ANFETAMINA, METANFETAMINA Y SUS ANÁLOGOS CON ANILLO SUSTITUIDO
75
NACIONES UNIDAS OFICINA CONTRA LA DROGA Y EL DELITO MÉTODOS RECOMENDADOS PARA LA IDENTIFICACIÓN Y EL ANÁLISIS DE ANFETAMINA, METANFETAMINA Y SUS ANÁLOGOS CON ANILLO SUSTITUIDO EN MATERIALES DECOMISADOS (Revisados y actualizados) MANUAL PARA USO DE LABORATORIOS NACIONALES DE ENSAYO DE ESTUPEFACIENTES V.05-86469
description
MANUAL PARA USODE LABORATORIOS NACIONALES DE ENSAYODE ESTUPEFACIENTES
Transcript of ANFETAMINA, METANFETAMINA Y SUS ANÁLOGOS CON ANILLO SUSTITUIDO

NACIONES UNIDAS OFICINA CONTRA LA DROGA Y EL DELITO MÉTODOS RECOMENDADOS PARA LA IDENTIFICACIÓN Y EL ANÁLISIS DE
ANFETAMINA, METANFETAMINA Y SUS ANÁLOGOS CON ANILLO SUSTITUIDO EN MATERIALES DECOMISADOS (Revisados y actualizados) MANUAL PARA USO DE LABORATORIOS NACIONALES DE ENSAYO DE ESTUPEFACIENTES V.05-86469


Sección de Laboratorio y Asuntos Científicos OFICINA DE LAS NACIONES UNIDAS CONTRA LA DROGA Y EL DELITO Viena MÉTODOS RECOMENDADOS PARA LA IDENTIFICACIÓN Y EL ANÁLISIS DE
ANFETAMINA, METANFETAMINA Y SUS ANÁLOGOS CON ANILLO SUSTITUIDO
EN MATERIALES DECOMISADOS (Revisados y actualizados) MANUAL PARA USO DE LABORATORIOS NACIONALES DE ENSAYO DE ESTUPEFACIENTES
NACIONES UNIDAS Nueva York, 2005

La mención de nombres de empresas y productos comerciales no implica opinión alguna de parte de las Naciones Unidas. La presente publicación no ha pasado por los servicios de edición.
ST/NAR/34

iii
AGRADECIMIENTOS La Sección de Laboratorio y Asuntos Científicos de la ONUDD desea expresar su agradecimiento a los expertos que participaron en la Reunión de Consulta sobre “El examen de los métodos para la identificación y el análisis de estimulantes de tipo anfetamínico y sus análogos con anillo sustituido en material decomisado” por su contribución al contenido del presente manual. Sra. Rosa Alis Rodríguez, Laboratorio de Drogas y Sanidad de Baleares, Palma de Mallorca (España) Dr. Hans Bergkvist, SKL — Laboratorio Nacional de Ciencias Forenses, Linköping (Suecia) Sra. Warank Boonchuay, División de Análisis de Estupefacientes, Departamento de Ciencias Médicas, Ministerio de Salud Pública, Nonthaburi (Tailandia) Dr. Rainer Dahlenburg, Bundeskriminalamt / KT34, Wiesbaden (Alemania) Sr. Adrian V. Kemmenoe, The Forensic Science Service, Birmingham Laboratory, Birmingham (Reino Unido) Dr. Tohru Kishi, Instituto Nacional de Investigación de Ciencia Policial, Chiba, Japón Dr. Waldemar Krawczyk, Laboratorio Forense Central de Policía, Ministerio del Interior y Administración, Varsovia, Polonia Sr. Ira Lurie, Special Testing and Research Laboratory, Drug Enforcement Administration, McLean, Virginia (Estados Unidos de América) Dr. Yukiko Makino, Departamento de Fiscalización de Estupefacientes, Kanto-Shin’etsu Oficina de Salud y Bienestar, Ministerio de Salud, Trabajo y Bienestar, Tokio (Japón) Sr. Tim McKibben, Special Testing and Research Laboratory, Drug Enforcement Administration, McLean, Virginia (USA) Sra. Anneke Poortman, Laboratorio de Ciencias Forenses, Ministerio de Justicia, Rijswijk (Países Bajos) Sra. Jana Skopec, Australian Government Analytical Laboratories, Pymble, NSW (Australia) Sr. Takahiro Terasaki, Kanto-Shin’etsu Oficina Regional de Fiscalización de Estupefacientes, Ministerio de Salud, Trabajo y Bienestar, Tokio (Japón) La Sección de Laboratorio y Asuntos Científicos de la ONUDD desea también expresar su agradecimiento a la Sra. Jana Skopec por la revisión, actualización y finalización del manuscrito, también con contribuciones adicionales de los participantes en la reunión1.
1 También se agradece mucho la revisión del proyecto del manual que hizo el Dr. Ken Tanaka, de la Agencia Nacional de Policía del Japón.


v
Índice
I. INTRODUCCIÓN.......................................................................................................... 1II. EMPLEO DEL MANUAL............................................................................................... 2III. CLASIFICACIÓN / DEFINICIONES.............................................................................. 3IV. DESCRIPCIÓN DE LOS COMPUESTOS PUROS ...................................................... 4 A. Estereoquímica .................................................................................................... 4 B. Características físicas .......................................................................................... 5V. FABRICACIÓN ILÍCITA DE ETA .................................................................................. 6 A. Síntesis de la anfetamina..................................................................................... 7 B. Síntesis de la metanfetamina............................................................................... 8 C. Síntesis de ETA con anillo sustituido................................................................... 10VI. ANÁLISIS CUALITATIVO Y CUANTITATIVO DE ETA................................................ 13 A. Ensayos presuntivos ............................................................................................ 13 1. Ensayos cromáticos..................................................................................... 13 2. Ensayo de aniones ...................................................................................... 17 3. Ensayos de microcristales ........................................................................... 19 B. Cromatografía de capa delgada (CCD) .............................................................. 19 C. Cromatografía en fase gaseosa (CG) — detector de ionización por llama (FID) 25 1. Análisis cualitativo ....................................................................................... 26 2. Análisis cuantitativo ..................................................................................... 27 i. Método A: método estándar único........................................................ 27 ii. Método B: método estándar múltiple sin derivatización ...................... 29 iii. Método B: método estándar múltiple con derivatización ..................... 31 D. Cromatografía en fase gaseosa — espectrometría de masas (CG-EM) ............ 31 E. Cromatografía líquida de gran rendimiento (CLGR) ........................................... 33 F. Espectrometría infrarroja transformada de Fourier (FTIR) ................................. 35 G. Análisis de isómeros ópticos................................................................................ 37 1. Punto de fusión............................................................................................ 38 2. Ensayos de microcristales ........................................................................... 38 3. Técnicas instrumentales .............................................................................. 40 i. Técnicas de CG .................................................................................... 40 ii. Técnicas de CLGR................................................................................ 42 iii. Técnicas de EC..................................................................................... 42 iv. Técnicas IR ........................................................................................... 43VII. OTRAS TÉCNICAS ANALÍTICAS PARA EL ANÁLISIS DE ETA................................. 45 A. Técnicas de resonancia magnética nuclear 1H (RMN) ....................................... 45 B. Electroforesis capilar (EC) .................................................................................. 46 C. Cromatografía en fase sólida — microextracción en fase sólida (CG-SPME) 46

vi
1. Método de análisis de cabeza .................................................................... 47 2. Análisis (de trazas) de muestras acuosas.................................................. 47 D. Cromatografía en fase gaseosa — espectroscopía infrarroja transformada de
Fourier (CG-FTIR) ............................................................................................... 47
ANEXOS................................................................................................................................ 49

1
I. INTRODUCCIÓN En el plano internacional la atención se centra cada vez más en una cuestión de importancia creciente: los estimulantes de tipo anfetamínico (ETA). En particular durante los últimos 10 a 15 años, el uso indebido de ETA relacionado con las anfetaminas (anfetamina y metanfetamina) y sustancias del grupo “éxtasis” (MDMA, MDA, MDEA, etc.), ha pasado a ser un problema mundial. Aunque hay diferencias regionales, en la actualidad ningún país está a salvo de una de las múltiples facetas de la fabricación, el tráfico o el uso indebido de ETA. Esta nueva situación, que con frecuencia abarca ETA nuevos y desconocidos, o combinaciones, y las tendencias del tráfico plantean un desafío tanto a las autoridades nacionales encargadas de hacer cumplir la ley como al personal científico de los laboratorios forenses. Hoy en día, los analistas deben estar en condiciones de analizar una amplia gama de sustancias y preparados, y deben utilizar métodos más precisos y científicos de identificación y análisis a fin de hacer frente al mayor número de análisis necesarios y a los requerimientos de las leyes nacionales más estrictas contra las drogas. Además, el carácter internacional del tráfico de drogas requiere el intercambio oportuno de datos analíticos entre laboratorios y los servicios de represión en los planos nacional, regional e internacional. Por estas razones, la Sección de Laboratorio y Asuntos Científicos de la ONUDD comenzó a principios de los años 80 a ejecutar un programa de armonización y establecimiento de métodos de ensayo recomendados para laboratorios nacionales de ensayo de drogas. En septiembre de 1998, la Sección de Laboratorio y Asuntos Científicos de la ONUDD, en cooperación con el Servicio de Ciencias Forenses del Reino Unido, organizó una reunión consultiva con la participación de 13 expertos para examinar métodos para la identificación y el análisis de estimulantes de tipo anfetamínico (ETA) y sus análogos con anillo sustituido en material decomisado. El presente manual refleja las conclusiones de esa reunión, que fueron revisadas y actualizadas nuevamente en 2004/2005. Ofrece asistencia práctica a las autoridades nacionales describiendo métodos recomendados para su empleo en laboratorios de ensayo de drogas con miras a identificar y analizar estimulantes de tipo anfetamínico (ETA) y sus análogos con anillo sustituido. El presente manual forma parte de una serie de publicaciones similares que tratan de la identificación y el análisis de diversos grupos de drogas sometidas a fiscalización internacional. Combina y reemplaza manuales publicados anteriormente sobre “Métodos recomendados para el ensayo de anfetamina y metanfetamina” (ST/NAR/9, 1987) y “Métodos recomendados para el ensayo de los derivados anfetamínicos ilícitos con anillo sustituido” (ST/NAR/12, 1988). El presente manual y los anteriores proponen enfoques que pueden ayudar a los analistas de drogas a seleccionar métodos apropiados a la muestra que se examina, con la posibilidad también de adaptarlos al nivel de adelanto de los diferentes laboratorios. Por primera vez en esta serie de publicaciones, el presente manual tiene también anexos con métodos validados seleccionados. La mayoría de los métodos que se describen han sido publicados en la bibliografía científica, y han sido utilizados durante un cierto número de años por laboratorios de reputación conocida. Al identificar esos métodos, la Reunión Consultiva tuvo presente que había varios otros métodos publicados en la literatura de las ciencias forenses que también producían resultados aceptables. El presente manual se limita a los métodos analíticos para los ETA. Un manual separado sobre técnicas analíticas más generales, y sus características y empleos prácticos para el análisis de drogas, complementa esta serie de manuales sobre métodos recomendados.

2
II. EMPLEO DEL MANUAL No todos los métodos descritos en el presente manual deben aplicarse a todas las muestras sospechosas de ser o contener anfetamina, metanfetamina u otros ETA. La elección del método y el enfoque de su análisis quedan a la discreción de los analistas y dependen del tipo de droga de que se trate, la disponibilidad de los instrumentos y los materiales de referencia apropiados, así como del nivel de prueba jurídicamente aceptable en la jurisdicción en que trabajan los analistas. Por consiguiente, si bien se reconoce que los requisitos exclusivos de diferentes jurisdicciones pueden determinar las prácticas que aplican los diferentes laboratorios, las “Buenas Prácticas de Laboratorio” exigen que un enfoque analítico para determinar la identidad de una sustancia sometida a fiscalización en material sospechoso comprenda, como mínimo, la determinación de por lo menos dos parámetros no relacionados entre sí. La selección de esos parámetros en cualquier caso particular deberá tener en cuenta la droga de que se trate y los recursos de laboratorio de que dispone el analista. Cuando sea posible, se deben utilizar tres técnicas analíticas totalmente diferentes: ensayos cromáticos, cromatografía (por ejemplo, CCD, CG o CLGR) y espectroscopía (por ejemplo, IR o UV). Las técnicas compuestas, como CG-EM, cuentan como dos parámetros, siempre que se utilice la información de ambas técnicas (por ejemplo, el tiempo de retención y las características del espectro de masa). Se hace hincapié también en la importancia fundamental de que los analistas de drogas dispongan de libros de referencia sobre drogas de uso indebido y técnicas analíticas. Además, el analista debe necesariamente estar al corriente de las tendencias del momento en el análisis de drogas, ajustándose coherentemente a la literatura sobre ciencias forenses y análisis de actualidad. La ONUDD ayuda a los laboratorios a este respecto proporcionando, previa solicitud, artículos seleccionados de la literatura científica. La Sección de Laboratorio y Asuntos Científicos de la ONUDD agradecerá todas las observaciones que se le hagan llegar sobre el contenido y la utilidad del presente manual. Los comentarios y las sugerencias se pueden dirigir a:
Sección de Laboratorio y Asuntos Científicos Oficina de las Naciones Unidas Contra la Droga y el Delito Centro Internacional de Viena P. O. Box 500 A-1400 Viena (Austria) Fax: +43-1-26060-5967 Correo-e: [email protected]
Todos los manuales, así como las directrices y otras publicaciones científicas y técnicas se pueden solicitar a la dirección mencionada más arriba.

3
III. CLASIFICACIÓN / DEFINICIONES Los estimulantes de tipo anfetamínico (ETA) son un grupo de sustancias, en su mayoría de origen sintético, estructuralmente derivadas de la β-fenetilamina (β-PEA, gráfico 1). Los ETA por lo general estimulan el sistema nervioso central. Por lo tanto son considerados, en diverso grado, como prototipos de estimulantes del sistema nervioso central con un potencial de toxicidad
sicótica cuando se usan en dosis excesivas o por largos períodos. Los ETA pueden producir uno o más síntomas relacionados con la dosis, incluido un estado de mayor alerta y euforia, pulsaciones aceleradas, mayor presión arterial, respiración y temperatura corporal. El uso indebido crónico puede producir agitación, temblores, hipertensión, pérdida de memoria, alucinaciones, episodios sicóticos, ilusiones paranoicas y comportamientos violentos. La abstinencia después de haber tomado dosis altas de ETA puede provocar depresión grave. Los
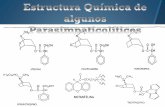
ETA se producen ilícitamente en una variedad de preparados (polvo, tabletas o cápsulas), y se pueden inyectar, ingerir oralmente, esnifar o fumar. La modificación química en los puntos R1 a R9 (gráfico 2)2 produce un número prácticamente ilimitado de compuestos farmacológicamente activos, algunos de los cuales son estimulantes más potentes que otros. Aunque hay varias posibilidades para la modificación de la cadena lateral, la sustitución en el anillo aromático aporta la mayor contribución a diferencias cualitativas sustanciales en los efectos farmacológicos. En cuanto a las características estructurales, los ETA se pueden dividir en tres grandes subgrupos, que corresponden en gran parte a las siguientes pautas de sustitución en el anillo aromático:
i. Ninguna sustitución en el anillo aromático (por ejemplo, anfetamina, metanfetamina, fenetilina);
ii. Grupo metilendioxi: sustitución en el anillo aromático (por ejemplo, MDA, MDMA, MBDB);
iii. Otras pautas de sustitución, por lo general incluyendo uno o más de los grupos alkiloxi (por ejemplo, 2C-B, STP/DOM).
Por razones de orden práctico, el presente manual contiene datos específicos sólo para una selección de los ETA más comunes. En particular, incluye ETA sometidos a fiscalización internacional y ETA seleccionados sometidos a fiscalización nacional. Los analistas deben tener presente que se pueden encontrar otros análogos estrechamente relacionados. En la mayoría de los casos, la metodología presentada se podrá aplicar también a esos análogos. Las estructuras químicas de los ETA seleccionados, junto con los nombres comunes y de la nomenclatura de la Unión Internacional de Química Pura y Aplicada (Nomenclatura IUPAC) figuran en el anexo I.
2 No se muestran todos los otros sustituyentes para saturar las valencias, ya que por lo general se utiliza hidrógeno (H).
NH2
Gráfico 1
R3R5
R9
R8
R7
R6
NR2
R1R4
Gráfico 2

4
IV. DESCRIPCIÓN DE LOS COMPUESTOS PUROS Los ETA se decomisan normalmente en forma de sales, en particular como clorhidratos, sulfatos, fosfatos o sales de bromuro. No obstante, no es difícil que en las investigaciones de laboratorios clandestinos se encuentren esos compuestos también en forma de bases (por lo general, un líquido aceitoso de color pardo). Las sales son sustancias cristalinas o en polvo, que varían en coloración desde el blanco (similar a los productos de calidad farmacéutica) hasta el rosado, el amarillo o el pardo. Con frecuencia se encuentran húmedos y despiden un olor desagradable característico, debido a la presencia de residuos de disolventes y/o precursores. Los ETA también se pueden encontrar en forma de tabletas. Además de los ETA activos, las tabletas suelen contener diferentes adulterantes, agentes reductores, colores de alimentos comunes y/o diferentes excipientes y agentes aglutinantes.
Anfetamina La anfetamina ilícita se suele encontrar en forma de polvo como sal sulfatada, y rara vez como tableta. En los laboratorios clandestinos se suele decomisar anfetamina base, normalmente como líquido aceitoso pardo con un olor desagradable característico de 1-fenil-2-propanona (P-2-P) y/o residuos de disolvente.
Metanfetamina La metanfetamina fabricada en forma ilícita está disponible en diferentes formas, según la región geográfica. Estas formas incluyen el polvo, los cristales (comúnmente conocidos como “Cristal”, “Ice” o “Shabu”) y las tabletas (comúnmente conocidas como “Yaba”). La forma de sal más común es el clorhidrato.
ETA con anillo sustituido con un grupo metilendioxi MDMA, MDA y MDEA se encuentran comúnmente como tabletas que pueden estar marcadas con un emblema. Rara vez se encuentran en forma de polvos, pero éstos contienen concentraciones elevadas de sustancias activas. Las tabletas suelen tener colores brillantes y con frecuencia varían en tamaño. El contenido de droga suele oscilar entre 40 y 140 mg. Se sabe de diferencias regionales en el contenido de droga y cambios que se producen con el tiempo. En Europa, por ejemplo, el contenido medio de MDMA en las tabletas de éxtasis ha bajado a unos 60-70 mg (en comparación con unos 100 mg a mediados de los años 90). La forma de sal de ETA de tipo metilendioxi que se encuentra con más frecuencia es el clorhidrato, pero también se han visto sales de bromuro y fosfatos.
A. ESTEREOQUÍMICA La mayoría de los ETA tienen por lo menos un centro quiral y, por lo tanto, pueden encontrarse como mezcla racémica o como enantiómeros individuales3. En los mercados ilícitos, la mayoría de los ETA se encuentran en un compuesto estereoquímico típico. La anfetamina, por ejemplo, y la mayoría de los ETA con anillo sustituido, se encuentran normalmente como racematos, mientras que la metanfetamina se suele encontrar como enantiómero S- o dextro (también
3 Los términos (d) o (+), (l) o (–) y (d, l) o (±) se utilizan normalmente para designar la rotación óptica de sustancias quirales. (R) y (S) describen la configuración estérica absoluta de sustituyentes en centros quirales individuales, y se prefieren sobre todo en el caso de los diastereómeros.

5
conocido como “Ice” o “Shabu”), además del racemato. En el capítulo VI.G., infra, se describe el análisis de isómeros ópticos.
B. CARACTERÍSTICAS FÍSICAS Puntos de fusión y de ebullición
Se conocen los puntos de fusión y/o ebullición de los ETA que se encuentran con más frecuencia. Los analistas deben tener presente, sin embargo, que esos datos se refieren a sustancias puras4. Con excepción de los ETA de alta pureza, como la metanfetamina cristalina (“Ice”), los puntos de fusión se deben utilizar, por lo tanto, sólo como ensayo presuntivo (respecto del uso de los puntos de fusión para la diferenciación de isómeros, véase el capítulo VI.G.1, infra).
Solubilidades Las solubilidades de ETA seleccionados y sus sales se proporcionan en la sección sobre ensayo de aniones (página 17, infra). La recristalización selectiva basada en las diferencias de solubilidad se puede utilizar para la separación de algunas sales de ETA (véase el capítulo VI.F. sobre FTIR).
Datos espectroscópicos Los datos del análisis de espectro de masa (EM), infrarrojo (IR) y de resonancia magnética nuclear (RMN) de los ETA más comunes aparecen en ediciones anteriores de los dos manuales de las Naciones Unidas relacionados con el análisis de ETA, es decir, “Métodos recomendados para el ensayo de anfetamina y metanfetamina” (ST/NAR/9) y “Métodos recomendados para el ensayo de los derivados anfetamínicos ilícitos con anillo sustituido” (ST/NAR/12). Los datos también se pueden encontrar en la página web de la Sección de Laboratorio y Asuntos Científicos.
4 Los analistas también deben tener presente que los puntos de fusión de algunos ETA pueden variar en función del disolvente que se utilice para la cristalización.

6
V. FABRICACIÓN ILÍCITA DE ETA El conocimiento del método utilizado en la fabricación ilícita de drogas de uso indebido puede cumplir una función importante en la interpretación de los resultados de análisis, especialmente en casos en que se han realizado análisis más a fondo de impurezas y subproductos de la fabricación, denominados estudios de perfil de impurezas. El uso de métodos de síntesis obtenidos o publicados ilícitamente (“literatura subterránea” o Internet), los “químicos” clandestinos sin experiencia, el equipo de laboratorio inapropiado y la falta de control de calidad en los laboratorios suelen dar por resultado productos impuros y de calidad inferior, y variaciones en la calidad y la potencia. En consecuencia, las drogas fabricadas ilícitamente suelen contener subproductos e intermedios derivados de materias primas con impurezas, reacciones incompletas y purificación inadecuada de los intermedios y el producto sintético final. Los tipos y las cantidades de impurezas que se encuentran en muestras de ETA ilícitos (el “perfil de impurezas”) dependen en gran parte de los métodos de síntesis, las proporciones, la fuente y la pureza de las materias primas, las condiciones de la reacción y los procesos de purificación, si los hubiere. La presencia o ausencia de impurezas específicas (marcadores) puede ser útil para determinar el método de síntesis empleado y las materias primas (precursores) utilizadas. El análisis de disolventes puede aumentar el cuerpo de información y, de esa forma, puede constituir un instrumento útil para la comparación y caracterización de muestras de ETA. Si bien los estudios de perfil de impurezas no son el tema del presente manual, algunos de los métodos descritos se pueden adaptar para ese propósito5. En la literatura se encuentran descripciones de varios métodos de síntesis de ETA que utilizan los fabricantes ilícitos o clandestinos. Los métodos de síntesis de uso más común para la fabricación ilícita de anfetamina se pueden modificar también para producir metanfetamina o anfetaminas con anillo sustituido. Esto se logra frecuentemente sustituyendo la fuente amina o la fuente del anillo aromático, respectivamente, durante el proceso de reacción. En general, la disponibilidad de precursores determina en gran medida la elección del método de síntesis utilizado en las operaciones ilícitas. A continuación se presentan descripciones breves de los métodos de síntesis más comúnmente utilizados para la anfetamina, la metanfetamina y la MDMA6. Los métodos de síntesis se clasifican sobre la base de los tipos de reducción utilizados en la reacción y del mecanismo de reducción. En la práctica, muchas de esas reacciones se conocen por nombres populares como método “Leuckart”, o métodos del ácido hidriódico/fósforo rojo, oxima, nitrostireno, Birch o “Emde”. Esos nombres populares se basan en el químico que descubrió el método, o en los reactivos característicos o los intermedios importantes. Siempre que es posible se incluyen los nombres populares.
5 Para una introducción general a este tema, se remite al lector al manual de las Naciones Unidas “Caracterización de drogas y elaboración de perfiles de impurezas” (ST/NAR/32/Rev.1, 2001); para métodos y enfoques más específicos para la elaboración de perfiles de impurezas de la metanfetamina, véase también ONUDD, publicación científica y técnica No. 17 (SCITEC/17), 2000. 6 Para más detalles, se remite al lector al manual de las Naciones Unidas “Clandestine manufacture of substances under internacional control” (ST/NAR/10/Rev. 2, 1998).

7
A. SÍNTESIS DE LA ANFETAMINA
La reacción central de todos los métodos utilizados para la síntesis de la anfetamina se basa en la reducción catalítica del 1-fenil-2-propanona (P-2-P, bencil metil ketona, BMK, fenilacetona), en presencia de amoníaco o metilamina. Los métodos de reducción más populares de la actualidad son el método Leuckart (reducción no metálica) y el método de reducción metálica catalítica (aminación reductiva, hidrogenación catalítica o hidrogenólisis).
Reacción Leuckart (reducción no metálica) En razón de su sencillez, la reacción “Leuckart” sigue siendo uno de los métodos de síntesis más populares para la fabricación ilícita de anfetaminas. La síntesis Leuckart es una reducción no metálica que normalmente se realiza en tres etapas. Para la síntesis de la anfetamina, se calienta una mezcla de P-2-P y formamida (algunas veces en presencia de ácido fórmico), o formiato de amonio hasta que se obtiene una reacción de condensación en el producto intermedio N-formilanfetamina. En la segunda etapa, la N-formilanfetamina se hidroliza, normalmente utilizando ácido clorhídrico (véase el gráfico 3). La mezcla de la reacción se hace básica, se aísla y se destila (vapor). En la etapa final, el producto se precipita de la solución, normalmente como sal sulfatada. La anfetamina base es un líquido aceitoso con un olor a “pescado y amina” característico.
Gráfico 3. Reacción Leuckart utilizada en la fabricación ilícita de anfetamina
El método Leuckart es uno de los más estudiados. En la literatura se identifican y describen varias impurezas específicas del método. Las impurezas más prominentes son el intermedio N-formilanfetamina (por lo general, arrastrado al producto final) y 4-metil-5-fenil pirimidina. Otros métodos sintéticos no dan tantas impurezas específicas como el método Leuckart.
Aminación reductiva (reducción metálica catalítica) La aminación reductiva es un proceso de reducción química o catalítica de aldehídos y cetonas en presencia de amoníaco, o una amina primaria o secundaria, que resulta en la amina relacionada de orden superior. El mecanismo de reacción pasa por la formación de una imina o producto intermedio de iminio por la reacción de un compuesto de carbonilo con un compuesto amino apropiado, seguida de la reducción. En la síntesis de la anfetamina que utiliza métodos de aminación reductiva se emplea P-2-P y un catalizador preferido. Los métodos utilizados con

8
más frecuencia se pueden dividir en tres tipos diferentes basados en las especies de reducción:
a) Reducción catalítica heterogénea utilizando óxido de platino, paladio o níquel Raney; b) Reducción metálica por disolución utilizando aluminio, zinc o amalgamas de
magnesio; c) Reducción metálica utilizando hidruro de aluminio-litio (LiAlH4), borohidruro de sodio
(NaBH4) o, con menos frecuencia, cianoborohidruro de sodio (NaCNBH3). La reducción catalítica heterogénea se logra normalmente utilizando una mezcla de P-2-P y gas de amoníaco cargado con hidrógeno en presencia de un catalizador seleccionado. El paladio en carbón (Pd/C) o el óxido de platino son los catalizadores de uso más común, seguidos por el níquel Raney. Las reducciones se logran normalmente a baja presión y baja temperatura. En raras ocasiones, también se han encontrado aminaciones a alta presión en un reactor a presión Parr (“presión” o “bomba de tubo”). La reducción metálica por disolución utilizando aluminio, zinc o amalgamas de magnesio es uno de los métodos de aminación reductiva de uso más común. El procedimiento más popular emplea amalgama de aluminio y mercurio (Al-Hg). El mecanismo de reducción de la amalgama se produce mediante la reducción de un aducto de base Schiff de P-2-P y la amina apropiada. En condiciones de clandestinidad difíciles, este método utiliza un recolector de aluminio o arenisca, y cloruro de mercurio (HgCl2). Las impurezas más características de las aminaciones reductivas son las bases Schiff, que se supone son formadas por la condensación de P-2-P y anfetamina, pero no son impurezas específicas del método y pueden estar presentes en cualquier procedimiento de síntesis que incluya P-2-P. Los compuestos de tipo P-2-P e imina también pueden aparecer como impurezas. Las impurezas inorgánicas resultantes del empleo de un catalizador determinado pueden servir de marcadores. Otros métodos de aminación reductiva de uso menos frecuente son las reducciones de hidruro metálico, como el método del “nitropropano” y el método de la “oxima”, denominados en función de los intermedios característicos (fenil-nitropropeno y oxima, respectivamente), que se forman durante la reacción. El método de la oxima es una reacción de P-2-P con hidroxilamina. El intermedio de oxima es posteriormente hidrogenado para obtener anfetamina. Un intermedio de oxima suele ser hidrolizado por reducción metálica utilizando Pd/H2, o por reducción de hidruro metálico utilizando LiAlH4. El método del “nitropropeno” comprende la condensación de benzaldehído con nitroetano, que produce 1-fenil-2-nitropropeno. La hidrogenación subsiguiente del doble enlace y la reducción del grupo nitro produce anfetamina. La fase de reducción se completa por lo general utilizando Pd/H2 o LiAlH4.
B. SÍNTESIS DE LA METANFETAMINA
La metanfetamina también se puede sintetizar utilizando los métodos mencionados más arriba pero sustituyendo el amoníaco con metilamina. Ahora bien, los métodos de síntesis de la metanfetamina más populares emplean efedrina o seudoefedrina como precursor, en lugar de P-2-P. Las reacciones se logran por lo general mediante una de las reacciones siguientes (véase el gráfico 4):
a) Reducciones no metálicas, como el método “'ácido hidriódico/fósforo rojo”; b) La reducción metálica por disolución, como la “reducción Birch”; o

9
c) La reducción catalítica heterogénea utilizando tionilcloruro y paladio u óxido de platino como catalizador (método Emde).
Es raro encontrar otras reacciones, como el método del “nitropropeno” o el de la “oxima”.
Gráfico 4. Métodos comunes para la fabricación ilícita de metanfetamina La efedrina y la seudoefedrina están presentes en muchos preparados farmacéuticos contra la tos, muchos de los cuales se pueden obtener sin receta. La hierba china Ma-huang, que se encuentra en varios suplementos alimenticios y productos de estilo de vida, es otra fuente de esos precursores. A diferencia del P-2-P, la efedrina y la seudoefedrina son sustancias quirales. Son diastereómeros y existen en una dos formas enantioméricas cada una, (d- y l-efedrina, y d- y l-seudoefedrina), además de dos racematos. El análisis quiral de los isómeros de efedrina o seudoefedrina también puede ayudar a determinar el proceso de fabricación de la metanfetamina ilícita. Todos los métodos de fabricación que comienzan con la producción de I-efedrina o d-seudoefedrina producen (+)-(S)-metanfetamina como único isómero óptico, que es por lo menos

10
dos veces más potente que la mezcla racémica producida por reacciones a partir de P-2-P. El método del “ácido hidriódico-fósforo rojo” se realiza normalmente calentando efedrina o seudoefedrina con fósforo rojo y ácido hidriódico. La mezcla de la reacción se filtra, se hace básica y se extrae en un disolvente. La metanfetamina base resultante es un líquido aceitoso, comúnmente denominado “meth oil”. De este líquido se cristalizan sales de clorhidratos utilizando éter/acetona y ácido clorhídrico. Alternativamente, un gas de cloruro de hidrógeno (de un cilindro, una solución acuosa o generado utilizando ácido sulfúrico y cloruro sódico) se bulle en el “meth oil” para que la sal clorhídrica precipite fuera de la solución. El Hl y el fósforo rojo se pueden sustituir por yodo y ácido hipofosfórico (hipofosfato sódico), o por agua y yodo. Raras veces, la mezcla de la reacción, a veces denominada “ox-blood”, se utiliza sin más purificación, principalmente por inyección. La mezcla de color rojo, causada por un exceso de yodo, contiene “meth oil” y diferentes impurezas provenientes del método Hl/fósforo rojo. Las impurezas típicas encontradas en muestras producidas por reducciones con Hl/fósforo rojo o yodo/ácido hipofosfórico son efedrina o seudoefedrina, aziridinas y dimetilnaftalinas. Las aziridinas no se pueden considerar como impurezas específicas del método, dado que también se producen a partir de la clorefedrina por eliminación alógena y cierre de los anillos (método Emde), o de un intermedio de oxima y N-hidroximetanfetamina. El proceso de la reducción Birch se realiza mediante una reducción metálica por disolución de efedrina o seudoefedrina en presencia de amoníaco. La reacción comprende mezclar efedrina o seudoefedrina con gases de amoníaco anhidro y metal de sodio o de litio. La mezcla se deja reposar hasta que se evapora el amoníaco. La separación del “meth oil” se realiza mediante extracción directa por disolvente y filtración. El producto de la reacción se purifica luego mediante la formación de la sal de clorhidrato y la recristalización. En la práctica ilícita, la reducción Birch se realiza normalmente en una reacción de un paso utilizando amoníaco, que está disponible en grandes cantidades, y cintas de litio de baterías. Pese a esto, la reducción Birch produce normalmente un producto final muy “limpio”. En la literatura se informa de varias impurezas específicas del método, como el N-metil-1-(1,4-ciclohexadienil))-2-propanamina. La reacción que incluye amoníaco anhidro es peligrosa y las explosiones en los laboratorios clandestinos no son raras. En el método Emde, la efedrina o la seudoefedrina se hacen reaccionar normalmente con tionilcloruro para obtener el intermedio clorefedrina, que luego es hidrogenado sobre un catalizador de platino o paladio para obtener metanfetamina. En los procedimientos analíticos basados en la CG, el intermedio clorefedrina se encuentra raras veces como impureza, porque se descompone durante el análisis para formar aziridinas. La clorefedrina también se descompone rápidamente durante la extracción básica de metanfetamina.
C. SÍNTESIS DE ETA CON ANILLO SUSTITUIDO
El estimulante anfetamínico de tipo metilendioxi que se encuentra más comúnmente es MDMA, y ocasionalmente MDEA y MDA. En general, los métodos de síntesis utilizados para la MDMA se aplican, con ligeras modificaciones, a otros análogos metilendioxi con anillo sustituido. El principal precursor utilizado para esa síntesis es 3,4-metilendioxifenil-2-propanona (también conocido como 3,4-MDP-2-P, 3,4-metilendioxi-fenilacetona, piperonil metil cetona, o PMK). El 3,4-MDP-2P está disponible en el comercio como precursor sometido a fiscalización internacional. La MDMA se puede sintetizar también a partir del safrol (3,4-metilendioxialilbenceno), ya sea directamente o utilizando isosafrol obtenido por isomerización del safrol. El safrol está disponible

11
en el comercio, o se puede extraer del aceite de sasafrás u otras partes de plantas o aceites esenciales con alto contenido de safrol. El piperonal (heliotropin, 3,4-metilendioxibenzaldehído), un producto químico industrial de gran uso, es otro precursor alternativo para la síntesis de 3,4-MDP-2-P. El método más directo para la síntesis de la MDMA es la aminación reductiva de 3,4-MDP-2-P con metilamina y gas de hidrógeno sobre un catalizador de platino (reducción metálica catalítica). La reducción se puede lograr también mediante una reducción metálica disolvente utilizando amalgama de aluminio (hoja de aluminio y mercuriocloro), o por reducción de hidruro metálico utilizando cianoborohidruro sódico (véase el gráfico 5). Sustituyendo la metilamina por etilamina se obtiene MDE; el gas de amoníaco produce MDA, y la dimetilamina produce MDDMA. Un método análogo para la fabricación ilícita de anfetamina comúnmente utilizado para la fabricación ilícita de MDMA es el método Leuckart. Se reducen 3,4-MDP-2-P y N-metilformamida utilizando ácido fórmico. El intermedio resultante, N-formil-MDMA se hidroliza por reflujo con un ácido o una base fuerte para producir MDMA.
Gráfico 5. Aminación reductiva utilizada en la fabricación ilícita de MDMA
El método del “nitropropeno” adquirió popularidad a principios del decenio de 1990. Comprende la reacción de piperonal con nitroetano en presencia de un catalizador básico, normalmente n-butilamina. En la literatura se informa de diversos procedimientos para la producción del intermedio fenil-nitropropeno, pero normalmente se deja reposar una mezcla de piperonal y nitroetano durante unos pocos días en la oscuridad. Alternativamente, se produce un reflujo de piperonal y nitroetano en ácido acético y acetato amónico. Los intermedios formados en estas reacciones son muy característicos en cuanto a apariencia, y por lo general precipitan de la solución de la reacción como cristales amarillos brillantes. Para producir MDA, el intermedio se suele reducir con hidruro de aluminio-litio. Para la síntesis de MDMA, MDEA u otros ETA, el nitropropeno intermedio se convierte en 3,4-MDP-2-P, que luego se reduce mediante uno de los métodos de aminación reductiva descritos más arriba. Los intermedios nitropropeno y nitroestireno son sustancias cristalinas de color amarillo o naranja brillante.

12
La MDMA también se fabrica normalmente utilizando el denominado método del “bromosafrol”. La reacción se produce mediante la brominación del safrol con ácido hidrobrómico a baja temperatura, seguida de un tratamiento con metilamina para formar el producto final. El rendimiento dependerá del contenido de agua de la mezcla de la reacción. La sustitución de la metilamina con otras aminas produce diferentes productos de tipo MDMA (por ejemplo, MDA, MDEA, o MDDMA). Las drogas de tipo metoxianfetamina se sintetizan normalmente utilizando aldehído y nitroetano con anillos sustituidos apropiados, mientras que para la mescalina, el 2C-B y otras fenetilaminas con anillo sustituido se utiliza nitrometano. El 2C-B se fabrica haciendo reaccionar 2,5 dimetoxibenzaldehído y nitroetano, seguida de una reducción LiALH4 para formar 2,5-dimetoxifenetilamina, que es luego brominada para obtener el producto final.

13
VI. ANÁLISIS CUALITATIVO Y CUANTITATIVO DE ETA
Por lo general, cuando se intenta determinar la identidad de una sustancia sometida a fiscalización en el material sospechoso, el enfoque analítico debe comprender la determinación de por lo menos dos parámetros no correlacionados. Se reconoce que la selección de estos parámetros en cualquier caso particular tendrá en cuenta la droga de que se trate y los recursos de laboratorio de que dispone el analista. También se acepta que los requisitos especiales establecidos en diferentes jurisdicciones pueden determinar las prácticas que se sigan en un laboratorio particular.
A. ENSAYOS PRESUNTIVOS Los ensayos presuntivos son procedimientos de clasificación rápidos que por lo general consisten en dos o tres ensayos independientes. Están diseñados para dar una indicación de la presencia o ausencia de tipos de drogas en la muestra ensayada y eliminar rápidamente las muestras negativas. Las buenas técnicas de ensayos presuntivos, como todas las técnicas analíticas, maximizan la probabilidad de un resultado “verdadero” y reducen al mínimo la probabilidad de un positivo falso. Ahora bien, los ensayos presuntivos no se consideran suficientes para identificar drogas y los resultados deben ser confirmados mediante otros ensayos de laboratorio. En los últimos tiempos, los ensayos presuntivos se utilizan con más frecuencia en pruebas de campo, aunque también se realizan en laboratorios como primer procedimiento de clasificación. Para la clasificación de los ETA, se usan normalmente ensayos cromáticos, o pruebas rápidas, aunque también se dispone de ensayos de inmunoanálisis y varias técnicas rápidas y con instrumentos portátiles. Los métodos de clasificación con instrumentos, como el espectrómetro de movilidad de iones (escaneo de iones), el espectrómetro de masas portátil y los espectrómetros FTIR o Raman, han obtenido popularidad en los últimos años. Se dispone también de muchos estuches de ensayos comerciales para la clasificación de ETA, aunque deben ser evaluados internamente en función de su especificidad y sensibilidad.
1. Ensayos cromáticos Los ensayos cromáticos suelen ser los ensayos clínicos más sencillos y más rápidos que un analista puede aplicar a una muestra. Los ensayos cromáticos son muy sensibles; por lo tanto, sólo se necesitan cantidades muy pequeñas de la muestra para completar con éxito el ensayo, y con frecuencia los mejores resultados se obtienen con las cantidades de muestras más pequeñas, generalmente menos de un miligramo. Dado que las muestras pueden variar en pureza (concentración de ETA) y pueden contener sustancias ajenas, los colores que aparecen en estos ensayos deben interpretarse con cuidado. Además, hay que tener presente el aspecto subjetivo de la evaluación del color.
Técnicas de ensayos cromáticos Normalmente se utilizan varios reactivos diferentes en los ensayos cromáticos de sustancias de tipo anfetamínico y sus análogos con anillo sustituido. Los más importantes para estas sustancias son los ensayos de Marquis, de Simon y de Chen. El ensayo de Marquis permite distinguir entre la anfetamina y sus análogos con anillo sustituido. El ensayo de Simon se utiliza generalmente para aminas secundarias, por ejemplo, la metanfetamina y las anfetaminas secundarias con anillo sustituido, incluidas la MDMA y la MDE. Ahora bien, otras aminas secundarias, por ejemplo la dietilamina y la piperidina, pueden dar colores similares. En general,

14
los colores son intensos pero pueden desaparecer rápidamente en presencia de algunas impurezas. Por estas razones, es esencial que el analista confirme los resultados del ensayo de Simon realizando un ensayo suplementario, por ejemplo, un ensayo de Marquis. El ensayo de Chen se utiliza para distinguir la efedrina, la seudoefedrina, la norefedrina, la fenilpropanolamina y la metcatinona de la anfetamina y la metanfetamina, que no reaccionan con el reactivo del ensayo de Chen. Un cuarto ensayo, el ensayo del ácido gálico, proporciona un medio simple de distinguir MDMA, MDA y MDEA de la anfetamina o la metanfetamina, porque reacciona específicamente con los compuestos aromáticos de metilendioxi sustituido. También reaccionan los precursores que contienen la subestructura metilendioxi, como el safrol y el isosafrol. MÉTODOS En el anexo II se describe la preparación de reactivos. Los reactivos se deben preparar en el momento.
Ensayo de Marquis 1) Colocar una pequeña cantidad (1-2 mg de polvo, o 1-2 gotas de líquido) del material
sospechoso en una ranura de una placa de ensayos. 2) Agregar una gota de reactivo de Marquis 1, luego una gota de reactivo 2, y mezclar. 3) Observar el color de la mezcla.
Ensayo de Simon 1) Colocar una pequeña cantidad (1-2 mg de polvo, o 1-2 gotas de líquido) del material
sospechoso en una ranura de una placa de ensayos. 2) Agregar una gota de reactivo de Simon 1, y mezclar. 3) Agregar una gota de reactivo de Simon 2, luego una gota de reactivo 3. 4) Observar el color de la mezcla.
Ensayo de Chen 1) Colocar una pequeña cantidad (1-2 mg de polvo, o 1-2 gotas de líquido) del material
sospechoso en una ranura de una placa de ensayos. 2) Agregar 2 gotas de reactivo de Chen 1. 3) Agregar 2 gotas de reactivo de Chen 2, y luego 2 gotas de reactivo 3 y mezclar. 4) Observar el color de la mezcla.
Ensayo del ácido gálico 1) Colocar una pequeña cantidad (1-2 mg de polvo, o 1-2 gotas de líquido) del material
sospechoso en una ranura de una placa de ensayos. 2) Agregar una gota de reactivo de ácido gálico. 3) Observar el color de la mezcla.
RESULTADOS En el cuadro 1 se proporcionan resultados de los tres principales ensayos cromáticos para la mayoría de los estimulantes de tipo anfetamínico y sus análogos con anillo sustituido que se encuentran más comúnmente. Dado que el ensayo de ácido gálico se utiliza principalmente para identificar, por lo general, el sustitutivo metilendioxi del anillo y no un ETA individual con esa subestructura, en este cuadro los resultados no se incluyen separadamente para sustancias individuales. Un color verde oscuro

15
brillante indica la presencia de MDA, MDMA, MDE, N-hidroxi-MDA o MMDA. En algunos casos, por ejemplo MDE y N-hidroxi-MDA, el color verde puede cambiar a pardo durante el ensayo. Cuadro 1. RESULTADOS DE LOS ENSAYOS CROMÁTICOS
Compuesto Reactivo de Marquis
Reactivo de Simon
Reactivo de Chen
Anfetamina Naranja, lentamente pasando a pardo
SR * SR *
Catinona SR SR * Pasa gradualmente del amarillo al naranja
Dimetilanfetamina Naranja SR * SR * Efedrina Seudoefedrina SR SR * Púrpura N-Etilanfetamina Naranja, pasando a
pardo
Metanfetamina Naranja, lentamente pasando a pardo
Azul intenso SR *
Metcatinona SR Azul pálido, precipitado como mancha o anillo
Pasa gradualmente del amarillo al naranja
Norefedrina SR SR Púrpura 2C-B Amarillo--> verde SR * SR * 2C-T-2 Rosa pálido, naranja SR * 2C-T-7 SR SR *
DMA Verde-->verde oscuro
SR *
DOB Amarillo--> verde SR * SR *
DOET Pardo amarillento SR * FLEA Azul oscuro/negro MBDB Azul oscuro/negro Azul intenso SR * MDA Azul oscuro/negro SR * SR * MDDM Azul oscuro/negro
MDEA Azul oscuro/negro Azul (intenso)--> pardo
SR *
MDMA Azul oscuro/negro Azul intenso SR * MDOH Azul oscuro/negro MMDA Púrpura SR * SR * 4-MTA SR SR *
PMA SR--> verde pálido SR *
STP / DOM Amarillo SR * SR * TMA Naranja SR * SR = sin reacción. * El color del reactivo se debe considerar negativo.

16
Notas analíticas a) ETA específicos Dado que los ETA, en especial la metanfetamina y los análogos con anillo sustituido en Asia sudoriental, se encuentran con frecuencia en forma de tabletas de colores brillantes, el resultado de los ensayos cromáticos puede quedar oculto, o se puede producir un cambio en el resultado de esos ensayos.
Aunque se utiliza una gran variedad de colores para producir tabletas de ETA, la mayoría de los colores son solubles en agua, y su solubilidad se puede manipular cambiando el pH de la solución. En esos casos, por lo tanto, el análisis se debe ajustar al procedimiento de extracción y se debe eliminar el color totalmente antes de proceder al ensayo cromático propiamente dicho.
En algunos casos, cuando el ensayo cromático no se puede interpretar claramente debido a la presencia de un colorante en la tableta, el procedimiento siguiente suele producir resultados aceptables:
Colocar una pequeña cantidad (aproximadamente 10 mg) de la muestra en un tubo de ensayo pequeño. Agregar aproximadamente 1 ml de metanol (o una mezcla de 4:1 metanol:cloruro de metileno). Después de filtrar la mezcla por lana de vidrio, se la evapora hasta que quede seca. La muestra se reconstituye en una pequeña cantidad de agua y luego se procede a realizar el ensayo cromático depositando cuidadosamente la solución de la muestra acuosa en una placa de ensayos y agregando el reactivo de color. Dado que la muestra ya está diluida, bastarán 3 gotas de reactivo por una gota de muestra.
La presencia de disolventes y adulterantes también puede perturbar las reacciones de coloración y dar resultados negativos falsos. El ensayo de Simon puede dar resultados negativos falsos en muestras de ETA con adulterantes o disolventes; el ensayo de Marquis es menos sensible a las muestras adulteradas y, por lo tanto, es preferible cuando la concentración de ETA en la muestra es muy baja.
b) General Los colores descritos son juicios subjetivos debido a que la percepción de los colores es individual. Por esta razón, es decir, el aspecto subjetivo de la evaluación del color, es necesario que cada analista ensaye estándares de referencia apropiados para asegurar que puede reconocer cada resultado del ensayo cromático. Asimismo, es conveniente realizar un ensayo con una muestra en blanco sin la sustancia de que se trata para asegurar la familiaridad con el color del reactivo.
Si se realiza adecuadamente, un ensayo de coloración negativo es por lo general bastante fidedigno para establecer la ausencia de un compuesto determinado; no obstante, los resultados positivos son sólo indicaciones presuntivas de la posible presencia de un compuesto. Muchos otros compuestos, con frecuencia benignos y no sometidos a fiscalización por las leyes nacionales o los tratados internacionales, pueden dar colores similares con un reactivo de ensayo cromático determinado.
Por lo tanto, es obligatorio que el analista confirme un ensayo cromático positivo de cualquier compuesto sometido a fiscalización por ley utilizando otros ensayos de laboratorio.
Para eliminar la posibilidad de que una placa de ensayo contaminada dé un resultado positivo falso, conviene colocar el reactivo en la placa y luego agregar una cantidad pequeña de la muestra al reactivo.

17
Lecturas recomendadas Naciones Unidas (1995), Métodos para el ensayo inmediato de drogas de uso indebido: Manual para uso del personal de laboratorios nacionales de estupefacientes y de los organismos de represión (ST/NAR/13/Rev.1)
S. A. Johns, A. A. Wist y A. R.Najam (1979), Spot tests: a colour chart reference for forensic chemists, J. Forensic Sci., vol. 24, págs.631 a 649.
E. Jungreis (1985), Spot test analysis, Clinical, environmental, forensic and geochemical applications, John Wiley & Sons, Nueva York-Chichester-Brisbane-Toronto-Singapur.
A. C. Moffat, M. D. Osselton y B. Widdop (eds.) (2004), Clarke’s Analysis of Drugs and Poisons, 3ra. ed., Pharmaceutical Press, Londres-Chicago.
C. L. O’Neil y otros (2000), Validation of twelve chemical spot tests for the detection of drugs of abuse, Forensic Sci. Int., vol.109, págs.189 a 201.
R. A. Velapoldi y S. A. Wicks (1974), The use of chemical spot test kits for the presumptive identification of narcotics and drugs of abuse, J. Forensic Sci., vol. 19, págs. 636 a 654.
2. Ensayo de aniones El ensayo de aniones con fines forenses utiliza normalmente las solubilidades combinadas con reacciones selectivas en que los resultados están determinados por la presencia o ausencia, y la solubilidad, de un precipitado. A continuación se describe la solubilidad de diferentes ETA y sus sales en agua y en varios sistemas de disolventes comunes.
Anfetamina y sus sales Base Clorhidrato Fosfato Sulfato Agua Ligeramente
soluble Soluble Soluble Soluble
Metanol o etanol Soluble Soluble Ligeramente soluble
Ligeramente soluble
Éter dietílico Soluble Insoluble Insoluble Insoluble Cloroformo Soluble Soluble Insoluble Insoluble
Metanfetamina y sus sales Base Clorhidrato Agua Ligeramente
soluble Soluble
Metanol o etanol Soluble Soluble
Éter dietílico Soluble Insoluble Cloroformo Soluble Soluble

18
ETA con anillo sustituido y sus sales Las bases libres de ETA con anillo sustituido son por lo general insolubles en agua y solubles en etanol, éter dietílico, cloroformo y otros disolventes orgánicos. Sus sales clorhídricas son solubles en agua y etanol, ligeramente solubles en cloroformo, e insolubles en éter dietílico. Las solubilidades de las diferentes sustancias de este grupo dependen del ETA con anillo sustituido específico de que se trate.
MÉTODOS
Todos los reactivos se deben preparar con arreglo a un procedimiento establecido. En el anexo II se describe detalladamente la preparación de los reactivos.
Ensayo del nitrato de plata La muestra del ETA desconocido se disuelve en varias gotas de agua desionizada y se trata con 1-2 gotas de solución de AgNO3. A continuación se dan los resultados para los aniones comunes. Dado que otros aniones pueden dar resultados similares, se deben realizar ensayos adicionales o trabajos de familiarización con las limitaciones del ensayo. Cloruro — Forma un precipitado cremoso que es insoluble en ácido nítrico concentrado. Después de lavarlo con agua, el precipitado es soluble en una solución de amoníaco diluida, a partir de la cual se lo puede volver a precipitar añadiendo ácido nítrico. Bromuro — Forma un precipitado de color amarillo pálido a crema que es insoluble en ácido nítrico. Después de lavarlo con agua, el precipitado se disuelve muy lentamente en soluciones de amoníaco diluidas y es soluble en soluciones de amoníaco concentradas. Se lo puede volver a precipitar a partir de ambas soluciones agregando ácido nítrico. Yoduro — Forma un precipitado amarillo brillante que es ligeramente soluble en una solución de amoníaco concentrada pero soluble en una solución de tiosulfato. Sulfato — Forma un precipitado cristalino ligeramente coloreado que se puede identificar fácilmente colocando la solución de ensayo en una placa de microscopio y buscando los característicos cristales de sulfato de plata en forma de “diamantes”. Fosfato — Forma un precipitado amarillo pálido, que ese soluble en una solución de amoníaco diluida, o en ácido nítrico frío. Para las sales sulfatadas y fosfatadas se pueden realizar otros ensayos específicos:
Ensayo de sal sulfatada La muestra del ETA desconocido (unos 100 mg) se disuelve en agua y se trata con una solución de cloruro de bario. Un precipitado blanco, que es insoluble en ácido clorhídrico, indica la presencia de una sal sulfatada.
Ensayo de sal fosfatada La muestra del ETA desconocido (unos 100 mg) se disuelve en una solución compuesta de volúmenes iguales (por ejemplo, 1 ml cada uno) de solución de ácido nítrico (10% v/v) y solución de molibdato de amonio (10% p/v). Al calentarla lentamente, la formación de un precipitado de color amarillo canario brillante, que es soluble en una solución de amoníaco, indica la presencia de una sal fosfatada.

19
Notas analíticas Dado que todos los ensayos de aniones se realizan en soluciones acuosas, la solubilidad en agua de las sales de ETA es una condición para obtener resultados significativos.
Es esencial lavar el precipitado con agua antes de realizar el ensayo de disolución del precipitado, a fin de remover cualquier anión soluble (no precipitado).
Lecturas recomendadas McKibben, T., Chappell, J. S., Evans, H., Mausolf, N., Analyses of Inorganic Components Found in Clandestine Drug Laboratory Evidence, J. Cland. Lab. Invest. Chem. Ass., 5(4), 1995, 19 a 33.
3. Ensayos de microcristales Los ensayos de microcristales son rápidos, sencillos y extremadamente sensibles para identificar sustancias y sus isómeros ópticos. Comprenden la formación de cristales a partir de la reacción del compuesto objetivo con un reactivo químico, seguida del análisis de los cristales resultantes utilizando un microscopio de polarización y comparándolos con los materiales de referencia, por lo general fotografías de cristales conocidos. MÉTODOS La forma más simple del ensayo consiste en agregar una gota de un reactivo adecuado a la sustancia que se ensaya, y observar y analizar luego los cristales formados en el microscopio de polarización. A fin de mantener un registro preciso, se deben describir las características principales de los cristales. El registro más preciso de los resultados del ensayo son las fotografías. Si no se dispone de fotografías, un esbozo de las formas de los cristales puede resultar útil. En el capítulo “Análisis de isómeros ópticos” (página 37) se describen métodos y procedimientos detallados para el ensayo de microcristales de ETA. En el anexo III se dan términos descriptivos para las formas de los cristales y fotografías de cristales de los principales ETA, así como los requisitos en materia de equipo e instrumentos básicos.
Lecturas recomendadas Cunningham, M. D. (1973), Rapid and sensitive technique for the differentiation of the optical isomeric forms of methamphetamine and amphetamine, Microgram, vol. 6, No.6, págs.87 a 95.
Fulton, C. C. (1969), Modern Microcrystal Tests for Drugs, Wiley-Interscience, A Division of John Wiley & Sons, Nueva York-Londres-Sydney-Toronto.
Ōno, M., Microcrystal Test, Japón, 1996 Proporciona una introducción amplia a la técnica de los ensayos de microcristales, con fotografías de resultados de ensayos de microcristales de 39 ETA y sus precursores.
B. CROMATOGRAFÍA DE CAPA DELGADA (CCD) La cromatografía de capa delgada (CCD) es una de las técnicas usadas con más frecuencia para separar e identificar drogas fabricadas ilícitamente. Es rápida (un análisis rara vez toma

20
más de 30 minutos), sensible (se requieren cantidades inferiores a 1 mg del analito), extremadamente flexible en las fases estacionaria y móvil y, por lo tanto, adecuada para una amplia variedad de sustancias en forma de bases o de sales, que van desde los materiales más polares hasta los más no polares. También se presta a una variedad de técnicas de visualización, y es poco costosa. Pese a las ventajas obvias de la CCD, en muchos países no se la acepta como técnica única de identificación de drogas. Placas de CCD (fases estacionarias) Recubrimiento: Gel de sílica G con un espesor de capa de 0,25 mm, y con un
indicador inerte, que fluorece bajo luz UV de longitud de onda de 254 nm
Nota: Las placas preparadas por el analista debe ser activadas antes de utilizarlas, colocándolas en un horno por lo menos durante 10 a 30 minutos a 120º C. Las placas se almacenan luego en un desecador libre de grasas sobre gel de sílica azul. La activación por calor normalmente no se requiere para capas enlazadas químicamente (placas comerciales).
Tamaños de placa típicos: 20x20 cm, 20x10 cm, 10x5 cm (la placa de 10x5 cm se debe utilizar únicamente con el lado de 10 cm en posición vertical en el tanque de CCD.
Sistemas de disolventes (fases móviles) Sistema A: Metanol 100 Amoníaco concentrado 1,5 Sistema B: Acetato etílico 85 Metanol 10 Amoníaco concentrado 5 Sistema C: Cicloexano 75 Tolueno 15 Dietilamina 10
En relación con sistemas de CCD más normalizados, véase:
Thin-layer chromatographic Rf values of toxicologically relevant substances on standardized systems, second, revised and enlarged edition, Report XVII of the DFG Commission for clinical-toxicological analysis, volumen especial del boletín TIAFT, VCH Verlagsgesellschaft mbH, 1992.
MÉTODOS
A continuación se proporcionan métodos recomendados y resultados seleccionados, pero el analista deberá familiarizarse con los resultados específicos para ETA individuales.
Sistemas de disolventes Preparar sistemas de disolventes de desarrollo en la forma más precisa posible utilizando pipetas y cilindros de medición. Dejar el sistema de disolvente en el tanque de CCD durante un tiempo suficiente para permitir la saturación de la fase de vapor antes del análisis (con tanques revestidos con papel absorbente, esto toma aproximadamente 5 minutos).

21
Preparación de muestras y soluciones estándar de ETA La forma de los estándares y las muestras (en sales o bases), no tiene importancia. Cualquiera de las dos es satisfactoria. Debido a la naturaleza básica de los disolventes de desarrollo, los compuestos emigran como bases libres.
Soluciones estándar de ETA Las soluciones estándar de ETA se deben preparar a una concentración de aproximadamente 2 mg/ml en metanol. Deben almacenarse a oscuras y en un lugar frío.
Soluciones de muestras de ETA (muestra de ETA desconocido) Las soluciones estándar de ETA se deben preparar a una concentración de aproximadamente 5 mg/ml en metanol. En algunos casos, cuando se sospecha que la pureza del ETA es demasiado baja debido a que ha sido adulterado, quizá sea necesario preparar soluciones de muestras más concentradas (se recomiendan soluciones 10 veces más concentradas como punto de partida). Para las muestras de ETA en otras formas distinta del polvo, las soluciones de muestra se deben preparar de la siguiente manera:
Tabletas: Triturar un número representativo de tabletas (siguiendo planes generales de muestreo) hasta obtener un polvo fino y preparar una solución con ese polvo.
Cápsulas: Extraer el contenido de una muestra representativa de las cápsulas (siguiendo planes generales de muestreo) y preparar una solución con ese polvo.
Soluciones acuosas: Aplicar directamente sobre la placa, o el equivalente de 5 mg/ml, si se conoce la concentración del ETA.
Aplicación y desarrollo Aplicar sobre la placa de CCD 1 µl y 5 µl de una solución de la muestra, junto con 2 µl de la solución o soluciones estándar (de preferencia, aplicar también a la placa un disolvente como control negativo). La aplicación se debe hacer con cuidado para no dañar la superficie de la placa. El punto de partida del análisis, es decir, la “línea de aplicación”, debe ser 2 cm desde el fondo de la placa. El espacio entre aplicaciones de la muestra (puntos de aplicación) debe ser de por lo menos 1 cm, y las aplicaciones no se deben colocar a menos de 1,5 cm del borde lateral de la placa. Para evitar aplicaciones difusas durante el desarrollo, el tamaño de la aplicación de la muestra debe ser lo más pequeño posible (>2 mm). Dejar que la aplicación se seque, y colocar la placa en el tanque de CCD saturado de disolvente (la saturación de la fase de vapor se logra utilizando almohadillas o papel de filtro saturados con disolvente como revestimiento del tanque). Extraer la placa del tanque de revelado tan pronto como el disolvente alcance la línea de revelado marcada de antemano; de otra forma, se producirán puntos difusos.
Visualización/detección Las placas se deben secar antes de la visualización: el disolvente se puede dejar secar a temperatura ambiente, o extraer con un secador de aire caliente. Si se utiliza aire caliente, hay que tener cuidado en razón de la volatilidad de las bases libres de ETA. Para el desarrollo del color apropiado, es importante que se eliminen de la placa todas las trazas de amoníaco.

22
La siguiente es una selección de métodos de visualización. Sobre la base de los resultados de los ensayos presuntivos, los ETA previstos se deben seleccionar utilizando uno de los siguientes métodos y reactivos, o una combinación de ellos: Método/ reactivo de visualización Analitos objetivo y resultados A. Luz UV a 254 nm Método universal. Muchas sustancias, incluidos los ETA,
producen manchas púrpura en una placa normalmente de color verde fluorescente.
B. Reactivo de ninhidrina Muchas aminas primarias y secundarias, adheridas a un átomo de carbono alifático, como la anfetamina y la metanfetamina, dan manchas violeta o rosa.
C. Reactivo de yodoplatinato de potasio acidificado
Reactivo general sensible. Muchas aminas primarias y secundarias dan manchas de color azul pálido.
D. Reactivo negro sólido de potasio
Las aminas primarias y secundarias dan manchas de diversos colores, desde el violeta (aminas primarias) hasta el naranja o el rojo-naranja (aminas secundarias).
E. Reactivo de Marquis Distinción entre ETA con y sin anillo sustituido. F. Reactivo de
fluorescamina (Fluram) Reactivo sensible para aminas primarias. Recomendado para la detección de concentraciones de aminas primarias.
G. Reactivo de Simon Reactivo general para aminas secundarias (la efedrina y la seudoefedrina no reaccionan).
H. Reactivo de Dragendorff Reactivo general para alcaloides y bases nitrogenadas. A. Luz UV
Observar la placa seca bajo luz UV a 254 nm y 366 nm.
B. Reactivo de ninhidrina Preparar una solución en etanol al 10%.
Rociar la placa con el reactivo de ninhidrina y calentar en un horno a 120º C por lo menos durante 15 minutos. Las aminas primarias, como la anfetamina, y las aminas secundarias, como la metanfetamina, producen manchas violetas o rosas. La efedrina también produce una mancha violeta.
C. Reactivo de yodoplatinato de potasio acidificado Disolver 0,25 g de cloruro platínico y 5 g de cloruro potásico en agua y preparar hasta 100 ml. Agregar 2 ml de ácido clorhídrico concentrado.
Rociar la placa con la solución de yodoplatinato de potasio acidificado y observar las manchas de colores. La anfetamina y la metanfetamina producen manchas difusas de color gris-violeta-pardo sobre un fondo rosa.
La solución también se puede utilizar para rociar las superficies de las placas que anteriormente habían sido rociadas con ninhidrina.

23
D. Reactivo negro sólido de potasio Solución A: Preparar una solución de sal de reactivo negro sólido de potasio en agua al 1% [2,5-dimetoxi-4-((4-nitrofenil)azo)benceno diazonio tetraclorozincato (2:1)]
Solución B: 1N NaOH
Rociar la placa con la solución A y observar las manchas de colores. Las aminas secundarias como la metanfetamina y la MDMA producen manchas inmediatamente. El nuevo rociado con solución B produce manchas de colores para la anfetamina y otros ETA con anillo sustituido. Secar las placas con aire y rociar una vez más con la solución A. Esto produce manchas de colores más intensos. Los colores varían del violeta para las aminas primarias al naranja o el naranja-rojo para las aminas secundarias como la metanfetamina y el MDMA.
E. Reactivo de Marquis Agregar de 8 a 10 gotas de solución de formaldehído al 40% a 10 ml de ácido sulfúrico concentrado. Preparar fresco antes de cada uso.
No se recomienda rociar con el reactivo de Marquis debido a la presencia de ácido sulfúrico concentrado. No obstante, proporciona información adicional útil para diferenciar entre los ETA, por ejemplo, después de la detección con ninhidrina. A tal fin, verter el reactivo de Marquis con una pipeta Pasteur sobre las manchas ya detectadas.
F. Reactivo de fluorescamina (Fluram) Disolver 10 mg de fluorescamina en 50 ml de acetona. Preparar diariamente.
Rociar la placa con el reactivo de fluorescamina. Secar utilizando aire caliente. Observar la placa con una luz UV a 366 nm. La anfetamina da una mancha fluorescente amarilla brillante. La metanfetamina no se detecta. (Para la estabilización en 366 nm, rociar con una solución de trietilamina al 10% v/v en diclorometano).
G. Reactivo de Simon7 Disolver 100 mg de nitroprusiato de sodio y 2 g de carbonato de sodio en 10 ml de agua (es decir, una solución acuosa que contiene nitroprusiato de sodio a una concentración del 1% y carbonato de sodio al 20%). Preparar el reactivo fresco antes de usarlo.
Rociar la placa con el reactivo de Simon. Colocar la placa en un tanque de desarrollo vacío junto con un vaso de precipitación con acetaldehído. Cubrir el tanque. El vapor de acetaldehído hará que una mancha de metanfetamina se vuelva de color azul intenso.
H. Reactivo de Dragendorff Solución madre: disolver 0,85 g de subnitrato de bismuto (básico) en 10 ml de ácido acético. Disolver 50 ml con agua y agregar 8 g de yoduro de potasio en 20 ml de agua.
Rociar la placa con una solución preparada de 1 ml de solución madre de Dragendorff, 2 ml de ácido acético y 10 ml de agua. Los colores varían del naranja al violeta.
7 Este método modificado mejora la sensibilidad para la metanfetamina a 0,1 µ (LOD). Las aminas primarias no se pueden detectar en razón de la baja sensibilidad del sistema a este grupo de sustancias, y a la interferencia con el color de fondo cuando se utiliza amoníaco en el disolvente de desarrollo.

24
Interpretación Después de la visualización, marcar las manchas (por ejemplo, con lápiz), y calcular los valores del factor de retardación (Rf):
Distancia de migración: del origen al centro de la zona del analito (mancha) Rf = Distancia de desarrollo: del origen al frente del disolvente Es muy común expresar factores de retención como Rf x 100, denominado hRf. RESULTADOS Comparar los colores y los valores Rf de la muestra del ETA desconocido con los de normas de referencia de ETA auténticos que se analizaron simultáneamente en la misma placa. En el cuadro 2 se dan los valores Rf de algunos de los ETA más comunes.
Cuadro 2. Valores Rf de los ETA y adulterantes que se encuentran con mayor frecuencia
Sistema de CCD* Nombre del ETA A B C Anfetamina 0,48 (0,43)** 0,37 (0,43) (0,20) Catinona 0,66 0,56 DOB 0,37 0,32 (0,13)
DOET 0,36 0,32 (0,24) DMA 0,37 0,33 (0,19) N-Etilanfetamina 0,47 0,37 (0,47)
Metanfetamina 0,35 (0,31) 0,22 (0,42) (0,28) MDA 0,36 (0,39) 0,33 (0,42) (0,18)
MDMA 0,31 (0,33) 0,21 (0,39) (0,24) MMDA 0,40 0,31 PMA 0,41 (0,73) 0,33 (0,43) (0,23) STP/DOM 0,35 (0,51) 0,31 (0,41) (0,15) TMA 0,35 0,20
Efedrina (0,30) (0,25) (0,05)
Cafeína (0,52) (0,52) (0,03) *Sistemas de disolventes: A, B o C; placa de CCD: Gel de sílica G con espesor de capa de 0,25mm Sistema A: metanol : amoníaco concentrado (100:1,5) Sistema B: acetato etílico: metanol : amoníaco concentrado (85:10:5) Sistema C: ciclohexano : tolueno : dietilamina (75:15:10) ** Los valores Rf entre paréntesis se obtuvieron utilizando placas de sílica impregnadas con KOH metanólico (0,1mol/l).

25
Notas analíticas Debido a que pequeños cambios en la composición y activación de la placa de CCD, los sistemas de disolventes, la saturación del tanque o la distancia de desarrollo pueden resultar en cambios significativos en los valores Rf, los valores proporcionados deben considerarse sólo como una indicación del comportamiento cromatográfico de los ETA y adulterantes enumerados. Es esencial que los patrones de referencia de los ETA se analicen simultáneamente en la misma placa. Alternativamente, se puede aumentar significativamente la posibilidad de reproducción utilizando compuestos de referencia y valores Rf corregidos (Rf
c).
A los fines de la identificación, siempre se deben considerar tanto los valores Rf como los colores de las manchas después del rociado con reactivos de visualización
Lecturas recomendadas Fried y J. Sherma (Eds.), Practical Thin-Layer Chromatography, A Multidisciplinary Approach, Boca Raton Press, 1995.
Jork y otros, Thin-Layer Chromatography, Reagents and Detection Methods, Weinheim, VCH, vol.1 (1990), Vol.2 (1992).
Neumann, H. (1987), Nachweis und Identifizierung von Feniletilaminen (Stimulantien und Halluzinogene), Sci. Pharm., 55, 1 a 11.
Ojanperä, I., Wähälä, K., y Hase, T. A. (1990): Fast black K salt: a versatile thin-layer chromatographic visualization reagent for the differentiation of aliphatic amines, Analyst, vol. 115, págs. 263 a 267.
Stead, A. H., Gill, R., Wright, T., Gibbs, J. P., Moffat, A. C. (1982), Standardized thin-layer chromatographic systems for the identification of drugs and poisons, Analyst, vol. 107, págs. 1106-1168.
C. CROMATOGRAFÍA EN FASE GASEOSA (CG) — DETECTOR DE IONIZACIÓN POR LLAMA (FID)
Para el análisis de ETA por CG, los principios generales de las técnicas son válidos. En la actualidad el instrumento preferido de CG para trabajos analíticos de rutina es el cromatógrafo de columna capilar de calibre menor, utilizando columnas capilares con diámetros internos entre 0,2 y 0,32 mm. Se reconoce que, por diversas razones, hay laboratorios que quizá deseen mantener un sistema de columna rellena. Para esos laboratorios, en una edición anterior del presente manual se describe un método que utiliza columnas rellenas. Los procedimientos de CG que utilizan una columna capilar de calibre mayor (0,53 mm de diámetro interno) representan un medio de mejorar el poder de resolución en comparación con las columnas rellenas, y son más robustos que los sistemas de columna capilar de menor calibre. Los sistemas de CG más antiguos, que fueron diseñados para columnas rellenas, se pueden adaptar para utilizarlos con columnas de mayor calibre.

26
MÉTODOS
1. Análisis cualitativo Preparación de muestras y soluciones estándar de ETA
a) Soluciones estándar de ETA
Poner unos 25 mg de sales estándar de ETA8 (debidamente pesados), en una probeta graduada de 25 ml y añadir agua hasta la marca. Colocar con una pipeta una alícuota de 1 a 5 ml de esta solución en un tubo de ensayo de vidrio con tapón de rosca de 10 ml. Añadir por goteo una solución de hidróxido de sodio al 5% hasta obtener un pH 10. Luego añadir 5 ml de disolvente de extracción9. Tapar e invertir el tubo de ensayo por lo menos 10 veces o mezclar en un mezclador Vortex durante 1 minuto y dejar reposar hasta que se separen las capas10. Utilizando una pipeta Pasteur, transferir la capa de disolvente (por ejemplo, el cloroformo) a través de la capa de sulfato de sodio anhidro a un vial de CG. Inyectar 1-2 µl de la capa de disolvente en el CG.
b) Soluciones de muestras de ETA (muestra de ETA desconocido) Pesar de 25 a 150 mg de la muestra, según la pureza anticipada, para obtener una concentración final de aproximadamente 1 mg/ml de sal del analito, y colocarla en una probeta graduada y añadir agua hasta la marca. La pureza prevista de la muestra se determina empíricamente11. Colocar con una pipeta una alícuota de 1 a 5 ml de esta solución en un tubo de ensayo de vidrio con tapón de rosca de 10 ml. Añadir por goteo una solución de hidróxido de sodio al 5% hasta obtener un pH 10. Añadir luego 5 ml de disolvente de extracción (por ejemplo, cloroformo). Tapar e invertir el tubo de ensayo por lo menos 10 veces o mezclar en un mezclador Vortex durante 1 minuto y dejar reposar hasta que se separen las capas. Utilizando una pipeta Pasteur, transferir la capa de disolvente a través de la capa de sulfato de sodio anhidro a un vial de CG. Inyectar 1-2 µl de la capa de disolvente en el CG. Si se requiere una muestra rápida del producto, se pueden tomar muestras directamente en etanol/amoníaco acuoso (99:1), e inyectarlas en el cromatógrafo de fase gaseosa. Con el empleo de este método, la condición del inyector y la columna pueden deteriorarse más rápidamente que
8 Ocasionalmente, los estándares de ETA se pueden obtener en forma de base. En esos casos, no se requiere extracción. En general, sin embargo, es importante que la forma de los estándares y las muestras sea siempre la misma. 9 Todos los analitos deben ser completamente solubles en el disolvente de extracción, y éste último debe ser inmiscible con una capa acuosa. Los disolventes adecuados son, entre otros, n-hexano, cloroformo, cloruro de metileno y acetato de butilo. 10 Cuando se emplea cloroformo como disolvente de extracción, se pueden formar emulsiones. En esos casos, la adición de NaCI mejora la taza de extracción al romper la emulsión. Si se utilizan agitadores modernos y luego se centrifuga la muestra para la separación de las dos capas, normalmente no se forman emulsiones. 11 La pureza de la muestra se puede determinar en base a la experiencia del químico, o utilizando el siguiente método aproximativo: disolver 100 mg de la muestra en 2-3 ml de cloroformo; filtrar la solución, recoger los insolubles, secar y pesar. La cantidad soluble (es decir, el peso original menos los insolubles secos) es la pureza prevista de la muestra. Hay que tener presente, sin embargo, que éste método puede resultar en una pureza anticipada de la muestra inferior a la real para las sales que no son solubles en cloroformo (véase el capítulo VI.A.2, sobre el ensayo de aniones, supra)

27
con las muestras extraídas. (Nota: Este método no es adecuado para análisis cuantitativos dado que es difícil producir una buena cromatografía en presencia de amoníaco.) Condiciones de trabajo con un CG Detector: FID (o NPD, si está disponible o es conveniente) Columna: DB-5 (5% fenil, 95% dimetilpolisiloxano), DB-1 (100% dimetil-
polisiloxano), o equivalente Longitud: 10-30 m, ID 0,20-0,53 mm Espesor de la película: 0,10-0,50 µm Gas portador12: Nitrógeno, helio o hidrógeno, a aprox. 0,8ml/min (N2) o 1-1,2 ml/min
(He o H2) Tasa de desdoblamiento:
20:1 a 50:1
Temperatura de la columna:
La temperatura inicial debe ser suficientemente baja (por ejemplo, 60-90°C) en razón de la alta volatilidad de los ETA bases, por ejemplo, 60°C, mantener por 0,5 min, a 280°C, a una tasa de 12°C/min, mantener la temperatura final durante 30 min.
Temperatura de inyección:
210-250°C
Temperatura del detector:
310°C
RESULTADOS La identificación se realiza comparando el tiempo de retención del analito con el de una norma de referencia. El orden de elución es como sigue: anfetamina < metanfetamina < seudoefedrina < efedrina < PMA < PMMA < MDA < MDMA < 4-MTA < MDEA < MBDB < 2C-B. En las condiciones descritas, la cafeína y la ketamina, que se encuentran con frecuencia en las muestras de ETA en algunas regiones, elucidan después de 2C-B.
2. Análisis cuantitativo A continuación se presentan tres métodos para el análisis cuantitativo de ETA por CG-FID: Un método estándar único (A) y dos métodos estándar múltiples (B y C). Los métodos A y B no requieren derivatización, mientras que el método C requiere sililación. Los tres métodos se describen para uso general. En el anexo IV hay ejemplos de métodos de CG validados para la cuantificación de ETA seleccionados.
i. Método A: método estándar único El método A es adecuado para la cuantificación de la mayoría de los ETA. Comprende la preparación de sólo una solución estándar de ETA en una concentración similar a la concentración prevista del analito.
Preparación de la solución estándar interna (SI) Pesar con precisión unos 25 mg de la solución estándar interna seleccionada (por ejemplo, n-tetradecano u otros n-alcanos con un número par de átomos de carbono, o difenilamina) y disolver en 25 ml de disolvente de extracción (por ejemplo, cloroformo). Si la solución estándar
12 El flujo de gas depende de la columna ID; se puede utilizar la programación del gradiente de presión, si está disponible.

28
interna se utiliza para la extracción, la concentración objetivo se debe preparar dentro del intervalo lineal del instrumento (no más de 0,5 mg/ml).
Preparación de muestras y soluciones estándar de ETA13
Las muestras y soluciones estándar de ETA se deben preparar siguiendo los procedimientos esbozados más arriba para el análisis cualitativo, utilizando la solución estándar interna para la extracción de muestras y estándares de ETA, de la siguiente manera:
a) Soluciones estándar de ETA
Pesar con precisión unos 25 mg de sales estándar14 del ETA de interés en una probeta graduada y añadir agua hasta la marca. Transferir con precisión una alícuota de 1 a 5 ml de esta solución a un tubo de ensayo de vidrio con tapón de rosca de 10 ml. Añadir por goteo una solución de hidróxido de sodio al 5% hasta obtener un pH 10. Añadir luego con precisión 5 ml de solución estándar interna. Tapar e invertir el tubo de ensayo por lo menos 10 veces o mezclar en un mezclador Vortex durante 1 minuto y dejar reposar hasta que se separen las capas. Utilizando una pipeta Pasteur, transferir la capa de disolvente a través de la capa de sulfato de sodio anhidro a un vial de CG. Inyectar 1-2 µl de la capa de disolvente en el CG. Analizar la solución estándar tres veces o más.
b) Soluciones de muestras de ETA (muestra de ETA desconocido)
Pesar con precisión de 25 a 150 mg de la muestra, según la pureza anticipada, para obtener una concentración final de aproximadamente 1 mg/ml de sal del analito, ponerla en una probeta graduada y añadir agua hasta la marca. La pureza prevista de la muestra se determina empíricamente. Con una pipeta, poner una alícuota de 1 a 5 ml de esta solución en un tubo de ensayo de vidrio con tapa de rosca de 10 ml. Añadir por goteo una solución de hidróxido de sodio al 5% hasta obtener un pH 10. Añadir luego con precisión 5 ml de la solución estándar interna. Tapar e invertir el tubo de ensayo por lo menos 10 veces o mezclar en un mezclador Vortex durante 1 minuto y dejar reposar hasta que se separen las capas. Utilizando una pipeta Pasteur, transferir la capa de disolvente a través de la capa de sulfato de sodio anhidro a un vial de CG. Inyectar 1-2 µl de la capa de disolvente en el CG. Analizar la solución de la muestra del ETA desconocido de preferencia dos veces o más. Condiciones de trabajo con CG Las mismas que para el análisis cualitativo por CG (pág. 26).
13 Las muestras y soluciones estándar de ETA y sus concentraciones están diseñadas para usar con columnas capilares y los procedimientos que se describen más abajo. El empleo de columnas y sistemas de CG alternativos puede requerir cambios en términos tanto de la composición relativa como de las concentraciones de los diferentes componentes. 14 Si se utilizan estándares de ETA en forma de base, no se requiere extracción. En esos casos, pesar no menos de unos 10 mg del estándar de ETA y disolver directamente en 10 ml de solución estándar interna dispensada con precisión. Para una cuantitación precisa, es importante que la forma del analito (sal o base) sea igual a la del estándar de ETA.

29
Cálculos De las inyecciones múltiples, calcular las tasas medias de las áreas del pico: 1) del estándar de ETA pertinente en relación con el estándar interno (ASt/IS), y 2) del ETA desconocido en relación con el estándar interno (AATS/IS). El porcentaje de contenido de droga de la muestra se puede calcular utilizando la siguiente fórmula:
CSt AATS/IS ATS (%) = CATS * ASt/IS * 100
donde ATS (%) = Contenido del ETA desconocido (como base o sal; véase la nota 14) en
el material de muestra original (= pureza de la muestra) CSt = Concentración de la solución estándar de ETA (mg/ml), preparada de
conformidad con a) supra (= peso del ETA estándar puro por mililitro de disolvente)
CATS = Concentración de la solución de la muestra del ETA desconocido (mg/ml), preparada de conformidad con b) supra (= peso de la muestra del ETA desconocido por mililitro de disolvente)
AATS/IS = Recuentos del área del pico del ETA desconocido dividido por el recuento del área del pico del estándar interno (de preferencia, una media de dos determinaciones)
ASt/IS = Recuentos del área del pico del ETA estándar dividido por el recuento del área del pico del estándar interno (de preferencia, una media de dos determinaciones)
El método A se puede modificar de único a múltiple diluyendo secuencialmente alícuotas de la solución estándar de ETA con la solución estándar interna utilizando probetas graduadas de 10 ml. De esta forma, se pueden preparar concentraciones estándar de ETA, en función de la concentración apropiada dentro del intervalo lineal del instrumento, y lograr una calibración de puntos múltiples.
ii. Método B: método estándar múltiple sin derivatización El que sigue es un método de calibración de puntos múltiples recomendado; en el anexo IV figuran métodos de CG validados para el análisis cuantitativo de ETA, con y sin derivatización.
Preparación de la solución estándar interna (SI) Pesar cuidadosamente de 0,3 a 0,4 g de la solución estándar interna seleccionada (n-tetradecano, otros n-alcanos, difenilamina o ETA estructuralmente relacionado en forma de base)15 en una probeta graduada de 500 ml y diluir al volumen con cloroformo para obtener una solución estándar interna de 0,6 a 0,8 mg/ml.
15 Si se utiliza un estándar interno en forma de sal (por ejemplo, la sal de un ETA estructuralmente relacionado), se requiere una extracción. En esos casos, pesar no menos de unos 10 mg de estándar de ETA y disolver directamente en 10 ml de solución estándar interna dispensada con precisión.

30
Preparación de soluciones estándar de ETA (soluciones de calibración de CG) Las soluciones madre estándar deben contener todos los compuestos de interés en concentraciones de aproximadamente 1000 mg/l. Se pueden mantener en una probeta cerrada en un refrigerador por un período de hasta un año. Para la preparación de soluciones madre:
a) Pesar con precisión unos 1000 mg de la sal o sales del ETA de interés y ponerlos en una probeta graduada de 1000 ml; añadir agua hasta la marca;
b) Transferir con precisión mediante una pipeta, 5 ml de esta solución a un tubo de ensayo de vidrio con tapa de rosca de 20 ml. Hacer básica al tornasol añadiendo unas pocas gotas de solución de amoníaco concentrada. Añadir con precisión 5 ml de cloroformo;
c) Cerrar y agitar bien, y dejar reposar hasta que se separen las capas. Utilizando una pipeta Pasteur, transferir aproximadamente 1 ml de la capa de cloroformo a través de sulfato de sodio anhidro a un pequeño vaso de precipitación. Esta solución estándar no debe permanecer en reposo más de media hora antes de utilizarla para la calibración;
d) Para la preparación de los estándares de calibración en una calibración de 5 puntos, preparar los diferentes niveles de la siguiente manera:
Preparación de estándares de calibración de puntos múltiples
Nivel de calibración
Solución estándar
de ETA (µl) Solución
SI (µl) CHCl3
(µl)
Concentración aproximada de sal
de ETA (mg/l) Nivel 1 20 100 880 20 Nivel 2 40 100 860 40 Nivel 3 60 100 840 60
Nivel 4 80 100 820 80
Nivel 5 100 100 800 100 Inyectar, por lo menos tres veces, 1-2 µl de cada nivel en el CG y utilizar los valores medios para establecer la curva de calibración.
Preparación de soluciones de muestras de ETA (muestra de ETA desconocido) En general, pero específicamente para los análisis cuantitativos, homogeneizar las muestras antes de iniciar cualquier ensayo o submuestreo.
a) Pesar con precisión una cantidad de muestra suficiente en una probeta graduada de 25 ml para obtener una concentración final de aproximadamente 0,2-1 mg/ml del analito. Agregar agua hasta la marca.
Nota: La cantidad de la muestra que se pese dependerá de la pureza anticipada, como se indica en el método de clasificación preliminar. Por ejemplo, si la pureza anticipada es de un 40%, la cantidad de muestra utilizada deberá ser aproximadamente 60 mg;
b) Transferir con precisión mediante una pipeta 5 ml de esta solución a un tubo de ensayo de vidrio con tapa de rosca de 20 ml. Hacer básica al tornasol añadiendo unas pocas gotas de solución de amoníaco concentrada. Añadir con precisión 5 ml de cloroformo;

31
c) Cerrar y agitar bien, y dejar reposar hasta que se separen las capas. Utilizando una pipeta Pasteur, transferir aproximadamente 1 ml de esta muestra a través de sulfato de sodio anhidro a un pequeño vaso de precipitación. Medir 100 µl de la solución de la muestra, 100 µl de la solución estándar interna y 800 µl de cloroformo y ponerlos en el vial de CG;
d) Inyectar 1-2 µl en el cromatógrafo de fase gaseosa. Analizar la solución de la muestra del ETA desconocido, de preferencia dos veces o más.
Condiciones de trabajo con un CG Para los análisis cuantitativos, es preferible utilizar un CG equipado con un instrumento de muestreo automático. Las condiciones de trabajo pueden ser las mismas que para el análisis cualitativo (pág. 26).
Cálculos El porcentaje de droga contenido en la muestra se calcula a partir de la concentración de la solución de la muestra del ETA desconocido y los correspondientes valores de la curva de calibración. Con instrumentos y programas informáticos de CG modernos, y después de conocer a través del operador las concentraciones de los diferentes estándares de calibración y la solución de la muestra desconocida, se establece la curva de calibración y se realizan los cálculos automáticamente para un punto único de la curva a la terminación del análisis. Normalmente, el resultado se expresará como contenido porcentual de la droga desconocida en el material de muestra original, es decir, como la pureza de la muestra (peso del analito en relación con el peso de la muestra).
iii. Método C: método estándar múltiple con derivatización Preparar muestras y soluciones estándar de ETA derivatizadas midiendo una alícuota (por ejemplo, 100 µl de base seca liberada (es decir, después de haber transferido las muestras y soluciones estándar a través de sulfato de sodio anhidro a un pequeño vaso de precipitación; pasos (c) supra) junto con una alícuota de 100 µl de solución estándar interna, 750 µl de cloroformo y 50 µl de BSTFA (o BSTFA + TMCS al 1%) a un vial de muestras de CG. Para la calibración de puntos múltiples, seguir el método B (véase el cuadro titulado “Preparación de estándares de calibración de puntos múltiples”), utilizando 50 µl de BSTFA en cada paso para la preparación de muestras y soluciones estándar, y añadiendo cloroformo hasta 1 ml.
D. CROMATOGRAFÍA EN FASE GASEOSA — ESPECTROMETRÍA DE MASAS (CG-EM)
La CG-EM es una de la técnicas más utilizadas para la identificación, y recientemente también para la cuantificación de muestras de drogas forenses. Como técnica compuesta, combina el poder de separación y la sensibilidad de la CG-FID con la especificidad por analito de una técnica espectroscópica, produciendo datos espectrales muy específicos de compuestos individuales en una mezcla compleja de compuestos sin separación previa.
Preparación de muestras y soluciones estándar de ETA: Se preparan las muestras en la forma descrita más arriba para los métodos de CG. Se mejoran la sensibilidad del análisis y la especificidad del espectro de masas del ETA mediante derivatización (véase el anexo VII). La preparación de derivativos es particularmente conveniente cuando el espectro de masas de la molécula no derivatizada tiene poco valor de

32
diagnóstico. La mayoría de los ETA no derivatizados tienen fragmentos de iones de bajo cociente masa/carga (m/z), baja intensidad y sólo un ion fragmentado más abundante (pico básico). La derivatización de ETA produce por lo general iones fragmentados de cociente m/z más alto y mayor abundancia. Los iones de gran masa son más específicos y tienen un mayor valor de diagnóstico, ya que no se ven afectados por iones de fondo que interfieren, como material de la purga de la columna u otros contaminantes. Igual que el análisis de CG descrito más arriba, si se requiere una muestra rápida, se puede tomar directamente en el etanol/amoníaco acuoso (99:1) e inyectar en el CG-EM. Ahora bien, en este caso, la condición del inyector y de la columna se puede deteriorar más rápidamente que con las muestras extraídas. Para utilizar en CG-EM, las muestras de aminas primarias también se pueden tomar directamente en disulfito de carbono (CS2), que resulta en la formación de una isotiocianato, un derivativo que da un espectro de masas más característico que los compuestos iniciales. Condiciones de trabajo con CS-EM Condiciones del horno de CG:
Igual que para el análisis de CG (se pueden utilizar las condiciones de los métodos A, B o C)
Columna: Igual que para el análisis de CG, por ejemplo: DB-5 (5% fenil, 95% dimetilpolisiloxano), DB-1 (100% dimetil-polisiloxano), 0,25mm x 30m x 0,25µm, o equivalente
Punto de inyección
Modalidad: Desdoblada/sin desdoblar Temperatura: 250°C Gas portador: Helio, 1 ml/min Modalidad: Flujo constante (o presión constante)
Detector
Modalidad de ionización:
Modalidad EI, 70 eV (Modalidad CI, si se prefiere)
Temperatura de la transferencia:
280°C
Temperatura de la fuente de iones:
230°C
Parámetros de escaneo:
Cámaras de escaneo de imágenes térmicas (microscopio de escaneo de iones, si se requiere), intervalo de escaneo: 35-450 (para sustancias como 2C-B se puede comenzar a m/z 29)
RESULTADOS La identificación se realiza comparando el tiempo de retención y el espectro de masas del analito con el de un estándar de referencia. Todos los compuestos identificados por CG-EM y comunicados por el analista se deben comparar con un espectro de masas actual del estándar de referencia apropiado, de preferencia obtenido con el mismo instrumento y utilizado en las mismas condiciones. Se cuenta con varias bibliotecas de referencia comerciales sobre espectro de masas. Respecto de la mayoría de los instrumentos de CG-EM, muchas de esas referencias están disponibles en línea. En general, sin embargo, es importante utilizar las bibliotecas de espectros de masas, ya sea de una fuente comercial o generada por el usuario, sólo con fines de referencia.

33
Lecturas recomendadas No se dispone de una fuente de bibliografía única para la interpretación del espectro de masas de los ETA, pero hay varias fuentes generales buenas, que en conjunto presentan datos razonablemente amplios, por ejemplo:
Karl Pfleger, Hans H. Maurer, Armin Weber (2000), Mass Spectral and GC Data of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites: Parts I-IV, Wiley. La ONUDD también tiene una colección limitada de espectros de masas relativos a los principales ETA. La colección se puede consultar por la Internet o, previa petición, también está disponible en un CD-ROM.
E. CROMATOGRAFÍA LÍQUIDA DE GRAN RENDIMIENTO (CLGR) Además de la CG, la CLGR es otra importante técnica de separación utilizada comúnmente en el análisis de drogas forense. Para facilitar la preparación de muestras y obtener una mejor reproducibilidad y estabilidad, se recomienda la cromatografía de fase inversa para el análisis de ETA y sus análogos con anillo sustituido. La columna más universal y versátil es una columna de sílica gel ligada con octadecilo (C18). La longitud y el diámetro de la columna, el tamaño de las partículas, el tamaño de los poros y la carga de carbono se deben examinar antes de la selección final de la columna. Dado que hay una gran variedad de fases estacionarias y móviles a disposición del analista, a continuación sólo se dan directrices. MÉTODO
Preparación de muestras y soluciones estándar de ETA Disolver una cantidad apropiada del estándar o la muestra en la fase móvil, apuntando a una concentración del componente activo entre 0,05-0,50 mg/ml. Las soluciones de la muestra se deben filtrar antes del análisis. Las soluciones madre y estándar se deben preparar a partir de estándares de referencia. Los estándares de trabajo deben estar comprendidos en el intervalo lineal del detector y aproximadamente 80%-120% de la concentración objetivo. Es conveniente realizar una calibración de puntos múltiples pero también es aceptable un método estándar único. Condiciones de trabajo Detector: Detector de ordenación de diodos, escaneo rápido o longitud
de onda variable, UV 200-210nm (utilizar también 280-290nm para las fenetilaminas con metilendioxi sustituido)
Fase estacionaria: C8 o C18 con partículas de 5 µm o más pequeñas Longitud de la columna: < 30 cm Diámetro de la columna: < 5,0 mm Columna preliminar: Diámetro: 2-4 mm, longitud: 25 mm, fase inversa C8 o C18 Temperatura de la columna: 15-35°C (se recomienda enérgicamente controlar la
temperatura en entornos sin termostato) Tampón de la fase móvil: Tampón de fosfato pH 2,0-3,2 (por ejemplo, 50 mM fosfato de
sodio monobásico ajustado con ácido fosfórico). Para los picos de los residuos, se pueden necesitar aditivos de fase móvil como sulfatos de alkilo o sulfonatos de alkilo o aminas.

34
Modificador orgánico de fase móvil: Metanol o acetonitrilo entre 2% y 20% (con grupos alkilos
más altos, sobre sulfatos de alkilo o sulfonatos de alkilo, se puedan requerir concentraciones orgánicas más altas).
Tasa de flujo: 0,1-2,0 ml/min Volumen de inyección: 1-100 µl Separación: Se debe realizar un análisis isocrático o de gradiente referido
a tiempos de retención de menos de 30 minutos. RESULTADOS La identificación se realiza comparando el tiempo de retención del analito con el del estándar de referencia y, si es posible, utilizando múltiples longitudes de onda UV o detección por ordenamiento de diodos o escaneo rápido. Para los análogos con anillo sustituido como MDA, MDMA y MDEA, se pueden utilizar también escaneos fluorescentes. La especificidad del método es importante, ya que siempre existe la posibilidad de que compuestos similares elucidan en el mismo tiempo de retención. Un orden de elución típico es el siguiente: norefedrina, efedrina, anfetamina, metanfetamina, MDA, MDMA y MDEA. La separación de los pares efedrina/seudoefedrina y norefedrina/ norseudoefedrina puede ser difícil, o puede requerir ligeros ajustes de las condiciones de la CLGR.
Cuantitación
Se puede utilizar una calibración estándar externa o interna (se recomienda la calibración estándar externa especialmente si se utiliza un inyector basado en válvulas en modalidad de sobrecarga). Se recomienda el uso del área del pico para la cuantitación CLGR, porque el aplastamiento del pico (disminución de la altura del pico) puede ocurrir como resultado del deterioro de la fase estacionaria, haciendo que la altura del pico no sea adecuada para la cuantitación. Se puede aumentar la especificidad de la detección utilizando métodos de detección fluorescente o de longitudes de onda UV múltiples. En el anexo V se describe uno de método de CLGR validado para la cuantitación de ETA.
Lecturas recomendadas Aalberg, L., DeRuiter, J., Noggle, F. T., Sippola, E. y Clark, C. R. (2000), Chromatographic and Mass Spectral Methods of Identification for the Side-Chain and Ring Regioisomers of Methylenedioxymethamphetamine, J. Chromatogr. Sci., 38, págs. 329 a 337.
Clark, C. R., DeRuiter, J., Valer, A. y Noggle, F. T. (1995), Gas Chromatographic-Mass Spectrometric and Liquid Chromatographic Analysis of Designer Butanamines Related to MDMA, J. Chromatogr. Sci., 33, págs. 328 a 337.
Lurie, I. S. (1995), Reversed-Phase High Performance Liquid Chromatographic Analysis of Drugs of Forensic Interest, cap. 4 en Analysis of Addictive and Misused Drugs, Adamovics, J. A., (Ed.), Marcel Dekker, Nueva York, NY, U.S.A., págs. 51 a 132.
Malone, J. V. (1998), HPLC Quantication of Clandestinely Manufactured Mixtures of Amphetamine and Methamphetamine, Microgram, 31, págs. 304 a 307.
Sadeghipour, F., Giroud, C., Rivier, L., y Veuthey, J.-L. (1997), Rapid Determination of anfetaminas by High-Performance Liquid Chromatography with UV Detection, J. Chromatogr., 761, págs.71 a 78.

35
Sadeghipour, F., y Veuthey, J.-L. (1997), Sensitive and Selective Determination of Methylenedioxylated Amphetamines by High-Performance Liquid Chromatography with Fluorimetric Detection, J. Chromatogr., 787, págs.137 a 143. Schneider, R. C., y Kovar, K. A. (2003), Analysis of Ecstasy with a Monolithic Reversed-Phase Column, Chromatographia, 57, págs. 287 a 291.
F. ESPECTROSCOPÍA INFRARROJA TRANSFORMADA DE FOURIER (FTIR) La espectroscopía infrarroja se utiliza mucho como método cualitativo, y desde hace poco también cuantitativo, para la determinación y el análisis estructural de ETA. El análisis FTIR puede ser particularmente útil para la clasificación rápida de tabletas y polvos de ETA, proporcionando indicaciones sobre la homogeneidad de las tabletas o la presencia de lotes mixtos.
Preparación de la muestra16 Los métodos preferidos para la preparación de muestras son la disolución de la muestra en un disolvente apropiado, o su suspensión en una pasta aceitosa. Una técnica menos conveniente es la del método del disco de haluro, en que la muestra se dispersa en un haluro de potasio finamente molido (KBr o KCl) que se imprime en un disco delgado. Ahora bien, la mayoría de los laboratorios forenses prefieren el método del disco de haluro por dos razones: i) los haluros de potasio son transparentes en IR en la denominada región de las huellas digitales ((2000-400 cm-1), y ii) el disco de haluro de potasio por lo general se puede volver a analizar muchas veces si se lo almacena sobre un desecante. El método del disco de haluro17 consiste en golpear una muestra seca hasta obtener un polvo muy fino, mezclar luego aproximadamente 1 mg del polvo de la muestra homogeneizado con 200 mg de haluro álcali molido y secado. Tras la molienda, la mezcla se imprime en un disco transparente delgado. El KCl y el KBr son igualmente apropiados. Ahora bien, el KCI es ligeramente menos higroscópico y en general se lo recomienda en lugar del KBr, especialmente cuando el analito es una sal clorhídrica (para eliminar el problema del intercambio de haluros). El KBr o el KCl que se utilicen deben ser de “pureza IR” y haber sido secado a 110ºC durante por lo menos una hora. Se lo puede almacenar sobre un desecante fuerte (por ejemplo, pentóxido de fósforo) en un desecador, o dejado en un horno y utilizado “cuando se necesite”. Esto puede ser importante en cualquier acción judicial subsiguiente. Asimismo, el material investigado se puede recuperar del disco de haluro para nuevos ensayos. El método de la pasta de Nujol requiere la mezcla de una muestra de polvo fino (2-3 mg) con una gota de Nujol (parafina líquida o perfluorinato alcano de cadena larga), y luego moler la mezcla en un mortero de ágata. La cantidad de Nujol añadido se ajusta para que la pasta final tenga la consistencia de una crema fina. La pasta resultante se distribuye en un disco de haluro álcali (por lo general, KBr) y un disco similar colocado encima. La película entre los discos de haluro no debe contener burbujas de aire. La desventaja evidente de este método es la interferencia del Nujol en el espectro. Un enfoque similar, la técnica de la película delgada, se 16 Un manual separado complementa la serie de manuales de las Naciones Unidas sobre métodos recomendados. Proporciona más detalles sobre las características y usos prácticos para análisis de drogas de diferentes técnicas analíticas. 17 Para sólidos, es importante i) reducir el tamaño de las partículas moliendo hasta una dimensión inferior a la longitud de onda analítica más corta (para evitar la dispersión), y ii) diluir la muestra a fin de reducir al mínimo la absorción en el disco preparado.

36
utiliza también para el análisis directo de bases libres de ETA, que por lo general son líquidos aceitosos. El método ATR de diamante de reflexión simple (por ejemplo, el dispositivo “Golden Gate”) es un método relativamente nuevo para líquidos y sólidos, que no requiere de preparación de muestras. Sin embargo, el espectro obtenido con este método no se puede comparar directamente con los obtenidos con cualquiera de los métodos descritos más arriba. Por último, el método de la célula de gas es un instrumento práctico para el análisis rápido de disolventes y algunos precursores de los laboratorios clandestinos. Aislamiento de la droga pura de una muestra a) Aislamiento de la base libre del ETA
Disolver 25-50 mg de la muestra en 1 ml de 0,1N de ácido tartárico. Agregar 4-5 gotas de hidróxido de amoníaco y extraer con cloroformo. Pasar la capa de cloroformo por una columna pequeña tapada con un algodón para eliminar las partículas en suspensión. Dejar que una parte de la solución de cloroformo se evapore directamente en un disco de KBr y registrar el espectro infrarrojo de la base libre, por ejemplo, utilizando la técnica de la capa delgada en discos de KBr.
b) Aislamiento de las sales del ETA
Triturar una porción de 20-50 mg de la muestra con 1-2 ml de cloroformo. Filtrar, recoger el extracto y el concentrado utilizando un chorro suave de nitrógeno. Inducir la cristalización, filtrar, secar los cristales y analizar el espectro infrarrojo de la sal de ETA resultante utilizando uno de los métodos que se describen a continuación. MÉTODOS El espectro de las sales del ETA se registra utilizando muestras preparadas por los siguientes métodos:
• Disco de haluro KBr (1-1,5%) • Método del aceite de Nujol • Método directo, por ejemplo, reflectancia difusa ATR • Método de la reflectancia difusa
El espectro de bases de ETA se registra utilizando muestras preparadas por los siguientes métodos:
• Método de la película delgada • Tarjetas IR • Método directo, por ejemplo, la reflectancia difusa ATR
Métodos alternativos de preparación de muestras IR para la metanfetamina Método de extracción seca en serie
Colocar 200 mg de muestra de metanfetamina en polvo en una pipeta Pasteur descartable con un tapón de lana de vidrio al final. Lavar la muestra con dos partes de 1 ml de acetona y recoger la fracción de acetona. Después de secarla con aire, lavar la columna con dos partes de 1 ml de cloroformo y recoger la fracción de cloroformo. Dejar que la columna se vuelva a secar y luego enjuagarla con dos alícuotas de 1 ml de metanol y recoger la fracción de metanol. El material insoluble se puede retirar luego de la pipeta. Todas las fracciones se examinan por espectroscopía infrarroja con fines de identificación.
Separación física/recristalización selectiva

37
Los procedimientos que se explican a continuación funcionan bien para el tipo de mezclas indicado; no funcionan tan bien con hidrocloruro de anfetamina.
Una mezcla con contenido de metanfetamina
Mezclar en un vaso de precipitación 25 mg de una muestra con 20 ml de una mezcla 2:1 de cloroformo y hexano, y concentrar la solución hasta aproximadamente la mitad del volumen original. Añadir éter dietílico al precipitado de metanfetamina. Filtrar, secar y obtener el espectro IR (hidrocloruro de metanfetamina).
Una mezcla con contenido de metanfetamina, seudoefedrina y efedrina
Colocar 100 mg de muestra en un pedazo de papel de filtro y lavar con 40 ml de una mezcla 3:1 de cloroformo y hexano. Lavar la parte insoluble con cloroformo, secar y recuperar con fines de examen (hidrocloruro de efedrina). Evaporar la solución original hasta que se seque y dividirla en dos partes iguales. Disolver la mitad de esta muestra en 20 ml de una mezcla 2:1 de cloroformo y hexano, concentrar hasta aproximadamente la mitad del volumen original y añadir éter dietílico para precipitar la metanfetamina. Filtrar, secar y obtener el espectro IR (hidrocloruro de metanfetamina). Disolver la otra mitad de la muestra en 2 ml de cloroformo y añadir 1,6 ml de hexano para precipitar la seudoefedrina. Filtrar, secar y obtener el espectro IR (hidrocloruro de seudoefedrina).
Lecturas recomendadas (métodos alternativos de preparación de muestras IR para la metanfetamina)
Ely, R. A. (1993), Dry Serial Extraction of Illicit metanfetamina Powders For the Identification of Adulterants and Diluents By Infrared Spectroscopy, Journal of the Clandestine Laboratory Investigating Chemists Association (JCLIC), 3(1), págs. 21 a 25.
Oulton, S. R. (1997), Separation and Identification of Ephedrine, Pseudoephedrine and Methamphetamine Mixtures, Microgram, 30(12), págs. 289 a 296.
Stinson, S. B. y Berry, M. R. (1974), Separation and identification of amphetamine or methamphetamine in combination with ephedrine or caffeine, 7(4), pág. 51.
RESULTADOS
La identificación se logra comparando el espectro del analito con el del estándar de referencia, o de una biblioteca espectral.
Lecturas recomendadas Laboratory Methods in Vibrational Spectroscopy (third ed.), editado por Willis, H. A., van der Maas J. H. y Miller, R. G. J., John Wiley & sons, Chichester, 1987
G. ANÁLISIS DE ISÓMEROS ÓPTICOS La mayoría de los ETA tiene un átomo de carbono asimétrico resultante en un par de enantiómeros para cada ETA. En función de la fuente, por lo tanto, se pueden encontrar formas enantioméricas diferentes de anfetamina, metanfetamina y otros ETA en muestras incautadas sometidas a análisis De conformidad con el Convenio sobre Sustancias Sicotrópicas de 1971 de las Naciones Unidas,

38
cada isómero óptico ( d- y I-), así como la mezcla racémica (dl) de anfetamina y metanfetamina, están sometidos a fiscalización. Con respecto a la mayoría de los ETA con anillo sustituido, sólo los racematos están sometidos a fiscalización, y los enantiómeros genéricos se incluyen mediante una frase genérica. Los isómeros ópticos difieren en cierta medida en cuanto a la actividad farmacológica y en ciertos países están sujetos a diferentes medidas de reglamentación. En los países en que la legislación nacional exige la identificación del isómero óptico específico presente, se pueden utilizar los siguientes procedimientos analíticos.
1. Punto de fusión La comparación del punto de fusión de la mezcla de la muestra con el estándar de referencia enantioméricamente puro es un ensayo rápido y sencillo para distinguir isómeros ópticos. Por ejemplo, d- y l-hidrocloruro de metanfetamina tienen el mismo punto de fusión (170-175°C), pero una mezcla de cantidades iguales de ambos isómeros ópticos (mezcla racémica) tiene un punto de fusión más bajo (130-135°C). Este método requiere muestras bastante puras. En general, los puntos de fusión se deben determinar utilizando muestras secas.
2. Ensayos de microcristales (Véase también la sección sobre el ensayo de microcristales como ensayo presuntivo, supra.) Para los ETA, normalmente se utiliza la técnica de la “gota en suspensión”, o ensayo de volatilidad. Esta técnica requiere una placa con cavidades, una cubierta de vidrio, el reactivo de ensayo y un reactivo de volatilización. En el anexo III se describen los reactivos de ensayo y de volatilización.
Ensayos de microcristales para isómeros ópticos de anfetamina Ensayo del cloruro de oro [HAuCl4 al 5% en H3PO4]
Transferir una pequeña cantidad del polvo de la muestra a la depresión de la placa de cavidad, y añadir una o dos gotas del reactivo de volatilización (solución de NaOH al 5%). Esto libera la amina libre en forma de base libre volátil, que emerge de la solución como un vapor. Transferir inmediatamente una gota del reactivo de ensayo (HAuCl4 al 5% en H3PO4) a la placa de vidrio e invertir y cruzar la placa sobre la cavidad de muestra. El reactivo reacciona entonces con el vapor de aminas presente en la cavidad. Tras un intervalo apropiado, volver a invertir la placa del reactivo y examinar los cristales en el reactivo o en el borde de la cuota de reactivo.
RESULTADOS
La d- y la l-anfetamina dan microcristales idénticos, que se asemejan a largas varillas amarillas o agujas ásperas y largas hojas angostas. La forma de distinguirlas es formar el racemato, que produce cristales diferentes. El racemato de dl-anfetamina da al principio gotas “aceitosas” y luego cristales escamosos coloreados. Estos cristales se forman en general después de la inversión.
Distinción de d- y l-anfetamina
Si el ensayo explicado más arriba indica que la muestra es de d- o l-anfetamina, se debe determinar cuál está presente. A tal fin, añadir parte del polvo de la muestra desconocida a una pequeña cantidad de sal de d-anfetamina conocida en una cavidad y a una pequeña cantidad de

39
sal de l-anfetamina en otra. Repetir el ensayo. La mezcla que sea (d+d) o (l+l) producirá largas varillas amarillas, etcétera. La mezcla que sea (d+l) dará los cristales escamosos del racemato descritos más arriba.
Metanfetamina
La d-metanfetamina ensayada con HAuCl4 al 5% en H3PO4 da hojas “V” con un lado más corto que el otro. Éstas se desarrollan muy rápidamente si el ensayo se realiza directamente sobre la muestra seca, o más lentamente si la muestra se diluye o el ensayo se realiza utilizando la técnica de la gota en suspensión La l-metanfetamina da hojas cruzadas singulares y hojas “V”, que forman extremos en forma de cigarros característicos (afiladas a ambos lados en el extremo de la hoja). La d,l-metanfetamina forma hojas singulares y “X”, que a veces tienen forma de “cuchillo”. Los extremos de la hoja sólo están afilados en un lado, igual que un cuchillo.
MDMA
La MDMA ensayada con HAuCl4 al 5% en H3PO4 da cristales blancos en forma de “X” de doble refacción con conglomerados en forma de estrella bajo luz polarizadora. Estos cristales son similares a los de la d,l-metanfetamina con cloruro de oro, pero con un poco de práctica se pueden diferenciar. Nota: Dado que la MDMA es muy sensible a la reacción con el reactivo de cloruro de oro, sólo basta una parte muy pequeña para obtener buenos resultados. El ensayo del cloruro de oro es también útil para los precursores efedrina y seudoefedrina, y puede ser útil para la nicotinamida y la cafeína. Se deben utilizar muestras de referencia de estas sustancias para obtener cristales de referencia.
Ensayos de microcristales para isómeros ópticos de la metanfetamina [H3BiI6 en H2SO4]
Utilizar el procedimiento de la gota en suspensión descrito más arriba para la anfetamina pero utilizando el reactivo de ensayo H3BiI6 en H2SO4 (véase el anexo III en relación con la preparación del reactivo). RESULTADOS
La d-metanfetamina da agujas largas de color naranja. La dl-Metanfetamina da varillas naranja-rojizas características con extremos sesgados.
Lecturas recomendadas Fulton, C.C. (1969), Modern Microcrystal Tests for Drugs, Wiley-Interscience, Nueva York
Ōno, M., Microcrystal Test, Japón, 1996 (presenta una introducción amplia a la técnica de los ensayos de microcristales, e incluye fotografías de resultados de ensayo de microcristales de 39 ETA y sus precursores).
Ruybal, R. (1986), Microcrystalline test for MDMA, Microgram, 19 (6), págs. 79 y 80.

40
American Society of Analytical Chemistry: AOAC Official Methods of Analysis (1984), págs. 704 a 714. Stall, W. J. (1981), The separation of methamphetamine and procaine utilizing the volatility test / hanging drop method for amphetamines, Microgram, 14 (10), pág. 148.
3. Técnicas instrumentales Hay varios métodos instrumentales directos (CG quiral, CLGR o EC) y métodos de derivatización indirectos que se pueden utilizar en el análisis de los isómeros ópticos de ETA. La elección del método depende del alcance del análisis y la disponibilidad de equipo y otros instrumentos de laboratorio. Los métodos directos e indirectos se deben considerar como complementarios, ya que ninguno ofrece una solución universal para la separación quiral. Las ventajas y desventajas de ambos criterios se deben examinar con cuidado. Los métodos instrumentales directos permiten analizar los isómeros ópticos sin derivatización, utilizando fases estacionarias quirales y/o aditivos quirales como tampón de corrida (EC). Los métodos indirectos se basan en la derivatización del analito con un reactivo quiral para producir dos diastereoisómeros diferentes con propiedades físicas y químicas diferentes que se pueden separar en una fase estacionaria aquiral. La elección del reactivo quiral depende de varios factores, como el poder de separación, la sensibilidad, la eficiencia de la derivatización y sus compatibilidades con la técnica instrumental. Los métodos que emplean la derivatización quiral son normalmente menos costosos y no requieren columnas o equipo especializado. El uso de columnas aquirales normales facilita la integración de las separaciones quirales en programas de análisis de rutina.
i. Técnicas de CG La cromatografía de fase gaseosa es una técnica probada de separación quiral. Se puede realizar utilizando métodos directos, empleando columnas capilares quirales, o métodos indirectos, que logran la separación utilizando reactivos quirales y fases estacionarias aquirales.
a) Cromatografía en fase gaseosa quiral (método directo de CG)
Las fases estacionarias disponibles en el comercio para la separación de isómeros ópticos por CG se producen normalmente añadiendo macromoléculas de ciclodextrina a una fase estacionaria común. La fase estacionaria quiral más común utilizada para los ETA es la beta-ciclodextrina hecha base. MÉTODO
La preparación (extracción) de muestras para análisis por CG quiral es igual a la de la CG normal (véase supra). Condiciones de trabajo con CG Columna: Dexcst, película de 0,25 mm x 30m x 0,25 micrones, o equivalente Gas portador: Helio a aprox, 1,2 ml/min Temperatura del horno: 120°C durante 1 min, luego 1,5°C/min a 175°C, mantener durante
1,5 min Volumen de inyección: 1 µl Temperatura del inyector: 190° C Temperatura del detector: 280° C

41
Nota: Se pueden utilizar otras condiciones de trabajo con la CG, pero hay que ensayarlas para la separación óptima de compuestos quirales. Él empleo de nitrógeno como gas portador requiere tasas de flujo más bajas (aprox. 0,8 ml/min) para la velocidad óptima, lo que da lugar a picos más amplios.
b) Derivatización quiral (método de CG indirecto)
Se pueden preparar diastereoisómeros de ETA utilizando diferentes reactivos como acilcloruros, alkilsulfonatos, isotiocianatos, cloroformatos. Los más populares son el ácido de Mosher [R(+)- o S(-)-α-metoxi-a-(trifluorometil)-ácido fenilacetico], cloruro de ácido de Mosher’s, y N-trifluoroacetil-1-prolil cloruro (TPC, también conocido como TFAP-Cl). El ácido de Mosher y el ácido clorhídrico dan lugar a una derivatización cuantitativa de la mayoría de las aminas, con excepción de la efedrina y la seudoefedrina. Se pueden utilizar como reactivos para análisis de CG y CLGR. Se sabe que el TPC produce de derivativos estables de casi todos los ETA, incluidas las efedrina. Se presta mejor al análisis por CG. En el anexo VII figuran detalles de la preparación de muestras de ETA utilizando ácido de Mosher para las derivatizaciones quirales. Las condiciones de trabajo con CG para análisis cualitativos (véase más arriba) también se pueden utilizar para el análisis de derivativos quirales.
Lecturas recomendadas Beckett, A. H. y Testa, B. (1972), Stereochemical separation and configural assignment by gas-liquid chromatography of N-trifluoroacetyl-l-prolyl amides of asymmetric l-fenilisopropyl-amines, J. Chromatogr., 69, pág. 285.
Cody, J. T., (1992), Determination of methamphetamine enantiomer ratios in urine by gas chromatography-mass spectrometry, J. Chromatogr., 580, págs. 77 a 95.
Jirovsk ý, D., y otros (1998), Methamphetamine—properties and analytical methods of enantiomer determination, Forensic Sci. Int., vol. 96, págs. 61 a 70.
Liu, J. H. y Ku, W. W. (1981), Determination of enantiomeric N-trifluoroacetyl-l-prolyl chloride amphetamine derivatives by capillary gas chromatography/mass spectrometry with chiral and achiral stationary phases, Anal. Chem., 53, pág. 2180.
Liu, J. H., Ku, W. W., Tsay, J. T., Fitzgerald, M. P., y Kim, S. (1982), Approaches to drug sample differentiation. III: A comparative study of the use of chiral and achiral capillary column gas chromatography/mass spectrometry for the determination of methamphetamine enantiomers and possible impurities, J. Forensic Sci., 27 (1), págs. 39 a 48.
Liu, J. H., Ku, W. W. y Fitzgerald, M. P. (1983), Separation and characterization of amine drugs and their enantiomers by capillary column gas chromatography—mass spectrometry, J. Assoc. Off Anal. Chem., 66, págs. 1443.
McKibben, T. (1992), Separation and Identification of Drug Enantiomers via N-TFA-(S)-Prolyl Chloride Derivatization, Journal of the Clandestine Laboratory Investigating Chemists Association, 2 (1), págs. 21 a 20. Esta referencia incluye la derivatización de enantiómeros de anfetamina, metanfetaminas, efedrina,
seudoefedrina, MDA, MDMA, DOB, DOM, 2,4,6-TMA, 3,4,5-TMA, propilhexedrina y PMA.

42
Moshers acid, R. Kaslauskas, G. Trouth and A. Lissi (AGAL), 16 Seminario de Colonia sobre análisis de presencia de drogas. Pastor-Navarro, M. D., Porras-Serrano, R., Herráez-Hernández, R., Campins-Falco, P. (1998), Automated determination of amphetamine enantiomers using a two-dimensional column-switching chromatographic system for derivatization and separation, Analyst, 123, pág. 319.
Shin, H. S. y Donike, M. (1996), Stereospecific derivatization of amphetamines, phenol alkylamines, and hidroxiamines and quantification of the enantiomers by capillary GLC/MS, Anal. Chem., 68, pág. 3015.
Wells, C. E. (1970), GLC determination of the optical isomers of anfetamina, J. Assoc. Off Anal. Chem., 53, pág.113.
ii. Técnicas de CLGR Para la CLGR se han utilizado varios criterios, incluida la derivatización con reactivos quirales, la incorporación de aditivos quirales en la fase móvil, y el empleo de fases estacionarias quirales. En el comercio hay una variedad de columnas para CLGR quirales. El comportamiento de las fases estacionarias quirales se ha mejorado mucho en los últimos años, aunque esos análisis siguen siendo costosos.
Lecturas recomendadas Lemr, K., Jirovsky, D. y Seveik, D. (1996), Effect of Some Parameters on Enantiomer Separation of Ephedrine, Methamphetamine and Selegiline using HPLC with β-Cyclodextrin Stationary Phase, J. Liq. Chrom. & Rel. Technol., 19, págs. 3173 a 3191.
Makino, Y., Suzuki, A., Shirota, T. Ogawa, Shirota, O. (1999), Direct analysis of methamphetamine enantiomers in urine with strong cation exchange pre-column and b-cyclodextrin-bonded semi-micro column, J. Chromatography B, 729, págs. 97 a 101.
Noggle, F. T., DeRuiter, J. y Clark, C. R. (1986), Liquid Chromatographic Determination of the Enantiomeric Composition of Methamphetamine Prepared from Ephedrine and Pseudoephedrine, Anal. Chem., 58, págs. 1643 a 1648.
Pihlainen, K. y Kostiainen, R. (2004), Effect of the Eluent on Enantiomer Separation of Controlled Drugs by Liquid Chromatography-Ultraviolet Absorbance Detection-Electrospray Ionisation Tandem Mass Spectrometry using Vancomycin and Native β-Cyclodextrin Chiral Stationary Phases, J. Chromatogr. A., 1033, págs. 91 a 99.
Rizzi, A. M., Hirz, R., Cladrowa-Runge, S. y Jonsson, H. (1994), Enantiomeric Separation of Amphetamine, Methamphetamine and Ring Substituted Amphetamines by Means of a β-Cyclodextrin-Chiral Stationary Phase, Chromatographia, 39, págs.131 a 137.
Sadeghipour, F. y Veuthey, J. L. (1998), Enantiomeric Separation of Four Methylenedioxylated Amphetamines on β-Cyclodextrin Chiral Stationary Phases, Chromatographia, 47, págs. 285 a 290.
Sellers, J. K., Duffitt, G. L., Gaines, M. L. y Liu, R. H. (1996), High Performance Liquid Chromatographic Analysis of Enantiomeric Composition of Abused Drugs, Forensic Sci. Rev., 8, págs. 92 a 108.
iii. Técnicas de EC La electroforesis de zona capilar es muy ventajosa para el análisis quiral ya que permite la separación sumamente eficiente de enantiómeros sin derivatización ni columnas especiales

43
(capilares). Para la separación de ETA, se utilizan normalmente aditivos quirales como tampones de corrida (como hidroxil-propil beta-ciclodextrin).
Lecturas recomendadas Iwata, Y. T., Garcia, A, Kanamori, T., Inoue, H., Kishi. T. y Lurie, I. S. (2002), The Use of Highly Sulfated Cyclodextrin for the Simultaneous Chiral Separation of Amphetamine-type Stimulants by Capillary Electrophoreis, Electrophoreis., 23, págs 1328 a 1334.
Lurie, I. S., Hays, P. A. y Parker, K. P. (2004), Capillary Electrophoreis Analysis of a Wide Variety of Seized Drugs Using the Same Capillary with Dynamic Coatings, Electrophoresis., 25, págs. 1580 a 1591.
Lurie, I. S., Klein, R. F. K., Dal Cason, T., LeBelle, M., Brenneisen y R., Weinberger, R. (1994), Chiral Resolution of Cationic Drugs of Forensic Interest by Capillary Electrophoresis with Mixtures of Neutral and Anionic Cyclodextrins, Anal. Chem., 66, págs. 4019 a 4026.
Varesio, E., Gauvrit, J. Y., Longeray, R., Lanteri, P., Veuthey, J. L. (1997), Central Composite Design in the Chiral Analysis of Amphetamines by Capillary Electrophoresis, Electrophoresis., 18, págs. 931 a 937.
iv. Técnicas IR Aunque los enantiómeros tienen el mismo espectro infrarrojo, la espectroscopia IR se puede utilizar para distinguir los enantiómeros de un compuesto determinado después de convertirlos en los correspondientes diastereómeros. Los ETA, como todas las bases orgánicas, se pueden hacer reaccionar fácilmente con ácidos orgánicos quirales para formar diastereómeros. Las d- y l-anfetamina, por ejemplo, se pueden acoplar con d-ácido mandélico para formar dos diastereómeros, d-anfetamina-d-mandelato y l-anfetamina-d-mandelato, que tiene un espectro IR diferente.
MÉTODO
Se hace básica una solución acuosa de sal de ETA (10-50 mg) y se extrae el ETA en cloruro de metileno. El cloruro de metileno se seca sobre sulfato de sodio anhidro y se concentra hasta aproximadamente 2 ml. Se añade una solución saturada de d-ácido mandélico en cloruro de metileno, varias gotas a la vez, hasta que la solución de ETA se neutraliza (papel pH). Se deja cristalizar la sal de d-mandelato-ETA, la solución se filtra por succión, y los cristales se lavan con una pequeña parte de cloruro de metileno. Después de secada, se prepara un disco de KBr con los cristales y se adquiere el espectro infrarrojo. El procedimiento se repite utilizando isómeros conocidos ópticamente puros de los ETA correspondientes.
RESULTADOS
La identificación de los isómeros ópticos se logra comparando el espectro resultante con uno de los obtenidos a partir de los estándares de referencia puros. Las bandas de IR en la región de 800-1600cm-1 proporciona la información analíticamente más diferenciada. El espectro IR de las sales de isómeros ópticos de anfetamina con ácido mandélico figuran en una edición anterior de manual de las Naciones Unidas sobre “Métodos recomendados para el ensayo de anfetamina y metanfetamina” (ST/NAR/9). Los datos se pueden consultar en la página web de la Sección de Laboratorio y Asuntos Científicos.

44
Lecturas recomendadas Chappell, J.S. (1997), Infrared discrimination of enantiomerically enriched and racemic samples of methamphetamine salts, Analyst, 122, 755 a 760.
Chappell, J.S. (1998), A novel infrared method for the determination of the enantiomeric composition of methamphetamine salts, Proceedings of the American Academy of Forensic Sciences, 4, 32.
Heagy, J. (1970), Infrared method for distinguishing optical isomers of amphetamine, Analytical Chemistry, 42 (1970), pág. 1459.

45
VII. OTRAS TÉCNICAS ANALÍTICAS PARA EL ANÁLISIS DE ETA
Hay otras técnicas analíticas adecuadas para la identificación forense y/o la cuantitación de ETA, entre ellas:
• Electroforesis capilar (EC) • Cromatografía en fase gaseosa — espectroscopía infrarroja transformada de Fourier
(CG-FTIR) • CL-EM y CG-EM • Espectroscopía casi infrarroja (NIR) • Espectroscopía de resonancia magnética nuclear (RMN) (cualitativa y cuantitativa) • FTIR cuantitativa • CCD cuantitativa • Espectroscopía FTIR Raman • Cromatografía en fase gaseosa-microextracción en fase sólida (CG-SPME)
La descripción de la mayoría de estas técnicas escapa al alcance del presente manual sobre métodos recomendados para el análisis de ETA, por lo que el analista debe remitirse a un manual complementario de carácter general sobre técnicas analíticas, sus características y sus usos prácticos para el análisis de drogas. A continuación se describen brevemente cuatro técnicas cualitativas: RMN, EC, CG-SPME y CG-FTIR porque constituyen opciones específicas atractivas para el análisis de ETA.
A. TÉCNICAS DE RESONANCIA MAGNÉTICA NUCLEAR 1H (RMN) La disponibilidad de un gran número de isómeros posicionales de ETA estructuralmente relacionados, especialmente ETA con anillo sustituido, requiere instrumentos eficaces que proporcionen la información estructural necesaria para su diferenciación. La RMN permite al analista distinguir inequívocamente entre diferentes derivativos de anfetamina con anillo sustituido, aun en presencia de diluyentes y otros adulterantes. Aunque ciertas pautas de sustitución se asemejan entre sí en el área correspondiente a los protones de la cadena lateral alkil, el espectro integrado y la pauta de las señales de protones aromáticos permite distinguir unos de otros. Aunque se trata de un poderoso instrumento para la identificación de análogos, el costo de la espectroscopía de RMN y la experiencia técnica necesaria impide su aplicación en gran escala a los análisis de rutina.
MÉTODO
Disolver unos 20 mg de la muestra de la droga en 1 ml de D2O. Si hay materiales insolubles, centrifugar; en caso contrario, transferir directamente el supernatante a un tubo de RMN. Registrar el espectro de esta solución que contiene el ETA en forma de sal. Liberar la base libre del ETA in situ añadiendo 20-30 mg de K2CO3 sólido y 0.5 ml de CDCl3 y registrar el espectro de la base libre. Comparar el espectro de la sustancia desconocida con el espectro de referencia, que se registró en un instrumento de transformación de Fourier a 90 MHz utilizando un ángulo de 18º (1 µsec) sin demora tras la adquisición de los datos. El espectro de RMN de referencia del ETA seleccionado figura en una edición anterior de los dos manuales de las Naciones Unidas relativos al análisis de ETA, es decir, “Métodos recomendados para el ensayo de anfetamina y metanfetamina” (ST/NAR/9) y “Métodos recomendados para el ensayo de los derivados anfetamínicos ilícitos con anillo sustituido” ((ST/NAR/12). Los datos se pueden consultar también en la página web de la Sección de Laboratorio y Asuntos Científicos.

46
Lecturas recomendadas Dawson, B. A. (1991), The use of Nuclear Magnetic Resonance Spectroscopy for the detection and quantitation of abused drugs, en: The analysis of drugs of abuse, T. A. Gough (ed.), Wiley & Sons Ltd, págs. 284 a 296.
Lee G. S. H, Craig D. C., Kannangar G. S. K., Dawson, M., Conn C., Robertson J., Wilson M. A. (1999), Analysis of 3,4-methylenedioxy-N-methylamphetamine (MDMA) in “ecstasy” tablets by 13C solid state nuclear magnetic resonance (NMR) spectroscopy, J. Forensic Sci., 44 (4), 761 a 771.
Chew, S. L., y Meyers, J. A. (2003), Identification and quantitation of gamma-hidroxibutyrate (NaGHB) by nuclear magnetic resonance spectroscopy, J. Forensic Sci., 48 (2), 292.
Rothchild, R. (2003), Identification of heroin diluent: one- and two-dimensional proton and carbon-13 NMR studies of procaine hydrochloride: computational studies of procaine and its conjugate acid, Spectroscopy Letters, 36 (1 & 2), 35 a 42.
B. ELECTROFORESIS CAPILAR (EC) La EC, igual que la CLGR, no requiere pasos de derivatización o extracción y, por lo tanto, es más conveniente que la CG para el análisis de compuestos de ETA. En contraste con la CLGR, la EC ofrece un mayor poder de resolución para el análisis de estos solubles, lo que confiere mayor rapidez al análisis. El empleo de capilares revestidos dinámicamente constituye una importante mejora en la precisión, la eficiencia de los picos y/o los tiempos de separación para los compuestos de ETA en comparación con métodos semejantes que utilizan capilares no revestidos. Además, el enfoque de los capilares dinámicamente revestidos puede facilitar el análisis quiral rápido de una muestra utilizando el mismo capilar y el mismo vial de muestra pero con una corrida de tampón diferente. En el anexo VI se incluye un método de EC para la cuantitación de ETA seleccionados y para la metodología de diferenciación de isómeros ópticos.
Lecturas recomendadas Lurie, I. S., Hays, P. A. y Parker, K. P. (2004), Capillary Electrophoreis Analysis of a Wide Variety of Seized Drugs Using the Same Capillary with Dynamic Coatings, Electrophoresis., 25, págs 1580 a 1591.
Lurie, I. S., Bethea, M. J., McKibben, T. D., Hays, P. A., Pellegrini, P., Sahai, R., Garcia, A. G. y Weinberger R. (2001), Use of Dynamically Coated Capillaries for the Routine Analysis of Methamphetamine, Amphetamine, MDA, MDMA, MDEA and Cocaine using Capillary Electrophoresis, J. Forensic Sci., 46, págs. 1025 a 1032.
Piette, V. y Parmentier, F. (2002), Analysis of Illicit Amphetamine Seizures by Capillary Zone Electrophoreis, J. Chromatogr. A., 979, págs. 345 a 352.
C. CROMATOGRAFÍA EN FASE GASEOSA — MICROEXTRACCIÓN EN FASE SÓLIDA (CG-SPME)
La SPME (microextracción en fase sólida) es una técnica de preparación de muestras libres de disolventes, que se puede utilizar para análisis de cabeza sobre soluciones o directamente sobre polvos de ETA, o que se puede utilizar para análisis (de trazas) de soluciones acuosas que contienen ETA. La SPME se realiza normalmente con una jeringa especial con una fibra de silicio colocada en la punta del pistón de la jeringa. La fibra está revestida con una fase polimérica como las que se utilizan en las columnas capilares. Durante el muestreo, el revestimiento de fibra absorbe los compuestos de la fase gaseosa en la cabeza, o directamente

47
de la fase acuosa. Hay muchos revestimientos de fibra diferentes disponibles para el análisis de ETA y otras sustancias. Uno de los más utilizados es una fibra revestida de polidimetilsiloxano (PDMS).
1. Método de análisis de cabeza Una muestra acuosa de ETA se hace alcalina para convertir las aminas en bases libres, aumentando de esta forma su volatilidad. La muestra se puede también calentar para aumentar la cantidad de aminas en la fase gaseosa sobre la solución de la muestra. La jeringa con la fibra expuesta se coloca en el espacio superior sobre la solución y las bases libres del ETA son absorbidas en la fibra. En la situación de equilibrio, la cantidad extraída de cada compuesto absorbido en la fibra es proporcional a su concentración en la solución, aunque con diferentes coeficientes de distribución. Una vez completada la extracción, la jeringa se transfiere al instrumento de análisis escogido. Cuando la fibra se inserta en el punto calentado del inyector de CG, las aminas extraídas son desorbidas en forma térmica. En CLGR, CEC y EC, la mezcla disolvente produce la elución de las aminas de la fibra.
2. Análisis (de trazas) de muestras acuosas La fibra se sumerge directamente en la solución de la muestra acuosa, que se hizo alcalina a fin de liberar las bases libres de ETA. La solución de la muestra se revuelve para aumentar el intercambio de los compuestos entre la solución y la fibra, a fin de acelerar la extracción. El análisis de la muestra se realiza en la misma forma que se describió para el método del análisis de cabeza.
Lecturas recomendadas Battu, C., Marquet, P., Fauconnet, A. L., Lacassie, E. y Lachâtre, G. (1998), Screening procedure for 21 amphetamine-related compounds in urine using solid-phase microextration and gas chromatography-mass spectrometry, J. Chromatogr. Sci., vol. 36, págs. 1 a 7.
Centini, F., Masti, A. y Barni Comparini, I. (1996), Quantitative and Qualitative analysis of MDMA, MDEA, MA and amphetamine in urine by head-space/solid phase micro-extraction (SPME) and GC-MS, Forensic Science International, vol. 83, págs. 161 a 166.
Pawliszyn, J., Solid Phase Microextraction, Wiley-VCH, Nueva York, 1997.
Yashiki, M., Kojima, T., Miyazaki, T., Nagasawa, N., Iwasaki, Y. y Hara, K. (1995), Detection of amphetamines in urine using head space-solid phase microextration and chemical ionization selected ion monitoring, Forensic Science International, vol. 76, págs. 169 a 177.
Zhang, Z., Yang, M. J. y Pawliszyn, J. (1994), Solid-Phase Microextraction, Analytical Chemistry, vol. 66, No.17, págs. 844A a 855A.
D. CROMATOGRAFÍA EN FASE GASEOSA — ESPECTROSCOPÍA INFRARROJA TRANSFORMADA DE FOURIER (CG-FTIR)
La CG-FTIR, otra técnica compuesta, combina las ventajas de la técnica de separación por CG con la alta especificidad por analito de la IR. La CG-FTIR con una célula de flujo ligera no tiene limitaciones en cuanto a la corriente de gas portador, por lo que es ideal cuando se la utiliza con columnas cortas de gran calibre, que permiten análisis eficientes y rápidos. Por ejemplo, los ETA más comunes se pueden analizar en menos de cinco minutos. Sin embargo, como esta técnica es más bien insensible, hay que insertar grandes cantidades de sustancia, y el espesor de la película de la fase estacionaria es fundamental para no sobrecargar la columna.

48
MÉTODO
Preparación de muestras de ETA La preparación de la muestra es similar a la del análisis por CG cualitativo descrito más arriba, pero las concentraciones del analito objetivo deben ser de 1-10 mg/ml.
Condiciones de trabajo con CG Se pueden utilizar las condiciones de trabajo con CG indicadas más arriba (por ejemplo, métodos CG-FID or CG-EM), con las siguientes modificaciones: Columna del CG: Columna de calibre mayor 0,32-0,53 mm ID Longitud de la columna: 3-10 m Espesor de la película: >1,0 µm Temperaturas de la línea de transferencia: 300°C Resolución espectral: 8 cm-1
RESULTADOS
La identificación se realiza comparando el tiempo de retención y el espectro FTIR del analito con el de un estándar de referencia.
Lecturas recomendadas Bergkvist, H., Eyem. J. y Lundberg, L. en: Sandra, P. (Ed), Proceedings of the Thirteenth International Symposium of Capillary Chromatography, Riva del Garda, 13 a 16 de mayo de 1991, Huertig, Heidelberg, págs. 1160 a 1170.
Duncan, W. y Soine, W. H., Identification of Amphetamine Isomers by GC/IR/MS, J. Chromatogr. Sci., 26, 1988, 521 a 526.
Griffiths, P. R. y de Haseth, J. A., Fourier Transform Infrared Spectrometry, John Wiley & sons, Nueva York, 1986.

49
ANEXOS Anexo I. Estructuras químicas de ETA seleccionados
Cuadro 3. ANFETAMINAS SIN ANILLO SUSTITUIDO
R3
NR2
R1R4
Nota: A menos que se indique específicamente, los nombres no se refieren a enantiómeros individuales
Nombre común Nombre de la IUPAC R1 R2 R3 R4 Anfetamina 1-metil-2-feniletilamina H H CH3 H
Metanfetamina N-metil-N-(1-metil-2-feniletil)amina CH3 H CH3 H N-etilanfetamina N-etil-N-(1-metil-2-feniletil)amina C2H5 H CH3 H Dimetilanfetamina N,N-dimetil-N-(1-metil-2-feniletil)amina CH3 CH3 CH3 H N-hidroxianfetamina N-(1-metil-2-feniletil)hidroxilamina H OH CH3 H N-hidroximetanfetamina N-metil-N-(1-metil-2-feniletil)hidroxilamina CH3 OH CH3 H
Catina (1S,2S)-2-amino-1-fenilpropano-1-ol H H CH3 OH Catinona 2-amino-1-fenilpropano-1-ona H H CH3 =O Metcatinona 2-(metilamino)-1-fenilpropano-1-ona CH3 H CH3 =O Fenetillina 1,3-dimetil-7-{2-[(1-metil-2-feniletil)amino] etil}-3,7-
dihidro-1H-purina-2,6-diona H teofillina CH3 H
Fenilpropilmetilamina (PPMA) N-metil-N-(2-fenilpropil)amina CH3 H CH3 CH3

50
Cuadro 4. ANFETAMINAS SUSTITUIDAS CON METILENEDIOXI
N
R3
R2
R1
R6
O
O
Nota: A menos que se indique específicamente, los nombres no se refieren a enantiómeros individuales
Nombres comunes (acrónimos) Nombre IUPAC R1 R2 R3 R6 3,4-metilenedioxianfetamina (MDA, tenanfetamina)
1-(1,3-benzodioxol-5-il)propan-2-amina H H CH3 H
3,4-metilenedioximetanfetamina (MDMA) N-[2-(1,3-benzodioxol-5-il)-1-metiletil]-N-metilamina CH3 H CH3 H 3,4-metilenedioxietilanfetamina (MDE, MDEA) N-[2-(1,3-benzodioxol-5-il)-1-metiletil]-N-etilamina C2H5 H CH3 H 3,4-metilenedioxi-N,N-dimetilanfetamina (MDDM)
N-[2-(1,3-benzodioxol-5-il)-1-metiletil]-N,N-dimetilamina
CH3 CH3 CH3 H
N-hidroxi-3,4-metilenedioxianfetamina (N-hidroxi-MDA, N-hidroxitenanfetamina)
N-[2-(1,3-benzodioxol-5-il)-1-metiletil]hidroxilamina H OH CH3 H
N-hidroxi-N-metil-3,4-metilenedioxianfetamina (N-hidroxi-MDMA, FLEA)
N-[2-(1,3-benzodioxol-5-il)-1-metiletil]-N-metilhidroxilamina
CH3 OH CH3 H
N-metil-1-(3,4-metilenedioxifenil)-2-butanamina (MBDB)
N-[1-(1,3-benzodioxol-5-ilmetil)propil]-N-metilamina CH3 H C2H5 H
1-(3,4-metilenedioxifenil)-2-butanamina (BDB) 1-(1,3-benzodioxol-5-il)butan-2-amina H H C2H5 H 5-metoxi-3,4-metilenedioxianfetamina (MMDA) 1-(7-metoxi-1,3-benzodioxol-5-il)propan-2-amina H H CH3 OCH3
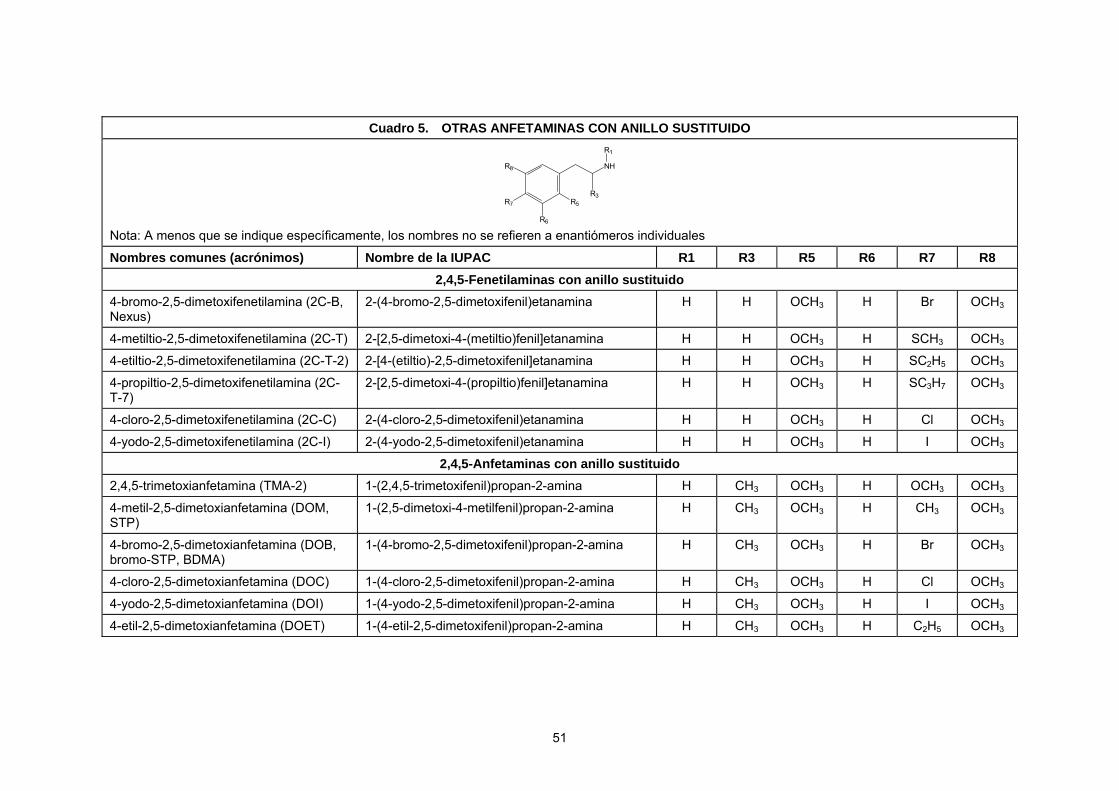
51
Cuadro 5. OTRAS ANFETAMINAS CON ANILLO SUSTITUIDO
R3
NH
R1
R6
R5R7
R8
Nota: A menos que se indique específicamente, los nombres no se refieren a enantiómeros individuales
Nombres comunes (acrónimos) Nombre de la IUPAC R1 R3 R5 R6 R7 R8 2,4,5-Fenetilaminas con anillo sustituido
4-bromo-2,5-dimetoxifenetilamina (2C-B, Nexus)
2-(4-bromo-2,5-dimetoxifenil)etanamina H H OCH3 H Br OCH3
4-metiltio-2,5-dimetoxifenetilamina (2C-T) 2-[2,5-dimetoxi-4-(metiltio)fenil]etanamina H H OCH3 H SCH3 OCH3 4-etiltio-2,5-dimetoxifenetilamina (2C-T-2) 2-[4-(etiltio)-2,5-dimetoxifenil]etanamina H H OCH3 H SC2H5 OCH3 4-propiltio-2,5-dimetoxifenetilamina (2C-T-7)
2-[2,5-dimetoxi-4-(propiltio)fenil]etanamina H H OCH3 H SC3H7 OCH3
4-cloro-2,5-dimetoxifenetilamina (2C-C) 2-(4-cloro-2,5-dimetoxifenil)etanamina H H OCH3 H Cl OCH3 4-yodo-2,5-dimetoxifenetilamina (2C-I) 2-(4-yodo-2,5-dimetoxifenil)etanamina H H OCH3 H I OCH3
2,4,5-Anfetaminas con anillo sustituido 2,4,5-trimetoxianfetamina (TMA-2) 1-(2,4,5-trimetoxifenil)propan-2-amina H CH3 OCH3 H OCH3 OCH3
4-metil-2,5-dimetoxianfetamina (DOM, STP)
1-(2,5-dimetoxi-4-metilfenil)propan-2-amina H CH3 OCH3 H CH3 OCH3
4-bromo-2,5-dimetoxianfetamina (DOB, bromo-STP, BDMA)
1-(4-bromo-2,5-dimetoxifenil)propan-2-amina H CH3 OCH3 H Br OCH3
4-cloro-2,5-dimetoxianfetamina (DOC) 1-(4-cloro-2,5-dimetoxifenil)propan-2-amina H CH3 OCH3 H Cl OCH3
4-yodo-2,5-dimetoxianfetamina (DOI) 1-(4-yodo-2,5-dimetoxifenil)propan-2-amina H CH3 OCH3 H I OCH3 4-etil-2,5-dimetoxianfetamina (DOET) 1-(4-etil-2,5-dimetoxifenil)propan-2-amina H CH3 OCH3 H C2H5 OCH3

52
Otras pautas de sustitución de anillos (fenetilaminas y anfetaminas) 3,4,5-trimetoxifenetilamina (mescalina) 2-(3,4,5-trimetoxifenil)etanamina H H H OCH3 OCH3 OCH3
para-metoxianfetamina (PMA) 3-(4-metoxifenil)-1-metilpropilamina H CH3 H H OCH3 H para-metoximetanfetamina (PMMA) N-[2-(4-metoxifenil)-1-metiletil]-N-metil- amina CH3 CH3 H H OCH3 H 2,5-dimetoxianfetamina (DMA) 1-(2,5-dimetoxifenil)propan-2-amina H CH3 OCH3 H H OCH3 3,4,5-trimetoxianfetamina (TMA) 1-(3,4,5-trimetoxifenil)propan-2-amina H CH3 H OCH3 OCH3 OCH3 4-metiltioanfetamina (4-MTA) 1-[4-(metiltio)fenil]propan-2-amina H CH3 H H SCH3 H

53
Anexo II. Preparación de reactivos para ensayos cromáticos y de aniones Todos los reactivos se deben preparar de conformidad con un procedimiento establecido. Ensayo de Chen
Reactivo 1: Añadir 1 ml de ácido acético glacial a 100 ml de agua (= 1% (v/v) solución de ácido acético acuosa)
Reactivo 2: Disolver 1 g de sulfato de cobre (II) en 100 ml de agua (= solución acuosa (v/v) de CuSO4 al 1%)
Reactivo 3: Disolver 8 g de hidróxido de sodio en 100 ml de agua (= 2N solución acuosa de hidróxido de sodio).
Ensayo del ácido gálico
Reactivo : Disolver 0,1 g de ácido gálico en 20 ml de ácido sulfúrico concentrado (= 0,5% (p/v) solución)
Ensayo de Marquis
Reactivo 1: Añadir de 8 a 10 gotas (aproximadamente 0,25 ml) de solución de formaldehído al 37% a 10 ml de ácido acético glacial
Reactivo 2: Ácido sulfúrico concentrado Ensayo del fosfato
Molibdato amónico: Disolver 10 g de molibdato amónico [(NH4)6Mo7024 x 4H20] en 100 ml
de agua (=solución acuosa (p/v) de molibdato amónico al 10%) Ácido nítrico: Añadir cuidadosamente 10 ml de ácido nítrico a 90 ml de agua
(= solución de ácido nítrico (v/v) al 10%) Ensayo del nitrato de plata (también conocido como el ensayo del cloruro) Disolver 1,7 g de nitrato de plata en 100ml de agua (= solución acuosa de nitrato de plata
al 1,7%). Ensayo de Simon
Reactivo 1: Disolver 2 g de carbonato de sodio en 100 ml de agua (= solución acuosa de carbonato de sodio al 2%)
Reactivo 2: Disolver 0,9 g de nitroprusiato de sodio en 90 ml de agua (= solución acuosa de nitroprusiato de sodio al 1%)
Reactivo 3: Mezclar 10 ml de solución de acetaldehído y 10 ml de etanol (= solución etanólica de acetaldehído (v/v) al 50%)
Ensayo del sulfato
Disolver 5 g de cloruro de bario dihidrato en 100 ml de agua (= solución acuosa de cloruro de bario aprox. al 5%).
Hay también otros procedimientos establecidos para la preparación de reactivos para ensayos cromáticos, por ejemplo, el método de Clarke, que muestra una ligera variación en las recetas.

54
Anexo III. Ensayos de microcristales Microcristales típicos clasificados Las formas típicas de los microcristales se pueden clasificar en nueve grupos, empleando los términos descriptivos que se indican a continuación. A fin de describir todos los tipos de microcristales, se pueden añadir adjetivos como irregular, fino o cuadrangular a los términos básicos
Fuente: Ōno, M., Microcrystal Test, Japón, 1996 Reactivos Reactivo de ensayo — HAuCl4 en H3PO4 al 5%
Disolver 1 g de ácido tetracloro áurico (HAuCl4.4H2O) en 20 ml de una solución que contenga un volumen de H3PO4 concentrado y dos volúmenes de agua.
Reactivo de ensayo — H3BiI6 in H2SO4
Disolver 1,25 g de yoduro de potasio en 2,0 ml de agua. Añadir 2,5 ml de una solución de H2S04 diluida al 1:7 con agua, 0,5 ml de solución de Bi(NO3)3 concentrada y 0,1 g de hipofosfito de sodio. La solución madre de Bi(NO3)3 concentrada se prepara disolviendo 50 g de subnitrato de bismuto en 70 ml de una solución de HNO3 (diluida al 1:1 con agua) y completada a 100 ml con agua. El reactivo de ensayo se puede mantener durante varios meses.

55
Reactivo de volatilización
Preparar una solución acuosa de NaOH al 5%.

56
Anexo IV. Métodos de CG validados para la cuantitación de ETA seleccionados
A continuación se proporcionan ejemplos de métodos de CG validados para el análisis cuantitativo de ETA seleccionados. El método B no requiere derivatización; el método C requiere sililación. Las soluciones estándar y muestras de ETA descritas y sus concentraciones están diseñadas para usar con columnas capilares y con los procedimientos que se describen más abajo. El empleo de columnas y sistemas de CG alternativos puede requerir cambios tanto en la composición relativa como en las concentraciones de componentes individuales. Equipo y reactivos
• Contenedores de cristal graduados de grado B o mejores. • Cromatógrafo de gas con detector de llama de ionización. • Balanza analítica capaz de pesar con una exactitud de ±0,0001 g. • Todos los reactivos deben ser de calidad analítica.
MÉTODO B: Método de calibración de puntos múltiples sin derivatización El método B es un método validado para el análisis cuantitativo por CG de ETA no derivatizados, específicamente los siguientes: anfetamina, metanfetamina, MDA, MDMA, MDEA y MBDB.
Preparación de la solución estándar interna (SI): fenilbencilamina (PBA) Pesar con exactitud 0,3 a 0,4 g de PBA en una probeta graduada de 500 ml y diluir a volumen con cloroformo para obtener una solución estándar interna de 0,6 a 0,8 mg/ml.
Preparación de soluciones estándar de ETA (soluciones de calibración de CG) Las soluciones madre estándar deben contener todos los compuestos de interés en concentraciones de aproximadamente 1000 mg/l. Se pueden mantener en una probeta cerrada en un refrigerador hasta un año. Para la preparación de soluciones madre:
a) Medir con exactitud unos 1000 mg del compuesto o los compuestos de interés en una probeta graduada de 1000 ml y completar hasta la marca con agua.
b) Con una pipeta, transferir exactamente 5 ml de esta solución a un tubo de ensayo de vidrio con tapa de rosca de 20 ml. Hacer básica a tornasol añadiendo unas pocas gotas de solución de amoníaco concentrada. Añadir exactamente 5 ml de cloroformo.
c) Tapar y batir bien, luego dejar reposar hasta que se separen las capas. Utilizando una pipeta de Pasteur, transferir aproximadamente 1 ml de la capa de cloroformo a través de sulfato de sodio anhidro a un vial pequeño. Esta solución estándar no se debe dejar reposar más de media hora antes de utilizarla en la calibración.
d) Para la preparación de estándares de calibración para una calibración de 5 puntos, preparar los diferentes niveles de la siguiente manera:

57
Preparación de estándares para una calibración de 5 puntos
Nivel de calibración
Solución estándar
de ETA (µl) Solución
SI (µl) CHCl3
(µl)
Concentración aproximada de sal
de ETA (mg/l)
Nivel 1 20 100 880 20 Nivel 2 40 100 860 40
Nivel 3 60 100 840 60 Nivel 4 80 100 820 80 Nivel 5 100 100 800 100
Preparación de soluciones de muestras de ETA (muestra de ETA desconocido) En general, pero específicamente para análisis cuantitativos, homogeneizar las muestras antes de iniciar cualquier ensayo o submuestreo.
a) Pesar exactamente una cantidad de muestra suficiente en una probeta graduada de 25 ml para obtener una concentración final de aproximadamente 0,2-1 mg/ml del analito. Completar con agua hasta la marca. Nota: La cantidad de muestra que se pese dependerá de la pureza anticipada en función del método de clasificación preliminar. Por ejemplo, si la pureza anticipada es del 40%, la cantidad de muestra debe ser aproximadamente 60 mg.
b) Con una pipeta, transferir exactamente 5 ml de esta solución a un tubo de ensayo de vidrio con tapa de rosca de 20 ml. Hacer base a tornasol añadiendo unas pocas gotas de solución de amoníaco concentrada. Agregar exactamente 5 ml de cloroformo.
c) Cerrar y batir bien, y luego dejar reposar hasta que se separen las capas. Utilizando una pipeta de Pasteur, transferir aproximadamente 1 ml de esta solución de muestra a través de sulfato de sodio anhidro a un vial pequeño. Colocar 100 µl de la solución de muestra, 100 µl de la solución estándar interna y 800 µl de cloroformo en un vial de muestra de CG.
d) Inyectar en el cromatógrafo de gas.
Condiciones de trabajo con CG Para el análisis cuantitativo, es preferible un CG equipado con un extractor de muestras automático. Se reconoce que el empleo de instrumentos diferentes puede requerir ajustes en las condiciones de trabajo. Columna: HP-5, 0,32mm x 30m x 0,5µm Portador de gas: Helio a aprox. 1,2 ml/min (presión de cabeza 12 psi) Temperatura del horno: 100°C durante 4 min, luego 10°C /min a 270°C, y mantener 1 minuto Volumen de inyección: 1 µl Temperatura del inyector: 190°C Detector: Detector de llama de ionización a 270°C
Tiempos de retención aproximados Anfetamina 7,18 min Metanfetamina 8,25 min
MDA 13,16 min

58
MDMA 13,93 min
MDEA 14,58 min MBDB 15,17 min Fenilbencilamina (IS) 16,33 min
Cafeína 17,92 min Ketamina 18,33 min
Cálculos En los análisis de rutina, el programa informático realizará los cálculos una vez terminado el ensayo analítico. El resultado se imprimirá automáticamente en un informe expresado como %w/w del analito como una base (es decir, peso del analito en relación con el peso de la muestra).
MÉTODO C: Método de calibración utilizando BSTFA como un reactivo de derivatización (calibración de puntos múltiples o de punto único)
El método C es un método validado para el análisis cuantitativo por CG de ETA derivatizados, específicamente los siguientes: efedrina, seudoefedrina, BDMA (4-bromo-2,5-dimetoxianfetamina) y 2C-B El empleo del método C se recomienda específicamente para muestras de ETA que contienen efedrina y/o seudoefedrina, que frecuentemente no se resuelven con otro tipo de analitos y producen picos amplios. Véase el anexo VII, que contiene detalles sobre derivatización.
Preparación de la solución estándar interna (SI): fenilbencilamina (PBA) Igual que para el método B, supra.
Preparación de soluciones estándar de ETA (soluciones de calibración para CG) Para las calibración de puntos múltiples utilizando BSTFA, preparar los diferentes niveles igual que para el método B supra, de la siguiente manera: tomar 20, 40, 60, 80 y 100 µl de soluciones madre estándar de ETA, para cada nivel de calibración, añadir 100 µl de la solución estándar interna, y 50 µl de BSTFA. Añadir cloroformo hasta completar 1 ml.
Preparación de soluciones de la muestra (muestra de ETA desconocido) a) Pesar con precisión la muestra en una probeta graduada de 25 ml hasta obtener una
concentración final de aproximadamente 0,2-1 mg/ml de analito. Completar con agua hasta la marca. Nota: La cantidad de la muestra dependerá de la pureza anticipada en función del método de clasificación preliminar. Por ejemplo, si la pureza anticipada es de un 40%, la cantidad de muestra debe ser aproximadamente 60 mg.
b) Con una pipeta, transferir exactamente 5 ml de esta solución a un tubo de ensayo de vidrio con tapa de rosca de 20 ml. Hacer base a tornasol añadiendo unas pocas gotas de solución de amoníaco concentrada. Añadir exactamente 5 ml de cloroformo.
c) Cerrar y batir bien, y luego dejar reposar hasta que se separen las capas. Utilizando una pipeta de Pasteur, transferir aproximadamente 1 ml de esta solución a través de sulfato de sodio anhidro a un vial pequeño. Colocar 100 µl de la solución de muestra, 750 µl de cloroformo y 50 µl de BSTFA en un vial de muestra de CG.

59
d) Inyectar en el cromatógrafo de gas.
Condiciones de trabajo con CG Columna: HP-5, 0,32 mm x 30m x 0,5 µm Portador de gas: Helio a aprox. 1,2 ml/min (presión de cabeza 12 psi) Temperatura del horno: 100°C 4 min., luego 5°C/min a 200°C, luego 10°C/min a 270°C, y
mantener 1 minuto Volumen de inyección: 1 µl Temperatura del inyector: 190° C Detector (FID): 270°C
Tiempos de retención aproximados Seudoefedrina-TMS 14,99 min Efedrina-TMS 15,16 min Fenilbencilamina-TMS (SL) 23,49 min Cafeína 26,22 min
Ketamina-TMS 26,75 min 2C-B-TMS 27,91 min
Cálculos En los análisis de rutina, el programa informático realizará los cálculos una vez terminado el ensayo analítico. El resultado se imprimirá automáticamente en un informe expresado como %w/w del analito como una base (es decir, peso del analito en relación con el peso de la muestra).

60
Anexo V. Método CLGR para la cuantitación de ETA seleccionados A continuación se examina un método validado para la cuantitación por CLGR de ETA seleccionados solubles, incluidas la anfetamina, la metanfetamina, la fentermina y la MDMA. MÉTODO CLGR PARA LA CUANTITACIÓN DE ETA Preparación de muestras y soluciones estándar de ETA
Solución estándar de ETA
Pesar una cantidad apropiada de estándar de ETA en una probeta graduada hasta obtener una concentración final de aproximadamente 0,50 mg/ml. Diluir a volumen con metanol.
Solución de la muestra del ETA Pesar una cantidad apropiada de muestra en una probeta graduada para obtener una concentración final de fenetilamina aproximadamente igual a la de la solución estándar. Diluir a volumen con metanol.
Condiciones de trabajo con CLGR Columna: 5 µm Luna C18 (Phenomenex, Torrance, CA, USA) 150 x 3,0 mm Temperatura de la columna: 35°C Inyección: 5 µl Fase móvil: 10% acetonitrilo, 90% (50 mM fosfato + 50 mM trietanolamina,
pH 2,2)*; tasa de flujo 1,0 ml/min
Longitud de onda UV: 210 nm
*El tampón se prepara disolviendo 22,5 ml de ácido fosfórico concentrado en 4 l de agua de calidad para CLGR. Añadir lentamente unos 25 ml de trietanolamina para ajustar la solución a un pH de 2,2.
Tiempos de migración relativos aproximados Nicotinimida 0,28
Fenetilamina 0,55 Fenilpropanolamina 0,56 Doxilamina 0,56
Procaina 0,62 Efedrina 0,64 Seudoefedrina 0,65
Anfetamina 0,82 Acetominifen 0,93
metanfetamina 1,00 (2,7 min) MDA 1,00 Quinina 1,04 Cloroquina 1,09 Dimetilanfetamina 1,12

61
MDMA 1,18
Cafeína 1,48 Lidocaina 1,50 MDEA 1,55
Ketamina 1,95 Clorofeniramina 1,99
P2P 3,17 Safrol 3,50 Quaifenesin 3,61 Aspirina 5,09
Cálculos El porcentaje de contenido de ETA de la muestra se calcula a partir del área de pico del ETA y el área de pico y la concentración del estándar de ETA pertinente.
Lecturas recomendadas Malone, J. V. (1998), HPLC Quantification of Clandestinely Manufactured Mixtures of Amphetamine and Methamphetamine, Microgram, 31, págs. 304 a 307

62
Anexo VI. Método de EC validado para cuantitación de ETA seleccionados A continuación se explica un método validado para la cuantitación por EC de ETA seleccionados solubles, incluidas la anfetamina, la metanfetamina, MDA, MDMA y MDEA en un instrumento de EC Agilent HP3D. Hay que tener presente que algunas condiciones, como la longitud capilar, el voltaje, los tiempos de lavado y las presiones y parámetros de sección pueden cambiar con otros fabricantes de instrumentos. MÉTODO DE CAPILARES DINÁMICAMENTE REVESTIDOS PARA CUANTITACIÓN DE ETA Preparación de muestras y soluciones estándar de ETA
Disolvente de inyección
Pesar 1034 mg de fosfato de sodio monobásico en una probeta graduada de 100 ml. Diluir a volumen con agua de calidad para CLGR (ajustar el pH a aproximadamente 2,6 utilizando ácido fosfórico y añadir por goteo). Transferir el contenido a una probeta graduada de 2000 ml con ayuda de agua de calidad para CLGR. Diluir a volumen con agua de calidad para CLGR. Esta solución final contiene 3,75 mM de fosfato, con un pH de 3,2. Alternativamente, transferir todo el contenido (con ayuda de agua de calidad para CLGR) de la botella de 250 ml de concentrado de disolvente de inyección (MicroSolv, Eatontown, NJ, USA) a una probeta de 5 litros. Diluir a volumen con agua de calidad para CLGR.
Solución estándar interna de ETA Pesar una cantidad apropiada de N-butilanfetamina HCl (o un estándar interno apropiado a una probeta graduada) y colocar en una probeta graduada para obtener una concentración final de aproximadamente 1,0 mg/ml. Diluir a volumen con disolvente de inyección.
Solución estándar de ETA Pesar una cantidad apropiada de estándar de ETA en una probeta graduada para obtener una concentración final de aproximadamente 0,08 mg/ml. Con una pipeta, transferir una cantidad apropiada de solución estándar interna para obtener una concentración final de 0,1 mg/ml. Diluir a volumen con disolvente de inyección.
Solución de muestra de ETA Pesar una cantidad apropiada de muestra en una probeta graduada para que la concentración final de fenetilamina sea aproximadamente igual a la de la solución estándar. Con una pipeta, transferir una cantidad apropiada de solución estándar interna para obtener una concentración final de 0,1 mg/ml. Diluir a volumen con disolvente de inyección. Condiciones de trabajo para EC (aquiral) Capilaridad: Sílica pura 32 cm (23,5 cm a la ventana del detector) por 50 µm i.d. Temperatura capilar: 15°C Acondicionamiento: 1 min 0,1N hidróxido de sodio; 1 min agua; 1 min CElixir A (MicroSolv);
2 min CElixir B, pH 2,5 (MicroSolv). Inyección: 50 milibares x 2 s de muestra seguida de 35 milibares x 1 s de agua Corrida de tampón: CElixir B, pH 2,5 Voltaje: 10 kV Longitud de onda UV: 195 nm

63
Tiempos de migración relativa aproximados Doxilamina 0,76
Clorfeniramina 0,78 Quinina 0,80
Beta-fenetilamina 0,81 Cloroquina 0,81 Nicotinimida 0,84 Anfetamina 0,87 Metanfetamina 0,88
Procaína 0,88 MDA 0,90 Norseudoefedrina 0,91 MDMA 0,91 Norefedrina 0,92 Seudoefedrina 0,92
Tetracaína 0,93 Efedrina 0,93 Fenilefrina 0,95 MDEA 0,96 Ketamina 0,96
Feniltoxilamina 0,97 N-butilanfetamina (SI) 1,00 (4,6 min)
Metorfán 1,00 Lidocaína 1,03 Benzocaína 1,25
Acetominofén 2,11 Cafeína 2,14 Quaifenesina 2,14 P2P 2,24
DMSO (marcador neutral) 2,40 Aspirina 2,71
Cálculos El contenido (%) del ETA desconocido se calcula a partir de su área de pico, y el área de pico y la concentración del estándar de ETA, en relación con el área de pico del estándar interno (estándar y muestra).

64
Condiciones de trabajo para EC (quiral) Capilaridad: Sílica pura 32 cm (23,5 cm a la ventana del detector) por 50 µm i.d. Temperatura capilar: 15°C Acondicionamiento: 1 min 0,1N hidróxido de sodio; 1 min agua; 1 min CElixir A (MicroSolv);
2 min CElixir B, pH 2,5 + 50 mM 2-hidroxipropil-Β-Ciclodextrina (MicroSolv).Inyección: 50 milibares x 2 s de muestra seguido de 35 milibares x 1 s de agua Voltaje: 20kV Longitud de onda UV: 195 nm (longitud de banda 10 nm)
Tiempos de migración relativa aproximados l-norseudoefedrina 0,81 d-norefedrina 0,83
l-norefedrina 0,83 l-seudoefedrina 0,83 l-anfetamina 0,85 d-efedrina 0,86
d-anfetamina 0,86 l-efedrina 0,87
l-metanfetamina 0,87 d-norseudoeferina 0,88 d-metanfetamina 0,89 d-seudoefedrina 0,90 N-butilanfetamina* 1,00 (3,75 min)
N-butilanfetamina* 1,02 MDA* 1,03 MDA* 1,04 MDMA* 1,05 MDMA* 1,07 MDEA* 1,10
MDEA* 1,12
* d- o l-enantiómero.
Lecturas recomendadas Lurie, I. S., Hays, P. A. y Parker, K. P. (2004), Capillary Electrophoresis Analysis of a Wide Variety of Seized Drugs Using the Same Capillary with Dynamic Coatings, Electrophoresis., 25, págs. 1580 a 1591.
Lurie, I. S., Bethea, M. J., McKibben, T. D., Hays, P. A., Pellegrini, P., Sahai, R., Garcia, A. G. y Weinberger R. (2001), Use of Dynamically Coated Capillaries for the Routine Analysis of Methamphetamine, Amphetamine, MDA, MDMA, MDEA and Cocaine using Capillary Electrophoresis, J. Forensic Sci., 46, págs. 1025 a 1032.

65
Anexo VII. Derivatizaciones La derivatización de ETA no es obligatoria, ya que la mayoría se presta a los análisis por CG y son termoestables. No obstante, la derivatización de ETA (aminas primarias o secundarias) mejora sus propiedades cromatográficas al reducir la absorción en columnas no deseable y no específica, así como las interferencias con la matriz. Los métodos de derivatización que se recomiendan más abajo funcionan bien para la mayoría de los ETA que se encuentran con más frecuencia; ahora bien, en raras ocasiones, algunas condiciones de la reacción, como el tiempo o la temperatura de reacción, deben ajustarse.
Nota analítica Si se escoge la derivatización como método de preparación de muestras, la extracción de la muestra se debe realizar en la forma descrita en las secciones pertinentes más arriba. Después de la extracción, el disolvente debe secarse por evaporación bajo una ligera corriente de nitrógeno a temperatura ambiente, o alternativamente a 30ºC. A fin de evitar pérdidas del analito por evaporación, este paso se debe realizar con mucho cuidado, especialmente para el análisis cuantitativo de ETA. La forma más eficiente de prevenir la evaporación del analito es evaporar el disolvente hasta aproximadamente 1 ml y luego añadir unas pocas gotas (50 µl) de un disolvente con alto punto de ebullición (conservador de disolvente), por ejemplo, dimetilformamida. Tras añadir el conservador de disolvente, se puede continuar la evaporación hasta que quede sólo una delgada capa de disolvente. La muestra está ahora lista para la derivatización utilizando uno de los métodos recomendados.
Procedimientos de derivatización Acetilaciones
Anhídrido heptafluorobutírico (HFBA) Procedimiento A (acetilación con anhídrido)
Añadir 50 µl de HFBA al residuo seco del ETA extraído en un vial de reacción18. Tapar el vial, batir por 30 segundos e incubar durante 20 min a 75ºC. Evaporar el exceso de reactivo. Reconstituir el residuo seco en 50 µl de acetato etílico, e insertar 1-2 µl en la columna del CG.
Procedimiento B (acetilación con anhídrido en presencia de un catalizador básico)
Añadir 50 µl de 0,5M hidróxido de potasio al residuo seco de ETA extraído y seguidamente 500 µl de tolueno. Después de mezclar y centrifugar, transferir la capa orgánica a un tubo de ensayo limpio, y añadir 50 µl de HFBA. Mezclar cuidadosamente y en seguida añadir 500 µl de bicarbonato de sodio p/v al 10% mezclando continuamente. Centrifugar el tubo de ensayo hasta que se separe la capa superior de tolueno. Inyectar 1-2 µl de la capa de tolueno en la columna del CG.
Anhídrido pentafluoroacético (PFAA) Añadir 50 µl de PFAA al residuo seco del ETA extraído en un vial de reacción. Tapar el vial, batir durante 30 segundos e incubar durante 20 min a 75ºC. Evaporar el exceso de reactivo. Reconstituir el residuo seco en 50 µl de acetato etílico, e inyectar 1-2 µl en la columna del CG.
18 Los viales de reacción son viales con tapa de rosca de vidrio grueso resistente a las temperaturas, por lo general con un fondo cónico dentro del vial. Si no se dispone de viales especializados, la derivatización se puede hacer en cualquier tubo de ensayo revestido de Teflon con tapa de rosca.

66
Anhídrido trifluoroacético (TFAA) Añadir 100 µl de acetato etílico y 50 µl de TFAA al residuo seco del ETA extraído en un vial de reacción. Tapar el vial, batir durante 30 segundos e incubar durante 20 min a 60ºC. Evaporar el exceso de reactivo. Reconstituir el residuo seco en 50 µl de acetato etílico, e inyectar 1-2 µl en la columna del CG.
N-metilbis-trifluoroacetamida (MBTFA) Añadir 500 µl de MBTFA al residuo seco del ETA extraído en un vial de reacción. Tapar el vial, batir durante 30 segundos e incubar durante 30 min a temperatura ambiente. Evaporar el exceso de reactivo. Reconstituir el residuo seco en 50 µl de acetato etílico, e inyectar 1-2 µl en la columna del CG. Dado que eluce temprano en el análisis de CG, la evaporación del exceso de reactivo puede no ser necesaria si se prevé que la concentración de analito será suficientemente grande para la inyección directa de la mezcla de derivatización.
Lecturas recomendadas M. Doneke, J. Chromatography, 78, 273 (1973).
Sililación
N-metil-N-tert.-butil-dimetilsilil trifluoroacetamida (MTBSTFA) Añadir 100 µl de acetonitrilo y 150 µl de MTBSTFA al residuo seco del ETA extraído. Tapar el vial y calentar durante 15 min a 90ºC. Dejar la muestra a temperatura ambiente por otras 2 horas, o más, especialmente si se han de sililizar grupos amino secundarios. Añadir 500 µl de acetonitrilo, mezclar bien e inyectar 1-2 µl en la columna del CG (si se prevén concentraciones bajas del analito, la muestra se puede inyectar directamente sin dilución con acetonitrilo). Alternativamente, para evitar un pico temprano de MTBSTFA en el cromatograma de CG, combinar el residuo seco del ETA extraído con 100 µl de acetonitrilo y 100 µl de MTBSTFA en un vial de 3 ml. Sellar el vial y dejar reposar la mezcla durante 2 horas. Añadir 100 µl de agua para hidrolizar cualquier parte de MTBSTFA que no haya reaccionado. Añadir 250 µl de n-hexano, mezclar vigorosamente y centrifugar. Decantar la capa superior de hexano e inyectar 1-2 µl en la columna del CG.
Lecturas recomendadas R. Melgar, R. C. Kelly, J. Anal. Toxicol., 17 (nov./dec., 1993), pág. 399.
N,O-bis-[(trimetilsilil)trifluoroacetamida] (BSTFA) Añadir 50 µl de BSTFA y 100 µl de acetonitrilo al residuo seco del ETA extraído en un vial de reacción. Tapar el vial, batir e incubar durante 15 min a 70ο C. Inyectar 1-2 µl en la columna del CG.

67
Derivatización quiral Se puedan preparar sustratos de diastereoisómero de ETA utilizando muchos reactivos diferentes, como los acilcloruros, alkilsulfonatos, isotiocianatos, cloroformatos, pero los dos que se indican a continuación son los más populares.
1. Ácido R(+)/S(-)-a-metoxi-a-(trifluorometil)-fenilacético (ácido de Mosher), o cloruro de ácido R(+)/S(-)-a-metoxi-a-(trifluorometil)-fenilacético (cloruro de ácido de Mosher)
Disolver el residuo seco de la muestra del ETA extraído en 1 ml de THF y mezclar con 0,5 ml de 0,2 M solución de ácido de Mosher en THF en un vial de reacción revestido de Teflon con tapa de rosca. Añadir 0,5 ml de solución de bicarbonato de sodio p/v al 10%, tapar el vial y calentar a 65°C durante 1 hora. Extraer la fase acuosa con cloroformo, secar con un sulfato de magnesio y secar por evaporación. Reconstituir el residuo en un disolvente adecuado para el análisis por CG (por ejemplo, cloroformo; véase la sección sobre análisis cualitativo por CG, supra). En lugar del ácido de Mosher, se puede utilizar el cloruro correspondiente. Está disponible en el comercio o se puede preparar por reflujo del ácido con cloruro de tionilo. Los cloruros ácidos suelen ser más reactivos, aunque el propio ácido de Mosher produce la derivatización cuantitativa de la mayoría de las aminas, con excepción de la efedrina y la seudoefedrina. El ácido de Mosher o sus derivados se pueden utilizar como reactivos para análisis tanto por CG como por CLGR.
2. Cloruro de N-trifluoroacetil-L-prolil (TPC, o TFAP-Cl) Disolver el residuo seco de la muestra del ETA extraído en 2 ml de cloroformo seco. Añadir 4 ml de reactivo de TPC. Revolver la mezcla, y luego añadir 40 mg de trietilamina seca. Continuar revolviendo durante 15 minutos. Lavar con 3 ml de 6 N HCl y luego con agua. Añadir MgSO4 y dejar reposar la mezcla durante 15 minutos. Inyectar 1-2 ml en la columna del CG. Se sabe que el TPC produce derivativos estables de casi todos los ETA, incluidas las efedrinas. Se presta mejor al análisis por CG.