![SISTEMA INMUNITARIO [TEORIA + IMÁGENES]](https://static.fdocuments.ec/doc/165x107/5571f3ec49795947648ec445/sistema-inmunitario-teoria-imagenes.jpg)
Enfermedades del sistema inmunitario
105
Enfermedades del sistema inmunitario Brian chavarro Alexander Díaz
-
Upload
alexander-diaz -
Category
Health & Medicine
-
view
58 -
download
2
Transcript of Enfermedades del sistema inmunitario

Enfermedades del sistema inmunitario
Brian chavarro
Alexander Díaz

Respuesta inmunitaria normal
los mecanismos de protección frente a las infecciones se encuentran en dos categorías:
1. La inmunidad innata (natural o nativa)
2. Inmunidad adaptativa (adquirida o especifica)

Inmunidad innata
Los principales componentes son:
1. Las barreras epiteliales
2. Células fagociticas ( neutrófilos y macrófagos)
3. Células dendríticas
4. Los linfocitos citoliticos naturales (NK, natural killer)
5. Varias proteínas plasmáticas ( proteínas del sistema del complemento)
Las 2 reacciones mas importantes de la inmunidad innata son la inflamación y la defensa antivírica
Reconocimiento celular (toll o TLR) ( sensores microbianos) (NF-kB)

Inmunidad adaptativa
Esta conformado por los linfocitos y sus productos
Hay dos tipos de inmunidad adaptativa
1. Inmunidad humoral: mediada por LT B
2. Inmunidad celular: mediada por LT T

Células del sistema inmunitario

Linfocito T• se forman a partir de precursores en el timo• Las células maduras se encuentran en la
sangre donde constituyen un 60% al 70% de los LT
• RLT Y COMPLEJO RLT• Hay otras proteínas que colaboran con el
complejo RLT como el CD4, CD8, CD2, integrinas, y el CD28
• LT cooperadores CD4 pueden reconocer y responder al CPH clase 2
• LT citotóxicos CD8 reconoce y responde al CPH clase 1
edgar alexander dìaz erazo

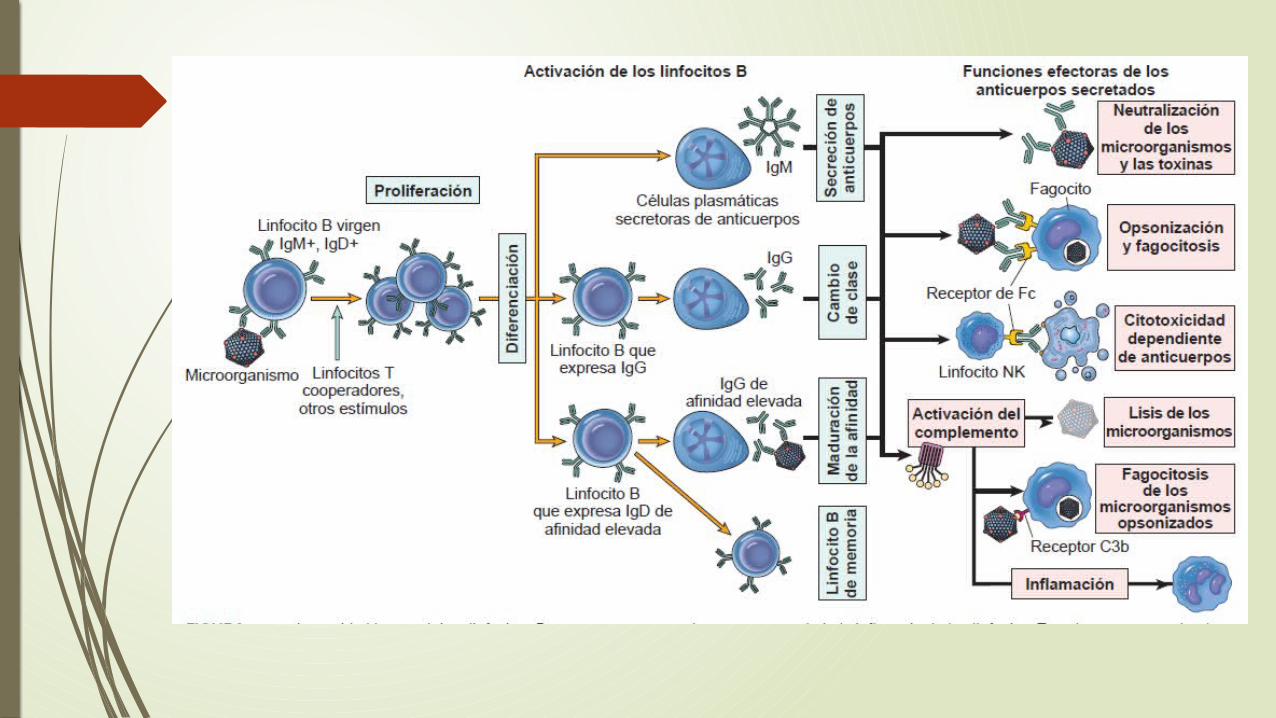
Linfocitos B
Se desarrollan en la medula osea
Constituyen del 10 al 20% de la población de LT periféricos circundantes
Después de la estimulación por un antígeno los LT B se transforman a células plasmáticas
Diferentes moléculas para el reconocimiento de antígenos Fc y CD40, CR2 o CD21
Virus Epstein-barr VEB

Células dendríticas
Hay 2 tipos de células con morfología dendrítica que son funcionalmente diferentes
Células dendríticas interdigitales
Principales presentadoras de antígeno CPA
Están situadas debajo de los epitelios y en los intersticios de los tejidos
Las células dendríticas inmaduras en la epidermis se llaman células de Langerhans
En respuesta, son atraídas hasta las zonas de LT T
Células dendríticas foliculares
Se encuentran en los centros germinales de los folículos linfáticos del bazo y de los ganglios linfáticos
Pueden atrapar antígenos a los anticuerpos o a proteínas del complemento
Presentan los antígenos a los LT B

macrófagos
Los macrófagos que han fagocitado microorganismos y antígenos proteicos, procesan los antígenos y presentan fragmentos peptídicos a los LT T ( CPA)
Son células efectoras de la inmunidad celular, fagocitando de forma eficiente y destruyen microorganismos que están optimizados por igG Y C3b

Linfocitos citoliticos naturales (NK 10 al 15% de los linfocitos de la sangre
periférica
gránulos azurófilos
No expresan RLT ni Ig
capacidad de destruir células infectadas y tumorales, sin exposición previa ni activación por estos microorganismos o tumores.
CD16 y CD56.
CD16 es un receptor de Fc para la IgG, destruir células diana recubiertas por IgG.
su capacidad funcional está regulada por un equilibrio entre señales de receptores activadores e inhibidores
IFN- ɣ. regulados por IL-2, IL-15 e IL-12.

Tejidos del sistema inmune
Los órganos linfáticos periféricos (primarios o centrales)
• Timo y médula ósea
Los órganos linfáticos periféricos (secundarios)
• los ganglios linfáticos, el bazo y los tejidos linfoides mucosos y cutáneos.

Recirculación de los linfocitos

Moléculas del complejo principal dehistocompatibilidad (CPH): fundamentales para el reconocimiento de los antígenos por los linfocitos T
y están asociadas a muchas enfermedades autoinmunitarias.
La función fisiológica de las moléculas del CPH es presentar fragmentos peptídicos de proteínas para su reconocimiento por linfocitos T específicos de antígeno.
En los seres humanos, los genes que codifican las principales moléculas de histocompatibilidad están agregados en un pequeño segmento del cromosoma 6, el complejo principal de histocompatibilidad, o el complejo de los antígenos leucocitos humanos (HLA)

Clasificación de los genes CPH CPH de clase I
• se expresan en todas las células nucleadas y en las plaquetas.
• Se codifican por tres loci HLA-A, HLA-B y HLA-C
• presentan péptidos derivados de proteínas, como antígenos víricos, que están localizados en el citoplasma y habitualmente se producen en la célula, y los péptidos asociados a la clase I son reconocidos por los linfocitos T CD8+
CPH de clase II
• son codificadas por una región llamada HLA-D, que tiene tres subregiones: HLA-DP, HLA-DQ y HLA-DR.
• presentan antígenos que son interiorizados hacia el interior de vesículas, y que típicamente proceden de microorganismos extracelulares y proteínas solubles, es reconocido por los linfocitos T CD4+
• se expresan principalmente en células que presentan antígenos ingeridos y responden a la cooperación de los linfocitos T (macrófagos, linfocitos B y células dendríticas).

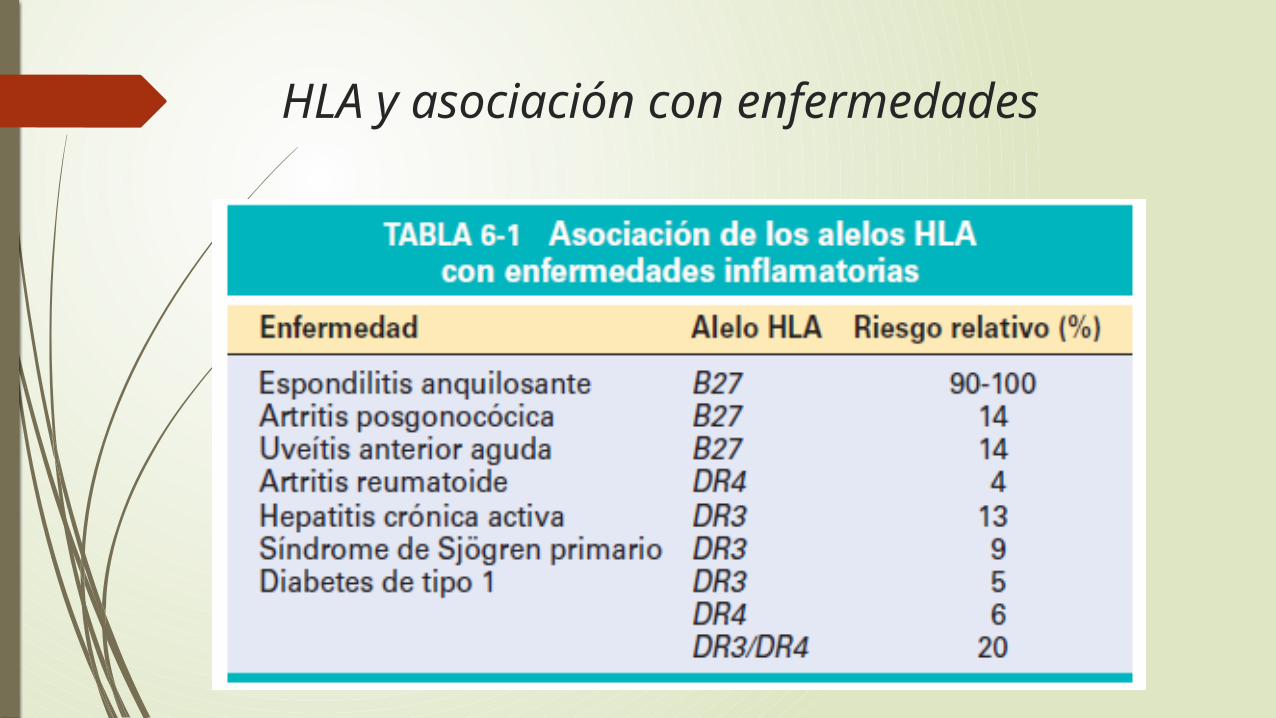
HLA y asociación con enfermedades
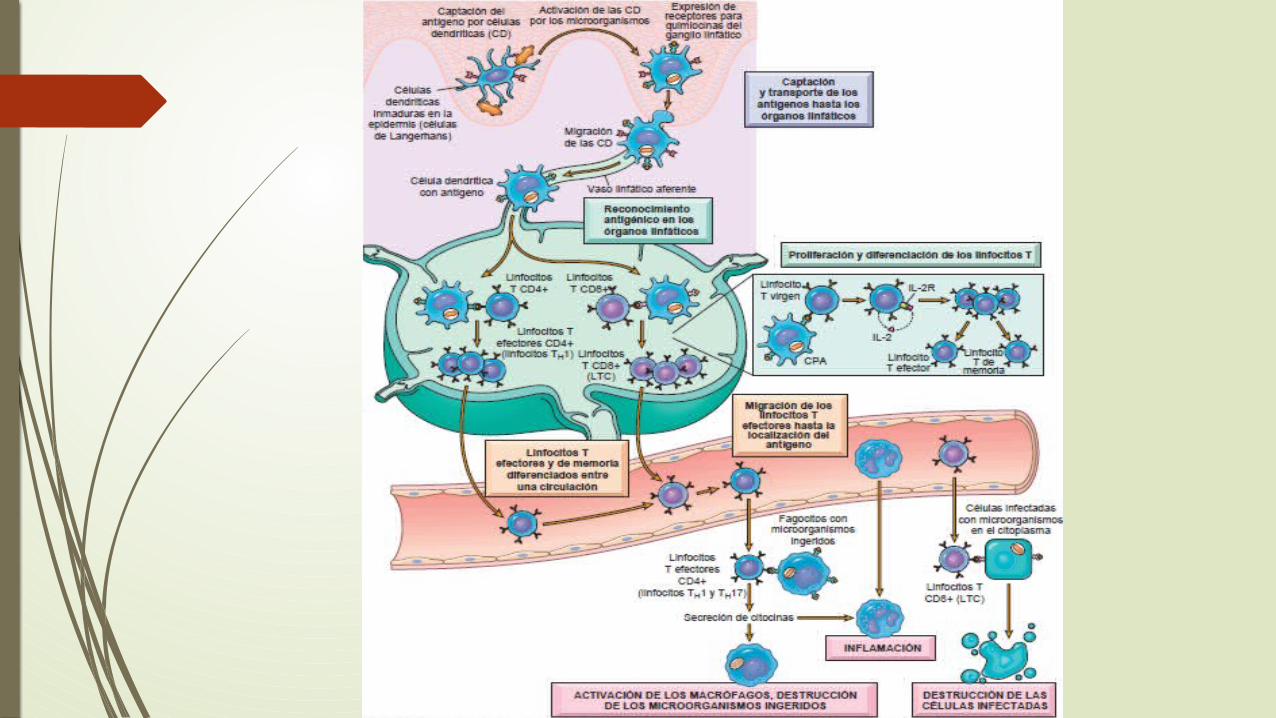


Hipersensibilidad y trastornosautoinmunitarios
MECANISMOS DE LAS REACCIONES DE HIPERSENSIBILIDAD
• los individuos que han estado expuestos previamente a un antígeno están sensibilizados
• En ocasiones, la exposición repetida al mismo antígeno desencadena una reacción patológica; estas reacciones se definen como hipersensibilidad, lo que implica una respuesta excesiva a un antígeno.
Antígenos tanto exógenos como endógenos pueden desencadenar reacciones de hipersensibilidad.
La aparición de enfermedades por hipersensibilidad (trastornos tanto alérgicos como autoinmunitarios) con frecuencia se asocia a la herencia de determinados genes de susceptibilidad.
Se ha planteado el principio general de que la hipersensibilidad refleja un desequilibrio entre los mecanismos efectores de las respuestas inmunitarias y los mecanismos de control que normalmente limitan dichas respuestas.


Hipersensibilidad inmediata (tipo I) es una reacción inmunitaria rápida que se produce pocos
minutos después de la combinación de un antígeno con un anticuerpo unido a los mastocitos en pacientes sensibilizados previamente al antígeno
se denominan alergia y los antígenos que las provocan son alérgenos.
se puede producir como un trastorno sistémico o como una reacción local
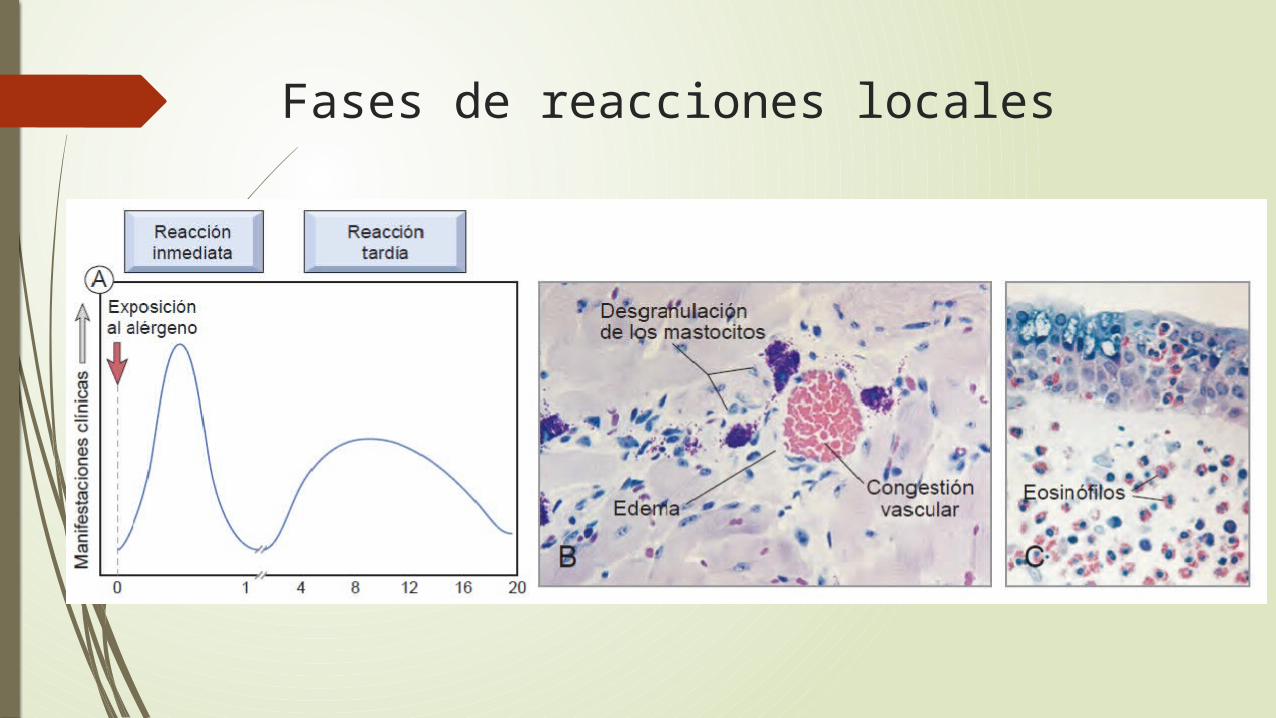
Fases de reacciones locales

mediadas por la activación de los mastocitos y de otros leucocitos dependientes de los anticuerpos IgE
Los mastocitos son células derivadas de la médula ósea que están ampliamente distribuidas en los tejidos
Son abundantes cerca de los vasos sanguíneos y los nervios y en tejidos subepiteliales
los mastocitos (y los basófi los) son activados por la reticulación de los receptores de afinidad elevada para el Fc de la IgE
También son activados por componentes del complemento C5a y C3a, Quimiocinas, fármacos como codeína y morfina, adenosina, melitina (presente en el veneno de abeja) y estímulos físicos (p. ej., calor, frío, luz solar).


Los linfocitos T H 2 tienen una participación central en el inicio y la propagación de las reacciones de hipersensibilidad inmediata mediante la estimulación de la producción de IgE y el fomento de la inflamación
IL-4, IL-5 e IL-13
IL-4 actúa sobre los linfocitos B para estimular el cambio a IgE y favorece la aparición de linfocitos T H 2 adicionales.
IL-5 participa en el desarrollo y la activación de los eosinófilos, son efectores importantes de la hipersensibilidad de tipo I.
IL-13 potencia la producción de IgE y actúa sobre las células epiteliales para estimular la secreción de moco



La susceptibilidad a las reacciones de hipersensibilidad inmediata está determinada genéticamente.
El término atopia se refiere a la predisposición a presentar reacciones de hipersensibilidad inmediata localizadas ante diversos alérgenos inhalados e ingeridos.
Los pacientes atópicos tienen mayores concentraciones séricas de IgE y más linfocitos T H 2 productores de IL-4
antecedente familiar positivo de alergia en el 50% de los pacientes atópicos.
Una proporción significativa de reacciones de hipersensibilidad inmediata está desencadenada por extremos de temperatura y por el ejercicio, y no están implicados los linfocitos T H 2 ni la IgE

Anafilaxia sistémica
se caracteriza por shock vascular, edema generalizado y dificultad respiratoria
Puede aparecer en individuos sensibilizados en contextos hospitalarios después de la administración de proteínas extrañas (p. ej., antisueros), hormonas, enzimas, polisacáridos y fármacos (como los antibióticos del grupo de la penicilina), o en el contexto comunitario después de la exposición a alérgenos alimentarios (p. ej., cacahuetes, marisco) o a toxinas de insectos (p. ej., los antígenos del veneno de abeja).
Dosis muy pequeñas de antígeno pueden desencadenar la anafilaxia
el paciente puede entrar en estado de shock e incluso morir en un plazo de 1 hora

Reacciones de hipersensibilidad inmediata locales
10 al 20% de la población tiene alergias que suponen reacciones localizadas a alérgenos ambientales
como polen, caspa de animales, polvo de casa, alimentos y otros similares
Las enfermedades específicas incluyen urticaria, angioedema, rinitis alérgica (fiebre del heno) y asma bronquial

Hipersensibilidad mediada por anticuerpos (tipo II)
Este tipo de hipersensibilidad está producido por anticuerpos que reaccionan con antígenos presentes en las superficies celulares o en la matriz extracelular
Los determinantes antigénicos pueden ser intrínsecos a la membrana celular o la matriz, o pueden adoptar la forma de un antígeno exógeno, como el metabolito de un fármaco, que queda adsorbido a la superficie celular o a la matriz.



Hipersensibilidad mediada por inmunocomplejos (tipo III)
Los complejos antígeno-anticuerpo (inmunocomplejos) producen lesión tisular principalmente generando inflamación en los lugares de depósito.
La reacción patológica se inicia cuando el antígeno se combina con anticuerpos dentro de la circulación (inmunocomplejos circulantes) se depositan típicamente en las paredes vasculares.
En ocasiones, los complejos se forman en localizaciones extravasculares en las que los antígenos pueden haberse «sembrado» previamente (llamados inmunocomplejos in situ ).




Hipersensibilidad mediada por linfocitos T (tipo IV)
se inicia por linfocitos T activados por el antígeno (sensibilizados), como linfocitos T CD4+ y CD8+
La hipersensibilidad mediada por linfocitos T CD4+ inducida por antígenos ambientales y por antígenos propios puede ser una causa de enfermedad inflamatoria crónica.
En infecciones víricas, los linfocitos CD8+ pueden ser las células efectoras dominantes.

Reacciones de linfocitos T CD4+: hipersensibilidadretardada e inflamación inmunitaria
se caracterizaron por la presencia de una hipersensibilidad retardada (HSR) contra antígenos administrados por vía exógena.
Los linfocitos T H 1 y T H 17 contribuyen a enfermedades específicas de órgano en las que la inflamación es un aspecto prominente de la anatomía patológica.
Proliferación y diferenciación de los linfocitos T CD4+ .




Reacciones mediadas por linfocitos T CD8+:citotoxicidad celular
los LTC CD8+ destruyen células diana portadoras de antígenos.
diabetes de tipo 1
importante en el rechazo de los injertos
participan en reacciones contra virus
La destrucción de las células infectadas lleva a la eliminación de la infección, y es responsable de la lesión celular que acompaña a la infección
participan en el rechazo tumoral
secretan un complejo formado por perforina, granzimas y una proteína llamada serglicina, que entra en las células diana mediante endocitosis
también producen citocinas, fundamentalmente IFN- ɣ , y participan en reacciones inflamatorias similares a la HSR

Enfermedades Autoinmunitarias

Definición
Una enfermedad autoinmune es una enfermedad causada por el sistema inmunitario, que ataca las células del propio organismo. En este caso, el sistema inmunitario se convierte en el principal agresor y ataca partes del organismo, en vez de protegerlas.

GENERALIDADES
Las enfermedades inmunitarias frente a los antígenos (autoinmunidad) son una causa importante de determinadas enfermedades en los seres humanos, y se estima que afectan al menos al 1-2% de toda la población estadounidense.
Se pueden encontrar autoanticuerpos en el suero de personas aparentemente normales, particularmente en grupos de mayor edad.
También se pueden generar autoanticuerpos inocuos después de una lesión tisular.
La autoinmunidad se debe a la perdida de la autotolerancia.

Las manifestaciones clínicas de los trastornos autoinmutarios son muy variadas.
Por un lado están las enfermedades en las que las respuestas inmunitarias se dirigen frente a un único órgano o tejido (enfermedades especificas de un órgano)
Ej: Diabetes mellitus tipo 1, en la que los linfocitos T y los anticuerpos autoarreactivos son específicos para células B de islotes pancreáticos.
En el otro extremo están enfermedades en las que las reacciones inmunitarias se dirigen a antígenos generalizados (enfermedades sistémicas generalizadas)
Ej: LES, en que los diversos anticuerpos dirigidos contra el DNA, las plaquetas, los eritrocitos, y complejos proteinas-fosfolipidos dan lugar a lesiones generalizadas.

Requisitos para considerar una enfermedad autoinmune
1. Presencia de una reacción inmunitaria especifica para algún antígeno o tejido propio.
2. Datos de que dicha reacción no es secundaria a la lesión tisular, si no que tiene un significado patogénico primario.
3.Ausencia de otra causa bien definida de la enfermedad.

Tolerancia Inmunitaria
la tolerancia inmunitaria es el fenómeno de ausencia de repuesta a un antígeno como consecuencia de la exposición de los linfocitos al mismo.
Autolerancia se refiere a la ausencia de respuesta a los antígenos propios de un individuo, subyace a nuestra capacidad de vivir en armonía con nuestras células y tejidos.
Los mecanismos de autorolerancia se pueden clasificar en dos grandes grupos: tolerancia central y tolerancia periférica.
TOLERANCIA CENTRAL: En este proceso los clones de linfocitos T y B autoarreactivos inmaduros que reconocen antígenos propios durante su maduración en los órganos linfáticos centrales (el timo para los linfocitos T) y (medula ósea para lo linfocitos B) son destruidos o se vuelven inofensivos.

TOLERANACIA PERFIFERICA: Varios mecanismos silencian a los linfocitos T y B potencialmente auotoarreactivos en los tejidos periféricos; estos mecanismos se han definido mejor para los linfocitos T e incluyen:
ANERGIA: Se refiere a la inactivación funcional prolongada o irreversible de los linfocitos, inducida por el contacto con antígenos en determinadas condiciones, este proceso se da ya que las segundas señales que proceden de determinadas moléculas asociadas a linfocitos T, como CD28, que se unen con sus ligandos (los estimuladores B7-1 y B7-2) sobre las CPA, si el antígeno es presentado por células que no tienen los coestimuladores, se presenta una señal negativa y la célula hace Anergia.
Causantes de anergia :
Activación de las ubicuitinas ligasas.
señal inhibidora procedente de receptores homólogos a CD28 producidos por
CTLA-4.


Supresión por los Linfocitos T reguladores: Tienen una función importante en la prevención de las respuesta inmunitarias
frente a los antígenos propios.
Los linfocitos T mejor definidos son los linfocitos CD4 que expresan constitutivamente CD25, la cadena alfa del receptor de la IL-2 y un factor de transcripción de la familia forkhead llamado Foxp3, las mutaciones de este gen producen autoinmunidad grave en seres humano y ratones en. En los humanos produce una enfermedad llamada (alteración de la regulacion inmunitaria poliendocrinopatia, enteropatía, ligando a X).

Mecanismos de la autoinmunidad la autoinmunidad se origina por una combinación de herencia de genes de
susceptibilidad, que pueden contribuir a la desaparición de la autotolerancia, y desencadenante ambientales, como infecciones y lesión tisular, que favorecen la activación de linfocitos autorreactivos.

Factores genéticos que contribuyen a la aparición de autoinmunidad
Polimorfonucleares del gen PTPN-22:
ARTRITIS REUMATOIDEA
DIABETES TIPO 1
Pilomorfonucleares del gen NOD-2
Enfermedad de CROHN ( enfermedad intestinal inflamatoria)

Enfermedades autoinmunitarias asociadas a infecciones
Se han propuesto dos mecanismos para explicar la asociación entre las infecciones y la autoinmunidad.
1. Las infecciones pueden activar la expresión de coestimuladores en las CPA.
2. Algunos microorganismos pueden expresar antígenos que tienen las mismas secuencia de aminoácidos que los antígenos propios.
Las respuestas inmunitarias frente a los antígenos microbianos pueden llevar a la activación de linfocitos autorreactivos, este fenómeno se llama mimetismo molecular. Ej: cardiopatía reumática, en la que los anticuerpos frente a las proteínas estreptocócicas dan reacción cruzada con proteínas miocárdicas y producen miocarditis.


Características de las enfermedades autoinmunitarias
Una vez que se ha incluido una enfermedad autoinmunitaria, tiende a ser progresiva, en ocasiones con recaídas y remisiones esporádicas, y la lesión se hace inexorable.
Las manifestaciones clínicas y anatomopatologicas de las enfermedades autoinmunitarias están determinadas por la naturaleza de las respuesta inmunitaria subyacente.
Diferentes enfermedades autoinmunitarias tienen importantes superposiciones clínicas, por ese motivo con frecuencias es difícil una clasificación fenotípica precisa de estos trastornos.

Lupus Eritematoso Sistémico(LES)
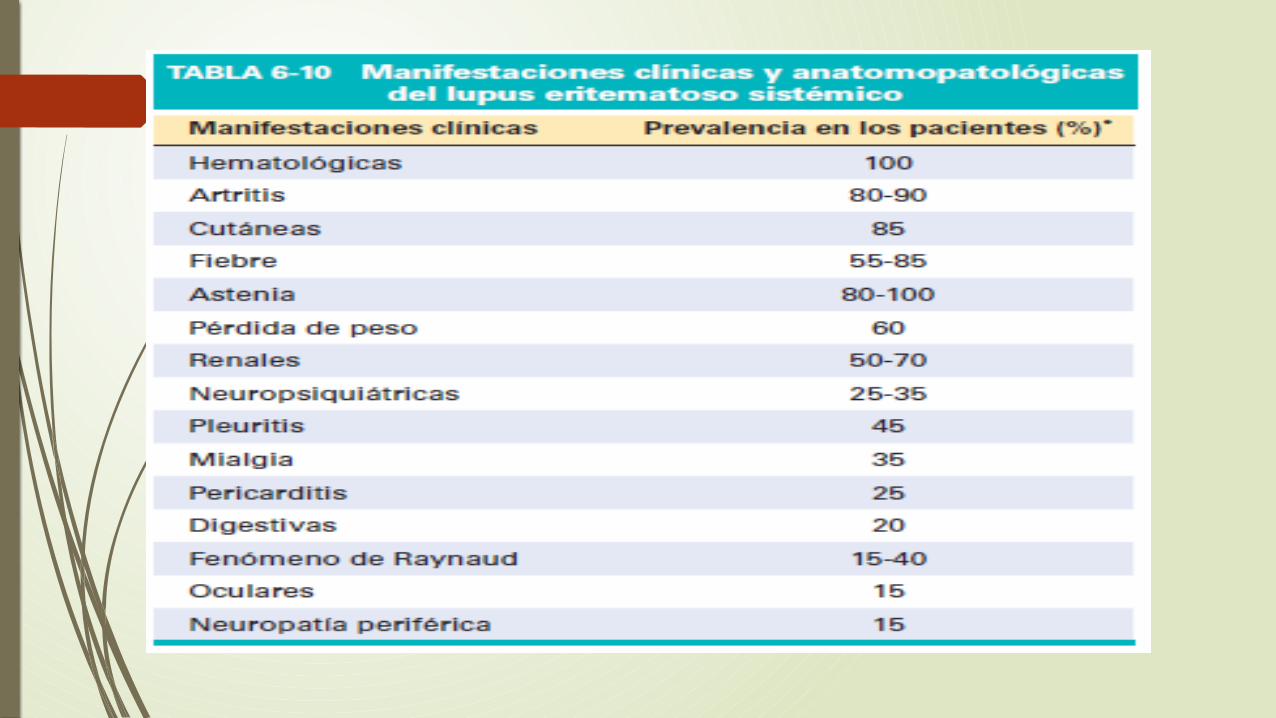
Es el prototipo de una enfermedad multisistémica de origen autoinmunitario, que se caracteriza por una gran variedad de anticuerpos atinucleares (ANA).
Enfermedad de inicio insidioso
Evolución crónica
Con remisiones y recaídas
Afecta:
Lesión de la piel
Las articulaciones
El riñón
Membranas serosas
Múltiples órganos.

Prevalencia
1 de cada 2500 en algunas poblaciones
Afecta predominantemente mujeres, con una frecuencia de 1 de cada 700 mujeres edad fértil
Una relación mujer-hombres de 9:1
La prevalencia de la enfermedad es 2-3 veces mayor en negros e hispanos que en blancos.
El LES comienza generalmente alrededor de la tercera y cuarta década de la vida, pero se puede manifestar en cualquier edad e incluso en la infancia.


Etología y patogenia del LES
La patogenia esta directamente implicada con el fallo de los mecanismos que mantienen la autotolerancia.
Factores tanto genéticos como ambientales participan en la patogenia del LES.
Genéticos:
El LES es una enfermedad con contribuciones de los genes CPH y de los genes no asociados a dicho complejo.
Ambientales:
La exposición a la luz ultra violeta (UV) empeora la enfermedad en muchas personas, ya que la exposición a este efecto puede inducir a la apoptosis de las células y puede alterare el ADN de tal forma que se haga inmunógeno, además puede estimular a lo queratinocitos para que produzca IL-1.


Manifestaciones clínicas
Golemerulonefritis lúpica mesangial: se evidencia en el 10-15 % de los pacientes, y se caracteriza por la proliferación de las células mesangiales y deposito de inmunocomplejos sin afectación de los capilares glomerulares.
Glomerulonefritis proliferativa focal: Se evidencia en el 20-35% de los pacientes, y se define por una afección menor del 50% de todos los glomérulos.
Glomerulonefritis proliferativa difusa: Es la forma mas grave de nefritis lúpica y parece en el 35-60% de los pacientes. Los cambios anatomapatologicos glomerulares pueden ser idénticos a los de la nefritis lúpica focal.
Glomerulonefritis membranosa: se caracteriza por engrosamiento difuso de las paredes capilares, aparece en el 10-15% de pacientes con nefritis lúpica, esta se acompaña de proteinuria grave.

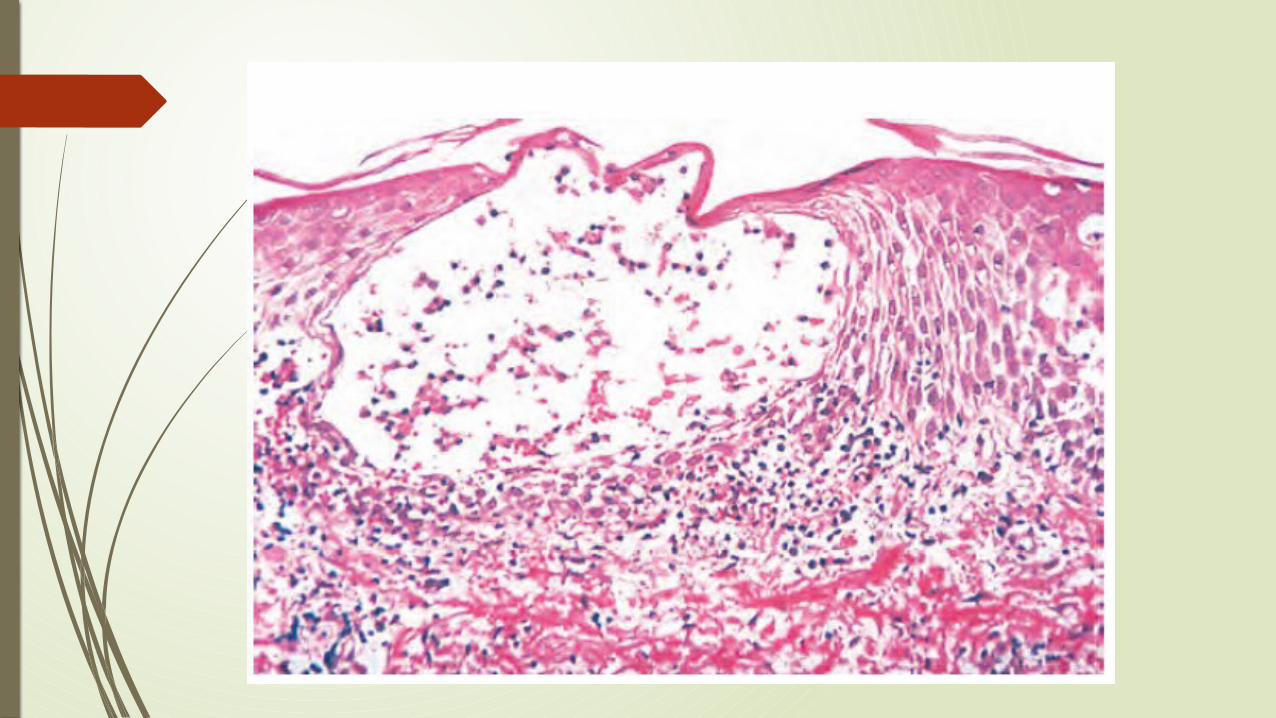
Piel: Se evidencia un eritema característico, que afecta el área facial, en forma de mariposa(malar) (puente de la nariz y mejillas), aproximadamente en el 50 de los pacientes, aunque también se puede ver un exantema similar en las extremidades y el tronco. También puede ver urticaria, ampollas, lesiones maculopapulares y ulceraciones.
Articulaciones: La afectación articular es típicamente una sinovitis no erosiva con poca deformidad, lo que contrasta con la artritis reumatoide.
Sistema nervioso central: Vasculitis aguda
Pericarditis y afectación de otras cavidades serosas: La afectación de las membranas serosas pueden ser agudas, subagudas o crónicas. La afectación del sistema cardiovascular se puede manifestar como lesión de cualquier capa del corazón, lo que puede producir una taquicardia en reposo y alteraciones electrocardiográficas.

Bazo: La esplenomegalia, el engrosamiento capsular y la hiperplasia folicular son datos frecuentes. Las arterias peniciliadas centrales pueden tener hiperplasia concéntrica de la intima y de células musculares lisas, dando lugar a las denominadas lesiones de piel de cebolla.
Pulmones: Las pleuritis y los derrames pleurales son las manifestaciones pulmonares mas frecuentes.

ARTRITIS REUMATOIDE
Es una enfermedad inflamatoria crónica que afecta principalmente a las articulaciones, aunque puede afectar a los tejidos extraarticulares, como piel vasos sanguíneos pulmones y corazón.
SINDROME DE SJOGREN: enfermedad crónica que se caracteriza por la sequedad ocular, este trastorno esta asociado a la artritis reumatoide este fenómeno es debido a una destrucción de mecanismo inmunitario de as glándulas lagrimales y saliva.
Patogenia : la disminución de lagrimas y saliva se debe a la infiltración linfocitaria y fibrosis de las glándulas lagrimales y salivares. El infiltrado contiene predominantemente linfocitos T cooperadores CD4 activados y algunos linfocitos B, además de células plasmáticas. Aproximadamente el 75% de los pacientes tienen factor reumatoide. ( un anticuerpo que reacciona con igG propia). Además se detectan AC específicos de la enfermedad SS-A (Ro) y SS-B(La)

Esclerosis sistémica (Esclerodermia)
Es una enfermedad crónica que se caracteriza por :
1. Inflamación crónica que se piensa que se debe a autoinmunidad
2. Lesión generalizada de vasos sanguíneos pequeños
3. Fibrosis intersticial y perivascular progresiva en la piel y múltiples órganos.
Categorías.
Esclerosis difusa: que se caracteriza por afectación cutánea generalizada al inicio, con progresión rápida y afectación visceral temprana.
Esclerodermia limitada: la afección cutánea con frecuencia esta limitada a los dedos de la manos . Los antebrazos y la cara, y la afectación visceral es tardía.

Patogenia
Respuestas autoinmunitarias anormales: se ha propuesto q los linfocitos CD4 responden a un Ag aun no identificado y hacen que se acumulen en la piel, y liberan citocinas que activan las células inflamatorias y a lo fibroblastos.
Fibrosis : característica de la enfermedad puede ser la acumulación de múltiples alteraciones, como las acciones de las citocinas por los leucocitos de los infiltrados y cicatrización después de la lesión isquémica producidas por las lesiones vasculares.
piel: presentan atrofia esclerótica difusa, que habitualmente comienza en los dedos de las manos en regiones distales y se extiende en dirección proximal hasta afectar brazos, hombro, cuello y cara.
Tubo digestivo: este se encuentra afectado en el 90% de los casos. Puede aparecer atrofia progresiva y sustitución por tejido fibroso colagenoso de la capa muscular a nivel del tubo digestivo.

Sistema osteomucular: la inflamación de la membrana sinovial, asociada a hipertrofia e hiperplasia de los tejidos blandos sinoviales, es frecuente en las fases tempranas.
Riñones: se producen alteraciones a nivel renal
Pulmones: los pulmones están afectados en mas del 50% de los pacientes con esclerosis sistémica, produciendo una hipertensión pulmonar y fibrosis intersticial.
Corazón: aparece pericarditis con derrame y fibrosis miocárdica, junto con engrosamiento de las arteriolas intramiocárdicas.

Miopatías inflamatorias : son un grupo infrecuente y heterogéneo de trastornos que se caracterizan por lesión e inflamación de los músculos principalmente esquelético, probablemente de mecanismos inmunitarios, dentro de estos se encuentran: dermatomiosistis, poliomiositis y miosotis por cuerpos de inclusión.
Poliartritis nudosa y otras vasculitis : la poliartritis nudosa pertenece a un grupo de enfermedades que se caracterizan por la inflamación necrosante de las paredes de los vasos sanguíneos y en las que hay datos sólidos a favor de un mecanismo patogénico inmunitario.
Rechazo de los trasplantes de órganos : El rechazo de lo trasplantes se analiza aquí porque están implicadas varias de las reacciones inmunitarias que subyacen a las enfermedades inflamatorias de mecanismo inmunitario. Esto esta dado debido a que el receptor reconoce el injerto como extraño y lo ataca.

Linfocitos T del receptor del trasplante reconoce las moléculas de las CPA del injerto
Los linfocitos T del anfitrión entran en contacto con las células dendríticas del donante
Los linfocitos T CD8 reconocen las moléculas del CPH de clase 1 y se diferencian en LTC activos, que pueden destruir las células del injerto.

Rechazo hiperagudo: esta forma de rechazo se produce de minutos a horas después del trasplante.
Rechazo agudo: esta forma de rechazo se puede producir a los pocos días del trasplante en un receptor no tratado o puede aparecer en varios meses e incluso en años.
Rechazo agudo celular: es el que se ve con mas frecuencia en los meses iniciales después del trasplante y esta precedido por signos clínicos y bioquímicos de insuficiencia renal.

Síndromes de inmunodeficiencia
se pueden clasificar en.
1. Primarias: están determinadas por factores genéticos. Una de las causas mas común es la agammaglobulinemia ligada a X, que se caracteriza por la ausencia de la maduración de los linfocitos B.
2. Secundarias: pueden generarse por complicaciones de canceres, infecciones, malnutrición o efectos adversos de inmunosupresión.
Inmunodeficiencia variable común:
Representa un grupo heterogéneo de trastornos, que se caracteriza por una hipogammaglobulinemia, que generalmente afecta a todas las clases de AC aunque en ocasiones solo a la IgG. Los pacientes por lo general presentan infecciones piógenas sinopulmonares recurrentes.

Deficiencia aislada de IgA: Es una inmunodeficiencia frecuente en la que las personas afectadas tienen una concentraciones muy bajas de IgA sérica y secretora, asociada a toxoplasmosis , parotiditis o alguna infección vírica, algunos pacientes con deficiencia de IgA también tienden a tener una deficiencia en la IgG2, IgG3, además puede estar asociada a LES y Artritis.
Síndrome de hiper-IgM: Los pacientes afectados sintetizan AC IgM, pero tienen una deficiencia de la capacidad para producirá AC IgG, IgA e IgE. Debido a estos afecta la capacidad de los linfocitos T cooperadores de ofrecer señales activadoras a los linfocitos B y macrófagos.

Síndrome de DiGeorge (hiperplasia tímica) : es un trastorno que se debe a la deleción de un gen, que esta localizado en el cromosoma 22q11.es una deficiencia de linfocitos T que se debe a la ausencia de desarrollo de la tercera y cuarta bolsa faríngea, provocando una perdida de la inmunidad mediada por linfocitos T. (debido a la hipoplasia o ausencia del timo), y malformaciones congénitas del corazón y los grandes vasos:
Inmunodeficiencia combinada grave: representan una constelación de síndromes diferentes desde el punto de vista genético, que tienen todos ellos en común defectos de las respuestas inmunitarias humorales y celulares. En su defecto produce Candidiasis oral .

Inmunodeficiencia con trombocitopenia y eccema( síndrome de wiskoot-aldrich): Es una enfermedad recesiva ligada a X, que se caracteriza por trombicitopenia, edema y una marcada vulnerabilidad a infecciones de repetición, que lleva a la muerte precoz, esto debido a mutaciones del gen que codifica la proteína del síndrome de Wiskoot-Aldrich, que esta localizado en el cromosoma Xp 11.23
Deficiencias congénitas del sistema del complemento:
C2 es la mas frecuente y ocasiona un aumento escaso o nulo de la susceptibilidad a las infecciones.
La deficiencia de (properdina y factor D) se asocia a infecciones piogenas.
La deficiencia de la C3 esta asociada a susceptibilidad a infecciones piógenas graves y recurrentes.
Los (C5,6,7.8 Y 9) son componentes esenciales para el ataque a la membrana, por tanto una disminución de estos componentes ocasionan infecciones recurrentes por neisseria.

INMUNODEFICIENCIAS SECUNDARIAS
en pacientes con cáncer, diabetes y otras enfermedades metabólicas, malnutrición, infección crónica y nefropatía.
en pacientes que reciben quimioterapia o radioterapia por el cáncer, o fármacos inmunodepresores para prevenir el rechazo de injertos o para tratar enfermedades autoinmunitarias
maduración defectuosa de los linfocitos
pérdida de inmunoglobulinas
síntesis inadecuada de Ig
depleción linfocítica
origen genético primario

SÍNDROME DE INMUNODEFICIENCIAADQUIRIDA (SIDA) es una enfermedad producida por el retrovirus llamado virus de
inmunodeficiencia humana (VIH)
se caracteriza por una profunda inmunodepresión que da lugar a infecciones oportunistas, neoplasias secundarias y manifestaciones neurológicas.
Al final de 2006, se habían notificado más de un millón de casos de sida en los EE. UU., en los que el sida es la segunda causa principal de muerte en hombres de 25 a 44 años, y la tercera causa principal de muerte en mujeres de este grupo de edad.
Actualmente se ha descrito en más de 190 países de todo el mundo, y el número de personas infectadas por VIH en África y Asia es elevado y está aumentando.

Epidemiología
han identificado cinco grupos de adultos con riesgo de presentar sida.
1. Los hombres homosexuales o bisexuales
2. Los pacientes que consumen drogas por vía intravenosa sin antecedentes de homosexualidad
3. Los hemofílicos
4. Los receptores de sangre y hemoderivados que no son hemofílicos, pero que recibieron transfusiones de sangre entera o de hemoderivados
5. Los contactos heterosexuales de los miembros de otros grupos de riesgo elevado
La epidemiología del sida es bastante diferente en niños menores de 13 años de edad.

Las tres principales vías de transmisión son contacto sexual , inoculación parenteral y paso del virus desde las madres infectadas a sus hijos recién nacidos
La transmisión sexual es responsable de más del 75% de los casos de transmisión del VIH.
Se ha descrito la transmisión parenteral de VIH en tres grupos de personas: consumidores de drogas por vía intravenosa, hemofílicos que reciben concentrados de factor VIII y de factor IX, y receptores aleatorios de transfusiones sanguíneas.
la transmisión de madre a hijo
no se puede transmitir por un contacto personal casual en el hogar, en el lugar de trabajo o en la escuela.

Etiología: propiedades del VIH
El sida está producido por el VIH
retrovirus humano no transformante que pertenece a la familia de los lentivirus
dos formas genéticamente diferentes llamadas VIH-1 y VIH-2 .
El VIH-1 es el tipo más frecuente asociado al sida en los EE. UU., Europa y África central
el VIH-2 produce una enfermedad similar principalmente en África occidental y la India
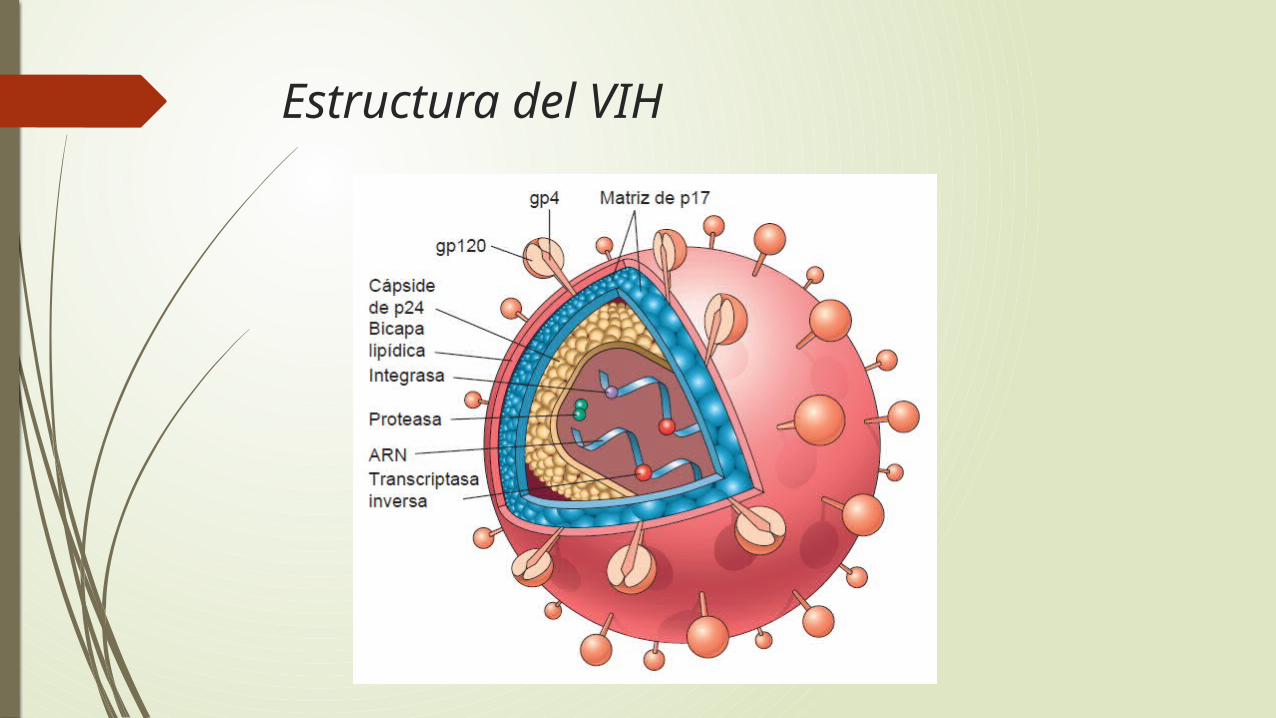
Estructura del VIH


Patogenia de la infección por VIH en el sida
hay dos dianas principales para la infección: el sistema inmunitario y el sistema nervioso central
Una inmunodeficiencia profunda
la infección y la pérdida grave de linfocitos T CD4+
los macrófagos y las células dendríticas son también dianas
El VIH entra en el cuerpo a través de los tejidos mucosos y la sangre
La infección se establece en los tejidos linfáticos, en los que el virus puede permanecer latente durante períodos prolongados
La replicación activa del virus se asocia a una mayor infección de las células y a progresión hasta sida.
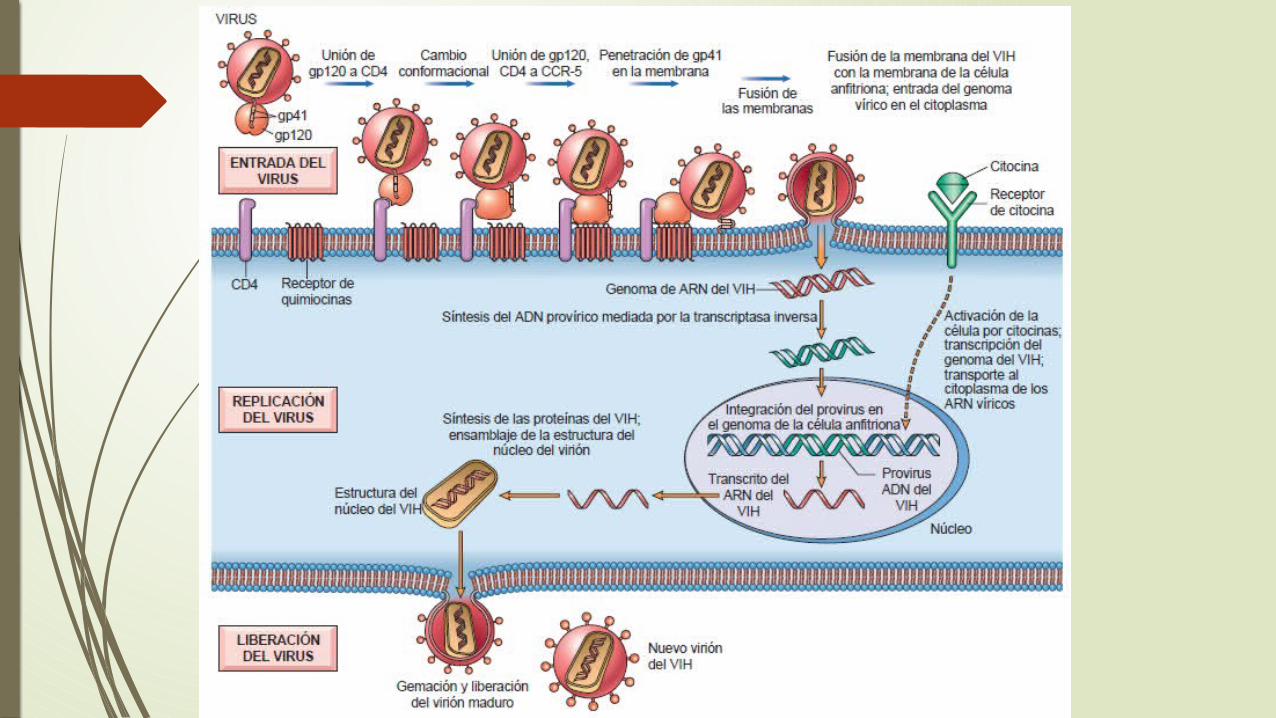

Mecanismo de la inmunodeficiencia de linfocitos T en la infección por el VIH


Patogenia de la afectación del sistemanervioso central
Los macrófagos y la microglía, células del sistema nervioso que pertenecen a la línea de los macrófagos, son los tipos celulares predominantes en el encéfalo que son infectados por el VIH
el VIH es transportado hasta el encéfalo por los monocitos infectados.
las neuronas no son infectadas por VIH
la alteración neurológica está producida indirectamente por productos víricos y por factores solubles producidos por la microglía infectada como IL-1, TNF e IL-6.
se ha implicado al óxido nítrico inducido en las células neuronales por gp41.





Tumores
Los pacientes con sida tienen una incidencia elevada de algunos tumores, especialmente sarcoma de Kaposi (SK) , linfoma no hodgkiniano de linfocitos B, cáncer cervical en mujeres y cáncer anal en hombres.
del 25 al 40% de los pacientes infectados por VIH y no tratados finalmente presentarán una neoplasia maligna
Sarcoma de Kaposi es la neoplasia más frecuente en pacientes con sida.

Enfermedad del sistema nervioso central
es una manifestación frecuente e importante del sida.
En algunos pacientes, las manifestaciones neurológicas pueden ser la única manifestación, o la manifestación más temprana, de la infección por el VIH
meningoencefalitis autolimitada
meningitis aséptica
mielopatía vacuolar
neuropatías periféricas
y, con más frecuencia, encefalopatía progresiva, denominada clínicamente complejo de sida-demencia

Efecto del tratamiento con fármacos antirretrovíricossobre la evolución clínica de la infección por VIH
tratamiento antirretrovírico de gran actividad (TARGA) o tratamiento antirretrovírico combinado
( < 50 copias de ARN por mililitro).
se detiene la pérdida progresiva de linfocitos T CD4+.
Esto se ha denominado síndrome inflamatorio de la reconstitución inmunitaria
lipoatrofia (pérdida de la grasa facial), lipohipertrofia (exceso de depósito central de grasa), elevación de los lípidos, resistencia insulína, neuropatía periférica, enfermedad cardiovascular prematura, nefropatía y disfunción hepática.

Amiloidosis
es una sustancia proteinácea patológica que se deposita en el espacio extracelular de diversos tejidos y órganos del cuerpo en una amplia variedad de situaciones clínicas.
el amiloide aparece como una sustancia extracelular hialina, eosinófila y amorfa, que cuando se acumula de forma progresiva envuelve las células adyacentes y produce atrofia por presión de las mismas.
Para diferenciar el amiloide de otros depósitos hialinos (p. ej., colágeno y fibrina) se utilizada la tinción de rojo Congo
microscopio de luz polarizada se observa la birrefringencia verde manzana del amiloide teñido



Naturaleza química del amiloide
el 95% del material del amiloide está formado por proteínas fibrilares, y el 5% restante es el componente P y otras proteínas.
1. El amiloide AL (cadena ligera del amiloide) procede de las cadenas ligeras de las Ig producidas por las células plasmáticas
2. El amiloide AA (asociada al amiloide) procede de una proteína diferenciada distinta a las Ig que es sintetizada por el hígado
3. El amiloide Aβ es producido a partir de la proteína precursora del amiloide y se encuentra en las lesiones cerebrales de la enfermedad de Alzheimer
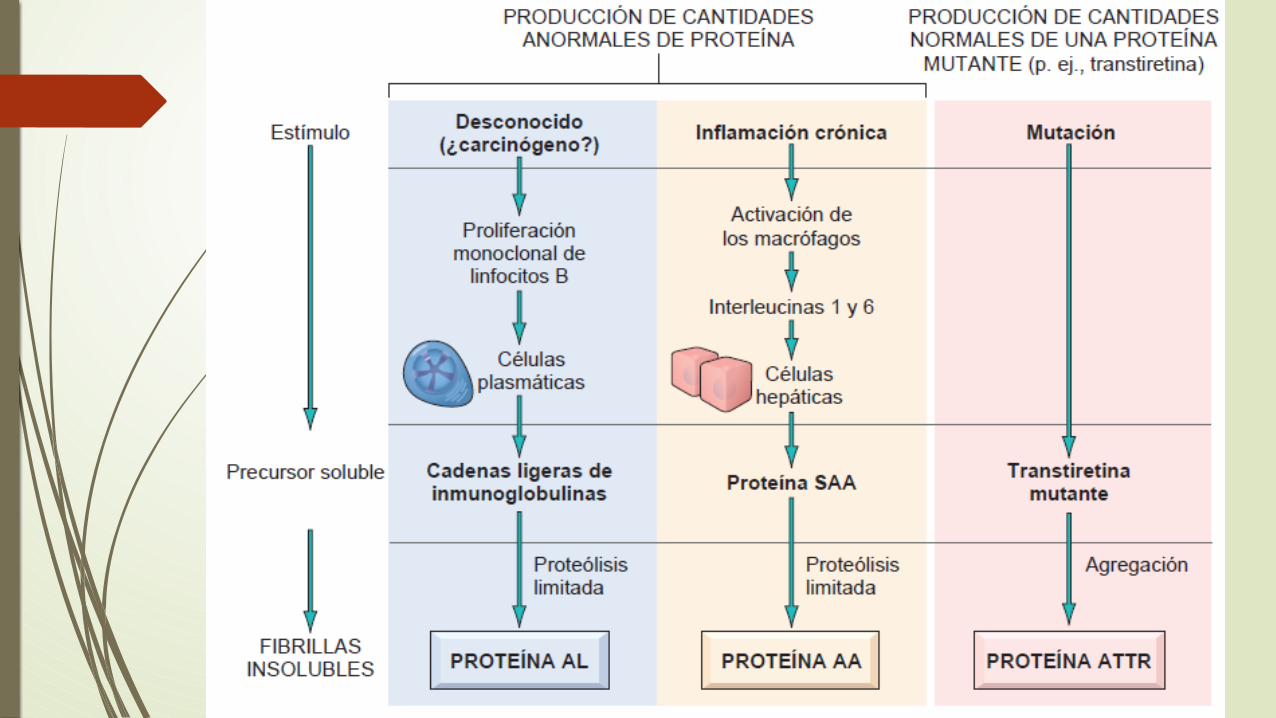


Características clínicas .
se puede encontrar como una alteración anatómica no sospechada, que no ha producido manifestaciones clínicas, o puede producir la muerte.
Los síntomas dependen de la magnitud de los depósitos y de las localizaciones u órganos particulares afectados.
debilidad, pérdida de peso, mareo o síncope. Posteriormente, aparecen hallazgos algo más específicos, que en la mayoría de los casos se relacionan con la afectación renal, cardíaca y digestiva.
El pronóstico de los pacientes con amiloidosis generalizada es malo.
Los que tienen amiloidosis derivada de inmunocitos tienen una mediana de supervivencia de 2 años después del diagnóstico.


















