
13 neurologia y neurocirugia by medikando
165
Manual CTO de Medicina y Cirugía 8. a edición
-
Upload
diego-carrion -
Category
Documents
-
view
3.342 -
download
10
Transcript of 13 neurologia y neurocirugia by medikando

Manual CTO de Medicina y Cirugía
8. a ed ic ión

0 1 . Introducción: anatomía, semiología y fisiología del sistema nervioso 1
1.1 Breve recuerdo anatómico 1 1.2. A l terac iones de las f unc i ones super iores 2 1.3. Trastornos d e la función m o t o r a 4 1.4. Trastornos de la sens ib i l idad 6 1.5. Trastornos de la coordinación. Ataxias 8 1.6. Alteración de los pares craneales 9 1.7. Trastornos camplmétricos y pupi lares 11 1.8. Síndromes lobares 12 1.9. Síndromes troncoencefálicos 13 1.10. Reflejos y síndromes medu la res 14 1.11. Sección y shock medu l a r 16 1.12. Fisiología de l s istema nerv ioso 17 1.13. Transmisión sináptica 18
02. Coma. Muerte encefálica 19 2.1. C o m a 19 2.2. S ignos de va lor local izados 19 2.3. M u e r t e encefálica 21
03. Demencias 22 3.1. C o n c e p t o y clasificación 22 3.2. En fe rmedad de A lzhe imer 23 3.3. Demenc i a f r o n t o - t e m p o r a l
( En fe rmedad de Pick) 26 3.4. Demenc i a vascular 26 3.5. Demenc i a po r cue rpos de Lewy 27
04. Enfermedades vasculares cerebrales 28
4 .1 . Terr i tor ios vasculares cerebrales 28 4.2. Clasificación y factores de r iesgo 29 4.3. Enfermedades cerebrovasculares
isguémicas 30 4.4. Hemor rag ia i n t r apa renqu ima tosa 37 4.5. Ma l fo rmac iones vasculares 39 4.6. Hemor rag ia subaracno idea 40
05. Trastornos del movimiento 45
5.1. Temb lo r 46 5.2. Distonías 47 5.3. Mioclonías 48 5.4. Tics 48 5.5. Síndrome de piernas Inquietas 48 5.6. Corea. En fe rmedad de H u n t i n g t o n 49 5.7. En fe rmedad de Parkinson idiopátlca 50 5.8. Otros síndromes park lnson ianos 54 5.9. Atrof ias multisistémlcas (AMS) 55
06. Enfermedades por alteración de la mielina 57
6.1. Esclerosis múltiple (EM) 57 6.2. Otras en f e rmedades desmie l in izantes 61

07. Epilepsia 63 7.1. Clasificación 63 7.2. Fisiopatología 65 7.3. Diagnóstico d i ferenc ia 65 7.4. Etiología 66 7.5. A l g u n o s síndromes epilépticos específicos 66 7.6. T ra tamiento . Fármacos ant i comic ia les 67 7.7. Epilepsia y e m b a r a z o 69
08. Enfermedades degenerativas del sistema nervioso 70
8.1. Ataxias he redodegenera t i vas 70 8.2. En fermedades de la m o t o n e u r o n a 72
09. Enfermedades virales y priónicas del sistema nervioso 75
9.1. Encefalitis herpética y otras encefal i t is virales 75 9.2. Paraparesia espástica t rop ica l 77 9.3. Leucoencefalopatía mu l t i foca l
progresiva (LMP) 77 9.4. Panencefal it is esclerosante
subaguda (PESS) 78 9.5. En fermedades priónicas 78
10. Enfermedades nutricionales y metabólicas del sistema nervioso 81
10.1. Enfermedades neurológicas deb idas a déficits nut r i c iona les 81
10.2. En fermedades metabólicas secundar ias 83
11. Neuropatías 86 11.1. Cons iderac iones generales 86 11.2. S índrome d e Guillaln-Barré 88 11.3. Polineuropatía desmie l i n i zan te
in f l amator i a crónica (PDIC) 89 11.4. Neuropatía diabética 90 11.5. Neuropatías en la infección por VIH 91 11.6. Neuropatías disproteinémicas 91 11.7. Neuropatías hereditar ias sin base
metabólica c o n o c i d a 92 11.8. M o n o n e u r i t i s múltiple 92 11.9. Mononeuropatías 93
12. Enfermedades de la placa motora 94
12.1. Miastenia gravis 94 12.2. Ot ros síndromes mias ten i fo rmes 97
V

13. Miopatías 100 16. Hidrocefalia 114 13.1. Distrofias musculares 100 16.1. C o n c e p t o y clasificación 114 13.2. Miopatías congénitas 102 16.2. E t iopa togen ia 115 13.3. Miopatías metabólicas 103 16.3. Clínica 115 13.4. Miopatías m i tocondr i a l e s 104 16.4.
16.5. T r a t am ien to Hidrocefa l ia crónica de l adu l t o
116 117
14. Cefaleas 105 17. Tumores intracraneales 118
14.1. Cons iderac iones genera les 105 14.2. Cefalea tens iona l 106 17.1. Cons iderac iones generales 118 14.3. Migraña 106 17.2. Metástasis cerebrales 119 14.4. Cefalea en cluster.histamínica o de H o r t o n 107 17.3. G l lomas 120 14.5. Otras cefaleas pr imar las 108 17.4.
17.5. Tumores de l p l exo c o r o i d e o Tumores embr iona r i os . Tumores neuroectodérmicosprlmitivos (PNET)
123
123
15. Síndrome de hipertensión intracraneal 109
17.6. 17.7. 17.8.
Tumores neurona les y neurogl ia les m ix tos M e n i n g i o m a N e u r i n o m a del VIII par ( s c h w a n n o m a vest ibular )
124 124
125 15.1. Flslopatología 109 17.9. Tumores de la región pinea l 126 15.2. Etiología de l síndrome 17.10. Tumores h ipof isar ios 126
de hipertensión intracraneal 110 17.11. Tumores de o r i g e n disembrioplásico 127 15.3. Clínica 110 17.12. L i n foma cerebral p r ima r i o 128 15.4. Síndromes de herniación cerebral 110 17.13. H e m a n g i o b l a s t o m a 128 15.5. Diagnóstico 111 17.14. Tumores de la base craneal 128 15.6. T r a t am ien to 111 15.7. Síndrome de hipertensión intracraneal
b e n i g n a ( p s e u d o t u m o r cerebrí) 112 15.8. Edema cerebral 113

* 1
18. Traumatismos craneoencefálicos 130
18.1. Escala de c o m a de G lasgow 130 18.2. Mane jo de l TCE en Urgencias 131 18.3. Fracturas craneales 131 18.4. Conmoción cerebral 133 18.5. H e m a t o m a ep idura l 134 18.6. H e m a t o m a subdura l 134 18.7. Contusión cerebral hemorrágica 135 18.8. Lesión axonal difusa 135 18.9. Neumoencéfalo 135 18.10. Comp l i cac iones y secuelas
del n e u r o t r a u m a centra l 135
19. Absceso cerebral y empiema subdural 137
19.1. Absceso cerebral 137 19.2. Emp iema subdura l 139
20. Patología raquimedular 140 20.1 . Do lor l u m b a r 140 20.2. Lumbociática. Hernia discal l u m b a r 141 20.3. Cerv icobraqu ia lg ia . Hernia discal cervical 145 20.4. Espondi losis cervical 146 20.5. Estenosis de l canal l u m b a r 146 20.6. Espondilol lstesis 147 20.7. Espondi lodisc i t is 147 20.8. Lesiones medulares traumáticas 148 20.9. Tumores intrarraquídeos 150 20.10. Absceso ep idura l espinal 152 20.11 . S i r ingomie l ia 152 20.12. Anomalías de la unión craneocerv ica l 153
2 1 . Anomalías del desarrollo 155 21.1 . Craneosinostosis 155 21.2. Malformación de Chiari 156 21.3. Quistes a racno ideos 157 21.4. Disraf ismo espinal 157 21.5. Encefalocele 158 21.6. Sinus pericranii (variz espuria) 159
22. Neurocirugía funcional 160 22.1 . Neuralg ia de l trigémino 160 22.2. Neuralg ia de l glosofaríngeo 161 22.3. Cirugía de l do lo r in t ra tab le 161 22.4. Cirugía de la e n f e r m e d a d de Parkinson 161 22.5. Cirugía de la epi lepsia 161 22.6. Neurocirugía de los t ras tornos menta les
(Psicocirugía) 161
Bibliografía 162
Vi l

Neurología y neurocirugía
o í . INTRODUCCIÓN: ANATOMÍA, SEMIOLOGÍA Y FISIOLOGÍA DEL SISTEMA NERVIOSO
Orientación
MIR r
Aspectos esenciales
En los últimos años, ha disminuido la importancia de este tema en el MIR, pero una lectura atenta es fundamental porque ayudará a entender los demás temas y, por tanto, a responder las preguntas de tipo caso clínico en las que los datos semiológicos son la clave diagnóstica.
f J J La asociación de aprax ia de la marcha , i n con t i nenc i a u r ina r i a y d e m e n c i a aparece característicamente en la h id roce fa l i a no rmotens i va y en las lesiones f ronta les b i latera les .
j~2~] El haz co r t i coesp ina l y el haz co r t i c onuc l e a r (fascículo gen i cu l ado ) son los dos tractos p r inc ipa les que f o r m a n el s istema p i r a m i d a l . En este sistema existe una p r imera m o t o n e u r o n a cuya lesión se caracter iza por afectación de amp l i o s grupos muscu lares , ref le jos osteotendinosos exa l tados , s igno de Babinski e hipertonía en " h o j a de nava ja " c o n ausenc ia de f ib r i l a c iones y fasc i cu lac iones . La lesión de la segunda m o t o n e u r o n a (en el asta anter ior de la médula espinal ) se caracter iza por afectación de músculos ais lados o pequeños grupos , ref lejos miotáticos d i s m i n u i d o s o ausentes y fasc icu lac iones y f ib r i l a c iones .
["3") Existen fundamenta lmente dos vías sensitivas: el sistema co lumna dorsal-lemnisco media l (sensibil idad epicrítica, se decusa a nivel del bu lbo y sus fibras hacen sinapsis en los núcleos de Go l l y Burdach) y el sistema anterolateral (sensibil idad protopática: dolor, temperatura y tacto grosero; las fibras cruzan al lado opuesto a nivel medular) .
["4"] La distribución de l déficit sensorial es i n d i c a t i v o de la localización de la lesión. N i ve l suspend ido para sensi b i l i d a d do lo rosa y térmica, c o n conservación de la táctil y p r o p i o c e p t i v a (déficit d i soc i ado de la sens ib i l idad ) , se ve en lesiones cen t romedu la res , c o m o la s i r i ngomie l i a .
Qfj En el síndrome cerebe loso v e r m i a n o , hay atax ia de la marcha y escasa o nu la atax ia de los m i e m b r o s ; en el síndrome cerebe loso hemisférico, l leva asoc iada ataxia de los m i e m b r o s y, c o n mayo r f recuenc ia q u e en el anter ior , dismetría, as inergia, d i sd i adococ ines i a y discronometría.
[~6"j Hay que pensar en lesión del t ronco encefálico siempre que aparezcan asociadas lesiones de pares craneales ip-silaterales con "vías largas" (motoras o sensitivas) contralaterales. Los pares craneales indican el nivel de la lesión.
["7") Los síndromes d isar t r ia-mano to rpe y ataxia-hemipares ia se pueden dar t an to en lesiones de la cápsula in ter na cont ra la tera l c o m o en las de p ro tube ranc i a .
f g ] En las lesiones bu lbares , se d i s t i nguen dos síndromes: lateral o de W a l l e n b e r g (oclusión de la arteria ver tebra l o cerebelosa postero in fe r io r ) y med ia l (oclusión de la arter ia espinal anter ior o vertebra l ) .
La neurología se caracteriza por una correlación precisa entre los signos y síntomas que presenta el paciente y
las estructuras anatómicas dañadas, de manera que la historia clínica, con una correcta exploración neurológica,
sigue siendo la base del diagnóstico.
Exploración neurológica
La exploración neurológica básica debe constar de los siguientes apartados: 1 . Nive l de consc iencia . 2. Explo
ración de las func iones superiores ( lenguaje, praxias, gnosias). 3. Pares craneales y campo visual . 4. Función
motora . 5. Función sensitiva. 6. Coordinación, estática y marcha.
(T) Preguntas
-MIR 09-10, 222, 224 • MIR 07-08, 52 • MIR 05-06, 53,54 - MIR 04-05, 54 -MIR 03-04, 28 -MIR 02-03, 132, 141,204 MIR 01-02, 221
- MIR 00-01, 238, 246 - MIR 00-01F, 66,213 -MIR 99-00, 197, 198 - MIR 98-99, 58, 229 - MIR 98-99F, 228 -MIR 97-98, 251
1.1. Breve recuerdo anatómico (Figura 1)
Hemisferios cerebrales
En cada hemisfer io se d is t inguen:
La corteza cerebral o sustancia gris, de unos 2 o 3 m m de espesor. Presenta numerosos pliegues que fo rman
las c i r cunvo luc iones cerebrales, surcos y fisuras y de l im i tan áreas con func iones determinadas, d iv id idas
1

Manual CTO de Medicina y Cirugía, 8. a edición
en cuatro lóbulos, que se denominan f ronta l , parietal , tempora l y occ ip i t a l . Los lóbulos frontal y parietal están separados por la cisura de Rolando. La cisura par ie toocc ip i ta l separa el lóbulo parietal del occ ip i t a l , y el lóbulo tempora l se encuentra por debajo de la cisura de Si lv io. La sustancia b lanca, fo rmada por sistemas de fibras que conectan entre sí diferentes puntos de la corteza cerebral o la corteza con los distintos núcleos del neuroeje, f o rmando la cápsula interna, la cápsula externa y la cápsula extrema. Cuerpo cal loso, fo rmado por fibras que interconectan ambos hemisferios.
Figura 1. Anatomía del sistema nerv ioso centra l
Diencéfalo
• Tálamo: núcleo de sustancia gris loca l izado en la zona media l del cerebro, a ambos lados del tercer ventrículo. Es el centro de integración de las señales sensoriales en su camino hacia la corteza.
• Hipotálamo: encargado de la regulación de las func iones viscerales: homeostasis, c i c l o sueño-vigilia, cont ro l endocr ino , etc.
Ganglios de la base
Los núcleos grises del cerebro son formaciones de sustancia gris situadas en la p rox im idad de la base del cerebro. Son el núcleo caudado, putamen y pálido (los dos últimos const i tuyen juntos el núcleo lent i cular) . Entre estos núcleos se encuentran interpuestas dos láminas de sustancia blanca, l lamadas cápsula interna y cápsula externa.
Tronco del encéfalo
El t ronco del encéfalo está d i v i d i do anatómicamente en: mesencéfalo, protuberanc ia y bu lbo raquídeo. • Mesencéfalo: en él se pueden encontrar los núcleos de los pares
craneales III y IV, además de los tubérculos cuadrigéminos, el núc leo ro jo , y la sustancia nigra.
• Protuberancia o puente: donde se loca l izan los núcleos de los pares craneales V motor , V I , VI I y VI I I , y los pedúnculos cerebelosos me dios, que conectan el t ronco del encéfalo con el cerebelo.
• Bulbo raquídeo: en el que se pueden local izar los núcleos de los pares craneales IX, X, XI y XII, así c o m o los centros de cont ro l de las func iones cardíacas, vasoconstrictoras y respiratorias, y otras ac t i v i dades reflejas c o m o el vómito.
Cerebelo
Posterior al t ronco del encéfalo, se encarga de la vía motora indirecta, secuenciando las actividades motoras, rea l izando las correcciones necesarias en su realización y regulando el t ono postural y el equ i l i b r i o (Figura 2).
Hemisferios cerebelosos
Figura 2. Partes anatómicas del cerebelo
1.2. Alteraciones de las funciones superiores
Las alteraciones de las func iones superiores traducen afectación de la sustancia gris cor t i ca l .

Neurología y neurocirugía
Trastornos del lenguaje
Los trastornos del lenguaje son los siguientes: Disartria. Es un trastorno específico de la articulación del lenguaje en el que las bases del m ismo (gramática, comprensión y elección de la palabra) están intactas.
• Disprosodia. Es una alteración en las i n f lexiones y r i tmo del habla, resultando un discurso monótono.
• Afasia. Es una pérdida o deter ioro del lenguaje causado por daño cerebral , con integridad de las estructuras neuromusculares formadoras del mismo. Responde a lesiones en el hemisfer io dominante . En cuanto a la dominancia cerebral, aprox imadamente el 9 0 % de la población es diestra; de ellos, más del 9 9 % presentan una fuerte dominanc ia hemisférica izquierda para las funciones lingüísticas. Por e l lo , en pacientes diestros, prácticamente sólo las lesiones hemisféricas izquierdas causarán afasia. Las personas zurdas presentan un patrón de dominanc i a hemisférica dist into, de fo rma que las func iones lingüísticas se encuentran representadas en ambos hemisferios; en ellos, lesiones en cualquier hemisfer io pueden dar lugar a afasia, pero son menos graves que las que aparecen con las mismas lesiones en pacientes diestros y t ienen mayor grado de recuperación func iona l . El área anterior del lenguaje está re lac ionada con la producción lingüística, corresponde al área 44 de Brodmann y se conoce c o m o "área de Broca". El área de Broca corresponde a la pars opercular o pars tr iangular de la circunvolución frontal infer ior. El área posterior del lenguaje, t rad ic iona lmente refer ido c o m o "área de Wern i cke " , es el área cort ica l re lac ionada con la comprensión del lenguaje hab lado. El área de Wern i cke corresponde al gyrus supramarginales o parte posterior de la circunvolución tempora l superior.
Fluencia Comprensión Nominación Repetición
BROCA No Sí No No
WERNICKE Sí No No No
CONDUCCIÓN Sí Sí No No
GLOBAL No No No No
TRANSCORTICAL MOTORA No Sí No Sí
TRANSCORTICAL SENSITIVA Sí No No Sí
Tabla 1. Diagnóstico diferencial de las afasias
Estos c inco t ipos de afasia se d i ferenc ian a tendiendo a los conceptos de f luencia , comprensión, nominación y repetición (Tabla 1). • La fluencia es la producción verbal durante la conversación. La afasia
no f luente se caracteriza por una escasa producción verbal (menos de 50 palabras por minuto) , pobre articulación, tendencia a frases cortas (a menudo una única palabra) y agramatismo (una organización anómala de las frases). La afasia fluente se caracteriza por una producción verbal normal o excesiva (100-200 palabras por m i n u to), ausencia de disartria, longitud normal de la frase y una ausencia de conten ido lingüístico en lo que se está hablando; es frecuente la sustitución de unas palabras por otras (parafasias), neologismos, o incluso presentar un lenguaje del todo in inte l ig ib le (jergafasia). Casi sin excepciones, las afasias no fiuentes se deben a lesiones anteriores a la cisura de Rolando, y las fiuentes son posteriores a dicha cisura.
• La comprensión se refiere al en tend imiento del lenguaje hablado, y se valora por órdenes verbales (cerrar los ojos, abrir la boca, etc.) o por preguntas que requieran la contestación sí o no.
• La nominación es la capac idad del paciente para reproduc i r los nombres de objetos, una parte de los mismos o su color , cuando son presentados por el examinador . Se pierde en todas las afasias.
• La repetición es la capac idad para repetir el lenguaje hablado, bien sean palabras o frases. Se conserva en las afasias transcorticales.
Tipos de afasia
Se pueden dist inguir c inco t ipos de afasia (Figura 3): afasia de Broca o motora , afasia de Wern i cke o sensitiva, afasia de conducción, afasia transcort ical motora y afasia transcort ical sensitiva, que corresponden a lesiones en distintas áreas cerebrales.
Afasia transcortical motora Afasia de conducción
Afasia motora o de Broca Afasia sensitiva Afasia transcortical o de Wernicke sensitiva
Figura 3. Localización anatómica de las pr incipales t i pos de afasia
Los pacientes con afasia de Broca presentan incapac idad para emit i r lenguaje, (tanto hablado c o m o escrito, supr imir esta frase), con c o m prensión conservada. Se produce por lesión en el área de Broca en el lóbulo frontal dominante , y puede ser aislada o bien impl icar hemipa-resia derecha u otros signos de lesión f ronta l , además de depresión o frustración, al ser conscientes de su déficit. El pronóstico de recuperación es generalmente más favorable que en aquéllos con afasia g loba l .
Las afasias de Wernicke no suelen asociarse a otras focal idades neuro log ías . Los pacientes no comprenden , y a su vez, presentan aumento de la f luenc ia , inc luso verborrea, con abundantes parafasias. No son conscientes de su prob lema lingüístico. T ienen peor pronóstico de recuperación, aún con terapia intensiva.
La afasia de conducción puede darse con lesiones del fascículo arcuato, y la que afecta a la ínsula, córtex auditivo adyacente y sustancia blanca subyacente. La comprensión está conservada, pero el paciente presenta d i f i cu l tad para nominar y repetir, con lenguaje fluente y abundantes parafasias.
Las afasias transcorticales motora o sensitiva t ienen las mismas características que las afasias motoras o sensitivas puras correspondientes, pero se caracterizan por conservar la capacidad de repetición. Se producen por infartos extensos en las zonas de vascularización frontera de las grandes arterias cerebrales. Las causas más frecuentes de afasias transcorticales son: anoxia secundaria a parada cardiorrespiratoria, obstrucción o estenosis signif icativa de la arteria carótida, intoxicación por monóxido de carbono o demencia .
3

Manual CTO de Medicina y Cirugía, 8. a edición
RECUERDA H a b i t u a l m e n t e sólo se afecta el lenguaje en lesiones del hemisfer ic m i n a n t e ( i zqu i e rdo , en pac ientes diestros) : el de recho es un hemis fe r io más "práct ico" (apraxia de la construcción y el ves t ido , recuerda m e l o días), mientras q u e el i z q u i e r d o es más " i n t e l e c t u a l " (habla, lee, escr ibe, suma, interpreta part i turas) .
La afasia global es la f o rma más grave y f recuente de afasia, secundaria a grandes lesiones que afectan a las áreas anter iores y poster iores del lenguaje y con l l e van déficit motores graves. Tales lesiones suelen responder a oc lus iones de la arter ia carótida interna o arter ia cerebral med ia i zqu ie rda en su o r igen . El pronóstico de recuperación es ma lo .
Agnosias
La agnosia es la incapac idad para reconocer un estímulo v isual , táctil o aud i t i vo cuando no hay alteración en la compresión ni defectos en las sensibi l idades pr imarias visuales, sensitivas o audit ivas. Reflejan un prob lema a nivel cor t i ca l .
Tipos de agnosias
La agnosia visual se def ine c o m o la incapacidad para conocer los objetos o estímulos que se presentan en el campo visual de un paciente alerta, atento, no disfásico y con una percepción visual normal . Una variante es la prosopagnosia o incapacidad para reconocer rostros humanos previamente conocidos o aprender nuevos. La simultanagnosia es la incapaci dad para percibir dos estímulos de forma simultánea. Ambas responden a lesiones occipitales bilaterales en las áreas de asociación.
La agnosia táctil es, análogamente a la v isual , la incapac idad para reconocer el s igni f icado de estímulos táctiles cuando la sensibi l idad táctil pr imar ia es normal en un paciente alerta y no disfásico. El paciente será incapaz de reconocer un objeto por el tacto con ojos cerrados, aunque sí describirá sus características de forma, tamaño o consistencia: es la astereognosia, que habi tua lmente responde a lesiones en la porción anter ior del lóbulo parietal contralateral .
La atopognosia es la impos ib i l i dad para local izar un estímulo táctil, y la agrafoestesia es la incapac idad para reconocer una determinada f igura trazada sobre la superf ic ie corpora l .
Otras formas de agnosia son la asomatognosia, o falta de reconoc imiento de partes del cuerpo como propias (generalmente hemicuerpo izquierdo), y la anosognosia, o incapacidad para reconocer su enfermedad; en estos casos, el paciente no reconoce su hemiparesia u otro defecto neurológico que acontece (p. e j . : síndrome de Antón en pacientes con afectación occipi ta l bilateral que niegan su ceguera cortical) . Ambas suelen responder a lesiones parietales no dominantes (derechas), aunque también pueden observarse en lesiones izquierdas.
Apraxias
La apraxia es la incapac idad para llevar a cabo actos motores ante una orden verbal o imitación en un paciente con una adecuada c o m p r e n sión y sin déficit motores o sensitivos pr imar ios que interf ieran con el desarrol lo del mov im ien to .
Tipos de apraxias
Las más importantes son las siguientes: • Apraxia ideomotora: es el t ipo más común de apraxia. Consiste en
la incapac idad para desarrollar un acto motor prev iamente aprend i do en respuesta a una orden verbal . Aparece con lesiones en áreas frontales y parietales izquierdas.
• Apraxia ideatoria: es la incapac idad para llevar a cabo una secuencia ordenada de actos motores (p.ej.: encender un c igarr i l lo ) a pesar de poder realizar cada acto por separado de fo rma correcta. Genera lmente, se ve en enfermedades cerebrales bilaterales. Puede o b servarse en demencias t ipo A lzhe imer y en estados confusionales.
• Apraxia constructiva: es la incapac idad para d ibu jar o construir f i guras o formas b id imensionales o t r id imensionales . Se explora sol i c i tando al paciente que efectúe sobre un papel la cop ia de varios d ibujos mode lo (círculo, cubo , etc.). Aparece más frecuentemente con lesiones hemisféricas derechas. Los pacientes con lesiones de rechas pierden las relaciones espaciales entre las distintas partes del mode lo , y aquéllos con lesiones izquierdas, los s impl i f i can notab le mente.
• Apraxia del vestido: es la incapac idad del paciente para vestirse de forma correcta cuando se le entregan las distintas piezas del vestuar io. Se ve más frecuentemente en lesiones par ietooccipi ta les derechas o bilaterales.
• Un t i p o especial es la apraxia de la marcha , en donde el pac i en te en posición bípeda no es capaz de in ic ia r la deambulación por haber pe rd i do los patrones motores aprend idos para caminar . Sin embargo , en decúbito, puede realizar la dinámica de deambulación. Característicamente, aparece en la hidrocefalia normo-tensiva (junto a incontinencia urinaria y demencia) y en lesiones frontales bilaterales.
• Apraxia bucolinguofacial: es la incapac idad para abrir o cerrar la boca o los ojos cuando se lo indica el examinador , aunque lo puede hacer de forma espontánea.
1.3. Trastornos de la función motora
Fisiología de la función motora
La función motora está sometida a un cont ro l muy estrecho en el que interv ienen distintas partes del Sistema Nerv ioso Central (SNC). Entre ellas, es necesario menc ionar la corteza motora , los ganglios básales, el cerebelo y la médula espinal .
Sistema piramidal
Las neuronas de la capa cort ica l V de la corteza motora pr imar ia (área 4 de Brodmann) , del área motora suplementar ia (en la cara media l del hemisfer io) , de la corteza premotora (rostral a la corteza motora p r ima ria) y de la corteza somatosenslt iva poscentral , emiten axones para el sistema p i r amida l . Este sistema está fo rmado por dos tractos pr incipales, el haz cort icoespinal y el haz cor t i conuc lear o fascículo gen icu lado.
Ambos están compuestos por dos neuronas motoras: la pr imera moto-neurona, que se or ig ina en la corteza y cuyas fibras descienden por la cápsula interna (MIR 09-10, 224) hasta el asta anter ior de la médula o
4

Neurología y neurocirugía
hasta los núcleos motores de los pares craneales, respect ivamente, y la segunda motoneurona , que se ext iende hasta la f ibra muscular.
Fascículo geniculado: recibe este nombre por su situación en la cápsula interna (genus = rodi l la ) y se encarga del contro l vo lun ta r io de la musculatura inervada por los pares craneales.
• Haz corticoespinal: discurre por el brazo posterior de la cápsula i n terna, y a su vez pueden diferenciarse dos tractos a partir del bu lbo raquídeo: - Tracto corticoespinal lateral (TCEL) : es c ruzado y discurre por la
región dorsal del cordón lateral de la médula. Sus axones t e r m i nan pr inc ipa lmente en las neuronas motoras del asta anter ior de la cara dorsolateral (que proyectarán, a su vez, a la musculatura distal).
- Tracto corticoespinal anterior o ventral (TCEV) : ipsi lateral, d is curre por la región media l del cordón anterior. Sus axones term inan en las de la cara vent romedia l (musculatura axial) (MIR 0 0 - 0 1 , 238) .
La lesión de las neuronas motoras cort icales o del haz p i ramida l , tras una fase de shock medular in ic ia l con parálisis f lacc ida, te rmina en una parálisis espástica con h iperact iv idad de los reflejos tendinosos. La espasticidad depende de la pérdida de la inhibición de las p royecc io nes bulboespinales (acompañantes del haz cort icoespinal ) , ya que en animales de experimentación se ha c o m p r o b a d o que la sección exc lu siva del haz p i ramida l no conduce a espasticidad (MIR 98-99F, 228) . Cuando ésta se encuentra establecida, se puede abol i r por la sección de las raíces dorsales al in ter rumpir el arco miotático. La lesión de las neuronas motoras del asta anterior cursa con una parálisis f lacc ida e h ipoac t i v idad de los reflejos tendinosos.
Fisiología de la placa motora (Figura 4)
La acet i lcol ina es el neurotransmisor empleado en la placa motora (unión neuromuscular) , ya que ésta no es sino un mode lo de sinapsis química. La acet i lcol ina se sintetiza en el citosol de la segunda motoneurona y se almacena en vesículas en el terminal presináptico. En reposo, se l iberan espontáneamente pequeñas cantidades, dando lugar a potenciales de placa en miniatura. Cuando un potencial de acción recorre el axón y alcanza la terminación presináptica, la despolariza, abriéndose canales de calc io regulados por voltaje y entrando grandes cantidades de d i cho ion a la terminación. El ca lc io atrae vesículas de acet i lcol ina y provoca su exocitosis (salen unas 125 vesículas). La acet i lcol ina se une a sus receptores, situados en la membrana muscular subyacente a la terminación axonal y cuya estructura es la de canales iónicos.
Célula de Schwann
Figura 4. Placa m o t o r a
Tras la apertura de los canales, se produce la entrada masiva de sodio a favor de gradiente (en el inter ior de la membrana muscular, el potencia l es de unos -80 mV) . De este m o d o , hay un c amb io local en el potencia l (pasa de -80 a + 6 0 mV) , denominado potencia l de placa motora , que se transmite a la f ibra muscular generando un potencia l de acción muscular y la contracción muscular.
La acet i lco l ina desaparece con rapidez de la hendidura sináptica por la presencia de la enz ima aceti lcol inesterasa, y por difusión fuera de d icha hendidura . Se considera que la sinapsis ha f ina l izado cuando se recapta el neurotransmisor l iberado.
Cerebelo y ganglios básales
Ambos fo rman parte de la vía motora indirecta, no consciente, reciben estímulos cort icales y modu l an la función del tracto p i ramida l a través del tálamo. Son sistemas comple jos y multisinápticos.
Básicamente, el cerebelo ayuda a secuenciar las act iv idades motoras y a efectuar las adaptaciones correctoras de estas actividades según se real izan. Además, interviene en la regulación de la postura y del equ i l i b r i o .
Los gangl ios básales, sin embargo, cont r ibuyen a p lani f icar y regular los patrones comple jos de mov im i en to muscular , mediante el cont ro l de la intensidad relativa de mov imientos , de la dirección y de la secuencia de mov imien tos necesarios.
Trastornos motores
Una vez vista la fisiología de la función motora , el siguiente paso es el estudio de los trastornos motores def ic i tar ios (paresias o parálisis). El déficit de fuerza se cuant i f ica c o m o f igura en la Tabla 2.
No contracción ni m o v i m i e n t o . Parálisis comp le t a
Contracción pero no m o v i m i e n t o
M o v i m i e n t o pero no contra gravedad
M o v i m i e n t o contra gravedad pero no contra resistencia
M o v i m i e n t o contra resistencia pero meno r que la ex t r em idad contra-lateral o la esperada para la edad
Fuerza no rma l
Tabla 2. Cuantlficación del déficit de la fuerza motora
Otras alteraciones de la función motora , c o m o los trastornos extrapi-ramidales, las crisis comic ia les motoras, los trastornos de la c o o r d i nación, así c o m o las ataxias y la apraxia o trastorno no paralítico del mov im i en to serán tratadas más adelante en este capítulo.
Las parálisis pueden derivar de: • Lesiones de la vía p i ramida l a nivel cort icoespinal o cor t i cobu lbar
(pr imera motoneurona) . • Lesiones de la motoneurona del asta anter ior medular y de los nú
cleos motores troncoencefálicos (segunda motoneurona) .
5

Manual CTO de Medicina y Cirugía, 8. a edición
Lesiones del nerv io periférico. Lesiones de la placa neuromuscular (miastenia gravis, síndrome miasténico de Eaton-Lambert, bo tu l i smo, etc.). Miopatías (Figura 5).
1. a motoneurona (piramidal)
Vía corticoespinal medial (10%)
Decusación
Vía corticoespinal lateral (90%)
Nervio periférico
2. a motoneurona (asta anterior)
Placa motora
Músculo
Figura 5. Vías motoras
A nivel clínico es de gran impor tanc ia la diferenciación entre lesión de pr imera y de la segunda motoneurona (Tabla 3).
1 . a MOTONEURONA 2 . a MOTONEURONA
Reflejos o s t e o t e n d i n o s o s
Vivos D isminu idos o ausentes
Respuesta c u t a n e o p l a n t a r Extensora (Babinski) Flexora
Músculo • Amp l i os g rupos
musculares • Atrof ia por desuso
• Músculos aislados o pequeños g rupos
• Amio t ro f i a precoz • Fasciculaciones • Fibri laciones
Tono A u m e n t a d o (parálisis espástica) D i sm inu ido (parálisis f laccida)
Tabla 3. Diagnóstico diferencial de las lesiones de primera y segunda motoneurona
Las lesiones de primera motoneurona (córtex cerebral, sustancia b lanca subcortical, cápsula interna, vía piramidal troncoencefálica y medular) producen parálisis de amplios grupos musculares, sin afectar nunca a músculos individuales. No suelen cursar con amiotrof ia i m portante, salvo la derivada del desuso en fases muy evolucionadas. No hay fasciculaciones ni f ibri laciones. Los reflejos miotáticos están exal tados y la respuesta cutaneoplantar es extensora (signo de Babinski).
Hay un aumento del tono muscular en "hoja de navaja", consistente en una mayor resistencia a la movilización pasiva de los miembros, máxima al iniciar el desplazamiento y que cede progresivamente, d i ferente de la rigidez secundaria a lesiones extrapiramidales, donde la resistencia a la movilización pasiva es constante a lo largo de todo el rango de mov imiento (rigidez plástica o en barra de plomo) . En las lesiones corticales, la distribución del déficit motor más co mún es la hemiparesia fac iobraqu iocrura l contralateral a la lesión. Las lesiones bilaterales de la vía p i ramida l cor t icoespina l , crónicamente establecidas, conducen al cuadro de parálisis pseudobulbar , caracter izado por disartria, disfagia, disfonía, parálisis facial b i la te ral y lab i l idad emoc iona l (risa y l lanto inapropiados) . Las lesiones troncoencefálicas asocian a la hemiparesia contra la teral clínica de pares craneales del lado de la lesión (hemiplejías cruzadas) (MIR 05-06, 54) . Las lesiones medulares cursan con paraparesia o tetraparesia, según la localización lesional. Cuando la paraplej ia se instaura de forma aguda, la etiología más frecuente son los traumat ismos; otras causas son isquemia y sangrado medular por mal formac iones . Las m i e l i tis desmiel in izantes (enfermedad de Devic) , abscesos y hematomas epidurales, t ienden a desarrollarse más lentamente, a lo largo de horas o días. Las paraplejias subagudas o crónicas suelen deberse a espondilosis cervical en personas mayores, o a esclerosis múltiple en jóvenes; otras causas son la degeneración subaguda comb inada de la médula, tumores medulares, men ingomie l i t i s sifilítica o lesiones frontales bilaterales parasagitales. Las lesiones de segunda motoneurona p roducen parálisis que puede afectar a pequeños grupos musculares, e inc luso músculos aislados. Cursa con amiotrof ia importante , fasciculaciones y f ibr i lac iones. El tono muscular está d i sm inu ido y los reflejos miotáticos son hipoac-tivos o ausentes. La respuesta cutaneoplantar es f lexora. La clínica de segunda motoneurona no acompañada de trastornos sensitivos habla a favor de lesión central (p. e j . : en la fo rma de atrofia muscu lar espinal de la ELA); la asociación de trastornos sensitivos apunta a lesión del nerv io periférico. Una de las causas más frecuentes de tetraparesia f lacc ida y arrefléxica, sin apenas alteraciones sensit i vas y con posib le afectación de pares craneales, es el síndrome de Guillain-Barré.
RECUERDA Hay q u e cons iderar si hay síntomas asociados para or ientar el diagnóst i c o , por e j e m p l o : • 1 . a m o t o n e u r o n a + afasia = cor teza m o t o r a . • 1 . a m o t o n e u r o n a + pares craneales contra latera les (síndrome c ruzado )
= t r o n c o del encéfalo. • 2 . a m o t o n e u r o n a aislada = médula o raíz moto ra . • 1 . ' m o t o n e u r o n a + déficit sensi t ivo = ne rv io periférico. • 1." motoneurona + 2. a motoneurona = esclerosis lateral amiotrófica (ELA).
1.4. Trastornos de la sensibilidad ( F igura6)
Sensibilidad somática
Los sentidos somáticos son los mecanismos nerviosos que recogen i n formación sensorial del cuerpo y se d i ferenc ian de los sentidos especiales, que son: vista, oído, o l fa to , gusto y equ i l i b r i o , cuya fisiología se estudia en otros apartados de esta obra. El resto de los sentidos somáticos se pueden clasif icar en tres:
6

Neurología y neurocirugía
Termoalgésica (dolor y temperatura ) Sensibilidad vibratoria, posicional y propiocept iva
Figura 6. Vías sensitivas
• Sentidos mecanorreceptores somáticos. Sensaciones táctiles y de posición, que se est imulan por el desplazamiento mecánico de a l gún te j ido corpora l .
• Sentidos termorreceptores. Detectan frío y calor. • Sentido algésico o del dolor.
Receptores sensoriales
Para detectar los estímulos, existen una serie de receptores sensoriales que son los encargados de captar estos estímulos, que poster iormente se interpretarán en el SNC para dar lugar a la percepción.
Los receptores sensoriales pueden ser: • Receptores sensoriales primarios. En este caso, son las propias ter
minac iones nerviosas las que actúan c o m o sensores. Receptores sensoriales secundarios. Const i tuidos por células especial izadas neurales o no neurales, que actúan c o m o transductoras del estímulo a la neurona sensorial pr imar ia a través de mecanismos sinápticos.
Entre las propiedades de los receptores sensoriales, cabe destacar dos pr inc ipales : la descarga repetit iva y la adaptab i l idad o fatiga. La p r ime ra hace mención al hecho de que cuanto mayor es la intensidad del estímulo, mayor es la f recuencia de descarga de los potenciales de acción (ya que no pueden aumentar en intensidad; recuérdese la ley del
todo o nada). La segunda (adaptabi l idad) imp l i ca que cuando se apl ica un estímulo de fo rma constante, los receptores se adaptan de forma total o parc ia l , pasado c ierto t i empo , de forma que responden con una frecuencia de descarga cada vez más lenta hasta que, f ina lmente , se reduce al mínimo o desaparece.
Vías sensitivas del SNC
Los estímulos del cuerpo se detectan en los diversos receptores espec ia l izados (corpúsculos de Pacini , de Meissner, t e rminac iones de Ruf f in i , amielínicas, etc.) y l legan a la médula por las raíces dorsales de los nervios raquídeos. Desde allí, pueden seguir f u n d a m e n t a l m e n te dos vías: • El s is tema co lumna dorsal - lemnisco media l . C o n d u c e n i m p u l
sos l l amados epicríticos o de discriminación f ina y v i b r a t o r i a . Suben por las c o l u m n a s poster iores de la médula ips i l a te ra l , ha c i e n d o su p r i m e r a s inapsis en los núcleos bu lbares de C o l l y Bu rdach , y c r u z a n d o a n i ve l de l b u l b o al l ado opues to , f o r m a n d o el l e m n i s c o med i a l y a c a b a n d o en el tálamo (núcleo vent ra l pos te ro la te ra l ) . Es una vía de conducc ión m u y rápida y presenta un a l to g rado de orientación espac ia l c on respecto al o r i g e n del estímulo. El sistema anterolateral. La sensibi l idad que conduce se denomina protopática, con capac idad de diversas modal idades: do lor , t emperatura y sensaciones de tacto grosero Tiene su pr imera sinapsis en las astas dorsales de la sustancia gris medular y, tras cruzar al lado opuesto de la médula, asciende por las co lumnas blancas anteriores y laterales (fascículo espinotalámico lateral), para terminar en todos los niveles del t ronco , y también en el núcleo ventral posterolateral del tálamo. Es un sistema más lento, con menor grado de or ienta ción espacial (MIR 97-98, 251) .
Desde el tálamo, se d i s t r ibuyen hacia la cor teza sensorial (tercera neurona que p royec ta al córtex par ieta l ) , d o n d e existe una repre sentación sensit iva de l cue rpo , el l l a m a d o homúnculo sens i t ivo de Penf ie ld .
La sensibi l idad de la cara es t ransmit ida por el V par craneal (trigémino) . La segunda neurona cruza la línea media en el t ronco y se i n corpora a la vía espinotalámica en posición media l , para encontrarse con la tercera neurona en el tálamo (núcleo ventral posteromedial ) y proyectar hacia el córtex par ieta l .
Clínica
La disfunción sensitiva se clasif ica en dos grupos (Figura 7): • Síntomas positivos: parestesias (percepc iones de sensaciones
anómalas sin apl icación de un estímulo aparente) y disestesias (sensación anómala tras apl icación de un estímulo). En estos ca sos, no se suele ob je t i va r en la exploración un déficit sensoria l demos t rab le .
• Síntomas negativos: con la demostración en la exploración de hi-poestesia (disminución de la percepción) o anestesia (ausencia comple ta de percepción).
La distribución de los déficit sensoriales es indicat iva de la localización lesional dentro del sistema nervioso.
La aparición de hipoestesia, disestesias y parestesias a nivel distal en miembros , con distribución en guante y calcetín, es indicat iva de po li neuropatía.
7

Manual CTO de Medicina y Cirugía, 8. a edición
Cuando aparece un déficit (generalmente un nivel suspendido) para la sensibi l idad dolorosa y térmica con conservación de la táctil y pro-piocept iva , se habla de déficit disociado de la sensibilidad; es típico de lesiones centromedulares (s ir ingomiel ia) pero también puede aparecer en algunas formas de po l i neuropatía leprosa, ami lo ide , diabética y neuropatía sensitiva hereditar ia. Las lesiones medulares también dan niveles sensitivos cuya d i s t r ibu ción es indicat iva del nivel lesional.
Las lesiones talámicas afectan a todas las sensibil idades del hemicuer-po contralateral , inc lu idas las de la cara. A veces, estas lesiones talámicas evo luc ionan para p roduc i r un cuadro de do lor o hiperpatía en el hemicuerpo afecto (síndrome de Déjerine-Roussy).
Las lesiones corticales parietales o de las proyecciones ta lamocort i-cales producen una afectación de las denominadas sensibil idades combinadas , con conservación relativa de las pr imarias (tacto, do lor y temperatura) . Hay pérdida de la discriminación entre dos puntos, ato-pognosia, extinción parietal (frente a dob le estimulación simultánea en áreas corporales simétricas, no se perc ibe la del lado afectado, genera l mente el hemicuerpo izqu ierdo , ya que responden a lesiones parietales derechas), agrafoestesia y astereognosia.
ATAXIA
SENSITIVA CEREBELOSA VESTIBULAR
a
01 o
HEMISFÉRICA VERMIANA CENTRAL
\ • Neuropatí periférica
• S. tabético
Tumoral
Vasular
• Postinfeccioso • Deg. subaguda combinada medular Tóxico (alcohol)
• Espondilosis cervical
Marcha y miembros
Romberg (+)
• Fármacos (fenitoína)
Y t Miembros Marcha
• Ictus vertebrobasilar
• Esclerosis múltiple
•Tumores ángulo pontocerebeloso
PERIFÉRICO
J • Posicional
• Neuronitis
• Laberintitis
• Méniére
Romberg
Bipedestación y marcha
Romberg (+)
Figura 8.Tipos de ataxia y etiología
Médula Nervio Corteza o tálamo
Figura 7. Anatomía de las alteraciones de las sensib i l idad
1.5. Trastornos de la coordinación. Ataxias (Figura 8)
• Sistema de la sensibilidad propioceptiva consciente (nervio periférico - raíz posterior - cordones posteriores - l emnisco me d ia l - tálamo - corteza) .
• Sistema de la sensibilidad propioceptiva inconsciente (haces espinocerebelosos poster ior y anter ior - pedúnculos cerebelo-sos - cerebelo) .
• Cerebelo (vérmis - hemisfer ios cerebelo-sos).
• Sistema vestibular (canales semic i rculares - utrículo - sáculo).
Los dos pr imeros hacen llegar información sensorial a la corteza y cerebelo.
El cerebelo part ic ipa en la coordinación auto mática del mov im ien to , la regulación del t ono muscular y el manten im ien to del equ i l i b r i o . El sistema vest ibu lar está i m p l i c a d o en el m a n t e n i m i e n t o del e q u i l i b r i o , el t o n o mus
cu lar y la orientación en el espac io . Las ace lerac iones l ineales son registradas por las máculas de utrículo y sáculo, y las ace lerac iones angulares por las crestas ampular is de los canales semic i rcu lares (para más detal les véase la Sección de Otorrinolaringología).
Se def ine la ataxia c o m o t o d o trastorno de la coordinación que, sin de b i l i dad motora y en ausencia de apraxia, altera la dirección y amp l i t ud del m o v i m i e n t o vo lunta r io , la postura y el equ i l i b r i o .
Consideraciones anatómicas
Los sistemas neurológicos impl i cados en la coordinación motora son fundamenta lmente cuatro:
Tipos sindrómicos de ataxia
Atend iendo a los distintos sistemas neuronales descritos prev iamente, se pueden diferenciar tres t ipos sindrómicos de ataxia (véase Figura 8): 1) ataxia sensitiva, 2) ataxia cerebelosa y 3) ataxia o desequ i l ib r io vestibular. • Ataxia sensitiva. Se produce c o m o consecuencia de trastornos que
afectan a la vía prop iocept iva consciente a n ive l de nervio periférico, raíces posteriores, cordones posteriores o lemnisco media l en el t ronco encefálico. Afecta p redominantemente a la marcha y miembros inferiores de
8

Neurología y neurocirugía
forma simétrica. Es característica de la ataxia sensitiva la ausencia de vértigo, nistagmo o disartria, y prácticamente diagnóstico el c laro empeoramien to cuando el paciente cierra los ojos o ejecuta m o v i mientos en situaciones con escasa luminos idad . En posición bípeda, con ojos abiertos, hay un aumento de la base de sustentación, y el paciente puede llegar a caer si cierra los ojos (signo de Romberg). El signo de Romberg no aparece en las lesiones cerebelosas. Ataxia cerebelosa. La ataxia cerebelosa puede afectar a la bipedes-tación, marcha y miembros y, a di ferencia de la ataxia sensitiva, persiste aún con ayuda visual y no se agrava tan intensamente con el c ierre de los ojos. Se asocia a hipotonía, disartria, t emblor cinético y nistagmo (MIR 99-00, 198) . Se dist inguen dos síndromes cerebelosos: 1) síndrome vermiano, caracter izado por ataxia de la marcha y escasa o nula ataxia de m i e m bros, con infrecuente presencia de hipotonía, nistagmo, disartria o temblor . Es característico de la degeneración cerebelosa alcohólica y del medulob las toma en niños. 2) Síndrome hemisférico, que cursa con ataxia de los miembros ipsilaterales a la lesión e imp l i ca h ipo tonía, disartria, n istagmo y temblor . Ataxia vestibular. La ataxia o desequi l ibr io vestibular se caracteriza por un trastorno del equ i l i b r i o durante la bipedestación y marcha, sin incoordinación en los mov imien tos de los miembros cuando el paciente es exp lorado en decúbito. El vértigo y el nistagmo están típicamente asociados, y no hay disartria. Para diferenciar entre síndrome vertiginoso periférico (nervio vestibular y sistema laberíntico) y central (núcleos vestibulares y vías de conexión), se valorarán los síntomas asociados y la armonios idad o congruencia del trastorno del equ i l ib r io objet ivado en la exploración. El síndrome periférico se caracteriza por vértigo o sensación de giro de objetos, generalmente in f luenc iab le con los mov imien tos cefálicos y de corta duración, síntomas vegetativos intensos, acúfenos e hipoacusia uni la tera l . Hay nistagmo espontáneo hor izontorota tor io hacia un lado de la mirada e i nh ib ido por la fijación v isual . La desviación en el test de Romberg y en la marcha es en la misma d i rec ción y co inc ide con la dirección del componen te lento del nistagmo (hacia el lado que presenta hipoacusia) . El síndrome vestibular central se caracteriza por la disarmonía de las respuestas, y a menudo es incomple to (no conl leva todos los componentes) . El vértigo no se inf luenc ia tan marcadamente con los mov imientos de la cabeza, su intensidad es menos pronunc iada que el trastorno del equ i l i b r i o , los síntomas vegetativos son moderados, no hay hipoacusia ni acúfenos, y el nistagmo espontáneo suele ser
bi lateral y, a menudo , puro (puramente hor izonta l , rotator io o ver t i cal). En el test de Romberg, la caída es hacia atrás o hacia los lados y atrás. La inclinación durante la marcha no co inc ide con la dirección del componen te lento del nistagmo ni con la dirección de caída en el test de Romberg. Además, es frecuente la coexistencia de otros signos o síntomas de disfunción neurológica troncoencefálica.
1.6. Alteración de los pares craneales ( F i g u r a d
Parálisis de los pares craneales oculomotores
Las parálisis o paresias de los pares craneales ocu lomotores (nervios motor ocular común (III), patético (IV) y motor ocu lar externo (VI)) p ro ducen diplopía binocular. La diplopía monocu la r se observa en la luxa ción del cr ista l ino.
Localización de lesiones del III par craneal (núcleo motor ocular común) «MIR 99-00F, 62)
La lesión puede establecerse a nivel del núcleo, del fascículo, de la porción subaracnoidea, del seno cavernoso o de la fisura orb i tar ia . Clín icamente, cursa con deb i l idad de los músculos inervados (constrictor pupi lar , recto superior, inferior, interno y ob l i cuo menor) y ptosis (elevador del párpado ipsilateral), p roduc i endo diplopía vert ical u ob l i cua b inocular . La causa más frecuente es la mononeuropatía diabética (MIR 99-00F, 62) . • Las lesiones nucleares aisladas del III par son muy raras. Se m a n i
fiestan clínicamente por deb i l idad de todos los músculos inervados por el III par ipsilateral (constrictor pupi lar , recto superior, inferior, interno y ob l i cuo menor) , más deb i l idad del recto superior contra la teral y ptosis bi lateral incompleta . La porción subaracnoidea puede afectarse especialmente por lesiones compresivas (aneurismas y herniación uncal) , isquémicas (diabetes y vasculitis) o aracnoidit is básales. Las lesiones compresivas se
LOCALIZACIÓN CLINICA DE LA LESIÓN
III PC Mesencéfalo
Protubera
Midriasis arreactiva Alteración de la mirada vertical
Alteración de la mirada horizontal (los ojos se desvían al lado contrario a la lesión)
VII PC Protuberancia
XII PC Bulbo
Parálisis facial: (boca se desvía al lado sano) • Periférica: completa (superior + inferior)
• Central: respeta porción superior.
Paresia, amiotrofias y fasciculaciones de la lengua, que se desvía hacia el lado de la lesión
v p c Núcleo principal (protuberancia) Núcleo espinal (prot. y bulbo) Hemihipoestesia facial ipsilateral
Figura 9. Localización d e los pares craneales e n el t r o n c o d e l encéfalo
9

Manual CTO de Medicina y Cirugía, 8.a edición
caracterizan in ic ia lmente por midriasis arreactiva de la pupi la , seguida de deb i l idad de la musculatura extraocular. Las lesiones isquémicas respetan la pupi la , ya que están confinadas a la porción central del nervio, y las fibras pupi lomotoras se local izan periféricamente. En el seno cavernoso, la lesión del III par se suele asociar a lesión de otros pares craneales (IV y V I : o f ta lmop le j i a comple ta , la p r ime ra y segunda ramas del trigémino). A este n ive l , la pup i l a puede ser norma l , pero la asociación de un síndrome de Horner y paresia ocu lomoto ra comb inada es patognomónica de lesión en el seno cavernoso (MIR 02-03, 141).
• Por la fisura orbitar ia superior discurren los pares III, IV y VI y pr imera rama del V (oftálmica) y la vena oftálmica. A este n ive l , el III par se d i v ide en dos ramas: superior (para el recto superior y
elevador del párpado superior) e infer ior (para el recto infer ior, recto interno, ob l i cuo menor y gangl io c i l iar [fibras pup i lomotoras ] ) . Las lesiones a este nivel no afectan a la segunda rama del trigémino.
Localización de lesiones del IV par craneal (núcleo troclear)
RECUERDA Ptosis: • M ias ten ia gravis. • Síndrome d e Horner . • Lesión del III PC
tracraneal (secundaria a t umor o hipertensión intracraneal benigna) pueden cursar con una paresia del V I par. La afectación a nivel de la punta del peñasco del tempora l p roduce el síndrome de Gradenigo (paresia del VI par, do lor facial ipsilateral por afectación del trigémino y sordera) (MIR 99-00, 197).
RECUERDA El IV PC es el más largo y de lgado y además abandona el t r o n c o del encéfalo po r su cara poster ior . Por e l l o , la causa más f recuente de su lesión son los t raumat i smos craneoencefálicos. El V I PC rea l iza u n largo r e co r r i do a través del espac io subaracno ideo , de ahí q u e sea suscept ib le de lesionarse ante e levac iones de la presión in t racranea l .
Dado su trayecto dentro del seno cavernoso en relación con las fibras oculosimpáticas que rodean a la carótida interna, es de gran valor loca l izador la asociación de un síndrome de Horner y paresia ipsilateral del VI par (por aneurismas de carótida interna, fístulas carot idocavernosas, etc.).
Lesión del nervio trigémino o V par craneal
El núcleo del IV par se loca l iza en el mesencéfalo dorsal inferior. Su porción fascicular se decusa y emerge del t ronco en la línea media poster ior, para dir igirse hacia adelante recorr iendo el mesencéfalo lateral en la cisterna perimesencefálica. Penetra en la pared lateral del seno cavernoso y alcanza la órbita a través de la fisura orbi tar ia superior para inervar el músculo ob l i cuo mayor contralateral .
La parálisis del IV par produce clínica de diplopía vert ical que aumen ta al mirar hacia abajo y al lado opuesto de la lesión. Los pacientes presentan, característicamente, desviación de la cabeza hacia el lado opuesto a la lesión, ya que la inclinación hacia el m i smo lado aumenta la diplopía (test de la inclinación cefálica de Bielschowsky).
La causa más frecuente de afectación uni lateral o bilateral del IV par son los traumat ismos craneales, especia lmente frontales. La segunda causa en frecuencia es la neuropatía isquémica por enfermedad de pequeño vaso (diabetes, mononeur i t i s múltiple, etc.).
Localización de lesiones del VI par craneal (núcleo motor ocular externo)
El núcleo del VI par se local iza en la protuberanc ia inferior, en íntima relación con la rod i l l a del fac ia l . Este núcleo presenta dos porc iones. De una de ellas se or ig ina el fascículo longitudinal medial, interneu-ronas que cruzan la línea media y ascienden para hacer sinapsis en el subnúcleo del recto interno del III par contra latera l , pe rmi t i endo de esta fo rma la mirada conjugada en el p lano hor izonta l .
La otra porción da lugar a las fibras del VI par p rop iamente dichas, que se d i r igen hacia adelante en la protuberanc ia y salen del t ronco para introducirse en el inter ior del seno cavernoso e inervar f ina lmente el recto externo, tras pasar por la fisura orb i tar ia superior. • La lesión del fascículo long i tud ina l media l p roduce la l lamada of
talmoplejia internuclear (parálisis de la aducción de un o jo con nistagmo en el o jo abducente) . Sus causas más frecuentes son la esclerosis múltiple y las lesiones vasculares. La porción subaracnoidea es muy susceptible de lesionarse por su largo recorr ido. Procesos tumorales o un aumento de la presión in-
El nervio trigémino inerva los músculos de la masticación y recoge la sensibil idad de la hemicara ipsilateral. Se compone de tres ramas: oftálmica, maxi lar y mandibular . La manifestación clínica más frecuente es el dolor en la hemicara ipsilateral. También puede cursar con hipoestesia de la hemicara ipsilateral, desviación de la mandíbula hacia el lado en fermo con debi l idad para la masticación y abolición del reflejo corneal .
Las causas más frecuentes son la infección por herpes zóster, la esclerosis múltiple y también la neuralgia idiopática.
Lesión del nervio facial o VII par craneal
El nerv io facial inerva los músculos de la mímica facia l , las glándulas lagr imal , submaxi lar y subl ingual , y los 2/3 anteriores de la lengua. La lesión periférica o nuclear produce deb i l idad de los músculos de la hemicara ipsilateral comple ta , de manera que al intentar elevar ambas comisuras, la boca se desvía hacia el lado sano, el paciente presenta frente lisa y d i f i cu l tad para cerrar el párpado ipsi lateral .
La lesión supranuclear (cortical) p roduce parálisis únicamente de la parte infer ior de la hemicara contralateral (la inervación de la parte inferior es contralateral , mientras que la inervación de la parte superior es bi lateral y, por tanto, está preservada). La parálisis facial bi lateral puede aparecer en el síndrome de Gulllain-Barré, en la enfermedad de Lyme y en la sarcoidosis.
Lesión del nervio estatoacústico u VIII par craneal
Está, a su vez, const i tu ido por dos nervios, el coclear y el vestibular. El nervio coclear es sensorial y transmite los estímulos audit ivos. El nervio vestibular interviene en la regulación del equ i l i b r i o y en la orientación en el espacio. La lesión del nerv io coclear produce tinnitus o acúfenos, así c o m o disminución de la agudeza audi t iva .
10

Neurología y neurocirugía
Lesión del nervio glosofaríngeo o IX par craneal
Inerva los músculos constr ictor superior de la far inge y estilofaríngeo, la sensibi l idad del terc io posterior de la lengua y de la orofar inge. Su lesión produce leve disfagia, pérdida de la sensibi l idad del terc io poster ior de la lengua, pérdida del reflejo faríngeo y desviación de la pared posterior hacia el lado sano (signo de la cort ina de Vernet) . Es muy rara su lesión aislada.
Lesión del nervio vago o X par craneal
Su lesión intracraneal p roduce disfagia, disartria, disfonía y anestesia laríngea. Es muy rara su lesión aislada.
Lesión del nervio espinal o XI par craneal
Es un nervio motor puro que inerva los músculos esternocleidomastoideo y trapecio. Su lesión produce debi l idad muscular ipsilateral a este nive l .
Lesión del nervio hipogloso o XII par craneal
Es un nerv io motor puro que inerva la hemi lengua contralateral (múscu lo geniogloso) . Su lesión produce hemiatrof ia ipsilateral de la lengua y desviación de ésta hacia el lado de la lesión.
1.7. Trastornos campimétricos y pupilares
gruentes, mientras que las lesiones próximas a la corteza occ ip i ta l p roducen defectos congruentes. La lesión de c int i l las ópticas, además de hemianopsia homónima contralateral , puede produc i r alteraciones en la react iv idad pupi lar .
Defectos campimétricos
Véase la Sección de Oftalmología.
• Las lesiones retinianas y del nervio óptico conducen a la aparición de escotomas. Las lesiones maculares producen escotomas centrales. La retinit is p igmentar ia característicamente produce una reducción concéntrica del c ampo v isual . Los defectos arcuatos responden a lesiones isquémicas del nervio óptico anterior, g laucoma y papi-ledema. Los escotomas centrales y cecocentrales son un signo de neuropatía óptica.
• Las lesiones quiasmáticas, hab i tua lmente compresivas por tumores hipof isar ios, craneofar ingiomas o aneurismas, dan lugar general mente a hemianopsias heterónimas o bi temporales. Más raras son las cuadrantanopsias bi temporales superiores o inferiores y la hemianops ia tempora l monocu la r .
• Las lesiones retroquiasmáticas (c inti l las, cuerpos geniculados, radiac iones ópticas y lóbulo occ ip i ta l ) dan lugar a defectos campimétricos homónimos cuya congruenc ia (s imi l i tud en cuanto al defecto campimétrico en cada ojo) está en función de lo anterior o posterior de la lesión. Las lesiones anteriores dan lugar a defectos incon-
UERDA La cuadran tanops ia b i t e m p o r a l super ior se p r o d u c e po r la compresión de las f ibras infer iores de l qu i a sma , y una de sus causas suelen ser los t u mores h ipof i sar ios . En c a m b i o , los c raneo fa r ing iomas , q u e c o m p r i m e n p r i m e r o las f ibras super iores, p r o v o c a n una cuadran tanops ia b i t e m p o r a l in fer ior .
Las lesiones de radiaciones ópticas no producen alteraciones pupila-res. La afectación de las radiaciones ópticas parietales produce una cuadrantanopsia homónima contralateral inferior, y cuando se afectan las temporales, se produce una cuadrantanopsia homónima superior. La lesión occ ip i ta l a nivel de la cisura calcar ina, genera lmente secundar ia a oclusión embólica de la arteria cerebral posterior, p ro duce una hemianopsia homónima contralateral congruente con respeto de la visión macular .
Alteraciones pupilares
Anisocoria esencial. Un 1 5-30% de la población normal t iene una di ferenc ia en el tamaño pupi lar de 0,4-1 m m con una react iv idad norma l a la luz. Defecto pupilar aferente relativo. Consiste en una disminución de la respuesta pupi la r constr ictora frente a un estímulo luminoso d i recto, con una respuesta normal si se est imula el o jo contralateral (respuesta consensual normal ) , e ind ica lesión del nervio óptico ip silateral (MIR 07-08, 52). Síndrome de Horner. Se produce por afectación de las fibras p u pilares simpáticas. La inervación simpática que di lata la pup i l a se or ig ina a nivel hipotalámico y desciende por el tegmento lateral troncoencefáiico hasta el núcleo intermediolatera l de la médula en los segmentos C8-D2. Desde aquí, pasa al gangl io cervical superior de la cadena simpática paravertebral y asciende con el p lexo pe-ricarotídeo, para incorporarse a la rama oftálmica del trigémino y alcanzar la pup i l a a través de los nervios cil iares largos. La lesión a cua lquiera de estos niveles puede produc i r un síndrome de Horner , que cursa con la tríada de ptosis, miosis y enofta lmos. A veces se suma anhidrosis facial (esto último cuando la lesión es previa a la bifurcación carotídea; si la lesión es posterior a la bifurcación, no hay anhidrosis) . La pup i l a responde adecuadamente a la luz y a los estímulos cercanos. La anisocor ia es mayor en la oscur idad y la p u pi la responde tanto a midriáticos c o m o a mióticos (MIR 03-04, 28). Lesión de las fibras pupilares parasimpáticas. Se or ig inan en el núc leo de Edinger-Westphal, loca l izado en la porción superior del núc leo del III par. Desde allí, discurren con las fibras del III par craneal hasta el gangl io ci l iar , loca l izado a nivel in t raorb i tar io y, a través de los nervios cil iares cortos, a lcanza el músculo constr ictor de la pup i l a . Las fibras parasimpáticas discurren en la periferia del III par, por lo que son muy sensibles a la patología compresiva (aneurismas, herniación uncal) . La lesión a cua lqu iera de estos niveles da l u gar a dilatación pupi lar sin respuesta a la luz. Cuando la dilatación pupi la r arreactiva se acompaña de una relativa preservación de la mot i l i dad ocular , la etiología suele ser compresiva en el espacio subaracnoideo. Las lesiones isquémicas del III par respetan la pup i l a in i c ia lmente (ya que la isquemia suele afectar a las fibras internas y, c o m o se ha comentado , las parasimpáticas se sitúan en la porción externa del III par).
11

Manual CTO de Medicina y Cirugía, 8.a edición
Pupila tónica de Adié. Se produce secundar iamente a lesión del gangl io c i l iar por causas locales (inflamación, infección o t raumat ismo) o c o m o parte de una neuropatía periférica o autonómica (síndrome de Cuillain-Barré, síndrome de Fisher, síndrome de Shy-Drager, ami lo idos is , neuropatía sensitiva hereditar ia, enfermedad de Charcot-Marie-Tooth, diabetes, a l coho l i smo o síndrome para-neoplásico). Es una pup i l a midriática, generalmente uni latera l , que no responde a la luz, y cuya respuesta frente a la visión cercana es lenta y tónica. La anisocor ia se hace más patente en condic iones de luminos idad . Responde tanto a midriáticos c o m o a mióticos. Puede acompañarse de mov imien tos vermiformes de los bordes del iris. Pupila de Argyll-Robertson. Es una afectación pupi la r bi lateral con pupi las pequeñas e irregulares que responden escasamente a la luz, pero conservan la acomodación para la visión cercana (disociación cerca-luz o DCL). Presenta respuesta adecuada a mióticos y escasa a midriáticos. Parece ser secundaria a una lesión mesencefálica rostral y característicamente se ve en pacientes con neurolúes. Otras causas de DCL son: sarcoidosis, diabetes, ami lo idos is fami l ia r , sínd rome de Adié, distrofia miotónica, h idrocefa l ia y tumores de la región p inea l . La DCL uni lateral se observa en lesiones del nervio óptico y retinianas ipsilaterales.
Figura 11 . Organización somatotópica de las áreas cort icales motoras y sensitivas.
1.8. Síndromes lobares (Figura k »
Lóbulo frontal ( M I R 0 4 - 0 5 , 5 4 )
• Las áreas motoras y premotoras están específicamente relacionadas con los mov imientos vo luntar ios y su lesión produce parálisis es-pástica contralateral (pr imera motoneurona) . Las áreas motoras p r i marias, al igual que las sensitivas, se organizan somatotópicamente de forma que áreas cort icales se corre lac ionan con áreas corporales específicas (Figura 11).
En el lóbulo f ronta l , se sitúa un centro de la mirada conjugada. Su lesión produce desviación oculocefálica conjugada hacia el lado de la lesión. Sin embargo, su irritación (crisis comicia les) desvía los ojos y la cabeza hacia el lado opuesto. La lesión del área motora suplementar ia dominan te ¡nicialmente produce mut i smo, para poster iormente evo luc ionar a afasia motora transcort ical . Cuando se afecta el área de Broca, aparece la afasia motora o no f luente. Lesiones más ampl ias en esta zona conducen al desarrol lo de agrafía y apraxia buco l inguofac ia l . La afectación bi latera l de las áreas frontales media les parasagita-
les conduce a un cuadro de apraxia de la marcha e i ncon t inenc i a ur inar ia .
• Las áreas prefrontales t ienen una función menos específica. Su lesión se ha re lac ionado con una ausencia de in ic iat iva y espontane idad (estado apático o abúlico), disminución de las relaciones
Área motora y premotora (parálisis espástica contralateral)
Centro de la mirada conjugada (desviación hacia la lesión)
Corteza prefronta l (mut ismo, abulia, mona,
reflejos arcaicos)
Área de Broca (afasia motora)
Corteza audit iva (alucinaciones audit ivas;
sordera cortical)
Radiaciones ópticas inferiores (cuadrantanopsia superior
contralateral)
Área de Wernicke (afasia sensitiva)
Corteza somatosensorial (hipoestesia contralateral)
Radiaciones ópticas superiores (cuadrantanopsia
inferior contralateral)
Corteza visual primaria (hemianopsia homónima contralateral con respeto macular; ceguera cortical)
Figura 10. Alteraciones de las funciones superiores y síndromes lobares
12

Neurología y neurocirugía
interpersonales, cambios en la personal idad (a veces con evidente desinhibición social , inestabi l idad e impu ls i v idad , especia lmente con lesiones frontales básales) y l igero deter ioro inte lectual , con au sencia de atención y concentración, incapac idad para anal izar los problemas y perseveración.
Lóbulo parietal
La afectación occ ip i ta l bi lateral produce : A) ceguera cort ica l por afectación de las áreas visuales pr imarias (cisuras calcarinas). Los pacientes con lesiones occipi ta les mediales extensas de carácter agudo y bilaterales con ceguera cort ica l pueden negar su ceguera (anosognosia visual) y confabular sobre lo que están v i endo ; es el síndrome de Antón; B) prosopagnosia; C) s imultanagnosia; D) sínd rome de Balint, que imp l i ca apraxia óptica (fal lo para d i r ig i r la m i rada en una dirección ante una orden , pudiéndolo hacer de forma espontánea).
• Las alteraciones sensitivas que aparecen c o m o consecuencia de la lesión del lóbulo parietal han sido descritas prev iamente (véase el apartado Síndromes sensitivos y agnosias) e inc luyen astereognosia, atopognosia, pérdida de la discriminación entre dos puntos, ex t in ción parietal , anosognosia y asomatognosia.
• El defecto campimétrico por lesión parietal es una hemianopsia homónima contralateral congruente , con c laro p r edomin i o en los campos inferiores (cuadrantanopsia homónima inferior por afectación de las radiaciones ópticas superiores).
• La apraxia construct iva y la del vest ido, así c o m o la anosognosia y la negl igencia hemicorpora l (asomatognosia), se observan más frecuentemente con lesiones parietales derechas, aunque también pueden aparecer en lesiones izquierdas. La lesión del lóbulo parietal dominan te conduce a la aparición de alexia, síndrome de Gerstmann (agrafía, alexia, aca lcul ia , agnosia digi ta l y desorientación derecha-izquierda), astereognosia b imanua l (agnosia táctil) y apraxia ideatoria e ideomotora (también pueden aparecer en lesiones frontales).
Lóbulo temporal
• Las lesiones del lóbulo tempora l dominan te producen cuadranta nopsia homónima superior por afectación de las radiaciones ópticas inferiores, afasia de Wern i cke o f luente, amusia ( incapac idad para leer y escribir música) y alteración en el aprendizaje del material verbal presentado por vía audi t iva .
• La lesión del lóbulo tempora l no dominan te produce el m ismo defecto campimétrico, alteración en las relaciones espaciales, deter io ro en el aprendizaje del material no verbal presentado por vía visual y una incapac idad para reconocer melodías.
• La lesión de cualquiera de los lóbulos temporales puede dar lugar a a luc inaciones e i lusiones audit ivas y compor tamien to psicótico con agresividad.
• La afectación tempora l bi lateral puede conduc i r a un síndrome amnésico de Korsakoff, síndrome de Klüver-Bucy (apatía, p lac idez, incremento en la act iv idad sexual y falta de reconoc imien to de o b jetos comestibles) y sordera cor t i ca l .
Lóbulo occipital
1.9. Síndromes troncoencefálicos
De manera general , hay que pensar en una lesión a nivel del t ronco del encéfalo siempre que aparezcan asociadas a lesiones de pares craneales ipsilaterales con "vías largas" (motor o sensitivo) contralaterales. Los pares craneales nos dan el nivel de la lesión (MIR 05-06, 54).
Dadas las numerosas vías y núcleos que con fo rman esta área encefál ica, conv iene d i v id i r los síndromes clínicos según las local izaciones anatómicas de la fo rma más exacta posible (Figura 12) (MIR 05-06, 53).
Síndromes mesencefálicos
• Síndrome de Weber: es un síndrome anterior que afecta a la vía p i ramida l y III par craneal, dando lugar a hemiparesia contralateral ( inc lu ida la cara) y paresia del III par del lado de la lesión con pupi la di latada arreactiva.
• Síndrome de Claude y Benedikt: afectan a III par y núcleo ro jo . Cursan con paresia del III par y temblor o ataxia.
• Síndrome mesencefálico dorsal o síndrome de Parinaud: suele ser secundario a tumores de la pineal o h idrocefa l ia (MIR 98-99, 58). El dato más característico es una parálisis de la mirada con jugada hacia arr iba, con pupi las genera lmente di latadas y acomodación c o n servada (fenómeno de disociación cerca-luz). Con los mov imien tos oculares en el p lano hor izonta l , el o jo que abduce puede moverse más lentamente que el adducente (pseudoparálisis del VI par).
Síndromes pontinos anteriores o ventrales
• Síndrome de locked-in o cautiverio: puede ser secundar io a mie l i-nólisis central pont ina (h iponatremia rápidamente recuperada), i n farto (trombosis de la basilar), tumor , hemorragia o t raumat ismo. Cursa con tetraplej ia y afectación de la mot i l i dad ocular hor izonta l . Sólo conservan la mot i l i dad ocular en el p lano vert ical y el parpadeo.
La lesión uni lateral p roduce una hemianopsia homónima contra-lateral congruente con respeto de la visión macular y puede cursar con a luc inac iones visuales elementales. Síndromes pontinos posteriores o dorsales
Q RECUERDA Las representac iones v isual y aud i t i va son bi latera les . Por eso, a u n q u e puede haber parálisis e hipoestesias de un so lo hemis fe r io , para que exista ceguera o sordera comp l e t a de o r igen co r t i c a l , son necesarias lesiones de a m b o s .
Síndrome de Foville: consiste en hemiplejía contralateral , parálisis facial ipsilateral y desviación conjugada de los ojos al lado opuesto de la lesión, con incapac idad para mirar hacia el lado de la lesión (ojos m i rando a la hemiplejía).
13

M a n u a l C T O d e M e d i c i n a y C i rugía , 8.a ed i c i ón
Síndromes bulbares
MESENCÉFALO • Parálisis de la mirada conjugada hacia arriba Síndrome de Parinaud • Dificultad para la convergencia y acomodado.
• Anisocoria y midriasis
Síndrome de Claude Ataxia contralateral (NR) Hipar
Síndrome de Benedikt ipsilateral Mov. anormales contralaterales (corea.temblor y balismo) (NR)
Síndrome de Weber • III par ipsilateral • Hemiparesia contralateral (VP)
PROTUBERANCIA
Síndrome de Millard-Gubler ' Hemiplejía contralateral respetando la cara (VP)
• Paresia del VI y VII pares ipsilaterales
BULBO Síndrome de Wallenberg • Hemihipoestesia facial ipsilateral (V par) • Hemihipoestesia corporal contralateral (ET)
(sd. sensitivo cruzado)
Además: síndrome vertiginoso, disartria y disfagia, diplopia, síndrome Horner ipsilateral y ataxia cerebelosa ipsilateral
Síndrome bulbar medial • XII par ipsilateral • Hemiplejía contralateral que respeta la cara (VP) • Ataxia sensitiva contralateral (LM)
VP: vía piramidal; LM; lemnisco medial; ET: vía espinotalámica; HR: núcleo rojo
Figura 12. Síndromes del t r o n c o de l encéfalo
Síndrome bulbar lateral o síndrome de Wallenberg (MIR 02-03, 204) : es secundar io a oclusión de la arteria vertebral o cerebelosa posteroinfer ior (PICA). Clínicamente, se caracter iza por : 1) sínd r o m e ver t ig inoso co n náuseas y vómitos por afectación de los núcleos ves t ibu la res; 2) disartr ia y disfagia por paresia de la cuerda voca l , far inge y ve lo del paladar ips i latera l , t o d o e l lo secundar io a lesión del núcleo a m b i g u o ; 3) diplopía, quizás secundar ia a la extensión de la lesión a la p ro tuberanc ia infer ior , donde se loca l iza el V I par; 4) hipoestesia fac ia l ips i la teral por afectación del núcleo t r igemi-na l ; 5) hipoestesia corpora l contra latera l por afectación del t racto espinotalámi-co ; 6) síndrome de Horner ips i la tera l ; 7) ataxia cerebelosa ipsi lateral secundar ia a la afectación del pedúnculo cerebelo-so infer ior y cerebe lo (MIR 02-03, 204 ) .
RECUERDA Síndrome bu lba r med i a l = M o t o r , a d i ferenc ia de Síndrome bu lba r lateral = W a l l e n b e r g (sensit ivo).
Síndrome bulbar medial: es consecuencia de la oclusión de la arteria espinal anterior o de la arteria vertebral . Cursa con : 1) paresia, amiotrof ias y fasci culac iones de la lengua por afectación del XII par craneal (la lengua protru ida se desvía hacia el lado de la lesión); 2) hemip l e jía contralateral con respeto de la cara; 3) ataxia sensitiva contralateral por afectación del lemnisco media l .
RECUERDA Los pares craneales nos dan el n ive l de la lesión. N o hay que o l v i da r la regla 2-2-4-4: los dos p r imeros pares " n o l legan al t r o n c o " , el III y el IV l legan al mesencéfalo; los pares V, V I , V I I y VI I I a la p ro tube ranc i a , y los cua t ro últimos al b u l b o .
1.10. Reflejos y síndromes medulares
Las neuronas motoras del asta anterior de la médula se d i v iden en las motoneuronas a, que inervan el músculo estriado, y las moto-neuronas y, que inervan el huso muscular.
14

Neurología y neurocirugía
Existen, además, en la sustancia gris m e d u lar, las interneuronas, con muchas c o n e x i o nes entre sí y con las motoneuronas , s iendo responsables de muchas de las func iones ¡ntegradoras de la médula. Así, el haz cor t i coesp ina l t e rmina casi to ta lmente en estas interneuronas, y sólo una vez que éstas han integrado el c o n j u n t o de señales procedentes de otros lugares, convergen f i na lmente en las motoneuronas anter iores.
U n t i p o especial de estas interneuronas son las células de Renshaw (MIR 01-02, 221 ) , que son excitadas por las propias motoneuronas , y cuya función es inh ib i r las motoneuronas vecinas (inhibición recurrente) de fo rma s i mi la r a c o m o ocur re en el sistema sensit ivo, para conseguir un cont ro l más f i no del m o v i m ien to y supr imi r la tendenc ia de las señales eléctricas a d i fund i rse a las neuronas adya centes.
Los pr incipales reflejos medulares son los s i guientes: • Reflejo miotático o de estiramiento mus
cular (Figura 13): la excitación de los husos (al aumentar la long i tud de la f ibra muscu lar) produce una contracción refleja de las grandes fibras esqueléticas que los rodean. Este reflejo se produce por una vía mono-sináptica (no part ic ipan interneuronas) en la que una f ibra sensitiva t i po la, que t iene su or igen en el huso, penetra por el asta posterior y realiza una sinapsis directa con las neuronas del asta anter ior que inervan las fibras del m ismo músculo del que p ro cede el estímulo. La cuantificación de los reflejos se expone en la Tabla 4.
REFLEJO FLEXOR Inhibición recíproca REFLEJO EXTENSOR CRUZADO
\ t /
Excitada
Inhibida
Estímulo doloroso de la mano
Figura 14. Reflejo f lexor
Nervio proplorreceptor
t
Médula espinal
Nervio motor
Figura 13. Reflejo miotático
0 A r r e f l e x i a
+ H i p o r r e f l e x i a
++ Ref le jos n o r m a l e s
+++ H i p e r r e f l e x i a
++++ Clonus
Tabla 4. Cuantificación de los reflejos os teotend inosos
Reflejo tendinoso: se produce cuando se excita el órgano tendinoso de Go lg i , capaz de detectar la tensión muscular. El estímulo llega a la médula a través de fibras t ipo Ib, que exci tan interneuronas inhib idoras que conectan con el asta anterior. Así, un aumento de tensión muscular inh ibe d i rectamente el músculo ind i v idua l , sin afectar a los músculos adyacentes.
• Reflejo flexor o de retirada (Figura 14): ante un estímulo sensorial cutáneo de cua lqu ier t i po , pero sobre todo do loroso (por esto se ha denominado también reflejo noc icept i vo o de dolor ) , se produce una contracción de los músculos flexores de la ext remidad y una relajación de los extensores. Reflejos medulares que producen espasmo muscular: b ien sea por una fractura ósea, por irritación del per i toneo parietal en una per i toni t is , etc.
• Reflejos autónomos: comprenden múltiples funciones, c o m o c a m bios en el tono vascular según la temperatura local , sudoración, reflejos intestinales y vesicales. Este t i po de reflejos suelen ser segmentar ios, pero en ocasiones se desencadenan de forma simultánea, en grandes porc iones de la médula, ante un estímulo noc i cept ivo fuerte o la repleción excesiva de una viscera. Es el l l amado reflejo en masa.
Es preciso recordar las pr incipales vías que recorren la médula (Figura 15 y Tabla 5) para poder reconocer los síndromes clínicos.
15

Manual CTO de Medicina y Cirugía, 8. a edición
apatía t ransversa Idiopática (mecan ismo inmunoalérgico), vírica, EM, LES, Sjógren
Déficit moto r . Paraplejia o te t rap le j ia in i c i a lmente flaccida y arrefléxica {shock medu la r ) ; pos t e r i o rmen te aparecen s ignos de afectación de pr imera m o t o n e u r o n a . Reflejos os teo tend inosos exal tados por deba jo de la lesión Déficit sensit ivo. Se afectan todas las moda l idades Trastornos autonómicos. Disfunción esf inter iana vesical (urgencia micc iona l lo más típico) y rectal (estreñimiento) Otros síntomas autonómicos son anhidrosis , cambios cutáneos tróficos y disfunción sexual ( impotenc ia )
Hemisección medular (S índrome de Brown-Séquard)
Traumat ismos penetrantes , lesiones ext ramedulares compres ivas
Pérdida de sensib i l idad do lorosa y térmica contra latera l (lesión de l t r a c to espinotalámico cruzado) Pérdida de sensib i l idad p rop iocep t i va ipsi lateral con ataxia sensit iva (interrupción d e los cordones poster iores) Parálisis espástica ipsi lateral (lesión de la vía p i ramida l cruzada)
Síndrome medular central
Lesión de las co lumnas posterolaterales
S i r ingomie l ia , h id romie l i a y t u m o r e s cen t romedu la res
Déficit sensit ivo suspend ido bi lateral con conservación de la sens ib i l idad táctil (déficit sensorial d isoc iado)
Degeneración subaguda c o m b i n a d a de la médula (déficit d e B12), mielopatía vacuolar asociada ai SIDA, compresión medu la r
Ataxia sensitiva con pérdida de sens ib i l idad p rop iocept i va y conservación de la sensib i l idad do lorosa y térmica La disfunción cor t i coesp ina l bi lateral p r o d u c e espast ic idad, hiperref lexia en m i e m b r o s infer iores y respuesta cu taneop lan ta r extensora (lesión de pr imera mo toneu rona )
Síndrome cordonal posterior
Z¿ Neurosífilis
S índrome d e la arteria espinal anterior
Ataxia sensitiva Impl ica do lores lancinantes en piernas, incont inenc ia ur inar ia y arreflexia rotu l iana y aqui lea La disfunción de los cordones poster iores en la región cervical da lugar a una sensación de "descarga eléctrica" descendente con la flexión de l cue l lo (s igno de Lhermi t te )
Disección aórtica, aterosclerosis, cirugía d e la aor ta a b d o m i n a l
Paraplejia o tetraple j ia aguda con disfunción vesical e intest ina l y anestesia do lorosa y térmica po r deba jo d e la lesión No hay afectación p rop iocep t i va
Tabla 5. Principales síndromes medulares
Cordón posterior Vía corticoespinal
Vía espinotalámica
Figura 15. Principales vías moto ras y sensitivas de la médula espinal
1.11. Sección y shock medular
Cuando se produce la sección repentina de la médula, se supr imen
todas las func iones medulares inferiores a la zona del t raumat ismo,
ya que la act iv idad normal de las neuronas medulares depende de la
estimulación tónica fac i l i tadora de los sistemas cor t icoesp ina l , reticu-
loespinal y vest ibuloespinal .
Ya se ha comentado anter iormente cómo, tras una fase de parálisis
f lacc ida, se llega a la espasticidad, según las neuronas medulares recu
peran gradua lmente su exc i tab i l idad .
En cuanto a los reflejos medulares, se recuperan gradualmente en or
den de comp l e j i d ad : los pr imeros en recuperarse son los reflejos de
est i ramiento y, poster iormente, los f lexores, los posturales antigravita-
torios y el resto de los reflejos de la marcha.
16

Neurología y neurocirugía
1.12. Fisiología del sistema nervioso Conducción nerviosa (Figura 16)
Las señales nerviosas se transmiten mediante potenciales de acción, que son cambios rápidos del potencia l de membrana .
Un potencia l de acción no se produce hasta que la elevación in ic ia l del potenc ia l de membrana sea lo bastante grande c o m o para alcanzar el denominado " u m b r a l " para la estimulación. Una vez a lcanzado el umbra l , se produce la siguiente secuencia de acontec imientos : • Fase de despolarización. El aumento de voltaje hace que se abran
canales de sodio, con lo cual se p roduce la entrada del m ismo al inter ior celular y el potencia l de membrana se hace posi t ivo.
• Fase de repolarización. Se cierran los canales de sodio y se abren los canales de potasio, pe rmi t i endo volver al potencia l basal. D u rante un pequeño lapso de t i empo , el potencia l de membrana se hace más negativo que durante el reposo; es una pequeña fase de hiperpolarización l lamada pospotencia l posi t ivo. Fase de reposo. Se recupera el e q u i l i b r i o iónico n o r m a l a ambos lados de la m e m b r a n a , grac ias a la b o m b a Na + /K + ATP d e p e n d i en t e .
Nodulo de Ranvier
Vaina de mielina
Figura 16. Conducción nerviosa e n una fibra mielínica
Propagación del potencial de acción
Un potenc ia l de acción que sucede en un pun to cua lqu ie ra de una membrana exc i tab le suele exc i tar porc iones adyacentes de la misma, lo que provoca la propagación del po tenc ia l de acción.
Este potenc ia l de acción puede viajar en ambas d i recc iones a través de la membrana exc i tada y c u m p l e la ley del t o d o o nada, es dec i r , o se propaga por toda la membrana (si ésta se ha l la en buen estado) o no lo hace en abso lu to .
Fibras mielínícas y amielínicas
La m i e l i n a está f o r m a d a f u n d a m e n t a l m e n t e por la e s f i ngomie l i na , un fosfolípido ais lante que d e p r i m e el f l u j o iónico a través de la m e m brana. En las fibras m ie l i n i zadas , ésta cons t i tuye una va ina que rodea al axón, i n t e r rump ida cada 1-3 m m por los nodu los de Ranvier. Los iones no pueden f lu i r a través de las gruesas vainas de m ie l i n a , pero sí lo pueden hacer a través de los nodu los de Ranvier. Por tanto , los potenc ia les de acción sólo pueden suceder en los nodu los y se d i r igen de n o d u l o a n o d u l o , en un patrón que se conoce c o m o c o n ducción saltator ia.
Ésta t iene impor tanc ia por tres razones: • Aumenta la ve loc idad de transmisión nerviosa entre 5 y 50 veces en
las fibras mie l in izadas. • Se conserva la energía del axón, porque sólo se despolar izan los
nodulos , por lo que la pérdida de iones es muchísimo menor que si la conducción sucediese de otro m o d o y, por tanto, se necesita menor metabo l i smo.
• El a is lamiento suministrado por la mie l ina permite que la repolar ización suceda con una transferencia mínima de iones y rápidamente.
Velocidad de conducción
Ésta depende de varios factores: Mielina. Es mayor en las fibras mie l in izadas que en las amielínicas.
• Diámetro de la fibra. Mayor a mayor diámetro.
Por tanto, la ve loc idad de conducción varía entre 0,5 m/s en las fibras amielínicas más pequeñas y 120 m/s en las fibras mie l in izadas muy grandes. En las fibras nerviosas mie l in izadas, la ve loc idad aumenta aprox imadamente con el diámetro de las mismas, y en las amielínicas, lo hace con la raíz cuadrada de su diámetro.
Tipos de fibras nerviosas
Existen dos clasif icaciones: una general , en la que están comprendidas las fibras motoras, sensoriales y autónomas, y otra referida sólo a las sensitivas. Aquí se hará referencia a la general (fibras de t ipos A, B y C), i nc luyendo la clasificación sensorial (tipos I, II, III y IV) j un to a cada clase de f ibra sensitiva. • Fibras A: corresponden a fibras mie l in izadas gruesas de los nervios
espinales. Existen diversas clases: - Fibras A a: poseen un diámetro entre 10 y 20 mieras y una ve lo
c idad de conducción de 60-120 m/s. - Fibras A B: diámetro de 8-9 mieras y ve loc idad de 30-70 m/s. - Fibras A y: fibras motoras del huso muscular, de 1 a 8 mieras de
diámetro y hasta 50 m/s. - Fibras A 8: entre 3 y 8 mieras y hasta 50 m/s. Engloba las fibras
t ipo III de la clasificación sensorial, dedicadas a la transmisión del do lor agudo, la temperatura fría y el tacto-presión groseros.
• Fibras B: diámetro de 3 mieras y ve loc idad de hasta 15 m/s. Corresponde a fibras levemente mie l in izadas, encargadas de la in fo rma ción autonómica pregangl ionar.
• Fibras C: no mie l in izadas y finas (0,5-2 mieras), son las más lentas (0,5-2 m/s). Componen aprox imadamente el 5 0 % de los nervios periféricos. Son las fibras sensitivas t ipo IV, relacionadas con el do lor sordo con t inuo , el prur i to , la temperatura cal iente y el tacto grosero. También son fibras C las autonómicas posganglionares.
17

Manual CTO de Medicina y Cirugía, 8. a edición
Neurotransmisores 1.13. Transmisión sináptica La transmisión sináptica es la fo rma de comunicación entre neuronas dentro del sistema nervioso, o entre una neurona y otra célula situada en estrecho contacto con el la .
Existen dos tipos pr incipales de sinapsis, las sinapsis eléctricas y las sinapsis químicas. • Sinapsis eléctricas: en este t i p o de sinapsis, el po tenc ia l de ac
ción presináptico se t ransmi te a la célula postsináptica a través de unos canales interce lu lares de baja resistencia eléctrica l l amados un iones comun i can tes o un iones en hend idu ra (gap junction o nexus).
• Sinapsis químicas: es el t i po de sinapsis p redominante en el sistema nervioso centra l . En las sinapsis químicas, la transmisión es u n i d i recc ional y más lenta que en las sinapsis eléctricas.
La transmisión f ina l iza al descender la concentración del neurotransmisor (NTS) en la hendidura sináptica, b ien por la acción de enzimas específicas que destruyen el NTS, b ien por difusión o recaptación del m ismo (MIR 98-99, 229) .
Se ha demostrado la existencia de mul t i tud de sustancias químicas que realizan la función de NTS. Pueden clasificarse en dos grupos principales: • Transmisores pequeños de acción rápida (noradrenal ina [ N A ] , do-
pamina [DA ] , g lutamato, 5 H T , acet i l co l ina , óxido nitroso [ N O ] , CABA, etc.). La mayoría se sintet izan en el c i tosol de la termina l presináptica a través de reacciones bioquímicas, y no suele existir un A R N m específico para su síntesis (MIR 00-01 F, 213). Or ig inan la mayor parte de las respuestas inmediatas del sistema nervioso, c o m o la transmisión de señales sensoriales al cerebro y de las señales motoras desde éste a los músculos.
• Neuropéptidos. Se sintetizan c o m o partes integrantes de grandes moléculas, que poster iormente son escindidas para dar lugar al neu-ropéptido de f in i t i vo . Dado que su síntesis es más laboriosa, se l ibe ran cantidades mucho menores, aunque este hecho se compensa en parte porque los neuropéptidos son m u c h o más potentes (VIP, sustancia P, diversas hormonas, encefal inas, etc.). Además, se d i fe rencian de los pequeños NTS en que su acción es más lenta y p ro longada, incluso con cambios a largo p lazo en el número y tamaño de sinapsis o de receptores (MIR 02-03, 132; MIR 0 0 - 0 1 , 246) .
18

Neurología y neurocirugía
0 2 . COMA. MUERTE ENCEFÁLICA
Orientación
MIR Aspectos esenciales
Este t e m a es m u y p o c o i m p o r t a n t e para el M IR . H a y que prestar atención a ios c o n c e p t o s destacados en los Aspectos esenciales y repasar los s ignos c o n va lo r l o c a l i z a d o r d e lesión, e s p e c i a l m e n t e la exploración p u p i l a r y los ref le jos troncoencefál icos.
ÜD m
m
tu m
El c o m a es el g rado más p r o f u n d o de disminución del n ive l de consc ienc i a .
La causa más f recuente de c o m a son los trastornos metabóllcos.
El n ive l de consc i enc i a se va lo ra en la exploración neurológica a través de la escala in te rnac iona l de G las g o w (véase Capítulo 18 . Traumatismos craneoencefálicos).
Los signos c o n va lo r l o ca l i zador en el pac ien te en c o m a son: el patrón resp i ra tor io (véase Figura 17), las a l terac iones pup i la res , los m o v i m i e n t o s ocu lares ref lejos y las posturas reflejas.
La presenc ia de los ref lejos oculocefálicos ( m o v i m i e n t o c o n j u g a d o de los o jos en dirección opuesta a la rotación de la cabeza) i nd i ca la in tegr idad f u n c i o n a l de l t r o n c o del encéfalo.
2.1. Coma
Fisiopatología
El nivel normal de consc iencia depende de la activación de los hemisferios cerebrales por grupos neuronales local izados en el sistema reticular act ivador (SRA) del t ronco del encéfalo. El SRA se local iza en la formación reticular comprend ida entre la porción rostral de la protuberanc ia y la parte caudal del diencéfalo, y t iene una impor tanc ia básica para el manten imien to del estado de v ig i l i a . Pequeñas lesiones local izadas en esta zona pueden determinar estados de coma.
Las lesiones hemisféricas también pueden causar coma por a lguno de los siguientes mecanismos: 1) lesiones estructurales general izadas o bilaterales, 2) lesiones unilaterales que c o m p r i m e n el hemisfer io contralateral , y 3) compresión troncoencefálica secundaria a herniación.
Los trastornos metabólicos son la causa más frecuente de coma sin signos de foca l idad con función t roncoen cefálica intacta.
2.2. Signos de valor localizados (Figura 17)
Patrón respiratorio
El patrón respiratorio de un paciente en coma puede ser útil para local izar el nivel de disfunción estructural en el neuroeje, pero las alteraciones metabólicas pueden afectar a los centros respiratorios de la protuberanc ia y
S M P M P P P B bu lbo , dando lugar a patrones similares a los produc idos por lesiones estructurales. Por tanto, la interpretación • a H H B Í H n É H de los cambios respiratorios de un paciente comatoso debe acompañarse de una evaluación completa y cuida-- MIR 04-05, 53 dosa del estado metabólico del paciente.
19

Manual CTO de Medicina y Cirugía, 8. a edición
PATRÓN RESPIRATORIO
Otras causas DESVIACIÓN PUPILAS
Hemisferios cerebrales
Diencéfalo (tálamo e
hipotálamo)
Mesencéfalo
CHEYNE-STOKES
HIPERVENTILACIÓN NEURÓGENA CENTRAL
Uremia Anoxia
ICC
Protuberancia
NO HAY OJOS DE MUÑECA
REFLEJO CORNEAL ABOLIDO
Midriáticas arreactivas
<3><3
BOBBING OCULAR
Puntiformes reactivas
Bulbo raquídeo
CLUSTER
ATÁXICA DE BIOT (AGÓNICA)
REFLEJO NAUSEOSO ABOLIDO
POSTURAS REFLEJAS Decorticación Descerebración
Figura 17. Signos de valor localizador en un paciente en coma
Respiración de Cheyne-Stokes. Representa una situación en la que los centros respiratorios se hacen más dependientes de las f luc tua ciones de P C O r Se alternan breves per iodos de hiperventilación con per iodos más cortos de apnea. Puede producirse en c o n d i c i o nes fisiológicas (ancianos durante el sueño, elevadas altitudes) o por lesiones estructurales (lesiones corticales bilaterales, disfunción talámica bi latera l , herniación) y trastornos metabólicos (uremia, anoxia , insuf ic iencia cardíaca congestiva). Hiperventilación neurógena central. Consiste en respiraciones regulares rápidas y profundas. Se produce en lesiones estructurales en mesencéfalo y protuberanc ia o por procesos metabólicos (cetoaci-dosis diabética, acidosis láctica, h ipoxemia ) . Cuando un cuadro de hiperventilación rítmica aparece en un paciente con acidosis, se habla de respiración de Kussmaul (MIR 04-05, 53) .
Pupilas
Los reflejos luminosos pupi lares son m u y resistentes a la disfunción metabólica, por lo que alteraciones de los mismos, fundamenta lmente si son unilaterales, ind ican lesión estructural , si se exceptúa: 1) uso de atropínicos en instilación, ingesta o resucitación ca rd iopu lmonar , cuando se ha usado atropina (midriasis arreactiva a la administración tópica de colinérgicos); 2) altas dosis de barbitúricos, succ in i l co l ina , lidocaína, fenotiacinas o aminoglucósidos. La presencia de pupi las fijas arreactivas es un signo de mal pronóstico, y puede observarse en encefalopatías metabólicas graves y en lesiones mesencefálicas.
/entilación RECUERDA El patrón resp i ra to r io de Cheyne-Stokes (períodos de h i p e r v e n t i k
c o n pausas d e apnea) p u e d e aparecer también en la u r e m i a y en la i n su f i c i enc i a ca rd i aca conges t i va . El patrón r esp i r a to r i o de Kussmaul (hiperventi lación rítmica c o n resp i rac iones p ro fundas o ba t ipnea ) apa rece en estados de ac idos is . A m b o s pat rones p u e d e n aparecer en la h i p o x i a .
Respiración apnéustica. Es una inspiración manten ida , seguida de espiración y pausa, y se produce por lesiones en el tegmento lateral de la protuberanc ia inferior. Respiración atáxica. Patrón comple tamente irregular, presente en pacientes agónicos. Precede al fa l lo respiratorio y se produce por lesión a nivel bu lbar dorsomedia l .
RECUERDA Pupilas midriáticas arreactivas: lesión mesencefálica. Pupilas punt i fo rmes reactivas: lesión pon t ina . Alteración pup i l a r un i la tera l : lesión estructura l .
La forma, tamaño, simetría y respuesta a la luz son de valor loca l izador en la función troncoencefálica o del III par.
Movimientos oculares
Reflejo corneal. Vía aferente por la pr imera rama del trigémino y vía eferente por el fac ia l . En cond ic iones normales, al est imular la córnea suavemente, se produce parpadeo bi latera l . Su alteración imp l i ca disfunción de t ronco a nivel protuberancia ! .
2 0

Neurología y neurocirugía
Reflejos oculocefálicos. En el paciente inconsciente, el ref lejo es normal si los ojos se mueven en las órbitas en dirección opuesta a la rotación de la cabeza, ind i cando integr idad del t ronco del encéfalo. La respuesta oculocefálica es anormal cuando, al mover la cabeza, los globos oculares no se mueven o lo hacen de forma desconjugada, s iendo entonces sugestivo de lesión estructural a nivel ponto-mesencefálico. El envenenamiento por barbitúricos también puede inh ib i r este ref lejo. Reflejos oculovestibulares. Son mov imientos oculares reflejos en respuesta a la irrigación de la membrana timpánica con agua fría. La respuesta normal en el paciente consciente es un nistagmo con desviación tónica de los ojos hacia el lado est imulado, seguido de un m o v i m i e n t o de corrección rápida hacia el lado contrar io ("los ojos huyen del agua fría"). Hay varios t ipos de respuesta en pac ien tes comatosos: - Si la fase lenta está ausente, s ignif ica lesión de t ronco . - Si la fase lenta es norma l , pero no se objet iva fase rápida, en ton
ces existe lesión hemisférica. - Si las fases lenta y rápida son normales, se debe pensar en un
coma histérico.
Movimientos oculares espontáneos. "Roving ocu la r " . O jos l igeramente divergentes, desplazándose lentamente de un lado a ot ro . Impl i ca t ronco cerebral intacto. "Bobbing ocular" . Mov im ien tos oculares conjugados rápidos hacia abajo con retorno lento a la posición pr imar ia . Se asocia a lesiones pont inas, pero también a encefalopatías toxicometabólicas.
O umga RECUERDA Oftaimoplejía in ternuc lear : lesión de l fascículo l ong i t ud ina l med i a l (en jóvenes hay q u e sospechar en f e rmedad desmie l i n i zan te mient ras que , en mayores , i squemia en el t r o n c o del encéfalo).
Desviación conjugada de la mirada. En las lesiones hemisféricas estructurales, los ojos se desvían con jugadamente hacia el lado de la lesión. Las lesiones irritativas los desvían al lado opuesto.
RECUERDA Las lesiones hemisféricas estructura les desvían los o jos hac ia ips i la tera l , mient ras q u e las lesiones hemisféricas irr i tat ivas y las lesiones t roncoen-cefálicas los desvían hac ia cont ra la te ra l .
Las lesiones a n ive l de la p ro tuberanc i a p roducen una desv ia ción de los ojos hacia el lado con t r a r i o de la lesión. Las lesiones hemisféricas pro fundas (tálamo) desvían los ojos hacia abajo y adent ro o hacia el lado con t r a r i o de la lesión (desviación ocu la r paradójica). Trastornos desconjugados de la mirada. Es la o f ta lmop le j i a in te rnu clear por lesión del fascículo long i tud ina l media l .
Posturas reflejas
Postura de descerebración. Cursa con extensión, aducción y ro tación interna de brazos y extensión de las piernas. Aparece por lesiones entre núcleo ro jo y núcleos vestibulares. Postura de decorticación. Presenta flexión de codo , aducción de h o m b r o y brazos, pronación e hiperflexión de muñecas. Las piernas están extendidas. Responde a lesiones hemisféricas profundas o hemisféricas bilaterales.
RECUERDA Las posturas ref le jas de decort icac ión y de descerebrac ión co r r e s p o n d e n a unas p u n t u a c i o n e s de 3 y 2, r e s p e c t i v a m e n t e , en la v a loración de la respuesta m o t o r a en la escala de l c o m a de G l a s g o w (en esta escala , la respuesta m o t o r a es, a su vez , el parámetro más i m p o r t a n t e ) .
Estados de pseudocoma
Falta de respuesta psicógena. El paciente aparece sin respuesta, pero está fisiológicamente despierto. La exploración es normal y la respuesta oculovest ibular está intacta. Mutismo acinético. Estado de v ig i l i a sin pos ib i l idad de elaborar respuesta. Puede ser deb ido a daño cerebral bi lateral (cuadro apálico), lesión en porción superior de mesencéfalo y diencéfalo o h idrocefa lia aguda.
2.3. Muerte encefálica
El Real Dec re to 2070/1 9 9 9 , de 30 de d i c i e m b r e , p u b l i c a d o el martes 4 de enero del 2 0 0 0 , establece que el diagnóstico y certificación de la muer te de una persona podrá real izarse tras la confirmación de l cese i r revers ib le de las func iones encefálicas (muer te encefálica) o de las func iones card ior resp i ra tor ias (muerte por parada cardiorres-p i ra tor ia ) .
Se ent iende por muerte encefálica la situación de coma arreact ivo de etiología estructural conoc ida (requiere ev idenc ia clínica o por neuro-imagen de lesión destructiva en el sistema nervioso centra l , compat ib l e con la situación de muerte encefálica) y carácter irreversible.
El diagnóstico de muerte encefálica exige: • Una exploración neurológica sistemática, comple ta y rigurosa. • El paciente debe encontrarse en situación de estabi l idad hemodiná-
mica , oxigenación y ventilación adecuadas, con una temperatura corpora l superior 32 °C , y no estar bajo los efectos de fármacos de presores del sistema nervioso centra l , b loqueantes neuromusculares ni presentar alteraciones metabólicas.
• Se recomienda repetir la exploración a las seir horas en lesiones destructivas y a las 24 horas en casos de encefalopatía anóxica, aun que el per iodo de observación se deja a cr i ter io médico, en función de las pruebas instrumentales que puedan realizarse. Las pruebas instrumentales no se consideran obligatorias, e inc luyen: - Pruebas que evalúan la función neurona l : EEG y potenciales
evocados. Pruebas que evalúan el f lu jo sanguíneo cerebral : arteriografía de cuatro vasos, angiografía cerebral por sustracción d ig i ta l , angio-gammagrafía y Dopp le r transcraneal.
El diagnóstico de muerte por parada cardiorrespirator ia se basa en la constatación inequívoca de ausencia de lat ido cardíaco (demostrada por ECG o ausencia de pulso central) y de respiración espontánea por un per iodo de t i empo no inferior a c inco minutos .
La i r revers ib i l idad del cese de las func iones cardiorrespiratorias se establece tras la aplicación de maniobras de reanimación ca rd iopu lmonar avanzada durante un per iodo de t i empo adecuado a la edad y c i rcunstancias que provocaron la parada cardiorrespirator ia.
2 1

DEMENCIAS
Orientación
MIR r
Aspectos esenciales L
En los últimos años, ha d i s m i n u i d o la i m p o r t a n c i a de este t e m a en el MIR, s in e m b a r g o , es c l a ve la d i f e r enc i a en t re c a d a t i p o de d e m e n c i a y c o n o c e r las características de la d e m e n c i a más f r e cuen te , el A l z h e i m e r .
Q~J Se de f ine d e m e n c i a c o m o el de te r io ro progres ivo de las func iones super iores, a d q u i r i d o y c o n preservación del n ive l de consc ienc i a . La preva lenc ia de la d e m e n c i a aumen ta c o n la edad .
[~2~¡ Las demenc ias se c las i f i can en irreversibles (la mayoría) o reversibles, y en cor t i ca les o subcor t ica les (véanse las tablas).
["3") La en f e rmedad de A l z h e i m e r es la causa más f recuente de d e m e n c i a en O c c i d e n t e . Es una d e m e n c i a co r t i c a l , de p r e d o m i n i o t e m p o r o p a r i e t a l . Su i n i c i o es ins id ioso y su progresión lenta, y la edad avanzada es el p r i n c ipa l fac tor de riesgo para su desar ro l lo . En su t r a t am ien to se e m p l e a n inh ib ido res de la acet i l co l inesterasa (donepez i l o , r i vas t igmina , ga lan tamina ) en las fases leve y mode rada , y antagonistas no compe t i t i vo s de los receptores glutamatérgicos N M D A (memant ina ) en fases avanzadas.
CJJ La d e m e n c i a f r o n t o t e m p o r a l o de Pick es también una d e m e n c i a co r t i c a l . Cursa c o n afasias, apatía, abu l i a y otras a l terac iones conduc tua l es , pe ro sin amnes ia , ni apraxias ni agnosias.
["5"] Las demenc ias de causa vascu lar son las segundas en f recuenc ia . Destacan la d e m e n c i a m u l t i i n f a r t o por embo l i as bi latera les rec id ivantes ( i n i c io brusco y c o n fo ca l i dad neurológica) y la en f e rmedad de B inswanger o encefalopatía aterosclerótica subcor t i ca l , en la q u e es típica la leucoara ios is o desmielinización per iven-t r i cu la r .
3.1. Concepto y clasificación
La demenc ia const i tuye la causa pr inc ipa l de incapac idad a largo p lazo en la tercera edad. Afecta al 2 % de la
población entre 65-70 años y al 2 0 % de los mayores de 80 años.
Se def ine c o m o un deter ioro crónico de las func iones superiores, adqu i r i do (a di ferencia del retraso mental ) y en
presencia de un nivel de consc ienc ia y atención normales (a di ferencia del delirium).
RECUERDA La p r i n c ipa l d i f e renc ia ent re d e m e n c i a y delirium es que , en este último, está d i s m i n u i d o el n ive l de consc i enc i a y está a l terada la m e m o r i a i nmed i a t a (depend ien te de la atención).
Preguntas
• MIR 06-07, 63 - MIR 05-06, 59, 233 - MIR 03-04, 250 -MIR 01-02, 56 - MIR 00-01, 51 - MIR 97-98, 37
La pérdida de una única función intelectual no es c r i
ter io sufic iente para el diagnóstico de demenc ia . La
demenc ia suele afectar a todas las func iones inte lec
tuales, aunque en las fases iniciales se puede establecer
el diagnóstico por el deter ioro de tres de las siguientes
áreas: lenguaje, memor ia , destreza v isuoespacia l , afec
to, personal idad o intelecto. Las causas más frecuentes
de demenc ia progresiva se inc luyen en la Tabla 6.
La mayor parte de las demencias se deben a procesos
degenerativos d iseminados y/o mult i foca les . Sin e m
bargo, la masa cerebral no es un buen ind icador del
grado de func iona l idad intelectual y, por tanto, la exis
tencia de una atrof ia cerebral general izada en las prue
bas de imagen no siempre es indicat iva de demenc ia .
Aunque la mayor parte de las demencias son ¡rreversi-
Enfermedad de A lzhe imer (50-90%) Infartos cerebrales múltiples (5-10%) A lcoho l (5-10%) Trastornos endoc r i no metabólicos: - H ipo t l ro id i smo - Deficiencia de v i t amina B1 2
Neoplasias intracraneales Hema toma subdura l crónico Hidrocefal ia a presión no rma l Otras enfermedades degenerat ivas : - Enfermedad de Pick - Enfermedad de Parkinson - Enfermedad de H u n t i n g t o n - Parálisis supranuclear progresiva Infecciones de l SNC: - VIH - Sífilis - Creutzfeldt-Jakob
Tabla 6. Causas más frecuentes de demencia
2 2

HE Neurología y neurocirugía _ ^ j " .
bles ( 7 0 % ) y no t ienen t ratamiento, salvo el sintomático, es importante ident i f icar aquéllas que son potenc ia lmente tratables (MIR 03-04, 250) (Tabla 7). Cerca del 1 0 % de las demencias son reversibles si se actúa a t i empo ; en otro 1 0 % , aunque irreversibles, se puede detener la progresión e l im inando los factores de riesgo; por último, un 1 0 % obedecen a causas psiquiátricas (pseudodemencias) .
TRATABLES
ers ib les
Demencias vasculares Demencias postraumáticas Demenc ia alcohólica
Enfermedades metabol icocarenc ia les - Tiroideas - Adrenales - Pelagra - Déficit de Bu y fo l a to - Déficit de B, - Uremia - Wi lson - Porfiria - Encefalopatía hepática - Tr .de calcio
Enfermedades inf lamator ias e infecciosas - Sífilis - Men ing i t i s - Encefalitis - Vasculitis (LES) Procesos intracraneales - Neoplaslas - H e m a t o m a subdura l - Hidrocefal ia no rmotens i va Depresión
NO TRATABLES E IRREVERSIBLES
Enfermedades degenerat ivas : - A lzhe imer - Pick - Parkinson - H u n t i n g t o n
Enfermedades infecciosas: - VIH - Creutzfeldt-Jakob Otras: - Esclerosis múltiple - Demenc ia dialítica
Tabla 7. Clasificación pronostica de las demencias
RECUERDA Las pseudodemenc i a s son deter io ros cogn i t i vos reversibles q u e pueden aparecer en trastornos depres ivos . A d i fe renc ia de las demenc ias , m e j o ran c o n la ag r ipn i a o privación de sueño.
Diagnóstico de demencias
El diagnóstico de las demencias es eminentemente clínico: una historia clínica detal lada es fundamenta l . As imismo, el desarrol lo de nume rosas técnicas neuropsicológicas ha permi t ido desarrollar patrones de afectación característicos de cada ent idad.
Entre los estudios neuropsicológicos, el más ex tend ido es el minimen-tal test, que de fo rma rápida permi te estudiar la memor i a , la o r i en ta ción temporoespac ia l , el lenguaje, la escr i tura, la lectura, el cálculo y las praxis v isuoespaciales e ideomotoras . Se puntúa de 0 a 30 puntos , considerándose norma l de 2 7 a 30 puntos , de ter io ro cogn i t i vo l igero de 24 a 27 y demenc i a por deba jo de los 24 puntos (MIR 06-07, 63) (Tabla 8).
PUNTUAC IÓN M Á X I M A
Or ientac ión: ¿Que año, estación, fecha, día de la semana y mes es? ¿Cuál es su nación, región, c iudad , hospi ta l y piso?
5 5
Rememorac ión : N o m b r e tres ob je tos (1 s cada uno) y pregúntelos después al pac iente (repetir los ob je tos otras veces hasta q u e los aprenda)
3
Atenc ión y cálculo: Debe deletrear al revés una palabra de cinco letras (por ej.: lápiz) o enumera r los siete pr imeros números (deteniéndolo en el 5)
5
Repet ic ión : Preguntar los tres ob je tos n o m b r a d o s antes 3
L e n g u a j e : • Señale un lápiz. El pac iente debe nombra r ese ob j e to • El paciente debe repet ir palabras sencillas c o m o : "no",
"siempre", " cuando" o "pe ro " • Dar al paciente las s iguente órdenes
(dar tres indicaciones) : "Tome un papel con la m a n o derecha" "Dob le el papel por la m i t a d " "Ponga el papel en el suelo"
• El pac iente debe escribir una frase a su gus to (que t enga sent ido)
• El pac iente debe copiar, con ángulos y cuadrádangulos de intersección dos pentágonos d ibu jados
2
1
3
1
1
1
Total 30
Tabla 8.Test de minimental
Pueden diferenciarse dos t ipos de demenc ia , en función de la loca l iza ción de las lesiones: cort icales y subcort icales (Tabla 9).
CORTICALES SUBCORTICALES
Anatomía patológica
• Corteza de lóbulos frontales, parietales y t empora les
• H i p o c a m p o
Núcleos grises p ro fundos del encéfalo
Clínica
• Afasia • Apraxia • Agnosia • Acalculia
• Retardo ps i comoto r • M o v i m e n t o s anormales • Disartria • Al terac iones posturales • Depresión
E jemplos
• A lzhe imer • Pick • Creutzfeldt-Jakob • Men lngoencefa l i t i s • Hipoxia • Vascular • Neoplasias • Postraumática
• H u n t i n g t o n • Parkinson y Parkinson Plus • Wi lson • VIH • Vascular • Neoplaslas • Postraumáticas
Tabla 9. Correlación anatomoclínica en las demencias
3.2. Enfermedad de Alzheimer
Epidemiología En los últimos años, se han ap l i cado técnicas radiológicas al diagnóstico de las demencias; fundamenta lmente se han real izado estudios con resonancia magnética y SPECT/PET. Es característica la atrofia tempora l y las disfunciones temporopar ieta les en la fase in ic ia l de la enfermedad de A lzhe imer , o la atrofia y disfunción frontal en la demenc ia fronto-tempora l .
La Enfermedad de A lzhe imer (EA) es la causa más frecuente de d e m e n cia en Occ idente . La mayoría de los pacientes in ic ia los síntomas de la enfermedad a partir de los 65 años, aunque un debut temprano , antes de los 4 0 años, también puede ocurr i r , especia lmente en aquel los casos afectados de una forma hereditaria de la enfermedad.
23

Manual CTO de Medicina y Cirugía, 8.a edición
La prevalencia de la enfermedad se dob la cada c inco años a partir de los 60 , de forma que afecta a un 1 % de los pacientes a los 60 años, a un 2 % a los 65 años, y a un 4 % a los 70 años.
Anatomía patológica
Se caracteriza por una degeneración progresiva y selectiva de pob la ciones neuronales en el córtex entorr inal , h ipocampo, cortezas de asociación temporal , frontal y parietal, núcleos subcorticales y núcleos del t ronco (/ocu5 coeruleus y núcleos del rafe). No se afectan las cortezas p r i marias motoras y sensitivas, los ganglios básales ni el cerebelo (Figura 18).
Figura 18.TC de pac iente con en f e rmedad de Alzheimer. Demuest ra u n a u m e n t o marcado del sistema ventr icu lar y de los surcos. La cisura de Silvio
y astas tempora les de los ventrículos laterales son los más severamente afectados.
A nivel macroscópico, la pérdida de neuronas se t raduce en una atrofia general izada, más grave en los lóbulos temporales, que se acompaña de dilatación secundaria del sistema ventr icular .
Q RECUERDA Las lesiones histológicas típicas de la en f e rmedad de A l z h e i m e r son los depósitos intrace lu lares de t h ipe r fos fo r i l ada y las placas de p-ami lo ide . El depósito de esta última se p r o d u c e también en cerebros anc ianos y en otras patologías c o m o el síndrome de D o w n , la angiopatía congófila y la mios i t i s por cuerpos de inclusión.
Histológicamente, pueden encontrarse ov i l los o madejas neurof ibr i-lares compuestos por pares de f i l amentos he l ico ida les , y en d o n d e es pos ib le ident i f i car dos proteínas: la proteína x en estado de hiperfos-forilación y la u b i q u i t i n a (MIR 05-06, 233) .
Sin embargo , el da to más característico de la en fe rmedad de A l z h e i mer son las placas de a m i l o i d e (placas seniles o neuríticas) que c o n t ienen f ragmentos neuronales degenerados, rodeados por una densa estructura de mater ia l a m i l o i d e compues to básicamente por proteína P-amiloide (Tablas 10 y 11).
• Enfermedad de A lzhe imer • Parkinson - demenc ia - ELA • Enfermedad de Creutzfe ldt - Jakob • Demenc ia pugilística • Síndrome de D o w n
• Parálisis supranuclear progresiva • Park insonismo postencefalítico • Enfermedad de Gerstmann-Straussler • Enfermedad por cuerpos de Lewy
di fusos • Enve jec imiento no rma l
Tabla 10. Procesos asociados con madejas neurofibrilares
Estas dos alteraciones, ov i l los neurof ibr i lares y placas seniles, no son patognomónicas y se pueden encontrar en otras formas de demenc ia y en cerebros sanos de pacientes ancianos, aunque en menor número. En la enfermedad de A lzhe imer , son especia lmente frecuentes en el h i pocampo y en el lóbulo tempora l .
- Familiar:
- Enfermedad de A lzhe imer fami l ia r : > C romosoma 21 > C romosoma 19 > C romosoma 14 > C romosoma 1
- Hemorrag ia cerebral hereditar ia con ami lo idos is : > C romosoma 21
• Enfermedad de A lzhe imer esporádica • Síndrome de D o w n • A c u m u l o cerebral focal del péptido p-A4 d e p e n d i e n t e de la edad • Angiopatía congófila esporádica • Miosit is por cuerpos d e inclusión
Tabla 11 . Enfermedades con depósito de proteína precursora de la P-amiloide
Alteración de neurotransmisores
La somatostatina es el neurotransmisor que con más frecuencia aparece d i sm inu ido , aunque la acet i l co l ina es el que parece más re lac ionado con el grado de deter ioro cogn i t i vo .
El núcleo basal de Meynert , pr inc ipa l fuente de inervación colinérgica de la corteza cerebral , se afecta precozmente en el curso de la enfermedad de A lzhe imer , conduc i endo a un déficit marcado de col inaace-tiltransferasa (CAT) y de la síntesis de acet i l co l ina (Ach). La reducción de CAT puede alcanzar el 6 0 - 9 0 % , especia lmente en los lóbulos t e m porales, exist iendo una correlación entre el grado de reducción y el grado de demenc ia . La aceti lcolinesterasa, enz ima que degrada la Ach , se encuentra también reducida en esta enfermedad.
Otros neurotransmisores afectados en la enfermedad de A lzhe imer son el ácidogammaaminobutírico (GABA), la serotonina (por afectación de los núcleos del rafe) y la noradrenal ina (por afectación del locus coeruleus).
Genética y factores de riesgo
La edad es el pr inc ipa l factor de riesgo para el desarrol lo de la enfermedad de A lzhe imer . Ap rox imadamente en un 2 5 % de los casos la historia clínica revela antecedentes famil iares. La enfermedad de A lzhe imer se hereda en un 5 - 1 0 % de los casos con carácter autosómico d o m i n a n te y, en algunos casos, con una edad de debut precoz (4. a -5. a década).
Se han imp l i c ado tres locus cromosómicos cuyas mutac iones se han asociado a EA de in i c io precoz: • Gen de la proteína precursora am i lo ide en el c romosoma 2 1 . • Gen de la presenilina I en el c romosoma 14. Es el locus más fre
cuentemente imp l i c ado en los casos de A lzhe imer de in i c io precoz ( 7 0 % ) .
• Gen de la presenilina 2 en el c romosoma 1, con una inc idenc ia muy baja.
24

Neurología y neurocirugía
En todos los casos, las mutac iones conducen a un incremento en la producción de p-amiloide (part icularmente la fo rma de 42 aminoácidos), que se deposita f o rmando las placas seniles.
Sólo un 5 % de los casos de EA se deben a mutac iones. La mayoría de estos casos son esporádicos, o bien ocur ren en fami l ias sin un patrón de herencia autosómico dominan te y debutan tardíamente. Los fac to res de riesgo asociados a EA esporádica son: • Vulnerabilidad genética. La presencia del a le lo E4 de la apo l ipopro-
teína E (c romosoma 19) conf iere vu lnerab i l idad para desarrollar la EA sin tener una asociación obl igator ia con la misma.
• Edad. La inc idenc ia y prevalencia se dup l i ca cada c inco años a partir de los 60; tras los 65 años, la prevalencia se sitúa en un 1 0 % .
• Sexo. Más frecuente en mujeres. • Historia de traumatismo craneal previo.
Factores protectores
Su conoc im ien to es importante para la planificación racional del tratamiento de esta enfermedad:
• Genotipo apo-E2. Este genot ipo se corre lac iona con una d i s m i n u ción del riesgo para presentar EA, y con un in i c io más tardío.
• Antiinflamatorios no esteroideos (AINE). El uso de los AINE está asociado con un riesgo más bajo para EA y con un deter ioro cogni-t ivo más lento en pacientes con la enfermedad. Este efecto podría deberse a una acción ant i in f lamator ia a nivel de las placas seniles. La atención actua lmente está en el desarrol lo de fármacos i n h i b i d o res de la c ic looxigenasa t i po II.
• Terapia estrogénica. Se ha demostrado que la terapia estrogénica en mujeres posmenopáusicas d i sminuye el riesgo de EA. Los es-trógenos tendrían varias acciones potencia l mente útiles: funciones neurotróficas, efecto neuroprotector y benefic ios sobre el f l u j o sanguíneo cerebral .
• Nivel educativo. Varios estudios han demostrado que los niveles educat ivos más altos están asociados con un riesgo más bajo de desarrollar la enfermedad.
Clínica
Se trata de una enfermedad de in i c io insidioso y progresión lenta, con una evolución media de unos ocho o d iez años desde el in i c io hasta la muerte.
La EA suele presentarse con un per iodo preclínico caracter izado por los errores puntuales de memor i a , sin que existan otros déficit. Poster iormente se establece una alteración de la memor ia reciente (capaci dad para almacenar nueva información y recuperarla después de un per iodo de t iempo) y de la capac idad de aprendizaje. In ic ia lmente la memor ia remota se mant iene intacta, pero en el trascurso de la enfermedad, el paciente presentará di f icul tades con la recuperación de los episodios lejanos.
In ic ia lmente pueden presentarse alteraciones del lenguaje: d i f i cu l tad para nominar objetos o para comprender órdenes comple jas o encadenadas. A medida que la enfermedad progresa, aparecen alteraciones francas de la nominación, ausencia de un lenguaje espontáneo, que en numerosas ocasiones se encuentra parco en palabras, fa l to de sign i f i cado y con errores gramaticales. En la fase f ina l , el paciente t iene una alteración grave de la formación y comprensión del lenguaje. Las
alteraciones v isuoconstruct ivas son muy frecuentes. Desde el i n i c io , se ev idenc ia una d i f i cu l tad en la realización de d ibujos (test del reloj a l terado desde el in ic io ) , construcciones t r id imensionales o en la capac i dad para orientarse en espacios abiertos. Con la progresión, el paciente pierde la capac idad para reconocer objetos, personas o lugares, a pesar de que las func iones visuales pr imarias se encuentran intactas (agnosia visual). Las manifestaciones apráxicas son raras en las fases iniciales de la enfermedad, aunque a medida que ésta avanza se establece una apraxia ideomotora .
Desde las fases iniciales, se pueden demostrar síntomas de disfunción ejecut iva, con d i f i cu l t ad para p lani f icar tareas o el razonamiento abstracto. Igualmente, las alteraciones de conducta también pueden presentarse en una fase media , s iendo la apatía el síntoma más frecuente, aunque la pérdida de interés por las relaciones sociales, la abul ia o bien agitación, i r r i tab i l idad , reacciones agresivas o conductas des inh i bitorias pueden presentarse. Los síntomas psiquiátricos pueden aparecer, s iendo la depresión la más frecuente. Las ideas y conductas paranoides son habituales en la fase in ic ia l-media de la enfermedad, y están asociadas a un peor pronóstico. Sólo en fases muy evoluc ionadas pueden aparecer signos extrapi-ramidales, c o m o marcha torpe, postura encorvada, bradic inesia general izada y r igidez. Genera lmente, la causa de la muerte suele ser una enfermedad intercurrente, sobre todo , infecciones.
Tratamiento farmacológico
Los potenciales objet ivos del t ra tamiento farmacológico son: 1) mejoría cogni t iva ; 2) en lentec imiento en la progresión; y 3) retraso en la apar i ción de la enfermedad.
• Inhibidores de la acetilcolinesterasa (MIR 01-02, 56) . Indicados en las fases leve y moderada de la enfermedad; no mod i f i can a largo p lazo la progresión de la enfermedad, pero producen una mejoría de las func iones cognit ivas durante los pr imeros meses de tratamiento . - Tacr ina: su v ida media corta ob l iga a una administración fre
cuente, y t iene frecuentes efectos adversos colinérgicos, además de una potenc ia l hepatotox ic idad grave que obl iga a controles regulares. N o se emplea en la actua l idad y ya no se comerc ia l i za .
- Donepezi lo: puede administrarse en una sola dosis diar ia y t iene mejor to leranc ia que la tacr ina, sin alteración de la act iv idad hepática.
- Rivastigmina: i nh ib idor de la aceti lcol inesterasa y de la but i r i l-colinesterasa, por lo que podría tener un dob le mecan ismo de acción. Se administra cada 12 horas, y c o m o efectos secundarios son destacables las alteraciones digestivas y la pérdida de peso.
- Galantamina: i nh ib idor de la aceti lcol inesterasa y modu lador de los receptores nicotínicos de acet i l co l ina , con lo que modificaría la transmisión colinérgica por dos vías diferentes. Efectos adversos digestivos. Administración cada 12 horas.
• La memantina es un antagonista no compet i t i vo de los receptores de N-metil-D-aspartato ( N M D A ) del g lutamato, ind icado en las fa ses moderadas y avanzadas de la enfermedad de A lzhe imer .
• Otros tratamientos con antiinflamatorios, estrógenos, an t i ox idan tes y factores neurotrópicos, una vez establecida la enfermedad, no han dado resultados posit ivos hasta el m o m e n t o . Recientemente, se ha ensayado en humanos con factores de riesgo la generación de ant icuerpos contra la proteína P-amiloide, dado que en ratones transgénicos que producen exceso de ami lo ide se ha demostrado
25

M a n u a l C T O d e M e d i c i n a y Cirugía , 8.a ed i c i ón
una reducción en la formación de placas, pero hasta el m o m e n t o los efectos secundarios han l im i tado el desarrol lo de estas nuevas terapias.
3.3. Demencia frontotemporal (enfermedad de Pick)
Es un trastorno degenerat ivo caracter izado por una marcada pérdida asimétrica de neuronas en las regiones anteriores de los lóbulos f r o n tales y temporales, con norma l idad del resto del cerebro (Figura 19).
Figura 19. Enfermedad de Pick
Histológicamente, hay dos datos característicos: 1) neuronas de Pick: son neuronas tumefactas, pálidas, que no se tiñen con las t inc iones habituales y local izadas en los lóbulos frontales; 2) cuerpos de Pick: son inclusiones citoplasmáticas local izadas en las regiones temporales anteriores. No se observan ov i l los neurof ibr i lares ni placas seniles.
Q RECUERDA La d e m e n c i a d e Pick se d i f e renc ia de l A l z h e i m e r en que : aparece en más jóvenes; n o cursa c o n amnes ia , n i apraxias n i agnosias, pese a ser una d e m e n c i a co r t i c a l ; las a l te rac iones conduc tua l e s y de l l engua je son más precoces ; y n o aparecen ov i l l o s n i placas neuríticas.
Afecta a pacientes de mediana edad, s iendo una de las demencias más frecuentes entre los pacientes de 45 a 65 años. Se manif iesta c o m o una demenc ia lentamente progresiva, donde las alteraciones de la persona l idad son los síntomas más l lamat ivos: d i f icul tades en las relaciones sociales, en la emoción, en el insight y con pérdida de las capacidades ejecutivas.
A med ida que avanza la enfermedad, la apatía y la abu l ia d o m i n a n el cuadro . Con juntamente a estos síntomas, los fal los en la memor i a reciente y la capac idad de aprendiza je son muy habituales. El lenguaje se ve afectado desde las fases inic ia les, pud i endo presentarse c o m o el pr imer síntoma (las l lamadas afasias pr imar ias progresivas). No aparecen alteraciones de t i po agnosia o apraxia, c o m o en la EA.
3.4. Demencia vascular
Demencia multiinfarto
Es aquel la que se produce c o m o consecuencia de múltiples áreas de infarto cerebral . Hay que sospecharla cuando la demenc ia t iene un in ic io brusco, sobre todo, si existen antecedentes de cualquier t ipo de enfermedad vascular cerebral y se acompaña de signos de foca l idad neurológica (MIR 97-98, 37). La causa más frecuente es la embo l i a cerebral bi lateral rec idivante.
Enfermedad de Binswanger
También denominada encefalopatía subcort ica l arteriosclerótica, es una forma de demenc ia vascular asociada a HTA y aterosclerosis. Se caracteriza por una desmielinización difusa de la sustancia b lanca subcort ica l con aumento del tamaño ventr icular subyacente (Figura 20).
Cursa c o m o una demenc ia subcort ica l , con marcha típica a pequeños pasos y base de sustentación ampl ia , parálisis pseudobulbar y signos cort icoespinales.
RECUERDA Las d e m e n c i a s d e causa v a s c u l a r son las segundas en f r e c u e n c i a . P u e d e n ser c o r t i c a l e s o s u b c o r t i c a l e s , y se c a r a c t e r i z a n p o r su a p a r ic ión b r u s c a , c o n f o c a l i d a d neurológica , y su c u r s o c l í n i co t'luc-t u a n t e .
La leucoaraiosis es un término neurorradiológico que describe las áreas hipodensas en la tomografía computar izada o hiperintensas en la RM, de distribución per iventr icu lar y en centro semiova l , que reflejan la desmielinización. Es típica de esta enfermedad, pero no patognomó-nica.
Figura 20. Resonancia magnética cerebral . Paciente con en f e rmedad d e B inswanger; en la RM se aprecia h iper in tens idad di fusa per iventr icu lar
co r respond ien te al concep to de leucoaraiosis
2 6

Neurología y neurocirugía
3.5. Demencia por cuerpos de Lewy Es la tercera causa de demenc ia en el anc iano, después de la EA y de la
demenc ia vascular. El estudio anatomopatológico revela un p r e d o m i
n io de los cuerpos de Lewy a nivel neocort ica l . Los pacientes presentan
un deter ioro cogn i t i vo lentamente progresivo de t ipo f rontosubcor t i ca l .
Las f luctuac iones cognit ivas son muy frecuentes, con variaciones no
tables en la atención y el estado de alerta. Las a luc inac iones visuales
o presenciales son características, así c o m o las alteraciones del sueño
REM (en la fase de atonía muscular, la presencia de un act iv idad física
incesante apoya el diagnóstico).
Se acompaña habi tua lmente de un park insonismo que, aunque fre
cuentemente t iene un p r edomin i o de la clínica rigidoacinética, con es
caso temb lo r y mala respuesta a la L-DOPA, puede ser indi ferenc iable
del de la Enfermedad de Parkinson (MIR 05-06, 59). Es f recuente la
elevada suscept ib i l idad a los neurolépticos, con empeoramien to motor
y cogn i t i vo con su uso (MIR 0 0 - 0 1 , 51).
Casos clínicos representativos
Un hombre de 77 años es traído a la consulta por su esposa para evaluación. Ella refiere que, durante los últimos seis meses, su marido ha experimentado fuertes alucinaciones visuales y auditivas e ideas delirantes paranoides. Asimismo, durante el último año, los déficit cognitivos progresivos se han vuelto cada vez más evidentes para ella y para otros miembros de la familia. Estos déficit, todavía en una fase leve, implican la memoria, las habilidades matemáticas, la orientación y la capacidad de aprender nuevas habilidades. Aunque se han observado fluctuaciones en la capacidad cognitiva día a día o semana a semana, es evidente un curso en declive definido. El paciente no ha estado tomando ningún medicamento. El examen físico revela un temblor en reposo de la rigidez en rueda dentada. La marcha del paciente se caracteriza por pasos cortos y arrastrados, y una disminución del balanceo de los brazos. iQué síndrome clínico es más compatible con los síntomas de este hombre?
1) E n f e r m e d a d d e P a r k i n s o n . 2) D e m e n c i a d e Creu tz fe ld t- Jacob . 3) D e m e n c i a c o n c u e r p o s d e L e w y . 4) D e m e n c i a v ascu l a r . 5) D e m e n c i a d e l lóbulo f r o n t a l .
M I R 0 5 - 0 6 , 5 9 ; RC 3
Ante una historia progresiva de 8 años de evolución, a partir de los 60 años, de deterioro intelectual, errores inexplicables en la actividad cotidiana, descuido en la higiene personal, que lleva al enfermo a una dependencia absoluta de sus familiares, con inmovil idad total, incontinencia de esfínteres, pérdida de peso, convulsiones, mioclonías y muerte, podremos establecer un diagnóstico de:
1) E n f e r m e d a d de P a r k i n s o n . 2) Degenerac ión h e p a t o l e n t i c u l a r o e n f e r m e d a d d e W i l s o n . 3) Encefalopat ía e s p o n g i f o r m e d e Creu tz fe ld t- J acob p o r "prote ína pr ión" . 4 ) D e m e n c i a vascu la r . 5) D e m e n c i a de l t i p o A l z h e i m e r .
RC 5
27

Neurología y neurocirugía
04 ENFERMEDADES
VASCULARES CEREBRALES
Orientación
MIR r
Aspectos esenciales k.
Este t e m a es el más i m p o r t a n t e de t o d a la neurología, y po r e l l o se d e b e es tud ia r en p r o f u n d i d a d . D e n t r o del m i s m o , es f u n d a m e n t a l c o n o c e r los t i pos d e acc iden tes ce rebrovascu la res , las d is t intas etiologías y factores d e r iesgo, la c l ín ica en función d e los te r r i to r ios vasculares a fectos , los métodos diagnósticos, el t r a t a m i e n t o en fase a g u d a y la p r o f i l a x i s . H a y q u e gu ia rse po r los Aspectos Esenciales de l t e m a y n o o l v i d a r las l l amadas de atención i n c l u i da s en e l t e x t o .
[T~| Los acc identes cerebrovasculares (ACV) pueden ser isquémicos ( 8 0 - 8 5 % de los casos) o hemorrágicos (15-2 0 % de los casos). Los factores de riesgo son en gran m e d i d a c o m u n e s a los de la patología isquémica car díaca. Destaca la H T A c o m o p r i n c i p a l fac tor de riesgo en los A C V tan to ateroscleróticos c o m o hemorrágicos, y la fibrilación aur i cu la r , en el caso de los embólicos.
Qf) Los A C V embólicos p r o d u c e n un déficit c o m p l e t o desde el i n i c i o y c o n mayo r t endenc i a a una t rans fo rma ción hemorrágica.
QTJ Los A C V isquémicos por afectación de la arter ia carótida interna se deben f u n d a m e n t a l m e n t e a ateroscle-rosis. Su clínica típica es la amauros is fugax. Si ésta se acompaña de d o l o r ce rv i ca l y síndrome de H o r n e r ipsi laterales, hay q u e sospechar una disección carotídea.
[~4~] Los A C V isquémicos por afectación de la arter ia cerebra l anter ior son infrecuentes . Su etiología suele ser embólica, y su cl ínica típica consiste en hemipares ia y hemih ipoestes ia contra latera les de p r e d o m i n i o c ru r a l , ref lejos arca icos y desinhibición c o n d u c t u a l .
Qf| Los A C V isquémicos por afectación de la arteria cerebral media son los más frecuentes. Su clínica típica consiste en hemiparesia y hemihipoestesia contralaterales de p r e d o m i n i o fac iobraqu ia l , hemianops ia homónima contra-lateral, afasias (si se afecta el hemisfer io dominante ) , agnosias, a lexia con agrafía y desviación ocular ipsi lateral .
(~o~] Los A C V isquémicos por afectación de la arter ia cerebra l poster ior pueden ser distales (hemianops ia homón ima contra la tera l c o n respeto macu la r , a lex ia y aca lcu l ia ) o p rox ima l e s (síndrome talámico: hemianestes ia g loba l cont ra la te ra l , hiperpatía en el h e m i c u e r p o afecto y m o v i m i e n t o s anormales ) .
|"T~] Los A C V isquémicos por afectación vertebrobasi lar p roducen los síndromes cruzados : hemipares ia y h e m i h i poestesia contralaterales y lesión ipsilateral de pares craneales (el par craneal de te rmina el lado de la lesión).
QTJ Los síndromes d isar t r ia-mano to rpe y ataxia-hemipares ia se pueden dar tanto en lesiones de la cápsula in ter na cont ra la tera l c o m o en las de p ro tube ranc i a .
|~g~j D e n t r o de los síndromes lacunares, hay q u e saber q u e se p r o d u c e n por afectación de pequeños vasos ( l ipo-hia l inosis ) y q u e el más f recuente es el ictus m o t o r p u r o (por lesión de l b razo poster ior de la cápsula in terna o de la p ro tube ranc i a anter ior ) .
QJjj Las t rombos i s venosas dura les pueden estar p roduc idas por muchas causas sistémicas y locales (véase tabla ) y suelen debutar por hipertensión in t rac ranea l . Es típico el s igno de la del ta vacía en la TC.
QTJ La p r ime ra p rueba diagnóstica q u e se real iza ante un A C V es la TC craneal para va lorar la presencia de he morragias (los ha l lazgos de la i squemia p u e d e n no v isual izarse en las pr imeras 24-72 horas).
| l 2 | El t r a t am ien to en fase aguda de los A C V isquémicos consiste en adopta r med idas de sopor te (con especia l con t ro l de la tensión arter ia l ) , fibrinólisis c o n rt-PA (salvo con t ra ind i cac iones ) , antiagregación y an t i coagu l a c ión (si c o n fibrilación aur i cu la r , disección carotídea).
[T3] La p ro f i l ax i s y el t r a t am ien to en fase crónica de los A C V isquémicos consiste en el c o n t r o l de los factores de riesgo ca rd iovascu la r (sobre t o d o de la HTA ) , antiagregación, h i p o l i p em ian t e s ( inc luso en normocolesterolé-micos ) , anticoagulación (en mayores de 65 años, si c o n fibrilación aur i cu la r o factores de r iesgo ca rd iovas cular ) y endarterectomía carotídea, si la estenosis es s ign i f i ca t i va .
Q~4~j Las hemorrag ias in t raparenqu imatosas p r o d u c e n cl ínica de hipertensión int racranea l y de te r io ro del n ive l de consc ienc i a . Se c las i f i can en hemorrag ias h ipertensivas (profundas) y hemorrag ias lobares espontáneas (causa más f recuente en anc ianos por angiopatía a m i l o i d e , y en jóvenes por ma l f o rmac i ones vasculares) .
P regun tas
- MIR -MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR - MIR
09-10, 63 08-09, 62 07-08, 64, 153 06-07, 55 05-06, 53 03-04, 241 , 249 02-03, 204, 213 01-02, 52 00-01, 53 00-01F, 71 99-00, 199 99-00F, 60, 63, 70 98-99, 61 98-99F, 70, 80, 81 97-98, 52, 53, 113
Es la tercera causa de muerte tras las cardiopatías y el cáncer. Su inc idenc ia aprox imada es de 0,5-1/100.000 habitantes. En personas mayores de 75 años, esta inc idenc ia aumenta a 20-30/1.000 habitantes.
4.1. Territorios vasculares cerebrales
Los terr i torios vasculares cerebrales están descritos en la Figura 2 1 .
2 8

Neurología y neurocirugía
Área mortora y premotora MMSS y cara
Figura 2 1 . Territorios vasculares cerebrales
Arteria cerebral anterior. Se or ig ina de la arteria carótida interna en la parte anter ior del polígono de W i l l i s . Irriga el córtex motor y sensit ivo de las piernas y pies, córtex frontal motor suplementar io , centros corticales de la micción en los lóbulos paracentrales, por ción anteroinfer ior del brazo anter ior de la cápsula interna, porción anteroinfer ior de la cabeza del núcleo caudado, porción anterior del g lobo pálido (globus pallidus) y putamen e hipotálamo anterior. Los cuatro últimos terr i tor ios se irrigan a través de las arterias lenticuloestr iadas, donde la más importante es la arteria recurrente de Heubner , que se or ig ina de la arteria cerebral anterior a nivel o distal a la unión de la comun ican te anterior. Arteria coroidea anterior. Se or ig ina de la porción suprac l inoidea de la arteria carótida interna. Irriga la porción anter ior del h i p o c a m po, uncus, amígdala, g lobo pálido, co la del núcleo caudado, tálamo lateral y cuerpo gen icu lado, y brazo posterior de la cápsula interna. Arteria cerebral media. Es el vaso más frecuentemente afectado en los ictus isquémicos. Irriga gran parte del córtex motor y sensitivo f rontopar ieta l , áreas frontales para los mov imien tos oculocefálicos conjugados, radiaciones ópticas, córtex sensorial aud i t i vo y áreas del lenguaje (hemisferio dominante ) . Por med io de las arterias len t iculoestr iadas, irr iga el putamen, cabeza y cuerpo del núcleo cau dado, g lobo pálido lateral, brazo anter ior de la cápsula interna y porción superior del brazo posterior de la cápsula interna. Arteria cerebral posterior. T iene su or igen en la circulación verte-brobasi lar , a n ive l distal de la arteria basilar, y comp le t a por detrás el polígono de W i l l i s . Irriga la superf ic ie infer ior del lóbulo t e m pora l , lóbulo occ ip i t a l , núcleo ro jo , sustancia negra, parte media l de los pedúnculos cerebrales, núcleos del tálamo, h i p o c a m p o e hipotálamo poster ior . Irrigación troncoencefálica. Las arterias vertebrales, que se or ig inan de las subclavias, conf luyen a nivel de la unión bu lboprotuberanc ia l para formar la arteria basilar. Previamente, la arteria vertebral da lugar a dos pequeñas arterias mediales que conf luyen para formar la
arteria espinal anterior. Lateralmente, de la arteria vertebral se o r ig i na la cerebelosa posteroinfer ior , que irriga la parte posterolateral del bu lbo e infer ior del cerebelo. La arteria basilar se dir ige hacia arriba y delante, en la zona media l anter ior de la protuberanc ia . Da lugar a pequeñas arterias mediales perforantes, c ircunferenciales cortas y a las arterias cerebelosas anteroinfer ior y anterosuperior. Termina en la unión pontomesencefálica, bifurcándose en las arterias cerebrales posteriores.
4.2. Clasificación y factores de riesgo
Se dist inguen dos grandes grupos de lesiones vasculares: isquémicas y hemorrágicas. • Las lesiones isquémicas representan el 8 0 - 8 5 % de los casos. Pueden
ser focales (por obstrucción arterial o venosa) o difusas (parada cardíaca, anoxia o hipoperfusión). También pueden clasificarse c o m o trombóticas o embólicas.
• La hemorragia intracraneal representa aprox imadamente un 15-2 0 % de todos los accidentes vasculares cerebrales, s iendo la hiper tensión arterial (HTA) el pr inc ipa l factor asociado ( 5 0 - 7 0 % de los casos). La mayoría de estas hemorragias están local izadas p ro funda mente en los hemisferios cerebrales.
Los mecanismos por los que se puede produc i r un accidente vascular cerebral son básicamente cuatro: • Por patología intrínseca de los vasos secundaria a aterosclerosis,
l ipohia l inos is , vascul it is, depósito de ami lo ide , mal formac iones vasculares, etc.
2 9

Manual CTO de Medicina y Cirugía, 8.a edición
• Por obstrucción vascular secundaria a material embólico o r ig inado a nivel cardíaco o en los vasos extracraneales (embol i smo arterioar-ter ia l , generalmente con or igen en la arteria carótida interna). Por hipoperfusión secundaria a hipotensión o incremento de la v is cos idad sanguínea.
• Por ruptura de un vaso sanguíneo dentro del espacio subaracnoideo o dentro del parénquima cerebral .
Los factores de riesgo para la enfermedad cerebrovascular son los mis mos que para la cardiovascular, pero en nuestro caso sin dudar lo el pr inc ipa l factor de riesgo es la HTA. Según el t ipo de enfermedad cerebrovascular los pr incipales factores de riesgo son:
• Enfermedad vascular aterosclerótica: hipertensión, hipercolestero-lemia, D M y tabaquismo.
• Embotica: fibrilación aur icular e infarto de m ioca rd io reciente (generalmente anterior) .
• Hemorrágica: la hipertensión es el pr inc ipa l factor de riesgo para la hemorragia cerebral pr imar ia .
• También la hipertensión es el factor de riesgo más importante para la l ipohia l inos is , que es la base de los infartos lacunares.
Q RECUERDA La H T A es un fac tor de r iesgo tanto para la en f e rmedad vascular cere bra l c o m o para la cardiopatía isquémica, pero el r iesgo re la t i vo es m a yor para la p r i m e r a . Además, el t r a t am ien to de la H T A ha demos t r ado mayo r efecto en la prevención de la en f e rmedad vascular cerebra l que en la prevención de la cardiopatía isquémica.
4.3. Enfermedades cerebrovasculares isquémicas
Etiología
Infarto aterotrombótico (Figura 22)
La mayor parte de las enfermedades cerebrales vasculares isquémicas son or iginadas por la aterosclerosis y sus compl i cac iones trombóticas y tromboembólicas. La aterosclerosis puede afectar a los vasos extracraneales e intracraneales y puede produc i r patología por embolización arterioarterial o por estenosis u oclusión in situ, dando lugar a s i tuac iones de bajo gasto. La localización preferente de las placas de ateroma es la bifurcación de la carótida y el or igen de la arteria carótida interna.
Figura 22. Angiografía carotídea d o n d e se aprecia afectación aterosclerótica grave en el o r igen de la arteria carótida in terna
Clasificación
Las enfermedades cerebrovasculares isquémicas se clasif ican en : • Accidente isquémico transitorio (AIT). Déficit neurológico con una
duración menor de 24 horas. En general , la duración es menor de una hora; si es mayor, la TC suele mostrar lesiones isquémicas. Ictus o stroke. Déficit neurológico que dura más de 24 horas, cau sado por disminución del f lu jo sanguíneo en un terr i tor io .
• Ictus progresivo. Es un déficit neurológico de instauración súbita que progresa o fluctúa mientras el pac iente permanece ba jo observación. Puede ser deb ido a estenosis trombótica progresiva de una arteria, desarro l lo de edema cerebra l , obliteración progresiva de ramas colaterales o hipotensión arter ia l . O t r a causa es el sangrado post in fa r to , o conversión de un infar to " b l a n c o " en un infar to " r o j o " , que aparece en el 4 0 % de los casos, y es más f recuente en ictus cardioembólicos extensos por reperfusión tras un pe r i odo de i squemia que ha dañado el endo-te l i o vascular . El sangrado post in far to rara vez p roduce nuevos síntomas.
RECUERDA U n ictus m a l i g n o es un ictus de l t e r r i t o r i o de la arter ia cerebra l med i a q u e se c o m p l i c a c o n edema cerebra l c o n d e s p l a z a m i e n t o de la línea m e d i a ( swe l l ing ) y c o n disminución de l n i ve l de c o n c i e n c i a . Se rea l iza una craneotomía descompres i va sólo si es de r e cho y el pac i en t e es j o v e n .
Infarto cardioembólico
Const i tuyen aprox imadamente un 2 0 % de los accidentes de t i po isquémico. La causa más frecuente de embo l i smo cerebral cardiogénico es la fibrilación aur icular paroxística o persistente (lo más frecuente es que sea una fibrilación aur icular (FA) en un corazón no reumático (véase Sección de Cardiología y cirugía cardiovascular), lo cual es lo más prevalente. No obstante, la FA en el contexto de cardiopatía reumática t iene más riesgo pero es más infrecuente).
Otros factores de riesgo son los siguientes: • Trombos murales.
- A partir de áreas discinéticas secundarias a infarto de miocar d io ; el que más frecuentemente embo l i za es el infarto agudo de mioca rd io ( IAM) anterior o septal. El 3 5 % de los IAM anteriores desarrol lan t rombos murales, y de ellos el 4 0 % , si no se anticoa-gula, p roduce embo l i smo sistémico en los cuatro meses s iguientes, prec isando anticoagulación profiláctica durante seis meses.
- A partir de cardiomiopatías, fundamenta lmente la di latada. Em-bol izan en un 1 5 % , con una mayor inc idenc ia si hay fibrilación aur icular . Ac tua lmente es indicación para ant icoagular si se asoc ian ambas, no siendo indicación absoluta de ant icoagular la presencia de cardiomiopatía aislada.
• Enfermedad valvular. Especialmente frecuente en pacientes con f i brilación aur icular y estenosis mi t ra l . Ot ra causa es la endocardi t is infecciosa o no infecciosa, esta última en asociación a procesos tu-morales de base.
30

Neurología y neurocirugía
Otras. Aumen to del tamaño ventr icular izqu ierdo , foramen oval permeable y aneurismas ventr iculares.
RECUERDA Los A C V isquémicos embólicos suelen p roduc i r se en el t e r r i t o r io de la arter ia cerebral anter ior . Cursan c o n un déficit c o m p l e t o desde el i n i c i o y t i enen mayor r iesgo de transformación en hemorrágicos (sobre t o d o tras la reperfusión, por daño endote l i a l ) .
Ilón, en leucocitosis mayor de 150 .000 células/pl y en macrog-lobul inemias o mie loma múltiple (la IgM es la ¡nmunoglobulina que produce un mayor síndrome de hiperviscosidad). Síndrome de hipercoagulabilidad, c o m o el que se produce en pacientes con tumores (adenocarcinomas), durante el embarazo o puerper io o durante el t ratamiento con ant iconcept ivos orales. En asociación a anticuerpos antifosfolípidos o anticardiolipinas. Se deben sospechar en pacientes con abortos de repetición y antecedentes de trombosis venosas.
Los cuadros embólicos se presentan con el déficit c o m p l e t o desde el i n i c i o (MIR 02-03, 213 ) . Con f recuenc ia , el e m b o l i s m o cerebral se p roduce sin objet ivarse una fuente obv ia . Se habla de e m b o l i s m o de etiología desconoc ida cuando la monitorización cardíaca, eco-cardiografía y eco-Doppler de t roncos supraaórticos no cons iguen demostrar una fuente de émbolos. El 4 0 % de los ictus isquémicos se sitúan en esta categoría; a veces, en estos casos, la eco-Doppler transesofágica ha demost rado placas de aterosclerosis embolígenas en la aorta ascendente.
Infarto lacunar (Figura 23)
Secundario a arteriopatía o l ipohial inosis de pequeño vaso, son menores de 15 m m de tamaño y están local izados en el terr i tor io de distribución de las pequeñas arterias perforantes procedentes del polígono de Wi l l i s , arteria cerebral media o sistema vertebrobasilar. Los infartos lacunares representan el 2 0 % de toda la patología vascular. La hipertensión arterial es el factor de riesgo más importante, observándose en un 6 5 % de pacientes. Otros diagnósticos etiológicos incluyen la diabetes mell i tus, cardiopatía, etc. Suelen ocasionar un déficit neurológico de más de 24 horas de duración. Hasta un 5 0 % de los pacientes presentan acc identes isquémicos transitorios previos, de una duración aproximada de 30 minutos y una latencia entre el AIT y el infarto lacunar de 24-72 horas.
Figura 23. Localización de los síndromes lacunares más frecuentes
• Arteriopatía no arteriosclerótica: disección arterial , enfermedad de moyamoya , displasia f ibromuscular .
• Enfermedad sistémica: conectivopatía, síndrome mie lopro l i fe ra t i vo , metabolopatía.
• Trombosis venosa cerebral.
Infarto de etiología indeterminada
Tras un exhaust ivo estudio diagnóstico, no se ha encontrado el mecan ismo etiopatogénico subyacente.
Síndromes vasculares
La localización de los síndromes vasculares más frecuentes está representada en la Figura 24 .
Arteria carótida interna
La bifurcación y el or igen de la arteria carótida interna (pared posterior) es el lugar de mayor inc idenc ia de aterotrombosis. Produce síntomas básicamente por embol i smos arterioarteriales, y menos frecuentemente por bajo f lu jo , pero su oclusión puede ser asintomática, si se establece de forma progresiva gracias a la circulación colatera l .
Los síntomas pueden simular, en muchos casos, los de afectación de la arteria cerebral media . La clínica más típica es la amaurosis fuga (MIR 0 0 - 0 1 , 53 ; MIR 99-00F, 63) por oclusión de la arteria oftálmica, que consiste en una pérdida uni lateral de la visión que se instaura en 10-15 s y dura escasos minutos .
Comienza c o m o visión borrosa indo lora que evo luc iona hasta la ceguera monocu la r comple ta , con resolución total posterior. En el fondo de o jo pueden observarse en ocasiones émbolos de colesterol en vasos retiñíanos.
Q RECUERDA En la exploración de l f o n d o de o j o de la amauros i s fugax p u e d e n o b servarse cr ista les de co les te ro l en los vasos retiñíanos. Por o t ra par te , en el f o n d o de o j o de la oclusión de la arter ia cen t ra l de la re t ina , lo característico es la p a l i d e z r e t i n i ana c o n la " m a n c h a c e r eza " a n i ve l macu l a r .
Infarto de causa inhabitual
• Causas hematológicas. - Hemoglobinopatías. La anemia de células fa lc i formes es la he-
moglobinopatía más frecuentemente relacionada con ictus. - Síndrome de hiperviscosidad. Se produce en pol ic i temias con
hematocr i to superior al 5 0 % , en t romboci tos is mayor de un mi-
La asociación de amaurosis fugaz, do lor cervical y síndrome de Horner es típica de la disección de arteria carótida.
Q RECUERDA Otras causas de síndrome de Horner , además de la disección carotídea,
son : el síndrome bu lba r lateral (Wa l l enberg ) , la s i r i ngomie l i a ce r v i c a l , la dilatación aur i cu l a r y el t u m o r de Pancoast.
31

Manual CTO de Medicina y Cirugía, 8.a edición
ACA: arteria cerebral anterior ACM: arteria cerebral media ACP: arteria cerebral poster ior
Área mortora y premotora MMII (parálisis espástica contralateral)
Área mortora y premotora MMSS y cara (parálisis espástica contralateral)
Centro de la mirada conjugada (desviación hacia la lesión)
ACA Corteza prefontral
(mut ismo, abulia, moria, reflejos arcaicos)
Área de Broca (afasia motora )
Corteza audit iva (alucinaciones auditivas,
sordera cortical)
ACM Área de Wernicke (afasia sensitiva)
Radiaciones ópticas (hemianopsia homónima
contralateral)
Corteza somatosensorial MMII (hipoestesia contralateral)
Corteza somatosensorial MMSS y cara
(hipoestética contralateral)
Corteza visual pr imaria (hemianopsia homónima contralateral con respeto macular; ceguera cort ical )
Tálamo (síndrome talámico)
Figura 24 . Localización de los síndromes vasculares más f recuentes
Arteria cerebral anterior
El infarto de la arteria cerebral anterior (ACA) es muy poco habitual, sea por la causa que sea, sin embargo, cuando se producen, la causa más probable del mismo es un embol ismo de origen cardíaco, no la aterotrom-bosis. No se debe olvidar que si se produce un émbolo cardíaco, la arteria que se afecta con más frecuencia es la A C M , no la ACA (MIR 97-98, 52).
La oclusión distal a la arteria comun i can te anterior da lugar a: Hemiparesia y hemihipoestesia contralaterales de p redomin io crural .
• Disminución de la act iv idad ps icomotora y del lenguaje espontáneo secundar io a afectación de áreas prefrontales.
• Reflejo de prensión, succión y r igidez paratónica por lesión de las áreas motoras suplementarias frontales.
• Apraxia de la marcha y, a veces, incont inenc ia ur inar ia por afectación del lóbulo frontal parasagital (en lesiones bilaterales).
i de H a k i m RECUERDA En lesiones f ronta les b i latera les , puede aparecer la tríada
A d a m s (apraxia de la marcha , i n con t i nenc i a ur inar ia y de te r io ro cogn i t i vo ) , característica de la h id roce fa l i a no rmotens i va .
Puede haber también asomatognosia (heminegl igenc ia corpora l ) , anosognosia y desorientación espacial en lesiones del hemisfer io no dominan te .
RECUERDA La afasia nos loca l i za la i squemia a n ive l co r t i ca l en el t e r r i t o r i o vascular de la arteria cerebra l med i a de l hemis fe r io d o m i n a n t e .
Arteria coroidea anterior
Cursa con hemiparesia y hemihipoestesia contralaterales, i nc luyendo la cara, y a veces hemianopsia contralateral homónima. El diagnóstico diferencia l con afectación de la arteria cerebral media a nivel clínico suele ser difícil.
Arteria cerebral posterior
Por lesión occ ip i t a l , da lugar a hemianops ia contralateral que suele respetar la visión macular . Los reflejos pupilares están conservados. Impl ica a veces alexia y acalcul ia (MIR 01-02, 52) .
Arteria cerebral media
Es el síndrome vascular más frecuente. Cursa con : • Hemiparesia y hemihipoestesia contralaterales, de p r edomin i o fa-
c iobraqu ia l (MIR 99-00, 199). • Hemianops ia homónima contralateral . • Desviación oculocefálica hacia el lado de la lesión, con conserva
ción de los reflejos oculocefálicos y oculovest ibulares. • Afasia de Broca, We rn i cke o g loba l , depend iendo de la localización
y extensión de la afectación (en lesiones del hemisfer io dominante ) .
RECUERDA Si existe a lex ia c o n agrafía, la afectación vascular está en el t e r r i t o r i o de la cerebra l medía; mientras q u e si existe a lexia sin agrafía, el t e r r i t o r i o a fec tado es el de la cerebra l poster ior .
Si se afecta la circulación p rox ima l , y dado que la mayor parte de la irrigación talámica depende de esta arteria, aparecerá un síndrome talámico caracter izado por hemianestesia contralateral extensa y para todos los t ipos de sensibi l idades, hiperpatía o do lor en el hemicuer-
32

Neurología y neurocirugía
po afectado, mano con mov imien tos pseudoatetoides secundarios a la pérdida de la sensibi l idad prop iocept iva (mano talámica), coreoatetosis y hemiba l i smo por extensión lesional a las áreas subtalámicas, aste-rixis contralateral y déficit en la supraducción y convergencia ocu la res. Otros signos y síntomas infrecuentes de la lesión talámica son la alteración en la memor ia , sueño y regulación de la temperatura, afasia subcort ical y defecto transi tor io homónimo del c a m p o visual .
Q RECUERDA Una f o rma senci l la de ident i f i car el te r r i to r io arterial a fectado en la enfer m e d a d isquémica cerebral es valorar si existe hemipares ia y hemianops ia : el síndrome de la arteria cerebral anter ior cursa c o n hemipares ia crural contra latera l y sin hemianops ia ; el de cerebral med ia (el más frecuente) cursa c o n hemipares ia fac iobraqu ia l y hemianops ia homónima cont ra l a terales; y el de cerebral poster ior cursa sin hemipares ia , pe ro c o n h e m i a nopsia homónima congruente contra la tera l , y c o n respeto macular .
Sistema vertebrobasilar
Los procesos isquémicos a este nivel p roducen los l lamados "síndromes cruzados" , caracterizados por alteraciones de vías largas contralaterales (hemiparesia, hemihipoestesia) y signos ipsilaterales cerebelosos o de pares craneales.
Q RECUERDA La patología vascu lar del s istema ver tebrobas i la r p r o d u c e los síndromes c ruzados : hemipares ia y hemih ipos tes ia contra latera les y afectación ips i latera l de pares craneales (el par cranea l de te rmina el l ado de la lesión).
• • • ^ • • • • • • • n H i i H
La isquemia vertebrobasi lar puede produc i r una pérdida brusca de la consciencia , con o sin recuperación posterior, precedida de síntomas de disfunción troncoencefálica (diplopía, vértigo, ataxia, etc.) (MIR 05-06 , 53). Los síndromes más importantes se han descrito prev iamente en la sección de Semiología, dentro del Capítulo Síndromes mesencefáli-cos, pontinos y bulbares (MIR 09-10, 63).
Infartos lacunares
Los síndromes lacunares más frecuentes son: • Ictus motor puro: es el síndrome lacunar más frecuente, con una
topografía lesional habi tua lmente local izada en el brazo posterior de la cápsula interna, aunque también puede local izarse en la porción anterior de la protuberanc ia . Consiste en la paresia o parálisis hemicorpora l f ac iobraqu iocrura l , generalmente proporc ionada (simi lar afectación en cara, brazo y pierna), sin afectación de otras áreas ni del nivel de consc ienc ia o las func iones superiores. Cuando se loca l izan en la rodi l la de la cápsula interna, puede cursar con paresia facial aislada, y cuando afectan a la parte más posterior de la misma, pueden dar lugar a una paresia crural aislada.
• Ictus sensitivo puro: resulta de un infarto lacunar a nivel del núcleo ventral posterolateral del tálamo. La clínica consta de un déficit sensit ivo que afecta a un hemicuerpo , inc lu ida la cara, s iendo menos frecuente la distribución quei roora l (afectación per ibucal y de la mano ipsilateral).
• Ataxia-hemiparesia: es también expresión, generalmente, de un i n farto lacunar loca l izado en el brazo anter ior de la cápsula interna o en la protuberanc ia . Cursa con hemiparesia contralateral (más grave en la ext remidad inferior) y ataxia (habi tua lmente en los miembros con déficit motor ) .
Disartria-mano torpe: cursa con paresia facial y torpeza de la mano ipsi lateral, sin afectación sensitiva. La causa más frecuente es un infarto lacunar en el brazo anterior o la rodi l la de la cápsula interna contralateral al hemicuerpo afectado, aunque puede producirse también por lesiones en la protuberanc ia (MIR 03-04, 249).
Figura 25. RM cerebral . Se observa pequeño in fa r to lacunar p r o f u n d o izqu ie rdo marcado por la f lecha
La existencia de múltiples infartos lacunares puede conduc i r a un sínd rome pseudobulbar (disartria, disfagia, disfonía, parálisis facial b i la te ral y lab i l idad emoc iona l ) .
Estudio diagnóstico (Figura 26)
La prueba diagnóstica inicial en cualquiera de los t ipos de infarto cerebral es la TC craneal, para establecer el diagnóstico de ictus isquémico o hemorrágico, así c o m o para descartar etiologías que pueden cursar como un proceso vascular (tumores, sangrados, metástasis, etc.) e infor ma sobre la extensión de la lesión isquémica. Durante las primeras 24-72 horas, pueden no observarse lesiones isquémicas (MIR 97-98, 53), aunque es posible detectar signos indirectos, c o m o asimetrías de surcos corticales por edema, desplazamiento de estructuras o aumento de den sidad de la arteria cerebral media , en su trayecto basal. Es de escasa ut i l idad para la visualización de infartos vertebrobasilares deb ido a los artefactos óseos que genera la fosa posterior. La TC supera a la resonancia magnética en la detección de sangrados, aunque ésta es más sensible para la visualización de lesiones de fosa posterior (procedimiento de elección) véase (Figura 25). En los infartos lacunares, la prueba in ic ia l de elección en la fase aguda es igualmente la TC craneal (para el diagnóstico diferencial isquemicohemorrágico), pero para su estudio posterior es necesario realizar una RMN craneal, ya que la TC no detecta los infartos menores de 5 m m y los situados en fosa posterior.
Profilaxis y tratamiento
Tratamiento en fase aguda
• Medidas generales: evitar hipertermias, h iperglucemias y elevación excesiva de la tensión arterial , así c o m o descensos bruscos de esta última.
3 3

Manual CTO de Medicina y Cirugía, 8.a edición
u < u
< u
< 9 o
o
s o uj u
o o
o u a
Sospecha clínica ACV AIT (< 24 horas)
ICTUS (> 24 horas)
Normal, pero clínica evidente: repetir en 72 horas
Descarta J ACV
ISQUÉMICO (80%)
< 24 horas > 24 horas Borramiento estructuras Hipodensidad focal
Efecto masa
TA ECG Analítica Rx tórax
TC craneal: elección en fase aguda. Se hará RM si clínica de fosa posterior
o síndrome lacunar o sospecha de trombosis venosa
HEMORRÁGICO (20%) Hiperdensidad
EMBÓLICO INFARTO LACUNAR
Aterosclerosis bifurcación carotídea
(HTA, tabaco, colesterol)
Fibrilación auricular Trombo mural (IAM)
Valvulopatía (EM) Suelen ir a cerebral media (80%)
HTA (Lipohialinosis)
Valorar fibrinólisis según t iempo de evolución
ECO Doppler carotídeo Angiografía
ECO transesofágico ECG
TROMBOSIS VENOSA
Procesos sépticos Embarazo y puerperio Deshidratación (ancianos) Anticonceptivos orales Traumatismos craneales Procesos hematológicos
RM Angiografía
OTROS
RM
Antiagregación con AAS (ticlopidina o clopidogrel de 2. a
elección) Estenosis carotídea
Sintomática Asintomática > 70%Tromboendarterectomía 50-70%?? < 50% AAS
Arteritis: temporal , Takayasu... Disección arterial Enf. moyamoya (volutas de humo) Estados de hipercoagulabilidad Enf. Binswanger Displasia fibromuscular Vasospasmo Anticonceptivos orales
Anticoagulación (diferida si infarto extenso)
Control estricto de la TA
AAS
Signo de la 8 vacía enTC con contraste
Anticoagulación
Figura 26. A l g o r i t m o diagnóstico y terapéutico de los accidentes cerebrovasculares
• Fibrinólisis con rt-PA: está indicada la administración de rt-PA en pacientes con : - Ictus isquémico de menos de tres horas desde la instauración de
los síntomas (MIR 08-09, 62). - Paciente menor de 80 años. - Puntuación en la escala N1HSS (escala internacional de grave
dad clínica del ictus) menor de de 25 puntos (Tabla 12). - Ausencia de a lguno de los criterios de exclusión que aparecen
en la Tabla 13 (MIR 07-08, 153).
• Antiagregación: en pacientes que no cump len criterios de fibrinólisis: el uso de ácido acetilsalicílico (AAS) 300 m g en las primeras 48 horas tras el ictus isquémico reduce el riesgo de recurrencia y la tasa de mor ta l idad a med io p lazo .
La inc idenc ia de ictus isquémico es de 1-2% por año tras infarto de mioca rd io , s iendo el riesgo mayor durante el pr imer mes post infarto ( 3 0 % ) . En estos pacientes, la prof i laxis pr imar ia inc luye : • Anticoagulación oral. Mantener el índice internacional no rma l i za
do (INR) entre 2 y 3, si el paciente asocia fibrilación aur icular (MIR 99-00F, 60).
• Estatinas ( inhibidores de la HMC-CoA reductasa), incluso en pa cientes con niveles de colesterol normales. Su efecto profiláctico parece estar al margen de su efecto h ipo l ipemian te y se debe a una estabilización del endote l io y placa aterosclerótica, efectos an t i i n f lamator ios e inhibición de la adhesión y agregación plaquetar ia.
La anticoagulación a largo plazo con dicumarínicos constituye la terapéutica de elección en la prevención primaria de la patología vascular cerebral, en los casos de fibrilación auricular asociada a patología valvular.
Prevención primaria
El t ra tamiento de la HTA reduce sustancialmente el riesgo de ictus. Una disminución en la presión arterial diastólica de 5-6 m m H g reduce el riesgo en un 4 2 % . El t ra tamiento de la HTA diastólica aislada en el anc iano d i sminuye el riesgo en un 3 6 % .
Q RECUERDA Las ind i cac iones de anticoagulación en fase aguda son: fibrilación a u
r icu lar , disección carotídea, ictus isquémico progres ivo o t rombos i s de senos venosos dura les . En fase crónica, se an t i coagu la si se c u m p l e n dos de los s iguientes c r i te r ios : edad super ior a 65 años, fibrilación a u r i cu la r , presencia de factores de r iesgo ca rd iovascu la r o demostración de t r o m b o s ¡ntracardíacos.
34

Neurología y neurocirugía
Estuporoso 0 No c laudica. BM 5 0 Coma 1
PIERNA IZQUIERDA Claudica. BM 4 Algún esfuerzo contra gravedad. BM 3 Sin esfuerzo contra gravedad. BM 2 Ningún m o v i m i e n t o . BM 0
1 2 3 4
••••••̂••Xc.-.:V. Responde ambas cor rec tamente 0 No claudica. BM 5 0 Responde una cor rec tamente 1 Claudica. BM 4 1
PREGUNTAS LOC Incorrecto PIERNA DERECHA Algún esfuerzo contra gravedad. BM 3 Sin esfuerzo contra gravedad. BM 2 Ningún m o v i m i e n t o . BM 0
2 3 4
Responde ambas co r rec tamente 0 Ausente 0 Responde una co r rec tamente 1 Presente en una ex t r em idad 1 Incorrecto 2 Presente en dos ext remidades 2
Ó R D E N E S LOC ATAXIA D E MIEMBROS Si está presente, se localiza en :
Brazo derecho (1 : sí; 0: no) Barzo i zqu ie rdo (1 : sí; 0: no) Pierna derecha ( 1 : sí; 0: no) Pierna izquierda ( 1 : sí; 0: no)
Norma l 0 Norma l 0 MIRADA Parálisis parcial de la mirada 1 SENSIB IL IDAD Hipoestesia l igera a moderada 1
Deviación oculocefálica 2 Hipoestesia severa o anestesia 2
Sin déficit campimétricos 0 Normal , sin afasia 0
CAMPOS V ISUALES Cuadrantanopsia Hemianops ia homónima
1 2 LENGUAJE
Afasia l igera a moderada Afasia grave. Broca, Wernicke...
1 2
Hemianopsia homónima bi lateral , ceguera 3 Afasia g loba l o m u t i s m o 3
Mov im ien tos normales y simétricos 0 Articulación no rma l Ligera a moderada Grave o nartr ia
0
PARÁL IS IS FACIAL Paresia l igera Parálisis parcial
1 2
DISATRIA Articulación no rma l Ligera a moderada Grave o nartr ia
1 2
Parálisis comp le t a 3
Articulación no rma l Ligera a moderada Grave o nartr ia
No claudica. BM 5 0 Sin amorma l i dad 0 Claudica. BM 4 1 Parcial (sólo una moda l i dad afectada) 1
BRAZO IZQUIERDO Algún esfuerzo cont ra gravedad. BM 3 Sin esfuerzo contra gravedad. BM 2 Ningún m o v i m i e n t o . BM 0
2 3 4
EXTINCIÓN Comple ta (más de una moda l idad ) 2
No c laudica. BM 5 0 Claudica. BM 4 1
BRAZO D E R E C H O Algún esfuerzo cont ra gravedad. BM 3 Sin esfuerzo contra gravedad. BM 2 Ningún m o v i m i e n t o . BM 0
2 3 4
BM Balance motor
Tabla 12. Escala NIHSS (escala internacional de gravedad clínica del ictus)
• Presencia de hemorrag ia en TC craneal previa a la administración del fármaco | • Presentación clínica sugestiva de HSA, inc luso conTC no rma l
• Déficit neurológico escaso o síntomas q u e me jo ran rápidamente • Escala NIH > 25 pun tos • Convuls iones al in ic io del ictus
! • Existencia de diátesis hemorrágica ( t r ombopen i a < 100.000, t r a t am ien to actual con ant icoagulantes , o t r a t a m i e n t o con hepar ina du ran te 48 horas previas y TTPA aumen tado )
• TA > 185/110 o necesidad de mane jo i.v. agresivo para reducir la • Glucosa sanguínea > 400 o < 50 mg/d l • Historia de ictus prev io y D M concomi t an t e • Ictus en los tres últimos meses • Hemorrag ia grave o pel igrosa manif iesta o reciente • An teceden te de lesión de SNC: hemorrag ia , neoplasla, aneur isma, cirugía... • Patología grave c o n c o m i t a n t e (endocardi t is , pancreatit is , gastropatía
ulcerat iva reciente, aneur ismas arteriales, neoplasias con riesgo hemorrágico, hepatopatía grave)
• Cirugía mayor o t r a u m a t i s m o i m p o r t a n t e en los tres últimos meses
Tabla 13. Criterios de exclusión para la fibrinólisis i.v.
Prevención secundaria
• Patología vascular cerebral con origen en territorio carotídeo o
vertebrobasilar:
- Antiagregación. Se ha demostrado que el AAS, en dosis máxi
mas de 300 mg/día, reduce el riesgo de nuevos ictus en ap rox i
madamente un 2 0 - 3 0 % . Actúa a nivel p laquetar io , p roduc i endo
una inhibición irreversible de la c ic looxigenasa y el t r o m b o x a n o
A2 .
La ticlopidina es otro antiagregante p laquetar io , con efect iv idad
igual o superior al AAS, que inh ibe la agregación plaquetar ia
mediada por difosfato de adenosina (ADP). No actúa sobre la
c ic looxigenasa ni fosfodiesterasa, por lo que no mod i f i ca los n i
veles de t r omboxano ni de monofosfato de adenosina cíclico
(AMP c).
Entre los efectos adversos de la t i c l op id ina , destaca la aparición
de neutropenia reversible ( 1 % de los pacientes), por lo que se
deben realizar controles hematológicos periódicos durante los
tres pr imeros meses de t ratamiento. Los trastornos gastrointesti
nales ( fundamenta lmente la diarrea) const i tuyen los efectos i n
deseables más frecuentes.
El ciopidogrel, de la misma fami l i a que la t i c l op id ina a la que
prácticamente ha desplazado, pero con menos efectos secunda
rios a nivel hematológico, está considerado el antiagregante de
segunda elección cuando existe contraindicación para admin i s
trar AAS. Puede administrarse en una sola dosis diar ia .
El dipiridamol actúa por inhibición de la fosfodiesterasa p laque
taria, encargada de degradar el A M P c. Se emplea asociado a
AAS c o m o antiagregante.
35

Manual CTO de Medicina y Cirugía, 8.a edición
< 5 0 % (no signif icativa)
> 7 0 % (signif icativa)
Ant iagregac ión Indiv idual ización de l caso
Endarterectomía
Figura 27. Prevención secundaria de la estenosis carotídea sintomática
Endarterectomía carotídea (Figura 28).
Es el t ra tamiento de elección en pacientes con estenosis carotídea sintomática que supera el 7 0 % de la luz (MIR 97-98, 113). Si la estenosis es infer ior al 5 0 % , hay que administrar al paciente antiagregantes (MIR 98-99F,70). Cuando la estenosis se encuen tra entre el 50-69% , la decisión terapéutica depende de los factores de riesgo vascular (podría indicarse en varones, y síntomas hemisféricos recientes).
En estenosis carotídeas asintomáticas, se ha recomendado la antiagregación. Sin embargo, cuando la estenosis es hemodi-námicamente signif icat iva y evolut iva en el t i empo , puede ser benefic iosa la endarterectomía carotídea, s iempre que la morb i-mor ta l idad operator ia no supere el 5% .
RECUERDA Si la estenosis de la carótida sintomática es del 5 0 - 6 9 % , se opta por la endarterectomía carotídea, en lugar de por la antiagregación, sólo si se trata de un varón joven , no diabético, con esperanza de v ida super ior a c i n c o años y c o n un riesgo quirúrgico infer ior al 6 % . Si la estenosis de la carótida sintomática es comp le ta ( 1 0 0 % ) , no se realiza cirugía, s ino antiagregación.
- Anticoagulación. La ut i l idad de la anticoagulación para la pre vención secundaria de la patología vascular carotídea es con t ro vert ida. Se puede ut i l izar t ransi tor iamente, en pacientes con AIT o ictus minor de repetición, cuando se objet iva una estenosis grave de la carótida interna y mientras se prepara la cirugía elect iva ; también en lesiones estenóticas no accesibles quirúrgicamente (arteria basilar) o cuando la cirugía está contra ind icada.
Patología vascular cardioembólica: si se ev idencia una fuente car-dioembólica, la prof i laxis secundaria de elección es la an t i coagu lación ora l . Cuando el área de isquemia cerebral es ampl ia , no se recomienda la anticoagulación en fase aguda, dado el a l to riesgo de transformación hemorrágica del infarto. En estos casos, se recomienda realizar una anticoagulación di fer ida.
Trombosis venosas
En el 2 5 - 4 0 % de los casos se desconoce la causa. Se han descrito asociaciones con procesos sépticos sistémicos o locales (meningitis) en aprox i madamente un 1 5 % de las trombosis venosas. Otras etiologías asociadas aparecen reflejadas en las Tablas 14 y 1 5. Es un proceso de difícil d iagnóstico debido a la var iabi l idad de sus manifestaciones clínicas y a la ¡nespecificidad de los hallazgos en las pruebas complementar ias.
La clínica es muy variada, desde las formas asintomáticas a las que cursan con cefalea o coma. Suele debutar con un síndrome de hipertensión intracraneal , s iendo la cefalea el síntoma más frecuente. Puede seguirse de un cuadro de foca l idad neurológica con crisis focales o general izadas, hemiparesia, afectación de pares craneales, etc. En la exploración, se puede observar edema de papi la .
COMUNES POCO FRECUENTES
• Infección • Embarazo-puerper io • Enf. in f lamator ia intest ina l • Deshidratación (ancianos) • Síndrome de Behcet • Ant i concept i vos orales • An t i coagu lan te lúpico • Coagulopatías ( t romboc i tos is . • Abuso de drogas
t r o m b o c i t o p e n i a ) • Síndromes • Hematológicos (anemia cel. fa lc i formes, HPN) paraneoplásicos • Tumor (invasión local , por ej.: men ing iomas ) • Traumat ismos
Tabla 14. Causas de trombosis venosa cerebral
Hemorrag ia h iper tens iva Aneur ismas arteriales Ma l fo rmac iones arter iovenosas Vasculopatías (ami lo ide , Moyamoya , vasculitis) Coagulopatías Hemorrag ia i n t r a tumora l Abuso de drogas (cocaína, simpaticomiméticos, anfetaminas) Secundaria a in fa r to venoso
Tabla 15. Causas de hematoma intraparequimatoso cerebral
3 6

Neurología y neurocirugía
En cuanto a las pruebas complementar ias , son útiles: • Líquido cefalorraquídeo: el dato más habitual es un aumento de
presión, que se acompañará de reacción meníngea inf lamator ia si hay un proceso séptico de base.
• T C : puede ser no rma l o mostrar datos ind i rectos de edema cerebral con disminución de los surcos cor t ica les . Puede observarse un in far to co r t i cosubcor t i ca l con hemorragización m u l t i f o c a l . El s igno de la "8 vac ía" (Figura 29) es m u y característico, y consiste en un re fo rzamien to de las paredes del seno t r ombosado rodean do a una zona centra l isodensa que , teóricamente, cor responde al t r o m b o .
Figura 29. TC craneal de pac iente con t rombos i s de seno l ong i tud ina l superior, d o n d e se observa ident i f i cado por la f lecha el s igno del de l ta vacío, que mues
tra la presencia de t r o m b o en el in ter ior de l seno l ong i tud ina l super ior
Resonancia magnética: es la técnica de elección, aunque no exc lu ye la realización de angiografía cerebral . Angiografía cerebral: es la técnica diagnóstica que permite asegurar la existencia de obstrucción venosa, aunque la RMN ha demostrado una buena correlación con la imagen angiográfica y gran f i ab i l idad diagnóstica (Figura 30).
Figura 30. TC ( imagen de la izquierda) y RM ( imagen de la derecha) que muest ran t rombos i s del seno l ong i tud ina l super ior "s igno de la 5 vacía"
en pac iente d iagnos t i cado de ot i t is med ia
4.4. Hemorragia intraparenquimatosa
Los procesos vasculares hemorrágicos representan aprox imadamente el 2 0 % de los ACV. Dado que la hemorragia en el espacio subaracnoi-deo o en el parénquima cerebral p roduce menos daño tisular que la isquemia, los pacientes que sobreviven a la fase aguda pueden mostrar una marcada recuperación func iona l .
La Figura 31 muestra las causas más frecuentes de hemorragia intracraneal espontánea. En este apartado se tratará solamente la hemorragia i ntraparenqu i matosa.
Las mal formac iones vasculares y la hemorragia subaracnoidea por ro tura de aneurisma se revisarán más deten idamente en los dos apartados siguientes.
ACV HEMORRÁGICO (20%)
HEMORRAGIA SUBARACNOIDEA
TC craneal: hiperdensidad
HEMORRAGIA HIPERTENSIVA
• Rotura microaneurismas de Charcot Bouchard
• Déficit neurológico focal de comienzo brusco y curso progresivo
HEMORRAGIA LOBAR ESPONTÁNEA
Sustancia blanca subcortical
PUTAMEN
Hemiparesia y hemihipoestesia contralaterales. Desv. ojos lado lesión. Dism. nivel consciencia.
TÁLAMO Sd. talámico + hemiplejía contralaterales
CEREBELO
Consciencia preservada inicial Cefalea occipital, ataxia, vómitos Hidrocefalia por compr. IV ventr.
PUENTE Véase esquema del coma
Anciano Joven Otras
ANGIOPATIA MALFORMACION AMILOIDE |i VASCULAR
Causa más frec. de hem. no HTA en el anciano. Recidivante
Tratamiento: control TA (evitar cambios bruscos).
Tratamiento de la HTIC | Cerebelosa (> 3 cm de diámetro)
Cirugía si Herniación
Angioma venoso: la más frec. asintomática Malf. arteriovenosa: la más frec. sintomática
Angioma capilar (telangiectasias)
Angioma cavernoso
Hay que sospecharlas en paciente
joven con hemorragia en un lugar atípico e historia de cefalea unilateral y pulsátil
con convulsiones. En estos casos debe hacerse
una angiografía.
Figura 31 . Hemorragia intraparenquimatosa
La hemorragia intraparenquimatosa o intracerebral es preferentemente causada por la ruptura de arterias situadas pro fundamente en el cerebro.
A di ferencia de los ictus isquémicos, de instauración súbita, los hemo rrágicos suelen evo luc ionar en el transcurso de varios minutos , y suelen acompañarse de cefalea, náuseas y vómitos. La sintomatología neurológica dependerá de la localización y del tamaño de la hemorragia.
La prueba diagnóstica de elección cuando se sospecha una hemorragia es la TC craneal (Figura 32).
3 7

Manual CTO de Medicina y Cirugía, 8. a edición
Figura 32. Hema toma in t rapa renqu imatoso p r o f u n d o
Hemorragia intracerebral focal hipertensiva. Las local izaciones anatómicas más frecuentes de la hemorragia cerebral focal de o r i gen hipertensivo son: pu tamen , tálamo, protuberanc ia y cerebelo (Tabla 16). Por tanto, la mayoría de las veces se trata de hemorra gias profundas, que se producen por rotura de microaneur ismas de Charcot-Bouchard, local izados en las pequeñas arterias perforantes. Con menor f recuencia , las hemorragias superficiales son de causa hipertensiva, en las que hab i tua lmente existe algún ot ro proceso patológico subyacente.
PUTAMEN (35-50 % )
Hemiparesia y hemih ipostes la contralaterales, de te r io ro del nivel de consciencia, desviación oculocefálica hacia el lado de la hemorrag ia con preservación de reflejos del t r o n c o
T Á L A M O ( 1 0 - 1 5 % ) Deter ioro del nivel de consciencia, síndrome talámico y hemiplejía contralaterales
C E R E B E L O ( 1 0 - 3 0 % )
Preservación Inicial de nivel de consciencia, cefalea occ ip i ta l , ataxia, vómitos, hidrocefal ia obs t ruc t i va (por compresión del IV ventrículo)
P R O T U B E R A N C I A ( 1 0 - 1 5 % ) Estado de coma, pronóstico infausto
Tabla 16. Clínica y localización de las hemorragias intracerebrales
• Malformaciones vasculares. Las mal formac iones vasculares (aneurismas y mal formac iones arteriovenosas) son la segunda causa de hematoma intraparenquimatoso espontáneo, y deben sospecharse fundamenta lmente en pacientes jóvenes no hipertensos con hemo rragias superficiales. Aunque la rotura de un aneurisma intracraneal característicamente produce una hemorragia subaracnoidea, a lgu nos pueden produc i r hematomas intracerebrales, f undamen ta lmen te los de la bifurcación carotídea, arteria comun i can te posterior y cerebral media . Se diagnost ican en la angiografía y requieren c i r u gía. Debe sospecharse una malformación arteriovenosa ante una hemorragia superf ic ial en un paciente joven con antecedentes de cefalea o crisis epilépticas.
• Angiopatía amiloide o congófila. Es la causa más frecuente de hemorragia espontánea no hipertensiva en pacientes ancianos, y suelen ser de localización lobar subcort ica l . Se presentan clínicamente c o m o hematomas espontáneos recurrentes (MIR 03-04, 241) . A menudo aparecen asociados a la enfermedad de A lzhe imer . El d iag nóstico de certeza sólo se obt iene con la necropsia, ident i f i cando material am i lo ide rojo congo posi t ivo en las arterias cerebrales.
RECUERDA An te una hemor rag ia lobar espontánea y recurrente en un a n c i a n o no h iper tenso, hay q u e sospechar una angiopatía congófila. En ésta, al igual q u e en el A l zhe ime r , se deposi ta p-ami lo ide .
Otras causas de sangrado cerebral focal. Las distintas coagu lopa tías, el t ratamiento con ant icoagulantes y los trombolíticos son otras causas frecuentes de hemorragia intracerebral de cua lquier l oca l i zación.
RECUERDA Las neoplasias intracraneales c o n mayor tendenc ia al sangrado son co r l o ca r c i noma , me l anoma , pulmón, riñon y t i ro ides. "Co-me pu-ri-to") .
Aunque potenc ia lmente cua lquier t umor cerebral puede sangrar, la hemorragia int ratumora l suele ser un signo de ma l ign idad . Es espec ia lmente frecuente en las metástasis cerebrales del me lanoma (las que con más frecuencia sangran), co r ioca rc inoma , carc inoma bron-cogénico, ca rc inoma de células renales y ca rc inoma de t iroides. Entre los tumores pr imar ios, suelen sangrar el g l ioblastoma mul t i fo r me en los adultos y el medu lob las toma en los niños. No obstante, también se han descrito hemorragias en tumores benignos: menin-g ioma, o l i godendrog l ioma , adenoma de hipófisis y hemangiobtas-toma , entre otros.
El abuso crónico de drogas simpaticomiméticas, entre ellas las anfetaminas y la cocaína, también se ha asoc iado a hemorrag ias i n t raparenqu ¡matosas. F ina lmente , en ocasiones puede verse la transformación hemorrá-gica de un infar to cuando se p roduce la reperfusión de vasos que habían s ido dañados por la i squemia . El r iesgo aumenta con el tamaño del in far to , y es mayor en infar tos extensos de or igen cardioembólico, por lo que en estos casos es d i scu t ida la anticoagulación en fase aguda, que puede favore cer la transformación hemorrágica.
Tratamiento
El t ratamiento médico se basa en el cont ro l de la tensión arterial y en la utilización de man i to l y otros agentes osmóticos para reducir la presión intracraneal .
La indicación quirúrgica en los hematomas i n t r a p a r e n q u i m a t o s o s es un t ema e n o r m e m e n t e c o n t r o v e r t i d o en la l i t e ra tu ra , y debe cons idera rse de f o r m a i n d i v i d u a l i z a d a en función de la edad y s i tuación neurológica del pac i en te , el tamaño y la local ización de l h e m a t o m a .
En general, se acepta que la cirugía no está indicada en el caso de hematomas profundos (ganglios de la base y t ronco del encéfalo), y se recomienda en pacientes con hemorragia cerebelosa aguda de 3-4 c m o más de diámetro con deterioro del nivel de consciencia (si el paciente permanece alerta y el hematoma es de pequeño tamaño, puede no necesitar cirugía) y signos radiológicos de herniación transtentorial inversa.
RECUERDA El h e m a t o m a cerebe loso es el único h e m a t o m a p r o f u n d o int racranea l q u e puede evacuarse quirúrgicamente (sólo si es mayo r de 3 c m y p r o duce de te r io ro cl ínico o herniación).
3 8
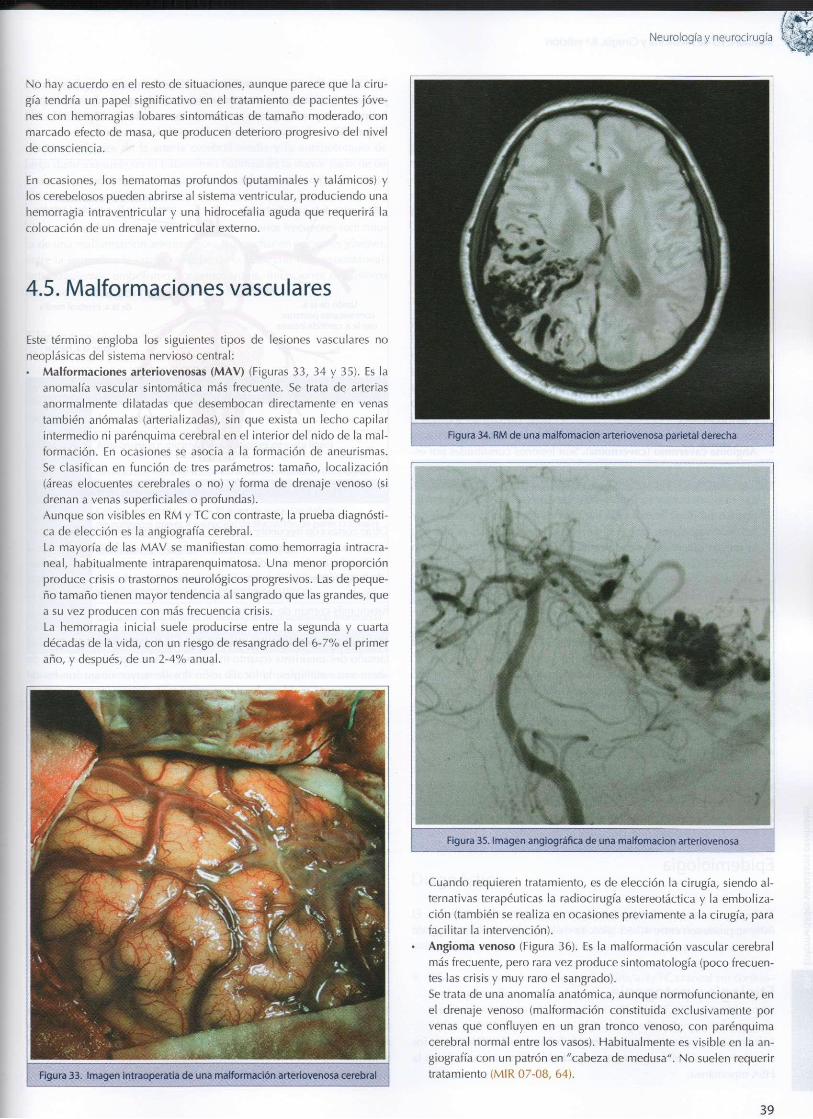
Neurología y neurocirugía
No hay acuerdo en el resto de situaciones, aunque parece que la c i r u gía tendría un papel s igni f icat ivo en el t ra tamiento de pacientes jóvenes con hemorragias lobares sintomáticas de tamaño moderado, con marcado efecto de masa, que producen deter ioro progresivo del nivel de consciencia .
En ocasiones, los hematomas profundos (putaminales y talámicos) y los cerebelosos pueden abrirse al sistema ventr icular , p roduc i endo una hemorragia intraventr icular y una hidrocefa l ia aguda que requerirá la colocación de un drenaje ventr icu lar externo.
4.5. Malformaciones vasculares
Este término engloba los siguientes t ipos de lesiones vasculares no neoplásicas del sistema nervioso centra l : • Malformaciones arteriovenosas (MAV) (Figuras 33 , 34 y 35). Es la
anomalía vascular sintomática más frecuente. Se trata de arterias anormalmente dilatadas que desembocan di rectamente en venas también anómalas (arterializadas), sin que exista un lecho capi lar in termedio ni parénquima cerebral en el inter ior del n ido de la m a l formación. En ocasiones se asocia a la formación de aneurismas. Se clasif ican en función de tres parámetros: tamaño, localización (áreas elocuentes cerebrales o no) y fo rma de drenaje venoso (si drenan a venas superficiales o profundas). Aunque son visibles en RM y TC con contraste, la prueba diagnóstica de elección es la angiografía cerebral . La mayoría de las M A V se manif iestan c o m o hemorragia intracraneal, habi tua lmente intraparenquimatosa. Una menor proporción produce crisis o trastornos neurológicos progresivos. Las de pequeño tamaño t ienen mayor tendencia al sangrado que las grandes, que a su vez producen con más frecuencia crisis. La hemorragia in ic ia l suele producirse entre la segunda y cuarta décadas de la v ida , con un riesgo de resangrado del 6 - 7 % el pr imer año, y después, de un 2 - 4 % anual .
Figura 33. Imagen in t raoperat ia de una malformación ar ter iovenosa cerebral
Figura 34. RM de una ma l fomac ion arter iovenosa par ieta l derecha
Figura 35. Imagen angiográfica de una ma l fomac ion arter iovenosa
Cuando requieren t ratamiento, es de elección la cirugía, s iendo a l ternativas terapéuticas la radiocirugía estereotáctica y la embo l i z a ción (también se realiza en ocasiones prev iamente a la cirugía, para faci l i tar la intervención). Angioma venoso (Figura 36) . Es la malformación vascular cerebral más frecuente, pero rara vez p roduce sintomatología (poco f recuentes las crisis y muy raro el sangrado). Se trata de una anomalía anatómica, aunque normofunc ionan te , en el drenaje venoso (malformación const i tu ida exc lus ivamente por venas que conf luyen en un gran t ronco venoso, con parénquima cerebral normal entre los vasos). Hab i tua lmente es v is ib le en la an giografía con un patrón en "cabeza de medusa" . No suelen requerir t ra tamiento (MIR 07-08, 64).
3 9

Manual CTO de Medicina y Cirugía, 8. a edición
I CLINICA Cefalea brusca e intensa Rigidez nuca Vómitos Fotofobia
DIAGNÓSTICO HSA
IAGNÓSTICO ETIOLÓGICO Angiofrafía cerebral 4 vasos
Figura 38. Algoritmo diagnóstico de la hemorragia subaracnoidea espontánea
Punción lumbar . Es la p rueba más sensible, pero de segunda e lec c ión. Está i nd i cada c u a n d o la TC es negat iva y existe una fuerte sospecha clínica (MIR 06-07 , 55) . La presencia de xantocromía se detecta en todos los casos a part i r de las 12 horas tras la hemor rag ia subaracno idea , si b ien puede ev idenc ia rse a part i r de las 4-6 horas; además, puede verse inc luso tres semanas después de haber t e n i d o el cuad ro . Las proteínas pueden estar elevadas y la g lucosa l igera mente d i s m i n u i d a . La punción traumática se d i f e renc ia de la HSA por a c l a r am ien to del LCR en la " p r u e b a de los tres t u b o s " (en la HSA, los tres t i enen el m i s m o aspecto hemático) y por la formación de coágulo.
R E C U E R D A El TC cerebral es la prueba inicial a realizar en el caso de que se sospeche una hemorragia subaracnoidea. Sin embargo, la prueba más sensible es la punción lumbar.
Diagnóstico etiológico La distribución de la hemorrag ia subaracnoidea en la TC sin contraste puede sugerir la localización del aneur isma: la hemorrag ia int raparenquimatosa es más f recuente , con aneurismas de la arteria cerebral media y arteria cerebral anter ior ; la hemorrag ia ¡nterhemisférica es característica de aneurismas de la c o m u n i c a n t e anter ior y de la arteria cerebral anter ior ; la extensión int raventr icu lar nos debe hacer sospechar un aneur isma de arteria c o m u n i c a n t e anter ior o aneurismas de la circulación poster ior ; la presencia de sangre a nive l de la cisura de Si lv io nos hace pensar en la pos ib i l i dad de un aneur isma en cerebral med ia o en c o m u n i c a n t e poster ior (Tabla 1 9 y Figura 39) .
D E S C R I P C I Ó N DE HALLAZGOS T O M O G R Á F I C O S
G rado 1
G rado l l
Sin sangre en la TC Grado 1
G rado l l Sangre difusa pero no lo bastante densa como para formar coágulos > 1 m m en sistemas verticales
Grado III
Sangre abundante en forma de coágulos densos de > 1 mm de grosor en el plano vertical (cisura interhemisférica, cisterna insular, cisterna ambiens) o más de 3 x 5 m m en el plano horizontal (cisterna silviana, supraselar o interpendular)
Grado IV Hematoma intracerebral y/o intraventricular con o sin sangrado subaracnoideo difuso
Tabla 1 9 . Escala radiológica de F isherde la hemorragia subaracnoidea. Gradación según los hallazgos de la tomografía computarizada
Figura 39. TC craneal donde se observa hemorragia subaracroidea
Angiografía de cuatro vasos. Se debe realizar tan pronto c o m o sea posible y debe inc lu ir los sistemas carotídeos y vertebrobasilar, dada la elevada incidencia de aneurismas múltiples. Sus objet ivos son def in ir la localización y morfología del aneurisma, identif icar otros posibles aneurismas no rotos, del inear los vasos adyacentes al aneurisma y valorar el grado de vasospasmo. Si la angiografía no revela ningún aneurisma, debería ser repetirse en 2-3 semanas, dado que la existencia de t rombos dentro del aneurisma o la existencia de vasos-pasmo pueden interferir la visualización angiográfica (Figura 40).
Figura 40. Ateriografía cerebral con aneurisma sacular
4 2

Neurología y neurocirugía
Complicaciones
Médicas
La h i p o n a t r e m i a supone la compl icac ión médica más f recuente de la hemor rag ia subaracno idea ; se p r o d u c e hab i t ua lmen te ent re el 4 ° y 10° día, y suele ser deb ida a un síndrome pierde-sal po r l i be ra ción del péptido natriurético. A causa de una exces iva estimulación simpática, pueden p roduc i r se ar r i tmias cardíacas en casi todos los pac ientes (s iendo la t aqu i c a rd i a s inusal la más f recuente ) . También se p r o d u c e i squemia subendocárdica y áreas de necrosis miocárdi-ca f o ca l , c on los cons igu ientes c amb ios electrocardiográficos, de te r io ro de la función cardíaca y edema p u l m o n a r . En estos casos se pueden u t i l i za r n i t ratos y antagonistas de l c a l c i o . La hipertensión arter ia l se puede con t ro l a r c on R-bloqueantes, que además reducen el r iesgo de a r r i tmias .
Otras compl i cac iones médicas pueden ser edema pu lmonar no cardio-génico, t r o m b o e m b o l i s m o pu lmonar , neumonías, hemorragia gastrointest inal , etc.
Neurológicas
La hidrocefa l ia , el resangrado por ruptura del aneurisma y el Vasospas-m o cerebral con isquemia son las tres pr incipales compl i cac iones neu rológicas de la hemorragia subaracnoidea. Si un paciente con sangrado subaracnoideo sufre un deter ioro clínico, no sólo se deben investigar las mencionadas compl i cac iones , sino que es preciso descartar h i p o tensión, h ipox ia o alteraciones electrolíticas. • Hidrocefalia. Puede desarrollarse de forma aguda en las primeras
24 horas, deb ido a que la sangre dentro de las cisternas básales o en el sistema ventr icular imp ide la normal circulación de líquido cefalorraquídeo. En estos casos, la colocación de un drenaje ventr icular externo puede mejorar espectacularmente la situación neurológica del paciente, aunque un descenso rápido de la presión intracraneal está asociado con un mayor riesgo de resangrado. La h idrocefa l ia también puede aparecer semanas después del sangrado. Se trata de una hidrocefa l ia comun ican te que se manifiesta clínicamente por deter ioro cogn i t i vo , incont inenc ia ur inar ia y trastornos de la marcha (MIR 00-01 F, 71). El t ra tamiento en este caso es la derivación vent r i cu loper i tonea l .
• Resangrado. Se postula que es deb ido a la ruptura del coágulo pe-rianeurismático. El 2 0 % de los pacientes presenta resangrado en las primeras dos semanas, un terc io en el pr imer mes y el 5 0 % dentro de seis meses, si el aneurisma no es abordado quirúrgicamente an tes. Después, el riesgo anual de resangrado de un aneurisma no tratado es de aprox imadamente un 3 % . Existen dos picos de inc idenc ia de resangrado, que t ienen lugar en las primeras 24-48 horas (en las primeras 24 horas, pueden resan-grar un 4 % de los aneurismas) y a la semana. El resangrado tiene una mor ta l idad del 7 5 % , y es más frecuente en mujeres y en pac ien tes con peor situación neurológica in i c ia l . La clínica es la misma que en el pr imer episodio , aunque pueden aparecer nuevos déficit neurológicos. Se han ut i l i zado antifibrinolíticos (ácido tranexámico y e-aminoca-proico) para prevenir el resangrado mediante un retraso en la lisis del coágulo, pero estos agentes se asocian con un aumento en el riesgo de vasospasmo cerebral y de h idrocefa l ia . La mejor fo rma de evitar el resangrado es exc lu i r el aneur isma de la circulación general por vía endovascular (embolización) o mediante cirugía.
• Vasospasmo. Es la pr inc ipa l causa de morb imor ta l i dad en pacientes que han sufr ido una hemorragia subaracnoidea. A di ferencia del resangrado, el Vasospasmo se desarrolla lentamente en horas o días y, aunque se aprecia angiográficamente en el 7 0 % de los pacientes, sólo es sintomático en el 3 6 % de los mismos. Se presenta entre el 4.°-12.° día postsangrado (máxima inc idenc ia entre 6 ° y 8.° día) y la clínica corresponde a un déficit del terr i tor io vascular afectado (por isquemia) o un empeoramien to neurológico no exp l i cab le por otras causas. La cant idad de sangre en la TC se corre lac iona con la gravedad del Vasospasmo. En la prof i laxis del Vasospasmo se ut i l iza un antagonista del ca lc io , el n i m o d i p i n o . El diagnóstico del mismo pasa por descartar otras causas de empeo ramiento neurológico (h iponatremia, edema cerebral , resangrado, infecciones, etc.) y con f i rmar lo mediante una angiografía cerebral (en la que se verá una estenosis de un vaso cerebral) ; últimamente, también puede detectarse con una eco-Doppler transcraneal, en la que se observa un aumento de la ve loc idad del f lu jo de la arteria cerebral media . Una vez establecido el Vasospasmo, la pr inc ipa l línea de t ra tamien to es la denominada terapia " t r i p l e H " (hemodilución-hipervolemia-hipertensión). Los objet ivos de esta terapia son aumentar la presión de perfusión cerebral (e levando la presión sistólica sanguínea y el vo lumen intravascular) y mejorar la microcirculación cerebral por med io de una disminución de la v iscosidad sanguínea.El pr inc ipa l inconveniente de este t ratamiento es que aumenta el riesgo de re-sangrado del aneurisma, si éste no ha sido exc lu ido de la circulación cerebral , ya sea mediante t ratamiento quirúrgico o endovascular. En el caso de que fracase la t r ip le H, se puede emplear la angioplast ia t rans lumina l percutánea y la administración intraarterial de sustancias vasodilatadoras, c o m o la papaver ina.
Q RECUERDA La pro f i l ax i s del vasospasmo se rea l iza c o n n i m o d i p i n o ; sin e m b a r g o , una vez q u e se ha es tab lec ido , se debe ap l i ca r la t r i p l e H y o l v idarse de l n i m o d i p i n o .
Tratamiento
A la hora de manejar a estos pacientes, se debe di ferenciar entre el trat amiento de la hemorragia subaracnoidea y el de la causa subyacente (generalmente un aneurisma).
Tratamiento de la hemorragia subaracnoidea
Los ob je t i vos pr inc ipa les del t ra tamiento de la hemorrag ia subaracno idea son preveni r el resangrado y el Vasospasmo. Para c u m p l i r el p r imer c o m e t i d o , el pac iente debe ser co lo cado en una habitación t ranqu i l a , con reposo abso luto en cama y la cabeza elevada 30° sobre la ho r i zon ta l , para fac i l i tar el drenaje venoso intracraneal . Hay que mantener un cont ro l estr icto de la tensión arterial (ni muy alta ni muy baja). Se deben evitar el estreñimiento y los vómitos. El pac iente debe rec ib i r una analgesia impor tan te , ya que el do lo r con l l eva una descarga simpática impor tan te que eleva la tensión arter ia l . Si fuera necesario, se puede conseguir la sedación del pac iente con d i azepam. Si hay crisis, el fármaco pre fe r ido es la fenitoína (no dep r ime el n ive l de consc ienc ia ) . La u t i l i dad de la dexametasona en estas s i tuaciones es cont rover t ida , aunque suele usarse para reduc i r la sintomatología do lorosa . Debe asociarse n i m o d i p i n o para real izar prof i lax is del Va sospasmo cerebra l . Se debe cu idar la función p u l m o n a r (para evitar atelectasias y neumonías).
4 3

Manual CTO de Medicina y Cirugía, 8.a edición
R E C U E R D A Tan to en el diagnóstico c o m o en el t r a t am ien to , se debe d i fe renc ia r la hemor rag i a subaracno idea de l aneur i sma cerebra l .
Tratamiento del aneurisma
En el m o m e n t o actual , existen dos proced imientos cuya f ina l idad últi
ma es exc lu i r el aneurisma de la circulación cerebral : la embolización
por vía endovascular y la craneotomía con c l ipaje quirúrgico (Figura
41). En los pacientes con buena situación neurológica (grados l-lll), los
aneurismas suelen tratarse de manera precoz (dentro de los pr imeros 4
días); sin embargo, si la situación neurológica es desfavorable (grados
IV-V), a veces es aconsejable manejar los de manera difer ida (a partir de
los 10 días). El t ra tamiento precoz del aneurisma, por cua lquiera de los
dos métodos, fac i l i ta el mane jo posterior de las compl i cac iones , sobre
todo , del Vasospasmo, al d i sminu i r el riesgo de resangrado con la t r ip le
H. En líneas generales, tanto la embolización c o m o el c l ipaje t ienen un
pronóstico a corto p lazo simi lar . En estos últimos años, la colocación
de stent y balones está fac i l i t ando la embolización aneurismática.
Casos clínicos representativos
Hombre de 85 años, con antecedentes de hemorragia cerebral hace dos años. Ingresa por cuadro agudo de hemiparesia derecha y somnolencia. En ela TC urgente, se objetiva un gran hematoma intracerebral lobar frontoparietal izquierdo. El paciente no es hipertenso. ¿Cuál, entre las siguientes, es la etiología más probable de la hemorragia del paciente?
1) Metástasis. 2 | A n e u r i s m a . 3 ) T r a u m a t i s m o . 4 ) Tóx icos o m e d i c a m e n t o s . 5} Angiopat ía a m i l o i d e .
M I R 0 3 - 0 4 , 2 4 1 ; RC: 5
Un hombre de 62 años acude a Urgencias por presentar, de forma brusca, mareo e inestabilidad. En la exploración se encuentra un nistagmo horizontal, un síndrome de Horner derecho, una pérdida de la sensibilidad dolorosa en la hemicara derecha y braquiocrural izquierda, una ataxia de miembros derecha y disfagia. ¿Cuál sería su sospecha diagnóstica?
1) I n f a r t o d e la a r t e r i a bas i l a r . 2) I n f a r t o d e la p r o t u b e r a n c i a . 3 ) I n f a r t o d e la a r t e r i a v e r t e b r a l i z q u i e r d a . 4 ) I n f a r t o d e la a r t e r i a c e r e b r a l d e r e c h a . 5) I n f a r t o la tera l b u l b a r d e r e c h o .
M I R 0 2 - 0 3 , 2 0 4 ; RC: 5
Un varón de 58 años, fumador de 2 cajetillas/día, bebedor habitual, hipertenso controlado irregularmente, ha notado en los últimos días dos episodios bruscos, de 15 y 45 minutos de duración, de visión borrosa en el ojo izquierdo y parestesias en mano derecha. La exploración neurológica es normal. Entre los siguientes, ¿cuál es el diagnóstico más probable?
1) J aqueca a compañada . 2) Cr i s i s pa r c i a l e s c o m p l e j a s . 3) Neuropatía óptica alcohólico-tabáquica. 4 ) i s q u e m i a c e r e b r a l t r ans i t o r i a e n t e r r i t o r i o carot ídeo. 5) B ro tes d e e n f e r m e d a d d e s m i e l i n i z a n t e r e c u r r e n t e - r e m i t e n t e .
M I R 0 0 - 0 1 , 5 3 ; RC: 4
Un paciente de 65 años, con antecedentes de HTA e hipercolesterolemia, sufre un accidente isquémico transitorio en territorio carotídeo derecho. La valoración clínica y el ECG no muestran evidencia de cardiopatía. Se realiza arteriografía cerebral, que muestra estenosis de la arteria carótida interna derecha del 3 0 % . ¿Qué medida terapéutica estaría indicada en este paciente?
1) Ant i coagu lac íón . 2) Cirugía carot ídea. 3) A n g i o p l a s t i a carot ídea. 4) A n t i a g r e g a n t e s p l a q u e t a r i o s . 5) N i n g u n a .
M I R 98-99F , 7 0 ; RC: 4
Varón de 50 años, con episodios repetidos de isquemia cerebral transitoria consistente en pérdida de fuerza y paresias en brazo y pierna derechas, y amaurosis fugaz en ojo izquierdo. Presenta estenosis del 7 5 % en inicio de carótida interna izquierda. ¿Cuál es la actitud correcta?
1) Ant i coagu lac ión c o n d icumar ín icos , 6-12 meses. 2) Ant i coagu lac ión c o n h e p a r i n a , 1 s e m a n a . 3) Ant i coagu lac íón c o n h e p a r i n a y an t i ag regan tes p l a q u e t a r i o s . 4) Endarterectomía d e carótida i n t e r n a i z q u i e r d a . 5) By pass aortocarot ídeo c o n v e n a safena autóloga.
RC: 4
Paciente de 30 años que acude al servicio de Urgencias de un hospital por presentar de forma aguda amaurosis transitoria del ojo derecho y cefalea con dolorimiento en región cervical derecha. En la exploración, se objetiva un síndrome de Horner derecho. ¿Cuál es el diagnóstico más probable, entre los siguientes?
1) Estenosis carot ídea d e r e c h a . 2) H e m a t o m a s u b d u r a l t raumát ico. 3) D i secc ión carot ídea d e r e c h a . 4 ) T r o m b o s i s d e la a r te r i a c en t r a l d e la r e t i na . 5) S índrome d e H o r t o n .
RC: 3
4 4

TRASTORNOS DEL MOVIMIENTO
Orientación
MIR r
Aspectos esenciales
Se trata de u n t e m a i m p o r t a n t e q u e i n c l u y e dos en f e rmedades m u y p reguntadas en el e x a m e n : la e n f e r m e d a d d e Pa rk inson y la corea d e H u n t i n g t o n . H a y c o n o c e r sus características y saber d i f e renc ia r l as d e otros c u a d r o s e x t r a p i r a m i d a l e s .
ÜD
LH
S
tu
[71
CE
La clínica de los trastornos del m o v i m i e n t o depende del gang l io basal les ionado: el núcleo subtalámico de Luys p r o d u c e h e m i b a l i s m o ; el núcleo caudado , corea; la sustancia negra compac t a , pa rk inson i smo ; y las lesiones pal idales bi laterales, brad ic ines ia .
El t emb lo r cerebeloso es el más lento (2,5-4 Hz) y aparece con el mov im ien to . El t emb lo r fisiológico exacerbado es el más rápido (8-12 Hz) y es postural , y el temblor del Parkinson está entre ambos (4,5-6,5 Hz) y es de reposo.
El t e m b l o r esencia l es el más f recuente de los temblores sintomáticos. C o m p a r t e características c o n el del Park inson: v e l o c i d a d i n t e rmed ia , asimétrico, coex is tenc ia c o n otros trastornos del m o v i m i e n t o (distonías, r ig idez en rueda dentada) , pe ro típicamente es postura l , presenta h is tor ia f am i l i a r y me jo ra c o n el a l c o h o l .
Las distonías pueden tratarse c o n benzod iacep inas , otros relajantes musculares y anticolinérgicos, según su gravedad .
La t o x i n a botulínica es el t r a t amien to de elección de las distonías foca les .
Los t ics se s u p r i m e n c o n la v o l u n t a d y a u m e n t a n c o n el estrés. La f o r m a más grave de tics múltiples es el síndrome de Gil íes de la Touret te , que se asocia a eco l a l i a y cop ro l a l i a . Se trata c o n neurolépticos.
La Corea de H u n t i n g t o n l leva asoc iado: t rastornos del m o v i m i e n t o , de te r io ro c o g n i t i v o y trastornos psiquiát r icos . Se hereda de f o r m a autosómica d o m i n a n t e y presenta el fenómeno de anticipación (por expansión del t r ip le te de nucleótidos CAC ) . Es característica la atrof ia del núcleo c audado , y el t r a t amien to sintomático se hace c o n neurolépticos.
El t e m b l o r de reposo, la brad ic ines ia , la r ig idez y la ines tab i l idad postura l carac ter izan el síndrome park in-son iano . La en fe rmedad de Park inson idiopática es la f o r m a más común de síndrome pa rk inson i ano . En el la p r e d o m i n a la clínica del t e m b l o r ( forma de presentación más f recuente) y la brad ic ines ia (manifestación más incapac i t an te de la en fe rmedad ) , s iendo la mejoría c o n L-dopa s ign i f i ca t i va .
La parálisis supranuc lear progres iva es u n o de los diagnósticos d i fe renc ia les de l Parkinson idiopático. C o n l leva distonías, p i r a m i d a l i s m o , parálisis de la infraversión de la m i r ada y d e m e n c i a .
Las atrof ias multisistémicas se carac ter izan por clínica pa rk inson iana c o n p r e d o m i n i o de la r ig idez y datos de afectación de otras estructuras nerviosas, c o m o la vía p i r a m i d a l , el ce rebe lo o el sistema vegetat ivo , desde estadios in ic ia les de la e n f e r m e d a d . La d e m e n c i a no f o r m a parte de l cuad ro .
P r egun tas
- MIR 09-10, 69 - MIR 08-09, 57 - MIR 06-07, 57, 64 - MIR 05-06, 58 - MIR 04-05, 58, - MIR 03-04, 239, 242 - MIR 02-03, 205, 209 -MIR 01-02, 57 - MIR 00-01, 253 - MIR 00-01F, 64, 67 - MIR 99-00, 194, 195
243 254
201,203 - MIR 98-99F, 72
Los trastornos del m o v i m i e n t o t ienen
su sustrato patológico p r i n c i pa lmen
te en los ganglios básales (Figura 42) .
Aunque son núcleos motores, no pro
yectan di rectamente sobre la médula
espinal , sino que reciben estímulos
corticales y proyectan de nuevo ha
cia la corteza, a través del tálamo,
para regular la amp l i t ud y ve loc idad
de los mov imien tos y part ic ipar en la
iniciación de los mismos (Figura 43) .
No es posible ident i f icar un t ipo
específico de mov im i en to p r o d u c i
do por los ganglios básales, pero sí
puede establecerse una correlación
entre lesiones de estos y la clínica
asociada. Así, la lesión del núcleo
subtalámico se asocia a hemiba l i smo
Putamen
Pálido lateral
Pálido medial
Caudado
Tálamo
Subtálamo
Figura 42. Anatomía de los ganglios básales
4 5

Manual CTO de Medicina y Cirugía, 8.a edición
y corea, la lesión del caudado y putamen a corea, la lesión de la por ción compacta de la sustancia negra a park insonismo y, las lesiones palidales bilaterales (encefalopatía anóxica) a bradic inesia grave. En muchas ocasiones, no es posib le determinar lesión estructural alguna en pacientes con manifestaciones extrapiramidales.
CORTEX
Glut, Aspart
STR IADO
GABA
D A X .
GABA GABA sust. P sust. P
SUSTANCIA NEGRA
P Á L I D O
Figura 43. Fisiología de los gangl ios básales
Temblor de reposo. Se produce en ausencia de act iv idad muscular vo luntar ia . El e j emp lo más típico es el t emblor observado en la e n fermedad de Parkinson. Temblor de acción. Se p roduce con la contracción muscular v o l u n tar ia , y se subd iv ide en t emb lo r postural y cinético o de m o v i m i e n to . El p r imero es p rovocado con el man ten im ien to de la postura, y son e jemplos el t emb lo r fisiológico, el t emb lo r fisiológico exacerbado, el t emb lo r esencial y el t emb lo r postural que puede aparecer en la enfermedad de Parkinson y otros trastornos del m o v i m i e n to . Son e jemplos típicos de t emb lo r postural el que se p roduce al beber, comer , abrocharse un botón o escribir . El t emb lo r cinético aparece con cua lqu ier fo rma de m o v i m i e n t o , y puede ocurr i r al i n i c io ( temblor in ic ia l ) , durante ( temblor de transición) o al f ina l del m o v i m i e n t o ( temblor te rmina l o in tenc iona l ) . El t emb lo r cinético es característico de patología cerebelosa o troncoencefálica (esclerosis múltiple, vascular, t u m o r a l , patología degenerat iva) .
RECUERDA El t e m b l o r t ípico de la e n f e r m e d a d de Park inson es de reposo , pe ro también es f r e cuen te el t e m b l o r pos tu ra l .
Clínica (Tabla 21)
Los trastornos del mov im i en to extrap i ramida l se d i v iden en h ipe rc i nesias ( temblor, distonía, corea, atetosis, ba l i smo, mioclonus, acatisia, piernas inquietas, etc.) e hipocinesias (parkinsonismos).
5.1. Temblor
El t emblor se def ine c o m o la presencia de osci laciones rítmicas de una parte del cuerpo, secundarias a contracc iones alternantes o sincrónicas de grupos musculares opuestos. Puede resultar de procesos fisiológicos o patológicos, y afecta más f recuentemente a manos, cabeza, piernas y voz.
Clasificación
La frecuencia de los temblores patológicos es re lat ivamente estable y fácil de medir mediante acelerometría. La Tabla 20 presenta una c las i ficación del t emblor t omando c o m o base su frecuencia.
FRECUENCIA T IPO DE TEMBLOR
2,5 - 4 Hz • Cerebeloso • Disfunción troncoencefálica
4 - 4,5 Hz • Enfermedad de Parkinson (reposo) • Temblor rubr ico o mesencefálico
5,5 - 7 Hz • Temblor esencial • Enfermedad de Parkinson (postural )
8- 1 2 H z • Fisiológico • Fisiológico exacerbado
Tabla 20. Clasificación del t emb lo r según su frecuencia
Atend iendo a la situación func iona l en la que aparece, el t emblor se puede clasif icar en temblor de reposo o t emb lo r de acción.
Cinético
F I S IOLÓGICO E X A G E R A D O - ++ +
ESENCIAL -(+) ++++ + (+)
PARKINSONIANO ++++ ++ +
MESENCEFÁL ICO ++ +++ +++
C E R E B E L O S O - + +++
Tabla 2 1 . Correlación cllnico-etlológica del t emb lo r
Temblor fisiológico exacerbado . Es un t e m b l o r fisiológico de f r e c u e n c i a n o r m a l (8-12 Hz ) , pe ro de m a y o r a m p l i t u d . Está ausente en reposo y presente con el m a n t e n i m i e n t o de la pos tu ra . Resulta de un i n c r e m e n t o de la a c t i v i d a d periférica (3-adrenérgica asoc iada a un a u m e n t o de l n i ve l de c a t e c o l a m i -nas c i r cu l an t e s . Es común en estados de ans iedad y en a q u e l los t ras tornos metabólicos que c o n l l e v a n una sob rea c t i v i d ad B-adrenérgica ( t i r o tox i cos i s , f e o c r o m o c i t o m a , h i p o g l u c e m i a ) . También aparece c o n la ingesta de a lgunos fármacos (catecola-minas , me t i l xan t i nas ) o c o n la re t i rada de ot ros (B-bloqueantes, m o r f i n a y a l c o h o l ) . Puede con t ro l a r se a d e c u a d a m e n t e con B-bloqueantes ( p r o p r a n o l o l ) . Temblor esencial (Tabla 22). Es la forma más común de t e m blor sintomático (MIR 03-04, 242 ; MIR 02-03, 205) y el trastorno del mov imien to más frecuente. Se hereda con carácter autosó-mico dominante y alta penetrancia. Se describe historia famil iar en un 3 0 % de los pacientes, aunque también existe una forma esporádica. Puede comenzar a cualquier edad y persiste duran te toda la v ida. A l in ic io es unilateral e intermitente, pero una vez establecido es bilateral y asimétrico, pudiéndose afectar cua l quier parte del cuerpo. Típicamente, produce oscilaciones flexo-extensoras a nivel de la muñeca o aproximación-separación de los dedos cuando los brazos están al frente. Su frecuencia es de 4-12 Hz, y se puede asociar a tareas específicas (escribir, mantener un objeto en una postura determinada, etc.). Se exacerba con el estrés, ansiedad y fatiga. Característicamente, mejora con el a l coho l .
46

Neurología y neurocirugía
CRITERIOS DE INCLUS IÓN (MIR 99-00, 203)
Presencia de t e m b l o r postura l v is ible y pers istente, a fec tando a las manos o antebrazos, q u e puede o no acompañarse de t e m b l o r cinético. Puede ser asimétrico y afectar a otras partes del cue rpo Pro longada duración (más de c inco años)
CRITERIOS DE EXCLUSION
Presencia de otras alteraciones neurológicas, con la excepción de r ig idez en rueda den tada (signo de Froment ) Existencia de causas de t e m b l o r fisiológico exacerbado (p. ej.: h lpe r t i ro id l smo) Exposición a fármacos tremorígenos o ret irada de fármacos antitremorígenos Historia de t r a u m a t i s m o craneal en los tres meses previos al in ic io de los síntomas Evidencia clínica de t e m b l o r pslcógeno Inicio súbito
Tabla 22, Criterios diagnósticos del t emb lo r esencial
Casi el 5 0 % de los pacientes con temb lo r esencial t ienen alguna fo rma de distonía asociada. Son variantes el t emb lo r cefálico aislado, de la voz , l ingua l , ortostático, etc. No hay datos de patología ext rap i ramida l o cerebelosa (aunque la presencia de r ig idez en rueda dentada ¡unto al t emb lo r no es cr i ter io de exclusión).
Fármacos q u e actúan sobre los sistemas colinérgicos
Acet i lco l ina, nicotínicos, muscaríni-cos, anticolinesterásicos
Fármacos q u e actúan sobre los sistemas monoaminérglcos
Neurolépticos, fen i l e t i l amina , Índoles
Fármacos q u e actúan sobre los sistemas adrenérgicos
Agonistas B-adrenérgicos, anfe tami-nas, epinefr inas, l i t io , cafeína
Otros fármacos p roduc to res de t e m b l o r de acción
Ant icomic ia les (valproato, fenitoína, carbamazepina)
Tabla 23. Fármacos que pueden producir t emb lo r
5.2. Distonías Concepto
Son mov imien tos involuntar ios sostenidos que producen desviación o torsión de un área corpora l . No se supr imen con la vo lun tad y pueden desencadenarse por mov imien tos o acciones específicas. Genera lmente cesan durante el sueño. Con frecuencia coexisten con temblor , básicamente de t ipo esencial. Hay también un " t emb lo r distónico", que aparece cuando el paciente intenta mover un segmento corpora l en dirección opuesta a la fuerza de la distonía.
RECUERDA El t e m b l o r esenc ia l es el más f r e cuen te de los t e m b l o r e s sintomátic Puede ser asimétrico y l levar asoc iada r i g idez en rueda den tada de f o r m a análoga al t e m b l o r de un pac i en te c o n Pa rk inson .
El t ra tamiento se realiza con p roprano lo l o p r i m i d o n a , habiéndose u t i l i zado en casos refractarios la tox ina botulínica. Casos excepc io nales son tratados con estimulación talámica. Temblor neuropático. El t emb lo r es una de las manifestaciones de neuropatía periférica y puede observarse en algunos pacientes con neuropatía desmie l in izante inf lamator ia aguda o crónica, neuropa tía sens i t ivomotora hereditar ia (síndrome de Lévy-Roussy) y neuro patías paraproteinémicas IgM. Es menos frecuente en la neuropatía asociada a diabetes, uremia y por f i r ia . Característicamente, es un temb lo r s imi lar al esencial , aunque pue de aparecer un componen te de reposo ind is t ingu ib le del presente en la enfermedad de Parkinson. La respuesta farmacológica a p ro p rano lo l , p r i m i d o n a o benzodiacep inas es imprev is ib le . Temblor rubrico (mesencefálico o de Holmes). Las lesiones en la vía de proyección desde el núcleo dentado del cerebelo al núcleo ventral posterolateral del tálamo, en las prox imidades del núcleo ro jo , pueden p roduc i r un t emb lo r característico. Está presente en reposo, empeora con la postura y llega a ser i ncon t ro l ado con el m o v i m i e n t o . Es común ver lo asociado a esclerosis múltiple o pato logía vascular de t ronco . Su cont ro l terapéutico es ma lo . Temblor cerebeloso. El t emb lo r cinético y su var iante, el t emb lo r in tenc iona l , se cons ideran característicos de patología cerebelosa. Puede llevar asociados signos de afectación cerebelosa (ataxia, dis-metría). El t ra tamiento sintomático es in f ructuoso y el ob je t i vo es tratar la causa etiológica subyacente. Temblor farmacológico. El t emb lo r es un efecto secundar io común de un gran número de fármacos. A u n q u e pueden produc i r cua l qu ier t i po de temblor , lo más frecuente es que sea de carácter postural y con grados variables de incapac idad (Tabla 23) .
RECUERDA El t e m b l o r fisiológico y esencia l se t ratan con p-bloqueantes; s in e m bargo , el t e m b l o r de reposo del Parkinson se trata c o n antlcolinérgicos.
Patogenia
No se ha observado n inguna alteración morfológica consistente en cerebros de pacientes con distonía pr imar ia y hay muy poca información sobre los posibles cambios bioquímicos subyacentes. Con la PET se ha encont rado una disminución en el metabo l i smo cerebral en el núcleo lent icular. Es posible que los sistemas dopaminérgicos y noradrenérgi-cos jueguen un papel en la patogénesis de la distonía pr imar ia .
Clasificación
Etiológicamente, se d iv iden en distonías pr imarias y secundarias. Las formas primarias pueden ser esporádicas (generalmente de in i c io en el adulto) o hereditarias (suelen comenzar en la infancia, asociadas a diferentes /ocu5 genéticos denominados DYT). Las secundarias suelen ser de in i c io brusco o rápidamente progresivo, y se asocian a otros síntomas neurológicos o generales. Además, se ha descrito un grupo denominado "distonía p lus" , donde se inc luyen enfermedades con distonía, que también presentan otros mov imien tos anormales, que las di ferencian de las pr imarias ( como la distonía que responde a levodopa o la distonía mioclónica).
Los mov imien tos distónicos pueden aparecer durante el reposo o con ciertas actividades musculares voluntar ias (distonía de acción). Dentro de este último grupo se inc luyen las distonías ocupacionales : espasmo del escribiente, del jugador de golf, del mecanógrafo, etc.
A tend iendo a su distribución anatómica, las distonías se clasif ican en : Distonías focales. Afectan a una única parte del cuerpo. Son esporádicas, no progresivas y suelen aparecer en la v ida adulta. Inc luyen la tortícolis o distonía cerv ica l ( forma más frecuente en este g ru po), blefarospasmo, hemiespasmo fac ia l , etc. Suelen ser idiopáticas, aunque pueden ser secundarias a patología vascular, esclerosis múlt ip le , encefal i t is, etc.
4 7

Manual CTO de Medicina y Cirugía, 8.a edición
Distonías segmentarias. Aparecen mov imientos distónicos en áreas corporales contiguas. Inc luye el síndrome de Meige , que cursa con blefarospasmo y distonía o romand ibu la r .
R E C U E R D A Ei síndrome de M e i g e se asoci d i b u l a r .
distonía o r o m a n -
5.4. Tics Son movimientos estereotipados, sin objetivo, que se repiten ¡rregularmente. Se caracterizan porque se suprimen con la voluntad y aumentan con el estrés. Pueden persistir durante el sueño. Se clasifican en tics primarios (esporádicos o hereditarios) y secundarios, motores y vocales, en simples y complejos.
Distonía multifocal. Afecta a músculos de más de dos regiones no cont iguas. Hemidistonías. Se asocian con lesiones estructurales en los ganglios básales contralaterales, par t icu larmente el pu tamen . Distonías generalizadas. Se caracter izan por distonía crural segmentaria y distonía en al menos una parte corpora l ad ic iona l . Las formas pr imarias pueden ser esporádicas o hereditarias, suelen debutar en las primeras décadas de la v ida y son de carácter progresivo.
Tratamiento
En el t ratamiento sintomático de la distonía leve se ut i l izan benzo-diacepinas (d iazepam, c lonazepam, lorazepam) y otros relajantes musculares, c o m o el bac lofeno o la t i zan id ina . La levodopa es efectiva en la distonía con f luctuac iones diurnas y en la asociada a park insonismo. En casos de distonía moderada o grave, se ut i l i zan anticolinérgicos ( t r ihex i fen id i l , b iper ideno) . Se ut i l iza la toxina botulínica c o m o me dicación de elección en el t ratamiento de las distonías focales. Son fármacos de segunda elección el baclofén, la carbamazepina o el val-proato. En los casos con mal contro l farmacológico se puede realizar t ratamiento quirúrgico con resultados más favorables en el caso de que las distonías sean primarias. Los sitios " d i ana " son el tálamo y, sobre todo , el segmento interno del g lobo pálido. Las técnicas que se ut i l izan son la lesión quirúrgica o bien la estimulación cerebral profunda.
R E C U E R D A La t o x i n a botulínica es de e lecc ión en el de las distonías foca les .
5.3. Mioclonías
Síndrome de Gilíes de la Tourette
Es la forma más grave de tics múltiples. Su herencia se presume autosó-mica dominante , en algunos casos asociada al c romosoma 18 (18q22.1) , aunque no puede excluirse una herencia ligada al c romosoma X.
Son cr iter ios diagnósticos de esta ent idad (MIR 06-07, 64): 1) Múltiples tics motores y uno o más tics fónicos. 2) Los tics ocurren muchas veces al día, casi todos los días a lo largo
de más de un año. 3) El t ipo , gravedad y comp le j i dad de los tics cambia con el t i empo . 4 ) In ic io antes de los 21 años. 5) Los mov imien tos involuntar ios y ruidos no pueden ser just i f icados
por otros medios. 6) Se asocian a ecola l ia y copro la l i a .
Es característica la asociación con trastornos obsesivo-compulsivos y trastorno por déficit de atención.
| J J R E C U E R D A H a y q u e tener presente la asociación entre el síndrome de Gil íes de la Toure t te , el t ras torno obses i vo-compu ls i vo y el t ras torno por déficit de atención.
El t ratamiento se realiza con neurolépticos (ha loper ido l , p imoz ida ) , c lon id ina y otros antidopaminérgicos.
5.5. Síndrome de piernas inquietas ( M I R 0 3 - 0 4 , 2 3 9 ,
Son mov imien tos invo luntar ios , súbitos y de escasa duración, causados por contracción muscular act iva. Se d i ferenc ian de la asterixis en que estas últimas son también mov imien tos rápidos y arrítmicos, pero p ro duc idos por pausas breves de la act iv idad muscular que causan pérdida del t ono postural (s i lencio eléctrico en el e lec t romiograma) .
Según su or igen, pueden clasificarse en corticales, subcort icales, espi nales o periféricas.
Trastorno del mov im i en to que se caracteriza por disestesias de predom i n i o en miembros inferiores, que aparecen preferentemente en reposo y que se a l iv ian con el mov im ien to . Puede asociarse con mov imien tos periódicos durante el sueño.
La etiología más frecuente es idiopática, deb iendo descartarse la po-lineuropatía sensitiva (urémica, diabética,...), anemia ferropénica o la coexistencia de otra patología.
Por su distribución, se clasif ican en focales ( impl i can un grupo de músculos discreto), segmentarias o general izadas (muchas veces de causa progresiva y asociadas a epilepsia).
R E C U E R D A La fer ropenia puede manifestarse en f o rma de síndrome de piern inquietas.
Por la fo rma de presentación, pueden ser espontáneas, de acción o reflejas.
En el t ra tamiento sintomático de las mioclonías, resultan muy efectivos el c lonazepam, va lproato, p i razetam, p i r im idona y 5-hidroxitriptófano.
El t ratamiento de las formas idiopáticas se basa en el uso de agonistas dopaminérgicos o levodopa, así c o m o benzodiazepinas u opiáceos.
Se debe realizar el diagnóstico di ferencia l con cuadros de acatisia.
4 8

Neurología y neurocirugía
5.6. Corea. Enfermedad de Huntington El término corea ("bai le") hace referencia a mov imientos arrítmicos, rápidos, irregulares, incoord inados e incesantes que pueden afectar a cua lquier parte del cuerpo. La Tabla 24 muestra una clasificación de los síndromes coreicos.
COREAS HEREDITARIOS
• Enfermedad de H u n t i n g t o n • Neuroacantoc i tos is • Síndrome de Fahr (calcificación de los gang l ios
básales)
COREAS METABÓL ICOS Y ENDOCRINOS
• H iperpara t i ro id i smo • H ipopa ra t i ro id i smo con calcificación
de los gang l ios básales • H ipe r t i ro id i smo • Degeneración hepatocerebra l adqu i r ida
VASCULIT IS • Lupus e r i t ematoso sistémico • Panarterit is nodosa
ICTUS DE GANGL IOS BASALES
FARMACOLÓGICO Discinesias tardías secundarias al t r a t a m i e n t o crónico con neurolépticos
Tabla 24. Clasificación de los síndromes coreicos
La enfe rmedad de H u n t i n g t o n (EH) es la fo rma más común de corea hered i tar io . Puede debutar a cua lqu ie r edad , aunque la mayor i n c i denc ia se sitúa entre la cuarta y la qu in ta décadas, e v o l u c i o n a n d o lentamente hacia la muerte en un pe r iodo de 10 a 25 años. La superv i venc ia es más corta entre i nd i v iduos con i n i c i o j u ven i l de la enfermedad . La neumonía y otras infecc iones intercurrentes son la causa más f recuente de muer te .
Genética
Hay antecedentes famil iares en prácticamente todos los pacientes. Se hereda con carácter autosómico dominan te y penetrancia comple ta , y es el resultado de un defecto genético loca l izado en el brazo cor to del c romosoma 4. Cada i nd i v iduo con un padre afectado t iene un 5 0 % de probabi l idades de heredar el gen y eventualmente manifestar la enfermedad. El sexo del padre afectado inf luye en la edad de in i c io , de fo rma que si el afectado era el padre o el abuelo, la enfermedad debutará antes. En la EH se observa el fenómeno de anticipación, en v i r tud del cual el debut se produce a edades más precoces en sucesivas generaciones. Esto es deb ido al progresivo aumento de un tr ip lete de nucleótidos (CAG) en el c romosoma 4. Los homocigóticos son raros, pero no es letal intraútero, y las manifestaciones clínicas y edad de in ic io pueden ser análogas a los pacientes heterocigóticos.
Anatomía patológica
Es característica de la enfermedad de Hunt ing ton la atrofia del núcleo caudado, con dilatación secundaria de las astas frontales de los ven trículos laterales. Puede haber también atrofia de otros ganglios de la base (putamen y pálido) y pérdida neuronal y gliosis en el córtex cerebral (especialmente en las áreas frontales). Se baraja la hipótesis de la neurotox ic idad induc ida por g lutamato c o m o base de la muerte de neuronas estriatales. La atrofia del núcleo caudado genera un déficit
de acet i l co l ina , lo que cont r ibuye a una h iperact iv idad dopaminérgica relativa, que es la base patológica de esta enfermedad (Figura 44) .
Figura 44 . TC craneal de pac iente con en fe rmedad de H u n t i n g t o n , en la q u e se aprecia la marcada atrof ia de la cabeza de a m b o s núcleos caudados, lo q u e
provoca la dilatación de las astas f ronta les de los ventrículos laterales
Clínica
Se caracteriza por la tríada: trastornos del mov im ien to , deter ioro c o g n i t i vo y clínica psiquiátrica (MIR 02-03, 209) .
• Trastornos del movimiento. En la forma que debuta en la edad adulta, el trastorno de mov imien to más característico es el corea, que en fases iniciales puede ser supr imido por la vo luntad. Posteriormente, puede ser tan v io lento que imposibi l i te al paciente sentarse sin riesgo de caer. Es frecuente la asociación con distonía y clínica parkinsoniana. Las alteraciones en los mov imien tos oculares a veces son los signos más precoces. La pérdida de los mov imien tos oculares sacádicos rápidos que permi ten la refijación en distintos objetos const i tuye el déficit más común. El habla es hipercinética, disprosódica y puede llegar a ser in in te l ig ib le .
En un 5-10% de los casos, la enfermedad se manif iesta antes de los 20 años (variante de Westphal ) ; en estos casos de in ic io j u ven i l , la r igidez p redomina sobre el corea y pueden asociar crisis comic ia les y ataxia cerebelosa. El 9 0 % de los mismos hereda el gen del padre afectado.
• Deterioro cognitivo. Aparece desde las fases iniciales de la enfermedad y se re lac iona con la patología de los gangl ios básales. Su curso es generalmente parale lo a la alteración motora . El trastorno de la memor ia es común, pero se trata de una demenc ia subcort ical y, a d i ferencia de la enfermedad de A lzhe imer , es rara la aparición de afasias, apraxias y agnosias.
• Trastornos psiquiátricos y de comportamiento. Aparecen en un 3 5 - 7 5 % de los pacientes, y generalmente en fases iniciales de la enfermedad. La manifestación más frecuente son los trastornos afectivos, i nc luyendo depresión unipolar o b ipolar , que afecta a un 5 0 %
4 9

Manual CTO de Medicina y Cirugía, 8.a edición
de los casos. El riesgo de su ic id io es mayor que en la población general . Trastornos psicóticos t ipo esquizofrenia aparecen entre un 5 - 1 0 % de los casos; de hecho, los pacientes con EH pueden haber estado diagnosticados de esquizofrenia años antes de comenzar con los trastornos de mov im ien to .
Diagnóstico
Puede establecerse con la historia clínica, la exploración y los antecedentes famil iares (MIR 98-99F, 72), o mediante el hal lazgo de un número excesivo de tripletes CAG (más de 40 repeticiones) en el c r o m o soma 4, lo que es diagnóstico per se (MIR 99-00, 194). La TC muestra atrofia de la cabeza del núcleo caudado con dilatación selectiva de las astas frontales de los ventrículos laterales. La RMN permite cuant i f icar el grado de pérdida de vo lumen del caudado y putamen, lo que se ha corre lac ionado con la progresión de la enfermedad. La tomografía por emisión de positrones (PET) demuestra trastornos metabólicos en gan glios básales y en algunas áreas cort icales.
Diagnóstico diferencial
El diagnóstico di ferencia l se realiza con : Neuroacantocitosis. Se hereda con carácter autosómico recesivo, aunque también hay formas esporádicas. Debuta en la edad adu l ta (3 . a -4. a década) con corea, demenc ia , distonía o ro l ingua l , auto-mut i lac iones con mordeduras en lab io y lengua, crisis comic ia les , amiotrof ias y neuropatía periférica. La CPK está aumentada. El d iag nóstico se establece demostrando los acantocitos en sangre periférica. Las B-lipoproteínas plasmáticas son normales, a di ferencia de la enfermedad de Bassen-Kornzweig, donde los acantocitos se asocian con abeta l ipoprote inemia , retinit is p igmentar ia y déficit neurológico progresivo, fundamenta lmente ataxia.
n R E C U E R D A • * En la neu roacan toc i tos i s se p r o d u c e corea c o n hematíes esp i cu i ados
(acantoc i tos ) , a u t o m u t i l a c i o n e s y cr is is c o m i c i a l e s .
Discinesias tardías, en pacientes en t ratamiento crónico con neurolépticos.
• Otros síndromes coreicos, c o m o el corea hereditario benigno, que es un corea no progresivo con in i c io en la infancia ; el corea senil, un comp l e j o sintomático raro que comienza después de los 60 años, sin trastornos del compor tamien to ni historia fami l iar ; el corea de Sydenham, asociado a la f iebre reumática o las distintas formas de corea asociadas a alteraciones metabólicas (véase la Tabla 24).
Tratamiento
• Terapia de reposición. El compor tamien to neurofarmacológico de la EH es, en c ier to sentido, inverso al que se da en la enfermedad de Parkinson. Hay una ligera h iperact iv idad dopaminérgica y, secundar iamente, una hipofunción colinérgica estriatal. Los receptores de dopamina , acet i l co l ina y serotonina en el estriado están d i s m i n u i dos. Además, se ha demostrado una importante disminución del GABA en esta enfermedad. Sin embargo, la terapia de sustitución con colinomiméticos o gabaérgicos ha resultado infructuosa.
• Neuroprotección. La hipótesis sobre si la exc i to tox i c idad mediada por g lutamato pudiera conduc i r a degeneración neuronal en esta enfermedad ha conduc ido a la utilización, sin ningún éxito, de fármacos c o m o el bac lofeno (gabaérgico que reduce la liberación de g lu tamato de las fibras corticoestriatales) o el dextrometorfan ( inh i b idor del receptor de g lutamato N M D A ) .
Puesto que no existe ningún t ratamiento patogénico ef icaz, funda menta lmente se realiza t ratamiento sintomático. El corea se trata s in tomáticamente con bloqueantes de receptores dopaminérgicos (neurolépticos) o con deplectores presinápticos de dopamina (reserpina o tetrabenacina), pero en bajas dosis y durante per iodos breves, dada la pos ib i l idad de discinesias tardías y park insonismo c o m o efectos secundarios. Respecto a las alteraciones psiquiátricas, la depresión se trata con antidepresivos tricíclicos o ISRS, y en casos concretos con i n h i b i dores de la M A O ; la psicosis se trata con neurolépticos (la c lozap ina , neuroléptico atípico, t iene menor efecto noc i vo sobre los trastornos motores, dada su baja interferencia sobre el sistema dopaminérgico, pero se debe recordar que es necesario una v ig i lanc ia hematológica por la pos ib i l idad de alteraciones a este n ive l , de las cuales la más grave es la agranulocitosis) .
5.7. Enfermedad de Parkinson idiopática
Es el síndrome park insoniano más común. Afecta más f recuentemente a varones, con una edad media de com ienzo de 55 años. Sólo un 5-10% debuta antes de los 40 años. El p romed io de inc idenc ia anual varía entre 7-19 casos por cada 100.000 habitantes, y su prevalencia es ampl iamente var iable en función de la edad y el área geográfica.
Patogenia
Es desconoc ida . El park insonismo es más común en el anc iano, y la edad avanzada es el factor de riesgo más importante en la etiología de esta enfermedad. Se han postu lado otros factores de riesgo con interés patogénico: genéticos, ambientales, t raumatismos, etc. • Factores genéticos. Aunque habi tua lmente t iene carácter esporádi
co, se han descr i to famil ias con enfermedad de Parkinson (EP) heredada con carácter autosómico dominante , penetrancia incompleta y edad de in i c io más precoz (45 años). Sin embargo, la inc idenc ia s imi lar entre gemelos monoc igotos y d ic igotos hace pensar que los factores genéticos no juegan un papel p r imord i a l . El A D N m i t o c o n dr ia l ha sido imp l i cado a través del déficit detectado en el comp l e j o I de la cadena respiratoria en la sustancia negra y plaquetas de pacientes parkinsonianos. Se han ident i f i cado diferentes genes en la patogenia de la EP fami l iar , c o m o el gen de la a-sinucleína y el gen de la parkina.
• Ambientales. La intoxicación accidental de drogadictos por la auto-inyección de MPTP (met i l fen i l te t rah idropi r id ina) da lugar a un cuadro de park insonismo muy similar al que presenta la fo rma idiopát ica, pero con alteraciones anatomopatológicas diferentes. La MPTP es un tóxico que, una vez ox idado por la MAO-B a su metabo l i to más act ivo MPP+, b loquea la función mi tocondr ia l ( inhibe el c o m p le jo I de la cadena respiratoria) y produce degeneración del sistema nígrico. El estudio de este mecan ismo de lesión ha ayudado a conocer la patogenia de la forma idiopática.
5 0

Neurología y neurocirugía
Otras sustancias c o m o el manganeso, el a l um in io , el arsénico, el-mercur io , el z inc , los pesticidas o los herbicidas se han imp l i c ado en la patogenia. Los niveles de hierro en los gangl ios básales se han descrito elevados en pacientes con enfermedad de Parkinson, postulándose que un exceso en los procesos oxidat ivos puede conduc i r a t ox i c i dad celular sufic iente para dar lugar a la enfermedad.
Anatomía patológica
En la enfermedad de Parkinson, hay una pérdida neuronal con despigmentación y gliosis preferentemente en la porción compacta de la sustancia negra (MIR 0 0 - 0 1 , 253) , aunque también pueden afectarse otros núcleos c o m o locus coeruleus, núcleos del rafe, núcleo basal de Meynert , co lumnas intermediolaterales de la médula y ganglios simpáticos y parasimpáticos (Figura 45) .
c ia negra se cor re lac iona con la acinesia y r ig idez (en park inson is mos uni laterales, la sustancia negra afectada más intensamente es la contra latera l ) . No hay área lesional c lara re lac ionada con el t emb lo r . La clínica autonómica se exp l i ca por la afectación de las co lumnas intermedio latera les de la médula espinal y los gangl ios simpáticos y parasimpáticos.
Los déficit cogni t ivos se re lac ionan con la lesión del núcleo basal de Meynert , locus coeruleus, y probab lemente por la afectación neocor-tícal directa. La escasa respuesta de la inestabi l idad postural al tratamiento con levodopa imp l i ca la participación lesional de estructuras no dopaminérgicas. Los fenómenos de congelación se han re lac ionado con defectos noradrenérgicos.
Clínica
El marcador anatomopatológico más característico son los cuerpos de Lewy, inclusiones intracitoplasmáticas eosinófilas rodeadas por un halo periférico menos densamente teñido que se loca l izan especial mente en las neuronas de la sustancia negra, locus coeruleus, núcleo dorsal del vago, núcleo basal de Meyner t y, con menor densidad, a nivel neocort ica l .
Los cuerpos de Lewy der ivan de elementos del c i toesqueleto neuronal alterado y se tiñen con anticuerpos frente a ub iqu i t ina . Sin embargo, a di ferencia de los ov i l los neurof ibr i lares de la enfermedad de A l z h e i mer, no se tiñen con ant icuerpos frente a la proteína x. A pesar de e l lo, son inmunorreact ivos con ant icuerpos frente al am i lo ide encontrado en la ami lo idos is fami l ia r t ipo f inlandesa ( re lac ionado con una mutación puntua l en el gen que cod i f i ca la gelsol ina, una proteína citoplasmática modu ladora de la actina).
n R E C U E R D A Los cuerpos de Lewy y la pérdida neuronal en la porcii de la sustancia negra son el marcador anatomopatológico de la enfermedad de Parkinson.
Se pueden establecer corre lac iones entre los lugares anatómicos afectados y los hal lazgos clínicos. La pérdida ce lu lar en la sustan-
Es un síndrome clínico caracter izado por temblor de reposo, brad ic ine sia, r igidez e inestabi l idad postural (MIR 99-00, 201) . Los dos pr imeros son los más típicos (Figura 46) .
Limitación en la supraelevación
de la mirada
Rigidez (fenómeno
de rueda dentada)
Hipomimia facial
Bradicinesia • Marcha festinante • Inestabilidad postural
Figura 46. Paciente con en fe rmedad de Parkinson
El temblor de reposo es un mov im i en to osc i lator io distal a 4-6 H z que afecta preferentemente a las manos, pero también puede afectar a labios, lengua, mandíbula y miembros inferiores. Rara vez afecta a la cabeza o cuerdas vocales. Típicamente, es asimétrico al in i c io (MIR 01 -02, 57). Const i tuye la fo rma de presentación más frecuente ( 6 0 - 7 0 % de los pacientes) y puede permanecer c o m o única manifestación de la enfermedad durante varios años. El t emb lo r postural está presente en aprox imadamente un 6 0 % de los pacientes, asociado o no a temblor de reposo.
51

Manual CTO de Medicina y Cirugía, 8.a edición
o R E C U E R D A El t e m b l o r de reposo en cuen ta de m o n e d a s es t ípicamente asimétrico al i n i c i o de la e n f e r m e d a d .
La bradicinesia consiste en una ralentización general izada de los m o v i mientos. Es la manifestación más incapaci tante de la enfermedad (MIR 02-03, 205). Resulta de la pérdida de los mecanismos dopaminérgicos inh ib i tor ios del estriado e h ipoac t i v idad de las neuronas del g l obo pál ido externo. Hay h i p o m i m i a fac ia l , disminución de la f recuencia de parpadeo (MIR 05-06, 58), lenguaje monótono e hipófono con fácil fa t igab i l idad, micrografía, d i f i cu l tad para levantarse de la silla y girarse en la cama. La marcha es típica, con flexión anterior del t ronco , a pequeños pasos, arrastrando los pies y con pérdida del braceo (marcha festinante) (MIR 03-04, 243).
La rigidez es un incremento de la resistencia a la movilización pasiva que predomina en la musculatura f lexora. Es constante a lo largo del mov im i en to (rigidez plástica), aunque se produce el fenómeno de r ig i dez en rueda dentada, que se considera c o m o la interferencia del t e m blor sobre la r igidez plástica durante la movilización pasiva del m i embro (se trata de una explicación parc ia l , dada la pos ib i l idad de rueda den tada en pacientes sin temblor de reposo). Se produce por desinhibición pal idal con incremento de la activación suprasegmentaria de los me canismos reflejos espinales normales y, por tanto, un incremento en la descarga de las a-motoneuronas.
R E C U E R D A En las fo rmas secundar ias de p a r k i n s o n i s m o sue le p r e d o m i n a r la r ig i d ez , y n o el t e m b l o r .
La inestabilidad postural se puede manifestar como propulsión (tendencia a desplazarse hacia delante) o retropulsión (desplazamiento hacia atrás).
Los hallazgos oculares inc luyen limitación en la supraelevación de la mirada y reflejo glabelar inagotable.
La disfunción autonómica se manif iesta por sialorrea, disfagia, estreñimiento , tendencia a la hipotensión, hipersudoración, nictur ia y urgencia m i c c i ona l . La nictur ia es el síntoma más precoz y frecuente de la clínica ur inar ia .
1) Dos de los s iguientes signos o síntomas: • Temblor de reposo • Rigidez • Bradicinesia • Inestabi l idad postura l
2) Mejoría s ignif icat iva con L-dopa
3) Descartar los park inson ismos secundar ios
4) Ausencia de signos incompat ib les con la E. de Parkinson: Oftalmoplejía supranuclear con parálisis en la intraversión de la mi rada Afectación cor t icoespina l Afectación de asta anter ior Signos cerebelosos Polineuropatía Mioclonías Crisis oculogiras
Tabla 25. Criterios diagnósticos de la enfermedad de Parkinson
R E C U E R D A La limitación en la supraversión de la m i r ada es típica del Park inson y la l imitación en la infraversión de la parálisis supranuc lear progres iva (MIR 04-05, 58) .
Tratamiento
Tratamiento farmacológico
En la enfermedad de Parkinson, existe un desequi l ibr io entre la d o pamina y la acet i l co l ina en los ganglios básales. C o m o consecuencia de la lesión de las vías nigroestriadas dopaminérgicas, se p roduce una caída de los niveles de dopamina estriatal, con el consiguiente predo m i n i o func iona l de los sistemas colinérgicos. Por tanto, la actuación farmacológica irá or ientada a potenciar los sistemas dopaminérgicos ( levodopa y/o agonistas dopaminérgicos) y d i sminu i r la act iv idad col i-nérgica (anticolinérgicos). • La levodopa (L-dopa) asociada a un inh ib idor de la dopadecarboxi-
lasa periférica (carbidopa-benseracida) sigue siendo el t ra tamiento de pr imera línea, y es especia lmente útil en el t ra tamiento de la bradic inesia y la r igidez; la falta de respuesta a levodopa habla en favor de síndrome park insoniano no idiopático. La carb idopa y ben-seracida, al inh ib i r la metabolización periférica de la levodopa, au mentan la b iod i spon ib i l i dad de la misma para su paso a través de la barrera hematoencefálica, pe rmi t i endo reducir la dosis y, por tanto, los efectos secundarios. En cua lquier caso, casi todos los pacientes que in ic ia lmente mejoran pierden su respuesta a la levodopa en 3-8 años, aparec iendo f luctuaciones motoras (fenómeno wearing off o f in de dosis, hipercinesia p i co de dosis, distonías f in de dosis, d i sc i nesias bifásicas, fenómenos on-off, fal los erráticos de dosis), y d i sc i nesias (pico de dosis, bifásicas) (MIR 99-00, 200). La asociación de levodopa con agonistas dopaminérgicos permite un contro l parcial de las mismas y la reducción de dosis de levodopa.
Los trastornos no motores en la enfermedad de Parkinson inc luyen cambios en la personal idad, deter ioro de func iones superiores (en fases avanzadas de la enfermedad (MIR 99-00, 195), al contrar io de lo que ocurre en los parkinsonismos secundarios, donde es más frecuente su aparición precoz), depresión y trastornos del sueño.
Diagnóstico
El diagnóstico de la enfermedad de Parkinson es clínico. Los criterios diagnósticos se resumen en la Tabla 25. El diagnóstico di ferencia l de la enfermedad de Parkinson se inc luye en la Tabla 26 (MIR 08-09, 57; MIR 06-07, 57).
R E C U E R D A Es p r o p i a de los p a r k i n s o n i s m o s secundar ios la p o c a respuesta a le v o d o p a .
Inhibidores de la C O M T : los inhib idores de la catecol-O-metil-transferasa (entacapona, to lcapona) aumentan también la b iod i spo n ib i l i dad de la levodopa, i nh ib i endo su metabol ismo, por lo que pueden administrarse asociados a levodopa. Existen formulac iones en las que se asocia levodopa + carb idopa + entacapona. Agonistas dopaminérgicos: se ut i l izan en monoterap ia cuando existe afectación leve-moderada, especia lmente en pacientes jóvenes y asociados a levodopa en fases avanzadas. Se clasif ican en ergóticos (cabergol ina, pergol ina, b romocr ip t ina , l isurida) y no ergóticos (pra-m i p e x o l , rop in i ro l , apomorf ina ) , estos últimos más ut i l izados en la actua l idad (MIR 09-10, 69).
52

Neurología y neurocirugía
I. PARKINSONISMO PRIMARIO
O IDIOPATICO
II. PARKINSONISMO SECUNDARIO, ADQUIR IDO
O S INTOMÁTICO
III. PARKINSONISMO HEREDODEGENERATIVO
IV. DEGENERAC IONES MULT IS ISTÉMICAS
Enfermedad de Parkinson Park insonismo juven i l
Infecciones
Drogas
Toxinas
Vascular
T raumat i smo
Otros
Enfermedad difusa por cuerpos de Lewy (A.D.) Enfermedad de H u n t i n g t o n Enfermedad de Wi lson Enfermedad de Hallervorden-Spatz Calcificación fami l iar de los gangl ios básales Parkinsonismo familiar con neuropatía periférica Neuroacantoc i tos is Comp le jo Parkinson-Demencia-ELA Parálisis supranuclear progresiva
Síndrome de Shy-Drager Degeneración estrionígrica Atrof ia ol ivo-ponto-cerebelosa Fallo autonómico progres ivo
Postencefalit is. Neurolúes. Neurobrucelos is E. de Creutzfeldt-Jakob. SIDA. Abscesos micó-ticos
B loqueantes de receptores dopaminérgicos: - Neurolépticos (ha loper ldo l , p imoz ide ,
etc.) - Depletores presinápticos de d o p a m i n a :
> Reserpina. tetrabenacina - Falsos neurotransmisores :
> a-met i ldopa > a-met i lparat i ros ina
- L i t io - Antagonis tas del calcio:
> Flunaricina, c inaric lna - Amioda rona - Isoniacida
MPTP, CO, M n , me tano l , e tano l
Mu l t i i n f a r to Shock h ipotens ivo
Encefalopatía pugilístíca (boxeador"sonado")
Alteraciones parat iroideas Hipotlroídismo Degeneración hepatocerebra l Tumor cerebral Hidrocefal ia normotens i va S lr ingomesencefal ia
Tabla 26. Diagnóstico diferencial de la enfermedad de Parkinson
Los anticolinérgicos ( t r ihex i fen id i l , b iper ideno) son útiles para el t ratamiento de pacientes jóvenes con p redomin io clínico del t e m blor de reposo, pero se debe evitar su uso en personas mayores, dados sus efectos secundarios confusionales y de alteración de la memor ia (MIR 00-01 F, 67). A menudo , su utilización se ve l imi tada por los efectos secundarios antimuscarínicos periféricos, que i n c l u yen boca seca, visión borrosa, estreñimiento, náuseas, retención ur inar ia , trastornos en la sudoración y taquicard ia .
to que aumenta la síntesis y l i beración de dopamina y puede d i sminu i r su recaptación en la hendidura sináptica. As imismo, se le atr ibuyen propiedades an-ticolinérgicas. Me jora la b rad i c i nesia, r igidez y temblor , pero en aprox imadamente un año de tratamiento se precisa añadir otros fármacos, por la pérdida de e f i cacia.
En la actual idad, se prefiere, en las formas iniciales del parkinson y en las formas leves, ut i l izar los agonistas dopaminérgicos c o m binándolos en ocasiones con la seligi l ina, mientras que en las formas sintomáticas (moderado-grave), se ut i l iza c o m o tratamiento de elección la levodopa.
Tratamiento quirúrgico de la enfermedad de Parkinson
El tratamiento quirúrgico de la EP comenzó en los años cuarenta, antes de que se dispusiera de la levodopa. Con la aparición de esta última, en los años sesenta, decayó el interés por la cirugía en esta enfermedad. La pérdida de efectividad a largo plazo de la levodopa, j un to a los efectos secundarios asociados con su administración, sobre todo, las discinesias tardías, ha mot ivado un resurgimiento de las técnicas q u i rúrgicas destinadas al control de
la sintomatología de esta enfermedad, fac i l i tado por los avances técnicos en el campo de la estereotaxia y neurofisiología, que permiten localizar las dianas con precisión.
R E C U E R D A En el t r a t am ien to quirúrgico, son de elección las técnicas de es t imu la ción sobre las ablat ivas. La estimulación bi la tera l de l núcleo subtalámi co es la de m e j o r resul tado.
R E C U E R D A El t e m b l o r de reposo se trata c o n anticolinérgicos El t e m b l o r postura l c o n p r o p a n o l o l .
El t ratamiento quirúrgico puede plantearse en pacientes relat ivamente jóvenes, con sintomatología incapacitante, que no responden a la me dicación o que presentan intolerancia a la misma o efectos secundarios importantes que l imi tan su uso.
El deprenil o selegilina, un inh ib idor selectivo de la MAO-B, ha demostrado en algunos estudios ralentizar el desarrol lo de la discapac idad motora y d i sminu i r el índice de progresión de la enfermedad cuando se usa en estadios tempranos de la enfermedad (efecto neuroprotector ) . Además, puede ofrecer una mejoría sintomática, deb ido a que incrementa las concentrac iones estriatales de d o p a m i na al b loquear su catabol ismo. La amantadina es débilmente efectiva para contro lar los síntomas. Su mecan ismo de acción es desconoc ido, aunque se ha propues-
Esencialmente, existen dos t ipos de técnicas para el t ra tamiento q u i rúrgico de la enfermedad de Parkinson: las técnicas ablativas (lesión mediante termocoagulación por radiofrecuencia que destruye las células y fibras nerviosas en el lugar de la lesión) y la estimulación cerebral profunda (estimulación crónica del núcleo a alta f recuencia , que p ro duce un efecto s imi lar al de la lesión, pero reversible). Ambos p roced i mientos t ienen c o m o diana ciertos núcleos de los gangl ios de la base (globo pálido media l y núcleo subtalámico) y el tálamo (núcleo ventral in termedio o V IM) .
53

Manual CTO de Medicina y Cirugía, 8.a edición
En la actua l idad, puede decirse que la técnica quirúrgica de elección es la estimulación bi lateral del núcleo subtalámico. En los casos en que p redomina el t emblor , la d iana de elección puede ser el V I M (lesión o estimulación). Para la bradic inesia y la r igidez, las dianas preferidas son el núcleo subtalámico y el g lobo pálido.
5.8. Otros síndromes parkinsonianos ( F ¡ g u r a 4 7 )
en extensión cerv ica l . La marcha es rígida y con base ampl ia . Hay escasa o nula respuesta al t ra tamiento con levodopa.
• Distonía que afecta p r inc ipa lmente al cue l lo (dato característico). Ot ra fo rma de distonía presente es el blefarospasmo.
• Disfunción corticobulbar o cort icoespinal con aumento de reflejos miotáticos, signo de Babinski , disartria, disfagia y lab i l idad e m o c i o nal (síndrome pseudobulbar ) .
• Parálisis de la mirada conjugada en el p lano vert ica l , especia lmente de la infraversión de la mirada en fases inic ia les. Se considera el signo clínico más característico de esta enfermedad.
• Demencia.
P a r á l i s i s s u p r a n u c l e a r p r o g r e s i v a ( e n f e r m e d a d n RECUERDA El paciente con enfermedad de Parkinson tiende a caerse hacia delante,
d e S t e e l e - R Í C h a r d S O n - O I S Z e W S k Í ) mientras que en la PSP, las caídas son hacia atrás y más frecuentes.
Se trata de una ent idad clínica que afecta a ancianos en el m ismo per io do de edad que la enfermedad de Parkinson, y que se caracteriza por: • Síndrome parkinsoniano con bradicinesia, r ig idez, escaso temblor
e inestabi l idad postural , s iendo frecuentes las caídas, especia lmente hacia atrás (MIR 02-03, 254) . La r igidez es más evidente en la musculatura axial que en los miembros , lo que conduce a una postura
ENFERMEDAD DE PARKINSON
IDIOPATICA
• Forma más común de sd. parkinsoniano
• Clínica de presentación: temblor de reposo
• Marcha típica: flexión anterior del tronco y pequeños pasos arrastrando los pies sin braceo
• Parálisis supraelevación de la mirada
• Buena respuesta a L-DOPA • AP: lesión de la porción compacta de la sustancia negra cuerpos de Lewy troncoencefálicosy en ganglios básales.
•Tratamiento: Fases iniciales: deprenil o
selegilina SI no responde, añadir agonistas DOPA El siguiente paso es L-DOPA + carbidopa (con deterioro funcional significativo) En jóvenes con predominio de temblor : anticolinérgicos (nunca en ancianos)
NO-
• Distonía cervical • Parálisis infraversión de la mirada
• Frecuentes caídas hacía atrás • Demencia y síndrome pseudobulbar
• Escaso temblor • Mala respuesta a L-DOPA • AP: madejas neurofibrilares, degeneración gránulo-vacuolar
Síntomas asociados
SIN demencia inicial
Sd. PARKINSONIANO
Bradicinesia + Rigidez +
Inestabilidad postural
+/-Temblor de reposo
PARÁLISIS SUPRANUCLEAR
PROGRESIVA (enf. de Steele-
Richardson-Olszewski)
CON demencia inicial
ATROFIAS MULTISISTÉMICAS
ENF. DIFUSA POR CUERPOS DE LEWY
Debe considerarse el diagnóstico en pacientes anc ianos con f r e cuen tes caídas, signos ext rap i ramida les , r ig idez cerv ica l y parálisis de la mirada ver t i ca l . Los fármacos ant ipark inson ianos p roducen escasos benef ic ios . • Degeneración corticobasal gangliónica. Se trata de un parkinsoni-
m o rigidoacinético progresivo, sin respuesta a la levodopa. Predomina en varones, a partir de los 50 años. Suele ser asimétrico, con
presencia de distonía, mioclonías, pudien-do conl levar temblor postural de acción, con pérdida sensorial cort ica l (heminegl i-gencía sensorial), apraxias ( ideomotora) y el fenómeno de " m a n o ajena o alienígena" , s iendo la demenc ia un signo tardío. Pueden aparecer trastornos de la mot i l i dad ocular , p i ramida l i smo y deter ioro bulbar sin ataxia.
• Enfermedad de Hallervorden-Spatz. Es una entidad con herencia autosómica recesiva que suele debutar en la adolescencia y que se caracteriza clínicamente por la presencia de demencia, alteraciones de la postura y tono muscular, clínica extrapiramidal (corea, distonía) y ataxia. En el estudio anatomopatológico se demuestra un acumulo de hierro en los ganglios básales, concretamente en pálido y porción reticular de la sustancia negra, que ofrece un aspecto característico en resonancia magnética conoc ido como "signo del o jo de t igre" . No hay trastorno sistémico del metabol ismo del hierro, por lo que la utilización terapéutica de quelantes de este metal es inútil (Figura 48).
Q RECUERDA " La HTA es un factor de riesgo tanto para la
enfermedad vascular cerebral como para la cardiopatía isquémica, pero el riesgo relativo es mayor para la primera.
• Aparición más precoz • Escaso temblor • Mala respuesta a L-DOPA • AP: no cuerpos de Lewy
ni madejas neurofibrilares. Pérdida neuronal y gliosis en diversos núcleos
Con predominio de: Disautonomía: Shy-Drager Ataxia: Atrofia olivopontocerebelosa esporádica Parkinsonismo: Degeneración estrionígrica
• Predominio de rigidez • Demencia, alt. psiquiátricas, mioclonías
• Escaso temblor • Mala respuesta a L-DOPA • AP: cuerpos de Lewy a nivel neocortical
Figura 47 Diagnóstico diferencial de los síndromes parkinsonianos
Las atrofias multisistémicas también pue den produc i r clínica park insoniana (se t ra tarán en el siguiente apartado). Otros síndromes parkinsonianos (véase Figura 47 . Diagnóstico di ferencia l de la enfermedad de Parkinson) se tratarán en otros temas de esta Sección o en otras partes de esta obra.
54

Neurología y neurocirugía
Figura 48 . RM con la imagen de "o jo de t igre". Se puede apreciar" la cara de t igre", con la boca abajo, la nariz en m e d i o y la imagen en o jo de t ig re
es la par te negra de arr iba, q u e es lo patológico
R E C U E R D A El signo del ojo de tigre en la RMN cerebral es característico de la enfermedad de Hallervorden-Spatz.
Las atrofias multisistémicas también pueden producir clínica parkinsonia-na (se tratarán en el siguiente apartado). Otros síndromes parkinsonianos (véase Figura 47. Diagnóstico diferencial de la enfermedad de Parkinson) se tratarán en otros temas de esta Sección o en otras partes de esta obra.
mente sin temblor de reposo que, si existe, no suele ser en "cuenta de monedas" . Suele asociarse a clínica cerebelosa y p i ramida l i smo. La respuesta a levodopa es mala. Piramidalismo. Se considera c o m o tal una clara respuesta cutaneoplantar extensora y la presencia de franca hiperref lexia. Es frecuente la combinación de espasticidad y r ig idez. Signos y síntomas cerebelosos. La ataxia de la marcha es el signo cerebeloso más común. Inc luye también habla escandida, h ipofo-nía, dismetría o d isd iadococ ines ia . Una acinesia grave puede en mascarar estos hal lazgos. Signos y síntomas autonómicos. Suelen preceder al resto de los síntomas. Inc luyen hipotensión, impotenc i a , i ncon t inenc i a u r ina ria, etc. La demenc i a y las crisis no son datos integrantes de esta ent idad y, a efectos prácticos, su presencia descarta una AMS no comp l i c ada .
J R E C U E R D A No es propio de las atrofias multisistémicas la demencia y las crisis comiciales.
El p r edomin i o clínico puede variar en un mismo paciente a lo largo de su evolución. Cuando p redomina la clínica parkinsoniana, se habla de degeneración estrionígrica; cuando predomina la clínica autonómica , de síndrome de Shy-Drager (MIR 00-01 F, 64) y, cuando lo hace la ataxia y el p i ramida l i smo, se habla prop iamente de OPCA (forma esporádica).
A nivel anatomopatológico, las AMS presentan pérdida neuronal y glio-sis (sin cuerpos de Lewy) que puede afectar a las siguientes estructuras: sustancia negra, caudado y pu tamen , pálido, olivas inferiores, p ro tube rancia, cerebelo y co lumnas intermediolaterales medulares.
5.9. Atrofias Multisistémicas (AMS)
Bajo esta denominación, se inc luye un grupo heterogéneo de pa to lo gías degenerativas del sistema nervioso referidas a lo largo de la historia con distintas denominac iones : atrofia ol ivo-ponto-cerebelosa (OPCA), degeneración estrionígrica, síndrome de Shy-Drager y fa l lo autonómico progresivo, depend iendo de la combinación de signos clínicos, las manifestaciones predominantes y los hal lazgos anatomopatológicos en la autopsia. En la actua l idad, se prefiere agrupar estas entidades bajo la común denominación de degeneraciones multisistémicas.
Clínicamente, pueden presentarse con una combinación de: • Clínica parkinsoniana. Suele debutar antes que la enfermedad de
Parkinson idiopática, t iende a ser simétrica en su distribución, con p redomin io de acinesia, r igidez y afectación postura l , pero general-
La ausencia de cuerpos de Lewy las dist ingue de la enfermedad de Parkinson idiopática, y la ausencia de madejas neurof ibr i lares las d i ferencian de la parálisis supranuclear progresiva y del park insonismo postencefalítico. Recientemente se ha demostrado la presencia de i n clusiones intranucleares e intracitoplasmáticas a nive l neuronal y ol igo-dendrog l ia l , en los casos de AMS esporádicas.
R E C U E R D A Ante un parkinsonismo con clínica disautonómica precoz y falta de respuesta a L-dopa, dse debe pensar en el síndrome de Shy-Drager. I
En cuanto al t ratamiento, los agonistas dopaminérgicos no son más efectivos que la levodopa, aunque algunos pacientes los toleran mejor . Son características las discinesias inducidas por levodopa crónicamente administrada: espasmos distónicos mantenidos de la musculatura fa c ia l , distonías cervicales o antecol is desproporc ionado. Se han descrito respuestas ocasionales a la amantadina .
55

Manual CTO de Medicina y Cirugía, 8.a edición
r
Casos clínicos representativos
U n p a c i e n t e d e 6 0 años r e f i e r e q u e , desde h a c e años, le t i e m b l a n las m a n o s a l sost e n e r la c u c h a r a , e l vaso o el bol ígrafo, s o b r e t o d o si está n e r v i o s o o f a t i g a d o , y es tos s íntomas m e j o r a n c o n pequeñas c a n t i d a d e s d e v i n o . Su p a d r e , ya f a l l e c i d o , había p r e s e n t a d o t e m b l o r en las m a n o s y la c a b e z a . La exp lorac ión neuro lóg ica sólo m u e s t r a t e m b l o r d e a c t i t u d s imétr ico en a m b a s m a n o s . Este c u a d r o c l ín i co es p r o b a b l e m e n t e c o n s e c u e n c i a d e :
1) U n h i p o t i r o i d i s m o f a m i l i a r . 2 ) U n a e n f e r m e d a d d e Pa rk inson i n c i p i e n t e . 3) S íntomas d e depr ivac ión etí l ica. 4) U n t e m b l o r e s e n c i a l . 5) U n a neuros i s d e a n s i e d a d orgánica f a m i l i a r .
M I R 0 3 - 0 4 , 2 4 2 RC: 4
H o m b r e d e 7 0 años , c o n t e m b l o r d e r e p o s o d e 4 h e r z i o s y t o r p e z a en e x t r e m i d a d s u p e r i o r d e r e c h a , desde hace u n año . A l c a m i n a r , el b r a c e o está d i s m i n u i d o en el l a d o d e r e c h o . Se in ic ió t r a t a m i e n t o c o n 7 5 0 m i l i g r a m o s d e l e v o d o p a y 75 m i l i g r a m o s d e c a r b i d o p a a l d ía , c o n desapar ic ión de los s íntomas. ¿ Q u é e n f e r m e d a d y evo luc ión son las más p r o b l a b l e s ?
1) C o r e a d e H u n t i n g t o n c o n d e t e r i o r o c o g n i t i v o p r o g r e s i v o .
2) Parálisis s u p r a n u c l e a r p rog res i va c o n apar ic ión tardía d e l imitac ión en la m i r a d a v e r t i c a l , t a n t o s u p e r i o r c o m o i n f e r i o r .
3) E n f e r m e d a d d e Pa rk i son c o n extensión de l t e m b l o r a la p i e r n a d e r e c h a . 4) E n f e r m e d a d d e Creu tz fe ld t- J akob c o n rápido d e t e r i o r o c o g n i t i v o y f r e c u e n t e s
mioc lon ías . 5) T e m b l o r e senc i a l f a m i l i a r c o n apar ic ión d e t e m b l o r en e x t r e m i d a d s u p e r i o r
i z q u i e r d a y c a b e z a .
M I R 0 1 - 0 2 , 5 7 ; RC: 3
U n h o m b r e d e 4 5 años d e e d a d es t ra ído a la c o n s u l t a p o r d e t e r i o r o c o n g n i t i v o p r o g r e s i v o . La exp lorac ión m u e s t r a m o v i m i e n t o i n v o l u n t a r i o s i r r e g u l a r e s de las e x t r e m i dades . El p a d r e d e l p a c i e n t e fa l lec ió a los 6 0 años d e e d a d e n u n c e n t r o ps iquiátr ico, y t ambién p r e s e n t a b a d i c h o s m o v i m i e n t o s i n v o l u n t a r i o s d e e x t r e m i d a d e s . ¿Cuá l es e l d iagnóst ico más p r o b a b l e ?
1) C o r e a d e S y d e n h a m . 2) E n f e r m e d a d d e H u n t i n g t o n . 3) E n f e r m e d a d d e La fora . 4) E n f e r m e d a d de H a l l e r v o r d e n - S p a t z . 5) Parálisis s u p r a n u c l e a r p rog res i va .
RC: 2
56

Neurología y neurocirugía
06 ENFERMEDADES POR ALTERACIÓN DE LA MIELINA
MIR T e m a i m p o r t a n t e y r en tab l e . H a y q u e centrarse en la esclerosis múltiple, c o n o c i e n d o sus d is t intas f o rmas e vo lu t i v a s y el t r a t a m i e n t o a d e c u a d o pa ra cada una . t u
Aspectos esenciales
La esclerosis múltiple ( ) afecta p r e d o m i n a n t e m e n t e a mujeres jóvenes, s i endo la causa más f recuente de d i s capac idad neurológica en adu l tos jóvenes.
Se p r o d u c e una reacción inmunológica con t ra los o l i godend roc i t o s , células fo rmadoras de la m i e l i n a a n ive l del SNC. N u n c a se afecta el SNP.
Se de tec tan bandas o l i goc lona l e s de IgG en el LCR de prácticamente todos los pac ientes c o n EM, a u n q u e n o es un s igno específico (también se encuen t r an en la neurolúes, infección por sarampión,...).
La f o r m a e v o l u t i v a remitente-recurrente q u e cursa a brotes es la f o r m a más f recuente y la q u e m e j o r responde al t r a t am ien to i n m u n o m o d u l a d o r .
["5"] El t r a t am ien to i n m u n o m o d u l a d o r q u e d i s m i n u y e el número de brotes es interferón p y acetato de g la t i ramer .
El t r a t am ien to sintomático de l b ro t e se rea l iza c o n co r t i co ides vía ora l (si el b ro t e es leve) o vía int ravenosa (si es moderado-grave ) .
["7"] Los síntomas sensit ivos (hipoestesias y parestesias) son la clínica de presentación más f recuente , seguidos de la neur i t i s óptica.
( j j j El s igno de Lhermi t te y la oftalmoplejía in te rnuc lea r en un a d u l t o j o v e n debe hacer pensar en la EM.
["g~| El diagnóstico d e EM es cl ínico, y requ iere la ex is tenc ia de cr i ter ios de diseminación t e m p o r a l (episodios de déficit neurológico separados en el t i empo ) y de diseminación espacial ( lesiones en dist intas zonas del SNC); ambos cr i ter ios p u e d e n ser eva luados en una única exploración: la resonanc ia magnética.
(TQ[ La corrección rápida de una h i p o n a t r e m i a es la causa más f recuente de mielinólisis centra l p o n t i n a .
P r egun tas
- M I R 09-- M I R 08-- M I R 07-- M I R 0 6 - M I R 05-- M I R 0 3 - M I R 02-- M I R 0 1 - M I R 0 1 -- M I R 00-- M I R 9 9 - M I R 99-- M I R 97-
1 0 , 6 4 , 2 2 8 0 9 , 5 8 0 8 , 61 0 7 , 5 9 0 6 , 5 5 , 5 6 0 4 , 2 4 0 , 2 4 4 0 3 , 2 0 7 0 2 , 5 3 0 2 , 5 4 0 1 , 5 5 0 0 , 1 9 7 O0F, 6 6 , 2 5 6 9 8 , 4 1 , 4 2 , 4 8
Las enfermedades des-
mielinizantes son un
conjunto de enferme
dades neurológicas que
tienden a afectar a adul
tos jóvenes.
Se caracter izan por
una inflamación y des
trucción selectiva de
la mie l ina del sistema
nervioso centra l , res
petando en general el
sistema nervioso pe r i
férico (MIR 01-02, 53)
(Tabla 27) .
ENFERMEDADES DESMI ELI N IZANTES
• Esclerosis múltiple • Síndrome de Devic • Enfermedad de Balo • Enfermedad de Marchiafava-Bignami • Mielinólisis centra l pon t i na - Encefalomiel i t is d i seminada aguda • Encefalomiel i t is hemorrágica necrot izante aguda
ENFERMEDADES DISMIELINIZANTES
• Leucodistrof ia metacromática. Alteración func iona l de arilsulfatasa A. Herencia autosómica recesiva
- Leucodistrof ia sudanófila • Adreno leucod is t ro f ia . Herencia l igada al c r o m o s o m a X • Enfermedad de Pelizaeus-Merzbacher. Herencia recesiva l igada
al c r omosoma X • Leucodistrof ia de células g lobo ides 0 en f e rmedad de Krabbe • Deficiencia de galactocerebrosidasa • A c u m u l o de galactos i lceramida • Herencia autosómica recesiva
Tabla 27. Enfermedades por alteración de la miel ina
6.1. Esclerosis múltiple (EM)
Es una enfermedad de etiología desconocida y patogenia au to inmun i ta r i a caracterizada por presentar, en el
7 5 % de los casos, un curso ondu lante con exacerbaciones y remisiones en su sintomatología. Exceptuando los
traumatismos, es la causa más frecuente de discapacidad neurológica en adultos jóvenes y const i tuye la fo rma
más frecuente de enfermedad por alteración de la mie l ina en el sistema nervioso centra l .
57

Manual CTO de Medicina y Cirugía, 8.a edición
Epidemiología
Afecta preferentemente a pacientes entre los 20-45 años, fundamenta l mente a mujeres ( 6 0 % de los casos). La prevalencia es mayor con fo rme uno se aleja del ecuador en ambos hemisferios (la inc idenc ia en zonas geográficas de c l ima t emp lado es superior a la que se observa en las zonas tropicales) y p redomina en la población de raza b lanca. Distintas evidencias apoyan la participación de factores ambientales en la apar i ción de esta enfermedad.
n R E C U E R D A La EM es mucho más frecuente en los países nórdicos, y fueron los vikingos quienes facilitaron su extensión.
Genética
Existe una clara suscept ib i l idad genética para el desarrol lo de EM. Son evidencias a favor la mayor f recuencia en gemelos univ i te l inos y la suscep t ib i l i dad en pacientes con los antígenos del comp le jo mayor de his-tocompa t ib i l i dad HLA-DR2 y HLA-DQ. No obstante, parece necesaria la unión de varios factores ambientales inc id iendo sobre un paciente con predisposición genética.
Anatomía patológica
La alteración más característica es la aparición de áreas o placas de desmielinización b ien del imitadas a nivel del SNC, local izadas pre ferentemente a nivel per iventr icu lar y subpia l , así c o m o en el t ronco encefálico, médula espinal y nervio óptico. En ellas hay un inf i l t rado de células T (CD4+) y macrófagos, con práctica ausencia de l infoci tos B y células plasmáticas. Cuando la placa se croni f ica , la población l infoci-taria p redominante es la de células B y T con fenot ipo supresor (CD8+) .
Estas lesiones son típicamente más numerosas de lo que pudiera an t iciparse por criterios clínicos, y no hay n inguna correlación entre el número de placas y su tamaño con los síntomas clínicos. En un 3 5 - 4 0 % de casos son clínicamente silentes y se ev idenc ian en la autopsia.
| H H [ ^ H H B H H B | ^ H B H H | J | | [ ^ H | n R E C U E R D A ™ * Nunca se afecta el SNP, porque la célula que forma la mielina a este
nivel es la célula de Schwann, y no el oligodendrocito.
El fenómeno patogénico p r imar io podría ser la lesión de los o l igoden-droci tos, células formadoras de mie l ina en el sistema nervioso centra l . El sistema nervioso periférico nunca se afecta.
Inmunología
Uno de los aspectos más importantes en esta patología es la presencia, hasta en un 9 5 % de los pacientes con EM, de bandas ol igoclonales de IgG en el LCR, que no están presentes en suero, y que se traducen en la activación de un número reducido de clones de l infocitos B con au mento de la síntesis intratecal (dentro de la barrera hematoencefálica) de anticuerpos, sin saberse con cierta precisión contra qué antígenos están
dir igidos. Las bandas ol igoclonales no son específicas de la esclerosis múltiple, pud iendo aparecer en otros trastornos. También en el LCR es posible objetivar una respuesta inespecífica contra distintos virus, entre los que destacan el del sarampión, herpes zoster, HTLV-1 y rubéola. Especial atención ha rec ibido también la presencia de l infocitos T inmuno-rreactivos frente a proteínas de la miel ina (proteína básica de la mie l ina, proteína proteolipídica y lipoproteína asociada a la miel ina) .
Curso clínico
Las manifestaciones clínicas son muy variables, distinguiéndose cuatro formas evolut ivas (Figura 49) .
E M CURSO CL ÍN ICO
ZfSpzf / V W F O R M A REMITENTE -RECURRENTE (RR)
F O R M A S E C U N D A R I A M E N T E PROGRESIVA (SP)
A
'"J^f j F O R M A PR IMAR IAMENTE PROGRESIVA (PP)
F O R M A PROGRESIVA RECURRENTE (PR)
Figura 49. Formas clínicas de la esclerosis múltiple
• Forma remitente en brotes (recurrente remitente o RR): el 8 5 % de los pacientes presentan ep isod ios de ep isodios o brotes de d is función neurológica, más o menos reversibles, que recurren en el t i e m p o y que , a med ida que se rep i ten , van de j ando secuelas neurológicas. Se cons idera un brote la aparición de síntomas o signos de déficit neurológico, de más de 24 horas de duración. Para cons iderar dos brotes d is t intos , t ienen que afectar a dist intas partes del SNC, y con un in terva lo entre e l los de, al menos, un mes.
• Forma secundariamente progresiva (SP): con el paso de los años, los pacientes con formas RR presentan un deter ioro lentamente p ro gresivo sin claros brotes. En los 10-15 pr imeros años de evolución de las formas RR, el 5 0 % de los pacientes se transforman en una forma SP, con lo que, en fases tardías, es la fo rma evo lut iva más frecuente. No existen indicadores exactos que predigan cuándo se producirá el paso de forma RR a SP, o si lo hará.
• Forma primaria progresiva (PP): el 1 0 % de los pacientes presentan un curso progresivo desde el com ienzo de la enfermedad, sin b ro tes. La forma más frecuente de com ienzo es con una paraparesia espástica progresiva. Son las formas de más difícil diagnóstico, dada
58

Neurología y neurocirugía
la ausencia de diseminación tempora l , que se considera una de las características de la enfermedad.
• Forma progresiva recurrente (PR): el 5 % de los pacientes presentan deter ioro progresivo desde el comienzo , pero en el curso de la en fermedad aparecen brotes.
En las primeras fases no hay claros factores que puedan determinar el curso que adoptará la enfermedad, por lo que el compor tamien to y gravedad de la misma es, en un pr imer momen to , impredec ib le . Existen, no obstante, algunos factores que pueden considerarse pronósticos de una evolución más grave, ident i f icables en el debut y los pr imeros brotes, y que están descritos en la Tabla 28 .
Paciente varón Debut en edad avanzada Enfermedad progresiva desde el inicio de los síntomas Signos motores y cerebelosos en el debut Escasa recuperación de un brote Corto intervalo entre los dos primeros brotes Múltiples lesiones en RM en el debut
Tabla 28. Marcadores pronósticos que predicen una evolución más grave de la EM
Síntomas
Es f recuente la lesión de la vía piramidal , con la cl ínica co r r e spon d ien te de p r imera m o t o n e u r o n a . La distribución del déficit de fuerza es va r i ab l e según la localización de la lesión: hemipares ia , para-paresia, tetraparesia , etc. En las lesiones medulares, es f recuente la asociación c o n urgenc ia m i c c i o n a l , i m p o t e n c i a y pérdida de la sens ib i l i dad co rdona l poster ior que c o n d u c e a ataxia sensit iva y s igno de Romberg . Si la lesión co rdona l es a n ive l ce r v i ca l , puede aparecer una especie de descarga eléctrica descendente al f l ex iona r el c u e l l o ; es el signo de Lhermitte, que también puede aparecer en otros t rastornos con c o m p r o m i s o de co rdones poster iores a n ive l ce r v i ca l , c o m o la espond i los i s ce r v i ca l , la tabes dorsal y la mie lopa-tía por radiación. Las fo rmas medu la res son las de peor pronóstico, d a d o que co n e levada f r ecuenc i a se hacen progresivas.
R E C U E R D A En un anc iano con signo de Lhermitte, debes descartar espondi losis cerv ica l más que EM.
Es f recuente en estos pac ientes la cl ínica de diplopía, genera lmente secundar ia a lesión del fascículo l o n g i t u d i n a l m e d i a l , que o r i g i na una oftalmoplejía internuclear. Su aparición en un pac iente j o ven nos debe hacer sospechar EM, mientras que en mayores de 50 años, la etiología suele ser vascular (MIR 99-00 , 197) . También puede o b servarse parálisis del V I par, pero son raras las de l III y IV.
La mayoría de los sistemas funcionales del SNC se verán afectados a lo largo de la evolución de la enfermedad, presentando el paciente una ampl ia var iedad de síntomas. La Tabla 29 muestra los síntomas neuro-lógicos más frecuentes al in i c io de la enfermedad (MIR 09-10, 64 ; MIR 09-10, 228 ; MIR 05-06, 56; MIR 99-00F, 66) .
Síntomas sensitivos (61%) - Hipoestesia (37%) - Parestesias (24%) Visión borrosa por neuritis óptica (36%) Debilidad y otros síntomas motores (35%) Diplopía (15%) Ataxia (11%)
Tabla 29. Clínica de presentación
Los síntomas sensitivos son los más frecuentes, e inc luyen parestesias o hipoestesias de var iable distribución. Es característica la sensibi l idad al calor, con reaparición o empeoramien to de los síntomas con el a u m e n to de la temperatura corpora l (MIR 02-03, 207) .
La afectación del cerebelo o de sus vías de conexión a nivel t roncoen-cefálico conduce a la aparición de ataxia, disartria cerebelosa (palabra escandida), nistagmo y temblor cinético.
La disfunción cogni t i va es común en casos avanzados, s iendo la pérdida de memor ia la manifestación más frecuente. La depresión aparece react ivamente al conocer que se padece la enfermedad o con la evo lución.
También en fases avanzadas, es característica la sintomatología frontal con eufor ia y compor tamien to des inh ib ido .
O t r o s síntomas son la fa t iga intensa c o n la m a r c h a o e j e r c i c i o m o d e r a d o y síntomas paroxísticos, c o m o cr is is c o m i c i a l e s ( 1 - 4 % ) , distonía, vértigo, acúfenos o neu ra lg i a de l trigémino (MIR 97-98 , 4 8 ) .
Diagnóstico La neuritis óptica (NO) es una manifestación muy frecuente en el curso de la enfermedad. Es generalmente uni la tera l , y su expresiv idad clínica varía desde ligera visión borrosa y pérdida de la saturación del co lor a amaurosis. Es más frecuente la neurit is retrobulbar ( fondo de o jo normal ) que la papi l i t is (tumefacción papilar en el f ondo de ojo) . El paciente con neurit is óptica presenta do lor con la movilización ocular (MIR 97-98, 41) y el examen campimétrico demuestra escotoma ceco-centra l . En fases crónicas, puede evo luc ionar a pa l idez papi lar (atrofia óptica). Cuando un paciente debuta con N O , la presencia de bandas o l igoc lonales (BOC) en LCR o la existencia de una RM patológica son signos de mal pronóstico para el desarrol lo de EM. En cond ic iones normales, el riesgo de desarrollar EM tras N O oscila entre un 3 5 - 7 5 % .
R E C U E R D A Escotoma cecocentra l y dolor con la movi l ización ocular sugieren neuritis óptica. En un adulto joven es indicativo de EM.
Aún hoy sigue s iendo un diagnóstico por exclusión, dada la ausencia de pruebas diagnósticas de certeza, y exige un diagnóstico di ferencia l exhaust ivo. La base para el diagnóstico sigue s iendo la clínica (MIR 99-00F, 256) .
Existen dist intos cr i ter ios diagnósticos, s iendo los más ut i l i zados los de Poser (1983) y los propuestos rec ientemente por M c D o n a l d (2001) , pero en todos , el diagnóstico clínico de la EM requiere la existencia de cr i ter ios de diseminación tempora l (dos o más episodios de déficit neurológico, separados entre sí por al menos un mes sin nuevos síntomas) y diseminación espacial (síntomas y signos que ind i can al menos dos lesiones independientes en el SNC). De estas lesiones se puede tener ev idenc ia clínica (algún s igno anorma l en la exploración) o en pruebas complementa r i as (demostración por m e d i o de potencia les evocados o prueba de imagen , aunque no hayan dado lugar a síntomas clínicos).
59

Manual CTO de Medicina y Cirugía, 8. a edición
Pruebas complementarias Tratamiento
LCR: aparece una ligera elevación de los l infocitos y de las proteínas totales en el 4 0 % de los pacientes; aumento de las gammaglobul inas en el 7 0 % ; elevación de IgG en el 8 0 % , y bandas ol igoclonales en algo más del 9 0 % (MIR 07-08, 6 1 ; MIR 01-02, 54), aunque n inguno de estos datos es patognomónico. Las bandas ol igoclonales reflejan la existencia de act iv idad inmunológica pr imaria en el SNC, y pueden aparecer en otras enfermedades que cursen con inflamación del SNC, c o m o neurolúes, SIDA o panencefalit is esclerosante subaguda. Potenciales evocados: se trata de estudiar los potenciales generados en el SNC tras la estimulación de un órgano sensorial periférico. La detección de un enlentecimiento en la conducción de alguna vía sensorial sugiere lesión desmielinizante, aún en ausencia de clínica. Se valoran los potenciales somatosensoriales, auditivos y visuales, pero su rendi miento, en comparación con el de las pruebas de imagen, es muy bajo, por lo que actualmente se emplean casi exclusivamente los visuales.
R E C U E R D A Las bandas o l i g o c l o n a l e s de IgG en el LCR son un h a l l a z g o presente en var ias en fe rmedades (EM, panence fa l i t i s esc lerosante subaguda , neurolúes, S IDA) .
• Neuroimagen: la RM es la prueba más sensible en la EM (MIR 03-04 , 244) (Figura 50). Permite determinar en un solo estudio la d i seminación espacial (demostrando distintas lesiones) y tempora l (demostrando lesiones agudas [captadoras de contraste] y crónicas), por lo que se ha conver t ido en la prueba complementa r i a más útil. La RM cerebral convenc iona l detecta lesiones en el 9 5 % de los pacientes. La administración de gado l in io permite valorar c o m o recientes las lesiones captantes. Las nuevas técnicas de RM (espectroscopia , RMN de difusión, RMN func iona l ) permi ten detectar el daño axonal y la atrofia cerebral en los pacientes con EM. Hay dist intos criterios radiológicos para aumentar la sensibi l idad y especifi-dad diagnósticas de la R M N , que se basan en el número de lesiones, su localización (más frecuentemente periventr iculares) , su tamaño y morfología, y la captación o no de contraste.
Q R E C U E R D A La R M es la m e j o r p rueba diagnóstica, ya q u e p u e d e ob j e t i v a r la d i se minac ión espac ia l y t e m p o r a l (las les iones agudas cap tan cont ras te ) .
Figura 50. RM cerebral (a) y cervical (b) donde se observan placas de desmielinización afectando a sustancia blanca típico de la EM
No existe en este m o m e n t o t ratamiento con capac idad para curar la enfermedad (MIR 97-98, 42) (Figura 51).
EM REMITENTE-RECURRENTE
Síntoma neurológico agudo
Tratamiento de base
Pseudobrote
Moderado Leve grave (sensitivo)
Brotes frecuentes
Brotes esporádicos
f
Interferón p¡ o glatiramer
acetato Observación
y y Corticoterapia Corticoterapia
i.v. oral
Buena respuesta
Intolerancia o no respuesta
y y Continuar Cambio de
tratamiento tratamiento
Figura 51. Manejo terapéutico de la esclerosis múltiple
Tratamiento sintomático del brote: aunque no hay consenso a propósito de la dosis más adecuada y la me jor vía de admin i s t r a ción, se acepta que los brotes de intens idad suf ic iente c o m o para alterar las act iv idades de la v ida d iar ia requieren la administración de cor t i co ides en alta dosis por vía intravenosa, durante 3-7 días (MIR 05-06, 55) , con o sin reducción progresiva poster ior por vía o ra l . Los brotes de intens idad leve (sintomatología exc lus ivamente sensitiva) pueden tratarse con cor t i co ides por vía ora l , con reduc ción progresiva de la dosis durante un mes. Cuando un pac iente con esclerosis múltiple sufre un deter io ro brusco, hay que valorar si es un nuevo brote, con nueva ac t i v idad de la en fe rmedad , o se trata de un empeo ram ien to t rans i tor io (pseudobrote) deb ido a f i e bre, infección concomi t an te , aumento de temperatura ambienta l o efecto adverso al t r a tamien to ; en este caso, está cont ra ind i cada la administración de cor t i co ides .
RECUERDA Los brotes sens i t ivos leves se t ra tan c o n c o r t i c o i d e s ora les ; el resto de brotes , c o n c o r t i c o i d e s in t ravenosos . Los c o r t i c o i d e s no d i s m i n u y e n el número de brotes de la e n f e r m e d a d .
60

Neurología y neurocirugía
• Tratamiento para modificar el curso de la enfermedad: tratamiento inmunomodu lador . En función de las formas clínicas, estaría indica do en: - Forma remitente-recurrente: en t o d o paciente con act iv idad clí
nica demostrada por haber padec ido al menos dos brotes en los tres últimos años.
- Forma secundar iamente progresiva: en aque l los pac ientes que hayan t e n i d o una fo rma secundar i amente progres iva con brotes en los últimos años, s iempre que todavía puedan deambu la r .
- Forma primariamente progresiva: no hay pruebas de que se o b tenga benef ic io con los i nmunomodu ladores .
- Primer episodio sugerente de esclerosis múltiple: se puede plantear INF p 1a en pacientes con un pr imer ep isod io , si la RM ind ica a l to riesgo de padecer la en fe rmedad (nueve o más lesiones).
Los fármacos existentes son los siguientes (MIR, 08-09, 58 ; MIR 06-07, 59): - Acetato de glat i ramer: es un análogo antigénico de la p r o
teína básica de la m i e l i n a . Su administración subcutánea d i a r i a r educe en un 3 0 % el número de brotes en las fo rmas RR. Sus efectos secundarios son reacciones locales a la inyección ( 9 0 % ) y un cuadro de disnea, palpi tac iones, do lor torácico y en ro jec imiento fac ia l , que dura menos de 30 minutos , en relación con la inyección, que aparece de forma idiosincrásica en el 1 5 % de los pacientes.
- Interferón B (1 a y 1 b): r educen también en un 3 0 % el número de brotes en los pac ientes con EM t i p o RR (MIR 03-04 , 2 4 0 ; MIR 0 0 - 0 1 , 55 ) ; su vía de administración es parente ra l , i n t r amuscu la r ( INF p 1a) o subcutánea (INF p 1 b), y sus efectos secundar ios más frecuentes son los cutáneos (más en el 1b), y un síndrome pseudogr ipa l en relación con la inyección, que responde a los A INE. Entre un 5 y un 2 5 % de los pac ientes t ratados con INF p 1 a y un 4 0 % con INF p 1b desar ro l l an , en el p r imer año de t r a t am ien to , an t i cuerpos neut ra l izantes que restan e f icac ia al fármaco.
- Otros : existen otros fármacos que podrían mod i f i ca r el curso natural de la en fe rmedad , en casos se lecc ionados: > Azatioprina: su administración por vía oral reduce el número
de brotes, pero precisa administrarse durante dos o más años para obtener esos resultados, y son necesarios controles he-matológicos y hepáticos periódicos por sus potenciales efectos secundarios.
> Mi toxantrona: ¡nmunodepresor de l i n f o c i t o s T y B. Su administración es in t r avenosa ; sus efectos secundar ios son a l opec i a , l eucopen ias y una cardiopatía dos i s-depen d i en t e .
Podría ser ef icaz en formas progresivas.
Q R E C U E R D A
" El t e m b l o r f isiológico y e senc i a l se t r a t an c o n B-b loqueantes ; s in e m b a r g o , el t e m b l o r d e r e p o s o d e l P a rk in son se t ra ta c o n ant icol inér g i c o s .
> Metotrexato: también precisa administración durante largo t i empo , aunque con la ventaja de una administración semanal . Compar te los efectos secundar ios de otros inmunosu-presores.
• Tratamiento sintomático de las secuelas: véase la Tabla 30 .
SECUELAS FÁRMACO
Espast ic idad Baclofeno Benzodiacepinas
Fat iga Aman tad ina Pemol lna
Síntomas paroxísticos (dolor , distonías, t e m b l o r )
Carbamazepina Gabapent lna
Disfunción eréctil S i ldenafi lo
H ipe r r e f l ex i a vesical (u rgenc ia m i c c i ona l , i n con t i nenc i a )
Anticolinérgicos (ox ibut ina , to l t e rod ina )
Atonía ves ica l (retención) Colinomiméticos (betanecol )
Depresión Inh ib idores de la recaptación de serotonina
Tabla 30. T ra tamiento sintomático de las secuelas
Esclerosis múltiple y embarazo
Las pacientes embarazadas exper imentan menor número de brotes d u rante la gestación y mayor número en los pr imeros tres meses posparto.
Este empeoramien to se atr ibuye a los niveles altos de pro lact ina que pueden generar una estimulación del sistema inmun i ta r io .
6.2. Otras enfermedades desmielinizantes
• Síndrome de Devic . Se considera una variante de la EM, aunque también puede producirse este síndrome en la sarcoidosis y la t u berculosis. Se asocia a neurit is óptica bi lateral y miel i t is transversa. Los dos síntomas pueden presentarse simultáneamente o separados por un intervalo de pocos días o semanas. La mielitis transversa puede conduc i r a un b loqueo mielográfico comple to , s iendo entonces sombrío el pronóstico de recuperación.
• Enfermedad de Marburg. Es un proceso fu lm inan te que se cons idera una var iedad monofásica aguda de esclerosis múltiple. Cursa con una desmielinización del t ronco encefálico que lleva a la muerte en el curso de meses, sin que responda al t ratamiento. El diagnóstico es postmortem.
• Enfermedad de Balo. Es una enfermedad desmiel in izante monofásica, de mal pronóstico, que se caracteriza a nivel anatomopatológico por áreas concéntricas de desmielinización en la sustancia blanca subcort ical . Su diagnóstico es estr ictamente histológico y por resonancia magnética.
• Enfermedad de Marchiafava-Bignami. Consiste en una degeneración pr imar ia del cuerpo cal loso (zona med ia l , con bordes dorsal y ventral conservados), que se presentó in i c ia lmente con especial f recuencia en varones i tal ianos de mediana edad o ancianos hab i tuados al consumo de a l coho l (vino). También aparece en pacientes desnutr idos y se desconoce si su patogenia es tóxica o metabólica. La alteración anatomopatológica es s imi lar a la encontrada en la tox i c idad por a l coho l metílico, arsénico o c ianuro . La presentación más frecuente es la demenc ia inespecífica. Los síntomas mentales están casi s iempre presentes y t ienen características de estados maníacos, depresivos, paranoides, etc. Las convuls iones son bastante frecuentes y se suelen asociar a afasias, apraxias o he-miparesias.
61

Manual CTO de Medicina y Cirugía, 8. a edición
R E C U E R D A Degeneración de l cuerpo ca l loso en un consumidor de v i no or ienta a en fe rmedad de Marchiaíava-Bignami.
Mielinólisis central pontina. Es una enfermedad desmie l in izante del t ronco cerebral , caracter izada por signos de parálisis pseudobulbar (disartria, disfagia), paraparesia o tetraparesia, conservando el parpadeo y los mov imien tos oculares verticales. Genera lmente aparece 2-6 días después de la corrección rápida de estados de h iponat re mia , pero también se ha descrito asociada a a l coho l i smo crónico y a trasplante hepático. Tiene un pobre pronóstico, y no hay t ra tamien to efect ivo. Encefalomielitis diseminada aguda. Es una enfermedad desmie l i n i zante, de in i c io súbito y curso monofásico, genera lmente asociada a inmunización previa (encefalomiel i t is posvacunal) o antecedente de enfermedad infecciosa exantemática (encefalomiel i t is postinfec-ciosa). Las vacunas más impl icadas eran las de la rabia y la v i ruela , pero su inc idenc ia es cada vez menor . El sarampión es el agente
infeccioso más frecuente, s iendo otras causas var icela, rubéola, Influenza o Mycoplasma. La gravedad clínica es variable, y cursa con fiebre, cefalea, meningis-mo y deterioro progresivo del nivel de consciencia. Las crisis son co munes, así c o m o la clínica motora (hemiparesia, tetraparesia) y cerebelosa. En el LCR, hay pleocitosis l infocitaria y ligera proteinorraquia. C o m o tratamiento, se u t i l i zan los cort icoides en altas dosis por vía intravenosa. La mor ta l idad es del 5-20% , y la mayoría de los pacientes quedan con secuelas neurológicas permanentes.
• Encefalomielitis hemorrágica necrotizante aguda. Es un cuadro clínico, de i n i c io brusco, varios días después de una infección de vías respiratorias altas, con una evolución clínica similar a la de la encefa lomie l i t is aguda d iseminada, pero más explosiva.
Anatomopatológicamente, hay destrucción intensa de la sustancia blanca subcort ica l y es característica la presencia de múltiples h e m o rragias de pequeño tamaño en disposición per ivenular , con intensa reacción inf lamator ia de las meninges.
Casos clínicos representativos
U n h o m b r e de 2 8 a n o s a c u d e a consu l t a , re f i r i endo desde h a c e d i ez días u n c u a d r o de a l te rac ión d e la sens ib i l idad de h e m i c u e r p o q u e inc luye la c a r a . T i e n e c o m o a n t e ceden te s haber p a d e c i d o u n a v is ión bo r rosa por e l o jo i z q u i e r d o h a c e u n a ñ o , que r e cupe ró por c o m p l e t o en u n m e s . En la exp lo rac ión a c t u a l , se ob je t i va u n a h e m i h i poes tes ia i zqu i e rda c o n s igno de Babinsk i de ese l ado . ¿ Q u é p rueba d iagnóst ica es la más a p r o p i a d a p a r a c o n o c e r la et iología más f r ecuen te d e es te p roceso ?
1) T C c e r e b r a l c o n c o n t r a s t e . 2 ) Es tud io r u t i n a r i o d e l LCR. 3) Es tud ios serológicos d e v i r u s . 4 ) R e s o n a n c i a magnét ica c e r e b r a l . 5 ) Po tenc i a l e s e v o c a d o s v i sua les .
M I R 0 3 - 0 4 , 2 4 4 ; RC: 4
1
62

Neurología y neurocirugía
07. EPILEPSIA
Orientación
MIR r
Aspectos esenciales
Es r e c o m e n d a b l e c o n o c e r la clasif icación d e la ep i l eps i a y en t ende r las d i f e renc ias en t re crisis parc ia les (s imples y co m p l e j a s ) y genera l i zadas . H a y a lgunas características m u y p rop i a s d e c u a d r o s f recuentes , c o m o las ausenc ias , q u e se han p r e g u n t a d o c o n f r e c u e n c i a . H a y q u e tener presente la etiología más f r e cuen t e de las cr is is , según los g rupos de e d a d y las i n d i c a c i o n e s y efectos secundar ios de los p r i n c i p a l e s a n t i c o n v u l s i v o s .
Q~J El EEC pe rm i t e d i fe renc ia r ent re crisis parc ia les y genera l izadas : las pr imeras son aquéllas q u e sólo ac t i van una región concre ta del córtex, mientras que en las últimas se p r o d u c e una ac t i v i dad eléctrica simultánea en ambos hemisfer ios , activándose t o d o el córtex.
r j " ) Las crisis parc ia les s imples no a l teran la conc i enc i a , y las crisis parc ia les comp le j a s sí.
f j j Las crisis parc ia les s imples pueden p r o d u c i r síntomas motores , sensit ivos, autónomos, sensoriales o psíquicos, d e p e n d i e n d o de l área co r t i ca l afecta.
[~4~] Las crisis parc ia les comp le j a s p r o d u c e n alteración de l n ive l de consc i enc i a y au tomat i smos c o n p e r i o d o de confusión tras las cr is is.
j~5~] Las crisis generalizadas más importantes son las ausencias, o pequeño ma l , y las crisis tónico-clónicas, o gran ma l .
fJTJ Es típico de las ausencias los ep isod ios bruscos y repet i t ivos de desconexión de l m e d i o c o n descargas gene ra l izadas y simétricas de punta-onda a 3 H z en el EEG. Aparecen en niños, resolviéndose a m e n u d o en la ado lescenc ia , y se con t ro l an adecuadamen te c o n fármacos.
["7"] Las crisis tónico-clónicas son el t i p o de crisis más frecuentes en los trastornos metabólicos.
f~3~| La causa más f recuente de convu l s iones según edad es: neonatos , encefalopatía hipóxica; lactantes y niños, crisis febr i les ; adolescentes y adu l tos jóvenes, t r aumat i smos ; y en mayores , la en f e rmedad cerebrovascu lar . Los tumores serían la causa más f recuente en adu l tos de edad med ia .
fg~j El síndrome de Wes t aparece en el p r ime r año de v ida , y se caracter iza por la tríada: espasmos infant i les , alteración del desar ro l lo p s i c o m o t o r e h ipsa r r i tm ia .
El síndrome de Lennox-Castaut es la evolución que presentan los pac ientes c o n Wes t a los 2-4 años.
El ácido v a l p r o i c o es el an t i convu l s i vo más u t i l i z a d o por su a m p l i o espectro. Produce a lopec i a y hepato-t o x i c i d a d .
La c a rbamazep ina es de elección en las crisis parc ia les , pe ro no debe admin is t rarse en las ausencias. Produce hepa to tox i c i dad , anem ia aplásica y síndrome de Stevens-Johnson.
La A C T H es de elección en el t r a t am ien to de l síndrome de West .
ou tm
Una convulsión o crisis epiléptica es un fenómeno paroxístico or ig inado por una act iv idad anorma l , excesiva y
sincrónica de un grupo de neuronas del SNC y que puede cursar clínicamente de distintas formas.
Epilepsia es la existencia de crisis epilépticas recurrentes debidas a un proceso crónico subyacente. La existencia de una
convulsión aislada, o de crisis recurrentes debidas a factores corregibles o evitables no es necesariamente una epilepsia.
Un síndrome epiléptico es una epilepsia con un conjunto de síntomas y signos que habitualmente se presentan j u n
tos, sugiriendo un mecanismo subyacente común.
Se habla de estatus epiléptico cuando una crisis dura más de 30 minutos o cuando existen crisis repetidas, entre
las cuales el paciente no recupera la consciencia.
7.1. Clasificación o a b i a 3 i )
Las convuls iones parciales (focales) son aquéllas en las que la act iv idad eléctrica queda c i rcunscr i ta a un área
concreta de la corteza cerebral , con independenc ia de que durante la crisis la consc ienc ia esté conservada (par-
- M I R 09-10, 68, 163 - M I R 07-08, 55, 154 - M I R 06-07, 53 - M I R 05-06, 61 - M I R 04-05, 61 - M I R 03-04, 248 - M I R 00-01, 54 - M I R 99-00,215 - M I R 99-00F, 71 - M I R 98-99F, 67, 69, 7.
63

Manual CTO de Medicina y Cirugía, 8 . a edición
cíales simples) o alterada (parciales complejas) . La sintomatología con la que cursa la crisis dependerá del área cort ical donde se sitúan las neu ronas causantes de la misma. Las crisis parciales complejas se or ig inan en el lóbulo tempora l en un 6 0 % de los casos, y en el frontal en el 3 0 % .
frecuencia del trazado anómalo.
CRISIS PARCIALES
• Simples (con síntomas motores, sensitivos, autónomos o psíquicos)
• Complejas • Con generalización secundaria
CRISIS GENERAL IZADAS
• Ausencias • Tónico-clónicas • Tónicas • Atónicas • Mioclónicas
CRISIS NO CLAS IF ICADAS • Convulsiones neonatales • Espasmos infantiles
Tabla 31. Clasificación de las crisis epilépticas (Liga Internacional de la Epilepsia, 1981)
Las crisis parciales simples pueden produc i r síntomas motores, sensitivos, autónomos (sudoración, piloerección), visuales (destellos simples o a lucinaciones complejas) , audit ivos (sonidos simples o elaborados), olfat ivos (olores intensos y poco habituales) o psíquicos (miedo, despersonalización, deja vu). Las crisis motoras pueden comenzar en un área muy pequeña y extenderse gradualmente (en segundos o minutos) a un área hemicorpora l más extensa (progresión jacksoniana). En ocasiones, tras una crisis motora , puede persistir una deb i l idad del área afectada (parálisis de Todd) , auto l imi tada en minutos u horas. Durante las crisis parciales complejas, el paciente t iene d i f i cu l tad para mantener un contacto normal con el med io , jun to con alteración del compor tamiento que puede ir desde la i nmov i l i dad o au tomatismos básicos (chupeteo, deglución), hasta comportamientos más elaborados; tras la crisis, existe característicamente un per iodo de confusión (MIR 06-07, 53 ; MIR 05-06, 6 1 ; MIR 98-99F, 69).
R E C U E R D A Las crisis parciales complejas son más frecuentes en adultos, suelen experimentar sensaciones extrañas y su origen es el lóbulo temporal, en la mayoría de las ocasiones.
Las crisis generalizadas se or ig inan simultáneamente en ambos hemisferios, aunque es difícil descartar por comp le to la existencia de una act iv idad focal in ic ia l que se propague con rapidez y que, en ocasiones, es reconoc ib le por la existencia de síntomas focales previos a la pérdida de la consciencia (aura). Las ausencias (pequeño mal) se compor tan c o m o breves episodios de pérdida brusca del nivel de consciencia, sin alteración del c o n trol postural ; característicamente, duran segundos y pueden repetirse muchas veces al día, suelen acompañarse de pequeños signos moto res bilaterales (parpadeo, masticación) y se recupera la consciencia de forma igualmente brusca, sin confusión posterior ni memor ia del episodio (MIR 09-10, 163; MIR 03-04, 248). La edad de comienzo suele estar entre los 4 años y el in i c io de la adolescencia, siendo la causa más frecuente de crisis en este rango de edad; no se acompañan de otros problemas neurológicos, responden de forma favorable al t ratamiento farmacológico, y entre un 60 y un 7 0 % de los casos remiten durante la adolescencia. Los hallazgos en el EEG son típicamente descargas generalizadas y simétricas de punta-onda a 3 Hz (MIR 99-OOF, 67) co inc id i endo con las crisis, aunque en el EEG interictal existen más periodos de act iv idad anormal que los visibles clínicamente. La hiperventilación incrementa la
R E C U E R D A La pérdida de consc ienc ia y los automatismos pueden aparecer en las crisis parciales complejas y en las ausencias. Cl ínicamente se diferenc ian en la presencia de periodo confusional tras la crisis en las parciales complejas, y no en las ausencias.
Existen las denominadas ausencias atípicas, con pérdida de consc iencia de mayor duración, con in ic io y f in menos brusco y generalmente signos focales. El EEG muestra trazados de punta-onda a frecuencias menores de 3 Hz , y suelen responder peor al t ratamiento que las ausencias típicas (Tabla 32).
R E C U E R D A Las ausencias atípicas muestran punta-onda a frecuencias menores de 3 H z y tienen un peor pronóstico porque no responden a la medicación y suelen ser expresión de una lesión estructural.
CRISIS PARCIAL COMPLE JA
VS AUSENC IA T ÍP ICA
Genera l izada Parcial
Sin aura Puede tener aura
< M a ñ o s Adultos
Segundos Minutos
Varias al día, incluso varias al minuto
Variable
Automat ismos escasos Automatismos frecuentes
No per iodo poscrítico Periodo poscrítico
VALPROATO ETOSUXAMIDA
CARBAMAZEPINA
Tabla 32. Crisis parcial compleja (CPC) versus ausencia típica
Las convulsiones tónico-clónicas (gran mal) suelen tener un co m ienzo brusco, sin aviso prev io , aunque algunos pacientes refieren síntomas poco def in idos en las horas previas, que no deben ser confund idos con auras causadas por un or igen focal de la crisis. La fase in ic ia l es una contracción tónica general izada, acompañada de cianosis, aumento de f recuencia cardíaca y de la presión arterial , y midriasis. En 10-20 s generalmente comienza la fase clónica, de duración var iable. En el poscrítico, existe ausencia de respuesta a estímulos externos, f lac idez muscular e hipersalivación que pue den compromete r la vía aérea, seguido de una fase de lenta recuperación del nivel de consc iencia (minutos-horas) acompañada de confusión. El paciente refiere cansancio, cefalea y mialgias durante varias horas tras la crisis. El EEG muestra distintos trazados a lo largo de la crisis: existe una act iv idad rápida de bajo voltaje, con descargas generalizadas y po-lipuntas de alto voltaje en la fase tónica; en la fase clónica aparece una punta-onda a baja frecuencia; y en el poscrítico, hay un enlente-c im ien to global que va resolviéndose jun to con la recuperación del nivel de consciencia. Son el t ipo de crisis más frecuentes en el contexto de trastornos metabólicos. Existen convulsiones tónicas puras y clónicas puras; son similares a las anteriores, con ausencia de alguna de las fases.
6 4

• Crisis atónicas: se caracter izan por la repentina pérdida del t ono muscular de escasos segundos de duración, con breve alteración del nivel de consc iencia , sin confusión posterior. Suelen presentarse en el contexto de síndromes epilépticos conoc idos .
• Las mioclonías son contracc iones breves de los músculos, que pue den estar or iginadas en distintos niveles (cort ical , subcort ica l , me dular) . Cuando existe or igen cor t i ca l , son consideradas fenómenos epilépticos, mostrando el EEG descargas de punta-onda bilaterales y sincrónicas. Suelen coexist ir con otros t ipos de crisis, aunque son la pr inc ipa l manifestación de algunos síndromes epilépticos.
7.2. Fisiopatología
Las crisis son la consecuencia del desequi l ibr io entre los mecanismos excitator ios e inh ib i tor ios del SNC. El mecan ismo básico de p roduc ción de las crisis, aunque no bien conoc idos , sería el siguiente: • Existe in i c ia lmente una act iv idad de descarga generada por la entra
da de Ca 2 + y Na + al inter ior de la neurona, causando una despolar i zación pro longada de la membrana . Esto generaría una punta en el EEG. En cond ic iones normales, esta act iv idad es frenada mediante una hiperpolarización mediada por los receptores GABA y los canales de K+.
• Las descargas repetidas or ig inan un aumento del K+ extracelular, del Ca 2 + extracelular y de la activación mediada por los receptores N M D A , con lo que se evita que tenga lugar la hiperpolarización norma l .
Existen muchos mecanismos que pueden alterar la tendencia de las neuronas a realizar descargas paroxísticas; en ocasiones se produce una transformación de toda un área neuronal que se convier te en hipe-rexcitable de forma crónica, convirtiéndose en un foco epiléptico: este proceso se conoce c o m o epileptogénesis.
7.3. Diagnóstico diferencial
• El pr imer paso es diferenciar las crisis de otros síntomas transitorios. El síncope y las pseudocrisis son las entidades más frecuentemente confundidas con epi lepsia. En la Tabla 33 aparecen los puntos clave para di ferenciar los de las crisis. Otros diagnósticos diferenciales son accidentes isquémicos transitorios, migraña, narcolepsia e h ipoglu-cemia (MIR 09-10, 68).
Q R E C U E R D A U n o de los diagnósticos diferenciales más difíciles son las crisis neuróticas o conversivas, que suelen estar desencadenadas por una emoción intensa.
El electroencefalograma (Figura 52) sigue siendo el método comp l e mentar io de elección para demostrar el carácter epiléptico de una crisis, y es esencial para def in i r algunos síndromes epilépticos. Sin embargo, no es un test que permita diagnosticar o exc lu i r epi lepsia por sí m ismo. U n EEG convenc iona l suele mostrar alteraciones epi-leptiformes en un 5 0 % de pacientes epilépticos, pero es importante conocer que entre un 1 0 - 1 5 % de la población normal puede tener un EEG patológico. De esta fo rma, un EEG anormal en ausencia de
Neurología y neurocirugía
síntomas nunca debe ser tratado con ant icomic ia les . Es infrecuente poder registrar la act iv idad EEG crítica, por lo que la información del EEG se realiza por las alteraciones intercríticas presentes.
CARACTERÍST ICAS CRISIS EP ILÉTICA S INCOPE
Factores desencadentes inmediatos
Habitualmente no Estrés, maniobra de Valsalva bipedestación
Síntomas previos No o aura Sudoración, náuseas...
Postura al inicio Indiferente + frecuente en bipedestación
Paso a inconsciencia Brusco + Frecuente progresivo
Duración de inconsciencia
Minutos + Frecuente segundos
Duración de movimientos tónico-clónicos
30-60 segundos Menos de 15 segundos
Aspecto facial Espuma por boca y cianosis
Palidez
Bajo nivel de consciencia posterior
Minutos hasta horas Pocos minutos
Dolor muscular posterior Frecuente Infrecuente
Mordedura de lengua Algunas veces Infrecuente
Incontinencia Algunas veces Infrecuente
Tabla 33. Diagnóstico diferencial de la epilepsia
Punta y onda (focales) Punta y onda (generalizadas)
Puntas (generalizadas/focales) Gran mal (generalizadas)
Paroxísticas (generalizadas/focales)
FORMAS INTERMITENTES
8 (< 3/s), generalizada o focal
0 (4-7/s)
a (8-13/s) Normal: 10/s (occipital)
B(15-30/s)
Figura 52. Clasificación de Gray-Walter del EEG

Manual CTO de Medicina y Cirugía, 8 . a edición
Estudios neurorradiológicos. La TC y la RMN son las técnicas de elección, s iendo la RMN más sensible para detectar alteraciones estructurales del sistema nervioso centra l .
7.4. Etiología
En la Tabla 34 se inc luyen las causas más frecuentes de crisis epilépticas, según la edad de aparición. En numerosas ocasiones, la etiología es desconoc ida (idiopática), aunque existen algunas causas de ep i l ep sia descritas a continuación:
EDAD ETIOLOGÍA
Neona tos (< 1 mes)
• Hipoxia per inata l • Hemorrag ia intracraneal • Infecciones de l SNC • Trastornos metabólicos • Abst inenc ia de tóxicos • Alteraciones genéticas • Alteraciones del desarrol lo
Lac tantes y niños (1 mes - 1 2 años)
• Crisis febri les • Alteraciones genéticas • Infecciones de l SNC • Alteraciones del desarrol lo • Traumat ismos - Idiopáticas
Ado lescen tes ( 1 2 - 1 8 años)
• Traumat ismos • Idiopáticas • Alteraciones genéticas • Tumores • C o n s u m o de tóxicos
A d u l t o s jóvenes ( 1 8 - 3 5 años)
• Traumat ismos • Abst inenc ia de a lcoho l • C o n s u m o de tóxicos • Tumores (MIR 98-99F, 67) • Idiopáticas
(> 35 años)
• E. cerebrovascular • Tumores • Abst inenc ia de a lcoho l • Trastornos metabólicos • Enfermedades degenerat ivas del SNC • Idiopáticas
Tabla 34. Etiología de las crisis epilépticas según la edad de inicio
Genética: cada vez se están ident i f i cando más genes causantes de epi lepsia; en varios casos, la alteración pr imar ia es un mal f unc io namiento de algún canal iónico (canalopatías).
R E C U E R D A Las cris is febr i les s imples son genera l izadas , du r an menos de 15 m i n u tos y no t i enen un mayo r r iesgo de presentar ep i leps ia en el f u t u r o .
Fiebre: las crisis febriles son un proceso típico de la edad infant i l (entre los tres meses y los c inco años), que se relaciona más f recuentemente con el aumento de la temperatura, lo que da lugar a una crisis el pr imer día de un proceso febr i l , independientemente del or igen del mismo. Las crisis febriles simples son general izadas, d u ran menos de 15 minutos , presentan buena recuperación posterior y los hallazgos en el per iodo intercrítico son normales o negativos; con frecuencia existen antecedentes famil iares de crisis febriles o de epi lepsia; son recurrentes en un terc io de los casos (aunque sólo el 1 0 % de los pacientes sufre más de dos episodios), de fo rma más probable si la crisis se p roduce en el pr imer año de v ida; no se rela
c ionan con un mayor riesgo de presentar epi lepsia. Las crisis febriles comple jas son las que t ienen signos focales, una duración superior a 15 minutos , o se repiten en el curso del m ismo episodio febr i l ; se re lac ionan con un 2 - 5 % de incremento del riesgo de sufrir epi lepsia con poster ior idad. Las crisis febriles pueden tratarse con d iazepam vía rectal o intravenosa, aunque dado que ceden espontáneamente, el mane jo más adecuado es el contro l de la temperatura, prefer ib lemente con pa-racetamol (MIR 99-00, 215) . En pacientes con crisis febriles típicas recurrentes, puede administrarse d iazepam oral o rectal en s i tuac iones de ascenso térmico. N o está ind i cado el t ratamiento con t inuado con ant icomic ia les c o m o prof i laxis de crisis febriles (MIR 07-08, 154).
• Traumatismo craneoencefálico (TCE): la p robab i l i dad de presentar epi lepsia tras un TCE está re lac ionada con la intensidad del mismo; las heridas abiertas, las fracturas con hund im ien to o con hemorra gia asociada t ienen entre un 40 y un 5 0 % de probabi l idades de padecer epi lepsia, mientras que en los TCE leves, el riesgo es de un 5 a un 2 5 % . Las crisis que aparecen en la pr imera hora tras el TCE (inmediatas) no suelen conl levar riesgo de epi lepsia a largo p lazo y suelen ser crisis general izadas. Las crisis precoces (entre la pr imera hora y el séptimo día tras el TCE) suelen ser más frecuentes en los niños, se asocian a lesiones traumáticas signif icativas y, a di ferencia de las inmediatas, con l levan riesgo de epi lepsia tardía y suelen ser crisis parciales; la utilización de medicación antiepiléptica se ha demostrado útil c o m o prevención pr imar ia de estas crisis (MIR 99-00F, 71). Por último, las crisis tardías (aquéllas que aparecen tras la pr imera semana) son más frecuentes en los adultos, y se trata de c r i sis parciales con tendencia a la generalización; no se recomienda la utilización de medicación antiepiléptica para prevenir la aparición de estas crisis.
Q R E C U E R D A Las crisis q u e aparecen en la p r ime ra hora post-TCE (crisis inmediatas) no c o n l l e v a n mayo r r iesgo de ep i leps ia .
Patología cerebrovascular: es la responsable del 5 0 % de los casos nuevos de epi lepsia en los mayores de 65 años. Las convuls iones en la fase aguda son menos frecuentes y acompañan generalmente a la patología embólica, mientras que las crisis más frecuentes co mienzan meses o años tras el proceso agudo, y se re lac ionan con cua lqu ier t ipo de patología cerebrovascular.
R E C U E R D A En genera l , las crisis parc ia les suelen ind i ca r una lesión estructura l de l parénquima nerv ioso , s i endo por e l l o más rec id ivantes y difíciles de con t ro l a r . Las a l terac iones metabólicas sue len manifestarse c o m o crisis genera l izadas .
7.5. Algunos síndromes epilépticos específicos
La ILAE (Liga Internacional Contra la Epilepsia) clasif ica varias decenas de síndromes epilépticos, de los que sólo se mencionarán aquí algunos de forma puntua l : • Epilepsias parciales benignas de la infancia:
- Epilepsia rolándica benigna.
66

Neurología y neurocirugía
Epilepsias de la infancia con mala respuesta al tratamiento: - Síndrome de West. - Síndrome de Lennox-Gastaut. Epilepsias generalizadas del adulto: - Epilepsia mloclónica j u ven i l .
Múltiples tipos de convulsiones, especia lmente tónicas (las más frecuentes y características), atónicas y ausencias atípicas. Afectación psicomotriz, con afectación ¡nvolutiva del desarrol lo o trastornos conductuales . No aparece en todos los casos, pero sí en la mayoría.
Epilepsias parciales benignas de la infancia
Se caracter izan por ser dependientes de la edad, con com ienzo después de los 18 meses, con crisis generalmente poco frecuentes, sin deter ioro neurológico asociado y con norma l idad en las pruebas c o m plementar ias, salvo el EEG, que presenta una act iv idad de fondo normal con comple jos focales, de localización más frecuentemente en la región tempora l media . Más de la mi tad de los pacientes con epi lepsia parcial benigna de la infancia t ienen una epi lepsia rolándica.
R E C U E R D A La misma lesión cerebral a distintas edades se manifiesta de distinta forma (en < 1 año con síndrome de West y en > 1 año con síndrome de Lennox-Gastaut), debido a los cambios madurativos que sufre el sistema nervioso central.
Alteraciones en el EEG : en v ig i l i a aparecen comple jos de punta-onda lentos, sobre un r i tmo de fondo lento; durante el sueño aparecen salvas de puntas. El registro crítico depende del t i po de crisis.
La epi lepsia rolándica debuta sobre todo entre los siete y los d iez años, remi t i endo en el 9 8 % de los casos en to rno a los 14 años. El 8 0 % de las crisis aparecen durante el sueño y suelen presentar foca l idad fac ia l . No suele requerir t ratamiento, dada su evolución espontánea.
Epilepsias de la infancia con mala respuesta al tratamiento
Existe un grupo de epilepsias que se caracter izan por impl i car alteraciones neurológicas jun to a crisis con mal contro l terapéutico.
En muchas ocasiones, el patrón clínico depende de la fase madurat iva cerebral , con lo que es frecuente que el mismo paciente pase por dis t intos síndromes según su edad. En el per iodo neonatal , generalmente aparece una encefalopatía mioclónica neonata l ; en la lactancia y p r i mera infancia, la epi lepsia mioclónica grave de la infancia y el síndrome de West, y en la segunda infancia, el síndrome de Lennox-Gastaut.
El síndrome de West aparece en el pr imer año de v ida , más f recuentemente entre el 4.° y 7.° mes, y p redomina en varones (1,5:1). Puede ser de or igen criptogénico o sintomático (más frecuente); de hecho, cua lqu ier daño cerebral importante que pueda generar epi lepsia a esta edad, probab lemente lo hará en la forma de este síndrome, con lo que el l istado de posibles causas es muy extenso. La tríada que def ine el síndrome consta de: • Espasmos infantiles: contracciones musculares breves (1 -3 s), en salvas,
más frecuentemente generalizadas, y de predominio en musculatura flexora, que aparecen al despertar, no persistiendo durante el sueño. Detención del desarrollo psicomotor: puede producirse un estancamiento o una regresión de lo adqu i r ido , depend iendo de la etiología subyacente; suele ser menos importante en los criptogénicos.
• Hipsarritmia intercrítica: es un cr i ter io impresc ind ib le para el d iag nóstico del síndrome de West. Es una act iv idad basal desorganizada, con ondas lentas de alto voltaje, intercalándose ondas agudas. Durante las crisis aparecen distintos t ipos de ondas, seguidos de una atenuación del vol ta je .
R E C U E R D A La hipsarritmia es interictal, es decir, entre las crisis, y es criterio imprescindible para el diagnóstico de West.
Epilepsias generalizadas del adulto
La epi lepsia mioclónica j uven i l es el p ro to t ipo de epi lepsia genera l iza da idiopática. Supone el 1 0 % de todas las epi lepsias, y es la epi lepsia mioclónica más frecuente. La edad de i n i c i o es entre los 8 y 25 años. La mayoría de los pacientes presentan dist intos t ipos de crisis, además de las mioclónicas: un 9 0 % padece crisis tónico-clónicas y, el 3 0 % , ausencias típicas. Las crisis se presentan c o m o sacudidas musculares breves, hab i tua lmente en m iembros superiores, característicamente al despertar, y favorecidas por la privación previa de sueño y el consumo de a l coho l . Se mant iene el n ive l de consc ienc ia , excepto en las crisis graves.
R E C U E R D A La presencia de mioclonías y otros tipos de crisis en un adolescente después de irse de fiesta (consumo de alcohol, privación del sueño) es típico de la EMJ.
El EEG muestra ac t i v idad paroxística punta-onda en la mayoría de los casos, y s iempre es patológico durante el sueño. Un terc io de los pacientes presenta act iv idad paroxística fotosensible, aunque no es f re cuente que tengan un corre lato clínico.
7.6. Tratamiento. Fármacos anticomiciales
El síndrome de Lennox-Gastaut t iene una edad de in i c io var iable entre uno y siete años, con p ico máximo entre los 2-4 años. Las posibi l idades etiológicas son superponibles a las del síndrome de West. Se caracteriza por la tríada de:
Inicio de tratamiento
La indicación de comenzar un t ratamiento epiléptico tras el diagnóst i co de epi lepsia (Figura 53) v iene marcada por el riesgo de presentar nuevas crisis, que a su vez depende de la etiología (mayor riesgo si existe lesión estructural) , la edad (mayor en menores de 16 años y ma yores de 60), el t ipo de crisis (recurren más las parciales que las generalizadas) y el EEG (mayor riesgo si aparecen descargas punta-onda en el p r imero real izado).
Una vez establecida la conven ienc ia de inic iar t ratamiento ant i comi-c ia l , debe iniciarse una escalada de dosis lenta, con cont ro l de los n ive les séricos del fármaco, si es pos ib le , hasta alcanzar las dosis máximas
67

Manual C T O de Medicina y Cirugía, 8.a edición
toleradas sin efectos secundarios. Si un pr imer fármaco no contro la las crisis, es preciso ut i l izar un segundo an t i comic i a l , generalmente m a n ten iendo el t ra tamiento prev io hasta que se a lcanzan niveles terapéuticos, hecho lo cual se retira el p r imero de forma progresiva.
Ap rox imadamente un terc io de los pacientes no se cont ro lan con mo-noterapia y precisan la combinación de diversos fármacos. No existe en la actua l idad una guía terapéutica única para el uso de pol i terapia , pero generalmente se comb inan dos fármacos de pr imera línea a los que se puede añadir un tercero de forma coadyuvante .
Fármacos antiepilépticos según el tipo de crisis
N o ex is ten reglas absolutas sobre cuál es el me jo r t r a t a m i e n t o , d a d o que no es pos ib l e d e t e r m i n a r s i empre cuál será el más e fec t i v o para cada pac i en te . N o obs tan te , pueden establecerse unas p r i o r i dades ent re los fármacos, d e p e n d i e n d o del t i p o de cr is is (Tablas 35 y 36) (MIR 07-08 , 55 ) .
Tratamiento en monoterapia
Buen control 7 0 % Control insatisfactori
Buen control 1 5 % Control insatisfactorio 1 5 %
Totalmente refractarias al Cirugía de la tratamiento 1 0 % epilepsia 5%
Figura 53. Control de las crisis de reciente comienzo
En un 7 0 % de los niños y un 6 0 % de los adultos epilépticos con buen contro l terapéutico puede suspenderse el t ra tamiento tras una t empora da sin crisis. No hay unos criterios comunes, pero se consideran signos favorables para permanecer sin crisis tras la retirada de medicación la presencia de un EEG norma l , una exploración neurológica sin alteraciones, un único t i po de crisis y un per iodo de uno a c inco años sin crisis. La mayor parte de las recidivas sucede en los tres pr imeros meses tras la retirada del t ratamiento.
Mecanismo de acción de los fármacos anticomiciales
A u n q u e la mayor ía posee m e c a n i s m o s de acc ión múlt ip les, e x i s t en u n o s m e c a n i s m o s básicos c o m p a r t i d o s p o r d i s t i n t o s fármacos : • Inhibición de los canales de Na + : fenitoína, carbamazepina , topira-
mato. • Inhibición de los canales de C a 2 + : fenitoína, va lproato e tosux imida . • Disminución de liberación de glutamato: lamotr ig ina. • Potenciación de la función de los receptores GABA: benzodiacepi-
nas, barbitúricos. • Aumento de la disponibilidad del GABA: gabapent ina, t iagabina,
v igabatr ina.
TÓNICO-CLÓNICAS GENERAL IZADAS
I nd i s t in tamente : fenitoína, carbamazepina, f enobarb i ta l o va lproato
PARCIALES 1.° Carbamazepina 2 ° Fenitoína
MIOCLÓNICAS 1. ° C lonazepam 2. ° Va lproato
AUSENCIAS 1. ° E tosuximida (típicas) 2. » Va lproato (atípicas) (MIR 00-01,54)
STATUS EP ILÉPT ICO
1 D i a z e p a m i.v., fenitoína i.v, f enobarb i ta l i.v. 2.° Anestesia con p ropo fo l y m idazo l am
Tabla 35. Tratamiento de las epilepsias
FÁRMACO E F E C T O S SECUNDARIOS
Difenilhidantoina Hlrsut ismo, hiperplasia g ing iva l
Carbamazep ina Hepáticos y hematológlcos
Topiramato Somnolenc ia . Litiasis renal
Etosuximida
Síndrome park inson iano y hematológicos Fenobarbital Sedación. Niños: h iperac t i v ldad
Benzodiazepinas Ver en Psiquiatría
Ac. valproico Hepáticos, hematológicos, pancreat i t is , a lopecia
Lamotr ig ina Exantema
Vigabatr ina Trastorno del c o m p o r t a m i e n t o y del c a m p o visual
Gabapent ina Intolerancia gastro intest ina l
Felbamato Hematológicos
Tabla 36. Efectos adversos de los principales antiepilépticos
RECUERDA La c a r b a m a z e p i n a está i nd i c ada en las ausencias.
• Crisis parciales simples o complejas: carbamazepina, fenitoína, va lproato, l amo tr ig ina.
• Crisis tónico-clónicas generalizadas: va lproato, fenitoína, carbamazepina.
• Ausencias: e tosux imida , va lproato. • Ausencias atípicas, crisis tónicas, crisis clónicas, crisis mioclónicas:
va lproato (MIR 0 0 - 0 1 , 54). • Síndrome de West: ACTH , cort icoides, c lonazepam, va lproato, v i
gabatr ina. • Estatus epiléptico: la pr imera opción es perfusión de d iazepam i.v. a
2 mg/min j un to con fenitoína i.v. 20 mg/kg; si esto no es suf ic iente, añadir fenobarbi ta l 20 mg/kg i.v.; y si esto fal la, anestesia con m ida zo lam o p ropo fo l .
Existe un fármaco antiepiléptico de reciente uso que es el levetirace-tam, extensamente usado en la clínica diar ia por ser útil tanto en las
6 8

Neurología y neurocirugía
crisis parciales c o m o general izadas, por su perf i l farmacodinámico con menos interacc iones y efectos adversos. Además también está d i spon ib l e para vía intravenosa.
segu imiento . C o m o comp l i cac iones más frecuentes en las lobecto-mías tempora les dominantes se han descr i to disfasia ( 6 % ) y trastornos de la m e m o r i a ( 2 % ) .
Tratamiento quirúrgico de la epilepsia 7.7. Epilepsia y embarazo
La intervención quirúrgica se planteará en aquel los pacientes con c r i sis mal contro ladas , a pesar de una correcta medicación durante al menos un año. Estas crisis deben tener la suf ic iente estab i l idad a lo largo del t i empo , en cuanto a su semiología y su f recuenc ia . Pr imero hay que hacer un diagnóstico de localización del f oco epileptógeno. Para e l l o se real izan pruebas de imagen (la resonancia magnética es de elección, aunque también se u t i l i zan TC, PET, SPECT y magneto-encefalografía) y electrocorticografía (con electrodos superf ic ia les y e lectrodos esfenoidales). En ocasiones, para detectar el foco , es necesario recurr i r a e lectrodos invasivos, subdurales, o inc luso e lectrodos pro fundos co locados mediante guía estereotáctica.
Se han u t i l i zado diversas técnicas quirúrgicas: • Epilepsia les ional .
- Epilepsia del lóbulo t empora l : resección estándar del lóbulo t empora l .
- Epilepsia ex t ra tempora l : resección de la lesión.
• Cirugía de desconexión: transecciones subpiales múltiples (ep i lepsias parciales c u y o foco se loca l iza en áreas elocuentes) y cal lo-sotomías totales o subtotales (múltiples focos irresecables, crisis tónico-clónicas secundar iamente general izadas, convu ls iones tónicas o atónicas, con caídas). Estimulación del nerv io vago: convu ls iones parciales intratables. Estimulación cerebral p ro funda , en diversas partes del cerebro . Hemisferectomía o hemisferotomía: síndromes panhemisféricos con convu ls iones intratables.
En el 5 0 % de los casos se cons igue erradicar las crisis, aunque debe mantenerse la fa rmacoterap ia ; en el 8 0 % de los casos se ob t i ene una reducción mayor del 5 0 % en la f recuenc ia de crisis a los dos años de
Durante el embarazo de las gestantes epilépticas, se mant iene la f re cuenc ia de las crisis en el 5 0 % , con mejoría en el 2 0 % y un e m p e o ramiento en el 3 0 % , que no es predec ib le , ya que no depende del t i p o de epi lepsia , ni del número de crisis en los últimos meses, ni del c o m p o r t a m i e n t o durante embarazos previos.
Durante el embarazo se ob je t i va un descenso en las concent rac iones plasmáticas de los dist intos an t i comic ia les , que no se t raduce en un mayor número de crisis. D a d o que no hay un fármaco de elección durante el embarazo , se debe mantener el t ra tamiento p rev io (MIR 98-99F, 75) a la dosis mínima ef icaz.
Q RECUERDA Ante una embarazada controlada con un fármaco anticonvulsivo, no se deben realizar modificaciones del tratamiento, ya que una crisis convulsiva puede ser fatal para el feto.
Se debe recomendar a la pac iente epiléptica que programe el emba razo en una época de buen con t ro l de las crisis, pre ferentemente en monote rap ia y en la mínima dosis pos ib le . Si el t ra tamiento es con va lproa to o ca rbamazep ina , hay que comenza r un t ra tamiento con ácido fólico desde que se abandonen los métodos ant i concept i vos , para preven i r los defectos de c ierre del t u b o neural (espina bífida). En gestantes con buen cont ro l con monote rap i a , el riesgo de aparición de defectos congénitos es del 5-6%, mientras que es del 2 - 3 % en mujeres sanas. En el 5 0 % de los recién nac idos se p roduce un déficit t rans i tor io y reversible de los factores de coagulación v i t am ina I nde pendientes, por lo que debe hacerse prof i l ax is en las últimas semanas del embarazo y en el recién nac ido .
Casos clínicos representativos
Un episodio caracterizado por sensación epigástrica que asciende hacia el tórax, seguido por dificultad para conectar con el entorno, movimientos de masticación, distonía de una mano y falta de respuesta, de un minuto de duración, con amnesia postcrítica, es una crisis:
1) Parc ia l s i m p l e . 2) Parc ia l s e c u n d a r i a m e n t e g e n e r a l i z a d a . 3) Parc ia l c o m p l e j a . 4) A u s e n c i a típica. 5) A u s e n c i a atípica.
M I R 0 5 - 0 6 , 6 1 ; RC: 3
Un paciente de 40 años, sin antecedentes relevantes, es traído a Urgencias por haber presentado desviación de la cabeza hacia la izquierda, convulsiones que se iniciaron en miembros izquierdos y se generalizaron enseguida a los cuatro miembros, con pérdida de consciencia, incontinencia vesical y estado confusional de una media hora de duración. Independientemente de los hallazgos de la exploración clínica y la analítica clínica de rutina, debería realizarse con premura como primera medida:
1) T C c e r e b r a l .
2 ) Determinac ión d e a l c o h o l e m i a . 3) Determinac ión d e opiáceos en sangre y o r i n a . 4) E l e c t roence f a l og rama . 5) Punc ión l u m b a r .
M I R 0 4 - 0 5 , 6 1 ; RC: 1
N iño d e 3 años q u e c o m i e n z a c o n síntomas catarra les y, unas horas después, presenta u n e p i s o d i o d e pérdida d e c o n o c i m i e n t o , m o v i m i e n t o s tonicoc lónicos d e e x t r e m i d a d e s y revulsión o c u l a r , d e una durac ión a p r o x i m a d a d e dos m i n u t o s . En la explorac ión, presenta T 3 9 ° C , exploración neurológica n o r m a l , e x c e p t o t e n d e n c i a al sueño, f a r i n g e m u y conges t i v a c o n amígdalas hipertróficas y t ímpanos hiperémicos. ¿ Q u é a c t i t u d , en t r e las s igu ientes , hay q u e adop ta r en ese m o m e n t o ?
1) I n i c i a r t r a t a m i e n t o c o n antitérmicos y v i g i l a n c i a pos te r io r . 2) Rea l izar una punción l u m b a r pa ra análisis de l l íquido cefalorraquídeo. 3) So l i c i ta r u n e l e c t r o e n c e f a l o g r a m a u rgen te . 4) In i c ia r t r a t a m i e n t o c o n d i a c e p a m in t r a venoso . 5) So l i c i ta r u n T C c r a n e a l .
M I R 9 9 - 0 0 , 2 1 5 ; RC: 1
J
69

Neurología y neurocirugía
08 ENFERMEDADES DEGENERATIVAS
DEL SISTEMA NERVIOSO
MIR T e m a difícil y p o c o ren tab le , en el q u e se debe p r i o r i z a r el e s tud io d e las en f e rmedades d e la m o t o n e u r o n a . Es r e c o m e n d a b l e i den t i f i c a r el caso c l ín ico y, po r e l l o , l o más i m p o r t a n t e es c o n o c e r la c l ínica d e la ELA.
Aspectos esenciales
[T"| Dentro de las ataxias heredodegenerativas, encontramos la ataxia de Friedreich (AF), que es la más frecuente.
(~2~1 La AF se caracteriza por degeneración neuronal de los ganglios dorsales medulares (ataxia de la marcha) y cerebelosa, junto con cifoescoliosis, pies cavos, diabetes mellitus y miocardiopatía hipertrófica.
(YJ La afectación cardíaca es la principal causa de muerte en estos pacientes.
("4"] En la ataxia-telangiectasia (AT) hay un déficit en los mecanismos de reparación del A D N . Presentan atrofia cerebelosa, de timo y ganglios linfáticos.
P f ] La cl ínica de la AT es ataxia cerebelosa, telangiectasias cutáneas e infecciones de repetición. T ienen un riesgo mayor de neoplasias.
[ g j La ELA puede ser hereditaria ( 1 0 % ) o esporádica ( 9 0 % ) , y se caracter iza por la degeneración selectiva de la 1 , a y 2 . a motoneurona, con sensibi l idad, capac idad cognitiva y función autonómica indemnes.
¡Y] La clínica es una combinación de datos de primera y segunda motoneurona, con un inicio asimétrico y una supervivencia media de tres años.
QTJ El único tratamiento disponible es el riluzol (bloqueo de los receptores N M D A del glutamato) con un beneficio discreto.
Agrupan un con jun to de enfermedades caracterizadas por: Etiología desconoc ida . Una parte impor tan te t iene un or igen genético, aunque también hay casos esporád icos .
• Muer te neuronal de evolución gradual y progresiva. • Se puede afectar de forma selectiva un sistema neuronal concreto , mientras que los demás permanecen i n
tactos. Los síntomas y signos suelen ser bilaterales y simétricos, aunque el in i c io sea asimétrico.
• Muchas evo luc ionan sin responder a las medidas terapéuticas.
En este capítulo se estudiarán las ataxias hereditarias y las enfermedades de motoneurona . Otras enfermedades degenerativas se explicarán en diferentes partes de la obra. Las facomatosis se tratan en la Sección de Dermatología.
8.1. Ataxias heredodegenerativas
Son un grupo comp le jo de enfermedades, muchas de las cuales están genéticamente determinadas. En este apartado únicamente se estudiarán las más significativas (Tabla 37).
Ataxias congénitas 0 P r egun tas
Se producen por anomalías del desarrol lo embr ionar io , c o m o disgenesia o agenesia del vermis, hemisferios cerebelosos o t ronco encefálico. Cursan con disfunción cerebelosa, desarrol lo motor anorma l , retraso mental y espasticidad. La coordinación puede mejorar con la edad.
- MIR 05-06, 234 - MIR 04-05, 55 - MIR 02-03, 203
7 0

Neurología y neurocirugía
A. ATAXIAS CONGENITAS
B. ATAXIAS HEREDITARIAS CON D E F E C T O METABÓL ICO CONOC IDO
B-1. Intermitentes
Hiperamon iemlas Aminoac ldur ias (E. de Har tnup ) Al terac iones del láctico y pirúvico. (E. de Leigh)
Déficit de hexosamlnidasa Leucodistrof las Encefalomiopatías ml tocondr ia les Déficit de v i tamina E E. de Wi lson Déficit de hlpoxantina-guanina-fosforr ibosi l-transferasa Ceroldol ipofusc inosis E. de Niemann-PIck ( l ipidosls por a c u m u l o de esf lngomie l ina ) Abe ta l i popro te inemias (E. de Bassen-Kornzweig)
C. ATAXIAS ASOC IADAS A UN TRASTORNO DEFECT IVO EN LA REPARACIÓN DEL ADN
C-1. Ataxia-telangiectasia
C-2. Xeroderma p igmentosum
C-3. S índrome de Cockayne
D. ATAXIAS HEREDITARIAS DE INICIO PRECOZ (< 20 AÑOS ) Y ET IOLOGIA DESCONOCIDA
D-1. Ataxia de Fríedreich
D-2. Ataxia cerebelosa de inicio precoz con :
Reflejos miotáticos conservados H i p o g o n a d i s m o Mioclonías (síndrome de Ramsay-Hunt) Sordera Atrof ia óptica y retraso menta l (síndrome de Behr) Cataratas y retraso men ta l (síndrome de Marinesco-Sjogren)
E. ATAXIAS HEREDITARIAS DE INICIO TARDÍO (>20 AÑOS)
Tabla 37. Clasificación de las ataxias hereditarias
R E C U E R D A Es típica la asociación de h ipor re f l ex ia (dato de 2 a mo toneu rona ) c o n Babinsk i (dato de 1 * mo toneu rona ) , además de la a tax ia .
La lesión de cordones posteriores mot iva una pérdida de la sensibi l idad profunda (artrocinética, posic ional y v ibrator ia) . En algunos casos hay amiotrof ia distal e hipoestesia en guante o calcetín por afectación del nervio periférico. Puede haber nistagmo, sordera y atrofia óptica. No hay afectación de funciones superiores. Se asocia también cifoescoliosis y pies cavos. Un 1 0 - 2 0 % de los pacientes cursa con diabetes mel l i tus.
R E C U E R D A Es f recuente la asociación de c i foesco l ios is , pies cavos, diabetes tus y miocardiopatía hipertrófica.
La pr inc ipa l causa de muerte es la insuf ic iencia cardíaca secundaria a miocardiopatía hipertrófica. No existe un t ratamiento eficaz.
Diagnóstico
Es fundamenta lmente clínico. En el EMG hay signos de de-nervación en músculos distales. El LCR es norma l . En la TC puede observarse una modera da atrofia cerebelosa en fases
avanzadas. Es importante descartar en estos pacientes un déficit de v i tamina E subyacente.
R E C U E R D A El déficit de v i t a m i n a E pu p rovoca r degeneración nerv iosa y a tax ia .
Ataxia-telangiectasia
Ataxia de Friedreich
Es el t i po más frecuente de ataxia hereditaria. Se hereda de forma au tosómica recesiva y el gen anómalo se local iza en el brazo cor to del c romosoma 9.
Es una enfermedad hereditaria multisistémica que afecta a la p ie l , sistema nervioso y sistema inmunológico. Se hereda de forma autosómica recesiva. La mutación se local iza en el brazo largo del c romosoma 1 1 y c o m o consecuencia hay un defecto en los mecanismos de reparación del A D N , que conduce a una elevada frecuencia de roturas y t ranslocaciones cromosómicas.
Anatomía patológica Anatomía patológica
El ha l lazgo más característico es la pérdida neuronal en los gangl ios de las raíces dorsales. Secundar iamente , hay degeneración retrógrada de fibras nerviosas de los cordones posteriores, t racto espinocerebe-loso y nervios periféricos. La mayoría de los casos imp l i c a también degeneración de la vía p i r amida l (cort icoespinal ) . La médula espinal es atrófica. A nivel sistémico, destaca la alteración cardíaca, con f ibro-sis intersticial crónica e h ipertrof ia ventr icular .
R E C U E R D A En la A T se detecta una d i s m i n u c ión de l in foc i tos T e Ig A y E.
A nivel neurológico, hay una grave pérdida celular en el córtex cerebeloso, núcleos denta-dos y ol ivas inferiores. A nivel sistémico, destaca la h ipopla-
sia del t i m o y ganglios linfáticos, que se ha re lac ionado con la alteración inmuni ta r ia que presentan estos pacientes. Las células T periféricas están d isminuidas .
Clínica
Debuta en la infanc ia o adolescencia con ataxia progresiva de la mar cha. Se sigue de ataxia de m iembros y disartr ia cerebelosa. Es muy característica la asociación de h ipor re f lex ia miotática con respuestas plantares extensoras (signo de Babinski ) . La pérdida de la capac idad de deambulación se p roduce ap rox imadamen te 15 años después.
Laboratorio
En esta patología, es característico el aumento de los niveles de cc-fetoproteína y antígeno ca rc inoembr iona r io en casi todos los casos, mientras que las i nmunog lobu l inas A y E séricas están d isminu idas . Es frecuente la aparición de endocrinopatías c o m o la diabetes.
71

Manual CTO Medicina y Cirugía 8 a Edición
ESCLEROS IS LATERAL AMIOTRÓFICA ESPORÁD ICA (80-90%)
ENFERMEDADES HEREDITARIAS DE LA NEURONA MOTORA
Esclerosis lateral pr imaria Parálisis bulbar progresiva Atrofia muscular espinal
ELA familiar ( 10% ) - Autosómica d o m i n a n t e - Autosómica recesiva
Atrofia muscular espinal recesiva - T ipo I: Werdn ig-Hof fmann in fant i l - T ipo II: Werdn ig-Hof fmann juven i l - T ipo III: Kugelberg-Welander - T ipo IV: de Inicio en la edad adu l ta
> T ipo p rox ima l > Fac loescapulohumeral > Escapuloperoneal > T ipo distal
Tabla 38. Clasificación de las enfermedades degenerativas de neurona motora
Clínica
Debuta a los pocos años de edad, con ataxia cerebelosa progresiva, coreoatetosis y apraxia ocu lomoto ra . A nivel cutáneo, aparecen telan-giectasias venosas en conjunt ivas , oídos y cara. C o m o consecuencia del defecto inmunológico, son frecuentes las infecciones bacterianas de repetición del tracto respiratorio (sinusitis y neumonías) y hay una alta inc idenc ia de neoplasias l inforret iculares (10-20% ) .
La tríada cl ínica: te langiectasias mucocutáneas, a tax ia cerebelosa e i n fecc iones bacter ianas de repetición.
El deter ioro neurológico e inmunológico es progresivo, y hab i tua l mente fa l lecen antes de los 20 años a consecuencia de infecciones incontrolables, insuf ic iencia respiratoria o neoplasias. No existe un tratamiento eficaz.
Ataxia cerebelosa de inicio en el adulto
Son un grupo de enfermedades con herencia autosómica dominante . A nivel anatomopatológico, hay degeneración espinocerebelosa y de cor dones posteriores, aunque la patología es tan variada c o m o la clínica.
Una forma especial es la enfermedad de Machado-Joseph, con pre servación del córtex cerebeloso y ol ivas, y degeneración nigroestriada y de núcleos dentados. A nive l clínico, debutan entre los 20-50 años con ataxia de la marcha y miembros , así c o m o disartria. Otros datos son pérdida de sensibi l idad profunda, paraparesia espástica y clínica ext rap i ramida l . En la enfermedad de Joseph, p redomina la espasticidad sobre los síntomas parkinsonianos.
8.2. Enfermedades de la motoneurona
Se caracter izan por una lesión selectiva de los sistemas neuronales que cont ro lan los mov imien tos vo luntar ios , con una l lamat iva ausencia de afectación de otras vías (se conservan indemnes la sensib i l idad, la capac idad cogni t iva , la función autonómica y el resto de órganos y sistemas) (Tabla 38).
Esclerosis lateral amiotrófica (ELA)
Es la fo rma más frecuente de enfermedad progresiva de la neurona m o tora, s iendo su inc idenc ia g lobal de 1-3 casos por cada 100 .000 hab i tantes/año. Suele ser más frecuente en varones (1,5/1). Se d ist inguen fundamenta lmente dos formas:
Famil iar ( 1 0 % de los casos). Con herenc ia f u n d a m e n t a l m e n t e autosómica d o m i n a n t e ( i n i c i o a los 11 años; p r e d o m i n i o de la espast ic idad) . A lgunas formas recesivas ( i n i c i o más p recoz , a los c i n c o años; p r e d o m i n i o bu lbar ) se han asoc iado a déficit de hexosamin idasa .
• Esporádica (80-90%) . Característicamente, afecta a pacientes mayo res de 50 años.
Excepto por una media de edad de i n i c io infer ior, los pacientes con ELA fami l ia r son indist inguibles de aquel los con enfermedad esporádica.
Anatomía patológica
Se habla genéricamente de esclerosis lateral amiotrófica (ELA) para referirse a un proceso degenerat ivo que afecta, en distintos grados, a la pr imera y segunda motoneuronas, p roduc i endo secundar iamente at rofia de las fibras musculares (MIR 05-06, 234).
A veces es posible encontrar pacientes con afectación selectiva de p r i mera motoneurona (esclerosis lateral pr imar ia ) , de neuronas de núcleos troncoencefálicos (parálisis bu lbar progresiva) o segunda motoneurona (atrofia muscular espinal) (Figura 54).
Esc leros is l a t e r a l amiot ró f i ca 1. a
Figura 54. Formas de esclerosis lateral amiotrófica
Característicamente, no hay afectación de los núcleos ocu lomotores ni de las neuronas parasimpáticas en la médula espinal sacra, que confor man el núcleo de Onuf , y que inervan los esfínteres vesical e intest inal .
72

Neurología y neurocirugía
Etiopatogenia
Es desconocida. Entre las distintas hipótesis propuestas, destacan: 1) def ic ienc ia de un factor de c rec imiento nervioso, y 2) exceso de g luta mato extracelular en el SNC deb ido a un defecto en la recaptación de g lutamato.
Diversas evidencias intentan impl i car al sistema inmun i ta r io en la patogenia de la enfermedad: el antígeno HLA-A3 se presenta en pac ien tes con ELA rápidamente progresiva, mientras que el HLA-A12 se ha encont rado preferentemente en aquéllos con progresión más lenta. Se han descubierto ant icuerpos frente a gangliósidos de la mie l ina en un porcentaje var iable de pacientes. As imismo, se han descrito en el suero de pacientes con ELA ant icuerpos d i r ig idos frente a canales del ca lc io voltaje-dependientes.
Ap rox imadamente el 1 0 % de pacientes describen una historia fami l ia r de ELA, habiéndose documentado de fo rma clara un patrón de herencia autosómica dominan te en algunas fami l ias . En un subgrupo de estas fami l ias, se ha ident i f i cado un defecto genético sobre el c romosoma 21 que imp l i ca al gen S O D 1 ; este gen codi f i ca la enz ima superóxido-dismutasa, que actúa reduc iendo la concentración de radicales libres.
Clínica
Cursa con deb i l idad muscular lentamente progresiva con signos de afectación de pr imera y/o segunda motoneurona .
El com ienzo suele ser insidioso y asimétrico, afectándose in ic ia lmente la musculatura distal de los miembros o la de los pares craneales inferiores con disartria y disfagia. Los hal lazgos explorator ios son una c o m binación de datos de pr imera motoneurona (debi l idad, espasticidad, hiperref lexia y signo de Babinski) y segunda motoneurona (debi l idad, amiot rof ia y fasciculaciones). En fases evoluc ionadas, es característica la aparición de un síndrome pseudobulbar con lab i l idad emoc iona l .
ga a la de enfermedad de motoneurona y no t ienen el fatal pronóstico de la ELA (Tabla 40) .
R E C U E R D A D e b i l i d a d muscu la r progres iva , de i n i c i o asimétrico y d is ta l , c o n afi tación de pares craneales bajos y p o c a afectación de la muscu la tu ra ex t raocu la r es sugestiva de ELA.
La musculatura extraocular no se suele afectar, excepto en los casos de parálisis bulbar progresiva de larga evolución. No hay trastornos sensitivos, ni disfunción vesical, intelectual o de la función sexual (MIR 02-03, 203) .
R E C U E R D A N o hay de te r io ro c o g n i t i v o , cl ínica sensit iva n i t rastornos de esfínteres.
La media de supervivencia es de tres años desde el in i c io de los síntomas. La deb i l idad de la musculatura respiratoria ocurre en casi todos los pacientes y, con o sin neumonía, es generalmente la causa responsable de la muerte. La Tabla 39 muestra los criterios diagnósticos de ELA.
PRESENCIA DE
• Signos de neurona motora inferior (incluyendo hallazgos electromiográficos en músculos normales clínicamente)
• Signos de neurona motora superior - Carácter progresivo
AUSENCIA DE
• Síntomas sensitivos • Trastornos esfinterianos • Trastornos visuales • Disfunción autonómica • Enfermedad de Parkinson • Demencia
EL D IAGNÓSTICO SE APOYA EN
• Fasciculaciones en una o más regiones • Cambios neurogénicos en el EMG • Velocidades de conducción motora y sensitiva normales • Ausencia de bloqueos de conducción
Tabla 39. Criterios diagnósticos de ELA
MIELOPATÍA COMPRES IVA
• Espondilosis cervical • Tumor extramedular
TOXIC IDAD POR METALES PESADOS
• Plomo • Mercurio
PROCESOS METABÓL ICOS
• Hipertiroidismo • Hiperparatiroidismo • Déficit de vitamina B 1 2 o E • Hipoglucemia
TRASTORNOS PARANEOPLÁS ICOS
• Linfoma • Carcinoma pulmonar • Tumor de células B
INFECCIONES
• Lyme • Poliomielitis • Síndrome postpolio • Mielopatía retroviral
DEFECTOS METABÓL ICOS HEREDITARIOS
• Déficit de hexosaminidasa A • Déficit de a-glucosidasa • Hiperlipidemia • Hiperglicinuria • Superóxido dismutasa • Defecto del receptor androgénico
(Síndrome de Kennedy)
Tabla 40. Diagnóstico diferencial de la ELA
Tratamiento
A c t u a l m e n t e sólo está c o m e r c i a l i z a d o el r i l u z o l c o m o t r a t am ien to para esta en f e rmedad .
El r i l u z o l i nh ibe la liberación presináptica de g l u t ama to , inter f iere a n ive l postsináptico con la acción de los aminoácidos exc i tadores med ian te un b l o q u e o de los receptores N M D A , y actúa d i r e c t a m e n te sobre los canales de sod io depend ien tes de vo l t a j e .
R E C U E R D A El único t r a t am ien to para la ELA es el r i l u z o l .
Diagnóstico diferencial Parece tener un benef ic io discreto sobre el aumento de la superv ivencia, en especial en pacientes con in i c io bulbar de la clínica.
Dado que no hay un t ratamiento ef icaz, es fundamenta l descartar en fermedades potenc ia lmente tratables que cursan con una clínica análo-
Otros ensayos con inmunosupresores, factores neurotróficos y otros i n hibidores del g lutamato no han dado resultado.
73

Manual CTO Medicina y Cirugía 8 a Edición
Enfermedades hereditarias de la neurona motora
Atrofia muscular espinal hereditaria
Se trata de un grupo de enfermedades famil iares, de herencia autosómica recesiva y com ienzo precoz, caracterizadas por deb i l idad muscular progresiva y atrofia por denervación secundarias a degeneración selectiva de las neuronas motoras de la médula espinal, sin afectación de la motoneurona superior.
La muerte se p roduce por deb i l idad de la musculatura respiratoria.
La enfermedad de Werdnig-Hoffmann infantil (tipo I) se mani f ies ta inc luso intraútero por disminución de los mov im i en tos fetales y e vo luc iona rápidamente, p r o d u c i e n d o la muerte en el p r imer año de v ida .
La enfermedad de Werdnig-Hoffmann juvenil (tipo II) se in ic ia más tarde y evo luc iona más lentamente, sobrev iv iendo el paciente hasta la preadolescencia y, en ocasiones, hasta la v ida adulta .
Por último, la enfermedad de Kugelberg-Welander (t ipo III) debuta al f i nal de la infancia y evo luc iona de forma lenta y crónica, con p redomin io de la afectación de la musculatura p rox ima l y, en algunos casos, pseu-dohipertrof ia de los músculos de la pantorr i l la s imulando una miopatía.
Atrofia muscular espinobulbar ligada a X
También es conoc ida c o m o enfermedad de Kennedy, l igada al c r o m o soma X. Se trata de un cuadro progresivo de deb i l i dad de la muscu la tura de las extremidades, que además afecta a la musculatura bulbar. Comienza en la edad adulta y se asocia a una insensibi l idad del receptor androgénico que se manif iesta por ginecomast ia e in fer t i l idad. A l igual que en la enfermedad de Hunt ing ton , se observa fenómeno de anticipación por expansión del tr ip lete de nucleótidos CAG.
r
Casos clínicos representativos L.
M u j e r de 64 años que consu l t a por c l ín ica progres iva , e n los últ imos cua t ro meses , de deb i l i dad en la p i e rna d e r e c h a . En la exp lorac ión se ob je t i va u n a pares ia c o n a m i o t rof ia de m i e m b r o infer ior d e r e c h o y u n a h iper re f l ex ia miotá t i ca de d i c h o m i e m b r o . ¿ C u á l es su d iagnóst ico?
1) H e r n i a d i s c a l l u m b a r d e f i c i t a r i a . 2 ) S índrome d e Gui l la in-Barré . 3) Esclerosis l a te ra l amiotróf ica. 4 ) Neuropat ía p o r e n f e r m e d a d d e L y m e . 5) Esclerosis múlt ip le .
M I R 0 4 - 0 5 , 5 5 ; RC: 3
Pac ien te de 5 0 años que p resen ta , de f o rma ins id iosa , deb i l i dad y c a l a m b r e s e n m i e m b r o super io r d e r e c h o . En la exp lorac ión neuro lógica se objet iva espas t i c idad e n e l h e m i c u e r p o d e r e c h o , a t rof ia de l p r imer interóseo de la m a n o d e r e c h a , c o n r e f le jos os teotend inosos presentes y s in t rastornos sensor ia les . ¿Cuá l es el d iagnóst ico más p robab le en este pac ien te , en t re los s iguientes?
1) S i r i n g o m i e l i a . 2 ) T u m o r a n i v e l d e l f o r a m e n m a g n o . 3) Esclerosis la tera l amiotróf ica. 4 ) M ie lopa t í a c e r v i c a l d e o r i g e n v a s cu l a r . 5) A s t r o c i t o m a m e d u l a r c e r v i c a l .
RC: 3
74

Neurología y neurocirugía
09 ENFERMEDADES VIRALES Y PRIÓNICAS DEL SISTEMA NERVIOSO
Orientación
MIR r
Aspectos esenciales
Es un t e m a d e i m p o r t a n c i a m e n o r , p e r o p u e d e serv i r para repasar la encefa l i t i s herpética, p r e g u n t a d a también en Enfermedades infecciosas. Las otras en f e rmedades v i ra les son cuad ros m u y característicos, d e los q u e s o l a m e n t e hay q u e c o n o c e r lo más t ípico. La e n f e r m e d a d de Creutz fe ld t-Jacob (ECJ) es la e n f e r m e d a d priónica p o r exce l enc i a .
fT~] Las encefalitis epidémicas están causadas por arbovirus y enterovirus.
[~2~j La encefa l i t i s esporádica más f recuente es la causada por el v i rus herpes s imp le t i p o 1 .
["3] La clínica de las encefalit is es similar a las meningit is (fiebre, cefalea y meningismo) con deter ioro cogni t i vo y fo ca l i dad neurológica.
[~4~| El LCR mostrará las características de las meningit is virales: h iperproteinorraquia, pleocitosís l infocitar ia y g lucosa n o r m a l .
Qn La encefa l i t i s herpética muestra f o c a l i d a d f r o n t o t e m p o r a l , s iendo la R M N la p rueba de imagen de elección.
[~p~| La PCR del A D N del virus herpes en LCR es la técnica diagnóstica de elección, sustituyendo a la biopsia cerebra l .
["7"] El t r a t am ien to c o n ac i c l o v i r es seguro y e fec t i vo ; por e l l o , debe instaurarse rápidamente ante la sospecha de encefa l i t i s .
[~8~| La paraparesia espástica tropical se ha re lac ionado con la infección por el HTLV-1 en zonas tropicales. Puede s imu lar una esclerosis múltiple con presencia de bandas ol igoclonales en LCR y placas de desmielinización en la RM.
[9"] La leucoencefalopatía mu l t i f o ca l progres iva se caracter iza por d e m e n c i a y hemianops ias en pacientes c o n SIDA. Se debe a la infección por el papovav i rus JC, q u e p r o d u c e desmielinización s imi la r a la esclerosis múltiple. El diagnóstico requ iere b iops ia ce rebra l .
J TQ ] La panencefa l i t i s esclerosante subaguda se presenta en niños de 5 - 1 5 años c o n h is tor ia de sarampión en los pr imeros años de v ida . El EEG muestra un patrón característico, y en LCR se detec tan bandas o l i goc lona l e s y a u m e n t o del título de Ac anti-sarampión.
[11 | Las enfermedades priónicas son un c o n j u n t o de enfermedades q u e t i ene en común la presenc ia de una p r o teína c o m o agente patógeno. La ECJ clásica se caracter iza por d e m e n c i a rápidamente progres iva , mioclonías y ataxia . El EEG muestra un patrón típico, a u n q u e el diagnóstico es anatomopatológico.
9.1. Encefalitis herpética y otras encefalitis virales
En las meningi t is , el proceso infeccioso in f lamator io está l im i tado a las meninges; en la encefal it is hay, además,
afectación del parénquima cerebral .
Etiología
Son causa frecuente de encefalitis epidémica los arbovirus y enterovirus. El virus herpes s imple t i po I es la causa
más frecuente de encefalitis esporádica.
0 3 P r egun tas Clínica • M I R 0 9 - 1 0 , 6 2 , 1 1 4 , 2 0 8 • M I R 0 6 - 0 7 , 5 8 • M I R 0 4 - 0 5 , 2 3 2 • M I R 0 2 - 0 3 , 52 • M I R 0 0 - 0 1 , 5 2 , 2 4 2 • M I R 98-99F , 2 5 2 • M I R 9 7 - 9 8 , 5 0
Cursa con f iebre, cefalea, signos meníngeos y, lo que es más característico de encefal i t is, deter ioro del nivel de
consciencia , que puede oscilar desde el estado confus ional al coma p ro fundo y signos y síntomas de foca l idad
neurológica. Los signos más frecuentes de foca l idad son: afasia, ataxia, hemiparesia espástica, trastornos del
m o v i m i e n t o (generalmente mioclonías) y paresia de pares craneales.
7 5

Manual CTO de Medicina y Cirugía, 8.a edición
Puede haber a luc inac iones, agitación, cambios en la personal idad e, inc luso, clínica psicótica. Hasta un 5 0 % de los pacientes t ienen crisis focales o general izadas.
R E C U E R D A Meningismo con deterioro cognitivo y focal ¡dad neurológica es indicativo de encefalitis.
Pruebas complementarias
Líquido cefalorraquídeo. En todo paciente con sospecha de encefalit is se debe realizar una punción lumbar, una vez descartada la existencia de un proceso expansivo intracraneal , hab i tua lmente me diante la realización de una prueba de imagen. Típicamente, hay pleocitosis l infoc i tar ia (> 9 5 % de los casos) con hiperprote inorra-qu ia y glucosa norma l . La ce lu lar idad puede ser normal en fases m u y iniciales o cuando el paciente está i n m u n o d e p r i m i d o . Pueden verse l infocitos atípicos en encefal it is por virus de Epstein-Barr. En algunos casos, hay un aumento en la cifra de hematíes (encefalitis necrohemorrágicas, c o m o la herpética). Si la glucosa está baja, de bemos pensar en otros diagnósticos (infección por bacterias, h o n gos, tuberculosis , parásitos, leptospiras, sífilis, sarcoidosis o m e n i n gitis carc inomatosa) , aunque a veces la glucosa está baja en fases tardías de la encefal i t is herpética (MIR 09-10, 62) .
R E C U E R D A La presencia de abundantes hematíes en LCR orienta a encefalitis nec hemorrágica por herpes simple.
Cult ivo de LCR. La pos ib i l i dad de cu l t i var v i rus es exce lente en casos de virus Coxsackie , Echo, v irus de la co r i omen ing i t i s l i n f o c i tar ia y v i rus de la parot id i t i s , pero los cu l t i vos son invar iab lemente negativos en casos de v irus herpes t i po I. En genera l , el r e n d i m i e n to es escaso. Estudios serológicos. En la mayoría de los casos, el diagnóstico de f in i t i vo depende de la documentación de una seroconversión en la titulación de ant icuerpos frente a un dete rminado virus, entre el m o m e n t o in ic ia l de la infección y la fase de conva lecenc ia , 2-4 semanas después. Esto es importante para el diagnóstico de forma retrospectiva, pero t iene un escaso valor en el diagnóstico y tratamien to en fase aguda. T C , RM y E EG . Se ut i l izan para intentar establecer la existencia de una encefal i t is focal o difusa y descartar otros diagnósticos alternat ivos. La existencia de foca l idad a nivel f rontotempora l en el EEG es sugestiva de encefal it is herpética. En estos casos, el EEG demuestra, a nivel preferentemente tempora l y sobre una act iv idad de fondo lenta y de baja amp l i t ud , puntas periódicas focales. En la TC, la encefal it is herpética puede dar lugar a zonas de hipo-densidad y efecto de masa en regiones f rontotempora les que pue den captar contraste. La RM es la técnica radiológica de elección para detectar cambios de señal en el parénquima cerebral en las encefal i t is (Figura 55).
R E C U E R D A La encefalitis herpética afecta predominantemente a la zona frontotemporal.
Biopsia cerebral. Su sensibi l idad es mayor del 9 5 % y la espec i f i c idad se acerca al 1 0 0 % . Sin embargo, su aplicación ha
Figura 55. RM que muestra afectación de región temporal derecha, que en un contexto de fiebre y meningismo
s altamente sugerente de encefalitis herpética e s ;
d i sm inu ido desde la introducción del ac ic lov i r c o m o agente terapéutico para laencefal itis herpética (gran ef i cac iacon escasosefec-tos secundarios). En general , se acepta que, en pacientes mayores de 30 años, la presencia de alteraciones focales en las pruebas de imagen y pleocitosis l infoc i tar ia en LCR son criterios suficientes para just i f icar una terapia empírica con ac ic lov i r sin necesidad de biopsia . Sin embargo, pacientes jóvenes sin evidencias de lesiones focales en TC, RM o EEG podrían ser candidatos a biopsia, ya que es poco probable que se trate de una encefal it is herpética, y podrían detectarse otros procesos potencia l mente tratables. Reacción en cadena de la polimerasa (PCR). La PCR del A D N del virus herpes s imple ha sust i tuido a la biopsia cerebral c o m o técnica diagnóstica de elección en la encefal it is herpética (MIR 09-10, 208 ; MIR 0 0 - 0 1 , 242) .
Diagnóstico diferencial
Hay que di ferenciar causas no virales de encefal i t is , i nc luyendo otros agentes infecciosos, los accidentes vasculares cerebrales, tumores, encefalopatías toxicometabólicas, etc. Dentro de las virales, es funda mental d i ferenciar la encefal it is herpética del resto, dado que sólo para la pr imera hay terapia efectiva.
Tratamiento ( M I R O 9 - I O , 1 1 4 )
El ac i c lov i r es la medicación de elección en la encefal it is por virus herpes s imple, y también es efect ivo para el t ra tamiento del virus de Epstein-Barr y varicela-zoster. Debe ser admin is t rado ante la s imple sospecha clínica, sin demora , d i l u i do por vía intravenosa y en infusión lenta, para evitar la disfunción renal. Su paso a través de la barrera hematoencefálica es excelente. El efecto secundar io más frecuente es gastrointestinal, con diarrea, náuseas y vómitos. Otras compl i cac iones del t ra tamiento son: elevación del B U N y creat in ina, t romboc i topen ia y tox i c idad neurológica (obnubilación, desorientación, a luc inac iones, temblor , convuls iones) .
76

Neurología y neurocirugía
R E C U E R D A La PCR del LCR es la técnica diagnóstica de elección en la encefa l i t i s herpética.
Pronóstico
Es variable, depend iendo del agente etiológico. Las encefalit is por virus de Epstein-Barr t ienen un excelente pronóstico, y los pacientes sobrev i ven sin secuelas. En la encefalit is herpética, la morta l idad alcanza un 2 0 % , y hasta un 4 0 % de los pacientes que sobreviven t ienen discapacidades graves. La inc idencia y gravedad de las secuelas está directamente relacionada con la edad del paciente, demora del t ratamiento y nivel de consciencia en el momen to de comenzar el t ratamiento; en pacientes jóvenes menores de 30 años, con buena función neurológica en el momen to del t ratamiento, la supervivencia puede ser del 1 0 0 % y, en más del 6 0 % no hay secuelas o son mínimas.
9.2. Paraparesia espástica tropical
Pruebas complementarias
La RM puede demostrar desmielinización medular y de la sustancia blanca hemisférica periventr icular . Los estudios neurofisiológicos pue den objet ivar disfunción de cordones posteriores y una neuropatía per i férica desmiel in izante. El estudio anatomopatológico medular demuestra cambios en la vía cort icoespinal (piramidal ) , cordones posteriores, haces espinocerebelosos y espinotalámicos.
Tratamiento
No hay tratamiento efectivo. Los pacientes pueden beneficiarse de tratamiento esteroideo.
9.3. Leucoencefalopatía multifocal progresiva (LMP)
Etiología Epidemiología
Es mul t i fac tor ia l , incluyéndose factores tóxicos y nutr ic ionales. El virus HTLV-1 resulta estar imp l i cado en un gran número de estos pacientes. Los casos asociados a HTLV-1 presentan anticuerpos específicos en LCR y bandas ol igoclonales en la mayoría de ellos (Tabla 41) .
VIRUS ASOC IADO
CLÍNICA CARACTERÍST ICA
Paraparesia espástica tropical
HTLV-1 Hiperref lexia y espast ic idad progresivas de MMI I
Leucoencefalopatía multifocal progresiva
Virus JC Tr. visuales (hemianopsia homónima), demenc i a y tr. persona l idad
Panencefalit is esc lerosante subaguda
Sarampión Mal r end im ien to escolar y t ras to rno de persona l idad Después, de te r io ro neurológico y tetraparesia espástica
Tabla 41. Enfermedades de or igen vírico
Aparece habitualmente en pacientes con trastornos ¡nmunológicos. En el momento actual, más del 6 0 % de los pacientes diagnosticados de LMP presentan SIDA. Otras causas posibles son trastornos linfoproliferativos, mieloproliferativos y enfermedades infecciosas crónicas y granulomatosas.
Anatomía patológica
La LMP se caracteriza por una desmielinización mul t i foca l que afecta al sistema nervioso central , con lesiones de tamaño variable. Los o l i-godendroci tos cont ienen inclusiones cristalinas que son partículas del virus JC (papovavirus).
Clínica
Clínica
Los pacientes suelen presentarse con trastornos visuales, generalmente en forma de hemianopsia homónima y deter ioro de funciones superiores con demenc ia y trastornos de la personal idad. En su evolución, es habitual la presencia de déficit motores.
Debuta en la 3. a-4. a década de la v ida, y es más frecuente en mujeres. Su distribución geográfica es típica, estando la mayoría de los casos en Japón, Caribe, Sudamérica y África occ identa l . Cursa con una paraparesia espástica lentamente progresiva, con signos de pr imera motoneurona y escasa clínica sensitiva (parestesias, disestesias, do lor y deter ioro de la sensibi l idad propiocept iva en miembros inferiores).
R E C U E R D A La p a r a p a r e s i a espást ica t r o p i c a l p r e sen t a caracter íst icas s i m i l a r e s a la e s c l e ros is múl t ip le . La c l ín i ca d e p a r a p a r e s i a espást ica c o n p o c a mani festac ión sens i t i v a y la p r e s e n c i a d e neuropat ía perifér ica son da tos d i s t i n t i v o s .
Pruebas complementarias
En la TC hay lesiones hipodensas en sustancia blanca que no captan contraste ni presentan edema asociado o efecto de masa. Se local izan a nivel per iventr icular , en centro semioval , regiones parietooccipitales y cerebelo. La RM es más sensible para detectar estas lesiones.
El EEG demuestra en lentec imiento focal o difuso. El LCR es típicamente normal , con ocasional prote inorraquia y pleocitosis mononuc lear (< 25 células/pl).
77

Manual CTO de Medicina y Cirugía, 8 . a edición
El diagnóstico def in i t i vo precisa de la identificación de los cambios anatomopatológicos típicos en la biopsia. Puesto que un 8 0 - 9 0 % de la población adulta normal es seroposit iva para el virus JC, la ident i f i ca ción del antígeno o genoma del virus no es diagnóstica de LMP si no se acompaña de los cambios patológicos típicos. La reacción en cadena de la pol imerasa (PCR) no es útil con fines diagnósticos en esta ent idad (MIR 06-07, 58).
Q R E C U E R D A En un paciente con S IDA y clínica neurológica, se debe descartar la toxoplasmosis. Si en la T C las lesiones no captan contraste y no presentan efecto masa, hay que sospechar la LMP como primera opción.
Pronóstico
La regla es la evolución fatal en meses. No existe t ratamiento efect ivo, aunque algunos casos mejoran con la reconstitución del sistema inmu-nitar io .
9.4. Panencefalitis esclerosante subaguda (PESS)
Epidemiología
Los pacientes generalmente t ienen una historia de sarampión en los dos pr imeros años de v ida (véase Tabla 41) (MIR 98-99F, 252) , desarrollándose el trastorno neurológico tras un per iodo de latencia de 5-10 años, con una edad máxima de inc idenc ia entre los 5-15 años. Su inc idenc ia ha d i sm inu ido en los últimos años, tras la vacunación sistemática frente al sarampión.
Clínica
Debuta con trastornos de la personal idad y mal rend imien to escolar. Posteriormente aparece un deter ioro neurológico progresivo, con crisis comic ia les , mioclonías, coreoatetosis, ataxia y trastornos visuales. En fases avanzadas, el paciente desarrolla un cuadro de tetraparesia espást ica y estado vegetat ivo.
Pruebas complementarias
En el EEG hay típicamente un patrón periódico con descargas de ondas lentas de gran voltaje, a 2-3 Hz y cada 5-8 s, que se siguen de un per io do de ap lanamiento de la act iv idad eléctrica cerebral .
El LCR puede ser diagnóstico y muestra ace lu la r idad con proteínas normales o l igeramente elevadas; las gammag lobu l inas están m u y elevadas y es pos ib le demostrar bandas o l igoc lona les contra el virus del sarampión y elevación de los niveles de ant icuerpos específicos.
La TC y la RM muestran múltiples lesiones en sustancia b lanca , atrof ia cor t i ca l y, secundar iamente , aumento del tamaño vent r i cu la r .
Tratamiento
No existe un t ratamiento def in i t i vo . El uso de la isoprinosina es c o n t rover t ido, habiéndose descrito supervivencias prolongadas y mejorías clínicas en algunos casos. Se postula la posible u t i l idad del interferón P en algunos pacientes.
9.5. Enfermedades priónicas (Tabla 42 y Figura 56)
ENFERMEDADES PR IÓNICAS INFECCIOSAS
Kuru ECJ iatrógeno
Canibalismo Trasplante de córnea
Injertos de duramadre Extractos hipofisarios Procedimientos neuroquirúrgicos
ENFERMEDADES PR IÓNICAS HEREDITARIAS
E G GSS IFF
Mutaciones en el gen de la PrP en el cromosoma 20 Mutaciones en el gen de la PrP en el cromosoma 20 Mutaciones en el gen de la PrP en el cromosoma 20
ENFERMEDADES PR IÓNICAS E SPORÁD ICAS
ECJ (85%) Mutación somática Conversión espontánea de PrPc a PrPsc Susceptibilidad a factores ambientales
Tabla 42. Categorías etiológlcas de las enfermedades priónicas
Forma normal Forma patológica (PrPc) o prión (PrPSc)
Figura 56. Configuración espacial de una proteína priónica
Bajo esta denominación, se i n c l u ye un c o n j u n t o de enfermedades c u y o agente patógeno es una proteína in fec t i va carente de ácido nuc l e i co , a la que se ha d a d o el n o m b r e de prión (PrP o proteína priónica). En c o n d i c i o n e s norma les , la proteína priónica ( i so forma no rma l o PrPc) es cod i f i c ada en un gen l o ca l i z ado en el b r azo co r to de l c r o m o s o m a 20 y parece desempeñar un pape l est ructura l en la m e m b r a n a ce lu la r sináptica y en la transmisión i n t e rneu rona l . Su i so fo rma patógena (PrPsc) d i f i e re de la va r i an te no rma l por su a l to c o n t e n i d o en p l egamien tos p, i n s o l u b i l i d a d en detergentes y
7 8

Neurología y neurocirugía
re lat iva resistencia a la proteólisis. La conversión de PrPc en PrPsc parece i m p l i c a r la desaparición de las hélices B y su conversión en 6, c on el cons igu i en te depósito in t r aneurona l en f o r m a de proteína a m i l o i d e . Las enfermedades priónicas humanas (véase Tab la 42 ) i n c luyen la en fe rmedad de Creutzfeldt-Jakob (ECJ), kurú, en f e rmedad de Cerstmann-Stráussler-Scheinker (GSS) y el i n s o m n i o f am i l i a r fatal (IFF) (MIR 98-99F, 252 ) .
R E C U E R D A Las bandas oligoclonales en LCR están presentes en múltiples patologías y sólo indican la activación de un pequeño pool de linfocitos B en el SNC.
Enfermedad de Creutzfeldt-Jakob (ECJ)
ondas agudas bilaterales y síncronas (cada 0,5-2,5 s y con una d u ración de 200-600 ms). Este patrón aparece en la mayoría de los casos, aunque puede no estar presente en fases iniciales o muy evo lucionadas. El EEG suele ser normal en la nueva variante.
R E C U E R D A La ECJ clásica tiene un patrón típico en el EEG, mientras que en la nueva variante el EEG suele ser normal.
Estudio anatomopatológico. Es el método diagnóstico de elección (ya sea en la biopsia o en la necropsia). Se caracteriza por una degeneración espongiforme máxima en la corteza cerebral, pero también pro minente en ganglios básales, tálamo y cerebelo. No suelen afectarse ni el t ronco cerebral ni la médula espinal. La detección de placas de ami lo ide compuestas de PrPsc es diagnóstica (MIR 04-05, 232).
Epidemiología
La mayoría de los casos son esporádicos, pero en un 5 - 1 5 % se ha demos t r ado un patrón de herenc ia autosómico d o m i n a n t e , en relación con mutac iones en el gen de la PrP en el c r o m o s o m a 2 0 , e inc luso transmisión de persona a persona en p r o c e d i m i e n t o s neuro-quirúrgicos con mater ia l no d e s c o n t a m i n a d o , trasplantes de córnea o in jer tos de d u r a m a d r e h u m a n a y med ian te extractos h ipof i sa r ios de h o r m o n a del c r e c i m i e n t o , ob ten idos de cadáveres afectados por la en f e rmedad . Los casos esporádicos no están b i en exp l i c ados .
R E C U E R D A La variante asociada a la "enfermedad de las vacas locas" difiere de la forma clásica de ECJ en que aparece en pacientes más jóvenes y predominan los síntomas psiquiátricos y la ataxia sobre la demencia y mioclonías.
Kuru
Esta en fe rmedad fue descr i ta entre los m i e m b r o s de una t r i b u de Nueva Gu inea que p rac t i caban el r i to del c a n i b a l i s m o y se ha suger i do que la transmisión de la en fe rmedad se p r o d u j o por la ingesta de ce rebro . El da to cl ínico f u n d a m e n t a l es el desar ro l lo subagudo de ataxia cerebelosa grave asoc iada a m o v i m i e n t o s i nvo lun ta r i os t i p o mioclonías, t e m b l o r o coreoatetos is . Poster iormente aparece un de te r i o ro de func iones super iores .
Enfermedad de Gerstmann-Stráussler-Scheinker (GSS)
Clínica
Se caracteriza por una demenc ia rápidamente progresiva que asocia mioclonías en un 9 0 % de los pacientes y ataxia cerebelosa (MIR 97-98, 50) Otros síntomas extrapiramidales inc luyen temblor , coreoatetosis y park insonismo ( 6 0 % ) . En el 5 0 % hay signos de afectación p i r amida l . La evolución es invar iab lemente fatal . Los pacientes con ECJ secundaria a extractos hipofisarios son típicamente más jóvenes y pueden desarrol lar una clínica con p r edomin i o de la ataxia sobre la demenc ia (a di ferencia de la ECJ clásica). Recientemente se ha descrito una nueva variante de esta enfermedad asociada epidemiológicamente al brote de encefalopatía espongi forme bov ina ( "enfermedad de las vacas locas"). Debuta en pacientes más jóvenes (16-40 años), cursa más lentamente y no se encuentran mutaciones en el c romosoma 20 . La clínica se i n i c ia con trastornos psiquiátricos, parestesias en miembros inferiores y ataxia. La superv ivencia media es de 15 meses, comparado con los 5 meses en la ECJ esporádica (MIR 0 0 - 0 1 , 52).
Los estudios de genética molecu lar han demostrado mutaciones en el gen de la PrP en el c romosoma 20 . Es un trastorno espinocerebeloso hereditar io que debuta en la v ida adulta, caracter izado por signos y síntomas de disfunción cerebelosa progresiva ( inestabi l idad, i n c o o r d i nación, trastorno progresivo para la deambulación, ataxia, disartria y nistagmo). A diferencia de la ECJ, no produce demenc ia ni mioclonías o estas son mínimas. Otros síntomas son clínica parkinsoniana, p i r a m i da l ismo, sordera, ceguera y parálisis de la mirada conjugada.
Q R E C U E R D A Clínica de ataxia cerebelosa sin demencia ni mioclonías es propia de GSS.
Insomnio familiar fatal (IFF)
Pruebas complementarias
• LCR. Gene ra lmen te no rma l o con l igero a u m e n t o de proteínas. Apoya el diagnóstico el ha l l azgo de proteína 14-3-3, a u n q u e p re senta falsos pos i t i vos y negat ivos .
• TC y RM. Pueden mostrar atrofia cort ica l general izada, pero de i n tensidad menor que la esperable por la gravedad de la demenc ia .
• EEG. Un patrón típico puede sugerir el diagnóstico. Sobre una act i v idad de base lent i f icada, es habitual la aparición de brotes de
Es una en f e rmedad autosómica d o m i n a n t e , rápidamente progres iva , que aparece en edades medias o avanzadas de la v ida . Se carac ter iza por i n s o m n i o in t ra tab le , h i pe r a c t i v i dad simpática, t rastornos endoc r i nos (pérdida de l r i t m o c i r c a d i a n o en la liberación de melato-n ina , p ro l a c t i na y h o r m o n a de l c r e c i m i e n t o , secreción de A C T H d is m i n u i d a , a u m e n t o en la secreción de cor t i so l ) , d isar t r ia y trastornos motores (mioclonías, t e m b l o r , ataxia , h iper re f l ex ia y espast ic idad) . Pueden presentar t rastornos de m e m o r i a y atención, pero no una demenc i a impo r t an t e . A veces hay a luc inac iones comp le j a s .
7 9

M a n u a l C T O d e M e d i c i n a y Cirugía , 8 . a ed i c i ón
r
Casos clínicos representativos
Pac i en te de 6 0 años , s in n ingún an teceden te de interés, que en un per íodo de seis meses desar ro l l a un c u a d r o de intenso de te r io ro cogni t i vo . En la exp lorac ión des ta c a , además de la ex i s tenc ia de un s índrome r ígido-acinético, u n a a tax ia de la m a r c h a y mioc lon ías . Ante este c u a d r o , ¿qué diagnóst ico, entre los s iguientes, e s t amos ob l i gados a p lan tearnos en p r imer lugar?
1) U n a leucoencefa lopat ía m u l t i f o c a l p r o g r e s i v a .
2) U n a p a n e n c e f a l i t i s e sc l e rosan te s u b a g u d a . 3) U n a f o r m a esporádica d e Creu tz fe ld t- J akob . 4 ) U n a a t r o f i a multisistémica. 5) U n a encefa lopat ía d e W e r n i c k e .
M I R 0 2 - 0 3 , 5 2 ; RC 5
8 0

Neurología y neurocirugía
10. ENFERMEDADES NUTRICIONALES Y METABÓLICAS DEL SISTEMA NERVIOSO
Orientación
MIR r
Aspectos esenciales L.
T e m a d e i m p o r t a n c i a i n t e r m e d i a en el M IR . Es r e c o m e n d a b l e estudiar b i e n la encefalopatía de W e r n i c k e y la degeneración s u b a g u d a c o m b i n a d a d e la médula. El resto es bastante p r e s c i n d i b l e .
pj~j La encefalopatía de W e r n i c k e aparece p r i n c i p a l m e n t e en alcohólicos, por hiperémesis y po r malnutrición.
[ Y ] La encefalopatía de W e r n i c k e se caracter iza por la tríada de o f ta lmopares ia (sobre t o d o b i la te ra l y asimétrica del V I PC), ataxia y síndrome con fus iona l q u e aparecen y desaparecen c o n el t r a t am ien to en este o r d e n . El síndrome con fus iona l puede e v o l u c i o n a r a un síndrome amnésico anterógrado: síndrome de Korsakoff .
El t r a t am ien to de la encefalopatía de W e r n i c k e es v i t a m i n a B r L3J
0 La degeneración subaguda c o m b i n a d a de la médula se p r o d u c e por un déficit de v i t a m i n a B 1 2, y se d i agnos t ica m e d i a n t e la determinación de los niveles de la m i s m a y por el test de Sch i l l i ng .
La degeneración subaguda c o m b i n a d a de la médula cursa c o n pérdida de sens ib i l idad v ib ra to r i a y p r o p i o cept i va , y clínica de p r imera m o t o n e u r o n a por afectación de los co rdones poster iores y laterales medu lares , respect i vamente . Recuérdese también q u e puede p r o d u c i r anemia megaloblástica. Su t r a t am ien to es v i t a m i na B ] 2 parentera l .
La pelagra se p r o d u c e por déficit de n iac ina . Es la en f e rmedad de las 3 " D " : dermat i t i s , d iarrea y demenc i a .
["y"! Las áreas cerebrales más sensibles a la anox i a son los gang l ios básales, ce rebe lo , h i p o c a m p o y regiones f rontera pa r i e toocc ip i t a l es .
10.1. Enfermedades neurológicas debidas a déficit nutricionales
Encefalopatía de Wernicke
Etiología y patogenia
Aparece en pacientes alcohólicos y malnutr idos (por hiperémesis, cáncer, inanición, etc.) deb ido a un déficit de t i amina o v i tamina B1 . El déficit de t i amina produce un deter ioro en el metabo l i smo cerebral de la glucosa y se ha postu lado c o m o mecan ismo patogénico la neuro tox ic idad mediada por g lutamato.
Anatomía patológica
Las lesiones se loca l izan preferentemente a nivel per iventr icu lar y son de carácter simétrico: se afecta tálamo,
hipotálamo, cuerpos mamilares, sustancia gris per iacueducta l mesencefálica, suelo del cuarto ventrículo y ver-
mis cerebeloso. Los núcleos ocu lomotores y vestibulares se afectan en menor grado, pero casi invar iab lemente.
CQ P r egun tas
• M I R 0 9 - 1 0 , 53 - M I R 0 7 - 0 8 , 5 4 , 6 5 • M I R 01 -02 , 5 2 , 55 •MIR 00-01F , 70 -MIR 9 9 - 0 0 , 4 3 , 1 9 8 • M I R 99-00F , 6 4 , 6 5 • M I R 98-99F , 6 4 , 6 8 , 73 - M I R 9 7 - 9 8 , 4 3 , 4 9
Clínica
Se caracteriza por la tríada de ofta lmoparesia , ataxia y síndrome confus ional (MIR 07-08, 54 ; MIR 07-08, 65; MIR 99-00, 4 3 ; MIR 99-00F, 65) .
R E C U E R D A En la tríada clásica (o f ta lmopares ia , a tax ia y síndrome con fus iona l ) , las mani fes tac iones aparecen y desaparecen en el m i s m o o r d e n .
81

Manual CTO de Medicina y Cirugía, 8.a edición
• Síntomas oculares. Si no están presentes, hay que dudar del d i ag nóstico. La afectación más f recuente es una paresia b i la tera l y as i métrica del V I par. Otras mani festac iones son n is tagmo, parálisis de la mi rada con jugada (más f recuente en el p l ano ho r i zon ta l que en el vert ica l ) o a l terac iones en la convergenc ia . La admin i s t r a ción parenteral de t i am ina p roduce una rápida mejoría de la m o t i l i d a d ocu lar , aunque puede persistir el n is tagmo. La no respuesta a la administración de t i am ina nos debe hacer dudar del diagnóst i co .
• Ataxia. Afecta con preferencia a la bipedestación y a la marcha . La t i am ina también me jo ra esta clínica, aunque el pac iente puede quedar con una base de sustentación amp l i a y una marcha inestab le . La ataxia de los m iembros es inf recuente , y si aparece, es más grave en los m iembros infer iores.
• Trastorno de funciones superiores. Aparece en casi todos los pa cientes, y la f o r m a más común de presentación es c o m o un cuadro con fus iona l carac ter izado por inatención, ind i fe renc ia , desor ien tación y escaso lenguaje espontáneo. En un 2 0 % de pacientes puede haber un cuadro de agitación s imi lar al d e l i r i u m tremens. Es m u y raro el de ter io ro del n ive l de consc ienc ia . En pac ientes somet idos a nutrición parente ra l , la h ipomagnese-mia puede dar un c u a d r o c l ínico s im i l a r a la encefalopatía de W e r n i c k e .
Pruebas complementarias
El LCR es norma l o muestra una mínima elevación de proteínas. En el EEG se encuentra una ac t i v idad lent i f i cada de fo rma difusa. Las p rue bas de función vest ibular están alteradas en todos los casos, b i latera l y simétricamente en fases in ic ia les . En casos no tratados, hay una e le vación del p i ruva to sérico con una disminución de la transcetolasa.
Evolución
A u n q u e todos estos síntomas pueden aparecer simultáneamente, lo hab i tua l es que la o f t a lmop l e j i a y/o la ataxia precedan al cuad ro c o n fus iona l en días o semanas.
Tras instaurar el t r a tamien to vitamínico, la secuencia de recupera ción es clásica. Pr imero mejora la o f ta lmopares ia y, poster iormente , la ataxia . Es pos ib le que queden c o m o secuelas un nistagmus h o r i zon ta l , aumento de la base de sustentación y marcha inestable.
Lo último que me jo ra es el cuadro con fus iona l , y según lo hace, puede aparecer un trastorno amnésico con incapac idad para retener nueva información. Es el síndrome de Korsakoff, c u y o pronóstico de recuperación varía; la mayor parte mant iene déficit de m e m o r i a va riables, y menos del 2 0 % presenta recuperación comp le t a .
Un 1 5 - 2 0 % de los pacientes con en fe rmedad de W e r n i c k e muere , genera lmente d e b i d o a infecc iones intercurrentes o insuf ic ienc ia he pática.
Q R E C U E R D A Antes de admin i s t ra r so luc iones g lucosadas a un pac iente alcohól ico, hay q u e admin i s t r a r t i a m i n a . De no hacer lo , existe riesgo de desenca denar o agravar una encefalopatía de W e r n i c k e .
Se debe tener especial cu idado a la hora de administrar soluciones g l u cosadas intravenosas a pacientes alcohólicos, embarazadas y otros con predisposición para estar desnutr idos, dado que pueden desencadenar una encefalopatía de Wern i cke o agravar notablemente la misma en sus fases iniciales (por consumo de las reservas de v i tamina B). Es pre ciso, por tanto, administrar t i amina antes de las soluciones glucosadas.
Degeneración subaguda combinada de la médula
Se produce por una def ic ienc ia de v i tamina B l 2, generalmente secundaria a una incapac idad para absorber la v i tamina de la dieta deb ido a la ausencia de factor intrínseco en la secreción gástrica (gastritis crónica atrófica). Raramente hay sintomatología neurológica en pacientes con enfermedad de íleon termina l (enfermedad de Crohn , l in foma o tras resección quirúrgica). Desde el punto de vista hematológico, se m a n i fiesta c o m o una anemia megaloblástica.
Clínica
La sintomatología neurológica está presente en la mayoría de los pa cientes con déficit de esta v i tamina y t iende a ser simétrica. Los síntomas iniciales son parestesias distales en los miembros . Posteriormente aparece la clínica secundaria a afectación de los cordones posteriores (el s igno más característico es la pérdida de la sensibi l idad v ibrator ia , sobre todo , en miembros inferiores) y de los cordones laterales medu la res (paraparesia espástica en miembros inferiores con signos de pr imera motoneurona) .
La marcha in i c ia lmente es atáxica y, poster iormente, se asocia una espast ic idad (MIR 99-OOF, 64).
Los síntomas mentales son frecuentes, con i r r i t ab i l idad , apatía, somno lencia y, a veces, cuadro confus ional y psicosis depresiva. También se ha descr i to neuropatía óptica, con deter ioro de agudeza visual y escotomas cecocentrales. La demenc ia es una manifestación rara.
El diagnóstico se conf i rma mediante la determinación de niveles de v i tamina B 1 2 y t e s t d e Schi l l ing (MIR 01-02, 55; MIR 98-99F, 73).
Q R E C U E R D A Para el diagnóstico de déficit de B 1 2, no es necesar io q u e existan a l tera c iones en sangre periférica.
Tratamiento
Se debe instaurar con carácter i nmed ia to y consiste en la adm in i s tración de v i t am ina B 1 , i n i c i a lmen te por vía parenteral y poster ior mente por vía oral (MIR 09-10 , 5 3 ; MIR 98-99F, 6 8 ; MIR 97-98, 49) . La administración precoz puede evitar el poster ior desar ro l lo de un síndrome amnésico.
Tratamiento
Administración de vitamina B | 2 parenteral (intramuscular). La recuperación puede ser completa, si el tratamiento se administra pocas semanas después del in ic io de los síntomas. En caso contrario, la recuperación será parcial e incompleta. Por tanto, el factor que más condic iona el grado de respuesta al tratamiento es la duración de los síntomas neurológicos.
82

Neurología y neurocirugía
Pelagra
Es una enfermedad nutr ic ional produc ida por def ic iencia de niacina o ácido nicotínico. Sus manifestaciones sistémicas son dermatit is, diarrea y demencia . La dermatit is es bilateral, simétrica y aparece en zonas expuestas a la luz solar deb ido a fotosensibi l idad.
^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^
Q R E C U E R D A El déficit de niacina produce pelagra, la enfermedad de las 3 "D " : diarrea, demencia y dermatitis.
Las manifestaciones neurológicas de la pelagra son diversas, p redomi nando la clínica de encefalopatía; otras manifestaciones son la mielopa-tía y neuropatía periférica: • Encefalopatía. Los síntomas iniciales pueden ser confundidos con un
trastorno psiquiátrico: insomnio, fatiga, ansiedad, i r r i tabi l idad, depresión, etc. Posteriormente aparece un enlentec imiento de los procesos mentales y trastornos de la memor ia .
• Mielopatía. Hay afectación de cordones posteriores y laterales, preferentemente de los primeros, con paraparesia atáxica y espástica. La neuropatía que puede acompañar a la pelagra no dif iere de otras neuropatías nutricionales como la alcohólica, la secundaria a déficit de p i r idoxina , v i tamina B12, ácido fólico, etc.
Síndrome de Strachan
Es un síndrome de probable or igen nutr ic iona l , pero en el que no se ha descrito el déficit vitamínico responsable. In ic ia lmente conoc ido como "neurit is jamaicana" , afecta esencialmente a los nervios periféricos y nervios ópticos. Cursa con neuropatía sensitiva (parestesias dolorosas de los pies, pérdida de la sensibil idad profunda y ataxia), deterioro de agudeza visual que puede evoluc ionar a la ceguera y dermatitis orogenita l .
10.2. Enfermedades metabólicas secundarias nabia43)
MIOCLONÍAS EDEMA CEREBRAL
Anóxico-lsquémica Hipercápnica
Hepática adquirida Hepática aguda
Demencia dialítica Síndrome de Reye
Urémlca aguda Síndrome de desequilibrio
Tabla 43. Clínica diferencial de encefalopatías metabólicas
Encefalopatía anoxicoisquémica
Etiología
Puede ser secundaria a cualquier causa que cond ic ione un déficit de oxigenación o perfusión cerebral (entre otras, el infarto de miocard io , parada cardiorrespiratoria, hipotensión o shock secundarios a hemorragia periférica, insuficiencia respiratoria grave por debi l idad de la musculatura respiratoria, intoxicación por monóxido de carbono, etc.) (Figura 57).
Figura 57. Encepalopatía anóxica por parada cardiorrespiratoria
Clínica
Si la anox ia persiste más de 3-5 minutos , se establece un daño cerebral irreversible. Las áreas cerebrales más sensibles a la anox ia son los gangl ios básales, cerebelo, h i p o c a m p o y regiones frontera parietooc-cipi ta les. Esto cond i c i ona las posibles secuelas de una encefalopatía anóxica: 1) clínica ex t rap i ramida l , f undamenta lmente mioclonías; 2) ataxia cerebelosa; 3) síndromes amnésicos t ipo Korsakoff, y 4) agnosia v isual .
En casos de anoxia pro longada, la afectación difusa de la corteza cerebral conducirá a un cuadro de demenc ia o, en su fo rma más grave, a un estado vegetativo persistente (MIR 00-01 F, 70).
Los pacientes que muestran una función troncoencefálica respetada (respuestas pupi lares normales, respuestas oculocefálicas y oculovest i-bulares conservadas) t ienen un mejor pronóstico de recuperación.
En los momentos siguientes a la recuperación de la función cardiorrespirator ia y la restauración de la perfusión cerebral , pueden aparecer crisis generalizadas y mioclonías mult i foca les .
Tras una mejoría in ic ia l (que puede ser completa ) , y pasados varios días, se puede produc i r un empeoramien to con apatía, confusión, i r r i t ab i l idad y agitación, que a veces evo luc iona a r igidez difusa, espast i c idad , coma y muerte ; es la denominada encefalopatía postanóxica retardada. El estudio anatomopatológico demuestra desmielinización difusa.
Tratamiento
Se basa en la restauración de la función cardiorrespirator ia de forma inmediata .
Encefalopatía hipercápnica
Etiología
La f ibrosis pu lmona r y el enf isema crónico son las causas más f re cuentes.
83

Manual CTO de Medicina y Cirugía, 8.a edición
Clínica
El C 0 2 es un vasodi latador, y gran parte de la clínica se manif iesta c o m o un cuadro de hipertensión intracraneal : in i c ia lmente somno len cia y después cefalea general izada, pap i ledema, confusión, estupor y coma. Conl leva temblor rápido de acción, mioclonías y asterixis. Puede p roduc i r edema cerebral .
El LCR se encuentra a presión elevada, y en el EEG hay en lentec imiento di fuso con ondas delta (8) y theta (9).
A di ferencia de la encefalopatía anóxica, la hipercápnica rara vez p ro duce un coma pro longado y no suele produc i r un daño cerebral i rreversible.
Tratamiento
Ventilación forzada con respirador a presión posit iva intermitente. Se debe actuar enérgicamente sobre la insuf ic iencia cardíaca, si ésta existe (cor pulmonale).
Encefalopatía hipoglucémica
Etiología
Las causas más frecuentes son: sobredosis de insul ina o antidiabéticos orales, insul inomas y otros tumores retroperitoneales, intoxicación por etanol y síndrome de Reye.
Clínica
Suele c o m e n z a r c u a n d o los niveles de g lucosa están a l r ededor de 30 mg/d l . I n i c i a lmen te hay una h ipe r a c t i v i dad adrenérgica, con ce fa lea, pa lp i t ac iones , sudoración, t e m b l o r y ans iedad. Si la g l u c e m i a sigue d i s m i n u y e n d o , aparece s o m n o l e n c i a , confusión, y puede evo l uc iona r al c o m a , con posturas de descerebración. En esta fase pue den aparecer cr is is y mioclonías. Con g lucemias de 10 mg/d l , hay c o m a p r o f u n d o , pup i l as f i jas, b rad i ca rd i a e hipotonía genera l i zada (fase bu lba r ) .
La distribución de las lesiones es s imi lar a la que se produce en la anoxia , pero en la h ipog lucemia no se afecta el córtex cerebeloso. A diferencia de la anoxia , se requieren periodos más prolongados de h ipog lucemia (60-90 minutos) para que se produzcan lesiones irreversibles. Sin embargo, la recuperación y las secuelas son muy parecidas. Las h ipoglucemias graves recidivantes pueden conduc i r a un estado de demenc ia .
Aunque los trastornos metabólicos generalmente no producen datos de foca l idad neurológica, la h ipog lucemia grave puede debutar con hemiparesia y otros signos neurológicos focales.
Tratamiento
Consiste en la corrección de la h ipog lucemia con soluciones glucosa-das. Si la glucosa se administra prev iamente a la fase bulbar , la recuperación suele ser comple ta . Una vez en fase bulbar , y más si ésta es pro longada, la recuperación será parc ia l .
Encefalopatía hepática. Degeneración hepatocerebral adquirida
Etiología
Se p roduce en situaciones de insuf ic ienc ia hepática con cor toc i rcu i tos entre la circulación portal y la circulación genera l .
El síndrome de Reye es una fo rma especial de encefalopatía hepática no ictérica, que afecta a niños en el contexto de infecc iones virales (generalmente var icela) , y que se puede desencadenar por la a d m i nistración de sal ic i latos. Estos pacientes presentan encefalopatía, síntomas de hipertensión intracraneal , h ipog lucemia , h ipe ramon iemia y elevación de transaminasas con b i l i r r ub ina no rma l .
Clínica
Es un cuadro de evolución subaguda en días o semanas que se in ic ia c o m o un síndrome confus iona l , con aumento o disminución de la ac t i v idad ps i comotora y asterixis, y que evo luc iona hacia un deter ioro progresivo del n ive l de consc ienc ia . Con f recuenc ia puede conduc i r a la muerte del paciente. Hay edema cerebral d i fuso.
En el EEG hay paroxismos de ondas 8 trifásicas que p redominan en las regiones frontales. En relación con la elevación del amoníaco plasmático, en las formas graves puede encontrarse un aumento de los niveles de g lu tamato en LCR.
Si el trastorno metabólico persiste durante meses o años, se puede establecer un cuadro de demenc ia y clínica ext rap i ramida l con temb lo r , disartr ia, ataxia y corea; es la degeneración hepatocerebral adqu i r ida o encefalopatía hepática crónica (MIR 97-98, 43) .
A nivel anatomopatológico, tanto en la fo rma aguda c o m o en la crónica, se observa un inc remento en el número y tamaño de los astrocitos protoplásmicos (astrocitos t i po II de A lzhe imer ) . En la fo rma crónica, se afectan además las neuronas de manera característica, rec ib iendo entonces la denominación de células de Opa l sk i . Esta combinación de glía t i po II de A l zhe ime r y células de Opa lsk i es también el sustrato anatomopatológico de la degeneración hepatocerebral fami l i a r (enfermedad de W i l son ) , que se trata en la Sección de Digestivo y cirugía general.
Tratamiento
Debe reducirse la cant idad de proteínas de la dieta. Se admin is t ran lactulosa y antibióticos orales para e l im ina r los mic roorgan ismos i n testinales productores de ureasa. El trasplante hepático ha s ido ef icaz en a lgunos pacientes.
Encefalopatía urémica
• Encefalopatía urémica aguda. In i c ia lmente hay apatía, inatención, fatiga e i r r i t ab i l i dad , y luego evo luc iona hacia un deter ioro progre sivo del nivel de consc ienc ia , que comprende a luc inac iones , m i o clonías, asterixis y convuls iones . No hay edema cerebral asociado. La corrección de la función renal restaura la función neurológica,
8 4

Neurología y neurocirugía
pero en aquel los casos con daño renal i rreversible y progres ivo, el t ra tamiento debe ser la diálisis o el trasplante renal . Síndrome de desequilibrio. Aparece tres o cuatro horas después de real izar hemodiálisis o diálisis per i tonea l , y es secundar io a un paso excesivo de agua desde el compa r t imen to plasmático al sistema nervioso central (puede p roduc i r edema cerebral ) . El síntoma más frecuente es la cefalea ( 7 0 % ) , que puede acompañarse de náuseas, i r r i t ab i l i dad , convu ls iones y , en ocasiones, de un síndrome de hipertensión intracraneal .
Encefalopatía dialítica o demencia dialítica. Es ac tua lmente una complicación poco f recuente de la diálisis crónica, y se cons ide ra la manifestación de una intoxicación crónica por a l u m i n i o , sin descartarse la participación de otros e lementos traza. Comienza con disartr ia, afasia de p r e d o m i n i o motor , mioclonías, crisis foca les o general izadas, trastornos de la personal idad y deter ioro in te lectua l . A l p r i n c i p i o , estos episodios aparecen de fo rma transi tor ia después de la diálisis, pero poster iormente se instauran de fo rma permanente .
r
Casos clínicos representativos
H o m b r e de 59 años , ex bebedor y gas t r ec tomizado por hemor rag ia d igest iva hace 15 años . N o sigue t ra tamiento a lguno . C o n s u l t a por un c u a d r o ins id ioso de d i f i cu l tad pa ra c am ina r , que e m p e o r a en la o s cu r i dad . En la exp lorac ión , se objet iva una a tax ia de la m a r c h a , c a y e n d o a l sue lo en la p rueba de Romberg , respuestas p lantares ex tensoras y conservac ión de la sens ib i l idad algésica, es tando abo l idas la v ibra tor ia y pos i c i ona l . Seña le , en t re las s iguientes, la a f i rmac ión c o r r e c t a :
1) El diagnóst ico es degenerac ión c e r ebe l o sa a l cohó l i ca .
2) Se t ra ta d e una degenerac ión c o m b i n a d a s u b a g u d a d e la médula e s p i n a l . 3) H a y q u e descar ta r una lesión c e n t r o m e d u l a r . 4 ) El diagnóst ico es esc le ros is múlt iple d e f o r m a p r i m a r i a p rog re s i v a . 5) El t r a t a m i e n t o d e e lecc ión es t i a m i n a i n t r a venosa .
M I R 99-00F , 6 4 ; RC: 2
85

Neurología y neurocirugía
11. NEUROPATÍAS
Orientación
MIR r
Aspectos esenciales L.
T e m a d e i m p o r t a n c i a i n t e r m e d i a en el M IR . H a y q u e centrarse en es tud ia r la neuropatía diabética, la neuropatía po r V I H y el S índrome d e Guil lain-Barré. Es r e c o m e n d a b l e revisar también las c o n s i d e r a c i o n e s genera les , p o r q u e p u e d e n fac i l i t a r la compresión de l t e m a . El resto es p r e s c i n d i b l e .
[~¡~| Las polineuropatías afectan a múltiples t roncos nerv iosos, las m o n o n e u r i t i s múltiples a t roncos nerv iosos no con t iguos , y las mononeuropatías son afectac iones foca les de un único t r o n c o nerv ioso .
[~2~] En el síndrome de Guillain-Barré, 2/3 de los pac ientes presentan el an tecedente de una infección respi rator ia o gastro intest ina l p rev ia . Cursa c o n una tetraparesia flácida y arrefléxica, simétrica y ascendente . Puede asociar parálisis fac ia l b i la tera l en la m i t a d de los casos y síntomas autonómicos.
("3"] En el síndrome de Guillain-Barré, es típico un LCR c o n disociación albuminocitológica. El t r a t amien to es el de sopor te de las func iones card ior resp i ra tor ias . Puede usarse también la plasmaféresis o las i n m u n o g l o b u l i -nas intravenosas. Los esferoides no son útiles.
("4] La neuropatía diabética puede ser simétrica (sensitiva d is ta l , autonómica, do lo rosa aguda y la am io t ro f i a d i a bética) o asimétrica (craneales, s iendo el par craneal más afectado el tercero , por a t r apamien to y de t ronco ) .
Qf) La f o r m a más f recuente de polineuropatía diabética es la sensit iva d is ta l .
0
0
La causa más f recuente de disautonomía es la neuropatía diabética.
En la infección por V I H pueden aparecer las s iguientes neuropatías: simétrica distal y m o n o n e u r i t i s múltiple (en fases avanzadas) , polineuropatías desmie l in izan tes agudas o crónicas (en fases precoces de la infección) o po l i r r ad i cu l i t i s ( lo más f recuente es q u e sea por C M V ) .
La f o r m a más f recuente de neuropatía en pacientes c o n S IDA es la simétrica d is ta l .
11.1. Consideraciones generales
Se ut i l iza el término general de neuropatía periférica para referirse a aquel los trastornos de los nervios periféricos, sea cual sea su causa (Figura 58).
DESMIELINIZANTE • Disminución de la velocidad de condución
• Aumento de las latencias distales
• Si no homogénea: - Dispersión - Bloqueos
• A m p l i t u d normal
8 6

Neurología y neurocirugía |
Se habla de polineuropatía para referirse, en general , a procesos de instauración gradual , que afectan a múltiples troncos nerviosos y que se caracter izan por ser simétricos y general izados, con afectación preferentemente distal .
La mononeuritis múltiple es una afectación simultánea o consecut iva de troncos nerviosos no cont iguos, con evolución de días o años. A veces presentan carácter conf luente , con difícil diagnóstico di ferencia l con las polineuropatías.
Las mononeuropatías son afectaciones focales de un único t ronco nervioso. Son consideradas con más detalle en la Sección de Traumatología. • Las radiculopatías son trastornos que afectan a las raíces nerviosas.
El término polirradiculopatía se refiere a la afectación de múltiples raíces nerviosas de forma consecut iva.
• Las plexopatías son trastornos que afectan al p lexo nervioso, de or igen diverso, y pueden ser traumáticas, compresivas o d i s inmu-nitarias.
Clínica
Trastornos sensitivos. Suele ser la pr imera manifestación clínica. Hay disestesias t ipo hormigueo , quemazón o p inchazo que in ic ia lmente aparecen a nivel distal en los miembros y con carácter simétrico, aun que en fases iniciales puede ser asimétrico. Posteriormente hay una extensión centrípeta con distribución de los déficit en guante o calcetín. La afectación del componen te p rop iocept i vo determinará una inestab i l idad en la marcha superior a la esperada por los déficit motores (ataxia sensitiva).
Trastornos motores. En las enfermedades del sistema nerv ioso pe riférico, el pac iente presentará d e b i l i d a d f l acc ida de los m i embros afectados, con h ipor re f l ex i a o arref lex ia . El ref le jo cutáneo plantar es f lexor . En las polineuropatías, i n i c i a lmen te , se ob je t i va pérdida de los reflejos aquíleos y d e b i l i d a d para la dorsiflexión del pie y, poste r i o rmen te , desaparece el ref le jo ro tu l i ano y se hace más ev idente el p ie péndulo.
La presencia de atrofia muscular depende en gran grado del t ipo de neuropatía, s iendo m u y importante en las afectaciones axonales, y poco habitual en las enfermedades desmiel in izantes.
Trastornos autonómicos. Los síntomas autonómicos inc luyen h ipo ten sión ortostática, retención ur inar ia , estreñimiento, diarrea, impotenc ia , etcétera.
Diagnóstico
Se deben valorar antecedentes de procesos virales previos, enfermedades sistémicas (diabetes mel l i tus, uremia, por f i r ia , déficit v i ta mínicos de B,, B6, fólico, ácido pantoténico, B l 2, hepatopatía crónica, ami lo idos is , h ipo t i ro id i smo, neumopatía crónica, acromegal ia , malabsorción, ca rc inoma, l in foma, po l i c i t emia vera, m ie loma , ga-mmapatía monoc l ona l , etc.), consumo de fármacos (amiodarona, c isplat ino, dapsona, h idra lac ina, isoniac ida, met ron idazo l , di feni-Ihidantoína, p i r idox ina , t a l i domida , v incr is t ina, nitrofurantoína), exposición a tóxicos (disolventes, pesticidas o metales pesados), ingesta de alcohol (MIR 99-00, 11). Evolución (Figura 59).
EVOLUCION Lentamente progresivas
Predominio motor: genéticas
Sensitivo-motora: • DM • Paraproteinémica
Subagudas o crónicas: tóxico-metabólicas Suelen ser: simétricas y distales en MMII
sensitivo-motoras axonales
Agudas: son tóxico-metabólicas, pero agudas: uremia, porfirias y arsénico
Figura 59. Clasificación de las polineuropatías por su evolución
Distribución (véase Figura 60) .
DISTRIBUCIÓN
- Generalmente simétrica, distal con inicio en MMII
-Excepcione: Inicio en • Plomo (mano péndula bilateral) MMSS: • Tangier (amígdalas hipertróficas
anaranjadas) • Porfirias (dolor abdominal , crisis
comiciales y psicosis)
Inicio proximal: . Diabetes • Porfirias • Talidomida
Figura 60. Clasificación de las polineuropatías por su distribución
Palpación de los troncos nerviosos. El signo de Tinnel (sensación eléctrica con la percusión del nervio) es característico de las neuropatías compresivas. El engrosamiento fusiforme del nervio se puede objet ivar en la neuritis por lepra y en la polineuropatía ami lo ide . Puede haber engrosamiento un i forme en algunas neuropatías genéticas.
R E C U E R D A Las polineuropatías desmiel inizantes cursan con una velocidad de con ducción enlentecida.
Neurofisiología. Los estudios de ve loc idad de conducción son f u n damentales para di ferenciar entre procesos axonales y desmie l i n i zantes. Los trastornos axonales se caracter izan por una pérdida de la amp l i t ud del potenc ia l de acción, mientras que las desmi l in izantes presentan aumento de latencias, disminución de las veloc idades de conducción, pud i endo aparecer fenómenos de b loqueo de la c o n ducción y de dispersión del potencia l nervioso (Figura 61).
NEUROFISIOLOGÍA Desmielinizante -
• Guillain-Barré • Genéticas: Charcot-Marie-ti| Déjerine-Sottas Refsum
Disminución de la velocidad de conducción Aumento de latencias distales
Si desmielinización no homogénea
Dispersión del potencial Bloqueos de conducción
Axonal Velocidad de conducción normal Disminución de la ampl i tud del potencial
• Metabólicas y tóxicas (excepto las incluidas en mixtas)
• Charcot-Marie-Tooth t ipo II
- Mixtas • Diabética • Asociada a l infoma • Asociada a mieloma
Figura 61. Clasificación de las polineuropatías según el estudio neurofisiológico
8 7

Manual CTO de Medicina y Cirugía, 8 . a edición
Los dos últimos se deben a una afectación desmie l in izante var iable dentro del m i smo nervio, y dist ingue esta ent idad de las neuropatías desmiel in izantes hereditarias, en las que todas las fibras se afectan de forma homogénea y no hay b loqueos ni dispersión.
• Biopsia de nervio. Se ut i l iza generalmente el nerv io sural. Se indica en casos de mononeur i t i s múltiple y en el diagnóstico de algunos trastornos infanti les determinados genéticamente ( leucodistrof ia metacromática, enfermedad de Krabbe, etc.) (Tabla 44) .
Polineuropatías asociadas a enfermedades sistémicas Polineuropatías asociadas a fármacos y tóxicos Polineuropatías desmielinizantes inflamatorias
- Aguda o Síndrome de Guillain-Barré - Crónica
Polineuropatías hereditarias con defecto metabóllco conocido Polineuropatías determinadas genéticamente (neuropatías sensitivomotoras hereditarias) Neuropatías asociadas a infección por VIH
Tabla 44. Clasificación de las polineuropatías por su etiología
Clínica
Cursa con un cuadro de tetraparesia flácida y arrefléxica con escasos síntomas sensitivos. No suele haber afectación esfinteriana (MIR 04-05, 60). En la mayoría de los casos, la deb i l i dad se in ic ia en los miembros inferiores y asciende progresivamente, para afectar a la to ta l idad cor pora l . La progresión de la paresia es muy var iable, y en casos graves se puede llegar a la plej ia comple ta con incapac idad para respirar, por deb i l idad de la musculatura diafragmática o de los intercostales, hablar o deglut i r , por deb i l idad de la musculatura faríngea. La afectación es simétrica y la atrofia infrecuente (MIR 99-00F, 68).
Puede haber paresia facial bi lateral en el 5 0 % de los casos; otros pares craneales que pueden afectarse son los que inervan la lengua y la musculatura deglutor ia , pero no afecta a los ocu lomotores .
A nivel sensitivo, puede haber parestesias distales especia lmente al i n i c io del cuadro, pero no existe un déficit de sensibi l idad marcado. Es frecuente la presencia de do lor en la zona lumbar o en las ext remida des inferiores al in i c io de la clínica.
11.2. Síndrome de Guillain-Barré Los síntomas autonómicos inc luyen taquicard ia , hipotensión postural , hipertensión y síntomas vasomotores. La Tabla 45 muestra los hal lazgos que deben hacer dudar o descartar el diagnóstico.
Se trata de una polirradiculoneuropatía desmie l in izante aguda de o r i gen inmunológico y que afecta preferentemente a adultos jóvenes va rones.
n R E C U E R D A C. jejuni se trata con eritromicina y puede producir cuadros clínica e histológicamente similares a la enfermedad Inflamatoria intestinal.
En más de 2/3 partes de los casos hay antecedente de infección viral respiratoria o gastrointest inal . Los virus más f recuentemente impl i cados son los del grupo herpes (c i tomegalov i rus, virus de Epstein-Barr). Más recientemente, el Campylobacter jejuni ha sido descrito en pacientes con Guillain-Barré y antecedente de gastroenterit is. También se ha asoc iado con el antecedente de procedimientos quirúrgicos, l infomas y lupus er i tematoso sistémico.
Patogenia
Es au to inmun i ta r i a . La desmielinización se produce por un dob le me can ismo: mediada por l infoci tos y por ant icuerpos circulantes. Recientemente se ha descr i to la presencia de ant icuerpos antigangliósido, c o m o el an t i -GM1 , en este síndrome, si b ien su papel etiopatogénico no es del todo conoc ido .
Anatomía patológica
Se caracteriza por la presencia de inflamación, desmielinización y de generación axona l , restringida al sistema nervioso periférico. La desmielinización es segmentaria y mu l t i foca l , y afecta con mayor select iv i dad a nivel p rox ima l en las raíces nerviosas. Puede haber degeneración axonal secundaria al proceso de desmielinización en las zonas de más intensa inflamación.
Existen múltiples variantes del Síndrome de Guillain-Barré clásico, s iendo el síndrome de Miller-Fisher el más hab i tua l . Se trata de una variante que imp l i ca ataxia, arref lexia y ofta lmoparesia, con posibles alteraciones pupi lares y escasa deb i l idad de miembros . Se ha demos trado una asociación de este síndrome con los ant icuerpos antigangliósido GQ-1b.
Recientemente se han descrito formas axonales, de patogenia poco co noc ida, en las que predomina la destrucción del axón sobre la m i e l i na. Se denominan A M A N (neuropatía axonal motora aguda) y A M S A N (neuropatía axonal sensi t ivomotora aguda).
Curso
Es característica la rápida progresión de la deb i l i dad , que alcanza su máximo en cuatro semanas en el 9 0 % de los casos. La recuperación suele comenzar en 2-4 semanas después de cesar la progresión, y pue de durar meses.
HALLAZGOS QUE PONEN EN DUDA EL D IAGNÓSTICO
• Debilidad asimétrica de forma marcada y persistente
• Nivel sensorial franco • Disfuncíón intestinal o vesical al inicio • Disfunción intestinal o vesical persistente • Pleocitosis mononuclear > a 50 cel/mm 3
• Pleocitosis de polimorfonucleares
H A L L A Z G O S QUE DESCARTAN EL D IAGNÓSTICO
• Síndrome sensitivo puro • Historia reciente de contactos con solventes
(hexacarbonos) • Metabolismo anormal de las porfirinas • Infección diftérica reciente • Evidencia de intoxicación por plomo • Diagnóstico definitivo de:
- Poliomielitis - Botullsmo - Neuropatía tóxica (dapsona, organofosforados)
Tabla 45. Síntomas y signos no compatibles con síndrome de Guillain-Barré
88

Neurología y neurocirugía
Pronóstico
Aunque la mayoría de los pacientes t iene una excelente recuperación func iona l , hay un 5 % de mor ta l idad y en el 5 0 % queda alguna secuela. Son factores predict ivos de pobre pronóstico en la recuperación la edad avanzada, el in i c io rápido, la necesidad de ventilación art i f ic ia l y, fundamenta lmente , el componen te axonal , va lorado en función de la amp l i t ud de los potenciales de acción.
Parálisis hipopotasémica Mielitis aguda Botulismo Poliomielitis Porfiria Difteria Neuropatías tóxicas (dapsona, talio, nitrofurantoína) Neuroborreliosis o enfermedad de Lyme
Tabla 46. Diagnóstico diferencial del síndrome de Guillain-Barré
Pruebas complementarias Tratamiento
R E C U E R D A En la miastenia gravis, se emplea la electromiografía; en el Gui l la in-Barré, la electroneurografía.
LCR. Es típica la disociación albuminocitológica (proteínas altas sin células). Las proteínas son generalmente normales durante los pr imeros días de la enfermedad, elevándose consistentemente tras la pr imera semana y manteniéndose así durante varios meses, incluso después de la recuperación clínica. El contaje de células mononucleares en LCR es menor de 10 células/ m m 3 ; la presencia de una pleocitosis mayor es espec ia lmente común en casos de síndrome de Guillain-Barré asociados a V IH (MIR 06-07, 52) .
R E C U E R D A Si el LCR presenta pleocitosis importante, hay que pensar en un síndrome de Guillain-Barré asociado a la infección por V IH .
Estudios neurofisiológicos. En las fases inic ia les, las ve loc idades de conducción motoras distales suelen ser normales y es de mayor valor la abolición de la onda F, que va lora la conducción motora p r o x i m a l , s iendo el pr imer s igno diagnóstico (MIR 07-08, 57 ; MIR 01-02, 59) .
En el 8 0 % de los pacientes existe ralentización en la ve loc idad de conducción y aumento de las latencias distales (desmielinización). No todos los nervios quedan afectados, ya que la desmielinización es par-cheada.
Consiste en el soporte de las func iones cardiorrespiratorias, con pre vención de las infecciones intercurrentes (MIR 99-00, 191).
Se han real izado múltiples estudios con esferoides que no han demos trado su efect iv idad en esta enfermedad.
El t ra tamiento con plasmaféresis o la administración de i nmunog lobu l i -nas intravenosas es el t ra tamiento de elección para aquel los pacientes que han perd ido la capac idad de deambular de forma autónoma, la combinación de ambos fármacos no parece ser mejor que la admin i s tración aislada de cua lquiera de ellos.
Consiguen acortar el t i empo de recuperación de la enfermedad, el número de pacientes con recuperación comple ta al año y, en los pac ien tes gravemente afectados, el t i empo de conexión al respirador.
11.3. Polineuropatía desmielinizante inflamatoria crónica (PDIC)
Tiene un p i co de inc idenc ia en la 5. a-6. a década, y los varones se afectan más frecuentemente.
En un terc io de los pacientes hay un antecedente séptico prev io . Es importante descartar infección por V IH (el in i c io de la neuropatía suele anteceder al desarrol lo comp le to de un SIDA).
Diagnóstico diferencial Anatomía patológica
Los criterios diagnósticos se exponen a continuación. Requeridos: - Deb i l i dad progresiva en uno o más miembros deb ido a neuropa
tía. - Arref lexia . - Curso de la enfermedad < 4 semanas. - Exclusión de otras causas.
• Sugestivos: - Deb i l i dad simétrica relativa. - Leve afectación sensorial. - Alteración de cua lquier par craneal . - Ausencia de f iebre.
- Evidencia electrofisiológica de desmielinización.
Se deben descartar las entidades inc lu idas en la Tabla 46 .
La biopsia de nervio periférico demuestra desmielinización, formación de "bu lbos de cebo l l a " (remielinización ineficaz), edema endoneura l e inf i l trados mononucleares mult i foca les con predilección por los nervios proximales y las raíces espinales.
Clínica
Tiene un debut clínico similar al síndrome de Guillain-Barré, pero con una instauración de los síntomas más gradual , superando a veces los 2 meses de progresión. Posteriormente, toma un curso crónico progresi vo o con recaídas intermitentes, por lo que algunos lo consideran c o m o un trastorno "esclerosis múltiple-Mce" que afecta al sistema nervioso periférico.
8 9

Manual CTO de Medicina y Cirugía, 8 . a edición
Hay mayor afectación sensitiva que en el síndrome de Guillain-Barré. La deb i l i dad está presente en los músculos proximales y distales m ien tras que las parestesias son de p redomin io distal . Conl leva afectación de pares craneales bajos y de músculos intercostales. En casi todos los casos hay hiporref lex ia miotática.
R E C U E R D A La polineuropatía desmie l in izante inflamatoria crónica es similar al síndrome de Guil lain-Barré, pero se diferencia de éste porque: • Afecta a pacientes de edad más avanzada (50-60 años). • La instauración es más lenta y el cuadro es crónico, pudiendo presentar
recurrencias. • El componente axonal y la afectación axonal son mayores. • Los cort icoides sí son útiles en su tratamiento.
Pronóstico
Los pacientes con curso recurrente t ienen mejor pronóstico que las for mas progresivas iniciales.
Pruebas complementarias
• LCR. Presenta hallazgos análogos al síndrome de Guillain-Barré. • Neurofisiología. Los estudios de ve loc idad de conducción son bá
sicos para establecer el diagnóstico de esta ent idad . La afectación nerviosa es mu l t i foca l . Hay disminución de la ve loc idad de conduc ción, dispersión de los potenciales de acción y b loqueos de c o n ducción. Los dos últimos se deben a una afectación desmie l in izante var iable dentro del m ismo nerv io, y dist ingue esta ent idad de las neuropatías desmiel in izantes hereditarias, en las que todas las fibras se afectan de forma homogénea y no hay bloqueos ni dispersión.
C o m o con todas las neuropatías desmiel in izantes, hay una pobre corre lación entre la ve loc idad de conducción y la deb i l i dad . La afectación axonal es más l lamat iva que en el síndrome de Guillain-Barré.
Diagnóstico diferencial
Las entidades que hay que descartar se inc luyen en la Tabla 47 .
• Porfiria • Neuropatías desmielinizantes asociadas a:
- Mieloma osteoesclerótico - Gammapatía monoclonal - Linfoma
• Neuropatías desmielinizantes hereditarias - Charcot-Marie-Tooth tipo I (NSMHI) - Dejerine-Sottas tipo III (NSMH III)
Tabla 47. Diagnóstico diferencial de la PDIC
Tratamiento
En esta fo rma crónica sí son efectivos los cort icoides, que son el t ra tamiento de elección, j un to a la plasmaféresis y las inmunog lobu l inas intravenosas, que se añaden en las formas más graves.
En casos refractarios, pueden ser ut i l izados inmunosupresores c o m o la azat iopr ina, c ic lo fosfamida y c ic lospor ina .
11.4. Neuropatía diabética
En la diabetes mel l i tus , pueden ocur r i r un a m p l i o rango de trastornos del sistema nerv ioso periférico que, en genera l , se c las i f ican en dos t ipos : polineuropatías simétricas y asimétricas, aunque lo habi tua l es que los pacientes presenten manifestac iones clínicas de varias de ellas.
Es característica la presencia de do lor en muchas de ellas.
Polineuropatías simétricas
La neuropatía está presente en menos de un 1 0 % de los diabéticos en el m o m e n t o del debut de la enfermedad, pero afecta a un 5 0 % de los que t ienen más de 25 años de evolución. Es más frecuente en diabéticos con mal cont ro l metabólico, aunque también puede aparecer en pacientes con buen cont ro l .
Se caracter izan por una combinación de degeneración axonal (preferentemente distal) y desmielinización segmentaria. • Polineuropatía sensitiva distal. Es la fo rma más frecuente de pol i-
neuropatía diabética (MIR 98-99F, 88). Cuando se afectan preferentemente las fibras gruesas, cursa con parestesias e hipoestesia en guante y calcetín, pérdida de la sensibi l idad v ibrator ia y arreflexia distal . Cuando se afectan las fibras de pequeño cal ibre, p redomina la clínica de do lor con sensación quemante en pies, que empeora nota b lemente por las noches. En la forma pseudosiringomiélica hay pérdida de la sensibi l idad dolorosa y térmica, y se asocia a clínica disautonómica. La fuerza, reflejos miotáticos y sensibi l idad táctil, v ibrator ia y posic ional están respetados. La forma pseudotabética con signo de Romberg, arref lexia en miembros inferiores, pérdida de la sensibi l idad profunda y ulceraciones en pies con de fo rmidad art icular (artropatía de Char-cot) es muy rara.
• Neuropatía autonómica. Genera lmente se asocia a neuropatía sensitiva y cursa con clínica cardiovascular (hipotensión ortostática, ta qu icard ia en reposo), geni tour inar ia (vejiga neurógena, impotenc ia , eyaculación retrógrada) y gastrointestinal (disfunción motora esofágica, gastroparesia, vómitos, estreñimiento o diarrea). La diarrea se considera el síntoma intestinal más frecuente. La diabetes es la causa más frecuente de disautonomía. Para valorar el grado de afectación card ioc i rcu la tor ia , se examina la respuesta de la f recuencia cardíaca y la tensión arterial a las man io bras de Valsalva y a la bipedestación. En presencia de neuropatía autonómica, la f recuencia cardíaca no aumenta con las maniobras de Valsalva (respuesta abol ida) . Otros síntomas atr ibuibles a la neuropatía autonómica son la hipo-g lucemia inadvert ida, la dishidrosis, sudoración gustativa, etc.
• Neuropatía dolorosa aguda. Aparece tras pérdida de peso y se caracteriza por do lor " queman te " muy intenso en las plantas de los pies, acompañado de gran hipersensibi l idad cutánea. La pérdida sensitiva es de escasa magn i tud en comparación al grado de h i perestesia. No se afectan los miembros superiores y no hay déficit motor .
9 0

Neurología y neurocirugía
Neuropatía motora próxima! de miembros inferiores (Síndrome de Garland). Se denomina "amio t ro f i a diabética". Aparece en diabéticos de larga evolución y consiste en do lor lumbar bajo y de áreas glúteas, seguido de deb i l idad progresiva de cuadríceps e i l iopsoas con eventual atrofia y pérdida de los reflejos rotul ianos. No hay terapia específica más allá del contro l de la g lucemia . Evoluciona hacia la recuperación espontánea, aunque puede recidivar.
R E C U E R D A Las neuropatías simétricas son más frecuentes en pacientes diabéticos con mal control metabólico.
Polineuropatías asimétricas
NEUROPATÍA S IMÉTRICA DISTAL
• VIH • Citomegalovirus • ddl y ddC (antirretrovirales) • Isoniacida • Alcaloides de la vinca • Déficit de vit. B 1 2
MONONEURIT IS MÚLT IPLE
• VIH • Citomegalovirus
POL IRRADICUL IT IS
• Citomegalovirus • VIH • Varicela zóster • Tuberculosis • Sífilis
NEUROPATÍAS DESMIEL IN IZANTES INFLAMATORIAS
• Aguda (tipo Guillain-Barré) • Crónica
Tabla 4 8 . Causas de alteración del nervio periférico en la infección por VIH según el patrón de afectación
Son menos comunes, ocurren más frecuentemente en ancianos y pue den aparecer antes en el curso de la enfermedad que las po l ineuropa tías simétricas. Su patogenia es con frecuencia vascular. • Neuropatías craneales. Pueden ser la pr imera manifestación de una
diabetes. El par III craneal es el más f recuentemente afectado (MIR 06-07, 72; MIR 97-98, 130). El in i c io es brusco, se asocia a intenso do lor retroorbi tar io , y hab i tua lmente respeta la mot i l i dad pupi lar , a di ferencia de los III pares compresivos (MIR 99-00F, 62). Suele recuperarse espontáneamente en varias semanas. Otros pares craneales que se afectan con frecuencia son el IV, VI y facial (VII).
R E C U E R D A La neuropatía diabética del tercer par craneal respeta la motilidad pupilar. Los terceros pares compresivos no la respetan
Neuropatías por atrapamiento. Cua lqu ier nerv io periférico puede afectarse (mediano, cub i ta l , radia l , peroneo lateral, etc.) y la e t io lo gía más frecuente es compresiva. La recuperación suele ser satisfactor ia si la lesión se local iza dista lmente. Neuropatía de tronco. Consiste en la afectación aguda y dolorosa uni lateral de uno o más nervios torácicos, y es más frecuente en mayores de 50 años. Cursa con do lo r y disestesias unilaterales en tórax y abdomen , que pueden controlarse con ami t r ip t i l i na . Puede confundirse con una afectación herpética en la fase previa a la e rup ción cutánea.
Tratamiento
El tratamiento de la neuropatía diabética es poco satisfactorio. Inc lu ye un buen control metabólico y tratamiento sintomático del do lor con analgésicos habituales y, si no cede, con carbamazepina, amit r ip t i l ina , fenitoína o c lonazepam. Las neuropatías por atrapamiento, c o m o el síndrome del túnel carpiano, pueden necesitar descompresión quirúrgica.
Neuropatía simétrica distal. Es la neuropatía más frecuente en pacientes con SIDA. Aparece más frecuentemente en estadios avanzados de la enfermedad y se suele asociar a mielopatía vacuolar o demenc ia . Es una neuropatía axona l , de p r edomin i o sensitivo, que cursa con parestesias dolorosas simétricas a nivel distal en m i e m bros inferiores. El t ra tamiento es sintomático del dolor , con ami t r i p t i l ina , carbamazepina o fenitoína, y se han descr i to respuestas al AZT. Mononeuritis múltiple. Es menos frecuente que la anterior. Apa rece también en fases evoluc ionadas de la enfermedad y, a nivel anatomopatológico, cursa con una vasculit is necrot izante s imi lar a la panarterit is nodosa. Polirradiculitis. La pol i r rad icu l i t i s por C M V en el V I H es la más frecuente y mejor caracter izada, aunque se puede produc i r también por el p rop io V I H . Afecta in ic ia lmente a las raíces lumbosacras y se presenta de fo rma aguda o subaguda, con pérdida de fuerza en los miembros inferiores, parestesias sacras y retención ur inar ia . Puede asociarse a encefal it is y retinit is, ambas también por CMV . En el LCR hay pleocitosis po l inuc lear , h iperprote inorraquia y glucosa normal o baja. La afectación es axonal . El t ra tamiento con gancic lo-vir y foscarnet, que debe iniciarse de fo rma empírica ante la sospecha clínica, ha sido benef ic ioso en algunos pacientes (dejada a su evolución natural , es casi s iempre morta l ) . Polineuropatías desmielinizantes inflamatorias. Tanto la fo rma aguda (síndrome de Guillain-Barré) c o m o la crónica aparecen exc lus i vamente en fases precoces de la infección, cuando la i n m u n i d a d está aún conservada. La clínica es análoga a la descrita para pacientes seronegativos y, en ambos casos, la patogenia se considera auto inmuni ta r i a . En el LCR, a di ferencia de los pacientes seronegativos, suele existir pleocitosis no superior a 50 células/mm 3. Las posibi l idades terapéuticas son las mismas que para seronegativos.
11.6. Neuropatías disproteinémicas
11.5. Neuropatías en la infección por VIH
La neuropatía periférica es muy frecuente en la infección por V I H . Puede aparecer en todas las fases de la infección, en múltiples ocasiones de forma subclínica. Los patrones de afectación son diversos (Tabla 48) .
El 5 % de los pacientes con m ie loma múltiple osteolítico desarrol lan una polineuropatía sensi t ivomotora de carácter axona l , a veces grave, que no revierte tras el t ra tamiento del m ie loma .
Más frecuente es la asociación de polineuropatía al m ie loma múltiple osteoesclerótico (MIR 98-99, 254) . En estos casos, hay respuesta al tratamiento del m ie loma , la polineuropatía es desmie l in izante y se asocia a diferentes proteínas monoc lona les y cadenas ligeras ( fundamenta l mente cadenas X). Es frecuente su asociación con otros trastornos sis-
91

Manual CTO de Medicina y Cirugía, 8.a edición
témicos (síndrome POEMS: polineuropatía, organomegal ia , endocr ino-patía, proteína M y alteraciones cutáneas).
La gammapatía monoc lona l IgM, generalmente con cadenas ligeras K, se asocia a una polineuropatía desmie l in izante con debut a los 60-70 años, y afectación sensi t ivomotora, distal y simétrica de lenta evolución. En 2/3 partes de los casos se detecta en sangre periférica la presencia de ant icuerpos ant i-MAC (glicoproteína asociada a la mie l ina) . Responde de forma pobre al t ratamiento, sin que los cort icoides, plasmaféresis o las i nmunog lobu l inas sean efectivas. Trabajos recientes sugieren que el r i tux imab puede ser de ut i l idad en este t ipo de neuropatía.
11.7. Neuropatías hereditarias sin base metabólica conocida nab i a49 )
Neuropat ías sensi t ivo-motoras hered i tar ias (NSMH) • Tipo I - Charcot-Marie-Tooth desmielinizante - AD • Tipo II - Charcot-Marie-Tooth axonal - AD • Tipo III - Déjerine-Sottas - desmielinizante - AR
Neuropat ías sens i t i vas y autonómicas hered i ta r ias • Disautonomía familiar (síndrome de Riley-Day) - AR
Tabla 49. Neuropatías hereditarias sin base metabólica conocida
Neuropatía sensitivomotora hereditaria tipo I
Inc luye los casos prev iamente denominados c o m o forma desmie l i n i zante de Charcot-Marie-Tooth (también l lamada forma hipertrófica). Se hereda preferentemente con carácter autosómico dominante , l igado generalmente al c romosoma 1 7 (t ipo la; la más frecuente), aunque en algunas fami l ias el imp l i c ado es el c romosoma 1 (t ipo Ib).
Es una neuropatía lentamente progresiva que debuta en la 1 . a - 2 . a década, con un franco p r edomin i o de la clínica motora : deb i l idad y a m i o trof ia que afecta a nive l distal a los miembros inferiores, j u n t o con de fo rmidad de los pies (pies cavos). La clínica sensitiva es muy escasa. El temblor esencial puede acompañar a la neuropatía (casos prev iamente conoc idos c o m o síndrome de Lévy-Roussy).
Los estudios neurofisiológicos demuestran una intensa ralentización de las ve loc idades de conducción motoras y sensitivas, con a m p l i t u des de los potencia les de acción normales . A nivel anatomopatológ ico , se encuentra desmielinización, remielinización y formación de "bu lbos de c e b o l l a " en los nervios periféricos. No existe t ra tamiento específico.
Neuropatía sensitivomotora hereditaria tipo II
Es menos frecuente que la t i po I, e inc luye los casos axonales de Charcot-Marie-Tooth. Se hereda con carácter autosómico dominan te l igado al c romosoma 22 .
Su in i c io es más tardío (4. a -5. a década), y se di ferencia del t i po I en que es una neuropatía pr imar iamente axona l , con disminución en la amp l i t ud de los potenciales, velocidades de conducción normales o l igeramente reducidas y ausencia de cambios hipertróficos.
Neuropatía sensitivomotora hereditaria tipo III
Inc luye los casos prev iamente denominados c o m o Déjerine-Sottas. Se trata de una neuropatía desmie l in izante con cambios hipertróficos, que se hereda con carácter autosómico recesivo y que evo luc iona más ráp idamente y peor que la t i po I.
Comienza en la infancia con deb i l idad en miembros inferiores y, a diferencia de las anteriores, hay afectación precoz de los miembros superiores, con grave pérdida sensorial, pero respetando la función au tonómica.
Se asocia con frecuencia a anomalías esqueléticas (talla corta, c i foescol iosis, deformidades en manos y pies).
Síndrome de Riley-Day
Es un trastorno autosómico recesivo, caracter izado por la ausencia congénita de neuronas autonómicas en las astas intermediolaterales de la médula y de células ganglionares sensoriales.
Cursa en la infancia con pobre succión, l lanto sin lágrimas, crisis de vómitos y f luctuaciones inexpl icadas de la temperatura corpora l . La sudoración suele ser norma l .
En niños mayores aparece hipotensión ortostática. Hay déficit sensorial distal en un patrón d isoc iado. La fuerza es norma l . Es frecuente la c i foescoliosis y estatura corta. Los potenciales de acción sensitivos están gravemente afectados.
El pronóstico es ma lo , con disminución de la expectat iva de v ida . Las causas de muerte están en relación con la disfunción autonómica: aspiración, grave hipotensión postural , anormal respuesta a la h ipox ia o deshidratación secundaria al vómito. Es l imi tante la lab i l idad autonómica y el déficit sensorial.
11.8. Mononeuritis múltiple
Es la afectación secuencial y asimétrica de múltiples nervios periféricos no cont iguos. Un terc io de los casos son desmiel in izantes, y el resto son de carácter axona l .
En el 5 0 % de las formas axonales se detecta una patogenia vascu-lítica. Entre las vascul i t is , la panarter i t is nodosa es la etiología más f recuente .
Otras causas son las afecciones del t e j i do c o n j u n t i v o (artritis reuma-to ide , lupus y en fe rmedad m ix t a del t e j i do con jun t i vo ) , c r i og l obu l i -
92

Neurología y neurocirugía
nemia , Sjógren, Wegener , etc. Son también causa de mononeur i t i s múltiple: lepra, V I H , sarcoidosis, ami lo idos i s y diabetes.
Q R E C U E R D A Ante una clínica de mononeuritis múltiple y datos de vasculitis (nodulos subcutáneos con necrosis, por ejemplo), piensa en una panarteritis nodosa.
Sobre una población no seleccionada, la diabetes es la causa más f re cuente de mononeur i t i s múltiple.
11.9. Mononeuropatías Se definen como la afectación focal de un t ronco nervioso único. Hab i tualmente se producen tras traumatismos directos, atrapamiento o c o m presión. Las más frecuentes son la compresión del nervio mediano en la muñeca (síndrome del túnel carpiano) y la neuropatía cubital (generalmente a nivel del canal epitrócleo-olecraneano en el codo). Otros e jemplos son la parálisis del "sábado noche" (radial), fractura de la cabeza del peroné (peroneo), patología pelv iana o del músculo psoas (femoral), etc. Se describen detenidamente en la Sección de Traumatología.
Casos clínicos
U n pac ien te d iabét ico , de 6 9 años, consu l t a por apar i c ión b r u s c a de do lor ocu la r d e r e c h o y visión dob le . En la exp lorac ión , hay ptosis d e r e c h a y parálisis de todos los mov im ien tos de ese o jo , excep to la abducc ión . Las pupi las son no rma les , así c o m o la a g u d e z a v i sua l . El diagnóst ico más p robab le es :
1) A n e u r i s m a d e a r te r i a c o m u n i c a n t e pos t e r i o r . 2) O f t a l m i t i s t u n g i c a diabét ica. 3) Mononeuropat í a diabét ica d e l III par . 4) P roceso e x p a n s i v o d e l seno c a v e r n o s o . 5) O f t a l m o p l e j i a ¡nternuclear.
M I R 99-00F , 6 2 ; RC: 3
U n pac i en t e de 2 8 años consu l t a por un c u a d r o , i n i c i ado h a c e 4 8 ho ras , de do lor l umba r y pares tes ias en c a r a poster ior de mus lo s y p ie rnas . P rog res i vamente , i m p o
s ib i l idad pa r a c a m i n a r . En la exp lo rac ión d e s t a c a parál is is de m i e m b r o s in fe r io res y deb i l i dad p rox ima l de m i e m b r o s supe r i o r e s . Exp lo rac ión sensor ia l y pa res c r a n e a les n o r m a l e s . Ref le jos miotá t i cos un i v e r s a lmen te abo l idos y respues tas p lan ta res ausentes . N o ref iere an t e ceden tes de interés, sa l vo gast roenter i t i s aguda h a c e 15 días. Seña l e , ent re las s igu ientes , la ac t i tud más impor tan te en el m a n e j o de este pac i en te :
1) V i g i l a n c i a es t recha d e la func ión r esp i r a to r i a y vent i lac ión mecán i ca en caso d e d e t e r i o r o .
2 ) Descompres ión quirúrgica i n m e d i a t a d e la médula c e r v i c a l . 3) Resonanc i a magnética d e la c o l u m n a c e r v i c a l desde C3 hac i a a b a j o . 4) Punc ión l u m b a r i n m e d i a t a , para desca r ta r h i p e r p r o t e i n o r r a q u i a . 5) T r a t a m i e n t o c o n 1 mg/kg/día d e p r e d n i s o n a , d u r a n t e una s e m a n a .
M I R 9 9 - 0 0 , 1 9 1 ; RC: 1
93

Neurología y neurocirugía
12. ENFERMEDADES DE LA PLACA MOTORA
MIR T e m a de i m p o r t a n c i a i n t e r m e d i a en el M IR , p e r o r e l a t i v a m e n t e fácil ; po r t a n t o , m u y r en tab l e . Es r e c o m e n d a b l e centrarse , sobre t o d o , en la m ias t en i a gravis y en saber rea l izar el diagnóstico d i f e r enc i a l c o n el s índrome d e Eaton-Lambert y el b o t u l i s m o .
Aspectos esenciales
pj~] La mias ten ia gravis se p r o d u c e por el b l o q u e o de los receptores colinérgicos postsinápticos por ant i cuerpos d i r i g idos f rente a e l los (aunque éstos no se encuen t r an en todos los pacientes) . El 7 5 % de los pacientes l levan asociada una alteración del t i m o (h iperplas ia o t i m o m a ) .
[2"] La mias ten ia gravis se caracter iza por d e b i l i d a d muscu l a r ex t raocu la r y p r o x i m a l de los m i e m b r o s q u e me jo ra c o n el reposo. N o hay a l terac iones sensit ivas, autonómicas, pup i la res n i de los ROT.
QjJ El diagnóstico de la miasten ia gravis se rea l iza c o n el test de e d r o f o n i o y la determinación de los ac ant i r re-ceptor de A c h (la p rueba más específica para el diagnóstico).
["4] En la miasten ia neonata l , hay ant i cuerpos ant i r receptor procedentes de la madre ; en la miastenia congénita no hay ant i cuerpos ant i r receptor .
[~5~¡ El t ratamiento básico de la miastenia gravis son los anticolinesterásicos. La timectomía se realizará en las formas genera l izadas en pac ientes entre la pube r t ad y los 55 años.
["o""] El síndrome de Eaton-Lambert y el bo tu l i smo son trastornos presinápticos que cursan c o n ROT y respuesta pup i l a r d i sm inu ida , así c o m o con disautonomía, a d i fe renc ia de la miastenia gravis.
["7") Hay que pensar en lesión del t ronco encefálico siempre que aparezcan asociadas lesiones de pares craneales ip-silaterales con "vías largas" (motoras o sensitivas) contralaterales. Los pares craneales indican el nivel de la lesión.
QTJ Los síndromes d isar t r ia-mano to rpe y ataxia-hemipares ia se pueden dar tan to en lesiones de la cápsula in ter na cont ra la tera l c o m o en las de p ro tube ranc i a .
[~9~] En las lesiones bu lbares , se d i s t inguen dos síndromes: lateral o de W a l l e n b e r g (oclusión de la arter ia ver tebra l o cerebelosa postero in fe r io r ) y med i a l (oclusión de la arter ia espinal anter ior o vertebra l ) .
[ [ ] P r egun tas
- M I R 0 7 - 0 8 , 58 - M I R 0 5 - 0 6 , 6 0 - M I R 0 3 - 0 4 , 2 4 5 - M I R 0 2 - 0 3 , 2 0 8 - M I R 00-01 F, 65 - M I R 9 9 - 0 0 , 2 0 4 , 2 5 3 - M I R 9 8 - 9 9 , 6 0 , 6 3 , 2 4 3 - M I R 9 8 - 9 9 F , 231 - M I R 9 7 - 9 8 , 1 4 9
Las enfermedades de la placa m o
tora inc luyen fundamenta lmente la
miastenia gravis (trastorno postsi-
náptico) (Figura 62) y el síndrome
miasténico de Eaton-Lambert y bo
tu l i smo (trastornos presinápticos)
(Tabla 50).
12.1. Miastenia gravis
Se trata de un trastorno au to inmu-
nitar io que cursa con deb i l idad y
fa t igabi l idad de la musculatura es
quelética.
G loba lmente afecta más f recuen
temente a mujeres, puede darse en
todos los grupos de edad, con un
p i co de inc idenc ia en las mujeres
entre la segunda y tercera décadas,
y algo más tardío en los hombres
(cuarta-quinta décadas).
Placa motora
Vaina de mielina
Músculo 1^-—
Axór 1
Miastenia gravis
Receptores de Ach
Figura 62. Unión neuromuscu la r no rma l (a). Miastenia gravis (b).
9 4

Neurología y neurocirugía
MIASTENIA GRAVIS S. EATON-LAMBERT BOTULISMO
Patogenia Ac-antiReceptor de Ach AUTOINMUNITARIA 7 5 % Alt.Tímlcas: 6 5 % Hiperplasla, 10%T¡moma
Sexo y edad Predominio MUJERES Cualquier edad: 20-30 a
50-60 a
Debilidad Musculatura extraocular Proximal MMII (85%) Asimétrica
Reflejos miotáticos Normales
Pupilas Normales
Disautonomía NO
Mejora REPOSO, sueño Anticolinesterásicos (TEST TENSILÓN)
Empeora Ejercicio Estrés Embarazo
Primer potencial (estímulo único) Normal (aumento del jitter en fibra única)
Estimulación repetida 2-3 Hz
E .R .>10Hz
Tratamiento
Anticolinesterásicos: - PIRIDOSTIGMINA - Neostigmina Corticoides Timectomía Plasmaféresis
Ac-anticanal de calcio (presináptico) Paraneoplásico
Predominio VARONES > 40 a
• PROXIMAL MMII • Extraocular (70%)
SI
EJERCICIO
Tubocurarina Exametonio
Tratamiento del tumor subyacente
Bloqueo liberación Ach TOXINA BOTULÍNICA Cl. botulinum
• Sexo indiferente • Cualquier edad • Forma más frecuente en lactantes
• BULBAR (m. extraocular) • Descendente SIMÉTRICA
Respuesta disminuida
Si
NO mejora tras ejercicio
i -X-
Antitoxina equina (no útil en forma infantil) Soporte vital
Tabla 50. Diagnóstico diferencial de los síndromes miasténicos
Patogenia
Es la en f e rmedad a u t o i n m u n i t a r i a me jo r ca rac te r izada . En un 85-9 0 % de los casos ex is ten an t i cue rpos d i r i g i d o s con t r a los receptores nicotínicos de a c e t i l c o l i n a (Ach) (MIR 98-99F, 231 ) . Estos an t i cue r pos actúan de tres maneras : 1) b l o q u e a n el receptor de a c e t i l c o l i n a ; 2) p r o m u e v e n su endoc i tos i s y pos te r io r destrucción, y 3) ac t i van el depósito de c o m p l e m e n t o sobre la m e m b r a n a postsináptica, con la cons igu i en te destrucción de los receptores y el a p l a n a m i e n t o a largo p l a z o de los p l iegues de l receptor postsináptico (MIR 99-00 , 204 ) . Estos c a m b i o s d e t e r m i n a n el he cho de que , pese a que la liberación de A c h en la terminación nerv iosa en respuesta a un po tenc i a l de acción es n o r m a l , ésta no es capaz de generar c o n tracción muscu la r .
El t i m o parece jugar un pape l impo r t an t e en la génesis de la respuesta a u t o i n m u n i t a r i a , d a d o que es ano rma l en el 7 5 % de los pac ientes (en el 6 5 % es hiperplásico, y en el 1 0 % hay t i m o m a ) (MIR 99-00, 253 ) .
Clínica
Cursa con debilidad y fatigabilidad muscular de distribución típica, sin alteración de otras func iones neurológicas.
Tres características marcan el diagnóstico de esta enfermedad: • Carácter fluctuante de la deb i l i dad , con empeoramien to tras el ejer
c i c io y mejoría con el reposo o el sueño. Los pacientes se quejan de mayor deb i l idad por las tardes.
• Afectación de la musculatura craneal, preferentemente la extraocular, con ptosis y diplopía. Puede simular una oftalmoplej ia internuclear (MIR 02-03, 208). Otros síntomas son disartria, disfagia y debi l idad de musculatura cervical. En la mayoría de los pacientes ( 85% ) , la deb i l i dad se generaliza a los músculos de los miembros, siendo de carácter proximal y asimétrica, con preservación de los reflejos miotáticos y sin amiotrofias. No hay alteraciones sensitivas, autonómicas ni pupilares.
• Respuesta clínica a los fármacos colinérgicos (anticolinesterásicos). Se denomina miastenia ocular a aquel la fo rma en la que únicamente existe deb i l idad de la musculatura ocular después de dos años del i n i c io de los síntomas. La miastenia general izada es la forma en la que existe una afectación de musculatura diferente a la ocular , bien en los miembros , b ien bulbar , con o sin afectación ocular . Se habla de crisis miasténica cuando la deb i l idad muscular respiratoria produce insuf ic iencia respiratoria o la deb i l idad bulbar imp ide la deglución, con necesidad de instaurar una sonda de alimentación por el riesgo de aspiración. Las crisis miasténicas pueden estar p ro vocadas por infecciones intercurrentes (lo más frecuente) u otros trastornos sistémicos acompañantes.
En la miastenia gravis, los ROT, las pupilas y el SNA están intactos, a diferencia del botulismo y el síndrome de Eaton-Lambert.
95

Manual CTO de Medicina y Cirugía, 8. a edición
Diagnostico
Para establecer el diagnóstico de f in i t i vo , se ut i l izan las siguientes pruebas complementar ias (MIR 05-06, 60) : • Test de Tensilon® (edrofonio) (MIR 98-99 , 2 4 3 ) . Debe r ea l i
zarse c u a n d o existe la sospecha cl ínica. El e d r o f o n i o es un fárm a c o que i n h i b e la ace t i l co l ines terasa a n i ve l de la h e n d i d u ra sináptica, a u m e n t a n d o así la d i s p o n i b i l i d a d de a ce t i l c o l i n a para ¡nteractuar con los receptores postsinápticos. Tras fat igar al pac i en te , la administración in t ravenosa de e d r o f o n i o p r o d u c e una mejoría i nmed i a t a y t r ans i to r i a . Los efectos secundar ios que pueden aparecer son : náuseas, d ia r rea , sal ivación, f a s c i c u l a c i o nes o síncopes (síntomas colinérgicos que pueden antagon izarse co n a t rop ina ) .
• Demostración de los ant icuerpos ant irreceptor de acet i lco l ina . Aparece en un 8 5 - 9 0 % de los pac ientes con miasten ia gene ra l i zada y en un 5 0 % de las miastenias ocu lares (MIR 07-08 , 58 ) . Su presenc ia es diagnóstica (MIR 03-04 , 245 ) , pe ro su ausenc ia no e x c l u y e el diagnóstico; no son patognomónicos de mias ten ia gra vis, ya que pueden aparecer en miastenias farmacológicas c o m o la generada por p e n i c i l a m i n a . Su titulación no se co r responde co n la gravedad de la en f e rmedad , pero s irve c o m o m o n i t o r i -zación de la evolución y respuesta al t r a t am ien to en pac ientes a is lados. En un 1 0 - 1 5 % de los casos no es pos ib le demost rar la presenc ia de estos an t i cuerpos en sangre; a este g rupo de p a c i e n tes se les d e n o m i n a mias ten ia seronegat iva . Rec ientemente se ha descub ie r to un nuevo an t i cue rpo presente únicamente en estos pac ientes seronegat ivos . Se trata de los an t i cue rpos an t i-MuSK. Este t i p o de an t i cue rpos se aprec ia rara vez en pac ientes con la f o r m a ocu la r a is lada.
• Estudios neurofisiológicos. Las ve loc idades de conducción nerv iosa son norma les . La a m p l i t u d de l po tenc i a l de acción ante un estímulo único es n o r m a l . Sin emba rgo , la estimulación nerv iosa repet i t i va a bajas f recuenc ias (3-5 Hz) p r o d u c e un dec r emen to progres i vo de la a m p l i t u d de los potenc ia les de acción evocados , máxima al 4.°-5.° po t enc i a l , que para ser s ign i f i ca t i va debe ser m a y o r de l 1 0 - 1 5 % de la a m p l i t u d del p r ime r po tenc i a l (respuesta dec rementa l ) (Figura 63 ) .
Decrement Test An. Pemneus Profundus
2mV/D . •j.-fj á¡-. ftVj'- -•• i - ! • 2ms/D ANALYSE
Figura 63. Respuesta decremental en paciente con miastenia gravis durante la estimulación nerviosa repetitiva a 3 Hz
La electromiografía de f ibra aislada muestra un incremento del Rit-ter. El jitter representa la var iab i l idad del interva lo interpotenc ia l , esto es, la var iab i l idad en las latencias entre dos fibras musculares pertenecientes a la misma un idad motora . En la miastenia gravis, la estimulación repetit iva a altas frecuencias incrementa el jitter (Figura 64) , mientras que en el síndrome de Eaton-Lambert y en el botu l i smo, el jitter se incrementa a bajas frecuencias y d i sminuye a altas frecuencias.
1
Figura 64. Electromiografía de fibra única de un paciente con miastenia gravis que demuestra una prolongación del jitter o variabilidad del itinerario
interponencial
• Radiología. Se debe rea l izar TC o RM torácica para detectar a l t e rac iones tímicas (h ipe rp las i a o t i m o m a ) . T o d o a u m e n t o de l t i m o en mayores de 4 0 años es a l t amen te sospechoso de t i m o ma.
• Otros. Debe hacerse un estudio de hormonas t i roideas, puesto que puede asociarse h ipe r t i ro id i smo en un 5 % de los pacientes y agravar la deb i l i dad miasténica. Dada la asociación con otros trastornos au to inmun i ta r ios (LES, artrit is reumato ide , t i ro id i t i s , vitíligo, pénfigo), se debe sol ic i tar factor reumato ide y ant icuerpos an t i nu cleares.
Formas clínicas
Las formas clínicas son las siguientes: • Miastenia neonatal . Apa rece en el 1 5 % de los h i jos de madres
miasténicas y se p r o d u c e por transmisión p lacen ta r i a de a n t i cue rpos de la madre miasténica al fe to . La cl ínica se i n i c i a el segundo o tercer día p o s n a c i m i e n t o . Cursa c o n hipotonía gene r a l i zada , d i f i c u l t a d resp i ra tor ia , d is fag ia y paresia diafragmática. T iene carácter t r ans i t o r i o y los síntomas desaparecen en pocas semanas. Si por la in tens idad de los síntomas fuera necesar io el t r a t am ien to , hay respuesta a fármacos anticolinesterásicos.
• Miastenia congénita. Ag rupa un c o n j u n t o de ent idades he red i t a rias de pa togen ia no a u t o i n m u n i t a r i a (no presentan an t i cue rpos an t i r receptor ) caracter izadas por d is t in tos trastornos de la unión neu romuscu l a r ( receptor de A c h a n o r m a l con p r o l o n g a d o t i e m p o de aper tura , de f i c i enc i a de acet i l co l inesterasa , t e rm ina l presináp-t i co pequeño co n escasa liberación de A c h , etc. ) . Representan el 1 % de los casos de mias ten ia . La cl ínica c o m i e n z a en la in fanc ia y progresa l en tamente hasta la edad adu l ta . Gene ra lmen te hay afectación de la muscu l a tu ra ex t raocu la r y progresa l en tamente a pesar de l t r a t am ien to .
Tratamiento
En genera l , los med ios terapéuticos d i spon ib l e s son cua t ro : me jo ra r la transmisión neu romuscu l a r con anticolinesterásicos, i n m u n o s u -presión con esferoides o citostáticos, plasmaféresis para d i s m i n u i r la titulación sérica de an t i cue rpos an t i r r e cep to r y timectomía para e l i m i n a r el pos ib l e o r igen de los m i smos (Figura 65) (MIR 00-01 F, 65 ) .
96

T R A T A M I E N T O D E L A M I A S T E N I A GRAVIS
Neurología y neurocirugía
F o r m a ocu l a r e x c l u s i v a
Anticol inesterásico ( p i r i d o s t i g m i n a )
F o r m a g e n e r a l i z a d a
Anticol inesterásico ( p i r i d o s t i g m i n a )
I n d i c a c i o n e s d e t imec tomía : • T i m o m a • F o r m a g e n e r a l i z a d a
Cr is is mias tén ica
T r a t a m i e n t o d e s o p o r t e (vent i lac ión, l íquidos)
Plasmaféresis o I n m u n o g l o b u l i n a i.v.
I n s u f i c i e n t e No m e j o r a
Plasmaféresis o i n m u n o g l o b u l i n a i.v.
s T imectomía
I Eva luac ión d e l e s t a d o cl ínico y si el p a c i e n t e
lo p rec i sa I I nmunosupres ión
( p r e d n i s o n a , a z a t r i o p i n a , c i c l o s p o r i n a )
Figura 65. Tratamiento de las distintas formas de miastenia gravis
Fármacos anticolinesterásicos. I nh iben la destrucción de A c h dent ro de la hend idu ra sináptica, a u m e n t a n d o su d i s p o n i b i l i d a d . Se u t i l i z a n la p i r i d o s t i g m i n a (oral) y la neos t igmina (parentera l ) , sobre t o d o la p r ime ra , por tener menos efectos muscarínicos a dosis terapéuticas (MIR 98-99 , 60 ) . Se usan c o m o t r a t am ien to sintomático, en mono te r ap i a en las formas ocu lares puras para cor reg i r la ptosis (y en meno r med ida la diplopía) y asoc iados a otros fármacos en las fo rmas genera l izadas . Su sobredosificación c o n d u c e a la aparición de síntomas muscarínicos c o m o , a u m e n to de la secreción b r o n q u i a l , d iar rea , sal ivación, náuseas, do lo r a b d o m i n a l y a u m e n t o de la d e b i l i d a d (crisis colinérgica). Corticoides. Se u t i l i zan : 1) cuando fal la la medicación anticol ines-terásica, en combinación con ésta (la mayoría de los pacientes son tratados con esteroides a menos que tengan una contraindicación mayor) ; 2) para mejorar la fuerza preoperator ia del paciente pre-timectomía; 3) cuando no hay remisión tras timectomía; y 4) raramente en la miastenia ocular pura. La mejoría comienza meses después de inic iar el t ra tamiento, y es común el empeoramien to en los pr imeros días de t ratamiento esteroideo. Inmunosupresores. Su uso se indica en combinación con los co r t i coides para reducir las dosis de los mismos. Además, están ind ica dos en aquel los casos que no responden al t ratamiento cor t i co ideo , o cuando éstos están contra ind icados . Se ut i l i zan micofeno la to , azat iopi r ina , c ic lospor ina y tacro l imus. La c ic lo fosfamida actua l mente se considera terapia de segunda línea, reservada a pacientes que no responden a los tratamientos previos. La azat iopr ina es la de uso más común y sus efectos secundarios inc luyen un síndrome febr i l con malestar general , depresión me dular y alteraciones de la función hepática (debe suspenderse si los leucocitos descienden por debajo de 3.000 o los l infocitos por de bajo de 1.000). Un tercer inmunosupresor que puede ut i l izarse es la c i c lospor ina , con cont ro l estricto de la función renal, aunque la
mayor inc idenc ia de efectos secundarios l imi ta su uso a casos muy concretos. En general , los inmunosupresores se ut i l izan cuando no dan resultado otras medidas. Plasmaféresis/inmunoglobulinas. A lgunos pacientes con miastenia gravis grave general izada, resistente a otras modal idades terapéuticas, pueden mejorar t ransi tor iamente con estos tratamientos. Su efecto es rápido, pero de corta duración, por lo que se usa de forma puntua l en las crisis miasténicas y en la preparación a pret imecto-mía cuando el resto de fármacos no consiguen una buena situación func iona l prequirúrgica. En el caso de la plasmaféresis, su efecto benef ic ioso se corre lac iona con una caída de la titulación sérica de ant icuerpos. Timectomía. Si existe un t i m o m a , la extirpación quirúrgica es necesaria, dada la pos ib i l idad de extensión local del tumor , aunque la mayoría de ellos son benignos. En ausencia de tumor , hasta un 8 5 % de los pacientes mejora tras timectomía, y en un 3 5 % se consigue la remisión sin necesidad de t ratamiento farmacológico. La t imec tomía puede estar indicada en todas las formas general izadas en pacientes entre la pubertad y los 55 años (MIR 00-01 F, 65). En las formas oculares puras, no se ha demostrado f i rmemente la ef icacia de la timectomía (MIR 97-98, 149).
R E C U E R D A Los ant icol inesterásicos en m o n o t e r a p i a sólo se e m p l e a n en las f o r mas o c u l a r e s p u r a s ; en los demás casos es n e c e s a r i o añadir o t ras m e d i d a s terapéuticas.
12.2. Otros síndromes miasteniformes
Síndrome miasténico de Eaton-Lambert
Es un trastorno presináptico de la transmisión neuromuscular causado por ant icuerpos que b loquean los canales de ca lc io dependientes de voltaje del termina l presináptico, i m p i d i e n d o de esta forma la l iberación de A c h .
Aparece más frecuentemente en varones (4/1). En un 7 0 % de los varones y un 2 5 % de las mujeres t iene carácter paraneoplásico, s iendo el tumor más f recuentemente asociado a este síndrome el ca rc inoma p u l monar de células pequeñas ( 5 0 % ) , por lo que ante la sospecha clínica de este proceso, está indicada la realización de una prueba de imagen del tórax (Rx o TC) (MIR 98-99, 63) . Puede asociarse a otras enfermedades, generalmente auto inmuni tar ias : t i rotoxicosis , h ipo t i ro id i smo , vitíl igo, anemia pernic iosa, artritis reumatoide, enfermedad celíaca, col i t is ulcerosa, esc lerodermia, etc.
Clínica
La deb i l idad afecta con preferencia a la musculatura p rox ima l de los miembros inferiores, con escasa afectación de la musculatura bulbar , aunque aparece ptosis y diplopía en un 7 0 % de los pacientes. Es característica la existencia de un incremento transi tor io de la fuerza tras unos segundos de e jerc ic io vo lun ta r io . Los reflejos miotáticos son hipoact i-vos o están abol idos, y cursa con clínica disautonómica: sequedad de boca, impotenc ia , visión borrosa, estreñimiento, etc.
97

Manual CTO de Medicina y Cirugía, 8.a edición
R E C U E R D A N o hay q u e o l v i d a r ped i r una radiografía de tórax si sospechas un síndrome de Eaton Lamber t : podría ser s e c u n d a r i o a un c a r c i n o m a microcít ico de pu lmón.
Diagnóstico
Se basa en el estudio neurofisiológico y en las pruebas serológicas. El estudio neurofisiológico demuestra unas velocidades de conducción normales. El potenc ia l de acción ante un estímulo único es de escasa amp l i t ud . La estimulación repetit iva a bajas frecuencias p roduce una respuesta decrementa l s imi lar a la observada en la miastenia gravis; a altas frecuencias (20-30 Hz) aparece un incremento progresivo en la amp l i t ud del potenc ia l (respuesta incremental ) .
La prueba más sensible para el diagnóstico es la detección de los an t icuerpos ant icanal de ca lc io que se encuentran en un 9 5 % de los pacientes.
NIÑOS Alimentos
TOXINA
ADULTOS Heridas
Alimentos
BOTULISMO. FISIOPATOLOGIA Y CLÍNICA
Parálisis bulbar descendente simétrica
Hipotonía (disminución del tono muscular)
Figura 6 6 . Fisiopatología y clínica del botulismo
Tratamiento
Hay respuesta a la plasmaféresis y terapia inmunosupresora, aunque hay que recordar que los mejores resultados se logran con el t ra tamien to del t umor subyacente. El fármaco u t i l i zado con preferencia para me jorar la transmisión neuromuscular es la 3-4 d i am inop i r i d i na .
Diagnóstico
El estudio neurofisiológico muestra hallazgos similares al síndrome de Eaton-Lambert, aunque el grado de potenciación frente a la es t imula ción repetit iva a altas frecuencias es de menor intensidad.
Tratamiento
Botulismo
Es también un trastorno presináptico. La tox ina botulínica b loquea la liberación de Ach mediada por ca lc io . Entre los 6 t ipos de tox ina bo tulínica, los t ipos patógenos humanos son el A, B y E (los t ipos A y B suelen contaminar conservas de vegetales y el t ipo E se encuentra en pescados). La mor ta l idad es mayor para los t ipos A o E.
Puede aparecer a cua lquier edad, s iendo la forma más frecuente la del lactante.
Exige medidas de soporte vital y la administración de la ant i tox ina equina, aunque esta última no es efectiva en las formas infanti les.
Miastenia inducida por fármacos y tóxicos
El t ratamiento con fármacos c o m o kanamic ina , neomic ina , antibióticos aminoglucósidos, proca inamida , pen ic i l amina o la exposición a tóxicos c o m o los organofosforados puede produc i r síndromes miastenifor-mes o exacerbación de la deb i l idad en pacientes miasténicos.
Clínica
Los síntomas aparecen uno o dos días tras la ingesta del a l imento c o n taminado , y uno o dos semanas después, cuando se trata de c o n t a m i nación de heridas. La disfunción gastrointestinal precede al in i c io de la clínica neurológica, que está marcada por la aparición de oftalmoplejía externa y ptosis. Los pacientes pueden tener pupi las dilatadas arreactivas, pérdida de la convergencia , disartr ia, disfagia y d i f i cu l tad en la masticación. Los reflejos miotáticos son hipoact ivos o están abol idos, y también cursa con clínica autonómica. Los músculos de las ex t remi dades se afectan poster iormente de forma general izada y con carácter agudo o subagudo (Figura 66).
A diferencia del síndrome de Eaton-Lambert, la fuerza no mejora tras el e jerc ic io in ic ia l .
R E C U E R D A La afectación muscular es: • En la miastenia: asimétrica. • En el bo tu l i smo: simétrica y descendente.
En el síndrome de Guillain-Barré: simétrica y ascendente.
También pueden agravar la deb i l idad miasténica los B-bloqueantes, e r i t romic ina , lidocaína, l i t io , magnesio, morf ina , benzodiazepinas, contrastes yodados, etc. Genera lmente, la deb i l idad es moderada y la recuperación comienza progresivamente una vez ret irado el fármaco responsable. El t ratamiento consiste en v ig i lanc ia de la función venti-laroria, y en ident i f icar la sustancia responsable para no re int roduc i r la al paciente, así c o m o para detectar otras con estructura bioquímica similar.
9 8

Neurología y neurocirugía
Casos clínicos representativos
Una paciente de 22 años consulta por presentar, desde una semana antes, ptosis palpebral izquierda, sin dolor, con diplopía en la mirada lateral izquierda. En la exploración física se comprueba la existencia de una ptosis izquierda, así como una paresia de la abducción del ojo izquierdo, con pupilas isocóricas y normorreactivas a la luz. ¿Qué enfermedad es más probable que padezca la paciente?
1) U n a neur i t i s ópt ica i z q u i e r d a en re lac ión c o n esc leros is múlt iple. 2) U n s índrome d e H o r n e r . 3) U n a m i a s t e n i a gravis. 4) U n a parálisis de l t e r ce r par i z q u i e r d o .
5) U n a miopatía h i p e r t i r o i d e a c o n afectación d e la m u s c u l a t u r a e x t r a o c u l a r .
M I R 0 2 - 0 3 , 2 0 8 ; RC: 3
La miastenia gravis se produce por:
1) D e c r e m e n t o d e la a c t i v i d a d eléctr ica presináptica. 2) B l o q u e o de los r ecep to res col inérgicos p o r n i c o t i n a . 3) D i sminuc ión d e la síntesis d e a c e t i l c o l i n a .
4) P resenc ia d e a n t i c u e r p o s pa ra r e cep to res col inérgicos. 5) M igrac ión d e los r e cep to res f u e r a d e la h e n d i d u r a sináptica.
M I R 9 8 - 9 9 F , 2 3 1 ; RC: 4
Varón de 55 años que padece, desde hace TRES meses, debilidad muscular a nivel proximal de las extremidades, sequedad de boca, dolores musculares y parestesias en los cuatro miembros. Durante la exploración, se comprueba debilidad de los músculos proximales. La sensibilidad está conservada y los reflejos osteotendinosos están disminuidos en miembros superiores y abolidos en los inferiores. ¿Qué prueba complementaria de las siguientes ayudaría a establecer el diagnóstico?
1) Es tud io de l LCR. 2) B iops i a d e l n e r v i o a f e c to . 3) B i ops i a de l múscu lo a f e c t o . 4) Rx d e tórax. 5) R M m e d u l a r .
M I R 9 8 - 9 9 , 6 3 ; RC: 4
J
99

Neurología y neurocirugía
13. MIOPATÍAS
Orientación
MIR r
Aspectos esenciales
T e m a t o t a l m e n t e p r e s c i n d i b l e para el M IR , ya q u e en los últimos años sólo ha h a b i d o dos preguntas . Es r e c o m e n d a b l e reco rda r al t ípico p a c i e n t e c o n d i s t ro f i a miotónica de Steinert .
["T"] La D M D es de herencia recesiva ligada al X. Es característica la pseudohipertrofia de pantorrillas y la manio bra de Cowers (el paciente trepa sobre sí mismo para levantarse desde el suelo).
[2~| La distrofia miotónica de Steinert es autosómica dominante. Es característico el bloqueo A-V, la ca lv ic ie frontal, las cataratas subcapsulares, resistencia a la insulina y el fenómeno miotónico en manos, lengua y párpados (dificultad para la relajación muscular) .
13.1. Distrofias musculares
Los t ipos de distrofias musculares están enumerados en la Tabla 5 1 .
• Distrofinopatías: - Distrofia muscular de Duchenne (MIR 98-99F, 76) - Distrofia muscular de Becker
• Distrofia facioescapulohumeral • Distrofia miotónica de Steinert • Distrofia muscular de las cinturas
Tabla 51. Distrofias musculares
Distrofinopatías
La distrofina es una proteína cod i f i cada por un gen s i tuado en el brazo cor to del c romosoma X (Xp21). Se loca l iza en la cara interna de la membrana plasmática de dist intos te j idos (músculo liso, esquelético, cardíaco y sistema nerv ioso central) y es necesaria para asegurar un buen f unc i onam ien to de la contracción muscular .
El grupo de las distrofinopatías engloba la distrof ia muscular progresiva t ipo Duchenne y la distrofia muscular t i po Becker. Ambas afectan casi exc lus ivamente a varones, actuando las mujeres c o m o portadoras. Se transmiten con herencia recesiva l igada al cromosoma X.
La terapia está todavía en fase de experimentación, con dos vías fundamentales de estudio: el trasplante de mio-blastos normales en el músculo afectado y la terapia génica, que in t roduce el gen normal de la distrof ina en el músculo de un paciente mediante un vector.
Se ha descrito otra proteína íntimamente relacionada con la distrof ina, con una secuencia homologa del 8 0 % y cod i f i cada en el c romosoma 6; es la utrof ina. En músculos normales, la utrofina se local iza p redominantemente en la unión neuromuscular , mientras que la distrof ina se encuentra en la inferíase sarcolémica. La pos ib i l idad de que la utrof ina pueda ser usada para corregir el defecto muscular en las distrofias musculares ligadas al X está siendo considerada.
Distrofia muscular de Duchenne (DMD: la clínica comienza a los 3-5 años, con trastornos en la marcha y debi l idad progresiva de la musculatura prox imal de los miembros y f lexora del cuel lo , estando los miembros inferiores más gravemente afectados que los superiores. Es característica la pseudohiper t rof ia de panto-
- M I R 0 2 - 0 3 , 2 1 2 - M I R 98-99F , 76
100

Neurología y neurocirugía
rri l las, deb ido al r eemplazamiento del músculo por grasa y te j ido con jun t i vo . Si el paciente intenta levantarse desde el suelo, desarro l la la man iobra de Gowers (trepa sobre sí m i smo para levantarse) (Figura 67).
Distrofia muscular de Becker: es una variante alélica de la D M D , de comienzo más tardío, evolución más benigna y frecuencia menor. La distribución de la afectación muscular es la misma que en la D M D , siendo también un hal lazgo precoz y p rominente la pseudo
hipertrof ia muscular, par t icu larmente gemelar. La edad de debut es a los 5-15 años, con deambulación mantenida más allá de los 15 años. La expectativa de v ida se sitúa en la cuarta y qu inta década, y es menos frecuente la asociación de retraso menta l . La CPK y el EMG son análogos a los de la D M D . En la biopsia se detecta distrof ina en escasa cant idad y de menor tamaño. No se conoce adecuadamente el resultado del t ratamiento con prednisona.
Distrofia facioescapulohumeral
Frecuentemente se asocia con deterioro intelectual no progresivo y escoliosis progresiva. Hacia los 12 años, la mayoría de los pacientes ut i l iza silla de ruedas. Suelen fal lecer en la segunda década de la v ida por infecciones pulmonares intercurrentes. La causa cardíaca de muerte es poco común, a pesar de la existencia de miocardiopatía en casi todos los casos. Hay importante aumento de la CPK sérica, incluso desde el nac i miento, y su valoración es fundamental en la detección de portadoras ( 5 0 % de ellas t ienen CPK alta). El electromiograma (EMG) demuestra hallazgos miopáticos con act iv idad espontánea y potenciales polifásicos breves de escasa a m p l i tud . La biopsia muscular establece el diagnóstico def in i t ivo ; muestra una total ausencia de distrofina, que puede detectarse mediante inmuno-histoquímica, western-blot o microscopía electrónica (Figura 68). El tratamiento con prednisona puede alterar el curso de la enfermedad.
Se denomina también distrofia de Landouzy-Dejerine. T iene una herencia autosómica dominante , con penetrancia casi comple ta y una afectación similar por sexos. Se ha loca l izado la anomalía genética en el brazo largo del cromosoma 4.
Su espectro clínico es muy a m p l i o , y suele debutar en edad j uven i l . La afectación facial suele ser la manifestación in ic ia l , con incapac idad para reír o cerrar comple tamente los ojos. La deb i l idad de la c i n t u ra escapular imp ide elevar los brazos, presentando escápula alada. La musculatura b ic ip i ta l y t r ic ip i ta l puede estar gravemente afectada, con respeto relat ivo del deltoides. En un 2 0 % hay también afectación de la c intura pelv iana.
No se afectan otros órganos, aunque puede haber hipertensión y sordera neurosensorial . La función intelectual y la esperanza de v ida son normales y la CPK puede ser norma l o levemente elevada.
Figura 68. Patrón de inmunotinción normal con el anticuerpo antidistrofina (izquierda)
Distrofia miotónica de Steinert
Es una enfermedad con herencia auto sómica dominante , t ransmit ida a través de un gen anómalo loca l izado en el brazo largo del cromosoma 19, que cod i f i ca la miotonina proteincinasa. El defecto genético es la repetición de un trinucleótido expand ido (CTG) en d i cho c romosoma, por lo que se produce fenómeno de anticipación.
La mutación es inestable, lo que expl ica el a m p l i o rango de gravedad clínica de la distrofia miotónica; algunos pac ien tes están gravemente afectados desde el
101

Manual CTO Medicina y Cirugía, 8 . a edición
nac imiento , sobre todo , cuando descienden de madre afectada; otros están mínimamente afectados hasta la v ida adulta.
Clínica
La enfermedad debuta en la segunda o tercera década, con deb i l idad en la musculatura fac ia l , f lexora del cue l lo y distal de los miembros y atrofia de musculatura fac ia l , maseteros y músculo tempora l . La afectación de lengua, faringe y paladar conduce a voz nasal y disfagia. Es característico el fenómeno miotónico en las manos, párpados y lengua, con una d i f i cu l tad para la relajación muscular que típicamente mejora con el e jerc ic io repet ido y empeora con el frío.
Se asocia a deter ioro inte lectual , h ipersomnia , ca lv ic ie f ronta l , cataratas subcapsulares, atrofia gonada l , insuf ic iencia respiratoria por deb i l idad de la musculatura respiratoria, resistencia a la insul ina, alteraciones gastrointestinales y cardiopatía con alteración del sistema de conduc ción (b loqueo A-V) (Figura 69) (MIR 02-03, 212).
Calvicie frontal
Cataratas subcapsulares
Bloqueo A-V
Debilidad muscular distal de los miembros
Figura 69. Distrofia miotónica de Steinert
Pruebas complementarias
La CPK puede ser normal o discretamente elevada y el EMC demuestra descargas miotónicas (Figura 70).
Q R E C U E R D A Otras enfermedades que presentan fenómeno de anticipación son la corea de Huntington (triplete CAG) y el síndrome X frágil (triplete CGG).
En la biopsia hay atrofia muscular, preferentemente de fibras t ipo I, pero a diferencia de otras distrofias musculares, no hay necrosis de éstas.
Tratamiento
El t ratamiento de elección de la miotonía, si se precisa, es la fenitoína. Los trastornos de conducción pueden requerir colocación de marca-pasos.
Distrofia muscular de las cinturas
Engloba un grupo amp l i o de distrofias musculares de herencia auto sómica dominan te (AD) o recesiva (AR). En los últimos años se han descrito un gran número de estas enfermedades, exist iendo en la actual idad 7 distrofias de cinturas A D y 11 AR.
Las enfermedades afectan por igual a ambos sexos y debutan entre la pr imera y la cuarta década, caracterizadas por deb i l i dad de la muscu latura p rox ima l de miembros inferiores y superiores. Pueden presentar insuf ic iencia respiratoria, aunque la gravedad y grado de afectación es muy var iable entre distintas enfermedades.
En España, se ha descrito una alta inc idenc ia en la etnia vasca de dis trof ia de cinturas t i po 2A. Se trata de una enfermedad con herencia au tonómica recesiva, y alteración de la proteína calpaína. Los pacientes pueden debutar en la infancia o en la adolescencia, aunque hay casos descritos de debut hasta los 40 años. Existe deb i l idad de la musculatura pelv iana, especia lmente del grupo posterior, y de la c intura escapular, pud iendo cursar con escápula alada.
13.2. Miopatías congénitas
Son un con jun to de enfermedades caracterizadas por la presencia de alteraciones anatomopatológicas e histoquímicas específicas en el músculo. Están presentes al nac imiento y su evolución suele ser be nigna, escasamente progresiva o no progresiva, aunque se han descrito casos graves de evolución fatal . No existe p roced imien to que evite la progresión de la enfermedad, limitándose a t ratamiento de soporte y rehabilitación.
La clínica miopática es muy simi lar en todas ellas y suelen impl i car alteraciones esqueléticas t ipo cifoescol iosis, luxación de caderas o pie cavo. Inc luyen: • Miopatía central-core. Se hereda, en general , con carácter autosó
m i c o dominan te l igado al c romosoma 19, aunque se han descrito
1 0 2

Neurología y neurocirugía
casos esporádicos. Asocia cifoescol iosis, lordosis, pie cavo y luxa ción congénita de caderas. La deb i l idad afecta preferentemente a la musculatura p rox ima l de miembros inferiores. La CPK es norma l . En la biopsia muscular, la parte central de las fibras musculares no muestra react iv idad histoquímica con enzimas oxidat ivas a causa de la v i r tual ausencia de mi tocondr ias (central-core). Es característica la predisposición de estos pacientes a sufrir hiper-termia mal igna cuando se someten a anestesia general . Miopatía nemalínica (Figura 71). Enfermedad autosómica d o m i n a n te con penetrancia incompleta y existencia de casos esporádicos. Conl leva facies e longada, paladar o j i va l , tendencia al prognat ismo, pectus excavatum, pie cavo, cifoescol iosis y, a veces, distribución escapuloperoneal de la deb i l i dad . Con la tinción de C o m o r i , las fibras, fundamenta lmente las de t ipo I, muestran múltiples cuerpos en forma de bastón (cuerpos de nema-lina) que cont ienen material idéntico a las líneas Z.
Figura 71. Cuerpos nemalínicos
Miopatía miotubular o centronuclear. Presenta heterogenic idad genética. Se di ferencia de las otras miopatías congénitas por la presencia de ptosis y grados variables de o f ta lmople j i a externa. Si se real izara una biopsia, se observarían agrupamientos de núcleos centrales. Desproporción congénita del tipo de fibras. En músculos normales, las fibras t ipo II representan el 6 0 % y el t i po I, el 3 0 - 4 0 % . Una relación inversa caracteriza este t i po de miopatía. La enfermedad puede estar genéticamente determinada de forma A D .
13.3. Miopatías metabólicas o a b i a s ^
- Por al teración de l me tabo l i smo h id roca rbonado • Por al teración de l me tabo l i smo l ípídico - E n e l s eno de en f e rmedades sistémica:
- Hipertiroidismo e hipotiroidismo - Hiperparatiroidismo e hipoparatlroidismo - Trastornos suprarrenales - Acromegalia - Diabetes - Deficiencia de vitaminas E y D
Tabla 52. Miopatías metabólicas
Enfermedad de Pompe o glucogenosis tipo II (deficiencia de maltasa acida)
La maltasa es un e n z i m a que degrada el glucógeno para la o b t e n ción de g lucosa . Su déficit c o n d u c e a la g lucogenos i s t i p o I I , una e n t i d a d autosómica recesiva c u y o gen está l o c a l i z a d o en el b r azo largo de l c romosoma 17 . Es pos ib l e el diagnóstico p r ena t a l . La e n f e r m e d a d de Pompe representa la f o r m a más grave de g l u c o g e nosis.
Clínica
Existen tres f o rmas cl ínicas. La forma infantil es la más común, y debu t a a los tres meses de v i d a . Cursa con d e b i l i d a d muscu l a r grave , c a r d i o m e g a l i a , h e p a t o m e g a l i a , m a c r o g l o s i a e i n su f i c i enc i a resp i ra to r i a , produciéndose la mue r t e g e n e r a l m e n t e du r an t e el p r i mer año.
La forma juvenil puede confundi rse clínicamente con una distrof ia muscular . La clínica se restringe al músculo, con deb i l i dad p rox ima l , aumento del tamaño de las pantorr i l las e insuf ic iencia respiratoria. El corazón puede estar afectado, pero no hay afectación hepática. La muerte se produce en la segunda década.
La forma del adulto debuta entre la tercera y la cuarta década, cursa con insuf ic iencia respiratoria y afectación p rox ima l de los miembros , pero no hay afectación cardíaca o hepática.
La CPK está elevada. El EMC muestra, j un to a un patrón miopático típico, descargas miotónicas y trenes de fibrilación y ondas positivas. El diagnóstico de f in i t i vo se establece con la determinación enzimática a nivel muscular.
Enfermedad de McArdle o glucogenosis tipo V (deficiencia de miofosforilasa)
Muestra una herencia autosómica recesiva l igada al brazo largo del cromosoma 11. Afecta p redominantemente a varones y suele debutar en la adolescencia.
Clínica
Es característica la i n to l e ranc i a al e j e r c i c i o , c on ca lambres m u s c u lares y fa t iga . Si reposan b revemente tras e j e r c i c i o , pueden c o n t i nuar la a c t i v i d a d . Los pac ientes están asintomáticos en reposo, y no hay afectación de otros órganos. El sobree je r c i c io puede c o n d u c i r a rabdomiólisis y m i o g l o b i n u r i a con f a l l o rena l . Sin emba rgo , la causa más común de m i o g l o b i n u r i a recurrente es una miopatía por t ras torno de l m e t a b o l i s m o lipídico: el déficit de c a r n i t i n a p a l m i t o i l -transferasa.
La CPK puede estar e levada, aún co n el pac ien te asintomático. El EMC es no rma l en fases asintomáticas. El e j e r c i c i o ¡sométrico del an tebrazo en cond i c i ones de i squemia no i nduce a u m e n t o del ácido láctico.
103

M a n u a l C T O M e d i c i n a y Cirugía, 8.a ed i c ión
13.4. Miopatías mitocondriales Son un grupo heterogéneo de enfermedades caracterizadas a nivel anatomopatológico por la presencia de las denominadas fibras ragged-red, que resultan del a cumu lo de mi tocondr ias anormales en el músculo, y que se ponen de manif iesto por med io de la tinción de tricrómico de G o m o r i (Figura 72). Por microscopía electrónica, la m i tocondr i a muestra inclusiones cristal inas.
Figura 72. Fibras ragged-red (miopatía mitocondrial)
Presentan herenc ia m i t o c o n d r i a l . Puesto que el A D N m i to cond r i a l se hereda d i rec tamente desde el c i top lasma del o voc i t o , los genes mi tocondr ia l es der i van casi exc lus i vamente de la madre (herencia materna) . • Síndrome de Kearn-Sayre. Se trata de una enfe rmedad esporádica,
sin histor ia f ami l i a r . Debuta antes de los 20 años, y se carac ter i za por la tríada de o f t a lmop le j i a externa progresiva, degeneración p igmentar ia de la retina y b l o q u e o de la conducción cardíaca. La mayoría de los pacientes con ptosis progresiva insidiosa no fluc-tuante y o f t a lmop le j i a t i enen una miopatía m i t o c o n d r i a l . Otros datos clínicos son: retraso sexual , sordera, estatura corta , ataxia y demenc i a .
• MERRF. Es el acrónimo inglés de epi lepsia mioclónica y fibras ragged-red. Es una miopatía mi tocondr ia l que cursa con crisis com i c i a les, mioclonías y deter ioro inte lectual , pero no hay o f ta lmople j i a . Se produce por una mutación puntua l del ARNt de la lisina en el A D N mi tocondr i a l .
• MELAS. Es el acrónimo inglés de miopatía, encefalopatía, acido-sis láctica y stroke o acc idente vascular cerebra l . Comienza en la infanc ia y cursa con miopatía, acc identes vasculares cerebrales recurrentes (hemiparesia, hemianops ia , ceguera cor t i ca l , etc.), vó mitos secundarios a lactacidosis, crisis comic ia les , epi lepsia m i o clónica y demenc ia . La muerte sobreviene antes de los 20 años. Se p roduce por una mutación en el ARNt de la leuc ina del A D N m i t o c o n d r i a l .
R E C U E R D A Las miopatías mitocondriales son de herencia materna o mitocondrial. Anatomopatológicamente, se caracterizan por las l lamadas fibras rojo-rasgadas (ragged-red).
En una exploración rutinaria de un paciente de 34 años de edad, se encuentra una glucemia de 160 mg/dl, una CPK de 429 U/I, GTP 62 U/l, GOT 43 U/l y GGT 32 U/l. En el electrocardiograma, presenta un bloqueo AV de primer grado. En la exploración física, se aprecian opacidades corneales incipientes y dificultad para relajar un músculo después de una contracción intensa, siendo muy evidente en las manos. jQué enfermedad padece el paciente?
Casos clínicos representativos
1) U n a miopat ía m i t o c o n d r i a l . 2) U n a d i s t r o f i a m u s c u l a r d e c i n t u r a s . 3) U n a d i s t r o f i a m u s c u l a r d e D u c h e n n e . 4) U n a d i s t r o f i a m u s c u l a r d e Ste inert . 5) U n a d i s t r o f i a m u s c u l a r d e Becker .
M I R 0 2 - 0 3 , 2 1 2 ; RC: 4
104

Neurología y neurocirugía |
14. CEFALEAS
Orientación
MIR r
Aspectos esenciales
T e m a d e i m p o r t a n c i a i n te rmed ia-ba ja en el M IR , p e r o q u e está c a y e n d o c o n s t a n t e m e n t e en las últimas c o n v o c a t o r i a s . Es r e c o m e n d a b l e centrarse en la migraña, e s p e c i a l m e n t e en su t r a t a m i e n t o y p r o f i l a x i s , y en la ce fa lea en c luster . El resto es p r e s c i n d i b l e .
p¡~] El tratamiento de los ataques migrañosos leves-moderados son los A INE. En el caso de ataques moderados-graves, se usan los triptanes. La ergotamina es una alternativa, pero su uso crónico puede producir una cefalea ergotamín-dependiente.
f j j Para la prevención de los ataques de migraña se usan p-bloqueantes, calcioantagonistas, antidepresivos tricíclicos, antiepilépticos y antagonistas de la serotonina. Recuérdese que la metisergida (antagonista de la serotonina) puede provocar fibrosis pleural, pericárdica o retroperitoneal.
La cefalea en cluster se presenta en hombres de 20-50 años. D a episodios de cefalea unilateral periocular, típicamente nocturna. Puede implicar lagrimeo, rinorrea, inquietud o síndrome de Horner, entre otros.
|"4~] El a lcohol es un desencadenante de la cefalea en cluster.
["5"] El tratamiento de elección de la cefalea en cluster es el sumatriptán subcutáneo. La segunda opción es oxígeno a alto flujo. C o m o profilaxis, se usa el verapamilo.
Qf ) La arteritis de la temporal es una causa de cefalea que debe ser sospechada ante pacientes mayores de 50 años con cefalea hemicraneal de reciente comienzo , asociada a c laudicación mandibular y a una arteria temporal sin pulso.
14.1. Consideraciones generales
CEU P regun tas
•MIR 0 9 - 1 0 , 6 7 • M I R 0 5 - 0 6 , 6 3 • M I R 0 4 - 0 5 , 6 3
M IR 0 3 - 0 4 , 2 3 8 • M I R 0 2 - 0 3 , 2 1 0 •MIR 9 9 - 0 0 , 1 9 2 •MIR 00-01 F, 6 8 , 1 5 3 •MIR 9 9 - 0 0 , 1 9 2 , 2 0 2
Se trata de unos de los mo t i vos de consu l ta más f recuentes en nuestro m e d i o . La cefalea es genera lmente un síntoma ben igno , y sólo ocas i ona lmen te es manifestación de una en f e rmedad seria, c o m o men ing i t i s , hemor rag ia subaracno idea , tumores o arter i t is de la t e m p o r a l . El p r ime r o b j e t i v o ante un pac ien te c o n ce falea es descartar estas patologías. Los cr i ter ios de gravedad de una cefa lea quedan resumidos en la Tabla 53 . En el diagnóstico, hay que tener en cuenta la edad , el sexo y la ocupación labora l del pac iente , la edad de c o m i e n z o de los síntomas, antecedentes personales y fami l i a res , f r ecuenc ia de los ep i sod ios , i n tens idad , duración, m o d o de instauración, cua l i dad del do l o r , local ización, factores modu l ado re s , síntomas genera les y neurológicos asoc iados, y hábitos tóxicos y c o n s u m o de fármacos (Tabla 54) . • Hemorragia subaracnoidea. Se mani f ies ta
con cefalea intensa, de c o m i e n z o súbito, que suele acompañarse de r ig idez de nuca, náuseas y vómitos.
• Meningitis. Cefa lea, f i ebre , más signos m e níngeos.
• Tumores . P roducen cefa lea de carácter p r o gres ivo , de días o semanas de evolución. Una cefa lea de estas características en un pac ien te sin cefa lea prev ia (o de dist intas ca racterísticas a la hab i tua l en pac ientes con antecedentes de cefalea) nos debe hacer pensar en la ex is tenc ia de un proceso e x p a n s ivo c o m o p r ime ra p o s i b i l i d a d diagnóstica. La cefa lea por hipertensión in t racranea l e m peora por la mañana y puede despertar al pac ien te del sueño, aumenta con la tos y los
Cefalea intensa de comienzo súbito Empeoramiento reciente de una cefalea crónica Cefalea de frecuencia y/o intensidad creciente Localización unilateral, siempre en el mismo lado (excepto cefalea en racimos, hemicránea paroxística o continua, neuralgia occipital, del trigémino, y otras cefaleas primarias unilaterales Manifestaciones acompañantes: - Alteración psíquica progresiva - Crisis epiléptica - Alteración neurológica focal - Papiledema - Fiebre - Náuseas y vómitos no explicables por una cefalea
primaria, ni por una enfermedad sistémica - Presencia de signos meníngeos
Cefalea precipitada por esfuerzo físico, tos o cambio postural
Tabla 53. Manifestaciones de alarma en una cefalea
105

Manual CTO Medicina y Cirugía 8 a Edición
vómitos, me jo ra i n i c i a l m e n t e con parace tamol o ácido acet i lsa l i-cí l ico (a d i f e renc ia de la cefalea psicógena) y se asocia a náuseas, vómitos, pap i l edema y signos neurológicos foca les .
14.3. Migraña
CEFALEAS PRIMARIAS
• Migraña • Cefalea tensional • Cefalea en racimos y otras cefalalgias
trigémino-autonómicas • Otras cefaleas primarias
CEFALEAS SECUNDARIAS
• Atribuidas a traumatismo craneal o cervical, a trastorno vascular craneal o cervical, a trastorno intracraneal no vascular, a una sustancia o a su supresión, a infección, a trastorno de la homeostasis, atribuido a estructuras faciales o craneales, o a trastorno psiquiátrico.
NEURALGIAS CRANEALES , DOLOR FACIAL CENTRAL
Y PRIMARIO Y OTRAS CEFALEAS
• Entre otras: neuralgia del trigémino, del glosofaríngeo, del occipital, síndrome deTolosa-Hunt
Tabla 54. Clasificación internacional de las cefaleas (IHS, 2004)
Arterit is de la temporal . Hay que sospechar la ante una cefa lea hemic ranea l en pac ientes mayores de 60 años. Son datos asoc iados : p o l i m i a l g i a reumática, c laudicación m a n d i b u l a r , d o l o r y tensión a la palpación del t rayec to arter ia l y ausencia de pu lso . La pérdida de agudeza v isua l por oclusión de la arter ia oftálmica ( 5 0 % de pacientes) es un dato o r i en t a t i vo en un pac iente mayor con cefa lea.
La mayoría de los pac ientes presenta el p r ime r ep i sod io de migraña entre los 10-30 años y en el 6 0 - 7 5 % de los casos son mujeres . Existe una predisposición hered i ta r ia .
Fisiopatología
La patogénesis de la migraña puede ser cons ide rada en tres fases: • Génesis troncoencefálica con pos ib le participación de los nú
cleos del rafe m e d i o (serotoninérgicos). • Act ivación vasomoto ra co n contracción vascular i n i c i a l , que jus
tificaría la f o c a l i d a d neurológica en la migraña con aura , y una segunda fase de vasodilatación.
• Act ivación de neuronas de l núcleo t r i g em ina l a n ive l bu lba r , c o n poster ior liberación de neuropéptidos vasoact ivos en las t e r m i nac iones vasculares de l ne r v io trigémino. Esta fase c o n d i c i o n a la tumefacción t i su lar y tensión de los vasos sanguíneos duran te el e p i s o d i o de migraña.
Du ran t e el e p i s o d i o de migraña se ha demos t r ado , por estudios de f l u j o sanguíneo reg iona l , una hipoperfusión co r t i c a l que c o m i e n z a en el córtex v isua l y se ex t i ende hac ia de lan te a una v e l o c i d a d de 2-3 m m / m i n . A veces, la hipoperfusión persiste a pesar de haber c e d i d o los síntomas.
R E C U E R D A Ante una neuritis óptica acompañada de sintomatología de la arteritis temporal en un paciente anciano, está indicado el tratamiento con corticoides, ya que es fundamental evitar la bilateralización y las compl i cac iones sistémicas.
Los cr i te r ios diagnósticos para la arter i t is de la t empora l son : - Edad super ior a 50 años. - Cefalea loca l i zada de rec iente c o m i e n z o . - Tensión sobre la arter ia t e m p o r a l o disminución del pu lso . - VSG mayo r a 50 mm/h (MIR 00-01 F, 68 ) . - B iopsia ar ter ia l mos t r ando arter i t is nec ro t i zan te .
Es pos ib l e que la migraña represente una perturbación hered i ta r ia de la neurotransmisión serotoninérgica. La se ro ton ina parece jugar un pape l p r i m o r d i a l en la pa togen ia de la migraña. Son ev idenc ias a favor : • La met i serg ida es un b l oquean t e de receptores serotoninérgicos
que se ha demos t r ado útil para p reven i r los ep isod ios de m i graña. El sumatriptán, c u y o m e c a n i s m o de acción es agonista de receptores serotoninérgicos 1 B, y espec ia lmente 1 D, es a l ta men te ef icaz en el t r a t am ien to , en fase aguda , de los ep isod ios migrañosos. Los n ive les p laque ta r ios de se ro ton ina desc ienden al i n i c i o de la cefa lea. Los ep isod ios migrañosos pueden ser desencadenados por fármacos que l ibe ran se ro ton ina .
14.2. Cefalea tensional Subtipos clínicos
Es el t i po de cefalea más frecuente y p redomina en la mujer . Se d is t in guen tres formas de cefalea de tensión: episódica infrecuente, episódica frecuente, y crónica. C o m o criterios diagnósticos, destacan episodios de cefalea que duren entre 30 minutos y siete días, de ca l idad opresiva, intensidad leve o moderada, localización bi lateral , no agravada por esfuerzos físicos o no asociada a náuseas ni vómitos.
El t r a t am ien to de l do lo r se rea l iza con A INE, pa race t amo l , o ana l gésicos comunes .
El t r a t am ien to p reven t i vo , según f recuenc ia , duración e in tens idad de los do lores , se basa en el uso de ant idepres ivos tricíclicos e i n h i b idores select ivos de la recaptación de la se ro ton ina .
La Tab la 55 recoge las características y los c r i te r ios diagnósticos de esta e n f e r m e d a d . • Migraña con aura o migraña clásica. Representa el 2 0 % de las
migrañas. Es una cefalea recurrente, de p r e d o m i n i o hemicranea l y carácter pulsátil, que puede acompañarse de náuseas, vómitos, f o to fob i a y sonofob ia , que dura entre 4 y 72 horas. Se precede de clínica de foca l i dad neurológica (aura), s iendo las man i fes tac io nes visuales las más frecuentes (escotomas centel leantes, visión borrosa, defectos hemianópsicos, espectro de fortificación, etc.), aunque también puede haber síntomas motores o sensit ivos. Preceden a la cefalea en 15-30 minutos , y hab i tua lmente desaparecen minutos antes de comenzar la cefalea.
• Migraña sin aura o migraña común. Representa el 7 5 % de los casos de migrañas.
1 0 6

Neurología y neurocirugía
Al menos 5 episodios, cumpliendo los siguientes criterios Duración del episodio de 2-72 horas (sin tratamiento o tratada sin éxito) Al menos 2 de los siguientes datos: - Unilateral (30-40 % son bilaterales) - Pulsátil (50 % de los casos son no pulsátiles) - Moderada a severa (interfiriendo o evitando
las tareas cotidianas) - Agravada por el movimiento (caminar o subir
escaleras)
Al menos un síntoma asociado: - Náuseas o vómitos - Fotofobia - Sonofobia
El dolor no se atribuye a otra enfermedad
A los criterios descritos anteriormente se añaden los siguientes: • Uno o más síntomas focales neurológicos
transitorios ( 9 0 % visuales) antes o durante la cefalea
• Duración del aura de 5-60 minutos • La cefalea acompaña o sigue al aura dentro
de los siguientes 60 minutos • El dolor no se atribuye a otra enfermedad
Tabla 55. Criterios diagnósticos simplificados para la migraña con y sin aura
Consiste en cefaleas de análogas características a las descritas en la migraña con aura, pero sin cl ínica de f o ca l i d ad neurológica p reced iendo o acompañando a la cefalea.
A m b o s t ipos de migraña pueden ocu r r i r en el m i s m o pac iente . Entre los ep isodios de migraña, el pac iente puede tener cefaleas tensiona-les. Las crisis se pueden desencadenar por diversos factores dietéticos, ambienta les , psicológicos, ho rmona les y farmacológicos.
Infarto migrañoso o migraña compl icada: c u a n d o los síntomas del aura migrañosa persisten más allá de la duración de la ce falea, y se asoc ian a una lesión isquémica cerebra l de l m i s m o te r r i to r io vascular, demos t rado por imagen (MIR 00-01 F, 1 53) .
Tratamiento
Las pr incipales opc iones de t ratamiento de las migrañas se expone en la Tabla 56.
14.4. Cefalea en cluster (o en racimos), histamínica o de Horton
Predomina en varones (10:1) y debuta a cua lquier edad, aunque preferentemente entre los 20-50 años. Se dist ingue una forma episódica y otra crónica (cuando hay ausencia de fases de remisión durante un año o más, o con remisiones que duran menos de un mes).
Se caracteriza por presentar episodios diarios de cefalea uni la tera l , lo cal izada preferentemente a nivel per iocular y con irradiación a la f ren te o a la mandíbula, de gran gravedad, y cuya duración puede variar entre 15-180 minutos , desde una vez cada dos días, hasta 8 veces al día. Aparece característicamente por la noche, aprox imadamente una hora después de conc i l i a r el sueño, y puede recurrir durante el día, a menudo a la misma hora. En muchos casos, se acompaña de lagr imeo, r inorrea, congestión ocular y obstrucción nasal ipsilateral al do lor , su-doración frontal y fac ia l , edema palpebral ipsi lateral, miosis-ptosis ips i lateral, e inqu ie tud motora y desasosiego (MIR 09-10, 67). En el 2 5 % de
Migraña basilar
Los síntomas neurológicos que preceden a la cefalea son característicos de disfunción t ronco-encefálica: vértigo, d isartr ia , diplopía, ataxia o síndrome con fus iona l , persisten durante 20-30 m inu tos y se s iguen de cefalea occ ip i t a l pulsátil. Suele aparecer en adul tos jóvenes.
Migraña hemipléjica
Se trata de una migraña con aura, que inc luye hemipares ia . Puede ser f ami l i a r , si hay un pa r iente de pr imer o segundo grado con ataques idénticos, o esporádica.
Complicaciones de la migraña
Migraña crónica: más de 15 ep isod ios al mes, por un t i e m p o super ior a los tres meses. Estado de mal migrañoso: más de 72 horas de duración, a pesar del t r a tamien to .
TIPO DE TRATAMIENTO
I. Ataques leves moderados
Preventivo (si la frecuencia es superior a dos episodios al mes)
FÁRMACOS COMENTARIOS
AINE (AAS, naproxeno o ibuprofeno)
Se deben administrar inmediatamente después del inicio de la cefalea, repitiendo la dosis cada 4-6 h. La administración de metoclopramida o domperidona no sólo mejora las náuseas o los vómitos, sino que además facilita la absorción de los analgésicos. Junto al tratamiento analgésico, se debe descansar, si es posible, en lugar oscuro y silencioso
TRIPTANOS (sumatriptán, nartriptán, zolmitriptán, rizatriptán almotriptán, eletriptán yfrovatriptan)
Son agonistas de receptores serotonlnérgico (5HT1B y 1D) con acción vasoconstrictora y reductora de la Inflamación alrededor de los vasos Contraindicaciones: cardiopatía isquémica o claudicación intermitente
P-BLOQUEANTES (propranolol)
El mecanismo por el que ejercen su acción profiláctica resulta desconocido, aunque se postula un efecto bloqueante de receptores serotoninérgicos 5-HT2 (MIR 99-00, 202)
CALCIOANTAGONISTAS (flunarlcina, cinaricina, verapamilo)
Los dos primeros se deben usar con precaución en pacientes con enfermedad de Parkinson, enfermedades depresivas previas o trastornos extrapiramidales de otro tipo
ANTIDEPRESIVOS TRIClCLICOS (amitriptilina, nortriptillna)
Especialmente indicados en pacientes con migraña asociada a cefalea tensional. Su acción parece Independiente de su actividad antidepresiva (MIR 05-06, 63)
ANTAGONISTAS DE LA SEROTONINA (clproheptadina, plzotifen, metisergida)
Muy eficaces; deben ser administrados con precaución debido a sus efectos secundarlos importantes, aunque reversibles, tras su uso prolongado: fibrosis pleural, pericárdlca y retroperitoneal.
Tabla 56. Principales opciones en el tratamiento de la migraña
1 0 7

Manual CTO Medicina y Cirugía 8 a Edición
los casos se acompaña de un síndrome de Horner que ocas ionalmente puede persistir (MIR 03-04, 238 ; MIR 02-03, 210 ; MIR 99-00, 192).
La cefalea aparece d iar iamente durante per iodos (cluster) que osci lan entre 1 -4 meses, quedando poster iormente asintomático durante largos per iodos de t i empo (1-2 años). No se acompaña de aura, náuseas ni historia fami l iar . • Tratamiento preventivo. Evitando factores desencadenantes, si es
tos existen, c o m o el a lcohol y otros vasodilatadores. • Tratamiento sintomático. La terapia de elección es el sumatriptán
subcutáneo, por su rapidez y ef icacia. La segunda medida más efect iva es la inhalación de oxígeno a f lu jo e levado (MIR 04-05, 63).
• Tratamiento profiláctico. Se considera el ve rapami lo c o m o el fármaco de elección. Si no hay respuesta, se puede intentar con cursos breves de cort icoides, top i ramato , la ergotamina en dosis única noc turna, o el l i t io .
14.5. Otras cefaleas primarias • Hemicránea paroxística. Predomina en la mujer , de in i c io en la
edad adulta. Existe una fo rma episódica y otra crónica. Se trata de una cefalea trigémino-autonómica, con episodios de do lor similares a la cefalea en racimos, pero con una duración más breve (2-30 min. ) , y una frecuencia mayor (5-30 episodios al día). La buena respuesta a la indometac ina es un cr i ter io diagnóstico.
• S U N C T (short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing). Se trata de crisis breves de do lor neura lg i forme uni latera l , acompañado de inyección con junt i va l y lagr imeo. Entre 3-200 crisis al día, en ocasiones desencadenados por ciertos estímulos orofaciales.
• Neuralgia del trigémino (véase el Capítulo de Neurocirugía funcional)-
r
Casos clínicos representativos
M u j e r de 34 años , d i agnos t i cada de migraña s in au ra , que consu l t a por ep isod ios de sus ce fa leas hab i tua les , en número de 4-5 al m e s . ¿Cuá l de estos t ra tamientos N O estaría ind i cado?
1) T o m a r t r i p t anes d u r a n t e t o d o s los a taques . 2) U t i l i z a r dos is ba jas d i a r i as d e e r g o t a m i n a . 3) A d m i n i s t r a r c o m o p r o f i l a x i s p r o p a n o l o l . 4 ) T ra ta r t o d o s los a t aques a g u d o s c o n n a p r o x e n o . 5) U t i l i z a r c o m o p r o f i l a x i s f l u n a r i c i n a .
M I R 0 5 - 0 6 , 6 3 ; RC; 2
U n pac i en te de 54 años ref iere , desde h a c e d iez días, u n a o dos c r i s i s de do lo r de o jo d e r e c h o , c o n lagr imeo, gran ne rv ios i smo, que le desp ier ta por la n o c h e y le ob l iga a sal ir de la c a m a , durándo le unas dos horas . ¿Cuá l de las s iguientes med idas ent iende que es más e f i c az pa ra c a l m a r el do lo r ?
1) Ox ígeno in t r anasa l . 2 ) Sumatr iptán subcutáneo. 3) I b u p r o f e n o o r a l . 4 ) T r a m a d o l o r a l . 5) M e t a m i z o l i n t r a m u s c u l a r .
M I R 0 4 - 0 5 , 6 3 : RC: 2
1 0 8

Neurología y neurocirugía
15. SÍNDROME DE HIPERTENSIÓN INTRACRANEAL
MIR D e n t r o d e este t e m a , se d e b e c o n o c e r m u y b i e n el p s e u d o t u m o r c e r e b r a l , e s p e c i a l m e n t e los c r i t e r i o s diagnósticos y las c o n d i c i o n e s asoc iadas a esta patología. Además , es i m p o r t a n t e c o n o c e r la c l ínica d e la herniac ión u n c a l y el t r a t a m i e n t o gene ra l d e la hipertensión i n t r a c r a n e a l .
Aspectos esenciales
Q J El f l u j o sanguíneo cerebra l se debe mantener den t ro de unos límites precisos para un co r rec to f u n c i o n a m i e n to del sistema nerv ioso cent ra l . El f l u j o sanguíneo depende de manera d i rec ta de la presión de perfusión cerebral y, de manera ind i rec ta , de las resistencias vasculares cerebra les .
[ J ] C o m o el f l u j o sanguíneo se debe mantener en valores constantes, las m o d i f i c a c i o n e s de la presión de per fusión cerebra l c o n l l e v a n a l terac iones de las resistencias vasculares cerebrales en la m i sma dirección. Es l o que se d e n o m i n a autorregulación cerebrovascu lar .
|~3~] Los factores que i n f l u yen en la presión int racranea l son la sangre, el parénquima y el líquido cefalorraquídeo. La hipótesis de Monro-Ke l l l e establece q u e la suma de los volúmenes de sangre, líquido cefalorraquídeo y parénquima se han de mantener constantes.
[~4~| La hiperventilación, q u e i m p l i c a h i p o c a p n i a , c o n l l e v a una vasoconstricción, lo q u e p rovoca q u e la presión int racranea l desc ienda . Sin emba rgo , puede p r o d u c i r también i squemia ce rebra l .
j j Q La hipertensión in t rac ranea l , q u e se de f ine c o m o valores por e n c i m a de 15 m m H g en el adu l t o , puede p r o duc i r se por a u m e n t o del v o l u m e n de parénquima, líquido cefalorraquídeo o sangre.
[ j j j Las hern iac iones cerebrales son desp lazamien tos del ce rebro desde el s i t io de mayo r al de m e n o r presión. La herniación unca l , q u e cl ínicamente se mani f ies ta c o m o una tríada consistente en midr ias is arreact iva en un o j o , hemipares ia contra la tera l a la midr ias is y disminución del n ive l de c o n c i e n c i a , cons t i tuye una urgenc ia neuroquirúrgica. La herniación ce rebe loamigda l a r se puede p rovoca r por una punción l umba r .
|~7~] En el t r a t am ien to de la hipertensión in t racranea l , se debe in ic ia r c o n med idas de p r ime r n ive l (que pers iguen f u n d a m e n t a l m e n t e d i s m i n u i r el v o l u m e n de sangre, parénquima o líquido cefalorraquídeo) y, ante el f racaso de las mismas, med idas de segundo n i ve l , q u e i m p l i c a n una alta tasa de c o m p l i c a c i o n e s .
QTJ Existen una serie de factores asoc iados al p s e u d o t u m o r ce rebra l , ent re los que destacan los fármacos y de te rminadas metabolopatías. Recuérdese q u e el sexo f e m e n i n o , la obes idad , la edad fértil y los trastornos menstruales son los q u e t i enen una confirmación epidemiológica.
[ 9 ] Los cr i ter ios diagnósticos de l p s e u d o t u m o r cerebral son seis: cl ínica de hipertensión in t racranea l , ausenc ia de f o c a l i d a d neurológica, n ive l de consc i enc i a n o r m a l , estudios de imagen normales , presión de líquido cefalorraquídeo e levada y análisis de líquido n o r m a l .
[Tp] Las med idas terapéuticas fundamenta l e s en el p s e u d o t u m o r cerebral son: e l i m i n a r los factores asociados, a ce t azo l am ida , derivación l u m b o p e r i t o n e a l y la descompresión del nerv io óptico.
Q T j El edema cerebra l vasogénico ( tumores, absceso) cede c o n cor t i co ides , pe ro no es el caso del edema c i to-tóxico ( infar to cerebra l ) .
15.1. Fisiopatología
El Flujo Sanguíneo Cerebral (FSC) puede calcularse d i v i d i endo la Presión de Perfusión Cerebral (PPC) entre las Resistencias Vasculares (RV), s iendo la PPC la di ferencia entre la tensión arterial media (TAM) y la presión intracraneal (PIC).
FSC = PPC / RV = (TAM - PIC) / RV
• M I R 0 8 - 0 9 , 2 3 4 •MIR 0 2 - 0 3 , 2 1 4 • M I R 0 0 - 0 1 , 5 6 •MIR 9 9 - 0 0 , 1 9 0
M I R 9 8 - 9 9 , 2 3 6
El grado óptimo de act iv idad cerebral depende de la existencia de un adecuado f l u jo sanguíneo que propor
c ione al te j ido nervioso un aporte suf ic iente de oxígeno y glucosa. Por tanto, es necesario que d i cho FSC se
mantenga estable frente a variaciones de la presión arterial sistémica o de las resistencias intracraneales. Existen
unos mecanismos de autorregulación cerebrovascular que consiguen que, dentro de un a m p l i o rango, grandes
variaciones de la presión arterial p roduzcan solamente pequeños cambios en el FSC.
1 0 9

M a n u a l CTO d e M e d i c i n a y Cirugía, 8.a ed i c i ón
La hipótesis de Monro-Kel l ie establece que el v o l u m e n total del conte n ido intracraneal (parénquima, sangre y líquido cefalorraquídeo) debe ser constante. Puesto que éstos se encuentran en el inter ior de una cav idad no distensible, c o m o es el cráneo, un incremento en el vo lumen de a lguno de estos componentes (p. e j . : un tumor cerebral , un hematoma ep idura l , subdural o una hidrocefal ia ) hará que, de manera compensator ia , se p roduzcan d isminuc iones en el vo lumen de los otros componentes . Si los mecanismos de compensación se saturan, se pro duce un aumento de la presión intracraneal (cuyos valores normales en los adultos osci lan entre 5 y 15 m m H g ) .
El aumento de la PIC se manif iesta clínicamente con una s in tomato logía característica agrupada bajo la denominación de síndrome de hipertensión intracraneal , común a un gran número de procesos patológicos intracraneales que se describirán en los próximos apartados, algunas de los cuales se recogen en la Tabla 57 .
15.2. Etiología del síndrome de hipertensión intracraneal
La etiología se recoge en la Tabla 57.
Traumat ismo craneoencefálico: - Hematoma epidural - Hematoma subdural - Contusión hemorrágica - Swelling Hidrocefalia Tumores Infecciones: - Absceso cerebral - Empiema subdural Procesos vasculares: - Infarto cerebral - Trombosis venosa - Hematoma intraparenquimatoso Encefalopatías que pueden cursar con edema cerebral : - Hipercápnica - Hepática - Síndrome de desequilibrio (diálisis)
Tabla 57. Causas más frecuentes de elevación de la presión intracraneal
15.3. Clínica
La clínica característica del síndrome de hipertensión intracraneal (HTIC) es: • Cefalea. Genera lmente es más grave durante la noche deb ido a la
h ipercapnia nocturna, que produce vasodilatación cerebral , sobre todo , en la fase REM del sueño. Puede despertar al paciente y e m peora por la mañana. Característicamente aumenta con las man io bras de Valsalva.
• Vómitos. De p r edomin i o matut ino , muy típicos en "escopetazo" . Edema de papila. Es el signo explorator io que traduce la existencia de HTIC. En los lactantes puede no encontrarse, pero se apreciará abombamien to de la fontanela y separación de las suturas (diástasis).
• También aparece frecuentemente diplopía, por lo general secundaria a lesión del VI par craneal. Los niños no suelen quejarse de visión
dob le porque e l iminan más fácilmente la imagen del o jo afectado, pero es frecuente que inc l inen la cabeza para hacer co inc id i r las dos imágenes.
• Alteración del nivel de consciencia.
En fases de HTIC moderada o avanzada, con compromi so en el FSC, puede observarse la tríada de Cushing: hipertensión arterial (lo más constante), bradicardia y alteraciones del r i tmo respiratorio, aunque sólo en un 3 0 % de los pacientes se observa la tríada comple ta .
Otros datos clínicos que pueden acompañar al síndrome de HTIC son las erosiones o úlceras gástricas de Cushing o signos de foca l idad neu rológica en relación con la localización de la lesión responsable de la elevación de la PIC.
15.4. Síndromes de herniación cerebral
El a u m e n t o de la PIC secundar ia a cua lqu i e ra de las ent idades descritas en la Tab la 57 , puede generar gradientes de presión ent re los c o m p a r t i m e n t o s del endocráneo, d a n d o lugar a desp lazamientos de algunas porc iones de l encéfalo cont ra estructuras rígidas óseas o durales, p r o v o c a n d o nuevos déficits neurológicos, de te r io ro de l n i vel de consc i enc i a o inc luso la muer te del pac iente . Estos desp laza mientos se c o n o c e n c o m o hern iac iones cerebrales o enc l avamien tos (Figura 73).
Figura 73. Herniaciones del sistema nervioso central
Se describen varios síndromes de herniación cerebral : • Herniación uncal (Figura 74). Más frecuente en lesiones temporales.
El uncus del lóbulo tempora l se hernia a través de la hendidura ten-tona ! y puede c o m p r i m i r el III par craneal , p roduc i endo midriasis ipsilateral c o m o signo más precoz, y el mesencéfalo, con hemip l e jía contralateral y disminución progresiva del n ive l de consc iencia (MIR 08-09, 234 ; MIR 99-00, 190).
1 1 0

Neurología y neurocirugía
Cerebelo
Tienda del cerebelo
Nervio III PC A. basilar
Figura 74. Herniación uncal
Herniación subfacial. Desplazamiento del parénquima cerebral por debajo de la hoz del cerebro (falx). Puede compr imi r la arteria cerebral anterior. Puede ser un aviso previo a una herniación transtentorial. Herniación central, transtentorial o tentorial. Se produce un desp lazamiento hacia abajo de los hemisferios cerebrales y ganglios básales, c o m p r i m i e n d o sucesivamente: diencéfalo, mesencéfalo, protuberanc ia y bu lbo .
R E C U E R D A La tríada de la herniación uncal: midriasis arreactiva en un ojo, hemiparesia contralateral a la midriasis y disminución del nivel de consciencia. Es imperativo realizar en este caso una TC cerebral urgente.
Herniación transtentorial inversa. Estructuras de la fosa posterior se hernian hacia arriba a través de la hendidura tentor ia l .
R E C U E R D A Antes de realizar una punción lumbar, se debe descartar mediante una TC cerebral que el paciente no tiene una causa que provoque una elevación de la presión dentro del cráneo y genere un gradiente entre el cráneo y la región lumbar.
Herniación cerebelo-amigdalar. Las amígdalas cerebelosas se hernian a través del foramen magno, p roduc iendo compresión bulbar y ocas ionando rápidamente un cuadro dramático con alteración del patrón vent i lator io , trastornos vasomotores y cardíacos e incluso muerte súbita. Puede estar provocada por la realización de una p u n ción lumbar cuando, en el espacio intracraneal, existe una causa que sea capaz de elevar la presión intracraneal y generar un gradiente de presión entre el cráneo y el espacio subaracnoideo lumbar.
La onda de presión intracraneal depende de osci laciones normales de la tensión arterial (en la sístole cardíaca se produce una elevación de la presión intracraneal , al llegar la sangre al cerebro) y del r i tmo respirator io (la inspiración conl leva un descenso de la presión intracraneal , ya que fac i l i ta el retorno venoso al corazón).
En una situación de hipertensión intracraneal , además de obtener en el registro cifras elevadas de presión, se pueden detectar var iaciones en la morfología de la onda normal que t ienen diferentes interpretaciones clínicas (ondas de Lundberg): • Ondas A meseta o plateau: e l e vac iones man ten idas de la PIC
(> 50 m m H g durante 5-20 minutos) que indican compromi so en la autorregulación cerebrovascular. Su morfología se recoge en la Figura 75.
• Ondas B: se han re lac ionado con alteraciones en el r i tmo respirator io, p r inc ipa lmente con la respiración de Cheyne-Stokes.
• Ondas C : probablemente en relación con variaciones en la presión arterial .
Espiración
100 -80 -60
40
20 -0
Ondas plateau
J I L 0 15 30 45 60 75 90 115
Minutos
Figura 75. Onda normal de PIC. Ondas plateau
Los estudios de imagen (TC o RM) ayudan a diagnost icar la causa de la HTIC y las posibles compl i cac iones asociadas (edema, desplazamientos o herniac iones, dilatación ventr icu lar , "swelling" o vasoplej ia venosa cerebral que produce "hinchazón" de los hemisfer ios cerebrales, etc.).
15.6. Tratamiento
15.5. Diagnóstico
El diagnóstico de certeza de un paciente con sospecha clínica de hiper tensión intracraneal se establece mediante la monitorización de la PIC, u t i l i zando sensores que pueden colocarse a nivel int raparenquimatoso, intraventr icular , ep idura l o subdural .
Siempre que sea posible , hay que tratar el p rob lema p r imar io responsable de la HTIC. Por e jemplo , en situaciones en las que haya una h i drocefal ia aguda, habrá que proceder a co locar un drenaje ventr icu lar externo.
En cuanto al t ratamiento general de la hipertensión intracraneal , independientemente de cuál sea la causa que la esté provocando , son útiles las siguientes medidas (Tabla 58) :
111

Manual CTO de Medicina y Cirugía, 8 . a edición
MEDIDAS DE PRIMER NIVEL
MEDIDAS DE S EGUNDO NIVEL
Elevación cabecera 30° Sedación y relajación Drenaje ventricular externo Manitol 2 0 % Suero hipertónico Hiperventilación
Craniectomía descompresiva Coma barbitúrico Hipotermia
Tabla 58. Tratamiento general de la hipertensión intracraneal
Elevar la cabeza del enfermo unos 30° para favorecer el retorno venoso. Evitar la hipotensión arterial , la h ipertermia y la h iperg lucemia . Sedación y relajación, si es necesario. Drenaje ventr icu lar externo. Se trata de una med ida que se emplea en el t ratamiento de la hipertensión intracraneal , aún no ten iendo hidrocefa l ia . Consiste en evacuar líquido cefalorraquídeo a un re-servorio exterior. Man i to l al 2 0 % . Es un diurético osmótico de alta ef icacia y de me can ismo de acción más rápido que los cort icoides, pero es impor tante no superar una osmolar idad plasmática de 320 mOsm/l (MIR 98-99, 236) . Suero hipertónico, que lo que provoca es un paso de líquido desde el espacio intersticial al espacio intravascular deb ido a sus prop ie dades osmóticas. Hiperventilación controlada, para disminuir la p C 0 2 hasta 30-35 m m H g (niveles inferiores t ienen riesgo de isquemia cerebral por vasoconstricción). En casos refractarios a estas medidas, puede ser necesario recurrir al coma barbitúrico, h ipotermia o craniectomías descompresivas.
enumerados en la tabla, los únicos en los que ha habido una confirmación epidemiológica de asociación a este cuadro son: mujer, edad reproductiva, trastornos mensuales, obesidad o aumento reciente de peso.
• Alteraciones del drenaje venoso • Enfermedad de Behcet • Embarazo y anticonceptivos orales - Fármacos: • Obesidad - Vitamina A y derivados (retinoides) • Hipo e hipertiroidismo - Tetraciclinas • Hipoparatiroidismo - Ácido nalidíxico • Insuficiencia suprarrenal - Nitrofurantoína • Síndrome de Cushing - Sulfamidas • Anemia ferropénica grave - Litio • Sarcoidosis - Indometacina • Lupus eritematoso sistémico - Fenitoína
Tabla 60. Etiología del pseudotumor cerebri
Se trata de una enfermedad generalmente auto l imi tada pero recurrente, cuyo riesgo pr inc ipa l es la pérdida de visión por edema de papi la . El t ratamiento t iene c o m o ob je t i vo la prevención de déficit visuales, me diante la disminución del v o l u m e n de LCR.
La pr imera medida terapéutica (Figura 76) debe ser la supresión de los fármacos responsables o el t ratamiento de la enfermedad asociada, si se conoce , y la pérdida de peso, si el paciente es obeso. Después de ben intentarse medidas conservadoras, c o m o el emp leo de diuréticos, especia lmente los inhib idores de la anhidrasa carbónica (acetazolami-da). En la mayoría de los pacientes, el pronóstico es bueno (el 8 0 % responde al t ratamiento conservador) . Otras medidas no quirúrgicas que se pueden emplear son la restricción hidrosal ina, la utilización de dexametasona (especialmente, si hay enfermedades cuyo t ratamiento es la cort icoterapia) y la furosemida.
15.7. Síndrome de hipertensión intracraneal benigna (pseudotumor cerebri)
Se define como la existencia de clínica de hipertensión intracraneal (la cefalea es el síntoma más frecuente y el papi ledema, el signo más constante), sin disminución del nivel de consciencia y sin foca l idad neurológica (salvo la d ip lop ia por afectación del VI par). En las pruebas de ima gen a realizar (resonancia magnética y ang ioRM de fase venosa) no se ev idencia causa just i f icable del cuadro. La punción lumbar presenta, de forma invariable, un incremento de presión de líquido cefalorraquídeo (MIR 02-03, 214) con un estudio analítico dentro de los parámetros normales, excepto ocas ionalmente un descenso de las proteínas (Tabla 59).
Clínica de hipertensión intracraneal No disminución del nivel de consciencia Ausencia de focalidad neurológica Pruebas de imagen normales Presión elevada de líquido cefalorraquídeo Analítica de líquido cefalorraquídeo normal
Tabla 59. Criterios diagnósticos de pseudotumor cerebri
Afecta con más frecuencia a mujeres en época reproductiva. La mayor parte son idiopáticas, aunque hay que descartar distintas condiciones asociadas a este cuadro que se recogen en la Tabla 60. De entre todos los factores
Eliminar factores asociados: obesidad, fármacos, tratamiento metabolopatías latías
A c e t a z o l a m i d a
Der ivac ión l u m b o p e r i t o n e a l
Descompres ión n e r v i o ópt ico
Figura 76. Manejo terapéutico general del pseudotumor cerebral
R E C U E R D A Se deben cumpl i r todos los criterios para diagnosticar un pseudotumor cerebral .
En pacientes con deterioro visual progresivo que no responden al tratamiento médico, el paso siguiente consiste en colocar una derivación l umboperitoneal (también se han indicado la derivación ventriculoperitoneal y las punciones lumbares de repetición) (MIR 00-01, 56). En algunos casos es necesario recurrir a otras técnicas, como la fenestración de la vaina del nervio óptico o la craniectomía descompresiva subtemporal.
112

Neurología y neurocirugía
15.8. Edema cerebral
Se def ine c o m o edema cerebral la acumulación de líquido en el espac io intersticial o ¡ntracelular (neuronas y glía). En función del mecanis m o patogénico resultante, se reconocen tres t ipos de edema cerebral : • Edema cerebral vasogénico: es el edema causado por una apertura
de la barrera hematoencefálica, por lo que se acumula líquido en el espacio interst ic ia l . Es el edema que acompaña a los procesos tumorales y al absceso cerebral . D i sminuye con la administración de cort ico ides (producen una disminución de la permeab i l idad de la barrera hematoencefálica).
Edema citotóxico: se trata de un a c u m u l o de líquido en el espac i o ¡ntracelular que se p r o d u c e po r malfunción de las bombas de sod io y potas io de la m e m b r a n a ce lu l a r d e b i d o a una h i p o x i a o i squemia ce lu l a r . Es el edema que se o r i g i n a en los infartos cerebra les . N o d i s m i n u y e con la administración de co r t i co i des . El t r a t a m i e n t o cons iste en la reversión de la i squemia causante del m i s m o . Edema intersticial: consiste en un exudado de líquido cefalorraquídeo a través del epéndimo c o m o consecuencia de un aumento de la presión intraventr icular . Se da en las hidrocefal ias agudas e hicro-cefalias crónicas con presión elevada. Me jo ra con la derivación del líquido cefalorraquídeo (drenaje ventr icu lar externo o derivación ventr icu loper i tonea l ) .
Casos clínicos representativos
U n a mu je r de 34 años , obesa , p resenta desde h a c e var ias s e m a n a s ce f a l ea y ep isod ios de pérdida de v is ión b inocu la r t rans i tor ios , pa r t i cu l a rmente al levantarse de la c a m a . En la exp lorac ión , t iene c o m o ún i co s igno u n pap i l edema b i la tera l . U n a r e sonan c i a magnét ica c r anea l y una angiograf ía ce reb ra l por r e sonanc i a resu l tan no rma l e s . ¿ Q u é p rueba indicar ía?
1) D o p p l e r d e t r o n c o s supraaórticos. 2) EEG y e s t u d i o d e sueño. 3) Punc ión l u m b a r y med ic ión d e la presión de l LCR. 4) Iniciaría t r a t a m i e n t o a n t i a g r e g a n t e antes d e rea l i za r más p r u e b a s . 5) Po tenc ia l es e v o c a d o s v i sua les .
M I R 0 2 - 0 3 , 2 4 ; RC: 3
Muje r de 2 4 años que , en los úl t imos 2 meses , p resen ta ep isod ios matut inos de ce f a lea a compañada de náuseas y v is ión bo r rosa ; en el ú l t imo ep isod io , presentó además diplopía. En la exp lorac ión , sólo c a b e des taca r pap i l edema bi latera l y obes idad . La r e sonanc i a magnét ica ce reb ra l es n o r m a l , y el es tud io de l íquido ce fa lor raqu ídeo obten ido por punc ión l umbar es n o r m a l , a excepc ión de un a u m e n t o de pres ión. ¿Cuá l de las s iguientes med idas terapéut icas N O sue le estar i n d i c a d a en el c u r s o de la en f e rmedad de esta pac ien te ?
1) P u n c i o n e s l u m b a r e s r epe t idas . 2) A c e t a z o l a m i d a . 3) De r i vac ión l u m b o p e r i t o n e a l d e LCR. 4) Esferoides.
M I R 0 0 - 0 1 , 5 6 ; RC: 5
I ¡5

Neurología y neurocirugía
16. HIDROCEFALIA
Orientación
MIR r
Aspectos esenciales
Lo más i m p o r t a n t e d e este t e m a es la h i d roce f a l i a crónica del a d u l t o . Sobre t o d o , hay q u e r e co rda r la tríada clásica de la m i s m a y los resu l tados de las d i fe rentes p ruebas q u e se u t i l i z a n para e l diagnóstico.
[~¡~| El líquido cefalorraquídeo (LCR) se p r o d u c e en los p lexos co ro ideos . D e allí c i r cu l a a través de los ventrículos y pasa al espac io subaracno ideo , y se reabsorbe a n ive l de los capi lares cerebrales (según las teorías más recientes).
[2"] Clásicamente, la h id roce fa l i a se d i v i d e en obs t ruc t i va o no c o m u n i c a n t e , si el p r o b l e m a se loca l i za a n ive l vent r i cu la r , y no obs t ruc t i va o c o m u n i c a n t e , si la alteración en la circulación de LCR se ub i ca a n ive l de l espac io subaracno ideo .
r j ] La causa más f recuente de h id roce fa l i a congénita es la estenosis del a cueduc to de S i lv io . El t r a t am ien to de elección es la ventriculostomía endoscópica.
[~4~] La h id roce fa l i a crónica de l a d u l t o puede ser p r imar i a (propia de anc ianos) o secundar ia a otros procesos, c o m o la hemor rag ia subaracno idea , los t raumat i smos y las men ing i t i s .
[~jf] En el diagnóstico de la h id roce fa l i a crónica del adu l t o , no existe n i nguna prueba q u e p r o p o r c i o n e datos patognomónicos.
["5] La tríada característica de la h id roce fa l i a crónica es: aprax ia de la marcha , d e m e n c i a e i n con t i nenc i a esfinte-r iana. La marcha cons t i tuye el síntoma más f recuente y la clínica de presentación más hab i tua l .
["7"] M e d i a n t e el test de infusión, se detecta un a u m e n t o de la resistencia a la sal ida de LCR, q u e es el m e c a n i s m o etiopatogénico i m p l i c a d o en la h id roce fa l i a crónica.
[ Q Preguntas
- M I R 0 5 - 0 6 , 5 4 , 5 7 , 6 4 - M I R 0 2 - 0 3 , 211 - M I R 0 1 - 0 2 , 52 - M I R 00-01 F, 6 6 - M I R 9 9 - 0 0 , 1 9 8 - M I R 9 8 - 9 9 , 6 7 , 6 8
16.1. Concepto y clasificación
El líquido cefalorraquídeo (LCR)
se p roduce en los plexos c o r o i
deos, f undamen ta lmen te a n ive l
de los ventrículos laterales y IV
ventrículo, a razón de a p r o x i
madamente 500 mi d iar ios .
Desde los ventrículos laterales,
a lcanza el tercer ventrículo a
través de los agujeros de M o n -
ro, y por el a cueduc to de S i lv io
llega al cuarto ventrículo en la
fosa poster ior, para salir a las
cisternas del espacio subarac
no ideo por los agujeros de Lus-
chka y Magend i e (MIR 02-03,
1 5 1 ) . Luego c i r cu l a por los es
pacios subaracnoideos y, según
las teorías más recientes, parece
ser que se reabsorbe a n ive l de
las granulac iones aracnoideas
en la convex idad dura l (Figura
7 7 ) .
Seno l o n g i t u d i n a l supe r i o r
D u r a m a d r e
P lexo c o r o i d e o ventrículo la tera l
A racno ldes
Plexo c o r o i d e o de l III ventrículo
A g u j e r o d e Luschka
P lexo c o r o i d e o de l IV ventrículo
Espacio s u b a r a c n o i d e o
Granu lac iones a racno ideas
Cisterna m a g n a
A g u j e r o d e M a g e n d i e
Figura 77. Circulación de l líquido cefalorraquídeo
1 1 4

Neurología y neurocirugía
La h idrocefa l ia puede definirse c o m o un disbalance entre la formación y absorción de LCR, de magn i tud suficiente c o m o para p roduc i r un a cumu lo neto del m i smo dentro de los ventrículos cerebrales, con el consecuente aumento del tamaño de parte o la tota l idad del sistema ventr icu lar demostrable en las pruebas de imagen (TC o RM).
Clásicamente, se han d is t ingu ido dos t ipos de h idrocefa l ia : • Hidrocefalia no comunicante u obstructiva. El LCR no puede a l
canzar el espacio subaracnoideo por la existencia de un obstáculo a nivel del sistema ventr icular .
• Hidrocefalia comunicante o no obstructiva. El LCR alcanza el espac io subaracnoideo, pero a este nivel encuentra di f icul tades para su circulación. También se engloban en este apartado las hidrocefal ias debidas a di f icul tades para la reabsorción de líquido cefalorraquídeo (hidrocefal ias arreabsortivas).
16.2. Etiopatogenia
Los mecanismos por los que puede producirse una hidrocefa l ia son los siguientes: • Hipersecreción de LCR. M u y raro, aunque puede ocurr i r en algunos
tumores del p lexo coro ideo (pap i loma o carc inoma) . • Trastornos del tránsito l icuoral. Es el mecanismo fundamenta l . El
obstáculo puede encontrarse a nivel del sistema ventr icular , resultando hidrocefal ias no comunicantes , c o m o en el caso de la estenosis del acueducto de Si lv io (la más frecuente de las hidrocefal ias congénitas (MIR 98-99, 67), atresia de los agujeros de Luschka y Magendie , tumores intraventr iculares, hemorragias intraventr icula-res, infecciones (ventricul it is) , etc. Otras veces, la d i f i cu l tad de la circulación se produce a nivel del espacio subaracnoideo (hidrocefal ias comunicantes) . Este es el me can ismo de las hidrocefal ias secundarias a meningi t is , hemorragia subaracnoidea, carc inomatosis o l infomatosis meníngea.
• Alteraciones del drenaje venoso intracraneal, que di f icul tan la reabsorción de LCR hacia el torrente sanguíneo, c o m o en el caso de la trombosis de los senos venosos durales, o vac iamientos radicales del cue l lo , síndrome de vena cava superior, etc.
16.3. Clínica
El a c u m u l o de LCR en el sistema ventr icu lar p roduce un síndrome de hipertensión intracraneal (HTIC). Los síntomas son diferentes en lactantes (con fontanelas abiertas), que en niños mayores y adultos, en los que el cráneo no es distensible al haberse cerrado las fontanelas.
Hidrocefalia del lactante
La h idroce fa l i a en los lactantes se manif iesta con un aumento del perímetro craneal (macrocefa l ia ) , dilatación de las venas epicraneales, a b o m b a m i e n t o de fontanelas, s igno de M a c e w e n (sonido típico a la percusión del cráneo sobre las zonas de dilatación ventr icu lar ) y t r an siluminación posi t iva de la cabeza. Son frecuentes el l lanto y la i r r i t ab i l i d ad . En la exploración puede ev idenciarse, en casos avanzados, o jos en "sol p o n i e n t e " y a l teraciones de l r i tmo respirator io (Figuras 78 y 79).
Figura 78. Resonancia magnética practicada a una embarazada que permitió diagnosticar una hidrocefalia en el feto
Figura 79. Hidrocefalia en un lactante
La causa más frecuente de hidrocefa l ia en recién nacidos es la estenosis congénita del acueducto de Si lv io.
El diagnóstico se realiza mediante medición del perímetro craneal (método más sensible) y pruebas de imagen (ecografía transfontanela o RM, c o m o técnicas más específicas).
La radiología s imple de cráneo puede ev idenciar diástasis de suturas, y en los casos crónicos, marcadas impresiones d ig i t i formes y agranda-miento , erosión o descalcificación de la si l la turca.
115

M a n u a l C T O d e M e d i c i n a y Cirugía, 8 . a ed i c i ón
Hidrocefalia en niños mayores y adultos
• Aguda . Clínica de HTIC de rápida instauración, inc luyendo cefalea, náuso cis y vómitos, edema de papi la , paresia del VI par y/o trastornos de la marcha. La dilatación aguda del tercer ventrículo, genera lmente secundaria a tumores de la región p inea l , puede produc i r el síndrome de Parinaud.
• Crónica. Clínica más insidiosa de HTIC, con edema de papi la y a veces incluso atrofia óptica. En enfermos m u y crónicos, es posible la aparición de alteraciones de la marcha, paraparesia espástica, dismetría en miembros superiores, e inc luso alteraciones endoc r i nas por distorsión de la hipófisis o de las proyecc iones hipotalámi-cas por un tercer ventrículo d i la tado.
16.4. Tratamiento
El t ratamiento de la h idrocefa l ia es quirúrgico y su ob je t i vo es reducir la PIC para conseguir una buena función neurológica, lo que no imp l i ca necesariamente lograr un tamaño ventr icu lar no rma l .
ventrículo-peritoneal
Figura 80. Tratamiento de la hidrocefalia. Derivaciones de LCR
Las técnicas quirúrgicas que pueden uti l izarse son (Figura 80): • Drenaje ventricular externo. Se trata de una solución tempora l para
hidrocefal ias agudas en las que se prevea que, tras el t ra tamiento correcto de la causa, no va a ser necesaria una derivación permanente de LCR. Resulta especia lmente útil en el caso de las hemorra gias intraventr iculares.
Derivaciones (shunt o válvulas). Son disposit ivos que der ivan de forma permanente el LCR desde los ventrículos cerebrales a otras cavidades del organismo. La más empleada es la ventr icu loper i to-neal (Figura 81), pero también pueden implantarse ventr iculoatr ia-les o ventr iculopleura les . Se ut i l izan en el caso de hidrocefal ias crónicas o en hidrocefal ias agudas en las que no se espera resolución de la h idrocefa l ia tras t ratamiento de la causa.
Figura 81. Derivación ventriculoperltoneal (Izquierda). El mismo paciente un año después (derecha)
Existen una serie de compl i cac iones (Tabla 61) relacionadas con los disposit ivos de derivación de líquido cefalorraquídeo, tales c o m o la obstrucción del shunt, la infección, el sobre func ionamiento de la derivación y la nefritis del shunt. La obstrucción es la complicación más frecuente; el paciente va a tener un cuadro clínico de h iper ten sión intracraneal y, al real izarle una TC cerebral , se va a ev idenciar un aumento del sistema ventr icu lar (hidrocefal ia) ; el t ra tamiento consistirá en cambiar el sistema der ivat ivo.
OBSTRUCCIÓN DEL SHUNT Hidrocefalia
INFECCIÓN S. epidermidis
HIPERFUNCIÓN • Cefalea ortostática • Higromas-hematomas subdurales. • Ventrículos pequeños
NEFRITIS SHUNT • Glomerulonefrítis complemento bajo
Tabla 61. Complicaciones de los sistemas derivativos
El Staphylococcus epidermidis es el germen más frecuentemente imp l i cado en las infecciones del shunt; suelen ser pacientes que acuden por f iebre, acompañada en muchas ocasiones por un cuadro de hipertensión intracraneal ; el t ratamiento consistirá en exte r io r i zar el shunt (con posterior recambio) y ant ib ioterapia intravenosa (de manera empírica, se empezará por vancomic ina o te icoplani-na). La hiperfunción del shunt puede provocar cuadros de cefalea ortostática (debidos a hipotensión de LCR), hematomas o higromas subdurales o el denominado síndrome de los ventrículos pequeños). Por último, una rara complicación es la nefritis del shunt, que es más propia de las derivaciones ventr iculoatr ia les y que, de forma característica, cursa c o m o una glomerulonefrítis con cifras de c o m p lemento bajo. Ventriculostomía premamilar endoscópica. Se trata de una técnica en auge en la que, con ayuda de un neuroendoscopio , se crea una comunicación directa entre el III ventrículo y el espacio subaracnoi deo, pe rmi t i endo prescindir de las der ivaciones y, por tanto, redu c iendo el riesgo de compl i cac iones relacionadas con el shunt. Está
116

Neurología y neurocirugía
indicada en el caso de hidrocefal ias obstructivas. Ac tua lmente , se considera la técnica de elección para el t ratamiento de la estenosis del acueducto de Si lv io.
16.5. Hidrocefalia crónica del adulto
También l lamada hidrocefa l ia normotensiva o hidrocefa l ia a presión norma l . Desde un pun to de vista etiológico, se pueden encontrar for mas idiopáticas ( 4 0 - 6 0 % de los casos) y secundarias a otros trastornos neurológicos c o m o la hemorragia subaracnoidea (lo más frecuente dentro de este grupo) , tras t raumat ismos craneoencefálicos (MIR 05-06, 64), posmeningíticas o tras tumores. La forma idiopática es una h id ro cefal ia que se presenta en pacientes de edad avanzada (> 60 años) y afecta l igeramente más a varones.
La clínica es muy característica, aunque no patognomónica, y se def ine por la tríada de Hakim-Adams: demenc ia (una de las pocas causas reversibles de demencia ) , incont inenc ia ur inar ia y trastorno de la marcha, que suele ser el signo más precoz y la clínica más frecuente (su ausencia debe hacernos dudar del cuadro) (MIR 05-06, 54 ; MIR 98-99, 68). A veces se acompaña de trastornos extrapiramidales (parkinsonismo).
R E C U E R D A P r e g u n t a MIR f r e cuen t e : Tr íada de Hak im-Adams : aprax ia de la m a r c h a , d e n * c ia e i n con t i nenc i a es f in te r i ana .
en-
Figura 82. En esta imagen se puede observar una dilatación del sistema ventr icu lar en ausencia de surcos p rominen tes en la convex idad,
t o d o el lo compa t i b l e con hidrocefa l ia no rmotensa
Resonancia magnética oTC cerebral RM de f l u j o Test de infusión Tap test o test de evacuación Monitorízación de la PIC
Tabla 62. Pruebas diagnósticas empleadas en la hidrocefalia crónica del adu l to
El diagnóstico se sospecha ante el hal lazgo de una hidrocefa l ia c o m u nicante en las pruebas de imagen (TC o RM) en un paciente con clínica compat ib l e , aunque no hay datos patognomónicos (MIR 02-03, 211) (Figura 82) . Debe realizarse diagnóstico di ferencia l con la h idrocefa l ia ex vacuo o secundaria a una atrofia cerebral , que es un aumento c o m pensador del tamaño del sistema ventr icu lar que aparece frecuentemente en ancianos con importante atrofia cerebral cor t i cosubcor t i ca l , y que no requiere t ra tamiento. A di ferencia de esta última hidrocefa l ia , en la crónica del adu l to existen signos de reabsorción transependimaria (h ipodensidad per iventr icu lar en la TC), balonización del tercer ven trículo y ausencia de surcos de la convex idad . Ac tua lmente se están emp leando estudios de RM de f l u jo de líquido cefalorraquídeo, en los que se muestra un aumento de la ve loc idad de f lu jo de LCR a nivel del acueducto de Si lvio (Tabla 62).
El diagnóstico se comp lementa mediante una monitorízación cont inua de la presión intracraneal , en la que puede observarse un aumento de la presión y/o la existencia de ondas patológicas de hipertensión intracraneal . También se realiza un test de infusión: se in t roduce suero en el espacio intratecal mediante una punción lumbar a una ve loc idad determinada y se registra la presión en el espacio subaracnoideo durante un t i empo ; con los datos obtenidos, se valora la resistencia a la salida de líquido cefalorraquídeo, que en el caso de la h idrocefa l ia crónica, se va a encontrar aumentada. Otra man iobra diagnóstica útil es la punción lumbar evacuadora para comprobar si existe mejoría clínica s igni f icat i va tras la extracción de LCR.
El t ra tamiento de elección es la derivación de LCR (habi tua lmente ven-tr icu loper i tonea l ) (MIR 05-06, 57).
r
Casos clínicos representativos
H o m b r e de 70 años que consu l ta por un t ras torno de la m a r c h a y un de te r io ro cog nit ivo sabagudo. Nos ind ican que ei d iagnóst ico de presunc ión de l pac i en te es h i d roce fa l i a a presión n o r m a l . En este c a so , ¿ cuá l de los s iguientes datos N O esperaría encon t r a r ?
1) U n a h i d r o c e f a l i a c o m u n i c a n t e c o n a c u e d u c t o d e S i l v io , p e r m e a b l e en la r e s o n a n c i a c e r e b r a l .
2) U n t r a s t o r n o d e la m a r c h a t i p o apráxico. 3) U n LCR c o n l eve e l evac ión d e la presión d e ape r tu r a , y c o n u n a u m e n t o d e células
y proteínas. 4) La real ización d e u n a punc ión l u m b a r e v a c u a d o r a ( 30 m i d e LCR) p u e d e m e j o r a r
la m a r c h a de l p a c i e n t e . 5) A u s e n c i a d e s ignos d e a t ro f i a c o r t i c a l c e r e b r a l .
U n pac ien te de 7 3 años sufrió un a c c iden te de t ráf ico c o n t r aumat i smo c r a n e a l , del que se recuperó . A los tres meses , in i c i a de fo rma progres i va a l te rac ión de func iones super io res , i n con t i nenc i a u r ina r i a ocas iona l y su c a m i n a r es torpe . P robab lemente presenta :
1) H e m a t o m a i n t r a p a r e n q u i m a t o s o c e r eb ra l tardío. 2) H e m o r r a g i a s u b a r a c n o i d e a . 3) H i d r o c e f a l i a a r r eabso r t i v a . 4) A t r o f i a c e r e b r a l postraumática. 5) T u m o r c e r e b r a l .
M I R 0 5 - 0 6 , 6 4 : RC: 3
M I R 0 5 - 0 6 , 5 7 ; RC: 3
1 1 7

TUMORES INTRACRANEALES
Orientación
MIR r
Aspectos esenciales
C o n s t i t u y e u n o d e los temas más i m p o r t a n t e s en Neurocirugía de cara al M IR . Lo más práctico es e l a b o r a r listas d o n d e se mues t re l o más f r e cuen t e y característico de c a d a u n o de los t u m o r e s , ya q u e c o n e l l o se contes ta sin m u c h a d i f i c u l t a d la g ran mayoría d e las preguntas de l M IR .
pj~| En los adultos, los tumores cerebrales más frecuentes son las metástasis cerebrales, mientras que el primario más frecuente es el glioblastoma multiforme.
p j ] Sin embargo, en los niños, el tumor más frecuente es el astrocitoma pilocítico y el tumor maligno más frecuente es el meduloblastoma. Las metástasis son muy raras.
[~3~| El síntoma de presentación más frecuente de los tumores cerebrales es la cefalea. Suele ser más intensa por la mañana y puede llegar a despertar a los pacientes por la noche.
["4") La metástasis más frecuente es la del carc inoma microcítico de pulmón. Sin embargo, el tumor con mayor tendencia a producir metástasis cerebrales es el melanoma.
|~5~| El tratamiento de elección de una metástasis cerebral única, en lugar acces ib le y con la enfermedad primaria controlada, es la cirugía y posterior radioterapia holocraneal .
Los astrocitomas constituyen los tumores primarios más frecuentes del sistema nervioso central.
[~7~] Los astrocitomas difusos se caracterizan por una tendencia a degenerar hacia formas más malignas con el paso del tiempo.
JjTJ El tratamiento de los astrocitomas difusos de alto grado (astrocitoma anaplásico y glioblastoma multiforme) es paliativo.
[~g~| El astrocitoma gigantocelular subependimario se asocia a la esclerosis tuberosa.
|Yo~] El tumor primario más epileptógeno es el ol igodendroglioma.
|l 11 Los tumores embrionarios se caracterizan por tener tendencia a diseminar a través del LCR.
Q Y j Los meningiomas son tumores que tienen una influencia hormonal evidente: afectan más a las mujeres y se han empleado antagonistas de progesterona en su tratamiento.
[131 El tratamiento de elección de los meningiomas es la cirugía. El grado de resección influye en la posibil idad de recidivas del tumor.
(T4] La cl ínica más frecuente de presentación de los tumores de la región pineal es la hipertensión intracraneal secundaria a una hidrocefalia. Sin embargo, la clínica carcterística es el síndrome de Parinaud, por afectación del área tectal del mesencéfalo.
[-[ 31 El tumor más frecuente de la región pineal es el germinoma, cuyo tratamiento de elección es la radioterapia.
QjQ Los adenomas hipofisarios pueden ser funcionantes o no funcionantes. Suelen manifestarse como una hemianopsia bitemporal de predominio en campos superiores, junto con la clínica derivada de la hiperfunción hormonal (si son funcionantes) y/o del hipopituitarismo producido.
|l 7| En ocasiones, los adenomas de hipófisis se manifiestan en forma de apoplejía hipofisaria, con cefalea súbita, oftalmoplejia, náuseas y vómitos, y pérdidas súbitas del campo visual. Se deben a una hemorragia o infarto dentro del tumor.
jJJTj Un tumor selar con quistes y ca lc io orienta al diagnóstico de craneofaringioma.
[T9] Un tumor cerebeloso asociado a policitemia es un hemangioblastoma.
GD P regun tas
- M I R 0 9 - 1 0 , 70 - M I R 0 7 - 0 8 , 2 3 5 - M I R 0 6 - 0 7 , 5 4 , 6 0 , 7 4 - M I R 0 5 - 0 6 , 5 4 , 6 4 - M I R 0 4 - 0 5 , 64 - M I R 0 3 - 0 4 , 2 4 6 - M I R 0 2 - 0 3 , 2 1 5 - M I R 0 1 - 0 2 , 61 - M I R 00-01 F, 73 - M I R 0 0 - 0 1 , 61 - M I R 9 9 - 0 0 , 1 9 6 , 2 5 2 - M I R 9 8 - 9 9 , 5 8 , 7 0 , - M I R 98-99F , 7 1 , 7 8 , 79 - M I R 9 7 - 9 8 , 4 4
17.1. Consideraciones generales
Epidemiología
Los t u m o r e s i n t r a c r a n e a l e s m á s f r e cuen t e s en e l adu l to s o n los metas tás i cos . Entre los t u m o r e s c e r e b r a l e s p r i m a
r ios, d e s t a c a n los g l i o m a s (el g l i o b l a s t o m a m u l t i f o r m e es e l t u m o r c e r e b r a l p r i m a r i o más f r e c u e n t e en m a y o r e s
d e 2 0 años ) (MIR 97-98, 44) .
1 1 8

Neurología y neurocirugía
Las neoplasias intracraneales son, después de las leucemias, los procesos malignos más frecuentes en la edad infant i l , y suponen la neoplasia sólida más frecuente en este grupo de edad. El mayor porcentaje lo const i tuyen los gl iomas (astrocitomas), seguidos por el meduloblastoma y el craneofar ingioma, siendo excepcionales las metástasis. En adultos, los tumores cerebrales son, en su mayoría, supratentoriales ( 80% ) , mientras que en niños hay una distribución más o menos homogénea entre el compar t imento supratentorial y el infratentorial , si bien en los dos pr imeros años predominan los que se sitúan por enc ima del tentor io.
Clínica
neoplasias con especial predilección por el lóbulo tempora l (ganglio-ci toma) y las metástasis cerebrales. Los tumores infratentoriales suelen debutar con síntomas de HTIC y no producen crisis; los supratentoriales suelen hacerlo con crisis y signos de foca l idad neurológica. En niños, es frecuente que la pr imera manifestación sea una alteración de la personal idad con mal rend imien to escolar, que puede preceder en semanas o meses al descubr imiento del tumor . En los tumores de fosa posterior, puede haber nistagmus (hor izonta l , que aumenta al mirar hacia el lado de la lesión en los de hemisferios cerebelosos; en todas las direcc iones son los que se loca l izan en vermis posterior o cuarto ven trículo; y hor izonta l , vert ical o rotator io en los de t ronco) .
El síntoma más frecuente de presentación de los tumores cerebrales es la cefalea. La cefalea tumora l se describe clásicamente c o m o más i n tensa por la mañana. Puede despertar al enfermo por la noche. Si existe HTIC, puede asociarse a náuseas y vómitos. Una cefalea de estas características, sobre todo , si va asociada a signos de foca l idad neurológica (afasia, hemiparesia, etc.) o crisis, debe hacer pensar en un tumor c o m o pr imer diagnóstico di ferencia l (Figura 83) .
Los tumores cerebrales suponen la pr imera causa de epi lepsia entre los 35 y 50 años. Son especia lmente epileptógenos los astrocitomas de bajo grado en hemisferios cerebrales, el o l i godendrog l ioma , algunas
17.2. Metástasis cerebrales
Las metástasis son los tumores cerebrales más frecuentes en el adul to , pero son excepcionales en niños. Se loca l izan generalmente a nivel de la unión cort icosubcort ica l de los hemisferios cerebrales ( 8 0 % ) y, menos f recuentemente, en los hemisferios cerebelosos (1 5% , procedentes sobre todo del tracto digest ivo y aparato geni tour inar io ) . Aún así, representan el tumor más frecuente en la fosa posterior del adul to . Pueden ser lesiones solitarias (mayor tendencia en el caso de las de mama y riñon) o múltiples (como en el caso de las metástasis de pulmón o
HEMISFÉRICOS Crisis + focalidad neurológica FOSA POSTERIOR
METÁSTASIS GLIOMAS
- Más frec. de tumor supradia-fragmático que infradiafragmático
- Unión corticosubcortical
LINFOMA 1.° DELSNC
- Inmunodeprimidos (VIH)
- Multicéntricos - Buena respuesta
a corticoides - RT de elección
Mejor pronóstico, el pilocítico y el subependimario de células gigantes (esclerosis tuberosa)
El de peor pronóstico
- Sust. blanca subcortical lóbulo frontal
- Crisis y calificaciones
Niños Adultos
Línea media Sust. blanca (cerebelo, subcortical tronco, n. óptico lóbulo frontal (NFMI)) y temporal Raro en hemisferios
MENINGIOMA
- Mujeres - Convexidad
parasagital - Hiperóstosis cráneo y olistering
-Aumento vascularización (único intradural vascularizado por la art. carótida externa)
- Hormonodependencia - Si infancia o múltiples (NFMII)
CEREBELO
Cefalea Hidrocefalia Sd. cerebeloso
TRONCOENCE-FÁLICOS
Gliomas Niños
IVVENTRÍCULO
Hidrocefalia
ANGULO PONTOCEREBELOSO
Astrocitoma Glioblastoma oligodendroglioma multiforme
Ependimoma El mixopapilar en filum termínale
Papiloma de plexos coroideos. En niños en ventrículos laterales
Astrocitoma hemisférico
Meduloblastoma vermiano Posible diseminación por LCR y/o metástasis
Hemangioblastoma Hemisférico Poliglobulia Von Hippel-Lindau
Niños
REGIÓN QUIASMÁTICA
Adultos
I.» Neurinoma
Meníngioma
3.° Colesteatoma
(quiste epidermoide
o tumor perlado)
Adenoma de hipófisis Más frec. el prolactinoma Hemianopsia bitemporal + clínica endocrinológica
Craneofaringioma Misma clínica campimétrica Calcificaciones supraselares
Coristoma Neurohípófisis
REGIÓN PINEAL
- Más frecuente el germinoma
- Hidrocefalia + sd. Parinaud
- El más radiosensible
Adultos Niños
Figura 83. Diagnóstico diferencial por localización
119

Manual CTO de Medicina y Cirugía, 8 . A edición
de las del melanoma) . El mayor porcentaje son de or igen pu lmonar (aprox imadamente un 5 0 % ) , s iendo más habituales en el ca rc inoma de células pequeñas u oat cel l que en ei resto de tumores broncogénicos. Otras fuentes frecuentes son mama (15-20% ) , riñon, me lanoma y tracto digest ivo. Hasta un 1 0 % son de or igen desconoc ido. El t umor que t iene más tendenc ia a metastatizar en el cerebro es el me lanoma.
La superv ivencia media de los pacientes con metástasis cerebrales t ra tadas es de unos seis meses.
17.3. Gliomas Radiológicamente, suelen observarse en la TC c o m o lesiones h ipoden-sas cuya pared se realza de forma importante tras la administración de contraste intravenoso (captación en an i l l o o imagen en "donu t " ) (Figura 84) (MIR 04-05, 64). Hab i tua lmente están rodeadas de profuso edema vasogénico d ig i t i fo rme. El diagnóstico di ferencia l de las lesiones que captan contraste en an i l l o debe establecerse entre metástasis, g l i o blastoma mu l t i f o rme , l i n foma cerebral p r imar io y abscesos ( inc lu ida la toxoplasmosis cerebral).
Figura 84. Metástasis cerebrales múltiples de carcinoma de pulmón con captación de contraste en anillo
Existen algunas metástasis con especial tendencia a sangrar (por tanto, hiperdensas en la TC): cor iocarc inoma, melanoma, carc inoma de t i r o i des, h ipernefroma (riñon) y carc inoma broncogénico. El tratamiento de elección para las metástasis cerebrales queda resumido en la Figura 85 (MIR 09-10, 70).
METASTASIS
Primario no controlado
No tratar/ RT holocraneal paliativa
Primario controlado
Única
T Múltiple
Cirugía (*) < 3 > 3
Cirugía (*)
I ) R a d i o c i r u g í a RT holocraneal
Figura 85. Tratamiento de elección en las metástasis cerebrales
Los g l iomas son las neoplasias cerebrales que der ivan de las células gliales. Son los tumores pr imar ios del sistema nervioso central más predominantes , especia lmente los tumores astrocitarios más agresivos (gl ioblastoma mul t i fo rme) (MIR 97-98, 44) . En la Tabla 63 se resume la clasificación actual de los g l iomas (OMS 2007) .
TUMORES ASTROCITARIOS
Astrocitoma difuso (fibrilar, protoplásmico, gemistocítico): - Astrocitoma de bajo grado - Astrocitoma anaplásico - Gliobastoma multiforme
> Gliobastoma de células gigantes > Gllosarcoma
Astrocitomas localizados: - Astrocitoma pilocítico - Xantoastrocítoma pleomórfico - Astrocitoma gigantocelular subependimario
TUMORES OL IGODENDROG L IALES
GL IOMAS MIXTOS
TUMORES EPENDIMARIOS
T U M O R E S GL IALES DE OR IGEN INCIERTO
Oligodendroglioma Oligodendroglioma anaplásico
Oligoastrocitoma Ollgoastrocitoma anaplásico
Ependimoma Ependimoma anaplásico Ependimoma mixopapilar Subependlmoma
Gliomatosis cerebri Astroblastoma Glloma cordoide de III ventrículo
Tabla 63. Clasificación de los tumores gliales (OMS 2007)
Astrocitomas
Los astrocitomas son tumores derivados de los astrocitos. Const i tuyen el g rupo más numeroso de tumores pr imar ios del sistema nervioso cen tra l . Son neoplasias que expresan proteína g l io f ibr i la r acida (CFAP). Según la clasificación actual de la Organización M u n d i a l de la Salud (OMS), se reconocen dos grandes grupos de astrocitomas:
Astrocitomas difusos o infiltrantes
Los tumores pertenecientes a esta categoría se caracter izan por el ca rácter inf i l t rante local y su capac idad de dispersión hacia lugares lejanos respecto a la localización in i c ia l . Además, t ienen la capac idad de degenerar hacia formas más malignas con el paso del t i empo .
En función de una serie de datos anatomopatológicos, los astrocitomas se clasif ican en tres grados: grado II, si el t umor muestra at ipia nuclear; grado III, si además de la at ipia nuclear, se ev idencia act iv idad mitóti-ca; y grado IV, si a t odo lo anter ior se le suma hiperplasia microvascular (proliferación endotel ia l ) o necrosis.
120

Neurología y neurocirugía
Al igual que el as t roc i toma anaplásico, el t ra tamiento consiste en cirugía, rad ioterapia y qu im io t e r ap i a ( local o sistémica). A pesar de toda la d i s p o n i b i l i d a d terapéutica, la med iana de superv i ven cia es de un año. Por esta razón, en muchas ocasiones se dec ide la abstención terapéutica por la pos ib i l i dad de dejar secuelas al pac iente , sobre t o d o , si está loca l i zado en áreas e locuentes.
Según la OMS, existen tres t ipos de astrocitomas difusos (Tabla 64):
Astrocitoma
ACTIV IDAD PROL IFERACION NUCLEAR MITOTICA ENDOTEL IAL
NECROSIS
Astrocitoma anaplásico
Gl ioblastoma mult i fome
Tabla 64. Características anatomopatológicas de los astrocitomas difusos
Astrocitomas (bajo grado): corresponden a tumores de grado II. Son tumores que t ienden a darse en niños y jóvenes adultos ( inc idenc ia máxima en la cuarta década) y los varones son los más afectados. Los lóbulos tempora l y frontal const i tuyen su localización más hab i tua l , y el cuadro clínico más frecuente en su presentación son las crisis epilépticas. En la RM, son tumores que no captan contraste o, si lo hacen, lo es en forma débil; en un estudio se comprobó que si el tumor captaba contraste con el t i empo , era un ind ica t i vo de progresión hacia formas más mal ignas. El t ra tamiento consiste en cirugía y radioterapia, si b ien , en algunas ocasiones solamente se adopta una act i tud expectante de seguimiento, sobre todo si afecta a áreas elocuentes; la qu imio te rap ia se reserva para recurrencias tumorales. La mediana de superv ivencia se sitúa en 5-10 años. Astrocitomas anaplásicos (tumores grado III): a estos tumores, j un to con el g l ioblastoma mul t i fo rme que se cita a continuación, se les denomina astrocitomas de alto grado. La inc idencia máxima de presentación se sitúa en torno a los 40 años y, de manera similar a los anteriores, son más frecuentes en varones. También los lóbulos frontal y tempora l son los más afectados. En la RM, son tumores menos circunscritos que los anteriores y no suelen captar contraste, si bien pueden hacerlo en más ocasiones que los astrocitomas grado II. El t ratamiento consiste en la resección quirúrgica, radioterapia y qu imioterap ia , ya sea sistémica o, en los últimos años, local (car-mustina), es decir, implantada en lecho quirúrgico tras la extirpación quirúrgica. La mediana de supervivencia se sitúa entre 2,5 y 3 años. Glioblastoma multiforme (tumores grado IV) (Figura 86) : son los tumores pr imar ios más frecuentes en los adultos. La edad media de presentación se sitúa en to rno a los 53 años, y son más frecuentes en los hombres. De manera característica, muestra realce en an i l lo tras la administración de contraste (Figura 87) (MIR 07-08, 235) .
Figura 86. Glioblastoma multiforme temporal derecho
Figura 87. Captación de contraste en anillo en paciente con OBH temporal derecho
Astrocitomas localizados
Son tumores que se caracter izan por ser re la t ivamente c i rcunscr i tos y tener una mínima capac idad de diseminación a través del sistema nerv ioso. Suelen ser más comunes en niños y en jóvenes adul tos . En esta categoría, se inc luyen tres t ipos de tumores : • Astrocitoma pilocítico (Figuras 88 y 89) (grado I de la OMS) : cons
t i tuye la neoplasia cerebral más f recuente en los niños, con una inc idenc ia máxima en la segunda década de la v ida (10-12 años c o m o p i co de inc idenc ia ) . Las fibras de Rosenthal const i tuyen un dato anatomopatológico característico de estos tumores . Se loca l izan sobre t o d o a n ive l de los hemisfer ios cerebelosos, y en la RM se muestran c o m o una lesión quística con un nodu lo captante en su inter ior . La extirpación quirúrgica total de este t u mor cons igue curar a este t i p o de pacientes, sin necesidad de más terapias complementa r i as . El pronóstico es excelente en casos en los que se cons igue la resección comp le t a del t umor .
• Xantoastrocitoma pleomórfico (grados II y III de la OMS) : son tumores que se dan en adultos de unos 20 años y se loca l izan f undamen ta lmen te a n ive l del lóbulo t empora l . Clínicamente, los pacientes se caracter izan por tener una histor ia de convu ls iones . Desde un pun to de vista anatomopatológico, se muestra un p l eomor f i smo celular , células cargadas de lípidos y un estroma con re t i cu l ina .
• Astrocitoma gigantocelular subependimario (grado I de la OMS) : es un t umor que se asocia a la esclerosis tuberosa. Se loca l iza a nive l de las paredes de los ventrículos laterales.
121

Manual CTO de Medicina y Cirugía, 8 . a edición
Es un t umor p rop io de la edad adulta, con un p ico en la 5. a década de la v ida . Afecta con más frecuencia a varones. Su localización más frecuente es el lóbulo f ronta l . Es típico que debute clínicamente con crisis epilépticas, s iendo el t umor cerebral p r imar io más epileptógeno.
R E C U E R D A An te un t u m o r en lóbulo f ron ta l , hay q u e pensar f undamen ta lmen te en tres pos ib i l idades : metástasis, g l iob las toma y o l i g o d e n d r o g l i o m a . Este ú l t i m o se caracter iza por la presencia de ca l c io , q u e los dos anter iores no suelen manifestar.
Figura 88. Astrocitoma pilocítico cerebeloso
Figura 89. (a) Glioma de tronco; (b) Glioma del nervio óptico derecho
Oligodendroglioma
Es un tumor raro, que representa menos del 1 0 % de todos los g l iomas. Su característica microscópica más l lamat iva es la existencia de células redondeadas que cont ienen núcleos hipercromáticos y citoplasmas de escasa apetencia t in tor i a l , con aspecto de "huevo fr i to " (Figura 90). Son frecuentes los quistes, las ca lc i f icac iones y las hemorragias espontáneas. No expresan GFAP, a di ferencia de los tumores astrocitarios. Se dist ingue una variante anaplásica de peor pronóstico.
Figura 90. Oligodendroglioma con la imagen típica de células en "huevo frito"
La TC ev idenc ia una lesión hipodensa con áreas quísticas y de ca l cificación, que no suele captar contraste intravenoso. El t ra tamiento de elección es la resección quirúrgica más qu imioterap ia (PCV: pro-carbacina, C C N U y v incr ist ina) ; en el caso de los o l igodendrog l iomas anaplásicos, se puede asociar radioterapia. La pérdida de los brazos cromosómicos 1 p y 19q se asocia a una mejor respuesta a la q u i m i o t e rapia y a una mayor superv ivencia.
Glioma mixto (oligoastrocitoma)
El o l igoast roc i toma, y su var iante, el o l igoast roc i toma anaplásico, son tumores guales compuestos por dos t ipos celulares neoplásicos d is t in tos que recuerdan a los presentes en el astrocitoma (con expresión de GAFP) y el o l i godendrog l ioma , por lo que se denominan también g l i o mas mixtos . Su pronóstico es in termedio entre ambas entidades.
Ependimoma
Es un t umor que der iva de los ependimoc i tos , que son las células que recubren las cavidades ventr iculares y el canal central medular . Su característica histológica más típica son las formaciones en «roseta». Genera lmente son benignos, aunque se ha descrito una variante anaplásica. Pueden local izarse a lo largo de todo el neuroeje. A nivel i n tracraneal , representan un 5-6% de los g l iomas, y crecen típicamente en el suelo del cuarto ventrículo (Figura 91) , p roduc i endo hidrocefa l ia (MIR 98-99, 70). Afectan generalmente a niños.
Figura 91. Ependimoma del cuarto ventrículo
122

Neurología y neurocirugía
Sin embargo, son m u c h o más frecuentes a nivel espinal , donde son más propios de adultos y son de mejor pronóstico. La co l umna cervical const i tuye el segmento donde se encuentra este t umor con mayor frecuencia . A nivel del f i l um termínale, se local iza de forma específica un subt ipo de epend imoma , que es la variante mixopap i la r .
El t ra tamiento de elección es la cirugía más radioterapia. Responden peor los de localización supratentor ia l , de mayor ve loc idad de c rec i miento . Pueden presentar siembras a través del LCR, en cuyo caso debe realizarse radioterapia de todo el neuroeje.
Gliomatosis cerebri
diferenciación neuronal o g l ia l . Los PNET se clasif ican en infratentoria l o medu lob las toma, que es el tumor embr iona r io más frecuente, y los PNET supratentoriales.
• Meduloepitelioma • Ependimoblastoma • Meduloblastoma (PNET infratentorial) • PNET supratentoriales:
- Neuroblastoma - Ganglioneuroblastoma
• Tumor rabdoide/teratoide atípico
Tabla 65. Clasificación de los tumores embrionarios según la WHO
Es un tumor glial d i fuso, de histogénesis incierta y controver t ida , que i n fi l tra el cerebro de forma extensa, afectando a más de dos lóbulos, con frecuencia bi lateral mente, y a menudo extendiéndose hacia estructuras de la fosa posterior, e inc luso a la médula. Se trata con radioterapia.
17.4. Tumores del plexo coroideo
Suelen ser tumores benignos (papi lomas del p lexo coro ide) , pero se han descr i to también formas malignas (carcinomas). Son más f recuentes en niños (2/3 de los casos), local izados generalmente a nivel de los ventrículos laterales. En adultos (1/3 de los casos), se loca l izan preferentemente a nivel infratentor ia l , en el cuarto ventrículo. La prealbúmi-na (transtirretina) es un marcador inmunohistoquímico de los tumores de plexos.
La mayoría se presentan con clínica de hipertensión intracraneal deb i do a hidrocefa l ia obstruct iva. A veces secretan una cant idad excesiva de LCR, dando lugar a hidrocefa l ia por hiperproducción (mecanismo casi exc lus ivo de estos tumores) . T ienden a diseminar a través del LCR. El t ratamiento es quirúrgico.
17.5. Tumores embrionarios. Tumores Neuroectodérmicos Primitivos (PNET)
Los tumores embr ionar ios son un grupo de neoplasias pr imit ivas (Tabla 65) clínicamente agresivas que se desarrol lan hab i tua lmente en la p r i mera década de la v ida . Se caracter izan por la propensión que t ienen a d iseminar a través del líquido cefalorraquídeo (MIR 02-03, 215) . Por este mot i vo , su diagnóstico exige RM de todo el eje craneoespinal y punción lumbar para realizar citología, y tras la cirugía, está indicada la radioterapia profiláctica craneoespinal . Excepcionalmente, pueden metastatizar fuera del sistema nervioso centra l .
Este grupo de tumores deriva de células inmaduras o pr imit ivas , que son las precursoras de células gliales, neuronales y ependimar ias del sistema nervioso.
Dent ro de los tumores embr ionar ios , destacan los tumores neuroectodérmicos pr imi t i vos (PNET), cuyas células muestran tendenc ia a la
Meduloblastoma (PNET infratentorial)
Se trata del tumor encefálico más frecuente en niños menores de c inco años y la neoplasia intracraneal maligna más frecuente en la edad infanti l .
Histológicamente, son características las formac iones en "roseta de Homer-Wr ight " , aunque no son patognomónicas porque también pueden aparecer en el resto de tumores embr ionar ios (Figura 92). En una tercera parte de los casos se ha demostrado una pérdida de material genético a nivel del brazo cor to del c romosoma 1 7. Se ha asociado a enfermedades hereditarias, c o m o el síndrome del ca rc inoma basoce-lular nevo ide (síndrome de Cor l in ) y el síndrome de Turcot (poliposis colónica y tumor cerebral) .
R E C U E R D A El meduloblastoma se origina en el techo del cuarto ventrículo, mientras que el ependimoma parte del suelo del cuarto ventrículo.
• I f t i - 1 * * v i < '
Figura 92. Rosetas de Homer-Wright, típicas de meduloblastoma
Radiológicamente, suele manifestarse c o m o un tumor sólido de la fosa posterior, que capta contraste de forma homogénea, hab i tua lmente l o ca l izado en la línea media a nivel del vermis cerebeloso (Figura 93) y techo del cuarto ventrículo, por lo que generalmente debuta con síntomas de hipertensión intracraneal por h idrocefa l ia obstruct iva y signos de disfunción cerebelosa (ataxia de t ronco) . En adultos, se loca l iza más frecuentemente a nivel hemisférico.
123

Manual CTO de Medicina y Cirugía, 8 . a edición
El t ratamiento de elección es la cirugía, seguida de radioterapia craneoespinal y qu imio te rap ia .
Figura 93. (a) Meduloblastoma en vermis cerebeloso. (b) Diseminación espinal
PNET supratentoriales
Dentro de los PNET supratentoriales, existen dos t ipos de tumores: el neuroblastoma cerebral y el gangl ioneuroblastoma. Se caracter izan porque el neuroblastoma muestra diferenciación neuroblástica y el gangl ioneuroblastoma, diferenciación neuronal .
En general , los PNET supratentoriales t ienen peor pronóstico y d i ferentes alteraciones genéticas que el medulob las toma.
17.6.Tumores neuronales y neurogliales mixtos
Dentro de este grupo, destacan dos t ipos:
Gangliocitoma y ganglioglioma
Son tumores bien di ferenciados, de c rec imiento lento, compuestos por células neuronales neoplásicas solas (gangl ioc i toma) o en combinación con células gliales atípicas (gangl iogl ioma) .
Son propios de la infancia o adultos jóvenes, muy epileptogénicos, con tendencia a las ca lc i f icac iones, y su localización más frecuente es el lóbu lo t empora l , aunque pueden aparecer en otras local izac iones c o m o la médula espinal , el t ronco del encéfalo, el cerebelo, la región pineal , etcétera. La enfermedad de Lhermitte-Duclos consiste en un gang l ioc i toma difuso de cerebelo.
Muestran pos i t iv idad para enolasa neuronal específica y proteína de los neurof i lamentos.
El t ra tamiento de elección es la cirugía, ten iendo buen pronóstico, sobre t o d o en relación con la resección quirúrgica comple ta .
Neurocitoma central
Se trata de un tumor que se or ig ina hab i tua lmente en el septum pellu-cidum y se local iza generalmente en el sistema ventr icular lateral en la región del foramen de M o n r o o en el tercer ventrículo. Es p rop io de adultos jóvenes.
Se observan a m e n u d o ca lc i f icac iones, y en el estudio anatomopatológ ico , rosetas similares a las de Borit. Presenta pos i t iv idad inmunohis to-química para sinaptofis ina y enolasa neuronal específica. El t ra tamiento de elección es la cirugía.
17.7. Meningioma
Epidemiología
El men ing ioma sigue en frecuencia a los gl iomas dentro de los tumores intracraneales pr imar ios en adultos ( 2 0 % ) , pero es el más frecuente de los tumores intracraneales extraparenquimatosos. Afecta p r i n c i pa lmen te a mujeres durante la quinta y sexta décadas de la v ida. Puede expre sar receptores para progesterona (lo que les conf iere mejor pronóstico) y, menos frecuentemente, para estrógenos. Se ha descrito una mayor f recuencia en mujeres que padecen cáncer de mama. Se han asociad o a traumas craneales previos y a radioterapia. Cuando se asocian a neurof ibromatosis t i po II, aparecen en la infancia y, con frecuencia , en forma de lesiones múltiples.
Anatomía patológica
Son tumores extraaxiales (extraparenquimatosos), de lento c rec imiento , generalmente benignos. Crecen a partir de la aracnoides ( leptomenin-ge), no de la duramadre . Su localización más frecuente es a nivel de la convex idad (Figura 94), pero pueden aparecer en cualquier lugar donde existan células aracnoideas: hoz cerebral , convex idad cerebral lateral, surco o l fa tor io (Figura 95) , ala mayor del esfenoides, tubérculo selar, c l ivus, ángulo pontocerebeloso, ventrículos cerebrales, etc.
Figura 94. Meningioma de la convexidad parasagital
124

Neurología y neurocirugía
Figura 95. M e n i n g i o m a del surco o l fa tor io
Q R E C U E R D A El m e n i n g i o m a se a soc i a a t r o m b o s i s v enosa p r o f u n d a y a t u m o r e s de m a m a .
Tratamiento
La cirugía es el t ra tamiento de elección para los mening iomas s intomáticos, y la resección comple ta puede ser curat iva (MIR 99-00, 196). El pr inc ipa l factor en la prevención de la rec id iva es la extensión de la resección quirúrgica (grados de Simpson). No se recomienda radioterapia postoperatoria en los mening iomas benignos, pero debe asociarse en el caso de los mal ignos o atípicos, en las resecciones incompletas o en casos de tumores recurrentes múltiples. La embolización arterial preoperator ia puede faci l i tar la cirugía, al obstruir las arterias nutricias del tumor .
Se han real izado ensayos terapéuticos con antagonistas de la progeste-rona (mifepristona) o, más recientemente, con agentes quimioterápicos c o m o la h idrox iurea (MIR 99-00, 252) .
Se reconocen diferentes t ipos histológicos: s incit ia l o meningote l ia l (es la fo rma más frecuente), t rans ic iona l , fibroblástico, microquístico, psamomatoso, co rdo ide , secretor, de células claras, l in fop lasmoc i to i-de, angiomatoso, papi lar, atípico y ma l igno (estos dos últimos, muy raros y más agresivos, con tendencia a la recidiva).
Tienen tendencia a la calcificación. Los cuerpos de psamoma son un hal lazgo anatomopatológico característico. La v iment ina y el EMA (an-tígeno epite l ia l de membrana) son dos marcadores inmunohistoquími-cos del men ing ioma .
Clínica
La clínica depende de la localización. Hay algunas presentaciones clínicas peculiares: los de la hoz cerebral frontal pueden s imular una clínica de h idrocefa l ia normotensiva con deter ioro cogn i t i vo , trastorno de la marcha e incont inenc ia ; los de foramen magno recuerdan, en ocasiones, a la clínica de una esclerosis lateral amiotrófica; los del surco o l fa tor io pueden produc i r un síndrome de Foster-Kennedy (anosmia, atrofia óptica ipsilateral y pap i ledema contralateral ) . Se asocian a una mayor frecuencia de trombosis venosa profunda.
Diagnóstico
Son tumores hipervascular izados, que muestran un aspecto homogéneo redondeado y bien de l im i t ado en la TC y la RM. Tras la admin is tración de contraste, se produce un marcado realce del tumor y, en ocasiones, se detecta la denominada "co la du ra l " , que es un hal lazgo característico de este tumor y que corresponde a la duramadre adyacente al anclaje del tumor .
Pueden presentar ca lc i f icac iones (cuerpos de psamoma) visibles en la TC y la RX de cráneo. A veces producen hiperostosis y fenómeno blis-ter ing en el hueso del cráneo vec ino . La angiografía permite conocer los aportes arteriales (la mayor parte se vascular izan a través de ramas meníngeas de la arteria carótida externa).
17.8. Neurinoma del VIII par (schwannoma vestibular)
Es un tumor benigno, de c rec imiento lento, que se or ig ina en la vaina de mie l ina de la rama vestibular del VIII par craneal (Figura 96) . Es el tumor más frecuente del ángulo pontocerebeloso (el segundo es el me n ing ioma y, en tercer lugar, el ep ide rmo ide o colesteatoma) y el t umor p r imar io más frecuente en la fosa posterior de los adultos (MIR 06-07, 60; MIR 03-04, 246) .
Figura 96. Neur inoma de l VIII par craneal
Pueden ser bilaterales y, en este caso, son patognomónicos de neuro-f ibromatosis t ipo II.
Se descr iben dos subtipos histológicos: uno más compac to , con células bipolares en empal izada (t ipo A de Anton i ) y otro más laxo, con células espumosas (tipo B de Anton i ) . La proteína S-100 es un marcador inmu-nohistoquímico del neur inoma (células de Schwann).
125

Manual CTO de Medicina y Cirugía, 8.a edición
Produce hipoacusia neurosensorial , acúfenos y vértigo (lesión del VIII par). En su c rec imiento puede c o m p r i m i r los pares V y V I I , dando l u gar a hipoestesia t r igeminal con abolición del ref lejo corneal y paresia fac ia l . Cuando es muy grande, puede llegar a c o m p r i m i r el t ronco en cefálico y otros pares craneales, dando lugar a ataxia, diplopía, afectación de pares bajos e inc luso trastornos respiratorios y coma, si no son diagnosticados antes.
El p roced im ien to diagnóstico de elección es la RM y, en segundo lugar, la TC con contraste.
El t ratamiento puede ser qu i rú rg i ca y/o mediante radiocirugía (terapia con rayos gamma procedentes de varias fuentes (Co-60) d i r ig idos al área c i rcunscr i ta del tumor , que se realiza en una única sesión). Se descr ibe con más detal le en la Seccón de Otorrinolaringología.
17.9. Tumores de la región pineal
Los tumores de esta región (Tabla 66) son, en genera l , más frecuentes en niños que en adul tos . H a b i t u a l m e n t e p roducen h id roce fa l i a por estenosis del a cueduc to de S i lv io y dan clínica de HTIC sin f o c a l i dad neurológica. U n ha l lazgo exp lo r a to r i o típico es el síndrome de Par inaud, por lesión de la porción más dorsal y rostral mesencefálica (tubérculos cuadrigéminos superiores y área pretecta l ) : parálisis de la elevación de la m i rada , ausencia de ref le jo f o t o m o t o r , conservando el ref le jo de acomodación a la d is tanc ia , con pupi las en midr ias is med ia y f i ja (a veces an isocor ia ) , parálisis de la convergenc ia , nistag-mus retráctil y, en ocasiones, pseudoparálisis de l V I par (MIR 98-99, 58).
ORIGEN GERMINAL OR IGEN NO GERMINAL
• Germinoma Astrocitoma • Coñocarcinoma • Pineoblastoma • Tumor seno endodérmico • Pineocitoma • Teratoma • Carcinoma embrionario
Tabla 66. Tumores de la región pineal
Tumores de células germinales
El ge rm inoma es el t i po histológico más frecuente (MIR 98-99F, 78) y const i tuye el t umor más frecuente de la región p inea l . Es más predominante en varones durante la infancia-adolescencia (los tumores de células germinales en la mujer se loca l izan más f recuentemente en la región supraselar que en la región pineal ) . Puede acompañarse de d ia betes insípida o pubertad precoz.
Frecuentemente es invasor, con citología posit iva en LCR. Hab i tua l mente, los marcadores tumorales en LCR o suero son negativos, pero puede presentar una elevación moderada de a-fetoproteína (aFP), fos-fatasa a lcal ina placentar ia o gonadot rop ina coriónica (HCG). Cuando son posit ivos, su determinación seriada permite evaluar la respuesta al t ratamiento y diagnosticar precozmente las recidivas.
Se descr iben también otros tumores de células germinales en esta región, también l lamados tumores no germinomatosos de la región p i neal: t umor del seno endodérmico (marcador: aFP), co r ioca rc inoma
(marcador: HCG), ca rc inoma embr iona r io y teratomas. Todos ellos, salvo los teratomas benignos, son tumores mal ignos y pueden metas-tatizar por LCR. El ge rm inoma es extraord inar iamente radiosensible y puede tratarse con radioterapia. El resto requiere cirugía. El pronóstico es m u c h o mejor en el caso del ge rm inoma que en los tumores no germinomatosos.
Tumores que no derivan de células germinales
Dentro de este grupo, destacan tres tumores : el astroci toma, el p i neoc i toma y el p ineoblastoma. El más frecuente entre el los es el astroc i toma. El p ineoc i toma es un tumor bien d i ferenc iado, der i vado de las células del parénquima p inea l . No t iene predilección por n inguna edad o sexo determinado. Son frecuentes las ca lc i f icac iones y las denominadas " r o setas de Bor i t " . La enolasa neuronal específica es un marcador inmu-nohistoquímico de este tumor . El p ineoblastoma es un tumor ma l igno que se considera un tumor neuroectodérmico p r im i t i vo (PNET). Tanto el p ineoc i toma c o m o el p ineoblastoma son tumores que se pueden d i seminar a través del líquido cefalorraquídeo. El t ra tamiento de elección de todas las neoplasias de este grupo es la cirugía.
17.10.Tumores hipofisarios
Adenoma
Son tumores benignos del lóbulo anterior de la hipófisis (adenohipó-fisis). Pueden ser funcionantes o secretores, que se dan en el 7 0 % de los casos (el más frecuente, el pro lac t inoma) , y no func ionantes . A ve ces son mixtos (más frecuentemente productores de G H y prolact ina) . Se clasif ican en función del tamaño c o m o microadenomas (menor de 1 cm) y macroadenomas (de tamaño mayor o igual a 1 cm) (Figura 97). Su inc idenc ia es s imi lar en ambos sexos, y son más frecuentes en las décadas tercera y cuarta de la v ida .
Figura 97. Macroadenoma de hipófisis
126

Neurología y neurocirugía
Pueden p roduc i r clínica endocr ino log ía por hipoperfusión o hiper-función (acromegal ia , Cush ing, amenorrea-galactorrea, etc.). Los f u n c ionantes suelen debutar más p recozmente con síntomas der ivados del exceso de la h o r m o n a que p roducen , pero los no func ionantes no suelen manifestarse hasta a lcanzar suf ic iente tamaño c o m o para p roduc i r efecto de masa; en estos casos, los pr imeros síntomas suelen ser a l teraciones campimétricas por compresión del qu iasma óptico desde abajo (lo más f recuente, hemianops ia heterónima b i t empora l (MIR 01-02, 61) y cuadrantanops ia super ior ) . Puede produc i rse hiper-p ro l ac t i nemia d e b i d o a la compresión del ta l lo que pueden provocar estas neoplasias. Los tumores grandes de cua lqu ie ra de las variantes pueden or ig inar panh ipop i tu i t a r i smo . Una fo rma rara de presentación es la apoplejía h ipof isar ia , que consiste en un deter ioro neurológico rápido que se manif iesta genera lmente por cefalea, de ter io ro visual ( i nc lu ida amaurosis súbita), o f t a lmop l e j i a y reducción del n ive l de consc ienc ia d e b i d o a una hemorrag ia , necrosis o infar to den t ro del t umor y la glándula adyacente (MIR 06-07, 54). Rara vez pueden ser causa de h idroce fa l i a .
Hay que hacer diagnóstico di ferencia l con otras lesiones de la región quiasmática: c raneofar ing ioma, quistes de la bolsa de Rathke, g l ioma hipotalámico, tumores de células germinales, aneurismas t rombosados de la arteria carótida o de la comun i can te anterior, tuberculosis , sar-coidosis, etc.
El t ra tamiento quirúrgico de elección es la resección por vía transesfe-no ida l . Dent ro de los tratamientos médicos, destacan la b romocr ip t ina para el p ro lac t inoma y el octreótido o análogos para los secretores de G H . La radioterapia posquirúrgica es a veces muy eficaz en el contro l de las recidivas.
En la Sección de Endocrinología, metabolismo y nutrición se desarrolla este tema con mayor p ro fund idad .
Carcinoma
Son tumores mal ignos muy raros, con capac idad de diseminación me-tastásica intraneural o extraneural .
Tumor de células granulares
También muy infrecuentes, der ivan de los p i tu ic i tos granulares del lóbu lo posterior de la hipófisis (neurohipófisis).
17.11. Tumores de origen disembrioplásico
Craneofaringioma
Es un t umor disembrioplásico or ig inado a partir de restos de la bolsa de Rathke, de localización supraselar, que afecta pr inc ipa lmente a niños y adolescentes (Figura 98) . Desde un punto de vista anatomopatológico, se ven dos variantes: adamant inomatosa (g lobalmente, la más f recuente) y la escamosa papi lar (se da en los adultos).
Figura 98. Craneofaringioma
Suelen tener un importante componente quístico de conten ido aceitoso y una pared parcia lmente calc i f icada (se describen las calci f icaciones en paréntesis en la RX lateral de cráneo) (MIR 00-01F, 73; MIR 98-99F, 79).
Produce clínica de disfunción neuroendocr ina y campimétrica por compresión del quiasma (hemianopsia b i tempora l o cuadrantanopsia inferior) . Puede produc i r ta l la baja y obesidad por afectación hipotála-mo-hipofisar ia.
El t ra tamiento de elección es la resección quirúrgica, pero se han u t i l i zado también la evacuación estereotáctica del quiste, terapia intracavi-taria con radioisótopos c o m o el ¡trio y el fósforo, b l eomic ina ¡ntralesio-nal , IFN-a, radioterapia convenc iona l y radiocirugía.
Quiste coloide
Es un t umor de adultos, clásicamente descrito c o m o der ivado de la paráfisis, que se loca l iza en la parte anter ior del tercer ventrículo (Figura 99) . Está bien encapsulado por te j ido epite l ia l y cont iene material g lucopro te i co PAS posi t ivo.
Figura 99. Quiste coloide del tercer ventrículo
127

Manual CTO de Medicina y Cirugía, 8.a edición
Característicamente, produce hidrocefa l ia aguda intermitente con los cambios posturales, por b loqueo de los agujeros de M o n r o . Aunque es de c rec imiento lento, se describe c o m o una de las causas de muerte súbita (más frecuente en la era pre-TC).
El tratamiento de elección es la cirugía, siendo útiles las técnicas neuroendoscópicas. También se han uti l izado la aspiración este-reotáctica y las derivaciones
ventriculoperitoneales (deben ser bilaterales, dada la obstrucción de ambos agujeros de Monro) .
R E C U E R D A El quiste colo ide se caracteriza por producir una hidrocefalia postural.
Suelen ser l infomas de células B y presentar distribución perivascular. Se local izan con más frecuencia en ganglios de la base, sustancia blanca periventricular y cuerpo calloso. Un dato anatomopatológico característico de estos tumores son los infi ltrados perivasculares de l infocitos. En la TC craneal captan contraste homogéneamente, con frecuencia en an i l lo . Puede asociarse a un l in foma ocular, que se manifiesta hab i tua lmente c o m o una disminución indolora de la agudeza v isual . Es característica la importante disminución o desaparición de las lesiones en la TC, tras un c i c l o de varias semanas con cort ico ides en dosis elevadas ( tumor " fantasma") . El t ra tamiento más eficaz es la radioterapia, que actua l mente suele combinarse con qu imio te rap ia con metotrexato.
Lipoma
Se localiza preferentemente en el cuerpo calloso. Puede asociarse a anomalías en el desarrollo del sistema nervioso (displasia de línea media) o a crisis epilépticas. En general, es asintomático y no requiere tratamiento.
Tumores dermoide y epidermoide (colesteatoma)
Son tumores benignos, hab i tua lmente quísticos, procedentes de restos embr ionar ios de or igen ectodérmico que quedan inc lu idos durante el cierre del t ubo neural . Aparecen pr inc ipa lmente en línea media , a nivel del ángulo pontocerebeloso, cisterna prepont ina y cuarto ventrículo en adultos jóvenes.
Son de c rec imiento lento y dan síntomas por efecto de masa, según la localización. Se han descrito cuadros de meningi t is aséptica rec id ivan te (por liberación de cristales de colesterol) y una variante de la misma: la meningi t is de Mo l l a re t (con células de probable estirpe macrofágica) por rotura del quiste.
El t ratamiento es la resección quirúrgica, j un to a la cápsula, para evitar recidivas.
Hamartoma neuronal
Son acúmulos de neuronas adultas fuera de su localización habi tual . Los de la región mesial (parahipocampal ) del lóbulo tempora l son cau sa de epi lepsia. Los hipotalámicos son propios de niños y producen pubertad precoz.
17.12. Linfoma cerebral primario
17.13. Hemangioblastoma
Es un t umor ben igno que aparece con más frecuencia en la fosa posterior, a nivel de los hemisferios cerebelosos, pero también se detecta a lo largo de t o d o el neuroeje. Es el más frecuente de los tumores pr imar ios intraaxiales de la fosa posterior en el adu l to . Puede ser sólido, aunque suele ser quístico, con un nodu lo mural h ipercaptante.
Aunque la mayoría son esporádicos, hasta un 2 0 % ocurren en el c o n texto de la enfermedad de V o n Hippel-L indau. En estos casos, con frecuencia son múltiples y se acompañan de hemangioblastomas retiñíanos y otras lesiones viscerales (habi tua lmente tumores o quistes, sobre todo , a nivel de páncreas y riñon). Es típica la relación de esta enfermedad con el f eoc romoc i toma (MIR 06-07, 74). En la Tabla 67 se descr iben las asociaciones más frecuentes entre facomatosis y tumores del sistema nervioso central .
ESCLEROSIS TUBEROSA Astrocitoma gigantocelular subependimario
NEUROFIBROMATOSIST IPO 1 Glioma de vías ópticas
NEUROFIBROMATOSIS TIPO II Neurinoma bilateral del VIII par. Meningiomas
STURGE-WEBER Angiomas leptomeníngeos
VON HIPPEL-LINDAU Hemangioblastoma cerebeloso
KLIPPEL-TRENAUNAY Angioma cavernoso de la médula espinal
Tabla 67. Facomatosis y tumores del sistema nervioso central
Q R E C U E R D A U n quiste con un nodulo captante en su interior en el seno de un hemisferio cerebeloso es un astrocitoma pilocít ico, si el paciente es un niño, y un hemangioblastoma si es adulto.
Pueden producir eritropoyetina y es característica la presencia de polici-temia en los estudios de laboratorio (MIR 00-01, 61). El tratamiento de elección es la cirugía (vaciamiento del quiste y exéresis del nodulo mural).
Hab i tua lmente , se observa en enfermos con defectos ¡nmunológicos mixtos (humoral y celular) , aunque su f recuencia en ind iv iduos inmu-nocompetentes está aumentando . En pacientes con SIDA es la neopla-sia cerebral más frecuente y la segunda lesión intracerebral en f recuencia en estos pacientes, después del absceso por toxoplasma. También puede verse en algunas afecciones del t e j ido con jun t i vo . Se asocia a infecciones por el virus de Epstein-Barr.
17.14.Tumores de la base craneal
• Tumor glómico yugular. Se local iza a nivel del agujero rasgado posterior, afectando a los pares craneales IX, X y XI. También hay tumores glómicos en el oído med io (neoplasia más frecuente en esta región).
128

Neurología y neurocirugía
El diagnóstico se realiza por RM ( imagen en sal y p imienta) y ang io grafía. Está ind icada la detección de catecolaminas en or ina (ácido vanilmandélico, metanefrinas y noradrenal ina) . El t ra tamiento es quirúrgico, con o sin embolización previa o rad ioterapia . El mane jo prequirúrgico de los tumores con secreción activa de catecolaminas es similar al del f eoc romoc i toma , admin i s t rando agentes al fabloqueantes y betabloqueantes.
• Cordoma. Es un tumor der ivado de restos de la notocorda , histológ icamente ben igno, pero loca lmente agresivo. Afecta a adultos, y puede local izarse en cl ivus ( 6 0 % ) o en la región sacrococcígea. Las células fisalíforas son características de esta neoplasia.
Se trata con cirugía y radioterapia, pero son frecuentes las recidivas.
En la Tabla 68 se resumen las características anatomopatológicas más típicas de los tumores que han aparec ido en el texto.
r
U n pac i en te de 62 años p resen ta , de f o rma b rus ca , u n a hemih ipoes tes i a termo-algésica del h e m i c u e r p o d e r e c h o , así c o m o h ipoes tes ia de la h e m i c a r a i zqu i e rda , hemia tax i a i zqu ie rda y deb i l idad de los múscu los de la mast i cac ión . ¿ D ó n d e loca l i z a r e m o s la lesión?
1) Mesencé fa lo la tera l d e r e c h o . 2 ) Mesencé fa lo m e d i a l i z q u i e r d o . 3) P r u t u b e r a n c i a la tera l i z q u i e r d a . 4 ) P r o t u b e r a n c i a m e d i a l d e r e c h a . 5) B u l b o m e d i a l d e r e c h o .
M I R 0 5 - 0 6 , 5 4 ; RC: 3
U n pac ien te de 73 años sufrió un a c c i d e n t e de t ráf ico c o n t r aumat i smo c r a n e a l , del que se recuperó . A los t res meses , i n i c i a de f o rma progres iva a l te rac ión de func iones super io res , i n con t i nenc i a u r ina r i a ocas iona l y su c a m i n a r es torpe. P robab lemente p resen ta :
1) H e m a t o m a i n t r a p a r e n q u i m a t o s o c e r eb ra l tardío. 2) H e m o r r a g i a s u b a r a c n o i d e a . 3) H i d r o c e f a l i a a r r e a b s o r t i v a . 4 ) A t r o f i a c e r eb ra l postraumática. 5) T u m o r c e r e b r a l .
M I R 0 5 - 0 6 , 6 4 ; RC: 3
V a r ó n de 2 9 años aque j ado de m a r e o s y t o r p e z a en m i e m b r o s i zqu ie rdos , en los úl t imos 6 meses . En la exp lo rac ión , p resenta dismetr ía y d i sd i adococ ines i a de m i e m bro super io r i zqu ie rdo . La T A C c r a n e a l mues t ra u n a lesión quíst ica, c o n nodu lo hi-percap tan te s i tuado en hemis fer io ce rebe loso i zqu ie rdo . L a anal í t ica es n o r m a l , a excepc ión de un hematoc r i t o de 5 8 % , y u n a T A C to racoabdomina l de tec tó quistes e n páncreas y r iñon . La na tu r a l eza más p robab le de la lesión in t r ac ranea l sería:
1) M e d u l o b l a s t o m a . 2) Metástasis d e c a r c i n o m a p u l m o n a r . 3 ) H e m a n g i o b l a s t o m a . 4 ) A s t r o c i t o m a pi loc í t íco. 5) G l i o b l a s t o m a m u l t i f o r m e .
M I R 0 0 - 0 1 , 6 1 ; RC: 3
V
A S T R O C I T O M A P I LOC IT ICO Fibras de Rosenthal
O L I G O D E N D R O G L I O M A Células en "huevo frito"
T U M O R E S EMBR IONAR IOS Rosetas de Homer-Wright
MEN ING IOMA Cuerpos de psammoma
SCHWANNOMA V E S T I B U L A R Fibras de Antoni
P INEOC ITOMA Rosetas de Borit
Q U I S T E C O L O I D E Material PAS+
L INFOMA Infiltrados linfocitarios perivasculares
C O R D O M A Células fisalíforas
Tabla 68. Datos anatomopatológicos característicos de los tumores del sistema nervioso central
Casos clínicos representativos
U n pac ien te de 60 años , c o n an teceden tes de cánce r de pu lmón , p resenta u n a c r i s is epi lépt ica. Se r ea l i za R M ce reb ra l , que mues t ra una lesión ún ica sugerente de metástasis. No hay e v i d e n c i a de metástasis ex t race rebra l es . ¿Cuá l de las s iguientes a f i rmac iones es F A L S A respec to a l t r a tamiento de l pac ien te ?
1) D e b e a d m i n i s t r a r s e med i cac ión antiepi léptica. 2) Los c o r t i c o i d e s son útiles pa ra d i s m i n u i r el e d e m a vasogénico. 3) Si la lesión es a c ce s i b l e , p u e d e estar i n d i c a d a la cirugía. 4) La r a d i o t e r a p i a c r a n e a l n o está i n d i c a d a . 5) La q u i m i o t e r a p i a p u e d e estar i n d i c a d a .
M I R 9 8 - 9 9 F , 7 1 ; RC: 4
En un va rón de 75 años, t ras la apar i c ión b rusca de un s índrome neurops iquiát r ico , u n a T C c r anea l demues t r a la ex i s tenc ia de u n a metástasis c e r e b r a l . ¿Cuá l es la l o c a l izac ión más p robab le de l t umor p r imar io metas ta t izante?
1) Gástr ica. 2) Rena l . 3) T i r o i d e a . 4) B r o n c o p u l m o n a r . 5) Tes t i cu l a r .
RC: 4
Ante un n iño de 5 años c o n un c u a d r o de hipertensión e n d o c r a n e a l , a l t e rac iones v isua les e h ipota lámicas , que presenta en una radiograf ía lateral de c ráneo ca l c i f i c a c i o n e s en f o rma de paréntesis, a n ive l suprase la r , ¿cuá l será su diagnóst ico p re sunt ivo? :
1) A d e n o m a h i p o f i s a r i o . 2 ) G l i o m a d e l n e r v i o ópt ico. 3) C r a n e o f a r i n g i o m a . 4) M e d u l o b l a s t o m a . 5) P i n e a l o m a p r o d u c t o r d e h i f r o c e f a l i a .
RC: 3
1 2 9

Neurología y neurocirugía
18. TRAUMATISMOS CRANEOENCEFÁLICOS
Los aspectos más p r e g u n t a d o s d e este t e m a son el h e m a t o m a e p i d u r a l y las f rac turas d e base d e cráneo. Es i m p o r t a n t e q u e se t engan c laros los c o n c e p t o s de cada una d e [as les iones q u e se p r o d u c e n c o m o c o n s e c u e n c i a d e u n t r a u m a t i s m o craneoencefál ico. Se d e b e d i f e r enc i a r u n h e m a t o m a e p i d u r a l de u n subdu ra l a g u d o , y es tud ia r la escala de G l a s g o w .
Aspectos esenciales
Q~J La escala de G l a sgow va lora el n i ve l de consc i enc i a de un pac iente t r a u m a t i z a d o en función de tres parámetros : respuesta ve rba l , m o t o r a y ocu la r . La puntuación mínima es de tres pun tos y la máxima (nivel de consc i enc i a no rma l ) , de 15.
¡"2~] Los t r aumat i smos craneoencefálicos (TCE) se d i v i d e n en leves (14-15 puntos en la escala de G lasgow) , m o derados (9 y 13 puntos) y graves (3 y 8 puntos ) .
ÍJ~J M e d i a n t e la h is tor ia clínica y la exploración, los pac ientes que han suf r ido un TCE deben ser cons iderados c o m o de ba jo , m o d e r a d o o a l to r iesgo de tener una lesión in t racranea l , ya q u e el mane jo es d i fe rente en cada uno de los tres g rupos .
[~4~""j Una f rac tura abier ta i m p l i c a un desgarro de la d u r a m a d r e y con l l e va un riesgo de infección in t rac ranea l . En las fracturas compuestas , la d u r a m a d r e está íntegra, por lo q u e hay un riesgo ba jo de infección in t racranea l .
r j j En las fracturas de base de cráneo, puede exist i r una afectación de pares craneales, espec ia lmente el I y II en fracturas de fosa cranea l anter ior , el VI I y VI I I en fracturas de peñasco y el V I en fracturas de c l i vus .
["5] La sa l ida de LCR a través de l oído o de la nar iz es un ha l l azgo característico, pero p o c o f recuente , de las fracturas de base de cráneo.
[Y] El h e m a t o m a ep idu ra l suele deberse a un desgarro de la arteria meníngea med i a por una f ractura ósea. El h e m a t o m a subdura l suele ser de or igen venoso , d e b i d o a una lesión de la cor teza ce rebra l .
[~8~| La cl ínica más f recuente de presentación de un h e m a t o m a ep idu ra l es la de r i vada de una herniación u n c a l ; sin emba rgo , lo más característico es la ex is tenc ia de un in terva lo lúcido.
|~9~] La edad avanzada , el a l c o h o l i s m o y las a l terac iones de la coagulación cons t i tuyen factores de r iesgo para el h e m a t o m a subdura l crónico. En la m i t a d de los casos se reconoce un antecedente traumático.
Los t raumat ismos craneoencefálicos suponen una importante causa de morb imor ta l i dad en jóvenes occ identa les, fundamenta lmente varones, en relación muy estrecha con los accidentes de tráfico. Se consideran la pr imera causa de pérdida de conoc im ien to ( inc luyendo desde la conmoción cerebral hasta las diferentes fases de coma) en la población general (MIR 98-99F, 206) y el factor etiológico más frecuente de epi lepsia entre los 18 y los 35 años.
18.1. Escala de coma de Glasgow
Para hacer una aproximación al nivel de consc ienc ia de los pacientes que han sufr ido un t raumat ismo craneo
encefálico (TCE), se ut i l iza la escala de coma de G lasgow (GCS) (Tabla 69) , que valora tres parámetros clínicos
(apertura de ojos, respuesta motora y respuesta verbal) , pun tuando entre un mínimo de 3 y un máximo de 15
(MIR 09-10, 230 ; MIR 05-06, 92).
[T ] P r egun tas
•MIR 0 9 - 1 0 , 2 3 0 - M I R 0 6 - 0 7 , 61 - M I R 0 5 - 0 6 , 9 2 , 9 4 •MIR 0 0 - 0 1 , 60 •MIR 9 9 - 0 0 , 193 - M I R 99-00F , 7 2 , 73 • M I R 9 8 - 9 9 , 66 - M I R 9 8 - 9 9 F , 7 7 , 2 0 6
R E C U E R D A La respuesta m o t o r a es el parámetro de mayor va lo r en la escala de G lasgow.
El nivel de consc iencia , va lorado según la
puntuación en esta escala, es el pr inc ipa l
factor pronóstico en el TCE. Se def ine c o m o
TCE leve el que t iene una puntuación de 14
o 15; TCE moderado es aquel que puntúa
entre 9 y 13; una puntuación total menor o igual a 8 indica TCE grave, de mal pronóstico. La evolución en ex
p loraciones repetidas de la puntuación en la escala de coma de G lasgow también t iene valor pronóstico; un
descenso de tres o más puntos se corre lac iona con alta pos ib i l idad de lesión grave.
130
Neurología y neurocirugía
PUNTUACIÓN APERTURA RESPUESTA RESPUESTA
PUNTUACIÓN DE OJOS MOTORA VERBAL
6 - Obedece órdenes -
5 - Localiza el dolor Orientado
4 Espontánea Retira el dolor Confuso
3 A la voz Flexora
(decorticación) Inapropiado
2 Al dolor Extensión
(descerebración) Incomprensible
1 No No No
Tabla 69. Escala de coma de Glasgow
Además, debe realizarse una exploración comple ta en busca de signos de foca l idad neurológica. En pacientes con bajo nivel de consc iencia o inconscientes, son de gran va lor el tamaño pupi la r (la asimetría pupi lar es un signo de urgencia, pero no el edema de papila) y los reflejos tron-coencefálicos (corneal, oculocefálicos, oculovest ibular , nauseoso, tusígeno, etc.). En el apartado 2 . 1 , ded icado al Coma, se detal la la exp lora ción neurológica de estos pacientes. En general , la prueba radiológica de elección para el diagnóstico de las lesiones intracraneales asociadas al t raumat ismo craneoencefálico es la TC craneal (MIR 0 0 - 0 1 , 60).
18.2. Manejo del TCE en urgencias
Todo paciente que llega o es trasladado al servic io de urgencias tras sufrir un t raumat ismo craneoencefálico debe ser cata logado con bajo, moderado o alto riesgo de tener una lesión intracraneal , ya que las recomendaciones dependerán del grupo en que se encuentre el m ismo. • Pacientes con riesgo bajo. Se engloba en esta situación a aquel los
pacientes asintomáticos, con cefalea, mareo o con una contusión o abrasión del cuero cabe l ludo. En este grupo de pacientes se recomienda la observación d o m i c i l i a ria, sin indicar n inguna prueba de imagen, s iempre y cuando haya una persona responsable que pueda v igi lar los. Se les proporcionará un catálogo en el que se reflejan una serie de hallazgos de modera do o alto riesgo de tener una lesión intracraneal ; se les advertirá que deben volver al hospital en el caso de que aparezca a lguno de ellos. Se recomienda la realización de una TC cerebral a los pacientes inc lu idos en este grupo, pero con factores de riesgo tales como : coagulopatías, eno l i smo, abuso de drogas, antecedentes neuroqui-rúrgicos, ancianos con incapac idad y epi lepsia.
• Pacientes de moderado riesgo. Dentro de este g rupo se inc luye a pacientes que hayan ten ido una previa disminución transitoria del nivel de consc iencia , pacientes con amnesia postraumática, que han ten ido convuls iones, pacientes que están vomi tando , pacientes con tumefacción signif icat iva subgaleal, cefalea progresiva, m e n o res de dos años o historia de ingesta de drogas. En este t ipo de pacientes, se recomienda la realización de una TC cerebral y, en la mayor parte de los casos, observación hospitalaria durante unas horas.
• Pacientes de alto riesgo. Son pacientes que t ienen un nivel de consc iencia dep r im ido , GCS < 14 o aquel los en los que se observa una disminución progresiva del nivel de consc iencia , pacientes que muestran foca l idad neurológica, TCE penetrantes o fracturas-hundimiento . Este grupo debe ser somet ido a la realización de una TC cerebral y valoración por el servicio de neurocirugía.
18.3. Fracturas craneales Según el patrón de fractura, pueden clasificarse c o m o se descr ibe en los apartados siguientes.
Lineal
La existencia de una fractura demuestra que el cráneo ha sufr ido un impacto de gran energía, pero el pronóstico del paciente dependerá de la posib le lesión encefálica subyacente, no de la fractura. Existe una pobre correlación entre lesión ósea y daño cerebral , de m o d o que un paciente puede tener una fractura sin afectación encefálica y, a la inversa, un daño encefálico masivo sin fractura.
El hal lazgo de una fractura en la RX s imple de cráneo es indicación de TC craneal urgente para valorar las posibles lesiones intracraneales asociadas.
En la RX s imple de cráneo, las fracturas lineales se d i ferenc ian de las impresiones vasculares porque son rectilíneas, no se rami f i can , t ienen un grosor de lgado y un i fo rme en todo su trayecto y por su localización (Figura 100).
Figura 100. Fractura lineal frontal
Por lo general , no requieren t ratamiento, pero es necesario mantener al paciente en observación, pues sugieren que el t raumat ismo ha sido importante . Se def ine c o m o abierta aquel la fractura l ineal que está en comunicación con una laceración de la duramadre .
El riesgo de infección intracraneal con fracturas lineales de la convex i dad es muy bajo, y sólo está aumentado cuando son abiertas (MIR 99-00F, 73).
Cuando la energía se apl ica sobre un área re lat ivamente pequeña, pue de produci rse una f ractura-hundimiento (Figura 101), que es aquel la en que la tabla externa se hunde por debajo del límite anatómico de la tabla interna.
En ocasiones son fracturas conminutas (con varios fragmentos). Suelen acompañarse de laceración del cuero cabe l ludo y de la duramadre .
131

Manual C T O de Medicina y Cirugía, 8 . A edición
Figura 101 . F rac tura-hundimiento parietal
Aunque pueden diagnosticarse en la RX simple de cráneo, la TC craneal es la prueba diagnóstica de elección, ya que permite determinar el grado de hund imiento y la existencia de lesiones intracraneales asociadas.
El t ra tamiento depende en gran medida de dichas lesiones pero, en general, requieren cirugía para elevar el f ragmento hund ido . Si son abiertas, debe extirparse este f ragmento óseo y se realizará una craneoplas-tia d i fer ida varios meses después, para reducir el riesgo de infecciones intracraneales que puedan aparecer. En estas fracturas, está aumentado el riesgo de crisis postraumáticas; se acepta que la reparación quirúrgica no reduce este riesgo ni mejora los posibles déficit neurológicos asociados, que dependen de la lesión parenquimatosa in i c ia l .
Q R E C U E R D A U n a f r a c t u r a - h u n d i m i e n t o es u n f a c t o r d e r i esgo para el d e s a r r o l l o d e cr is is e p i lépticas p r e c o c e s .
Compuesta
La fractura compuesta es una fractura craneal asociada a una herida en cuero cabe l ludo o en con t inu idad con una fractura en pared de senos paranasales, celdas mastoideas o cav idad del oído med io .
En este caso, hay que desbridar la herida y administrar t ratamiento an tibiótico para prevenir osteomiel i t is e infecciones del cuero cabe l ludo (más f recuentemente por Staphylococcus aureus).
Diastática
Son aquellas en las que el t razo de fractura co inc ide con una sutura craneal . Son más frecuentes en niños pequeños (menores de tres años).
Creciente o evolutiva
Son típicas de niños y se caracter izan porque la fractura desgarra la duramadre , pe rmi t i endo que la aracnoides se hernie a través de la línea de fractura, de m o d o que las pulsaciones de LCR la van agrandando progresivamente. Se l laman también quistes leptomeníngeos postrau-máticos y requieren cirugía para cerrar el defecto meníngeo.
Fractura en "ping-pong"
En lactantes, por la plast ic idad del cráneo, los hund imien tos cerrados suelen produc i r este t ipo de fractura característica en ta l lo verde. En ausencia de daño parenquimatoso, la reparación quirúrgica suele ser necesaria solamente en las frontales por mot ivos estéticos (en otras l o cal izaciones, suelen desaparecer con el c rec imiento) (Figura 102).
Figura 102. Fractura en "p ing-pong" en un lactante
Fracturas de la base del cráneo
Las fracturas de la base del cráneo (fracturas basilares) se sospechan cuando un paciente que ha sufr ido un TCE presenta determinados signos explorator ios : hemotímpano, equimosis retroauricular (signo de Battle), equimosis per iorbi tar ia ("ojos de mapache" ) , lesión de pares craneales que discurren por la base (anosmia por lesión del I par craneal en las fracturas f rontoetmoida les , lesión del VI I y VIII par en las de peñasco y del VI par en las de cl ivus) (MIR 98-99, 66) y, con menos frecuencia (aunque diagnósticas), otorrea o r inorrea l icuorales o hemá-ticas (MIR 99-00F, 72) (Figura 103).
O t o r r e a
H e m a t o m a p e r i o r b i t a r i o b i l a t e r a l ( o j o s d e m a p a c h e )
Figura 103. Signos clínicos de fractura de la base del cráneo
132

Neurología y neurocirugía
Las local izac iones más frecuentes son la región f rontoe tmoida l y el peñasco.
Las que interesan al peñasco se clasif ican según la dirección del trazo de fractura en longitudinales o fracturas timpánicas (oído med io - t r o m pa - caja timpánica - CAE) y transversales o fracturas neurosensoriales (oído interno - cápsula laberíntica - CAI y agujero rasgado posterior) . Sus características clínicas se descr iben en el Capítulo de ORL. Un tercer t i po serían las fracturas obl icuas o timpanolaberínticas, que s i guen el eje neurosensorial (CAE - caja timpánica - oído interno - CAI) y p roducen parálisis facial en todos los casos, sordera mixta y otorrea (Tabla 70).
LONGITUDINAL TRANSVERSAL
Frecuencia 70-90% 12-20%
Perforación Frecuente Rara
Otorrea Frecuente Rara
Hemot ímpano Rara Frecuente
Otol icuorrea Frecuente Rara
Hipoacusia Transmisiva Perceptiva (cófosis)
Parálisis facial 2 0 % . Transitoria 5 0 % . Permanente
Vértigo Raro y leve (posicional) Frecuente y servero
Radiología Schüller Stenvers
Tabla 70. Diagnóstico diferencial de fracturas del peñasco
Las fracturas de la base del cráneo son difícilmente evidenciables en la RX s imple, por lo que la prueba radiológica de elección es la TC craneal con ventana ósea. Un signo ind i recto es la presencia de aire intracraneal (pneumoencéfalo). Existen ciertas proyecciones radiológicas clásicas, ahora en desuso, para diagnosticar fracturas de peñasco: Schüller para las longi tudinales y Stenvers para las transversales.
Q R E C U E R D A El TC c o n v e n t a n a ósea es la p r u e b a radiológica diagnóstica d e e lecc ión en las f r ac tu ras d e base d e cráneo. El t r a t a m i e n t o d e estas f rac turas es c o n s e r v a d o r en la m a y o r p a r t e d e los casos.
La mayor parte de las fracturas basilares no precisan t ratamiento por sí mismas. Sin embargo, pueden asociarse con determinadas c o m p l i caciones que sí requieren un manejo específico: aneurisma carotídeo traumático, fístula carot idocavernosa postraumática, fístula de LCR, meningi t is ( incluso en ausencia de fístula de LCR), parálisis fac ia l , etc. El uso de antibióticos profilácticos es cont rover t ido . La fístula de LCR t iene especial t ranscendencia clínica por su posib le complicación con meningi t is recurrente, hab i tua lmente neumocócica. El diagnóstico de esta fístula se realiza al observar salida de líquido c laro y pulsátil por las fosas nasales o por el oído, que aumenta con las maniobras de Valsal va. Se conf i rma en laborator io , al presentar el líquido un con ten ido de glucosa > 30 mg/dl y detectar B-transferrina en la electroforesis.
El t ratamiento es pos ic ional (reposo en cama y cabecero l igeramente elevado) con restricción de líquidos y acetazolamida. Si no cede, se co loca un drenaje lumbar ( importante descartar prev iamente su asociación con hematomas intracraneales traumáticos). Cuando existe infección, se administ ran antibióticos. Si no desaparece la fístula, hay que plantear cirugía reparadora, previa localización de la fístula mediante cisternografía isotópica o RM potenciada en 12.
18.4. Conmoción cerebral Es la lesión traumática cerebral más frecuente y de menor t rascendencia. Se def ine c o m o una alteración del nivel de consc iencia , t ransi tor ia y de duración var iable, c o m o consecuencia de un t raumat ismo no penetrante p rovocado en el cerebro. Se pueden produc i r diversas alteraciones en el compor tamien to del paciente, amnesia del episod io , incoordinación, etc., que se recuperan en un t i empo var iable y generalmente, breve. No se ha c o m p r o b a d o ningún t ipo de alteración anatomopatológica ni radiológica en el encéfalo, únicamente una leve disfunción bioquímica (descenso de ATP mi tocondr i a l o alteración de neurotransmisores excítatenos). No precisa t ratamiento específico.
18.5. Hematoma epidural (Figuram
Aparece en un 1-3% de los traumat ismos craneales. Es más común en la segunda y tercera décadas, sobre todo, en varones. Los accidentes de tráfico son la causa más frecuente. El 8 5 % de los casos son de or igen arterial , p r inc ipa lmente por desgarro de la arteria meníngea media tras una fractura de hueso tempora l o parietal . En frecuencia, le siguen los de localización f ronta l y de fosa posterior.
La presentación clínica clásica es pérdida de consciencia, seguida de un periodo de lucidez (intervalo lúcido). Posteriormente se produce un deterioro neurológico de rápida evolución, en general debido a herniación uncal por el importante efecto de masa de la colección hemática (MIR 99-0 0 , 1 9 3 ; MIR 98-99F, 77). Sin embargo, menos del 3 0 % se presenta con la secuencia completa (con frecuencia, no hay pérdida de consciencia inicial o la hay sin intervalo lúcido posterior).
El diagnóstico se real iza mediante TC, que demuestra una imagen hi-perdensa por debajo de la tabla interna del cráneo con morfología de " lente b i convexa " , que c o m p r i m e el parénquima cerebral subyacente (efecto de masa) (Figura 104).
H e m a t o m a
Figura 104. Hemorrragias intracaneales
La mayoría precisan evacuación quirúrgica urgente por craneotomía. La mor ta l idad con t ratamiento precoz es de aprox imadamente el 1 0 % .
133

Manual CTO de Medicina y Cirugía, 8. a edición
18.6. Hematoma subdural nabia 71)
HEMATOMA EP IDURAL
HEMATOMA SUBDURAL
Origen
Lesión sarénquima
Mortal idad
Sangrado arterial (85%) Lo más frecuente, rotura de la a. meníngea media
• Rotura de vv. corticales • Agudo: primera semana • Subagudo: 7-10 días postTCE • Crónico: TCE trivial o no identificado en
5 0 % . Típico de ancianos y alcohólicos
Conmoción cerebral 4-
Intervalo lúcido 1
herniación uncal (coma de rápida
evolución) Aunque < 3 0 % se presentan con la clínica clásica
• Agudo: clínica de herniación uncal progresiva de rápida evolución
• Crónico: cefalea y demencia progresivas (parecido a ACV isquémico, perofluctuante)
Hiperdensidad en forma de lente biconvexa Frecuentemente efecto de masa
• Agudo: hiperdensidad en forma de semiluna
• Subagudo: isodenso • Crónico: hipodensidad en forma de
semiluna
En general, menor y más tardía (por compresión)
En general, mayor y desde el principio (la sangre está en contacto con el parénquima cerebral)
Con diagnóstico y tratamiento precoz, la mortalidad esaprox. 1 0 %
Las formas agudas tienen una mortalidad del 50-90%
Evacuación quirúrgica mediante craneotomía
• Agudo: evacuación quirúrgica mediante craneotomía
• Crónico: evacuación quirúrgica mediante trépano, con o sin drenaje subdural
Tabla 68. Hematomas epidural y subdural
Suele ser consecuencia de una hemorragia venosa causada por la ro tura de las venas puente cort icales o una laceración del parénquima cerebral . Se c lasi f ican, en función del t i empo de evolución desde el impacto , en agudos (tres pr imeros días tras el t raumat ismo) , subagudos (entre tres días y tres semanas) y crónicos (a partir de las tres semanas). • Hematoma subdural agudo. Supone una de las lesiones traumáticas
con mayor morb imorta l idad (50-90% a pesar de la cirugía). General mente, la magni tud del impacto es mayor que en el hematoma ep i dura l , y suele acompañarse de daño del parénquima subyacente, por lo que tienen peor pronóstico. Cursan con deterioro neurológico de rápida evolución. Se diagnostica en la TC por una imagen hiperdensa en forma de "semi luna" (véase Figura 105). Se recomienda la utilización de antiepilépticos por el riesgo de crisis epilépticas precoces. El tratamiento requiere la evacuación quirúrgica urgente por craneotomía.
. Hematoma subdural subagudo. Suelen ser isodensos con el parénq u i m a cerebral , aunque rara vez es preciso recurrir a la RM para su diagnóstico.
• Hematoma subdural crónico. Aparece sobre todo en pacientes de edad avanzada y alcohólicos crónicos, que suelen presentar c ierto grado de atrofia cerebral (con el consecuente aumento del espacio subdural ) , y en pacientes ant icoagulados. El t raumat ismo desencadenante es a menudo tan tr iv ia l que el paciente y la fami l ia no lo recuerdan.
R E C U E R D A La i m a g e n en s e m i l u n a es p r o p i a de l h e m a t o m a s u b d u r a l a g u d o , y la i m a g e n en f o r m a d e " l e n t e b i c o n v e x a " es característica de l h e m a t o m a e p i d u r a l .
Figura 105. (a) Hematoma epidural con forma de lente biconvexa; (b) hematoma subdural agudo con forma de semiluna. Ambos producen gran desplazamiento de
estructuras de línea media
Los síntomas y signos del hematoma subdural crónico son muy he terogéneos y pueden simular la clínica de otras entidades, c o m o un accidente vascular cerebral , tumores, encefalopatías metabólicas, demenc ia o psicosis. Predominan la alteración del estado menta l , la hemiparesia, las caídas frecuentes y la cefalea. En la TC cerebral son hipodensos (densidad líquido), también en forma de " semi luna " . Si son sintomáticos, requieren evacuación quirúrgica, pero al estar evo luc io nados, pueden evacuarse a través de agujeros de trépano, de jando o no un drenaje con t inuo .
18.7. Contusión cerebral hemorrágica
Son lesiones necroticohemorrágicas intraparenquimatosas traumáticas (hiperdensas en la TC) (Figura 106) cuya localización más frecuente es el lóbulo f ronta l (polo y superf ic ie orbi tar ia ) , la porción anterobasal del lóbulo tempora l y el po lo occ ip i t a l , zonas más sensibles al daño por cont rago lpe ocas ionado por el m o v i m i e n t o brusco del encéfalo dentro de la caja craneal , ya que se go lpean contra rebordes óseos. La indicación quirúrgica dependerá de la localización, tamaño y estado neurológico del paciente. Suelen precisar t ra tamiento antiepiléptico con fenitoína (mayor porcentaje de crisis focales precoces o tardías asociadas).
1 mí i
Figura 106. Contusiones cerebrales hemorrágicas múltiples
134

Neurología y neurocirugía
18.8. Lesión axonal difusa Es una lesión pr imar ia del parénquima cerebral que se produce en pa cientes que sufren un TCE con mecan ismo rotac ional de aceleración-deceleración. Anatomopatológicamente, se detectan lesiones difusas en los axones. Produce un deter ioro precoz y manten ido del nivel de consc iencia , sin que haya en la TC cerebral una lesión que just i f ique el cuadro (MIR 06-07, 61) .
La gravedad del daño axonal v iene determinada por la localización de hemorragias punt i formes en la TC craneal : grado I si están en la unión cor t i cosubcor t i ca l ; grado II, a nivel de cuerpo cal loso, y grado III, en la porción dorsolateral del t ronco encefálico. Son lesiones que, en general , con l levan un mal pronóstico.
Q R E C U E R D A Importante disminución del nivel de consciencia tras TCE de manera mantenida, sin hallazgos relevantes en la TC hace pensar en lesión axonal difusa.
18.9. Neumoencéfalo
Se def ine c o m o la existencia de aire intracraneal . Es más frecuente en fracturas en las que se imp l i can también los senos paranasales (frontal o e tmoida l ) , inc lu idas las fracturas de la base del cráneo, o en fracturas de la convex idad con desgarro dura l .
Otras causas no traumáticas frecuentes de neumoencéfalo son la c i r u gía (poscraneotomía, trépano, ventriculostomía, etc.), punción lumbar, barotrauma, algunos defectos congénitos del cráneo e infecciones por gérmenes productores de gas. El síntoma más frecuente de presentación es la cefalea.
El neumoencéfalo a tensión se caracteriza porque el aire se encuentra a elevada presión y produce clínica de hipertensión intracraneal, con efecto de masa y, en ocasiones, con rápido deterioro del nivel de consciencia (se debe a un efecto de válvula que deja entrar aire, pero no salir).
El aire puede observarse en la RX de cráneo, pero es de elección la TC craneal . Si no ejerce efecto de masa, sólo requiere observación, pero
cuando se trata de un neumoencéfalo a tensión, es necesaria la evacuación urgente, c o m o si se tratara de un hematoma intracraneal . Cuando se asocia a fístula de LCR, debe tratarse ésta.
18.10. Complicaciones y secuelas del neurotrauma central
• Infecciones tardías en traumatismos abiertos (meningit is postrau-mática recurrente, empiema, absceso, t rombof leb i t i s , laberintit is purulenta) .
• Fístula de líquido cefalorraquídeo. Descritas en el apartado 18.2. • Crisis epilépticas postraumáticas. Ya fueron tratadas en el apartado
Capítulo 7. Epilepsia. • Fístula carotidocavernosa. Más frecuente en traumat ismos de la
base o penetrantes. También puede aparecer de manera espontánea. Se produce por rotura parcial del sifón carotídeo dentro del seno cavernoso y cursa con exofta lmos uni lateral o bi lateral pulsát i l , soplo aud ib le por el p rop io paciente dentro de la cabeza (suele ser el síntoma inic ia l ) , quemosis con junt i va l importante y, a veces, lesión de pares craneales ocu lomotores (más frecuente del VI par, que es el único loca l izado en el inter ior del seno cavernoso) o de las ramas tr igeminales del seno cavernoso ( 1 . a y 2. a). El diagnóstico se conf i rma por angiografía y el t ra tamiento de elección es la embolización, que debe realizarse en las de al to f lu jo o si existen alteraciones visuales.
• Síndrome postraumático. Aparece típicamente días o meses después de traumat ismos craneoencefálicos leves y cursa con cefaleas muy variadas, mareos, i r r i tab i l idad , ansiedad, déficit de concent ra ción o síntomas pseudopsicóticos, con exploración neurológica generalmente no rma l .
• Hidrocefalia postraumática. Este cuadro se caracteriza por la tríada de Hakim-Adams (MIR 05-06, 64). U n 4 % de los traumat ismos craneoencefálicos graves pueden compl icarse con este t ipo de h id ro cefalia comun icante . El mane jo diagnóstico y terapéutico es similar a la fo rma idiopática vista anter iormente.
• Encefalopatía traumática crónica. Es una secuela crónica que c o m bina trastornos de personal idad, cogni t ivos (bradipsíquia y déficit memorísticos) y motores (disfunción cerebelosa, park insonismo, a l teraciones de la vía p i ramida l ) .
• Demenc ia postraumática.
r
Casos clínicos representativos
Pac i en te de 25 años , que sufrió un t r aumat i smo c raneoence fá l i co de a l ta energía, ingresó en el hospi ta l en c o m a c o n u n a va lo rac ión en la e s c a l a de G l a s g o w de 5 puntos . Se r ea l i za ron d iversas T C ce rebra les que resu l ta ron repe t idamente no rma les . U n a R M rea l i zado al c a b o de u n a s e m a n a del a c c i d e n t e de tec tó una z o n a de c o n tusión hemorrág ica a n ive l de l e sp l en io de l c u e r p o ca l loso . A l c a b o de un m e s de l t r aumat i smo , la s i tuac ión del pac i en te persistía i nmod i f i c ada , c o n u n a puntuac ión de 5 puntos de G l a s g o w , p resen tando d iversos ep isod ios de h iperh idros is e h ipe rp i rex ia , y no detectándose otras les iones que la c i t ada e n suces i vos con t ro l es radio lógicos:
1) La causa más frecuente de coma mantenido en un traumático craneal es el estatus epiletico, y debería iniciarse tratamiento para ello.
2) Creo que la situación clínica del paciente obedece a causas no neurológicas. 3) Creo que el paciente presenta una lesión axonal difusa. 4) Es imposible que un paciente en coma presente una TC normal. 5) Debería precederse a la evacuación quirúrgica de la lesión del cuerpo calloso.
MIR 06-07, 6 1 ; RC: 3
135

Manual CTO de Medicina y Cirugía, 8. a edición
Casos clínicos representativos
U n n iño de 12 años sufre pérdida de c o n c i e n c i a breve , tras cae rse de una b i c i c l e ta . A l l legar a U r g e n c i a s , está o r i en tado y p resen ta signos de impac to en región p a r ietal d e r e c h a . D o s horas más tarde , le aque j a ce fa l ea de intens idad ráp idamente c r e c i en t e , seguida de a l terac ión del n ivel de c o n s c i e n c i a . La c a u s a más probab le de l de te r io ro , entre las s iguientes, es :
1) Contusión c e r e b r a l pa r i e t a l d e r e c h a . 2) Hipertensión i n t r a c r anea l a g u d a s e c u n d a r i a a e d e m a ce r eb ra l vasogénico. 3) H e m a t o m a e x t r a d u r a l a g u d o . 4) M e n i n g i t i s neumocóc i ca f u l m i n a n t e , p o r b r e c h a meníngea s e c u n d a r i a a f r a c tu ra
d e la base c r a n e a l .
5) H i d r o c e f a l i a a g u d a s e c u n d a r i a a h e m o r r a g i a s u b a r a c n o i d e a postraumática.
M I R 9 9 - 0 0 , 1 9 3 ; RC: 3
U n pac ien te que h a sufr ido un t r aumat i smo c r anea l l lega consc i en t e al se rv i c io de U rgenc i a s . Rad io lóg icamente , se a p r e c i a u n a f r ac tu ra l ineal de la bóveda c r a n e a l . A las 12 horas del a c c iden te , c o m i e n z a a r educ i r se de f o r m a progres iva el n ive l de c o n s c i e n c i a , observándose asimetr ía pupi lar . ¿ Q u é diagnóst ico de Eos s iguientes debe hace r se en p r imer lugar?
1) U n h e m a t o m a s u b d u r a l . 2) U n h e m a t o m a e p i d u r a l . 3) U n a cr is is epi lépt ica postraumática. 4) U n a m e n i n g i t i s . 5) U n c o m a metaból ico yatrogénico.
M I R 98-99F , 7 7 ; RC: 2
136

ABSCESO CEREBRAL Y EMPIEMA SUBDURAL
Orientación
MIR r
Aspectos esenciales
Lo más i m p o r t a n t e d e este t e m a es c o n o c e r los aspectos básicos de l absceso ce reb ra l .
[~T"| La extensión por contigüidad constituye el mecanismo patogénico más frecuente del absceso cerebral.
["2~] El S. aureus es el germen que se aisla con más frecuencia en los abscesos cerebrales.
["3"] Existe una tríada clínica típica de presentación: clínica de hipertensión intracraneal, fiebre y focal idad neu rológica.
["4"] En la T C es una de las lesiones que captan contraste en anil lo.
["5"] El tratamiento fundamental consiste en antibioterapia y evacuación quirúrgica de la lesión. Los corticoides se utilizan para el efecto masa, si bien pueden favorecer la infección.
19.1. Absceso cerebral
Se trata de un proceso supurat ivo focal en el inter ior del parénquima cerebral . En su formación, pasa inic ia l-mente por una fase de cerebrit is a lrededor del foco necrótico y , poster iormente se forma una cápsula de te j ido colágeno con gliosis pericapsular (Figura 107).
Figura 107. Abscesos cerebrales múltiples con captación en anillo
Etiopatogenia
CD P regun tas N o hay p r e g u n t a s M I R representa t i vas .
Se dist inguen los siguientes mecanismos patogénicos (Tabla 72): • Extensión por contigüidad desde un foco infeccioso próximo. Se trata del mecan ismo patogénico más fre
cuente. Suelen ser abscesos únicos. Los secundarios a sinusitis se loca l izan preferentemente en el lóbulo f ronta l . Los otógenos y los de origen mastoideo son más frecuentes en lóbulo tempora l y cerebelo. La menin-
137

Manual CTO de Medicina y Cirugía, 8 a . Edición
gitis bacteriana no suele ser causa de absceso cerebral . Por último, serían destacables los de or igen dentar io . Diseminación hematógena desde un foco infeccioso lejano ( también l lamados abscesos metastásicos). Hab i tua lmente son abscesos múltiples y, en ocasiones, no se ident i f ica el foco embolígeno. En los adultos, los de origen pu lmonar son los más frecuentes (bron-quiectasias, emp iema y, sobre todo , los abscesos). En niños, son más frecuentes los secundarios a cardiopatías congénitas con cor to c i r cu i to de derecha a izquierda (por e j emp lo , la tetralogía de Fallot). Son raros c o m o complicación de la endocardi t is infecciosa que, por el contrar io , suele ocasionar encefal it is embólica focal , infartos o aneurismas micóticos.
Extensión por contigüidad: - Sinusitis - Otitis - Mastoiditis - Osteomielitis
Diseminación hematógena: - Pulmonar (absceso, empiema, bronquiectasias) - Cardiopatías con cortocircuito derecha-izquierda (Fallot) - Fístulas AV pulmonares (Rendu-Osler-Weber) - Osteomielitis - Infecciones intraabdominales (diverticulitis, colecistitis) - Infecciones dentales - Otras causas de bacteriemia
Posquirúrgicos Postraumáticos
- Traumatismos penetrantes - Fracturas de la base del cráneo
Tabla 72. Etiopatogenia de los abscesos cerebrales
Clínica
El absceso cerebral se manif iesta con la típica tríada de clínica de hipertensión intracraneal (la cefalea es el síntoma más frecuente, dándose en el 8 0 % de los casos), f iebre y un cuadro de foca l idad neuro lógica (ya sea en fo rma de déficit o b ien en fo rma de crisis epiléptica). Sin embargo, en muchos casos no se presenta de fo rma comple ta . La hemiparesia y las crisis convuls ivas se p roducen en el 3 0 - 5 0 % de las ocasiones, y la f iebre, en el 5 0 % de los casos.
Diagnóstico
La sensib i l idad de la TC con contraste es cercana al 1 0 0 % (lesión hi-podensa que capta contraste en an i l l o y se rodea de un área de edema, que plantea diagnóstico d i ferenc ia l con metástasis, l i n foma cerebral p r imar io y g l iob las toma mu l t i fo rme ) . La RM es más sensible que la TC en fase de cerebrit is .
R E C U E R D A Las c inco lesiones cerebrales que captan contraste en anil lo: metástasis, glioblastoma multiforme, absceso cerebral, toxoplasmosis y l infoma.
En sangre, puede haber leucocitosis , elevación de VSG y de proteína C reactiva. La punción lumbar t iene bajo r end im ien to y, en general , no está ind icada por el riesgo de herniación cerebral .
Q R E C U E R D A El origen más frecuente de los abscesos cerebra lesa partir dediseminación hematógena es el absceso pulmonar en los adultos y la tetralogía de Fallot en los niños.
• Posquirúrgicos y postraumáticos. Los abscesos cerebrales pueden ser una complicación rara de proced imientos neuroquirúrgicos (craneotomía). Con más frecuencia , pueden comp l i ca r t raumat ismos craneoencefálicos penetrantes, en relación con fragmentos óseos contaminados retenidos.
Microbiología
En términos generales, los gérmenes más frecuentemente impl icados en los abscesos cerebrales son los de la fami l ia de los Streptococcus. Sin embargo, en el caso de abscesos or ig inados a partir de t raumat is mos craneoencefálicos o a partir de diversos procedimientos quirúrgicos, es el Staphylococcus aureus, el que se aisla con más frecuencia .
En la cuarta parte de los casos, los cul t ivos son estériles, mientras que en un porcentaje m u y var iable (según las series, puede oscilar entre un 1 0 % a un 9 0 % ) , pueden crecer múltiples gérmenes.
Hay otros gérmenes que se deben considerar en algunos casos par t i cu lares: cuando el paciente ha rec ib ido un trasplante, se debe tener en cuenta las infecciones fúngicas (Aspergillus fumigatus); en el caso de niños pequeños, los gérmenes gramnegativos deben considerarse; por último, en pacientes i nmunocomprome t i dos , Nocardia y Toxoplasma pueden ser or igen de los abscesos cerebrales.
Tratamiento
El t ra tamiento del absceso cerebral debe comb ina r ant ib ioterapia y cirugía evacuadora.
El antibiótico se el ige empíricamente en base a la etiología sospechada. Una buena combinación es pen i c i l i na C o una cefa lospor ina de tercera generación (cefotaxima) con me t ron idazo l , durante 4-6 semanas. Si se sospecha Staphylococcus aureus, debe añadirse vancomi-cina. El t ra tamiento de la toxoplasmosis se realiza con sul fadiac ina y p i r imetamina .
La cirugía evacuadora está ind icada en lesiones únicas, accesibles, con sintomatología de HTIC, efecto de masa o edema impor tante . Ac tua lmente se u t i l i zan la punción-aspiración con aguja (guiada o no con estereotaxia) y la resección quirúrgica (de elección en los secundarios a TCE penetrante, para e l im inar los cuerpos extraños y te j idos desvital izados) .
Los cort ico ides (dexametasona) pueden retrasar la formación de la cápsula y reducir la penetración de los antibióticos, pero están i n d i cados cuando hay deter ioro clínico del paciente deb ido al efecto de masa de la lesión.
A l ser una lesión epileptógena, el t ra tamiento puede complementarse con la administración de antiepilépticos, aunque no están indicados de forma profiláctica si no ha ten ido crisis.
La mor ta l idad es de un 1 0 % y las secuelas neurológicas frecuentes (hemiparesia y crisis persistentes).
1 3 8

Neurología y neurocirugía
19.2. Empiema subdural Es un proceso supurat ivo loca l izado en el espacio subdural craneal . El 7 5 % son unilaterales y habi tua lmente la infección se or ig ina por c o n t i güidad desde los senos frontal o e tmoida l o del oído med io (el m i c r o organismo más frecuente es el estreptococo). Es rara la diseminación hematógena. También puede ser posquirúrgico y postraumático, con frecuencia por Staphylococcus aureus.
Se trata de una enfe rmedad grave y progresiva que , en genera l , requ iere un t ra tamiento urgente. Hay que sospechar lo en pacientes con f iebre alta, cefalea, signos meníngeos y foca l i dad neurológica un i l a te ral , con los antecedentes descr i tos. Puede p roduc i r también síntomas de HTIC. Se diagnost ica en la TC con contraste. El t ra tamiento requiere la evacuación quirúrgica urgente del material puru len to mediante craneotomía ampl ia . Debe completarse con ant ib ioterapia .
La mor ta l idad es de un 2 0 % y son frecuentes las secuelas neurológicas.
139

Neurología y neurocirugía
20. PATOLOGÍA RAQUIMEDULAR
Orientación
MIR r
Aspectos esenciales
Se t ra ta d e u n o d e los t emas f u n d a m e n t a l e s para el MIR, p o r l o q u e se d e b e r ea l i z a r u n e s t u d i o p o r m e n o r i z a d o de l m i s m o . Sobre t o d o , hay q u e t ra ta r de pres tar espec i a l a tenc ión a las s igu ien tes par tes d e este t e m a : a c t i t u d diagnóstico-terapéutica a a d o p t a r an t e una l u m b a l g i a , lumbociát ica o c e r v i c o b r a q u i a l g i a , explorac ión d e las d i f e r en tes raíces b r a q u i a l e s y c ru r a l e s ( m u y i m p o r t a n t e s los c u a d r o s de l Manual), la estenosis de l c a n a l l u m b a r y la s i r i n g o m i e l i a .
[ Q P r egun tas
- MIR 0 8 - 0 9 , 52 - MIR 0 6 - 0 7 , 5 6 , 7 8 - M IR 05-06 , 6 2 , 9 3
MIR 0 4 - 0 5 , 8 7 , 9 4 -M IR 0 3 - 0 4 , 1 4 , 2 4 7 -M IR 0 2 - 0 3 , 2 1 9 , 2 2 8 -M IR 0 1 - 0 2 , 6 2 , 8 7 - MIR 0 0 - 0 1 , 5 8 , 78 , 8 8 - MIR 00-01 F, 72 , 76 - MIR 99-00 , 2 0 7 - MIR 9 9 - 0 0 F , 6 9 , 74 -M IR 9 8 - 9 9 , 6 4 , 6 9 , 9 1 , 9 3 - M I R 9 8 - 9 9 F , 6 6 , 1 1 1 , 2 3 6 - MIR 9 7 - 9 8 , 5 4
A n t e un paciente con lumbalgia, lumbociática o cervicobraquialgia sin factores de riesgo de etiología grave, sin déficit motor ni signos de síndrome de cola de cabal lo (en el caso de lumbociática) o mielopatía (en el caso de cervicobraquialgia), se debe iniciar un tratamiento sintomático sin realizar ninguna prueba diagnóstica; si a las seis semanas el cuadro no cede, hay que empezar a plantearse realizar estudios con fines diagnósticos.
["2~| El reposo en cama no ha demostrado ser útil en el tratamiento de la lumbalgia y la lumbociática.
("J") La intensidad de la lumbociática aumenta con las maniobras de Valsalva. El Laségue es una prueba que se con sidera positiva si la lumbociática se desencadena angulando la pierna menos de 6 0 ° respecto a la horizontal.
|~4J La radiculopatía L4 afecta a la cara anteromedial de la pierna, al reflejo rotuliano y a la extensión de la rodilla. La radiculopatía L5 afecta a la cara anterolateral de la pierna y dorso del pie y a la flexión dorsal del pie. La radiculopatía S1 afecta a la cara posterior de la pierna y planta del pie, al reflejo aquíleo y a la flexión plantar del pie.
Son indicaciones de intervención quirúrgica de una lumbociática el fracaso del tratamiento conservador y la que se acompaña de déficit motor o síndrome de co la de cabal lo.
("fr"] La radiculopatía C 6 afecta a la cara lateral del antebrazo y dedos primero y segundo, los reflejos bicipital y estilorradial y la flexión del codo y extensión de la muñeca. La radiculopatía C 7 afecta al tercer dedo, el reflejo tricipital y la extensión del codo y flexión de la muñeca. La radiculopatía C 8 afecta a la cara medial del antebrazo y dedos cuarto y quinto, y a movil idad intrínseca de los dedos de la mano.
j"7~j Son indicaciones de intervención quirúrgica de una cervicobraquialgia el fracaso del tratamiento conservador y la que se acompaña de déficit motor o mielopatía.
QTJ La estenosis de canal lumbar es un proceso típico de personas de edad avanzada . Se local iza con mayor frecuencia a nivel de L4-L5.
QTJ La c laudicación neurógena es típica de la estenosis de canal lumbar. La sintomatología aumenta con el ejerc ic io y cede al flexionar el tronco.
Q75] En los traumatismos medulares, es prioritario establecer el nivel de la lesión, mediante la exploración motora y sensitiva fundamentalmente, y determinar si la lesión es completa o incompleta, ya que el pronóstico es muy diferente.
QJJ Los traumatismos medulares se pueden asociar a un shock espinal, que es un periodo de parálisis f laccida y arrefléxica, y un shock neurogénico, que es un shock hemodinámico con bradicardia por lesión de la vía simpática.
QYj N ° se recomienda la realización de una RX cervical a un paciente traumático que cumple T O D O S los requisitos siguientes: G lasgow 15, sin ev idencia de tóxicos, sin dolor a la movil ización cerv ical , sin contractura cerv ical , sin lesiones distractoras y con mecan ismo lesional de bajo riesgo.
{-] 3 ] El tumor intrarraquídeo más frecuente son las metástasis y el primario más frecuente, el neurinoma. Las metástasis constituyen la causa más frecuente de compresión medular.
| l4 | El 5. aureus constituye el germen más frecuentemente aislado del absceso epidural espinal.
| 1 5 | La cl ínica de un absceso epidural espinal es progresiva: se inicia con dolor en la región afectada, y en el transcurso de pocos días se va estableciendo la clínica neurológica.
| l D | La clínica típica de la siringomielia es la disociación de la sensibi l idad de manera suspendida, ya que suele afectar a las extremidades superiores y respetar las inferiores.
20.1. Dolor lumbar
El do lor de espalda es la causa más frecuente de incapac idad en pacientes mayores de 45 años, lo que supone un prob lema médico de máxima relevancia en términos económicos y sociales.
140

Neurología y neurocirugía
Se suele clasif icar, en función de la duración, c o m o do lor lumbar agudo (duración inferior a seis semanas), subagudo (entre seis semanas y tres meses) y crónico (más de tres meses).
La mayor parte de las lumbalgias corresponden a un sobreesfuerzo y son autol imitadas ( lumbalgias mecánicas). En la mayoría de los casos ( 8 5 % ) no es posible establecer un diagnóstico específico. La valoración in ic ia l debe encaminarse a la exclusión de aquellas etiologías graves de do lor lumbar que, aunque son infrecuentes, pueden requerir trat amiento inmedia to (traumatismos, infecciones, tumores, síndrome de cola de cabal lo) . Para e l lo , se realizará historia clínica y exploración física, pon iendo especial atención en la presencia de factores de riesgo que hagan sospechar un or igen grave del do lor , según las guías euro peas del mane jo en atención pr imar ia de las lumbalgias agudas (Tabla 73) (MIR 01-02, 87).
Q R E C U E R D A En la lumbalgia, debemos descartar tres procesos etiológicos: infecc ión, tumor y traumatismo.
Tabla 73. Factores de riesgo de etiología grave del dolor de espalda
Aunque los datos disponibles para establecer normas de práctica clínica son incompletos , por la escasez de estudios bien diseñados, en ausencia de sospecha de una etiología grave del do lor no se r ecomien da la realización de estudios de laborator io (hemograma, ve loc idad de sedimentación, bioquímica y analítica de orina) , pruebas de imagen (RX, RM o TC) ni otras técnicas diagnósticas durante el pr imer mes de evolución, inc luso en pacientes con sospecha clínica de hernia discal .
La mayoría de los pacientes con do lor de espalda mejorará en el p lazo de un mes, con o sin t ratamiento, por lo que el mane jo in ic ia l de un paciente con do lor lumbar agudo sin factores de riesgo debe ser c o n servador, con objeto de conseguir un a l i v io sintomático (Tabla 743). El paciente debe ser in formado sobre la naturaleza del cuadro que padece (MIR 04-05, 87 ; MIR 0 0 - 0 1 , 78; MIR 98-99, 93).
• Dar información positiva y tranquilizadora al paciente • No se recomienda reposo en cama • Recomendar a los pacientes que estén activos y que continúen
con las actividades diarias normales, incluyendo el trabajo • Fármacos de primera línea, si la Intensidad del dolor lo requiere:
- Inicio con paracetamol - SI no mejora, AINE pautados (máximo 3 meses) - Si no mejora, añadir un ciclo de miorrelajant.es (menos de 1 semana)
• Prescribir ejercicio a partir de las 2-6 semanas • Neurorreflexoterapia en casos rebeldes
Tabla 74. Opciones terapéuticas en el tratamiento del dolor de espalda
La med ida p r i nc ipa l durante la fase aguda ha s ido t r ad i c i ona lmen-te el reposo abso lu to en cama ; sin embargo , estudios recientes han demost rado que el reposo en cama de más de dos días con l l e va peo res resultados, en cuan to a d o l o r y a i ncapac idad f u n c i o n a l , que la ac t i tud act iva , por lo que se r e comienda la reanudación precoz de la deambulación y las act iv idades físicas habi tua les (con excepción de los trabajos manuales pesados). Es útil la reeducación postura l , i n t en tando evitar aquel las ac t iv idades y posturas que desencadenan el do lo r .
El t ratamiento farmacológico se basa en los fármacos analgésicos (paracetamol ) , ant i in f lamator ios ( fundamenta lmente AINE) y relajantes musculares (estos últimos, no más de una semana).
No se recomiendan , en el t ratamiento de la lumbalg ia , las siguientes medidas: cort icoides, AINE tópicos, inh ib idores de la recaptación de serotonina, láser, ultrasonidos, masajes, fajas, acupuntura , in f i l t rac io nes facetarías o epidurales, ozonoterap ia .
Si la sintomatología persiste más allá de 2-6 semanas a pesar del tratamiento conservador o la intensidad aumenta durante el m ismo, es necesario volver a valorar al paciente de forma comple ta , rea l izando pruebas diagnósticas y tratamientos específicos, si lo precisa.
Cuando el do lor persiste más allá de 12 semanas (tres meses), se establece el diagnóstico de do lor lumbar crónico. En estos casos, una vez descartada patología grave en la exploración física, no se recomienda ningún método diagnóstico, salvo que se sospeche una causa específ ica.
En estos pacientes, es necesario un programa rehabi l i tador mu l t i d i s c i p l inar (unidades del do lor , programas educat ivos, e jerc ic io , t ratamiento psicológico) j un to con la administración de analgésicos (antidepresivos tricíclicos, parches de capsaicina, opiáceos en casos de fracaso con los tratamientos previos). C o m o última opción de t ratamiento conservador, se puede indicar la neuroestimulación percutánea. Si el do lo r es intenso e inva l idante y persiste durante más de dos años, puede estar indicada la cirugía (artrodesis o fusión vertebral) .
En la Figura 108 se esquematiza un a lgor i tmo de mane jo del do lo r l u m bar, con o sin síntomas radiculares, en el paciente adul to .
20.2. Lumbociática. Hernia discal lumbar
El término lumbociática se ut i l iza para describir el do lo r lumbar i r radiado hacia al m i embro inferior, sugir iendo una compresión de una raíz nerviosa. La causa más frecuente de lumbociática es la hernia discal lumbar . Cuando la lumbalg ia es el único síntoma, debe pensarse en otros diagnósticos; sin embargo, la presencia de ciática t iene gran sens ib i l idad para el diagnóstico de hernia discal (solamente uno de cada 1.000 hernias de disco significativas cursan sin ciática).
Bases anatómicas
Las raíces nerviosas abandonan el canal vertebral a través de los agujeros de conjunción. Las raíces cervicales t ienen un trayecto intrarraquí-
• Primer episodio en menores de 20 años o mayores de 55 años • Déficit neurológico difuso (incluyendo síndrome de cola de caballo) • Dolor no Influido por posturas, movimientos o esfuerzos, sin mejorar con el
descanso • Dolor exclusivamente dorsal • Deformidad estructural de aparición reciente • Tratamiento prolongado con glucocorticoides • Antecedentes de traumatismo reciente • Mal estado general • Pérdida de peso no explicada • Diagnóstico previo de cáncer • Fiebre • Historia de inmunosupresión (trasplante, VIH, etc.) • Consumo de drogas por vía parenteral
141

Manual CTO de Medicina y Cirugía, 8. a edición
DOLOR LUMBAR AGUDO con o sin ciática
No
H a clínica y exploración
Factores de riesgo de etiología grave? (Tabla 73)
• Fracturas •Tumores • Infecciones • Déficit neurológico progresivo
No solicitar pruebas diagnósticas
Solicitar pruebas diagnósticas según la etiología sospechada
Tratamiento médico (Tabla 74 )
- No ¿Signos de enfermedad grave?
Consulta al especialista Tratamiento específico
Dolor lumbar agudo+/- ciática de más de cuatro semanas de evolución resistente al trata
miento
DOLOR LUMBAR CRÓNICO • Tratamientos a largo plazo • Unidades del Dolor Crónico opcionales
• Repetir estudio, si precisa
Consulta al especialista Solicitar pruebas diagnósticas
•RM,TComielo-TC • EMG, si nivel radicular dudoso
Duración > 1 2 semanas
No
Patología que requiere No requiere cirugía Tratamiento sintomático tratamiento quirúrgico o rechaza la cirugía Rehabilitación opcional ¿Mejoría?
É Cirugía Reincorporación a
actividades normales
Figura 108. Algoritmo del manejo del dolor lumbar agudo
deo cor to , ya que salen del canal raquídeo prácticamente a nivel del segmento medular del mismo nombre , un nivel por enc ima del cuerpo vertebral que lleva el nombre de la raíz (la raíz C6 sale por el agujero C5-C6). Por el cont rar io , las raíces dorsales, lumbares y sacras salen del canal un nivel por debajo de la vértebra correspondiente (la raíz L4 sale por el agujero L4-L5).
Puesto que la médula espinal te rmina generalmente a nivel del cuerpo vertebral de L1 o L2, las raíces lumbosacras deben realizar un largo recorr ido por el canal raquídeo hasta alcanzar el agujero intervertebral correspondiente, y pueden ser afectadas por diversas patologías en distintos puntos de este trayecto, no necesariamente co inc identes con el nivel por el que abandonan el canal vertebral .
Patogenia
La hernia discal es la patología neuroquirúrgica más frecuente. Resulta de la degeneración del núcleo pulposo y del ani l lo f ibroso del disco intervertebral, de modo que el pr imero sobresale por el ani l lo (herniación) o incluso puede salir del espacio intervertebral, convirtiéndose en un fragmento libre en el interior del canal raquídeo (extrusión). Se suele asociar a espondilosis, sobreesfuerzo físico o traumatismos, y es más frecuente en edades medias de la vida (pico de incidencia sobre la cuarta década).
El disco hern iado c o m p r i m e los elementos nerviosos que discurren por el canal , y puede dar lugar a una radiculopatía (por compresión de la
142

Neurología y neurocirugía
raíz nerviosa) o una mielopatía (por compresión de la médula espinal, sólo en los niveles cervicodorsales, no en los lumbares bajos).
La localización más frecuente de las hernias discales es la co l umna lumbar , p r inc ipa lmente en los espacios L4-L5 y, sobre todo , el L5-S1. El l igamento vertebral común posterior es muy potente en su porción centra l . Por este mot i vo , la mayoría de las hernias discales se loca l izan más lateralmente (hernias discales posterolaterales), si bien se descr i ben también hernias centrales y hernias foraminales (en el agujero de conjunción) menos frecuentes.
Clínica
nerviosa c o m p r i m i d a por la hernia d isca l . En las hernias discales posterolaterales (las más frecuentes), la radiculopatía es genera l mente uni la tera l , y la raíz afectada suele ser la que lleva el nombre de la vértebra infer ior del espacio d isca l . Sin embargo , las hernias foraminales pueden c o m p r i m i r la raíz que sale por ese espacio (la de nombre igual a la vértebra superior) , y en las hernias centrales, la afectación puede ser bi lateral (Figura 110).
Q R E C U E R D A El Laségue suele ser pos i t i vo para radiculopatías L5 y S I . El Laségue i nve r t i do suele ser pos i t i vo para radiculopatías lumbares super iores, y el c r u z a d o , para radiculopatías p rovocadas por hernias centra les.
Puede comenzar con dolor lumbar paravertebral ( lumbalgia) y do lor a la percusión de apófisis espinosas, acompañado de contractura de la musculatura paravertebral . El do lor aumenta con la flexión de la co lumna , suele empeorar con la bipedestación y mejorar en decúbito. Es posib le la existencia de una escoliosis func iona l antiálgica. Lo característico de la hernia discal lumbar es que el do lor se irradia al m i embro inferior (ciática) deb ido a la compresión de la raíz nerviosa. En las radiculopatías compresivas, el do lor aumenta típicamente con las maniobras de Valsalva (tos, defecación) (MIR 0 0 - 0 1 , 58). Puede reproducirse con distintas maniobras explorator ias. La ma niobra de Laségue (también conoc ida c o m o man iobra de elevación de la pierna recta) consiste en la elevación pasiva de la pierna extendida con el paciente en decúbito supino, y es posit iva si aparece do lo r con una angulación menor a 60 grados (Figura 109). La maniobra de Bragard es igual que la de Laségue, pero además con dorsiflexión pasiva del pie. Ambas maniobras estiran fundamenta l mente las raíces L5 y S1. Para valorar las raíces lumbares superiores (L2 a L4), se ut i l iza la man iobra de elevación de la pierna recta invert ida (Laségue inver t i do), con el paciente en decúbito p rono . Se denomina Laségue c ru zado cuando la elevación de una pierna produce do lor en la otra. La lesión de la raíz nerviosa es s iempre del lado en que se produce el do lor . El Laségue c ruzado suele ser posi t ivo en el caso de hernias en posición centra l .
Radiculopatía. El paciente puede presentar trastornos sensitivos (hipoestesia, parestesias) o alteración de reflejos y, menos f recuentemente , déficits motores en el te r r i tor io correspondiente a la raíz
Hernia discal
Anillo fibroso
Raíz cervical
Núcleo pulposo
Vértebra L4
Hernia discal posterolateral
L4-L5
Raíz L5
Raíz L4
Hernia discal cervical (arriba): comprime la raíz que sale en el espacio. Hernias discales lumbares (abajo): la hernia
posterolateral comprime la raíz que sale por el espacio inferior. La hernia foramínal comprime la que sale por el mismo espacio
Figura 110. Hernia discal cervical (arriba) y hernias discales lumbares (abajo)
Las manifestaciones típicas de la afectación de cada raíz, en el caso de hernias posterolaterales, son (Tabla 75):
143

Manual CTO de Medicina y Cirugía, 8. a edición
NIVEL DE LA HERNIA D ISCAL
Raíz habi tua lmente afectada
L1-L2 L2-L3 | L3-L4 L4-L5 | L5-S1 I I l
Raíz habi tua lmente afectada
L2 L3 L4 L5 SI
Reflejo alterado - Rotuliano Aquíleo (a veces) Aquíleo
Déficit motor Flexión cadera (psoas)
• Flexión cadera (psoas)
• Extensión rodilla (cuadríceps)
• Extensión rodilla (cuadríceps)
• Dorsiflexión del piel (tibial anterior)
• Extensión del dedo gordo (1. "dedo)
Flexión plantar del pie (gastrocnemio y soleo)
Déficit sensit ivo Cara anterior muslo
Cara anterior muslo y rodilla
• Rodilla y cara interna de la pierna
• Maléolo medial • Cara medial del pie
Cara anterolateral de la pierna. Dorso del pie hasta 1 d e d o
• Maléolo externo • Planta y borde lateral
del pie hasta 5° dedo
Tabla 75. Exploración de las raíces nerviosas del plexo lumbosacro
• Hernia discal L1-L2 (raíz L2): do lor y/o alteración de la sensibi l idad en cara anter ior del mus lo (tercio prox imal ) , con deb i l idad para la flexión de la cadera (psoas).
• Hernia discal L2-L3 (raíz L3): do lor y/o alteración de la sensibi l idad en cara anterior del mus lo (tercio medio ) y una zona pequeña de ro d i l l a , con deb i l idad para la flexión de la cadera (psoas) y extensión de la rodi l la (cuadríceps).
• Hernia discal L3-L4 (raíz L4): do lor y/o alteración de la sensibi l idad en el terc io distal del mus lo , la rod i l l a y cara interna de la pierna. Puede cursar con abolición del ref lejo ro tu l i ano y d i f i cu l tad para la extensión de la rod i l la , con atrofia del cuadríceps.
• Hernia discal L4-L5 (raíz L5): do lor i r radiado por cara posterolateral del mus lo y lateral de la pierna hasta el dorso del p ie y pr imer dedo. Disminución de fuerza para la dorsiflexión del píe (t ibial anterior) y pr imer dedo, sin abolición de reflejos osteotendinosos (excepcio-nalmente se ha descrito leve disminución del aquíleo) (MIR 04-05, 94; MIR 98-99F, 236; MIR 97-98, 54).
• Hernia discal L5-S1 (raíz S1): do lor i r radiado por cara posterior del muslo y pierna hasta la planta y borde lateral del pie y qu in to dedo, con alteraciones de la sensibi l idad en el m ismo terr i tor io . Abolición del ref lejo aquíleo y deb i l idad para la flexión plantar del p ie (gemelos y soleo) (MIR 06-07, 78; MIR 01-02, 62; MIR 00-01F, 72; MIR 99-00F, 69; MIR 98-99, 91).
Diagnóstico
La correlación entre los hal lazgos de imagen y la clínica dolorosa no suele ser muy buena. Se ha c o m p r o b a d o que más de un terc io de los adultos asintomáticos a los que se realiza una RM lumbar pueden mostrar signos radiológicos de patología discal , hal lazgo que podría c o n duc i r al t ra tamiento quirúrgico de pacientes que no lo precisan, y que inc luso pueden empeorar si la cirugía añade inestabi l idad.
Por este m o t i v o , las pruebas de imagen sólo deben sol ic i tarse en pa cientes con sospecha clínica que no responden adecuadamente a t r a tamien to médico durante un pe r i odo de t i e m p o suf ic iente y sean cand idatos potenc ia les a cirugía. La prueba de elección es la RM (Figura 111), que en la ac tua l idad está sust i tuyendo a la TC y mie lo-grafía-TC, por su me jo r capac idad para va lorar los te j idos b landos . Las normas de práctica clínica actuales no r e comiendan la rea l iza ción de las mismas durante el p r imer mes y m e d i o de sintomatología, en ausencia de factores de riesgo que hagan sospechar una etiología grave (véase Tabla 72), déficit moto r progres ivo o síndrome de co la de caba l lo .
Figura 111. RM de hernia discal lumbar
Cuando, por los datos clínicos, existen dudas sobre el nivel radicular afectado, deben realizarse estudios neurofisiológicos que conf i rmen la existencia de una radiculopatía. Para valorar el nivel y el grado de la lesión radicular , se ut i l iza la electromiografía (EMC).
Tratamiento
El t ratamiento in ic ia l de la hernia discal debe ser conservador. Todas las medidas descritas para el t ra tamiento del do lor lumbar (véase Tabla 74) son apl icables al t ra tamiento sintomático de la lumbociática y c o n siguen mejoría en el 9 0 % de los casos (MIR 98-99F, 111). En pacientes con ciática, el reposo en cama no ha demostrado efect iv idad a la hora de mejorar el do lor o la incapac idad func iona l (MIR 05-06, 62).
Cuando estas medidas no resultan eficaces, o cuando hay signos cl ínicos que sugieran lesión radicular importante (pérdida de fuerza ob je t iva o síndrome de cola de cabal lo) , está ind icado el t ratamiento q u i rúrgico (Tabla 76). La técnica quirúrgica de elección es la flavectomía con extirpación del disco afectado (discectomía o microdiscectomía). En casos de inestabi l idad vertebral asociada, debe realizarse una artro-desis de los niveles impl icados . Dada la posible mala correlación entre los hal lazgos radiológicos y el dolor , la correcta indicación quirúrgica se establecerá con mayor seguridad cuando exista concordanc ia entre
1 4 4

Neurología y neurocirugía
la historia clínica, los hal lazgos explorator ios, las técnicas de imagen y los estudios neurofisiológicos.
Lesión de la raíz que produce una pérdida aguda o progresiva de fuerza, objetivable clínicamente o por EGM. Es indicación de cirugía urgente Signos clínicos sugestivos de síndrome de cola de caballo o lesión medular (disfunción de esfínteres, anestesia perineal en silla de montar, etc.). Es indicación de cirugía urgente Fracaso del tratamienteo conservador, es decir, dolor incapacitante, de características radiculares, que no responde a tratamiento médico durante un periodo mínimo de cuatro semanas Incapacidad recidivante a pesar del tratamiento médico
Tabla 76. Indicaciones de cirugía en la hernia discal lumbar
Hernia discal C5-C6 (raíz C6 ) : do lo r y/o alteración de la sens ib i l i dad en la cara lateral del antebrazo i rradiado hasta los dedos pulgar e índice de la mano, con disminución o abolición de los reflejos b ic ip i ta l y/o esti lorradial (más específico) y deb i l i dad para la flexión del codo (bíceps braquial ) y pronación. Hernia discal C6-C7 (raíz C7 ) : do lo r y/o alteración de la sensibi l idad en cara extensora de m iembro superior, tercer dedo y borde radial del cuarto dedo de la mano. Abolición del reflejo t r i c ip i ta l y deb i l i dad de la musculatura extensora del codo y f lexora de la m u ñeca (MIR 99-00, 207). Hernia discal C7-D1 (raíz C8 ) : do lo r y/o alteración de la sensib i l i dad en la cara media l del antebrazo, i r radiado al qu in to dedo y borde cubi ta l del cuarto dedo de la mano . Deb i l i dad en la musculatura intrínseca de la mano y, a veces, ref lejo t r ic ip i ta l abo l ido .
Otras técnicas quirúrgicas que se han ut i l i zado son la quimionucleó-lisis (con quimíopapaína inyectada intradiscalmente) , la nucleotomía percutánea y la discectomía endoscópica percutánea. Recientemente, se está ap l i cando la ozonoterap ia , con resultados muy discut ibles.
20.3. Cervicobraquialgia. Hernia discal cervical
El término cerv icobraquia lg ia se ut i l iza para describir el do lor cervical i r radiado por el m i e m b r o superior. A nive l cerv ica l , las hernias discales se desarrol lan preferentemente en los espacios C5-C6 y C6-C7 (hernia cervical más frecuente) y, c o m o las lumbares, suelen ser de loca l iza ción posterolateral . La patogenia es la misma que a nivel lumbar .
En la exploración de la cerv icobraquia lg ia , se describe el signo de Spurl ing (el examinador hace presión sobre el vértex craneal con la cabeza extendida y rotada hacia el lado sintomático; es pos i t ivo si se desencadena el do lor ) . La abducción del h o m b r o ( l levando las manos sobre la cabeza) suele al iv iar el do lor radicular.
Las manifestaciones de radiculopatía cervical son (Tabla 76):
NIVEL DE LA HERNIA D ISCAL
C4-C5 C5-C6 C6-C7 C7-D1
Raíz habi tualmente afectada
C5 C6 C7 C8
Reflejo alterado Bicipital
Bicipital Estilorradial
Tricipital Tricipital (a veces)
Déficit motor
Separación y flexión del hombro
• Flexión del codo
• Extensión de la muñeca
• Extensión del codo
• Flexión de la muñeca
• Flexión dedos • Musculatura
intrínseca de la mano
Déficit sensit ivo
Hombro y cara lateral del brazo
Cara lateral del antebrazo hasta 1.™ y 2.° dedos
Cara dorsal de MS hasta 3.° y borde radial del 4.° dedo
5.° dedo y cara cubital del 4.° dedo. Cara medial del antebrazo
Tabla 77. Exploración de las raíces nerviosas del plexo braquial
Hernia discal C4-C5 (raíz C5 ) : do lor y/o alteración de la sensibi l idad en el h o m b r o y cara lateral del brazo, con deb i l idad de la musculatura p rox ima l (flexión y separación del brazo).
La prueba de imagen de elección en la patología cervical es la RM. El EMC puede ayudar a establecer la raíz afectada.
El esquema de mane jo de la cerv icobraquia lg ia es el m ismo que el de la lumbociática (véase Figura 1 0 8 ) . La RM se indicará en casos en los que no haya mejora con el t ratamiento conservador, en casos de déficit neurológico o en casos en los que exista una mielopatía.
O R E C U E R D A En un espac io intervertebral lumbar, sale la raíz de la vértebra superior, y la raíz de la vértebra inferior sue le ser la afectada en el caso de una hernia d isca l a ese nivel ; sin embargo, en un espac io intervertebral cerv ica l , sale y se afecta la raíz de la vértebra inferior.
La mayoría de los pacientes con cerv icobraquia lg ia ( 9 5 % ) mejora con t ratamiento conservador (analgésicos, ant i inf lamator ios y relajantes musculares) y no es necesario realizar pruebas de imagen in ic ia lmente (MIR 02-03, 228). Pueden ser útiles las tracciones cervicales.
Las indicac iones de cirugía de la hernia discal cervical son similares a la lumbar ; se reserva para aquel los casos con do lo r rebelde al tratamiento médico, mielopatía o afectación radicular importante que i m pl ica un déficit motor . La técnica quirúrgica de elección es la discectomía anter ior con injerto ¡ntersomático óseo o metálico (técnicas de C lowa rd y de Smith-Robinson) (Figura 11 2).
Figura 112. Discectomía cervical con injerto intersomático (Cloward)
145
Manual C T O de Medicina y Cirugía, 8. a edición
Sí existen signos de mielopatía cerv ica l , en relación con espondi losis cervical o con uno o varios discos herniados, se puede plantear la cor-porectomía con in jerto y placa cervical anterior, la laminoplast ia o la laminectomía posterior.
20.4. Espondilosis cervical
El término de espondilosis cervical engloba todos los procesos que t ie nen lugar a nivel de la co lumna cerv ica l , c o m o consecuencia de los cambios degenerativos que se producen con la edad. D icha situación comprende :
Estenosis cerv ica l . Degeneración de discos cervicales, con pos ib i l idad de que se pro duzcan hernias de disco. Formación de osteofitos. H iper t rof ia , engrosamiento y osificación de l igamentos y uniones articulares. Alteraciones en las curvas fisiológicas. Subluxaciones.
Tratamiento quirúrgico: se indicará t ratamiento quirúrgico en las siguientes circunstancias: - Do lo r cervical discapacitante que no mejora con t ratamiento
sintomático. - Do lo r radicular discapacitante, refractario al t ratamiento conser
vador, o deb i l idad muscular progresiva. - Mielopatía aguda.
El abordaje quirúrgico de la espondi losis cervical puede realizarse por vía anter ior o posterior, depend iendo de una serie de factores c o m o la edad del paciente, la curva de la co l umna o el número de niveles vertebrales afectados.
R E C U E R D A La causa más f recuente de mielopatía en personas de más de 55 años es la espondi los is ce rv i ca l . La causa más f recuente de compresión medu l a r es la metástasis.
20.5. Estenosis del canal lumbar
Se trata de una patología muy común en los adultos. De hecho, pueden presentarse signos radiológicos de espondilosis cervical en el 2 5 - 5 0 % de las personas de 50 años, y en el 7 5 - 8 5 % de la población de 65 años.
Clínica
La espondilosis cervical puede manifestarse c o m o dolor cervical que aumenta con los mov imientos , radiculopatía cervical por la existencia de una hernia discal o de osteofitosis, o por un cuadro clínico de m ie lopatía (la espondilosis cervical es la causa pr inc ipa l de mielopatía en personas por enc ima de los 55 años).
Diagnóstico
El diagnóstico se comp lementa con estudios radiológicos simples (MIR 05-06, 62), para estudiar el tamaño del canal y la parte ósea de la co l umna cervical (búsqueda de osteofitos, ca lc i f icac iones anormales, alineación de la co lumna) , y la resonancia magnética, para observar los l igamentos, los discos intervertebrales, las raíces y la médula cervical (datos de mielopatía). También puede considerarse la mielografía-TC.
Q R E C U E R D A La espond i l o s i s ce rv i ca l e n g l o b a d iversos t ras tornos de la c o l u m n a ce r v i c a l de r i v ados de la degeneración p r o p i a de la c o l u m n a .
Tratamiento
La raquiestenosis es una reducción del diámetro anteroposter ior del canal vertebral , que puede produc i r compresión o compromi so vascular de las raíces de la co la de cabal lo . Por tanto, es un diagnóstico anatóm i co que se establece por imagen (RM, TC o mielografía-TC) (Figura 113).
Figura 113.TC d e raquiestenosis l umba r
Puede ser congénita o adqui r ida (espondilosis, espondilol istesis, Paget, acromegal ia , postraumática, etc.), aunque lo más habitual es que sea adqui r ida sobre una estenosis congénita previa. Es una patología cuya inc idenc ia aumenta con la edad. Es más frecuente a nivel del espacio L4-L5.
El t ra tamiento de los pacientes con espondi losis cervical sintomática se basa en una serie de medidas conservadoras y un tratamiento q u i rúrgico. • Tratamiento conservador: consiste en la administración de anal
gésicos y ant i inf lamator ios , j un to con la colocación de un collarín cervical durante un breve per iodo de t i empo y la realización de una serie de ejercic ios cervicales.
La raquiestenosis lumbar produce típicamente dolor lumbar, lumbociáti-cas (con frecuencia bilaterales), y es la causa más frecuente de c laudica ción neurogénica de extremidades inferiores (dolor lumbar, en nalgas y en piernas al caminar o en bipedestación, que cede con el reposo) (MIR, 08-09, 52 ; MIR 03-04, 14; MIR 02-03, 219). Hay que establecer d iag nóstico diferencial con la claudicación de origen vascular (Tabla 77). El dolor aumenta con la hiperextensión de la co lumna y, a diferencia de la
146

Neurología y neurocirugía
hernia discal, cede al sentarse (con la flexión de la co lumna) ; por e l lo, los pacientes t ienden a adoptar una postura antropoide o también denomi nada postura "de carr i to de supermercado" (MIR 06-07, 56).
CLAUDICACIÓN NEUROGÉN ICA
CLAUDICACIÓN VASCULAR
Distribución del dolor
Territorio de un nervio (dermatoma)
Grupo muscular con irrigación común
Factores desencadenantes
• Ejercicio de intensidades variables
• Mantenimiento prolongado de una postura
• Al ponerse en pie, antes de comenzar la marcha
• Ejercicio con intensidad constante, menor confome progresa la enfermedad
• Raro en pie sin caminar
Distancia al caminar para aparición
Variable Constante
Alivio con el reposo
• Lento • Dependiente
de la postura (mejor en flexión de la columna)
Inmediato • No depende
de la postura
Pulsos periféricos Conservados Disminuidos o ausentes
Palidez cutánea al elevar los MMII
No Marcada
Temperatura en los MMII
Normal Disminuida
Tabla 77. Diagnóstico diferencial entre claudicación neurogénica y vascular
El t ratamiento in ic ia l es conservador. La cirugía se indicará de manera urgente en casos de paresia relevante bi lateral y síndrome de cola de caba l lo y de manera programada cuando la claudicación persiste más allá de seis meses a pesar del t ratamiento conservador. La cirugía c o n sistirá en descompr imi r el canal mediantelaminectomía (Figura 114).
Figura 114. Laminectomía lumbar
20.6. Espondilolistesis
Se def ine c o m o un desplazamiento hacia delante de la vértebra super ior sobre su inmediatamente inferior. En función del porcentaje de des l izamiento, se ha c las i f icado en c inco grados: grado I, cuando el des l izamiento es menor del 2 5 % ; grado II, entre 2 5 - 5 0 % ; grado III, 5 0 - 7 5 % ; grado IV, 7 5 - 1 0 0 % , y grado V o espondi loptosis , cuando la
vértebra superior supera en toda su long i tud a la inferior y, por tanto, bascula, t end iendo a la verticalización. Las espondilol istesis de grado I y II se denominan de bajo grado, mientras que las de grados lll-V, espondi lol istesis de alto grado. En función del mecan ismo patogénico, la espondilol istesis puede clasificarse en c inco tipos (Tabla 78):
Tipo I o displásicas: hay una def ic ienc ia congénita en las uniones facetarías L5-S1, que provoca el des l izamiento de L5 sobre S1. Tipo II, ístmicas o espondilólisis: se p roduce por una alteración en la pars interarticularis (fractura o elongación). Son las espon di lol istesis más frecuentes, y se dan sobre t o d o a nive l L5-S1. Su inc idenc ia aumenta con la edad . La clínica consiste en lumbalg ias y síntomas radiculares. En los casos en los que los síntomas no me jo ren c o n t ra tamiento conservador , o b ien se p roduzca una p r o gresión en la d e f o r m i d a d , se procederá a plantear un t ra tamiento quirúrgico. Tipo III o degenerativa (MIR 05-06; 93) : son debidas a procesos degenerativos discales y de otras estructuras del segmento vertebra l , c o m o el l igamento amar i l lo . Son más frecuentes en mujeres y a partir de los 50 años. El nivel más afectado es el L4-L5. Desde un punto de vista clínico, pueden provocar claudicación neurógena, por la estenosis que produce el des l izamiento, lumbalg ia mecánica y radicula lg ia , por la compresión de la raíz a nivel del foramen de conjunción. Si estos síntomas persisten más allá de tres meses a pesar del t ratamiento médico e interf ieren con la v ida del paciente, o se establece un déficit neurológico progresivo o el paciente ref iere síntomas esfinterianos, se indicará el t ratamiento quirúrgico, que consiste en una laminectomía descompresiva con fusión ¡ntersomá-tica que, en ocasiones, se acompaña de instrumentación.
Q R E C U E R D A La espondilolistesis más frecuente es la de tipo II o espondilólisis o espondilolistesis ístmica, que afecta sobre todo a L5-S1. La degenerativa (tipo III) afecta sobre todo a L4-L5.
••Hl i^Hl i l^HHIHni
• Tipo IV o traumática: el mecan ismo patogénico imp l i ca una fractura aguda en la vértebra a nivel de una zona diferente a la pars interart icular is.
• Tipo V o patológica: es debida a una enfermedad ósea que afecta a la pars interart icular is o al pedículo.
20.7. Espondilodiscitis
Se def ine c o m o espondi lodisc i t is una infección del disco y de la vértebra adyacente. La infección suele iniciarse en la región metafisaria de la vértebra y poster iormente diseminarse al disco intervertebral . Puede
T I P O I D I S P L Á S I C A Deficiencia congénita facetaría
T I PO II Í STM ICA
O E S P O N D I L Ó L I S I S Fractura o elongación de pars interarticularis
T I PO III DEGENERAT IVA
Degeneración estructuras columna
T I PO IV T R A U M Á T I C A
Fractura que no afecta a pars
T I P O V PATOLÓGICA Enfermedad ósea, afectando a pars o pedículo
Tabla 78. Espondilolistesis lumbar
1 4 7

Manual CTO de Medicina y Cirugía, 8 . a edición
ser espontánea (más frecuente) o posquirúrgica (habi tualmente, varias semanas después de una discectomía). La espondi lodisc i t is es más co mún en la región lumbar. El germen más f recuentemente imp l i c ado en la infección del disco ¡ntervertebral es el Staphylococcus aureus. En pacientes usuarios de drogas por vía parenteral , puede observarse i n fección discal por Pseudomonas aeruginosa. Entre las formas crónicas, destaca la espondi lodisc i t is tuberculosa (enfermedad de Pott), c o m o más frecuente, y brucelósica (en fases tardías de la infección).
Puede debutar de fo rma aguda o presentar un in i c io larvado. El síntoma más frecuente de presentación es el do lo r lumbar , que aumenta con cualquier mov im ien to , se a l iv ia con el reposo, y hab i tua lmente está bien loca l izado en el nivel afectado. Suele acompañarse de contractura de la musculatura paravertebral y puede impl i car irradiación radicular. La f iebre es un síntoma inconstante. Es raro encontrar anomalías en la exploración neurológica.
Hay escasos hallazgos de laborator io que ind iquen infección; en general son normales, salvo una elevación persistente de la VSG y de la proteína C reactiva y, en ocasiones, una discreta leucocitosis.
Los hal lazgos en la radiología s imple (disminución de altura del espacio discal , áreas líticas en plat i l los vertebrales) tardan en aparecer entre 2 y 4 semanas. Se pueden emplear estudios gammagráficos; sin embargo, el diagnóstico de imagen de elección es la RM (Figura 115). El diagnóstico de f in i t i vo se establece mediante estudio microbiológico o histopatológico de material discal , ob ten ido por punción-aspiración con aguja o por biopsia del espacio afectado, aunque en muchas oca siones no se consigue aislar ningún mic roorgan ismo.
Figura 115. RM típica de espondilodiscitis, con afectación del espacio intervertebral y edema óseo, implicando
los cuerpos vertebrales superior e inferior.
El t ra tamiento consiste en inmovilización (reposo en cama y después inmovilización con un corsé) y ant ib io terap ia intravenosa pro longada (durante 4-6 semanas), seguida de ot ro pe r iodo s imi lar de antibióticos orales. Genera lmente , se in ic ia t ra tamiento empírico con vancomi-cina y r i f amp ic ina , y se corr ige según el estudio microbiológico, si los cu l t i vos son posi t ivos. Hay que asociar t ra tamiento sintomático del do lo r con analgésicos, an t i in f l amator ios y relajantes musculares. (MIR 00-01 F, 76). En casos refractarios al t ra tamiento médico, o en casos en los que se ha p r o d u c i d o una de fo rm idad s igni f icat iva en la c o l u m n a , será necesaria la intervención quirúrgica. Genera lmente , el pronóstico es bueno .
20.8. Lesiones medulares traumáticas Las lesiones traumáticas óseas de la c o l u m n a vertebral (fracturas vertebrales), así c o m o su evaluación y manejo , se descr iben ampl iamente en el Capítulo de Traumatología. En este capítulo se recoge la semiología neurológica de aquel los t raumat ismos raquídeos que afectan a la f u n ción de la médula espinal (lesiones medulares traumáticas).
Cualquier paciente que haya sufr ido un t raumat ismo grave, los pac ien tes con pérdida de consciencia , cua lqu ier ev idencia de abuso de d ro gas, y aquel los en los que existe sintomatología que sugiera daño a la co l umna vertebral (dolor cerv ica l o de espalda) o a la médula espinal (anestesia, hormigueo , acorchamiento , deb i l i dad o parálisis en una ext remidad , respiración abdomina l , pr iap ismo, etc.) deben considerarse y tratarse, desde el p rop io lugar del acc idente, c o m o si tuv ieran una lesión ver tebromedular hasta que se demuestre lo contrar io con las pruebas diagnósticas. La inmovilización precoz en el lugar del acc idente antes de realizar n inguna otra maniobra , j un to con la colocación de un collarín cervical rígido y adecuado contro l hemodinámico y respiratorio, formarán parte muy importante del mane jo prehospita lar io de estos pacientes.
El segmento cervical de la co l umna vertebral es el más frecuentemente afectado por las lesiones traumáticas, seguido del segmento lumbar . Debe tenerse en cuenta que hasta un 2 0 % de los pacientes con una lesión espinal traumática grave presenta una segunda lesión en otro nivel espinal , y un alto porcentaje sufre lesiones graves de otros órganos (traumatismos craneoencefálicos, torácicos y abdominales) que deben descartarse en la evaluación in i c ia l , y que en ocasiones resultan enmascaradas por la lesión espinal .
Evaluación hospitalaria
Todo paciente que llega al hospital con sospecha de lesión medular debe ser eva luado de la manera que se describe a continuación.
Semiología de la lesión medular traumática
En la valoración neurológica de un t raumat ismo ver tebromedular , es pr ior i tar io establecer el nivel de la lesión medular , s iendo éste el nivel más bajo en el que existe función neurológica norma l . Es importante te ner en cuenta que, deb ido a la desproporción entre el c rec imiento de la co l umna vertebral y la médula espinal durante el desarrol lo, la médula no ocupa todo el canal vertebral . Por tanto, no existe una equiva lenc ia exacta entre el nivel de la fractura y el nivel de la lesión medular (en general , se cump l e que, de C2 a D10 , el nivel medular se corresponde con uno o dos niveles más que la apófisis espinosa de la vértebra afectada; e j emp lo : vértebra C4-médula C5, vértebra D3-médula D5) , y en el segmento vertebral de D11 a L1 se loca l izan muy próximos todos los niveles medulares lumbares, sacros y coccígeos, con el cono medular a nivel L1-L2 en la mayoría de los pacientes. El resto del canal vertebral cont iene las raíces de la cola de cabal lo .
Para determinar el nivel lesional, deben valorarse la función motora y sensitiva, según la escala ASIA (MIR 0 0 - 0 1 , 88). El nivel sensit ivo se establece en base a la distribución de la inervación cutánea en derma-tomas, s iendo algunos puntos c lave los que se recogen en la Tabla 79.
148

Neurología y neurocirugía
Para la evaluación de la función motora , deben explorarse una serie de mov imien tos que se recogen en la Tabla 80, ten iendo en cuenta que las lesiones por enc ima de C4 producen tetraplej ia, y las lesiones D1 o más bajas conducen a la aparición de paraplej ia , al preservar la inervación del p lexo braquia l . La función respiratoria puede verse compromet ida en lesiones por enc ima de C4, por ser éste el nivel del que surge la inervación para el músculo diafragma (nervios frénicos).
NIVEL DERMATOMA
C4 Hombro
C6 Dedo pulgar ( 1 ° mano)
C7 Dedo corazón (3.° mano)
C8 Dedo meñique (5.° mano)
D5 Pezón (mamila)
DIO Ombligo
L1 Ingle
L3 Rodilla
L4 Maléolo interno
L5 Dorso del pie y 1.° dedo pie
SI Maléolo externo
S4-S5 Perianal
Tabla 79. Exploración del nivel sensitivo
NIVEL M Ú S C U L O FUNCIÓN MOTORA
Extremidades . . . . . •. . . . . . . . . • •
C5 • Deltoides • Bíceps braquial
• Separación del hombro • Flexión del codo
C6 • Bíceps braquial • Extensores de la muñeca
• Flexión del codo • Extensión de la muñeca
C7 Tríceps Extensión del codo
C8 • Flexor profundo de los dedos • Mm. intrínsecos de la mano
Apretar la mano
L2 lleo-psoas Flexión de la cadera
L3 • lleo-psoas • Cuadríceps
• Flexión de la cadera • Extensión de la rodilla
L4 Cuadríceps • Extensión de la rodilla
L5 Tibial anterior • Dorsiflexión del pie • Dorsiflexión del 1 5 dedo del pie
S1 Gastrocnemio/sóleo Flexión plantar del pie
Hg ¡§§jtai Muscuiat
ura axial
C4 Diafragma Parálisis diafragmática
D2-D9 Músculos intercostales Respiración abdominal
D9-D10 Musculatura abdominal superior
D11-D12 Musculatura abdominal inferior
Tabla 80. Exploración de la función motora
NIVEL DERMATOMA
D8-D9 Cutáneo-abdominales superiores
D10-D12 Cutáneo-abdominales inferiores
L1-L2 Cremastérico
S3-S4 • Bulbocavernoso • Cutáneo-anal
Tabla 81. Exploración de los reflejos cutáneos
El examen motor y sensitivo permite establecer si la lesión medular es completa o incompleta . Se def ine c o m o incompleta aquélla en la que existe algún grado de función residual motora o sensitiva más de tres segmentos por debajo del nivel de la lesión; se clasif ican en este grupo el síndrome centromedular , el síndrome de Brown-Séquard, el síndrome espinal anterior y el síndrome espinal posterior, que se describen en el apartado 1.9 del tema de Semiología de este capítulo. Se define c o m o completa aquel la en la que no existe n inguna función neurológica conservada más de tres segmentos por debajo del nivel de lesión. Solamente un 3 % de los pacientes con lesión medular completa en la pr imera exploración tendrán alguna mejoría en las primeras 24 horas; la persistencia de una lesión comple ta más allá de 24 horas indica que no se producirá n inguna recuperación de la función neurológica. La persistencia de los reflejos cutaneoabdominales , el reflejo cremastérico, ref le jo cutaneoanal y bulbocavernoso se ut i l iza c o m o ind icat ivo de lesión medular incompleta . El pr iap ismo en presencia de una lesión traumática espinal es ind icat ivo de lesión medular por falta de tono simpático.
En el seno de un traumatismo medular, pueden evidenciarse dos tipos de shock. En pr imer lugar, se define como shock espinal un per iodo de parálisis flaccida y arrefléxica que habitualmente se resuelve en el transcurso de unas 48 horas; de manera gradual, se va estableciendo posteriormente la parálisis espástica e hiperrefléxica. El hecho de que in ic ia lmente se establezca la hiperreflexia y la espasticidad es poco común, pero imp l i ca ría un mal pronóstico. El establecimiento nuevamente del reflejo bulbo-cavernoso suele indicar el f in de esta fase de shock espinal. En segundo lugar, puede producirse un shock neurogénico, caracterizado por una situación de colapso cardioc i rculator io , con hipotensión y bradicardia, debido a la interrupción de la vía simpática dentro de la médula espinal, lo que hace que predomine el sistema parasimpático en el organismo. Se asocia a lesiones cervicales y torácicas altas.
R E C U E R D A El nivel lesional v iene determinado por el último nivel con función neurológica normal.
Exploración radiológica
• Exploración cervical inicial. No se recomienda la evaluación rad io lógica cervical a todo paciente que cump la todos los requisitos que v ienen reflejados en la Tabla 82.
R E C U E R D A Hasta un 2 0 % de los pacientes con traumatismo medular tienen otra lesión espinal. No olvides que debemos buscar lesiones en otras regiones del organismo.
También deben explorarse los reflejos de est i ramiento y cutáneos (Tabla 81), especia lmente de va lor en pacientes comatosos en los que no se pueden evaluar las func iones motoras y sensitivas.
Nivel de consciencia normal Ausencia de tóxicos No contractura a la palpación cervical No dolor cervical en todos los rangos de movimiento No lesiones distractoras No mecanismo lesional de alto riesgo: fracturas múltiples, caídas de gran altura, accidente de tráfico
* Se han d e c u m p l i r t o d o s los c r i te r ios
Tabla 82. Criterios para no realizar exploración cervical*
149

Manual CTO de Medicina y Cirugía, 8 . A edición
En el caso de que se encuentre algún cr i ter io de los señalados en la tabla, se debe realizar una serie de proyecciones radiológicas simples de la c o l u m n a cervical (cogiendo hasta la vértebra D1) . En el caso de que haya áreas sospechosas o el área correspondiente al déficit neurológico o que no hayan pod ido ser visual izadas, se procederá a realizar una TC cervical (Figura 11 6).
Figura 116.TC cervical normal, donde se observa la correcta alineación de los cuerpos vertebrales, así como la ausencia de líneas de fractura en los mismos
Exploración torácica y lumbosacra inicial. Todo paciente inconsc iente, con do lor de espalda o prec ip i tado desde más de dos metros, debe someterse a una exploración radiológica s imple, que se c o m plementará con una TC del área de la anorma l idad ósea, o bien del déficit neurológico. Resonancia magnética. Se reservará para aquel los casos en los que, con la exploración radiológica anterior, no se aclare la causa del déficit neurológico.
Tratamiento de la lesión medular
Administración de corticoides. Es opc iona l administrar met i lpred-nisolona en altas dosis en las primeras ocho horas tras haberse p ro d u c i d o un t raumat ismo medular , manteniéndola durante 24 horas si ésta se administra en las primeras tres horas, o 48 horas, si se in ic ia entre las tres y las ocho horas (protoco lo NASCIS III). Sin embargo, según la ev idencia reciente, la administración de cort ico ides no ha demostrado benef ic io clínico s igni f icat ivo en estos pacientes. Ade más, se ha c o m p r o b a d o que la inc idenc ia de neumonía y sepsis grave se incrementa, especia lmente cuando se mant ienen durante 48 horas. Tracción cervical. En pacientes conscientes con fractura-dislocación cerv ical , se recomienda la reducción de la lesión mediante tracción cerv ical , salvo en una serie de circunstancias determinadas. Descompresión quirúrgica. Está especia lmente indicada en aque
llos pacientes con deter ioro neurológico progresivo, s iendo con t ro ver t ido el m o m e n t o más adecuado para realizar d i cho p roced im i en to quirúrgico (precoz o tardío). Los benef ic ios de la descompresión son muy dudosos en casos de déficit comp le to sin disrupción anatómica de la médula.
Sciwora (lesión de médula espinal sin alteración radiológica)
Se trata de una mielopatía traumática sin ev idenc ia de fracturas o d is lo caciones de la co l umna vertebral que puedan estudiarse a través de la radiología convenc iona l y la TC. Es una patología más común en niños menores de nueve años, en los que existe una falta de madurez de los tej idos de sostén de la co l umna vertebral que hace que la médula sea más propensa a lesionarse en casos de traumat ismos espinales. El segmento más afectado en este proceso es el cerv ica l . En la mi tad de los casos, hay un intervalo de t i empo entre el m o m e n t o de producirse la lesión y el in i c io de las manifestaciones clínicas.
La RM podrá revelar cambios en los tej idos de sostén, c o m o rupturas l i-gamentarias y hemorragias, al igual que cambios en la médula espinal , con impl icac iones pronosticas. El t ra tamiento se basa en la i n m o v i l i zación y la controvert ida administración de cort icoides. El pronóstico dependerá de la situación neurológica in i c ia l .
R E C U E R D A El S C I W O R A es una lesión típica de niños en la que no se justificable en la RX y en el T C . El tratamiento es conservador
20.9.Tumores ¡ntrarraquídeos
Representan un 1 5 % de los tumores pr imar ios del sistema nervioso centra l . Aunque los tumores espinales más frecuentes son metastásicos, la mayoría de los tumores espinales pr imar ios son benignos, a d i fe ren cia de los tumores craneales, y suelen dar clínica por compresión más que por invasión.
Los tumores ¡ntrarraquídeos se clasif ican en tres grupos (Tabla 83) (Figura 11 7):
E X T R A D U R A L E S • Metástasis • Cordoma
I N T R A D U R A L E S • Neurinoma E X T R A M E D U L A R E S • Meningioma
I N T R A D U R A L E S • Astrocitoma I N T R A M E D U LARES • Ependimoma
Tabla 83. Tumores ¡ntrarraquídeos
Extradurales (los más frecuentes, 5 5 % ) . Crecen en el cuerpo vertebral y/o el espacio ep idu ra l . A u n q u e las metástasis pueden encontrarse en cua lqu ie ra de los tres grupos, suelen ser de l o ca l i zación ext radura l . O t ro t u m o r encuadrado en este g rupo es el cor-d o m a sacrococcígeo (células "fisalíforas" típicas en la anatomía patológica).
1 5 0

Neurología y neurocirugía
Figura 117. (a) Metástasis epidural; (b) Neurinoma cervical; (c) Meningioma dorsal; (d) Astrocitoma cervical
• Intradurales extramedulares ( 4 0 % ) . En su mayoría, crecen a partir de las raíces nerviosas (neurinomas) o las leptomeninges (men ing io mas) (Figura 11 8).
Figura 118. Meningioma intrarraquídeo
Intramedulares ( 5 % ) . Crecen inf i l t rando y destruyendo la sustancia gris y blanca medular (astrocitomas y ependimomas) .
Metástasis espinales
Las metástasis extradurales suelen tener or igen en carc inomas bronco-génicos, seguidos por neoplasias hematológicas ( l infomas, mie lomas) y carc inomas de mama y de próstata (estas dos últimas pueden ser oste-oblástícas). Son el tumor intrarraquídeo más frecuente y la causa más frecuente de compresión medular (MIR 98-99, 69) .
Su distribución es proporc iona l a la long i tud del segmento (más frecuentes a nivel dorsal). Deben sospecharse en todo paciente con antecedentes de cáncer y do lor de espalda, sobre todo, si se asocia a déficit neurológico.
El t ra tamiento no pro longa la superv ivencia, y t iene c o m o ob je t i vo c o n trolar el do lor e intentar preservar la función neurológica. Las pos ib i l idades de recuperación func iona l dependen d i rectamente del estado neurológico del paciente cuando se in ic ia el t ra tamiento, que puede ser quirúrgico o mediante radioterapia loca l , con resultados similares (se prefiere la cirugía cuando el t umor pr imar io es radiorresistente, cuando no hay ev idencia de tumor p r imar io o se desconoce su histología, o cuando existen dudas diagnósticas sobre la naturaleza tumora l de la lesión; p. e j . : absceso epidura l espinal) (MIR 99-00F, 74).
Q R E C U E R D A El tratamiento de una metástasis intrarraquídea (ya sea quirúrgico o mediante radioterapia) es paliativo, buscando una descompresión medular.
Tumores intradurales extramedulares
Los neur inomas son los tumores pr imar ios ¡ntrarraquídeos más f recuentes. Suelen local izarse a nivel dorsal o cerv ica l , y provocan do lor y déficit neurológico en el ter r i tor io de la raíz de la que crecen. Se d iag nostican por RM y el t ra tamiento de elección es la cirugía, que puede ser curat iva (Figura 11 9 ) .
Los men ing iomas p redominan en mujeres y en región torácica. O r i g i nan clínica de do lor ( radicula lgia intercostal) y compresión medular . El t ratamiento de elección es la cirugía, que es curat iva si la resección es comple ta .
Un grupo raro de tumores intradurales lo const i tuyen las tumorac iones mal format ivas o disembriogénicas: teratomas, quistes dermoides, epi-dermoides y l ipomas, de situación habi tua lmente intraduralextramedu-lar aunque pueden tener un componen te intramedular .
Suelen debutar en niños, se loca l izan generalmente a nivel lumbosa-cro, en cono medular y cauda equina, y pueden asociarse a disraf ismo espinal (espina bífida ocu l ta , estigmas cutáneos, etc.). Provocan clínica de radiculalgias, incont inenc ia de esfínteres, o incluso c l aud ica ción neurógena por anclaje medular en posición infant i l (L4-L5), que a veces puede resultar en deformidades de los pies (equino, zambo) o escoliosis en niños.
Tumores intradurales intramedulares
Los astrocitomas son los tumores intramedulares más frecuentes fuera del f i l u m termínale. Suelen ser de bajo grado (pilocíticos). Predominan
151

Manual CTO de Medicina y Cirugía, 8 . a edición
a nivel cerv ica l , pud iendo produc i r un síndrome siringomiélico (disociación termoalgésica de la sensib i l idad, de p redomin io en ext remida des superiores). Const i tuyen los tumores intramedulares más frecuentes en la edad pediátrica.
Los epend imomas son los tumores intramedulares más frecuentes en el cono medular y filum termínale (única localización de la variante mixo-papi lar) . T ienen una relativa tendencia a sangrar. Const i tuyen el grupo de tumores intramedulares más numeroso en la edad adulta.
R E C U E R D A El t u m o r ¡ntramedular más f recuente en los niños es el as t roc i toma, mient ras que en los adu l tos es el e p e n d i m o m a .
En ambos casos, el t ra tamiento es quirúrgico (microcirugía), aunque no siempre es posible la extirpación comple ta .
20.10. Absceso epidural espinal
Se trata de una colección purulenta en el espacio epidura l espinal . Se loca l iza con mayor f recuencia a nivel dorsal ( 5 0 % ) , seguido del segmento lumbar ( 3 5 % ) y cervical ( 1 5 % ) . Con frecuencia se asocia a osteomie l i t i s o discit is.
El mecan ismo patogénico más frecuente es la siembra hematógena desde focos infecciosos cutáneos, ur inar ios, respiratorios, cardíacos, etc. Otros posibles mecanismos son la extensión por contigüidad (abscesos de psoas, mediast init is , etc.), los traumatismos penetrantes y los p ro cedimientos quirúrgicos recientes sobre la co l umna vertebral . En un porcentaje e levado de casos ( 5 0 % ) , no se encuentra un or igen.
Son factores de riesgo la diabetes, consumo de drogas por vía parenteral, a l coho l i smo e insuf ic iencia renal crónica.
Figura 119. Imagen de absceso epidural cervical, donde se observa una masa (señalada con flecha blanca) que ocupa el espacio epidural cervical y que
comprime la médula, sobre todo por la parte posterior
R E C U E R D A La espond i lod i sc i t i s y el absceso ep idu ra l suelen asociarse c o n m u c h a f recuenc ia . La V S G y la proteína C react iva son útiles marcadores de segu im ien to de la infección.
El mic roorgan ismo más frecuentemente imp l i cado en las formas agudas es el Staphylococcus aureus; en las formas crónicas, Mycobacte-rium tuberculosis.
20.11. Siringomielia
Genera lmente se presenta con f iebre elevada, do lor y r igidez de espalda. Suelen impl i car síntomas radiculares, y evo luc ionan progresiva y rápidamente hacia una compresión medular con disfunción de esfínteres y paraparesia o tetraparesia (MIR 99-00F, 74; MIR 98-99, 64) .
La prueba de imagen de elección es la RM (masa epidura l que c o m pr ime el saco dural ) (Figura 119). Suelen presentar leucocitosis y elevación de la ve loc idad de sedimentación; este último parámetro puede servir c o m o marcador de seguimiento en estos pacientes. Puede obte nerse pus para cu l t i vo mediante una punción lumbar cuidadosa, aun que es prefer ible evitarla y tomar las muestras microbiológicas durante la cirugía.
El t ra tamiento consiste en inmovilización, ant ib io te rap ia (de manera empírica, se recomienda empezar con una cefa lospor ina de 3. a generación más v a n c o m i c i n a más r i f ampic ina ) durante 6-8 semanas, y cirugía en caso de déficit neurológico (si b ien otros autores r e c o m i e n dan cirugía, aún no ex is t iendo déficit motor ) . El pronóstico es ma lo con e levada mor ta l i dad y secuelas neurológicas frecuentes (más, cuanto peor es la situación neurológica del pac iente en el m o m e n t o del diagnóstico).
Se def ine c o m o la existencia de cavidades quísticas, también l lamadas sir inx, en la médula espinal, que pueden comun i ca r o no con el canal epend imar io centra l . Genera lmente se loca l izan a nivel cervical o dor sal (Figura 120). En algunos casos se ext ienden rostralmente y a lcanzan el bu lbo raquídeo, denominándose entonces s i r ingobulb ia .
Se asocia con frecuencia a mal formac iones congénitas (sobre todo, malformación de Chiar i t i po I), neoplasias medulares ( fundamenta l mente, astrocitomas), aracnoidi t is y t raumat ismos espinales (s ir ingomie l ia postraumática).
Da lugar a un cuadro clínico típico que se caracteriza por un síndrome centromedular , con un déficit suspendido y d isoc iado de la sensib i l i dad (abolición de la sensibi l idad termoalgésica, respetando los co rdo nes posteriores).
Es característico que el paciente refiera heridas o quemaduras cutáneas indoloras, sin haberse dado cuenta de cuándo se p roducen . Puede acompañarse de deb i l i dad , trastornos tróficos, arref lexia y fasc icu lac io nes en las extremidades superiores (resultado de la lesión de segunda motoneurona) .
152

Neurología y neurocirugía
Figura 120. Siringomielia. Se aprecia la cavidad siringomiélica cervicodorsal
enfermedades c o m o la artritis reumato ide (la más frecuente), síndrome de D o w n o enfermedad de M o r q u i o .
El síntoma fundamenta l es do lor subocc ip i ta l . En ocasiones se acompa ña de déficit neurológico. Puede causar muerte súbita por mov imien tos bruscos de flexión cerv ica l .
El diagnóstico es radiológico, al encontrar una distancia entre la odonto ides y el arco anter ior de C1 mayor de 5 m m en niños o mayor de 3 m m en adul tos.
Los pacientes asintomáticos con pequeñas luxaciones se tratan con c o llarín cerv ica l y controles clinicoradiológicos. Si están sintomáticos o asintomáticos con luxación importante (> 8 mm) , se recurre a la c i r u gía (fijación cervical posterior C1-C2 u occ ip i tocerv ica l ) . En casos con luxaciones irreductibles o compresión medular por el pannus in f l amator io , puede estar indicada la odontoidectomía transoral .
Impresión basilar
Es la malformación más frecuente de la charnela occ ip i tocerv ica l y la segunda anomalía cervical asociada a la artritis reumatoide. La base craneal aparece descendida respecto al límite superior de la o d o n t o i des.
En el caso de que sea sintomática, deberá ser tratada. In i c i a lmente , se recomienda una tracción. Si me jora , se realizará una artrodesis o c c ip i t o ce r v i c a l . Si no mejora , se procederá a la extirpación de la odonto ides .
La prueba diagnóstica de elección es la RM (MIR 98-99F, 66) . Cuan do es c laramente sintomática, o el cuadro clínico progresa en sucesivos controles, se puede optar por t ratamiento quirúrgico. En los casos asociados a malformación de Chiar i , el t ratamiento de elección es la craniectomía descompresiva subocc ip i ta l con plastia de dura, para a m pliar el tamaño de la fosa posterior. En otros casos se real izan der ivaciones sir ingosubaracnoideas o sir ingoperitoneales. El ob je t i vo funda mental del t ratamiento es evitar la progresión del déficit neurológico.
Platibasia
Supone una apertura anómala del "ángulo basal del cráneo" (mayor de 145°), con el consiguiente ap lanamiento de la base craneal (MIR 03-04, 247 ) .
Enfermedad de Klippel-Feil
20.12. Anomalías de la unión craneocervical
Luxación atloaxoidea
Se debe a alteraciones del l igamento transverso que fi ja el atlas a la odonto ides . Su or igen puede ser congénito, aunque con mayor frecuenc ia es secundar io a traumatismos o se presenta en el contexto de
Es un trastorno en el desarrol lo óseo con fusión congénita de dos o más vértebras cervicales, que se debe a un fa l lo en la segmentación normal de los somitas cervicales durante el desarrol lo embr ionar io . Se caracteriza por un descenso de la línea posterior de implantación del cabel lo , cue l lo cor to y limitación de la mov i l i dad cerv ica l .
R E C U E R D A La malformación craneocerv ica l más frecuente es la impresión basilar. El síndrome de D o w n y la artritis reumatoide pueden causar alteraciones a nivel de la charnela craneocerv ica l .
153

Manual CTO de Medicina y Cirugía, 8.a edición
Casos clínicos representativos
U n h o m b r e de 8 0 años ref iere , desde hace n u e v e meses , do lo r in termitente en p ier nas y parestes ias que a p a r e c e n después de c a m i n a r 100-200 met ros . Los s íntomas c o m i e n z a n en las z o n a s dista les de las ex te rmidades infer iores , a s c i enden a los glúteos y se a c o m p a ñ a n de do lor lumbar . Los ep isod ios son más f recuentes c u a n d o c a m i n a cues t a aba jo que cues t a a r r i ba , y se a l i v ian a l sentarse o ponerse en cuc l i l l as , o f lex ionándose h a c i a de lante mient ras s igue c a m i n a n d o . L a exp lorac ión neuro lógica es n o r m a l . ¿Cuá l de los s iguientes diagnóst icos es el más p robab le ?
1) D i s c o torác ico h e r n i a d o . 2 ) Estenosis e s p i n a l l u m b a r . 3) Estenosis d e la a r te r i a i l i a c a . 4 ) M i a s t e n i a g rav i s .
5) Neuropat ía periférica d e s m i e l i n i z a n t e .
M I R 0 6 - 0 7 , 5 6 ; RC: 2
U n pac ien te de 62 años p resen ta una h is tor ia de ce r v i ca lg i a i r rad iada a hombros . D e s d e h a c e un a ñ o , p resenta d i f i cu l tad progres i va pa ra c a m i n a r , añad iéndose do lor en b r a z o d e r e c h o . En la exp lo rac ión , p resen ta un ref le jo b ic ip i ta l abo l ido y unos ref le jos os teotend inosos pol ic inét icos en p ie rnas .
1) C r e o q u e t i e n e u n t u m o r m e d u l a r , y le solicitaría u n a R M c e r v i c a l . 2) C r e o q u e t i e n e u n a h e r n i a d i s c a l c o n e s p o n d i l o s i s , y le solicitaría R M c e r v i c a l . 3) P r o b a b l e m e n t e t i e n e u n a s i r i n g o m i e l i a , y le solicitaría u n a R M . 4) C r e o q u e t i e n e e s p o n d i l o s i s c e r v i c a l , y le solicitaría u n T A C de c o l u m n a c e r v i c a l . 5) C r e o q u e u n e s t u d i o radio lógico s i m p l e d e c o l u m n a y u n t r a t a m i e n t o c o n A I N E es
l o a d e c u a d o .
M I R 0 5 - 0 6 , 6 2 ; RC: 2
U n a lbañi l suf re un a c c iden te l abora l , prec ip i tándose desde 6 met ros de a l tura . P re senta un impor tante do lo r a n ive l l umbar y déf ic i t de extensión con t r a g ravedad de los dedos del p ie d e r e c h o . H a b r á que pensar que puede tener :
1) U n a lesión d e la raíz L-3. 2) U n a lesión d e la raíz L-4. 3) U n a lesión d e la raíz S-1. 4) U n a lesión d e la raíz L-5. 5) U n a lesión d e la raíz S-2.
M I R 0 4 - 0 5 , 9 4 ; RC: 4
M u j e r de 35 años , s in an teceden tes de interés, que a c u d e c o n un do lo r in tenso en c o l u m n a ce r v i c a l de dos días de e vo luc ión , q u e le imp ide la rea l izac ión de su t rabajo de aux i l i a r admin is t ra t i vo . El do lo r se i r rad ia hac i a h o m b r o d e r e c h o , a u m e n t a c o n los mov im ien tos de f lexión y ro tac ión de l cue l l o , y no se a compaña de otras m a n i fes tac iones c l ín icas . L a exp lorac ión genera l y la neuro lógica son no rma les , si bien la va lo rac ión de la f u e r z a de los m i e m b r o s super io res se h a y a inter fer ida po r ei do lor . ¿ Q u é ac t i tud es la más a d e c u a d a ?
1) Reposo a b s o l u t o y co l lar ín rígido d u r a n t e 3 0 días. 2 ) Resonanc i a magnét ica n u c l e a r u r g e n t e , y a c t u a r e n c o n s e c u e n c i a . 3) P a r a c e t a m o l c o n code ína , educac ión p o s t u r a l , e j e r c i c i o s suaves y segu i r e v o l u
c i ón en s ie te días. 4 ) Radiología s i m p l e y, e n caso d e e n c o n t r a r s e rect i f icación d e la c u r v a t u r a f isiológi
ca d e la c o l u m n a , t r a c c i o n e s c e r v i c a l e s . 5) Radio logía s i m p l e y, e n a u s e n c i a d e h a l l a z g o s s i g n i f i c a t i v o s , r ea l i za r e l e c t r o m i o -
g r a m a .
M I R 0 2 - 0 3 , 2 2 8 ; RC: 3
H o m b r e de 3 4 años , sin an teceden tes de interés. P resenta desde h a c e u n a s e m a n a do lo r en z o n a l umba r ba ja , que no le h a imped ido r ea l i za r su ac t i v idad l abora l . En las ú l t imas 2 4 ho ras , el do lo r h a a u m e n t a d o , hasta conve r t i r se en seve ro e i n c a p a c i tante , d i f i cu l tándole ta reas c o m o d e a m b u l a r o levantarse de la c a m a . El pac ien te a c u d e a U rgenc i a s , donde se objet iva u n a exp lorac ión física genera l es t r i c t amente n o r m a l , una exp lorac ión neuro lógica d i f i cu l tada por e l do lo r , s in a l t e rac iones en la sens ib i l idad , c o n Laségue a 60 ° , B ragard negat ivo y c o n ref le jos os teotend inosos conse r v ados y s imétr icos en las cua t ro ex t remidades . ¿ Q u é ac t i tud es la más i nd i cada en el estudio y t ra tamiento de este pac ien te ?
1) Se r ea l i z an Rx s imp l e s d e c o l u m n a l u m b a r , q u e son n o r m a l e s , se d i agnos t i c a d e l u m b a l g i a aguda , se p resc r iben analgésicos n i ve l I I , re la jantes muscu la res , r eposo en c a m a d u r a n t e dos semanas, y se va lo ra evo luc ión al f i n a l de l p e r i o d o d e r eposo .
2) Se d i a g n o s t i c a d e l u m b a l g i a a g u d a , n o se r e a l i z a ningún e s t u d i o c o m p l e m e n t a r i o , se i n f o r m a al p a c i e n t e y su f a m i l i a s o b r e e l c u a d r o q u e p a d e c e , se p r e s c r i b e n a n a l gésicos n i v e l I I , r e la jan tes m u s c u l a r e s , mov i l i zac ión p r e c o z , y se v a l o r a e vo luc ión e n u n a s emana .
3) Se d i a g n o s t i c a d e h e r n i a d i s c a l , y se ingresa pa ra cirugía. 4) Se rea l iza TC u rgen te d e c o l u m n a l u m b a r , q u e es n o r m a l , se ingresa al pa c i en t e para
c o m p l e t a r el e s tud io c o n R M e Isótopos, c o n sospecha de neop las i a o infección. 5) Se r e a l i z a n T C y R M urgen tes , q u e son e s t r i c t a m e n t e n o r m a l e s , se c o n s u l t a a
Psiquiatría pa ra desca r ta r c o m p o n e n t e f u n c i o n a l .
M I R 0 0 - 0 1 , 7 8 ; RC: 2
Si se r e c i be a un motor i s ta que se h a es t re l lado c o n t r a un á rbo l , está p l enamente consc i en te , no presenta les iones ex te rnas re levantes , man t i ene la vent i lac ión e s p o n tánea y no puede m o v e r ni sent ir las ex t r emidades , ¿en qué rango de n ive l s e g m e n tar io esperar ía encon t r a r u n a impor tan te lesión medu la r ?
C e r v i c a l C1 - C 4 . C e r v i c a l C 6 - C 8 . Torác i co T I - T 3 . Torác i co T 4 - T 8 . Torác i co T 9 - T 1 2 .
M I R 0 0 - 0 1 , 8 8 ; RC: 2
U n va rón adul to p resenta u n c u a d r o de do lo r y r ig idez de cue l l o , c o n i r rad iac ión del do lor a ex t r em idad super ior d e r e c h a a través de la c a r a dorsa l del an t eb r azo y del te rce r d e d o , comprobándose , al m i s m o t i empo, deb i l idad de los f lexores de la m u ñ e c a y d isminuc ión de l ref le jo t r ic ip i ta l . T ras la radiología c o n v e n c i o n a l y r e sonanc i a magnét ica , se es tab lece el d iagnóst ico de he rn i a d i sca l c e r v i c a l . El d i s co he rn i ado es e l c o m p r e n d i d o ent re :
C1-C2 . C2-C3 . C 3 - C 4 . C 4 - C 5 . C 6 - C 7 .
M I R 9 9 - 0 0 , 2 0 7 ; RC: 5
Pac iente de 6 0 años que presenta f i ebre e l evada , do lo r de e spa lda y pa rapa res i a . Se le r ea l i za u n a r e sonanc i a nuc l ea r magnét ica de c o l u m n a que mues t r a m a s a e p i d u ral que c o m p r i m e la médu la do r sa l . ¿ C u á l , de los s iguientes, es el diagnóst ico más p robab le ?
1) M e n i n g i o m a d o r s a l . 2) Metástasis e p i d u r a l . 3) H e m a t o m a e p i d u r a l . 4) A b s c e s o e p i d u r a l . 5) I n f a r t o m e d u l a r .
M I R 9 8 - 9 9 , 6 4 ; RC: 4
Ante u n a pac ien te de 32 años que , dos horas antes de su admis ión , sufre ce fa l ea b rusca e in tensa m ien t ras m o n t a b a en b i c i c l e ta , y que presenta exp lorac ión neuro ló g i ca y T C c r anea l no rma l e s , ¿ cuá l sería la ac t i tud más co r r e c t a ?
1) So l i c i t a EEG ( e l e c t r o e n c e f a l o g r a m a ) . 2) Pautar t r a t a m i e n t o pa ra migraña y da r el a l t a . 3) Reeva lua r c o n T C c r a n e a l a las 2 4 horas . 4) Rea l i za r punc ión l u m b a r , pasadas unas horas . 5) A c o n s e j a r la supresión d e e j e r c i c i o físico en l o suces i vo .
RC: 4
154

21.
Aspectos esenciales
ANOMALÍAS DEL DESARROLLO
Orientación
MIR El t e m a d e las anomal ías de l desa r ro l l o t i ene m u y p o c a i m p o r t a n c i a pa ra el M IR . So l amen te basta c o n tener ¡deas c laras y c o n c e p t o s básicos de las d i fe rentes f o r m a s d e c raneos inos tos i s , d e las m a l f o r m a c i o n e s d e Ch i a r i y d e los d i s ra f i smos esp ina les .
pj~| La escafocefaíia es la craneosinostosis más f recuente . La b raqu i ce fa l i a puede asociarse a d ismorf ias fac ia les .
^2] La malformación de Chiar i t i p o I es la más f recuente . Es un descenso de las amígdalas cerebelosas a través del f o r a m e n magnum. Puede asociarse a la s i r i gomie l i a .
["3"] La malformación de Chiar i t i p o I! se asocia al m i e l o m e n i n g o c e l e y a h id roce fa l i a .
¡~4~] El d is ra f ismo espinal puede ser abier to o quístico, cuando no está cub ie r to de p ie l , u ocu l t o , c u a n d o hay recu b r im ien to cutáneo.
[~5~] El m i e l o m e n i n g e c e l e es un d is ra f i smo espina l quístico. Se asocia c o n el déficit de ácido fólico y la t o m a de va lp roa to duran te el emba razo . El diagnóstico prenata l se establece med ian te la determinación de cifras elevadas de oc-fetoproteína en suero mate rno y líquido amniótico y la ecografía.
21.1. Craneosinostosis
En la Figura 121 se puede ver la configuración anatómica craneal , en lo que a suturas y fontanelas se refiere.
155
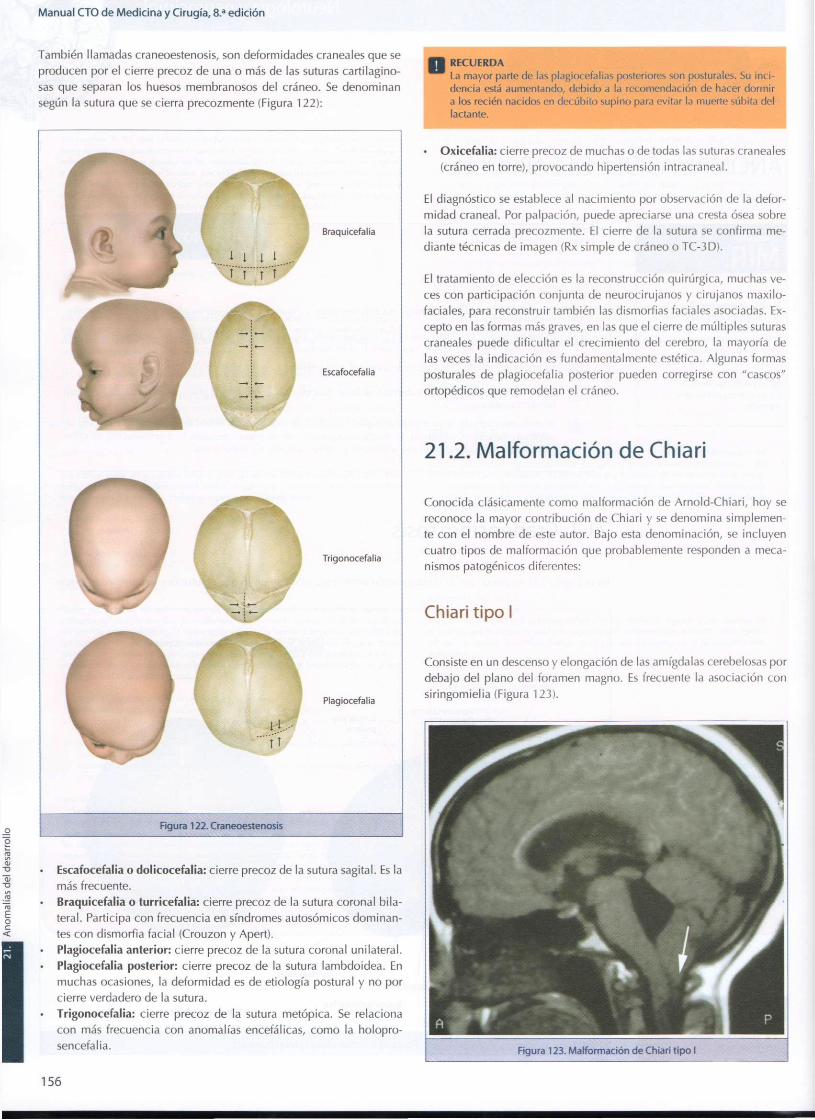
Manual CTO de Medicina y Cirugía, 8.a edición
También l lamadas craneoestenosis, son deformidades craneales que se producen por el c ierre precoz de una o más de las suturas car t i l ag inosas que separan los huesos membranosos del cráneo. Se denominan según la sutura que se cierra precozmente (Figura 122) :
B r a q u i c e f a l i a
Escafocefalia
Trigonocefalia
Plagiocefalia
Figura 122. Craneoestenosis
Escafocefalia o dolicocefalia: c ierre precoz de la sutura sagital. Es la más frecuente. Braquicefalia o turricefalia: c ierre precoz de la sutura corona l b i l a teral . Participa con frecuencia en síndromes autosómicos d o m i n a n tes con d ismorf ia facial (Crouzon y Apert ) . Plagiocefalia anterior: c ierre precoz de la sutura coronal uni latera l . Plagiocefalia posterior: c ierre precoz de la sutura l ambdoidea . En muchas ocasiones, la de fo rmidad es de etiología postural y no por c ierre verdadero de la sutura. Trigonocefalia: c ierre precoz de la sutura metópica. Se re lac iona con más frecuencia con anomalías encefálicas, c o m o la holopro-sencefalia.
ali
R E C U E R D A La mayor parte de las plagiocefalias posteriores son posturales. Su inc i dencia está aumentando, debido a la recomendación de hacer dormir a los recién nacidos en decúbito supino para evitar la muerte súbita del lactante.
• Oxicefal ia: c ierre precoz de muchas o de todas las suturas craneales (cráneo en torre), p rovocando hipertensión intracraneal .
El diagnóstico se establece al nac imiento por observación de la deformidad craneal . Por palpación, puede apreciarse una cresta ósea sobre la sutura cerrada precozmente . El cierre de la sutura se con f i rma me diante técnicas de imagen (Rx s imple de cráneo o TC-3D).
El t ratamiento de elección es la reconstrucción quirúrgica, muchas ve ces con participación con junta de neuroc i ru janos y c i rujanos maxi lo-faciales, para reconstruir también las dismorf ias faciales asociadas. Excepto en las formas más graves, en las que el cierre de múltiples suturas craneales puede d i f icu l tar el c rec imiento del cerebro, la mayoría de las veces la indicación es fundamenta lmente estética. Algunas formas posturales de plagiocefa l ia posterior pueden corregirse con "cascos" ortopédicos que remodelan el cráneo.
21.2. Malformación de Chiari
Conoc ida clásicamente c o m o malformación de Arnold-Chiar i , hoy se reconoce la mayor contribución de Chiar i y se denomina s imp lemen te con el nombre de este autor. Bajo esta denominación, se inc luyen cuatro t ipos de malformación que probab lemente responden a meca nismos patogénicos diferentes:
Chiari tipo
Consiste en un descenso y elongación de las amígdalas cerebelosas por debajo del p lano del foramen magno. Es frecuente la asociación con s i r ingomie l ia (Figura 123).
1 5 6

Neurología y neurocirugía
Suele debutar en la adolescencia y edad adulta (edad media : 40 años) y es un poco más frecuente en mujeres. El síntoma más habitual de presentación es la cefalea subocc ip i ta l , que aumenta con las maniobras de Valsalva, y que se acompaña en ocasiones de síntomas de afectación t ronca l , cerebelosa o cent romedular (con p redomin io en miembros superiores). El nistagmus vertical es un hal lazgo típico.
La técnica diagnóstica de elección es la RM. Últimamente, se han ven i do u t i l i zando los estudios de f lu jo de líquido cefalorraquídeo mediante RM, en los que se detecta problemas de circulación del m ismo a nivel del foramen magno.
n R E C U E R D A U n a cefalea suboccipital en personas de unos 30 años, que aumenta con las maniobras de Valsa lva, es sugestiva de malformación de Chiari tipo I.
ción del parénquima nervioso próximo. Son más frecuentes en la cisura de Silvio. Los únicos de localización extradural son los intraselares.
En general , son asintomáticos (hal lazgo casual), si b ien pueden cursar con crisis epilépticas, signos focales o HTIC.
La tendenc ia actual es no tratar aquel los que no causan efecto de masa o síntomas. El t ipo III de Calassi (gran quiste de la cisura de Si lv io con desplazamiento de línea media) requiere t ratamiento quirúrgico. Puede realizarse aspiración con aguja, craneotomía con fenestración o ext i r pación de la pared del quiste, o derivación cistoper i toneal (técnica que probablemente consigue los mejores resultados).
21.4. Disrafismo espinal
El t ratamiento de elección para los pacientes con Chiari t i po I s intomáticos o asociados a s i r ingomie l ia es la craniectomía descompresiva subocc ip i ta l (que suele ampl iarse con una laminectomía de C1 y C2) con apertura de la duramadre y colocación de una plastia de dura, para ampl iar el espacio de la fosa posterior. Cuando se asocia a h idrocefa l ia , debe implantarse una derivación de LCR. Los pacientes asintomáticos deben ser v ig i lados y operados sólo en caso de deter ioro.
Chiari tipo II
Es un descenso del vermis cerebeloso, cuarto ventrículo, protuberanc ia y bu lbo por debajo del p lano del foramen magno. Se asocia f recuentemente con mie lomen ingoce le e h idrocefa l ia . Suele debutar en la i n fancia. Las manifestaciones clínicas se deben a disfunción de t ronco y pares craneales bajos. Cursa con estridor respiratorio, apnea episódica, aspiraciones frecuentes, retrocol l i y/o signos cerebelosos. Se d iagnost i ca por RM. Debe colocarse una derivación ventr icu loper i tonea l para la h idrocefa l ia y realizar una descompresión ampl ia de la fosa posterior. La d i f i cu l tad respiratoria es la pr inc ipa l causa de la elevada morb imor-ta l idad de la malformación de Chiari t ipo II.
Chiari tipo III
Consiste en un descenso de las estructuras de la fosa posterior (vermis, hemisferios cerebelosos y t ronco) dentro de un encefa lomeningoce le cervical a l to. Es la forma más grave, generalmente incompat ib l e con la v ida.
Chiari tipo IV
Es una hipoplas ia cerebelosa sin herniación.
21.3. Quistes aracnoideos
Son divertículos aracnoideos, ocupados por LCR, que permanecen como vestigio embr ionar io y que ocasionalmente pueden comprometer la fun-
El disraf ismo espinal const i tuye una serie de anomalías congénitas espinales que se caracter izan por un defecto del cierre de las estructuras en la línea media (Tabla 84) . Si la lesión inc luye defectos de los arcos óseos vertebrales posteriores, se denomina espina bífida. La espina bí-fida s imple (sólo inc luye defectos de los arcos vertebrales posteriores) es un hal lazgo radiográfico frecuente, sobre todo a nive l de L5-S1, que no conl leva ningún s igni f icado patológico. El disraf ismo espinal puede ser de dos t ipos: espina bífida ocu l ta y espina bífida quística o abierta (Tabla 85).
ESPINA B ÍF IDA Mielomeningocele QUÍST ICA (ABIERTA) Mielomeningocele
• Lipomielomeningocele • Filum terminale hipertrofiado • Seno dérmico congénito
ESPINA B ÍF IDA • Quiste neuroentérico OCULTA • Agenesia del sacro
• Diastematomiella • Mielocistocele • Meningocele sacro anterior
Tabla 85. Disrafísmos espinales
CRANEOENCEFÁLICOS
RAQU IMEDULARES (ESPINAS B ÍF IDAS)
Anencefalía Iniencefalia Craneorra quisquisis totalis
Encefafaloce* Aplasia cutis congénita
* (Son realmente defectos en la Inducción de las cubiertas)
Espina bífida oculta
Espina bífida manifiesta
Simple Seno dérmico congénito Síndrome de cono anclado Dlastematomielia Lipoma lumbosacro Extasia dural Menlngoceles sacro anterior e intrasaco Quistes: neuroentérico, aracnoideo, perineural (Tarlov) Teratoma sacrocoxígeo Síndrome de regresión caudal o agenesia del sacro (hijos de madre diabética)
Mielomeningocele (más frecuente) Meningocele (mejor pronóstico) Raquisquisis o mielocele (peor pronóstico)
Tabla 84. Defectos en el tubo neural (disrafísmos)
1 5 7

Manual CTO de Medicina y Cirugía, 8 . a edición
Espina bífida (disrafismo espinal) oculta
Es aquel disraf ismo que está cubier to por p ie l . Puede acompañarse de algunos estigmas cutáneos en el nivel afectado (mechón de pelo , angio-mas capilares, etc.). Ocas iona lmente puede asociarse a l ipomas, t u m o res dermoides, senos dérmicos o d iastematomie l ia (dos hemimédulas). Cuando es sintomática deb ido a alguna de estas anomalías asociadas, suele cursar c o m o un síndrome de médula anclada (cono medular por debajo de L1-L2), con deb i l idad y atrofia de miembros inferiores, trastornos de la marcha, trastornos del cont ro l de esfínteres, do lor vago a nivel de genitales, periné y parte anter ior del muslo , hipoestesia en periné, deformidades en los pies y escoliosis.
O R E C U E R D A El disrafismo espinal oculto puede acompañarse de estigmas cutáneos y dar lugar a un síndrome de médula anclada, que se asocia a sintomatología motora, esfinteriana y deformidades ortopédicas.
El seno dérmico congénito es una forma de espina bífida ocu l ta . Se trata de un tracto revestido por ep i te l io escamoso estratif icado que aparece en o muy cerca de la línea media , en cua lquier punto desde el nasión al cóccix (localización más frecuente, lumbosacro) . Comienza en la p ie l , y puede terminar en el te j ido subcutáneo o llegar al canal medular ( 3 0 - 5 0 % de los casos, más frecuente en los lumbares, en general anc lado a un quiste de rmo ide o un teratoma).
En la exploración, j u n t o al pun to d e p r i m i d o en la p i e l , se suele apreciar hipertr icosis , a lguna lesión angiomatosa o un l ipoma. Pueden infectarse o dar lugar a un síndrome de médula anc lada. Exige d iag nóstico d i ferenc ia l con el sinus pilonídal, que suele tener un trayecto más cor to (casi nunca penetra en el sistema nervioso central ) , a nivel sacrococcígeo, con reacción in f lamator ia granulomatosa, y que no suelen requerir t ra tamiento , al ser una lesión benigna. A u n q u e sean asintomáticos, se recomienda la extirpación del tracto y el con ten ido int radura l , antes de que se p roduzcan déficit neurológicos o cuadros infecciosos. Si hubiese infección, debe tratarse, en pr imer lugar, con las medidas adecuadas.
Espina bífida quística o abierta (disrafismo espinal abierto)
Si la piel no cubre las mal formac iones de la médula espinal , se habla de disraf ismo espinal abierto o manif iesto.
El m ie lomen ingoce le es la fo rma pro to t ipo de espina bífida abierta (Figura 124), y debe ser d is t ingu ido del meningoce le , el cual es un defecto congénito de los arcos vertebrales posteriores con herniación en forma de quiste de las meninges, pero sin anomalías del te j ido nervioso subyacente; en un terc io de los casos puede acompañarse de s in tomato lo gía neurológica.
El m i e l omen ingoce l e se caracter iza por la exposición poster ior del canal central de la médula espinal al exter ior , estando los bordes de la médula anclados a la superf ic ie cutánea. El líquido cefalorraquídeo se acumularía justo por delante del defecto, lo que hace que éste se vea e m p u j a d o hacia atrás. Esto último es lo que d i ferenc ia el m i e l o men ingoce le del mie losquis is , en el que el líquido no se acumu la . Sin embargo , hay autores que recomiendan evitar el término de mie los
quisis y referirse s iempre a m ie lomen ingoce l e . Se loca l iza con más f recuenc ia a nivel lumbar . En un 7 5 - 8 0 % de los casos se asocia con h idrocefa l ia , y muchos de el los t ienen también una malformación de Chiar i t i p o II.
Figura 124. Lipomielomeningocele
Se ha re lac ionado con déficit de ácido fólico en la madre, y con déficit de z inc , h iperv i taminosis A, viriasis y administración de ácido va lproi-co durante el embarazo. Se aconseja su prevención mediante la a d m i nistración de ácido fólico a la madre, desde al menos uno o dos meses previos a la gestación.
Puede hacerse diagnóstico prenatal mediante la determinación de c i fras elevadas de cc-fetoproteína en suero materno y líquido amniótico (determinación a la 14-18 semanas de gestación) y, más específicamente, por ecografía.
Requiere cirugía precoz, generalmente en las primeras 48-72 horas tras el nac imiento , para cerrar el defecto y tratar de reconstruir la anatomía normal en varias capas. Si se acompaña de h idrocefa l ia , debe i m p l a n tarse una derivación de LCR simultáneamente. La cirugía precoz no mejora la función neurológica, pero reduce el riesgo de infecciones. En la actua l idad, existen más de 400 casos que ya han sido operados in útero.
21.5. Encefalocele
Es un defecto del c ierre del cráneo en la línea media , más frecuente a nivel occ ip i t a l , que puede acompañarse de una herniación de las meninges y LCR (meningocele) o además, de un prolapso de te j ido cerebral o cerebeloso fuera de los límites del cráneo (encefalocele [Figura 126]) . Cuando, además de te j ido nervioso, inc luyen parte del ventrículo, se l laman cistoencefaloceles.
Se dist ingue entre: • Encefaloceles de la convexidad: son los más frecuentes, especial
mente los occipita les ( 7 0 % ) , en la línea medía y, generalmente, pe-dicu lados . Básales: son los únicos que no producen una masa vis ible al exterior, y se manif iestan c o m o fístulas de LCR o meningi t is recurrentes. Toda masa po l ipo idea intranasal en un recién nac ido debe cons iderarse un encefalocele hasta que se demuestre lo contrar io .
• Sincipitales o frontoetmoidales: se abren a la cara. El más frecuente de estos es el nasofrontal , que se suele asociar a h iper te lor ismo. Se descr iben también las formas nasoetmoidal y nasoorbitar ia.
• Fosa posterior: inc luyen cerebelo y, generalmente, cuarto ventrículo.
158

Neurología y neurocirugía
Figura 125. Encefalocele
En cuanto al t ratamiento, debe tratarse la infección, si existe (encefa-loceles abiertos) y ext irpar el saco con su conten ido (tej ido func iona l-mente inv iable , en la mayoría de las ocasiones).
21.6. Sinus pericranii (variz espuria)
Consiste en un c o n j u n t o de vasos venosos encapsulados en la tabla externa, a través de un pequeño defecto óseo, que se comun i can d i rec tamente con un seno venoso dura l (en general , el seno sagital superior). Aparece c o m o una tumoración epicraneal b landa, que desaparece al ser compr im ida . Aumenta de tamaño al bajar la cabeza y no pulsa
1 5 9

Neurología y neurocirugía
22. NEUROCIRUGÍA FUNCIONAL
Orientación
MIR r
Aspectos esenciales L ;
En este t e m a , hay q u e es tud ia r c o n p r o f u n d i d a d la neu ra lg i a de l tr igémino. El resto d e los apar tados n o t i ene i m p o r t a n c i a d e ca ra al e x a m e n .
[~T"j La neuralgia del trigémino se caracter iza por un dolor neuropático, episódico y recidivante, que no DESPIERTA al paciente por la noche y que se distribuye por las ramas del trigémino. Puede desencadenarse por ciertas maniobras o por estimulación de determinadas zonas faciales.
["2~| Las ramas más afectadas son la segunda y tercera, es decir, la infraorbitaria y mandibular, respectivamente.
["3] Desde un punto de vista etiológico, puede ser esencial o secundario a diversos procesos intracraneales, como tumores o esclerosis múltiple.
Q El tratamiento de elección es la carbamazepina . En caso de fracaso o intolerancia al tratamiento médico, se planteará un tratamiento neuroquirúrgico.
22.1. Neuralgia del trigémino
Es un síndrome do loroso de la cara, hab i tua lmente uni latera l , de presentación súbita, carácter lancinante y localización en el terr i tor io cutáneo de una o más ramas del nervio trigémino (segunda y tercera, con más frecuencia) . Las crisis dolorosas son de escasa duración y recidivantes, con una intensidad tal que incapacitan al paciente, e inc luso lo l levan a conductas suicidas, y no despiertan al paciente por la noche.
Se presentan espontáneamente o tras estímulos sensoriales en las denominadas "áreas ga t i l l o " (roce de la cara, bostezo, masticación, l impiarse los dientes, con la deglución o al hablar). En el caso de que haya un déficit neurológico asociado al do lor o cuando la presentación no sea episódica, sino cont inua , se debe sospechar la pos ib i l idad de que se esté ante casos de neuralgia secundaria a otros procesos (MIR 06-07, 6 2 ; 00-01F, 74 ; MIR 99-00F, 61) .
Se clasif ican en : • Neuralgias esenciales. Es el grupo más numeroso. Suele afectar a mujeres mayores de 4 0 años, con carácter
cíclico. • Neuralgias secundarias a inf lamaciones, anomalías vasculares, tumores del ángulo pontocerebeloso, infec
ciones o enfermedades desmiel in izantes (esclerosis múltiple) que afectan al V par craneal en su trayecto.
El t ra tamiento in ic ia l de elección es la carbamazepina , en dosis crecientes, pero s iempre con un riguroso cont ro l hematológico (riesgo de neutropenia) . En menor grado son de ut i l idad la fenitoína, el bac lofeno, el c lonazepam, la gabapent ina o la ami t r ip t i l i na .
(TJ P r egun tas
- M I R 0 6 - 0 7 , 62 - M I R 0 0 - 0 1 F , 7 4 - M I R 99-00F , 61
Si fracasa el t ratamiento médico, puede estar ind icado el t ratamiento neuroquirúrgico: las dos técnicas más ut i l izadas son la rizotomía percutánea (destruir las fibras nocicept ivas mediante termocoagulación por radiofre cuencia o creando un trauma mecánico a través de un balón hinchable ) y la descompresión microquirúrgica (separar una arteria que se encuentra sobre el gangl io de Casser y que, a través del lat ido, lo está i rr i tando). Otras técnicas que existen son alcoholización del gangl io de Casser, fenolización de fibras sensitivas y la rizotomía retrogaseriana.
R E C U E R D A Una neuralgia del trigémino con exploración patológica y/o con dolor de manera continua, es decir, no episódica, obliga a descartar causa secundaria.

Neurología y neurocirugía
22.2. Neuralgia del glosofaríngeo Se trata de un dolor lancinante y grave en el terr itorio de distribución de los nervios glosofaríngeo y vago (fundamentalmente en la garganta y base de la lengua, con irradiación al oído y, en ocasiones, al cuel lo) . Puede acompañarse de salivación, tos y, rara vez, de hipotensión y síncope. No hay claras áreas gat i l lo. Con frecuencia, requiere tratamiento quirúrgico.
22.3. Cirugía del dolor intratable
El tratamiento quirúrgico del do lor intratable es apropiado para ind iv i duos en los que el tratamiento conservador no ha proporc ionado una mejoría adecuada del dolor o en situaciones en las que los tratamientos conl levan efectos secundarios indeseables.
Los procedimientos empleados se clasifican en los dos grandes grupos s i guientes: técnicas neuromodulat ivas (consideradas actualmente de elección) y técnicas ablativas.
Técnicas neuromodulativas
Dentro de estos procedimientos, a su vez, existen dos grandes grupos: • Infusión de drogas en el sistema nervioso (intratecal o intraventr icu
lar): la pr incipal indicación de esta terapia es el dolor nocíceptivo (por e jemplo, el relacionado con el cáncer). Sin embargo, también se puede uti l izar en el síndrome de la espalda fal l ida.
• Técnicas de estimulación - Estimulación de la médula espinal y del nervio periférico: ind ica
da en el do lor radicular persistente asociado al síndrome de espalda fal l ida o en la distrofia simpática refleja.
- Estimulación cerebral profunda del tálamo somatosensorial y de la sustancia gris periacueductalventricular (Figura 126): indicada en el dolor de origen no maligno(síndrome de espalda fa l l ida, do lor neuropático tras lesión del sistema nervioso central o periférico o do lor tr igeminal) .
Figura 126. Guía estereotáctica utilizada en neurocirugía funcional y en la obtención de biopsias cerebrales. En este paciente se está realizando una
estimulación cerebral profunda
- Estimulación de la corteza motora: mismas indicaciones que la estimulación talámica.
Técnicas ablativas
En general, son las técnicas de último recurso cuando todos los proced i mientos han fracasado. Suelen ser más apropiadas para el dolor nocícept ivo que para el neuropático. Existen tres grandes grupos: • Intervenciones periféricas (sobre el nervio):
- Simpatectomía: do lor visceral asociado con el cáncer o asociado con trastornos vasospásticos.
- Neurectomía: dolor tras lesión de un nervio periférico (amputación de un miembro , por ejemplo) .
- Rizotomía dorsal y ganglioneurectomía: do lor en el t ronco o abdomen relacionado con alguna neoplasia.
• Intervenciones espinales: - Lesión de DREZ (zona de entrada de la raíz dorsal): dolor neuropá
t ico tras avulsión de la raíz. - Cordotomía y mielotomía: dolor relacionado con el cáncer.
• Intervenciones supraespinales: mesencefalotomía, talamotomía, cin-gulotomía e hipofisectomía.
22.4. Cirugía de la enfermedad de Parkinson
Se desarrolla en el apartado 5.6. Tratamiento de la enfermedad de Parkinson.
22.5. Cirugía de la epilepsia
Se desarrolla en el apartado 7.6. Tratamiento de la epilepsia.
22.6. Neurocirugía de los trastornos mentales (psicocirugía)
M u y poco ut i l izada en la actual idad, se l imi ta a casos seleccionados y especialmente rebeldes a los diversos psicofármacos disponibles. Con sigue, si no la curación, alcanzar en ocasiones una notable mejoría en la ca l idad de vida de estos pacientes. Se deben calcular, previamente a la intervención, determinados objet ivos (targets o dianas) estereotáxicos. Existen cuatro procedimientos psicoquirúrgicos en la actual idad: • Cingulotomía anterior: indicada en casos de depresión mayor, ansie
dad crónica y trastorno obsesivo-compulsivo. • Tractotomía subcaudada: t iene las mismas indicaciones que la c ingu
lotomía anterior. • Leucotomía límbica: combina los dos procedimientos anteriores, por
lo que posee las mismas indicaciones. • Capsulotomía anterior: en casos de trastorno obsesivo-compulsivo.
161

Manual CTO de Medicina y Cirugía, 8. a edición
B IBL IOGRAFÍA
Neurología y neurocirugía
• Bradley W G, D M FRCP, Robert B. Neurology in Clinical Practice. 4th Edit ion. But terwor th-Heinemann, 2003 . • Brazis, P W , Masdeu J C, Biller J. Localization in Clinical Neurology. 5th Edition L ipp incot t W i l l i ams and W i l k ins , 2006 . • Engel J JR. Epilepsy. A comprehensive textbook. 2nd Edit ion. Editorial L ippincott , 2008 . • Greenberg M S. Handbook of Neurosurgery. 7th Edit ion. N e w York. Editorial Th ieme Med ica l Publishers N e w York, 2 0 1 0 . • Laner A.J. A dictionary of Neurological signs. 2 n d Edit ion. Editorial Springer, 2006 . • Lindsay K W . Neurology and Neurosurgery illustrated. 3rd Edit ion. Editorial Church i l l Livingstone, 2 0 0 7 . • Moo re A J. Neurosurgery. Principies and Practice. 2nd Edit ion. Editorial Springer, 2005 . • Pahwa R. Handbook of Parkinson's Disease. 4 th Edit ion. Informa Healthcare USA, Inc., 2 0 0 7 . • Patten J P. Neurological Differential Diagnosis. 2 n d Edit ion. Springer, 1996. • Ropper A H. Adams and Victor's Principies of Neurology. 8th Edit ion. McGraw-H i l l , 2005 • Rowland L P. Merrit's Textbook of Neurology. 9th Edit ion. L ipp incot t W i l l i ams and W i l k ins , 1997.
162


















