal de plata del indicador, pese a que enmuchos casos sea poco soluble, ya que no sesupera su producto de solubilidad.
Como ya se ha comentado, los indicadoresaniónicos (fluoresceína y derivados) son ade-cuados para la valoración de haluros con plata;un factor limitante es el pH, ya que el indicadordebe encontrarse en su forma disociada. Por otraparte, los indicadores catiónicos del grupo de lasrodaminas, sólo funcionan si están cargados posi-tivamente y pueden ser útiles para la determi-nación de cationes (fundamentalmente plata)capaces de formar precipitados coloidales conlos haluros. En la figura 8.5 se muestran los indi-cadores de adsorción más comunes y en el cua-
290 Equilibrios iónicos y sus aplicaciones analíticas
Color 1 Color 2
FIGURA 8.4. Fundamento del modo de operación de los indicadores de adsorción.
a) Antes del punto de equivalencia b) Después del punto de equivalencia

dro 8.4 sus características y aplicaciones más rele-vantes.
8.4. Aplicaciones
Las volumetrías de precipitación más impor-tantes desde el punto de vista práctico, utilizanel ion plata como valorante. Habitualmente sepreparan disoluciones de nitrato de plata de con-centración 0,1 mol L–1, bien por disolución de lasal o bien a partir del ataque con ácido nítrico dela plata metálica (patrón primario).
Es conveniente que tanto el nitrato de pla-ta sólido, como sus disoluciones sean protegi-
das del polvo, la materia orgánica y la luz, yaque la presencia de estos agentes cataliza lareacción de reducción del ion plata a platametálica.
Las disoluciones de nitrato de plata usual-mente deben ser estandarizadas frente a unpatrón primario adecuado, en general se utilizacloruro de sodio como patrón primario, que debesecarse previamente, en una estufa, a 120 ºCdurante dos horas aproximadamente.
En la estandarización, la detección del puntofinal se lleva a cabo con los indicadores visualescomentados, o bien mediante detección poten-ciométrica.
Capítulo 8: Valoraciones de precipitación 291
CUADRO 8.4Características de los indicadores de adsorción más utilizados en valoraciones de precipitación
Indicador Cambio de color Aplicaciones
AniónicosFluoresceinato de sodio Verde amarillento Determinación de Cl– (Br– posible)
rosa vino en medio neutro o ligeramente básico2,7-DCF Verde amarillento Determinación de Cl– (Br– posible)
rosa vino en medio ligeramente ácidoEosinato de sodio Rosa – rojo intenso Determinación de Cl– y de I–
en medio ácido moderado
CatiónicosRodamina 6G Rojo – violeta Determinación de Ag+ con Br–
Fenosafrina Rojo – azul Determinación de Ag+ con Br–
2,7-DCF : 2,7-diclorofluoresceinato de sodio
FIGURA 8.5. Estructura química de losindicadores de adsorción más utilizadosen valoraciones de precipitación.

292 Equilibrios iónicos y sus aplicaciones analíticas
Cuestiones
8.1. En la valoración de Cl– con Ag+ el valor de pCl en el punto de equivalencia, depende de:
[ ] Concentración de Ag+ y valor de KS[ ] Sólo del valor de KS[ ] Concentración de iones cloruro y KS[ ] Concentraciones de iones cloruro y plata y de KS
8.2. En la valoración de un ion metálico M+ con un agente precipitante X– por formación de una especie insolubleMX, el punto final vendrá dado por:
[ ] Aparición de M+
[ ] Aparición de X–
[ ] Aparición de MX[ ] Desaparición de M+
8.3. La valoración directa de iones cloruro con Ag+ mediante el método de Mohr se lleva a cabo en medio neu-tro, ya que:
a) en medio ácido:
b) en medio básico:
8.4. ¿En qué procesos se fundamenta el uso de indicadores de adsorción en valoraciones de precipitación?
[ ] Retención superficial del indicador en el punto final[ ] Coagulación del precipitado en el punto final[ ] Retención superficial del exceso de reactivo después del punto final[ ] Cambio de color al retenerse superficialmente a un precipitado que cambia de características en
el punto final[ ] Disolución de un precipitado formado con el analito en el punto final[ ] Formación de un precipitado coloreado en el punto final con un ligero exceso de valorante
8.5. El salto en el punto de equivalencia en una valoración de precipitación es más nítido a medida que:
[ ] Se incrementa la concentración de valorante[ ] Se incrementa el valor de KS[ ] Se incrementa la temperatura[ ] Se incrementa la fuerza iónica de la disolución
8.6. En la valoración de Ba2+ con SO42– por formación de la especie poco soluble BaSO4, ¿qué especies se encon-
trarán en la disolución problema en el punto de equivalencia?
[ ] Ba2+ [ ] SO42–
[ ] Ninguna [ ] Ba2+ y SO42–

Capítulo 8: Valoraciones de precipitación 293
Seminarios: problemas numéricos
• Objetivo pedagógicoDesarrollo de problemas tipo sobre trazado de curvasde valoración y aplicaciones de estas valoracioneshaciendo uso del método de Mohr.
1. Representar la curva de valoración que se obtiene alvalorar NaCl 0,1000 mol L–1 con una disoluciónpatrón de AgNO3.AgCl (KS = 10–9,8).Sol.:
F 0,00 0,50 0,90 0,99pCl 1,0 1,3 2,0 3,0F 1,00 1,01 1,50 2,00pCl 4,9 6,8 8,5 8,8
2. Representar la curva de valoración obtenida al valorar50 mL de una disolución de NaI, NaBr y NaCl, cadauno de ellos 0,0400 mol L–1, con AgNO3 0,4 mol L–1.AgCl (KS = 10–9,8); AgBr (KS = 10–12,3);AgI (KS = 10–16,1).Sol.:
VValorante, mL 0,1 1,0 2,0 3,0 4,0 5,0pAg 14,7 14,6 14,5 14,3 14,0 10,9VValorante, mL 6,0 7,0 8,0 9,0 10,0 11,0pAg 10,8 10,7 10,5 10,2 8,4 8,3VValorante, mL 12,0 13,0 14,0 15,0 16,0 17,0pAg 8,2 8,0 7,7 4,9 2,1 1,8VValorante, mL 18,0 19,0 20,0pAg 1,6 1,5 1,4
3. Dibujar la curva de valoración obtenida al valorar50,00 mL de Na2SO4 0,0250 mol L–1 con BaCl2 0,500mol L–1. Despreciar el efecto de la dilución.BaSO4 (KS = 10–9,9).Sol.:
F 0,00 0,50 0,90 0,99pSO4
2– 1,6 1,9 2,6 3,6F 1,00 1,01 1,10 2,00pSO4
2– 4,9 6,3 7,3 8,3
4. Una disolución 0,1 mol L–1 de IO3– se valora con Ag+
0,1 mol L–1 utilizando CrO42– como indicador. La con-
centración de CrO42–en el punto final es 10–2,69 mol L–1.
Calcular la concentración de IO3– en el punto final.
AgCrO4 (KS = 10–11,9); AgIO3 (KS = 10–7,5).
Esta valoración, que se fundamenta en el métodode Mohr, consiste en las siguientes reacciones:
Reacción de valoración:
IO3– + Ag+ →← AgIO3 (S)
KS = [Ag+] [IO3– ] = 10–7,5
Reacción indicadora del punto final:
CrO42–+ 2Ag+ →← Ag2CrO4 (S)
KS = [Ag+]2 [CrO42–] = 10–11,9
La concentración de ion yodato en el punto final vie-ne delimitada por la correspondiente de ion plata eneste punto, que a su vez depende de la concentraciónde indicador añadida, esto es de ion cromato. En estascondiciones, y de acuerdo con la expresión del productode solubilidad del cromato de plata, la concentraciónde ion plata en el punto final viene dada por:
A partir de esta concentración de plata se puedecalcular fácilmente la concentración de ion yodatoen este punto haciendo uso de la expresión del pro-ducto de solubilidad del yodato de plata:
Esta concentración de ion yodato se puede compa-rar con la correspondiente al punto de equivalencia:
lo que permite estimar si se comete error por exceso opor defecto en la valoración. En este caso:
[ ( ),
,
IO ]
mol L
3 Punto equivalencia AgIO
–1
3− −
−
= = =
=
KS 10
10
7 5
3 75
[[
( )
,
,,
IO ]Ag ]
mol L
3 Punto finalAgIO
Punto final
–
−+
−
−−
= =
= =
KS 3
1010
107 5
4 62 9 1
[ ][ ]
( )
,
,,
AgCrO
mol L
Punto finalAg CrO
–
2+−
−
−−
= =
= =
KS 4
42
11 9
2 694 6 110
1010

[IO3– ]Punto final = 10–2,9 > [IO3
– ]Punto equivalencia = 10–3,75
por lo que el error será por defecto.
5. Se valora una disolución de Cl– 0,01 mol L–1 (VCl =100,0 mL) con Ag+ 0,1 mol L–1 y CrO4
2– como indica-dor, [CrO4
2–]= 10–2,79 mol L–1. Calcular cuál será elexceso de valorante (expresado en mL) que habráque añadir para que precipite el indicador. Despre-ciar la variación de volumen.AgCrO4 (KS = 10–11,9); AgCl (KS = 10–9,8).Sol.: 0,028 mL.
6. Se valoran 25 mL de una disolución 0,1 mol L–1 deNaCl con AgNO3 0,1 mol L–1 usando K2CrO4 comoindicador. Si la concentración de ion CrO4
2– libre enel punto final es de 5 × 10–5 mol L–1, explicar si secomete error por exceso o bien por defecto en estavaloración.AgCrO4 (KS = 10–11,9); AgCl (KS = 10–9,8).Sol.: Error por exceso.
7. Se valoran 50 mL de una disolución 0,100 mol L–1 decloruro con nitrato de plata, 0,100 mol L–1 según la téc-nica de Mohr, usando K2CrO4 como indicador. Si laconcentración analítica de ion cromato es 0,02 mol L–1
y el pH = 4,0, ¿cuál es el error de valoración expresa-do en términos de mL de disolución de plata añadidosen exceso o por adicionar para alcanzar el punto final?AgCrO4 (KS = 10–11,9); AgCl (KS = 10–9,8); H2CrO4(pKa1 = 1,0, pKa2 = 6,3).
La valoración de ion cloruro que se aborda en esteproblema es un ejemplo típico del método de Mohr,aunque en este caso se ha de considerar el pH delmedio ya que éste influye apreciablemente en la con-centración de ion cromato libre en disolución queactúa como sistema indicador. Se tienen, por tanto,los siguientes reacciones:
Reacción de valoración:
AgCl (S)→← Cl– + Ag+
KS = [Ag+] [Cl–] = 10–9,8
Reacción indicadora del punto final:
CrO42– + 2Ag+ →← Ag2CrO4 (S)
KS = [Ag+]2 [CrO42–] = 10–11,9
Reacciones de protonación del ion cromato:
CrO42– + H+ →← HCrO4
–
CrO42– + 2 H+ →← H2CrO4
En estas condiciones, el producto de solubilidadcondicional viene dado por:
siendo αCrO4(H) el coeficiente de reacción secundariadel ion cromato con relación al H+. Sustituyendo losvalores dados en la expresión anterior se tiene:
A partir de este producto de solubilidad condicio-nal se puede calcular la concentración de ion plataen el punto final:
Para conocer si estamos en una situación de exce-so o defecto de valorante (ion plata), se ha de calcu-lar la concentración de este ion en el punto de equi-valencia:
Dado que la concentración de ion plata en el pun-to final es superior a la del punto de equivalencia, elerror en la valoración será por exceso.
Al valorarse 50 mL de una disolución de ion clo-ruro 0,1 mol L–1 con disolución de nitrato de plata 0,1mol L–1, el volumen en el punto de equivalencia seráde 50 + 50 = 100 mL. En estas condiciones los mmolde ion plata en exceso serán:
[
,
( ),Ag ]
mol L
Punto equivalencia AgCl
–1
+ −
−
= = =
= ×
KS 10
1 26 10
9 8
5
[Ag ]CrO
mol L
Punto finalAg CrO
42
–1
2+−
−
−
=′
= × =
= ×
KS ( )
[ ],
,
,
42 3 10
0 02
1 12 10
10
4
′ = + × + × =
= ×
− − −
−
KS 10 1 10 10 10 10
2 53 10
11 9 6 3 4 7 3 8
10
, , ,( )
,
′ = × = + ++ +K K KS S Sα β βCrO (H)
H2H
4H [H( [ ] ] )1 1
2
β2 22 1
6 3 17 3
1
110 10
10
H 2 4
42
H CrOCrO ][H
= = =
=×
=
− +
− −
[ ][ ]
,,
K Ka a
β14
42
2
6 36 3
1
110
10
H HCrOCrO H
= = =
= =
−
− +
−
[ ][ ][ ]
,,
Ka
294 Equilibrios iónicos y sus aplicaciones analíticas

o bien:
Al sustituir los valores calculados e igualar ambasexpresiones se tiene:
(1,12 × 10–4 – 1,26 × 10–5) × 100 = VAg+ × 0,1
De donde VAg+ = 0,1 mL.
8. El contenido de ion cloruro de una muestra de aguase determina por el método de Mohr. Se valora unaalícuota de 100 mL y se gastan 1,68 mL de una diso-lución de AgNO3 0,0976 mol L–1.¿Cuál es la concen-tración, en mg mL–1 o partes por millón, de ion Cl–
en la muestra?AgCrO4 (KS = 10–11,9); AgCl (KS = 10–9,8);Peso atómico: Cl = 35,45.Sol.: 58,13 ppm.
9. Calcular los µg de ion bromuro que quedan sin pre-cipitar en el punto final de la valoración de 50 mL deuna disolución 0,1 mol L–1 de este ion con nitrato deplata 0,1 mol L–1, según la técnica de Mohr, supo-niendo que la concentración de ion cromato en elpunto final es 0,02 mol L–1.AgCrO4 (KS = 10–11,9); AgBr (KS = 10–12,3);Peso atómico: Br = 79,92.Sol.: 0,504 µg.
10. Una disolución contiene KCN y KCl, una alícuota de50 mL consume 15,00 mL de AgNO3 0,1000 mol L–1
hasta la aparición de una ligera turbidez. A conti-nuación se añaden 28,10 mL de AgNO3 a la mismadisolución y se filtra. El exceso de Ag+ en el filtradose determina por valoración con KSCN 0,0833 molL–1, utilizando Fe3+ como indicador. Hay que añadir2,20 mL de KSCN para observar la aparición del colorrojo. Calcular la concentración de KCN y KCl en ladisolución inicial en g L–1.Pesos atómicos: Cl = 35,45; K = 39,1; C = 12,01; N = 14,00.Sol.: 3,907 g L–1 KCN; 1,678 g L–1 KCl.
mmol Ag (mL) 0,1exceso Ag Ag Ag
+ = × = ×+ + +V C V
([Ag ] [Ag ] ) 100Puntofinal Puntoequivalencia+ += − ×
mmol Ag exceso
+ =
Capítulo 8: Valoraciones de precipitación 295


9
9.1. Introducción9.2. Factor gravimétrico y sensibilidad9.3. Reactivos orgánicos y gravimetrías9.4. Aspectos experimentales de interés
en gravimetrías9.5. Métodos gravimétricos de calcinación9.6. Métodos gravimétricos de desecaciónCuestionesSeminarios: problemas numéricosGRAVIMETRÍAS

9.1. Introducción
La gravimetría es una técnica analítica clási-ca que permite cuantificar la masa y/o la con-centración de un analito por pesada. Común-mente se utiliza un producto de reacción oderivado estequiométrico del mismo. De acuer-do con esta definición, es evidente que la instru-mentación necesaria para el desarrollo de losmétodos gravimétricos es muy simple: una balan-za analítica es suficiente, cuya calibración se lle-va a cabo con estándares no analíticos, tales comolas pesas de referencia. Esta característica dife-rencial de las gravimetrías frente a las valora-ciones, que requieren además el uso de estánda-res químico-analíticos primarios (o secundariosestandarizados) para preparar las correspon-dientes disoluciones valoradas, hace que losmétodos gravimétricos presenten una cadena detrazabilidad más corta y ofrezcan en teoría unamayor exactitud y precisión que las valoraciones.Sin embargo, la bondad de estas propiedadesanalíticas está estrechamente relacionada con lainfluencia de diversos factores experimentales,esto es, con las propiedades analíticas comple-mentarias. Desafortunadamente, los métodosgravimétricos son lentos, tediosos, requieren deuna gran intervención humana y son difíciles deautomatizar y por tanto, en su conjunto, no sontan recomendables como las valoraciones. Apesar de todo, no se puede decir hoy por hoy quela gravimetría sea una técnica obsoleta ya quepara la resolución de determinados problemasanalíticos puede ser la alternativa a seguir.
Se pueden considerar tres tipos de métodos gra-vimétricos: 1) Los denominados métodos gravi-métricos por volatilización, en los que el compo-
nente a determinar presenta carácter volátil y sesepara de la muestra por destilación. Los vaporesse recogen sobre un sorbente adecuado, cuyoaumento de peso permite cuantificar la cantidadde especie absorbida, o bien se pesa la muestra quequeda tras la separación del componente volátil yse determina éste por diferencia. 2) Los métodosgravimétricos que hacen uso de la corriente eléc-trica, electrogravimetrías. Se basan en el estableci-miento de una diferencia de potencial entre unánodo y un cátodo (generalmente una rejilla deplatino) en el cual se lleva a cabo la reducción deun ion metálico a su estado elemental, que se depo-sita en el cátodo. La diferencia de peso de la rejilladel cátodo antes y después del proceso de elec-trolisis total, permite la determinación cuantitati-va del analito. 3) Los métodos gravimétricos basa-dos en la precipitación química son los querealmente se conocen con el nombre genérico degravimetrías. Estos métodos junto con las valora-ciones de precipitación constituyen el campo deaplicación cuantitativa de los equilibrios hetero-géneos de precipitación.
En la figura 9.1 se esquematiza el proceso ana-lítico en el que se basa una determinación gravi-métrica. A una alícuota de la muestra que contie-ne al analito, M, se le añade el reactivo precipitante,X, en unas condiciones experimentales dadas. Trasla formación cuantitativa del precipitado MX, éstese separa de la disolución mediante un proceso defiltración. Este precipitado constituye la denomi-nada forma de precipitación del analito. Esta for-ma de precipitación, tras un tratamiento térmicoa una temperatura más o menos elevada a tenorfundamentalmente de su pureza y estabilidad, ori-gina la correspondiente forma de pesada, base delmétodo gravimétrico, la cual puede ser igual, MX,
298 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Describir desde un punto de vista general los aspec-tos más relevantes de los métodos gravimétricos.
• Exponer aquellos métodos gravimétricos más repre-sentativos para la determinación de iones metálicos,
con el uso de agentes precipitantes inorgánicos yorgánicos.
• Estudio sistemático de estos métodos en base a su fun-damento y detalles experimentales más relevantes.

o diferente, MR, a la forma de precipitación segúnel tratamiento térmico realizado.
FIGURA 9.1. Esquema general parala determinación gravimétrica.
La forma de precipitación del analito es el pre-cipitado originado a partir del equilibrio de precipitación en el que se basa la determinacióngravimétrica. Se deben cumplir una serie de requi-sitos para su utilización en gravimetrías. La reac-ción ha de ser: 1) cuantitativa (valor de KS sufi-cientemente pequeño). Se recomienda que lasolubilidad de la forma de precipitación sea infe-rior a 10–6 mol L–1 y 2) selectiva. Se ha de traba-jar en unas condiciones experimentales tales quepermitan conseguir una forma de precipitación
lo más libre de impurezas posible. Además, laforma de precipitación ha de ser estable y pose-er una buenas propiedades mecánicas con obje-to de favorecer el proceso de filtración.
Cuando la forma de precipitación del analitocumple los requisitos indicados anteriormente,un tratamiento térmico a baja temperatura (seca-do entre 80-120 °C), con objeto de eliminar lahumedad, la transforma en la forma de pesada.En este caso, ambas formas son idénticas y pre-sentan una composición química definida y per-fectamente conocida, base de los cálculos gravi-métricos. Sin embargo, en el caso de que la formade pesada no sea estable debido a posibles inte-racciones con agentes atmosféricos tales comoH2O, CO2 y O2, esta forma no será adecuada parasu uso en gravimetría.
Cuando la forma de precipitación no presen-ta una estequiometría definida, se somete a untratamiento térmico a elevadas temperaturas (cal-cinación) con objeto de conseguir una especiederivada de la misma que posea una composiciónquímica definida y sea estable. A veces es nece-sario hacer uso del proceso de calcinación paraconseguir una forma de pesada estable, a pesarde que la forma de precipitación tenga una pure-za adecuada para su uso como forma de pesada.
En este capítulo, sólo se van a considerar losmétodos gravimétricos basados en la precipita-ción química y en concreto aquellas determina-ciones más representativas de iones metálicos conel uso de agentes precipitantes tanto inorgánicoscomo orgánicos, con objeto de mostrar una visiónglobal del potencial analítico de las técnicas gra-vimétricas. En cada caso, se considera el funda-mento de la metodología y aquellos detalles expe-rimentales más relevantes que justifican lapropuesta del procedimiento analítico recomen-dado para llevar a cabo la determinación.
9.2. Factor gravimétrico y sensibilidad
La cuantificación de la masa o bien de la con-centración de un analito a partir de la pesadaobtenida en una determinación gravimétrica, se
Capítulo 9: Gravimetrías 299

basa en cálculos sencillos en los que se utiliza eldenominado factor gravimétrico, G, el cual expre-sa la relación estequiométrica que debe existirentre el analito y su forma de pesada. En estesentido, el factor gravimétrico se define como elcociente entre el peso fórmula del analito y elpeso molecular de la forma de pesada:
[9.1]
Así, por ejemplo, en la determinación gravi-métrica de bario basada en la precipitación delión Ba2+ como BaSO4, la relación estequiomé-trica entre ambas especies: 1 mol de ion Ba2+ ori-gina 1 mol de BaSO4, dada por el factor gravi-métrico, se expresa como:
[9.2]
De acuerdo con la definición de G, se puedeestablecer la siguiente relación para el caso de ladeterminación gravimétrica del contenido de unanalito (Pa) en una muestra, a partir de su pesa-da gravimétrica (Pg):
[9.3]
Por tanto, el factor gravimétrico también sepuede definir como aquel número adimensionalpor el que hay que multiplicar la pesada gravi-métrica para obtener el peso de analito en lamuestra. Este factor es un parámetro caracterís-tico para cada método gravimétrico y es de vitalimportancia a la hora de evaluar su sensibilidad.
Dado que G define la relación estequiomé-trica que existe entre el analito y su forma de
pesada, se ha de tener sumo cuidado en aque-llos casos en los que se presenta una diferentecomposición estequiométrica entre ambas. Así,por ejemplo, en la determinación gravimétricade magnesio por precipitación con fosfato deamonio:
Mg2+ + (NH4)3PO4→←
→← MgNH4PO4 . 6 H2O(S)[9.4]
la forma de precipitación se ha de calcinar a ele-vadas temperaturas para obtener la forma depesada Mg2P2O7, que contiene dos átomos demagnesio, por lo que el factor gravimétrico eneste caso vendrá dado por:
[9.5]
Una situación similar se presenta en la deter-minación gravimétrica de potasio con ácido hexa-cloroplatínico:
2 K+ + H2Cl6Pt →← K2Cl6Pt (S) [9.6]
En este caso, la forma de pesada es Pt0, porlo que G viene dado por:
[9.7]
Dado que en este caso la forma de pesada nocontiene el analito a determinar, la relación este-quiométrica entre el potasio y el platino para calcular el valor de G se establece a partir de lacomposición de la forma de precipitación.
La sensibilidad en un método gravimétrico sesuele definir, en términos de límite de cuantifi-cación, como la mínima cantidad de analito quese puede determinar con un error aceptable. Estelímite de cuantificación está directamente rela-cionado con G y con la pesada gravimétrica Pgde acuerdo con la expresión:
Pa = G × Pg [9.3]
G = 2 K
Pt
G = 2 Mg
Mg P O2 2 7
Pa Pg
G Pg
= × =
= ×
Peso fórmula analitoPeso molecular forma de pesada
G = = =
= =
Peso atómico BaPeso molecular BaSO
BaBaSO
137, 36233, 43
0, 5884
4 4
G = Peso fórmula analito
Peso molecular forma de pesada
300 Equilibrios iónicos y sus aplicaciones analíticas

Si se considera que el error en la pesada gra-vimétrica es del 0,1%, de forma similar a la defi-nición de la región de equivalencia en las valo-raciones y que la incertidumbre de la balanzaanalítica se puede estimar en ±0,1 mg, el valormínimo de la pesada gravimétrica correspon-diente al límite de cuantificación será de 100 mg.Si se sustituye este valor mínimo de Pg en laexpresión [9.3] se tiene:
Pmin = G × 100 [9.8]
siendo Pmin la mínima cantidad de analito, expre-sada en mg, que permite determinar el métodogravimétrico en estudio, esto es, su límite decuantificación.
La expresión [9.8] pone de manifiesto que ellímite de cuantificación de un determinado méto-do gravimétrico, está directamente relacionadocon su factor gravimétrico. Así, cuanto menor
sea G más sensible será el método gravimétrico,lo que de acuerdo con la expresión [9.1] implicaen definitiva un mayor peso molecular de la for-ma de pesada. En el cuadro 9.1 se muestran loslímites de cuantificación de algunos métodos gra-vimétricos, obtenidos en las condiciones indica-das, en función del valor de G, esto es, del reac-tivo precipitante empleado en la determinación.
Se observa, por ejemplo, en el caso de ladeterminación de aluminio, que el empleo de unreactivo orgánico precipitante como la oxina ori-gina una sensibilidad de casi un orden de mag-nitud superior que empleando la precipitacióncomo hidróxido, debido básicamente a la grandiferencia entre el peso molecular de ambas for-mas de pesada. Este incremento de la sensibili-dad, debido al empleo de un reactivo orgánicocomo agente precipitante, es una de las causaspor las que se recomienda el uso de reactivosorgánicos.
Capítulo 9: Gravimetrías 301
CUADRO 9.1Sensibilidad alcanzada en la determinación gravimétrica de algunos iones metálicos
en función de su forma de pesada
Analito Reactivo Forma Factor Límite dede pesada gravimétrico cuantificación, mg
K+ ClO4̄ KClO4 0,2222 22,22B(C6H5)4̄ KB(C6H5)4 0,1091 10,91
Mg2+ PO43¯ Mg2P2O7 0,2185 21,85
Oxina Mg(Ox)2 0,0778 7,78Al3+ OH¯ Al2O3 0,5292 52,92
Oxina Al(Ox)3 0,0587 5,87
9.3. Reactivos orgánicos y gravimetrías
El uso de compuestos orgánicos constituyeuna interesante vía para la determinación gravi-métrica de iones metálicos. La mejora de pro-piedades analíticas básicas como la sensibilidady la selectividad cuando se hace uso de este tipode reactivos en lugar de reactivos inorgánicos,justifica plenamente su empleo.
Dado que el reactivo orgánico suele tener unpeso molecular elevado, éste también lo será para
la forma de pesada, de forma que el factor gra-vimétrico será muy pequeño, lo que hará que lasensibilidad sea elevada. Véase el ejemplocomentado en el apartado 9.2 correspondiente alincremento de la sensibilidad que se origina en ladeterminación gravimétrica de aluminio cuandose utiliza oxina como agente precipitante orgáni-co frente a su precipitación como óxido hidratado.
En general la precipitación de iones metáli-cos con reactivos orgánicos es bastante selecti-va, debido a que la estructura de la molécula

orgánica puede “manipularse” mediante la intro-ducción de grupos reactivos, que provoquen efec-tos estéricos, etc., de tal manera que se consigaun alto grado de selectividad en la reacción deprecipitación del ion metálico.
Las aplicaciones más significativas de estos rea-tivos orgánicos dan lugar a precipitados que se hande secar a bajas temperaturas. El precipitado obte-nido con estos reactivos ha de poseer unas carac-terísticas similares a las que se comentan en elapartado 9.4, pero en el procedimiento gravimé-trico se deben considerar aspectos concretos rela-cionados con la naturaleza orgánica de estos com-puestos, tales como:
1. El reactivo orgánico debe ser soluble enagua. Aunque en algunos casos se puedenutilizar disolventes orgánicos miscibles conagua (etanol, acetona, etc.) para poder pre-parar sus disoluciones. Como contraparti-da, el precipitado se origina en un medioacuo-orgánico que favorece su solubilidady en consecuencia, el medio de precipita-ción no debe contener una elevada pro-porción de disolvente orgánico. Además,no se pueden utilizar estos disolventes enel lavado de los precipitados obtenidos. Porotra parte, al rebajar la proporción de disol-vente orgánico en el medio de reacción sefavorece la co-precipitación de reactivo pre-cipitante en exceso.
2. El precipitado obtenido debe ser establefrente al secado e inerte frente a la hume-dad y otros agentes atmosféricos. En gene-ral, en estos métodos gravimétricos la for-ma de precipitación coincide con la formade pesada, lo que reduce el número de ope-raciones a realizar y el tiempo de análisis.
9.4. Aspectos experimentalesde interés en gravimetrías
Como ya se ha indicado, los métodos gravi-métricos son tediosos y requieren de una granintervención humana, por tanto, es de interés
considerar desde un punto de vista general, yantes de pasar a la descripción de los métodosgravimétricos más significativos, los aspectosprácticos más relevantes de las gravimetrías.
El primer paso de cualquier método gravi-métrico consiste en la precipitación del analitopor adición del reactivo precipitante. Este pro-ceso de precipitación debe ser lo más lento posi-ble con objeto de obtener precipitados más purosy con mejores propiedades mecánicas. En efec-to, las propiedades físicas de un precipitadodependen en gran medida del modo de forma-ción del mismo, en el que se pueden diferenciartres aspectos: nucleación, crecimiento cristalinoy envejecimiento o maduración.
La nucleación es el proceso que da lugar a laformación de partículas muy pequeñas y que soncapaces de crecer originando otras mayores.Cuando la precipitación se lleva a cabo rápida-mente, y especialmente con precipitados muyinsolubles, se favorece la etapa de nucleación(formación de micropartículas) en detrimentodel crecimiento cristalino. El precipitado en estascondiciones se encuentra muy disperso, en for-ma de gel, presentando una gran superficie espe-cífica, lo que acarrea dos graves inconvenientes:1) malas propiedades mecánicas, lo que dificul-ta considerablemente su posterior filtración, y2) alto grado de contaminación, generalmentepor adsorción de especies interferentes sobre susuperficie. Para minimizar estos inconvenientes,el proceso de precipitación ha de ser lo más len-to posible con objeto de favorecer la etapa denucleación.
Se pueden llevar a cabo diversas acciones conobjeto de ralentizar el proceso de precipitación.Además de añadir el agente precipitante sobrela disolución del analito gota a gota (se suelehacer uso de una bureta) y en continua agitación,una norma general consiste en llevar a cabo laprecipitación en caliente (60-80 °C) con objetode conseguir la máxima solubilidad para la for-ma de precipitación. En el caso de la precipita-ción de iones metálicos como hidróxidos (óxidoshidratados), se han de utilizar bases débiles, talescomo NH3, disolución reguladora NH4Cl/NH3, etc.,
302 Equilibrios iónicos y sus aplicaciones analíticas

como reactivos precipitantes con objeto de queel cambio de pH por adición de las mismas nosea muy acusado y se favorezca el proceso denucleación.
Una de las alternativas que da lugar a la for-mación de precipitados con buenas propiedadesmecánicas y con un mínimo grado de contami-nación es la denominada precipitación en diso-lución homogénea. Este modo de precipitaciónconsiste en provocar la precipitación en el senode la disolución del analito mediante una reac-ción química. Un ejemplo representativo de laprecipitación homogénea en gravimetrías, es elempleo de la hidrólisis de la urea en lugar de laadición de NH3 para conseguir un incrementogradual y controlado del pH en la precipitaciónde óxidos hidratados. Inicialmente a la disolu-ción que contiene el ion metálico a precipitar sele añade cloruro de amonio y urea. En estas con-diciones no se origina ningún precipitado; sinembargo, al calentar la disolución, y después deun tiempo, aparece el precipitado de óxido hidra-tado, ya que la urea se hidroliza de forma con-trolada con el tiempo y la temperatura, según lareacción:
H2N–CO–NH2 + H2O ∆T CO2 + NH3
[9.9]
Esta reacción origina NH3 en toda la masalíquida, lo que favorece la etapa de nucleación yal enfriar la disolución se produce la interrupciónde la hidrólisis, y por tanto, el incremento del pH.Hay que indicar finalmente que en este modo deprecipitación, la velocidad de generación del reac-tivo precipitante se puede disminuir hasta el pun-to de que sean necesarias varias horas para con-seguir que ésta sea cuantitativa, lo que permiteobtener precipitados con propiedades físicas yquímicas muy favorables.
Una vez obtenido el precipitado, es conve-niente someterlo a un período de digestión antesde proceder a la etapa de filtración. General-mente la digestión se realiza en caliente y en con-tacto con sus aguas madres. Durante este proce-so de digestión del precipitado, más o menos
prolongado según la composición del mismo, selogra una notable reducción de su superficie espe-cífica (las partículas se aglutinan reduciendo elgrado de dispersión del precipitado) y no sólo seproducen fenómenos de disolución–precipitaciónen su superficie, sino que casi siempre se origi-nan cambios químicos, tal como la formación depolímeros. En resumen, este proceso de enveje-cimiento del precipitado mejora su pureza y fun-damentalmente su propiedades mecánicas.
Existen dos procedimientos básicos para lafiltración de un precipitado, esto es su separaciónde la disolución, que dependen fundamental-mente del tratamiento térmico a realizar poste-riormente:
1. Si el precipitado obtenido presenta unapureza que permite utilizarlo como formade pesada, un tratamiento térmico a bajatemperatura (secado entre 80-120 °C) essuficiente para eliminar su humedad. Eneste caso, la filtración se realiza en una pla-ca filtrante mediante succión. Estas placasde cristal, porcelana o cuarzo tienen el fon-do poroso de vidrio sinterizado, cuya poro-sidad suele variar entre 5 y 100 micras.Cuando son de vidrio no pueden calentarsea temperaturas superiores a 150-160 °C.
2. Si el precipitado obtenido se ha de calci-nar, o sea someter a temperaturas eleva-das para conseguir la forma de pesada, elproceso de filtración se lleva a cabo sobrepapel de filtro sin cenizas. Este papel es decelulosa pura, sin impurezas minerales, asícuando se calcina no deja ningún residuoponderable. Este papel de filtro se sumi-nistra con un diferente grado de porosi-dad: grande, media y muy fina.
Una vez retenido el precipitado en el filtro yantes de someterlo al tratamiento térmico ade-cuado, es conveniente proceder a su lavado conobjeto de eliminar impurezas adsorbidas o rete-nidas, ya que alterarían la pesada gravimétricaoriginando errores por exceso. Las disolucionesde lavado son específicas para cada precipitado.
Capítulo 9: Gravimetrías 303
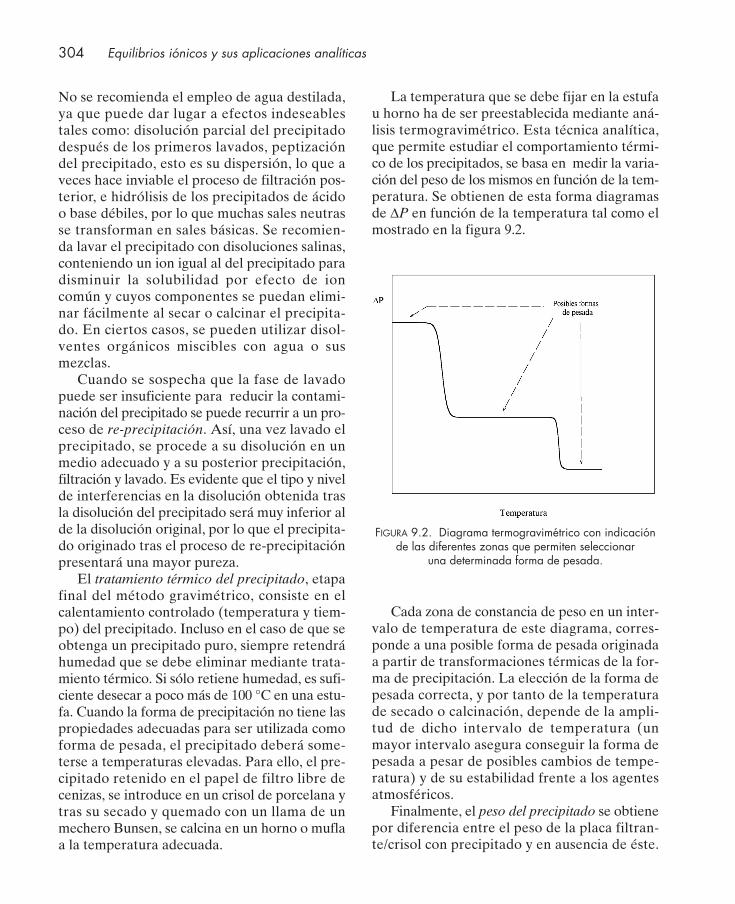
No se recomienda el empleo de agua destilada,ya que puede dar lugar a efectos indeseablestales como: disolución parcial del precipitadodespués de los primeros lavados, peptizacióndel precipitado, esto es su dispersión, lo que aveces hace inviable el proceso de filtración pos-terior, e hidrólisis de los precipitados de ácidoo base débiles, por lo que muchas sales neutrasse transforman en sales básicas. Se recomien-da lavar el precipitado con disoluciones salinas,conteniendo un ion igual al del precipitado paradisminuir la solubilidad por efecto de ioncomún y cuyos componentes se puedan elimi-nar fácilmente al secar o calcinar el precipita-do. En ciertos casos, se pueden utilizar disol-ventes orgánicos miscibles con agua o susmezclas.
Cuando se sospecha que la fase de lavadopuede ser insuficiente para reducir la contami-nación del precipitado se puede recurrir a un pro-ceso de re-precipitación. Así, una vez lavado elprecipitado, se procede a su disolución en unmedio adecuado y a su posterior precipitación,filtración y lavado. Es evidente que el tipo y nivelde interferencias en la disolución obtenida trasla disolución del precipitado será muy inferior alde la disolución original, por lo que el precipita-do originado tras el proceso de re-precipitaciónpresentará una mayor pureza.
El tratamiento térmico del precipitado, etapafinal del método gravimétrico, consiste en elcalentamiento controlado (temperatura y tiem-po) del precipitado. Incluso en el caso de que seobtenga un precipitado puro, siempre retendráhumedad que se debe eliminar mediante trata-miento térmico. Si sólo retiene humedad, es sufi-ciente desecar a poco más de 100 °C en una estu-fa. Cuando la forma de precipitación no tiene laspropiedades adecuadas para ser utilizada comoforma de pesada, el precipitado deberá some-terse a temperaturas elevadas. Para ello, el pre-cipitado retenido en el papel de filtro libre decenizas, se introduce en un crisol de porcelana ytras su secado y quemado con un llama de unmechero Bunsen, se calcina en un horno o muflaa la temperatura adecuada.
La temperatura que se debe fijar en la estufau horno ha de ser preestablecida mediante aná-lisis termogravimétrico. Esta técnica analítica,que permite estudiar el comportamiento térmi-co de los precipitados, se basa en medir la varia-ción del peso de los mismos en función de la tem-peratura. Se obtienen de esta forma diagramasde ∆P en función de la temperatura tal como elmostrado en la figura 9.2.
FIGURA 9.2. Diagrama termogravimétrico con indicaciónde las diferentes zonas que permiten seleccionar
una determinada forma de pesada.
Cada zona de constancia de peso en un inter-valo de temperatura de este diagrama, corres-ponde a una posible forma de pesada originadaa partir de transformaciones térmicas de la for-ma de precipitación. La elección de la forma depesada correcta, y por tanto de la temperaturade secado o calcinación, depende de la ampli-tud de dicho intervalo de temperatura (unmayor intervalo asegura conseguir la forma depesada a pesar de posibles cambios de tempe-ratura) y de su estabilidad frente a los agentesatmosféricos.
Finalmente, el peso del precipitado se obtienepor diferencia entre el peso de la placa filtran-te/crisol con precipitado y en ausencia de éste.
304 Equilibrios iónicos y sus aplicaciones analíticas

Dado que la placa filtrante/crisol constituyen lamayor parte del peso, se ha de conocer su pesocon gran exactitud, así se han de someter vacíosal mismo tratamiento térmico que cuando con-tienen el precipitado y realizar pesadas sucesivashasta obtener un peso constante de los mismos.
9.5. Métodos gravimétricosde calcinación
Bajo esta denominación se agrupan todasaquellas gravimetrías en las que la forma de pre-cipitación del analito no presenta una estequio-metría definida o bien no es estable, por lo quedebe someterse a un tratamiento térmico a tem-peraturas elevadas o calcinación, con objeto detransformarla en una especie derivada estable yque posea una composición química definida.Existen un gran número de métodos gravimétri-cos de calcinación para la determinación de ionesmetálicos y no metales que se pueden clasificaren dos grandes grupos de acuerdo con las carac-terísticas químicas de la forma de precipitación:óxidos hidratados y compuestos salinos. A con-tinuación, se describe de forma detallada cadauno de estos tipos de métodos gravimétricos conejemplos significativos de los mismos.
9.5.1. Determinaciones por formaciónde óxidos hidratados
La determinación de iones metálicos, talescomo Fe3+, Al3+, Ti4+, Be2+, Cr3+, etc., y no meta-les (SiO3
2¯) se basa en la formación de un preci-pitado de sal básica (óxido hidratado) al aumen-tar el pH de la disolución. En efecto, cuando seeleva el pH de una disolución que contiene estosiones metálicos, los precipitados obtenidos mues-tran una estequiometría variable que no secorresponde con M(OH)n, dado que precipitanfrecuentemente como sales básicas de fórmula:Mx(OH)yXz. a H2O, siendo X un anión presenteen la disolución, tal como Cl¯, NO3̄, SO4
2¯, entreotros. La composición de las sales básicas es
variable y depende de las condiciones de preci-pitación y de la concentración de OH–, lo quehace inviable su empleo como forma de pesada.A pesar de ello, la precipitación del ion metáli-co es cuantitativa debido a los pequeños valoresde sus productos de solubilidad. La posteriortransformación térmica de este precipitado (seca-do y calcinación) conduce a la formación del óxi-do anhidro, M2On, que se utiliza como forma depesada.
La precipitación como óxido hidratado confines gravimétricos es un proceso poco selecti-vo, ya que al precipitar el analito (Mn+), si en ladisolución existen otros iones metálicos diva-lentes, trivalentes o tetravalentes se incorporanal precipitado, bien por co-precipitación (sealcanza su valor de KS) o bien por fenómenosde adsorción superficial. Los iones metálicosdivalentes quedan frecuentemente incorpora-dos en la superficie del precipitado en la deno-minada “doble capa”. Como se muestra en lafigura 9.3, un precipitado de óxido hidratado sesuele rodear, debido a fenómenos de adsorción,de una primera capa de iones OH– por la ele-vada concentración de los mismos en el seno dela disolución; si además existen otros ionesmetálicos divalentes en esta disolución, se pue-den incorporar en una segunda capa si se alcan-za puntualmente su producto de solubilidad. Alcalcinar este precipitado el óxido del ion metá-lico adsorbido, originado en este tratamientotérmico, contamina al precipitado originandoerrores por exceso.
Este tipo de interferencia se puede eliminarmediante un proceso denominado adsorción porintercambio, para ello se adiciona una cantidadelevada de ion NH4
+ a la disolución que contieneel analito y los iones M2+ interferentes y se pro-duce en la segunda capa del precipitado forma-do un intercambio entre el ion metálico divalen-te y el ion amonio debido a su elevadaconcentración (como se muestra en la figura 9.3).El nuevo precipitado puede considerarse purodesde un punto de vista gravimétrico ya que alcalcinarse los iones OH– y NH4
+ se volatilizancomo H2O y NH3, respectivamente.
Capítulo 9: Gravimetrías 305

Sin embargo, cuando el precipitado presentauna gran actividad superficial, es difícil suprimircompletamente las interferencias por este pro-ceso, a pesar de que a elevadas concentracionesde ion amonio también decrece la adsorción pri-maria de iones OH–. En tal caso, se pueden aña-dir aniones orgánicos voluminosos, tales comoacetato o benzoato, a la disolución problema, conobjeto de que los mismos formen parte del pre-cipitado de sal básica en lugar de los típicos ionesCl–, NO3̄, o SO4
2¯, con vistas a reducir la superfi-cie específica del precipitado. En estas condicio-nes, el precipitado adsorbe un menor número deiones OH– en la primera capa, lo que originaráuna menor adsorción de iones metálicos diva-lentes interferentes, que se pueden eliminar total-mente mediante adsorción por intercambio. Estosaniones orgánicos no originan errores en la deter-minación gravimétrica ya que se eliminan comoCO2 y H2O en la etapa de calcinación.
Es interesante resaltar el importante papel quejuega el par ácido-base NH4
+/NH3 en la determi-nación gravimétrica de iones metálicos por preci-pitación como óxidos hidratados. El amoniaco ejer-ce una doble función, ya que se utiliza como agenteprecipitante y ligando enmascarante, pues dismi-nuye la adsorción de iones metálicos divalentestales como Ni2+, Co2+, Zn2+, etc., por formación de
sus correspondientes complejos amoniacales. Porotra parte, el ion amonio a elevadas concentra-ciones reduce el pH al que se lleva a cabo la pre-cipitación, con lo que decrece la adsorción prima-ria de iones OH– y elimina la interferencia de ionesM2+ mediante adsorción por intercambio.
La determinación de hierro es uno de los ejem-plos más representativos de los métodos gravi-métricos basados en la formación de óxidos hidra-tados. El precipitado de sal básica de Fe3+ es muyinsoluble y por tanto comienza a precipitar a pHácido: si se parte de una disolución 10–2 mol L–1
de Fe3+ la precipitación es completa a partir de pH = 4. En líneas generales, el procedimientoexperimental para llevar a cabo la determinaciónha de considerar los siguientes factores:
1. La precipitación debe hacerse en presen-cia de sales amónicas y en caliente paraflocular al precipitado. Si la disolución eslo bastante ácida, las sales amónicas seforman in situ al adicionar amoniaco, encaso contrario se añaden uno o dos gramosde cloruro de amonio.
2. El precipitado se deja una media hora enreposo, se filtra en caliente sobre papel defiltro de poro grande y se lava con aguacaliente o con disolución de NH4NO3 has-
306 Equilibrios iónicos y sus aplicaciones analíticas
FIGURA 9.3. Fundamento delproceso de adsorción porintercambio en determinacio-nes gravimétricas basadas enla precipitación como óxidoshidratados.

ta reacción negativa de iones Cl– en lasaguas de lavado, ya que si éstos quedanretenidos en el precipitado se origina alcalcinar FeCl3 volátil, dando lugar a erro-res por defecto.
3. El precipitado se seca dentro de un crisolde porcelana previamente tarado a pesoconstante, se calienta para quemar el papelcon un mechero Bunsen y posteriormentese calcina a unos 900° en un horno o mufla.Tras este tratamiento se deja enfriar en undesecador y se pesa hasta peso constante.
4. La diferencia entre ambas pesadas corres-ponde al peso de Fe2O3 (PFe2O3
) y median-te el factor gravimétrico se puede determi-nar el peso de hierro (PFe) presente en lamuestra haciendo uso de la expresión [9.3]
[9.10]
de donde:
[9.11]
A pesar de seguir las indicaciones expuestas enel apartado anterior con relación a evitar la con-taminación del precipitado, esta determinación
gravimétrica no está exenta de ciertas interfe-rencias, tales como: la contaminación debida aiones M3+ y M4+ que no puede evitarse en lapráctica, y la interferencia de aniones que reac-cionan con el ion Fe3+, que puede originar erro-res por exceso y por defecto. Así, los anionesque precipitan al ion Fe3+, especialmente el ionPO4
3¯, se incorporan al precipitado y originanerrores por exceso. Los aniones que formancomplejos estables con el ion Fe3+ (F–, citrato,tartrato, etc.) provocan su precipitación parcialy, por tanto, originan errores por defecto.
9.5.2. Determinaciones por formaciónde compuestos salinos
En este tipo de determinaciones gravimétri-cas, la forma de precipitación es el resultado deun proceso de asociación iónica, dando lugar aprecipitados de compuestos salinos:
Mn+ + Am¯ →← MmAn(s) [9.12]
En estos casos el analito a determinar puedeser tanto un ion metálico Mn+ como el anión Am–.Desde un punto de vista formal, la precipita-ción como compuestos salinos no se diferencia
P PFe Fe O0,6994
2 3= ×
P G P PFe Fe O
2 3Fe O2 3 2 3
2 FeFe O
= × = ×
Capítulo 9: Gravimetrías 307
CUADRO 9.2Determinaciones gravimétricas más relevantes por precipitación como compuestos salinos
Analito Forma Forma Tratamiento Factor de precipitación de pesada térmico, °C gravimétrico
Cloro (Cl–) AgCl AgCl 1.110 0,2474Azufre (SO4
2¯) BaSO4 BaSO4 1.800 0,4115Fósforo (PO4
3¯) MgNH4PO4 Mg2P2O7 1.000 0,4267Arsénico (AsO4
3¯) MgNH4AsO4 Mg2As2O7 1.900 0,4474Estaño (Sn4+) SnO2 SnO2 1.000 0,7876Plomo (Pb2+) PbSO4 PbSO4 1.500 0,6832Mercurio (Hg2+) HgS HgS 1.105 0,8622Calcio (Ca2+) CaC2O4 . H2O CaCO3 1.500 0,4004Magnesio (Mg2+) MgNH4PO4 Mg2P2O7 1.000 0,2185Estroncio (Sr2+) SrSO4 SrSO4 1.300 0,4770

de la de óxidos hidratados, Am– juega el papel delos iones OH–; sin embargo, sus característicasgravimétricas son bien diferentes. En el cuadro9.2 se muestran los métodos gravimétricos mássignificativos basados en la precipitación comocompuestos salinos.
Por lo general, la forma de precipitación pre-senta las características gravimétricas adecuadaspara su empleo como forma de pesada. Sinembargo, en algunos casos el tratamiento térmi-co se debe realizar a temperaturas superiores ala de un simple secado (105-110 °C), como en elcaso de los sulfatos de bario, estroncio y plomo,debido a que estos precipitados retienen gran can-tidad de humedad. Otros precipitados no tienenuna composición perfectamente definida y portanto se deben someter a un proceso de calcina-ción como en el caso del MgNH4PO4, forma deprecipitación en la determinación gravimétricade Mg2+ y PO4
3¯, que debe calcinarse a Mg2P2O7.A continuación, y como ejemplo represen-
tativo de los métodos gravimétricos por preci-pitación como compuestos salinos, se describedetalladamente la determinación gravimétricade calcio.
Esta determinación se fundamenta en la pre-cipitación del ion Ca2+ con oxalato de amoniooriginando un precipitado de oxalato de calciomonohidrato:
Ca2+ + C2O42¯ →← CaC2O4 . H2O(S) [9.13]
El pH influye decisivamente en este equili-brio de precipitación y en la pureza del preci-pitado obtenido. Dado que el anión oxalato pro-cede de un ácido débil, sus reaccionessecundarias de protonación originarán unaumento de la solubilidad del precipitado deoxalato de calcio en medio ácido, como se hamostrado en el capítulo 7. Los valores de lasconstantes de disociación del ácido oxálico sonpKa1 = 1,27 y pKa2 = 4,47 y se puede afirmar quea valores de pH inferiores a 2 el precipitado sesolubilizará totalmente, mientras que a valoresde pH superiores a 6, el equilibrio de precipita-ción, ecuación [9.13], estará totalmente despla-
zado hacia la derecha. En algunos casos, y conobjeto de eliminar la interferencia de ionesmetálicos que precipitan con el oxalato, se reco-mienda llevar a cabo la precipitación a pH = 3y en presencia de un exceso de reactivo preci-pitante. Por lo general, la precipitación se llevaa cabo a un valor de pH ligeramente superior a 6, que viene dado por el viraje del indicadorrojo de metilo (4,4-6,2), que se añade inicial-mente a la disolución que contiene el ion Ca2+.
El procedimiento experimental para llevar acabo esta determinación gravimétrica consiste en:
1. A la disolución de Ca2+ en medio ácido sele añade oxalato de amonio en exceso yunas gotas del indicador rojo de metilo. Secalienta a unos 80 °C y se añade NH3 1:1hasta viraje del indicador (color amarillo).
2. Se deja una hora en reposo y se filtra a tra-vés de un papel de porosidad media y selava varias veces con disolución de oxala-to de amonio al 1%.
3. El precipitado se seca dentro de un crisolde porcelana previamente tarado, secalienta para quemar el papel con unmechero Bunsen y posteriormente sesomete al correspondiente tratamiento tér-mico de calcinación en una mufla.
Según el tratamiento térmico la forma de pre-cipitación obtenida en esta determinación gravi-métrica, será distinta, ya que se pueden conse-guir diversas formas de pesada al aparecer variaszonas de constancia de peso con la temperaturaen el correspondiente diagrama termogravimé-trico, como se aprecia en la figura 9.4.
El oxalato de calcio monohidratado es un pre-cipitado higroscópico y por tanto no es aptocomo forma de pesada. A 200 °C se transformaen CaC2O4 , que podría utilizarse como forma depesada aunque como en el caso anterior, tambiénes algo higroscópico. Entre 475 y 525 °C se ori-gina la transformación del oxalato de calcio a car-bonato, por desprendimiento de CO:
CaC2O4 → CaCO3 + CO ↑ [9.14]
308 Equilibrios iónicos y sus aplicaciones analíticas

Ésta es la mejor forma de pesada con finesgravimétricos ya que es estable frente a los agen-tes atmosféricos. A temperaturas elevadas se pro-duce otra constancia de peso en el diagrama ter-mogravimétrico debido a la formación del óxidode calcio:
CaCO3 → CaO + CO2 ↑ [9.15]
que es también higroscópico y retiene CO2atmosférico (se carbonata fácilmente).
FIGURA 9.4. Comportamiento termogravimétricodel precipitado de oxalato de calcio.
A tenor de que la forma de pesada más ade-cuada es CaCO3 y en base a unos cálculos simi-lares a los descritos para la determinación gra-vimétrica de hierro, el contenido de calcio (PCa)en la muestra a analizar, se calcula de acuerdocon:
[9.16]
de donde:
[9.17]
El Mg2+es sin duda una de las interferenciasde mayor importancia en esta determinación. Elprocedimiento indicado es adecuado cuando enla muestra existen cantidades aproximadamen-te iguales de ambos iones metálicos, mientras quecuando el contenido de ion Mg2+ es superior alde Ca2+, se origina la co-precipitación parcial delMgC2O4 y se debe recurrir a la re-precipitaciónpara eliminar esta interferencia.
9.6. Métodos gravimétricosde desecación
Tal como se ha comentado anteriormente, enciertos casos la forma de precipitación inicial pre-senta ya características gravimétricas adecuadaspara su empleo como forma de pesada. En estoscasos el tratamiento térmico que se debe realizares un simple secado a 105-110 ºC. Se habla enton-ces de métodos gravimétricos de desecación, enellos podemos tener como forma de precipitaciónun compuesto salino o bien un compuesto for-mado por un ion inorgánico y un reactivo orgáni-co. Para ilustrar este tipo de gravimetrías, se des-cribe a continuación un ejemplo característico decada uno de estos métodos gravimétricos.
9.6.1. Determinación de plomocomo cromato de plomo
El plomo forma una sal poco soluble, de coloramarillo, con el ion cromato en medio modera-damente ácido, según la reacción:
Pb2+ + CrO42¯ →← PbCrO4(S) [9.18]
La precipitación es óptima a un pH aproxima-damente igual a 4; si es menor, el plomo puede pre-cipitar en forma de dicromato de plomo, mientrasque si es mayor aumenta la solubilidad del cro-mato de plomo. Otros iones pueden formar cro-matos insolubles, pero no suponen una interfe-rencia, ya que el cromato de plomo es el único queprecipita en medio relativamente ácido.
P PCa CaCO0,40043
= ×
P G P PCa = × = ×CaCO
3CaCO3 3
CaCaCO
Capítulo 9: Gravimetrías 309

Se puede llevar a cabo una precipitación homo-génea, en la que se genera “in situ” el cromatomediante la oxidación del ion Cr3+ con broma-to de potasio, o bien seguir el método conven-cional, si bien el pequeño tamaño de las partícu-las del precipitado obtenido hace necesario unproceso de digestión antes de la filtración.
El procedimiento experimental para llevar acabo esta determinación gravimétrica consiste en:
1. A la disolución de Pb2+ (muestra disuelta)se le ajusta el pH a 4 y se le añade una diso-lución de cromato de potasio al 4%, gota agota y con agitación, hasta que no se obser-ve la aparición de más precipitado y segui-damente un exceso de 2 mL. Finalmente,se digiere el precipitado durante una hora.
2. Se filtra sobre un crisol de placa filtrantedel número 4, previamente tarado. Antesde filtrar toda la disolución es convenien-te asegurarse de que la precipitación hasido completa; para ello basta con añadirunas gotas de cromato de potasio a las pri-meras gotas del filtrado y comprobar queno precipita más cromato de plomo.
3. Se lava con un mínimo de 10 porciones de3-4 mL de ácido nítrico al 0,1% y se secaa 110-120 °C hasta peso constante.
La expresión a utilizar para calcular el pesode plomo (PPb) en la muestra es:
[9.19]
9.6.2. Determinación de níquelcon dimetilglioxima
La dimetilglioxima (DMG) forma con el ionNi2+ un quelato insoluble de color rojo-rosadoen medio amoniacal de estequiometría 1:2,metal:ligando, que sirve de base para la deter-minación gravimétrica de níquel.
Para conseguir las características deseablesdesde un punto de vista gravimétrico en el pre-cipitado, se han de conjugar una serie de facto-res experimentales, tales como:
1. Al ser la DMG insoluble en agua, debeañadirse disuelta en etanol, aunque con-trolando estrictamente la relación finaletanol/agua en el medio de precipitación,que debe ser inferior a 1/2 para evitar ladisolución parcial del precipitado.
2. El exceso de reactivo tiende a co-precipi-tar con el quelato de níquel. Para contro-lar este exceso se podría añadir la DMG auna disolución amoniacal de Ni2+; pero elprecipitado formado en estas condicionespresenta malas propiedades mecánicas y, por tanto, el amoniaco debe añadirsedespués de la DMG.
3. Un gran exceso de amoniaco hace más difí-cil la precipitación.
En estas condiciones, el procedimiento expe-rimental, en líneas generales, contempla lassiguientes etapas:
1. La disolución de Ni2+ se calienta a ebulli-ción, se le añade exceso de amoniaco dilui-do y se neutraliza con HCl. Se añade unexceso de disolución al 1% de DMG enetanol y a continuación un ligero excesode NH3. Se deja en reposo durante unahora.
2. Se filtra el precipitado en placa filtrantepreviamente tarada y se lava con aguacaliente. Se seca a 110 °C hasta peso cons-tante del quelato Ni(DMG)2.
P G P P
P
Pb PbCrO4
PbCrO
PbCrO
4 4
4
PbPbCrO
0, 6431
= × = × =
= ×
310 Equilibrios iónicos y sus aplicaciones analíticas

La expresión a utilizar para calcular el pesode níquel (PNi) en la muestra es:
[9.20]
La dimetilglioxima forma quelatos solublese insolubles con diversos iones metálicos que
originan interferencias. La interferencia origi-nada por iones metálicos trivalentes, tales comoFe3+, Al3+ y Cr3+, se puede eliminar por adiciónde tartrato, que forma complejos con los mis-mos, mientras que la de iones metálicos diva-lentes, como por ejemplo Co2+ y Zn2+ que for-man quelatos solubles con la DMG, se sueleevitar añadiendo un ligero exceso de reactivoprecipitante, aunque siempre con precuacióndada la insolubilidad de la DMG en el mediode reacción.
P G P
P P
Ni Ni(DMG)
2Ni(DMG Ni(DMG)
2
NiNi(DMG)
= × =
= × = ×) ,2 2
0 2032
Capítulo 9: Gravimetrías 311
Cuestiones
9.1. Para que un precipitado sea adecuado como forma de pesada en una determinación gravimétrica, ha de poseer:
[ ] Estructura cristalina[ ] Alta pureza[ ] Bajo peso molecular[ ] Estabilidad frente al O2
[ ] Composición definida en un intervalo de temperatura
9.2. La digestión de un precipitado permite:
[ ] Conseguir que la forma de precipitación sea igual a la forma de pesada[ ] Disminuir los fenómenos de contaminación[ ] Favorecer la adsorción por intercambio[ ] Facilitar su posterior filtración
9.3. Si un precipitado presenta una estequiometría definida y alta pureza, ¿por qué razón(es) no puede serempleado como forma de pesada en una determinación gravimétrica?
9.4. En las determinaciones gravimétricas basadas en la formación de óxidos hidratados, interesa que la velocidadde precipitación sea:
[ ] Lenta[ ] Rápida
ya que se favorece el/la
[ ] Nucleación del precipitado[ ] Crecimiento cristalino del precipitado
9.5. ¿Por qué está prohibido el uso de NaOH como agente precipitante en los métodos gravimétricos basados enla precipitación como óxido hidratado?

9.6. ¿Qué es la adsorción por intercambio?
[ ] Un proceso que reduce las interferencias en las determinaciones gravimétricas basadas en la precipitacióncomo óxidos hidratados
[ ] Una forma de preconcentrar un analito antes de una gravimetría[ ] Una forma de mejorar las propiedades mecánicas de los precipitados
9.7. En las determinaciones gravimétricas con el uso de reactivos orgánicos interesa que la forma de precipita-ción sea igual a la forma de pesada, ya que:
[ ] Se incrementa la sensibilidad[ ] Se incrementa la selectividad[ ] El procedimiento gravimétrico es más rápido
9.8. Indicar cuál es la forma de pesada en las siguientes determinaciones gravimétricas:
Analito Reactivo precipitante Forma de pesada
Pb2+ K2CrO4
Fe3+ NH3
Ca2+ (NH4)2C2O4
Ni2+ Dimetilglioxima
312 Equilibrios iónicos y sus aplicaciones analíticas
Seminarios: problemas numéricos
• Objetivo pedagógicoDesarrollo de diversos problemas sobre las determina-ciones gravimétricas abordadas en este capítulo, mos-trando en algunos de ellos las fuentes de error más comu-nes que se presentan en los métodos gravimétricos.
1. Una muestra de 1,000 g de un mineral que contieneun 8,30% de aluminio se disuelve y se precipita selec-tivamente obteniéndose un precipitado de óxido dealuminio que pesa 167,2 mg. ¿Qué error se ha come-tido por higroscopicidad de la forma de pesada?Pesos atómicos: Al = 26,98; O = 16,00.
Dado que se conoce el contenido de aluminio enel mineral se puede calcular el peso del precipitadode óxido de aluminio que se emplea como forma depesada, y evaluar así el error cometido por higrosco-picidad. El peso de aluminio será:
1.000 × 8,3100 = 83 mg de aluminio
De acuerdo con la expresión [9.3] se tiene:
Pa = G × Pg
y al sustituir:
de donde se obtiene un peso de óxido de aluminio,Pg, de 156,8 mg.
Este valor es inferior al obtenido experimental-mente, por tanto, se ha cometido un error por exce-so que viene determinado por:
2. Una mezcla de AgCl y AgBr, que pesa 1,000 g con-tiene 0,6635 g de Ag. Calcular el porcentaje de ionbromuro en la muestra.Pesos atómicos: Ag = 107,88; Br = 79,92; Cl = 35,46.Sol.: 21,27%.
Error167,2 156,8
156,8100= + − × = +6 63, %
83
2AlAl O
2 26,98101,962 3
= × =×
×Pg Pg

3. El azufre contenido en cinco comprimidos de sacari-na (C7H5NO3S) con un peso total de 0,2140 g se oxi-da a sulfato y se precipita en forma de BaSO4. Si elpeso de precipitado de BaSO4 es de 0,2070 g, calcu-lar el contenido de sacarina en cada tableta.Pesos atómicos: S = 32,07; H = 1,00; O = 16,00;N = 14,00; C = 12,01; Ba = 137,36.Sol.: 0,0325 g.
4. Una muestra de Na2SO4 con impurezas, origina unprecipitado de BaSO4 de peso igual a la muestra.¿Cuál es el porcentaje de sulfato de sodio en lamuestra?Pesos atómicos: S = 32,07; O = 16,00; Ba = 137,36;Na = 22,99.
Si se denomina Pmuestra al peso de la muestra yPNa2SO4
al contenido de la misma en sulfato de sodio,se puede establecer la siguiente relación:
Pmuestra PNa2SO4
100 % % Na2SO4
de donde:
Por otra parte, la determinación de sulfato de sodioen la muestra se lleva a cabo por precipitación delmismo como sulfato de bario, con lo cual de acuerdocon la ecuación [9.3] se tiene:
Al igualar las expresiones obtenidas para el con-tenido de sulfato de sodio en la muestra se llega a:
y al aplicar la condición dada por el problema de quePmuestra = PBaSO4
se tiene:
de donde:
5. Para determinar el contenido de aluminio en un mine-ral se disuelven 1,5000 g del mismo y el Al3+ forma-do se precipita con oxina y se obtiene una pesada de1584,3 mg. ¿Cuál es el factor gravimétrico si el por-centaje de aluminio en la muestra es del 6,2%? ¿Quépesada gravimétrica se obtendría si se precipitase otramuestra idéntica como óxido hidratado y se calcina-se hasta óxido de aluminio?Pesos atómicos: Al = 26,98; O = 16,00.Sol.: 1) 0,0587; 2) 175,7 mg.
6. 2,000 gr de un mineral que contiene el 3,2% de cal-cio se trata convenientemente y se obtiene una diso-lución de 250 mL. Una alícuota de 100 mL se preci-pita con oxalato y el precipitado se calcina a 800 °Cformándose óxido de calcio que es higroscópico yretiene CO2 atomosférico. Si se pesan 40,2 mg de óxi-do, ¿qué error relativo por exceso o por defecto seha cometido en la pesada?Pesos atómicos: Ca = 40,08; O = 16,00; C = 12,01.
Inicialmente se ha de calcular el contenido de ionCa2+ presente en la alícuota de 100 mL que se some-te a la determinación gravimétrica con oxalato. Deacuerdo con el porcentaje de calcio en el mineral ylas diluciones realizadas se puede establecer:
Esta muestra de calcio se precipita como oxalato,se calcina a 800 °C y se pesa como CaO. La cantidadque se debería pesar de este óxido si no existen losproblemas de higroscopicidad y retención de CO2indicados en el problema se calcula a partir de:
de donde:
Se obtiene debido a los problemas indicados unapesada de CaO de 40,2 mg, lo que originará errores
P PCaO Ca
CaOCa
56,0840,08
25,6 35,8 mgCaO= × = × =
P PCa CaO
CaCaO
= ×
2.000 mg
3,2100
100250
25,6 mgCa× × =
%Na SO
Na SOBaSO
100142,05233,43
100 60,85%2 42 4
4
= × = × =
%Na SO100
Na SOBaSO
2 4 2 4
4
=
PPmuestra 2 4 2 4
4BaSO
%Na SO100
Na SOBaSO 4
×= ×
P PNa SO
2 4
4BaSO2 4 4
Na SOBaSO
= ×
P
PNa SO
muestra 2 42 4
%Na SO100
=×
Capítulo 9: Gravimetrías 313

por exceso en esta determinación gravimétrica. Elporcentaje de error vendrá dado por:
7. Una muestra de 2,000 g que sólo contiene K2CO3 yPbCO3 da un cierto peso de CO2 cuando se trata conexceso de ácido fuerte. Una muestra de 1,000 g deMgCO3 puro da exactamente el mismo peso de CO2cuando se trata de la misma manera. ¿Cuál es el por-centaje de K2CO3 en la mezcla?Pesos atómicos: C = 12,01; O = 16,00; K = 39,10;Pb = 207,21; Mg = 24,32.Sol.: 62,62%.
8. El magnesio contenido en una muestra, 0,9400 g, sedetermina gravimétricamente como MgNH4PO4. Elprecipitado contiene 0,0150 g de (NH4)3PO4. Calcu-lar el error cometido en la determinación de magne-sio, si en el proceso de calcinación, para pasar aMg2P2O7, el (NH4)3PO4 queda como HPO3.Pesos atómicos: P = 30,98; Mg = 24,32; N = 14,00; H = 1,00; O = 16,00.Sol.: 0,186%.
9. Se disuelve un mineral de plomo y el Pb2+ formadose precipita como PbCrO4 (forma de pesada). ¿Quépeso de muestra debe tomarse para que 5 mg dePbCrO4 represente el 2% de plomo en el mineral?Pesos atómicos: Cr = 51,01; O = 16,00; Pb = 207,21.Sol.: 160,8 mg.
10. Se trituran y se mezclan perfectamente 25 tabletasde un complemento alimentario de hierro, siendo lamasa total 22,1318 g. Se toman 1,1066 g de muestra,se disuelven en ácido nítrico y se calienta la disolu-ción para oxidar todo el hierro a Fe3+. Por adiciónde amoniaco se precipita el hierro cuantitativamentecomo óxido hidratado, y tras calcinar se obtiene unprecipitado de Fe2O3 que pesa 0,2641 g. ¿Cuál es elcontenido de FeSO4.7H2O en cada tableta?Pesos atómicos: Fe = 55,86; S = 32,07; O = 16,00; H = 1,00.
Para determinar el contenido de ion Fe3+ en lamuestra de 1,1066 g se lleva a cabo su precipitacióncon amoniaco y pesada como Fe2O3. De acuerdo conla expresión [9.3] se tiene:
Dado que este contenido de hierro procede de lasal FeSO4.7H2O, el contenido de la misma en la mues-tra analizada será:
Para determinar el contenido de esta sal por table-ta, basta con considerar la proporción entre la mues-tra analizada y la inicial así como del número de table-tas que conforman la misma:
11. Una muestra de BaCl2 contiene K2CO3 como impu-reza. Al disolver una muestra en agua se origina unprecipitado de BaCO3, el cual una vez filtrado, lava-do y seco pesa 65 mg. A la disolución resultante se leañade exceso de Na2SO4 y el precipitado formado deBaSO4 una vez filtrado, lavado y seco pesa 900 mg.Calcular el % de BaCl2 y K2CO3 en la muestra.Pesos atómicos: Ba = 137,36; Cl = 35,46; K = 39,10; C = 12,01; O = 16,00; Na = 22,99; S = 32,07.Sol.: 4,96 % K2CO3 y 95,04 % BaCl2.
12. Para determinar el contenido de níquel en un ace-ro inoxidable, se pesa una muestra de 0,2175 g y setrata con 10 mL de ácido nítrico concentrado.Cuando se ha disuelto completamente, se diluye aun volumen final de 200 mL. A continuación se leañade ácido tartárico, dimetilglioxima (DMG) y seneutraliza con NH3. El precipitado obtenido deNi(DMG)2 se filtra en una placa filtrante previa-mente tarada (peso de tara = 25,8218 g), y se secaen una estufa a 120 ºC. El peso final obtenido es de26,0092 g. ¿Qué efecto tiene la adición de ácido tar-tárico y amoniaco? ¿Cuál es el porcentaje de níquelen el acero?Peso atómico: Ni = 58,71.Peso molecular: DMG = 116,04.Sol.: 17,52%.
0, 9190
22,13181,1066
125
0, 7352 g sal/tableta× × =
0,1847FeSO .7 H O
Fe0,1847
277, 9355, 86
0, 9190 g FeSO .7 H O
4 2
4 2
× = × =
=
P PFe
2 3Fe O
2 FeFe O
2 55,86159,72
0,2641 0,1847 g Fe2 3
= × =×
× =
% Error
40, 2 35, 835, 8
100 12, 29 %= − × =
314 Equilibrios iónicos y sus aplicaciones analíticas

10
10.1. Introducción10.2. Estudio de reacciones redox: celdas
electroquímicas10.3. Potencial y carácter químico redox:
ecuación de Nernst10.4. Medida del potencial redox:
electrodos de referencia10.5. Potencial estándar10.6. Constante de equilibrio de una
reacción redox10.7. Potencial de equilibrio10.8. Sistemas poliredox10.9. Disoluciones reguladoras redox
10.10. Sistemas redox del agua10.11. Representación gráfica del equilibrio
redox10.12. Equilibrios concurrentes: potencial
condicionalCuestionesSeminarios: problemas numéricos
EQUILIBRIOS DE OXIDACIÓN-
REDUCCIÓN

10.1. Introducción
Una reacción de oxidación-reducción, gene-ralmente conocida como reacción redox, es unproceso en el que se produce una transferenciao intercambio de electrones entre las especiesimplicadas en la misma. En todo intercambiosiempre existe una especie dadora y otra acep-tora.
Se denomina reductor (Red) a la especie quecede electrones y oxidante (Ox) a la que los acep-ta. Dado que el reductor se oxida al ceder elec-trones, una reacción de oxidación es aquella enla que la especie pierde electrones: Red ←→ Ox+n e, mientras que en una reacción de reducciónlos gana: Ox + n e ←→ Red.
Un sistema formado por un oxidante y su for-ma reducida se conoce con el nombre de parredox (Ox/Red), que viene caracterizado por elnúmero de electrones (n) intercambiados entreambas especies y por su potencial normal (E 0).Éste permite evaluar el carácter redox del parcomo se mostrará posteriormente. En definitiva,una reacción redox consiste en una transferen-cia de electrones entre las especies implicadas endos pares redox:
Par 1 Ox1/Red1 n1 e Ox1 + n1 e ←→ Red1 [10.1]
Par 2 Ox2/Red2 n2 e Ox2 + n2 e ←→ Red2 [10.2]
Como en una reacción redox el número deelectrones que gana un sistema es el mismo queel que pierde el otro, el número global de elec-trones que intervienen en la reacción será nT =n1 × n2, excepto en los casos en que n1 = n2, yaque en estos el número de electrones involucra-
dos es nT = n1 = n2. Así, la reacción redox vendrádada, tras el correspondiente ajuste electrónico:
n2 Ox1 + n1 Red2←→ n2 Red1 + n1 Ox2 [10.3]
Un ejemplo de este tipo de reacciones es laoxidación de Sn2+ por el Fe3+:
2 Fe3+ + Sn2+ ←→ 2 Fe2+ + Sn4+ [10.4]
constituida por los siguientes pares redox:
Fe3+ + e ←→ Fe2+ (reducción del Fe3+) [10.5]
Sn2+ ←→ Sn4+ + 2 e (oxidación del Sn2+) [10.6]
en la que el Fe3+ actúa como oxidante y el Sn2+
como reductor. El Fe3+ se reduce a Fe2+ mientrasque el Sn2+ se oxida a Sn4+.
Cuando una de las formas del par redox con-tiene oxígeno (MnO4
–/Mn2+) o bien cuando exis-te una diferente proporción de oxígeno entre lasmismas (NO3
–/NO2–), la semireacción redox corres-
pondiente contiene protones y moléculas de aguaen proporción estequiométrica:
+ 8 H+ + 5 e ←→ Mn2+ + 4 H2O [10.7]
+ 2 H+ + 2 e ←→ NO2– + H2O [10.8]
En estos casos se pone de manifiesto la directadependencia de estos sistemas redox con el pH dela disolución. Así, el permanganato será oxidanteen medio ácido, disminuyendo esta capacidad amedida que se incrementa el pH, e igual compor-tamiento se puede aducir para el ion nitrato.
Desde un punto de vista formal, las reaccionesredox presentan un cierto paralelismo con las reac-
NO3−
MnO4−
316 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Descripción de los aspectos fundamentales de las reac-ciones redox: celdas electroquímicas, constante deequilibrio, ecuación de Nernst, tipos de electrodos,potencial estándar, sistemas poliredox y representa-ciones gráficas.
• Estudio de las diversas alternativas de equilibrios con-currentes que se originan al combinar los diferentestipos de equilibrios iónicos considerados en este libro,mediante el uso de potenciales condicionales y coefi-cientes de reacción secundaria.

ciones ácido-base. En efecto, en ambos casos exis-ten dos formas conjugadas (ácido/base y oxidan-te/reductor) entre las que se intercambian unaspartículas (protones en ácido-base y electrones enredox); sin embargo, a pesar de esta similitudambos equilibrios son esencialmente diferentes. Alo largo de este capítulo se establecerán los para-lelismos y divergencias que vayan surgiendo sobreambos equilibrios a medida que se desarrollen losaspectos básicos de los equilibrios redox.
10.2. Estudio de reacciones redox: celdas electroquímicas
El intercambio de electrones entre oxidantey reductor necesario para que tenga lugar la reac-ción redox, se puede llevar a cabo tanto en mediohomogéneo como heterogéneo. En el primercaso, el equilibrio de transferencia electrónica sealcanza directamente mediante la mezcla de dosdisoluciones: una contiene la forma oxidada deun par redox y la otra la forma reducida del otropar. Este proceso se denomina reacción químicaredox. Si tomamos como ejemplo la oxidación deiones Fe2+ por el permanganato de potasio
+ 5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O[10.9]
la reacción química redox se desarrolla, como seaprecia en la figura 10.1 mezclando las disolucio-nes de Fe2+ y MnO4
– en medio ácido. A medidaque se va adicionando Fe2+ se observa la decolo-ración de la disolución violeta de MnO4
–, llegan-do a desaparecer completamente su color cuan-do se añade la cantidad estequiométrica de Fe2+
o bien se supera.La transferencia de electrones también se pue-
de llevar a cabo en un medio heterogéneo, porvía electroquímica, en la que el oxidante y elreductor están físicamente separados uno delotro. El dispositivo experimental denominadocelda electroquímica, consta de dos electrodos,habitualmente de platino, sumergidos en dosvasos (celdas) que contienen las disoluciones de
oxidante y reductor en el medio de reacción ade-cuado, un puente salino que permite la conexióneléctrica y no química entre ambas celdas y final-mente un dispositivo eléctrico (voltímetro o fuen-te de energía externa) que completa el circuitoeléctrico, como se muestra en la figura 10.2.
FIGURA 10.1. Reacción química redox entre los ionese Fe2+.
En las celdas electroquímicas, se denominacátodo al electrodo sumergido en la celda en laque tiene lugar la reacción de reducción y se deno-mina ánodo al electrodo sumergido en la celda enla que se desarrolla el proceso de oxidación. Asípor ejemplo, son reacciones catódicas:
Ag+ + e ←→ Ag0 Ce4+ + e ←→ Ce3+
Fe3+ + e ←→ Fe2+ [10.10]
y reacciones anódicas:
Sn2+ ←→ Sn4+ + 2 e 2 Cl– ←→ Cl2 + 2e Cu0 ←→ Cu2+ + 2 e [10.11]
En la reacción redox que se utiliza como ejem-plo en la figura 10.2, las reacciones correspon-dientes son:
Reacción catódica:
MnO4– + 8 H+ + 5 e ←→ Mn2+ + 4 H2O [10.12]
Reacción anódica:
Fe2+ ←→ Fe3+ + e [10.13]
MnO4−
MnO4−
Capítulo 10: Equilibrios de oxidación-reducción 317
Fe2+
MnO4– Mn2+

FIGURA 10.2. Tipos de celda electroquímica utilizada parael desarrollo de la reacción redox entre el MnO4
– y el Fe2+. a) Celda galvánica y b) Celda electrolítica.
El puente salino es otro elemento de las cel-das electroquímicas. Generalmente consiste enun tubo, en forma de U invertida, relleno de ungel (agar-agar) que contiene KCl o cualquier otroelectrolito fuerte que no participe de manera acti-va en la reacción redox.
El dispositivo eléctrico que completa el cir-cuito puede ser un elemento distinto según el tipode celda electroquímica. Cuando la reacciónredox se produce de manera espontánea, esto es,según su sentido termodinámico, lo que intere-sa es medir la diferencia de potencial que se esta-
blece entre ambos electrodos debida al flujo deelectrones entre el ánodo y el cátodo, el disposi-tivo eléctrico adecuado en este caso es un voltí-metro. La celda electroquímica así constituidarecibe el nombre de celda galvánica, o tambiéncelda o pila voltáica, o electrocelda (figura 10.2a).Sin embargo, si el propósito es manipular la reac-ción redox, e incluso invertir su sentido termo-dinámico, el circuito debe completarse con unafuente externa de energía eléctrica. En estas con-diciones, la celda electroquímica se denominacelda electrolítica (figura 10.2b).
10.2.1. Celdas galvánicas
En las celdas galvánicas se origina inicial-mente una diferencia de potencial, ECelda, que amedida que transcurre la reacción redox vadecreciendo hasta llegar a cero una vez que sealcanza el equilibrio. Esta diferencia de poten-cial viene dada por:
[10.14]
Sin embargo, para que esta ecuación sea váli-da, es necesario respetar el convenio de signos,que fundamentalmente consiste en lo siguiente:
1. Las semirreacciones se escriben siemprecomo reducciones, independientementedel sentido que tengan en el electrodo.
2. El signo del potencial indica en qué senti-do transcurre un proceso. Un signo positi-vo significa que éste está desplazado haciala derecha, mientras que un signo negativosignifica que lo está hacia la izquierda.
Por ejemplo, para una celda formada por unelectrodo con una mezcla de Fe3+ y Fe2+ y el otrocon una mezcla de Sn4+ y Sn2+, en condiciones están-dar en todos los casos, se cumple lo siguiente:
Proceso catódico: Fe3+ + e ←→ Fe2+
[10.15]EFe /Fe0
3 2 0,771 V+ + =
E E ECelda Cátodo Ánodo= −
318 Equilibrios iónicos y sus aplicaciones analíticas
a)Voltímetro
Fe3+ Fe2+ Mn2+ MnO4–
Puente salino
Fuente de potencial
Fe3+ Fe2+ Mn2+ MnO4–
Puente salino
ÁNODO CÁTODO
CÁTODO ÁNODO
b)

Proceso anódico:
Sn4+ + 2 e ←→ Sn2+
[10.16]
Reacción global:
2 Fe3+ + Sn2+ ←→ 2 Fe2+ + Sn4+ [10.4]
De acuerdo con la ecuación [10.14] el poten-cial de la celda vendrá dado por:
ECelda + 0,771 – 0,154 = 0,617 V [10.17]
lo que pone de manifiesto que la reacción estádesplazada hacia la derecha, es decir, que el Fe3+
es capaz de oxidar el Sn2+.En este tipo de celdas, el cátodo (electrodo
sumergido en la disolución que contiene el oxi-dante) se considera el polo positivo de la misma,mientras que el ánodo corresponde al polo nega-tivo. Desde su introducción por Alessandro Vol-ta, han tenido una amplia repercusión desde elpunto de vista práctico, dado que abrieron elcamino para el aprovechamiento de la energíaeléctrica (ver recuadro 10.1).
ESn /Sn0
4 2 ,154 V+ + = 0
Capítulo 10: Equilibrios de oxidación-reducción 319
RECUADRO 10.1
Las celdas galvánicas, también denominadas pi-las eléctricas, han experimentado un importante desarro-llo desde su propuesta por Volta sobre el año 1800. Ini-cialmente se emplearon las conocidas pila voltaica y pilade gravedad de Daniell, basadas ambas en la reacciónredox:
Cu2+ + Zn0 ←→ Cu0 + Zn2+
en la que se utiliza un cátodo de cobre y un ánodo de cinc, aunque con diferente diseño experimental. Estaspilas conocidas también como pilas primarias experi-mentaron un importante auge gracias a la pila Leclanchéo pila seca, desarrollada por el químico francés GeorgesLeclanché (MnO2 + NH4
+ + e ←→ MnO(OH) + NH3, Zn0
←→ Zn2+ + 2 e).
En la actualidad, con un diseño similar al original, seutiliza profusamente la pila de cinc-óxido de mercurio. Estapila está formada por un ánodo de cinc:
Zn2+ + 2 e ←→ Zn(Hg)
y un cátodo de óxido de mercurio:
HgO(S) + H2O + 2 e ←→ Hg0 + 2OH–
actuando como electrolito una disolución de hidróxido depotasio, dada la influencia de los iones OH– sobre este parredox. De ahí que también se conozcan estas pilas comopilas alcalinas. Esta pila origina un potencial de unos 1,34V y puede tener forma de disco pequeño utilizándose enrelojes eléctricos, audífonos, etc.
En la reacción redox que se ha tomado comoejemplo, mediante la celda galvánica que se muestra en la figura 10.2a, se consigue llevara cabo la reacción entre el MnO–
4 y el Fe2+, ya que los electrones necesarios para que elMnO–
4 se reduzca a Mn2+ se los proporciona elFe2+ (que se oxida a Fe3+) a través de los elec-trodos.
Es común utilizar una notación simplificadapara describir este tipo de celdas, en la que sesitúa siempre el cátodo a la derecha, el puentesalino se identifica con dos trazos verticales,
mientras que la interfase electrodo/disoluciónse simboliza por un solo trazo vertical. Confor-me a esta notación, una celda galvánica se repre-senta por:
Electrodo Disolución Disolución Electrodo(Ánodo) Reductor Oxidante (Cátodo)
y en el caso de la pila de Volta por:
Zn | ZnSO4 (mol L–1) || CuSO4 (mol L–1) | Cu

Un tipo interesante de celda galvánica sonlas denominadas pilas de concentración, en lasque la diferencia de potencial entre comparti-mentos no se origina por una reacción entre distintos pares redox, sino que obedece a la dife-rencia de concentración de uno de los compo-nentes de un mismo par redox introducido encada una de las celdas. Estas pilas pueden ser degran interés, ya que permiten evaluar la influen-cia de otros equilibrios (ácido-base, precipita-ción y complejación) sobre el propio equilibrioredox.
10.2.2. Celdas electrolíticas
Como ya se ha indicado, en las celdas gal-vánicas la reacción química redox se producede manera espontánea, sin embargo, si paracerrar el circuito eléctrico se hace uso de unafuente de energía, esto es, una fuente de po-tencial en sentido opuesto al paso de corrienteoriginado por la celda galvánica, se puede ma-nipular el sentido de la reacción redox consi-derando que ésta sea reversible. En estas con-diciones la celda galvánica se ha transformadoen una celda electrolítica. Conforme a la mag-nitud relativa del potencial impuesto por lafuente de energía frente al suministrado por
la celda galvánica, se pueden originar tres situa-ciones:
1. Si el potencial impuesto es inferior, la reac-ción química redox no cambia de sentidoaunque se alcanza un estado de equilibriodiferente.
2. Si ambos potenciales son iguales la reac-ción redox no progresa.
3. Si el potencial de la fuente es superior, se in-vierte el sentido de la reacción química redox.
En este tipo de celdas, el cátodo y ánodo tie-nen el mismo significado químico que en las cel-das galvánicas. El cátodo (se produce la reduc-ción) tiene signo negativo al estar conectado alpolo de igual signo de la fuente, mientras queal ánodo (se lleva a cabo la oxidación) se le asig-na el signo positivo. Como se muestra en la figu-ra 10.2b cuando la reacción redox entre el MnO–
4y el Fe2+ se lleva a cabo en la celda electrolíticaes posible conseguir en el cátodo la reduccióndel Fe3+ a Fe2+, mientras que en el ánodo el Mn2+
se oxida a MnO–4. Estas celdas presentan un gran
interés para el estudio del comportamiento elec-troquímico de diferentes especies y compuestosy desde un punto de vista práctico constituyenla base de las baterías o acumuladores, como sedescribe en el recuadro 10.2.
320 Equilibrios iónicos y sus aplicaciones analíticas
RECUADRO 10.2
Una batería o acumulador, también denominada pilasecundaria, es un tipo de celda electrolítica desarrollada en1859 por el físico francés Gaston Planté. Esta batería, quecontiene de tres a seis pilas conectadas en serie, se usa enautomóviles, aviones y en otros vehículos. Está constituidapor un ánodo de placas de plomo
PbSO4 (S) + 2 e ←→ Pb0 + SO2–4
y un cátodo de dióxido de plomo
PbO2 (S) + 4 H+ + SO2–4 + 2 e ←→ PbSO4 (S) + 2 H2O
siendo el electrólito una disolución diluida de ácido sulfúri-co. Cuando suministra energía (celda galvánica), el dióxi-do de plomo es capaz de oxidar al plomo para produciriones Pb2+, que precipitan como sulfato debido a la pre-sencia de ácido sulfúrico, reduciéndose también a Pb2+, pro-duciendo nuevamente sulfato de plomo.
Esta batería de plomo y ácido, se agota porque el ácidosulfúrico se transforma gradualmente en agua y en sulfato deplomo. Al recargar la batería (celda electrolítica), las reac-ciones químicas descritas anteriormente revierten hasta su posi-ción original. Esta batería, que origina unos 2 V por elemen-to, tiene una vida útil de unos cuatro años.

10.3. Potencial y carácter químicoredox: ecuación de Nernst
Como ya se ha indicado, una reacción redoximplica una transferencia de electrones entre dosespecies; por tanto, cuanto mayor sea la tenden-cia de una especie a captar electrones mayor serásu carácter oxidante, y cuanto mayor sea su capa-cidad para cederlos mayor será su carácter reduc-tor. Este enfoque puramente cualitativo requie-re de una visión cuantitativa que permitaestablecer una relación entre la fuerza oxidante(o reductora) de una especie (su carácter quími-co) y una propiedad física susceptible de medi-da, el potencial, que esté directamente relacio-nado con la corriente eléctrica generada por estatransferencia (desplazamiento) de electrones. Endefinitiva, el potencial redox es una propiedadfísica (expresada en voltios) que permite evaluarel carácter químico redox de una sustancia.
Para establecer esta relación, se hace uso dela expresión propuesta por Walther HermannNernst (físico-químico alemán que contribuyóconsiderablemente al desarrollo de la Electro-química) que relaciona el potencial, E, con el flu-jo de electrones, ed, puesto en juego en la diso-lución:
[10.18]
siendo R la constante de los gases (8,314 J K–1
mol–1), T la temperatura en grados Kelvin y F elFaraday (96.485 culombios). A 25 °C está ecua-ción se transforma en:
[10.19]
Hay que indicar que aunque ed tiene un signi-ficado físico de concentración de electrones, care-ce de sentido práctico dado que los electrones notienen existencia libre en disolución. Sin embar-go, es un apoyo didáctico interesante para dedu-cir “teóricamente” la relación que nos ocupa.
De manera general, para el par redox a Ox +n e ←→ b Red, se puede establecer la siguiente“constante de equilibrio”:
[10.20]
Al tomar logaritmos en esta expresión y rea-lizar las transformaciones oportunas se llega a laexpresión:
[10.21]
y al sustituir la misma en la ecuación [10.19] setiene:
[10.22]
que finalmente se transforma en la conocidaecuación de Nernst para una semireacción o parredox:
[10.23]
al designar al primer término de la ecuación[10.22] como E0, constante característica del parredox conocida como potencial normal o están-dar y considerar que en las disoluciones relati-vamente diluidas habitualmente utilizadas enQuímica Analítica puede aceptarse que la acti-vidad es aproximadamente igual a la concentra-ción.
A la vista de la ecuación [10.23] se puedededucir que el potencial de una disolución quecontiene un par redox depende directamente dela naturaleza del propio par, reflejada en el valorde E0, y de la temperatura (la expresión se hadeducido para 25 °C). Por otra parte, este poten-cial aumenta a medida que lo hace la concentra-ción de la forma oxidada y disminuye a medidaque se incrementa la concentración de la formareducida.
En este punto es interesante realizar algunasmatizaciones sobre el empleo de esta expresiónde Nernst, cuando la naturaleza del par redox es
E En
a
b= +0 0 059,
log[ ][ ]
OxRed
En
kn
a
a
a
b= +0 059 0 059,
log,
logparOx
Red
log log 1 1e n
ka
adpar
a
b=
Ox
Red
k
a
a epar
b
adn
= Red
Ox
Eed
= 0 0591
, log
ERTF ed
= ln1
Capítulo 10: Equilibrios de oxidación-reducción 321

diferente a la que se ha considerado para sudeducción.
1. Así, cuando una de las formas del par redoxcontiene oxígeno o bien cuando existe unadiferente proporción de oxígeno entre lasmismas, la semireacción redox, como ya seha indicado, está influenciada por la con-centración de protones y el potencial depen-de del pH. Este es el caso de la reduccióndel permanganato a Mn2+ (ecuación [10.7])cuyo potencial redox dado por:
[10.24]
muestra claramente la influencia del pHsobre el mismo. Un aumento de pH origi-na una disminución del potencial redox yconsecuentemente de la capacidad oxi-dante del permanganato.
2. En otros casos, una de las especies del parredox puede no estar presente en la diso-lución, esto es, se puede encontrar en esta-do gaseoso o bien en estado sólido. Comoejemplo del primer caso se puede consi-derar la reducción de los iones H+ a hidró-geno conforme a la semireacción redox:
2 H+ + 2 e ←→ H2 (g) [10.25]
En este sistema la concentración dehidrógeno se expresa en términos de su pre-sión parcial (pH2
) y dado que generalmentese trabaja a una presión de 1 atmósfera, laexpresión del potencial se transforma en:
[10.26]
Se observa, a partir de esta expresión,cómo en medio ácido fuerte (valores bajosde pH) se incrementa el poder oxidantedel H+, llegando incluso a reaccionar conreductores fuertes originando desprendi-miento de hidrógeno.
3. Cuando una de las formas del par redox seencuentra en estado sólido, como porejemplo en el sistema Ag+/Ag0, al consi-derarse por convenio que la actividad deun sólido es la unidad, la expresión delNernst queda como sigue:
[10.27]
donde el potencial del sistema es directa-mente proporcional a la concentración deAg+ en disolución.
10.4. Medida del potencial redox: electrodos de referencia
Hasta ahora se ha utilizado la expresión deNernst para el cálculo del potencial de una semi-reacción o par redox, en función de las concen-traciones de las especies implicadas en el mismo.Sin embargo, no es posible medir el valor abso-luto de un potencial, sino únicamente su dife-rencia respecto a otro, por lo que, formalmente,lo más correcto es utilizar el término “diferenciade potencial”. Esta circunstancia obliga a esta-blecer un estado o nivel de referencia. Si a esteestado de referencia se le asigna, por convenio,un valor cero, las diferencias de potencial medi-das experimentalmente serán idénticas a las quecabría esperar según la ecuación de Nernst.
La medida de la diferencia de potencial entredos pares redox se puede llevar a cabo fácilmentecon una celda galvánica, y si en una de las semi-celdas se sitúa el par que se utiliza como refe-rencia (un electrodo estándar de hidrógeno, alque por convenio se asigna un E0 = 0) y en la otraaquel cuyo potencial se desea determinar, la lec-tura del voltímetro proporciona directamente elpotencial del par redox objeto de estudio.
E E= + +0 0 059, log[ ]Ag
E Ep
E E
= +=
=
= + = −
+
+
0
0 0
0 0592
0 059 0 059
,log
, log[ ,
[H ]1
H ] pH
2
H2
E E
E
= + =
= + −
− +
+
−
+
0
0
0 0595
0 0595
0 0595
8
,log
,
log,
[MnO ][H ][Mn ]
[MnO ][Mn ]
pH
48
2
42
322 Equilibrios iónicos y sus aplicaciones analíticas

Es preciso, por tanto, disponer de una semi-celda de referencia, denominada electrodo dereferencia, que presente las siguientes caracte-rísticas: que sea aceptada universalmente, fácilde construir, reversible, que cumpla la ecuaciónde Nernst, que sea reproducible y que propor-cione un potencial constante en unas determi-nadas condiciones.
10.4.1. Electrodo estándar de hidrógeno
Este electrodo cumple los requisitos indica-dos para ser utilizado como electrodo de refe-rencia y así ha sido usado durante muchos años.Está constituido por un conductor de platino,recubierto con negro de platino finamente divi-dido (para incrementar su superficie específicay asegurar así la reversibilidad de la semireac-ción redox), sumergido en una disolución acuo-sa ácida en la que la actividad del ion hidróge-no se mantiene constante. La disolución semantiene saturada de hidrógeno, burbujeandoel gas a presión constante sobre la superficie delelectrodo.
El potencial, de acuerdo con la ecuación deNernst, para la semireacción redox en la que sefundamenta este electrodo, ecuación [10.25] , vie-ne dado a 25 °C por:
[10.28]
y depende de la temperatura, de la actividad delos iones H+ y de la presión parcial del hidrógeno.Para que este electrodo pueda actuar como elec-trodo de referencia, es necesario trabajar en unascondiciones estándar tales como: actividad unidadpara los iones H+ y presión parcial del hidrógeno1 atmósfera. En estas condiciones E = E0, y si porconvenio al potencial normal, E0, de este par redoxse le asigna el valor cero a 25 °C, el potencial delelectrodo estándar de hidrógeno tomado comoreferencia es también cero.
Una vez establecida la referencia, el poten-cial de la disolución de un par redox se determi-
na fácilmente haciendo uso de la siguiente celdagalvánica:
Pt pH2= 1 atm | aH+ = 1 || [Ox]/[Red] | Pt
La reversibilidad que presenta este electrodode referencia, que puede actuar como ánodo (elhidrógeno se oxida a iones H+) o como cátodo(los iones H+ se reducen a hidrógeno), permitela medida del potencial del par redox con el quese acopla para formar la celda galvánica.
Uno de los principales inconvenientes de esteelectrodo, es su difícil utilización en laboratoriosde control o de rutina, debido a la peligrosidadque entraña el empleo de gas H2. No es extraño,por tanto, que se hayan diseñado y propuestootros electrodos de referencia, tal como el elec-trodo de calomelanos saturado, consistente enmercurio en contacto con una disolución satura-da de Hg2Cl2 (calomelanos), en presencia de unadisolución saturada de KCl. A pesar de sus bue-nas prestaciones, se ha dejado de comercializardebido a la elevada toxicidad que presenta elmercurio.
10.4.2. Electrodo de Ag/AgCl
En la actualidad, es éste el electrodo de refe-rencia más utilizado, consistente en un hilo deplata sumergido en una disolución de cloruro depotasio saturada de cloruro de plata. El poten-cial de este electrodo se fundamenta en la semi-reacción redox:
AgCl (S) + e ←→ Ag0 + Cl– [10.29]
y depende de la actividad del ion cloruro segúnla expresión:
[10.30]
cuya deducción se llevará a cabo posteriormen-te cuando se aborde la influencia de los equili-brios de precipitación sobre los equilibrios redox.
E E K aS= + − −
0 0 059 0 059, log , log(AgCl) Cl
E E
a
p= +0
20 0592
,log H
H
+
2
Capítulo 10: Equilibrios de oxidación-reducción 323

Para que este electrodo presente un potencialconstante y pueda ser considerado como electro-do de referencia se debe fijar la concentración(actividad) del ion cloruro. Es práctica comúnfijar la concentración de KCl en 3,5 mol L–1 o bienutilizar una disolución saturada (∼ 4,6 mol L–1) enesta sal. En estas condiciones y a 25 °C, el poten-cial de este electrodo, determinado frente al elec-trodo normal de hidrógeno, es de 0,205 y 0,199 Vpara una concentración de cloruro de potasio de3,5 mol L–1 y saturada, respectivamente.
El diseño comercial de este electrodo constade un tubo interno que contiene un hilo de pla-ta recubierto de una capa de cloruro de plata,sumergido en una disolución de cloruro de pota-sio que está saturada con cloruro de plata. Estetubo se pone en contacto con la disolución de unelectrolito (cloruro de potasio de la misma con-centración), mediante un pequeño orificio en suparte inferior y finalmente con la disolución pro-blema, a través de un pequeño disco de vidrioporoso (fritado). Este diseño incorpora el pro-pio puente salino y así permite el uso directo deeste electrodo por simple inmersión en la diso-lución del par redox objeto de estudio.
10.5. Potencial estándar
El potencial estándar o potencial normal, E 0,es una constante física característica de cada parredox. De acuerdo con la ecuación de Nernst:
[10.31]
el potencial redox o potencial de electrodo coin-cide con E0 si el término logarítmico es igual acero, lo que ocurre en los casos siguientes:
1. Si la forma oxidada y la forma reducida sonsolubles, cuando el cociente entre ambas esigual a la unidad. (En sentido estricto, las
condiciones estándar corresponden a unaactividad igual a la unidad.)
2. Si la forma oxidada o la forma reducidason insolubles, cuando la actividad de laforma soluble es igual a la unidad.
3. Si la forma oxidada o reducida son gaseo-sas, cuando su presión es igual a 1 atm.
4. Si en la semirreacción intervienen pro-tones, cuando su actividad es igual a 1.
Este potencial se puede determinar fácilmentehaciendo uso de una celda galvánica, en la quese utilice como electrodo de referencia el elec-trodo normal de hidrógeno, situando en el otrocompartimento los componentes del par redoxcuyo E 0 se desea calcular:
Pt pH2 = 1 atm | [H+] = 1 M || [Ox] / [Red] | Pt
En función de las características del par redoxestudiado, el electrodo de hidrógeno (que hacede referencia) podría actuar como ánodo o comocátodo.
Conforme al proceso de medida llevado acabo, el potencial estándar obtenido correspon-de a la semireacción de reducción indicada. Rea-lizando esta operación para los diferentes paresredox existentes se obtienen los correspondien-tes valores de E0, cuya ordenación constituye ladenominada serie electroquímica. En el cuadro10.1 se muestra de forma resumida esta serie,conforme a un orden decreciente en los valoresde los potenciales normales.
A partir de la observación de este cuadro, obien de la propia serie electroquímica, se puedenextraer las siguientes conclusiones:
1. Consta de dos partes separadas por lasemirreacción redox que se ha tomadocomo referencia y cuyo E0 = 0 [2 H+ + 2 e←→ H2(g)].
2. La forma oxidada de cualquier par redoxserá capaz de oxidar a la forma reducidade un par cuyo E0 sea menor (o, dicho deotra manera, la forma reducida de cual-
E En
En
E
a
b= +
+ =
0
0 0
0 059
0 0591 0
,log
[ ][ ]
,log( , )
OxRed
=
=
324 Equilibrios iónicos y sus aplicaciones analíticas
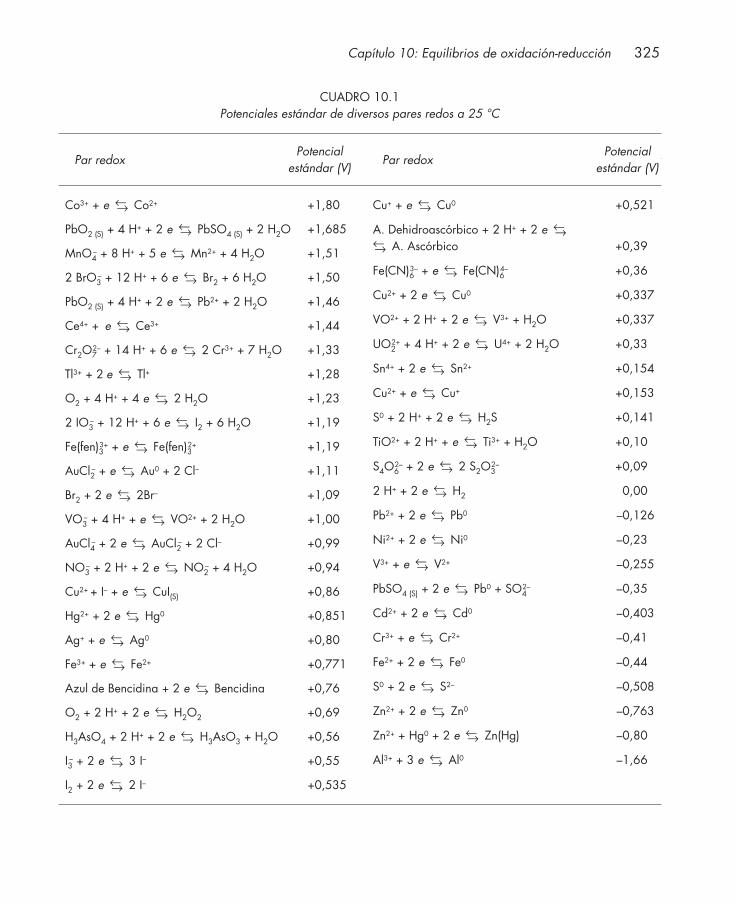
Capítulo 10: Equilibrios de oxidación-reducción 325
Co3+ + e ←→ Co2+ +1,80
PbO2 (S) + 4 H+ + 2 e ←→ PbSO4 (S) + 2 H2O +1,685
MnO4– + 8 H+ + 5 e ←→ Mn2+ + 4 H2O +1,51
2 BrO3– + 12 H+ + 6 e ←→ Br2 + 6 H2O +1,50
PbO2 (S) + 4 H+ + 2 e ←→ Pb2+ + 2 H2O +1,46
Ce4+ + e ←→ Ce3+ +1,44
Cr2O72– + 14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O +1,33
Tl3+ + 2 e ←→ Tl+ +1,28
O2 + 4 H+ + 4 e ←→ 2 H2O +1,23
2 IO3– + 12 H+ + 6 e ←→ I2 + 6 H2O +1,19
Fe(fen)33+ + e ←→ Fe(fen)32+ +1,19
AuCl2– + e ←→ Au0 + 2 Cl– +1,11
Br2 + 2 e ←→ 2Br– +1,09
VO3– + 4 H+ + e ←→ VO2+ + 2 H2O +1,00
AuCl4– + 2 e ←→ AuCl2– + 2 Cl– +0,99
NO3– + 2 H+ + 2 e ←→ NO2
– + 4 H2O +0,94
Cu2+ + I– + e ←→ CuI(S) +0,86
Hg2+ + 2 e ←→ Hg0 +0,851
Ag+ + e ←→ Ag0 +0,80
Fe3+ + e ←→ Fe2+ +0,771
Azul de Bencidina + 2 e ←→ Bencidina +0,76
O2 + 2 H+ + 2 e ←→ H2O2 +0,69
H3AsO4 + 2 H+ + 2 e ←→ H3AsO3 + H2O +0,56
I3– + 2 e ←→ 3 I– +0,55
I2 + 2 e ←→ 2 I– +0,535
Cu+ + e ←→ Cu0 +0,521
A. Dehidroascórbico + 2 H+ + 2 e ←→←→ A. Ascórbico +0,39
Fe(CN)63– + e ←→ Fe(CN)64– +0,36
Cu2+ + 2 e ←→ Cu0 +0,337
VO2+ + 2 H+ + 2 e ←→ V3+ + H2O +0,337
UO22+ + 4 H+ + 2 e ←→ U4+ + 2 H2O +0,33
Sn4+ + 2 e ←→ Sn2+ +0,154
Cu2+ + e ←→ Cu+ +0,153
S0 + 2 H+ + 2 e ←→ H2S +0,141
TiO2+ + 2 H+ + e ←→ Ti3+ + H2O +0,10
S4O62– + 2 e ←→ 2 S2O3
2– +0,09
2 H+ + 2 e ←→ H2 0,00
Pb2+ + 2 e ←→ Pb0 –0,126
Ni2+ + 2 e ←→ Ni0 –0,23
V3+ + e ←→ V2+ –0,255
PbSO4 (S) + 2 e ←→ Pb0 + SO42– –0,35
Cd2+ + 2 e ←→ Cd0 –0,403
Cr3+ + e ←→ Cr2+ –0,41
Fe2+ + 2 e ←→ Fe0 –0,44
S0 + 2 e ←→ S2– –0,508
Zn2+ + 2 e ←→ Zn0 –0,763
Zn2+ + Hg0 + 2 e ←→ Zn(Hg) –0,80
Al3+ + 3 e ←→ Al0 –1,66
CUADRO 10.1Potenciales estándar de diversos pares redos a 25 °C
Par redox Potencial
estándar (V) Par redox
Potencial estándar (V)

quier par será capaz de reducir a la formaoxidada de otro cuyo E0 sea mayor). Porejemplo, las reacciones:
Cr2O72– + Fe2+ + 14 H+ ←→
←→ 2 Cr3+ + Fe3+ + 7 H2O [10.32]
Cd2+ + Zn0 ←→ Cd0 + Zn2+ [10.33]
son espontáneas y en las correspondientesceldas galvánicas originarían unos poten-ciales de +0,559V (1,33 – 0,771= 0,599) yde 0,36 V (–0,403 – (–0,763) = 0,36), res-pectivamente.
3. Un caso particular, de gran importanciapráctica, es el de la reacción con el siste-ma de referencia. Así, las formas oxida-das de los pares redox con E0 > 0 soncapaces de oxidar el H2 a ion H+, mientrasque las formas reducidas de los paresredox con E0 < 0 reducen los iones H+ aH2. Por ejemplo, tanto la reacción entreel Cu2+ (E0 = 0,337 V) y el hidrógeno
Cu2+ + H2←→ Cu0 + 2 H+ [10.34]
como la reacción entre Zn e iones H+
(E0 = –0,763 V)
Zn0 + 2 H+ ←→ Zn2+ + H2 [10.35]
son espontáneas, ya que el potencial de cel-da es positivo en ambos casos (+0,337 y+0,763 V, respectivamente), aunque el elec-trodo de hidrógeno actúa como ánodo en elprimero y como cátodo en el segundo.
Este diferente comportamiento frenteal ion H+ explica el hecho experimental deque algunos metales (los que tienen E0 < 0)se disuelvan fácilmente en ácidos no oxi-dantes, mientras que otros (los que tienenE0 > 0) sean insolubles en ellos y requieranla acción de ácidos oxidantes para su diso-lución.
10.6. Constante de equilibrio de una reacción redox
De lo expuesto en el apartado anterior sedesprende que el potencial normal, caracterís-tico de cada par redox, permite evaluar la fuer-za relativa como oxidante o como reductor delas diferentes especies implicadas en una reac-ción redox, lo que, en principio, hace posiblepredecir el sentido de la misma. Así, de mane-ra general, para la reacción expresada por laecuación [10.3]:
n2 Ox1 + n1 Red2←→ n2 Red1 + n1 Ox2 [10.3]
en la que están implicados los pares redoxOx1/Red1 (n1, E1
0) y Ox2/Red2 (n2, E20), el sentido
dependerá de los valores de los potenciales nor-males (E1
0 y E20) y de las concentraciones de las
especies implicadas. Esta visión cualitativa delequilibrio redox debe concretarse con un enfo-que cuantitativo basándose en el empleo de cons-tantes de equilibrio, de manera similar a loexpuesto en los otros equilibrios estudiados encapítulos precedentes.
Para la reacción indicada la constante de equi-librio viene dada por:
[10.36]
Cuando se alcanza el equilibrio en una reac-ción redox, el potencial del sistema es único y sedenomina potencial de equilibrio (Eeq). Estepotencial se puede calcular haciendo uso de cual-quiera de las ecuaciones de Nernst correspon-dientes a los pares redox implicados en la reac-ción:
[10.37]
E E E En
En
eq
OxRed
OxRed
= = = + =
= +
1 2 10
1
1
1
20
2
2
2
0 059
0 059
,log
[ ][ ]
,
log[ ][ ]
Kn n
n n= [ ] [ ]
[ ] [ ]
Red Ox
Ox Red1 2
1 2
2 1
2 1
326 Equilibrios iónicos y sus aplicaciones analíticas

Si se multiplica y divide el término 0,059/n1por n2 y se realiza una operación similar para eltérmino 0,059/n2 aunque en este caso con n1 setiene:
[10.38]
y operando
[10.39]
de donde finalmente se tiene la expresión:
[10.40]
en la que E10 corresponde al potencial normal del
par redox, que actúa como oxidante, y E20 al del
par que actúa como reductor. Como ya se habíacomentado en el apartado 10.1, el término n1 ×n2 indica el número total de electrones implica-dos en la reacción, nT; si n1 = n2 es evidente quenT = n1 = n2, mientras que si n1 ≠ n2 se puedecomprobar que nT corresponde al mínimo comúnmúltiplo, en general, nT = n1 × n2.
La ecuación [10.40] permite establecer unarelación cuantitativa entre el valor de la cons-
tante de equilibrio, los potenciales normales delos pares redox implicados en la reacción y elnúmero de electrones puestos en juego en cadauna de las semireacciones redox. Cuanto mayordiferencia exista entre los potenciales normalesy mayor número de electrones se intercambienentre las especies implicadas en la misma, mayorserá la constante de equilibrio y, por lo tanto, másdesplazada estará la reacción hacia la derecha.En el recuadro 10.3 se muestra a modo de ejem-plo el cálculo de la constante de equilibrio parala batería de plomo y ácido comentada anterior-mente.
La constante de equilibrio es el parámetroque indica en qué extensión y sentido transcurreuna reacción. Sin embargo, a diferencia de losrestantes tipos de equilibrio iónico, los datos quehabitualmente se encuentran tabulados en oxi-dación-reducción no son las constantes de equi-librio, sino los potenciales de los pares redoximplicados en la reacción. Además, en QuímicaAnalítica surge otro problema: por lo generalinteresa conocer no sólo el sentido de la reac-ción sino también si es cuantitativa o no, es decir,si transcurre en una extensión igual o superioral 99,9%. Puede, por tanto, ser útil conocer ladiferencia de potencial mínima, que debería exis-tir entre los potenciales de los pares redox queintervienen en una reacción, para garantizar queésta sea cuantitativa.
Si se considera en primer lugar el caso mássencillo posible, en el que la estequiometría de
log
,K
E En n= −1
020
1 20 059
E En n
n n
n n10
20
1 21 2
1 20 059
2 1
2 1
− =,
log[ ] [ ]
] [ ]
Red Ox
[Ox Red
En n
En n
n
n
n
n
10
1 2
1
1
20
2 1
2
2
0 059
0 059
2
2
1
1
+ =
= +
,log
[ ]
[ ]
,log
[ ]
[ ]
Ox
Red
Ox
Red
Capítulo 10: Equilibrios de oxidación-reducción 327
RECUADRO 10.3
La batería o acumulador de plomo y ácido se basa enlos siguientes pares redox
PbO2 (S) + 4 H+ + 2 e ←→ PbSO4 (S) + 2 H2O (E10 = 1,685 V)
PbSO4 (S) + 2 e ←→ Pb0 + SO42– (E2
0 = –0,35 V)
dando lugar a la reacción redox:
PbO2 (S) + Pb0 + 4 H+ + 2 SO42– ←→ 2 PbSO4 (S) + 2 H2O
cuando actúa como fuente de energía. Conforme a la ecua-ción [10.40], su constante de equilibrio viene dada por:
de donde K ≈ 1069. Este elevado valor de la constante deequilibrio pone de manifiesto claramente el gran despla-zamiento hacia la derecha que se observa en esta reacciónredox, y por tanto su empleo como batería.
log,
, ( , ),
,K E E n= − = − − =10
20
0 0591 685 0 35
0 0592 68 98

328 Equilibrios iónicos y sus aplicaciones analíticas
la reacción es 1:1 entre los dos pares redoxOx1/Red1 y Ox2/Red2, siendo las concentracio-nes totales C1 y C2, respectivamente, la reacciónredox:
Ox1 + Red2←→ Red1 + Ox2 [10.41]
será cuantitativa cuando se cumpla que:
[10.42]
[10.43]
y dado que la correspondiente constante de equi-librio se puede expresar como:
[10.44]
es evidente que para que la reacción sea cuanti-tativa debe cumplirse que:
[10.45]
Introduciendo esta condición en la ecuación[10.40], que relaciona el valor de la constante deequilibrio con los potenciales de los pares redoximplicados en la reacción y el número total deelectrones:
[10.40]
se puede calcular la diferencia de potencial míni-ma que debe existir entre los potenciales de lospares redox que intervienen en la reacción paraque ésta sea cuantitativa. En el cuadro 10.2 semuestran los valores obtenidos para distintasreacciones en función de su estequiometría ynúmero total de electrones.
CUADRO 10.2Diferencia de potencial mínima necesaria para
garantizar que una reacción redox sea cuantitativa
Estequiometría Kmin nT E10 – E2
0
1:1 106 1 + 0,36 V 1:1 106 2 + 0,18 V 1:1 106 3 + 0,12 V 2:1 109 2 + 0,27 V 3:2 1015 6 + 0,15 V 5:1 1018 5 + 0,21 V 5:2 1021 10 + 0,13 V
A la vista de estos resultados, es fácil llegar alas conclusiones siguientes:
1. La diferencia de potencial que, como míni-mo, debe existir entre el par que se reducey el que se oxida depende de la estequio-metría de la reacción y del número de elec-trones que intervienen en ella, aunque, parauna estequiometría concreta, el potencialnecesario es tanto menor cuanto mayor esel número de electrones implicados.
2. En el caso más desfavorable (estequime-tría 1:1 y nT = 1), basta una diferencia de0,36 V entre los potenciales de los paresredox implicados en la reacción paragarantizar la cuantitatividad. En no pocasocasiones, una diferencia inferior a 0,3 Ves suficiente.
10.7. Potencial de equilibrio
Un parámetro analítico que frecuentemente esimportante conocer es el potencial de una disolu-ción que contiene dos pares redox. Cabe distinguirdos posibilidades: a) uno de los compuestos seencuentra en exceso, y b) los compuestos seencuentran en proporciones estequiométricas.Ambas situaciones son de gran interés en la cons-trucción de las curvas de valoración redox, comose mostrará en el capítulo siguiente.
log
,K
E En n= −1
020
1 20 059
KC C
C C≥
×
×≥
99 9100
99 9100
0 1100
0 1100
101 2
1 2
6
, ,
, ,
K = [ ] [ ][ ] [ ]Red OxOx Red
1 2
1 2
[ ],
Red2 2
0 1100
≥ C[ ],
Ox2 2
99 9100
≥ C
[ ],
Ox1 1
0 1100
≥ C[ ],
Red1 1
99 9100
≥ C

Uno de los compuestos se encuentra en exce-so. En este caso resulta fácil determinar las con-centraciones de su forma oxidada y reducida ycalcular el potencial mediante la correspondienteecuación de Nernst. Por ejemplo, si se mezclan50 mL de una disolución que contiene 0,2 molL–1 de MnO4
– (E10 = +1,51 V) con 50 mL de otra
que contiene 0,2 mol L–1 de Fe2+ (E20 = + 0,771 V)
a pH = 0, es evidente que la reacción
MnO4– + 5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O
[10.9]
está totalmente desplazada hacia la derecha(Ecelda = +1,51 – 0,771 = +0,739 V) y, por tanto,es posible efectuar las siguientes hipótesis(teniendo en cuenta la variación de volumen):
— [Fe3+] = 0,1 mol L–1.— [Fe2+] desconocida, pero despreciable fren-
te a [Fe3+].
— 0,02 mol L–1.
— 0,08 mol L–1.
Al sustituir en la ecuación de Nernst corres-pondiente al par :
[10.24]
se obtiene el valor del potencial de equilibrio,que en este caso al ser a pH = 0, toma el valor de+ 1,514 V.
Los compuestos se encuentran en proporcio-nes estequiométricas. En este caso no es posiblededucir las concentraciones en el equilibrio delas formas oxidada y reducida de ninguno de losdos pares redox implicados en la reacción, lo queimpide utilizar la ecuación de Nernst correspon-diente a uno de ellos.
Por ejemplo, si se mezclan 50 mL de una diso-lución que contiene 0,04 mol L–1 de MnO4
–
(E10 = +1,51 V) con 50 mL de otra que contiene
0,1 mol L–1 de Sn2+ (E20 = +0,154 V) a pH = 0, es
evidente que, en función de la estequiometría dela reacción:
2 MnO4– + 5 Sn2+ + 16 H+ ←→ 2 Mn2+ +
+ 5 Sn4+ + 8 H2O [10.45]
es posible llegar a las siguientes conclusiones:
— [Sn4+] = 0,05 mol L–1.— [Sn2+] desconocida, pero despreciable fren-
te a [Sn4+].
— 0,02 mol L–1.
— desconocida, pero despreciablefrente a [Mn2+].
Sin embargo, la estequiometría de la reacciónpermite llegar a otras relaciones útiles. Así, secumple que:
[10.42]
pero, además y única y solamente en el caso deque existan proporciones estequiométricas deambos compuestos, se cumplirá también que:
[10.43]
Por otra parte, en el equilibrio, el potencialde ambos pares redox será idéntico, por lo que
[10.24]
y asimismo:
[10.48]
Si se multiplica la ecuación [10.24] por 2 y la[10.48] por 5, de manera que se eliminen losdenominadores, y seguidamente se suman, seobtiene:
E Eeq
4
2
[Sn ][Sn ]
= ++
+20 0 059
2,
log
E Eeq4
8
2
[MnO ][H ][Mn ]
= +− +
+10 0 059
5,
log
[Sn ]52
[MnO ]24
+ −=
[Sn ]
52
[Mn ]4 2+ +=
[MnO ]4−
[Mn ]25
[Sn ]2 4+ += =
E Eeq4
8
2log
[MnO ][H ][Mn ]
= +− +
+10 0 059
5,
MnO /Mn42− +
[MnO ] 0, 1 [Mn ]42− += − =
[ ]Mn ]15
[Fe2 3+ += =
Capítulo 10: Equilibrios de oxidación-reducción 329

[10.49]
que, una vez ordenada, queda en la forma:
[10.50]
A pH = 0 y a tenor de las ecuaciones [10.46]y [10.47] es evidente que el término logarítmicoes igual a cero, por lo que:
[10.51]
En general, para una reacción del tipo:
n2Ox1 + n1Red2←→ n2Red1 + n1Ox2 [10.3]
en la que están implicadas las semirreaccionesOx1 + n1 e ←→ Red1 y Ox2 + n2 e ←→ Red2, esposible deducir que el potencial de equilibriode una disolución que contiene cantidades este-quiométricas de ambos reactivos viene dado porla ecuación:
[10.52]
Esta ecuación general es válida para condi-ciones estándar, en las que la actividad del ionhidrógeno es igual a la unidad. Si esta condiciónno se cumple, el potencial de la disolución depen-derá del pH. Por otra parte, hay ocasiones en quela forma oxidada o la forma reducida de uno delos pares que intervienen en la reacción puedeser polinuclear y en este caso el potencial depen-derá de la concentración de uno de los paresredox. Por ejemplo, al mezclar cantidades este-quiométricas de K2Cr2O7 y de FeSO4, que pre-sentan la reacción:
Cr2O72– + Fe2+ + 14 H+ ←→ 2 Cr3+ + Fe3+ + 7 H2O
[10.32]
el potencial de equilibrio de la disolución obte-nida viene dado por la ecuación:
[10.53]
donde E01 y E0
2 son los potenciales estándar corres-pondientes a los pares y Fe3+/Fe2+,respectivamente.
10.8. Sistemas poliredox
Un sistema poliredox es aquel en el que unamisma especie puede presentar varios estados deoxidación, existiendo un intercambio escalonadode electrones entre ellos. Estos sistemas presentanuna gran analogía con los ácidos y bases polipró-ticos estudiados en los equilibrios ácido-base, mos-trando interesantes analogías con los mismos. Elmanganeso y el vanadio son unos de los ejemplosmás significativos de estos sistemas poliredox.
Manganeso:
[10.54]
Vanadio:
[10.55]
En estos sistemas se pueden distinguir lassiguientes especies:
1. Polioxidante. Especie que puede reducir-se a diferentes estados de reducción. En elcaso del manganeso, el ion permanganatoes un ejemplo típico de polioxidante ya quepodría estar implicado en las siguientesreacciones de reducción:
VO VO V V3− + + +← → ← → ← →e e e2 3 2
Mn Mn+ +← → ← →e e3 2
MnO MnO MnO4 42− −← → ← → ← →e e e2
2
Cr O /2 Cr2 72 3− +
EE E
eq
14
3
[H ]2[Cr ]
= + ++
+
67
0 0597
10
20 ,
log
En E n E
n neq = ++
1 10
2 20
1 2
EE E
eq = +5 27
10
20
E
E Eeq
44 8
2 2
[MnO ][Sn ][H ][Mn ][Sn ]
= + +− + +
+ +
5 27
0 0597
10
20 ,
log
+ +
+
+2 0 0592
0E , log[Sn ][Sn ]
4
2
7 5 0 05910E Eeq
48
2
[MnO ][H ][Mn ]
= +− +
+, log
330 Equilibrios iónicos y sus aplicaciones analíticas

+ e ←→ [10.56]
+ 4 H+ + 3 e ←→ MnO2 (S) + 2 H2O [10.57]
+ 8 H+ + 4 e ←→ Mn3+ + 4 H2O[10.58]
+ 8 H+ + 5 e ←→ Mn2+ + 4 H2O[10.7]
2. Polireductor. Especie que puede oxidarsey pasar a varios estados de oxidación. Elestado de oxidación +2 del vanadio seríaun ejemplo típico de polireductor ya quepuede experimentar las siguientes reac-ciones de oxidación:
V2+ ←→ V3+ + e [10.59]V2+ + H2O ←→ VO2+ + 2 H+ + 2 e [10.60]
V2+ + 3 H2O ←→ + 6 H+ + 3 e [10.61]
3. Especies intermedias Pueden actuar comooxidantes y como reductores y por tanto secaracterizan por ser la forma oxidada de unpar redox y la forma reducida de otro. Denuevo se presenta en este caso un parale-lismo entre estos equilibrios y los equilibriosácido-base, ya que el comportamiento dela especie intermedia de un polioxidante esequivalente al que presenta un anfolito enácido-base. Por ejemplo, el ion V3+, quepuede reducirse de acuerdo con:
V3+ + e ←→ V2+ [10.62]
y oxidarse según la reacción:
V3+ + H2O ←→ VO2+ + 2 H+ + e [10.63]
sería el equivalente en redox al hidroge-nocarbonato de sodio en ácido-base, porejemplo.
4. Dismutación. Es un proceso de auto oxi-dación-reducción, en el que un mismocompuesto actúa como oxidante y como
reductor y que teóricamente puede pro-ducirse en el caso de las formas intermediasde los polioxidantes, aunque el hecho de quela reacción tenga lugar o no depende de lospotenciales estándar y de las condicionesexperimentales.
Por ejemplo, la reacción:
2 V3+ + H2O ←→ V2+ + VO2+ + 2 H+ [10.64]
no está favorecida, ya que, en función delos correspondientes potenciales estándar:
V3+ + e ←→ V2+ [10.65]
VO2+ + e ←→ V3+ [10.66]
puede calcularse la constante de equilibrio,expresada como:
[10.67]
cuyo valor indica que la reacción [10.64]está desplazada hacia la izquierda y que,por lo tanto, no tan sólo el V3+ no tiene ten-dencia a dismutarse sino que el VO2+ y elV2+ reaccionarían entre sí para dar V3+.
Si se considera otro caso, tal como laposible dismutación de los iones Cu+:
2 Cu+ ←→ Cu2+ + Cu0 [10.68]
en la que están implicados los siguientespares redox:
Cu+ + e ←→ Cu0 = 0,521 V [10.69]
Cu2+ + e ←→ Cu+ = 0,153 V [10.70]
y se calcula la constante de equilibrio:
[10.71] log
, ,,
,K = − =0 521 0 1530 059
6 23
E20
E10
log,
, ,,
,KE E= − = − − = −1
020
0 0590 255 0 337
0 05910 03
E20 0 337= + , V
E10 0 25= − , 5 V
VO3−
MnO4−
MnO4−
MnO4−
MnO42−
MnO4−
Capítulo 10: Equilibrios de oxidación-reducción 331

puede comprobarse que la reacción estádesplazada hacia la derecha y que, por tan-to, en condiciones estándar el Cu+ se dis-muta para dar Cu2+ y Cu0. Sin embargo,cabe mencionar que, como se comentarácon detalle en el apartado 10.12.2 la pre-sencia de iones CN– en la disolución esta-biliza los iones Cu+ hasta el punto de queen estas condiciones no sólo se produce ladismutación, sino que el Cu2+ pasa a ser unoxidante fuerte.
A la vista de estos ejemplos se puede concluirque siempre que E0
1 (potencial de la semireac-ción en la que la especie intermedia del polioxi-dante actúa como oxidante) sea mayor que E0
2(potencial de la semireacción en la que la espe-cie intermedia del polioxidante actúa comoreductor), se produce la reacción de dismutación,aunque es conveniente recalcar de nuevo que sise usan los potenciales estándar esta conclusiónsólo es válida para las condiciones estándar.
El potencial de una disolución que contiene unaespecie intermedia de un polioxidante puede calcu-larse fácilmente mediante la ecuación [10.52], dedu-cida para el caso en que existen proporciones este-quiométricas de oxidante y de reductor.
10.9. Disoluciones reguladoras redox
En los capítulos dedicados al estudio del equi-librio ácido-base se han descrito las disolucionesreguladoras como aquellas que contienen canti-dades relativamente elevadas de un ácido débily de su base conjugada y que se caracterizan por-que su valor de pH no varía de forma significa-tiva al añadir una cantidad moderada de ácido ode base. Este concepto, sin embargo, no es exclu-sivo del ácido-base y puede extenderse a los res-tantes tipos de equilibrio.
En el caso concreto del equilibrio redox, seconsideran reguladoras aquellas disoluciones queson capaces de mantener el potencial práctica-mente constante al añadir cantidades moderadasde oxidante o de reductor. Estas disoluciones
están constituidas por los componentes de un parredox, esto es, un oxidante y su forma reduciday la característica que las define, su capacidadreguladora, al igual que las disoluciones regula-doras ácido-base, depende de la concentraciónglobal de las formas oxidada y reducida, así comode su concentración relativa. La capacidad regu-ladora es máxima cuando las concentraciones deoxidante y reductor son iguales y además la sumade ambas es relativamente elevada. En estas con-diciones y de acuerdo con la ecuación de Nernst,es fácil deducir que el potencial correspondien-te a esta máxima capacidad reguladora coincidecon el potencial normal del par redox.
Un ejemplo típico de este tipo de disolucio-nes es la formada por el par redox Fe3+/Fe2+
(E0 = 0,771 V) preparada, por ejemplo, a partirde la mezcla de sales de ambas especies a unamisma concentración final, tal como 1,0 mol L–1.En este caso, el potencial inicial de la disoluciónreguladora es igual a E0, esto es, 0,771 V.
¿Qué ocurre cuando se adiciona a la mismaun oxidante fuerte como es el ion Ce4+ hasta unaconcentración final de 0,1 mol L–1, si se suponeque no hay variación de volumen? Dado sucarácter oxidante, el Ce4+ reacciona con el Fe2+
conforme a la reacción:
Ce4+ + Fe2+ ←→ Ce3+ + Fe3+ [10.72]
siendo al final 1,1 y 0,9 mol L–1 las concentracio-nes de Fe3+ y Fe2+, respectivamente, por lo queel potencial de la disolución reguladora será:
[12.73]
incrementándose en tan sólo 0,005 V, lo quedemuestra la capacidad reguladora de este siste-ma redox.
Si se añade un reductor como Cr2+, en lugar deCe4+, también a una concentración final de 0,1 molL–1, se reducirá en este caso una cantidad equiva-
E E= + =
= + =
+
+0 0 059
0 771 0 0591 10 9
0 776
, log[ ][ ]
, , log,,
,
FeFe
V
3
2
332 Equilibrios iónicos y sus aplicaciones analíticas

lente de Fe3+ y el cociente [Fe3+]/[Fe2+] pasará a0,9/1,1 y el potencial final a 0,766 V, lo que suponeuna disminución de 0,005 V. Estos resultados ponende manifiesto, también en este caso, la capacidadreguladora del sistema Fe3+/Fe2+.
10.10. Sistemas redox del agua
Los aspectos básicos descritos sobre los equi-librios redox: carácter oxidante o reductor dediferentes especies, interacción entre ellas, etc.,se refieren en todos los casos al uso de disolu-ciones acuosas, considerando que el agua no esun elemento activo en estos procesos. Para eva-luar si esta suposición es válida o no es necesa-rio estudiar con cierta profundidad los sistemasredox de este disolvente.
De hecho, el agua puede actuar como reduc-tor y como oxidante. En el primer caso se oxidaa oxígeno según la reacción:
←→ [10.74]
mientras que en el segundo se reduce a hidrógeno
←→ [10.75]
En condiciones estándar, los potenciales sonlos siguientes:
←→[10.76]
←→ [10.25]
Dado que el agua puede intervenir en las reac-ciones redox, es necesario considerar tres casosclaramente diferenciados.
A) >
En este caso, la forma oxidada del sistema encuestión es inestable en disolución acuosa, ya quees capaz de oxidar al agua a oxígeno. La forma
reducida, en cambio, es estable. Por ejemplo,para el sistema Co3+/Co2+ el potencial estándares de 1,80 V y para el sistema MnO4
–/Mn2+ es de1,51 V. Como consecuencia, en condiciones están-dar, las reacciones
→E = 1,80 – 1,23 = 0,57 V [10.77]
→E = 1,51 – 1,23 = 0,28 V [10.78]
están totalmente desplazadas hacia la derechadesde el punto de vista termodinámico. Sinembargo, en no pocas ocasiones, las reaccionesredox tienen una cinética extremadamente len-ta, especialmente cuando en ellas se encuentraimplicada el agua, lo que justifica el hecho deque, pese a la conclusión anterior, el permanga-nato pueda existir en disolución acuosa.
Hay que mencionar que, en la práctica, lasdisoluciones de permanganato de potasio se sue-len preparar en medio neutro y que en estas con-diciones la reacción con el agua sería realmente:
4MnO4– + 2H2O → 4MnO2(S) + 3O2 + 4OH–
[10.79]
aunque este hecho no invalida la conclusión ante-rior, ya que la reacción [10.79] se caracterizaigualmente por estar totalmente desplazada haciala derecha y por ser lenta.
B)
En este caso, tanto la forma oxidada como lareducida del sistema son estables en disoluciónacuosa, ya que no reaccionan con el disolvente.Sin embargo, es posible que la forma reducida seoxide por acción del oxígeno atmosférico, aun-que éste sea un proceso diferente. Por ejemplo,en el caso del sistema Fe3+/Fe2+, donde:
Fe3+ + e ←→ Fe2+ E0 = + 0,771 V [10.15]
E E EO /H O0
sistema0
2H /H0
2 2+
21,23 V > > 0 V= =
4Mn 5O 6H O22 2
+ + + 4MnO 12H4− ++
4Co 4H O22
+ ++ +4Co 2H O32
+ +
EO /H O
02 2
1, 23 V= Esistema0
E2 H /H0
2V+ = 0 0, H22H 2+ + e
EO /H O
02 2
1, 23 V= 2H O2 O 4H 42 + ++ e
H 2 OH2 + − 2 H O 2 2 + e
O 4H 42 + ++ e 2 H O2
Capítulo 10: Equilibrios de oxidación-reducción 333

es evidente que las reacciones:
4 Fe3+ + 2 H2O ← 4 Fe2+ + O2 + 4 H+
E = 0,771 – 1,23 = – 0,459 V [10.80]
2 Fe2+ + 2 H+ ← 2 Fe3+ + H2 E = 0 – 0,771 = – 0,771 V [10.81]
están totalmente desplazadas hacia la izquierda.No obstante, es posible la reacción:
4 Fe2+ + O2 + 4 H+ → 4 Fe3+ + 2 H2OE = 1,23 – 0,771 = 0,459 V [10.82]
que al estar desplazada hacia la derecha, justifi-ca el hecho experimental de que las disolucio-nes de Fe2+ se oxidan lentamente a Fe3+, a menosque previamente se haya eliminado el oxígenodisuelto y se conserven en una atmósfera exen-ta de oxígeno.
C) >
En este caso la forma reducida del sistema esinestable en disolución acuosa, ya que es capazde reducir el agua a hidrógeno. Por su parte, laforma oxidada es estable.
Por ejemplo, para el sistema Cr3+/Cr2+
Cr3+ + e ←→ Cr2+ E0 = – 0,41 V [10.83]
se produce la reacción
2 Cr2+ + 2 H+ → 2 Cr3+ + H2E = 0 – (–0,41) = 0,41 V [10.84]
aunque tampoco puede excluirse la posibilidadde que reaccione con el oxígeno atmosférico:
4 Cr2+ + O2 + 4 H+ → 4 Cr3+ + 2 H2O E = 1,23 – (–0,41) = 1,64 V [10.85]
Esta característica justifica el comportamien-to de los metales frente al agua o a los ácidos.
Los metales con potencial estándar mayor quecero, como el cobre ( V) requie-ren ácidos oxidantes, como el ácido nítrico, parasu disolución, mientras que los metales conpotencial estándar negativo, como el sodio( ), se disuelven perfectamenteen agua, a menos que otro factor impida la reac-ción. Por ejemplo, el cinc (se disuelve en ácido clorhídrico, ya que la reac-ción:
Zn0 + 2 H+ → Zn2+ + H2 [10.86]
está desplazada hacia la derecha. Por otra parte,es bien conocido que este metal no es soluble enagua. La razón es que, aunque la reacción que seproduce en medio neutro:
Zn0 + 2 H2O → Zn(OH)2 (S) + H2 [10.87]
está desplazada hacia la derecha, provoca la apa-rición de un precipitado de hidróxido de cinc,que recubre el metal e impide que la reacciónprosiga; este fenómeno se conoce con el nombrede pasivado. En cambio, el cinc se disuelve per-fectamente en medio fuertemente básico, ya queen estas condiciones se forman hidroxocomple-jos solubles:
Zn0 + 2 H2O + 2 OH– → + H2 [10.88]
Evidentemente, todas las conclusiones de esteapartado se refieren a condiciones estándar. Enotras condiciones serán los potenciales condi-cionales los factores que realmente determina-rán el sentido del equilibrio y, por lo tanto, lasconclusiones pueden ser distintas. Sin embargo,se puede concluir indicando que el agua es gene-ralmente inerte en los procesos redox desde elpunto de vista práctico, debido a la lentitud dela cinética de las reacciones en las que está inmer-sa (producción de gases, hidrógeno y oxígeno,según el caso) y sólo en contadas ocasiones mani-fiesta una cierta actividad, especialmente en pre-sencia de catalizadores.
Zn(OH)42−
EZn /Zn0
2 0 V)+ = −0 763,
EaN /Na
00 V+ = −2 71,
ECu /Cu0
2 0 0, 337+ =
Esistema0
E
2H /H0
2V+ = 0
334 Equilibrios iónicos y sus aplicaciones analíticas

10.11. Representación gráfica del equilibrio redox
El empleo de las representaciones gráficas enel estudio de los equilibrios redox, permite visua-lizar el comportamiento de las diferentes espe-cies implicadas en los mismos conforme a lavariación de una magnitud experimental comoes el potencial redox. En la situación más sim-ple, en la que estas especies están sólo implica-das en un equilibrio redox, las representacionesgráficas son similares a las desarrolladas para losequilibrios ácido-base en el capítulo 3. Así, porejemplo, se puede hacer uso de diagramas loga-rítmicos de concentración, en los que se repre-sentan los logarítmos de las concentraciones delas especies que existen en disolución en funcióndel potencial (o, en algunos casos, del pe, que sedefine, a 25 ºC, como el potencial dividido por0,059) que se representa en abscisas. Estos dia-gramas permiten, en definitiva, evaluar la pro-porción relativa entre oxidante y reductor paraun determinado par redox a un determinadovalor de potencial.
Se considera como ejemplo para el desarro-llo de estos diagramas una disolución acuosa conuna concentración total de hierro CFe = [Fe3+] +[Fe2+]. El sistema redox implicado es
Fe3+ + e ←→ Fe2+ [10.5]
y el potencial viene dado por:
[10.89]
Evidentemente el diagrama puede obtenersea base de calcular las concentraciones de Fe3+ yde Fe2+ para una serie de valores de potencial. Sinembargo, se puede optar por conseguir la expre-sión que delimita en este diagrama la existenciade la especie Fe3+ en función del potencial, es decirla representación log [Fe3+] = ϕ(E). Para ello, sehan de llevar a cabo una serie de operaciones sen-cillas en la ecuación [10.89], que se resumen a con-
tinuación, considerando que la concentración totalde hierro es 0,1 mol L–1:
[10.90]
y al operar y reagrupar términos se tiene:
[10.91]
A partir de esta ecuación, se puede deducirfácilmente la [Fe3+] para cada valor del potencial(E) y llevar a cabo posteriormente la represen-tación de log [Fe3+] en función del potencialredox, que se muestra en la figura 10.3. La líneaen este diagrama correspondiente al Fe2+ se obtie-ne mediante un procedimiento similar.
FIGURA 10.3. Diagrama logarítmico de concentración del sistema redox Fe3+/Fe2+.
Hay que indicar, que resulta más cómododibujar el diagrama como en el caso de los dia-gramas logarítmicos ácido-base. Para ello se divi-de toda la ecuación de Nernst por 0,059:
[ ][ ]
Fe0, 1 Fe
103
3
0,7710,059
+
+
−
−=
E
E = +
−
+
+0, 771 0, 059 log
[Fe0, 1 Fe
3
3
][ ]
E E= ++
+0
3
20, 059 log
FeFe
[ ][ ]
Capítulo 10: Equilibrios de oxidación-reducción 335

[10.92]
y tras definir pe y pe0 por las expresiones:
[10.93]
la ecuación de Nernst queda en la forma:
[10.94]
Haciendo uso de esta expresión, se puedenconsiderar tres zonas claramente diferenciadasen el diagrama logarítmico de concentración:
Zona 1: E >> E0 ⇒ pe >> pe0
En esta zona prácticamente todo el hierro seencontrará en la forma oxidada, por lo que pue-de aceptarse que [Fe3+] ≈ CFe >> [Fe2+], y en con-secuencia, log [Fe3+] = log CFe, que correspondea una recta horizontal. Haciendo uso de la expre-sión [10.90] se tiene:
pe = pe0 + log CFe – log [Fe2+] [10.95]
de donde se deduce que:
log [Fe2+] = pe0 + log CFe – pe [10.96]
que corresponde a la ecuación de una recta dependiente –1.
Zona 2: punto del sistema:E = E0 ⇒ pe = pe0
En este punto se cumple que:
[Fe3+] = [Fe2+] = [10.97]
y por tanto:
log [Fe3+] = log [Fe2+] = log CFe – 0,3 [10.98]
lo que significa que las líneas que representan loslogaritmos de la forma oxidada y de la forma redu-cida se cortan en un punto situado 0,3 unidadeslogarítmicas por debajo del punto del sistema.
Zona 3: E0 >> E ⇒ pe0 >> pe
En esta zona prácticamente todo el hierro seencontrará en la forma reducida, por lo que pue-de aceptarse que [Fe2+] ≈ CFe >> [Fe3+], y en con-secuencia, log [Fe2+] = log CFe, que correspondea una recta horizontal. Mediante la expresión[10.94] se tiene:
pe = pe0 – log CFe + log [Fe3+] [10.99]
de donde se deduce que:
log [Fe3+] = log CFe – pe0 + pe [10.100]
ecuación que corresponde a una recta de pen-diente +1.
El diagrama que se obtiene, considerandoestas ecuaciones, se muestra en la figura 10.3, don-de se observa un punto singular que correspon-de al punto de corte de las líneas que represen-tan log[Fe3+] y log [Fe2+]. En este punto [Fe3+] =[Fe2+] y por tanto E = E0 = 0,771 V. A valores depotencial significativamente superiores puedeaceptarse que la concentración de los iones Fe3+
es constante, mientras que a valores de potencialsignificativamente inferiores es la concentraciónde Fe2+ la que permanece constante. Para obte-ner la concentración de ambas especies en diso-lución, a un determinado valor de potencial, bas-ta con trazar una línea vertical a este valor de Ey leer las ordenadas correspondientes a los pun-tos de intersección de esta línea con las que repre-sentan log[Fe3+] y log [Fe2+]. Puede comprobarseque este diagrama es idéntico al diagrama ácido-base de un protolito monoprótico, por lo que noes necesario repetir el procedimiento para dibu-jarlo ni insistir en cómo se utilizan.
Pese a la analogía de estos diagramas con losde ácido-base, es conveniente comentar que, sibien el diagrama de un sistema simple, como el
CFe
2
pe pe= +
+
+0
3
2log
FeFe
[ ][ ]
peE0
0
0,059=pe
E=0,059
E E0 059 0 059, ,
][ ]
= ++
+
0 3
2log
[FeFe
336 Equilibrios iónicos y sus aplicaciones analíticas

del Fe3+/Fe2+, es fácil de deducir y de dibujar, lossistemas se complican, especialmente, cuandouna de las especies es un sólido (caso delZn2+/Zn0). Pueden darse reacciones distintas enfunción del potencial (sistema Fe3+/Fe2+/Fe0) ouna de las formas es polinuclear (sistemas /2Cr3+ o I2/2I–). En consecuencia, su deducción yconstrucción no tiene demasiado sentido, ya quela información que proporcionan puede obte-nerse sin dificultades mediante un cálculo senci-llo; en cambio, sí que es conveniente saber inter-pretarlos si se encuentran ya dibujados.
10.12. Equilibrios concurrentes: potencial condicional
Los equilibrios concurrentes se originan cuan-do coexisten en una misma disolución dos o másequilibrios, ya sean homogéneos o heterogéneos.Esta concurrencia genera diversos tipos de influen-cia de mayor o menor importancia entre las espe-cies implicadas en los diferentes equilibrios. En elcaso de los equilibrios redox, de acuerdo con elenfoque jerarquizado sobre el estudio de los equi-librios iónicos en disolución expuesto en el capítu-lo 5, se pueden considerar diversos tipos de inte-racciones con un grado creciente de complejidad,tales como:
1. Binarias: redox/ácido-base, redox/forma-ción de complejos y redox/precipitación.
2. Ternarias: redox/ácido-base/formación decomplejos, redox/ácido-base/precipitacióny redox/formación de complejos/precipita-ción.
3. Cuaternarias: redox/ácido-base/formaciónde complejos/precipitación.
Estas influencias pueden ser tan importantesque en ocasiones llegan a cambiar el sentido de lareacción redox, lo que es de gran interés en eldesarrollo de numerosos métodos analíticos.
En este contexto, y al igual que en los equili-brios de formación de complejos y de precipita-ción, es conveniente hacer uso de “constantes”
condicionales que consideren la influencia de estasreacciones secundarias o parásitas. En este casose define un potencial condicional, E0′, que, comoocurre con todas las constantes condicionales, noes una auténtica constante ya que depende de lascondiciones experimentales, pero indica el autén-tico carácter oxidante o reductor de un par redoxen unas condiciones concretas.
La comparación entre los valores de E0 y E0′permite establecer de forma cuantitativa lainfluencia de las reacciones secundarias sobre elpar redox en cuestión, esto es, si se incrementa odisminuye su carácter oxidante o reductor, y enconsecuencia, cómo afecta este hecho a la reac-ción redox en que esté implicado. En concreto,puede afirmarse que en condiciones diferentes alas estándar la condición para que una reacción:
Ox1 + Red2←→ Red1 + Ox2 [10.41]
tenga lugar es que E0′1 – E0′
2 > 0, donde E0′1 y E0′
2 co-rresponden a los potenciales condicionales delpar que se reduce y se oxida, respectivamente.
Aunque seguidamente se comentarán estosaspectos con mayor detalle, puede afirmarse queel potencial condicional de un par redox corres-ponde al potencial que dicho par presentaría res-pecto al electrodo estándar de hidrógeno en unascondiciones concretas y siempre que se cumplanlos requisitos siguientes:
1. Si la forma oxidada y la forma reducidason solubles, que el cociente entre las con-centraciones condicionales de ambas seaigual a la unidad.
2. Si la forma oxidada o la forma reducida soninsolubles, que la concentración condicio-nal de la forma soluble sea igual a la unidad.
10.12.1. Equilibrios concurrentes redox/ácido-base
La influencia del pH sobre los equilibriosredox se puede atribuir básicamente a dos pro-cesos de distinta naturaleza:
Cr O2 72−
Capítulo 10: Equilibrios de oxidación-reducción 337

1. Cuando a pesar de que ni la forma oxida-da ni la forma reducida presentan carác-ter ácido o básico, los iones H+ intervienenen la semirreacción redox. Esto ocurrecuando la proporción de oxígeno varía enfunción del grado de oxidación, como esel caso de las reacciones siguientes:
+ 8 H+ + 5 e ←→ Mn2+ + 4 H2O[10.7]
←→[10.101]
←→[10.102]
2. Cuando la forma oxidada, la forma redu-cida, o ambas, presentan carácter ácido obásico y en consecuencia están implicadasen una reacción secundaria de disociacióno de protonación, independientemente deque, además, los iones H+ puedan interve-nir en la semirreacción redox. A pH = 0,que corresponde al estado estándar, los pro-tolitos débiles están totalmente protona-dos; un aumento del pH originará su diso-ciación y, por tanto, una disminución de laconcentración de la especie que aparece enla ecuación de Nernst. Algunos ejemplosde este tipo de pares redox son:
2 CO2 + 2 H+ + 2 e ←→ H2C2O4 [10.103]
S0 + 2 H+ + 2 e ←→ H2S [10.104]
HBrO + H+ + 2 e ←→ Br– + H2O [10.105]
H3PO4 + 3 H+ + 3 e ←→ P0 + 3 H2O [10.106]
En sentido estricto, en este apartado sedeberían incluir las reacciones secundarias,o parásitas, de hidrólisis de los iones metá-licos, ya que son reacciones ácido-base. Sinembargo, se ha preferido no hacerlo porlas razones siguientes: la mayoría las reac-ciones redox se llevan a cabo en medio áci-do y salvo algunos casos particulares (Ce4+
y Fe3+ fundamentalmente), puede aceptar-se que en estas condiciones las reaccionesde hidrólisis son despreciables; y, en mediobásico, la mayoría de los iones metálicos(excepto los alcalinos y alcalinotérreos) pre-cipitan inicialmente en forma de hidróxi-do, aunque algunos pueden redisolverse enpresencia de un exceso de iones OH– for-mando los correspondientes complejoshidroxilados. En consecuencia, esta influen-cia se tratará posteriormente como casosconcretos de los equilibrios concurrentesredox/ácido-base/precipitación o bien redox/ácido-base/precipitación/formación de com-plejos.
A) Influencia del pH originada por la diferenteproporción de oxígeno entre la forma oxidada y reducida del par redox
En este caso, la influencia del pH se debe aque la semireacción redox correspondiente con-tiene protones y moléculas de agua en propor-ción estequiométrica para ajustar la diferenteproporción de oxígeno entre ambas formas delpar.
De manera general, la semireacción corres-pondiente a un par redox con diferente propor-ción de oxígeno entre la forma oxidada y redu-cida, se puede escribir como:
←→ [10.107]
De acuerdo con la expresión de Nernst, elpotencial vendrá dado por:
[10.108]
que al operar queda como:
[10.109] E E
n n= + × ++0 0 059 0 059,
log[ ],
loga H[Ox][Red]
E E
n
a
= ++
0 0 059,log
[Ox][H ][Red]
Red
a2
H O2+Ox aH+ ++ ne
2 Cr 7 H O32
+ +Cr O 14 H2 72− ++ + 6 e
VO 2H O22
+ +VO 4H3− ++ + e
MnO4−
338 Equilibrios iónicos y sus aplicaciones analíticas
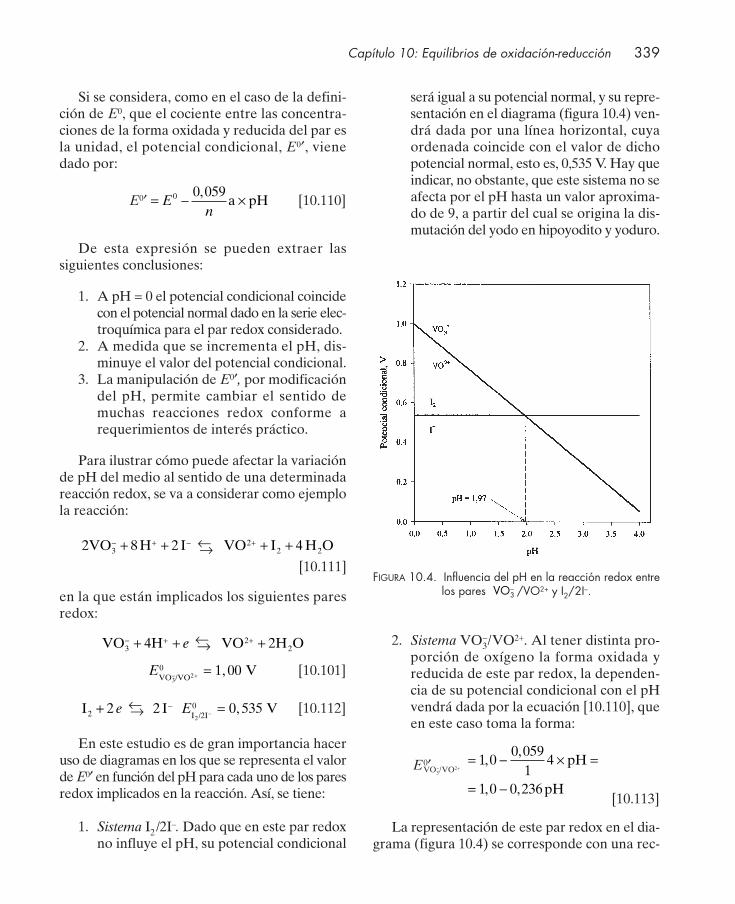
Si se considera, como en el caso de la defini-ción de E0, que el cociente entre las concentra-ciones de la forma oxidada y reducida del par esla unidad, el potencial condicional, E0′, vienedado por:
E0′ [10.110]
De esta expresión se pueden extraer lassiguientes conclusiones:
1. A pH = 0 el potencial condicional coincidecon el potencial normal dado en la serie elec-troquímica para el par redox considerado.
2. A medida que se incrementa el pH, dis-minuye el valor del potencial condicional.
3. La manipulación de E0′, por modificacióndel pH, permite cambiar el sentido demuchas reacciones redox conforme arequerimientos de interés práctico.
Para ilustrar cómo puede afectar la variaciónde pH del medio al sentido de una determinadareacción redox, se va a considerar como ejemplola reacción:
←→[10.111]
en la que están implicados los siguientes paresredox:
←→
[10.101]
←→ [10.112]
En este estudio es de gran importancia haceruso de diagramas en los que se representa el valorde E0′ en función del pH para cada uno de los paresredox implicados en la reacción. Así, se tiene:
1. Sistema I2/2I–. Dado que en este par redoxno influye el pH, su potencial condicional
será igual a su potencial normal, y su repre-sentación en el diagrama (figura 10.4) ven-drá dada por una línea horizontal, cuyaordenada coincide con el valor de dichopotencial normal, esto es, 0,535 V. Hay queindicar, no obstante, que este sistema no seafecta por el pH hasta un valor aproxima-do de 9, a partir del cual se origina la dis-mutación del yodo en hipoyodito y yoduro.
FIGURA 10.4. Influencia del pH en la reacción redox entrelos pares /VO2+ y I2/2I–.
2. Sistema VO3–/VO2+. Al tener distinta pro-
porción de oxígeno la forma oxidada yreducida de este par redox, la dependen-cia de su potencial condicional con el pHvendrá dada por la ecuación [10.110], queen este caso toma la forma:
E0′VO3–/VO2+
[10.113]
La representación de este par redox en el dia-grama (figura 10.4) se corresponde con una rec-
pH
pH
= − × =
= −
1 00 059
14
1 0 0 236
,,
, ,
VO3−
E
I /2I0
20, 535 V− = 2 I−
I2 2+ e
E
VO /VO0
32 1, 00 V− + =
VO 2H O22
+ + VO 4H3− ++ + e
VO I 4 H O22 2
+ + + 2VO 8 H 2 I3− + −+ +
En
0 0 059a pH= − ×,
Capítulo 10: Equilibrios de oxidación-reducción 339

ta de pendiente negativa (–0,236) y cuya orde-nada en el origen (a pH = 0) coincide con el valorde su potencial normal (1,0 V).
El pH al cual se produce la intersección entreambas rectas es un punto singular ya que en estascondiciones la reacción redox se encuentra enequilibrio. En este caso, los potenciales condicio-nales de ambos sistemas son iguales y por tanto:
E0′I2/2I– = E0′VO3–/VO2+ = 0,535 = 1,0 – 0,236 pH
[10.114]
de donde pH = 1,97.De este diagrama se deduce, que a un deter-
minado valor de pH, la forma oxidada del parsituado por encima en la representación, es capazde oxidar a la forma reducida del otro par, y vice-versa. En consecuencia:
1. A valores de pH inferiores a 1,97 el ionVO3
– oxida al ion yoduro a yodo y la reacción se encuentra desplazada hacia la derecha.
2. Por el contrario, a pH superiores a 1,97 esel yodo la especie que es capaz de oxidar alion VO2+ a VO3
–, por lo que en este caso lareacción está desplazada hacia la izquierda.
Por regla general, basta con los potencialescondicionales para decidir el sentido de una reac-ción redox y evaluar su extensión aunque evi-dentemente también es posible calcular la cons-tante condicional de equilibrio. La expresióndeducida en el apartado 10.6 para calcular laconstante de equilibrio, ecuación [10.40], depen-de de los potenciales normales de los pares redoximplicados en la misma y del número de elec-trones intercambiados en cada uno de ellos:
[10.40]
Dado que dichos potenciales normales, extraí-dos de la serie electroquímica, corresponden a unvalor de pH = 0, cuando en la expresión anteriorse trabaje con potenciales condicionales (a valo-
res de pH distintos de cero) ésta se transforma enla ecuación que permite el cálculo de la constan-te condicional del equilibrio redox.
Por ejemplo, si en una determinada reacciónredox, se considera que sólo depende del pH elpar que actúa como oxidante, E 0′
1 (la situaciónmás común en la práctica), al sustituir la ecua-ción [10.110] en la [10.40] se tiene:
[10.115]
o bien:
[10.116]
y finalmente:
[10.117]
A partir de esta expresión se observa, una dis-minución del valor de la constante condicional dela reacción redox al aumentar el pH del medio,influyendo además de forma importante en estecomportamiento el número de iones H+ implica-dos en el par redox que actúa como oxidante, a1,así como el número de electrones intercambia-dos en el par que actúa como reductor, n2.
Para la reacción entre el vanadio y el yodoque se ha tomado como ejemplo, la expresión[10.116] se transforma en:
[10.118]
De esta expresión se deduce fácilmente quea valores de pH superiores a 1,97 (cuando eltérmino 8 × pH es superior a 15,76) cambia el
log,
,,
′ = − × − × × =
= − ×
K1 0 535
0 0591 2 2 4
15 76 8
pH
pH
log log′ = − × ×K K n2 1a pH
log,
′ = − − × ×KE E
n n n01
02
1 2 2 10 059a pH
log,
,
,
′ =′ − =
=− × −
KE E
n n
En
En n
10
20
1 2
10
11 2
0
1 2
0 0590 059
0 059
a pH
log
,K
E En n= −1
020
1 20 059
340 Equilibrios iónicos y sus aplicaciones analíticas

sentido de la reacción redox tal cómo se demos-tró anteriormente haciendo uso del diagramaE0′ .
B) Reacciones secundarias de disociación de las formas oxidada o reducida
Cuando alguna de las formas del par redox esun protolito débil, su concentración y, por tanto,su influencia en el equilibrio del par dependedirectamente del pH del medio. Si se considera,por ejemplo, que la forma reducida de un parredox es un ácido poliprótico, HmRed, el siste-ma redox vendrá dado por:
←→[10.119]
Las reacciones secundarias, o parásitas, quese consideran en este caso son las correspon-dientes a la disociación del ácido poliprótico queconstituye la forma reducida del par:
HmRed ←→ Hm–1Red– + H+ [10.120]
Hm–1Red– ←→ Hm–2Red2– + H+ ... [10.121]
Si se denomina [HmRed]′ a la concentraciónde la forma reducida del par, independientementede su grado de protonación, se puede establecerel siguiente balance de materia:
[10.122]
El coeficiente de reacción secundaria para laespecie HmRed puede definirse como:
[10.123]
y puede calcularse mediante la ecuación:
[10.124]
donde Ka1, Ka2, ..., Kam son las constantes de diso-ciación de HmRed.
En medio fuertemente ácido (pH << pKa1) setiene que [HmRed] = [HmRed]′ y, por tanto, αHmRed
= 1; si esta condición no se cumple, [HmRed] <[HmRed]′ y, en consecuencia, αHmRed > 1.
Al sustituir el valor de [HmRed], dado por laexpresión anterior, en la ecuación de Nernst, setiene:
[10.125]
Si se define el potencial condicional de estepar redox como el que proporcionaría cuando[Ox] = [HmRed]′, éste vendrá dado finalmentepor:
[10.126]
expresión que no permite evaluar de forma inme-diata la influencia del pH, ya que cuando ésteaumenta también lo hace αHmRed, pero disminuye[H+]. Sin embargo, si se sustituye αHmRed por laecuación [10.124] se obtiene:
E Enm m m
mOx/H Red0
Ox/H Red0
H RedH′ = + +0 059,log[ ] α
En
n
m m
m
m
Ox/H Red0
H Red[H
[Ox][H Red]
+ +
+′
+0 059
0 059
,log ]
,log
α
Enm
m
m
mOx/H Red
H Red[Ox][H ]
[H Red]0 0 059+
′=
+,
logα
E Enm
m
m
= + =+
Ox/H Red0 [Ox][H ]
[H Red]0 059,
log
αH Red H H Hm
K K K K K Ka a a a a amm
= + + + ++ + +1 1 1 22
1 2
[ ] [ ] [ ]�
�
αH Red
1
[H Red][H Red][H Red] [H Red ] [Red ]
[H Red]
m
m
m
m mm
m
= ′ =
= + + +−− −�
[H Red] [H Red] [H Red ] [Red ]1m m mm′ = + + +−
− −�
E
mOx/H Red0H RedmOx m H+ ++ n e
pH)= ϕ(
Capítulo 10: Equilibrios de oxidación-reducción 341
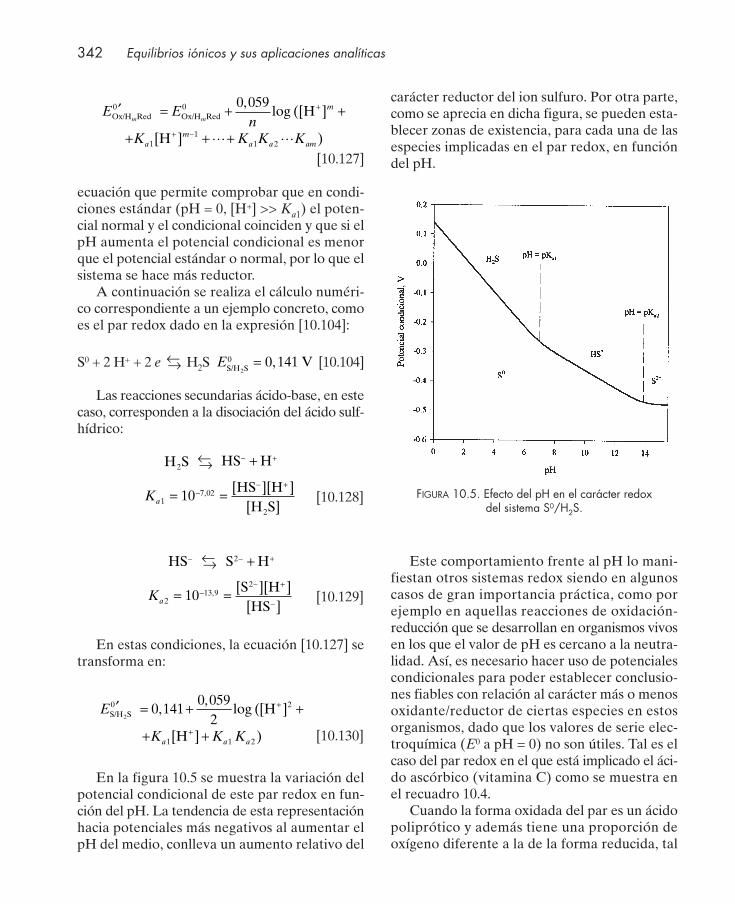
[10.127]
ecuación que permite comprobar que en condi-ciones estándar (pH = 0, [H+] >> Ka1) el poten-cial normal y el condicional coinciden y que si elpH aumenta el potencial condicional es menorque el potencial estándar o normal, por lo que elsistema se hace más reductor.
A continuación se realiza el cálculo numéri-co correspondiente a un ejemplo concreto, comoes el par redox dado en la expresión [10.104]:
S0 + 2 H+ + 2 e ←→ H2S [10.104]
Las reacciones secundarias ácido-base, en estecaso, corresponden a la disociación del ácido sulf-hídrico:
←→
[10.128]
←→
[10.129]
En estas condiciones, la ecuación [10.127] setransforma en:
[10.130]
En la figura 10.5 se muestra la variación delpotencial condicional de este par redox en fun-ción del pH. La tendencia de esta representaciónhacia potenciales más negativos al aumentar elpH del medio, conlleva un aumento relativo del
carácter reductor del ion sulfuro. Por otra parte,como se aprecia en dicha figura, se pueden esta-blecer zonas de existencia, para cada una de lasespecies implicadas en el par redox, en funcióndel pH.
FIGURA 10.5. Efecto del pH en el carácter redox del sistema S0/H2S.
Este comportamiento frente al pH lo mani-fiestan otros sistemas redox siendo en algunoscasos de gran importancia práctica, como porejemplo en aquellas reacciones de oxidación-reducción que se desarrollan en organismos vivosen los que el valor de pH es cercano a la neutra-lidad. Así, es necesario hacer uso de potencialescondicionales para poder establecer conclusio-nes fiables con relación al carácter más o menosoxidante/reductor de ciertas especies en estosorganismos, dado que los valores de serie elec-troquímica (E0 a pH = 0) no son útiles. Tal es elcaso del par redox en el que está implicado el áci-do ascórbico (vitamina C) como se muestra enel recuadro 10.4.
Cuando la forma oxidada del par es un ácidopoliprótico y además tiene una proporción deoxígeno diferente a la de la forma reducida, tal
E
K K Ka a a
S/H S0
20, [H
H ]
′ = + +
+ +
+
+
1410 059
22
1 1 2
,log ( ]
[ )
Ka 2
13 910= =−− +
−, [S ][H ]
[HS ]
2
S H2− ++ HS−
Ka1
7 0210= =−− +
, [HS ][H ][H S]2
HS H− ++ H S2
ES/H S
02
0, 141 V=
E En
K K K K
m m
m
am
a a am
Ox/H Red0
Ox/H Red0
1
H ]
H ]
′ = + +
+ + +
+
+ −
0 059
1 1 2
,log ([
[ )� �
342 Equilibrios iónicos y sus aplicaciones analíticas

como en los pares redox dados en las expresio-nes [10.105] y [10.106], la sistemática a seguir paraconseguir una expresión válida que relacione elpotencial condicional con el pH es similar a laexpuesta anteriormente. Se obtienen expresio-nes que son combinaciones de las deducidas encada uno de los apartados anteriores.
10.12.2. Equilibrios concurrentes redox/formación de complejos
Estos equilibrios concurrentes se ponen demanifiesto cuando una o ambas formas del parredox están implicadas en un equilibrio secun-dario de formación de complejos, lo que originauna disminución de la concentración efectiva dela especie electroactiva, con el consiguiente cam-bio de potencial. La situación más común, esaquella en la que un ligando reacciona con unao ambas formas del par redox, constituido éstepor dos estados de oxidación de un ion metáli-co. Sin embargo, también puede considerarse lasituación opuesta en la que el ligando forma par-te del par redox y la adición de un ion metálicoprovoca el equilibrio secundario de formaciónde complejos.
Sea el par redox Ox + n e ←→ Red, al que sele añade un ligando L que forma complejos, porejemplo, con la forma oxidada y reducida. Lasreacciones secundarias, o parásitas, que se con-sideran en este tratamiento son las siguientes:
Complejación de la forma oxidada:
←→ [10.131]
←→ [10.132]
. . . . . . . . . . .
←→ [10.133]
Complejación de la forma reducida:
←→ [10.134]
←→ [10.135]
. . . . . . . . . . . .
←→ [10.136]
Si independientemente de que se encuentrenlibres o complejadas las concentraciones de las
Red L zRed L+ z
Red L2Red 2 L+
Red L Red L+
OxLmOx L+ m
OxL2 Ox 2 L+
OxL Ox L+
Capítulo 10: Equilibrios de oxidación-reducción 343
RECUADRO 10.4
Los ácidos L-ascórbico (H2A) o vitamina C y L-dehidro-ascórbico (DA) forman parte del siguiente par redox:
←→ = 0,390 V
A partir del valor del potencial normal a pH = 0 de estasemireacción redox, no se infiere el conocido carácter reduc-tor del ácido ascórbico. Sin embargo, como tal ácido suexistencia depende del pH del medio, siendo sus constan-tes de disociación: Ka1 = 10–4,1 y Ka2 = 10–11,8, por lo quese ha de considerar su potencial condicional.
Para este sistema redox, el potencial condicional con-forme con la expresión [10.127] viene dado por:
Al sustituir los valores de las constantes de disociación,potencial normal y considerar un valor de pH = 7, se tiene:
= –0,062 V
Este valor de potencial pone de manifiesto el carác-ter reductor del ácido ascórbico en un medio fisiológico.Por otra parte y dado que este carácter disminuye apre-ciablemente en medio ácido, es por lo que es recomen-dable utilizar este pH para la conservación de alimentosricos en esta vitamina con objeto de evitar su deterioropor oxidación.
10 10 10 104 1 7 4 1 118 ), , ,− − − −+ × + ×
0 0592
10 14, log ( − +EDA/H A0
2′ = +0 390,
0 0592
21 1 2
, log ([ ] [ ] )H H+ ++ +K K Ka a a E EDA/H A0
DA/H A0
2 2′ = +
EDA/H A0
2H A H O2 2+DA 2H+ ++ 2e

formas oxidada y reducida se designan como[Ox]′ y [Red]′, respectivamente, es posible plan-tear los siguientes balances de materia:
[10.137]
[10.138]
Al dividir estos balances de materia por [Ox]y [Red], según el caso, se tiene:
[10.139]
[10.140]
Estas expresiones se corresponden con los res-pectivos coeficientes de reacción secundaria dela forma oxidada y reducida con respecto al ligan-do L, esto es, αOx(L) y αRed(L) . De acuerdo con loexpuesto en el capítulo 5, estas expresiones setransforman en:
[10.141]
[10.142]
siendo y las constantes de formaciónglobal respectivas de cada uno de los comple-jos implicados en estos equilibrios concurren-tes.
En estas condiciones, la expresión de Nernstpara este par redox responde a la expresión:
[10.143]
Si [Ox]′ = [Red]′ (condición para definir alpotencial condicional), éste vendrá dado por:
E0′ [10.144]
De esta ecuación se pueden extraer lassiguientes conclusiones:
1. La adición de un ligando que forme com-plejos con la forma oxidada (αOx(L) aumen-ta) provoca una disminución del potencialdel sistema.
2. La adición de un ligando que forme com-plejos con la forma reducida (αRed(L) aumen-ta) provoca un aumento del potencial con-dicional del sistema.
3. La adición de un ligando que forme com-plejos con ambas formas podrá provocarun aumento o una disminución del poten-cial del sistema, en función de cuál de lasformas tenga los complejos más estables.
En efecto, es evidente que para el equilibrio
Ox + n e ←→ Red↓↑ ↓↑OxLm RedLz
una disminución de la [Ox] por formación deOxLm desplazará el equilibrio hacia la izquier-da, con lo que el sistema se hará menos oxidan-te, mientras que una disminución de la [Red]por formación de RedLz desplazará el equlibriohacia la derecha, con el consiguiente aumentodel poder oxidante del sistema. A continuación,
En
= +0 0 059,log
αα
Red(L)
Ox(L)
E En
En
En n
= + =
= +′
′=
= + + ′′
0
0 Red(L)
Ox(L)
Red(L)
Ox(L)
0,059log
[Ox][Red]
0,059log
[Ox]
[Red]
[Ox][Red]
αα
αα
0 0 059 0 059,log
,log
βRedβOxL
α β
β β
Red(L) 1RedL
2RedL2
RedL
[Red][Red]
1 [L]
[L] [L]
= ′ = + +
+ + +... zz
α β
β β
Ox(L) 1OxL
2OxL2
OxL
[Ox][Ox]
1 [L]
[L] [L]
= ′ = + +
+ + +... mm
[Red][Red]
[Red] [RedL] [RedL ] [RedL ][Red]
2′ =+ + + +� z
[Ox][Ox]
[Ox] [OxL] [OxL ] [OxL ][Ox]
2′ = + + + +� m
[Red] [Red] [RedL] [RedL ] [RedL ]2′ = + + + +� z
[Ox] [Ox] [OxL] [OxL ] [OxL ]2′ = + + + +� m
344 Equilibrios iónicos y sus aplicaciones analíticas

se considerarán algunos casos concretos carac-terísticos.
En primer lugar, se aborda como ejemplo lamodificación del carácter redox de un par en elque sólo forma complejos la forma oxidada delmismo, tal como el par redox:
Hg+2 + 2 e ←→ Hg0 = 0,851 V [10.145]
en presencia de ion yoduro.Se originan las siguientes reacciones de com-
plejación:
Hg2+ + I– ←→ HgI+ = 1012,8 [10.146]
Hg2+ + 2 I– ←→ HgI2 (S) = 10–23,7 [10.147]
Hg2+ + 3 I– ←→ = 1027,5 [10.148]
Hg2+ + 4 I– ←→ = 1029,7 [10.149]
Con estos datos, se calcula el coeficiente dereacción secundaria para el ion Hg2+ de acuerdocon la ecuación [10.141] y teniendo en cuenta queHgI2 es insoluble:
[10.150]
Al sustituir este valor en la expresión del poten-cial condicional, ecuación [10.144], se tiene:
[10.151]
Si se trabaja con una concentración de ionyoduro de 0,1 mol L–1, al sustituir en la expre-sión anterior, resulta un potencial condicionalde 0,092 V.
La acusada disminución del potencial conrelación al valor en condiciones estándar, E0 =0,851 V, pone de manifiesto la pérdida del carác-ter oxidante del sistema Hg2+/Hg0 en presencia
de un ligando que compleja la forma oxidadadel par.
Finalmente, se considera el caso concreto deun par redox al que se le añade un ligando, L,que forma complejos con la forma oxidada yreducida del par. Sea, por ejemplo, el par redox:
Fe3+ + e ←→ Fe2+ [10.5]
que en presencia de un exceso del ligando 1,10-fenantrolina (fen), forma mayoritariamente lossiguientes complejos:
Fe3+ + 3 fen ←→ = 1014,1
[10.152]
Fe2+ + 3 fen ←→ = 1021,2
[10.153]
La expresión general [10.144] se transforma eneste caso en:
[10.154]
dado que se puede despreciar 1 frente a los valo-res de las constantes de formación global. Al sus-tituir los datos numéricos dados en la expresiónanterior, resulta un valor de potencial condicio-nal de 1,19 V, por lo que el sistema Fe3+/Fe2+
incrementa considerablemente su carácter oxi-dante en presencia de este ligando debido a lamayor estabilidad del complejo Fe(fen)3
2+. Estesistema redox tiene un gran interés analítico,dado que presenta las características adecuadaspara su empleo como indicador visual en valo-raciones redox: un elevado potencial, apto parasu uso en los casos en que se valora con un oxi-dante y una apreciable diferencia de color entreambas especies para proporcionar un cambiobrusco y nítido en el punto final de la valoración:el complejo Fe(fen)3
2+ es de color azul pálido
′ = + ×++
=
= + ×
+
+
+
+
E E
E
0 03
3
0
3
0 0591
1
0 0592
3
, log
, log
ββ
ββ
3Fe
3Fe
3Fe
Fe
2
3
[fen]
[fen]
β3Fe2+ Fe(fen)32+
β3Fe3+Fe(fen)33+
EFe /Fe0
3 2 0, 771 V+ + =
′ = − + +
+ +
+ +−
− −
E EHg /Hg0
Hg /Hg0
1Hg(I)
3Hg(I) 4Hg(I)
2 0 2 0 [I ]
[I ] [I ]
0 0592
1
3 4
,log(
)
β
β β
α β β βHg(I 1Hg(I) 3Hg(I)3
4Hg(I)[I ] [I ] [I) ]= + + +− − −1 4
β4Hg(I) HgI42−
β3Hg(I)HgI3−
KS( )HgI2
β1Hg(I)
EHg /Hg0
2 0+
Capítulo 10: Equilibrios de oxidación-reducción 345

mientras que el complejo Fe(fen)32+ es de color
rojo intenso. En el capítulo siguiente, corres-pondiente a valoraciones redox, se profundizarásobre el uso de este sistema como indicador visualen este tipo de valoraciones.
Como se ha puesto de manifiesto, el poten-cial de un sistema redox se puede variar a volun-tad introduciendo en el medio de reacción unasustancia que forme complejos con una o ambasformas del par. Sin embargo, este equilibriosecundario de formación de complejos ofrece, enocasiones, un interés adicional ya que permiteestabilizar el estado de oxidación de ciertos ionesmetálicos en disolución. Así, por ejemplo, en elapartado 10.8 se ha mostrado que el ion Cu+ esinestable en disolución ya que sufre un procesode dismutación. Sin embargo, en presencia deion CN– se estabiliza dicho estado de oxidacióndebido a la formación de complejos cianurados.Así, para el par redox:
Cu2+ + e ←→ Cu+ E0 = 0,153 V [10.61]
en presencia de ion cianuro, el cual origina lassiguientes reacciones de formación de com-plejos:
Cu+ + 2 CN– ←→ Cu(CN)2–; β2 = 1024 [10.155]
Cu+ + 3CN– ←→ Cu(CN)22–; β3 = 1028,6 [10.156]
Cu+ + 4CN– ←→ Cu(CN)23–; β4 = 1030,3 [10.157]
la expresión para el cálculo del potencial condi-cional, de acuerdo con la ecuación general[10.144], viene dada por:
[10.158]
De acuerdo con esta expresión, el valor delpotencial condicional para el par redox Cu2+/Cu+
en presencia de ion cianuro es de 1,714 V, si seconsidera que la concentración de ion cianuroes 0,1 mol L–1 y se desprecian los complejos cia-
nurados del Cu2+, menos estables. Se observacómo la presencia de ion cianuro incrementade manera considerable el poder oxidante delCu2+, que en estas condiciones pasa a ser su-perior al de oxidantes fuertes como permanga-nato y dicromato en condiciones estándar. Elcomportamiento del sistema del cobre en pre-sencia de cianuro es de interés en análisis cua-litativo, dado que permite disolver el sulfuro decobre una de las especies más insolubles de esteion:
→[10.159]
debido a la estabilización del ion cuproso porformación del correspondiente complejo cianu-rado y oxidación del cianuro a cianato gracias alelevado poder oxidante del Cu2+ en presencia decianuro. El ion cianuro, por tanto, presenta unadoble función en esta reacción: reductor y ligan-do formador de complejos.
Otra aplicación de interés del incremento delpotencial redox del sistema Cu2+/Cu+ es la iden-tificación de cianuros. En condiciones normales,el Cu2+ (E1
0 = +0,153 V) no es capaz de oxidar ala bencidina (E2
0 = +0,76 V). En cambio, en pre-sencia de CN–, el potencial condicional del siste-ma Cu2+/Cu+ aumenta hasta el punto de que lareacción:
Cu2++ benred (incolora) + 4 CN– ←→
←→ + benox (azul) [10.160]
está totalmente desplazada hacia la derecha. Elprocedimiento experimental es muy sencillo: ala disolución problema se le añade NaHCO3, queproporciona un pH suficiente para formar HCNvolátil y el recipiente se cubre con un papel defiltro sobre el que se han depositado unas gotasde disolución de Cu2+ y de bencidina. Si la mues-tra contiene CN–, aparece una mancha de colorazul intenso en el papel.
Cu(CN)43−
2 Cu(CN) 2 S CNO H O43 2
2− − −+ + +
2 CuS 9CN 2 OH(S) + +− −
′ = + × + ++ +
+ + + +−
− −
E ECu /Cu
0Cu /Cu0
2 2 [CN ]
CN CN
0 059 1 22
33
44
, log(
[ ] [ ] )
ββ β
346 Equilibrios iónicos y sus aplicaciones analíticas

10.12.3. Equilibrios concurrentes redox/precipitación
Estos equilibrios concurrentes se originancuando una de las formas del par redox seencuentra en equilibrio con una especie insolu-ble tras la adición de un agente precipitante a ladisolución. Así, si se considera el equilibrio hete-rogéneo de precipitación:
MmAz (S)←→ mM + zA KS = [M]m[A]z [10.161]
en el que A es el agente precipitante y M es laespecie electroactiva, se pueden originar dossituaciones según sea M la forma oxidada o redu-cida del par redox. En ambos casos, se produci-rá una modificación del potencial redox, refleja-da en el valor del potencial condicional, tal comose ha estudiado en secciones precedentes sobrela influencia de otros equilibrios sobre el poten-cial redox.
Si se considera que en el equilibrio heterogé-neo de precipitación dado por la expresión[10.161], la especie M es la forma oxidada del parredox, M + n e ←→ Mred, siendo E0 su potencialnormal, el potencial redox vendrá dado por:
[10.162]
Si se sustituye en esta ecuación la concentra-ción de la especie M a partir de la expresión delproducto de solubilidad, se tiene:
[10.163]
de donde el potencial condicional, tras conside-rar que la concentración de Mred es 1,0 mol L–1,responde a la expresión:
[10.164]
A partir de esta expresión se deduce que cuan-do la forma oxidada del par redox está involucra-da en un equilibrio de precipitación el potencialcondicional será normalmente inferior al poten-cial normal, dado que por lo general KS << [A].La variación será tanto más acusada cuanto másinsoluble sea el precipitado formado (menor valorde KS) y cuanto mayor sea la concentración deagente precipitante A. La estequiometría del pre-cipitado (valores de m y z), como se aprecia en laexpresión [10.164], también afecta al valor de estepotencial condicional.
Cuando es la forma reducida del par redox,la que participa en el equilibrio de precipitaciónMox + n e ←→ M, la expresión correspondiente alpotencial condicional se deduce fácilmentesiguiendo un tratamiento similar al realizadoanteriormente, resultando:
[10.165]
De esta expresión se pueden extraer con-clusiones similares a las consideradas para laecuación [10.164], aunque en este caso referi-das al aumento del potencial condicional del parredox al disminuir la solubilidad del precipita-do (menor valor de KS) y al aumentar la con-centración de agente precipitante A. De igualforma, la estequiometría del precipitado tam-bién ejerce su influencia sobre el valor delpotencial condicional.
Un ejemplo significativo de la influencia delos equilibrios de precipitación sobre el poten-cial redox lo constituye el fundamento del elec-trodo de referencia más utilizado hoy en día: elelectrodo de Ag/AgCl. Tal como se indicó en elapartado 10.4.2, el potencial de este electrododeriva de la semireacción redox:
AgCl(S) + e ←→ Ag0 + Cl– [10.29]
E E
n mK
zn mS
0 0 0 059 0 059′ =×
+ ××
–,
log,
log[ ]A
E En m
Kz
n mS0 0 0 059 0 059′ = +
×− ×
×,
log,
log[ ]A
E En
K
En m
Kz
n m
n
Sz
m
S
= + × =
= +×
××
0
0
0 059 1
0 059 0 059
0 059
,log
[ ] [
,log –
,log[ ] –
–,
log[ ]
A M ]
A
M
red
red
E E
n= +0 0 059,
log[M]
[M ]red
Capítulo 10: Equilibrios de oxidación-reducción 347

cuyo potencial condicional de acuerdo con laexpresión [10.164] viene dado por:
[10.166]
dado que m = z = 1.Si se sustituye en esta expresión el valor de
E0 (0,800 V para el sistema Ag+/Ag0) y del pro-ducto de solubilidad (10–9,8), se tiene:
[10.167]
y se fija, como es común, la concentración de clo-ruro de potasio en 3,5 mol L–1, el potencial sereduce aproximadamente a 0,2 V (potencial dereferencia de este electrodo). Se observa cómo aladicionar ion cloruro al sistema Ag+/Ag0, dismi-nuye considerablemente su potencial y por ende
su carácter oxidante. Este valor de potencial, nomuy diferente al del electrodo normal de hidró-geno, junto con su estabilidad, facilidad de mani-pulación y carencia de toxicidad, ha generalizadosu uso como electrodo de referencia en sustitu-ción del electrodo normal de hidrógeno.
Finalmente, en el recuadro 10.5 se muestraun ejemplo sobre la aplicación práctica de estosequilibrios concurrentes de oxidación-reduccióny precipitación.
10.12.4. Equilibrios concurrentes con variasinteracciones
En estos sistemas se originan interaccionesentre más de dos equilibrios iónicos, siendo eldenominador común de todos ellos el equilibrioredox. Así, se abordará en primer lugar la influen-
E0 0 222 0 059′ = − −, , log[Cl ]
E E KS0 0 0 059 0 059′ = + − −, log , log[(AgCl) Cl ]
348 Equilibrios iónicos y sus aplicaciones analíticas
RECUADRO 10.5
Un ejemplo del aprovechamiento práctico de estos equi-librios concurrentes lo constituyen las denominadas bate-rías de plomo utilizadas en muchos vehículos (ver recuadro10.3). Estas baterías cuando suministran energía son cel-das galvánicas en las que están implicados los siguientespares redox:
PbSO4 (S) + 2 e ←→ Pb0 +
PbO2 (S) + 4 H+ + + 2 e ←→ PbSO4 (S) + 2 H2O
que actúan como ánodo y cátodo, respectivamente.En ausencia de ion sulfato, los potenciales normales
de los pares redox Pb2+/Pb0 y PbO2/Pb2+ son –0,126 y1,46 V, respectivamente. Conforme a estos potenciales, elpotencial suministrado por la celda galvánica será:
Si se añade ion sulfato a ambos compartimentos de estacelda galvánica, por ejemplo ácido sulfúrico 0,5 mol L–1, seorigina la precipitación del sulfato de plomo (KS = 10–7,8),siendo los potenciales de los compartimentos catódicos yanódicos, conforme a las ecuaciones [10.164] y [10.165]:
y el potencial de la celda galvánica (batería):
Se observa cómo la adición de ion sulfato origina unincremento apreciable del potencial suministrado por estacelda, hasta 2,0 V aproximadamente, dado que la forma-ción de la especie insoluble PbSO4 incrementa tanto el poderoxidante del PbO2 como el reductor del Pb0. El empleo deseis celdas galvánicas en serie (12 V) constituye la bateríageneralmente utilizada en los diversos tipos de vehículo.
E E ECelda Cátodo Ánodo 8 V= ′ − ′ = − − =1681 0 347 2 02, ( , ) ,
′ = − +
+ =
−EAnodo
1V
1 46 0 0592
10
0 0592
0 5 168
7 8, , log
, log , ,
,
′ = − + −
= −
−ECátodo
V
0 126 0 0592
10
0 0592
0 5 0 347
7 8, , log
– , log , ,
,
E E ECelda Cátodo Ánodo 6 V= − = − − =1 46 0 126 1 58, ( , ) ,
SO42−
SO42−

Capítulo 10: Equilibrios de oxidación-reducción 349
cia de los equilibrios ácido-base sobre el equilibrioconcurrente redox/formación de complejos debi-do a la disociación del ligando formador de com-plejos al variar el pH del medio de reacción. Pos-teriormente, se considerará el efecto de reaccionessecundarias o parásitas de hidrólisis de iones metá-licos que forman parte de un par redox, lo que dalugar a dos tipos de equilibrios concurrentes: el ionmetálico sólo forma el correspondiente hidróxidocon lo que se tiene el equilibrio mixto triple:redox/acidez/precipitación, y el ion metálico for-ma además del hidróxido un complejo soluble conlos iones OH– cuando el pH del medio es franca-mente básico, y por tanto, en este sistema partici-pan los cuatro equilibrios iónicos estudiados eneste libro, esto es, redox/acidez/precipitación/for-mación de complejos.
A) Influencia del pH en el equilibrio concurrente redox/formación de complejos
A tenor del tratamiento realizado en el aparta-do 10.12.2, el potencial condicional para el par redoxOx + n e ←→ Red, si ambas formas forman com-plejos con un ligando L, viene dado por la expre-sión:
[10.168]
Si L es un anión procedente de un ácido débil,HxL, el coeficiente de reacción secundaria de esteligando con el ion hidrógeno, αL(H), de acuerdocon lo expuesto en el capítulo 5 vendrá dado por:
[10.169]
y la constante condicional de formación globalpor:
[10.170]
En estas condiciones, la expresión del poten-cial condicional será similar a la expuesta en laecuación [10.168], sin más que sustituir los valo-res de constante de formación global por loscorrespondientes a sus constantes condiciona-les en función del pH al cual se lleve a cabo lareacción.
El ejemplo que se describe a continuaciónmuestra cómo se modifica el carácter redox delsistema Ag+/Ag0 en presencia de amoniaco porformación de los correspondientes complejosaminados de plata, en función del pH.
En este sistema concurrente se pueden con-siderar los siguientes equilibrios con sus respec-tivas constantes:
Ag+ + e ←→ Ag0 [10.171]
Ag+ + NH3←→ Ag(NH3)
+ β1 = 103,32 [10.172]
Ag+ + 2 NH3←→ β2 = 107,24 [10.173]
←→ NH3 + H+ pKa = 9,24 [10.174]
En este caso, la expresión [10.168] se trans-forma en:
[10.175]
siendo:
[10.176]
las constantes condicionales de formación de loscomplejos de la Ag+ con el amoniaco y
[10.177]
la constante de protonación del NH3.
β1
H = 1Ka
′ =
+ +β ββi i i
( [ )1
1 1H H ]
′ = − + ′ + ′+ +E EAg /Ag
0Ag /Ag0
30 0 NH NH0 059 1 1 3 22, log( [ ] [ ] ) β β
NH4+
Ag(NH )3 2+
EAg /Ag0
0 0, 8 V+ =
β βαi i i
( )′ = 1
L(H)
α β β βL(H) 1H
2H
xH[H ] [H ] H= + + + ++ + +1 2 ... [ ]x
E En
zz
mm
0 0
2
2
0 059
1
1
′ = +
+ + + ++ + + +
,
log]
[
β β ββ β β
1RedL 2RedL RedL
1OxL 2OxL OxL
[L] [L] [L
[L] [L] L]
��
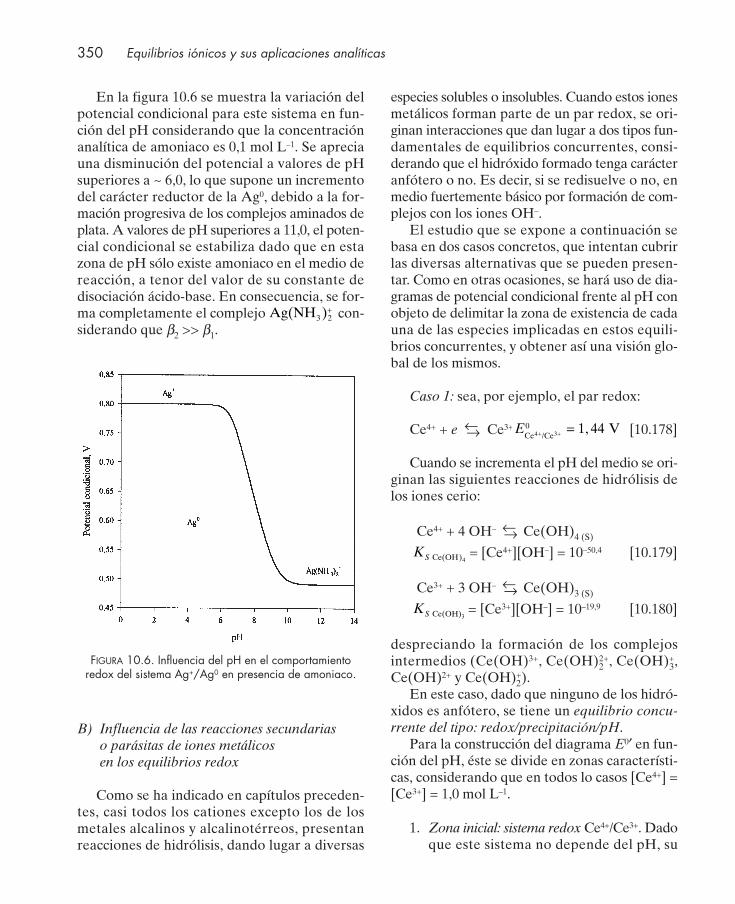
En la figura 10.6 se muestra la variación delpotencial condicional para este sistema en fun-ción del pH considerando que la concentraciónanalítica de amoniaco es 0,1 mol L–1. Se apreciauna disminución del potencial a valores de pHsuperiores a ~ 6,0, lo que supone un incrementodel carácter reductor de la Ag0, debido a la for-mación progresiva de los complejos aminados deplata. A valores de pH superiores a 11,0, el poten-cial condicional se estabiliza dado que en estazona de pH sólo existe amoniaco en el medio dereacción, a tenor del valor de su constante dedisociación ácido-base. En consecuencia, se for-ma completamente el complejo con-siderando que β2 >> β1.
FIGURA 10.6. Influencia del pH en el comportamientoredox del sistema Ag+/Ag0 en presencia de amoniaco.
B) Influencia de las reacciones secundarias o parásitas de iones metálicos en los equilibrios redox
Como se ha indicado en capítulos preceden-tes, casi todos los cationes excepto los de losmetales alcalinos y alcalinotérreos, presentanreacciones de hidrólisis, dando lugar a diversas
especies solubles o insolubles. Cuando estos ionesmetálicos forman parte de un par redox, se ori-ginan interacciones que dan lugar a dos tipos fun-damentales de equilibrios concurrentes, consi-derando que el hidróxido formado tenga carácteranfótero o no. Es decir, si se redisuelve o no, enmedio fuertemente básico por formación de com-plejos con los iones OH–.
El estudio que se expone a continuación sebasa en dos casos concretos, que intentan cubrirlas diversas alternativas que se pueden presen-tar. Como en otras ocasiones, se hará uso de dia-gramas de potencial condicional frente al pH conobjeto de delimitar la zona de existencia de cadauna de las especies implicadas en estos equili-brios concurrentes, y obtener así una visión glo-bal de los mismos.
Caso 1: sea, por ejemplo, el par redox:
Ce4+ + e ←→ Ce3+ [10.178]
Cuando se incrementa el pH del medio se ori-ginan las siguientes reacciones de hidrólisis delos iones cerio:
Ce4+ + 4 OH– ←→ Ce(OH)4 (S)
= [Ce4+][OH–] = 10–50,4 [10.179]
Ce3+ + 3 OH– ←→ Ce(OH)3 (S)
= [Ce3+][OH–] = 10–19,9 [10.180]
despreciando la formación de los complejosintermedios (Ce(OH)3+, Ce(OH)2
2+, Ce(OH)3+,
Ce(OH)2+ y Ce(OH)2+).
En este caso, dado que ninguno de los hidró-xidos es anfótero, se tiene un equilibrio concu-rrente del tipo: redox/precipitación/pH.
Para la construcción del diagrama E0′ en fun-ción del pH, éste se divide en zonas característi-cas, considerando que en todos lo casos [Ce4+] =[Ce3+] = 1,0 mol L–1.
1. Zona inicial: sistema redox Ce4+/Ce3+. Dadoque este sistema no depende del pH, su
KS Ce(OH)3
KS Ce(OH)4
ECe /Ce0
4 3 1, 44 V+ + =
Ag(NH )3 2+
350 Equilibrios iónicos y sus aplicaciones analíticas

potencial condicional será igual al poten-cial normal, E0′ = E0 = 1,44 V, lo que serefleja en el diagrama mediante una líneahorizontal. Este valor de potencial se man-tiene constante hasta que comienza la pre-cipitación del Ce(OH)4, hidróxido másinsoluble.
2. Valores de pH para la precipitación de cadauno de los hidróxidos. De acuerdo con lasexpresiones de los productos de solubili-dad se tiene:
Ce4+:
mol L–1 ⇒ pH = 1,4 [10.181]
Ce3+:
mol L–1 ⇒ pH = 7,37 [10.182]
3. Variación del potencial por precipitaciondel Ce(OH)4. Esta zona está comprendidaentre los valores de pH 1,4 y 7,37 (comien-zo de la precipitación de ambos hidróxi-dos). Dado que es la forma oxidada del parla que está implicada en el equilibrio hete-rogéneo de precipitación, se puede haceruso de la expresión [10.164]
[10.164]
que se transforma en este caso en:
[10.183]
ya que n = 1, m = 1 y z = 4. Al operar y sus-tituyendo el valor de la concentración deion hidroxilo por Kw/[H+] esta ecuaciónqueda como:
E0′ = 1,77 – 0,236 pH [10.184]
Esta expresión corresponde en el dia-grama a una recta de pendiente –0,236 yde ordenada 1,77. En esta zona, el poten-cial disminuye a medida que se incremen-ta el pH del medio.
4. Variación del potencial por la precipitaciónde ambos hidróxidos. A valores de pHsuperiores a 7,37 comienza la precipitacióndel Ce(OH)3, lo que supone la precipita-ción conjunta de ambos hidróxidos. Si sesustituye en la expresión de Nernst, las con-centraciones de los iones Ce4+ y Ce3+ obte-nidas a partir de sus respectivos productosde solubilidad, se tiene:
[10.185]
Esta expresión, que depende del valor delpotencial normal del sistema, de los respectivosproductos de solubilidad y del pH, se corres-ponde con la del potencial condicional de estepar redox, y tras operar queda como:
[10.186]
Al sustituir en esta ecuación los datos numé-ricos, se tiene:
E0′ = 0,467 – 0,059 pH [10.187]
En el diagrama, esta expresión viene repre-sentada por una recta de pendiente –0,059 y deordenada 0,467.
El diagrama se completa con líneas verticalestrazadas a partir de los puntos de intersección que
E E
K
KKS
S
w0 0 059 0 0593
′ = + −+ + +Ce /Ce0 Ce(OH)
Ce(OH)
4 34
H, log , log
[ ]
E
K
K
S
S
= ++ +
−
−
Ce /Ce0
Ce(OH)
Ce(OH)
4 3
OH
OH
0 059
4
3
4
3
, log [ ]
[ ]
E E= + =+ +
+
+Ce /Ce0
4
34 3
[Ce ][Ce ]
0 059, log
E E KS0 0 059
1 10 059 4
1 1
4′ = +
×−
××
+ +
−
Ce /Ce0
Ce(OH)4 3
OH
,log
–,
log[ ]
E En m
Kz
n mS0 0 0 059 0 059′ = +
×− ×
×,
log,
log ][A
,−= 6 6310 [ ]
[ ] ,
,
OHCe
Ce(OH)3−+
−
= = =KS
43
19 9
310
1 0
−= ,12 610 [OH ]
CeCe(OH)
44−
+
−
= = =KS
[ ] ,
,4
50 4
410
1 0
Capítulo 10: Equilibrios de oxidación-reducción 351
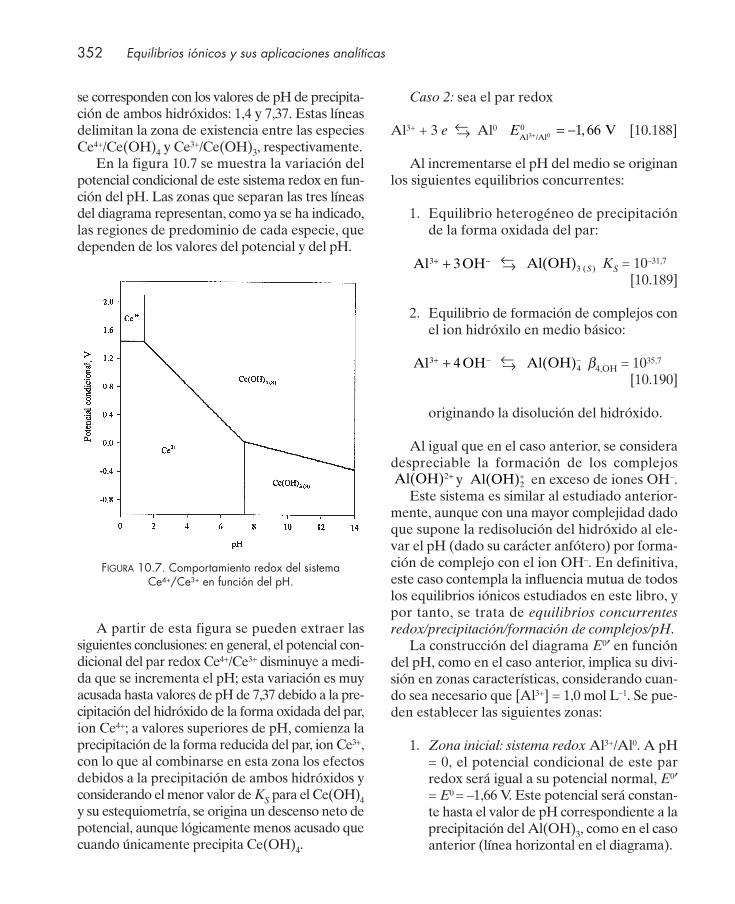
se corresponden con los valores de pH de precipita-ción de ambos hidróxidos: 1,4 y 7,37. Estas líneasdelimitan la zona de existencia entre las especiesCe4+/Ce(OH)4 y Ce3+/Ce(OH)3, respectivamente.
En la figura 10.7 se muestra la variación delpotencial condicional de este sistema redox en fun-ción del pH. Las zonas que separan las tres líneasdel diagrama representan, como ya se ha indicado,las regiones de predominio de cada especie, quedependen de los valores del potencial y del pH.
FIGURA 10.7. Comportamiento redox del sistemaCe4+/Ce3+ en función del pH.
A partir de esta figura se pueden extraer lassiguientes conclusiones: en general, el potencial con-dicional del par redox Ce4+/Ce3+ disminuye a medi-da que se incrementa el pH; esta variación es muyacusada hasta valores de pH de 7,37 debido a la pre-cipitación del hidróxido de la forma oxidada del par,ion Ce4+; a valores superiores de pH, comienza laprecipitación de la forma reducida del par, ion Ce3+,con lo que al combinarse en esta zona los efectosdebidos a la precipitación de ambos hidróxidos yconsiderando el menor valor de KS para el Ce(OH)4y su estequiometría, se origina un descenso neto depotencial, aunque lógicamente menos acusado quecuando únicamente precipita Ce(OH)4.
Caso 2: sea el par redox
Al3+ + 3 e ←→ Al0 [10.188]
Al incrementarse el pH del medio se originanlos siguientes equilibrios concurrentes:
1. Equilibrio heterogéneo de precipitaciónde la forma oxidada del par:
←→ KS = 10–31,7
[10.189]
2. Equilibrio de formación de complejos conel ion hidróxilo en medio básico:
←→ β4,OH = 1035,7
[10.190]
originando la disolución del hidróxido.
Al igual que en el caso anterior, se consideradespreciable la formación de los complejos
y en exceso de iones OH–.Este sistema es similar al estudiado anterior-
mente, aunque con una mayor complejidad dadoque supone la redisolución del hidróxido al ele-var el pH (dado su carácter anfótero) por forma-ción de complejo con el ion OH–. En definitiva,este caso contempla la influencia mutua de todoslos equilibrios iónicos estudiados en este libro, ypor tanto, se trata de equilibrios concurrentesredox/precipitación/formación de complejos/pH.
La construcción del diagrama E0′ en funcióndel pH, como en el caso anterior, implica su divi-sión en zonas características, considerando cuan-do sea necesario que [Al3+] = 1,0 mol L–1. Se pue-den establecer las siguientes zonas:
1. Zona inicial: sistema redox Al3+/Al0. A pH= 0, el potencial condicional de este parredox será igual a su potencial normal, E0′= E0 = –1,66 V. Este potencial será constan-te hasta el valor de pH correspondiente a laprecipitación del Al(OH)3, como en el casoanterior (línea horizontal en el diagrama).
Al(OH)2+
Al(OH)2+
Al(OH)4−
Al 4 OH3+ −+
Al(OH) ( )3 S Al 3OH3+ −+
EAl /Al0
3 0 V+ = −1 66,
352 Equilibrios iónicos y sus aplicaciones analíticas

2. Valor de pH para la precipitación delAl(OH)3. De acuerdo con la expresión delproducto de solubilidad se tiene:
= 10–10,57 mol L–1 ⇒ pH = 3,43[10.191]
De acuerdo con este valor de pH, lazona inicial (línea horizontal en el diagra-ma) está comprendida entre los valores depH 0,0 y 3,43.
3. Variación del potencial por precipitacion delAl(OH)3. A partir de pH = 3,43 comienzala precipitación del hidróxido de aluminio,y considerando que es la forma oxidada delpar redox la que está implicada en el equi-librio heterogéneo de precipitación, se pue-de hacer uso de la expresión [10.164]:
[10.164]
que en este caso se puede escribir como:
[10.192]
ya que n = 3, m = 1 y z = 3. Al operar, estaecuación queda como:
[10.193]
Esta expresión se corresponde en eldiagrama con una recta de pendiente–0,059 y de ordenada –1,457 V. En estazona, el potencial disminuye a medida quese incrementa el pH del medio.
4. Variación del potencial por formación deespecies hidroxiladas. En medio básico seorigina la redisolución del hidróxido de alu-minio por formación de la especie hidroxi-lada dada en la ecuación [10.190]. El poten-cial condicional en esta zona, conforme a laexpresión [10.144], viene dado por:
[10.194]
Al sustituir los datos numéricos en estaexpresión, y si se considera que β4,OH[OH–] >> 1, dado que se trabaja en mediobásico, ésta se transforma en:
[10.195]
que se corresponde con la expresión deuna recta de pendiente –0,079 y ordenada–1,261 V.
5. Valor de pH correspondiente a la redisolu-ción del Al(OH)3. Este valor de pH es elcorrespondiente al punto de intersecciónde las rectas correspondientes a la preci-pitación del hidróxido y formación de laespecie hidroxilada, dadas en las ecuacio-nes [10.193] y [10.195], respectivamente.En estas condiciones se tiene:
–1,457 – 0,059 pH = –1,261 – 0,079 pH [10.196]
de donde se obtiene un valor de pH de 9,85.
FIGURA 10.8. Influencia de las reacciones secundarias deformación de compuestos hidroxilados del ion Al3+ en el sis-
tema redox Al3+/Al0.
E0 1 261 0 079′ = − −, , pH
E E0 0 0593
1′ = − ++−
Al /Al0
4,OH4
3 0 [OH ] ),
log( β
E0 1 457 0 059′ = − −, , pH
E E KS0 0 059
3 10 059 3
3 1′ = +
×− ×
×+−
Al /Al0
3 0 [OH ],
log,
log
E En m
Kz
n mS0 0 0 059 0 059′ = +
×− ×
×,
log,
log[ ]A
[[ ] ,
,
OH ]Al3
−+
−
= = =KS3
31 7
310
1 0
Capítulo 10: Equilibrios de oxidación-reducción 353

Finalmente, y como en el caso anterior, el dia-grama se completa con líneas verticales trazadasa partir de los puntos de intersección, y que deli-mitan las zonas de existencia entre las especies:Al3+/Al(OH)3 y Al(OH)3/Al(OH)–
4.En la figura 10.8 se muestra el diagrama del
potencial condicional frente al pH para este sis-
tema redox. Como en el caso anterior, el parredox incrementa su carácter reductor a medidaque se incrementa el pH del medio, bien por laformación del hidróxido insoluble (pH = 3,43 –9,85) o por la redisolución del mismo debido a laformación de la especie Al(OH)–
4, a valores depH superiores a 9,85.
354 Equilibrios iónicos y sus aplicaciones analíticas
10.1. ¿En qué tipo de celda electroquímica la reacción redox transcurre espontáneamente?
[ ] Celda electrolítica[ ] Celda galvánica[ ] Reacción química en disolución
10.2. Indicar sin son verdaderas (V) o falsas (F) las siguientes afirmaciones:
[ ] En una celda electrolítica el sentido de la reacción redox es igual al que se obtiene cuando se desarrollaen disolución
[ ] El cátodo pasa a ser ánodo cuando se pasa de una celda galvánica a electrolítica[ ] En una celda electroquímica el sentido de la reacción redox siempre es diferente al que se obtiene en
disolución
10.3. El potencial en una pila de concentración se establece en base a:
[ ] Potencial estándar de los pares redox implicados en la misma[ ] Presencia de un equilibrio concurrente en una de las celdas[ ] Constante de equilibrio de la reacción redox
10.4. Indicar sin son verdaderas (V) o falsas (F) las siguientes afirmaciones:
[ ] En el ánodo de una celda galvánica se origina un proceso de oxidación[ ] En el ánodo de una celda electrolítica se origina un proceso de reducción[ ] El paso de Fe3+ a Fe2+ en una celda galvánica se origina en el ánodo[ ] El paso de a Mn2+ en una celda electrolítica se origina en el cátodo
10.5. ¿De qué depende la constante de equilibrio de una reacción redox?
[ ] Concentración de las especies implicadas[ ] Potenciales estándar o condicionales de los pares involucrados en la reacción[ ] Capacidad reguladora redox de la disolución resultante[ ] Número de electrones intercambiados por ambos pares redox
10.6. Si se consideran tres especies de un mismo ion con diferentes estados de oxidación: M+, M2+ y M3+, indicarcuál es la especie: 1) más oxidante; 2) más reductora; 3) el anfolito. Escribir la reacción de dismutación.
10.7. El vanadio presenta los siguientes estados de oxidación en disolución: V2+, V3+,VO2+ y . Indicar lasespecies que actúan como: 1) polioxidantes; 2) polireductores; 3) anfolitos.
VO3−
MnO4−
Cuestiones

10.8. Escribir el sistema redox del agua en medio alcalino frente a oxidantes muy potentes.
10.9. ¿Qué es un diagrama potencial–pH?
10.10. Si a una disolución de un par redox se añaden las especies que se indican a continuación, indicar (marcan-do con una X en la casilla correspondiente) en cada caso cómo varía el potencial de la disolución:
Potencial
Especie añadida al par Aumenta Disminuye
Precipita con la forma oxidada del par
Forma complejos con la forma reducida del par
Ácido fuerte (en el caso de que exista diferente proporción de oxígeno entre las formas del par)
Precipita con la forma reducida del par
10.11. La capacidad oxidante de un par redox se incrementa cuando
[ ] Precipita la forma reducida[ ] Forma complejos la forma oxidada[ ] Al subir el pH (H+ implicados en el par redox)[ ] Al bajar el pH (H+ implicados en el par redox)
10.12. Indicar, marcando con una X, en qué condiciones el potencial condicional de un par redox será superior asu potencial estándar.
[ ] La forma oxidada forma complejos con un ligando[ ] En presencia de un ligando que forma complejos con la forma reducida[ ] La forma oxidada participa en un equilibrio heterogéneo de precipitación[ ] Se añade a la disolución un reactivo que precipita con la forma reducida
Capítulo 10: Equilibrios de oxidación-reducción 355
• Objetivo pedagógicoEjercitar al alumno en el manejo de los conceptosrelacionados con el cálculo del potencial de dife-rentes tipos de celda electroquímica, de constantesde equilibrio de reacciones redox, concentracionesde especies implicadas en el mismo y potencialescondicionales, derivadas de la influencia de di-versas reacciones secundarias sobre el equilibrioredox.
Potencial de celdas electroquímicas
1. Calcular el potencial que adquiere una lámina de pla-ta metálica introducida en una disolución de sulfatode plata 10–2 mol L–1.E0
Ag+/Ag0 = 0,80 V.
El sulfato de plata es una sal que se disocia total-mente en agua conforme al equilibrio:
Seminarios: problemas numéricos

356 Equilibrios iónicos y sus aplicaciones analíticas
Ag2SO4 → + 2 Ag+
originando iones Ag+, a concentración 2 × 10–2 mol L–1
dada la concentración inicial de la disolución. Al intro-ducir una lámina de plata en esta disolución se tieneel sistema redox Ag+/Ag0 cuyo potencial viene dadopor la expresión de Nernst:
E = E0 + 0,059 log [Ag+] = = 0,80 + 0,059 log (2 × 10–2) = 0,70 V
2. Calcular el potencial que adquiere un electrodo deplatino al sumergirlo en una disolución: a) de sulfa-to de cadmio 10–3 mol L–1 y b) preparada al mezclar10 mL de cloruro de Fe3+ 10–3 mol L–1 con 30 mL desal de Mohr, Fe(NH4)2 (SO4)2, 2,0 × 10–3 mol L–1.E0
Cd2+/Cd0 = – 0,403 V; E0Fe3+/Fe2+ = 0,771 V.
Sol.: a) – 0,491 V; b) 0,725 V.
3. Calcular el potencial estándar del par redoxCr2O7
2–/2Cr2+ a pH = 0, si los potenciales estándar delos sistemas Cr2O7
2–/2Cr3+ y Cr3+/Cr2+ son 1,33 y –0,41 V,respectivamente.
La reacción de la que se desea calcular el E0 es:
Cr2O72– + 14 H+ + 8 e ←→ 2 Cr2+ + 7 H2O
y la correspondiente ecuación de Nernst puede escri-birse como:
Los datos disponibles son las reacciones:
Cr2O72– + 14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O
Cr3+ + e ←→ Cr2+
y sus potenciales estándar correspondientes son:
y
No hay ningún inconveniente en suponer que lareacción de reducción del ion dicromato a Cr2+ es elresultado de una combinación de las reacciones ante-riores que transcurren de forma consecutiva. Es decir:
+ 14 H+ + 6 e ←→ 2 Cr3+ + 7 H2OCr3+ + e ←→ Cr2+
+ 14 H+ + 8 e ←→ 2 Cr2+ + 7 H2O
La solución, por tanto, consiste en combinar losdatos que se conocen: los valores del potencial están-dar E0
Cr2O72–/2Cr3+, E0
Cr3+/Cr2+ y las correspondientes ecua-ciones de Nernst de manera que se obtenga unaexpresión equivalente a la indicada para el potencialdel sistema Cr2O7
2–/2Cr2+. Así, en el equilibrio sepodrá escribir:
Si se multiplica por 6 la ecuación de Nernst corres-pondiente al sistema :
y por 2 la ecuación de Nernst del sistema Cr3+/Cr2+:
y se suman estas ecuaciones, resulta:
o bien:
Simplificando esta ecuación y comparándola conla correspondiente a la ecuación de Nernst para elsistema es evidente que:
EE E
Cr O /2Cr
Cr O /2Cr0
Cr /Cr
2 72 2
2 72 3 3
V
− +
− + + +
=+
=
= × − × =
0
06 2
86 1 33 2 0 41
80 895
2
, ,,
Cr O /2 Cr2 72 2− +
EE E
=+
+
+ ×
− + + +
− +
+
+
+
6 2
80 059
8
0Cr O /2Cr Cr /Cr
0
2 72 14
3 2
3 2
2 2
2 72 3 3 2
[Cr O ][H ][Cr ]
[Cr ][Cr ]
,
log
8 6 0 059
2
E E
E
= + +
+ +
− +
+ +
− +
+
+
+
Cr O /2Cr0 2 7
2 14
3 2
Cr /Cr0
3 2
2 2
2 72 3
3 2
[Cr O ][H ][Cr ]
0,059log[Cr ][Cr ]
, log
2 0 059E= ++ +
+
+Cr / Cr0
3 2
2 23 2
[Cr ][Cr ]
, log
2 2 0 059 20E E= + × =+ +
+
+Cr / Cr
3
23 2
[Cr ][Cr ]
, log
6 6 0 059E E= +− +
− +
+Cr O /2 Cr0 2 7
2 14
3 22 72 3
[Cr O ][H ][Cr ]
, log
Cr O /2 Cr2 72 3− +
E E= ++ +
+
+Cr / Cr0
3
23 2
[Cr ][Cr ]
0 059, log
E E= +− +
− +
+Cr O /2 Cr0 2 7
2 14
3 22 72 3
0, 0596
log[Cr O ][H ]
[Cr ]
Cr O2 72−
Cr O2 72−
ECr /Cr0
3 2 0, 41 V+ + = −ECr O /2 Cr0
2 72 3 1, 33 V− + =
E E= +− +
− +
+Cr O /2 Cr0 2 7
2 14
2 22 72 2
[Cr O ][H ][Cr ]
0 0598
,log
SO42−

4. Calcular el potencial de un electrodo de platino brillantesumergido en una disolución que es 5,0 × 10–5 mol L–1
en KMnO4 y 10–2 mol L–1 en Mn(NO3)2 a pH = 0.E0
MnO4–/Mn2+ = 1,51 V.
Sol.: 1,48 V.
5. Se mezclan 50 mL de una disolución 0,1 mol L–1 dedicromato de potasio con 30 mL de disolución 0,2 molL–1 de sulfato de Fe2+. La mezcla se diluye a 100 mL,se ajusta a pH = 0 con ácido clorhídrico y se introdu-ce un electrodo de platino brillante. ¿Qué potencialadquiere este electrodo?
1,33 V; 0,771 V.
Cuando se mezclan las disoluciones indicadas seorigina la siguiente reacción redox conforme a lospares redox implicados en la misma:
Cr2O72– + 14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O
Fe2+ ←→ Fe3+ + e
Cr2O72– + 6 Fe2+ + 14 H+ ←→ 2 Cr3+ + 6 Fe3+ + 7 H2O
Inicialmente se adicionan 50 × 0,1 = 5,0 mmoles dedicromato y 30 × 0,2 = 6,0 mmoles de sal de Fe2+. Deacuerdo con la estequiometría de la reacción redox,6,0 mmoles de Fe2+ reaccionan con 1,0 mmol deCr2O7
2–, y, por tanto, en las condiciones del problemase tiene en el equilibrio 5,0 – 1,0 = 4,0 mmoles deCr2O7
2– sin reaccionar y se forman 2,0 mmoles de Cr3+.Al diluir esta disolución a 100 mL, las concentracio-nes respectivas de estas especies son:
0,04 mol L–1
[Cr3+] = 0,02 mol L–1
El potencial que adquiere el electrodo de platinoal introducirlo en esta disolución viene dado por la expresión de Nernst aplicada al sistema redoxCr2O7
2–/2 Cr3+:
quedando finalmente al considerar la condiciónimpuesta de que pH = 0:
= 1,35 V
6. Se mezclan 100 mL de una disolución 0,2 mol L–1
de sulfato de Fe2+ con 25 mL de una disolución 0,1
mol L–1 de permanganato de potasio. La mezcla sediluye a 200 mL y la acidez se ajusta a cero. Calcularel potencial que adquiere un electrodo de platino bri-llante al introducirlo en la disolución resultante.E0
MnO4–/Mn2+ = 1,51 V; E0
Fe3+/Fe2+ = 0,771 V.Sol.: 0,784 V.
7. Calcular el valor del potencial redox de las disolu-ciones obtenidas al mezclar volúmenes iguales de lassiguientes disoluciones: 1) Cr2+ 1,0 mol L–1 + Fe3+ 0,2mol L–1; 2) Fe2+ 0,5 mol L–1 + Tl3+ 1,0 mol L–1.E0
Cr3+/Cr2+ = –0,41 V; E0Fe3+/Fe2+ = 0,771 V; E0
Tl3/Tl+ = 1,28 V.Sol.: 1) –0,445 V; 2) 1,294 V.
8. ¿Qué relación existe entre las concentraciones de Fe2+
y Cu2+ cuando la celda galvánica
Fe | Fe2+ (1,0 mol L–1) || Cu2+ (1,0 mol L–1) | Cu
disminuye su potencial inicial a la mitad?E0
Fe2+/Fe0 = – 0,440 V; E0Cu2+/Cu0 = 0,337 V.
El potencial de esta celda galvánica, conforme a laecuación [10.14], viene dado por: ECelda = ECátodo –EÁnodo actuando como cátodo el sistema del cobre ycomo ánodo el del hierro. En estas condiciones:
ECátodo = E0 + log [Cu2+] = 0,337 +
+ log (1,0) = 0,337 V
EÁnodo = E0 + log [Fe2+] = –0,440 +
= log (1,0) = –0,440 V
ECelda = ECátodo – EÁnodo = 0,337 – (–0,440) = 0,777 V
Al disminuir este potencial a la mitad de su valor,se tiene:
= 0,337 + log [Cu2+] –
–
y al operar:
– 0,777 =
0 0592
,log
[Cu ][Fe ]
2
2
+
+
0 7772
,
− +
+0 440
0 0592
,,
log[Fe ]2
0 0592
,
0 7772
,
0 0592
,
0 0592
,
0 0592
,
0 0592
,
E = +1 33
0 0596
0 040 02 2
,,
log[ , ][ , ]
E E= +
− +
+0 0, 059
6log
[Cr O ][H ][Cr ]
2 72 14
3 2
2 mmoles100 ml
=
[Cr O ]4 mmoles
100 ml2 72− = =
EFe /Fe0
3 2+ + =ECr O /2Cr0
2 72 3− + =
Capítulo 10: Equilibrios de oxidación-reducción 357

de donde finalmente:
= 1,48 × 1013
9. Calcular la variación que experimenta el potencialde la celda galvánica:
Pt pH2 | (1 atm) aH+ = 1 || CuSO4 (100 mL 0,1 mol L–1) | Cu
cuando se añade al compartimento catódico 50 mLde disolución de Sn2+ 10–2 mol L–1.E0
Cu2+/Cu0 = 0,337 V; E0Sn4+/Sn2+ = 0,154 V.
Sol.: el potencial disminuye en 1,92%.
10. En el compartimento anódico de una celda electro-química se añaden 25 mL de nitrato de plomo 0,1 molL–1, mientras que en el comportamiento catódico seadicionan 50 mL de nitrato de cinc 0,2 mol L–1. Indi-car el sentido de la reacción Pb0 + Zn2+ ←→ Pb2+ + Zn0
y calcular el potencial que hay que aplicar a esta cel-da para conseguir invertir de la misma.E0
Pb2/Pb0 = – 0,126 V; E0Zn2+/Zn0 = – 0,763 V.
Conforme a la ecuación [10.14] el potencial de estacelda viene dado por:
ECelda = –0,763 + log (0,2) –
– = – 0,629 V
De acuerdo con el valor de potencial negativo obte-nido para esta celda, la reacción:
Pb0 + Zn2+ ←→ Pb2+ + Zn0
se encuentra desplazada hacia la izquierda y parainvertir el sentido de la misma se ha de hacer uso deuna celda electrolítica aplicando un potencial positi-vo de 0,629 V o superior.
11. En una celda electrolítica el compartimento catódi-co contiene una disolución de nitrato de cinc 0,1 molL–1, mientras que en el anódico se sitúa una disolu-ción de nitrato de cadmio 0,2 mol L–1. Calcular elpotencial que hay que aplicar a esta celda para queproduzca la reacción: Cd2+ + Zn0 ←→ Cd0 + Zn2+.E0
Cu2+/Cu0 = – 0,403 V; E0Zn2+/Zn0 = – 0,763 V.
Sol.: + 0,369 V.
Constantes de equilibrio y especies implicadas
12. Calcular el valor de X en la reacción que se indica enestado de equilibrio:
Cu0 + 2 Ag+ (X mol L–1) ←→ Cu2+ (0,1 mol L–1) + 2 Ag0
E0Ag+/Ag0 = 0,80 V; E0
Cu2+/Cu0 = 0,337 V.
Cuando se alcanza el equilibrio en una reacciónredox, el potencial de los sistemas redox implica-dos en la misma son lógicamente iguales. Por tanto:Eequilibrio = Ecobre = Eplata. En estas condiciones se tiene:
+ log [Cu2+]equilibrio =
= + 0,059 log [Ag+]equilibrio
0,337 + log (0,1) = 0,80 + 0,059 log X
de donde X = 4,5 × 10–9 mol L–1.
13. Calcular la concentración de Fe2+ presente en una diso-lución obtenida al mezclar volúmenes iguales de diso-luciones de Fe2+ 0,1 mol L–1 y de Ce4+ 0,3 mol L–1.E0
Fe3+/Fe2+ = 0,771 V; E0Ce4+/Ce3+ = 1,44 V.
Sol.: 1,14 × 10–13 mol L–1.
14. Calcular la constante de equilibrio de la reacción apH = 0:
2 + 5 Sn2+ + 16 H+ ←→ 2 Mn2+ + 5 Sn4+ + 8 H2O
E0MnO4
–/Mn2+ = 1,51 V; E0Sn4+/Sn2+ = 0,154 V.
La constante de equilibrio de una reacción redoxviene dada por la ecuación [10.40]:
en la que el subíndice 1 corresponde a la especie queactúa como oxidante y el 2 a la que actúa como reduc-tor. A tenor de los potenciales normales de los sistemasredox implicados en esta reacción, el sistema del man-ganeso actúa como oxidante ( → Mn2+ ganando5 electrones) y el del estaño como reductor (Sn2+ → Sn4+
cediendo 2 electrones). En estas condiciones:
log
, ,,
,K =−
× =1 51 0 154
0 0595 2 229 8
MnO4−
log,
KE E
n n=−01 02
1 20 059
MnO4−
0 0592
,
EAg /Ag0
0+
0 0592
, ECu /Cu
02 0+
− +
0 126
0 0592
0 1,,
log( , )
0 0592
,
[Fe ][Cu ]
2
2
+
+
358 Equilibrios iónicos y sus aplicaciones analíticas

de donde K = 6,3 × 10229. Este elevado valor de K nosindica la gran tendencia que tiene el permanganatopara oxidar a las disoluciones de Sn2+.
15. Calcular la constante de equilibrio de las siguientesreacciones:
1) 2 Fe3+ + 2 I– ←→ 2 Fe2+ + I22) 2 Fe3+ + Sn2+ ←→ 2 Fe2+ + Sn4+
E0Fe3+/Fe2+ = 0,771 V; E0
I4/2I– = 0,535 V; E0Sn4+/Sn2+ = 0,154 V.
Sol.: 1) log K = 8,0; 2) log K = 20,91.
16. A una disolución de sulfato de cadmio 10–2 mol L–1
se le añade polvo de hierro en exceso. Una vez quese alcanza el equilibrio en la reacción:
Fe0 + Cd2+ ←→ Fe2+ + Cd0
calcular las concentraciones de las especies Fe2+ y Cd2+
en disolución.E0
Fe2+/Fe0 = – 0,440 V; E0Cd2+/Cd0 = – 0,403 V.
La constante de equilibrio de la reacción redoxen estudio se calcula haciendo uso de la ecuación[10.40], considerando que en este caso n1 = n2 = 2.Así, se tiene:
o bien:
de donde:
= 17,8
Para este sistema se puede establecer el siguientebalance de materia tras alcanzarse el equilibrio:
[Cd2+]Inicial = 10–2 = [Cd2+]Equilibrio + [Fe2+]Formado
y al sustituir en la expresión anterior se tiene:
de donde:
[Cd2+ ] = 5,3 × 10–4 mol L–1
[Fe2+] = 9,47 × 10–3 mol L–1
17. Se añade aluminio metal en exceso a una disolución0,2 mol L–1 de sulfato de níquel y se origina la reac-ción:
2 Al0 + 3 Ni2+ ←→ 2 Al3+ + 3 Ni0
Calcular la concentración de Al3+ en la disolucióncuando se alcanza el equilibrio.E0
Al3+/Al0 = – 1,66 V; E0Ni2+/Ni0 = – 0,23 V.
Sol.: [Al3+] = 0,133 mol L–1.
18. Calcular el sentido y la constante de equilibrio de lareacción:
Tl3+ + 2 Fe2+ ←→ Tl+ + 2 Fe3+
y la concentración del ion Fe2+ cuando se alcanza elequilibrio en una disolución formada por la mezclade 25 mL de una disolución 2 × 10–2 mol L–1 de Fe2+
y 25 mL de disolución 10–2 mol L–1 de Tl3+.E0
Fe3+/Fe2+ = 0,771 V; E0Tl3+/Tl+ = 1,28 V.
Sol.: la reacción está desplazada hacia a la derecha;K = 1017,25; [Fe2+] = 1,78 × 10–8 mol L–1.
19. Se añade un exceso de aluminio metal a una diso-lución de Cu2+ 0,3 mol L–1; a) ¿Cuál sera la concen-tración de Cu2+ cuando se alcanza el equilibrio?, b)¿Cuál será la constante de equilibrio de la reacción?:
2 Al0 + 3 Cu2+ ←→ 2 Al3+ + 3 Cu0
E0Al3+/Al0 = – 1,66 V; E0
Cu2+/Cu0 = 0,337 V.Sol: a) 6,76 × 10–69 mol L–1; b) log K = 203,08.
20. Calcular la constante de equilibrio de la reacción
2 Cu+ ←→ Cu2+ + Cu0
y las concentraciones de las especies Cu2+ y Cu+ enuna disolución 10–2 mol L–1 de ion Cu+.E0
Cu2+/Cu+ = 0,153 V; E0Cu+/Cu0 = 0,521 V.
Se trata de la reacción de dismutación de un anfo-lito como es el ion cuproso. De acuerdo con la ecua-ción [10.40], la constante de equilibrio viene dada por:
de donde K = 1,7 × 106.Si se parte de una disolución 10–2 mol L–1 de Cu+,
al dismutarse en este ion en disolución acuosa, se pue-
log, ,
,,K =
−=
0 521 0 1530 059
6 2310 [Cd ]
[Cd ]
2 2
2
− +
+
−= 17 8,
[Fe ][Cd ]
2
2
+
+
log , logK = =+
+1 25
[Fe ][Cd ]
2
2
log, ( , )
,,K =
− − −=
0 403 0 4400 059
2 1 25
Capítulo 10: Equilibrios de oxidación-reducción 359

de establecer el siguiente balance de masas, si se deno-mina X a la concentración de Cu+ que reacciona:
[Cu+]Equilibrio = 10–2 – X [Cu2+]Equilibrio = X/2
Sustituyendo estas concentraciones en la expresiónde la constante de equilibrio se tiene:
de donde: [Cu2+] ≈ 5,0 × 10–3 mol L–1 y [Cu+] = 5,4 ×10–5 mol L–1.
21. Calcular la constante de dismutación del complejoAuCl2
– en HCl 2,0 mol L–1 y la concentración del com-plejo AuCl2
– en equilibrio con una disolución 5,0 ×10–4 mol L–1 de AuCl2
– en HCl 2,0 mol L–1 y en exce-so de oro metálico.E0
AuCl2–/Au0 = 1,11 V; E0
AuCl4–/AuCl2
– = 0,99 V.Sol.: K = 1,1 × 104; [AuCl4
–] = 3,4 × 10–7 mol L–1.
Equilibrios concurrentes
22. Calcular el potencial redox de las disoluciones obte-nidas al mezclar volúmenes iguales de las siguientesdisoluciones: a) Fe2+ 0,1 mol L–1 y MnO4
– 0,02 mol L–1
(pH = 1); b) Fe2+ 0,12 mol L–1 y Cr2O72– 0,02 mol L–1
(pH = 1).E0
MnO4–/Mn2+ = 1,51 V; E0
Cr2O72–/2Cr3+ = 1,33 V; E0
Fe3+/Fe2+ = 0771.
a) Al mezclar disoluciones de Fe2+ y se ori-gina la siguiente reacción redox:
Fe2+ ←→ Fe3+ + e (× 5)MnO4
– + 8 H+ + 5 e ←→ Mn2+ + 4 H2O
MnO4– + 5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O
Dado que se mezclan volúmenes iguales deambas disoluciones, las concentraciones inicialesde ambas especies serán:
[Fe2+] = mol L–1
[ ] = mol L–1
cuya relación es justamente la estequiométrica.En estas condiciones cuando se alcanza el equili-brio se cumple que:
[Fe2+] = 5 [MnO4–]
[Fe3+] = 5 [Mn2+]
En el equilibrio, el potencial, E, viene dado por:
Si se multiplica por 5 la expresión del potencialcorrespondiente al par redox /Mn2+ y sesuma con la del sistema Fe3+/Fe2+, se tiene:
Al sustituir en esta expresión, la condición deequilibrio dada anteriormente, ésta se transfor-ma en:
El cálculo del potencial a partir de esta expre-sión se realiza fácilmente sin más que sustituir losvalores de los respectivos potenciales estándar y considerar que se trabaja a pH = 1, esto es [H+]= 10–1 mol L–1. Así, se tiene:
b) En este caso, al mezclar volúmenes iguales de lasdisoluciones de Fe2+ y Cr2O7
2– se origina la reac-ción redox:
Fe2+ ←→ Fe3+ + e (× 6)Cr2O7
2– + 14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O
Cr2O72– + 6 Fe2+ + 14 H+ ←→ 2 Cr3+ + 6 Fe3+ + 7 H2O
cuyas concentraciones iniciales son:
[Fe2+] = mol L–1
[ ] = mol L–1
0 022
0 01,
,= Cr O2 72−
0 122
0 06,
,=
E =
× ++ × × =−5 1 51 0 771
60 059
68 10 1 31, , ,
log( ) , 08 V
E
E E=
++ × ×
− + + + +5
60 059
68MnO /Mn Fe /Fe
04
2 3 2H
,log[ ]
6 5 0 059 8E E E= + +− + + +
+MnO /Mn0
Fe /Fe0
42 3 2 H ], log[
6 5 059E E E= + +− + + +
− + +
+ +MnO /Mn0
Fe /Fe0 4
8 3
2 242 3 2 0
[MnO ][H ] [Fe ][Mn ][Fe ]
, log
MnO4−
E E= ++ +
+
+Fe /Fe0
3
23 2 0,059 log[Fe ][Fe ]
E E= +− +
− +
+MnO /Mn0 4
8
242
0, 0595
log[MnO ][H ]
[Mn ]
0 022
0 01,
,=MnO4−
0 12
0 05,
,=
MnO4−
K
XX
= × = =−
+
+ −1 7 10
210
62 2
,/
( )[Cu ][Cu ]
2
2
360 Equilibrios iónicos y sus aplicaciones analíticas

y como en el caso anterior, se encuentran en rela-ción estequiométrica, por tanto, la reacción estáen equilibrio. En estas condiciones, se tiene:
[Fe2+] = 6 [ ] [Fe3+] = [Cr3+]
De manera similar al apartado anterior, el poten-cial, E, en el equilibrio se determina como sigue:
Si se multiplica por 6 la expresión del potencialcorrespondiente al par redox /2Cr3+ y sesuma con el del sistema Fe3+/Fe2+, se tiene:
Al sustituir las concentraciones de los iones Fe3+
y Fe2+, de acuerdo con la condición de equilibriodada anteriormente, en el término logarítmico deesta expresión, ésta se transforma en:
y el potencial vendrá dado por:
o bien:
dado que se trabaja a pH = 1,0.A diferencia del apartado anterior, la expresión
del potencial en el equilibrio depende de la con-centración de ion Cr3+. Si se denomina X a la con-centración de ion dicromato que queda sin reac-cionar tras alcanzarse el equilibrio, se puedeestablecer:
[Fe2+]equilibrio = 6X
[ ]equilibrio = X[Fe3+]equilibrio = 6(0,01 – X)[Cr3+]equilibrio = 2(0,01 – X)
Si se admite que esta reacción redox presentauna alta constante de equilibrio, lo que es presu-mible a tenor de la gran diferencia entre los poten-ciales estándar de ambos pares redox, X se pue-de despreciar frente a 0,01 en la expresión de laconcentración de ion Cr3+ en el equilibrio, y portanto, el potencial en el equilibrio será:
23. Calcular el potencial (frente al electrodo estándarnormal de hidrógeno) de un electrodo de platinobrillante sumergido en una disolución en la que[MnO4
–] = 5,0 × 10–5 mol L–1, [Mn2+] = 1,0 × 10–2 molL–1 y [H+] = 4,0 × 10–2 mol L–1.E0
MnO4–/Mn2+ = 1,51 V.
Sol.: 1,35 V.
24. El potencial del par redox , a pH = 0, esde 1,33 V. ¿Oxidará el dicromato al ion yoduro a pH6,0?E0
I2/2I– = 0,535 V.Sol.: no.
25. Se mezclan disoluciones de los iones yodato y yodu-ro, de tal forma que su concentración analítica sea 0,01mol L–1, con una cantidad limitada de ácido fuerte 10–3
mol L–1. ¿Cuál será el pH final de esta disolución?E0
I2/2I– = 0,535 V; E02IO3
–/I2= 1,19 V.
Al mezclar disoluciones de yodato y yoduro tienelugar la reacción redox:
2 + 12 H+ + 10 e ←→ I2 + 6 H2O2 I– ←→ I2 + 2 e (× 5)
2 + 10 I– + 12 H+ ←→ 6 I2 + 6 H2O
El potencial de esta disolución cuando se alcanzael equilibrio, viene dado por:
E E E= + = +− −− −I /2I
0 22 I /2I
0 25
102 2
[I ][I ]
[I ][I ]
0 0592
0 05910
,log
,log
E E= +−
− +
2IO /I0 3
2 12
263 2
[IO ] [H ][I ]
0 05910,
log
IO3−
IO3−
Cr O /2Cr2 72 3− +
E = + ×
×=−
−1 25 8 42 10
102 0 02
1314
, , log,
,144 V
Cr O2 72−
= + × −−
+1 25 8 42 10 3, , log
102[Cr ]
14
3
E =
× ++ =
+
+
6 1 33 0 7717
0 0597
, , ,log
[H ]2[Cr ]
14
3
E
E E=
++
− + + + +
+
6
70 059
7Cr O /2Cr Fe /Fe
0 14
32 7
2 3 3 2 [H ]2[Cr ]
,log
[Cr O ][H ] 3[Cr ][Cr ] 6[Cr O ]
[H ]2[Cr ]
2 72 14 3
3 22 7
2
14
3
− + +
+ −
+
+=
[Cr O ][H ] [Fe ][Cr ] [Fe ]
2 72 14 3
3 2 2
− + +
+ +=
7 6 0 0590E E E= + +− + + +
− + +
+ +Cr O /2Cr Fe /Fe0 2 7
2 14 3
3 2 22 72 3 3 2
[Cr O ][H ] [Fe ][Cr ] [Fe ]
, log
Cr O2 72−
E E= ++ +
+
+Fe /Fe0
3
23 2 0,059 log[Fe ][Fe ]
E E= +− +
− +
+Cr O /2Cr0 2 7
2 14
3 22 72 3
0, 0596
log[Cr O ][H ]
[Cr ]
62
Cr O2 72−
Capítulo 10: Equilibrios de oxidación-reducción 361

Al combinar ambas expresiones se llega a:
En esta expresión se conocen los valores de lospotenciales estándar y se pueden calcular las con-centraciones de las diferentes especies en el equi-librio, salvo la [H+] que es la incógnita del proble-ma.
Si se parte de disoluciones de los iones yodato yyoduro 0,01 mol L–1 en un medio ácido, donde la con-centración de iones H+ es 10–3 mol L–1, de acuerdocon la estequiometría de la reacción redox, los ionesH+ están en defecto y por tanto controlan el desa-rrollo de la reacción. En estas condiciones se puedeestablecer:
9,83 × 10–3 mol L–1
9,16 × 10–3 mol L–1
5,0 × 10–4 mol L–1
Al sustituir estas concentraciones en la expresiónanterior, se tiene:
de donde pH = 8,86.El valor obtenido tiene sólo interés matemático ya
que para que la reacción se produzca es necesario quela concentración de protones sea mayor, es decir esnecesario pH ácido.
26. Se disuelven 0,2942 g de dicromato de potasio en 100mL de agua y se ajusta el pH a un valor de 0,6. Si seborbotea sobre esta disolución una corriente de H2S,¿Cuál será el pH una vez que todo el dicromato seha reducido a ion Cr3+?E0
Cr2O72–/2Cr3+ = 1,33 V; E0
S0/H2S= 0,141 V.
Pesos atómicos: Cr = 51,0; O = 16,0; K = 39,1.Sol.: 0,76.
27. Calcular el potencial de un electrodo de cobre intro-ducido en la disolución que resulta al mezclar: 1) 25mL de CuSO4 0,05 mol L–1 con 50 mL de NH3 0,5 mol
L–1; 2) 50 mL de CuSO4 0,05 mol L–1 con 20 mL deNH3 0,1 mol L–1.E0
Cu2+/Cu0 = 0,337 V; (log β4 = 12,6).Sol.: 1) – 0,019 V; 2) 0,291 V.
28. Calcular el potencial de un electrodo de plata in-troducido en la disolución que resulta al mezclar 50 mL de AgNO3 0,1 mol L–1 y 100 mL de NH3 0,5mol L–1.E0
Ag+/Ag0 = 0,80 V; (log β2 = 7,24).Sol.: 0,353 V.
29. Se tiene una disolución que es 10–2 mol L–1 en Fe3+ y1,0 mol L–1 en SCN–. ¿Qué potencial habrá que apli-car a la misma para que desaparezca la coloraciónroja del complejo FeSCN2+, si éste es perceptible aconcentraciones superiores a 10–5,5 mol L–1?E0
Fe3+/Fe2+ = 0,771 V; FeSCN+ (log β = 2,3).
En este sistema se tienen los siguientes equilibriosconcurrentes:
Fe3+ + e ←→ Fe2+
Fe3+ + SCN– ←→ FeSCN2+
Para hacer desaparecer la coloración roja del com-plejo FeSCN2+ su concentración debe ser inferior a10–5,5 mol L–1. Por tanto, al sustituir esta concentra-ción en la expresión de la constante, se puede calcu-lar la concentración de Fe3+ en equilibrio que cum-ple esta condición, ya que la concentración de ionSCN– está en exceso. Así:
de donde [Fe3+] = 10–7,8 mol L–1, por lo que todo elFe3+ inicial se ha transformado prácticamente en Fe2+
debido al cambio de potencial.Si se sustituye esta concentración en el balance de
materia para el hierro:
CFe = 10–2 = [Fe3+] + [Fe2+] = 10–7,8 + [Fe2+]
se tiene que [Fe2+] ≅ 10–2 mol L–1.En estas condiciones se puede calcular el potencial
que hay que aplicar a la disolución para hacer desa-parecer la coloración roja del complejo, sin más quehacer uso de la ecuación de Nernst:
102 3, = =×
+
+ −
−
+
[FeSCN ][Fe ][SCN ]
10[Fe ] 1, 0
2
3
5,5
3
β12 30= =
+
+ −
[FeSCN ][Fe ][SCN ]
12
3,
EFe /Fe0
3 2 0, 771 V+ + =
Ag(NH )3 2+
Cu(NH )3 42+
( , , ),
log( , )
( , ) ( , ) [1 19 0 535 10
0 0595 0 10
9 16 10 9 83 10
4 6
3 10 3 2 12
− ×=
×× ×
−
− − +H ]
[I ]10
22 formado
3
= =−
[I ]Equilibrio−
−
= − × =0 015 10
6
3
,
[IO ] 0,0110
63 Equilibrio
3−
−
= − =
( )
,log
E E2IO /I0
I /2I0
26
103
2 123 2 2 [I ]
[I ] [IO ] [H ]
− −− ×=
− − +
10
0 059
362 Equilibrios iónicos y sus aplicaciones analíticas

Habrá que aplicar, por tanto, un potencial inferiora 0,429 V.
30. Calcular el potencial de un electrodo de níquel intro-ducido en la disolución que resulta al mezclar volú-menes iguales de nitrato de níquel 0,001 mol L–1 y eti-lendiamina (en) 1,0 mol L–1.E0
Ni2+/Ni0 = – 0,23 V; Ni(en)x2+ (log β1 = 7,7; log β2 = 14,0;
log β3 = 18,6).Sol.: –0,849 V.
31. El potencial condicional del sistema Ni2+/Ni0 en pre-sencia de ion cianuro 0,1 mol L–1 es –0,749 V. Calcularla constante de formación del complejo .E0
Ni2+/Ni0 = – 0,23 V.Sol.: 1021,6.
32. El reconocimiento del ion cianuro se basa en el des-prendimiento de HCN por adición de NaHCO3 a ladisolución y posterior oxidación de la bencidina (B)a azul de bencidina (AB), tras la adición de Cu2+ almedio de reacción. Justificar el fundamento de esteensayo de reconocimiento del ion cianuro.E0
Cu2+/Cu+ = 0,153 V; E0AB/B = 0,76 V; log αCu2+/(CN–) = 1,5;
log αCu+/(CN–) = 15,4.
Este ensayo, de reconocimiento del ion cianuro, sebasa en la oxidación de la bencidina por el ion Cu2+
de acuerdo con la siguiente reacción redox:
bencidina + 2 Cu2+ ←→ azul de bencidina + 2 Cu+
que no es espontánea debido al escaso carácter oxi-dante del Cu2+, de acuerdo con los respectivos poten-ciales estándar:
= 0,153 – 0,76 = –0,607 V
Sin embargo, en presencia de ion cianuro se incre-menta considerablemente el carácter oxidante delCu2+, debido a que el ion Cu+ forma complejos muchomás estables con dicho ligando. La ecuación delpotencial condicional para el equilibrio concurrentede complejación se puede expresar como:
y en este caso concreto, por:
Al sustituir en la expresión anterior los datos numé-ricos dados por el problema, el potencial condicionaldel sistema Cu2+/Cu+ toma el valor de:
En estas condiciones, la reacción de oxidación dela bencidina es espontánea, ya que el valor del poten-cial condicional es superior al potencial estándar delsistema azul de bencidina/bencidina, 0,76 V. Dadoque la reacción se origina debido a la presencia deion cianuro, se puede utilizar como ensayo de reco-nocimiento para esta especie.
33. El potencial estándar del par redox Fe3+/Fe2+ se incre-menta en 0,419 V en presencia de 1,10-fenantrolina (fen)en exceso. Calcular la constante de formación globaldel complejo .E0
Fe3+/Fe2+ = 0,771 V; (log β3 = 21,2).Sol.: 1014,1.
34. Se mezclan 50 mL de una disolución 0,2 mol L–1 deyoduro con 50 mL de una disolución 0,02 mol L–1 deCu2+. Calcular las concentraciones de las especies for-madas cuando se alcanza el equilibrio.E0
Cu2+/CuI = 0,86 V; E0I2/2I– = 0,535 V; CuI (KS = 10–12).
Cuando se mezclan disoluciones de yoduro y Cu2+
se origina la reacción redox:
2 Cu2+ + 4 I– ←→ 2 CuI(S) + I2
en la que están implicados los pares redox:
Cu2+ + I– + e ←→ CuI(S)
I2 + 2 e ←→ 2 I–
Al mezclar 50 mL de una disolución 0,2 mol L–1 deyoduro con 50 mL de una disolución 0,02 mol L–1
de Cu2+ se tiene:
[I–]inicial = mol L–1
[Cu2+]inicial= mol L–1
50 0 02100
0 01×
=,
,
50 0 2100
0 1×
=,
,
E
I /2I02
0, 535 V− = ECu /CuI
02 0, 86 V+ =
Fe(fen)32+
Fe(fen)33+
E0 0 153 0 059 15 4 1 5 0 97′ = + − =, , ( , , ) , 3V
E E0 0 059′ = ++ +
+ −
+ −Cu /Cu0 Cu (CN )
Cu (CN )
2
2
, logαα
E E
n Lz
z
mm
0 0 1 22
1 22
0 059 1
1′ = +
+ + + ++ + + +
,log
[ ] [ ]
[ ] [ ] [ ]
β β ββ β β
RedL RedL RedL
OxL OxL OxL
[L] L L
L L
��
∆E E E= −+ +Cu /Cu0
AB/B0
2
Ni(CN)42−
+ =−
−9 V0 059
1010
0 427 8
2, log ,
,
E E= + = ++ +
+
+Fe /Fe0
3
23 2
[Fe ][Fe ]
0 059 0 771, log ,
Capítulo 10: Equilibrios de oxidación-reducción 363

por lo que el ion yoduro se encuentra en exceso deacuerdo con la estequiometría de la reacción.
Si se denomina X a la concentración de ion Cu2+
una vez que se alcanza el equilibrio, se pueden esta-blecer los siguientes balances de materia:
[I2]equilibrio = mol L–1
[I–]equilibrio = 0,1 – 2(0,01 – X) ≅ 0,08 mol L–1
Para poder calcular el valor de X, [Cu2+]equilibrio, seha de conocer el valor de la constante de equilibriode esta reacción:
y sustituir en ella las concentraciones de yoduro yyodo, ya calculadas. De acuerdo con la expresión[10.40]:
el valor de dicha constante será:
11,02
Una vez conocido el valor de esta constante, sepuede calcular fácilmente la concentración de ionCu2+ en el equilibrio. Así,
y, por tanto, [Cu2+] = 3,41 × 10–5 mol L–1.
35. El sistema redox formado por la reacción M2+ + 2 e←→ M(S) tiene un potencial estándar de 0,0118V. Lacelda galvánica constituida por:
M | MX2 (S) , X– (0,4 mol L–1) || αΗ+ =1 | pΗ2(1 atm) Pt
presenta un potencial de 0,205V. Calcular el valor delproducto de solubilidad del compuesto poco solubleMX2.Sol.: 7,15 × 10–9.
36. Representar gráficamente la variación del poten-cial del sistema Ag+/Ag0 en función del pH para
una disolución que inicialmente es 0,1 mol L–1 en elion Ag+.E0
Ag+/Ag0 = 0,80 V; Ag(OH) (KS = 10–7,7).
37. Los productos de solubilidad de los sulfuros de mer-curio y plata son 10–53,5 y 10–48,2, respectivamente, sien-do el ion sulfuro oxidable (S/S2–) con un potencialestándar de 0,508 V en medio básico. 1) ¿Se puedenseparar ambos sulfuros por oxidación sucesiva, siestán presentes a una concentración de 10–2 mol L–1?2) ¿Cuál es el intervalo de potencial que permite oxi-dar entre el 1% y el 100% de cada uno de ellos?
En este sistema se tienen los siguientes equilibrios:
HgS(S)←→ Hg2+ + S2– KS = [Hg2+][S2–] = 10–53,5
Ag2S(S)←→ 2 Ag+ + S2– KS = [Ag+]2[S2–] = 10–48,2
S0 + 2 e ←→ S2–
Si se considera en primer lugar el sulfuro de mer-curio, dada su mayor insolubilidad, la concentraciónde ion Hg2+ en disolución cuando se ha disuelto poroxidación el 1% y 100%, será:
Se disuelve el 1% el HgS. La concentración de ionHg2+ en disolución en este caso será:
[Hg2+] = mol L–1
y por tanto:
E = − − =
−
−0 508
0 0592
1010
0 9553 5
4,
,log ,
,
2 V
E E
KS= − = − −−−
+S/S0 2
22 S ]Hg
0 0592
0 5080 059
2,
log[ ,,
log[ ]
10
1100
102 4− −=
ES/S0
2 0, 508 V− = −
K = = =
×+ −
−
+
[I ][Cu ] [I ]
5, 0 10[Cu ] (0, 08)
22 2 4
3
2 2 41011 02,
log, ,
,K =
−× =
0 86 0 5350 059
1 2
log,
KE E
n n=−1
020
1 20 059
K =+ −
[I ][Cu ] [I ]
22 2 4
0 012
0 012
5 0 10 3, ,,
−≈ = × −X
364 Equilibrios iónicos y sus aplicaciones analíticas

Se disuelve el 100% el HgS. De forma similar al casoanterior se tiene:
[Hg2+] = mol L–1
De acuerdo con estos valores de potenciales, sepuede indicar que variando el potencial entre 0,952y 1,011 V, se consigue la total disolución del HgS poroxidación del ion sulfuro.
En el caso del sulfuro de plata, se sigue un trata-miento similar, que se resume a continuación:
Se disuelve el 1% el Ag2S:
[Ag+] = 2 × mol L–1
Se disuelve el 100% del Ag2S:
[Ag+] = mol L–1
En este caso, variando el potencial en el intervalo0,696 – 0,814 V se consigue la total disolución del sul-furo de plata. Hay que hacer notar que por la propiaestequiometría del sulfuro, [Ag+] = 2,0 × 10–2 mol L–1.
En resumen, ambos sulfuros se pueden separar conun control adecuado del potencial. Inicialmente enel intervalo 0,696 – 0,814 se disuelve el Ag2S, y pos-teriormente se eleva el potencial entre 0,952 y 1,011para conseguir la total disolución del HgS.
38. La concentración analítica de los iones Fe3+ e Fe2+ enuna disolución a pH = 4,0 es 0,1 mol L–1. ¿Será posi-ble oxidar con esta disolución otra 0,1 mol L–1 de ionyoduro?E0
Fe3+/Fe2+ = 0,771 V; E0I2/2I– = 0,535 V; Fe(OH)3 (KS =
10–39,5); Fe(OH)2 (KS 10–15,1).Sol.: no.
39. La determinación yodométrica de cobre se basa en laadición de yoduro de potasio a una disolución de Cu2+
y posterior valoración con tiosulfato de sodio. Demos-
trar que la presencia de Fe3+ no interfiere en la deter-minación de cobre si la disolución contiene además ionfluoruro a una concentración de 1,0 mol L–1.E0
Cu2+/CuI = 0,86 V; E0I2/2I– = 0,535 V; E0
Fe3+/Fe2+ = 0,771 V; log αFe3+/(Fe–) = 0,86.
Como se ha indicado en el problema 34, la determi-nación yodométrica de cobre se basa en la reacción:
2 Cu2+ + 4 I– ←→ 2 CuI(S) + I2
Sin embargo, si existe Fe3+ en la disolución, se pue-de originar la siguiente reacción:
2 Fe3+ + 2 I– ←→ I2 + 2 Fe2+
en la que están implicados los pares redox:
Fe3+ + e ←→ Fe2+
I2 + 2 e ←→ 2 I–
Es evidente que la presencia de Fe3+ es una inter-ferencia en esta determinación: reacciona con el ionyoduro y origina errores por defecto.
Cuando se añade ion fluoruro a la disolución, se for-man complejos florurados de diferente estequiometría,sólo con el ion Fe3+. En estas condiciones, el potencialcondicional del sistema Fe3+/Fe2+ vendrá dado por:
de acuerdo con la expresión [10.144].Al sustituir los valores dados por el problema en
la expresión anterior se tiene:
Este potencial es mucho menor que el potencialestándar del sistema I2/2I– (0,535 V), por lo que lareacción entre los iones Fe3+ y yoduro no es espon-tánea, y en consecuencia el ion Fe3+ no interfiere enla determinación yodométrica de Cu2+ si existe ionfluoruro en el medio de reacción.
40. Demostrar que el yodo en disolución acuosa se dis-muta totalmente en presencia de ion Ag+ por forma-ción de AgI y AgIO3 poco solubles. ¿Cómo influyeel pH en esta reacción de dismutación?E0
I2/2I– = 0,535 V; E02IO3
–/I2= 1,19 V; AgI (KS = 10–16,1);
AgIO3 (KS = 10–7,5).Sol.: Kdismutación = 10–32,5. Se favorece en medio ácido.
E0 0 771 0 059 15 52 0 14′ = − × = −, , , , 5V
E E0 059′ = − ×+ + + −Fe /Fe0
Fe (F )3 2 30, logα
E
I /2I02
0, 535 V− =
EFe /Fe0
3 2 0, 771 V+ + =
E = − −×
=−
−0 5080 059
210
2 100 814
48 2
2 2,
,log
( ),
,
V
2 10100100
2 102 2× = ×− −
E =×
=– , –,
log( )
,– ,
–0 508
0 0592
102 10
0 69648 2
4 2 V
10
1100
2 102 4− −= ×
E = − − =
−
−0 508
0 0592
1010
1 01153 5
2,
,log ,
,
V
10
100100
102 2− −=
Capítulo 10: Equilibrios de oxidación-reducción 365

41. Calcular el potencial de un electrodo de cobre metalintroducido en una disolución 0,1 mol L–1 de sulfatode cobre que es 0,3 mol L–1 en EDTA y cuyo pH semantiene en 5,0.E0
Cu2+/Cu0 = 0,337 V; CuY2– (log K = 16,3); EDTA (log β 1H
= 10,2; log β2H = 16,3; log β3
H = 19,0; log β4H = 21,0).
Se trata de un equilibrio múltiple que contemplala influencia del pH y formación de complejos sobreun sistema redox. El tratamiento, de acuerdo con loexpuesto en el apartado 10.12.4 implica, inicialmen-te, el cálculo de la constante condicional de forma-ción del complejo CuY2–. Esta constante condicional,viene dada por la expresión:
siendo el coeficiente de reacción secundaria delEDTA con el ion hidrógeno, αY(H), de acuerdo conlo expuesto en el capítulo 5:
Al sustituir los valores dados en el problema, setiene:
y, por tanto:
En estas condiciones, el problema se simplifica alconsiderar la influencia de un equilibrio de forma-ción de complejos sobre el sistema redox Cu2+/Cu0,con las siguientes características:
[EDTA] = 0,3 mol L–1 = 1010
[Cu2+] = 0,1 mol L–1
Antes de añadir EDTA a la disolución de Cu2+, elpotencial de acuerdo con la expresión de Nernst, vie-ne dado por:
Cuando se adiciona EDTA, se puede establecer elsiguiente balance de materia:
CEDTA = 0,3 = [EDTA] + [CuY2–]
CCu = 0,1 = [Cu2+] + [CuY2–]
A tenor del valor de la constante condicional delcomplejo CuY2–, se puede despreciar [Cu2+] en elbalance de materia y, por tanto, [CuY2–] = 0,1 mol L–1.Si se sustituye este valor en el balance de materia delEDTA, se tiene:
CEDTA = 0,3 = [EDTA] + 0,1
de donde [EDTA] = 0,2 mol L–1.Al sustituir estas concentraciones en la expresión
de la constante de formación del complejo CuY2–, sepuede calcular la concentración de ion Cu2+ en elequilibrio y a partir de aquí, el potencial que adquie-re la disolución:
de donde [Cu2+] = 5,0 × 10–11 mol L–1. Al aplicar laecuación de Nernst:
De acuerdo con estos resultados, se puede indicarque el potencial disminuye en 0,307 – 0,003 = 0,274 V.
42. A una disolución de la sal de Cu2+ se le añade exce-so de yoduro de potasio y se forma yodo y un preci-pitado de yoduro de Cu+. A continuación se le aña-de EDTA. Explicar los fenómenos que tienen lugarde forma cualitativa y cuantitativa, calculando en esteúltimo caso la constante de la reacción global.E0
Cu2+/Cu+ = 0,153 V; E0I2/2I– = 0,535 V; CuI (KS = 10–12);
CuY2– (log K = 16,3).Sol.: 1010,8.
43. Calcular la constante de disociación del ácido débilHA si el potencial suministrado por la siguiente pilade concentración es de 0,346 V.
+ Pt pΗ2(1 atm) | HCl (0,1 mol L–1) ||
|| HA (10–3 mol L–1) | pΗ2(1 atm) Pt –
= + × =− 3 V0 3370 059
25 0 10 0 0311,
,log( , ) ,
E E= + =+
+Cu /Cu0 2
2 0 Cu ]0 059
2,
log[
KCuY
2
210
22
[CuY ][Cu ][EDTA]
100,1
[Cu ] 0, 2− = = =
×
−
+ +
E E= + =
= + =
++
Cu /Cu0 2
2 0 Cu
7 V
0 0592
0 3370 059
20 1 0 30
,log[ ]
,,
log ( , ) ,
ECu /Cu0
2 0 0, 337 V+ =
KCuY2−
KCuY
16,36,32 10
110
10−′ = = 10
+ × + × ≅− −10 10 10 10 1019 15 21 20 6 33,
αY(H) = + × + × +− −1 10 10 10 1010 2 5 16 3 10, ,
α β β β βY(H) 1H
2H
3H
4H[H [H ] [H [H= + + + ++ + + +1 2 3 4] ] ]
K KCuY CuY
Y(H)
2 2− −′ = 1
α
366 Equilibrios iónicos y sus aplicaciones analíticas

La pila de concentración que se utiliza para deter-minar la constante de acidez del protolito débil HA,contiene en cada uno de sus compartimentos el parredox 2H+/H2, correspondiente al electrodo de refe-rencia, y por tanto su potencial estándar es cero. Elpotencial de esta pila de concentración (celda gal-vánica) responde a la expresión:
ECelda = E+ – E–
siendo:
De acuerdo con el potencial suministrado por estapila se tiene:
0,346 = –0,059 – 0,059 log [H+]
de donde [H+] = 10–6,86 mol L–1.Este valor de pH es el que presenta la disolución
10–3 mol L–1 del ácido débil HA debido a su disocia-ción:
HA ←→ A– + H+
En estas condiciones se pueden establecer lossiguientes balances de materia:
[A–] = [H+] = 10–6,86 mol L–1
[HA] = [HA]inicial – [H+] = 10–3 – 10–6,86 ≅ 10–3 mol L–1
Al sustituir estas concentraciones en la expresiónde la constante de disociación se tiene:
44. Calcular el cambio de potencial que se produce en lasiguiente pila de concentración:
+ Pt pΗ2(1 atm) | HCl (50 mL 0,1 mol L–1) ||
|| HA (50 mL 0,1 mol L–1) | pΗ2(1 atm) Pt –
cuando se adicionan 20 mL de una disolución deNaOH 0,1 mol L–1 al compartimento que contiene elprotolito débil.HA (pKa = 3,0).Sol.: 48,6 mV.
45. ¿Cuál es el potencial que presenta la siguiente pilade concentración:
+ Pt pΗ2(1 atm) | HCl (100 mL 0,1 mol L–1) ||
|| HA (50 mL 0,1 mol L–1) | pΗ2(1 atm) Pt –
si se añaden 50 mL de disolución de NaOH 0,1 mol L–1
al compartimento que contiene al protolito fuerte.HA (pKa = 5,0).Sol.: 89,9 mV.
46. La pila de concentración formada por
+ Ag | AgNO3 (5 × 10–2 mol L–1) |||| AgNO3 (5×10–2 mol L–1), NH3 (0,2 mol L–1) | Ag –
presenta un potencial de 0,309 V. Calcular la cons-tante de formación del complejo .E0
Ag+/Ag0 = 0,80 V.
El potencial de esta pila de concentración, dadopor la expresión, ECelda = E+ – E–, se basa en el parredox Ag+/Ag0, y por tanto en ambos compartimen-tos responde a la concentración de ion Ag+ presenteen la disolución, de acuerdo con la expresión deNernst:
Cuando se añade amoniaco, la concentración deion Ag+ es la que queda libre tras la formación delcomplejo aminado de plata:
Ag+ + 2 NH3←→
cuya constante de formación viene dada por:
De acuerdo con las concentraciones iniciales de ionplata y amoniaco, se pueden establecer los siguien-tes balances de materia:
Si se considera que toda la plata inicial forma prác-ticamente complejo con el amoniaco, se puede admi-
CNH 3 3 230, 2 [NH ] 2[Ag(NH ) ]= = + +
CAg 3 20, 05 [Ag ] [Ag(NH ) ]+ = = ++ +
β2 =
+
+
[Ag(NH ) ][Ag ][NH ]
3 2
32
Ag(NH )3 2+
E E= + =
= ++
+
+Ag /Ag0
0 [Ag ]
0,8 0,059 log [Ag ]
0 059, log
Ag(NH )3 2+
Ka = = =
+[A ][H ][HA]
– ( )– ,
–– ,10
1010
6 86 2
310 72
Ka =
− +[A ][H ][HA]
E E
E E
++
−+
= + =
= + = −= +
+
+
2H /H0
2H /H0
2
2
0,059 log[H ]
9 V
0,059 log[H ]
, , log( , ) ,
0 0 0 059 0 1 0 05
Capítulo 10: Equilibrios de oxidación-reducción 367

tir que [Ag(NH3)2+] ≅ = 0,05 mol L–1. Al sustituir
esta condición en el balance de materia del amonia-co se tiene:
de donde [NH3] = 0,1 mol L–1.Para poder calcular la constante de formación de
este complejo es necesario determinar la concentra-ción de ion Ag+ en el equilibrio. Como ya se ha indi-cado, esto se lleva a cabo mediante el uso de la pilade concentración.
De acuerdo con el potencial de esta pila se tiene:
ECelda = 0,309 = E+ – E–E+ = 0,80 + 0,059 log (5 × 10–2) = 0,723 V
y de aquí:
E– = 0,80 – 0,309 = 0,414 = 0,8 + 0,059 log [Ag+]
por tanto [Ag+] = 2,88 × 10–7 mol L–1.Para el cálculo de la constante de formación del
complejo aminado de plata, basta con sustituir losvalores de concentraciones calculados para cada unade las especies implicadas en la misma. Así resulta:
47. Calcular la constante de formación global del com-plejo Cu(NH3)4
2+ si la pila de concentración forma-da por:
+ Cu | CuSO4 (0,1 mol L–1) |||| CuSO4 (0,1 mol L–1), NH3 (5,0 mol L–1) | Cu –
proporciona un potencial de 449 mV.E0
Cu2+/Cu0 = 0,337 V.Sol.: 3,98 × 1012.
48. Calcular el potencial que presenta la pila de concen-tración formada por:
+ Pt pΗ2(1 atm) | HCl (10–2 mol L–1) ||
|| M(OH)2 (S) , [M2+] (10–3 mol L–1) | pΗ2(1 atm) Pt –
M(OH)2 (KS = 10–12,2).
En esta pila de concentración, el potencial de lacelda se basa en el sistema redox 2H+/H2, y por tan-to, responde a la concentración de iones H+ en cadauno de los compartimentos. De acuerdo con la expre-sión de Nernst para este sistema redox, se tiene:
y de acuerdo con las condiciones dadas en el proble-ma:
Para el cálculo de E– se hace uso de la misma expre-sión, aunque en este caso la [H+] corresponde a la queexiste en el equilibrio de precipitación del hidróxidoM(OH)2:
M(OH)2 (S)←→ M2+ + 2 OH– KS = [M2+] [OH–]2
Sustituyendo la concentración de M2+ dada en elproblema y expresando la concentración de ionesOH– en función de Kw y [H+] , en el producto de solu-bilidad:
se tiene que [H+] = 10–9,4 mol L–1. De acuerdo con esteresultado, el valor de E– será:
y finalmente:
ECelda = E+ – E– = –0,118 – (–0,555) = 0,437 V
49. Calcular el potencial que presenta la pila de concen-tración formada por:
+ Pt pΗ2(1 atm) | HCl (0,1 mol L–1) ||
|| Cu(OH)2 (S), NH3 (0,1 mol L–1) | pΗ2(1 atm) Pt –
E0Cu2+/Cu0 = 0,337 V; Cu(OH)2 (KS = 10–18,8); Cu(NH3)4
2+
(β4 = 1012,6).Sol.: 0,608 V.
E−+ −= + = = −0 0 059 0 059 10 0 5559 4, log , log( ) ,,[H ] V
KS = = ×
− −
−
+10 1012 2 3
2
, 10[H ]
14
E++ −= + = = −0 0 059 0 059 10 0 12, log , log ,[H ] 18V
E Ep
= ++
+
2H /H0
2
H2
2
0,0592
log[H ]
β2
71 74 10= =×
= ×+
+ −
[Ag(NH ) ][Ag ][NH ]
0, 05(2, 88 10 )(0,1)
3 2
32 7 2
,
0 2, = + ×[NH ] 2 0, 053
CAg+
368 Equilibrios iónicos y sus aplicaciones analíticas

11
11.1. Introducción11.2. Curvas de valoración11.3. Indicadores redox11.4. Normalidad versus molaridad
en valoraciones redox11.5. AplicacionesCuestionesSeminarios: problemas numéricos
VALORACIONES DE OXIDACIÓN-
REDUCCIÓN

11.1. Introducción
Las valoraciones de oxidación-reducción, basede la aplicación cuantitativa de los equilibriosredox, conforman un conjunto de métodos clá-sicos de gran interés para la determinación de unelevado número de sustancias con propiedadesredox, tanto inorgánicas como orgánicas. En unavaloración redox se produce la interacción de dospares redox, donde uno corresponde al analito yel otro al valorante. Por ejemplo, si se considerala valoración de una especie con carácter reduc-tor con una disolución patrón o valorante, decarácter oxidante, la reacción de valoración
Ox1 + Red2←→ Red1 + Ox2 [11.1]
permite determinar la cantidad de valorante, Ox1,químicamente equivalente a la cantidad de ana-lito, Red2, presente en la muestra.
La reacción debe cumplir una serie de requi-sitos para su uso adecuado en valoraciones redox,los cuales se especifican a continuación.
Ser cuantitativa. La reacción de valoracióndebe tener una constante de equilibrio alta, loque implica fundamentalmente, de acuerdo conla expresión deducida para dicha constante en elcapítulo anterior:
[11.2]
una diferencia apreciable entre los potencialesestándar o condicionales de los pares redox impli-cados en la misma, aunque la diferencia mínima
necesaria depende de la estequiometría de lareacción (ver cuadro 10.2).
Ser rápida. La lentitud de algunas reaccionesredox priva a la valoración de una de sus carac-terísticas más importante desde un punto de vis-ta práctico. De ahí que sea frecuente el uso decatalizadores, el control de la temperatura y elempleo de las valoraciones por retroceso parasubsanar este importante inconveniente. Dadoque la tabla de potenciales estándar no es másque una guía sobre las condiciones de equilibriotermodinámico, ya que no proporciona informa-ción sobre los aspectos cinéticos de las reaccio-nes redox, el cálculo de la constante de equilibriosegún la expresión [11.2] es un indicativo nece-sario, pero no suficiente para asegurar el empleode dicha reacción como base del método volu-métrico. En definitiva, se han de considerar tan-to los aspectos termodinámicos como cinéticos ala hora de establecer si una determina reacciónredox es adecuada para su uso como reacción devaloración en valoraciones redox.
Ser estequiométrica. Deben evitarse los pro-blemas relacionados con la estequiometría de lasreacciones redox cuando el analito o el valoran-te pueden presentar diversos estados de oxida-ción, lo que podría conducir a que se produjeransimultáneamente varias reacciones redox. Unconocimiento, lo más completo posible, del meca-nismo de estas reacciones, permite ajustar lascondiciones experimentales (los aspectos cinéti-cos juegan un papel de gran importancia en estecontexto) de tal forma que la reacción de valo-ración presente una estequiometría definida, ytambién es importante asegurar que el analito se
log
,K
E En n= −1
020
1 20 059
370 Equilibrios iónicos y sus aplicaciones analíticas
OBJETIVOS
• Describir las características de las valoraciones redox:influencia de variables en el perfil de la curva de valo-ración y estudio y selección de indicadores visualesadecuados.
• Discutir los aspectos teóricos y prácticos, implicadosen los diferentes tipos de valoraciones redox, en fun-ción del reactivo valorante utilizado.
• Mostrar las aplicaciones más importantes de esta téc-nica analítica cuantitativa.

encuentre en un único estado de oxidación, loque en ocasiones requiere un tratamiento pre-vio.
Disponer de un sistema indicador. La princi-pal dificultad en el desarrollo de las valoracio-nes redox fue la disponibilidad de sistemas dedetección visual del punto final. En la actuali-dad, el desarrollo de sistemas de detección delpunto final, tanto en forma de indicadores visua-les como de sistemas instrumentales, ha permi-tido superar este inconveniente. No obstante,son pocos los indicadores visuales que presen-tan un adecuado funcionamiento, si se compara,por ejemplo, con la gran cantidad de indicado-res ácido-base disponibles hoy en día.
A tenor de estas características, se podría pen-sar que las valoraciones redox tienen un campode aplicación algo limitado, sin embargo, esto noes así ya que se pueden determinar una granvariedad de analitos, tanto inorgánicos comoorgánicos, mediante el empleo de diferentes tiposde valorantes y modos de valoración.
Es necesario tener en cuenta que si bien lapreparación y utilización de disoluciones patrónde sustancias oxidantes no presenta grandes pro-blemas, no sucede lo mismo con las de sustanciasreductoras. En primer lugar, estas disolucionesson poco estables, ya que se oxidan por contac-to con el oxígeno atmosférico y su concentracióndisminuye apreciablemente con el tiempo, lo querequiere el uso de dispositivos especiales para suconservación. En segundo lugar, deberían extre-marse las precauciones en el propio proceso devaloración, lo que significa que es aconsejableoperar en atmósfera inerte, con los inconve-nientes que ello acarrea. Finalmente, existenpocos indicadores visuales adecuados y por logeneral se han de utilizar sistemas potenciomé-tricos para la detección del punto final. La con-secuencia es que raramente se utilizan valoran-tes de carácter reductor, por ello su estudio nose contempla en este libro (a excepción del tio-sulfato de sodio o de amonio, empleados enyodometría). Sin embargo, la escasa utilizaciónde valorantes de carácter reductor no significaque no sea posible la determinación de analitos
de carácter oxidante, ya que ello puede realizar-se fácilmente mediante valoraciones por retro-ceso.
11.2. Curvas de valoración
Una curva de valoración redox es la repre-sentación gráfica de la variación del potencialde la disolución, que contiene la especie a valo-rar, en función del volumen de valorante aña-dido, o de la fracción valorada. De acuerdo conla expresión de Nernst, esta variación de poten-cial es función logarítmica del cambio de con-centración de los componentes de la reacciónde valoración, por tanto, como en otras valora-ciones, la curva de valoración tiene forma sig-moidal. Así, durante una gran parte de la valo-ración el potencial de la disolución cambialentamente, pero en la región del punto de equi-valencia el cambio de potencial es muy acusa-do. Como en otros casos, la construcción de lacurva de valoración permite evaluar la exacti-tud con la que, teóricamente, puede llevarse acabo la valoración y también seleccionar el indi-cador más adecuado.
Se describe a continuación la construcción dela curva de valoración teórica basada en la reac-ción redox:
n2 Ox1 + n1 Red2←→ n2 Red1 + n1 Ox2 [11.3]
siendo Red2 el analito y Ox1 el valorante, los cua-les forman parte de los siguientes pares redox:
Ox1 + n1 e ←→ Red1 [11.4]
Ox2 + n2 e ←→ Ox2 [11.5]
Para construir la curva de valoración redox deuna disolución de la especie Red2 de concentra-ción Canalito, de la que se valora un determinadovolumen, Vanalito, con una disolución de valoranteOx1 de concentración Cvalorante, se representa elpotencial de la disolución a valorar E en funciónde la fracción valorada (F = Vvalorante /Vequivalencia),
E20
E10
Capítulo 11: Valoraciones de oxidación-reducción 371

que se hace variar, como en otros casos entre F = 0y F = 2. Para ello, se han de considerar:
1. Los balances de materia del analito y valo-rante en cualquier punto de la valoración:
Canalito = [Red2] + [Ox2] [11.6]
Cvalorante = F × Canalito = [Ox1] + [Red1]
[11.7]
2. La expresión de Nernst para ambos paresredox:
[11.8]
[11.9]
Hay que indicar que por la propia naturalezade estas expresiones, en ellas no influyen los fenó-menos de dilución, ya que el potencial obtenidomediante la expresión de Nernst responde a uncociente de concentraciones.
Para abordar el trazado de una curva de valo-ración redox, se ha seguido un procedimientosimilar al descrito en capítulos anteriores, estoes, descomponer la curva de valoración en variaszonas y desarrollar las ecuaciones adecuadas paracada una de ellas. Así, se consideran zonas deinterés de la curva de valoración:
— Punto inicial (antes de añadir disoluciónde valorante).
— Antes del punto de equivalencia (zona condefecto de valorante).
— Punto de equivalencia.— Después del punto de equivalencia (zona
con exceso de valorante).
En cada una de estas zonas se consideran lasespecies predominantes y se establecen las expre-siones para el cálculo del potencial a partir de la
fracción valorada en cada momento. Dado queen cualquier punto de la valoración, después decada adición de valorante, la reacción transcu-rre hasta que los potenciales de los dos paresredox implicados en la misma se igualan en elequilibrio, el potencial podría calcularse median-te la expresión de Nernst de cualquiera de lospares redox implicados en la reacción de valo-ración, aunque en la práctica sólo es posiblehacerlo mediante la expresión de Nernst corres-pondiente al par redox de la especie predomi-nante en cada zona de la curva de valoración:analito antes del punto de equivalencia y valo-rante después del punto de equivalencia. Final-mente, se discuten ejemplos concretos así comolos factores que influyen sobre el perfil de la cur-va de valoración.
Zona 1: punto inicial(F = 0; Vvalorante = 0)
Antes de iniciar la adición de valorante, sóloestá presente la cantidad inicial de reductor avalorar y puede aceptarse que Canalito ≈ [Red2].La ecuación de Nernst correspondiente a estepar quedaría en la forma:
[11.10]
Si se supone que [Red2] = Canalito, se tiene que[Ox2] = 0, por lo que resulta un potencial igual a– ∞. Es evidente que este resultado carece de sen-tido. En la práctica existirá una pequeña canti-dad de Ox2 en disolución, debido a la reacciónde Red2 con el oxígeno atmosférico. Sin embar-go, es imposible evaluar la [Ox2] y, como conse-cuencia, no se puede calcular el potencial corres-pondiente al punto inicial de una valoraciónredox. Sin embargo, esto no supone un inconve-niente grave, ya que, de ser necesario, podría uti-lizarse el procedimiento que a continuación seindica, utilizable en la zona anterior al punto deequivalencia, para calcular el potencial de unpunto infinitamente próximo al inicial.
E E
n C= +2
0
2
0 059,log
[ ]Ox2
analito
E E
n2 20
2
0 059= + ,log
[Ox ][Red ]
2
2
E E
n1 10
1
0 059= + ,log
[Ox ][Red ]
1
1
nn
2
1
372 Equilibrios iónicos y sus aplicaciones analíticas

Zona 2: antes del punto de equivalencia(0 < F < 1; 0 < Vvalorante< Vequivalencia)
Las cantidades de valorante añadidas son infe-riores a la necesaria para alcanzar el punto deequivalencia y, por tanto, se cumplen las siguien-tes relaciones:
[Ox2] = [Red1] [Ox1] ≈ 0 [11.11]
y dado que los balances de materia se transfor-man en:
Canalito = [Red2] + [Ox2] [11.6]
Cvalorante = F × Canalito =
= [Red1]+[Ox1] ≈ [Red1] = [Ox2]
[11.12]
al combinar las expresiones [11.11], [11.6] y[11.12] se obtiene que
[Ox2] = F × Canalito [11.13]
[Red2] = (1 – F) × Canalito [11.14]
y al sustituir en la expresión de Nernst:
[11.15]
Zona 3: Punto de equivalencia(F = 1; Vvalorante = Vequivalencia)
En este punto la cantidad de valorante aña-dido es estequiométricamente equivalente a lacantidad de analito. El potencial se determinacomo se ha indicado en el apartado 10.7 para elcaso en que existen cantidades estequiométricasde oxidante y de reductor. En el caso más sen-cillo, cuando los iones H+ no intervienen en la
reacción (o cuando su actividad es igual a la uni-dad) y ninguna de las especies implicadas es poli-nuclear, el potencial del punto de equivalenciase calcula mediante la ecuación:
[11.16]
que indica que este potencial depende única-mente de los potenciales estándar de los paresredox implicados y del número de electrones queintervienen y que es independiente del volumende la disolución.
Zona 4: exceso de valorante(F > 1; Vvalorante > Vequivalencia)
Después del punto de equivalencia, práctica-mente todo el analito está oxidado, por lo que secumplen las relaciones siguientes:
[Ox2] = [Red1] [Red2] ≈ 0 [11.17]
por lo que los balances de materia se transfor-man en:
Canalito = [Red2] + [Ox2] ≈ [Ox2] = [Red1]
[11.18]
Cvalorante = F × Canalito = [Ox1] + [Red1]
[11.7]
y al combinar estas expresiones se obtiene:
[Red1] = [Ox2] = Canalito [11.19]
[Ox1] = Canalito × (F – 1) [11.20]
y al sustituir en la expresión [11.8] se tiene:
[11.21] E E
nF= + −1
0
1
0 0591
,log( )
nn
2
1
nn
2
1
nn
2
1
nn
2
1
nn
1
2
nn
1
2
En E n E
n nequivalencia = ++
1 10
2 20
1 2
E E
nF
F= +
−20
2
0 0591
,log
nn
2
1
nn
2
1
nn
1
2
Capítulo 11: Valoraciones de oxidación-reducción 373

En el recuadro 11.1 se desarrollan las expre-siones a utilizar para el trazado de la curva devaloración en dos ejemplos concretos de dife-rente complejidad: valoración de una disoluciónde Fe2+ con Ce4+ y con KMnO4.
En la figura 11.1 se representan las curvascorrespondientes a los ejemplos tratados en el
recuadro 11.1. En ella puede comprobarse que,mientras que en el caso de la curva de valoraciónde Fe2+ con Ce4+ (donde n1 = n2), el punto deequivalencia coincide con el punto de inflexión(como sucedía en todas las curvas vistas hastaahora), esto no ocurre en el caso de la curva devaloración de Fe2+ con MnO–
4 (n1 ≠ n2), donde el
374 Equilibrios iónicos y sus aplicaciones analíticas
RECUADRO 11.1
La determinación de Fe2+ con Ce4+, en la que están impli-cados los siguientes pares redox:
Fe3+ + e ←→ Fe2+
Ce4+ + e ←→ Ce3+
y donde la reacción que tiene lugar es:
Fe2+ + Ce4+ ←→ Fe3+ + Ce3+
constituye el caso más sencillo de una valoración redox,dado que n1 = n2 = 1.
Debido a la estequiometría de la reacción redox, se pue-den establecer los siguientes balances de materia:
Canalito = [Fe2+] + [Fe3+]Cvalorante = F × Canalito = [Ce4+] + [Ce3+]
Antes del punto de equivalencia se puede establecer:
[Fe3+] = [Ce3+] [Ce4+] = 0
Al sustituir en los balances de materia se tiene:
Cvalorante = F × Canalito = [Fe3+] ⇒ [Fe3+] = F × Canalito
Canalito = [Fe2+] + F × Canalito ⇒ [Fe2+] = (1 – F ) Canalito
quedando finalmente la expresión del potencial como:
En el punto de equivalencia el potencial se calculamediante la expresión:
Después del punto de equivalencia se tiene:
[Ce3+] = [Fe3+] [Fe2+] = 0
Al sustituir estas relaciones en los respectivos balancesde materia, se obtiene:
Canalito = [Ce3+]
Cvalorante = F × Canalito = [Ce4+] + Canalito ⇒
⇒ [Ce4+] = (F – 1) Canalito
y la expresión del potencial queda como:
La valoración de una disolución de Fe2+ con perman-ganato de potasio a pH = 0, constituye un ejemplo concretode una valoración redox en la que n1 ≠ n2. En ella estánimplicados los siguientes pares redox:
Fe3+ + e ←→ Fe2+
+ 8 H+ + 5 e ←→ Mn2+ + 4 H2O
siendo la reacción de valoración:
+ 5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2OMnO4−
EMnO /Mn0
42 1, 51V (pH 0)− + = =
MnO4−
EFe /Fe0
3 2 0, 771V+ + =
F= + × −1 44 0 059 1, , log ( )
E EF C
CCe Ce= + ×
− ×=+ +4 3
0 0 0591
/, log
( ) analito
analito
EE E
equivalenciaCe /Ce0
Fe /Fe0
4 3 3 2V=
+= + =
+ + + +
21 44 0 771
21105, , ,
FF
= + ×−
0 771 0 0591
, , log( )
E EF C
F CFe Fe= + ×
×−
=+ +3 20 0 059
1/, log
( )analito
analito
ECe /Ce0
4 3 1, 44 V+ + =
EFe /Fe0
3 2 V+ + = 0 771,
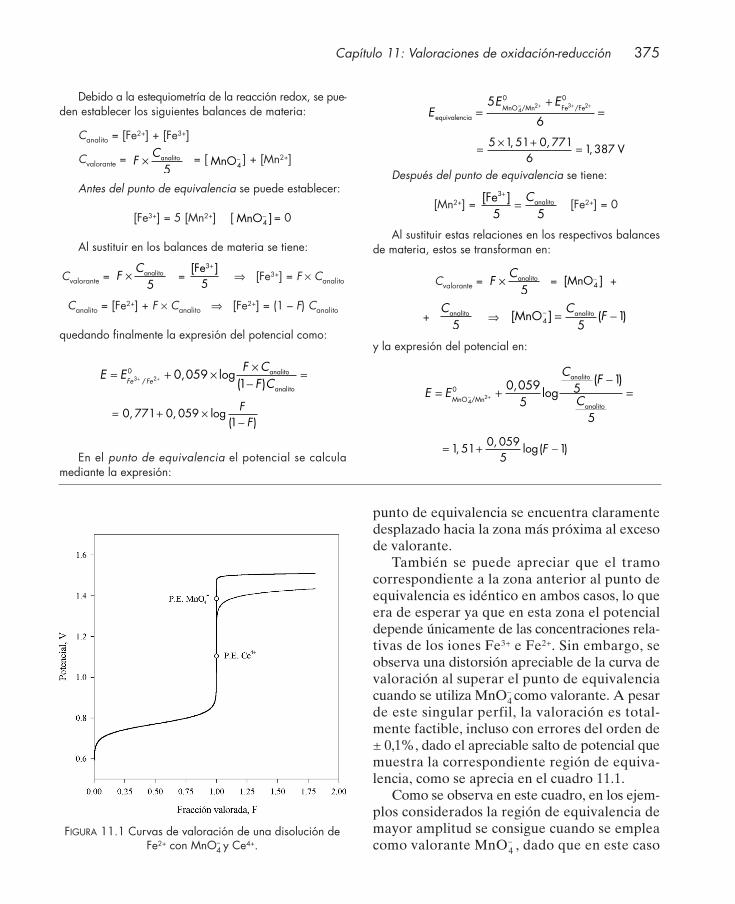
Capítulo 11: Valoraciones de oxidación-reducción 375
Debido a la estequiometría de la reacción redox, se pue-den establecer los siguientes balances de materia:
Canalito = [Fe2+] + [Fe3+]
Cvalorante = = [ ] + [Mn2+]
Antes del punto de equivalencia se puede establecer:
[Fe3+] = 5 [Mn2+] [ = 0
Al sustituir en los balances de materia se tiene:
Cvalorante = = ⇒ [Fe3+] = F × Canalito
Canalito = [Fe2+] + F × Canalito ⇒ [Fe2+] = (1 – F) Canalito
quedando finalmente la expresión del potencial como:
En el punto de equivalencia el potencial se calculamediante la expresión:
Después del punto de equivalencia se tiene:
[Mn2+] = [Fe2+] = 0
Al sustituir estas relaciones en los respectivos balancesde materia, estos se transforman en:
Cvalorante = = +
+ ⇒
y la expresión del potencial en:
F= + −1 51 0 0595
1, , log( )
E E
C F
C= +
−=− +MnO /Mn
0
analito
analito4
25
5
0 0595
1, log( )
[MnO ]54
analito− = −C
F( )1Canalito
5
[MnO ]4−F
C× analito
5
[Fe ]5
3analito
+
=C
5
= × + =5 1 51 0 7716
1 387 V, , ,
EE E
equivalenciaMnO /Mn0
Fe /Fe0
42 3 2
=+
=− + + +5
6
FF
= + ×−
0 771 0 0591
, , log( )
E EF C
F CFe Fe= + ×
×−
=+ +3 20 0 059
1/, log
( )analito
analito
[Fe ]5
3+
FC
× analito
5
MnO ]4−
MnO4−F
C× analito
5
punto de equivalencia se encuentra claramentedesplazado hacia la zona más próxima al excesode valorante.
También se puede apreciar que el tramocorrespondiente a la zona anterior al punto deequivalencia es idéntico en ambos casos, lo queera de esperar ya que en esta zona el potencialdepende únicamente de las concentraciones rela-tivas de los iones Fe3+ e Fe2+. Sin embargo, seobserva una distorsión apreciable de la curva devaloración al superar el punto de equivalenciacuando se utiliza MnO–
4 como valorante. A pesarde este singular perfil, la valoración es total-mente factible, incluso con errores del orden de± 0,1%, dado el apreciable salto de potencial quemuestra la correspondiente región de equiva-lencia, como se aprecia en el cuadro 11.1.
Como se observa en este cuadro, en los ejem-plos considerados la región de equivalencia demayor amplitud se consigue cuando se empleacomo valorante MnO–
4 , dado que en este casoFIGURA 11.1 Curvas de valoración de una disolución de
Fe2+ con MnO–4 y Ce4+.

existe una mayor diferencia entre los potencia-les estándar de los pares redox implicados en lavaloración, lo que implica una mayor constantede equilibrio para la reacción de valoración deacuerdo con la ecuación [11.2].
CUADRO 11.1Valoración de una disolución de Fe2+ utilizando
diferentes oxidantes como valorantes
Potencial (V)
% Valoración MnO–4 Ce4+
0 -- -- 10 0,715 0,715 50 0,771 0,771 90 0,827 0,827 99 0,889 0,889 99,9 0,948 0,948
100 1,386 1,105 100,1 1,475 1,263 101 1,486 1,322 110 1,498 1,381 150 1,506 1,422 200 1,510 1,440
11.2.1. Influencia de reacciones secundarias
Hasta ahora, el trazado de las curvas de valo-ración se ha realizado a partir de los potencialesestándar, es decir, suponiendo que ni el analitoni el valorante presentan reacciones secundariasy que los iones H+ no ejercen ninguna influenciasobre la misma. Sin embargo, es convenienteplantear lo que ocurriría en un caso general ycomentar cómo afectarían la presencia de posi-bles reacciones secundarias (parásitas) la valo-ración redox.
Sea la reacción:
n2 Ox1 + n1 Red2 + x H+ ←→←→ n2 Red1 + n1 Ox2 + H2O [11.22]
resultante de las semireacciones:
Ox1 + n1 e + x H+ ←→ Red1 + H2O
[11.23]
Ox2 + n2 e ←→ Red2 [11.5]
en las que además existe la posibilidad de que lasformas oxidadas o reducidas del valorante (Ox1) ydel analito (Red2) presenten reacciones secundarias.
Como en otros casos, el trazado de la curvade valoración contempla las siguientes zonas:
A) Zona anterior al punto de equivalencia
Como ya se ha comentado, el potencial enesta zona, en la que existe defecto de valorante,se calcula mediante la ecuación de Nernst corres-pondiente al par redox del analito:
[11.9]
que, en el caso de que existan reacciones secun-darias de una de estas especies –o de ambas– ysi se tiene en cuenta que
[11.24]
queda en la forma:
[11.25]
y el potencial condicional correspondiente a estepar puede expresarse como:
[11.26]
A la vista de estas ecuaciones se pueden obte-ner las siguientes conclusiones:
1. Las reacciones secundarias de la forma oxi-dada del analito (aumento de αOx2
) pro-
E En2
020
2
0 059
2
′ = + ,log
αα
Red
Ox
2
E En
= +′′2
0
2
0 059,log
αα
Red 2
Ox 2
2
2
[Ox ]
[Red ]
αRed2
22
[Red ][Red ]
= ′αOx2
22
[Ox ][Ox ]
= ′
E E
n= +2
0
2
0 059,log
[Ox ][Red ]
2
2
E20
E10
x2
x2
376 Equilibrios iónicos y sus aplicaciones analíticas

vocarán una disminución del potencial con-dicional del par redox y, en consecuencia,los potenciales medidos en la zona dedefecto de valorante de la curva de valo-ración serán más bajos que los que seobtendrían en ausencia de reacciones pará-sitas. Este efecto es positivo, ya que pro-voca un ampliación de la zona de equiva-lencia de la curva.
2. Las reacciones parásitas de la forma redu-cida del analito (aumento de αRed2
) pro-ducirán un aumento del potencial condi-cional y, por lo tanto, los potenciales en lazona anterior al punto de equivalenciaserán más elevados que en ausencia dereacciones secundarias. Este efecto esnegativo, ya que disminuye la amplitud dela zona de equivalencia.
3. En el caso de que tanto la forma oxidadacomo la reducida presenten reaccionessecundarias, el efecto neto dependerá decuál de dichas formas las presente enmayor extensión.
B) Zona posterior al punto de equivalencia
En esta zona, en la que hay exceso de valoran-te, el potencial se calcula mediante la ecuación deNernst correspondiente a este compuesto:
[11.27]
Si una de las especies, o ambas, presentanreacciones secundarias, se cumple que
[11.28]
y al sustituir estas expresiones en la ecuación deNernst se tiene:
[11.29]
lo que permite obtener la ecuación del potencialcondicional de este par redox:
[11.30]
A partir de esta ecuación se pueden obtenerlas siguientes conclusiones:
1. Un aumento del pH provocará una dismi-nución del potencial condicional del par([H+] disminuye) y, en consecuencia, lospotenciales en la zona posterior al puntode equivalencia serán menores que los que se obtendrían en condiciones estándar(pH = 0). Este efecto es negativo, ya quereduce la amplitud de la zona de equiva-lencia.
2. Las reacciones secundarias de la forma oxi-dada del valorante ejercen también unainfluencia negativa, ya que si αOx1
aumen-ta, el potencial condicional disminuye.
3. Las reacciones parásitas de la forma redu-cida del valorante ejercen un efecto posi-tivo, ya que si αRed2
aumenta, también lohace el potencial condicional y, por tanto,se incrementa la amplitud de la zona deequivalencia.
4. Si tanto la forma oxidada como la formareducida tienen reacciones secundarias, elefecto neto depende de cuál de ellas lastenga en mayor extensión.
C) Punto de equivalencia
Cuando existen reacciones secundarias, elpotencial en el punto de equivalencia se calculamediante la ecuación similar a la [11.16]:
[11.31]
por lo que el potencial de este punto podrá sersuperior o inferior al obtenido en ausencia de
En E n E
n nequivalencia =′ + ′
+1 1
02 2
0
1 2
E En
x
10
10
1
0 059
1
′ = ++
,log
αα
Red
Ox
1[H ]
E En
x
= +′
′
+
10
1
0 059,log
αα
Red 1
Ox 1
1
1
[Ox ] [H ]
[Red ]
αRed1
11
[Red ][Red ]
= ′
αOx
1
11
[Ox ][Ox ]
= ′
E En
x
= ++
10
1
0 059,log
[Ox ][H ][Red ]
1
1
Capítulo 11: Valoraciones de oxidación-reducción 377

estas reacciones, en función de la influencia quelas mismas ejerzan sobre los potenciales condi-cionales de los pares redox implicados en la reac-ción analítica.
Todo lo anterior puede resumirse en losiguiente: la constante condicional de una reac-ción redox se calcula a partir de la ecuación:
[11.32]
y, por tanto, es evidente que cualquier factor queincremente la diferencia entre los potencialescondicionales, ya sea porque E0′
1 aumente o por-que E0′
2 disminuya, originará un aumento de laconstante condicional, es decir, a una reacciónmás completa y, consecuentemente, a una mayoramplitud de la zona de equivalencia de la curvade valoración. También queda claro que cual-quier factor que disminuya la diferencia entre lospotenciales condicionales tendrá un efecto nega-tivo y puede llegar a provocar que la reacción nosea cuantitativa o, incluso, que no se produzcade forma apreciable.
Esta influencia de las reacciones secundariasy de la acidez en la cuantitatividad de la reaccióny, consecuentemente, en la amplitud de la zonade equivalencia, es lo que justifica, entre otrasrazones, el hecho de que, como se verá en lasaplicaciones, muchas valoraciones redox se lle-van a cabo en medio fuertemente ácido y, en oca-siones, en presencia de ligandos que complejanla forma oxidada del analito.
11.3. Indicadores redox
Un indicador visual redox es, generalmente,una sustancia orgánica que presenta dos formas,una oxidada, InOx, y otra reducida, InRed, de dis-tinto color, en equilibrio reversible:
InOx + n e ←→ InRed [11.33]
El cambio de color del indicador se debe, eneste caso, a cambios en el valor del potencial de
la disolución. Antes de pasar a estudiar en pro-fundidad las características de estos indicadoresredox (intervalo de transición, selección del indi-cador, etc.) es interesante comentar que en lasvaloraciones redox se utilizan, en ciertos casos,indicadores visuales que no están regidos estric-tamente por el equilibrio reversible dado en laecuación [11.33]. Se pueden constatar los casosque se indican a continuación:
Sistemas autoindicadores. En este caso el pun-to final de la valoración se pone de manifiesto sinnecesidad de añadir otra sustancia (el indicador)a la disolución. Esta situación se origina cuandoel reactivo valorante (normalmente la forma oxi-dada de un par redox) es coloreado y el produc-to de su reacción (por lo general, la forma redu-cida) es prácticamente incoloro. Así, por ejemplo,en las valoraciones con KMnO4, de intenso colorvioleta, una gota de valorante en exceso es sufi-ciente para proporcionar a la disolución un colorrosa claramente apreciable, lo que permite dete-ner la valoración en este punto.
Sustancias que reaccionan con una sola formadel par redox del analito. Este tipo de indicación delpunto final en valoraciones redox, se basa en unequilibrio adicional de formación de complejosentre una de las formas del par redox del analito yun ligando. El complejo coloreado formado actúarealmente como sistema indicador. Así, en las yodo-metrías se puede hacer uso del color amarillo delyodo como base para la detección del punto finalde la valoración. Sin embargo, en las cercanías delpunto final, este color es muy tenue, lo que da lugara errores significativos en la valoración. La adiciónde almidón, que forma un complejo de color azulcon el yodo, permite detectar con gran exactitud elpunto final de esta valoración.
Indicadores redox reversibles. Estos indica-dores, constituidos por sustancias orgánicas oquelatos metálicos, responden al equilibrio redoxmostrado en la ecuación [11.33]. Su estudio seabordará con detalle posteriormente.
Indicadores redox pseudo-reversibles. El meca-nismo de acción de estos indicadores se basa en dosetapas, una irreversible y otra reversible, tales como:
log
,′ =
′ − ′K
E EnT
10
20
0 059
378 Equilibrios iónicos y sus aplicaciones analíticas

InRed → + n1 e [11.34]
←→ + n2 e [11.35]
El cambio de color se produce generalmenteentre las formas InOx1
e InOx2. El ácido N-feni-
lantranílico o la difenilamina, son ejemplos carac-terísticos de este tipo de indicadores.
Indicadores irreversibles. Este tipo de indica-dores se destruyen de forma irreversible cuandoreaccionan con el exceso de oxidante, que se ori-gina en el punto final de la valoración:
InRed → InOx + n e [11.36]
siendo el ejemplo más característico el rojo demetilo. El color rojo de este indicador, muy uti-lizado en las bromometrías (valoraciones conbromo), desaparece por reacción con el bromoen el punto final de la valoración.
A pesar de que se recomienda el uso de indi-cadores redox reversibles, no siempre son losmás adecuados. Así, además de mostrar un inter-valo de transición adecuado y ofrecer un cambiobrusco de color en el punto final, un indicadorredox debe presentar otras dos características degran importancia: rapidez en el proceso de cam-bio de color y solubilidad en agua, ya que no sepuede hacer uso de disolventes orgánicos dadoel carácter reductor de los mismos. La combina-ción más adecuada de estas propiedades facultala elección del indicador a utilizar en la valora-ción.
De todas estas propiedades, el intervalo detransición o zona de viraje del indicador es la demayor significación a la hora de conocer si elindicador es adecuado o no para una determi-nada valoración. Este intervalo se define, paraun indicador redox, como la región de la escalade potencial en que el indicador manifiesta elcambio de color. Para establecer este intervalode transición se acepta, de forma aproximada,que en el caso de una mezcla de colores, el ojohumano sólo detecta un cambio de tonalidadcuando la relación entre las concentraciones de
las dos especies coloreadas varía entre 10 y 0,1.Es decir, si
⇒ [11.37]
se observará el color de la forma oxidada del indi-cador, y si
⇒ [11.38]
se observará el color de la forma reducida delmismo. En el intervalo entre ambas condiciones,esto es, si
⇒
[11.39]
se observará un color que será mezcla de los colo-res correspondientes a ambas formas del indicador.
Al sustituir la condición dada en la ecuación[11.39] en la expresión de Nernst para el sistemaindicador,
[11.40]
se obtiene:
[11.41]
donde E0In es el potencial estándar del sistema
indicador y n el número de electrones intercam-biado por el mismo. Si existen equilibrios con-currentes, caso muy habitual, ya que es frecuen-te que los iones H+ intervengan en la reacciónredox del indicador y además no es raro que unade sus formas, o ambas, tenga carácter ácido, elintervalo de transición se expresa en función delpotencial condicional:
∆ E E
n= ±In
0 0 059,
E E
n= +In
0 Ox
Red
[In ][In ]
0 059,log
–1 log[In ][In ]
1Ox
Red
< <
0, 1[In ][In ]
10Ox
Red
< <
log
[In ][In ]
1Ox
Red
≥ −
[In ][In ]
0, 1Ox
Red
≤
log
[In ][In ]
1Ox
Red
≥
[In ]
[In ]10Ox
Red
≥InOx2
InOx1
InOx1
Capítulo 11: Valoraciones de oxidación-reducción 379

[11.42]
A partir de esta expresión se pueden deducirlas siguientes conclusiones:
1. El intervalo de potencial en que se pro-duce el viraje de un indicador depende desu potencial condicional, por lo que cual-quier factor que modifique éste provoca-rá un desplazamiento de dicho intervalo.
2. El número de electrones intercambiadosafecta a la amplitud del intervalo de tran-sición del indicador. Cuanto mayor sea n,más estrecho será este intervalo.
El tratamiento realizado para establecer elintervalo de transición de un indicador es pura-mente teórico, dado que, como ya se ha indica-do, la sensibilidad del ojo humano no es la mis-ma para distinguir todos los colores. Enconsecuencia, el intervalo de viraje de un indi-cador determinado puede ser ligeramente supe-rior o inferior al establecido por la expresión
[11.42] y puede no ser perfectamente simétricoalrededor del valor del potencial condicional delindicador, en función del color concreto de susformas oxidada y reducida. Este hecho se ponede manifiesto, por ejemplo, en el caso de la ferroí-na, ( , siendo fen: 1,10-fenan-trolina) con E0
In = 1,06 V y n = 1, cuya zona deviraje, que teóricamente debería estar compren-dida entre 1,00-1,12 V, se desplaza a 1,04-1,18 Ven la práctica, debido a la diferente cromaticidadentre la forma oxidada (azul pálido) y reducida(roja) del indicador. Así, para apreciar el colorazul en presencia del rojo la concentración deforma oxidada debe ser al menos 100 veces supe-rior a la de forma reducida, mientras que en elcaso contrario una concentración de forma redu-cida doble a la de forma oxidada es suficientepara diferenciar claramente ambos colores.
En el cuadro 11.2 se muestran algunos de losindicadores redox más utilizados, junto con elvalor de su potencial estándar (o, en algunoscasos, del potencial condicional), los coloresimplicados en el cambio y la concentración ópti-ma de la disolución de indicador a utilizar.
Fe(fen) /Fe(fen)32
33+ +
∆ E En
= ′ ±In0 0 059,
380 Equilibrios iónicos y sus aplicaciones analíticas
CUADRO 11.2Valores de E0′In cambio de color y concentración óptima de uso de los indicadores más utilizados en valoraciones redox
Indicador E0′In Cambio de color Disolución de indicador(Red – Ox) (InRed)
Difenilamina 0,76 Incoloro – Violeta 0,1% H2SO4 concentrado Ácido p-difenilamino sulfónico 0,85 Incoloro – Rojo violeta 0,2% sal de bario en agua Ferroína 1,06 Rojo – Azul pálido 0,025 mol L–1 en agua Acido N-fenilantranílico 1,08 Incoloro – Azul violeta 0,005 mol L–1 en Na2CO3 0,5% 5-Nitroferroína 1,25 Rojo – Azul pálido 0,025 mol L–1 en agua
Estos indicadores cubren prácticamente todoel intervalo de potencial adecuado para llevar acabo los diversos tipos de volumetrías redox y,en consecuencia, los diferentes tipos de valora-ciones redox que se discutirán en el apartadoAplicaciones de este capítulo. Desde un puntode vista estructural, estos indicadores se puedenclasificar en dos grupos o familias importantes:
indicadores del grupo de difenilamina y deriva-dos, e indicadores del tipo ferroína.
La importancia de la difenilamina como indi-cador redox se debe, más bien, a razones histó-ricas, ya que son sus derivados los que realmen-te se utilizan con este fin. En la figura 11.2a semuestra el sistema redox de esta sustancia, quese puede extender a sus correspondientes deri-

vados. La oxidación de este indicador conduce,en una primera etapa irreversible, a la formaciónde difenilbencidina incolora, que se transformamediante un cambio redox reversible, en el queestán implicados dos electrones, en una estruc-tura quinoidea conocida como violeta de dife-nilbencidina.
Esta sustancia presenta una serie de incon-venientes, que limitan su empleo como indica-dor redox en valoraciones redox que utilizancomo valorante un oxidante. Así, presenta unpotencial estándar relativamente bajo, tan sólo0,76 V, y es poco soluble en agua. Con objeto depaliar estos inconvenientes se han desarrolladodiversos derivados, entre ellos cabe destacar losácidos p-difenilamino sulfónico y N-fenilantra-nílico (figura 11.2a). La introducción en la molé-
cula de difenilamina de un grupo sulfónico y car-boxílico, respectivamente, incrementa de formaapreciable su solubilidad en agua, así como elvalor del potencial estándar del sistema redox(cuadro 11.2).
Los indicadores del tipo ferroína constituyenun caso especial de indicadores redox, ya que elindicador no es una molécula orgánica sino unquelato que cambia de color debido a un proce-so reversible de intercambio de electrones. Enconcreto se trata de quelatos formados por losiones Fe3+ e Fe2+ con ligandos orgánicos que po-seen la agrupación diimina. En la figura 11.2b semuestra el ejemplo más característico de estosindicadores, que corresponde a los quelatos for-mados con el ligando 1,10-fenantrolina (fen). Elcomplejo con el ion Fe2+ recibe el nombre de
Capítulo 11: Valoraciones de oxidación-reducción 381
FIGURA 11.2. Indicadoresvisuales más utilizados en valo-raciones redox con valorantes
oxidantes.
a)
b)

ferroína, Fe(fen)2+3
, y tiene un color rojo intenso.La forma oxidada, Fe(fen)3+
3, es de color azul páli-
do. Se ha comprobado que el intercambio deelectrones entre ambas formas del indicador nose realiza a través de los iones Fe3+ e Fe2+, pro-cedentes de la posible disociación de los corres-pondientes quelatos, sino que obedece a un pro-ceso de oxidación-reducción interna a través delsistema de electrones π de ambos quelatos. Estesistema indicador es muy apropiado para suempleo en volumetrías redox ya que: posee unpotencial estándar elevado, el sistema es perfec-tamente reversible, presenta un cambio bruscode color y el complejo es muy soluble en agua.La propuesta de diferentes derivados se ha rea-lizado con idea de modificar el potencial de estesistema redox. Así, los derivados metilados ori-ginan una disminución del potencial, mientrasque los nitrados o sulfonados lo incrementan. Setiene de esta forma un abanico de indicadorescuyos potenciales estándar oscilan entre aproxi-madamente 0,80 y 1,26 V.
Finalmente, la selección del indicador ade-cuado para una determinada valoración se basa,como en otras valoraciones, en el concepto deregión de equivalencia. Si se admite un error del0,1% en la valoración, dicha región de equiva-lencia está comprendida entre el 99,9% y el100,1% de la misma, y por tanto, cualquier indi-cador que vire dentro de esta zona de potencialse podrá utilizar a priori en dicha valoración.Así, por ejemplo, para la valoración de una diso-lución de Fe2+ con Ce4+ o MnO–
4, la región deequivalencia del 0,1% está comprendida entre0,948-1,263 y 0,948-1,475 V, respectivamente (vercuadro 11.1). De acuerdo con los potenciales delos indicadores mostrados en el cuadro 11.2 ycon el número de electrones intercambiados porlos mismos (figura 11.2), la ferroína y el ácidoN-fenilantranílico son adecuados cuando seemplea Ce4+ como valorante, dado que el inter-valo de transición de los mismos (1,001-1,119 y1,051-1,109 V, respectivamente) se sitúa en laregión de equivalencia del 0,1%. Es fácil dedu-cir que estos indicadores junto con la 5-nitrofe-rroína son también adecuados para la valoración
de disoluciones de Fe2+ cuando se utiliza MnO–4
como valorante, dado el mayor intervalo depotencial que presenta su región de equivalen-cia del 0,1%, aunque en este caso no seríaimprescindible el uso de un indicador, ya que,salvo que se use a concentraciones muy bajas, elpermanganato de potasio puede actuar comoautoindicador.
11.4. Normalidad versus molaridad en valoraciones redox
A lo largo de todo este libro, se ha utilizadola molaridad (mol L–1), de acuerdo con las reco-mendaciones dadas por la IUPAC, para expre-sar la concentración de las disoluciones utiliza-das tanto en el estudio de los diferentesequilibrios iónicos, como en las respectivas valo-raciones. Sin embargo, en las valoraciones redoxpresenta un gran interés, desde un punto de vis-ta práctico, el uso de los conceptos de factor deequivalencia, equivalente y normalidad, dadoque facilitan enormemente los cálculos a reali-zar en las mismas, ya que las reacciones secorresponden equivalente a equivalente o, lo quees lo mismo, disoluciones de igual normalidad secorresponden volumen a volumen. Aunque eltérmino normalidad está recogido en el capítulo6 del texto Compendium of Analytical Nomen-clature. Definitive Rules, editado por la IUPACen 1997, su uso no es recomendado por este orga-nismo. A pesar de ello, es común hoy en díaexpresar la concentración de las disolucionesvaloradas, utilizadas en valoraciones redox, entérminos de normalidad por las ventajas que pre-senta. A la vista de esta situación, se ha optadopor emplear en este capítulo y especialmente ensu apartado correspondiente a Seminarios: pro-blemas numéricos, ambas formas (molaridad ynormalidad) para expresar la concentración delas diversas disoluciones implicadas en el proce-so de valoración.
En general y de acuerdo con la IUPAC, el fac-tor de equivalencia de una sustancia X, feq (X),implicada en una determinada reacción de valo-
382 Equilibrios iónicos y sus aplicaciones analíticas

ración, es un número derivado de la estequio-metría de la misma. En el caso de una reacciónredox, el factor de equivalencia, para cada com-ponente, debe estar relacionado con los electro-nes intercambiados en la reacción. Así, para lareacción:
Mn+ + 2 e ←→ M(n–2)+ [11.43]
el factor de equivalencia viene dado por:
feq (M2+) = 1/2 [11.44]
por lo que se puede definir más concretamentecomo el inverso del número de electrones inter-cambiados en el par redox. Así, por ejemplo, losfactores de equivalencia para las especies impli-cadas en los pares redox:
8 H+ + 5 e ←→ Mn2+ + 4 H2O[11.45]
14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O [11.46]
son:
= 1/5 feq (Mn2+) = 1/5 [11.47]
= 1/6 feq (2 Cr3+) = 1/6 [11.48]
Por otra parte, el equivalente de una sustan-cia en una reacción redox se define como la enti-dad que, en una reacción concreta, se combina-ría, o sería equivalente, a una entidad deelectrones. El equivalente de una sustancia sepuede establecer a partir del factor de equiva-lencia, feq(X), y del peso molecular del compuesto(X) y será igual a feq(X)X.
Este factor de equivalencia permite estable-cer la relación entre las dos formas consideradaspara expresar la concentración de una disolución.Así, se tiene:
[11.49]
por lo que el valor de la normalidad será n vecessuperior al de la correspondiente molaridad, sien-do n el número de electrones intercambiados enel par redox. Así, por ejemplo, si se prepara unadisolución de concentración 1,0 mol L–1 de laespecie Mn+, de acuerdo con la expresión [11.49]su concentración expresada en términos de nor-malidad será de 2,0 eq L–1. Por tanto, de formageneral se puede establecer que:
equivalentes = moles × n [11.50]
o bien:
moles = equivalentes × feq [11.51]
Aunque la IUPAC no recomienda el uso deltérmino normalidad, sí indica que cuando se expre-se la concentración de una disolución de esta for-ma se ha de acompañar siempre del correspon-diente factor de equivalencia. Así, al preparar unadisolución de KMnO4 de concentración 0,1 eq L–1,si ésta se ha estandarizado mediante una valora-ción redox en la cual el permanganato de potasiose reduce a Mn2+ (se intercambian 5 electrones),dicha disolución debe etiquetarse como: KMnO40,1 eq L–1; feq (KMnO4) = 1/5.
11.5. Aplicaciones
Como se ha indicado en la introducción deeste capítulo, las valoraciones redox ocupan unlugar importante dentro de los métodos de aná-lisis cuantitativos clásicos, dado su amplio cam-po de aplicación cuando se utiliza un reactivo oxi-dante como valorante.
En el contexto de las valoraciones con oxi-dantes, es interesante reseñar las característicasdeseables de las sustancias empleadas en la pre-paración de las correspondientes disolucionesvaloradas. Así, es conveniente que posean losrequisitos exigibles a un patrón primario conobjeto de que sus disoluciones se puedan prepa-rar directamente por pesada y que además estasdisoluciones sean estables con el tiempo. Por otra
× feq Normalidad (equivalenteL )1−
Molaridad(mol L )1− =
feq 2 72(Cr O )−
feq 4(MnO )−
CrO72− +
MnO4− +
Capítulo 11: Valoraciones de oxidación-reducción 383

parte, es recomendable que tengan un potencialestándar alto para así oxidar a un gran númerode sustancias, lo que conlleva un mayor campode aplicación, aunque ello redunda en una menorselectividad. Finalmente, han de ser compatiblescon los sistemas indicadores visuales de puntofinal reseñados en el apartado anterior.
Estas propiedades son difíciles de reunir alcompleto en una sustancia, como se mostrará pos-teriormente en el estudio detallado de aquellosreactivos oxidantes más utilizados en valoracio-nes redox, como son el permanganato de potasioy dicromato de potasio En este estudio se abor-dará además el empleo del sistema yodo/yoduroen valoraciones redox, dado su amplio campo deaplicación debido a las singulares característicasde este par redox. El esquema general que se vaa utilizar para el estudio de estas valoracionesconsta de los siguientes puntos:
1. Estudio de las características redox delreactivo oxidante: par o pares redox impli-cados, potenciales condicionales, aspectoscinéticos (catalizadores) y posibilidad dereacciones colaterales (que en algunoscasos pueden ser no deseables, pero enotros pueden resultar útiles).
2. Preparación, estandarización y conserva-ción de disoluciones valoradas.
3. Tipos de valoraciones.4. Aplicaciones más relevantes.
11.5.1. Tratamiento previo en volumetrías redox
Antes de pasar a la descripción del uso ana-lítico de estas valoraciones, se ha de recordar queen ciertos casos puede ser necesario llevar a caboun tratamiento previo a la determinación, conobjeto de asegurar que la especie a determinarse encuentre en un estado de oxidación perfec-tamente definido y adecuado para su posteriorvaloración. La reacción redox en la que se basaeste tratamiento previo ha de cumplir una seriede requisitos y por tanto ha de ser cuantitativa,rápida y selectiva.
Además de cumplir estos requisitos, una eta-pa de tratamiento previo no es recomendabledesde un punto de vista práctico si no es posiblela completa eliminación del exceso de reactivo uti-lizado en la misma, dado que éste reaccionaríacon el valorante utilizado posteriormente.
A) Agentes reductores previos
Aunque se han descrito diversos tipos deagentes reductores previos, tanto sólidos, líqui-dos, como gaseosos (se considera además en estegrupo aquellos reactivos que se pueden eliminarpor volatización, aunque se adicionen en formade disolución), sólo se tratarán a continuaciónaquellos agentes reductores previos sólidos mássignificativos, dada la gran facilidad que presen-ta su eliminación de la disolución tras el trata-miento previo. Se incluye además el cloruro deestaño, debido a su uso generalizado en la reduc-ción de Fe3+ a Fe2+ como paso previo a su valo-ración con permanganato o con dicromato.
Metales puros. Metales tales como cinc, pla-ta, plomo, cadmio, etc., se han utilizado comoreductores en diversas formas físicas, tales como,lámina, hilo, polvo, grano, etc., bien por adicióndirecta de éstos a la disolución que contiene elanalito (el exceso de reductor se puede eliminarpor filtración) o mediante las denominadascolumnas reductoras, en cuyo caso la disoluciónque contiene la especie de interés se hace pasara través de la columna que contiene al reductorprevio. Es evidente que, desde un punto de vis-ta práctico, es recomendable el uso de columnasreductoras. La selectividad que se puede conse-guir en este tratamiento depende en gran medi-da del potencial estándar del sistema Mn+/M0.
De estos reductores cabe destacar el cinc,reductor muy poderoso ya que el potencial están-dar del sistema Zn2+/Zn0 es de –0,763 V. Debidoa este valor tan negativo de potencial, el Zn0
reduce a la mayor parte de los elementos a suestado inferior de oxidación, lo que va en detri-mento de la selectividad del proceso. Así, porejemplo, este metal da lugar a reducciones pre-
384 Equilibrios iónicos y sus aplicaciones analíticas

vias tales como: Fe3+ → Fe2+, Cr3+ → Cr2+, Ti4+ →Ti3+, etc., e incluso muy drásticas como la reduc-ción de NO–
3 y NO–2 a NH4
+. El principal inconve-niente para su empleo es que las reducciones pre-vias se llevan a cabo en medio ácido y en estascondiciones también se produce la reacción:
Zn0 + 2 H+ → Zn2+ + H2 [11.52]
que provoca no sólo un desprendimiento dehidrógeno, sino también un gran consumo dereductor auxiliar, una considerable contamina-ción de la disolución (la concentración de Zn2+
acostumbra a ser elevada después de una reduc-ción previa con Zn0) y una importante modifica-ción de la concentración de iones H+.
Metales amalgamados. El empleo de metalesamalgamados tiene como finalidad evitar el des-prendimiento de hidrógeno, cuando las reduc-ciones se llevan a cabo en medio ácido, debido ala elevada sobretensión que presenta el mercu-rio en dicho proceso. Se pueden preparar diver-sas amalgamas cuyo poder reductor varía entreamplios límites según el metal amalgamado.Dependiendo del contenido de mercurio puedenser tanto líquidas como sólidas.
Las amalgamas sólidas se emplean, general-mente, en columna y presentan, por tanto, unmayor interés práctico. La más utilizada es laamalgama de cinc (contiene normalmente 5% demercurio) denominada reductor de Jones. La adi-ción de mercurio no afecta al potencial estándardel sistema Zn2+/Zn0, aunque sí a la cinética delproceso de reducción, cuya velocidad dependedel contenido de cinc en la superficie de la amal-gama. Con esta columna reductora se logranreducciones intensas y muchos iones quedan enestado de oxidación inestable al aire, por sucarácter reductor. Este problema se evita reco-giendo el eluyente de la columna en una disolu-ción de Fe3+. Frente a un reductor enérgico, elFe3+ se reduce a la cantidad equivalente de Fe2+,lo suficientemente estable para permitir su valo-ración. Una ventaja importante del reductor deJones es que no reduce el H+ a H2, con lo que seevitan los inconvenientes mencionados para éste.
Cloruro de estaño. Este reactivo tiene unaaplicación limitada, pero de gran importanciapráctica, ya que se emplea para la reducción deFe3+ a Fe2+ como paso previo para su valoracióncon MnO–
4 o con Cr2O2–7. Al añadir un ligero exce-
so de disolución de SnCl2 (para asegurar la cuan-titatividad del proceso) sobre una disolución deFe3+ a ebullición se produce la reacción:
2 Fe3+ + Sn2+(exceso) → 2 Fe2+ + Sn4+ + Sn2+
[11.53]
El Sn2+ sobrante se elimina añadiendo rápi-damente a la disolución fría un exceso de HgCl2.En estas condiciones tiene lugar la reacción:
Sn2+ + 2 Hg2+(exceso) + 2 Cl– →
→ Sn4+ + Hg2Cl2 (S) + Hg2+ [11.54]
El Sn4+ y el Hg2+ se encuentran en su estadode oxidación máximo, por lo que no interfierenen la valoración de Fe2+ con un oxidante y la inso-lubilidad del Hg2Cl2 (de color blanco, tambiéndenominado calomelanos) impide la reacción delHg2+
2 con los oxidantes. Una ventaja adicional de este procedimiento es
que resulta fácil comprobar que la reducción pre-via ha sido completa: en efecto, la ausencia del pre-cipitado blanco de Hg2Cl2 indica que no se habíaañadido exceso de Sn2+. Asimismo, la presencia deun precipitado negro implica que se habría añadi-do un gran exceso de Sn2+, suficiente para pasar elmercurio a Hg0, especie que no debe obtenerse yaque sí reacciona con el valorante oxidante.
B) Agentes oxidantes previos
Aunque no es un caso tan frecuente como elde las reducciones previas, en ocasiones puedeser necesario llevar el analito a su estado máxi-mo de oxidación. Para ello se pueden utilizarvarios agentes oxidantes.
Peroxodisulfato de amonio, (NH4)2S2O8. Estecompuesto, también conocido como persulfatode amonio, se reduce a sulfato, en presencia deAg+ como catalizador, según la reacción:
Capítulo 11: Valoraciones de oxidación-reducción 385

+ 2 e ←→ 2 [11.55]
cuyo exceso se elimina fácilmente por ebullición:
2 + 2 H2O 4 + O2 ↑ + 4 H+
[11.56]
Este reactivo permite la oxidación de Cr3+ →Cr6+, Mn2+ → Mn7+, Fe2+ → Fe3+, Ce3+ → Ce4+, etc.
Bismutato de sodio, NaBiO3. Este compues-to es un oxidante enérgico, reduciéndose a Bi3+
en medio ácido según la reacción:
NaBiO3 + 6 H+ + 2 e ←→←→ Bi3+ + Na+ + 3 H2O [11.57]
y el exceso de oxidante se elimina por filtración.Es capaz de oxidar Cr3+ → Cr6+ y Mn2+ → Mn7+.
Peróxido de hidrógeno. Es un oxidante efi-ciente, particularmente en medio básico,
H2O2 + 2 H+ + 2 e ←→ 2 H2O [11.58]
cuyo exceso se destruye por ebullición, ya queeste reactivo se descompone a temperatura ele-vada:
2 H2O2 O2 ↑ + 2 H2O [11.59]
Esta reacción de descomposición se encuen-tra catalizada por numerosos iones metálicos.Ejemplos de reacciones de oxidación son las deCr3 → Cr6+ en disolución de NaOH y de Co2+ aCo3+ en disolución de NaHCO3, entre otras.
11.5.2. Valoraciones con permanganato (permanganimetrías)
El permanganato de potasio es el reactivo oxi-dante más utilizado en valoraciones redox, dadoel elevado valor de potencial estándar que poseeel sistema MnO–
4/Mn2+. No es sustancia patrón,
por lo que sus disoluciones valoradas se han deestandarizar frente a un patrón primario, gene-ralmente oxalato de sodio. Estas disoluciones sonbastante inestables y, por tanto, se han de extre-mar las precauciones para su conservación. Laspermanganimetrías no precisan de un sistema indi-cador visual externo para detectar el punto finalde la valoración, ya que, como se ha indicado enel apartado 11.3, el propio color violeta del per-manganato pone de manifiesto el mismo.
A) Características redox
Las características redox del permanganatoestán directamente relacionadas con el pH delmedio en el cual se lleva a cabo la valoración. Enmedio ácido, la reducción del MnO–
4 transcurresegún la reacción:
8 H+ + 5 e ←→ Mn2+ + 4 H2O
feq = 1/5
[11.45]
mientras que en disolución débilmente ácida,neutra o moderadamente alcalina (3 < pH < 12)se reduce cuantitativamente a dióxido de man-ganeso:
4 H+ + 3 e ←→ MnO2 (S) + 2 H2O
feq = 1/3[11.60]
Si bien el potencial estándar es más oxidanteque el correspondiente a la reducción a Mn2+, debi-do a que la reacción [11.60] tiene lugar desde pHdébilmente ácidos a moderadamente básicos y queel potencial condicional depende del pH, este valordisminuirá al aumentar el pH. Por ejemplo, a pH= 7 el potencial condicional es 1,14 V.
En disolución fuertemente alcalina se reducea manganato:
e ←→ feq = 1,0 [11.61]
E0 0 6= , 0 V MnO42−MnO4
− +
E0 1 692= , V MnO4
− +
E0 1= , 51 V MnO4
− +
ebullición →
E0 1 7= , 8 V
E0 1= , 60 V
SO42− ebullición →S O2 8
2−
E0 2 00= , V SO4
2− S O2 8
2−
386 Equilibrios iónicos y sus aplicaciones analíticas

De estos sistemas redox, la reacción [11.45]constituye la base de la mayoría de las aplica-ciones del KMnO4 en valoraciones redox.
Algunas reacciones redox con MnO–4 exhi-
ben un comportamiento cinético lento y, portanto, es frecuente en las permanganimetrías eluso de catalizadores y el control de la tempe-ratura, con objeto de acelerar las mismas y parapoder utilizarlas como base de una reacción devaloración. Así, la típica reacción de estandari-zación de disoluciones de permanganato conoxalato de sodio:
2 5 + 16 H+ ←→←→ 2 Mn2+ + 10 CO2 + 8 H2O [11.62]
que en frío prácticamente no tiene lugar, es rápi-da en caliente y está catalizada por el ion Mn2+
(producto de la reacción), lo que es de vitalimportancia a la hora de establecer el procedi-miento experimental para llevar a cabo dichaestandarización.
Un inconveniente del MnO–4 es que su poten-
cial es lo suficientemente elevado como para poderoxidar el ion cloruro a cloro, según la reacción:
2 10 Cl– + 16 H+ ←→←→ 2 Mn2+ + 5 Cl2 + 8 H2O [11.63]
y ello supone un problema, ya que, en principio,impediría la valoración de muestras que contuvie-ran iones Cl– y obligaría a emplear ácidos distintosdel HCl para la disolución. Sin embargo, el incon-veniente no es grave, ya en muchos casos esta reac-ción virtualmente no se produce, hasta el punto deque en la práctica sólo supone una dificultad parala determinación de Fe2+ y aun en este caso es fácilevitar que tenga lugar (ver apartado 11.5.2c).
B) Preparación, conservación y estandarización de disoluciones de KMnO4
La preparación de las disoluciones de per-manganato se ha de llevar a cabo en unas condi-
ciones experimentales tales que permitan su pos-terior conservación por largos períodos de tiem-po. El KMnO4, que se expende con una purezadel orden del 99,5%, es soluble en agua, pero susdisoluciones no son muy estables, ya que puedeproducirse la reacción:
4 2 H2O → 4 MnO2 (S) + 3 O2 + 4 OH–
[11.64]
desplazada hacia la derecha, aunque con unacinética muy lenta. Sin embargo la reacción estácatalizada por la luz, el calor, los ácidos, las bases,el Mn2+ y el MnO2. Los primeros factores nosuponen un problema serio, ya que la influenciade la luz y el calor se elimina al conservar lasdisoluciones de permanganato de potasio en reci-pientes de color topacio y a temperatura ambien-te y prepararlas en agua destilada (sin adiciónde ácidos ni bases); por otra parte, la concen-tración de Mn2+ será virtualmente nula. El fac-tor que fundamentalmente reduce la estabilidadde estas disoluciones es la presencia de MnO2,que no sólo se encuentra al nivel de trazas en elKMnO4 comercial, sino que se forma cuando estecompuesto reacciona con las impurezas orgáni-cas que contiene el agua utilizada para disolverlo.Para evitar este inconveniente basta con hervirlas disoluciones de permanganato recién prepa-radas durante un par de horas, dejar reposar yfiltrar el MnO2 mediante una placa de vidrioporoso.
Si se toman las precauciones descritas en elpárrafo anterior, las disoluciones de permanga-nato de potasio son estables durante varias sema-nas. Sin embargo, deben ser filtradas y estanda-rizadas nuevamente en el momento que seaprecie la formación de MnO2.
En la elección del patrón primario adecuadopara estandarizar las disoluciones de permanga-nato de potasio no sólo se han de considerar lapureza y estabilidad del mismo, sino que ademáses necesario conocer el mecanismo en el que sefundamenta la reacción de valoración. Tanto elAs2O3 como el Na2C2O4 se han utilizado paraestandarizar las disoluciones de KMnO4 y en
MnO4− +
MnO4− +
C O2 42−
MnO4− +
Capítulo 11: Valoraciones de oxidación-reducción 387

ambos casos la reacción es lenta, aunque puedeacelerarse por la presencia de catalizadores. Deellos, el oxalato de sodio se considera como elpatrón primario más adecuado, a pesar de que lacinética de esta reacción es algo compleja.
El Na2C2O4 se encuentra en el comercio conuna pureza de 99,95%. Para poder utilizarlocomo patrón primario se ha de desecar en estu-fa entre 105–110 °C. Esta sustancia forma partedel siguiente par redox:
2 CO2 + 2 e ←→ feq = 1/2 [11.65]
Para llevar a cabo la estandarización de unadisolución de KMnO4 0,1 eq L–1 con este patrónprimario, conforme con la reacción de valora-ción:
2 5 + 16 H+ ←→←→ 2 Mn2+ + 10 CO2 + 8 H2O [11.62]
una cantidad exactamente pesada (con exacti-tud de hasta la décima de mg) del mismo (entre0,25 y 0,30 g) se disuelve en 60 mL de agua des-tilada en un erlermeyer de 300 mL. Se añadenunos 15 mL de H2SO4 2,0 mol L–1, se calienta aunos 80 °C y se comienza la valoración median-te la adición de la disolución de KMnO4 desdela bureta de 50 mL. Al comienzo de la valora-ción, la adición de valorante se lleva a cabo gotaa gota ya que la decoloración del MnO–
4 se pro-duce lentamente. Durante el curso de la valora-ción, la velocidad de la reacción se incrementaprogresivamente debido al efecto catalítico delion Mn2+ producido en la reacción. El punto finalviene dado por la aparición de un color rosaapreciable durante 20 o 30 segundos. El color noes permanente, ya que se produce la reacción:
2 3 Mn2+ + 2 H2O ←→←→ 5 MnO2 (S) + 4 H+ [11.66]
que provoca la desaparición del ligero exceso dereactivo que se toma como punto final de la valo-ración.
Como se ha expuesto en otras valoraciones,es común expresar la concentración de la diso-lución de valorante de acuerdo con:
Creal = Cnominal × f [11.67]
donde Cnominal es la concentración que, en princi-pio, debería tener la disolución de KMnO4, Crealsu concentración real y f el factor volumétrico.
C) Tipos de valoraciones con KMnO4
La metodología más utilizada en las valoracio-nes redox con KMnO4 es la valoración directa, enla que la disolución valorante es una disoluciónpatrón de KMnO4, y la disolución a valorar con-tiene el analito, a la que previamente se le añadeácido para conseguir la acidez necesaria para elproceso de valoración. Sin embargo, en ciertoscasos se llevan a cabo valoraciones indirectas quepermiten resolver problemas analíticos concretos,tal como la valoración de sustancias con un carác-ter reductor acusado o incluso la valoración de oxi-dantes. Este modo de operación permite, en el pri-mer caso, obviar los problemas inherentes atrabajar en atmósfera inerte necesaria para evitarla oxidación al aire de las sustancias muy reducto-ras antes y durante la valoración, mientras que, enel segundo caso, la valoración indirecta es una alter-nativa útil, sencilla y práctica a la correspondien-te valoración con una sustancia reductora.
En estas valoraciones indirectas juega unpapel muy importante el sistema redox:
Fe3+ + e ←→ Fe2+ E0 = 0,776 V feq = 1,0 [11.68]
debido a su valor de potencial estándar, que leconfiere una gran reactividad tanto frente a sus-tancias oxidantes como reductoras. Como se mos-trará a continuación, estas determinaciones sebasan en su etapa final en la valoración de unadisolución de Fe2+ con KMnO4.
La valoración directa de especies con un fuer-te carácter reductor, como por ejemplo Ti3+, Cr2+,
MnO4− +
C O2 42−
MnO4− +
C O2 42−
388 Equilibrios iónicos y sus aplicaciones analíticas

V2+, etc., requiere trabajar, como se ha indicado,en unas condiciones experimentales de ausenciade oxígeno. Realmente, estas especies no estánpresentes como tales tras la puesta en disoluciónde una muestra real, como por ejemplo un ace-ro. En efecto, tras disolver el acero en medio áci-do se tendrían los iones Ti4+, Cr3+ y VO+
2, que-dando el hierro como Fe3+. La determinación deestos iones por valoración redox es complicadapor la estabilidad que presentan los indicadosestados de oxidación de estos iones metálicos.Sin embargo, combinando la acción de un trata-miento previo y valoración indirecta con KMnO4el problema se resuelve fácilmente.
El procedimiento consiste, como se muestraen la figura 11.3, en pasar la muestra disuelta poruna columna reductora (reductor de Jones) conlo cual dichos iones pasarían a su estado inferiorde oxidación y recogida de los mismos sobre unadisolución de Fe3+, dando lugar a las siguientesreacciones:
Ti3+ + → Ti4+ + Fe2+ +
[11.69]
Cr2+ + → Cr3+ + Fe2+ +
[11.70]
V2+ + → VO2+ + Fe2+ +
[11.71]
Posteriormente, el Fe2+ formado se valora conuna disolución patrón de KMnO4 de acuerdo conla reacción:
5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O[11.72]
Dado que las reacciones se correspondenequivalente a equivalente, el número de equiva-lentes de KMnO4 consumidos en la valoracióndebe ser igual al de Fe2+ valorados.
FIGURA 11.3. Dispositivo instrumental utilizado para el tra-tamiento previo de una muestra de acero en una columnareductora (reductor de Jones) para la determinación poste-
rior de su contenido en titanio, cromo y vanadio.
La valoración de sustancias oxidantes con diso-luciones patrón de KMnO4 se basa en la acciónintermedia del sistema Fe3+/Fe2+. Se añade unexceso de disolución estandarizada de Fe2+ a ladisolución del analito (oxidante) y se espera has-ta que se complete la reacción, calentando si esnecesario. De forma general, para un oxidanteimplicado en el par redox Ox + n e ←→ Red feq =1/n, esta reacción vendría dada por:
Ox + n ←→ Red + n Fe3+ +
[11.73]
En este caso, la valoración posterior de ladisolución de ion Fe2+ que queda sin reaccionarcon una disolución patrón de KMnO4 permite
Fesin reaccionar2+
Feexceso2+
MnO4− +
Fesin reaccionar3+
Feexceso3+
Fesin reaccionar3+
Feexceso3+
Fesin reaccionar3+
Feexceso3+
Capítulo 11: Valoraciones de oxidación-reducción 389
Ti4+ Cr3+ VO2+
Fe3+ + Fe2+
Fe3+
Ti4+ Cr3+, VO2+
Ti3+ Cr3+ VO2+
(Sensibles al aire)
REDUCTOR JONES
(Amalgama de Zn)
Valoración con KMnO4

determinar mediante cálculos sencillos la con-centración de Ox en la muestra. Así, se puedeestablecer que:
eq Ox = eq Fe2+ añadidos – eq [11.74]
Hay que indicar que en este proceso la espe-cie Red no debe presentar actividad redox en lavaloración con KMnO4.
D) Aplicaciones más relevantes
La valoración de disoluciones de Fe2+ con diso-lución patrón de KMnO4 es una de las valora-ciones de mayor relieve en el ámbito de las per-manganimetrías. Esta valoración es rápida ycuantitativa en medio ácido:
5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O
[11.72]
pero no podría llevarse a cabo en un medio quecontenga iones Cl–, ya que la reacción:
2 10 Cl– + 16 H+ ←→←→ 2 Mn2+ + 5 Cl2 + 8 H2O
[11.63]
que frecuentemente tiene lugar en una extensióndespreciable (por ejemplo, la valoración de As3+ yde Sb3+ con MnO–
4 se lleva a cabo en un medio deHCl 1 o 2 mol L–1, respectivamente), es importan-te en presencia de hierro y conduce a errores porexceso.
El inconveniente se evita fácilmente por adi-ción de la denominada disolución de Zimmer-mann-Reinhardt, que consiste en una mezcla deH2SO4 concentrado, H3PO4 concentrado yMnSO4 y que ejerce varias funciones:
1. El H2SO4 contribuye a asegurar el mediofuertemente ácido necesario para llevar acabo la valoración.
2. El Mn2+ disminuye el potencial del MnO–4,
ya que aumenta la concentración de laforma reducida del par redox.
3. El H3PO4 compleja el Fe3+ (de color ama-rillo), para formar FeH2PO2+
4 (incoloro) loque, por una parte, disminuye el potencialde la disolución antes del punto de equi-valencia (aumenta la exactitud de la deter-minación) y por otra, elimina el color ama-rillo del Fe3+, mejorando la nitidez delpunto final de la valoración.
Hay que indicar que al disolver una muestraque contenga hierro, la disolución siempre con-tiene una cierta cantidad de Fe3+, ya que aún enel caso de que la muestra contenga exclusiva-mente Fe2+ o Fe0 y se disuelva en un ácido no oxi-dante, el Fe3+ se forma rápidamente por la accióndel oxígeno atmosférico. En consecuencia, antesde valorar hierro con permanganato, o cualquierotro oxidante, es necesario proceder a una reduc-ción previa mediante uno de los reactivos indi-cados en el apartado 11.5.1A, preferentementeel SnCl2 o, si es necesario analizar una gran can-tidad de muestras, el reductor de Jones.
La determinación de peróxido de hidrógenoes otra de las aplicaciones de interés de las valo-raciones directas con KMnO4. La reacción devaloración que sirve de base a esta determina-ción es la siguiente:
2 5 H2O2 + 6 H+ ←→←→ 2 Mn2+ + 5 O2 + 8 H2O
[11.75]
en la que el factor de equivalencia para el peró-xido de hidrógeno es 1/2, dado que en el medioácido en el cual se lleva a cabo la misma estáimplicado el siguiente par redox:
O2 + 2 H+ + 2 e ←→ H2O2 [11.76]
Como en otras valoraciones con KMnO4 laacidez necesaria se consigue con H2SO4. Hay queindicar que un apreciable número de iones inor-gánicos catalizan la descomposición del peróxi-
MnO4− +
MnO4− +
MnO4− +
MnO4−
390 Equilibrios iónicos y sus aplicaciones analíticas

do de hidrógeno, por lo que su presencia en elmedio de reacción originaría errores por defec-to. Para evitar este inconveniente se recomiendaañadir ligandos complejantes tales como EDTAy oxina, entre otros. También hay que indicar quealgunas disoluciones comerciales de peróxido dehidrógeno contienen pequeñas cantidades deagentes conservantes, de carácter orgánico, quepodrían oxidarse por la acción del permangana-to y, en consecuencia, provocar resultados con unligero error por exceso.
La determinación del contenido de mangane-so, MnO2, en un mineral tal como la pirolusita esotra de las aplicaciones de las valoraciones conKMnO4. En este caso el ion oxalato actúa comoun agente reductor previo del MnO2, ya que éstees capaz de oxidarlo en medio ácido y en calien-te, a unos 80 °C.
El procedimiento consiste en la adición de unvolumen exactamente medido de disoluciónpatrón de oxalato a la muestra de pirolusita, demanera que se tiene un exceso conocido dereductor. En estas condiciones, se origina lasiguiente reacción:
MnO2 (S) + + 4 H+ →
→ Mn2+ + 2 CO2 + 2 H2O +
[11.77]
A continuación se valora el exceso de oxala-to con la disolución patrón de KMnO4 en basea la reacción [11.62]. Dado que en la reaccióncon el ion oxalato, el MnO2 se reduce a ion Mn2+
y se intercambian 2 e, su factor de equivalenciaserá de 1/2.
11.5.3. Valoraciones con dicromato (dicromatometrías)
El campo de aplicación del dicromato de pota-sio en valoraciones redox es más restringido queel del permanganato debido a su menor carácteroxidante. Sin embargo, presenta la importante
ventaja frente al KMnO4 de que es patrón pri-mario, lo que le permite mantener su vigencia eimportancia como valorante. A pesar del colornaranja intenso que presentan las disolucionesde K2Cr2O7, el propio reactivo no puede utili-zarse como indicador del punto final de la valo-ración, pues su forma reducida, el ion Cr3+, tieneun color verde intenso que dificulta dicha detec-ción. Se usan, por tanto, indicadores visualesexternos del tipo difenilamina o ferroína.
A) Características redox
El par redox en el que se basa el empleo deldicromato como reactivo oxidante en valoracio-nes redox, viene dado por:
14 H+ + 6 e ←→ 2 Cr3+ + 7 H2O E0′ = 1,33 V feq = 1/6
[11.78]
El valor del potencial condicional para estesistema redox, y por consiguiente el carácter oxi-dante del dicromato, depende de la naturaleza yconcentración del ácido empleado. El valor indi-cado en la reacción [11.78] se corresponde a unmedio H2SO4 6,0 mol L–1; si la concentración deeste ácido se reduce a 1,0 mol L–1 el valor de E0′desciende a 1,03 V y si la acidez se ajusta con HCl1,0 mol L–1, el potencial condicional es de 1,0 V.
B) Preparación y conservación de disoluciones de K2Cr2O7
El dicromato de potasio es muy soluble enagua y como ya se ha indicado es patrón prima-rio, por lo que sus disoluciones patrón se puedenpreparar por pesada directa, tras un proceso desecado de la sal a 140-150 °C. Las disolucionesacuosas de K2Cr2O7 son extremadamente esta-bles y se ha comprobado que la concentración deuna disolución 0,1 eq L–1 permanece práctica-
CrO72− +
C O2 42
sin reaccionar−
C O2 42
exceso medido−
Capítulo 11: Valoraciones de oxidación-reducción 391
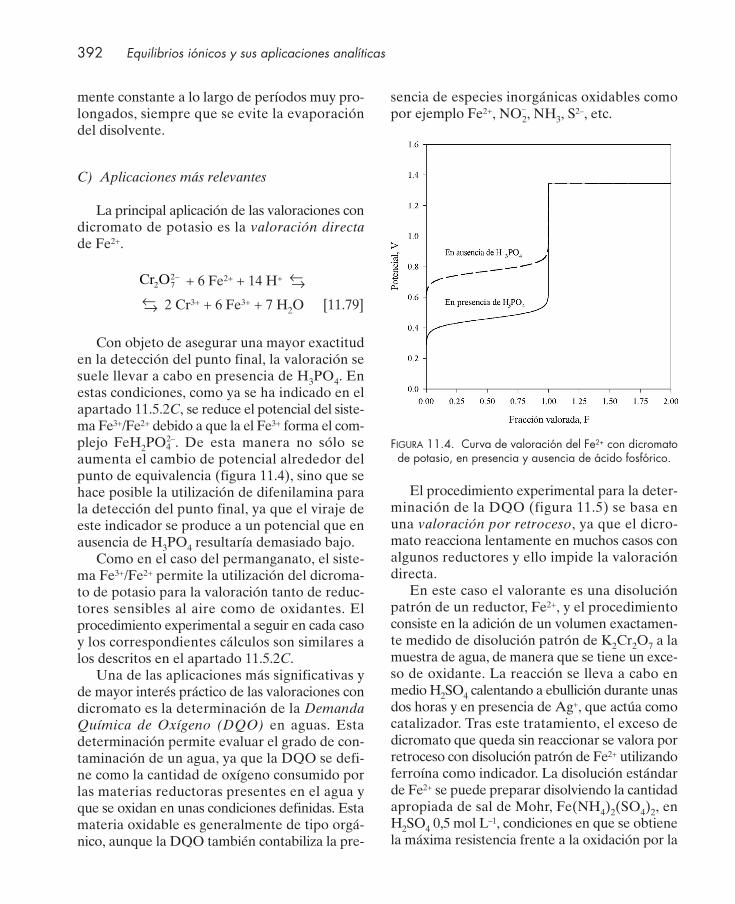
mente constante a lo largo de períodos muy pro-longados, siempre que se evite la evaporacióndel disolvente.
C) Aplicaciones más relevantes
La principal aplicación de las valoraciones condicromato de potasio es la valoración directade Fe2+.
+ 6 Fe2+ + 14 H+ ←→←→ 2 Cr3+ + 6 Fe3+ + 7 H2O [11.79]
Con objeto de asegurar una mayor exactituden la detección del punto final, la valoración sesuele llevar a cabo en presencia de H3PO4. Enestas condiciones, como ya se ha indicado en elapartado 11.5.2C, se reduce el potencial del siste-ma Fe3+/Fe2+ debido a que la el Fe3+ forma el com-plejo FeH2PO4
2–. De esta manera no sólo seaumenta el cambio de potencial alrededor delpunto de equivalencia (figura 11.4), sino que sehace posible la utilización de difenilamina parala detección del punto final, ya que el viraje deeste indicador se produce a un potencial que enausencia de H3PO4 resultaría demasiado bajo.
Como en el caso del permanganato, el siste-ma Fe3+/Fe2+ permite la utilización del dicroma-to de potasio para la valoración tanto de reduc-tores sensibles al aire como de oxidantes. Elprocedimiento experimental a seguir en cada casoy los correspondientes cálculos son similares alos descritos en el apartado 11.5.2C.
Una de las aplicaciones más significativas yde mayor interés práctico de las valoraciones condicromato es la determinación de la DemandaQuímica de Oxígeno (DQO) en aguas. Estadeterminación permite evaluar el grado de con-taminación de un agua, ya que la DQO se defi-ne como la cantidad de oxígeno consumido porlas materias reductoras presentes en el agua yque se oxidan en unas condiciones definidas. Estamateria oxidable es generalmente de tipo orgá-nico, aunque la DQO también contabiliza la pre-
sencia de especies inorgánicas oxidables comopor ejemplo Fe2+, NO–
2, NH3, S2–, etc.
FIGURA 11.4. Curva de valoración del Fe2+ con dicromatode potasio, en presencia y ausencia de ácido fosfórico.
El procedimiento experimental para la deter-minación de la DQO (figura 11.5) se basa enuna valoración por retroceso, ya que el dicro-mato reacciona lentamente en muchos casos conalgunos reductores y ello impide la valoracióndirecta.
En este caso el valorante es una disoluciónpatrón de un reductor, Fe2+, y el procedimientoconsiste en la adición de un volumen exactamen-te medido de disolución patrón de K2Cr2O7 a lamuestra de agua, de manera que se tiene un exce-so de oxidante. La reacción se lleva a cabo enmedio H2SO4 calentando a ebullición durante unasdos horas y en presencia de Ag+, que actúa comocatalizador. Tras este tratamiento, el exceso dedicromato que queda sin reaccionar se valora porretroceso con disolución patrón de Fe2+ utilizandoferroína como indicador. La disolución estándarde Fe2+ se puede preparar disolviendo la cantidadapropiada de sal de Mohr, Fe(NH4)2(SO4)2, enH2SO4 0,5 mol L–1, condiciones en que se obtienela máxima resistencia frente a la oxidación por la
Cr O2 72−
392 Equilibrios iónicos y sus aplicaciones analíticas

acción del oxígeno atmosférico, aunque aún así laestabilidad de estas disoluciones no supera un día;de hecho lo más práctico es preparar una disolu-ción de Fe2+ de concentración aproximada y estan-darizarla con dicromato de potasio justo antes desu utilización.
La DQO se expresa en términos de mg de oxí-geno consumido por litro de agua. Si se conside-ra que se parte de un volumen de agua, Vagua, yque el factor de equivalencia para el oxígeno enesta reacción es 1/4 a tenor del par redox:
O2 + 4 H+ + 4 e ←→ 2 H2O [11.80]
en el que encuentra implicado, los cálculos a rea-lizar para la determinación de la DQO son lossiguientes:
meq – meq Fe2+valoración =
= meq = meq Materia oxidable =
= meq Oxígeno [11.81]
[11.82]
de donde queda finalmente:
[11.83]
En esta expresión, los volúmenes de dicro-mato y de Fe2+ se expresan en mL y las respecti-vas concentraciones en eq L–1.
11.5.4. Valoraciones en las que interviene el yodo
Este tipo de volumetrías presenta un graninterés en el contexto de las valoraciones redoxdado el valor intermedio del potencial estándardel sistema yodo/yoduro, E0 = 0,535 V, que per-mite que el ion I– sea oxidado por gran cantidadde sustancias y también que el I2 pueda oxidar aalgunas. Este comportamiento dual ha permiti-do el desarrollo de dos tipos de volumetrías.
Métodos directos, o yodimetrías (valoracionesCON yodo): se utiliza una disolución patrón deyodo como valorante:
I2 + Analito reductor ←→←→ 2 I– + Producto oxidación [11.84]
DQO
( ) 8mg O LCr O Cr O Fe Fe
agua2
12 72
2 72 2 2
=× − × ×− − + + −
V C V C
V L
× × ×
−
1 meqO1 meqCr O
1mmol O4 meqO
32 mg O1mmol O
2
2 72
2
2
2
2
DQO
( ) meq Cr O
V LCr O Cr O Fe Fe 2 7
2
agua
2 72
2 72 2 2
=× − ×
×− − + +
−V C V C
Cr O2 7
2que reaccionan
−
Cr O2 72
añadidos−
Capítulo 11: Valoraciones de oxidación-reducción 393
FIGURA 11.5. Fundamento de la determinación de la Demanda Química de Oxígeno (DQO) en una muestra de agua contaminada.
TRATAMIENTO DE LA MUESTRA DE AGUA
VALORACIÓN POR RETROCESO

De las diferentes especies que se puedendeterminar mediante esta valoración directa conyodo cabe citar AsO3–
3 , Sb3+, Sn2+, H2S, SO2–3 ,
hidracina, etc., entre otras.Métodos indirectos o yodometrías (valoracio-
nes DE yodo). En este caso se aprovecha elcarácter reductor suave del ion yoduro para ladeterminación indirecta de numerosos oxidan-tes. El proceso consta de dos etapas:
1. Adición de un exceso de ion yoduro a ladisolución que contiene el analito de carác-ter oxidante:
2 I–exceso + Analito oxidante ←→
←→ I2 + Producto reducción [11.85]
2. Valoración del yodo producido en la reac-ción anterior con una disolución patrón decarácter reductor, generalmente tiosulfa-to de sodio:
I2 + 2 → 2 I– + [11.86]
El campo de aplicación se extiende a todas lassustancias cuyo potencial estándar sea superioral del sistema I2/2I–. Entre ellas se pueden men-cionar: AsO3–
4 , IO–3, BrO2
3, ClO–3, Cl2, NO–
2, Sb5+,Fe3+, H2O2, Cu2+, etc.
A) Características redox
El par yodo/yoduro se caracteriza por la semi-reacción redox
I2 + 2 e ←→ 2 I– E0 = 0,535 V feq (I2) = 1/2 feq (I–) = 1,0 [11.87]
la cual, como ya se ha indicado, muestra que elyodo y el yoduro tienen un carácter redox rela-tivamente débil. Sobre este sistema redox influ-ye de manera apreciable la concentración de ionyoduro presente en el medio de reacción, debi-do a la formación del complejo triyoduro:
I2 + I– ←→ [11.88]
A pesar de que el valor de β1 no es muy ele-vado, la formación de este complejo es de granimportancia, ya que por una parte hace que elyodo sea soluble en agua (el I2 es una moléculacovalente y, por lo tanto, muy poco soluble endisolventes polares) y por otra reduce su volati-lidad, lo que aumenta la estabilidad del yodo endisolución.
Otra importante característica de este siste-ma redox es que su potencial estándar no se afec-ta apreciablemente por el pH. En efecto, a par-tir de la semireacción redox dada en la expresión[11.87] no se infiere la influencia de esta varia-ble. Sin embargo, a valores extremos de pH seproducen reacciones colaterales, que se han decontrolar con objeto de evitar errores indesea-bles en el proceso de valoración.
En medio básico (pH > 9,0), el yodo se dis-muta en yoduro e hipoyodito:
I2 + 2 OH– ←→ I– + IO– + H2O [11.89]
La manera en que esta reacción puede afec-tar a los resultados no está clara. Es evidente queprovoca una disminución de la cantidad de yodopresente en disolución, aunque ello no tendríapor qué ser especialmente grave, ya que el hipo-yodito también tiene carácter oxidante y el fac-tor de equivalencia sería idéntico al del yodo. Sinembargo, en el caso de la reacción con tiosulfa-to de sodio:
I2 + 2 → 2 I– + [11.86]
4 IO– + + H2O → 4 I– + 2 + 2 H+
[11.90]
es evidente que la presencia de hipoyodito en ladisolución provocará un considerable error pordefecto.
SO42−
S O2 32−
S O4 62−
S O2 32−
β1
3
2
[I ][I ][I ]
= =−
−710 I3
−
S O4 62−
S O2 32−
394 Equilibrios iónicos y sus aplicaciones analíticas

En medio ácido, debido a que se incrementael potencial redox del oxígeno, éste es capaz deoxidar las disoluciones de yoduro a yodo:
4 I– + O2 + 4 H+ ←→ 2 I2 + 2 H2O [11.91]
Esta reacción está catalizada por iones metá-licos de número de oxidación variable, particu-larmente el cobre, y además es de naturale-za fotoquímica. Este es una de los motivos porlos que en las yodometrías, que generalmentese llevan a cabo en medio ácido, es aconsejablevalorar lo antes posible el yodo formado. (Otromotivo es la ya comentada volatilidad delyodo.)
B) Disoluciones valoradas
Para las yodimetrías se utiliza, por lo general,una disolución patrón de yodo, mientras que enlas yodometrías el yodo formado se valora habi-tualmente con una disolución patrón de tiosul-fato de sodio (Na2S2O3).
Las disoluciones de yodo se suelen prepararen presencia de un exceso de yoduro de potasio(se forma el complejo I–
3), con objeto de favore-cer su solubilidad en agua y la estabilidad de lasmismas. Aunque es relativamente fácil obteneryodo de calidad patrón primario (para ello bas-ta con sublimar yodo comercial), el sólido se eva-pora mientras se pesa y, además, las disolucionesson poco estables, por lo que lo más común espreparar una disolución de yodo de concentra-ción aproximada y seguidamente estandarizarlacon un patrón primario adecuado, por lo gene-ral el As2O3. Este compuesto se disuelve fácil-mente en medio básico
As2O3 + 3 H2O → 2 + 2 H+ [11.92]
y, aunque en estas condiciones se oxida con rela-tiva facilidad por la acción del oxígeno, basta conacidificar la disolución para obtener ácido arse-nioso, H3AsO3, estable durante largos períodosde tiempo.
El yodo reacciona con el ácido arseniososegún la reacción:
I2 + H3AsO3 + H2O ←→ 2 I– + H3AsO4 + 2 H+
[11.93]
en la que está implicado el sistema redoxH3AsO4/H3AsO3, siendo el factor de equivalen-cia para el As2O3 de 1/4.
Esta reacción depende en gran medida del pHdel medio. Así, el par redox As5+/As3+ se encuen-tra doblemente influido por el pH debido: a ladiferente proporción de oxígeno entre la formaoxidada y reducida del par, y a la posible diso-ciación de los ácidos H3AsO4 y H3AsO3 al variarel pH de la valoración. Así, se tiene:
pH < 2,3 H3AsO4 + 2 H+ + 2 e ←→ H3AsO3 + H2O
[11.94]
2,3 < pH < 4,4+ 3 H+ + 2 e ←→ H3AsO3 + H2O
[11.95]
4,4 < pH < 9,2+ 4 H+ + 2 e ←→ H3AsO3 + H2O
[11.96]
pH > 9,2+ 4 H+ + 2 e ←→ + H2O
[11.97]
Este comportamiento se ha de conjugar conel que manifiesta el sistema I2/2I– en función delpH, comentado anteriormente. Tras un estudioen profundidad de estos sistemas redox, se reco-mienda llevar a cabo la valoración a valores depH comprendidos entre 4,0 y 9,0 con objeto deasegurar la rapidez y cuantitividad de la reacción[11.93]. Para su ajuste se pueden emplear diso-luciones reguladoras preparadas a partir deNa2HPO4 o bórax, pero lo más habitual es aña-dir hidrogenocarbonato de sodio a la disoluciónácida de H3AsO3, con lo que se obtiene un pHaproximadamente igual a 8.
H AsO2 3−AsO4
3−
HAsO42−
H AsO2 4−
H AsO2 3−
Capítulo 11: Valoraciones de oxidación-reducción 395

C) Preparación, conservación y estandarizaciónde disoluciones de Na2S2O3
Aunque el As2O3 es patrón primario, no es habi-tual utilizarlo en yodometría y en su lugar se empleael tiosulfato de sodio – que no es patrón primario–para valorar el yodo formado por la reacción deyoduro con un oxidante. La razón fundamental esque, como ya se ha comentado, la valoración delyodo con ácido arsenioso debe llevarse a cabo a unpH superior a 4, mientras que muchas yodometríasse realizan en medio francamente ácido. Otra razónes la peligrosidad del arsénico.
La reacción redox entre el yodo y el tiosulfa-to en la que éste se oxida a tetrationato:
I2 + 2 → 2 I– + feq ( ) = 1,0
[11.86]
es peculiar, ya que la mayoría de los oxidantesoriginan SO2–
4 o SO2–3 . Esta reacción irreversible
es rápida y cuantitativa a valores de pH infe-riores a 7,0, en medio básico el yodo se dismu-ta de acuerdo con la reacción [11.89] y el hipo-yodito formado oxida parcialmente el tiosulfatoa ion sulfato (ver reacción [11.90]). Como sis-tema indicador del punto final de esta valora-ción se utiliza engrudo de almidón, que formaun complejo con el yodo de intenso color azul.La desaparición de este color pone de mani-fiesto el final de la misma.
Las disoluciones de tiosulfato se preparangeneralmente por disolución de tiosulfato desodio pentahidrato, Na2S2O3. 5 H2O [en sentidoestricto, el nombre sería tiosulfato de sodio-agua(1:5)], que no es patrón primario. Para ello serequiere agua de elevada pureza, preferiblementehervida recientemente, ya que el posible CO2disuelto es capaz de dar la acidez necesaria paraque se produzca la reacción:
2 + H+ → + S(S) [11.98]
Pese a la circunstancia de que sus disolucio-nes sean inestables en medio ácido, es posible
utilizar tiosulfato para valorar yodo en medio áci-do, ya que la reacción con el yodo es mucho másrápida que la reacción anterior.
Por otra parte, la presencia de impurezas demetales pesados origina la oxidación catalíticadel tiosulfato por el aire:
+ 1/2 O2 → S (S) + [11.99]
y la presencia de microorganismos (bacterias)favorece esta reacción de oxidación, dado queconsumen azufre; por esta razón es recomenda-ble agregar una pequeña cantidad de clorofor-mo, Hg2I o benzoato de sodio (bactericidas queinhiben el crecimiento microbiano) a las disolu-ciones de tiosulfato de sodio.
Otro aspecto importante en la conservaciónde las disoluciones de Na2S2O3 es el pH de lasmismas, ya que este compuesto se descomponeen medio ácido (pH < 4,5). La adición de unapequeña cantidad de Na2CO3 a las disolucionesde tiosulfato, de forma que el pH se sitúe entre9 y 10, favorece enormemente su estabilidad, aun-que una alcalinidad excesiva las hace de nuevoinestables. En resumen, si se origina una turbi-dez en la disolución, es mejor desecharla y pre-parar otra nueva.
Para estandarizar las disoluciones de tiosul-fato se suele utilizar como patrón primario yoda-to de potasio. Esta sustancia se puede encontraren el comercio con una pureza del 99,99%, y parapoder utilizarla como patrón primario se ha dedesecar en estufa a una temperatura de 180 °Cdurante unas 12 horas. El uso analítico de estepatrón primario para estandarizar disolucionesde tiosulfato, se basa en la generación cuantita-tiva de yodo a partir de su reacción con excesode yoduro de potasio en medio ácido:
+ 5 I– + 6 H+ ←→ 3 I2 + 3 H2O [11.100]
Dado que un mol de yodato de potasio origi-na tres moles de yodo y que por cada mol deyodo en su valoración con tiosulfato se consu-men dos electrones:
IO3−
SO42−
S O2 32−
HSO3−
S O2 32−
S O2 32−
S O4 62−
S O2 32−
396 Equilibrios iónicos y sus aplicaciones analíticas

I2 + 2 → 2 I– + [11.86]
se tiene que en la valoración de yodato con tio-sulfato a través del yodo generado se intercam-bian 6 electrones, por lo que el factor de equi-valencia para el yodato de potasio será 1/6. Lareacción [11.100] presenta un gran interés analí-tico, como se mostrará en el apartado corres-pondiente a problemas numéricos.
Para llevar a cabo la estandarización de unadisolución de Na2S2O3 0,1 eq L–1 con yodato depotasio, una cantidad exactamente pesada (conprecisión de hasta la décima de mg) del mismodel orden de 0,15 g, se disuelve en 25 mL de aguadestilada en un enlermeyer de 300 mL. Se aña-den 2 g de KI exentos de iodato, pesados en gra-natario y 10 mL de H2SO4 0,5 mol L–1. El ordende adición de los reactivos indicado se ha de cum-plir estrictamente con objeto de evitar en lo po-sible pérdidas de yodo. Se comienza la valora-ción mediante la adición de forma rápida de unos10 mL de la disolución de Na2S2O3 desde la bure-ta de 50 mL. Se continúa valorando gota a gotahasta que la disolución de color marrón, pasa aamarillo pálido. En este instante se añade engru-do de almidón como indicador y se continúa has-ta la decoloración del color azul violáceo forma-do tras la adición del almidón. El cambio de colorocurre bruscamente.
D) Aplicaciones más relevantes
Las valoraciones en que interviene el yodopresentan un importante y amplio campo de apli-cación, que se extiende tanto a la determinaciónde especies inorgánicas como de compuestosorgánicos. En concreto, la determinación dereductores se lleva a cabo mediante las yodime-trías, debido al carácter oxidante débil del yodo.Es posible determinar algunas especies inorgá-nicas, tales como As3+, Sn2+, S2–, etc., medianteeste método, pero su verdadero campo de apli-cación es la determinación de compuestos orgá-nicos, como por ejemplo fenoles, ácido ascórbi-co, mercaptanos, etc.
Dado que el campo de aplicación de las yodi-metrías está orientado básicamente hacia la valo-ración de compuestos orgánicos, cuya descrip-ción cae fuera de los objetivos de este libro, comoaplicación más significativa del uso de las valo-raciones directas con disolución patrón de yodose comentará la determinación de agua por elmétodo de Karl Fischer.
Este método es el recomendado para ladeterminación del contenido de agua en una granvariedad de muestras, como por ejemplo en ali-mentos, fármacos y disolventes orgánicos, sien-do particularmente útil cuando dicho contenidoes inferior al 0,1%. También se puede emplearen muchas determinaciones indirectas basadasen reacciones cuantitativas que consumen o pro-ducen agua. El método se basa en la valoracióndel agua, disuelta en metanol, con el denomina-do reactivo de Karl-Fischer, una disolución quecontiene dióxido de azufre y yodo disueltos enpiridina (C5H5N) y metanol.
La reacción que tiene lugar es la siguiente:
C5H5N·I2 + C5H5N·SO2 + C5H5N + H2O →→ 2 C5H5N · HI + C5H5N·SO3
[11.101]
A continuación, en presencia de una cantidadelevada de metanol se produce la reacción:
C5H5N·SO3 + CH3OH → C5H5N(H)SO4CH3
[11.102]
que impide la reacción del complejo piridina-dióxido de azufre con el agua
C5H5N·SO3 + H2O → C5H5NHSO4H[11.103]
que también consumiría agua. Por tanto, la reacción global es la que se indi-
ca a continuación:
I2 + SO2 + H2O + CH3OH + 3 C5H5N →→ 2 C5H5N · HI + C5H5N(H)SO4CH3
[11.104]
S O4 62−
S O2 32−
Capítulo 11: Valoraciones de oxidación-reducción 397

Esta reacción implica una estequiometría de1 mol de yodo, 1 mol de dióxido de azufre y tresmoles de piridina por cada mol de agua. En lapráctica el reactivo utilizado contiene exceso depiridina y de dióxido de azufre, por lo que el fac-tor que determina su capacidad de reacción conel agua es la cantidad de yodo que contiene.
El reactivo de Karl-Fischer evidentemente noes patrón primario, por lo que debe ser estanda-rizado. Además es poco estable (aunque la des-composición más importante se produce en lashoras posteriores a su preparación; transcurridas24 horas su estabilidad aumenta) por lo que esconveniente llevar a cabo dicha estandarizacióninmediatamente antes de utilizarlo. Como patrónprimario es habitual emplear agua disuelta enmetanol, aunque también es posible hacer usode una sal con agua de cristalización, asimismodisuelta en metanol.
Para llevar a cabo la determinación de agua,las muestras se disuelven en metanol, si es posi-ble. En caso de que sean insolubles, el agua quecontienen se extrae con metanol anhidro, dejan-do un tiempo de maceración para que la extrac-ción sea completa. La disolución de agua enmetanol se valora con el reactivo de Karl-Fischer,adicionado desde la bureta. El punto final sedetecta visualmente por el color marrón produ-cido debido al exceso de yodo o bien potencio-métricamente mediante la técnica de dead-stopcon electrodos de platino. Se debe tener una pre-caución especial con la propia humedad atmos-férica, tanto en la conservación de los reactivoscomo en el desarrollo de la valoración. El límitede detección para esta metodología se sitúa en0,3 mg de agua. En la actualidad existen diver-sos dispositivos automatizados para llevar a caboesta determinación, la mayoría de ellos con detec-ción amperométrica.
La determinación de especies con carácteroxidante se lleva a cabo mediante las yodome-trías, en las que se aprovecha el carácter débil-mente reductor del ion yoduro. Aunque se po-drían comentar una gran cantidad de métodosbasados en este tipo de valoraciones, se ha opta-do por la descripción pormenorizada de sólo dos
de ellos: determinación de cobre, y determina-ción de compuestos clorados, dado su gran inte-rés práctico.
La determinación yodométrica de cobre sebasa en la reacción:
2 Cu2+ + 4 I– ←→ 2 CuI(S) + I2 feq (Cu2+) = 1,0[11.105]
y en la posterior valoración del yodo generadocon una disolución patrón de tiosulfato.
Esta reacción es un ejemplo claro de la apli-cación práctica de los equilibrios iónicos con-currentes, en este caso en valoraciones redox. Enefecto, de acuerdo con los valores de los poten-ciales estándar de los pares redox I2/2I– (E0 = 0,535V) y Cu2+/Cu+ (E0 = 0,153 V) es imposible que elCu2+ pueda oxidar el ion yoduro a yodo. Sinembargo, debido a la formación de la especie inso-luble CuI, el potencial condicional del sistema delcobre se incrementa hasta 0,86 V aproximada-mente, que es suficiente para llevar a cabo la oxi-dación del ion yoduro. Este ion, por tanto, pre-senta una doble función en esta reacción, ya queactúa como reductor y agente precipitante.
A pesar de que los iones H+ no aparecen enla reacción [11.105], su efecto sobre la misma esmuy importante. Así, no es recomendable tra-bajar a pH > 4,0 ya que se origina la hidrólisis delion Cu2+ o la precipitación de su hidróxido y lareacción se hace muy lenta. Como límite inferiorde pH se suele tomar el valor de 0,5, puesto queuna mayor acidez favorece la oxidación del ionyoduro por el aire, de acuerdo con la reacción[11.91], dando lugar a errores por exceso. Unamanera sencilla de obtener un pH adecuado con-siste en añadir un exceso de NH4HF2 a la diso-lución; este compuesto presenta la ventaja adi-cional de que compleja al Fe3+, con lo que seelimina la interferencia de las trazas de este ionque pudieran estar presentes en la disolución.
La adsorción del yodo por el CuI, que se apre-cia porque el precipitado es crema en lugar deblanco, provoca errores por defecto, aunque porlo general no suelen ser muy elevados. En cual-quier caso, para obtener resultados exactos es
398 Equilibrios iónicos y sus aplicaciones analíticas

necesario valorar primero todo el yodo presen-te en la disolución con una disolución patrón detiosulfato y una vez se produce la decoloracióndel indicador (almidón) se añade KSCN oNH4SCN sólido: el CuSCN es más insoluble queel CuI, con lo que el yodo adsorbido en éste que-da liberado y pasa a la disolución. Seguidamen-te se prosigue la valoración hasta nueva decolo-ración del indicador.
La determinación del contenido de cloro enlejías y otros agentes blanqueantes o en produc-tos para piscinas, es una interesante aplicaciónde las valoraciones redox y en concreto de lasyodometrías. El procedimiento se basa en la reac-ción:
Cl2 + 2 I– ←→ 2 Cl– + I2 [10.106]
Sin embargo, los productos mencionados con-sisten por lo general en una disolución alcalinade hipoclorito o de clorito, o bien en una mezclade hipoclorito y cloruro. Ello no es inconveniente,ya que basta con acidificar para que se produz-can las reacciones siguientes:
ClO– + 2 I– + 2 H+ ←→ I2 + Cl– + H2O
[11.107]
ClO–2 + 4 I– + 4 H+ ←→ 2 I2 + Cl– + 2 H2O
[11.108]
ClO– + Cl– + 2 I– + 2 H+ ←→ I2 + 2 Cl– + H2O[11.109]
A la vista de estas reacciones se pueden extraerlas siguientes conclusiones:
1. El cloro como tal en disolución da lugar a:
1 mol Cl2 → 1 mol I2
2. Si el cloro se hidroliza parcialmente dan-do lugar a hipoclorito, se tiene:
1 mol Cl2 → 1 mol ClO– → 1 mol I2
3. Si el cloro se hidroliza parcialmente dan-do lugar a clorito, se tiene:
2 mol Cl2 → 1 mol ClO–2 → 2 mol I2
Se observa que sea cual sea la especie que exis-ta en disolución y el grado de hidrólisis que puedapresentar el cloro, siempre se cumple la equiva-lencia de que 1 mol Cl2 → 1 mol I2, por lo que elresultado de la valoración se expresa como conte-nido de cloro útil o cloro disponible en la muestraanalizada. El factor de equivalencia para el cloroen esta valoración es 1/2. La valoración se suele lle-var a cabo en medio H2SO4 con objeto de asegu-rar que la reacción [11.108] sea cuantitativa. Si noexiste ClO–
2 en la muestra, la valoración se puededesarrollar en medio ácido acético.
Capítulo 11: Valoraciones de oxidación-reducción 399
Cuestiones
11.1. El potencial en el punto de equivalencia de una valoración redox depende de:
[ ] La concentración del analito a valorar[ ] El pH del medio de reacción[ ] El indicador redox utilizado en la misma[ ] El número de electrones intercambiados entre ambos sistemas redox

11.2. El salto de potencial en una valoración redox depende de:
[ ] Valores relativos de los potenciales normales de los pares redox implicados[ ] Concentración de las especies reaccionantes[ ] Indicador redox empleado en la valoración[ ] pH del medio de reacción
11.3. Un indicador redox tiene un potencial condicional de 0,15 V e intercambia dos electrones. ¿Cuál sería suzona de viraje?
Zona de viraje: ................................................................................................................
Indicar si es adecuado para:
[ ] Valoraciones con un oxidante[ ] Valoraciones con un reductor[ ] Ambos tipos de valoraciones
11.4. ¿Qué causas de error de las siguientes, sólo se originan en las valoraciones redox?
[ ] Error de valoración[ ] Cinética lenta[ ] Bureta mal calibrada[ ] Pesada incorrecta del patrón[ ] Interacción de O2 con la disolución valorante [ ] Diferencia entre el punto final y el punto de equivalencia[ ] Eliminación incompleta del reactivo del tratamiento previo[ ] Indicador en mal estado
11.5. ¿Por qué las valoraciones redox se llevan a cabo preferentemente en medio ácido?
11.6. Los tratamientos previos en valoraciones redox se utilizan para:
[ ] Transformar al analito a un estado de oxidación adecuado[ ] Preparar la disolución estandarizada del valorante[ ] Asegurar que todo el analito se encuentre en un único estado de oxidación[ ] Preparar la disolución del indicador para que vire correctamente en el punto final
11.7. Indicar los factores que afectan a la estabilidad de una disolución de KMnO4.
11.8. ¿Por qué es necesario utilizar como tratamiento previo el reductor de Jones u otro pre-reductor en la deter-minación del contenido de hierro en un mineral por valoración con KMnO4?
11.9. Si se disuelve una muestra en HCl y se pretende valorar su contenido de hierro con un reactivo oxidante,el tratamiento previo deberá ser:
[ ] Una reducción[ ] Una oxidación
El reactivo oxidante:
[ ] K MnO4[ ] K2Cr2O7
400 Equilibrios iónicos y sus aplicaciones analíticas

y el pH:
[ ] Ácido[ ] Básico
11.10. ¿Qué es la demanda química de oxígeno?
[ ] Un indicador global de la contaminación química del agua[ ] Un indicador de la capacidad oxidante de una muestra[ ] Una medida del oxígeno necesario para una oxidación[ ] La concentración de O2 (mg L–1) necesaria para oxidar a los reductores de un litro de agua[ ] La cantidad de oxígeno requerida para la respiración de un ser vivo
11.11. En la reacción redox originada entre los pares redox Cr2O72–/2Cr3+ e Fe3+/Fe2+, ¿en qué condiciones de pH
oxidaría el Fe3+ al Cr2+?
[ ] Ácido[ ] Neutro[ ] Básico
¿Cuál es el factor de equivalencia del Cr6+ y Cr3+ en esta reacción redox?
11.12. ¿Cuál es el factor de equivalencia del KIO3 en la siguiente reacción:
IO–3+ 5 I– + 6 H+ ←→ 3 I2 + 3 H2O
11.13. ¿Qué factores afectan la estabilidad de una disolución de tiosulfato?
[ ] Acidez[ ] CO2 atmosférico[ ] O2 atmosférico[ ] Humedad ambiental[ ] Presencia de microorganismos
11.14. Indicar (marcando con una X ) los parámetros que afectan a la estabilidad de las siguientes disoluciones:
Acidez O2 Atmosférico Luz
KMnO4
KI
Na2S2O3
11.15. El par redox Fe3+/Fe2+ tiene una gran utilidad en valoraciones redox, y así, por ejemplo, está implicado en la:
[ ] Determinación de sustancias muy reductoras con KMnO4[ ] Determinación de pirolusita con KMnO4[ ] Determinación de oxidantes con K2Cr2O7
Capítulo 11: Valoraciones de oxidación-reducción 401

• Objetivo pedagógicoEjercitar al alumno en el manejo de aspectos relacio-nados con las curvas de valoración redox y selecciónde indicadores, así como en las aplicaciones más rele-vantes de estas valoraciones, haciendo uso de valo-rantes oxidantes como permanganato y dicromato depotasio. Finalmente se abordan problemas numéricossobre las aplicaciones de las valoraciones en las queinterviene el yodo.
Curvas de valoración e indicadores
1. Se valora una disolución de Fe2+ 10–2 eq L–1 con Cr2O2–7
0,1 eq L–1 en un medio en el que [H+] = 1 mol L–1. Cal-cular el valor del potencial cuando la fracción valo-rada toma los valores de 90%, 99%, 100%, 101% y110% (despreciar el efecto de la dilución en la valo-ración).
= 1,33 V; = 0,771 V
La reacción de valoración en la que se fundamen-ta esta valoración redox es:
+ 6 Fe2+ + 14 H+ ←→ 2 Cr3+ + 6 Fe3+ + 7 H2O
Cuando la fracción valorada toma los valores de90 y 99% el proceso de valoración se encuentra en lazona anterior al punto de equivalencia, queda Fe2+
sin valorar, y en estas condiciones, de acuerdo con laexpresión:
el potencial vendrá dado por:
90% Valoración:
F = 0,90 0,827 V
99% Valoración:
F = 0,99 0,889 V
Para calcular el potencial en el punto de equiva-lencia de esta valoración no se puede hacer uso de laexpresión general, dado que la relación entre la for-ma oxidada y reducida del par redox que actúa comooxidante, Cr2O
2–7 /2Cr3+, no es 1:1, como en la gran
mayoría de sistemas redox.En este caso, se tienen las siguientes relaciones de
concentración en el punto de equivalencia, de acuer-do con la reacción de valoración:
[Fe2+] = 6 [ ]
2 [Fe3+] = 6 [Cr3+] ⇒ [Fe3+] = 3 [Cr3+]
Con objeto de obtener una expresión útil para cal-cular el potencial en el punto de equivalencia, se sigueun razonamiento similar al expuesto en el apartado10.7, esto es, se aplica la expresión de Nernst paracada sistema redox:
Si se multiplica por seis ambos miembros de laexpresión del potencial para el sistema que actúacomo valorante, y en ella se hace [H+]14 = 1,0 dadoque se trabaja a pH = 0, y después se suman ambasexpresiones, se tiene:
Si se sustituye en esta expresión la relación de con-centraciones dadas en el punto de equivalencia,resulta:
o bien en función de la concentración de Fe3+:
7 8 751 0 059E = +
+, , log
32[Fe ]3
7 8 751 0 059E = +
+, , log
12[Cr ]3
7 8 751 0 059E = ++ −
+ +, , log
[Fe ][Cr O ][Fe ][Cr ]
32 7
2
2 3 2
E = +− +
+1 33
0 0596
,,
log[Cr O ][H ]
[Cr ]2 7
2 14
3 2
E = ++
+0 771 0 059, , log
[Fe ][Fe ]
3
2
Cr O2 72−
E = +
−=0 771 0 059
0 991 0 99
, , log,
,
E = +
−=0 771 0 059
0 91 0 9
, , log,
,
E En
FF
= +−2
0
2
0 0591
,log
Cr O2 72−
EFe /Fe0
3 2+ +ECr O /2 Cr0
2 72 3− +
402 Equilibrios iónicos y sus aplicaciones analíticas
Seminarios: problemas gráficos

Dado que se parte de una disolución de Fe2+ 10–2
eq L–1 (feq = 1,0), ésta será, por tanto, 10–2 mol L–1, yen el punto de equivalencia [Fe3+] = 10–2 mol L–1. Alsustituir en la expresión anterior se obtiene un poten-cial de 1,268 V.
Tras el punto de equivalencia, esto es, para valoresde fracción valorada del 101 y 110%, se ha de aplicarla expresión de Nernst correspondiente al sistemaCr2O
2–7 /2Cr3+ a pH = 0, esto es:
Las concentraciones de Cr2O2–7 y Cr3+, a sustituir en
esta expresión, se calculan considerando el exceso devalorante adicionado tras el punto de equivalencia. Eneste punto se puede establecer:
10–2 mol L–1 = [Fe2+] = 6 [Cr2O2–7 ] ⇒
⇒ mol L–1
10–2 mol L–1 = [Fe3+] = 3 [Cr3+] ⇒
⇒ mol L–1
La concentración de ion Cr3+ es la misma sea cualsea el exceso de valorante, dado que se ha valoradotodo el Fe2+ presente en la muestra; sin embargo, laconcentración de ion dicromato sí que cambia cuan-do se adiciona un exceso más o menos elevado delmismo. De acuerdo con los valores del problema, setiene:
101% Valoración
= 1,67 × 10–5 mol L–1
110% Valoración
=1,67 × 10–4 mol L–1
de donde los valores de potencial son 1,332 y 1,341V, respectivamente.
2. a) ¿Es cuantitativa en el punto de equivalencia lavaloración de ácido arsenioso con Ce4+ en medioH2SO4 si [H+] = 1 mol L–1? b) ¿Podría emplearseferroína como indicador del punto final de esta valo-ración con un error del 0,1%?
= 0,56 V; = 1,44 V; =1,10 V.Sol.: a) sí; b) sí.
3. El método recomendado para la determinación del con-tenido de hierro en una muestra es su valoración redoxcon KMnO4. Calcular: a) el valor del pH para que estareacción sea cuantitativa al 99,9%. b) La región de equi-valencia (± 0,1%) de la curva de valoración de Fe2+ 0,1mol L–1 con KMnO4 0,02 mol L–1 a pH = 1. Indicarcómo influiría en la curva de valoración un aumentode pH y una disminución de la concentración de ana-lito.
= 0,771 V; = 1,51 V.
La valoración de hierro con permanganato se basaen la siguiente reacción redox:
+ 5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O
cuya constante de equilibrio viene dada por:
y se puede calcular teniendo en cuenta la expresión[10.40]:
Para que esta reacción sea cuantitativa al 99,9%,se ha de cumplir para el analito:
[Fe3+] = mol L–1
[Fe2+] = mol L–1
de donde:
y para el valorante:
5 mol L–1
5 mol L–1
[Mn ] [Fe ]2 3+ += = ×
99 9100
0 1,
,
[MnO ] [Fe ]4
2− += = ×0 1100
0 1,
,
[Fe ][Fe ]
3
2
+
+= ≈
99 90 1
103,,
0 1100
0 1,
,×
99 9100
0 1,
,×
log
,, ,
,,K
E En n=
−=
−× =1
020
1 20 0591 51 0 771
0 0595 1 62 63
K =
+ +
− + +
[Mn ][Fe ][MnO ][Fe ] [H ]
2 3 5
42 5 8
MnO4−
E
MnO /Mn0
42− +E
Fe /Fe0
3 2+ +
Eferroína0
ECe /Ce0
4 3+ +EH AsO /H AsO0
3 4 3 3
10100
10100
106
2
× = × =−−
[ ]Cr O2 72
1100
× = × =−−
[Cr O ]1
10010
62 72
2
[Cr ]
103
32
+−
=
[Cr O ]10
62 72
2−
−=
E = +
−
+1 33
0 0596
,,
log[Cr O ][Cr ]
2 72
3 2
Capítulo 11: Valoraciones de oxidación-reducción 403

por tanto:
Al sustituir los valores de estas relaciones de con-centraciones en la expresión de la constante de equi-librio se llega a:
Para calcular el valor de pH al cual esta reacciónes cuantitativa al 99,9% se sustituye el valor de laconstante de equilibrio en la expresión anterior:
de donde pH = 5,58. Se ha de ajustar el pH de la valo-ración, por tanto, a un valor de pH igual o inferior a5,58, ya que a valores inferiores se incrementa elpoder oxidante del permanganato y la reacción sehace aún más cuantitativa. En la práctica es aconse-jable trabajar a un pH más ácido para evitar otrasreacciones, tales como la precipitación del Fe(OH)3o la reducción del permanganato a MnO2.
Cuando se lleva a cabo la valoración a pH = 1, lasexpresiones a utilizar son idénticas a las deducidas enel apartado 11.2.1, sin más que sustituir el potencialestándar del sistema MnO–
4/Mn2+ por su potencial con-dicional a pH = 1,0, calculado de acuerdo con laexpresión:
1,416 V
Para delimitar la región de equivalencia del±0,1%, en este caso, basta con hacer uso de lasexpresiones:
99,9% Valoración:
F = 0,999
0,948 V
100,1% Valoración:
F = 1,001
1,381 V
Por tanto, la región de equivalencia está compren-dida entre 0,948 y 1,381 V.
Un aumento de pH originaría un menor salto en elpunto de equivalencia de esta valoración, ya que elpotencial condicional del par redox MnO–
4/Mn2+ seríacada vez más pequeño. Por otra parte, la curva devaloración no se afecta por una disminución de la con-centración de analito, ya que la expresión del poten-cial depende de una relación de concentraciones, deacuerdo con la expresión de Nernst.
4. La figura muestra la curva de valoración de una diso-lución que contiene los iones U4+ 0,005 mol L–1 e Fe2+
0,01 mol L–1 con Ce4+, en presencia de ácido sulfúri-co. Justificar: a) la forma de la curva de valoraciónindicando las especies que se determinan en cadapunto de equivalencia; y b) mediante los corres-pondientes cálculos numéricos, cuál o cuáles de lossiguientes indicadores se podrían utilizar en estavaloración para detectar cada punto de equivalen-cia con un error inferior al 0,1%: ferroína (E0′ = 1,06V, n = 1); ácido p-difenilamino sulfónico (E0′ = 0,85 V,n = 2); difenilamina (E0′ = 0,76 V, n = 2); azul de me-tileno (E0′ = 0,53 V, n = 2); índigo tetrasulfonado (E0′ = 0,36 V, n = 2).
E = + − =1 4160 059
51 001 1,
,log ( , )
E En
F= + −10
1
0 0591
,log ( )
E = +
−=0 771 0 059
0 991 0 99
, , log,
,
E E
nF
F= +
−20
2
0 0591
,log
E En
a0 0 0 0591 51
0 0595
8 1′ = − × = − × =,,
,pH
1062 63, =
+
10[H ]
18
8
K = =
×=
+ +
+ + + +
[Mn ][Fe ][MnO ][Fe ] [H ]
10 (10 )[H ]
10[H ]
2 3 5
4– 2 5 8
3 3 5
8
18
8
[Mn ][MnO ]
99, 90,1
102
4
3+
−= ≈
404 Equilibrios iónicos y sus aplicaciones analíticas

= 0,334 V; = 0,771 V; =1,44 V.Sol.: a) En primer lugar se valora el ion U4+ y después
el Fe2+; b) para el primer salto (0,422-0,594 V)se utiliza azul de metileno y para el segundo(0,948-1,263 V) la ferroína.
5. Un indicador redox tiene un potencial estándar de0,52 V. El potencial varía según la reacción: Indox +2H+ + 2 e ←→ Indred. Calcular el potencial condicionaldel indicador a pH = 2 y la zona de viraje de pH = 0.Sol.: 1) 0,402 V; 2) 0,490 – 0,549 V
6. Calcular el potencial estándar de un indicador emple-ado en la valoración de Sn2+ con K2Cr2O7 0,001 molL–1 en H2SO4 a pH = 0. El cambio de color se produ-ce cuando se ha oxidado el 75% del indicador. El cam-bio de oxidación del indicador es de un electrón pormol. El error de valoración es de 0,1% por defecto.
= 0,154 V.
Las disoluciones de Sn2+ se pueden valorar condicromato de potasio siguiendo la reacción redox:
+ 3 Sn2+ + 14 H+ ←→ 2 Cr3+ + 3 Sn4+ + 7 H2O
detectando el punto final de la misma con diversossistemas indicadores visuales. En el problema se uti-liza el sistema indicador:
InOx + e ←→ InRed
Si el error de valoración es del 0,1% por defecto,se ha valorado el 99,9% del Sn2+ presente en la diso-lución y queda un 0,1% sin valorar. Si se supone quela concentración inicial de ion Sn2+ en la disolucióna valorar es CSn, en este punto de la valoración setiene:
[Sn2+] = [Sn4+] =
de donde:
Al sustituir esta relación de concentraciones en laexpresión de Nernst para este par redox, se tiene el
potencial en el punto final de la valoración asumien-do un error del 0,1% por defecto:
A este valor de potencial se ha de producir el cam-bio de color del indicador, cuando se ha oxidado el75% del mismo. Si se sustituyen estas condiciones enla expresión de Nernst para el sistema indicador:
se tiene:
de donde 0,214 V.
7. ¿Cuál debe ser el potencial estándar de un indicadorredox que intercambia dos electrones y presenta elcambio de color cuando se ha oxidado el 40% del mis-mo, si se desea utilizarlo en la valoración de[Fe(CN)6]
4– con Ce4+ en medio H2SO4?= 0,36 V; = 1,44 V.
Sol.: 0,905 V.
8. Se quiere valorar una disolución de Fe2+ con K2Cr2O70,1 mol L–1 en medio H2SO4 a pH = 0. Se utiliza paraello un indicador cuyo cambio de color se producecuando se ha oxidado el 80% del indicador. Si elpotencial estándar del indicador es de 0,83 V a pH =0 y el cambio de oxidación del indicador es de 2 elec-trones por mol, calcular el error de valoración.
= 0,771 V.Sol.: – 4,72%. El indicador no es adecuado.
Valoraciones con permanganato y dicromato
9. Se toman 10 mL de una disolución de H2O2 y se dilu-yen a 250 mL en un matraz aforado. Una alícuota de5,0 mL consume 22,50 mL de KMnO4 0,1569 eq L–1
para su valoración en medio ácido. Calcular la con-
EFe /Fe0
3 2+ +
ECe /Ce0
4 3+ +EFe(CN) /Fe(CN)0
63
64− −
EIn0 =
0 242 0 059
7525
, , log= +EIn0
E E= +In
0 Ox
Red
[In ][In ]
0 059, log
= + =0 1540 059
210 0 2423,
,log , V
E = + =+
+0 154
0 0592
,,
log[Sn ][Sn ]
4
2
[Sn ][Sn ]
4
2
+
+= ≈
0 990 1
103,,
0 99100, × CSn
0 1100
, × CSn
Cr O2 72−
ESn /Sn0
4 2+ +
ECe /Ce0
4 3+ +EFe /Fe0
3 2+ +EUO /U0
22 4+ +
Capítulo 11: Valoraciones de oxidación-reducción 405

centración de la disolución inicial de peróxido dehidrógeno y expresarla en eq L–1 y en peso/volumen.Pesos atómicos: H = 1,00; O = 16,00.
La determinación de H2O2 se suele llevar a cabomediante valoración redox con permanganato depotasio:
2 + 5 H2O2 + 6 H+ ←→ 2 Mn2+ + 5 O2 + 8 H2O
siendo el factor de equivalencia para el H2O2 en estareacción, de 1/2.
Dado que las reacciones se relacionan equivalen-te a equivalente, se puede establecer que:
meq = 22,5 × 0,1569 = 3,53 meq = meq H2O2
Considerando que se toman 10 mL de disoluciónde H2O2, se diluyen a 500 mL y se lleva a cabo la valo-ración con una alícuota de 5,0 mL; su concentraciónexpresada en eq L–1 será:
3,53 meq H2O2 = 17,65 eq L–1
y en % peso/volumen:
3,53 meq H2O2
= 30,0%
10. 25 mL de una disolución de 7,300 g L–1 de KHC2O4 .H2C2O4 . 2H2O (tetraoxalato de potasio) se valorancon: 1) NaOH 0,100 mol L–1 y fenolftaleína como indi-cador; y 2) con KMnO4 0,100 eq L–1 (feq = 1/5) enmedio H2SO4. ¿Qué volúmenes de NaOH y KMnO4se consumen en estas valoraciones?Peso molecular: sal = 254,1.Sol.: 1) 21,64 mL de NaOH; 2) 28,78 mL de KMnO4.
11. Un volumen de 50 mL de una disolución de KMnO4,que es 0,100 eq L–1 (feq = 1/5) se hace reaccionar fren-te a MnSO4. ¿Cuántos mL de NaOH 0,1 mol L–1 seconsumen para valorar el H+ producido?Sol.: 20 mL.
12. Para determinar el contenido de MnO2 en una piro-lusita se toman 0,500 g de la muestra y se disuelvenen 50,0 mL de una disolución de oxalato 0,1 eq L–1 en
medio ácido. La disolución resultante se valora conuna disolución de permanganato 0,050 eq L–1 gas-tándose 32,5 mL. Calcular el porcentaje de MnO2 enel mineral.Pesos atómicos: Mn = 54,94; O = 16,00.Sol.: 29,34%.
13. Se quiere conocer el contenido en peróxido de hidró-geno de una disolución acuosa de desinfectante. Paraello de toman 50,0 mL de la disolución y se valorancon una disolución de permanganato de la que se con-sumen 41,5 mL. 1,0 mL de la disolución de valoran-te equivale a 30,0 mg de Fe2+. Calcular el porcentajede peróxido de hidrógeno en el desinfectante.Pesos atómicos: Fe = 55,85; O = 16,00; H = 1,00.Sol.: 0,75%.
14. Una disolución de KMnO4 tiene una concentracióntal que valorando con ella 1,000 g de un mineral dehierro, el número de mL gastados es igual al porcen-taje de Fe2O3 en la muestra. Se toman 0,200 g de unamuestra de pirolusita impura y después de disuelta yreducido todo el manganeso a Mn2+, se valora éstecon la citada disolución de KMnO4 (todo el manga-neso queda como MnO2). Se consumen 38,3 mL dela disolución de KMnO4. Calcular el porcentaje deMnO2 en la muestra.Pesos moleculares: MnO2 = 86,94; Fe2O3 = 159,7.
En un principio, se ha de calcular la concentra-ción de la disolución de permanganato de potasioque se empleará posteriormente para la determina-ción del contenido de MnO2 en la pirolusita. Estaestandarización se basa en la reacción redox:
5 Fe2+ + 8 H+ ←→ Mn2+ + 5 Fe3+ + 4 H2O
en la que el factor de equivalencia para el hierro esla unidad. Dado que meq MnO4
– = meq Fe2+, paracalcular la normalidad de la disolución de perman-ganato se han de sustituir en esta igualdad las condi-ciones dadas por el problema. Así, se puede estable-cer:
mg Fe2O3 muestra =
mg Fe2O3 muestra
× × =2 mmol Fe
1 mmol Fe O1 meq Fe
1 mmol Femeq Fe
2 3
× ×
1 mmol Fe O159, 7 mg Fe O
2 3
2 3
1000 % Fe O100
2 3×
MnO4− +
× × ×34 mg H O
1 mmol H O1g
1000 mg100 mL10 mL
2 2
2 2
× × ×
2505
1 mmol H O2 meqH O
2 2
2 2
× ×
2505
110 mL
MnO4−
MnO4−
406 Equilibrios iónicos y sus aplicaciones analíticas

Al combinar ambas expresiones se tiene:
Esta expresión se puede igualar a:
=
= meq =
de donde al considerar la condición dada por el pro-blema: % Fe2O3 = VMnO–
4(mL), se tiene:
Con esta disolución de permanganato se valora elcontenido de MnO2 en una pirolusita, una vez queéste se ha reducido a Mn2+. La reacción de valoraciónes:
2 3 Mn2+ + 2 H2O ←→ 5 MnO2 (S) + 4 H+
en la que el factor de equivalencia para el MnO4– es
1/3 (se reduce a MnO2) y para el ion Mn2+ de 1/2 (seoxida a MnO2).
Dado que el permanganato presenta un factor deequivalencia en esta reacción diferente al de su valo-ración con Fe2+, su concentración expresada en eq L–1
variará, y vendrá dada por:
En estas condiciones, se puede establecer que:
meq MnO4– = 38,3 × 0,0751 =
= 2,8763 meq = meq MnO2
Finalmente, el porcentaje de MnO2 en la pirolusi-ta se calcula a partir de estos meq, su factor de equi-valencia, peso molecular (86,94) y peso de muestra(0,2 g):
2,8763 me MnO2
15. Una muestra de 0,8000 g de un mineral que contie-ne hierro y titanio se disuelve en medio ácido, se redu-ce con una disolución de SnCl2 y se valora conKMnO4 0,0856 eq L–1, del que se consumen 26,0 mL.Otra muestra, también de 0,8000 g, se disuelve enmedio ácido y se pasa a través del reductor de Jones,la disolución reducida se recoge sobre una disoluciónde Fe3+ y se valora con KMnO4, y se consumen 48,0mL. Calcular el porcentaje de hierro y titanio en lamuestra.Pesos atómicos: Fe = 55,85; Ti = 47,90.Sol.: 15,54% Fe; 11,27% Ti.
16. Un mineral de hierro contiene el 0,20% de titanio.Se toma 1,000 g de este mineral, se disuelven enH2SO4 y se reduce con el reductor de Jones y el elu-yente se recoge sobre una disolución de Fe3+. Se valo-ra con KMnO4 y el resultado se expresa en tanto porciento de hierro. ¿Qué error se comete en el porcen-taje de hierro debido a la presencia de titanio en elmineral?Pesos atómicos: Fe = 55,85; Ti = 47,90.Sol.: + 0,033%.
17. Para determinar el contenido de calcio en una mues-tra se pesan 0,6890 g de la misma, se disuelve y el cal-cio se precipita con oxalato de amonio. El precipita-do se filtra y una vez lavado se disuelve en ácidosulfúrico y se valora con KMnO4 0,1025 eq L–1, delque se consumen 43,20 mL. Calcular el tanto por cien-to de carbonato de calcio presente en la muestra.Pesos atómicos: C = 12,01; O = 16,00; Ca = 40,08.Sol.: 32,16%.
18. Una muestra de 2 g de un mineral de plomo se disuel-ve en medio ácido y el plomo se precipita comoPbC2O4 por adición de 100 mL de Na2C2O4 0,1 molL–1. El precipitado se filtra, se lava y se disuelve enácido sulfúrico consumiendo 20,0 mL de KMnO4 0,1mol L–1 para su valoración. Calcular el porcentaje deplomo en el mineral.Peso atómico: Pb = 207,21.
Esta determinación de plomo se basa en la reac-ción de precipitación previa del ion Pb2+ con el oxa-lato, para su separación de forma selectiva de lamatriz de la muestra:
Pb2+ + ←→ PbC2O4 (S)
Una vez separado este precipitado, se disuelve enmedio ácido y se valora el ion oxalato presente en el
C O2 42−
× × =86,94 mgMnO1 mmolMnO
100200 mg
62,54%2
2
× ×
1 mmol MnO2 meqMnO
2
2
C f
MnO1
eq4
1 eqL− = × = =−0 125235
0 075 1 3, , ( / )
MnO4− +
C fMnO
1eq
40,1252 eqL− = =− ( / )1 5
V CMnO MnO
1
4 4(mL) (eq L )− −× −
MnO4−
meq Fe mg Fe O muestra2
159, 72 3= ×
meq Fe mg Fe O muestra2
159, 72 3= ×
Capítulo 11: Valoraciones de oxidación-reducción 407

mismo, equivalente al plomo contenido en la mues-tra, con una disolución estandarizada de permanga-nato:
2 5 + 16 H+ ←→ 2 Mn2+ + 10 CO2 + 8 H2O
De acuerdo con estas reacciones, la relación este-quiométrica entre el permanganato y el ion Pb2+ es2:5, MnO4
– : Pb2+. Dado que en la valoración se con-sumen 20 × 0,1 = 2,0 mmol MnO4
– , de acuerdo conesta relación, se han valorado 5,0 mmol de Pb2+. Elporcentaje de plomo en la muestra se determina deacuerdo con:
5,0 mmol Pb
19. Una muestra de Mn3O4 que pesa 0,3120 g se disuel-ve y se reduce el manganeso a Mn2+. Se valora conKMnO4 0,100 eq L–1 (feq = 1/5) , en presencia de unaelevada concentración de fluoruros, de forma que elmanganeso queda en forma compleja trivalente. Segastan 22,7 mL de KMnO4. Calcular el tanto por cien-to de Mn3O4 en la muestra.Peso molecular: Mn3O4 = 228,82.Sol.: 44,39%.
20. Se quiere determinar el contenido de ácido fórmico(HCOOH) en una muestra mediante valoración conKMnO4 en medio básico y en presencia de un exce-so de ion Ba2+. Para ello se estandariza previamentela disolución de KMnO4 frente a Na2C2O4 en medioácido. Se pesan 100,0 mg de oxalato de sodio, sedisuelven en ácido y se consumen 25 mL de la diso-lución de KMnO4. A continuación se toman 5,0 mLde una muestra de ácido fórmico y se valora en lascondiciones indicadas consumiéndose 12,5 mL de estadisolución de permanganato (el MnO4
– se reduce aMnO2
4– y el HCOO– se oxida a CO2). Calcular la con-
centración de la disolución de ácido fórmico expre-sándola en mol L–1.Pesos atómicos: Na = 22,99; C = 12,01; O = 16,00; K = 39,10; Mn = 54,94; H = 1,00.Sol.: 1,49 × 10–2 mol L–1.
21. Una disolución de KMnO4, A , tiene una concentra-ción de 0,0151 g de sal por mL. Otra disolución, B, también de KMnO4 tiene una concentración tal que20,0 mL de la misma oxidan a 0,120 g de hierro. ¿Enqué proporción deben mezclarse para preparar un litrode disolución, para que su poder oxidante en medio
ácido sea el mismo que el de un litro de disolución deK2Cr2O7 0,333 eq L–1?Pesos atómicos: Mn = 54,94; O = 16,00; K = 39,10; Fe = 55,85.
Si se considera que el factor de equivalencia paralas disoluciones A y B de permanganato es 1/5, casomás general, las concentraciones de ambas disolu-ciones se pueden calcular a partir de:
Disolución A:
Disolución B:
meq Fe2+ = meq
de donde 0,1074 eq L–1.
Si se desea mezclar ambas disoluciones para con-seguir un litro de una disolución de KMnO4 que ten-ga el mismo poder oxidante que 1,0 L de una disolu-ción de dicromato de potasio 0,333 eq L–1, se puedeestablecer:
1.000 × 0,333 = (1.000 – X) 0,4775 + 0,1074 X
siendo X el volumen a tomar de la disolución B.Resolviendo esta ecuación se tiene que X = 390,4
mL, por lo que se tomaría para preparar la mezcla:
1.000 – 390,4 = 609,6 mL de la disolución A y 390,4 mL de la disolución B
esto es, en la proporción VA/VB = 1,56.
22. Una disolución de ácido fosfórico se trata con exce-so de (NH4)2MoO4. El precipitado obtenido tieneuna composición anómala: X [(NH4)3PO4].Y MoO3.1,000 g de este precipitado se disuelve en amoniacoy se diluye a 500 mL con agua destilada. Se toman50,0 mL de esta disolución, se acidifican con ácido
CMnO4
− =
120 mg Fe1 mmol Fe
55, 85 mg Fe1 meq Fe
1 mmol Fe20 mL CMnO4
× × = × −
MnO4−
× × = −5 eqKMnO1 molKMnO
1.000 mL1 L
0,4775 eq L4
4
1
× ×1 molKMnO158,04 gKMnO
4
4
0, 0151 g KMnO1 mL
4
× × =207,19 mgPb1 mmolPb
1002.000 mg
51,8%
C O2 42−
MnO4− +
408 Equilibrios iónicos y sus aplicaciones analíticas

sulfúrico y se reduce con cinc metal. El ion Mo3+ for-mado se reoxida a Mo6+ con FeCl3 en exceso y el ionFe2+ formado se valora con KMnO4 0,125 eq L–1,consumiéndose 15,4 mL en esta valoración. Cal-cular los valores de X e Y en la fórmula del preci-pitado.Pesos moleculares: MoO3 = 143,95; (NH4)3PO4 =148,98.Sol.: X = 2; Y = 25.
23. Una disolución de FeCl3 en HCl 1,0 mol L–1, se redu-ce haciéndola pasar a través de una columna de pla-ta metálica. Para valorar la sal de Fe2+ formada se gas-tan 23,45 mL de una disolución 0,1067 eq L–1 dedicromato de potasio. ¿Qué cantidad de plata se hatransformado en AgCl?Peso atómico: Ag = 107,88.Sol.: 0,27 g.
24. Se emplea (NH4)2Cr2O7 creyendo que es K2Cr2O7para preparar una disolución valorada 0,1188 eq L–1
(feq = 1/6). Esta disolución se emplea para determi-nar el contenido de hierro en un mineral, propor-cionando un valor de 45,7% de Fe. Calcular el ver-dadero porcentaje de hierro en el mineral. Pesos moleculares: (NH4)2Cr2O7 = 250,02; K2Cr2O7= 292,22.Sol.: 53,39%.
25. Para determinar la demanda química de oxígeno(DQO) de un agua residual contaminada se pipete-an 25,00 mL de muestra, se acidifica con H2SO4 con-centrado, se añade Ag+ como catalizador y se tratacon 10,00 mL de K2Cr2O7 0,2526 eq L–1 a reflujodurante dos horas. Una vez realizado este trata-miento, el exceso de K2Cr2O7 se valora con una diso-lución patrón de Fe2+, utilizando ferroína como indi-cador, consumiéndose 22,60 mL de disolución devalorante. Una disolución blanco, tratada de formasimilar, consume 24,92 mL de la disolución de Fe2+.Para establecer la concentración de la disolución deFe2+, ésta se valora con K2Cr2O7; 25 mL de Fe2+ con-sumen 9,35 mL de la disolución de dicromato de pota-sio. Calcular el valor de la DQO de la muestra deagua, expresado en mg O2 por litro de agua. Peso molecular: O2 = 32,00.
En este proceso, en primer lugar se ha de calcularel valor de la concentración de la disolución de Fe2+
que se utiliza tanto para la valoración del blanco comode la muestra. Para ello, ésta se estandariza frente adicromato de potasio:
+ 6 Fe2+ + 14 H+ ←→ 2 Cr3+ + 6 Fe3+ + 7 H2O
Dado que meq = meq Fe2+, se tiene:
9,35 × 0,2526 = 25 ×
de donde 0,0945 eq L–1.Una vez conocida la concentración de la disolución
de Fe2+, la diferencia entre los volúmenes de la mis-ma consumidos en la valoración del blanco y de lamuestra, permite determinar los meq de sustanciaoxidable presentes en la muestra de agua. Así:
(24,92 – 22,6) × 0,0945 = 0,2192 meq Fe2+ = = meq sustancia oxidable = meq O2
Considerando que la DQO se expresa en términosde mg de oxígeno consumido por litro de agua, y si setiene en cuenta que el factor de equivalencia para eloxígeno en esta reacción es 1/4 a tenor del par redox:
O2 + 4 H+ + 4 e ←→ 2 H2O
se puede establecer:
de donde DQO = 70,14.
26. Se analiza una disolución de clorato de sodio porreducción a cloruro, con exceso de FeSO4. Se toman25,0 mL de la disolución de clorato de sodio, se tra-tan con 10,00 mL de disolución de FeSO4 0,2517 eqL–1, y el exceso de Fe2+ consume 9,12 mL de disolu-ción patrón de dicromato de potasio 0,1024 eq L–1.Calcular la concentración de la disolución de clora-to de sodio expresándola en mg mL–1.Peso molecular: NaClO3 = 106,45.Sol.: 1,12 mg mL–1.
Valoraciones en las que interviene el yodo
27. El contenido de agua de una muestra de acetona sedetermina yodométricamente mediante el método
1.000 mL1 L
70,14 mgO L21× = −
0, 2192 meqO25 mL
1 mmol O4 meqO
32 mg O1 mmol O
2 2
2
2
2
× × ×
CFe2+ =
CFe2+
Cr O2 72−
Cr O2 72−
Capítulo 11: Valoraciones de oxidación-reducción 409

de Karl-Fischer. Se pesa una alícuota de 10 mL dela muestra (7,1420 g), se le añade 10 mL de meta-nol y se valora con el reactivo de Karl-Fischer, delque se gastan 7,35 mL. Para la estandarización delreactivo de Karl-Fischer, se pesan 0,2105 g de aguay se mezcla con metanol hasta un volumen final de100 mL. 5 mL de esta disolución diluida a 10 mLcon metanol consumen 10,20 mL del reactivo. Cal-cular el contenido de agua en la muestra de aceto-na, expresándolo en % peso/volumen (g H2O/100mL) y % peso/peso (g H2O/100 g), si 20 mL demetanol consumen 2,75 mL del reactivo de Karl-Fischer.
Como en otras ocasiones, inicialmente se ha deestandarizar el reactivo valorante que sirve de basea la determinación final, en este caso una disoluciónde yodo (reactivo de Karl-Fischer) que permite deter-minar el contenido de agua en una muestra de ace-tona. Para ello, se prepara una disolución patrón deacuerdo con:
0,2105 g de agua se llevan a un volumen final 100 mL con metanol
Si se considera que la densidad del agua es 1,00 gmL–1, esta disolución contiene 0,21 mL de agua y(100 – 0,21) = 99,79 mL de metanol. Si se toma parasu estandarización una alícuota de 5,0 mL de estadisolución y se le añade 10,0 mL de metanol, se ten-drá:
Dado que 20 mL de metanol consumen 2,75 mL dereactivo de Kar-Fischer, en la estandarización del mis-mo se gastarán para el metanol que acompaña alagua:
Karl-Fischer
por tanto de los 10,20 mL gastados en la valoración,sólo corresponden (10,20 – 2,06) = 8,14 mL al conte-nido de agua en la muestra patrón. En estas condi-ciones, la concentración del reactivo de Karl-Fischerse puede expresar como:
Con esta disolución de reactivo valorante se llevaa cabo la determinación del contenido de agua en lamuestra de acetona. Por diluir la muestra con 10 mLde metanol, se consumen 2,75/2 = 1,375 mL de reac-tivo de Karl-Fischer. Dado que en la valoración segastan 7,35 mL, el agua en la muestra de acetona real-mente consume (7,35 – 1,375) = 5,975 mL de reacti-vo, y por tanto, su contenido será:
5,975 × 1,29 × 10–3 = 0,0077 g de H2O
Este resultado, de acuerdo con el problema se hade expresar como:
peso/volumen
peso/peso
28. Una muestra de una aleación de cadmio, que pesa0,4020 g, se disuelve en medio ácido y el Cd2+ se sepa-ra como sulfuro. El CdS se disuelve en una disolu-ción ácida que contiene 40,0 mL de yodo 0,128 eq L–1,y esta disolución consume 11,6 mL de NaH2AsO30,108 eq L–1. Calcular el porcentaje de cadmio en lamuestra.Peso atómico: Cd = 112,41.Sol.: 54,07%.
29. Una muestra de un mineral de antimonio (sulfuro deantimonio, Sb2S3) se valora con una disolución de yodo0,050 eq L–1. ¿Qué peso de muestra debe tomarse paraque el % de Sb2S3 en el mineral sea igual a 1,5 veceslos mL gastados en la valoración?Peso molecular: Sb2S3 = 339,73.Sol.: 188,7 mg.
30. Para determinar el contenido de agua en una grasavegetal se toman 0,500 g de la misma, se disuelven enmetanol y se someten al método de Karl-Fischer. Seconsumen 24,2 mL de disolución del reactivo de KarlFischer 0,100 eq L–1. ¿Cuál es el porcentaje de aguaen la grasa?Pesos atómicos: H = 1,00; O = 16,00.Sol.: 4,36%.
0,0077 gH O7,142 g muestra
12 × =00 0 108, %
0 0077, gH O100 mL10 mL
0,077%2 × =
10 525 108 14
1 29 103
3,,
,×
= ×−
− −g H O mL21
2 7514 9895
202 06,
,,× = mL
99 79
5100
10 14 9895, , mL metanol× + =
0 21055
100, × = × −10, 525 10 g H O3
2
410 Equilibrios iónicos y sus aplicaciones analíticas

31. Una muestra que sólo contiene K2SO4 y KHSO4 y quepesa 1,000 g se disuelve en agua. A esta disolución seañade un exceso de KIO3-KI y el yodo liberado con-sume 40 mL de Na2S2O3 0,120 eq L–1. Calcular el por-centaje de K2SO4 en la mezcla.Pesos atómicos: K = 39,1; S = 32,07; O = 16,00; H = 1,00.
Al añadir a la muestra disuelta en agua un excesode KIO3-KI se origina la siguiente reacción redox:
+ 5 I– + 6 H+ ←→ 3 I2 + 3 H2O
controlada por la acidez que proporciona la disolu-ción de la sal KHSO4 presente en la muestra. El yodoliberado en esta reacción se valora con una disolu-ción estándar de tiosulfato de sodio:
I2 + 2 → 2 I– +
En la reacción anterior, el factor de equivalencia parael ion H+ es la unidad, de acuerdo con lo indicado enel apartado 10.5.4C, por lo que se puede establecer:
de donde:
4,8 meq KHSO4
Dado que la muestra es pura, el porcentaje deK2SO4 será (100 – 65,36) = 34,64%.
32. Una muestra de polvo de gas que pesa 0,200 g se ana-liza yodométricamente. Se disuelve, acidifica y se aña-de exceso de IK. El yodo producido consume 40,0mL de Na2S2O3 0,100 eq L–1. 250 g de estos polvos degas se añaden al agua de una piscina hasta obteneruna disolución homogénea. Se valora el cloro activoen el agua mediante fotometría con o-toluidina, y seencuentra una concentración de 2,0 µg mL–1. Calcu-lar el volumen de agua de piscina.Peso molecular: Cl2 = 70,92.
Inicialmente, se ha de conocer la actividad de lospolvos de gas en términos de meq de cloro activo,para ello se estandarizan de acuerdo con la reacción:
Cl2 + 2 I– ←→ I2 + 2 Cl–
Dado que el yodo producido consume 40 mL dedisolución de tiosulfato de sodio 0,100 eq L–1, se tiene:
meq = 40 × 0,1 = 4,0 meq = meq I2 = meq Cl2
o bien:
Cuando se adicionan 250 g de estos polvos de gasa una piscina, se añaden realmente 250 × 20 = 5.000meq Cl2 = 5 eq Cl2. Si se tiene en cuenta que el fac-tor de equivalencia del cloro es 1/2 en esta reacción setiene:
5 eq Cl2
Una vez que es homogéneo el contenido de cloroen la piscina, éste se determina fotométricamente yse encuentra una concentración de 2,0 µg mL–1, quetambién se puede expresar como 2,0 g de Cl2 por cadam3 de la piscina. Conforme a estos datos, el volumende la piscina será de:
33. Una muestra de 1,5000 g que contiene As2O3, As2O5y materia inerte se disuelve en medio NaOH, se neu-traliza y se diluye a 100 mL con agua destilada. 25 mLde esta disolución consumen 19,45 mL de I2 0,0500eq L–1 para su valoración. A otra alícuota de 25 mL,previamente acidificada, se le añade un exceso de KIy el yodo liberado se valora con Na2S2O3 0,100 eq L–1,del cual se gastan 23,10 mL. Calcular la composiciónde la muestra.Pesos moleculares: As2O3 = 197,82; As2O5 = 229,82.Sol.: 12,83% As2O3 y 35,39% As2O5.
34. Se quiere determinar el contenido de un ácido orgá-nico (H2A) en una muestra. Para ello, se toman 0,5000g de muestra, se disuelven de forma adecuada y setratan con exceso de KIO3-KI. El yodo liberado con-sume 28,5 mL de una disolución de Na2S2O3, cuyaestandarización se ha llevado a cabo previamente uti-lizando KIO3 como patrón primario de acuerdo conel siguiente procedimiento: 0,1430 g de KIO3 sedisuelven en medio ácido, se añade exceso de KI, y
177, 32, 0
88, 65 m3=
× × =
1 mol Cl2 eqCl
70, 92 g Cl1 mol Cl
177, 3 g Cl2
2
2
22
4, 0 meqCl0, 2 g muestra
20 meqCl g22
1= −
S O2 32−
× =136,17 mgKHSO1 mmolKHSO
1001.000 mg
65,36%4
4
× ×
1 mmol KHSO1 meqKHSO
4
4
meq S O 40 0,12 4,8 meqmeq I meq H
2 32
2
−
+
= × = == =
S O4 62−S O2 3
2−
IO3−
Capítulo 11: Valoraciones de oxidación-reducción 411

el yodo liberado consume 41,5 mL de la disoluciónde tiosulfato. Calcular el porcentaje de ácido orgáni-co en la muestra.Pesos moleculares: H2A = 120; KIO3 = 214,01.Sol.: 32,97%.
35. El plomo contenido en una muestra de 0,4770 g seprecipita como PbCrO4. El precipitado se filtra, selava, y se disuelve en ácido y a la disolución resul-tante se le añade un exceso de KI. El yodo liberadoconsume 22,30 mL de Na2S2O3. Para estandarizar ladisolución de tiosulfato de sodio se pesan 0,9390 g deKIO3, se disuelven en agua y se enrasa con agua des-tilada a 250 mL. Una alícuota de 20 mL de esta diso-lución consume después de acidificarla y adicionarleun exceso de KI, 23,00 mL de la disolución de tio-sulfato. Calcular el tanto por ciento de plomo en lamuestra.Pesos atómicos: Pb = 207,21; K = 39,1; I = 126,91; O = 16,00.Sol.: 29,57%.
36. Una muestra de hipoclorito de sodio, de 15,0 mL, setrata con exceso de yoduro y el yodo liberado se valo-ra con tiosulfato de sodio 0,1174 eq L–1, gastándose22,53 mL. Calcular la concentración de hipocloritode sodio en la muestra expresada en mol L–1.Sol.: 0,088 mol L–1.
37. Una muestra de 0,9350 g que contiene KHSO4 sedisuelve en 100 mL de agua. Se toma una alícuota de10,0 mL y se le añade exceso de KIO3/KI. El yodoliberado consume 20,18 mL de arsenito de sodio0,0113 eq L–1 para su valoración. Calcular el porcen-taje de KHSO4 en la muestra.Peso molecular: KHSO4 = 136,17.Sol.: 33,21%.
38. Se tienen dos disoluciones, una de NaClO y otra deNaClO2. Para su análisis se toman alícuotas de lasmismas y se valoran con yodo de la siguiente forma:a) A 10,0 mL de la disolución de NaClO en medioácido se le añade exceso de ioduro de potasio y elyodo liberado consume 33,45 mL de Na2S2O3 0,0853eq L–1. b) A 5,0 mL de la disolución de NaClO2 tam-bién en medio ácido se le añade exceso de ioduro depotasio y el yodo liberado consume 47,23 mL de lamisma disolución de tiosulfato. Calcular las concen-traciones de NaClO y NaClO2 en ambas disolucionesexpresadas en mol L–1.Sol.: a) 0,143 mol L–1 NaOCl; b) 0,201 mol L–1
NaClO2.
39. Se quiere determinar el contenido de cobre en unmineral. Para ello, se toman 0,518 g de muestra, sedisuelven en medio ácido y se diluye a 100 mL conagua destilada. 10,0 mL de esta disolución se tratancon exceso de ioduro de potasio en medio ácido, y elyodo liberado consume 39,24 mL de Na2S2O3 0,0105eq L–1 para su valoración. Calcular el porcentaje decobre en el mineral.Peso atómico: Cu = 63,54.
La determinación yodométrica de cobre (métodode Haen-Low) se basa en las siguientes reacciones:
2 Cu2+ + 4 I– ←→ 2 CuI(S) + I2
I2 + 2 → 2 I– +
en las que el cobre tiene un factor de equivalenciaigual a la unidad.
Como en otros casos, se puede establecer:
meq = 39,24 × 0,0105 = 0,412 meq =
= meq I2 = meq Cu
siendo el contenido de cobre:
0,412 meq Cu
40. Una muestra de ácido sulfámico puro se disuelve enagua. Una alícuota de 10,0 mL consume 24,3 mL deNaOH 0,1 mol L–1 para su valoración con fenolftale-ína como indicador. A otra alícuota de 5,0 mL se leañade exceso de KIO3-KI y el yodo liberado consu-me 20,2 mL de una disolución de tiosulfato de sodio.¿Cuál es la concentración de esta disolución de tio-sulfato de sodio expresada en mol L–1?Sol.: 0,06 mol L–1.
41. Para comprobar si un preparado de vitamina C, quecontiene teóricamente 100 mg de ácido ascórbico porcomprimido, cumple las especificaciones, se toman 10comprimidos (peso total: 9,7465 g), se trituran y seobtiene una muestra homogénea. Se pesan 0,9632 g,se disuelven en agua, se le añaden 50 mL de KBrO30,0488 eq L–1 y un exceso de KBr y se deja reaccionar.A continuación se le adiciona un exceso de KI y elyodo liberado se valora con Na2S2O3 0,0504 eq L–1,
× =100518 mg
50,54%
× × ×
1 mmol Cu1 meqCu
63, 54 mg Cu1 mmol Cu
S O2 32−
S O4 62−S O2 3
2−
412 Equilibrios iónicos y sus aplicaciones analíticas

consumiéndose 24,33 mL. Calcular el contenido deácido ascórbico en cada comprimido.Peso molecular: ácido ascórbico = 176.Sol.: 108,1 mg.
42. El índice de yodo (gramos de yodo retenidos por cada100 g de muestra) de un aceite de oliva se determinapor el método de Hanus. Se pesan 0,1304 g de aceite,se le añaden 25 mL de CCl4 y 25 mL de IBr en mediode ácido acético. Se deja reaccionar durante una horay a continuación se le añade agua y un exceso de KI yel yodo formado se valora con Na2S2O3 0,1086 eq L–1,del cual se consumen 21,35 mL. En un ensayo en blan-co del reactivo se comprueba que el yodo liberado por20 mL de la disolución de IBr reacciona con 23,48 mLde la disolución de tiosulfato de sodio. Calcular el índi-ce de yodo en la muestra de aceite.Peso molecular: I2 = 253,82.
El método de Hanus para la determinación del índi-ce de yodo de un aceite se basa en la adición de IBral doble enlace C = C, y posterior determinación delexceso de reactivo mediante las reacciones redox:
IBr + I– ←→ Br– + I2
I2 + 2 → 2 I– +
La diferencia entre el yodo liberado en el ensayoen blanco y la muestra, permite calcular los meq deIBr y, en su caso, de yodo que ha reaccionado con lamuestra de aceite. Así, se tiene:
meq IBr consumidos por 25 mL de muestra:
21,35 × 0,1086 = 2,3186 meq = = meq I2 = meq IBr
meq IBr consumidos por 25 mL de disolución blanco:
23,48 × 0,1086 = 3,1874 meq =
= meq I2 = meq IBr
La diferencia entre ambos: (3,1874 – 2,3186) =0,8688 son los meq de IBr o bien de yodo que hanreaccionado con 0,1304 g de muestra de aceite. A par-tir de estos datos, el índice de yodo será:
esto es, se retienen 84,55 g I2 por cada 100 g muestra.
43. Una disolución de los iones IO–3 y IO–
4 se analiza dela siguiente forma: a) A una alícuota de 50,0 mL dela misma se le ajusta el pH a 9,0 con tampón borax,se le añade exceso de KI y el yodo liberado consume18,4 mL de disolución patrón de tiosulfato de sodio0,100 eq L–1 (el ion IO–
4 se reduce a IO–3). b) Otra alí-
cuota de 10 mL se pone en medio ácido fuerte, se leañade exceso de KI y se gastan 48,7 mL de la disolu-ción de tiosulfato para la valoración del yodo libera-do. Calcular las concentraciones de IO–
3 y IO–4 en la
disolución original en mol L–1.Sol.: a) 0,0567 mol L–1 IO–
3 y b) 0,0184 mol L–1 IO–4.
Otras valoraciones
44. Una muestra de As2O3 purísimo, que pesa 0,2325 g,se disuelve en HCl 6,0 mol L–1 en un enlermeyer de300 mL. Se añaden 5 mL de Cl4C y se valora esta faseorgánica con KIO3 gastándose 48,96 mL de la diso-lución de yodato de potasio. ¿Cuál es la concentra-ción de la disolución de KIO3 expresada en eq L–1?Peso molecular: As2O3 = 197,82.Sol.: 0,096 eq L–1.
45. Los nitroderivados se reducen a aminas primarias porel TiCl3. Una muestra de nitroglicerina que pesa 1,000g se disuelve en metanol y se diluye a 100 mL. 10 mLde la disolución anterior se tratan con 25 mL de TiCl30,0509 eq L–1. El exceso de la sal de Ti3+ consume 10,6mL de disolución de Fe3+ 0,0421 eq L–1. Calcular elporcentaje de nitroglicerina en la muestra.Peso molecular: nitroglicerina = 227,03.
La reducción de nitroderivados por el ion Ti3+ seutiliza para la determinación de explosivos como lanitroglicerina. En esta valoración, están implicadoslos siguientes pares redox:
R-NO2 + 6 H+ + 6 e ←→ R-NH2 + 2 H2OTi4+ + e ←→ Ti3+
Fe3+ + e ←→ Fe2+
Dado que la nitroglicerina posee tres grupos nitro,se intercambiarán 18 electrones en su reducción a
1 g1.000mg
1000,1304 g
84,55× × =
0, 8688 meqI
1 mmol I2 meqI
253, 82 mg I1 mmol I2
2
2
2
2
× × ×
S O2 32−
×
2520
S O2 32−
S O4 62−S O2 3
2−
Capítulo 11: Valoraciones de oxidación-reducción 413

amina primaria, por lo que su factor de equivalenciaen esta reacción será de 1/18.
En las condiciones dadas en el problema, los meqde nitroglicerina presentes en la muestra serán:
meq nitroglicerina
y su contenido expresado en porcentaje:
46. Se pesan 2,2346 g de una muestra que contiene NaNO2,se disuelven en agua y se diluyen a 500 mL. A una alí-cuota de 25 mL de esta disolución se le añade 50 mL deCe4+ 0,100 eq L–1. El exceso de Ce4+ consume 21,32 mL
de Fe2+ 0,0932 eq L–1. Calcular el porcentaje de nitritode sodio en la muestra. Peso molecular: NaNO2 = 68,99.Sol.: 93,02%.
47. La hidroquinona se oxida a quinona por sales de Ce4+
reaccionando un mol de quinona con 2 moles de Ce4+.Una muestra de hidroquinona que pesa 0,1930 g conimpurezas inertes, consume 33,2 mL de disolución deCe(SO4)2 0,0998 eq L–1 para su valoración. Calcularla pureza de la muestra de hidroquinona.Peso molecular: hidroquinona = 110,06.Sol.: 94,47%.
48. El ion Zn2+ contenido en 100 mL de una disolución, seprecipita con 60 mL de K4Fe(CN)6 0,1 mol L–1 dandolugar a K2Zn3[Fe(CN)6]2. El filtrado y los líquidos delavado del precipitado consumen 18,2 mL de disolu-ción de Ce4+ 0,100 eq L–1 para su valoración. Calcularla concentración de la disolución original de cinc expre-sándola en mol L–1.Sol.: 0,0273 mol L–1.
227, 03 g Nitro1 mmol Nitro
1001000 g
10, 42%× × =
8, 262 meq Nitro1 mmol Nitro18 meq Nitro
× ×
,8 262= ( , ) ( , , )25 0 0509 10 6 0 0421
10010
× − × × =
414 Equilibrios iónicos y sus aplicaciones analíticas




















