Absorción distribución y eliminación de los fármacos.PDF
26
I. PRINCIPIOS GENERALES 1. Relación entre la dosis, la concentración plasmática y el efecto Para que un fármaco produzca sus efectos terapéuti- cos o tóxicos, debe alcanzar un intervalo preciso de con- centraciones en la biofase, es decir, el medio en que inter- actúa con sus receptores. Debajo de este intervalo, no se observará ningún efecto farmacológico o éste será sub- terapéutico; por encima, el efecto puede ser excesivo o pueden aparecer otros efectos no deseados. La concentración de un fármaco que se alcanza en su lugar de acción es la consecuencia de los siguientes pro- cesos (fig. 4-1): a) Absorción, es decir, la entrada del fármaco en el organismo que incluye los procesos de liberación de su forma farmacéutica, disolución y absorción propiamente dicha. b) Distribución del fármaco para que llegue primero del lugar de absorción a la circulación sistémica y desde ella hasta los tejidos. Para que el fármaco alcance desde su lugar de absorción su lugar de acción, debe atravesar diversas membranas para llegar a la sangre y para pasar de ésta al líquido intersticial y, en su caso, al interior de las células e, incluso, de estructuras intracelulares. El paso del fármaco de la sangre a los tejidos depende de la fija- ción del fármaco a las proteínas del plasma, ya que sólo el fármaco libre difunde libremente a los tejidos. c) Eliminación del fármaco, sea por metabolismo principalmente hepático o por excreción del fármaco inal- terado por la orina, bilis, etc. En algunos casos, este me- tabolismo puede producir metabolitos activos cuya pre- sencia también deberá tenerse en cuenta. La intensidad de los procesos de absorción, distribu- ción y eliminación varía con el tiempo; por este motivo, la cantidad de fármaco que hay en el organismo no per- manece estática sino que varía con el tiempo. El curso temporal de la cantidad de fármaco que hay en el orga- nismo depende de la influencia conjunta de los procesos de absorción, distribución y eliminación. El de los meta- bolitos dependerá de los procesos de formación y elimi- nación (fig. 4-2). En la práctica resulta difícil medir la concentración de los fármacos en su lugar de acción y puesto que en mu- chos casos el curso temporal de las concentraciones tisu- lares depende de las concentraciones plasmáticas, suele utilizarse el curso temporal de las concentraciones plas- máticas para predecir los efectos. Por ejemplo, tras la ad- ministración de un fármaco por vía oral aumenta la con- centración plasmática, mientras la absorción predomina sobre la eliminación, alcanza un máximo cuando la en- 47 4 Absorción, distribución y eliminación de los fármacos J. A. Armijo Fijación a tejidos inactivos Plasma Tejidos activos Metabolismo Excreción FP MP F M D·f F FP M F + M F M F M Fex Mex D Fig. 4-1. Procesos farmacocinéticos.
Transcript of Absorción distribución y eliminación de los fármacos.PDF

I. PRINCIPIOS GENERALES
1. Relación entre la dosis, la concentraciónplasmática y el efecto
Para que un fármaco produzca sus efectos terapéuti-cos o tóxicos, debe alcanzar un intervalo preciso de con-centraciones en la biofase, es decir, el medio en que inter-actúa con sus receptores. Debajo de este intervalo, no seobservará ningún efecto farmacológico o éste será sub-terapéutico; por encima, el efecto puede ser excesivo opueden aparecer otros efectos no deseados.
La concentración de un fármaco que se alcanza en sulugar de acción es la consecuencia de los siguientes pro-cesos (fig. 4-1):
a) Absorción, es decir, la entrada del fármaco en elorganismo que incluye los procesos de liberación de suforma farmacéutica, disolución y absorción propiamentedicha.
b) Distribución del fármaco para que llegue primerodel lugar de absorción a la circulación sistémica y desdeella hasta los tejidos. Para que el fármaco alcance desdesu lugar de absorción su lugar de acción, debe atravesardiversas membranas para llegar a la sangre y para pasarde ésta al líquido intersticial y, en su caso, al interior delas células e, incluso, de estructuras intracelulares. El pasodel fármaco de la sangre a los tejidos depende de la fija-ción del fármaco a las proteínas del plasma, ya que sóloel fármaco libre difunde libremente a los tejidos.
c) Eliminación del fármaco, sea por metabolismoprincipalmente hepático o por excreción del fármaco inal-terado por la orina, bilis, etc. En algunos casos, este me-tabolismo puede producir metabolitos activos cuya pre-sencia también deberá tenerse en cuenta.
La intensidad de los procesos de absorción, distribu-ción y eliminación varía con el tiempo; por este motivo,la cantidad de fármaco que hay en el organismo no per-manece estática sino que varía con el tiempo. El cursotemporal de la cantidad de fármaco que hay en el orga-nismo depende de la influencia conjunta de los procesosde absorción, distribución y eliminación. El de los meta-
4Absorción, distribución y elim
J. A. Armijo
bolitos dependerá de los procesos de formación y elimi-nación (fig. 4-2).
En la práctica resulta difícil medir la concentración delos fármacos en su lugar de acción y puesto que en mu-chos casos el curso temporal de las concentraciones tisu-lares depende de las concentraciones plasmáticas, sueleutilizarse el curso temporal de las concentraciones plas-máticas para predecir los efectos. Por ejemplo, tras la ad-ministración de un fármaco por vía oral aumenta la con-centración plasmática, mientras la absorción predominasobre la eliminación, alcanza un máximo cuando la en-
47
inación de los fármacos
Fijación a tejidos inactivos
Plasma Tejidos activos
Metabolismo Excreción
FP MPF M
D · f F
FP M
F
+
M
F M F M
Fex Mex
D
Fig. 4-1. Procesos farmacocinéticos.

48 Farmacología humana
a = Constante de disposición rápida del modelo bi-compartimental.
ATP = Ácido trifosfórico.AUC = Área bajo la curva de concentraciones plasmá-
ticas.AUCev = Área bajo la curva obtenida tras la administra-
ción extravascular.AUCiv = Área bajo la curva obtenida por vía intravenosa.b = Constante de disposición lenta del modelo bi-
compartimental y tricompartimental.BHE = Barrera hematoencefálica.CC = Compartimiento central en el modelo bicom-
partimental.Cl = Aclaramiento corporal total del fármaco.ClH = Aclaramiento hepático.Cli = Aclaramiento intrínseco.ClR = Aclaramiento renal.CME = Concentración mínima eficaz.CMT = Concentración mínima tóxica.Cmáx = Concentración plasmática máxima.CmáxE = Concentración plasmática máxima en equili-
brio.CmínE = Concentración plasmática mínima en equili-
brio.Cp = Concentración plasmática del fármaco.C p
0 = Concentración plasmática teórica en el tiem-po 0.
CpE = Concentración plasmática en equilibrio.CP = Compartimiento periférico en el modelo bi-
compartimental.CPP = Compartimiento periférico profundo en el mo-
delo tricompartimental.CPS = Compartimiento periférico superficial en el mo-
delo tricompartimental.Cu = Concentración urinaria.D = Dosis administrada.D · f = Cantidad de fármaco absorbida.DI = Dosis inicial.DM = Dosis de mantenimiento.Dmáx = Dosis máxima del metabolismo (máxima canti-
dad de fármaco que puede eliminarse).F = Fármaco libre.f = Fracción de absorción biodisponible.f’ = Fluctuación de los niveles plasmáticos tras la ad-
ministración de una dosis en un régimen de do-sis múltiples.
fc = Fracción de cambio tras una infusión continuao tras dosis múltiples (cambio que se ha produ-cido respecto al total que debe producirse).
APÉNDICE. ABREVIA
Fex = Fármaco excretado.fls = Fracción libre del fármaco en sangre.flt = Fracción libre del fármaco en los tejidos.FP = Fármaco unido a proteínas.h = Horas.g = Constante de disposición ultralenta del modelo
tricompartimental.IE = Intensidad del efecto.Ka = Constante de absorción.Ke = Constante de eliminación.Km = Constante de metabolismo (concentración para
la que está saturado el sistema biotransformanteen el 50 %).
K12 = Constante de distribución del compartimientocentral al periférico.
K21 = Constante de distribución del compartimientoperiférico al central.
kg = Kilo.LCR = Líquido cefalorraquídeo.M = Metabolito activo libre.Mex = Metabolito activo excretado.MP = Metabolito activo unido a proteínas.pHl = pH de la leche.PL = Período de lactancia.Q = Velocidad de infusión continua intravenosa.QH = Flujo sanguíneo hepático.SNC = Sistema nervioso central.t = Tiempo.t' = Tiempo en el que se alcanza la concentración
máxima tras una dosis en la fase de nivel esta-ble = TmáxEE.
T = Duración de una infusión continua intravenosacorta.
TE = Tiempo eficaz.tmáx = Tiempo en el que se alcanza la concentración
máxima tras una dosis única.t1/2a = Semivida de absorción.t1/2e = Semivida de eliminación.t = Intervalo de administración en un régimen de
dosis múltiples.Vc = Volumen de distribución en el compartimiento
central.Vd = Volumen de distribución en el modelo mono-
compartimental.Vs = Volumen sanguíneo.Vss = Volumen aparente de distribución en equili-
brio.Vt = Volumen tisular.Vu = Volumen de orina.
TURAS Y SÍMBOLOS
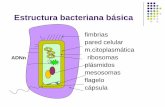
4. Absorción, distribución y eliminación de los fármacos 49
trada iguala a la salida y desciende cuando la absorcióndisminuye por debajo de la eliminación (fig. 4-2). En losfármacos en los que el efecto depende directamente dela concentración alcanzada en el lugar de acción y en losque esta concentración en la biofase está en equilibriocon la concentración plasmática, es posible establecer unarelación entre el curso temporal de las concentracionesplasmáticas y el de los efectos mediante unos parámetrosque conviene definir:
a) Concentración mínima eficaz (CME): aquélla porencima de la cual suele observarse el efecto terapéutico.
b) Concentración mínima tóxica (CMT): aquélla porencima de la cual suelen observarse efectos tóxicos. Elcociente entre la CMT y la CME definen el índice tera-péutico del fármaco: cuanto mayor sea este índice, másfácil será conseguir efectos terapéuticos sin producir efec-tos tóxicos.
c) Período de latencia (PL): tiempo que transcurredesde la administración hasta el comienzo del efecto far-macológico, es decir, hasta que la concentración plasmá-tica alcanza la CME. Como se comenta más adelante hayfármacos en los que el efecto puede aparecer más tarde.
d) Intensidad del efecto:para muchos fármacos guardarelación con la concentración máxima que se alcance, perola concentración en los tejidos puede variar en función de
Tiempo
100
75
50
25
0
Por
cent
aje
de
la d
osis
Fármaco en el lugar de absorción
Metabolito excretado
Fármaco en el cuerpo
Fármaco excretado
Metabolito en el cuerpo
A
Fig. 4-2. A) Curso temporal de la cantidad de fármaco en el lugfármaco y su metabolito excretados tras la administración de una
ción plasmática de un fármac
la unión a las proteínas del plasma, el flujo sanguíneo re-gional o la afinidad del fármaco por un determinado te-jido. Además, hay fármacos cuya respuesta es de tipo«todo o nada» y otros en los que, al haberse alcanzado elefecto máximo, el aumento de las concentraciones plas-máticas no aumenta la intensidad del efecto sino su dura-ción (v. cap. 6). Si la concentración plasmática supera laCMT, se producirán efectos tóxicos cuya intensidad tam-bién dependerá de la concentración máxima alcanzada.
e) Duración de la acción: también llamado tiempoeficaz (TE), es el tiempo transcurrido entre el momentoen que se alcanza la CME y el momento en que desciendepor debajo de ésta. Hay fármacos, como los que se acu-mulan en los tejidos y aquellos que tienen acción diferidao irreversible, en los que el efecto se prolongará más alláde sus niveles plasmáticos.
2. Variabilidad individual
La administración de la misma dosis de un fármaco aun grupo de pacientes produce el efecto esperado en lamayor parte de ellos, pero en algunos pacientes resultaineficaz y en otros se observan efectos tóxicos. Esta va-riabilidad en la respuesta a los fármacos principalmentedepende de factores farmacocinéticos que alteran los pro-cesos de absorción, distribución y eliminación y, por lo
Tiempo
CME
CMT
Cmáx
Tmáx
Con
cent
raci
ón p
lasm
átic
a
TEPL
IE
AUC
B
ar de absorción, del fármaco y su metabolito en el cuerpo, y del dosis por vía extravascular. B) Curso temporal de la concentra-o y relación con sus efectos.

50 Farmacología humana
tanto, la relación entre la dosis que se administra y el ni-vel plasmático que se alcanza. Además, la variabilidad enla respuesta depende de factores farmacodinámicos quealteran la sensibilidad del organismo al fármaco y, por lotanto, la relación entre los niveles plasmáticos y los efec-tos. Los factores más importantes son los siguientes:
a) Factores fisiológicos, como el patrón genético, laedad, los hábitos dietéticos, la ingesta de alcohol o el há-bito de fumar. Son particularmente importantes las dife-rencias entre el niño, el adulto y el anciano, así como lainfluencia del embarazo.
b) Factores patológicos, como la existencia de alte-raciones de la función renal, hepática o cardíaca.
c) Factores yatrógenos, es decir, las interacciones en-tre fármacos administrados simultáneamente que puedanalterar la respuesta.
3. Concepto de farmacocinética
El conocimiento de los procesos de absorción, distri-bución y eliminación de los fármacos y de los factores quelos alteran es esencial para la adecuada selección del pre-parado farmacéutico, la vía de administración, la dosis yla pauta de administración más adecuados para conseguirla máxima eficacia con el menor riesgo en un pacienteconcreto.
La farmacocinética estudia el curso temporal de lasconcentraciones y cantidades de los fármacos, y de susmetabolitos, en los líquidos biológicos, tejidos y excretas,así como su relación con la respuesta farmacológica, yconstruye modelos adecuados para interpretar estos da-tos. La farmacocinética clínica se marca como objetivo al-canzar y mantener la concentración plasmática necesariapara conseguir el efecto terapéutico sin llegar a producirefectos tóxicos. Estudia el curso temporal de las concen-traciones plasmáticas de los fármacos en el ser huma-no, su relación con los efectos y la influencia que tienensobre ellos diversos factores fisiológicos, patológicos oyatrógenos. Basándose en estos conocimientos puedepredecirse el curso temporal de las concentraciones plas-máticas y de los efectos, y diseñarse pautas especiales parasubgrupos de pacientes, llegando a individualizar el tra-tamiento en un paciente concreto en función de sus ca-racterísticas fisiológicas, enfermedad y tratamiento.
II. MECANISMOS DE TRANSPORTE
Todos los procesos farmacocinéticos de absorción, dis-tribución y eliminación requieren el paso de las moléculasdel fármaco a través de membranas biológicas formadaspor una doble capa de lípidos en la que se intercalan pro-teínas. Aunque las proteínas son las responsables de lamayor parte de las funciones de la membrana, incluyendoalgunos procesos de transporte de fármacos, los lípidoscondicionan en mayor grado el paso de los fármacos.
Los lípidos pueden ser fosfolípidos (p. ej., fosfatidil-colina, esfingomielina, fosfatidilserina o fosfatidiletanol-amina), colesterol y glucolípidos, y determinan la estruc-tura básica de la membrana. Los fosfolípidos se orientanespontáneamente de forma perpendicular al plano de lamembrana, dejando los grupos polares hacia fuera y lascadenas hidrofóbicas de ácidos grasos hacia dentro.
Las proteínas son las responsables de la mayor partede las funciones de la membrana. Dispersas irregular-mente, pueden ocupar la parte externa o interna de lamembrana, o atravesarla. Su región hidrófoba interactúacon la de los lípidos, mientras que la hidrófila está en con-tacto con el agua fuera o dentro de la membrana. Estasproteínas pueden actuar como ionóforos que permiten elacceso de iones y otras pequeñas moléculas polares ocomo canales que, tras un cambio de conformación, seabren o cierran influyendo en la polaridad de la mem-brana. También pueden comportarse como transporta-dores activos, como receptores específicos de ligandosendógenos y como enzimas reguladoras que respondena estímulos fisiológicos y farmacológicos.
Los principales mecanismos de transporte quedan ex-puestos en el capítulo 3 (v. fig. 3-2). Las moléculas de pe-queño tamaño atraviesan las membranas por difusiónpasiva, por difusión facilitada o por transporte activo. Lasde gran tamaño lo hacen por procesos de pinocitosis yexocitosis. La velocidad de difusión a través de la bicapalipídica depende del tamaño de la molécula, de su lipo-solubilidad y de su grado de ionización: las moléculas pe-queñas y no polares son las que difunden con mayor ra-pidez; las moléculas polares sin carga eléctrica difundencon rapidez si son pequeñas y con lentitud si son mayo-res; las moléculas ionizadas, por pequeñas que sean, noatraviesan la barrera lipídica. Las moléculas que pasanmal a través de la bicapa lipídica utilizan proteínas espe-cíficas que actúan como canales o como sistemas trans-portadores: los canales dejan pasar moléculas de un ta-maño y una carga determinadas a favor de un gradienteelectroquímico; las proteínas transportadoras fijan la mo-lécula y la transfieren a través de la membrana. Cuandoeste transporte es a favor del gradiente electroquímico,no requiere energía y se denomina difusión facilitada;cuando se realiza contra un gradiente electroquímico,consume energía y se denomina transporte activo.
1. Difusión pasiva
Es el mecanismo de transporte más habitual. La ma-yor parte de los fármacos tienen un tamaño pequeño-me-diano que permite su paso a través de las membranas pordifusión pasiva a favor de un gradiente de concentracióncuando no están ionizados. La velocidad, según la ley deFick, será tanto mayor cuanto mayor sea el gradiente deconcentración, menor sea el tamaño de la molécula y ma-yor sea su liposolubilidad. A su vez, la liposolubilidad de-pende del grado de ionización: la forma ionizada no di-funde a través de la membrana, mientras que la forma no

4. Absorción, distribución y eliminación de los fármacos 51
ionizada difundirá hasta que se equilibre la concentracióna ambos lados de ella. La mayoría de los fármacos sonelectrólitos débiles que están más o menos ionizados de-pendiendo de su pKa (fig. 4-3), es decir, del logaritmo ne-gativo de la constante de ionización de un ácido y del pHdel medio según la fórmula de Henderson-Hasselbach:
pH = pKa + log ([base]/[ácido])
Para ácidos:
pH = pKa + log ([ionizado]/[no ionizado])
Para bases:
pH = pKa + log ([no ionizado]/[ionizado])
La forma no ionizada difundirá libremente hasta quese equilibre a ambos lados de la membrana, mientras quela forma ionizada, por su riqueza en grupos hidrofílicos,no pasará. Cuando la membrana separa dos medios condistinto pH (p. ej., la sangre respecto a la luz intestinal,orina, leche, saliva o líquido prostático), se producirá unaacumulación del fármaco en el lado en que haya mayorgrado de ionización: las bases en el medio ácido y los áci-dos en el medio básico. En los procesos de absorción, elfármaco absorbido es retirado constantemente por la san-gre, que lo transporta al resto del organismo, por lo queno llega a alcanzarse un equilibrio y el proceso continúahasta que la absorción es completa.
2. Transporte activo
De esta forma se transportan los fármacos contra ungradiente electroquímico. Requiere consumo de energíaprocedente del metabolismo celular, por lo que está ínti-mamente acoplado a una fuente de energía, como la hi-drólisis de ATP. Con frecuencia, el transporte de la mo-lécula se asocia al de iones (como H+ o Na+), que puedenser transportados en la misma dirección o en direccióncontraria (v. cap. 3, I, C). Esta modalidad de transporte:
a) Debe ser saturable a una concentración que ocupetodos los puntos de fijación de la proteína transportadora.
b) Debe permitir la posibilidad de una inhibicióncompetitiva con sustancias afines.
c) Puede ser inhibida por mecanismos o sustanciasque interfieran con la producción de energía (cambios detemperatura, atmósfera anaerobia o inhibidores meta-bólicos como el cianuro), por sustancias que interfierancon las proteínas transportadoras (la uabaína inhibe laATPasa de la bomba de Na+) y por la carencia de sus-tancias necesarias para la síntesis o funcionamiento de lasproteínas transportadoras.
Este tipo de transporte activo de fármacos se ha ob-servado en el túbulo proximal renal, el tubo digestivo, el
tracto biliar, el paso del LCR a la sangre y el paso de lasangre a la saliva.
3. Otros sistemas de transporte
La filtración a favor de hendiduras intercelulares se ob-serva en la pared de algunos capilares sanguíneos, dondelos fármacos pasan del intersticio a los capilares, de loscapilares al intersticio o de los capilares al túbulo proxi-
Ácidos Bases
0
1
2
3
4
5
6
7
8
9
10
11
12
Fuerte Débil
FuerteDébil
Penicilinas
Acetilsalicílico
Furosemida
Fenilbutazona
Warfarina
Fenobarbital
Tiopental
Fenitoína
Teofilina
Nitrazepam
Oxazepam
Cafeína
Oxazepam
Nitrazepam
Diazepam
Quinidina
Clordiazepóxido
Propoxifeno
Cimetidina
Trimetoprima
Lidocaína
Quinidina
Petidina
Procainamida
Anfetamina
Nortriptilina
pKa
Fig. 4-3. pKa de algunos fármacos.

52 Farmacología humana
mal renal a través de hendiduras existentes entre las cé-lulas. La velocidad de filtración depende del tamaño delas hendiduras y de las partículas (por ello no pasa el fár-maco unido a proteínas), del gradiente de concentracióny de las presiones hidrostática, en la parte arterial del ca-pilar, y coloidosmótica, en su parte venosa.
En la difusión facilitada se utiliza una proteína trans-portadora, como en el transporte activo, pero en estecaso el transporte se realiza a favor de un gradiente deconcentración y no se consume energía; esta difusiónpuede saturarse e inhibirse competitivamente, como su-cede con el transporte de glucosa en la membrana de loshematíes.
Las macromoléculas se transportan mediante exoci-tosis o endocitosis. En la exocitosis, las vesículas intra-celulares se fusionan con la membrana expulsando sucontenido al exterior. En la endocitosis se forma una in-vaginación, pequeña en la pinocitosis y grande en la fa-gocitosis, que engloba las macromoléculas del exterior dela membrana; estas invaginaciones se rompen en el inte-rior de la célula, formando vesículas que contienen lasmacromoléculas.
Los ionóforos son pequeñas moléculas sintetizadas pormicroorganismos, que se disuelven en la capa lipídica dela membrana, aumentando su permeabilidad. Pueden sertransportadores móviles de iones y formadores de canal.Por ejemplo, la valinomicina y el A23187 son transpor-tadores móviles que aumentan la permeabilidad de lamembrana al K+ y al Ca2+, respectivamente, mientras quela gramicidina A es un ionóforo formador de canal quefacilita el paso de diversos cationes.
Los liposomas se utilizan también para favorecer elacceso de fármacos a diversas células. Los liposomas sonestructuras sintéticas formadas por una o más bicapasconcéntricas de fosfolípidos que acomodan en su interiorfármacos hidrosolubles o liposolubles y macromoléculas(como enzimas, hormonas, antígenos, material genéticoy otros agentes), que de esta forma consiguen acceder acélulas con capacidad de atrapar estos liposomas.
III. ABSORCIÓN
1. Concepto de absorción
El proceso de absorción comprende los procesos de li-beración del fármaco de su forma farmacéutica, su diso-lución, la entrada de los fármacos en el organismo desdeel lugar de administración, los mecanismos de transportey la eliminación presistémica, así como las característicasde cada vía de administración, la velocidad y la cantidadcon que el fármaco accede a la circulación sistémica y losfactores que pueden alterarla. El conocimiento de las ca-racterísticas de absorción de un fármaco es útil para se-leccionar la vía de administración y la forma farmacéu-tica óptimas para cada caso, así como para conocer lasrepercusiones que pueden tener sobre la respuesta la exis-
tencia de factores que alteran la velocidad de absorcióno la cantidad absorbida.
La absorción de un fármaco depende de las siguientescaracterísticas:
a) Características fisicoquímicas del fármaco. Com-prenden el peso molecular que condiciona el tamaño dela molécula, la liposolubilidad y su carácter ácido o alca-lino, que junto con su pKa, condicionan el grado de ioni-zación. De estos factores depende el mecanismo por elcual se produce la absorción (difusión pasiva, filtración ytransporte activo) y la velocidad a la que se realiza.
b) Características de la preparación farmacéutica.Para que el fármaco se absorba, debe estar disuelto. Lapreparación farmacéutica condiciona la velocidad conque el fármaco se libera, se disgrega y se disuelve. Algu-nas características son: la formulación (solución, polvo,cápsulas o comprimidos), el tamaño de las partículas, lapresencia de aditivos y excipientes, y el propio procesode fabricación.
c) Características del lugar de absorción. Dependende la vía de administración (oral, intramuscular o subcu-tánea). En general, la absorción será tanto más rápidacuanto mayor y más prolongado sea el contacto con lasuperficie de absorción. Algunas de estas característicasson: la superficie y el espesor de la membrana, el flujosanguíneo que mantiene el gradiente de concentración;en la administración oral, el pH del medio y la motilidadgastrointestinal, y en la administración intramuscular osubcutánea, los espacios intercelulares.
d) Eliminación presistémica y fenómeno «primer pa-so». Por cualquier vía que no sea la intravenosa puede ha-ber absorción incompleta porque parte del fármaco ad-ministrado sea eliminado o destruido antes de llegar a lacirculación sistémica. Por ejemplo, por vía oral, un fár-maco puede eliminarse por las heces antes que se com-plete su absorción, puede ser quelado, degradado por laacción del pH ácido del estómago o de las enzimas di-gestivas y metabolizado por las bacterias de la luz intes-tinal; una vez absorbido, puede metabolizarse en el epi-telio intestinal en el hígado (primer paso hepático) o en lospulmones antes de llegar a la circulación sistémica.
Se entiende por primer paso hepático la metaboliza-ción del fármaco absorbido en el tracto gastrointestinalque llega al hígado a través de la vena porta y que se me-taboliza en él antes de llegar a la circulación sistémica. Lafracción de extracción hepática es la fracción del fármacoque hay en el cuerpo que se metaboliza en un solo pasopor el hígado. Los fármacos con primer paso hepático (ta-bla 4-1) poseen una fracción de extracción alta, mayor de0,7, lo que significa que menos del 30 % de la dosis ab-sorbida alcanzará la circulación sistémica. El primer pasohepático explica que la biodisponibilidad de algunosß-bloqueantes sea inferior al 30 % a pesar de que su ab-sorción gastrointestinal es completa; también explica quela dosis por vía oral deba ser notablemente mayor queaquélla por vía intravenosa.

4. Absorción, distribución y eliminación de los fármacos 53
2. Vías de administración
2.1. Vías enterales
La absorción del fármaco por vía oral depende deforma muy importante de la preparación farmacéutica,que condiciona los procesos de disgregación y disolución.La absorción se produce en el estómago y especialmenteen el duodeno, principalmente por difusión. En algunoscasos puede haber transporte activo (p. ej., metildopa olevodopa) y filtración a través de poros intercelulares (p.ej., furosemida o atenolol). La vía oral es cómoda, baratay unipersonal, adecuada para el tratamiento crónico. Re-quiere voluntad y capacidad de deglución, y no debe uti-lizarse cuando el fármaco irrite la mucosa o el pacienteesté inconsciente, se haya sometido a una intervenciónquirúrgica o presente vómitos que contraindiquen estavía. Los preparados con cubierta entérica evitan la ab-sorción en el estómago y retrasan el comienzo de laabsorción, pero no su velocidad. Los preparados de libe-ración mantenida enlentecen la absorción y permitenreducir las fluctuaciones de las concentraciones plasmá-ticas o el número de tomas al día para mejorar el cum-plimiento terapéutico. Habitualmente están formadospor una matriz a la que se une fuertemente el fármaco yde la que se va liberando lentamente conforme se pro-duce la absorción. También hay cápsulas cuya pared dejaentrar osmóticamente el agua desde la luz intestinal, em-pujando lentamente el fármaco hacia el exterior a travésde un fino orificio (p. ej., de indometazina y ß-bloquean-tes).
En la vía sublingual, el fármaco depositado debajo dela lengua se absorbe por la mucosa sublingual accediendo
Tabla 4-1. Ejemplos de fármacos con una fracción de absor-ción menor de 0,5 por primer paso hepático
Alprenolol LorcainidaAmitriptilina 6-MercaptopurinaCitarabina MetilfenidatoClormetiazol MetoprololClorpromazina MorfinaDextropropoxifeno NalbufinaDihidroergotamina NaloxonaDiltiazem NaltrexonaDinitrato de isosorbida NeostigminaDoxepina NicardipinoDoxorubicina NicotinaEncainida NifedipinoEscopolamina NitroglicerinaEstradiol Papaverina5-Fluorouracilo PentazocinaHidralazina PetidinaImipramina PrazosinaIsoprenalina PropranololKetamina TestosteronaLabetalol VerapamiloLidocaína
por la vena cava a la aurícula derecha. Al evitar su pasointestinal y hepático se consigue un efecto más rápido eintenso, que es útil en situaciones agudas, como el trata-miento de una crisis anginosa con nitroglicerina o de unacrisis hipertensiva con nifedipino.
La vía rectal es más incómoda que la vía oral y la ab-sorción puede ser errática, lenta e incompleta. Se utilizapara administrar fármacos que producen irritación gas-trointestinal, son destruidos por el pH ácido del estómagoo las enzimas digestivas, tienen un olor o sabor desagra-dables, o para evitar parcialmente el primer paso hepá-tico. También se utiliza como una alternativa a la vía oralen pacientes con vómitos, inconscientes o quirúrgicos.La absorción rectal de algunos fármacos administradoscomo supositorios puede ser lenta e irregular, mejorandocuando se administra como solución rectal (p. ej., diaze-pam), mientras que la absorción rectal de otros fármacoses mejor que la oral (p. ej., metoclopramida o propra-nolol).
2.2. Vías parenterales
La vía intravenosa es de elección en situaciones agu-das. Sus ventajas son la rapidez de la acción y la precisiónde las concentraciones plasmáticas que se alcanzan, al nodepender de los procesos de absorción ni de los factoresque pueden alterarlos. También permite reducir los efec-tos irritantes y administrar grandes volúmenes. Sus in-convenientes son la dependencia de personal especiali-zado, la posibilidad de reacciones graves (especialmentecuando la administración es muy rápida y se alcanzan al-tas concentraciones) y el peligro de embolias e infeccio-nes. La vía intraarterial se utiliza para realizar arterio-grafías y para alcanzar altas concentraciones locales (p.ej., de un fibrinolítico, como la estreptocinasa).
La vía intramuscular se emplea para la administraciónde fármacos que por vía oral se absorben mal (p. ej., ami-noglucósidos), son degradados por vía oral (p. ej., peni-cilina G) o tienen un primer paso hepático muy impor-tante (p. ej., lidocaína). También puede utilizarse paraasegurar el cumplimiento terapéutico o como una opcióna la vía oral y/o rectal en pacientes quirúrgicos o con vó-mitos. Los preparados de absorción mantenida (p. ej., depenicilinas u hormonas) liberan el fármaco lentamente,consiguiendo un efecto más prolongado. También puedeutilizarse para conseguir un efecto más rápido ya que larica vascularización del músculo permite una rápida ab-sorción en 10-30 min, pero en algunos casos la absorciónintramuscular es lenta (p. ej., fenobarbital o diazepam) yalgunos fármacos como la fenitoína precipitan al pH fi-siológico del tejido muscular, por lo que no deben admi-nistrarse por esta vía. La absorción intramuscular puedeser distinta en los músculos glúteos y en el deltoides (p.ej., la lidocaína se absorbe con mayor rapidez del deltoi-des) y puede resultar lenta e incompleta cuando hay hi-poperfusión (p. ej., la morfina, cuando hay insuficienciacardíaca) o estasis (p. ej., en el embarazo).

54 Farmacología humana
En la vía subcutánea, el flujo sanguíneo es menor queen la vía intramuscular, por lo que la absorción es máslenta. Disminuye cuando hay hipotensión, vasoconstric-ción por frío o administración simultánea de vasocons-trictores y aumenta cuando hay vasodilatación producidapor el calor o cuando los fármacos se administran juntocon hialuronidasa. Puede estar muy reducida cuando hayhipotensión, como sucede con la morfina subcutánea enel edema agudo de pulmón. Existen preparados de ab-sorción lenta, como las bombas osmóticas comentadaspara la vía oral, que pueden implantarse bajo la piel y pro-porcionar niveles mantenidos durante tiempo muy pro-longado (p. ej., de anticonceptivos). También se puedenutilizar bombas de infusión (p. ej., de insulina o morfina),cuya velocidad se puede adaptar a las necesidades del pa-ciente.
2.3. Otras vías
La vía dérmica se utiliza en forma de cremas y po-madas para el tratamiento local de afecciones de la piel.Los fármacos liposolubles difunden bien, pero si el fár-maco es hidrosoluble y la afección está en las capas pro-fundas de la piel llegará mejor por otras vías (v. cap. 75).También se emplea para la administración sistémicamantenida de fármacos de forma aguda (p. ej., esco-polamina y fentanilo) o crónica (p. ej., nitratos, estró-genos y nicotina). La administración cutánea evita elprimer paso hepático y las fluctuaciones de las concen-traciones plasmáticas, permite terminar rápidamente laabsorción del fármaco, reduce la variabilidad interindi-vidual en la absorción, prolonga la duración de la ac-ción y mejora el cumplimiento terapéutico. La absor-ción cutánea es menor cuando la piel es gruesa o estáexpuesta a la intemperie. Por la piel se absorben tam-bién diversos tóxicos, como solventes orgánicos o pre-parados organofosforados. La vía nasal es útil para eltratamiento local de la rinitis alérgica y la congestiónnasal, pero también se emplea para la administraciónsistémica de fármacos, como las hormonas peptídicas,el fentanilo o el propranolol.
Las vías epidural, intratecal e intraventricular se utili-zan para hacer llegar al SNC fármacos que atraviesan malla BHE (p. ej., algunos antibióticos o antineoplásicos) ypara conseguir concentraciones elevadas (p. ej., de anes-tésicos o analgésicos) en áreas localizadas, como las raí-ces espinales. En la administración epidural se inyecta elfármaco en el espacio epidural que queda entre el liga-mento amarillo y la duramadre, de donde difunde al es-pacio subaracnoideo, la vaina de las raíces nerviosas y losvasos. En la intratecal se inyecta el fármaco en el espaciosubaracnoideo donde se encuentra el LCR y de donde di-funde al SNC o a los espacios y las vainas de las raícesnerviosas. Por vía intraventricular se consiguen mayoresconcentraciones cerebrales que por vía intratecal. Apartela mayor dificultad técnica, estas vías conllevan un riesgode neurotoxicidad e infecciones. A pesar de estos riesgos
se utiliza para la administración de opiáceos y de baclo-feno en situaciones especiales.
La vía inhalatoria se utiliza principalmente para la ad-ministración de fármacos que deban actuar localmenteen las vías respiratorias como ß2-adrenérgicos, cromogli-cato sódico, corticoides o anticolinérgicos inhalatorios(v. cap. 42). El acceso al lugar de acción depende de latécnica utilizada (inhaladores o nebulizadores), del ta-maño de las partículas (por encima de 20 µ se deposita enla orofaringe y vías respiratorias altas, y por debajo de1 µ no se deposita) y de la existencia de obstrucción bron-quial que dificulte el acceso del aerosol. Algunos fárma-cos administrados por esta vía pueden provocar bronco-constricción (p. ej., el cromoglicato o la N-acetilcisteína)y el uso de nebulizadores conlleva un riesgo de infeccio-nes. La vía inhalatoria se utiliza también para adminis-trar gases (p. ej., oxígeno) y anestésicos volátiles. Por estavía acceden al organismo tóxicos como el tabaco, líqui-dos volátiles, contaminantes y alergenos.
Las vías conjuntival, uretral, vesical y vaginal se utili-zan para actuar localmente sobre las respectivas mucosasy la vía intraperitoneal, para diálisis en casos de insufi-ciencia renal e intoxicaciones.
3. Cinética de absorción
La cinética de absorción cuantifica la entrada de fár-maco en la circulación sistémica y engloba los procesosde liberación del fármaco de su forma farmacéutica, di-solución, absorción propiamente dicha y eliminación pre-sistémica. Incluye el estudio de la velocidad de absorción,de la cantidad absorbida y de los factores que la alteran.
3.1. Velocidad de absorción y cantidad absorbida
La velocidad de absorción, es decir, el número de mo-léculas de un fármaco que se absorbe en la unidad detiempo, depende de la constante de absorción y del nú-mero de moléculas que se encuentren en solución en ellugar de absorción. La constante de absorción (Ka) puedeexpresarse como la probabilidad que tiene una moléculade absorberse en la unidad de tiempo. Por ejemplo, unaKa de 0,03 h-1 indica que en 1 hora se absorberá aproxi-madamente el 3 % de las moléculas en disolución que es-tán disponibles para absorberse. La semivida de absor-ción (t1/2a) es el tiempo que tarda en reducirse a la mitadel número de moléculas que quedan por absorberse y esla inversa de la constante de absorción:
t1/2a = 0,693/Ka
Por lo tanto, cuanto más rápida sea la absorción de unfármaco, mayor será su constante de absorción y menorsu semivida de absorción.
Tipos de cinética de absorción. La absorción puede serde orden 1 (o de primer orden) y de orden 0. En la ab-sorción de orden 1, la velocidad de absorción disminuye
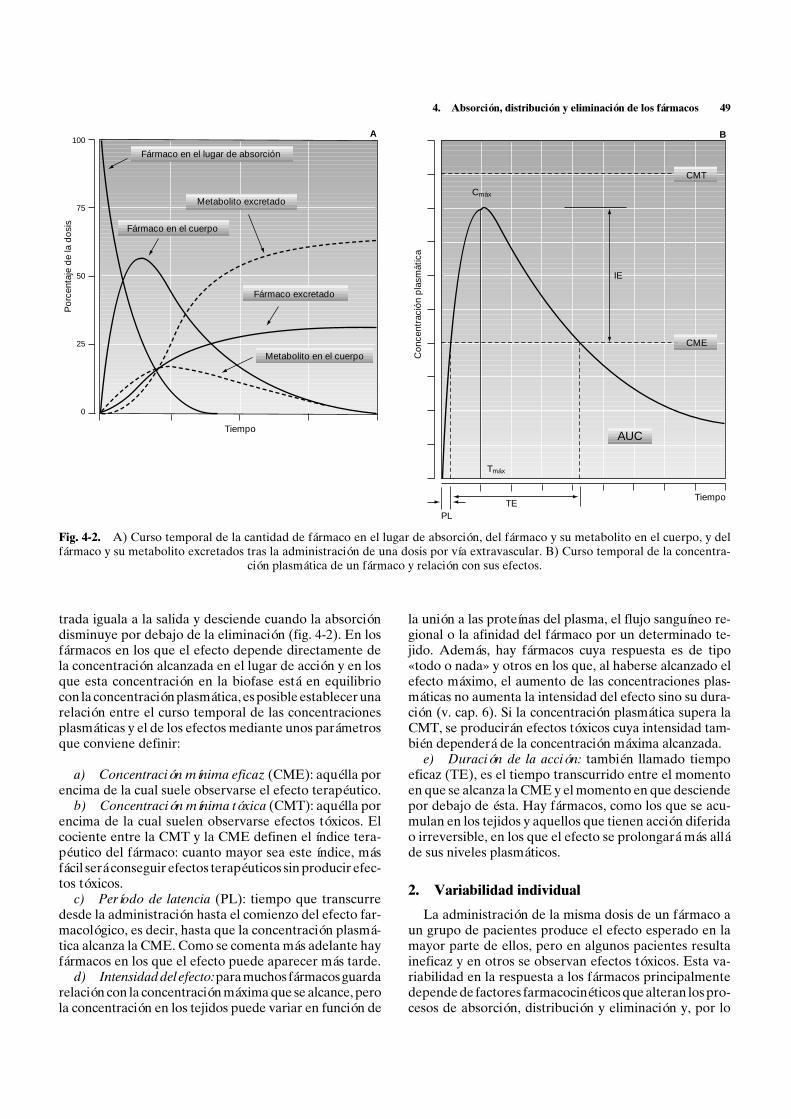
4. Absorción, distribución y eliminación de los fármacos 55
con la cantidad de fármaco que queda por absorberse y,por lo tanto, el número de moléculas que se absorbe enla unidad de tiempo disminuye con el tiempo de formaexponencial. Dicha curva exponencial puede represen-tarse como una recta si se representan las concentracio-nes en una escala semilogarítmica, siendo la constante deabsorción la pendiente de dicha recta (fig. 4-4). Es ca-racterística de la mayor parte de las formas farmacéuti-cas en las que la totalidad de las moléculas administradasestán inicialmente disponibles para absorberse, disminu-yendo a medida que se van absorbiendo.
En la absorción de orden 0, el número de moléculasque se absorbe en la unidad de tiempo permanece cons-tante durante todo o la mayor parte del proceso de ab-sorción. Es característica de formas de administración,como la perfusión intravenosa continua, la administra-ción de gases anestésicos, los preparados de absorciónmantenida intramusculares, subcutáneos o dérmicos ylos preparados orales de liberación lenta, en las que el nú-mero de moléculas disponibles no disminuye con eltiempo, ya que las moléculas absorbidas son respuestasdesde el depósito.
La cantidad absorbida se considera que es igual a la ad-ministrada cuando el fármaco se administra por vía in-travascular y suele expresarse mediante el área bajo lacurva (AUC) de concentraciones plasmáticas. Este áreasuele calcularse por el método trapezoidal a partir de lasconcentraciones plasmáticas obtenidas a diferentes tiem-pos. Por cualquier otra vía es posible que la cantidad ab-sorbida sea inferior a la dosis administrada debido a lapreparación farmacéutica y a la eliminación presistémica.La fracción de absorción biodisponible (f) es la fracciónde la dosis administrada que llega a la circulación sisté-mica en una forma inalterada, y se obtiene dividiendo elárea bajo la curva obtenida tras la administración extra-vascular (AUCev) por la obtenida por vía intravenosa(AUCiv), teniendo en cuenta la dosis administrada (D)por cada vía y el aclaramiento (Cl) del individuo:
AUCev Div/(peso · Cliv)f = ———— · ————————
AUCiv Dev/(peso · Clev)
100
80
60
40
20
0Por
cent
aje
del
fárm
aco
que
que
da
por
ab
sorb
er Orden 0
Orden 0
Orden 1
Orden 1
Tiempo Tiempo
100
50
10
5
1
Fig. 4-4. Cinética de absorción de orden 1 (línea continua) yde orden 0 (línea discontinua) en representación numérica (iz-
quierda) y semilogarítmica (derecha).
Cuando la comparación de las áreas se realiza en losmismos pacientes y la dosis por vía extravascular es iguala la intravenosa la fórmula se simplifica a:
AUCevf = ————
AUCiv
Así pues, la cantidad absorbida por vía extravascularserá el producto de la dosis administrada por la fracciónde absorción correspondiente a la forma farmacéutica ya la vía de administración utilizadas:
Cantidad absorbida = D · f
3.2. Concepto de biodisponibilidad
La biodisponibilidad de un fármaco indica la veloci-dad y la cantidad de la forma inalterada de un fármacoque accede a la circulación sistémica y, por lo tanto, estádisponible para acceder a los tejidos y producir un efecto.La cantidad absorbida suele valorarse mediante el áreabajo la curva (AUC) de concentraciones plasmáticas o lafracción de absorción biodisponible (f), y la velocidad deabsorción por la forma de esa curva expresada por la con-centración máxima (Cmáx) y el tiempo en que se alcanza(tmáx). La biodisponibilidad de un fármaco depende nosólo de los procesos de absorción, sino también de los dedistribución y eliminación. Ahora bien, cuando la distri-bución y la eliminación se mantienen constantes, las va-riaciones en la biodisponibilidad reflejan diferencias enla absorción del fármaco, sea en la velocidad de absor-
Tiempo
CME
a
b
d
cCon
cent
raci
ón p
lasm
átic
a
Fig. 4-5. Influencia de las variaciones en la biodisponibilidadde un fármaco sobre la intensidad y la duración de sus efectos.En a, b y c, la cantidad absorbida es completa (las áreas bajo lacurva son iguales), pero la velocidad de absorción es distinta in-fluyendo en el comienzo, intensidad y duración del efecto. Enc, la absorción es tan lenta que no llega a alcanzarse la CME.En d, la velocidad de absorción es igual que la de a, pero la ab-sorción es incompleta (el área bajo la curva es menor), con res-
puesta rápida, pero poco intensa y fugaz.

56 Farmacología humana
ción, en la cantidad absorbida o en ambas (fig. 4-5). Labiodisponibilidad de los fármacos, como expresión de suabsorción, depende críticamente de la vía de administra-ción y de la forma farmacéutica utilizadas, pero puede va-riar de unos individuos a otros, especialmente cuandohaya factores que alteren la absorción.
3.3. Factores que alteran la absorción
Las diferencias en la absorción de los fármacos dependen princi-palmente de la preparación farmacéutica y la vía de administración (fig. 4-6), pero también puede ser alterada por otros factores:
a) Factores fisiológicos. En el recién nacido, especialmente en elprematuro, durante el embarazo y en el anciano puede haber altera-ciones de la absorción tanto por vía oral (debido a alteraciones en el pHy en la motilidad intestinal), como por vía intramuscular o subcutáneapor alteraciones del flujo sanguíneo (v. cap. 7). La absorción de los fár-macos por vía oral puede alterarse con alimentos o con algún tipo con-creto de alimentos, como las grasas. Los alimentos pueden reducir lavelocidad de absorción y la cantidad absorbida, pero también puedenno alterarla e incluso aumentarla (tabla 4-2). La importancia de esta in-fluencia es muy variable y con frecuencia clínicamente irrelevante, porlo que se prefiere administrar los medicamentos con las comidas paramejorar el cumplimiento terapéutico, con la excepción de algunos fár-macos como la isoniazida, la rifampicina, la penicilina o las tetracicli-nas, que deben administrarse 2 horas antes de las comidas.
b) Factores patológicos. La absorción oral puede alterarse cuandohay vómitos, diarrea y enfermedades digestivas que alteren el vacia-miento gástrico, el tránsito intestinal o la superficie de absorción. Porvía intramuscular y subcutánea son importantes las alteraciones queproduce la insuficiencia cardíaca y el choque hemodinámico por re-ducción del flujo sanguíneo (v. cap. 8).
c) Factores yatrógenos. Hay numerosas interacciones que puedenafectar la absorción, directamente por formación de precipitados que
Tabla 4-2. Influencia de los alimentos
Disminuye Retrasa
Ácido acetilsalicílico Amoxicilina BAmoxicilina Aspirina CAmpicilina Bumetanida DCiprofloxacinaa Cefaclor DEritromicina base Cefalexina DFluorouracilo Cefradina EHidroclorotiazida Cimetidina EIsoniazidaa Cinoxacina GKetoconazol Diflunisal GLevodopa Digoxina IPenicilina Va Eritromicina MPivampicilinaa Furosemida MPropantelina Indoprofeno ORifampicinaa Nitrofurantoína PSotalol Paracetamol PTeofilina Potasio PTetraciclinaa Sulfadiazina PTrazodona Sulfisoxazol S
Teofilina SValproato T
T
a Influencia que puede ser clínicamente importante, por lo que no deben adminisb El aumento en la absorción se debe a que los alimentos disminuyen su metaboli
impiden la absorción o indirectamente por producir cambios en el pH,la motilidad gastrointestinal o el flujo sanguíneo (v. cap. 10).
Lo más frecuente es que estos factores reduzcan la velocidad de ab-sorción, disminuyendo la concentración máxima y alargando el tiempoen que ésta se alcanza, lo que puede reducir los efectos de dosis únicas(fig. 4-5). Sin embargo, cuando se administran dosis múltiples, las alte-raciones en la velocidad de absorción no reducen el nivel estable, porlo que no influyen en los efectos de los fármacos con una semivida larga,aunque pueden reducir los efectos de los fármacos con una semivida deeliminación muy corta en los que el efecto dependa del máximo alcan-zado. Los factores que reducen la cantidad absorbida también reducenla concentración máxima tras dosis únicas, pero no el tiempo en que sealcanza (fig. 4-5); tras dosis múltiples reducen el nivel estable y, por lotanto, los efectos (v. cap. 6).
sobre la absorción oral de los fármacos
No cambia Aumenta
endroflumetiazida Carbamazepinalorpropamida Clorotiazidaiazepam Diazepamigoxina Dicumaroloxiciclina Eritromicina estearatoritromicina estolato Eritromicina etilsuccinatospiramicina Espiromicinalibenclamida Fenitoínaa
lipizida Griseofulvinandoprofeno Hidralazinab
etronidazol Hidroclorotiazidaa
inociclina Labetalolxazepam Litioaracetamol Mebendazola
enicilinas Metoprololb
rednisona Nitrofurantoínaa
ropiltiouracilo Propoxifenoulfamidas Propranololb
ulfonilureaseofilinaolbutamida
trarse con los alimentos.smo en la pared intestinal.
Tiempo Tiempo
Con
cent
raci
ón p
lasm
átic
a
Con
cent
raci
ón p
lasm
átic
aIV IV
IM
Oral
Rectal
Solución oral
Cápsulas
Comprimidos
Fig. 4-6. Influencia de la vía de administración y de la prepa-ración farmacéutica sobre la curva de concentraciones plasmá-
ticas de un fármaco.

4. Absorción, distribución y eliminación de los fármacos 57
IV. DISTRIBUCIÓN
La distribución de los fármacos permite su acceso a losórganos en los que debe actuar y a los órganos que losvan a eliminar y condiciona las concentraciones que al-canzan en cada tejido. Tiene especial importancia en laelección del fármaco más adecuado para tratar enferme-
Porcentaje de unión a las proteínas
del plasma Fármaco
– Atenolol, litio
– Procainamida, gentamicina
– Digoxina
– Penicilina G
– Teofilina
– Fenobarbital
– Carbamazepina
– Quinidina
– Verapamilo
– Alfentanilo
– Fenitoína
– Diazóxido, propranolol
– Clindamicina, dicloxacilina
– Nifedipino
– Amiodarona
– Digitoxina
– Clorpromazina
– Oxazepam
– Furosemida
– Ketoprofeno
– Diazepam
– Warfarina, ibuprofeno
– Fenilbutazona
– Naproxeno
– Flurbiprofeno
0
50
80
90
95
98
99
99,5
99,8
99,9
Fig. 4-7. Variabilidad en la unión de los fármacos a las pro-teí-
dades localizadas en áreas especiales, como el SNC, y enla valoración del riesgo de los fármacos durante el em-barazo y la lactancia.
1. Transporte en la sangre y unión a proteínasplasmáticas
Las moléculas de un fármaco son transportadas en lasangre disueltas en el plasma, fijadas a las proteínas plas-máticas o unidas a las células sanguíneas. La unión de losfármacos a las proteínas del plasma es muy variable, ha-ciendo que el porcentaje de fármaco libre que pasa a lostejidos fluctúe desde el 100 % del atenolol al 0,1 % delflurbiprofeno (fig. 4-7). La fijación a la albúmina es la másfrecuente e importante. Aunque la carga de la albúminaa pH de 7,4 es negativa, fija tanto fármacos ácidos comobases mediante enlaces iónicos y, ocasionalmente, enla-ces covalentes. Los fármacos ácidos suelen fijarse a la al-búmina en el sitio I (tipo warfarina) o II (tipo diazepam)(tabla 4-3). Las bases débiles y las sustancias no ioniza-bles liposolubles suelen unirse a las lipoproteínas, y lasbases débiles, además, a la albúmina y a la a-glucopro-teína, no siendo infrecuente que una base débil se una si-multáneamente a varias proteínas (tabla 4-4).
Tabla 4-3. Sitios de fijación de los fármacos ácidos a la albú-mina del plasma
Sitio I (warfarina) Sitio II (diazepam)Acenocumarol Ácido clofíbricoÁcido nalidíxico Ácido etacrínicoÁcido salicílicoa Ácido flufenámicoBilirrubina Ácido salicílicoa
Bumetanida BenzodiazepinasClorotiazida CloxacilinaClorpropamida DicloxacilinaDicumarol (1) Dicumarol (2)Diflunisala Diflunisala
Fenilbutazona Flucloxacilina (2)Fenitoína Flurbiprofeno (1)Flucloxacilina (1) Glibenclamidaa
Flurbiprofeno (2) Ibuprofeno (1)Furosemida Indometazinaa
Glibenclamidaa Ketoprofenoa
Indometazinaa Naproxenoa
Ketoprofeno (2) ProbenecidaNaproxenoa SulfobromoftaleínaSulfamidas Tamoxifeno (2)Sulfinpirazona TolazamidaTolbutamidaa Tolbutamidaa
ValproatoWarfarina
Sitio digitoxina Sitio tamoxifenoAcetildigitoxina ClomifenoDigitoxina Tamoxifeno (1)
a Se unen al sitio I y al sitio II; 1: sitio de unión preferente; 2: sitio de unión se-cundario.

58 Farmacología humana
La fijación a proteínas es reversible y sigue la ley deacción de masas. La cantidad de fármaco unido a pro-teínas (FP) depende de la concentración de fármaco li-bre (F), de la constante de asociación (K1/K2), del númerode sitios de fijación libres por mol de proteína y de la con-centración molar de proteína:
K1
F + sitios libres FP
K2
Tabla 4-4. Características de algunos fármacos con alta unión a las proteínas del plasma
Unión a Tipo de Fracción de Volumen deproteínas proteína extracción distribución
Acetilsalicílicoa # I, II • 11Amitriptilina > 90 • P 1.085Anfotericina B 96 A, a G 280Clindamicina 94 • • 56Clofibrato # • P 8Clorotiazida 95 I P —Clorpromazina > 95 • G 1.470Diazepamb 99 II P 140Diazóxido 90 • • 15Dicloxacilinaa 94 II P 14Dicumarolb 99 I, II • 11Diflunisal 98 I, II • 8Digitoxina 90 d P 32Disopiramida # A, a • 182Doxiciclina 90 • P 49Fenilbutazonaa # I P 12Fenitoínab 90 I P 56Flurbiprofeno 100 II, I • 7Furosemida 96 I P 21Glibenclamidab 99 I, II • 11Heparina 95 L • 5Ibuprofeno 99 II • 10Imipramina > 90 A, a • 1.470Indometazinab 90 I, II • 14Ketoprofeno 92 II • 8Lorazepam 93 II P 105Naproxenoa 98 II, I • 7Nortriptilina 95 A, a • 1.470Prazosina 93 • • 35Propranolol 93 A, a, L S 196Sulfisoxazola 90 I P 25Tolbutamidab 93 I P 11Valproatoa # I P 11Warfarinab 99 I P 11
Unión a proteínas: #: saturable. Tipo de proteína: A: albúmina; I: sitio I de laalbúmina; II: sitio II de la albúmina; d: sitio digitoxina de la albúmina; L: lipo-proteína; a: a-glucoproteína.
Fracción de extracción: G próxima a 1,0 y P próxima a 0,1.a Suele ser causa de desplazamiento.b Suele ser objeto de desplazamiento.
Habitualmente, el porcentaje de la concentración to-tal del fármaco que se encuentra unido a proteínas per-manece constante dentro de un intervalo amplio de con-centraciones, pero hay fármacos, como el valproatosódico, que, cuando se utilizan concentraciones altas, sa-turan los puntos de fijación, aumentando la proporciónde fármaco libre.
2. Distribución en los tejidos
2.1. Distribución regional
El fármaco disuelto en la sangre pasa de los capilaresa los tejidos a favor del gradiente de concentración. Estepaso depende de las características del fármaco (tamañode la molécula, liposolubilidad y grado de ionización), desu unión a las proteínas plasmáticas, del flujo sanguíneodel órgano, de la luz capilar, del grado de turgencia y delas características del endotelio capilar.
Un fármaco muy liposoluble accederá más fácilmentea los órganos muy irrigados, como el cerebro, el corazón,el hígado o los riñones, más despacio al músculo y conmayor lentitud a la grasa y otros tejidos poco irrigados,como las válvulas cardíacas. Incluso puede haber dife-rencias dentro de un órgano, por ejemplo, entre la cor-teza y la médula renales, o entre el hueso cortical y es-ponjoso. Un fármaco menos liposoluble llegará bien a lostejidos cuyos capilares son ricos en hendiduras intercelu-lares, como es el caso de los sinusoides hepáticos cuyasabundantes fenestraciones y hendiduras intercelularespermiten el paso de sustancias con elevado peso molecu-lar, pero tendrá dificultad para acceder a los tejidos quecarecen de ellas, como el SNC. La inflamación producevasodilatación y aumento de la permeabilidad capilar, loque puede aumentar la concentración alcanzada en al-gunos tejidos. Cuando la concentración plasmática dis-minuye, el fármaco pasa de nuevo de los tejidos a los ca-pilares a favor del gradiente de concentración.
Dentro de un órgano, el fármaco puede estar disueltoen el líquido intersticial y en el agua intracelular (cuandoaccede al interior de la célula) o fijado a diversos com-ponentes, como proteínas o lípidos. La forma del fármacoque accede al líquido intersticial del tejido celular subcu-táneo, a las cavidades peritoneal, pleural y articular, a losalvéolos y bronquios es la forma libre, por lo que dependede las variaciones en la unión a proteínas. El acceso al in-terior de las células y a las estructuras intracelulares serealiza por difusión pasiva y depende de la liposolubili-dad y del grado de ionización que puede variar en cir-cunstancias patológicas (p. ej., la acidosis).
La mayoría de los fármacos tienen la capacidad de fi-jarse a determinados tejidos en los que alcanzan con-centraciones más altas que en el resto del organismo, in-cluso aunque estén poco irrigados, como sucede con laacumulación de los fármacos liposolubles en la grasa, lastetraciclinas en el hueso o la griseofulvina en la piel. Lafijación intensa a ciertos tejidos puede reducir la con-

4. Absorción, distribución y eliminación de los fármacos 59
centración del fármaco en su lugar de acción; por ejem-plo, la acción anestésica del tiopental termina cuando elfármaco abandona el SNC para pasar al músculo y lagrasa.
2.2. Distribución a áreas especiales
El acceso a áreas especiales, como el SNC y el ojo, elpaso a la circulación fetal y el acceso a secreciones exo-crinas como lágrimas, saliva, leche o líquido prostático,presentan características peculiares, ya que la filtración através de hendiduras intercelulares en estas áreas estámuy limitada. Por ello, el transporte de fármacos en es-tas áreas ha de realizarse por difusión pasiva o por trans-porte activo. Además, en algunas de estas áreas haydiferencias de pH que pueden generar un efecto de atra-pamiento ya comentado.
a) Barrera hematoencefálica (BHE). Está formadapor un conjunto de estructuras que dificultan notable-mente el paso de las sustancias hidrófilas desde los capi-lares hacia el SNC (fig. 4-8): 1) las células endoteliales delos capilares sanguíneos del SNC están íntimamente ado-sadas sin dejar espacios intercelulares; 2) entre una y otracélula existen bandas o zónulas ocludens que cierran her-méticamente el espacio intercelular; 3) hay una mem-brana basal que forma un revestimiento continuo alre-dedor del endotelio; 4) los pericitos forman una capadiscontinua de prolongaciones citoplasmáticas que ro-dean el capilar y 5) las prolongaciones de los astrocitosde la glía perivascular forman un mosaico que cubre el85 % de la superficie capilar.
Como consecuencia, no hay ni filtración ni pinocitosis,por lo que los fármacos sólo pueden pasar por difusiónpasiva. La velocidad de paso depende críticamente de laliposolubilidad y del grado de ionización, por lo que esalterada por los cambios de pH en el plasma y en el es-pacio extracelular. La permeabilidad de la BHE puedealterarse por la isquemia y la anoxia de origen vascular uotras causas, traumatismos, neoplasias, sustancias citolí-ticas, soluciones hiperosmóticas, infecciones, enferme-dades autoinmunes y por pérdida de autorregulación,como la que tiene lugar en la encefalopatía hipertensiva,la hipertensión intracraneal, los estados de mal convulsi-vos o la hipercapnia. En algunos casos hay transporte ac-tivo (hexosas, grandes aminoácidos neutros, aminoácidosbásicos y ácidos monocarboxílicos de cadena corta), quepuede saturarse y ser inhibido farmacológicamente. Enotros, la célula endotelial puede metabolizar el fármaco(p. ej., la dopa a dopamina). Algunos fármacos pasan laBHE como un precursor liposoluble que se transformaen el SNC en el principio activo más hidrófilo (p. ej., laheroína liposoluble se metaboliza a morfina poco liposo-luble). Algunos núcleos cerebrales, como la eminenciamedia, el área postrema, el órgano subfornical, la glán-dula pineal y el órgano subcomisural carecen de estaBHE, lo que permite un mejor acceso de los fármacos.
Los fármacos que llegan hasta el líquido cefalorraquí-deo (LCR) a partir de los capilares de los plexos coroi-deos o por administración intratecal e intraventricularsólo están separados del SNC por la pía-aracnoides o lamembrana ependimaria, por lo que puede considerarseel LCR una prolongación del espacio intersticial cerebral.Sólo accede el fármaco que no está unido a las proteínasplasmáticas, por lo que la concentración en LCR sueleser similar a la concentración libre en plasma. Sin em-bargo, la dinámica del LCR puede producir diferenciasen la concentración de los fármacos entre la región lum-bar y los ventrículos cerebrales o la cisterna magna. Laconcentración cerebral de los fármacos no siempre esigual a la que alcanzan en el LCR ya que algunos fárma-cos, como los antiepilépticos, se fijan al cerebro alcan-zando concentraciones varias veces superiores a las delLCR.
La salida de los fármacos y de sus metabolitos del SNCtiene la misma dificultad que su entrada; también se handescrito mecanismos de transporte activo desde el LCRhacia la sangre que pueden ser inhibidos con inhibidoresdel transporte, como la probenecida.
b) Barrera placentaria. Separa y une a la madre conel feto. Para atravesarla, los fármacos y sus metabolitostienen que salir de los capilares maternos, atravesar unacapa de células trofoblásticas y mesenquimáticas, y en-trar en los capilares fetales. Los fármacos pasan princi-
Neurona
Glía
Glía
LCR
d
c
e
b
a
f
Seno venoso
Sangre
San
gre
Fig. 4-8. Esquema de los compartimientos intracraneales. Lasflechas continuas indican la dirección del flujo del LCR. Las fle-chas discontinuas indican los sitios donde existe difusión de aguay solutos: a) a través de la BHE (de capilar a espacio intersti-cial); b) a través del epitelio de los plexos coroideos; c) a travésde la membrana ependimaria entre el espacio ventricular y elespacio intersticial; d) a través de la piamadre entre el espaciointersticial y el espacio subaracnoideo; e) a través de la mem-brana neuronal, y f) a través de la membrana de células gliales.

60 Farmacología humana
palmente por difusión pasiva y su velocidad de pasodepende del gradiente de concentración, de la liposolu-bilidad, del grado de ionización y del pH de la sangre ma-terna y fetal. La fijación a proteínas limita el paso cuandoel fármaco difunde con dificultad. Cuando es muy lipó-filo y no polar no depende de la unión a proteínas sinodel flujo sanguíneo placentario. La unión a proteínas y elpH fetales son menores que en la madre. La placenta tieneenzimas que pueden metabolizar los fármacos y los me-tabolitos que pasan de la madre al feto, y viceversa. Labarrera placentaria es particularmente acentuada en elprimer trimestre del embarazo y disminuye en el tercertrimestre debido al progresivo aumento en la superficiey la reducción de su grosor. Las características de la uni-dad materno-placento-fetal y las consecuencias del pasode los fármacos a través de la placenta se analizan conmás detalle en el capítulo 7.
3. Cinética de distribución
3.1. Compartimientos farmacocinéticos
El organismo humano está formado por múltiples com-partimientos reales y ficticios. Por una parte, existencompartimientos acuosos, como el agua plasmática, elagua intersticial y el agua intracelular. Por otra parte, haymedios no acuosos que pueden actuar como depósitos,como las proteínas plasmáticas y tisulares, los ácidos nu-cleicos y los lípidos intracelulares. Desde un punto devista cinético suelen considerarse tres compartimientos,atendiendo a la velocidad con que el fármaco los ocupay abandona:
a) El compartimiento central incluye el agua plas-mática, intersticial e intracelular fácilmente accesible; esdecir, la de los tejidos bien irrigados, como corazón, pul-món, hígado, riñón, glándulas endocrinas y SNC (si el fár-maco atraviesa bien la BHE).
b) El compartimiento periférico superficial está for-mado por el agua intracelular poco accesible; es decir, lade los tejidos menos irrigados, como piel, grasa, músculoo médula ósea, así como los depósitos celulares (proteí-nas y lípidos) a los que los fármacos se unen laxamente.
c) El compartimiento periférico profundo incluye losdepósitos tisulares a los que el fármaco se une más fuer-temente y de los que, por tanto, se libera con mayor len-titud.
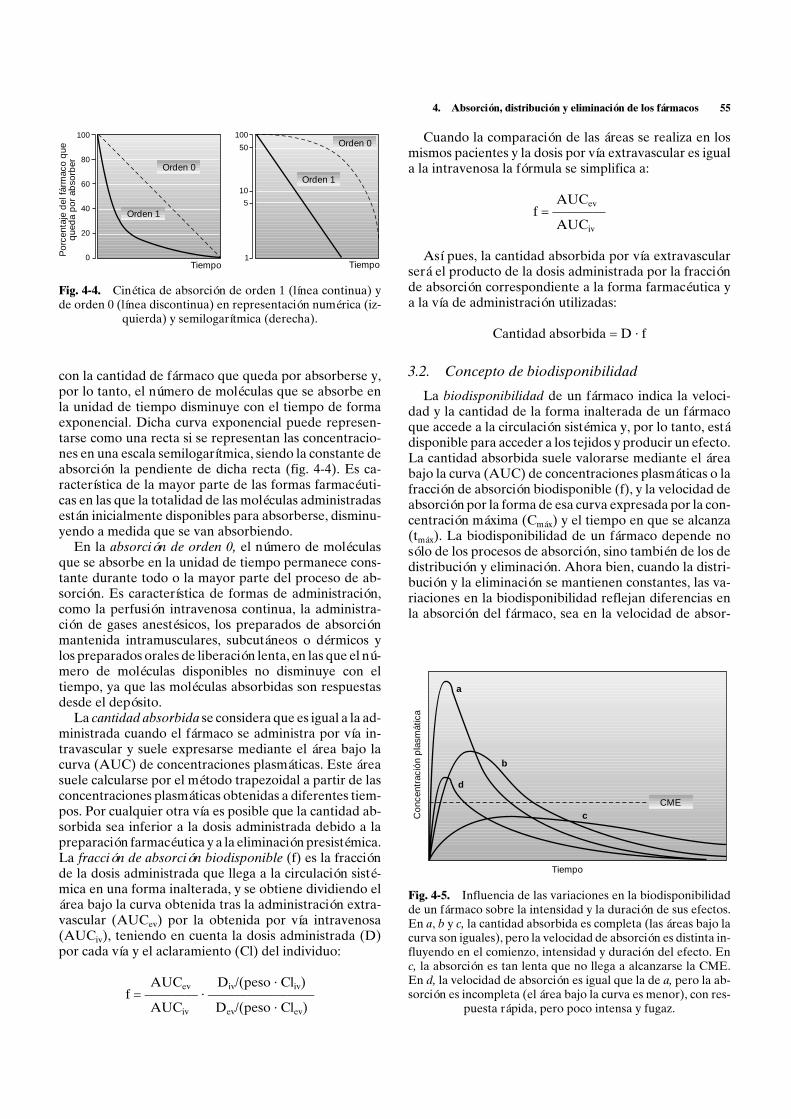
La distribución de un fármaco se considera mono-compartimental cuando se distribuye rápida y uniforme-mente por todo el organismo, es decir, cuando el orga-nismo se comporta como un único compartimientocentral. En el modelo de distribución bicompartimental,los fármacos administrados por vía intravenosa difundencon rapidez al compartimiento central y con más lenti-tud al compartimiento periférico. Los fármacos con dis-tribución tricompartimental se fijan fuertemente a deter-
minados tejidos en los que se acumulan y de los que seliberan con lentitud (figs. 4-9 y 4-10). La mayor parte delos fármacos se adaptan a un modelo bicompartimental,pero en algunos la distribución a los tejidos es tan pe-
a) b)
a) b) c)
a) b) c) d)
Modelo tricompartimental
Modelo bicompartimental
Modelo monocompartimental
Fig. 4-9. Modelos compartimentales. Modelo monocomparti-mental: a) antes de la administración y b) después de la adminis-tración, la distribución es rápida y uniforme. Modelo bicompar-timental: a) antes de la administración; b) inmediatamentedespués, el fármaco difunde a los órganos bien irrigados, y c)luego se equilibra con el resto del organismo. Modelo tricom-partimental: a) antes de la administración; b) inmediatamentedespués, el fármaco difunde a los órganos bien irrigados; c) luegose equilibra con el resto del organismo, y d) la acumulación con-
tinúa en los órganos a los que el fármaco se fija fuertemente.
4. Absorción, distribución y eliminación de los fármacos 61
K12 K21
K12 K21
K13 K31
B
B
B
A
A
A
Ka
Ka
Ka
Ke
K13
Ke
CP
CPS
CC
CPP
CC
D·f
D·f
D·f
Modelo monocomparti
Modelo bicompartim
Modelo tricompartime
Fig. 4-10. Modelos compartimentales. A) Características del mode un modelo físico aplicable a los modelos compartimentales, doperiférico en el modelo bicompartimental, y en el acceso y el retopartimental. C) Curso temporal de las concentraciones plasmáticas
noexponencial, biexponencial y triexponencial para los m
Fig. 4-11. Disociación entre concentraciones plasmáticas y tisulatimental hay una disociación durante la fase distributiva entre los producen en el compartimiento periférico). B) Tras dosis múltiplemomento en que se alcanzan las máximas concentraciones plasmá
timiento periférico profundo); tras la supresión, desapar
10,0
7,0
4,0
2,0
1,0
0,6
0,4
4 8 12 16 20 24
Horas
Digoxina 0,75 mg, IV
Tisular
Plasmática
Fase dedistribución
Fase deeliminaciónC
once
ntra
ción
de
dig
oxin
a (n
g/m
l)
A
Ln d
e co
ncen
trac
ión
Ln d
e co
ncen
trac
ión
Ln d
e co
ncen
trac
ión
C
C
C
Ke
Tiempo
Tiempo
Tiempo
a
a
b
b
C0p
Cp
g
Cu¥Cu
mental
ental
ntal
delo y constantes farmacocinéticas que intervienen. B) Esquemande puede observarse el retraso en el acceso al compartimientorno al compartimiento periférico profundo en el modelo tricom- tras una administración intravenosa: puede verse el carácter mo-odelos mono, bi y tricompartimental, respectivamente.
res. A) Tras una inyección intravenosa de un fármaco bicompar-niveles plasmáticos y los niveles tisulares (y los efectos cuando ses de un fármaco tricompartimental hay una disociación entre elticas y tisulares (y los efectos cuando se producen en el compar-
ece el fármaco del plasma mucho antes que de los tejidos.
B
0 1 2 3 4 5 6 7 8 9 10 11
100
10
1
0
Con
cent
raci
ón (e
n %
)
Concentración mínima eficaz
Concentración mínima detectable
Duración del efecto
Tiempo

62 Farmacología humana
queña que sólo se aprecia por vía intravenosa y no porvía oral, por lo que suelen tratarse cinéticamente, comosi fueran monocompartimentales (p. ej., aminoglucósi-dos o teofilina).
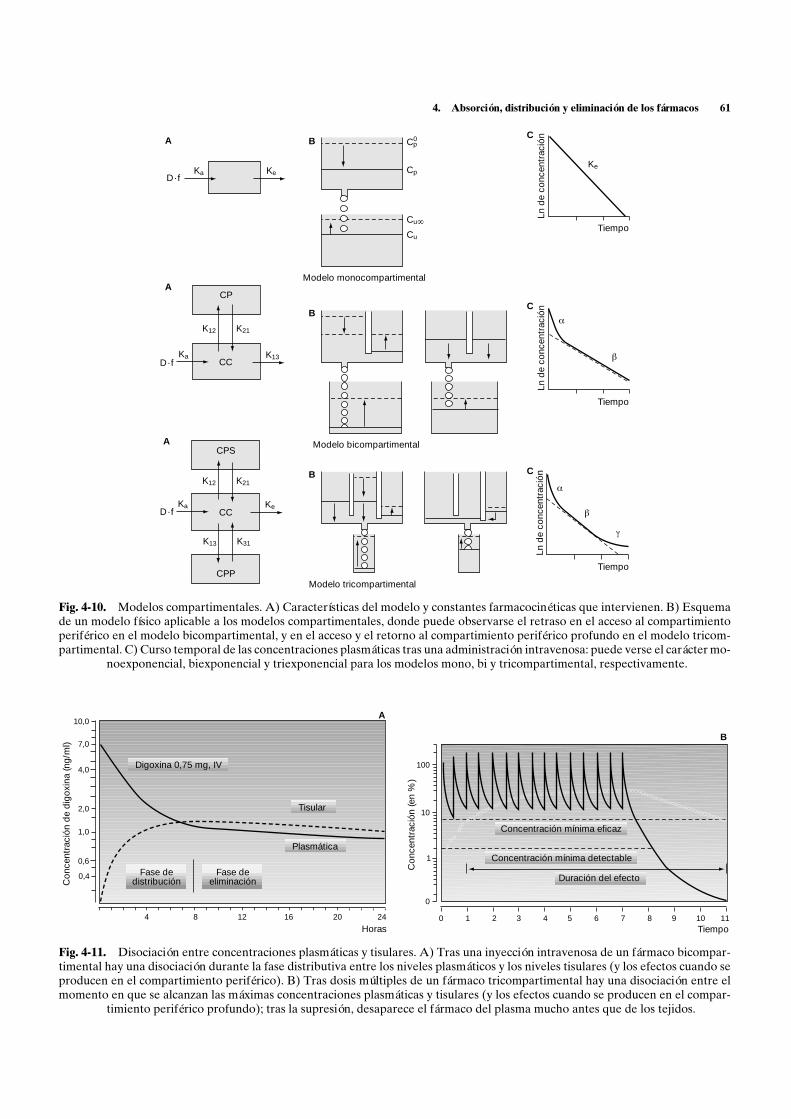
En el modelo monocompartimental hay un parale-lismo entre el curso temporal de las concentracionesplasmáticas y los efectos conseguidos. En el modelo bi-compartimental, también se observa este paralelismocuando el efecto es consecuencia de su acción en el com-partimiento central; pero, cuando se produce en el com-partimiento periférico, hay disociación entre las altasconcentraciones plasmáticas iniciales y las todavía ba-jas concentraciones tisulares, volviendo a ser paraleloscuando se alcanza el equilibrio entre ambos comparti-mientos, es decir, en la fase posdistributiva (fig. 4-11A).En el modelo tricompartimental, cuando el efecto tienelugar en el compartimiento periférico profundo, elefecto máximo tardará en aparecer y desaparecerá tam-bién más de lo que indican las concentraciones plasmá-ticas (fig. 4-11B).
3.2. Volumen aparente de distribución
Si el organismo estuviera organizado como un com-partimiento único en cuya agua se distribuye el fármacouniformemente, se podría calcular dicho volumen divi-diendo la cantidad administrada entre la concentraciónplasmática alcanzada, que sería la misma que en el restodel organismo. Pero, en realidad, el fármaco que hay enel organismo no sólo está disuelto en el agua corporal sinoque puede estar unido a las proteínas del plasma y a lostejidos. Por lo tanto, el volumen de distribución (Vd) noes un volumen real sino un volumen aparente que rela-ciona la cantidad total del fármaco que hay en el orga-nismo en un determinado momento con la concentraciónplasmática:
Cantidad de fármacoVd = ————————————
Concentración plasmática
Dicho de otra forma, el volumen aparente de distri-bución de un fármaco es el volumen en que tendría quehaberse disuelto la dosis administrada de un fármaco paraalcanzar la concentración plasmática observada. Este vo-lumen aparente dependerá del volumen real en que sedistribuya el fármaco, de su unión a las proteínas delplasma y de su unión a los tejidos (fig. 4-12).
El volumen real en que se distribuyen los fármacos de-pende de: a) sus características fisicoquímicas que condi-cionan su paso a través de las membranas y, por lo tanto,que queden confinados al plasma (unos 3 l en el adulto),que lleguen también al espacio intersticial (unos 12 l), oque accedan además al agua intracelular (unos 40 l); b)el peso del individuo; de aquí la conveniencia de expre-sar la dosis en dosis/kg en lugar de en dosis total, y c) laproporción de agua por kilogramo de peso, que en el re-
cién nacido es del 85 % y en el adulto es del 65 %, lo quedetermina que en algunos casos sea mejor expresar la do-sis por unidad de superficie corporal que por unidad depeso.
Por otra parte, la unión a las proteínas del plasma au-menta la concentración plasmática total del fármaco, laque se mide habitualmente, dando la impresión de que elfármaco se ha distribuido en un volumen menor del real.Por ejemplo, un fármaco con un volumen de distribuciónreal de unos 40 l (0,6 l/kg) que se una fuertemente a lasproteínas plasmáticas dará la impresión de distribuirse entan sólo 7 l (0,1 l/kg).
Por el contrario, la unión a los tejidos producirá bajasconcentraciones en plasma, dando la impresión de que elfármaco se ha distribuido en un volumen mayor del real.Por ejemplo, el gran volumen de distribución de la digo-xina de unos 400 l (6 l/kg) se debe a su fuerte fijación alos tejidos (fig. 4-13). Cuando el fármaco se une simultá-neamente a las proteínas del plasma y a los tejidos, habráuna influencia contrapuesta, de forma que el volumen de
Plasma Tejidos
D—–Cp
= VdD—–
Cp= Vd
D—–Cp
= Vd
D—–Cp
= VdD—–
Cp= Vd
A B
C
D E
Fig. 4-12. Influencia del volumen real, la unión a proteínas yla unión a los tejidos sobre la concentración plasmática totaly sobre el volumen aparente de distribución. El volumen realde un fármaco que no se une a las proteínas plasmáticas ni a lostejidos y se distribuye por todo el agua corporal (A) es mayorque el de un fármaco confinado al compartimiento vascular (B);al aumentar el tamaño del individuo aumenta el volumen dedistribución real y disminuye la concentración plasmática (C);cuando el fármaco se une mucho a proteínas plasmáticas au-menta la concentración plasmática total, dando la impresión deque su volumen de distribución es pequeño (D); cuando el fár-maco se une mucho a los tejidos, disminuye la concentraciónplasmática total, dando la impresión de que su volumen de dis-tribución es grande (E). Obsérvese que si el fármaco se uniesemucho a las proteínas plasmáticas y a los tejidos, su volumen de
distribución se parecería al volumen real de A.

4. Absorción, distribución y eliminación de los fármacos 63
distribución de un fármaco puede ser similar al volumenreal (unos 0,5 l/kg) tanto porque no se fije ni a las pro-teínas plasmáticas ni a los tejidos como sucede con la eto-suximida, como porque se una por igual a ambas comosucede con la fenitoína (fig. 4-13).
En los fármacos con distribución monocompartimen-tal se considera un único volumen aparente de distribu-ción (Vd) que se obtiene dividiendo la cantidad absorbida(D · f) entre la concentración en el tiempo cero (Cp
0).
D · fVd = ———
Cp0
El volumen aparente de distribución se utiliza para cal-cular la dosis inicial que debe administrarse para alcan-zar con rapidez niveles terapéuticos en situaciones ur-gentes.
Cuando la distribución es bicompartimental, existe un volumen apa-rente de distribución del compartimiento central y un volumen total ovolumen en equilibrio. El volumen central se calcula, como en el mo-delo monocompartimental, a partir de la concentración inicial:
D · fVc = ——–—
Cp0
El volumen en equilibrio (Vss) depende del volumen sanguíneo (Vs),del volumen tisular (Vt) y de la fracción libre sanguínea (fls) y tisular(flt):
flsVss = Vs + (Vt · —– )
flt
Con frecuencia se utiliza en su lugar el volumen de distribución du-rante la fase ß, que se calcula a partir del aclaramiento (Cl) o del áreabajo la curva (AUC) y la constante de disposición ß que se comenta másadelante:
Cl D · fVb = —— = —–———
b AUC · b
El volumen central se utiliza para calcular la dosis inicial en los fár-macos que actúan en el compartimiento central (p. ej., tiopental, dia-zepam o lidocaína), mientras que el volumen en equilibrio se utilizapara calcular la dosis inicial de los fármacos que actúan en el compar-timiento periférico (p. ej., digoxina).
3.3. Factores que alteran la distribución
Los factores que alteran el volumen de distribución influyen sobrela concentración máxima que se alcanza tras una dosis única o una do-sis inicial, por lo que deberá aumentarse ésta cuando haya factores queaumenten el volumen de distribución y reducirla cuando lo disminuyan.Por el contrario, los cambios en el volumen de distribución no afectanel nivel estable que se alcanza tras dosis múltiples ni, por lo tanto, lasdosis de mantenimiento (v. cap. 6). Los factores que alteran el volumende distribución pueden ser fisiológicos, patológicos y yatrógenos (v.caps. 7, 8 y 10).
a) Factores que alteran el volumen real. Las variaciones de peso in-fluyen en el volumen de distribución total, pero no alteran el volu-
men/kg. Los edemas y los derrames pleurales y ascíticos aumentan elvolumen de distribución de los fármacos hidrosolubles y reducen el delos liposolubles. Por el contrario, la obesidad reduce el volumen de dis-tribución de los fármacos hidrosolubles y aumenta el de los liposolu-bles. La insuficiencia cardíaca reduce el fármaco que llega a los tejidosy, por lo tanto, el volumen de distribución de los fármacos tanto hidro-solubles como liposolubles. La acidosis aumenta el acceso al SNC y alinterior de las células de los ácidos débiles (lo que aumenta su volumende distribución) y reduce el de las bases débiles. Diversas circunstan-cias patológicas pueden alterar el acceso de los fármacos a áreas con-cretas, como sucede cuando hay inflamación de las meninges, en un abs-ceso, en la artrosis u osteomielitis y en la enfermedad renal.
b) Factores que alteran la unión a las proteínas plasmáticas. Di-versos factores fisiológicos (recién nacido o anciano), patológicos (hi-poalbuminemia o hiperbilirrubinemia) y numerosas interacciones pue-den alterar la unión de los fármacos a las proteínas del plasma, pero notodos afectan por igual las diversas proteínas (tabla 4-5). Por ejemplo,en la uremia suele estar reducida la albúmina, pero puede estar au-mentada la a-glucoproteína. Además, puede estar alterada la capaci-dad funcional de la proteína, como sucede en la uremia en la que dis-minuye la capacidad de la albúmina para fijar los fármacos. Para que lainfluencia de estos factores sea relevante se precisa que:
Volumen aparente de distribución
l/70 kg l/ kgFármaco
Quinacrina
Cloroquina
AmiodaronaDoxorubicina
ClorpromazinaNortriptilinaMinoxidilo
DigoxinaDiltiazem
VerapamiloMorfina
Ranitidina
DiazepamEritromicina
FenitoínaTeofilina
Ampicilina
IbuprofenoWarfarina
50.000
20.000
10.000
5.000
2.000
1.000
500
200
100
50
20
10
5
3
500
200
100
50
20
10
5
2
1
0,5
0,2
0,1
0,05
Fig. 4-13. Volumen aparente de distribución de algunos fár-macos. Con líneas discontinuas se indica el volumen real delplasma (unos 3 l), el líquido intersticial (unos 12 l) y el agua in-tracelular (unos 40 l). Obsérvese que el volumen de distribu-ción de algunos fármacos es mucho mayor que el real debido a
su fuerte fijación a los tejidos.

64 Farmacología humana
1) El factor afecte la proteína a la que se fija el fármaco. Por ejem-plo, en la uremia está reducida la unión de los fármacos que se fijana la albúmina, pero no la de los fármacos que se fijan a la a-glucopro-teína.
2) El factor afecte el mismo lugar de fijación. Por ejemplo, el val-proato desplaza a la fenitoína porque ambos se unen al sitio I, pero noal diazepam que se une al sitio II. Cuando dos fármacos se fijan al mismolugar de la albúmina, se comportará como desplazante no el que tengamayor afinidad, sino el que tenga mayor producto de concentración li-bre por la afinidad. Por ello suelen comportarse como desplazantes losfármacos, como el ácido valproico o la fenilbutazona, cuya unión a laalbúmina se satura a dosis terapéuticas, mientras que son desplazadosla warfarina o la fenitoína cuyas concentraciones habituales están muylejos de la de saturación (tabla 4-4). También explica por qué las sulfa-midas con baja afinidad desplazan a la bilirrubina con alta afinidad porel sitio I de la albúmina.
Tabla 4-5. Factores fisiológicos y patológicos que alteranlas proteínas plasmáticas
Disminuyen Aumentan
AlbúminaAbscesos hepáticos EjercicioCirrosis hepática ¿Enfermedades neurológicas?Cirugía EsquizofreniaEdad (neonato o anciano) HipotiroidismoEmbarazo NeurosisEnfermedades gastro- Paranoia
intestinales PsicosisFibrosis quística Tumores benignosHistoplasmosisInsuficiencia renalLepraMalnutrición graveMieloma múltipleNeoplasias malignasNeumonía bacterianaPancreatitis agudaQuemadurasSíndrome nefróticoTraumatismos
a-GlucoproteínaAnticonceptivos orales Artritis reumatoideaFeto CirugíaSíndrome nefrótico Edad (anciano)
Enfermedad celíacaEnfermedad de CrohnEstrésInfarto de miocardioInsuficiencia renalTraumatismos
Lipoproteínas¿Enfermedad hepática? DiabetesHipertiroidismo ¿Enfermedad hepática?Traumatismos Hipotiroidismo
Síndrome nefrótico
3) Que la unión del fármaco a las proteínas plasmáticas sea mayordel 80 %. Debe tenerse en cuenta que la cantidad de un fármaco quehay en el plasma, respecto al total, es de:
7 % si no se une nada a las proteínas del plasma.12 % si se une en el 50 %.19 % si se une en el 70 %.26 % si se une en el 80 %.42 % si se une en el 90 %.59 % si se une en el 95 %.88 % si se une en el 99 %.
y que el aumento de la concentración libre en los tejidos al aumentar aldoble la concentración libre en plasma sería:
De 1 a 1,8 si pasa la unión a proteínas del plasma del 99 al 98 % y laconcentración libre en plasma del 1 al 2 %.
De 1 a 1,4 si pasa la unión a proteínas del 95 al 90 % y la concen-tración libre del 5 al 10 %.
De 1 a 1,3 si pasa la unión a proteínas del 90 al 80 % y la concen-tración libre del 10 al 20 %.
De 1 a 1,14 si pasa la unión a proteínas del 80 al 60 % y la concen-tración libre del 20 al 40 %.
De 1 a 1,11 si pasa la unión a proteínas del 70 al 40 % y la concen-tración libre del 30 al 60 %.
De 1 a 1,06 si pasa la unión a proteínas del 50 al 0 % y la concen-tración libre del 50 al 100 %.
4) Que el volumen de distribución del fármaco sea pequeño (me-nor de 0,15 l/kg).
Las consecuencias de una disminución en la unión a las proteínasplasmáticas sobre el volumen de distribución dependerá de su fija-ción a los tejidos: no cambia cuando se une poco a los tejidos y el vo-lumen de distribución del fármaco es pequeño (< 0,15 l/kg) y aumentacuando es grande (> 1,5 l/kg) y no disminuye simultáneamente launión a los tejidos (v. ecuación de Vss). Las consecuencias sobre elaclaramiento y las concentraciones total y libre dependen del tipo deeliminación y se comentan en el apartado sobre factores que alteranla eliminación.
c) Factores que alteran la unión a los tejidos. Cuando el fármaco seune fuertemente a las proteínas plasmáticas y a los tejidos, la disminu-ción en la unión a las proteínas plasmáticas sin cambios en su fijacióntisular aumenta el volumen de distribución (como ya se ha comentadoanteriormente), pero cuando disminuye simultáneamente la fijación alas proteínas del plasma y a los tejidos, puede no haber cambio en el vo-lumen de distribución.
V. ELIMINACIÓN
La concentración activa del fármaco en el organismohumano disminuye como consecuencia de dos mecanis-mos: la metabolización y la excreción. Los fármacos li-posolubles, aunque se filtren por el riñón, se reabsorbeny deben metabolizarse (principalmente en el hígado) ametabolitos más polares. Estos metabolitos, junto con losfármacos hidrosolubles, se excretan principalmente porel riñón y la bilis. La mayoría de los fármacos se elimi-nan, en mayor o menor proporción, por ambos mecanis-mos. Las características de eliminación de un fármaco sonimportantes en el momento de elegir el fármaco adecuadoen función de la duración del efecto y del número de to-mas deseadas, así como para valorar los factores que pue-den alterarlas.

4. Absorción, distribución y eliminación de los fármacos 65
La eliminación de un fármaco condiciona el tiempoque tarda en alcanzarse y en desaparecer su efecto cuandose administran dosis múltiples y, por lo tanto, el númerode tomas diarias que deben administrarse para evitar fluc-tuaciones excesivas de sus concentraciones plasmáticas.Las diferencias en la eliminación son la causa principalde la variabilidad individual en la respuesta a un fármacoy condicionan la necesidad de ajustar la dosis de mante-nimiento cuando haya factores que la alteren.
1. Metabolismo
La mayor parte de los fármacos se metabolizan en elorganismo humano a metabolitos, que pueden ser acti-vos o inactivos. La velocidad con que se metaboliza cadafármaco, la variedad de sus metabolitos y su concentra-ción dependen del patrón metabólico genéticamente es-tablecido de cada individuo y de la influencia de nume-rosos factores fisiológicos, patológicos y yatrogénicos quecondicionan notables diferencias de unos individuos aotros. De hecho, las diferencias en el metabolismo de losfármacos es el factor que más contribuye a que dosis igua-les den lugar a niveles plasmáticos distintos en diferentesindividuos. Los procesos metabólicos y los principalesfactores que los alteran se explican en el capítulo si-guiente.
2. Excreción
Los fármacos se excretan, por orden decreciente de im-portancia, por vía urinaria, vía biliar-entérica, sudor, sa-liva, leche y epitelios descamados. La excreción tiene in-terés en cuanto a que se trata de uno de los mecanismospor los que se eliminan del organismo los fármacos y susmetabolitos (excreción renal y biliar) y también por la po-sibilidad de tratar enfermedades localizadas en dichos ór-ganos de excreción (p. ej., infecciones urinarias). Indi-rectamente tiene interés para valorar el riesgo que puedarepresentar la excreción por la leche para el lactante ypara estudiar la cinética de algunos fármacos mediantelas determinaciones salivares de antiepilépticos, antipi-rina o teofilina.
2.1. Excreción renal
Es la vía más importante de excreción de los fármacos,siendo particularmente relevante cuando se eliminan deforma exclusiva o preferente por esta vía, en forma inal-terada o como metabolitos activos. Por el contrario, espoco importante en los fármacos que se eliminan princi-palmente por metabolismo, aun cuando una parte sus-tancial de sus metabolitos inactivos se eliminen por el ri-ñón. La cantidad final de un fármaco que se excreta porla orina es la resultante de la filtración glomerular y de lasecreción tubular, menos la reabsorción tubular.
La filtración glomerular se produce en los capilares delglomérulo renal, que poseen abundantes poros interce-
lulares por donde pasan todas las moléculas, excepto lasde gran tamaño y las unidas a las proteínas plasmáticas.Como consecuencia, la filtración aumenta cuando dismi-nuye la unión de los fármacos a las proteínas plasmáticas.La filtración glomerular, expresada por el aclaramientode inulina, es de 10 ml/min en el niño de un mes y medioy de 130 ml/min en el adulto.
La secreción tubular puede ser activa o pasiva. El trans-porte activo utiliza proteínas transportadoras de sustan-cias endógenas. Hay un sistema de transporte activo paraaniones orgánicos (p. ej., penicilina, probenecida, salici-latos o ácido úrico) que pueden competir entre sí y otropara cationes orgánicos que compiten igualmente entresí. La secreción pasiva se realiza en la parte más proximaldel túbulo renal a favor de un gradiente de concentra-ción. La suma de la filtración renal y de la secreción tu-bular, expresadas mediante el aclaramiento de ácido pa-raaminohipúrico, es de 25 ml/min en el niño de un mes ymedio y de 650 ml/min en el adulto.
La reabsorción tubular se produce principalmente pordifusión pasiva cuando la reabsorción de agua en el tú-bulo proximal aumenta la concentración de fármaco ensu luz, invirtiendo el gradiente de concentración. Lareabsorción pasiva depende de la liposolubilidad del fár-maco y, por lo tanto, del pH de la orina que condicionael grado de ionización. La alcalinización de la orina au-menta la eliminación de ácidos débiles, como barbitúri-cos o salicilatos, mientras que la orina ácida favorece laeliminación de bases débiles, como las anfetaminas o qui-nidina (fig. 4-3). La relación entre concentración urina-ria (Cu) y plasmática (Cp) puede deducirse de la fórmulade Henderson-Hasselbach a partir del pH plasmático(pHp) y urinario (pHu) y del pKa del fármaco.
Para ácidos:
1 + 10(pHu– pKa)
Cu/Cp = ———————1 + 10(pHp– pK
a)
y para bases:
1 + 10(pKa– pHu)
Cu/Cp = ———————1 + 10(pKa– pH
p)
La reabsorción tubular puede llevarse a cabo tambiénpor transporte activo ya que los mecanismos de trans-porte son bidireccionales. Por ejemplo, en el caso delácido úrico, su secreción activa es inhibida por los salici-latos a dosis bajas, mientras que su reabsorción activa esinhibida por los salicilatos a dosis altas.
2.2. Excreción biliar e intestinal: circulaciónenterohepática
Excreción biliar. Sigue en importancia a la excreciónurinaria y está muy relacionada con los procesos de bio-

66 Farmacología humana
transformación. Se produce principalmente por secreciónactiva con sistemas de transporte diferentes para sustan-cias ácidas, básicas y neutras. Se eliminan principalmentepor la bilis (tabla 4-6):
a) Sustancias con elevado peso molecular (al menosde 325 ± 50). La conjugación hepática, al añadir radica-les, eleva el peso molecular, facilitando la excreción bi-liar.
b) Sustancias con grupos polares, tanto anionescomo cationes, que pueden ser del fármaco (principal-mente, amonio cuaternario) o de los radicales suminis-trados por el metabolismo (glucuronatos o sulfatos).
c) Compuestos no ionizables con una simetría de gru-pos lipófilos e hidrófilos que favorece la secreción biliar(p. ej., digitoxina, digoxina y algunas hormonas).
d) Algunos compuestos organometálicos.
La excreción biliar de algunos fármacos, como ampi-cilina y rifampicina, puede ser útil en infecciones deltracto biliar y la de digoxina y oxazepam compensa enparte la disminución de la excreción renal en enfermosrenales.
Excreción intestinal. Los fármacos pueden pasar di-rectamente de la sangre a la luz intestinal, por difusiónpasiva, en partes distales en que el gradiente de concen-tración y la diferencia de pH lo favorezcan.
Circulación enterohepática. Los fármacos elimina-dos a la luz intestinal en forma activa a través de la biliso del epitelio intestinal pueden reabsorberse pasiva-mente en el intestino a favor de un gradiente de concen-tración. También los metabolitos pueden contribuir aesta reabsorción de fármaco mediante la acción de la floraintestinal. Por ejemplo, ciertas bacterias poseen glucu-ronidasas que liberan el fármaco original de su conjugadocon ácido glucurónico. Estos procesos dan origen a unacirculación enterohepática en que parte del fármaco quepasa a la luz intestinal es reabsorbido, lo que retrasa lacaída de las concentraciones plasmáticas y prolonga laduración del efecto. En caso de intoxicación, puede ace-lerarse la eliminación de los fármacos con circulación en-
Tabla 4-6. Ejemplos de fármacos con excreción biliar signifi-cativa
Acebutolol 5-FluorouraciloAmpicilina HidrocortisonaCarbenoxolona IndometacinaCefamandol MetronidazolCefoperazona NafcilinaCloranfenicol PivampicilinaClortetraciclina PractololDesmetilclortetraciclina RifampicinaDigitoxina TerbutalinaDigoxina TestosteronaDoxiciclina VincristinaEstradiol
terohepática, administrando carbón activado por víaoral, con el fin de atrapar en la luz intestinal el fármacoque pase a ella con la bilis o desde la sangre y eliminarlocon las heces.
2.3. Otras vías de excreción
La excreción a la leche puede hacer que los fármacoslleguen al lactante y originen reacciones idiosincrásicasy tóxicas (v. cap. 7). Los fármacos pasan a la leche sobretodo por difusión pasiva, por lo cual el cociente leche/plasma será tanto mayor cuanto mayor sea su liposolubi-lidad y menor sea su grado de ionización y unión a prote-ínas plasmáticas. La relación entre las concentraciones enla leche (Cl) y en el plasma (Cp) puede deducirse de la fór-mula de Henderson-Hasselbach a partir del pH de laleche (pHl) y del plasma (pHp) y del pKa del fármaco.
Para ácidos:
1 + 10(pHl– pKa)
Cl/Cp = ———————1 + 10(pHp– pK
a)
y para bases:
1 + 10(pKa– pHl)
Cl/Cp = ———————1 + 10(pKa– pH
p)
Dado que el pH de la leche es ligeramente más ácidoque el de la sangre materna, el cociente leche/plasma serámayor para los fármacos básicos, similar para los neutrosy menor para los ácidos.
La concentración en la leche depende también de launión del fármaco a las proteínas y lípidos de la leche, yalgunos fármacos pasan a la leche mediante transporteactivo.
La excreción salival es poco importante desde el puntode vista cuantitativo y, además, la mayor parte del fár-maco excretado por la saliva pasa al tubo digestivo, desdedonde puede reabsorberse de nuevo. Los fármacos pasana la saliva principalmente por difusión pasiva, por lo quela concentración salival es similar a la concentración li-bre del fármaco en el plasma. Este hecho permite valo-rar de una forma incruenta la velocidad de eliminaciónde fármacos como la antipirina o la cafeína, que sirvenpara valorar la función hepática. También permite mo-nitorizar indirectamente las concentraciones libres de al-gunos fármacos, como la fenitoína, la carbamazepina o lateofilina.
No obstante, debe tenerse en cuenta que hay fárma-cos, que pasan a la saliva por transporte activo, en los quela concentración salival es mayor que la plasmática (p. ej.,el litio) y otros cuyo paso a la saliva depende críticamentedel pH salival (p. ej., el fenobarbital). Además, la con-centración salival de los fármacos puede variar con el flujosalival, el volumen de saliva obtenido, el momento de ob-
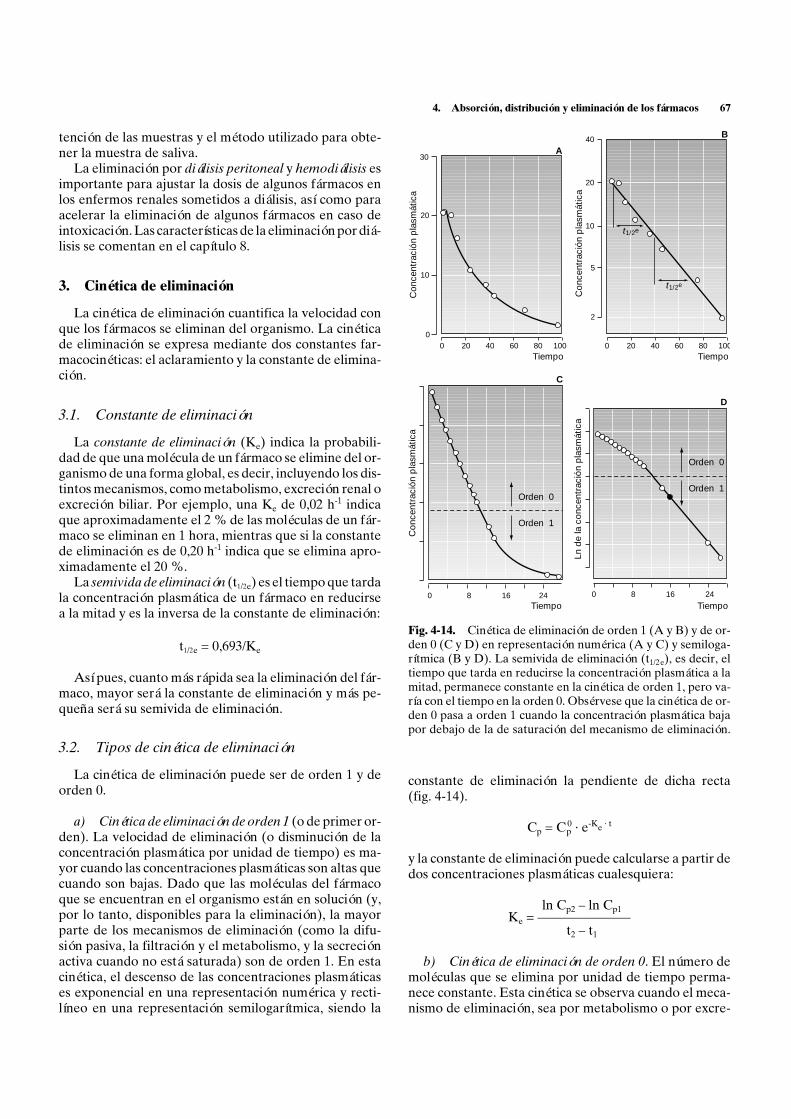
4. Absorción, distribución y eliminación de los fármacos 67
tención de las muestras y el método utilizado para obte-ner la muestra de saliva.
La eliminación por diálisis peritoneal y hemodiálisis esimportante para ajustar la dosis de algunos fármacos enlos enfermos renales sometidos a diálisis, así como paraacelerar la eliminación de algunos fármacos en caso deintoxicación. Las características de la eliminación por diá-lisis se comentan en el capítulo 8.
3. Cinética de eliminación
La cinética de eliminación cuantifica la velocidad conque los fármacos se eliminan del organismo. La cinéticade eliminación se expresa mediante dos constantes far-macocinéticas: el aclaramiento y la constante de elimina-ción.
3.1. Constante de eliminación
La constante de eliminación (Ke) indica la probabili-dad de que una molécula de un fármaco se elimine del or-ganismo de una forma global, es decir, incluyendo los dis-tintos mecanismos, como metabolismo, excreción renal oexcreción biliar. Por ejemplo, una Ke de 0,02 h-1 indicaque aproximadamente el 2 % de las moléculas de un fár-maco se eliminan en 1 hora, mientras que si la constantede eliminación es de 0,20 h-1 indica que se elimina apro-ximadamente el 20 %.
La semivida de eliminación (t1/2e) es el tiempo que tardala concentración plasmática de un fármaco en reducirsea la mitad y es la inversa de la constante de eliminación:
t1/2e = 0,693/Ke
Así pues, cuanto más rápida sea la eliminación del fár-maco, mayor será la constante de eliminación y más pe-queña será su semivida de eliminación.
3.2. Tipos de cinética de eliminación
La cinética de eliminación puede ser de orden 1 y deorden 0.
a) Cinética de eliminación de orden 1 (o de primer or-den). La velocidad de eliminación (o disminución de laconcentración plasmática por unidad de tiempo) es ma-yor cuando las concentraciones plasmáticas son altas quecuando son bajas. Dado que las moléculas del fármacoque se encuentran en el organismo están en solución (y,por lo tanto, disponibles para la eliminación), la mayorparte de los mecanismos de eliminación (como la difu-sión pasiva, la filtración y el metabolismo, y la secreciónactiva cuando no está saturada) son de orden 1. En estacinética, el descenso de las concentraciones plasmáticases exponencial en una representación numérica y recti-líneo en una representación semilogarítmica, siendo la
constante de eliminación la pendiente de dicha recta(fig. 4-14).
Cp = Cp0 · e-Ke · t
y la constante de eliminación puede calcularse a partir dedos concentraciones plasmáticas cualesquiera:
ln Cp2 – ln Cp1Ke = ———————
t2 – t1
b) Cinética de eliminación de orden 0. El número demoléculas que se elimina por unidad de tiempo perma-nece constante. Esta cinética se observa cuando el meca-nismo de eliminación, sea por metabolismo o por excre-
30
20
10
00 20 40 60 80 100
Con
cent
raci
ón p
lasm
átic
a
Tiempo
A
Con
cent
raci
ón p
lasm
átic
a
0 20 40 60 80 100Tiempo
B40
20
5
2
10 t1/2e
t1/2e
Con
cent
raci
ón p
lasm
átic
a
Tiempo
C
0 8 16 24
Orden 0
Orden 1
Tiempo
D
Orden 0
Orden 1
0 8 16 24
Ln d
e la
con
cent
raci
ón p
lasm
átic
a
Fig. 4-14. Cinética de eliminación de orden 1 (A y B) y de or-den 0 (C y D) en representación numérica (A y C) y semiloga-rítmica (B y D). La semivida de eliminación (t1/2e), es decir, eltiempo que tarda en reducirse la concentración plasmática a lamitad, permanece constante en la cinética de orden 1, pero va-ría con el tiempo en la orden 0. Obsérvese que la cinética de or-den 0 pasa a orden 1 cuando la concentración plasmática bajapor debajo de la de saturación del mecanismo de eliminación.

68 Farmacología humana
ción renal, es saturable y las concentraciones plasmáticasalcanzan valores que saturan estos mecanismos. En la ci-nética de orden 0, el descenso de los niveles plasmáticoses lineal en una representación numérica y se mantendráhasta que la concentración plasmática del fármaco des-cienda por debajo de la de saturación, en cuyo momentopasará a ser de orden 1 (fig. 4-14). En este tipo de ciné-tica mixta, denominada de Michaelis-Menten, que se co-menta con más detalle en el capítulo siguiente, el des-censo de las concentraciones plasmáticas con el tiempodepende de la dosis máxima del proceso (Dmáx) y de laconstante de metabolismo o concentración para la que elproceso se encuentra saturado en el 50 % (Km):
dCp Dmáx · Cp– —— = —————
dt Km + Cp
3.3. Constantes de disposición
En el modelo monocompartimental, el descenso de los niveles plas-máticos tras una administración intravenosa depende de la constantede eliminación (fig. 4-10).
En el modelo bicompartimental, el descenso de las concentracionesplasmáticas tras una administración intravenosa depende tanto de losprocesos de distribución como de eliminación que se consideran con-juntamente como procesos de disposición. Este descenso es biexpo-nencial con dos constantes de disposición a y b, que dependen de losprocesos de distribución del compartimiento central al periférico (K12),de retorno del compartimiento periférico al central (K21) y de elimina-ción (Ke). La caída rápida a depende principalmente del paso de losfármacos del compartimiento central al periférico (fase distributiva),pero también del retorno y de la constante de eliminación. La caídalenta b se inicia cuando se ha establecido el equilibrio entre el compar-timiento central y periférico (fase posdistributiva) y depende princi-palmente de los procesos de eliminación (aunque también intervienenel paso de los fármacos a los tejidos y su retorno), cumpliéndose que:
a + b = K12 + K21 + Ke
La constante de disposición ß y su inversa, la semivida de elimina-ción ß, desempeñan el papel de Ke en el modelo monocompartimental,rigiendo el tiempo que tarda en alcanzarse el nivel estable o en desa-parecer los efectos (fig. 4-10). Como ya se ha descrito en el apartado so-bre modelos compartimentales, puede haber una disociación entre ni-veles y efectos durante la fase a cuando el lugar de acción se encuentraen el compartimiento periférico.
En el modelo tricompartimental, la caída de las concentracionesplasmáticas es de tipo triexponencial, es decir, además de las fases dedisposición a y b, hay una tercera fase de disposición ultralenta (deno-minada p o g), que depende principalmente del retorno del comparti-miento periférico profundo al central. Esta intensa fijación tisular es laresponsable de que, cuando se inicia el tratamiento, se alcance el efectomáximo más tarde que el nivel estable y que, cuando se suprime, dureel efecto más que las concentraciones plasmáticas.
3.4. Aclaramiento
El aclaramiento (Cl)de un fármaco por un órgano in-dica la capacidad de ese órgano para eliminarlo. Se ex-presa mediante el número de mililitros de plasma que elórgano aclara (es decir, de los que elimina totalmenteel fármaco) en la unidad de tiempo. Habitualmente, no
es posible calcular el aclaramiento de cada uno de los ór-ganos que contribuyen a eliminar el fármaco del orga-nismo, por lo que es más práctico estimar el aclaramientocorporal total (Cl) a partir de la dosis absorbida (D · f) ydel área bajo la curva (AUC) de concentraciones plas-máticas:
D · fCl = ———
AUC
El aclaramiento es una constante no compartimental,es decir, independiente del comportamiento monocom-partimental, bicompartimental o tricompartimental delfármaco.
a) Aclaramiento hepático (ClH). Depende del flujosanguíneo hepático (QH), de la fracción libre del fármacoen sangre (Fls) y de la capacidad metabólica del hepato-cito o aclaramiento intrínseco (Cli).
QH · Fls · CliClH = —————–—
QH + (Fls · Cli)
En función de su fracción de extracción hepática y de su unión a lasproteínas plasmáticas, los fármacos pueden clasificarse en tres grupos(fig. 4-15 y tabla 4-7):
1) Fármacos dependientes del flujo sanguíneo hepático. Tienen unaalta fracción de extracción hepática, mayor de 0,8 (tabla 4-8), por lo queel aclaramiento intrínseco es mucho mayor que el flujo sanguíneo he-pático, por lo que:
ClH = QH
Por lo tanto, su aclaramiento hepático depende críticamente delflujo sanguíneo hepático y es relativamente independiente de los cam-bios en la capacidad metabólica y en la unión a las proteínas del plasma,por lo que se los denomina de eliminación no restrictiva. Debe tenerseen cuenta que aunque sólo el fármaco libre pasa al hepatocito, el tiempoque tarda en liberarse nuevo fármaco desde las proteínas del plasma esde unos pocos milisegundos, reponiendo continuamente el fármaco quese va metabolizando.
2) Fármacos dependientes de la capacidad metabólica. Tienen unabaja fracción de extracción (menor de 0,2), por lo que el aclaramientointrínseco es mucho menor que el flujo sanguíneo hepático, y una po-bre unión a las proteínas del plasma (menor del 20 %), por lo que:
ClH = Cli
Por lo tanto, su aclaramiento hepático depende de la capacidad me-tabólica del hepatocito, pero es relativamente independiente de los cam-bios en el flujo sanguíneo hepático y en la unión a las proteínas plas-máticas.
3) Fármacos dependientes de la capacidad metabólica y de la unióna las proteínas plasmáticas. Tienen una baja fracción de extracción y unaalta unión a proteínas (mayor del 80 %), por lo que:
ClH = Fls · Cli
Por lo tanto, su aclaramiento hepático depende de los cambios en lacapacidad metabólica y de la mayor o menor unión a las proteínas plas-máticas, por lo que se les denomina de eliminación restrictiva.

4. Absorción, distribución y eliminación de los fármacos 69
Dependientes del flujo sanguíneo hepático
Dependientesde la capacidad
(proteín-insensibles)
Dependientesde la capacidad
(proteín-sensibles)
0 20 50 80 100
0,2
0,4
0,6
0,8
Unión a proteínas plasmáticas
Frac
ción
de
extr
acci
ón
Fig. 4-15. Clasificación de los fármacos en función de su frac-ción de extracción y de su unión a las proteínas plasmáticas. Lascaracterísticas de los tres grupos que se describen en el textoson aplicables a los fármacos que se eliminan exclusivamentepor metabolismo hepático y quedan próximos a los vértices del
triángulo.
Tabla 4-7. Ejemplos de fármacos cuyo metabolismo dependedel flujo sanguíneo hepático, de la capacidad metabólica y dela
unión a proteínas del plasma
Fracción Uniónde extracción a proteínas
Fármaco hepática del plasma
Dependientes del flujo sanguíneo hepáticoClormetiazol 0,90 64Labetalol 0,70 50Lidocaína 0,70 45-80Pentazocina 0,80 60-70Petidina 0,50 65-75Propranolol 0,64 93
Dependientes de la capacidad metabólicaAmilobarbital 0,03 61Antipirina 0,07 10Cloranfenicol 0,28 60-80Paracetamol 0,43 < 5Teofilina 0,09 50-60Tiopental 0,28 72-86
Dependientes de la capacidad metabólica y de la unión a proteínasClindamicina 0,23 93Clorpromazina 0,22 98Diazepam 0,03 98Digitoxina 0,005 97Fenitoína 0,03 90Quinidina 0,27 82-9Tolbutamida 0,02 95
b) Aclaramiento renal (ClR). Se calcula a partir de laorina recogida durante un período mayor de 5 semividasde eliminación del fármaco, dividiendo la cantidad defármaco eliminada por la orina (concentración del fár-maco en la orina por el volumen de orina) por la con-centración plasmática media durante ese período (Cp) ypor el tiempo durante el cual se ha recogido la orina (t):
Cu · VuClR = ————
Cp · t
A su vez, la concentración plasmática media duranteese período se calcula dividiendo el área bajo la curva en-tre el tiempo de recogida de la orina, por lo que:
Cu · VuClR = ————
AUC
La cantidad de fármaco eliminada en la orina es la sumade la cantidad filtrada más la cantidad segregada, menosla cantidad reabsorbida. La cantidad filtrada depende dela unión a proteínas, pero la secreción tubular activa no.
Tabla 4-8. Ejemplos de fármacos con fracción de extracción hepática o renal baja, intermedia y alta
Baja Intermedia Alta(< 0,3) (de 0,3 a 0,7) (> 0,7)
HepáticaÁcido salicílico Ácido acetilsalicílico AlprenololÁcido valproico Codeína CocaínaCarbamazepina Nifedipino LidocaínaDiazepam Nortriptilina MorfinaFenitoína Quinidina NicotinaFenobarbital NitroglicerinaIndometazina PentazocinaNaproxeno PetidinaNitrazepam PropoxifenoProcainamida PropranololTeofilina VerapamiloWarfarina
RenalAmoxicilina Amilorida Glucurónidos Atenolol Cefalotina (muchos)Cefazolina Cimetidina HipuratosCiprofloxacina Ranitidina PenicilinasDigoxina (algunas)Furosemida SulfatosGentamicina (muchos)LitioTetraciclina

70 Farmacología humana
Por ello, al igual que en el aclaramiento hepático, los fár-macos con una alta fracción de extracción renal son re-lativamente insensibles a los cambios en la unión a lasproteínas plasmáticas, es decir, tienen eliminación no res-trictiva, mientras que los que se eliminan solamente porfiltración dependen de la mayor o menor unión a las pro-teínas del plasma, es decir, tienen eliminación restrictiva(tabla 4-8). En este caso, puede calcularse el aclaramientorenal del fármaco libre:
Cu · VuClR libre = —————
AUC libre
Si conocemos el aclaramiento total del fármaco y suaclaramiento renal, podemos valorar la importancia delaclaramiento extrarrenal restando ambos:
Cl extrarrenal = Cl total – ClR
Y dado que la mayoría de los fármacos se eliminan pormetabolismo y excreción renal, el aclaramiento extrarre-nal nos permite estimar de forma indirecta el aclara-miento hepático:
ClH = Cl total – ClR
3.5. Relación entre constante de eliminación,aclaramiento y volumen de distribución
La velocidad de eliminación de un fármaco dependede la capacidad excretora de los órganos de excrecióny de la concentración en el plasma que accede a estos ór-ganos. A su vez, para una misma cantidad de fármaco enel organismo, la concentración plasmática depende delvolumen aparente de distribución. Por ello, la constantede eliminación puede considerarse la resultante secun-daria de dos procesos primarios: la capacidad de elimi-nación del organismo, expresada por el aclaramiento, y
Tabla 4-9. Relación entre aclaramiento, volumen de distribu-ción, constante de eliminación y semivida de eliminación
Fármaco distribuido en
Agua Agua Aguaplasmática extracelular corporal total
Aclaramiento (3 l) (12 l) (40 l)
Reabsorción parcial 0,01 min–1 0,0025 min–1 0,00073 min–1
(p. ej., 30 ml/min) (69 min) (277 min) (947 min)Filtración glomerular 0,043 min–1 0,011 min–1 0,0032 min–1
(130 ml/min) (16 min) (64 min) (219 min)Secreción tubular 0,22 min–1 0,054 min–1 0,016 min–1
(p. ej., 650 ml/min) (3 min) (13 min) (44 min)
Las cifras son los valores de Ke. Los valores entre paréntesis corresponden ala semivida de eliminación.
la distribución del fármaco, expresada por su volumenaparente de distribución:
Ke = Cl/Vd
y también:
t1/2e = 0,693 · Vd/Cl
Por lo tanto, la constante de eliminación de un fármacoaumenta con el aclaramiento y disminuye con el volumende distribución, sucediendo lo contrario con la semividade eliminación (tabla 4-9).
Si se conoce la constante de eliminación y el volumenaparente de distribución de un fármaco, puede calcularsesu aclaramiento:
Cl = Ke · Vd
y en el modelo bicompartimental:
Cl = Ke · Vc
y también:
Cl = b · Vss
Sin embargo, no debe confundirse esta relación mate-mática con una relación de causalidad ya que el aclara-miento no depende de la constante de eliminación y delvolumen de distribución, sino que es la constante de eli-minación, la que depende del aclaramiento y del volumende distribución.
3.6. Factores que alteran la eliminación
En la tabla 4-10 se resumen los factores individuales, ambientales,patológicos y yatrogénicos que pueden influir en la eliminación de losfármacos, que se comentan con más detalle en los capítulos 7 y 8.
Los factores que reducen la función renal y/o hepática, sea por in-madurez, involución, enfermedad o interacciones, reducen el aclara-miento de los fármacos y por ello se alcanzan niveles estables más al-tos que pueden ser tóxicos. Para evitarlo deberán utilizarse dosis demantenimiento menores y/o intervalos de administración más prolon-gados.
La influencia de estos factores sobre la constante de eliminación de-pende de que afecten o no simultáneamente el volumen de distribución:si no lo alteran, la reducción del aclaramiento se acompaña de una dis-minución proporcional de la constante de eliminación, pero si alteranel volumen de distribución, los cambios en la semivida de eliminaciónserán la resultante de los cambios en el aclaramiento y en el volumende distribución. Por ejemplo, en la insuficiencia renal moderada, en queno varía el volumen de distribución de la digoxina, la disminución delaclaramiento renal de digoxina se acompaña de un alargamiento de susemivida de eliminación. Sin embargo, en la insuficiencia cardíaca, enque también está reducido el volumen de distribución, la disminucióndel aclaramiento se acompaña tan sólo de un ligero aumento de sus se-mividas de eliminación (p. ej., de la lidocaína o la procainamida).
Una reducción en la unión a proteínas repercutirá en el aclaramientode un fármaco y, por lo tanto, en sus concentraciones plasmáticas enfunción de sus características de distribución y eliminación:
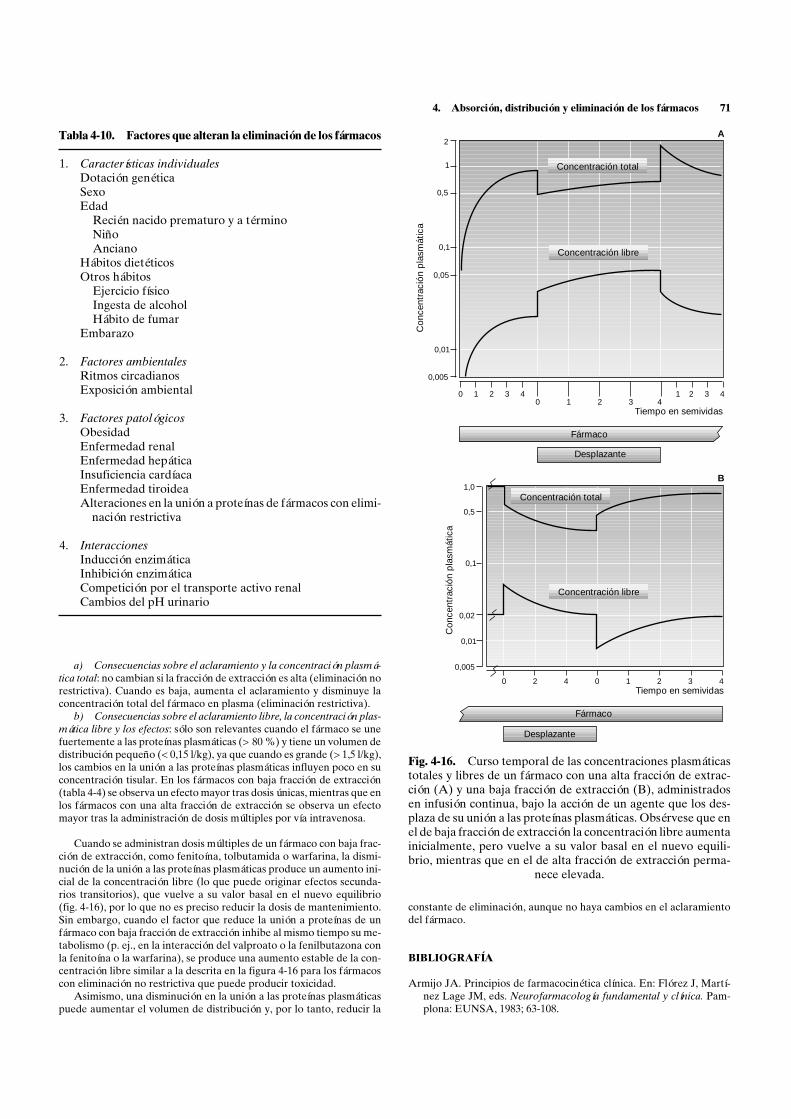
4. Absorción, distribución y eliminación de los fármacos 71
a) Consecuencias sobre el aclaramiento y la concentración plasmá-tica total: no cambian si la fracción de extracción es alta (eliminación norestrictiva). Cuando es baja, aumenta el aclaramiento y disminuye laconcentración total del fármaco en plasma (eliminación restrictiva).
b) Consecuencias sobre el aclaramiento libre, la concentración plas-mática libre y los efectos: sólo son relevantes cuando el fármaco se unefuertemente a las proteínas plasmáticas (> 80 %) y tiene un volumen dedistribución pequeño (< 0,15 l/kg), ya que cuando es grande (> 1,5 l/kg),los cambios en la unión a las proteínas plasmáticas influyen poco en suconcentración tisular. En los fármacos con baja fracción de extracción(tabla 4-4) se observa un efecto mayor tras dosis únicas, mientras que enlos fármacos con una alta fracción de extracción se observa un efectomayor tras la administración de dosis múltiples por vía intravenosa.
Cuando se administran dosis múltiples de un fármaco con baja frac-ción de extracción, como fenitoína, tolbutamida o warfarina, la dismi-nución de la unión a las proteínas plasmáticas produce un aumento ini-cial de la concentración libre (lo que puede originar efectos secunda-rios transitorios), que vuelve a su valor basal en el nuevo equilibrio(fig. 4-16), por lo que no es preciso reducir la dosis de mantenimiento.Sin embargo, cuando el factor que reduce la unión a proteínas de unfármaco con baja fracción de extracción inhibe al mismo tiempo su me-tabolismo (p. ej., en la interacción del valproato o la fenilbutazona conla fenitoína o la warfarina), se produce una aumento estable de la con-centración libre similar a la descrita en la figura 4-16 para los fármacoscon eliminación no restrictiva que puede producir toxicidad.
Asimismo, una disminución en la unión a las proteínas plasmáticaspuede aumentar el volumen de distribución y, por lo tanto, reducir la
Tabla 4-10. Factores que alteran la eliminación de los fármacos
1. Características individualesDotación genéticaSexoEdad
Recién nacido prematuro y a términoNiñoAnciano
Hábitos dietéticosOtros hábitos
Ejercicio físicoIngesta de alcoholHábito de fumar
Embarazo
2. Factores ambientalesRitmos circadianosExposición ambiental
3. Factores patológicosObesidadEnfermedad renalEnfermedad hepáticaInsuficiencia cardíacaEnfermedad tiroideaAlteraciones en la unión a proteínas de fármacos con elimi-
nación restrictiva
4. InteraccionesInducción enzimáticaInhibición enzimáticaCompetición por el transporte activo renalCambios del pH urinario
constante de eliminación, aunque no haya cambios en el aclaramientodel fármaco.
BIBLIOGRAFÍA
Armijo JA. Principios de farmacocinética clínica. En: Flórez J, Martí-nez Lage JM, eds. Neurofarmacología fundamental y clínica. Pam-plona: EUNSA, 1983; 63-108.
0 1 2 3 4 1 2 3 40 1 2 3 4
2
1
0,5
0,1
0,05
0,01
0,005
Tiempo en semividasC
once
ntra
ción
pla
smát
ica
Concentración total
Concentración libre
A
Fármaco
Desplazante
Tiempo en semividas
Con
cent
raci
ón p
lasm
átic
a
B1,0
0,5
0,1
0,02
0,01
0,005
Fármaco
Desplazante
0 2 4 0 1 2 3 4
Concentración total
Concentración libre
Fig. 4-16. Curso temporal de las concentraciones plasmáticastotales y libres de un fármaco con una alta fracción de extrac-ción (A) y una baja fracción de extracción (B), administradosen infusión continua, bajo la acción de un agente que los des-plaza de su unión a las proteínas plasmáticas. Obsérvese que enel de baja fracción de extracción la concentración libre aumentainicialmente, pero vuelve a su valor basal en el nuevo equili-brio, mientras que en el de alta fracción de extracción perma-
nece elevada.

72 Farmacología humana
Benet LZ, Massoud N, Gambertoglio JG, eds. Pharmacokinetic basisfor drug treatment. Nueva York: Raven Press, 1984.
Evans WE, Schentag JJ, Kusko WJ. Applied pharmacokinetics: princi-ples of therapeutic drug monitoring, 3.ª ed. Vancouver: Applied The-rapeutics, 1992.
Gillespie WR. Noncompartmental versus compartmental modelling inclinical pharmacokinetics. Clin Pharmacokin 1991; 20: 253-262.
Ludden TM. Nonlinear pharmacokinetics: Clinical implications. ClinPharmacokin 1991; 20: 429-446.
Ritschel WA. Handbook of basic pharmacokinetics including clinicalapplications, 3.ª ed. Hamilton: Drug Intelligence, 1986.
Rowland M, Sheiner LB, Steimer JL. Variability in drug therapy: Des-cription, estimation and control. Nueva York: Raven Press, 1985.
Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts andapplications, 3.ª ed. Filadelfia: Lea and Febiger, 1995.
Wagner JG: Farmacocinética clínica. Barcelona: Reverté, 1983.Winter ME. Farmacocinética clínica básica, 2.ª ed. Madrid: Díaz de San-
tos, 1994.