
“EFECTOS DEL OZONO EN UN MODELO … · Nfkb e incrementó significativo de IL-6, IL-1, Col4 y...
114
Título del Programa de Doctorado: Clínica Veterinaria e Investigación Terapéutica. Mención de Calidad Otorgada por el Ministerio de Ciencia e Innovación del Gobierno de España (2008). Centro/Departamento Responsable del Programa: Dpto. de Patología Animal, Bromatología y Tecnología de los Alimentos, Campus Universitario de Arucas, Facultad de Veterinaria. Unidad de Investigación. Entidades: Universidad de Las Palmas de Gran Canaria. Hospital Universitario de Gran Canaria Dr. Negrín. “EFECTOS DEL OZONO EN UN MODELO EXPERIMENTAL DE FIBROSIS PULMONAR INDUCIDA CON BLEOMICINA” Tesis Doctoral presentada por: Dña. CHARLÍN MÉNDEZ CORDOVEZ Dirigida por los Doctores: Dr. Bernardino Clavo Varas Dr. Norberto Santana Rodríguez Dr. Pedro R. Llontop Santisteban Dra. Mª Dolores Fiuza Pérez Director, Director, Director, Director, Doctoranda (Firma) (Firma) (Firma) (Firma) (Firma) Las Palmas de Gran Canaria, a 9 de Noviembre de 2015.
Transcript of “EFECTOS DEL OZONO EN UN MODELO … · Nfkb e incrementó significativo de IL-6, IL-1, Col4 y...

Título del Programa de Doctorado: Clínica Veterinaria e Investigación Terapéutica. Mención de Calidad Otorgada por el Ministerio de Ciencia e Innovación del Gobierno de España (2008). Centro/Departamento Responsable del Programa: Dpto. de Patología Animal, Bromatología y Tecnología de los Alimentos, Campus Universitario de Arucas, Facultad de Veterinaria. Unidad de Investigación. Entidades: Universidad de Las Palmas de Gran Canaria. Hospital Universitario de Gran Canaria Dr. Negrín.
“EFECTOS DEL OZONO EN UN MODELO EXPERIMENTAL DE FIBROSIS PULMONAR INDUCIDA CON BLEOMICINA”
Tesis Doctoral presentada por:
Dña. CHARLÍN MÉNDEZ CORDOVEZ
Dirigida por los Doctores:
Dr. Bernardino Clavo Varas
Dr. Norberto Santana Rodríguez
Dr. Pedro R. Llontop Santisteban
Dra. Mª Dolores Fiuza Pérez
Director, Director, Director, Director, Doctoranda
(Firma) (Firma) (Firma) (Firma) (Firma)
Las Palmas de Gran Canaria, a 9 de Noviembre de 2015.

BERNARDINO CLAVO VARAS, Doctor en Medicina por la Universidad Autónoma de Madrid, Médico Especialista en Oncología Radioterápica e Investigador en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín. CERTIFICA: Que el trabajo de investigación titulado “Efectos del ozono en un modelo experimental de fibrosis pulmonar inducida con bleomicina” ha sido realizado por Dña. Charlín Méndez Cordovez, en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín, bajo mi dirección y asesoramiento, una vez revisada la presente memoria la encuentro apta para su defensa como Tesis Doctoral, para optar al grado de doctor. Y para que así conste y surta los efectos oportunos, extiendo el presente certificado, en Las Palmas de Gran Canaria a 9 de Noviembre de dos mil quince.
Fdo. Bernardino Clavo Varas.
Hospital Universitario de Gran Canaria Dr.Negrín. Unidad de Investigación.
Barranco de la Ballena, Sn 35010 – Las Palmas de Gran Canaria
Telf.: 630751093

NORBERTO SANTANA RODRIGUEZ, Doctor en Medicina por la Universidad de Las Palmas de Gran Canaria, Médico Especialista en Cirugía Torácica y Responsable de Cirugía Experimental e Investigador en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín.
CERTIFICA:
Que el trabajo de investigación titulado “Efectos del ozono en un modelo experimental de fibrosis pulmonar inducida con bleomicina” ha sido realizado por Dña. Charlín Méndez Cordovez, en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín, bajo mi dirección y asesoramiento, una vez revisada la presente memoria la encuentro apta para su defensa como Tesis Doctoral, para optar al grado de doctor. Y para que así conste y surta los efectos oportunos, extiendo el presente certificado, en Las Palmas de Gran Canaria a 9 de Noviembre de dos mil quince.
Fdo. Norberto Santana Rodríguez.
Hospital Universitario de Gran Canaria Dr.Negrín. Unidad de Investigación.
Barranco de la Ballena, Sn 35010 – Las Palmas de Gran Canaria
Telf.: 630751093

PEDRO ROLANDO LLONTOP SANTISTEBAN, Doctor en Veterinaria por la Universidad de Las Palmas de Gran Canaria e Investigador en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín.
CERTIFICA:
Que el trabajo de investigación titulado “Efectos del ozono en un modelo experimental de fibrosis pulmonar inducida con bleomicina” ha sido realizado por Dña. Charlín Méndez Cordovez, en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín, bajo mi dirección y asesoramiento, una vez revisada la presente memoria la encuentro apta para su defensa como Tesis Doctoral, para optar al grado de doctor.
Y para que así conste y surta los efectos oportunos, extiendo el presente certificado, en Las Palmas de Gran Canaria a 9 de Noviembre de dos mil quince.
Fdo. Pedro Rolando LLontop Santisteban.
Hospital Universitario de Gran Canaria Dr.Negrín. Unidad de Investigación.
Barranco de la Ballena, Sn 35010 – Las Palmas de Gran Canaria
Telf.: 630751093

Mª DOLORES FIUZA PÉREZ, Doctora en Medicina por la Universidad de Las Palmas de Gran Canaria, Médico Especialista en Medicina Preventiva y Salud Pública y Epidemióloga Clínica del Hospital Universitario de Gran Canaria Dr. Negrín.
CERTIFICA:
Que el trabajo de investigación titulado “Efectos del ozono en un modelo experimental de fibrosis pulmonar inducida con bleomicina” ha sido realizado por Dña. Charlín Méndez Cordovez, en la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín, bajo mi dirección y asesoramiento, una vez revisada la presente memoria la encuentro apta para su defensa como Tesis Doctoral, para optar al grado de doctor.
Y para que así conste y surta los efectos oportunos, extiendo el presente certificado, en Las Palmas de Gran Canaria a 9 de Noviembre de dos mil quince.
Fdo. Mª. Dolores Fiuza Pérez
Hospital Universitario de Gran Canaria Dr.Negrín. Unidad de Investigación.
Barranco de la Ballena, Sn 35010 – Las Palmas de Gran Canaria
Telf.: 630751093

Financiación

La presente Tesis Doctoral ha recibido financiación del Gobierno de Canarias, Consejería de Sanidad, a través de la convocatoria pública de la Fundación Canaria de Investigación y Salud (FUNCIS) del año 2008, de ayudas para Personal Investigador en Formación otorgada a la doctoranda.
El Proyecto de investigación “Efectos del Ozono en un modelo experimental de fibrosis pulmonar inducida con bleomicina” ha sido financiado por el propio grupo investigador.
Investigador principal: Dr. Norberto Santana Rodríguez.

A mis padres, Aida y Jesús.
La distancia que nos ha separado estos años no ha sido inconveniente para sentir sus ánimos, apoyo y cariño que me han permitido seguir adelante para cumplir mis sueños.

Agradecimientos

Después de un largo tiempo de trabajo, quiero agradecer en primer lugar a las instituciones que han hecho posible la realización de esta Tesis Doctoral tanto por su ayuda económica, formación, así como por todos los medios puestos a mi alcance: a la Fundación Canaria de Investigación y Salud, a la Facultad de Veterinaria (ULPGC), a la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín, sería muy difícil lograrlo si no es con el apoyo y estímulo de todas las personas que forman parte de ellas
En segundo lugar, mi más sincero agradecimiento a mis directores de Tesis, por su ayuda incondicional y por acogerme en su Grupo de Investigación de “Cirugía Experimental”, en especial al Dr. Santana y a mi Tutor el Dr. Bernardino Clavo, por su predisposición y aliento desde el principio, e impagable generosidad intelectual; a la Dra. Fiuza, por formarme en el área de la Metodología de la Investigación en Medicina y por su ayuda incondicional para realizar el análisis estadístico de este proyecto; a mi compañero de trabajo, el Dr. LLontop por romper la rutina al brindarme su apoyo “Verás …Charlín…verás” , etc.
Agradezco a los doctores de la Facultad de Veterinaria, donde di mis primeros pasos como investigadora, su formación en el Área de la Investigación Clínica y Terapéutica Veterinaria, donde desarrolle el “gusanillo” este de la investigación, en especial al Dr. Alberto Montoya, a la Dra. Maite Tejedor, Dr. Carlos Gutiérrez, al Dr. Pablo, a la Dra. Candelaria Juste, y al Dr. Antonio Espinosa de los Monteros y Zayas.
Agradecer al Dr. Camacho, Jefe de Servicio de Anatomía Patológica, y a la Dra. Ana Bordes del Servicio de Microbiología del Hospital Universitario Dr. Negrín, su predisposición y asesoramiento en el procesado y análisis de las muestras de este proyecto.
A todos los compañeros que he tenido en los Servicios por los que he rotado, en mi formación como investigadora, de la mano del Dr. Carlos Rodríguez Santana (Servicio de Inmunología) y la Dra. Teresa Gómez (Servicio de Hematología).
A mis compañeros de la Unidad de Investigación, en mi corazón solo puede haber agradecimiento hacia ustedes porque son como una familia para mí: Ángela, Rosa, Miguel, Fran, José Luis, Lidia, Tatiana, y en especial a Yanira, por su ayuda técnica en el laboratorio.
Agradecer al Servicio de Ilustración, a D. Juan y a D. Ramón por estar siempre que los he necesitado en la elaboración del diseño e imagen de las presentaciones a congresos, a los que he asistido y su ayuda en la presente Tesis.
Agradecer a toda mi familia, en especial a mi madre, mi mayor fuente de inspiración, fortaleza, lucha, dedicación y amor.

RESUMEN

INTRODUCCIÓN: La fibrosis pulmonar (FP) idiopática es la forma más frecuente y la de peor
pronóstico de FP, con media de supervivencia de 3-5 años. Las opciones terapéuticas son muy
limitadas. El modelo experimental de FP inducida por instilación orotraqueal (IOT) de bleomicina (BLM)
se ha mostrado útil en el estudio del desarrollo de la FP, condicionada por la perpetuación de estrés
oxidativo e inflamación. Múltiples trabajos han evidenciado que el tratamiento con ozono (O3) puede
modular esos factores.
OBJETIVOS: Estudiar en un modelo de FP inducida mediante IOT de BLM en ratas, los posibles
efectos protectores del O3, algo que, hasta la fecha, ningún estudio ha evaluado previamente.
MATERIAL Y MÉTODO: se emplearon 21 ratas distribuidas en cuatro grupos. A): Control, 3 animales
(analizados 6 pulmones). Resto de grupos con 6 ratas: B) Sham; C) BLM; D) Bleomicina+O3. Al grupo
Control no se le realizó instilación IOT. Al grupo Sham se le realizó IOT de suero fisiológico. A los
grupos BLM y BLM+O3 se les indujo FP mediante IOT de BLM. Al grupo de BLM+03, además, se le
administró O3 vía rectal, desde el 2º día pos-IOT hasta el momento del sacrificio a los 28 días pos-IOT.
RESULTADOS: El grado de FP fue mayor en el grupo Sham que en el grupo Control (p= 0.002), y en
el grupo BLM respecto al grupo SHAM (p= 0.002). El grupo BLM+O3 mostró menos fibrosis que el de
BLM (p= 0.026). El estudio de expresión génica mostró alteraciones en el grupo Sham respecto al
grupo Control. Respecto al grupo Sham, el grupo BLM mostró un incremento significativo de la fibrosis
y de la expresión de Hsp27, Nfkb, IL-6, IL-1, Col4 y Tgfb-1. El tratamiento con O3 disminuyó
significativamente el grado de fibrosis y la expresión de Hsp27, con normalización de la expresión de
Nfkb e incrementó significativo de IL-6, IL-1, Col4 y Tgfb-1.
CONCLUSIONES: Nuestro modelo experimental de FP mediante IOT de BLM se mostró útil en la
producción de la enfermedad. La administración de O3 vía rectal, disminuyó significativamente el grado
de fibrosis y de expresión de Hsp27 y normalizó Nfkb, lo que muestra su capacidad para influir en la

evolución de la enfermedad. Desde el punto de vista traslacional, el O3 se plantea como una nueva
opción potencialmente aplicable en la FP, enfermedad grave y con limitadas opciones terapéuticas.

ÍNDICE GENERAL
ÍNDICE DE TABLAS.
ÍNDICE DE FIGURAS.
ABREVIATURAS.
I.- INTRODUCCIÓN………………………………………………………………………………. 1
1.- Fibrosis Pulmonar…………………………………………………………………………... 2
1.1.- Definición……………………………………………………………………………………. 2
1.2.- Epidemiología………………………………………………………………………………. 2
1.3.- Factores de riesgo…………………………………………………………………………. 3
1.3.1.- Factores genéticos hereditarios…………………………………………………………. 3
1.3.2.- Factores ambientales…………………………………………………………………….. 3
1.3.3.- Enfermedad por reflujo gastroesofágico……………………………………………….. 3
1.3.4.- Infecciones virales………………………………………………………………………… 4
1.3.5.- Autoinmunidad…………………………………………………………………………….. 4
1.3.6.- Exposición a fármacos………………………………………………………………….... 4
1.3.7.- Otros factores a tener en cuenta………………………………………………………... 5
1.4.- Patogénesis………………………………………………………………………………… 5
1.4.1.- Daño y activación de las células epiteliales alveolares (CEA)………………………. 5
1.4.2.- Efectos profibróticos de la activación anómala de las CEA en el microambiente pulmonar…………………………………………………………………………………………….
5
1.4.3.- Diferenciación de los fibroblastos en miofibroblastos………………………………… 6
1.4.4.- La ausencia de neumocitos tipo I………………………………………………………. 6
1.4.5.- Metaloproteinasas de la matriz (MMPs) y la anormal remodelación pulmonar……. 7

1.4.6.- Angiogénesis y remodelación microvascular…………………………………………. 7
1.4.7.- Relación entre FPI y envejecimiento…………………………………………………… 7
1.4.8.- Los pericitos y las células mesoteliales pleurales…………………………………….. 8
1.5.- Prevención...………………………………………………………………………………... 8
1.6.- Diagnóstico 8
1.6.1.- Manifestaciones clínicas y analíticas…………………………………………………. 9
1.6.2. Diagnóstico radiológico……………………………………………………………………. 10
1.6.2.1.- Radiografía de toráx……………………………………………………………. 10
1.6.2.2.- Tomografía Computadorizada de Alta Resolución (TCAR)……………….. 11
1.6.3.- Pruebas de función pulmonar…………………………………………………………… 13
1.6.4.- Lavado broncoalveolar (LBA)……………………………………………………………. 13
1.6.5.- Biopsia pulmonar………………………………………………………………………….. 14
1.6.6.- Estudio histológico………………………………………………………………………... 14
1.7.- Tratamiento…………………………………………………………………………………. 15
1.7.1.- Terapia farmacológica……………………………………………………………………. 16
1.7.1.1.- Pirfenidona……………………………………………………………………… 16
1.7.1.2.- Nintedanib………………………………………………………………………. 17
1.7.1.3.- N-Acetilcisteína………………………………………………………………… 18
1.7.1.4.- Otros fármacos…………………………………………………………………. 18
1.7.2.- Rehabilitación respiratoria……………………………………………………………….. 19
1.7.3.- Trasplante pulmonar……………………………………………………………………… 19
1.8.- Pronóstico…………………………………………………………………………………... 20
2.- Bleomicina y Modelo Experimental de Fibrosis Pulmonar………………………….. 21
2.1.- Bleomicina…………………………………………………………………………………... 21
2.1.1.- Estructura de la bleomicina……………………………………………………………… 21
2.1.2.- Mecanismos de acción de la bleomicina……………………………………………….. 22

2.1.3.- Farmacocinética y metabolismo………………………………………………... 22
2.1.4.- Toxicidad pulmonar por bleomicina…………………………………………… 23
2.2- Modelo Experimental de Fibrosis Pulmonar…………………………………………. 23
3.- Ozono…………………………………………………………………………………………... 25
3.1.- Historia………………………………………………………………………………………. 25
3.2.- Definición y propiedades………………………………………………………………… 26
3.3.- Mecanismos de acción…………………………………………………………………… 27
3.4.- Vías de administración…………………………………………………………………… 31
3.5.- Efectos secundarios y contraindicaciones de la ozonoterapia………………… 32
II.- JUSTIFICACIÓN………………………………………………………………………………. 33
III.- OBJETIVOS…………………………………………………………………………………… 35
IV.- MATERIAL Y MÉTODOS…………………………………………………………………… 37
4.1. Material……………………………………………………………………………………….. 38
4.1.1.- Animales de experimentación…………………………………………………………… 38
4.1.2.- Salas, equipos y material inventariable………………………………………………… 39
4.1.3.- Material quirúrgico………………………………………………………………………… 40
4.1.4.- Medicación anestésica y analgésica…………………………………………………… 41
4.1.5- Material para conservación del tejido pulmonar………………………………………. 42
4.2.- Método……………………………………………………………………………………….. 42
4.2.1.- Diseño……………………………………………………………………………………… 42
4.2.2.- Inducción de fibrosis pulmonar por BLM……………………………………………… 43
4.2.2.1.- Anestesia……………………………………………………………………….. 44
4.2.2.2.- Intubación y ventilación………………………………………………………. 44
4.2.2.3.- Administración de BLM………………………………………………………. 44
4.2.2.4- IOT de solución salina isotónica 0,9% (solución fisiológico) y BLM……. 45
4.2.3.- Tratamiento con ozono………………………………………………………………….. 45

4.2.4.- Evolución clínica en el periodo postoperatorio……………………………………….. 46
4.2.5.- Obtención y recogida de muestras…………………………………………………….. 46
4.2.6.- Mediciones………………………………………………………………………………… 49
4.2.6.1- Análisis microbiológico………………………………………………………….. 49
4.2.6.2.-Análisis histológico……………………………………………………………….. 49
4.2.6.2.1.- Procesamiento de las muestras……………………………………………. 49
4.2.6.2.2.- Lectura de las muestras…………………………………………………….. 50
4.2.6.3. Análisis de la expresión genómica……………………………………………… 51
4.2.6.3.1- Extracción de RNA y síntesis de cDNA……………………………………. 51
4.2.6.3.2- Análisis de la expresión genómica mediante qRT-PCR………………… 52
4.2.7.- Análisis estadístico……………………………………………………………………….. 53
V.- RESULTADOS……………………………………………………………………………….. 54
5.1.- Resultados y datos clínicos de los animales………………………………………… 55
5.2.- Descripción macroscópica………………………………………………………………. 56
5.3. Estudio histológico. Cuantificación del grado de fibrosis según escala Ashcroft……………………………………………………………………………………………
57
5.4.- Análisis microbiológico………………………………………………………………….. 59
5.5.- Análisis de Expresión génica……………………………………………………………. 59
VI.- DISCUSIÓN…………………………………………………………………………………… 70
VII.- CONCLUSIONES……………………………………………………………………………. 76
VIII.- BIBLIOGRAFÍA……………………………………………………………………………... 79

ÍNDICE DE TABLAS
Tabla 1. Pruebas de laboratorio ante sospecha de FPI……………………………………………... 10
Tabla 2. Criterios de patrón de neumonía intersticial usual (NIU) en la TCAR…………………… 12
Tabla 3. Recomendaciones basadas en la evidencia para el tratamiento de la FPI……………. 16
Tabla 4. Resumen de algunos de los efectos tópicos y sistémicos del ozono médico…………. 30
Tabla 5. Diseño de grupos para el estudio histológico, microbiológico y de expresión génica…. 43
Tabla 6. Volumen de IOT de los grupos de estudio………………………………………………….. 43
Tabla 7. Criterio para valorar el grado de fibrosis pulmonar………………………………………… 51
Tabla 8. Genes y condiciones de la PCR a tiempo real…………………………………………….. 52
Tabla 9. Evolución del peso de los animales…………………………………………………………. 55
Tabla 10. Días empleados en recuperar el peso inicial del estudio según grupos……………….. 55
Tabla 11. Grado de fibrosis pulmonar según la escala de Ashcroft y su significación estadística.. 59
Tabla 12. Expresión de los genes analizados entre el grupo Control va SHAM………………….. 60
Tabla 13. Expresión de los genes analizados entre el grupo Control vs BLM……………………… 61
Tabla 14. Expresión de los genes analizados entre el grupo Control vs BLM+O3………………… 62
Tabla 15. Expresión de los genes analizados entre el grupo SHAM vs BLM……………………… 63
Tabla 16. Expresión de los genes analizados entre el grupo SHAM vs BLM+O3…………………. 64
Tabla 17. Expresión de los genes analizados entre el grupo BLM y BLM+O3……………………. 65

ÍNDICE DE FIGURAS
Figura 1. Algoritmo diagnóstico de la fibrosis pulmonar idiopática (FPI)………………………….. 9
Figura 2. Hallazgos en imágenes con opacidades panal de abeja con una disminución periférica y basal……………………………………………………………………………….
11
Figura 3. Hallazgos en imágenes con opacidades panal de abeja con una disminución periférica y basal……………………………………………………………………………….
11
Figura 4. La tomografía computerizada de alta resolución (TCAR) puede mostrar la estructura en panal de abeja aparece como sombras anulares estrechamente aproximadas, el hallazgo implica enfermedad pulmonar en estado avanzado. Izquierda: imagen de un corte axial. Derecha: imagen de un corte coronal…………………………………………
12
Figura 5. Patrón de neumonía intersticial usual en la biopsia pulmonar: fibrosis periférica (A) con focos de actividad fibroblástica en áreas de interface (B) y focos de micropanal (C…………………………………………………………………………………………………
15
Figura 6. Estructura de la bleomicina (BLM)………………………………………………………….. 21
Figura 7. Esquema del modelo de fibrosis pulmonar inducida por bleomicina………………….. 24
Figura 8. Leyes de Ohms y de Poiseuille, donde F= Flujo, P= Presión de flujo, R= Resistencia, η= Viscosidad sanguínea, r= Radio y L= Longitud del vaso…………………………….
29
Figura 9. Esquema de la acción del O3 sobre la oxigenación y flujo de los tejidos. ROS: radicales libres; LPO: lipoperóxidos; A.O.: antioxidantes; Nrf2: factor nuclear eritroide-2; 2,3-DPG: 2,3 difosfoglicerato; NO: óxido nítrico; PG: prostaglandinas; Hb: hemoglobina……………………………………………………………………………………
29
Figura 10. Esquema de cómo la ozonoterapia sistémica (vía insuflación rectal o mediante autohemoterapia) y los lipoperóxidos a los que da lugar, pueden inducir una respuesta adaptativa en distintos órganos……...............................................................
30
Figura 11. Jaula 20 x 35 x 55 cm…………………………………………………………………………. 38
Figura 12. Sala del postoperatorio……………………………………………………………………….. 39
Figura 13. Instalaciones quirúrgicas…………………………………………………………………….. 39
Figura 14. Ventilador Servo 900C………………………………………………………………………… 40
Figura 15. Generador de O3………………………………………………………………………………. 40
Figura 16. Microscopio de pie Carl Zeiss………………………………………………………………... 40
Figura 17. Congelador Jouan – 80º C…………………………………………………………………… 40

Figura 18. Instrumental quirúrgico………………………………………………………………………... 41
Figura 19. Anestésicos y analgésicos empleados en las ratas……………………………………….. 41
Figura 20. Muestra el proceso de IOT…………………………………………………………………… 45
Figura 21. Insuflación rectal de O3 en la rata……………………………………………………………. 45
Figura 22. Esternotomía media. Acceso a bloque cardio-pulmonar………………………………….
46
Figura 23. Rata en posición de decúbito supino, traqueotomía y ventilación mecánica…………… 47
Figura 24. Esternotomía media…………………………………………………………………………… 48
Figura 25. Extracción del bloque cardio-pulmonar……………………………………………………… 48
Figura 26. Procesador de tejidos Leica TP 1020……………………………………………………….. 50
Figura 27. Dispensador de parafina Leica EG 1160……………………………………………………. 50
Figura 28. Microtomo de rotación Thermoscientific HM 340 E………………………………………... 50
Figura 29. Microscopio óptico Olympus BX40………………………………………………………….. 50
Figura 30. Gráfica de evolución del peso de los animales durante los 28 días del estudio……….. 56
Figura 31. Pulmón grupo Control y grupo Sham……………………………………………………….. 56
Figura 32. Pulmón grupo BLM……………………………………………………………………………. 56
Figura 33. Pulmón grupo BL+03…………………………………………………………………………... 57
Figura 34. Estudio histológico grupo Control……………………………………………………………. 57
Figura 35. Estudio histológico grupo SHAM…………………………………………………………….. 58
Figura 36. Estudio histológico grupo BLM…………………………………………………………………………………………………….. 58
Figura 37. Estudio histológico grupo BLM+03................................................................................... 58
Figura 38. Expresión de los genes analizados entre control vs SHAM……………………………… 60
Figura 39. Expresión de los genes analizados entre control vs BLM………………………………… 61
Figura 40. Expresión de los genes analizados entre control vs BLM+O3.…………………………... 62
Figura 41. Expresión de los genes analizados entre grupo SHAM vs BLM + O3…………………... 64
Figura 42. Expresión de los genes analizados entre BLM vs BLM+O3………………………………. 65

Figura 43. Expresión de HSP27 entre Sham vs BLM vs BLM+O3……………………………………. 66
Figura 44. Expresión de Vegfa entre Sham vs BLM vs BLM+O3……………………………………... 67
Figura 45. Expresión de Nfkb entre Sham vs BLM vs BLM+O3………………………………………………………………. 67
Figura 46. Expresión de IL6 entre Sham vs BLM vs BLM+O3………………………………………….. 68
Figura 47. Expresión de Col4 entre Sham vs BLM vs BLM+O3………………………………………. 68
Figura 48 Expresión de IL1 entre Sham vs BLM vs BLM+O3……………………………………....... 69
Figura 49. Expresión de Tgfb entre Sham vs BLM vs BLM+O3………………………………………... 69

ÍNDICE DE ABREVIATURAS
AHT: Autohemoterapia.
ALAT: Asociación Latinoamericana del Tórax.
AO: Sistemas antioxidantes.
AOS: Apnea obstructiva del sueño.
AP-1: Proteína activadora 1.
AR: Artritis reumatoide.
ATP: Adenosina trifosfato.
ATS: American Thoracic Society.
BP: Biopsia pulmonar.
BPA: Biopsia pulmonar abierta.
BPQ: Biopsia pulmonar quirúrgica.
BR – EPI: Bronquiolitis respiratoria asociada a enfermedad pulmonar intersticial.
BTB: Biopsia transbronquial.
BTS: Sociedad Británica del Tórax.
CE: Células epiteliales.
CEA: Células epiteliales alveolares.
(CIO- ): Anión hipoclorito.
CO: Monóxidos de carbono.
CVF: Capacidad vital forzada.
CPT: Capacidad pulmonar total.
DLCO: Capacidad de “difusión del monóxido de carbono”.
DM: Diabetes mellitus.
2,3- DPG: 2,3 difosfoglicerato.
EAC: Enfermedad de las arterias coronarias.

EDPP: Enfermedad difusa del parénquima pulmonar.
EMC: Estatus Microbiológico Convencional.
EPID: Enfermedades pulmonares intersticiales difusas.
ERGE: Enfermedad por reflujo gastroesofágico.
ERS: European Respiratory Society.
F: Flujo sanguíneo.
FELASA: Federaciones de Asociaciones Europeas de las Ciencias del Animal de Laboratorio.
FP: Fibrosis pulmonar.
FPF: Fibrosis pulmonar familiar.
FPI: Fibrosis pulmonar idiopática.
FPIF: Fibrosis pulmonar idiopática familiar.
GITC: Tiaocinato de guanidino.
GSH: Glutatión.
4-HNE: 4-hidroxinonenal.
HO: Hemo oxigenasa.
HP: Hipertensión pulmonar.
HP secundaria: Hipertensión pulmonar secundaria.
HPS: Síndrome de Hermansky- Pudlak.
HSP-70: Proteínas de shock térmico de 70 KDa.
HE: Hematoxilina- eosina.
IgG: Inmunoglobulinas G.
IP: Vía intraperitoneal.
IOT: Instilación orotraqueal.
IR: Insuflación rectal.
JRS: Sociedad Respitaroria Japonesa.
LBA: Líquido del lavado brocoalveolar.

LPO: Lipoperóxidos.
MALT: Tejido linfoide asociado a mucosas.
NADPH: Nicotinamida adenina dinucleótido fosfato.
NFAT: Factor nuclear de las células T activadas.
NH: Neumonitis por hipersensibilidad.
NII: Neumonía intersticial idiopática.
NIU: Neumonía intersticial usual.
NID: Neumonía intersticial descamativa.
NIL: Neumonía intersticial linfoide.
NINE: Neumonía intersticial no específica.
Nrf2: Factor de transcripción nuclear.
N2O2: Peroxinitrilos.
O2: Oxígeno atmósferico.
O3: Ozono.
(0=N00- ): Peroxinitrilo.
PCR: Proteína c reactiva.
PTEN: Gen de la fosfatasa y tensina homólogo.
PLA2: Fosfolipasa A2.
P (A-a) O2: Diferencia alvéolo-arterial de oxígeno en reposo.
PUFA: Grasos poliinsaturados.
R: Resistencia vascular.
RCP: Resucitación cardio-pulmonar.
SEPAR: Sociedad Española de Patología del Aparato Respiratorio.
Shn: Gen Sonic hedgehog.
SOD: Superóxido dismutase.
TCAR: Tomografía axial computarizada de alta resolución.

TEM: Transición epitelio mesenquimal.
TEV: Tromboembolismo venoso.
TP: Trasplante pulmonar.
UIP: Neumonía interstical usual.
RL: Radicales libres.
RXT: Radiografía de tórax.
UV: Radiación ultravioleta.
UVB: Ultravioleta de onda media.
UVC: Ultravioleta de onda corta.
VEB: Virus de Epstein-Barr.
VHC: Virus de la hepatitis C.
VEMS: Volumen espirado máximo en el primer segundo de la espiración forzada.
VSG: Velocidad de sedimentación.

I. INTRODUCCIÓN.

Tesis Doctoral Charlín Méndez Cordovez
2
1.- Fibrosis Pulmonar Idiopática (FPI).
1.1.- Definición.
La fibrosis pulmonar idiopática (FPI) pertenece a una familia de aproximadamente 200
enfermedades relacionadas conocidos como enfermedades pulmonares intersticiales (EPID) que se
caracterizan por la afección difusa del parénquima pulmonar. Existen EPID en las que predominan los
procesos inflamatorios, como la sarcoidosis o la neumonitis por hipersensibilidad. En otras EPID
predominan los procesos de fibrosis, y entre ellas destaca por su mayor prevalencia la FPI.
La FPI se define como una forma específica de neumonía intersticial fibrosante, crónica,
progresiva, de causa desconocida, que ocurre generalmente en adultos de edad media o avanzada, y está
limitada a los pulmones. Está asociada a un patrón radiológico y/o histológico de la neumonía intersticial
usual (NIU). Se caracteriza por deterioro progresivo de disnea y función pulmonar y se asocia con mal
pronóstico y escasas opciones terapéuticas, con una supervivencia media de aproximadamente tres años
desde el momento del diagnóstico (1-3).
1.2.- Epidemiología.
La FPI es una enfermedad huérfana que afecta a un creciente número de personas en los Estados
Unidos (EE.UU.) y en Europa.
En EE.UU., los datos más fiables estiman que la prevalencia de la FPI varía en unos 14 a 27,9
casos por cada 100.000 habitantes utilizando definiciones estricta de casos y alrededor de 42,7 a 63 casos
por cada 100.000 habitantes utilizando definiciones de casos más amplia, dependiendo de la población
estudiada (4). La definición estrecha de casos cumple con todos los criterios mayores y menores de la
American Thoracic Society (ATS) y la European Respiratory Society (ERS) (5) y requiere patrones de
neumonía intersticial usual (NIU) definidas en tomografía computarizada de alta resolución pulmonar
(TCAR), mientras que la definición de casos generales incluye que los pacientes respondan a la definición
estrecha, así como aquellos con TCAR que ofrecen alguna característica posible de NIU. La incidencia
reportada fue de unos 6,8 a 8,8 casos por cada 100.000 habitantes.
En Europa la prevalencia de esta enfermedad varía de 1,25 a 23,4 casos por 100.000 habitantes
(6, 7) y su incidencia anual se estima entre 0,22 y 7,4 por 100.000 habitantes (6, 8).
De acuerdo a la normativa sobre diagnóstico y tratamiento de la FPI de la Sociedad Española de
Patología del Aparato Respiratorio (SEPAR), se estima que en España la FPI puede estar afectando a unas
7.500 personas (9). Se desconoce si la incidencia y prevalencia están influidas por factores étnicos, raciales
o geográficos. En los últimos años se ha observado un incremento en la prevalencia, probablemente debido
a la optimización de los métodos diagnósticos y al aumento de la esperanza de vida (8, 10).

Tesis Doctoral Charlín Méndez Cordovez
3
1.3.- Factores de riesgo.
Los factores de riesgo potenciales para el desarrollo de la FP incluyen: las exposiciones
ambientales y ocupacionales, el consumo de tabaco, las comorbilidades y los polimorfismos genéticos.
Conocer los factores de riesgo potenciales en el desarrollo de la FPI permitiría optimizar estrategias de
prevención, diagnóstico precoz y tratamiento más adecuadas, incluyendo el trasplante pulmonar (TP).
1.3.1.- Factores genéticos y hereditarios.
Las principales alteraciones genéticas relacionadas con FP son: mutaciones en los genes que
mantienen la longitud de los telómeros (TERT, TERC) y que son más frecuentes en las formas familiares,
en la proteína C del surfactante, en la región promotora de la mucina 5B (MUC5B) y polimorfismos de los
genes que codifican citoquinas como interleuquinas, factor de necrosis tumoral α (TNFα), factor de
crecimiento transformante β1 (TGF β1) o metaloproteinasas de la matriz extracelular (MMPs).
La presentación familiar supone menos del 5% de los casos de FPI, siendo éstos clínica e
histológicamente indistinguibles de los casos de presentación esporádica. La forma más probable de
transmisión genética de FPI familiar es a través de una herencia autosómica dominante con penetrancia
variable.
Entre las enfermedades hereditarias relacionadas con mayor riesgo de aparición de FP están: el
síndrome de Hermansky- Pudlak, la enfermedad de la disqueratosis congénita o el síndrome de insuficiencia
hereditaria de la médula ósea (1, 11).
Aproximadamente el 15% de los casos de FPI familiar y el 5% de los casos esporádicos portan la
mutación heterocigótica en alguno de sus genes, las células de los pacientes retienen el 50% de la actividad
de la polimerasa.
1.3.2.- Factores ambientales.
Múltiples líneas evidencian que la exposición a los agentes ambientales inhalados (tales
como el polvo de metales, piedras, maderas y textiles, asbesto o sílice), tabaquismo (≥ 20
paquetes/año) y actividades en la agricultura y la ganadería, se asocian a un riesgo aumentado de
FPI.
1.3.3.- Enfermedad por reflujo gastroesofágico (ERGE).
La ERGE ha sido asociada a la FPI debido a la presencia de ácidos de pepsina y / o biliares en el
líquido del lavado broncoalveolar (LBA) (12). El reflujo puede predisponer a la microaspiración, iniciando y/o
conduciendo a la inflamación pulmonar, que puede progresar a FP en un individuo susceptible, y la
evidencia acumulada ha vinculado ERGE con aspiración como factor para la predisposición y la progresión
de la FPI (13). El procedimiento quirúrgico de la funduplicatura de Nissen es la técnica de elección para
el tratamiento de la ERGE, mejorando significativamente la supervivencia de los pacientes con FPI (14).

Tesis Doctoral Charlín Méndez Cordovez
4
1.3.4.- Infecciones virales.
La FPI se caracterizada por una acumulación de macrófagos alveolares y neutrófilos en el tracto
respiratorio inferior, lesión del parénquima y fibrosis intersticial. Aunque no existe evidencia suficiente para
considerar que las infecciones virales sean factores etiológicos de la FPI, su potencial contribución sigue
siendo objeto de estudio. Además del herpes virus y de los adenovirus, cabría destacar:
El virus de Epstein-Barr (VEB), que normalmente infecta el tracto respiratorio superior, pero que
también se ha demostrado que infecta y se replica en el tracto respiratorio inferior (15). Se han identificado
tanto la proteína VEB y la expresión de ADN en el tejido pulmonar de pacientes con FPI. (16). El supuesto
papel de VEB en el desarrollo de la FPI se ha ampliado ante los hallazgos de que la expresión de la proteína
de la membrana EBV latente 1 en las células del epitelio alveolar se asocia con un mal pronóstico en
pacientes con FPI (17).
EL virus de la hepatitis C (VHC), en especial en pacientes con trastornos inmunes, puede
desencadenar una alveolitis linfocitaria subclínica, aunque se necesitan más estudios para definir su
importancia en la etiología de la enfermedad pulmonar intersticial (18).
1.3.5.- Autoinmunidad.
Un posible factor autoinmune se basa en que las manifestaciones radiológicas y/o histológicas de
la NIU se asocian a la presencia de enfermedades del tejido conectivo, como lo es la artritis reumatoide, la
esclerodermia, el lupus eritematoso sistémico, la granulomatosis de Wegener y la sarcoidosis, aunque estas
suelen cursar con histología de neumonía intersticial no específica.
1.3.6.- Exposición a fármacos.
Ciertas investigaciones sugieren una asociación entre la exposición a antidepresivos y el riesgo de
desarrollar FPI. Un estudio de casos y controles analizó la hipótesis de que los pacientes con alveolitis
fibrosante criptogénica (AFC) tenían más probabilidades de haber sido expuestos a ciertos medicamentos.
Analizando todas las recetas hasta la fecha del diagnóstico, se encontró que un número significativamente
mayor de prescripciones de antidepresivos en el grupo de casos que en los sujetos control. La asociación
se observó con antidepresivos tricíclicos y compuestos relacionados, en particular con la imipramina, la
doxeprina, y la mianserina (19).
La FP puede ser una toxicidad tardía de algunos tratamientos oncológicos, como la exposición
pulmonar o torácica a radioterapia, o el empleo de algunos quimioterápicos como bleomicina o metotrexate,
pudiendo llegar a ser una toxicidad limitante de dosis.
De hecho, como veremos en el apartado (2.2.),uno de los modelos animales existentes de FP se
basa en su inducción mediante bleomicina.

Tesis Doctoral Charlín Méndez Cordovez
5
1.3.7.- Otros factores a tener en cuenta.
Las comorbilidades y ciertas enfermedades pulmonares fibróticas pueden tener un impacto
negativo en la evolución de la enfermedad comprometiendo la vida de los pacientes con FPI (20) (21).
Estos incluyen: hipertensión pulmonar secundaria, enfermedad coronaria (22, 23), tromboembolismo
venoso (24), apnea obstructiva del sueño, enfisema coexistente, osteoporosis, diabetes mellitus, ansiedad
y depresión.
1.4.- Patogénesis (20).
El concepto de FPI ha evolucionado desde el de una “enfermedad inflamatoria crónica” al de una
“enfermedad heterogénea y multifactorial caracterizada por un proceso de cicatrización aberrante” (25). En
su patogénesis vamos a destacar ocho puntos que se consideran fundamentales:
1.4.1.- Daño y activación de las células epiteliales alveolares (CEA).
Los factores de riesgo (genéticos y/o ambientales) pueden causar un aumento del estrés del
retículo endoplasmático en las células epiteliales alveolares (CEA) pudiendo inducir en ellas tanto apoptosis
como una transición epitelio-mesenquimal (TEM) (26). A pesar de la muerte de las CEA tipo I (encargadas
del intercambio de gases), las CEA tipo II (progenitoreas de las tipo I y responsables de la producción de
proteinas surfactantes) se activan y recubren los quistes de la lesión en forma de panal de abeja.
En la FP están alterados varios genes y proteínas con función morfogenética. Las CEA y los
fibroblastos sobre-expresan diferentes ligandos de la vía de señalización Wnt-β-catenina, encargada de la
producción de glucoproteínas necesarias en los procesos morfogenéticos. Por otra parte, una
sobreexpresión del gen Sonic hedgehog (Shh) y una disminución de la expresión del gen del homólogo de
la fosfatasa y tensina (PTEN) en las CEA sugieren un proceso de reparación defectuoso, que favorecerá
una disminución de la apoptosis en los fibroblastos y aumento de proliferación de miofibroblastos.
1.4.2.- Efectos profibróticos de la activación anómala de las CEA en el
microambiente pulmonar.
En la FPI, la activación anómala de las CEA permite la activación del factor X de la coagulación.
Este factor (de la superficie de las plaquetas) estimula los fibroblastos e induce activación de la protrombina
(factor II, también en la superficie plaquetaria) convirtiéndolo en trombina, que a su vez estimula la
diferenciación de fibroblastos a miofibroblastos vía “receptor-I activado por proteasa”. Los miofibroblastos
tienen mayor capacidad profibrótica que los fibroblastos, e inducen depósito de matriz extracelular y
destrucción de la arquitectura pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
6
Las CEA producen diversos mediadores como factor derivado de plaquetas (PDGF), factor de
crecimiento transformante beta (TGF-β), factor de necrosis tumoral (TNF-α) y endotelina-1, que contribuyen
a la migración, proliferación y diferenciación de las células mesenquimales. Por otra parte, en la FPI, las
CEA también presentan una marcada sobreexpresión del factor derivado de células del estroma-1
(CXCL12) que, activando el receptor de quimioquina CXC tipo-4 (CXCR4) presente en los fibrocitos
circulantes, lleva a un reclutamiento de fibrocitos desde la sangre al pulmón.
Por último, las CEA pueden aumentar directamente el número de fibroblastos y miofibroblastos
mediante TEM. En este proceso, las células epiteliales adquieren propiedades de las células
mesenquimales, por las que aumentan su capacidad para moverse y sintetizar matriz intersticial. En la FPI,
las células mesenquimáticas locales, los fibrocitos circulantes y la TEM participa en la expansión de los
fibroblastos y los miofibroblastos.
En resumen, a partir del daño y activación de las CEA se activa una vía de señalización
procoagulante, el aumento de fibroblastos y miofibroblastos y el reclutamiento de fibrocitos al sitio de la
lesión.
1.4.3.- La diferenciación de los fibroblastos en miofibroblastos.
Tres factores principales intervienen en la diferenciación de fibroblastos a miofibroblastos:
1. la elevada tensión mecánica (mecanotransducción) que induce la diferenciación a
protomiofibroblastos,
2. el incremento de la actividad del TGFβ1
3. y la presencia de proteínas especializadas de la matriz celular.
Posteriormente, los miofibroblastos inducirán la acumulación excesiva de matriz extracelular, la
rotura de la membrana basal y la muerte de células epiteliales.
La eliminación de los miofibroblastos mediante el mecanismo de apoptosis es esencial durante las
fases finales del proceso normal de cicatrización. Pero esto no parece producirse en los focos fibroblásticos
de la FPI. Se desconocen las razones por la que los fibroblastos y miofibroblastos sobreviven mientras que
las CEA mueren dentro del mismo microentorno. Una explicación posible sería la deficiencia de
prostaglandina E2 que se observa en la FPI (27), que aumenta la sensibilidad de las CEA a la apoptosis
pero disminuye la sensibilidad de los fibroblastos a la apoptosis.
1.4.4.- La ausencia de neumocitos tipo I.
En condiciones normales las CEA tipo I (neumocitos tipo I) cubren más del 90% de la superficie
alveolar, y su estrecha relación con los capilares pulmonares proporciona una superficie fácilmente
permeable para los gases. Los pacientes con FPI tienen una importante pérdida de neumocitos tipo I,
aunque este efecto no está claro. Por otra parte, la transdiferenciación de las CEA tipo II (neumocitos tipo
II) en neumocitos tipo I (indispensable para poder restablecer la función epitelio alveolar) está alterada en
la FPI, debido a las anormalidades severas de la matriz extracelular y la rotura de la membrana basal.

Tesis Doctoral Charlín Méndez Cordovez
7
La pérdida de las CEA tipo I podría provocar la reducción de algunas moléculas antifibróticas
importantes (caveolina-1). Sin embargo, se desconoce si en la patogénesis de la FPI está involucrada la
disminución de las concentraciones de caveolina-1, específicamente atribuible a la pérdida de los
neumocitos tipo I. Por otra parte, el receptor de los productos finales de la glicación avanzada (RAGE) está
también disminuido en la FPI. Este receptor es un miembro de la superfamilia de los receptores de
inmunoglobulinas de la superficie celular que son altamente expresados por las CEA tipo I normales. La
pérdida de este receptor podría resultar en una disminución de la unión de las CEA tipo I a la membrana
basal, lo que impide la correcta reepitelización de los alvéolos durante la fibrogénesis.
1.4.5.- Metaloproteinasas de la matriz (MMPs) y la anormal remodelación
pulmonar.
Los fibroblastos están repartidos por toda la estructura pulmonar y juegan un papel clave en la
regulación de la homeostasis. Los fibroblastos modulan la renovación de la matriz extracelular a través de
la expresión de diversas metaloproteinasas de la matriz extracelular (MMPs) (que degradan la matriz
extracelular) y de inhibidores tisulares de las MMPs.
La fibrosis es inducida por la activación, proliferación y migración de estas células en el sitio de la
lesión y el depósito de proteínas en la matriz.
1.4.6.- Angiogénesis y la remodelación microvascular.
Se desconoce el papel de la angiogénesis en el pulmón fibrótico experimental. Al igual que ocurre
con los procesos profibróticos, la neovascularización es esencial en la regeneración de un tejido después
de una lesión y depende del equilibrio entre diversos factores que promueven o inhiben la angiogénesis.
En los pulmones se produce una remodelación vascular anormal, pero las áreas fibróticas apena
presentan neovascularización, mientras que el tejido de las zonas no fibróticas la neovascularización es
abundante. Por tanto, parece que en la FPI el proceso fibrótico no necesita de neovascularización.
1.4.7.- Relación entre FPI y envejecimiento.
En la FPI, al igual que en el envejecimiento, se produce un acortamiento acelerado de los telómeros
después de cada división celular. Ambos procesos se relaciona también con un aumento del estrés oxidativo
o la disminución de los mecanismos antioxidantes, con un desequilibrio redox, que puede provocar daños
directos en el ADN y alterar el funcionamiento de enzimas.

Tesis Doctoral Charlín Méndez Cordovez
8
1.4.8.- Los pericitos y las células mesoteliales pleurales (28).
Los pericitos pulmonares son células derivadas del mesénquima que se localizan dentro de la
membrana basal de los vasos sanguíneos (29, 30) . Pueden dar lugar a miofibroblastos y están implicados
en la cicatrización de las heridas y la producción de colágeno. Sin embargo, se necesita estudiar mejor la
contribución específica de los pericitos en la FPI.
Por último, comentar que la pleura está constituida en gran parte por mesotelio, que se origina en
el mesodermo embrionario. Las células mesoteliales pleurales han sido implicadas en el proceso de
migración y transformación en miofibroblastos, tanto in vitro como in vivo, a través de un proceso
dependiente de TGF-β1 (30). Las células mesoteliales pleurales están presentes en los pulmones con FPI
grave (31).
1.5. - Prevención.
Las únicas medidas con las que se puede prevenir la FPI consisten en prevenir las posibles
factores de riesgo, enfermedades subyacentes e inhalaciones de gases irritantes, humos y sustancias
tóxicas en general.
Por otra parte, pueden resultar de gran importancia la prevención y tratamiento precoz de las
potenciales comorbilidades y complicaciones puede resultar de gran importancia para enlentecer, en lo
posible, la evolución de la enfermedad y el deterioro de la calidad de vida.
1.6.- Diagnóstico.
El primer consenso sobre criterios diagnósticos y recomendaciones para evaluar, la evolución de
la FPI se estableció en el año 2000 por la American Thoracic Society (ATS) y European Respiratory Society
(ERS) (32). Los nuevos hallazgos y ensayos clínicos llevaron a un nuevo consenso 2011 con la participación
de la ATS, la ERS, la Sociedad Respiratoria Japonesa (JRS) y la Asociación Latinoamericana del Tórax
(ALAT) (1). Estas recomendaciones han sido más recientemente adaptadas y publicadas en forma de
“normativa” por la Sociedad Española de Patología del Aparato Respiratorio (SEPAR), que se han resumido
a continuación (9).
El diagnóstico definitivo de FPI requiere (figura 1):
1.- La exclusión de otras EPID de causa conocida (enfermedades del tejido conectivo, toxicidad por
fármacos, exposición ambiental u ocupacional)
2.- La presencia de un patrón histológico de NIU en la tomografía axial computarizada de alta resolución
(TCAR) en individuos no sometidos a una biopsia pulmonar (BP).
3.- La presencia de un patrón histológico de NIU, o una combinación específica de patrones de TCAR y de
histología en los individuos sometidos a BP.

Tesis Doctoral Charlín Méndez Cordovez
9
El consenso de 2011 recomienda que el diagnóstico de la FPI se debe basar en el consenso entre
el clínico, el radiológico y el patólogo.
SI
NO NIU
NIU POSIBLE NIU NO CONCORDANTE CON NIU
NO NIU
NIU
PROBABLE NIU / POSIBLE NIU NO - CLASIFICABLE FIBROSIS
Figura 1. Algoritmo diagnóstico de la fibrosis pulmonar idiopática (FPI) (1).
1.6.1.- Manifestaciones clínicas y analíticas.
El cuadro clínico de la FPI empieza de manera insidiosa y se caracteriza por disnea de esfuerzo
progresiva de meses a años de evolución, tos seca persistente estertores crepitantes en el 90% de los
casos y acropaquias en el 20%-50%. En la fase avanzada es frecuente la aparición de cor pulmonale, con
signos de hipertensión pulmonar (HP) (acentuación del segundo tono pulmonar o desviación del impulso
apical a la derecha) y finalmente signos de insuficiencia cardiaca derecha. También se va instaurando
progresivamente un cuadro de hipoxemia y pruebas de función pulmonar restrictivas con una disminución
de la capacidad de difusión del monóxido de carbono (DLCO) (20).
En los análisis sanguíneos no existen determinaciones específicas para la FPI, aunque la velocidad
de sedimentación, la proteína C reactiva y las inmunoglobulinas G pueden estar alterados y los anticuerpos
antinucleares y el factor reumatoide son positivos en el 10%-20% de los casos. Se recomienda realizar
estudio de posibles conectivopatías y de Ig G frente a anticuerpos relacionados con neumonitis por
hipersensibilidad (ver tabla 1).
Sospecha de FPI
¿Causa identificable de enfermedad pulmonar intersticial difusa (EPID)?
TCAR
Biopsia pulmonar quirúrgica
Discusión multidisciplinar
FPI / NO FPI FPI
NO FPI

Tesis Doctoral Charlín Méndez Cordovez
10
Tabla 1. Pruebas de laboratorio ante sospecha de FPI.
Hemograma
Velocidad de Sedimentación
Perfil hepático
CPK
Enzima Convertidora de la Angiotensina (ECA)
Factor reumatoide
Análisis de orina
Prueba de hipersensibilidad
Anticuerpos Antinucleares (ANA)
1.6.2.- Diagnóstico radiológico:
1.6.2.1.- Radiografía de tórax.
Es la exploración básica e inicial en la mayoría de los pacientes con síntomas respiratorios. Para
una correcta valoración debería constar siempre de las proyecciones posteroanterior y lateral. Ambas se
realizan con el paciente de pie, en inspiración máxima y con técnica de alto kilovoltaje. Proporciona
información sobre el parénquima pulmonar, pleura, pared torácica, silueta cardíaca y mediastino. La
radiografía lateral es imprescindible para la localización de lesiones, confirmar imágenes y valorar zonas
ciegas o de difícil valoración en la proyección posteroanterior. La utilización de una técnica con alto
kilovoltaje permite una mejor visualización del parénquima pulmonar y del mediastino, teniendo como
inconveniente la disminución en la capacidad de detectar calcio en las lesiones y una pérdida en el detalle
óseo.
En la radiografía de tórax muestra opacidades reticulares asociadas o no a imágenes en panal, de
distribución basal y bilateral (figura 2 y figura 3) ante la presencia de imágenes alveolares debe plantearse
la posibilidad de un diagnóstico definitivo.

Tesis Doctoral Charlín Méndez Cordovez
11
1.6.2.2.- La Tomografía (Axial) Computarizada de Alta Resolución (TCAR).
Generalmente la TCAR se realiza tras la práctica de una radiografía de tórax, ya sea para estudiar
una alteración observada en la misma o por una sospecha clínica. La TCAR, se basa en la realización de
cortes de 1 a 2 mm, sin medio de contraste, tiene una sensibilidad del 90%, y puede ser útil para:
1. obtener el diagnóstico específico de FPI, cuantificar su extensión y valorar su evolución durante
el seguimiento.
2. Identificar un sitio apropiado para la biopsia pulmonar en caso de ser necesaria.
3. valorar la presencia de comorbilidades y/o complicaciones asociadas (enfisema, HP, cáncer de
pulmón)
4. descartar la presencia de otras EPID.
En el consenso de 2011 (1, 9), se estableció que en la TCAR, el diagnóstico de certeza de la NIU
se basa en la identificación de los 4 “hallazgos típicos” (tabla 2) (figura 4)
1. la afectación pulmonar debe tener un predominio basal y una localización subpleural
2. presencia de reticulación evidente
3. existencia de panalización con/sin bronquiectasias/ bronquiectasias de tracción
4. demostrar la ausencia de hallazgos considerados excluyentes de un patrón de NIU, como
micronódulos broncovasculares. La presencia del patrón de vidrio deslumbrado debe ser mínima
o inexistente.
La panalización (formada por espacios quísticos agrupados con paredes finas, habitualmente de localización subpleural y de 3-10 mm de diámetro, aunque pueden ser mayores) es un hallazgo imprescindible para el diagnóstico de certeza de NIU.
Figura 2 y Figura 3.- Hallazgos en imágenes con opacidades panal de abeja con una disminución periférica y basal
Tesis Doctoral Charlín Méndez Cordovez
12
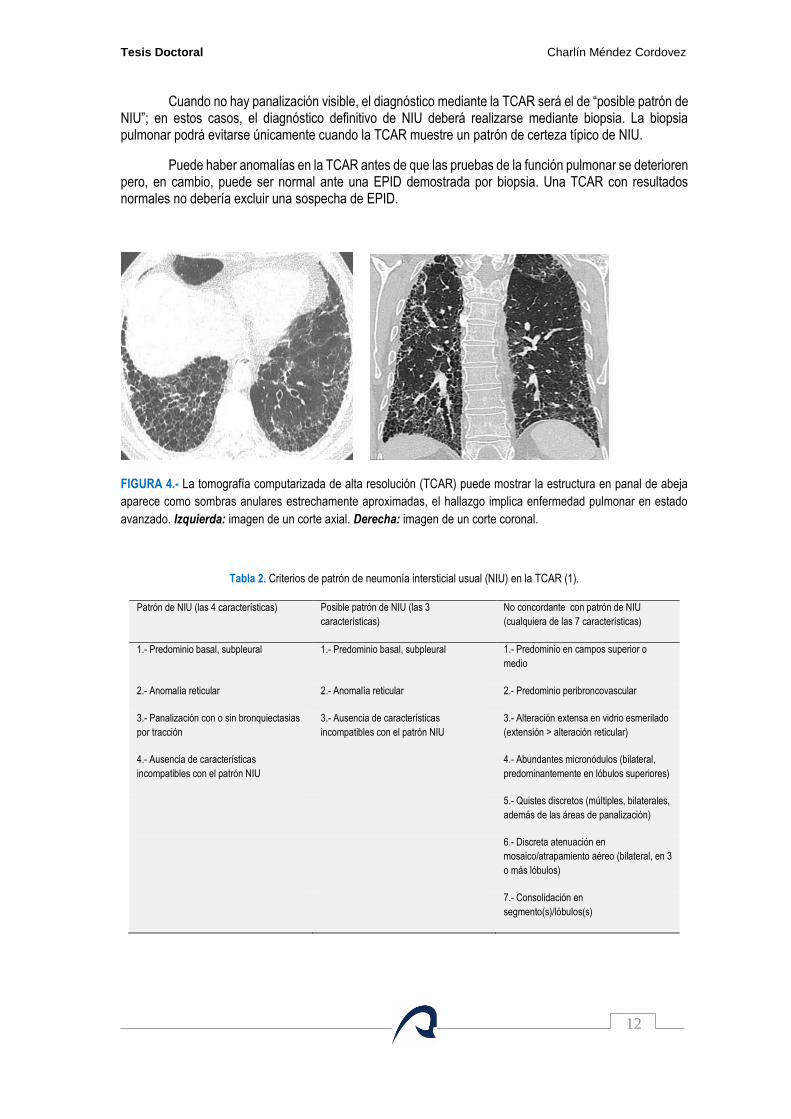
Cuando no hay panalización visible, el diagnóstico mediante la TCAR será el de “posible patrón de NIU”; en estos casos, el diagnóstico definitivo de NIU deberá realizarse mediante biopsia. La biopsia pulmonar podrá evitarse únicamente cuando la TCAR muestre un patrón de certeza típico de NIU.
Puede haber anomalías en la TCAR antes de que las pruebas de la función pulmonar se deterioren pero, en cambio, puede ser normal ante una EPID demostrada por biopsia. Una TCAR con resultados normales no debería excluir una sospecha de EPID.
FIGURA 4.- La tomografía computarizada de alta resolución (TCAR) puede mostrar la estructura en panal de abeja
aparece como sombras anulares estrechamente aproximadas, el hallazgo implica enfermedad pulmonar en estado
avanzado. Izquierda: imagen de un corte axial. Derecha: imagen de un corte coronal.
Tabla 2. Criterios de patrón de neumonía intersticial usual (NIU) en la TCAR (1).
Patrón de NIU (las 4 características) Posible patrón de NIU (las 3
características) No concordante con patrón de NIU
(cualquiera de las 7 características)
1.- Predominio basal, subpleural 1.- Predominio basal, subpleural 1.- Predominio en campos superior o
medio
2.- Anomalía reticular 2.- Anomalía reticular 2.- Predominio peribroncovascular
3.- Panalización con o sin bronquiectasias
por tracción 3.- Ausencia de características
incompatibles con el patrón NIU 3.- Alteración extensa en vidrio esmerilado
(extensión > alteración reticular)
4.- Ausencia de características
incompatibles con el patrón NIU 4.- Abundantes micronódulos (bilateral,
predominantemente en lóbulos superiores)
5.- Quistes discretos (múltiples, bilaterales,
además de las áreas de panalización)
6.- Discreta atenuación en
mosaico/atrapamiento aéreo (bilateral, en 3
o más lóbulos)
7.- Consolidación en
segmento(s)/lóbulos(s)

Tesis Doctoral Charlín Méndez Cordovez
13
1.6.3.- Pruebas de función pulmonar.
Las pruebas de función pulmonar ayudan a demostrar las anormalidades fisiológicas que
acompañan a las EPID, a precisar su gravedad y a valorar la evolución y la respuesta al tratamiento.
Las alteraciones fisiológicas en la EPID son la reducción de los volúmenes pulmonares (capacidad
vital forzada (CVF), capacidad pulmonar total, reducción de DLco y relación normal o alterada entre VEF1 y
CVF). La elasticidad pulmonar también se reduce (disminuye el volumen pulmonar para cualquier presión
transpulmonar) y la presión transpulmonar máxima aumenta (se debe generar una presión negativa muy
alta para abrir los alvéolos fibrosados).
Las excepciones de este cuadro clásico son histiocitosis X, linfangioleiomiomatosis,
neurofibromatosis, sarcoidosis y esclerosis tuberosa; en ellas, las alteraciones provocan aumento de la
capacidad pulmonar total y limitación del flujo de aire. En la neumonía organizada con bronquiolitis
obliterante (BOOP) se observa un patrón mixto tanto restrictivo como obstructivo.
Los gases arteriales traducen hipoxemia leve. Rara vez existe retención de dióxido de carbono,
incluso en las últimas fases de la enfermedad. La mayoría de los pacientes con EPID muestran un aumento
de la ventilación por minuto durante el reposo y el esfuerzo, lo que reduce la Pco2 y conlleva alcalosis
respiratoria compensada. Este incremento se logra al aumentar la frecuencia respiratoria en lugar del
volumen de ventilación pulmonar. La hiperventilación no se debe a anormalidades del equilibrio ácido-
básico, ni a hipoxemia, sino a un mayor estímulo del centro respiratorio por las señales neurales originadas
en los mecanorreceptores alterados del parénquima pulmonar desorganizado. En los pacientes con EPID,
la prueba de esfuerzo se acorta, y con el ejercicio la Po2 desciende y la Pco2 permanece constante.
La hipoxemia es consecuencia de una relación anormal entre la ventilación y la perfusión y
anormalidades en la difusión. En un principio se creía que estas últimas eran el resultado del engrosamiento
de las paredes alveolares, pero ahora se sabe que se deben a la pérdida del área capilar transversal, y a
que el paso de eritrocitos a través de los capilares pulmonares funcionales es tan rápido que permite una
saturación completa de la hemoglobina. El pH arterial suele ser normal, pero puede descender con el
ejercicio por efecto del metabolismo anaeróbico en los músculos carentes de oxígeno.
1.6.4.- Lavado broncoalveolar (LBA).
En la actualidad, y tras el último consenso 2011 se recomienda que el LBA “no se debe realizar”
en la evaluación diagnóstica de la FPI en la mayoría de los pacientes. (1). Sin embargo, puede ser apropiado
en una minoría de pacientes para diagnóstico diferencial de trastornos alternativos como neumonitis por
hipersensibilidad, neumonía intersticial no específica, sarcoidosis, infecciones o tumores (1, 33, 34).

Tesis Doctoral Charlín Méndez Cordovez
14
1.6.5- Biopsia pulmonar.
Siguiendo el consenso 2011 la biopsia pulmonar quirúrgica (abierta) está indicada cuando el
diagnóstico es dudoso o incierto después de revisar las pruebas diagnósticas, en especial si la TCAR no
muestra un patrón de certeza típico de NIU (1, 9). Sin embargo un gran porcentaje de los pacientes con
sospecha de FPI no pueden someterse a un procedimiento de BP quirúrgica, debido a contraindicaciones
(p.ej. condiciones comórbidas, gravedad de la enfermedad, edad avanzada, etc.) o porque el paciente
rechaza el procedimiento (35).
La toracoscopia y la minitoracotomía son preferibles a la cirugía convencional mediante
toracotomía. La toracoscopia, en especial, supone reducción del tiempo operatorio, de la estancia
hospitalaria y de las posibles complicaciones postquirúrgicas Deben tomarse muestras de al menos dos
lóbulos diferentes, incluyendo áreas con aspecto macroscópico patológico y otras con aspecto
macroscópico normal.
La BP transparietal con aguja no debe utilizarse, debido al pequeño tamaño de las muestras
obtenidas y a la gran incidencia de neumotórax secundario.
La BP transbronquial tampoco está indicada habitualmente. Sin embargo, en casos seleccionados
puede ser útil para confirmar/ descartar otros procesos específicos (sarcoidosis, tumores…).
1.6.6.- Estudio histopatológico
El patrón histológico de NIU en la FPI se asocia con un pronóstico significativamente peor y
mortalidad más precoz, que otros patrones histológicos de neumonía intersticial crónica (8, 36). La tasa de
supervivencia media es de 2-5 años, peor que la de varios tipos de cáncer (8).
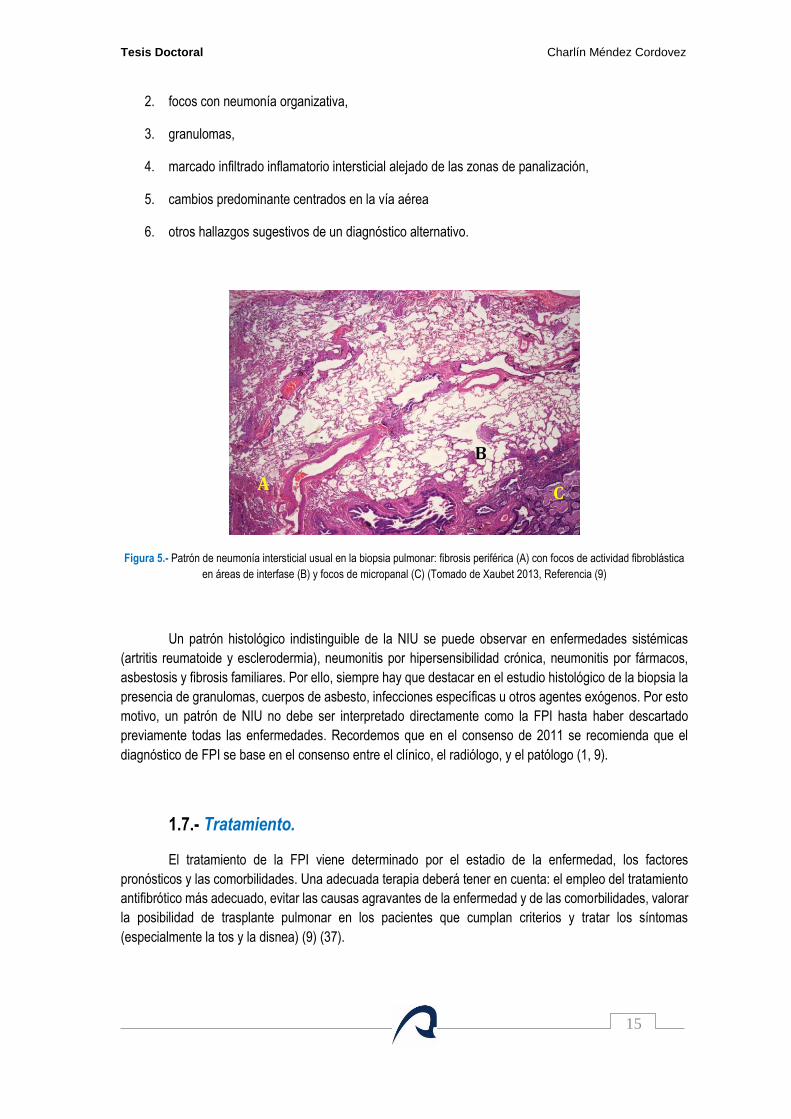
El patrón histológico típico de NIU (figura 5) debe cumplir los siguientes 4 criterios (1, 9):
1. Evidencia de marcada fibrosis/ de formación de la arquitectura pulmonar, asociada o no a
panalización con distribución predominante subpleural y paraseptal.
2. Afectación parcheada del parénquima pulmonar por fibrosis.
3. Presencia de focos fibroblásticos.
4. Ausencia de hallazgos histopatológicos incompatibles con diagnóstico de NIU, y que pudieran
sugerir un diagnóstico alternativo.
Entre las características “no compatibles” con el patrón de NIU estarían la presencia de alguno
de los siguientes 6 criterios (1) (9):
1. membranas hialinas,
Tesis Doctoral Charlín Méndez Cordovez
15
2. focos con neumonía organizativa,
3. granulomas,
4. marcado infiltrado inflamatorio intersticial alejado de las zonas de panalización,
5. cambios predominante centrados en la vía aérea
6. otros hallazgos sugestivos de un diagnóstico alternativo.
Figura 5.- Patrón de neumonía intersticial usual en la biopsia pulmonar: fibrosis periférica (A) con focos de actividad fibroblástica
en áreas de interfase (B) y focos de micropanal (C) (Tomado de Xaubet 2013, Referencia (9)
Un patrón histológico indistinguible de la NIU se puede observar en enfermedades sistémicas
(artritis reumatoide y esclerodermia), neumonitis por hipersensibilidad crónica, neumonitis por fármacos,
asbestosis y fibrosis familiares. Por ello, siempre hay que destacar en el estudio histológico de la biopsia la
presencia de granulomas, cuerpos de asbesto, infecciones específicas u otros agentes exógenos. Por esto
motivo, un patrón de NIU no debe ser interpretado directamente como la FPI hasta haber descartado
previamente todas las enfermedades. Recordemos que en el consenso de 2011 se recomienda que el
diagnóstico de FPI se base en el consenso entre el clínico, el radiólogo, y el patólogo (1, 9).
1.7.- Tratamiento.
El tratamiento de la FPI viene determinado por el estadio de la enfermedad, los factores
pronósticos y las comorbilidades. Una adecuada terapia deberá tener en cuenta: el empleo del tratamiento
antifibrótico más adecuado, evitar las causas agravantes de la enfermedad y de las comorbilidades, valorar
la posibilidad de trasplante pulmonar en los pacientes que cumplan criterios y tratar los síntomas
(especialmente la tos y la disnea) (9) (37).

Tesis Doctoral Charlín Méndez Cordovez
16
1.7.1.- Terapia Farmacológica.
El tratamiento farmacológico de la FPI busca evitar a al menos reducir la progresión de la
enfermedad a través del uso de antifibróticos, identificar y el tratar las complicaciones específicas de la
enfermedad y al alivio de los síntomas.
En la (tabla 3) se especifican las recomendaciones del tratamiento farmacológico específico de la fibrosis pulmonar idiopática, aunque como podemos ver, el arsenal terapéutico es muy limitado.
Tabla 3. Recomendaciones basadas en la evidencia para tratamiento de la FPI. (Modificada/actualizada de la Referencia. (9).
Agente
Mecanismo de acción
Recomendaciones
Recomendado en pacientes seleccionados Pirfenidona
Nintedanib
NAC c en monoterapia
Antifibrótico + antiinflamatorio + antioxidante + anti TGF β 1
Antifibrótico + antiangiogénico
Antioxidante
Sí, recomendación débil a Sí, recomendación débil a, b No, recomendación débil
No recomendados Esteroides + Azatioprina + NAC
Anticoagulación Bosentán
Esteroides en monoterapia Esteroides + terapia inmunomoduladora Colchicina
Ciclosporina A Etanercept Interferón gamma
Inmunosupresor + antioxidante + antiinflamatorio Anticoagulante Antagonismo dual del receptor de la endotelina Inmunosupresor Inmunosupresor Inhibidor proliferación/síntesis de colágeno Inmunosupresor Anti TNF alfa Antifibrótico e inmunomodulador
No utilizar
No utilizar No utilizar
No utilizar No utilizar No utilizar
No utilizar No utilizar No utilizar
ª En pacientes con FPI leve/moderada. b Aprobado en España el 11-2014. c N-acetil-cisteina.

Tesis Doctoral Charlín Méndez Cordovez
17
1.7.1.1.- Pirfenidona.
La pirfenidona (Esbriet®) fue el primer fármaco en demostrar eficacia contrastada en el tratamiento
de la FPI en ensayos clínicos controlados.
El papel de la pirfenidona en el tratamiento de la FPI se evaluó en 3 ensayos controlados
multicéntricos fase 3, aleatorizados, doble ciego, controlados con placebo en pacientes con FPI en Europa,
EEUU, Australia (estudios CAPACITY con 779 pacientes) y uno de dichos ensayos se llevó cabo en Japón
con 275 pacientes, donde el tratamiento con pirfenidona redujo la declinación de la CVF a la semana 52 y
mejoró la supervivencia libre de la enfermedad.
Sin embargo, y pese a ser el primer fármaco activo aprobado para el tratamiento de la FPI, no está
claro que la pirfenidona disminuya el grado de disnea o aumente la supervivencia global o la ligada a FPI,
habiéndose descrito los beneficios significativos en un metaanálisis de 3 ensayos clínicos pero no en cada
uno de los ensayos por separado (38).
La dosis recomendada de pirfenidona es de 2.403 mg/24 h tras un aumento gradual de la dosis.
Así, durante la primera semana de tratamiento se debe tomar una cápsula de 267 mg/8 h, en la segunda
semana 2 cápsulas/8 h y, a partir de la tercera semana 3 cápsulas/8h, durante 12 meses como mínimo. Si
el paciente responde adecuadamente debe continuarse con el tratamiento. Esto se traduce en una
reducción de la disminución de la CVF del 30%. Además, el intervalo libre de progresión y la distancia
recorrida en la prueba de marcha de 6 minutos (PM6M) fue significativamente mejor en pacientes con una
CVF > 50% y DLCO > 35% (39-41).
Las principales reacciones adversas asociadas con este fármaco son: náuseas (32%), erupción
cutánea (26%), diarrea (19%), cansancio (19%), dispepsia (16%), anorexia (11%), cefalea (10%) y,
reacciones de fotosensibilidad (9%). Estos efectos son dependientes de la dosis, y generalmente reducibles
al disminuir la dosis administrada (42) (Ficha técnica de la Agencia Europea del Medicamento (EMEA)
http://www.ema.europea.eu/docs/es_ES/document_library/EPAR_-
Product_Information/human/002154/WC500103049.pdf.
1.7.1.2.- Nintedanib.
Bajo el nombre comercial de Ofev®, nintedanib fue designado medicamento huérfano para el tratamiento de la FPI por la EMEA el 26 de abril del 2013. A finales del 2014, la Agencia Española del Medicamento y Productos Sanitarios (AEMPS) aprobó en España la indicación de este anticuerpo monoclonal para el tratamiento de la fibrosis pulmonar idiopática (FPI) en adultos (http:// www.aemps.gob.es/informa/boletinMensual/2014/noviembre/boletin-noviembre.htm#cambiosEspecial).
También conocido como (BIBF 1120), nintedanib es un inhibidor de la tirosina quinasa que bloquea la actividad quinasa de los receptores del (43) (44):
1. Factor de crecimiento endotelial vascular (VEGFR 1-3), que estimula la angiogénesis.
2. Factor de crecimiento derivado de plaquetas (PDGFR alfa y beta)
3. Y del factor de crecimiento de fibroblastos (FGFR 1-3) crucial para la proliferación y migración de los fibroblastos pulmonares.

Tesis Doctoral Charlín Méndez Cordovez
18
El primer ensayo randomizado, placebo, controlado, doble ciego, con nintedanib fue el estudio TOMORROW, un fase 2 con 432 pacientes. Aunque en él no se encontraron diferencias significativas en la variable principal, CVF, sí se encontraron diferencias significativas favorables en alguna de las variables secundarias: exacerbaciones agudas y disminución en la calidad de vida (44).
Los dos estudios fundamentales fueron 2 ensayos clínicos fase III doble ciego y aleatorizados (INPULSIS 1 y 2) que examinaron la eficacia y seguridad de nintedanib versus placebo en 1066 pacientes con diagnóstico de FPI, que emplearon dos dosis diarias de 150 mg de nintedanib frente a placebo, durante 52 semanas. El criterio primario de valoración fue la tasa anual de deterioro de la CVF en ml a las 52 semanas. El uso de nintedanib se asoció a un retraso significativo de la progresión de la FPI reflejado en una reducción del 50% en la tasa anual de declinación de la CVF en comparación con placebo en ambos ensayos, tanto el INPULSIS-1 (-114,7 ml nintedanib vs. -207,3 ml placebo, p < 0,001) como el INPULSIS-2 (-113,6 mL nintedanib vs. -207,3 mL placebo, p< 0,001).. El INPULSIS-2 mostró, además, una disminución significativa del tiempo transcurrido hasta la primera exacerbación aguda (p = 0,005) y un menor deterioro de la calidad de vida media con el St. George's Respiratory Questionnaire (2,80 vs 5,48 p = 0.02) (45).
Las reacciones adversas más comunes observadas durante los ensayos clínicos fueron los trastornos gastrointestinales, siendo el más frecuente la diarrea (3 veces más frecuente en los grupos de nintedanib que en los placebo), seguida de vómitos, náuseas, y aumento de las transaminasas.
1.7.1.3.- N-Acetilcisteína.
La N-Acetilcisteína (NAC) aumenta la síntesis de glutatión, un potente antioxidante, y disminuye la respuesta fibrótica en modelos animales de fibrosis pulmonar.
En el ensayo fase III IFIGENIA, se encontró que frente a un tratamiento con azatioprina + prednisona + placebo, la sustitución del placebo por NAC preservaba mejor la función pulmonar (46). Esto llevó a que la combinación de estos tres fármacos (prednisona, azatioprina y NAC) se convirtiera en el tratamiento de elección y recomendado como opción terapéutica en las guías de consenso hasta hace pocos años.
Más tarde, el estudio randomizado PANTHER comparó la combinación de 3 fármacos (prednisona, azatioprina y NAC) frente a NAC y frente a placebo, en pacientes con FP y compromiso funcional leve a moderado. Este estudio demostró que los pacientes con terapia triple presentaban mayor mortalidad e ingresos hospitalarios que los pacientes del grupo placebo o del grupo NAC. Por tanto, en la actualidad se recomienda “no utilizar” esta triple terapia (47). Posteriormente, se continuó sólo con los grupos de NAC y placebo. Pero frente al grupo placebo, la NAC no mostró beneficio en CVF, morbilidad ni en la frecuencia de exacerbaciones agudas (48). Por tanto, en la actualidad, no existe evidencia para su uso específico como tratamiento de la FPI.
1.7.1.4.- Otros fármacos antifibróticos.
Durante la última década se han realizado varios ensayos clínicos aleatorizados, fase II y III, con fármacos antifibróticos considerados experimentales, pero que no han demostrado ninguna eficacia para el tratamiento de la FPI (49). Entre ellos se incluyen:
1. Imatinib (Gleevec®), es un medicamento utilizado originalmente para el tratamiento de la leucemia mieloide crónica (LMC). Actúa como un inhibidor selectivo de la tirosina quinasa con actividad contra los receptores del factor de crecimiento derivado de

Tesis Doctoral Charlín Méndez Cordovez
19
plaquetas (PDGFR), c-kit, y c-Abl tirosina quinasa. Imatinib ha demostrado proteger contra la fibrosis en modelos de roedores de la lesión pulmonar (50-52). Sin embargo, un ensayo fase II controlado con placebo que incluyó a pacientes con FPI de leve a moderada, imatinib no mostró ningún efecto sobre la variable principal de evaluación, la supervivencia libre de progresión (definida por una disminución del 10% de la CVF) o la supervivencia durante un período de 96 semanas (50).
2. Anti-IL 13: La interleucina IL-13 es un potente estimulador de la proliferación de fibroblastos y de la síntesis de la matriz extracelular (53, 54), que induce citoquinas profibróticas como el TGF-β, PDGF, el factor de crecimiento similar a la insulina que se llaman FCI-1, la metaloproteinasa de la matriz 9 (MMP9), ligandos de quimiocinas CC (CCL18), el factor de crecimiento de tejido conectivo (FGTC), y la producción de fibronectina. En los pacientes con FPI (55), los niveles de IL-13 están aumentados en LBA. La sobreexpresión de IL-13 en ratones induce la FPI (56), mientras que la inmunoneutralización de la IL-13 en la lesión pulmonar inducida por bleomicina atenúa la fibrosis pulmonar. El anticuerpo monoclonal humano anti IL-13 (QAX 576) (57) se está evaluando actualmente en un ensayo clínico fase II aleatorizado, doble ciego, controlado con placebo y multicéntrico (NCT01266135).
3. Anticuerpo anti- LOXL2 o GS-6624 (AB0024): en el modelo de fibrosis pulmonar inducida por bleomicina en ratón , la regulación positiva LOXL2 es atenuada por un anticuerpo monoclonal anti-LOX2 (AB0024), se observa reducción del colágeno fibrilar, fibroblastos activados, citoquinas inflamatorias, y el TGF-β1 (58). Actualmente, están comenzando los primeros ensayos clínicos.
4. Otros en estudio: Interferón gamma 1-beta, antagonistas del factor de necrosis tumoral-α (etanercept), anticoagulantes (warfarina), antagonistas de la endotelina (bosentán, macitentán, ambrisentán) y sildenafilo se están evaluando en ensayos clínicos fase I o II, así como otras moléculas (www.clinical.gov).
Posiblemente, una adecuada terapéutica farmacológica contra la FPI debería incluir la asociación de fármacos que actúen sobre diferentes vías patogénicas de la enfermedad de forma sinérgica: contra la fibrosis (los aprobados hasta ahora), modulando el estrés oxidativo (¿NAC?) +/- modulando los procesos inflamatorios.
1.7.2.- Rehabilitación respiratoria.
Una reciente actualización de The Cochrane Collaboration en enfermedad pulmonar intersticial y en FPI, muestra que la rehabilitación pulmonar mejora la capacidad funcional ante el ejercicio, la disnea y la calidad de vida, si bien no se ha evidenciado impacto sobre la supervivencia a largo plazo (59).
1.7.3.- Trasplante pulmonar.
Actualmente, el trasplante de pulmón (TP) es el único tratamiento del que se dispone para incrementar la supervivencia en los pacientes afectados con FPI de una manera considerable. El momento más adecuado para realizar el TP está en función de los factores pronósticos del paciente.

Tesis Doctoral Charlín Méndez Cordovez
20
Lo más idóneo sería realizar el TP en el momento en el que está justificado por el mal pronóstico
de la enfermedad a corto o medio plazo, pero no tan tarde para que el deterioro físico del paciente haga
previsible que no soportará la intervención. Los beneficios del trasplante deben sopesarse con los
inconvenientes que supone el mismo. Estos inconvenientes incluyen: una limitada disponibilidad de órganos
y los riesgos quirúrgico, de infección, de rechazo del órgano trasplantado y de la terapia inmunosupresora.
Según las directrices de la Sociedad Internacional de Trasplante de Corazón y Pulmón (SITCP),
se recomienda que los pacientes con FPI deben ser indicados para el trasplante, si su DLco es < 40%, si
durante los 6 meses anteriores al trasplante han sufrido una disminución de la CVF > 10%, o si la oximetría
de pulso o pulsioximetría muestra una saturación < 88% durante la prueba de caminata en 6 minutos.
Los datos disponibles en el Reino Unido, sugieren que la FPI representa el 20% de todos los
trasplantes de pulmón. La tasa de supervivencia a los 5 años tras el trasplante de pulmón se sitúa en un
45% - 50% (60, 61).
1.8.- Pronóstico.
La FPI es una enfermedad de curso clínico variable, por lo que es importante identificar factores
que puedan ayudar a definir el pronóstico de los pacientes (9).
Entre los factores basales adversos (en el momento del diagnóstico) están: 1) Edad avanzada (>
70 años) y sexo masculino. 2) Grado de disnea basal, medido con la escala de MRC. (62). 3) DLCO < 40%
(porcentaje del valor predicho). 4) Saturación de oxígeno percutánea SpO2 ≤ 88% en la prueba de
marcha de 6 minutos (PM6M) (63). 5) Extensión del patrón en “panal de abeja” en la TCAR. 6)
Hipertensión pulmonar arterial 7) Biomarcadores, tanto a nivel histológico si hay biopsia (profusión de focos
de fibroblastos) como niveles séricos del péptido natriurético cerebral o de proteínas relacionadas con el
daño de las CEA, la inflamación o el estrés oxidativo.
Entre los factores adversos durante la evolución de la enfermedad cabe destacar: 1) El
aumento del grado de disnea > 5% (valor absoluto). 2) Descenso en 6-12 meses de la CVF ≥ 10
% o de la DLCO ≥ 15% (64), 3) Disminución en 6 meses de la distancia recorrida durante la
PM6M > 50 m (63). 4) Disminución en 6 meses de la SpO2 > 15%. 5) Extensión y progresión de la
fibrosis en la TCAR.
En pacientes con SpO2 ≤ 88%, la disminución > 15 % de la DLCO en 6 meses es el mejor
predictor de mortalidad, mientras que en aquellos con SpO2 > 88% el parámetro más significativo es
la disminución > 10% de la CVF. Se están desarrollando escalas multidimensionales para intentar predecir
el riesgo individual de mortalidad en pacientes con FPI, pero aún no existe suficiente evidencia científica
para su uso en la práctica clínica (65, 66).
Entre las comorbilidades y complicaciones que pueden empeorar el curso clínico y el pronóstico
de la FPI están: las exacerbaciones agudas, HTP, enfisema pulmonar, reflujo gastroesofágico, síndrome de
apnea/ hipopnea del sueño, carcinoma de pulmón (prevalencia 5-10%), neumotórax (incidencia 11%),
enfermedad coronaria o tromboembólica venosa.

Tesis Doctoral Charlín Méndez Cordovez
21
2.- Bleomicina y Modelo Experimental de Fibrosis Pulmonar.
Los estudios realizados en modelos animales juegan un papel importante en la investigación biomédica. Entre los modelos animales para inducir FP están el empleo de radiaciones ionizantes a nivel torácico en un animal susceptible y la administración de un agente fibrogénico bien vía sistémica (paraquat o la propia bleomicina (BLM)) o bien mediante instilación oro-traqueal (IOT) como BLM, amiodarona (67), sílice, cobalto o asbesto) (68-70). Pero entre todos ellos, el modelo más empleado para la inducción de FP experimental es la IOT de BLM (69).
2.1.- Bleomicina.
Las bleomicina (BLM) es un antibiótico antineoplásico del tipo glucopéptido aislado de una cepa
de Streptomices verticillus (71, 72). Tiene un efecto mínimo como mielosupresor e inmunosupresor;
destacando su toxicidad dermatológica, que puede llegar a ser grave. Sin embargo, su eficacia terapéutica
se ve limitada por su toxicidad pulmonar, que es “limitante en dosis” por su capacidad de inducir neumonitis
y fibrosis pulmonar (FP) en forma dosis-dependiente (73) (74) (69).
2.1.1.- Estructura de la bleomicina.
Las bleomicinas comparten la misma estructura central (figura 6), pero pueden diferir según el tipo
de carbohidratos y cadenas cargadas positivamente que contienen (75) (74).
Figura 6. Estructura central de la bleomicina (BLM).
La BLM presenta diferentes dominios funcionales:
1.- Dominio de unión a metales: este dominio contiene átomos de nitrógeno que reaccionan con el
metal, sobre todo con metales de transición como el cobre y el hierro. En presencia de oxígeno, esta unión
puede dar lugar a la rotura de cadenas de ADN (tanto simples como dobles cadenas) así como la formación

Tesis Doctoral Charlín Méndez Cordovez
22
de especies reactivas del oxígeno (ROS) o radicales libres (RL) que pueden potenciar o perpetuar el daño
(74, 76-81).
2.- Dominio de unión al DNA: dependiente de la pirimidina y el grupo tiazol de la BLM. Las cargas
positivas del grupo tiazol favorecen la unión electrostática de la BLM con el DNA (74, 79, 80, 82).
3.- Carbohidratos: la bleomicina puede estar glicosilada con α-D-manosa y L-gulosa. No se conoce
con precisión el papel de los carbohidratos, pero existen evidencias de que pueden modular la afinidad de
la BLM por el DNA (74, 83).
2.1.2.- Mecanismos de acción de la bleomicina.
Para activarse, la BLM requiere de la unión con un metal de transición reducido [Fe (II) o Cu (I)],
la presencia de una molécula de oxígeno y un agente reductante. La BLM activa ejerce su efecto citotóxico
por daño directo sobre el DNA y RNA, e indirectamente a través de la generación de RL (75, 79, 80, 82,
84).
Daño al DNA: estudios in vitro han mostrado que la BLM puede romper directamente tanto las
cadenas simples como las cadenas dobles de DNA, e inhibe la reparación de DNA mediante inhibición de
la DNA ligasa. El grado de lesión de la BLM sobre el DNA depende de la dosis y del ciclo celular. La BLM
se considera ciclo-celular específico de la fase 2, aunque también es activa (en menor cuantía) en la fase
G1 tardía y en la fase S y M. Así mismo, también depende del estado de activación transcripcional, siendo
más susceptible la eucromatina que la heterocromatina. La bleomicina no interfiere directamente con la
replicación del DNA, ni con la síntesis del RNA y de las proteínas (81, 82).
Daño al RNA: recientemente se ha demostrado in vitro que la BLM puede degradar el RNA
mediante hidrólisis directa o indirecta mediada por la generación de RL (84).
Radicales libres (o especies reactivas de oxígeno): la generación de RL por la BLM (como los
radicales hidroxilo, superóxido y peróxido de hidrógeno) dará lugar a un daño indirecto, mediado por la
acción de estas sustancias altamente reactivas con capacidad para oxidar rápidamente cualquier molécula
a su alcance, incluyendo proteínas, lípidos y el propio DNA y RNA (79).
2.1.3.- Farmacocinética y metabolismo de la bleomicina.
La BLM es un fármaco muy soluble en agua, lo que facilita su administración por múltiples vías, y
permite su utilización vía orotraqueal en el modelo animal que se describe más adelante. Apenas se une a
las proteínas plasmáticas, y tiene una vida media de unas 2 horas tras su administración intravenosa. Se
elimina por vía renal, el 50%-70% en las primeras 24 horas. La BLM tiene una amplia distribución por todos
los tejidos y es ampliamente degradada en todos los tejidos excepto en la piel y el pulmón, que carecen de
la enzima “hidrolasa de bleomicina” necesaria para su degradación. Esta enzima es una proteasa de
cisteína, perteneciente a un grupo de enzimas que incluye la familia de las caspasas, calpaína y papaína.

Tesis Doctoral Charlín Méndez Cordovez
23
Bajos niveles de esta enzima (como ocurre en piel y pulmón) dan lugar a toxicidad, mientras niveles
elevados en células tumorales se asocian a resistencia a BLM (78, 85-87).
2.1.4.- Toxicidad pulmonar por bleomicina.
La neumonitis por BLM puede llegar a ser mortal, y es una toxicidad limitante de dosis. Se
caracteriza por disnea, tos seca, estertores finos y húmedos en las bases, hipoxemia, hipocapnia y
radiología con patrón intersticial.
Entre los factores que aumentan el riesgo de toxicidad pulmonar por BLM están: la edad, la
presencia de enfermedad pulmonar previa, la irradiación pulmonar o torácica previa o concomitante, la
oxigenoterapia a altas concentraciones sobre todo la dosis acumulada de BLM.
El riesgo de FP e insuficiencia respiratoria es de un 1% para pacientes que reciben dosis
acumuladas de < 200 U/m2 y del 10 % para pacientes que reciben dosis > 200 U/m2. En su uso clínico se
recomienda no sobrepasar una dosis total acumulada de > 300-400 U/m2 (según distintos oncólogos).
Como ocurre con la FPI, no hay tratamiento curativo. El manejo es principalmente
sintomático, y sólo en ocasiones se ha descrito mejoría con corticoides, especialmente en casos con
reacciones de hipersensibilidad.
2.2.- Modelos Experimentales de Fibrosis Pulmonar..
La capacidad de la BLM para inducir FP experimental se observó inicialmente en perros, por
Fleischmann, en 1971. Más tarde se utilizaron hámsters y cobayos, y Snyder en 1978 describió su
administración vía intratraqueal. Los efectos de la toxicidad pulmonar por BLM en este modelo son
semejantes a los encontrados en humanos, y se ha mostrado de gran valor para profundizar en los
mecanismos que llevan a FP. Actualmente la IOT de BLM es el modelo más empleado para el estudio in
vivo de la FP.
Como se describe en los mecanismos de acción de la BLM, ésta es capaz de generar daño directo
en el DNA y generar RL en el interior de la célula, que indirectamente causarán daño adicional en el DNA y
en otros componentes celulares (RNA, proteínas, enzimas, mitocondrias…). A través de todos estos
mecanismos, la IOT de BLM generará un daño directo sobre las CEA mediado por el proceso de apoptosis
(muerte celular programada). Este daño, a su vez, inducirá la liberación de citocinas y factores de
crecimiento que a nivel local producirán un reclutamiento de células inflamatorias y fibroblastos (figura 7).

Tesis Doctoral Charlín Méndez Cordovez
24
Figura 7. Esquema del modelo de fibrosis pulmonar inducida por bleomicina. (Tomado de Referencia (75)).
A los tres días tras la IOT, se produce un aumento de celularidad en los septos alveolares debido
a la infiltración de células inflamatorias, principalmente macrófagos y neutrófilos. Entre el 6º día y un mes
tras la IOT, se produce un progresivo incremento de linfocitos que pueden liberar factores profibróticos, con
posterior inducción de la proliferación de fibroblastos y la síntesis de colágeno, liberando factores de
crecimiento profibrogénicos. A las 2 semanas tras la IOT, aparecen cambios difusos y multifocales como
hiperplasia epitelial y fibrosis intersticial e intraalveolar. Tras la 3ª semana de la IOT aparecen grandes focos
de fibrosis en forma difusa, caracterizados por la presencia de fibroblastos, miofibroblastos y depósito de
colágeno (75).
Los avances en los modelos experimentales han permitido estudiar patrones de susceptibilidad a
la fibrosis pulmonar, tales como alteraciones en la expresión de citoquinas y factores de crecimiento
específicos que pueden estar implicados en este proceso (enzimas, proteínas y otros componentes
intersticiales y del epitelio alveolar). Los animales transgénicos pueden portar un fragmento de ADN
exógeno en su genoma, lo cual permite el estudio del patrón de expresión de ese gen y las consecuencias
biológicas de la sobreexpresión de la proteína exógena codificada por el transgen en tejidos específicos.
Para decidir qué genes estudiar, es útil investigar previamente como influye la inducción del
proceso fibrótico en el cambio de la expresión génica. Este análisis se ha visto facilitado por la aparición de
la técnica de micromatrices (microarrays) (88).
En los últimos años se han cruzado ratones transgénicos para obtener modificaciones genéticas
combinadas que permiten evaluar varios factores implicados en el proceso fibrótico. Este modelo se ajusta
más a la realidad, dado que actualmente se cree que la FPI es una enfermedad poligénica. El inconveniente
básico de trabajar con ratones es que son pocos los laboratorios que trabajan en este campo.

Tesis Doctoral Charlín Méndez Cordovez
25
Probablemente la mayor accesibilidad al uso de estos animales sea una de las mejoras que se producirán
en los próximos años.
Sin embargo, se sabe que la historia natural de la FP en humanos difiere en varios aspectos de la
inducida en animales. Por una parte, no hay ningún modelo que siga una reproducción precisa de la
cronicidad o progresión de la enfermedad, probablemente por el desconocimiento de los factores que
provocan en el pulmón humano que un evento reparativo patológico lleve a la perpetuación de la fibrosis.
Además, en la mayoría de los modelos animales los cambios producidos por una inducción inicial
tienden a revertir, lo que no ocurre en humanos. Por este motivo, para extrapolar a la clínica los resultados
obtenidos en un modelo animal será primordial su correcta y cautelosa interpretación. La tendencia a
experimentar con fenotipos que reproduzcan lo más posible el proceso en humanos, es la base para mejorar
los modelos animales en el estudio de la fibrosis pulmonar en un futuro próximo.
3.- Ozono.
3.1.- Historia.
El Ozono tiene más de 200 años de historia, y la ozonoterapia más de 100
(http://www.aepromo.org/en/historia.php) (89).
El Ozono (O3) se detecta por primera vez en 1.785 por el físico Martinus Van Marum (1.750-1.837)
al percibir un olor peculiar detectado cerca de las máquinas eléctricas, pero no fue hasta 1839 que el químico
alemán Christian Friedrich Schönbein (1.799-1.868), lo produce y le da el nombre de ozono (del griego
“ozein”, oloroso).
En 1.857 Werner Von Siemens, construye el primer tubo de inducción superior, con el cual
Kleinmann realizó los primeros ensayos para destrucción de microorganismos y la primera insuflación del
gas en animales y humanos.
En 1.870, el médico alemán Lender realizó la primera publicación sobre efectos biológicos
prácticos, en relación a la desinfección de aguas.
En 1.873, Fox descubre la capacidad de este agente químico para la eliminación de
microorganismos. Existen evidencias como desinfectante a partir del 1.881, de acuerdo a lo mencionado
por el Dr. Kellogg en su libro sobre difteria. El descubrimiento de las propiedades antimicrobianas del
ozono revolucionó la medicina de la época, recordemos que la penicilina no se descubriría hasta 70 años
más tarde.
Nikola Tesla (1.856-1.943), patentó el primer generador de ozono (1.896), y en 1.900 funda la “The
Tesla Ozone Company”, empresa fabricante de generadores de uso médico.
En 1.911, el Dr. Noble Eberhart, jefe del departamento de Fisiología de la Universidad de Loyola Chicago, en el “Manual de Funcionamiento de alta frecuencia”, indica que utilizaba el ozono para tratar la

Tesis Doctoral Charlín Méndez Cordovez
26
tuberculosis, anemia, clorosis, zumbidos, tos ferina, asma, bronquitis, fiebre del heno, insomnio, pulmonía, diabetes, gota y sífilis.
En 1943, el Dr. Blass funda la primera asociación alemana de ozonoterapia.
En 1.935 el austriaco-alemán Payr presenta en el Congreso de la Sociedad Alemana de Cirugía sus resultados sobre la capacidad cicatrizante del ozono, y en 1.936 el francés Aubourg publica un artículo proponiendo la insuflación rectal de O3 en colitis crónica y fístulas.
Sin embargo, la ausencia en esos años de materiales plásticos resistentes al ozono limitó la aplicación y difusión de la ozonoterapia, hasta que aparecen y se incorporan nuevos materiales plásticos durante la segunda mitad del siglo XX.
En 1957, el Dr.Hansler fabrica en Alemania el primer generador de ozono moderno, en cuyo diseño se basan los generadores actuales.
En 1961, el Dr. Hans Wolf introdujo en su práctica médica la autohemoterapia mayor y menor.
3.2.- Definición y propiedades.
El O3 es una forma alotrópica del oxígeno, compuesto por tres átomos de oxígeno. El ozono tiene
un peso molecular de 48 g/mol y es veces más soluble en agua que el O2 (49,0 ml en 100 ml de agua a 0°
C, 0,02 M), obedeciendo a la Ley de Henry, lo que le confiere su capacidad de reaccionar rápidamente con
cualquier compuesto soluble y biomoléculas llevando a la generación de RL.
El O3 es un gas inestable y su vida media depende de la temperatura (40 min a 20° C y de unos
140 minutos a 0° y 3 meses a 50ºC bajo cero, lo que imposibilita su almacenamiento (89) (90-92). Se
conserva en estado líquido a la temperatura de -111,9°C con un peso específico de 1,571 g/ml.
En la naturaleza el O3 es el gas más importante de la estratosfera (más o menos de los 15 a 50
km de la superficie de la terrestre) produciéndose y destruyéndose continuamente en un equilibrio dinámico
que consume la mayor parte de la radiación ultravioleta (UV) de longitud de onda < 290 nm. Aquí están
incluidos los UV de onda corta (UVC: 100-280 nm) y parte de los UV de onda media (UVB: 280-315 nm),
siendo ambos los UV de capacidad más mutagénica e inductora de tumores cutáneos. Así, en la
estratosfera, la radiación solar convierte la molécula de oxígeno atmosférico (O2) en dos átomos de oxígeno
altamente reactivos, proceso causado por la radiación ultravioleta en consonancia con la teoría de
Chapman, cada uno de estos átomos de oxígeno se combina con la molécula de O2 para formar el oxígeno
triatómico (O3) en una reacción endotérmica. Posteriormente, la radiación UV convierte una molécula de
ozono en una de oxígeno diatómico y un átomo de oxígeno diatómico y un átomo de oxígeno.
O2 + UV (<290 nm) → O + O
O2 + O → O3
O3 + UV (<290 nm) → O2 + O

Tesis Doctoral Charlín Méndez Cordovez
27
Las principales repercusiones clínicas del O3 atmosférico dependen de su localización:
● En la estratosfera el O3 tiene efecto beneficioso, y su disminución (agujero de la capa de ozono)
se relaciona con el cambio climático y el aumento de la incidencia de los tumores de piel.
● En la troposfera (0,03 ppm), su aumento juega un papel negativo como contaminante ambiental.
Su capacidad de producir toxicidad pulmonar ha llegado a la generalización del dogma de que el
O3 es siempre tóxico, a cualquier concentración y en cualquier tejido. Esto no sólo “no es cierto”
sino que, en los últimos quince años, varios publicaciones relevantes han descrito que la
producción de O3 por el propio organismo podría jugar un importante papel en la función del sistema
inmune (93, 94).
Por tanto, para el uso de O3 con fines médicos, se debe evitar su toxicidad asegurándose que tanto
el paciente como el personal lo inhalen. La obtención de O3 para uso clínico debe realizarse exclusivamente
a partir de O2 medicinal, ya que el producido a partir del aire genera altos niveles de peroxinitrilos (N2O2).
Se debe emplear un generador de ozono preciso y equipado con un fotómetro bien estandarizado,
que permita; a) determinar la concentración de O3 en tiempo real; b) la obtención de un volumen de gas
preciso; c) la selección y producción de una concentración de ozono definida, que será variable en función
del efecto terapéutico deseado; d) recoger el O3 sobrante que no se utilice para destruirlo (reconvirtiéndolo
en O2 mediante un catalizador). Recientemente se ha publicado en detalle las características ideales que
deben poseer (95).
La concentración de ozono médico producido por el generador será inversamente proporcional al
flujo de O2 por unidad de tiempo. En la mezcla final de O3-O2, la concentración máxima de este último será
del 5%. El proceso de formación de O3 a partir de O2 medicinal en el equipo se consigue mediante un
proceso endotérmico que tiene lugar gracias a los gradientes de voltaje muy elevados que se consiguen
entre los electrodos de un tubo de Siemens (89).
3O2 ↔ 2O3 – 68400 cal
3.3.- Mecanismos de acción de la ozonoterapia.
El O3 no actúa sobre receptores celulares específicos y su principal mecanismo de acción es
indirecto, mediante la producción de un “moderado y controlado” estrés oxidativo.
El O3 tiene mayor capacidad oxidante que el O2, siendo el tercer agente oxidante en orden de
potencia, después del flúor y del persulfato. Esta propiedad, junto con su alta solubilidad, hará que reaccione
en cuestión de segundos con todos los compuestos que estén a su alcance en el plasma y las membranas
celulares, formando ozónidos y RL. Los diferentes sistemas antioxidantes (AO) intentarán eliminar todos
estos compuestos oxidados u ozonizados. El resultado final dependerá de la concentración y cantidad de
O3, del tipo los RL formados y de los sistemas AO presentes, siendo ambos variables en las distintas células,
tejidos y localizaciones del organismo.

Tesis Doctoral Charlín Méndez Cordovez
28
En condiciones fisiológicas, los RL actúan como reguladores de la transducción de señales,
mediadores de la defensa del huésped y en la respuesta inmune. Un exceso de RL puede ocasionar la
formación de compuestos como el peroxinitrilo (N2O2) y el anión hipoclorito (CIO-), de muy alta reactividad.
Aunque el tiempo de vida media de los RL es muy corto (< 1 segundo), son capaces de dañar distintos
componentes celulares.
A dosis y concentración adecuada, el estrés oxidativo producido por el O3 inducirá una respuesta
de adaptación en el organismo con modulación del sistema inmune y potenciación de los sistemas AO,
mediante mecanismos de acción bien descritos (96, 97). Por otra parte, hay que tener en cuenta que el
efecto del O3 no tiene una relación dosis/respuesta lineal, sino que sigue el concepto de hormesis: debe
alcanzar una dosis suficientemente alta para inducir una respuesta adaptativa, pero no tan alta como para
sobrepasar la capacidad de adaptación y ocasionar un efecto dañino (97).
Entrando un poco más en detalle, el O3 reacciona rápidamente sobre todo con:
● Los AO hidrosolubles (ácido ascórbico y ácido úrico), compuestos de tiol con grupos SH,
tales como la cisteína, glutatión reducido (GSH) o la albúmina. Estos serán los encargados
de eliminar en pocos segundos las RL.
● Los ácidos grasos poliinsaturados (PUFA), formando lipoperóxidos (LPO) que tienen una
vida media mucho mayor que los RL.
La peroxidación lipídica producida a nivel de las membranas celulares inducirá la producción de
segundos mensajeros a nivel intracelular, destacando entre ellos el peróxido de hidrógeno (H2O2), y los
alquenos (hidrocarburos insaturados que tienen uno o varios dobles enlaces carbono-carbono en su
molécula). Entre estos últimos, el más importante es el 4-hidroxinonenal (4-HNE) (98). Estos segundos
mensajeros llevan a la activación de factores de transcripción nuclear como el factor nuclear eritroide-2
(Nrf2) que a su vez induce la transcripción de elementos de respuesta antioxidante con la posterior
producción de enzimas AO como superóxido dismutasa (SOD), glutatión peroxidasa (GPX), proteínas de
shock térmico de 70 KDa (HSP-70) o hemo oxigenasa-1 (HO-1) (98-100).
A nivel del sistema inmune, Nrf2 es capaz de suprimir la activación del factor nuclear kappa B
(NFkB) el cual tiene efecto pro-inflamatorio. Esta acción inmunomoduladora del O3 se complementa
mediante la inducción de otros factores de transcripción nuclear como el factor nuclear de las células T
activadas (NFAT) o la proteína activadora 1 (AP-1) y la capacidad del O3 para modular la acción de
interferones e interleucinas (89).
En los eritrocitos, la peroxidación lipídica y los segundos mensajeros inducen:
● La activación del ciclo de las pentosas para asegurar así una cantidad suficiente de la sustancia
redox nicotinamida adenina dinucleótido fosfato (NADPH), necesaria para la regeneración del
glutatión (GSH). El metabolito más importante del efecto anterior es el 2,3-DPG (2,3
difosfoglicerato), que desplaza la curva de disociación de la hemoglobina hacia la derecha. Esto
confiere menor afinidad de la Hb por el O2 y, por consiguiente, mayor liberación de O2 hacia los
tejidos (89). Se ha descrito que este aumento podría ocurrir sólo en aquellos pacientes con niveles
basales (pre-O3) disminuidos de 2,3-DPG (101).

Tesis Doctoral Charlín Méndez Cordovez
29
Modificación de su carga eléctrica induciendo:
− Disminución del pH en el eritrocito, que producirá un desplazamiento hacia la derecha de la curva
de disociación de la hemoglobina (efecto Bohr) independiente de los niveles de 2,3-DPG (102).
− Aumento de la flexibilidad de la membrana del eritrocito, que disminuye la viscosidad sanguínea y
la resistencia vascular, mejorando finalmente las propiedades rheológicas de la sangre y el flujo
sanguíneo (103, 104). Según las leyes de Ohms y de Poiseuille, la disminución en la viscosidad
sanguínea (η) y/o aumento del radio (r) de los vasos, producirán una disminución de la resistencia
vascular (R), que llevará a un aumento del flujo sanguíneo (F) (figura 8).
Figura 8. Leyes de Ohms y de Poiseuille, donde F= Flujo, P= Presión
de flujo, R= Resistencia, η= Viscosidad sanguínea, r = Radio y L=
Longitud del vaso.
Sobre las células del endotelio vascular, los segundos mensajeros pueden inducir la producción
de prostaciclina, óxido nítrico, IL-8, produciendo vasodilatación a nivel de la micro-circulación, con la
consiguiente disminución de la resistencias vasculares y aumento del flujo sanguíneo. Esta acción se
complementa con el aumento de adenosina trifosfato (ATP) y modulación de prostaglandinas (figura 9).
Figura 9. Esquema de la acción del O3 sobre la oxigenación y flujo de los tejidos. ROS: radicales libres; LPO: lipoperóxidos; A.O.:
antioxidantes; Nrf2: factor nuclear eritroide-2; 2,3-DPG: 2,3 difosfoglicerato; NO: óxido nítrico; PG: prostaglandinas; Hb:
hemoglobina.
𝑹 =𝟖 ∗ 𝜼 ∗ 𝒍
𝝅 ∗ r4
𝑭 =∆𝑷
𝑹

Tesis Doctoral Charlín Méndez Cordovez
30
Nuestro grupo de investigación ha descrito el impacto de estos factores sobre varios parámetros
relacionados con el flujo sanguíneo y la oxigenación (105, 106) y el beneficio clínico que se puede obtener
de la ozonoterapia en pacientes con toxicidad crónica por radioterapia, la cual suele estar mediada por
procesos de fibrosis a nivel de la microvascularización y del tejido conectivo (107-111).
Por último, hay que destacar otro hecho de gran relevancia. Los variados productos de la
peroxidación producidos por el O3 en el plasma (o que llegan a él) serán capaces de alcanzar los distintos
órganos y tejidos. Las acciones de éstos sobre la médula ósea, hígado, sistema nervioso central o las
glándulas endocrinas, aunque no bien conocidas, justifican los múltiples y diversos efectos del O3 a nivel
sistémico (figura 10).
Figura10. Esquema de cómo la ozonoterapia sistémica (vía insuflación rectal o mediante autohemoterapia) y los lipoperóxidos a
los que da lugar, pueden inducir una respuesta adaptativa en distintos órganos. LPO: Lipoperóxidos, MALT: tejido linfoide asociado
a mucosas (modificado a partir de Ref. (96).
Tabla 4. Resumen de algunos de los efectos tópicos y sistémicos del ozono médico.
Efecto tópico Efecto sistémico por acción en los
eritrocitos y endotelio vascular Efecto sistémico por acción en
células inmunitarias
Efecto microbicida:
bactericida,fungicida,virustático Mejoría de las propiedades
rheológicas Activación moderada de células
mononucleares

Tesis Doctoral Charlín Méndez Cordovez
31
Efecto de limpieza de heridas Activación del metabolismo
eritrocitario: aumento de 2,3-DPG y
ATP
Liberación de citoquinas:IL-1,IL-2,
IFN-G, TNF-α y TGF-β
Desplazamiento del equilibrio
HbO2/Hb hacia la derecha, con
mejoría de la liberación de oxígeno.
Mejoría de la cicatrización de las
heridas Liberación de prostaciclinas (acción
vasodilatadora e inhibición de
agregación plaquetaria).
Inmunomodulación
Inmunoactivador eficiente Activación/potenciación de rutas
enzimáticas ligadas al metabolismo
de la glucosa (G-6pdh),y de los
mecanismos antioxidantes (GSH-
Px,GSHred, SOD)
Liberación de prostaciclinas (potencial
acción antimetastásica).
3.4.- Vías de administración.
La principal toxicidad del ozono es debida a la inhalación, por la limitada presencia de AO a nivel
pulmonar. Así, en el epitelio respiratorio el O3 induce un aumento de la fosfolipasa A2 y con ello de ácido
araquidónico y RL, lo que se relaciona con la toxicidad pulmonar. Sin embargo, la adecuada administración
de O3 por vías “no respiratorias” ha mostrado un efecto potenciador de AO y una posterior disminución de
RL, junto con una disminución de la fosfolipasa A2 y de la síntesis de prostaglandinas inflamatorias,
colaborando así en la acción antiinflamatoria.
Dado el gran número de patologías en las que se utiliza la Ozonoterapia, la vía de administración
(y la concentración) dependerá de la patología y características del paciente. En Junio de 2015 se ha
publicado la 2ª edición de la Declaración de Madrid sobre la Ozonoterapia, una guía clínica de consenso
internacional accesible en la página web del International Scientific Committee Of Ozone Therapy
(www.isco3.org), y en Octubre de 2015 se ha publicado una Revisión sobre ozonoterapia basada en la
evidencia accesible en la página web de la World Federation of Ozone Therapy. ttp://www.wfoot.org/wfots-
review-on-evidence-based-ozone-therapy/).
Puesto que el O3 es tóxico a muy pequeñas concentraciones sobre el epitelio pulmonar, es
indispensable durante la aplicación de ozonoterapia, evitar la exposición, tanto del paciente como del
personal sanitario que lo administra. Las principales rutas de administración del O3 para obtener efectos
sistémicos son la autohemoterapia y la insuflación rectal. Sus efectos pueden ser básicamente
superponibles, siendo la insuflación rectal la técnica más empleada en los experimentos con animales, y en
menor medida la vía intraperitoneal.
La insuflación rectal ha sido la vía de administración de ozono utilizada en la realización de esta
Tesis Doctoral.
En 1936, el Dr. P. Aubourg fue el primero en proponer las insuflaciones de ozono mediante el uso
de una cánula a través del recto para tratar la colitis crónica, fisuras y fístulas anales. Aquí, el O3 interactúa

Tesis Doctoral Charlín Méndez Cordovez
32
con la mucosa intestinal generando segundos mensajeros a ese nivel, y cuyos efectos se extenderán vía
vena porta al sistema linfático intestinal.
En humanos, generalmente se insuflan entre 100 y 300 ml de gas O3/O2 en el recto, a concentraciones entre 10 y 30 µg/ml. En cambio, en ratas suelen utilizarse unos 6-8 ml de gas y las concentraciones habitualmente empleadas en los estudios son de 50 µg/ml, las que se han empleado para la realización de esta Tesis Doctoral.
3.5.- Efectos secundarios y contraindicaciones de la ozonoterapia.
Si la ozonoterapia se realiza correctamente, no suele ocasionar efectos secundarios de
importancia. Los casos, anecdóticos, de toxicidad severa publicados en la literatura fueron debidos a un
procedimiento médico defectuoso (técnica de infiltración, de transfusión) o directamente a una mala praxis
médica, más que al efecto directo del Ozono en sí mismo. En cualquier caso, el especialista en ozonoterapia
debe ser capaz de superar cualquier emergencia porque un retraso en la intervención puede terminar con
la muerte del paciente. Debe saber todos los pasos de la resucitación cardio-pulmonar (RCP) y tener a la
mano el ambú, oxígeno médico, un desfibrilador automatizado y algunas ampollas de adrenalina, atropina
y corticoides.
Las principales contraindicaciones para el empleo de ozonoterapia son:
● Pacientes con deficiencia significativa de la enzima glucosa-6-fosfato-deshidrogenasa (G-6PD),
favismo. Esta enzima participa en los mecanismos AO de los eritrocitos, y por tanto estos
pacientes pudieran presentar una deficiente adaptación al estrés oxidativo generado por el ozono.
● Embarazo. Aunque estudios en ratas no han evidenciado efecto mutagénico en fetos, es preferible
no utilizarlo.
● Pacientes con tratamiento con fármacos inhibidores de la enzima convertidora de la angiotensina
(IECA) usados como antihipertensivos.
● Situaciones graves o no controladas de: hipertiroidismo, inestabilidad cardiaca, trombocitopenia o
estados de hipocoagulación (especialmente para la autohemoterapia).
● Alergia conocida al ozono.
En conclusión, no se puede evitar decir que el ozono puede ser tóxico como cualquier tipo de
fármaco o sustancia pero, hasta el momento, los datos experimentales y la evidencia clínica han demostrado
un gran margen de seguridad. Los efectos adversos severos prácticamente siempre han estado ligados a
una deficiente técnica médica o mala praxis. Por otro lado, a pesar de su famosa “toxicidad”, un estudio
realizado por Jacobs en 1982 sobre posibles efectos negativos de la ozonoterapia encontró que la incidencia
de toxicidad importante fue de 0,0007%, realmente baja para lo que habitualmente encontramos en
medicina.

Tesis Doctoral Charlín Méndez Cordovez
33
II.- JUSTIFICACIÓN.

Tesis Doctoral Charlín Méndez Cordovez
34
En la actualidad, la FPI es una enfermedad pulmonar progresiva que compromete de una forma
muy seria la vida del paciente. Las opciones terapéuticas son muy escasas. Sólo muy recientemente han
aparecido 2 fármacos capaces de ofrecer un claro beneficio, aunque es limitado en cuantía y duración, En
los estadios finales de la enfermedad, el trasplante pulmonar se mantiene como la única alternativa
terapéutica eficaz, pero sólo indicada en pacientes muy seleccionados.
Los procesos de óxido-reducción intracelular y las defensas frente a los mismos están presentes
y regulados genéticamente en la fibrosis pulmonar, y se inician desde las primeras fases de la lesión. De
forma que en el pulmón con lesiones fibróticas se produce una situación de estrés oxidativo celular crónico
responsable del daño tisular irreversible. La administración de un potente inductor de antioxidantes, como
el ozono, de forma continua y mantenida, no utilizado antes en modelos de fibrosis pulmonar podría paliar
esta situación de estrés celular, daño en el epitelio alveolar y, de esta forma, cambiar la evolución de la
fibrosis pulmonar.
Para inducir la fibrosis pulmonar en animales de experimentación se ha optado por el modelo de
fibrosis pulmonar mediante instilación orotratraqueal (IOT) de bleomicina provocado con nuestra técnica
la aparición de lesiones pulmonares similares a las observadas en los enfermos con FPI.
El pobre pronóstico de los pacientes con FPI y las escasas opciones terapéuticas demandan un
esfuerzo mayor en el campo de la investigación traslacional. Esta Tesis Doctoral se ha diseñado con
carácter traslacional, con el objetivo último de que si el tratamiento con ozono fuera capaz de reducir el
grado de fibrosis pulmonar, su efecto podría ser evaluado en humanos mediante el diseño de los pertinentes
ensayos clínicos.

Tesis Doctoral Charlín Méndez Cordovez
35
III.- OBJETIVOS.

Tesis Doctoral Charlín Méndez Cordovez
36
1. “Desarrollar un modelo experimental válido en ratas de fibrosis pulmonar
inducida con bleomicina”.
2. “Valorar si el tratamiento con ozono disminuye el daño pulmonar en un
modelo experimental de fibrosis pulmonar inducido por bleomicina”.
3. “Estudiar si la expresión de los genes analizados se relaciona con el daño
pulmonar y su respuesta al tratamiento”.

Tesis Doctoral Charlín Méndez Cordovez
37
IV.- MATERIAL Y MÉTODOS.

Tesis Doctoral Charlín Méndez Cordovez
38
4.1- Material.
4.1.1.- Animales de experimentación.
Este estudio se llevó a cabo con 21 ratas (Rattus norvegicus) de la cepa Sprague-Dawley, machos,
de 5 meses de edad y pesos comprendidos entre los 300-350g, con un Estatus Microbiológico Convencional
(EMC) y de acuerdo Normas Europeas vigentes en las recomendaciones de las Federaciones de
Asociaciones Europeas de las Ciencias del Animal de Laboratorio (FELASA) sobre el cuidado y protección
de los animales de experimentación, bajo estrictas condiciones sanitarias. Estos animales proceden del
bioterio de la Unidad de Investigación del Hospital Universitario de Gran Canaria Dr. Negrín. Las condiciones
de estabulación, bienestar y salud de los animales se cumplieron de acuerdo a las recomendaciones y
exigencias recogidas en el R.D. 1201/05 y la directiva 86/609/CEE.
En cuanto a las condiciones de estabulación, todos los animales fueron destetados a los 21 días
de edad, ubicándose en jaulas con cubeta de macrolón de 20 X 35 X 55 cm., tapa de rejilla de acero
inoxidable y provisto de biberón, agrupados de cuatro en cuatro e identificados individualmente. El lecho
que se utilizó fue de abeto (figura 11).
Figura 11. Jaula con cubeta de macrolón 20 x 35 x 55 cm.
La iluminación es regulada artificialmente mediante fotoperiodos con un ciclo de luz/ oscuridad de
12 horas y una intensidad de 270 lux. El sistema de ventilación de las salas permite mantenerlas en presión
positiva. El sistema de renovación del aire se realiza mediante un sistema de extracción que permite que la
humedad relativa se encuentre dentro de los límites establecidos (16 renovaciones/hora y una temperatura
ambiente de 21° ± 1°C).

Tesis Doctoral Charlín Méndez Cordovez
39
Figura 12. Sala del postoperatorio. Figura 13.- Instalaciones quirúrgicas.
Los animales se sometieron a una dieta ad libitum con la siguiente composición: vitamina A 6000
U.I/Kg.; vitamina D3 600 U.I/Kg; Fe 50 mg/kg.; Mn 44 mg/Kg.; Zn 31 mg/Kg.; Cu 7 mg/Kg.; I 6,2 mg/Kg.;
Co 0,5 mg/Kg.; proteína bruta 14,5%; materia grasa bruta 4 %; fibras brutas 4,5%; cenizas brutas 4,7%.;
P.O. Box 44220, Madison, Italy). El agua también se les suministró ad libitum con un pH 6,5-8,5.
En el postoperatorio, los animales se observaron durante los 28 días que duró el experimento,
monitoreándose la supervivencia de los mismos. Siguiendo las recomendaciones de FELASA, durante la
observación diaria de los animales, se registró el peso, aspecto externo, así como la cromodacriorrea y
aspecto del pelaje.
En cuanto a las condiciones de estabulación, todos los animales fueron destetados a los 21 días
de edad, ubicándose en jaulas con cubeta de macrolón de 20 X 35 X 55 cm., tapa de rejilla de acero
inoxidable y provisto de biberón, agrupados de cuatro en cuatro e identificados individualmente. El lecho
que se utilizó fue de abeto
4.1.2.- Salas, equipos y material inventariable.
Las salas de alojamiento (preoperatorio y postoperatorio) donde se ha llevado a cabo la
investigación experimental, dispone de los sistemas adecuados para mantener a una temperatura e
iluminación constante. (fig. 12), (fig. 13).
Las instalaciones quirúrgicas disponen de un quirófano experimental con un nivel de contención
de seguridad biológica tipo II y con equipamiento completo para microscopía quirúrgica.
Equipamiento utilizado ha sido un ventilador Servo 900 C® (Siemens-Elema, Suecia) (fig. 14), un
generador de ozono (Ozonosan alpha plus, Dr. Hänslerr, Alemania) (fig.15), microscopio de pie Carl Zeiss
OPMI® 9-12,5x (West, Alemania) (fig.16), congelador Jouan modelo VXE 490 de -80 ºC (fig. 17).

Tesis Doctoral Charlín Méndez Cordovez
40
Figura 14. Ventilador Servo 900C. Figura 15. Generador de ozono
Figura 16. Microscopio de pie Carl Zeiss. Figura 17. Congelador -80ºC
4.1.3.- Material quirúrgico.
Para garantizar la esterilidad se han utilizado paños quirúrgicos estériles, así como bisturís nº 22,
jeringas de 1ml y de 2 ml, agujas 21 G, tijeras de Metzembaum y Mayo, pinzas de disección, pinzas de
Adson, pinzas hemostáticas de mosquito, pinzas de microcirugía (relojero), tijeras de microcirugía, tijeras
de microcirugía Westcott, y seda negra 3/0 (Silkan®. B. Braun Surgical S.A. España), torundas y gasas
estériles (figura 18).

Tesis Doctoral Charlín Méndez Cordovez
41
Figura 18.- Instrumental quirúrgico.
4.1.4.- Medicación anestésica y analgésica.
Los fármacos que se han empleado en la anestesia de los animales han sido: Lidocaína 1%
(Xilonibsa®, Inibsa, Barcelona, España), Hidrocloruro de medetomidina (Dormilan® Vetpharma Animal
Health, Barcelona, España), Ketamina (Ketalar 500®, Pfizer, Madrid, España) y Atropina 1% (Atropina®,
Braun, Madrid, España). El efecto de la medetomidina se ha revertido con su antagonista, el Atipamezol
(hidrocloruro) (Antisedan®, Esteve, Barcelona, España.) Como analgésico postoperatorio se ha empleado
la Buprenorfina (Buprex®, Shering-Plug, Barcelona. España) (figura 19).
Figura 19.- Anestésicos y analgésicos empleados en las ratas.

Tesis Doctoral Charlín Méndez Cordovez
42
4.1.5.- Conservación del tejido pulmonar.
Las muestras de tejido fresco son fijadas en formaldehído ya que preserva la morfología histológica
del tejido pulmonar 3,7-4% pH7 y embebido en parafina (Leica, EG 1160, Microsystems, Nussloch Gmbh,
Alemania). La deshidratación se llevó a cabo con una batería de alcoholes y el aclarado posterior se realiza
en baños de xilol.
Para el procesamiento de muestras histológicas, se utiliza como técnica de corte para obtener las
secciones el micrótomo rotación (Thermoscientific Microm HM 340 E, Walldorf, Alemania), un calentador
de agua JP Selecta y láminas portaobjetos 76 x26 (HistoBond®SX, Knittel Glasser).
Además, las muestras se tiñeron rutinariamente con hematoxilina- eosina (HE), el material
empleado: xilol, alcohol absoluto (100°), alcoholes, agua corriente, agua destilada, hematoxilina alumínica
de Harris, alcohol ácido, sustancias alcalinas (agua amoniacal), solución de bicarbonato de sodio al 2% y
carbonato de litio al 1%.
Para purificar el RNA se usó el tiaocinato de guanidino (GITC)-fenol/cloroformo. El bromuro de
etidio (BrEt) (Pierce, Rockford, Ll). Par la síntesis del cDNA se usó el Kit iScrip cDNA síntesis (BioRad,
Barcelona-España). Se utilizó la herramienta on-line www.thermo.com para fabricar los cebadores. Para
realizar las PCRs en tiempo real se utilizó SYBR Green Master Mix (Foster, California). El resto de los
reactivos procedían de Sigma- Aldrich Chemical Co. (St. Louis, Mo).
4.2.- Método.
4.2.1.- Diseño.
El Comité Ético de Experimentación y Bienestar Animal del Hospital Universitario de Gran Canaria
Dr. Negrín evaluó favorablemente el procedimiento CEEBA-HUGCDN 013/2011 para la realización de este
trabajo de investigación.
Se han utilizado 21 ratas machos de la cepa Sprague- Dawley, para el desarrollo del modelo
experimental, con pesos comprendidos entre 300 y 350 g, y en condiciones óptimas de estabulación. Los
animales se han distribuido en cuatro grupos de la siguiente manera (tabla 5): A) Un grupo Control (n= 6
pulmones de 3 animales) no se les realizó ninguna manipulación terapéutica ni experimental. Se les extrajo
muestra de sangre y de pulmón derecho e izquierdo sanos (pulmones analizados = 6); B) Un grupo Sham
(n=6) se les realizó IOT de suero fisiológico y se les extrajo muestras de sangre y de pulmón derecho e
izquierdo a los 28 días de instilación; C) Un grupo Bleomicina (BLM) (n=6) a los que se le ha realizado IOT
de BLM (2,5 mg/Kg de peso). Se les ha extraído muestras de sangre y de pulmón derecho e izquierdo sano
a los 28 días de instilada la BLM; y D) Un grupo BLM + O3 (n=6). Se les ha instilado BLM de la misma forma
que el “Grupo BLM”. Además, los animales fueron tratados con O3 por vía rectal desde el segundo día de
la IOT de BLM hasta el final del estudio.
La IOT se ha realizado mediante un catéter intravenoso (i.v.) calibre 14G x 51mm (Abbocath®-T,
Abbott Ireland Ltd., Sligo, Irlanda). Tras la IOT, las ratas pasaron al postoperatorio donde se les ha
administrado H2O y comida ad líbitum. Transcurridos 28 días después de cada procedimiento, se han

Tesis Doctoral Charlín Méndez Cordovez
43
sacrificado los animales y se les ha extraído el árbol broncopulmonar. Se han tomado muestras tanto del
pulmón derecho como del izquierdo para su estudio histológico, microbiológico y expresión génica.
TABLA 5. Diseño de grupos para el estudio histológico, microbiológico y de expresión génica.
GRUPO
TRATAMIENTO
Nº ANIMALES
TOTAL
A
GRUPO CONTROL
6 pulmones de
3 animales
3
B
GRUPO SHAM
6
6
C
GRUPO BLM
6
6
D
GRUPO BLM + O3
6
6
Total
21
Tabla 6. Volumen de IOT de los grupos de estudio.
GRUPO
TRATAMIENTO
INSTILACIÓN
RATAS
A
GRUPO CONTROL
------
0
B
GRUPO SHAM
SUERO FISIOLÓGICO
(Volumen de 500 µl)
6
C
GRUPO BLM
BLEOMICINA
(2,5 mg/Kg en 500 µl de solución salina)
6
D
GRUPO BLM + O3
BLEOMICINA
(2,5 mg/Kg en 500 µl de solución salina)
6
4.2.2 – Inducción a la fibrosis pulmonar por BLM.

Tesis Doctoral Charlín Méndez Cordovez
44
4.2.2.1.- Anestesia.
En la inducción de la anestesia se utilizó la combinación de un α–2 agonista (medetomidina) y un
agente que produce disociación, la ketamina, vía parenteral (VP) (112). De este modo se procede a la
premedicación con 0,5 mg/Kg por vía subcutánea (s.c.) de medetomidina (Dormilan® Vetpharma Animal
Health, Barcelona, España), transcurridos cinco minutos y una vez comprobada la sedación de las ratas se
indujo una anestesia corta, inyectando al animal por vía intraperitoneal (i.p.) 25 mg/kg de ketamina (Ketolar
500®, Pfizer, Madrid, España). Posteriormente se le inyectó intramuscular (i.m.) 0,04 mg/Kg de atropina 1%
(Atropina® Braun, España) (figura 19).
4.2.2.2.- Intubación y ventilación.
Una vez que el animal es anestesiado, se realiza la maniobra de intubación orotraqueal (IOT)
según protocolo descrito (113). Para ello, se colocó al animal en decúbito dorsal fijado, seguidamente se le
aplicó una luz en la parte central del cuello para visualizar por transiluminación los pliegues de la laringe
junto con la entrada de la tráquea. Después de retirar la lengua con una pinza atraumática, y previa
aplicación de lidocaína 1% (Xilonibsa®, Inibsa, Barcelona, España), se introdujo la cánula 14G (Abbocath®-
T) por la boca en la epiglotis con la ayuda de una guía de teflón, con cuidado de no tocar la glotis, evitando
un posible edema o moco que dificulte la respiración. Una vez introducida la cánula se comprueba el la
estabilidad respiratoria, se fija al maxilar superior, evitando así su desplazamiento durante el procedimiento.
Para la ventilación mecánica de los animales, se ha utilizado un ventilador mecánico (Servo 900,
Siemens, Madrid, España) (figura 14) con una presión inspiratoria de 12 cmH2O, con una relación
inspiración/espiración de 1:2, una fracción inspirada de oxígeno (FiO2) de 0,2 y un volumen corriente (VC)
de 1ml/100g de peso vivo, una presión positiva al final de la expiración de 2 mbar, una fracción de O2
espirado (FiO2) de 0,21, se modificó la relación inspiración espiración a 1:2, y una frecuencia respiratoria
de 60 respiraciones por minuto (rpm).
4.2.2.3. Administración de BLM.
La BLM fue preparada en el Servicio de Farmacología del Hospital Universitario de Gran Canaria
Dr. Negrín, y consistió en una mezcla estéril de 2,5 mg/kg de BLM en un volumen total de 500 ul de solución
salina en jeringas de insulina.
Una vez anestesiado e intubado el animal, como previamente se ha descrito, inclinamos la mesa
quirúrgica, colocándola lo más vertical posible. Acto seguido, se procedió a la IOT con la ayuda de una
jeringa de insulina conectada a una aguja Nº 21G de 50 mm de largo, permitiendo de este modo llegar lo
más lejos posible hasta antes de la carina. Esta aguja se introdujo dentro de la cánula de 14 G, asegurando
de este modo no lesionar la tráquea y que la dosis a instilar sea más exacta. El vaciado de la jeringa coincidió
con el momento de la inspiración del animal, evitando de esta forma alguna resistencia por la espiración.
Para asegurarnos que la BLM se disperse a la mayor área pulmonar posible, y siempre con la
mesa quirúrgica inclinada, se procedió inmediatamente a intubar al animal, dejándolo con la ventilación
mecánica 5 min. Transcurrido ese tiempo se inició el proceso de revertir la anestesia con 0,5 mg/Kg (im) de
atipamezol (hidrocloruro) (Antisedan®, Esteve, Barcelona, España.).

Tesis Doctoral Charlín Méndez Cordovez
45
4.2.2.4- IOT de solución salina isotónica 0,9% (solución fisiológico).
Tras iniciar el procedimiento de intubación, se inclina la camilla del quirófano unos 60º colocando
los animales tal y como se indica en la imagen (figura 20). La instilación del suero fisiológico, en el grupo
Sham, se ha realizado de la misma forma y el mismo volumen (500 ul) que la BLM. Trascurrido 5 min de
ventilación mecánica, se han desentubado y, finalmente, revertida la anestesia con la administración de 0,5
mg/kg intramuscular (i.m) de atipamezol (Antisedan®, Pfizer, Madrid, España).
Figura 20. Muestra el proceso de IOT.
4.2.3.- Tratamiento con O3.
El O3 se ha administrado por vía rectal (insuflación rectal) con una sonda de silicona flexible 14G, por medio de un generador médico de ozono. La concentración de ozono se ha controlado por medio de espectrofotometría UV a 254 nm. El volumen de gas (mezcla de O3/O2) fue de 20 ml/kg. La administración de O3 se realizó de forma diaria durante 5 días a la semana, desde el segundo día tras la IOT de BLM, a concentraciones crecientes (de 5 en 5 µg/ml - µg de O3 por ml de O2 -) desde 20 µg/ml hasta 50 µg/ml. Posteriormente, la concentración se mantuvo constante en 50 µg/m, y en el mismo número de veces/semana hasta el momento del sacrificio. El gas se ha insuflado lentamente para evitar disconfort abdominal en la rata. El proceso suele durar de uno a dos minutos. Posteriormente, se presiona el ano durante 2 minutos después de cada insuflación, para evitar así la salida del gas.
Figura 21. Insuflación rectal de O3 en la rata.

Tesis Doctoral Charlín Méndez Cordovez
46
4.2.4.- Evolución clínica en el periodo postoperatorio.
Tras la IOT, los animales fueron llevados a áreas de postoperatorio de la Unidad de Investigación
del Hospital Universitario de Gran Canaria Dr. Negrín. Allí se les hizo un seguimiento clínico, donde se
recogieron datos de control del dolor (piloerección, dacriorrea, inmovilización, ojos cerrados) y presencia de
signos de depresión.
Hasta los 28 días tras IOT, las ratas estuvieron en sus respectivas jaulas (convencionales
Tipo1000, con rejilla y biberón de 500 ml), donde dispusieron de comida y agua ad libitum, provistas de
confort térmico (mantas calefactoras, lámparas de calor) para prevenir la hipotermia.
4.2.5.- Obtención y recogida de muestras.
El sacrificio de los animales, salvo el grupo Control, se ha realizado a los 28 días de instilarles la
BLM o el suero fisiológico. Para la obtención de muestras, los animales han sido profundamente
anestesiados con medetomidina (0,5 mg/Kg) SC y Ketamina (75 mg/Kg) IP y premedicados con Atropina
(0,04 mg/Kg) im. Una vez anestesiado, y tras comprobar la existencia de un plano profundo de la misma
antes de comenzar el procedimiento (falta de reflejos al pinchar almohadilla plantar, relajación y respiración
regular) (figura 22), se rasura la parte anterior del tórax.
Figura 22.- Esternotomía media. Acceso a bloque cardio-pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
47
Los animales fueron colocados en posición decúbito supino, y luego de ventilados, se les realizó
una traqueostomía, accediendo, de esta forma, a los órganos intratorácicos como pulmón, corazón,
esófago, mediastino, aorta y cava. Para la extracción del bloque cardio-pulmonar, se procedió a la ligadura
y sección de la tráquea con el pulmón en semiinsuflación y mediante tracción, se ha separado del esófago,
con sección de los troncos supraaórticos, aorta, venas cavas y ligamento pulmonar (figura 23) (figura 24).
Una vez extraído el bloque, se separan los pulmones y se extrajeron muestras para el estudio histológico,
genómico y análisis microbiológico.
Figura 23.- Rata en posición de decúbito supino,traqueostomía y ventilación mecánica.
La extracción del bloque cardio-pulmonar, se ha procedido a la ligadura y sección de la tráquea
con el pulmón en semiinsuflación y a la extracción del bloque cardiopulmonar mediante tracción,
separándolo del esófago, con sección de los troncos supraaórticos, aorta, venas cavas y ligamento
pulmonar. Una vez se extrajo el bloque (figura 25), se separaron los pulmones para el estudio histológico
y genómico.
Para el estudio histológico las muestras se depositaron en formaldehído a una concentración del
4%. Para el estudio genómico, las muestras de pulmón se depositaron en PBS fría, cuya composición es
NaCl (8 g/L), KCl (0,2g/L), Na2HPO4 (1,15 g/L), KH2PO4 (0,2 g/L) y glucosa (1g/L), congelándose de forma
inmediata en N2 líquido y posteriormente fueron almacenadas a -80º C hasta su análisis. Para el estudio
microbiológico, las muestras de tejido pulmonar fueron depositadas un bote de boca ancha estéril y se
añadió 2 gotas de suero fisiológico estéril (de modo que el tejido quede humedecido, pero no embebido).
y llevadas inmediatamente al Servicio de Microbiología del Hospital Universitario de Gran Canaria Dr. Negrín
para procesamiento inmediato (figura 24).

Tesis Doctoral Charlín Méndez Cordovez
48
Figura 24.- Esternotomía media.
Figura 25.- Extracción del bloque cardio-pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
49
4.2.6.- Mediciones.
4..2.6.1.- Análisis microbiológico.
Las muestras de biopsia de pulmón se remitieron en un contenedor estéril al laboratorio de
Microbiología para cultivo.
Se introdujo la biopsia en BD Fluid Thioglicollate Medium (Becton Dickinson) el cual se incubó
durante 7 días a 35º C y se visualizó diariamente para detección de turbidez que indicará crecimiento. Si se
detectaba turbidez antes de los 7 días de incubación se realizaba un subcultivo en agar sangre, agar
chocolate y agar McConkey (BBL, BD Diagnostic Systems) que se inocularon durante 48 horas a 35º C en
atmósfera con 5% de CO2 para investigación de bacterias aerobias y en agar Brucella suplementado con
vitamina K1 y hemina (BBL, BD Diagnostic Systems) que se incubó durante 4 días a 35º C en atmósfera
anaerobia para investigación de bacterias anaerobias. Para la identificación y test de susceptibilidad de las
bacterias aisladas se disponía del sistema ViteK2 (BioMérieux).
4.2.6.2.- Análisis histológico.
Tras la extracción del bloque cardiopulmonar, se ha tomado de cada muestra una biopsia
pulmonar.
Estas muestras han sido fijadas en formol tamponado al 3,7 - 4% a temperatura ambiente.
4.2.6.2.1.- Procesamiento de las muestras.
Posteriormente las muestras histológicas fueron sometidas a deshidratación, inclusión y corte de
las secciones.
Deshidratación: Mediante un procesador de tejidos (Leica, TP 1020, Leica Biosystems, Nussloch
Gmbh, Alemania) (figura 26).
Inclusión: Las muestras se incluyeron en parafina por medio de un dispensador de parafina (Leica,
EG 1160) Microsystems, Nussloch Gmbh, Alemania) (figura 27). Así, una vez deshidratadas las muestras
se depositaron en un medio que contenía parafina líquida a 60º C. Posteriormente, se procedió a enfriar la
parafina y se dejó reposar todo el material durante 12 horas.
Transcurrido este tiempo, se fundió la parafina y se extrajeron las muestras para luego
confeccionar los bloques colocados con la orientación correcta en cada una de las muestras. Se han dejado
solidificar los bloques, primero sobre una base fría. Los bloques se identificaron y se guardaron en un

Tesis Doctoral Charlín Méndez Cordovez
50
ambiente seco, oscuro y fresco hasta el momento de su corte sin identificar en los mismos el grupo de
estudio al que pertenece.
Corte de las secciones: Los cortes se efectuaron a 5μm de grosor con un micrótomo de rotación
(Thermoscientific Microm HM 340 E, Walldorf, Alemania) (figura 28). Estos se han dejado calentar en un
calefactor a agua, se han recogido las muestras y se han colocado en los portaobjetos. Se han secado y
dejado enfriar a temperatura ambiente. Posteriormente se han teñido las preparaciones histológicas con
hematoxilina y eosina y tricrómico.
A continuación se procedió al montaje de la lámina cubreobjetos con fosfato dibutil xileno (DPX).
Teñidor automático Citolab 1000.
Figura 26.- TP1020 Figura 27.- Leica EG 1160. Figura 28.- Microtomo de rotación
4.2.6.2.2.- Lectura de las muestras.
Las muestras histológicas del pulmón han sido estudiadas por el Especialista en Anatomía
Patológica, en forma ciega con un microscopio óptico Olympus BX40 (Olympus España S.A., Madrid,
España) (figura 29) y se han clasificado de modo didáctico teniendo en cuenta los fenómenos que
acontecen según la escala de Ashcroft, a los 28 días post IOT de la BLM.
Figura 29. Microscopio óptico Olympus BX40.
Se ha determinado el grado de fibrosis según la Escala Numérica de Ashcroft (tabla 7):

Tesis Doctoral Charlín Méndez Cordovez
51
Tabla 7. Criterios para valorar el grado de fibrosis pulmonar.
GRADO
HALLAZGOS HISTOLÓGICOS
0
Pulmón normal
1
Mínimo engrosamiento de pared sin daño de arquitectura
2 3
Moderado engrosamiento de pared definido de estructura pulmonar
4 5
Fibrosis con daño definido de estructura pulmonar
6 7
Distorsión severa de estructuras y áreas fibróticas.
8
Obliteración fibrosa total.
4.2.6.3. Análisis de la expresión genómica.
Las muestras de pulmón se homogeneizaron siguiendo el protocolo citrato sódico (Sigma Sigma,
Saint Louis, Missouri, USA) y cloroformo (Merck KGaA, Darmstadt, Germany). Posteriormente se ha
realizado la extracción de ARN total. Se ha determinado la concentración de ARN en las muestras mediante
espectrofotometría de absorbancia (NanoDrop 1000 Spectrophotometer V3,7, Thermo Scientific). La
síntesis del cDNA monocatenario se ha realizado con primers de hexanucleótidos al azar (Iscript cADN
Synhesis Kit, Biorad).
4.2.6.3.1.- Extracción de RNA y síntesis de cDNA.
La extracción de RNA se llevó a cabo utilizando el protocolo de Tri- Reagent de extracción de RNA
total. Las muestras conservadas a -80ºC, se homogenizaron con Tri-Reagen® (Sigma, Saint Louis,
Missouri, USA), utilizando un homogenizador Ultra-Turrax T- 10 Basic® (Instrumentación científico técnica,
España) y se centrifuga a 12.000 g, 10’ a 4º C.
La extracción se realizó con cloroformo al 100% (Sigma-aldrich, USA), posteriormente para
eliminar una posible contaminación de proteínas se realizó un lavado con fenol-cloroformo isoamílico
(sigma-aldrich, USA). No se realizó en ningún caso.

Tesis Doctoral Charlín Méndez Cordovez
52
Finalmente, para la purificación y aislamiento del RNA, se agregó citrato sódico/NaCl e isopropanol
para precipitar el RNA, se incubó a -20ºC durante overnight (24h), se centrifugó a 12.000g, 4ºC durante 10
minutos. Después se realizaron dos lavados con etanol al 75%, se eliminó lentamente el sobrenadante y
seco el pellet de RNA, se disolvió en agua DEPC.
Una vez que se obtuvo el RNA, y que se determinó su pureza química por medio del NanoDrop
1000 Spectrophotometer V3.7, Thermo Scientific) en un ratio de 28S/18S.
Para la síntesis de cDNA se ha utilizado el kit iScriptTM cDNA Synthesis de (Bio-Rad®) (REF#170-
8890). Se mezclaron 2µg de RNA total con 4µl, 5x de la mezcla de reacción conteniendo “random primers”
y la transcriptasa reversa en un volumen final de 20µl. la mezcla se mantuvo durante 5 minutos a 25ºC para
permitir el correcto anillamiento de los cebadores y durante 30 minutos a 42ºC para producirse la extensión.
Finalmente las muestras se mantuvieron a 85ºC durante 5 minutos. Disminuyendo la temperatura hasta 4º
de forma indeterminada. Con el propósito de inactivar la transcriptasa reversa para que no interfiera en la
PCR.
4.2.6.3.2.- Análisis de la expresión genómica mediante qRT-PCR.
Se ha utilizado un termociclador de placas de 96 pocillos (C1000 Thermal Cycler, CFX96TM Real
Time System) y se ha seguido el protocolo del fabricante.
Para realizar el diseño de cebadores se ha utilizado software online “Primer3”
(http://frodo.wi.mit.edu/), se ha optimizado la temperatura predicha por el programa informático y la
concentración de MgCl2. La especificidad de los cebadores ha sido visualizada mediante electroforesis en
gel de agarosa, para ello se ha realizado un pool de cada grupo experimental y con cada pareja de primers
de los genes estudiados, se ha hecho una PCR. El fragmento amplificado se ha analizado mediante una
curva de melting que permite la identificación y diferenciación de las variantes genéticas.
Los cebadores específicos que se han empleado y las condiciones de la RTPCR en tiempo real
han sido recogidos en la tabla 8. Los resultados han sido corregidos mediante el gen PPIA (peptidil-prolil
isomerase A, ciclofilina A) como housekeeping gene (114), ya que es el que presenta una expresión más
estable.
Tabla 8. Genes y condiciones de la PCR a tiempo real
GEN
DESCRIPCIÓN
PRIMER FORWARD
PRIMER REVERSE
Nfkβ
Factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas
GTATGGCTTCCCGCACTATGG
TCGTCACTCTTGGCACAATC
HSpb27
Heat shock protein-1 27KDa
GGCACACTCACGGTGGAGGC
GGGAGGGCTGGTGACGGCTA

Tesis Doctoral Charlín Méndez Cordovez
53
TGFβ1
Factor de crecimiento transformante beta 1
TGAGTGGCTGTCTTTTGACG
TGGGAGTGATCCCATTGATT
VEGF-A
Factor de crecimiento endotelial vascular
CCAGGCTGCACCCACGACAG
CGCACACCGCATTAGGGGCA
IL-1
Interleucina 1
GGAGGCCATAGCCCATGATT
TCCTTCAGCAACACAGGCTT
IL-6
Interleucina 6
CATTCTGTCTCGAGCCCACC
GCTGGAAGTCTCTTGCGGAG
Col4
Colágeno de tipo IV
GCCAAGTGTGCATGAGAAGA
AGCGGGGTGTGTTAGTTACG
PPIasa
Peptidil - prolil isomerasa A ( la ciclofilina A (CypA))
AGCACTGGGGAGAAAGGATT
AGCCACTCAGTCTTGGCAGT
4.2.7.- Análisis estadístico.
El análisis estadístico se ha realizado mediante el paquete estadístico SPSS v15,0 para Windows
(2005, SPSS Inc. Illinois, USA). La expresión de los genes la mostramos como media con su desviación
estándar (SD), salvo Vegfa que se ha valorado en base a la mediana con valores mínimo y máximo. Las
determinaciones histológicas se expresan en porcentajes. Para los contrastes de hipótesis hemos
considerado un error α= 0,05. Hemos aplicado el test de la U de Mann Whitney para dos muestras
independientes para verificar las diferencias en el peso y en la expresión de los genes, ya que estas no
siguen una distribución normal (Test de Kolmogorov- Smirnov). Se ha realizado el Test de Chi-Cuadrado
con la razón de verosimilitud (RV) para valorar la asociación de las determinaciones histológicas de los
tratamientos versus los controles.

Tesis Doctoral Charlín Méndez Cordovez
54
V.- RESULTADOS
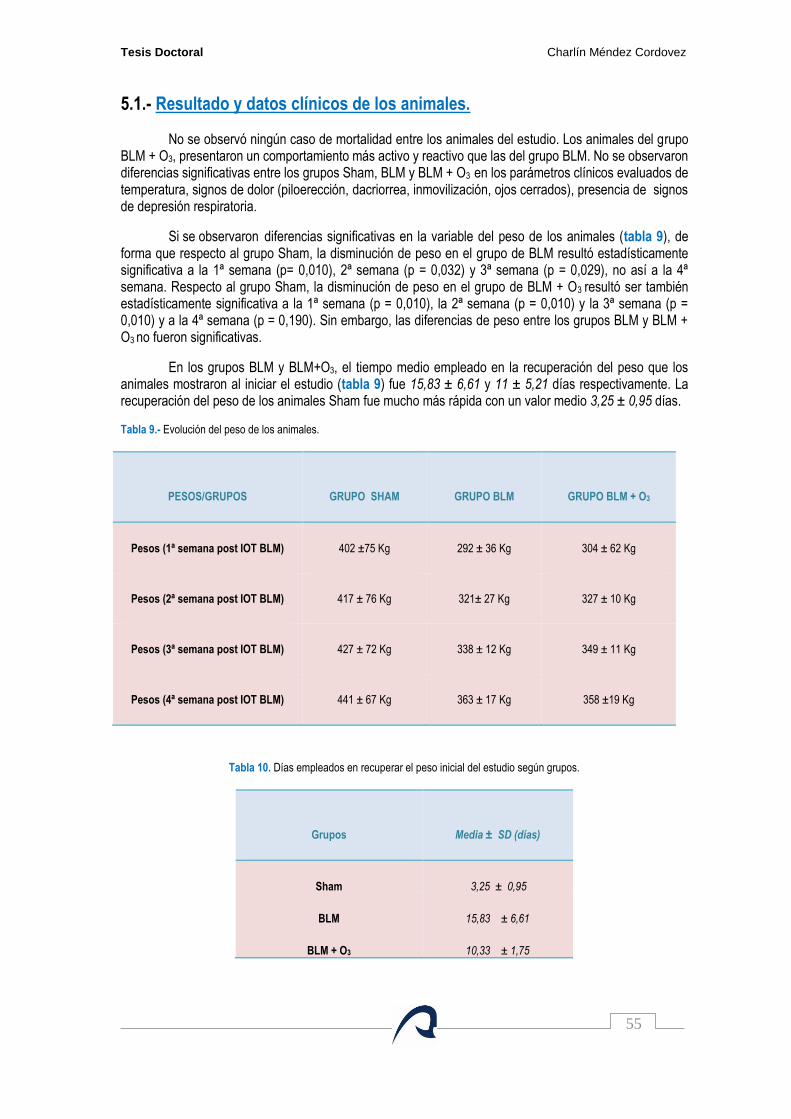
Tesis Doctoral Charlín Méndez Cordovez
55
5.1.- Resultado y datos clínicos de los animales.
No se observó ningún caso de mortalidad entre los animales del estudio. Los animales del grupo BLM + O3, presentaron un comportamiento más activo y reactivo que las del grupo BLM. No se observaron diferencias significativas entre los grupos Sham, BLM y BLM + O3 en los parámetros clínicos evaluados de temperatura, signos de dolor (piloerección, dacriorrea, inmovilización, ojos cerrados), presencia de signos de depresión respiratoria.
Si se observaron diferencias significativas en la variable del peso de los animales (tabla 9), de forma que respecto al grupo Sham, la disminución de peso en el grupo de BLM resultó estadísticamente significativa a la 1ª semana (p= 0,010), 2ª semana (p = 0,032) y 3ª semana (p = 0,029), no así a la 4ª semana. Respecto al grupo Sham, la disminución de peso en el grupo de BLM + O3 resultó ser también estadísticamente significativa a la 1ª semana (p = 0,010), la 2ª semana (p = 0,010) y la 3ª semana (p = 0,010) y a la 4ª semana (p = 0,190). Sin embargo, las diferencias de peso entre los grupos BLM y BLM + O3 no fueron significativas.
En los grupos BLM y BLM+O3, el tiempo medio empleado en la recuperación del peso que los animales mostraron al iniciar el estudio (tabla 9) fue 15,83 ± 6,61 y 11 ± 5,21 días respectivamente. La recuperación del peso de los animales Sham fue mucho más rápida con un valor medio 3,25 ± 0,95 días.
Tabla 9.- Evolución del peso de los animales.
PESOS/GRUPOS
GRUPO SHAM
GRUPO BLM
GRUPO BLM + O3
Pesos (1ª semana post IOT BLM)
402 ±75 Kg
292 ± 36 Kg
304 ± 62 Kg
Pesos (2ª semana post IOT BLM)
417 ± 76 Kg
321± 27 Kg
327 ± 10 Kg
Pesos (3ª semana post IOT BLM)
427 ± 72 Kg
338 ± 12 Kg
349 ± 11 Kg
Pesos (4ª semana post IOT BLM)
441 ± 67 Kg
363 ± 17 Kg
358 ±19 Kg
Tabla 10. Días empleados en recuperar el peso inicial del estudio según grupos.
Grupos
Media ± SD (días)
Sham
3,25 ± 0,95
BLM
15,83 ± 6,61
BLM + O3
10,33 ± 1,75
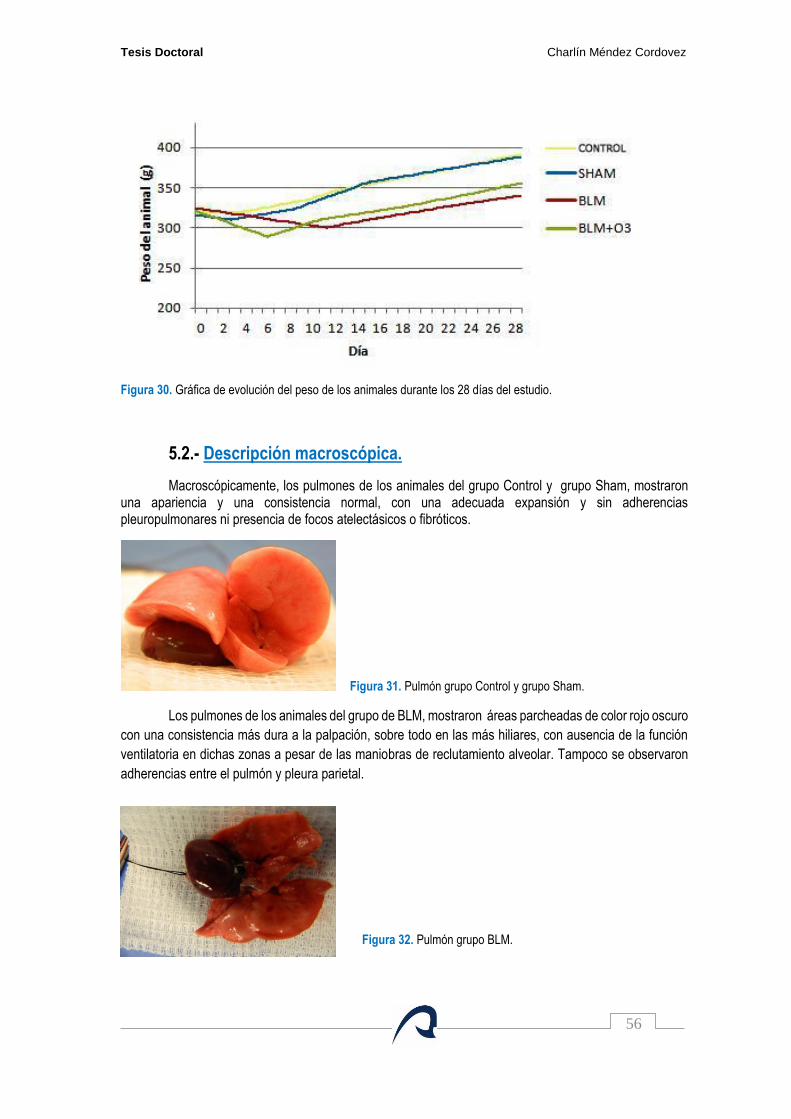
Tesis Doctoral Charlín Méndez Cordovez
56
Figura 30. Gráfica de evolución del peso de los animales durante los 28 días del estudio.
5.2.- Descripción macroscópica.
Macroscópicamente, los pulmones de los animales del grupo Control y grupo Sham, mostraron una apariencia y una consistencia normal, con una adecuada expansión y sin adherencias pleuropulmonares ni presencia de focos atelectásicos o fibróticos.
Figura 31. Pulmón grupo Control y grupo Sham.
Los pulmones de los animales del grupo de BLM, mostraron áreas parcheadas de color rojo oscuro
con una consistencia más dura a la palpación, sobre todo en las más hiliares, con ausencia de la función
ventilatoria en dichas zonas a pesar de las maniobras de reclutamiento alveolar. Tampoco se observaron
adherencias entre el pulmón y pleura parietal.
Figura 32. Pulmón grupo BLM.

Tesis Doctoral Charlín Méndez Cordovez
57
En el grupo de animales de grupo BLM+O3, los pulmones presentaron un aspecto y consistencia
más parecido al pulmón normal. Las lesiones fueron menores en tamaño y cantidad que las observadas en
el grupo BLM y el pulmón presentaba una adecuada expansión en su mayor parte. Tampoco se observaron
adherencias entre el pulmón y pleura parietal.
Figura 33. Pulmón grupo BLM+O3.
5.3.- Estudio histológico. Cuantificación del grado de fibrosis según
escala Ashcroft.
El análisis histológico del grupo Control mostró una arquitectura pulmonar normal. No se
observaron signos de fibrosis, siendo su cuantificación según el método de Ashcroft de 0.
Figura 34. Estudio histológico grupo Control.
En el grupo Sham se observó un leve engrosamiento de las paredes alveolares conservando la
normalidad del resto de las estructuras pulmonares. El valor medio de la cuantificación de la fibrosis según
el método de Ashcroft con respecto al grupo control fue de 3,5 ± 0,28 (p = 0,002).
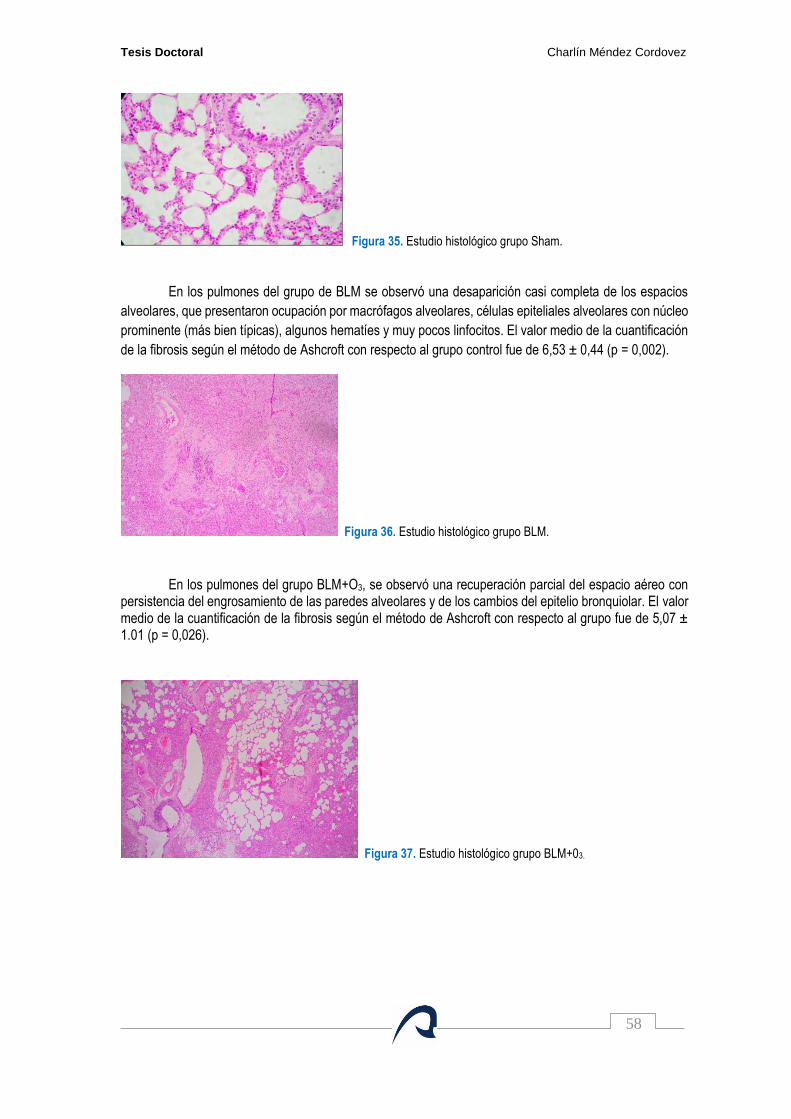
Tesis Doctoral Charlín Méndez Cordovez
58
Figura 35. Estudio histológico grupo Sham.
En los pulmones del grupo de BLM se observó una desaparición casi completa de los espacios
alveolares, que presentaron ocupación por macrófagos alveolares, células epiteliales alveolares con núcleo
prominente (más bien típicas), algunos hematíes y muy pocos linfocitos. El valor medio de la cuantificación
de la fibrosis según el método de Ashcroft con respecto al grupo control fue de 6,53 ± 0,44 (p = 0,002).
Figura 36. Estudio histológico grupo BLM.
En los pulmones del grupo BLM+O3, se observó una recuperación parcial del espacio aéreo con
persistencia del engrosamiento de las paredes alveolares y de los cambios del epitelio bronquiolar. El valor medio de la cuantificación de la fibrosis según el método de Ashcroft con respecto al grupo fue de 5,07 ± 1.01 (p = 0,026).
Figura 37. Estudio histológico grupo BLM+03.

Tesis Doctoral Charlín Méndez Cordovez
59
A continuación se representa gráficamente el valor medio de la cuantificación de la fibrosis según
el método de Ashcroft.
Tabla 11.- Grado de fibrosis pulmonar según la escala de Ashcroft y su significación estadística.
Control
GRUPO SHAM
p
GRUPO
BLM
p*
GRUPO
BLM + O3
p**
0
3,5 ± 0,28
p = 0,002
6,53 ± 0,44
p = 0,002
5,07 ± 1,01
p = 0,026
5.4.- Análisis microbiológico.
El cultivo de las muestras de pulmón de los grupos Control, Sham, BLM y BLM + O3 fue negativo
para bacterias aerobias y anaerobias
5.5.- Análisis de expresión génica.
El análisis de la expresión de RNA mensajero de las muestras de pulmón del grupo Control vs
SHAM, BLM y BLM+O3 mostró los siguientes resultados para cada uno de los genes analizados.

Tesis Doctoral Charlín Méndez Cordovez
60
Tabla 12. Expresión de los genes analizados entre el grupo Control vs Sham.
SHAM
Genes
Controles
SHAM
p
Hsp27
1 ± 0,09
0,94 ± 0,10
0,522
Vegfa
1 ± 0,05
0,98 ± 0,09
0,631
Nfkb
1 ± 0,03
0,97 ± 0,11
0,715
IL6
1 ± 0,22
2,02 ± 0,34
0,006*
Col4
0,99 ± 0,10
1,25 ± 0,19
0,016*
IL1
1 ± 0,14
3,08 ± 0,56
0,006*
Tgfb
1 ± 0,12
0,17 ± 0,03
0,006*
Figura 38. Expresión de los genes analizados entre Control vs SHAM.

Tesis Doctoral Charlín Méndez Cordovez
61
Tabla 13. Expresión de los genes analizados entre el grupo control vs BLM.
BLEOMICINA
Genes
Controles
BLEOMICINA
p
Hsp27
1 ± 0,09
1,24 ± 0,17
0,045*
Vegfa
1 ± 0,05
1 ± 0,53
0,855
Nfkb
1 ± 0,03
1,29 ± 0,12
0,006*
IL6
1 ± 0,22
2´,14 ± 0,47
0,006*
Col4
0,99 ± 0,10
2,21 ± 0,33
0,004*
IL1
1 ± 0,14
1,43 ± 0,26
0,010*
Tgfb
1 ± 0,12
2,54 ± 0,44
0,004*
Los valores se muestran en función de la media ± desviación típica. La significación estadística
entre ambos grupos (p < 0,005) se indica con un *.
Figura 39. Expresión de los genes analizados entre Control vs BLM.

Tesis Doctoral Charlín Méndez Cordovez
62
Tabla 14. Expresión de los genes analizados entre el grupo control vs BLM+O3.
OZONO
Genes
Controles
BLEOMICINA + O3
p
Hsp27
1 ± 0,09
0,97 ± 0,12
0,855
VEGFA
1 ± 0,05
1,06 ± 0,19
0,873
Nfkb
1 ± 0,03
1,07 ± 0,21
0,715
IL6
1 ± 0,22
3,63 ± 0,76
0,006*
Col4
0,99 ± 0,10
2,48 ± 0,59
0,006*
IL1
1 ± 0,14
3,16 ± 0,81
0,006*
Tgfb
1 ± 0,12
3,49 ± 0,78
0,004*
Los valores se muestran en función de la media ± desviación típica. La significación estadística
entre ambos grupos (p < 0,005) se indica con un *.
Figura 40. Expresión de los genes analizados entre Control vs BLM+O3.

Tesis Doctoral Charlín Méndez Cordovez
63
El análisis de la expresión de RNA mensajero de las muestras de pulmón del grupo Sham vs
BLM y BLM + O3 mostró los siguientes resultados para cada uno de los gens analizados.
Tabla 15. Expresión de los genes analizados entre el grupo SHAM vs BLM.
BLEOMICINA
Genes
CONTROLES
SHAM
BLEOMICINA
p
Hsp27
1,06 ± 0,11
1,40 ± 0,20
0,018*
Vegfa
1,03 ± 0,09
1,05 ± 0,05
0,885
Nfkb
1 ± 0,11
1,33 ± 0,13
0,009*
IL6
1,02 ± 0,17
1,08 ± 0,24
0,602
Col4
1,01 ± 0,15
1,78 ± 0,26
0,004*
IL1
1,06 ± 0,19
0,49 ± 0,09
0,006*
Tgfb
1 ± 0,20
14,44 ± 2,50
0,006*
Los valores se muestran en función de la media ± desviación típica. La
significación estadística entre ambos grupos (p < 0,005) se indica con un *.

Tesis Doctoral Charlín Méndez Cordovez
64
Tabla 16. Expresión de los genes analizados entre el grupo Sham vs BLM+O3.
OZONO
Genes
Controles
SHAM
BLEOMICINA + O3
p
HSP27
1,06 ± 0,11
1,10 ± 0,14
0,465
VEGFA
1,03 ± 0,09
1,12 ± 0,20
0,631
NFKB
1 ± 0,11
1,11 ± 0,22
0,465
IL6
1,02 ± 0,17
1,84 ± 0,38
0,009*
Col4
1,01 ± 0,15
2 ± 0,48
0,006*
IL1
1,06 ± 0,19
1,09 ± 0,28
0,917
TGFb
1 ± 0,20
19,81 ± 4,45
0,006*
Los valores se muestran en función de la desviación típica. La significación estadística entre ambos
grupos (p < 0,005) se indica con un *.
- Figura 41. Expresión de los genes analizados Sham vs BLM+O3.

Tesis Doctoral Charlín Méndez Cordovez
65
Tabla 17. Expresión de los genes analizados entre el grupo BLM y BLM + O3.
OZONO
Genes
Controles
BLEOMICINA
BLEOMICINA + O3
p
HSP27
0,96 ± 0,09
0,75 ± 0,09
0,047*
VEGFA
1,03 ± 0,05
1,09 ± 0,20
0,855
NFKB
0,96 ± 0,09
0,80 ± 0,16
0,117
IL6
1,12 ± 0,25
1,90 ± 0.39
0,016*
Col4
0,91 ± 0,13
1,02 ± 0,24
0,584
IL1
0,92 ± 0,17
2,03 ± 0,52
0,006
TGFb
0,97 ± 0,16
1,33 ± 0,30
0,025
Los valores se muestran en función de la media ± desviación típica. La significación estadística
entre ambos grupos (p < 0,005) se indica con un *.
- Figura 42. Expresión de los genes analizados BLM vs BLM+O3.
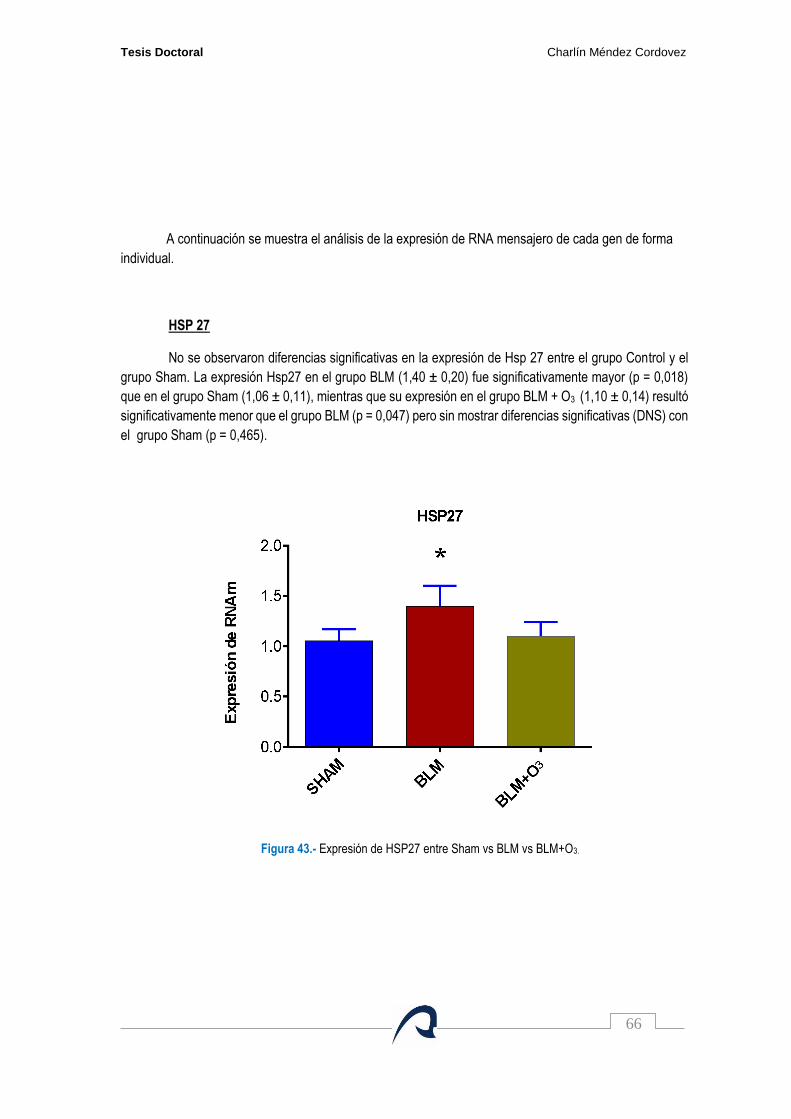
Tesis Doctoral Charlín Méndez Cordovez
66
A continuación se muestra el análisis de la expresión de RNA mensajero de cada gen de forma
individual.
HSP 27
No se observaron diferencias significativas en la expresión de Hsp 27 entre el grupo Control y el
grupo Sham. La expresión Hsp27 en el grupo BLM (1,40 ± 0,20) fue significativamente mayor (p = 0,018)
que en el grupo Sham (1,06 ± 0,11), mientras que su expresión en el grupo BLM + O3 (1,10 ± 0,14) resultó
significativamente menor que el grupo BLM (p = 0,047) pero sin mostrar diferencias significativas (DNS) con
el grupo Sham (p = 0,465).
Figura 43.- Expresión de HSP27 entre Sham vs BLM vs BLM+O3.

Tesis Doctoral Charlín Méndez Cordovez
67
Vegfa
No se observaron diferencias significativas en la expresión de Vegfa entre el grupo Control y el grupo Sham. No hubo diferencias significativas de la expresión de Vegfa entre los distintos grupos: BLM (1,05 ± 0,05), Sham (1,03 ± 0,09) y BLM + O3 (1,12 ± 0,20).
Nfkb
No se observaron diferencias significativas en la expresión de Nfkb entre el grupo Control y el grupo Sham. La expresión de Nfkb en el grupo BLM (1,33 + 0,13) fue significativamente mayor (p = 0,009) que en el grupo Sham (1 + 0,11). La expresión en el grupo BLM + O3 (1,11 + 0,22) mostró DNS con el grupo BLM y con el grupo Sham.
IL6
Figura 44.- Expresión de Vegfa entre Sham vs BLM vs BLM+O3.
Figura 45.- Expresión de Nfkb entre Sham vs BLM vs BLM+O3.
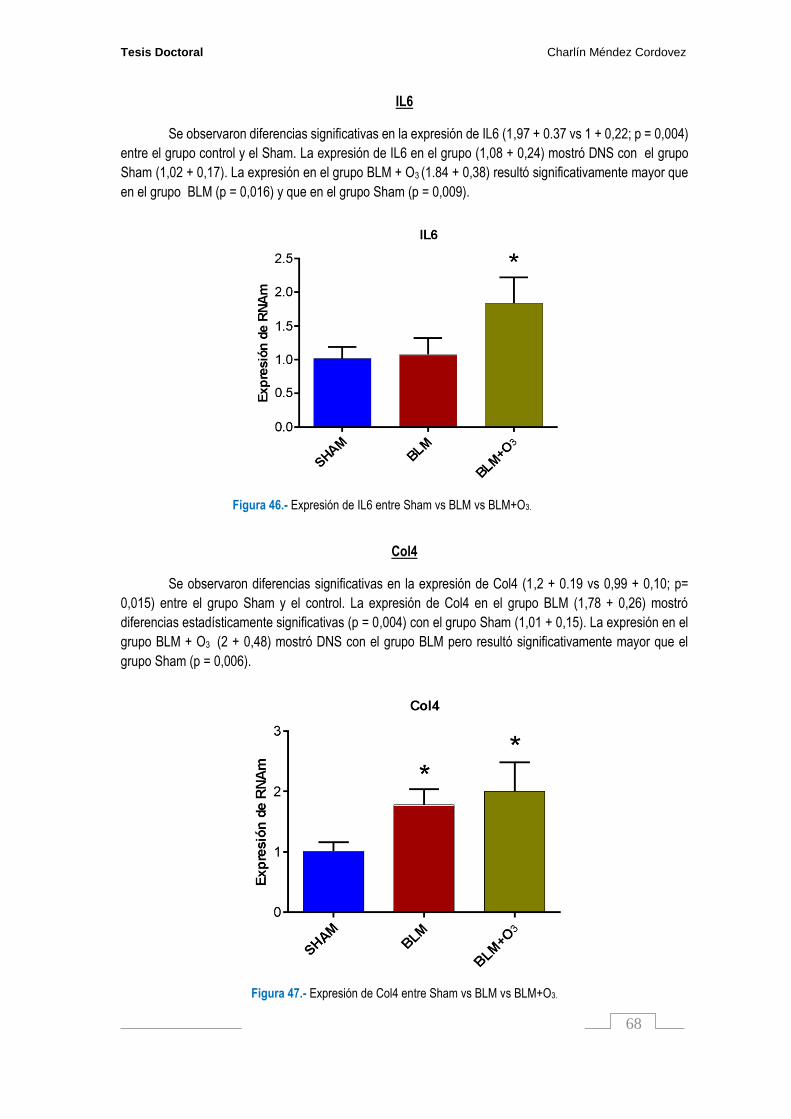
Tesis Doctoral Charlín Méndez Cordovez
68
IL6
Se observaron diferencias significativas en la expresión de IL6 (1,97 + 0.37 vs 1 + 0,22; p = 0,004)
entre el grupo control y el Sham. La expresión de IL6 en el grupo (1,08 + 0,24) mostró DNS con el grupo
Sham (1,02 + 0,17). La expresión en el grupo BLM + O3 (1.84 + 0,38) resultó significativamente mayor que
en el grupo BLM (p = 0,016) y que en el grupo Sham (p = 0,009).
Col4
Se observaron diferencias significativas en la expresión de Col4 (1,2 + 0.19 vs 0,99 + 0,10; p=
0,015) entre el grupo Sham y el control. La expresión de Col4 en el grupo BLM (1,78 + 0,26) mostró
diferencias estadísticamente significativas (p = 0,004) con el grupo Sham (1,01 + 0,15). La expresión en el
grupo BLM + O3 (2 + 0,48) mostró DNS con el grupo BLM pero resultó significativamente mayor que el
grupo Sham (p = 0,006).
Figura 46.- Expresión de IL6 entre Sham vs BLM vs BLM+O3.
Figura 47.- Expresión de Col4 entre Sham vs BLM vs BLM+O3.

Tesis Doctoral Charlín Méndez Cordovez
69
IL1
Se observaron diferencias significativas en la expresión de IL1 (2,88 + 0,40 vs 1+ 0,14; p= 0,004)
entre el grupo Sham y el grupo control. La expresión de IL1 en el grupo BLM (0,49 + 0,09) fue
significativamente menor (p = 0,006) que en el grupo Sham (1,06 + 0.19). La expresión en el grupo BLM +
O3 (1,09 + 0,28) resultó significativamente mayor que en el grupo BLM (p = 0,006) pero con diferencias no
significativas (DNS) con el grupo Sham.
Tgfb
Se observaron diferencias significativas en la expresión de Tgfb (0,17 + 0,04 vs 1 + 0,12; p = 0,004)
entre el grupo Sham y el Control. La expresión de Tgfb en el grupo BLM (14,44 + 2,50) fue significativamente
mayor (p = 0,006) que en el grupo Sham (1+ 0,20). La expresión en el grupo BLM + O3 (19,81+ 4,45) resultó
significativamente mayor que en el grupo BLM (p = 0,025) y que el grupo Sham (p = 0,006).
Figura 48.- Expresión de IL1 entre Sham vs BLM vs BLM+O3.
Figura 49.- Expresión de Tgfb entre Sham vs BLM vs BLM+O3.

Tesis Doctoral Charlín Méndez Cordovez
70
VI.- DISCUSIÓN

Tesis Doctoral Charlín Méndez Cordovez
71
La FPI es una enfermedad pulmonar progresiva, que afecta de forma importante a la cantidad y a
la calidad de vida de los pacientes considerándose un problema de “homeostasis tisular alterada” y
“comunicación aberrante” entre distintas células del pulmón (26). Existen limitadas opciones terapéuticas
en la actualidad, sin que se haya descrito un tratamiento médico eficaz que pueda frenar o cambiar el curso
evolutivo de la misma y evitar su fatal progresión. El pobre pronóstico de los pacientes con FPI y las escasas
opciones terapéuticas demandan un esfuerzo mayor en el campo de la investigación traslacional”.
Este trabajo recoge varias de las recomendaciones para investigación en FPI recientemente
propuestas en un workshop de National Heart Lung and Blood Institute – NHLBI (115) como son: 1) el
estudio de nuevos tratamientos durante las fases de fibrogénesis y de inicio de la fase inflamatoria, 2) el
estudio de los cambios en la matriz extracelular y la histología de la arquitectura pulmonar, 3) caracterizar
y explotar vías endógenas que inhiban la inflamación y la fibrosis y 4) realizar estudios génicos para
identificar nuevas dianas que influencien el desarrollo de la FPI.
En este sentido, los modelos experimentales de enfermedad son una gran herramienta de trabajo
para la investigación traslacional siempre que se realicen bajo la estricta regulación de la normativas
internacionales de uso de animales de experimentación y con el máximo cumplimiento de las conocidas 3
R, descrita por los biólogos ingleses, Russel y Burch, en su libro “The Principle of Humane Experimental
Technique” en la década de los 60 y que hacen referencia a los métodos de reemplazo, reducción y
refinamiento. Las alternativas de reemplazo aluden a métodos que eviten o sustituyan el uso de animales
que incluyen tanto los reemplazos absolutos (es decir, sustituir animales por modelos informáticos), como
los reemplazos relativos (es decir, sustituir vertebrados, por animales con una menor percepción del dolor,
como algunos invertebrados). Las alternativas de reducción aluden a cualquier estrategia que tenga como
resultado el uso de un menor número de animales para obtener datos suficientes que respondan a la
cuestión investigada, o la maximización de la información obtenida por animal, para así limitar o evitar
potencialmente el uso posterior de otros animales, sin comprometer el bienestar animal. Las alternativas
de refinamiento aluden a la modificación de la cría de animales o de los procedimientos para minimizar el
dolor y la angustia, así como para mejorar el bienestar de los animales utilizados en la ciencia desde su
nacimiento hasta su muerte. Así, siguiendo el segundo principio “alternativas de reducción”, en el grupo
control en vez de emplear 6 pulmones de 6 animales distintos, decidimos analizar por separado pulmones
derechos e izquierdos de 3 animales (6 pulmones igualmente) minimizando el número de animales. Por el
mismo motivo, aunque la mayoría de las publicaciones con modelos experimentales de ozonoterapia
incluyen un grupo control con oxígeno sin ozono, nosotros decidimos no incluirlo. En todos esos estudios,
el grupo de oxígeno muestra menor generación inicial de radicales libres y, por consiguiente, una ausente
(ocasionalmente mínima) inducción de los sistemas antioxidantes y una nula acción protectora en los muy
variados modelos estudiados.
El modelo de desarrollo de fibrosis pulmonar en ratas mediante la instilación orotratraqueal (IOT)
de bleomicina utilizado por nuestro grupo, ha demostrado ser un buen modelo experimental dada la: 1)
aparición de lesiones pulmonares similares a las observadas en los enfermos con FPI en un grado
significativamente mayor que el observado en el grupo SHAM, 2) pérdida de peso de los animales en
relación con la instauración de la enfermedad, 3) ausencia de mortalidad entre los animales de los grupos
tratados y 4) expresión significativa de los genes relacionados con la fibrosis pulmonar como son Hsp27,
Nfkb, IL6, IL1, Tgfb y Col4. Además, dicho modelo cumple con las 3 R descritas en cuanto que no existen
alternativas de reemplazo y que se han extremado las alternativas de reducción del número de animales y

Tesis Doctoral Charlín Méndez Cordovez
72
las técnicas de refinamiento durante la realización de los procedimientos y durante el sacrificio de los
animales. Por todo ello, podemos dar por cumplido el primer objetivo de nuestro trabajo de “desarrollar un
modelo experimental válido en ratas de fibrosis pulmonar inducida con bleomicina”. En este sentido,
a pesar de ser un buen modelo experimental pensamos que, desde un punto de vista traslacional, el modelo
puede ser mejorable a la hora de mantener de forma crónica el proceso de fibrosis pulmonar para que se
asemeje más a lo que ocurre en la FPI en humanos, de forma que permita ensayar diferentes alternativas
terapéuticas de forma más prolongada, modelo en el que ya estamos trabajando.
Por otra parte, se ha demostrado la capacidad de la ozonoterapia por vía sistémica (96-100) de
modular el sistema inmunológico, el equilibrio pro/antioxidante y los procesos de inflamación y reparación
del organismo, con un efecto general de “regulación” que tiende a la recuperación de la homeostasis del
individuo cuando se utiliza de forma progresiva y mantenida. Se sabe que los procesos de óxido-reducción
intracelular y las defensas frente a los mismos están presentes y regulados genéticamente en la fibrosis
pulmonar, y que se inician desde las primeras fases de la lesión. De esta forma, en el pulmón con lesiones
fibróticas se produce una situación de estrés oxidativo celular crónico que es responsable del daño tisular
irreversible.
La administración de forma progresiva y mantenida, de un potente inductor de los sistemas
antioxidantes como el O3, no utilizado antes en modelos de fibrosis pulmonar, ha paliado de forma
significativa en nuestro modelo esta situación de estrés celular y el daño pulmonar subsecuente asociado a
la fibrosis pulmonar. La disminución significativa de las lesiones pulmonares y del grado de fibrosis en el
grupo tratado con O3 sugiere que la ozonoterapia es capaz de modular los procesos de estrés oxidativo que
conducen a la aparición de la fibrosis pulmonar. Estos hechos adquieren un valor adicional teniendo en
cuenta que: 1) el análisis histológico se realizó de forma ciega respecto al grupo de procedencia de las
muestra y 2) los resultados negativos del análisis microbiológico en todas las muestras descarta que la
gradación del proceso de fibrosis observado entre los distintos grupos pudiera haberse debido a infecciones
intercurrentes. En este sentido, damos también por cumplido el segundo objetivo de nuestro trabajo de
“valorar si el tratamiento con ozono disminuye el daño pulmonar en un modelo experimental de
fibrosis pulmonar inducido por bleomicina”.
La presencia de cambios histológicos en el grupo SHAM con un leve engrosamiento de las paredes
alveolares y un grado de fibrosis pulmonar significativamente superior al de los animales controles, así como
la expresión significativa de los genes relacionados con la fibrosis, creemos que tiene relación con la
administración del suero fisiológico a través de la tráquea o con la propia ventilación mecánica. No es el
objeto de estudio de esta tesis doctoral, pero desde luego abre un interesante camino de estudio en este
campo.
Los resultados observados en los cambios de expresión génica, con aumento en el grupo de BLM
y significativa reducción en el grupo de BLM+O3 de la expresión de los genes descritos en la FPI, nos
permiten dar por cumplido también el tercer objetivo de nuestro trabajo de “estudiar si la expresión de los
genes analizados se relaciona con el daño pulmonar y su respuesta al tratamiento”.
En este sentido, Hsp27 es una pequeña proteína miembro de la familia de los Heat Shock Protein
capaz de formar largas cadenas que actúan como chaperonas involucradas en el plegado y la re-
naturalización de las proteínas y en la degradación de aquellas que se han desnaturalizado de manera

Tesis Doctoral Charlín Méndez Cordovez
73
irreversible. Entre sus acciones más destacadas se encuentran: 1) su capacidad para modular las ROS y
estimular la expresión de GPX actuando entonces como un citoprotector, 2) inhibir la apoptosis y proteger
a los filamentos de actina de la fragmentación, así como, 3) tener un papel esencial en la diferenciación de
los tejidos y en la termotolerancia y en la citoprotección bajo condiciones de estrés y oxidación. Sin embargo,
a pesar de sus propiedades protectoras Wettstein G et al. (116) describieron que esta proteína está
directamente relacionada con la FPI y que su inhibición bloquea el desarrollo en la fibrosis pulmonar. De
hecho, otros autores como Vidyasagar (117), la señalan como un biomarcador de enfermedad y una
potencial diana terapéutica. Una posible explicación a todos estos hallazgos podría ser la existencia de un
efecto diferencial de Hsp27, con mayor acción o sobre expresión en fibroblastos y miofibroblastos
(disminuyendo su apoptosis y favoreciendo su perpetuación más allá de acabado el proceso de reparación)
y con un menor efecto o sobreexpresión sobre las CEA (y por tanto con menor acción protectora sobre las
mismas). En nuestro estudio, hemos podido observar que la Hsp27 se expresa de forma significativa en el
grupo de BLM y que el tratamiento con O3 disminuye su expresión hasta niveles normales lo que indica que:
o bien esta proteína efectivamente está directamente implicada en el desarrollo de la FP y que el efecto
protector del O3 reduce su expresión contribuyendo a la disminución del grado de fibrosis pulmonar, o bien
Hsp27 se expresa ante el daño pulmonar inducido por fibrosis en un intento por proteger a las células del
daño oxidativo, de forma que su expresión se disminuiría con el O3 al disminuir éste el estrés oxidativo y la
lesión pulmonar. Esta segunda hipótesis, se ve apoyada por dos trabajos de nuestro grupo de investigación.
El primero corresponde a un estudio con DNA microarrays del rechazo crónico (RC) en el trasplante
pulmonar (118) en el que se concluyó que Hsp27 protege el citoesqueleto del RC e inhibe la apoptosis. El
segundo estudio, sobre los efectos del O3 sobre el rechazo crónico del trasplante pulmonar, fue financiado
por una beca del ISCIII (PI 10/01485) y motivo de una Tesis Doctoral Europea cuyos resultados están
pendientes de publicación; en él se observó que en los animales tratados con O3 la expresión de Hsp27
estaba también disminuida, como muestra del efecto antioxidante con carácter reparador sobre la
sobreexpresión de estas proteínas. Por estos motivos, pensamos que para averiguar cuál es el papel real
de Hsp27 en la FPI se requiere de más estudios, en los que nuestro grupo está en la actualidad trabajando.
Nfkb es un factor de transcripción nuclear con importante actividad proinflamatoria que, una vez
activado, se traslada al núcleo donde estimula la expresión de genes implicados en una amplia variedad de
funciones biológicas, incluida la síntesis de citocinas proinflamatorias como IFNg, TNFα e IL-8. Nfkb se
activa por numerosos estímulos intracelulares y extracelulares como son las citocinas, RLO, radiación
ultravioleta y productos bacterianos o víricos. Su activación se ha asociado con numerosas enfermedades
inflamatorias y se ha podido comprobar que existe un incremento de su activación en la fibrosis pulmonar
(119). Como se comentó en el apartado 3.3, el O3 (a través de segundos mensajeros) es capaz de activar
Nrf2, que a su vez induce la transcripción de elementos de respuesta AO con la posterior producción de
enzimas antioxidantes como SOD, GPX, HSP-70 o hemo oxigenasa-1 (HO-1 o HSP-32) (98-100). Por otra
parte, se ha descrito que la activación de Nrf2 está implicada en la reparación del DNA (120) y es capaz de
disminuir los efectos proinflamatorios de Nfkb (121). Así, el aumento de Nrf2 durante la ozonoterapia podría
justificar su efecto positivo sobre la FPI. En nuestro estudio observamos un incremento significativo de la
expresión de Nfkb en el grupo tratado con BLM respecto al grupo Sham, demostrando su relación
proinflamatoria en la fibrosis pulmonar. A su vez, el grupo tratado con BLM+O3 mostró menor expresión de
Nfkb. Y si bien esta disminución no fue estadísticamente significativa respecto al grupo de BLM, sí fue lo
suficiente como para que tampoco mostrara diferencias significativas con el grupo Sham. Esto evidencia
que el tratamiento con O3 ejerció una clara tendencia a la regulación (en este caso disminución) de la
sobreexpresión de Nfkb.

Tesis Doctoral Charlín Méndez Cordovez
74
El pulmón fibrótico se asemeja marcadamente a un órgano "supercicatrizado", sugiriendo que la
fibrosis podría visualizarse como un proceso de reparación "excesivo" probablemente como resultado de la
inflamación continuada. De hecho, un número considerable de estudios ha examinado la interacción entre
células inflamatorias y células del parénquima pulmonar, principalmente fibroblastos, ilustrando la
relevancia de estas interacciones en la patogénesis de la fibrosis pulmonar. Así, el incremento en el depósito
de fibras colágenas en el intersticio pulmonar que caracteriza a esta enfermedad, ocurriría como resultado
de la activación de los fibroblastos por moléculas liberadas por varias células inflamatorias (citoquinas),
particularmente por macrófagos alveolares y linfocitos.
En este sentido, la IL-6 es una glicoproteína de pequeño tamaño (21 KDa), una citoquina producida
por las células del sistema inmunológico innato como potente inductor de la fase de respuesta aguda. Su
acción proinflamatoria y de su acción profibrótica han sido descritas en modelos experimentales de fibrosis
pulmonar inducida con BLM (122). La IL-6 ha sido también conocida como interferon-R2 (IFN-R2) o B-cell
stimulating factor-2 (BSF-2) y es idéntica al factor estimulante de hepatocitos (HSF), lo que prueba su
capacidad para estimular los hepatocitos y producir proteínas de la fase aguda, como el fibrinógeno y la
alfa-1-antitripsina entre otros (123).
La IL-1 otra citoquina producida por los macrófagos activados, posee numerosas actividades
biológicas, como son la proliferación de linfocitos T a través de la liberación de IL-2, la proliferación y
diferenciación de los linfocitos B, actuar como factor de crecimiento de los fibroblastos, como pirógeno
endógeno, y liberar prostaglandinas y colagenasas. Se ha descrito que los macrófagos alveolares producen
IL-6 e IL-1 y los fibroblastos pulmonares producen IL-6. A su vez, la producción de IL-1 por los monocitos
regula la expresión de IL6 por los fibroblastos pulmonares, de forma que ambas están íntimamente
entrelazadas (124).
Se ha descrito que Tgfb-1, además, es capaz de estimular directamente a los fibroblastos y
miofibroblastos e inducir fibrosis pulmonar (125) por lo que sus actividades están íntimamente relacionadas
con la generación de enfermedad obstructiva pulmonar e hipertensión pulmonar. Es interesante destacar
que, en un gran número de células, el Tgfb-1 es capaz de provocar una respuesta de estrés oxidativo la
cual parece estar asociada con la actividad del factor de transcripción TIEG (Tgfb-1-inducible early-response
gene) (126).
El gen Col4a1 se localiza en el brazo largo del cromosoma 16 (16q12.5) y codifica una proteína de
la membrana basal que actúa como constituyente estructural de la matriz extracelular, el colágeno tipo IV.
Dicho colágeno se encuentra implicado en la diferenciación de las células epiteliales, la organización de la
membrana basal, la morfogénesis de los vasos sanguíneos y sus alteraciones se han asociado con múltiples
enfermedades: angiopatía hereditaria, nefropatía, aneurismas y calambres musculares, sistema nervioso
central con vasos de pequeño calibre y con hemorragia. Las ratas que recibieron un tratamiento previo con
un inhibidor específico para la enzima inducible que produce óxido nítrico (iNOS) mostraron menores niveles
de expresión de colágeno Tipo IV. Por otro lado, la señalización generada por el óxido nítrico potencia la
expresión de Tgfb1, TIMP-1 y HSP47 en fibroblastos pulmonares que juegan un papel importante en la
fibrosis pulmonar.
Conviene puntualizar que la capacidad del organismo para articular una reacción inflamatoria en
respuesta a agresiones tisulares es esencial para la supervivencia del organismo. De modo que para
comprender la patogénesis de la fibrosis pulmonar es fundamental entender las causas que alteran una
reacción, esencialmente homeostática, como es la inflamación. De hecho, la FPI es la expresión de la

Tesis Doctoral Charlín Méndez Cordovez
75
enfermedad como la continuación de los procesos fisiológicos más allá de los límites requeridos para una
función homeostática. Tan pronto como el pulmón es dañado, se inician mecanismos de reparación, donde
las mismas citoquinas y factores de crecimiento juegan un papel central en la orquestación de estos
mecanismos de inflamación-reparación, en constante tendencia al desequilibrio para producir o bien
enfermedad o bien reparación.
Es probable que el incremento en la expresión de IL-1, IL-6, Tgfb-1 y Col4 observado en los
animales tratados con O3 esté en relación con esa delicada línea de equilibrio entre inflamación y reparación
que hacen que el órgano dañado se recupere o muera; es decir, que el incremento en la expresión de estos
genes junto a la significativa disminución del grado de fibrosis y de lesión pulmonar inducido por el O3, no
descrito con anterioridad en un modelo de fibrosis, pueda tener relación con el desequilibrio que se inclina
hacia el efecto reparador del tejido, por varios caminos como podrían ser, 1) la inducción de apoptosis de
las células lesionadas a través del aumento de expresión de citoquinas, la activación de macrófagos para
la fagocitosis de los restos celulares, 3) la estimulación de la división y crecimiento celular y 4) la producción
de nuevo colágeno en aras de regenerar el tejido dañado. Este sistema de interacción entre citoquinas y
factores de crecimiento entre c inflamatorias y células estructurales es extraordinariamente complejo y,
hasta hoy, sólo se ha descifrado una mínima parte de su modus operandi. Es aparente que la instrucción
final que una célula recibe depende de cómo es articulado el mensaje en cuanto al número de citoquinas
participantes, las interacciones entre ellas, su secuencia de acción y modo de presentación.
La evolución del ser humano ha conllevado un precio biológico considerable. De forma que cuando
un ser de estirpe filogenética inferior pierde una extremidad, como el caso de la rana o del lagarto ésta se
regenera en su totalidad. Los humanos, como otros animales de estirpe filogenética superior, han perdido,
o reprimido, el aparato genético requerido para la regeneración de órganos; poseen, en su lugar, sólo
mecanismos de reparación. Estos mecanismos son eficaces frente a agresiones tisulares moderadas. Sin
embargo, son probablemente inadecuados en situaciones de agresión prolongada asociadas a considerable
destrucción tisular. Tal y como sugirió Shmuel Shoshan, la fibrosis podría ser no más que la segunda mejor
solución, y la única posible, en un organismo o microambiente determinado, al problema creado por la
disrupción de la integridad tisular generado por la fibrosis pulmonar, por cuanto la alternativa a este es la
muerte del individuo.
Finalmente, en nuestro estudio, el tratamiento con O3 ha disminuido de forma significativa el grado
de fibrosis y de lesión pulmonar inducido por la BLM debido a la inducción en el organismo de un efecto
antiinflamatorio, antioxidante y probablemente inmunosupresor a nivel local. Estos resultados abren una
nueva alternativa de tratamiento y/o prevención de la fibrosis pulmonar. La expresión de los genes
estudiados Hsp21, Nfkb, IL-1, IL-6, Col4 y Tgfb1 podrían servir como marcadores de respuesta al
tratamiento con O3, o como potenciales dianas para su modulación terapéutica. Dado que administrado en
concentración y vía apropiada el O3 no produce efectos adversos, es económico y fácil de administrar,
desde el punto de vista traslacional supone una nueva opción potencialmente aplicable en esta patología
donde los tratamientos no quirúrgicos son casi inexistentes hasta la fecha. Los futuros estudios
experimentales y los ensayos clínicos que devengan nos ayudarán a confirmar esta singular aplicabilidad
terapéutica del O3 en la fibrosis pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
76
VII.- CONCLUSIONES.

Tesis Doctoral Charlín Méndez Cordovez
77
Primera.- El modelo de desarrollo de fibrosis pulmonar en ratas mediante la instilación orotratraqueal de bleomicina utilizado por nuestro grupo, ha demostrado ser un buen modelo experimental de estudio de la enfermedad, dada: 1) la aparición de lesiones pulmonares similares a las observadas en los enfermos con FPI en un grado significativamente mayor que el observado en el grupo SHAM, 2) la pérdida de peso de los animales en relación con la instauración de la enfermedad, 3) la ausencia de mortalidad entre los animales de los grupos tratados y 4) la expresión significativa de los genes relacionados con la fibrosis pulmonar como son Hsp27, Nfkb, IL6, IL1, Tgfb y Col4 en el grupo de animales tratados con BLM.
Segunda.- La administración de ozonoterapia de forma progresiva y mantenida, no utilizada antes
en modelos experimentales de fibrosis pulmonar, ha disminuido de forma significativa
el grado de fibrosis pulmonar y el daño pulmonar subsecuente asociado a la fibrosis
pulmonar.
Tecera.- Estos hechos, adquieren un valor adicional teniendo en cuenta que: 1) el análisis
histológico se realizó de forma ciega respecto al grupo de procedencia de las muestra
y 2) los resultados negativos del análisis microbiológico en todas las muestras descarta
que la gradación del proceso de fibrosis pulmonar observado entre los distintos grupos
pudiera haberse debido a infecciones intercurrentes.
Cuarta.- La administración de O3, de forma progresiva y mantenida, ha disminuido de forma
significativa la expresión de Hsp27, ha reducido la sobreexpresión de Nfkb hasta
niveles cercanos a los del grupo Sham y ha incrementado significativamente la
expresión de IL6, IL1, Col4 y Tgfb1.
Quinta.- Hsp27 se expresa de forma significativa en el grupo de BLM y el tratamiento con O3
disminuye su expresión hasta niveles normales lo que indica que o bien este gen está
directamente implicado en el desarrollo de la FP y que el efecto protector del O3 reduce
su expresión contribuyendo a la disminución del grado de fibrosis pulmonar, o bien, que
la Hsp27 se expresa ante el daño pulmonar inducido por la fibrosis en un intento de
proteger a las células del daño oxidativo, de forma que su expresión se disminuiría con
el O3 al disminuir éste el estrés oxidativo y la lesión pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
78
Sexta.- En nuestro estudio observamos un incremento significativo de la expresión de Nfkb en
el grupo tratado con BLM, demostrando su relación proinflamatoria en la fibrosis
pulmonar. El grupo tratado con BLM+O3 mostró menor sobreexpresión de Nfkb y, si
bien las diferencias con el grupo de BLM no fueron significativas, tampoco lo fueron con
el grupo Sham, evidenciando una clara tendencia a la normalización de la expresión de
Nfkb
Séptima.- Es probable que el incremento en la expresión de IL-1, IL-6, Tgfb-1 y Col4 observado
en los animales tratados con O3 esté en relación con esa delicada línea de equilibrio
entre inflamación y reparación que hacen que el órgano dañado se recupere o muera.
Es decir, que el incremento en la expresión de estos genes junto a la significativa
disminución del grado de fibrosis y de lesión pulmonar inducida por el O3 (no descrito
con anterioridad en un modelo de fibrosis) pudiera tener relación con un desequilibrio
que se inclina más hacia el efecto reparador del tejido, por varios caminos como podrían
ser: 1) la inducción de apoptosis de las células lesionadas, o de los fibrocitos
excedentes, a través del aumento de expresión de citoquinas, 2) la activación de
macrófagos para la fagocitosis de los restos celulares, 3) la estimulación de la división
y crecimiento de las células epiteliales alveolares y 4) la producción de nuevo colágeno
en aras de regenerar el tejido dañado.
Octava.- La expresión de los genes estudiados Hsp21, Nfkb, IL-1, IL-6, Col4 y Tgfb-1 podría
servir como marcador tanto de la evolución de la fibrosis pulmonar, como de la
respuesta al tratamiento con O3.
Novena.- Dado que administrado en concentración y vía apropiada el O3 no produce efectos
adversos, es económico y fácil de administrar, desde el punto de vista traslacional
supone una nueva opción potencialmente aplicable en la fibrosis pulmonar, donde los
tratamientos no quirúrgicos son casi inexistentes y, hasta la fecha, sólo ofrecen una
mejoría limitada en cuantía y duración. Los futuros estudios experimentales y los
ensayos clínicos que devengan nos ayudarán a confirmar esta singular aplicabilidad
terapéutica del O3 en la fibrosis pulmonar.

Tesis Doctoral Charlín Méndez Cordovez
79
VIII. BIBLIOGRAFIA

Tesis Doctoral Charlín Méndez Cordovez
80
1. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An
official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based
guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011 Mar
15;183(6):788-824.
2. King TE, Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. Dec
3;378(9807):1949-61.
3. Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis.
2008;3:8.
4. Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE,
Bartholmai BJ, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary
fibrosis: a population-based study. Chest. Jan;137(1):129-37.
5. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of
idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012 Dec
1;21(126):355-61.
6. Thomeer M, Demedts M, Vandeurzen K. Registration of interstitial lung diseases
by 20 centres of respiratory medicine in Flanders. Acta Clin Belg. 2001 May-
Jun;56(3):163-72.
7. von Plessen C, Grinde O, Gulsvik A. Incidence and prevalence of cryptogenic
fibrosing alveolitis in a Norwegian community. Respir Med. 2003 Apr;97(4):428-35.
8. Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, Fogarty A, et al. The
rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011 Jun;66(6):462-
7.
9. Xaubet A, Ancochea J, Bollo E, Fernandez-Fabrellas E, Franquet T, Molina-
Molina M, et al. Guidelines for the diagnosis and treatment of idiopathic pulmonary
fibrosis. Sociedad Espanola de Neumologia y Cirugia Toracica (SEPAR) Research Group
on Diffuse Pulmonary Diseases. Arch Bronconeumol. 2013 Aug;49(8):343-53.
10. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and
prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006 Oct
1;174(7):810-6.
11. Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, et al. High
prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis.
Eur Respir J. 2006 Jan;27(1):136-42.
12. Savarino E, Carbone R, Marabotto E, Furnari M, Sconfienza L, Ghio M, et al.
Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients.
Eur Respir J. 2013 Nov;42(5):1322-31.
13. Raghu G, Meyer KC. Silent gastro-oesophageal reflux and microaspiration in IPF:
mounting evidence for anti-reflux therapy? Eur Respir J. 2012 Feb;39(2):242-5.
14. Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, et al.
Gastroesophageal reflux therapy is associated with longer survival in patients with
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011 Dec 15;184(12):1390-4.
15. Lung ML, Lam WK, So SY, Lam WP, Chan KH, Ng MH. Evidence that
respiratory tract is major reservoir for Epstein-Barr virus. Lancet. 1985 Apr
20;1(8434):889-92.
16. Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, et al. The detection
of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 1999 Apr;159(4 Pt 1):1336-41.

Tesis Doctoral Charlín Méndez Cordovez
81
17. Tsukamoto K, Hayakawa H, Sato A, Chida K, Nakamura H, Miura K.
Involvement of Epstein-Barr virus latent membrane protein 1 in disease progression in
patients with idiopathic pulmonary fibrosis. Thorax. 2000 Nov;55(11):958-61.
18. Manganelli P, Salaffi F, Pesci A. [Hepatitis C virus and pulmonary fibrosis].
Recenti Prog Med. 2002 May;93(5):322-6.
19. Hubbard R, Venn A, Smith C, Cooper M, Johnston I, Britton J. Exposure to
commonly prescribed drugs and the etiology of cryptogenic fibrosing alveolitis: a case-
control study. Am J Respir Crit Care Med. 1998 Mar;157(3 Pt 1):743-7.
20. King TE, Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011
Dec 3;378(9807):1949-61.
21. King C, Nathan SD. Identification and treatment of comorbidities in idiopathic
pulmonary fibrosis and other fibrotic lung diseases. Curr Opin Pulm Med. 2013
Sep;19(5):466-73.
22. Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM, et
al. Association between pulmonary fibrosis and coronary artery disease. Arch Intern Med.
2004 Mar 8;164(5):551-6.
23. Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J, et al.
Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir
Med. 2010 Jul;104(7):1035-41.
24. Sprunger DB, Olson AL, Huie TJ, Fernandez-Perez ER, Fischer A, Solomon JJ,
et al. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease.
Eur Respir J. 2012 Jan;39(1):125-32.
25. Park S, Lee EJ. Recent advances in idiopathic pulmonary fibrosis. Tuberc Respir
Dis (Seoul). 2013 Jan;74(1):1-6.
26. Ding Q, Luckhardt T, Hecker L, Zhou Y, Liu G, Antony VB, et al. New insights
into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs. 2011 May
28;71(8):981-1001.
27. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M.
Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have
a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J
Clin Invest. 1995 Apr;95(4):1861-8.
28. Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung
injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. Aug 18;380(9842):680-8.
29. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and
pathological perspectives, problems, and promises. Dev Cell. Aug 16;21(2):193-215.
30. Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung
injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012 Aug 18;380(9842):680-
8.
31. Mubarak KK, Montes-Worboys A, Regev D, Nasreen N, Mohammed KA, Faruqi
I, et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary
fibrosis. Eur Respir J. Jan;39(1):133-40.
32. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and
treatment. International consensus statement. American Thoracic Society (ATS), and the
European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000 Feb;161(2 Pt
1):646-64.
33. Ohshimo S, Bonella F, Cui A, Beume M, Kohno N, Guzman J, et al. Significance
of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir
Crit Care Med. 2009 Jun 1;179(11):1043-7.

Tesis Doctoral Charlín Méndez Cordovez
82
34. Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, et al.
Interstitial lung disease guideline: the British Thoracic Society in collaboration with the
Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax.
2008 Sep;63 Suppl 5:v1-58.
35. Hunninghake GW, Zimmerman MB, Schwartz DA, King TE, Jr., Lynch J, Hegele
R, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J
Respir Crit Care Med. 2001 Jul 15;164(2):193-6.
36. Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, et al.
Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J
Respir Crit Care Med. 1998 Jan;157(1):199-203.
37. Lee JS, McLaughlin S, Collard HR. Comprehensive care of the patient with
idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2011 Sep;17(5):348-54.
38. King TE, Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg
MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N
Engl J Med. 2014 May 29;370(22):2083-92.
39. Spagnolo P, Del Giovane C, Luppi F, Cerri S, Balduzzi S, Walters EH, et al. Non-
steroid agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev.
2010(9):CD003134.
40. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et
al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two
randomised trials. Lancet. 2011 May 21;377(9779):1760-9.
41. Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone
in idiopathic pulmonary fibrosis. Eur Respir J. 2010 Apr;35(4):821-9.
42. Jiang C, Huang H, Liu J, Wang Y, Lu Z, Xu Z. Adverse events of pirfenidone for
the treatment of pulmonary fibrosis: a meta-analysis of randomized controlled trials. PLoS
One. 2012;7(10):e47024.
43. Roth GJ, Heckel A, Colbatzky F, Handschuh S, Kley J, Lehmann-Lintz T, et al.
Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the
discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J
Med Chem. 2009 Jul 23;52(14):4466-80.
44. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al.
Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med.
2011 Sep 22;365(12):1079-87.
45. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al.
Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014
May 29;370(22):2071-82.
46. Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. High-
dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005 Nov
24;353(21):2229-42.
47. Raghu G, Anstrom KJ, King TE, Jr., Lasky JA, Martinez FJ. Prednisone,
azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012 May
24;366(21):1968-77.
48. Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Jr., Raghu G. Randomized
trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014 May
29;370(22):2093-101.
49. Luppi F, Spagnolo P, Cerri S, Richeldi L. The big clinical trials in idiopathic
pulmonary fibrosis. Curr Opin Pulm Med. 2012 Sep;18(5):428-32.

Tesis Doctoral Charlín Méndez Cordovez
83
50. Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR. Imatinib
treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results.
Am J Respir Crit Care Med. 2010 Mar 15;181(6):604-10.
51. Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, et al. Imatinib as a
novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir
Crit Care Med. 2005 Jun 1;171(11):1279-85.
52. Vuorinen K, Gao F, Oury TD, Kinnula VL, Myllarniemi M. Imatinib mesylate
inhibits fibrogenesis in asbestos-induced interstitial pneumonia. Exp Lung Res. 2007
Sep;33(7):357-73.
53. Richter A, Puddicombe SM, Lordan JL, Bucchieri F, Wilson SJ, Djukanovic R, et
al. The contribution of interleukin (IL)-4 and IL-13 to the epithelial-mesenchymal trophic
unit in asthma. Am J Respir Cell Mol Biol. 2001 Sep;25(3):385-91.
54. Phipps S, Ying S, Wangoo A, Ong YE, Levi-Schaffer F, Kay AB. The relationship
between allergen-induced tissue eosinophilia and markers of repair and remodeling in
human atopic skin. J Immunol. 2002 Oct 15;169(8):4604-12.
55. Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by
alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol. 1998
Jan;18(1):60-5.
56. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary
expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial
fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999
Mar;103(6):779-88.
57. Belperio JA, Dy M, Burdick MD, Xue YY, Li K, Elias JA, et al. Interaction of IL-
13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir
Cell Mol Biol. 2002 Oct;27(4):419-27.
58. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu
M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a
pathologic microenvironment. Nat Med. 2010 Sep;16(9):1009-17.
59. Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung
disease. Cochrane Database Syst Rev. 2014;10:CD006322.
60. Trulock EP, Christie JD, Edwards LB, Boucek MM, Aurora P, Taylor DO, et al.
Registry of the International Society for Heart and Lung Transplantation: twenty-fourth
official adult lung and heart-lung transplantation report-2007. J Heart Lung Transplant.
2007 Aug;26(8):782-95.
61. Orens JB, Estenne M, Arcasoy S, Conte JV, Corris P, Egan JJ, et al. International
guidelines for the selection of lung transplant candidates: 2006 update--a consensus report
from the Pulmonary Scientific Council of the International Society for Heart and Lung
Transplantation. J Heart Lung Transplant. 2006 Jul;25(7):745-55.
62. Mura M, Porretta MA, Bargagli E, Sergiacomi G, Zompatori M, Sverzellati N, et
al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year
prospective study. Eur Respir J. Jul;40(1):101-9.
63. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et
al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal
clinically important difference. Am J Respir Crit Care Med. 2011 May 1;183(9):1231-7.
64. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et
al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and
minimal clinically important difference. Am J Respir Crit Care Med. Dec
15;184(12):1382-9.

Tesis Doctoral Charlín Méndez Cordovez
84
65. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et
al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 2011 Aug 15;184(4):459-66.
66. Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A
multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern
Med. 2012 May 15;156(10):684-91.
67. Uhal BD, Wang R, Laukka J, Zhuang J, Soledad-Conrad V, Filippatos G.
Inhibition of amiodarone-induced lung fibrosis but not alveolitis by angiotensin system
antagonists. Pharmacol Toxicol. 2003 Feb;92(2):81-7.
68. Coin PG, Osornio-Vargas AR, Roggli VL, Brody AR. Pulmonary fibrogenesis
after three consecutive inhalation exposures to chrysotile asbestos. Am J Respir Crit Care
Med. 1996 Nov;154(5):1511-9.
69. Chua F, Gauldie J, Laurent GJ. Pulmonary fibrosis: searching for model answers.
Am J Respir Cell Mol Biol. 2005 Jul;33(1):9-13.
70. Dave NB, Kaminski N. Analysis of microarray experiments for pulmonary
fibrosis. Methods Mol Med. 2005;117:333-58.
71. Adamson IY. Pulmonary toxicity of bleomycin. Environ Health Perspect. 1976
Aug;16:119-26.
72. Umezawa H, Ishizuka M, Maeda K, Takeuchi T. Studies on bleomycin. Cancer.
1967 May;20(5):891-5.
73. Sleijfer S. Bleomycin-induced pneumonitis. Chest. 2001 Aug;120(2):617-24.
74. Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer. 2005
Feb;5(2):102-12.
75. Cabrera S. Bleomicina: un modelo de fibrosis pulmonar. Rev Inst Nal Enf Resp
Mex 2006;19 (1): 53-61.
76. Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of
bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002 Jun;83(3):111-9.
77. Kaminski N, Zuo F, Cojocaro G, Yakhini Z, Ben-Dor A, Morris D, et al. Use of
oligonucleotide microarrays to analyze gene expression patterns in pulmonary fibrosis
reveals distinct patterns of gene expression in mice and humans. Chest. 2002 Mar;121(3
Suppl):31S-2S.
78. Ramotar D, Wang H. Protective mechanisms against the antitumor agent
bleomycin: lessons from Saccharomyces cerevisiae. Curr Genet. 2003 Jul;43(4):213-24.
79. Bukowski MR, Zhu S, Koehntop KD, Brennessel WW, Que L, Jr.
Characterization of an Fe III-OOH species and its decomposition product in a bleomycin
model system. J Biol Inorg Chem. 2004 Jan;9(1):39-48.
80. Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor
antibiotics: bleomycin, enediynes, and mitomycin. Chem Rev. 2005 Feb;105(2):739-58.
81. Joshi N, Grant SG. DNA double-strand break damage and repair assessed by
pulsed-field gel electrophoresis. Methods Mol Biol. 2005;291:121-9.
82. Vorobjev PE, Smith JB, Pyshnaya IA, Levina AS, Zarytova VF, Wickstrom E.
Site-specific cleavage of RNA and DNA by complementary DNA--bleomycin A5
conjugates. Bioconjug Chem. 2003 Nov-Dec;14(6):1307-13.
83. Chen J, Stubbe J. Bleomycins: new methods will allow reinvestigation of old
issues. Curr Opin Chem Biol. 2004 Apr;8(2):175-81.
84. Abraham AT, Lin JJ, Newton DL, Rybak S, Hecht SM. RNA cleavage and
inhibition of protein synthesis by bleomycin. Chem Biol. 2003 Jan;10(1):45-52.

Tesis Doctoral Charlín Méndez Cordovez
85
85. O'Farrell PA, Gonzalez F, Zheng W, Johnston SA, Joshua-Tor L. Crystal structure
of human bleomycin hydrolase, a self-compartmentalizing cysteine protease. Structure.
1999 Jun 15;7(6):619-27.
86. Schwartz DR, Homanics GE, Hoyt DG, Klein E, Abernethy J, Lazo JS. The
neutral cysteine protease bleomycin hydrolase is essential for epidermal integrity and
bleomycin resistance. Proc Natl Acad Sci U S A. 1999 Apr 13;96(8):4680-5.
87. Nuver J, Lutke Holzik MF, van Zweeden M, Hoekstra HJ, Meijer C, Suurmeijer
AJ, et al. Genetic variation in the bleomycin hydrolase gene and bleomycin-induced
pulmonary toxicity in germ cell cancer patients. Pharmacogenet Genomics. 2005
Jun;15(6):399-405.
88. Arras M, Louahed J, Simoen V, Barbarin V, Misson P, van den Brule S, et al. B
lymphocytes are critical for lung fibrosis control and prostaglandin E2 regulation in IL-9
transgenic mice. Am J Respir Cell Mol Biol. 2006 May;34(5):573-80.
89. Bocci V. Ozone. A new medical drug. 2nd ed. Dordrecht, The Netherlands:
Springer Publ.; 2011.
90. Di Paolo N, Bocci V, Gaggiotti E. Ozone therapy. Int J Artif Organs. 2004
Mar;27(3):168-75.
91. Bocci V. Biological and clinical effects of ozone. Has ozone therapy a future in
medicine? Br J Biomed Sci. 1999;56(4):270-9.
92. Bocci V. Does ozone therapy normalize the cellular redox balance? Implications
for therapy of human immunodeficiency virus infection and several other diseases. Med
Hypotheses. 1996 Feb;46(2):150-4.
93. Wentworth P, Jr., Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, et al.
Evidence for ozone formation in human atherosclerotic arteries. Science. 2003 Nov
7;302(5647):1053-6.
94. Yamashita K, Miyoshi T, Arai T, Endo N, Itoh H, Makino K, et al. Ozone
production by amino acids contributes to killing of bacteria. Proc Natl Acad Sci U S A.
2008 Nov 4;105(44):16912-7.
95. Konrad H. Guidelines and Recommendations for Medical Professionals Planning
to Acquire a Medical Ozone Generator. Journal [serial on the Internet]. 2014 Date.
96. Bocci V, Borrelli E, Travagli V, Zanardi I. The ozone paradox: ozone is a strong
oxidant as well as a medical drug. Med Res Rev. 2009 Jul;29(4):646-82.
97. Bocci VA, Zanardi I, Travagli V. Ozone acting on human blood yields a hormetic
dose-response relationship. J Transl Med. 2011;9:66.
98. Pecorelli A, Bocci V, Acquaviva A, Belmonte G, Gardi C, Virgili F, et al. NRF2
activation is involved in ozonated human serum upregulation of HO-1 in endothelial cells.
Toxicol Appl Pharmacol. 2013 Feb 15;267(1):30-40.
99. Re L, Martinez-Sanchez G, Bordicchia M, Malcangi G, Pocognoli A, Morales-
Segura MA, et al. Is ozone pre-conditioning effect linked to Nrf2/EpRE activation
pathway in vivo? A preliminary result. Eur J Pharmacol. 2014 Nov 5;742:158-62.
100. Bocci V, Valacchi G. Nrf2 activation as target to implement therapeutic
treatments. Front Chem. 2015;3:4.
101. Mattassi R, D'Angelo F, Bisetti P, Colombo R, Vaghi M. Terapia con ozono per
via parenterale nelle arteripatie obliteranti periferiche: mecanismo biochimico e risultati
clinici. II Giornale Di Chirurgia. 1987;VIII:109-11.
102. Coppola L, Giunta R, Verrazzo G, Luongo C, Sammartino A, Vicario C, et al.
Influence of ozone on haemoglobin oxygen affinity in type-2 diabetic patients with
peripheral vascular disease: in vitro studies. Diabete Metab. 1995 Oct;21(4):252-5.

Tesis Doctoral Charlín Méndez Cordovez
86
103. Verrazzo G, Coppola L, Luongo C, Sammartino A, Giunta R, Grassia A, et al.
Hyperbaric oxygen, oxygen-ozone therapy, and rheologic parameters of blood in patients
with peripheral occlusive arterial disease. Undersea Hyperb Med. 1995 Mar;22(1):17-22.
104. Giunta R, Coppola A, Luongo C, Sammartino A, Guastafierro S, Grassia A, et al.
Ozonized autohemotransfusion improves hemorheological parameters and oxygen
delivery to tissues in patients with peripheral occlusive arterial disease. Ann Hematol.
2001 Dec;80(12):745-8.
105. Clavo B, Perez JL, Lopez L, Suarez G, Lloret M, Rodriguez V, et al. Effect of
ozone therapy on muscle oxygenation. J Altern Complement Med. 2003 Apr;9(2):251-6.
106. Clavo B, Catala L, Perez JL, Rodriguez V, Robaina F. Ozone Therapy on Cerebral
Blood Flow: A Preliminary Report. Evid Based Complement Alternat Med. 2004
Dec;1(3):315-9.
107. Clavo B, Gutierrez D, Martin D, Suarez G, Hernandez MA, Robaina F.
Intravesical ozone therapy for progressive radiation-induced hematuria. J Altern
Complement Med. 2005 Jun;11(3):539-41.
108. Clavo B, Suarez G, Aguilar Y, Gutierrez D, Ponce P, Cubero A, et al. Brain
ischemia and hypometabolism treated by ozone therapy. Forsch Komplementmed.
2011;18(5):283-7.
109. Clavo B, Santana-Rodriguez N, Lopez-Silva SM, Dominguez E, Mori M,
Gutierrez D, et al. Persistent PORT-A-CATH(R)-related fistula and fibrosis in a breast
cancer patient successfully treated with local ozone application. J Pain Symptom Manage.
2012 Feb;43(2):e3-6.
110. Clavo B, Ceballos D, Gutierrez D, Rovira G, Suarez G, Lopez L, et al. Long-term
control of refractory hemorrhagic radiation proctitis with ozone therapy. J Pain Symptom
Manage. 2013 Jul;46(1):106-12.
111. Clavo B, Santana-Rodriguez N, Llontop P, Gutierrez D, Ceballos D, Mendez C,
et al. Ozone Therapy in the Management of Persistent Radiation-Induced Rectal Bleeding
in Prostate Cancer Patients. Evid Based Complement Alternat Med. 2015;2015:480369.
112. Hedenqvist P, Roughan JV, Flecknell PA. Effects of repeated anaesthesia with
ketamine/medetomidine and of pre-anaesthetic administration of buprenorphine in rats.
Lab Anim. 2000 Apr;34(2):207-11.
113. Weksler B, Ng B, Lenert J, Burt M. A simplified method for endotracheal
intubation in the rat. J Appl Physiol. 1994 Apr;76(4):1823-5.
114. Radonic A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A. Guideline to
reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun.
2004 Jan 23;313(4):856-62.
115. Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, et al.
Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report.
Am J Respir Crit Care Med. 2014 Jan 15;189(2):214-22.
116. Wettstein G, Bellaye PS, Kolb M, Hammann A, Crestani B, Soler P, et al.
Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail
degradation. FASEB J. 2013 Apr;27(4):1549-60.
117. Vidyasagar A, Reese SR, Hafez O, Huang LJ, Swain WF, Jacobson LM, et al.
Tubular expression of heat-shock protein 27 inhibits fibrogenesis in obstructive
nephropathy. Kidney Int. 2012 Jan;83(1):84-92.
118. Santana-Rodriguez N, Garcia-Herrera R, Clavo B, Llontop P, Ponce-Gonzalez
MA, Villar J, et al. Searching for novel molecular targets of chronic rejection in an

Tesis Doctoral Charlín Méndez Cordovez
87
orthotopic experimental lung transplantation model. J Heart Lung Transplant. 2012
Feb;31(2):213-21.
119. Rahman I, MacNee W. Role of transcription factors in inflammatory lung
diseases. Thorax. 1998 Jul;53(7):601-12.
120. Villeneuve NF SZ, Chen W, Zhang DD. Nrf2 and p21 regulate the fine balance
between life and death by controlling ROS levels. Cell Cycle
2009;8:3255-3256.
121. Li W KT, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-
antioxidant signaling attenuates NF kappa B-inflammatory response and elicits apotosis
Biochem Pharmacol. 2008;76:1485-1489.
122. Saito F, Tasaka S, Inoue K, Miyamoto K, Nakano Y, Ogawa Y, et al. Role of
interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell
Mol Biol. 2008 May;38(5):566-71.
123. Bronconeumol JeaA. Fibrosis Pulmonar: Perspectivas actuales. . 1989:332-44. .
124. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta
causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine
lung. Am J Respir Cell Mol Biol. 2005 Apr;32(4):311-8.
125. Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of
growth factors on proliferation and matrix production of normal and fibrotic human lung
fibroblasts. Lung. 2005 Jul-Aug;183(4):225-37.
126. Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The transforming growth
factor beta(1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative
stress. Hepatology. 1999 Dec;30(6):1490-7.

Tesis Doctoral Charlín Méndez Cordovez
88

Tesis Doctoral Charlín Méndez Cordovez
89


















