
Determinación del orden de una reacción mediante ... · PDF...
13
Determinación del orden de una reacción mediante espectrofotometría Física Macroscópica 2011 Barros, Rodrigo; Núñez, Yanina; Pagliaro, Paula; Panelo, Mauro Germán Resumen Se determinó el orden de una reacción respecto a una de las especies involucradas en el régimen de exceso de las restantes analizando los cambios en su concentración como función del tiempo, evaluados mediante un espectrofotómetro calibrado a tal fin. Se determinó que la reacción es de primer orden respecto a la especie de interés y se realizó un análisis sobre las condiciones experimentales Introducción Por medio de la termodinámica es posible predecir la ocurrencia de reacciones, pero no se habla de un aspecto que es de suma importancia y que tiene relación con el tiempo en el cual se llevan a cabo estos procesos. La cinética química da cuenta de este aspecto y por tanto define la velocidad con que se lleva a cabo una reacción. En algunos casos, una reacción puede durar algunos segundos, mientras que en otros casos puede durar siglos. Un punto que se debe visualizar es que la cinética también es aplicable a cambios físicos y/o alotrópicos de la materia. En este sentido, se puede dar como ejemplo el caso del carbono. Dos posibles estructuras alotrópicas del carbono son el carbono grafito y el diamante. Sin embargo en condiciones normales, la termodinámica predice que solo debe existir el carbono grafito. La pregunta que es interesante es por qué motivo existe la joya del diamante, la cual tiene sentido a altas presiones según la termodinámica pero no a bajas. La respuesta se encuentra en la velocidad de transformación de diamante a grafito. Esta transformación es tan lenta en condiciones normales que puede demorar miles de años y si bien la termodinámica predice que el sistema debe evolucionar a la formación de grafito en estas condiciones, esto en la práctica no es apreciable. Factores que determinan la velocidad de reacción. La velocidad de una reacción depende de la composición de las sustancias reaccionantes así como también de una serie de otros factores. Entre ellos destacan, su forma física, del nivel de mezcla de los reactivos, del tamaño y concentración de los reactivos, de la temperatura, de la presión, de las concentraciones de los reactivos, de sustancias que afecten la reacción sin ser ellas un reactivo o producto (catalizadores) y de algunas condiciones especiales propias del sistema como pueden ser la radiación de luz visible, UV, RX, neutrones u otras. Definiciones. Sea la siguiente reacción química irreversible: ∑ ⇒∑
Transcript of Determinación del orden de una reacción mediante ... · PDF...

Determinación del orden de una reacción mediante
espectrofotometría
Física Macroscópica 2011 Barros, Rodrigo; Núñez, Yanina; Pagliaro, Paula; Panelo, Mauro Germán
Resumen
Se determinó el orden de una reacción respecto a una de las especies involucradas en el
régimen de exceso de las restantes analizando los cambios en su concentración como
función del tiempo, evaluados mediante un espectrofotómetro calibrado a tal fin. Se
determinó que la reacción es de primer orden respecto a la especie de interés y se realizó un
análisis sobre las condiciones experimentales
Introducción
Por medio de la termodinámica es posible predecir la ocurrencia de reacciones, pero no se
habla de un aspecto que es de suma importancia y que tiene relación con el tiempo en el
cual se llevan a cabo estos procesos. La cinética química da cuenta de este aspecto y por
tanto define la velocidad con que se lleva a cabo una reacción. En algunos casos, una
reacción puede durar algunos segundos, mientras que en otros casos puede durar siglos.
Un punto que se debe visualizar es que la cinética también es aplicable a cambios físicos
y/o alotrópicos de la materia. En este sentido, se puede dar como ejemplo el caso del
carbono. Dos posibles estructuras alotrópicas del carbono son el carbono grafito y el
diamante. Sin embargo en condiciones normales, la termodinámica predice que solo debe
existir el carbono grafito. La pregunta que es interesante es por qué motivo existe la joya
del diamante, la cual tiene sentido a altas presiones según la termodinámica pero no a bajas.
La respuesta se encuentra en la velocidad de transformación de diamante a grafito. Esta
transformación es tan lenta en condiciones normales que puede demorar miles de años y si
bien la termodinámica predice que el sistema debe evolucionar a la formación de grafito en
estas condiciones, esto en la práctica no es apreciable.
Factores que determinan la velocidad de reacción.
La velocidad de una reacción depende de la composición de las sustancias reaccionantes así
como también de una serie de otros factores. Entre ellos destacan, su forma física, del nivel
de mezcla de los reactivos, del tamaño y concentración de los reactivos, de la temperatura,
de la presión, de las concentraciones de los reactivos, de sustancias que afecten la reacción
sin ser ellas un reactivo o producto (catalizadores) y de algunas condiciones especiales
propias del sistema como pueden ser la radiación de luz visible, UV, RX, neutrones u otras.
Definiciones.
Sea la siguiente reacción química irreversible:
∑ ⇒∑

Donde Ai son los reactivos y Bi los productos de la reacción; ai y bi son los coeficientes
estequiométricos de la misma. Se define la velocidad de del siguiente modo, donde
∏[ ]
Donde [ ] son concentraciones de las especies reactivas y los exponentes son coeficientes
reales hallados experimentalmente para cada reacción, llamados orden respecto a la especie . Se
habla concentraciones ya que se asume fase acuosa. En caso de gases se habla en términos
de presión. es una constante de proporcionalidad y se designa como constante de
velocidad de la reacción en cuestión.
El término de velocidad ‘V’ se puede entender pensando en 2 posibles situaciones: Como
velocidad de desaparición de reactivos o bien como velocidad de aparición de productos.
Ya que ambas velocidades en magnitud son proporcionales ya que los productos son una
consecuencia de la desaparición de reactivos, se puede establecer la siguiente expresión
general que resume ambas posibilidades, válida para todo i y todo j
[ ]
[ ]
Se define además el orden global de la reacción como
∑
Si el orden global es igual a 1 se dice que la reacción es de primer orden. Si es de orden 2
se dice que es de segundo orden, etc. Pueden existir órdenes no enteros.
Efectos de la temperatura y la presión en la velocidad de una reacción.
En estudios de cinética se observa que la velocidad de reacción se ve afectada en la práctica
con variaciones de presión y de temperatura. Sin embargo dichos cambios son mucho más
significativos a las fluctuaciones de las temperaturas y por lo tanto se suele hacer el estudio
dando prioridad a esta variable y despreciando los cambios que experimenta a las
variaciones de presión (esto se apoya en el supuesto de que los cambios de presión
aplicados en el sistema son pequeños y por lo tanto se pueden considerar despreciables).
Sin embargo es muy sensible a la temperatura. La ecuación que relaciona la temperatura
con la velocidad de una reacción (constante de velocidad) es la ecuación de Arrhenius que
se expresa mediante:

Donde A es el factor de frecuencia, es la energía de activación, R es la constante de los
gases y T es la temperatura.
De acuerdo con Arrhenius, la ecuación anterior indica que las moléculas deben adquirir una
cierta energía antes de que puedan reaccionar, siendo el factor de Boltzmann la
fracción de moléculas que están en condiciones de alcanzar la energía necesaria. Se suele
suponer que el factor A es un valor constante.
En la presente experiencia se tuvo como objetivo determinar el orden de la reacción
siguiente respecto a la especie Cr2O72-
:
⇒
La ley general de velocidades para esta reacción corresponde a
[
] [ ] [ ] [ ]
[ ]
Donde a, b, c son los órdenes respecto a cada especie y k la constante de velocidad. Se
trabajó en un régimen donde se tuviera un gran exceso de ácido y alcohol, resultando en
una concentración aproximadamente constante de los mismos, simplificándose la expresión
a una en donde el orden global es
[
] [ ]
Se puede vincular la concentración de una especie y su capacidad para absorber radiación
electromagnética mediante una relación lineal a primer orden (pequeñas concentraciones)
entre absorbancia y concentración, dada por la ley de Lambert-Beer donde es la
absorbancia molar, b el camino óptico y [C] la concentración de la especie en cuestión:
[ ]
Lo que finalmente permite transformar la ecuación anterior, considerando que es
cosntante, en:
Ligando la absorbancia medida con el orden de la reacción de interés. Esta ecuación es
fácilmente integrable por separación de variables y su solución general se utilizó para
ajustar a los datos experimentales, excepto en el caso particular que no responde a la
solución general sin un paso al límite. Las soluciones corresponden respectivamente a
y
{√

Procedimiento Experimental
Experimentalmente, se siguió la concentración del dicromato en función del tiempo
mientras se producía la reacción y posteriormente se ajustó la solución general de la
ecuación diferencial anterior a los datos, con el fin de determinar a. Por otro lado, se ajustó
el caso particular 1, que no responde al término general sin un paso al límite, por lo que el
programa computacional es incapaz de obtenerlo exactamente.
La concentración del ión se siguió mediante un espectrofotómetro configurado para
detectar las variaciones en la absorbancia de la muestra, asociadas al cambio de la
concentración de dicromato, la cual es máxima para .
El 0 del instrumento se fijó utilizando en él una solución con los siguientes componentes:
1ml H2SO4, 9ml de agua destilada y 2.5 ml de etanol.
Seguidamente se determinó la absorbancia a t=0 a fin de tener un dato independiente con el
cual verificar el avance de la reacción, para ello se utilizó una muestra formada por 1.5ml
de K2Cr2O7, 1ml H2SO4 y 10ml de agua destilada.
Al prepararse la muestra de medición, se inició el cronómetro al momento de mezclar el
dicromato con etanol, a fin de no observar un corrimiento temporal en los datos. En la
misma se utilizaron 1.5ml de K2Cr2O7, 1ml H2SO4, 2.5 ml de etanol y 7.5 ml de agua
destilada.
Una vez ubicada la muestra en el espectrofotómetro, se recogieron datos cada 30 segundos
durante 20 minutos sobre los cuales se trabajó en el programa OriginPro, donde se realizó
el análisis anteriormente mencionado.

Resultados y discusión
En el siguiente gráfico (figura 1) se presentan los resultados obtenidos para la absorbancia
en función del tiempo, a lo largo de la reacción. El punto observado a t=0, corresponde a la
solución preparada a tal fin, que corresponde a un valor A=0.785
Inicialmente se realizó el ajuste propuesto con la función general, realizando un cambio de
variable y agregándose un parámetro d para dar cuenta de posibles
desplazamientos en la calibración del cero
( )
Para realizar el ajuste, se procedió inicialmente a estimar los parámetros del mismo
comparando simulaciones numéricas, que dependen fuertemente del valor de h, hecho que
permitió ubicarlo en el intervalo , suficiente para iniciar el proceso. En esta
región, la dependencia con u es despreciable pues decrece muy rápidamente con h hacia 1,
motivo por el cual se lo inició en ese valor. se estimó a partir del valor medido para la
solución sin etanol, se inició en 0 y en . Al iniciar el ajuste, el valor de h fue
rápidamente hacia 0 como se puede observar, lo que permitió fijar
independientemente del valor , pues el exponente los lleva a 1 irremediablemente. Esto
0 200 400 600 800 1000 1200
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8 Absorbancia
Ab
so
rban
cia
(U
A)
Tiempo (s)
Figura 1: Representación gráfica de Absorbancia en función del tiempo para la reacción analizada

Figura 2: Representación gráfica de Absorbancia en función del tiempo con un ajuste de la solución general
se hizo con el objetivo de mejorar la convergencia del algoritmo, sus estimaciones de error
y correlación entre variables.
0 200 400 600 800 1000 1200
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Absorbancia
General Solution Fit
Absorb
an
cia
(U
A)
Tiempo (s)
Model Cinetica (User)
Equation(-(h*k*v^h)*x+a^h) (̂1/h)+d
Reduced Chi-Sqr 6,069E-5
Adj. R-Square 0,99804Value Standard Error
Absorbancia a 0,703 0,005
Absorbancia h 2,897E-9 4E-6
Absorbancia k 0,00355 6E-5
Absorbancia v 1 0
Absorbancia d 0,05 0,003
La observación del ajuste en la figura 2, permite estimar que el modelo con es la
mejor representación del sistema ( está contenido en la incerteza del ajuste), por lo
que se procedió a ajustarlo. Cabe destacar la independencia de esta solución respecto a ,
consistente con la aproximación anterior.
Se presenta a modo ilustrativo un ajuste para orden 2 ( fijo, figura 3), en donde se
ve que la absorbancia inicial obtenida difiere en un 15% del valor considerado inicial y se
observan evidentes desviaciones de la función respecto a los datos experimentales. Debe
notarse la incerteza asociada a y , pues como su cociente es constante y ambas pesan
igualmente en el ajuste, no se las puede resolver unívocamente por este método sin conocer
alguna.
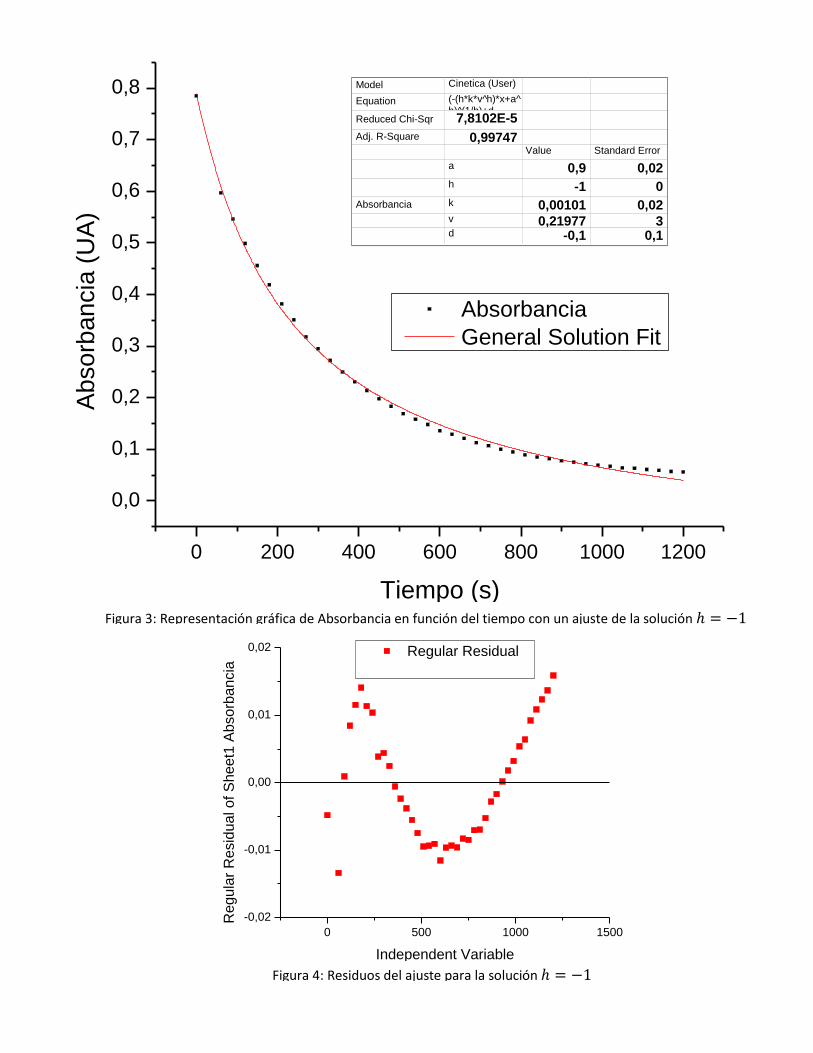
Figura 3: Representación gráfica de Absorbancia en función del tiempo con un ajuste de la solución
Figura 4: Residuos del ajuste para la solución
0 500 1000 1500
-0,02
-0,01
0,00
0,01
0,02
Re
gula
r R
esid
ua
l of
Sh
ee
t1 A
bso
rban
cia
Independent Variable
Regular Residual
0 200 400 600 800 1000 1200
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Absorbancia
General Solution Fit
Ab
so
rban
cia
(U
A)
Tiempo (s)
Model Cinetica (User)
Equation (-(h*k*v^h)*x+a^h) (̂1/h)+d
Reduced Chi-Sqr 7,8102E-5
Adj. R-Square 0,99747Value Standard Error
a 0,9 0,02h -1 0
Absorbancia k 0,00101 0,02v 0,21977 3d -0,1 0,1

Figura 5: Representación gráfica de Absorbancia en función del tiempo con un ajuste de la solución
Puede observarse comparando las figuras 2 y 4 que los coeficientes de ambos ajustes son
casi idénticos así como su representatividad de los datos, aclarando que y sus
valores coinciden. Asimismo, al realizar el paso al límite, se observa una ligera mejora en
los valores de y . El hecho de que exista un corrimiento constante, el cual no se
modifica en ninguno de los ajustes, habla de un corrimiento en la calibración del 0 del
aparato, la cual se realizó con sólo etanol.
0 200 400 600 800 1000 1200
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Absorbancia
Exponential Decay Fit
Ab
so
rban
cia
(U
A)
Tiempo (s)
Model ExpDec1
Equation y = A1*exp(-x/t1) + y0
Reduced Chi-Sqr 5,901E-5Adj. R-Square 0,99809
Value Standard Error
Absorbancia y0 0,05 0,003Absorbancia A1 0,703 0,005Absorbancia t1 282 5
A continuación se presentan gráficas de en función de (figura 6), gráfica que de no
existir el corrimiento de 0, sería lineal, pero aun así puede verse la correspondencia entre
modelo y datos experimentales

0 200 400 600 800 1000 1200
0,04979
0,13534
0,36788
Absorbancia
Exponential Decay Fit
ExpDecay y0=0
Absorb
ancia
(U
A)
Tiempo (s)
Model ExpDec1
Equation y = A1*exp(-x/t1) + y0
Reduced Chi-Sqr 5,901E-5Adj. R-Square 0,99809
Value Standard Error
Absorbancia y0 0,05 0,003Absorbancia A1 0,703 0,005Absorbancia t1 282 5
Model ExpDecay y0=0
Equationy=A1*exp(-x/t1)
Reduced Chi-Sqr 4,2309E-4
Adj. R-Square 0,98632Value Standard Error
Absorbancia A1 0,7 0,1
Absorbancia t1 363 9
Figura 6: Representación gráfica de Absorbancia en función del tiempo en escala y-logarítmica junto a los ajustes
con y sin corrimiento constante
En la figura 6 pueden observarse dos ajustes, uno es el de la figura 5 y el otro un
decaimiento exponencial sin corrimiento constante graficados en una escala y-logarítmica.
Puede observarse el desviamiento respecto a la linealidad por parte de los datos
experimentales y cómo el corrimiento constante da cuenta por la mayoría de la desviación,
si bien aún se observan diferencias en los extremos de los datos.
A partir de la última observación, se debe analizar las desviaciones de los puntos
experimentales respecto a las diferentes funciones de ajuste. Por lo pronto puede
establecerse que el orden de la reacción es 1 y restringir el análisis al caso exponencial. A
continuación se presentan los residuos de ambos ajustes (figuras 7 y 8) y se añade el
residuo de por completitud (figura 4).

Figura 8: Residuos del ajuste para con corrimiento cero
0 500 1000 1500
-0,2
0,0
0,2
0,4
Regula
r R
esid
ual of S
heet1
LnA
Independent Variable
Regular Residual of ExpDecay y0=0
0 500 1000 1500
-0,02
0,00
0,02
0,04 Regular Residual Exponential Decay
Regula
r R
esid
ual of S
heet1
Absorb
ancia
Independent Variable
Figura 7: Residuos del ajuste para el con corrimiento constante

Figura 9: Representación gráfica de Absorbancia en función del tiempo con un ajuste de la solución y punto 𝑡 excluido
Puede observarse a simple vista que para los tres casos, los residuos presentan tendencias
bien definidas y que además el punto t=0 presenta un comportamiento fuera de la tendencia
para todas las funciones. El análisis de residuos permite ver cuantitativamente dónde se
encuentran las regiones de baja correspondencia del modelo con los datos. Ahora puede
evidenciarse aún más la desviación de respecto a las medidas tomadas, teniendo
desviaciones más amplias respecto a . Una buena función de ajuste debería presentar
residuos distribuidos simétricamente alrededor de 0 y ningún punto especialmente diferente
al resto. Orígenes posibles para estas gráficas son modelos no representativos del fenómeno
u puntos que no se encuentran relacionados con el resto, en este caso el correspondiente a
t=0 presenta una diferencia respecto a la función en el orden del 12%, mucho mayor que el
resto de los datos, asimismo un comportamiento fuera de la tendencia observada. Cabe
aclarar que otro posible origen para este comportamiento es un problema con el inicio del
cronómetro, reportando menos tiempo que el correspondiente entre este punto y el primero
del resto, si bien la probabilidad existe, a la luz del análisis siguiente, se podrá ver que para
compatibilizar el punto, el corrimiento debería ser de orden de 30 segundos, lo cual es
experimentalmente improbable. A la luz de las consideraciones anteriores, se excluirá el
punto t=0 y se compararán los datos nuevamente con el modelo. No se tratará el caso sin
corrimiento, dado que representa el desplazamiento mayor de la linealidad.
0 200 400 600 800 1000 1200
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Absorbancia
Exponential Decay Fit
Ab
so
rban
cia
(U
A)
Tiempo (s)
Model ExpDec1
Equation y = A1*exp(-x/t1) + y0
Reduced Chi-Sqr 7,2379E-7Adj. R-Square 0,99997
Value Standard Error
Absorbancia y0 0,0428 4E-4Absorbancia A1 0,6756 7E-4Absorbancia t1 304,9 0,7

Figura 10: Representación gráfica de Absorbancia en función del tiempo en escala y-logarítmica con ajuste exponencial y punto
𝑡 excluido
Comparando los parámetros de ajuste entre las figuras 5 y 9, puede observarse que el
segundo conjunto reduce en dos órdenes de magnitud y aumenta ligeramente ,
además de reducirse la diferencia entre la función y los datos para bajo t
En la figura 10, gráfica con escala y-logarítmica, puede observarse que la exclusión del
punto no sólo mejora el ajuste para la porción inicial de los datos, sino que además la
función de ajuste se vuelve mucho más representativa para el resto de los puntos, incluida
la porción final de los datos con largos t. Este hecho puede observarse en la figura 11,
correspondiente a los residuos para el ajuste 10, donde los mismos se encuentran
distribuidos uniformemente alrededor de 0 y se observa una reducción de los mismos en
aproximadamente dos órdenes, compatible con la reducción de mencionada.
0 200 400 600 800 1000 1200
0,04979
0,13534
0,36788
Absorbancia
Exponential Decay Fit
Absorb
ancia
(U
A)
Tiempo (s)
Model ExpDec1
Equation y = A1*exp(-x/t1) + y0
Reduced Chi-Sqr 7,2379E-7Adj. R-Square 0,99997
Value Standard Error
Absorbancia y0 0,0428 4E-4Absorbancia A1 0,6756 7E-4Absorbancia t1 304,9 0,7

Figura 11: Residuos del ajuste para el ajuste exponencial con corrimiento constante
Puede verse que existe un punto a primera vista incompatible, pero dado que la diferencia
es del orden de en la región de 300 segundos (en el orden del tiempo característico de
la función), es atribuible a un par de segundos de demora entre la lectura del dato y el
instante correspondiente pues el cambio promedio entre los datos para dicha región es al
menos un orden mayor.
Conclusión
El método utilizado permitió determinar que la reacción analizada es de primer orden
respecto al ión dicromato, a pesar de la presencia de un error sistemático en la calibración
del cero. Asimismo, existe algún efecto adicional en la interacción etanol-dicromato,
resultando en la discrepancia observada entre la solución de t=0 y el resto de los datos pues
los volúmenes usados eran iguales, lo cual indica que no puede considerárselo en igualdad
de condiciones y por tanto no apto para el análisis.
0 500 1000 1500
-0,004
-0,002
0,000
0,002
Regula
r R
esid
ual of S
heet1
Absorb
ancia
Independent Variable
Regular Residual


















![s26bef4fb0b7c48da.jimcontent.com...a.La ecuación cinética de esa reacción es v = K [ A] [ B] . Indique el orden total de la reacción, así como las unidades de la constante cinética](https://static.fdocuments.ec/doc/165x107/5e5ae04e90a9fa59fa70756e/-ala-ecuacin-cintica-de-esa-reaccin-es-v-k-a-b-indique-el-orden.jpg)