
CINÉTICA EN SOLUCIÓN DE LA DESCOMPOSICIÓN … · DEDICATORIA A Rosmira Usuga, Mi madre por su...
43
CINÉTICA EN SOLUCIÓN DE LA DESCOMPOSICIÓN TÉRMICA DEL 2-PROPENAL DEISY MARITZA CASTAÑO USUGA MELISSA CAROLINA VARÓN BALLESTEROS UNIVERSIDAD NACIONAL DE COLOMBIA FACULTAD DE MINAS ESCUELA DE PROCESOS Y ENERGÍA SEDE MEDELLÍN 2009
Transcript of CINÉTICA EN SOLUCIÓN DE LA DESCOMPOSICIÓN … · DEDICATORIA A Rosmira Usuga, Mi madre por su...

CINÉTICA EN SOLUCIÓN DE LA DESCOMPOSICIÓN TÉRMICA DEL 2-PROPENAL
DEISY MARITZA CASTAÑO USUGA
MELISSA CAROLINA VARÓN BALLESTEROS
UNIVERSIDAD NACIONAL DE COLOMBIA
FACULTAD DE MINAS
ESCUELA DE PROCESOS Y ENERGÍA
SEDE MEDELLÍN
2009

CINÉTICA EN SOLUCIÓN DE LA DESCOMPOSICIÓN TÉRMICA DEL 2-PROPENAL
DEISY MARITZA CASTAÑO USUGA
MELISSA CAROLINA VARÓN BALLESTEROS
Trabajo de grado presentado para optar al título de
Ingeniero de Químico
Directores
Jairo Quijano Tobón, Doctor en Ciencias Químicas
Elizabeth Pabón Gelves, Doctora en Química
UNIVERSIDAD NACIONAL DE COLOMBIA
FACULTAD DE MINAS
ESCUELA DE PROCESOS Y ENERGÍA
SEDE MEDELLÍN
2009

DEDICATORIA
A Rosmira Usuga, Mi madre por su constante dedicación y apoyo para el logro de mis metas. Su valentía, trabajo incansable y acompañamiento han sido mis mejores alicientes.
Deisy Maritza Castaño Usuga
Por ser no solo el motor que me impulsó a alcanzar este logro en mí vida, sino quienes han hecho posible con su amor que cada sueño sea realizable. A mis tesoros más grandes…Mi madre Aideé Ballesteros y Mi hermanita Nataly Varón. A mi padre Marcos Varón, quien ha estado siempre presente en mí caminar.
Melissa Carolina Varón Ballesteros

AGRADECIMIENTOS
A ti Dios, por ser el mejor amigo, la fortaleza y no dejarnos caer nunca. Al profesor Jairo Quijano Tobón y Profesora Elizabeth Pabón Gelves, por su contribución a nuestra formación académica. A Ederley Vélez y Juliana Murillo, por su colaboración en el trabajo computacional. A Elkin Cataño, amigo y compañero incondicional por su apoyo en momentos difíciles y por compartir cada una de nuestras alegrías. A la universidad de la nación, por hacernos profesionales competentes y socialmente responsables.

CONTENIDO
Pág.
RESUMEN 5 ABSTRACT 6 INTRODUCCION 7 1. PLANTEAMIENTO DEL PROBLEMA 8 2. OBJETIVOS 9 2.1 OBJETIVO GENERAL 9 2.2 OBJETIVOS ESPECIFICOS 9
3. MARCO TEORICO 10 3.1 ANTECEDENTES 10 3.2 CINÉTICA QUÍMICA 10 3.3 ECUACION DE VELOCIDAD 11 3.3.1 Determinación experimental de la velocidad de reacción 11 3.3.2 Tiempo de vida media 12
3.4 MECANISMOS DE REACCIÓN 12
3.5 INFLUENCIA DE LA TEMPERATURA EN LAS CONSTANTES DE VELOCIDAD 14
3.5.1 Ecuación De Arrhenius 14 3.5.2 Teoría de las colisiones 15
3.5.3 Teoría del estado de transición 16
3.6 CARACTERIZACION DE LOS COMPUESTOS 18 3.6.1 Espectrometría de masas 18

3.7 QUÍMICA COMPUTACIONAL 19
3.7.1 Métodos utilizados en química computacional 21
3.7.1.1 Métodos ab initio 22
3.7.1.1.1 Método de Hartree-Fock 23 4. METODOLOGIA 25 4.1 TERMOLISIS DEL 2-PROPENAL 25 4.1.1 Sistema controlador de temperatura 25 4.1.2 Seguimiento de la cinética de reacción por medio de espectroscopia 25 gases-masas 4.2 CÁLCULOS PARA DETERMINAR CONSTANTES DE VELOCIDAD 26 Y GRÁFICO DE ARRHENIUS. 4.3 CÁLCULOS COMPUTACIONALES 27 5. RESULTADOS Y DISCUSIÓN 28 5.1 RESULTADOS DE ANÁLISIS INSTRUMENTAL 28 5.1.1 Análisis del estudio cinético 28 5.2 CÁLCULOS PARA DETERMINAR CONSTANTES DE VELOCIDAD 31 Y GRÁFICO DE ARRHENIUS 5.3 RESULTADOS DE ANÁLISIS COMPUTACIONAL 34 6. CONCLUSIONES 37 BIBLIOGRAFÍA 38

LISTA DE TABLAS
Pág.
Tabla 1 Constantes de velocidad k (s-1) para la descomposición 33
térmica de 2-propenal a varias temperaturas
Tabla 2 Constante cinética de 2-propenal 35
Tabla 3 Parámetros cinéticos del 2-propenal 36

LISTA DE FIGURAS
Pág.
Figura 1 Mecanismo 1 8 Figura 2 Mecanismo 2 8 Figura 3 Diferencia de energía entre reactivo y complejo activado Ea 13 Figura 4 Sistema controlador de temperatura 25 Figura 5 Cromatógrafo de Gases-Masas 26 Figura 6 Espectro de masas y estructura de 2- propenal (acroleína) 28 Figura 7 Cromatógrama integrado y ampliado del reactivo y estándar 29 de la termólisis del 2-propenal Figura 8 Cinética a 180 grados centígrados desaparición de la acroleína 30 Figura 9 Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica del 31 2 propenal a 453.15 K Figura 10 Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica de 31 2 propenal a 473.15 K Figura11. Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica del 32 2 propenal a 493.15 K Figura 12 Gráfico de Arrhenius 33
Figura 13 Mecanismo 1 de descomposición térmica del 2-propenal 34 Figura 14 Mecanismo 2 de descomposición térmica del 2-propenal 34 Figura 15 Gráfico de Arrhenius 35

5
RESUMEN
En este trabajo se realizó el estudio cinético del 2-propenal, cuyos productos de descomposición son CO y eteno en un intervalo de temperatura de 180 a 220 º C y una presión de 1 atm. El estudio se abordó según la teoría del estado de transición mediante los parámetros de activación derivados del estudio cinético en solución. Los resultados de dicho estudio mecanístico permitieron postular dos posibles mecanismos: un primer mecanismo a través de un estado de transición cíclico de tres miembros sin intermediario y el otro un mecanismo de dos pasos por medio de la formación de carbeno intermediario a través de una estructura de transición cíclica de cuatro miembros. La cinética y los parámetros termodinámicos obtenidos arrojaron que el mecanismo de reacción es el segundo. El cual se muestra a continuación:
C C O
H
C
H
H
H
C O+C C
CH O
H
H
H
C C
H
H
H
H
C C
HH
H
H
C
H
H C
H
H
La descomposición del 2-propenal es homogénea, unimolecular y obedece una cinética de primer orden.
Palabras Claves: Termólisis, cinética, mecanismos, 2- propenal

6
ABSTRACT
In this work the kinetic study of the 2-propenal was carried out, whose products of thermal decomposition are CO and etene in an interval of temperature from 180 to 220 º C and a pressure of 1 Atm. The study was undertaken according to the theory of the state of transition by means of the by-products activation parameters of the kinetic study in solution. The results of said study permitted to advance two possible mechanisms: a first mechanism through a state of cyclic transition of three members without intermediary and the other a mechanism of two steps through the formation of carbeno intermediary through a structure of cyclic transition of four members. The kinetic one and the thermodynamic parameters obtained threw that the mechanism of reaction is the second. Which is shown subsequently:
C C O
H
C
H
H
H
C O+C C
CH O
H
H
H
C C
H
H
H
H
C C
HH
H
H
C
H
H C
H
H
The decomposition of the 2-propenal is homogeneous, unimolecular and obeys a kinetic leading one. Keywords: Termólisis, kinetic, mechanisms, 2- propenal

7
INTRODUCCION
Los aldehídos son un tipo importante de moléculas orgánicas, la cinética en descomposición térmica de éstos ha sido poco estudiada; por ello se realizó el estudio de la termólisis del 2- propenal. Uno de los métodos para el estudio de los mecanismos de reacción es la experimentación. Por medio de este se logran determinar ciertos parámetros que permiten realizar una propuesta de desarrollo del proceso. Para muchas reacciones, en particular las elementales, la expresión de velocidad puede escribirse como el producto de un término dependiente de la temperatura por otro dependiente de la composición. En el caso de estas reacciones se ha encontrado que prácticamente en todos los casos, el término dependiente de la temperatura, así como la constante de velocidad de la reacción, están bien representados por la ecuación de Arrhenius1. . Debido a que no es posible determinar directamente la estructura en el estado de transición, se recurren a cálculos teóricos que permiten la interpretación del fenómeno experimental.
1 LEVENSPIEL, Octave. Ingeniería de las Reacciones Químicas. Editorial Reverte. 1987. 2ª ed. p. 27

8
1. PLANTEAMIENTO DEL PROBLEMA
En estudios realizados acerca del mecanismo de reacción de la descomposición térmica de aldehídos α-β insaturados, se ha encontrado que como productos primarios se obtienen CO y un alqueno. Basándose en dichos productos y en la cinética de la reacción se ha propuesto que dicho proceso puede ocurrir a través de dos mecanismos, así pues se presentaran dichos mecanismos para el 2-propenal. Figura 1. Mecanismo 1
C C O
H
C
H
H
H
C C O
H
H
H
HC
C O+C C
H
H
H
H
Figura 2. Mecanismo 2
C C O
H
C
H
H
H
C O+C C
CH O
H
H
H
C C
H
H
H
H
C C
HH
H
H
C
H
H C
H
H
El primer mecanismo procede a través de una etapa y sugiere que esta reacción de eliminación, procede a través de un estado de transición cíclico de tres miembros. El segundo mecanismo sugiere la formación de una estructura de formación cíclica de cuatro miembro implica la formación de un carbeno como intermediario. El propósito de este trabajo experimental y computacional es seleccionar uno de los mecanismos postulados, realizado la termólisis del 2- propenal y siguiendo la cinética por medio de cromatografía de gases acoplado a masas.

9
2. OBJETIVOS
2.1 OBJETIVO GENERAL
Contribuir al campo de la investigación, estudio y descripción de los mecanismos de reacciones químicas orgánicas aplicando metodologías experimentales y teóricas disponibles en dicha área.
2.2 OBJETIVOS ESPECIFICOS
Estudiar la cinética de descomposición térmica en solución del 2-propenal teórica y experimentalmente, con el fin de determinar su velocidad de reacción y sus parámetros de activación ΔH#, ΔG#, ΔS.
Realizar un análisis comparativo entre los resultados experimentales y teóricos con el fin de contribuir a la elucidación del mecanismo de reacción propuesto, por medio del cual procede la termólisis y establecer las relaciones existentes con los sistemas similares.
Explorar las superficies de energía potencial, geometría de puntos estacionarios (reactivos, productos y estados de transición) y la variación de cargas alrededor del centro de reacción empleando teorías ab-initio, en la reacción de descomposición
térmica de aldehídos α-β insaturados.

10
3. MARCO TEORICO
3.1 ANTECEDENTES
El estudio y descripción del mecanismo de la reacción de termólisis de compuestos orgánicos en fase gaseosa y/o solución en solventes de baja polaridad, se ha venido realizando desde hace algún tiempo por el grupo de Fisicoquímica Orgánica. Trabajos experimentales en fase gaseosa relacionados con los aldehídos han sido publicados anteriormente y han mostrado que proceden a través de reacciones con radicales libres, en el benzaldehído, por ejemplo, el grupo CHO ataca directamente el sistema π, también va por vía radicalaria para producir CO como producto primario y el bifenil e hidrogeno gaseoso como productos secundarios. No es de nuestro conocimiento que se hayan publicado trabajos de este tipo de compuesto en solución, es por esto que se realizó el estudio tanto experimental como computacional partiendo de lo planteado por Chuchani y colaboradores quienes postulan dos posibles mecanismos de eliminación: uno de ellos de una sola etapa a través de un estado de transición cíclico de 3 miembros para obtener CO y un alqueno, y otro de dos etapas a través de un estado de transición cíclico de cuatro miembros para obtener CO y un intermediario carbeno que se rearregla para formar el alqueno.
3.2 CINÉTICA QUÍMICA
En química se realiza el estudio de las reacciones químicas teniendo en cuenta el mecanismo de reacción, los cambios físicos y energéticos que tiene lugar, y la velocidad con la que se forman los productos. En una reacción química, una sustancia (o grupo de sustancias) se convierte en otras. Para este proceso, se requiere una reordenación de los electrones de enlace de los átomos que constituyen las sustancias2.
La cinética química estudia la velocidad y el mecanismo a través del cual ocurre la reacción química. La velocidad se expresa como la concentración de un producto formado o de un reactante consumido por unidad de tiempo. El mecanismo es la secuencia de eventos químicos individuales cuyo resultado global produce la reacción observada. La cinética química no sólo se refiere a los estudios de las velocidades de reacción y su dependencia con los parámetros relevantes, sino también a la comprensión de los factores que determinan con qué rapidez tienen lugar las reacciones Químicas. Es por esto que la cinética abarca tanto medidas experimentales como enfoques empíricos y teóricos de la interpretación de estas medidas.
2 LOGAN, S. R. Fundamentos de Cinética Química. 1 ed. España. : addison wesley Iberoamericana, 2000. p. 2

11
3.3 ECUACION DE VELOCIDAD Se expresará la velocidad de una reacción química de una manera cuantitativa utilizando la definición de Velocidad de variación de la concentración como consecuencia de la reacción. Considerando la reacción hipotética.
(1)
La cual tiene lugar en un espacio cerrado. La velocidad de esta reacción viene dada por la velocidad de disminución de la concentración de A con el tiempo o por la velocidad de aumento de la concentración de B con el tiempo.
Para expresar la velocidad de variación de una manera más precisa se hace necesario recurrir a cálculos matemáticos, para la reacción anterior sería:
dt
d
dt
dreaccióndeVelocidad (2)
Así la velocidad de una reacción química se puede expresar como la velocidad de descomposición de un reactivo o la velocidad de formación de un producto.
3.3.1 Determinación experimental de la velocidad de reacción Cuando se afronta el
estudio cinético de una reacción, el primer objetivo es establecer la ecuación de velocidad. Lo cual supone la determinación de los órdenes de reacción y de la constante de velocidad.
Como primera aproximación, podemos suponer una ley general del tipo:
mnBAkv (3)
Donde los exponentes se conocen como orden de la reacción con respecto a A y B. Estos ordenes son cantidades de tipo experimental y no necesariamente integrales, la suma de los ordenes parciales n+m......, se denomina orden de la reacción. Para determinar los órdenes de reacción se puede hacer uso del método de las velocidades iníciales, el cual se basa en la medida de la velocidad inicial, vo, para diversas mezclas de reacción en las que se mantienen constantes todas las concentraciones excepto una. En esas condiciones la velocidad aparece como una función de la única concentración variable y, por lo tanto, se puede determinar fácilmente el orden de reacción respecto a dicha especie. Experimentalmente, la determinación de la velocidad inicial se lleva a cabo tomando medidas de la concentración dentro de un intervalo de tiempo suficientemente pequeño al comienzo de la reacción.

12
3.3.2 Tiempo de vida media El tiempo necesario para que la concentración inicial
disminuya hasta la mitad se conoce como el tiempo de vida media de la reacción. Para una reacción dada, la vida media
21
t de un reactivo en particular es el tiempo necesario
para que su concentración alcance un valor entre sus valores inicial y final. El valor de la vida media es inversamente proporcional a la constante de velocidad; y en general depende de las concentraciones de los reactivos3. Las medidas de la concentración del reactivo se toman haciendo uso de un método analítico suficientemente sensible, rápido y selectivo. Estos pueden dividirse en 2 categorías, Químicos o Físicos. Químicamente los análisis implican la determinación directa de uno de los reactivos o productos por procedimientos volumétricos o gravimétricos. Los métodos de análisis físicos son más convenientes que los Métodos Químicos ya que aquellos permiten seleccionar una propiedad física de la mezcla de reacción que cambia sustancialmente en el proceso de reacción por ej: La presión, la cual se mide a medida que cambia el tiempo; Dilatometría, medida del cambio de volumen; técnicas ópticas como: Polarimetría, Refractometría, Colorimetría, Fluorometría, Espectroscopia. Medidas eléctricas como: Conductimetría, Potenciometría, Polarografia; también se hace uso de Espectroscopia de Masas, Resonancia Magnética nuclear (NMR), Resonancia de espín de electrón (ESR), Cromatografía de Gas - Líquido (GL); teóricamente son algunas de las técnicas que se pueden usar porque permiten medir los cambios para seguir el curso de una reacción. Teóricamente cualquier propiedad que cambie lo suficiente puede usarse para seguir el curso de la reacción. Conductividad térmica, temperaturas de solidificación, viscosidad, fuente de coagulación hacia coloides, y colores de reacción son otras de las propiedades utilizadas4.
3.4 MECANISMOS DE REACCIÓN
Aunque algunas reacciones químicas ocurren en una sola etapa, la gran mayoría se producen en varias etapas. La forma de combinar estas etapas de la reacción, determina como se observa la desaparición de un reactivo y la formación de un determinado producto. Por esta razón, el estudio de las velocidades de una reacción química puede dilucidar el mecanismo mediante el cual se lleva cabo dicha reacción.
El estudio de los mecanismos de reacción ha sido objetivo importante de la Química experimental5. Esta ha permitido obtener evidencias importantes sobre la geometría de los reaccionantes y productos a partir de métodos espectroscópicos. En una reacción química, los reactivos constituyen el estado inicial y los productos el estado final. Al pasar del estado inicial al estado final, se produce un cambio de la energía
3 LAIDLER, Keith. J; MEISER, John. H. Fisicoquímica. 2 ed. México : Continental, 1998. p. 362-363 4 MOORE, J. W. and PEARSON, R. G. Kinetics and Mechanis. 3 ed. New York. John wiley and sons, 1980. p. 37-38 5 CAREY, F. A., SUNDBERG, R. J. Advanced Organic Chemistry : Parte A structure and mechanisms. 3 Ed. New York :
Plenum Press, 1990.

13
libre. Cuando el cambio es negativo se dice que la reacción es espontánea y no existe ningún impedimento para que la reacción se produzca, sin embargo este enfoque sólo tiene en cuenta las propiedades de reactivos y productos aislados, no incluye los posibles obstáculos que puedan existir durante la reacción.
La formación de un nuevo enlace requiere que las moléculas de los reactivos se acerquen a distancias lo suficientemente cortas para permitir un solapamiento eficaz de sus orbítales. Sin embargo, la disminución de la distancia implica un aumento de la repulsión entre las nubes electrónicas. Para vencer la repulsión, las moléculas de los reactivos deben acercarse con suficiente energía cinética. Por encima de una determinada energía que permita el solapamiento eficaz, se empezaran a formar los nuevos enlaces de los productos, a la vez que se debilitaran los enlaces de los reactivos, formándose una especie integrada por todas las moléculas de reactivos y en la cual unos enlaces se están rompiendo mientras otros se están formando. Esta especie se conoce como complejo activado. Finalmente, se acabaran de romper los enlaces de los reactivos para conducir a la formación de los productos de la reacción.
Puesto que para formar el complejo activado los reactivos deben vencer las fuerzas de repulsión, la energía del complejo activado es más alta que las energías de los reactivos y de los productos. La diferencia entre la energía de los reactivos y la del complejo activado se denomina energía de activación, y puede considerarse como una barrera energética que deben sobrepasar los reactivos para transformarse en productos. Como puede observarse en la Figura 3. Las reacciones en las cuales se produce la formación de un solo complejo activado y se supera una sola barrera de energía de activación, se conocen como reacciones elementales. Una reacción puede transcurrir también mediante dos o más procesos
elementales. En tal caso, diremos que la reacción es compleja. El número de moléculas que toman parte como reactivos en un proceso elemental se denomina molecularidad. Se conocen reacciones elementales unimoleculares,
bimoleculares y trimoleculares, aunque estas últimas son muy escasas. Figura 3. Diferencia de energía entre reactivo y complejo activado Ea

14
3.5 INFLUENCIA DE LA TEMPERATURA EN LAS CONSTANTES DE VELOCIDAD
Las constantes de velocidad (k) dependen sustancialmente de la temperatura y aumentan normalmente con ella. Una regla aproximadamente válida para muchas reacciones en disolución, es que cerca de la temperatura ambiente, k se duplica o triplica por cada aumento de temperatura de 10°C. Para la mayoría de las reacciones químicas se ha encontrado que el factor dependiente de la temperatura se ajusta a la ecuación de Arrhenius6 3.5.1 Ecuación De Arrhenius En 1889, Arrhenius demostró que los datos de la constante k (T) para muchas reacciones podían ajustarse por la expresión.
k= Aexp[-Ea/RT] (4) Donde A y Ea son constantes características de la reacción. Ea es la energía de activación de Arrhenius. Las unidades de A son las mismas de k. Las unidades de Ea son las mismas de RT, es decir, energía por mol; Ea generalmente se expresa en Kcal/mol o KJ/mol. Si la ecuación de Arrhenius se cumple, una representación de log k en función de 1/T ha de dar una línea recta de pendiente - Ea / 2.303 R y ordenada log A. Esto nos permite obtener Ea y A. El error típico experimental en Ea es 1 Kcal/mol y en A un factor de 3 ó 4.
La ecuación resulta satisfactoria para casi todas las reacciones homogéneas y muchas reacciones complejas. Una interpretación simple de k =Aexp[-Ea/RT] es que dos moléculas que colisionan requieren una cantidad mínima en su energía cinética relativa
6 MOORE, J. W. and PEARSON, R. G.. Kinetics and Mechanis. 3 ed. New York. John wiley and sons, 1980. p. 31-34

15
para romper el enlace apropiado y que pueda formarse el nuevo compuesto. Esta energía es la energía de activación (Para una reacción unimolecular se necesita una energía mínima para isomerizar o descomponer la molécula; la fuente de esta energía son las colisiones). También se puede describir la energía de activación como una barrera de energía potencial. Solo reaccionarán aquellas moléculas que tengan suficiente energía para alcanzar la cima de la barrera y formar el complejo activado. Con el fin de predecir teóricamente la velocidad de una reacción, es necesario postular la configuración del complejo activado o estado de transición. Es fácil comprender que cuanto más baja sea esta barrera (o sea mientras más baja sea la energía de activación) mayor será el número de moléculas activadas, y tanto más alta la velocidad de reacción y que una energía de activación alta significa reacción lenta. El aumento rápido de k, a medida que T aumenta, se debe principalmente al aumento en el número de colisiones cuya energía excede la energía de activación. Teorías más complejas de la velocidad de reacción conducen a ecuaciones similares a esta expresión excepto que A y Ea dependen de la temperatura. Cuando Ea » RT (lo cual se cumple para la mayoría de las reacciones químicas), las dependencias con la temperatura de Ea y A son, generalmente demasiado pequeñas para ser detectadas a partir de los datos cinéticos inexactos disponibles, a menos que se haya estudiado un amplio intervalo de temperatura.
Las energías de activación empíricas caen en el intervalo de 0 a 80 Kcal/mol para la mayoría de las reacciones químicas elementales y tienden a ser más bajas para reacciones bimoleculares que unimoleculares. En reacciones unimoleculares, A suele tener un valor entre 1012 y 1015 s-1.
La teoría de Arrhenius es el primer paso para dar una interpretación molecular de los procesos que ocurren en una reacción química. Sin embargo, la teoría no conduce a resultados cuantitativos, por lo que se han desarrollado otras, que basándose en las ideas de Arrhenius, pueden predecir en casos sencillos los resultados experimentales. 3.5.2 Teoría de las colisiones La teoría de las colisiones considera que la velocidad
está regida por el número de colisiones intermoleculares. Su objetivo es evaluar el número de colisiones efectivas que tienen lugar por unidad de volumen y unidad de tiempo. Para que dos moléculas experimenten una reacción química no solo deben chocar con suficiente energía, sino que también deben tener una orientación adecuada de tal manera que se puedan romper o formar los enlaces necesarios. La teoría de las colisiones, al considerar a las moléculas que reaccionan como esferas duras, dice que cada colisión es igual de efectiva que cualquier otra; por otra parte, cuando las moléculas son complejas, sólo en una pequeña fracción de colisiones éstas se unirán de manera correcta para que se lleve a cabo la reacción química.

16
Algunos investigadores trataron de remediar la situación conservando la teoría anterior, pero introduciendo en el factor preexponencial un factor estérico P, que supuestamente
representa la fracción del número total de colisiones efectivas desde el punto de vista de la orientación mutua de las moléculas. Así la constante de velocidad se representa:
RTE
ABePZk (5)
Este proceso genera algunas mejoras, pero no permite evaluar P de una manera satisfactoria, además existen ciertos factores aparte de la orientación, que influyen en la magnitud del factor preexponencial, y es imposible estimar con facilidad este factor.
Por lo tanto se requiere un método alterno para resolver el problema, es así como surgió un tratamiento más satisfactorio conocido como teoría del complejo activado o teoría del estado de transición.
3.5.3 Teoría del estado de transición Eyring, Polanyi y cols. (1931)7,8 , aplicaron los
principios de la mecánica cuántica al estudio de las velocidades de reacción y el resultado se conoce como teoría del complejo activado o teoría del estado de transición. Esta teoría, supone:
Que todas las supermoléculas que cruzan la superficie crítica desde el lado de los reactivos dan lugar a productos. Esta suposición es razonable, ya que una vez que la supermolécula cruza la superficie crítica, se encuentra en una trayectoria descendente hacia los productos.
Que durante la reacción se mantiene la distribución de Boltzmann para las energías de las moléculas reactivas.
Que las supermoléculas que cruzan la superficie crítica desde el lado de los reactivos tienen una distribución de Boltzmann de energías correspondiente a la temperatura del sistema reactivo9.
A + B PR O D U C TO STS
El estado de transición se concibe como una especie totalmente inestable, cuya concentración será siempre despreciable con respecto a la de A y B. Así la suposición
de equilibrio puede hacerse sin necesidad de considerar la disminución de las concentraciones de A y B; ya que el estado de transición permanece en equilibrio con los reactivos, independientemente de la velocidad a la que se van formando los productos de la reacción.
7 LAIDLER, Keith. J; GLASSTONE, Samuel and EYRING, H. The Theory of Rate Processes. New York : McGraw-Hill
Book Company, 1941. Cap I 8 LEVINE, Ira. N. Físico Química. 3 ed. España : Mcgraw-Hill, 1991. p. 986-987. 9 LEVINE, Ira. N. Físico Química. 3 ed. España : Mcgraw-Hill, 1991. p. 986

17
La constante de equilibrio o de pseudo-equilibrio se puede expresar como:
BA
TSK (6)
La velocidad de la reacción estaría dada por:
TSkdt
productod (7)
Donde k = constante de velocidad del estado de transición
Despejando [TS] de la ecuación 1 quedaría:
BAKkdt
productod
(8)
Basados en la aplicación de la termodinámica estadística la constante de equilibrio y la
constante de velocidad se pueden relacionar mediante la expresión:
Kh
Tkk
b
(9)
Donde
bk es la constante de Boltzmann, h = es la constante de planck, T = es la
temperatura (Kelvin) y K = constante de equilibrio entre los reactivos y el estado de transición. La constante de equilibrio también se puede expresar en términos de la energía libre de
Gibbs ( G ):
RTLnKG . (10)
La energía libre de Gibbs puede ser expresada en términos de la entalpía y la entropía de activación así:
STHG (11)
Esta ecuación permite discutir los cambios que se observan en las constantes de velocidad a partir de los cambios en las entropías y entalpías de activación.
Así la ecuación (9) se puede expresar como:

18
RT
G
h
kk
bexp (12)
Reemplazando 11 en 12:
R
S
RT
H
h
kk
bexpexp (13)
Esta ecuación es conocida como la ecuación de Eyring10 El factor preexponencial para reacciones en solución unimoleculares es11:
RS
Be
h
TkeA
/
(14)
3.6 CARACTERIZACION DE LOS COMPUESTOS
Dentro de las técnicas más utilizadas para la caracterización de compuestos orgánicos se tienen:
3.6.1 Espectrometría de masas La espectrometría de masas (MS) da nombre a un
conjunto de técnicas utilizadas para la medida de la masa de los iones y su abundancia en la fase gaseosa. Cada uno de los pasos de la medida de la masa son:
Generación de las moléculas en fase gaseosa (y fragmentos de moléculas y átomos)
Ionización
separación según su masa
Detección del pico del ión Este proceso de identificación se lleva a cabo en el siguiente orden: a. Identificación del ión molecular a partir de la fórmula molecular utilizando los pesos
atómicos de los isótopos más abundantes en la naturaleza. b. Modelo para el estudio de la distribución de los isótopos atómicos presentes en una
determinada molécula o un fragmento.
10 EYRING, H. En : Journal Chemical Physic. Vol 3 (1935); 492 11 LAIDLER, Keith. J; MEISER, John. H. Fisicoquímica. 2 ed. México : Continental, 1998. p. 386

19
c. Explicación del modelo de fragmentación. Cuando las moléculas de la muestra son ionizadas en la fuente, parte de la energía introducida hace que la molécula, una vez ionizada, se fragmente. Los fragmentos de la ruptura ayudan a corroborar la identificación de la molécula.
Un espectrómetro de masas bombardea una sustancia con un haz de electrones y registra cuantitativamente el resultado a modo de un espectro de fragmentos de ion positivo. La separación de los fragmentos tiene como base la relación masa / carga (m/z) 12. En un espectro de masas, las masas son medidas a partir de la posición del pico en el eje horizontal. Teniendo en cuenta que la magnitud del eje no es la masa. La espectroscopia infrarroja, en combinación con la espectrometría resonancia magnética nuclear y la espectroscopia de masas, forman la base del análisis químico orgánico cualitativo contemporáneo: La identificación de la estructura molecular de compuestos y mezclas desconocidas.
3.7 QUÍMICA COMPUTACIONAL
La química computacional es una rama de la química que utiliza los resultados de la química teórica, incorporados en algún software para calcular las estructuras y las propiedades de moléculas y cuerpos sólidos. Mientras sus resultados normalmente complementan la información obtenida en experimentos químicos, pueden, en algunos casos, predecir fenómenos químicos no observados a la fecha.
Ejemplos de propiedades y estructuras calculables con la química computacional son: la energía absoluta y relativa, distribución de carga electrónica, dipolo eléctrico y momentos multipolares superiores, frecuencias vibratorias, reactividad u otras cantidades espectrales y secciones eficaces para la colisión con otras partículas.
Los métodos empleados cubren situaciones estáticas y dinámicas. Estos métodos, por lo tanto, se basan en teorías que van desde la alta precisión, pero apropiados para pequeños sistemas, a las buenas aproximaciones, pero apropiadas para grandes sistemas. Los métodos más precisos son llamados métodos ab initio, los cuales están basados totalmente en la teoría de los primeros principios. Los menos precisos son llamados empíricos o semi-empíricos, debido a que son obtenidos de resultados experimentales, a menudo de átomos o moléculas relacionadas, se usan en conjunto a la teoría.
Las moléculas están conformadas de núcleos y electrones, de manera que los métodos de la mecánica cuántica pueden ser aplicados. Químicos computacionales a menudo intentan resolver la no relativista ecuación de Schrödinger, con correcciones relativista
12 RUBINSON, Kenneth A y RUBINSON, Judith F. Análisis Instrumental. Madrid : Prentice may, 2001. p. 522-525

20
agregadas, aunque algunos progresos han sido hechos resolviendo la ecuación de Schrödinger relativista por completo. Teóricamente, es posible resolver la ecuación de Schrödinger, ya sea en su forma tiempo-dependiente o independiente de manera apropiada para poder ser manejada, pero esto en la práctica sólo es posible para sistemas muy pequeños. Por lo tanto, un gran número de métodos de aproximación se esfuerzan por alcanzar de mejor manera la disyuntiva entre precisión y costo computacional. La precisión puede siempre ser mejorada incrementando el costo computacional. La química computacional del presente puede calcular con exactitud de manera rutinaria las propiedades de las moléculas que contienen a lo más 40 electrones. Errores para la energía pueden estar bajo 1 Kcal/mol. Para las geometrías, el tamaño de las cadenas puede ser predecido a los pocos picómetros y los ángulos de las cadenas con 0.5o. El tratamiento de grandes moléculas que contienen algunas decenas de electrones es computacionalmente tratable mediante método de aproximación tales como la teoría del funcional de la densidad (DFT). Existe cierta controversia en el campo debido a si los últimos modelos son suficientes para describir reacciones químicas complejas, como las presentes en la bioquímica. Grandes moléculas pueden ser estudiadas por medio de métodos aproximados semi-empíricos. Incluso las moléculas más grandes son tratadas por métodos de la mecánica clásica que son llamados mecánica molecular.
En la química teórica, químicos, físicos y matemáticos desarrollan algoritmos y software para predecir propiedades atómicas y moleculares y para encontrar los caminos que llevan a las reacciones químicas. Por el contrario, los químicos computacionales pueden simplemente aplicar los programas y metodologías existentes para las preguntas
químicas específicas. Hay dos aspectos diferentes para la química computacional:
Estudios computacionales pueden llevarse a cabo con el fin de encontrar un punto de partida para la síntesis de laboratorio o para ayudar en el entendimiento de la data experimental, tales como la posición y fuentes espectroscópicas peaks.
Estudios computacionales pueden ser usados para predecir moléculas hasta la fecha totalmente desconocidas o explorar mecanismos de reacción que no han
sido fáciles de estudiar mediante experimentos.
Dentro de la química computacional se destacan importantes áreas:
La predicción de la estructura molecular de las moléculas por el uso de simulación de fuerzas, o más precisión en los métodos de la química cuántica, para encontrar puntos estacionarios sobre la hipersuperficie de energía como la posición de los núcleos que es variada.
Almacenamiento y búsqueda de datos en entidades químicas. Identificar correlaciones entre estructuras químicas y propiedades. Enfoques computacionales para ayudar a una eficiente síntesis de componentes. Enfoques computacionales para diseñar moléculas que interactúen de forma
específica con otras moléculas (diseño de fármacos).
3.7.1 Métodos utilizados en química computacional Una fórmula molecular dada puede representar un número de moléculas isómeras. Cada isómero es un mínimo local

21
de energía sobre la superficie (llamado energía potencial de superficie) creada de la energía total (energía de los electrones más la energía de repulsión de los núcleos) como una función de coordenadas de todos los núcleos. Un punto estacionario es una geometría tal que la derivada de la energía con respecto a todos los desplazamientos de los núcleos es cero. Un mínimo local (de energía) es un punto estacionario donde todos esos desplazamientos conducen a un aumento de energía. El más bajo mínimo local es llamado mínimo global y corresponde al isómero más estable. Si hay un cambio en una coordenada en particular que lleve a una disminución de la energía total en ambas direcciones el punto estacionario es un estado de transición y la coordenada es la coordenada de reacción. Este proceso de determinar los puntos estacionarios es llamado optimización geométrica.
La determinación de la estructura molecular vía optimización geométrica se convirtió en una rutina sólo cuando métodos eficientes para el cálculo de la primera derivada de la energía con respecto a todas las coordenadas atómicas estuviesen disponibles. La evaluación de las segundas derivadas permite la predicción de las frecuencias vibratorias suponiendo movimientos armónicos. Las frecuencias están relacionadas con los valores propios de la matriz de segundas derivadas (la matriz Hessiana). Si los valores propios son todos positivos, entonces las frecuencias son todas reales y el punto estacionario es un mínimo local. Si un valor propio es negativo (una frecuencia imaginaria), el punto estacionario es un estado de transición. Si más de un valor propio es negativo el punto estacionario es más complejo aún, y usualmente de poco interés. Cuando se encuentra, es necesario mover la búsqueda fuera de él, si se está buscando un mínimo local y un
estado de transición.
La energía total está determinada por una solución aproximada de la ecuación de Schrödinger dependiente del tiempo, usualmente incluyendo términos no relativistas, y haciendo uso de la aproximación de Born-Oppenheimer, la cual, está basada en las más altas velocidades de los electrones en comparación con los núcleos, permitiendo la separación de movimientos electrónicos y nucleares, simplificando la ecuación de Schrödinger. Esto conduce a la evaluación de la energía total como una suma de energía electrónica en las posiciones fijas del núcleo más la energía de repulsión del mismo. Una notable excepción la constituyen los enfoques llamados química cuántica directa, los cuales tratan a los electrones y los núcleos por igual. Métodos del funcional de densidad y métodos semi-empíricos son variantes del tema principal. Para cada sistema grande, la energía total relativa puede ser comparada utilizando mecánica molecular. En nuestro caso se utilizan métodos ab initio para la determinación de la energía total para predecir
la estructura molecular.
3.7.1.1 Métodos ab initio Los programas utilizados en química computacional están basados en diferentes métodos de la química cuántica que resuelven la ecuación de Schrödinger asociada al Hamiltoniano molecular. Métodos que no incluyen ningún parámetro empírico o semi-empírico en sus ecuaciones (siendo derivadas directamente de principios teóricos, sin la inclusión de datos experimentales), son llamados métodos ab initio. Esto no implica que la solución sea exactamente una; son todos cálculos
aproximados de mecánica cuántica. Esto significa que una aproximación está

22
rigurosamente definida en base a los primeros principios (teoría cuántica) y su resolución es con un margen de error que es cualitativamente conocido de antemano. Si métodos numéricos iterativos han sido empleados, la meta es iterar hasta obtener la máxima precisión que la máquina pueda dar.
El tipo más simple de cálculo de estructura electrónica ab initio es el método de Hartree-Fock (HF), una extensión de la teoría de órbita molecular, en la cual la correlacionada repulsión electrón-electrón no es específicamente tomada en cuenta; sólo su efecto promedio es incluido en los cálculos. Como el tamaño de las bases de conjunto es incrementado, la energía y la función de onda tienden a un límite llamado el límite Hartree-Fock. Muchos tipos de cálculos, conocidos como métodos pos Hartree-Fock, comienzan con un cálculo Hartree-Fock para corregir posteriormente la repulsión electrón-electrón, conocida también como correlación electrónica. Ya que estos métodos son llevados al límite, la solución entregada se aproxima a la solución exacta de la ecuación no relativista de Schrödinger. Con el fin de obtener un total acuerdo con los experimentos, se hace necesario incluir términos relativistas y la interacción órbita-espín, ambos solamente importantes para átomos pesados. En todos estos enfoques, además de la elección del método, es necesario elegir un conjunto base. Éste es un conjunto de bases, usualmente centrado alrededor de los diferentes átomos de la molécula, los cuales son usados para expandir las órbitas moleculares. Los métodos ab initio necesitan definir un
nivel de teoría (el método) y un conjunto base.
Dentro del modelo mono electrónico, la función de onda Hartree-Fock viene descrita por una única configuración electrónica o determinante de Slater. Dicha descripción es adecuada para representar la naturaleza del estado fundamental de las moléculas, en general cerca de las estructuras de equilibrio de los sistemas. A partir de dicha referencia única los métodos químico-cuánticos ab initio añaden los efectos de correlación
electrónica a través de procedimientos de interacción de configuraciones, perturbativos u otros. Existen muchos casos en los que la descripción uniconfiguracional HF es inadecuada para representar la estructura electrónica del sistema, o para estudiar la evolución del sistema químico en diferentes regiones de las superficies de energía potencial. Situaciones típicas son el estudio de la disociación del enlace químico, el cálculo del estado electrónico excitado, la descripción de situaciones degeneradas entre estados, casos birradicalarios, estados de transición complejos, estructura electrónica en metales de transición, lantánidos o actínidos, con muchas capas electrónicas cercanas en energía, etc. En esas situaciones es necesario describir los estados electrónicos con más de una sola configuración electrónica a través de los métodos multiconfiguracionales, el más conocido de los cuales es el método CASSCF. En ellos tanto los coeficientes de las configuraciones como los coeficientes de los orbitales moleculares se optimizan simultáneamente. Una vez más, será necesario incluir el resto de la correlación electrónica a partir de dicha función de onda multiconfiguracional. El empleo de métodos uniconfiguracionales en situaciones que requieren descripciones multiconfiguracionales
da lugar, en general, a graves defectos en los resultados.
La energía molecular total puede ser evaluada como una función de la geometría molecular, en otras palabras, la energía potencial de superficie. Esta superficie puede ser usada para una reacción dinámica. Los puntos estacionarios de la superficie nos llevan a la predicción de diferentes isómeros y la estructuras de transición para la conversión entre
http://es.wikipedia.org/w/index.php?title=Teor%C3%ADa_de_%C3%B3rbita_molecular&action=edit&redlink=1

23
isómeros, pero estos pueden ser determinados sin un conocimiento acabado de la
superficie completa.
Un objetivo especialmente importante, llamada termoquímica computacional, es para calcular cantidades termoquímicas tales como la entalpía de formación para la precisión de químicos. La precisión de químicos es la precisión requerida para hacer predicciones químicas realistas y se considera generalmente que es 1 Kcal/mol ó 4 Kj/mol. Para alcanzar tal precisión de manera económica es necesario utilizar una serie de métodos pos Hartree-Fock y combinar los resultados. Estos métodos son llamados métodos compuestos de química cuántica.
3.7.1.1.1 Método de Hartree-Fock El método de Hartree-Fock (HF) es una forma
aproximada de las ecuaciones de mecánica cuántica para fermiones, utilizada en física y química (donde también se conoce como método de campo autoconsistente). Esto se debe a que sus ecuaciones, basadas en orbitales de una partícula, son más accesibles computacionalmente que los métodos basados en funciones de onda de muchas partículas.
La aproximación de Hartree-Fock es el equivalente, en física computacional, a la aproximación de orbitales moleculares, de enorme utilidad conceptual para los físicos. Este esquema de cálculo es un procedimiento iterativo para calcular la mejor solución monodeterminantal a la ecuación de Schrödinger independiente del tiempo, para moléculas aisladas, tanto en su estado fundamental como en estado excitados. La interacción de un único electrón en un problema de muchos cuerpos con el resto de los electrones del sistema se aproxima promediándolo como una interacción entre dos cuerpos (tras aplicar la aproximación de Born-Oppenheimer). De esta forma, se puede obtener una aproximación a la energía total de la molécula. Como consecuencia, calcula la energía de intercambio de forma exacta, pero no tiene en absoluto en cuenta el efecto
de la correlación electrónica.
La base del método de Hartree-Fock es suponer que la función de onda de muchos cuerpos es un determinante de Slater de orbitales de una partícula. Esto garantiza la antisimetría de la función de onda y considera la energía de intercambio. Sin embargo, no considera efectos de correlación que no necesariamente son despreciables. A partir de esta suposición, se puede aplicar el principio variacional de mecánica cuántica, se encuentra una ecuación de autovalores para los orbitales de una partícula.
El punto de partida para el cálculo Hartree-Fock es un conjunto de orbitales aproximados. Para un cálculo atómico, estos son típicamente los orbitales de un átomo hidrogenoide (un átomo con una carga nuclear cualquiera pero con un sólo electrón). Para cálculos moleculares o cristalinos, las funciones de ondas iníciales son típicamente una combinación lineal de orbitales atómicos. Esto da una colección de orbitales mono electrónicos, que por la naturaleza fermiónica de los electrones, debe ser antisimétrica, lo que se consigue mediante el uso del determinante de Slater. El procedimiento básico fue diseñado por Hartree, y Fock añadió el antisimetrizado.

24
Una vez se ha construido una función de ondas inicial, se elige un electrón. Se resume el efecto de todos los demás electrones, que se usa para generar un potencial. (Por este motivo, se llama a veces a este método un procedimiento de campo promedio). Esto da un electrón en un campo definido, para el que se puede resolver la ecuación de Schrödinger, dando una función de ondas ligeramente diferente para este electrón. Entonces, el procedimiento se repite para cada uno de los otros electrones, hasta completar un paso del procedimiento. De esta forma, con la nueva distribución electrónica se tiene un nuevo potencial eléctrico. El procedimiento se repite, hasta alcanzar la convergencia (hasta que el cambio entre un paso y el siguiente es lo suficientemente
pequeño).

25
4. METODOLOGIA
La metodología seguida en el estudio cinético de la descomposición del 2-propenal en solución fue: 4.1 TERMOLISIS DEL 2-PROPENAL La termólisis del 2-propenal se realizó en capilares de vidrio sellados con un diámetro interno de 2 mm y una longitud de 4 cm. Cada cinética estaba formada de 6 a 10
capilares, los cuales contenían 30 l de la solución cinética. Dicha solución cinética se preparo así:
0,1 ml acroleína (2-propenal)
2 ml de tolueno correspondiente al solvente
0,1 ml de benceno deuterado como estándar interno 4.1.1 Sistema controlador de temperatura Para la termólisis del 2-propenal se utilizó un sistema controlador de temperatura (sistema térmico), en el cual se introducían los capilares y posteriormente eran retirados en intervalos regulares de tiempo (8 y 12h). El sistema térmico se compone de las siguientes partes: Figura 4. Sistema controlador de temperatura
4.1.2 Seguimiento de la cinética de reacción por medio de espectroscopia gases-masas El seguimiento cinético se realizó por Cromatografía Gaseosa a través de un
cromatógrafo de gases marca Agilent 6890N con un detector selectivo de masas marca Agilent 5973, acoplado a un Chemstation Hardware. La cinética se siguió por la aparición de los productos y/o por la desaparición del 2-propenal al medir las áreas bajo la curva en los cromatogramas obtenidos.
HORNO
TERMOCUPLA
RELÉ
CONTROLADOR

26
Figura 5. Cromatógrafo de Gases-Masas
Las condiciones cromatográficas fueron:
Columna: Modelo No Zebron de 30 metros de longitud, diámetro interno 0.25 mm,
espesor de película 0.25 y límite de temperatura 340 C, programada a una temperatura
inicial de 40 C durante 5 minutos, de 40 a 100 C durante un tiempo total de corrida de
25 minutos; temperatura del detector 280 C; temperatura del inyector 180 C; gas de arrastre Helio. 4.2 CÁLCULOS PARA DETERMINAR CONSTANTES DE VELOCIDAD Y GRÁFICO DE ARRHENIUS.
Las constantes de velocidad para la reacción de termólisis se determinaron a partir de un
gráfico de t
Ln s tiempo, donde la pendiente es – kobs.
Los datos de las constantes obtenidas se pueden ajustar a la expresión:
RTEa
Aek/
(Ecuación de Arrhenius) (15)

27
Mediante una representación de )(1
KTvskLnobs
, para obtener una línea recta.
4.3 CÁLCULOS COMPUTACIONALES
Todos los cálculos computacionales se llevaron a cabo utilizando el paquete Gaussian, pero previamente se dibujaron las moléculas de interés con el programa Spartan Pro, y se realizó el análisis conformacional de los puntos estacionarios con un método semiempírico para determinar las moléculas más estables en la superficie de energía potencial (mínimos globales). Los parámetros geométricos de los puntos estacionarios se optimizaron usando el método ab-initio. Todos los parámetros de la geometría fueron optimizados usando un método del gradiente de energía. Los cálculos de las frecuencias vibracionales se realizaron para confirmar los puntos estacionarios, incluyendo las estructuras del estado de transición. De igual forma se realizó el cálculo de la coordenada intrínseca de reacción (IRC) con el fin de verificar que la estructura localizada del estado de transición conecta los reaccionantes y los productos correspondientes. Los cálculos en disolución se realizaron con el Modelo Continuo Polarizado que más se ajuste a las reacciones de interés.
28
5. RESULTADOS Y DISCUSIÓN
5.1 RESULTADOS DE ANÁLISIS INSTRUMENTAL
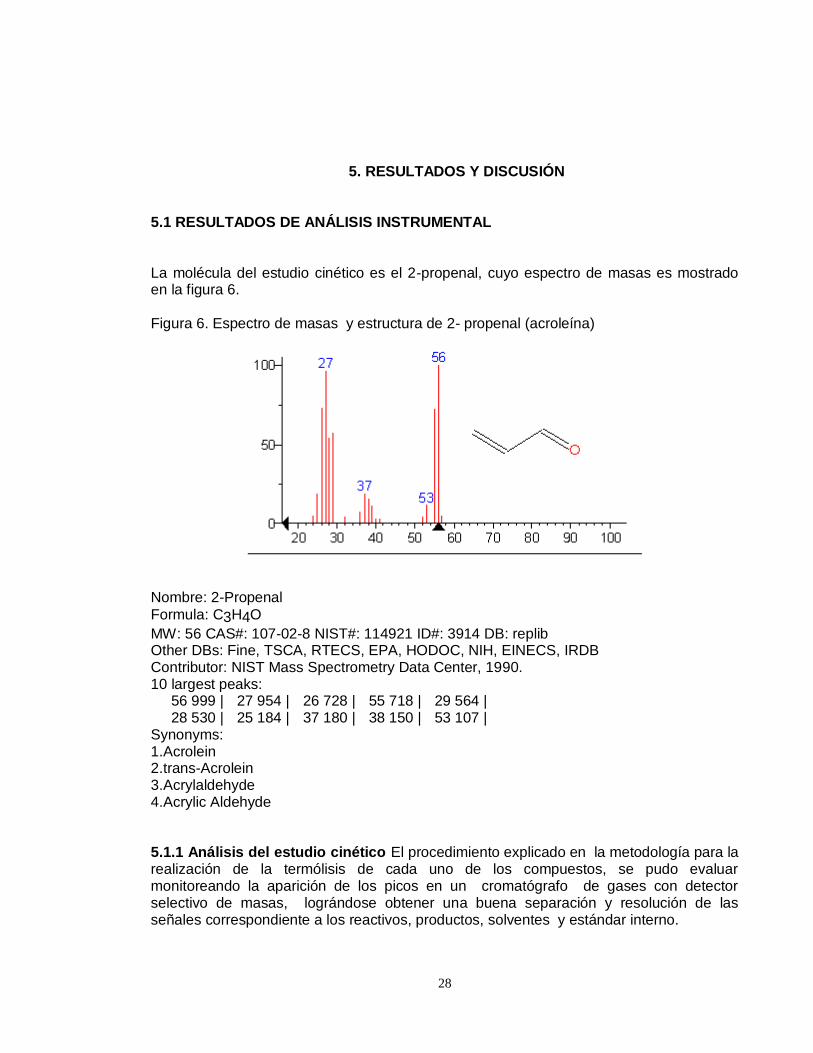
La molécula del estudio cinético es el 2-propenal, cuyo espectro de masas es mostrado en la figura 6.
Figura 6. Espectro de masas y estructura de 2- propenal (acroleína)
Nombre: 2-Propenal
Formula: C3H4O
MW: 56 CAS#: 107-02-8 NIST#: 114921 ID#: 3914 DB: replib Other DBs: Fine, TSCA, RTECS, EPA, HODOC, NIH, EINECS, IRDB Contributor: NIST Mass Spectrometry Data Center, 1990. 10 largest peaks: 56 999 | 27 954 | 26 728 | 55 718 | 29 564 | 28 530 | 25 184 | 37 180 | 38 150 | 53 107 | Synonyms: 1.Acrolein 2.trans-Acrolein 3.Acrylaldehyde 4.Acrylic Aldehyde 5.1.1 Análisis del estudio cinético El procedimiento explicado en la metodología para la realización de la termólisis de cada uno de los compuestos, se pudo evaluar monitoreando la aparición de los picos en un cromatógrafo de gases con detector selectivo de masas, lográndose obtener una buena separación y resolución de las señales correspondiente a los reactivos, productos, solventes y estándar interno.
29
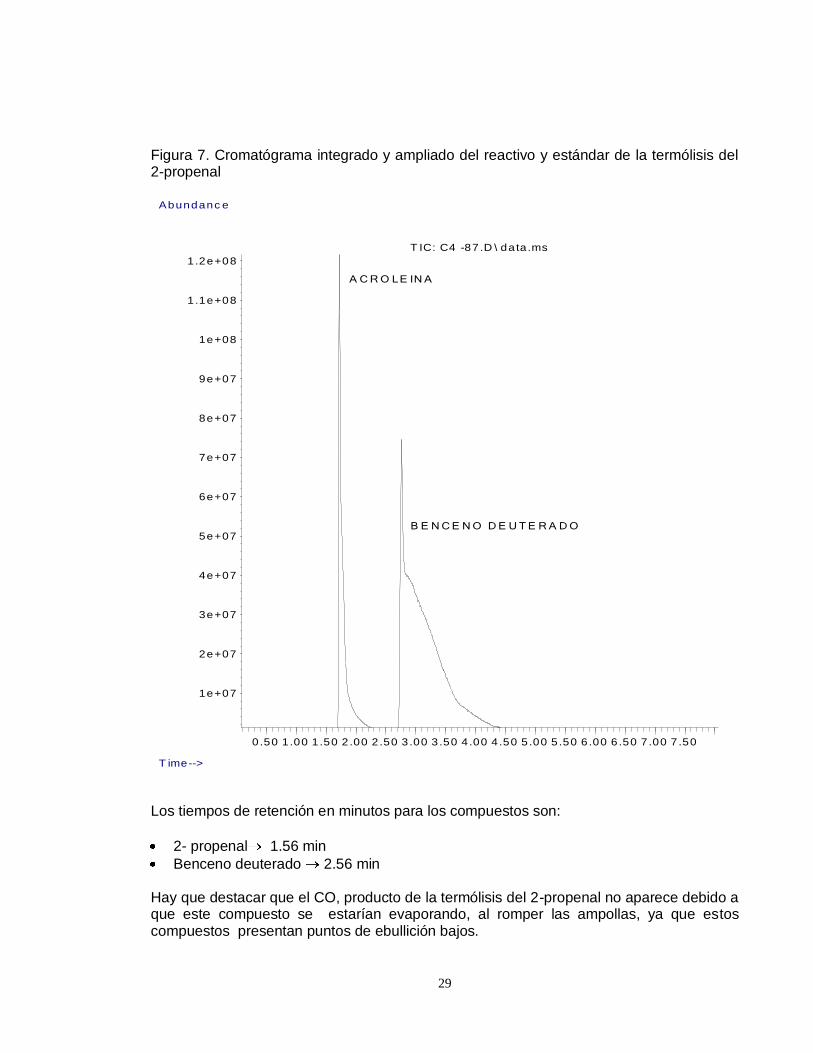
Figura 7. Cromatógrama integrado y ampliado del reactivo y estándar de la termólisis del 2-propenal
0.50 1.00 1.50 2.00 2.50 3.00 3.50 4.00 4.50 5.00 5.50 6.00 6.50 7.00 7.50
1e+07
2e+07
3e+07
4e+07
5e+07
6e+07
7e+07
8e+07
9e+07
1e+08
1 .1e+08
1 .2e+08
T ime-->
Abundanc e
T IC: C4 -87 .D \ da ta.ms
A C R O LE IN A
B E N C E N O D E U T E R A D O
Los tiempos de retención en minutos para los compuestos son:
2- propenal 1.56 min
Benceno deuterado 2.56 min Hay que destacar que el CO, producto de la termólisis del 2-propenal no aparece debido a que este compuesto se estarían evaporando, al romper las ampollas, ya que estos compuestos presentan puntos de ebullición bajos.

30
CO -191.5°C
Eteno 103.7 °C Se nota a una buena separación y resolución lograda en cada una de las señales. En la Figura 8, se presenta una superposición de los cromatógramas obtenidos para la reacción estudiada, observándose claramente el avance de la reacción en el transcurso del tiempo con la desaparición de la acroleína. Se nota claramente la disminución del reactivo a medida que pasa el tiempo. El avance de la reacción también puede ser determinado monitoreando la aparición los productos.
Figura 8. Cinética a 180 grados centígrados desaparición de la acroleína
1.68 1 .70 1 .72 1 .74 1 .76 1 .78 1 .80 1 .82 1 .84 1 .86 1 .88 1 .90 1 .92 1 .94 1 .96 1 .98
2000000
4000000
6000000
8000000
1e+07
1 .2e+07
1 .4e+07
1 .6e+07
1 .8e+07
2e+07
T ime-->
Abundanc e
T IC: C4 - 135 .D \ da ta .ms
T IC: C4 - 136 .D \ da ta .ms
T IC: C4 - 137 .D \ da ta .ms
T IC: C4 - 138 .D \ da ta .ms
T IC: C4 - 139 .D \ da ta .ms
T IC: C4 - 140 .D \ da ta .ms
T IC: C4 - 141 .D \ da ta .ms
5.2 CÁLCULOS PARA DETERMINAR CONSTANTES DE VELOCIDAD Y GRÁFICO DE ARRHENIUS

31
La cuantificación de la constante de velocidad experimental se realizó mediante la siguiente ecuación para una reacción de primer orden
kt
aexa (16)
Donde a representa la concentración inicial y x la concentración al tiempo t .
Cuando se utiliza la propiedad física , para seguir la reacción, la ecuación anterior se transforma en:
ktLnt
0
(17)
Donde es la relación de área Acroleína/Benceno ([A]/[B]) para los tiempos (t). Los datos obtenidos fueron ajustados al modelo de regresión lineal, de cuya pendiente se obtuvo el valor de la constante de velocidad. Figura 9. Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica del 2 propenal a 453.15
K
Figura 10. Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica del 2 propenal a
473.15 K

32
Figura11. Gráfico Ln ([A]/ [B]) vs t para la descomposición térmica del 2 propenal a 493.15
K
En general todas las cinéticas muestran una buena correlación lineal 97.0–99.9 %, lo cual confirma una reacción de primer orden.

33
En la tabla 1, se observa que los valores de las constantes de velocidad se hacen más grandes a medida que aumenta la temperatura Tabla 1. Constantes de velocidad k (s-1) para la descomposición térmica de 2-propenal a
varias temperaturas.
Temperatura k k promedio
453.15 K 4E-06 4E-06 4E-06
473.15 K 7E-06 8E-06 7,5E-06
493.15 K 1E-05 1E-05 1E-05
Con estas constantes se realizó el gráfico de Arrhenius para obtener el valor de la energía de activación.
Figura 12. Gráfico de Arrhenius
Con esto se obtiene finalmente el valor experimental de la energía de activación;
Ea= 42,76045 kJ.mol-1.
Este valor de la energía de activación indica la altura de la barrera energética durante el paso de los reactivos a los productos, implicando una participación energética en la formación y ruptura de enlaces.

34
5.3 RESULTADOS DE ANÁLISIS COMPUTACIONAL En Spartan pro se dibujaron las móleculas que participan en la reacción y posteriormente se exportaron Gaussian view. A continuación se muestran los mecanismos representados
a través del programa (figuras 13 y 14).
Figura 13. Mecanismo 1 de descomposición térmica del 2-propenal
Figura 14. Mecanismo 2 de descomposición térmica del 2-propenal

35
Mediante este análisis computacional se determinaron todos los parámetros cinéticos y termodinámicos del 2-propenal a la temperatura de 453,15; 473,15 y 493,15ºK. A continuación se presentan los resultados de la constante cinética obtenida para los dos mecanismos de reacción. Tabla 2. Constante cinética de 2-propenal
Teniendo en cuenta que los valores que más se asemejan a las constantes halladas experimentalmente, son las que se refieren al segundo mecanismo de reacción. Por lo tanto con estas constantes se realizó el gráfico de Arrhenius para obtener el valor de la energía de activación.
Figura 15. Gráfico de Arrhenius
Temperatura k Mecanismo 1 k Mecanismo 2
453,15ºK 1,86914E-38 1,46195E-06
473,15ºK 2,7713E-36
3,204E-06
493,15ºK 2,7436E-34
7,03171E-06

36
Con esto se obtiene finalmente el valor computacional de la energía de activación.
Ea= 72,909523 KJ.mol-1
El estudio de los parámetros cinéticos obtenidos experimentalmente y también computacionalmente nos permite suponer también algunos aspectos relacionados con el mecanismo de reacción del 2-propenal.
Tabla 3. Parámetros cinéticos del 2-propenal
PARÁMETROS EXPERIMENTAL COMPUTACIONAL
Ea (KJ.mol-1) 42.76 72.91
S (J.mol-1K-1) -14.75 -15.85
H (KJ.mol-1) 157.14 159.98
G (KJ.mol-1) 164.12 167.48
Las entropías de activación obtenida experimentalmente y computacionalmente en
solución son negativas S 0, esto nos indica que la entropía del estado de transición es más baja que la entropía de los reactivos lo cual coincide con un estado de transición más ordenado que los reactivos.
Los valores de H y los valores de G son altos, lo cual sugiere un estado de transición avanzado, muy parecido a los productos.

37
6. CONCLUSIONES
Se confirma que a medida que aumenta la temperatura para la termólisis del 2-propenal; aumenta la velocidad de reacción, tanto en el análisis experimental como en el computacional.
Posiblemente el mecanismo de reacción que sigue la descomposición térmica del 2-propenal es el mecanismo 2, el cual tiene un primer estado de transición de cuatro miembros para la formación de un carbeno.
Los valores de la entropía de activación negativa, confirman que el mecanismo de reacción se lleva a cabo como se había postulado, con un estado de transición cíclico de cuatro miembros con la formación de un carbeno intermediario.

38
BIBLIOGRAFÍA AUGUST, Ryan; McEWEN, Lan and TAYLOR, Roger. The mechanism of Thermal Eliminations. Part 22. En : Journal chemical of society Perkins Trans. II . (1987); p. 1683 – 1689. CAREY, F. A., Sundberg, R. J. Advanced Organic Chemistry : Parte A structure and mechanisms. 3 ed. New York : Plenum Press, 1990. CHUCHANI, G. et al. Kinetic and mechanism of the homogeneous, unimolecular elimination of α, β-unsaturated aldehydes in the gas phase. En : Journal of Physical Organical Chemistry. 2007;p. 307-312. INSTITUTO COLOMBIANO DE NORMAS TÉCNICAS. Normas colombianas para la presentación de tesis, trabajos de grado y otros trabajos de investigación. Sexta actualización. Santafé de Bogotá D.C. ICONTEC, 2008. 42p. NTC 1486. INSTITUTO COLOMBIANO DE NORMAS TÉCNICAS. Referencias bibliográficas. Contenido forma y estructura. Santafé de Bogotá D.C. ICONTEC, 2008. 38p. NTC 5613. LAIDLER, Keith. J; GLASSTONE, Samuel and EYRING, H. The Theory of Rate Processes. New York : McGraw-Hill Book Company, 1941. Cap I. LAIDLER, Keith. J; MEISER, John. H. Fisicoquimica. 2 ed. México : Continental, 1998. p. 362-363. LEVENSPIEL, Octave. Ingeniería de las Reacciones Químicas. Editorial Reverte. 1987. 2ª ed. LEVINE, Ira. N. Físico Química. 3 ed. España : Mcgraw-Hill, 1991. p. 986-987. LOGAN, S. R. Fundamentos de Cinética Química. 1 ed. España : addison wesley Iberoamericana, 2000. p. 2. MARCH, J. Advanced Organic Chemistry. 3 ed United States : John wiley & sons, 1985. p. 822.

39
MOORE, J. W. and Pearson, R. G. Kinetics and Mechanis. 3 ed. New York. John wiley and sons, 1980. p. 31-34, 37-38. RUBINSON, Kenneth A y RUBINSON, Judith F. Análisis Instrumental. Madrid : Prentice may, 2001. p. 446, 522-525.


















